WO2004050656A1 - 1,3−ジヒドロ−イミダゾール縮合環化合物 - Google Patents
1,3−ジヒドロ−イミダゾール縮合環化合物 Download PDFInfo
- Publication number
- WO2004050656A1 WO2004050656A1 PCT/JP2003/015402 JP0315402W WO2004050656A1 WO 2004050656 A1 WO2004050656 A1 WO 2004050656A1 JP 0315402 W JP0315402 W JP 0315402W WO 2004050656 A1 WO2004050656 A1 WO 2004050656A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- compound
- formula
- methyl
- atom
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D473/00—Heterocyclic compounds containing purine ring systems
- C07D473/02—Heterocyclic compounds containing purine ring systems with oxygen, sulphur, or nitrogen atoms directly attached in positions 2 and 6
- C07D473/18—Heterocyclic compounds containing purine ring systems with oxygen, sulphur, or nitrogen atoms directly attached in positions 2 and 6 one oxygen and one nitrogen atom, e.g. guanine
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D473/00—Heterocyclic compounds containing purine ring systems
- C07D473/26—Heterocyclic compounds containing purine ring systems with an oxygen, sulphur, or nitrogen atom directly attached in position 2 or 6, but not in both
- C07D473/32—Nitrogen atom
- C07D473/34—Nitrogen atom attached in position 6, e.g. adenine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P15/00—Drugs for genital or sexual disorders; Contraceptives
- A61P15/08—Drugs for genital or sexual disorders; Contraceptives for gonadal disorders or for enhancing fertility, e.g. inducers of ovulation or of spermatogenesis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
- A61P19/10—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease for osteoporosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/18—Antivirals for RNA viruses for HIV
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D473/00—Heterocyclic compounds containing purine ring systems
- C07D473/02—Heterocyclic compounds containing purine ring systems with oxygen, sulphur, or nitrogen atoms directly attached in positions 2 and 6
- C07D473/16—Heterocyclic compounds containing purine ring systems with oxygen, sulphur, or nitrogen atoms directly attached in positions 2 and 6 two nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D473/00—Heterocyclic compounds containing purine ring systems
- C07D473/02—Heterocyclic compounds containing purine ring systems with oxygen, sulphur, or nitrogen atoms directly attached in positions 2 and 6
- C07D473/24—Heterocyclic compounds containing purine ring systems with oxygen, sulphur, or nitrogen atoms directly attached in positions 2 and 6 one nitrogen and one sulfur atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D473/00—Heterocyclic compounds containing purine ring systems
- C07D473/26—Heterocyclic compounds containing purine ring systems with an oxygen, sulphur, or nitrogen atom directly attached in position 2 or 6, but not in both
- C07D473/32—Nitrogen atom
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D473/00—Heterocyclic compounds containing purine ring systems
- C07D473/40—Heterocyclic compounds containing purine ring systems with halogen atoms or perhalogeno-alkyl radicals directly attached in position 2 or 6
Definitions
- the present invention relates to novel compounds having DP PIV inhibitory activity, c relates useful 1, 3-dihydro one imidazole fused ring compound Particularly DPPIV inhibitor
- Dipeptidyl peptidase-IV is a serine protease that specifically hydrolyzes one X-Pro (X may be any amino acid) dipeptide from the free N-terminus of a polypeptide chain. It is one kind of.
- Glucose-dependent Peptide-1 and GIP Glucose-dependent Insulinotropic Polypeptide secreted from the intestinal tract after meals by this DPP IV 5) ⁇ ?, inactive '[4.
- DPP IV Glucose-dependent Insulinotropic Polypeptide
- the DPPP IV inhibitor can be a useful therapeutic or preventive agent for GLP-1, GIP-related diseases such as obesity and diabetes.
- Non-Patent Document 2 Prevention and treatment of AIDS (see Non-Patent Document 2)
- Non-patent Documents 5, 6 (4) Prevention and treatment of hyperlipidemia, diabetes and obesity (Non-patent Documents 5, 6)
- Non-Patent Document 8 Prevention and treatment of inflammatory diseases, autoimmune diseases, and rheumatoid arthritis.
- DPP IV inhibitors Although some DPP IV inhibitors are known (see Patent Documents 2 to 11), DPP IV inhibitors having a 1,3-dihydro-imidazole fused ring are not known.
- Non-Patent Document 7 Agents and actions, 32, 125-127, 1991.
- an object of the present invention is to search for and find a compound having a DPPIV inhibitory activity that is useful as a therapeutic or prophylactic agent for the above-mentioned diseases (particularly, diabetes diseases and the like). Disclosure of the invention
- the present inventors have conducted intensive studies to solve the above-mentioned object, and as a result, succeeded in synthesizing a novel 1,3-dihydro-imidazo / di-conjugated compound, and these compounds have excellent DPPIV inhibition.
- the present invention was found to have an effect, and the present invention was completed.
- the present invention includes the following.
- T 1 is 1 or 2 nitrogen atoms in the ring, and may have a substituent Means a 4- to 12-membered heterocyclic ring that is monocyclic or bicyclic
- X 3 is an oxygen atom, a sulfur atom or a formula
- X 4 is a hydrogen atom, which may have a substituent Ci-e alkyl group, optionally have a substituent C 3 - 8 cycloalkyl group, or may have a substituent group C 6 _ means one 0 Ariru Ji 6 alkyl group;
- X 1 is optionally substituted alkyl group, but it may also have a substituent C 2 - 6 alkenyl group which may have a substituent C 2 _ 6 alkyl group , which may have a substituent C 6 _ 10 Ariru group, Te to be or 5-10 membered substituted Roariru group which may have a substituent C 6 - i.
- Ariru CI- 6 alkyl group or 5 may have a substituent group: you mean Teroari Ichiru 0 6 alkyl group to the L 0 members;
- Z 2 and Z 3 each independently represent a nitrogen atom, a formula 1 CR 1 -carbonyl group or a formula 1 NR 2 —;
- the formula Means a double bond or a single bond
- R ⁇ RR 3 and X 2 each independently represent a hydrogen atom, a 4- to 8-membered heterocyclic group which may have a substituent, or a group represented by the formula: A ° —A 1 —A 2 To
- a G is selected from a single bond or the following substituent group A:! Means a C- 6 alkylene group which may have up to 3 groups;
- a 1 is a single bond, an oxygen atom, a sulfur atom, a sulfuryl group, a sulfonyl group, a carbonyl group, a formula—o—CO—, a formula—CO—O—, a formula—NR A— , and a formula—CO—NR A— , Formula 1 NR A — CO—, Formula 1 SO 2 — NR A — or Formula 1 NR A
- a 2 and R A each independently represent a hydrogen atom, a cyano group, ⁇ 6 alkyl group, C 3 - 8 cycloalkyl group, C 2 one 6 alkenyl, C 2 - 6 alkynyl group, Ji.
- Aryl means Ci- 6 alkyl group;
- a 2 and RA may each independently have 1 to 3 groups selected from the following substituent group A.
- Substituent group A includes a hydroxyl group, a mercapto group, a cyano group, a halogen atom,. ! DOO 6 alkyl le group, C 3 _ 8 cycloalkyl group, C 2 - 6 alkenyl group, C 2 - 6 alkyl groups, C 6 _ 0 Ariru group, 5: Teroariru group to L 0-membered, 4 8-membered heterocyclic group,.
- X 3 a means an oxygen atom or a sulfur atom
- T 1 a is 1 or 2 nitrogen atoms in the ring, means an amino group or a C _ 6 Arukirua amino group may have a monocyclic 4-8 membered heterocyclic ring;
- X la is a hydrogen atom, C 2 _ 6 alkenyl, C 2 - means a 6 Arukiniru group or a benzyl group;
- R 1 a and R 2 a are each independently a hydrogen atom, a halogen atom, an alkyl group, Shiano group, or a group of the formula one A Ga - refers to the group represented by A la;
- a ° a is an oxygen atom, a sulfur atom or a NA 2a - means a group represented by;
- a la is a hydrogen atom, C - 6 alkyl group, C 2 - 6 alkenyl group, C 2 _ 6 Arukini group, phenyl Group, cyanophenyl group, carpamoyl fuel group, benzyl group, pyridylmethyl group or pyridyl group;
- a 2a represents a hydrogen atom or an alkyl group
- X 2a is a hydrogen atom, C 2 _ 6 alkenyl, C 2 - 6 alkynyl group, cyclohexenyl group cyclohexylene, 1H-pyridin-one 2-Oniru group, 1-methyl-1H-pyridin-one-2-one Iru group, Substituent Group
- An alkyl group optionally having a group selected from B, a phenyl group optionally having a group selected from the following Substituent Group B, and a group selected from the following Substituent Group B and heteroaryl groups may be 5 or 6-membered, which may have a group selected group from a good phenyl C _ 6 alkyl group or the following substituent group B may have a selected from the following substituent group B Pyridyl — means a 6-alkyl group;
- Substituent group B a chlorine atom, a bromine atom, Shiano group, C - 6 alkyl group, C 2 - 6 an alkenyl group, C 2 - 6 alkynyl group, C 3 _ 8 cycloalkyl group, - 6 alkoxy group, Karupamoiru group refers to a group consisting of carboxyl group Contact Yopi C DOO 6 alkoxy Cal Poni Le group. ] The compound represented by these, its salt, or those hydrate.
- T lb represents a piperazine-11-yl group, 3-amino-1-piperidine-11-yl group or 3-methylamino-piperidine-11-inole group;
- X ib represents a 2- pentyl group, a 2-butul group, a 3-methyl-12-butur group, a 2-butul group or a benzyl group;
- R la and X 2a are the same as X la and X 2a described in [2]. ] The compound represented by these, its salt, or those hydrates.
- R la is a hydrogen atom, a chlorine atom, a cyano group, a methoxy group, an ethoxy group, an i-pyroxy group, a methylthio group, an aryloxy group, a 2-butynyloxy group, a phenyloxy group, or a cyanophenyloxy group
- X 2 a is a hydrogen atom, a methyl group, an ethyl group, an n-propyl group, a 2-methylpropyl group, a formula CH 2 —R 10 (where R 10 is a carpamoyl group, a carboxyl group, A xycarbonyl group, a cyano group, a cyclopropyl group or a methoxy group.), A 3-cyanopropyl group, an aryl group, a 2-propynyl group, a 2-butynyl group, a 2-methyl-1-propane group Nyl, 2-cyclohexyl, cyclopyridyl, methoxypyridyl, methoxypyrimidyl, pyridyl, furyl, phenyl, phenyl, pyridylmethyl, 1H-pyridin-2-one-5 —Inole group, 1-methyl — 1 H-pyridin-2-one-1-Ino
- the substituent group Y is a group consisting of a chlorine atom, a bromine atom, a methoxy group, a cyano group, a vinyl group, and a methyl group; (2) to (5), or a salt thereof, or a compound thereof. Hydrate.
- X 2a is a methyl group, an n-propyl group, an aryl group, a 2-propyl group, a 2-butyl group, a cyclopropylmethyl group, a pheninole group, a 3-pyridyl group, a 3-furyl group, a 3 1-Cheninole group, 2-methoxy-5-pyrimidinyl group, 2-methoxy-5-pyridinyl [2] to [5], a 2-chloro-4_pyridyl group or a 1H-pyridin-2-one-15-yl group, or a salt thereof or a hydration thereof. object.
- a medicament comprising the compound according to [1], a salt thereof, or a hydrate thereof.
- a dipeptidinolepeptidase IV inhibitor comprising the compound according to [1], a salt thereof, or a hydrate thereof.
- a pharmaceutical composition comprising the compound according to [1], a salt thereof, or a hydrate thereof, and a formulation auxiliary.
- a prophylactic or therapeutic agent for diabetes mellitus comprising the compound according to [1], a salt thereof, or a hydrate thereof.
- a method for treating or preventing a disease in which dipeptidyl peptidase IV inhibition is effective comprising administering to a patient a pharmacologically effective amount of the compound according to [1], a salt thereof, or a hydrate thereof.
- the structural formula of a compound may represent a certain isomer for convenience, but the present invention is based on all geometric isomers and asymmetric carbon generated in the structure of the compound. It includes isomers such as optical isomers, stereoisomers, and tautomers, and isomer mixtures, and is not limited to the description of formulas for convenience. Either one isomer or a mixture may be used. Therefore, a power having an asymmetric carbon atom in the molecule and possibly having an optically active or racemic form is not particularly limited in the present invention, and any of ⁇ is included.
- polymorphism may exist, but is not similarly limited, and any one of the crystal forms may be single or a mixture, and either an anhydride or a hydrate may be used. Good. Further, the compound of the present invention may be a solvate which has absorbed some other solvent.
- the present invention also includes compounds that undergo the metabolism such as oxidation, reduction, hydrolysis, and conjugation in the living body and still exhibit a desired activity, and the present invention further includes oxidation, reduction, hydrolysis, and the like in the living body. And the compounds which produce the compound of the present invention upon metabolism of the present invention.
- ⁇ 6 alkyl group is a monovalent group derived from an aliphatic hydrocarbon having from! To 6 aliphatic hydrocarbons by removing one hydrogen atom, and having 1 to 6 carbon atoms.
- the "C 2 _ 6 alkenyl group” means a straight or branched ⁇ Luque two Le group number 2 to 6 carbon atoms, Bulle groups as specific examples, Ariru group, 1 one pro Examples include a beer group, a 1-methylvinyl group, a 1-pteninole group, a 2-pthyl group, a 3-pthenyl group, a pentenyl group, and a hexenyl group.
- C 2 6 alkynyl group means a straight or branched ⁇ Rukiniru group number 2 to 6 carbon atoms, Echuru group Specific examples, 1 one prop - group, 2 —Propynyl group, butul group, penture group, hexyl group and the like.
- the - and "C 3 8 cycloalkyl group” means a cyclic aliphatic hydrocarbon group having 3 to 8 carbon atoms, and specific examples, cyclohexyl cyclopropyl group, Shikuropuchiru group, cyclopentyl group, cyclohexylene Group, cycloheptyl group, cyclooctyl group and the like.
