WO2000012564A1 - High-purity polysaccharide containing hydrophobic groups and process for producing the same - Google Patents
High-purity polysaccharide containing hydrophobic groups and process for producing the same Download PDFInfo
- Publication number
- WO2000012564A1 WO2000012564A1 PCT/JP1999/001683 JP9901683W WO0012564A1 WO 2000012564 A1 WO2000012564 A1 WO 2000012564A1 JP 9901683 W JP9901683 W JP 9901683W WO 0012564 A1 WO0012564 A1 WO 0012564A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- polysaccharide
- hydrophobic group
- purity
- producing
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08B—POLYSACCHARIDES; DERIVATIVES THEREOF
- C08B37/00—Preparation of polysaccharides not provided for in groups C08B1/00 - C08B35/00; Derivatives thereof
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08B—POLYSACCHARIDES; DERIVATIVES THEREOF
- C08B15/00—Preparation of other cellulose derivatives or modified cellulose, e.g. complexes
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08B—POLYSACCHARIDES; DERIVATIVES THEREOF
- C08B33/00—Preparation of derivatives of amylose
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08B—POLYSACCHARIDES; DERIVATIVES THEREOF
- C08B35/00—Preparation of derivatives of amylopectin
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08B—POLYSACCHARIDES; DERIVATIVES THEREOF
- C08B37/00—Preparation of polysaccharides not provided for in groups C08B1/00 - C08B35/00; Derivatives thereof
- C08B37/0006—Homoglycans, i.e. polysaccharides having a main chain consisting of one single sugar, e.g. colominic acid
- C08B37/0009—Homoglycans, i.e. polysaccharides having a main chain consisting of one single sugar, e.g. colominic acid alpha-D-Glucans, e.g. polydextrose, alternan, glycogen; (alpha-1,4)(alpha-1,6)-D-Glucans; (alpha-1,3)(alpha-1,4)-D-Glucans, e.g. isolichenan or nigeran; (alpha-1,4)-D-Glucans; (alpha-1,3)-D-Glucans, e.g. pseudonigeran; Derivatives thereof
- C08B37/0021—Dextran, i.e. (alpha-1,4)-D-glucan; Derivatives thereof, e.g. Sephadex, i.e. crosslinked dextran
Definitions
- the present invention relates to a high-purity hydrophobic group-containing polysaccharide and a method for producing the same.
- Water-soluble polymers include natural polymers, semi-synthetic polymers, and synthetic polymers.
- examples of the natural polymer include carbohydrates such as starch and seaweed; plant-based viscous substances such as gum arabic;
- Examples of semi-synthetic polymers include cellulosic polymers such as viscose.
- Synthetic polymers include polyvinyl alcohol, polyvinyl pyridine, polyglycerin and the like.
- polysaccharide-coated carriers in which drug carriers such as ribosome microcapsules, microspheres, OZW emollients, and erythrocyte guests are coated with hydrophobic group-containing polysaccharides are included in this carrier.
- drug carriers such as ribosome microcapsules, microspheres, OZW emollients, and erythrocyte guests are coated with hydrophobic group-containing polysaccharides
- a compound in which the water-soluble polymer is a polysaccharide and the hydrophobic group is a steryl group, that is, a polysaccharide-sterol derivative is a ribosome polysaccharide coating agent (Japanese Patent Application Laid-Open No. Sho 61-68901) and a fat emulsion. It was already disclosed as a coating agent (Japanese Patent Application Laid-Open No. 63-1991 / 46) and a polymer surfactant for preparing a polysaccharide-coated emulsion (Japanese Patent Application Laid-Open No. 2-144140). As one of the synthesizing methods, a technique disclosed in Japanese Patent Application Laid-Open No. Sho 61-69801 is disclosed.
- the three-step method has been adopted so far.
- the carboxyl group in step 2 tends to remain unreacted to the end, and the liposome or emulsion when coated with the polysaccharide is subjected to physicochemical treatment.
- the effects of the negative charge of the carboxy group cannot be prevented on stability, cell specificity, and compatibility.
- the number of steps in the synthesis is long.
- Japanese Patent Application Laid-Open No. 3-292301 discloses that a diisocyanate compound is reacted with sterol in the first step to form a steryl group at the ⁇ -position at one end of the alkane. After synthesizing a monoisocyanate compound having an isocyanate group at the ⁇ -position at the other end, the polyisocyanate is reacted with the monoisocyanate compound in the second step to give steryl to the polysaccharide.
- a synthesis method that can easily introduce a group is disclosed.
- polysaccharide-sterol derivative When the polysaccharide-sterol derivative is used as a drug carrier or coated on liposomes or the like, a high-purity polysaccharide-sterol derivative with less by-products is desired.
- a first object of the present invention is to provide a high-purity hydrophobic compound capable of easily and efficiently producing a high-purity hydrophobic group-containing polysaccharide having a low content of impurities such as unsubstituted polysaccharides and sterol dimers. It is to propose a method for producing a group-containing polysaccharide.
- a second object of the present invention is to provide a high-purity hydrophobic group-containing polysaccharide obtained by the above production method. Disclosure of the invention
- the present inventors have conducted intensive studies in view of the above-mentioned conventional problems.
- a ketone-based solvent is used as a reprecipitation solvent, and purification by ultracentrifugation or purification with an aprotic polar solvent is combined.
- a polysaccharide containing a high-purity hydrophobic group can be obtained, and have completed the present invention. That is, the present invention
- the following is a high-purity hydrophobic group-containing polysaccharide and a method for producing the same.
