Nucleosid-Derivate mit photolabilen Schutzgruppen
Beschreibung
Gegenstand der vorliegenden Erfindung sind Nucleosid-Derivate mit photolabilen Schutzgruppen und Verfahren zu deren Herstellung.
Photolabile Schutzgruppen für die Hydroxy- und Phosphatfunktionen in Nucleosiden bzw. Nucleotiden sind von besonderem Interesse, da sie bspw. für lichtgesteuerte Parallel-Synthesen von Oligonucleotiden auf einem festen Träger geeignet sind (vgl. S.P.A. Fodor et al. Science 1991, 251, S.767ff). Mit ihrer Hilfe können sogenannte DNA-Chips (d.h. Trägerplättchen, auf deren Oberfläche viele verschiedene Oligonucleotide angeordnet sind) hergestellt werden, die wiederum in der Molekularbiologie für eine schnelle DNA-Sequenz-Analyse benötigt werden.
Entsprechend dem Stand der Technik wurden bisher als photolabile Schutzgruppen in der Nucleosid- bzw. Nucleotidchemie vor allem die o-Nitrobenzyl-Gruppe und ihre Derivate eingesetzt (vgl. V.N.R. Pillai, Org. Photochem.
1987, 9 , S.225ff bzw. J.W. Walker et al. , J. Am. Chem. Soc.
1988, 110, S.7l70ff) . Als besonders nachteilig bei diesen Schutzgruppen hat sich die langsame und zum Teil nur unvollständige Entschützung der entsprechenden Nucleosid- bzw. Nucleotid-Derivate erwiesen. Außerdem entstehen bei der Abspaltung der o-Nitrobenzyl-Verbindungen z.T. unerwünschte Nebenprodukte in Form von toxischen
Nitrosophenylverbindungen.
Als weitere photolabile Schutzgruppe für Nucleoside wurde entsprechend dem Artikel von W. Pfleiderer et al. in "Biophosphates and Their Analogues - Synthesis, Structure, Metabolism and Activity", Elsevier Science Publishers B.V. (Amsterdam) 1987, S.133ff die 2- (o-Nitrophenyl)ethylgruppe empfohlen, die jedoch ausschließlich als Schutzgruppe im Basenteil, insbesondere in 06-Stellung des Guanosins, eingeführt wird. In derselben Publikation werden auch die p-Nitrophenylethoxycarbonyl (NPEOC) - und die 2,4- Dinitrophenylethoxycarbonyl(DNPEOC) -Gruppen sowohl als Schutzgruppen für die Aminofunk ion als auch für die Hydroxylfunktionen im Zuckerteil beschrieben, doch erfolgt die Abspaltung dieser Gruppen ausschließlich durch basenkatalysierte ß-Eliminierung.
Der vorliegenden Erfindung lag daher die Aufgabe zugrunde, Nucleosid-Derivate mit photolabilen Schutzgruppen für die 5'- OH-Funktion im Zuckerteil zu entwickeln, welche die genannten Nachteile entsprechend dem Stand der Technik nicht aufweisen, sondern sich vergleichsweise schnell, quantitativ und ohne Bildung unerwünschter Nebenprodukte entschützen lassen.
Diese Aufgabe wurde erfindungsgemäß durch Nucleosid-Derivate der allgemeinen Formel (I) entsprechend Anspruch 1 gelöst. Es hat sich nämlich überraschenderweise gezeigt, daß sich die erfindungsgemäßen Schutzgruppen wesentlich schneller und vollständiger abspalten lassen als z.B. die o-Nitrobenzylgruppen. Außerdem konnten bei der Entschützung bisher keine unerwünschten Nebenprodukte festgestellt werden, was ebenfalls nicht vorhersehbar war.
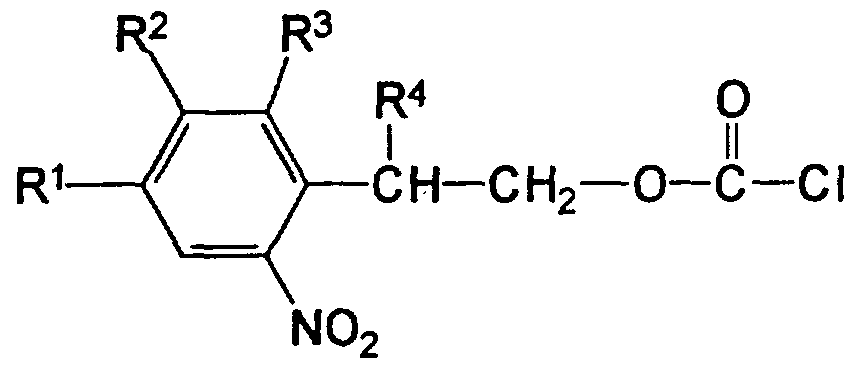
Die erfindungsgemäßen Nucleosid-Derivate weisen folgende allgemeine Formel (I) auf:
wobei die Reste R1, R2 und R3 am Phenylring folgende Bedeutung haben können:
R1 = H, N02, CN, OCH3, Halogen oder Alkyl oder
Alkoxyalkyl mit 1 bis 4 C-Atomen R2 = H, OCH3 R3 = H, F, Cl, Br, N02
Gemäß einer bevorzugten Ausführungsform bedeutet R3 = H, falls R2 = OCH3 ist.
Der Rest R4, der am C2~Atom der o-Nitrophenylethyl- Gruppierung sitzt, kann entweder H, Halogen, OCH3 oder ein Alkylrest mit 1 bis 4 C-Atomen sein. Der Alkylrest kann hierbei linear oder verzweigt, substituiert (insbesondere mit einem oder mehreren Halogenatomen) oder unsubstituiert sowie gesättigt oder ungesättigt sein; das gleiche gilt auch für die Alkyl- und Alkoxyalkylreste bei R . Vorzugsweise stellt R4 einen Methylrest dar. Im Falle von R4 ≠ H sind die Substituenten R1, R2 und R3 am Phenylring vorzugsweise Wasserstoffreste. Außerdem stellt im Falle von R2 = OCH3 R3 insbesondere einen Wasserstoffrest dar.
Halogen bedeutet in dieser Anmeldung durchwegs F, Cl, Br, I und vorzugsweise F, Cl oder Br.
Der Nucleosidteil der erfindungsgemäßen Verbindungen besteht aus den üblichen D-Ribofuranose- bzw. 2' -Desoxyribofuranose- Einheiten sowie den Pyrimidin- (B = Cytosin, Thymin, Uracil) bzw. Purinbasen (B = Adenin, Guanin) . Als Basen können auch 2, 6-Diaminopurin-9-yl-, Hypoxanthin-9-yl-, 5-Methylcytosin-1- yl-, 5-Amino-4-imidazolcarbonsäureamid-l-yl- oder 5-Amino-4-imidazolcarbonsäureamid-3-yl-Reste eingesetzt werden.
Die OH-Gruppe(n) im Ribofuranosid- bzw.
2' -Desoxyribofuranose-Teil können je nach Bedarf frei oder geschützt sein. Zum Schutz der 3' -Stellung haben sich hierbei die bekannten Phosphitamid-Gruppen bewährt, wie z.B.
NC—CH2—CH2—O—P—N(R7)2 oder
p-N02—C6H4—CH2—CH2—O—P—N(R7)2
wobei die R7-Gruppen gleich oder verschieden sein können und lineare oder verzweigte Alkylreste mit 1 bis 4 C-Atomen bedeuten. Vorzugsweise sind sie Ethyl- oder Isopropylreste.
In der 2' -Stellung des Ribofuranosid-Teiles (Position R ) kann neben einem Wasserstoff- oder Halogenatom (insbesondere F, Cl, Br) eine freie oder geschützte OH-Gruppe vorliegen, wobei eine beliebige in der Nucleotidchemie übliche o
Schutzgruppe (R ) verwendet werden kann. Insbesondere kann auf die üblichen Alkyl-, Alkenyl-, Acetal- oder Silylether- Schutzgruppen für Sauerstoffatome (X=0) zurückgegriffen werden. R6 kann auch eine S-Alkylgruppe darstellen (X=S, R8=Alkyl) . Bevorzugte Beispiele für O-Alkyl-Schutzgruppen sind O-Methyl- oder O-Ethylreste, für O-Alkenyl-Schutzgruppen O-Allylreste, für O-Acetal-Schutzgruppen O-Tetrahydropyranyl- bzw. O-Methoxytetrahydropyranyl-Reste sowie für O-Silylether- Schutzgruppen O-t-Butyldimethylsilyl-Reste.
Gemäß einer bevorzugten Ausführungsform können die Pyrimidin- bzw. Purinbasen mit primären Aminofunktionen (z.B. Adenin, Cytosin und Guanin) noch permanente Schutzgruppen vorzugsweise auf Carbonylbasis aufweisen. Bevorzugt sind hierbei vor allem Phenoxyacetyl- oder Dimethylfor amidino- Reste, die für alle drei genannten Basen in Frage kommen. Daneben gibt es noch spezielle Schutzgruppen, die nur bei bestimmten Basen eingeführt werden. Dies sind bspw. im Falle von Adenin Benzoyl- oder p-Nitrophenylethoxycarbonyl (p-NPEOC) -Reste. Für Guanin können neben den p-NPEOC-Resten auch Isobutyroyl-Schutzgruppen eingeführt werden. Schließlich eignen sich für Cytosin neben den p-NPEOC-Resten auch noch Benzoyl-Schutzgruppen.
Die Herstellung der erfindungsgemäßen Nucleosid-Derivate kann in zwei Stufen erfolgen. In der ersten Stufe a) wird ein Alkohol der allgemeinen Formel (II)
in der R1, R2, R3, R4 die oben angegebene Bedeutung besitzen, mit einem Phosgen-Derivat vorzugsweise in einem unpolaren organischen Lösemittel bei Temperaturen zwischen -20 und +25°C zur Umsetzung gebracht. Als Phosgen-Derivat kann neben dem bevorzugten Phosgen auch Diphosgen
(Chlorameisensäuretrichlormethylester) oder Triphosgen (Bis- trichlormethylcarbonat) verwendet werden.
Die Alkoholkomponente ist in den meisten Fällen bekannt oder kann in analoger Weise nach bekannten Verfahren hergestellt werden. Als unpolares organisches Lösemittel wird in Stufe a)
vorzugsweise Toluol oder THF verwendet. Die Reaktionskomponenten können zwar in annähernd stochiometrischem Verhältnis eingesetzt werden, doch wird das Phosgen-Derivat vorzugsweise in deutlichem Überschuß, bspw. in zwei- bis fünffachem molaren Überschuß, bezogen auf die Alkoholkomponente eingesetzt. Auch die Konzentration der Alkoholkomponente kann in weiten Grenzen variiert werden, doch hat es sich als besonders vorteilhaft erwiesen, diese Konzentration auf 0,1 bis 10,0 mmol pro 10 ml Solvens einzustellen.
Bei dieser Reaktion (Reaktionsdauer ca. 1 bis 6 Std.) entstehen in guter Reinheit und hoher Ausbeute (> 90 %) die entsprechenden Chlorkohlensäureester der allgemeinen Formel (IV)
Die Aufarbeitung der entsprechenden Produkte erfolgt vorzugsweise, indem man zunächst das überschüssige Phosgen sowie das Lösemittel im Vakuum abdeεtilliert. Der Chlorkohlensäureester (IV) kann dann ohne weitere Aufarbeitung in Stufe b) mit den Nucleosiden der allgemeinen Formel (III)
umgesetzt werden, wobei R
5, R
6 und B die oben angegebene Bedeutung besitzen.
Die Umsetzung erfolgt vorzugsweise in einem Lösemittelgemisch bestehend aus Dichlormethan und einem polaren organischen Lösemittel ggf. in Gegenwart einer Base bei Temperaturen zwischen -60 und +25°C. Als polares organisches Lösemittel wird hierbei bevorzugt DMF oder Pyridin eingesetzt, wobei im Falle von Pyridin keine zusätzliche Base erforderlich ist. Wird jedoch mit Dichlormethan/DMF-
Lösemittelgemischen gearbeitet, empfiehlt sich die Zugabe einer Base wie z.B. Pyridin, Triethylamin oder Ethyldiisopropylamin, um die bei der Reaktion freigesetzten Protonen abzufangen. Das Mischungsverhältnis Dichlormethan zu Pyridin bzw. DMF ist ebenfalls unkritisch, doch werden vorzugsweise 1 bis 3 Vol.-teile Dichlormethan pro Vol. -teil Pyridin bzw. DMF eingesetzt.
Gemäß einer bevorzugten Ausführungsform wird das entsprechende Nucleosid (III) , welches in Pyridin oder DMF/Base gelöst wurde, vorgelegt und eine Lösung des Chlorkohlensäureesters in Dichlormethan bei der jeweiligen Reaktionstemperatur zugetropft. Das Molverhältnis von Nucleosid zu Chlorkohlensäureester kann hierbei entsprechend der Stöchiometrie auf ca. 1:1 eingestellt werden. Vorzugsweise wird jedoch der Chlorkohlensäureester im Überschuß eingesetzt, und zwar in einer solchen Menge, daß das Molverhältnis Nucleosid zu Chlorkohlensäureester 1:1 bis 1:2 beträgt. Schließlich kann auch die Konzentration des Nucleosids im Lösemittelgemisch in weiten Grenzen variiert werden, doch wird es bevorzugt auf 0,1 bis 3,0 mmol pro 10 ml Solvens eingestellt.
Nach erfolgter Umsetzung (Reaktionszeit ca. 1 bis 5 Std.) können die erfindungsgemäßen Nucleosid-Derivate nach bekannten Methoden isoliert bzw. gereinigt werden, wie z.B.
Verdünnen mit Dichlormethan, Auswaschen aller Salze mit Wasser, Trocknen der organischen Phase, Einengen der Lösung bzw. Kristallisation und anschließende Kieselgel- Chromatographie. Auf diese Weise können die entsprechenden Nucleosid-Derivate mit hoher Reinheit und in guten Ausbeuten (60 bis 85 %) erhalten werden.
Gemäß einer bevorzugten Ausführungsform kann man im Anschluß an die Reaktionsstufe b) in die 3" -Stellung der Nucleosid- Derivate mit R5 ■= H die Phosphitamid-Gruppe
NC—CHa -CH. -O—P—N(R7)2 oder
p-N02—C6H4—CH2—CH2—O—P— (R7)2
nach bekannten Methoden einführen. Üblicherweise erfolgt diese Umsetzung mit den entsprechenden Phosphinen in Gegenwart von IH-Tetrazol als Aktivator in einem Lösemittelgemisch bestehend aus Dichlormethan und Acetonitril bei Temperaturen zwischen 0 und 25°C. Vorzugsweise wird das Phosphin in zwei- bis dreifachem molaren Überschuß eingesetzt, während das Molverhältnis Phosphin zu IH-Tetrazol auf 3:ca.1,0 eingestellt wird. Das Mengenverhältnis von Dichlormethan zu Acetonitril ist relativ unkritisch und beträgt vorzugsweise 1:1 bis 4:1. Nach erfolgter Umsetzung (ca. 10 bis 20 h) kann das entsprechende Nucleosid wie in Stufe b) beschrieben aufgearbeitet werden.
Wie Bestrahlungsversuche mit polychromatischem Licht mit Wellenlänge > 289 nm belegen, lassen sich die erfindungsgemäßen Nucleoside sehr rasch (t0/5 = 1 bis 7 Min.) und weitgehend entschützen (Ausbeuten bis zu 97 %) , sodaß die besonderen Anforderungen an die Photolabilität der Schutzgruppen in hervorragender Weise erfüllt werden.
Aufgrund dieser besonderen Eigenschaften eignen sich die erfindungsgemäßen Nucleoside sehr gut für die Herstellung von Oligonucleotiden durch lichtgesteuerte Schutzgruppenabspaltung insbesondere auf festen Trägerplatten.
Die nachfolgenden Beispiele sollen die Erfindung näher erläutern.
Beispiel 1
a) 2- (2-Nitrophenyl)ethanol [1, 2]
Ein Gemisch von o-Nitrotoluol (9,2 g, 67 mmol) und Paraformaldehyd (0,8 g, 25 mmol) in DMSO (10 ml, Synthese- Qualität, zusätzlich 2 d über Molekularsieb 4 Ä getrocknet) wurde mit KOH (21 mg, 0,37 mmol) versetzt und 2,5 h bei 95°C gerührt. Das Lösemittel wurde im Hochvakuum abdestilliert. Der Rückstand wurde durch Säulenchromatographie (Siθ2, 20 x 3 cm, Lösemittel: Toluol 750 ml, Toluol/EtOAc 10:1 500 ml, 7:1 500 ml, 5:1 500 ml) gereinigt. Man erhielt 2- (2-Nitrophenyl)ethanol (2,33 g, 21 %) als gelbes Öl.
