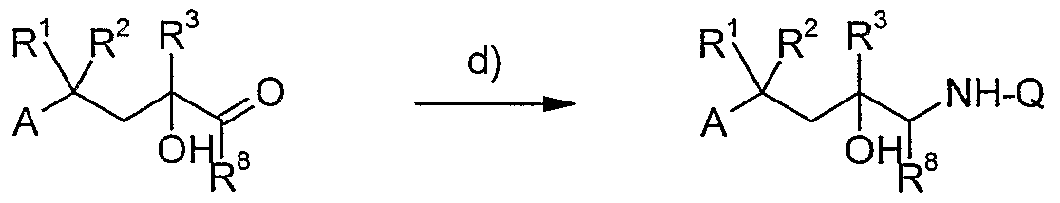Heterozyklisch substituierte Pentanol-Derivate, Verfahren zu ihrer Herstellung und ihre Verwendung als Entzündungshemmer
Die Erfindung betrifft heterozyklisch substituierte Pentanol-Derivate, insbeson- dere durch Chinazolin, Chinoxalin, Cinnolin, Indazol, Phthalazin, Naphthyridin, Benzothiazol, Dihydroindolon, Dihydroisoindolon, Benzimidazol oder Indol substituierte Pentanol-Derivate, Verfahren zu ihrer Herstellung und ihre Verwendung als Entzündungshemmer.
Aus dem Stand der Technik WO 00/32584, DE 100 38 639 A1und WO 02/10143 sind Entzündungshemmer der allgemeinen Formel
bekannt, wobei der Ar-Rest Phthalide, Thiophthalide, Benzoxazinone oder Phthalazinone umfasst. Diese Verbindungen zeigen im Experiment Wirkdissoziationen zwischen antiinflammatorischen und unerwünschten metabolischen Wirkungen und sind den bisher beschriebenen, nichtsteroidalen Glucocortico- iden überlegen oder weisen zumindest eine ebenso gute Wirkung auf.
Weiterhin sind aus WO03/059899 Verbindungen bekannt, worin Q einen aromatischen carbozyklischen Rest darstellt.
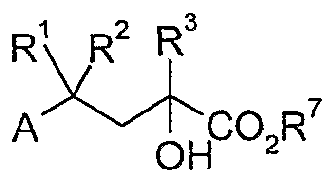
Die Selektivität der Verbindungen des Standes der Technik gegenüber den anderen Steroidrezeptoren ist jedoch noch verbesserungsbedürftig. Daher war es Aufgabe der vorliegenden Erfindung, Verbindungen zur Verfügung zu stellen, deren Selektivität gegenüber den anderen Steroidrezeptoren verbes- sert ist. Diese Aufgabe wird von den Verbindungen gemäß der Patentansprüche gelöst. Die vorliegende Erfindung betrifft daher Verbindungen der allgemeinen Formel I
worin
A für eine Aryl-, eine Benzyl- oder eine Phenethylgruppe steht, wobei die Aryl-, Benzyl- oder Phenethylgruppe gegebenenfalls substituiert sein kann durch einen oder mehrere Reste aus der Gruppe Cι-C5-A!kyl, C C5-Alkoxy, CrC5-Alkylthio, d-Cs-Perfluor- alkyl, Halogen, Hydroxy, Cyano, Nitro, -O-(CH2)n-O-, -O-(CH2)n-CH2-, -O-CH=CH-, -(CH2)n+2-, wobei n = 1 oder 2 ist und die endständigen Sauerstoffatome und/oder Kohlenstoffatome mit direkt benachbarten Ring-Kohlenstoffatomen verknüpft sind, oder NR4R5, wobei R4 und R5 unabhängig voneinander Wasserstoff, CrCs-Alkyl oder (COJ-CrCδ-Alkyl sein können, R1 und R2 unabhängig voneinander ein Wasserstoffatom, eine Methyl- oder Ethylgruppe oder gemeinsam mit dem Kohlenstoffatom der Kette einen C3-C5-Cycloalkylring, R3 eine gegebenenfalls unabhängig voneinander durch einen oder mehrere Gruppen ausgewählt aus Halogen, Hydroxy oder CrC3-Alkoxy substituierte C-i-Cs-Alkylgruppe, oder eine gegebenenfalls teilweise oder vollständig fluorierte CrC3-Alkylgruppe, eine gegebenenfalls substituierte Gruppe ausgewählt aus C2-C-6- Alkenyl, C2-C6-Alkinyl, C3-C8-Cycloalkyl, C3-C7-Heterocyclyl, Aryl, Heteroaryl, (C1-C8-Alkyl)C3-C8-Cycloalkyl, (C C8-Alkyl)Aryl, oder (C Cs-Alky