TW202000778A - 生物可降解聚合物 - Google Patents
生物可降解聚合物 Download PDFInfo
- Publication number
- TW202000778A TW202000778A TW108122410A TW108122410A TW202000778A TW 202000778 A TW202000778 A TW 202000778A TW 108122410 A TW108122410 A TW 108122410A TW 108122410 A TW108122410 A TW 108122410A TW 202000778 A TW202000778 A TW 202000778A
- Authority
- TW
- Taiwan
- Prior art keywords
- polymer
- group
- pva
- formula
- groups
- Prior art date
Links
Images






Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K49/00—Preparations for testing in vivo
- A61K49/04—X-ray contrast preparations
- A61K49/0433—X-ray contrast preparations containing an organic halogenated X-ray contrast-enhancing agent
- A61K49/0442—Polymeric X-ray contrast-enhancing agent comprising a halogenated group
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F16/00—Homopolymers and copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by an alcohol, ether, aldehydo, ketonic, acetal or ketal radical
- C08F16/02—Homopolymers and copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by an alcohol, ether, aldehydo, ketonic, acetal or ketal radical by an alcohol radical
- C08F16/04—Acyclic compounds
- C08F16/06—Polyvinyl alcohol ; Vinyl alcohol
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K49/00—Preparations for testing in vivo
- A61K49/04—X-ray contrast preparations
- A61K49/0433—X-ray contrast preparations containing an organic halogenated X-ray contrast-enhancing agent
- A61K49/0447—Physical forms of mixtures of two different X-ray contrast-enhancing agents, containing at least one X-ray contrast-enhancing agent which is a halogenated organic compound
- A61K49/0476—Particles, beads, capsules, spheres
- A61K49/048—Microparticles, microbeads, microcapsules, microspheres, i.e. having a size or diameter higher or equal to 1 micrometer
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/50—Microcapsules having a gas, liquid or semi-solid filling; Solid microparticles or pellets surrounded by a distinct coating layer, e.g. coated microspheres, coated drug crystals
- A61K9/5005—Wall or coating material
- A61K9/5021—Organic macromolecular compounds
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F8/00—Chemical modification by after-treatment
- C08F8/02—Alkylation
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F8/00—Chemical modification by after-treatment
- C08F8/12—Hydrolysis
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F8/00—Chemical modification by after-treatment
- C08F8/14—Esterification
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F8/00—Chemical modification by after-treatment
- C08F8/18—Introducing halogen atoms or halogen-containing groups
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F8/00—Chemical modification by after-treatment
- C08F8/26—Removing halogen atoms or halogen-containing groups from the molecule
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F8/00—Chemical modification by after-treatment
- C08F8/30—Introducing nitrogen atoms or nitrogen-containing groups
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F8/00—Chemical modification by after-treatment
- C08F8/34—Introducing sulfur atoms or sulfur-containing groups
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F8/00—Chemical modification by after-treatment
- C08F8/48—Isomerisation; Cyclisation
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J3/00—Processes of treating or compounding macromolecular substances
- C08J3/02—Making solutions, dispersions, lattices or gels by other methods than by solution, emulsion or suspension polymerisation techniques
- C08J3/03—Making solutions, dispersions, lattices or gels by other methods than by solution, emulsion or suspension polymerisation techniques in aqueous media
- C08J3/075—Macromolecular gels
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J3/00—Processes of treating or compounding macromolecular substances
- C08J3/12—Powdering or granulating
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J3/00—Processes of treating or compounding macromolecular substances
- C08J3/24—Crosslinking, e.g. vulcanising, of macromolecules
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09D—COATING COMPOSITIONS, e.g. PAINTS, VARNISHES OR LACQUERS; FILLING PASTES; CHEMICAL PAINT OR INK REMOVERS; INKS; CORRECTING FLUIDS; WOODSTAINS; PASTES OR SOLIDS FOR COLOURING OR PRINTING; USE OF MATERIALS THEREFOR
- C09D5/00—Coating compositions, e.g. paints, varnishes or lacquers, characterised by their physical nature or the effects produced; Filling pastes
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F2810/00—Chemical modification of a polymer
- C08F2810/20—Chemical modification of a polymer leading to a crosslinking, either explicitly or inherently
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F2810/00—Chemical modification of a polymer
- C08F2810/30—Chemical modification of a polymer leading to the formation or introduction of aliphatic or alicyclic unsaturated groups
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2300/00—Characterised by the use of unspecified polymers
- C08J2300/16—Biodegradable polymers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08J—WORKING-UP; GENERAL PROCESSES OF COMPOUNDING; AFTER-TREATMENT NOT COVERED BY SUBCLASSES C08B, C08C, C08F, C08G or C08H
