DERIVES DE L'UREE, LEUR PREPARATION
ET LES MEDICAMENTS LES CONTENANT
La présente invention concerne des composés de formule
leur préparation et les médicaments les contenant.
Dans la formule (I) :
-R1 représente un radical alkyle,
-R2 représente un radical phényle ou une chaîne -(CH2)m-CO- R4 dans laquelle m est égal à 0, 1 ou 2 et R4 représente un radical hydroxy, alcoxy ou amino,
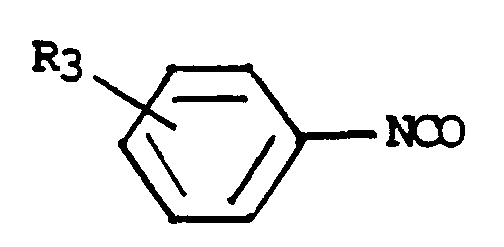
-R3 représente un atome d'hydrogène ou d'halogène ou un radical alkyle, alcoxy ou alkylthio,
-n est égal à 0 ou 1.
Dans les définitions qui précèdent et celles qui seront citées ci-après, sauf mention contraire, les radicaux alkyle et alcoxy contiennent 1 à 4 atomes de carbome en chaîne droite ou ramifiée.
Dans la formule (I), les atomes d'halogène sont de préférence des atomes de chlore, de brome ou de fluor.
Les racémiques et les énantiomères des composés de formule (I) font partie de l'invention.
Les composés de formule (I), pour lesquels R2 représente un radical phényle ou une chaîne -(CH2)m-CO-R4 dans laquelle m est égal à 0, 1 ou 2 et R4 représente un radical alcoxy, peuvent être obtenus par action d'une aminé de formule :
(II)
dans laquelle R
1 a les mêmes significations que dans la formule (I) , sur un acide de formule :
HO
2C - (CH
2)
n - - NH - CO - NH (III)
dans laquelle n et R
3 ont les mêmes significations que dans la formule (I) et R
2 a les mêmes significations que précédemment, ou un dérivé réactif de cet acide.
Lorsque l'on met en oeuvre l'acide, on opère en présence d'un agent de condensation peptidique tel qu'un carbodiimide (par exemple le dicyclohexylcarbodiimide) ou le N,N'-diimidazolecarbonyle dans un solvant inerte tel qu'un éther (par exemple THF, dioxanne), un amide (par exemple DMF) ou un solvant chloré (par exemple chlorure de méthylène, dichloro-1,2 éthane, chloroforme) à une température comprise entre 0°C et la température de reflux du mélange reacticnnel.
lorsque l'on met en oeuvre un dérivé réactif de l'acide, il est possible de faire réagir l'anhydride, un anhydride mixte, un halogénure d'acide ou un ester (qui peut être choisi parmi les esters activés ou non de l'acide).
On opère alors soit en milieu organique, éventuellement en présence d'un accepteur d'acide tel qu'une base organique azotée (par exemple une trialkylamine, une pyridine, le diaza-1,8 bicyclo[5.4.0] undécène-7 ou le diaza-1,5 bicyclo[4.3.0] nonène-5), dans un solvant tel que cité ci-dessus, ou un mélange de ces solvants, à une température comprise entre 0°C et la température de reflux du mélange reactionnel, soit en milieu hydroorganique biphasique en présence d'une base alcaline ou alcalino-terreuse
(soude, potasse) ou d'un carbonate ou bicarbonate d'un métal alcalin ou alcalino-terreux à une température comprise entre 0 et 40°C.
Les amines de formule (II) peuvent être obtenues par action d'aniline sur tin dérivé de formule :
Hal - CH2 - CO2R1 (IV) dans laquelle R1 a les mêmes significations que dans la formule (I) et Hal représente un atome d'halogène (de préférence chlore ou brome).