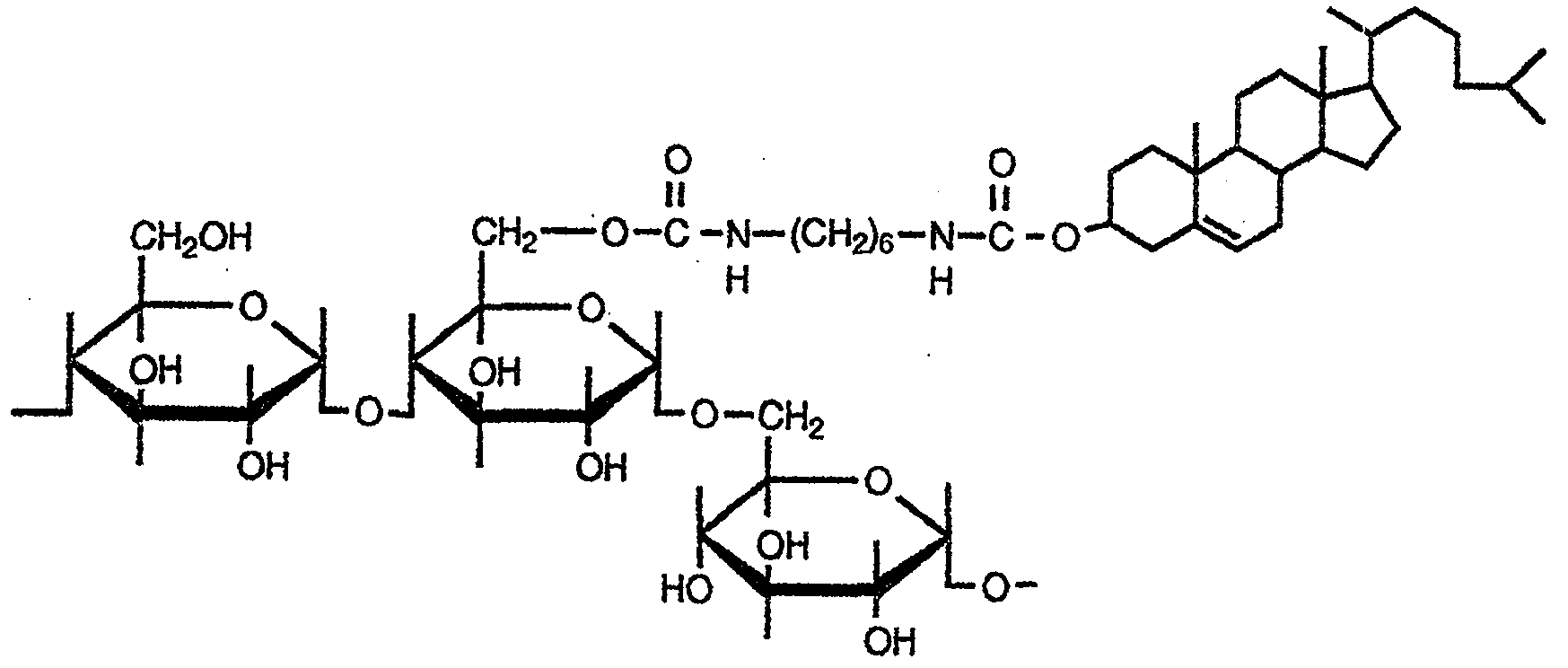
- a hydroxyl group-containing hydrocarbon or sterol having 12 to 50 carbon atoms and ⁇ CN—R 1 —NCO (where R 1 is a hydrocarbon group having from! To 50 carbon atoms)
- a diisocyanate compound represented by the formula (1) to produce a hydroxyl group-containing hydrocarbon having 12 to 50 carbon atoms or an isocyanate group-containing hydrophobic compound in which one molecule of a sterol has reacted.
- the isocyanate group-containing hydrophobic compound obtained in the first step reaction is further reacted with a polysaccharide to form a hydrocarbon group having 12 to 50 carbon atoms or a steryl group as a hydrophobic group.
- a polysaccharide to form a hydrocarbon group having 12 to 50 carbon atoms or a steryl group as a hydrophobic group.
- a method for producing a high-purity hydrophobic group-containing polysaccharide wherein the reaction product of the second step reaction is purified with a ketone-based solvent.
- the polysaccharides are pullulan, amylopectin, amylose, dextran, hydroxyxetinolesenorelose, hydroxyxetinolesdextran, mannan, leban, inulin, chitin, chitosan, xyloglucan, and water-soluble cellulose.
- the method for producing a high-purity hydrophobic group-containing polysaccharide according to the above (1) which is at least one selected from the group consisting of:
- ketone solvent is at least one selected from the group consisting of acetone, methyl ethyl ketone, getyl ketone, and diisopropyl ketone.
- the hydrophobic group-containing polysaccharide may be represented by one XH group [where X is an oxygen atom or a nitrogen-containing group represented by NY (where Y is a hydrogen atom or a hydrocarbon group having 1 to 10 carbon atoms) )
- X is an oxygen atom or a nitrogen-containing group represented by NY (where Y is a hydrogen atom or a hydrocarbon group having 1 to 10 carbon atoms)
- XH group an oxygen atom or a nitrogen-containing group represented by NY (where Y is a hydrogen atom or a hydrocarbon group having 1 to 10 carbon atoms)
- R 1 represents a hydrocarbon group having 1 to 50 carbon atoms
- R 2 represents a hydrocarbon group having 12 to 50 carbon atoms or a steryl group.
- the non-protonic polar solvent is one or more selected from the group consisting of N, N-dimethylformamide, N, N-dimethylacetamide and dimethylsulfoxide (13) Or (14) the method for producing a high-purity hydrophobic group-containing polysaccharide according to (14).
- the content of impurities reacted with a hydroxyl group-containing hydrocarbon or sterol of ⁇ 50 is 0.
- the isocyanate obtained in the first step reaction is further reacted with a polysaccharide to form a hydrophobic group containing a hydrocarbon group having 12 to 50 carbon atoms or a steryl group as a hydrophobic group.
- a polysaccharide containing a functional group Manufactures a polysaccharide containing a functional group
- a polysaccharide having an XH group [where X is an oxygen atom or a nitrogen-containing group represented by NY (where Y is a hydrogen atom or a hydrocarbon group having 1 to 10 carbon atoms)];
- the number of sugar units constituting the polysaccharide is 100, and 0.1 to 10 — XH groups are represented by the following formula (1)
- R 1 represents a hydrocarbon group having 1 to 50 carbon atoms
- R 2 represents a hydrocarbon group having 12 to 50 carbon atoms or a steryl group.
- the polysaccharides are pullulan, amylopectin, amylose, dextran, hydroxyxetinoresenorelose, hydroxyxechinoledextran, mannan, lenoquin, inulin, chitin, chitosan, xyloglucan
- FIG. 1 is a diagram showing a 1 H-NMR spectrum of a pullulan-cholesterol derivative (CHP) obtained in Example 1-2.
- FIG. 2 (a) shows the pullulan monocholesterol derivative (C) obtained in Example 1-2.
- FIG. 7 is a diagram showing the results of SEC analysis of a solution subjected to ultrasonic treatment. The vertical axis indicates the intensity of the differential refractometer (no unit) (the same applies hereinafter).
- FIG. 3 is a diagram showing a 1 H—NMR spectrum of a mannan-cholesterol derivative (C HM) obtained in Example 2-1.
- FIG. 4 is a diagram showing a 1 H-NMR spectrum of a mannan-cholesterol derivative (CHM) obtained in Example 2-2.
- FIG. 5 is a diagram showing the result of purulun-monocholesterol derivative (CHP) obtained in Examples 12 and 12 purified by a non-polar solvent, irradiated with ultrasonic waves, and then analyzed by SEC (size exclusion chromatography).
- CHP purulun-monocholesterol derivative
- “high purity” means that the content of the dimer obtained by reacting the hydrophobic group of the hydrocarbon group and the steryl group with the diisocyanate compound and the content of the unsubstituted polysaccharide are small.
- the hydroxyl group-containing hydrocarbon having 12 to 50 carbon atoms used in the present invention is used as a raw material for introducing a hydrophobic group.
- Examples of the hydroxyl group-containing hydrocarbon group having 12 to 50 carbon atoms used in the present invention include, for example, lauryl alcohol, myristyl alcohol, cetanolanolone, stearyl alcohol, araquinyl alcohol, docosanole, and pentacosanole.