- C08J2329/00—Characterised by the use of homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by an alcohol, ether, aldehydo, ketonic, acetal, or ketal radical; Hydrolysed polymers of esters of unsaturated alcohols with saturated carboxylic acids; Derivatives of such polymer
- C08J2329/02—Homopolymers or copolymers of unsaturated alcohols
- C08J2329/04—Polyvinyl alcohol; Partially hydrolysed homopolymers or copolymers of esters of unsaturated alcohols with saturated carboxylic acids
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Polymers & Plastics (AREA)
- General Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Epidemiology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Dispersion Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Materials Engineering (AREA)
- Wood Science & Technology (AREA)
- Pharmacology & Pharmacy (AREA)
- Medicinal Preparation (AREA)
- Addition Polymer Or Copolymer, Post-Treatments, Or Chemical Modifications (AREA)
- Biological Depolymerization Polymers (AREA)
- Polyesters Or Polycarbonates (AREA)
- Materials For Medical Uses (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Abstract
一種聚合物,其具有包含與C3至C8二酸交聯之多羥基化聚合物的主鏈。
Description
本發明係關於聚合物,且尤其關於可降解聚合物,關於包含此等聚合物之醫療裝置及使用該等裝置及聚合物之醫學治療方法。本發明尤其關於可降解微球體。
治療性栓塞係一種微創性程序,其中材料引入血管中以產生阻塞,從而減緩或中止血流。通常,此類材料經由導管遞送,自諸如腿或腕之邊緣點前往標靶部位。此方法適用於治療諸如胃腸出血、動靜脈畸形、多血管性惡性腫瘤(諸如肝細胞癌)、良性生長(諸如子宮纖維瘤)之病狀,且最近用於治療尤其良性前列腺增生(benign prostate hyperplasia,BPH)。
生物相容性微球體係適用之栓塞劑,因為其容易遞送至標靶部位且可在界定之尺寸範圍內提供,根據血管尺寸形成更可預測栓塞。
已提議多種可降解微球體。此等微球體可基於天然存在之聚合物,諸如白蛋白、明膠、幾丁聚糖或澱粉。然而,由於對聚合物本身或對污染物敏感,故此類聚合物可能在人類中激起被動反應(reactive response)。一些源自於動物來源之天然聚合物需要謹慎控制諸如病毒之污染物,或可能具有天然存在之變化性,引起聚合物及其性質在來源之間潛在的變化性。
亦已提議合成聚合物。最廣泛使用之合成之生物可降解聚合物係聚酯,諸如聚丙交酯、聚乙交酯或此等聚酯之共聚物,諸如聚丙交酯-共-乙交酯。此等聚合物通常為固體且堅硬,壓縮性較差,在用於製造微球體時,此可引起導管堵塞。雖然可例如藉由在製造期間將藥物混合至聚合物中而使此等材料併有藥物,但由於材料具有疏水性,故此通常限於親脂性藥物或固體調配物。此外,因為聚合物通常為整體的,所以藥物之釋放取決於聚合物之降解,因此可能相對緩慢。在合成後亦難以自材料移除溶劑殘餘物,因此材料中可能殘存微量。
合成之生物可降解聚合物之另一缺點為必須對分解產物之安全性及自身體之清除進行大範圍表徵,從而使得研發時間長且成本高。
本發明人已確定上述問題中之一或多個可藉由本發明之聚合物來解決。
在第一態樣中,因此,本發明提供一種具有包含多羥基化聚合物之主鏈的聚合物,該多羥基化聚合物與C3至C8二羧酸交聯。此態樣亦提供一種可藉由具有多羥基化聚合物主鏈之聚合物與C3至C8二羧酸交聯而獲得之聚合物。
多羥基化聚合物係包含載有一或多個側接羥基之重複單元之聚合物。較佳多羥基化聚合物包括包含以下之多羥基化聚合物:丙烯酸酯及甲基丙烯酸酯之多元醇酯;聚(羥基烷基丙烯酸酯)及聚(羥基烷基甲基丙烯酸酯),諸如聚(羥基乙基甲基丙烯酸酯);聚(羥基烷基丙烯醯胺)及聚(羥基烷基甲基丙烯醯胺),諸如三羥基甲基甲基丙烯醯胺;聚(PEG丙烯酸酯)及聚(PEG甲基丙烯酸酯);包含乙烯醇之聚合物,諸如聚(乙烯醇)或(乙烯-乙烯醇)共聚物;及多醣,諸如澱粉、幾丁聚糖、肝糖、諸如甲基纖維素之纖維素、海藻酸鹽及多醣樹膠,諸如角叉菜膠、瓜爾膠、黃原膠、結冷膠、槐豆膠及阿拉伯樹膠。
尤其較佳之聚合物為包含1,2-二醇或1,3-二醇基團之聚合物且更佳為包含乙烯醇之聚合物,諸如聚(乙烯醇) (PVA)或乙烯-乙烯醇(EtVA)聚合物及共聚物。最佳地,聚合物為PVA均聚物或共聚物,或多醣。
PVA之重量平均分子量(MW)較佳介於2000 Da與180,000 Da或200,000 Da之間,且尤其介於2000 Da或3000 Da與67,000 Da之間。重量平均分子量為67,000 Da或更少之PVA聚合物為較佳,因為在此重量範圍內之PVA能夠迅速地自體內清除,尤其經由腎排泄。更佳地,PVA具有2000至32,000或10,000至32,000之重量平均分子量(MW)。
本文中之生物可降解聚合物具有在體內藉由水解而裂解,由此聚合物分解之鍵。聚合物降解時段可藉由改變諸如主鏈之平均分子量、主鏈與交聯劑之莫耳比率及交聯劑之種類的參數而調整,如本文中進一步說明。較佳聚合物在1小時至1年時段內降解成可溶組分。
多羥基化聚合物可與多種C3、C4、C5、C6、C7或C8二酸、較佳C3至C6二酸交聯。此等二酸可為例如C3、C4、C5、C6、C7或C8飽和二酸、單不飽和二酸及在C6、C7及C8不飽和二酸之情況下為雙不飽和酸。在各情況下,未分支酸為較佳。
在聚合物與C3至C8飽和二酸交聯之情況下,此較佳選自丙二酸、丁二酸及戊二酸。
飽和二酸可經選自-OH、=O及-NH2
之基團取代。在酸經酮取代之情況下,其較佳為C4、C5、C6、C7或C8 α酮酸,較佳α酮戊二酸酯。可替代地,α酮酸為草醯乙酸酯。在酸經胺基取代之情況下,其較佳為天冬胺酸或麩胺酸。包含PVA之聚合物與此類二酸交聯的聚合物為一較佳實施例。
在聚合物與C4至C8不飽和二酸交聯之情況下,此較佳選自順丁烯二酸、反丁烯二酸或順式或反式半乳糖醛酸,反式異構體為較佳。
在所有情況下,較佳地,交聯二酸天然存在於人體中,因為此類化合物容易代謝及/或自體內清除。此類酸之實例包括(但不限於)丙二酸、丁二酸、戊二酸、反丁烯二酸、戊烯二酸、蘋果酸、天冬胺酸、麩胺酸、草醯乙酸酯及α酮戊二酸酯。
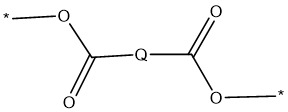
在本發明之一個實施例中,聚合物包含式1之基團: 1
其中
*為經由酯基與多羥基化聚合物附接之附接點;且
其中Q為式1a之基團:
1
其中
*為經由酯基與多羥基化聚合物附接之附接點;且
其中Q為式1a之基團: 其中n為1至5,較佳1至3;更佳1或2;
或Q為C1-6
伸烷基或C2-6
伸烯基;較佳C1-4
伸烷基或C2-4
伸烯基;其中伸烷基視情況經-OH或-NH2
取代。單個取代為較佳。
其中n為1至5,較佳1至3;更佳1或2;
或Q為C1-6
伸烷基或C2-6
伸烯基;較佳C1-4
伸烷基或C2-4
伸烯基;其中伸烷基視情況經-OH或-NH2
取代。單個取代為較佳。
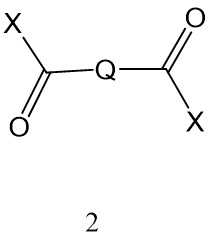
在另一實施例中,本發明提供一種可藉由使包含多羥基化聚合物之聚合物與式II化合物交聯而獲得的聚合物,其中在多羥基化聚合物與式2化合物之間形成酯鍵; 其中X為-OH或合適離去基且其中離去基係選自咪唑基、甲磺酸酯基、甲苯磺酸酯基、-O-烷基(諸如-O-(C1-6
)烷基)、氯基、溴基、氟基及-O-醯基,諸如-O-(C1-6
)醯基。
其中X為-OH或合適離去基且其中離去基係選自咪唑基、甲磺酸酯基、甲苯磺酸酯基、-O-烷基(諸如-O-(C1-6
)烷基)、氯基、溴基、氟基及-O-醯基,諸如-O-(C1-6
)醯基。
在X為-OH之情況下,氧可藉由使用諸如DCC (N,N'
-二環己基碳化二亞胺)、EDCI (N-(3-二甲基胺基丙基)-N′-乙基碳化二亞胺)及HOBt (羥基苯并三唑)之活化劑活化。
反應通常在諸如1-甲基-2-吡咯啶酮(NMP)、DMF或DMSO、其混合物之極性非質子溶劑中進行。反應可在高溫,亦即超過20℃下進行。
在聚合物主鏈為或包含PVA,諸如PVA均聚物及共聚物之情況下,聚合物包含與PVA交聯之式3之交聯基團, 。
3
。
3
本發明之第二態樣提供一種製造生物可降解聚合物之方法,其包括使多羥基化聚合物與式2化合物交聯以在多羥基化聚合物與式2化合物之間形成酯鍵,從而使聚合物交聯; 其中X為-OH或合適離去基。離去基可選自咪唑基、甲磺酸酯基、甲苯磺酸酯基、-O-烷基(諸如-O-(C1-6
)烷基)、氯基、溴基、氟基及-O-醯基,諸如-O-(C1-6
)-醯基,如上所述。
其中X為-OH或合適離去基。離去基可選自咪唑基、甲磺酸酯基、甲苯磺酸酯基、-O-烷基(諸如-O-(C1-6
)烷基)、氯基、溴基、氟基及-O-醯基,諸如-O-(C1-6
)-醯基,如上所述。
在一尤其較佳實施例中,聚合物為包含PVA之聚合物(諸如PVA均聚物或共聚物),其與α-酮戊二酸酯交聯。
聚合物可具備多種功能性。此等包括:經由藥物以物理方式併入聚合物中或藉由藥物與聚合物之帶電基團偶合,進行藥物溶離;放射性,諸如藉由放射性同位素併入聚合物(例如呈粉末),藉由包含同位素之部分與聚合物偶合,諸如含有放射性原子之化合物,或藉由放射性同位素經由離子相互作用與聚合物結合(諸如經由聚合物上之帶電基團或經由與結合於聚合物之螯合基團附接)。另外,可經由將可成像物質併入聚合物(例如呈粉末)中,藉由包含可成像物質之部分與聚合物偶合,或藉由可成像物質經由離子相互作用與聚合物結合(諸如經由聚合物上之帶電基團或經由與結合於聚合物之螯合基團附接),提供可成像性。
可成像物質使聚合物為諸如X射線、正電子發射成像(PET)或單光子發射電腦斷層攝影(SPECT)及MRI之醫學成像模態可見。
X射線可成像之物質包括例如碘、鋇及鉭。
正電子發射成像物質包括18
F (其可例如呈18
F-氟去氧葡萄糖併入,且與聚合物偶合)。SPECT可成像物質包括鉈-201、鍀-99m、碘-123及鎵-67。
MRI可成像物質包括釓、鐵(尤其呈超順磁性氧化鐵粒子)、鉑及錳。
可成像物質可與聚合物偶合(諸如藉由共價鍵、離子相互作用或藉由螯合作用),或其可以物理方式併入聚合物中,例如呈粉末(例如金屬粒子,諸如鐵或鉭,或粉末狀化合物,諸如硫酸鋇)。
聚合物亦可藉由回波描記術成像(例如藉由在聚合物內提供空隙或氣泡)。
本文所述之多羥基化聚合物可藉由在多羥基化主鏈上提供側基,以提供一或多種功能性,諸如藥物之可成像性、螯合作用或離子鍵結來改質。此類側基可經由羥基中之一或多個與主鏈偶合。側基可經由醚基、酯基、碳酸酯基、胺基甲酸酯基或環狀縮醛基,諸如1,3-二氧雜環戊二烯酮基及1,3-二噁烷基偶合。
此類側部分可在交聯之前或之後與主鏈偶合。在與預先形成之交聯聚合物偶合時經由單個羥基偶合為較佳,因為其避免無意之額外交聯的可能性,例如在側基經由羥基化聚合物主鏈之超過一個羥基連接的情況下。
在一個實施例中,側基可帶正電或負電,其能夠可逆地結合在生理pH值(pH 7.4)下帶相反電荷之化合物,諸如藥物。可使用多種帶電物質,包括磺酸酯基、磷酸酯基、銨基、鏻基及羧酸酯基;羧酸酯基及磺酸酯基為較佳。
示例帶電基團包括C1-6
分支或未分支烷基、C2-6
分支或未分支烯基或C5-7
芳基或雜芳基(較佳苯基或苯甲基),各獨立地經1至3個選自-COOH、-OPO3
H2
及-SO3
H之基團取代。在此等基團中,載有1至3個羧酸酯基或磺酸酯基之C1-6
分支或未分支烷基及C2-6
分支或未分支烯基為較佳。