- C 6 alkylene group means a divalent group derived by removing one arbitrary hydrogen atom from the above-defined “. ⁇ 6 alkyl group”, and specific examples thereof include a methylene group , 1,2-ethylene, 1,1-ethylene, 1,3-propylene, tetramethylene, pentamethylene, and hexamethylene.
- Bok 6 alkyl group is bonded to oxygen radicals defined above, specific examples, methoxy, ethoxy, 1-propylidene Ruokishi group, 2 —Propyloxy group, 2-methyl-1-propyloxy group, 2-methyl-2-propyloxy group, .1 monobutyloxy group, 2-butyloxy group, 1 ⁇ ° -ethyloxy group, 2-pentyloxy group, 3-pentyloxy group, 2 —Methyl-1-butyloxy group, 3-methyl-1-butyloxy group, 2-methyl-1-butyloxy group, 3-methyl-2-butyloxy group, 2,2-dimethyl-1-propyloxy group, 1-hexyloxy group, 2-hexyloxy group, 3-hexyloxy group, 2-methyl-1-pentyloxy group, 3-methyl-1-pentyloxy group, 4-methyl 1-pentyloxy group, 2-methyl
- 0 6 alkoxy Cal Poni Le group rc of the definitions - means that a 6-carbonyl group which alkoxy group "is bonded, as a specific example, Metokishikarupo - group, ethoxycarbonyl group, 1- Examples include a propyloxycarbonyl group, a 2-propyloxycarbonyl group, a 2-methyl-11-propyloxycarbonyl group, and a 2-methyl-21-propyloxycarbonyl group.
- the “0 ⁇ 6 alkylthio group” means a sulfur atom to which the above-mentioned “dialkyl group” is bonded, and specific examples thereof include a methylthio group, an ethylthio group, an 11-propylthio group, a 2-propylthio group, 2-methyl-1-propylthio group, 2-methyl-2-propylthio group, 1-butylthio group, 2-butylthio group, 1-pentylthio group, 2-pentylthio group, 3-pentylthio group, 2-methyl-1- Butylthio group, 3-methyl-1-pentynolethio group, 2-methyl-2-butylthio group, 3-methyl-2-butylthio group, 2,2-dimethyl-11-propylthio group, 1-hexylthio group, 2-hexylthio Group, 3-hexylthio group, 2-methyl-1-pentylthio group, 3-methyl-1-pentyl
- hetero atom means a sulfur atom, an oxygen atom or a nitrogen atom.
- the number of atoms constituting the ring of the cyclic group is 4 to 8
- the "4- to 8-membered hetero ring” include a pyrrolidine ring, a piperidine ring, an azepan ring, a tetrahydrofuran ring, a tetrahydrovirane ring, a morpholine ring, a thiomorpholine ring, a piperazine ring, a thiazolidine ring, and a dioxane.
- T 4 is a methylene group, an oxygen atom or a NT 5 wherein - (wherein, T 5 is the group represented by represents a hydrogen atom or an alkyl group). And the like) and the like.
- the above “4 to 8 membered heterocyclic group” means a monovalent group derived by removing one hydrogen atom at an arbitrary position from the above defined “4 to 8 membered heterocyclic group”.
- C 6 -i. Aryl group refers to an aromatic hydrocarbon cyclic group having 6 to 10 carbon atoms, and specific examples thereof include a phenyl group, a 1-naphthinole group, and a 2-naphthyl group. Can be exacerbated.
- a 5- to 10-membered heteroaryl ring refers to an aromatic ring having 5 to 10 atoms constituting a ring of a cyclic group and containing a hetero atom in the atoms constituting the ring of the cyclic group.
- Ring of family And specific examples thereof include a pyridine ring, a thiophene ring, a furan ring, a pyrrole ring, an oxazole ring, an isoxazole ring, a thiazole ring, an isothiazole ring, an imidazole ring, a triazole ring, a pyrazole ring, a flazane ring, and a thiadiazole ring.
- Examples include a lysine ring, a pyridine ring having a pyrrole, a pyrimidine ring having a pyrrole, and a pyrido
- the above “5- to 10-membered heteroaryl group” refers to a monovalent group that can be derived by removing one hydrogen atom at any position from the above-defined “5- to 10-membered heteroaryl ring”. I do. Any hydrogen atom in the above definition is a ". 6 _ 1 0 Ariru. 1 _ 6 Arukiru group”"shooting 6 alkyl group” means a defined group obtained by substituting at ".. Ariru group”, specifically Examples include benzyl, phenethyl, 3-phenyl-11-propyl and the like.
- 5- or 6-membered heteroaryl ring refers to a cyclic group in which the number of atoms constituting the ring is 5 or 6, and one or more heteroatoms are contained in the atoms constituting the ring of the cyclic group.
- aromatic ring contained therein, and specific examples thereof include a pyridine ring, a thiophene ring, a furan ring, a pyrrole ring, an oxazole ring, an isoxazono V, a thiazolyl ring, an isothiazole ring, an imidazole ring, a triazole ring, Examples include a pyrazole ring, a thiadiazole ring, an oxadiazole ring, a pyridazine ring, a pyrimidine ring, and a pyrazine ring.
- the “5- or 6-membered heteroaryl group” means a monovalent group derived from the “5- or 6-membered aromatic heteroaryl ring” by removing one hydrogen atom at any position.
- the above “pyridyl group” means a 2-pyridyl group, a 3-pyridyl group or a 4-pyridyl group.
- furyl group means a 2-furyl group or a 3-furyl group.
- Chenyl group means a 2-Chenyl group or a 3-Chenyl group.
- cyclohexeninole group means a 1-cyclohexeninole group, a 2-cyclohexenyl group or a 3-cyclohexenyl group.
- 1-methyl-1-H-pyridin-2-one-yl group is a monovalent group derived by removing one arbitrary hydrogen atom from “1-methyl-1-H-pyridin-2-one”.
- phenyl C i_ 6 alkyl group the definition arbitrary hydrogen atom of "0 ⁇ 6 alkyl group” in means substituted group phenyl group, specific examples base Njiru group, Hue Nechiru group And 3-phenyl-11-propyl group.
- pyridylalkyl group means a group in which any hydrogen atom in the above-defined “0 ⁇ 6 alkyl group” is substituted by the above-defined “pyridyl group”, and specific examples thereof include 2-pyridylmethyl Group, 3-pyridylmethyl group or 4-pyridylmethyl group.
- pyridylmethyl group means a 2-pyridylmethyl group, a 3-pyridylmethyl group or a 4-pyridylmethyl group.
- pyridyloxy group means a 2-pyridyloxy group, a 3-pyridyloxy group or a 4-pyridyloxy group.
- pyridylmethyloxy group means a 2-pyridylmethyloxy group, a 3-pyridylmethyloxy group or a 4-pyridylmethyloxy group.
- cyanophenyl group means a 2-cyanophenyl group, a 3-cyanophenyl group or a 4-cyanophenyl group.
- carboxy group means a 2-cyanophenyloxy group, a 3-cyanophenyl group or a 4-cyanophenyl group.
- carboxy group means a 2-carpamoyl phenyloxy group, a 3-carpamoyl phenyloxy group or a 4-carpamoyl phenyloxy group.
- cyclopyridyl group means a group in which any hydrogen atom in the above-defined “pyridyl group” is replaced by a chlorine atom, and specific examples thereof include 2-cyclopyridine-3-yl group, 2 —Cross pyridine-14-inole group or 6—Cross pyridine-13- ⁇ (
- methoxypyridyl group means a group in which an arbitrary hydrogen atom in the above-defined “pyridyl group” is substituted with a methoxy group, and specific examples thereof include 2-methoxypyridine-13-inole group and 2-methoxypyridine. And a 14-yl group or a 6-methoxypyridine-13-yl group.
- methoxypyrimidyl group means a group in which an arbitrary hydrogen atom in the above-defined “pyrimidinole group” is substituted with a methoxy group, and specific examples thereof include a 2-methoxypyrimidine-15-yl group and a 2-methoxypyrimidyl group. —Methoxypyrimidine-14-yl group and the like.
- the number of atoms constituting the ring of the cyclic group is 4 to 12,
- a non-aromatic ring that is monocyclic or bicyclic.
- T la means “a monocyclic 4- to 8-membered heterocyclic group which may have an amino group or a 6- alkylamino group having 1 or 2 nitrogen atoms in the ring”,
- the number of atoms constituting the ring of the cyclic group is 4 to 8,
- Ci- 6 alkylamino group means that the above-defined “0 ⁇ 6 alkyl group” is a nitrogen atom having one or two bonded atoms, and specific examples thereof include a methylamino group, an ethylamino group, and a propyl group. Examples include an amino group, a dimethylamino group, a getylamino group, and a dipyramino group.
- T la is preferably a piperazine-1 -yl group, [1.4] diazepan-11-yl group, [1.5] diazocan-11-yl group, an amino group or — 6 azetidine-11-yl group which may have alkylamino group, amino group or C- 6 pyrrolidine-11-yl group which may have alkylamino group, amino group or C ⁇ e alkylamino group A pyridine or an amino or alkylamino group which may have a pyridine or an amino group; Azocanyl group which may be (2) More preferably the formula
- R 5D represents an amino group or a methylamino group
- one of R 51 and R 52 represents an amino group or a methylamino group, and the other represents a hydrogen atom
- R 53 And one of R 54 represents an amino group or a methylamino group, and the other represents a hydrogen atom
- any one of R 55 to: 57 represents an amino group or a methylamino group; The remaining two are hydrogen atoms.
- T lb means piperazine-11-yl group, 3-amino-biperidine-11-yl group or 3-methylamino-1-piperidine-11-yl group, preferably piperazine-11-yl Group.
- x3a is a force meaning an oxygen atom or a sulfur atom, and is preferably an oxygen atom.
- X la is a hydrogen atom, C 2 _ 6 alkenyl, C 2 _6 alkynyl group or a benzyl group
- (1) preferably a hydrogen atom, a 2- ⁇ nthyl group, a 2-butynyl group, a 3-methynole-1-butul group, a benzyl group or a 2-ptul group,
- X lb represents a hydrogen atom, a 2-pentulyl group, a 2-butynyl group, a 3-methynole-1-butenyl group, a benzyl group or a 2-butulyl group,
- R la is "hydrogen atom, a halogen atom, an alkyl group, Shiano group, or a group of the formula one A 0 a - in A la (wherein, A ° a is an oxygen atom, a sulfur atom or a NA 2a - meaning a group represented by and, A la is a hydrogen atom, C ⁇ 6 alkyl group, C 2 - 6 alkenyl group, C 2 _ 6 alkynyl group, phenyl group, Shianofueniru group, Scarpa carbamoyl-phenylalanine group, a benzyl group, a pyridylmethyl group or a pyridyl group A 2a «means a hydrogen atom or a 6- alkyl group).
- X 2 a is a hydrogen atom, C 2 - 6 alkenyl group, C - 6 alkynyl group, cyclohexenyl group cyclohexylene, 1 H- pyridin one 2-Oniru group, 1-methyl-one 1 H- pyridin one-2-one Iru group,
- a 5- or 6-membered heteroaryl group, a phenylalkyl group optionally having a group selected from the following substituent group B, or a group selected from the following substituent group B means a pyridyl C _ 6 alkyl group (location substituent group B a chlorine atom, a bromine atom, Shiano group, - 6 alkyl group, C 2 - 6 alkenyl group, C 2
- a hydrogen atom a methyl group, an ethyl group, an n-propyl group, a 2-methylpropyl group, a formula CH 2 —R 10 (wherein R 10 is a carpamoyl group, a lipoxyl group , A methoxycarbonyl group, a cyano group, a cyclopropyl group or a methoxy group), a 3-cyanopropyl group, an aryl group, a 2-propyl group, a 2-butyr group, a 2-methyl-2- group.
- T la or T lb, X, Xla or XLB, R1a showed a suitable base in significance of X2a, T la or T lb, X 3 a, Xla or XLB, R1a, group or al suitably made of X
- X Compounds obtained by selecting any group and arbitrarily combining them can be mentioned as specific compounds.
- Formula 1 — 2 — ⁇ 3 (wherein ⁇ 2 is a single bond, — 6 alkylene group, oxygen atom, sulfur atom, sulfinyl group, sulfol group, carbonyl group, formula 1 O—CO—, Formula—CO—O—, Formula—NR T— , Formula—CO_NR T _, Formula—NR T —CO—, Formula—S0 2 —NR T —or Formula—NR T —S0 2— and, T 3 Oyopi 1 ⁇ are their respective independently a hydrogen atom, C - 6 alkyl group, - 6 alkoxy groups, C 3 one 8 Shikuroa alkyl group, C 2 - 6 alkenyl group, C 2 _ 6 alkynyl groups, C 6 -.
- Ariru group means a heterocyclic groups 5 to 0-membered Teroariru group, the 4-8 membered however, T 3 and R ⁇ may each independently have 1 to 3 groups selected from the group consisting of the following substituents ⁇ . Except where T 2 is a is T 3 is hydrogen atom single bonds.
- salts in the present invention include a salt with an inorganic acid, a salt with an organic acid, a salt with an inorganic base, a salt with an organic base, and a salt with an acidic or basic amino acid.
- pharmaceutically acceptable salts are preferred.
- salts with inorganic acids include, for example, salts with hydrochloric acid, hydrobromic acid, sulfuric acid, nitric acid, phosphoric acid, and the like.
- Preferred examples of salts with organic acids include, for example, acetic acid, succinic acid And salts with fumaric acid, maleic acid, tartaric acid, citric acid, lactic acid, stearic acid, benzoic acid, methanesulfonic acid, P-toluenesulfonic acid, and the like.
- Preferred examples of the salt with an inorganic base include an alkali metal salt such as a sodium salt and a potassium salt, an alkaline earth metal salt such as a calcium salt and a magnesium salt, an aluminum salt, and an ammonium salt.
- Preferred examples of the salt with an m-containing group include salts with getinoleamine, diethanolamine, meglumine, N, N, -dibenzylethylenediamine and the like.
- Preferred examples of the salt with an acidic amino acid include, for example, salts with aspartic acid and glutamic acid.
- Preferred examples of the salt with a basic amino acid include, for example, salts with arginine, lysine, orditin and the like. Salt.
