相關申請案的交叉參考 本申請案主張2012年11月2日提出申請之美國臨時專利申請案第61/721,622號、2012年11月20日提出申請之第61/728,328號、2013年2月28日提出申請之第61/770,668號、2013年5月16日提出申請之第61/824,005號及2013年6月28日提出申請之第61/840,668號之優先權,所有申請案之整體內容皆以全文引用方式併入本文中。
定義
如本文所使用,「CFTR」代表囊性纖維化跨膜傳導調控子。 如本文所使用,「ΔF508突變」或「F508-del突變」係CFTR蛋白內之特定突變。該突變係在位置508處缺失包含胺基酸苯丙胺酸之密碼子之三個核苷酸,從而導致CFTR蛋白缺乏此苯丙胺酸殘基。 如本文所使用,對於具體突變(例如ΔF508)為「同型合子」之患者在每一等位基因上具有相同突變。 如本文所使用,對於具體突變(例如ΔF508)為「異型合子」之患者在一個等位基因上具有此突變且在另一等位基因上具有不同突變。 如本文所使用,術語「CFTR校正劑」係指使至細胞表面之功能CFTR蛋白之量增加從而使得離子運輸增強之化合物。 如本文所使用,術語「CFTR增效劑」係指使位於細胞表面處之CFTR蛋白通道活性增加從而使得離子運輸增強之化合物。 如本文所使用,術語「活性醫藥成份」或「API」係指生物活性化合物。 術語「固體形式(solid form、solid forms)」及相關術語在本文中使用時係指呈具體固體形式(例如晶體、非晶形狀態及諸如此類)之化合物1或化合物2。 如本文所使用,術語「實質上非晶形」係指在分子位置中幾乎沒有或沒有長程有序之固體材料。例如,實質上非晶形材料具有小於約15%之結晶度(例如,小於約10%之結晶度或小於約5%之結晶度)。亦應注意,術語「實質上非晶形」包括闡述詞「非晶形」,其係指不具有結晶度(0%)之材料。 如本文所使用,術語「實質上結晶」(如在片語實質上結晶化合物1形式I中一般)係指在分子位置中主要具有長程有序之固體材料。例如,實質上結晶材料具有大於約85%之結晶度(例如,大於約90%之結晶度或大於約95%之結晶度)。亦應注意,術語「實質上結晶」包括闡述詞「結晶」,其係指具有100%結晶度之材料。 本文所使用術語「結晶」及相關術語當用於闡述物質、組份、產物或形式時,意指物質、組份或產物係實質上結晶的,如藉由X射線繞射所測定。(例如,參見Remington: The Science and Practice of Pharmacy,第21版,Lippincott Williams & Wilkins, Baltimore, Md. (2003);美國藥典(The United States Pharmacopeia),第23版,1843-1844 (1995))。 如本文所使用,「賦形劑」包括醫藥組合物中之功能及非功能成份。 如本文所使用,「崩解劑」係使醫藥組合物水合且有助於錠劑分散之賦形劑。如本文所使用,「稀釋劑」或「填充劑」係增加醫藥組合物膨松性之賦形劑。 如本文所使用,「表面活性劑」係增強醫藥組合物之溶解性及/或可濕性之賦形劑。 如本文所使用,「黏合劑」係增強醫藥組合物之內聚力或拉伸強度(例如,硬度)之賦形劑。 如本文所使用,「助流劑」係增強醫藥組合物之流動性質之賦形劑。 如本文所使用,「著色劑」係賦予醫藥組合物(例如錠劑)期望色彩之賦形劑。著色劑之實例包括市售顏料,例如FD&C藍1號鋁色澱、FD&C藍2號、其他FD&C藍顏料、二氧化鈦、氧化鐵及/或其組合。在一個實施例中,本發明所提供錠劑為粉色。 如本文所使用,「潤滑劑」係添加至壓製成錠劑之醫藥組合物中之賦形劑。潤滑劑有助於自模壓機將顆粒壓實成錠劑並頂出醫藥組合物錠劑。 如本文所使用,「立方公分」與「cc」可互換使用以表示體積單位。應注意,1 cc = 1 mL。 如本文所使用,「公斤力」與「kP」可互換使用且係指1 kP =大約9.8牛頓(Newton)之力之量度。 如本文所使用,「脆碎度」係指儘管存在外部壓力,但錠劑仍保持完整且保留其形式之性質。脆碎度可使用等式1中所呈現之數學表達來量化:
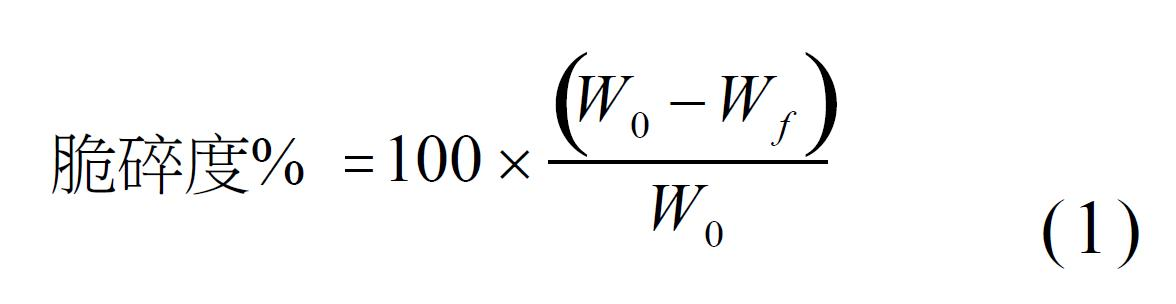
其中
W0
係錠劑之初始重量且
Wf
係在使錠劑經受脆碎度測定儀後之最終重量。脆碎度係使用標準USP測試裝置來量測,該裝置將實驗錠劑翻轉100轉或400轉。本發明之一些錠劑之脆碎度小於5.0%。在另一實施例中,脆碎度小於2.0%。在另一實施例中,在400轉後,目標脆碎度小於1.0%。 如本文所使用,「平均粒子直徑」係如使用諸如雷射光散射、影像分析或篩析等技術所量測之平均粒子直徑。在一個實施例中,用於製備本發明所提供醫藥組合物之顆粒的平均粒子直徑小於1.0 mm。 如本文所使用,「堆密度」係材料粒子之質量除以粒子所佔總體積。總體積包括粒子體積、粒子間空隙體積及內部孔隙體積。堆密度並非材料之固有性質;其可隨材料處理方式而變化。在一個實施例中,用於製備本發明所提供醫藥組合物之顆粒的堆密度為約0.5-0.7 g/cc。 本發明化合物之有效量或」治療有效量」可根據諸如個體之疾病狀態、年齡及重量以及本發明化合物引發個體之期望反應之能力等因素而改變。可對劑量方案進行調整以獲得最佳治療反應。有效量亦係本發明化合物之治療有益效應勝過其任何有毒或有害效應(例如,副效應)之量。 如本文所用,且除非另有說明,否則術語化合物之「治療有效量」及「有效量」係指足以在疾病或病症之治療或管控中提供治療益處或足以延遲或使與該疾病或病症相關之一或多種症狀降至最低之量。化合物之「治療有效量」及「有效量」意指治療劑單獨或與一或多種其他藥劑組合在疾病或病症之治療或管控中提供治療益處之量。術語「治療有效量」及「有效量」可涵蓋改良總體療法、減少或避免疾病或病症之症狀或病因或增強另一治療劑之治療功效的量。 「實質上純淨」如在片語「實質上純淨化合物1形式I」中所使用意指純度大於約90%。在另一實施例中,實質上純淨係指純度大於約95%。在另一實施例中,實質上純淨係指純度大於約98%。在另一實施例中,實質上純淨係指純度大於約99%。 關於化合物1形式I或包含實質上非晶形化合物2之固體分散體,術語「約」及「大約」當結合組合物或劑型之成份之劑量、量或重量%使用時,意指熟習此項技術者所識別欲提供等效於自所指定劑量、量或重量%所獲得者之藥理學效應的劑量、量或重量%。特定而言,術語「約」或「大約」意指如熟習此項技術者所測定之具體值的可接受誤差,其部分地取決於該值之量測或測定方式。在某些實施例中,術語「約」或「大約」意指在1、2、3或4個標準偏差內。在某些實施例中,術語「約」或「大約」意指在給定值或範圍之30%、25%、20%、15%、10%、9%、8%、7%、6%、5%、4%、3%、2%、1%、0.5%、0.1%或0.05%以內。
醫藥組合物
本發明提供包含化合物1形式I及包含實質上非晶形化合物2之固體分散體之醫藥組合物。在此態樣之一些實施例中,存於醫藥組合物中之化合物1形式I之量為100 mg、125 mg、150 mg、200 mg、250 mg、300 mg或400 mg。在此態樣之一些實施例中,存於醫藥組合物中之化合物1形式I之重量%為10%至75%。在該等及其他實施例中,化合物1形式I係作為實質上純淨化合物1形式I存在。在此態樣之一些實施例中,存於醫藥組合物中之實質上非晶形化合物2之量為100 mg、125 mg、150 mg、200 mg或250 mg。在此態樣之一些實施例中,存於醫藥組合物中之實質上非晶形化合物2之重量%為10%至75%。在該等及其他實施例中,實質上非晶形化合物2係作為實質上純淨且非晶形的化合物2存在。「實質上純淨」意指大於90%純淨;較佳地大於95%純淨;更佳地大於99.5%純淨。 因此,在一個態樣中,本發明提供包含以下之醫藥組合物: a. 化合物1形式I; b. 包含實質上非晶形化合物2之固體分散體; c. 填充劑; d. 崩解劑; e. 表面活性劑;及 f. 黏合劑。 在此態樣之一個實施例中,醫藥組合物包含25 mg化合物1形式I。在此態樣之另一實施例中,醫藥組合物包含50 mg化合物1形式I。在此態樣之另一實施例中,醫藥組合物包含100 mg化合物1形式I。在此態樣之另一實施例中,醫藥組合物包含125 mg化合物1形式I。在此態樣之另一實施例中,醫藥組合物包含150 mg化合物1形式I。在此態樣之另一實施例中,醫藥組合物包含200 mg化合物1形式I。在此態樣之另一實施例中,醫藥組合物包含250 mg化合物1形式I。在此態樣之另一實施例中,醫藥組合物包含400 mg化合物1形式I。 在此態樣之一個實施例中,醫藥組合物包含25 mg實質上非晶形化合物2。在此態樣之另一實施例中,醫藥組合物包含50 mg實質上非晶形化合物2。在此態樣之另一實施例中,醫藥組合物包含100 mg實質上非晶形化合物2。在此態樣之另一實施例中,醫藥組合物包含125 mg實質上非晶形化合物2。在此態樣之另一實施例中,醫藥組合物包含150 mg實質上非晶形化合物2。在此態樣之另一實施例中,醫藥組合物包含200 mg實質上非晶形化合物2。在此態樣之另一實施例中,醫藥組合物包含250 mg實質上非晶形化合物2。 在一些實施例中,醫藥組合物包含化合物1形式I,其中化合物1形式I係以組合物之重量計至少15 wt% (例如,至少20 wt%、至少30 wt%、至少40 wt%、至少50 wt%或至少60 wt%)之量存在。 在一些實施例中,醫藥組合物包含實質上非晶形化合物2,其中該實質上非晶形化合物2係以組合物之重量計至少15 wt% (例如,至少20 wt%、至少30 wt%、至少40 wt%、至少50 wt%或至少60 wt%)之量存在。 在一些實施例中,醫藥組合物包含化合物1形式I、包含實質上非晶形化合物2之固體分散體、填充劑、崩解劑、表面活性劑及黏合劑。在此實施例中,組合物包含以組合物之重量計約25 wt%至約55 wt% (例如,約30-50 wt%)之化合物1形式I,且更通常地,以組合物之重量計40 wt%至約45 wt%之化合物1形式I。在此實施例中,組合物包含以組合物之重量計約15 wt%至約40 wt% (例如,約20-35 wt%)之實質上非晶形化合物2,且更通常地,以組合物之重量計25 wt%至約30 wt%實質上非晶形化合物2。 組合物中化合物1形式I及實質上非晶形化合物2之濃度取決於若干因素,例如提供期望量之化合物1形式I及實質上非晶形化合物2所需醫藥組合物之量及醫藥組合物之期望溶解曲線。 在另一實施例中,醫藥組合物包含化合物1形式I,其中呈固體形式之化合物1形式I之平均粒子直徑為0.1微米至10微米,如藉由光散射(例如,使用購自Malvern Instruments (England)之Malvern Mastersizer)所量測。在另一實施例中,化合物1形式I之粒徑係1微米至5微米。在另一實施例中,化合物1形式I之粒徑D50為2.0微米。 如所指示,除化合物1形式I即實質上非晶形化合物2之固體分散體以外,在本發明之一些實施例中,為口服調配物之醫藥組合物亦包含一或多種賦形劑,例如填充劑、崩解劑、表面活性劑、稀釋劑、黏合劑、助流劑、潤滑劑、著色劑或芳香劑及其任一組合。 適於本發明之填充劑可與醫藥組合物之成份相容,即,其不會實質上降低醫藥組合物之溶解性、硬度、化學穩定性、物理穩定性或生物活性。例示性填充劑包括:纖維素、改質纖維素(例如羧甲基纖維素鈉、乙基纖維素、羥甲基纖維素、羥丙基纖維素)、乙酸纖維素、微晶纖維素、磷酸鈣、磷酸氫鈣、澱粉(例如玉米澱粉、馬鈴薯澱粉)、糖(例如,山梨醇、乳糖、蔗糖或諸如此類)或其任一組合。 因此,在一個實施例中,醫藥組合物包含至少一種以組合物之重量計至少5 wt%(例如,至少約20 wt%、至少約30 wt%或至少約40 wt%)之量的填充劑。例如,醫藥組合物包含以組合物之重量計約10 wt%至約60 wt%(例如,約20 wt%至約55 wt%、約25 wt%至約50 wt%或約27 wt%至約45 wt%)之填充劑。在另一實例中,醫藥組合物包含以組合物之重量計至少約20 wt%(例如,至少30 wt%或至少40 wt%)之微晶纖維素(例如MCC Avicel PH102)。在再一實例中,醫藥組合物包含以組合物之重量計約10 wt%至約60 wt%(例如,約20 wt%至約55 wt%或約25 wt%至約45 wt%)之微晶纖維素。 適於本發明之崩解劑可增強醫藥組合物之分散性且可與醫藥組合物之成份相容,即,其不會實質上降低醫藥組合物之化學穩定性、物理穩定性、硬度或生物活性。例示性崩解劑包括交聯羧甲基纖維素鈉、羥基乙酸澱粉鈉或其組合。 因此,在一個實施例中,醫藥組合物包含以組合物之重量計約10 wt%或更小(例如,約7 wt%或更小、約6 wt%或更小或約5 wt%或更小)之量的崩解劑。例如,醫藥組合物包含以組合物之重量計約1 wt%至約10 wt% (例如,約1.5 wt%至約7.5 wt%或約2.5 wt%至約6 wt%)之崩解劑。在另一實例中,醫藥組合物包含以組合物之重量計約10 wt%或更小(例如,7 wt%或更小、6 wt%或更小,或5 wt%或更小)之交聯羧甲基纖維素鈉。在再一實例中,醫藥組合物包含以組合物之重量計約1 wt%至約10 wt% (例如,約1.5 wt%至約7.5 wt%或約2.5 wt%至約6 wt%)之交聯羧甲基纖維素鈉。在一些實例中,醫藥組合物包含以組合物之重量計約0.1%至約10 wt% (例如,約0.5 wt%至約7.5 wt%或約1.5 wt%至約6 wt%)之崩解劑。在又一些實例中,醫藥組合物包含以組合物之重量計約0.5%至約10 wt% (例如,約1.5 wt%至約7.5 wt%或約2.5 wt%至約6 wt%)之崩解劑。 適於本發明之表面活性劑可增強醫藥組合物之可濕性且可與醫藥組合物之成份相容,即,其不會實質上降低醫藥組合物之化學穩定性、物理穩定性、硬度或生物活性。例示性表面活性劑包括月桂基硫酸鈉(SLS)、硬脂基富馬酸鈉(SSF)、聚氧乙烯20山梨醇酐單油酸酯(例如,Tween™)、其任一組合或諸如此類。 因此,在一個實施例中,醫藥組合物包含以組合物之重量計約10 wt%或更小(例如,約5 wt%或更小、約2 wt%或更小、約1 wt%或更小、約0.8 wt%或更小或約0.6 wt%或更小)之量的表面活性劑。例如,醫藥組合物包括以組合物之重量計約10 wt%至約0.1 wt% (例如,約5 wt%至約0.2 wt%或約2 wt%至約0.3 wt%)之表面活性劑。在另一實例中,醫藥組合物包含以組合物之重量計10 wt%或更小(例如,約5 wt%或更小、約2 wt%或更小、約1 wt%或更小、約0.8 wt%或更小或約0.6 wt%或更小)之月桂基硫酸鈉。在再一實例中,醫藥組合物包含以組合物之重量計約10 wt%至約0.1 wt% (例如,約5 wt%至約0.2 wt%或約2 wt%至約0.3 wt%)之月桂基硫酸鈉。 適於本發明之黏合劑可增強醫藥組合物之錠劑強度且可與醫藥組合物之成份相容,即,其不會實質上降低醫藥組合物之化學穩定性、物理穩定性或生物活性。例示性黏合劑包括聚乙烯吡咯啶酮、磷酸氫鈣、蔗糖、玉米澱粉、改質纖維素(例如,羥甲基纖維素)或其任一組合。 因此,在一個實施例中,醫藥組合物包含以組合物之重量計至少約0.1 wt% (例如,至少約1 wt%、至少約3 wt%、至少約4 wt%,或至少約5 wt%)之量的黏合劑。例如,醫藥組合物包含以組合物之重量計約0.1 wt%至約10 wt% (例如,約1 wt%至約10 wt%或約2 wt%至約7 wt%)之黏合劑。在另一實例中,醫藥組合物包含以組合物之重量計至少約0.1 wt% (例如,至少約1 wt%、至少約2 wt%、至少約3 wt%或至少約4 wt%)之聚乙烯吡咯啶酮。在再一實例中,醫藥組合物包含以組合物之重量計在約0.1 wt%至約10 wt%之範圍內(例如,約1 wt%至約8 wt%或約2 wt%至約5 wt%)之聚乙烯吡咯啶酮之量的助流劑。 適於本發明之稀釋劑可增加調配物之必要體積以製備期望大小之錠劑,且通常可與醫藥組合物之成份相容,即,其不會實質上降低醫藥組合物之溶解性、硬度、化學穩定性、物理穩定性或生物活性。例示性稀釋劑包括:糖(例如,糖粉(confectioner's sugar)、可壓製糖)、葡萄糖結合劑、糊精、右旋糖、乳糖、甘露醇、山梨醇、纖維素及改質纖維素(例如,粉末纖維素)、滑石粉、磷酸鈣、澱粉或其任一組合。 因此,在一個實施例中,醫藥組合物包含以組合物之重量計40 wt%或更小(例如,35 wt%或更小、30 wt%或更小或25 wt%或更小或20 wt%或更小或15 wt%或更小或10 wt%或更小)之量的稀釋劑。例如,醫藥組合物包含以組合物之重量計約40 wt%至約1 wt% (例如,約35 wt%至約5 wt%或約30 wt%至約7 wt%、約25 wt%至約10 wt%、約20 wt%至約15 wt%)之稀釋劑。在另一實例中,醫藥組合物包含以組合物之重量計40 wt%或更小(例如,35 wt%或更小、25 wt%或更小或15 wt%或更小)之甘露醇。在再一實例中,醫藥組合物包含以組合物之重量計約35 wt%至約1 wt% (例如,約30 wt%至約5 wt%或約25 wt%至約10 wt%)之甘露醇。 適於本發明之助流劑可增強醫藥組合物之流動性且可與醫藥組合物之成份相容,即,其不會實質上降低醫藥組合物之溶解性、硬度、化學穩定性、物理穩定性或生物活性。例示性助流劑包括膠態二氧化矽、滑石粉或其組合。 因此,在一個實施例中,醫藥組合物包含以組合物之重量計2 wt%或更小(例如,1.75 wt%、1.25 wt%或更小或1.00 wt%或更小)之量的助流劑。例如,醫藥組合物包含以組合物之重量計約2 wt%至約0.05 wt% (例如,約1.5 wt%至約0.07 wt%或約1.0 wt%至約0.09 wt%)之助流劑。在另一實例中,醫藥組合物包含以組合物之重量計2 wt%或更小(例如,1.75 wt%、1.25 wt%或更小或1.00 wt%或更小)之膠態二氧化矽。在再一實例中,醫藥組合物包含以組合物之重量計約2 wt%至約0.05 wt% (例如,約1.5 wt%至約0.07 wt%或約1.0 wt%至約0.09 wt%)之膠態二氧化矽。 在一些實施例中,醫藥組合物可包括口服固體醫藥劑型,其可包含可防止顆粒-珠粒混合物附著至表面(例如,混合缽、壓模及/或衝壓器之表面)之潤滑劑。潤滑劑亦可降低顆粒內之粒子間摩擦力並改良經壓製醫藥組合物自模壓機之壓製及頂出。潤滑劑亦可與醫藥組合物之成份相容,即,其不會實質上降低醫藥組合物之溶解性、硬度或生物活性。例示性潤滑劑包括硬脂酸鎂、硬脂酸鈣、硬脂酸鋅、硬脂酸鈉、硬脂酸、硬脂酸鋁、白胺酸、山崳酸甘油酯、氫化植物油或其任一組合。在一個實施例中,醫藥組合物包含以組合物之重量計5 wt%或更小(例如,4.75 wt%、4.0 wt%或更小或3.00 wt%或更小或2.0 wt%或更小)之量的潤滑劑。例如,醫藥組合物包含以組合物之重量計約5 wt%至約0.10 wt% (例如,約4.5 wt%至約0.5 wt%或約3 wt%至約1 wt%)之潤滑劑。在另一實例中,醫藥組合物包含以組合物之重量計5 wt%或更小(例如,4.0 wt%或更小、3.0 wt%或更小或2.0 wt%或更小或1.0 wt%或更小)之硬脂酸鎂。在再一實例中,醫藥組合物包含以組合物之重量計約5 wt%至約0.10 wt% (例如,約4.5 wt%至約0.15 wt%或約3.0 wt%至約0.50 wt%)之硬脂酸鎂。 本發明醫藥組合物可視情況包含一或多種著色劑、矯味劑及/或芳香劑以增強組合物之視覺吸引力、味道及/或氣味。適宜著色劑、矯味劑或芳香劑可與醫藥組合物之成份相容,即,其不會實質上降低醫藥組合物之溶解性、化學穩定性、物理穩定性、硬度或生物活性。在一個實施例中,醫藥組合物包含著色劑、矯味劑及/或芳香劑。在一個實施例中,本發明所提供醫藥組合物為紫色。 在一些實施例中,醫藥組合物包括或可製成錠劑且錠劑可經著色劑包覆並視情況使用適宜墨水標記標誌、其他影像及/或文字。在又一些實施例中,醫藥組合物包括或可製成錠劑且錠劑可經著色劑包覆,上蠟並視情況使用適宜墨水標記標誌、其他影像及/或文字。適宜著色劑及墨水可與醫藥組合物之成份相容,即,其不會實質上降低醫藥組合物之溶解性、化學穩定性、物理穩定性、硬度或生物活性。適宜著色劑及墨水可為任一色彩且以水為基礎或以溶劑為基礎。在一個實施例中,自醫藥組合物製得之錠劑經著色劑包覆且隨後使用適宜墨水標記標誌、其他影像及/或文字。例如,包含本文所述醫藥組合物之錠劑可經約3 wt% (例如,小於約6 wt%或小於約4 wt%)之包含著色劑之薄膜包衣包覆。經著色錠劑可使用適宜墨水標記標誌及文字,以指示錠劑中活性成份之強度。在另一實例中,包含本文所述醫藥組合物之錠劑可經約3 wt% (例如,小於約6 wt%或小於約4 wt%)之包含著色劑之薄膜包衣包覆。 在另一實施例中,自醫藥組合物製得之錠劑經著色劑包覆,上蠟且隨後使用適宜墨水標記標誌、其他影像及/或文字。例如,包含本文所述醫藥組合物之錠劑可經約3 wt% (例如,小於約6 wt%或小於約4 wt%)之包含著色劑之薄膜包衣包覆。經著色錠劑可經重約0.01% w/w之起始錠劑核心重量之量的巴西棕櫚蠟粉末上蠟。經上蠟錠劑可使用適宜墨水標記標誌及文字,以指示錠劑中活性成份之強度。在另一實例中,包含本文所述醫藥組合物之錠劑可經約3 wt% (例如,小於約6 wt%或小於約4 wt%)之包含著色劑之薄膜包衣包覆。經著色錠劑可經重約0.01% w/w之起始錠劑核心重量之量的巴西棕櫚蠟粉末上蠟。經上蠟錠劑可使用醫藥級墨水(例如黑墨水(例如,Opacode® S-1-17823,一種以溶劑為基礎之墨水,購自Colorcon公司,West Point, PA))標記標誌及文字,以指示錠劑中活性成份之強度。 一種例示性醫藥組合物包含以組合物之重量計約15 wt%至約70 wt% (例如,約15 wt%至約60 wt%,約15 wt%至約50 wt%,或約20 wt%至約70 wt%,或約30 wt%至約70 wt%)化合物1形式I;及以組合物之重量計約15 wt%至約40 wt% (例如,約20-35 wt%)實質上非晶形化合物2,且更通常,以組合物之重量計25 wt%至約30 wt%實質上非晶形化合物2。上述組合物亦可包括一或多種醫藥上可接受之賦形劑,例如,約20 wt%至約50 wt%填充劑;約1 wt%至約5 wt%崩解劑;約2 wt%至約0.3 wt%表面活性劑;及約0.1 wt%至約5 wt%黏合劑。 另一例示性醫藥組合物包含以組合物之重量計約15 wt%至約70 wt% (例如,約15 wt%至約60 wt%、約15 wt%至約50 wt%、或約15 wt%至約40 wt%、或約20 wt%至約70 wt%、或約30 wt%至約70 wt%、或約40 wt%至約70 wt%、或約50 wt%至約70 wt%)之化合物1形式I,以組合物之重量計約15 wt%至約40 wt% (例如,約20-35 wt%)之實質上非晶形化合物2,且更通常,以組合物之重量計25 wt%至約30 wt%實質上非晶形化合物2,及一或多種賦形劑,例如,約20 wt%至約50 wt%之填充劑;約1 wt%至約5 wt%之崩解劑;約2 wt%至約0.3 wt%之表面活性劑;約0.1 wt%至約5 wt%之黏合劑;及約2 wt%至約0.1 wt%之潤滑劑。 另一例示性醫藥組合物包含以組合物之重量計約15 wt%至約70 wt% (例如,約15 wt%至約60 wt%,約15 wt%至約50 wt%,或約15 wt%至約40 wt%或約20 wt%至約70 wt%,或約30 wt%至約70 wt%,或約40 wt%至約70 wt%,或約50 wt%至約70 wt%)之化合物1形式I,以組合物之重量計約15 wt%至約40 wt% (例如,約20-35 wt%)之實質上非晶形化合物2,且更通常以組合物之重量計25 wt%至約30 wt%之實質上非晶形化合物2,及一或多種賦形劑,例如,約20 wt%至約50 wt%之填充劑;約1 wt%至約5 wt%之崩解劑;約2 wt%至約0.3 wt%之表面活性劑;約0.1 wt%至約5 wt%之黏合劑;約2 wt%至約0.1 wt%之潤滑劑;約2 wt%至約4 wt%之著色劑;及約0.005 wt%至約0.015 wt%之蠟。 在一個實施例中,本發明係顆粒醫藥組合物,其包含: a. 以組合物之重量計約43 wt%之化合物1形式I; b. 以組合物之重量計約34 wt%包含實質上非晶形化合物2之固體分散體; c. 以組合物之重量計約17 wt%之微晶纖維素; d. 以組合物之重量計約2 wt%之交聯羧甲基纖維素鈉; e. 以組合物之重量計約1 wt%之月桂基硫酸鈉;及 f. 以組合物之重量計約3 wt%之聚乙烯吡咯啶酮。 在一個實施例中,本發明係錠劑,其包含: a. 以組合物之重量計約35 wt%之化合物1形式I; b. 以組合物之重量計約28 wt%包含實質上非晶形化合物2之固體分散體; c. 以組合物之重量計約26 wt%之微晶纖維素; d. 以組合物之重量計約6 wt%之交聯羧甲基纖維素鈉; e. 以組合物之重量計約3 wt%之聚乙烯吡咯啶酮; f. 以組合物之重量計約1 wt%之月桂基硫酸鈉;及 g. 以組合物之重量計約1 wt%之硬脂酸鎂。 在一個實施例中,本發明係錠劑,其包含: a. 以組合物之重量計約34 wt%之化合物1形式I; b. 以組合物之重量計約27 wt%包含實質上非晶形化合物2之固體分散體; c. 以組合物之重量計約26 wt%之微晶纖維素; d. 以組合物之重量計約6 wt%之交聯羧甲基纖維素鈉; e. 以組合物之重量計約2 wt%之聚乙烯吡咯啶酮; f. 以組合物之重量計約1 wt%之月桂基硫酸鈉; g. 以組合物之重量計約1 wt%之硬脂酸鎂; h. 以組合物之重量計約3 wt%之著色劑;及 i. 以組合物之重量計約0.010 wt%之蠟。 本發明之另一錠劑包含: a. 約150 mg至250 mg之化合物1形式I; b. 約100 mg至150 mg之實質上非晶形化合物2; c. 約125 mg至175 mg之微晶纖維素; d. 約20 mg至40 mg之交聯羧甲基纖維素鈉; e. 約10 mg至20 mg之聚乙烯吡咯啶酮; f. 約2 mg至6 mg之月桂基硫酸鈉;及 g. 約3 mg至7 mg之硬脂酸鎂。 本發明之另一錠劑包含: a. 約200 mg化合物1形式I; b. 約125 mg實質上非晶形化合物2; c. 約150 mg微晶纖維素; d. 約34 mg交聯羧甲基纖維素鈉; e. 約15 mg聚乙烯吡咯啶酮; f. 約4 mg月桂基硫酸鈉;及 g. 約6 mg硬脂酸鎂。 本發明之另一錠劑包含: a. 約200 mg化合物1形式I; b. 約125 mg實質上非晶形化合物2; c. 約150 mg微晶纖維素; d. 約34 mg交聯羧甲基纖維素鈉; e. 約15 mg聚乙烯吡咯啶酮; f. 約4 mg月桂基硫酸鈉; g. 約6 mg硬脂酸鎂; h. 約17 mg著色劑;及 i. 約0.06 mg蠟。 在一個實施例中,本發明係顆粒醫藥組合物,其包含: a. 以組合物之重量計約38 wt%之化合物1形式I; b. 以組合物之重量計約40 wt%包含實質上非晶形化合物2之固體分散體; c. 以組合物之重量計約16 wt%之微晶纖維素; d. 以組合物之重量計約2 wt%之交聯羧甲基纖維素鈉; e. 以組合物之重量計約1 wt%之月桂基硫酸鈉;及 f. 以組合物之重量計約3 wt%之聚乙烯吡咯啶酮。 在一個實施例中,本發明係錠劑,其包含: a. 以組合物之重量計約31 wt%之化合物1形式I; b. 以組合物之重量計約32 wt%包含實質上非晶形化合物2之固體分散體; c. 以組合物之重量計約26 wt%之微晶纖維素; d. 以組合物之重量計約6 wt%之交聯羧甲基纖維素鈉; e. 以組合物之重量計約3 wt%之聚乙烯吡咯啶酮; f. 以組合物之重量計約1 wt%之月桂基硫酸鈉; g. 以組合物之重量計約1 wt%之硬脂酸鎂;及 h. 以組合物之重量計約3 wt%之著色劑。 本發明之另一錠劑包含: a. 約100 mg至200 mg之化合物1形式I; b. 約100 mg至150 mg之實質上非晶形化合物2; c. 約100 mg至150 mg之微晶纖維素; d. 約20 mg至40 mg之交聯羧甲基纖維素鈉; e. 約10 mg至20 mg之聚乙烯吡咯啶酮; f. 約2 mg至6 mg之月桂基硫酸鈉;及 g. 約3 mg至7 mg之硬脂酸鎂。 本發明之另一錠劑包含: a. 約150 mg化合物1形式I; b. 約125 mg實質上非晶形化合物2; c. 約129 mg微晶纖維素; d. 約29 mg交聯羧甲基纖維素鈉; e. 約13 mg聚乙烯吡咯啶酮; f. 約4 mg月桂基硫酸鈉; g. 約5 mg硬脂酸鎂;及 h. 約15 mg著色劑。 本發明醫藥組合物可處理成適於經口投與之錠劑形式、膠囊形式、袋形式、菱形錠劑形式或其他固體形式。因此,在一些實施例中,醫藥組合物呈錠劑形式。 本發明之另一個態樣提供由錠劑組成之醫藥調配物,該錠劑包括化合物1形式I、包含實質上非晶形化合物2之固體分散體及賦形劑(例如,填充劑、崩解劑、表面活性劑、黏合劑、著色劑、潤滑劑或其任一組合),其每一者闡述於上文及下文實例中,其中錠劑在約30分鐘內之溶解率為至少約50% (例如,至少約60%、至少約70%、至少約80%、至少約90%或至少約99%)。 在一個實例中,醫藥組合物由錠劑組成,該錠劑包括化合物1形式I,其量在25 mg至400 mg範圍內,例如,25 mg,或50 mg,或75 mg,或100 mg,或150 mg、200 mg、250 mg、300 mg,或400 mg;實質上非晶形化合物2,其量在25 mg至250 mg範圍內,例如,25 mg,或50 mg,或75 mg,或100 mg,或150 mg、200 mg、250 mg;及一或多種賦形劑(例如,填充劑、崩解劑、表面活性劑、黏合劑、著色劑、潤滑劑,或其任一組合),其每一者闡述於上文及下文實例中,其中該錠劑在約30分鐘內之溶解率為約50%至約100% (例如,約55%至約95%或約60%至約90%)。 溶解率可利用標準USP II型裝置在約37℃之溫度下以約50-75 rpm攪拌來量測,該裝置採用0.1% CTAB溶解於900 mL去離子水中之溶解介質(用50 mM磷酸二氫鉀以pH 6.8緩衝)。在裝置之每一測試器皿中測試單一實驗錠劑。溶解率亦可利用標準USP II型裝置在約37℃之溫度下以約65 rpm攪拌來量測,該裝置採用0.7%月桂基硫酸鈉溶解於900 mL 50 mM磷酸鈉緩衝液(pH 6.8)中之溶解介質。在裝置之每一測試器皿中測試單一實驗錠劑。溶解率亦可利用標準USP II型裝置在約37℃之溫度下以約65 rpm攪拌來量測,該裝置採用0.5%月桂基硫酸鈉溶解於900 mL 50 mM磷酸鈉緩衝液(pH 6.8)中之溶解介質。在裝置之每一測試器皿中測試單一實驗錠劑。
製備化合物 1 形式 I 及包含實質上非晶形化合物 2 之固體分散體之方法 化合物 1
使用化合物1作為化合物1形式I之起點且可根據方案1至4藉由使醯氯部分與胺部分偶合來製備。
方案 1. 醯氯部分之合成。 方案1繪示1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷羰基氯之製備,其用於方案3中以產生化合物1之醯胺連接。 起始材料2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-甲酸購自Saltigo (Lanxess公司之分部)。將2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-甲酸中之羧酸部分還原為一級醇,之後使用亞硫醯氯(SOCl
2
)將其轉化為相應氯化物,得到5-(氯甲基)-2,2-二氟苯并[d][1,3]二氧雜環戊烯,隨後使用氰化鈉將其轉化為2-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)乙腈。用鹼及1-溴-2-氯乙烷處理2-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)乙腈,得到1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲腈。使用鹼將1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲腈中之腈部分轉化為羧酸,從而得到1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲酸,使用亞硫醯氯將其轉化為期望之醯氯。
方案 2. 醯氯部分之替代合成。 方案2繪示所需醯氯之替代合成。在鈀觸媒存在下使5-溴甲基-2,2-二氟-1,3-苯并二氧雜環戊烯與氰基乙酸乙酯偶合以形成相應α氰基乙基酯。將酯部分皂化為羧酸,得到氰基乙基化合物。在鹼存在下用1-溴-2-氯乙烷將氰基乙基化合物烷基化,得到氰基環丙基化合物。用鹼處理氰基環丙基化合物,得到羧酸鹽,藉由用酸處理將其轉化為羧酸。然後使用氯化劑(例如亞硫醯氯或諸如此類)將羧酸轉化為醯氯。
方案 3. 胺部分之合成。 方案3繪示所需3-(6-胺基-3-甲基吡啶-2-基)苯甲酸第三丁基酯之製備,其在方案3中與1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷羰基氯偶合,從而得到化合物1。在鈀催化下使2-溴-3-甲基吡啶與3-(第三丁氧基羰基)苯基
酸偶合,得到3-(3-甲基吡啶-2-基)苯甲酸第三丁基酯,隨後將其轉化為期望化合物。
方案 4. 3-(6-(1-(2,2- 二氟苯并 [d][1,3] 二氧雜環戊烯 -5- 基 ) 環丙烷甲醯胺基 )-3- 甲基吡啶 -2- 基 ) 苯甲酸之酸式鹽之形成。 方案4繪示使用三乙胺及4-二甲基胺基吡啶使1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷羰基氯與3-(6-胺基-3-甲基吡啶-2-基)苯甲酸第三丁基酯偶合,以初步得到化合物1之第三丁基酯。
化合物 1 形式 I
化合物1形式I係藉由將化合物1之鹽形式(例如HCl鹽)於合適溶劑中分散或溶解有效時間長度來製備。用諸如HCl等酸處理第三丁基酯,得到化合物1之HCL鹽,其通常為結晶固體。化合物1形式I亦可藉由用合適酸(例如甲酸)處理自第三丁基酯前體直接製備。 可使用3-(6-(1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲醯胺基)-3-甲基吡啶-2-基)苯甲酸之HCl鹽,藉由將3-(6-(1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲醯胺基)-3-甲基吡啶-2-基)苯甲酸之HCl鹽於合適溶劑中分散或溶解有效時間長度來製備形式I。可使用3-(6-(1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲醯胺基)-3-甲基吡啶-2-基)苯甲酸之其他鹽,例如,自其他礦物酸或有機酸衍生之鹽。可自第三丁基酯部分之酸介導之水解獲得其他鹽。衍生自其他酸之鹽可包括(例如)硝酸鹽、硫酸鹽、磷酸鹽、硼酸鹽、乙酸鹽、苯甲酸鹽及丙二酸鹽。端視所使用溶劑而定,3-(6-(1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲醯胺基)-3-甲基吡啶-2-基)苯甲酸之該等鹽形式可溶或不可溶,但缺少溶解性並不阻礙化合物1形式I之形成。例如,在一個實施例中,合適溶劑可係水或醇/水混合物,例如50%甲醇/水混合物,即使3-(6-(1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲醯胺基)-3-甲基吡啶-2-基)苯甲酸之HCl鹽形式僅微溶解於水中。在一個實施例中,合適溶劑為水。 自3-(6-(1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲醯胺基)-3-甲基吡啶-2-基)苯甲酸之鹽形成化合物1形式I的有效時間長度可介於2小時至24小時或更長之間之任一時間。應瞭解,所需時間長度與溫度成反比。亦即,溫度愈高,達成酸解離以形成化合物1形式I所需時間愈短。在溶劑為水時,將分散液在室溫下攪拌大約24小時可以大約98%之產率得到化合物1形式I。若出於處理目的需要3-(6-(1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲醯胺基)-3-甲基吡啶-2-基)苯甲酸鹽之溶液,則可使用升高溫度。在升高溫度下將溶液攪拌有效時間長度,之後使其冷卻並重結晶從而得到實質上純淨之化合物1形式I。在一個實施例中,實質上純淨係指純度大於約90%。在另一實施例中,實質上純淨係指純度大於約95%。在另一實施例中,實質上純淨係指純度大於約98%。在另一實施例中,實質上純淨係指純度大於約99%。所選溫度部分地取決於所用溶劑,且熟習此項技術者能夠確定。在一個實施例中,溫度介於室溫與約80℃之間。在另一實施例中,溫度介於室溫與約40℃之間。在另一實施例中,溫度介於約40℃與約60℃之間。在另一實施例中,溫度介於約60℃與約80℃之間。 亦可自3-(6-(1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲醯胺基)-3-甲基吡啶-2-基)-苯甲酸第三丁基酯(參照方案3) (其係化合物1之鹽之前體)直接形成化合物1形式I。因此,使3-(6-(1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲醯胺基)-3-甲基吡啶-2-基)-苯甲酸第三丁基酯經受在合適反應條件下與合適酸(例如,甲酸)之反應,從而得到化合物1形式I。 可藉由自有機溶劑重結晶來進一步純化化合物1形式I。有機溶劑之實例包括(但不限於)甲苯、異丙基苯、苯甲醚、1-丁醇、乙酸異丙酯、乙酸丁酯、乙酸異丁酯、甲基第三丁基醚、甲基異丁酮及1-丙醇-水混合物。溫度可如上所述。例如,在75℃下將化合物1形式I溶解於1-丁醇中直至其完全溶解。以0.2℃/min之速率將溶液冷卻至10℃,從而得到化合物1形式I之晶體,其可藉由過濾加以分離。 在一個實施例中,化合物1形式I之特徵在於在使用Cu K α輻射獲得之X射線粉末繞射中,在15.2度至15.6度、16.1度至16.5度及14.3度至14.7度處具有一或多個峰。在另一實施例中,化合物1形式I之特徵在於在15.4度、16.3度及14.5度處具有一或多個峰。在另一實施例中,化合物1形式I之特徵另外在於在14.6度至15.0度處具有峰。在另一實施例中,化合物1形式I之特徵另外在於在14.8度處具有峰。在另一實施例中,化合物1形式I之特徵另外在於在17.6度至18.0度處具有峰。在另一實施例中,化合物1形式I之特徵另外在於在17.8度處具有峰。在另一實施例中,化合物1形式I之特徵另外在於在16.4度至16.8度處具有峰。在另一實施例中,化合物1形式I之特徵另外在於在16.4度至16.8度處具有峰。在另一實施例中,化合物1形式I之特徵另外在於在16.6度處具有峰。在另一實施例中,化合物1形式I之特徵另外在於在7.6度至8.0度處具有峰。在另一實施例中,化合物1形式I之特徵另外在於在7.8度處具有峰。在另一實施例中,化合物1形式I之特徵另外在於在25.8度至26.2度處具有峰。在另一實施例中,化合物1形式I之特徵另外在於在26.0度處具有峰。在另一實施例中,化合物1形式I之特徵另外在於在21.4度至21.8度處具有峰。在另一實施例中,化合物1形式I之特徵另外在於在21.6度處具有峰。在另一實施例中,化合物1形式I之特徵另外在於在23.1度至23.5度處具有峰。在另一實施例中,化合物1形式I之特徵另外在於在23.3度處具有峰。在一些實施例中,化合物1形式I之特徵在於實質上類似於圖1之繞射圖案。在一些實施例中,化合物1形式I之特徵在於實質上類似於圖2之繞射圖案。 在一些實施例中,化合物1形式I之D90之粒徑分佈為約82 μm或更小。在一些實施例中,化合物1形式I之D50之粒徑分佈為約30 μm或更小。
化合物 2
化合物2係包含實質上非晶形化合物2之固體分散體之起始點且可藉由根據方案5至7使4-側氧基-二氫喹啉甲酸部分與胺部分偶合來製備。
方案 5 : 4- 側氧基 - 二氫喹啉甲酸部分之合成。 方案 6 : 胺部分之合成。 方案 7 : 4- 側氧基 - 二氫喹啉甲酸部分與胺部分之偶合。 包含實質上非晶形化合物 2 之固體分散體