Cette réaction s'effectue de préférence au sein d'un solvant inerte tel que l'acétonitrile, le diméthylformamide, le tétrahydrofuranne, à la température d'ébullition du solvant.
Les dérivés de formule (IV) peuvent être obtenus par applicaticn ou adaptation des méthodes décrites dans le Beilstein 2,213 et 2,197.
Les acides de formule (III) peuvent être préparés par action d'un phénylisccyanate de formule :
(V)
dans laquelle R
3 a les mêmes significations que dans la formule (I), sur un dérivé de formule :
HO
2C - (CH
2)
n - - NH
2 (VI)
dans laquelle n a les mânes significations que dans la formule (I)
et R2 représente un radical phényle ou une chaîne -(CH2)m-CO-R4 dans laquelle m est égal à 0, 1 eu 2 et R4 représente un radical alcoxy.
Cette réaction s'effectue généralement au sein d'un solvant inerte tel que le tétrahydrofuranne, le diméthylformamide, un solvant chloré (chloroforme, dichloro-1,2 éthane par exemple), un solvant aromatique (benzène, toluène par exemple), à une température comprise entre 10°C et la température d'ébullition du solvant.
Les isocyanates de formule (V) peuvent être obtenus par application ou adaptation de la méthode décrite par R. RICHIER et coll., The Chemistry of Cyanate and their thio derivatives, S. PATAI, part 2, Wiley New York (1977).
Les dérivés de formule (VI) pour lesquels R2 représente une chaîne -(CH2)m-CO-R4 dans laquelle R4 représente un radical alcoxy peuvent être obtenus par application ou adaptation de la méthode décrite par D. COLEMAN, J. Chem. Soc., 2294 (1951).
Les composés de formule (I) pour lesquels R2 représente une chaîne -(CH2)m-CO-R4 dans laquelle R4 représente un radical hydroxy peuvent être préparés par hydrolyse du composé correspondant de formule (I) pour lequel R4 représente un radical alcoxy.
Cette hydrolyse s'effectue généralement au moyen d'une base telle que la soude ou la potasse, dans un solvant inerte tel que l'eau, un alcool ou un mélange de ces solvants, à une température voisine de 20°C suivie de l'action d'un acide pour libérer le produit sous forme acide.
Les composés de formule (I) pour lesquels R2 représente une chaîne -(CH2)m-CO-R4 dans laquelle R4 représente un radical amino peuvent être obtenus par action d'ammoniac sur le composé correspondant pour lequel R4 représente un radical alcoxy.
Cette réaction s'effectue généralement au sein d'un alcool tel que le méthanol, l'éthanol, à une température voisine de 25°C.
Les ériantiomères des composés de formule (I) peuvent être
obtenus par dédoublement des racémiques par exemple par chromatographie sur colonne chirale selon W.H. PIRCKLE et coll., asymétrie synthesis, vol. 1, Academic Press (1983) ou par synthèse à partir des précurseurs chiraux.
Les composés de formule (I) peuvent être purifiés par les méthodes connues habituelles, par exemple, par cristallisation, chromatographie, extraction...
Les composés de formule (I) pour lesquels R4 représente un radicalhydroxy, peuvent être transformés en sels métalliques ou en sels d'addition avec les bases azotées selon des méthodes connues en soi. Ces sels peuvent être obtenus par action d'une base métallique (alcaline ou alcalinoterreuse par exemple), de l'ammoniac ou d'une amine sur un composé de formule (I), dans un solvant tel qu'un alcool, un éther ou l'eau ou par réaction d'échange avec un sel d'un acide organique. Le sel formé est séparé par les méthodes habituelles.
Ces sels font également partie de l'invention.
Corme exemples de sels pharmaceutiquement acceptables, peuvent être cités les sels avec les métaux alcalins (sodium, potassium, lithium) ou avec les métaux alcalinoterreux (calcium, magnésium), le sel d'ammonium, les sels de bases azotées (éthanolamine, triméthylamine, méthylamine, benzylamine, N-benzyl-β-phénéthylamine, choline, arginine, leucine, lysine, N-méthylglucamine).