- UU 2 is a leaving group (a chlorine atom, a bromine atom, an iodine atom, a methanesulfonyloxy group, a p-toluenes / refonyl group) Okishi group, one B (OH) 2 4, 4 5, 5- tetramethyl-1, 3 2 Jioki Saporan one 2-I group or the formula one Sn (R z) 3 (where, 1 ⁇ in. 1 over 6 means an alkyl group, such as a group represented by parentheses).
- Ha 1 represents a halogen atom such as a chlorine atom, a bromine atom, and an iodine atom.
- M 1 is a hydrogen atom, a sodium atom, a lithium atom, a lithium atom, —MgCl—MgBr, one Sn (R z ) 3 (where R z has the same meaning as defined above;), etc.
- Means Y represents a halogen atom such as a chlorine atom, a bromine atom and an iodine atom, or a hydrogen atom.
- PP 2 each independently represents a protecting group for an amino group such as a benzyl group, a piperyloxymethyl group, a t-toxycanolepodinole group, and a cyanoethyl group.
- T 2 b are you mean T 1 having an amino group attached the same meaning of group or coercive Mamorumoto (such as t-hept propoxycarbonyl Interview le group) and T 1.
- a substituent is introduced into the 7-position amino group of compound (la) by subjecting compound (1a) [CAS No. 1076-22-8] to a substitution reaction with compound (1a-2). This is a step of obtaining a compound (2a).
- the compound (la-2) is an electrophile represented by the formula X 1 —U 1 (wherein X 1 and U 1 have the same meanings as defined above), specifically, eodomethane, eodoethane, Alkyl halides such as eodopropane and benzylpromide, arylpromide,
- phenolic halide such as 1-promo-3-methyl-12-butene or alkynyl halide such as propanolegyl bromide or 1-bromo-2-butyne
- the reaction conditions for the substitution reaction are not particularly limited, but, for example, in a solvent such as dimethyl sulfoxide, N, N-dimethylformamide, N-methylpyrrolidone, dioxane, tetrahydrofuran, tonoleene, zK lithium oxide, hydroxide Sodium, potassium heptamate, lithium carbonate, sodium carbonate, potassium carbonate, cesium carbonate, lithium hydride, sodium hydride, 7_potassium borohydride, butyllithium, methyllithium, lithium bistrimethylsilylamide, sodium bistrimethylsilylamide,
- the reaction can be performed at a temperature of 0 ° C to 150 ° C in the presence of a base such as potassium bistrimethylsilylamide. In this case, it is preferable to use 1 to 2 equivalents of the base with respect to the compound (la).
- X 1 to be introduced may have a substituent C 6 — 10 aryl group or may have a substituent 5 to: L 0 member ⁇ 8 ⁇ compound of a heteroaryl group (1 As a-2), specifically, the reaction can be carried out using, for example, arylpoic acid or heteroarylpoic acid. In this case, it is preferable to use 1 to 10 equivalents of the compound (1a-2) with respect to the compound (la).
- a solvent such as dichloromethane, chloroform, 1,4-dioxane, 1,4-dioxane, tetrahydrofuran, toluene, pyridine, N, N-dimethylformamide, N-methylpyrrolidone, triethylamine, diisopropylethylamine, pyridine, N 0 ° in the presence of a base such as N, N-dimethylaminopyridine or a copper catalyst such as copper acetate (11), copper trifluoroacetate (I 1), copper chloride (11) or copper iodide (II).
- the reaction can be carried out at a temperature between C and 150 ° C. In this case, it is preferable to use 0.1 to 2 equivalents of the copper catalyst with respect to the compound (la).
- This step is a step of reacting compound (2a) with a halogenating agent to obtain compound (3a).
- halogenating agent examples include N-chlorosuccinimide, N-bromosuccinimide, N-odosuccinimide and the like. It is preferable to use 1 to 4 equivalents of such a halogenating agent with respect to the conjugate (2a).
- the reaction conditions for the halogenation are not particularly limited. However, in a solvent such as acetonitrile, N, N-dimethylformamide, N-methylpyrrolidone, 1,4-dioxane, tetrahydrofuran, dimethoxyethane, etc.
- a solvent such as acetonitrile, N, N-dimethylformamide, N-methylpyrrolidone, 1,4-dioxane, tetrahydrofuran, dimethoxyethane, etc.
- the reaction can be performed at temperatures between C and 150 ° C.
- This step is a step of chlorinating the compound (3a) to obtain the compound (4a).
- reaction conditions are not particularly limited, compound (3a) and oxychloride phosphorus, phosphorus pentachloride, or a mixture thereof in a solvent or without solvent may be used in the range of 0 to 15%.
- the reaction can be performed at a temperature of C.
- the solvent for example, toluene, acetonitrile, dichloroethane and the like can be used.
- Step A4 This step is a step of obtaining a compound (5a) by a hydrolytic reaction of the compound (4a).
- a base such as sodium acetate, potassium carbonate, sodium hydroxide, etc.
- a solvent such as dimethyl sulfoxide (hydrated), N-methylpyrrolidone (hydrated), tetrahydrofuran (hydrated) or water, or a mixed solvent thereof at 0 ° C
- the reaction can be performed at 150 ° C.
- the base is preferably used in an amount of 1 to: L 0 equivalent to the compound (4a).
- This step is a step for obtaining a compound (6a) by subjecting the compound (5a) to a substitution reaction with the compound (5a-2). If X 2 is a hydrogen atom, skip this step. Can be.
- Compound (5 a- 2) has the formula X 2 - U 2 (wherein, X 2 and U 2 are defined respectively the same meaning) electrophile represented by, specifically Yodometan, Yodoeta down
- alkyl halides such as thiopropane, benzyl bromide, etc., arylpromide, alkenyl halides such as 1-promo 3-methyl-2-butene, or alkynyl halides such as propargyl lobe amide, 1-promo 2-butyne, etc.
- the reaction is carried out under the following conditions. It can be performed. In this case, it is preferable to use 1 to 2 equivalents of the compound (5a-2) with respect to the compound (5a).
- the reaction conditions for the substitution reaction are not particularly limited, but, for example, in a solvent such as dimethylsulfoxide / N, N-dimethylformamide, ⁇ -methinolepyrrolidone, dioxane, tetrahydrofuran, toluene, etc.
- a solvent such as dimethylsulfoxide / N, N-dimethylformamide, ⁇ -methinolepyrrolidone, dioxane, tetrahydrofuran, toluene, etc.
- the reaction can be carried out at a temperature of 0 ° C. to 150 ° C. in the presence of a base such as noresylylamide and potassium bistrimethyl
- X 2 is introduced an optionally substituted C 6 _ also.
- the 5- to 10-membered heteroaryl group optionally having an aryl group or a substituent (5a-2) include, for example, arylboronic acid or The reaction can be carried out using heteroarylboronic acid or the like. In this case, it is preferable to use the compound (5a-2) in an amount of 1 to 10 equivalents to the compound (5a).
- reaction can be carried out at a temperature of 0 to 150 ° C. in the presence of a copper catalyst such as copper (I 1), copper chloride (11) or copper (II) iodide. In this case, it is preferable to use 0.1 to 2 equivalents of the copper catalyst based on the compound (5a).
- a copper catalyst such as copper (I 1), copper chloride (11) or copper (II) iodide. In this case, it is preferable to use 0.1 to 2 equivalents of the copper catalyst based on the compound (5a).
- This step is a step of reacting compound (6a) with compound (7a) to obtain compound (8a).
- the compound (7a) is preferably used in an amount of 1 to 4 equivalents based on the compound (6a).
- reaction conditions are not particularly limited, for example, in a solvent such as N, N-dimethylformamide, N-methylpyrrolidone, methanol, ethanol, 1,4-dioxane, acetonitrile, toluene, xylene, or no solvent.
- a solvent such as N, N-dimethylformamide, N-methylpyrrolidone, methanol, ethanol, 1,4-dioxane, acetonitrile, toluene, xylene, or no solvent.
- a solvent such as N, N-dimethylformamide, N-methylpyrrolidone, methanol, ethanol, 1,4-dioxane, acetonitrile, toluene, xylene, or no solvent.
- compound (6a) and compound (7a) are mixed in the presence or absence of a base such as triethylamine, sodium hydrogen carbonate, or potassium carbonate, and mixed at a temperature of 0
- a substituent is introduced into the 2-position of the compound (8a) by subjecting the compound (8a) to a substitution reaction with the compound (8a-2). This is the step of obtaining
- a compound (8a-2) the presence of a suitable base represented by the formula I ⁇ -M 1 (wherein R 1 and M 1 have the same meanings as defined above, respectively)
- a suitable base represented by the formula I ⁇ -M 1 (wherein R 1 and M 1 have the same meanings as defined above, respectively)
- Any compound that can be a nucleophilic reactant in the presence or absence of the compound may be used, but preferred examples include alkyl alcohols such as methanol, ⁇ -propanol, isopropanol and benzyl alcohol, and aryl alcohols such as phenol and salicylamide.
- Alkylamines such as ammonia, methylamine, dimethylamine, and getylamine; arylamines such as aniline; alkylmercaptans such as methanethiol and o-butylmercaptan; arylmercaptans such as thiophenol; and other organic lithium reactants; Grignard reagents, organocopper reagents and the like can be mentioned.
- the compound (8a-2) is 1 to L to the compound (8a): 0 equivalent or 5 to 10 by weight ratio. It is preferable to use 0 times.
- reaction solvent acetonitrile, N, N-dimethylformamide, N-methylpyrrolidone, 1,4-dioxane, tetrahydrofuran, dimethoxyethane, methanol / ethanol, etc. can be used.
- the reaction can be carried out in the presence of a base or in the absence of a base.
- a base lithium hydroxide, sodium hydroxide, hydroxide hydroxide, lithium carbonate, Sodium carbonate, potassium carbonate, cesium carbonate, lithium hydride, sodium hydride, potassium hydride, butyllithium, methyllithium, dimethylbistrimethylsilylamide, sodium bistrimethylsilylamide, potassium bistrimethylsilylamide, triethylamine, etc.
- the reaction can be carried out at a temperature of 0 ° C to 150 ° C.
- a compound (8 a- 2) M 1 a is M g C l, in M g B r, S n ( R z) 3 (wherein, the R z same as defined above
- the compound (8a) can be reacted with a compound (meaning meaning) to obtain a compound (9a).
- the compound (8a-2) is used in an amount of 1 to 50 equivalents to the compound (8a).
- reaction solvent acetonitrile, N, N-dimethylformamide, N-methylpyrrolidone, 1,4-dioxane, tetrahydrofuran, dimethoxyethane and the like can be used.
- the metal catalyst examples include a palladium catalyst and a copper catalyst.
- the palladium insect medium examples include tetrakistripheninolephosphine palladium, palladium acetate, dibenzylideneacetone palladium, and the like.
- the catalyst Iowidai copper and the like can be used.
- the metal catalyst is preferably used in an amount of 0.01 to 2 equivalents to the compound (8a).
- the reaction can be carried out in the presence of an organophosphorus ligand.
- the reaction is carried out in the presence of an organic phosphorus-based ligand such as ortho tolyl phosphine, diphenylphosphinophene or the like.
- the organic ligand is preferably used in an amount of 1 to 5 equivalents to the metal catalyst.
- the reaction can be carried out in the presence of a base or in the absence of a base, but the reaction is carried out in the presence of a base # ⁇ , and as the base, lithium 7 oxide, sodium hydroxide, 7R oxidizing room, lithium carbonate, Sodium carbonate, potassium carbonate, cesium carbonate, lithium hydride, sodium hydride, lithium hydride, potassium phosphate, lithium bistrimethylsilylamide, sodium bistrimethylsilylamide, potassium bistrimethylsilylamide, triethylamine and the like can be used.
- the reaction can be carried out at a reaction temperature of 0 to 150 ° C.
- deprotection can be performed using an anhydrous hydrogen chloride methanol solution, an anhydrous hydrogen chloride ethanol solution, an anhydrous hydrogen chloride dioxane solution, trifluoroacetic acid or formic acid.
- Step B 1 This step is a step of obtaining a compound (2b) by protecting the 9-position amino group of the compound (lb) (compound 5a in Production Method A).
- the reaction can be carried out under the reaction conditions for introducing a protecting group, which are generally used for the reagent, according to the reagent for protecting the amino group to be used.
- a reagent for protecting an amino group a reagent generally used for introducing an amino-protecting group can be used.
- chloromethylpiparate or the like can be used.
- the protecting reagent is for compound (lb) :! It is preferable to use an amount of ⁇ 2 equivalents.
- the reaction solvent the reaction can be carried out using acetate, N, N-dimethylformamide, N-methylpyrrolidone, 1,4-dioxane, tetrahydrofuran, dimethoxyethane, etc., and preferably N, N-dimethylformamide Amides can be used.
- the reaction can be performed in the presence of a base.
- a base in this case, cesium carbonate, lithium carbonate, sodium carbonate, potassium carbonate, sodium hydrogen hydride and the like can be used, and preferably, sodium hydride can be used.
- the base is preferably used in an amount of 1 to 5 equivalents based on compound (lb).
- the reaction can be carried out at a temperature of 0 ° C. to 150 ° C., preferably at room temperature.
- This step is a step of reacting the compound (2b) with the compound (2b-2) to obtain a compound (3b).
- the reaction conditions the same conditions as in Production method A [Step A6] can be applied.
- the reaction conditions vary depending on the protecting group used.
- the protecting group is a bivalyloxymethyl group
- sodium methoxide, sodium hydride, 1,8-diazabicyclo is used in methanol or a mixed solution of methanol and tetrahydrofuran.
- the reaction can be performed at 0 ° C to 150 ° C in the presence of a base such as 7-indene. This ⁇ ⁇ base is preferably used in an amount of 0.1 to 2 equivalents to compound (3b).
- the compound (4b) and the compound (4b-2) are subjected to a substitution reaction to introduce a substituent into the amino group at the 9-position of the compound (4b), thereby obtaining the compound (5b). It is a process of obtaining.
- the reaction conditions the same conditions as in Production Method A [Step A5] can be applied.
- the compound (5b) is subjected to a substitution reaction with the compound (5b-2) to introduce a substituent at the 2-position of the compound (5b), thereby obtaining a compound (6b). is there.