- alcohols having 12 to 35 carbon atoms, particularly 12 to 20 carbon atoms are preferred because they are easily available.
- the hydroxyl group-containing hydrocarbon having 12 to 50 carbon atoms can be used alone or in combination of two or more. Hydroxyl-containing carbon used as a raw material for introducing a hydrophobic group If the carbon number of the hydrogen hydride is less than 12, it is not preferable because the hydrophobic aggregation effect cannot be sufficiently exhibited. On the other hand, if the carbon number exceeds 50, it becomes difficult to obtain, which is not preferable.
- the sterol used in the present invention is used as a raw material for introducing a hydrophobic group.
- the sterol used in the present invention includes, for example, cholesterol, stigmasterol, / 8-sitosterol, lanosterol, ergosterol and the like. Among these, cholesterol is preferred from the viewpoint of availability and the like.
- Sterols can be used alone or in combination of two or more.
- the hydroxyl group-containing hydrocarbon group having 12 to 50 carbon atoms and a sterol may be used in combination.
- the diisocyanate compound used in the present invention is OCN—R 1 —NCO (wherein, R 1 is a carbon atom having 1 to 50 carbon atoms).
- diisocyanate compound represented by the following formula: If the carbon number of R 1 exceeds 50, it becomes difficult to obtain, which is not preferable.
- the diisocyanate compound include ethylene diisocyanate in which R 1 is an ethylene group, butylene diisocyanate in which a butylene group is present, hexamethylene diisocyanate in which a hexamethylene group is present, and diphenylmethane group. And diphenylmethane diisocyanate.
- polysaccharide used in the present invention for example, a natural or semi-synthetic polysaccharide can be used. Specifically, from pullulan, amylopectin, amylose, dextran, hydroxyxetinoresenolerose, hydroxyxetichi redextran, mannan, levan, inulin, chitin, chitosan, xyloglucan and water-soluble cellulose At least one selected from the group consisting of: Among these, pullulan, mannan, xylognole, amylopectin, amylose, dextran, and hydroxyxetyl cellulose are preferred.
- nitrogen-containing polysaccharides such as chitin, partially deacetylated chitin and chitosan.
- Polysaccharides can be used alone or in combination of two or more.
- the hydrophobic group-containing polysaccharide produced by the production method of the present invention is a single XH group [wherein X is an oxygen atom or a nitrogen-containing group represented by NY (where Y is a hydrogen atom or And 10 to 10.
- the -H group is a hydrophobic group-containing polysaccharide substituted with the hydrophobic group represented by the formula (1).
- R 1 is a group derived from the diisocyanate compound.
- R 2 is a hydroxyl group-containing hydrocarbon group having 12 to 50 carbon atoms and a group derived from Z or sterol.
- Specific examples of the group represented by R 2 include a lauryl group, a myristyl group, a cetyl group, a stearyl group, a cholesteryl group, a stigmasteryl group, a / 3-citosteryl group, a lanosteryl group, an ergosteryl group, and the like.
- Can be Preferable examples include a myristyl group, a stearyl group, and a cholesteryl group.
- monosaccharides constituting polysaccharides such as pullulan and mannan
- both the OH group of the CH 2 OH group and the OH group directly bonded to the monosaccharide have the hydrophobicity represented by the above formula (1).
- the proportion of OH groups in CH 2 OH groups is overwhelmingly large, and the proportion of OH groups directly bonded to monosaccharides is small.
- a polysaccharide having a CH 2 OH group and an NH 2 group bonded thereto such as chitosan
- the OH group of the CH 2 OH group, the NH 2 group, and the OH group directly bonded to the monosaccharide are used. All groups are substituted with the hydrophobic group represented by the above formula (1), but the substitution rate is predominantly OH and NH 2 groups of CH 2 OH groups, and they are directly bonded to the monosaccharide. The proportion of OH groups is small.
- the production method of the present invention includes the following steps 1 to 3 or steps 1 to 4.
- steps 1 to 3 or steps 1 to 4 In the following description of the production method, a case where pullulan is used as the polysaccharide and a steryl group is used as the hydrophobic group will be described. However, when other substances are used, the production can be similarly performed.
- the reaction according to the production method of the present invention is represented by the following reaction formula (I) And the reaction formula ( ⁇ ). Reaction equation (I) corresponds to step 1, and reaction equation ( ⁇ ) corresponds to step 2.
- R 2 steryl group
- steryl isocyanate having a steryl group at one end and an isocyanate group at the other end of the alkane used in the present invention is represented by the formula (4).
- the compound is obtained by the reaction between a diisocyanate compound represented by the formula (2) and a sterol represented by the formula (3).
- an isocyanate group at one end of a diisocyanate compound represented by the formula (2) in an organic solvent in the presence of a basic catalyst By reacting with the hydroxyl group of the sterol represented by (3), one end is bonded to the sterol by a urethane bond, and the other end is left unreacted as an isocyanate group.
- a sterol dimer represented by the formula (5) is usually produced as a by-product at about 10% by weight.
- a polysaccharide-sterol derivative polysaccharide containing a hydrophobic group represented by the formula (7)
- a hydroxyl group of the polysaccharide represented by the formula (6) is added to a steryl isocyanate represented by the formula (4) in an organic solvent in the presence of a basic catalyst.
- the reaction product obtained in the above step 2 is purified by reprecipitation in a ketone-based solvent (hereinafter, this purification is referred to as ketone purification).
- this purification a sterol dimer by-produced in step 1 is mainly removed, and a high-purity polysaccharide-sterol derivative (polysaccharide containing a hydrophobic group) can be obtained.
- a purified product obtained by ketone purification is referred to as a purified ketone product.
- the purified ketone can be further purified by a dialysis method to further remove the reaction solvent.
- Step 4 1) The purified ketone (including the purified dialyzed product) obtained in the above step 3 is further purified by ultracentrifugation. In this purification by ultracentrifugation, unsubstituted polysaccharides (unreacted polysaccharides) are mainly removed, and a highly pure polysaccharide containing a hydrophobic group can be obtained.
- purification can also be performed using a non-protonic polar solvent.
- the non-protonic polar solvent is added to the purified ketone (including dialyzed purified product) obtained in the above step 3 to dissolve the solution, and the solution is dissolved in the solution. Add water, mix well with a stirrer, etc., and remove the separated aqueous layer.
- Unpurified polysaccharides are mainly removed by the purification with the non-protonic polar solvent, so that a polysaccharide containing a hydrophobic group having higher purity can be obtained. This operation may be repeated many times, and by repeating it two or three times, the purity of the hydrophobic group-containing polysaccharide is further improved. Further, by removing the non-protonic polar solvent, the hydrophobic group-containing polysaccharide can be obtained as a powdery solid.
- the step of producing the compound represented by the formula (4) includes the step of preparing the compound represented by the formula (3) and the diisocyanate compound represented by the formula (2) in an organic solvent in the presence of a basic catalyst.
- a basic catalyst With the sterols to be reacted.
- the amount of the diisocyanate compound used is 1 to 30 molar equivalents, preferably 10 to 20 molar equivalents, based on sterol.
