WO1994002675A1 - Procede de stabilisation de l'hygrodilatation d'un produit de fibres proteiniques - Google Patents
Procede de stabilisation de l'hygrodilatation d'un produit de fibres proteiniques Download PDFInfo
- Publication number
- WO1994002675A1 WO1994002675A1 PCT/JP1993/001005 JP9301005W WO9402675A1 WO 1994002675 A1 WO1994002675 A1 WO 1994002675A1 JP 9301005 W JP9301005 W JP 9301005W WO 9402675 A1 WO9402675 A1 WO 9402675A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- protein fiber
- fiber product
- weight
- water
- derivative
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- D—TEXTILES; PAPER
- D06—TREATMENT OF TEXTILES OR THE LIKE; LAUNDERING; FLEXIBLE MATERIALS NOT OTHERWISE PROVIDED FOR
- D06M—TREATMENT, NOT PROVIDED FOR ELSEWHERE IN CLASS D06, OF FIBRES, THREADS, YARNS, FABRICS, FEATHERS OR FIBROUS GOODS MADE FROM SUCH MATERIALS
- D06M13/00—Treating fibres, threads, yarns, fabrics or fibrous goods made from such materials, with non-macromolecular organic compounds; Such treatment combined with mechanical treatment
- D06M13/10—Treating fibres, threads, yarns, fabrics or fibrous goods made from such materials, with non-macromolecular organic compounds; Such treatment combined with mechanical treatment with compounds containing oxygen
- D06M13/11—Compounds containing epoxy groups or precursors thereof
-
- D—TEXTILES; PAPER
- D06—TREATMENT OF TEXTILES OR THE LIKE; LAUNDERING; FLEXIBLE MATERIALS NOT OTHERWISE PROVIDED FOR
- D06M—TREATMENT, NOT PROVIDED FOR ELSEWHERE IN CLASS D06, OF FIBRES, THREADS, YARNS, FABRICS, FEATHERS OR FIBROUS GOODS MADE FROM SUCH MATERIALS
- D06M15/00—Treating fibres, threads, yarns, fabrics, or fibrous goods made from such materials, with macromolecular compounds; Such treatment combined with mechanical treatment
- D06M15/19—Treating fibres, threads, yarns, fabrics, or fibrous goods made from such materials, with macromolecular compounds; Such treatment combined with mechanical treatment with synthetic macromolecular compounds
- D06M15/37—Macromolecular compounds obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds
- D06M15/53—Polyethers
-
- D—TEXTILES; PAPER
- D06—TREATMENT OF TEXTILES OR THE LIKE; LAUNDERING; FLEXIBLE MATERIALS NOT OTHERWISE PROVIDED FOR
- D06M—TREATMENT, NOT PROVIDED FOR ELSEWHERE IN CLASS D06, OF FIBRES, THREADS, YARNS, FABRICS, FEATHERS OR FIBROUS GOODS MADE FROM SUCH MATERIALS
- D06M15/00—Treating fibres, threads, yarns, fabrics, or fibrous goods made from such materials, with macromolecular compounds; Such treatment combined with mechanical treatment
- D06M15/19—Treating fibres, threads, yarns, fabrics, or fibrous goods made from such materials, with macromolecular compounds; Such treatment combined with mechanical treatment with synthetic macromolecular compounds
- D06M15/37—Macromolecular compounds obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds
- D06M15/55—Epoxy resins
-
- D—TEXTILES; PAPER
- D06—TREATMENT OF TEXTILES OR THE LIKE; LAUNDERING; FLEXIBLE MATERIALS NOT OTHERWISE PROVIDED FOR
- D06M—TREATMENT, NOT PROVIDED FOR ELSEWHERE IN CLASS D06, OF FIBRES, THREADS, YARNS, FABRICS, FEATHERS OR FIBROUS GOODS MADE FROM SUCH MATERIALS
- D06M2101/00—Chemical constitution of the fibres, threads, yarns, fabrics or fibrous goods made from such materials, to be treated
- D06M2101/02—Natural fibres, other than mineral fibres
- D06M2101/10—Animal fibres
-
- D—TEXTILES; PAPER
- D06—TREATMENT OF TEXTILES OR THE LIKE; LAUNDERING; FLEXIBLE MATERIALS NOT OTHERWISE PROVIDED FOR
- D06M—TREATMENT, NOT PROVIDED FOR ELSEWHERE IN CLASS D06, OF FIBRES, THREADS, YARNS, FABRICS, FEATHERS OR FIBROUS GOODS MADE FROM SUCH MATERIALS
- D06M2101/00—Chemical constitution of the fibres, threads, yarns, fabrics or fibrous goods made from such materials, to be treated
- D06M2101/02—Natural fibres, other than mineral fibres
- D06M2101/10—Animal fibres
- D06M2101/12—Keratin fibres or silk
Definitions
- the present invention relates to a method for stabilizing a hygral expansion of a protein fiber product without impairing a soft feel.
