WO1994024042A1 - Katalysator zur herstellung von synthesegas - Google Patents
Katalysator zur herstellung von synthesegas Download PDFInfo
- Publication number
- WO1994024042A1 WO1994024042A1 PCT/DE1994/000513 DE9400513W WO9424042A1 WO 1994024042 A1 WO1994024042 A1 WO 1994024042A1 DE 9400513 W DE9400513 W DE 9400513W WO 9424042 A1 WO9424042 A1 WO 9424042A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- catalyst
- weight
- coating
- catalyst according
- synthesis gas
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J21/00—Catalysts comprising the elements, oxides, or hydroxides of magnesium, boron, aluminium, carbon, silicon, titanium, zirconium, or hafnium
- B01J21/06—Silicon, titanium, zirconium or hafnium; Oxides or hydroxides thereof
- B01J21/066—Zirconium or hafnium; Oxides or hydroxides thereof
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/38—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals
- B01J23/40—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals of the platinum group metals
- B01J23/42—Platinum
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/38—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals
- B01J23/54—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of noble metals combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36
- B01J23/56—Platinum group metals
- B01J23/63—Platinum group metals with rare earths or actinides
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/70—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper
- B01J23/74—Iron group metals
- B01J23/755—Nickel
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J23/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00
- B01J23/70—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper
- B01J23/76—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36
- B01J23/83—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group B01J21/00 of the iron group metals or copper combined with metals, oxides or hydroxides provided for in groups B01J23/02 - B01J23/36 with rare earths or actinides
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B3/00—Hydrogen; Gaseous mixtures containing hydrogen; Separation of hydrogen from mixtures containing it; Purification of hydrogen; Reversible storage of hydrogen
- C01B3/02—Production of hydrogen; Production of gaseous mixtures containing hydrogen
- C01B3/32—Production of hydrogen; Production of gaseous mixtures containing hydrogen by reaction of gaseous or liquid organic compounds with gasifying agents, e.g. water, carbon dioxide or air
- C01B3/34—Production of hydrogen; Production of gaseous mixtures containing hydrogen by reaction of gaseous or liquid organic compounds with gasifying agents, e.g. water, carbon dioxide or air by reaction of hydrocarbons with gasifying agents
- C01B3/38—Production of hydrogen; Production of gaseous mixtures containing hydrogen by reaction of gaseous or liquid organic compounds with gasifying agents, e.g. water, carbon dioxide or air by reaction of hydrocarbons with gasifying agents using catalysts
- C01B3/40—Production of hydrogen; Production of gaseous mixtures containing hydrogen by reaction of gaseous or liquid organic compounds with gasifying agents, e.g. water, carbon dioxide or air by reaction of hydrocarbons with gasifying agents using catalysts characterised by the catalyst
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B2203/00—Integrated processes for the production of hydrogen or synthesis gas
- C01B2203/02—Processes for making hydrogen or synthesis gas
- C01B2203/0205—Processes for making hydrogen or synthesis gas containing a reforming step
- C01B2203/0227—Processes for making hydrogen or synthesis gas containing a reforming step containing a catalytic reforming step
- C01B2203/0238—Processes for making hydrogen or synthesis gas containing a reforming step containing a catalytic reforming step the reforming step being a carbon dioxide reforming step
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B2203/00—Integrated processes for the production of hydrogen or synthesis gas
- C01B2203/10—Catalysts for performing the hydrogen forming reactions
- C01B2203/1041—Composition of the catalyst
- C01B2203/1047—Group VIII metal catalysts
- C01B2203/1052—Nickel or cobalt catalysts
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B2203/00—Integrated processes for the production of hydrogen or synthesis gas
- C01B2203/10—Catalysts for performing the hydrogen forming reactions
- C01B2203/1041—Composition of the catalyst
- C01B2203/1047—Group VIII metal catalysts
- C01B2203/1064—Platinum group metal catalysts
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B2203/00—Integrated processes for the production of hydrogen or synthesis gas
- C01B2203/10—Catalysts for performing the hydrogen forming reactions
- C01B2203/1041—Composition of the catalyst
- C01B2203/1047—Group VIII metal catalysts
- C01B2203/1064—Platinum group metal catalysts
- C01B2203/107—Platinum catalysts
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B2203/00—Integrated processes for the production of hydrogen or synthesis gas
- C01B2203/10—Catalysts for performing the hydrogen forming reactions
- C01B2203/1041—Composition of the catalyst
- C01B2203/1082—Composition of support materials
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/141—Feedstock
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/52—Improvements relating to the production of bulk chemicals using catalysts, e.g. selective catalysts
Definitions
- the invention relates to a catalyst for the production of synthesis gas in the form of CO and H 2 from CO 2 and CH 4 and / or other light
- Measured energy consumption released CO 2 Another way of reducing the release of CO 2 is to reuse this gas, for example to use it to manufacture other products.