自化合物2起始,化合物2之非晶型可藉由噴霧乾燥方法來製備。噴霧乾燥係將液體進料轉化成乾燥微粒形式之過程。視情況,可使用二級乾燥過程(例如流化床乾燥或真空乾燥)來將殘餘溶劑減少至醫藥上可接受之含量。通常,噴霧乾燥涉及使高度分散的液體懸浮液或溶液與充足體積之熱空氣接觸來蒸發並乾燥液滴。欲噴霧乾燥之製劑可為任一溶液、粗糙懸浮液、漿液、膠態分散液或膏糊,其可使用所選噴霧乾燥裝置來霧化。在標準程序中,將製劑噴霧至溫熱過濾的空氣流中,該空氣流使溶劑蒸發並將乾燥產物傳送至收集器(例如旋風器)。然後,用溶劑將消耗空氣耗盡,或另一選擇為,將消耗空氣運送至冷凝器以捕獲並潛在地再循環溶劑。可使用市售類型裝置來實施噴霧乾燥。例如,市售噴霧乾燥器係由Buchi有限公司及Niro製造(例如,由Niro製造之PSD系列噴霧乾燥器) (參見US 2004/0105820;US 2003/0144257)。 噴霧乾燥通常採用約3重量%至約30重量% (例如約4重量%至約20重量%、較佳地至少約10重量%)之材料(即,藥物及賦形劑)之固體加載。一般而言,固體加載之上限取決於所得溶液之黏度(例如,泵送之能力)及組份在溶液中之溶解性。通常,溶液之黏度可決定所得粉末產物中粒子之大小。 噴霧乾燥之技術及方法可參見Perry’s Chemical Engineering Handbook,第6版,R. H. Perry, D. W. Green及J. O. Maloney編輯), McGraw-Hill book公司(1984);及Marshall 「Atomization and Spray-Drying」 50, Chem. Eng. Prog. Monogr. Series 2 (1954)。一般而言,在約60℃至約200℃ (例如,約95℃至約185℃,約110℃至約182℃,約96℃至約180℃,例如,約145℃)之入口溫度下實施噴霧乾燥。通常,以約30℃至約90℃ (例如約40℃至約80℃,約45℃至約80℃,例如,約75℃)之出口溫度實施噴霧乾燥。霧化流速通常為約4 kg/h至約12 kg/h,例如,約4.3 kg/h至約10.5 kg/h,例如,約6 kg/h或約10.5 kg/h。進料流速通常為約3 kg/h至約10 kg/h,例如,約3.5 kg/h至約9.0 kg/h,例如,約8 kg/h或約7.1 kg/h。霧化比率通常為約0.3至1.7,例如,約0.5至1.5,例如,約0.8或約1.5。 溶劑之去除可能需要後續乾燥步驟,例如盤式乾燥、流體床乾燥(例如,約室溫至約100℃)、真空乾燥、微波乾燥、旋轉滾筒乾燥或雙錐真空乾燥(例如,約室溫至約200℃)。 在一個實施例中,噴霧乾燥分散體係經流體床乾燥的。 在一個製程中,溶劑包括揮發性溶劑,例如沸點小於約100℃之溶劑。在一些實施例中,溶劑包括溶劑之混合物,例如揮發性溶劑之混合物或揮發性溶劑與非揮發性溶劑之混合物。倘若使用溶劑之混合物,則混合物可包括一或多種非揮發性溶劑,例如,其中非揮發性溶劑係以小於約15% (例如,小於約12%,小於約10%,小於約8%,小於約5%,小於約3%或小於約2%)存於混合物中。 較佳溶劑係化合物2之溶解度為至少約10 mg/ml (例如,至少約15 mg/ml、20 mg/ml、25 mg/ml、30 mg/ml、35 mg/ml、40 mg/ml、45 mg/ml、50 mg/ml或更大)之溶劑。更佳溶劑包括化合物2之溶解度為至少約20 mg/ml者。 可測試之例示性溶劑包括丙酮、環己烷、二氯甲烷、N,N-二甲基乙醯胺(DMA)、N,N-二甲基甲醯胺(DMF)、1,3-二甲基-2-咪唑啉酮(DMI)、二甲基亞碸(DMSO)、二噁烷、乙酸乙酯、乙醚、冰乙酸(HAc)、甲基乙基酮(MEK)、N-甲基-2-吡咯啶酮(NMP)、甲基第三丁基醚(MTBE)、四氫呋喃(THF)、戊烷、乙腈、甲醇、乙醇、異丙醇、乙酸異丙基酯及甲苯。例示性共溶劑包括丙酮/DMSO、丙酮/DMF、丙酮/水、MEK/水、THF/水、二噁烷/水。在兩種溶劑的系統中,溶劑可以約0.1%至約99.9%存在。在一些較佳實施例中,水與丙酮係共溶劑,其中水係以約0.1%至約15% (例如約9%至約11%,例如,約10%)存在。在一些較佳實施例中,水與MEK係共溶劑,其中水係以約0.1%至約15% (例如約9%至約11%,例如,約10%)存在。在一些實施例中,溶劑溶液包括三種溶劑。例如,丙酮及水可與第三溶劑(例如DMA、DMF、DMI、DMSO或HAc)混合。在實質上非晶形化合物2係固體分散體之組份之情形下,較佳溶劑溶解化合物2及聚合物。適宜溶劑包括彼等闡述於上文者,例如,MEK、丙酮、水、甲醇及其混合物。 可修改粒徑及溫度乾燥範圍來製備最佳噴霧乾燥分散體。如熟習此項技術者所應瞭解,小粒徑可改良溶劑去除。然而,申請者已發現較小粒子可產生膨松粒子,其在一些情形下不能提供最佳噴霧乾燥分散體用於諸如製錠等下游製程。在較高溫度下,實質上非晶形化合物2可能發生結晶或化學降解。在較低溫度下,不能去除足夠量之溶劑。本文之方法提供最佳粒徑及最佳乾燥溫度。 一般而言,粒徑使得D10 (μm)小於約5 (例如,小於約4.5,小於約4.0,或小於約3.5),D50 (μm)通常小於約17 (例如,小於約16,小於約15,小於約14,小於約13),且D90 (μm)通常小於約175 (例如,小於約170,小於約170,小於約150,小於約125,小於約100,小於約90,小於約80,小於約70,小於約60,或小於約小於約50)。一般而言,噴霧乾燥粒子之堆密度為約0.08 g/cc至約0.20 g/cc,例如,約0.10至約0.15 g/cc,例如,約0.11 g/cc或約0.14 g/cc。噴霧乾燥粒子之敲緊密度通常在約0.08 g/cc至約0.20 g/cc範圍內,例如,約0.10 g/cc至約0.15 g/cc,例如,對於10次敲打約0.11 g/cc或約0.14 g/cc;0.10 g/cc至約0.25 g/cc,例如,約0.11至約0.21 g/cc,例如,對於500次敲打約0.15 g/cc、約0.19 g/cc或約0.21 g/cc;0.15 g/cc至約0.27 g/cc,例如,約0.18 g/cc至約0.24 g/cc,例如,對於1250次敲打約0.18 g/cc、約0.19 g/cc、約0.20 g/cc或約0.24 g/cc;及0.15 g/cc至約0.27 g/cc,例如,約0.18 g/cc至約0.24 g/cc,例如,對於2500次敲打約0.18 g/cc、約0.21 g/cc、約0.23 g/cc或約0.24 g/cc。
聚合物
本文亦包括包含非晶形化合物2及聚合物(或固態載劑)之噴霧乾燥分散體。例如,化合物2係作為固體非晶形分散體之組份以非晶形化合物形式而存在。固體非晶形分散體通常包括實質上非晶形化合物2及聚合物。例示性聚合物包括纖維質聚合物,例如含有HPMC或HPMCAS及吡咯啶酮之聚合物,例如PVP/VA。在一些實施例中,固體非晶形分散體包括一或多種其他賦形劑,例如表面活性劑。 在一個實施例中,聚合物能夠溶解於水性介質中。聚合物之溶解性可為pH獨立性或pH依賴性。後者包括一或多種腸溶聚合物。術語「腸溶聚合物」係指相對於胃之更酸環境優先在腸之較不酸環境中溶解之聚合物,例如,在酸性水性介質中不溶解但當pH超過5-6時溶解之聚合物。合適聚合物應為化學及生物學惰性。為改良噴霧乾燥分散體之物理穩定性,聚合物之玻璃轉變溫度(T
g
)應儘可能地高。例如,較佳聚合物之玻璃轉變溫度至少等於或大於藥物(即,化合物2)之玻璃轉變溫度。其他較佳聚合物之玻璃轉變溫度在藥物(即,化合物2)之約10℃至約15℃內。聚合物之適宜玻璃轉變溫度之實例包括至少約90℃、至少約95℃、至少約100℃、至少約105℃、至少約110℃、至少約115℃、至少約120℃、至少約125℃、至少約130℃、至少約135℃、至少約140℃、至少約145℃、至少約150℃、至少約155℃、至少約160℃、至少約165℃、至少約170℃,或至少約175℃ (如在乾燥條件下所量測)。不欲受限於理論,據信,根本機制係具有較高T
g
之聚合物在室溫下通常具有較低分子遷移率,此可係使非晶形噴霧乾燥分散體之物理穩定性穩定之至關重要因素。 另外,聚合物之吸濕力應儘可能低,例如,小於約10%。出於在此應用中比較之目的,聚合物或組合物之吸濕力之特徵在於處於約60%相對濕度下。在一些較佳實施例中,聚合物之水吸收小於約10%,例如水吸收小於約9%、小於約8%、小於約7%、小於約6%、小於約5%、小於約4%、小於約3%,或小於約2%。吸濕力亦可影響噴霧乾燥分散體之物理穩定性。通常,吸附於聚合物中之水分可大大降低聚合物以及所得噴霧乾燥分散體之T
g
,從而將進一步降低如上所闡述噴霧乾燥分散體之物理穩定性。 在一個實施例中,聚合物係一或多種水溶性聚合物或部分地水溶性聚合物。水溶性或部分地水溶性 聚合物包括(但不限於)纖維素衍生物(例如,羥基丙基甲基纖維素(HPMC)、羥丙基纖維素(HPC))或乙基纖維素;聚乙烯吡咯啶酮(PVP);聚乙二醇(PEG);聚乙烯醇(PVA);丙烯酸酯,例如聚甲基丙烯酸酯(例如,Eudragit® E);環糊精(例如,β-環糊精)及其共聚物及衍生物,包括(例如) PVP-VA (聚乙烯吡咯啶酮-乙酸乙烯基酯)。 在一些實施例中,聚合物係羥基丙基甲基纖維素(HPMC),例如HPMCAS、HPMC E50、HPMCE15或HPMC60SH50)。 如本文所論述,聚合物可為pH依賴性腸溶聚合物。該等pH依賴性腸溶聚合物包括(但不限於)纖維素衍生物(例如,乙酸鄰苯二甲酸纖維素(CAP))、鄰苯二甲酸羥基丙基甲基纖維素(HPMCP)、琥珀酸羥基丙基甲基乙酸纖維素(HPMCAS)、羧甲基纖維素(CMC)或其鹽(例如,鈉鹽,例如(CMC-Na));乙酸偏苯三酸纖維素(CAT)、乙酸鄰苯二甲酸羥基丙基纖維素(HPCAP)、乙酸鄰苯二甲酸羥基丙基甲基纖維素(HPMCAP)及乙酸鄰苯二甲酸甲基纖維素(MCAP)或聚甲基丙烯酸酯(例如,Eudragit® S)。在一些實施例中,聚合物係乙酸琥珀酸羥基丙基甲基纖維素(HPMCAS)。在一些實施例中,聚合物係HG級乙酸琥珀酸羥基丙基甲基纖維素(HPMCAS-HG)。 在又一實施例中,聚合物係聚乙烯吡咯啶酮共聚物,例如,乙烯基吡咯啶酮/乙酸乙烯基酯共聚物(PVP/VA)。 在化合物2與聚合物(例如與HPMC、HPMCAS或PVP/VA聚合物)形成噴霧乾燥分散體之實施例中,聚合物相對於噴霧乾燥分散體之總重量之量在約0.1重量%至99重量%範圍內。除非另有說明,否則如所闡述藥物、聚合物及其他賦形劑在分散體內之%係以重量%給出。聚合物之量通常為至少約20%,且較佳地為至少約30%,例如,至少約35%、至少約40%、至少約45%或約50% (例如,49.5%)。該量通常為約99%或更少,且較佳地為約80%或更少,例如約75%或更少、約70%或更少、約65%或更少、約60%或更少或約55%或更少。在一個實施例中,聚合物之量佔分散體之總重量為至多約50% (且甚至更特定而言,介於約40%與50%之間,例如約49%、約49.5%或約50%)。HPMC及HPMCAS係以各種等級購自ShinEtsu,例如,HPMCAS係以多種變化形式購得,包括AS-LF、AS-MF、AS-HF、AS-LG、AS-MG、AS-HG。該等等級中之每一者皆隨乙酸酯及琥珀酸酯之取代%而變化。 在一些實施例中,實質上非晶形化合物2及聚合物係以大致相等的量存在,例如聚合物及藥物中之每一者佔分散體之重量%的約一半。例如,聚合物係以約49.5%存在且藥物係以約50%存在。 在一些實施例中,在噴霧乾燥之前,實質上非晶形化合物2及所組合聚合物佔非噴霧乾燥分散體之總固體內容物之1% w/w至20% w/w。在一些實施例中,在噴霧乾燥之前,實質上非晶形化合物2及所組合聚合物佔非噴霧乾燥分散體之總固體內容物之5% w/w至15% w/w。在一些實施例中,在噴霧乾燥之前,實質上非晶形化合物2及所組合聚合物佔非噴霧乾燥分散體之總固體內容物之約11% w/w。 在一些實施例中,分散體進一步包括其他次要成份,例如表面活性劑(例如,SLS)。在一些實施例中,表面活性劑係以佔分散體小於約10% (例如小於約9%、小於約8%、小於約7%、小於約6%、小於約5%、小於約4%、小於約3%、小於約2%、約1%或約0.5%)存在。 在包括聚合物之實施例中,聚合物應以有效使噴霧乾燥分散體穩定之量存在。穩定包括抑制或防止實質上非晶形化合物2之結晶。該穩定可抑制化合物2自非晶形轉化成結晶形式。例如,聚合物可防止至少一部分(例如,約5%、約10%、約15%、約20%、約25%、約30%、約35%、約40%、約45%、約50%、約55%、約60%、約65%、約70%、約75%或更大)化合物2自非晶形轉化成結晶形式。穩定可藉由例如以下來量測:量測噴霧乾燥分散體之玻璃轉變溫度、量測非晶形材料之鬆弛速率,或量測化合物2之溶解性或生物利用度。 適宜與化合物2組合而(例如)形成噴霧乾燥分散體(例如非晶形噴霧乾燥分散體)之聚合物應具有以下性質中之一或多者: 聚合物之玻璃轉變溫度之溫度較實質上非晶形化合物2之玻璃轉變溫度應低不小於約10-15℃。較佳地,聚合物之玻璃轉變溫度大於實質上非晶形化合物2之玻璃轉變溫度,且一般而言較藥物產品之期望儲存溫度高出至少50℃。例如,至少約100℃、至少約105℃、至少約105℃、至少約110℃、至少約120℃、至少約130℃、至少約140℃、至少約150℃、至少約160℃、至少約160℃或更大。 聚合物應相對較不吸濕。例如,聚合物在於標準條件下儲存時吸收小於約10%之水,例如,小於約9%、小於約8%、小於約7%、小於約6%,或小於約5%、小於約4%或小於約3%之水。較佳地,聚合物在於標準條件下儲存時將實質上不含吸收水。 聚合物與化合物2相比應在適於噴霧乾燥過程之溶劑中具有類似或更好的溶解性。在較佳實施例中,聚合物將溶解於與化合物2相同之溶劑或溶劑系統中之一或多者中。較佳地,聚合物溶於至少一種含非羥基之溶劑(例如二氯甲烷、丙酮或其組合)中。 與化合物2在不存在聚合物下之溶解性相比,或與化合物2在與參考聚合物組合時之溶解性相比,聚合物在與實質上非晶形化合物2組合於(例如)噴霧乾燥分散體或液體懸浮液中時可增加化合物2在水性及生理上相關介質中之溶解性。例如,聚合物可藉由使自固體非晶形分散體或自液體懸浮液轉化成結晶化合物2之非晶形化合物2之量降低而增加非晶形化合物2之溶解性。 聚合物應使非晶形物質之鬆弛速率降低。 聚合物應使實質上非晶形化合物2之物理及/或化學穩定性增加。 聚合物應改良實質上非晶形化合物2之可製造性。 聚合物應改良實質上非晶形化合物2之處置、投與或儲存性質中之一或多者。 聚合物不應與其他醫藥組份(例如賦形劑)發生不利相互作用。 可使用本文闡述之噴霧乾燥方法(或其他方法)來測試候選聚合物(或其他組份)形成非晶形組合物之適宜性。候選組合物可在穩定性、耐形成晶體性或其他性質方面進行比較,且與參考製劑(例如,純淨非晶形化合物2或結晶化合物2之製劑)進行比較。例如,候選組合物可經測試來確定其是否抑制溶劑介導結晶之起始時間,或測定在既定時間在受控條件下之轉化率%,例如至少50%、75%、100%或110%,亦如此測試參考製劑,或候選組合物可經測試以確定其與結晶化合物2相比是否改良生物利用度或溶解性。
表面活性劑
噴霧乾燥分散體可包括表面活性劑。表面活性劑或表面活性劑混合物可通常使噴霧乾燥分散體與水性介質之間之界面張力降低。合適表面活性劑或表面活性劑混合物亦可增強來自噴霧乾燥分散體之化合物2之水性溶解性及生物利用度。結合本發明使用之表面活性劑包括(但不限於)山梨醇酐脂肪酸酯(例如,Spans®)、聚氧乙烯山梨醇酐脂肪酸酯(例如,Tweens®)、月桂基硫酸鈉(SLS)、十二烷基苯磺酸鈉(SDBS)、磺琥珀酸鈉二辛酯(多庫酯鈉(Docusate))、脫氧膽酸鈉鹽(DOSS)、山梨醇酐單硬脂酸酯、山梨醇酐三硬脂酸酯、十六烷基三甲基溴化銨(HTAB)、N-月桂醯基肌胺酸鈉、油酸鈉、肉豆蔻酸鈉、硬脂酸鈉、棕櫚酸鈉、Gelucire 44/14、乙二胺四乙酸(EDTA)、維生素E d-α生育酚聚乙二醇1000琥珀酸酯(TPGS)、卵磷脂、MW 677-692、麩胺酸單鈉單水合物、Labrasol、PEG 8辛酸/癸酸甘油酯、Transcutol、二乙二醇單乙基醚、Solutol HS-15、聚乙二醇/羥基硬脂酸酯、牛膽酸、Pluronic F68、Pluronic F108及Pluronic F127 (或任何其他聚氧乙烯聚氧丙烯共聚物(Pluronics®)或飽和聚乙二醇化甘油酯(Gelucirs®))。可結合本發明使用之該等表面活性劑之特定實例包括(但不限於) Span 65、Span 25、Tween 20、Capryol 90、Pluronic F108、月桂基硫酸鈉(SLS)、維生素E TPGS、pluronics及共聚物。SLS通常較佳。 表面活性劑(例如,SLS)相對於噴霧乾燥分散體之總重量之量可介於0.1%至15%之間。較佳地,其為約0.5%至約10%,更佳地約0.5%至約5%,例如,約0.5%至4%、約0.5%至3%、約0.5%至2%、約0.5%至1%或約0.5%。 在某些實施例中,表面活性劑相對於噴霧乾燥分散體之總重量之量為至少約0.1%,較佳地為約0.5%。在該等實施例中,表面活性劑可以不超過約15%且較佳地不超過約12%、約11%、約10%、約9%、約8%、約7%、約6%、約5%、約4%、約3%、約2%或約1%之量存在。表面活性劑之量為約0.5重量%之實施例較佳。 可以與針對測試聚合物所闡述類似之方式測試候選表面活性劑(或其他組份)於本發明中使用之適宜性。
製備醫藥組合物之方法
可藉由濕法製粒、在壓力下壓實或壓製混合物或組合物(例如,粉末或顆粒)以形成穩定三維形狀(例如,錠劑)來產生本發明之醫藥組合物。如本文所使用,「錠劑」包括所有形狀及大小之經壓製醫藥劑量單位形式,無論包覆與否。 如本文所使用,術語「錠劑」係指適於欲治療病患之藥劑之物理離散單元。一般而言,經壓實混合物之密度大於該混合物在壓實前之密度。本發明之劑量錠劑幾乎可具有任何形狀,包括凹面及/或凸面、圓角或有角角及約為直線之形狀。在一些實施例中,本發明之經壓製錠劑包含具有扁平面之圓形錠劑。可藉由為熟習形成經壓製固體醫藥劑型之技術者已知之任一壓實及壓製方法製備本發明之錠劑。在具體實施例中,可使用彼等熟習醫藥調配物領域者已知之習用方法來製備本文所提供調配物,如(例如)相關教科書中所闡述。例如,參見Remington: The Science and Practice of Pharmacy,第21版,Lippincott Williams & Wilkins, Baltimore, Md. (2003);Ansel等人,Pharmaceutical Dosage Forms And Drug Delivery Systems,第7版,Lippincott Williams & Wilkins, (1999);The Handbook of Pharmaceutical Excipients,第4版,Rowe等人編輯,American Pharmaceuticals Association (2003);Gibson, Pharmaceutical Preformulation And Formulation,CRC Press (2001),該等參考文獻以全文引用方式併入本文中。
製粒及壓製
在一些實施例中,根據本文所述配方稱量各成份。接下來,對所有顆粒內成份實施篩分並將其充分混合。可用適宜潤滑劑(例如,硬脂酸鎂)潤滑成份。下一步驟可包含壓實/擊壓粉末混合物及定大小之成份。接下來,將經壓實或擊壓摻合物研磨成顆粒並篩分以獲得期望大小。接下來,可用(例如)硬脂酸鎂進一步潤滑顆粒。接下來,可在適宜衝壓器上將本發明顆粒組合物壓製成本發明之各種醫藥調配物。視情況,錠劑可經薄膜、著色劑或其他包衣包覆。 本發明之另一態樣提供產生醫藥組合物之方法,其包含提供包含以下之組合物之混合物:化合物1形式I、包含實質上非晶形化合物2之固體分散體及一或多種選自填充劑、稀釋劑、黏合劑、表面活性劑、潤滑劑、崩解劑之賦形劑,及將該組合物壓製成在約30分鐘內之溶解率為至少約50%之錠劑。 在另一實施例中,對粉末及液體成份之混合物實施濕法製粒製程以得到本發明醫藥調配物。例如,根據本文所述配方稱量包含包括以下之組合物之混合物之醫藥組合物:化合物1形式I、包含實質上非晶形化合物2之固體分散體之組合物,及一或多種選自填充劑、黏合劑、表面活性劑或崩解劑之賦形劑。接下來,在高剪切或低剪切製粒機中使用水或具有表面活性劑之水或具有黏合劑之水或具有表面活性劑及黏合劑之水對所有顆粒內成份實施篩分並混合,以製粒粉末摻合物。非水流體亦可與表面活性劑及/或黏合劑一起使用或不與其一起使用,以製粒粉末摻合物。接下來,可視情況使用適宜研磨機研磨濕顆粒。隨後,可視情況藉由以任一適宜方式乾燥成份自混合物去除水。接下來,可視情況將乾燥顆粒研磨成所需大小。接下來,可藉由摻合添加顆粒外賦形劑(例如填充劑、稀釋劑及崩解劑)。接下來,可用硬脂酸鎂及崩解劑(例如,交聯羧甲基纖維素鈉)進一步潤滑定大小之顆粒。接下來,可將本發明之顆粒組合物篩分足夠時間以獲得正確大小,且然後在適宜衝壓器上壓製成本發明之各種醫藥調配物。視情況,錠劑可經薄膜、著色劑或其他包衣包覆。令人驚訝地,可實施濕法製粒且實質上不損失化合物1形式I或實質上非晶形化合物2之固態形式。 在尤其有利的實施例中,本發明醫藥組合物係藉由連續雙螺桿濕法製粒(TSWG)製程來製備。連續製造在線上監測及控制下產生高品質且高度一致的產品。連續製造亦因設計研發利用「富數據」設計空間且更易於瞭解上游變量對下游製程及最終產品品質之影響而有利於品質。此外,本發明醫藥組合物可早在商業規模設備上完結,從而避免在研發晚期擴大規模的風險及調配物變化。最後,連續製造具有商業製造優點,例如製程控制有所改良、產品處置有所減少及實時釋放效率。總體而言,產生具有較少製程檢查從而提高產品品質且因此提高患者安全性之更穩健、可控且規模可變的製程。該等優點解決了Janet Woodcock (藥物評估與研究中心(Center for Drug Evaluation and Research,CDER)的主任)對化學與製造控制不能趕上高度有效療法之迅速臨床研發之擔憂(「What we are seeing is that often the rate limiting step is going to be manufacturing」,2013年7月24日,癌症之友(Friends of Cancer)主持的會議簡要「Answering a Compelling Need: Expediting Life-Saving Treatments to Patients」,用以論述美國食品藥物管理局之突破性療法認定)。 例如,高剪切製粒(HSG)係常用製粒技術,眾所周知其具有過度製粒及差製程控制之風險。對於此製程擴大規模極具挑戰性且涉及顯著風險。從HSG製程變化為連續TSWG製程允許使用相同設備擴大規模以藉由運行更長時間來產生不同批量。此消除其他製粒製程所通常遇到之擴大規模的風險。另外,發現TSWG製程更為穩健,較不易於過度製粒。如在針對化合物1錠劑之圖3中可看出,HSG製程顯示溶解率隨水含量增加而顯著降低,而TSWG製程對於水添加之類似範圍未顯示變化。令人驚訝地發現,使用雙螺桿濕法製粒製程時包含介於45重量%至55重量%間之化合物1之錠劑調配物及包含介於60重量%至70重量%間之化合物1之錠劑調配物沒有性能變化。此與HSG製程之情形不同。另外,此連續且產品品質有所提高之製程解決了FDA關於有需要之患者缺乏藥物可用性之普遍抱怨。 在一個實施例中,連續製程起始於藉助失重式(loss-in-weight)進給將個別賦形劑、化合物1及化合物2進給至連續在線摻合機。自此摻合機連續傳送材料並藉助雙螺桿濕法製粒進行處理,乾燥,研磨,添加顆粒外賦形劑,摻合,壓製並包覆薄膜。 例如,在一個實施例中,可根據下文流程圖連續製備包含化合物1及化合物2之錠劑。

。 此例示性混合物之每一成份闡述於上文及下文實例中。此外,混合物可包含可選添加劑,例如一或多種著色劑、一或多種矯味劑及/或一或多種芳香劑,如上文及下文實例中所闡述。在一些實施例中,該等成份(及任何可選添加劑)中之每一者在混合物中之相對濃度(例如,wt%)亦呈現於上文及下文實例中。構成混合物之成份可依序或以添加之任一組合提供;且成份或成份之組合可以任一順序提供。在一個實施例中,潤滑劑係添加至混合物中之最後組份。 在另一實施例中,混合物包含化合物1形式I、實質上非晶形化合物2之固體分散體及任一或多種以下賦形劑之組合物:黏合劑、表面活性劑、稀釋劑、潤滑劑、崩解劑及填充劑,其中該等成份中之每一者係以粉末形式提供(例如,以平均(mean或average)直徑(藉由光散射量測)為250 μm或更小(例如,150 μm或更小、100 μm或更小、50 μm或更小、45 μm或更小、40 μm或更小或35 μm或更小)之粒子提供)。 在另一實施例中,將混合物壓製成錠劑係藉由用混合物填充模板(例如,模具)並對混合物施加壓力來達成。此可使用模壓機或其他類似裝置來達成。在一些實施例中,可首先將化合物1形式I、包含實質上非晶形化合物2之固體分散體及賦形劑之混合物處理成顆粒形式。然後,可根據醫藥技術內已知之方法將顆粒定大小並壓製成錠劑或經調配用於囊封。亦應注意,將壓力施加至模板中之混合物可在每一壓製期間使用相同壓力或在壓製期間使用不同壓力重複進行。在另一實例中,可使用施加足夠壓力之模壓機來壓製粉末成份或顆粒之混合物,以形成溶解率在約30分鐘下為約50%或更大(例如,在約30分鐘下為約55%或更大或在約30分鐘下為約60%或更大)之錠劑。例如,使用模壓機壓製混合物以產生硬度為至少約5 kP (至少約5.5 kP、至少約6 kP、至少約7 kP、至少約10 kP或至少15 kP)之錠劑。在一些情形下,壓製混合物以產生硬度介於約5 kP與20 kP之間之錠劑。 在一些實施例中,包含本文所述醫藥組合物之錠劑可經以錠劑重量計約3.0 wt%包含著色劑之薄膜包衣包覆。在某些情形下,用於包覆錠劑之著色劑懸浮液或溶液包含以著色劑懸浮液或溶液之重量計約20%w/w之固體。在其他情形下,經包覆錠劑可標記有標誌、其他影像或文字。 在另一實施例中,產生醫藥組合物之方法包含提供固體形式之混合物,例如粉末及/或液體成份之混合物,該混合物包含化合物1形式I、包含實質上非晶形化合物2之固體分散體及一或多種選自以下之賦形劑:黏合劑、稀釋劑、表面活性劑、潤滑劑、崩解劑及填充劑;混合該混合物直至混合物實質上均勻,及將混合物壓製或壓實成顆粒形式。然後,可將包含化合物1形式I及包含實質上非晶形化合物2之固體分散體之顆粒組合物壓製成錠劑或調配成膠囊,如上文及下文實例所闡述。另一選擇為,產生醫藥組合物之方法包含提供化合物1形式I、包含實質上非晶形化合物2之固體分散體及一或多種賦形劑(例如黏合劑、稀釋劑、表面活性劑、潤滑劑、崩解劑及填充劑)之混合物;混合該混合物直至混合物實質上均勻,及使用高剪切濕法顆粒壓實製程將該混合物壓製/壓實成顆粒形式,如下文實例所述。本文所闡述之醫藥調配物(例如錠劑)可使用除本文所闡述之所選賦形劑以外納入化合物1形式I及包含實質上非晶形化合物2之固體分散體製得之顆粒來製備。 在一些實施例中,可藉由使用手動混合、混合器、摻合機、其任一組合或諸如此類實施攪拌、摻合、振盪或諸如此類來混合混合物。當依序添加成份或成份之組合時,混合可在連續添加之間進行,在整個成份添加期間持續進行,在添加所有成份或成份之組合後進行,或其任一組合。對混合物實施混合直至其具有實質上均勻之組成。 在另一實施例中,本發明包含在適宜的習用研磨裝置中使用適於產生大部分粒徑介於0.1微米與50微米之間之粒子之空氣壓力來噴射研磨包含化合物1形式I及包含實質上非晶形化合物2之固體分散體之醫藥組合物。在另一實施例中,粒徑介於0.1微米與20微米之間。在另一實施例中,粒徑介於0.1微米與10微米之間。在另一實施例中,粒徑介於1.0微米與5微米之間。在再一實施例中,醫藥組合物之粒徑D50為2.0微米。 本發明調配物提供固定劑量之兩種API用於有效治療囊性纖維化,其係接受來自FDA之僅兩個突破性療法認定中之一者之組合,且與此同時具有令人驚訝的穩定性(如藉由化合物2之非晶形固體形式之少量損失所量測)。圖4繪示化合物2在PC-XVII中在60%相對濕度下預平衡後在50℃下隨時間之少量結晶度。即使在該等條件下接近1000小時後,亦只有小於5重量%之化合物2結晶。圖5顯示,PC-XVII在60%相對濕度下預平衡後在60℃之較高溫度下,在該等條件下接近1000小時時,亦只有小於10重量%之化合物2結晶。圖6及圖7針對PC-XX顯示類似結果。因此,本發明調配物便於在令人驚訝地穩定的醫藥組合物中提供固定劑量之兩種突破性API。該等調配物增加患者順從性,此與疾病之有效治療直接相關。 可根據Test 711 「Dissolution」,美國藥典29, United States Pharmacopeial Convention公司,Rockville, Md., 2005 (「USP」)對如上文所製備之劑型實施活體外溶解率評估,以測定活性物質自劑型釋放之速率。藉由諸如高效液相層析(HPLC)等技術來方便地量測活性物質之含量及雜質含量。 在一些實施例中,本發明包括包裝材料之應用,例如高密度聚乙烯(HDPE)、低密度聚乙烯(LDPE)及或聚丙烯之容器及封閉物及/或玻璃、玻璃紙箔、鋁袋、及由鋁或高密度聚氯乙烯(PVC)構成之泡鼓或條帶,其視情況包括乾燥劑、聚乙烯(PE)、聚二氯亞乙烯(PVDC)、PVC/PE/PVDC及諸如此類。在使用醫藥技術中常用之化學或物理滅菌技術對包裝及其內含物進行適當滅菌後,該等包裝材料可用於以無菌方式儲存各種醫藥組合物及調配物。
投與醫藥組合物之方法
在一個態樣中,可每天或約每24小時一次將本發明醫藥組合物投與患者。另一選擇為,可每天兩次將本發明醫藥組合物投與患者。另一選擇為,可約每十二小時投與本發明醫藥組合物。該等醫藥組合物係作為含有以下之口服調配物投與:約25 mg、50 mg、100 mg、125 mg、150 mg、200 mg、250 mg、300 mg或400 mg化合物1形式I;及約25 mg、50 mg、100 mg、125 mg、150 mg、200 mg或250 mg實質上非晶形化合物2。在此態樣中,除化合物1形式I及實質上非晶形化合物2以外,醫藥組合物包含填充劑、崩解劑、表面活性劑、黏合劑及潤滑劑(端視醫藥組合物為顆粒抑或錠劑而定)。例如,400 mg劑量之化合物1形式I可包含本發明之兩個各自含有200 mg化合物1形式I之錠劑。250 mg劑量之實質上非晶形化合物2可包含本發明之兩個各自含有125 mg實質上非晶形化合物2之錠劑。 亦應瞭解,本發明之化合物及醫藥上可接受之組合物及調配物可用於組合療法中,即,化合物1形式I及實質上非晶形化合物2之固體分散體及其醫藥上可接受之組合物可與一種或多種其他期望治療劑或醫療程序同時、在其之前或在其之後投與。 在一個實施例中,其他治療劑係選自溶黏蛋白劑、枝氣管擴張藥、抗生素、抗感染劑、消炎劑、除本發明化合物1形式I及實質上非晶形化合物2外亦包括CFTR活性劑之化合物或營養劑。 在一個實施例中,其他藥劑係(
R
)-1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)-N-(1-(2,3-二羥基丙基)-6-氟-2-(1-羥基-2-甲基丙-2-基)-1H-吲哚-5-基)環丙烷甲醯胺。在另一實施例中,其他藥劑係4-(3-(1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲醯胺基)異喹啉-1-基)苯甲酸。在另一實施例中,其他藥劑係選自表1:
表 1. 。 在另一實施例中,其他藥劑係上述藥劑之任一組合。例如,該組合可包含包括化合物1形式I及實質上非晶形化合物2之固體分散體之本發明醫藥組合物或錠劑,且其他治療劑係(
R
)-1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)-N-(1-(2,3-二羥基丙基)-6-氟-2-(1-羥基-2-甲基丙-2-基)-1H-吲哚-5-基)環丙烷甲醯胺。在另一實例中,該組合可包含包括化合物1形式I及實質上非晶形化合物2之固體分散體之本發明醫藥組合物或錠劑,且其他治療劑係4-(3-(1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲醯胺基)異喹啉-1-基)苯甲酸。在另一實例中,該組合可包含包括化合物1形式I及實質上非晶形化合物2之固體分散體之本發明醫藥組合物或錠劑,且其他治療劑係來自表1之化合物中之任一者,即表1之化合物1至14,或其任一組合。 在另一實施例中,其他藥劑係選自表1:
在另一實施例中,其他藥劑係選自表2:
在一個實施例中,其他治療劑係抗生素。本文所用例示性抗生素包括妥布黴素(tobramycin) (包括妥布黴素吸入式粉劑(TIP))、阿奇黴素(azithromycin)、胺曲南吸入劑(cayston)、胺曲南(aztreonam) (包括胺曲南之氣溶膠化形式)、阿米卡星(amikacin) (包括其脂質體調配物)、環丙沙星(ciprofloxacin) (包括其適於藉由吸入投與之調配物)、左氧氟沙星(levoflaxacin) (包括其氣溶膠化調配物),及兩種抗生素之組合(例如,磷黴素(fosfomycin)與妥布黴素)。 在另一實施例中,其他藥劑係溶黏蛋白劑。本文所用例示性溶黏蛋白劑包括Pulmozyme®。 在另一實施例中,其他藥劑係枝氣管擴張藥。例示性枝氣管擴張藥包括沙丁胺醇(albuterol)、硫酸奧西那林(metaproterenol sulfate)、乙酸吡布特羅(pirbuterol acetate)、沙美特羅(salmeterol)或硫酸特他布林(tetrabuline sulfate)。 在另一實施例中,其他藥劑可有效恢復肺氣道表面液體。該等藥劑可改良細胞內外鹽之移動,從而提高肺氣道中之黏液之水合程度並由此更易於清除。該等例示性藥劑包括高滲生理鹽水、地紐福索四鈉(denufosol tetrasodium) (([[(3S,5R)-5-(4-胺基-2-側氧基嘧啶-1-基)-3-羥基四氫呋喃-2-基]甲氧基-羥基磷醯基] [[[(2R,3S,4R,5R)-5-(2,4-二側氧基嘧啶-1-基)-3,4-二羥基四氫呋喃-2-基]甲氧基-羥基磷醯基]氧基-羥基磷醯基]磷酸氫酯)或布若尼妥(bronchitol) (甘露醇之吸入式調配物)。 在另一實施例中,其他藥劑係消炎藥,即,可減小肺中炎症之藥劑。本文所用該等例示性藥劑包括布洛芬(ibuprofen)、二十二碳六烯酸(DHA)、西地那非(sildenafil)、吸入式麩胱甘肽、吡格列酮(pioglitazone)、羥氯喹(hydroxychloroquine)或斯伐他汀(simavastatin)。 在另一實施例中,其他藥劑係除化合物1形式I或包含實質上非晶形化合物2之固體分散體以外加強或誘導CFTR活性之化合物,即,具有誘導或加強CFTR活性之效應之藥劑。該等例示性藥劑包括阿塔魯仁(ataluren) (「PTC124®」;3-[5-(2-氟苯基)-1,2,4-噁二唑-3-基]苯甲酸)、西那普肽(sinapultide)、蘭考韋泰(lancovutide)、地來司他(depelestat) (人類重組嗜中性白血球彈性蛋白酶抑制劑)及考前列酮(cobiprostone) (7-{(2R,4aR,5R,7aR)-2-[(3S)-1,1-二氟-3-甲基戊基]-2-羥基-6-側氧基八氫環戊[b]吡喃-5-基}庚酸)。 在另一實施例中,其他藥劑係營養劑。例示性營養劑包括胰脂肪酶(代替胰臟酶),包括Pancrease®、Pancreacarb®、Ultrase®或Creon®、Liprotomase®(先前之Trizytek®)、Aquadeks®或吸入式麩胱甘肽。在一個實施例中,其他營養劑係胰脂肪酶。 在另一實施例中,其他藥劑係選自以下之化合物:慶大黴素(gentamicin)、薑黃素(curcumin)、環磷醯胺、4-苯基丁酸酯、麥格司他(miglustat)、非洛地平(felodipine)、尼莫地平(nimodipine)、根皮紅B (Philoxin B)、染料木黃酮(geniestein)、芹菜素(Apigenin)、cAMP/cGMP加強劑或誘導劑(例如咯利普蘭(rolipram)、西地那非、米力農(milrinone)、他達拉非(tadalafil)、胺力農(amrinone)、異丙腎上腺素(isoproterenol)、沙丁胺醇及沙美特羅、去氧精胍菌素(deoxyspergualin)、HSP 90抑制劑、HSP 70抑制劑、蛋白酶體抑制劑(例如環氧甲酮四肽蛋白酶體抑制劑(epoxomicin)、乳胞素(lactacystin))等。 在另一實施例中,其他藥劑係選自以下之化合物:3-胺基-6-(4-氟-苯基)-5-三氟甲基-吡啶-2-甲酸(3,3,3-三氟-2-羥基-2-甲基-丙基)-醯胺;5-胺基-6′-甲基-3-三氟甲基-[2,3]聯吡啶基-6-甲酸(3,3,3-三氟-2-羥基-2-甲基-丙基)-醯胺;3-胺基-6-環丙基-N-(3,3,3-三氟-2-羥基-2-甲基丙基)-5-(三氟甲基)甲吡啶醯胺;3-胺基-6-甲氧基-N-(3,3,3-三氟-2-羥基-2-(三氟甲基)丙基)-5-(三氟甲基)甲吡啶醯胺;3-胺基-6-(4-氟-苯基)-5-三氟甲基-吡啶-2-甲酸((S)-3,3,3-三氟-2-羥基-2-甲基-丙基)-醯胺;3-胺基-6-甲氧基-5-三氟甲基-吡啶-2-甲酸((S-3,3,3-三氟-2-羥基-2-甲基-丙基)-醯胺;3-胺基-6-甲氧基-5-三氟甲基-吡啶-2-甲酸((R)-3,3,3-三氟-2-羥基-2-甲基-丙基)-醯胺;3-胺基-6-(2,4-二氯-苯基)-5-三氟甲基-吡啶-2-甲酸((S)-3,3,3-三氟-2-羥基-2-甲基-丙基)-醯胺;3-胺基-6-(2,4-二氯-苯基)-5-三氟甲基-吡啶-2-甲酸((R)-3,3,3-三氟-2-羥基-2-甲基-丙基)-醯胺;3-胺基-6-(4-氟-苯基)-5-三氟甲基-吡啶-2-甲酸(2-羥基-2-甲基-丙基)-醯胺;3-胺基-5,6-雙-三氟甲基-吡啶-2-甲酸((S)-3,3,3-三氟-2-羥基-2-甲基-丙基)-醯胺;3-胺基-5,6-雙-三氟甲基-吡啶-2-甲酸((R)-3,3,3-三氟-2-羥基-2-甲基-丙基)-醯胺;(S)-3-胺基-6-乙氧基-N-(3,3,3-三氟-2-羥基-2-甲基丙基)-5-(三氟甲基)甲吡啶醯胺;3-胺基-6-甲氧基-5-三氟甲基-吡啶-2-甲酸((S)-3,3,3-三氟-2-羥基-2-甲基-丙基)-醯胺;3-胺基-6-甲氧基-5-三氟甲基-吡啶-2-甲酸((R)-3,3,3-三氟-2-羥基-2-甲基-丙基)-醯胺;3-胺基-6-(4-氟-苯基)-5-三氟甲基-吡啶-2-甲酸(3,3,3-三氟-2-羥基-2-甲基-丙基)-醯胺;3-胺基-5,6-雙-三氟甲基-吡啶-2-甲酸((S)-3,3,3-三氟-2-羥基-2-甲基-丙基)-醯胺;3-胺基-5,6-雙-三氟甲基-吡啶-2-甲酸((R)-3,3,3-三氟-2-羥基-2-甲基-丙基)-醯胺,或其醫藥上可接受之鹽。在另一實施例中,其他藥劑係揭示於美國專利第8,247,436號及國際PCT公開案WO 2011113894中之化合物,該等案件係以全文引用方式併入本文中。 在另一實施例中,其他藥劑可為揭示於PCT公開案WO2012035158、WO2009074575、WO2011028740、WO2009150137、WO2011079087或WO2008135557中之上皮鈉通道(ENac)調節劑,該等案件係以全文引用方式併入本文中。 在其他實施例中,其他藥劑係揭示於WO 2004028480、WO 2004110352、WO 2005094374、WO 2005120497或WO 2006101740中之化合物。在另一實施例中,其他藥劑係展現CFTR誘導或加強活性之苯并[c]喹嗪鎓衍生物或展現CFTR誘導或加強活性之苯并吡喃衍生物。在另一實施例中,其他藥劑係揭示於美國專利第7,202,262號、美國專利第6,992,096號、US20060148864、US20060148863、US20060035943、US20050164973、WO2006110483、WO2006044456、WO2006044682、WO2006044505、WO2006044503、WO2006044502或WO2004091502中之化合物,該等案件係以全文引用方式併入本文中。在另一實施例中,其他藥劑係揭示於WO2004080972、WO2004111014、WO2005035514、WO2005049018、WO2006099256、WO2006127588或WO2007044560中之化合物,該等案件係以全文引用方式併入本文中。 在一個實施例中,可向有需要之個體投與400 mg化合物1形式I及250 mg實質上非晶形化合物2。在該等實施例中,可藉由投與一或多個本發明錠劑來達成劑量量。例如,400 mg化合物1形式I及250 mg實質上非晶形化合物2之投與可藉由投與兩個各自含有200 mg化合物1形式I及125 mg實質上非晶形化合物2之錠劑來達成。投與之持續時間可持續直至疾病改善或直至個體之醫師建議停止,例如,投與之持續時間可為短於1週、1週、2週、3週、4週(28天)或一個月或更長。在一個實施例中,可每天向患者投與兩個各自包含200 mg化合物1形式I及125 mg實質上非晶形化合物2之錠劑。在另一實施例中,該兩個錠劑可在一天期間同時或在不同時間投與。在另一實施例中,每12小時投與一個錠劑。 在一個實施例中,可向有需要之個體投與400 mg化合物1形式I及500 mg實質上非晶形化合物2。在該等實施例中,劑量量可藉由投與兩個各自含有200 mg化合物1形式I及250 mg實質上非晶形化合物2之錠劑來達成。在一個實施例中,每12小時一次投與錠劑。在另一實施例中,劑量量亦可藉由每12小時投與兩個各自含有100 mg化合物1形式I及125 mg實質上非晶形化合物2之錠劑來達成。在另一實施例中,劑量量亦可藉由以單獨錠劑投與化合物1形式I及實質上非晶形化合物2來達成。例如,劑量量可藉由投與兩個含有200 mg化合物1形式I之錠劑及四個含有125 mg實質上非晶形化合物2之錠劑或兩個含有150 mg實質上非晶形化合物2之錠劑及兩個含有100 mg實質上非晶形化合物2之錠劑來達成。投與之持續時間可持續直至疾病改善或直至個體之醫師建議停止,例如投與之持續時間可為短於1週、1週、2週、3週、4週(28天)或1個月或更長。在一個實施例中,可每天向患者投與兩個包含200 mg化合物1形式I之錠劑及四個包含125 mg實質上非晶形化合物2之錠劑。在一個實施例中,可每天向患者投與兩個包含200 mg化合物1形式I之錠劑,且可每天兩次向患者投與兩個包含150 mg及100 mg實質上非晶形化合物2之錠劑。在另一實施例中,該兩個錠劑可在一天期間同時或在不同時間投與。在另一實施例中,每12小時投與一個包含200 mg化合物1之錠劑,且每12小時投與兩個包含150 mg及100 mg實質上非晶形化合物2之錠劑。 在一個實施例中,可向有需要之個體投與300 mg化合物1形式I及250 mg實質上非晶形化合物2。在該等實施例中,可藉由投與一或多個本發明錠劑來達成劑量量。例如,300 mg化合物1形式I及250 mg實質上非晶形化合物2之投與可藉由投與兩個各自含有150 mg化合物1形式I及125 mg實質上非晶形化合物2之錠劑來達成。投與之持續時間可持續直至疾病改善或直至個體之醫師建議停止,例如,投與之持續時間可為短於1週、1週、2週、3週、4週(28天)或一個月或更長。在一個實施例中,可每天向患者投與兩個各自包含150 mg化合物1形式I及125 mg實質上非晶形化合物2之錠劑。在另一實施例中,該兩個錠劑可在一天期間同時或在不同時間投與。在另一實施例中,每12小時投與一個錠劑。 在一個實施例中,可向有需要之個體投與600 mg化合物1形式I及500 mg實質上非晶形化合物2。在該等實施例中,可藉由投與一或多個本發明錠劑來達成劑量量。例如,600 mg化合物1形式I及500 mg實質上非晶形化合物2之投與可藉由每12小時投與兩個各自含有150 mg化合物1形式I及125 mg實質上非晶形化合物2之錠劑來達成。投與之持續時間可持續直至疾病改善或直至個體之醫師建議停止,例如投與之持續時間可為短於1週、1週、2週、3週、4週(28天)或1個月或更長。在一個實施例中,可每天向患者投與4個各自包含150 mg化合物1形式I及125 mg實質上非晶形化合物2之錠劑。在另一實施例中,該4個錠劑可在一天期間同時或在不同時間投與。在另一實施例中,每12小時投與兩個錠劑。 在一個實施例中,可向有需要之個體投與800 mg化合物1形式I及500 mg實質上非晶形化合物2。在該等實施例中,可藉由投與一或多個本發明錠劑來達成劑量量。例如,800 mg化合物1形式I及500 mg實質上非晶形化合物2之投與可藉由投與4個各自含有200 mg化合物1形式I及125 mg實質上非晶形化合物2之錠劑來達成。投與之持續時間可持續直至疾病改善或直至個體之醫師建議停止,例如,投與之持續時間可為短於1週、1週、2週、3週、4週(28天)或一個月或更長。在一個實施例中,可每天向患者投與4個各自包含200 mg化合物1形式I及125 mg實質上非晶形化合物2之錠劑。在另一實施例中,該4個錠劑可在一天期間同時或在不同時間投與。在另一實施例中,每次投藥機會投與兩個錠劑,且每天有兩次投藥機會。在另一實施例中,藉由每天兩次(BID)投與兩個各自包含200 mg化合物1及125 mg化合物2之錠劑來向患者投與800 mg化合物1及500 mg化合物2。在另一實施例中,藉由每12小時(q12h)投與兩個各自包含200 mg化合物1及125 mg化合物2之錠劑來向患者投與800 mg化合物1及500 mg化合物2。 在一個實施例中,可向有需要之個體投與600 mg化合物1形式I及250 mg實質上非晶形化合物2。在該等實施例中,可藉由投與一或多個本發明錠劑來達成劑量量。例如,600 mg化合物1形式I及250 mg實質上非晶形化合物2之投與可藉由投與三個錠劑(各自含有200 mg化合物1形式I及83.3 mg實質上非晶形化合物2)來達成。投與之持續時間可持續直至達成改善疾病或直至個體之醫師告知為止,例如,投與之持續時間可為短於1週、1週、2週、3週、4週(28天)或一個月或更長。在一個實施例中,可每天向患者投與三個各自包含200 mg化合物1形式I及83.3 mg實質上非晶形化合物2之錠劑。在另一實施例中,該三個錠劑可在一天期間同時或在不同時間投與。在另一實施例中,同時投與三個錠劑。 在一個實施例中,可向有需要之個體投與600 mg化合物1形式I及500 mg實質上非晶形化合物2。在該等實施例中,可藉由投與一或多個本發明錠劑來達成劑量量。例如,600 mg化合物1形式I及500 mg實質上非晶形化合物2之投與可藉由以下來達成:投與三個各自含有200 mg化合物1形式I及83.3 mg實質上非晶形化合物2之錠劑,隨後投與兩個各自包含125 mg化合物2之其他錠劑。投與之持續時間可持續直至達成改善疾病或直至個體之醫師告知為止,例如,投與之持續時間可為短於1週、1週、2週、3週、4週(28天)或一個月或更長。在一個實施例中,每日(qd)投與600 mg化合物1且每天兩次(bid)投與250 mg化合物2可藉由每日(qd)投與三個各自包含200 mg化合物1形式I及83.3 mg實質上非晶形化合物2之錠劑且每12小時(q12h)投與兩個各自包含125 mg化合物2之錠劑來達成。在一個實施例中,每日(qd)投與600 mg化合物1且每12小時(q12h)投與250 mg化合物2可藉由每日(qd)投與三個各自包含200 mg化合物1形式I及83.3 mg實質上非晶形化合物2之錠劑且每12小時(q12h)投與兩個各自包含125 mg化合物2之錠劑來達成。 該等組合可用於治療本文所闡述疾病,包括囊性纖維化。該等組合亦可用於本文所闡述套組中。在另一態樣中,本發明係關於套組,其包含包括化合物1形式I及包含實質上非晶形化合物2之固體分散體本發明之醫藥組合物或錠劑以及其單獨其他治療劑或醫藥組合物。在另一實施例中,本發明之醫藥組合物或錠劑、其單獨其他治療劑或醫藥組合物係在單獨容器中。在另一實施例中,單獨容器係瓶。在另一實施例中,單獨容器係小瓶。在另一實施例中,單獨容器係泡鼓包裝。存在於本發明組合物中之其他治療藥劑之量將不超過在包含該治療劑作為唯一活性藥劑之組合物中正常投與之量。較佳地,本文所揭示組合物中其他治療藥劑之量將在包含該藥劑作為唯一治療活性藥劑之組合物中所正常存在量的約50%至100%之範圍內。
