PHENYLA INOPYRIMIDINE UND IHRE VERWENDUNG ALS RHO-KINASE INHIBITOREN
Die Erfindung betrifft Phenylaminopyrimidine, ein Verfahren zu ihrer Herstellung sowie ihre Verwendung zur Herstellung von Arzneimitteln zur Behandlung und/oder Prophylaxe von Krankheiten bei Menschen und Tieren, insbesondere von kardiovaskulären Erkrankungen.
Ein Anstieg der intrazellulären Calcium-Konzentration ist ein Hauptauslöser für die Kontraktion der Gefaßmuskulatur (Somlyo, A.P. und Himpens, B., FASEB J. 1989, 3, 2266-2276). Dies geschieht in erster Linie durch Agonisten wie z.B. Phenylephrin oder Thromboxan A2, die nach Stimulierung der Phosphatidylinositolkaskade die Freisetzung von Calcium aus dem sarkoplasmatischen Retikulum bewirken. Die Erhöhung des intrazellulären Calciums aktiviert die MLC-Kinase (Myosin-Leichte- Ketten-Kinase), die die MLC-Untereinheiten des Myosinmoleküls phosphoryliert (Kamm, K.H. und Stull, J.T., Annu. Rev. Pharmacol.Toxicol. 1985, 25, 593-603). MLC-Phosphorylierung induziert die Glattmuskelkontraktion, MLC-Dephosphory- lierung nach einer Reduktion der intrazellulären Calciumkonzentration resultiert in der Relaxation des Gefäßes.
Neben der Calcium-abhängigen MLC-Phosphorylierung existiert noch ein weiterer zentraler aber Calcium-unabhängiger Regulationsmechanismus des Gefäßtonus. Hierbei handelt es sich um den Rho/Rho-Kinase-Signalweg (Noda, M. et al., FEBS Lett. 1995, 367, 246-250; Uehata, M. et al, Nature 1997, 389, 990-994; Fukata, Y. et al., Trends in Pharmacological Sciences 2001, 22, 32-39). Binden Agonisten wie z.B. Phenylephrin oder Thromboxan A2 an ihre Rezeptoren, so führt dies zur Aktivierung der kleinen G-Proteine Rho, die dann mit der Rho-Kinase interagieren und diese aktivieren. Die aktivierte Rho-Kinase inhibiert die Myosin-Phosphatase, nachdem sie eine Untereinheit des Enzyms phosphoryliert hat. Gleichzeitig phosphoryliert Rho-Kinase MLC an der Stelle, die auch von der MLC-Kinase phosphoryliert wird. Eine Hemmung der Myosin-Phosphatase sowie der Phos-
phorylierung von MLC induziert die Kontraktion der Gefaßmuskulatur. Im Gegensatz dazu fuhrt eine Hemmung der Rho-Kinase zu einer Gefäßrelaxation. Inhibitoren der Rho-Kinase bewirken daher eine Senkung des Blutdruckes und eine Steigerung des koronaren Blutflusses.
Strukturell ähnliche Verbindungen sind in anderen Indikationen bzw. für andere Wirkmechanismen bekannt. So beschreiben beispielsweise US 3 478 030 und US 3 432493 substituierte Aminopyrimidine, die den koronaren Blutfluss steigern können, dabei aber als Carboanhydrase-Inhibitoren wirken (J. Chem. Inf. Comp. Sciences 2002, 42, 94-102). Andere Pyrimidin-Derivate sind als Anti-Krebs- und Anti-HIV-Mittel (Debi, M.; Indian J. Exp. Biol. 1997, 35, 1208-1213) oder als cdk2- Inhibitoren (WO-A 01/64654) beschrieben.
Aufgabe der vorliegenden Erfindung ist die Bereitstellung von Arzneimitteln für die Behandlung von Erkrankungen, insbesondere von kardiovaskulären Erkrankungen.
Diese Aufgabe wird durch die Verbindungen der Formel (I) gelöst, die als Rho- Kinase-Inhibitoren wirken.
Gegenstand der vorliegenden Erfindung sind Verbindungen der Formel (I)
R1 Amino oder Hydroxy bedeutet,
R2 Wasserstoff, (Cι-C6)-Alkyl oder (C3-C8)-Cycloalkyl bedeutet,
R ,3 und j R> unabhängig voneinander Cyano, Wasserstoff, Fluor oder Chlor bedeuten,
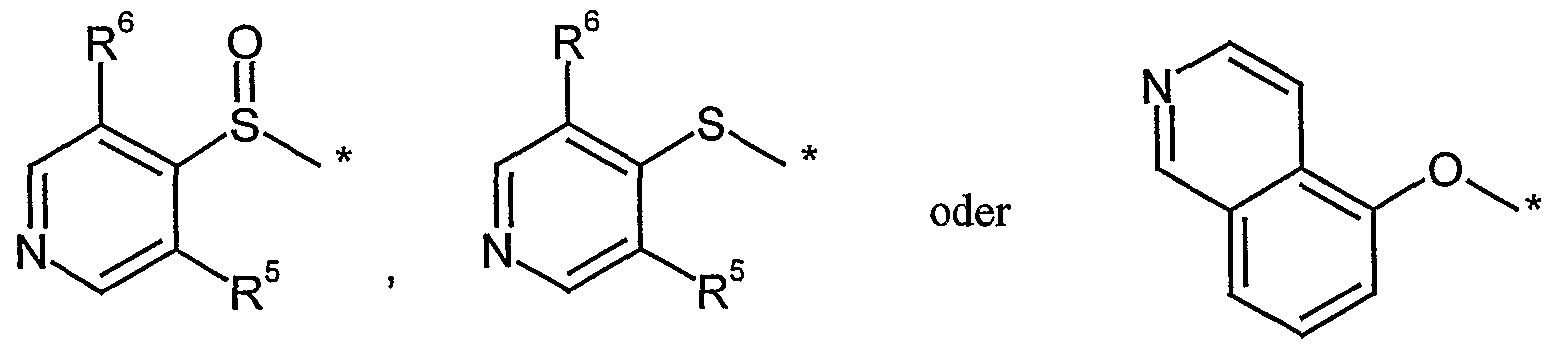
A einen Rest
R5 und R6 unabhängig voneinander Wasserstoff, Fluor oder Chlor bedeuten,
D (1) einen Rest bedeutet, der ausgewählt ist aus der Gruppe von
Phenyl, das seinerseits durch (Cι-C
4)-Alkylcarbonylamino, Hydroxy- methyl, Cyano,
oder 1,2-Dioxymethylen substituiert ist,
Chinolin, Isochinolin, Indol oder 6-gliedriges Heteroaryl mit 2 oder 3 Stickstoffatomen, wobei die Ringe jeweils über ein Kohlenstoffatom verknüpft sind,
Pyridylmethyl, 2-Oxo-2H-pyridin-l-yl, 4-Oxo-4H-pyridin-l-yl, die ihrerseits durch Fluor, Chlor oder (Cι-C4)-Alkyl substituiert sein können, und
Pyridyl, das seinerseits durch Fluor, Chlor oder (Cι-C4)-Alkyl substituiert ist,
oder
n
(2) einen Rest *-OR bedeutet,
worin
R7 Phenyl, das durch (Cι-C4)-Alkyl oder (CrC^-Alkoxy, die ihrerseits durch Hydroxy und/oder *-NR8R9 substituiert sein können, Trifluormethyl, Trifluormethoxy, Nitro, Cyano,
NR »8 Rr>9 , Fluor, Chlor oder 1,2-Dioxymethylen substituiert sein kann,
3- bis 7-gliedriges Heterocyclyl mit einem Stickstoffatom, das durch Wasserstoff, (Cι-C6)-Alkyl oder (C3-C8)-Cycloalkyl substituiert sein kann,
5- oder 6-gliedriges Heteroaryl mit bis zu drei Stickstoffatomen,
(Cι-C6)-Alkyl oder (C3-C7)-Cycloalkyl, die ihrerseits durch
Hydroxy oder *-NR8R9 substituiert sein können,
Thienyl, Furyl, Pyridylmethyl, Naphthyl oder Benzyl bedeutet,
worin
R und R unabhängig voneinander Wasserstoff oder (CrC4)- Alkyl, das seinerseits durch Hydroxy oder Amino substituiert sein kann, bedeuten oder
R und R gemeinsam mit dem Stickstoffatom, an das sie gebunden sind, einen 5- bis 7-gliedrigen Heterocyclus bilden, der noch ein Sauerstoffatom oder eine Gruppe N-H oder N-(Cι.-C4)-Alkyl im Ring aufweisen kann,
oder
(3) einen Rest *-NR 1ι0υRr. lxl l bedeutet,
worin
R10 Wasserstoff oder (Cι-C4)-Alkyl bedeutet und
R11 einen Rest *-(CH2)x-Phenyl, wobei Phenyl bis zu vierfach, unabhängig voneinander, durch Fluor, Chlor oder (CrC4)- Alkyl substituiert sein kann, oder *-(CH2)y-E bedeutet,
worin
1, 2 oder 3 bedeutet,
0, 1, 2 oder 3 bedeutet und
E Pyrrolidin oder Piperidin, die ihrerseits durch (Cι-C4)-
Alkyl substituiert sein können, oder Pyridyl, das bis zu vierfach, unabhängig voneinander, durch Fluor, Chlor oder ( -G^-Alkyl substituiert sein kann, bedeutet,
oder
R10 und R11 gemeinsam mit dem Stickstoffatom, an das sie gebunden sind, einen 5- oder 6-gliedrigen Heterocyclus bilden, der durch
*-NRI R13, 1,1-Dioxyethylen, 5- oder 6-gliedriges Hetero- cyclyl mit einem oder zwei Heteroatomen N und/oder O, das seinerseits durch (Cj.-C4)-Alkyl substituiert sein kann, (Cj.-C4)- Alkoxy, durch Hydroxy oder (Cι-C4)-Alkoxy substituiertes (C i -C4)- Alkyl, oder (C i -C4)- Alkoxycarbonyl substituiert ist,
worin
R12 und R13 unabhängig voneinander Wasserstoff, (Cι-C6)- Alkyl, (C_-C4)- Alkoxycarbonyl, (C3-C8)-Cycloalkyl oder (C].-C4)-Alkanoyl bedeuten
oder
R12 und R13 gemeinsam mit dem Stickstoffatom, an das sie gebunden sind, einen 5- oder 6-gliedrigen Heterocyclus bilden,
oder
R10 und R11 gemeinsam mit dem Stickstoffatom, an das sie gebunden sind, einen 7- bis 12-gliedrigen bicyclischen Heterocyclus bilden, der anneliert oder spirocyclisch ist und ein oder zwei weitere Heteroatomen aus der Reihe N und/oder O im Ring aufweisen kann und der durch (Cι-C )-Alkyl, (Cι-C4)-Alkoxy- carbonyl, (Cι-C4)-Alkanoyl oder Benzyl substituiert sein kann,
oder
R ° und R11 gemeinsam mit dem Stickstoffatom, an das sie gebunden sind, einen Rest
worin
R14 (C2-C6)-Alkenyl, (d-C^-Alkoxycarbonyl, oder *-(CH2)z-G bedeutet,
worin
0 oder 1 bedeutet und
G (C3-C8)-Cycloalkyl, Pyridyl, gegebenenfalls durch (Cι-C4)-Alkyl oder ( -G -Alkoxy sub- stituiertes Phenyl, Tetrahydrofuran oder 1,3-
Dioxolan bedeutet,
und
R15 Wasserstoff oder (C!-C4)-Alkyl bedeutet,
und ihre Salze, Hydrate, Hydrate der Salze und Solvate.
Die erfindungsgemäßen Verbindungen können in Abhängigkeit von. ihrer Struktur in stereoisomeren Formen (Enantiomere, Diastereomere) existieren. Die Erfindung betrifft deshalb die Enantiomeren oder Diastereomeren und ihre jeweiligen Mischungen. Aus solchen Mischungen von Enantiomeren und/oder Diastereomeren lassen sich die stereoisomer einheitlichen Bestandteile in bekannter Weise isolieren.
Die Erfindung betrifft in Abhängigkeit von der Struktur der Verbindungen auch Tauto- mere der Verbindungen.
Als Salze sind im Rahmen der Erfindung physiologisch unbedenkliche Salze der erfindungsgemäßen Verbindungen bevorzugt.
Physiologisch unbedenkliche Salze der Verbindungen (I) umfassen Säureadditionssalze von Mineralsäuren, Carbonsäuren und Sulfonsäuren, z.B. Salze der Chlorwasserstoff- säure, Bromwasserstoffsäure, Schwefelsäure, Phosphorsäure, Methansulfonsäure,
Ethansulfonsäure, Toluolsulfonsäure, Benzolsulfonsäure, Naphthalindisulfonsäure, Essigsäure, Propionsäure, Milchsäure, Weinsäure, Äpfelsäure, Zitronensäure, Fumar- säure, Maleinsäure und Benzoesäure.
Physiologisch unbedenkliche Salze der Verbindungen (I) umfassen auch Salze üblicher
Basen, wie beispielhaft und vorzugsweise Alkalimetallsalze (z.B. Natrium- und Kaliumsalze), Erdalkalisalze (z.B. Calcium- und Magnesiumsalze) und Ammoniumsalze, abgeleitet von Ammoniak oder organischen Aminen mit 1 bis 16 C- Atomen, wie beispielhaft und vorzugsweise Ethylamin, Diethylamin, Triethylamin, Ethyldiiso- propylamin, Monoethanolamin, Diethanolamin, Triethanolamin, Dicyclo-hexylamin,
Dimethylaminoethanol, Prokain, Dibenzylamin, N-Methylmorpholin, Dihydroabietyl- a in, Ärgüiin, Lysin, Ethylendiamin und Methylpiperidin.