- Synthetic gas (CO + H 2 ) is used in a number of chemical processes. Such synthesis gas can be produced in different ways. The most widely used method is methane reforming. This leads to a product with one
- the H 2 / CO ratio can be increased by an increase in the use of H 2 O compared to the amount of CH 4 used and by a subsequent one
- a synthesis gas produced in this way is particularly suitable for the synthesis of methanol, which in turn can be further converted into other petrochemical products.
- Fischer-Tropsch synthesis which requires an H 2 / CO ratio of about 1.7-2.5
- Oxo alcohols are made by
- CH 3 -CH 2 -CH 2 -CH CH 2 + CO + H 2 CH 3 -CH 2 -CH 2 -CH 2 -CH 2 -CHO
- the aldehyde is then hydrogenated to the desired alcohol.
- An H 2 / CO ratio of about 1 is required in this production process.
- Coal gasification provides one way of generating such a synthesis gas.
- Carrier material which consists of Al 2 O 3 or SiO 2 .
- the basic effectiveness of such tests can be demonstrated in laboratory tests Detect catalysts easily. However, they are unsuitable for commercial use. This is due to the fact that through the test
- DE 41 02 185 A1 describes a catalytic system for producing synthesis gas by reforming light hydrocarbons CO 2 described, in which a metal or a compound of metals of the platinum group is provided as the catalytically active coating material. Rhodium, ruthenium and iridium are mentioned as preferred metals. These metals are applied to an oxidic carrier material, which from the group AI, Mg, Zr, Si, Ce and / or La
- the exemplary embodiments are limited to the two carrier materials, MgO 2 and Al 2 O 3 .
- Coating material on the carrier material is silicified in a pretreatment.
- Coating material itself takes place a chemical conversion of the feed materials in the form of a heterogeneous solid-liquid reaction, whereby a CO atmosphere or an inert atmosphere
- Coking can be significantly reduced.
- the object of the invention is to find a catalyst which is not only sufficiently active in order to achieve a high yield of CO and H 2 , but which also avoids excessive coking, in particular over a sufficiently long time, and therefore remains active long enough if the amounts of CO 2 and CH used are relatively close to the stoichiometric values, i.e. the ratio of CO 2 / CH 4 (molecular weights) is approximately 1. It should be possible to dispense with the use of water vapor during the reaction.
- thermodynamic equilibrium values at the respective temperature At a reaction temperature of, for example, 500 ° C., the catalyst was deactivated by coking within a very short time. On the other hand, a corresponding Ni catalyst based on 2 £ ⁇ RL 2 ° 3 showed notable coking, but was considerably less effective because it only
- Ni / ZrO 2 catalyst has a CH 4 conversion rate of about 50% of the thermodynamic equilibrium values at the respective temperature and for example, showed no significant coking at 500 ° C for a long time. Even better CH 4 conversion rates, which are close to the thermodynamic equilibrium values (at 650 ° C
- the invention has shown that in particular Pt and Ni catalysts on a thermally stabilized consisting predominantly of ZrO 2
- Backing material (at least 80% by weight, preferably 90% by weight) not only provide good activity values, but also one at the same time
- the weight percentage of the coating can be max. 7% can be limited. In the case of Pt catalysts, the coating should make up a weight fraction of 0.1-5%, preferably 0.1-2.0%. For Ni catalysts, the weight fraction is preferably included
- catalyst materials also other elements of group VIII of the periodic table, e.g. Pd, Co
- Pt platinum
- Ni nickel
- a carrier material which consists largely of ZrO 2 .