Heteroaryl B eine gegebenenfalls durch eine Methyl- oder Ethylgruppe substituierte Methylengruppe oder eine Carbonylgruppe und Q eine über eine beliebige Position verknüpfte Chinazolinyl-, Chinoxalinyl-, Cinnolinyl-, Indazolyl-, Phthalazinyl-, Naphthyridinyl-, Benzothiazolyl-, Dihydroindolonyl-, Dihydroisoindolonyl-, Benzimidazol- oder Indolylgruppe bedeutet, die gegebenenfalls substituiert sein kann durch einen oder mehrere Reste aus der
Gruppe Cι-C5-A!kyl, Cι-C5-Alkoxy, CrC5-Alkylthio, Ct-C5-Perfluor- alkyl, Halogen, Hydroxy, Cyano, Nitro, oder NR4R5, wobei R4 und R5 unabhängig voneinander Wasserstoff, Cι-C5-Alkyl oder (CO)-Cι-Cδ-AIkyl sein können, wobei Phthalazinone ausgenommen sind, sowie deren Racemate oder getrennt vorliegenden Stereoisomeren, und gegebenenfalls deren physiologisch verträgliche Salze.
Phthalazinone wurden angesichts des Standes der Technik WO 98/54159 und WO 00/32584 ausgenommen. Sie ergeben sich durch die Definition in Anspruch 1 von Q = Phthalazin in Kombination mit dem möglichen Substituenten Hydroxy, da Hydroxyphthalazine in einem Tautomerengleichgewicht mit Phthalazinonen liegen.
Ein Aspekt der Erfindung sind Verbindungen der allgemeinen Formel I worin A für eine Aryl-, eine Benzyl- oder eine Phenethylgruppe steht, wobei die Aryl-, Benzyl- oder Phenethylgruppe gegebenenfalls substituiert sein kann durch einen oder mehrere Reste aus der Gruppe C-i-Cs-Alkyl, C-i- Cs-Alkoxy, Cι-C5-Alkylthio, Cι-C5-Perfluoralkyl, Halogen, Hydroxy, Cyano, Nitro, -O-(CH2)n-O-, -O-(CH2)n-CH2-, -O-CH=CH-, -(CH2)n+2- wobei n = 1 oder 2 ist und die endständigen Sauerstoffatome und/oder Kohlenstoffatome mit direkt benachbarten Ring-Kohlenstoffatomen verknüpft sind, oder NR4R5, wobei R4 und R5 unabhängig voneinander Wasserstoff, Cι-C5-AIkyl oder (CO)-C C5-AlkyI sein können,
R1 und R2 unabhängig voneinander ein Wasserstoffatom, eine Methyl- oder Ethylgruppe oder gemeinsam mit dem Kohlenstoffatom der Kette einen C3- C6-Cycloalkylring, R3 eine CrC3-Alkylgruppe oder eine gegebenenfalls teilweise oder vollständig fluorierte Cι-C3-Alkylgruppe,
B eine gegebenenfalls durch eine Methyl- oder Ethylgruppe substituierte Methylengruppe oder eine Carbonylgruppe und
Q eine über eine beliebige Position verknüpfte Chinazolinyl-, Chinoxalinyl-, Cinnolinyl-, Indazolyl-, Phthalazinyl-, Naphthyridinyl-, oder Benzothiazolyl- gruppe bedeutet, die gegebenenfalls substituiert sein kann durch einen oder mehrere Reste aus der Gruppe CrC5-Alkyl, Cι-C5-Alkoxy, C C5-Alkylthio, Cι-C5-Perfluoralkyl, Halogen, Hydroxy, Cyano, Nitro, oder NR4R5, wobei R4 und R5 unabhängig voneinander Wasserstoff, C-i-Cs-Alkyl oder (CO)-C1-C5-Alkyl sein können wobei Phthalazinone ausgenommen sind, sowie deren Racemate oder getrennt vorliegenden Stereoisomeren, und gege- benenfalls deren physiologisch verträgliche Salze.