Rf (Si0 , Toluol/EtOAc 10:1) 0,21
UV(MeOH), λmaχ [nm] (log ε) : 205 (4,07), 256 (3,65), 348
(Schulter, 2,56) ---H-NMR (250 MHz, CDC13): 7,93 (dd, 1 arom. H, ortho zu N02) ;
7,49 (m, 3 arom. H) ; 3,96 (t, α-CH2); 3,17 (t, ß-CH2) ,
1,72 (s, OH) C8H9N03 (167,2)
Literatur:
[1] G. M. Bennet, M. M. Hafez, J. Chem. Soc. 1941, 287 [2] E. Uhl ann, W. Pfleiderer, Helv. Chim. Acta 1981, 64, 1688
b) 2- (2-Nitrophenyl)ethoxycarbonylchlorid
In eine Lösung von 2- (2-Nitrophenyl)ethanol (5,2 g, 31 mmol; in THF (20 ml, dest. über CaH ) wurde bei Raumtemperatur unter Rühren Phosgen eingeleitet. Nach 1,5 h wurde das überschüssige Phosgen sowie das Lösemittel im Hochvakuum abdeεtilliert. Man erhielt 2- (2-
Nitrophenyl)ethoxycarbonylchlorid (6,69 g, 94 %) als gelbes
Öl.
Rf (Si02, CHC13) 0,84
UV(CH3CN), λrnax [nm] (log ε) : 202 (4,12), 218 (Schulter,
3,74); 256 (3,70); 298 (Schulter, 3,16);
346 (Schulter, 2,59) ---H-NMR (250 MHz, CDCI3) : 7,99 (dd, H-C(3)) ; 7,48 (m, 3 arom.
H) ; 4,62 (t, α-CH2) ; 3,31 (t, ß-CH2) Anal. ber. für C9H8C1N04 (229,62) : C 47,08, H 3,51, N 6,10; gef. : C 47,30, H 3,70, N 6,00
c) 5' -0- (2- (2-Nitrophenyl)ethoxycarbonyl)thy idin
Thymidin (1 g, 4,13 mmol) wurde mit Pyridin koevaporiert (2 x 10 ml pro analysi-Qualität, zusätzlich über Molekularsieb 4 Ä getrocknet), in Pyridin (10 ml, s.o.) gelöst und auf -30°C gekühlt. In 10 min wurde hierzu eine Lösung von 2- (2-Nitrophenyl) -ethoxycarbonylchlorid (1,45 g, 6,31 mmol) getropft. Nach weiteren 4 h 50 min Rühren unter i-Pr0H/N -Kühlung (-30 bis -15°C) wurde das Gemisch mit CH2C1 (150 ml) verdünnt und mit H 0 (150 ml) gewaschen. Die wäßrigen Phasen wurden mit CH C1 (2 x 150 ml) nachextrahiert. Die vereinigten organischen Phasen wurden über gSθ4 getrocknet, filtriert, einrotiert und mit Toluol (5 x 20 ml) und CH2C12 (2 x 20 ml) koevaporiert. Das Rohprodukt wurde durch Säulenchromatographie (Si02,
15 x 3,5 cm, Lösemittel: CH2C12 1300 ml, CH2Cl2/Aceton 20:1 1200 ml, 10:1 600 ml, 8:1 500 ml, 5:1 500 ml, 4:1 500 ml, 2:1 750 ml, 1:1 500 ml) gereinigt. Man erhielt 5'-0-(2-(2- Nitrophenyl)ethoxycarbonyl) thymidin (1,15 g, 64 %) als farblosen Feststoff.
Rf (Si02, Toluol/EtOAc/MeOH 5:4:1) 0,46
UV(MeOH), λmax [nm] (log ε) : 205 (4,31), 262 (4,12), 334 (Schulter, 2,64)
---H-NMR (250 MHz, CDC13): 8,92 (s (br) , NH) ; 7,96 (dd, (H-C(3) v. NPEOC) ; 7,55 (t, 1 arom. H v. NPEOC) ; 7,42 (m, H-C(6) v. Thymin, 2 arom. H v. NPEOC); 6,35 (t, H-C(l')); 4,44 (m, H-C(3'), 2 x H-C(5'), α-CH2 v. NPEOC); 4,15 (q, H-C(4'); 3,34 (m, ß-CH2 v. NPEOC); 2,89 (d, OH-C(3')); 2,41 (m, H-C(2')); ,22 (m, H-C(2')); 1,85 (s, CH3)
Anal. ber. für C19H21N309 (435,39): C 52,41, H 4,86, N 9,65; gef.: C 52,07, H 5,15, N 9,65
Beispiel 2
a) 2- (2, 6-Dinitrophenyl)ethanol [1]
Zu 2, 6-Dinitrotoluol (18,2 g, 0,1 mol, 3 d im Hochvakuum über Blaugel getrocknet) und Paraformaldehyd (3 g, 0,1 mol) in DMSO (50 ml, Synthese-Qualität, zusätzlich 2 d über Molekularsieb 4 Ä getrocknet) wurde eine Lösung von Kaliumtertiärbutylat (1,8 g, 8 mmol) in tert.-Butanol (20 ml, Synthese-Qualität, 99 %) gegeben. Nach der Zugabe der Kaliumtertiärbutylat-Lösung trat ein Farbumschlag von gelb nach tief-violett ein. Es wurde 5 min bei Raumtemperatur und 10 min bei 70°C (Ölbadtemperatur) gerührt. Danach ließ man auf Raumtemperatur abkühlen, neutralisierte das Gemisch mit konz. HC1, verdünnte mit H20 (300 ml) und versetzte es mit NaCl bis zur Sättigung. Die wäßrige Phase wurde mit EtOAc (3 x 500 ml) extrahiert. Die vereinigten organischen Phasen
wurden mit gesättigter NaCl Lösung (300 ml) gewaschen, über Na Sθ4 getrocknet, filtriert und einrotiert. Das Rohprodukt
(24,3 g) wurde in wenig EtOAc in der Siedehitze gelöst, mit Petrolether (nachfolgend PE genannt) (100 ml) versetzt und im Eisfach zur Kristallisation gebracht. Der Niederschlag wurde abgesaugt und durch Säulenchromatographie (20,7 g verunreinigtes Produkt, 300 g Si0 , 18 x 6,5 cm, Lösemittel: Toluol/EtOAc 5:1, 4:1, 3:1) gereinigt. Mischfraktionen wurden einrotiert und durch erneute Säulenchromatographie (200 g Si02, 20 x 5,3 cm, Lösemittel: Toluol/EtOAc 7:1) gereinigt. Man erhielt nach Einrotieren der reinen Produktfraktionen 2-
(2, 6-Dinitrophenyl)ethanol (13,6 g, 64 %) als gelben Feststoff.
Rf (Si02, Toluol/EtOAc 9:1) 0,21
Schmelzpunkt: 69 bis 70°C (Lit. [1]: 69°C)
UV(MeOH), λmax [nm] (log ε) : 203 (4,20), 231 (4,00), 280
(Schulter, 3,17), 327 (Schulter, 2,80) ---H-NMR (250 MHz, CDCI3): 8,00 (d, J = 8,1, H-C(3), H-C(5));
7,57 (t, J = 8,1, H-C(4)); 3,97 (q, α-CH2); 3,33 (t, ß-CH2) ; 1,69 (t, OH) Anal. ber. für C8H8N205 (212,16) : C 45,29, H 3,80, N 13,20; gef.: C 45,39, H 3,90, N 13,32
Literatur:
[1] N. S. Girgis, H. B. Cottam, R. K. Robins, J. Heterocycl. Chem. 1988, 25, 361
b) 2- (2, 6-Dinitrophenyl)ethoxycarbonylchlorid
Zu einer auf 0°C gekühlten Lösung von
Chlorameisensäuretrichlormethylester (3,81 g, 2,33 ml, 19,28 mmol) in THF (10 ml, dest. über CaH2) wurde in 20 min eine Lösung von 2- (2, 6-Dinitrophenyl)ethanol (4,08 g, 19,23 mmol) und Et N (2,7 ml, 19,28 mmol) in THF (30 ml,
s.o.) zugetropft. Es wurde 25 min unter Eisbadkühlung und 1 h 45 min bei Raumtemperatur gerührt. Die Mischung wurde über Celite filtriert. Nachwaschen des Filterkuchens mit THF, Abdestillieren des Lösemittels sowie des überschüssigen Reagenses von den vereinigten Filtraten und Trocknen im Hochvakuum ergaben 5,13 g 2- (2,6-Dinitrophenyl) - ethoxycarbonylchlorid (97 %) als hellbraunen Feststoff.
Rf (Si02, CH2C12) 0,76
Schmelzpunkt: 84 bis 85°C
UV(CH3CN), λmaχ [nm] (log ε) : 233 (4,02), 292 (Schulter,
3,11), 331 (Schulter, 2,82) ---H-NMR (250 MHz, CDC13) : 8,10 (d, J = 8,2, H-C(3), H-C.5));
7,67 (t, J = 8,1, H-C(4)); 4,67 (t, α-CH2); 3,50 (t, ß-CH2); Anal. ber. für C9H7C1N206 (274,616) : C 39,36, H 2,57, N
10,20; gef.: C 39,40, H 2,60, N 10,20
c) 5' -0- (2- (2,6-Dinitrophenyl)ethoxycarbonyl)thymidin
Thymidin (1 g, 4,13 mmol) wurde mit Pyridin (3 x 10 ml, pro analysi-Qualität, zusätzlich über Molekularsieb 4 Ä getrocknet) koevaporiert, in Pyridin (15 ml, s.o.) gelöst und auf -50°C gekühlt. Hierzu tropfte man in 1 h eine Lösung von 2- (2, 6-Dinitrophenyl)ethoxycarbonylchlorid (1,7 g, 6,19 mmol) in CH2C12 (15 ml, dest. über CaH2) . Nach weiteren 3,5 h Rühren unter i PrOH/N2-Kühlung (-50 bis -20°C) wurde das Gemisch mit CH2C12 (50 ml) verdünnt und mit H20 (50 ml) gewaschen. Die wäßrigen Phasen wurden mit CH2C1 (2 x 50 ml) nachextrahiert. Die vereinigten organischen Phasen wurden über a2Sθ4 getrocknet, filtriert, einrotiert und mit Toluol (3 x 50 ml) koevaporiert. Das Rohprodukt (2,66 g) wurde in CH2Cl2/MeOH 2:1 (60 ml) in der Siedehitze digeriert. Der erhaltene weiße Niederschlag wurde abgesaugt und aus MeOH (25 ml) umkristallisiert. Man erhielt 5'-0- (2- (2, 6-
Dinitrophenyl)ethoxycarbonyl)thymidin (855 mg, 43 %) als farblosen Feststoff. Die vereinigten Filtrate wurden zur Trockene einrotiert und durch Säulenchromatographie (1,4 g Rohprodukt, 56 g Si02 17 x 3 cm, CH2Cl2/MeOH 100:5 1240 ml, 100:7 105 ml, 100:10 330 ml) gereinigt. Man erhielt 5'-0-(2- (2, 6-Dinitrophenyl)ethoxycarbonyl)thymidin (691 mg, 34,8 %) als farblosen Feststoff. Die Ausbeute an 5' -0- (2- (2, 6- Dinitrophenyl)ethoxycarbonyl)thymidin betrug insgesamt 1,55 g (78 %) .
Rf (Si02, Toluol/EtOAc/MeOH 5:4:1) 0,41
UV(MeOH), λrπax [nm] (log ε) : 206 (4,28), 244 (Schulter,
4,07), 256 (4,08) 355 (Schulter, 2,72) ---H-NMR (250 MHz, DMSO-d6): 11,32 (s, NH) , 8,27 (d, H-C(3),
H-C(5)); 7,78 (t, H-C(4)); 7,40 (s, H-C(6) V. Thymin) ;
6,18 (t, H-C(l')); 5,44 (d, OH-CU*)); 4,37 (t, α-CH2);
4,23 (m, H-C(3*). 2 x H-C(5')); 3,91 (m, H-C(4')); 3,29
(t, ß-CH2); 2,15 (m, 2 x H-C(2')); 1,71 (s, CH3) Anal. ber. für C19H20N O11 (480,386): C 47,51, H 4,20, N
11,66; gef.: C 47,40, H 4,15, N 11,57
Beispiel 3
a) 2- (2-Fluor-6-nitrophenyl)ethanol [1]
Zu 2-Fluor-6-nitrotoluol (776 mg, 5 mmol) und Paraformaldehyd (150 mg, 5 mmol) in DMSO (2,5 ml, Synthese-Qualität, zusätzlich 2 d über Molekularsieb 4 Ä getrocknet) wurde eine Lösung von Kaliumtertiärbutylat (90 mg, 0,8 mmol) in tert. - Butanol (1 ml, Synthese-Qualität, 99 %) gegeben. Nach der Zugabe der Kaliumtertiärbutylat-Lösung trat ein Farbumschlag von gelb nach tief-violett ein. Es wurde 5 min bei Raumtemperatur und 30 min bei 70°C (Ölbadtemperatur) gerührt. Danach ließ man auf Raumtemperatur abkühlen und neutralisierte mit wenigen Tropfen konz. HC1. Das Gemisch
wurde mit EtOAc (30 ml) verdünnt und mit H20 (20 ml) gewaschen. Die wäßrige Phase wurde mit EtOAc (2 x 20 ml) nachextrahiert. Trocknen der organischen Phasen über Na2SÜ4, Filtrieren und Abrotieren des Lösemittels ergaben das Rohprodukt (1,11 g) , welches durch Säulenchromatographie (20 g Si02, 12 x 2 cm, Lösemittel: PE 40 ml, PE/EtOAc 10:1 110 ml, 8:1 270 ml, 6:1 210 ml, 5:1 60 ml, 4:1 50 ml) gereinigt wurde. Man erhielt 2- (2-Fluor-6-nitrophenyl)ethanol (653 mg, 71 %) als gelben Feststoff.
Rf (Si02, Toluol/EtOAc 9:1) 0,24
UV(MeOH), λ ax [nm] (log ε) : 205 (4,06), 251 (3,61), 294
(Schulter, 3,21) ---H-NMR (250 MHz, CDCI3): 7,73 (m, 1 arom. H) ; 7,36 (m, 2 arom. H) ; 3,94 (t, α-CH2) ,* 3,21 (dt, J = 2,2, 6,5, ß-CH2) ; 1,67 (s (br) , OH) Anal. ber. für C8H8FN03 (185,154): C 51,90, H 4,36, N 7,57; gef.: C 51,92, H 4,40, N 7,42
Literatur:
[1] Chem. Abstr. 1989, 110, P 75032 k
b) 2- (2-Fluor-6-nitrophenyl)ethoxycarbonylchlorid
Zu einer auf 0°C gekühlten Lösung von Chlorameisensäure- trichlormethylester (641 mg, 3,24 mmol) in THF (6,75 ml, deεt. über CaH2) wurde in 5 min eine Lösung von 2- (2-Fluor-6* nitrophenyl)ethanol (500 mg, 2,7 mmol) und Et3N (273 mg, 2,7 mmol, dest. über KOH) in THF (6,75 ml, s.o.) zugegeben. Man ließ 1 h unter Eisbadkühlung und 1 h bei Raumtemperatur rühren. Das Gemisch wurde über Celite filtriert. Nachwaschen des Filterkuchens mit THF und Abdestillieren des Lösemittels sowie des überschüssigen Reagenses von den vereinigten Filtraten bei 30°C im Hochvakuum ergaben 2- (2-Fluor-6-
nitrophenyl)ethoxycarbonylchlorid (620 mg, 93 %) als hellbraunes Öl.