- Chemically pure ZrO 2 shows an undesirably strong tendency to sinter at temperatures above 600 ° C.
- the carrier material is therefore by
- the catalyst according to the invention is prepared in such a way that the ZrO 2 is first calcined at a maximum of 670 ° C. and with the
- thermal stabilizer eg Y 2 O 3
- Y 2 O 3 thermal stabilizer
- Solvent present coating material instead of the carrier material.
- the solvent is then evaporated (e.g. by thermal drying under negative pressure). The so obtained
- the catalyst material is then calcined again at a maximum of 800 ° C.
- the catalyst according to the invention takes place at temperatures of 400-900 ° C., preferably at 700-900 ° C.
- the pressure during the reaction can be 1-30 bar and is preferably 10-20 bar.
- the amounts of CO 2 and CH 4 should be coordinated so that the
- the molar weight ratio of CO 2 / CH 4 is between 0.5 and 4, the
- Range 0.5 to 1.5 and in particular the value 1 are particularly preferred.
- Figures 1 to 6 each show a graphic representation
- Catalyst from Example 1 were determined for a feed with a CH 4 / CO 2 ratio of 1: 1.09, which is practically the
- Another catalyst was made using the dry impregnation method. For this purpose, 4 g of monoclinic ZrO 2 at 650 ° C 15 5td. Long calcined in air, thermally stabilized, pressed into pellets and then into grains with a grain size of 0.3
- This material had a BET surface area of
- Ni / ZrO 2 catalyst a granular 5 6% by weight Ni / ZrO 2 catalyst was obtained, of which 300 mg (0.8 cm 3 ) were investigated at a feed gas flow of 170 cm 3 / min in the temperature range of approximately 400-620 ° C.
- Feed gas had a CH 4 / CO 2 ratio of 1: 3.9 and flowed through the
- This catalyst is 550 ° C., as a comparison with Table 1 shows in its effectiveness even slightly superior to that of the first example. At higher temperatures, the situation is reversed.
- Lifetime test showed the result shown in Figure 2 under the conditions mentioned above at a test temperature of 501 ° C.
- the catalyst according to Example 6 was also examined under the condition that the CH 4 / CO 2 ratio in the feed gas was 1: 1.09.
- the catalyst showed, as can be seen from Table 7, in
- Ni catalyst on an AlO 3 carrier material that is deactivated much faster.
- Comparative Example 1 Similar to Example 1, a conventional Pt catalyst was produced. Instead of ZrO 2 5 g -Al 2 O 3 were used and in the same way to a granular carrier material with 0.3-0.6 mm
- Example 1 It is crucial, however, that this conventional catalyst was inactivated by coking within a very short time. This can be seen from the illustrations in FIGS. 3, 4 and 5, which show the conversion rates for CO 2 and CH 4 or the CO yield as a function of the temperature for a first and only a second temperature cycle. It follows that this
- a carrier material made of 5 g ⁇ -Al 2 O 3 with a grain size of 0.3-0.6 mm was produced in the same way as in Comparative Example 1. This material was then treated with 10 cm of an aqueous Ni (NO 3 ) ⁇ 6H 2 O solution at 60 ° C. in a rotary evaporator to obtain a 10% by weight Ni / y-Al 2 O 3 catalyst.
- the catalyst was dried at 110 ° C. for 4 hours as in Example 1 and then calcined at 650 ° C. for 15 hours.
- the results for the CO 2 , the CH 4 conversion rate and the CO yield for the first temperature cycle are shown in FIG. 6. At temperatures of about 500 ° C, a drastic deactivation was already evident in this first cycle, so that such a Ni catalyst is not useful for practical operation.
