WO1999064392A1 - Benzamidine derivative - Google Patents
Benzamidine derivative Download PDFInfo
- Publication number
- WO1999064392A1 WO1999064392A1 PCT/JP1999/003055 JP9903055W WO9964392A1 WO 1999064392 A1 WO1999064392 A1 WO 1999064392A1 JP 9903055 W JP9903055 W JP 9903055W WO 9964392 A1 WO9964392 A1 WO 9964392A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- carbon atoms
- formula
- carbonyl
- piperidine
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/04—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C257/00—Compounds containing carboxyl groups, the doubly-bound oxygen atom of a carboxyl group being replaced by a doubly-bound nitrogen atom, this nitrogen atom not being further bound to an oxygen atom, e.g. imino-ethers, amidines
- C07C257/10—Compounds containing carboxyl groups, the doubly-bound oxygen atom of a carboxyl group being replaced by a doubly-bound nitrogen atom, this nitrogen atom not being further bound to an oxygen atom, e.g. imino-ethers, amidines with replacement of the other oxygen atom of the carboxyl group by nitrogen atoms, e.g. amidines
- C07C257/18—Compounds containing carboxyl groups, the doubly-bound oxygen atom of a carboxyl group being replaced by a doubly-bound nitrogen atom, this nitrogen atom not being further bound to an oxygen atom, e.g. imino-ethers, amidines with replacement of the other oxygen atom of the carboxyl group by nitrogen atoms, e.g. amidines having carbon atoms of amidino groups bound to carbon atoms of six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D207/00—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D207/46—Heterocyclic compounds containing five-membered rings not condensed with other rings, with one nitrogen atom as the only ring hetero atom with hetero atoms directly attached to the ring nitrogen atom
- C07D207/48—Sulfur atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D211/00—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings
- C07D211/04—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom
- C07D211/06—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members
- C07D211/36—Heterocyclic compounds containing hydrogenated pyridine rings, not condensed with other rings with only hydrogen or carbon atoms directly attached to the ring nitrogen atom having no double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D211/40—Oxygen atoms
- C07D211/44—Oxygen atoms attached in position 4
- C07D211/46—Oxygen atoms attached in position 4 having a hydrogen atom as the second substituent in position 4
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/04—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member
- C07D233/20—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having one double bond between ring members or between a ring member and a non-ring member with substituted hydrocarbon radicals, directly attached to ring carbon atoms
- C07D233/26—Radicals substituted by carbon atoms having three bonds to hetero atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D401/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom
- C07D401/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings
- C07D401/06—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, at least one ring being a six-membered ring with only one nitrogen atom containing two hetero rings linked by a carbon chain containing only aliphatic carbon atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D417/00—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00
- C07D417/02—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings
- C07D417/04—Heterocyclic compounds containing two or more hetero rings, at least one ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for by group C07D415/00 containing two hetero rings directly linked by a ring-member-to-ring-member bond
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/28—Phosphorus compounds with one or more P—C bonds
- C07F9/38—Phosphonic acids [RP(=O)(OH)2]; Thiophosphonic acids ; [RP(=X1)(X2H)2(X1, X2 are each independently O, S or Se)]
- C07F9/40—Esters thereof
- C07F9/4003—Esters thereof the acid moiety containing a substituent or a structure which is considered as characteristic
- C07F9/4015—Esters of acyclic unsaturated acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/28—Phosphorus compounds with one or more P—C bonds
- C07F9/38—Phosphonic acids [RP(=O)(OH)2]; Thiophosphonic acids ; [RP(=X1)(X2H)2(X1, X2 are each independently O, S or Se)]
- C07F9/40—Esters thereof
- C07F9/4003—Esters thereof the acid moiety containing a substituent or a structure which is considered as characteristic
- C07F9/4056—Esters of arylalkanephosphonic acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/553—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom having one nitrogen atom as the only ring hetero atom
- C07F9/576—Six-membered rings
- C07F9/59—Hydrogenated pyridine rings
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07F—ACYCLIC, CARBOCYCLIC OR HETEROCYCLIC COMPOUNDS CONTAINING ELEMENTS OTHER THAN CARBON, HYDROGEN, HALOGEN, OXYGEN, NITROGEN, SULFUR, SELENIUM OR TELLURIUM
- C07F9/00—Compounds containing elements of Groups 5 or 15 of the Periodic Table
- C07F9/02—Phosphorus compounds
- C07F9/547—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom
- C07F9/6558—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing at least two different or differently substituted hetero rings neither condensed among themselves nor condensed with a common carbocyclic ring or ring system
- C07F9/65583—Heterocyclic compounds, e.g. containing phosphorus as a ring hetero atom containing at least two different or differently substituted hetero rings neither condensed among themselves nor condensed with a common carbocyclic ring or ring system each of the hetero rings containing nitrogen as ring hetero atom
Definitions
- the present invention provides a novel orally administrable benzamidine derivative which reversibly inhibits activated blood coagulation factor X and exhibits a potent anticoagulant effect, and is caused by a blood coagulation inhibitor or a thrombus or embolus containing them as an active ingredient. It is related to the prevention and treatment of diseases.
- Examples of the disease to be applied include diseases in cerebrovascular disorders such as cerebral infarction, cerebral thrombosis, cerebral embolism, transient ischemic attack ( ⁇ ⁇ ⁇ ), subarachnoid hemorrhage (vascular spasm), acute and chronic myocardial infarction , Unstable angina, diseases in ischemic heart disease such as coronary artery thrombosis, diseases in pulmonary vascular disorders such as pulmonary infarction, pulmonary embolism, peripheral arterial occlusion, deep vein thrombosis, disseminated intravascular coagulation, and artificial Thrombus formation after angioplasty and valve replacement, reocclusion and restenosis after coronary artery bypass surgery, percutaneous transluminal coronary angioplasty (PTCA) or percutaneous transluminal coronary revascularization (PTCR) Reocclusion and restenosis after revascularization, and thrombus formation during extracorporeal circulation.
- diseases in cerebrovascular disorders such as cerebral infarction, cerebral thro
- thromboembolic diseases such as myocardial infarction, cerebral thrombosis, and peripheral arterial thrombosis tend to increase year by year, and the importance of their treatment is increasing.
- Anticoagulant therapy together with fibrinolytic therapy and antiplatelet therapy, plays a part in medical treatment in the treatment and prevention of thrombosis.
- antithrombin drugs have been developed as thrombus formation inhibitors.Thrombin not only controls the activation of fibrinogen to fipurin, which is the final stage of the coagulation reaction, but also activates thrombocytes and platelets.
- Activated blood coagulation factor X is located at the confluence of extrinsic and intrinsic coagulation cascade reactions and is located upstream of thrombin, so inhibition of this factor is more efficient and specific than thrombin inhibition [Tidwell, R .; Webster, WP; Shaver, SR; Geratz, JD THROMBOSIS RESEARCH] Vol. 19, Vol. 0 years]. Disclosure of the invention
- An object of the present invention is to provide a compound having an excellent activated blood coagulation factor X inhibitory action.
- Another object of the present invention is to provide a compound having an inhibitory action specific to activated blood coagulation factor X which can be orally administered.
- Another object of the present invention is to provide an anticoagulant or a prophylactic / therapeutic agent for thrombus or embolus containing the above compound.
- the present invention provides a benzamidine derivative represented by the following general formula (1-1), (1-2), (1-3) or (1-4) or a pharmaceutically acceptable salt thereof, and It is an anticoagulant used as a component.
- L represents any organic group of the formula from (2) (5) c
- W represents a hydrogen atom, an alkyl group having 1 to 6 carbon atoms, an aryl group having 4 to 10 carbon atoms or an aralkyl group having 5 to 12 carbon atoms
- one of D and D ' represents a bond to ⁇ in the general formula (1), the other represents a hydrogen atom,
- X represents a hydrogen atom, a carboxyl group, an alkoxyl group having 1 to 3 carbon atoms, or an alkyl group having 1 to 3 carbon atoms which may have a substituent or a substituent.
- a benzyl group which may have a substituent, such as a carboxyl group, an alkoxycarbonyl group having 2 to 8 carbon atoms, an alkylsulfonyloxy group having 1 to 6 carbon atoms, a piperidyloxy group, or a carbon atom having 6 carbon atoms.
- X and W may combine to form a ring, and in this case, —W—X— represents an ethylene group, a trimethylene group, or a tetramethylene group.