- addition of amines as a basic catalyst is desirable, so that the reaction proceeds efficiently.
- Examples of the organic solvent used in the reaction include ether solvents, non-protonic polar solvents, halogen solvents, aliphatic and aromatic hydrocarbon solvents, and the like.
- Examples of the ether-based solvent include aliphatic ethers such as ethyl ether and heterocyclic ethers such as tetrahydrofuran.
- Non-protonic polar solvents include, for example, acetone, dimethylformamide (DMF), dimethylsulfoxide and the like. (DMSO).
- Examples of the halogen-based solvent include methylene chloride, chloroform and the like.
- Examples of the aliphatic hydrocarbon solvent include pentane and hexane.
- Examples of the aromatic hydrocarbon solvent include benzene, toluene, and the like. Of these, aromatic hydrocarbons are preferred.
- the amines used in the reaction of the reaction formula (I) include triethylamine, pyridine and the like.
- the amount of the amine used is 1 to 20 molar equivalents, preferably 1 to 3 molar equivalents, based on sterol.
- the reaction temperature and time vary depending on the diisocyanate compound used and the solvent used, and are set according to the progress of the reaction.
- the reaction temperature is preferably from room temperature to 100 ° C. Is preferably 3 to 24 hours.
- the reaction is preferably carried out using a dried solvent and a basic catalyst, and more preferably in an inert gas atmosphere.
- the inert gas include nitrogen, argon and the like.
- the step of producing the compound represented by the formula (7) is carried out in an organic solvent in the presence of a basic catalyst with the polysaccharide represented by the formula (6) and the compound produced in the step 1 (4) reacting with a steryl isocyanate compound represented by the formula:
- the charging ratio of the polysaccharide to the steryl isocyanate compound is set depending on the amount of the steryl group introduced to the polysaccharide, and is 0.1 to 1 per 100 monosaccharide units of the polysaccharide.
- Examples of the organic solvent used in the reaction include ether solvents, non-protonic polar solvents, halogen solvents, aliphatic and aromatic hydrocarbon solvents, and the like.
- Examples of the ether-based solvent include aliphatic ethers such as ethyl ether and heterocyclic ethers such as tetrahydrofuran.
- Examples of the aprotic polar solvent include acetone, dimethylformamide (DMF), dimethylsulfoxide (DMSO) and the like.
- Examples of the halogen-based solvent include methylene chloride, chloroform and the like.
- Examples of the aliphatic hydrocarbon solvent include pentane, 1 b hexane and the like.
- aromatic hydrocarbon solvent examples include benzene, toluene, and the like. Of these, nonprotonic polar solvents are preferred.
- Amines are preferred as the basic catalyst used in the reaction of the reaction formula (II), and examples thereof include triethylamine and pyridine.
- the amount of the amine used is 1 to 10 molar equivalents, preferably 1 to 3 molar equivalents, based on the polysaccharide.
- the reaction temperature and time vary depending on the polysaccharide and solvent used, and are set according to the progress of the reaction. The reaction temperature is preferably room temperature to 100 ° C, and the reaction time is preferably 30 ° C. Minutes to 24 hours.
- the reaction is preferably carried out using a dried solvent and a basic catalyst, and more preferably in an inert gas atmosphere.
- the inert gas include nitrogen, argon and the like.
- Examples of the ketone solvent used in the ketone purification in Step 3 include one or more selected from the group consisting of acetone, methyl ethyl ketone, getyl ketone, and diisopropyl ketone.
- the amount of the ketone solvent used is 4 to 50 times, preferably 8 to 20 times the weight of the reaction solution obtained in the step 2.
- the reaction product obtained in step 2 is added to a ketone solvent, the polysaccharide-sterol derivative (hydrophobic group-containing polysaccharide) precipitates, and the sterol dimer by-produced in step 1 is dissolved in the ketone solvent. Therefore, a high-purity polysaccharide-sterol derivative can be obtained by collecting the precipitate.
- the precipitate can be dried by freeze drying, vacuum drying, or other methods.
- Ketone purification has a higher sterol dimer removal rate than conventional purification methods such as dialysis, reprecipitation purification using ethanol, and column chromatography, and therefore high-purity polysaccharides containing hydrophobic groups. Can be easily obtained.
- the content of the hydrophobic group-containing polysaccharide in the purified ketone is at least 80% by weight, preferably at least 90% by weight.
- the content of unsubstituted polysaccharide is 20% by weight or less, preferably 10% by weight or less.
- two NCO groups in the diisocyanate compound the content of impurities that have reacted with the hydrophobic groups is 0.05% by weight or less, and preferably 0.01% by weight or less.
- water is added to the purified ketone product in the step 3 and ultrasonic irradiation is performed, followed by ultracentrifugation.
- water used here include distilled water and ion-exchanged water.
- the amount of water used is 5 to 100 times, preferably 30 to 60 times the weight of the purified ketone.
- the ultracentrifugation is carried out at 10,000 to 200,000 G, preferably 30,000 to 100,000 G, for 1 to 24 hours, preferably 3 to 15 hours. Ultracentrifugation separates the high molecular weight polysaccharide-sterol derivative into the lower layer and the low-molecular weight unsubstituted polysaccharide into the upper layer.
- a high-purity polysaccharide is obtained.
- a sterol derivative can be obtained.
- the content of the hydrophobic group-containing polysaccharide in the purified product obtained by ultracentrifugation is 98% by weight or more, preferably 99.9% by weight or more.
- the content of unsubstituted polysaccharide is 2% by weight or less, preferably 0.1% by weight or less.
- the content of impurities in which both two NCO groups in the diisocyanate compound have reacted with the hydrophobic group is 0.05% by weight or less, preferably 0.01% by weight or less.
- non-protonic polar solvent examples include N, N-dimethylformamide (DMF), N, N-dimethylacetamide (DMAc), 1,3-dimethyl-12-imidazolidinone (DMI) and dimethylsulfoxide (DMSO).
- the amount of the non-protonic polar solvent used is 3 to 50 times by weight, preferably 5 to 15 times by weight, based on the purified ketone obtained in the above step 3. If it is less than 3 times by weight, it is not enough to dissolve the purified ketone, and if it is more than 50 times by weight, it will not be separated into two layers when water is added later.
- the temperature at which the purified ketone product is dissolved in the non-protonic polar solvent is 0 to 150 ° C, preferably room temperature to 1 oo ° C.
- Solvents other than aprotic polar solvents include hydrophobic groups. 1 o It is not preferable because polysaccharides and unsubstituted polysaccharides do not dissolve.
- Examples of water that can be used in step 4-2) include distilled water, ion-exchanged water, and pure water.
- the amount of water used is at least 5 times by weight, and preferably from 10 to 100 times by weight, of the solution of the purified ketone in the nonprotonic polar solvent. If it is less than 5 times by weight, the two layers are not separated and are mixed, which is not preferable. On the other hand, if the amount is more than 100 times by weight, it is not preferable because remarkable improvement in removal efficiency of unsubstituted polysaccharide cannot be expected. It is preferable to add a predetermined amount of water at a time and then mix it with a stirrer or the like. If added with stirring, it is not preferable because water is mixed and two layers are difficult to separate. In the two-layer separation step, the mixture may be allowed to stand still or may be forcibly separated by a centrifuge or the like.