Landscapes
- Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Health & Medical Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Combustion & Propulsion (AREA)
- Inorganic Chemistry (AREA)
- Catalysts (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Hydrogen, Water And Hydrids (AREA)
Abstract
Description



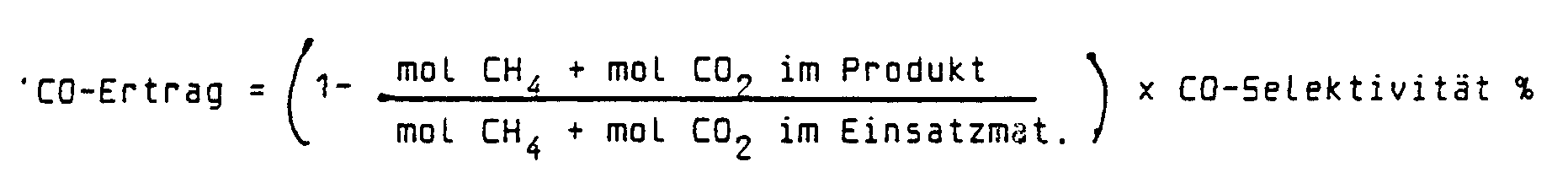








Claims
Priority Applications (10)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PL94311201A PL175047B1 (pl) | 1993-04-22 | 1994-04-20 | Sposób wytwarzania katalizatora do procesu wytwarzania gazu syntezowego |
| EP94914323A EP0695279B1 (de) | 1993-04-22 | 1994-04-20 | Verwendung eines katalysators zur herstellung von synthesegas |
| DE59403037T DE59403037D1 (de) | 1993-04-22 | 1994-04-20 | Verwendung eines katalysators zur herstellung von synthesegas |
| AU66759/94A AU6675994A (en) | 1993-04-22 | 1994-04-20 | Catalyst for producing synthesis gas |
| JP6522634A JPH09500054A (ja) | 1993-04-22 | 1994-04-20 | 合成ガスを生成するための触媒 |
| CA002161132A CA2161132C (en) | 1993-04-22 | 1994-04-20 | Catalyst for the production of synthesis gas |
| BR9406357A BR9406357A (pt) | 1993-04-22 | 1994-04-20 | Catalisador para a fabricaçao de gás de sintese |
| US08/537,791 US5989457A (en) | 1993-04-22 | 1994-04-20 | Process for the production of synthesis gas |
| NO953943A NO953943L (no) | 1993-04-22 | 1995-10-04 | Katalysator for fremstilling av syntesegass |
| GR970401922T GR3024273T3 (en) | 1993-04-22 | 1997-07-30 | Catalyst for producing synthesis gas |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE4313673 | 1993-04-22 | ||
| DEP4313673.7 | 1993-04-22 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1994024042A1 true WO1994024042A1 (de) | 1994-10-27 |
Family
ID=6486435
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/DE1994/000513 Ceased WO1994024042A1 (de) | 1993-04-22 | 1994-04-20 | Katalysator zur herstellung von synthesegas |
Country Status (16)
| Country | Link |
|---|---|
| US (1) | US5989457A (de) |
| EP (1) | EP0695279B1 (de) |
| JP (1) | JPH09500054A (de) |
| CN (1) | CN1042100C (de) |
| AT (1) | ATE153987T1 (de) |
| AU (1) | AU6675994A (de) |
| CA (1) | CA2161132C (de) |
| CZ (1) | CZ286018B6 (de) |
| DE (1) | DE59403037D1 (de) |
| DK (1) | DK0695279T3 (de) |
| ES (1) | ES2105701T3 (de) |
| GR (1) | GR3024273T3 (de) |
| HU (1) | HUT72430A (de) |
| NO (1) | NO953943L (de) |
| PL (1) | PL175047B1 (de) |
| WO (1) | WO1994024042A1 (de) |
Cited By (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1997040931A1 (de) * | 1996-04-26 | 1997-11-06 | Basf Aktiengesellschaft | Katalysator zur selektiven aromatisierung |
| RU2141383C1 (ru) * | 1998-07-20 | 1999-11-20 | Институт катализа им.Г.К.Борескова СО РАН | Способ приготовления катализатора конверсии углеводородов |
| EP0979799A4 (de) * | 1997-04-11 | 2000-05-24 | Chiyoda Chem Eng Construct Co | Verfahren zur herstellung von synthesegas mittels outothermer reformierung |
| EP0974550A4 (de) * | 1997-04-11 | 2000-05-24 | Chiyoda Chem Eng Construct Co | Katalysator für die herstellung von synthesegas und verfahren zur herstellung von kohlenmonoxid |