組合物之治療用途
在一個態樣中,本發明亦提供治療患者之疾病、減輕該疾病之嚴重程度或根據症狀治療該疾病之方法,該方法包含向患者、較佳地哺乳動物投與有效量之本發明之醫藥組合物或錠劑,其中該疾病係選自囊性纖維化、哮喘、吸煙誘導之COPD、慢性枝氣管炎、鼻竇炎、便秘、胰臟炎、胰臟功能不全、由輸精管先天性雙側缺失(CBAVD)引起之男性不育、輕度肺病、特發性胰臟炎、過敏性枝氣管肺麴菌病(ABPA)、肝病、遺傳性肺氣腫、遺傳性血色素沉著病、凝血-纖維蛋白溶解缺陷(例如蛋白質C缺陷)、1型遺傳性血管性水腫、脂質加工缺陷(例如家族性高膽固醇血症、1型乳糜微粒血症、無β脂蛋白血症)、溶酶體儲存病(例如I細胞疾病/假何勒氏病(pseudo-Hurler))、黏多糖累積病、桑德霍夫/泰-薩克斯病(Sandhof/Tay-Sachs)、II型克裏格勒-納賈爾病(Crigler-Najjar type II)、多內分泌腺病/高胰島素血症、糖尿病、拉倫侏儒症(Laron dwarfism)、髓過氧化物酶缺陷、原發性低甲狀旁腺功能症、黑色素瘤、1型糖鎖異常CDG (glycanosis CDG type 1)、先天性甲狀腺機能亢進症、成骨不全、遺傳性低纖維素原血症、ACT缺陷、尿崩症(DI)、神經生長性DI (neurophyseal DI)、腎源性DI、夏-馬-圖三氏症候群(Charcot-Marie Tooth syndrome)、佩-梅二氏病(Perlizaeus-Merzbacher disease)、神經變性疾病(例如阿茲海默氏病(Alzheimer’s disease)、帕金森氏病(Parkinson’s disease)、肌萎縮側索硬化、進行性核上性麻痹、皮克病(Pick’s disease))、若干多麩醯胺酸神經性病症(例如亨廷頓氏病(Huntington’s)、I型脊髓小腦性共濟失調、脊髓延髓肌肉萎縮症、齒狀紅核蒼白球肌萎縮症及肌強直性營養不良),以及海綿樣腦病變(例如遺傳性克雅氏病(Creutzfeldt-Jakob disease) (由朊病毒蛋白加工缺陷引起)、法布裏病(Fabry disease)、格斯特曼-施特勞斯納症候群(Straussler-Scheinker syndrome)、COPD、乾眼病或薛格連氏病(Sjogren’s disease))、骨質疏鬆症、骨質減少、骨癒合及骨生長(包括骨損傷、骨再生、骨吸收減少及骨沈積增多)、戈漢症候群(Gorham's Syndrome)、氯離子通道病變(例如先天性肌強直(湯姆森(Thomson)形式及貝克爾(Becker)形式)、III型巴特氏症候群(Bartter's syndrome type III)、登特氏病(Dent's disease)、過度驚嚇症、癲癇症、過度驚嚇症、溶酶體儲存病、安格曼氏症候群(Angelman syndrome)),及原發性纖毛運動障礙(Primary Ciliary Dyskinesia, PCD)、纖毛結構及/或功能之遺傳病症的術語,包括具有位置逆轉之PCD(亦稱作卡特金納氏症候群(Kartagener syndrome))、不具有位置逆轉之PCD及纖毛不發育。 在一個態樣中,本發明亦提供治療患者之疾病、減輕該疾病之嚴重程度或根據症狀治療該疾病之方法,該方法包含向患者、較佳地哺乳動物投與有效量之本發明之醫藥組合物或錠劑,其中該疾病係選自全面性癲癇伴熱性驚厥附加症(generalized epilepsy with ferbrile seizures plus,GEFS+)、全面癲癇伴熱性及非熱性驚厥、肌強直、先天性肌剛痙病、鉀加重肌強直、高血鉀週期性麻痹、LQTS、LQTS/布魯格達氏症候群(Brugada syndrome)、常染色體顯性LQTS伴耳聾、常染色體隱性LQTS、LQTS伴畸形特徵、先天性及獲得性LQTS、蒂姆症候群(Timothy syndrome)、嬰兒持續高胰島素性低血糖、擴張性心肌病、常染色體顯性LQTS、登特病(Dent disease)、骨石化病、III型巴特氏症候群、中央軸空病(central core disease)、惡性體溫過高及兒茶酚胺敏感性多形性室性心搏過速(catecholaminergic polymorphic tachycardia)。 在一個態樣中,本發明係關於治療患者之囊性纖維化、減輕該疾病之嚴重程度或根據症狀治療該疾病之方法,該方法包含向患者、較佳地哺乳動物投與有效量之本發明之醫藥組合物或錠劑,其中該患者具有CFTR遺傳突變N1303K、ΔI507或R560T。 在一個態樣中,本發明係關於治療患者之囊性纖維化、減輕該疾病之嚴重程度或根據症狀治療該疾病之方法,該方法包含向患者、較佳地哺乳動物投與有效量之本發明之醫藥組合物或錠劑,其中該患者具有CFTR遺傳突變G551D。在另一實施例中,患者係G551D同型合子的。在另一實施例中,患者係G551D異型合子的,其中另一CFTR遺傳突變係以下各項中之任一者:ΔF508、G542X、N1303K、W1282X、R117H、R553X、1717-1G->A、621+1G->T、2789+5G->A、3849+10kbC->T、R1162X、G85E、3120+1G->A、ΔI507、1898+1G->A、3659delC、R347P、 R560T、R334W、A455E、2184delA或711+1G->T。 在一個態樣中,本發明係關於治療患者之囊性纖維化、減輕該疾病之嚴重程度或根據症狀治療該疾病之方法,該方法包含向患者、較佳地哺乳動物投與有效量之本發明之醫藥組合物或錠劑,其中該患者具有CFTR遺傳突變ΔF508。在另一實施例中,患者係ΔF508同型合子的。在另一實施例中,患者係ΔF508異型合子的,其中另一CFTR遺傳突變係以下各項中之任一者:G551D、G542X、N1303K、W1282X、R117H、R553X、1717-1G->A、621+1G->T、2789+5G->A、3849+10kbC->T、R1162X、G85E、3120+1G->A、ΔI507、1898+1G->A、3659delC、R347P、 R560T、R334W、A455E、2184delA或711+1G->T。 在一個態樣中,本發明係關於治療患者之囊性纖維化、減輕該疾病之嚴重程度或根據症狀治療該疾病之方法,該方法包含向患者、較佳地哺乳動物投與有效量之本發明之醫藥組合物或錠劑,其中該患者具有選自以下各項之CFTR遺傳突變:G178R、G551S、G970R、G1244E、S1255P、G1349D、S549N、S549R、S1251N、E193K、F1052V、G1069R、R117C、D110H、R347H、R352Q、E56K、P67L、L206W、A455E、D579G、S1235R、S945L、R1070W、F1074L、D110E、D1270N、D1152H、1717-1G->A、621+1G->T、3120+1G->A、1898+1G->A、711+1G->T、2622+1G->A、405+1G->A、406-1G->A、4005+1G->A、1812-1G->A、1525-1G->A、712-1G->T、1248+1G->A、1341+1G->A、3121-1G->A、4374+1G->T、3850-1G->A、2789+5G->A、3849+10kbC->T、3272-26A->G、711+5G->A、3120G->A、1811+1.6kbA->G、711+3A->G、1898+3A->G、1717-8G->A、1342-2A->C、405+3A->C、1716G/A、1811+1G->C、1898+5G->T、3850-3T->G、IVS14b+5G->A、1898+1G->T、4005+2T->C及621+3A->G。 在一個態樣中,本發明係關於治療患者之囊性纖維化、減輕該疾病之嚴重程度或根據症狀治療該疾病之方法,該方法包含向患者、較佳地哺乳動物投與有效量之本發明之醫藥組合物或錠劑,其中該患者具有選自以下各項之CFTR遺傳突變:G178R、G551S、G970R、G1244E、S1255P、G1349D、S549N、S549R、S1251N、E193K、F1052V及G1069R。在此態樣之一個實施例中,本發明提供治療CFTR之方法,其包含向具有選自以下各項之人類CFTR突變之患者投與化合物1:G178R、G551S、G970R、G1244E、S1255P、G1349D、S549N、S549R及S1251N。在一個態樣中,本發明係關於治療患者之囊性纖維化、減輕該疾病之嚴重程度或根據症狀治療該疾病之方法,該方法包含向患者、較佳地哺乳動物投與有效量之本發明之醫藥組合物或錠劑,其中該患者具有選自以下各項之CFTR遺傳突變:E193K、F1052V及G1069R。在此態樣之一些實施例中,該方法使得氯離子運輸相對於基線氯離子運輸提高大於10倍。 在一個態樣中,本發明係關於治療患者之囊性纖維化、減輕該疾病之嚴重程度或根據症狀治療該疾病之方法,該方法包含向患者、較佳地哺乳動物投與有效量之本發明之醫藥組合物或錠劑,其中該患者具有選自以下各項之CFTR遺傳突變:R117C、D110H、R347H、R352Q、E56K、P67L、L206W、A455E、D579G、S1235R、S945L、R1070W、F1074L、D110E、D1270N及D1152H。在此態樣之一個實施例中,該方法使得氯離子運輸相對於基線氯離子運輸提高大於或等於10%。 在一個態樣中,本發明係關於治療患者之囊性纖維化、減輕該疾病之嚴重程度或根據症狀治療該疾病之方法,該方法包含向患者、較佳地哺乳動物投與有效量之本發明之醫藥組合物或錠劑,其中該患者具有選自以下各項之CFTR遺傳突變:1717-1G->A、621+1G->T、3120+1G->A、1898+1G->A、711+1G->T、2622+1G->A、405+1G->A、406-1G->A、4005+1G->A、1812-1G->A、1525-1G->A、712-1G->T、1248+1G->A、1341+1G->A、3121-1G->A、4374+1G->T、3850-1G->A、2789+5G->A、3849+10kbC->T、3272-26A->G、711+5G->A、3120G->A、1811+1.6kbA->G、711+3A->G、1898+3A->G、1717-8G->A、1342-2A->C、405+3A->C、1716G/A、1811+1G->C、1898+5G->T、3850-3T->G、IVS14b+5G->A、1898+1G->T、4005+2T->C及621+3A->G。在一個態樣中,本發明係關於治療患者之囊性纖維化、減輕該疾病之嚴重程度或根據症狀治療該疾病之方法,該方法包含向患者、較佳地哺乳動物投與有效量之本發明之醫藥組合物或錠劑,其中該患者具有選自以下各項之CFTR遺傳突變:1717-1G->A、1811+1.6kbA->G、2789+5G->A、3272-26A->G及3849+10kbC->T。在一個態樣中,本發明係關於治療患者之囊性纖維化、減輕該疾病之嚴重程度或根據症狀治療該疾病之方法,該方法包含向患者、較佳地哺乳動物投與有效量之本發明之醫藥組合物或錠劑,其中該患者具有選自2789+5G->A及3272-26A->G之CFTR遺傳突變。 在一個態樣中,本發明係關於治療患者之囊性纖維化、減輕該疾病之嚴重程度或根據症狀治療該疾病之方法,該方法包含向患者、較佳地哺乳動物投與有效量之本發明之醫藥組合物或錠劑,其中該患者具有選自以下各項之CFTR遺傳突變:G178R、G551S、G970R、G1244E、S1255P、G1349D、S549N、S549R、S1251N、E193K、F1052V、G1069R、R117C、D110H、R347H、R352Q、E56K、P67L、L206W、A455E、D579G、S1235R、S945L、R1070W、F1074L、D110E、D1270N、D1152H、1717-1G->A、621+1G->T、3120+1G->A、1898+1G->A、711+1G->T、2622+1G->A、405+1G->A、406-1G->A、4005+1G->A、1812-1G->A、1525-1G->A、712-1G->T、1248+1G->A、1341+1G->A、3121-1G->A、4374+1G->T、3850-1G->A、2789+5G->A、3849+10kbC->T、3272-26A->G、711+5G->A、3120G->A、1811+1.6kbA->G、711+3A->G、1898+3A->G、1717-8G->A、1342-2A->C、405+3A->C、1716G/A、1811+1G->C、1898+5G->T、3850-3T->G、IVS14b+5G->A、1898+1G->T、4005+2T->C及621+3A->G,及選自以下各項之人類CFTR突變:ΔF508、R117H及G551D。 在一個態樣中,本發明係關於治療患者之囊性纖維化、減輕該疾病之嚴重程度或根據症狀治療該疾病之方法,該方法包含向患者、較佳地哺乳動物投與有效量之本發明之醫藥組合物或錠劑,其中該患者具有選自以下各項之CFTR遺傳突變:G178R、G551S、G970R、G1244E、S1255P、G1349D、S549N、S549R、S1251N、E193K、F1052V及G1069R,以及選自以下各項之人類CFTR突變:ΔF508、R117H及G551D。在一個態樣中,本發明係關於治療患者之囊性纖維化、減輕該疾病之嚴重程度或根據症狀治療該疾病之方法,該方法包含向患者、較佳地哺乳動物投與有效量之本發明之醫藥組合物或錠劑,其中該患者具有選自以下各項之CFTR遺傳突變:G178R、G551S、G970R、G1244E、S1255P、G1349D、S549N、S549R及S1251N,以及選自以下各項之人類CFTR突變:ΔF508、R117H及G551D。在一個態樣中,本發明係關於治療患者之囊性纖維化、減輕該疾病之嚴重程度或根據症狀治療該疾病之方法,該方法包含向患者、較佳地哺乳動物投與有效量之本發明之醫藥組合物或錠劑,其中該患者具有選自以下各項之CFTR遺傳突變:E193K、F1052V及G1069R,以及選自以下各項之人類CFTR突變:ΔF508、R117H及G551D。在此態樣之一些實施例中,該方法使得氯離子運輸相對於基線氯離子運輸提高大於10倍。 在一個態樣中,本發明係關於治療患者之囊性纖維化、減輕該疾病之嚴重程度或根據症狀治療該疾病之方法,該方法包含向患者、較佳地哺乳動物投與有效量之本發明之醫藥組合物或錠劑,其中該患者具有選自以下各項之CFTR遺傳突變:R117C、D110H、R347H、R352Q、E56K、P67L、L206W、A455E、D579G、S1235R、S945L、R1070W、F1074L、D110E、D1270N及D1152H,以及選自以下各項之人類CFTR突變:ΔF508、R117H及G551D。在此態樣之一個實施例中,該方法使得氯離子運輸相對於基線氯離子運輸提高大於或等於10%。 在一個態樣中,本發明係關於治療患者之囊性纖維化、減輕該疾病之嚴重程度或根據症狀治療該疾病之方法,該方法包含向患者、較佳地哺乳動物投與有效量之本發明之醫藥組合物或錠劑,其中該患者具有選自以下各項之CFTR遺傳突變:1717-1G->A、621+1G->T、3120+1G->A、1898+1G->A、711+1G->T、2622+1G->A、405+1G->A、406-1G->A、4005+1G->A、1812-1G->A、1525-1G->A、712-1G->T、1248+1G->A、1341+1G->A、3121-1G->A、4374+1G->T、3850-1G->A、2789+5G->A、3849+10kbC->T、3272-26A->G、711+5G->A、3120G->A、1811+1.6kbA->G、711+3A->G、1898+3A->G、1717-8G->A、1342-2A->C、405+3A->C、1716G/A、1811+1G->C、1898+5G->T、3850-3T->G、IVS14b+5G->A、1898+1G->T、4005+2T->C及621+3A->G,以及選自以下各項之人類CFTR突變:ΔF508、R117H及G551D。在一個態樣中,本發明係關於治療患者之囊性纖維化、減輕該疾病之嚴重程度或根據症狀治療該疾病之方法,該方法包含向患者、較佳地哺乳動物投與有效量之本發明之醫藥組合物或錠劑,其中該患者具有選自以下各項之CFTR遺傳突變:1717-1G->A、1811+1.6kbA->G、2789+5G->A、3272-26A->G及3849+10kbC->T,以及選自以下各項之人類CFTR突變:ΔF508、R117H及G551D。在一個態樣中,本發明係關於治療患者之囊性纖維化、減輕該疾病之嚴重程度或根據症狀治療該疾病之方法,該方法包含向患者、較佳地哺乳動物投與有效量之本發明之醫藥組合物或錠劑,其中該患者具有選自2789+5G->A及3272-26A->G之CFTR遺傳突變及選自ΔF508、R117H之人類CFTR突變。 在一個態樣中,本發明係關於治療患者之囊性纖維化、減輕該疾病之嚴重程度或根據症狀治療該疾病之方法,該方法包含向患者、較佳地哺乳動物投與有效量之本發明之醫藥組合物或錠劑,其中該患者具有選自以下各項之CFTR遺傳突變:G178R、G551S、G970R、G1244E、S1255P、G1349D、S549N、S549R、S1251N、E193K、F1052V、G1069R、R117C、D110H、R347H、R352Q、E56K、P67L、L206W、A455E、D579G、S1235R、S945L、R1070W、F1074L、D110E、D1270N、D1152H、1717-1G->A、621+1G->T、3120+1G->A、1898+1G->A、711+1G->T、2622+1G->A、405+1G->A、406-1G->A、4005+1G->A、1812-1G->A、1525-1G->A、712-1G->T、1248+1G->A、1341+1G->A、3121-1G->A、4374+1G->T、3850-1G->A、2789+5G->A、3849+10kbC->T、3272-26A->G、711+5G->A、3120G->A、1811+1.6kbA->G、711+3A->G、1898+3A->G、1717-8G->A、1342-2A->C、405+3A->C、1716G/A、1811+1G->C、1898+5G->T、3850-3T->G、IVS14b+5G->A、1898+1G->T、4005+2T->C及621+3A->G,以及選自以下各項之人類CFTR突變:ΔF508、R117H及G551D。 在一個態樣中,本發明係關於治療患者之囊性纖維化、減輕該疾病之嚴重程度或根據症狀治療該疾病之方法,該方法包含向患者、較佳地哺乳動物投與有效量之本發明之醫藥組合物或錠劑,其中該患者具有選自以下各項之CFTR遺傳突變:G178R、G551S、G970R、G1244E、S1255P、G1349D、S549N、S549R、S1251N、E193K、F1052V及G1069R。在一個態樣中,本發明係關於治療患者之囊性纖維化、減輕該疾病之嚴重程度或根據症狀治療該疾病之方法,該方法包含向患者、較佳地哺乳動物投與有效量之本發明之醫藥組合物或錠劑,其中該患者具有選自以下各項之CFTR遺傳突變:G178R、G551S、G970R、G1244E、S1255P、G1349D、S549N、S549R及S1251N。在一個態樣中,本發明係關於治療患者之囊性纖維化、減輕該疾病之嚴重程度或根據症狀治療該疾病之方法,該方法包含向患者、較佳地哺乳動物投與有效量之本發明之醫藥組合物或錠劑,其中該患者具有選自以下各項之CFTR遺傳突變:E193K、F1052V及G1069R。在此態樣之一些實施例中,該方法使得氯離子運輸相對於基線氯離子運輸提高大於10倍。 在一個態樣中,本發明係關於治療患者之囊性纖維化、減輕該疾病之嚴重程度或根據症狀治療該疾病之方法,該方法包含向患者、較佳地哺乳動物投與有效量之本發明之醫藥組合物或錠劑,其中該患者具有選自以下各項之CFTR遺傳突變:R117C、D110H、R347H、R352Q、E56K、P67L、L206W、A455E、D579G、S1235R、S945L、R1070W、F1074L、D110E、D1270N及D1152H。在此態樣之一個實施例中,該方法使得氯離子運輸相對於基線氯離子運輸提高大於或等於10%。 在一個態樣中,本發明係關於治療患者之囊性纖維化、減輕該疾病之嚴重程度或根據症狀治療該疾病之方法,該方法包含向患者、較佳地哺乳動物投與有效量之本發明之醫藥組合物或錠劑,其中該患者具有選自以下各項之CFTR遺傳突變:1717-1G->A、621+1G->T、3120+1G->A、1898+1G->A、711+1G->T、2622+1G->A、405+1G->A、406-1G->A、4005+1G->A、1812-1G->A、1525-1G->A、712-1G->T、1248+1G->A、1341+1G->A、3121-1G->A、4374+1G->T、3850-1G->A、2789+5G->A、3849+10kbC->T、3272-26A->G、711+5G->A、3120G->A、1811+1.6kbA->G、711+3A->G、1898+3A->G、1717-8G->A、1342-2A->C、405+3A->C、1716G/A、1811+1G->C、1898+5G->T、3850-3T->G、IVS14b+5G->A、1898+1G->T、4005+2T->C及621+3A->G。在一個態樣中,本發明係關於治療患者之囊性纖維化、減輕該疾病之嚴重程度或根據症狀治療該疾病之方法,該方法包含向患者、較佳地哺乳動物投與有效量之本發明之醫藥組合物或錠劑,其中該患者具有選自以下各項之CFTR遺傳突變:1717-1G->A、1811+1.6kbA->G、2789+5G->A、3272-26A->G及3849+10kbC->T。在一個態樣中,本發明係關於治療患者之囊性纖維化、減輕該疾病之嚴重程度或根據症狀治療該疾病之方法,該方法包含向患者、較佳地哺乳動物投與有效量之本發明之醫藥組合物或錠劑,其中該患者具有選自2789+5G->A及3272-26A->G之CFTR遺傳突變。 在一個態樣中,本發明係關於治療患者之囊性纖維化、減輕該疾病之嚴重程度或根據症狀治療該疾病之方法,該方法包含向患者、較佳地哺乳動物投與有效量之本發明之醫藥組合物或錠劑,其中該患者具有選自以下各項之CFTR遺傳突變:G178R、G551S、G970R、G1244E、S1255P、G1349D、S549N、S549R、S1251N、E193K、F1052V、G1069R、R117C、D110H、R347H、R352Q、E56K、P67L、L206W、A455E、D579G、S1235R、S945L、R1070W、F1074L、D110E、D1270N、D1152H、1717-1G->A、621+1G->T、3120+1G->A、1898+1G->A、711+1G->T、2622+1G->A、405+1G->A、406-1G->A、4005+1G->A、1812-1G->A、1525-1G->A、712-1G->T、1248+1G->A、1341+1G->A、3121-1G->A、4374+1G->T、3850-1G->A、2789+5G->A、3849+10kbC->T、3272-26A->G、711+5G->A、3120G->A、1811+1.6kbA->G、711+3A->G、1898+3A->G、1717-8G->A、1342-2A->C、405+3A->C、1716G/A、1811+1G->C、1898+5G->T、3850-3T->G、IVS14b+5G->A、1898+1G->T、4005+2T->C及621+3A->G,以及選自以下各項之人類CFTR突變:ΔF508、R117H及G551D,及一或多種選自以下各項之人類CFTR突變:ΔF508、R117H及G551D。 在一個態樣中,本發明係關於治療患者之囊性纖維化、減輕該疾病之嚴重程度或根據症狀治療該疾病之方法,該方法包含向患者、較佳地哺乳動物投與有效量之本發明之醫藥組合物或錠劑,其中該患者具有選自以下各項之CFTR遺傳突變:G178R、G551S、G970R、G1244E、S1255P、G1349D、S549N、S549R、S1251N、E193K、F1052V及G1069R,以及一或多種選自以下各項之人類CFTR突變:ΔF508、R117H及G551D。在一個態樣中,本發明係關於治療患者之囊性纖維化、減輕該疾病之嚴重程度或根據症狀治療該疾病之方法,該方法包含向患者、較佳地哺乳動物投與有效量之本發明之醫藥組合物或錠劑,其中該患者具有選自以下各項之CFTR遺傳突變:G178R、G551S、G970R、G1244E、S1255P、G1349D、S549N、S549R及S1251N,以及一或多種選自以下各項之人類CFTR突變:ΔF508、R117H及G551D。在一個態樣中,本發明係關於治療患者之囊性纖維化、減輕該疾病之嚴重程度或根據症狀治療該疾病之方法,該方法包含向患者、較佳地哺乳動物投與有效量之本發明之醫藥組合物或錠劑,其中該患者具有選自以下各項之CFTR遺傳突變:E193K、F1052V及G1069R,以及一或多種選自以下各項之人類CFTR突變:ΔF508、R117H及G551D。在此態樣之一些實施例中,該方法使得氯離子運輸相對於基線氯離子運輸提高大於10倍。 在一個態樣中,本發明係關於治療患者之囊性纖維化、減輕該疾病之嚴重程度或根據症狀治療該疾病之方法,該方法包含向患者、較佳地哺乳動物投與有效量之本發明之醫藥組合物或錠劑,其中該患者具有選自以下各項之CFTR遺傳突變:R117C、D110H、R347H、R352Q、E56K、P67L、L206W、A455E、D579G、S1235R、S945L、R1070W、F1074L、D110E、D1270N及D1152H,以及一或多種選自以下各項之人類CFTR突變:ΔF508、R117H及G551D。在此態樣之一個實施例中,該方法使得氯離子運輸相對於基線氯離子運輸提高大於或等於10%。 在一個態樣中,本發明係關於治療患者之囊性纖維化、減輕該疾病之嚴重程度或根據症狀治療該疾病之方法,該方法包含向患者、較佳地哺乳動物投與有效量之本發明之醫藥組合物或錠劑,其中該患者具有選自以下各項之CFTR遺傳突變:1717-1G->A、621+1G->T、3120+1G->A、1898+1G->A、711+1G->T、2622+1G->A、405+1G->A、406-1G->A、4005+1G->A、1812-1G->A、1525-1G->A、712-1G->T、1248+1G->A、1341+1G->A、3121-1G->A、4374+1G->T、3850-1G->A、2789+5G->A、3849+10kbC->T、3272-26A->G、711+5G->A、3120G->A、1811+1.6kbA->G、711+3A->G、1898+3A->G、1717-8G->A、1342-2A->C、405+3A->C、1716G/A、1811+1G->C、1898+5G->T、3850-3T->G、IVS14b+5G->A、1898+1G->T、4005+2T->C及621+3A->G,以及一或多種選自以下各項之人類CFTR突變:ΔF508、R117H及G551D。在一個態樣中,本發明係關於治療患者之囊性纖維化、減輕該疾病之嚴重程度或根據症狀治療該疾病之方法,該方法包含向患者、較佳地哺乳動物投與有效量之本發明之醫藥組合物或錠劑,其中該患者具有選自以下各項之CFTR遺傳突變:1717-1G->A、1811+1.6kbA->G、2789+5G->A、3272-26A->G及3849+10kbC->T,以及一或多種選自以下各項之人類CFTR突變:ΔF508、R117H及G551D。在一個態樣中,本發明係關於治療患者之囊性纖維化、減輕該疾病之嚴重程度或根據症狀治療該疾病之方法,該方法包含向患者、較佳地哺乳動物投與有效量之本發明之醫藥組合物或錠劑,其中該患者具有選自2789+5G->A及3272-26A->G之CFTR遺傳突變,以及一或多種選自以下各項之人類CFTR突變:ΔF508、R117H及G551D。 在某些實施例中,包含化合物1形式I及實質上非晶形化合物2之固體分散體之本發明之醫藥上可接受之組合物或錠劑可用於治療在呼吸及非呼吸上皮之頂膜中展現殘餘CFTR活性之患者之囊性纖維化、減輕該疾病之嚴重程度或根據症狀治療該疾病。可使用業內已知之方法容易地檢測殘餘CFTR活性在上皮表面處之存在,例如標準電生理技術、生物化學技術或組織化學技術。該等方法使用活體內或離體電生理技術、對汗液或唾液Cl
-
濃度之量測或監測細胞表面密度之離體生物化學或組織化學技術來鑒定CFTR活性。可使用該等方法在各種不同突變之異型合子或同型合子患者中容易地檢測殘餘CFTR活性,包括最常見突變ΔF508以及其他突變(例如G551D突變或R117H突變)之同型合子或異型合子患者。在某些實施例中,包含化合物1形式I及包含實質上非晶形化合物2之固體分散體之醫藥上可接受之組合物或錠劑可用於治療展現極少殘餘CFTR活性至不展現殘餘CFTR活性之患者之囊性纖維化、減輕該疾病之嚴重程度或根據症狀治療該疾病。在某些實施例中,包含化合物1形式I及包含實質上非晶形化合物2之固體分散體之醫藥上可接受之組合物或錠劑可用於治療在呼吸上皮之頂膜中展現極少殘餘CFTR活性至不展現殘餘CFTR活性之患者之囊性纖維化、減輕該疾病之嚴重程度或根據症狀治療該疾病。 在另一實施例中,本發明之化合物及組合物可用於治療已使用藥理學方法誘導或加強殘餘CFTR活性之患者之囊性纖維化或減輕該疾病之嚴重程度。在另一實施例中,本發明之化合物及組合物可用於治療已使用基因療法誘導或加強殘餘CFTR活性之患者之囊性纖維化或減輕該疾病之嚴重程度。該等方法增加CFTR存於細胞表面處之量,藉此在患者中誘導一直不存在之CFTR活性或加強患者中殘餘CFTR活性之現有水準。 在一個實施例中,本發明之如本文所闡述包含化合物1形式I及包含實質上非晶形化合物2之固體分散體之醫藥組合物及錠劑可用於治療在某些基因型內展現殘餘CFTR活性(例如,I類突變(不可合成)、II類突變(錯誤摺疊)、III類突變(調控或門控受損)、IV類突變(傳導改變)或V類突變(合成降低))之患者之囊性纖維化或減輕該疾病之嚴重程度。 在一個實施例中,本發明之如本文所闡述包含化合物1形式I及包含實質上非晶形化合物2之固體分散體之醫藥組合物及錠劑可用於治療在某些臨床表型(例如,中等至輕度臨床表型,其通常與上皮頂膜中殘餘CFTR活性之量相關)內之患者之囊性纖維化、減輕該疾病之嚴重程度或根據症狀治療該疾病。該等表型包括展現胰臟功能不全之患者。 在一個實施例中,本發明之如本文所闡述包含化合物1形式I及包含實質上非晶形化合物2之固體分散體之醫藥組合物及錠劑可用於治療經診斷患有以下疾病之患者、減輕以下疾病之嚴重程度或根據症狀治療以下疾病:胰臟功能不全、特發性胰臟炎及輸精管先天性雙側缺失,或輕度肺病,其中該患者展現殘餘CFTR活性。 在一個實施例中,本發明之如本文所闡述包含化合物1形式I及包含實質上非晶形化合物2之固體分散體之醫藥組合物及錠劑可用於治療經診斷患有以下疾病之患者、減輕以下疾病之嚴重程度或根據症狀治療以下疾病:胰臟功能不全、特發性胰臟炎及輸精管先天性雙側缺失,或輕度肺病,其中該患者具有野生型CFTR。 除囊性纖維化以外,CFTR活性之調節可有益於並非由CFTR之突變直接引起之其他疾病,例如分泌疾病及其他由CFTR介導之蛋白摺疊疾病。該等疾病包括(但不限於)慢性阻塞性肺病(COPD)、乾眼病及休葛蘭氏症候群(Sjögren’s Syndrome)。COPD之特徵在於具有進展性且不完全可逆之氣流受限。氣流受限係因黏液分泌過多、肺氣腫及細枝氣管炎引起。突變或野生型CFTR之活化劑可治療在COPD中常見的黏液分泌過多及黏膜纖毛清除受損。特定而言,增加陰離子跨CFTR之分泌可有利於流體運輸至氣道表面液體中來水合黏液並最佳化纖毛周圍(periciliary)流體黏度。此可使得增強黏膜纖毛清除並減輕與COPD相關之症狀。乾眼病之特徵在於水性淚液產量減少及異常淚膜脂質、蛋白質及黏蛋白特徵。乾眼病具有許多起因,一些起因包括年齡、雷射角膜成塑術(Lasik)眼外科手術、關節炎、醫藥、化學/熱燒傷、過敏及諸如囊性纖維化及休葛蘭氏症候群等疾病。經由CFTR增加陰離子分泌可增強眼睛周圍之角膜內皮細胞及分泌腺之流體運輸以增加角膜水合。此將有助於減輕與乾眼病相關之症狀。休葛蘭氏症候群係自體免疫疾病,其中免疫系統攻擊整個身體內產生水分之腺,包括眼睛、口腔、皮膚、呼吸組織、肝、陰道及腸。症狀包括乾眼、乾口及乾陰道病以及肺病。該疾病亦與類風濕性關節炎、全身性紅斑狼瘡、全身性硬化及多發性肌炎/皮肌炎相關。據信,蛋白輸送缺陷可引起疾病,其治療選擇有限。CFTR活性之加強劑或誘導劑可使受疾病折磨之各個器官水合且有助於緩和相關症狀。 在一個實施例中,本發明係關於加強或誘導活體外或活體內陰離子通道活性之方法,其包含使通道與醫藥組合物
PC-I
至
PC-XXV
中之任一者接觸
。
在另一實施例中,陰離子通道係氯離子通道或碳酸氫根離子通道。在另一實施例中,陰離子通道係氯離子通道。 端視個體之物種、年齡及一般狀況、感染嚴重程度、具體藥劑、其投與模式及諸如此類,所需確切量將隨個體而變化。本發明化合物較佳地調配為劑量單位形式以便於投與及統一劑量。如本文所使用,表達「劑量單位形式」係指適於欲治療患者之藥劑之物理離散單位。然而,應瞭解,本發明化合物及組合物之總日用量將由主治醫師在合理的醫學判斷範圍內決定。任一具體患者或生物體之特定有效劑量量將取決於各種因素,包括所治療病症及病症之嚴重程;所用特定化合物之活性;所用特定組合物;患者之年齡、體重、一般健康狀況、性別及飲食;所用特定化合物之投與時間、投與途徑及排泄速率;治療持續時間;與所用特定化合物組合或同時使用之藥物;及醫療技術中熟知之類似因素。如本文所使用,術語「患者」意指動物,較佳地哺乳動物,且最佳地人類。倘若在本申請案中有任一處化合物之名稱不能正確闡述該化合物之結構,則應用結構代替名稱且以結構為準。
實例 XRPD (X 射線粉末繞射 )
用具有HI-STAR二維檢測器及平板石墨單色器之Bruker D8 DISCOVER粉末繞射計來收集化合物1之X射線繞射(XRD)數據。以40 kV,35 mA使用具有Kα輻射之Cu密封管。在25℃下將試樣置於零背景矽晶圓上。對於每一試樣而言,每120秒以2個不同θ
2
角度(8º及26º)收集兩個數據訊框。用GADDS軟體整合數據並用DIFFRACT
plus
EVA軟體進行歸併。所報告峰位置之不確定度為± 0.2度。
差示掃描量熱法 (DSC)
使用DSC Q100 V9.6 Build 290 (TA Instruments, New Castle, DE)來收集化合物1形式I之差示掃描量熱(DSC)數據。用銦校準溫度且用藍寶石校準熱容量。將3-6 mg試樣稱量至鋁盤中,使用具有1個針孔之蓋扣住(crimp)鋁盤。以1.0℃/min之加熱速率及50 ml/min之氮氣吹掃自25℃至350℃掃描試樣。藉由Thermal Advantage Q SeriesTM 2.2.0.248版軟體收集數據且藉由Universal分析軟體4.1D版(TA Instruments, New Castle, DE)進行分析。所報告數值代表單一分析。
化合物 1 形式 I 單晶結構確定
用配備有密封管Cu K-α源及Apex II CCD檢測器之Bruker Apex II繞射計獲得繞射數據。使用SHELX程式對該結構實施解析及精修(Sheldrick, G.M., Acta Cryst, (2008) A64, 112-122)。根據系統消光及強度統計學,對P2
1
/n空間群之結構進行解析及精修。 Vitride®(雙(2-甲氧基乙氧基)氫化鋁鈉[或NaAlH
2
(OCH
2
CH
2
OCH
3
)
2
],存於甲苯中之65 wgt%溶液)購自Aldrich Chemicals。 2,2-二氟-1,3-苯并二氧雜環戊烯-5-甲酸購自Saltigo (Lanxess公司之分部)。
化合物 1 之製備 (2,2- 二氟 -1,3- 苯并二氧雜環戊烯 -5- 基 )- 甲醇之製備。 在甲苯(10體積)中漿化市售2,2-二氟-1,3-苯并二氧雜環戊烯-5-甲酸(1.0當量)。以可將溫度維持在15-25℃之速率經由加料漏斗添加Vitride®(2當量)。在添加結束時使溫度升高至40℃並保持2小時(h),然後經由加料漏斗小心地添加10% (w/w) NaOH水溶液(4.0當量)並將溫度維持在40-50℃。在再攪拌30分鐘(min)後,在40℃下使各層分離。使有機相冷卻至20℃,然後用水(2 × 1.5體積)洗滌,乾燥(Na
2
SO
4
),過濾,並濃縮以提供粗(2,2-二氟-1,3-苯并二氧雜環戊烯-5-基)-甲醇,將其直接用於下一步驟中。
5- 氯甲基 -2,2- 二氟 -1,3- 苯并二氧雜環戊烯之製備。 將(2,2-二氟-1,3-苯并二氧雜環戊烯-5-基)-甲醇(1.0當量)溶解於MTBE(5體積)中。添加催化量之4-(N,N-二甲基)胺基吡啶(DMAP) (1 mol%)且經由加料漏斗添加SOCl