- ethylene glycol diglycidyl ether hereinafter referred to as EGDE
- PGDE propylene glycol diglycidyl ether
- a method for stabilizing the hygral expansion of high-grade wool fabrics using an acid or a salt thereof has been proposed (Japanese Patent Application Laid-Open No. 55-33634).
- the wool fabric is immersed in a weakly acidic treatment solution consisting of the above-mentioned EGDE or PGDE and the above-mentioned catalyst, squeezed, pre-dried, and heat-treated at 150 ° C to obtain the degree of moisture absorption and divergence of water.
- EGDE or PGDE is dissolved in water with a solvent of isopropyl alcohol having a solubility parameter of 11.15 (cal / cm 3 ) 1/2 and a boiling point of 100 ° C or less. Therefore, the prepared treatment liquid does not react much with the wool fabric, and the solvent film does not exist by the heat treatment at 150 ° C.
- the polyvalent carboxylic acid or its salt used as a catalyst for reacting EGDE or PGDE with woolen fabric has a low reaction rate, and the crosslinked structure reacted under this catalyst has The resistance to hydrolysis was poor, and as a result, the stabilizing effect of Hygral Expansion was not so high.
- the milk repellent of EGDE or PGDE remains on the woolen fabric, so that the water repellency of the woolen fabric is reduced.
- An object of the present invention is to provide a method for stably stabilizing a high-grade protein fiber product without impairing a soft feel. It is another object of the present invention to provide a method for stabilizing a hygral expansion capable of improving the quality of a protein fiber product by removing by-products that are hardly soluble in water generated by heat treatment of the protein fiber product. . Disclosure of the invention
- a method for stabilizing a hygral expansion of a protein fiber product of the present invention comprises preparing a polyoxirane derivative having a water solubility of 95% by weight or more with a solubility parameter of 13.0 to: L 0.1. (cal / cm 3 ) 1/2 and boiling point is 101-190.
- This is a method comprising a step of heat-treating the product to cause a cross-linking reaction of the polyoxirane type derivative with the protein fiber product, and a step of removing by-products from the heat-treated protein fiber product.
- the above polyoxirane type derivative is ethylene or It is an ethylene glycol diglycidyl ether derivative (hereinafter referred to as PEGDE) or a propylene or polypropylene glycol diglycidyl ether derivative represented by the following formula (2) (hereinafter referred to as PPGDE).
- PEGDE ethylene glycol diglycidyl ether derivative
- PPGDE propylene or polypropylene glycol diglycidyl ether derivative represented by the following formula (2)
- the protein fiber product of the present invention may be an animal hair fiber such as wool, cashmere hair, or alpaca hair, a cocoon fiber obtained from a cocoon such as a silkworm or a wild silkworm, or a wool, a silk thread, or a fiber made of these fibers or Woven, knitted and non-woven fabrics made from yarn.
- the protein fiber product also includes a blended product, a woven product, and a knitted product with other natural fibers or chemical fibers.
- the polyoxirane-type derivative of the present invention is PEGDE represented by the above formula (1) or PPGDE represented by the above formula (2).
- PEGDE or PPGD E has an addition mole number of ethylene dalicol or propylene dalicol in the range of 1 to 4, respectively, and has a water solubility of 95% by weight or more.