- base may have a substituent Nzoiru, benzenesulfonyl, 2-naphthoquinone evening Rensuruhoniru, piperidines Rajin Carbonyl, cinnamoyl, piperidinecarbonyl, 4-methyl-thiazol-5-carbonyl group, phenylacetyl, phenylthiocarbonyl or benzimidoyl group or a carbon number of 1 to 6 which may have a substituent Represents an alkanesulfonyl group of When L is an organic group represented by the formula (5), V, represents an aryl group having 4 to 10 carbon atoms which may have a substituent.
- the substituent may be a carboxyl group, an alkoxycarbonyl group having 2 to 7 carbon atoms, Rubamoyl group, mono- or dialkyl group having 2 to 7 carbon atoms Lubamoyl group, amidino group, mono- or dialkylamidino group having 2 to 7 carbon atoms, acyl group having 1 to 8 carbon atoms, halogeno group, amino group, 1 to carbon atoms 6 mono or dialkylamino group, 4 to 6 carbon atom arylamino group, 2 to 7 carbon atom alkoxycarbonylamino group, 1 to 3 carbon atom aminoalkyl group, 2 to 7 carbon atom mono or dialkylaminoalkyl Group, an N-alkyl-N-alkoxycarbonylaminoamino group having 4 to 10 carbon atoms, a piperidyloxy group, an iminoalkylpiperidyl
- alkoxycarbonylpyrrolidinyloxy groups charcoal Hydroxycarbonylalkyl group having 2 to 7 carbon atoms, alkoxycarbonylalkyl group having 3 to 8 carbon atoms, hydroxycarbonylalkenyl group having 3 to 7 carbon atoms, alkoxycarbonylalkenyl group having 4 to 8 carbon atoms, 4 to 10 carbon atoms
- Aryl alkenyl having 6 to 12 carbons, alkoxy having 1 to 10 carbons, nitro, trifluoromethyl, alkyl having 3 to 8 carbons, and 4 to 10 carbons
- ⁇ represents a halogeno group
- B represents a hydrogen atom, an alkyl group having 1 to 6 carbon atoms, a halogeno group, or an amino group.
- Y represents any of the following formulas (10) to (16). -(CH2) n — O- one S- CH2CH2 —— CH2 CH- m
- n represents an integer of 0 to 2
- R 1 is a hydrogen atom, a hydroxycarbonylalkyl group having 2 to 7 carbon atoms, and 3 to 8 carbon atoms. Represents an alkoxycarbonylalkyl group and a hydroxycarbonylalkenyl group having 3 to 7 carbon atoms.
- R 2 represents a hydroxyl group, Represents an alkoxyl group, a trifluoromethyl group, an amino group, or a mono- or dialkylamino group having 1 to 6 carbon atoms of Formulas 1 to 5.
- R 3 represents a hydrogen atom, a carbon atom of 1 to 6 carbon atoms.
- R 4 represents a hydrogen atom and an alkyl group having 1 to 6 carbon atoms
- R 5 represents a hydrogen atom And an alkyl group having 1 to 6 carbon atoms.
- R 15 represents a halogeno group.
- Z propositionally represents a carboxyl group, an ethoxycarbonylethyl group, a hydroxymethyl group or a hydroxypropyl group, and E represents an oil-soluble organic group.
- L represents an organic group of any of the formulas (2) to (5) in the general formula (1)
- W has the same meaning as in the above general formula (1)
- X has the same meaning as in the above general formula (1). Meaningful,
- V 2 is benzoyl having a substituent, benzenesulfonyl, 2-naphthene lensulfonyl, cinnamoyl, piperidinecarbonyl, phenylacetyl Represents a phenylthiocarbonyl or benzimidoyl group.
- V 2 represents an aryl group having 4 to 10 carbon atoms which may have a substituent.
- the substituent of V 2 may be a trialkylamidino group having 4 to 7 carbon atoms or a dialkoxybenzoyl group having 9 to 13 carbon atoms. And an alkylpyridino group having 6 to 9 carbon atoms.
- Y represents any one of the formulas (10) to (16) in the general formula (1-1), wherein n is an integer of 1 or 2, and R 1 is the same as described above.
- Z 2 represents a hydrogen atom, an alkyl group having 1 to 6 carbon atoms, a halogeno group, or the following formula (13-2).
- R 22 is a carboxyl group and has 2 carbon atoms.
- V Factory L 2 -Y 2 [In the general formula (1), L 2 represents any one of the organic groups of the formulas (2) to (4) in the above general formula (1), and in formulas (2) and (3), W and In the formula (2), X has the same meaning as in the above general formula U-1),
- L 2 is an organic group represented by any of the formulas (2) to (4), V, has the same meaning as in the above general formula (1), and when V t has a substituent, Examples of the substituent include the same ones as in the above general formula (1-1).
- Y 2 represents any one of the formulas (10) and (11) in the general formula (1), and R 3 represents a hydrogen atom, an alkyl group having 1 to 6 carbon atoms, or an acetyl group.
- the alkyl may include a branch or a ring.
- the alkyl group includes a cyclohexylmethyl group and the like.
- Aryl represents not only an aromatic hydrocarbon ring group but also a heteroaromatic ring group having 1 to 3 heteroatoms selected from 0, ⁇ and S.
- Specific examples of aryl groups include phenyl, pyridyl, imidazolyl, and pyrrolyl groups.
- aryl alkenyl groups include 2- (4-pyridyl) vinyl groups and the like.
- the dialkylamidino group refers to ⁇ , ⁇ -alkylamidino group and ⁇ , ⁇ , monodialkylamidino group.
- dialkylamino group Two alkyl groups in the dialkylamino group, dialkylamidino group, dialkylamino group, dialkylaminoalkyl group, dialkylaminosulfonyl group, and dialkylguanidino group may be bonded to form a ring.
- one of CH 2 may be substituted with ⁇ , NH, or S.
- a 1-pyrrolidinecarbonyl group or the like may be specifically mentioned as a dialkylamino group, or a 2-imidazoline-12-yl group or (pyrrolidine-11-yl) (imino) methyl may be mentioned as a dialkylamidino group.
- the dialkylguanidino group includes an imidazoline-12-amino group and the like.
- acyl is not only alkylcarbonyl, but also arylcarbonyl. Also contains Bonil.
- the acyl group having 1 to 8 carbon atoms includes a benzoyl group and the like.
- the alkoxy group includes a cyclohexyloxy group, a phenoxy group and the like, and the alkoxycarbonyl group includes a benzyloxycarbonyl group and the like.
- a 1-alkylpyridinio group having 6 to 9 carbon atoms do you have 6 or 9 carbon atoms? Any one of to 9 is suitable.
- the compound of the present invention may have an asymmetric carbon, and the compound of the present invention may be a mixture of various stereoisomers such as geometric isomers, tautomers, and optical isomers, or an isolated one. included.
- the amidino group in the compound of the present invention may be substituted by an appropriate substitution that is exchanged for an amidino group in a living body.
- a hydrogen atom bonded to a nitrogen atom having a double bond of an amidino group bonded to a benzene ring may be a hydroxyl group or an ethoxy group.
- 1 or 2 hydrogens such as alkoxyl groups, amino groups, carboxyl groups, alkenyl groups such as ethoxycarbonyl groups, alkylsulfonyl groups such as ethylsulfonyl groups, rubamoyl groups, and diethoxycarbamoyl groups.
- alkyl group such as an acyl group such as a rubamoyl group, a formyl group, and an acetyl group, and an alkylcarboxyl group such as an acetyl group.
- L is preferably the formulas (2) to (4), more preferably the formula (2) or (4), and particularly preferably the formula (2).
- W is preferably a hydrogen atom or an alkyl group having 1 to 6 carbon atoms, and particularly preferably a hydrogen atom.
- X is preferably a hydrogen atom, a carboxyalkyl group having 2 to 3 carbon atoms, or an alkoxycarbonylalkyl group having 3 to 10 carbon atoms, and particularly preferably a hydrogen atom, a carboxymethyl group or an ethoxycarbonylmethyl group.
- X is preferably a hydrogen atom, a carboxyl group, an optionally substituted alkyl group having 1 to 3 carbon atoms, or an optionally substituted benzyl group, particularly preferably a hydrogen atom, An alkyl group having 1 carbon atom having a group is preferred.