Les composés de formule (I) présentent des propriétés pharmacologiques intéressantes. Ces composés possèdent une forte affinité pour les récepteurs de la cholécystokinine (CCK) et de la gastrine et sent donc utiles dans le traitement et la prévention des désordres liés à la CCK et à la gastrine au niveau du système nerveux et de l'appareil gastrointestinal.
C'est ainsi que ces composés peuvent être utilisés pour le traitement ou la prévention des psychoses, des troubles anxieux, de la maladie de Parkinson, de la diskinésie tardive, du syndreme du colon irritable, de la pancréatite aiguë, des ulcères, des désordres de la motilité intestinale, de certaines tumeurs de
l'oesophage inférieur, du colon et de l'intestin, et comme régulateur de l'appétit.
Ces composés ont également un effet de potentialisaticn sur l'activité analgésique des médicaments narcotiques et non narcotiques.
L'affinité des composés de formule (I) pour les récepteurs de la CCK a été déterminée selon une technique inspirée de celle de A. SAITO et coll., J. Neuro. Chem., 37, 483-490 (1981) au niveau du cortex cérébral et au niveau du pancréas.
Dans ces tests, la CI50 des composés de formule (I) est généralement égale ou inférieure à 1000 nM.
Par ailleurs, il est connu que les produits qui reconnaissent les récepteurs centraux de la CCK ont une spécificité similaire pour les récepteurs de la gastrine dans le tractus gastrointestinal (BOCK et coll., J. Med. Chem., 32, 16-23 (1989), REYFΕLD et coll., Am. J. Physiol., 240, G255-266 (1981) ; BEINFELD et coll., Neuropeptides, 3, 411-427 (1983)).
Les composés de formule (I) présentent une toxicité faible. Leur DL50 est généralement supérieure à 40 mg/kg par voie sous-cutanée chez la souris.
Les composés préférés sent ceux pour lesquels R1 représente un radical tert-butyle.
D'un intérêt particulier, sont les composés suivants :
- {[(méthyl-3 phényl)-3 uréido]-3 phényl-3 N-phényl- proρicnamido}-2 acétate de tert-butyle-(RS).
- [(méthyl-3 phényl)-3 uréido]-3 N-(tert-butoxycarbonyl- méthyl) N-phàiyl-succinamate de méthyle-(RS).
- acide [(méthyl-3 phényl)-3 uréido]-3 N-(tert-butoxy- carbonylméthyl) N-phényl-succinamique-(RS).
- [(méthyl-3 phényl)-3 uréido]-2 N-(tert-butoxycarbonyl- méthyl) N-phényl-succinamide-(RS).
- [(méthyl-3 phényl)-3 uréido]-4 N-(tert-butoxycarbonyl- méthyl) N-phényl-glutaramate de méthyle-(RS).
- acide [(méthyl-3 phényl)-3 uréido]-4 N-(tert-butoxy- carbonylméthyl) N-phényl-glutaramique-(RS).
Les exemples suivants illustrent l' invention sans la limiter.
EXEMPLE 1
Une suspension de 4,1 g d'anilino acétate de tert-butyle et de 5,97 g d'acide[(méthyl-3 phényl)-3 uréido]-3 phényl-3 propionique-(R,S) dans 50 cm3 de dichloro-1,2 éthane anhydre est chauffée à reflux. On ajoute alors 1,45 cm3 de chlorure de thionyle en maintenant le reflux jusqu'à la fin du dégagement gazeux. Le mélange reacticnnel est alors versé dans 30 cm3 d'une solution aqueuse saturée d'hydrogénocarbonate de sodium puis on ajoute 50 cm3 de chlorure de méthylène. La phase organique est lavée par 50 cm3 d'eau, séchée sur sulfate de magnésium et concentrée à sec sous pression réduite (2,7 kPa) à 40°C. Après deux recristallisations successives d'abord dans un mélange d'oxyde de diéthyle et d'oxyde de diisopropyle (50-50 en volumes) puis dans l'oxyde de diéthyle, en obtient 5,4 g de {[(méthyl-3 phényl)-3 uréido]-3 phényl-3 N-phényl- propionamido}-2 acétate de tert-butyle-(RS).