類似於以下所詳述,此類基團可經由連接子X與聚合物主鏈偶合,或較佳可直接與聚合物主鏈鍵結。
帶電基團可經由羥基中之一或多個與聚合物主鏈偶合,但較佳經由單個基團,尤其在已交聯之後與聚合物偶合時。至於其他側基,合適偶合基團包括醚基、酯基、碳酸酯基、胺基甲酸酯基或環狀縮醛基,諸如1,3-二氧雜環戊二烯酮基及1,3-二噁烷基偶合。較佳偶合基團為醚基、酯基及1,3-二噁烷基。醚及酯為較佳,尤其在交聯後用於偶合側基時。
可能側基亦包括包含一或多個,諸如2個、3個或4個共價附接之碘的基團;較佳地,此類基團包含具有2個、3個或4個共價附接於芳族環,諸如苯環或苯甲基之碘的碘化芳族基。在一較佳方法中,聚合物包含載有2個至4個共價結合之碘的苯基。碘可為此類環之唯一取代基,或苯基可另外包含一或兩個基團W,下文進一步描述W。此類基團亦可經由諸如醚基、酯基、醯胺基、碳酸酯基、胺基甲酸酯基、1,3-二氧雜環戊二烯酮基及1,3-二噁烷基及尤其醚基、酯基及1,3-二噁烷基之偶合基團與聚合物主鏈偶合,下文進一步描述。醚及酯為較佳,尤其在交聯後使用時。
含碘側基用來為聚合物提供不透射線性。如下所詳述,此類基團可經由連接子X與聚合物主鏈偶合,或較佳可直接與聚合物主鏈鍵結。
在一個實施例中,碘化側基可為式4a或4b之基團:
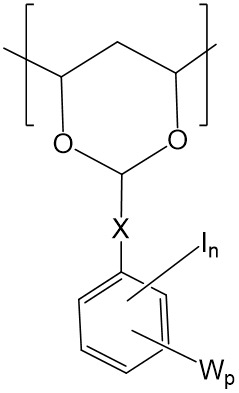 4a 4b
W係獨立地選自-OH、-COOH、-SO3
H、-OPO3
H2
、-O-(C1-4
烷基)、-O-(C1-4
烷基)OH、-O-(C1-4
烷基)R2
、-O-(C2
H5
O)q
R1
、-(C=O)-O-C1-4
烷基及-O-(C=O)C1-4
烷基;或者可替代地,W可為式-BZ之兩性離子基團,不過此類-BZ基團次佳;
其中-OH、COOH、-OPO3
H2
及-SO3
H可呈醫藥學上可接受之鹽形式;
X為一鍵或具有1至8個碳及視情況1至4個選自O、N及S之雜原子的鍵聯基團,不過S次佳;
G為式I之基團與該聚合物偶合所經由之偶合基團且係選自醚、酯、醯胺、碳酸酯、胺基甲酸酯、1,3-二氧雜環戊烯酮及1,3-二噁烷;尤其醚、酯、醯胺、碳酸酯、胺基甲酸酯,及最尤其醚或酯,R1
為H或C1-4
烷基;
R2
為-COOH、-SO3
H或-OPO3
H2
;較佳-COOH或-SO3
H;
q為整數1至4;
n為整數1至4;較佳2或3;
p為0、1或2;較佳1或2;且
其中-COOH、-OPO3
H2
及-SO3
H以及酚-OH可呈醫藥學上可接受之鹽形式;
在W為式-BZ之兩性離子基團之情況下:B為一鍵,或直鏈烷二基、氧基伸烷基、伸烷基氧雜伸烷基或伸烷基(寡氧雜伸烷基),視情況含有一或多個氟取代基;且Z為銨、鏻或鋶之磷酸酯或膦酸酯兩性離子基團。
4a 4b
W係獨立地選自-OH、-COOH、-SO3
H、-OPO3
H2
、-O-(C1-4
烷基)、-O-(C1-4
烷基)OH、-O-(C1-4
烷基)R2
、-O-(C2
H5
O)q
R1
、-(C=O)-O-C1-4
烷基及-O-(C=O)C1-4
烷基;或者可替代地,W可為式-BZ之兩性離子基團,不過此類-BZ基團次佳;
其中-OH、COOH、-OPO3
H2
及-SO3
H可呈醫藥學上可接受之鹽形式;
X為一鍵或具有1至8個碳及視情況1至4個選自O、N及S之雜原子的鍵聯基團,不過S次佳;
G為式I之基團與該聚合物偶合所經由之偶合基團且係選自醚、酯、醯胺、碳酸酯、胺基甲酸酯、1,3-二氧雜環戊烯酮及1,3-二噁烷;尤其醚、酯、醯胺、碳酸酯、胺基甲酸酯,及最尤其醚或酯,R1
為H或C1-4
烷基;
R2
為-COOH、-SO3
H或-OPO3
H2
;較佳-COOH或-SO3
H;
q為整數1至4;
n為整數1至4;較佳2或3;
p為0、1或2;較佳1或2;且
其中-COOH、-OPO3
H2
及-SO3
H以及酚-OH可呈醫藥學上可接受之鹽形式;
在W為式-BZ之兩性離子基團之情況下:B為一鍵,或直鏈烷二基、氧基伸烷基、伸烷基氧雜伸烷基或伸烷基(寡氧雜伸烷基),視情況含有一或多個氟取代基;且Z為銨、鏻或鋶之磷酸酯或膦酸酯兩性離子基團。
基團Z為兩性離子,且包含銨基、鏻基或鋶基作為陽離子部分。陽離子較佳為銨基。兩性離子之陰離子為磷酸基部分。一般為磷酸二酯或基於膦酸酯之部分。
一般在Z中,陰離子比陽離子更靠近B (非磷酸化甜菜鹼)。然而,在一些兩性離子中,陽離子比陰離子更靠近基團B (以下稱為磷酸化甜菜鹼)。
較佳在非磷酸化甜菜鹼中,Z為通式5之基團。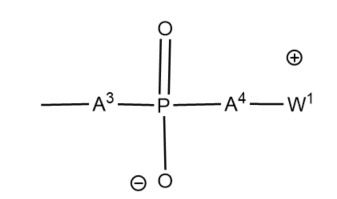 5
5
其中相同或不同之部分A3
及A4
為-O、-S、-NH-或價鍵;較佳-O-,且W+
為包含銨、鏻或鋶陽離子基團之基團及連接陰離子部分及陽離子部分之基團,較佳C1-12
烷二基,較佳其中W1+
為下式基團:
-W2
-N+
R4 3
、-W2
-P+
R5 3
、-W2
-S+
R5 2
或-W2
-Het+
;其中:
W2
為視情況含有一或多個乙烯系不飽和雙鍵或三鍵之具有1個或更多個,較佳2-6個碳原子之烷二基、二取代之芳基(伸芳基)、伸烷基伸芳基、伸芳基伸烷基或伸烷基芳基伸烷基、環烷二基、伸烷基環烷基、環烷基伸烷基或伸烷基環烷基伸烷基,該基團W1
視情況含有一或多個氟取代基及/或一或多個官能基;且任一基團R4
相同或不同且各為氫或具有1至4個碳原子之烷基,較佳甲基,或芳基,諸如苯基,或基團R4
中之兩者連同其附接之氮原子一起形成含有5至7個原子之脂族雜環,或三個基團R4
連同其附接之氮原子一起形成在各環中含有5至7個原子之稠環結構,且視情況基團R4
中之一或多個經親水性官能基取代;
基團R5
相同或不同且各為R4
或基團OR4
,其中R4
如上定義;且
Het為含氮、磷或硫,較佳含氮芳環,例如吡啶。
其中Z為其中W+
為W1
N+
R4 3
之通式的化合物可如WO9301221中所述製備。鏻及鋶類似物描述於WO9520407及WO9416749中。其中Z為其中W1+
為W2
N+
R4 3
之此通式的化合物為較佳。
一般,式5之Z-基團具有較佳通式6 6
6
其中基團R6
相同或不同且各為氫或C1-4
烷基,且m為1至4,其中較佳基團R6
相同,較佳甲基。此W基團之尤其較佳實例為磷醯基膽鹼基團: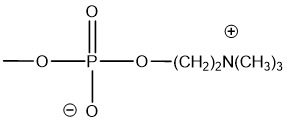
在基於磷酸化甜菜之基團中,Z可具有通式7: 7
其中
A5
為價鍵、-O-、-S-或-NH-,較佳-O-;
R7
為價鍵(連同A5
一起)或烷二基、-C(O)伸烷基-或-C(O)NH伸烷基,較佳烷二基,且較佳在烷二基鏈中含有1至6個碳原子;
W3
為S、PR8
或NR8
;
或各基團R8
為氫或具有1至4個碳原子之烷基或兩個基團R8
連同其附接之雜原子一起形成具有5至7個原子之雜環;
R9
為具有1至20個,較佳1至10個,更佳1至6個碳原子之烷二基;
A6
為一鍵、NH、S或O,較佳O;且
R10
為羥基、C1-12
烷基、C1-12
烷氧基、C7-18
芳氧基、C7-18
芳烷氧基、C6-18
芳基或C6-18
芳基氧基。
7
其中
A5
為價鍵、-O-、-S-或-NH-,較佳-O-;
R7
為價鍵(連同A5
一起)或烷二基、-C(O)伸烷基-或-C(O)NH伸烷基,較佳烷二基,且較佳在烷二基鏈中含有1至6個碳原子;
W3
為S、PR8
或NR8
;
或各基團R8
為氫或具有1至4個碳原子之烷基或兩個基團R8
連同其附接之雜原子一起形成具有5至7個原子之雜環;
R9
為具有1至20個,較佳1至10個,更佳1至6個碳原子之烷二基;
A6
為一鍵、NH、S或O,較佳O;且
R10
為羥基、C1-12
烷基、C1-12
烷氧基、C7-18
芳氧基、C7-18
芳烷氧基、C6-18
芳基或C6-18
芳基氧基。
包含通式7之基團之化合物可藉由如JP03031718B中所述之方法製備,其中經胺基取代之化合物與磷雜環戊烷反應。
在包含通式7之基團之化合物中,較佳地:
A5
為一鍵;
R7
為C2-6
烷二基;
W3
為NR8
,其中各R8
為C1-4
烷基;
R9
為C2-6
烷二基;
A6
為O;且
R10
為C1-4
烷氧基。
在諸如具有式5及6之基團之磷酸化甜菜的磷酸化甜菜及諸如具有式7之基團之未磷酸化甜菜的未磷酸化甜菜中,B較佳為一鍵、C1-6
分支或未分支烷二基,諸如亞甲基、伸乙基、伸丙基或伸丁基,或分支或未分支C1-6
氧基伸烷基,諸如氧基亞甲基、氧基伸乙基、氧基伸丙基或氧基伸丁基。
在存在情況下,W較佳獨立地選自-OH、-COOH、-SO3
H、-O-(C2
H5
O)q
R1
、-O-(C1-4
烷基)R2
、-(C=O)-O-C1-4
烷基及-O-(C=O)C1-4
烷基;且尤其選自-OH、-COOH、-SO3
H、-O-(C2
H5
O)q
R1
、-O-(C1-4
烷基)R2
;其中-SO3
H、-COOH及酚-OH可呈醫藥學上可接受之鹽形式;
在本文中之任何聚合物中,在W為-O-(C1-4
烷基)R2
之情況下,其較佳為-O-(C2-4
烷基)R2
,且更佳為-O-(C3
烷基)R2
或-O-(C4
烷基)R2
。
連接子X較佳為一鍵或為具有1至4個碳及視情況1個選自O及N之雜原子的鍵聯基團;且更佳選自一鍵、(C1-4
)伸烷基、(C1-4
)氧基伸烷基、胺基(C1-4
)伸烷基。具體實例包括一鍵、C1
、C2
或C3
伸烷基、氧基甲基或氧基乙基、胺基亞甲基及胺基伸乙基。在存在連接子之情況下其尤其為亞甲基、氧基亞甲基或胺基亞甲基。最佳環直接鍵結於基團G,使得X為一鍵。
q較佳為1、2或3;尤其為2或3;
n較佳為2或3且最佳為3;
R1
較佳為H或甲基;且
R2
較佳為-COOH或-SO3
H。
在一較佳實施例中,式4a之碘化側基為其中G為酯鍵且X為一鍵或(C1-4
)伸烷基之基團。在另一較佳實施例中,式4a之基團為其中G為醚鍵且連接子X為一鍵或(C1-4
)伸烷基、尤其(C1-4
)伸烷基之基團。在另一較佳實施例中,式4b之基團為其中連接子X為一鍵、(C1-4
)伸烷基、(C1-4
)氧化伸烷基、胺基(C1-4
)伸烷基之基團。在此實施例中,X之尤其較佳實例包括一鍵、C1
、C2
或C3
伸烷基、氧基甲基或氧基乙基、胺基亞甲基及胺基伸乙基。在此實施例中,在存在連接子之情況下,其尤其為亞甲基、氧基亞甲基或胺基亞甲基且在此實施例中最佳X為一鍵。在此等較佳實施例中之每一個中,聚合物較佳為或包含PVA。
出於以上給出之原因,在聚合物交聯之後形成側基的情況下,式IVa之側基為較佳,尤其其中G係選自醚、酯、醯胺、碳酸酯、胺基甲酸酯且最尤其醚或酯。
較佳地,碘化側基包含僅經2個、3個或4個碘、尤其僅2個或3個碘取代之苯環,或為以下列方式中之一或多個取代的苯基: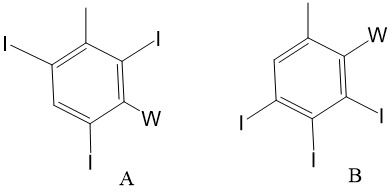

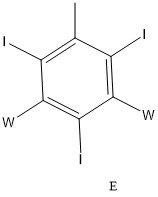 尤其,碘化側基包含僅經2個、3個或4個碘、尤其僅2個或3個碘取代之苯環,或為以下列方式中之一或多個取代的苯基:
尤其,碘化側基包含僅經2個、3個或4個碘、尤其僅2個或3個碘取代之苯環,或為以下列方式中之一或多個取代的苯基:




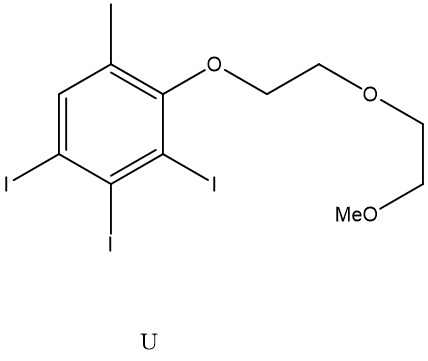
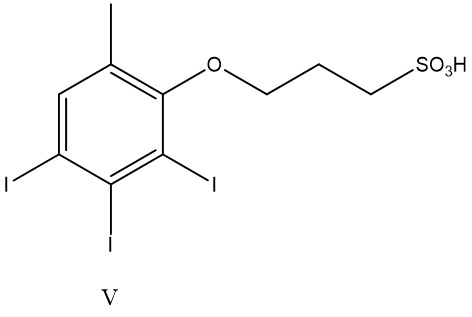 碘化苯基為
碘化苯基為 其中n為2至4,尤其2或3;且尤其:
其中n為2至4,尤其2或3;且尤其: 其中-COOH、-SO3
H及酚-OH可呈醫藥學上可接受之鹽形式,諸如包括鈉或鉀之金屬鹽。
其中-COOH、-SO3
H及酚-OH可呈醫藥學上可接受之鹽形式,諸如包括鈉或鉀之金屬鹽。
碘化苯甲醛及具有類似官能基之碘化苯基與PVA偶合之方法描述於WO2015/033093中。
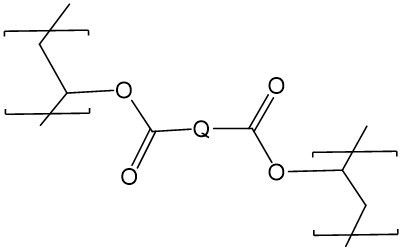
其中G為酯鍵之不透射線性生物可降解聚合物可藉由使諸如PVA之多羥基化聚合物與式8化合物反應來製備: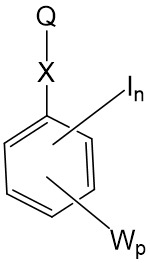 8
其中Q為羧酸、酸性鹵化物(諸如Cl或Br)或活化羧酸。
8
其中Q為羧酸、酸性鹵化物(諸如Cl或Br)或活化羧酸。
在Q為羧酸之情況下,反應通常在酸性條件(例如硫酸、三氟乙酸、三氟甲烷硫酸、含氫溴酸之乙酸、乙酸及甲烷磺酸)下在適當極性溶劑(例如DMSO、DMF、NMP)中進行。
在Q為酸性鹵化物之情況下,反應通常在輕度鹼性條件下在適當極性溶劑(例如DMSO、DMF、NMP)中例如在弱鹼(例如吡啶、三甲胺、二甲基吡啶、三甲基吡啶或咪唑)存在下進行。
在Q為活化羧酸之情況下,諸如碳化二亞胺及羰基二咪唑,例如DCC (N