- substituents include benzyloxycarbonyl, carboxyl, methoxycarbonyl, ethoxycarbonyl, ethanesulfonyloxy, butanesulfonyloxy, 4-piperidyloxy, -Acetimidoyl—4-piperidyloxy group, (1—acetoimidoyl—4-piperidyl) methyl group, 1—acetimidyl-1-3-pyrrolidinyloxy group, isopropyl group, 3-indolyl group, iodine atom, etc. Can be Of these, a carboxyl group is particularly preferred.
- V is preferably a benzoyl group which may have a substituent, a piperidinecarbonyl group which may have a substituent, or a pyridinecarbonyl group which may have a substituent, More preferred are a substituted benzoyl group and a substituted piperidinecarbonyl group.
- V has a substituent
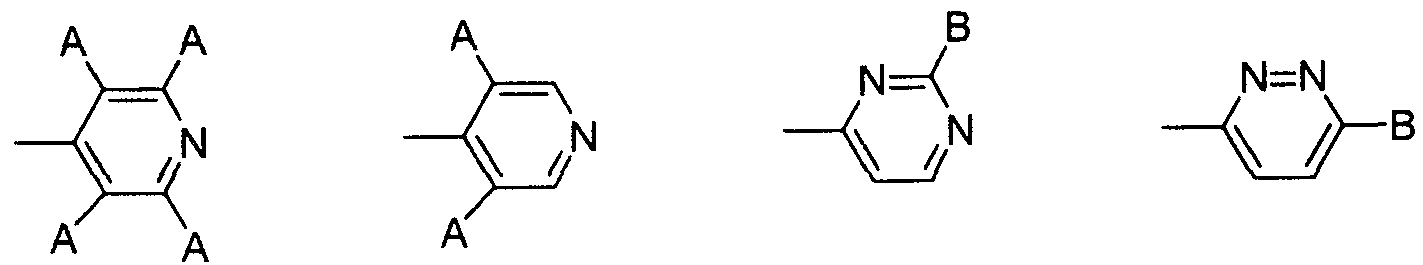
- substituents include 4-piperidyloxy, 1-acetimidyl-4-piperidyloxy, 4-pyridyl, tetrafluoropyridyl, 3,5-dichloropyridyl, —Cyclid pyridazyl group, pyridazyl group, 2-Cyclic pyrimidyl group, pyrimidyl group, 4-pyridine-4-ylmethyl group, and 4-pyridylcarbonyl group, and more preferably 1-acetimidyl-4-piperidyloxy group And 4-pyridyl groups.
- V is particularly preferably any one of a 1-acetimidyl-1-4-piperidyloxybenzoyl group and a 1- (4-pyridyl) -1piperidine-14-carbonyl group.
- Y is preferably an organic group represented by the formula (10), and in the formula (10), n is more preferably an integer of 1.
- Z is preferably represented by formulas (17), (19), (21) and (23), more preferably a carboxyl group, an ethoxycarbonylethyl group, a sulfoethyl group. , Phosphonoethyl, diethoxyphosphorylethyl, monoethoxyhydroxyphosphorylethyl, hydroxymethyl, hydroxypropyl, etc.
- a carboxyl group, an ethoxycarbonylethyl group, a hydroxymethyl group, and a hydroxypropyl group are particularly preferable.
- L represents an organic group of the formula (2)
- W represents a hydrogen atom
- X represents a hydrogen atom, a carboxymethyl group, or an ethoxycarbonylmethyl group.
- a derivative or a pharmaceutically acceptable salt thereof is preferred.
- a benzamidine derivative in which Y represents an organic group represented by the formula (10) and n represents an integer of 1 or 2 or a pharmaceutically acceptable salt thereof is preferable.
- V is 1-acetimidoyl-4-piperidyloxybenzoyl group, 1- (4-pyridyl) -piperidine-14-carbonyl group, 1- (2,3,5,6- Tetrafluoropyridine-4-yl) -piperidine-4-carbonyl group, 1- (3,5-dichloropyridine-4-yl) -piperidine-4-carbonyl group, 1_ (6-cyclopyridazine- 3-yl) -piperidine-4-carbonyl group, 1- (pyridazine-3-yl) -piperidine-4-carbonyl group, 1- (2-cyclopyrimidine-4-yl) -piperidine-4- Carbonyl group, 1- (pyrimidine-4-yl) -piperidine-4-force carbonyl group, 1- (4-pyridine-4-ylmethyl) -piperidine-4-carbonyl group,
- Benzamidine derivatives which are any of the 2-pyridyl-4-yl-thiazol-5-carbonyl groups or pharmaceutically acceptable salts thereof are preferred.
- a benzamidine derivative or a pharmaceutically acceptable salt thereof is preferred.
- L represents an organic group represented by the formula (2)
- Y represents an organic group represented by the formula (10)
- 1-acetimidoyl-4-piperidyloxybenzoyl represents a carboxyl group, an ethoxycarbonylethyl group, a sulfoethyl group, a hydroxymethyl group, a hydroxymethyl group, or a 1- (4-pyridyl) -piperidine-1-4-carbonyl group. Or a pharmaceutically acceptable salt thereof is preferred.
- L represents an organic group of the formulas (2) to (4)
- Y represents an organic group of the formulas (10) to (13) or a pharmaceutically acceptable benzamidine derivative thereof. Preferred salts are preferred.
- V is a hydrogen atom, benzoyl optionally having substituent (s), benzenesulfonyl, 2-naphthyllensulfonyl, cinnamoyl, ⁇ represents a lysinecarbonyl, phenylacetyl, phenylthiocarbonyl, or benzimidoyl group or an optionally substituted alkanesulfonyl group having 1 to 6 carbon atoms, and L represents an organic group represented by the formula (5).
- V represents an aryl group having 4 to 10 carbon atoms which may have a substituent
- the substituent may be a carboxyl group, an alkoxycarbonyl group having 2 to 7 carbon atoms, or a carbamoyl group.
- alkoxycarbonylpyrrolidinyloxy group C2-C7 hydroxycarbonylalkyl group, C3-C8 alkoxycarbonylalkyl group, C3-C7 hydroxycarbonylalkenyl group, carbon
- Y represents any one of the formulas (10) to (16), and in the formulas (10) and (11), n represents an integer of 1 or 2.
- Z represents one of the formulas (17) and (18), wherein m represents an integer of 1 to 3, R 2 represents a hydroxyl group, an alkoxyl group having 1 to 5 carbon atoms, an amino group, A benzamidine derivative or a pharmaceutically acceptable salt thereof, which represents a mono- or di-alkylamino group having 1 to 6 carbon atoms, is preferred.
- L represents an organic group of the formula (2)
- W represents a hydrogen atom
- X represents any one of a hydrogen atom, a carboxymethyl group and an ethoxycarbonylmethyl group.
- Y represents an organic group represented by the formula (10) and n represents an integer of 1.
- n represents an integer of 1.
- Y represents an organic group represented by the formula (10) and n represents an integer of 1.
- n represents an integer of 1.
- Z is any one of a carboxyethyl group, an ethoxycarbonylethyl group, a carboxyvinyl group, an ethoxycarbonylvinyl group, a carbamoylethyl group, and a carbamoylvinyl group.
- L represents the organic group of the formula (2)
- Y represents the organic group of the formula (10)
- V! But 1 _ Asetoimi Doyle one 4-piperidyl O carboxymethyl benzo I le group or one primary, (4 - pyridyl) indicates either one piperidine one 4-carbonyl group, Z t is Cal Bokishechiru group, ethoxycarbonyl E Chill Or a carbamoylethyl group.
- the benzamidine derivative represented by the general formula (II) or a pharmaceutically acceptable salt thereof has an inhibitory effect on activated blood coagulation factor X.
- E is an oil-soluble organic group, which, together with other groups in the general formula (II), forms a compound represented by the general formula (II). It imparts an inhibitory effect on activated blood coagulation factor X.
- the inhibitory effect on activated blood coagulation factor X can be determined based on the method described in Examples in the specification.
- E is a group containing a bonding group bonded to a benzene ring and a terminal aromatic group and / or a heterocyclic group, and includes the entire group, wherein the bonding group is an oxygen atom or a nitrogen atom.
- Aliphatic groups such as an alkylene group and an oxyalkylene group which may contain a substituent; and a terminal aromatic group and / or a heterocyclic group include a phenyl group and a naphthyl group.
- L represents an organic group represented by the formula (2)
- Y represents the group represented by the formula (10).
- Represents an organic group It is preferable to show either a 1-acetimidoyl-4-piperidyloxybenzoyl group or a 1- (4-pyridyl) -piperidine-14-carbonyl group.