L'acide [(méthyl-3 phényl)-3 uréido]-3 phényl-3 propionique-(RS) peut être préparé de la manière suivante : à une suspension de 6,6 g d'acide amino-3 phényl-3 propionique-(RS) et de 3,36 g d'hydrogénocarbonate de sodium dans 100 cm3 d'eau, en ajoute en 1 minute, 5,14 cm3 d'isocyanate de méthyl-3 phényle. Le mélange est agité pendant 20 heures à une température voisine de 20°C puis est extrait par 3 fois 75 cm3 d'acétate d'éthyle. La phase organique est lavée par 2 fois 50 cm3 d'eau, les extraits aqueux sont réunis et acidifiés jusqu'à pH 1 par une solution aqueuse 4N d'acide chlorhydrique. Le précipité est filtré, lavé à l'eau et séché sous pression réduite (0,07 kPa) à 40°C. On obtient ainsi 8,7 g d'acide [(méthyl-3 phényl)-3 uréido]-3 phényl-3 propionique-(RS) fondant à 164°C.
EXEMPLE 2
A une suspension de 7,5 g d'acide [(méthyl-3 phényl)-3 uréidol-2 méthoxycarbonyl-3 propionique-(RS) et de 5,55 g de N-phényl-glycinate de tert-butyle dans 500 cm3 de dichloro-1,2 éthane au reflux, on ajoute 3,2 g de chlorure de thionyle. Le milieu reacticnnel est encore agité pendant 15 minutes au reflux, puis refroidi à 50°C et versé dans 200 cm3 d'une solution aqueuse de bicarbonate de sodium à 10 %. La phase organique est décantée, lavée par 150 cm3 d'eau, séchée sur sulfate de magnésium puis concentrée sous pression réduite (2,7 kPa). Le résidu huileux est purifié par chromatographie sur 120 g de silice (0,063- 0,200 mm) contenus dans une colonne de 2,4 cm de diamètre [éluant : ehlorure de méthylène-méthanol (99,5-0,5 en volumes)] en recueillant des fractions de 40 cm3. Les fractions 20 à 56 sent réunies et concentrées à sec sous pression réduite (2,7 kPa) à 50°C. On obtient, après cristallisation dans l'oxyde de diéthyle, 2,6 g de [(méthyl-3 phényl)-3 uréido]-3 N-(tert-butoxycarbonyl- méthyl) N-phényl-succinamate de méthyle-(RS) fondant à 149°C.
L'acide [(méthyl-3 phényl)-3 uréido]-2 méthoxycarbonyl-3 propionique-(RS) peut être préparé de la manière suivante : à une solution de 16,1 g d'acide amino-2 méthoxycarbonyl-3 propienique- (RS) et de 8,4 g de bicarbonate de sodium dans 160 cm3 d'eau, en ajoute 13,3 g d'isocyanate de méthyl-3 phényle en 30 minutes à 17°C. Le milieu réactionnel est encore agité pendant 16 heures puis l'insoluble est filtré. Le filtrat est amené à pH 1 par de l'acide chlorhydrique 4N et extrait par trois fois 15 cm3 d'acétate d'éthyle. Les extraits sent réunis, lavés par deux fois 15 cm3 d'eau, séchés sur sulfate de magnésium puis amenés à sec sous pression réduite (2,7 kPa) à 70°C. Après cristallisation dans l'éther de pétrole, on obtient 24 g d'acide [(méthyl-3 phényl)-3 uréido]-2 méthoxycarbonyl-3 propionique-(RS).
