WO1996023764A1 - VERFAHREN ZUR RACEMATTRENNUNG VON 2-ARYL-2-ALKYL-φ-ALKYLAMINO-ALKANNITRILEN - Google Patents
VERFAHREN ZUR RACEMATTRENNUNG VON 2-ARYL-2-ALKYL-φ-ALKYLAMINO-ALKANNITRILEN Download PDFInfo
- Publication number
- WO1996023764A1 WO1996023764A1 PCT/EP1996/000236 EP9600236W WO9623764A1 WO 1996023764 A1 WO1996023764 A1 WO 1996023764A1 EP 9600236 W EP9600236 W EP 9600236W WO 9623764 A1 WO9623764 A1 WO 9623764A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- optically active
- aryl
- resolution
- alkylaminoalkane
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 CC(*)(C(CC(*1CC1)=C1OC)C=C1OC)C#N Chemical compound CC(*)(C(CC(*1CC1)=C1OC)C=C1OC)C#N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C253/00—Preparation of carboxylic acid nitriles
- C07C253/32—Separation; Purification; Stabilisation; Use of additives
- C07C253/34—Separation; Purification
Definitions
- Phenylacetonitriles differently substituted on the phenyl group and provided with a basic side chain have found application as active pharmaceutical ingredients.
- EP 0 271 013 describes phenylacetonitriles which carry an aliphatic side chain instead of the dimethoxyphenylethyl radical on the nitrogen as active substances against cardiovascular diseases and asthma diseases.
- the substituted phenylacetonitriles have a chiral carbon atom, so that they form two enantiomers (optical antipodes) in the absence of further chiral centers.
- both enantiomers are produced in equal amounts, so that a racemate is present.
- Enantiomers of verapamil and gallopamil are based on the stereoisomeric lactic acids and cannot be carried out on an industrial scale due to the numerous reaction stages and complicated chemical transformations of functionalities.
- Racemate resolution methods have been published more frequently.
- a special case is in WO 92/07821, where the racemate cleavage of the phenyl acetonitrile part takes place by linking to optically active ⁇ -methylaminotetralin, which becomes part of the active ingredient, and fractional crystallization of the diastereomers:
- Brucine is used to split III (DE 2059 985), which is disadvantageous because of the extreme toxicity and the high price of brucine.
- IV can be cleaved with chinchonidine, two functionalities have to be modified in order to obtain the desired product, which is technically difficult to carry out (H. Ramuz, Helv. Chim. Acta 58, 2050-2060 [1975] ).
- camphorsulfonic acid is not suitable for the racemate resolution of verapamil, it was surprising that the basic substituted phenylacetonitriles with camphorsulfone acid can be easily split into highly enantiomerically pure optical antipodes.
- the invention relates to a process for the resolution of 2-aryl-2-alkylene- ⁇ -alkylaminoalkannitriles of the formula I
- R 1 is hydrogen or methoxy
- R 2 and R 3 which are the same or different, mean C ⁇ - alkyl and
- n 2 or 3
- C 4 alkyl groups methyl, ethyl, n-propyl, isopropyl, cyclopropyl, n-butyl, isobutyl, sec-butyl, tert-butyl.
- the process according to the invention is preferably carried out as follows: 1 equivalent of racemic I is in an alcohol such as methanol, ethanol, n-propanol, isopropanol, glycol, diglycol or a mixture of the alcohols or a mixture of an alcohol with acetone or Ethyl acetate (preferably isopropanol) is introduced, and 0.5 to 1 (preferably 0.75) equivalent of capric sulfonic acid monohydrate are added. The mixture is then heated to 40 ° C. to 100 ° C. or to the boiling point of the solvent used (preferably 60 ° C.).
- an alcohol such as methanol, ethanol, n-propanol, isopropanol, glycol, diglycol or a mixture of the alcohols or a mixture of an alcohol with acetone or Ethyl acetate (preferably isopropanol) is introduced, and 0.5 to 1 (preferably 0.75) equivalent of capric sulfonic acid monohydrate are added.
- the mixture
- the salt is preferably recrystallized from the solvent or solvent mixture already used.
- dilute alkaline solution for example sodium hydroxide solution, potassium hydroxide solution, potassium carbonate solution
- the optically active compound I can be precipitated from these solutions or can be directly implemented further therein.
- camphorsulfonic acid is very cheap and can also be obtained in technical quantities and because the intermediates of formula I obtained produce numerous active ingredients, such as, for example, Verapamil, Gallopamil and the compounds described in EP 0 271 013 allow.