- L is V 2 and the substituents in the case of any of the organic groups of Formula (2) to (5) are as described above.
- Particularly preferred V 2 is a Benzoiru group having location substituent, the examples of the substituent tutorials Kiruamijino group of 4-7 carbon atoms, a dialkoxy benzo I le group having 9-13 carbon atoms, 1 to 6 to 9 carbon atoms A monoalkylpyridino group is exemplified.
- V 2 is a 4- (3,4-dimethoxybenzoyl) benzoyl group, a 1- (1-methylpyridinium-14-yl) piperidine-14-carbonyl group, a 4- (1-methyl-2 —Imidazoline— 2-yl) It is preferably one of the benzoyl groups.
- Y represents any one of the formulas (10) to (16) in the general formula (1-1), and Z 2 represents a hydrogen atom, an alkyl group having 1 to 6 carbon atoms, a halogeno group or a formula (13-2) Show.
- Preferred examples of Y are the same as those in the general formula (1).
- Preferred for Z 2 is formula (13-2), wherein R 22 is a carboxyl group.
- L represents an organic group represented by the formula (2)
- W represents a hydrogen atom
- V 2 represents a 4- (3,4-dimethoxybenzoyl) benzoyl group, 1- (1-methylpyridinium-14-yl) piperidine-14-carbonyl group, 4- (1-methyl-12-imidazo'phosphine-12-yl) benzoyl group
- Z 2 is preferably a hydrogen atom or a 2-carboxy-2-oxoethyl group.
- L represents an organic group represented by the formula (2)
- W represents a hydrogen atom
- X represents a hydrogen atom
- V 2 represents 4- (1-methyl-2-imidazoline).
- 2-yl) represents a benzoyl group
- Z 2 is preferably any one of a 2-carboxy_2-oxoethyl group.
- Y 2 is preferably represented by the formula (10), and R 3 is preferably a hydrogen atom, an alkyl group having 1 to 3 carbon atoms, or an acetyl group, and more preferably a hydrogen atom.
- L 2 represents an organic group of the formula (2)
- W represents a hydrogen atom
- X represents any one of a hydrogen atom, a carboxymethyl group and an ethoxycarbonylmethyl group.
- Y 2 represents an organic group represented by the formula (10), and ⁇ represents an integer of 1 or 2. In particular, ⁇ preferably represents an integer of 1.
- V represents 1-acetimidoyl-4-piperidyloxybenzoyl group, 1-1 (4-pyridyl) -1-piperidine-1-4-carbonyl group, 1- (2,3,5, 6-tetrafluoropyridine-4-yl) -piperidine-4-carbonyl group, 1- (3,5-dichloropyridine-4-yl) -piperidine-4-carbonyl group, 1- (6-chloro Mouth pyridazine-3-yl) -piperidine-4-carbonyl group, 1- (pyridazine-3-yl) -piperidine-4-carbonyl group, 1- (2-cyclopyrimidine-4-yl) -piperidine -4-carbonyl group, 1- (pyrimidine-4-yl) -piperidine-4 -force propyl group, 1- (4-pyridine-4-ylmethyl) -piperidine-4-carbonyl group, 1- (4 -Pyridine-4-carbon
- L 2 represents an organic group represented by the formula (2)
- Y represents an organic group represented by the formula (10).
- V represents either 1-acetimidoyl-1-4-piperidyloxybenzoyl group or 1_ (4-pyridyl) -1-piperidine-14-carbonyl group
- R 3 represents hydrogen It is preferably an atom.
- dimethylformamide or the like is used as a solvent on a nitrogen-protected aminoalkyl halide (25) protected with a benzyloxycarbonyl group or a t-butoxycarbonyl group in the presence of a base such as potassium carbonate.
- (27) can be obtained by acting hydroxy-4-benzobenzonitrile (26).
- the obtained (27) is converted to an acrylic acid derivative (28) by using, for example, dimethylformamide or the like as a solvent and condensing it with, for example, ethyl acrylate by a Heck reaction or the like.
- the amine (29) can be obtained by deprotecting the obtained protecting group on nitrogen in (28) in an acidic solution such as a dioxane solution of 4N hydrogen chloride.
- Prot has 7f protecting groups such as Boc group and Z group.
- Hal represents a halogen atom.
- the amide (29) is condensed with carboxylic acid by using a condensing agent in the presence of a base such as triethylamine, for example, using dimethylformamide or the like as a solvent. Can be obtained.
- the amide (30) is reacted with an alcohol such as ethanol containing hydrogen halide such as hydrogen chloride, and then reacted with an ammonium salt such as ammonium carbonate to convert the cyano group to an amidino group.
- an alcohol such as ethanol containing hydrogen halide such as hydrogen chloride
- an ammonium salt such as ammonium carbonate
- L is the formula (2)
- Y is the formula (10)
- Z can produce a benzamidine derivative (31) represented by the formula (18)
- V-H 2 The benzamidine derivative (31) is reacted under an atmosphere of hydrogen, for example, using an alcohol such as methanol as a solvent in the presence of a catalyst such as palladium ion, and then hydrolyzed with an acidic aqueous solution such as concentrated hydrochloric acid.
- a benzamidine derivative (32) in which L is represented by the formula (2), Y is represented by the formula (10), and Z is represented by the formula (17) can be produced.
- V- The compound of the present invention or a salt thereof thus produced can be isolated by a known separation and purification means, for example, extraction, concentration, concentration under reduced pressure, solvent extraction, crystallization, recrystallization, phase transfer, various chromatographies, etc. It can be purified.
- the salt of the benzamidine derivative of the present invention may be any pharmaceutically acceptable salt, for example, mineral acids such as hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid, formic acid, acetic acid, trifluoroacetic acid, lactic acid, and the like.
- mineral acids such as hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid, formic acid, acetic acid, trifluoroacetic acid, lactic acid, and the like.
- Salicylic acid, mandelic acid, citric acid, oxalic acid, maleic acid, fumaric acid, tartaric acid, tannic acid, malic acid, tosylic acid, methanesulfonic acid, benzene 'Acid addition salts with organic acids such as acids can be mentioned.
- the compound of the present invention or a salt thereof may be administered as it is or as various pharmaceutical compositions.
- Such pharmaceutical compositions may be in the form of tablets, powders, pills, granules, capsules, suppositories, solutions, dragées, or depots, for example, using ordinary formulation auxiliaries. And can be manufactured in accordance with ordinary methods.
- tablets may contain the active ingredient of the invention, the benzamidine derivative, as a known auxiliary substance, for example, an inert diluent such as lactose, calcium carbonate or calcium phosphate, a binder such as acacia, corn starch or gelatin.
- the administration route may be oral or parenteral, and the dosage varies depending on the age, weight, condition, and administration method of the patient.
- the daily dose to an adult is usually 0.01 to 100 mg, preferably 0.1 to 50 mg for oral administration, and 0.1 to 50 mg for parenteral administration. It is 1 ⁇ 10 mg, preferably 0.01-1 mg.
- 1-t-butoxycarbonyl obtained by subjecting 1.7 g (10.2 mmol) of 4-hydroxyethyl benzoate to t-butoxycarbonylation of 4-hydroxypiperidine using di-t-butyldicarbonate in a conventional manner.
- the crude product was obtained by treatment with ethyl acetate as an extraction solvent according to a conventional method. Subsequently, the residue was purified by silica gel column chromatography to obtain the title compound.
- N-t-butoxycarboxyl-D-aspartic acid mono-/?-Benzyl ester (15.0 g, 46.4 mmol) and triethylamine 6.47 ml (46.4 mmol) were dissolved in 230 ml of tetrahydrofuran. Under ice cooling, 4.4 ml (46.4 mmol) of ethyl chloroformate was added, and the mixture was stirred for 15 minutes. The resulting precipitate was removed by suction filtration, and 5 g of ice and 1.8 g (46.4 mmol) of sodium borohydride were added to the filtrate under ice cooling, and the mixture was stirred for 1.5 hours.
- ⁇ ⁇ ⁇ 3 ⁇ 4 (IO UIUI 'z) S ⁇ - ⁇ ( ⁇ ⁇ ⁇ (- ⁇ ⁇ ⁇ -C N' t N ' ⁇ C N ( ⁇ O UIUI ⁇ -z) S ⁇ ' ⁇ I ⁇ ⁇ ⁇ ⁇ : ⁇ , (T oram '' ⁇ ) s 9 '0 I (( ⁇ -, ⁇ .>
- the crude product obtained by evaporating the solvent was purified in the same manner as in Step 9 of Example 1 to obtain a purified product.