| EP0974551A4 (de) * | 1997-04-11 | 2000-05-24 | Chiyoda Chem Eng Construct Co | Verfahren zur herstellung von synthesegas |
| EP1124635A4 (de) * | 1998-09-21 | 2002-01-30 | Univ Queensland | Katalysator zur umwandlung von kohlenwasserstoffen |
| US6387843B1 (en) | 2001-04-05 | 2002-05-14 | Chiyoda Corporation | Method of preparing Rh- and/or Ru-catalyst supported on MgO carrier and reforming process using the catalyst |
| US6656978B2 (en) | 2001-04-05 | 2003-12-02 | Chiyoda Corporation | Process of producing liquid hydrocarbon oil or dimethyl ether from lower hydrocarbon gas containing carbon dioxide |
Families Citing this family (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE60044334D1 (de) | 1999-03-18 | 2010-06-17 | Panasonic Elec Works Co Ltd | Verwendung eines katalysators für die wasser-gas-umwandlungsreaktion, verfahren zur entfernung von krgieerzeugung aus einer brennstoffzelle |
| US6524550B1 (en) | 1999-05-03 | 2003-02-25 | Prashant S. Chintawar | Process for converting carbon monoxide and water in a reformate stream |
| JP4564645B2 (ja) * | 2000-11-27 | 2010-10-20 | 株式会社キャタラー | 排ガス浄化用触媒 |
| US20080028680A1 (en) | 2003-04-15 | 2008-02-07 | Wouter Detlof Berggren | Process to Prepare Synthesis Gas |
| ES2259535B1 (es) * | 2005-01-14 | 2007-11-01 | Greencell, S.A. | Catalizador para un proceso catalitico para la obtencion de hidrogeno a partir de bioetanol y/o etanol, procedimiento de preparacion del catalizador, y su uso en el proceso catalitico. |
| US20080260631A1 (en) | 2007-04-18 | 2008-10-23 | H2Gen Innovations, Inc. | Hydrogen production process |
| JP5910189B2 (ja) * | 2012-03-09 | 2016-04-27 | マツダ株式会社 | アルコールの合成方法 |
| CN106085526B (zh) * | 2016-06-22 | 2019-02-26 | 刘世超 | 一种合成氢燃料 |
| TWI611839B (zh) * | 2016-09-07 | 2018-01-21 | 國立清華大學 | 應用於低碳烴之低溫部分氧化產氫之觸媒 |
| CN113648988A (zh) * | 2021-09-08 | 2021-11-16 | 四川大学 | 一种高比表面积氧化锆基催化剂载体及其制备方法 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0033505A1 (de) * | 1980-02-01 | 1981-08-12 | The M. W. Kellogg Company | Verfahren und Katalysator zum Dampfreformieren |
| EP0333037A2 (de) * | 1988-03-12 | 1989-09-20 | Igarashi, Akira c/o Kohgakuin University | Verfahren zur Dampfreformierung von Kohlenwasserstoffen |
| EP0414573A1 (de) * | 1989-08-25 | 1991-02-27 | Tonen Corporation | Katalysator für die Dampfreformierung von Kohlenwasserstoffen und Verfahren für seine Herstellung |
Family Cites Families (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3830752A (en) * | 1968-09-20 | 1974-08-20 | Union Oil Co | Hydrocarbon conversion catalysts |
| US4065544A (en) * | 1970-05-11 | 1977-12-27 | Union Carbide Corporation | Finely divided metal oxides and sintered objects therefrom |
| US4255288A (en) * | 1979-08-13 | 1981-03-10 | Exxon Research & Engineering Co. | Zirconia-containing catalysts |
| US4539310A (en) * | 1980-02-01 | 1985-09-03 | The M. W. Kellogg Company | Steam reforming catalyst |
| US4729981A (en) * | 1981-10-13 | 1988-03-08 | Shell Internationale Research Maatschappij B.V. | ROR-activated catalyst for synthesis gas conversion |
| DE3340569A1 (de) * | 1983-11-09 | 1985-05-23 | Sued Chemie Ag | Katalysator zur herstellung von synthesegas bzw. von wasserstoff und verfahren zu dessen herstellung |