,N
'-二環己基碳化二亞胺)、EDCI (N-(3-二甲基胺基丙基)-N’-乙基碳化二亞胺)及HOBt (羥基苯并三唑)之活化劑可用於極性非質子溶劑,諸如DMSO、四氫呋喃、乙酸乙酯、丙酮、二甲基甲醯胺及乙腈中。反應通常在催化量之鹼存在下及在無水條件下進行以實現活化。鹼通常為中等強度(共軛酸之pKa約為10-13),且合適鹼包括多種吡啶、胺、含氮雜環、三乙胺、N
,N
-二異丙基乙胺、DMAP及其類似物。
例如在WO2011/110589 (例如其中製備實例1至6)、WO2014/152488及Mawad等人(2009) Biomaterials, 30, 5667-5674中論述及例示碘化苯基與PVA經由酯鍵偶合。
為形成醚鍵,諸如PVA之多羥基化聚合物可與其中Q為選自以下之基團的式8化合物起反應:鹵化物,諸如氟化物、氯化物、溴化物、碘化物;甲基磺酸酯、甲基甲苯磺酸酯、三氟甲烷-磺酸酯。Q可為例如溴。
在WO2011/110589 (參見其中實例1及2)中論述碘化苯基與PVA經由醚鍵偶合。
在聚合物為具有1,2-二醇基團(諸如許多多醣)或1,3-二醇基團(諸如PVA)之多羥基化聚合物之情況下,其中G為1,3-二氧戊環或1,3-二噁烷之不透射線性生物可降解聚合物可藉由使聚合物與其中Q為諸如醛、縮醛及半縮醛之官能基的式8化合物反應來製備。在WO2015/033092中描述碘化基團與PVA以此方式偶合。
其中G為碳酸酯鍵之聚合物可藉由多羥基化聚合物與其中Q為諸如式9之氯甲酸酯基之式IV化合物反應來製備。
同時其中G為胺基甲酸酯鍵之聚合物可藉由多羥基化聚合物與其中Q為諸如式10之胺基甲醯氯基團: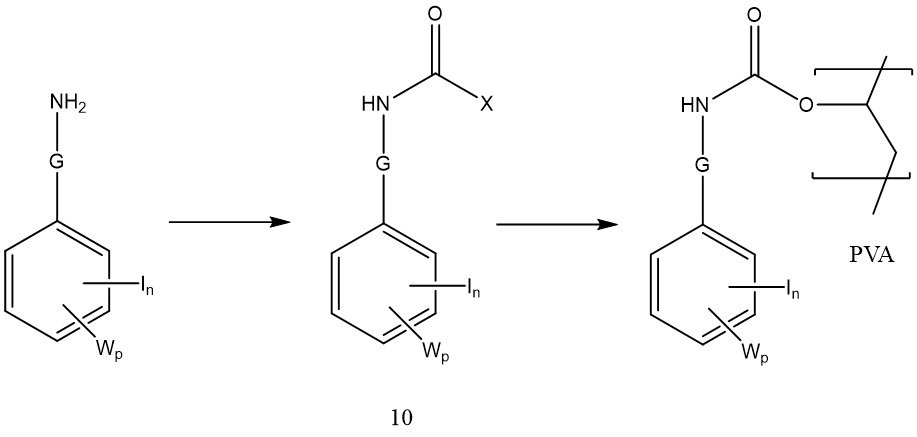 或諸如式11之異氰酸酯基:
或諸如式11之異氰酸酯基: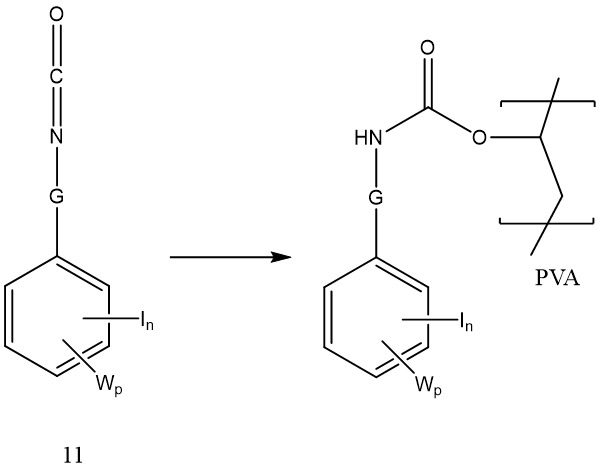 之式8化合物反應來製備。
之式8化合物反應來製備。
此兩個反應藉由諸如吡啶、三甲胺、二甲基吡啶、三甲基吡啶或咪唑之弱鹼介導。
不透射線性或放射密度可根據需要,藉由調整聚合物中碘之量而變化。此可藉由變化環上碘的數目或藉由變化側基與聚合物之比例來實現。
本發明之聚合物較佳每cm3
包含至少10 mg碘,較佳25 mg/cm3
,更佳至少50 mg/cm3
且尤其至少100 mg/cm3
。
聚合物中碘之量可為按乾重計至少10%,較佳至少20%,更佳至少30%且最佳至少35% wt/wt聚合物。可在此等聚合物中獲得高放射密度,其中碘超過40% wt/wt乾燥聚合物。
較佳地,本發明之聚合物具有至少1000 HU或2000 HU、更佳至少3000 HU且尤其至少4000 HU之放射密度。當在65kV下量測時,特別如根據實例13量測。
聚合物主鏈亦可包含其他側基,諸如一或多種螯合物質,諸如巰基乙醯基三甘胺酸(MAG-3)、EDTA、EGTA、BAPTA、DOTA、DTPA-單醯胺、DOTA-R、D03A-R、NOTA-BnR、NODASA-R及NODAGA-R。
接著螯合物質可用於螯合多種金屬或非金屬物質,該等金屬或非金屬物質可包括X射線可成像元素,諸如鉍;發射α、β或γ之醫用放射性同位素,諸如鍀-99 (Tc-99)、鈷-60、碘-131、銥-192、碘-125、鈀-103、鍶-89、釤-153、錸-186、鑥-177、鉍-213、鉛-212、釔-90、碘-131、銫-131、鈀-103、鐳-223、錒-225及鈥-166;正電子發射可成像元素,諸如Ga-68、Zr-89或Rb-82;或順磁物質,諸如鐵、鎂、鉬及鉭。
此類螯合基團及其與聚合物及尤其微球體偶合之方法的實例揭示於例如WO18093566A1、WO14159759A1及WO08034911A1中。
較佳地,聚合物為水凝膠,亦即聚合物係水可膨脹的但不溶於水。因此,在含水液體存在下,聚合物將形成水凝膠。其可包含按重量計超過50%且較佳多達98%水,較佳60%至95%或60%至85%。
根據本發明製備之α-酮戊二酸酯水凝膠可具有介於80%與98% w/w之間的水含量。當添加側基時可改變水含量。
本發明之另一態樣提供包含本文所述之交聯多羥基化聚合物之微粒。在本發明之此態樣之一實施例中,微粒適用於血管栓塞。通常此類微粒為微球體。聚合物微球體通常具有至多2000 um之平均最大直徑,不過所用實際尺寸範圍將尤其視臨床需要而定。此類粒子可例如藉由篩分來製備成所需之任何過篩範圍。典型尺寸範圍包括30-70 um、70-150 um、100-300 um、300-500 um、500-700 um及700-900 um,不過在一些情況下,較小尺寸範圍可為有利的,尤其歸因於其更遠端栓塞性質。此類較小尺寸範圍包括70-150 um或40至90 um。通常,避免尺寸小於20 um,因為穿過毛細血管床引起偏離標靶栓塞;因此,實際下限為約20-30 um。當前在40至700 um範圍內之尺寸最常用於臨床實踐中。所用聚合物可如本文所述帶電,以便微球體適合於藉由離子相互作用來負載藥物。
在一個特定實施例中,聚合物在生理pH值下具有淨電荷,較佳在生理pH值(7.4)下具有淨負電荷。
此實施例之聚合物可與合適之醫藥學上可接受之載劑或稀釋劑,諸如注射用水組合使用,且可直接用於使血管栓塞。因此,包含本文所述之聚合物之醫藥組合物形成本發明之另一態樣。
可替代地或另外,組合物中可包括有效量之一或多種生物活性劑。可能需要將活性劑從呈水凝膠形式之聚合物遞送。可能需要遞送之生物活性劑包括包含有機及無機分子及細胞之預防劑、治療劑及診斷劑(本文中統稱為「活性劑」、「治療劑」或「藥物」)。多種活性劑可併入聚合物中。藉由活性劑自聚合物擴散、聚合物降解及/或使活性劑與聚合物偶合之化學鍵的降解,實現所併入之活性劑自聚合物釋放。在此背景下,「有效量」係指獲得所需作用需要的活性劑之量。
活性劑較佳可逆地保持在聚合物內。活性劑藉由離子相互作用,諸如藉由與如本文所述之聚合物之帶正電或帶負電基團的相互作用而可逆地結合於聚合物內,可替代地,活性劑可藉由諸如沈澱之另一方式保持在聚合物內(參見例如WO207/085615或WO2007090897)。
活性劑可為化學治療劑、諸如西妥昔單抗(cetuximab)、曲妥珠單抗(trastuzimab)及納武單抗(nivolumab)之抗體、抗體片段、肽、低分子量蛋白質或其組合。
例示性化學治療劑包括蒽環類,諸如(但不限於)多柔比星(doxorubicin)、柔紅黴素(daunarubicin)、表柔比星(epirubicin)及伊達比星(idarubicin);喜樹鹼類,諸如(但不限於)伊立替康(irinotecan)、拓撲替康(topotecan)及依沙替康(exatecan);鉑類,諸如順鉑(cisplatin)、奧沙利鉑(oxaliplatin)、卡鉑(carboplatin)及米鉑(miriplatin);絲裂黴素C (mitomycin C);抗代謝物,諸如5-氟尿嘧啶(5-fluorouracil);多酪胺酸激酶抑制劑,諸如(但不限於)索拉非尼(sorafenib)、舒尼替尼(sunitinib)、瑞戈非尼(regorafenib)、博瑞維尼(brivinb)、達薩替你(dasetanib)、博舒替尼(bosutinib)、埃羅替尼(erlotinib)、吉非替尼(gefitinib)、伊馬替尼(imatinib)及凡得替尼(vandetinib)、雷帕黴素(rapamycin)或其任何組合。在此類化合物可電離之情況下,此類化合物可通常呈其離子形式使用。
本發明之另一態樣提供醫學治療方法,其包括向有需要之個體之血管遞送如本文所述之聚合物,諸如以形成栓塞。聚合物可呈微球體形式或其他微粒形式。聚合物可包含有效量之一或多種生物活性劑。聚合物可與X射線造影劑組合遞送,例如呈微球體形式之聚合物可在遞送前懸浮在造影劑中,或造影劑可在聚合物即將遞送時或在聚合物遞送後立即遞送。聚合物可藉由經導管途徑遞送,尤其在呈微球體形式時。
治療可為例如藉由經動脈栓塞或化療栓塞治療多血管性腫瘤,但聚合物亦可藉由局部注射例如微球體來遞送,該等微球體可例如提供藥物儲槽。
在另一態樣中,聚合物可用於製備可植入醫療裝置,諸如縫線、支架、基準標記物或組織分離器(例如藉由在將一個組織與另一個組合分離之程序期間位於兩個組織之間來使用),或用作醫療裝置之塗層。
在另一態樣中,本發明提供一種製造生物可降解聚合物微球體之方法,其包括:
提供第一液體,其為使以下溶解於其中之溶劑:(i)為或包含PVA之聚合物,及(ii)式2化合物,其中Q及X如本文所述: 提供與第一液體不混溶之第二液體;
使第一液體與第二液體接觸,使得第一液體在第二液體內形成不連續相;以及
在不連續相內使PVA與式2化合物交聯,從而形成微球體。
提供與第一液體不混溶之第二液體;
使第一液體與第二液體接觸,使得第一液體在第二液體內形成不連續相;以及
在不連續相內使PVA與式2化合物交聯,從而形成微球體。
使第一液體與第二液體接觸較佳形成乳液,其中第一相在第二相內之液滴具有適於提供所需尺寸微球體之尺寸。乳液通常將藉由用力混合來維持。
溶劑可為如上所述之極性非質子溶劑。第二液體可為油,諸如礦物油。反應可在乳化劑,諸如界面活性劑(例如SPAN 20)存在下進行。
在可在1小時至24小時之時間內進行的聚合物微球體形成之後,可回收微球體。
現將藉助於以下非限制性實例,參考圖式,進一步描述本發明。此等實例僅係出於例示之目的而提供,且熟習此項技術者按照此等實例將想到在申請專利範圍之範疇內之其他實例。所有本文中引用之參考文獻均以引用之方式整體併入本文中。參考文獻與本申請案之間的任何矛盾將以本申請案為準。
實例
實例
1.
合成呈微球體形式之
PVA-α-
酮戊二酸酯可降解聚合物
在惰性氛圍下在緩緩加熱至90℃下使PVA (10 kDa Mw,1.0 g,0.1 mmol,1 eq)溶於1-甲基-2-吡咯啶酮(4 ml)中,接著使之冷卻至室溫。分別使α-酮戊二酸(KGA 0.06 g,0.42 mmol,4.2 eq)及1,1′-羰基二咪唑(CDI,0.15 g,0.91 mmol,9.1 eq)溶於1-甲基-2-吡咯啶酮(2 ml)中,接著在周圍溫度下混合兩種溶液5分鐘,形成咪唑中間物。
在乾燥1L圓底燒瓶中,在氮氣層下在機械攪拌下混合重質或輕質礦物油(500 ml)及界面活性劑Span20 (6 ml),且將反應燒瓶加熱至70℃溫度。將PVA 1-甲基-2-吡咯啶酮溶液與CDI活化之α-酮戊二酸溶液混合。將混合物在周圍溫度下進行滾筒混合約20至30分鐘。接著在氮氣層下且在強力攪拌下將棕色溶液添加至礦物油溶液。在70℃下懸浮之微液滴經1至10小時之過程逐漸固化成微粒。
當中止反應時,使懸浮液沈積,且吸出礦物油且所得微粒依序用乙酸烷酯(2×500 ml)及乙醇(2×500 ml)洗滌。在pH 3下將經洗滌之粒子轉移至鹽水溶液,且篩分膨脹微粒,以收集不同尺寸範圍之部分:32-70 µm、70-150 µm、150-300 µm、300-500 µm及500至700 µm。將所收集之微粒置於丙酮中以移除水,接著在周圍溫度下真空乾燥24小時。元素分析結果顯示無法自背景偵測乾燥微粒之氮水準,此表明副產物咪唑洗淨。使用25 kGy之劑量對微粒進行γ殺菌。 實例 2. 合成生物可降解之 PVA- 反丁烯二酸聚合物
實例 2. 合成生物可降解之 PVA- 反丁烯二酸聚合物
在惰性氛圍下在緩緩加熱下使PVA (10 kDa Mw,1.0 g,0.1 mmol,1 eq)溶於1-甲基-2-吡咯啶酮(4 ml)中。分別使反丁烯二酸(0.08 g,0.7 mmol)及1,1′-羰基二咪唑(CDI,0.22 g,1.4 mmol)溶於DMSO (2 ml)中,接著在周圍溫度下混合兩種溶液10分鐘,形成咪唑中間物。
在乾燥1L圓底燒瓶中,在氮氣層下在機械攪拌下混合重質或輕質礦物油(500 ml)及界面活性劑Span20 (6 ml),且將反應燒瓶加熱至70℃溫度。將PVA之1-甲基-2-吡咯啶酮溶液與CDI活化之反丁烯二酸溶液混合。將混合物在周圍溫度下進行滾筒混合約20至30分鐘。接著在氮氣層下且在強力攪拌下將棕色溶液添加至礦物油溶液。在70℃下懸浮之微液滴經15小時之過程逐漸固化成微粒。
反丁烯二酸交聯之珠粒的處理與實例1中相同。實例 3. 合成 PVA- 丁二酸生物可降解聚合物
在惰性氛圍下在緩緩加熱下使PVA (10 kDa Mw,1.0 g,0.1 mmol,1 eq)溶於1-甲基-2-吡咯啶酮(5 ml)中。分別使丁二酸(0.08 g,0.7 mmol)及1,1′-羰基二咪唑(CDI,0.24 g,1.5 mmol)溶於1-甲基-2-吡咯啶酮(2 ml)中,接著在周圍溫度下混合兩種溶液5分鐘,形成咪唑中間物。