Ein Gegenstand der Erfindung sind Verbindungen der allgemeinen Formel I worin
A für eine Aryl-, eine Benzyl- oder eine Phenethylgruppe steht, wobei die Aryl-, Benzyl- oder Phenethylgruppe gegebenenfalls substituiert sein kann durch einen oder mehrere Reste aus der Gruppe C-i-Cs-Alkyl, C-i- C5-Alkoxy, CrC5-Alkylthio, Cι-C5-Perfluoralkyl, Halogen, Hydroxy, Cyano, Nitro, -O-(CH2)n-O-, -O-(CH2)n-CH2-, -O-CH=CH-, -(CH2)n+2- wobei n = 1 oder 2 ist und die endständigen Sauerstoffatome und/oder Kohlenstoffatome mit direkt benachbarten Ring-Kohlenstoffatomen verknüpft sind, oder NR4R5, wobei R4 und R5 unabhängig voneinander Wasserstoff, Cι-Cε-Alkyl oder (CO)-Cι-Cδ-Alkyl sein können, R1 und R2 unabhängig voneinander ein Wasserstoff atom, eine Methyl- oder Ethylgruppe oder gemeinsam mit dem Kohlenstoffatom der Kette einen C3- C6-Cycloalkylring, R3 eine C-ι-C3-Alkylgruppe oder eine gegebenenfalls teilweise oder vollständig fluorierte Cι-C3-Alkylgruppe, B eine gegebenenfalls durch eine Methyl- oder Ethylgruppe substituierte Methylengruppe oder eine Carbonylgruppe und Q eine über eine beliebige Position verknüpfte Chinazolinyl-, Chinoxalinyl-, Cinnolinyl-, Indazolyl-, Naphthyridinyl-, oder Benzothiazolylgruppe bedeutet,
die gegebenenfalls substituiert sein kann durch einen oder mehrere Reste aus der Gruppe Cι-C5-Alkyl, Cι-C5-Alkoxy, C C5-Alkylthio, d-Cs-Perfluor- alkyl, Halogen, Hydroxy, Cyano, Nitro, oder NR4R5, wobei R4 und R5 unabhängig voneinander Wasserstoff, Ci-Cs-Alkyl oder (CO)-CrC5-Alkyl sein können sowie deren Racemate oder getrennt vorliegenden Stereoisomeren, und gegebenenfalls deren physiologisch verträgliche Salze
Ein Gegenstand der Erfindung sind Verbindungen der allgemeinen Formel I, worin R3 eine gegebenenfalls unabhängig voneinander durch einen oder mehrere Gruppen ausgewählt aus Halogen, Hydroxy oder C-ι-C3-Alkoxy substituierte CrCs-Alkylgruppe, oder eine gegebenenfalls teilweise oder vollständig fluorierte CrC3-Alkylgruppe darstellt.
Ein besonderer Gegenstand der Erfindung sind Verbindungen der allgemeinen
Formel I worin R3eine Cι-C3-Alkylgruppe oder eine gegebenenfalls teilweise oder vollständig fluorierte d-Cs-Alkylgruppe ist. Bevorzugt sind die CF3-Gruppe und die C2F5-Gruppe.
Die CrC5-oder Ci-Ce-Alkylgruppen können geradkettig oder verzweigt sein und für eine Methyl-, Ethyl-, n-Propyl-, iso-Propyl-, n-Butyl, iso-Butyl, tert.-Butyl- oder n-Pentyl-, 2,2-Dimethylpropyl-, 2-Methylbutyl- oder 3-Methylbutylgruppe stehen. Eine Methyl- oder Ethylgruppe ist bevorzugt.
Für eine teilweise oder vollständig fluorierte Cι-C3-AIkylgruppe kommen beispielsweise die teilweise oder vollständig fluorierten geradkettigen oder verzweigten folgenden Gruppen in Betracht: Fluormethyl, Difluormethyl, Trifluormethyl, Fluorethyl, 1 ,1-Difluorethyl, 1 ,2-Difluorethyl, 1 ,1 ,1-Trifluorethyl, Tetra- fluorethyl, Pentafluorethyl. Von diesen bevorzugt sind die Trifluormethyl- oder die Pentafluorethylgruppe.