- racemic starting compounds of the formula VI used for the process according to the invention can be prepared according to the following scheme:
- the mixture was heated to an internal temperature of 60 ° C., giving a clear solution. Then it was slowly cooled. After inoculation with the salt at 30 to 35 ° C, the crystallization began. The mixture was cooled to 20 ° C. with stirring. The precipitate was filtered off with
- the dried diastereomeric salt was dissolved in 300 l of water with 30 kg of 50% sodium hydroxide solution and extracted twice with 100 1 20 each of methyl t-butyl ether.
- the product is in the form of an oil.
- the substance was produced analogously to Example 1.
- the hydrochloride melts at 172 to 173 ° C.
- the substance was produced analogously to Example 2.
- the hydrochloride melts at 172 to 175 ° C.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Description







Claims

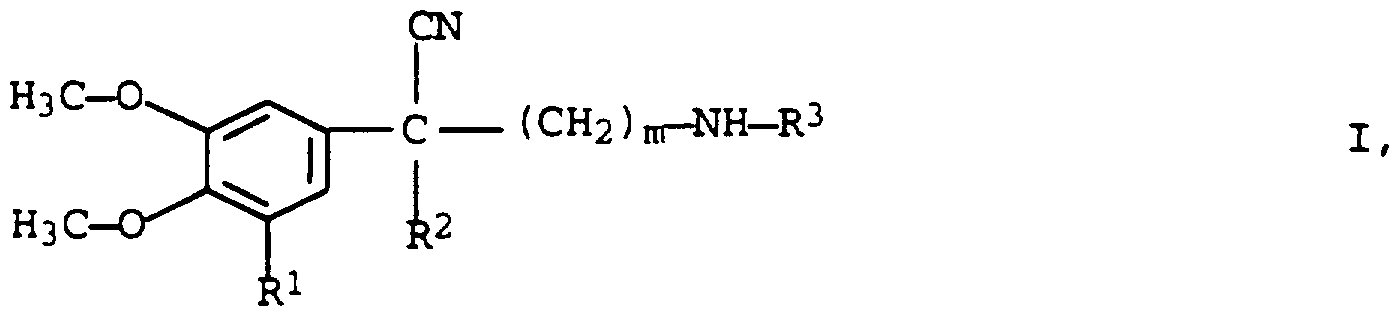
Priority Applications (9)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP96901746A EP0808302B1 (de) | 1995-01-31 | 1996-01-20 | Verfahren zur racemattrennung von 2-aryl-2-alkyl-omega-alkylamino-alkannitrilen |
| JP8523206A JPH10512890A (ja) | 1995-01-31 | 1996-01-20 | 2−アリール−2−アルキル−ω−アルキルアミノ−アルカンニトリルのラセミ化合物分割法 |
| AT96901746T ATE191464T1 (de) | 1995-01-31 | 1996-01-20 | Verfahren zur racemattrennung von 2-aryl-2-alkyl- omega-alkylamino-alkannitrilen |
| DK96901746T DK0808302T3 (da) | 1995-01-31 | 1996-01-20 | Fremgangsmåde til racematadskillelse af 2-aryl-2-alkyl-omega-alkylaminoalkannitriler |
| MX9705200A MX9705200A (es) | 1995-01-31 | 1996-01-20 | La resolucion de racematos de 2-aril-2-alquil-w-alquilaminoalcanonitrilos. |
| DE59604890T DE59604890D1 (de) | 1995-01-31 | 1996-01-20 | Verfahren zur racemattrennung von 2-aryl-2-alkyl-omega-alkylamino-alkannitrilen |
| CA002210337A CA2210337C (en) | 1995-01-31 | 1996-01-20 | Resolution of racemates of 2-aryl-2-alkyl-.omega.-alkylaminoalkane-nitriles |
| US08/860,664 US5786498A (en) | 1995-01-31 | 1996-01-20 | Resolution of racemates of 2-aryl-2-alkyl-ω-alkylaminoalkane-nitriles |
| GR20000400732T GR3033164T3 (en) | 1995-01-31 | 2000-04-06 | 2-aryl-2-alkyl-omega-alkylaminoalkane nitrile racemate separation process |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE19502967.4 | 1995-01-31 | ||
| DE19502967A DE19502967A1 (de) | 1995-01-31 | 1995-01-31 | Verfahren zur Racemattrennung von 2-Aryl-2-omega-alkylaminoalkannitrilen |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1996023764A1 true WO1996023764A1 (de) | 1996-08-08 |
Family
ID=7752720
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP1996/000236 Ceased WO1996023764A1 (de) | 1995-01-31 | 1996-01-20 | VERFAHREN ZUR RACEMATTRENNUNG VON 2-ARYL-2-ALKYL-φ-ALKYLAMINO-ALKANNITRILEN |
Country Status (13)
| Country | Link |
|---|---|
| US (1) | US5786498A (de) |
| EP (1) | EP0808302B1 (de) |
| JP (1) | JPH10512890A (de) |
| CN (1) | CN1068871C (de) |
| AT (1) | ATE191464T1 (de) |
| CA (1) | CA2210337C (de) |
| DE (2) | DE19502967A1 (de) |
| DK (1) | DK0808302T3 (de) |
| ES (1) | ES2145427T3 (de) |
| GR (1) | GR3033164T3 (de) |
| MX (1) | MX9705200A (de) |
| PT (1) | PT808302E (de) |
| WO (1) | WO1996023764A1 (de) |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6130302A (en) | 1996-08-19 | 2000-10-10 | Northwestern University | Synthesis and use of (polyfluoroaryl)fluoroanions of aluminum, gallium and indium |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE2059985A1 (de) * | 1970-12-05 | 1972-06-15 | Knoll Ag | Optisch aktive,basisch substituierte Phenylacetonitrile |