- Water is added to a solution of the purified ketone in an aprotic polar solvent, mixed with water, and separated into two layers, an aqueous layer and an aprotic polar solvent layer.
- the unsubstituted polysaccharide is transferred to the aqueous layer.
- the target hydrophobic group-containing polysaccharide is still dissolved in the aprotic polar solvent layer, the unsubstituted polysaccharide can be removed by removing the aqueous layer.
- a highly pure hydrophobic group-containing polysaccharide can be obtained.
- the removal of the non-protonic polar solvent includes methods such as chromatography, lyophilization or reprecipitation.
- a ketone-based or alcohol-based solvent is preferable, and examples thereof include acetone, methylethylketone, methanol, and ethanol.
- the amount of the poor solvent used is 4 to 50 times, preferably 8 to 20 times the weight of the solution to be reprecipitated.
- the precipitate can be dried by a freeze-drying method or a vacuum drying method.
- Purification with a non-protonic polar solvent can be carried out simply and efficiently, as compared with the purification by ultracentrifugation in step 4 1). This makes it possible to mass-produce high-purity hydrophobic group-containing polysaccharides required for use in living organisms such as pharmaceuticals.
- the content of hydrophobic group-containing polysaccharide in purified product purified with non-protonic polar solvent is 9 It is at least 8% by weight, preferably at least 99.9% by weight.
- the content of unsubstituted polysaccharide is 2% by weight or less, preferably 0.1% by weight or less.
- the content of impurities in which both of the two NCO groups in the diisocyanate compound have reacted with the hydrophobic group is not more than 0.02% by weight, preferably not more than 0.01% by weight.
- sterol dimers and unsubstituted polysaccharides which have remained in the target product and have been extremely difficult to completely remove, can be easily prepared. It can be efficiently removed, whereby a high-purity polysaccharide having a high-purity hydrophobic group can be obtained.
- the high-purity hydrophobic group-containing polysaccharide of the present invention is a high-purity hydrophobic group-containing polysaccharide obtained by the production method of the present invention, and has a hydrophobic group represented by the formula (1). However, it is a high-purity hydrophobic group-containing polysaccharide having a purity of 80% by weight or more, preferably 90% by weight or more.
- the high-purity hydrophobic group-containing polysaccharide of the present invention can form a solution in which the hydrophobic group represented by the formula (1) is coagulated and finely dispersed, and as a result, a core-shell type polymer micelle is obtained. Has the ability to form
- the high-purity hydrophobic group-containing polysaccharide of the present invention can be used as a medical material such as a coating material for coating a drug carrier containing a drug.
- a medical material such as a coating material for coating a drug carrier containing a drug.
- it can be used as a coating material for coating a drug carrier such as a ribosome microcapsule, a microsphere, an ozw emollination or an erythrocyte ghost.
- the high-purity hydrophobic group-containing polysaccharide of the present invention has a low content of by-products and unsubstituted polysaccharides and is highly pure, so that it can be safely used as a medical material.
- the method for producing a high-purity hydrophobic group-containing polysaccharide of the present invention is performed using a ketone solvent, the content of impurities such as unsubstituted polysaccharide and sterol dimer is reduced.
- a high-purity hydrophobic group-containing polysaccharide having a small amount can be easily and efficiently produced. Purification by ultracentrifugation or purification with nonprotonic polar solvents By performing the combination, it is possible to produce a higher-purity polysaccharide containing a hydrophobic group.
- the high-purity hydrophobic group-containing polysaccharide of the present invention can be obtained by the above production method, it has high purity and can be safely used as a medical material.
- the purified product purified by ascent was analyzed by 1 H-NMR, and the content of cholesterol dimer was calculated from the proton ratio. As a result, the presence of cholesterol dimer was not confirmed. Therefore, the content of the cholesterol dimer represented by the formula (5a) in the purified product purified by the ascent is 0% by weight.
- the amount of cholesterol dimer contained in 78 g (3.2 1 mmol) was 0. It is calculated to be 142 g (Example 11-1 confirms that the content of cholesterol dimer is 8% by weight; see Example 1-1). This from the data of The amount of sterol dimer removed was calculated. Table 1 shows the results.
- FIG. 1 shows the 1 H-NMR spectrum of the pullulan-co-resteromonole derivative represented by the formula (7a), which is the target substance, obtained as described above. From the integrated value of the 1 H-NMR spectrum, the rate of introduction of a cholesterol group into pullulan in the pullulan monocholesterol derivative was calculated by the following equation (A).
- a pullulan-cholesterol derivative was synthesized in the same manner as in Example 1-2. After completion of the reaction, the precipitate was purified by reprecipitation in ethanol to obtain a pullulan monocholesterol derivative.
- the purified product purified with ethanol was analyzed using preparative TLC in the same manner as in Examples 1-2. As a result, the presence of the cholesterol dimer (R f value: 0.65) represented by the above formula (5a) was confirmed.
- the purified product purified with ethanol was analyzed by 1 H-NMR in the same manner as in Example 1-2 to calculate the content of cholesterol dimer. As a result, the cholesterol dimer was contained in an amount of 0.4% by weight.
- the ethanol used in the purification was recovered, and the cholesterol dimer contained therein was quantified to be 0.016 g. there were. From this data, Examples 1-2 Similarly, the removal amount of the cholesterol dimer was calculated. Table 1 shows the results.
- CHP pullulan monocholesterol derivative
- the purified product purified by the ascent was analyzed using preparative TLC in the same manner as in Example 1-2. As a result, the presence of the cholesterol dimer (R i value: 0.65) represented by the above formula (5a) was not confirmed.
- the purified product purified by ascent was analyzed by 1 H-NMR, and the content of cholesterol dimer was calculated based on the proton ratio. As a result, the presence of cholesterol dimer was not confirmed. Therefore, the content of the cholesterol dimer represented by the formula (5a) in the purified product obtained by the ascent is 0% by weight.
- FIG. 3 shows the 1 H-NMR spectrum of the compound represented by the formula (7b) obtained as described above.
- the introduction rate of cholesterol groups into mannan in this compound was calculated in the same manner as in Examples 1-2. As a result, it was found that the degree of substitution of the cholesterol group was 1.1 per 100 monosaccharides.
- the purified product purified by the ascent was analyzed using preparative TLC in the same manner as in Examples 1-2. As a result, the presence of the cholesterol dimer (R f value: 0.65) represented by the above formula (5a) was not confirmed.
- the purified product purified by ascent was analyzed by 1 H-NMR, and the content of cholesterol dimer was calculated based on the proton ratio. As a result, the presence of cholesterol dimer was not confirmed. Therefore, the content of the cholesterol dimer represented by the above formula (5a) in the purified product purified by the ascent is 0% by weight.