L'acide amino-2 méthoxycarbonyl-3 propionique-(RS) peut être préparé selon la méthode décrite par D. COLEMAN, J. Chem. Soc., 2294 (1951).
EXEMPLE 3
A une solution de 3,5 g de [(méthyl-3 phényl)-3 uréido]-3 N-(tert-butoxycarbonylméthyl) N-phényl-succinamate de méthyle-(RS) dans 120 cm3 de méthanol, on ajoute 7,5 cm3 de soude normale. Le milieu reacticnnel est agité 24 heures à une température voisine de 20°C, puis on évapore le méthanol sous pression réduite (2,7 kPa) à 60°C. Le résidu est dilué par 210 cm3 de soude 0,005 N. La phase aqueuse est lavée par deux fois 70 cm3 d'acétate d'éthyle, amenée à pH acide par de l'acide chlorhydrique 4N et extraite par trois fois 80 cm3 d'acétate d'éthyle. Les extraits organiques sont réunis, séchés sur sulfate de magnésium et amenés à sec sous pression réduite 2,7 kPa) à 70°C. Après cristallisation dans l'oxyde de diéthyle, on obtient 1,6 g d'acide [(méthyl-3 phényl)-3 uréido]-3 N-(tert-butaxycarbonylméthyl) N-phényl-succinamique-(RS) fondant à 198°C.
EXEMPLE 4
Dans une solution de 4,7 g de [(méthyl-3 phényl)-3 uréido]-3 N-(tert-butoxycarbonylméthyl) N-phényl-succinamate de méthyle-(RS) dans 150 cm3 de méthanol, en fait passer un courant d'ammoniac pendant 11 heures à une température voisine de 25°C. La solution est ensuite dégazée par un courant d'azote et concentrée sous vide (2,7 kPa) à 60°C. Le résidu est cristallisé dans l'oxyde de diéthyle et le solide extrait par 40 cm3 de diehlorométhane. La solution ainsi obtenue est purifiée par chranatographie sur 25 g de silice (0,063-0,200 mm) contenus dans une colonne de 1,6 cm de diamètre [éluant : 1100 cm3 de chlorure de méthylène puis chlorure de méthylène-méthanol (98,5-1,5 en volumes)] en recueillant des fractions de 15 cm3. Les fractions 80 à 127 sont réunies et concentrées à sec sous pression réduite (2,7 kPa) à 60°C. On obtient, après cristallisaticn dans l'éther de pétrole 1,1 g de [(méthyl-3 phényl)-3 uréido]-2 N-(tert-butxoycarbonyl- méthyl) N-phényl-succinamide-(RS) fondant à 192°C.
EXEMPLE 5
On opère d'une manière analogue à celle décrite à l'exemple 2, mais à partir de 1,47 g d'acide [(méthyl-3 phényl)-3 uréido]-2 méthoxycarbonyl-4 butyrique-(RS), de 1,04 g de N-phényl-glycinate de tert-butyle et de 0,60 g de chlorure de thionyle. Le produit obtenu est purifié par chromatographie sur 50 g de silice (0,063-0,200 mm) contenus dans une colonne de 1,8 cm de diamètre [éluant : 225 cm3 de chloorure de méthylène puis chlorure de méthylène-méthanol (99-1 en volumes)] en recueillant des fractions de 15 cm3. Les fractions 36 à 40 sont réunies et concentrées à sec sous pression réduite (2,7 kPa) à 50°C. On obtient, après cristallisation dans l'oxyde de diéthyle puis recristallisation dans un mélange d'hexane et d'éthanol (85-15 en volumes), 1,9. g de [(méthyl-3 phényl)-3 uréido]-4 N-(tert-butoxycarbonylméthyl) N-phényl-glutaramate de méthyle-(RS) fendant à 139°C.