Rf (Si02, PE/EtOAc 19:1) 0,25
UV(MeOH), ax [nm] (log ε) : 204 (4,04), 251 (3,67), 293
(Schulter, 3,23) ---H-NMR (250 MHz, CDC13) : 7,83 (d, J = 7,7, 1 arom. H) ; 7,44 (m, 2 arom. H) ; 4,63 (t, α-CH2); 3,37 (dt, J = 1,6, 6,4 ß-CH2) Anal. ber. für C9H7C1FN04 (247,609): C 43,66, H 2,85, N 5,66; gef. : C 43,97, H 3,02, N 5,59
c) 5' -O- (2- (2-Fluor-6-nitrophenyl)ethoxycarbonyl)thymidin
Thymidin (200 mg, 0,83 mmol) wurde mit Pyridin (3 x 3 ml, pro analysi-Qualität, zusätzlich über Molekularsieb 4 Ä getrocknet) koevaporiert, in Pyridin (3 ml. s.o.) gelöst und auf -60°C (i-PrOH/N2) gekühlt. Hierzu tropfte man in 20 min eine Lösung von 2- (2-Fluor-6-nitrophenyl)ethoxycarbonyl¬ chlorid (280 mg, 1,13 mmol) in CH2C12 (3 ml, dest. über CaH2) . Man ließ 3 h 40 min unter i-PrOH/N2 Kühlung (-60 bis -15°C) und danach 1 h ohne Kältebad rühren, wobei die Temperatur gegen Ende bei 0°C lag. Die Reaktionsmischung wurde mit CH2C12 (10 ml) verdünnt und mit H20 (10 ml) gewaschen. Die wäßrige Phase wurde mit CH2C12 (3 x 10 ml) nachextrahiert. Trocknen der organischen Phasen über Na2S04, Filtrieren, Abrotieren des Lösemittels und Koevaporieren mit Toluol (3 x 10 ml) ergaben das Rohprodukt, welches durch Säulenchromatographie (20 g Si0 12 x 2 cm, Lösemittel: CH2Cl2/MeOH 100:1 50 ml, 100:2 102 ml, 100:3 206 ml, 100:3,5 103 ml, 100:4 208 ml) gereinigt wurde. Man eluierte zuerst 3 ' , 5' -Bis-O- (2- (2-Fluor-6-nitrophenyl)ethoxycarbonyl) - thymidin, dann 3' -0- (2- (2-Fluor-6-nitrophenyl) - ethoxycarbonyl) thymidin und zuletzt 5' -0- (2- (2-Fluor-6- nitrophenyl)ethoxycarbonyl) hymidin. Nach Abrotieren des
Lösemittels und Trocknen im Hochvakuum erhielt man 3 ',5'-Bis- O- (2- (2-Fluor-6-nitrophenyl)ethoxycarbonyl)thymidin (27 mg, 5 %) als hellgelben Schaum, 3' -0- (2- (2-Fluor-6- nitrophenyl)ethoxycarbonyl)thymidin (15 mg, 4 %) als farblosen Schaum und 5' -0- (2- (2-Fluor-6-nitrophenyl) - ethoxycarbonyl)thymidin (262 mg, 70 %) als farblosen Feststoff.
5' -0- (2- (2-Fluor-6-nitrophenyl)ethoxycarbonyl) thymidin:
Rf (Si02, Toluol/EtOAc/MeOH 5:4:1) 0,44
UV (MeOH) , λmax [nra] ( log ε) : 205 (4 , 29 ) , 261 ( 4 , 10 ) ,
---H-NMR (250 MHz , CDC13 ) : 8 , 09 ( s (br) , NH) ; 7 , 76 (m, 1 arom .
H v. FNPEOC) ; 7,41 (m, H-C(6) v. Thymin, 2 arom. H v.
FNPEOC) , 6,35 (t, H-C(l')); 4,45 (m, H-C.3-). 2 x
H-C(5'), α-CH2 v. FNPEOC); 4,14 (q, J = 3,2, H-C(4'));
3,36 (m, ß-CH2 v. FNPEOC); 2,40 (m, H-C(2')); 2,23 (m,
H-C(2'), OH-C(3»)); 1,85 (s, CH3) Anal. ber. für C19H20FN3O9 (453,379) : C 50,34, H 4,45, N
9,27; gef.: C 50,22, H 4,49, N 9,18
Beispiel 4
a) 2- (2-Chlor-6-nitrophenyl)ethanol [l, 2]
Zu einem Gemisch aus 2-Chlor-6-nitrotoluol (25 g, 146 mmol) und Paraformaldehyd (1,9 g, 60 mmol) in DMSO (20 ml, Synthese-Qualität, zusätzlich 2 d über Molekularsieb 4 Ä getrocknet) gab man Triton B (2 ml, 35 % in MeOH) und ließ bei 90°C rühren. Nach 2 h wurde das Reaktionsgemisch mit wenigen Tropfen konz. HC1 neutralisiert, mit H20 (50 ml) verdünnt und mit EtOAc (4 x 150 ml) extrahiert. Die vereinigten organischen Phasen wurden über MgSθ4 getrocknet, filtriert und einrotiert. Das Rohprodukt wurde durch Sublimation im Hochvakuum (p = 0,06 Torr, Ölbadtemperatur
95°C) gereinigt. Man erhielt 2- (2-Chlor-6-nitrophenyl) ethanol (8,01 g, 66 %) in Form von hellgelben Kristallen.
Rf (Si02, Toluol/EtOAc 10:1) 0,36
Schmelzpunkt: 59 bis 61°C
UV(MeOH) , λ ax [n ] (log ε) : 212 (4,13) , 248 (3,48) , 294
(Schulter, 3,03) , 336 (Schulter, 2,58) ---H-NMR (250 MHz, CDCI3) : 7,70 (dd, 1 arom. H) ; 7,62 (dd, 1 arom. H) ; 7,31 (t, H-C(4)) ; 3,94 (q, α-CH2) ; 3,26 (t, ß-CH2) ; 1,70 (t, OH) Anal. ber. für C8H8C1N03 (201,61) : C 47,66, H 4,00, N 6,95; gef . : C 47,79, H 4,06, N 6,92
Literatur:
[1] T. Morimoto, I. Hashimoto, H. Yamaoka, Che . Abstr.
1978, 88, 104880 v. [2] Y. Tsuji, S. Kotachi, K.-T. Huh, Y. Watanabe, J. Org.
Chem. 1990, 55, 580
b) 2- (2-Chlor-6-nitrophenyl)ethoxycarbonylchlorid
In eine Lösung von 2- (2-Chlor-6-nitrophenyl) ethanol (31 g, 154 mmol) in THF (190 ml, deεt. über CaH2) wurde bei Raumtemperatur unter Rühren Phosgen eingeleitet. Nach 2,5 h wurden das überschüssige Phosgen sowie das Lösemittel im Hochvakuum abdestilliert. Man erhielt 2- (2-Chlor-6- nitrophenyl)ethoxycarbonylchlorid (39,4 g, 97 %) als gelbes Öl.
Rf (Si02, CHCI3) 0,76
UV(CH3CN) , λ-nax [nm] (log ε) : 211 (4,14) , 253 (3,53) , 300
(Schulter, 3,02) ---H-NMR (250 MHz, CDCI3) : 7,81 (dd, 1 arom. H) ; 7,69 (dd, 1 arom. H) ; 7,41 (t, H-C(4)) ; 4,63 (t, α-CH2) ; 3,46 (t, ß-CH2)
Anal. ber. für C9H7C12N04 (264,06): C 40,94, H 2,67, N 5,30; gef.: C 41,05, H 2,73, N 5,00
c) 5' -0- (2- (2-Chlor-6-nitrophenyl)ethoxycarbonyl)thymidin
Thymidin (4 g, 16,5 mmol) wurde mit Pyridin (3 x 40 ml, pro analysi-Qualität, zusätzlich über Molekularsieb 4 Ä getrocknet) koevaporiert, in Pyridin (48 ml, s.o.) gelöst und auf -50°C (i-PrOH/N2) gekühlt. Hierzu tropfte man in 2 h 45 min eine Lösung von 2- (2-Chlor-6-nitrophenyl) - ethoxycarbonylchlorid (5,2 g, 19,8 mmol) in CH2C1 (48 ml, dest. über CaH2) . Man ließ weitere 30 min unter i-PrOH/N2 Kühlung (-50 bis -30°C) rühren. Die Reaktionsmischung wurde mit CH2C12 (100 ml) verdünnt und mit H 0 (100 ml) gewaschen. Die wäßrige Phase wurde mit CH C1 (2 x 100 ml) nachextrahiert. Trocknen der organischen Phasen über Na2Sθ4, Filtrieren, Abrotieren des Lösemittels und Koevaporieren mit Toluol (3 x 50 ml) ergaben das Rohprodukt, welches durch Säulenchromatographie (305 g Si02, 22 x 5,9 cm, Lösemittel: CH2C12 200 ml, CH2Cl2/MeOH 100:2 1020 ml, 100:3 515 ml, 100:4 1560 ml, 100:5 850 ml, 100:6 318 ml, 100:8 324 ml, 100:9 654 ml) gereinigt wurde. Man eluierte zuerst eine Mischfraktion (1,4 g) von 3' ,5' -Bis-0- (2- (2-Chlor-6- nitrophenyl)ethoxycarbonyl)thymidin und 3 ' -0- (2- (2-Chlor-6- nitro-phenyl)ethoxycarbonyl)thymidin, dann eine Mischfraktion (367 mg) von 3' -0- (2- (2-Chlor-6-nitrophenyl)ethoxycarbonyl) - thymidin und 5 ■ -0- (2- (2-Chlor-6-nitrophenyl)ethoxycarbonyl) - thymidin und zuletzt eine reine Fraktion von 5'-0-(2-(2- Chlor-6-nitrophenyl)ethoxycarbonyl)thymidin (6,21 g, 80 %, farbloser Feststoff) . Die Mischfraktionen wurden durch weitere Säulenchromatographie gereinigt. Man erhielt 3' , 5' -Bis-O- (2- (2-Chlor-6-nitrophenyl)ethoxycarbonyl)thymidin (1,175 g, 10 %) als hellgelben Schaum, 3* -0- (2- (2-Chlor-6- nitrophenyl)ethoxycarbonyl)thymidin (203 mg, 3 %) als farblosen Schaum und 5' -0- (2- (2-Chlor-6-
nitrophenyl)ethoxycarbonyl) thymidin (182 mg, 2 %) als farblosen Feststoff. Die Gesamtausbeute an 5 ' -0- (2- (2-Chlor- 6-nitrophenyl) ethoxycarbonyl) thymidin betrug somit 6,392 g (82 %) .
Rf (Si02, Toluol/EtOAc/MeOH 5:4:1) 0,53 UV(MeOH) , ^x [nm] (log ε) : 210 (4,36), 263 (4,05) -H-NMR (250 MHz, CDC13) : 8,11 (s, NH) ; 7,74 (d, J = 8,2, 1 arom. H v. C1NPE0C) ; 7,67 (d, J = 8,0, 1 arom. H v. C1NPE0C) , 7,39 (t, H-C(4) v. C1NPE0C) ; 7,38 (s, H-C(6) v. Thymin) ; 6,36 (t, H-C(l')); 4,44 (m, α-CH2 v. C1NPE0C, H-C.31) , 2 x H-C(5')) ; 4,15 (q, H-C(4')) ; 3,45 (t, ß-CH2 v. C1NPEOC) ; 2,38 (m, H-C(2')) ; 2,26 (m, H-C(2')) , 2,18 (d, J = 3,9, OH-C(3')) ; 1,86 (s, CH3) Anal. ber. für C19H20ClN3O9 (469,83) : C 48,57, H 4,29, N 8,94; gef.: C 48,53, H 4,34, N 8,91
Beispiel 5
a) 2- (2-Brom-6-nitrophenyl)ethanol [1]
Zu 2-Brom-6-nitrotoluol (1,08 g, 5 mmol) und Paraformaldehyd (150 mg, 5 mmol) in DMSO (2,5 ml, Synthese-Qualität, zusätzlich 2 d über Molekularsieb 4 Ä getrocknet) wurde eine Lösung von Kaliumtertiärbutylat (90 mg, 0,8 mmol) in tert.- Butanol (1 ml, Synthese-Qualität, 99 %) gegeben. Nach der Zugabe der Kaliumtertiärbutylat-Lösung trat ein Farbumschlag von gelb nach tief-violett ein. Es wurde 5 min bei Raumtemperatur und 30 min bei 70°C (Ölbadtemperatur) gerührt. Danach ließ man auf Raumtemperatur abkühlen und neutralisierte mit wenigen Tropfen konz. HC1. Das Gemisch wurde mit EtOAc (30 ml) verdünnt und mit H20 (20 ml) gewaschen. Die wäßrige Phase wurde mit EtOAc (2 x 20 ml) nachextrahiert. Trocknen der organischen Phasen über a2SÜ4, Filtrieren und Abrotieren des Lösemittels ergaben das
Rohprodukt (1,49 g) , welches durch Säulenchromatographie (20 g Si02, 20 x 2 cm, Lösemittel: PE 45 ml, PE/EtOAc 10:1 110 ml, 8:1 180 ml, 7,5:1 340 ml, .6:1 140 ml) gereinigt wurde. Man erhielt 2- (2-Brom-6-nitrophenyl)ethanol (867 mg, 70 %) als gelben Feststoff.
Rf (Si02, Toluol/EtOAc 9:1) 0,32
UV(MeOH), cix [nm] (log ε) : 204 (4,18), 210 (Schulter,
4,16), 251 (3,48), 293 (Schulter, 3,07) ---H-NMR (250 MHz, CDC13): 7,82 (dd, J = 1,1, 8,0, 1 arom. H) ;
7,74 (dd, J = 1,1, 8,2, 1 arom. H) ,* 7,26 (m, H-C(4));
3,96 (t, α-CH2); 3,30 (t, ß-CH2) ; 1,75 (s (br) , OH) Anal. ber. für C8H8BrN03 (246,06): C 39,05, H 3,28, N 5,69; gef.: C 39,09, H 3,26, N 5,56
Literatur:
[1] Chem. Abstr. 1989, 110, P 75032 k
b) 2- (2-Brom-6-nitrophenyl)ethoxycarbonylchlorid
Zu einer auf 0°C gekühlten Lösung von
Chlorameisensäuretrichlormethylester (442 mg, 2,23 mmol) in THF (5 ml, dest. über CaH2) wurde in 10 min eine Lösung von 2- (2-Brom-6-nitrophenyl)ethanol (500 mg, 2,03 mmol) und Et3N (206 mg, 2,03 mmol, dest. über KOH) in THF (5 ml, s.o.) zugegeben. Man ließ 5 min unter Eisbadkühlung und 1 h 45 min bei Raumtemperatur rühren. Das Gemisch wurde über Celite filtriert. Nachwaschen des Filterkuchens mit THF und Abdestillieren des Lösemittels sowie des überschüssigen Reagenses von den vereinigten Filtraten bei 30°C im Hochvakuum ergaben 2 - (2-Brom-6-nitrophenyl) - ethoxycarbonylchlorid (625 mg, 99,8 %) als hellbraunes Öl.
Rf (Si02, PE/EtOAc 19:1) 0,31
UV(MeOH), λ ax [nm] (log ε) : 205 (Schulter, 4,14), 211
(4,16) , 254 (3,52), 297 (Schulter, 3,05) ---H-NMR (250 MHz, CDC13) : 7,85 (2 arom. H) ; 7,33 (t, H-C(4)) ;
4,62 (t, α-CH2) ; 3,47 (t, ß-CH2) Anal. ber. für C9H7BrClN04 (308,515) : C 35,04, H 2,29, N
4,54; gef. : C 35,50, H 2,58, N 4,50
c) 5 ' -O- (2- (2-Brom-6-nitrophenyl) ethoxycarbonyl) thymidin
Thymidin (200 mg, 0,83 mmol) wurde mit Pyridin (3 x 3 ml, pro analysi-Qualität, zusätzlich über Molekularsieb 4 Ä getrocknet) koevaporiert, in Pyridin (3 ml, s.o.) gelöst und auf -60°C (i-PrOH/N ) gekühlt. Hierzu tropfte man in 10 min eine Lösung von 2- (2-Brom-6-nitrophenyl) ethoxycarbonylchlorid (354 mg, 1,15 mmol) in CH2C12 (3 ml, dest. über CaH2) . Man ließ 3 h 40 min unter i-PrOH/N2 Kühlung (-60 bis -20°C) rühren. Die Reaktionsmischung wurde mit CH2C1 (10 ml) verdünnt und mit H20 (10 ml) gewaschen. Die wäßrige Phase wurde mit CH C12 (2 x 10 ml) nachextrahiert. Trocknen der organischen Phasen über Na2SÜ4, Filtrieren, Abrotieren des Lösemittels und Koevaporieren mit Toluol (4 x 10 ml) ergaben das Rohprodukt (552 mg) . Durch Kristallisation aus wenig CH2C12 und MeOH konnte 5 ' -0- (2- (2-Brom-6-nitrophenyl) - ethoxycarbonyl) thymidin (163 mg, 38 %) als farbloser Feststoff isoliert werden. Der Rückstand wurde durch Säulenchromatographie (18 g Si02, 11 x 2 cm, Lösemittel: CH2Cl2/MeOH 100:1 50 ml, 100:3 103 ml, 100:4 208 ml, 100:5 52 ml) gereinigt. Man eluierte zuerst 3 ' , 5 ' -Bis-O- (2- (2-Brom- 6-nitrophenyl) ethoxycarbonyl) thymidin, dann 3 ' -O- (2- (2-Brom- 6-nitrophenyl) ethoxycarbonyl) thymidin und zuletzt 5'-0-(2-(2- Brom-6-nitrophenyl) ethoxycarbonyl) thymidin. Die Produktfraktionen wurden einrotiert und im Hochvakuum getrocknet. Man erhielt 3 ' , 5 ' -Bis-O- (2- (2-Brom-6- nitrophenyl) ethoxycarbonyl) hymidin (62 mg, 10 %) als farblosen Schaum, 3 ' -0- (2- (2-Brom-6-nitrophenyl) -
ethoxycarbonyl)thymidin (8 mg, 2 %) als farbloses Öl und 5 - 0- (2- (2-Brom-6-nitrophenyl)ethoxycarbonyl) hymidin (151 mg, 35 %) als farblosen Feststoff. Die Gesamtausbeute an 5'-0-(2- (2-Brom-6-nitrophenyl)ethoxycarbonyl)thymidin betrug somit 314 mg (73 %) .