- FIG. 4 shows the NMR spectrum.
- the introduction rate of cholesterol groups into mannan in this compound was calculated in the same manner as in Example 1-2. As a result, it was found that the degree of substitution of the cholesterol group was 0.8 per 100 monosaccharides.
- the amount of purification is not limited in principle, and can be easily increased.
- the high-purity hydrophobic group-containing polysaccharide obtained by the production method of the present invention contains a drug.
- ribosome microcapsules For example, ribosome microcapsules, microspheres, O / W emulsion
- the high-purity hydrophobic group-containing polysaccharide of the present invention is a by-product and an unsubstituted polysaccharide.
Landscapes
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Biochemistry (AREA)
- Materials Engineering (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Molecular Biology (AREA)
- General Health & Medical Sciences (AREA)
- Polysaccharides And Polysaccharide Derivatives (AREA)
- Steroid Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Description

















Claims

Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR1020007004490A KR100541752B1 (ko) | 1998-08-31 | 1999-03-31 | 고순도 소수성기함유다당류 및 그 제조방법 |
| EP99912075A EP1026174B1 (en) | 1998-08-31 | 1999-03-31 | High-purity polysaccharide containing hydrophobic groups and process for producing the same |
| US09/530,347 US6566516B1 (en) | 1998-08-31 | 1999-03-31 | High purity polysaccharide containing a hydrophobic group and process for producing it |
| DE69925355T DE69925355T2 (de) | 1998-08-31 | 1999-03-31 | Hydrophobierte und sehr reine polysaccharide und verfahren zu deren herstellung |
| AU30545/99A AU755283B2 (en) | 1998-08-31 | 1999-03-31 | High-purity polysaccharide containing hydrophobic groups and process for producing the same |
| JP54456199A JP3416951B2 (ja) | 1998-08-31 | 1999-03-31 | 高純度疎水性基含有多糖類およびその製造方法 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP10/244671 | 1998-08-31 | ||
| JP24467198 | 1998-08-31 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US09/530,347 A-371-Of-International US6566516B1 (en) | 1998-08-31 | 1999-03-31 | High purity polysaccharide containing a hydrophobic group and process for producing it |
| US10/091,992 Continuation US20020143160A1 (en) | 1998-08-31 | 2002-03-06 | High purity polysaccharide containing hydrophobic group and process for producing it |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2000012564A1 true WO2000012564A1 (en) | 2000-03-09 |
Family
ID=17122226
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP1999/001683 Ceased WO2000012564A1 (en) | 1998-08-31 | 1999-03-31 | High-purity polysaccharide containing hydrophobic groups and process for producing the same |
Country Status (7)
| Country | Link |
|---|---|
| US (2) | US6566516B1 (ja) |
| EP (1) | EP1026174B1 (ja) |
| JP (1) | JP3416951B2 (ja) |
| KR (1) | KR100541752B1 (ja) |
| AU (1) | AU755283B2 (ja) |
| DE (1) | DE69925355T2 (ja) |
| WO (1) | WO2000012564A1 (ja) |
Cited By (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2000057841A1 (en) * | 1999-03-31 | 2000-10-05 | Nof Corporation | Cosmetics containing polysaccharide-sterol derivatives |
| JP2002284693A (ja) * | 2001-03-29 | 2002-10-03 | Nof Corp | ゲル状外用剤組成物 |
| US6566516B1 (en) * | 1998-08-31 | 2003-05-20 | Nof Corporation | High purity polysaccharide containing a hydrophobic group and process for producing it |
| WO2003097672A1 (en) * | 2002-05-21 | 2003-11-27 | Japan Science And Technology Corporation | Method for controlling protein |
| JP2006117746A (ja) * | 2004-10-20 | 2006-05-11 | Kao Corp | 多糖誘導体 |
| WO2006049032A1 (ja) * | 2004-11-01 | 2006-05-11 | Tokyo Medical And Dental University | ナノゲル-アパタイト複合体の調製 |
| WO2007083643A1 (ja) | 2006-01-18 | 2007-07-26 | National University Corporation Tokyo Medical And Dental University | 骨形成促進物質とナノゲルを含有する骨形成用生体材料 |
| WO2008136536A1 (ja) | 2007-05-01 | 2008-11-13 | National University Corporation Tokyo Medical And Dental University | 化学架橋ヒアルロン酸誘導体を含むハイブリッドゲルおよびそれを用いた医薬組成物 |
| WO2010050578A1 (ja) | 2008-10-31 | 2010-05-06 | 国立大学法人東京医科歯科大学 | カチオン性ナノゲルを用いる粘膜ワクチン |
| WO2012017313A2 (en) | 2010-08-06 | 2012-02-09 | Chanel Parfums Beaute | Method for producing composition for external use containing physiologically acceptable salt of tranexamate |
| JP2012504697A (ja) * | 2008-10-06 | 2012-02-23 | アドシア | 疎水性アルコール誘導体により置換されたカルボキシル官能基を含有する多糖類 |
| JPWO2014054588A1 (ja) * | 2012-10-01 | 2016-08-25 | 国立大学法人京都大学 | ナノゲル/エキソソーム複合体とdds |
| US9833407B2 (en) | 2014-02-17 | 2017-12-05 | Intellectual Property Strategy Network, Inc. | Nasal vaccine for Streptococcus pneumoniae |
| WO2020027309A1 (ja) | 2018-08-03 | 2020-02-06 | 国立大学法人東京大学 | 細胞性免疫を誘導する経鼻ワクチン |
| WO2020027318A1 (ja) | 2018-08-03 | 2020-02-06 | 国立研究開発法人農業・食品産業技術総合研究機構 | ウシ乳房炎に対する粘膜ワクチン組成物 |
| WO2020203731A1 (ja) | 2019-03-29 | 2020-10-08 | 国立大学法人東京大学 | 肺炎球菌表層タンパク質 |
| WO2022210465A1 (ja) | 2021-03-30 | 2022-10-06 | 国立大学法人東京大学 | ナノゲル被覆型ワクチン |
| WO2023022141A1 (ja) | 2021-08-17 | 2023-02-23 | ユナイテッド・イミュニティ株式会社 | がん治療剤 |
| WO2024190865A1 (ja) | 2023-03-15 | 2024-09-19 | ユナイテッド・イミュニティ株式会社 | 脂質粒子 |
| WO2024248117A1 (ja) | 2023-06-01 | 2024-12-05 | ユナイテッド・イミュニティ株式会社 | 複合体 |
