WO1999033802A1 - Verfahren zur isolierung von carbazol sowie herstellung von 9,10-dihydroanthracen aus rohanthracen - Google Patents
Verfahren zur isolierung von carbazol sowie herstellung von 9,10-dihydroanthracen aus rohanthracen Download PDFInfo
- Publication number
- WO1999033802A1 WO1999033802A1 PCT/EP1998/008218 EP9808218W WO9933802A1 WO 1999033802 A1 WO1999033802 A1 WO 1999033802A1 EP 9808218 W EP9808218 W EP 9808218W WO 9933802 A1 WO9933802 A1 WO 9933802A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- weight
- anthracene
- carbazole
- hydrogenation
- crude
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C13/00—Cyclic hydrocarbons containing rings other than, or in addition to, six-membered aromatic rings
- C07C13/28—Polycyclic hydrocarbons or acyclic hydrocarbon derivatives thereof
- C07C13/32—Polycyclic hydrocarbons or acyclic hydrocarbon derivatives thereof with condensed rings
- C07C13/54—Polycyclic hydrocarbons or acyclic hydrocarbon derivatives thereof with condensed rings with three condensed rings
- C07C13/573—Polycyclic hydrocarbons or acyclic hydrocarbon derivatives thereof with condensed rings with three condensed rings with three six-membered rings
- C07C13/58—Completely or partially hydrogenated anthracenes
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C5/00—Preparation of hydrocarbons from hydrocarbons containing the same number of carbon atoms
- C07C5/02—Preparation of hydrocarbons from hydrocarbons containing the same number of carbon atoms by hydrogenation
- C07C5/10—Preparation of hydrocarbons from hydrocarbons containing the same number of carbon atoms by hydrogenation of aromatic six-membered rings
- C07C5/11—Partial hydrogenation
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2523/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group C07C2521/00
- C07C2523/16—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group C07C2521/00 of arsenic, antimony, bismuth, vanadium, niobium, tantalum, polonium, chromium, molybdenum, tungsten, manganese, technetium or rhenium
- C07C2523/24—Chromium, molybdenum or tungsten
- C07C2523/28—Molybdenum
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2523/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group C07C2521/00
- C07C2523/16—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group C07C2521/00 of arsenic, antimony, bismuth, vanadium, niobium, tantalum, polonium, chromium, molybdenum, tungsten, manganese, technetium or rhenium
- C07C2523/24—Chromium, molybdenum or tungsten
- C07C2523/30—Tungsten
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2523/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group C07C2521/00
- C07C2523/70—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group C07C2521/00 of the iron group metals or copper
- C07C2523/74—Iron group metals
- C07C2523/75—Cobalt
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2523/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group C07C2521/00
- C07C2523/70—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group C07C2521/00 of the iron group metals or copper
- C07C2523/74—Iron group metals
- C07C2523/755—Nickel
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2523/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group C07C2521/00
- C07C2523/70—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group C07C2521/00 of the iron group metals or copper
- C07C2523/76—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group C07C2521/00 of the iron group metals or copper combined with metals, oxides or hydroxides provided for in groups C07C2523/02 - C07C2523/36
- C07C2523/84—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group C07C2521/00 of the iron group metals or copper combined with metals, oxides or hydroxides provided for in groups C07C2523/02 - C07C2523/36 with arsenic, antimony, bismuth, vanadium, niobium, tantalum, polonium, chromium, molybdenum, tungsten, manganese, technetium or rhenium
- C07C2523/85—Chromium, molybdenum or tungsten
- C07C2523/88—Molybdenum
- C07C2523/882—Molybdenum and cobalt
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2523/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group C07C2521/00
- C07C2523/70—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group C07C2521/00 of the iron group metals or copper
- C07C2523/76—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group C07C2521/00 of the iron group metals or copper combined with metals, oxides or hydroxides provided for in groups C07C2523/02 - C07C2523/36
- C07C2523/84—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group C07C2521/00 of the iron group metals or copper combined with metals, oxides or hydroxides provided for in groups C07C2523/02 - C07C2523/36 with arsenic, antimony, bismuth, vanadium, niobium, tantalum, polonium, chromium, molybdenum, tungsten, manganese, technetium or rhenium
- C07C2523/85—Chromium, molybdenum or tungsten
- C07C2523/88—Molybdenum
- C07C2523/883—Molybdenum and nickel
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2523/00—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group C07C2521/00
- C07C2523/70—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group C07C2521/00 of the iron group metals or copper
- C07C2523/76—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group C07C2521/00 of the iron group metals or copper combined with metals, oxides or hydroxides provided for in groups C07C2523/02 - C07C2523/36
- C07C2523/84—Catalysts comprising metals or metal oxides or hydroxides, not provided for in group C07C2521/00 of the iron group metals or copper combined with metals, oxides or hydroxides provided for in groups C07C2523/02 - C07C2523/36 with arsenic, antimony, bismuth, vanadium, niobium, tantalum, polonium, chromium, molybdenum, tungsten, manganese, technetium or rhenium
- C07C2523/85—Chromium, molybdenum or tungsten
- C07C2523/888—Tungsten
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2603/00—Systems containing at least three condensed rings
- C07C2603/02—Ortho- or ortho- and peri-condensed systems
- C07C2603/04—Ortho- or ortho- and peri-condensed systems containing three rings
- C07C2603/22—Ortho- or ortho- and peri-condensed systems containing three rings containing only six-membered rings
- C07C2603/24—Anthracenes; Hydrogenated anthracenes
Definitions
- the present invention relates to a process for isolating carbazole and to producing 9, 10-dihydroanthracene from crude anthracene, comprising the partial hydrogenation of enriched crude anthracene and a subsequent solvent crystallization of the hydrogenation product of the enriched crude anthracene.
- anthracene oil In technical coal tar distillation, in addition to light oil, medium oil, heavy oil and pitch, the so-called anthracene oil is obtained in the boiling range of 270-400 ° C. This crude anthracene oil is cooled and the crystal mass which separates out is separated off by filtration or centrifugation. In this way two crude products are obtained, the crude anthracene and the filtered or centrifuged anthracene oil. In addition to anthracene, crude anthracene mainly contains carbazole, phenanthrene and fluorene.
- Carbazole is used in the manufacture of various dyes, insecticides and vinyl carbazole. Also of industrial importance is a partial hydrogenation product of anthracene, 9, 10-dihydroanthracene, which serves as a starting material for the production of anthraquinone. There is therefore increased interest in obtaining these two substances, carbazole and 9, 10-dihydroanthraquinone.