- the reaction conditions the same conditions as in Production method A [Step A7] can be applied.
- the reaction conditions are not particularly limited, for example, in a solvent such as N, N-dimethinoleformamide, N-methylpyrrolidone, methanol, ethanol, 1,4-dioxane, acetonitrile, toluene, xylene, or without solvent
- a solvent such as N, N-dimethinoleformamide, N-methylpyrrolidone, methanol, ethanol, 1,4-dioxane, acetonitrile, toluene, xylene, or without solvent
- a base such as triethylamine, sodium bicarbonate, or carbonated lime
- This step is a step of obtaining a conjugate (4c) by reducing the nitro group of the compound (3c).
- the reaction conditions are not particularly limited.
- the catalytic reduction can be performed using a metal catalyst in a hydrogen atmosphere or in the presence of 2 to 3 equivalents of hydrazine.
- a reaction solvent methanol, ethanol, N, N-dimethylformamide, tetrahydrofuran, 1,2-dimethoxyethane, 1,4-dioxane, water, or a mixed solvent thereof can be used.
- the metal catalyst palladium carbon, platinum oxide, Raney nickel or the like can be used.
- the metal catalyst is preferably used in an amount of 0.5 to 20% by mass relative to the compound (3c).
- the reaction can be carried out at a temperature of 0 ° C to 150 ° C.
- This step is a step of converting the compound (4c) into the compound (5c).
- the reaction conditions are not particularly limited, but include, for example, acetonitrile, tetrahydrofuran, ethanol, methanol / le, 1,4-dioxane, toluene, and toluene.
- a solvent such as silene
- a base such as triethylamine, sodium bicarbonate, or carbonated lime
- the reaction can be carried out at a temperature of 0 ° C. to 150 ° C. with Noresimidazonore. It is preferred to use ⁇ , 1 to 10 equivalents of N, N ; -disuccinimidyl carbonate.
- the compound (5c) is subjected to a substitution reaction with the compound (5c-2) to introduce a substituent into the amino group at the 7-position of the compound (5c).
- This is the step of obtaining.
- the reaction conditions the same conditions as in Production method A [Step A1] can be applied.
- This step is the 9-position Amino-protecting group P 2 of the compound (6 c) is deprotected to give compound compound (7 c).
- the reaction conditions vary depending on the protecting group used.
- a base such as sodium methoxide or sodium hydride in methanol or a mixture of methanol and tetrahydrofuran at 0 ° C to 150 ° C. It can be obtained by acting at temperature.
- This base is preferably used in an amount of 1 to 10 equivalents based on compound (6c).
- the compound (7c) is subjected to a substitution reaction with the compound (7c-2) to introduce a substituent into the amino group at the 9-position of the compound (7c).
- the reaction conditions the same conditions as in Production method A [Step A5] can be applied.
- This step is a step of reacting the compound (2c) with the amine (9c) to obtain the compound (10c). It is desirable to use 1 to 10 equivalents of this ⁇ 8 ⁇ amine (9c).
- the reaction conditions are not particularly limited, but the same conditions as in Production method C-1 [Step C2] can be applied.
- This step is for converting the compound (11c) into a cyclic urea (12c).
- the same conditions as in Production method C [Step C4] can be applied.
- the compound (12c) and the compound (12c-2) are subjected to a substitution reaction to introduce a substituent into the amino group at the 7-position of the compound (12c), thereby obtaining the compound (13c). It is about to get.
- the reaction conditions the same conditions as in Production method A [Step A1] can be applied.
- a protecting group such as T 2 b
- t-butoxide deer Lupo group carried out after [Step C11], followed by deprotection.
- the conditions for the deprotection reaction the same conditions as those for the deprotection shown in Production method A [Step A7] can be applied.
- This step is a step of reacting compound (Id) with compound (1d-2) to obtain compound (2d).
- reaction conditions the same conditions as in Production method A [Step A6] can be applied.
- the compound (2d) is substituted with the compound (2d-2) to introduce a substituent into the amino group at the 9-position of the compound (2d), thereby obtaining the compound (3d) It is a process.
- the reaction conditions the same conditions as in Production method A [Step A5] can be applied.
- This step is a step of reacting the compound (3d) with a halogenating agent to obtain a compound (4d).
- a halogenating agent for example, the same conditions as in Production method A [Step A2] can be applied.
- This step is a step of obtaining a compound (5d) by a hydrolysis reaction of the compound (4d).
- the reaction conditions the same conditions as in Production method A [Step A4] can be applied.
- a substituent is introduced into the 7-position amino group of the compound (5d) to obtain a compound (6d).
- the reaction conditions the same conditions as in Production method A [Step A1] can be applied.
- 4-ethoxy-3-nitropyridine hydrochloride (1e) [CAS No. 94602-04-7] is reacted with arylamine to obtain a disulfide compound (2e).
- the arylamine is preferably used in an amount of 1 to 20 equivalents based on the compound (1e).
- the reaction can be performed at a reaction temperature of 20 ° C to 150 ° C.
- a reaction solvent methanol, ethanol, water, a mixed solvent thereof or the like can be used.
- This step is a step of obtaining the compound (3e) by reducing it while chlorinating the compound (2e).
- Tin salts such as tin chloride can be used as the reducing agent.
- This reducing agent is preferably used in an amount of 4 to 20 equivalents based on compound (2e).
- Concentrated hydrochloric acid can be used as a solvent.
- the reaction can be performed at a reaction temperature of 20 ° C to 150 ° C.
- This step is for converting the compound (3e) into a cyclic urea (4e).
- the same conditions as in Production method C [Step C4] can be applied.
- This step is a step of reacting compound (4e) with compound (4e-2) to obtain compound (5e).
- reaction conditions the same conditions as in Production method A [Step A1] can be applied.
- reaction conditions are not particularly limited.
- osmium in a solvent such as tetrahydrofuran, 1,4-dioxane, 1,2-dimethoxyethane, 7 at 20 ° C. to 100 ° C.
- the compound (6e) can be obtained by the action of sodium periodate.
- a compound (6e) is reacted with (6e-2) to introduce a substituent into the amino group at the 1-position of the compound (6e).
- the same conditions as in Production method A [Step A5] can be applied.
- This step is a step of reacting the compound (1f) with the compound (1f-2) to obtain a compound (2f).
- the reaction can be performed at a reaction temperature of 20 ° C to 150 ° C.
- a reaction solvent methanol, ethanol, water, a mixed solvent thereof or the like can be used.
- the compound (1 f-2) is preferably used in an amount of 5 to 100 equivalents relative to the compound (If).
- This step is a step of obtaining a compound (3f) by reducing the compound (2f) while chlorinating the compound (2f).
- the same conditions as in Production method E [Step E2] can be applied.
- This step is a step of converting compound (3f) into compound (4f). Reaction conditions Therefore, the same conditions as in Production method C [Step C4] can be applied.
- a substituent is introduced into the 3-position amino group of the compound (4f) to obtain a compound (5f).
- the reaction conditions the same conditions as in Production method A [Step A1] can be applied.
- This step is a step of reacting the compound (5f) with the compound (5f-2) to obtain a compound (6f).
- the reaction conditions the same conditions as in Production method A [Step A6] can be applied.
- This step is a step of reacting the compound (6f) with a halogenating agent to obtain a compound (7f).
- a halogenating agent for example, the same conditions as in Production method A [Step A2] can be applied.
- This step is a step of reacting compound (7f) with a nucleophile in the presence of a catalyst and a base to obtain compound (8f).
- nucleophile a derivative of phenol or aniline can be used, and it is preferable to use 1 to 3 equivalents of the nucleophile with respect to the compound (7f).
- Cesium carbonate or the like can be used as the base, and the base is preferably used in an amount of 1 to 3 equivalents based on compound (7f).
- Copper catalyst and 2 of copper (I) chloride or the like as a catalyst, 2, 6, 6-tetramethyl ⁇ -. 3, can be used Putajion to 5, respectively 0.00 1-0 2 using the equivalent Is preferred.
- reaction solvent 1-methyl-1-pyrrolidone, N, N-dimethylformamide and the like can be used. The reaction can be performed at 20 ° C to 150 ° C.
- the above is a typical example of the method for producing the compound (I) according to the present invention.
- the solvent used also varies depending on the starting materials, reagents, etc., and it goes without saying that there is no particular limitation as long as it does not inhibit the reaction and dissolves the starting materials to some extent.
- the compound (I) according to the present invention is obtained as a free form, it can be converted into a salt or a hydrate thereof which may be formed by the compound (I) according to a conventional method.
- the compound (I) according to the present invention is obtained as a salt of the compound (I) or a hydrate of the compound (I): ⁇ It can be converted to a free form of the compound (I) according to a conventional method. .
- various isomers for example, geometric isomers, optical isomers based on asymmetric carbon, rotamers, stereoisomers, tautomers, etc.
- conjugated product (I) according to the present invention can be purified and purified by using ordinary separation means, for example, recrystallization, diastereomeric salt method, enzymatic resolution method, and various types of chromatography (for example, thin layer chromatography, column chromatography, gas chromatography, etc.). can do.
- the compound according to the present invention or a salt thereof or a hydrate thereof can be prepared by a commonly used method using tablets, powders, fine granules, granules, coated tablets, capsenoles, syrups, troches, inhalants, suppositories, etc. , Orchids, ointments, eye ointments, eye drops, nasal drops, ear drops, cataplasms, lotions and the like.
- Excipients can be used, and are generally formulated by mixing the components commonly used as raw materials for pharmaceutical preparations.
- the compound of the present invention or a pharmacologically acceptable After adding salts and excipients, binders, disintegrants, lubricants, coloring agents, flavoring agents, etc. as needed, powders, fine granules, granules, tablets, Coated tablets, capsules, etc.
- These components include, for example, animal and vegetable oils such as soybean oil, beef tallow, and synthetic dalyceride; hydrocarbons such as liquid paraffin, squalane, and solid paraffin; ester oils such as otatyldodecyl myristate and isopropyl myristate; setosteryl alcohol , Higher alcohols such as behenyl alcohol; silicone resins; silicone oils; polyoxyethylene fatty acid esters, sorbitan fatty acid esters, glycerin fatty acid esters, polyoxyethylene sorbitan fatty acid esters, polyoxyethylene hardened castor oil, and polyoxetylene.
- animal and vegetable oils such as soybean oil, beef tallow, and synthetic dalyceride
- hydrocarbons such as liquid paraffin, squalane, and solid paraffin
- ester oils such as otatyldodecyl myristate and isopropyl myristate
- setosteryl alcohol Higher alcohols such as behen
- Surfactants such as polyoxypropylene block copolymer; hydroxyethyl cenorose mouth, polyacrylic acid, canolepoxy vinyl polymer, polyethylene glycol, polyvinylpyrrolidone, methyl Water-soluble polymers such as rucellose; lower alcohols such as ethanol and isopropanol; polyhydric alcohols such as glycerin, propylene darico-one, dipropylene darico-one, and sonorebito / re; glucose and sucrose Sugars: inorganic powders such as silicic anhydride, magnesium aluminum silicate and aluminum silicate, and purified water.
- Excipients include, for example, lactose, corn starch, sucrose, dextrose, mannitol, sorbitol, crystalline cellulose, silicon dioxide, and the like
- binders include, for example, poly (vinylenoleanolone), poly (vinylinoleatenore), Ethylene 7 resenolerose, gum arabic, tragacanth, gelatin, shellac, hydroxypropylmethylcellulose, hydroxypropylcellulose, polyvinylpyrrolidone, polypropylene propylene glycol ethylene block polymer, medalmin, etc.
- starch starch, agar, gelatin powder, crystalline cellulose, calcium carbonate, sodium bicarbonate, calcium citrate, dextrin, pectin, carboxymethylcellulose 'calcium, etc.
- Lubricants such as magnesium stearate, tanolek, polyethylene glycol, silica, and hydrogenated vegetable oils are permitted to be added to pharmaceuticals as coloring agents.
- Cocoa is a flavoring agent. Powder, heart-shaped brain, aromasan, heart-shaped oil, dragon brain, cinnamon powder, etc. are used.
- these mallets and granules may be coated with sugar or other coatings as needed.
- a liquid preparation such as a syrup or an injection preparation
- the compound of the present invention or a pharmacologically acceptable salt thereof is required to contain a pH adjusting agent, a solubilizing agent, an isotonic agent and the like. Add a solubilizer, stabilizing agent, etc. according to the formula, and formulate it in a usual manner.
- the method for producing the external preparation is not limited, and it can be produced by an ordinary method. That is, as raw materials used in the formulation, various raw materials usually used for pharmaceuticals, quasi-drugs, cosmetics, and the like can be used. Use »J As raw materials, specifically, animal and vegetable oils, mineral oils, ester oils, waxes, higher alcohols, fatty acids, silicone oils, surfactants, phospholipids, alcohols, polyhydric alcohols , 7 soluble polymers, clay minerals, purified water, etc., and if necessary, add pH adjusters, antioxidants, chelating agents, antiseptic / antifungal agents, coloring agents, fragrances, etc. However, the raw material of the external preparation according to the present invention is not limited to these.
- the amount of the base material to be added is an amount that usually gives a concentration set in the production of an external preparation.
- a salt thereof, or a hydrate thereof When administering the compound according to the present invention, a salt thereof, or a hydrate thereof, the form is not particularly limited, and oral administration or parenteral administration may be performed by a commonly used method.
- oral administration or parenteral administration may be performed by a commonly used method.
- the dose of the medicament according to the present invention can be appropriately selected according to the degree of symptoms, age, sex, body weight, dosage form, type of salt, specific type of disease, and the like.
- the dosage depends on the type of disease, the degree of symptoms, the patient's age, gender, and sensitivity to the drug. Normally, as an adult, about 0.03-10 O Omg, preferably 0.1-50 Omg, and more preferably 0.1-1 O Omg per day as an adult Administer several divided doses.
- the injection is usually about l ⁇ g / kg-300O / zgZkg, preferably about 3 / zgZkg-1000 / ig / kg.
- the compound according to the present invention can be produced, for example, by the method described in the following examples. However, these are merely examples, and the conjugate according to the present invention is not limited to the following specific examples in any case.