2
(1.2當量)。以可將反應器中之溫度維持在15-25℃之速率添加SOCl
2
。使溫度升高至30℃並保持1 h,然後使其冷卻至20℃。經由加料漏斗添加水(4體積),同時將溫度維持在30℃以下。在再攪拌30分鐘後,使各層分離。攪拌有機層且添加10% (w/v) NaOH水溶液(4.4體積)。在攪拌15 min至20 min後,使各層分離。然後,乾燥(Na
2
SO
4
)有機相,過濾並濃縮以提供粗5-氯甲基-2,2-二氟-1,3-苯并二氧雜環戊烯,將其直接用於下一步驟中。
(2,2- 二氟 -1,3- 苯并二氧雜環戊烯 -5- 基 )- 乙腈之製備。 將5-氯甲基-2,2-二氟-1,3-苯并二氧雜環戊烯(1當量)存於DMSO (1.25體積)中之溶液添加至NaCN (1.4當量)存於DMSO (3體積)中之漿液中,同時將溫度維持在30-40℃之間。將混合物攪拌1 h,且然後依次添加水(6體積)與甲基第三丁基醚(MTBE) (4體積)。在攪拌30 min後,分離各層。用MTBE(1.8體積)萃取水層。用水(1.8體積)洗滌合併之有機層,乾燥(Na
2
SO
4
),過濾並濃縮以提供粗(2,2-二氟-1,3-苯并二氧雜環戊烯-5-基)-乙腈(95%),將其直接用於下一步驟中。
(2,2- 二氟 -1,3- 苯并二氧雜環戊烯 -5- 基 )-1- 乙酸乙酯 - 乙腈之合成。 用氮吹掃反應器且用900 mL甲苯裝填。經由氮吹掃將溶劑脫氣不短於16 h。然後,向反應器中依次裝填Na
3
PO
4
(155.7 g, 949.5 mmol)、雙(二亞苄基丙酮)鈀(0) (7.28 g, 12.66 mmol)。在23℃下經10 min自經氮吹掃加料漏斗裝填第三丁基膦存於己烷中之10% w/w溶液(51.23 g, 25.32 mmol)。將混合物攪拌50 min,此時經1 min添加5-溴-2,2-二氟-1,3-苯并二氧雜環戊烯(75 g, 316.5 mmol)。在再攪拌50 min後,經5 min向混合物裝填氰基乙酸乙酯(71.6 g, 633.0 mmol),隨後一次性裝填水(4.5 mL)。經40 min將混合物加熱至70℃且每1 - 2 h藉由HPLC分析反應物至產物之轉化百分比。在觀察到完全轉化(通常在5 - 8 h後100%轉化)後,使混合物冷卻至20 - 25℃並藉助矽藻土墊過濾。用甲苯(2 × 450 mL)沖洗矽藻土墊且在真空下在60 - 65℃下將經合併有機物濃縮至300 mL。向濃縮物裝填225 mL DMSO且在真空下在70 - 80℃下將其濃縮,直至有效的溶劑蒸餾結束為止。使溶液冷卻至20 - 25℃且用DMSO稀釋至900 mL以準備用於步驟2。
1
H NMR (500 MHz, CDCl
3
) δ 7.16 - 7.10 (m, 2H), 7.03 (d,
J
= 8.2 Hz, 1H), 4.63 (s, 1H), 4.19 (m, 2H), 1.23 (t,
J
= 7.1 Hz, 3H)。
(2,2- 二氟 -1,3- 苯并二氧雜環戊烯 -5- 基 )- 乙腈之合成。 經20 min向來自上文之(2,2-二氟-1,3-苯并二氧雜環戊烯-5-基)-1-乙基乙酸酯-乙腈之DMSO溶液裝填3 N HCl (617.3 mL, 1.85 mol),同時將內部溫度維持在<40℃。然後,經1 h將混合物加熱至75℃且每1 - 2 h藉由HPLC分析轉化%。當觀察到轉化率> 99%(通常在5 - 6 h後)後,使反應物冷卻至20 - 25℃並用MTBE (2 × 525 mL)萃取,在萃取期間進行足夠時間以使相完全分離。用5% NaCl (2 × 375 mL)洗滌經合併有機萃取物。然後,將溶液轉移至適於1.5 - 2.5托(Torr)真空蒸餾之設備,其配備有冷卻接收燒瓶。在<60℃下在真空下濃縮溶液以去除溶劑。隨後在125 - 130℃(烘箱溫度)及1.5 - 2.0托下自所得油蒸餾出(2,2-二氟-1,3-苯并二氧雜環戊烯-5-基)-乙腈。自5-溴-2,2-二氟-1,3-苯并二氧雜環戊烯分離產率為66%之澄清油(2,2-二氟-1,3-苯并二氧雜環戊烯-5-基)-乙腈(2個步驟),且HPLC純度為91.5% AUC (對應於95%之w/w分析)。
1
H NMR (500 MHz, DMSO) δ 7.44 (br s, 1H), 7.43 (d,
J
= 8.4 Hz, 1H), 7.22 (dd,
J
= 8.2, 1.8 Hz, 1H), 4.07 (s, 2H)。
(2,2- 二氟 -1,3- 苯并二氧雜環戊烯 -5- 基 )- 環丙烷甲腈之製備。 將(2,2-二氟-1,3-苯并二氧雜環戊烯-5-基)-乙腈(1.0當量)、50 wt% KOH水溶液(5.0當量)、1-溴-2-氯乙烷(1.5當量)與Oct
4
NBr(0.02當量)之混合物在70℃下加熱1 h。冷卻反應混合物,然後用MTBE及水處理。用水及鹽水洗滌有機相。去除溶劑以提供(2,2-二氟-1,3-苯并二氧雜環戊烯-5-基)-環丙烷甲腈。
1-(2,2- 二氟 -1,3- 苯并二氧雜環戊烯 -5- 基 )- 環丙烷甲酸之製備。 在80℃下使用存於乙醇(5體積)中之6 M NaOH(8當量)過夜水解(2,2-二氟-1,3-苯并二氧雜環戊烯-5-基)-環丙烷甲腈。使混合物冷卻至室溫且在真空下蒸發乙醇。用水及MTBE吸收殘餘物,添加1 M HCl且分離各層。然後,用二環己胺(DCHA) (0.97當量)處理MTBE層。使漿液冷卻至0℃,過濾並用庚烷洗滌以獲得對應DCHA鹽。用MTBE及10%檸檬酸吸收該鹽且將其攪拌直至所有固體皆溶解。分離各層且用水及鹽水洗滌MTBE層。將溶劑改為庚烷,隨後過濾,且在真空烘箱中於50℃下乾燥過夜後獲得1-(2,2-二氟-1,3-苯并二氧雜環戊烯-5-基)-環丙烷甲酸。
1-(2,2- 二氟 -1,3- 苯并二氧雜環戊烯 -5- 基 )- 環丙烷羰基氯之製備。 在甲苯(2.5體積)中漿化1-(2,2-二氟-1,3-苯并二氧雜環戊烯-5-基)-環丙烷甲酸(1.2當量)且將混合物加熱至60℃。經由加料漏斗添加SOCl
2
(1.4當量)。在30分鐘後自反應混合物蒸餾出甲苯及SOCl
2
。添加另一甲苯(2.5體積)且將所得混合物再次蒸餾,剩下呈油狀物之醯氯產物,其不進一步純化即使用。
3-(3- 甲基吡啶 -2- 基 ) 苯甲酸第三丁基酯之製備。 使2-溴-3-甲基吡啶(1.0當量)溶解於甲苯(12體積)中。依次添加K
2
CO
3
(4.8當量)及水(3.5體積)。在N
2
流下將所得混合物加熱至65℃並保持1小時。然後,添加3-(第三丁氧基羰基)苯基
酸(1.05當量)及Pd(dppf)Cl
2 ∙
CH
2
Cl
2
(0.015當量)且將混合物加熱至80℃。在2小時後,關閉加熱器,添加水(3.5體積),且使各層分離。然後,用水(3.5體積)洗滌有機相並用10%甲磺酸水溶液(2當量MsOH,7.7體積)萃取。用50% NaOH水溶液(2當量)使水相呈鹼性並用EtOAc(8體積)萃取。濃縮有機層以提供粗3-(3-甲基吡啶-2-基)苯甲酸第三丁基酯(82%),將其直接用於下一步驟中。
2-(3-( 第三丁氧基羰基 ) 苯基 )-3- 甲基吡啶 -1- 氧化物之製備。 使3-(3-甲基吡啶-2-基)苯甲酸第三丁基酯(1.0當量)溶解於EtOAc(6體積)中。依次添加水(0.3體積)及過氧化氫脲(3當量)。然後,以可將反應器中之溫度維持在45℃以下之速率將鄰苯二甲酸酐(3當量)逐份添加至呈固體形式之混合物中。在鄰苯二甲酸酐添加完成後,將混合物加熱至45℃。在再攪拌4小時後,關閉加熱器。經由加料漏斗添加10% w/w Na
2
SO
3
水溶液(1.5當量)。在Na
2
SO
3
添加完成後,將混合物再攪拌30 min並分離各層。攪拌有機層並添加10% wt/wt Na
2
CO
3
水溶液(2當量)。在攪拌30分鐘後,使各層分離。用13% w/v NaCl水溶液洗滌有機相。然後,過濾並濃縮有機相以提供粗2-(3-(第三丁氧基羰基)苯基)-3-甲基吡啶-1-氧化物(95%),將其直接用於下一步驟中。
3-(6- 胺基 -3- 甲基吡啶 -2- 基 ) 苯甲酸第三丁基酯之製備。 將2-(3-(第三丁氧基羰基)苯基)-3-甲基吡啶-1-氧化物(1當量)及吡啶(4當量)存於MeCN (8體積)中之溶液加熱至70℃。經50 min經由加料漏斗添加甲磺酸酐(1.5當量)存於MeCN (2體積)中之溶液,同時將溫度維持在75℃以下。在添加完成後將混合物再攪拌0.5小時。然後,使混合物冷卻至環境溫度。經由加料漏斗添加乙醇胺(10當量)。在攪拌2小時後,添加水(6體積)並使混合物冷卻至10℃。在攪拌3小時後,藉由過濾收集固體並用水(3體積)、2:1乙腈/水(3體積)及乙腈(2 × 1.5體積)洗滌。在真空烘箱中於50℃及輕微N
2
吹掃下將固體乾燥至恒重(差異<1%)以提供呈紅-黃色固體形式之3-(6-胺基-3-甲基吡啶-2-基)苯甲酸第三丁基酯(產率53%)。
3-(6-(1-(2,2- 二氟苯并 [d][1,3] 二氧雜環戊烯 -5- 基 ) 環丙烷甲醯胺基 )-3- 甲基吡啶 -2- 基 )- 苯甲酸第三丁基酯之製備。 使上文所述粗醯氯溶解於甲苯(以醯氯計,2.5體積)中,且經由加料漏斗將其添加至3-(6-胺基-3-甲基吡啶-2-基)苯甲酸第三丁基酯(1當量)、DMAP (0.02當量)與三乙胺(3.0當量)存於甲苯(以3-(6-胺基-3-甲基吡啶-2-基)苯甲酸第三丁基酯計,4體積)之混合物中。在2小時後,將水(以3-(6-胺基-3-甲基吡啶-2-基)苯甲酸第三丁基酯計,4體積)添加至反應混合物中。在攪拌30分鐘後,分離各層。然後,過濾並濃縮有機相以提供3-(6-(1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲醯胺基)-3-甲基吡啶-2-基)-苯甲酸第三丁基酯之濃稠油(定量粗產率)。添加乙腈(以粗產物計,3體積)且蒸餾直至發生結晶。添加水(以粗產物計,2體積)且將混合物攪拌2 h。藉由過濾收集固體,用1:1 (體積比)乙腈/水(以粗產物計,2 × 1體積)洗滌,且在真空下於過濾器上進行部分乾燥。在真空烘箱中於60℃及輕微N
2
吹掃下將固體乾燥至恒重(差異<1%)以提供呈褐色固體形式之3-(6-(1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲醯胺基)-3-甲基吡啶-2-基)-苯甲酸第三丁基酯。
3-(6-(1-(2,2- 二氟苯并 [d][1,3] 二氧雜環戊烯 -5- 基 ) 環丙烷甲醯胺基 )-3- 甲基吡啶 -2- 基 ) 苯甲酸 • HCL 鹽之製備。 向3-(6-(1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲醯胺基)-3-甲基吡啶-2-基)-苯甲酸第三丁基酯(1.0當量)存於MeCN(3.0體積)中之漿液中依次添加水(0.83體積)及濃HCl水溶液(0.83體積)。將混合物加熱至45 ± 5℃。在攪拌24 h至48 h後,反應完成,且使混合物冷卻至環境溫度。添加水(1.33體積)並攪拌混合物。藉由過濾收集固體,用水(2 × 0.3體積)洗滌,且在真空下於過濾器上進行部分乾燥。在真空烘箱中於60℃及輕微N
2
吹掃下將固體乾燥至恒重(差異<1%)以提供呈灰白色固體形式之3-(6-(1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲醯胺基)-3-甲基吡啶-2-基)苯甲酸• HCl。 化合物1之
1
HNMR光譜顯示於圖8中且圖9繪示呈HCl鹽形式之化合物1之
1
HNMR光譜。下表2列述化合物I之
1
HNMR數據。
表 2 。 化合物 1 形式 I 之製備 化合物 1 形式 I 之製備 , 方法 A 。 在環境溫度下攪拌3-(6-(1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲醯胺基)-3-甲基吡啶-2-基)苯甲酸• HCl (1當量)存於水(10體積)中之漿液。在攪拌24小時後取樣。過濾試樣且用水(2次)洗滌固體。對固體試樣實施DSC分析。當DSC分析指示完全轉化為形式I時,藉由過濾收集固體,用水(2 × 1.0體積)洗滌,且在真空下於過濾器上進行部分乾燥。然後,在真空烘箱中於60℃及輕微N
2
吹掃下將固體乾燥至恒重(差異<1%)以提供呈灰白色固體形式之化合物1形式I (產率98%)。
1
H NMR (400 MHz, DMSO-d6) 9.14 (s, 1H), 7.99-7.93 (m, 3H), 7.80-7.78 (m, 1H), 7.74-7.72 (m, 1H), 7.60-7.55 (m, 2H), 7.41-7.33 (m, 2H), 2.24 (s, 3H), 1.53-1.51 (m, 2H), 1.19-1.17 (m, 2H)。
化合物 1 形式 I 之製備 , 方法 B 。 將3-(6-(1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲醯胺基)-3-甲基吡啶-2-基)-苯甲酸第三丁基酯(1.0當量)存於甲酸(3.0體積)中之溶液在攪拌下加熱至70 ± 10℃並保持8 h。當藉由層析方法確定3-(6-(1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲醯胺基)-3-甲基吡啶-2-基)-苯甲酸第三丁基酯保持不超過1.0% AUC時,認為反應完成。使混合物冷卻至環境溫度。將溶液添加至水(6體積)中,在50℃下加熱,並攪拌混合物。然後,將混合物加熱至70 ± 10℃直至3-(6-(1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲醯胺基)-3-甲基吡啶-2-基)-苯甲酸第三丁基酯之量不超過0.8% (AUC)。藉由過濾收集固體,用水(2 × 3體積)洗滌,且在真空下於過濾器上進行部分乾燥。然後在真空烘箱中於60℃及輕微N
2
吹掃下將固體乾燥至恒重(差異<1%)以提供呈灰白色固體形式之化合物1形式I。化合物1形式I之DSC跡線顯示於圖10中。化合物1形式I在約204℃下發生熔化。自化合物1形式I之單晶結構計算X射線繞射圖案且顯示於圖1中。表3列示圖1中之計算峰。
表 3 。
化合物1形式I之實際X射線粉末繞射圖案顯示於圖2中。表4列示圖2中之實際峰。
表 4 。
藉由以0.2℃/min之速率使濃1-丁醇溶液自75℃冷卻至10℃來獲得化合物1形式I之無色晶體。選擇尺寸為0.50 × 0.08 × 0.03 mm之晶體,用礦物油清潔,將其安放於MicroMount上且定位於Bruker
APEX
II系統中央。獲得在倒易空間中分離之3批40個訊框以提供取向矩陣及初始晶胞參數。獲得最終晶胞參數並根據全數據集合進行精修。 對於每一訊框以0.82 Å之解析度使用0.5°步長且使用30 s暴露獲得倒易空間之繞射數據集合。在100 (2) K下收集數據。使用APEXII軟體對強度進行積分並精修晶胞參數在數據收集之後,觀察到晶體並未顯示降格跡象。化合物1形式I基於單晶X射線分析之構象圖片顯示於圖11中。化合物1形式I係單斜晶P
2
1/n,且其具有以下單胞尺寸:a=4.9626(7) Å,b=12.299(2) Å,c=33.075 (4) Å,β=93.938(9)°,V=2014.0 Å
3
,Z=4。在100 K下自結構數據計算之化合物1形式I之密度為1.492 g/cm
3
。
化合物 2 之製備 4- 側氧基 -1,4- 二氫喹啉 -3- 甲酸 (26) 之合成 4- 側氧基 -1,4- 二氫喹啉 -3- 甲酸乙酯 (25) 之製備程序 經0.5小時在低於30℃下將化合物
23
(4.77 g, 47.7 mmol)逐滴添加至化合物
22
(10 g, 46.3 mmol)中,且利用表面下N
2
流逐出乙醇。然後,將溶液加熱至100-110℃並攪拌2.5小時。在將混合物冷卻至60℃以下後,添加二苯基醚。將所得溶液逐滴添加至已加熱至228-232℃並保持1.5小時之二苯基醚中且利用表面下N
2
流逐出乙醇。將混合物在228-232℃下再攪拌2小時,冷卻至100℃以下,且然後添加庚烷以沈澱產物。將所得漿液在30℃下攪拌0.5小時。然後,過濾固體,並用庚烷洗滌濾餅且在真空中乾燥以獲得呈褐色固體形式之化合物
25
。
1
H NMR (DMSO-d
6
;400 MHz) δ 12.25 (s), δ 8.49 (d), δ 8.10 (m), δ 7.64 (m), δ 7.55 (m), δ 7.34 (m), δ 4.16 (q), δ 1.23 (t)。
4- 側氧基 -1,4- 二氫喹啉 -3- 甲酸 (26) 之製備程序 方法 1
將化合物
25
(1.0當量)懸浮於HCl (10.0當量)及H
2
O (11.6體積)之溶液中。將漿液加熱至85 – 90℃,但替代溫度亦適於此水解步驟。例如,另一選擇為,水解可在約75℃至約100℃之溫度下實施。在一些情形下,水解係在約80℃至約95℃之溫度下實施。在其他情形下,水解步驟係在約82℃至約93℃ (例如,約82.5℃至約92.5℃或約86℃至約89℃)之溫度下實施。在85 – 90℃下攪拌大約6.5小時後,對反應物取樣以判斷反應是否完成。可在適於水解之任一溫度下實施攪拌。然後,將溶液冷卻至20 – 25℃並過濾。用H
2
O (2體積× 2)沖洗反應器/濾餅。然後,用2體積H
2
O洗滌濾餅直至pH ≥ 3.0。然後,在真空及60℃下乾燥濾餅以獲得化合物
26
。
方法 2
將化合物
25
(11.3 g, 52 mmol)添加至10% NaOH (水溶液) (10 mL)與乙醇(100 mL)之混合物中。將溶液加熱至回流16小時,冷卻至20-25℃,且然後,用8% HCl將pH調節至2-3。然後,將混合物攪拌0.5小時並過濾。用水(50 mL)洗滌濾餅,且然後在真空中乾燥以獲得呈褐色固體形式之化合物
26
。
1
H NMR (DMSO-d
6
;400 MHz) δ 15.33 (s), δ 13.39 (s), δ 8.87 (s), δ 8.26 (m), δ 7.87 (m), δ 7.80 (m), δ 7.56 (m)。
N-(2,4- 二 - 第三丁基 -5- 羥基苯基 )-4- 側氧基 -1,4- 二氫喹啉 -3- 甲醯胺 ( 化合物 2) 之總合成 碳酸 2,4- 二 - 第三丁基苯基酯甲基酯 (30) 之製備程序 方法 1
在0℃下向2,4-二-第三丁基酚
29
(10 g, 48.5mmol)於二乙醚(100 mL)及三乙胺(10.1 mL, 72.8 mmol)中之溶液中逐滴添加氯甲酸甲酯(7.46 mL, 97 mmol)。然後,使混合物升溫至室溫並再攪拌2小時。然後,添加另5 mL三乙胺及3.7 mL氯甲酸甲酯,並將反應物攪拌過夜。然後,過濾反應物,將濾液冷卻至0℃,且然後,添加另5 mL三乙胺及3.7 mL氯甲酸甲酯,並使反應物升溫至室溫,且然後,再攪拌1小時。在此階段,反應幾乎完全並藉由過濾處理,然後,依次用水(2×)及鹽水洗滌。然後,濃縮溶液以產生黃色油,並使用管柱層析純化以獲得化合物
30
。
1
H NMR (400 MHz, DMSO-
d 6
) δ 7.35 (d,
J
= 2.4 Hz, 1H), 7.29 (dd,
J
= 8.4, 2.4 Hz, 1H), 7.06 (d,
J
= 8.4 Hz, 1H), 3.85 (s, 3H), 1.30 (s, 9H), 1.29 (s, 9H)。
方法 2
向裝填有4-二甲基胺基吡啶(DMAP, 3.16 g, 25.7 mmol)及2,4-二第三-丁基酚(化合物
29
, 103.5 g, 501.6 mmol)之反應器器皿中添加二氯甲烷(415 g, 313 mL),並攪動溶液直至所有固體皆溶解。然後,添加三乙胺(76 g, 751 mmol),並將溶液冷卻至0 – 5℃。然後,經2.5 – 4小時逐滴添加氯甲酸甲酯(52 g, 550.3 mmol),同時使溶液溫度保持介於0至5℃之間。然後,將反應混合物緩慢加熱至23 – 28℃並攪拌20小時。然後,將反應物冷卻至10 – 15℃並用150 mL水裝填。將混合物在15 – 20℃下攪拌35 – 45分鐘,且然後,分離水層並用150 mL二氯甲烷萃取。合併有機層並在5 – 20℃之溫度下用2.5% HCl (水溶液)中和,以使最終pH為5 – 6。然後,用水洗滌有機層並在真空中在低於20℃之溫度下濃縮至150 mL,以獲得存於二氯甲烷中之化合物
30
。
碳酸 5- 硝基 -2,4- 二 - 第三丁基苯基酯甲基酯 (31) 之製備程序 方法 1
在0℃下向化合物
30
(6.77g, 25.6 mmol)之攪拌溶液中逐滴添加6 mL硫酸及硝酸之1:1混合物。使混合物升溫至室溫並攪拌1小時。使用液相層析(ISCO, 120 g, 0-7% EtOAc/己烷, 38 min)純化產物,從而產生化合物
31
之區域異構體之
呈
白色固體形式之約8:1 – 10:1混合物。
1
H NMR (400 MHz, DMSO-
d 6
) δ 7.63 (s, 1H), 7.56 (s, 1H), 3.87 (s, 3H), 1.36 (s, 9H), 1.32 (s, 9H)。HPLC滯留時間3.92 min,10-99% CH
3
CN,運行5 min;ESI-MS 310 m/z (MH)
+
。
方法 2
向化合物
30
(100g, 378 mmol)中添加DCM (540 g, 408 mL)。攪拌混合物直至所有固體皆溶解,且然後,冷卻至-5 – 0℃。然後,逐滴添加濃硫酸(163 g),同時維持反應物之初始溫度並將混合物攪拌4.5小時。然後,經2-4小時逐滴添加硝酸(62 g),同時維持反應物之初始溫度,且然後,在此溫度下再攪拌4.5小時。然後,將反應混合物緩慢添加至冷水中,將溫度維持在5℃以下。然後,將淬滅反應物加熱至25℃,並去除水層且用二氯甲烷萃取。用水洗滌經合併有機層,使用Na
2
SO
4
乾燥,並濃縮至124 – 155 mL。添加己烷(48 g),並將所得混合物再濃縮至124 – 155 mL。隨後,將更多己烷(160 g)添加至混合物中。然後,將混合物在23 – 27℃下攪拌15.5小時,且然後進行過濾。向濾餅中添加己烷(115 g),將所得混合物加熱至回流並攪拌2 – 2.5小時。然後,將混合物冷卻至3 – 7℃,再攪拌1 – 1.5小時,並過濾以獲得呈淺黃色固體形式之化合物
31
。
碳酸 5- 胺基 -2,4- 二 - 第三丁基苯基酯甲基酯 (32) 之製備程序 將碳酸2,4-二-第三丁基-5-硝基苯基酯甲基酯(1.00當量)及5% Pd/C (2.50 wt%乾基, Johnson-Matthey Type 37)依次裝填至適宜氫化反應器中。將MeOH (15.0體積)裝填至反應器中,並關閉系統。用N
2
(g)吹掃系統,且然後,用H
2
(g)加壓至2.0巴。在25℃ +/- 5℃之反應溫度下實施反應。當完成時,過濾反應物,並用MeOH (4.00體積)洗滌反應器/濾餅。將所得濾液在真空下在不超過50℃下蒸餾至8.00體積。在45℃ +/- 5℃下添加水(2.00體積)。將所得漿液冷卻至0℃ +/- 5。將漿液在0℃ +/- 5℃下保持不小於1小時,並過濾。用0℃ +/- 5℃ MeOH/H
2
O (8:2) (2.00體積)將濾餅洗滌一次。在真空(-0.90巴及-0.86巴)及35℃–40℃下乾燥濾餅,以獲得化合物
32
。
1
H NMR (400 MHz, DMSO-
d 6
) δ 7.05 (s, 1H), 6.39 (s, 1H), 4.80 (s, 2H), 3.82 (s, 3H), 1.33 (s, 9H), 1.23 (s, 9H)。 在完成反應後,用約5體積至10體積MeOH (例如,約6體積至約9體積MeOH,約7體積至約8.5體積MeOH,約7.5體積至約8體積MeOH,或約7.7體積MeOH)稀釋所得混合物,將其加熱至約35 ± 5℃之溫度,過濾,洗滌,並乾燥,如上文所闡述。
N-(2,4- 二 - 第三丁基 -5- 羥基苯基 )-4- 側氧基 -1,4- 二氫喹啉 -3- 甲醯胺 ( 化合物 2) 之製備。 將4-側氧基-1,4-二氫喹啉-3-甲酸
26
(1.0當量)及碳酸5-胺基-2,4-二-第三丁基苯基酯甲基酯
32
(1.1當量)裝填至反應器中。依次添加2-MeTHF (4.0體積,相對於酸)及T3P
®
於2-MeTHF中之50%溶液(1.7當量)。用2-MeTHF (0.6體積)洗滌裝填T3P之器皿。然後,添加吡啶(2.0當量),並將所得懸浮液加熱至47.5 +/- 5.0℃並在此溫度下保持8小時。採取試樣並藉由HPLC檢查完成情況。在完成後,將所得混合物冷卻至25.0℃ +/- 2.5℃。添加2-MeTHF (12.5體積)以稀釋混合物。用水(10.0體積)將反應混合物洗滌2次。添加2-MeTHF以使反應物之總體積達到40.0體積(裝填約16.5體積)。向此溶液中添加NaOMe/MeOH (1.7當量)以實施甲醇解。將反應物攪拌1.0小時以上,並藉由HPLC檢查完成情況。在完成後,用1 N HCl (10.0體積)淬滅反應物,並用0.1 N HCl (10.0體積)洗滌。將有機溶液精細過濾(polish filter)以去除任何微粒,並將其置於另一反應器中。在不大於35℃ (夾套溫度)且不小於8.0℃ (內部反應溫度)下在減壓下將過濾溶液濃縮至20體積。添加CH
3
CN至40體積,並在不大於35℃ (夾套溫度)且不小於8.0℃ (內部反應溫度)下將溶液濃縮至20體積。將CH
3
CN之添加及濃縮循環重複2次以上,總共3次添加CH
3
CN且4次濃縮至20體積。在最終濃縮至20體積後,依次添加16.0體積CH
3
CN及4.0體積H
2
O,從而使得10% H
2
O/CH
3
CN相對於起始酸最終濃縮為40體積。將此漿液加熱至78.0℃ +/- 5.0℃ (回流)。然後,將漿液攪拌5小時以上。經5小時將漿液冷卻至0.0℃ +/- 5℃,並過濾。用0.0℃ +/- 5.0℃ CH
3
CN (5體積)將濾餅洗滌4次。在真空烘箱中在50.0℃ +/- 5.0℃下乾燥所得固體(化合物2)。
1
H NMR (400 MHz, DMSO-
d 6
) δ 12.8 (s, 1H), 11.8 (s, 1H), 9.2 (s, 1H), 8.9 (s, 1H), 8.3 (s, 1H), 7.2 (s, 1H), 7.9 (t, 1H), 7.8 (d, 1H), 7.5 (t, 1H), 7.1 (s, 1H), 1.4 (s, 9H), 1.4 (s, 9H)。
N-(2,4- 二 - 第三丁基 -5- 羥基苯基 )-4- 側氧基 -1,4- 二氫喹啉 -3- 甲醯胺 ( 化合物 2) 之替代製備。 將4-側氧基-1,4-二氫喹啉-3-甲酸
26
(1.0當量)及碳酸5-胺基-2,4-二-第三丁基苯基酯甲基酯
32
(1.1當量)裝填至反應器中。依次添加2-MeTHF (4.0體積,相對於酸)及T3P
®
於2-MeTHF中之50%溶液(1.7當量)。用2-MeTHF (0.6體積)洗滌裝填T3P之器皿。然後,添加吡啶(2.0當量),並將所得懸浮液加熱至47.5 +/- 5.0℃並在此溫度下保持8小時。採取試樣,並藉由HPLC檢查完成情況。在完成後,將所得混合物冷卻至20℃ +/- 5℃。添加2-MeTHF (12.5體積)以稀釋混合物。用水(10.0體積)將反應混合物洗滌2次,並將2-MeTHF (16.5體積)裝填至反應器中。向此溶液中裝填30% w/w NaOMe/MeOH (1.7當量)以實施甲醇解。將反應物在25.0℃ +/- 5.0℃下攪拌1.0小時以上,並藉由HPLC檢查完成情況。在完成後,用1.2 N HCl/H
2
O (10.0體積)淬滅反應物,並用0.1 N HCl/H
2
O (10.0體積)洗滌。將有機溶液精細過濾以去除任何微粒,並將其置於另一反應器中。 在不超過35℃ (夾套溫度)且不小於8.0℃ (內部反應溫度)下在減壓下將過濾溶液濃縮至20體積。添加CH
3
CN至40體積,並在不超過35℃(夾套溫度)且不小於8.0℃(內部反應溫度)下將溶液濃縮至20體積。將CH
3
CN之添加及濃縮循環重複2次以上,總共3次添加CH
3
CN且4次濃縮至20體積。在最終濃縮至20體積後,依次裝填16.0體積CH
3
CN及4.0體積H
2
O,從而使得10% H
2
O/CH
3
CN相對於起始酸最終濃縮為40體積。將此漿液加熱至78.0℃ +/- 5.0℃ (回流)。然後,將漿液攪拌5小時以上。經5小時將漿液冷卻至20℃至25℃,並過濾。用加熱至20℃至25℃之CH
3
CN (5體積)將濾餅洗滌4次。在真空烘箱中在50.0℃ +/- 5.0℃下乾燥所得固體(化合物2)。
1
H NMR (400 MHz, DMSO-
d 6
) δ 12.8 (s, 1H), 11.8 (s, 1H), 9.2 (s, 1H), 8.9 (s, 1H), 8.3 (s, 1H), 7.2 (s, 1H), 7.9 (t, 1H), 7.8 (d, 1H), 7.5 (t, 1H), 7.1 (s, 1H), 1.4 (s, 9H), 1.4 (s, 9H)。
N-(2,4- 二 - 第三丁基 -5- 羥基苯基 )-4- 側氧基 -1,4- 二氫喹啉 -3- 甲醯胺 ( 化合物 2) 之重結晶程序 將化合物2 (1.0當量)裝填至反應器中。依次添加2-MeTHF (20.0體積)及0.1N HCl (5.0體積)。攪拌雙相溶液並分離,且用0.1N HCl (5.0體積)將上部有機相洗滌兩次以上。將有機溶液精細過濾以去除任何微粒,並將其置於另一反應器中。在不超過35℃ (夾套溫度)且不低於8.0℃ (內部反應溫度)下在減壓下將過濾溶液濃縮至10體積。添加乙酸異丙酯(IPAc) (10體積),並在不超過35℃ (夾套溫度)且不低於8.0℃ (內部反應溫度)下將溶液濃縮至10體積。將IPAc之添加及濃縮重複2次以上,總共3次添加IPAc且4次濃縮至10體積。在最終濃縮後,裝填10體積IPAc,並將漿液加熱至回流並維持在此溫度下達5小時。經5小時將漿液冷卻至0.0℃ +/- 5℃,並過濾。用IPAc (5體積)將濾餅洗滌一次。在真空烘箱中在50.0℃ +/- 5.0℃下乾燥所得固體。
包含實質上非晶形化合物 2 之固體分散體之製備
在配備有磁力攪拌器及熱循環之反應器中將MEK及DI水根據比率90 wt% MEK / 10 wt% DI水調配之溶劑系統加熱至20 - 30℃之溫度。根據比率19.5 wt%乙酸琥珀羥丙甲纖維素/ 0.5 wt% SLS / 80 wt%化合物2向此溶劑系統中添加乙酸琥珀羥丙甲纖維素聚合物(HPMCAS) (HG級)、SLS及化合物2。所得混合物含有10.5 wt%固體。生成此混合物之成份及溶劑之實際用量列示於下表5中:
表 5
:中間體F之固體噴霧分散體成份。
將混合物溫度調節至20 - 45℃之範圍並進行混合直至其實質上均勻且所有組份皆實質上溶解。 遵循列示於下表6中之乾燥噴霧製程參數,在正常噴霧乾燥模式下使用噴霧乾燥器Niro PSD4商業噴霧乾燥器,其安裝有壓力噴嘴(Spray Systems Maximum Passage系列SK-MFP,其具有孔口/核心大小54/21),該壓力噴嘴配備有抗倒流帽(anti-bearding cap)。
表 6
:用以生成中間體F之乾燥噴霧製程參數。
高效旋風器自噴霧氣體及溶劑蒸氣分離濕產物。該濕產物含有8.5 – 9.7% MEK及0.56 – 0.83%水且具有17 – 19 µm之平均粒徑及0.27 – 0.33 g/cc之堆密度。將濕產物轉移至4000 L不銹鋼雙錐形真空乾燥器中進行乾燥,以將殘餘溶劑降低之小於約5000 ppm之水準,且生成非晶形化合物2之乾燥噴霧乾燥分散體,該乾燥分散體含有<0.03% MEK及0.3%水。
自完全連續濕法製粒製程形成錠劑 設備 / 製程 設備
完全連續Development and Launch Rig (DLR)或類似類型設備。
篩選
可將化合物1形式I、包含實質上非晶形化合物2之固體分散體及賦形劑分配於單獨中間箱式容器(IBC)中。該等材料可使用「箱至箱(bin-to-bin)」篩選操作來篩選。合適網篩大小為20網目、40網目或60網目。
摻合
可將含有經篩選化合物1形式I、包含實質上非晶形化合物2之固體分散體及賦形劑之IBC對接至進料機系統,該系統可以受控方式(例如在重量進料機中使用體積或重量損失)將材料進給至連續摻合機中。個別組份之進料速率係由調配組合物及總體線路速率來界定。線路速率可為8 kg/hr至30 kg/hr。連續摻合機可具有不同的片組態以容許進行合適摻合,且該等片之旋轉速度可介於80 RPM與300 RPM之間。
濕法製粒
可藉由在不銹鋼容器中使用頂置攪拌器以700 RPM之攪拌速度將48 g月桂基硫酸鈉及159 g聚乙烯吡咯啶酮溶解於1,626 g水中來製備製粒溶液。可將製粒溶液置於容器中,可使用具有質量流量計及控制之蠕動幫浦使用適於製程之流速將溶液自該容器泵送至雙螺桿製粒機中。可使用雙螺桿製粒機(例如為DLR之一部分之製粒機)將摻合物製粒。可使用失重式進料機(例如DLR上之K-Tron進料機)以8 kg/hr至24 kg/hr之進料速率將摻合物添加至雙螺桿製粒機中。雙螺桿製粒機可以25℃之桶溫及200 RPM至950 RPM之螺桿轉速操作。製粒製程對於小批量可實施三分鐘或對於大批量可實施若干小時。
乾燥
可將濕顆粒直接進給至流體床乾燥器,例如DLR上之分段流體床乾燥器。可在排放期間在40℃至55℃範圍內之溫度下選擇產物乾燥終點,顆粒在該點下之水含量可為2.1 %w/w (「乾燥損失,LOD」)或更小。達到期望乾燥終點之乾燥時間可為12分鐘或更短或更長。
研磨
可研磨乾燥顆粒以減小顆粒之大小。此可使用錐形磨,例如整合式Quadro U10 CoMil。
摻合
可使用失重式進料機及連續摻合機將顆粒與顆粒外賦形劑(例如填充劑及潤滑劑)摻合。摻合速度可為80 – 300 RPM。
壓製
可使用為DLR系統之一部分之單工位或旋轉壓錠機(例如Courtoy Modul P壓機)使用大小合適的工具將壓製摻合物壓製成錠劑。對於200 mg化合物1形式I及125 mg實質上非晶形化合物2之劑量,錠劑之重量可為約500 mg或600 mg。
薄膜包覆
可使用創新型Omega薄膜包覆機對錠劑實施薄膜包覆,該包覆機為DLR系統之一部分。此包覆機使得能夠對1 kg至4 kg之亞批進行快速薄膜包覆從而容許連續製造。
印刷
可利用(例如) Ackley ramp印刷機在薄膜衣錠之一或兩個錠劑面上印刷交織字母。
自雙螺桿濕法製粒製程形成錠劑 設備 / 製程 設備
雙螺桿濕法製粒機:ConsiGma-1、ConsiGma-25或Leistritz nano。
篩選 / 稱量
可在稱量出之前或之後篩選化合物1形式I、包含實質上非晶形化合物2之固體分散體及賦形劑。合適網篩大小為20網目、40網目或60網目。化合物1形式I及/或包含實質上非晶形化合物2之固體分散體可與一或多種賦形劑預摻合以簡化篩選。
摻合
可將化合物1形式I、包含實質上非晶形化合物2之固體分散體及賦形劑以不同順序添加至摻合機中。可在Turbula摻合機、v-殼摻合機或箱式摻合機中實施摻合。可將組份摻合10分鐘。
濕法製粒
可藉由在不銹鋼容器中使用頂置攪拌器以700 RPM之攪拌速度將48 g月桂基硫酸鈉及159 g 聚乙烯吡咯啶酮溶解於1,626 g水中來製備製粒溶液。可使用雙螺桿製粒機(例如ConsiGma-1)將摻合物製粒。可使用蠕動幫浦(例如ConsiGma-1上之幫浦)以67 g/min之進料速率將製粒溶液添加至雙螺桿製粒機中。可使用失重式進料機(例如ConsiGma-1上之Brabender進料機)以10 kg/hr之進料速率將摻合物添加至雙螺桿製粒機中。雙螺桿製粒機可以25℃之桶溫及400 RPM之螺桿轉速操作。製粒製程可實施四分鐘。製粒製程可實施更短或更長之持續時間以產生更少量或更大量的濕顆粒。
乾燥
可將濕顆粒直接進給至流體床乾燥器中,例如ConsiGma-1上之乾燥室或CTL-25上之分段流體床乾燥器。可在43℃之產物溫度下選擇乾燥終點,顆粒在該點下之水含量可為1.6%w/w (「乾燥損失,LOD」)。達到期望乾燥終點之乾燥時間可為12分鐘或更短或更長。可以59 m
3
/min之空氣流量及60℃之入口溫度實施乾燥。另一選擇為,可將來自雙螺桿製粒機之濕顆粒收集至箱或容器中並保持一定時間段,此後將濕顆粒轉移至單獨獨立流體床乾燥器(例如Vector Multi 15)中。
研磨
可研磨乾燥顆粒以減小顆粒之大小。此可使用錐形磨,例如Quadro 194 CoMil。
摻合
可使用V-殼摻合機或箱式摻合機摻合顆粒與顆粒外賦形劑(例如填充劑及潤滑劑)。摻合時間可為5分鐘、3分鐘或1分鐘。
壓製
可使用單工位或旋轉壓錠機(例如Courtoy Modul P壓機)使用0.55’ × 0.33’橢圓形工具將壓製摻合物壓製成錠劑。對於200 mg化合物1形式I及125 mg實質上非晶形化合物2之劑量,錠劑之重量可為約500 mg或600 mg。
薄膜包覆
可使用鍋式包覆機(例如Thomas Engineering Compu-Lab包覆機)對錠劑實施薄膜包覆。可添加痕量巴西棕櫚蠟來改良錠劑外觀及製程能力。
印刷
可利用(例如) Hartnett Delta印刷機在薄膜衣錠之一或兩個錠劑面上印刷交織字母。
自連續雙螺桿濕法製粒製程形成錠劑 設備 / 製程 設備
製粒機:ConsiGma或Leistritz或Thermo Fisher雙螺桿製粒機。
篩選 / 稱量
可在稱量出之前或之後篩選化合物1及賦形劑。可能的網篩大小係20網目、40網目或60網目。化合物1可與一或多種賦形劑預摻合以簡化篩選。
摻合
可將化合物1及賦形劑以不同順序添加至摻合機中。可在Turbula摻合機、v-殼摻合機、箱式摻合機或連續摻合機中實施摻合。對於分批摻合機可將組份摻合10分鐘,或對於連續摻合機可連續摻合組份。
製粒操作
製粒流體-將SLS及黏合劑添加至純化水中並進行混合直至溶解。適宜比率係存於水中之2.5% w/w SLS及10.0% w/w PVP K30。 製粒–可使用失重式進料機以10 kg/hr之速率將含有化合物1及賦形劑之摻合物投配至雙螺桿製粒機中。可使用蠕動幫浦以3.5 kg/hr之速率添加製粒流體。製粒機可以400 RPM之速度運行。本發明雙螺桿濕法製粒製程之一個顯著優點係使用包含表面活性劑與黏合劑二者之製粒流體,從而可藉助增加之可濕性而更好地製粒。在一個實施例中,表面活性劑係SLS。另一顯著優點在於,由於製程係連續的且在任一時刻僅處理有限量的材料,故製程可充分受控且產生高品質產品。
研磨
可在乾燥之前或在乾燥之後或乾燥前後均使用篩磨或錐形磨來減小顆粒之大小。
乾燥
可使用真空烘箱、盤式乾燥器、雙錐形乾燥器或流體床乾燥器乾燥顆粒。
摻合
顆粒可與顆粒外賦形劑摻合。使用300公升箱式摻合機將顆粒摻合60轉。
壓製
使用Courtoy Modul P旋轉壓機將壓製摻合物壓製成錠劑。
薄膜包覆
可使用鍋式包覆機(例如O’Hara Labcoat)對錠劑實施薄膜包覆。
印刷
可利用(例如) Hartnett Delta印刷機在薄膜衣錠之一或兩個錠劑面上印刷交織字母。
分析 方案 1 檢測及量測化合物之 Δ F508-CFTR 增效性質之分析 分析化合物之 Δ F508-CFTR 調節性質之膜電勢光學方法
該分析利用螢光電壓敏感性染料以使用螢光讀板器(例如,FLIPR III, Molecular Devices公司)來量測膜電勢變化,作為NIH 3T3細胞中之功能ΔF508-CFTR增加之讀出值。反應之驅動力係藉由在預先利用化合物處理細胞且隨後使該等細胞加載有電壓敏感性染料後實施單一液體添加步驟所產生之氯離子梯度與通道活化。
增效劑化合物之鑑別
為鑑別ΔF508-CFTR之增效劑,實施雙加樣HTS分析格式。此HTS分析利用螢光電壓敏感性染料以在FLIPR III上量測膜電勢變化,作為溫度校正之ΔF508 CFTR NIH 3T3細胞中之ΔF508 CFTR之門控(傳導)增加之量度。反應之驅動力係在預先利用增效劑化合物(或DMSO媒劑對照)處理細胞且隨後使該等細胞加載有再分佈染料之後借助福司柯林(forskolin)在單一液體添加步驟中所產生之Cl
-
離子梯度與通道活化(使用螢光讀板器(例如FLIPR III)量測)。
溶液
1號浴液:(以mM計) NaCl 160、KCl 4.5、CaCl
2
2、MgCl
2
1、HEPES 10,用NaOH調節至pH 7.4。
無氯離子浴液
:用葡萄糖酸鹽取代1號浴液(上文)中之氯化物鹽。
細胞培養
使用穩定表現ΔF508-CFTR之NIH3T3小鼠成纖維細胞來對膜電勢實施光學量測。在存於175 cm
2
培養瓶中之補加有2 mM麩醯胺酸、10%胎牛血清、1 × NEAA、β-ME、1 ×青黴素/鏈黴素及25 mM HEPES之達爾伯克改良伊戈爾培養基(Dulbecco’s modified Eagle’s medium)中,將細胞維持在37℃及5% CO
2
及90%濕度下。對於所有光學分析而言,在384孔基質膠塗覆板中以約20,000/孔接種細胞且在37℃下培養2 hr,之後在27℃下培養24 hr以實施增效劑分析。對於校正分析而言,在27℃或37℃及存在或不存在化合物之情況下將細胞培養16小時至24小時。 分析化合物之ΔF508-CFTR調節性質之電生理分析。
尤斯室分析 (Ussing Chamber Assay)
對表現ΔF508-CFTR之極化氣道上皮細胞實施尤斯室實驗以進一步表徵在光學分析中鑑別之ΔF508-CFTR加強劑或誘導劑。枝氣管組織分離非CF及CF氣道上皮,如先前所闡述進行培養(Galietta, L.J.V., Lantero, S., Gazzolo, A., Sacco, O., Romano, L., Rossi, G.A.及Zegarra-Moran, O. (1998)
In Vitro Cell. Dev. Biol
.
