WO2003084923A1 - Acyl-3-carboxyphenyl-harnstoffderivate, verfahren zu deren herstellung und deren verwendung - Google Patents
Acyl-3-carboxyphenyl-harnstoffderivate, verfahren zu deren herstellung und deren verwendung Download PDFInfo
- Publication number
- WO2003084923A1 WO2003084923A1 PCT/EP2003/003254 EP0303254W WO03084923A1 WO 2003084923 A1 WO2003084923 A1 WO 2003084923A1 EP 0303254 W EP0303254 W EP 0303254W WO 03084923 A1 WO03084923 A1 WO 03084923A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- alkyl
- cycloalkyl
- alkynyl
- phenyl
- alkenyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C275/00—Derivatives of urea, i.e. compounds containing any of the groups, the nitrogen atoms not being part of nitro or nitroso groups
- C07C275/46—Derivatives of urea, i.e. compounds containing any of the groups, the nitrogen atoms not being part of nitro or nitroso groups containing any of the groups, X being a hetero atom, Y being any atom, e.g. acylureas
- C07C275/48—Y being a hydrogen or a carbon atom
- C07C275/54—Y being a carbon atom of a six-membered aromatic ring, e.g. benzoylureas
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/54—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with at least one nitrogen and one sulfur as the ring hetero atoms, e.g. sulthiame
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
Definitions
- a tablet can be produced by compressing or shaping a powder or granulate of the compound, optionally with one or more additional components.
- Compressed tablets can be prepared by tabletting the compound in free-flowing form, such as a powder or granules, optionally mixed with a binder, lubricant, inert diluent and / or a (several) surface-active / dispersing agent in a suitable machine.
- Shaped tablets can be shaped of the powdery compound moistened with an inert liquid diluent are made in a suitable machine.
- active ingredients for the combination preparations are also suitable as active ingredients for the combination preparations: all antidiabetic agents mentioned in the 2001 Red List, Chapter 12. They can be combined with the compounds of the formula I according to the invention in particular to improve the synergistic effect.
- the active ingredient combination can be administered either by separate administration of the active ingredients to the patient or in the form of combination preparations in which several active ingredients are present in a pharmaceutical preparation. Most of the active ingredients listed below are disclosed in the USP Dictionary of USAN and International Drug Names, US Pharmacopeia, Rockville 2001.
- the compounds of formula 1 are used in combination with a cholesterol absorption inhibitor, e.g. Ezetimibe, Tiqueside, Pamaqueside, administered.
- a cholesterol absorption inhibitor e.g. Ezetimibe, Tiqueside, Pamaqueside
- the compounds of the formula I are used in combination with a mixed PPAR alpha / gamma agonist, e.g. GW 1536, AVE 8042, AVE 8134, AVE 0847, or as described in PCT / US 11833, PCT / US 11490, DE10142734.4.
- a mixed PPAR alpha / gamma agonist e.g. GW 1536, AVE 8042, AVE 8134, AVE 0847, or as described in PCT / US 11833, PCT / US 11490, DE10142734.4.
- the compounds of the formula I in combination with a fibrate such as e.g. Fenofibrate, clofibrate, bezafibrate.