- PEGDE or PPGDE is added in an amount of 2.5 to 25% by weight, preferably 5 to 15% by weight, based on the protein fiber product. 2. If it is less than 5% by weight, it does not contribute to the stabilization of the hygral expansion, and if it exceeds 25% by weight, the texture of the protein fiber product tends to become coarse and hard.
- PGPDE polyglycerol polyglycidyl ether type derivative
- GPGDE glycerol polyglycidyl ether type derivative
- glycerol glycidyl One or more derivatives having a water solubility of 95% by weight or more may be contained.
- This solvent has a solubility parameter of 13.0 to: L O.1 (calZcm 3 ) 1/2 , a boiling point in the range of 101 to 190, and is arbitrarily soluble in water.
- this solvent include N, N-dimethyl-formamide, 1,4-dioxane, dimethyl sulfoxide and the like. These solvents may be used alone or as a mixture of two or more.
- the solvent is not limited to the exemplified solvents.
- an aprotic solvent is preferable because it stabilizes the solution of the polyoxysilane derivative and is suitable for the reaction between the protein fiber product and the polyoxirane derivative in an aqueous system.
- the catalyst for an oxysilane compound of the present invention comprises at least two kinds of catalysts selected from the group consisting of (1) dicyandiamid, (2) oxycarboxylic acid, (3) thiocyanate, and (4) L-cysteines. These are used in combination. It is preferable to include the L-cysteines of the above (4) in this combination because the reaction is sufficiently promoted. As used herein, the term "L-cysteines" refers to not only L-cysteine but also L-cysteine derivatives. In addition, if the above three kinds of catalysts (1), (2) and (3) are used in combination, it is not necessary to use the L-cysteines of the above (4). In addition, when any one of the above-mentioned catalysts (1) to () is used alone, the feeling of the protein fiber product becomes undesirably coarse and hard.
- Examples of the (2) oxycarboxylate include aliphatic metal salts of aliphatic acids such as citric acid, dalconic acid, lactic acid, malic acid and tartaric acid. Among them, potassium salts, particularly tripotassium citrate, are preferred.
- As an example of the thiocyanate of (3) there may be mentioned an alkali metal salt of thiocyanic acid, of which a potassium salt is preferred.
- examples of the L-cysteines of (4) include L-cysteine, L-cysteine hydrochloride monohydrate, and N-acetyl-L-cysteine.
- L-cystine and L-cystine hydrochloride When oxidized, L-cystine and L-cystine hydrochloride monohydrate precipitate as L-cystine and do not become a stable aqueous solution. Therefore, when used, a large amount of N-acetyl-L-cystine is used when used. They need to coexist.
- the dicyandiamide is 1 to 15.7% by weight (preferably 3 to 8% by weight)
- the carboxylic acid salt is 0.8 to 1%. 2.5% by weight (preferably 0.8 to 5% by weight)
- thiocyanate 0.75 to: L 1.8% by weight (preferably 0.75 to 5% by weight)
- L-cysteines 0.5 to 12% by weight (preferably 0.5 to 1.6% by weight).
- the amount of L-cystine is 30% by weight of L-cystine,
- a composition comprising 10% by weight of hydrochloride monohydrate and 60% by weight of 1-acetyl-L-cysteine is used.
- N-acetyl-L-cystine is used alone. Is preferred.
- a composition comprising 60 to 70% by weight of N-acetyl L and one cysteine of 40 to 30% by weight is preferable.
- the treatment solution for the protein fiber product is prepared by adding the aqueous solution containing the catalyst for the oxysilane compound of (1) to the water-soluble solution of the polyoxirane-type derivative (c).
- the polyoxirane-type derivative 10 Add 10 to 62.5% by weight of the catalyst for the oxysilane compound to 0% by weight, and if the amount is less than 10% by weight, the reaction is not sufficiently promoted, and if it exceeds 62.5% by weight, the hygral expansion is added. It contributes to the stabilization of the protein fiber product, but the hand feel exceeds the practical range of protein fiber products.