- 1.86 g (3.37 mmol) of the obtained purified product was dissolved in 30 ml of ethanol, and 1.27 g (10.29 mmol) of ethyl acetimide hydrochloride and 2.4 ml (17.15 mmol) of triethylamine were added, followed by stirring with stirring.
- the crude product obtained by distilling off the solvent was subjected to the same operation as in Step 1 of Example 1 to obtain the title compound.
- the mixture was treated according to a conventional method to obtain a crude product. Subsequently, the residue was purified by silica gel column chromatography to obtain the title compound.
- N- [2- (5-cyano 2-benzyloxyphenoxy) ethyl] —4— (1-t-butoxycarbonyl-1-piperidyloxy) benzamide 51 Omg (0.89mmo 1) 5 ml of a hydrogen chloride dioxane solution and 1 ml of ethanol were dissolved and stirred for 3 days.
- the crude product obtained by distilling off the solvent was dissolved in 10 ml of ethanol, 50 Omg of ammonium carbonate was added, and the mixture was stirred overnight.
- the crude product obtained by evaporating the solvent was dissolved in 1 Oml of ethanol, 5 Omg of 10% palladium on carbon was added, and the mixture was stirred overnight in the presence of hydrogen.
- the crude product obtained by evaporating the solvent was dissolved in 4 ml of a 4N hydrogen chloride dioxane solution and 4 ml of ethanol, and the mixture was stirred for 3 days.
- the crude product obtained by distilling off the solvent was dissolved in ethanol (20 ml), ammonium carbonate (100 Omg) was added, and the mixture was stirred overnight.
- the crude product obtained by distilling off the solvent was dissolved in 10 ml of 6N hydrochloric acid aqueous solution, and The mixture was stirred at 0 ° C for 2 hours.
- the title compound was obtained from the crude product obtained by evaporating the solvent by the same operation as in Step 9 of Example 1.
- Pencil fluoropyridine 1.lg (6.5 mmol), piperidine-1-4-ethyl carboxylate 1.lg (6.5 mmol), diisopropylethylamine 2.27 ml (13.7 mmol)
- the mixture was stirred at room temperature in 5 ml of ethanol for 24 hours.
- the crude product was obtained by treating according to a conventional method using ethyl acetate as an extraction solvent. Subsequently, the residue was purified by silica gel column chromatography to obtain the title compound.
- the crude product obtained by distilling off the solvent was dissolved in 5 ml of 4N hydrogen chloride dioxane solution and 1 ml of ethanol, and the mixture was stirred for 3 days.
- the crude product obtained by distilling off the solvent was dissolved in 20 ml of ethanol, 50 Omg of ammonium carbonate was added, and the mixture was stirred overnight.
- the crude product obtained by distilling off the solvent was dissolved in 10 ml of a 6 N aqueous hydrochloric acid solution and stirred at 50 ° C for 2 hours.
- the crude product obtained by distilling off the solvent was subjected to the same operation as in step 1 of Example 1 to obtain a title compound.
- the crude product obtained by evaporating the solvent was dissolved in DMF 1 Om 1 and 3- [2- (2-aminoethoxy) -14-cyanophenyl] ethyl ethyl propionate hydrochloride 70 Omg (2.5 mmol) To the mixture were added 634 mg (3.75 mmo1) of 2-chloro-1,3-dimethylimidazonium chloride and 1.74 ml of triethylamine (7.5 mmo1), and the mixture was stirred overnight.
- the crude product obtained by evaporating the solvent was dissolved in 5 ml of a 4N hydrogen chloride dioxane solution and 1 ml of ethanol, and the mixture was stirred for 3 days.
- the crude product obtained by distilling off the solvent was dissolved in 2 Oml of ethanol, 50 Omg of ammonium carbonate was added, and the mixture was stirred overnight.
- the crude product obtained by distilling off the solvent was dissolved in 10 ml of a 6N aqueous hydrochloric acid solution, and the mixture was stirred at 50 for 2 hours.
- the title compound was obtained from the crude product obtained by evaporating the solvent by the same operation as in Step 9 of Example 1.
- the crude product obtained by evaporating the solvent was dissolved in 10 ml of a 6N aqueous hydrochloric acid solution, and the mixture was stirred at 50 ° C for 2 hours.
- the crude product obtained by distilling off the solvent was subjected to the same operation as in Step 1 of Example 1 to obtain the title compound.
- the crude product obtained by evaporating the solvent was dissolved in 1 Oml of a 6 N aqueous hydrochloric acid solution, and the mixture was stirred at 50 ° C for 2 hours.
- the crude product obtained by distilling off the solvent was subjected to the same operation as in Step 1 of Example 1 to obtain the title compound.
- the crude product obtained by treatment with ethyl acetate as an extraction solvent according to a conventional method was dissolved in 10 ml of a 4N hydrogen chloride dioxane solution and 2 ml of ethanol, and the mixture was stirred for 3 days.
- the crude product obtained by evaporating the solvent was dissolved in 2 Oml of ethanol, 50 Omg of ammonium carbonate was added, and the mixture was stirred overnight.
- the crude product obtained by distilling off the solvent was dissolved in 1 Oml of 6N hydrochloric acid aqueous solution, and the mixture was stirred at 50 ° C for 2 hours.
- the crude product obtained by distilling off the solvent was used in Example 1 By the same operation as in Step 9, the title compound was obtained.
- the crude product obtained by evaporating the solvent was dissolved in ethanol (20 ml), ammonium carbonate (20 Omg) was added, and the mixture was stirred overnight.
- the crude product obtained by distilling off the solvent was dissolved in 10 ml of a 6N aqueous hydrochloric acid solution and stirred at 50 ° C for 2 hours.
- the crude product obtained by distilling off the solvent was subjected to the same operation as in Step 9 of Example 1 to obtain the title compound.
- 1,3-Dimethylimidazonium chloride 98 Omg (5.82 mm 01) and 1.6 ml of triethylamine (11.6 mmo 1) were added, and the mixture was stirred for 16 hours.
- ethyl acetate as an extraction solvent, the mixture was treated according to a conventional method to obtain a crude product. Dissolve the obtained crude product in 20 ml of 4N hydrogen chloride dioxane solution and 5 ml of ethanol And stirred for 3 days. The crude product obtained by evaporating the solvent was dissolved in 1 Oml of ethanol, 50 Omg of ammonium carbonate was added, and the mixture was stirred overnight.
- the crude product obtained by evaporating the solvent was dissolved in ethanol (10 ml), 10% palladium-carbon (10 Omg) was added, and the mixture was stirred overnight in the presence of hydrogen.
- the reaction solution was filtered through celite, the solvent was distilled off, and a crude product obtained was dissolved in 28 ml of a 6N aqueous hydrochloric acid solution and stirred at 60 ° C for 2 hours.
- the title compound was obtained from the crude product obtained by evaporating the solvent by the same operation as in Step 9 of Example 1.
- the crude product was obtained by treating according to a conventional method using black-mouthed form as an extraction solvent.
- the obtained crude product was dissolved in 4 ml of a 4N hydrogen chloride dioxane solution and 0.2 ml of ethanol, and the mixture was stirred at room temperature for 3 days.
- the residue obtained by evaporating the solvent was dissolved in 50 ml of ethanol, 0.14 g (2.45 mmo 1) of ammonium carbonate was added, and the mixture was stirred at room temperature overnight.
- the title compound was obtained from the crude product obtained by evaporating the solvent by the same operation as in Step 9 of Example 1.
- the solvent was distilled off, and the residue was treated according to a conventional method using ethyl acetate as an extraction solvent to obtain a crude product. Subsequently, the residue was purified by silica gel column chromatography to obtain the title compound.
- the obtained crude product is dissolved in 10 ml of N, N-dimethylformamide (dehydrated), and 1- (4-pyridyl) -piperidine-14-butane rubonic acid hydrochloride 1.25 g (5 lmmol) Then, 0.95 g (5.6 mmo 1) of 2-chloro-1,3-dimethylimidazolinium chloride and 4 ml (27.8 mmo 1) of triethylamine were added, and the mixture was stirred overnight at room temperature. Using a dichloromethane as an extraction solvent, the mixture was treated according to a conventional method to obtain a crude product.
- the obtained crude product was dissolved in 20 ml of a 4N hydrogen chloride dioxane solution and 2 ml of ethanol, and the mixture was stirred at room temperature for 3 days.