在乾燥1L圓底燒瓶中,在氮氣層下在機械攪拌下混合重質或輕質礦物油(500 mL)及界面活性劑Span20 (6 mL),且將反應燒瓶加熱至70℃溫度。將PVA之1-甲基-2-吡咯啶酮溶液與CDI活化之丁二酸溶液混合。將混合物在周圍溫度下進行滾筒混合約20至30分鐘。接著在氮氣層下在以約300 rpm強力攪拌下將混合物添加至礦物油溶液。在70℃下懸浮之微液滴經2至10小時之過程逐漸固化成微粒。反丁烯二酸交聯之珠粒的處理與實例1中相同。實例 4. 合成具有高 PVA 固體含量之生物可降解 PVA-KGA 聚合物
遵循實例1,PVA (Mw 10kDa,1.50 g,0.15 mmol,1 eq) α-酮戊二酸(0.15 g)、1,1′-羰基二咪唑(0.38 g)用於在500 mL重質礦物油中合成微粒。Span 20用於穩定懸浮液滴。在70℃下反應15小時,且根據實例1加工所產生之微粒。實例 5. 合成具有 3kDa PVA 之生物可降解 PVA-KGA 聚合物
遵循實例1中之程序,PVA (3kDa Mw,1.00 g,0.10 mmol,1 eq)、α-酮戊二酸(0.10 g,0.7 mmol,7 eq)、1,1′-羰基二咪唑(0.25 g,1.5 mmol,15.5 eq)用於在重質礦物油中合成生物可降解微粒。Span 20用於穩定懸浮液滴。在70℃下反應2至15小時,且所產生之微粒如實例1進行加工。實例 6. 聚合物降解研究
將三組尺寸範圍在60至300 um之0.1 g乾燥微粒預先稱重,且置於Duran®
瓶中100 mL磷酸鹽緩衝鹽水(PBS:NaCl 136.7 mM、KCl 2.7 mM、Na2HPO4 10.1 mM、KH2PO4 1.7 mM) (pH 7.4,各組中n = 3)中。在偶爾攪動下將PBS中之微粒在37℃下培育。藉由使用40 um篩過濾來週期性地收集微粒,真空乾燥且稱重。直接藉由凝膠滲透層析來分析經過濾之溶液。亦分析用於微粒合成之PVA聚合物原料之樣品作為對照物。樣品之凝膠滲透層析與PEG標準及PVA參考相比,以測定降解產物之分子量及分佈。圖1中展示在降解期間微粒之重量變化。實例 7. 固體含量、懸浮及導管遞送測試
測試具有四個尺寸範圍32至70 µm、70至125 µm、125至300 µm及300至500 µm之生物可降解微粒(PVA-KGA 8%)之固體含量。藉由準確地稱重乾燥微粒,接著微粒經鹽水水合至飽和,來進行測試。為獲得水合微粒之重量,藉由移液及棉紙芯吸來移除額外鹽水。表1中列出微粒之固體含量。表 1. 水合微粒之固體含量 
表2說明各種KGA水準對根據以上實例製備之微球體的固體含量的作用。表 2. 具有變化 KGA 含量之水合微粒之固體含量 (%w/w) 
對於懸浮測試,使50 mg乾燥微粒在5 mL鹽水中水合且與造影劑Omnipaque 350混合,以5分鐘過程內實現穩定懸浮液。發現Omnipaque 350:鹽水之最佳比率為約4-5:5 (v/v,mL)。藉由經2.4 Fr Progreat導管注射微粒懸浮液,進行微粒之導管遞送。所有四個尺寸範圍均經由導管遞送,無堵塞。微粒遞送之容易程度隨微粒尺寸減少而增加,亦即32至70 μm尺寸範圍最易遞送,接著70至125 μm微粒、125至300 μm微粒及300至500 μm微粒。在遞送之後,顯微鏡影像未顯示微粒破壞之跡象。實例 8. 碘化苯甲醛及醛衍生物與 PVA 偶合之通用方案
向在氮氣層下預乾燥之反應器添加PVA (通常5-10 g)及無水溶劑(通常DMSO或NMP,相對於PVA質量40體積)及催化劑(通常相對於PVA質量2.2體積,例如甲烷磺酸)。將攪拌懸浮液加熱至高溫(大約90℃)以溶解PVA。當獲得均質溶液時,使混合物冷卻至所需反應溫度(通常50-80℃),添加第一及第二側基之所需醛基質(通常相對於PVA二醇官能基0.01至0.6 eq)。第一及第二側基醛基質與PVA 1,3-二醇基團之實際比率及第一側基與第二側基之比率將視所需聚合物之親水性與疏水性的調整而定,但通常第一側基將處於比第二側基高的比率下。
接著在N2
層下攪拌反應,且藉由HPLC,針對基質之消耗,監測反應轉化。在預定時間(通常當基質停止消耗時),自滴液漏斗逐滴添加反溶劑(通常丙酮、DCM、MeCN或TBME,大約40體積)。藉由經濾膜抽吸來移除上清液,且饋入另外反應溶劑(通常40體積)且攪拌,直至固體完全溶解。此溶劑洗滌階段重複多達3次。接著固體再溶於反應溶劑中,且藉由緩慢添加水(通常多達100體積)而沈澱。自上清液移除所得聚集固體且在混合器中在水中均質化。將懸浮液過濾且再懸浮在水(典型地100體積)中,歷時長達30分鐘成漿,且過濾。重複水成漿,直至獲得中性pH值,接著濕固體在丙酮(100體積,攪拌30分鐘,重複2次)中成漿,過濾且在高真空烘箱中在30℃下乾燥長達24小時。實例 9. 5-((2,2- 二甲氧基乙基 ) 胺基 )-2,4,6- 三碘 - 間苯二甲酸之偶合 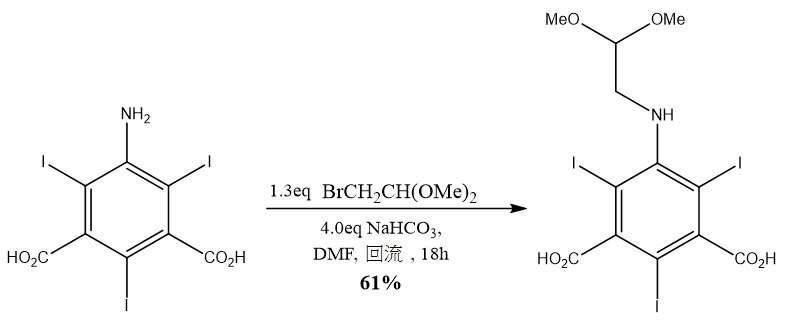
向在氮氣下之直火烘乾之500 ml圓底燒瓶經由套管添加固體5-胺基-2,4,6-三碘間苯二甲酸 (46.95 g,84.03 mmol,1.0 eq)、碳酸氫鈉(28.21 g,335.8 mmol,4.0 eq)及DMF (大約400 ml)。向所得棕色溶液逐滴添加2-溴-1,1-二甲氧基乙烷(13 ml,110.0 mmol,1.3 eq)且將所得溶液加熱至回流,保持18小時。冷卻至室溫後,藉由在真空(9 mBar,55℃)下旋轉蒸發移除大部分DMF,且用乙酸乙酯(1L)萃取所得橙色固體。此懸浮液用飽和氯化鋰溶液(7×400 ml)洗滌,移除殘餘DMF及鹽,經硫酸鎂乾燥,過濾且蒸乾。所得固體自乙酸乙酯再結晶,用異己烷洗滌且過濾。此過程重複總共3次且所得橙色固體在高真空下乾燥,得到標題化合物(33.04 g,61%,91.7% HPLC純度)。產物可經由矽膠管柱層析法(DCM中MeOH,0-15%)進一步純化(4.91 g,82%產率,96% HPLC純度);δH
(CDCl3
, 500.1 MHz)/ppm;8.01 (1H, s), 4.86 (2H, br s), 4.76 (1H, t, 5.5Hz), 4.37 (2H, d, 5.5Hz), 3.44 (6H, s);δC
NMR (CDCl3
, 125.8 MHz)/ppm;
將根據實例1 (0.50 g)製備之各種尺寸之乾燥微粒添加至攪拌之N,N-二甲基甲醯胺(40 mL)溶液中,以使微粒膨脹。將催化劑甲烷磺酸(2.2 mL)及5-((2,2-二甲氧基乙基)胺基)-2,4,6-三碘間苯二甲酸(7.46 g,11.5 mmol)添加至反應容器。在惰性氛圍下溫度升至70℃,保持24小時。冷卻至室溫(約15℃至25℃)後,吸出微粒,接著分別用二甲基甲醯胺(3×40 ml)、乙醇(2×50ml)及丙酮(3×50 ml)洗滌。在移除丙酮之後,將微粒在周圍溫度下真空乾燥18小時。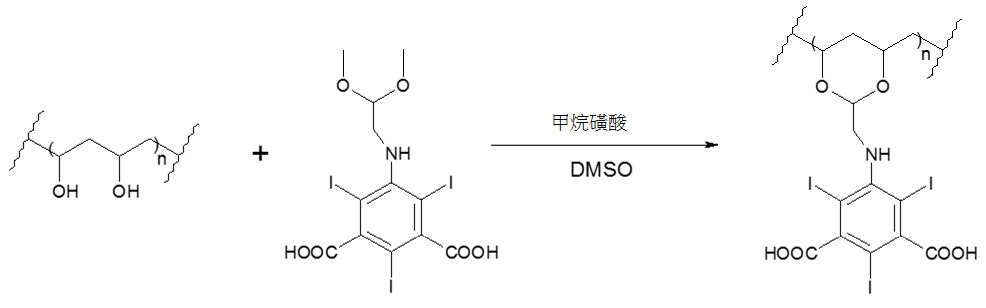
根據實例13測得此等微球體之放射密度為6288±450 HU。在約1分鐘內微球體懸浮在造影劑及鹽水混合物(2:0.5-2:1,v/v)中。珠粒成功經由2.4 Fr Progreat導管遞送。
圖3展示此等微球體之microCT影像。實例 10. 合成具有磺化及碘化苯基之聚合物:合成 3-(3- 甲醯基 -2,4,6- 三碘苯氧基 ) 丙烷 -1- 磺酸鈉鹽及 3-(1- 甲醯基 -3,4,5- 三碘苯氧基 ) 丙烷 -1- 磺酸鈉鹽 
在150 mL三頸圓底燒瓶中,藉由磁性攪拌器,使3-羥基-2,4,6-三碘苯甲醛(10 g,20 mmol)溶於50 mL無水四氫呋喃(THF)中。將2.47 g (22 mmol)第三丁醇鉀與20 mL THF混合且在室溫下在氮氣氛圍下慢慢添加懸浮液至燒瓶,接著將溫度增加至40℃,以使產物完全溶解。接著使15 g (120 mmol)磺內酯溶於15 mL THF中且慢慢添加混合物至反應燒瓶。幾乎立即出現沈澱。在40℃下反應3小時之後,將反應混合物傾倒至500 mL乙酸乙酯中,得到固體粗產物。經過濾之固體用100 mL乙酸乙酯洗滌,且在乙醇中再結晶。在真空乾燥24小時之後,得到10.7 g產物,產率80%。磺酸基TIBA質子核磁共振(NMR)分析,D2
O溶劑:δ (ppm) 2.24-2.34 (m, 2H), 3.12-3.25 (t, 2H), 3.88-4.02 (t, 2H), 8.18-8.25 (s, 1H), 9.42-9.50 (s, 1H)。元素分析結果: C18.56, H 2.22, S 5.66, I 52.31, K 6.27。計算值:C 18.20, H 1.22, S 4.85, I 57.68, K 5.92。
類似地自3,4,5-三碘水楊醛合成3-(1-甲醯基-3,4,5-三碘苯氧基)丙烷-1-磺酸鈉鹽(參見實例19)。用 3-(3- 甲醯基 -2,4,6- 三碘苯氧基 ) 丙烷 -1- 磺酸鈉 (STIBA) 改質 PVA
使6.56 g STIBA及3.98 g PVA (Mw 10 kDa)溶於反應燒瓶中40 mL二甲亞碸(DMSO)中。將8.8 mL催化劑甲基磺酸與20 mL DMSO混合且添加至燒瓶。在60℃下反應24小時之後,在攪拌下反應混合物在900 mL丙酮中沈澱兩次。使所收集之固體溶於去離子水中且置於滲析袋(MWCO:1000)中。聚合物針對水透析三天,以移除小分子雜質,接著凍乾,得到1.55公克聚合物。
如上製備之3-(1-甲醯基-3,4,5-三碘苯氧基)丙烷-1-磺酸鹽可與PVA以類似方式偶合。亦可藉由使用類似合成途徑,使2-磺酸基苯甲醛鈉鹽(Sigma Aldrich UK)、4-甲醯基苯1,3-二磺酸二鈉鹽(Sigma Aldrich UK)及4-甲醯基苯甲酸(Sigma Aldrich UK)與PVA偶合。
遵循實例1中之相同程序,藉由使用KGA交聯劑,合成珠粒。
使以上獲得之STIBA改質之PVA溶於1-甲基-2-吡咯啶酮(5 mL)。分別使α-酮戊二酸(0.06 g,0.42 mmol)及1,1′-羰基二咪唑(CDI,0.15 g,0.91 mmol)溶於2 mL 1-甲基-2-吡咯啶酮(2 mL)中,接著在周圍溫度下混合兩種溶液5分鐘,形成咪唑中間物。接著將PVA溶液與CDI活化之α-酮戊二酸溶液混合,接著在70℃下在以300 rpm機械攪拌下與500 mL礦物油及界面活性劑Span20混合。懸浮微滴逐漸隔夜固化成微粒。接著將所得珠粒用乙酸乙酯及乙醇洗滌,以移除殘餘油及反應物。將珠粒真空乾燥。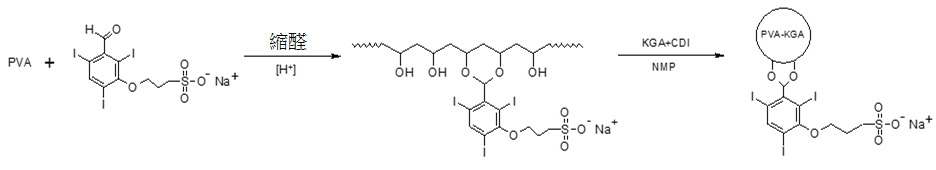 實例 11. 具有側接羧基之生物可降解聚合物
實例 11. 具有側接羧基之生物可降解聚合物
0.5 g根據實例1製備之PVA微粒分散至35 mL二甲基甲醯胺(DMF),接著添加順式-烏頭酸酐(0.442 g,2.8 mmol)及三乙胺(0.525 ml,3.8 mmol)。反應溫度保持在60℃下且在350 rpm下攪拌24小時。在中止反應之後,將微粒用30 mL DMF及PBS洗滌,接著用丙酮洗滌。接著在室溫(約15℃至25℃)下微粒真空乾燥隔夜。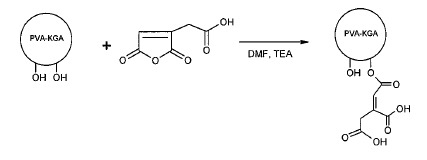 實例 12. 具有側接磺醯基之生物可降解聚合物
實例 12. 具有側接磺醯基之生物可降解聚合物
0.5 g PVA微粒分散至35 mL DMF中,接著添加氯磺乙醯氯(1.00 g,5.6 mmol)及三乙胺(1.65 ml,11.8 mmol)。反應溫度保持在60℃下且在350 rpm下攪拌反應混合物24小時。在中止反應之後,將微粒用30 mL DMF及PBS洗滌,以移除殘餘反應物。藉由用丙酮洗滌且接著真空乾燥,進一步加工微粒。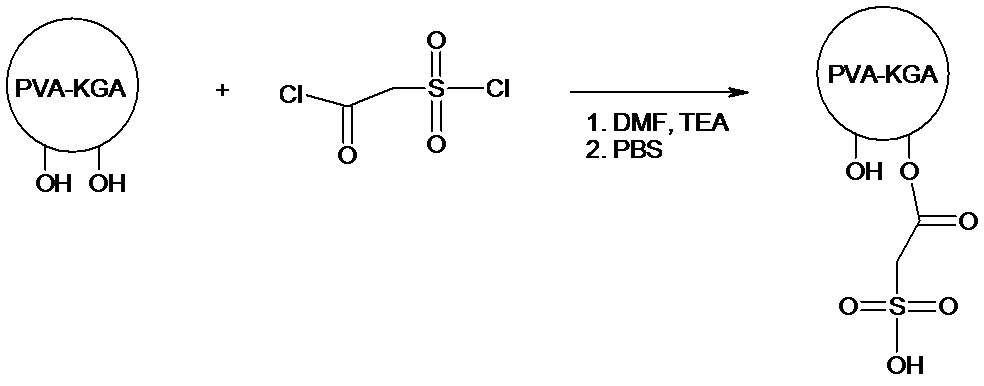 實例 13. 具有側接丙酸基團之微球體
實例 13. 具有側接丙酸基團之微球體
交聯PVA微粒分散至35 mL DMF中,添加3,3-二甲氧基丙酸及甲烷磺酸以與聚合物上之二醇基團反應。將微粒吸出且接著分別用二甲基甲醯胺、乙醇及丙酮洗滌。在移除丙酮之後,將微粒在周圍溫度下真空乾燥。實例 14. 測定放射密度
Micro-CT用於評估根據以上通用實例8製備之不透射線性栓塞珠粒之樣品的不透射線性(放射密度)。在Nunc冷凍管小瓶(Sigma-Aldrich產品編碼V7634,48 mm × 12.5 mm)中製備樣品。使珠粒懸浮在1%瓊脂糖凝膠(由Sigma-Aldrich產品編碼A9539製備)中。所得懸浮液一般稱為「珠粒幻影」。為製備此等珠粒幻影,首先將瓊脂糖溶液(1%)升高至約50℃之溫度。接著添加已知量之珠粒,且將兩者輕輕地混合在一起,直至溶液開始固化或膠凝。當溶液冷卻時,其膠凝且珠粒保持均勻分散及懸浮在瓊脂糖凝膠內。