- the above treatment liquid is stored in a predetermined liquid tank, and the protein fiber product is immersed in the treatment liquid and squeezed with a padding mangle or the like to dehydrate. It is preferable to repeat the immersion and dehydration twice to ensure the impregnation of the treatment liquid.
- the protein fiber product is preferably immersed in the treatment liquid at the time of washing if it is a pre-dyed product or a fabric product, or at the time of dyeing if it is a post-dyed product.
- wet and dry There are two types of heat treatment, wet and dry.
- wet heat treatment immerse the protein fiber product dehydrated in hot water at a temperature of 80 to 100 minutes for 40 to 20 minutes, or pass superheated steam through the protein fiber product and then dry it. It is performed by In the dry heat treatment, the dehydrated protein fiber product is pre-dried at a temperature of 80 to 100 minutes for 30 to 10 minutes, and then dried at a temperature of 120 to 165 for 20 to 1 minute. King.
- the temperature of the heat treatment depends on the boiling point of the solvent described in (c) above.
- the boiling point of the solvent of the present invention is higher than the boiling point of water, so that water is reduced by evaporation and contains a polyoxirane derivative and a catalyst.
- the solvent film is present in the protein fiber product.
- a 2 to 10% by weight aqueous solution of isopropyl alcohol is prepared, and the protein fiber product after heat treatment is repeatedly immersed in this aqueous solution to wash and dehydrate.
- the high-boiling solvent described in (c) above or the L-cysteines described in (d) remain unreacted, respectively. In some cases, these residues are also removed.
- the catalyst causes a cross-linking reaction of the polyoxirane type derivative to the protein fiber product prior to the inter-solution reaction. Since the polyoxysilane type derivative has a predetermined molecular length, it appropriately reacts with each fiber of the protein fiber product, and makes the protein fiber product a fiber structure having strong hydrolysis resistance.
- Denaseol EX-521 (m 3) (trade name, manufactured by Nagase Kasei Kogyo Co., Ltd.) was used.
- Denacol EX-313 (trade name, manufactured by Nagase Kasei Kogyo Co., Ltd.) was used as a GPGDE-based polyoxirane derivative.
- n or m in parentheses in the above (1) to (3) is the number of moles added in the above-mentioned formulas (1) to (3).
- An aqueous solution was prepared containing a total of 21% by weight of three types of catalysts, 1% by weight of dicyandiamide, 10% by weight of tripotassium citrate and 10% by weight of potassium thionate (hereinafter referred to as Cat-1).
- Cat-3 The average of 62.5% by weight of Cat-1 and 37.5% by weight of Cat-2 A mixed aqueous solution was prepared (hereinafter referred to as Cat-3).
- An aqueous solution was prepared by uniformly mixing 7.5% by weight of dicyandiamide, 40% by weight of the above Cat_2, 40% by weight of N, N-dimethylformamide and 12.5% by weight of water (hereinafter, referred to as Cat-4).
- the worsted yarn 2 Z 60 metric count as the warp, using worsted yarn 1Z60 metric count as weft in warp density is 48 ZCM, five harness satin tissue having a basis weight of 220 gZm 2 the weft density is woven with 38 present ZCM Fabric wool fabric was prepared. After dyeing and drying this woolen fabric, it is individually immersed in each of the four treatment solutions shown in Table 1 and squeezed with a two-roll budging mangle to uniformly disperse the treatment solution at a pickup rate of 90% by weight. Impregnated.
- the heat treatment was performed by a dry method. That is, the wool fabric was pre-dried at 100 eC for 5 minutes, and then baked at 165 eC for 1 minute. Next, the heat-treated woolen fabric was washed with a 2% by weight aqueous solution of isopropyl alcohol at 30 ° C. for 5 minutes, followed by dewatering and drying. The obtained wool fabric was used as a test cloth.
- Table 1 The processing solutions shown in Table 1 were all PEGDE-based polyoxirane derivatives conforming to Formula (1) or Formula (2), and used three or more catalysts as catalysts for oxysilane compounds. Applicable.
- Treatment solution 1 Treatment solution 2
- Treatment solution 3 Treatment solution 4
- Dyed wool fabric of the same type as in Example 1 was individually immersed in each of the six types of treatment liquids shown in Table 2, and treated in the same manner as in Example 1 to obtain a test cloth.