Unter einer Cι-C5-Perfluoralkylgruppe ist eine vollständig fluorierte geradkettige oder verzweigte Alkylgruppe zu verstehen wie z.B. CF3, C2F5, C3F7, C4F9, C5Fn.
Die Alkylreste R1 und R2 können zusammen mit dem Kohlenstoffatom der Kette einen 3 bis 6-gliedrigen Ring bilden. Bevorzugt für R1 und R2 ist die Methyl- oder die Ethylgruppe.
Als Alkylreste R4 und R5 sind d-Cs-Alkyl bevorzugt, wobei die Cι-C3-Alkyl- gruppe geradkettig oder verzweigt sein kann.
Die Cι-C5-Alkoxygruppen in A und Q können geradkettig oder verzweigt sein und zum Beispiel für eine Methoxy-, Ethoxy-, n-Propoxy-, iso-Propoxy-, n- Butoxy, iso-Butoxy, tert.-Butoxy- oder n-Pentoxy-, 2,2-Dimethylpropoxy-, 2- Methylbutoxy- oder 3-Methylbutoxygruppe stehen. Eine Methoxy- oder Ethoxy- gruppe ist bevorzugt.
Die Cι-C5-Alkylthiogruppen in A und Q können geradkettig oder verzweigt sein und für eine Methylthio-, Ethylthio-, n-Propylthio-, iso-Propylthio-, n-Butylthio, iso-Butylthio, tert.-Butylthio- oder n-Pentylthio-, 2,2-Dimethylpropylthio-, 2- Methylbutylthio- oder 3-Methylbutylthiogruppe stehen. Eine Methylthio- oder Ethylthiogruppe ist bevorzugt.
Die Bezeichnung Halogenatom oder Halogen bedeutet ein Fluor-, Chlor-, Bromoder lodatom. Bevorzugt ist ein Fluor-, Chlor- oder Bromatom. Die NR4R5-Gruppe kann beispielsweise NH2, N(H)CH3, N(CH3)2, N(H)(CO)CH3, N(CH3)(CO)CH3, N[(CO)CH3]2, N(H)CO2CH3, N(CH3)CO2CH3, N(CO2CH3)2, bedeuten.
Als Acylreste R4 und R5 sind (CO)-C-ι-C3-Alkyl bevorzugt, wobei der C C3-Alkyl- rest geradkettig oder verzweigt sein kann. Die C2-C6- oder C2-C5-Alkenylgruppe ist geradkettig oder verzweigt, es kommt beispielsweise Vinyl, Propenyl, isopropenyl, Butenyl, Isobutenyl in Frage. Die C2-C6- oder C2-C5-Alkinylgruppe ist geradkettig oder verzweigt, es kommt beispielsweise C C, Propinyl, Isopropinyl, Butinyl, Isobutinyl in Frage. Für eine Cycloalkylgruppe kommen beispielsweise Cyclopropyl, Cyclobutyl, Cyclopentyl, Cylohexyl, Cycloheptyl, Cyclooctyl, in Betracht. Die Cι-C8-Alkyl(C3-C8)Cycloalkylgruppe kann beispielsweise Cyclobutylmethyl, Cyclopentylmethyl, Cyclohexylmethyl, Cycloheptylmethyl sein. Die Verknüpfung mit der Kette erfolgt über die Alkylgruppe. Die Heterocyclylgruppe ist nicht aromatisch und kann beispielsweise Pyrrolidin, Imidazolidin, Pyrazolidin, Piperidin sein.
Für eine Arylgruppe kommen Phenyl und Naphthyl, und für (Cι-C8)Alkylaryl Benzyl und Homobenzyl in Betracht. Wenn die Arylgruppe für A steht, ist die Phenylgruppe bevorzugt .
Heteroaryl umfasst beispielsweise Furanyl, Thienyl, Thiazolyl, Oxazolyl, Imidazoyl, Triazolyl, Pyridyl und Pyrimidyl; .
(Cι-C8-Alkyl)Heteroaryl umfasst alle Kombinationen aus der oben gegebenen Definition von Alkyl mit monozyklischen aromatischen Heterozyklen, insbesondere den namentlich genannten Heterozyklen. Die Verknüpfung mit der Kette erfolgt über die Alkylgruppe, die ihrerseits an beliebiger chemisch möglicher Position des Heterozyklus verknüpft ist.