5' -0- (2- (2-Brom-6-nitrophenyl)ethoxycarbonyl)thymidin Rf (Si02, Toluol/EtOAc/MeOH 5:4:1) 0,38 UV(MeOH), ^ax [nm] (log ε) : 210 (4,37), 263 (4,06) ---H-NMR (250 MHz, CDCI3): 8,35 (s, NH) ; 7,85 (d, 1 arom. H v. BNPEOC) ; 7,76 (d, 1 arom. H v. BNPEOC) , 7,38 (s, H-C(6) v. Thy in) ; 7,30 (t, H-C(4) v. BNPEOC, teilweise unter CHC13-Signal verborgen); 6,37 (t, H-C(l')); 4,45 (m, H-C(3'), 2 x H-C(5'), α-CH2 V. BNPEOC)); 4,15 (m, H-C(4')); 3,47 (t, ß-CH2 v. BNPEOC); 2,41 (m, H-C(2'), OH-C(3')); 2,25 (m, H-C(2*)); 1,86 (s, CH3) Anal. ber. für C19H20BrN3O9 (514,285): C 44,37, H 3,92, N 8,17; gef.: C 44,31, H 3,96, N 8,11
Beispiel 6
a) 2- (4-Chlor-2-nitrophenyl)ethanol [1, 2, 3]
Ein Gemisch von 4-Chlor-2-nitrotoluol (50 g, 291 mmol) und Paraformaldehyd (3,8 g, 120 mmol) in DMSO (40 ml, Synthese- Qualität, zusätzlich 2 d über Molekularsieb 4 Ä getrocknet) wurde mit Triton B (4 ml, 35 % in MeOH) versetzt und 2,5 h bei 90°C gerührt. Man neutralisierte mit wenigen Tropfen konz. HC1, verdünnte mit H20 (100 ml) und extrahierte mit EtOAc (5 x 150 ml) . Die vereinigten organischen Phasen wurden über MgSÜ4 getrocknet, filtriert und einrotiert. Der ölige Rückstand wurde durch Sublimation im Hochvakuum (p = 0,1 Torr, Ölbadtemperatur 110°C) gereinigt. Man erhielt 2- (4- Chlor-2-nitrophenyl)ethanol (13,23 g, 55 %) in Form gelber Kristalle.
Rf (Si02, Toluol/EtOAc 10:1) 0,26
Schmelzpunkt: 61 bis 64°C
UV(MeOH) , λπax [nm] (log ε) : 213 (4,29) , 252 (3,61) , 294
(Schulter, 3,10) ---H-NMR (250 MHz, CDC13) : 7,94 (d, J = 2,2, H-C(3)) ; 7,53 (dd,
H-C(5)) ; 7,40 (d, H-C(6)) ; 3,93 (t, α-CH2) ; 3,14 (t, ß-CH2) ; 1,70 (S, OH) Anal. ber. für C8H8C1N03 (201,61) : C 47,66, H 4,00, N 6,95; gef. : C 47,69, H 4,01, N 6,76
Literatur:
[1] J. Bakke, Acta Chem. Scand. 1969, 23, 3055
[2] T. Morimoto, I. Hashimoto, H. Yamaoka, Chem. Abstr.
1978, 88, 104880 v [3] Y. Tsuji, S. Kotachi, K.-T. Huh, Y. Watanabe, J. Org.
Chem. 1990, 55, 580
b) 2- (4-Chlor-2-nitrophenyl) ethoxycarbonylchlorid
In eine Lösung von 2- (4-Chlor-2-nitrophenyl) ethanol (6,8 g, 34 mmol) in THF (50 ml, dest. über CaH2) wurde bei Raumtemperatur Phosgen eingeleitet. Nach 2,5 h wurde das überschüssige Phosgen sowie das Lösemittel im Hochvakuum abdestilliert. Man erhielt 2- (4-Chlor-2-nitrophenyl) - ethoxycarbonylchlorid (8,53 g, 95 %) als gelbes Öl.
Rf (Si02, CHCI3) 0,85
UV(CH3CN) , ^x [nm] (log ε) : 213 (4,26) ; 254 (3,63) ; 300
(Schulter, 3,14) ---H-NMR (250 MHz, CDCI3) : 8,03 (d, H-C(3)) ; 7,59 (dd, H-C(5)) ;
7,36 (d, H-C(6)) ; 4,62 (t, α-CH2) ; 3,32 (ß-CH2) Anal. ber. für C9H7C12N04 (264,06) : C 40,94, H 2,67, N 5,30; gef. : C 40,97, H 2,69, N 5,00
c) 5' -0- (2- (4-Chlor-2-nitrophenyl)ethoxycarbonyl) thymidin
Thymidin (150 mg, 0,62 mmol) wurde mit Pyridin (2 x 5 ml, pro analysi-Qualität, zusätzlich über Molekularsieb 4 Ä getrocknet) koevaporiert, in Pyridin gelöst und auf -30°C gekühlt. Hierzu tropfte man in 5 min eine Lösung von 2- (4- Chlor-2-nitrophenyl)ethoxycarbonylchlorid (255 mg, 0,97 mmol) in CH2C12 (3 ml, dest. über CaH2) . Nach 4 h Rühren unter i-PrOH/N2 Kühlung (-30 bis -15°C) wurde das Gemisch mit CH2C12 (30 ml) verdünnt und mit H20 (30 ml) gewaschen. Die wäßrige Phase wurde mit CH2C1 (2 x 30 ml) nachextrahiert. Die vereinigten organischen Phasen wurden über MgS04 getrocknet, filtriert und mit Toluol (4 x 10 ml) und CH2C12 (20 ml) koevaporiert. Das Rohprodukt wurde durch Säulenchromatographie (Si02 15 x 3 cm, Lösemittel: CH2Cl2/MeOH 100:7 700 ml) gereinigt. Man erhielt 5*-0-(4- Chlor-2-nitrophenyl)ethoxycarbonyl)thymidin (219 mg, 75 %) als farblosen Feststoff.
Rf (Si02, Toluol/EtOAc/MeOH 5:4:1) 0,36
UV(MeOH), ^x [nm] (log ε) : 212 (4,43), 261 (4,08), 310 (Schulter, 3,07)
---H-NMR (250 MHz, CDC13): 8,75 (s (br) , NH) ; 7,94 (d, H-C(3) v. CNPEOC) ; 7,52 (dd, H-C(5)); 7,31 (m, H-C(6) v. CNPEOC, H-C(6) von Thymin) ,* 6,32 (t, H-C(l')); 4,41 (m, H-C(3'), 2 x H-C(5'), α-CH2 v. CNPEOC); 4,12 (q, H-C(4')); 3,27 (t, ß-CH2 v. CNPEOC); 2,75 (s, (br) , OH-C.31)); 2,39 (m, H-C(2')), 2,19 (m, H-C(2*)); 1,86 (s, CH3)
Anal. ber. für C19H20C1N309 (469,83): C 48,57, H 4,29, N 8,94; gef.: C 48,87, H 4,56, N 8,64
Beispiel 7
a) 2- (5-Methoxy-2-nitrophenyl) ethanol [1, 2]
Ein Gemisch aus 5-Methoxy-2-nitrotoluol (25 g, 150 mmol) und Paraformaldehyd (2,3 g, 73 mmol) in DMSO (20 ml, Synthese- Qualität, zusätzlich 2 d über Molekularsieb 4 Ä getrocknet) wurde bei 80°C mit Triton B (2 ml, 35 % in MeOH) versetzt. Nach 2,5 h Rühren bei dieser Temperatur ließ man auf Raumtemperatur abkühlen und neutralisierte mit wenigen Tropfen konz. HCl. Das Gemisch wurde mit H20 (50 ml) verdünnt und mit EtOAc (5 x 100 ml) extrahiert. Die organischen Phasen wurden über MgSÜ4 getrocknet, filtriert und einrotiert. Der Rückstand wurde einer Destillation im Hochvakuum (p = 0,1 Torr) unterworfen, wobei bei 70QC eine Fraktion mit Edukt (12,1 g) erhalten wurde. Das restliche Rohprodukt wurde durch Säulenchromatographie (240 g Si02, Lösemittel: Toluol/EtOAc 8:1 1400 ml, 7:1 1600 ml, 6:1 400 ml, 5:1 400 ml, 3:1 400 ml, 2:1 600 ml und 1:1 600 ml) gereinigt. Man erhielt 2- (5- Methoxy-2-nitrophenyl)ethanol (6,87 g, 48 %) als gelbes Öl.
Rf (Si02, Toluol/EtOAc 1:1) 0,43
UV(MeOH) , λrηax [nm] (log ε) : 204 (4,00) , 231 (3,82) ; 301
(3,84) ---H-NMR (250 MHz, CDC13) : 8,04 (dd, H-C{4) ; 6,82 (m, H-C(3) ,
H-C(6)) ; 3,94 (q, α-CH2) ; 3,87 (s, OCH3 ) ; 3,20 (t, ß-CH2) ; 1,64 (s (br) , OH) Anal. ber. für C9H11N04 (197,19) : C 54,82, H 5,62, N 7,10; gef. : C 54,78, H 5,86, N 7,00
Literatur:
[1] T. Morimoto, I. Hashimoto, H. Yamaoka,
Chem. Abstr. 1978, 88, 104880 v [2] Y. Tsuji, S. Kotachi, K.-T. Huh, Y. Watanabe,
J. Org. Chem. 1990, 55, 580
b) 2- (5-Methoxy-2-nitrophenyl)ethoxycarbonylchlorid
In eine Lösung von 2- (5-Methoxy-2-nitrophenyl) ethanol (3,0 g, 15 mmol) in THF (40 ml, dest. über CaH2) wurde bei Raumtemperatur unter Rühren Phosgen eingeleitet. Nach 2,5 h wurden das überschüssige Phosgen sowie das Lösemittel im Hochvakuum abdestilliert. Man erhielt 2- (5-Methoxy-2- nitrophenyl)ethoxycarbonylchlorid (3,72 g, 96 %) als gelbes Öl.
Rf (Si02, CHC13) 0,79
UV(CH3CN) , λmax [nm] (log ε) : 222 (Schulter, 3,87), 230
(3,90) , 303 (3,87) ---H-NMR (250 MHz, CDCI3) : 8,12 (d, H-C(3)) ; 6,88 (dd, H-C(4)) ;
6,79 (d, J = 2,8, H-C(6)) , 4,63 (t, α-CH2) ; 3,89 (s,
OCH3) ; 3,35 (t, ß-CH2) Anal. ber. für C10H10ClNO5: C 46,26, H 3,88, -N 5,39; gef. : C
46,42, H 4,00, N 5,50
c) 5 ' -0- (2- (5-Methoxy-2-nitrophenyl) ethoxycarbonyl) thymidin
Thymidin (150 mg, 0,62 mmol) wurde mit Pyridin (2 x 5 ml, pro analysi-Qualität, zusätzlich über Molekularsieb 4 Ä getrocknet) koevaporiert, in Pyridin (5 ml, s.o.) gelöst und auf -30°C (i-PrOH/N2) gekühlt. Hierzu tropfte man in 5 min eine Lösung von 2- (5-Methoxy-2-nitrophenyl) - ethoxycarbonylchlorid (250 mg, 0,96 mmol) in CH2C12 (5 ml, dest. über CaH ) . Nach insgesamt 4 h Rühren unter i-PrOH/N Kühlung (-30 bis -15°C) wurde das Gemisch mit CH2C1 (30 ml) verdünnt und mit H20 (30 ml) gewaschen. Die wäßrige Phase wurde mit CH2C12 (2 x 30 ml) nachextrahiert. Trocknen der organischen Phasen über Na2Sθ4, Filtrieren, Abrotieren des Lösemittels und Koevaporieren mit Toluol (4 x 10 ml) und CH2C1 (10 ml) ergaben das Rohprodukt, welches durch
Säulenchromatographie (Si02, 16 x 3 cm, CH2Cl2/MeOH 100:6 800 ml) gereinigt wurde. Man erhielt 5 ' -0- (2- (5-Methoxy-2- nitrophenyl)ethoxycarbonyl) thymidin (205 mg, 71 %) als farblosen Feststoff.
Rf (Si02, Toluol/EtOAc/MeOH 5:4:1) 0,32
UV(MeOH) , ^x [nm] (log ε) : 205 (4,30) , 234 (Schulter,
3,94) , 270 (4,09), 302 (Schulter, 3,84) -•-H-NMR (250 MHz, CDC13) : 8,79 (s, NH) ; 8,06 (H-C(3) v.
MNPEOC) ; 7,34 (s, H-C(6) v. Thymin) ; 6,85 (dd, H-C(4) v.
MNPEOC) ; 6,77 (d, J = 2,7, H-C(6) v. MNPEOC) ; 6,32 (t,
H-C(l')) ; 4,43 (m, H-C(3') , 2 x H-C(5') , α-CH2 v.
MNPEOC) ; 4,12 (q, H-C(4')) ; 3,85 (ε, OCH3) ; 3,33 (m, ß-CH2 v. MNPEOC) ; 2,75 (d, 0H-C(3')) ; 2,38 (m, H-C(2')) ;
2,18 (m, H-C(2')); 1,83 (s, CH3) Anal. ber. für C20H23N3010 (465,42) : C 51,61, H 4,98, N 9,03; gef. : C 51,31, H 5,09, N 8,63
Beispiel 8
a) 2- (2, 4-Dichlor-6-nitrophenyl) ethanol
Zu einem Gemisch von 2,4-Dichlor-6-nitrotoluol (1,03 g,
5 mmol) und Paraformaldehyd (150 mg, 5 mmol) in DMSO (2,5 ml,
Synthese-Qualität, zusätzlich 2 d über Molekularsieb 4 Ä getrocknet) wurde eine Lösung von Kaliumtertiärbutylat
(90 mg, 0,8 mmol) in tert. -Butanol (1 ml, Synthese-Qualität,
99 %) gegeben. Nach der Zugabe der Kaliumtertiärbutylat-
Lösung trat ein Farbumschlag von gelb nach tief-violett ein.
Es wurde 5 min bei Raumtemperatur und 30 min bei 70 bis 80°C
(Ölbadtemperatur) gerührt. Danach ließ man auf Raumtemperatur abkühlen und neutralisierte mit wenigen Tropfen konz. HCl.
Das Gemisch wurde mit EtOAc (30 ml) verdünnt und mit H 0
(30 ml) gewaschen. Die wäßrige Phase wurde mit EtOAc
(2 x 30 ml) nachextrahiert. Trocknen der organischen Phasen
über Na2Sθ4, Filtrieren und Abrotieren des Lösemittels ergaben das Rohprodukt, welches durch Säulenchromatographie (20 g Si02, 12 x 2 cm, Lösemittel: PE 30 ml, PE/EtOAc 10:1 110 ml, 9:1 100 ml, 8:1 360 ml, 7:1 80 ml) gereinigt wurde. Man erhielt 2- (2,4-Dichlor-6-nitrophenyl)ethanol (769 mg, 65 %) als gelben Feststoff.