- the polyoxirane type derivatives were PEGDE-based, 0-based and ? It is a GDE system, and three or more types of catalysts were used as catalysts for the oxysilane compound.
- the number of moles added is about 13 for PEGDE-EX-841 in treatment solution 5
- the polyoxirane-type derivative of PGPDE-based EX-521 or GPGDE-based EX-313 is used alone in reaction volume. Therefore, not all treatment liquids fall under the present invention.
- Table 2 Table 2
- Dyed wool fabric of the same type as in Example 1 was individually immersed in each of the six types of treatment liquids shown in Table 3, and then treated in the same manner as in Example 1 to obtain a test cloth.
- the polyoxirane-type derivatives were PEGDE-based, PPGDE-based and GPGDE-based derivatives, and one type of catalyst was used as the catalyst for the oxysilane compound. The case where there is only one type of this catalyst does not fall under the present invention.
- the worsted yarn 2Z56 metric count as the warp, using worsted yarn 2Z48 metric count as weft, with the warp density of 46 ZCM, Giyabajin of 1 Z 3 having a basis weight of 250 g Zm 2 the weft density is woven with 25 present ZCM Fabric wool fabric of the tissue was prepared. After the fabric was dyed and dried, the fabric was individually immersed in each of the four treatment solutions shown in Table 4, and treated in the same manner as in Example 1 to obtain a test cloth.
- the treatment liquids shown in Table 4 are all applicable to the present invention because the polyoxirane-type derivatives are PPGDE-based and PEGDE-based, and three or more catalysts are used as catalysts for oxysilane compounds.
- the worsted yarn 2Z48 metric count as the warp yarns, using as weft a catcher yarn to 1Z32 metric count of Mo, in warp density is 38 present ZCM, five sheets having a basis weight of 250 gZm 2 the weft density is woven in 24 Zc m satin Fabric wool fabric of the tissue was prepared. After the fabric was dyed and dried, it was individually immersed in each of the four treatment solutions shown in Table 4 in the same manner as in Example 2, and then treated in the same manner as in Example 1 to obtain a test cloth.
- weft density is mass per unit area 260 GZm 2 which is woven with 36 present ZCM five harness satin tissue Fabric wool fabric was prepared. After dyeing and drying the woven fabric, it was individually immersed in each of the five treatment solutions shown in Table 5, and treated in the same manner as in Example 1 to obtain a test cloth.
- the treating solution shown in Table 5 is a composition in which the polyoxirane-type derivative is a mixture of PEGDE and GPGDE, and two or more catalysts are used as the catalyst for the oxysilane compound. .
- Table 5
- Example 1 The 28 types of test cloths obtained in Example 1, Comparative Example 1, Comparative Example 2, Example 2, Example 3 and Example 4 were subjected to the Higlare Extension test, hand measurement and appearance inspection. Was.
- the test was conducted according to the old law of the Hygral Explosion Test set by I. W. S. (International Bureau of Wool). That is, a test cloth of about 25 cm x 25 cm is marked at intervals of 20 cm, and this test cloth is folded without being folded into a 70 ° C aqueous solution containing 0.1% non-ionic surfactant. Soak for 30 minutes to fully impregnate the aqueous solution. Next, remove the test cloth, sandwich it with a dry cloth and press it to remove water, and measure the length between marks (hereinafter referred to as Lw). Next, after drying the test cloth at 80 ° C for 4 hours or more, measure the length between marks (hereinafter referred to as Ld) again.
- the value of the hygral expansion (hereinafter referred to as HG (%)) is expressed by the following equation (4).
- HG (%) ((Lw-L d) / L d) X 100 (4)
- Tables 6 and 7 show the values of the 28 types of HG (%).
- Treatment liquid 1 8. 4 4. 2 ⁇ Treatment 2 6. 6 3. 4 ⁇
- the method of this invention stabilizes the hygral expansion of a protein fiber product more reliably, without impairing a soft feeling.