- crude anthracene is first separated from the anthracene oil and then this anthracene oil is separated to obtain carbazole and anthracene.
- the crude anthracene is separated from the anthracene oil by a crystallization step using methanol and hydrocarbons in the form of paraffins, aromatic alkyl hydrocarbons and naphthenes as solvents.
- the crude anthracene crystals formed during the crystallization can be separated from the mother liquor by conventional separation methods such as filtering or centrifuging.
- the crude anthracene is then extracted with N-methylpyrrolidone and optionally 10% water in two stages at temperatures so low that only a little anthracene dissolves in the N-methylpyrrolidone, while the other components of the crude anthracene, especially carbazole and phenanthrene, with the N- Form methylpyrrolidone complexes and can be separated.
- the carbazole can be obtained from the extractant in two ways. On the one hand, a carbazole / phenanthrene fraction can first be separated off by adding water, from which crude carbazole is subsequently extracted with pyrolytic oil as the extractant and finally recrystallized.
- the N-methylpyrrolidone can first be driven off from the N-methylpyrrolidone extract and the distillation residue can then be subjected to a fractional crystallization from pyrolytic oil in order to separate carbazole and phenanthrene.
- the main disadvantage of this multi-stage process is its complexity. Since the carbazole content in the crude anthracene used for the extraction is also relatively low and is, for example, only 10% by weight large quantities of solvents are required in the various extraction steps, which reduces the economic attractiveness of the process.
- the object of the present invention is to provide a new and more efficient process which makes it possible to provide both carbazole and 9, 10-dihydroanthracene from crude anthracene in a simple manner with high selectivity and, at the same time, good overall yield.
- This object is achieved by a process for isolating carbazole and producing 9, 10-dihydroanthracene from crude anthracene which contains anthracene, carbazole and phenanthrene, comprehensively
- step (3) working up the hydrogenation product from step (3) by solvent crystallization to form a second crystal product which contains at least 90% carbazole.
- the crude anthracene used in the process according to the invention usually originates from the working up of carbo-derived crude tar and contains anthracene, carbazole and phenanthrene and optionally fluorene.
- Preference is raw anthracene, which contains 20-50% by weight, in particular 30-40% by weight, anthracene, 10-40% by weight, preferably 20-30% by weight. and in particular 20-25% by weight of carbazole, 15-30% by weight, in particular 20-30% by weight of phenanthrene and 0-15% by weight, in particular 3-6% by weight of fluorene.
- the crude anthracene may also contain methylfluorenes, methylphenanthrenes, methylanthracenes.
- small amounts of sulfur e.g. in the form of diphenylene sulfide, usually in the range from 1000 to 5000 ppm, corresponding to 0.1 to 0.5%.
- naphthalene drip oil The residual oil from pure naphthalene production used in the suspension crystallization together with the crude anthracene is referred to as so-called naphthalene drip oil and preferably contains 30-60% by weight, in particular 40-50% by weight, naphthalene, 10-30% by weight, in particular 20-30 % By weight, methylindene, 1-10% by weight. , in particular 1-5% by weight, methylnaphthalene and 1-10% by weight, in particular 4-8% by weight, indene.
- the suspension crystallization of the crude anthracene together with the residual oil of the pure naphthalene production according to step 1 of the process according to the invention is carried out by first heating the residual oil of the pure naphthalene production in a stirred vessel to a temperature of 100-180 ° C., then adding the raw anthracene and at the temperature of 100 - Stirring 180 ° C until the raw anthracene has dissolved. The solution is then cooled to a temperature of 50-100.degree. C., preferably 60-90.degree. C. over a period of 3-6 hours, the enriched crude anthracene crystallizing out as crystal material.
- step (2) of the process according to the invention crystal material is removed from the mother liquor, i.e. the supernatant
- the isolated crystal contains essentially carbazole and anthracene.
- the crystal material of the suspension crystallization is therefore also referred to as enriched raw anthracene.
- the crystal of enriched crude anthracene preferably contains 35-60% by weight, preferably 50-60% by weight and in particular 50-55% by weight of anthracene, 15-40% by weight, preferably 30-40% by weight.
- the enriched raw anthracene can consist of up to 95% carbazole and anthracene.
- the carbazole yield of the suspension crystallization is 65-75%, based on the carbazole in the crude anthracene.
- the crystal material separated from enriched crude anthracene separated in step (2) is subjected in step (3) to a partial hydrogenation in the presence of sulfur-resistant hydrogenation catalysts.
- Preferred sulfur-resistant hydrogenation catalysts are sulfide-activated catalysts which, in addition to oxides of metals from group VIII of the periodic table, contain tungsten or molybdenum oxide. These catalysts are preferably on a support material such as aluminum oxide, silica or mixtures thereof applied. Furthermore, the catalyst can contain basic promoter oxides, which are selected from groups IA, IIA, the lanthanides and the actinides of the periodic table. Sulfidated Co / Mo, Ni / Mo or Ni / W catalysts are particularly suitable. The sulfidation of the catalysts before use in the hydrogenation is preferably carried out using dimethyl disulfide. The sulfided catalyst is usually used as a fixed bed in the subsequent hydrogenation experiments.
- Suitable catalysts are described, for example, in US Pat. No. 4,720,477, EP-A-0 389 119, EP-A-0 082 726 and EP-A-0 046 634. The disclosure of these catalysts in the cited documents is hereby specifically referred to ("incorporation by reference").
- the hydrogenation of the enriched crude anthracene can be carried out continuously or batchwise. Work is preferably carried out batchwise.
- the enriched crude anthracene is placed in a reaction vessel, for example a stirred autoclave, mixed with the suspended form of the sulfided catalyst and the mixture is melted while heating to 170-220 ° C., preferably 190-210 ° C.
- the sulfidated hydrogenation catalyst is used in an amount of 5-10%, based on the entire reaction mixture.
- the hydrogenation is carried out at a reaction temperature of 150-300 ° C., preferably 240-290 ° C. and a reaction pressure of 40-100 bar, preferably 50-70 bar hydrogen and a reaction time of 2-8 h, preferably 4-5 h .
- the anthracene portion of the crude anthracene is selectively and largely completely hydrogenated to 9, 10-dihydroanthracene, while the carbazole portion of the crude anthracene remains unchanged.