在RSSL Laboratories (Reading, Berkshire, UK)使用裝有鎢陽極之Bruker Skyscan 1172 Micro-CT掃描器,使用微型電腦斷層攝影術(Micro-CT)測試珠粒幻影之不透射線性。使用相同儀器組態分析每個幻影,其中鎢陽極在64 kV之電壓及155 μA之電流下操作。使用鋁製濾光板(500 μm)。表 1 :採集參數: 
將少量純化MilliQ®
水小心地傾析至每個樣品管中。接著藉由X射線微型電腦斷層攝影術,使用單次掃描來分析每個樣品,包括水參考及珠粒。接著使用NRecon重建樣品,且針對淨化水參考之感興趣體積(VOI)進行校準。空氣及水之感興趣區(ROI)在校準之後進行分析以證實亨氏校準(Hounsfield calibration)。
自跨越珠粒之線掃描投影,以亨氏單位報導放射密度。NRecon (閾值處理)中用於所有樣品之動態範圍的值:-0.005、0.13 (最小及最大衰減係數)。根據此通用方案量測之來自實例5的微球體具有6288±450 HU之放射密度。在約1分鐘內珠粒懸浮在造影劑及鹽水混合物(2:0.5-2:1,v/v)中。珠粒成功經由2.4 Fr Progreat導管遞送。實例 15 :合成 3,5- 二碘 -2-(2-(2- 甲氧基乙氧基 ) 乙氧基 ) 苯甲醛 
向裝有回流冷凝器及懸浮式磁力攪拌器之HEL PolyBlock8平行合成125 ml反應器添加3,5-二碘水楊醛(13.9011 g,37.72 mmol,1.0 eq)及TBAI (2.7481 mg,0.802 mmol,0.2 eq)。向其中添加水,且用1M NaOH (總水溶液體積97 ml)調節pH值至9.5。反應器設定為500 rpm攪拌,直至完全溶解,得到嫩黃色溶液,且添加1-溴-2-(2-甲氧基乙氧基)乙烷(5.00 ml,37.17 mmol,1.0 eq)。反應器區設定為加熱至120℃。藉由TLC (i-hex中30% EA)監測反應,且在2小時之後添加額外溴化物(2.50 ml,18.59 mmol,0.5 eq)。又0.5小時之後,由於消耗溴化物,pH值再調至9.5。又2小時之後,添加額外溴化物(1.25 ml,9.29 mmol,0.25 eq),且反應器調低至50℃且攪拌隔夜。在19小時之後,將所得懸浮液再加熱至回流,保持1小時,冷卻至室溫且轉移至分液漏斗中之乙酸乙酯(400 ml)中。將有機物用飽和碳酸氫鈉洗滌兩次,經硫酸鎂乾燥,自甲苯熱過濾,且自甲苯/異己烷再結晶,在過濾及高真空乾燥後得到呈黃色粉末狀之所需產物(15.2909 g,86.4%產率);δH
(CDCl3
, 500.1 MHz)/ppm;10.31 (1H, s), 8.31 (1H, d, 2.2Hz), 8.09 (1H, d, 2.2Hz), 4.26 (2H, app. t, 4.5Hz), 3.89 (2H, app. t, 4.5Hz), 3.67 (2H, app. t, 4.6Hz), 3.55 (2H, app. t, 4.6Hz), 3.38 (3H, s);δC
NMR (CDCl3
, 125.8 MHz)/ppm;188.71 (CH), 161.55 (q), 152.43 (CH), 137.57 (CH), 131.75 (q), 94.07 (q), 89.19 (q), 75,56 (CH2), 71.90 (CH2), 70.79 (CH2), 70.06 (CH2), 59.13 (CH3)。實例 16 :合成 3- 羥基 -2,4,6- 三碘苯甲醛 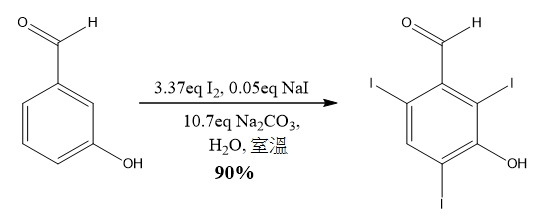
向具有大橢圓形攪拌棒之2L 3頸圓底燒瓶添加3-羥基苯甲醛(10.007 g,81.89 mmol)、碘化鈉(0.614 g,4.09 mmol,0.05 eq)及碳酸鈉(93.028 g,877.44 mmol,10.7 eq),用總共750 ml去離子水沖洗。當苯甲醛溶解得到嫩黃色攪拌溶液時,經30分鐘分2部分添加碘球(70.008 g,275.80 mmol,3.37 eq),每次用225 ml水沖洗。反應藉由TLC (i
-hex中60% DCM)追蹤,且在3小時內,碘幾乎完全溶解,產生暗黃色/橙色沈澱。固體藉由布氏過濾(Büchner filtration)分離且用異己烷洗滌,以移除任何殘餘碘。分離之固體再溶於溫水(2L,45℃)中,得到清澈棕色溶液,向其中添加100 ml飽和硫代硫酸鈉溶液以還原任何殘存碘。使用1M HCl (小心,因為放出CO2
),小心地將溶液之pH值自10.2減少至3.26。固體藉由過濾分離,用水(2×500 ml)洗滌,且在高真空烘箱中在30℃下乾燥,得到呈黃色固體狀之所需化合物(37.002 g,90.3%產率,97.2% HPLC純度);δH
(CDCl3
, 500.1 MHz)/ppm; 9.65 (1H, s), 8.35 (1H, s), 6.42 (1H, s);δC
NMR (CDCl3
, 125.8 MHz)/ppm; 194.90 (CH), 155.12 (q), 149.77 (CH), 135.69 (q), 88.78 (q), 87.66 (q), 85.70 (q)。實例 17 :合成 2,4,6- 三碘 -3-(2-(2- 甲氧基乙氧基 ) 乙氧基 ) 苯甲醛 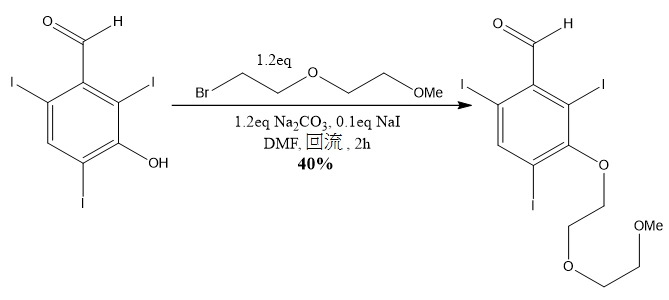
向在氮氣氛圍下含有攪拌棒且裝有回流冷凝器的直火烘乾之250 ml 3頸圓底燒瓶添加3-羥基-2,4,6-三碘苯甲醛(15.627 g,31.3 mmol,1.0 eq)、碘化鈉(469 mg,3.13 mmol,0.1 eq)、無水碳酸鈉(3.981 g,37.6 mmol,1.2 eq)及無水DMF (160 ml)。攪拌懸浮液,直至醛完全溶解,接著藉由注射器添加1-溴-2-(2-甲氧基乙氧基)乙烷(6.87 g,37.5 mmol,1.2 eq)且將反應加熱至回流。在2小時之後,TLC分析(i
-hex中10% EA)指示起始物質消耗且使反應冷卻至室溫,轉移至250 ml圓底燒瓶且在高真空下蒸乾。將所得懸浮液用500 ml乙酸乙酯稀釋,用3×100 ml 1M NaOH、2×100 ml飽和鹽水洗滌,用活性木炭漂白且經硫酸鎂乾燥。將所得溶液濃縮至乾,且藉由二氧化矽管柱層析法(異己烷中2%-20%乙酸乙酯)來純化且在高真空下乾燥,得到呈黃色粉末狀之所需化合物(7.556 g,40.1%);δH
(CDCl3
, 500.1 MHz)/ppm;9.65 (1H, s), 8.44 (1H, s), 4.20 (2H, t, 6.4Hz), 4.01 (2H, t, 6.4Hz), 3.79 (2H, app. t, 5.8Hz), 3.60 (2H, app. t, 5.8H), 3.41 (3H, s);δC
NMR (CDCl3
, 125.8 MHz)/ppm;194.97 (CH), 159.10 (q), 150.83 (CH), 138.27 (q), 97.06 (q), 95.70 (q), 90.40 (q), 72.47 (CH2), 72.04 (CH2), 70. 89 (CH2), 68.89 (CH2), 59.19 (CH3)。實例 18 :合成 2,4,6- 三碘 -3-(2-(2-(2- 甲氧基乙氧基 ) 乙氧基 ) 乙氧基 ) 苯甲醛 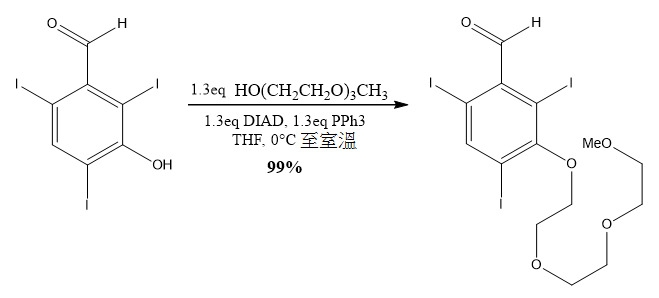
向在氮氣層下含有攪拌器之直火烘乾之100 ml 3頸圓底燒瓶添加三苯基膦(1.7216 g,6.502 mmol,1.3 eq)及無水THF (35 ml)。開始攪拌且在PPh3完全溶解之後,反應器在冰浴中冷卻至大約0℃。經由注射器向無色溶液逐滴添加DIAD (1.28 ml,6.502 mmol,1.3 eq),得到持久黃色溶液。在攪拌5分鐘之後,藉由注射器逐滴添加三乙二醇單甲醚(1.04 ml,6.502 mmol,1.3 eq)。在又攪拌5分鐘之後,一次性添加3-羥基-2,4,6-三碘苯甲醛(2.5077 g,5.002 mmol,1.0 eq),立即引起變色。藉由TLC (甲苯中5% Et2
O)監測反應且攪拌隔夜。將溶液用乙醚稀釋,以使三苯基氧化膦沈澱,接著濃縮至乾。藉由管柱層析法(甲苯中2-10% Et2
O)來純化所得稠油狀物,在濃縮及高真空乾燥之後得到呈黃色粉末狀之所需產物(3.2077 g,99%產率,94.4% HPLC純度);δH
(DMSO-D6
, 500.1 MHz)/ppm;9.58 (1H, s), 8.47 (1H, s), 4.08 (2H, t, 4.9Hz), 3.57-3.53 (4H, m), 3.44 (2H, app. t, 4.8Hz), 3.24 (3H, s)。實例 19 :合成 3,4,5- 三碘水楊醛 
向含有大橢圓形攪拌器之3頸2L圓底燒瓶添加4-碘-水楊醛(25.01 g,100.86 mmol,1.0 eq)及乙酸(300 ml)。在攪拌5分鐘以使固體溶解之後,將預溫熱之液體一氯化碘(39.11 g,2.4 eq)用AcOH (100 ml)稀釋且轉移至圓底燒瓶上之滴液漏斗。經10分鐘添加此溶液。接著將反應器置於裝有1L滴液漏斗、溫度計及冷凝器的大批量矽油中且設定加熱至80℃。在加熱期間,水(700 ml)慢慢添加至溶液,引起黃色/橙色沈澱。在80℃下20分鐘之後,關閉加熱。又過一段時間,移除加熱浴,且使黑色溶液/黃色懸浮液冷卻至室溫且攪拌65小時;藉由TLC (iHex中20% EA)分析反應。藉由布氏過濾分離固體且用水(2×500 ml)洗滌。為移除殘餘碘晶體,將固體用異己烷(200 ml)重複形成漿液,直至異己烷上清液不再為紫色。分離之固體在高真空烘箱中乾燥,得到呈黃色結晶固體狀之所需產物(40.84 g,81%產率,藉由HPLC分析93.2%純)。產物可自丙酮:水(9:1)進一步再結晶至較高純度;δH
(CDCl3
, 500.1 MHz)/ppm;12.15 (1H, s), 9.67 (1H, s), 8.09 (1H, s);δC
NMR (CDCl3
, 125.8 MHz)/ppm;194.53 (CH), 159.58 (C), 142.24 (CH), 133.39 (C), 120.87 (C), 101.68 (C), 94.02 (C)。實例 20 :合成 3,4,5- 三碘 -2-(2-(2- 甲氧基乙氧基 ) 乙氧基 ) 苯甲醛 
(5 g規模):藉由注射器向在正壓氮氣下含有小八角形攪拌棒之直火烘乾之250 ml 3頸圓底燒瓶添加三苯基膦(2.76 g,10.5 mmol,1.05 eq)及無水THF (70 ml)。將圓底燒瓶置於裝有低溫溫度計之Dewer浴中且用EtOH/液氮浴冷卻至-68℃。經1分鐘,藉由注射器逐滴添加偶氮二甲酸二乙酯(1.65 ml,10.5 mmol,1.05 eq),且攪拌5分鐘,得到黃色懸浮液。接著逐滴添加二乙二醇單甲醚(1.77 ml,15 mmol,1.5 eq)且攪拌5分鐘。向其中一次性添加固體3,4,5-三碘水楊醛(5.00 g,10.0 mmol,1.0 eq)。初始暗橙色/紅色懸浮液變淡,得到淺黃色溶液,攪拌2小時,藉由TLC分析(甲苯中20%乙醚)監測且升溫至室溫,隔夜。TLC指示醛起始物質完全消耗,具有乾淨反應概況。所得溶液轉移至500 ml圓底燒瓶,用乙醚(200 ml)稀釋且在冷凍機中冷卻。所得懸浮液經短二氧化矽塞過濾以移除三苯基氧化膦且用另外乙醚(200 ml)清洗。將所得溶液濃縮至乾,且藉由管柱層析法,用甲苯中乙醚(2-20%)溶離來純化,其中產物部分濃縮至乾且在高真空下乾燥,得到呈黃色非晶固體狀之所需產物(4.91 g,82%產率,96% HPLC純度);δH
(CDCl3
, 500.1 MHz)/ppm;10.26 (1H, s), 8.34 (1H, s), 4.22 (2H, t, 4.5Hz), 3.90 (2H, t, 4.5Hz), 3.90 (2H, t, 4.6Hz), 3.55 (2H, t, 4.6Hz), 3.38 (3H, s);δC
NMR (CDCl3
, 125.8 MHz)/ppm;實例 21. 具有醚鍵鍵結之羧酸藥物結合物質的生物可降解聚合物