- the residue obtained by distilling off the solvent was dissolved in 50 ml of ethanol, 1.3 g (23.15 mmol) of ammonium carbonate was added, and the mixture was stirred at room temperature overnight.
- the title compound was obtained from the crude product obtained by evaporating the solvent by the same operation as in Step 9 of Example 1.
- the solvent was distilled off, and the resulting crude product was dissolved in 10 ml of dioxane and 10 ml of 4N hydrogen chloride dioxane, and the mixture was stirred at room temperature for 4 hours. After evaporating the solvent, dissolve in 12 ml of ethanol, add 0.87 g (7.05 mmol) of ethylacetimidate hydrochloride and 1.64 ml (1.75 mmol) of triethylamine, and stir at room temperature overnight did. The solvent was distilled off, and dissolved in 20 ml of a 4N hydrogen chloride dioxane solution and 3 ml of ethanol, followed by stirring at room temperature for 3 days.
- the solvent was distilled off and the crude product was dissolved in 10 ml of ethanol, and 0.67 g (ll. 75 mmo 1) of ammonium carbonate was added, followed by stirring overnight.
- the crude product obtained by distilling off the solvent was subjected to the same operation as in step 1 of Example 1 to obtain the title compound.
- Example 39 4 _Amidino-1 2- [2- (4- [l- (1-acetimidyl)-4 —Piperidyloxy] benzoylamino) ethoxy j Synthesis of ethyl benzoate ditrifluoroacetate The title compound was obtained as a by-product of Step 2 of Example 38.
- Example 37 3- (2- (t-butoxycarbonylamino) ethoxy) 1-4-hydroxymethylbenzonitrile obtained by the same operation as in Step 1 0.3 g (1.
- Example 4 2- (4-amidino-2- [2-[(1- (1-pyridine-1-4-yl) piperidine_4_carbonyl) amino] ethoxy] phenyl obtained from step 3 7) 72.2 mg (0.103 mmo 1) of vinylsulfonic acid ditrifluoroacetate is dissolved in 20 ml of ethanol, and 30 mg of 10% palladium-carbon (50% water-containing) is added in the presence of argon and at room temperature in the presence of hydrogen. Stirred overnight. The crude product obtained by celite filtration was subjected to the same operation as in step 1 of Example 1 to obtain the title compound.
- the product obtained by distilling off the solvent was dissolved in 5 ml of N, N-dimethylformamide (dehydrated), and 1- (4-pyridyl) -piperidine-4-carboxylic acid hydrochloride 440 mg (1.8 Ommo) 1), 2-chloro-1,3-dimethylimidazolidum 33 Omg (1.97 mmo 1), triethylamine 1.4 ml (9.84 mmo 1) were added, and the mixture was stirred at room temperature overnight.
- the crude product obtained by evaporating the solvent was treated according to a conventional method using dichloromethane as an extraction solvent to obtain a crude product.
- the obtained crude product was dissolved in 4N hydrogen chloride dioxane solution (1 Oml) and ethanol (1ml) and stirred at room temperature for 3 days.
- the residue obtained by evaporating the solvent was dissolved in 1 Oml of ethanol, 0.46 g (8.2 mmo 1) of ammonium carbonate was added, and the mixture was stirred at room temperature overnight.
- the crude product obtained by removing the solvent was subjected to the same operation as in step 1 of Example 1 to obtain the title compound.
- Example 44 2 (4-Amidino 2 — [2 — [(1— (1-pyridine-14-yl) piperidine-14-carbonyl) amino] ethoxy] phenyl) vinyl phosphoric acid ethyl ester ditrifluoroacetate
- Step 1 2 [(2— ( Synthesis of 2-t-butoxycarbonylamino) ethoxy) —4-cyanophenyl] vinyl phosphate getyl ester
- Example 45 [2- (4-Amidino-1-2- [2-[(1- (1-pyridine-14-yl) piperidine-1-4-carbonyl) amino] [Ethoxy] phenyl) vinyl] phosphate monoethyl ester ditrifluoroacetate It is a by-product obtained in Step 2 of Example 44.
- the solvent was distilled off, and the residue was washed with ethyl acetate.
- the obtained crude product was dissolved in 20 ml of ethanol, 1.52 g (20.6 mmol) of N-methylethylenediamine was added, and the mixture was heated under reflux for 6 hours. After distilling off the solvent, use dichloromethane as extraction solvent The resulting crude product was dissolved in 1 Oml of concentrated hydrochloric acid and stirred at 50 ° C overnight. After evaporating the solvent, a crude product of the title compound was obtained.
- reaction solution was poured into 1N hydrochloric acid and 5 g of ice, and treated with chloromethane as an extraction solvent according to a conventional method.
- the residue obtained by evaporating the solvent was washed with ethyl acetate and dichloromethane to obtain the title compound.
- Tris-HCl buffer adjusted to 118.4, and then human activated blood coagulation factor X (Enzyme Research) was added to pH 8.4 Tris-HCl Apply 10 1 solution prepared to 0.5 units / m 1 with buffer! ], And incubated at room temperature for 10 minutes. Then, N-benzoyl-L-iso-isyl-L-glucamyl-glycyl-L-arginyl-l-p-to-n-anilide hydrochloride (manufactured by Peptide Research Laboratories) was dissolved in Tris-HCl buffer (pH 8.4).
- Tris-hydrochloric acid buffer adjusted to 118.4 was added to 10 ⁇ 1 of the aqueous solution of the evaluation compound, and then thrombin (produced by SIGMA) was added to the cells at pH 8.4 using Tris-HCl buffer (2 units). Then, 10 ⁇ 1 of the solution adjusted to / ml was added, and the mixture was incubated at room temperature for 10 minutes. Next, D-phenylalanil L-pipecolyl-L-arginyl-P-nitronitrolide hydrochloride (Daiichi Pure Chemicals, S-2238) was adjusted to 0.4 mM with Tris-HCl buffer (pH 8.4).
- Table 1 below shows the inhibitory activity of tomon binbin on typical compounds.
- Example 56 Measurement of anticoagulant activity
- Anticoagulant activity was determined using the prothrombin time (PT) assay.
- PT measurement was performed as shown below. That is, blood was collected from a healthy person, and 1/10 volume of a 3.8% aqueous solution of trisodium citrate was added thereto, and plasma was separated by centrifugation. 5/1 DMS 0 solution containing the compound to be evaluated was added to plasma 451, and the mixture was incubated at room temperature for 2 minutes. After placing the test tube containing the plasma solution on a Sysinex CA-3000 fully automatic blood coagulation analyzer (Toa Medical Electronics), incubate at 37 ° C for 3 minutes, and Sysmex PT II (Toa Medical Electronics, Egret brain tissue thromboplastin).
- Sysinex CA-3000 fully automatic blood coagulation analyzer
- Sysmex PT II Toa Medical Electronics, Egret brain tissue thromboplastin.
- the anticoagulant containing the compound of the present invention or a salt thereof as an active ingredient is excellent in activated blood coagulation.