Als Solvate werden im Rahmen der Erfindung solche Formen der Verbindungen bezeichnet, welche in festem oder flüssigem Zustand durch Koordination mit Lösungs-
mittelmolekülen einen Komplex bilden. Hydrate sind eine spezielle Form der Solvate, bei denen die Koordination mit Wasser erfolgt.
Im Rahmen der vorliegenden Erfindung haben die Substituenten, soweit nicht anders spezifiziert, die folgende Bedeutung:
Alkyl per se und "Alk" und "Alkyl" in Alkoxy, Alkatioyl, Alkylcarbonylamino. Alkoxycarbonyl und Alkoxymethyl stehen für einen linearen oder verzweigten Alkyl- rest mit in der Regel 1 bis 6, vorzugsweise 1 bis 4, besonders bevorzugt 1 bis 3 Kohlen- Stoffatomen, beispielhaft und vorzugsweise für Methyl, Ethyl, n-Propyl, Isopropyl, tert-Butyl, n-Pentyl und n-Hexyl.
Alkoxy steht beispielhaft und vorzugsweise für Methoxy, Ethoxy, n-Propoxy, Iso- propoxy, tert-Butoxy, n-Pentoxy und n-Hexoxy.
Alkanoyl steht beispielhaft und vorzugsweise für Acetyl und Propanoyl.
Alkoxycarbonyl steht beispielhaft und vorzugsweise für Methoxycarbonyl, Ethoxy- carbonyl, n-Propoxycarbonyl, Isopropoxycarbonyl, tert.-Butoxycarbonyl, n-Pentoxy- carbonyl und n-Hexoxycarbonyl.
Alkoxycarbonylamino steht beispielhaft und vorzugsweise für Methoxycarbonylamino, Emoxyc-irbonylamino, n-Propoxycarbonylamino, Isopropoxycarbonylamino, tert.- Butoxycarbonylamino, n-Pentoxycarbonylamino und n-Hexoxycarbonylamino.
Alkenyl steht für einen linearen oder verzweigten Alkenylrest mit in der Regel 1 bis 6 Kohlenstoffatomen. Bevorzugt ist ein geradkettiger oder verzweigter Alkenykest mit 2 bis 4, besonders bevorzugt mit 2 oder 3 Kohlenstoffatomen. Beispielsweise und vorzugsweise seien genannt: Vinyl, Allyl, n-Prop-1-en-l-yl und n-But-2-en-l-yl.
Cycloalkyl steht für eine Cycloalkylgruppe mit in der Regel 3 bis 8, bevorzugt 5 bis 7 Kohlenstoffatomen, beispielhaft und vorzugsweise für Cyclopropyl, Cyclobutyl, Cyclopentyl, Cyclohexyl und Cycloheptyl.
Heteroaryl steht für einen aromatischen, mono- oder bicyclischen Rest mit in der
Regel 5 bis 10, vorzugsweise 5 oder 6 Ringatomen und bis zu 5, vorzugsweise bis zu 4 Heteroatomen aus der Reihe S, O und N, beispielhaft und vorzugsweise für Thienyl, Furyl, Pyrrolyl, Thiazolyl, Oxazolyl, Imidazolyl, Pyridyl, Pyrimidyl, Pyridazinyl, Indolyl, k dazolyl, Benzofuranyl, Benzothiophenyl, Chinolinyl, Isochinolinyl.
Heterocyclyl steht für einen mono- oder polycyclischen, vorzugsweise mono- oder bicyclischen heterocyclischen Rest mit in der Regel 4 bis 12, vorzugsweise 5 bis 8 Ringatomen und bis zu 3, vorzugsweise bis zu 2 Heteroatomen und/oder Hetero- gruppen aus der Reihe N, O, S, SO, SO . Die Heterocyclyl-Reste können gesättigt oder teilweise ungesättigt sein. Im Fall von polycyclischen, beispielsweise bicyclischen Resten kann ein Ring aromatisch sein. Bevorzugt sind 5- bis 8-gliedrige, monocyclische gesättigte Heterocyclylreste mit bis zu zwei Heteroatomen aus der Reihe O, N und S, wie beispielhaft und vorzugsweise Tetrahydrofuran-2-yl, Pyrro- lidin-2-yl, Pyrrolidin-3-yl, Pyrrolinyl, Piperidinyl, Morpholinyl, Perhydroazepinyl.
Ein Symbol * an einer Bindung bedeutet die Verknüpfungsstelle im Molekül.
Wenn Reste in den erfindungsgemäßen Verbindungen substituiert sind, können die Reste, soweit nicht anders spezifiziert, ein- oder mehrfach gleich oder verschieden substituiert sein. Eine Substitution mit bis zu drei gleichen oder verschiedenen
Substituenten ist bevorzugt. Ganz besonders bevorzugt ist die Substitution mit einem Substituenten.
Bevorzugt sind Verbindungen der Formel (I),
worin
R1 Amino bedeutet,
R Wasserstoff bedeutet,
R )3 . u.„ndJ r R>4 unabhängig voneinander Wasserstoff, Fluor oder Chlor bedeuten,
A einen Rest
R5 und R6 Wasserstoff bedeuten,
D (1) einen Rest bedeutet, der ausgewählt ist aus der Gruppe von
Phenyl, das durch (Cι-C4)-Alkylcarbonylamino, Hydroxymethyl, ( - C4)-Alkoxymethyl oder 1,2-Dioxymethylen substituiert ist,
Chinolin, Indol oder 6-gliedriges Heteroaryl mit 2 oder 3 Stickstoffatomen, wobei die Ringe jeweils über ein Kohlenstoffatom verknüpft sind,
Pyridylmethyl, das durch (C1-C4)- Alkyl substituiert sein kann,
und
Pyridyl, das durch (CrC4)- Alkyl substituiert ist,
oder
(2) einen Rest *-OR7 bedeutet,
worin
R7 Phenyl, das durch Fluor, Chlor, (C C4)- Alkyl, (Cι-C4)-Alkoxy oder 1 ,2-Dioxymethylen substituiert sein kann,
(C Ce)- Alkyl oder (C3-C8)-Cycloalkyl, die ihrerseits durch Hydroxy oder *-NR8R9 substituiert sein können,
Naphthyl oder Benzyl bedeutet,
worin
R8 und R9 unabhängig voneinander Wasserstoff oder (C1-C4)- Alkyl bedeuten
oder
R8 und R9 gemeinsam mit dem Stickstoffatom, an das sie gebunden sind, einen 5- bis 7-gliedrigen Heterocyclus bilden, der noch ein Sauerstoffatom oder eine Gruppe N-H oder N-(Cι-C4)-Alkyl im Ring aufweisen kann,
oder
(3) einen Rest *-NR 1ι0υRr. ll bedeutet
worin
R10 Wasserstoff oder (Cι-C )-Alkyl bedeutet und
R11 einen Rest *-(CH2)x-Phenyl, wobei Phenyl bis zu vierfach, unabhängig voneinander, durch Fluor, Chlor oder (Cι-C4)- Alkyl substituiert sein kann, oder *-(CH__)y-E bedeutet,
worin
x 1 oder 2 bedeutet,
y 0, 1 oder 2 bedeutet und
E Pyrrolidin oder Piperidin, die ihrerseits durch (Cι-C4)- Alkyl substituiert sein können, oder Pyridyl, das bis zu vierfach, unabhängig voneinander, durch Fluor, Chlor oder (C1-C4)- Alkyl substituiert sein kann, bedeutet,
oder
R10 und R11 gemeinsam mit dem Stickstoffatom, an das sie gebunden sind, einen 5- oder 6-gliedrigen Heterocyclus bilden, der durch *-NR12R13, 1,1-Dioxymethylen, 5- oder 6-gliedriges Hetero- cyclyl mit einem oder zwei Heteroatomen N und/oder O, das seinerseits durch (Ci-C^-Alkyl substituiert sein kann, oder (Cι-C4)-Alkoxymethyl substituiert ist,
worin
R12 und R13 unabhängig voneinander Wasserstoff, (d-C6)- Alkyl, (C3-C8)-Cycloalkyl oder (Cι-C4)-Alkanoyl bedeuten oder
R und R gemeinsam mit dem Stickstoffatom, an das sie gebunden sind, einen 5- oder 6-gliedrigen Heterocyclus bilden,
oder
R10 und R11 gemeinsam mit dem Stickstoffatom, an das sie gebunden sind, einen 8- bis 10-gliedrigen bicyclischen Heterocyclus bilden, der anneliert oder spirocyclisch ist und ein oder zwei weitere Heteroatomen aus der Reihe N und/oder O im Ring aufweisen kann und der durch (C1-C4)-Alkyl, (C1-C4)- Alkoxycarbonyl, (Cι-C4)-Alkanoyl oder Benzyl substituiert sein kann,
oder
R10 und R11 gemeinsam mit dem Stickstoffatom, an das sie gebunden sind, einen Rest
R14 (C3-C8)-Cycloalkyl, (C2-C6)-Alkenyl, (C_-C4)- Alkoxycarbonyl oder Tetrahydrofuran-2-yl-methyl bedeutet,
und
R15 Wasserstoff oder (Cι-C4)-Alkyl bedeutet,
und ihre Salze, Hydrate, Hydrate der Salze und Solvate.
Besonders bevorzugt sind Verbindungen der Formel (I),
worin
R Amino bedeutet,
R Wasserstoff bedeutet,
R >3 . u„nd R unabhängig voneinander Wasserstoff, Fluor oder Chlor bedeuten,
A einen Rest
R5 und R6 Wasserstoff bedeuten,
D (1) einen Rest bedeutet, der ausgewählt ist aus der Gruppe von
Chinolin, Indol, Pyrazin, Pyridazin und Triazin, wobei die Ringe jeweils über ein Kohlenstoffatom verknüpft sind,
oder
(2) einen Rest *-OR7 bedeutet,
worin
R7 Phenyl, das durch Fluor, Chlor, (Cι-C4)-Alkyl, (Cι-C4)-Alkoxy oder 1,2-Dioxymethylen substituiert sein kann,
(Cι-C6)-Alkyl oder (C3-C8)-Cycloalkyl, die ihrerseits durch Hydroxy oder *-NR8R9 substituiert sein können, bedeutet,
worin
R und R unabhängig voneinander Wasserstoff oder (C1-C4)-
Alkyl bedeuten
oder
R8 und R9 gemeinsam mit dem Stickstoffatom, an das sie gebunden sind, einen Morpholin- oder Piperazinring bilden, wobei das zweite Stickstoffatom des Pipera- zinringes durch (Cι-C4)-Alkyl substituiert sein kann,
oder
(3) einen Rest *-NR10Rn bedeutet,
worm
R10 Wasserstoff oder (C C4)- Alkyl bedeutet und
R11 einen Rest *-(CH2)y-E bedeutet,
worin
y 0 oder 1 bedeutet und
E Pyrrolidin oder Pyridyl, die ihrerseits durch (C1-C4)- Alkyl substituiert sein können, bedeutet,
oder
R10 und R11 gemeinsam mit dem Stickstoffatom, an das sie gebunden sind, einen Pyrrolidin- oder Piperidinring bilden, die durch
*-NR R , 1,1-Dioxymethylen, 5- oder 6-gliedriges Hetero- cyclyl mit einem oder zwei Heteroatomen N und/oder O, das seinerseits durch (Q-G -Alkyl substituiert sein kann, oder
(Cι-C )-Alkoxymethyl substituiert ist,
worin
R
12 und R
13 unabhängig voneinander Wasserstoff,
Alkyl, (C3-C8)-Cycloalkyl oder (Cι-C4)-Alkanoyl bedeuten
oder
R12 und R13 gemeinsam mit dem Stickstoffatom, an das sie gebunden sind, einen 5- oder 6-gliedrigen Heterocyclus bilden,
oder
R10 und R11 gemeinsam mit dem Stickstoffatom, an das sie gebunden sind, einen 8- bis 10-gliedrigen bicyclischen Heterocyclus bilden, der anneliert oder spirocyclisch ist und ein oder zwei weitere Heteroatomen aus der Reihe N und/oder O im Ring aufweisen kann und der durch (Cι-C )-Alkyl, (Cι~C4)- Alkoxycarbonyl, (Cι-C4)-Alkanoyl oder Benzyl substituiert sein kann,
und ihre Salze, Hydrate, Hydrate der Salze und Solvate.
Ganz besonders bevorzugt sind Kombinationen von zwei oder mehreren der oben genannten Vorzugsbereiche.
Gegenstand der vorliegenden Erfindung ist auch ein Verfahren zur Herstellung der Verbindungen der Formel (I), das dadurch gekennzeichnet ist, dass man entweder
[A] Verbindungen der Formel (II)
worin
A, R
1, R
2, R
3 und R
4 die oben angegebene Bedeutung aufweisen,
mit Verbindungen der Formel (III)
D— X1 (ffl), worin
D die oben angegebene Bedeutung aufweist und
X1 für Wasserstoff oder *-B(OH)2 steht,
oder
[B] Verbindungen der Formel (IN)
D die oben angegebene Bedeutung aufweist,
mit Verbindungen der Formel (V)
A, R2, R3 und R4 die oben angegebene Bedeutung aufweisen,
zu Verbindungen der Formel (I) umsetzt.