Rf (Si02, Toluol/EtOAc 9:1) 0,41
UV(MeOH), ^x [nm] (log ε) : 205 (4,32), 218 (4,20), 254
(Schulter, 3,40), 292 (3,08) ---H-NMR (250 MHz, CDC13): 7,73 (d, J = 2,0, 1 arom H) ; 7,65
(d, J = 2,0, 1 arom. H) ,* 3,92 (m, α-CH2) ; 3,26 (t, ß-CH2) ; 1,70 (S (br) , OH) Anal. ber. für C8H7C12N03 (236,054): C 40,71, H 2,99, N 5,93; gef.: C 40,36, H 2,96, N 5,85
b) 2- (2,4-Dichlor-6-nitrophenyl)ethoxycarbonylchlorid
Zu einer auf 0°C gekühlten Lösung von
Chlorameisensäuretrichlorτnethylester (503 mg, 2,5 mmol) in THF (5 ml, dest. über CaH ) wurde in 5 min eine Lösung von 2- (2,4-Dichlor-6-nitrophenyl)ethanol (500 mg, 2,12 mmol) und Et3N (214 mg, 2,12 mmol, dest. über KOH) in THF (5 ml, s.o.) zugegeben. Man ließ 1 h unter Eisbadkühlung und 2 h bei Raumtemperatur rühren. Das Gemisch wurde dann über Celite filtriert. Nachwaschen des Filterkuchens mit THF und Abdestillieren des Lösemittels und des überschüssigen Reagenses von den vereinigten Filtraten bei 30°C im Hochvakuum ergaben 2- (2,4-Dichlor-6-nitrophenyl) - ethoxycarbonylchlorid (597 mg, 94 %) als hellgelben Feststoff.
Rf (Si02, PE/EtOAc 19:1) 0,57
UV(MeOH), λmax [nm] (log ε) : 204 (4,35), 217 (4,25), 252 (Schulter, 3,47), 295 (3,10)
---H-NMR ( 250 MHz , CDC13 ) : 7 , 82 (d , J = 1 , 8 , 1 arom . H) ; 7 , 70 (d, J = 1 , 8 , 1 arom . H) ; 4 , 59 ( t , α- CH2 ) ; 3 , 42 ( t , ß- CH2 )
Anal. ber. für C9H6C13N0 (298,509) : C 36,21, H 2,03, N 4,69; gef. : C 36,37, H 2,30, N 4,60
c) 5 ' -0- (2- (2,4-Dichlor-6-nitrophenyl)ethoxycarbonyl) thymidin
Thymidin (200 mg, 0,83 mmol) wurde mit Pyridin (3 x 3 ml, pro analysi-Qualität, zusätzlich über Molekularsieb 4 Ä getrocknet) koevaporiert, in Pyridin (3 ml, s.o.) gelöst und auf -60°C (i-PrOH/N2) gekühlt. Hierzu tropfte man in 15 min eine Lösung von 2- (2, -Dichlor-6-nitrophenyl) - ethoxycarbonylchlorid (320 mg, 1,07 mmol) in CH2C12 (3 ml, dest. über CaH2) . Nach insgesamt 6 h Rühren unter i-PrOH/N Kühlung (-60 bis -15°C) wurde das Gemisch mit CH2C12 (10 ml) verdünnt und mit H 0 (10 ml) gewaschen. Die wäßrige Phase wurde mit CH C12 (2 x 10 ml) nachextrahiert. Trocknen der organischen Phasen über Na2S0 , Filtrieren, Abrotieren des Lösemittels und Koevaporieren mit Toluol (4 x 10 ml) ergaben das Rohprodukt (543 mg) , welches durch Säulenchromatographie (20 g Si02, 12,5 x 2,1 cm, Lösemittel: CH2Cl2/MeOH 100:1 50 ml, 100:2 204 ml, 100:3 360 ml) gereinigt wurde. Man eluierte zuerst 3 ' , 5 ' -Bis-O- (2- (2, 4-Dichlor-6- nitrophenyl) ethoxycarbonyl) thymidin, dann 3 ' -0- (2- (2, 4- Dichlor-6-nitrophenyl)ethoxycarbonyl) hymidin und 5'-0-(2- (2, 4-Dichlor-6-nitrophenyl)ethoxycarbonyl) thymidin. Die Produktfraktionen wurden einrotiert und im Hochvakuum getrocknet. Man erhielt 3 ' , 5 ' -Bis-O- (2- (2, 4-Dichlor-6- nitrophenyl) ethoxycarbonyl) thymidin (53 mg, 8 %) als hellgelben Schaum sowie 3 ' -0- (2- (2,4-Dichlor-e- nitrophenyl) ethoxycarbonyl) thymidin (14 mg, 3 %) und 5'-0-(2- (2, 4-Dichlor-6-nitrophenyl) ethoxycarbonyl) thymidin (339 mg, 81 %) jeweils als farblosen Schaum.
5 ' -0- (2- (2, 4-Dichlor-6-nitrophenyl)ethoxycarbonyl) thymidin:
Rf (Si02, Toluol/EtOAc/MeOH 5:4:1) 0,40
UV(MeOH) , λmax [nm] (log ε) : 203 (4,51), 214 (Schulter,
4,38), 263 (4,02), 304 (Schulter, 3,06) ---H-NMR (250 MHz, CDC13) : 8,91 (s, NH) ; 7,75 (d, J = 2,1, 1 arom. H v. DCNPEOC) ; 7,68 (d, J = 2,1, 1 arom. H v.
DCNPEOC) ; 7,37 (s, H-C(6) v. Thymin) ; 6,37 (t, H-C(l')) ;
4,63 (m, H-C(3') , 2 x H-C(5 ) , α-CH2 v. DC1NPEOC) ; 4,16
(m, H-C(4')) ; 3,40 (t, ß-CH2 v. DC1NPEOC) ; 2,86 (d,
OH-C(3')) ; 2,42 (m, H-C(2')) ; 2,24 (m, H-C(2')) ; 1,89
(s, CH3) Anal. ber. für C19H19C12N309 (504,279) : C 45,25, H 3,80, N
8,33, gef. : C 45,02, H 3,89, N 8,04
Beispiel 9
a) 2- (4, 5-Dimethoxy-2-nitrophenyl) ethanol [1]
Zu einer auf -10°C (Viehsalz/Eis) gekühlten Mischung aus Homoveratrylalkohol (3,02 g, 16,6 mmol) in Eisessig (30 ml) wurde in 6 min unter Rühren konz. HNO3 (4,8 ml, 65 %, d = 1,4) zugetropft. Anschließend ließ man in 30 min auf 23°C erwärmen. Nach 1 h Rühren bei dieser Temperatur wurde das Gemisch mit H20 (30 ml) verdünnt, mit NaHCÜ3 neutralisiert und mit EtOAc (3 x 30 ml) extrahiert. Die vereinigten organischen Phasen wurden über Na2Sθ4 getrocknet, filtriert und einrotiert. Der Rückstand (3,25 g) wurde durch Säulenchromatographie (90 g Si02 Toluol/EtOAc 4:1 500 ml, Toluol/EtOAc 3:1 400 ml, Toluol/EtOAc 2:1 300 ml) gereinigt, Man erhielt 2- (4, 5-Dimethoxy-2-nitrophenyl) ethanol (2,13 g, 56 %) als gelben Feststoff.
Rf (Si02, Toluol/EtOAc 2:1) 0,24
UV(MeOH) , λroax [nm] (log ε) : 202 (4,18) , 216 (4,06), 242 (3,97), 297 (3,63), 340 (3,70)
---H-NMR-Spektrum (250 MHz, CDC13) : 7,61 (s, H-C(3)) ; 6,80 (s, H-C(6)) ; 3,97 (s, OCH3) ; 3,96 (m, α-CH2, versteckt) ; 3,95 (s, OCH3) ; 3,21 (t, ß-CH2) , 1,70 (s (br) , OH)
Anal. ber. für C10H13NO5 (227.216) : C 52,86, H 5,77, N 6,16; gef. : C 52,87, H 5,82, N 6,12
Literatur
[1] E. McDonald, R. D. Wylie, Tetrahedron 1979, 35, 1415
b) 2- (4, 5-Dimethoxy-2-nitrophenyl) ethoxycarbonylchlorid
Chlorameisensäuretrichlormethylester (0,26 ml, 2,2 mmol) in THF (5 ml, dest. über CaH2) wurde mittels eines Eisbades auf 0°C gekühlt. Hierzu tropfte man in 10 min eine Lösung von 2- (4, 5-Dimethoxy-2-nitrophenyl) ethanol (500 mg, 2,2 mmol) und Et3N (218 mg, 0,3 ml, 2,2 mmol, dest. über CaH2) in THF (15 ml, dest. über CaH2) . Anschließend entfernte man das Eisbad und ließ bei Raumtemperatur weiterrühren. Nach 30 min Rühren versetzte man die Reaktionsmischung mit einer Spatelspitze Aktivkohle. Das Reaktionsgemisch wurde nach weiteren 2,5 h Rühren bei Raumtemperatur über Celite abgesaugt. Man destillierte das Lösemittel sowie überschüssiges Reagens im Hochvakuum vom Filtrat ab und erhielt 2- (4, 5-Dimethoxy-2-nitrophenyl) ethoxycarbonylchlorid (624 mg, 98 %) als gelbbraunen Feststoff.
Rf (Si02, Toluol/EtOAc 2:1) 0,77
UV(MeOH) , λmax [nm] (log ε) . 203 (4,18), 217 (4,08), 242
(4,01) , 297 (3,67) , 339 (3,73) ---H-NMR (250 MHz, CDCI3) : 7,67 (ε, H-C(3)) ; 6,74 (s, H-C(6)) ;
4,66 (t, α-CH2) ; 3,99 (s, OCH3) ; 3,96 (s, OCH3) ; 3,36
(t, ß-CH2) Anal. ber. für C11H12N06C1 (289,671) : C 45,61, H 4,18, N
4,84; gef. : C 45,59, H 4,26, N 4,94
c) 5 ' -0- (2- (4, 5-Dimethoxy-2-nitrophenyl)ethoxycarbonyl) - thymidin
Thymidin (200 mg, 0,83 mmol) wurde mit Pyridin (2 x 2 ml, pro analysi-Qualität, zusätzlich über Molekularsieb 4 Ä getrocknet) koevaporiert, in Pyridin (2,4 ml, s.o.) gelöst und auf -60°C (i-PrOH/N2) gekühlt. Hierzu tropfte man in 15 min eine Lösung von 2- (4, 5-Dimethoxy-2-nitrophenyl) - ethoxycarbonylchlorid (361 mg, 1,25 mmol) in CH2C12 (2,4 ml, dest. über CaH2) . Nach insgesamt 6,5 h Rühren unter i-PrOH/N2 Kühlung (-40 bis -15°C) wurde das Gemisch mit CH2C12 (10 ml) verdünnt und mit H20 (10 ml) gewaschen. Die wäßrige Phase wurde mit CH2C12 (2 x 10 ml) nachextrahiert. Trocknen der organischen Phasen über Na2SÜ4, Filtrieren, Abrotieren des Lösemittels und Koevaporieren mit Toluol (4 x 20 ml) ergaben das Rohprodukt (530 mg) , welches durch Säulenchromatographie (20 g Si02, CH2Cl2/MeOH 100:3 309 ml, 100:4 104 ml, 100:5 160 ml) gereinigt wurde. Man eluierte zuerst 3 ' , 5' -Bis-O- (2- (4, 5-Dimethoxy-2-nitrophenyl)ethoxycarbonyl)thymidin und dann 5' -0- (2- (4,5-Dimethoxy-2-nitrophenyl)ethoxycarbonyl)thymidin. Die Produktfraktionen wurden einrotiert und im Hochvakuum getrocknet. Man erhielt 3 ' ,5' -Bis-O- (2- (4,5-Dimethoxy-2- nitrophenyl)ethoxycarbonyl)thymidin (154 mg, 25 %) und 5'-0- (2- (4,5-Dimethoxy-2-nitrophenyl)ethoxycarbonyl)thymidin (272 mg, 66 %) jeweils als hellgelbe Feststoffe.
5' -0- (2- (4,5-Dimethoxy-2-nitrophenyl)ethoxycarbonyl) - thymidin:
Rf (Si02, Toluol/EtOAc/MeOH 5:4:1) 0,23
UV(MeOH), λmax [nm] (log ε) : 203 (4,33), 212 (Schulter,
4,30), 247 (4,13), 267 (4,03), 343 (3,69) ---H-NMR (250 MHz, CDC13): 9,10 (s, NH) ; 7,62 (S, H-C(3)); 7,37
(d, J = 1,2, H-C(6) v. Thymin) ,* 6,73 (s, H-C(6)); 6,35
(t, H-C(l')); 4,44 (m, H-C(3'), 2 x H-C(5'), α-CH2 v.
DMNPEOC) ; 4,15 (m, H-C.4')); 3,96 (ε, OCH3); 3,94 (s,
OCH3) ; 3,33 (dt, ß-CH2 v. DMNPEOC) ; 3,06 (s (br) , OH-C.3')) ; 2,42 (m, H-C(2')) ; 2,20 (m, H-C(2')) ; 1,88 (d, J = 0,9, CH3) Anal. ber. für C21H25N3011 (495,441) : C 50,91, H 5,09, N 8,48, gef. : C 50,94, H 5,09, N 8,32
Beispiel 10
a) 2- (2-Nitrophenyl)propanol [1, 2]
Zu 2-Nitroethylbenzol (3,02 g, 20 mmol) und Paraformaldehyd (600 mg, 20 mmol) in DMSO (10 ml, Synthese-Qualität, zusätzlich 2 d über Molekularsieb 4 Ä getrocknet) wurde eine Lösung von Kaliumtertiärbutylat (360 mg, 3,2 mmol) in tert. - Butanol (4 ml, dest. über CaH2) gegeben. Es wurde 15 min bei Raumtemperatur und 1 h 45 min bei 70°C (Ölbadtemperatur) gerührt. Danach ließ man auf Raumtemperatur abkühlen und neutralisierte mit wenigen Tropfen konz. HCl. Das Gemisch wurde mit EtOAc (100 ml) verdünnt und mit gesättigter NaCl- Lsg. (60 ml) gewaschen. Die wäßrige Phase wurde mit EtOAc (2 x 100 ml) nachextrahiert. Trocknen der organischen Phasen über a Sθ4, Filtrieren und Abrotieren des Lösemittels ergaben das Rohprodukt (5,06 g) , welches durch Säulenchromatographie (80 g Si02, 19 x 3,4 cm, Lösemittel: Toluol 150 ml, Toluol/EtOAc 8:1 270 ml, 7:1 240 ml, 6:1 280 ml, 5:1 180 ml) gereinigt wurde. Man erhielt 2- (2- Nitrophenyl)propanol (2,539 g, 70 %) als hellgelbes Öl.
Rf (Si02, Toluol/EtOAc 9:1) 0,25
UV(MeOH) , λ^ax [nm] (log ε) : 206 (4,08), 220 (Schulter,
3,75) , 254 (3,53), 285 (Schulter, 3,27) ---H-NMR (250 MHz, CDCI3) : 7,75 (dd, J = 1,2, 8,1, 1 arom. H, ortho zu N02) ; 7,55 (m, 2 arom. H) ; 7,36 (m, 1 arom. H) ;
3,80 (m, α-CH2) ; 3,52 (sextett, ß-CH) ; 1,67 (ε (br) ,
OH) ; 1,33 (d, J = 6,9, CH3)
Anal. ber. für C9H1:LNθ3 (181,191) : C 59,66, H 6,12, N 7,73; gef. : C 59,55, H 6,12, N 7,90
Literatur:
[1] J. Org. Chem. 1986, 3143
[2] Chem. Abstr. 1989, 110, P 75032 k.
b) 2- (2-NitrophenylJpropoxycarbonylchlorid
Zu einer auf 0°C gekühlten Lösung von
Chlorameisensäuretrichlormethylester (655 mg, 3,3 mmol) in THF (6,75 ml, dest. über CaH2) wurde in 5 min eine Lösung von 2- (2-Nitrophenyl)propanol (500 mg, 2,76 mmol) und Et3 (279 mg, 0,385 ml, 2,76 mmol, dest. über KOH) in THF (6,75 ml, s.o.) zugegeben. Man ließ 1 h unter Eisbadkühlung und 1 h bei Raumtemperatur rühren. Das Gemisch wurde über Celite filtriert. Nachwaschen des Filterkuchens mit THF und .Abdestillieren des Lösemittels sowie des überschüssigen Reagenses von den vereinigten Filtraten bei 30°C im Hochvakuum ergaben 2- (2-Nitrophenyl)propoxycarbonylchlorid (644 mg, 96 %) als hellbraunes Öl.