Landscapes
- Engineering & Computer Science (AREA)
- Textile Engineering (AREA)
- Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Treatments For Attaching Organic Compounds To Fibrous Goods (AREA)
- Chemical Or Physical Treatment Of Fibers (AREA)
- Peptides Or Proteins (AREA)
Description
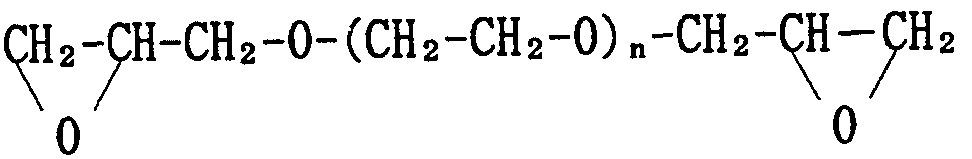






Claims


Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU45841/93A AU653295B2 (en) | 1992-07-17 | 1993-07-19 | Method of stabilizing hygral expansion of protein fiber product |
| EP93916188A EP0617158B1 (en) | 1992-07-17 | 1993-07-19 | Method of stabilizing hygral expansion of protein fiber product |
| KR1019940700868A KR940702575A (ko) | 1992-07-17 | 1993-07-19 | 단백섬유품의 흡습팽창의 안정화법(method for stabilizing the hygral expansion behavior of protein fiber products) |
| DE69302672T DE69302672T2 (de) | 1992-07-17 | 1993-07-19 | Verfahren zur stabilisierung der hygrothermischen ausdehnung eines proteinfaserprodukt |
| KR94700868A KR960008846B1 (en) | 1992-07-17 | 1994-03-17 | Method for stabilizing the hygral expansion behavior of protein products |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP4/213713 | 1992-07-17 | ||
| JP4213713A JP2598206B2 (ja) | 1992-07-17 | 1992-07-17 | 蛋白繊維品のハイグラル・エクスパンションの安定化法 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1994002675A1 true WO1994002675A1 (fr) | 1994-02-03 |
Family
ID=16643760
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP1993/001005 Ceased WO1994002675A1 (fr) | 1992-07-17 | 1993-07-19 | Procede de stabilisation de l'hygrodilatation d'un produit de fibres proteiniques |
Country Status (9)
| Country | Link |
|---|---|
| US (1) | US5494487A (ja) |
| EP (1) | EP0617158B1 (ja) |
| JP (1) | JP2598206B2 (ja) |
| KR (2) | KR940702575A (ja) |
| AU (1) | AU653295B2 (ja) |
| CA (1) | CA2118914C (ja) |
| DE (1) | DE69302672T2 (ja) |
| NZ (1) | NZ254240A (ja) |
| WO (1) | WO1994002675A1 (ja) |
Families Citing this family (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2937677B2 (ja) * | 1993-01-06 | 1999-08-23 | ツヤック 株式会社 | 蛋白繊維品の耐久的形状固定法 |
| TW406020B (en) * | 1993-09-29 | 2000-09-21 | Bristol Myers Squibb Co | Stabilized pharmaceutical composition and its method for preparation and stabilizing solvent |
| JPH1112945A (ja) * | 1997-06-16 | 1999-01-19 | Kurabo Ind Ltd | 獣毛繊維のピリング防止方法および抗ピリング性獣毛繊維 |
| EP1722880B2 (en) * | 2004-02-17 | 2018-10-03 | Donaldson Company, Inc. | Air cleaner arrangements; serviceable filter elements; and, methods |
| US7905936B2 (en) | 2004-04-30 | 2011-03-15 | Donaldson Company, Inc. | Filter arrangements; housing; assemblies; and, methods |
| WO2007044677A1 (en) | 2005-10-11 | 2007-04-19 | Donaldson Company, Inc. | Air filter arrangement; assembly; and, methods |
| DE102006001126A1 (de) | 2006-01-09 | 2007-07-12 | Kettenbach Gmbh & Co. Kg | Dentalabformmassen, daraus hergestellte gehärtete Produkte und Verwendung von Tensiden zur Herstellung von Dentalabformmassen |
| EP1986761A2 (en) * | 2006-01-20 | 2008-11-05 | Donaldson Company, Inc. | Air cleaner configured for receipt of various sized filter cartridges; components thereof; and, methods |