Im Verfahrensschritt [ A ] für den Fall, dass X1 für Wasserstoff steht, erfolgt die Umsetzung in inerten Lösungsmitteln oder in Substanz, gegebenenfalls in Gegenwart einer Base, bevorzugt in einem Temperaturbereich von 20°C bis zum Rückfluss der Lösungsmittel oder in der Schmelze bei Normaldruck.
Inerte Lösungsmittel sind beispielsweise Alkohole wie Methanol, Ethanol, Propanol, Isopropanol oder Butanol, N-alkylierte Carbonsäureamide wie Dimethylformamid oder Dimethylacetamid, Alkylsulfoxide wie Dimethylsulfoxid, oder andere Lösungs- mittel wie Acetonitril oder Pyridin, bevorzugt sind Ethanol oder Dimethylformamid.
Basen sind beispielsweise Alkalihydroxide wie Natrium- oder Kaliumhydroxid, oder Alkalicarbonate wie Cäsiumcarbonat, Natrium- oder Kaliumcarbonat, oder Amide wie Lithiumdiisopropylamid, oder andere Basen wie DBU, Triethylamin oder Diiso- propylethylamin, bevorzugt Diisopropylethylamin oder Triethylamin.
Im Verfahrensschritt [A] für den Fall, dass X1 für *-B(OH)2 steht, erfolgt die Umsetzung zu Verbindungen der Formel (I) im Allgemeinen in inerten Lösungsmitteln, in Gegenwart eines Übergangsmetallkatalysators, in Gegenwart einer Base, bevor- zugt in einem Temperaturbereich von 70°C bis 110°C bei Normaldruck.
Inerte Lösungsmittel sind beispielsweise Ether wie Dioxan, Tetrahydrofuran oder 1,2-Dimethoxyethan, Kohlenwasserstoffe wie Benzol, Xylol oder Toluol, Nitroaro- maten wie Nitrobenzol, gegebenenfalls N-alkylierte Carbonsäureamide wie Di- methylformamid, Dimethylacetamid, Alkylsulfoxide wie Dimethylsulfoxid oder cyclische Lactame wie N-Methylpyrrolidon. Die Lösungsmittel finden gegebenenfalls
unter Zusatz von Ethanol Verwendung. Bevorzugt sind Lösungsmittel aus der Reihe Dimethylformamid, 1,2-Dimethoxyethan und Toluol/Ethanol.
Als Übergangsmetallkatalysatoren werden bevorzugt Palladium(O)- oder Palladium- (II)-verbindungen, insbesondere Bis-(diphenylphosphanferrocenyl)-palladium(II)- chlorid, Dichlorbis(triphenylphosphin)-palladium oder Tetrakis(triphenylphosphin)- palladium(O) verwendet.
Als Basen werden Kalium-tert.-butylat, oder Alkalihydroxide oder -salze wie Kaliumacetat, Natriumhydroxid, Natriumhydrogencarbonat, Natriumcarbonat oder
Kaliumcarbonat, gegebenenfalls in Form ihrer wässrigen Lösungen, bevorzugt.
Im Verfahrensschritt [B] erfolgt die Umsetzung zu Verbindungen der Formel (I) in konzentrierter Salzsäure, bevorzugt in einem Temperaturbereich von 70°C bis 110°C bei Normaldruck. Bei dieser Umsetzung kann die Amino-Gruppe am Pyrimidin gegebenenfalls zur Hydroxy-Gruppe hydrolysiert werden.
Zur Herstellung der Verbindungen der Formel (II) aus Verfahrensschritt [A] setzt man Verbindungen der Formel (V) mit der Verbindung der Formel (VI)
unter Reaktionsbedingungen, wie für den Verfahrensschritt [B] beschrieben, um.
Bei dieser Umsetzung kann die Amino-Gruppe am Pyrimidin gegebenenfalls zur entsprechenden Hydroxy-Gruppe hydrolysiert werden.
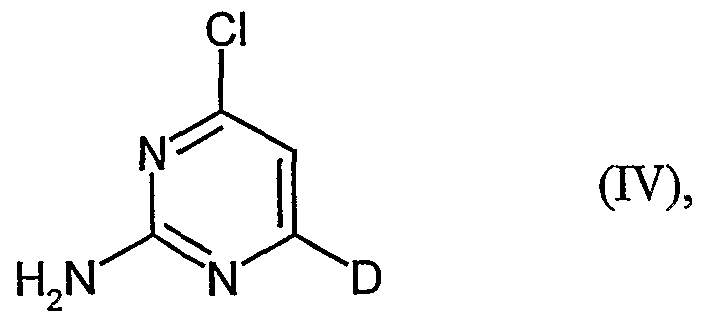
Zur Herstellung der Verbindungen der Formel (IN) aus Verfahrensschritt [B] setzt man Verbindungen der Formel (VII)
D die oben angegebene Bedeutung aufweist,
mit Phosphorylchlorid in N,N-Dimethylanilin, bevorzugt in einem Temperatur- bereich von 70°C bis 110°C bei Normaldruck um.
In einer anderen Verfahrensvariante setzt man zur Herstellung der Verbindungen der Formel (IN) Verbindungen der Formel (VI) mit Verbindungen der Formel (III) unter Reaktionsbedingungen, wie für den Verfahrensschritt [A] beschrieben, um.
Zur Herstellung der Verbindungen der Formel (Nil) setzt man Verbindungen der Formel (VIII)
D die oben angegebene Bedeutung aufweist und
X für Alkyl, bevorzugt für Methyl oder Ethyl, steht,
mit der Verbindung der Formel (IX)
um.
Die Umsetzung der Verbindungen der Formel (VIII) und (IX) erfolgt zunächst mit konzentrierter Salzsäure in Ethanol, bevorzugt in einem Temperaturbereich von 50°C bis zum Rückfluss der Lösungsmittel bei Normaldruck, und anschließend mit wässriger Natronlauge, bevorzugt in einem Temperaturbereich von 50°C bis zum Rückfluss der Lösungsmittel bei Normaldruck.
Zur Herstellung von Verbindungen der Formel (Va) aus Verfahrensschritt [B], in welcher R2 für (Cι-C6)-Alkyl oder (C3-C8)-Cycloalkyl steht, setzt man Verbindungen der Formel (Vb), in welcher R2 für Wasserstoff steht,
mit Verbindungen der Formel (X)
R2-X4 (X), worin
R2 für (Cι-C6)-Alkyl oder (C3-C8)-Cycloalkyl und
X4 für Halogen, bevorzugt Brom oder Chlor, steht,
um.
Die Umsetzung erfolgt im Allgemeinen in inerten Lösungsmitteln, gegebenenfalls in Gegenwart einer Base, bevorzugt in einem Temperaturbereich von Raumtemperatur bis zum Rückfluss der Lösungsmittel bei Normaldruck.
Inerte Lösungsmittel sind beispielsweise Halogenkohlenwasserstoffe wie Methylenchlorid, Trichlormethan oder 1,2-Dichlorethan, Ether wie Dioxan, Tetrahydro- furan oder 1,2-Dimethoxyethan, oder andere Lösemittel wie Aceton, Dimethylformamid, Dimethylacetamid, 2-Butanon oder Acetonitril, bevorzugt Tetrahydro- furan, Methylenchlorid, Aceton, 2-Butanon, Acetonitril, Dimethylformamid oder 1,2- Dimethoxyethan.
Basen sind beispielsweise Alkalicarbonate wie Cäsiumcarbonat, Natrium- oder Kaliumcarbonat, oder Natrium- oder Kaliummethanolat, oder Natrium- oder Kalium- ethanolat oder Kalium-tert.-butylat, oder Amide wie Natriumamid, Lithium-bis- (trimethylsilyl)amid oder Lithiumdiisopropylamid, oder metallorganische Verbindungen wie Butyllithium oder Phenyllithium, oder andere Basen wie Nattiumhydrid, DBU, bevorzugt Kalium-tert.-butylat, Cäsiumcarbonat, DBU, Natriumhydrid, Kaliumcarbonat oder Natriumcarbonat.
Zur Herstellung der Verbindungen der Formel (Vb) aus Nerfahrensschritt [B], in welcher R2 für Wasserstoff steht, setzt man Verbindungen der Formel (XI),
A, R ,3 und j r R>4 die oben angegebene Bedeutung aufweisen,
mit Reduktionsmitteln um.
Die Umsetzung erfolgt im Allgemeinen in inerten Lösungsmitteln, gegebenenfalls in Gegenwart von Hydrazinhydrat, bevorzugt in einem Temperaturbereich von Raumtemperatur bis zum Rückfluss der Lösungsmittel bei Normaldruck bis 3 bar.
Reduktionsmittel sind beispielsweise Palladium auf Aktivkohle und Wasserstoff,
Platinoxid auf Aktivkohle und Wasserstoff, Zinndichlorid oder Titantrichlorid, bevorzugt ist Palladium auf Aktivkohle und Wasserstoff in Gegenwart von Hydrazinhydrat oder Platinoxid auf Aktivkohle und Wasserstoff.
Inerte Lösungsmittel sind beispielsweise Ether wie Diethylether, Methyl-tert.- butylether, 1,2-Dimethoxyethan, Dioxan, Tetrahydrofuran, Glykoldimethylether oder Diethylenglykoldimethylether, Alkohole wie Methanol, Ethanol, n-Propanol, iso- Propanol, n-Butanol, tert. -Butanol oder 2-Ethylhexanol, Kohlenwasserstoffe wie Benzol, Xylol, Toluol, Hexan, Cyclohexan oder Erdölfraktionen, oder andere Lösungsmittel wie Dimethylformamid, Dimethylacetamid, Acetonitril oder Pyridin, als Lösungsmittel sind bevorzugt Ethanol, n-Butanol oder 2-Ethylhexanol.
Zur Herstellung der Verbindungen der Formel (XI) setzt man Verbindungen der Formel (XU),
R3 und R4 die oben angegebene Bedeutung aufweisen und
X r5 für Halogen, bevorzugt Fluor oder Chlor, steht,
mit Verbindungen der Formel (XIII)
A-H (XIII),
worin
A die oben angegebene Bedeutung aufweist,
um.
Die Umsetzung erfolgt im Allgemeinen in inerten Lösungsmitteln, gegebenenfalls in Gegenwart einer Base, bevorzugt in einem Temperaturbereich von Raumtemperatur bis zum Rückfluss der Lösungsmittel bei Normaldruck.
Inerte Lösungsmittel sind beispielsweise Halogenkohlenwasserstoffe wie Methylenchlorid, Trichlormethan oder 1,2-Dichlorethan, Ether wie Dioxan, Tetrahydrofuran oder 1,2-Dimethoxyethan, oder andere Lösemittel wie Aceton, Dimethylformamid, Dimethylacetamid, 2-Butanon oder Acetonitril, bevorzugt Acetonitril, Dimethylformamid oder 1,2-Dimethoxyethan.
Basen sind beispielsweise Alkalicarbonate wie Cäsiumcarbonat, Natrium- oder Kaliumcarbonat, oder Natrium- oder Kaliummethanolat, oder Natrium- oder Kaliumethanolat oder Kalium-tert.-butylat, oder Amide wie Natriumamid, Lithium- bis-(trimethylsilyl)amid oder Lithiumdiisopropylamid, oder metallorganische Verbin- düngen wie Butyllithium oder Phenyllithium, oder andere Basen wie Natriumhydrid,
DBU, bevorzugt Kalium-tert.-butylat, Cäsiumcarbonat, Kaliumcarbonat oder Natriumcarbonat.
Die Verbindungen der Formel (III), (VI), (VIII), (K), (X), (XII) und (XΩI) sind dem Fachmann an sich bekannt oder lassen sich nach üblichen literaturbekannten Verfahren herstellen.
Die Verbindungen der Formel (I) lassen sich beispielsweise durch Umsetzung mit Oxidationsmitteln weiter derivatisieren.
Die Herstellung der erfindungsgemäßen Verbindungen kann durch folgende Syntheseschemata verdeutlicht werden.
(XIII) (XII) (XI)
Hydrazinhydrat,
(Vb) (Va)
(il)
[ B ]
( I )
Die erfindungsgemäßen Verbindungen zeigen ein nicht vorhersehbares, wertvolles pharmakologisches und pharmakokinetisches Wirkspektrum. Sie eignen sich daher zur Verwendung als Arzneimittel zur Behandlung und/oder Prophylaxe von Krankheiten bei Menschen und Tieren.
Die pharmazeutische Wirksamkeit der erfindungsgemäßen Verbindungen der Formel (I) lässt sich durch ihre Wirkung als Rho-Kmase-Mύbitoren erklären.
Die erfindungsgemäßen Verbindungen der Formel (I) können aufgrund ihrer pharma- kologischen Eigenschaften allein oder in Kombination mit anderen Wirkstoffen ein-
gesetzt werden zur Behandlung und/oder Prophylaxe von Ertoankungen, insbesondere von kardiovaskulären Erkrankungen.