在90℃下使1.0 g PVA粉末(10 kDa)溶於35 mL 1-甲基-2-吡咯啶酮。接著溫度降低至50℃,接著添加3-溴丙酸(0.42 g,2.7 mmol)及氫氧化鈉粉末(0.22 g,5.4 mmol)。反應溫度維持在50℃下,且在350 rpm下繼續攪拌隔夜。在中止反應之後,將溶液逐滴添加至丙酮(200 ml),以沈澱析出聚合物。接著將聚合物固體用甲醇(100 ml)洗滌,以移除氫氧化鈉,且聚合物真空乾燥(24小時)。接著根據實例1使用官能化之PVA聚合物合成生物可降解珠粒。 實例 22. 具有酯鍵鍵結之不透射線性基團的生物可降解聚合物
實例 22. 具有酯鍵鍵結之不透射線性基團的生物可降解聚合物
在100 mL三頸圓底燒瓶中2,3,5-三碘苯甲酸(5 g,10 mmol)溶於25 ml NMP中。將亞硫醯氯(1.3 g,11 mmol)用5 ml NMP溶液稀釋且添加至反應容器。將反應混合物加熱至70℃,持續3小時。在反應之後,溶液置於旋轉蒸發器以移除過量亞硫醯氯及氣體副產物。
在90℃下使1.0 g PVA粉末(10 kDa)溶於35 mL 1-甲基-2-吡咯啶酮。接著溫度降低至50℃,接著添加2,3,5-三碘苯甲醯氯中間物溶液(7 ml,2.3 mmol)至PVA溶液以及三乙胺催化劑溶液(1 ml)。將反應在350 rpm下攪拌隔夜。在中止反應之後,將溶液逐滴添加至丙酮(200 ml),以沈澱析出聚合物。接著此聚合物再溶於NMP且再次在丙酮中沈澱析出,以純化聚合物。接著聚合物真空乾燥(24小時)。接著根據實例1使用官能化之PVA聚合物合成生物可降解珠粒。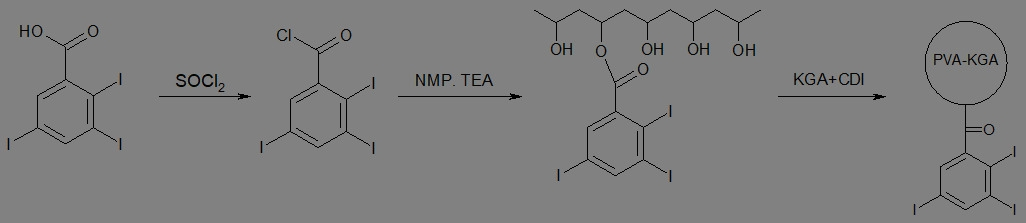 實例 23 :經改質之微球體之載藥率
實例 23 :經改質之微球體之載藥率
在恆定攪動下,使1 mL來自實例9、10、11及12之微球體懸浮在1.5 mL多柔比星溶液(濃度25 mg/mL)中。在預定時間點,對上清液取樣,且相對於已知參考,在UV下在483 nm下測定多柔比星濃度。
圖4中給出負載曲線。
圖 1
展示根據實例6量測之生物可降解聚合物之降解。
圖 2
展示乾燥狀態(A)、在鹽水中水合(B)及在導管遞送之後(C)生物可降解聚合物(125-300 μm)之影像。
圖 3
展示根據實例8製備之微球體之microCT影像。
圖 4
示出4種微球體製劑之多柔比星負載的載藥率曲線。
Claims (38)
- 一種聚合物,其具有包含與C3至C8二酸交聯之多羥基化聚合物的主鏈。
- 如申請專利範圍第1項之聚合物,其中該C3至C8二酸係選自飽和二酸、單不飽和二酸且在C6至C8不飽和二酸之情況下為雙不飽和二酸。
- 如申請專利範圍第1項或第2項中任一項之聚合物,其中該不飽和二酸經選自-OH、=O及-NH2 之基團取代。
- 如申請專利範圍第1項至第3項中任一項之聚合物,其中該聚合物與較佳選自丙二酸、丁二酸及戊二酸之C3至C8飽和二酸交聯。
- 如申請專利範圍第4項之聚合物,其中該聚合物與C4或C5 α酮酸,較佳α酮戊二酸酯交聯。
- 如申請專利範圍第1項至第3項中任一項之聚合物,其中該聚合物與較佳選自順丁烯二酸、反丁烯二酸或順式或反式半乳糖醛酸之C4至C6不飽和二酸交聯。
- 如前述申請專利範圍中任一項之聚合物,其中該等交聯基團具有式1:1 其中 *為與該多羥基化聚合物之附接點;且 其中Q為式1a之基團:
 其中 n為1至5,較佳1、2或3;或 Q為C1-6 伸烷基或C2-6 伸烯基。
其中 n為1至5,較佳1、2或3;或 Q為C1-6 伸烷基或C2-6 伸烯基。
- 一種聚合物,其藉由使包含多羥基化聚合物之聚合物與式II化合物交聯而形成,其中在該多羥基化聚合物與該式2化合物之間形成酯鍵;其中X為-OH或合適離去基;且 Q為式Ia之基團
 其中 n為1至5,較佳1至3;較佳1或2;或Q為C1-6 伸烷基或C2-6 伸烯基,其中伸烷基視情況經-OH或-NH2 取代。
其中 n為1至5,較佳1至3;較佳1或2;或Q為C1-6 伸烷基或C2-6 伸烯基,其中伸烷基視情況經-OH或-NH2 取代。
- 如申請專利範圍第8項之聚合物,其中該合適離去基係選自咪唑基、甲磺酸酯基、甲苯磺酸酯基、-O-烷基、氯基、溴基、氟基及-O-醯基。
- 如前述申請專利範圍中任一項之聚合物,其中該多羥基化聚合物為或包含PVA。
- 如前述申請專利範圍中任一項之聚合物,其中該多羥基化聚合物為或包含PVA且包含與該PVA交聯之式3之交聯基團,3 其中 Q為式1a之基團
 其中 n為1至5,較佳1至3;較佳1或2; 或Q為C1-6 伸烷基或C2-6 伸烯基,其中伸烷基視情況經-OH或-NH2 取代。
其中 n為1至5,較佳1至3;較佳1或2; 或Q為C1-6 伸烷基或C2-6 伸烯基,其中伸烷基視情況經-OH或-NH2 取代。
- 如申請專利範圍第10項或第11項之聚合物,其中該多羥基化聚合物為或包含PVA且該PVA與經選自-OH、=O及-NH2 之基團取代之C6至C8不飽和二酸交聯。
- 如申請專利範圍第12項之聚合物,其中該聚合物與C4或C5 α酮酸、較佳α酮戊二酸酯交聯。
- 如申請專利範圍第10項至第13項中任一項之聚合物,其中該PVA具有2000 kDa至180,000 kDa之重量平均分子量。
- 如申請專利範圍第10項至第13項中任一項之聚合物,其中該PVA具有10,000 MW至32,000 MW之重量平均分子量。
- 如申請專利範圍第10項至第13項中任一項之聚合物,其中該PVA具有2,000 kDa至32,000 kDa之重量平均分子量。
- 如前述申請專利範圍中任一項之聚合物,其中該聚合物為水凝膠。
- 如前述申請專利範圍中任一項之聚合物,其中該聚合物進一步包含可成像劑。
- 如申請專利範圍第18項之聚合物,其中該可成像劑與該聚合物偶合。
- 如申請專利範圍第18項或第19項之聚合物,其中該可成像劑可藉由X射線、正電子發射成像、SPECT或磁共振成像進行成像。
- 如申請專利範圍第18項之聚合物,其中該可成像劑係選自碘及溴。
- 如申請專利範圍第19項之聚合物,其中該可成像劑包含碘化苯基。
- 如申請專利範圍第19項之聚合物,其中該聚合物包含碘化苯基,其中該苯基另外包含一或兩個其他基團W,其中W係獨立地選自-OH、-COOH、-SO3 H、-OPO3 H2 、-O-(C1-4 烷基)、-O-(C1-4 烷基)OH、-O-(C1-4 烷基)R2 、-O-(C2 H5 O)q R1 、-(C=O)-O-C1-4 烷基及-O-(C=O)C1-4 烷基;或W為式-BZ之兩性離子基團; 其中: R1 為H或C1-4 烷基; R2 為-COOH、-SO3 H或-OPO3 H2 ;較佳-COOH或-SO3 H, q為整數1至4; B為一鍵,或直鏈烷二基、氧基伸烷基、伸烷基氧雜伸烷基或伸烷基(寡氧雜伸烷基),視情況含有一或多個氟取代基; Z為銨、鏻或鋶之磷酸酯或膦酸酯兩性離子基團;且 其中-COOH、-OPO3 H2 、-SO3 H及酚-OH可呈醫藥學上可接受之鹽形式。
- 如申請專利範圍第22項或第23項之聚合物,其中該碘化苯基經由醚基、酯基、醯胺基、碳酸酯基、胺基甲酸酯基、1,3-二氧雜環戊烯酮基或1,3-二噁烷基,尤其醚基、酯基或1,3-二噁烷基與該聚合物主鏈偶合。
- 如前述申請專利範圍中任一項之聚合物,其中該聚合物在pH 7.4下帶離子電荷。
- 如前述申請專利範圍中任一項之聚合物,其中該聚合物包含在pH 7.4下離子化之共價結合之離子基團。
- 如申請專利範圍第25項或第26項之聚合物,其中該離子基包含選自以下之帶電物質:磺酸酯基、磷酸酯基、銨基、鏻基及羧酸酯基;較佳羧酸酯基或磺酸酯基。
- 如申請專利範圍第25項或第26項之聚合物,其中該帶電基團係選自C1-6 分支或未分支烷基、C2-6 分支或未分支烯基或C5-7 芳基或雜芳基,各獨立地經1至3個選自由-COOH、-OPO3 H2 及-SO3 H組成之群的基團取代。
- 如申請專利範圍第25項或第26項之聚合物,其中該離子基經由醚基、酯基、醯胺基、碳酸酯基、胺基甲酸酯基、1,3-二氧雜環戊烯酮基或1,3-二噁烷基,尤其醚基、酯基或1,3-二噁烷基與該聚合物主鏈偶合。
- 如申請專利範圍第25項或第26項之聚合物,其中該聚合物具有總負電荷。
- 如申請專利範圍第25項至第30項之聚合物,其中該聚合物與帶相反電荷之藥物以靜電作用締合。
- 如前述申請專利範圍中任一項之聚合物,其係呈微粒或微球體形式。
- 一種微粒或微球體,其包含如申請專利範圍第1項至第31項中任一項之聚合物。
- 如申請專利範圍第32項或第33項中任一項之微粒或微球體,其用於使血管栓塞。
- 一種醫藥組合物,其包含一或多個如申請專利範圍第32項或第33項中任一項之微粒或微球體及醫藥學上可接受之載劑或稀釋劑。
- 一種製造生物可降解聚合物之方法,該方法包括: 使多羥基化聚合物與式2化合物交聯其中X為-OH或合適離去基;且其中Q為式1a之基團:
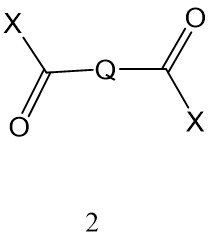 其中n為1至5; 或Q為C1-6 伸烷基或C2-6 伸烯基,其中伸烷基視情況經-OH或-NH2 取代; 從而在該多羥基化聚合物與該式2化合物之間形成酯鍵,從而使該聚合物交聯。
其中n為1至5; 或Q為C1-6 伸烷基或C2-6 伸烯基,其中伸烷基視情況經-OH或-NH2 取代; 從而在該多羥基化聚合物與該式2化合物之間形成酯鍵,從而使該聚合物交聯。 - 如申請專利範圍第36項之方法,其中該合適離去基係選自咪唑基、甲磺酸酯基、甲苯磺酸酯基、-O-烷基、氯基、溴基、氟基及-O-醯基。
- 一種製造生物可降解聚合物微球體之方法,該方法包括: 提供第一液體,其為使以下溶解於其中之溶劑:(i)包含PVA之聚合物,及(ii)式2化合物;其中 Q為式1a基團其中n為1至5; 或Q為C1-6 伸烷基或C2-6 伸烯基,其中伸烷基視情況經-OH或-NH2 取代;且 X為-OH或合適離去基; 提供與該第一液體不混溶之第二液體; 使該第一液體與該第二液體接觸,使得該第一液體在該第二液體內形成不連續相;及 在該不連續相內使該PVA與該式2化合物交聯,從而形成微球體。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GBGB1810788.8A GB201810788D0 (en) | 2018-06-29 | 2018-06-29 | Biodegradable polymer |
| GB1810788.8 | 2018-06-29 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| TW202000778A true TW202000778A (zh) | 2020-01-01 |
Family
ID=63143624
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| TW108122410A TW202000778A (zh) | 2018-06-29 | 2019-06-26 | 生物可降解聚合物 |
Country Status (8)
| Country | Link |
|---|---|
| US (1) | US12049525B2 (zh) |
| EP (3) | EP3814407B1 (zh) |
| JP (1) | JP7322071B2 (zh) |
| CN (1) | CN112313267A (zh) |
| CA (1) | CA3098650C (zh) |
| GB (1) | GB201810788D0 (zh) |
| TW (1) | TW202000778A (zh) |
| WO (1) | WO2020003152A2 (zh) |
Families Citing this family (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN111476856B (zh) * | 2020-04-08 | 2023-06-06 | 中北大学 | 一种多谱ct成像方法 |
| CN113461851B (zh) * | 2021-06-11 | 2022-05-20 | 华中科技大学 | 一种聚合物的应用及包含该聚合物的光刻胶 |
| US20230021742A1 (en) * | 2021-07-12 | 2023-01-26 | Boston Scientific Scimed, Inc. | Radiopaque compositions |