L'acide [(méthyl-3 phényl-3 uréido]-2 méthoxcycarbonyl-4 butyrique-(RS) peut être préparé d'une manière analogue à celle décrite à l'exemple 2 pour la préparation de l'acide [(méthyl-3 phényl)-3 uréido]-2 méthoxycarbonyl-3 proρionique-(RS), mais à partir de 16,1 g d'acide amino-2 méthoxycarbonyl-4 butyrique-(RS) et de 13,3 g d'isocyanate de méthyl-3 phényle. On obtient ainsi 23,5 g d'acide [(méthyl-3 phényl)-3 uréido]-2 méthoxycarbonyl-4 butyrique-(RS) fondant à 127°C.
EXEMPLE 6 On opère comme à l'exemple 3, mais à partir de 4,7 g de [(méthyl-3 phényl)-3 uréido]-4 N-(tert-butoxycarbonylméthyl) N-phényl-glutaramate de méthyle-(RS) et de 9,7 cm3 de soude normale. Le produit obtenu est purifié par diromatographie sur 60 g de silice (0,063-0,200 mm) contenus dans une colonne de 2,4 cm de diamètre [éluant : 1000 cm3 de chlorure de méthylène- méthanol
(99-1 en volumes) puis chlorure de méthylène-méthanol (97-3 en volumes)] en recueillant des fractions de 25 cm3. Les fractions 42 à 105 sent réunies et concentrées à sec sous pression réduite (2,7 kPa) à 60°C. On obtient, après cristallisation dans l'oxyde de diéthyle 1,45 g d'acide [(méthyl-3 phényl)-3 uréido]-4 N-(tert-butoxycarbonylméthyl) N-phényl-glutaramique-(RS) fondant à 191°C.
La présente invention concerne également les médicaments constitués par au moins un composé de formule (I) à l'état pur ou sous forme d'une composition dans laquelle il est associé à tout autre produit pharmaceutiquement compatible, pouvant être inerte ou physiologiquement actif. Les médicaments selon l'invention peuvent être employés par voie orale, parentérale, rectale ou topique.
Comme compositions solides pour administration orale, peuvent être utilisés des comprimés, des pilules, des poudres (capsules de gélatine, cachets) ou des granulés. Dans ces compositions, le principe actif selon l'invention est mélangé à un ou plusieurs diluants inertes, tels que amidon, cellulose, saccharose, lactose ou silice. Ces compositions peuvent également comprendre des substances autres que les diluants, par exemple un ou plusieurs lubrifiants tels que le stéarate de magnésium ou le talc, un colorant, un enrobage (dragées) ou un vernis.
Comme cempositions liquides pour administration orale, on peut utiliser des solutions, des suspensions, des émulsiens, des sirops, des élixirs pharmaceutiquement acceptables contenant des diluants inertes tels que l'eau, l'éthanol, le glycérol, les huiles végétales ou l'huile de paraffine. Ces compositicns peuvent comprendre des substances autres que les diluants, par exemple des produits mouillants, édulcorants, épaississants, aromatisants ou stabilisants.
Les compositiens stériles pour administration parentérale, peuvent être de préférence des solutions aqueuses ou des solutions non aqueuses, des suspensions ou des émulsions. Gomme solvant ou véhicule, on peut employer l'eau, le propylèneglycol, un polyéthylèneglycol, des huiles végétales, en particulier l'huile
d'olive, des esters organiques injectables, par exemple l'oléate d'éthyle ou autres solvants organiques convenables. Ces compositions peuvent également contenir des adjuvants, en particulier des agents mouillants, isotonisants, émulsifiants, dispersants et stabilisants. La stérilisation peut se faire de plusieurs façons, par exemple par filtration aseptisante, en incorporant à la composition des agents stérilisants, par irradiation ou par chauffage. Elles peuvent également être préparées sous forme de compositions solides stériles qui peuvent être dissoutes au moment de l'emploi dans un milieu stérile injectable.