| CA1301964C (en) * | 1986-08-22 | 1992-05-26 | Kiichiro Mitsui | Method for treatment of waste water |
| US5015617A (en) * | 1988-04-14 | 1991-05-14 | Nippon Shokubai Kagaku Kogyo Co., Ltd. | Catalyst for purifying exhaust gas and method for production thereof |
| US5061464A (en) * | 1988-11-14 | 1991-10-29 | Johnson Matthey, Inc. | Oxidation process and catalyst for use therefor |
| US5232890A (en) * | 1990-01-02 | 1993-08-03 | Ganguli Partha S | Precious metal catalysts with oxygen-ion conducting support |
| GB9028034D0 (en) * | 1990-12-24 | 1991-02-13 | Isis Innovation | Improved processes for the conversion of methane to synthesis gas |
| GB9108656D0 (en) * | 1991-04-23 | 1991-06-12 | Shell Int Research | Process for the preparation of a catalyst or catalyst precursor |
| CA2103876A1 (en) * | 1992-08-27 | 1994-02-28 | Stuart Leon Soled | Group viii metal containing tungsten oxide silica modified zirconia as acid catalyst |
| FR2701471B1 (fr) * | 1993-02-10 | 1995-05-24 | Rhone Poulenc Chimie | Procédé de synthèse de compositions à base d'oxydes mixtes de zirconium et de cérium, compositions ainsi obtenues et utilisations de ces dernières. |
| US5395813A (en) * | 1993-05-11 | 1995-03-07 | Exxon Research And Engineering Company | Particulate solids for catalyst supports and heat transfer materials |
| CO4370053A1 (es) * | 1993-11-29 | 1996-10-07 | Shell Int Research | Proceso para la oxidacion parcial catalitica de hidrocarbu- ros |
| US5741440A (en) * | 1994-02-28 | 1998-04-21 | Eastman Chemical Company | Production of hydrogen and carbon monoxide |
-
1994
- 1994-04-20 WO PCT/DE1994/000513 patent/WO1994024042A1/de not_active Ceased
- 1994-04-20 CZ CZ952761A patent/CZ286018B6/cs not_active IP Right Cessation
- 1994-04-20 AU AU66759/94A patent/AU6675994A/en not_active Abandoned
- 1994-04-20 CA CA002161132A patent/CA2161132C/en not_active Expired - Lifetime
- 1994-04-20 AT AT94914323T patent/ATE153987T1/de not_active IP Right Cessation
- 1994-04-20 DK DK94914323.4T patent/DK0695279T3/da active
- 1994-04-20 PL PL94311201A patent/PL175047B1/pl unknown
- 1994-04-20 ES ES94914323T patent/ES2105701T3/es not_active Expired - Lifetime
- 1994-04-20 CN CN94191860A patent/CN1042100C/zh not_active Expired - Lifetime
- 1994-04-20 EP EP94914323A patent/EP0695279B1/de not_active Expired - Lifetime
- 1994-04-20 JP JP6522634A patent/JPH09500054A/ja active Pending
- 1994-04-20 DE DE59403037T patent/DE59403037D1/de not_active Expired - Lifetime
- 1994-04-20 HU HU9503026A patent/HUT72430A/hu unknown
- 1994-04-20 US US08/537,791 patent/US5989457A/en not_active Expired - Lifetime
-
1995
- 1995-10-04 NO NO953943A patent/NO953943L/no unknown
-
1997
- 1997-07-30 GR GR970401922T patent/GR3024273T3/el unknown
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0033505A1 (de) * | 1980-02-01 | 1981-08-12 | The M. W. Kellogg Company | Verfahren und Katalysator zum Dampfreformieren |
| EP0333037A2 (de) * | 1988-03-12 | 1989-09-20 | Igarashi, Akira c/o Kohgakuin University | Verfahren zur Dampfreformierung von Kohlenwasserstoffen |