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Diabetes (AREA)
- Medicinal Chemistry (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Hematology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Obesity (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Endocrinology (AREA)
- Emergency Medicine (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Acyclic And Carbocyclic Compounds In Medicinal Compositions (AREA)
Abstract
Description





Claims

Priority Applications (15)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| BR0309130-9A BR0309130A (pt) | 2002-04-11 | 2003-03-28 | Derivados de acil-3-carboxifenil-uréia, processo para a sua preparação e sua aplicação |
| EP03745772A EP1497263B1 (de) | 2002-04-11 | 2003-03-28 | Acyl-3-carboxyphenyl-harnstoffderivate, verfahren zu deren herstellung und deren verwendung |
| KR10-2004-7016149A KR20040097332A (ko) | 2002-04-11 | 2003-03-28 | 아실-3-카복시페닐우레아 유도체, 이의 제조방법 및 용도 |
| DE50310320T DE50310320D1 (de) | 2002-04-11 | 2003-03-28 | Acyl-3-carboxyphenyl-harnstoffderivate, verfahren zu deren herstellung und deren verwendung |
| IL16424803A IL164248A0 (en) | 2002-04-11 | 2003-03-28 | Acyl-3-carboxyphenylurea derivatives, processes for preparing them and their use |
| HU0500203A HUP0500203A2 (hu) | 2002-04-11 | 2003-03-28 | Acil-3-karboxi-fenil-karbamid-származékok, eljárás ezek előállítására és ezeket tartalmazó gyógyszerkészítmények és alkalmazásuk |
| AU2003232192A AU2003232192A1 (en) | 2002-04-11 | 2003-03-28 | Acyl-3-carboxyphenylurea derivatives, method for production and use thereof |
| YUP-887/04A RS88704A (sr) | 2002-04-11 | 2003-03-28 | Derivati acil-3-karboksifenil- karbamida, postupak za njihovu proizvodnju i njihova primena |
| MXPA04009239A MXPA04009239A (es) | 2002-04-11 | 2003-03-28 | Derivados de acil-3-carboxifenilurea, procedimientos para su preparacion y su utilizacion. |
| NZ535833A NZ535833A (en) | 2002-04-11 | 2003-03-28 | Acyl-3-carboxyphenylurea derivatives, processes for preparing them and their use |
| JP2003582122A JP4328212B2 (ja) | 2002-04-11 | 2003-03-28 | アシル−3−カルボキシフェニル尿素誘導体、その製造方法およびその使用 |
| CA002481819A CA2481819A1 (en) | 2002-04-11 | 2003-03-28 | Acyl-3-carboxyphenylurea derivatives, method for production and use thereof |
| HK05107763.5A HK1075650B (en) | 2002-04-11 | 2003-03-28 | Acyl-3-carboxyphenylurea derivatives, method for production and use thereof |
| HR20040931A HRP20040931A2 (en) | 2002-04-11 | 2003-03-28 | Acyl-3-carboxyphenylurea derivatives, method for production and use thereof |
| NO20044881A NO20044881L (no) | 2002-04-11 | 2004-11-09 | Acyl-3-karboksyfenylureaderivater, fremstillingsfremgangsmate og anvendelse derav |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| DE10215908.4 | 2002-04-11 | ||
| DE10215908A DE10215908B4 (de) | 2002-04-11 | 2002-04-11 | Acyl-3-carboxyphenyl-harnstoffderivate und deren Verwendung als Arzneimittel |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2003084923A1 true WO2003084923A1 (de) | 2003-10-16 |