- the term “purification by reversed-phase high-performance liquid chromatography” described in this Example means “reverse-phase using acetonitrile-aqueous mobile phase (containing 0.1% trifluoroacetic acid)” unless otherwise specified. Means high performance liquid chromatography purification.
- Example 1 7- (2-butyninole) -1,2-methoxy-19-methyl-6-1 (piperazin-1-yl) -17,9-dihydropurine-18-one trifluoroacetate
- the obtained organic layer was washed sequentially with water and a saturated diet, and then dried over anhydrous magnesium sulfate.
- Example 3 4- [7- (2-Petinole) 12-chloro-9-methyl-8-oxo-8,9-dihydro-1 7H-purine 6-yl] pidazine-11-canolepo
- the same treatment as in Example 3 was carried out using 10 mg of t-butyl acid acid and dimethylamine 301 in place of getylamine to obtain 5.96 mg of the title compound.
- Example 4 The same procedure as in Example 4 was carried out except that 30 ⁇ l of aqueous ammonia (28 to 30%) was used in place of gentinoleamine to obtain 0.84 mg of the title compound.
- Example 7 7- (2-Petinole) 1-2-isopropoxy-19-methyl-16- (piperazine-11-yl) -7,9-dihydropurin-8-one-trifluoroacetate
- Example 8 7- (2-butyninole) -12-hydroxy-19-methyl-16- (piperazin-11-yl) -1,7-9-dihydropurine-18-one trifluoroacetate
- Example 9 7- (2-butulyl) -1-methylsulfanyl-1-9-methyl-6- (piperazine-11-yl) -17,9-dihydropurine-1-8-one-trifluoroacetic acid
- Example 10 7- (2-Petinole) 1-2-ethoxy-1 9-methinole-6- (piperazin-1-yl) -17, —9-dihydropurine-18-one—trifluoroacetate
- Example 1 1 2—Benzyloxy 7— (2-ptur) —9—Methynole 6— (pi Perazine (11-yl)-7, _9-dihydropurine-1-one _ trifluoroacetate.
- Example 10 The same procedure was followed as in Example 10 except that 30 ⁇ l of benzyl alcohol was used in place of ethanol to give 11.28 mg of the title compound.
- Example 1 2 7— (2-petit-norre) 1-9-methyl-2-phenoxy-1 6— (piperazine-1 1 ⁇ T) 1-7, —9-dihydropurine-18-one-trifluoroacetate
- Example 1 3 [7— (2-Petinole) —9-methyl-8-oxo-16- (piperazine-1-inole) -8,9-dihydro-7H-purine-1-yloxy] benzodi Tolyl trifluoroacetate
- Example 14 The same procedure as in Example 14 was carried out except that salicylamide was replaced with allylic alcohol 301 to obtain 1.2 Omg of the title compound.
- Example 1 7- (2-butulyl) -1-2- (2-butynyloxy) -19-methinole 6- (piperazine-11-yl) -7,9-dihydropurine-1-8-one trifrenole mouth acetate
- Example 14 The same procedure as in Example 14 was repeated, except that 30 ⁇ l of 2-butyn-1-ol was used in place of salicylamide, to give 1.20 mg of the title compound.
- Example 1 7 2 1-Pendinoleamino-7- (2-butulyl) -19-methyl-16- (piperazine-11-yl) -7, _9-dihydropurine-18-one-trifluoroacetate
- Example 18e The same procedure was followed as in Example 18e), except that 1-bromo-1-methyl-2-butene (15 ⁇ l) was used in place of 1-bromo-12-pentyne to give the title compound 1 25 mg were obtained.
- Example 20 7- (2-Ptul) -12-chloro-9-methyl-6- (piperazine-11-yl) -1,7,9-dihydropurine-18-one trifluoroacetate
- Example 18e The same treatment as in Example 18e) was carried out in the same manner as in Example 18e) except that 1-promo 2-peptene was used instead of 1-promo 2-pentyne, to give 1.84 mg of the title compound. .
- Example 2 1-7 Venginole 2—Black mouth 9—Methinole 61 (Piperazine 1-1) -7), 9-dihydropurine 8-one-trifluorate
- Example 27 In a), (piperidin-3-yl) carboxamic acid t-butyl The same treatment as in Example 27 was carried out in the same manner as in Example 27, except that methyl (piperidine-13-inole) t-butyl carbamic acid (compound 26) was used instead of the ester to obtain 4.16 mg of the title compound.
- Example 30 7- (2-Putinyl) -1-2-chloro-1-6- (piperazine-11-yl) -1-9-propyl-1 7 ⁇ 9-dihydropurine-8-one-trifluoroacetate
- Example 3 [7- (2-Petitinole) 12-chloro-1-8-oxo-16- (piperazin-11-yl) -17,8-dihydropurine-19-yl] acetic acid-trifluoro Acetate
- Example 29 d the same treatment as in Example 29 d) was performed using 20 ⁇ l of bromoacetic acid t -butyl ester instead of 2- (bromomethinole) benzonitrile, to give 3.55 mg of the title compound. Obtained.
- Example 29 d This was treated in the same manner as in Example 29 d), except that 5 mg of 2-bromoacetamide was used in Example 29 d) instead of 2- (bromomethyl) benzonitrile, to give 4.7 lmg of the title compound.
- Example 29 In Example 9d), the same treatment as in Example 29 d) was carried out except that (2-promoethyl) benzene 20/1 was used instead of 2- (bromomethyl) benzonitrile, to give 5.12 mg of the title compound. Got.
- Example 29 In 9d), 2- (4-bromophenyl) ethyl ester 1 Omg of methanesulfonic acid was used instead of 2- (bromomethyl) benzonitrile in the same manner as in Example 29d) to give the title compound 1 .56 mg were obtained.
- Example 29 In Example 9d), the same treatment as in Example 29 d) was performed, except that benzylbutamide 201 was used instead of 2- (bromomethyl) benzonitrile, to obtain 1.23 mg of the title compound. .
- Example 29 d The same procedure was followed as in Example 29 d), except that 4-chlorobutyronitrile 201 was used in place of 2- (bromomethyl) benzonitrile, to give 5.8 Omg of the title compound.
- 4-chlorobutyronitrile 201 was used in place of 2- (bromomethyl) benzonitrile, to give 5.8 Omg of the title compound.
- Example 3 8 7- (2-Putinyl) 1-2-chloro-1-9-cyclopropylmethyl-6- (piperazine-11-yl) -7,9-dihydropurin-1-8_one_triphnoleo
- the residue was dissolved in 0.25 ml of methanol and 0.25 ml of tetrahydrofuran, and 0.5 ml of aqueous ammonia and 0.3 ml of 30% aqueous hydrogen peroxide were added thereto. Stir the reaction solution for 12 hours at room temperature. After stirring, the reaction mixture was concentrated. The residue was dissolved in trifluoroacetic acid, and the reaction solution was stirred at room temperature for 5 minutes and concentrated. The residue was purified by reverse phase high performance liquid chromatography to obtain 1.15 mg of the title compound.
- Example 40 [7- (2-Petinole) -12-chloro-8-oxo-16- (piperazin-1-inole) -17,8-dihydropurine-19-yl] acetic acid methinooleestenole trifluoroacetic acid salt
- the obtained organic layer was concentrated, the residue was dissolved in methanol (0.5 ml), and cesium carbonate (1 Omg) was added thereto. 70. After stirring the reaction solution at C for 18 hours, the reaction was concentrated. The residue was dissolved in trifluoroacetic acid, and the reaction solution was stirred at room temperature for 5 minutes and then concentrated. The residue was purified by reversed-phase high performance liquid chromatography to obtain 2.85 mg of the title compound.
- Example 41 7 (2-petit-nore) 1-2-chloro-9-pheninole 6— (piperazin-1-1-inole) 17,9-dihydropurine-18-one-trifluoroacetate
- Example 41 The same procedure as in Example 41 was repeated, except that 1-mg of 41-cyanophenylporonic acid was used in place of phenylporonic acid in Example 41 to give 1.57 mg of the title compound.
- Example 41 The same procedures as in Example 41 were repeated, except that 1-Omg of 3-furanboronic acid was used in place of phenylporonic acid, to give 1.23 mg of the title compound.
- Example 41 The same procedure was followed as in Example 41, except that 10 mg of 3-thiophenboronic acid was used in place of phenylporonic acid, to give 3.57 mg of the title compound.
- Example 41 The same procedure was followed as in Example 41, except that pyridine-13-boronic acid (10 mg) was used in place of phenylporonic acid, to give the title compound (3.44 mg).
- Example 47 The same procedure as in Example 47 was repeated, except that 20 ⁇ l of 1-bromo-2-butyne was used in place of alipromide, to give 1.99 mg of the title compound.
- Example 51 7- (2-butul) -1-9-cyclopropylmethyl-2-methoxy-6- (piperazine-1-yl) -17,9-dihydropurine-18-one trifnoreo mouth acetate
- Example 47 was treated in the same manner as in Example 47 except that bromomethylcyclopropane 25 ⁇ 1 was used instead of arylpromide, to give 2.46 mg of the title compound. W> mle (ESI) 357 (M + H) +
- Example 52 7- (2-Putinyl) -12-methoxy-6- (piperazine-11-yl) -19-propyl-17-9-dihydropurine-18-one-trifluoroacetate
- Example 47 The same procedures as in Example 47 were repeated, except that, instead of aryl bromide, 11-propane 251 was used, the title compound was obtained in an amount of 3.9 Omg.
- Example 47 The same procedure was followed as in Example 47, except that propargyl promide (25 ⁇ l) was used instead of aryl promide, to give 2.63 mg of the title compound.
- Example 54 7- (2-Putinyl) -2-methoxy-19-phenyl-6- (piperazine-1-inole) -17, —9-dihydropurine-18-one-trifluoroacetate
- the obtained organic layer was concentrated, the residue was dissolved in 0.5 ml of methanol, and 10 mg of cesium carbonate was added thereto. After stirring the reaction mixture at 70 ° C for 18 hours, the reaction mixture was concentrated. The residue was dissolved in trifluoroacetic acid, and the reaction solution was stirred at room temperature for 5 minutes and concentrated. The residue was purified by reverse phase high performance liquid chromatography 1 to obtain 2.88 mg of the title compound.
- Example 54 The same procedure was followed as in Example 54, except that pyridine-13-polonic acid (10 mg) was used instead of phenylboronic acid, to give 2.29 mg of the title compound.
- Example 56 7- (2-butchur) -19- (furan-3-yl) -12-methoxy-6- (piperazine-11-yl) -7,9-dihydropurine-1-8-one trifrenole mouth acetate
- Example 5 7- (2-butul) -1-9- (thiophene-3-yl) -2-methoxy 6- (piperazine-11-yl) -17,9-dihydropurine-8-one trif Luo p acetate
- Example 6 1f the same treatment as in Example 6 1f) was carried out except that 1-mg of phenylpropoic acid was used instead of pyridine-13-poronic acid, to give 6.94 mg of the title compound.
- Example 6 1f the same treatment as in Example 6 1f) was carried out except that 1-mg of phenylpropoic acid was used instead of pyridine-13-poronic acid, to give 6.94 mg of the title compound.
- Example 6 If), 1 Omg of furan-3-polonic acid was used in place of pyridine-13-boronic acid, and the reaction was carried out at a reaction temperature of 50 ° C in the same manner as in Example 6 1f). 1.28 mg of the compound was obtained.
- Example 6 1 f 10 mg of 2-methoxy-15-pyrimidineporonic acid was used in place of pyridine-1-boronic acid, the reaction temperature was set to 50 ° C., and the reaction time was set to 48 hours. The same treatment as in f) was performed to obtain 2.52 mg of the title compound.
- Example 65 7- (2-Petinole) -19- (2-chloro-pyridine-14-yl) -6- (piperazine-11-yl) -17,9-Dihydropurin-8-one trifrenole Mouth acetate W
- Example 61 f 1-Omg of 2-chloropyridin-4-boronic acid was used in place of pyridine-13-polonic acid, the reaction temperature was 90 ° C, and the reaction time was 48 hours. ) To give 4.48 mg of the title compound.
- Example 6 lb 5-amino-1-methyl-1H-pyridin-2-one was used in place of 3-aminopropio-tolyl, and treated in the same manner as in Examples 6 lb) to d) to give the title compound. Obtained.
- Example 69 The same procedure as in Example 69 was repeated, except that propargylbumid 251 was used in place of arylpromide, to give 3.7 lmg of the title compound.
- Example 69 2-bromoacetamide (20 mg) was used in place of arylpromide, and treated in the same manner as in Example 69 to obtain 7.55 mg of the title compound.
- Example 72 7- (2-Petinole) -19-cyclopropylmethyl-6-1 (piperazin-1-yl) -17, —9-dihydropurine-18-one-trifluoroacetate 0 1
- Example 69 bromomethylcyclopropane 251 was used in place of arylpromide, and the same treatment was carried out as in Example 69 to obtain 7.28 mg of the title compound.
- Example 6 9 using 4-Xia no benzylbromide Ma id 2 0 mg instead of ⁇ Lil Puromaido, was treated in the same manner as in Example 6-9, the title compound 9. 5 6 mg obtained c M / e ( ESI) 388 (M + H) +
- Example 7 4 7— (2-butynyl) 19-phenethyl-16- (piperazine-11-yl) -17,9-dihydropurine-18-one trifluoroacetate
- Example 69 phenethylpromide 251 was used in place of aryl bromide, and the same treatment as in Example 69 was carried out to obtain 7.14 mg of the title compound.
- Example 75 7- (2-Petinole) -6- (piperazine-11-yl) -17,9-dihydropurine 8-triene-trinole-chloroacetate
- 6-Black mouth purine [CAS No. 87-42-3] 7.73 g of ethanol 10 Oml solution, diisopropylethylamine 26.1 ml and piperazine-1 1-carboxylic acid t-butyl ester 11.16 g was calorie, and the mixture was heated under reflux for 16 hours. The solvent was concentrated under reduced pressure, and the residue was suspended in 200 ml of water. The precipitate was collected by filtration, and washed twice with 50 ml of water and twice with 50 ml of t-butyl methinooleate to obtain 13.99 g of the title compound.