| EP0495534A2 (de) * | 1988-03-12 | 1992-07-22 | Igarashi, Akira c/o Kohgakuin University | Katalysator für die Dampfreformierung von Kohlenwasserstoffen |
| EP0414573A1 (de) * | 1989-08-25 | 1991-02-27 | Tonen Corporation | Katalysator für die Dampfreformierung von Kohlenwasserstoffen und Verfahren für seine Herstellung |
Cited By (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1997040931A1 (de) * | 1996-04-26 | 1997-11-06 | Basf Aktiengesellschaft | Katalysator zur selektiven aromatisierung |
| EP0979799A4 (de) * | 1997-04-11 | 2000-05-24 | Chiyoda Chem Eng Construct Co | Verfahren zur herstellung von synthesegas mittels outothermer reformierung |
| EP0974550A4 (de) * | 1997-04-11 | 2000-05-24 | Chiyoda Chem Eng Construct Co | Katalysator für die herstellung von synthesegas und verfahren zur herstellung von kohlenmonoxid |
| EP0974551A4 (de) * | 1997-04-11 | 2000-05-24 | Chiyoda Chem Eng Construct Co | Verfahren zur herstellung von synthesegas |
| US6312660B1 (en) | 1997-04-11 | 2001-11-06 | Chiyoda Corporation | Process for preparing synthesis gas |
| US6340437B1 (en) | 1997-04-11 | 2002-01-22 | Chiyoda Corporation | Process for preparing synthesis gas by autothermal reforming |
| US6376423B2 (en) | 1997-04-11 | 2002-04-23 | Chiyoda Corporation | Catalyst for preparation of synthesis gas and process for preparing carbon monoxide |
| RU2141383C1 (ru) * | 1998-07-20 | 1999-11-20 | Институт катализа им.Г.К.Борескова СО РАН | Способ приготовления катализатора конверсии углеводородов |
| EP1124635A4 (de) * | 1998-09-21 | 2002-01-30 | Univ Queensland | Katalysator zur umwandlung von kohlenwasserstoffen |
| US6387843B1 (en) | 2001-04-05 | 2002-05-14 | Chiyoda Corporation | Method of preparing Rh- and/or Ru-catalyst supported on MgO carrier and reforming process using the catalyst |
| US6656978B2 (en) | 2001-04-05 | 2003-12-02 | Chiyoda Corporation | Process of producing liquid hydrocarbon oil or dimethyl ether from lower hydrocarbon gas containing carbon dioxide |
| US6806296B2 (en) | 2001-04-05 | 2004-10-19 | Chiyoda Corporation | Process of producing liquid hydrocarbon oil or dimethyl ether from lower hydrocarbon gas containing carbon dioxide |
Also Published As
| Publication number | Publication date |
|---|---|
| EP0695279B1 (de) | 1997-06-04 |
| NO953943D0 (no) | 1995-10-04 |
| CZ286018B6 (cs) | 1999-12-15 |
| ATE153987T1 (de) | 1997-06-15 |
| PL175047B1 (pl) | 1998-10-30 |
| GR3024273T3 (en) | 1997-10-31 |
| DK0695279T3 (da) | 1997-10-27 |
| DE59403037D1 (de) | 1997-07-10 |
| HUT72430A (en) | 1996-04-29 |
| CA2161132C (en) | 2003-10-07 |
| US5989457A (en) | 1999-11-23 |
| NO953943L (no) | 1995-10-04 |
| JPH09500054A (ja) | 1997-01-07 |
| CN1042100C (zh) | 1999-02-17 |
| CA2161132A1 (en) | 1994-10-27 |
| EP0695279A1 (de) | 1996-02-07 |
| ES2105701T3 (es) | 1997-10-16 |
| AU6675994A (en) | 1994-11-08 |
| CN1121701A (zh) | 1996-05-01 |
| PL311201A1 (en) | 1996-02-05 |
| CZ276195A3 (en) | 1996-03-13 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP0695279B1 (de) | Verwendung eines katalysators zur herstellung von synthesegas | |
| DE3882886T2 (de) | Verfahren und Katalysator für die Umwandlung von Synthesegas zu Kohlenwasserstoffen. | |