34
, 478-481),並將其平鋪至預塗覆NIH3T3調節之培養基之Costar® Snapwell
TM
過濾器上。在四天後,去除頂端培養基,並使細胞在使用前在空氣液體界面處生長>14天。此產生單層完全分化的柱狀細胞,其具纖毛,該等特性屬於氣道上皮細胞之特徵。自不具有任一已知肺病之非吸煙者分離非CF HBE。自ΔF508同型合子患者分離CF-HBE。 將在Costar® Snapwell™細胞培養插件上生長之HBE安放於尤斯室(Physiologic Instruments公司, San Diego, CA)中,並使用電壓鉗系統(Department of Bioengineering, University of Iowa, IA)量測基底外側至頂端之Cl
-
梯度(I
SC
)存在下之跨上皮電阻及短路電流。簡言之,在電壓鉗記錄條件(V
保持
= 0 mV)在37℃下檢驗HBE。基底外側溶液含有(以mM計) 145 NaCl、0.83 K
2
HPO
4
、3.3 KH
2
PO
4
、1.2 MgCl
2
、1.2 CaCl
2
、10葡萄糖、10 HEPES (用NaOH調節至pH 7.35),且頂端溶液含有(以mM計) 145葡萄糖酸Na、1.2 MgCl
2
、1.2 CaCl
2
、10葡萄糖、10 HEPES (用NaOH調節至pH 7.35)。
增效劑化合物之鑑別
典型方案利用基底外側至頂膜之Cl
-
濃度梯度。為設定此梯度,在基底外側膜上使用正常林格氏溶液(ringers),而用等莫耳葡萄糖酸鈉(用NaOH滴定至pH 7.4)來替代頂端NaCl,以獲得較大跨上皮Cl
-
濃度梯度。將福斯柯林(10 μM)及所有測試化合物添加至細胞培養插件頂側。比較假定ΔF508-CFTR增效劑與已知增效劑染料木黃酮之功效。
膜片鉗記錄
如先前文獻所述使用穿孔膜片記錄組態監測∆F508-NIH3T3細胞中之總Cl
-
電流(Rae, J., Cooper, K., Gates, P.及Watsky, M. (1991)
J. Neurosci. Methods 37
, 15-26)。在22℃下使用Axopatch 200B膜片鉗放大器(Axon Instruments公司,Foster City, CA)實施電壓鉗記錄。吸管溶液含有(以mM計) 150
N
-甲基-D-還原葡糖胺(NMDG)-Cl、2 MgCl
2
、2 CaCl
2
、10 EGTA、10 HEPES及240 µg/mL兩性黴素B (amphotericin-B) (用HCl調節至pH 7.35)。細胞外培養基含有(以mM計) 150 NMDG-Cl、2 MgCl
2
、2 CaCl
2
、10 HEPES (用HCl調節至pH 7.35)。使用配備有Digidata 1320 A/D介面以及Clampex 8(Axon Instruments公司)之PC來實施脈衝生成、數據採集及分析。為活化ΔF508-CFTR,將10 µM福司柯林及20 µM染料木黃酮添加至浴液中,且每30秒監測電流-電壓關係。
增效劑化合物之鑑別
亦使用穿孔膜片記錄技術來研究ΔF508-CFTR增效劑在穩定表現ΔF508-CFTR之NIH3T3細胞中增大巨觀ΔF508-CFTR Cl
-
電流(I
Δ F508
)之能力。自光學分析鑑別之增效劑可引起IΔ
F508
之劑量依賴性增大,且效能及功效與在光學分析中所觀察到者類似。在所有經檢驗細胞中,在施加增效劑之前及期間之逆轉電勢為-30 mV左右,其為計算之E
Cl
(-28 mV)。
細胞培養
使用穩定表現ΔF508-CFTR之NIH3T3小鼠成纖維細胞來實施全細胞記錄。在存於175 cm
2
培養瓶中之補加有2 mM麩醯胺酸、10%胎牛血清、1 × NEAA、β-ME、1 ×青黴素/鏈黴素及25 mM HEPES之達爾伯克改良伊戈爾培養基中,將細胞維持在37℃及5% CO
2
及90%濕度下。對於全細胞記錄而言,將2,500 - 5,000個細胞接種於塗覆有聚L-離胺酸之玻璃蓋玻片上且在27℃下培養24 - 48 hr,之後使用其來測試增效劑之活性;且在37℃及存在或不存在校正化合物之情形下培育以量測校正劑之活性。
單通道記錄
如先前文獻中所闡述使用Axopatch 200B膜片鉗放大器(Axon Instruments公司)且使用經切除內面向外式膜片記錄觀察NIH3T3細胞中表現之wt-CFTR及溫度校正之∆F508-CFTR之門控活性(Dalemans, W., Barbry, P., Champigny, G., Jallat, S., Dott, K., Dreyer, D., Crystal, R.G., Pavirani, A., Lecocq, J-P., Lazdunski, M. (1991)
Nature
354, 526 – 528)。吸管含有(以mM計):150 NMDG、150天冬胺酸、5 CaCl
2
、2 MgCl
2
及10 HEPES (用Tris鹼調節至pH 7.35)。浴液含有(以mM計):150 NMDG-Cl、2 MgCl
2
、5 EGTA、10 TES及14 Tris鹼(用HCl調節至pH 7.35)。在切除後,藉由添加1 mM Mg-ATP、75 nM cAMP依賴性蛋白激酶催化亞單位(PKA;Promega公司Madison, WI)及10 mM NaF以抑制蛋白磷酸酶來活化wt-CFTR及∆F508-CFTR二者,從而防止電流減少。使吸管電勢維持在80 mV。自含有≤ 2個活性通道之膜片分析通道活性。在實驗過程期間同時開啟之最大數量決定活性通道數。為確定單通道電流幅值,以100 Hz「離線」過濾根據120秒之ΔF508-CFTR活性記錄之數據,且然後使用其來構築所有點之幅值直方圖,使用Bio-Patch分析軟體(Bio-Logic公司,France)將該等圖與複高斯函數(multigaussian function)擬合。根據120秒之通道活性測定總微觀電流及開放概率(P
o
)。使用Bio-Patch軟體或根據方程P
o
= I/i(N)來測定P
o
,其中I =平均電流,i =單通道電流幅值,且N =膜片中之活性通道數。
細胞培養
使用穩定表現ΔF508-CFTR之NIH3T3小鼠成纖維細胞來實施切除膜片鉗記錄。在存於175 cm
2
培養瓶中之補加有2 mM麩醯胺酸、10%胎牛血清、1 × NEAA、β-ME、1 ×青黴素/鏈黴素及25 mM HEPES之達爾伯克改良伊戈爾培養基中,將細胞維持在37℃及5% CO
2
及90%濕度下。對於單通道記錄而言,在塗覆有聚L-離胺酸之玻璃蓋玻片上接種2,500 - 5,000個細胞且在使用前於27℃下培育24 - 48 hr。
方案 2 檢測及量測化合物之 Δ F508-CFTR 校正性質之分析
分析化合物之ΔF508-CFTR調節性質之膜電勢光學方法 光學膜電勢分析利用Gonzalez及Tsien闡述之電壓敏感性FRET感測器(參見Gonzalez, J. E.及R. Y. Tsien (1995),「Voltage sensing by fluorescence resonance energy transfer in single cells」
Biophys J
69(4): 1272-80,及Gonzalez, J. E.及R. Y. Tsien (1997),「Improved indicators of cell membrane potential that use fluorescence resonance energy transfer」
Chem Biol
4(4): 269-77)以及量測螢光變化之儀錶,例如電壓/離子探針讀取器(VIPR) (
參見
Gonzalez, J. E., K. Oades等人(1999),「Cell-based assays and instrumentation for screening ion-channel targets」
Drug Discov Today
4(9): 431-439)。 該等電壓敏感性分析係基於膜溶性電壓敏感性染料DiSBAC
2
(3)與螢光磷脂CC2-DMPE (其附著至質膜外葉且用作FRET供體)之間螢光共振能量轉移(FRET)之變化。膜電勢(V
m
)之變化導致帶負電之DiSBAC
2
(3)在質膜兩側重新分配,且導致來自CC2-DMPE之能量轉移量發生相應變化。使用VIPR
TM
II來監測螢光發射變化,該儀器係經設計在96孔或384孔微量滴定板中實施細胞基篩選之整合液體處置器及螢光檢測器。
校正化合物之鑑別
為鑑別可校正與ΔF508-CFTR相關之輸送缺陷之小分子,實施單加樣HTS分析格式。於37℃下在存在或不存在測試化合物之(陰性對照)之無血清培養基中將細胞培育16 hr。作為陽性對照,在27℃下將平鋪於384孔板中之細胞培養16 hr以對ΔF508-CFTR實施「溫度校正」。隨後用克萊伯林格氏溶液(Krebs Ringers solution)將細胞沖洗3次並加載電壓敏感性染料。為活化ΔF508-CFTR,將10 μM福斯柯林及CFTR增效劑染料木黃酮(20 μM)連同無Cl
-
培養基一起添加至各孔中。添加無Cl
-
培養基可促進因應ΔF508-CFTR活化之Cl
-
外向通量,且使用FRET基電壓感測器染料以光學方式監測所導致之膜去極化。
增效劑化合物之鑑別
為鑑別ΔF508-CFTR之增效劑,實施雙加樣HTS分析格式。在第一次加樣期間,將含有或不含有測試化合物之無Cl
-
培養基添加至各孔中。在22秒後,添加含有2 μM至10 μM福司柯林之無Cl
-
培養基之第二次添加物以活化ΔF508-CFTR。在兩次添加後細胞外Cl
-
濃度為28 mM,此可促進因應ΔF508-CFTR活化之Cl
-
外向通量,且使用FRET基電壓感測器染料以光學方式監測所導致之膜去極化。
溶液
1號浴液: (以mM計):NaCl 160、KCl 4.5、CaCl
2
2、MgCl
2
1、HEPES 10,用NaOH調節至pH 7.4。 無氯離子浴液: 用葡萄糖酸鹽取代1號浴液(上文)中之氯化物鹽。 CC2-DMPE: 製備為存於DMSO中之10 mM原液並儲存在-20℃下。 DiSBAC
2
(3): 製備為存於DMSO中之10 mM原液並儲存在-20℃下。
細胞培養
使用穩定表現ΔF508-CFTR之NIH3T3小鼠成纖維細胞來對膜電勢實施光學量測。在存於175 cm
2
培養瓶中之補加有2 mM麩醯胺酸、10%胎牛血清、1 × NEAA、β-ME、1 ×青黴素/鏈黴素及25 mM HEPES之達爾伯克改良伊戈爾培養基中,將細胞維持在37℃及5% CO
2
及90%濕度下。對於所有光學分析而言,在384孔基質膠塗覆板中以30,000/孔接種細胞且在37℃下將其培養2 hr,之後在27℃下培養24 hr以實施增效劑分析。對於校正分析而言,在27℃或37℃及存在及不存在化合物之情況下將細胞培養16小時至24小時。 分析化合物之ΔF508-CFTR調節性質之電生理分析
尤斯室分析
對表現ΔF508-CFTR之極化上皮細胞實施尤斯室實驗以進一步表徵在光學分析中鑑別之ΔF508-CFTR加強劑或誘導劑。將在Costar Snapwell細胞培養插件上生長之FRT
ΔF508-CFTR
上皮細胞安放在尤斯室(Physiologic Instruments公司,San Diego, CA)中,且使用電壓鉗系統(Department of Bioengineering, University of Iowa, IA及Physiologic Instruments公司,San Diego, CA)來使單層持續短路。藉由施加2-mV脈衝來量測跨上皮電阻。在該等條件下,FRT上皮顯示4 KΩ/ cm
2
或更大之電阻。將溶液維持在27℃且用空氣鼓泡。使用無細胞插件來校正電極偏移電勢及流體電阻。在該等條件下,電流反映Cl
-
經由在頂膜中表現之ΔF508-CFTR之流動。使用MP100A-CE介面及AcqKnowledge軟體(3.2.6版;BIOPAC Systems,Santa Barbara, CA)以數位方式獲得I
SC
。
校正化合物之鑑別
典型方案利用基底外側至頂膜之Cl
-
濃度梯度。為設定此梯度,在基底外側膜上使用正常林格氏溶液,而用等莫耳葡萄糖酸鈉(用NaOH滴定至pH 7.4)來替代頂端NaCl,以獲得較大跨上皮Cl
-
濃度梯度。所有實驗皆係用完整單層來實施。為充分活化ΔF508-CFTR,施加福司柯林(10 μm)及PDE抑制劑IBMX (100 μm),之後添加CFTR增效劑染料木黃酮(50 μm)。 如在其他細胞類型中所觀察到者,在低溫下培育穩定表現ΔF508-CFTR之FRT細胞可提高功能CFTR在質膜中之密度。為確定校正化合物之活性,在37℃下將細胞與10 μM測試化合物一起培育24小時且隨後將其洗滌3次,之後記錄。相對於27℃及37℃對照標準化經化合物處理之細胞中之cAMP-及染料木黃酮介導之I
SC
且將其表示為活性百分比。將細胞與校正化合物一起預培育可相對於37℃對照顯著提高cAMP-及染料木黃酮介導之I
SC
。
增效劑化合物之鑑別
典型方案利用基底外側至頂膜之Cl
-
濃度梯度。為設定此梯度,在基底外側膜上使用正常林格氏溶液且用製黴菌素(nystatin) (360 µg/ml)對其實施可滲透化處理,而用等莫耳葡萄糖酸鈉(用NaOH滴定至pH 7.4)來替代頂端NaCl,以獲得較大跨上皮Cl
-
濃度梯度。所有實驗皆係在製黴菌素可滲透化處理30 min後實施。將福斯柯林(10 μM)及所有測試化合物添加至細胞培養插件兩側。比較假定ΔF508-CFTR增效劑與已知增效劑染料木黃酮之功效。
溶液
基底外側溶液(以mM計): NaCl (135)、CaCl
2
(1.2)、MgCl
2
(1.2)、K
2
HPO
4
(2.4)、KHPO
4
(0.6)、N-2-羥乙基六氫吡嗪-N’-2-乙磺酸(HEPES) (10)及右旋糖(10)。用NaOH將溶液滴定至pH 7.4。 頂端溶液(以mM計): 與基底外側溶液相同,只是用葡萄糖酸Na (135)替代NaCl。
細胞培養
使用表現ΔF508-CFTR之Fisher大鼠上皮(FRT)細胞(FRT
Δ F508-CFTR
)針對自吾人之光學分析鑑別之假定ΔF508-CFTR加強劑或誘導劑來實施尤斯室實驗。在Costar Snapwell細胞培養插件上培養細胞且在37℃及5% CO
2
下於補加有5%胎牛血清、100 U/ml青黴素及100 µg/ml鏈黴素之庫恩改良漢姆氏F-12培養基(Coon’s modified Ham’s F-12 medium)中培養5天。在用於表徵化合物之增效劑活性之前,在27℃下將細胞培育16 hr至48 hr以校正ΔF508-CFTR。為確定校正化合物之活性,在27℃或37℃及存在及不存在化合物之情況下將細胞培育24小時。
全細胞記錄
在穩定表現ΔF508-CFTR之溫度-及測試化合物校正的NIH3T3細胞中使用穿孔膜片全細胞記錄來監測巨觀ΔF508-CFTR電流(I
Δ F508
)。簡言之,在室溫下使用Axopatch 200B膜片鉗放大器(Axon Instruments公司,Foster City, CA)實施I
ΔF508
之電壓鉗記錄。所有記錄皆係以10 kHz之採樣頻率來獲得且以1 kHz實施低通濾波。在填充有細胞內溶液時吸管具有5 MΩ至6 MΩ之電阻。在該等記錄條件下,在室溫下Cl
-
之計算逆轉電勢(E
Cl
)為-28 mV。所有記錄皆具有> 20 GΩ之密封電阻及< 15 MΩ之串聯電阻。使用配備有Digidata 1320 A/D介面以及Clampex 8(Axon Instruments公司)之PC來實施脈衝生成、數據採集及分析。浴液含有< 250 μl之鹽水且使用重力驅動灌注系統以2 ml/min之速率持續灌注。
校正化合物之鑑別
為確定校正化合物提高功能ΔF508-CFTR在質膜中之密度之活性,在用校正化合物處理24 hr後使用上述穿孔膜片記錄技術來量測電流密度。為充分活化ΔF508-CFTR,將10 μm福司柯林及20 μM染料木黃酮添加至細胞中。在吾人之記錄條件下,在27℃下培育24 hr後之電流密度高於在37℃下培育24 hr後所觀察到者。該等結果與低溫培育對ΔF508-CFTR在質膜中之密度之已知效應一致。為確定校正化合物對CFTR電流密度之效應,在37℃下將細胞與10 μM測試化合物一起培育24小時,且將該電流密度與27℃及37℃對照進行比較(活性%)。在記錄前,用細胞外記錄培養基將細胞洗滌3次以去除任何殘餘測試化合物。與10 μM校正化合物一起預培育與37℃對照相比可顯著提高cAMP-及染料木黃酮依賴性電流。
增效劑化合物之鑑別
亦使用穿孔膜片記錄技術來研究ΔF508-CFTR增效劑在穩定表現ΔF508-CFTR之NIH3T3細胞中增大巨觀ΔF508-CFTR Cl
-
電流(I
Δ F508
)之能力。自光學分析鑑別之增效劑可引起I
ΔF508
之劑量依賴性增大,且效能及功效與在光學分析中所觀察到者類似。在所有經檢驗細胞中,在施加增效劑之前及期間之逆轉電勢為-30 mV左右,其為計算之E
Cl
(-28 mV)。
溶液
細胞內溶液(以mM計): 天冬胺酸Cs (90)、CsCl (50)、MgCl
2
(1)、HEPES (10)及240 μg/ml兩性黴素B (用CsOH調節至pH 7.35)。 細胞外溶液(以mM計): N-甲基-D-還原葡糖胺(NMDG)-Cl (150)、MgCl
2
(2)、CaCl
2
(2)、HEPES (10) (用HCl調節至pH 7.35)。
細胞培養
使用穩定表現ΔF508-CFTR之NIH3T3小鼠成纖維細胞來實施全細胞記錄。在存於175 cm
2
培養瓶中之補加有2 mM麩醯胺酸、10%胎牛血清、1 × NEAA、β-ME、1 ×青黴素/鏈黴素及25 mM HEPES之達爾伯克改良伊戈爾培養基中,將細胞維持在37℃及5% CO
2
及90%濕度下。對於全細胞記錄而言,將2,500 - 5,000個細胞接種於塗覆有聚L-離胺酸之玻璃蓋玻片上且在27℃下培養24 - 48 hr,之後使用其來測試增效劑之活性;且在37℃及存在或不存在校正化合物之情形下培育以量測校正劑之活性。
單通道記錄
使用經切除內面向外式膜片來觀察在NIH3T3細胞中穩定表現之溫度校正ΔF508-CFTR之單通道活性及增效劑化合物之活性。簡言之,在室溫下用Axopatch 200B膜片鉗放大器(Axon Instruments公司)來實施單通道活性之電壓鉗記錄。所有記錄皆係以10 kHz之採樣頻率來獲得且以400 Hz實施低通濾波。膜片吸管製自Corning Kovar Sealing 7052號玻璃(World Precision Instruments公司,Sarasota, FL)且在填充有細胞外溶液時具有5 - 8 MΩ之電阻。在切除後藉由添加1 mM Mg-ATP及75 nM cAMP依賴性蛋白激酶催化亞單位(PKA;Promega公司,Madison, WI)來活化ΔF508-CFTR。在通道活性穩定後,使用重力驅動微灌注系統對膜片實施灌注。流入物經放置毗鄰膜片,從而在1 - 2秒內完成溶液交換。為在快速灌注期間維持ΔF508-CFTR活性,將非特異性磷酸酶抑制劑F
-
(10 mM NaF)添加至浴液中。在該等記錄條件下,在整個膜片記錄過程(最長60 min)期間通道活性保持恆定。正電荷自細胞內溶液移動至細胞外溶液(陰離子以相反方向移動)所產生之電流顯示為正電流。使吸管電勢(V
p
)維持在80 mV。 自含有≤2個活性通道之膜片分析通道活性。在實驗過程期間同時開啟之最大數量決定活性通道數。為確定單通道電流幅值,以100 Hz「離線」過濾根據120秒之ΔF508-CFTR活性記錄之數據,且然後使用其來構築所有點之幅值直方圖,使用Bio-Patch分析軟體(Bio-Logic公司,France)將該等圖與複高斯函數擬合。根據120秒之通道活性測定總微觀電流及開放概率(P
o
)。使用Bio-Patch軟體或根據方程P
o
= I/i(N)來測定P
o
,其中I = 平均電流,i =單通道電流幅值,且N =膜片中之活性通道數。
溶液
細胞外溶液(以mM計): NMDG (150)、天冬胺酸(150)、CaCl
2
(5)、MgCl
2
(2)及HEPES (10) (用Tris鹼調節至pH 7.35)。 細胞內溶液(以mM計): NMDG-Cl (150)、MgCl
2
(2)、EGTA (5)、TES (10)及Tris鹼(14) (用HCl將pH調節至7.35)。
細胞培養
使用穩定表現ΔF508-CFTR之NIH3T3小鼠成纖維細胞來實施切除膜片鉗記錄。在存於175 cm
2
培養瓶中之補加有2 mM麩醯胺酸、10%胎牛血清、1 × NEAA、β-ME、1 ×青黴素/鏈黴素及25 mM HEPES之達爾伯克改良伊戈爾培養基中,將細胞維持在37℃及5% CO
2
及90%濕度下。對於單通道記錄而言,在塗覆有聚L-離胺酸之玻璃蓋玻片上接種2,500 - 5,000個細胞且在使用前於27℃下培育24 - 48 hr。 本發明之化合物1及化合物2可用作CFTR活性之加強劑或誘導劑。下表5說明化合物1及化合物2之EC50及相對功效。在下表5中,以下含義適用。EC50:「+++」意味著<10 uM;「++」意味著介於10 uM至25 uM之間;「+」意味著介於25 uM至60 uM之間。功效%:「+」意味著< 25%;「++」意味著介於25%至100%之間;「+++」意味著> 100%。
表 5 。 其他實施例
本發明中所提及之所有出版物及專利皆以引用方式併入本文中,其併入程度如同明確地及單獨地指出將每一個別出版物或專利申請案皆以引文方式併入一般。倘若以引用方式所併入任一專利或出版物中術語之含義與本發明中所用術語衝突,則意欲以本發明中術語之含義為準。此外,上文論述僅揭示並闡述本發明之例示性實施例。熟習此項技術者根據該論述且根據附圖及申請專利範圍將易於瞭解,可對其作出各種變化、修改及改變,此並不背離本發明之精神及範圍,如以下申請專利範圍中所定義。











 化合物1;及 具有以下結構之實質上非晶形N-(5-羥基-2,4-二第三-丁基-苯基)-4-側氧基-1H-喹啉-3-甲醯胺化合物2之固體分散體:
化合物1;及 具有以下結構之實質上非晶形N-(5-羥基-2,4-二第三-丁基-苯基)-4-側氧基-1H-喹啉-3-甲醯胺化合物2之固體分散體: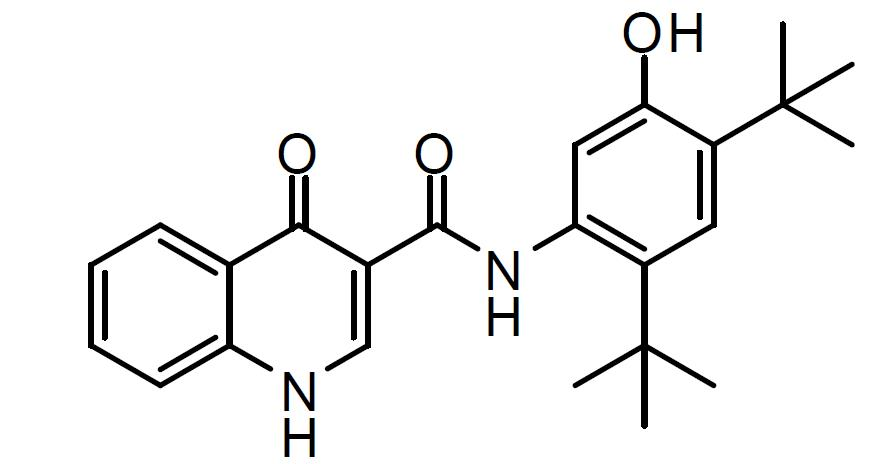 化合物2; 該等醫藥組合物之治療方法、製造方法、投與方法及套組。 在一個態樣中,本發明係關於醫藥組合物,其包含: a. 化合物1形式I; b. 包含實質上非晶形化合物2之固體分散體; c. 填充劑; d. 崩解劑; e. 表面活性劑;及 f. 黏合劑; 其稱作PC-I
。 在一個實施例中,本發明醫藥組合物包含30重量%至55重量%之化合物1形式I,及10重量%至45重量%之包含實質上非晶形化合物2之固體分散體。 在一個實施例中,填充劑係選自纖維素、改質纖維素、羧甲基纖維素鈉、乙基纖維素羥甲基纖維素、羥丙基纖維素、乙酸纖維素、微晶纖維素、磷酸氫鈣、蔗糖、乳糖、玉米澱粉、馬鈴薯澱粉或其任一組合。在另一實施例中,填充劑係微晶纖維素,且係以10重量%至20重量%範圍內之量存在。 在一個實施例中,崩解劑係選自瓊脂、藻膠、碳酸鈣、羧甲基纖維素、纖維素、羥丙基纖維素、低取代羥丙基纖維素、黏土、交聯羧甲基纖維素鈉、交聯聚維酮(crospovidone)、樹膠、矽酸鎂鋁、甲基纖維素、波拉克林鉀(polacrilin potassium)、海藻酸鈉、羥基乙酸澱粉鈉、玉米澱粉、馬鈴薯澱粉、木薯澱粉或其任一組合。在另一實施例中,崩解劑係交聯羧甲基纖維素鈉,且係以1重量%至3重量%範圍內之量存在。 在一個實施例中,表面活性劑係選自月桂基硫酸鈉、硬脂基硫酸鈉、聚氧乙烯20山梨醇酐單油酸酯或其任一組合。在另一實施例中,表面活性劑係月桂基硫酸鈉,且係以0.5重量%至2重量%範圍內之量存在。 在一個實施例中,黏合劑係選自聚乙烯吡咯啶酮、磷酸氫鈣、蔗糖、玉米澱粉、改質纖維素或其任一組合。在另一實施例中,黏合劑係聚乙烯吡咯啶酮,且係以0至5重量%範圍內之量存在。 在一個實施例中,本發明係關於具有以下配方之醫藥組合物:
化合物2; 該等醫藥組合物之治療方法、製造方法、投與方法及套組。 在一個態樣中,本發明係關於醫藥組合物,其包含: a. 化合物1形式I; b. 包含實質上非晶形化合物2之固體分散體; c. 填充劑; d. 崩解劑; e. 表面活性劑;及 f. 黏合劑; 其稱作PC-I
。 在一個實施例中,本發明醫藥組合物包含30重量%至55重量%之化合物1形式I,及10重量%至45重量%之包含實質上非晶形化合物2之固體分散體。 在一個實施例中,填充劑係選自纖維素、改質纖維素、羧甲基纖維素鈉、乙基纖維素羥甲基纖維素、羥丙基纖維素、乙酸纖維素、微晶纖維素、磷酸氫鈣、蔗糖、乳糖、玉米澱粉、馬鈴薯澱粉或其任一組合。在另一實施例中,填充劑係微晶纖維素,且係以10重量%至20重量%範圍內之量存在。 在一個實施例中,崩解劑係選自瓊脂、藻膠、碳酸鈣、羧甲基纖維素、纖維素、羥丙基纖維素、低取代羥丙基纖維素、黏土、交聯羧甲基纖維素鈉、交聯聚維酮(crospovidone)、樹膠、矽酸鎂鋁、甲基纖維素、波拉克林鉀(polacrilin potassium)、海藻酸鈉、羥基乙酸澱粉鈉、玉米澱粉、馬鈴薯澱粉、木薯澱粉或其任一組合。在另一實施例中,崩解劑係交聯羧甲基纖維素鈉,且係以1重量%至3重量%範圍內之量存在。 在一個實施例中,表面活性劑係選自月桂基硫酸鈉、硬脂基硫酸鈉、聚氧乙烯20山梨醇酐單油酸酯或其任一組合。在另一實施例中,表面活性劑係月桂基硫酸鈉,且係以0.5重量%至2重量%範圍內之量存在。 在一個實施例中,黏合劑係選自聚乙烯吡咯啶酮、磷酸氫鈣、蔗糖、玉米澱粉、改質纖維素或其任一組合。在另一實施例中,黏合劑係聚乙烯吡咯啶酮,且係以0至5重量%範圍內之量存在。 在一個實施例中,本發明係關於具有以下配方之醫藥組合物:



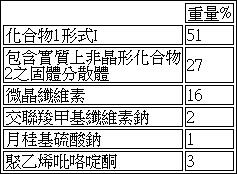


















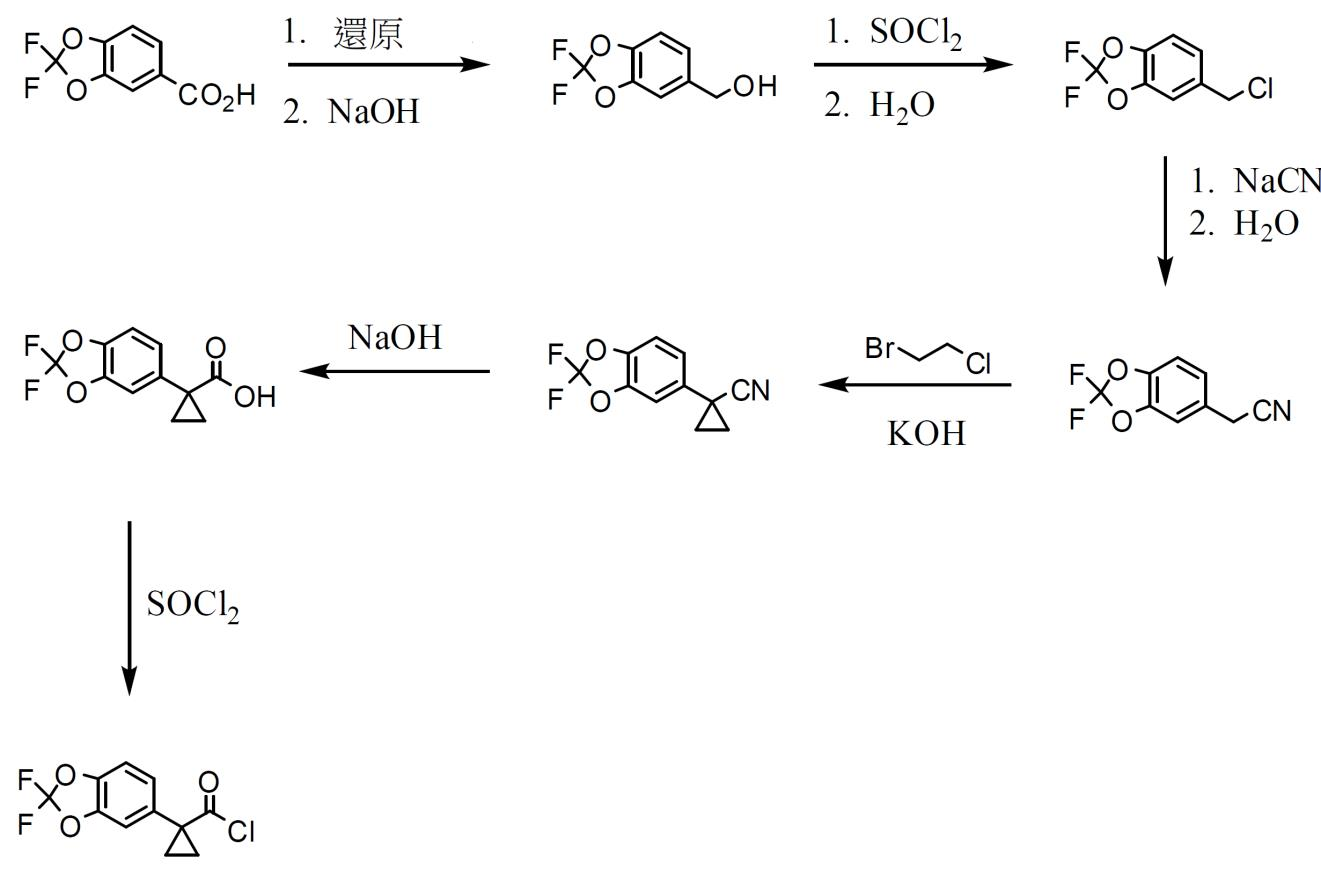 方案1繪示1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷羰基氯之製備,其用於方案3中以產生化合物1之醯胺連接。 起始材料2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-甲酸購自Saltigo (Lanxess公司之分部)。將2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-甲酸中之羧酸部分還原為一級醇,之後使用亞硫醯氯(SOCl2
)將其轉化為相應氯化物,得到5-(氯甲基)-2,2-二氟苯并[d][1,3]二氧雜環戊烯,隨後使用氰化鈉將其轉化為2-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)乙腈。用鹼及1-溴-2-氯乙烷處理2-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)乙腈,得到1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲腈。使用鹼將1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲腈中之腈部分轉化為羧酸,從而得到1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲酸,使用亞硫醯氯將其轉化為期望之醯氯。方案 2. 醯氯部分之替代合成。
方案1繪示1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷羰基氯之製備,其用於方案3中以產生化合物1之醯胺連接。 起始材料2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-甲酸購自Saltigo (Lanxess公司之分部)。將2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-甲酸中之羧酸部分還原為一級醇,之後使用亞硫醯氯(SOCl2
)將其轉化為相應氯化物,得到5-(氯甲基)-2,2-二氟苯并[d][1,3]二氧雜環戊烯,隨後使用氰化鈉將其轉化為2-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)乙腈。用鹼及1-溴-2-氯乙烷處理2-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)乙腈,得到1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲腈。使用鹼將1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲腈中之腈部分轉化為羧酸,從而得到1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲酸,使用亞硫醯氯將其轉化為期望之醯氯。方案 2. 醯氯部分之替代合成。  方案2繪示所需醯氯之替代合成。在鈀觸媒存在下使5-溴甲基-2,2-二氟-1,3-苯并二氧雜環戊烯與氰基乙酸乙酯偶合以形成相應α氰基乙基酯。將酯部分皂化為羧酸,得到氰基乙基化合物。在鹼存在下用1-溴-2-氯乙烷將氰基乙基化合物烷基化,得到氰基環丙基化合物。用鹼處理氰基環丙基化合物,得到羧酸鹽,藉由用酸處理將其轉化為羧酸。然後使用氯化劑(例如亞硫醯氯或諸如此類)將羧酸轉化為醯氯。方案 3. 胺部分之合成。
方案2繪示所需醯氯之替代合成。在鈀觸媒存在下使5-溴甲基-2,2-二氟-1,3-苯并二氧雜環戊烯與氰基乙酸乙酯偶合以形成相應α氰基乙基酯。將酯部分皂化為羧酸,得到氰基乙基化合物。在鹼存在下用1-溴-2-氯乙烷將氰基乙基化合物烷基化,得到氰基環丙基化合物。用鹼處理氰基環丙基化合物,得到羧酸鹽,藉由用酸處理將其轉化為羧酸。然後使用氯化劑(例如亞硫醯氯或諸如此類)將羧酸轉化為醯氯。方案 3. 胺部分之合成。  方案3繪示所需3-(6-胺基-3-甲基吡啶-2-基)苯甲酸第三丁基酯之製備,其在方案3中與1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷羰基氯偶合,從而得到化合物1。在鈀催化下使2-溴-3-甲基吡啶與3-(第三丁氧基羰基)苯基
方案3繪示所需3-(6-胺基-3-甲基吡啶-2-基)苯甲酸第三丁基酯之製備,其在方案3中與1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷羰基氯偶合,從而得到化合物1。在鈀催化下使2-溴-3-甲基吡啶與3-(第三丁氧基羰基)苯基 酸偶合,得到3-(3-甲基吡啶-2-基)苯甲酸第三丁基酯,隨後將其轉化為期望化合物。方案 4. 3-(6-(1-(2,2- 二氟苯并 [d][1,3] 二氧雜環戊烯 -5- 基 ) 環丙烷甲醯胺基 )-3- 甲基吡啶 -2- 基 ) 苯甲酸之酸式鹽之形成。
酸偶合,得到3-(3-甲基吡啶-2-基)苯甲酸第三丁基酯,隨後將其轉化為期望化合物。方案 4. 3-(6-(1-(2,2- 二氟苯并 [d][1,3] 二氧雜環戊烯 -5- 基 ) 環丙烷甲醯胺基 )-3- 甲基吡啶 -2- 基 ) 苯甲酸之酸式鹽之形成。  方案4繪示使用三乙胺及4-二甲基胺基吡啶使1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷羰基氯與3-(6-胺基-3-甲基吡啶-2-基)苯甲酸第三丁基酯偶合,以初步得到化合物1之第三丁基酯。化合物 1 形式 I
化合物1形式I係藉由將化合物1之鹽形式(例如HCl鹽)於合適溶劑中分散或溶解有效時間長度來製備。用諸如HCl等酸處理第三丁基酯,得到化合物1之HCL鹽,其通常為結晶固體。化合物1形式I亦可藉由用合適酸(例如甲酸)處理自第三丁基酯前體直接製備。 可使用3-(6-(1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲醯胺基)-3-甲基吡啶-2-基)苯甲酸之HCl鹽,藉由將3-(6-(1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲醯胺基)-3-甲基吡啶-2-基)苯甲酸之HCl鹽於合適溶劑中分散或溶解有效時間長度來製備形式I。可使用3-(6-(1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲醯胺基)-3-甲基吡啶-2-基)苯甲酸之其他鹽,例如,自其他礦物酸或有機酸衍生之鹽。可自第三丁基酯部分之酸介導之水解獲得其他鹽。衍生自其他酸之鹽可包括(例如)硝酸鹽、硫酸鹽、磷酸鹽、硼酸鹽、乙酸鹽、苯甲酸鹽及丙二酸鹽。端視所使用溶劑而定,3-(6-(1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲醯胺基)-3-甲基吡啶-2-基)苯甲酸之該等鹽形式可溶或不可溶,但缺少溶解性並不阻礙化合物1形式I之形成。例如,在一個實施例中,合適溶劑可係水或醇/水混合物,例如50%甲醇/水混合物,即使3-(6-(1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲醯胺基)-3-甲基吡啶-2-基)苯甲酸之HCl鹽形式僅微溶解於水中。在一個實施例中,合適溶劑為水。 自3-(6-(1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲醯胺基)-3-甲基吡啶-2-基)苯甲酸之鹽形成化合物1形式I的有效時間長度可介於2小時至24小時或更長之間之任一時間。應瞭解,所需時間長度與溫度成反比。亦即,溫度愈高,達成酸解離以形成化合物1形式I所需時間愈短。在溶劑為水時,將分散液在室溫下攪拌大約24小時可以大約98%之產率得到化合物1形式I。若出於處理目的需要3-(6-(1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲醯胺基)-3-甲基吡啶-2-基)苯甲酸鹽之溶液,則可使用升高溫度。在升高溫度下將溶液攪拌有效時間長度,之後使其冷卻並重結晶從而得到實質上純淨之化合物1形式I。在一個實施例中,實質上純淨係指純度大於約90%。在另一實施例中,實質上純淨係指純度大於約95%。在另一實施例中,實質上純淨係指純度大於約98%。在另一實施例中,實質上純淨係指純度大於約99%。所選溫度部分地取決於所用溶劑,且熟習此項技術者能夠確定。在一個實施例中,溫度介於室溫與約80℃之間。在另一實施例中,溫度介於室溫與約40℃之間。在另一實施例中,溫度介於約40℃與約60℃之間。在另一實施例中,溫度介於約60℃與約80℃之間。 亦可自3-(6-(1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲醯胺基)-3-甲基吡啶-2-基)-苯甲酸第三丁基酯(參照方案3) (其係化合物1之鹽之前體)直接形成化合物1形式I。因此,使3-(6-(1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲醯胺基)-3-甲基吡啶-2-基)-苯甲酸第三丁基酯經受在合適反應條件下與合適酸(例如,甲酸)之反應,從而得到化合物1形式I。 可藉由自有機溶劑重結晶來進一步純化化合物1形式I。有機溶劑之實例包括(但不限於)甲苯、異丙基苯、苯甲醚、1-丁醇、乙酸異丙酯、乙酸丁酯、乙酸異丁酯、甲基第三丁基醚、甲基異丁酮及1-丙醇-水混合物。溫度可如上所述。例如,在75℃下將化合物1形式I溶解於1-丁醇中直至其完全溶解。以0.2℃/min之速率將溶液冷卻至10℃,從而得到化合物1形式I之晶體,其可藉由過濾加以分離。 在一個實施例中,化合物1形式I之特徵在於在使用Cu K α輻射獲得之X射線粉末繞射中,在15.2度至15.6度、16.1度至16.5度及14.3度至14.7度處具有一或多個峰。在另一實施例中,化合物1形式I之特徵在於在15.4度、16.3度及14.5度處具有一或多個峰。在另一實施例中,化合物1形式I之特徵另外在於在14.6度至15.0度處具有峰。在另一實施例中,化合物1形式I之特徵另外在於在14.8度處具有峰。在另一實施例中,化合物1形式I之特徵另外在於在17.6度至18.0度處具有峰。在另一實施例中,化合物1形式I之特徵另外在於在17.8度處具有峰。在另一實施例中,化合物1形式I之特徵另外在於在16.4度至16.8度處具有峰。在另一實施例中,化合物1形式I之特徵另外在於在16.4度至16.8度處具有峰。在另一實施例中,化合物1形式I之特徵另外在於在16.6度處具有峰。在另一實施例中,化合物1形式I之特徵另外在於在7.6度至8.0度處具有峰。在另一實施例中,化合物1形式I之特徵另外在於在7.8度處具有峰。在另一實施例中,化合物1形式I之特徵另外在於在25.8度至26.2度處具有峰。在另一實施例中,化合物1形式I之特徵另外在於在26.0度處具有峰。在另一實施例中,化合物1形式I之特徵另外在於在21.4度至21.8度處具有峰。在另一實施例中,化合物1形式I之特徵另外在於在21.6度處具有峰。在另一實施例中,化合物1形式I之特徵另外在於在23.1度至23.5度處具有峰。在另一實施例中,化合物1形式I之特徵另外在於在23.3度處具有峰。在一些實施例中,化合物1形式I之特徵在於實質上類似於圖1之繞射圖案。在一些實施例中,化合物1形式I之特徵在於實質上類似於圖2之繞射圖案。 在一些實施例中,化合物1形式I之D90之粒徑分佈為約82 μm或更小。在一些實施例中,化合物1形式I之D50之粒徑分佈為約30 μm或更小。化合物 2
化合物2係包含實質上非晶形化合物2之固體分散體之起始點且可藉由根據方案5至7使4-側氧基-二氫喹啉甲酸部分與胺部分偶合來製備。方案 5 : 4- 側氧基 - 二氫喹啉甲酸部分之合成。
方案4繪示使用三乙胺及4-二甲基胺基吡啶使1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷羰基氯與3-(6-胺基-3-甲基吡啶-2-基)苯甲酸第三丁基酯偶合,以初步得到化合物1之第三丁基酯。化合物 1 形式 I