Families Citing this family (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE19962272A1 (de) | 1999-12-23 | 2001-06-28 | Basf Ag | Isocyanatgruppen aufweisende Bausteine sowie ihre Verwendung zur Funktionalisierung oder Modifizierung von Verbindungen oder Oberflächen |
| GB0015981D0 (en) * | 2000-06-29 | 2000-08-23 | Glaxo Group Ltd | Novel process for preparing crystalline particles |
| EP1627070A4 (en) * | 2003-04-21 | 2008-05-07 | Univ Georgia Res Found | XYLOGLUCANKONE JUGATES SUITABLE FOR THE MODIFICATION OF CELLULOSE-TEXTILE TEXTILES |
| DE102004022897A1 (de) * | 2004-05-10 | 2005-12-08 | Bayer Cropscience Ag | Azinyl-imidazoazine |
| KR100578382B1 (ko) * | 2004-07-16 | 2006-05-11 | 나재운 | 항암제의 전달체용 수용성 키토산 나노입자 및 그 제조방법 |
| MX2007013725A (es) | 2005-05-05 | 2008-04-09 | Sensient Flavors Inc | Produccion de beta-glucanos y mananos. |
| US20070042970A1 (en) * | 2005-08-22 | 2007-02-22 | Chemical Soft R&D Inc. | Folate-modified cholesterol-bearing pullulan as a drug carrier |
| JP4866173B2 (ja) * | 2006-01-25 | 2012-02-01 | 大日精化工業株式会社 | ヒドロキシアルキル化キトサン溶液 |
| US8932858B2 (en) * | 2008-03-07 | 2015-01-13 | Corning Incorporated | Modified polysaccharide for cell culture and release |
| US8426382B2 (en) * | 2008-10-06 | 2013-04-23 | Adocia | Polysaccharides comprising carboxyl functional groups substituted by a hydrophobic alcohol derivative |
| US11173106B2 (en) | 2009-10-07 | 2021-11-16 | Johnson & Johnson Consumer Inc. | Compositions comprising a superhydrophilic amphiphilic copolymer and a micellar thickener |
| US8258250B2 (en) * | 2009-10-07 | 2012-09-04 | Johnson & Johnson Consumer Companies, Inc. | Compositions comprising superhydrophilic amphiphilic copolymers and methods of use thereof |
| US8399590B2 (en) * | 2009-10-07 | 2013-03-19 | Akzo Nobel Chemicals International B.V. | Superhydrophilic amphiphilic copolymers and processes for making the same |
| FR2954325B1 (fr) | 2009-12-23 | 2012-02-03 | Flamel Tech Sa | Polymere amphiphile fonctionnalise par la methionine |
| FR2975912B1 (fr) | 2011-05-30 | 2013-06-14 | Flamel Tech Sa | Composition a liberation controlee de buprenorphine |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS6169801A (ja) * | 1984-09-12 | 1986-04-10 | Junzo Sunamoto | 天然由来多糖誘導体およびその製造方法 |
| JPS63319046A (ja) * | 1987-06-24 | 1988-12-27 | Eisai Co Ltd | 被覆脂肪乳剤 |
| JPH02144140A (ja) * | 1988-11-25 | 1990-06-01 | Nippon Oil & Fats Co Ltd | 多糖誘導体によって安定化された脂肪乳剤 |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6120536A (en) * | 1995-04-19 | 2000-09-19 | Schneider (Usa) Inc. | Medical devices with long term non-thrombogenic coatings |
| FR2745014B1 (fr) * | 1996-02-20 | 1998-04-03 | Rhone Poulenc Chimie | Procede de traitement antisalissure des articles a base de coton tisse |
| DE19613990A1 (de) * | 1996-04-09 | 1997-10-16 | Wolff Walsrode Ag | Thermoplastischer Werkstoff bestehend aus aliphatischen Carbamidsäurederivaten von Polysacchariden und niedermolekularen Harnstoffderivaten sowie ein Verfahren zu dessen Herstellung und Verwendung desselben |
| US6566516B1 (en) * | 1998-08-31 | 2003-05-20 | Nof Corporation | High purity polysaccharide containing a hydrophobic group and process for producing it |
| JP3742984B2 (ja) * | 2000-03-28 | 2006-02-08 | 株式会社資生堂 | 油中水型乳化組成物 |
-
1999
- 1999-03-31 US US09/530,347 patent/US6566516B1/en not_active Expired - Lifetime
- 1999-03-31 KR KR1020007004490A patent/KR100541752B1/ko not_active Expired - Fee Related
- 1999-03-31 JP JP54456199A patent/JP3416951B2/ja not_active Expired - Lifetime
- 1999-03-31 WO PCT/JP1999/001683 patent/WO2000012564A1/ja not_active Ceased
- 1999-03-31 EP EP99912075A patent/EP1026174B1/en not_active Expired - Lifetime
- 1999-03-31 AU AU30545/99A patent/AU755283B2/en not_active Ceased
- 1999-03-31 DE DE69925355T patent/DE69925355T2/de not_active Expired - Lifetime
-
2002
- 2002-03-06 US US10/091,992 patent/US20020143160A1/en not_active Abandoned
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS6169801A (ja) * | 1984-09-12 | 1986-04-10 | Junzo Sunamoto | 天然由来多糖誘導体およびその製造方法 |
| JPS63319046A (ja) * | 1987-06-24 | 1988-12-27 | Eisai Co Ltd | 被覆脂肪乳剤 |
| JPH02144140A (ja) * | 1988-11-25 | 1990-06-01 | Nippon Oil & Fats Co Ltd | 多糖誘導体によって安定化された脂肪乳剤 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP1026174A4 * |
Cited By (24)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6566516B1 (en) * | 1998-08-31 | 2003-05-20 | Nof Corporation | High purity polysaccharide containing a hydrophobic group and process for producing it |
| WO2000057841A1 (en) * | 1999-03-31 | 2000-10-05 | Nof Corporation | Cosmetics containing polysaccharide-sterol derivatives |
| JP2002284693A (ja) * | 2001-03-29 | 2002-10-03 | Nof Corp | ゲル状外用剤組成物 |
| WO2003097672A1 (en) * | 2002-05-21 | 2003-11-27 | Japan Science And Technology Corporation | Method for controlling protein |
| JP2006117746A (ja) * | 2004-10-20 | 2006-05-11 | Kao Corp | 多糖誘導体 |
| WO2006049032A1 (ja) * | 2004-11-01 | 2006-05-11 | Tokyo Medical And Dental University | ナノゲル-アパタイト複合体の調製 |
| WO2007083643A1 (ja) | 2006-01-18 | 2007-07-26 | National University Corporation Tokyo Medical And Dental University | 骨形成促進物質とナノゲルを含有する骨形成用生体材料 |
| WO2008136536A1 (ja) | 2007-05-01 | 2008-11-13 | National University Corporation Tokyo Medical And Dental University | 化学架橋ヒアルロン酸誘導体を含むハイブリッドゲルおよびそれを用いた医薬組成物 |