Family
ID=28684936
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2003/003254 Ceased WO2003084923A1 (de) | 2002-04-11 | 2003-03-28 | Acyl-3-carboxyphenyl-harnstoffderivate, verfahren zu deren herstellung und deren verwendung |
Country Status (23)
| Country | Link |
|---|---|
| EP (1) | EP1497263B1 (de) |
| JP (1) | JP4328212B2 (de) |
| KR (1) | KR20040097332A (de) |
| CN (1) | CN1263734C (de) |
| AR (1) | AR039402A1 (de) |
| AT (1) | ATE404532T1 (de) |
| AU (1) | AU2003232192A1 (de) |
| BR (1) | BR0309130A (de) |
| CA (1) | CA2481819A1 (de) |
| DE (2) | DE10215908B4 (de) |
| HR (1) | HRP20040931A2 (de) |
| HU (1) | HUP0500203A2 (de) |
| IL (1) | IL164248A0 (de) |
| MA (1) | MA27195A1 (de) |
| MX (1) | MXPA04009239A (de) |
| NO (1) | NO20044881L (de) |
| NZ (1) | NZ535833A (de) |
| PL (1) | PL371317A1 (de) |
| RS (1) | RS88704A (de) |
| RU (1) | RU2004133046A (de) |
| TW (1) | TW200401765A (de) |
| WO (1) | WO2003084923A1 (de) |
| ZA (1) | ZA200407361B (de) |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2004075815A2 (de) | 2003-02-27 | 2004-09-10 | Aventis Pharma Deutschland Gmbh | Diarylcycloalkylderivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| US7148246B2 (en) | 2003-02-27 | 2006-12-12 | Sanofi-Aventis Deutschland Gmbh | Cycloalkyl derivatives having bioisosteric carboxylic acid groups, processes for their preparation and their use as pharmaceuticals |
| WO2006131231A1 (de) | 2005-06-09 | 2006-12-14 | Sanofi-Aventis | Azolopyridin-2-on-derivate als inhibitoren von lipasen und phospholipasen |
| US7220876B2 (en) | 2003-02-27 | 2007-05-22 | Sanofi-Aventis Deutschland Gmbh | Arylcycloalkyl derivatives having branched side chains, processes for their preparation and their use as pharmaceuticals |
| US7241787B2 (en) | 2004-01-25 | 2007-07-10 | Sanofi-Aventis Deutschland Gmbh | Substituted N-cycloexylimidazolinones, process for their preparation and their use as medicaments |
| US7259177B2 (en) | 2003-02-27 | 2007-08-21 | Sanofi-Aventis Deutschland Gmbh | Cycloalkylmethoxy-substituted acetic acid derivatives, processes for their preparation and their use as pharmaceuticals |
| WO2007128761A2 (de) | 2006-05-04 | 2007-11-15 | Boehringer Ingelheim International Gmbh | Verwendungen von dpp iv inhibitoren |
| EP2083006A1 (de) | 2004-04-01 | 2009-07-29 | Sanofi-Aventis Deutschland GmbH | Oxadiazolone, Verfahren zu ihrer Herstellung und ihre Verwendung als Arzneimittel |
| EP1497262B1 (de) * | 2002-04-11 | 2010-08-04 | Sanofi-Aventis Deutschland GmbH | Acyl-4-carboxy-phenyl-harnstoffderivate, verfahren zu deren herstellung und deren verwendung |