- the hydrogenation product of the enriched crude anthracene from step (3) is worked up in step (4) by solvent crystallization.
- the hydrogenation product is dissolved in an aromatic solvent, preferably by raising the temperature to 70-130 ° C.
- BTX aromatics benzene / toluene / xylene
- the solution obtained is then cooled to a temperature of 0-30 ° C. with stirring.
- the second crystal material formed in the solvent crystallization is separated off by filtration, optionally with a subsequent washing step, and dried.
- the selectivity of this solvent crystallization with respect to the carbazole is excellent.
- the crystal contains at least 90% by weight, preferably at least 95% by weight and in particular at least 96% by weight of carbazole.
- the suspension crystallization is characterized in that the substances contained in addition to carbazole and anthracene in the raw anthracene and also referred to as ballast components are removed in a targeted manner, with a simultaneous increase in the carbazole and anthracene content in the enriched raw anthracene. Since at least some of the ballast components would be accessible for hydrogenation, their absence or only a low concentration in the enriched crude anthracene means that a smaller amount of hydrogen is required in the hydrogenation. At the same time, a higher mass throughput of the target compound carbazole can be achieved in the hydrogenation. Both Effects make a positive contribution to the economy of the overall process.
- the subsequent solvent crystallization is only so excellent because the hydrogenation product of the enriched crude anthracene which occurs contains, apart from carbazole and 9, 10-dihydroanthracene, only small amounts of other hydrogenated or non-hydrogenated dietary fibers which could negatively influence the quality of the crystallization.
- Co / Mo catalysts with 4% cobalt oxide and 14.5% molybdenum oxide are used on an aluminum oxide support. These are commercial products from BASF AG with the designations BASF M8-12 and M8-14.
- a Ni / Mo catalyst with 1-10% cobalt oxide and less than 25% molybdenum on an alumina / silica support is used.
- the hydrogenation catalysts Before the hydrogenation catalysts are used, they are subjected to sulfidation in the presence of dimethyl disulfide subject.
- the catalyst placed in a 2 L reaction tube with 1.5 kg of a 67% solution of dimethyl disulfide in 38 kg mineral oil at 225 to 300 ° C under a hydrogen atmosphere (240 L exhaust gas / h) at a throughput of 1.0 V / Vh ( Volume of liquid educt (enriched raw anthracene) / (volume of catalyst xh)) sulfurized over 15 hours.
- the temperature gradient between 225 and 300 ° C is 3 ° C per hour.
- the hydrogenation of the enriched crude anthracene is carried out batchwise in a stirred autoclave.
- the composition of the enriched crude anthracene used as the starting material is shown in Table 2.
- the enriched crude anthracene used in Example II-1 is the product obtained in the suspension crystallization described under point I.
- the enriched raw anthracene present as crystal is introduced into the stirred autoclave.
- the hydrogenation catalyst is then added in suspended form and the mixture is melted while heating to 200.degree.
- the amount of catalyst used in the respective experiments and the other reaction conditions, such as reaction temperature, reaction pressure and reaction time, are shown in Tables 3, 4 and 5.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Indole Compounds (AREA)
Abstract
Das vorliegende Verfahren ermöglicht die Isolierung von Carbazol sowie die gleichzeitige Herstellung von 9,10-Dihydroanthracen aus Rohanthracen, welches Anthracen, Carbazol und Phenanthren enthält. Das Verfahren umfasst eine Suspensionskristallisation des Rohanthracens unter Bildung von angereichertem Rohanthracen, welches nach Abtrennung hydriert sowie abschließend einer Lösemittelkristallisation unterworfen wird. Das erhaltene Kristallgut enthält mindestens 90% Carbazol.
Description
Verfahren zur Isolierung von Carbazol sowie Herstellung von 9, 10-Dihydroanthracen aus Rohanthracen
Die vorliegende Erfindung betrifft ein Verfahren zur Isolierung von Carbazol sowie Herstellung von 9, 10-Dihydro- anthracen aus Rohanthracen umfassend die partielle Hydrierung von angereichertem Rohanthracen und eine anschließende Lösungsmittelkristallisation des Hydrierpro- duktes des angereicherten Rohanthracens.
Bei der technischen Steinkohlenteerdestillation fällt neben Leichtöl, Mittelöl, Schweröl und Pech im Siedebereich von 270-400°C das sogenannte Anthracenöl an. Dieses rohe Anthracenöl wird ausgekühlt und die sich dabei ausscheidende Kristallmasse durch Filtrieren oder Zentrifugieren abgetrennt. Man erhält auf diese Weise zwei Rohprodukte, das Rohanthracen und das filtrierte bzw. abgeschleuderte Anthracenöl. Das Rohanthracen ent- hält neben Anthracen vor allem Carbazol, Phenanthren und Fluoren.
Carbazol findet in der Herstellung von verschiedenen Farbstoffen, Insektiziden sowie Vinylcarbazol Anwendung. Von industrieller Bedeutung ist auch ein partielles Hydrierungsprodukt des Anthracens, 9, 10-Dihydroanthracen, das als Ausgangsmaterial für die Herstellung von Anthra- chinon dient. An einer Gewinnung dieser beiden Substanzen, Carbazol und 9, 10-Dihydroanthrachinon, besteht daher ein gesteigertes Interesse.
Die Gewinnung von Carbazol aus Rohanthracen über einen Kristallisationsschritt bereitet Probleme, da Carbazol und Anthracen immer gemeinsam in einem bestimmten Ver- hältnis zueinander auskristallisieren.
Aus der Literatur sind extraktive Verfahren zur Abtrennung von Carbazol aus Anthracenölen bekannt .