Rf (Si02, PE/EtOAc 19:1) 0,24
UV(MeOH) , λrnax [nm] (log ε) : 205 (4,07); 218 (Schulter,
3,75), 251 (3,59) ---H-NMR (250 MHz, CDC13): 7,84 (dd, J = 1,2, 8,0, 1 arom. H, ortho zu N02) ; 7,62 (m, 1 arom. H) ; 7,44 (m, 2 arom. H) ;
4,50 (d, J = 6,3, α-CH2) ; 3,80 (sextett ß-CH2) ; 1,42 (d,
J = 7,0, CH3) Anal. ber. für C10H10ClNO4 (243,646) : C 49,30, H 4,14, N
5,75; gef.: C 49,71, H 4,32, N 5,70
c) 5' -0- (2- (2-Nitrophenyl)propoxycarbonyl)thymidin
Thymidin (1 g, 4,1 mmol) wurde mit Pyridin (3 x 15 ml, pro analysi-Qualität, zusätzlich über Molekularsieb 4 Ä getrocknet) koevaporiert, in Pyridin (15 ml, s.o.) gelöst und auf -60°C (i-PrOH/N2) gekühlt. Hierzu tropfte man in 30 min eine Lösung von 2- (2-Nitrophenyl)propoxycarbonylchlorid (1,31 g, 5,38 mmol) in CH2C12 (18 ml, dest. über CaH2) . Man ließ weitere 6 h unter i-PrOH/N2 Kühlung (-60 bis -20°C) rühren. Die Reaktionsmischung wurde mit CH2C1 (50 ml) verdünnt und mit H20 (50 ml) gewaschen. Die wäßrige Phase wurde mit CH2C12 (3 x 50 ml) nachextrahiert. Trocknen der organischen Phasen über a2S04, Filtrieren, Abrotieren des Lösemittels und Koevaporieren mit Toluol (4 x 15 ml) ergaben das Rohprodukt (2,22 g) , welches durch Säulenchromatographie (80 g Si02, 19 x 3,4 cm, Lösemittel: CH2Cl2/MeOH 100:1 101 ml, 100:2 204 ml, 100:3 206 ml, 100:3,5 207 ml, 100:4 520 ml, 100:5 53 ml, 100:6 53 ml) gereinigt wurde. Man eluierte zuerst 3',5' -Bis-O- (2- (2-
NitrophenyDpropoxycarbonyl)thymidin, dann 3'-0-(2-(2- NitrophenyDpropoxycarbonyl)thymidin und zuletzt 5'-0-(2-(2- Nitrophenyl)propoxycarbonyl)thymidin. Nach Einrotieren der Produktfraktionen und Trocknen im Hochvakuum erhielt man 3 ' , 5 ' -Bis-O- (2- (2-Nitrophenyl)propoxycarbonyl)thymidin (145 mg, 5 %) als hellgelben Schaum sowie 3*-0-(2-(2- Nitrophenyl)propoxycarbonyl)thymidin (71 mg, 4 %) und 5 ' -0- (2- (2-Nitrophenyl)propoxycarbonyl)thymidin (1,307 g, 71 %) jeweils als farblosen Schaum.
5' -0- (2- (2-Nitrophenyl)propoxycarbonyl) hymidin: Rf (Si02, Toluol/EtOAc/MeOH 5:4:1) 0,43 UV(MeOH) , ^ax t1™] (log ε) : 207 (4,30), 263 (4,07) ---H-NMR (250 MHz, CDC13) : 8,66 (s, NH, Diaεtereomer) ; 8,64 (ε, NH, Diaεtereomer); 7,77 (m, 1 arom. H v. NPPOC) ; 7,59 (t, 1 arom. H v. NPPOC); 7,43 (m, 2 arom. H v. NPPOC); 7,33 (s, H-C(6) v. Thyτnin, Diaεtereomer); 7,30 (s,
H-C(6) v. Thymin, Diastereomer) , 6,34 (t, H-C(l'), Diastereomer) ; 6,32 (t, H-C(l'), Diastereomer) ; 4,29 (m, H-C(3') , H-C(4'), 2 X H-C(5'), α-CH2 v. NPPOC) ; 3,80 (m, ß-CH V. NPPOC) ; 2,62 (d, J = 4,2, OH-C.31), Diastereomer) ; 2,60 (d, J = 4,4, OH-C(3') , Diastereomer) ; 2,39 (m, H-C(2')) ; 2,18 (m, H-C(2')) ; 1,86 (s, CH3 v. Thymin, Diastereomer) ; 1,75 (s, CH3 v. Thymin, Diastereomer) ; 1,38 (d, J = 7,0, CH3 v. NPPOC, Diastereomer) ; 1,37 (d, J = 7,0, CH3 v. NPPOC, Diastereomer) Anal. ber. für C20H23N3O9 (449,416) : C 53,45, H 5,16, N 9,35; gef. : C 53,14, H 5,21, N 9,16
Beispiel 11
a) 2-Chlor-2- (2-nitrophenyl)ethanol
Eine Lösung von 2-NitrobenzylChlorid (4,3 g, 25 mmol) in DMSO (3,5 ml, Synthese-Qualität, zusätzlich 2 d über Molekularsieb 4 Ä getrocknet) wurde mit Paraformaldehyd (750 mg, 25 mmol) und DBU (0,5 ml, 3,3 mmol) versetzt und bei Raumtemperatur 20 min gerührt. Der pH-Wert der Reaktionsmischung wurde mit wenigen Tropfen konz. AcOH auf etwa pH 3 eingestellt. Das Gemisch wurde mit CH2C1 (120 ml) verdünnt, mit H20 gewaschen (80 ml) und mit CH2C12 (2 x 100 ml) nachextrahiert. Die organischen Phasen wurden über a2Sθ4 getrocknet, filtriert und einrotiert. Das Rohprodukt (5,543 g) wurde durch Säulenchromatographie (150 g Si02, 22 x 4,1 cm, Lösemittel: Toluol 150 ml, Toluol/EtOAc 25:1 260 ml, 15:1 320 ml, 10:1 330 ml, 5:1 720 ml, 4:1 250 ml) gereinigt. Man eluierte zuerst das Edukt (2,587 g, 60 %) , dann eine Mischfraktion (267 mg) und zuletzt 2-Chlor-2- (2-nitrophenyl)ethanol (1,178 g, 23 % bzw. 61 % bezüglich des umgesetzten Edukts) . Die erhaltene Mischfraktion (267 mg) wurde durch eine weitere Säulenchromatographie (8 g Si02, 15 x 1,2 cm,- Lösemittel:
Toluol 50 ml, Toluol/EtOAc 100:1 50 ml) gereinigt. Es wurde zuerst weiteres Edukt (44 mg, 1 %) und dann 2-Nitrostyrolepoxid (68 mg, 2 % bzw. 4 % bzgl . deε umgesetzten Edukts) eluiert. Die Gesamtmenge an zurückisoliertem Edukt betrug 2,631 g (61 %) .
2-Chlor-2- (2-nitrophenyl) ethanol (gelber Feststoff) :
Rf (Si02, CH2C12) 0,23
Schmelzpunkt: 49 bis 50°C
UV(MeOH), λmax [nm] (log ε) : 209 (4,11), 254 (3,64) , 332
(Schulter, 2,70) ---H-NMR (250 MHz, CDC13) : 7,94 (dd, J = 1,3, 8,3, 1 arom. H) ;
7,89 (dd, J = 1,3, 7,9, 1 arom. H) ; 7,69 (m, 1 arom. H) ;
7,51 (m, 1 arom. H) ; 5,73 (dd, J = 4,6, 6,8, ß-CH) ; 4,11 (dd, J = 4,6, 12,1, α-CH) ; 4,00 (dd, J = 6,8, 12,0, -CH) ; 2,14 (s, OH) -•-H-NMR (250 MHz, DMSO-dg) : 7,94 (dd, J = 1,2, 8,1, 1 arom.
H) ; 7,86 (dd, J = 1,5, 7,9, 1 arom. H) ; 7,77 (dt, J =
1,2, 7,4, 1 arom. H) ; 7,60 (dt, J = 1,6, 8,3, 1 arom.
H) ,- 5,43 (t, J = 6,4, ß-CH; s (br) , teilweise versteckt,
OH), 3,86 (d, J = 6,4, α-CH2) Anal. ber. für C8H8C1N03 (201,609) : C 47,66, H 4,00, N 6,95; gef. : C 48,01, H 4,11, N 7,00
b) 2-Chlor-2- (2-nitrophenyl) ethoxycarbonylchlorid
Eine Lösung von Diphosgen (1,7 ml, 14 mmol) in THF (10 ml, dest. über CaH2) wurde mittels eines Eisbades auf etwa 0°C gekühlt. Hierzu wurde eine Lösung von 2-Chlor-2- (2- nitrophenyl) ethanol (705 mg, 3,5 mmol) und Et3 (354 mg, 0,485 ml, 3,5 mmol) in THF (10 ml, deεt . über CaH2) in ca. 30 min gegeben. Nach einer weiteren Stunde Rühren wurde das Eisbad entfernt. Man ließ noch 3 h bei Raumtemperatur rühren. Das Gemisch wurde über Celite abfiltriert. Der Niederschlag wurde mit THF (10 ml, dest. über CaH2) nachgewaschen. Die
vereinigten Filtrate wurden einrotiert und anschließend im Hochvakuum bei Raumtemperatur getrocknet. Man erhielt 2-Chlor-2- (2-nitrophenyl)ethoxycarbonylchlorid (896 mg, 97 %) als hellbraunes Öl.
Rf (Si02, CH2C12) 0,82
UV(MeOH), λmax [nm] (log ε) : 208 (4,11); 253 (3,66)
---H-NMR (250 MHz, CDCI3): 8,01 (dd, J = 1,1, 8,1, 1 arom. H) ; 7,93 (dd, J = 1,1, 7,9, 1 arom. H) ; 7,74 (dt, J = 1,1, 8,2, 1 arom. H) ; 7,57 (m, 1 arom. H) ; 5,89 (dd, J = 4,9, 6,4, ß-CH); 4,80 (dd, J = 6,5, 11,4, α-CH); 4,73 (dd, J = 4,9, 11,5, α-CH)
Anal. ber. für C9H7C12N04 (264,064): C 40,94, H 2,67, N 5,30; gef.: C 41,39, H 2,89, N 5,38
c) 5' -0- (2-Chlor-2- (2-nitrophenyl)ethoxycarbonyl)thymidin
Thymidin (250 mg, 1,03 mmol) wurde mit Pyridin (2 x 3 ml, pro analysi-Qualität, zusätzlich über Molekularsieb 4 Ä getrocknet) koevaporiert, in Pyridin (3,5 ml, s.o.) gelöst und auf ca. -40°C (i-PrOH/N2) gekühlt. Hierzu wurde in ca. 30 min eine Lösung von 2-Chlor-2- (2-nitrophenyl) - ethoxycarbonylchlorid (356 mg, 1,35 mmol) in CH2C12 (3,5 ml, dest. über CaH ) gegeben. Man ließ weitere 4 h 45 min unter i-PrOH/N2 Kühlung (-40 bis -10°C) rühren. Die Reaktionsmischung wurde mit H20 (10 ml) verdünnt und mit CH2C12 (4 x 10 ml) extrahiert. Die organischen Phasen wurden über Na2Sθ4 getrocknet, abfiltriert, einrotiert und mit Toluol (4 x 10 ml) koevaporiert. Das Rohprodukt (665 mg) wurde durch Säulenchromatographie (20 g Si02, 12 x 2,1 cm, Lösemittel: CH2C12 100 ml, CH2Cl2/Me0H 100:1 101 ml, 100:2 102 ml, 100:2,5 102 ml, 100:3 206 ml, 100:4 104 ml, 100:5 105 ml) gereinigt. Man eluierte zuerst Bis- (2-Chlor-2- (2- nitro-phenyl)ethyl)carbonat (60 mg, 10 %) als gelbes Öl, anschließend 3 ' ,5' -Bis-O- (2-Chlor-2- (2-
nitrophenyl) ethoxycarbonyl) thymidin (48 mg) als hellgelben Schaum, dann 3 ' -0- (2-Chlor-2- (2-nitrophenyl) - ethoxycarbonyl) thymidin (16 mg, 3 %) als farblosen Schaum und zuletzt 5 ' -0- (2-Chlor-2- (2-nitrophenyl) ethoxycarbonyl) - thymidin (394 mg, 81 %) als farblosen Schaum.
5'-0- (2-Chlor-2- (2-nitrophenyl) ethoxycarbonyl) thymidin: Rf (Si02, Toluol/EtOAc/MeOH 5:4:1) 0,34 UV(MeOH) , λmax [nm] (log ε) : 209 (4,34), 262 (4,10) ---H-NMR (250 MHz, CDC13) : 8,37 (s, (br) , NH) ; 7,97 (d, J = 8,1, 1 arom. H v. CNPEOC) ; 7,91 (dd, J = 1,0, 7,9, 1 arom. H v. CNPEOC) ; 7,72 (t, J = 7,3, 1 arom. H v. CNPEOC) ; 7,55 (m, 1 arom. H v. CNPEOC); 7,34 (m, H-C(6) v. Thymin) , 6,35 (dd, J = 4,0, 6,7, H-C(l'), Diastereomer) ; 6,32 (dd, J = 3,9, 6,5, H-C(l'), Diastereomer) ; 5,89 (m, ß-CH v. CNPEOC); 4,57 (m, α-CH2 v. CNPEOC) , H-C(3') , 2 X H-C(5')); 4,15 (q, J = 3,2, H-C(4-)) ; 2,41 (m, H-C(2-)) ; 2,22 (m, H-C(2-)) ; 1,90 (s, CH3 v. Thymin, Diastereomer); 1,86 (s, CH3 v. Thymin, Diastereomer) ; 1,61 (s (br) , OH-C(3')) Anal. ber. für C19H20C1N309 (469,834) : C 48,57, H 4,29, N 8,94; gef.: C 48,17, H 4,40, N 8,47
Beispiel 12
a) 2-Methoxy-2- (2-nitrophenyl) ethanol
Zu einer Lösung von 2-Nitrobenzylmethylether (4,18 g, 25 mmol) in DMSO (3,5 ml, Synthese-Qualität, zusätzlich 2 d über Molekularsieb 4 Ä getrocknet) gab man Paraformaldehyd (750 mg, 25 mmol) und DBU (1,85 ml, 12,4 mmol) und ließ bei Raumtemperatur 4 h reagieren. Der pH-Wert wurde mit wenigen Tropfen konz. AcOH von pH 9 auf etwa pH 3, 5 eingestellt. Das Gemisch wurde mit CH2C12 (120 ml) verdünnt und mit H20 (80 ml) gewaschen. Die wäßrige Phase wurde mit CH C12
(2 x 80 ml) nachextrahiert. Die vereinigten organischen Phasen wurden über Na2Sθ4 getrocknet, filtriert und einrotiert. Das Rohprodukt (5,56 g) wurde durch Säulenchromatographie (140 g Si02, 21 x 4,2 cm, Lösemittel: Toluol 300 ml, Toluol/EtOAc 20:1 210 ml, 15:1 160 ml, 10:1 220 ml, 15:2 170 ml, 6:1 210 ml, 5:1 180 ml, 4:1 400 ml, 3:1 240 ml, 2:1 240 ml) gereinigt. Zuerst wurde 2-Nitrobenzylmethylether (2,442 g, 58 %) eluiert und dann 2-Methoxy-2- (2-nitrophenyl)ethanol (1,696 g, 34 % bzw. 82 % bezüglich des umgesetzten Edukts) .