Die Verbindungen der Formel (I) sind geeignet für die Prophylaxe und/oder Behand- lung von kardiovaskulären Erlαankungen wie beispielsweise Bluthochdruck und Herzinsuffizienz, stabiler und instabiler Angina pectoris, peripheren und kardialen Gefaßerkrankungen, von Archytlimien, von thromboembolischen Erlα-ankiingen und Ischämien wie Myokardinfarkt, Hirnschlag, transitorischen und ischämischen Attacken, peripheren Durchblutungsstörungen, Subarachnoidalblutungen, Verhinderung von Restenosen wie beispielsweise nach Thrombolysetherapien, percutanen transluminalen
Angioplastien (PTA), percutanen ttansluminalen Koronarangioplastien (PTCA), Bypass sowie zur Prophylaxe und/oder Behandlung von Arteriosklerose, asthmatischen Erkrankungen, COPD und Krankheiten des Urogenitalsystems wie beispielsweise Prostatahypertrophie, erektiler Dysfunktion, weiblicher sexueller Dysftinktion, Osteo- poröse, Gastroparese und Inkontinenz.
Weiterhin können die Verbindungen der Formel (I) zur Prophylaxe und/oder Behandlung von Krebserkrankungen, insbesondere von Tumoren eingesetzt werden.
Im Rahmen der vorliegenden Erfindung umfasst die Definition von Tumoren sowohl benigne, wie auch maligne Tumore und damit beispielsweise auch benigne Neo- plasien, Dysplasien, Hyperplasien, wie auch Neoplasien mit Metastasenbildung. Weitere Beispiele für Tumore sind Karzinome, Sarkome, Karzinosarkome, Tumore der blutbildenden Organe, Tumore des Nervengewebes z.B. des Gehirns oder Tumore von Hautzellen. Bei der Tumorbildung kommt es zur unkontrollierten oder unzureichend kontrollierten Zellteilung. Der Tumor kann örtlich begrenzt sein, er kann aber auch das umliegende Gewebe infiltrieren und sich dann durch das lymphatische System oder durch den Blutstrom an einem neuen Ort festsetzen. Somit gibt es primäre und sekundäre Tumore. Primäre Tumore sind ursprünglich in dem Organ entstanden, in dem sie gefunden werden. Sekundäre Tumore haben sich durch
Metastasenbildung in einem anderen Organ festgesetzt und sich dann an ihrem neuen Ort ausgebreitet.
Weiterer Gegenstand der vorliegenden Erfindung ist die Verwendung der Verbin- düngen der Formel (I) zur Herstellung von Arzneimitteln zur Prophylaxe und/oder
Behandlung der zuvor genannten Krankheitsbilder.
Weiterer Gegenstand der vorliegenden Erfindung ist ein Verfahren zur Prophylaxe und/oder Behandlung der zuvor genannten Krankheitsbilder mit den Verbindungen der Formel (I).
Weiterer Gegenstand der vorliegenden Erfindung sind Arzneimittel, die mindestens eine erfindungsgemäße Verbindung, vorzugsweise zusammen mit einem oder mehreren pharmakologisch unbedenklichen Hilfs- oder Trägerstoffen enthalten, sowie deren Verwendung zu den zuvor genannten Zwecken.
Der Wirkstoff kann systemisch und/oder lokal wirken. Zu diesem Zweck kann er auf geeignete Weise appliziert werden, wie z.B. oral, parenteral, pulmonal, nasal, sublin- gual, lingual, buccal, rectal, transdermal, conjunctival, otisch, als Stents oder als Implantat.
Für diese Applikationswege kann der Wirkstoff in geeigneten Applikationsformen verabreicht werden.
Für die orale Applikation eignen sich bekannte, den Wirkstoff schnell und/oder modifiziert abgebende Applikationsformen, wie z.B. Tabletten (nicht überzogene sowie überzogene Tabletten, z.B. mit magensaftresistenten Überzüge versehene Tabletten oder Filmtabletten), Kapseln, Dragees, Granulate, Pellets, Pulver, Emulsionen, Suspensionen, Lösungen und Aerosole.
Die parenterale Applikation kann unter Umgehung eines Resorptionsschrittes geschehen (intravenös, intraarteriell, intrakardial, intraspinal oder intralumbal) oder unter Einschaltung einer Resorption (intramuskulär, subcutan, intracutan, percutan, oder intraperitoneal). Für die parenterale Applikation eignen sich als Applikations- formen u.a. Injektions- und Infusionszubereitungen in Form von Lösungen, Suspensionen, Emulsionen, Lyophilisaten und sterilen Pulvern.
Für die sonstigen Applikationswege eignen sich z.B. Inhalationsarzneiformen (u.a. Pulverinhalatoren, Nebulizer), Nasentropfen / -lösungen, Sprays; lingual, sublingual oder buccal zu applizierende Tabletten oder Kapseln, Suppositorien, Ohren- und
Augenpräparationen, Vaginalkapseln, wässrige Suspensionen (Lotionen, Schüttelmixturen), lipophile Suspensionen, Salben, Cremes, Milch, Pasten, Streupuder oder Implantate.
Die Wirkstoffe können in an sich bekannter Weise in die angeführten Applikationsformen überführt werden. Dies geschieht unter Verwendung inerter nichttoxischer, pharmazeutisch geeigneter Hilfsstoffe. Hierzu zählen u.a. Trägerstoffe (z.B. mikrokristalline Cellulose), Lösungsmittel (z.B. flüssige Polyethylenglycole), Emulgatoren (z.B. Natriumdodecylsulfat), Dispergiermittel (z.B. Polyvinylpyrrolidon), syntlie- tische und natürliche Biopolymere (z.B. Albumin), Stabilisatoren (z.B. Antioxi- dantien wie Ascorbinsäure), Farbstoffe (z.B. anorganische Pigmente wie Eisenoxide) oder Geschmacks- und / oder Geruchskorrigentien.
Im Allgemeinen hat es sich sowohl in der Human- als auch in der Veterinärmedizin als vorteilhaft erwiesen, den erfindungsgemäßen Wirkstoff in Gesamtmengen von etwa
0,01 bis etwa 700, vorzugsweise 0,01 bis 100 mg/kg Körpergewicht je 24 Stunden, gegebenenfalls in Form mehrerer Einzelgaben, zur Erzielung der gewünschten Ergebnisse zu verabreichen. Eine Einzelgabe enthält den erfindungsgemäßen Wirkstoff vorzugsweise in Mengen von etwa 0,1 bis etwa 80, insbesondere 0,1 bis 30 mg/kg Körper- gewicht.
Trotzdem kann es gegebenenfalls erforderlich sein, von den genannten Mengen abzuweichen, und zwar in Abhängigkeit von Körpergewicht, Applikationsweg, individuellem Verhalten gegenüber dem Wirkstoff, Art der Zubereitung und Zeitpunkt bzw. Intervall, zu welchem die Applikation erfolgt. So kann es in einigen Fällen ausreichend sein, mit weniger als der vorgenannten Mindestmenge auszukommen, während in anderen Fällen die genannte obere Grenze überschritten werden muss. Im Falle der Applikation größerer Mengen kann es empfehlenswert sein, diese in mehreren Einzelgaben über den Tag zu verteilen.
Die Prozentangaben in den folgenden Tests und Beispielen sind, sofern nicht anders angegeben, Gewichtsprozente; Teile sind Gewichtsteile. Lösungsmittelverhältnisse, Verdünnungsverhältnisse und Konzentrationsangaben von flüssig/flüssig-Lösungen beziehen sich jeweils auf das Volumen.
Beispiele
Abkürzungen:
DC Dünnschichtchromatographie
DCI direkte chemische Ionisation (bei MS)
DCM Dichlormethan
DIEA N, N-Diisopropylethylamin
DMSO Dimethylsulfoxid
DMF N, N-Dimethylformamid d. Th. der Theorie
EE Ethylacetat (Essigsäureethylester)
EI Elektronenstoß-Ionisation (bei MS)
ESI Elektrospray-Ionisation (bei MS)
Fp. Schmelzpunkt ges. gesättigt h Stunde
HPLC Hochdruck-, Hochleistungsflüssigchromatographie konz. konzentriert
LC-MS Flüssigchromatographie-gekoppelte Massenspektroskopie
LDA Lithium-Diisopropylamid
MPLC Mitteldruck-, Mittelleistungsflüssigchromatographie
MS Massenspektroskopie
NMR Kemresonanzspektroskopie proz. prozentig
RP-HPLC Reverse Phase HPLC
RT Raumtemperatur
Rf Retentionsindex (bei DC)
Rt Retentionszeit (bei HPLC)
THF Tetrahydrofuran
HPLC-, LCMS- und GCMS-Methoden:
Methode 1 (HPLC):
Instrument: HP 1100 mit DAD-Detelction; Säule: Kromasil RP-18, 60 mm x 2 mm, 3.5 μm; Eluent: A=5 ml HCIO4/I Wasser, B=Acetonitril; Gradient: 0 min 2 % B,
0.5 min 2 % B, 4.5 min 90 % B, 6.5 min 90 % B; Fluss: 0.75 ml/min; Temp.: 30°C; Detektion UV 210 nm.
Methode 2 (HPLC): Instrument: HP 1100 mit DAD-Detektion; Säule: Kromasil RP-18, 60 mm x 2 mm,
3.5 μm; Eluent: A=5 ml HCIO4/I Wasser, B=Acetonitril; Gradient: 0 min 2 % B, 0.5 min 2 % B, 4.5 min 90 % B, 9 min 90 % B; Fluss: 0.75 ml/min; Temp.: 30°C; Detektion UV 210 nm.
Methode 3 (HPLC):
Instrument: Finnigan MAT 900S, TSP: P4000,AS3000,UV3000HR; Säule: Symmetry C 18, 150 mm x 2.1 mm, 5.0 μm; Eluent C: Wasser, Eluent B: Wasser + 0.3 g 35 %ige Salzsäure, Eluent A: Acetonitril; Gradient: 0.0 min 2 % A -_> 2.5 min 95 % A -_» 5 min 95 % A; Ofen: 70°C; Fluss: 1.2 ml/min; UV-Detektion: 210 um.
Methode 4 (LCMS):
Instrument: Micromass Quattro LCZ, HP1100; Säule: Symmetry C18, 50 mm x 2.1 mm, 3.5 μm; Eluent A: Acetonitril + 0.1 % Ameisensäure, Eluent B: Wasser + 0.1 % Ameisensäure; Gradient: 0.0 min 10 % A -> 4.0 min 90 % A -_ 6.0min 90 % A; Ofen: 40°C; Fluss: 0.5 ml/min; UV-Detektion: 208-400 um.
Methode 5 (LCMS):
Instrument: Micromass Platform LCZ, HP1100; Säule: Symmetry C18, 50 mm x 2.1 mm, 3.5 μm; Eluent A: Acetonitril + 0.1 % Ameisensäure, Eluent B: Wasser + 0.1 % Ameisensäure; Gradient: 0.0 min 10 % A - 4.0 min 90 % A - 6.0min 90 % A; Ofen: 40°C; Fluss: 0.5 ml/min; UV-Detektion: 208-400 um.
Methode 6 (LCMS :
Gerätetyp MS: Micromass ZQ; Gerätetyp HPLC: Waters Alliance 2790; Säule: Symmetry C 18, 50 mm x 2.1 mm, 3.5 μm; Eluent B: Acetonitril + 0.05 % Ameisensäure, Eluent A: Wasser + 0.05 % Ameisensäure; Gradient: 0.0 min 10 % B
-> 3.5 min 90 % B - 5.5 min 90 % B; Ofen: 50°C; Fluss: 0.8 ml/min; UV- Detektion: 210 um.
Methode 7 (LCMS): Instrument: Micromass Platform LCZ, HP1100; Säule: Symmetry C18, 50 mm x
2.1 mm, 3.5 μm; Eluent A: Wasser + 0.05 % Ameisensäure, Eluent B: Acetonitril + 0.05 % Ameisensäure; Gradient: 0.0 min 90 % A -» 4.0 min 10 % A -» 6.0 min 10 % A; Ofen: 40°C; Fluss: 0.5 ml/min; UN-Detektion: 208-400 um.
Methode 8 (LCMS :
Instrument: Micromass Quattro LCZ, HP 1100; Säule: Symmetry C18, 50 mm x 2.1 mm, 3.5 μm; Eluent A: Wasser + 0.05 % Ameisensäure, Eluent B: Acetonitril + 0.05 % Ameisensäure; Gradient: 0.0 min 90 % A -_> 4.0 min 10 % A -» 6.0 min 10 % A; Ofen: 40°C; Fluss: 0.5 ml/min; UV-Detektion: 208-400 um.
Methode 9 (GCMS):
Säule: HP-5 30 m x 320 μm x 0.25 μm (Filmdicke); Trägergas: Helium; Temperaturgradient: 14°C/min bis 300°C, dann 1 min konst. 300°C; Fluss: 1.5 ml/min; Anfangstemperatur: 60°C; Anfangszeit: 2 min; Frontinjektor-Temp.: 250°C.
Methode 10 (HPLC):
Instrument: Waters Alliance 2790 LC; Säule: Symmetry C18, 50 mm x 2.1, 3.5 μm; Eluent A: Wasser + 0.1 % Ameisensäure, Eluent B: Acetonitril + 0.1 % Ameisensäure; Gradient: 0.0 min 5 % B - 5.0 min 10 % B -» 6.0 min 10 % B; Temperatur: 50°C; Fluss: 1.0 ml/min; UV-Detektion: 210 um.
Methode 11 (chirale HPLC):
Säule: chirale stationäre Phase, basierend auf dem optisch aktiven Monomer N- Methacrylacyl-L-leucin-dicyclopropylmethylamid; Eluent A: iso-Hexan, Eluent B: Essigsäureethylester; Gradient: A:B -_ 20:80; Fluss: 15 ml/min.