| US8048901B2 (en) | 2003-02-27 | 2011-11-01 | Aventis Pharma Deutschland Gmbh | 1,3-substituted cycloalkyl derivatives having acidic, mostly heterocyclic groups, processes for their preparation and their use as pharmaceuticals |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE10306502B4 (de) * | 2003-02-17 | 2005-03-17 | Aventis Pharma Deutschland Gmbh | Substituierte 3-(Benzoylureido)-thiophenderivate und sie enthaltende Arzneimittel |
Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001094300A1 (de) * | 2000-06-09 | 2001-12-13 | Aventis Pharma Deutschland Gmbh | Acylphenylharnstoffderivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ZA867420B (en) * | 1985-09-30 | 1987-05-27 | Union Carbide Corp | Pesticidal 1-(4-aryloxyphenyl)-3-benzoyl urea compounds and process for preparation |
| GB9313268D0 (en) * | 1993-06-28 | 1993-08-11 | Zeneca Ltd | Chemical compounds |
| FI974437A7 (fi) * | 1995-06-06 | 1997-12-05 | Pfizer | Substituoituja N-(indoli-2-karbonyyli)amideja ja johdannaisia glykogee nifosforylaasi-inhibiittoreina |
| WO2000056707A1 (en) * | 1999-03-24 | 2000-09-28 | Sepracor, Inc. | Diaryl thioethers, compositions and uses thereof |
| US20010053791A1 (en) * | 2000-03-16 | 2001-12-20 | Babcock Walter C. | Glycogen phosphorylase inhibitor |
| GB0021831D0 (en) * | 2000-09-06 | 2000-10-18 | Astrazeneca Ab | Chemical compounds |
| PE20021091A1 (es) * | 2001-05-25 | 2003-02-04 | Aventis Pharma Gmbh | Derivados de fenilurea sustituidos con carbonamida y procedimiento para su preparacion |
-
2002
- 2002-04-11 DE DE10215908A patent/DE10215908B4/de not_active Expired - Fee Related
-
2003
- 2003-03-28 WO PCT/EP2003/003254 patent/WO2003084923A1/de not_active Ceased
- 2003-03-28 CA CA002481819A patent/CA2481819A1/en not_active Abandoned
- 2003-03-28 AU AU2003232192A patent/AU2003232192A1/en not_active Abandoned
- 2003-03-28 MX MXPA04009239A patent/MXPA04009239A/es active IP Right Grant
- 2003-03-28 PL PL03371317A patent/PL371317A1/xx not_active Application Discontinuation
- 2003-03-28 AT AT03745772T patent/ATE404532T1/de not_active IP Right Cessation
- 2003-03-28 EP EP03745772A patent/EP1497263B1/de not_active Expired - Lifetime
- 2003-03-28 RU RU2004133046/04A patent/RU2004133046A/ru not_active Application Discontinuation
- 2003-03-28 HU HU0500203A patent/HUP0500203A2/hu unknown
- 2003-03-28 JP JP2003582122A patent/JP4328212B2/ja not_active Expired - Fee Related
- 2003-03-28 IL IL16424803A patent/IL164248A0/xx unknown
- 2003-03-28 RS YUP-887/04A patent/RS88704A/sr unknown
- 2003-03-28 HR HR20040931A patent/HRP20040931A2/hr not_active Application Discontinuation
- 2003-03-28 DE DE50310320T patent/DE50310320D1/de not_active Expired - Lifetime
- 2003-03-28 CN CNB038082241A patent/CN1263734C/zh not_active Expired - Fee Related
- 2003-03-28 KR KR10-2004-7016149A patent/KR20040097332A/ko not_active Withdrawn
- 2003-03-28 NZ NZ535833A patent/NZ535833A/xx unknown
- 2003-03-28 BR BR0309130-9A patent/BR0309130A/pt not_active IP Right Cessation
- 2003-04-09 TW TW092108055A patent/TW200401765A/zh unknown