- Example 76 e 5 ⁇ l of 1-promo 3-methyl-12-butene was used in place of 1-promo 2-putin, and treated in the same manner as in Example 76 e) to obtain the title compound 4.3m g was obtained.
- Example 76e the same treatment as in Example 76e) was carried out using 5 ⁇ l of benzylbutamide instead of 1-promo 2-putin to obtain 4.8 mg of the title compound.
- the solid collected by filtration was dissolved in 500 ml of dichloromethane, washed successively with 200 ml of 5% aqueous sodium hydrogen carbonate and 200 ml of water, combined with an organic layer of ethyl acetate, and dried over anhydrous magnesium sulfate. The organic layer was filtered, concentrated under reduced pressure, and the residue was recrystallized from toluene to obtain 2.04 g of the title compound.
- the reaction solution was cooled, t-butyl methyl ether was added, and the mixture was filtered through celite.
- the celite was washed with 25 ml of ethyl acetate, and the organic layers were combined and washed sequentially with 10 ml of 2N hydrochloric acid, 10 ml of 0.5N hydrochloric acid, 10 ml of 1N aqueous sodium hydroxide solution and 10 ml of a saturated aqueous solution of sodium salt and sodium chloride. It was dried over anhydrous magnesium sulfate, filtered, and concentrated under reduced pressure. The residue was purified by reversed-phase high performance liquid chromatography to obtain 0.023 g of the title compound.
- Methyl- (3-nitropyridine-41-yl) amine 7. Add 90 ml of a 150 ml solution of concentrated hydrochloric acid in O Og. C., 52.2 g of tin (II) chloride dihydrate was added, and the mixture was ripened at 90 ° C for 30 minutes. The reaction solution was cooled to 0 ° C, 700 ml of ice / water was added, and the mixture was stirred for 30 minutes. ⁇ was concentrated under reduced pressure, 700 ml of a saturated methanol solution of ammonia was added to the residue, and the mixture was stirred at 5 ° C. for 15 hours.
- reaction solution was cooled, 2 ml of dichloromethane and 3 ml of trifluoroacetic acid were added, and the mixture was stirred for 2 hours. After the solvent was concentrated under reduced pressure, the residue was purified by reversed-phase high-performance liquid chromatography to obtain 0.007 g of the title compound.
- DPP-IV obtained from porcine kidney was dissolved to 1 OmUZmL, and this was dissolved in 110 mL. ⁇ 1 was added. Further, after adding 15 ⁇ 1 of the drug obtained in the above Example, the mixture was incubated at room temperature for 20 minutes, and G 1 yPron itroanilide dissolved in 2 mM was added to 25 1 (final concentration 0.33). In addition, the enzymatic reaction was started. The reaction time was 20 minutes, and 25 1 of a 1 N phosphoric acid solution was added to stop the reaction. The absorbance at 405 iim was measured to determine the enzyme reaction inhibition rate, and the IC 50 was calculated. Table 1 shows the results.
- Test compounds were suspended in 0.5% methylcellulose (MC) solution at the doses shown in Table 2 below.
- a suspension of this test compound and NVP DPP 728 (US Patent No. 6011 155) or a vehicle control group of 0.5% MC ⁇ was orally administered at a volume of 10 mL / kg, and 30 minutes later.
- the glucose solution was orally administered in a volume of 1 OmLZkg.
- Glucose was administered orally at a dose of 2 g / kg.
- test substance just before administration of NVP DPP 728, and immediately before and after administration of glucose, and at 30, 60, and 120 minutes after the administration, the tail vein of the mouse is injured with a razor blade and bled slightly under anesthesia.
- Collect 10 // L of blood and immediately mix with 0.6M perchloric acid 140 // L.
- 1,3-dihydro-1-midazole condensate showing DPPIV inhibitory action A fused compound could be provided.
- the 1,3-dihydro-imidazole fused ring compound in the present invention is, for example, a therapeutic agent for diabetes, obesity, hyperlipidemia, AIDS, osteoporosis '', a therapeutic agent for gastrointestinal disorders, angiogenesis. It is useful as a therapeutic agent, infertility treatment agent, anti-inflammatory agent, anti-allergic agent, immunomodulator, hormonal modulator, anti-rheumatic agent, cancer treatment agent, etc., and is useful as a prophylactic agent.
Landscapes
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Diabetes (AREA)
- Hematology (AREA)
- Obesity (AREA)
- Endocrinology (AREA)
- Immunology (AREA)
- Physical Education & Sports Medicine (AREA)
- Rheumatology (AREA)
- Reproductive Health (AREA)
- Virology (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Tropical Medicine & Parasitology (AREA)
- Communicable Diseases (AREA)
- Oncology (AREA)
- Pain & Pain Management (AREA)
- Heart & Thoracic Surgery (AREA)
- Biomedical Technology (AREA)
- Child & Adolescent Psychology (AREA)
- Molecular Biology (AREA)
- Emergency Medicine (AREA)
- Gynecology & Obstetrics (AREA)
- Pregnancy & Childbirth (AREA)
- Pulmonology (AREA)
- AIDS & HIV (AREA)
Description






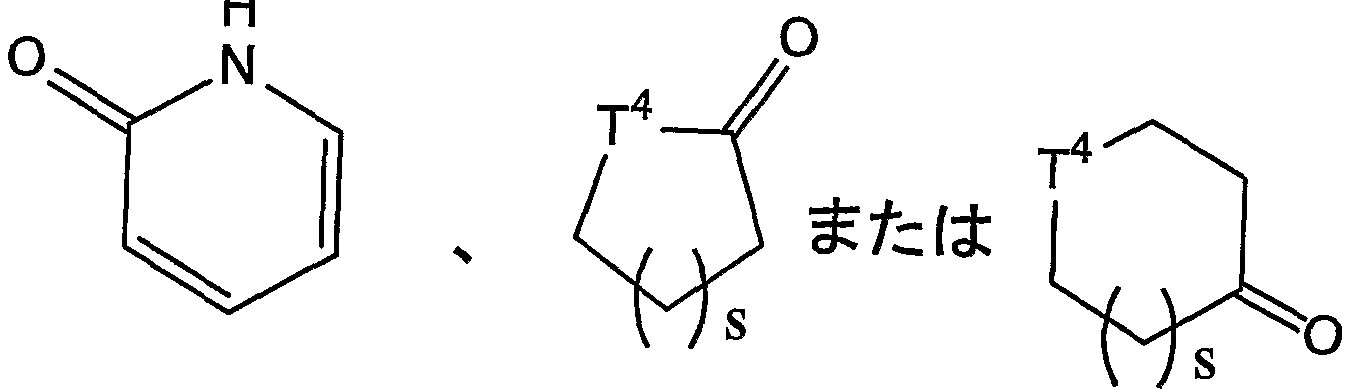




















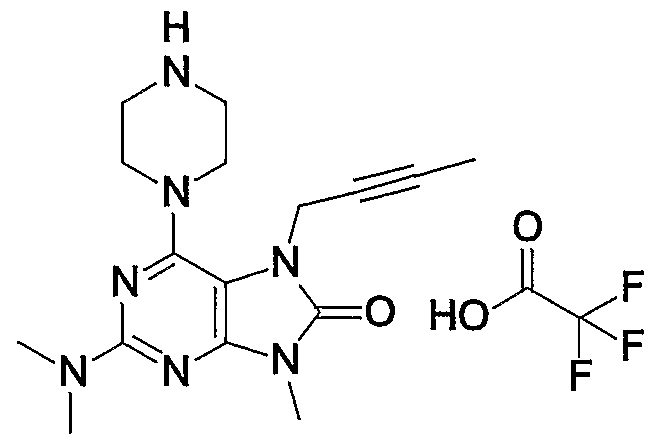
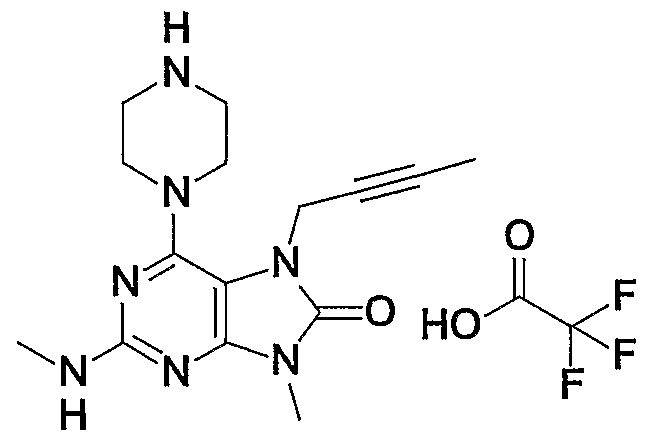
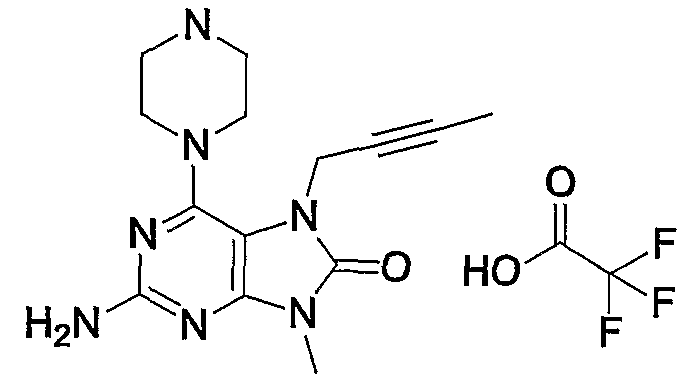







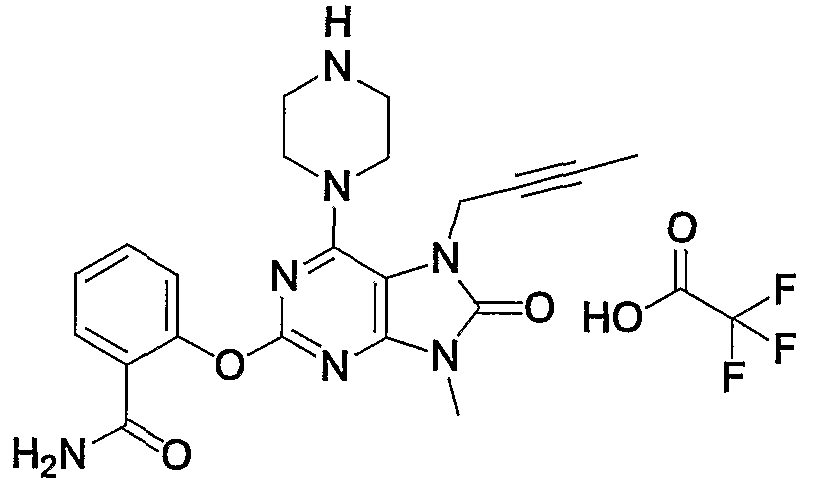
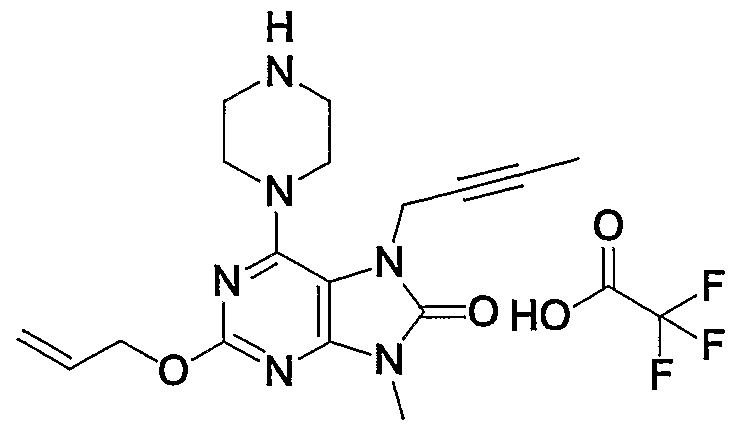


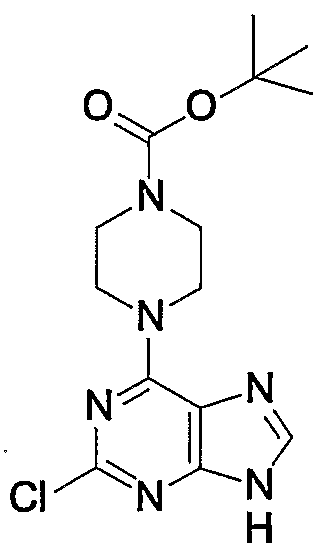












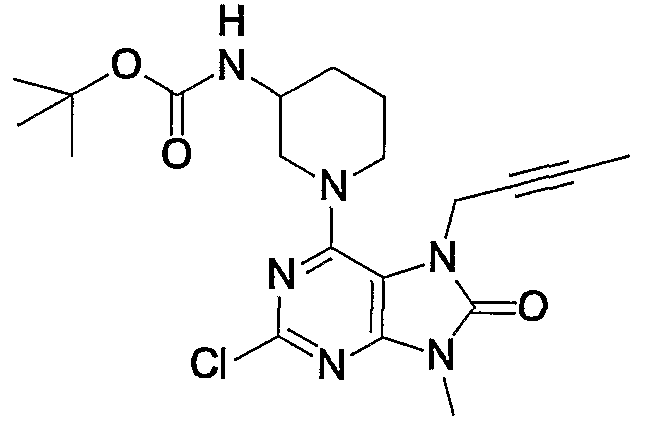












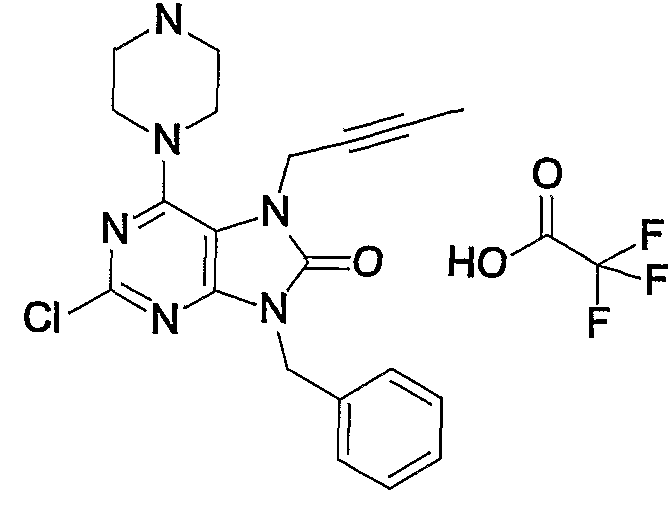




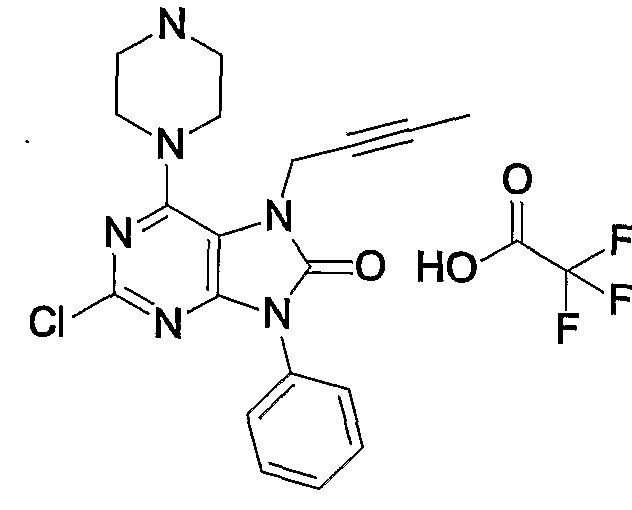
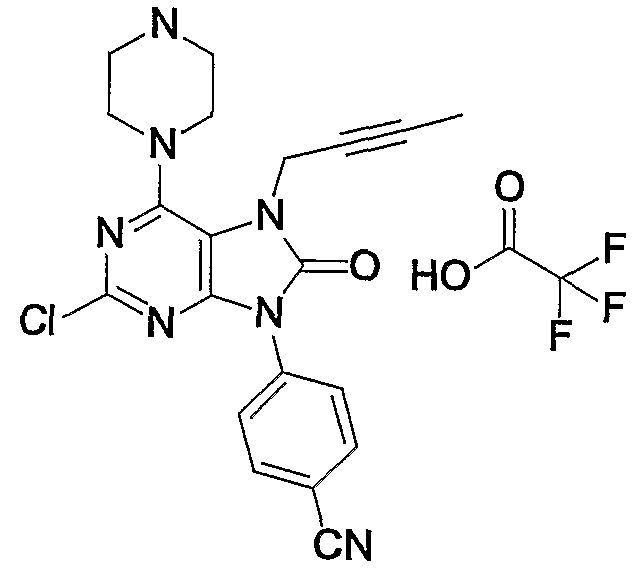












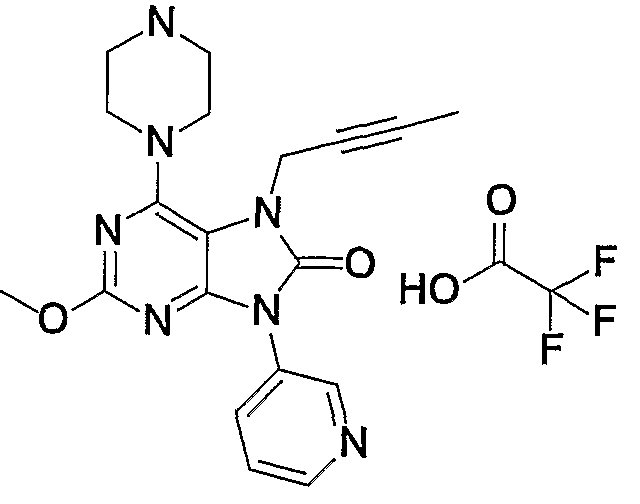






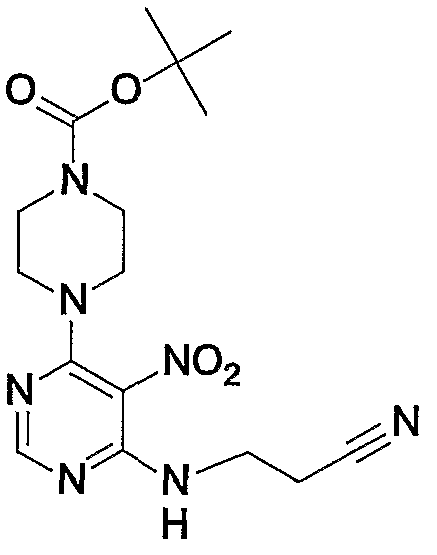

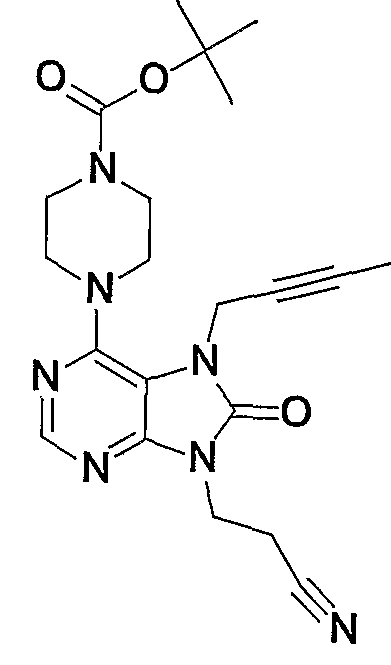
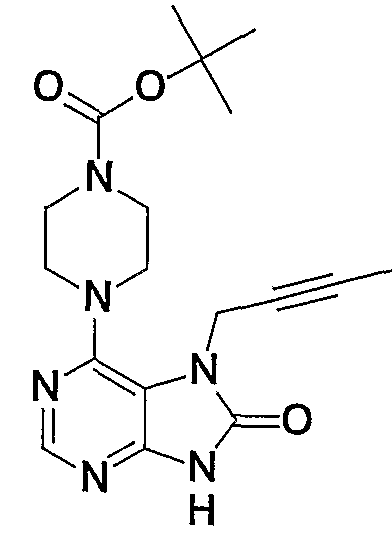
















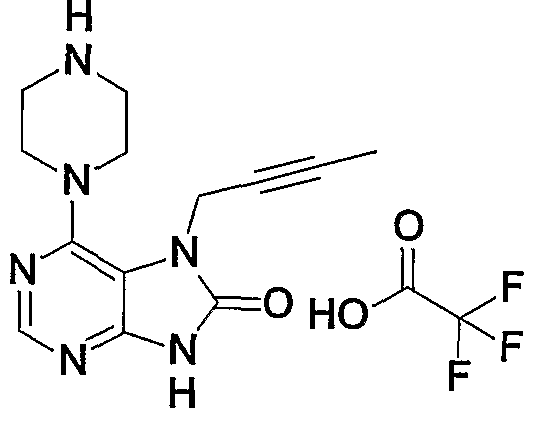





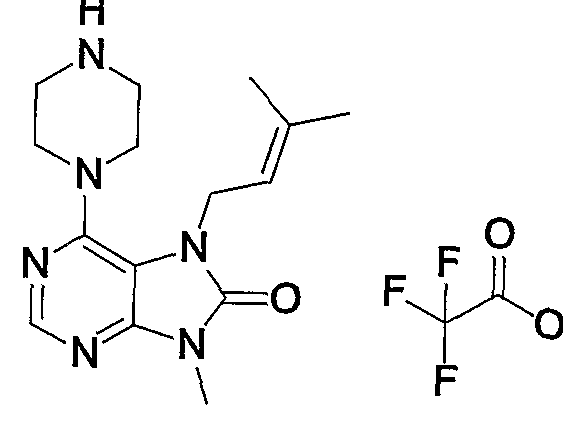































Claims





Priority Applications (11)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| HK06106475.5A HK1086556B (en) | 2002-12-04 | 2003-12-02 | Fused 1,3-dihydro-imidazole ring compounds |
| NZ540495A NZ540495A (en) | 2002-12-04 | 2003-12-02 | 1,3-Dihydro-imidazole fused-ring compound |
| BR0316991-0A BR0316991A (pt) | 2002-12-04 | 2003-12-02 | Compostos de anel de 1,3-diidro-imidazol fundido |
| JP2004556886A JP4279784B2 (ja) | 2002-12-04 | 2003-12-02 | 1,3−ジヒドロ−イミダゾール縮合環化合物 |
| MXPA05005983A MXPA05005983A (es) | 2002-12-04 | 2003-12-02 | Compuestos de anillo fusionado 1,3-dihidroimidazol. |
| CA002507763A CA2507763A1 (en) | 2002-12-04 | 2003-12-02 | Fused 1,3-dihydro-imidazole ring compounds |
| EP03812368A EP1568699A4 (en) | 2002-12-04 | 2003-12-02 | 1,3-DIHYDROIMIDAZOLE CONNECTION WITH CONDENSED RING |
| US10/537,227 US7524847B2 (en) | 2002-12-04 | 2003-12-02 | Fused 1,3-dihydro-imidazole ring compounds |
| AU2003302657A AU2003302657B8 (en) | 2002-12-04 | 2003-12-02 | Fused 1,3-dihydroimidazole ring compounds |
| IL168784A IL168784A (en) | 2002-12-04 | 2005-05-25 | Fused 1,3-dihydroimidazole ring compounds and pharmaceutical compositions comprising the same |
| NO20053246A NO20053246L (no) | 2002-12-04 | 2005-07-01 | Kondensert 1,3-dihydroimidazol-ringforbindelse. |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2002-352186 | 2002-12-04 | ||
| JP2002352186 | 2002-12-04 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2004050656A1 true WO2004050656A1 (ja) | 2004-06-17 |
Family
ID=32463216