So wird in Koks, Smola, Gaz (1986), 31(4), 65-7 beschrieben, zunächst Rohanthracen aus dem Anthracenöl abzutrennen und dieses Anthracenöl dann unter Gewinnung von Carbazol und Anthracen aufzutrennen. Die Abtrennung des Rohanthracens aus dem Anthracenöl erfolgt dabei durch einen Kristallisationsschritt unter Verwendung von Methanol und Kohlenwasserstoffen in Form von Paraffinen, aromatischen Alkylkohlenwasserstoffen sowie Naphthenen als Lösungsmitteln. Die bei der Kristallisation gebildeten Rohanthracenkristalle können durch übliche Separationsme- thoden wie Filtieren oder Abschleudern von der Mutterlauge abgetrennt werden. Das Rohanthracen wird anschließend mit N-Methylpyrrolidon und gegebenenfalls 10% Wasser in zwei Stufen bei so niedrigen Temperaturen extrahiert, daß sich nur wenig Anthracen im N-Methylpyrrolidon löst, während die übrigen Komponenten des Rohanthracens, vor allem Carbazol und Phenanthren, mit dem N-Methylpyrrolidon Komplexe bilden und so abgetrennt werden können. Das Carbazol kann aus dem Extraktionsmittel auf zwei Wegen gewonnen werden. Zum einen kann zuerst durch Zusatz von Wasser eine Carbazol/Phenanthren Fraktion abgeschieden werden, aus der nachfolgend mit pyrolytischem Öl als Extraktionsmittel Rohcarbazol extrahiert und abschließend rekristallisiert wird. Zum anderen kann aus dem N-Methyl- pyrrolidon-Extrakt zunächst das N-Methylpyrrolidon abge- trieben und der Destillationsrückstand zur Trennung von Carbazol und Phenanthren dann einer fraktionierenden Kristallisation aus pyrolytischem Öl unterworfen werden. Nachteilig an diesem in der Summe vielstufigen Verfahren ist vor allem die Komplexität. Da der Carbazol -Anteil in dem zur Extraktion eingesetzten Rohanthracen zudem relativ niedrig sein und beispielsweise nur 10 Gew.% betragen
kann, werden große Mengen Lösungsmittel in den verschiedenen Extraktionsschritten benötigt, was die wirtschaftliche Attraktivität des Verfahrens mindert.
Die Aufgabe der vorliegenden Erfindung besteht darin, ein neues und leistungsfähigeres Verfahren zur Verfügung zu stellen, das die Bereitstellung sowohl von Carbazol als auch von 9, 10-Dihydroanthracen aus Rohanthracen auf einfachem Weg mit hoher Selektivität und gleichzeitig guter Gesamtausbeute ermöglicht.
Gelöst wird diese Aufgabe durch ein Verfahren zur Isolierung von Carbazol sowie Herstellung von 9, 10-Dihydroan- thracen aus Rohanthracen, welches Anthracen, Carbazol und Phenanthren enthält, umfassend
(1) den Einsatz des Rohanthracens zusammen mit einem Rückstandsöl aus der Reinnaphthalinerzeugung in einer Suspensionskristallisation unter Bildung eines Kristallguts aus angereichertem Rohanthracen, (2) Abtrennung dieses Kristallguts aus angereichertem Rohanthracen von der Mutterlauge,
(3) partielle Hydrierung des abgetrennten Kristallguts aus Schritt (2) in Gegenwart eines Schwefel -resisten- ten Katalysators unter Erhalt eines Hydrierproduktes und
(4) Aufarbeitung des Hydrierproduktes aus Schritt (3) durch eine Lösemittelkristallisation unter Bildung eines zweiten Kristallguts, welches mindestens 90% Carbazol enthält.
Das im erfindungsgemäßen Verfahren eingesetzte Rohanthracen stammt üblicherweise aus der Aufarbeitung von carbo- stämmigem Rohteer und enthält Anthracen, Carbazol und Phenanthren sowie gegebenenfalls Fluoren. Bevorzugt ist Rohanthracen, welches 20 - 50 Gew.%, insbesondere 30 - 40 Gew.% Anthracen, 10 - 40 Gew.%, bevorzugt 20 - 30 Gew.%
und insbesondere 20 - 25 Gew.% Carbazol, 15 - 30 Gew.%, insbesondere 20 - 30 Gew.% Phenanthren und 0 - 15 Gew.%, insbesondere 3 - 6 Gew.% Fluoren enthält. Gegebenenfalls enthält das Rohanthracen ferner Methylfluorene, Methylphenanthrene , Methylanthracene .
Zudem können auch geringe Mengen an Schwefel, z.B. in Form von Diphenylensulfid, enthalten sein, üblicherweise im Bereich von 1000 - 5000 ppm, entsprechend 0.1 bis 0.5%.
Das in der Suspensionskristallisation zusammen mit dem Rohanthracen eingesetzte Rückstandsöl der Reinnaphthalinerzeugung wird als sogenanntes Naphthalintropföl bezeichnet und enthält bevorzugt 30 - 60 Gew.%, ins- besondere 40 - 50 Gew.%, Naphthalin, 10 - 30 Gew.%, insbesondere 20 - 30 Gew.%, Methylindene, 1 - 10 Gew% . , insbesondere 1 - 5 Gew.%, Methylnaphthalin und 1 - 10 Gew.%, insbesondere 4 - 8 Gew.%, Inden.
Die Suspensionskristallisation des Rohanthracens zusammen mit dem Rückstandsöl der Reinnaphthalinerzeugung gemäß Schritt 1 des erfindungsgemäßen Verfahrens wird durchgeführt, indem man zunächst das Rückstandsöl der Reinnaphthalinerzeugung in einem Rührbehälter auf eine Temperatur von 100 - 180°C erwärmt, darauf das Rohanthracen hinzugibt und bei der Temperatur von 100 - 180°C rührt, bis das Rohanthracen in Lösung gegangen ist. Anschließend wird die Lösung innerhalb eines Zeitraums von 3 - 6 h auf eine Temperatur von 50 - 100°C, bevorzugt 60 - 90°C abgekühlt, wobei das angereicherte Rohanthracen als Kristallgut auskristallisiert.
Das bei der Suspensionskristallisation entstandene
Kristallgut wird gemäß Schritt (2) des erfindungsgemäßen Verfahrens von der Mutterlauge, d.h. der überstehenden
Lösung abgetrennt. Üblicherweise erfolgt diese Abtrennung
in einer Zentrifuge, die bevorzugt beheizt wird. Die abgetrennte Mutterlauge kann ohne weitere Behandlung als Mischkomponente zur Herstellung technischer aromatischer Öle eingesetzt werden.