| EP0357565A2 (de) * | 1988-07-12 | 1990-03-07 | Ministero Dell' Universita' E Della Ricerca Scientifica E Tecnologica | Verfahren zur Herstellung von Levodopa |
| WO1995009150A1 (en) * | 1993-09-27 | 1995-04-06 | Chiroscience Limited | Chiral nitriles, their preparation and their use for the manufacture of verapamil and analogues |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE3642331A1 (de) * | 1986-12-11 | 1988-06-23 | Basf Ag | Basisch substituierte phenylacetonitrile, ihre herstellung und diese enthaltende arzneimittel |
| DE3723684A1 (de) * | 1987-07-17 | 1989-01-26 | Basf Ag | Verfahren zur herstellung der enantiomeren von verapamil |
| SE8803012L (sv) * | 1988-08-29 | 1990-03-01 | Skf Nova Ab | Anordning foer aastadkommande av bromsning av ett vridmomentoeverfoerande organ |
| WO1992007821A1 (en) * | 1990-11-01 | 1992-05-14 | G.D. Searle & Co. | Diastereoisomers of bicyclo-substituted phenylacetonitrile derivatives |
| DE4203547A1 (de) * | 1992-02-07 | 1993-08-12 | Knoll Ag | Verfahren zur racemattrennung von verapamil |
-
1995
- 1995-01-31 DE DE19502967A patent/DE19502967A1/de not_active Withdrawn
-
1996
- 1996-01-20 ES ES96901746T patent/ES2145427T3/es not_active Expired - Lifetime
- 1996-01-20 US US08/860,664 patent/US5786498A/en not_active Expired - Lifetime
- 1996-01-20 PT PT96901746T patent/PT808302E/pt unknown
- 1996-01-20 JP JP8523206A patent/JPH10512890A/ja active Pending
- 1996-01-20 AT AT96901746T patent/ATE191464T1/de not_active IP Right Cessation
- 1996-01-20 WO PCT/EP1996/000236 patent/WO1996023764A1/de not_active Ceased
- 1996-01-20 DE DE59604890T patent/DE59604890D1/de not_active Expired - Fee Related
- 1996-01-20 EP EP96901746A patent/EP0808302B1/de not_active Expired - Lifetime
- 1996-01-20 CA CA002210337A patent/CA2210337C/en not_active Expired - Fee Related
- 1996-01-20 CN CN96191705A patent/CN1068871C/zh not_active Expired - Fee Related
- 1996-01-20 DK DK96901746T patent/DK0808302T3/da active
- 1996-01-20 MX MX9705200A patent/MX9705200A/es not_active IP Right Cessation
-
2000
- 2000-04-06 GR GR20000400732T patent/GR3033164T3/el not_active IP Right Cessation
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE2059985A1 (de) * | 1970-12-05 | 1972-06-15 | Knoll Ag | Optisch aktive,basisch substituierte Phenylacetonitrile |
| EP0357565A2 (de) * | 1988-07-12 | 1990-03-07 | Ministero Dell' Universita' E Della Ricerca Scientifica E Tecnologica | Verfahren zur Herstellung von Levodopa |
| WO1995009150A1 (en) * | 1993-09-27 | 1995-04-06 | Chiroscience Limited | Chiral nitriles, their preparation and their use for the manufacture of verapamil and analogues |
Also Published As
| Publication number | Publication date |
|---|---|
| CA2210337A1 (en) | 1996-08-08 |
| CA2210337C (en) | 2004-04-27 |
| US5786498A (en) | 1998-07-28 |
| MX9705200A (es) | 1997-10-31 |
| JPH10512890A (ja) | 1998-12-08 |
| EP0808302B1 (de) | 2000-04-05 |
| PT808302E (pt) | 2000-09-29 |
| ES2145427T3 (es) | 2000-07-01 |
| ATE191464T1 (de) | 2000-04-15 |
| DK0808302T3 (da) | 2000-07-17 |
| DE59604890D1 (de) | 2000-05-11 |
| EP0808302A1 (de) | 1997-11-26 |
| GR3033164T3 (en) | 2000-08-31 |
| CN1068871C (zh) | 2001-07-25 |
| CN1172470A (zh) | 1998-02-04 |
| DE19502967A1 (de) | 1996-08-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO1997043244A1 (de) | Racemattrennung von ketamin | |
| WO1997011927A1 (de) | Verfahren zur racematspaltung | |
| AT396252B (de) | Enzymatisches verfahren zur enantioselektiven herstellung optisch aktiver cyanhydrine | |