| CN114031933A (zh) * | 2021-11-30 | 2022-02-11 | 江苏芬茂新材料科技有限公司 | 一种基于汽车零部件生产用高韧性塑料颗粒及其制备方法 |
| CN118434411A (zh) | 2021-12-23 | 2024-08-02 | 波士顿科学医疗设备有限公司 | 化学栓塞组合物及其治疗方法 |
| CN114262279B (zh) * | 2021-12-30 | 2022-12-16 | 上海汇禾医疗科技有限公司 | 一种x射线可显影分子、栓塞微球及其制备方法 |
| EP4215224A1 (en) * | 2022-01-25 | 2023-07-26 | Biotronik Ag | Tissue protection |
| JP2025537730A (ja) * | 2022-11-04 | 2025-11-20 | エッチ ジェー ハインツ カンパニー ブランズ エルエルシー | 酵素カスケードを用いたポリビニルアルコールおよびその誘導体の分解 |
| CN116554510A (zh) * | 2023-05-10 | 2023-08-08 | 天津市胸科医院 | 一种碘基纳米造影剂的制备方法 |
| CN117586440B (zh) * | 2024-01-17 | 2024-04-09 | 苏州美创医疗科技有限公司 | 一种碘化多羟基聚合物、制备方法及其制备的液体栓塞剂 |
| CN118725426B (zh) * | 2024-08-05 | 2025-09-30 | 株洲时代新材料科技股份有限公司 | 一种生物基高阻尼橡胶材料及其制备方法 |
Family Cites Families (22)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE480866C (de) | 1924-07-20 | 1929-08-15 | Consortium Elektrochem Ind | Verfahren zur Darstellung von Derivaten des polymeren Vinylalkohols |
| JPS59199696A (ja) | 1983-04-28 | 1984-11-12 | Oki Electric Ind Co Ltd | リン脂質類似構造を有する化合物及びポリマ−並びにその製造方法 |
| WO1993001221A1 (en) | 1991-07-05 | 1993-01-21 | Biocompatibles Limited | Polymeric surface coatings |
| GB9204918D0 (en) * | 1992-03-06 | 1992-04-22 | Nycomed As | Chemical compounds |
| GB9301701D0 (en) | 1993-01-28 | 1993-03-17 | Biocompatibles Ltd | New zwitterionic materials |
| GB9415926D0 (en) | 1994-08-04 | 1994-09-28 | Biocompatibles Ltd | New materials |
| US6699920B1 (en) * | 2002-02-13 | 2004-03-02 | Nicholas Andros | Methods of manufacturing polishing substrates |
| US20080305176A1 (en) | 2006-01-24 | 2008-12-11 | Biocompatibles Uk Limited | Process For Loading Polymer Particles With Drug |
| EP2520287B1 (en) | 2006-02-10 | 2025-03-26 | Boston Scientific Medical Device Limited | Loading of hydrophobic drugs into hydrophilic polymer delivery systems |
| FR2906253A1 (fr) | 2006-09-22 | 2008-03-28 | Biosphere Medical Sa | Nouveaux materiaux polymeres bio-compatibles, leur procede d'obtention et leurs utilisations, notamment en imagerie medicale par resonnance magnetique |
| FR2927629B1 (fr) * | 2008-02-14 | 2011-07-29 | Bostik Sa | Composition adhesive thermofusible biodegradable. |
| EP2365009A1 (en) | 2010-03-10 | 2011-09-14 | Universite Claude Bernard Lyon 1 (UCBL) | Radiopaque, non-biodegradable, water-insoluble iodinated benzyl ethers of poly(vinyl alcohol), preparation method thereof, injectable embolizing compositions containing thereof and use thereof |
| EP2522695A1 (de) | 2011-05-10 | 2012-11-14 | Basf Se | Biologisch abbaubare Polyesterfolie |
| US9591853B2 (en) * | 2013-01-07 | 2017-03-14 | Ramot At Tel-Aviv University Ltd. | Jellyfish-derived polymer |
| WO2014159759A1 (en) | 2013-03-13 | 2014-10-02 | Biosphere Medical, Inc. | Compositions and associated methods for radioisotope-binding microparticles |
| JP6420817B2 (ja) | 2013-03-15 | 2018-11-07 | バイオコンパティブルズ ユーケー リミテッド | 結像可能な塞栓性微小球 |
| GB2521997A (en) | 2013-09-06 | 2015-07-15 | Biocompatibles Uk Ltd | Radiopaque polymers |
| GB2519738A (en) | 2013-09-06 | 2015-05-06 | Biocompatibles Uk Ltd | Radiopaque polymers |
| DE102016210898A1 (de) * | 2016-06-17 | 2017-12-21 | Tesa Se | Biologisch abbaubare Haftklebmasse |
| US11382990B2 (en) | 2016-11-16 | 2022-07-12 | The Usa, As Represented By The Secretary, Dhhs | Imageable polymers, methods of making and methods of use thereof |
| JP2018145328A (ja) | 2017-03-07 | 2018-09-20 | 株式会社クラレ | 吸水性樹脂およびその製造方法 |
| CN107899066B (zh) * | 2017-12-01 | 2021-02-09 | 苏州恒瑞迦俐生生物医药科技有限公司 | 阳离子多羟基聚合物栓塞微球及其制备方法 |
-
2018
- 2018-06-29 GB GBGB1810788.8A patent/GB201810788D0/en not_active Ceased
-
2019
- 2019-06-26 JP JP2020565303A patent/JP7322071B2/ja active Active
- 2019-06-26 EP EP19765550.9A patent/EP3814407B1/en active Active
- 2019-06-26 CN CN201980040696.1A patent/CN112313267A/zh active Pending
- 2019-06-26 TW TW108122410A patent/TW202000778A/zh unknown
- 2019-06-26 US US17/252,552 patent/US12049525B2/en active Active
- 2019-06-26 EP EP25193319.8A patent/EP4617292A3/en active Pending
- 2019-06-26 WO PCT/IB2019/055392 patent/WO2020003152A2/en not_active Ceased
- 2019-06-26 EP EP22195184.1A patent/EP4130105A1/en not_active Withdrawn
- 2019-06-26 CA CA3098650A patent/CA3098650C/en active Active
Also Published As
| Publication number | Publication date |
|---|---|
| GB201810788D0 (en) | 2018-08-15 |
| JP7322071B2 (ja) | 2023-08-07 |
| US12049525B2 (en) | 2024-07-30 |
| US20210115171A1 (en) | 2021-04-22 |
| EP4617292A2 (en) | 2025-09-17 |
| EP3814407A2 (en) | 2021-05-05 |
| JP2021524876A (ja) | 2021-09-16 |
| CN112313267A (zh) | 2021-02-02 |
| EP4617292A3 (en) | 2025-11-05 |
| EP4130105A1 (en) | 2023-02-08 |
| CA3098650A1 (en) | 2020-01-02 |
| WO2020003152A3 (en) | 2020-07-30 |
| WO2020003152A2 (en) | 2020-01-02 |
| EP3814407B1 (en) | 2025-08-13 |
| CA3098650C (en) | 2023-04-04 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| TW202000778A (zh) | 生物可降解聚合物 | |
| JP6723312B2 (ja) | 結像可能な塞栓性微小球 | |
| JP7728813B2 (ja) | 放射線不透過性ポリマーを含む放射線不透過性ミクロスフェア | |
| BR112012019754B1 (pt) | éteres benzílicos iodados de poli (álcool vinílico) radiopacos, não biodegradáveis, insolúveis em água, processo de preparação, composições embolizantes injetáveis contendo o mesmo, utilização do mesmo e partículas radiopacas | |
| JP7095112B2 (ja) | 放射線不透過性ポリマー | |
| US11992575B2 (en) | Radioactive liquid embolic | |
| US20230355833A1 (en) | Radiopaque polymers | |
| EP3541368A1 (en) | Imageable particles, methods of making and methods of use thereof |