Methode 12 (LCMS):
Instrument: Micromass Platform LCZ mit HPLC Agilent Serie 1100; Säule: Grom- SIL120 ODS-4 HE, 50 mm x 2.0 mm, 3 μm; Eluent A: 1 1 Wasser + 1 ml 50 %ige Ameisensäure, Eluent B: 1 1 Acetonitril + 1 ml 50 %ige Ameisensäure; Gradient: 0.0 min 100 % A -» 0.2 min 100 % A ■» 2.9 min 30 % A - 3.1 min 10 % A -i> 4.5 min 10 % A; Ofen: 55°C; Fluss: 0.8 ml/min; UV-Detektion: 208-400 um.
Methode 13 (LCMS): Gerätetyp MS: Micromass ZQ; Gerätetyp HPLC: Waters Alliance 2790; Säule:
Uptisphere C 18, 50 mm x 2.0 mm, 3.0 μm; Eluent B: Acetonitril + 0.05 % Ameisensäure, Eluent A: Wasser + 0.05 % Ameisensäure; Gradient: 0.0 min 5 % B ■» 2.0 min 40 % B -» 4.5 min 90 % B- 5.5 min 90 % B; Ofen: 45 °C; Fluss: 0.0 min 0.75 ml/min -> 4.5 min 0.75 ml/min-_> 5.5 min 1.25 ml/min; UV-Detektion: 210 um.
Methode 14 (LCMS):
Instrument: Micromass Quattro LCZ mit HPLC Agilent Serie 1100; Säule:
UPTISPHERE HDO, 50 mm x 2.0 mm, 3 μm; Eluent A: 1 1 Wasser + 1 ml 50 %ige Ameisensäure, Eluent B: 1 1 Acetonitril + 1 ml 50 %ige Ameisensäure; Gradient: 0.0
min 100 % A -_ 0.2 min 100 % A -» 2.9 min 30 % A -^ 3.1 min 10 % A ■» 4.5 min 10 % A; Ofen: 55°C; Fluss: 0.8 ml/min; UV-Detektion: 208-400 nm.
Methode 15 (LCMS): Gerätetyp MS: Micromass ZQ; Gerätetyp HPLC: Waters Alliance 2790; Säule:
Symmetry C 18, 50 mm x 2.1 mm, 3.5 μm; Eluent B: Acetonitril + 0.05 % Ameisensäure, Eluent A: Wasser + 0.05 % Ameisensäure; Gradient: 0.0 min 5 % B - 4.5 min 90 % B - 5.5 min 90 % B; Ofen: 50 °C; Fluss: 1.0 ml/min; UV- Detektion: 210 nm.
Ausgangsverbindungen
Beispiel I
4-Chlor-6-( 1 H-indol-5-yl)-2-pyrimidinamin
380 mg (2.33 mmol) 2-Amino-4,6-dichlorpyrimidin werden in 20 ml Toluol und 10 ml Ethanol suspendiert und mit 80 mg (0.07 mmol) Tetrakis(triphenylphos- phin)palladium(O) versetzt. Nach 20 Minuten Rühren bei Raumtemperatur werden 450 mg (2.80 mmol) 5-Indolboronsäure und 3.90 ml einer 2M Natriumcarbonat- lösung zugegeben. Man lässt 20 Stunden bei 120°C rühren. Nach dem Abkühlen wird die Reaktionslösung mit IN Salzsäure neutral gestellt und dreimal mit je 50 ml Ethylacetat extrahiert. Die organische Phase wird über Natriumsulfat getrocknet, abfiltriert und im Vakuum eingeengt. Das Rohprodukt wird über Kieselgel 60 chromatographisch gereinigt (Laufmittel: Cyclohexan - Cyclohexan-Ethylacetat
1:1).
Man erhält 32 mg (4 % d. Th.) Produkt.
1H-NMR (400 MHz, DMSO-d6): δ = 6.47 (s, 1H), 7.33 (s, 1H), 7.50 (dd, 2H), 7.98
(s, 1H), 11.13 (s, 1H) MS (ESIpos): m/z = 245 (M+H)+ HPLC (Methode 1): Rt = 4.12 min
Das in der folgenden Tabelle aufgeführte Beispiel kann analog der für Beispiel I beschriebenen Vorschrift aus den entsprechenden Ausgangsverbindungen hergestellt werden.
Beispiel III
3 -Oxo-3 -(4-pyridinyl)propansäureethylester
25 g (203 mmol) Isonicotinsäure, 35.12 g (243.7 mmol) 2,2-Dimethyl-l,3-dioxolan- 4,6-dion und 49.6 g (406 mmol) 4-Dimethylaminopyridin werden in 300 ml Dichlor- methan vorgelegt und auf 0°C gekühlt. Es wird eine IN Lösung von 46.1 g
(223.4 mmol) 1,3-Dicyclohexylcarbodiimid in Dichlormethan zugetropft. Es wird 2 Stunden bei Raumtemperatur nachgerührt. Der entstandene Niederschlag wird abfiltriert und mit Dichlormethan nachgewaschen. Das Filtrat wird im Vakuum eingeengt. Der Rückstand wird in 1200 ml Ethanol gelöst und mit einer Lösung aus 96.6 g (507.7 mmol) p-Toluolsulfonsäure-monohydrat in 300 ml Ethanol versetzt und eine Stunde unter Rückfluss gerührt. Nach dem Abkühlen wird das Ethanol im Vakuum abgezogen. Der Rückstand wird in 1000 ml Ethylacetat und 900 ml Wasser
aufgenommen und in der Hitze gelöst. Die organische Phase wird abgetrennt, mit 600 ml gesättigter Natriumhydrogencarbonatlösung und gesättigter Natriumchlorid- lösung gewaschen und über Natriumsulfat getrocknet. Es wird im Vakuum eingeengt. Das Rohprodukt wird über eine Kieselgel-Fritte mit Dichlormethan-Methanol 10:1 filtriert. Da die wässrige Phase noch Produkt enthält, wird diese mit Dichlormethan extrahiert, über Natriumsulfat getrocknet und im Vakuum eingeengt. Das Rohprodukt wird über eine Kieselgel-Fritte mit Dichlormethan-Methanol 10:1 filtriert.
Insgesamt erhält man 25.9 g (42 % d. Th.) Produkt.
1H-NMR (300 MHz, DMSO-d6): δ = 1.17 (t, 3H), 4.12 (q, 2H), 4.25 (s, 2H), 7.82
(dd, 2H), 8.83 (dd, 2H)
LC-MS (Methode 3): Rt = 2.40 min
MS (ESIpos): m z = 194 (M+H)+
Beispiel IV
2-Amino-6-(4-pyridinyl)-4-pyrimidinol
25 g (81.52 mmol) der Verbindung aus Beispiel III und 13.22 g (73.37 mmol) Guanidiniumcarbonat werden in 250 ml Ethanol gelöst, mit konzentrierter Salzsäure versetzt und über Nacht unter Rückfluss geriihrt. Nach dem Abkühlen wird der Niederschlag abgesaugt, mit Ethanol nachgewaschen und im Hochvakuum ge- trocknet. Der Feststoff wird mit 250 ml IN Natriumhydroxidlösung versetzt und
2 Stunden unter Rückfluss gerührt. Nach dem Abkühlen wird mit konzentrierter
Essigsäure sauer gestellt, das ausgefallene Produkt abgesaugt und mit Diethylether nachgewaschen.
Nach dem Trocknen im Hochvakuum erhält man 12.52 g (82 % d. Th.) Produkt.
1H-NMR (300 MHz, DMSO-d6): δ = 6.23 (s, 1H), 6.89 (br.s, 2H), 7.86 (dd, 2H), 8.64 (dd, 2H), 11.65 (br.s, 1H). LC-MS (Methode 4): Rt = 0.30 min MS (ESIpos): m z = 189 (M+H)+
Beispiel V 4-Chlor-6-(4-pyridinyl)-2-pyrimidinamin
10.4 g (55.26 mmol) der Verbindung aus Beispiel IN werden in 28.33 ml (303.95 mmol) Phosphorylchlorid gelöst. Es werden 0.88 g (7.18 mmol) Ν,Ν- Dimethylanilin langsam zugetropft und eine Stunde bei 100°C gerührt. Anschließend wird die Reaktionslösung noch 2 Stunden bei Raumtemperatur gerührt. Das Phos- phorylchlorid wird im Vakuum abrotiert. Der Rückstand wird mit Wasser-Di- chlormethan 9:1 versetzt und für 5 Minuten aufgekocht. Dann wird mit gesättigter Natriumhydrogencarbonatlösung neutralisiert, das Produkt abgesaugt und im Hochvakuum getrocknet.
Es werden 5.9 g (49 % d. Th.) Produkt erhalten.
1H-NMR (300 MHz, DMSO-d6): δ = 7.31 (br.s, 2H), 7.38 (s, 1H), 8.00 (dd, 2H),
8.74 (dd, 2H)
LC-MS (Methode 4): Rt = 1.08 min
MS (ESIpos): m/z = 207 (M+H)+
Beispiel VI l-Chlor-2,3- difluor-5-nitrobenzol
Die Verbindung ist zugänglich durch Oxidation des in JP 05059067 beschriebenen 3- Chlor-4,5-difluoranilins mit Wasserstoffperoxid in Trifluoressigsäure gemäß einem Verfahren, das zur Herstellung analoger Derivate beschrieben ist in Heaton, A. et al., J. Fluorine Chem. 1997, 81 (2), 133-138 und Krapcho, A. P. et al., J. Org. Chem. 1990, 55 (21), 5662-5664.
Beispiel VII
4-[(2-Fluor-4-nitrophenyl)sulfanyl]pyridin
21 g (188.9 mmol) 4-Mercaptopyridin, 30.05 g (188.9 mmol) 3,4-Difluornitrobenzol und 60.05 g (434.5 mmol) Kaliumcarbonat werden in Dimethylformamid gelöst und 3 Stunden bei 40°C gerührt. Anschließend wird die Reaktionslösung mit 500 ml
43
Ethylacetat und 300 ml Wasser verdünnt. Die wässrige Phase wird iiünfmal mit je 100 ml Ethylacetat extrahiert. Die vereinigten organischen Phasen werden mit 200 ml gesättigter Natriumchloridlösung gewaschen, über Natriumsulfat getrocknet und im Vakuum einrotiert. Der Rückstand wird über eine MPLC gereinigt (Laufmittel: Ethylacetat-Cyclohexan 1:1)
Es werden 37.3 g (79 % d. Th.) Produkt erhalten.
1H-NMR (300 MHz, DMSO-d6): δ = 7.28 (dd, 2H), 7.79 (t, 1H), 8.15 (dd, 1H), 8.30 (dd, 1H), 8.50 (dd, 2H)
LC-MS (Methode 4): Rt = 2.68 min MS (ESIpos): m/z = 251 (M+H)+
Die in der folgenden Tabelle aufgeführten Beispiele können analog der für Beispiel VII beschriebenen Vorschrift aus den entsprechenden Mercapto- bzw. Hydroxy- heterocyclen und deren entsprechenden 4-Fluor- bzw. 4-Chloπύtrobenzol-Derivaten hergestellt werden.
Beispiel XI
3-Fluor-4-(4-pyridinylsulfanyl)anilin
37 g (147.9 mmol) der Verbindung aus Beispiel VII werden in 1000 ml Ethanol gelöst und mit 143.86 ml (2.95 mol) Hydrazinhydrat und 4 g Palladium auf Kohle versetzt. Das Reaktionsgemisch wird über Nacht unter Rückfluss gerührt. Nach dem Abkühlen wird der Ansatz über Kieselgel abgesaugt und mit Ethanol nachgewaschen. Das Filtrat wird im Vakuum einrotiert. Der Rückstand wird mit Diethylether aufgeschlämmt und abgesaugt. Anschließend wird der Niederschlag mit Wasser aufgeschlämmt und abgesaugt. Es wird noch zweimal mit wenig Wasser nachgewaschen.
Nach dem Trocknen im Hochvakuum erhält man 27.3 g (84 % d. Th.) Produkt.
1H-NMR (300 MHz, DMSO-d6): δ = 6.02 (br.s, 2H), 6.49-6.54 (m, 2H), 6.93 (dd, 2H), 7.23 (t, 1H), 8.32 (dd, 2H) LC-MS (Methode 4): Rt = 0.96 min MS (ESIpos): m/z = 221 (M+H)+
Das in der folgenden Tabelle aufgeführte Beispiel kann analog der für Beispiel XI beschriebenen Vorschrift aus der Verbindung aus Beispiel VIII hergestellt werden.
Beispiel XIII
3-Chlor-5-fluor-4-(4-pyridinylsulfanyl)anilin
3.19 g (11.205 mmol) der Verbindung aus Beispiel IX werden in 200 ml Ethanol gelöst. Anschließend werden 638 mg (2.81 mmol) Platin(IV)oxid hinzugefügt und der Ansatz wird 2 Stunden bei RT und Normaldruck unter Wasserstoffatmosphäre gerührt. Zur Aufarbeitung wird die Reaktionslösung über Kieselgur abgesaugt und gut mit Ethanol nachgewaschen. Das Filtrat wird im Vakuum eingeengt.
Es werden 2.755 g (81 % d. Th.) Produkt erhalten.