化合物1形式I係藉由將化合物1之鹽形式(例如HCl鹽)於合適溶劑中分散或溶解有效時間長度來製備。用諸如HCl等酸處理第三丁基酯,得到化合物1之HCL鹽,其通常為結晶固體。化合物1形式I亦可藉由用合適酸(例如甲酸)處理自第三丁基酯前體直接製備。 可使用3-(6-(1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲醯胺基)-3-甲基吡啶-2-基)苯甲酸之HCl鹽,藉由將3-(6-(1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲醯胺基)-3-甲基吡啶-2-基)苯甲酸之HCl鹽於合適溶劑中分散或溶解有效時間長度來製備形式I。可使用3-(6-(1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲醯胺基)-3-甲基吡啶-2-基)苯甲酸之其他鹽,例如,自其他礦物酸或有機酸衍生之鹽。可自第三丁基酯部分之酸介導之水解獲得其他鹽。衍生自其他酸之鹽可包括(例如)硝酸鹽、硫酸鹽、磷酸鹽、硼酸鹽、乙酸鹽、苯甲酸鹽及丙二酸鹽。端視所使用溶劑而定,3-(6-(1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲醯胺基)-3-甲基吡啶-2-基)苯甲酸之該等鹽形式可溶或不可溶,但缺少溶解性並不阻礙化合物1形式I之形成。例如,在一個實施例中,合適溶劑可係水或醇/水混合物,例如50%甲醇/水混合物,即使3-(6-(1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲醯胺基)-3-甲基吡啶-2-基)苯甲酸之HCl鹽形式僅微溶解於水中。在一個實施例中,合適溶劑為水。 自3-(6-(1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲醯胺基)-3-甲基吡啶-2-基)苯甲酸之鹽形成化合物1形式I的有效時間長度可介於2小時至24小時或更長之間之任一時間。應瞭解,所需時間長度與溫度成反比。亦即,溫度愈高,達成酸解離以形成化合物1形式I所需時間愈短。在溶劑為水時,將分散液在室溫下攪拌大約24小時可以大約98%之產率得到化合物1形式I。若出於處理目的需要3-(6-(1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲醯胺基)-3-甲基吡啶-2-基)苯甲酸鹽之溶液,則可使用升高溫度。在升高溫度下將溶液攪拌有效時間長度,之後使其冷卻並重結晶從而得到實質上純淨之化合物1形式I。在一個實施例中,實質上純淨係指純度大於約90%。在另一實施例中,實質上純淨係指純度大於約95%。在另一實施例中,實質上純淨係指純度大於約98%。在另一實施例中,實質上純淨係指純度大於約99%。所選溫度部分地取決於所用溶劑,且熟習此項技術者能夠確定。在一個實施例中,溫度介於室溫與約80℃之間。在另一實施例中,溫度介於室溫與約40℃之間。在另一實施例中,溫度介於約40℃與約60℃之間。在另一實施例中,溫度介於約60℃與約80℃之間。 亦可自3-(6-(1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲醯胺基)-3-甲基吡啶-2-基)-苯甲酸第三丁基酯(參照方案3) (其係化合物1之鹽之前體)直接形成化合物1形式I。因此,使3-(6-(1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲醯胺基)-3-甲基吡啶-2-基)-苯甲酸第三丁基酯經受在合適反應條件下與合適酸(例如,甲酸)之反應,從而得到化合物1形式I。 可藉由自有機溶劑重結晶來進一步純化化合物1形式I。有機溶劑之實例包括(但不限於)甲苯、異丙基苯、苯甲醚、1-丁醇、乙酸異丙酯、乙酸丁酯、乙酸異丁酯、甲基第三丁基醚、甲基異丁酮及1-丙醇-水混合物。溫度可如上所述。例如,在75℃下將化合物1形式I溶解於1-丁醇中直至其完全溶解。以0.2℃/min之速率將溶液冷卻至10℃,從而得到化合物1形式I之晶體,其可藉由過濾加以分離。 在一個實施例中,化合物1形式I之特徵在於在使用Cu K α輻射獲得之X射線粉末繞射中,在15.2度至15.6度、16.1度至16.5度及14.3度至14.7度處具有一或多個峰。在另一實施例中,化合物1形式I之特徵在於在15.4度、16.3度及14.5度處具有一或多個峰。在另一實施例中,化合物1形式I之特徵另外在於在14.6度至15.0度處具有峰。在另一實施例中,化合物1形式I之特徵另外在於在14.8度處具有峰。在另一實施例中,化合物1形式I之特徵另外在於在17.6度至18.0度處具有峰。在另一實施例中,化合物1形式I之特徵另外在於在17.8度處具有峰。在另一實施例中,化合物1形式I之特徵另外在於在16.4度至16.8度處具有峰。在另一實施例中,化合物1形式I之特徵另外在於在16.4度至16.8度處具有峰。在另一實施例中,化合物1形式I之特徵另外在於在16.6度處具有峰。在另一實施例中,化合物1形式I之特徵另外在於在7.6度至8.0度處具有峰。在另一實施例中,化合物1形式I之特徵另外在於在7.8度處具有峰。在另一實施例中,化合物1形式I之特徵另外在於在25.8度至26.2度處具有峰。在另一實施例中,化合物1形式I之特徵另外在於在26.0度處具有峰。在另一實施例中,化合物1形式I之特徵另外在於在21.4度至21.8度處具有峰。在另一實施例中,化合物1形式I之特徵另外在於在21.6度處具有峰。在另一實施例中,化合物1形式I之特徵另外在於在23.1度至23.5度處具有峰。在另一實施例中,化合物1形式I之特徵另外在於在23.3度處具有峰。在一些實施例中,化合物1形式I之特徵在於實質上類似於圖1之繞射圖案。在一些實施例中,化合物1形式I之特徵在於實質上類似於圖2之繞射圖案。 在一些實施例中,化合物1形式I之D90之粒徑分佈為約82 μm或更小。在一些實施例中,化合物1形式I之D50之粒徑分佈為約30 μm或更小。化合物 2
化合物2係包含實質上非晶形化合物2之固體分散體之起始點且可藉由根據方案5至7使4-側氧基-二氫喹啉甲酸部分與胺部分偶合來製備。方案 5 : 4- 側氧基 - 二氫喹啉甲酸部分之合成。 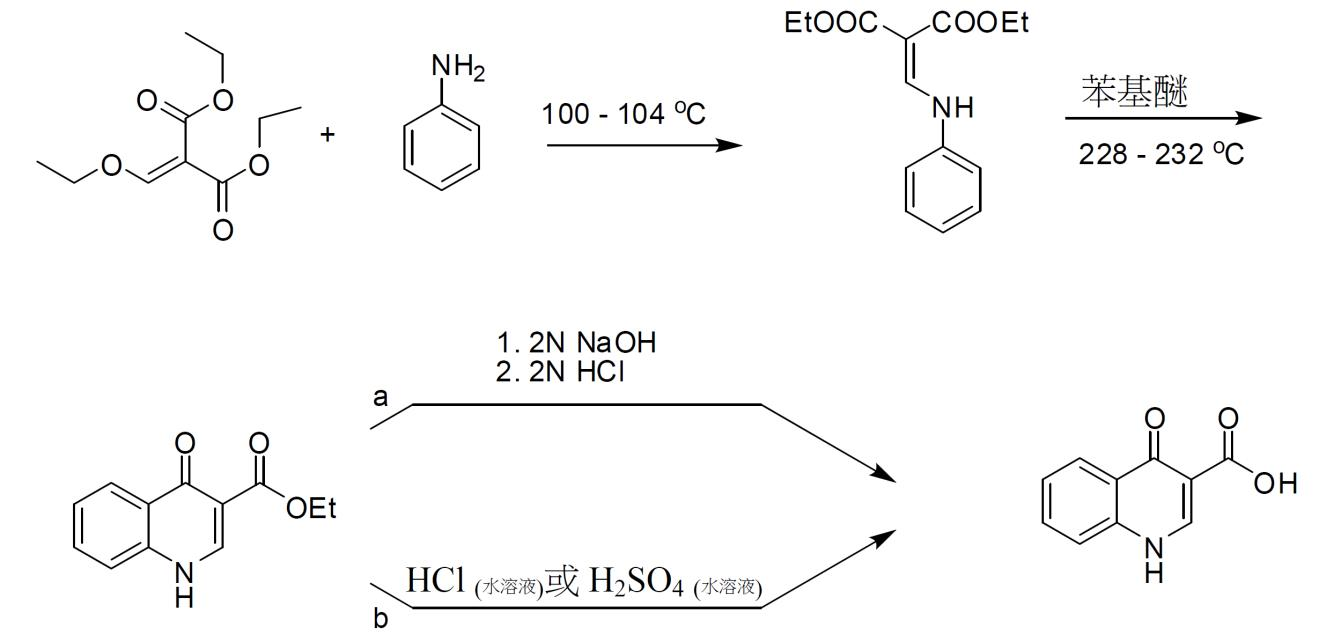 方案 6 : 胺部分之合成。
方案 6 : 胺部分之合成。  方案 7 : 4- 側氧基 - 二氫喹啉甲酸部分與胺部分之偶合。
方案 7 : 4- 側氧基 - 二氫喹啉甲酸部分與胺部分之偶合。 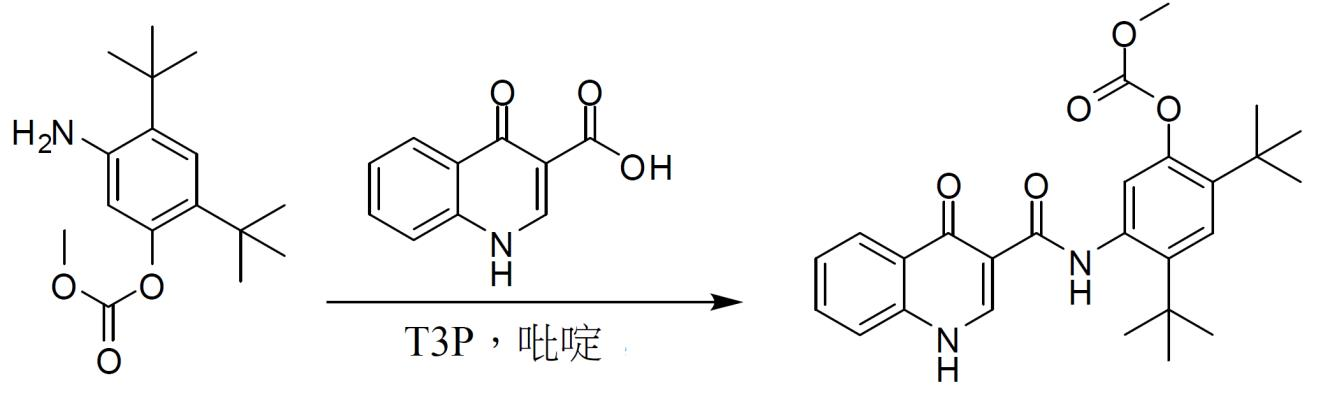
 包含實質上非晶形化合物 2 之固體分散體
自化合物2起始,化合物2之非晶型可藉由噴霧乾燥方法來製備。噴霧乾燥係將液體進料轉化成乾燥微粒形式之過程。視情況,可使用二級乾燥過程(例如流化床乾燥或真空乾燥)來將殘餘溶劑減少至醫藥上可接受之含量。通常,噴霧乾燥涉及使高度分散的液體懸浮液或溶液與充足體積之熱空氣接觸來蒸發並乾燥液滴。欲噴霧乾燥之製劑可為任一溶液、粗糙懸浮液、漿液、膠態分散液或膏糊,其可使用所選噴霧乾燥裝置來霧化。在標準程序中,將製劑噴霧至溫熱過濾的空氣流中,該空氣流使溶劑蒸發並將乾燥產物傳送至收集器(例如旋風器)。然後,用溶劑將消耗空氣耗盡,或另一選擇為,將消耗空氣運送至冷凝器以捕獲並潛在地再循環溶劑。可使用市售類型裝置來實施噴霧乾燥。例如,市售噴霧乾燥器係由Buchi有限公司及Niro製造(例如,由Niro製造之PSD系列噴霧乾燥器) (參見US 2004/0105820;US 2003/0144257)。 噴霧乾燥通常採用約3重量%至約30重量% (例如約4重量%至約20重量%、較佳地至少約10重量%)之材料(即,藥物及賦形劑)之固體加載。一般而言,固體加載之上限取決於所得溶液之黏度(例如,泵送之能力)及組份在溶液中之溶解性。通常,溶液之黏度可決定所得粉末產物中粒子之大小。 噴霧乾燥之技術及方法可參見Perry’s Chemical Engineering Handbook,第6版,R. H. Perry, D. W. Green及J. O. Maloney編輯), McGraw-Hill book公司(1984);及Marshall 「Atomization and Spray-Drying」 50, Chem. Eng. Prog. Monogr. Series 2 (1954)。一般而言,在約60℃至約200℃ (例如,約95℃至約185℃,約110℃至約182℃,約96℃至約180℃,例如,約145℃)之入口溫度下實施噴霧乾燥。通常,以約30℃至約90℃ (例如約40℃至約80℃,約45℃至約80℃,例如,約75℃)之出口溫度實施噴霧乾燥。霧化流速通常為約4 kg/h至約12 kg/h,例如,約4.3 kg/h至約10.5 kg/h,例如,約6 kg/h或約10.5 kg/h。進料流速通常為約3 kg/h至約10 kg/h,例如,約3.5 kg/h至約9.0 kg/h,例如,約8 kg/h或約7.1 kg/h。霧化比率通常為約0.3至1.7,例如,約0.5至1.5,例如,約0.8或約1.5。 溶劑之去除可能需要後續乾燥步驟,例如盤式乾燥、流體床乾燥(例如,約室溫至約100℃)、真空乾燥、微波乾燥、旋轉滾筒乾燥或雙錐真空乾燥(例如,約室溫至約200℃)。 在一個實施例中,噴霧乾燥分散體係經流體床乾燥的。 在一個製程中,溶劑包括揮發性溶劑,例如沸點小於約100℃之溶劑。在一些實施例中,溶劑包括溶劑之混合物,例如揮發性溶劑之混合物或揮發性溶劑與非揮發性溶劑之混合物。倘若使用溶劑之混合物,則混合物可包括一或多種非揮發性溶劑,例如,其中非揮發性溶劑係以小於約15% (例如,小於約12%,小於約10%,小於約8%,小於約5%,小於約3%或小於約2%)存於混合物中。 較佳溶劑係化合物2之溶解度為至少約10 mg/ml (例如,至少約15 mg/ml、20 mg/ml、25 mg/ml、30 mg/ml、35 mg/ml、40 mg/ml、45 mg/ml、50 mg/ml或更大)之溶劑。更佳溶劑包括化合物2之溶解度為至少約20 mg/ml者。 可測試之例示性溶劑包括丙酮、環己烷、二氯甲烷、N,N-二甲基乙醯胺(DMA)、N,N-二甲基甲醯胺(DMF)、1,3-二甲基-2-咪唑啉酮(DMI)、二甲基亞碸(DMSO)、二噁烷、乙酸乙酯、乙醚、冰乙酸(HAc)、甲基乙基酮(MEK)、N-甲基-2-吡咯啶酮(NMP)、甲基第三丁基醚(MTBE)、四氫呋喃(THF)、戊烷、乙腈、甲醇、乙醇、異丙醇、乙酸異丙基酯及甲苯。例示性共溶劑包括丙酮/DMSO、丙酮/DMF、丙酮/水、MEK/水、THF/水、二噁烷/水。在兩種溶劑的系統中,溶劑可以約0.1%至約99.9%存在。在一些較佳實施例中,水與丙酮係共溶劑,其中水係以約0.1%至約15% (例如約9%至約11%,例如,約10%)存在。在一些較佳實施例中,水與MEK係共溶劑,其中水係以約0.1%至約15% (例如約9%至約11%,例如,約10%)存在。在一些實施例中,溶劑溶液包括三種溶劑。例如,丙酮及水可與第三溶劑(例如DMA、DMF、DMI、DMSO或HAc)混合。在實質上非晶形化合物2係固體分散體之組份之情形下,較佳溶劑溶解化合物2及聚合物。適宜溶劑包括彼等闡述於上文者,例如,MEK、丙酮、水、甲醇及其混合物。 可修改粒徑及溫度乾燥範圍來製備最佳噴霧乾燥分散體。如熟習此項技術者所應瞭解,小粒徑可改良溶劑去除。然而,申請者已發現較小粒子可產生膨松粒子,其在一些情形下不能提供最佳噴霧乾燥分散體用於諸如製錠等下游製程。在較高溫度下,實質上非晶形化合物2可能發生結晶或化學降解。在較低溫度下,不能去除足夠量之溶劑。本文之方法提供最佳粒徑及最佳乾燥溫度。 一般而言,粒徑使得D10 (μm)小於約5 (例如,小於約4.5,小於約4.0,或小於約3.5),D50 (μm)通常小於約17 (例如,小於約16,小於約15,小於約14,小於約13),且D90 (μm)通常小於約175 (例如,小於約170,小於約170,小於約150,小於約125,小於約100,小於約90,小於約80,小於約70,小於約60,或小於約小於約50)。一般而言,噴霧乾燥粒子之堆密度為約0.08 g/cc至約0.20 g/cc,例如,約0.10至約0.15 g/cc,例如,約0.11 g/cc或約0.14 g/cc。噴霧乾燥粒子之敲緊密度通常在約0.08 g/cc至約0.20 g/cc範圍內,例如,約0.10 g/cc至約0.15 g/cc,例如,對於10次敲打約0.11 g/cc或約0.14 g/cc;0.10 g/cc至約0.25 g/cc,例如,約0.11至約0.21 g/cc,例如,對於500次敲打約0.15 g/cc、約0.19 g/cc或約0.21 g/cc;0.15 g/cc至約0.27 g/cc,例如,約0.18 g/cc至約0.24 g/cc,例如,對於1250次敲打約0.18 g/cc、約0.19 g/cc、約0.20 g/cc或約0.24 g/cc;及0.15 g/cc至約0.27 g/cc,例如,約0.18 g/cc至約0.24 g/cc,例如,對於2500次敲打約0.18 g/cc、約0.21 g/cc、約0.23 g/cc或約0.24 g/cc。聚合物
本文亦包括包含非晶形化合物2及聚合物(或固態載劑)之噴霧乾燥分散體。例如,化合物2係作為固體非晶形分散體之組份以非晶形化合物形式而存在。固體非晶形分散體通常包括實質上非晶形化合物2及聚合物。例示性聚合物包括纖維質聚合物,例如含有HPMC或HPMCAS及吡咯啶酮之聚合物,例如PVP/VA。在一些實施例中,固體非晶形分散體包括一或多種其他賦形劑,例如表面活性劑。 在一個實施例中,聚合物能夠溶解於水性介質中。聚合物之溶解性可為pH獨立性或pH依賴性。後者包括一或多種腸溶聚合物。術語「腸溶聚合物」係指相對於胃之更酸環境優先在腸之較不酸環境中溶解之聚合物,例如,在酸性水性介質中不溶解但當pH超過5-6時溶解之聚合物。合適聚合物應為化學及生物學惰性。為改良噴霧乾燥分散體之物理穩定性,聚合物之玻璃轉變溫度(Tg
)應儘可能地高。例如,較佳聚合物之玻璃轉變溫度至少等於或大於藥物(即,化合物2)之玻璃轉變溫度。其他較佳聚合物之玻璃轉變溫度在藥物(即,化合物2)之約10℃至約15℃內。聚合物之適宜玻璃轉變溫度之實例包括至少約90℃、至少約95℃、至少約100℃、至少約105℃、至少約110℃、至少約115℃、至少約120℃、至少約125℃、至少約130℃、至少約135℃、至少約140℃、至少約145℃、至少約150℃、至少約155℃、至少約160℃、至少約165℃、至少約170℃,或至少約175℃ (如在乾燥條件下所量測)。不欲受限於理論,據信,根本機制係具有較高Tg
之聚合物在室溫下通常具有較低分子遷移率,此可係使非晶形噴霧乾燥分散體之物理穩定性穩定之至關重要因素。 另外,聚合物之吸濕力應儘可能低,例如,小於約10%。出於在此應用中比較之目的,聚合物或組合物之吸濕力之特徵在於處於約60%相對濕度下。在一些較佳實施例中,聚合物之水吸收小於約10%,例如水吸收小於約9%、小於約8%、小於約7%、小於約6%、小於約5%、小於約4%、小於約3%,或小於約2%。吸濕力亦可影響噴霧乾燥分散體之物理穩定性。通常,吸附於聚合物中之水分可大大降低聚合物以及所得噴霧乾燥分散體之Tg
,從而將進一步降低如上所闡述噴霧乾燥分散體之物理穩定性。 在一個實施例中,聚合物係一或多種水溶性聚合物或部分地水溶性聚合物。水溶性或部分地水溶性 聚合物包括(但不限於)纖維素衍生物(例如,羥基丙基甲基纖維素(HPMC)、羥丙基纖維素(HPC))或乙基纖維素;聚乙烯吡咯啶酮(PVP);聚乙二醇(PEG);聚乙烯醇(PVA);丙烯酸酯,例如聚甲基丙烯酸酯(例如,Eudragit® E);環糊精(例如,β-環糊精)及其共聚物及衍生物,包括(例如) PVP-VA (聚乙烯吡咯啶酮-乙酸乙烯基酯)。 在一些實施例中,聚合物係羥基丙基甲基纖維素(HPMC),例如HPMCAS、HPMC E50、HPMCE15或HPMC60SH50)。 如本文所論述,聚合物可為pH依賴性腸溶聚合物。該等pH依賴性腸溶聚合物包括(但不限於)纖維素衍生物(例如,乙酸鄰苯二甲酸纖維素(CAP))、鄰苯二甲酸羥基丙基甲基纖維素(HPMCP)、琥珀酸羥基丙基甲基乙酸纖維素(HPMCAS)、羧甲基纖維素(CMC)或其鹽(例如,鈉鹽,例如(CMC-Na));乙酸偏苯三酸纖維素(CAT)、乙酸鄰苯二甲酸羥基丙基纖維素(HPCAP)、乙酸鄰苯二甲酸羥基丙基甲基纖維素(HPMCAP)及乙酸鄰苯二甲酸甲基纖維素(MCAP)或聚甲基丙烯酸酯(例如,Eudragit® S)。在一些實施例中,聚合物係乙酸琥珀酸羥基丙基甲基纖維素(HPMCAS)。在一些實施例中,聚合物係HG級乙酸琥珀酸羥基丙基甲基纖維素(HPMCAS-HG)。 在又一實施例中,聚合物係聚乙烯吡咯啶酮共聚物,例如,乙烯基吡咯啶酮/乙酸乙烯基酯共聚物(PVP/VA)。 在化合物2與聚合物(例如與HPMC、HPMCAS或PVP/VA聚合物)形成噴霧乾燥分散體之實施例中,聚合物相對於噴霧乾燥分散體之總重量之量在約0.1重量%至99重量%範圍內。除非另有說明,否則如所闡述藥物、聚合物及其他賦形劑在分散體內之%係以重量%給出。聚合物之量通常為至少約20%,且較佳地為至少約30%,例如,至少約35%、至少約40%、至少約45%或約50% (例如,49.5%)。該量通常為約99%或更少,且較佳地為約80%或更少,例如約75%或更少、約70%或更少、約65%或更少、約60%或更少或約55%或更少。在一個實施例中,聚合物之量佔分散體之總重量為至多約50% (且甚至更特定而言,介於約40%與50%之間,例如約49%、約49.5%或約50%)。HPMC及HPMCAS係以各種等級購自ShinEtsu,例如,HPMCAS係以多種變化形式購得,包括AS-LF、AS-MF、AS-HF、AS-LG、AS-MG、AS-HG。該等等級中之每一者皆隨乙酸酯及琥珀酸酯之取代%而變化。 在一些實施例中,實質上非晶形化合物2及聚合物係以大致相等的量存在,例如聚合物及藥物中之每一者佔分散體之重量%的約一半。例如,聚合物係以約49.5%存在且藥物係以約50%存在。 在一些實施例中,在噴霧乾燥之前,實質上非晶形化合物2及所組合聚合物佔非噴霧乾燥分散體之總固體內容物之1% w/w至20% w/w。在一些實施例中,在噴霧乾燥之前,實質上非晶形化合物2及所組合聚合物佔非噴霧乾燥分散體之總固體內容物之5% w/w至15% w/w。在一些實施例中,在噴霧乾燥之前,實質上非晶形化合物2及所組合聚合物佔非噴霧乾燥分散體之總固體內容物之約11% w/w。 在一些實施例中,分散體進一步包括其他次要成份,例如表面活性劑(例如,SLS)。在一些實施例中,表面活性劑係以佔分散體小於約10% (例如小於約9%、小於約8%、小於約7%、小於約6%、小於約5%、小於約4%、小於約3%、小於約2%、約1%或約0.5%)存在。 在包括聚合物之實施例中,聚合物應以有效使噴霧乾燥分散體穩定之量存在。穩定包括抑制或防止實質上非晶形化合物2之結晶。該穩定可抑制化合物2自非晶形轉化成結晶形式。例如,聚合物可防止至少一部分(例如,約5%、約10%、約15%、約20%、約25%、約30%、約35%、約40%、約45%、約50%、約55%、約60%、約65%、約70%、約75%或更大)化合物2自非晶形轉化成結晶形式。穩定可藉由例如以下來量測:量測噴霧乾燥分散體之玻璃轉變溫度、量測非晶形材料之鬆弛速率,或量測化合物2之溶解性或生物利用度。 適宜與化合物2組合而(例如)形成噴霧乾燥分散體(例如非晶形噴霧乾燥分散體)之聚合物應具有以下性質中之一或多者: 聚合物之玻璃轉變溫度之溫度較實質上非晶形化合物2之玻璃轉變溫度應低不小於約10-15℃。較佳地,聚合物之玻璃轉變溫度大於實質上非晶形化合物2之玻璃轉變溫度,且一般而言較藥物產品之期望儲存溫度高出至少50℃。例如,至少約100℃、至少約105℃、至少約105℃、至少約110℃、至少約120℃、至少約130℃、至少約140℃、至少約150℃、至少約160℃、至少約160℃或更大。 聚合物應相對較不吸濕。例如,聚合物在於標準條件下儲存時吸收小於約10%之水,例如,小於約9%、小於約8%、小於約7%、小於約6%,或小於約5%、小於約4%或小於約3%之水。較佳地,聚合物在於標準條件下儲存時將實質上不含吸收水。 聚合物與化合物2相比應在適於噴霧乾燥過程之溶劑中具有類似或更好的溶解性。在較佳實施例中,聚合物將溶解於與化合物2相同之溶劑或溶劑系統中之一或多者中。較佳地,聚合物溶於至少一種含非羥基之溶劑(例如二氯甲烷、丙酮或其組合)中。 與化合物2在不存在聚合物下之溶解性相比,或與化合物2在與參考聚合物組合時之溶解性相比,聚合物在與實質上非晶形化合物2組合於(例如)噴霧乾燥分散體或液體懸浮液中時可增加化合物2在水性及生理上相關介質中之溶解性。例如,聚合物可藉由使自固體非晶形分散體或自液體懸浮液轉化成結晶化合物2之非晶形化合物2之量降低而增加非晶形化合物2之溶解性。 聚合物應使非晶形物質之鬆弛速率降低。 聚合物應使實質上非晶形化合物2之物理及/或化學穩定性增加。 聚合物應改良實質上非晶形化合物2之可製造性。 聚合物應改良實質上非晶形化合物2之處置、投與或儲存性質中之一或多者。 聚合物不應與其他醫藥組份(例如賦形劑)發生不利相互作用。 可使用本文闡述之噴霧乾燥方法(或其他方法)來測試候選聚合物(或其他組份)形成非晶形組合物之適宜性。候選組合物可在穩定性、耐形成晶體性或其他性質方面進行比較,且與參考製劑(例如,純淨非晶形化合物2或結晶化合物2之製劑)進行比較。例如,候選組合物可經測試來確定其是否抑制溶劑介導結晶之起始時間,或測定在既定時間在受控條件下之轉化率%,例如至少50%、75%、100%或110%,亦如此測試參考製劑,或候選組合物可經測試以確定其與結晶化合物2相比是否改良生物利用度或溶解性。表面活性劑
噴霧乾燥分散體可包括表面活性劑。表面活性劑或表面活性劑混合物可通常使噴霧乾燥分散體與水性介質之間之界面張力降低。合適表面活性劑或表面活性劑混合物亦可增強來自噴霧乾燥分散體之化合物2之水性溶解性及生物利用度。結合本發明使用之表面活性劑包括(但不限於)山梨醇酐脂肪酸酯(例如,Spans®)、聚氧乙烯山梨醇酐脂肪酸酯(例如,Tweens®)、月桂基硫酸鈉(SLS)、十二烷基苯磺酸鈉(SDBS)、磺琥珀酸鈉二辛酯(多庫酯鈉(Docusate))、脫氧膽酸鈉鹽(DOSS)、山梨醇酐單硬脂酸酯、山梨醇酐三硬脂酸酯、十六烷基三甲基溴化銨(HTAB)、N-月桂醯基肌胺酸鈉、油酸鈉、肉豆蔻酸鈉、硬脂酸鈉、棕櫚酸鈉、Gelucire 44/14、乙二胺四乙酸(EDTA)、維生素E d-α生育酚聚乙二醇1000琥珀酸酯(TPGS)、卵磷脂、MW 677-692、麩胺酸單鈉單水合物、Labrasol、PEG 8辛酸/癸酸甘油酯、Transcutol、二乙二醇單乙基醚、Solutol HS-15、聚乙二醇/羥基硬脂酸酯、牛膽酸、Pluronic F68、Pluronic F108及Pluronic F127 (或任何其他聚氧乙烯聚氧丙烯共聚物(Pluronics®)或飽和聚乙二醇化甘油酯(Gelucirs®))。可結合本發明使用之該等表面活性劑之特定實例包括(但不限於) Span 65、Span 25、Tween 20、Capryol 90、Pluronic F108、月桂基硫酸鈉(SLS)、維生素E TPGS、pluronics及共聚物。SLS通常較佳。 表面活性劑(例如,SLS)相對於噴霧乾燥分散體之總重量之量可介於0.1%至15%之間。較佳地,其為約0.5%至約10%,更佳地約0.5%至約5%,例如,約0.5%至4%、約0.5%至3%、約0.5%至2%、約0.5%至1%或約0.5%。 在某些實施例中,表面活性劑相對於噴霧乾燥分散體之總重量之量為至少約0.1%,較佳地為約0.5%。在該等實施例中,表面活性劑可以不超過約15%且較佳地不超過約12%、約11%、約10%、約9%、約8%、約7%、約6%、約5%、約4%、約3%、約2%或約1%之量存在。表面活性劑之量為約0.5重量%之實施例較佳。 可以與針對測試聚合物所闡述類似之方式測試候選表面活性劑(或其他組份)於本發明中使用之適宜性。製備醫藥組合物之方法
可藉由濕法製粒、在壓力下壓實或壓製混合物或組合物(例如,粉末或顆粒)以形成穩定三維形狀(例如,錠劑)來產生本發明之醫藥組合物。如本文所使用,「錠劑」包括所有形狀及大小之經壓製醫藥劑量單位形式,無論包覆與否。 如本文所使用,術語「錠劑」係指適於欲治療病患之藥劑之物理離散單元。一般而言,經壓實混合物之密度大於該混合物在壓實前之密度。本發明之劑量錠劑幾乎可具有任何形狀,包括凹面及/或凸面、圓角或有角角及約為直線之形狀。在一些實施例中,本發明之經壓製錠劑包含具有扁平面之圓形錠劑。可藉由為熟習形成經壓製固體醫藥劑型之技術者已知之任一壓實及壓製方法製備本發明之錠劑。在具體實施例中,可使用彼等熟習醫藥調配物領域者已知之習用方法來製備本文所提供調配物,如(例如)相關教科書中所闡述。例如,參見Remington: The Science and Practice of Pharmacy,第21版,Lippincott Williams & Wilkins, Baltimore, Md. (2003);Ansel等人,Pharmaceutical Dosage Forms And Drug Delivery Systems,第7版,Lippincott Williams & Wilkins, (1999);The Handbook of Pharmaceutical Excipients,第4版,Rowe等人編輯,American Pharmaceuticals Association (2003);Gibson, Pharmaceutical Preformulation And Formulation,CRC Press (2001),該等參考文獻以全文引用方式併入本文中。製粒及壓製
在一些實施例中,根據本文所述配方稱量各成份。接下來,對所有顆粒內成份實施篩分並將其充分混合。可用適宜潤滑劑(例如,硬脂酸鎂)潤滑成份。下一步驟可包含壓實/擊壓粉末混合物及定大小之成份。接下來,將經壓實或擊壓摻合物研磨成顆粒並篩分以獲得期望大小。接下來,可用(例如)硬脂酸鎂進一步潤滑顆粒。接下來,可在適宜衝壓器上將本發明顆粒組合物壓製成本發明之各種醫藥調配物。視情況,錠劑可經薄膜、著色劑或其他包衣包覆。 本發明之另一態樣提供產生醫藥組合物之方法,其包含提供包含以下之組合物之混合物:化合物1形式I、包含實質上非晶形化合物2之固體分散體及一或多種選自填充劑、稀釋劑、黏合劑、表面活性劑、潤滑劑、崩解劑之賦形劑,及將該組合物壓製成在約30分鐘內之溶解率為至少約50%之錠劑。 在另一實施例中,對粉末及液體成份之混合物實施濕法製粒製程以得到本發明醫藥調配物。例如,根據本文所述配方稱量包含包括以下之組合物之混合物之醫藥組合物:化合物1形式I、包含實質上非晶形化合物2之固體分散體之組合物,及一或多種選自填充劑、黏合劑、表面活性劑或崩解劑之賦形劑。接下來,在高剪切或低剪切製粒機中使用水或具有表面活性劑之水或具有黏合劑之水或具有表面活性劑及黏合劑之水對所有顆粒內成份實施篩分並混合,以製粒粉末摻合物。非水流體亦可與表面活性劑及/或黏合劑一起使用或不與其一起使用,以製粒粉末摻合物。接下來,可視情況使用適宜研磨機研磨濕顆粒。隨後,可視情況藉由以任一適宜方式乾燥成份自混合物去除水。接下來,可視情況將乾燥顆粒研磨成所需大小。接下來,可藉由摻合添加顆粒外賦形劑(例如填充劑、稀釋劑及崩解劑)。接下來,可用硬脂酸鎂及崩解劑(例如,交聯羧甲基纖維素鈉)進一步潤滑定大小之顆粒。接下來,可將本發明之顆粒組合物篩分足夠時間以獲得正確大小,且然後在適宜衝壓器上壓製成本發明之各種醫藥調配物。視情況,錠劑可經薄膜、著色劑或其他包衣包覆。令人驚訝地,可實施濕法製粒且實質上不損失化合物1形式I或實質上非晶形化合物2之固態形式。 在尤其有利的實施例中,本發明醫藥組合物係藉由連續雙螺桿濕法製粒(TSWG)製程來製備。連續製造在線上監測及控制下產生高品質且高度一致的產品。連續製造亦因設計研發利用「富數據」設計空間且更易於瞭解上游變量對下游製程及最終產品品質之影響而有利於品質。此外,本發明醫藥組合物可早在商業規模設備上完結,從而避免在研發晚期擴大規模的風險及調配物變化。最後,連續製造具有商業製造優點,例如製程控制有所改良、產品處置有所減少及實時釋放效率。總體而言,產生具有較少製程檢查從而提高產品品質且因此提高患者安全性之更穩健、可控且規模可變的製程。該等優點解決了Janet Woodcock (藥物評估與研究中心(Center for Drug Evaluation and Research,CDER)的主任)對化學與製造控制不能趕上高度有效療法之迅速臨床研發之擔憂(「What we are seeing is that often the rate limiting step is going to be manufacturing」,2013年7月24日,癌症之友(Friends of Cancer)主持的會議簡要「Answering a Compelling Need: Expediting Life-Saving Treatments to Patients」,用以論述美國食品藥物管理局之突破性療法認定)。 例如,高剪切製粒(HSG)係常用製粒技術,眾所周知其具有過度製粒及差製程控制之風險。對於此製程擴大規模極具挑戰性且涉及顯著風險。從HSG製程變化為連續TSWG製程允許使用相同設備擴大規模以藉由運行更長時間來產生不同批量。此消除其他製粒製程所通常遇到之擴大規模的風險。另外,發現TSWG製程更為穩健,較不易於過度製粒。如在針對化合物1錠劑之圖3中可看出,HSG製程顯示溶解率隨水含量增加而顯著降低,而TSWG製程對於水添加之類似範圍未顯示變化。令人驚訝地發現,使用雙螺桿濕法製粒製程時包含介於45重量%至55重量%間之化合物1之錠劑調配物及包含介於60重量%至70重量%間之化合物1之錠劑調配物沒有性能變化。此與HSG製程之情形不同。另外,此連續且產品品質有所提高之製程解決了FDA關於有需要之患者缺乏藥物可用性之普遍抱怨。 在一個實施例中,連續製程起始於藉助失重式(loss-in-weight)進給將個別賦形劑、化合物1及化合物2進給至連續在線摻合機。自此摻合機連續傳送材料並藉助雙螺桿濕法製粒進行處理,乾燥,研磨,添加顆粒外賦形劑,摻合,壓製並包覆薄膜。 例如,在一個實施例中,可根據下文流程圖連續製備包含化合物1及化合物2之錠劑。
包含實質上非晶形化合物 2 之固體分散體
自化合物2起始,化合物2之非晶型可藉由噴霧乾燥方法來製備。噴霧乾燥係將液體進料轉化成乾燥微粒形式之過程。視情況,可使用二級乾燥過程(例如流化床乾燥或真空乾燥)來將殘餘溶劑減少至醫藥上可接受之含量。通常,噴霧乾燥涉及使高度分散的液體懸浮液或溶液與充足體積之熱空氣接觸來蒸發並乾燥液滴。欲噴霧乾燥之製劑可為任一溶液、粗糙懸浮液、漿液、膠態分散液或膏糊,其可使用所選噴霧乾燥裝置來霧化。在標準程序中,將製劑噴霧至溫熱過濾的空氣流中,該空氣流使溶劑蒸發並將乾燥產物傳送至收集器(例如旋風器)。然後,用溶劑將消耗空氣耗盡,或另一選擇為,將消耗空氣運送至冷凝器以捕獲並潛在地再循環溶劑。可使用市售類型裝置來實施噴霧乾燥。例如,市售噴霧乾燥器係由Buchi有限公司及Niro製造(例如,由Niro製造之PSD系列噴霧乾燥器) (參見US 2004/0105820;US 2003/0144257)。 噴霧乾燥通常採用約3重量%至約30重量% (例如約4重量%至約20重量%、較佳地至少約10重量%)之材料(即,藥物及賦形劑)之固體加載。一般而言,固體加載之上限取決於所得溶液之黏度(例如,泵送之能力)及組份在溶液中之溶解性。通常,溶液之黏度可決定所得粉末產物中粒子之大小。 噴霧乾燥之技術及方法可參見Perry’s Chemical Engineering Handbook,第6版,R. H. Perry, D. W. Green及J. O. Maloney編輯), McGraw-Hill book公司(1984);及Marshall 「Atomization and Spray-Drying」 50, Chem. Eng. Prog. Monogr. Series 2 (1954)。一般而言,在約60℃至約200℃ (例如,約95℃至約185℃,約110℃至約182℃,約96℃至約180℃,例如,約145℃)之入口溫度下實施噴霧乾燥。通常,以約30℃至約90℃ (例如約40℃至約80℃,約45℃至約80℃,例如,約75℃)之出口溫度實施噴霧乾燥。霧化流速通常為約4 kg/h至約12 kg/h,例如,約4.3 kg/h至約10.5 kg/h,例如,約6 kg/h或約10.5 kg/h。進料流速通常為約3 kg/h至約10 kg/h,例如,約3.5 kg/h至約9.0 kg/h,例如,約8 kg/h或約7.1 kg/h。霧化比率通常為約0.3至1.7,例如,約0.5至1.5,例如,約0.8或約1.5。 溶劑之去除可能需要後續乾燥步驟,例如盤式乾燥、流體床乾燥(例如,約室溫至約100℃)、真空乾燥、微波乾燥、旋轉滾筒乾燥或雙錐真空乾燥(例如,約室溫至約200℃)。 在一個實施例中,噴霧乾燥分散體係經流體床乾燥的。 在一個製程中,溶劑包括揮發性溶劑,例如沸點小於約100℃之溶劑。在一些實施例中,溶劑包括溶劑之混合物,例如揮發性溶劑之混合物或揮發性溶劑與非揮發性溶劑之混合物。倘若使用溶劑之混合物,則混合物可包括一或多種非揮發性溶劑,例如,其中非揮發性溶劑係以小於約15% (例如,小於約12%,小於約10%,小於約8%,小於約5%,小於約3%或小於約2%)存於混合物中。 較佳溶劑係化合物2之溶解度為至少約10 mg/ml (例如,至少約15 mg/ml、20 mg/ml、25 mg/ml、30 mg/ml、35 mg/ml、40 mg/ml、45 mg/ml、50 mg/ml或更大)之溶劑。更佳溶劑包括化合物2之溶解度為至少約20 mg/ml者。 可測試之例示性溶劑包括丙酮、環己烷、二氯甲烷、N,N-二甲基乙醯胺(DMA)、N,N-二甲基甲醯胺(DMF)、1,3-二甲基-2-咪唑啉酮(DMI)、二甲基亞碸(DMSO)、二噁烷、乙酸乙酯、乙醚、冰乙酸(HAc)、甲基乙基酮(MEK)、N-甲基-2-吡咯啶酮(NMP)、甲基第三丁基醚(MTBE)、四氫呋喃(THF)、戊烷、乙腈、甲醇、乙醇、異丙醇、乙酸異丙基酯及甲苯。例示性共溶劑包括丙酮/DMSO、丙酮/DMF、丙酮/水、MEK/水、THF/水、二噁烷/水。在兩種溶劑的系統中,溶劑可以約0.1%至約99.9%存在。在一些較佳實施例中,水與丙酮係共溶劑,其中水係以約0.1%至約15% (例如約9%至約11%,例如,約10%)存在。在一些較佳實施例中,水與MEK係共溶劑,其中水係以約0.1%至約15% (例如約9%至約11%,例如,約10%)存在。在一些實施例中,溶劑溶液包括三種溶劑。例如,丙酮及水可與第三溶劑(例如DMA、DMF、DMI、DMSO或HAc)混合。在實質上非晶形化合物2係固體分散體之組份之情形下,較佳溶劑溶解化合物2及聚合物。適宜溶劑包括彼等闡述於上文者,例如,MEK、丙酮、水、甲醇及其混合物。 可修改粒徑及溫度乾燥範圍來製備最佳噴霧乾燥分散體。如熟習此項技術者所應瞭解,小粒徑可改良溶劑去除。然而,申請者已發現較小粒子可產生膨松粒子,其在一些情形下不能提供最佳噴霧乾燥分散體用於諸如製錠等下游製程。在較高溫度下,實質上非晶形化合物2可能發生結晶或化學降解。在較低溫度下,不能去除足夠量之溶劑。本文之方法提供最佳粒徑及最佳乾燥溫度。 一般而言,粒徑使得D10 (μm)小於約5 (例如,小於約4.5,小於約4.0,或小於約3.5),D50 (μm)通常小於約17 (例如,小於約16,小於約15,小於約14,小於約13),且D90 (μm)通常小於約175 (例如,小於約170,小於約170,小於約150,小於約125,小於約100,小於約90,小於約80,小於約70,小於約60,或小於約小於約50)。一般而言,噴霧乾燥粒子之堆密度為約0.08 g/cc至約0.20 g/cc,例如,約0.10至約0.15 g/cc,例如,約0.11 g/cc或約0.14 g/cc。噴霧乾燥粒子之敲緊密度通常在約0.08 g/cc至約0.20 g/cc範圍內,例如,約0.10 g/cc至約0.15 g/cc,例如,對於10次敲打約0.11 g/cc或約0.14 g/cc;0.10 g/cc至約0.25 g/cc,例如,約0.11至約0.21 g/cc,例如,對於500次敲打約0.15 g/cc、約0.19 g/cc或約0.21 g/cc;0.15 g/cc至約0.27 g/cc,例如,約0.18 g/cc至約0.24 g/cc,例如,對於1250次敲打約0.18 g/cc、約0.19 g/cc、約0.20 g/cc或約0.24 g/cc;及0.15 g/cc至約0.27 g/cc,例如,約0.18 g/cc至約0.24 g/cc,例如,對於2500次敲打約0.18 g/cc、約0.21 g/cc、約0.23 g/cc或約0.24 g/cc。聚合物
本文亦包括包含非晶形化合物2及聚合物(或固態載劑)之噴霧乾燥分散體。例如,化合物2係作為固體非晶形分散體之組份以非晶形化合物形式而存在。固體非晶形分散體通常包括實質上非晶形化合物2及聚合物。例示性聚合物包括纖維質聚合物,例如含有HPMC或HPMCAS及吡咯啶酮之聚合物,例如PVP/VA。在一些實施例中,固體非晶形分散體包括一或多種其他賦形劑,例如表面活性劑。 在一個實施例中,聚合物能夠溶解於水性介質中。聚合物之溶解性可為pH獨立性或pH依賴性。後者包括一或多種腸溶聚合物。術語「腸溶聚合物」係指相對於胃之更酸環境優先在腸之較不酸環境中溶解之聚合物,例如,在酸性水性介質中不溶解但當pH超過5-6時溶解之聚合物。合適聚合物應為化學及生物學惰性。為改良噴霧乾燥分散體之物理穩定性,聚合物之玻璃轉變溫度(Tg
)應儘可能地高。例如,較佳聚合物之玻璃轉變溫度至少等於或大於藥物(即,化合物2)之玻璃轉變溫度。其他較佳聚合物之玻璃轉變溫度在藥物(即,化合物2)之約10℃至約15℃內。聚合物之適宜玻璃轉變溫度之實例包括至少約90℃、至少約95℃、至少約100℃、至少約105℃、至少約110℃、至少約115℃、至少約120℃、至少約125℃、至少約130℃、至少約135℃、至少約140℃、至少約145℃、至少約150℃、至少約155℃、至少約160℃、至少約165℃、至少約170℃,或至少約175℃ (如在乾燥條件下所量測)。不欲受限於理論,據信,根本機制係具有較高Tg
之聚合物在室溫下通常具有較低分子遷移率,此可係使非晶形噴霧乾燥分散體之物理穩定性穩定之至關重要因素。 另外,聚合物之吸濕力應儘可能低,例如,小於約10%。出於在此應用中比較之目的,聚合物或組合物之吸濕力之特徵在於處於約60%相對濕度下。在一些較佳實施例中,聚合物之水吸收小於約10%,例如水吸收小於約9%、小於約8%、小於約7%、小於約6%、小於約5%、小於約4%、小於約3%,或小於約2%。吸濕力亦可影響噴霧乾燥分散體之物理穩定性。通常,吸附於聚合物中之水分可大大降低聚合物以及所得噴霧乾燥分散體之Tg