- the compound of the present invention is useful for diseases in cerebrovascular disorders such as cerebral infarction, cerebral thrombosis, cerebral embolism, transient cerebral ischemic attack (TIA), subarachnoid hemorrhage (vascular spasm), acute and chronic myocardial infarction, unstable narrowing Heart disease, diseases in ischemic heart diseases such as coronary artery thrombosis, diseases in pulmonary vasculopathy such as pulmonary infarction, pulmonary embolism, peripheral arterial occlusion, deep vein thrombosis, disseminated intravascular coagulation, and artificial angioplasty and artificial heart Thrombus formation after valve replacement, reocclusion and restenosis after coronary artery bypass surgery, percutaneous transluminal coronary angioplasty (PTCA) or percutaneous transluminal coronary artery revascularization (PTCR) It can be used as a therapeutic agent for preventing and treating restenosis and restenosis and
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Biochemistry (AREA)
- General Health & Medical Sciences (AREA)
- Molecular Biology (AREA)
- Hydrogenated Pyridines (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Plural Heterocyclic Compounds (AREA)
Description


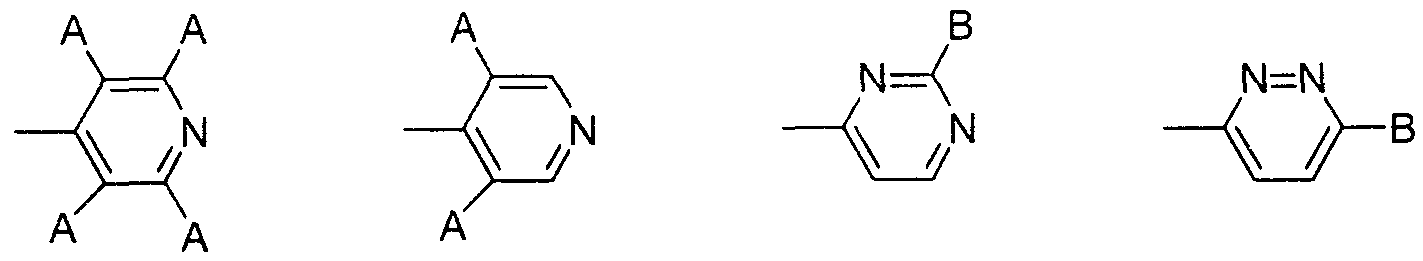

























































Claims










Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR1020007013949A KR20010052694A (ko) | 1998-06-08 | 1999-06-08 | 벤즈아미딘 유도체 |
| AU40604/99A AU758567B2 (en) | 1998-06-08 | 1999-06-08 | Benzamidine derivative |
| EP99923959A EP1086946A4 (en) | 1998-06-08 | 1999-06-08 | BENZAMIDINE DERIVATIVE |
| CA002334476A CA2334476A1 (en) | 1998-06-08 | 1999-06-08 | Benzamidine derivative |
| US09/731,729 US6410538B2 (en) | 1998-06-08 | 2000-12-08 | Benzamidine derivatives |
| US10/073,985 US6812231B2 (en) | 1998-06-08 | 2002-02-14 | Benzamidine derivative |
Applications Claiming Priority (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP15962798 | 1998-06-08 | ||
| JP10/159627 | 1998-06-08 | ||
| JP15962898 | 1998-06-08 | ||
| JP10/159628 | 1998-06-08 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US09/731,729 Continuation US6410538B2 (en) | 1998-06-08 | 2000-12-08 | Benzamidine derivatives |
| US10/073,985 Continuation US6812231B2 (en) | 1998-06-08 | 2002-02-14 | Benzamidine derivative |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO1999064392A1 true WO1999064392A1 (en) | 1999-12-16 |
Family
ID=26486362
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP1999/003055 Ceased WO1999064392A1 (en) | 1998-06-08 | 1999-06-08 | Benzamidine derivative |
Country Status (7)
| Country | Link |
|---|---|
| US (2) | US6410538B2 (ja) |
| EP (1) | EP1086946A4 (ja) |
| KR (1) | KR20010052694A (ja) |
| CN (1) | CN1311771A (ja) |
| AU (1) | AU758567B2 (ja) |
| CA (1) | CA2334476A1 (ja) |
| WO (1) | WO1999064392A1 (ja) |
Cited By (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001042254A1 (en) * | 1999-12-08 | 2001-06-14 | Ajinomoto Co., Inc. | Benzamidine derivatives |
| WO2001042199A1 (en) * | 1999-12-06 | 2001-06-14 | Ajinomoto Co.,Inc. | Amidinophenylpyruvic acid derivative |
| WO2002022575A1 (en) * | 2000-09-11 | 2002-03-21 | Genentech, Inc. | Amidine inhibitors of serine proteases |
| US6410538B2 (en) | 1998-06-08 | 2002-06-25 | Ajinomoto Co., Inc. | Benzamidine derivatives |
| WO2002053534A1 (en) * | 2000-12-28 | 2002-07-11 | Daiichi Pharmaceutical Co., Ltd. | Vla-4 inhibitors |
| WO2006083003A1 (ja) | 2005-02-02 | 2006-08-10 | Ajinomoto Co., Inc. | 新規ベンズアミジン化合物 |
| WO2010005087A1 (ja) | 2008-07-11 | 2010-01-14 | 味の素株式会社 | アミジン誘導体 |
| US7691894B2 (en) | 2003-07-24 | 2010-04-06 | Daiichi Pharmaceutical Co., Ltd. | Cyclohexanecarboxylic acid compound |
| WO2010119877A1 (ja) * | 2009-04-13 | 2010-10-21 | 味の素株式会社 | アミジン誘導体の製造方法 |
| US8778925B2 (en) | 2008-10-06 | 2014-07-15 | Cancer Research Technology Ltd. | Pyridine and pyrimidine based compounds as Wnt signaling pathway inhibitors for the treatment of cancer |
| EP2982668A2 (en) | 2002-12-03 | 2016-02-10 | Pharmacyclics LLC | 2-(2-hydroxybiphenyl-3-yl)-1h-benzoimidazole-5-carboxamidine derivatives as factor viia inhibitors for the treatment of thromboembolic disorders |
Families Citing this family (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TW542822B (en) * | 1997-01-17 | 2003-07-21 | Ajinomoto Kk | Benzamidine derivatives |
| WO2002089800A2 (en) * | 2001-05-05 | 2002-11-14 | Smithkline Beecham P.L.C. | N-aroyl cyclic amine derivatives as orexin receptor antagonists |
| US6790977B2 (en) * | 2002-07-30 | 2004-09-14 | Sinon Corporation | Process for preparing 3,4-dihydroxy-benzonitrile |
| CN104910049B (zh) * | 2015-06-16 | 2016-06-29 | 苏州明锐医药科技有限公司 | Azd9291中间体及其制备方法 |
| CN110523540B (zh) * | 2019-08-14 | 2021-05-11 | 江西理工大学 | 一种新型表面活性剂在氧化锌矿浮选上的应用方法 |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1996005189A1 (de) * | 1994-08-09 | 1996-02-22 | Pentapharm Ag | Neue inhibitoren vom benzamidintyp |
| WO1996040744A1 (en) * | 1995-06-07 | 1996-12-19 | Cor Therapeutics, Inc. | KETOHETEROCYCLIC INHIBITORS OF FACTOR Xa |
| WO1998031661A1 (en) * | 1997-01-17 | 1998-07-23 | Ajinomoto Co., Inc. | Benzamidine derivatives |
Family Cites Families (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5239113A (en) * | 1991-10-15 | 1993-08-24 | Monsanto Company | Substituted β-amino acid derivatives useful as platelet aggregation inhibitors and intermediates thereof |
| IL112795A (en) * | 1994-03-04 | 2001-01-28 | Astrazeneca Ab | Peptide derivatives as antithrombic agents their preparation and pharmaceutical compositions containing them |
| JPH07330695A (ja) * | 1994-04-11 | 1995-12-19 | Green Cross Corp:The | 新規カルボン酸誘導体またはその塩、およびその医薬用途 |
| AU723338B2 (en) * | 1996-01-02 | 2000-08-24 | Aventis Pharmaceuticals Inc. | Substituted n-{(aminoiminomethyl or aminomethyl)phenyl}propyl amides |
| JPH101467A (ja) * | 1996-06-13 | 1998-01-06 | Banyu Pharmaceut Co Ltd | ビフェニルアミジン誘導体 |
| DE19632772A1 (de) * | 1996-08-14 | 1998-02-19 | Basf Ag | Neue Benzamidine |
| US6740682B2 (en) * | 1997-08-29 | 2004-05-25 | Tularik Limited | Meta-benzamidine derivatives as serine protease inhibitors |
| JPH11140040A (ja) | 1997-11-06 | 1999-05-25 | Kissei Pharmaceut Co Ltd | 3−アミジノフェニルエーテル誘導体、活性化血液凝固第x因子阻害剤およびそれらの製造中間体 |
| AU753675B2 (en) | 1998-03-19 | 2002-10-24 | Ajinomoto Co., Inc. | Aminoisoquinoline derivatives |
| CA2334476A1 (en) * | 1998-06-08 | 1999-12-16 | Ajinomoto Co., Inc. | Benzamidine derivative |
| US6037365A (en) * | 1998-09-25 | 2000-03-14 | G.D. Searle & Co. | Aminobenzamidinosuccinyl lactone derivatives useful as inhibitors of platelet aggregation |
-
1999
- 1999-06-08 CA CA002334476A patent/CA2334476A1/en not_active Abandoned
- 1999-06-08 CN CN99809391A patent/CN1311771A/zh active Pending