| DE69905467T2 (de) | Katalytische teiloxidation mit einem rhodium-iridium legierungskatalysator | |
| DE69927032T2 (de) | Verfahren für autotherme reformierung einer kohlenwasserstoffeinspeisung | |
| DE69808983T2 (de) | Methanolsynthese- und Reformierungskatalysator bestehend aus Kupfer, Zink und Aluminium | |
| DE3103171C2 (de) | Fester Katalysator zur Synthese von Methan und Verfahren zu seiner Herstellung | |
| DE69006772T2 (de) | Verfahren zur Dampfreformierung von Kohlenwasserstoffen. | |
| EP2490804B1 (de) | Katalysator für die wasserdampfreformierung von methanol | |
| DE69006802T2 (de) | Katalysator für die Dampfreformierung von Kohlenwasserstoffen und Verfahren für seine Herstellung. | |
| DE3872777T2 (de) | Dehydrierungskatalysator. | |
| DE3540152A1 (de) | Katalysator und verfahren zur herstellung von alkanolen | |
| DE2417862A1 (de) | Verfahren zur herstellung eines wasserstoffreichen gases | |
| DE1263716B (de) | Verfahren zur Umsetzung von Kohlenwasserstoffen mit Wasserdampf | |
| DE2546513A1 (de) | Hydrierungskatalysator und dessen verwendung zum hydrieren von kohlenwasserstoff | |
| DE3586630T2 (de) | Ruthenium-rhenium-titaniumoxyd-katalysatoren und ihre verwendung zur fischer-tropsch-synthese. | |
| DD216447A5 (de) | Verfahren zur herstellung von methanol | |
| DE69312607T2 (de) | Hydroumwandlungskatalysator | |
| DE69103573T2 (de) | Verfahren zur Umwandlung von Methanol zu flüssigen Kohlenwasserstoffen. | |
| DE69318056T2 (de) | Katalysatorzusammensetzung mit einem modifizierten Siliciumdioxid-Aluminiumdioxid-Träger | |
| DE2628958C3 (de) | Verfahren zur Herstellung von Äthan durch selektive, katalytische Hydrogenolyse von Alkanen | |
| WO2010076126A1 (de) | Heterogener katalysator für die fischer-tropsch-synthese und ein verfahren zu dessen herstellung | |
| DE2650425A1 (de) | Verfahren zur umsetzung von kohlenmonoxid | |
| DE2818308A1 (de) | Verfahren zur herstellung von kohlenwasserstoffen | |
| DE2153475A1 (de) | Verfahren zum katalytischen reformieren von benzinen | |
| DE3853619T2 (de) | Chemische Reaktion in Anwesenheit eines Kupfer-Trägerkatalysators. |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AU BB BG BR BY CA CN CZ FI GE HU JP KG KP KR KZ LK LV MD MG MN MW NO NZ PL RO RU SD SI SK TJ UA US UZ VN |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): AT BE CH DE DK ES FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN ML MR NE SN TD TG |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 1994914323 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2161132 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PV1995-2761 Country of ref document: CZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 08537791 Country of ref document: US |
|
| WWP | Wipo information: published in national office |
Ref document number: 1994914323 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: PV1995-2761 Country of ref document: CZ |
|
| WWG | Wipo information: grant in national office |
Ref document number: 1994914323 Country of ref document: EP |
|
| WWG | Wipo information: grant in national office |
Ref document number: PV1995-2761 Country of ref document: CZ |