1H-NMR (200 MHz, DMSO-d6): δ = 6.37 (s, 2H), 6.49 (dd, 1H), 6.72-6.74 (m, 1H), 6.93 (dd, 2H), 8.34 (dd, 2H) HPLC (Methode 1): Rt = 3.68 min
46
MS (ESIpos): m/z = 255 (M+H)+
Das in der folgenden Tabelle aufgeführte Beispiel kann analog der für Beispiel XIII beschriebenen Vorschrift aus der Verbindung aus Beispiel X hergestellt werden.
Beispiel XV 3-Fluor-N-methyl-4-(4-pyridinylsulfanyl)anilin
440.5 mg (2 mmol) der Verbindung aus Beispiel XI werden in 2 ml Methanol gelöst und mit 2 ml einer 21 %igen Natriumethanolatlösung versetzt. Es werden 84 mg (2.8 mmol) Paraformaldehyd zugegeben und über Nacht bei Raumtemperatur ge- rührt. Das Reaktionsgemisch wird mit 75.7 mg (2 mmol) Natriumborhydrid versetzt und 1.5 Stunden unter Rückfluss erhitzt. Nach dem Abkühlen wird mit IM Kaliumhydroxidlösung vorsichtig hydrolysiert. Nachdem keine Reaktion mehr zu erkennen ist, wird der anfallende Feststoff abgesaugt und mit viel Wasser nachgewaschen. Der feste Rückstand wird in Diethylether gelöst, über Natriumsulfat getrocknet und im Vakuum einrotiert.
Es werden 387 mg (83 % d. Th.) Produkt erhalten.
1H-NMR (200 MHz, DMSO-d6): δ = 2.73 (d, 3H), 6.45-6.52 (m, 2H), 6.59-6.69 (m, 1H), 6.92 (dd, 2H), 7.29 (t, 1H), 8.32 (dd, 2H) LC-MS (Methode 4): Rt = 2.35 min
MS (ESIpos): m/z = 235 (M+H)+
Beispiel XVI
N-(2- Amino-6-chlor-4-pyrimidinyl)-N- [3 -fluor-4-(4-pyridinylsulfanyl)phenyl] amin
2.98 g (18.16 mmol) 2-Amino-4,6-dichlorpyrimidin werden in 300 ml Wasser suspendiert und dann mit 4 g (18.16 mmol) der Verbindung aus Beispiel XI und 1.82 ml konzentrierter Salzsäure versetzt. Über Nacht wird bei 100°C gerührt. Zur
Aufarbeitung lässt man die Reaktionslösung abkühlen und stellt mit gesättigter Nattiumhydrogencarbonatlösung basisch. Das dabei ausgefallene Produkt wird abgesaugt und im Vakuumtrockenschrank bei 40°C getrocknet.
Es werden 6.04 g (74 % d. Th.) Produkt erhalten.
1H-NMR (200 MHz, DMSO-d6): δ = 6.09 (s, 1H), 6.99 (dd, 4H), 7.41 (dd, 1H), 7.56 (dd, 1H), 8.27 (dd, 1H), 8.36 (dd, 2H), 9.89 (s, 1H) HPLC (Methode 1): Rt = 3.69 min. MS (ESIpos): m/z = 348 (M+H)+
Die in der folgenden Tabelle aufgeführten Beispiele können analog der oben für Beispiel XVI beschriebenen Vorschrift aus den entsprechenden Ausgangsverbindungen hergestellt werden.
Beispiel XX
5-Benzyl-l-oxa-5-azaspiro[4.2]heptan
Die Verbindung kann nach einer Vorschrift von E.J. Corey et al. gemäß US 4,508,724 aus N-Benzyl-3-pyrrolidinon hergestellt.
Beispiel XXI 1 -Benzyl-3 -hydroxy-3 -(2-hydroxyethylaminomethyl)-pyrrolidin
Man tropft 32.7 g (0.17 mol) von Beispiel XX zu 31 g (0.52 mol) Ethanolamin in 250 ml Wasser und riihrt über Nacht bei Raumtemperatur. Man extrahiert mit
Diethylether, engt die wässrige Phase ein und destilliert den Rückstand im Hochvakuum.
Es werden 42.1g (96 % d. Th.) Produkt erhalten. Siedepunkt: 180 - 190°C/ 0.1 mbar
Beispiel XXII
7-Benzyl-l-oxa-4,7-diazaspiro[5.4]dekan
Man löst 85 g (340 mmol) von Beispiel XXI in einem Gemisch von 280 ml konzentrierter Schwefelsäure und 140 ml Wasser und erhitzt über Nacht auf 180°C. Man stellt mit 45%iger Natronlauge alkalisch, löst ausgeschiedene Salze mit Wasser und extrahiert fünfmal mit je 200 ml Chloroform. Man trocknet die organischen Phasen über Kaliumcarbonat, trennt das Trockenmittel ab und engt die Lösung ein. Der Rückstand wird im Hochvakuum destilliert.
Es werden 60 g (76 % d.Th.) des Produktes erhalten. Siedepunkt: 125°C/0.08 mbar
Beispiel XXIII
7-Benzyl-l-oxa-4,7-diazaspiro[5.4]dekan-4-carbonsäure-tert.-butylester
Zu 10.3 g (47 mmol) Beispiel XXII in 30 ml tert. -Butanol gibt man 2 g Natriumhydroxidplätzchen in 25 ml Wasser und tropft 11 g (50 mmol) Pyrokohlensäuredi- tert-butylester hinzu. Man rührt über Nacht bei Raumtemperatur, versetzt mit 50 ml Wasser, extrahiert dreimal mit Chloroform, trocknet über Kaliumcarbonat, saugt das Trockenmittel ab, engt das Filtrat ein und destilliert den Rückstand im Hochvakuum.
Es werden 13.8 g (88 % d. Th.) des Produktes erhalten. Siedepunkt: 160°C/ 0.3 mbar
Beispiel XXIV l-Oxa-4,7-diazaspiro[5.4]dekan-4-carbonsäure-tert.-butylester
Man löst 13.7 g (41 mmol) von Beispiel XXIII, setzt 3 g 10 %ige Palladium- Aktivkohle hinzu und hydriert bei 100°C und 100 bar. Man saugt den Katalysator ab, engt das Filtrat ein und destilliert den Rückstand im Hochvakuum.
Es werden 7.6 g (75 % d.Th.) des Produktes erhalten.
Siedepunkt: 113°C/ 0.07 mbar
Beispiel XXV
9-Methyl-6-oxa-2,9-diazaspiro[4.5]dekan
Die Verbindung kann aus Beispiel XXII durch reduktive Alkylierung nach der zur Herstellung von Beispiel XV beschriebenen Methode und anschließender hydro- genolytischer Abspaltung der Benzyl-Gruppe nach der Methode, die zur Herstellung von Beispiel XXJV beschrieben wurde, hergestellt werden.
Beispiel XXVI
3 ,7-Dibenzyl-3 ,7-diazabicyclo [3.3.0] oktan-2,4-dion
Man erhitzt 18.7 g (0.1 mol) N-Benzylmaleinimid mit 25 g (0.15 mol) N-Benzyl- glycin und 5 g (0.157 mol) Paraformaldehyd in 500 ml Toluol bis zum Ende der CO -Entwicklung unter Rückfluss. Man engt ein und verwendet das Rohprodukt ohne weitere Reinigung.
Beispiel XXVII
3 -Benzyl-3 ,7-diazabicyclo [3.3.0] oktan-2,4-dion
Man hydriert 150 g (0.4 mol) des Rohproduktes aus Beispiel XXVI in 750 ml Ethanol an 15 g 10 %iger Palladium-Aktivkohle bei 100°C und 100 bar. Man filtriert den Katalysator ab, engt die Lösung ein und filtriert mit Dichlormethan über 1.2 kg Kieselgel.
Es werden 38 g (42% d.Th.) des Produktes erhalten.
Rf Wert: 0.35 (Laufmittel: Dichlormethan 96 % / Methanol 4 %).
Beispiel XXVIII
3 -Benzyl-3 ,7-diazabicyclo [3.3.0] oktan
Man legt 16 g (0.42 mol) Limiumaluminiumhydrid in 350 ml absolutem Tetrahydrofiiran vor und tropft 38 g (0.165 mol) von Beispiel XXVII in 150 ml absolutem Tetrahydrofiiran hinzu. Man erhitzt noch fünf Stunden unter Rückfluss, tropft je 16 ml Wasser, 16 %ige Kalilauge und wieder Wasser hinzu, saugt die anorganischen Salze ab und kocht sie zweimal mit Tetrahydrofiiran aus. Die Filtrate werden eingeengt und im Membranpumpenvakuum destilliert.
Es werden 31.4 g (94 % d. Th.) des Produktes erhalten. Siedepunkt: 140°C/4 mbar
Beispiel XXIX
7-Benzyl-3 ,7-diazabicyclo [3.3.0] oktan-3 -carbonsäure-tert.-butylester
Man legt 6 g (29.7 mmol) Beispiel XXVIII vor, setzt 1.4 g (35 mmol) Natriumhydroxid in 30 ml Wasser hinzu und tropft dann 8 g (36.7 mmol) Pyrokohlensäure- tert-butylester hinzu. Man rührt über Nacht bei Raumtemperatur, extrahiert mit Chloroform, trocknet über Kaliumcarbonat, filtriert das Trockenmittel ab, engt das Filtrat ein und destilliert den Rückstand.
Es werden 7.6 g (75 % d.Th.) des Produktes erhalten. Siedepunkt: 153-156°C/0.2 mbar
Beispiel XXX 3,7-Diazabicyclo[3.3.0]oktan-3-carbonsäure-tert.-butylester
7.6 g (25.3 mmol) von Beispiel XXIX werden in 100 ml Ethanol an 1.5 g 10 %iger Palladium- Aktivkohle bei 100°C und 100 bar hydriert. Man filtriert den Katalysator ab, engt das Filtrat ein und destilliert den Rückstand.
Es werden 3.6 g (76 % d. Th.) des Produktes erhalten. Siedepunkt: 92°C/0.08 mbar
Beispiel XXXI
3 ,7-Diazabicyclo [3.3.0]oktan
Die Verbindung kann aus Beispiel XXX durch Abspaltung der tert.-Butoxycarbonyl-
Gruppe mit Chlorwasserstoff (4 M in Dioxan) oder Trifluoressigsäure/Dichlormethan (1:1) hergestellt werden.
Beispiel XXXII
(4aR,7aS)-4-Benzyloktahydropyrrol[3,4-b][l,4]oxazin
Die Herstellung der Verbindung ist in US 6 004 956 beschrieben.
Beispiel XXXIII
5-Methyloktahydropyrrol [3 ,4-b]pyrrol
Die Herstellung der Verbindung ist in DE-A 4 032 560 beschrieben.
Beispiel XXXIV
[S,S]-8-Benzyl-2,8-diazabicyclo[4.3.0]nonan
3 g (20 mmol) D(-)- Weinsäure werden in 10 ml Dimethylformamid durch Erwärmen auf 80°C gelöst und mit einer Lösung von 2.16 g (10 mmol) cis-8-Benzyl-2,8- diazabicyclo-[4.3.0]-nonan in 3 ml Dimethylformamid versetzt. Es wird 1 Stunde bei
0°C nachgerührt, dann wird abgesaugt und mit Dimethylformamid und Methoxyethanol gewaschen. (Ausbeute: 1.93 g) Schmelzpunkt: 146-151°C
[α]D 24*.= -19.3° (c= l, H2O)
Durch einmaliges Umkristallisieren aus Methoxyethanol wird diastereomeremeines [S,S]-8-Benzyl-2,8-diazabicyclo[4.3.0]nonan-D-tartrat erhalten. Schmelzpunkt: 148-154°C [ ]D 24= -22.7° (c= l, H2O)
40 g des Salzes werden in 250 ml Wasser gelöst und mit 32 g 45 %iger Natronlauge versetzt. Das ausgefallene Öl wird in 150 ml tert.-Butyl-methylether aufgenommen, die wässrige Phase wird nochmals mit 150 ml tert.-Butyl-methylether extrahiert. Die organischen Phasen werden vereinigt und nach dem Trocknen über Natriumsulfat eingeengt. Dann wird im Vakuum destilliert.
Es werden 18.5 g Produkt erhalten. Siedepunkt: 107-109°C/ 0.1 mbar [α]D 24= 17.3° (unverdünnt)
Beispiel XXXV
[S,S]-2,8-Diazabicyclo[4.3.0]nonan
28.4 g (0.131 mol) [S,S]-8-Benzyl-2,8-diazabicyclo[4.3.0]nonan werden in 190 ml
Methanol über 5.8 g Palladium auf Aktivkohle (5%) bei 90°C und 90 bar innerhalb von 5 Stunden hydriert. Dann wird der Katalysator abgesaugt, mit Methanol ge-
waschen und das Filtrat im Vakuum eingeengt. Der Rückstand wird ohne zu fraktionieren destilliert.