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2003/015402 Ceased WO2004050656A1 (ja) | 2002-12-04 | 2003-12-02 | 1,3−ジヒドロ−イミダゾール縮合環化合物 |
Country Status (16)
| Country | Link |
|---|---|
| US (1) | US7524847B2 (ja) |
| EP (1) | EP1568699A4 (ja) |
| JP (1) | JP4279784B2 (ja) |
| KR (1) | KR100867484B1 (ja) |
| CN (1) | CN100376573C (ja) |
| AU (1) | AU2003302657B8 (ja) |
| BR (1) | BR0316991A (ja) |
| CA (1) | CA2507763A1 (ja) |
| IL (1) | IL168784A (ja) |
| MX (1) | MXPA05005983A (ja) |
| NO (1) | NO20053246L (ja) |
| NZ (1) | NZ540495A (ja) |
| PL (1) | PL377187A1 (ja) |
| RU (1) | RU2328498C9 (ja) |
| WO (1) | WO2004050656A1 (ja) |
| ZA (1) | ZA200504973B (ja) |
Cited By (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2005118555A1 (en) * | 2004-06-04 | 2005-12-15 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US7074798B2 (en) | 2002-02-25 | 2006-07-11 | Eisai Co., Ltd | Xanthine derivative and DPPIV inhibitor |
| US7470700B2 (en) | 2003-08-13 | 2008-12-30 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US7550590B2 (en) | 2003-03-25 | 2009-06-23 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US7638638B2 (en) | 2003-05-14 | 2009-12-29 | Takeda San Diego, Inc. | Dipeptidyl peptidase inhibitors |
| US7772226B2 (en) | 2002-06-06 | 2010-08-10 | Eisai R&D Management Co., Ltd. | Condensed imidazole derivatives |
| US7960384B2 (en) | 2006-03-28 | 2011-06-14 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US8084605B2 (en) | 2006-11-29 | 2011-12-27 | Kelly Ron C | Polymorphs of succinate salt of 2-[6-(3-amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethy]-4-fluor-benzonitrile and methods of use therefor |
| US8222411B2 (en) | 2005-09-16 | 2012-07-17 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| JP2012520269A (ja) * | 2009-03-13 | 2012-09-06 | カトリーケ ユニバーシテイト ルーヴェン、ケー.ユー. ルーヴェン アール アンド ディー | 免疫抑制剤としてのチアゾロピリミジン調節因子 |
| JP2013517225A (ja) * | 2010-01-15 | 2013-05-16 | 山東軒竹医薬科技有限公司 | 縮環ピリジン誘導体 |
| JP2014520763A (ja) * | 2011-07-09 | 2014-08-25 | 山東軒竹医薬科技有限公司 | ジペプチジルペプチダーゼ−iv阻害剤の塩の結晶形i、及びその製造方法並びにその応用 |
| US8906901B2 (en) | 2005-09-14 | 2014-12-09 | Takeda Pharmaceutical Company Limited | Administration of dipeptidyl peptidase inhibitors |
Families Citing this family (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| MY179032A (en) * | 2004-10-25 | 2020-10-26 | Cancer Research Tech Ltd | Ortho-condensed pyridine and pyrimidine derivatives (e.g.purines) as protein kinase inhibitors |
| UY29177A1 (es) | 2004-10-25 | 2006-05-31 | Astex Therapeutics Ltd | Derivados sustituidos de purina, purinona y deazapurina, composiciones que los contienen métodos para su preparación y sus usos |
| JP2009534454A (ja) * | 2006-04-25 | 2009-09-24 | アステックス、セラピューティックス、リミテッド | 医薬化合物 |
| DK3421471T3 (da) | 2006-04-25 | 2021-06-14 | Astex Therapeutics Ltd | Purin- og deazapurinderivater som farmaceutiske forbindelser |
| PL2201012T3 (pl) | 2007-10-11 | 2014-11-28 | Astrazeneca Ab | Pochodne pirolo[2,3-d]pirymidyny jako inhibitory kinazy białkowej b |
| BRPI0917540A2 (pt) * | 2008-08-05 | 2015-11-17 | Daiichi Sankyo Co Ltd | composto, sal farmacologicamente aceitavél, composição farmacêutica, e, uso de um composto ou sal farmacologicamente aceitável |
| CN102153538B (zh) * | 2010-02-11 | 2013-12-11 | 山东轩竹医药科技有限公司 | 苯并环衍生物 |
| CN102276627B (zh) * | 2010-04-29 | 2013-07-31 | 山东轩竹医药科技有限公司 | 吡啶并杂环衍生物 |
| CN102234281B (zh) * | 2010-04-29 | 2013-03-27 | 山东轩竹医药科技有限公司 | 嘧啶并环衍生物 |
| EA027506B1 (ru) | 2011-04-01 | 2017-08-31 | Астразенека Аб | Способ лечения рака молочной железы |
| HRP20191982T4 (hr) | 2011-11-30 | 2023-01-06 | Astrazeneca Ab | Kombinacijsko liječenje raka |
| US8501724B1 (en) * | 2012-01-31 | 2013-08-06 | Pharmacyclics, Inc. | Purinone compounds as kinase inhibitors |
| AU2013204533B2 (en) | 2012-04-17 | 2017-02-02 | Astrazeneca Ab | Crystalline forms |
| WO2015078799A1 (en) * | 2013-11-27 | 2015-06-04 | Bayer Pharma Aktiengesellschaft | Glucose transport inhibitors |
| CN110872297B (zh) * | 2018-09-04 | 2023-02-14 | 上海再极医药科技有限公司 | 氨基嘧啶并五元杂环化合物、其中间体、制备方法、药物组合物及应用 |
| EP3877371A4 (en) | 2018-11-07 | 2022-07-27 | Dana-Farber Cancer Institute, Inc. | IMIDAZOPYRIDINE DERIVATIVES AND AZA-IMIDAZOPYRIDINE DERIVATIVES AS JANUS KINASE 2 INHIBITORS AND USES THEREOF |
| US12522583B2 (en) | 2018-11-07 | 2026-01-13 | Dana-Farber Cancer Institute, Inc. | Benzimidazole derivatives and aza-benzimidazole derivatives as Janus kinase 2 inhibitors and uses thereof |
| EP3876939A4 (en) | 2018-11-07 | 2022-08-10 | Dana-Farber Cancer Institute, Inc. | Benzothiazole derivatives and 7-aza-benzothiazole derivatives as janus kinase 2 inhibitors and uses thereof |
| CN109608462B (zh) * | 2018-12-15 | 2021-11-23 | 华南理工大学 | 一种7-烃基-9-烷氧/硫基嘌呤-8-酮类化合物及其合成方法与在药物中的应用 |
| US11691963B2 (en) | 2020-05-06 | 2023-07-04 | Ajax Therapeutics, Inc. | 6-heteroaryloxy benzimidazoles and azabenzimidazoles as JAK2 inhibitors |
| US12043632B2 (en) | 2020-12-23 | 2024-07-23 | Ajax Therapeutics, Inc. | 6-heteroaryloxy benzimidazoles and azabenzimidazoles as JAK2 inhibitors |
| US12162881B2 (en) | 2021-11-09 | 2024-12-10 | Ajax Therapeutics, Inc. | Forms and compositions of inhibitors of JAK2 |
| WO2023086319A1 (en) | 2021-11-09 | 2023-05-19 | Ajax Therapeutics, Inc. | 6-he tero aryloxy benzimidazoles and azabenzimidazoles as jak2 inhibitors |
Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2000086663A (ja) * | 1998-09-09 | 2000-03-28 | Taisho Pharmaceut Co Ltd | アリールテトラヒドロピリジン誘導体 |
| JP2001151777A (ja) * | 1999-03-11 | 2001-06-05 | Taisho Pharmaceut Co Ltd | カルバモイルテトラヒドロピリジン誘導体 |
Family Cites Families (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB8515934D0 (en) * | 1985-06-24 | 1985-07-24 | Janssen Pharmaceutica Nv | (4-piperidinomethyl and-hetero)purines |
| US5057517A (en) * | 1987-07-20 | 1991-10-15 | Merck & Co., Inc. | Piperazinyl derivatives of purines and isosteres thereof as hypoglycemic agents |
| NZ225447A (en) | 1987-07-20 | 1991-12-23 | Merck & Co Inc | Piperazinyl derivatives of purine and purine isosteres and pharmaceutical compositions |
| EP1176146B1 (en) | 1999-03-11 | 2005-06-08 | Taisho Pharmaceutical Co., Ltd | Carbamoyl tetrahydropyridine derivatives |
| EP1338595B1 (en) | 2002-02-25 | 2006-05-03 | Eisai Co., Ltd. | Xanthine derivatives as DPP-IV inhibitors |
| AU2003241960B2 (en) | 2002-06-06 | 2009-07-30 | Eisai R&D Management Co., Ltd. | Novel fused imidazole derivative |
| MXPA05003252A (es) | 2002-09-26 | 2005-07-05 | Eisai Co Ltd | Farmaco de combinacion. |
| JP4768265B2 (ja) * | 2002-10-15 | 2011-09-07 | シンタ ファーマシューティカルズ コーポレーション | 新規化合物 |
| EP1677791A4 (en) | 2003-10-31 | 2007-08-15 | Takeda Pharmaceutical | NITROGENIC CONDENSED HETEROCYCLIC COMPOUNDS |
| CA2554809C (en) * | 2004-02-05 | 2014-04-29 | Probiodrug Ag | Novel n-alkyl thiourea- and thioamide-substituted imidazolyl inhibitors of glutaminyl cyclase |
-
2003
- 2003-12-02 CN CNB2003801095193A patent/CN100376573C/zh not_active Expired - Fee Related
- 2003-12-02 MX MXPA05005983A patent/MXPA05005983A/es unknown
- 2003-12-02 PL PL377187A patent/PL377187A1/pl not_active Application Discontinuation
- 2003-12-02 BR BR0316991-0A patent/BR0316991A/pt not_active IP Right Cessation
- 2003-12-02 KR KR1020057010179A patent/KR100867484B1/ko not_active Expired - Fee Related
- 2003-12-02 US US10/537,227 patent/US7524847B2/en not_active Expired - Fee Related