,從而將進一步降低如上所闡述噴霧乾燥分散體之物理穩定性。 在一個實施例中,聚合物係一或多種水溶性聚合物或部分地水溶性聚合物。水溶性或部分地水溶性 聚合物包括(但不限於)纖維素衍生物(例如,羥基丙基甲基纖維素(HPMC)、羥丙基纖維素(HPC))或乙基纖維素;聚乙烯吡咯啶酮(PVP);聚乙二醇(PEG);聚乙烯醇(PVA);丙烯酸酯,例如聚甲基丙烯酸酯(例如,Eudragit® E);環糊精(例如,β-環糊精)及其共聚物及衍生物,包括(例如) PVP-VA (聚乙烯吡咯啶酮-乙酸乙烯基酯)。 在一些實施例中,聚合物係羥基丙基甲基纖維素(HPMC),例如HPMCAS、HPMC E50、HPMCE15或HPMC60SH50)。 如本文所論述,聚合物可為pH依賴性腸溶聚合物。該等pH依賴性腸溶聚合物包括(但不限於)纖維素衍生物(例如,乙酸鄰苯二甲酸纖維素(CAP))、鄰苯二甲酸羥基丙基甲基纖維素(HPMCP)、琥珀酸羥基丙基甲基乙酸纖維素(HPMCAS)、羧甲基纖維素(CMC)或其鹽(例如,鈉鹽,例如(CMC-Na));乙酸偏苯三酸纖維素(CAT)、乙酸鄰苯二甲酸羥基丙基纖維素(HPCAP)、乙酸鄰苯二甲酸羥基丙基甲基纖維素(HPMCAP)及乙酸鄰苯二甲酸甲基纖維素(MCAP)或聚甲基丙烯酸酯(例如,Eudragit® S)。在一些實施例中,聚合物係乙酸琥珀酸羥基丙基甲基纖維素(HPMCAS)。在一些實施例中,聚合物係HG級乙酸琥珀酸羥基丙基甲基纖維素(HPMCAS-HG)。 在又一實施例中,聚合物係聚乙烯吡咯啶酮共聚物,例如,乙烯基吡咯啶酮/乙酸乙烯基酯共聚物(PVP/VA)。 在化合物2與聚合物(例如與HPMC、HPMCAS或PVP/VA聚合物)形成噴霧乾燥分散體之實施例中,聚合物相對於噴霧乾燥分散體之總重量之量在約0.1重量%至99重量%範圍內。除非另有說明,否則如所闡述藥物、聚合物及其他賦形劑在分散體內之%係以重量%給出。聚合物之量通常為至少約20%,且較佳地為至少約30%,例如,至少約35%、至少約40%、至少約45%或約50% (例如,49.5%)。該量通常為約99%或更少,且較佳地為約80%或更少,例如約75%或更少、約70%或更少、約65%或更少、約60%或更少或約55%或更少。在一個實施例中,聚合物之量佔分散體之總重量為至多約50% (且甚至更特定而言,介於約40%與50%之間,例如約49%、約49.5%或約50%)。HPMC及HPMCAS係以各種等級購自ShinEtsu,例如,HPMCAS係以多種變化形式購得,包括AS-LF、AS-MF、AS-HF、AS-LG、AS-MG、AS-HG。該等等級中之每一者皆隨乙酸酯及琥珀酸酯之取代%而變化。 在一些實施例中,實質上非晶形化合物2及聚合物係以大致相等的量存在,例如聚合物及藥物中之每一者佔分散體之重量%的約一半。例如,聚合物係以約49.5%存在且藥物係以約50%存在。 在一些實施例中,在噴霧乾燥之前,實質上非晶形化合物2及所組合聚合物佔非噴霧乾燥分散體之總固體內容物之1% w/w至20% w/w。在一些實施例中,在噴霧乾燥之前,實質上非晶形化合物2及所組合聚合物佔非噴霧乾燥分散體之總固體內容物之5% w/w至15% w/w。在一些實施例中,在噴霧乾燥之前,實質上非晶形化合物2及所組合聚合物佔非噴霧乾燥分散體之總固體內容物之約11% w/w。 在一些實施例中,分散體進一步包括其他次要成份,例如表面活性劑(例如,SLS)。在一些實施例中,表面活性劑係以佔分散體小於約10% (例如小於約9%、小於約8%、小於約7%、小於約6%、小於約5%、小於約4%、小於約3%、小於約2%、約1%或約0.5%)存在。 在包括聚合物之實施例中,聚合物應以有效使噴霧乾燥分散體穩定之量存在。穩定包括抑制或防止實質上非晶形化合物2之結晶。該穩定可抑制化合物2自非晶形轉化成結晶形式。例如,聚合物可防止至少一部分(例如,約5%、約10%、約15%、約20%、約25%、約30%、約35%、約40%、約45%、約50%、約55%、約60%、約65%、約70%、約75%或更大)化合物2自非晶形轉化成結晶形式。穩定可藉由例如以下來量測:量測噴霧乾燥分散體之玻璃轉變溫度、量測非晶形材料之鬆弛速率,或量測化合物2之溶解性或生物利用度。 適宜與化合物2組合而(例如)形成噴霧乾燥分散體(例如非晶形噴霧乾燥分散體)之聚合物應具有以下性質中之一或多者: 聚合物之玻璃轉變溫度之溫度較實質上非晶形化合物2之玻璃轉變溫度應低不小於約10-15℃。較佳地,聚合物之玻璃轉變溫度大於實質上非晶形化合物2之玻璃轉變溫度,且一般而言較藥物產品之期望儲存溫度高出至少50℃。例如,至少約100℃、至少約105℃、至少約105℃、至少約110℃、至少約120℃、至少約130℃、至少約140℃、至少約150℃、至少約160℃、至少約160℃或更大。 聚合物應相對較不吸濕。例如,聚合物在於標準條件下儲存時吸收小於約10%之水,例如,小於約9%、小於約8%、小於約7%、小於約6%,或小於約5%、小於約4%或小於約3%之水。較佳地,聚合物在於標準條件下儲存時將實質上不含吸收水。 聚合物與化合物2相比應在適於噴霧乾燥過程之溶劑中具有類似或更好的溶解性。在較佳實施例中,聚合物將溶解於與化合物2相同之溶劑或溶劑系統中之一或多者中。較佳地,聚合物溶於至少一種含非羥基之溶劑(例如二氯甲烷、丙酮或其組合)中。 與化合物2在不存在聚合物下之溶解性相比,或與化合物2在與參考聚合物組合時之溶解性相比,聚合物在與實質上非晶形化合物2組合於(例如)噴霧乾燥分散體或液體懸浮液中時可增加化合物2在水性及生理上相關介質中之溶解性。例如,聚合物可藉由使自固體非晶形分散體或自液體懸浮液轉化成結晶化合物2之非晶形化合物2之量降低而增加非晶形化合物2之溶解性。 聚合物應使非晶形物質之鬆弛速率降低。 聚合物應使實質上非晶形化合物2之物理及/或化學穩定性增加。 聚合物應改良實質上非晶形化合物2之可製造性。 聚合物應改良實質上非晶形化合物2之處置、投與或儲存性質中之一或多者。 聚合物不應與其他醫藥組份(例如賦形劑)發生不利相互作用。 可使用本文闡述之噴霧乾燥方法(或其他方法)來測試候選聚合物(或其他組份)形成非晶形組合物之適宜性。候選組合物可在穩定性、耐形成晶體性或其他性質方面進行比較,且與參考製劑(例如,純淨非晶形化合物2或結晶化合物2之製劑)進行比較。例如,候選組合物可經測試來確定其是否抑制溶劑介導結晶之起始時間,或測定在既定時間在受控條件下之轉化率%,例如至少50%、75%、100%或110%,亦如此測試參考製劑,或候選組合物可經測試以確定其與結晶化合物2相比是否改良生物利用度或溶解性。表面活性劑
噴霧乾燥分散體可包括表面活性劑。表面活性劑或表面活性劑混合物可通常使噴霧乾燥分散體與水性介質之間之界面張力降低。合適表面活性劑或表面活性劑混合物亦可增強來自噴霧乾燥分散體之化合物2之水性溶解性及生物利用度。結合本發明使用之表面活性劑包括(但不限於)山梨醇酐脂肪酸酯(例如,Spans®)、聚氧乙烯山梨醇酐脂肪酸酯(例如,Tweens®)、月桂基硫酸鈉(SLS)、十二烷基苯磺酸鈉(SDBS)、磺琥珀酸鈉二辛酯(多庫酯鈉(Docusate))、脫氧膽酸鈉鹽(DOSS)、山梨醇酐單硬脂酸酯、山梨醇酐三硬脂酸酯、十六烷基三甲基溴化銨(HTAB)、N-月桂醯基肌胺酸鈉、油酸鈉、肉豆蔻酸鈉、硬脂酸鈉、棕櫚酸鈉、Gelucire 44/14、乙二胺四乙酸(EDTA)、維生素E d-α生育酚聚乙二醇1000琥珀酸酯(TPGS)、卵磷脂、MW 677-692、麩胺酸單鈉單水合物、Labrasol、PEG 8辛酸/癸酸甘油酯、Transcutol、二乙二醇單乙基醚、Solutol HS-15、聚乙二醇/羥基硬脂酸酯、牛膽酸、Pluronic F68、Pluronic F108及Pluronic F127 (或任何其他聚氧乙烯聚氧丙烯共聚物(Pluronics®)或飽和聚乙二醇化甘油酯(Gelucirs®))。可結合本發明使用之該等表面活性劑之特定實例包括(但不限於) Span 65、Span 25、Tween 20、Capryol 90、Pluronic F108、月桂基硫酸鈉(SLS)、維生素E TPGS、pluronics及共聚物。SLS通常較佳。 表面活性劑(例如,SLS)相對於噴霧乾燥分散體之總重量之量可介於0.1%至15%之間。較佳地,其為約0.5%至約10%,更佳地約0.5%至約5%,例如,約0.5%至4%、約0.5%至3%、約0.5%至2%、約0.5%至1%或約0.5%。 在某些實施例中,表面活性劑相對於噴霧乾燥分散體之總重量之量為至少約0.1%,較佳地為約0.5%。在該等實施例中,表面活性劑可以不超過約15%且較佳地不超過約12%、約11%、約10%、約9%、約8%、約7%、約6%、約5%、約4%、約3%、約2%或約1%之量存在。表面活性劑之量為約0.5重量%之實施例較佳。 可以與針對測試聚合物所闡述類似之方式測試候選表面活性劑(或其他組份)於本發明中使用之適宜性。製備醫藥組合物之方法
可藉由濕法製粒、在壓力下壓實或壓製混合物或組合物(例如,粉末或顆粒)以形成穩定三維形狀(例如,錠劑)來產生本發明之醫藥組合物。如本文所使用,「錠劑」包括所有形狀及大小之經壓製醫藥劑量單位形式,無論包覆與否。 如本文所使用,術語「錠劑」係指適於欲治療病患之藥劑之物理離散單元。一般而言,經壓實混合物之密度大於該混合物在壓實前之密度。本發明之劑量錠劑幾乎可具有任何形狀,包括凹面及/或凸面、圓角或有角角及約為直線之形狀。在一些實施例中,本發明之經壓製錠劑包含具有扁平面之圓形錠劑。可藉由為熟習形成經壓製固體醫藥劑型之技術者已知之任一壓實及壓製方法製備本發明之錠劑。在具體實施例中,可使用彼等熟習醫藥調配物領域者已知之習用方法來製備本文所提供調配物,如(例如)相關教科書中所闡述。例如,參見Remington: The Science and Practice of Pharmacy,第21版,Lippincott Williams & Wilkins, Baltimore, Md. (2003);Ansel等人,Pharmaceutical Dosage Forms And Drug Delivery Systems,第7版,Lippincott Williams & Wilkins, (1999);The Handbook of Pharmaceutical Excipients,第4版,Rowe等人編輯,American Pharmaceuticals Association (2003);Gibson, Pharmaceutical Preformulation And Formulation,CRC Press (2001),該等參考文獻以全文引用方式併入本文中。製粒及壓製
在一些實施例中,根據本文所述配方稱量各成份。接下來,對所有顆粒內成份實施篩分並將其充分混合。可用適宜潤滑劑(例如,硬脂酸鎂)潤滑成份。下一步驟可包含壓實/擊壓粉末混合物及定大小之成份。接下來,將經壓實或擊壓摻合物研磨成顆粒並篩分以獲得期望大小。接下來,可用(例如)硬脂酸鎂進一步潤滑顆粒。接下來,可在適宜衝壓器上將本發明顆粒組合物壓製成本發明之各種醫藥調配物。視情況,錠劑可經薄膜、著色劑或其他包衣包覆。 本發明之另一態樣提供產生醫藥組合物之方法,其包含提供包含以下之組合物之混合物:化合物1形式I、包含實質上非晶形化合物2之固體分散體及一或多種選自填充劑、稀釋劑、黏合劑、表面活性劑、潤滑劑、崩解劑之賦形劑,及將該組合物壓製成在約30分鐘內之溶解率為至少約50%之錠劑。 在另一實施例中,對粉末及液體成份之混合物實施濕法製粒製程以得到本發明醫藥調配物。例如,根據本文所述配方稱量包含包括以下之組合物之混合物之醫藥組合物:化合物1形式I、包含實質上非晶形化合物2之固體分散體之組合物,及一或多種選自填充劑、黏合劑、表面活性劑或崩解劑之賦形劑。接下來,在高剪切或低剪切製粒機中使用水或具有表面活性劑之水或具有黏合劑之水或具有表面活性劑及黏合劑之水對所有顆粒內成份實施篩分並混合,以製粒粉末摻合物。非水流體亦可與表面活性劑及/或黏合劑一起使用或不與其一起使用,以製粒粉末摻合物。接下來,可視情況使用適宜研磨機研磨濕顆粒。隨後,可視情況藉由以任一適宜方式乾燥成份自混合物去除水。接下來,可視情況將乾燥顆粒研磨成所需大小。接下來,可藉由摻合添加顆粒外賦形劑(例如填充劑、稀釋劑及崩解劑)。接下來,可用硬脂酸鎂及崩解劑(例如,交聯羧甲基纖維素鈉)進一步潤滑定大小之顆粒。接下來,可將本發明之顆粒組合物篩分足夠時間以獲得正確大小,且然後在適宜衝壓器上壓製成本發明之各種醫藥調配物。視情況,錠劑可經薄膜、著色劑或其他包衣包覆。令人驚訝地,可實施濕法製粒且實質上不損失化合物1形式I或實質上非晶形化合物2之固態形式。 在尤其有利的實施例中,本發明醫藥組合物係藉由連續雙螺桿濕法製粒(TSWG)製程來製備。連續製造在線上監測及控制下產生高品質且高度一致的產品。連續製造亦因設計研發利用「富數據」設計空間且更易於瞭解上游變量對下游製程及最終產品品質之影響而有利於品質。此外,本發明醫藥組合物可早在商業規模設備上完結,從而避免在研發晚期擴大規模的風險及調配物變化。最後,連續製造具有商業製造優點,例如製程控制有所改良、產品處置有所減少及實時釋放效率。總體而言,產生具有較少製程檢查從而提高產品品質且因此提高患者安全性之更穩健、可控且規模可變的製程。該等優點解決了Janet Woodcock (藥物評估與研究中心(Center for Drug Evaluation and Research,CDER)的主任)對化學與製造控制不能趕上高度有效療法之迅速臨床研發之擔憂(「What we are seeing is that often the rate limiting step is going to be manufacturing」,2013年7月24日,癌症之友(Friends of Cancer)主持的會議簡要「Answering a Compelling Need: Expediting Life-Saving Treatments to Patients」,用以論述美國食品藥物管理局之突破性療法認定)。 例如,高剪切製粒(HSG)係常用製粒技術,眾所周知其具有過度製粒及差製程控制之風險。對於此製程擴大規模極具挑戰性且涉及顯著風險。從HSG製程變化為連續TSWG製程允許使用相同設備擴大規模以藉由運行更長時間來產生不同批量。此消除其他製粒製程所通常遇到之擴大規模的風險。另外,發現TSWG製程更為穩健,較不易於過度製粒。如在針對化合物1錠劑之圖3中可看出,HSG製程顯示溶解率隨水含量增加而顯著降低,而TSWG製程對於水添加之類似範圍未顯示變化。令人驚訝地發現,使用雙螺桿濕法製粒製程時包含介於45重量%至55重量%間之化合物1之錠劑調配物及包含介於60重量%至70重量%間之化合物1之錠劑調配物沒有性能變化。此與HSG製程之情形不同。另外,此連續且產品品質有所提高之製程解決了FDA關於有需要之患者缺乏藥物可用性之普遍抱怨。 在一個實施例中,連續製程起始於藉助失重式(loss-in-weight)進給將個別賦形劑、化合物1及化合物2進給至連續在線摻合機。自此摻合機連續傳送材料並藉助雙螺桿濕法製粒進行處理,乾燥,研磨,添加顆粒外賦形劑,摻合,壓製並包覆薄膜。 例如,在一個實施例中,可根據下文流程圖連續製備包含化合物1及化合物2之錠劑。
 。 在另一實施例中,其他藥劑係上述藥劑之任一組合。例如,該組合可包含包括化合物1形式I及實質上非晶形化合物2之固體分散體之本發明醫藥組合物或錠劑,且其他治療劑係(R
)-1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)-N-(1-(2,3-二羥基丙基)-6-氟-2-(1-羥基-2-甲基丙-2-基)-1H-吲哚-5-基)環丙烷甲醯胺。在另一實例中,該組合可包含包括化合物1形式I及實質上非晶形化合物2之固體分散體之本發明醫藥組合物或錠劑,且其他治療劑係4-(3-(1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲醯胺基)異喹啉-1-基)苯甲酸。在另一實例中,該組合可包含包括化合物1形式I及實質上非晶形化合物2之固體分散體之本發明醫藥組合物或錠劑,且其他治療劑係來自表1之化合物中之任一者,即表1之化合物1至14,或其任一組合。 在另一實施例中,其他藥劑係選自表1:
。 在另一實施例中,其他藥劑係上述藥劑之任一組合。例如,該組合可包含包括化合物1形式I及實質上非晶形化合物2之固體分散體之本發明醫藥組合物或錠劑,且其他治療劑係(R
)-1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)-N-(1-(2,3-二羥基丙基)-6-氟-2-(1-羥基-2-甲基丙-2-基)-1H-吲哚-5-基)環丙烷甲醯胺。在另一實例中,該組合可包含包括化合物1形式I及實質上非晶形化合物2之固體分散體之本發明醫藥組合物或錠劑,且其他治療劑係4-(3-(1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲醯胺基)異喹啉-1-基)苯甲酸。在另一實例中,該組合可包含包括化合物1形式I及實質上非晶形化合物2之固體分散體之本發明醫藥組合物或錠劑,且其他治療劑係來自表1之化合物中之任一者,即表1之化合物1至14,或其任一組合。 在另一實施例中,其他藥劑係選自表1:

 在甲苯(10體積)中漿化市售2,2-二氟-1,3-苯并二氧雜環戊烯-5-甲酸(1.0當量)。以可將溫度維持在15-25℃之速率經由加料漏斗添加Vitride®(2當量)。在添加結束時使溫度升高至40℃並保持2小時(h),然後經由加料漏斗小心地添加10% (w/w) NaOH水溶液(4.0當量)並將溫度維持在40-50℃。在再攪拌30分鐘(min)後,在40℃下使各層分離。使有機相冷卻至20℃,然後用水(2 × 1.5體積)洗滌,乾燥(Na2
SO4
),過濾,並濃縮以提供粗(2,2-二氟-1,3-苯并二氧雜環戊烯-5-基)-甲醇,將其直接用於下一步驟中。5- 氯甲基 -2,2- 二氟 -1,3- 苯并二氧雜環戊烯之製備。
在甲苯(10體積)中漿化市售2,2-二氟-1,3-苯并二氧雜環戊烯-5-甲酸(1.0當量)。以可將溫度維持在15-25℃之速率經由加料漏斗添加Vitride®(2當量)。在添加結束時使溫度升高至40℃並保持2小時(h),然後經由加料漏斗小心地添加10% (w/w) NaOH水溶液(4.0當量)並將溫度維持在40-50℃。在再攪拌30分鐘(min)後,在40℃下使各層分離。使有機相冷卻至20℃,然後用水(2 × 1.5體積)洗滌,乾燥(Na2
SO4
),過濾,並濃縮以提供粗(2,2-二氟-1,3-苯并二氧雜環戊烯-5-基)-甲醇,將其直接用於下一步驟中。5- 氯甲基 -2,2- 二氟 -1,3- 苯并二氧雜環戊烯之製備。  將(2,2-二氟-1,3-苯并二氧雜環戊烯-5-基)-甲醇(1.0當量)溶解於MTBE(5體積)中。添加催化量之4-(N,N-二甲基)胺基吡啶(DMAP) (1 mol%)且經由加料漏斗添加SOCl2
(1.2當量)。以可將反應器中之溫度維持在15-25℃之速率添加SOCl2
。使溫度升高至30℃並保持1 h,然後使其冷卻至20℃。經由加料漏斗添加水(4體積),同時將溫度維持在30℃以下。在再攪拌30分鐘後,使各層分離。攪拌有機層且添加10% (w/v) NaOH水溶液(4.4體積)。在攪拌15 min至20 min後,使各層分離。然後,乾燥(Na2
SO4
)有機相,過濾並濃縮以提供粗5-氯甲基-2,2-二氟-1,3-苯并二氧雜環戊烯,將其直接用於下一步驟中。(2,2- 二氟 -1,3- 苯并二氧雜環戊烯 -5- 基 )- 乙腈之製備。
將(2,2-二氟-1,3-苯并二氧雜環戊烯-5-基)-甲醇(1.0當量)溶解於MTBE(5體積)中。添加催化量之4-(N,N-二甲基)胺基吡啶(DMAP) (1 mol%)且經由加料漏斗添加SOCl2
(1.2當量)。以可將反應器中之溫度維持在15-25℃之速率添加SOCl2
。使溫度升高至30℃並保持1 h,然後使其冷卻至20℃。經由加料漏斗添加水(4體積),同時將溫度維持在30℃以下。在再攪拌30分鐘後,使各層分離。攪拌有機層且添加10% (w/v) NaOH水溶液(4.4體積)。在攪拌15 min至20 min後,使各層分離。然後,乾燥(Na2
SO4
)有機相,過濾並濃縮以提供粗5-氯甲基-2,2-二氟-1,3-苯并二氧雜環戊烯,將其直接用於下一步驟中。(2,2- 二氟 -1,3- 苯并二氧雜環戊烯 -5- 基 )- 乙腈之製備。  將5-氯甲基-2,2-二氟-1,3-苯并二氧雜環戊烯(1當量)存於DMSO (1.25體積)中之溶液添加至NaCN (1.4當量)存於DMSO (3體積)中之漿液中,同時將溫度維持在30-40℃之間。將混合物攪拌1 h,且然後依次添加水(6體積)與甲基第三丁基醚(MTBE) (4體積)。在攪拌30 min後,分離各層。用MTBE(1.8體積)萃取水層。用水(1.8體積)洗滌合併之有機層,乾燥(Na2
SO4
),過濾並濃縮以提供粗(2,2-二氟-1,3-苯并二氧雜環戊烯-5-基)-乙腈(95%),將其直接用於下一步驟中。(2,2- 二氟 -1,3- 苯并二氧雜環戊烯 -5- 基 )-1- 乙酸乙酯 - 乙腈之合成。
將5-氯甲基-2,2-二氟-1,3-苯并二氧雜環戊烯(1當量)存於DMSO (1.25體積)中之溶液添加至NaCN (1.4當量)存於DMSO (3體積)中之漿液中,同時將溫度維持在30-40℃之間。將混合物攪拌1 h,且然後依次添加水(6體積)與甲基第三丁基醚(MTBE) (4體積)。在攪拌30 min後,分離各層。用MTBE(1.8體積)萃取水層。用水(1.8體積)洗滌合併之有機層,乾燥(Na2
SO4
),過濾並濃縮以提供粗(2,2-二氟-1,3-苯并二氧雜環戊烯-5-基)-乙腈(95%),將其直接用於下一步驟中。(2,2- 二氟 -1,3- 苯并二氧雜環戊烯 -5- 基 )-1- 乙酸乙酯 - 乙腈之合成。  用氮吹掃反應器且用900 mL甲苯裝填。經由氮吹掃將溶劑脫氣不短於16 h。然後,向反應器中依次裝填Na3
PO4
(155.7 g, 949.5 mmol)、雙(二亞苄基丙酮)鈀(0) (7.28 g, 12.66 mmol)。在23℃下經10 min自經氮吹掃加料漏斗裝填第三丁基膦存於己烷中之10% w/w溶液(51.23 g, 25.32 mmol)。將混合物攪拌50 min,此時經1 min添加5-溴-2,2-二氟-1,3-苯并二氧雜環戊烯(75 g, 316.5 mmol)。在再攪拌50 min後,經5 min向混合物裝填氰基乙酸乙酯(71.6 g, 633.0 mmol),隨後一次性裝填水(4.5 mL)。經40 min將混合物加熱至70℃且每1 - 2 h藉由HPLC分析反應物至產物之轉化百分比。在觀察到完全轉化(通常在5 - 8 h後100%轉化)後,使混合物冷卻至20 - 25℃並藉助矽藻土墊過濾。用甲苯(2 × 450 mL)沖洗矽藻土墊且在真空下在60 - 65℃下將經合併有機物濃縮至300 mL。向濃縮物裝填225 mL DMSO且在真空下在70 - 80℃下將其濃縮,直至有效的溶劑蒸餾結束為止。使溶液冷卻至20 - 25℃且用DMSO稀釋至900 mL以準備用於步驟2。1
H NMR (500 MHz, CDCl3
) δ 7.16 - 7.10 (m, 2H), 7.03 (d,J
= 8.2 Hz, 1H), 4.63 (s, 1H), 4.19 (m, 2H), 1.23 (t,J
= 7.1 Hz, 3H)。(2,2- 二氟 -1,3- 苯并二氧雜環戊烯 -5- 基 )- 乙腈之合成。
用氮吹掃反應器且用900 mL甲苯裝填。經由氮吹掃將溶劑脫氣不短於16 h。然後,向反應器中依次裝填Na3
PO4
(155.7 g, 949.5 mmol)、雙(二亞苄基丙酮)鈀(0) (7.28 g, 12.66 mmol)。在23℃下經10 min自經氮吹掃加料漏斗裝填第三丁基膦存於己烷中之10% w/w溶液(51.23 g, 25.32 mmol)。將混合物攪拌50 min,此時經1 min添加5-溴-2,2-二氟-1,3-苯并二氧雜環戊烯(75 g, 316.5 mmol)。在再攪拌50 min後,經5 min向混合物裝填氰基乙酸乙酯(71.6 g, 633.0 mmol),隨後一次性裝填水(4.5 mL)。經40 min將混合物加熱至70℃且每1 - 2 h藉由HPLC分析反應物至產物之轉化百分比。在觀察到完全轉化(通常在5 - 8 h後100%轉化)後,使混合物冷卻至20 - 25℃並藉助矽藻土墊過濾。用甲苯(2 × 450 mL)沖洗矽藻土墊且在真空下在60 - 65℃下將經合併有機物濃縮至300 mL。向濃縮物裝填225 mL DMSO且在真空下在70 - 80℃下將其濃縮,直至有效的溶劑蒸餾結束為止。使溶液冷卻至20 - 25℃且用DMSO稀釋至900 mL以準備用於步驟2。1
H NMR (500 MHz, CDCl3
) δ 7.16 - 7.10 (m, 2H), 7.03 (d,J
= 8.2 Hz, 1H), 4.63 (s, 1H), 4.19 (m, 2H), 1.23 (t,J
= 7.1 Hz, 3H)。(2,2- 二氟 -1,3- 苯并二氧雜環戊烯 -5- 基 )- 乙腈之合成。 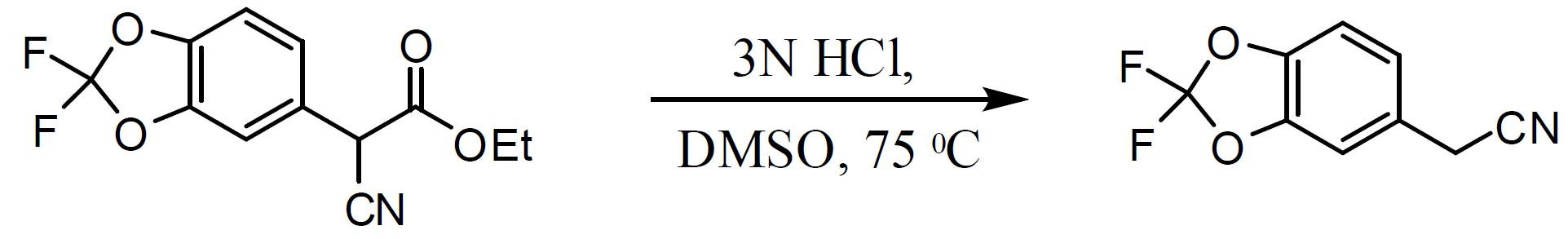 經20 min向來自上文之(2,2-二氟-1,3-苯并二氧雜環戊烯-5-基)-1-乙基乙酸酯-乙腈之DMSO溶液裝填3 N HCl (617.3 mL, 1.85 mol),同時將內部溫度維持在<40℃。然後,經1 h將混合物加熱至75℃且每1 - 2 h藉由HPLC分析轉化%。當觀察到轉化率> 99%(通常在5 - 6 h後)後,使反應物冷卻至20 - 25℃並用MTBE (2 × 525 mL)萃取,在萃取期間進行足夠時間以使相完全分離。用5% NaCl (2 × 375 mL)洗滌經合併有機萃取物。然後,將溶液轉移至適於1.5 - 2.5托(Torr)真空蒸餾之設備,其配備有冷卻接收燒瓶。在<60℃下在真空下濃縮溶液以去除溶劑。隨後在125 - 130℃(烘箱溫度)及1.5 - 2.0托下自所得油蒸餾出(2,2-二氟-1,3-苯并二氧雜環戊烯-5-基)-乙腈。自5-溴-2,2-二氟-1,3-苯并二氧雜環戊烯分離產率為66%之澄清油(2,2-二氟-1,3-苯并二氧雜環戊烯-5-基)-乙腈(2個步驟),且HPLC純度為91.5% AUC (對應於95%之w/w分析)。1
H NMR (500 MHz, DMSO) δ 7.44 (br s, 1H), 7.43 (d,J
= 8.4 Hz, 1H), 7.22 (dd,J
= 8.2, 1.8 Hz, 1H), 4.07 (s, 2H)。(2,2- 二氟 -1,3- 苯并二氧雜環戊烯 -5- 基 )- 環丙烷甲腈之製備。
經20 min向來自上文之(2,2-二氟-1,3-苯并二氧雜環戊烯-5-基)-1-乙基乙酸酯-乙腈之DMSO溶液裝填3 N HCl (617.3 mL, 1.85 mol),同時將內部溫度維持在<40℃。然後,經1 h將混合物加熱至75℃且每1 - 2 h藉由HPLC分析轉化%。當觀察到轉化率> 99%(通常在5 - 6 h後)後,使反應物冷卻至20 - 25℃並用MTBE (2 × 525 mL)萃取,在萃取期間進行足夠時間以使相完全分離。用5% NaCl (2 × 375 mL)洗滌經合併有機萃取物。然後,將溶液轉移至適於1.5 - 2.5托(Torr)真空蒸餾之設備,其配備有冷卻接收燒瓶。在<60℃下在真空下濃縮溶液以去除溶劑。隨後在125 - 130℃(烘箱溫度)及1.5 - 2.0托下自所得油蒸餾出(2,2-二氟-1,3-苯并二氧雜環戊烯-5-基)-乙腈。自5-溴-2,2-二氟-1,3-苯并二氧雜環戊烯分離產率為66%之澄清油(2,2-二氟-1,3-苯并二氧雜環戊烯-5-基)-乙腈(2個步驟),且HPLC純度為91.5% AUC (對應於95%之w/w分析)。1
H NMR (500 MHz, DMSO) δ 7.44 (br s, 1H), 7.43 (d,J
= 8.4 Hz, 1H), 7.22 (dd,J
= 8.2, 1.8 Hz, 1H), 4.07 (s, 2H)。(2,2- 二氟 -1,3- 苯并二氧雜環戊烯 -5- 基 )- 環丙烷甲腈之製備。  將(2,2-二氟-1,3-苯并二氧雜環戊烯-5-基)-乙腈(1.0當量)、50 wt% KOH水溶液(5.0當量)、1-溴-2-氯乙烷(1.5當量)與Oct4
NBr(0.02當量)之混合物在70℃下加熱1 h。冷卻反應混合物,然後用MTBE及水處理。用水及鹽水洗滌有機相。去除溶劑以提供(2,2-二氟-1,3-苯并二氧雜環戊烯-5-基)-環丙烷甲腈。1-(2,2- 二氟 -1,3- 苯并二氧雜環戊烯 -5- 基 )- 環丙烷甲酸之製備。
將(2,2-二氟-1,3-苯并二氧雜環戊烯-5-基)-乙腈(1.0當量)、50 wt% KOH水溶液(5.0當量)、1-溴-2-氯乙烷(1.5當量)與Oct4
NBr(0.02當量)之混合物在70℃下加熱1 h。冷卻反應混合物,然後用MTBE及水處理。用水及鹽水洗滌有機相。去除溶劑以提供(2,2-二氟-1,3-苯并二氧雜環戊烯-5-基)-環丙烷甲腈。1-(2,2- 二氟 -1,3- 苯并二氧雜環戊烯 -5- 基 )- 環丙烷甲酸之製備。 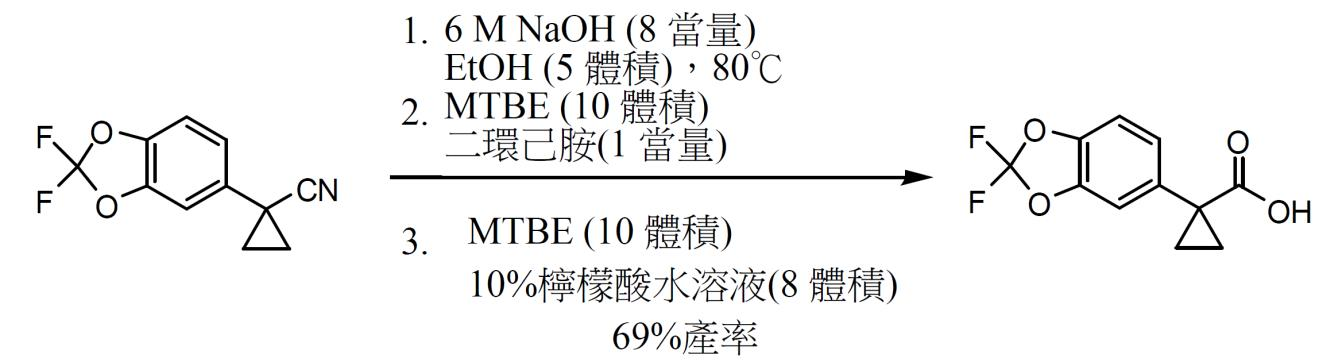 在80℃下使用存於乙醇(5體積)中之6 M NaOH(8當量)過夜水解(2,2-二氟-1,3-苯并二氧雜環戊烯-5-基)-環丙烷甲腈。使混合物冷卻至室溫且在真空下蒸發乙醇。用水及MTBE吸收殘餘物,添加1 M HCl且分離各層。然後,用二環己胺(DCHA) (0.97當量)處理MTBE層。使漿液冷卻至0℃,過濾並用庚烷洗滌以獲得對應DCHA鹽。用MTBE及10%檸檬酸吸收該鹽且將其攪拌直至所有固體皆溶解。分離各層且用水及鹽水洗滌MTBE層。將溶劑改為庚烷,隨後過濾,且在真空烘箱中於50℃下乾燥過夜後獲得1-(2,2-二氟-1,3-苯并二氧雜環戊烯-5-基)-環丙烷甲酸。1-(2,2- 二氟 -1,3- 苯并二氧雜環戊烯 -5- 基 )- 環丙烷羰基氯之製備。
在80℃下使用存於乙醇(5體積)中之6 M NaOH(8當量)過夜水解(2,2-二氟-1,3-苯并二氧雜環戊烯-5-基)-環丙烷甲腈。使混合物冷卻至室溫且在真空下蒸發乙醇。用水及MTBE吸收殘餘物,添加1 M HCl且分離各層。然後,用二環己胺(DCHA) (0.97當量)處理MTBE層。使漿液冷卻至0℃,過濾並用庚烷洗滌以獲得對應DCHA鹽。用MTBE及10%檸檬酸吸收該鹽且將其攪拌直至所有固體皆溶解。分離各層且用水及鹽水洗滌MTBE層。將溶劑改為庚烷,隨後過濾,且在真空烘箱中於50℃下乾燥過夜後獲得1-(2,2-二氟-1,3-苯并二氧雜環戊烯-5-基)-環丙烷甲酸。1-(2,2- 二氟 -1,3- 苯并二氧雜環戊烯 -5- 基 )- 環丙烷羰基氯之製備。 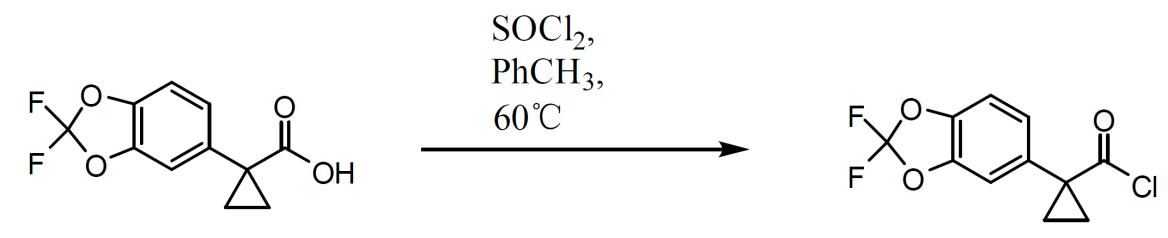 在甲苯(2.5體積)中漿化1-(2,2-二氟-1,3-苯并二氧雜環戊烯-5-基)-環丙烷甲酸(1.2當量)且將混合物加熱至60℃。經由加料漏斗添加SOCl2
(1.4當量)。在30分鐘後自反應混合物蒸餾出甲苯及SOCl2
。添加另一甲苯(2.5體積)且將所得混合物再次蒸餾,剩下呈油狀物之醯氯產物,其不進一步純化即使用。3-(3- 甲基吡啶 -2- 基 ) 苯甲酸第三丁基酯之製備。
在甲苯(2.5體積)中漿化1-(2,2-二氟-1,3-苯并二氧雜環戊烯-5-基)-環丙烷甲酸(1.2當量)且將混合物加熱至60℃。經由加料漏斗添加SOCl2
(1.4當量)。在30分鐘後自反應混合物蒸餾出甲苯及SOCl2
。添加另一甲苯(2.5體積)且將所得混合物再次蒸餾,剩下呈油狀物之醯氯產物,其不進一步純化即使用。3-(3- 甲基吡啶 -2- 基 ) 苯甲酸第三丁基酯之製備。  使2-溴-3-甲基吡啶(1.0當量)溶解於甲苯(12體積)中。依次添加K2
CO3
(4.8當量)及水(3.5體積)。在N2
流下將所得混合物加熱至65℃並保持1小時。然後,添加3-(第三丁氧基羰基)苯基
使2-溴-3-甲基吡啶(1.0當量)溶解於甲苯(12體積)中。依次添加K2
CO3
(4.8當量)及水(3.5體積)。在N2
流下將所得混合物加熱至65℃並保持1小時。然後,添加3-(第三丁氧基羰基)苯基 使3-(3-甲基吡啶-2-基)苯甲酸第三丁基酯(1.0當量)溶解於EtOAc(6體積)中。依次添加水(0.3體積)及過氧化氫脲(3當量)。然後,以可將反應器中之溫度維持在45℃以下之速率將鄰苯二甲酸酐(3當量)逐份添加至呈固體形式之混合物中。在鄰苯二甲酸酐添加完成後,將混合物加熱至45℃。在再攪拌4小時後,關閉加熱器。經由加料漏斗添加10% w/w Na2
SO3
水溶液(1.5當量)。在Na2
SO3
添加完成後,將混合物再攪拌30 min並分離各層。攪拌有機層並添加10% wt/wt Na2
CO3
水溶液(2當量)。在攪拌30分鐘後,使各層分離。用13% w/v NaCl水溶液洗滌有機相。然後,過濾並濃縮有機相以提供粗2-(3-(第三丁氧基羰基)苯基)-3-甲基吡啶-1-氧化物(95%),將其直接用於下一步驟中。3-(6- 胺基 -3- 甲基吡啶 -2- 基 ) 苯甲酸第三丁基酯之製備。
使3-(3-甲基吡啶-2-基)苯甲酸第三丁基酯(1.0當量)溶解於EtOAc(6體積)中。依次添加水(0.3體積)及過氧化氫脲(3當量)。然後,以可將反應器中之溫度維持在45℃以下之速率將鄰苯二甲酸酐(3當量)逐份添加至呈固體形式之混合物中。在鄰苯二甲酸酐添加完成後,將混合物加熱至45℃。在再攪拌4小時後,關閉加熱器。經由加料漏斗添加10% w/w Na2
SO3
水溶液(1.5當量)。在Na2
SO3
添加完成後,將混合物再攪拌30 min並分離各層。攪拌有機層並添加10% wt/wt Na2
CO3
水溶液(2當量)。在攪拌30分鐘後,使各層分離。用13% w/v NaCl水溶液洗滌有機相。然後,過濾並濃縮有機相以提供粗2-(3-(第三丁氧基羰基)苯基)-3-甲基吡啶-1-氧化物(95%),將其直接用於下一步驟中。3-(6- 胺基 -3- 甲基吡啶 -2- 基 ) 苯甲酸第三丁基酯之製備。  將2-(3-(第三丁氧基羰基)苯基)-3-甲基吡啶-1-氧化物(1當量)及吡啶(4當量)存於MeCN (8體積)中之溶液加熱至70℃。經50 min經由加料漏斗添加甲磺酸酐(1.5當量)存於MeCN (2體積)中之溶液,同時將溫度維持在75℃以下。在添加完成後將混合物再攪拌0.5小時。然後,使混合物冷卻至環境溫度。經由加料漏斗添加乙醇胺(10當量)。在攪拌2小時後,添加水(6體積)並使混合物冷卻至10℃。在攪拌3小時後,藉由過濾收集固體並用水(3體積)、2:1乙腈/水(3體積)及乙腈(2 × 1.5體積)洗滌。在真空烘箱中於50℃及輕微N2
吹掃下將固體乾燥至恒重(差異<1%)以提供呈紅-黃色固體形式之3-(6-胺基-3-甲基吡啶-2-基)苯甲酸第三丁基酯(產率53%)。3-(6-(1-(2,2- 二氟苯并 [d][1,3] 二氧雜環戊烯 -5- 基 ) 環丙烷甲醯胺基 )-3- 甲基吡啶 -2- 基 )- 苯甲酸第三丁基酯之製備。
將2-(3-(第三丁氧基羰基)苯基)-3-甲基吡啶-1-氧化物(1當量)及吡啶(4當量)存於MeCN (8體積)中之溶液加熱至70℃。經50 min經由加料漏斗添加甲磺酸酐(1.5當量)存於MeCN (2體積)中之溶液,同時將溫度維持在75℃以下。在添加完成後將混合物再攪拌0.5小時。然後,使混合物冷卻至環境溫度。經由加料漏斗添加乙醇胺(10當量)。在攪拌2小時後,添加水(6體積)並使混合物冷卻至10℃。在攪拌3小時後,藉由過濾收集固體並用水(3體積)、2:1乙腈/水(3體積)及乙腈(2 × 1.5體積)洗滌。在真空烘箱中於50℃及輕微N2
吹掃下將固體乾燥至恒重(差異<1%)以提供呈紅-黃色固體形式之3-(6-胺基-3-甲基吡啶-2-基)苯甲酸第三丁基酯(產率53%)。3-(6-(1-(2,2- 二氟苯并 [d][1,3] 二氧雜環戊烯 -5- 基 ) 環丙烷甲醯胺基 )-3- 甲基吡啶 -2- 基 )- 苯甲酸第三丁基酯之製備。 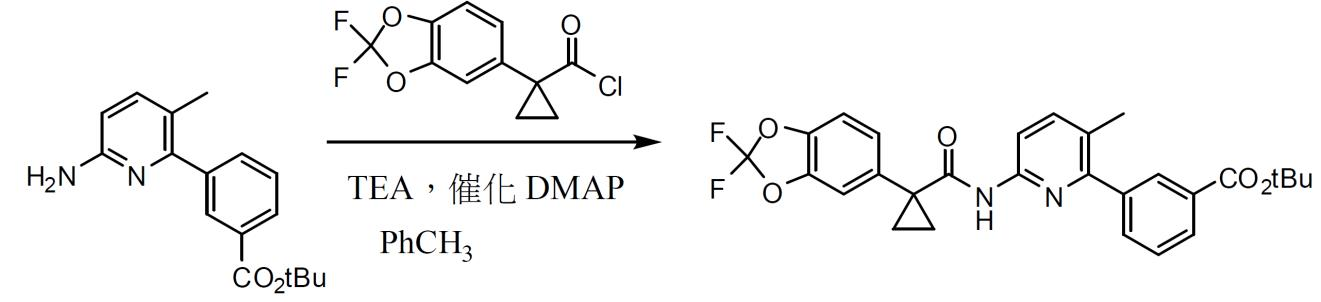 使上文所述粗醯氯溶解於甲苯(以醯氯計,2.5體積)中,且經由加料漏斗將其添加至3-(6-胺基-3-甲基吡啶-2-基)苯甲酸第三丁基酯(1當量)、DMAP (0.02當量)與三乙胺(3.0當量)存於甲苯(以3-(6-胺基-3-甲基吡啶-2-基)苯甲酸第三丁基酯計,4體積)之混合物中。在2小時後,將水(以3-(6-胺基-3-甲基吡啶-2-基)苯甲酸第三丁基酯計,4體積)添加至反應混合物中。在攪拌30分鐘後,分離各層。然後,過濾並濃縮有機相以提供3-(6-(1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲醯胺基)-3-甲基吡啶-2-基)-苯甲酸第三丁基酯之濃稠油(定量粗產率)。添加乙腈(以粗產物計,3體積)且蒸餾直至發生結晶。添加水(以粗產物計,2體積)且將混合物攪拌2 h。藉由過濾收集固體,用1:1 (體積比)乙腈/水(以粗產物計,2 × 1體積)洗滌,且在真空下於過濾器上進行部分乾燥。在真空烘箱中於60℃及輕微N2
吹掃下將固體乾燥至恒重(差異<1%)以提供呈褐色固體形式之3-(6-(1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲醯胺基)-3-甲基吡啶-2-基)-苯甲酸第三丁基酯。3-(6-(1-(2,2- 二氟苯并 [d][1,3] 二氧雜環戊烯 -5- 基 ) 環丙烷甲醯胺基 )-3- 甲基吡啶 -2- 基 ) 苯甲酸 • HCL 鹽之製備。
使上文所述粗醯氯溶解於甲苯(以醯氯計,2.5體積)中,且經由加料漏斗將其添加至3-(6-胺基-3-甲基吡啶-2-基)苯甲酸第三丁基酯(1當量)、DMAP (0.02當量)與三乙胺(3.0當量)存於甲苯(以3-(6-胺基-3-甲基吡啶-2-基)苯甲酸第三丁基酯計,4體積)之混合物中。在2小時後,將水(以3-(6-胺基-3-甲基吡啶-2-基)苯甲酸第三丁基酯計,4體積)添加至反應混合物中。在攪拌30分鐘後,分離各層。然後,過濾並濃縮有機相以提供3-(6-(1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲醯胺基)-3-甲基吡啶-2-基)-苯甲酸第三丁基酯之濃稠油(定量粗產率)。添加乙腈(以粗產物計,3體積)且蒸餾直至發生結晶。添加水(以粗產物計,2體積)且將混合物攪拌2 h。藉由過濾收集固體,用1:1 (體積比)乙腈/水(以粗產物計,2 × 1體積)洗滌,且在真空下於過濾器上進行部分乾燥。在真空烘箱中於60℃及輕微N2