| EP0312726A1 (de) | Optisch aktive Salze aus einem substituierten Thiazolidin-4-carboxylat und 3-Chlor-2-hydroxypropyltrimethylammonium, deren Herstellung und Verwendung | |
| EP0808302B1 (de) | Verfahren zur racemattrennung von 2-aryl-2-alkyl-omega-alkylamino-alkannitrilen | |
| DE69409001T2 (de) | Chirale nitrile, ihre herstellung und ihre verwendung in der herstellung von verapamil und dessen analogen | |
| DE10032254B4 (de) | Nukleotidsequenz kodierend für eine Benzaldehyd-Lyase und Verfahren zur stereoselektiven Synthese von (R)-2-Hydroxyketonen | |
| EP0865500B1 (de) | Verfahren zur herstellung von optisch aktiven aminen | |
| DE10348674B4 (de) | Racemisierung von optisch aktiven 2-substituierten Phenylglycinestern | |
| EP1413565B1 (de) | Verfahren zur Racematsspaltung von 3-Aminopentannitril | |
| EP1589019B1 (de) | Stereoselektives Verfahren zur Herstellung von Clopidogrel | |
| DE3734219C2 (de) | ||
| DE2901537C2 (de) | ||
| DE3880904T2 (de) | Weinsäureamidderivate und Verfahren zu ihrer Herstellung. | |
| DE2721265A1 (de) | Verfahren zur herstellung von di- n-propylacetonitril | |
| DE10237246B3 (de) | (S)-3-Methylamino-1-(2-thienyl)-1-propanol-(-)-2,3,4,6-Di-O-isopropyliden-2-keto-L-gulonsäuresalz, Verfahren zu dessen Herstellung, dessen Verwendung sowie ein Verfahren zur Enantiomerenanreicherung von (S)-3-Methylamino-1-(2-thienyl)-1-propanol | |
| DE19536658C2 (de) | Verfahren zur kontinuierlichen Herstellung von basischen, cyclischen, optisch aktiven alpha-Aminosäuren | |
| DE3342524C2 (de) | ||
| EP0001821B1 (de) | Verfahren zur Racematspaltung von DL-alpha-Aminocarbonsäuren und dafür verwendete Salze | |
| DE1518029C (de) | Stereospezifisches Verfahren zur Her stellung von L() 3,4 Dimethoxyphenyl ace tyl carbinol | |
| DE952715C (de) | Verfahren zur Herstellung neuer antihistaminwirksamer basischer AEther | |
| WO2005005375A1 (de) | Verfahren zur herstellung von 4-cyano-3-hydroxybuttersäureestern | |
| EP0046950A1 (de) | Optisch aktive Isomere von trans-3-(2-Chlor-2-(4-chlor-phenyl)-vinyl)-2,2-dimethyl-cyclopropan-1-carbonsäure-(alpha-cyano-4-fluor-3-phenoxy-benzyl)-ester, Verfahren zu deren Herstellung und deren Verwendung als Ektoparasitizide | |
| DE3737341A1 (de) | Neue fluormethoxyphenyl-dihydropyridine, verfahren zur herstellung und ihre verwendung in arzneimitteln | |
| DE2928022A1 (de) | Verfahren zur herstellung des 2-(n- benzyl-n-methylamino)-aethylesters der 2,6- dimethyl-4-(3-nitrophenyl)-3-methoxycarbonyl-1,4-dihydropyridin-5-carbonsaeure |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 96191705.9 Country of ref document: CN |
|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): CA CN JP MX US |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): AT BE CH DE DK ES FR GB GR IE IT LU MC NL PT SE |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 1996901746 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 08860664 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PA/a/1997/005200 Country of ref document: MX |
|
| ENP | Entry into the national phase |
Ref document number: 2210337 Country of ref document: CA Ref document number: 2210337 Country of ref document: CA Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 1996 523206 Country of ref document: JP Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 1996901746 Country of ref document: EP |
|
| WWG | Wipo information: grant in national office |
Ref document number: 1996901746 Country of ref document: EP |