- 2003-12-02 RU RU2005120780/04A patent/RU2328498C9/ru not_active IP Right Cessation
- 2003-12-02 JP JP2004556886A patent/JP4279784B2/ja not_active Expired - Fee Related
- 2003-12-02 AU AU2003302657A patent/AU2003302657B8/en not_active Ceased
- 2003-12-02 WO PCT/JP2003/015402 patent/WO2004050656A1/ja not_active Ceased
- 2003-12-02 NZ NZ540495A patent/NZ540495A/en not_active IP Right Cessation
- 2003-12-02 EP EP03812368A patent/EP1568699A4/en not_active Withdrawn
- 2003-12-02 CA CA002507763A patent/CA2507763A1/en not_active Abandoned
-
2005
- 2005-05-25 IL IL168784A patent/IL168784A/en not_active IP Right Cessation
- 2005-06-20 ZA ZA200504973A patent/ZA200504973B/en unknown
- 2005-07-01 NO NO20053246A patent/NO20053246L/no not_active Application Discontinuation
Patent Citations (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2000086663A (ja) * | 1998-09-09 | 2000-03-28 | Taisho Pharmaceut Co Ltd | アリールテトラヒドロピリジン誘導体 |
| JP2001151777A (ja) * | 1999-03-11 | 2001-06-05 | Taisho Pharmaceut Co Ltd | カルバモイルテトラヒドロピリジン誘導体 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP1568699A4 * |
Cited By (16)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7074798B2 (en) | 2002-02-25 | 2006-07-11 | Eisai Co., Ltd | Xanthine derivative and DPPIV inhibitor |
| US7772226B2 (en) | 2002-06-06 | 2010-08-10 | Eisai R&D Management Co., Ltd. | Condensed imidazole derivatives |
| US7550590B2 (en) | 2003-03-25 | 2009-06-23 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US7638638B2 (en) | 2003-05-14 | 2009-12-29 | Takeda San Diego, Inc. | Dipeptidyl peptidase inhibitors |
| US7470700B2 (en) | 2003-08-13 | 2008-12-30 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US7579357B2 (en) | 2003-08-13 | 2009-08-25 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| WO2005118555A1 (en) * | 2004-06-04 | 2005-12-15 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US8906901B2 (en) | 2005-09-14 | 2014-12-09 | Takeda Pharmaceutical Company Limited | Administration of dipeptidyl peptidase inhibitors |
| US8222411B2 (en) | 2005-09-16 | 2012-07-17 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US7960384B2 (en) | 2006-03-28 | 2011-06-14 | Takeda Pharmaceutical Company Limited | Dipeptidyl peptidase inhibitors |
| US8084605B2 (en) | 2006-11-29 | 2011-12-27 | Kelly Ron C | Polymorphs of succinate salt of 2-[6-(3-amino-piperidin-1-yl)-3-methyl-2,4-dioxo-3,4-dihydro-2H-pyrimidin-1-ylmethy]-4-fluor-benzonitrile and methods of use therefor |
| JP2012520269A (ja) * | 2009-03-13 | 2012-09-06 | カトリーケ ユニバーシテイト ルーヴェン、ケー.ユー. ルーヴェン アール アンド ディー | 免疫抑制剤としてのチアゾロピリミジン調節因子 |
| JP2015028088A (ja) * | 2009-03-13 | 2015-02-12 | カトリーケ ユニバーシテイト ルーヴェン、ケー.ユー. ルーヴェン アール アンド ディー | 免疫抑制剤としてのチアゾロピリミジン調節因子 |
| JP2013517225A (ja) * | 2010-01-15 | 2013-05-16 | 山東軒竹医薬科技有限公司 | 縮環ピリジン誘導体 |
| US8680089B2 (en) | 2010-01-15 | 2014-03-25 | Xuanzhu Pharma Co., Ltd. | Fused pyridine derivatives |
| JP2014520763A (ja) * | 2011-07-09 | 2014-08-25 | 山東軒竹医薬科技有限公司 | ジペプチジルペプチダーゼ−iv阻害剤の塩の結晶形i、及びその製造方法並びにその応用 |
Also Published As
| Publication number | Publication date |
|---|---|
| NZ540495A (en) | 2007-09-28 |
| KR20050087825A (ko) | 2005-08-31 |
| RU2328498C9 (ru) | 2008-12-10 |
| AU2003302657A2 (en) | 2004-06-23 |
| PL377187A1 (pl) | 2006-01-23 |
| CA2507763A1 (en) | 2004-06-17 |
| AU2003302657A1 (en) | 2004-06-23 |
| HK1086556A1 (zh) | 2006-09-22 |
| EP1568699A1 (en) | 2005-08-31 |
| NO20053246L (no) | 2005-08-30 |
| CN100376573C (zh) | 2008-03-26 |
| CN1745080A (zh) | 2006-03-08 |
| KR100867484B1 (ko) | 2008-11-06 |
| AU2003302657B2 (en) | 2009-09-24 |
| AU2003302657B8 (en) | 2009-12-03 |
| JPWO2004050656A1 (ja) | 2006-03-30 |
| EP1568699A4 (en) | 2007-08-15 |
| IL168784A (en) | 2010-12-30 |
| US20060111362A1 (en) | 2006-05-25 |
| RU2005120780A (ru) | 2006-01-20 |
| US7524847B2 (en) | 2009-04-28 |
| MXPA05005983A (es) | 2005-08-18 |
| JP4279784B2 (ja) | 2009-06-17 |
| RU2328498C2 (ru) | 2008-07-10 |
| ZA200504973B (en) | 2006-04-26 |
| BR0316991A (pt) | 2005-10-25 |
| NO20053246D0 (no) | 2005-07-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2004050656A1 (ja) | 1,3−ジヒドロ−イミダゾール縮合環化合物 | |
| US12304897B2 (en) | 15-PGDH inhibitors | |
| JP5366349B2 (ja) | ホスホジエステラーゼ阻害剤として有効な置換ピロロピリジノン誘導体 | |
| AU2024227155A1 (en) | Carboxy-benzimidazole GLP-1R modulating compounds. | |
| AU2002230618B2 (en) | Heterocyclic ether substituted imidazoquinolines | |
| JP7761588B2 (ja) | 新規大環状lrrk2キナーゼインヒビター | |
| US20090270443A1 (en) | 1-amino imidazo-containing compounds and methods | |
| AU2006232377A1 (en) | Pyrazolopyridine-1,4-diamines and analogs thereof | |
| EP1968587A1 (en) | Substituted imidazoquinolines, imidazonaphthyridines, and imidazopyridines, compositions, and methods | |
| US20230027198A1 (en) | Inhibitors of enl/af9 yeats | |
| JP2008531716A (ja) | 化合物 | |
| EP3921322A1 (en) | Phosphorus imidazoquinoline amine derivatives, pharmaceutical compositions and therapeutic methods thereof | |
| CN115003383B (zh) | 干扰素基因刺激物sting的双环激动剂 | |
| JP2025536295A (ja) | ムスカリン性アセチルコリン受容体m1のポジティブアロステリックモジュレーター | |
| HK1086556B (en) | Fused 1,3-dihydro-imidazole ring compounds |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A1 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A1 Designated state(s): BW GH GM KE LS MW MZ SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LU MC NL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| DFPE | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed before 20040101) | ||
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 1-2005-501067 Country of ref document: PH |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2004556886 Country of ref document: JP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 168784 Country of ref document: IL |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2507763 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 540495 Country of ref document: NZ |
|
| WWE | Wipo information: entry into national phase |
Ref document number: PA/a/2005/005983 Country of ref document: MX Ref document number: 377187 Country of ref document: PL Ref document number: 1020057010179 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2003812368 Country of ref document: EP |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 200504973 Country of ref document: ZA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2003302657 Country of ref document: AU |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1470/CHENP/2005 Country of ref document: IN |
|
| ENP | Entry into the national phase |
Ref document number: 2005120780 Country of ref document: RU Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 1200500920 Country of ref document: VN |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 20038A95193 Country of ref document: CN |
|
| WWP | Wipo information: published in national office |
Ref document number: 2003812368 Country of ref document: EP Ref document number: 1020057010179 Country of ref document: KR |
|
| ENP | Entry into the national phase |
Ref document number: PI0316991 Country of ref document: BR |
|
| ENP | Entry into the national phase |
Ref document number: 2006111362 Country of ref document: US Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 10537227 Country of ref document: US |
|
| WWP | Wipo information: published in national office |
Ref document number: 10537227 Country of ref document: US |