- 2003-04-09 AR ARP030101246A patent/AR039402A1/es unknown
-
2004
- 2004-09-14 ZA ZA200407361A patent/ZA200407361B/xx unknown
- 2004-10-08 MA MA27903A patent/MA27195A1/fr unknown
- 2004-11-09 NO NO20044881A patent/NO20044881L/no unknown
Patent Citations (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2001094300A1 (de) * | 2000-06-09 | 2001-12-13 | Aventis Pharma Deutschland Gmbh | Acylphenylharnstoffderivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
Cited By (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1497262B1 (de) * | 2002-04-11 | 2010-08-04 | Sanofi-Aventis Deutschland GmbH | Acyl-4-carboxy-phenyl-harnstoffderivate, verfahren zu deren herstellung und deren verwendung |
| US7259177B2 (en) | 2003-02-27 | 2007-08-21 | Sanofi-Aventis Deutschland Gmbh | Cycloalkylmethoxy-substituted acetic acid derivatives, processes for their preparation and their use as pharmaceuticals |
| US7148246B2 (en) | 2003-02-27 | 2006-12-12 | Sanofi-Aventis Deutschland Gmbh | Cycloalkyl derivatives having bioisosteric carboxylic acid groups, processes for their preparation and their use as pharmaceuticals |
| US7160911B2 (en) | 2003-02-27 | 2007-01-09 | Sanofi-Aventis Deutschland Gmbh | Diarylcycloalkyl derivatives, process for their preparation and their use as pharmaceuticals |
| US7220876B2 (en) | 2003-02-27 | 2007-05-22 | Sanofi-Aventis Deutschland Gmbh | Arylcycloalkyl derivatives having branched side chains, processes for their preparation and their use as pharmaceuticals |
| US8048901B2 (en) | 2003-02-27 | 2011-11-01 | Aventis Pharma Deutschland Gmbh | 1,3-substituted cycloalkyl derivatives having acidic, mostly heterocyclic groups, processes for their preparation and their use as pharmaceuticals |
| WO2004075815A2 (de) | 2003-02-27 | 2004-09-10 | Aventis Pharma Deutschland Gmbh | Diarylcycloalkylderivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel |
| US7872034B2 (en) | 2003-02-27 | 2011-01-18 | Sanofi-Aventis Deutschland Gmbh | Arylcycloalkyl-substituted alkanoic acid derivatives, processes for their preparation and their use as pharmaceuticals |
| US7335671B2 (en) | 2003-02-27 | 2008-02-26 | Sanofi-Aventis Deutschland Gmbh | Arylcycloalkyl-substituted alkanoic acid derivatives, processes for their preparation and their use as pharmaceuticals |
| US7365084B2 (en) | 2003-02-27 | 2008-04-29 | Sanofi-Aventis Deutschland Gmbh | Cycloalkyl-substituted amino acid derivatives, processes for their preparation and their use as pharmaceuticals |
| US7241787B2 (en) | 2004-01-25 | 2007-07-10 | Sanofi-Aventis Deutschland Gmbh | Substituted N-cycloexylimidazolinones, process for their preparation and their use as medicaments |
| EP2083006A1 (de) | 2004-04-01 | 2009-07-29 | Sanofi-Aventis Deutschland GmbH | Oxadiazolone, Verfahren zu ihrer Herstellung und ihre Verwendung als Arzneimittel |