- 1999-06-08 WO PCT/JP1999/003055 patent/WO1999064392A1/ja not_active Ceased
- 1999-06-08 EP EP99923959A patent/EP1086946A4/en not_active Withdrawn
- 1999-06-08 AU AU40604/99A patent/AU758567B2/en not_active Ceased
- 1999-06-08 KR KR1020007013949A patent/KR20010052694A/ko not_active Withdrawn
-
2000
- 2000-12-08 US US09/731,729 patent/US6410538B2/en not_active Expired - Lifetime
-
2002
- 2002-02-14 US US10/073,985 patent/US6812231B2/en not_active Expired - Fee Related
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1996005189A1 (de) * | 1994-08-09 | 1996-02-22 | Pentapharm Ag | Neue inhibitoren vom benzamidintyp |
| WO1996040744A1 (en) * | 1995-06-07 | 1996-12-19 | Cor Therapeutics, Inc. | KETOHETEROCYCLIC INHIBITORS OF FACTOR Xa |
| WO1998031661A1 (en) * | 1997-01-17 | 1998-07-23 | Ajinomoto Co., Inc. | Benzamidine derivatives |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP1086946A4 * |
Cited By (24)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6410538B2 (en) | 1998-06-08 | 2002-06-25 | Ajinomoto Co., Inc. | Benzamidine derivatives |
| US6812231B2 (en) | 1998-06-08 | 2004-11-02 | Ajinomoto Co., Inc. | Benzamidine derivative |
| EP1086946A4 (en) * | 1998-06-08 | 2003-03-05 | Ajinomoto Kk | BENZAMIDINE DERIVATIVE |
| EP1236712A4 (en) * | 1999-12-06 | 2005-04-20 | Ajinomoto Kk | Amidinophenylpyruvic acid DERIVATIVE |
| US6710056B2 (en) | 1999-12-06 | 2004-03-23 | Ajinomoto Co., Inc. | Amidinophenylpyruvic acid derivatives |
| WO2001042199A1 (en) * | 1999-12-06 | 2001-06-14 | Ajinomoto Co.,Inc. | Amidinophenylpyruvic acid derivative |
| WO2001042254A1 (en) * | 1999-12-08 | 2001-06-14 | Ajinomoto Co., Inc. | Benzamidine derivatives |
| US6784191B2 (en) | 1999-12-08 | 2004-08-31 | Ajinomoto Co., Inc. | Benzamidine derivatives |
| US6838479B2 (en) | 2000-09-11 | 2005-01-04 | Genentech, Inc. | Amidine inhibitors of serine proteases |
| US6410733B1 (en) | 2000-09-11 | 2002-06-25 | Genentech, Inc. | Amidine inhibitors of serine proteases |
| WO2002022575A1 (en) * | 2000-09-11 | 2002-03-21 | Genentech, Inc. | Amidine inhibitors of serine proteases |
| RU2290403C2 (ru) * | 2000-12-28 | 2006-12-27 | Дайити Фармасьютикал Ко., Лтд. | Ингибиторы vla-4 |
| JPWO2002053534A1 (ja) * | 2000-12-28 | 2004-04-30 | 第一製薬株式会社 | Vla−4阻害薬 |
| WO2002053534A1 (en) * | 2000-12-28 | 2002-07-11 | Daiichi Pharmaceutical Co., Ltd. | Vla-4 inhibitors |
| US7157487B2 (en) | 2000-12-28 | 2007-01-02 | Daiichi Pharmaceutical Co., Ltd. | Vla-4 inhibitors |
| EP2982668A2 (en) | 2002-12-03 | 2016-02-10 | Pharmacyclics LLC | 2-(2-hydroxybiphenyl-3-yl)-1h-benzoimidazole-5-carboxamidine derivatives as factor viia inhibitors for the treatment of thromboembolic disorders |
| US7691894B2 (en) | 2003-07-24 | 2010-04-06 | Daiichi Pharmaceutical Co., Ltd. | Cyclohexanecarboxylic acid compound |
| US7893279B2 (en) | 2003-07-24 | 2011-02-22 | Daiichi Pharmaceutical Co., Ltd. | Cyclohexanecarboxylic acid compound |
| WO2006083003A1 (ja) | 2005-02-02 | 2006-08-10 | Ajinomoto Co., Inc. | 新規ベンズアミジン化合物 |
| JP4900238B2 (ja) * | 2005-02-02 | 2012-03-21 | 味の素株式会社 | 新規ベンズアミジン化合物 |
| US8227506B2 (en) | 2005-02-02 | 2012-07-24 | Ajinomoto., Inc. | Benzamidine compound |
| WO2010005087A1 (ja) | 2008-07-11 | 2010-01-14 | 味の素株式会社 | アミジン誘導体 |
| US8778925B2 (en) | 2008-10-06 | 2014-07-15 | Cancer Research Technology Ltd. | Pyridine and pyrimidine based compounds as Wnt signaling pathway inhibitors for the treatment of cancer |
| WO2010119877A1 (ja) * | 2009-04-13 | 2010-10-21 | 味の素株式会社 | アミジン誘導体の製造方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP1086946A1 (en) | 2001-03-28 |
| AU4060499A (en) | 1999-12-30 |
| US6812231B2 (en) | 2004-11-02 |
| US20020107290A1 (en) | 2002-08-08 |
| US6410538B2 (en) | 2002-06-25 |
| CN1311771A (zh) | 2001-09-05 |
| KR20010052694A (ko) | 2001-06-25 |
| AU758567B2 (en) | 2003-03-27 |
| CA2334476A1 (en) | 1999-12-16 |
| US20010056123A1 (en) | 2001-12-27 |
| EP1086946A4 (en) | 2003-03-05 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO1999064392A1 (en) | Benzamidine derivative | |
| WO1999040075A1 (en) | Sulfonamide derivatives, process for producing the same and utilization thereof | |
| JP4103147B2 (ja) | ベンズアミジン誘導体 | |
| RU2332412C2 (ru) | Производные пиразола | |
| CA2416384A1 (en) | Sulfone derivatives, their production and use | |
| KR20010042606A (ko) | 아미딘 화합물 | |
| JP6986972B2 (ja) | 置換4−ベンジル及び4−ベンゾイルピペリジン誘導体 | |
| CN102325754B (zh) | 含杂原子的环状化合物 | |
| ES2211559T3 (es) | Derivados de indolilpiperidina como agentes anhistaminicos y antialergicos. | |
| AU753675B2 (en) | Aminoisoquinoline derivatives | |
| BRPI0509256B1 (pt) | Derivados de pirrolidina-3,4-dicarboxamida que inibem o fator xa de coagulação | |
| EP1188755A1 (en) | Acylhydrazine derivatives, process for preparing the same and use thereof | |
| JP4446145B2 (ja) | ベンズアミジン誘導体 | |
| JP3283485B2 (ja) | アミジン化合物 | |
| JPWO1999064392A1 (ja) | ベンズアミジン誘導体 | |
| JPWO2001042254A1 (ja) | ベンズアミジン誘導体 | |
| HUP0200223A2 (en) | Alkynyl containing hydroxamic acid derivatives, their preparation and their use as matrix metalloproteinase (mmp) inhibitors/tnf-alpha converting enzyme (tace) inhibitors and pharmaceutical compositions containing them | |
| JPWO1999047503A1 (ja) | アミノイソキノリン誘導体 | |
| JP2005298343A (ja) | ベンゾジアゼピン誘導体 | |
| EP1455788A1 (en) | 1-glycinyl-4-aminomethyl-piperidine derivatives as factor xa inhibitors |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 99809391.2 Country of ref document: CN |
|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AL AM AT AU AZ BA BB BG BR BY CA CH CN CU CZ DE DK EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MD MG MK MN MW MX NO NZ PL PT RO RU SD SE SG SI SK SL TJ TM TR TT UA UG US UZ VN YU ZA ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW SD SL SZ UG ZW AM AZ BY KG KZ MD RU TJ TM AT BE CH CY DE DK ES FI FR GB GR IE IT LU MC NL PT SE BF BJ CF CG CI CM GA GN GW ML MR NE SN TD TG |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| ENP | Entry into the national phase |
Ref document number: 2334476 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 40604/99 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020007013949 Country of ref document: KR Ref document number: 09731729 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1999923959 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 1999923959 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020007013949 Country of ref document: KR |
|
| WWG | Wipo information: grant in national office |
Ref document number: 40604/99 Country of ref document: AU |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1020007013949 Country of ref document: KR |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 1999923959 Country of ref document: EP |