Es werden 15 g (91 % d. Th.) Produkt erhalten. Siedepunkt: 44-59°C/ 0.18 mbar
[α]D 24= -2.29° (unverdünnt) ee > 99% (gaschromatographisch nach Derivatisierung mit Moshe s Reagenz bestimmt)
Beispiel XXXVI
[R,R]-8-Benzyl-2,8-diazabicyclo[4.3.0]nonan
75 g (0.5 mol) L(+)- Weinsäure werden bei 80°C in 250 ml Dimethylformamid gelöst und 54.1 g (0.25 mol) cis-8-Benzyl-2,8-diazabicyclo[4.3.0]nonan als Lösung in 75 ml Dimethylformamid zugetropft. Es wird langsam auf 20°C abgekühlt und die Kristallsuspension 1 Stunde nachgerührt. Die Kristalle ([R,R]-8-Benzyl-2,8-diaza- bicyclo[4.3.0]nonan-L-tartrat) werden abgesaugt. (Das Filtrat kann weiter verarbeitet werden, um [S,S]-8-Benzyl-2,8-diazabicyclo[4.3.0]nonan zu erhalten). Diese
Kristalle werden mit Dimethylformamid und Methoxyethanol gewaschen (Rohausbeute: 49.2 g) und aus 300 ml Methoxyethanol umkristallisiert. Man erhält 45.6 g enantiomerenreines [R,R]-8-Benzyl-2,8-diazabicyclo[4.3.0]nonan-L-tartrat (Enantio- merenreinheit gaschromatographisch nach Derivatisierung mit Chlorameisensäure- menthylester bestimmt).
Schmelzpunkt: 121-124°C [α]D 24= +22.3° (c= 1, H2O)
Das erhaltene Salz wird jetzt in die freie Base überfuhrt. Dazu werden 44.5 g in 280 ml Wasser gelöst und mit 35.6 g 45 %iger Natronlauge versetzt. Das ausgefallene Öl wird in 170 ml tert.-Butyl-methylether aufgenommen, die wässrige Phase wird nochmals mit 170 ml tert.-Butyl-methylether extrahiert. Die organischen Phasen werden vereinigt und nach dem Trocknen über Natriumsulfat eingeengt. Dann wird im Vakuum destilliert. Siedepunkt: 107-111°C/ 0.04 mbar [ ]D 24= -17.5° (unverdünnt)
Beispiel XXXVII
[R,R]-2,8-Diazabicyclo[4.3.0]nonan
19.4 g (0.09 mol) [R,R]-8-Benzyl-2,8-diazabicyclo[4.3.0]nonan werden in 130 ml Methanol über 3.96 g Palladium auf Aktivkohle (5 %) bei 90°C und 90 bar innerhalb von 5 Stunden hydriert. Dann wird der Katalysator abgesaugt, mit Methanol gewaschen und das Filtrat im Vakuum eingeengt. Der Rückstand wird ohne zu fraktionieren destilliert.
Es werden 9.61 g (85 % d. Th.) Produkt erhalten.
Siedepunkt: 45-58°C/ 0.08 mbar [ ]D 24= +2.30° (unverdünnt)
Beispiel XXXVIII
N-[2-Amino-6-(4-pyridinyl)-4-pyrimidinyl]-N-[3-fluor-4-(4-pyridinylsulfanyl)- phenyljamin
4.32 g (20.9 mmol) Beispiel V werden zusammen mit 4.61 g (20.9 mmol) Beispiel XI mit 29.6 ml Wasser versetzt. Die Mischung wird mit 9.25 ml konzentrierter Salzsäure versetzt und anschließend über Nacht bei 100°C gerührt. Nach Abkühlen auf Raumtemperatur wird mit Methanol verdünnt und mit gesättigter wässriger Natrium- hydrogencarbonat-Lösung neutralisiert. Die Mischung wird auf Kieselgel aufgezogen und durch Säulenchromatographie an Kieselgel 60 mit DicWormethan/Methanol 95:5 - Methanol gereinigt. Ausbeute: 3.87 g (47 % d. Th.) 1H-NMR (300 MHz, DMSO-d6): δ = 6.65 (s, 1H), 6.71 (s, 2H), 7.02 (dd, 2H), 7.47-
7.58 (m, 2H), 7.87 (dd, 2H), 8.30-8.41 (m, 3H), 8.72 (d, 2H), 9.90 (s, 1H) LC-MS (Methode 1): Rt = 3.34 min MS (ESIpos): m/z= 391 (M+H)+
Beispiel XXXIX
3,4,5- Trifluornitrobenzol
Die Verbindung ist zugänglich durch Oxidation von 3,4,5-Trifluoranilin mit Wasserstoffperoxid in Trifluoressigsäure gemäß einem Verfahren, das zur Herstellung analoger Derivate beschrieben ist in Heaton, A. et al., J Fluorine Chem. 1997, 81 (2), 133-138 und Krapcho, A. P. et al., J Org. Chem. 1990, 55 (21), 5662-5664.
Das in der folgenden Tabelle aufgeführte Beispiel kann analog der für Beispiel VII beschriebenen Vorschrift aus 4-Mercaptopyridin und 3,4,5-Trifluornitrobenzol hergestellt werden.
Das in der folgenden Tabelle aufgeführte Beispiel kann analog der für Beispiel XIH beschriebenen Vorschrift aus Beispiel XL hergestellt werden.
Das in der folgenden Tabelle aufgeführte Beispiel kann analog der für Beispiel XVI beschriebenen Vorschrift aus den entsprechenden Ausgangsverbindungen hergestellt werden.
Beispiel XLIII tert.-Butylhexahydropyrano[3,4-c]pyrrol-3a(4H)-ylmethylcarbamat
Die Verbindung kann nach einem in DE- A 4 032 560 beschriebenen Verfahren hergestellt werden.
Beispiel XLIV tert.-Butylhexahydropyrrolo[3,4-b]pyrrole-5(lH)-carboxylat
Die Verbindung kann nach einem in DE- A 4 032 560 beschriebenen Verfahren hergestellt werden.
Beispiel XLV
2,3-Dihydro- 1 H-pyrrolo [3 ,4-c]pyridin
Die Verbindung kann nach einem in der Literatur beschriebenen Verfahren hergestellt werden: Gabriel Colman, Chem. Ber. 1902, 35, 2845.
Beispiel XLVI 4-(3 -Pyrrolidinyl)morpholin
Die Verbindung kann nach einem in der Literatur beschriebenen Verfahren herge- stellt werden: J Org. Chem. 1998, 63 (23), 8266 - 8275.
Ausführungsbeispiele
Beispiel 1
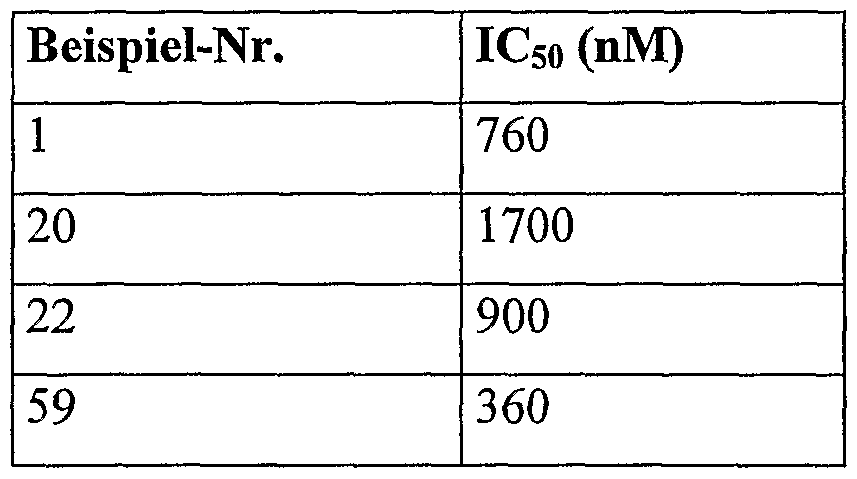
N-[2-Amino-6-(6-chinolinyl)-4-pyrimidinyl]-N-[3-fluor-4-(4-pyridinylsulfanyl)- phenyl]amin
1.50 g (5.84 mmol) der Verbindung aus Beispiel II und 1.29 g (5.84 mmol) der Verbindung aus Beispiel XI werden zusammen in 70 ml Wasser vorgelegt und mit
10 Tropfen konzentrierter Salzsäure (37 %ig) versetzt. Die Suspension wird über Nacht bei 100°C gerührt. Nach dem Abkühlen wird das Reaktionsgemisch mit konzentrierter Natriumhydrogencarbonat-Lösung basisch gestellt und im Vakuum bis zur Trockene eingeengt.
Der Rückstand wird über eine präparative HPLC gereinigt und anschließend durch Flash-Chromatographie an Kieselgel mit Dichlormethan/Methanol 30/1 als Eluens gereinigt.
Es werden 330 mg (12 % d. Th.) Produkt erhalten.
1H-NMR (300 MHz, DMSO-d6): δ = 6.66 (br.s, 2H), 6.74 (s, 1H), 7.01 (d, 2H), 7.53 (d, 1H), 7.56-7.62 (m, 2H), 8.13 (d, 1H), 8.32-8.38 (m, 4H), 8.50 (d, 1H), 8.58 (d, 1H), 8.95 (dd, 1H), 9.87 (s, 1H) HPLC (Methode 1): Rt=4.22 min
Das in der folgenden Tabelle aufgeführte Beispiel kann analog der für Beispiel 1 beschriebenen Vorschrift aus den entsprechenden Ausgangsverbindungen hergestellt werden.
Beispiel 3
N-[2-Amino-6-(6-chinolinyl)-4-pyrimidinyl]-N-[3-fluor-4-(5-isochinolinyloxy)- phenyljamin
110 mg (0.29 mmol) der Verbindung aus Beispiel XVII werden in 12 ml eines Lösungsmittelgemisches aus Toluol und Ethanol im Verhältnis 2:1 suspendiert. 10 mg (0.01 mmol) Tetrakis(triphenylphosphin)palladium(0) werden zugegeben und 20 Minuten bei Raumtemperatur gerührt. Dann wird die Suspension mit 60 mg
(0.35 mmol) 6-Chinolinboronsäure und 1 ml einer 2 M Natriumcarbonatlösung versetzt und über Nacht unter Rückfluss gerührt. Nach dem Abkühlen wird das Reaktionsgemisch im Vakuum eingeengt. Der Rückstand wird über Kieselgel 60 chromatographisch gereinigt (Laufmittel: Dichlormethan-Methanol 30:1 - 10:1).
Es werden 82 mg (60 % d. Th.) des Produktes erhalten.
1H-NMR (200 MHz, DMSO-d6): δ = 6.57 (br.s, 2H), 6.70 (s, IH), 6.99 (d, IH), 7.92 (t, IH), 7.41-7.47 (m, IH), 7.55-7.63 (m, 2H), 7.85 (d, IH), 8.13 (t, 2H), 8.28-8.37 (m, 2H), 8.50 (br.d, IH), 8.58-8.62 (m, 2H), 8.95 (dd, IH), 9.39 (s, IH), 9.66 (br.s,
IH)
HPLC (Methode 1): Rt = 3.49 min. MS (ESIpos): m/z = 475 (M+H)+
Die in der folgenden Tabelle aufgeführten Beispiele können analog der für Beispiel 3 beschriebenen Vorschrift aus den entsprechenden Ausgangsverbindungen hergestellt werden.
N-{2-Arnino-6-[3-(dimethylamino)-l-pyrrolidinyl]-4-pyrimidinyl}-N-[3-fluor-4-(4- pyridinyl-sulfanyl)phenyl] amin
600 mg (1.73 mmol) der Verbindung aus Beispiel XVI werden in 40 ml Ethanol gelöst und mit 788 mg (6.9 mmol) 3-(Dimethylamino)-pyrrolidin und 3 ml (17.25 mmol) N,N-Diisopropylethylamin versetzt. Es wird über Nacht bei 80°C gerührt. Nach dem Abkühlen wird die Reaktionslösung über eine MPLC gereinigt
(Laufmittel: Dichlormethan-Methanol 5:1 + 1% konzentrierter Ammoniaklösung).
Es werden 475 mg (58 % d. Th.) Produkt erhalten. (Enantiomerengemisch)
1H-NMR (300 MHz, DMSO-d6): δ = 1.69-1.83 (m, IH), 2.06-2.15 (m, IH), 2.20 (s,
6H), 2.69-2.80 (m, IH), 3.05 (t, IH), 3.43-3.70 (br.m, 2H), 4.05 (q, IH), 5.16 (s, IH), 5.95 (br.s, 2H), 6.97 (d, 2H), 7.36-7.47 (m, 2H), 8.21 (dd, IH), 8.34 (dd, 2H), 9.23 (s, IH) LC-MS (Methode 7): Rt = 0.44 min MS (ESIpos): m/z - 426 (M+H)+
Die folgenden beiden Enantiomere werden aus Beispiel 9 durch Enantiomeren- trennung mittels chiraler HPLC (Methode 11) gewonnen.
Die in der folgenden Tabelle aufgeführten Beispiele können analog der für Beispiel 9 beschriebenen Vorschrift aus den entsprechenden Ausgangsverbindungen hergestellt werden.
Beispiel 16
N-[ 1 -(2- Amino-6- { [3 -fluor-4-(4-pyridinylsulfanyl)phenyl] amino } -4-pyrimidinyl)- 3 -pyrrolidinyl] -N-methylamin
46 mg (0.09 mmol) der Verbindung aus Beispiel 12 werden mit 8 ml 4 M Salzsäure in Dioxan versetzt und 4 Stunden bei Raumtemperatur gerührt. Anschließend wird im Vakuum eingeengt. Der Rückstand wird mit wenig konzentrierter Ammoniaklösung versetzt, eimotiert und dann mit 2 ml Wasser aufgeschlämmt. Der Niederschlag wird abgesaugt und mit 2 ml Dichlormethan nachgewaschen.