| WO2007149561A2 (en) * | 2006-06-22 | 2007-12-27 | Donaldson Company, Inc. | Air cleaner arrangements; components thereof; and, methods |
| US7713321B2 (en) * | 2006-06-22 | 2010-05-11 | Donaldson Company, Inc. | Air cleaner arrangements; components thereof; and, methods |
| US8728193B2 (en) | 2007-09-07 | 2014-05-20 | Donaldson Company, Inc. | Air filter assembly; components thereof and methods |
| KR101410960B1 (ko) * | 2012-12-05 | 2014-06-23 | 동일방직주식회사 | 섬유처리제, 섬유처리제로 처리된 섬유 및 그 처리방법 |
| CN112697958B (zh) * | 2020-12-24 | 2023-09-05 | 新疆畜牧科学院畜牧业质量标准研究所(新疆维吾尔自治区种羊与羊毛羊绒质量安全监督检验中心) | 一种检测牛羊肉新鲜度的方法 |
Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS469426B1 (ja) * | 1968-01-25 | 1971-03-10 | ||
| JPS5031194A (ja) * | 1973-06-21 | 1975-03-27 | ||
| JPS52987A (en) * | 1975-06-24 | 1977-01-06 | Mitsuboshi Belting Ltd | Method for adhering an aromatic polyamide fiber with chloroprene rubbe r |
| JPH01266276A (ja) * | 1988-04-19 | 1989-10-24 | Agency Of Ind Science & Technol | 絹繊維製品の改質加工方法 |
| JPH0345780A (ja) * | 1989-07-12 | 1991-02-27 | Minoru Ban | 絹繊維製品の化学的加工法 |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2829071A (en) * | 1954-04-26 | 1958-04-01 | Shell Dev | Treatment of wool containing textiles |
| US3102774A (en) * | 1961-10-05 | 1963-09-03 | Nathan H Koenig | Treatment of wool with epoxides in the presence of dimethylformamide |
| JPS5536343A (en) * | 1978-09-02 | 1980-03-13 | Kiichi Nagai | Controlling of expansion of high grade wool fabric |
| JPS6420380A (en) * | 1987-07-14 | 1989-01-24 | Minoru Ban | Chemical processing of silk knitted fabric |
| JPH0641668B2 (ja) * | 1988-10-12 | 1994-06-01 | 京都府 | 絹繊維製品の樹脂加工方法 |
-
1992
- 1992-07-17 JP JP4213713A patent/JP2598206B2/ja not_active Expired - Fee Related
-
1993
- 1993-07-19 DE DE69302672T patent/DE69302672T2/de not_active Expired - Fee Related
- 1993-07-19 AU AU45841/93A patent/AU653295B2/en not_active Ceased
- 1993-07-19 NZ NZ254240A patent/NZ254240A/en not_active IP Right Cessation
- 1993-07-19 KR KR1019940700868A patent/KR940702575A/ko not_active Expired - Lifetime
- 1993-07-19 CA CA002118914A patent/CA2118914C/en not_active Expired - Fee Related
- 1993-07-19 WO PCT/JP1993/001005 patent/WO1994002675A1/ja not_active Ceased
- 1993-07-19 EP EP93916188A patent/EP0617158B1/en not_active Expired - Lifetime
-
1994
- 1994-03-07 US US08/204,254 patent/US5494487A/en not_active Expired - Lifetime
- 1994-03-17 KR KR94700868A patent/KR960008846B1/ko not_active Expired - Lifetime
Patent Citations (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS469426B1 (ja) * | 1968-01-25 | 1971-03-10 | ||
| JPS5031194A (ja) * | 1973-06-21 | 1975-03-27 | ||
| JPS52987A (en) * | 1975-06-24 | 1977-01-06 | Mitsuboshi Belting Ltd | Method for adhering an aromatic polyamide fiber with chloroprene rubbe r |
| JPH01266276A (ja) * | 1988-04-19 | 1989-10-24 | Agency Of Ind Science & Technol | 絹繊維製品の改質加工方法 |
| JPH0345780A (ja) * | 1989-07-12 | 1991-02-27 | Minoru Ban | 絹繊維製品の化学的加工法 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP0617158A4 * |