2-Methoxy-2- (2-nitrophenyl)ethanol (gelber Feststoff):
Rf (Si02, CH2Cl2/MeOH 100:3) 0,31
Schmelzpunkt: 61 bis 63°C
UV(MeOH), ax [nm] (log ε) : 206 (4,09), 256 (3,66)
---H-NMR (250 MHz, CDC13): 8,00 (m, 1 arom. H) ,* 7,70 (m, 2 arom. H) ; 7,48 (m, 1 arom. H) ; 4,95 (dd, J = 3,2, 7,2, ß-CH); 3,94 (ddd, J = 3,2, 8,8, 11,8, α-CH); 3,67 (ddd, J = 4,4, 7,2, 11,7, α-CH); 3,31 (s, OCH3); 2,29 (dd, J 4,4, 8,8, OH)
Anal. ber. für C9H11N0 (197,19): C 54,82, H 5,62, N 7,10; gef.: C 55,14, H 5,57, N 7,15
b) 2-Methoxy-2- (2-nitrophenyl)ethoxycarbonylchlorid
Zu einer auf 0°C gekühlten Lösung von Diphosgen (1,2 ml, 10 mmol) in THF (15 ml, dest. über CaH2) wurde in 1 h eine Lösung von 2-Methoxy-2- (2-nitrophenyl)ethanol (986 mg, 5 mmol) und Et3N (506 mg, 0,693 ml, 5 mmol) in THF (10 ml, dest. über CaH2) gegeben. Nach weiteren 15 min Rühren bei 0°C wurde das Eisbad entfernt. Man ließ noch 1 h 30 min bei Raumtemperatur rühren. Das Gemisch wurde über Celite abfiltriert und der Niederschlag wurde mit THF (30 ml, dest. über CaH2) nachgewaschen. Die vereinigten Filtrate wurden einrotiert und im Hochvakuum bei Raumtemperatur getrocknet.
Man erhielt 2 -Methoxy- 2 - ( 2 -nitrophenyl ) ethoxycarbonylchlorid ( 1 , 264 g , 97 % ) al s hellbraunes Öl .
Rf (Si02, CH2C12) 0,73
UV(MeOH) , λmax [nm] (log ε) : 205 (4,12) , 253 (3,71), 348 (Schulter, 2,74)
---H-NMR (250 MHz, CDC13) : 8,07 (dd, J = 1,1, 8,2, 1 arom. H) ; 7,81 (dd, J = 1,6, 7,8, 1 arom. H) ; 7,73 (dt, J = 1,1, 7,6, 1 arom. H) ; 7,54 (m, 1 arom. H) ; 5,13 (dd, J = 3,2, 6,3, ß-CH) ; 4,60 (dd, J = 3,2, 11,2, α-CH) ; 4,54 (dd, J = 6,4, 11,3, α-CH)
Anal. ber. für C10H10ClNO5 (259,645) : C 46,26, H 3,88, N 5,39; gef. : C 46,74, H 3,88, N 5,40
c) 5 ' -O- (2-Methoxy-2- (2-nitrophenyl) ethoxycarbonyl) - thymidin
Thymidin (250 mg, 1,03 mmol) wurde mit Pyridin (2 x 3 ml, pro analysi-Qualität, zusätzlich über Molekularsieb 4 Ä getrocknet) koevaporiert, in Pyridin (3,5 ml, s.o.) gelöst und auf ca. -50°C (i-PrOH/N2) gekühlt. Hierzu wurde in ca. 1,5 h eine Lösung von 2-Methoxy-2- (2- nitrophenyl) ethoxycarbonylchlorid (348 mg, 1,34 mmol) in CH2C12 (3,5 ml, dest. über CaH2) gegeben. Man ließ weitere 3,5 h unter i-PrOH/N Kühlung (-40 bis -10°C) rühren. Die Reaktionsmischung wurde mit H 0 (10 ml) verdünnt und mit CH C12 (3 x 10 ml) extrahiert. Die organischen Phasen wurden über Na2SÜ4 getrocknet, abfiltriert, einrotiert und mit Toluol (4 x 10 ml) koevaporiert. Das Rohprodukt (578 mg) wurde durch Säulenchromatographie (20 g Si02, 12 x 2,1 cm, Lösemittel: CH2C12 100 ml, CH2Cl2/Me0H 100:1 202 ml, 100:2 102 ml, 100:3 103 ml, 100:4 104 ml, 100:5 105 ml, 100:6 106 ml) gereinigt. Man eluierte zuerst Bis (2-Methoxy-2- (2- nitrophenyl) ethyl) carbonat (88 mg, 16 %) alε hellgelben Feststoff und dann eine Mischfraktion von 3 ' -0- (2-Methoxy-2-
(2-nitrophenyl)ethoxycarbonyl)thymidin und 5' -0- (2-Methoxy-2- (2-nitrophenyl)ethoxycarbonyl)thymidin (323 mg) . Die Mischfraktion wurde durch erneute Säulenchromatographie (7 g Si02, 13 x 1,2 cm, Lösemittel: CH2C12 30 ml, CH2Cl2/MeOH 100:1 50 ml, 100:2 51 ml, 100:2,5 51 ml, 100:3 51 ml, 100:4 52 ml, 100:5 52 ml) gereinigt. Es wurde zuerst 3'-0-(2- Methoxy-2- (2-nitrophenyl)ethoxycarbonyl)thymidin (11 mg, 2 %) und dann 5' -0- (2-Methoxy-2- (2-nitrophenyl)ethoxycarbonyl) - thymidin (290 mg, 60 %) jeweils als farbloser Schaum eluiert.
5• -0- (2-Methoxy-2- (2-nitrophenyl)ethoxycarbonyl)thymidin:
Rf (Si02, Toluol/EtOAc/MeOH 5:4:1) 0,40
UV(MeOH), λmax [nm] (log ε) : 207 (4,33), 263 (4,12)
---H-NMR (250 MHz, CDC13): 8,67 (s (br) , NH) ; 8,03 (d, J = 5,0,
1 arom. H v. MNPEOC); 7,78 (m, 1 arom. H v. MNPEOC);
7,70 (t, J = 7,5, 1 arom. H v. MNPEOC); 7,52 (m, 1 arom.
H v. MNPEOC); 7,43 ( , H-C(6) v. Thymin); 6,38 (dd, J =
3,8, 6,7, H-C(l-), Diastereomer); 6,35 (dd, J = 3,7,
6,4, H-C(l'), Diastereomer); 5,14 (m, ß-CH v. MNPEOC);
4,42 (m, α-CH2 V. MNPEOC, H-C(3*), 2 X H-C(5*)); 4,15
(q, J = 3,2, H-C(4-)); 3,28 (s, OCH3, Diastereomer);
3,27 (ε, OCH3, Diastereomer); 2,64 (s (br) , OH-C(3'));
2,42 (m, H-C.2')); 2,23 (m, H-C(2')); 1,94 (s, CH3 v.
Thymin, Diastereomer); 1,92 (s, CH3 v. Thymin,
Diastereomer) Anal. ber. für C2oH23N3010 (465,415) : C 51,51, H 4,98, N
9,03; gef.: C 51,65, H 5,18, N 8,94
Beispiel 13
5' -0- (2- (2-Chlor-6-nitrophenyl)ethoxycarbonyl) -N4- (2- (4- nitrophenyl)ethoxycarbonyl) -2' -desoxycytidin
N4- (2- (4-Nitrophenyl)ethoxycarbonyl) -2' -desoxycytidin (5 g, 11,9 mmol) wurde mit Pyridin (2 x 60 ml, pro analysi-
Qualität, zusätzlich über Molekularsieb 4 Ä getrocknet) koevaporiert, in Pyridin (60 ml, s.o.) gelöst und auf -60°C (i-PrOH/N2) gekühlt. Hierzu tropfte man in 75 min eine Lösung von 2- (2-Chlor-6-nitrophenyl) -ethoxycarbonylchlorid (4,7 g, 17,8 mmol) in CH C12 (60 ml, dest. über CaH ) und ließ 1 h 45 min unter i-PrOH/N2 Kühlung (-40 bis -25°C) rühren. Das Gemisch wurde mit CH2C12 (150 ml) verdünnt und mit H20 (100 ml) gewaεchen. Die wäßrigen Phasen wurden mit CH2C12 (100 ml) nachextrahiert. Trocknen der organ. Phasen über Na S04, Filtrieren, Abrotieren des Lösemittels und Koevaporieren mit Toluol (3 x 50 ml) ergaben das Rohprodukt (9,8 g) , welches durch Säulenchromatographie (440 g Si02, 32 x 5,9 cm, Lösemittel: CH2Cl2/MeOH 100:4) gereinigt wurde. Man erhielt 5 ' -0- (2- (2-Chlor-6-nitrophenyl) ethoxycarbonyl) - N4- (2- (4-nitrophenyl) ethoxycarbonyl) -2 ' -desoxycytidin (5,247 g, 68 %) als farblosen Schaum.
Rf (Si02, CH2Cl2/MeOH 5:1) 0,82
UV(MeOH) , λmax [nm] (log ε) : 205 (Schulter, 4,56), 211 (4,58) , 242 (4,28) , 275 (4,17)
---H-NMR (250 MHz, CDCI3) : 8,18 (m, 2 arom. H v. NPEOC, ortho zu N02) : 8,04 (d, H-C(6) v. Cytosin) ; 8,03 (s (br) , NH, teilweise verborgen) ; 7,73 (dd, 1 arom. H v. CNPEOC) ; 7,66 (dd, 1 arom. H v. CNPEOC) ; 7,41 (m, 2 arom. H v. NPEOC, meta zu N02); 7,37 (t, H-C(4) v. CNPEOC, teilweise verborgen) ; 7,22 (d, H-C(5) v. Cytosin), 6,34 (t, H-C(l')) ; 4,43 (m, H-C(3*) , 2 x H-C(5*) f α-CH2 v. CNPEOC, α-CH2 v. NPEOC); 4,26 (m, H-C(4')) ; 3,76 (s (br) , 0H-C(3')) ; 3,43 (t, ß-CH2 v. CNPEOC) ; 3,12 (t, ß-CH2 v. NPEOC) ; 2,73 (m, H-C(2')) ; 2,14 (m, H-C(2'))
Anal. ber. für C27H26N5012C1 (647,981) : C 50,05, H 4,04, N 10,81; gef. : C 49,87, H 4,17, N 10,74
Beispiel 14
5' -O- (2- (2-Chlor-6-nitrophenyl)ethoxycarbonyl) -N6- (2- (4- nitrophenyl)ethoxycarbonyl) -2* -desoxyadenosin
N6- (2- (4-Nitrophenyl)ethoxycarbonyl) -2' -desoxyadenosin (5 g, 11,3 mmol) wurde mit Pyridin (2 x 60 ml, pro analysi- Qualität, zusätzlich über Molekularsieb 4 Ä getrocknet) koevaporiert, in Pyridin (60 ml, s.o.) gelöst und auf -60°C (i-PrOH/N ) gekühlt. Hierzu tropfte man in 2 h eine Lösung von 2- (2-Chlor-6-nitrophenyl)ethoxycarbonylchlorid (4,17 g, 15,8 mmol) in CH2C12 (60 ml, dest. über CaH2) . Nach weiteren 2 h Rühren unter i-PrOH/N Kühlung (-40 bis -25°C) wurde das Gemisch mit CH2C12 (150 ml) verdünnt und mit H20 (100 ml) gewaschen. Die wäßrigen Phasen wurden mit CH2C12 (100 ml) nachextrahiert. Trocknen der organ. Phasen über Na2SÜ4, Filtrieren, Abrotieren des Lösemittels und Koevaporieren mit Toluol (3 x 50 ml) ergaben das Rohprodukt (8,73 g) , welches durch Säulenchromatographie (400 g Si02, 28 x 5,7 cm, Lösemittel: CH2Cl2/MeOH 100:3) gereinigt wurde. Man erhielt 5 ' -0- (2- (2-Chlor-6-nitrophenyl)ethoxycarbonyl) -N6- (2- (4- nitrophenyl)ethoxycarbonyl) -2' -desoxyadenosin (6,318 g, 83 %) als farblosen Schaum.
Rf (Si02, Toluol/EtOAc/MeOH 5:4:1) 0,33 UV(MeOH) , λmax [nm] (log ε) : 209 (4,67), 266 (4,46) ---H-NMR (250 MHz, CDC13) : 8,71 (ε, H-C(8)) ; 8,53 (s (br) , NH, teilweise verborgen), 8,23 (s, H-C(2)); 8,17 (m, 2 arom. H v. NPEOC, ortho zu N02) ; 7,73 (dd, 1 arom. H v. CNPEOC); 7,65 (dd, 1 arom. H v. CNPEOC); 7,44 (m, 2 arom. H v. NPEOC, meta zu N02) ; 7,36' (t, H-C(4) v. CNPEOC, teilweise verborgen); 6,54 (t, H-C(l')); 4,77 (m, H-C(3')); 4,55 (t, α-CH2 v. CNPEOC); 4,41 ( , α-CH2 v. NPEOC, 2 x H-C(5')); 4,29 (q, H-C(4')); 3,43 (t, ß-CH2 v. CNPEOC); 3,16 (t, ß-CH2 v. NPEOC); 3,15 (s
(br) , OH-C(3") , teilweise verborgen) ; 2,89 (m, H-C(2')) ; 2,60 (m, H-C(2')) Anal. ber. für C28H26N7011C1 (672,007) : C 50,05, H 3,90, N 14,59; gef. : C 49,62, H 3,96, N 14,33
Beiεpiel 15
5 ' -0- (2- (2-Chlor-6-nitrophenyl)ethoxycarbonylthymidin-3 ' -0- ( (ß-cyanoethyl) (N,N-diisopropylamino)phosphitamid)
In einem mit Argon gespülten Kolben wurden 5 ' -0- (2- (2-Chlor- 6-nitrophenyl) ethoxycarbonyl) thymidin (1,9 g, 4 mmol) und IH-Tetrazol (140 mg, 2 mmol) in CH2C12 (20 ml, dest. über CaH2) und CH3CN (8 ml, pro analysi-Qualität) suspendiert und mit Bis (diiεopropylamino) (ß-cyanoethoxy)phosphin (1,87 g, 6,2 mmol) versetzt. Nach 16,5h Rühren bei Raumtemperatur wurde das Gemisch mit CH2C12 (50 ml) verdünnt und mit gesättigter NaHCθ3~Lsg. (25 ml) gewaschen. Die wäßrige Phase wurde mit CH2C12 (2 x 25 ml) nachextrahiert. Trocknen der organ. Phasen über Na2S04, Filtrieren und Abrotieren des Lösemittel ergaben das Rohprodukt (3,4 g) , welches durch Säulenchromatographie (40 g Si02, 12 x 3,1 cm, Lösemittel: Toluol/EtOAc 5:1 160 ml, 4:1 150 ml, 3:1 120 ml, 2:1 150 ml, 1:1 100 ml, 1:2 150 ml, 1:3 160 ml, jeweils unter Zugabe von 1 Vol.-% Et3N) gereinigt wurde. Man erhielt 5 ' -0- (2- (2-Chlor- 6-nitrophenyl) ethoxycarbonyl) thymidin-3 ' -0-
( (ß-cyanoethyl) (N,N-diisopropylamino)phosphitamid) (2,012 g, 75 %) als farblosen Schaum.
Rf (Toluol/EtOAc/MeOH 5:4:1) 0,64 und 0,70
(Diastereomere) UV(MeOH) , λmaχ [nm] (log ε) : 205 (4,45) , 262 (4,06) ---H-NMR (250 MHz, CDC13) : 8,40 (s, NH) ; 7,74 (td, 1 arom. H v. CNPEOC; 7,66 (dd, 1 arom. H v. CNPEOC) ; 7,38 (m, H-C(4) v. CNPEOC) ; 6,37 (m, H-C(l')) ; 4,61 bis 4,35 (m,
H-C(3'), 2 x H-C(5') , α-CH2 v. CNPEOC) ; 4,28 (m,
H-C(4'), Diastereomer) ; 4,22 (m, H-C(4') , Diastereomer) ;
3,90 - 3,66 ( , α-CH2 v. Cyanoethoxy) ; 3,57 (m, 2 x
NCH) ; 3,43 (td, ß-CH2 V. CNPEOC); 2,66 (t, ß-CH2 V.