Das isolierte Kristallgut enthält im wesentlichen Carbazol und Anthracen. Für den erfolgreichen, weiteren Verlauf des Gesamtverfahrens ist es entscheidend, daß durch die Suspensionskristallisation der Anteil aller übrigen im Rohanthracen enthaltenen Komponenten, wie Phenanthren, Fluoren, Methylfluorene, Methylphenanthrene und Methylan- thracene erheblich verringert wird. Das Kristallgut der Suspensionskristallisation wird daher auch als angereichertes Rohanthracen bezeichnet. Bevorzugt enthält das Kristallgut aus angereichertem Rohanthracen 35 - 60 Gew.%, bevorzugt 50 - 60 Gew.% und insbesondere 50 - 55 Gew.% Anthracen, 15 - 40 Gew.%, bevorzugt 30 - 40 Gew.%. und insbesondere 30 - 35 Gew.% Carbazol, 1 - 25 Gew.%, bevorzugt 1 - 10 Gew.% und insbesondere 1 - 5 Gew.% Phenanthren und 1 - 8 Gew.%, bevorzugt 1 - 5 Gew.% und insbesondere 1 - 3 Gew.% Fluoren. Dies bedeutet, daß das angereicherte Rohanthracen in der Summe bis zu 95% aus Carbazol und Anthracen bestehen kann. Die Carbazol - Ausbeute der Suspensionskristallisation liegt bei 65 - 75%, bezogen auf das Carbazol im Rohanthracen.
Das in Schritt (2) abgetrennte Kristallgut aus angereichertem Rohanthracen wird in Schritt (3) des Verfahrens einer partiellen Hydrierung in Gegenwart Schwefel- resistenter Hydrierkatalysatoren unterworfen.
Bevorzugt kommen als Schwefel-resistente Hydrierkatalysatoren sulfidaktivierte Katalysatoren in Frage, die neben Oxiden von Metallen der Gruppe VIII des Periodensystems Wolfram- oder Molybdänoxid enthalten. Diese Katalysatoren sind bevorzugt auf einem Trägermaterial wie Alumini-
umoxid, Kieselsäure oder Gemischen davon aufgebracht. Ferner kann der Katalysator basische Promotoroxide enthalten, die ausgewählt sind aus den Gruppen IA, IIA, den Lanthaniden und den Actiniden des Periodensystems. Beson- ders geeignet sind sulfidierte Co/Mo, Ni/Mo oder Ni/W- Katalysatoren. Die Sulfidierung der Katalysatoren vor dem Einsatz in der Hydrierung erfolgt dabei bevorzugt unter Verwendung von Dimethyldisulfid. Der sulfidierte Katalysator wird in den nachfolgenden Hydrierversuchen üb- licherweise als Festbett eingesetzt.
Geeignete Katalysatoren sind beispielsweise in der US-A- 4,720,477, der EP-A-0 389 119, der EP-A-0 082 726 und der EP-A-0 046 634 beschrieben. Auf die Offenbarung dieser Katalysatoren in den genannten Schriften wird hiermit konkret Bezug genommen ( "incorporation by reference") .
Die Hydrierung des angereicherten Rohanthracens kann kontinuierlich oder diskontinuierlich erfolgen. Bevorzugt wird diskontinuierlich gearbeitet. Hierzu wird das ange- reicherte Rohanthracen in einem Reaktionsgefäß, beispielsweise einem Rührautoklaven vorgelegt, mit der suspendierten Form des sulfidierten Katalysators versetzt und das Gemisch unter Erwärmung auf 170-220°C, bevorzugt 190-210°C, aufgeschmolzen. Der sulfidierte Hydrierkatalysator wird in einer Menge von 5 - 10%, bezogen auf das gesamte Reaktionsgemisch, eingesetzt. Die Hydrierung wird bei einer Reaktionstemperatur von 150-300°C, bevorzugt 240-290°C und einem Reaktionsdruck von 40-100 bar, bevorzugt 50-70 bar Wasserstoff sowie einer Reaktionszeit von 2-8 h, bevorzugt 4-5 h, durchgeführt.
Bei dieser partiellen Hydrierung wird der Anthracenanteil des Rohanthracens selektiv und weitgehend vollständig zu 9, 10-Dihydroanthracen hydriert, während der Carbazolan- teil des Rohanthracens unverändert bleibt .
Das Hydrierprodukt des angereicherten Rohanthracens aus Schritt (3) wird in Schritt (4) durch eine Lösemittelkristallisation aufgearbeitet. Hierzu wird das Hydrierprodukt in einem aromatischen Lösungsmittel, bevorzugt unter Temperaturerhöhung auf 70 - 130°C, aufgelöst. Als aromatische Lösungsmittel werden bevorzugt BTX-Aromaten (Benzol/Toluol/Xylol) eingesetzt. Die erhaltene Lösung wird anschließend unter Rühren auf eine Temperatur von 0 - 30°C abgekühlt. Das bei der Lösemittelkristallisa- tion gebildete zweite Kristallgut wird durch Filtration gegebenenfalls mit anschließendem Waschschritt abgetrennt und getrocknet . Die Selektivität dieser Lösungsmittelkristallisation im Hinblick auf das Carbazol ist ausgezeichnet. Im Kristallgut sind mindestens 90 Gew.%, bevorzugt mindestens 95 Gew.% und insbesondere mindestens 96 Gew.% Carbazol enthalten.
Für das Gesamtverfahren ergibt sich eine Carbazol-Ausbeute von 63-72%, bezogen auf das Carbazol im einge- setzten Rohanthracen.
Nur durch die Hintereinanderschaltung der Schritte (1) bis (4) im erfindungsgemäßen Verfahren gelingt es, das Carbazol in den genannten Ausbeuten zu isolieren. Die Suspensionskristallisation zeichnet sich dadurch aus, daß die neben Carbazol und Anthracen im Rohanthracen enthaltenen und auch als Ballastkomponenten bezeichneten Substanzen gezielt entfernt werden, bei gleichzeitiger Erhöhung des Carbazol- und Anthracen-Anteils im ange- reicherten Rohanthracen. Da zumindest einige der Ballast- komponenten einer Hydrierung zugänglich wären, hat ihre Abwesenheit bzw. nur geringe Konzentration im angereicherten Rohanthracen zur Folge, daß in der Hydrierung eine geringere Wasserstoffmenge benötigt wird. Gleichzei- tig kann in der Hydrierung ein höherer Massendurchsatz der Zielverbindung Carbazol erreicht werden. Beide
Effekte tragen zur Wirtschaftlichkeit des Gesamtverfahrens positiv bei.
Ensprechend gelingt auch die nachfolgende Lösemittel - kristallisation nur deshalb so exzellent, weil das eintretende Hydrierprodukt des angereicherten Rohanthracens außer Carbazol und 9, 10-Dihydroanthracen nur geringe Mengen anderer hydrierter oder nicht hydrierter Ballaststoffe enthält, die die Güte der Kristallisation negativ beeinflussen könnten.