Les cαipositicns pour administraticn rectale sont les suppositoires ou les capsules rectales qui contiennent, outre le produit actif, des excipients tels que le beurre de cacao, des glycérides semi-synthétiques ou des polyéthylèneglycols.
Les appositions pour administration topique peuvent être par exeπple des crèmes, des ppmmades, des lotions, des collyres, des collutoires, des gouttes nasales ou des aérosols.
En thérapeutique humaine, les composés selon l'invention sont particulièrement utiles dans le traitement et la prévention des désorctres liés à la CCK et à la gastrine au niveau du système nerveux et de l'appareil gastrointestinal. Ces composés peuvent dcnc être utilisés dans le traitement et la prévention des psychoses, des troubles anxieux, de la maladie de Parkinson, de la diskinésie tardive, du syndrome du colon irritable, de la pancréatite aiguë, des ulcères, des désordres de la motilité intestinale et de certaines tumeurs de l'oesophage inférieur, du colon et de l'intestin, comme potentialisateur de l'activité analgésique des médicaments analgésiques narcotiques et non narcotiques et comme régulateur de l'appétit.
Les doses dépendent de l'effet recherché, de la durée du traitement et de la voie d'administration utilisée ; elles sont généralement comprises entre 0,05 g et 1 g par jour par voie orale pour un adulte avec des doses unitaires allant de 10 mg à 500 mg de substance active.
D'une façon générale, le médecin déterminera la posologie appropriée en fonction de l'âge, du poids et de tous les autres facteurs propres au sujet à traiter.
Les exemples suivants illustrent des compositions selon l'invention :
EXEMPLE A
On prépare, selon la technique habituelle, des gélules dosées à 50 mg de produit actif ayant la composition suivante :
- {[(méthyl-3 phényl)-3 uréido]-3 phényl-3 N-phényl
propionamido}-2 acétate de tert-butyle-(RS)...... 50 mg
- Cellulose ............................................................... 18 mg
- Lactose .................................................................... 55 mg
- Silice colloïdale ..................................................... 1 mg
- Carboxyméthylamidon sodique .............. ........................ 10 mg - Talc .................... ....................................... ................... 10 mg
- Stéarate de magnésium ........................................ 1 mg
EXEMPTA B
On prépare, selon la technique habituelle, des comprimés dosés à 50 mg de produit actif ayant la composition suivante : - {[(méthyl-3 phényl)-3 uréido]-3 N-(tert-butoxy- carbonylméthyl) N-phényl-succinamate de méthyle-
(RS) ...................................................................... 50 mg
- Lactose ................................................................ 104 mg
- Cellulose ............................................................ 40 mg - Polyvidone .................................................... 10 mg
- Carboxymêthylamidon sodique ....................................... 22 mg
- Talc .............................................................................. 10 mg
- Stéarate de magnésium ......................................... 2 mg
- Silice colloïdale ................................................. 2 mg
- Mélange d'hydroxyméthylcellulose, glycérine,
oxyde de titane (72-3, 5-24, 5) ..................q.s.p. 1 comprimé pellicule terminé à 245 mg
EXEMPLE C
On prépare une solution injectable contenant 10 mg de produit actif ayant la composition suivante : - acide [(méthyl-3 phényl)-3 uréido]-3 N-(tert- butoxycarbonylméthyl) N-phényl-succinamique-
(RS) .......................................................................... 10 mg
- Acide benzoïque .................................................. 80 mg
- Alcool benzylique ................................................ 0, 06 cm3 - Benzoate de sodium .................................................... 80 mg
- Ethanol à 95 % ............................................................ 0,4 cm3
- Hydroxyde de sodium ..................................................... 24 mg
- Propylène glycol .................................................................... 1,6 cm3
- eau ................................................................... q.s.p. 4 cm3