Cyanoethoxy); 2,48 (m, H-C(2')); 2,23 (m, H-C(2-)); 1,86
(d, CH3) ; 1,23 (m, 2 x NC(CH3)2) 31P-NMR (161,7 MHz, CDC13) : 149,73 und 149,85
(Diastereomere) Anal. ber. für C28H37N5O10PCl (670,056): C 50,19, H 5,57, N
10,45; gef.: C 50,43, H 5,90, N 10,43
Beispiel 16
5' -O- (2- (2-Chlor-6-nitrophenyl)ethoxycarbonyl) -N4- (2- (4- nitrophenyl)ethoxycarbonyl) -2' -desoxycytidin-3' -0- ( (ß-cyanoethyl) (N,N-diisopropylamino)phosphitamid)
In einem mit Argon gespülten Kolben wurden 5' -0- (2- (2-Chlor- 6-nitrophenyl)ethoxycarbonyl) -N4- (2- (4-nitrophenyl) - ethoxycarbonyl) -2' -desoxycytidin (1,9 g, 2,9 mmol) und IH-Tetrazol (102 mg, 1,45 mmol) in CH2C1 (20 ml, dest. über CaH2) und CH3CN (8 ml, pro analysi-Qualität) mit Bis (diisopropylamino) (ß-cyanoethoxy)phosphin (1,32 g, 4,38 mmol) versetzt und bei Raumtemperatur gerührt. Nach 13,5 h wurde die Lösung mit CH2C12 (50 ml) verdünnt und mit gesättigter NaHCθ3-Lsg. (25 ml) gewaschen. Die wäßrige Phase wurde mit CH C12 (2 x 25 ml) nachextrahiert. Die vereinigten organischen Phasen wurden über Na2Sθ4 getrocknet, filtriert und einrotiert. Das Rohprodukt (2,99 g) wurde durch Säulenchromatographie (61 g Si02, 18 x 3,2 cm, Lösemittel: PE/Aceton 5:1 170 ml, 4:1 200 ml, 3:1 200 ml, 2:1 600 ml, 3:2 150 ml, 1:1 80 ml, 2:3 300 ml, 1:2 90 ml) gereinigt. Man erhielt 5' -O- (2- (2-Chlor-6-nitrophenyl)ethoxycarbonyl) -N4- (2- (4-nitrophenyl)ethoxycarbonyl) -2' -desoxycytidin-3■ -0-
( (ß-cyanoethyl) (N,N-diisopropylamino)phosphitamid) (2,29 g, 93 %) als farblosen Schaum.
Rf (Si02, Toluol/EtOAc/MeOH 5:4:1) 0,61 und 0,67
(Diastereomere) UV(MeOH) , λj ax [nm] (log ε) : 203 (4,66), 209 (Schulter,
4,64) , 241 (4,30), 276 (Schulter, 4,18) ---H-NMR (250 MHz, CDC13) : 8,18 (m, 2 arom. H v. NPEOC) , ortho zu N02) ; 8,02 (m, H-C(6) v. Cytosin) ; 7,74 (td, 1 arom.
H v. CNPEOC) ; 7,66 (dd, 1 arom. H v. CNPEOC) ; 7,40 (m, 2 arom. H v. NPEOC, meta zu N02) ; 7,39 (m, H-C(4) v.
CNPEOC, teilweise verborgen) ; 7,17 (d, H-C(5) v.
Cytosin) ; 6,28 (m, H-C(l')) ; 4,47 bis 4,29 (m, α-CH2 v.
NPEOC) , α-CH2 v. CNPEOC), H-C(3') , H-C(4*) , 2 x
H-C(5*)) ; 3,90 bis 3,53 (m, α-CH2 v. Cyanoethoxy, 2 x
NCH) ; 3,43 (t, ß-CH2 v. CNPEOC) ; 3,12 (t, ß-CH2 v.
NPEOC) ; 2,73 (m, H-C(2') teilweise verborgen) ; 2,65 (t, ß-CH2 v. Cyanoethoxy) ; 2,17 (m, H-C(2*)) ; 1,23 (m, 2 x
NC(CH3)2) 31p_NMR (161,7 MHz, CDCI3) : 149,67 und 150,02
(Diastereomere) Anal. ber. für C36H43N7013PC1 (848,203) : C 50,98, H 5,11, N
11,56; gef.: C 50,88, H 5,18, N 11,36
Beispiel 17
5 ' -0- (2- (2-Chlor-6-nitrophenyl)ethoxycarbonyl) -N6- (2- (4- nitrophenyl) ethoxycarbonyl) - 2 ' -desoxyadenosin-3 ' -0- ( (ß-cyanoethyl) (N,N-diisopropylamino)phosphitamid)
In einem mit Argon gespülten Kolben wurde ein Gemisch von 5' 0- (2- (2-Chlor-6-nitrophenyl) ethoxycarbonyl) -N6- (2- (4- nitrophenyl) ethoxycarbonyl) -2 ' -desoxyadenosin (2 g, 2,98 mmol) und IH-Tetrazol (104 mg, 1,49 mmol) in CH2C12 (20 ml, dest. über CaH2) und CH3CN (8 ml, pro analysi-
Qualität) mit Bis (diisopropylamino) (ß-cyanoethoxy)phosphin (1,36 g, 4,51 mmol) versetzt und bei Raumtemperatur 13,5 h gerührt. Die Lösung wurde mit CH2C12 (50 ml) verdünnt und mit gesättigter NaHCθ3~Lsg. (25 ml) gewaschen. Die wäßrige Phase wurde mit CH2C12 (2 x 25 ml) nachextrahiert. Die vereinigten organischen Phasen wurden über Na2Sθ4 getrocknet, filtriert und einrotiert. Das Rohprodukt wurde durch Säulenchromatographie (92 g Si02, 13 x 4,2 cm, Lösemittel: PE/Aceton 6:1 140 ml, 5:1 180 ml, 4:1 200 ml, 3:1 240 ml, 2:1 750 ml, 3:2 150 ml, 1:1 400 ml, 2:3 600 ml, jeweils unter Zugabe von 1 Vol.-% Et3N) gereinigt. Man erhielt 5'-0-(2-(2- Chlor-6-nitrophenyl)ethoxycarbonyl) -N6- (2- (4- nitrophenyl)ethoxycarbonyl) -2* -desoxyadenosin-3' -0- ( (ß-cyanoethyl) (N,N-diisopropylamino)phosphitamid) (2,146 g, 83 %) als farblosen Schaum.
Rf (Si02, Toluol/EtOAc/MeOH 5:4:1) 0,71 und 0,76 (Diastereomere)
UV(MeOH), λmax [nm] (log ε) : 205 (4,68), 266 (4,43)
---H-NMR (250 MHz, CDCI3): 8,74 (d, H-C(8) v. Adenin); 8,23 (ε, H-C(2) v. Adenin); 8,18 (m, 2 arom. H v. NPEOC, ortho zu N02) ; 7,73 (m, 1 arom. H v. CNPEOC); 7,65 (m, 1 arom. H v. CNPEOC); 7,44 (m, 2 arom. H v. NPEOC, meta zu N02); 7,36 (t, H-C(4) v. CNPEOC); 6,52 (m, H-C(l')); 4,77 (m, H-C(3*)); 4,54 (t, α-CH2 v. CNPEOC); 4,39 (m, α-CH2 v. NPEOC, H-C(4'), 2 x H-C.5')); 3,93 biε 3,59 (m, 2 x NCH, α-CH2 v. Cyanoethoxy); 3,41 (td, ß-CH2 v. CNPEOC); 3,16 (t, ß-CH2 v. NPEOC); 2,90 (m, H-C.21)); 2,71 (m, H-C(2'), teilweise verborgen); 2,67 (m, ß-CH2 v. Cyanoethoxy); 1,24 (m, 2 x NC(CH3) )
31P-NMR (161,7 MHz, CDCI3) : 149,70 und 149,79 (Diastereomere)
Anal. ber. für C37H43N9012PC1 (872,229) : C 50,95, H 4,97, N 14,45; gef.: C 50,92, H 5,11, N 14,21
Zusammenfassung der Herstellungsbeispiele
Bsp. Rl R2 R R4 R5 R6 B
1 H H H H H H Thymin
2 H H N02 H H H Thymin
3 H H F H H H Thymin
4 H H Cl H H H Thymin
5 H H Br H H H Thymin
6 Cl H H H H H Thymin
7 H OCH3 H H H H Thymin
8 Cl H Cl H H H Thymin
9 OCH3 OCH3 H H H H Thymin
10 H H H CH3 H H Thymin
11 H H H Cl H H Thymin
12 H H H OCH3 H H Thymin
13 H H Cl H H H N4- (2-pNPEOC) - Cytosin
14 H H Cl H H H N6- (2-pNPEOC) - Adenin
15 H H Cl H H Thymin
16 H H Cl H H N4- (2-pNPEOC) - Cytosin
17 H H Cl H H N
6- (2-pNPEOC) - Adenin
Bestrahlungsexperimente
1. Durchführung
Die entsprechend geschützten Thymidin-Nucleoside wurden mittels einer Bestrahlungsapparatur bestrahlt, die aus einer Hg-Höchstdrucklampe (OSRAM HBO, 200 W) , einer Sammellinse, einem Verschluß mit einem elektronischen Steuergerät zur Einstellung der Verschlußzeiten sowie einem temperierbaren Küvettenhalter bestand. Es war außerdem ein Wärmefilter (0,032 molare CuSθ4-Lsg.) zwischen Lampe und Probe eingebaut. Es wurden 0,2 mM Lösungen der Nucleoside in MeOH/H20 1:1 bei 20 bis 30°C mit dem gesamten Lampenspektrum (polychromatisches Licht mit Wellenlängen λ > 289 nm) belichtet.
Die Abspaltung der Schutzgruppen wurde durch HPLC quantitativ verfolgt. Die HPLC-Anlage bestand aus folgenden Geräten: Merck-Hitachi L-6200 Intelligent Pump, Merck-Hitachi Intelligent Auto Sampler AS 4000, Merck-Hitachi Interface, Merck Lichrosorb-Säule RP 18, 125 x 5 mm. Das UV/VIS- Spektrophotometer war ein UVIKON 730 LC der Fa. Kontron. Die Detektion erfolgte bei 260 nm. Die Integration der Chromatogramm-Signale erfolgte mittels Software der Fa. Merck: D-6000 HPLC-Manager.
Es wurde grundsätzlich in MeOH/H 0-Gemischen chromatographiert. Dabei wurde folgender Gradient verwendet (Flow: 1 ml/min) :
Zeit (min) MeOH/H20 1:1 H20 MeOH
0 10 90 0
5 10 90 0
15 90 10 0
30 50 0 50
35 10 90 0
Für Thymidin und die geschützten Nucleoside wurden durch Einspritzen von Verdünnungεreihen in den Chromatographen Eichkurven erstellt.
Aus den belichteten Lösungen wurden nach bestimmten Zeitintervallen Proben (Schleifen-Volumen: 20 μl) entnommen. Jede Probe wurde zweimal in den Chromatographen eingespritzt. Für die weitere Auswertung wurden die Mittelwerte dieser Doppelbestimmungen genommen. Die Peakflachen der Chromatogramme wurden mittels der oben erstellten Eichkurven in die Konzentrationen der geschützten Nucleoside bzw. von Thymidin umgerechnet. Die so erhaltenen Konzentrationswerte wurden durch die maximal erreichbare Konzentration von 0,2 mM (= Konzentration der geschützten Nucleoside zu Beginn der Belichtung) dividiert und als "Relative Menge" in Prozent gegen die Bestrahlungszeit aufgetragen. Die in Abschnitt 2.3 angegebene Tabelle zeigt ein Resultat einer solchen Messung mit der Verbindung 5' -0- (2- (2-Chlor-6-nitrophenyl) - ethoxycarbonyl)thymidin.
Beispiele
2.1 Eichung Thymidin
Für die Eichung von Thymidin wurde eine Verdünnungsreihe mit folgenden Konzentrationen erstellt: 0,2 mM, 0,16 mM, 0,12 mM, 0,08 mM und 0,04 mM. Hiervon wurden je Konzentration drei
Einspritzungen in den Chromatographen gemacht. Aus den resultierenden Mittelwerten sowie dem Nullpunktswert (0 Peakfläche = 0 Konzentration) wurden mittels linearer Regression die Eichgerade berechnet (siehe Tabelle) .
Eichung Thymidin
Peakfläche Konzentration Thymidin (mM)
516,893 0,20
414,730 0,16
307,575 0,12
208,952 0,08
99,5080 0,04
0,00000 0,00
Lineare Regression ergab: y = 4,96777-10"4 + 3, 85757•10~7*x mit x = Peakfläche und y = Konzentration Thymidin,* r = 0,9999.
2.2 Eichung 5• -0- (2- (2-Chlor-6-nitrophenyl) - ethoxycarbonyl)thymidin
Es wurde eine Verdünnungsreihe mit folgenden drei Konzentrationen erstellt: 0,2 mM, 0,14 mM und 0,08 mM. Die Mittelwerte aus drei Einspritzungen je Konzentration sowie der Nullpunktswert wurden zur Berechnung der Regresεionsgerade eingesetzt (siehe Tabelle) :
Eichung CNPEOC-T
Peakfläche Konzentration CNPEOC-T (mM)
617,057 0,20 435,070 0,14 238,624 0,08 0,00000 0, 00
Lineare Regression ergab: y = 9,27999*10'4 + 3, 22516 • 10"7 *x mit x = Peakfläche und y = Konzentration CNPEOC-T; r = 0,9998.
2.3 Ergebnisse
(a) 5' -O- (2- (2-Chlor-6-nitrophenyl) -ethoxycarbonyl) thymidin
Bestrahlung von 5• -0- (2- (2-Chlor-6- nitrophenyl)ethoxycarbonyl)thymidin
Bestrahlungszeit Peakfläche Konzentration Relative Menge (min) CNPEOC-T CNPEOC-T (mM) CNPEOC-T (%)
1,00000 458911 0,1489 74,5
2,00000 253292 0,0826 41,3
3,00000 250989 0,0819 40,9
5,00000 84081,0 0,0281 14,0
15,0000 24181,0 0,0088 4,4
30,0000 0 0 0
60,0000 0 0 0
120,000 0 0 0
Bestrahlungszeit Peakfläche Konzentration Relative Menge (min) Thymidin Thymidin (mM) Thymidin (%)
1,00000 83460,0 0,0327 16,3
2,00000 221149 0,0858 42,9
3,00000 228893 0,0888 44,4
5, 00000 330106 0,1278 63,9
15, 0000 432514 0,1673 83,7
30,0000 437588 0,1693 84,6
60, 0000 442230 0,1711 85,5
120,000 407543 0,1577 78,9
Diese Tabelle zeigt deutlich, daß die Photolyse über einen sehr großen Zeitraum der Kinetik einer ersten Ordnung folgt, was auf einen eindeutigen Abspaltungsmechanismus ohne ausbeutemindernde Nebenreaktionen schließen läßt.
(b) Weitere erfindungsgemäße Verbindungen
Die nach der oben angegebenen Methode erhaltenen und bezüglich Halbwertszeit und Ausbeute (Prozentsatz entεchützteε Nucleosid-Derivat, bezogen auf die maximal erreichbare Konzentration) ausgewerteten Ergebnisse einiger weiterer erfindungsgemäßen Derivate und der zwei Vergleichsverbindungen VI (5' -0- (2-Nitrobenzyloxycarbonyl) - thymidin) und V2 (5' -0- (2,4-Dinitrobenzyloxycarbonyl) - thymidin) werden in der nachstehenden Tabelle zusammengefaßt
Verbindung Nr. to,5 ( in) Ausbeute ( % cmax-
1 2,6 79
2 1,37 92
4 1,71 86
5 1,71 89
6 2,3 70
8 1,68 77
9 7,2 77
10 0,7 89
11 1,46 97
VI 2,5 55
V2 3,3 38
Wie aus der Tabelle ersichtlich ist, sind die erfindungεgemäßen Nucleosid-Derivate hinsichtlich der photolytiεchen Abspaltung der 5' -Schutzgruppe in Bezug auf Schnelligkeit und hohe Ausbeuten (vgl. insbesondere Verbindungen 10 und 11) den Schutzgruppen entsprechend dem Stand der Technik (vgl. VI und V2) deutlich überlegen.
2.4 Anwendungsbeispiel
Die Verbindung 5' -0- (2- (2-Chlor-6-nitrophenyl) - ethoxycarbonyl)thymidin und andere erfindungsgemäße Nucleosid-Derivate wurden nach einer Methodik gemäß S.P.A. Fodor et al. Science 1991, 251, S.767ff zur Darstellung von Oligonucleotiden auf einem DNA-Chip eingesetzt. Es zeigte sich, daß die erfindungsgemäßen Verbindungen eine unkomplizierte Oligonucleotidsynthese mit sehr hohen Ausbeuten erlaubten, so daß sie in der Praxis für lichtgesteuerte Parallel-Synthesen von Oligonucleotiden geeignet sind.