Also Published As
| Publication number | Publication date |
|---|---|
| KR960008846B1 (en) | 1996-07-05 |
| EP0617158B1 (en) | 1996-05-15 |
| NZ254240A (en) | 1996-02-27 |
| KR940702575A (ko) | 1994-08-20 |
| AU4584193A (en) | 1994-02-14 |
| CA2118914A1 (en) | 1994-02-03 |
| DE69302672D1 (de) | 1996-06-20 |
| US5494487A (en) | 1996-02-27 |
| EP0617158A4 (en) | 1995-05-03 |
| JPH0657631A (ja) | 1994-03-01 |
| DE69302672T2 (de) | 1996-10-10 |
| AU653295B2 (en) | 1994-09-22 |
| JP2598206B2 (ja) | 1997-04-09 |
| EP0617158A1 (en) | 1994-09-28 |
| CA2118914C (en) | 1998-06-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO1994002675A1 (fr) | Procede de stabilisation de l'hygrodilatation d'un produit de fibres proteiniques | |
| JP3848621B2 (ja) | 再生コラーゲン繊維の製造方法およびセット方法 | |
| KR20100014793A (ko) | 섬유 재료의 난연 가공법 | |
| JP4559680B2 (ja) | 臭気が抑制され、セット性の改良された再生コラーゲン繊維およびその製造方法、ならびにセット方法 | |
| US20090044347A1 (en) | Process for Finishing Textiles | |
| JP3770373B2 (ja) | セルロース系繊維含有繊維構造物 | |
| Kottes Andrews et al. | How Mixed Catalysts Differ' | |
| JP4262927B2 (ja) | 撥油性、防汚性、吸水性に優れたセルロース系繊維 | |
| JP2989131B2 (ja) | 蛋白繊維製品の蛍光染色化剤及びこれを用いた染色法 | |
| JP2000096442A (ja) | セルロース系繊維布帛の加工方法 | |
| US7087093B2 (en) | Modification of fibers or fabrics | |
| DE1802187A1 (de) | Hochmolekulares Polyaethylenglykol als Mittel zur Erhoehung des Aufnahmevermoegens von cellulosehaltigen Geweben | |
| JP2852495B2 (ja) | セルロース系織物の形態安定加工法 | |
| US2562161A (en) | Stabilization of regenerated cellulose fabric with glyoxal-amide reaction product | |
| JP2937677B2 (ja) | 蛋白繊維品の耐久的形状固定法 | |
| JP2852494B2 (ja) | セルロース系織物の形態安定加工方法 | |
| SU1523604A1 (ru) | Способ антимикробной отделки целлюлозного текстильного материала | |
| US4039282A (en) | Durable-press finishing of cellulose-containing textiles with aluminum chlorhydroxide-hydrogen peroxide catalyst system | |
| US4208463A (en) | Non-durable flame-repellent finish for synthetic fabrics and synthetic-cotton blends | |
| JP5861231B2 (ja) | 絹繊維品のプリーツ加工方法及び絹繊維品のプリーツ加工品 | |
| Chance et al. | Imparting Wrinkle Resistance to Cotton with 1, 1-Carbonylbisaziridine | |
| JP2852492B2 (ja) | セルロース系織物の加工法 | |
| JP2005256193A (ja) | 吸湿発熱性獣毛繊維製品及びその製造方法 | |
| JPS6366947B2 (ja) | ||
| JP3601632B2 (ja) | 木綿繊維含有繊維製品 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AU CA KR NZ US |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): DE GB IT |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 08204254 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2118914 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 254240 Country of ref document: NZ Ref document number: 1019940700868 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1993916188 Country of ref document: EP |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWP | Wipo information: published in national office |
Ref document number: 1993916188 Country of ref document: EP |
|
| WWG | Wipo information: grant in national office |
Ref document number: 1993916188 Country of ref document: EP |