| WO2006131231A1 (de) | 2005-06-09 | 2006-12-14 | Sanofi-Aventis | Azolopyridin-2-on-derivate als inhibitoren von lipasen und phospholipasen |
| WO2007128761A2 (de) | 2006-05-04 | 2007-11-15 | Boehringer Ingelheim International Gmbh | Verwendungen von dpp iv inhibitoren |
| EP2351568A2 (de) | 2006-05-04 | 2011-08-03 | Boehringer Ingelheim International GmbH | Verwendungen von dpp iv Inhibitoren |
Also Published As
| Publication number | Publication date |
|---|---|
| DE10215908A1 (de) | 2003-11-06 |
| BR0309130A (pt) | 2005-02-01 |
| CN1263734C (zh) | 2006-07-12 |
| AU2003232192A1 (en) | 2003-10-20 |
| TW200401765A (en) | 2004-02-01 |
| ATE404532T1 (de) | 2008-08-15 |
| DE10215908B4 (de) | 2005-08-18 |
| RU2004133046A (ru) | 2005-04-20 |
| ZA200407361B (en) | 2006-02-22 |
| DE50310320D1 (de) | 2008-09-25 |
| RS88704A (sr) | 2007-02-05 |
| AR039402A1 (es) | 2005-02-16 |
| EP1497263A1 (de) | 2005-01-19 |
| NO20044881L (no) | 2005-01-07 |
| MA27195A1 (fr) | 2005-01-03 |
| JP2005522481A (ja) | 2005-07-28 |
| PL371317A1 (en) | 2005-06-13 |
| IL164248A0 (en) | 2005-12-18 |
| HRP20040931A2 (en) | 2005-02-28 |
| CA2481819A1 (en) | 2003-10-16 |
| HUP0500203A2 (hu) | 2005-06-28 |
| NZ535833A (en) | 2006-03-31 |
| JP4328212B2 (ja) | 2009-09-09 |
| HK1075650A1 (en) | 2005-12-23 |
| CN1646486A (zh) | 2005-07-27 |
| KR20040097332A (ko) | 2004-11-17 |
| EP1497263B1 (de) | 2008-08-13 |
| MXPA04009239A (es) | 2005-01-25 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP1497262B1 (de) | Acyl-4-carboxy-phenyl-harnstoffderivate, verfahren zu deren herstellung und deren verwendung | |
| EP1713803B1 (de) | 7-phenylamino-4-chinolon-3-carbonsäure-derivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel | |
| EP1513800B1 (de) | N-benzoylureido-zimts urederivate, verfahren zu deren herste llung und deren verwendung | |
| EP1523471B1 (de) | Harnstoff- und urethan-substituierte acylharnstoffe, verfahren zu deren herstellung und deren verwendung als arzneimittel | |
| DE10215908B4 (de) | Acyl-3-carboxyphenyl-harnstoffderivate und deren Verwendung als Arzneimittel | |
| DE102004004972B3 (de) | Heterocyclisch substituierte 7-Amino-4-chinolon-3-carbonsäure-Derivate, Verfahren zu ihrer Herstellung und ihre Verwendung als Arzneimittel | |
| EP1713804B1 (de) | Cycloalkyl substituierte 7-amino-4-chinolon-3-carbonsäure-derivate, verfahren zu ihrer herstellung und ihre verwendung als arzneimittel | |
| EP1603895B1 (de) | Substituierte benzoylureidopyridyl-piperidin- und -pyrrolidin-carbons urederivate, verfahren zu deren herstellung und deren v erwendung | |
| DE10306502B4 (de) | Substituierte 3-(Benzoylureido)-thiophenderivate und sie enthaltende Arzneimittel | |
| DE10302452B4 (de) | Carbonylamino-substituierte Acyl-phenyl-harnstoffderivate, Verfahren zu deren Herstellung und deren Verwendung | |
| EP1556339B1 (de) | Carboxyalkoxy-substituierte acyl-carboxyphenyl-harnstoffderivate, verfahren zu ihre herstellung und ihre verwendung als arzneimittel | |