Beispiele;
I Suspensionskristallisation von Rohanthracen mit Naphthalintropföl
1000 g Naphthalintropföl der in Tabelle la angegebenen Zusammensetzung werden in einem 4 L Rührbehälter vorgelegt und auf 170°C erwärmt. Anschließend werden portions- weise insgesamt 1000 g Rohanthracen der in Tabelle lb angegebenen Zusammensetzung hinzugegeben und bei 170°C vollständig in Lösung gebracht. Die Lösung wird dann unter Rühren während einer Zeitdauer von ca. 4 Stunden auf 80°C abgekühlt, wobei ein angereichertes Rohanthracen auskristallisiert. Die gesamte Kristallsuspension wird in eine auf 80°C geheizte Zentrifuge überführt, in der das Kristallgut aus angereichertem Rohanthracen von der Mutterlauge abgetrennt wird. Hierbei erhält man 457 g Kristallgut der in Tabelle 1 angegebenen Zusammensetzung.
Tabelle 1a:

Tabelle l :
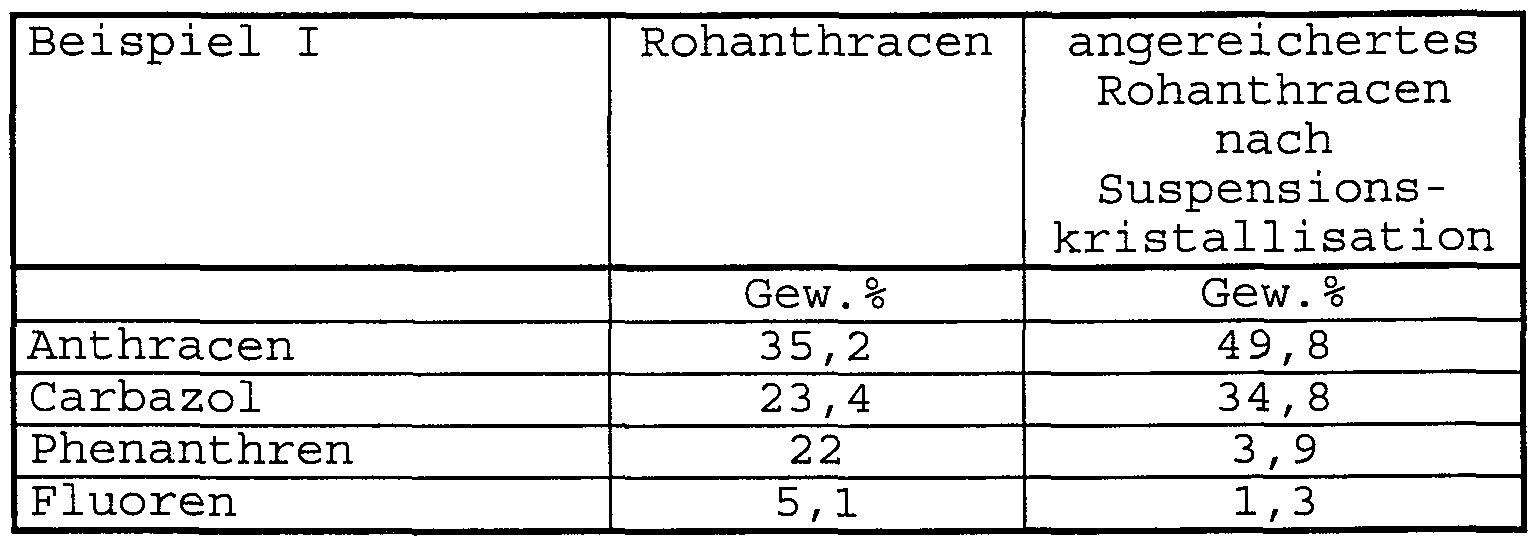
Rohanthracen gemäß Schritt 3 des erfindungsgemäßen Verfahrens a) Sulfidierung des Hydrierkatalysators
In den nachfolgenden Beispielen II- 3,7 und 8 betreffend den Hydrierungsschritt werden Co/Mo-Katalysatoren mit 4% Kobaltoxid und 14.5% Molybdänoxid auf einem Aluminiumoxid-Träger eingesetzt. Es handelt sich hierbei um Handelsprodukte der BASF AG mit den Bezeichnungen BASF M8-12 und M8-14. In den Beispielen II- 1, 2, 4, 5 und 6 wird ein Ni/Mo-Katalysator mit 1-10% Kobaltoxid und weniger als 25% Molybdän auf einem Aluminiumoxid/Kieselsäure- Träger eingesetzt .
Vor dem Einsatz der Hydrierkatalysatoren werden diese zuvor einer Sulfidierung in Gegenwart von Dimethyldisulfid
unterworfen. Hierbei wird der in ein 2 L Reaktionsrohr eingebrachte Katalysator mit 1.5 kg einer 67%igen Lösung von Dimethyldisulfid in 38 kg Mineralöl bei 225 bis 300°C unter Wasserstoff-Atmosphäre (240 L Abgas/h) bei einem Durchsatz von 1.0 V/Vh (Volumen flüssiges Edukt (angereichertes Rohanthracen) / (Volumen Katalysator x h) ) über 15 Stunden geschwefelt. Der Temperaturgradient zwischen 225 und 300°C beträgt 3°C pro Stunde.
b) Hydrierung
Die Hydrierung des angereicherten Rohanthracens erfolgt diskontinuierlich in einem Rührautoklaven. Die Zusammensetzung des als Ausgangsmaterial eingesetzten, angerei- cherten Rohanthracens ist Tabelle 2 zu entnehmen. Bei dem in Beispiel II -1 eingesetzten angereicherten Rohanthracen handelt es sich um das in der unter Punkt I beschriebenen Suspensionskristallisation erhaltene Produkt.
Zunächst wird das als Kristallgut vorliegende, angereicherte Rohanthracen in den Rührautoklaven eingebracht . Anschließend wird der Hydrierkatalysator in suspendierter Form hinzugegeben und das Gemisch unter Erwärmung auf 200°C aufgeschmolzen. Die in den jeweiligen Versuchen eingesetzte Katalysatormenge und die übrigen Reaktionsbedingungen wie Reaktionstemperatur, Reaktionsdruck und Reaktionszeit sind in den Tabellen 3, 4 und 5 zusammengestellt.