| US8987230B2 (en) | 2007-05-01 | 2015-03-24 | National University Corporation Tokyo Medical And Dental University | Hybrid gel comprising chemically crosslinked hyaluronic acid derivative and pharmaceutical composition comprising the same |
| JP2012504697A (ja) * | 2008-10-06 | 2012-02-23 | アドシア | 疎水性アルコール誘導体により置換されたカルボキシル官能基を含有する多糖類 |
| WO2010050578A1 (ja) | 2008-10-31 | 2010-05-06 | 国立大学法人東京医科歯科大学 | カチオン性ナノゲルを用いる粘膜ワクチン |
| WO2012017313A2 (en) | 2010-08-06 | 2012-02-09 | Chanel Parfums Beaute | Method for producing composition for external use containing physiologically acceptable salt of tranexamate |
| JPWO2014054588A1 (ja) * | 2012-10-01 | 2016-08-25 | 国立大学法人京都大学 | ナノゲル/エキソソーム複合体とdds |
| US9833407B2 (en) | 2014-02-17 | 2017-12-05 | Intellectual Property Strategy Network, Inc. | Nasal vaccine for Streptococcus pneumoniae |
| WO2020027309A1 (ja) | 2018-08-03 | 2020-02-06 | 国立大学法人東京大学 | 細胞性免疫を誘導する経鼻ワクチン |
| WO2020027318A1 (ja) | 2018-08-03 | 2020-02-06 | 国立研究開発法人農業・食品産業技術総合研究機構 | ウシ乳房炎に対する粘膜ワクチン組成物 |
| KR20210040387A (ko) | 2018-08-03 | 2021-04-13 | 고쿠리츠다이가쿠호우진 도쿄다이가쿠 | 세포성 면역을 유도하는 경비 백신 |
| US11564993B2 (en) | 2018-08-03 | 2023-01-31 | The University Of Tokyo | Intranasal vaccine that induces cellular immunity |
| WO2020203731A1 (ja) | 2019-03-29 | 2020-10-08 | 国立大学法人東京大学 | 肺炎球菌表層タンパク質 |
| WO2022210465A1 (ja) | 2021-03-30 | 2022-10-06 | 国立大学法人東京大学 | ナノゲル被覆型ワクチン |
| KR20230163443A (ko) | 2021-03-30 | 2023-11-30 | 고쿠리츠다이가쿠호우진 도쿄다이가쿠 | 나노겔 피복형 백신 |
| WO2023022141A1 (ja) | 2021-08-17 | 2023-02-23 | ユナイテッド・イミュニティ株式会社 | がん治療剤 |
| WO2024190865A1 (ja) | 2023-03-15 | 2024-09-19 | ユナイテッド・イミュニティ株式会社 | 脂質粒子 |
| WO2024248117A1 (ja) | 2023-06-01 | 2024-12-05 | ユナイテッド・イミュニティ株式会社 | 複合体 |
Also Published As
| Publication number | Publication date |
|---|---|
| US20020143160A1 (en) | 2002-10-03 |
| US6566516B1 (en) | 2003-05-20 |
| AU755283B2 (en) | 2002-12-05 |
| DE69925355D1 (de) | 2005-06-23 |
| EP1026174A1 (en) | 2000-08-09 |
| AU3054599A (en) | 2000-03-21 |
| EP1026174A4 (en) | 2003-01-29 |
| DE69925355T2 (de) | 2006-01-12 |
| EP1026174B1 (en) | 2005-05-18 |
| KR20010031457A (ko) | 2001-04-16 |
| JP3416951B2 (ja) | 2003-06-16 |
| KR100541752B1 (ko) | 2006-01-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2000012564A1 (en) | High-purity polysaccharide containing hydrophobic groups and process for producing the same | |
| JPWO2000012564A1 (ja) | 高純度疎水性基含有多糖類およびその製造方法 | |
| EP3974462B1 (en) | Polyethylene glycol derivative, preparation method therefor, and polyethylene glycol hydrogel enabling fast crosslinking reaction | |
| JP4248189B2 (ja) | ホスホリルコリン基含有多糖類及びその製造方法 | |
| JPH0425505A (ja) | シクロデキストリンポリマー及びシクロデキストリン膜の製造方法 | |
| Toomari et al. | Fabrication of biodendrimeric β-cyclodextrin via click reaction with potency of anticancer drug delivery agent | |
| EP1550688A1 (en) | Organopolysiloxane-modified polysaccharide and process for producing the same | |
| Tirino et al. | Synthesis of chitosan–PEO hydrogels via mesylation and regioselective Cu (I)-catalyzed cycloaddition | |
| EP2159250A1 (en) | POLYROTAXANE HAVING MAIN CHAIN BACKBONE ESSENTIALLY COMPOSED OF -Si-O- AND METHOD FOR PRODUCING THE SAME, AND CROSSLINKED POLYROTAXANE OBTAINED BY CROSSLINKING THE POLYROTAXANE AND METHOD FOR PRODUCING THE SAME | |
| CN103450369A (zh) | 聚乙二醇单甲醚-壳聚糖衍生物的制备方法 | |
| JP2017520674A (ja) | ポリエーテルアミンをグラフトすることにより多糖類を修飾する方法、その方法により修飾された多糖類、およびその多糖類を備え、温度感受性のレオロジー特性を有する製剤 | |
| Kop et al. | Polysaccharide-fullerene supramolecular hybrids: Synthesis, characterization and antioxidant activity | |
| JPH03292301A (ja) | 多糖類―ステロール誘導体とその製造法 | |
| EP4180464B1 (en) | Cyclodextrin derivative having polymerizable unsaturated group | |
| CN112194741A (zh) | 一种聚乙二醇衍生物改性β-环糊精及其制备方法和应用 | |
| JP3371429B2 (ja) | 疎水性基含有多糖類の集合体の形成方法 | |
| CN103694377B (zh) | 一种两亲性c-6-(4-(甲基氨基)-1,2,3-三氮唑)脱氧菊糖衍生物及其制备和应用 | |
| NO312634B1 (no) | Fremgangsmåte for fremsilling av et polyoksyalkylenderivat som er substituert med suksinimidylgruppe | |
| JP3604390B2 (ja) | 疎水性化合物を溶解するためおよびエナンチオマの純度を照査するためのモノ−3,6−アンヒドロシクロデキストリンの使用およびこれらのシクロデキストリンの調製方法 | |
| JP5463541B2 (ja) | 修飾擬ポリロタキサンおよび修飾ポリロタキサン、ならびにそれらの製造方法 | |
| JPH069709A (ja) | シクロデキストリン誘導体及びその製造方法 | |
| JPH1143447A (ja) | キラル化合物、その合成および担体におけるその使用 | |
| JP3453278B2 (ja) | 胆汁酸吸着性樹脂 | |
| JPH11510429A (ja) | 相間移動触媒反応での触媒または共補助剤としてのデンドリマー型の高分子の用途 | |
| WO2024014395A1 (ja) | ロタキサン化合物 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AU JP KR US |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): BE CH DE FR GB IT NL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 09530347 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1020007004490 Country of ref document: KR |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 30545/99 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1999912075 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 1999912075 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020007004490 Country of ref document: KR |
|
| WWG | Wipo information: grant in national office |
Ref document number: 30545/99 Country of ref document: AU |
|
| WWG | Wipo information: grant in national office |
Ref document number: 1999912075 Country of ref document: EP |
|
| WWG | Wipo information: grant in national office |
Ref document number: 1020007004490 Country of ref document: KR |