| DE102004004973A1 (de) | 7-Phenylamino-4-chinolon-3-carbonsäure-Derivate, Verfahren zu ihrer Herstellung und ihre Verwendung als Arzneimittel | |
| DE10335092B3 (de) | Substituierte Benzoylureido-o-benzoylamide, Verfahren zu deren Herstellung und deren Verwendung | |
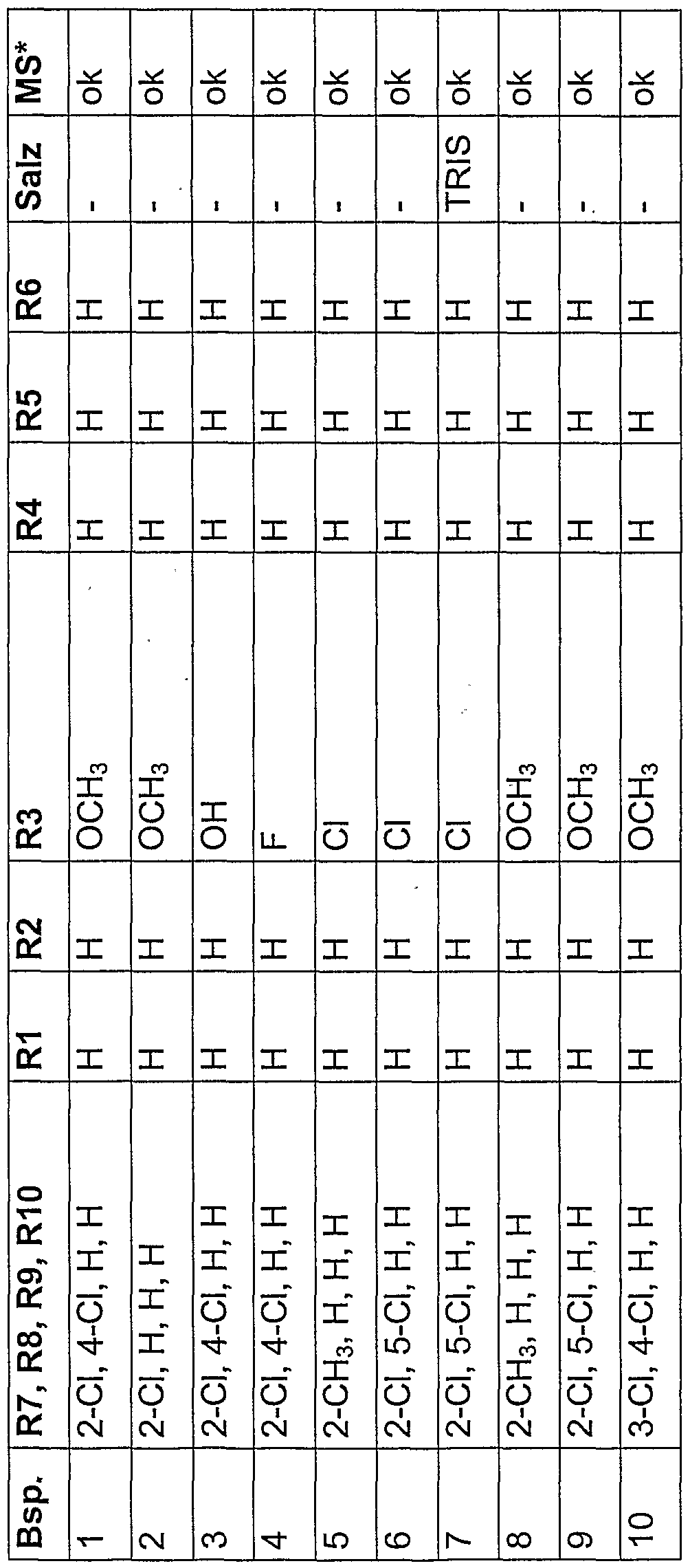
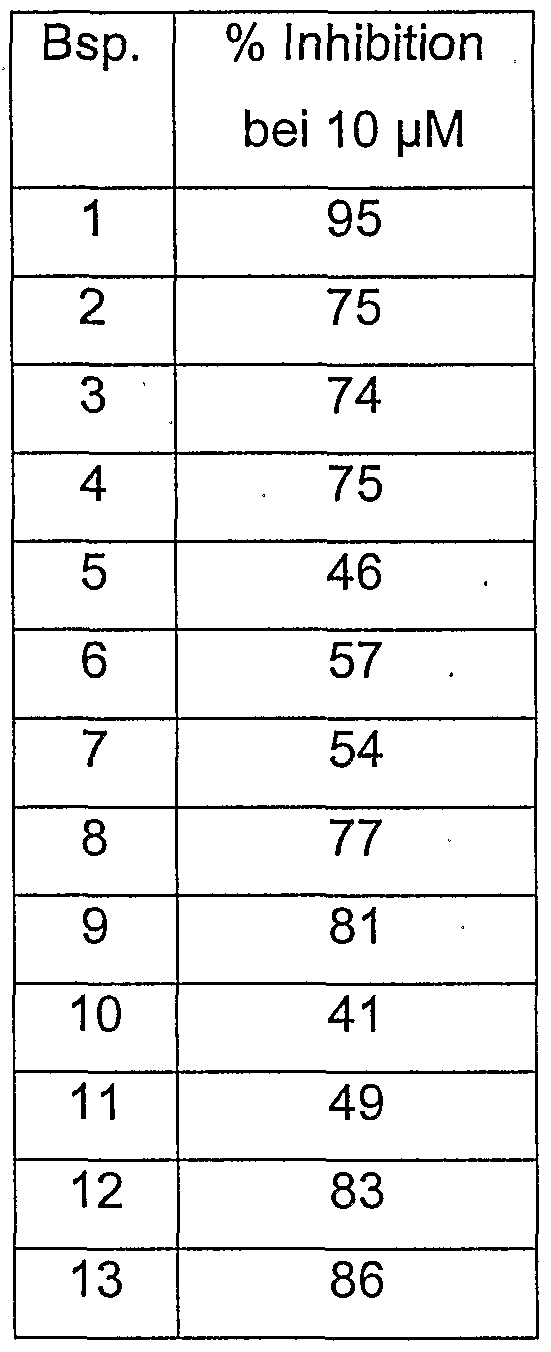
| US20030216370A1 (en) | Acyl-3-carboxyphenylurea derivatives, processes for preparing them and their use | |
| DE10306503A1 (de) | Substituierte Benzoylureidophenyl-piperidin- und pyrrolidin-carbonsäurederivate, Verfahren zu deren Herstellung und deren Verwendung | |
| DE102004033405A1 (de) | 7-Phenylamino-4-chinolon-3-carbonsäure-Derivate, Verfahren zu ihrer Herstellung und ihre Verwendung als Arzneimittel |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: P-887/04 Country of ref document: YU |
|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NO NZ OM PH PL PT RO RU SC SD SE SG SK SL TJ TM TN TR TT TZ UA UG UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): GH GM KE LS MW MZ SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LU MC NL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 200407361 Country of ref document: ZA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PA/a/2004/009239 Country of ref document: MX Ref document number: 164248 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2003745772 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 02236/CHENP/2004 Country of ref document: IN Ref document number: 2236/CHENP/2004 Country of ref document: IN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: P20040931A Country of ref document: HR Ref document number: 2481819 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 535833 Country of ref document: NZ Ref document number: 1020047016149 Country of ref document: KR Ref document number: 2003232192 Country of ref document: AU Ref document number: 2003582122 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1-2004-501413 Country of ref document: PH |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 20038082241 Country of ref document: CN |
|
| ENP | Entry into the national phase |
Ref document number: 2004133046 Country of ref document: RU Kind code of ref document: A |
|
| WWP | Wipo information: published in national office |
Ref document number: 1020047016149 Country of ref document: KR |
|
| WWP | Wipo information: published in national office |
Ref document number: 2003745772 Country of ref document: EP |
|
| REG | Reference to national code |
Ref country code: DE Ref legal event code: 8642 |
|
| WWP | Wipo information: published in national office |
Ref document number: 535833 Country of ref document: NZ |
|
| WWG | Wipo information: grant in national office |
Ref document number: 2003745772 Country of ref document: EP |