Tabelle 3 -. Beispiel II-l

Tabe.1 3 ; Beispiele II- 2 und 3 (Variation des Katalysators)


Tab l 1 e : Beispiel II-7 und 8 (Variation der Reaktionszeit)

Hydrierprodukts des angereicherten Rohanthracens
100 g des Hydrierprodukts des angereicherten Rohanthracens aus Beispiel II-l mit der in Tabelle 6 angegebenen Zusammensetzung werden bei 100°C in 400 mL Toluol gelöst. Anschließend wird die Lösung unter Rühren auf 20°C abgekühlt. Das ausgefallene Kristallgut wird durch Filtration von der Mutterlauge abgetrennt, mit 40 mL frischem Toluol bedeckt und anschließend getrocknet. Die Zusammensetzung des Kristallguts ist in Tabelle 6 angegeben. Enthalten sind 97,4% Carbazol.
Tabelle 6

Claims
1. Verfahren zur Isolierung von Carbazol sowie Herstellung von 9 , 10 -Dihydroanthracen aus Rohanthracen, welches Anthracen, Carbazol und Phenanthren enthält, umfassend
(1) den Einsatz des Rohanthracens zusammen mit einem Rückstandsöl aus der Reinnaphthalinerzeugung in einer Suspensionskristallisation unter Bildung eines Kristallguts aus angereichertem Rohanthracen , (2) Abtrennung dieses Kristallguts aus angereichertem Rohanthracen von der Mutterlauge,
(3) partielle Hydrierung des abgetrennten Kristallguts aus Schritt (2) in Gegenwart eines Schwefel- resistenten Katalysators unter Erhalt eines Hydrierproduktes und
(4) Aufarbeitung des Hydrierproduktes aus Schritt (3) durch eine Lösemittelkristallisation unter Bildung eines zweiten Kristallguts, welches mindestens 90% Carbazol enthält.
2. Verfahren nach Anspruch 1, dadurch gekennzeichnet, daß das Rohanthracen 20 - 50 Gew.%, insbesondere 30 - 40 Gew.% Anthracen, 10 - 40 Gew.%, bevorzugt 20 - 30 Gew.% und insbesondere 20 - 25 Gew.% Carbazol, 15-30 Gew.%, insbesondere 20 - 30 Gew.% Phenanthren und 0 - 15 Gew.%, insbesondere 3 - 6 Gew.% Fluoren enthält.
3. Verfahren nach Anspruch 1 oder 2 , dadurch gekennzeichnet, daß das in der Suspensionskristallisation zusammen mit dem Rohanthracen eingesetzte Rückstandsöl der Reinnaphthalinerzeugung 30 - 60 Gew.%,
insbesondere 40 - 50 Gew.%, Naphthalin, 10 - 30 Gew.%, insbesondere 20 - 30 Gew.%, Methylindene, 1 - 10 Gew.%, insbesondere 1 - 5 Gew.%, Methylnaphthalin und 1 - 10 Gew.%, insbesondere 4 - 8 Gew.%, Inden enthält .
4. Verfahren nach einem oder mehreren der Ansprüche 1 bis 3, dadurch gekennzeichnet, daß in der Suspensionskristallisation zunächst das Rückstandsöl der Reinnaphthalinerzeugung in einem Rührbehälter auf eine Temperatur von 100 - 180°C erwärmt, darauf das Rohanthracen hinzugegeben und bei der Temperatur von 100 - 180 °C gerührt wird, bis das Rohanthracen in Lösung gegangen ist und anschließend die Lösung innerhalb eines Zeitraums von 3 - 6 h auf eine Temperatur von 50 - 100°C, bevorzugt 60 - 90°C abgekühlt wird.
5. Verfahren nach einem oder mehreren der Ansprüche 1 bis 4, dadurch gekennzeichnet, daß das Kristallgut aus angereichertem Rohanthracen 35 - 60 Gew.%, bevorzugt 50 - 60 Gew.% und insbesondere 50 - 55 Gew.% Anthracen, 15 - 40 Gew.%, bevorzugt 30 - 40 Gew.% und insbesondere 30 - 35 Gew.% Carbazol, 1 - 25 Gew.%, bevorzugt 1 - 10 Gew.% und insbesondere 1
- 5 Gew.% Phenanthren und 1 - 8 Gew.%, bevorzugt 1
- 5 Gew.% und insbesondere 1 - 3 Gew.% Fluoren enthält .
6. Verfahren nach einem oder mehreren der Ansprüche 1 bis 5, dadurch gekennzeichnet, daß im Hydrierschritt (3) sulfidaktivierte Katalysatoren verwendet werden, die neben Oxiden von Metallen der Gruppe VIII des Periodensystems Wolfram- oder Molybdänoxid enthalten.
7. Verfahren nach Anspruch 6, dadurch gekennzeichnet, daß die Katalysatoren auf Aluminiumoxid, Kieselsäure oder Gemischen davon als Trägermaterial aufgebracht sind und basische Promotoroxide enthalten, die ausge- wählt sind aus den Gruppen IA, IIA, den Lanthaniden und den Actiniden des Periodensystems.
8. Verfahren nach Anspruch 6 oder 7 , dadurch gekennzeichnet, daß sulfidierte Co/Mo, Ni/Mo oder Ni/W-Ka- talysatoren eingesetzt werden.
9. Verfahren nach einem oder mehreren der Ansprüche 1 bis 8, dadurch gekennzeichnet, daß die Hydrierung des angereicherten Rohanthracens in Schritt (3) kontinu- ierlich oder diskontinuierlich und bei einer Reaktionstemperatur von 150 - 300°C, bevorzugt 240 - 290°C, und einem Reaktionsdruck von 40 - 100 bar, bevorzugt 50 - 70 bar, Wasserstoff sowie einer Reaktionszeit von 2 - 8 h, bevorzugt 4 - 5 h, durchgeführt wird.