Man erhält 23 mg (62 % d. Th.) Produkt.
1H-NMR (300 MHz, DMSO-d6): δ = 1.40 (s, 3H), 2.13-2.40 (m, 2H), 2.60 (s, 3H), 3.51-3.90 (m, 6H), 5.29 (s, IH), 7.00 (d, IH), 7.16 (s, IH), 7.33-7.52 (m, 3H), 8.18
(br.d, IH), 8.36 (d, IH) LC-MS (Methode 1): Rt = 0.68 min MS (ESIpos): m/z = 412 (M+H)+
Beispiel 17
N- [2- Amino-6-(4-cyclopentyl- 1 -piperazinyl)-4-pyrimidinyl] -N- [3 -fluor-4-(4- pyridinyl-sulfanyl)phenyl] amin
300 mg (0.86 mmol) der Verbindung aus Beispiel XVI werden in 8 ml 2-Ethyl-l- hexanol suspendiert und mit 266 mg (1.73 mmol) 1-Cyclopentylpiperazin und 0.75 ml (4.31 mmol) N,N-Diisopropylethylamin versetzt. Es wird über Nacht bei 150°C gerührt. Nach dem Abkühlen wird die Reaktionslösung über eine MPLC gereinigt (Laufmittel: Dichlormethan-Methanol 10:1 + 1% konzentrierter Ammo- niaklösung).
Es werden 216 mg (52 % d. Th.) Produkt erhalten.
1H-NMR (400 MHz, DMSO-d6): δ = 1.28-1.41 (m, 2H), 1.44-1.55 (m, 2H), 1.57-
1.68 (m, 2H), 1.70-1.85 (m, 2H), 2.43 (br.s, 5H), 3.41 (br.s, 4H), 5.39 (s, IH), 6.04 br.s, 2H), 6.97 (d, 2H), 7.36 (dd, IH), 7.45 (t, IH), 8.23 (dd, IH), 8.34 (d, 2H), 9.28
(s, IH)
LC-MS (Methode 7): Rt = 2.38 min
MS (ESIpos): m/z = 466 (M+H)+
Die in der folgenden Tabelle aufgeführten Beispiele können analog der für Beispiel 17 beschriebenen Vorschrift aus den entsprechenden Ausgangsverbindungen hergestellt werden.
Beispiel 39
N-(2- Amino-4, 5 '-bipyrimidin-6-yl)-N- [3 -fluor-4-(4-pyridinylsulfanyl)phenyl] amin
100 mg (0.253 mmol) der Verbindung Beispiel XI werden in 10 ml Dimethylformamid vorgelegt und mit 350 mg (2.53 mmol) Kaliumcarbonat versetzt. Es werden 52.13 mg (2.53 mmol) 5-(4,4,5,5-Tetramethyl-l,3,2-dioxaborolan-2-yl)pyri- midin und 20.7 mg (0.025 mmol) l,r-Bis(diphenylphosphin)ferrocen-dichlor- palladium(II)-komplex mit Dichlormethan im Argongegenstrom zugegeben. Die Reaktionslösung färbt sich nach kurzer Zeit schwarz. Es wird über Nacht bei 120°C
gerührt. Nach dem Abkühlen wird das Reaktionsgemisch mit ca. 30 ml Ethylacetat verdünnt und mit Wasser extrahiert. Die wässrige Phase wird noch zweimal mit 20 ml Ethylacetat extrahiert. Die organische Phase wird einmal mit gesättigter Natriumchloridlösung gewaschen, über Natriumsulfat getrocknet und im Vakuum eingeengt. Der Rückstand wird über Kieselgel 60 (Laufmittel: Dichlormethan-
Methanol 20:1) und anschließend über eine präparative HPLC gereinigt.
Man erhält 9 mg (8% d. Th.) des Produktes.
1H-NMR (400 MHz, DMSO-d6): δ = 6.66 (s, IH), 7.33 (d, 2H), 7.55-7.65 (m, 2H),
8.40 (dd, IH), 8.50 (br.s, 2H), 9.25 (s, 2H), 9.32 (s, IH), 10.26 (br.s, IH) MS (ESIpos): m/z = 392 (M+H)+ HPLC (Methode 1): Rt= 3.53 min
Beispiel 40
N- {2- Amino-6- [4-(2-pyridinyl)- 1 -piperazinyl] -4-pyrimidinyl} -N- [3 -fluor-4-(4- pyridinyl-sulfanyl)pheny 1] amin
34.78 mg (0.1 mmol) der Verbindung aus Beispiel XVI werden in 0.5 ml Dimethylformamid gelöst und mit 32.6 mg (0.2 mmol) l-(2-Pyridinyl)piperazin und 25.8 mg (0.2 mmol) N,N-Diisoρropylethylamin versetzt. Es wird über Nacht bei 120°C
gerührt. Zur Aufarbeitung wird die Reaktionslösung zuerst über eine LCMS gereinigt, anschließend noch einmal über eine UV/HPLC. Es werden 13.1 mg (20 % d. Th.) Produkt erhalten. LC-MS (Methode 4): Rt = 1.30 min MS (ESIpos): m/z = 475 (M+H)+
Die in der folgenden Tabelle aufgeführten Beispiele können analog der für Beispiel 40 beschriebenen Vorschrift aus Beispiel XVI und den entsprechenden Ausgangsverbindungen hergestellt werden.
Beispiel 53
N- [2- Amino-6-(4-methylphenoxy)-4-pyrimidinyl] -N- [3 -fluor-4-(4-pyridinylsulfanyl) phenyl] amin
30 mg (0.086 mmol) der Verbindung aus Beispiel XVI werden mit 37.31 mg (0.345 mmol) p-Kresol und 9.679 mg (0.173 mmol) festem Kaliumhydroxid gut vermengt und unter Argon als Schmelze für 3 Stunden bei 150°C gerührt. Zur Aufarbeitung wird die Schmelze über eine Säulenchromatographie an Kieselgel 60 mit Dichlormethan/ Methanol 95:5 gereinigt.
Es werden 15 mg (41 % d. Th.) Produkt erhalten.
LC-MS (Methode ): Rt = 2.40 min MS (ESIpos): m/z = 420 (M+H)+
Die in der folgenden Tabelle aufgeführten Beispiele können analog der für Beispiel 53 beschriebenen Vorschrift aus den entsprechenden Ausgangsverbindungen hergestellt werden.
83
Die in der folgenden Tabelle aufgeführten Beispiele können analog der für Beispiel 17 beschriebenen Vorschrift aus den entsprechenden Ausgangsverbindungen hergestellt werden.
Die in der folgenden Tabelle aufgeführten Beispiele können analog der für Beispiel 53 beschriebenen Vorschrift aus den entsprechenden Ausgangsverbindungen hergestellt werden.
Beispiel 82
1 -(2-Amino-6- { [3 -fluor-4-(4-pyridinylsulfanyl)phenyl] amino } -4-pyrimidinyl)- octaliydropyrrolo [3 ,4-b]pyrrol-5-ium Trifluoracetat
35 mg (0.067 mmol) tert.-Butyl-l-(3-Amino-5-{[3-fluor-4-(4-pyridinylsulfanyl)- phenyl] amino }phenyl)hexahydropyrrolo [3 ,4-b]pyrrol-5 ( 1 H)-carboxylat (aus Beispiel 60) werden in 1.25 ml Dichlormethan/Trifluoressigsäure (1/1) 30 min gerührt. Die
Reaktionslösung wird im Vakuum zur Trockene eingeengt.
Man erhält 35 mg (97 % d. Th.) des Produktes.
HPLC (Methode 1): Rt = 3.20 min .
MS (ESIpos): m/z = 424 (M+H)+
Beispiel 83
1 -(2- Amino-6- { [3 -fluor-4-(4-pyridinylsulfanyl)phenyl] amino } -4-pyrimidinyl)- octahydropyrrolo [3 ,4-b]pyrrol
100 mg (0.86 mmol) l-(2-Amino-6-{[3-fluor-4-(4-pyridinylsulfanyl)phenyl]amino}- 4-pyrimidinyl)octahydropyrrolo[3,4-b]pyrrol-5-ium Trifluoracetat werden in 40 ml Dichlormethan/Methanol (20:1) gelöst und mit 40 ml IN Natriumhydroxid-Lösung ausgeschüttelt, die organische Phase wird über Natriumsulfat getrocknet und im
Vakuum einrotiert.
Man erhält 53 mg (67 % d. Th.) des Produktes.
LC-MS (Methodel3):Rt = 1.55 min
MS (ESIpos): m/z = 424 (M+H)+
B. Bewertung der physiologischen Wirksamkeit
In einem in vitro-Assay mit rekombinanter Rho-K_inase-II wird die Hemmung des Enzyms untersucht. Die gefäßrelaxierende Wirkung wird an Phenylephrin-indu- zierten Kontraktionen isolierter Ringe der Kaninchen-Arteria-Saphena bestimmt. Die Eignung der erfindungsgemäßen Verbindungen zur Behandlung von kardiovaskulären Erkrankungen kann durch Untersuchung der blutdrucksenkenden Wirkung an narkotisierten Ratten gezeigt werden.
Hemmung der rekombinanten Rho-Kinase II (ROKα)
Die Aktivität der Rho-Kinase wird durch den Einbau von P-Phosphat in ein Sub- stratpeptid bestimmt. Dazu wird kommerziell erhältliche Rho-Kinase II (Upstate Biotechnology) in Gegenwart des S6 Phosphate-Acceptor-Peptides mit den Testsub- stanzen oder einer Lösungsmittelkontrolle 10 min bei 37°C vorinkubiert. Anschlies- send wird die Kinase-Reaktion durch Zugabe von 33P-markiertem ATP gestartet. Nach 20 min bei 37°C wird die Reaktion durch Zugabe von H3PO4 gestoppt. Ali- quots werden auf Filter pipettiert, die Filter gewaschen und anschließend mit Scintil- lator beschichtet. Die Radioaktivität der am Filter gebundenen P-markierten Pep- tide wird in einem Micro-Beta-Counter gemessen. Der IC5o-Wert entspricht der Kon- zentration einer Testsubstanz bei welcher der Rho-Kinase katalysierte Einbau von P in das Peptid im Vergleich zu einer Lösungsmittelkontrolle um 50 % gehemmt ist. Die experimentellen Daten sind in der folgenden Tabelle zusammengestellt.
Gefäßrelaxierende Wirkung in vitro
3 mm breite Ringe der isolierten Kaninchen-Arteria-Saphena werden einzeln in 5 ml- Organbäder mit 37°C warmer, carbogenbegaster Krebs-Henseleit-Lösung gebracht. Der Gefäßtonus wird isometrisch erfasst und registriert. Kontraktionen werden durch Zugabe von 3x10"8 g/ml Phenylephrin induziert und nach 4 min wieder ausgewaschen. Nach mehreren Kontrollzyklen wird die zu untersuchende Substanz mit jedem weiteren Durchgang in steigender Dosierung vorinkubiert und die darauffolgende Kontraktion mit der Höhe der letzten Kontrollkontraktion verglichen. Es wird die Konzentration errechnet, die erforderlich ist, um die Höhe des Kontrollwertes um 50 % zu reduzieren (IC5o). Die experimentellen Daten sind in der folgenden Tabelle zusammengestellt.
Blutdruckmessung an narkotisierten Ratten
Männliche Wistar-Ratten mit einem Körpergewicht von 300 - 350 g werden mit Thiopental (100 mg/kg i.p.) anästhesiert. Nach der Tracheotomie wird in die Femoralarterie ein Katheter zur Blutdruckmessung eingeführt. Die zu prüfenden Substanzen werden als Lösungen entweder oral mittels Schlundsonde oder über die Femoralvene intravenös verabreicht.
C. Ausführungsbeispiele für pharmazeutische Zusammensetzungen
Die erfindungsgemäßen Verbindungen können folgendermaßen in pharmazeutische
Zubereitungen überführt werden:
Tablette:
Zusammensetzun :
100 mg der Verbindung von Beispiel 1, 50 mg Lactose (Monohydrat), 50 mg Maisstärke (nativ), 10 mg Polyvinylpyrolidon (PVP 25) (Fa. BASF, Ludwigshafen,
Deutschland) und 2 mg Magnesiumstearat.
Tablettengewicht 212 mg. Durchmesser 8 mm, Wölbungsradius 12 mm.
Herstellung:
Die Mischung aus Wirkstoff, Lactose und Stärke wird mit einer 5%-igen Lösung (m/m) des PVPs in Wasser granuliert. Das Granulat wird nach dem Trocknen mit dem Magnesiumstearat für 5 min. gemischt. Diese Mischung wird mit einer üblichen Tablettenpresse verpresst (Format der Tablette siehe oben). Als Richtwert für die Verpressung wird eine Presskraft von 15 kN verwendet.
Oral applizierbare Suspension:
Zusammensetzung:
1000 mg der Verbindung von Beispiel 1, 1000 mg Ethanol (96 %), 400 mg Rhodigel (Xanthan gum der Fa. FMC, Pennsylvania, USA) und 99 g Wasser.
Einer Einzeldosis von 100 mg der erfindungsgemäßen Verbindung entsprechen 10 ml orale Suspension.
Herstellung:
Das Rhodigel wird in Ethanol suspendiert, der Wirkstoff wird der Suspension zugefügt. Unter Rühren erfolgt die Zugabe des Wassers. Bis zum Abschluss der Quellung des Rhodigels wird ca. 6h gerührt.