吹掃下將固體乾燥至恒重(差異<1%)以提供呈褐色固體形式之3-(6-(1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲醯胺基)-3-甲基吡啶-2-基)-苯甲酸第三丁基酯。3-(6-(1-(2,2- 二氟苯并 [d][1,3] 二氧雜環戊烯 -5- 基 ) 環丙烷甲醯胺基 )-3- 甲基吡啶 -2- 基 ) 苯甲酸 • HCL 鹽之製備。 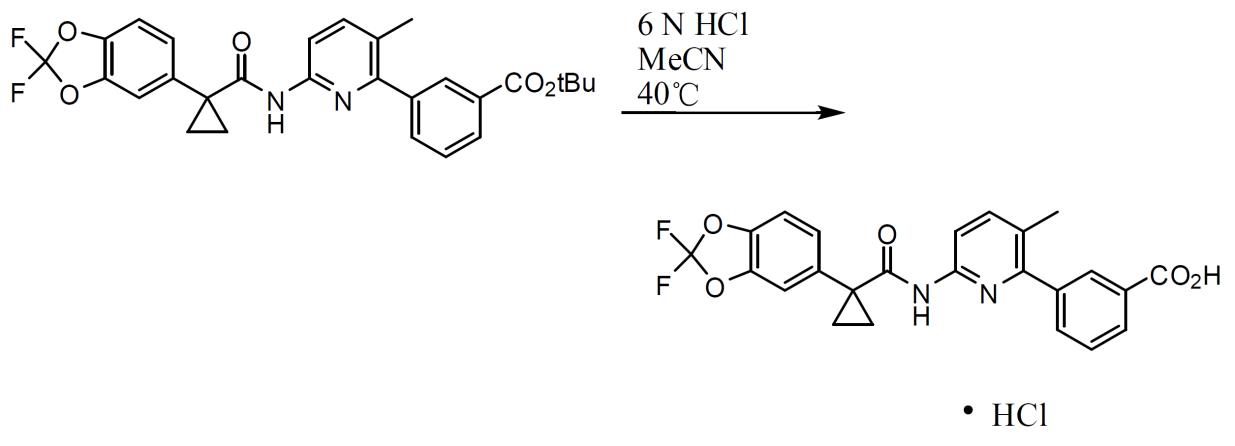 向3-(6-(1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲醯胺基)-3-甲基吡啶-2-基)-苯甲酸第三丁基酯(1.0當量)存於MeCN(3.0體積)中之漿液中依次添加水(0.83體積)及濃HCl水溶液(0.83體積)。將混合物加熱至45 ± 5℃。在攪拌24 h至48 h後,反應完成,且使混合物冷卻至環境溫度。添加水(1.33體積)並攪拌混合物。藉由過濾收集固體,用水(2 × 0.3體積)洗滌,且在真空下於過濾器上進行部分乾燥。在真空烘箱中於60℃及輕微N2
吹掃下將固體乾燥至恒重(差異<1%)以提供呈灰白色固體形式之3-(6-(1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲醯胺基)-3-甲基吡啶-2-基)苯甲酸• HCl。 化合物1之1
HNMR光譜顯示於圖8中且圖9繪示呈HCl鹽形式之化合物1之1
HNMR光譜。下表2列述化合物I之1
HNMR數據。表 2 。
向3-(6-(1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲醯胺基)-3-甲基吡啶-2-基)-苯甲酸第三丁基酯(1.0當量)存於MeCN(3.0體積)中之漿液中依次添加水(0.83體積)及濃HCl水溶液(0.83體積)。將混合物加熱至45 ± 5℃。在攪拌24 h至48 h後,反應完成,且使混合物冷卻至環境溫度。添加水(1.33體積)並攪拌混合物。藉由過濾收集固體,用水(2 × 0.3體積)洗滌,且在真空下於過濾器上進行部分乾燥。在真空烘箱中於60℃及輕微N2
吹掃下將固體乾燥至恒重(差異<1%)以提供呈灰白色固體形式之3-(6-(1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲醯胺基)-3-甲基吡啶-2-基)苯甲酸• HCl。 化合物1之1
HNMR光譜顯示於圖8中且圖9繪示呈HCl鹽形式之化合物1之1
HNMR光譜。下表2列述化合物I之1
HNMR數據。表 2 。 
 在環境溫度下攪拌3-(6-(1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲醯胺基)-3-甲基吡啶-2-基)苯甲酸• HCl (1當量)存於水(10體積)中之漿液。在攪拌24小時後取樣。過濾試樣且用水(2次)洗滌固體。對固體試樣實施DSC分析。當DSC分析指示完全轉化為形式I時,藉由過濾收集固體,用水(2 × 1.0體積)洗滌,且在真空下於過濾器上進行部分乾燥。然後,在真空烘箱中於60℃及輕微N2
吹掃下將固體乾燥至恒重(差異<1%)以提供呈灰白色固體形式之化合物1形式I (產率98%)。1
H NMR (400 MHz, DMSO-d6) 9.14 (s, 1H), 7.99-7.93 (m, 3H), 7.80-7.78 (m, 1H), 7.74-7.72 (m, 1H), 7.60-7.55 (m, 2H), 7.41-7.33 (m, 2H), 2.24 (s, 3H), 1.53-1.51 (m, 2H), 1.19-1.17 (m, 2H)。化合物 1 形式 I 之製備 , 方法 B 。
在環境溫度下攪拌3-(6-(1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲醯胺基)-3-甲基吡啶-2-基)苯甲酸• HCl (1當量)存於水(10體積)中之漿液。在攪拌24小時後取樣。過濾試樣且用水(2次)洗滌固體。對固體試樣實施DSC分析。當DSC分析指示完全轉化為形式I時,藉由過濾收集固體,用水(2 × 1.0體積)洗滌,且在真空下於過濾器上進行部分乾燥。然後,在真空烘箱中於60℃及輕微N2
吹掃下將固體乾燥至恒重(差異<1%)以提供呈灰白色固體形式之化合物1形式I (產率98%)。1
H NMR (400 MHz, DMSO-d6) 9.14 (s, 1H), 7.99-7.93 (m, 3H), 7.80-7.78 (m, 1H), 7.74-7.72 (m, 1H), 7.60-7.55 (m, 2H), 7.41-7.33 (m, 2H), 2.24 (s, 3H), 1.53-1.51 (m, 2H), 1.19-1.17 (m, 2H)。化合物 1 形式 I 之製備 , 方法 B 。  將3-(6-(1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲醯胺基)-3-甲基吡啶-2-基)-苯甲酸第三丁基酯(1.0當量)存於甲酸(3.0體積)中之溶液在攪拌下加熱至70 ± 10℃並保持8 h。當藉由層析方法確定3-(6-(1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲醯胺基)-3-甲基吡啶-2-基)-苯甲酸第三丁基酯保持不超過1.0% AUC時,認為反應完成。使混合物冷卻至環境溫度。將溶液添加至水(6體積)中,在50℃下加熱,並攪拌混合物。然後,將混合物加熱至70 ± 10℃直至3-(6-(1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲醯胺基)-3-甲基吡啶-2-基)-苯甲酸第三丁基酯之量不超過0.8% (AUC)。藉由過濾收集固體,用水(2 × 3體積)洗滌,且在真空下於過濾器上進行部分乾燥。然後在真空烘箱中於60℃及輕微N2
吹掃下將固體乾燥至恒重(差異<1%)以提供呈灰白色固體形式之化合物1形式I。化合物1形式I之DSC跡線顯示於圖10中。化合物1形式I在約204℃下發生熔化。自化合物1形式I之單晶結構計算X射線繞射圖案且顯示於圖1中。表3列示圖1中之計算峰。表 3 。
將3-(6-(1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲醯胺基)-3-甲基吡啶-2-基)-苯甲酸第三丁基酯(1.0當量)存於甲酸(3.0體積)中之溶液在攪拌下加熱至70 ± 10℃並保持8 h。當藉由層析方法確定3-(6-(1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲醯胺基)-3-甲基吡啶-2-基)-苯甲酸第三丁基酯保持不超過1.0% AUC時,認為反應完成。使混合物冷卻至環境溫度。將溶液添加至水(6體積)中,在50℃下加熱,並攪拌混合物。然後,將混合物加熱至70 ± 10℃直至3-(6-(1-(2,2-二氟苯并[d][1,3]二氧雜環戊烯-5-基)環丙烷甲醯胺基)-3-甲基吡啶-2-基)-苯甲酸第三丁基酯之量不超過0.8% (AUC)。藉由過濾收集固體,用水(2 × 3體積)洗滌,且在真空下於過濾器上進行部分乾燥。然後在真空烘箱中於60℃及輕微N2
吹掃下將固體乾燥至恒重(差異<1%)以提供呈灰白色固體形式之化合物1形式I。化合物1形式I之DSC跡線顯示於圖10中。化合物1形式I在約204℃下發生熔化。自化合物1形式I之單晶結構計算X射線繞射圖案且顯示於圖1中。表3列示圖1中之計算峰。表 3 。 

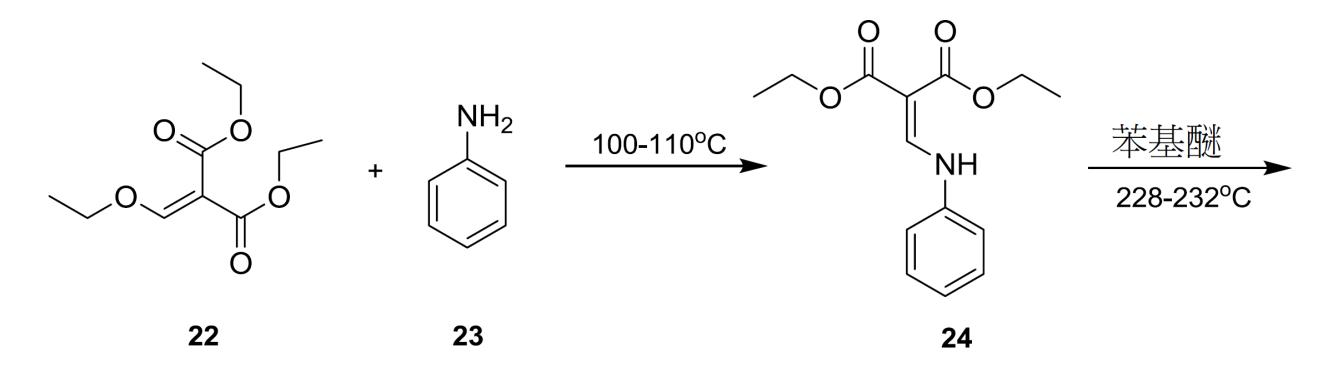
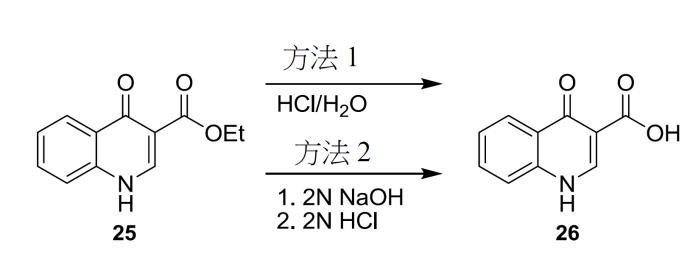 4- 側氧基 -1,4- 二氫喹啉 -3- 甲酸乙酯 (25) 之製備程序
4- 側氧基 -1,4- 二氫喹啉 -3- 甲酸乙酯 (25) 之製備程序  經0.5小時在低於30℃下將化合物23
(4.77 g, 47.7 mmol)逐滴添加至化合物22
(10 g, 46.3 mmol)中,且利用表面下N2
流逐出乙醇。然後,將溶液加熱至100-110℃並攪拌2.5小時。在將混合物冷卻至60℃以下後,添加二苯基醚。將所得溶液逐滴添加至已加熱至228-232℃並保持1.5小時之二苯基醚中且利用表面下N2
流逐出乙醇。將混合物在228-232℃下再攪拌2小時,冷卻至100℃以下,且然後添加庚烷以沈澱產物。將所得漿液在30℃下攪拌0.5小時。然後,過濾固體,並用庚烷洗滌濾餅且在真空中乾燥以獲得呈褐色固體形式之化合物25
。1
H NMR (DMSO-d6
;400 MHz) δ 12.25 (s), δ 8.49 (d), δ 8.10 (m), δ 7.64 (m), δ 7.55 (m), δ 7.34 (m), δ 4.16 (q), δ 1.23 (t)。4- 側氧基 -1,4- 二氫喹啉 -3- 甲酸 (26) 之製備程序
經0.5小時在低於30℃下將化合物23
(4.77 g, 47.7 mmol)逐滴添加至化合物22
(10 g, 46.3 mmol)中,且利用表面下N2
流逐出乙醇。然後,將溶液加熱至100-110℃並攪拌2.5小時。在將混合物冷卻至60℃以下後,添加二苯基醚。將所得溶液逐滴添加至已加熱至228-232℃並保持1.5小時之二苯基醚中且利用表面下N2
流逐出乙醇。將混合物在228-232℃下再攪拌2小時,冷卻至100℃以下,且然後添加庚烷以沈澱產物。將所得漿液在30℃下攪拌0.5小時。然後,過濾固體,並用庚烷洗滌濾餅且在真空中乾燥以獲得呈褐色固體形式之化合物25
。1
H NMR (DMSO-d6
;400 MHz) δ 12.25 (s), δ 8.49 (d), δ 8.10 (m), δ 7.64 (m), δ 7.55 (m), δ 7.34 (m), δ 4.16 (q), δ 1.23 (t)。4- 側氧基 -1,4- 二氫喹啉 -3- 甲酸 (26) 之製備程序 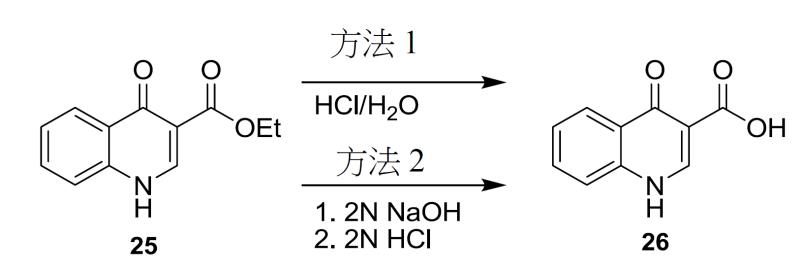 方法 1
將化合物25
(1.0當量)懸浮於HCl (10.0當量)及H2
O (11.6體積)之溶液中。將漿液加熱至85 – 90℃,但替代溫度亦適於此水解步驟。例如,另一選擇為,水解可在約75℃至約100℃之溫度下實施。在一些情形下,水解係在約80℃至約95℃之溫度下實施。在其他情形下,水解步驟係在約82℃至約93℃ (例如,約82.5℃至約92.5℃或約86℃至約89℃)之溫度下實施。在85 – 90℃下攪拌大約6.5小時後,對反應物取樣以判斷反應是否完成。可在適於水解之任一溫度下實施攪拌。然後,將溶液冷卻至20 – 25℃並過濾。用H2
O (2體積× 2)沖洗反應器/濾餅。然後,用2體積H2
O洗滌濾餅直至pH ≥ 3.0。然後,在真空及60℃下乾燥濾餅以獲得化合物26
。方法 2
將化合物25
(11.3 g, 52 mmol)添加至10% NaOH (水溶液) (10 mL)與乙醇(100 mL)之混合物中。將溶液加熱至回流16小時,冷卻至20-25℃,且然後,用8% HCl將pH調節至2-3。然後,將混合物攪拌0.5小時並過濾。用水(50 mL)洗滌濾餅,且然後在真空中乾燥以獲得呈褐色固體形式之化合物26
。1
H NMR (DMSO-d6
;400 MHz) δ 15.33 (s), δ 13.39 (s), δ 8.87 (s), δ 8.26 (m), δ 7.87 (m), δ 7.80 (m), δ 7.56 (m)。N-(2,4- 二 - 第三丁基 -5- 羥基苯基 )-4- 側氧基 -1,4- 二氫喹啉 -3- 甲醯胺 ( 化合物 2) 之總合成
方法 1
將化合物25
(1.0當量)懸浮於HCl (10.0當量)及H2
O (11.6體積)之溶液中。將漿液加熱至85 – 90℃,但替代溫度亦適於此水解步驟。例如,另一選擇為,水解可在約75℃至約100℃之溫度下實施。在一些情形下,水解係在約80℃至約95℃之溫度下實施。在其他情形下,水解步驟係在約82℃至約93℃ (例如,約82.5℃至約92.5℃或約86℃至約89℃)之溫度下實施。在85 – 90℃下攪拌大約6.5小時後,對反應物取樣以判斷反應是否完成。可在適於水解之任一溫度下實施攪拌。然後,將溶液冷卻至20 – 25℃並過濾。用H2
O (2體積× 2)沖洗反應器/濾餅。然後,用2體積H2
O洗滌濾餅直至pH ≥ 3.0。然後,在真空及60℃下乾燥濾餅以獲得化合物26
。方法 2
將化合物25
(11.3 g, 52 mmol)添加至10% NaOH (水溶液) (10 mL)與乙醇(100 mL)之混合物中。將溶液加熱至回流16小時,冷卻至20-25℃,且然後,用8% HCl將pH調節至2-3。然後,將混合物攪拌0.5小時並過濾。用水(50 mL)洗滌濾餅,且然後在真空中乾燥以獲得呈褐色固體形式之化合物26
。1
H NMR (DMSO-d6
;400 MHz) δ 15.33 (s), δ 13.39 (s), δ 8.87 (s), δ 8.26 (m), δ 7.87 (m), δ 7.80 (m), δ 7.56 (m)。N-(2,4- 二 - 第三丁基 -5- 羥基苯基 )-4- 側氧基 -1,4- 二氫喹啉 -3- 甲醯胺 ( 化合物 2) 之總合成  碳酸 2,4- 二 - 第三丁基苯基酯甲基酯 (30) 之製備程序
碳酸 2,4- 二 - 第三丁基苯基酯甲基酯 (30) 之製備程序 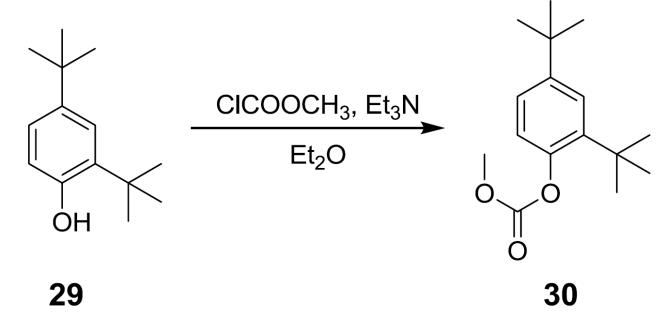 方法 1
在0℃下向2,4-二-第三丁基酚29
(10 g, 48.5mmol)於二乙醚(100 mL)及三乙胺(10.1 mL, 72.8 mmol)中之溶液中逐滴添加氯甲酸甲酯(7.46 mL, 97 mmol)。然後,使混合物升溫至室溫並再攪拌2小時。然後,添加另5 mL三乙胺及3.7 mL氯甲酸甲酯,並將反應物攪拌過夜。然後,過濾反應物,將濾液冷卻至0℃,且然後,添加另5 mL三乙胺及3.7 mL氯甲酸甲酯,並使反應物升溫至室溫,且然後,再攪拌1小時。在此階段,反應幾乎完全並藉由過濾處理,然後,依次用水(2×)及鹽水洗滌。然後,濃縮溶液以產生黃色油,並使用管柱層析純化以獲得化合物30
。1
H NMR (400 MHz, DMSO-d 6
) δ 7.35 (d,J
= 2.4 Hz, 1H), 7.29 (dd,J
= 8.4, 2.4 Hz, 1H), 7.06 (d,J
= 8.4 Hz, 1H), 3.85 (s, 3H), 1.30 (s, 9H), 1.29 (s, 9H)。方法 2
向裝填有4-二甲基胺基吡啶(DMAP, 3.16 g, 25.7 mmol)及2,4-二第三-丁基酚(化合物29
, 103.5 g, 501.6 mmol)之反應器器皿中添加二氯甲烷(415 g, 313 mL),並攪動溶液直至所有固體皆溶解。然後,添加三乙胺(76 g, 751 mmol),並將溶液冷卻至0 – 5℃。然後,經2.5 – 4小時逐滴添加氯甲酸甲酯(52 g, 550.3 mmol),同時使溶液溫度保持介於0至5℃之間。然後,將反應混合物緩慢加熱至23 – 28℃並攪拌20小時。然後,將反應物冷卻至10 – 15℃並用150 mL水裝填。將混合物在15 – 20℃下攪拌35 – 45分鐘,且然後,分離水層並用150 mL二氯甲烷萃取。合併有機層並在5 – 20℃之溫度下用2.5% HCl (水溶液)中和,以使最終pH為5 – 6。然後,用水洗滌有機層並在真空中在低於20℃之溫度下濃縮至150 mL,以獲得存於二氯甲烷中之化合物30
。碳酸 5- 硝基 -2,4- 二 - 第三丁基苯基酯甲基酯 (31) 之製備程序
方法 1
在0℃下向2,4-二-第三丁基酚29
(10 g, 48.5mmol)於二乙醚(100 mL)及三乙胺(10.1 mL, 72.8 mmol)中之溶液中逐滴添加氯甲酸甲酯(7.46 mL, 97 mmol)。然後,使混合物升溫至室溫並再攪拌2小時。然後,添加另5 mL三乙胺及3.7 mL氯甲酸甲酯,並將反應物攪拌過夜。然後,過濾反應物,將濾液冷卻至0℃,且然後,添加另5 mL三乙胺及3.7 mL氯甲酸甲酯,並使反應物升溫至室溫,且然後,再攪拌1小時。在此階段,反應幾乎完全並藉由過濾處理,然後,依次用水(2×)及鹽水洗滌。然後,濃縮溶液以產生黃色油,並使用管柱層析純化以獲得化合物30
。1
H NMR (400 MHz, DMSO-d 6
) δ 7.35 (d,J
= 2.4 Hz, 1H), 7.29 (dd,J
= 8.4, 2.4 Hz, 1H), 7.06 (d,J
= 8.4 Hz, 1H), 3.85 (s, 3H), 1.30 (s, 9H), 1.29 (s, 9H)。方法 2
向裝填有4-二甲基胺基吡啶(DMAP, 3.16 g, 25.7 mmol)及2,4-二第三-丁基酚(化合物29
, 103.5 g, 501.6 mmol)之反應器器皿中添加二氯甲烷(415 g, 313 mL),並攪動溶液直至所有固體皆溶解。然後,添加三乙胺(76 g, 751 mmol),並將溶液冷卻至0 – 5℃。然後,經2.5 – 4小時逐滴添加氯甲酸甲酯(52 g, 550.3 mmol),同時使溶液溫度保持介於0至5℃之間。然後,將反應混合物緩慢加熱至23 – 28℃並攪拌20小時。然後,將反應物冷卻至10 – 15℃並用150 mL水裝填。將混合物在15 – 20℃下攪拌35 – 45分鐘,且然後,分離水層並用150 mL二氯甲烷萃取。合併有機層並在5 – 20℃之溫度下用2.5% HCl (水溶液)中和,以使最終pH為5 – 6。然後,用水洗滌有機層並在真空中在低於20℃之溫度下濃縮至150 mL,以獲得存於二氯甲烷中之化合物30
。碳酸 5- 硝基 -2,4- 二 - 第三丁基苯基酯甲基酯 (31) 之製備程序 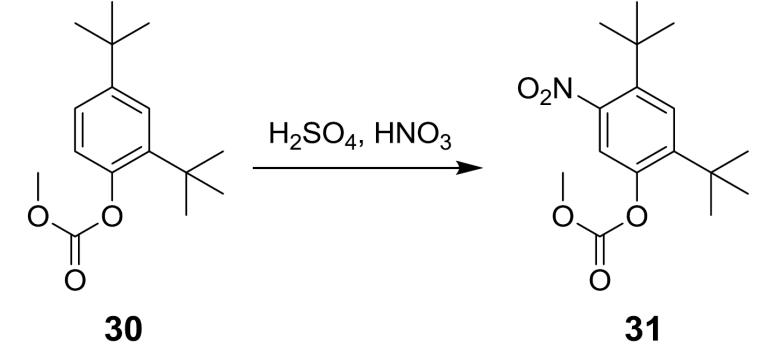 方法 1
在0℃下向化合物30
(6.77g, 25.6 mmol)之攪拌溶液中逐滴添加6 mL硫酸及硝酸之1:1混合物。使混合物升溫至室溫並攪拌1小時。使用液相層析(ISCO, 120 g, 0-7% EtOAc/己烷, 38 min)純化產物,從而產生化合物31
之區域異構體之呈
白色固體形式之約8:1 – 10:1混合物。1
H NMR (400 MHz, DMSO-d 6
) δ 7.63 (s, 1H), 7.56 (s, 1H), 3.87 (s, 3H), 1.36 (s, 9H), 1.32 (s, 9H)。HPLC滯留時間3.92 min,10-99% CH3
CN,運行5 min;ESI-MS 310 m/z (MH)+
。方法 2
向化合物30
(100g, 378 mmol)中添加DCM (540 g, 408 mL)。攪拌混合物直至所有固體皆溶解,且然後,冷卻至-5 – 0℃。然後,逐滴添加濃硫酸(163 g),同時維持反應物之初始溫度並將混合物攪拌4.5小時。然後,經2-4小時逐滴添加硝酸(62 g),同時維持反應物之初始溫度,且然後,在此溫度下再攪拌4.5小時。然後,將反應混合物緩慢添加至冷水中,將溫度維持在5℃以下。然後,將淬滅反應物加熱至25℃,並去除水層且用二氯甲烷萃取。用水洗滌經合併有機層,使用Na2
SO4
乾燥,並濃縮至124 – 155 mL。添加己烷(48 g),並將所得混合物再濃縮至124 – 155 mL。隨後,將更多己烷(160 g)添加至混合物中。然後,將混合物在23 – 27℃下攪拌15.5小時,且然後進行過濾。向濾餅中添加己烷(115 g),將所得混合物加熱至回流並攪拌2 – 2.5小時。然後,將混合物冷卻至3 – 7℃,再攪拌1 – 1.5小時,並過濾以獲得呈淺黃色固體形式之化合物31
。碳酸 5- 胺基 -2,4- 二 - 第三丁基苯基酯甲基酯 (32) 之製備程序
方法 1
在0℃下向化合物30
(6.77g, 25.6 mmol)之攪拌溶液中逐滴添加6 mL硫酸及硝酸之1:1混合物。使混合物升溫至室溫並攪拌1小時。使用液相層析(ISCO, 120 g, 0-7% EtOAc/己烷, 38 min)純化產物,從而產生化合物31
之區域異構體之呈
白色固體形式之約8:1 – 10:1混合物。1
H NMR (400 MHz, DMSO-d 6
) δ 7.63 (s, 1H), 7.56 (s, 1H), 3.87 (s, 3H), 1.36 (s, 9H), 1.32 (s, 9H)。HPLC滯留時間3.92 min,10-99% CH3
CN,運行5 min;ESI-MS 310 m/z (MH)+
。方法 2
向化合物30
(100g, 378 mmol)中添加DCM (540 g, 408 mL)。攪拌混合物直至所有固體皆溶解,且然後,冷卻至-5 – 0℃。然後,逐滴添加濃硫酸(163 g),同時維持反應物之初始溫度並將混合物攪拌4.5小時。然後,經2-4小時逐滴添加硝酸(62 g),同時維持反應物之初始溫度,且然後,在此溫度下再攪拌4.5小時。然後,將反應混合物緩慢添加至冷水中,將溫度維持在5℃以下。然後,將淬滅反應物加熱至25℃,並去除水層且用二氯甲烷萃取。用水洗滌經合併有機層,使用Na2
SO4
乾燥,並濃縮至124 – 155 mL。添加己烷(48 g),並將所得混合物再濃縮至124 – 155 mL。隨後,將更多己烷(160 g)添加至混合物中。然後,將混合物在23 – 27℃下攪拌15.5小時,且然後進行過濾。向濾餅中添加己烷(115 g),將所得混合物加熱至回流並攪拌2 – 2.5小時。然後,將混合物冷卻至3 – 7℃,再攪拌1 – 1.5小時,並過濾以獲得呈淺黃色固體形式之化合物31
。碳酸 5- 胺基 -2,4- 二 - 第三丁基苯基酯甲基酯 (32) 之製備程序 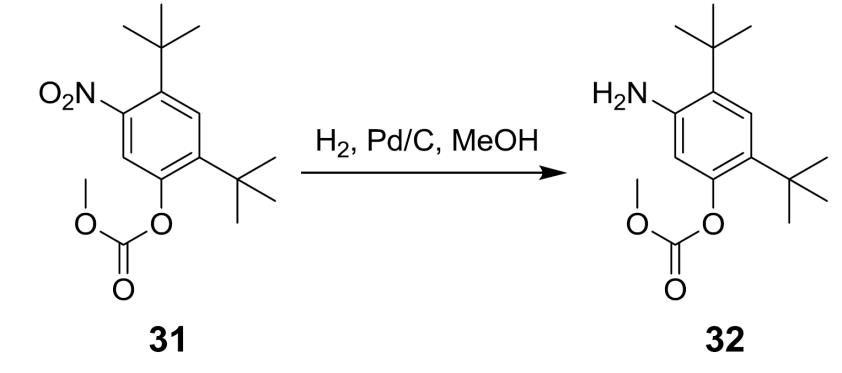 將碳酸2,4-二-第三丁基-5-硝基苯基酯甲基酯(1.00當量)及5% Pd/C (2.50 wt%乾基, Johnson-Matthey Type 37)依次裝填至適宜氫化反應器中。將MeOH (15.0體積)裝填至反應器中,並關閉系統。用N2
(g)吹掃系統,且然後,用H2
(g)加壓至2.0巴。在25℃ +/- 5℃之反應溫度下實施反應。當完成時,過濾反應物,並用MeOH (4.00體積)洗滌反應器/濾餅。將所得濾液在真空下在不超過50℃下蒸餾至8.00體積。在45℃ +/- 5℃下添加水(2.00體積)。將所得漿液冷卻至0℃ +/- 5。將漿液在0℃ +/- 5℃下保持不小於1小時,並過濾。用0℃ +/- 5℃ MeOH/H2
O (8:2) (2.00體積)將濾餅洗滌一次。在真空(-0.90巴及-0.86巴)及35℃–40℃下乾燥濾餅,以獲得化合物32
。1
H NMR (400 MHz, DMSO-d 6
) δ 7.05 (s, 1H), 6.39 (s, 1H), 4.80 (s, 2H), 3.82 (s, 3H), 1.33 (s, 9H), 1.23 (s, 9H)。 在完成反應後,用約5體積至10體積MeOH (例如,約6體積至約9體積MeOH,約7體積至約8.5體積MeOH,約7.5體積至約8體積MeOH,或約7.7體積MeOH)稀釋所得混合物,將其加熱至約35 ± 5℃之溫度,過濾,洗滌,並乾燥,如上文所闡述。N-(2,4- 二 - 第三丁基 -5- 羥基苯基 )-4- 側氧基 -1,4- 二氫喹啉 -3- 甲醯胺 ( 化合物 2) 之製備。
將碳酸2,4-二-第三丁基-5-硝基苯基酯甲基酯(1.00當量)及5% Pd/C (2.50 wt%乾基, Johnson-Matthey Type 37)依次裝填至適宜氫化反應器中。將MeOH (15.0體積)裝填至反應器中,並關閉系統。用N2
(g)吹掃系統,且然後,用H2
(g)加壓至2.0巴。在25℃ +/- 5℃之反應溫度下實施反應。當完成時,過濾反應物,並用MeOH (4.00體積)洗滌反應器/濾餅。將所得濾液在真空下在不超過50℃下蒸餾至8.00體積。在45℃ +/- 5℃下添加水(2.00體積)。將所得漿液冷卻至0℃ +/- 5。將漿液在0℃ +/- 5℃下保持不小於1小時,並過濾。用0℃ +/- 5℃ MeOH/H2
O (8:2) (2.00體積)將濾餅洗滌一次。在真空(-0.90巴及-0.86巴)及35℃–40℃下乾燥濾餅,以獲得化合物32
。1
H NMR (400 MHz, DMSO-d 6
) δ 7.05 (s, 1H), 6.39 (s, 1H), 4.80 (s, 2H), 3.82 (s, 3H), 1.33 (s, 9H), 1.23 (s, 9H)。 在完成反應後,用約5體積至10體積MeOH (例如,約6體積至約9體積MeOH,約7體積至約8.5體積MeOH,約7.5體積至約8體積MeOH,或約7.7體積MeOH)稀釋所得混合物,將其加熱至約35 ± 5℃之溫度,過濾,洗滌,並乾燥,如上文所闡述。N-(2,4- 二 - 第三丁基 -5- 羥基苯基 )-4- 側氧基 -1,4- 二氫喹啉 -3- 甲醯胺 ( 化合物 2) 之製備。 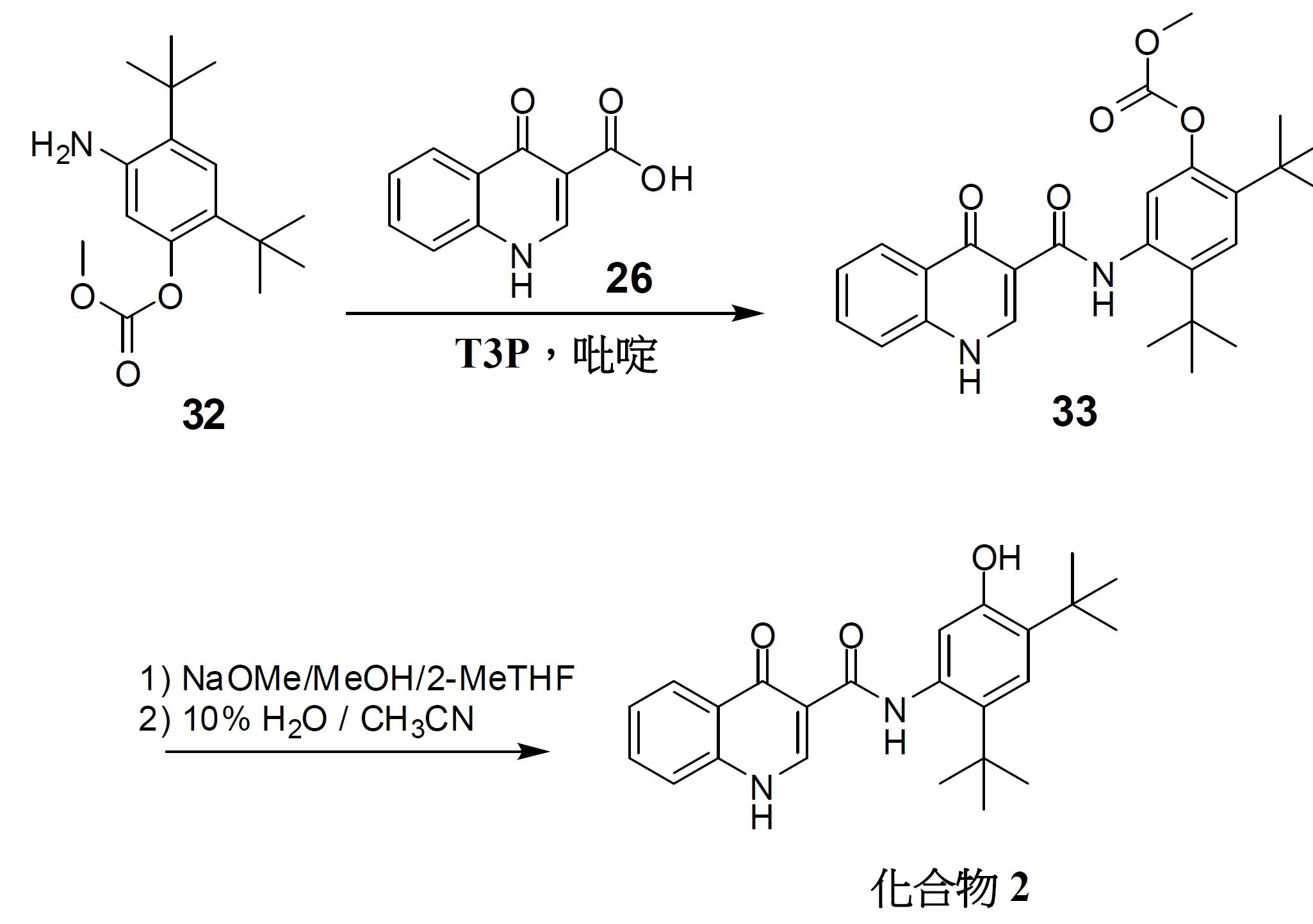 將4-側氧基-1,4-二氫喹啉-3-甲酸26
(1.0當量)及碳酸5-胺基-2,4-二-第三丁基苯基酯甲基酯32
(1.1當量)裝填至反應器中。依次添加2-MeTHF (4.0體積,相對於酸)及T3P®
於2-MeTHF中之50%溶液(1.7當量)。用2-MeTHF (0.6體積)洗滌裝填T3P之器皿。然後,添加吡啶(2.0當量),並將所得懸浮液加熱至47.5 +/- 5.0℃並在此溫度下保持8小時。採取試樣並藉由HPLC檢查完成情況。在完成後,將所得混合物冷卻至25.0℃ +/- 2.5℃。添加2-MeTHF (12.5體積)以稀釋混合物。用水(10.0體積)將反應混合物洗滌2次。添加2-MeTHF以使反應物之總體積達到40.0體積(裝填約16.5體積)。向此溶液中添加NaOMe/MeOH (1.7當量)以實施甲醇解。將反應物攪拌1.0小時以上,並藉由HPLC檢查完成情況。在完成後,用1 N HCl (10.0體積)淬滅反應物,並用0.1 N HCl (10.0體積)洗滌。將有機溶液精細過濾(polish filter)以去除任何微粒,並將其置於另一反應器中。在不大於35℃ (夾套溫度)且不小於8.0℃ (內部反應溫度)下在減壓下將過濾溶液濃縮至20體積。添加CH3
CN至40體積,並在不大於35℃ (夾套溫度)且不小於8.0℃ (內部反應溫度)下將溶液濃縮至20體積。將CH3
CN之添加及濃縮循環重複2次以上,總共3次添加CH3
CN且4次濃縮至20體積。在最終濃縮至20體積後,依次添加16.0體積CH3
CN及4.0體積H2
O,從而使得10% H2
O/CH3
CN相對於起始酸最終濃縮為40體積。將此漿液加熱至78.0℃ +/- 5.0℃ (回流)。然後,將漿液攪拌5小時以上。經5小時將漿液冷卻至0.0℃ +/- 5℃,並過濾。用0.0℃ +/- 5.0℃ CH3
CN (5體積)將濾餅洗滌4次。在真空烘箱中在50.0℃ +/- 5.0℃下乾燥所得固體(化合物2)。1
H NMR (400 MHz, DMSO-d 6
) δ 12.8 (s, 1H), 11.8 (s, 1H), 9.2 (s, 1H), 8.9 (s, 1H), 8.3 (s, 1H), 7.2 (s, 1H), 7.9 (t, 1H), 7.8 (d, 1H), 7.5 (t, 1H), 7.1 (s, 1H), 1.4 (s, 9H), 1.4 (s, 9H)。N-(2,4- 二 - 第三丁基 -5- 羥基苯基 )-4- 側氧基 -1,4- 二氫喹啉 -3- 甲醯胺 ( 化合物 2) 之替代製備。
將4-側氧基-1,4-二氫喹啉-3-甲酸26
(1.0當量)及碳酸5-胺基-2,4-二-第三丁基苯基酯甲基酯32
(1.1當量)裝填至反應器中。依次添加2-MeTHF (4.0體積,相對於酸)及T3P®
於2-MeTHF中之50%溶液(1.7當量)。用2-MeTHF (0.6體積)洗滌裝填T3P之器皿。然後,添加吡啶(2.0當量),並將所得懸浮液加熱至47.5 +/- 5.0℃並在此溫度下保持8小時。採取試樣並藉由HPLC檢查完成情況。在完成後,將所得混合物冷卻至25.0℃ +/- 2.5℃。添加2-MeTHF (12.5體積)以稀釋混合物。用水(10.0體積)將反應混合物洗滌2次。添加2-MeTHF以使反應物之總體積達到40.0體積(裝填約16.5體積)。向此溶液中添加NaOMe/MeOH (1.7當量)以實施甲醇解。將反應物攪拌1.0小時以上,並藉由HPLC檢查完成情況。在完成後,用1 N HCl (10.0體積)淬滅反應物,並用0.1 N HCl (10.0體積)洗滌。將有機溶液精細過濾(polish filter)以去除任何微粒,並將其置於另一反應器中。在不大於35℃ (夾套溫度)且不小於8.0℃ (內部反應溫度)下在減壓下將過濾溶液濃縮至20體積。添加CH3
CN至40體積,並在不大於35℃ (夾套溫度)且不小於8.0℃ (內部反應溫度)下將溶液濃縮至20體積。將CH3
CN之添加及濃縮循環重複2次以上,總共3次添加CH3
CN且4次濃縮至20體積。在最終濃縮至20體積後,依次添加16.0體積CH3
CN及4.0體積H2
O,從而使得10% H2
O/CH3
CN相對於起始酸最終濃縮為40體積。將此漿液加熱至78.0℃ +/- 5.0℃ (回流)。然後,將漿液攪拌5小時以上。經5小時將漿液冷卻至0.0℃ +/- 5℃,並過濾。用0.0℃ +/- 5.0℃ CH3
CN (5體積)將濾餅洗滌4次。在真空烘箱中在50.0℃ +/- 5.0℃下乾燥所得固體(化合物2)。1
H NMR (400 MHz, DMSO-d 6
) δ 12.8 (s, 1H), 11.8 (s, 1H), 9.2 (s, 1H), 8.9 (s, 1H), 8.3 (s, 1H), 7.2 (s, 1H), 7.9 (t, 1H), 7.8 (d, 1H), 7.5 (t, 1H), 7.1 (s, 1H), 1.4 (s, 9H), 1.4 (s, 9H)。N-(2,4- 二 - 第三丁基 -5- 羥基苯基 )-4- 側氧基 -1,4- 二氫喹啉 -3- 甲醯胺 ( 化合物 2) 之替代製備。  將4-側氧基-1,4-二氫喹啉-3-甲酸26
(1.0當量)及碳酸5-胺基-2,4-二-第三丁基苯基酯甲基酯32
(1.1當量)裝填至反應器中。依次添加2-MeTHF (4.0體積,相對於酸)及T3P®
於2-MeTHF中之50%溶液(1.7當量)。用2-MeTHF (0.6體積)洗滌裝填T3P之器皿。然後,添加吡啶(2.0當量),並將所得懸浮液加熱至47.5 +/- 5.0℃並在此溫度下保持8小時。採取試樣,並藉由HPLC檢查完成情況。在完成後,將所得混合物冷卻至20℃ +/- 5℃。添加2-MeTHF (12.5體積)以稀釋混合物。用水(10.0體積)將反應混合物洗滌2次,並將2-MeTHF (16.5體積)裝填至反應器中。向此溶液中裝填30% w/w NaOMe/MeOH (1.7當量)以實施甲醇解。將反應物在25.0℃ +/- 5.0℃下攪拌1.0小時以上,並藉由HPLC檢查完成情況。在完成後,用1.2 N HCl/H2
O (10.0體積)淬滅反應物,並用0.1 N HCl/H2
O (10.0體積)洗滌。將有機溶液精細過濾以去除任何微粒,並將其置於另一反應器中。 在不超過35℃ (夾套溫度)且不小於8.0℃ (內部反應溫度)下在減壓下將過濾溶液濃縮至20體積。添加CH3
CN至40體積,並在不超過35℃(夾套溫度)且不小於8.0℃(內部反應溫度)下將溶液濃縮至20體積。將CH3
CN之添加及濃縮循環重複2次以上,總共3次添加CH3
CN且4次濃縮至20體積。在最終濃縮至20體積後,依次裝填16.0體積CH3
CN及4.0體積H2
O,從而使得10% H2
O/CH3
CN相對於起始酸最終濃縮為40體積。將此漿液加熱至78.0℃ +/- 5.0℃ (回流)。然後,將漿液攪拌5小時以上。經5小時將漿液冷卻至20℃至25℃,並過濾。用加熱至20℃至25℃之CH3
CN (5體積)將濾餅洗滌4次。在真空烘箱中在50.0℃ +/- 5.0℃下乾燥所得固體(化合物2)。1
H NMR (400 MHz, DMSO-d 6
) δ 12.8 (s, 1H), 11.8 (s, 1H), 9.2 (s, 1H), 8.9 (s, 1H), 8.3 (s, 1H), 7.2 (s, 1H), 7.9 (t, 1H), 7.8 (d, 1H), 7.5 (t, 1H), 7.1 (s, 1H), 1.4 (s, 9H), 1.4 (s, 9H)。N-(2,4- 二 - 第三丁基 -5- 羥基苯基 )-4- 側氧基 -1,4- 二氫喹啉 -3- 甲醯胺 ( 化合物 2) 之重結晶程序
將4-側氧基-1,4-二氫喹啉-3-甲酸26
(1.0當量)及碳酸5-胺基-2,4-二-第三丁基苯基酯甲基酯32
(1.1當量)裝填至反應器中。依次添加2-MeTHF (4.0體積,相對於酸)及T3P®
於2-MeTHF中之50%溶液(1.7當量)。用2-MeTHF (0.6體積)洗滌裝填T3P之器皿。然後,添加吡啶(2.0當量),並將所得懸浮液加熱至47.5 +/- 5.0℃並在此溫度下保持8小時。採取試樣,並藉由HPLC檢查完成情況。在完成後,將所得混合物冷卻至20℃ +/- 5℃。添加2-MeTHF (12.5體積)以稀釋混合物。用水(10.0體積)將反應混合物洗滌2次,並將2-MeTHF (16.5體積)裝填至反應器中。向此溶液中裝填30% w/w NaOMe/MeOH (1.7當量)以實施甲醇解。將反應物在25.0℃ +/- 5.0℃下攪拌1.0小時以上,並藉由HPLC檢查完成情況。在完成後,用1.2 N HCl/H2
O (10.0體積)淬滅反應物,並用0.1 N HCl/H2
O (10.0體積)洗滌。將有機溶液精細過濾以去除任何微粒,並將其置於另一反應器中。 在不超過35℃ (夾套溫度)且不小於8.0℃ (內部反應溫度)下在減壓下將過濾溶液濃縮至20體積。添加CH3
CN至40體積,並在不超過35℃(夾套溫度)且不小於8.0℃(內部反應溫度)下將溶液濃縮至20體積。將CH3
CN之添加及濃縮循環重複2次以上,總共3次添加CH3
CN且4次濃縮至20體積。在最終濃縮至20體積後,依次裝填16.0體積CH3
CN及4.0體積H2
O,從而使得10% H2
O/CH3
CN相對於起始酸最終濃縮為40體積。將此漿液加熱至78.0℃ +/- 5.0℃ (回流)。然後,將漿液攪拌5小時以上。經5小時將漿液冷卻至20℃至25℃,並過濾。用加熱至20℃至25℃之CH3
CN (5體積)將濾餅洗滌4次。在真空烘箱中在50.0℃ +/- 5.0℃下乾燥所得固體(化合物2)。1
H NMR (400 MHz, DMSO-d 6
) δ 12.8 (s, 1H), 11.8 (s, 1H), 9.2 (s, 1H), 8.9 (s, 1H), 8.3 (s, 1H), 7.2 (s, 1H), 7.9 (t, 1H), 7.8 (d, 1H), 7.5 (t, 1H), 7.1 (s, 1H), 1.4 (s, 9H), 1.4 (s, 9H)。N-(2,4- 二 - 第三丁基 -5- 羥基苯基 )-4- 側氧基 -1,4- 二氫喹啉 -3- 甲醯胺 ( 化合物 2) 之重結晶程序  將化合物2 (1.0當量)裝填至反應器中。依次添加2-MeTHF (20.0體積)及0.1N HCl (5.0體積)。攪拌雙相溶液並分離,且用0.1N HCl (5.0體積)將上部有機相洗滌兩次以上。將有機溶液精細過濾以去除任何微粒,並將其置於另一反應器中。在不超過35℃ (夾套溫度)且不低於8.0℃ (內部反應溫度)下在減壓下將過濾溶液濃縮至10體積。添加乙酸異丙酯(IPAc) (10體積),並在不超過35℃ (夾套溫度)且不低於8.0℃ (內部反應溫度)下將溶液濃縮至10體積。將IPAc之添加及濃縮重複2次以上,總共3次添加IPAc且4次濃縮至10體積。在最終濃縮後,裝填10體積IPAc,並將漿液加熱至回流並維持在此溫度下達5小時。經5小時將漿液冷卻至0.0℃ +/- 5℃,並過濾。用IPAc (5體積)將濾餅洗滌一次。在真空烘箱中在50.0℃ +/- 5.0℃下乾燥所得固體。包含實質上非晶形化合物 2 之固體分散體之製備
在配備有磁力攪拌器及熱循環之反應器中將MEK及DI水根據比率90 wt% MEK / 10 wt% DI水調配之溶劑系統加熱至20 - 30℃之溫度。根據比率19.5 wt%乙酸琥珀羥丙甲纖維素/ 0.5 wt% SLS / 80 wt%化合物2向此溶劑系統中添加乙酸琥珀羥丙甲纖維素聚合物(HPMCAS) (HG級)、SLS及化合物2。所得混合物含有10.5 wt%固體。生成此混合物之成份及溶劑之實際用量列示於下表5中:表 5
:中間體F之固體噴霧分散體成份。
將化合物2 (1.0當量)裝填至反應器中。依次添加2-MeTHF (20.0體積)及0.1N HCl (5.0體積)。攪拌雙相溶液並分離,且用0.1N HCl (5.0體積)將上部有機相洗滌兩次以上。將有機溶液精細過濾以去除任何微粒,並將其置於另一反應器中。在不超過35℃ (夾套溫度)且不低於8.0℃ (內部反應溫度)下在減壓下將過濾溶液濃縮至10體積。添加乙酸異丙酯(IPAc) (10體積),並在不超過35℃ (夾套溫度)且不低於8.0℃ (內部反應溫度)下將溶液濃縮至10體積。將IPAc之添加及濃縮重複2次以上,總共3次添加IPAc且4次濃縮至10體積。在最終濃縮後,裝填10體積IPAc,並將漿液加熱至回流並維持在此溫度下達5小時。經5小時將漿液冷卻至0.0℃ +/- 5℃,並過濾。用IPAc (5體積)將濾餅洗滌一次。在真空烘箱中在50.0℃ +/- 5.0℃下乾燥所得固體。包含實質上非晶形化合物 2 之固體分散體之製備
在配備有磁力攪拌器及熱循環之反應器中將MEK及DI水根據比率90 wt% MEK / 10 wt% DI水調配之溶劑系統加熱至20 - 30℃之溫度。根據比率19.5 wt%乙酸琥珀羥丙甲纖維素/ 0.5 wt% SLS / 80 wt%化合物2向此溶劑系統中添加乙酸琥珀羥丙甲纖維素聚合物(HPMCAS) (HG級)、SLS及化合物2。所得混合物含有10.5 wt%固體。生成此混合物之成份及溶劑之實際用量列示於下表5中:表 5
:中間體F之固體噴霧分散體成份。