10. Verfahren nach einem oder mehreren der Ansprüche 1 bis 9, dadurch gekennzeichnet, daß das Hydrierprodukt zur Lösemittelkristallisation in einem aromatischen Lösungsmittel , bevorzugt unter Temperaturerhöhung auf 70 - 130°C, aufgelöst und anschließend unter Rühren auf eine Temperatur von 0 - 30°C abgekühlt wird.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE19757530A DE19757530C2 (de) | 1997-12-23 | 1997-12-23 | Verfahren zur Isolierung von Carbazol sowie Herstellung von 9,10-Dihydroanthracen aus Rohanthracen |
| DE19757530.7 | 1997-12-23 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1999033802A1 true WO1999033802A1 (de) | 1999-07-08 |
Family
ID=7853181
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP1998/008218 Ceased WO1999033802A1 (de) | 1997-12-23 | 1998-12-15 | Verfahren zur isolierung von carbazol sowie herstellung von 9,10-dihydroanthracen aus rohanthracen |
Country Status (3)
| Country | Link |
|---|---|
| DE (1) | DE19757530C2 (de) |
| WO (1) | WO1999033802A1 (de) |
| ZA (1) | ZA9811724B (de) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN118851983A (zh) * | 2024-07-25 | 2024-10-29 | 大连理工大学 | 一种富蒽抽提液选择加氢制9,10-二氢蒽和咔唑的方法 |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE10103208A1 (de) * | 2001-01-24 | 2002-08-14 | Ruetgers Vft Ag | Gewinnung von Anthracen und Carbazol durch Schmelzkristallisation |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| NL74130C (de) * | 1952-05-07 | |||
| US2438148A (en) * | 1945-02-01 | 1948-03-23 | Koppers Co Inc | Crude anthracene separation |
| US4720477A (en) * | 1985-10-10 | 1988-01-19 | Ashland Oil, Inc. | Method for converting coal to upgraded liquid product |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR1401255A (fr) * | 1964-04-02 | 1965-06-04 | Expl Des Procedes Ab Der Halde | Procédé d'extraction de l'anthracène et du carbazol des huiles anthracéniques brutes de goudrons de houille |
| US3624174A (en) * | 1970-05-11 | 1971-11-30 | Chem Systems | Recovery of anthracene and carbazole |
-
1997
- 1997-12-23 DE DE19757530A patent/DE19757530C2/de not_active Expired - Fee Related
-
1998
- 1998-12-15 WO PCT/EP1998/008218 patent/WO1999033802A1/de not_active Ceased
- 1998-12-21 ZA ZA9811724A patent/ZA9811724B/xx unknown
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2438148A (en) * | 1945-02-01 | 1948-03-23 | Koppers Co Inc | Crude anthracene separation |
| NL74130C (de) * | 1952-05-07 | |||
| US4720477A (en) * | 1985-10-10 | 1988-01-19 | Ashland Oil, Inc. | Method for converting coal to upgraded liquid product |
Non-Patent Citations (1)
| Title |
|---|
| CHEMICAL ABSTRACTS, vol. 35, no. 15, 10 August 1941, Columbus, Ohio, US; KOSAKA ET AL.: "Separation of anthracene and carbazole from anthracene cake" column 5108; XP002099401 * |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN118851983A (zh) * | 2024-07-25 | 2024-10-29 | 大连理工大学 | 一种富蒽抽提液选择加氢制9,10-二氢蒽和咔唑的方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| ZA9811724B (en) | 1999-06-24 |
| DE19757530A1 (de) | 1999-07-08 |
| DE19757530C2 (de) | 2000-03-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| DE2526887C2 (de) | Verfahren zur Herstellung von aromatischen Kohlenwasserstoffen | |
| DE2424296C2 (de) | Verfahren zur Herstellung von Schmierölen | |
| DE1271292B (de) | Verfahren zur Herstellung von Schmieroelen oder Schmieroelbestandteilen | |
| DE2425982B2 (de) | Verfahren zur Herstellung von 2,6-Dimethylnaphthalin | |
| DE2459385A1 (de) | Verfahren zur herstellung von schmieroelen mit hohem viskositaetsindex | |
| EP0441195B1 (de) | Verfahren zur Herstellung von oxidationsstabilen und kältestabilen Grundölen und Mitteldestillaten | |
| DE69433800T2 (de) | Verfahren zur Kristallisierung von 2,6-Dimethylnaphthalen | |
| DE69417607T2 (de) | Verfahren zur Herstellung von Benzin mit hoher Oktanzahl | |
| DE2143972A1 (de) | Verfahren zur Herstellung von Schmier öl mit verbessertem Viskositätsindex | |
| DE3590067T (de) | Oberflächenaktives Mittel | |
| WO1999033802A1 (de) | Verfahren zur isolierung von carbazol sowie herstellung von 9,10-dihydroanthracen aus rohanthracen | |
| DE1949702A1 (de) | Verfahren zur Herstellung verbesserter Schmieroele | |
| DE69201824T2 (de) | Verfahren zur Raffinierung von Methylnaphtalen enthaltendes Öl. | |
| DD263288A5 (de) | Verfahren zur herstellung von 5-h-dibenzo[b,f]azepin oder iminostilben | |
| DE60102641T2 (de) | Verfahren zur Herstellung von 2,6-Dimethylnaphthalin | |
| DE69209934T2 (de) | Verfahren zur Herstellung von 2-Methylnaphthalin | |
| DE2952062A1 (de) | Verfahren zur herstellung von anthracen | |
| DE68905673T2 (de) | Verfahren zur Ausscheidung von 2,6-Dimethylnaphthalin. | |
| DE1959869C3 (de) | Verfahren zur Herstellung einer Schmierölfraktion mit erhöhtem Viskositätsindex | |
| EP0999210A1 (de) | Verfahren zur Reinigung von Isochinolin | |
| EP0921111B1 (de) | Verfahren zur Herstellung von substituierten Anthrachinonen | |
| EP0155610B1 (de) | Verfahren zur Herstellung von 2-Hydroxy-(9H)-carbazol | |
| DE701418C (de) | Gewinnung mehrkerniger cyclischer Verbindungen | |
| EP0014406B1 (de) | Verfahren zur Abtrennung von Selen, Selenverbindungen, Schwefel und/oder Schwefelverbindungen aus diese Elemente bzw. Verbindungen aufweisenden Urethanen | |
| DE1644935A1 (de) | Verfahren zur Herstellung von Schmieroelen |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): BR CA CN CZ ID IN JP KR MX PL RU UA US |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| NENP | Non-entry into the national phase |
Ref country code: KR |
|
| NENP | Non-entry into the national phase |
Ref country code: CA |
|
| 122 | Ep: pct application non-entry in european phase |