Die Substituenten der Gruppen in R3 können sein C-i-Cβ-Alkyl, C2-C6-Alkenyl, C2-C6-Alkinyl, Ci-Ce-Alkoxy, Halogen, Hydroxy, NR4R5.
Die substituierten Aryl, Benzyl- oder Phenethylgruppen tragen- am Ring 1-4 oder 1 - 3 Substituenten, bevorzugt 2 Substituenten. Die Substituenten für A können unabhängig voneinander ausgewählt werden aus der Gruppe bestehend aus C-ι-C5-Alkyl, Cι-C5-Alkoxy, d-Cs-Alkylthio, C-p C5-PerfluoraIkyl, Halogen, Hydroxy, Cyano, Nitro, -O-(CH2)n-O-, -O-(CH2)n-CH2- , -O-CH=CH-, -(CH2)n+2- (wobei n = 1 oder 2 ist und die endständigen Sauerstoffatome und/oder Kohlenstoffatome mit direkt benachbarten Ring- Kohlenstoffatomen verknüpft sind), oder NR4R5 (wobei R4 und R5 unabhängig voneinander Wasserstoff, Cι-C5-AIkyl oder (CO)-C-ι-C5-Alkyl sind). Bevorzugt werden d-C5-Alkyl, CrC5-Alkoxy, CrC5-Alkylthio, C-ι-C5-Perfluor- alkyl, Halogen, Hydroxy, -O-(CH2)n-O-, -O-(CH2)n-CH2-, -O-CH=CH- und - (CH2)n+2-. Besonders bevorzugt werden CrC5-Alkyl, Cι-C5-Alkoxy, Cι-C5-Alkylthio, C Cö- Perfluoralkyl, Halogen, Hydroxy, -O-(CH2)n-O-, und -O-(CH2)n-CH2-. Insbesondere sind Verbindungen, deren Substituenten von A ausgewählt sind aus der Gruppe Cι-C5-AIkyl, Cι-C5-Alkoxy, C C5-Alkylthio, d-Cs-Perfluoralkyl, Halogen und Hydroxy ein Gegenstand der Erfindung. Ein weiterer Gegenstand der Erfindung sind Verbindungen, deren Substituenten von A ausgewählt sind aus der Gruppe -O-(CH2)n-O-, -O-(CH2)n-CH2-, -O-CH=CH- und -(CH2)n+2-, bevorzugt -O-(CH2)n-O-, und -O-(CH2)n-CH2-.
Ein weiterer Gegenstand der Erfindung sind Verbindungen der Formel 1 , worin A einen Phenylrest bedeutet, dessen Substituenten ausgewählt sind aus der Gruppe Hydroxy, d-C5-Alkoxy und Halogen.
Folgende Definition und Substitutionsmuster am Ring A sind ein besonderer Gegenstand der Erfindung: 2,5-disubstituierte Phenylderivate und 2,4-disubsti- tuierte Phenylderivate.
Für den Rest B sind die unsubstituierte Methylengruppe und die Carbonylgruppe bevorzugt.
Ein besonderer Gegenstand der Erfindung sind Verbindungen der Formel 1 nach Anspruch 1 , worin B für eine gegebenenfalls durch eine Methyl- oder Ethylgruppe substituierte Methylengruppe steht.
Ein Gegenstand der Erfindung sind Verbindungen gemäß Anspruch 1 , worin Q eine über eine beliebige Position verknüpfte Benzothiazolyl-, Chinazolinyl-, Chinoxalinyl-, Cinnolinyl-, Phthalazinyl-, 1 ,7- oder 1 ,8-Naphthyridinyl-, Indazolyl-, Dihydroindolonyl-, Dihydroisoindolonyl-, Benzimidazolyl- oder Indolylgruppe bedeutet.
Ein weiterer Gegenstand der Erfindung sind Verbindungen der Formel I, worin Q eine über eine beliebige Position verknüpfte Benzothiazolyl-, Chinazolinyl-, Chinoxalinyl-, Cinnolinyl-, Indazolyl-, Phthalazinyl- oder eine 1 ,7- oder 1 ,8- Naphthyridinylgruppe bedeutet .
Bevorzugte Reste Q sind Chinazolin, Benzothiazol, Naphthyridin, Indazol, Indolon, Benzimidazol, und Isoindolon. Besonders bevorzugt sind Chinazolin, Indazol und Benzimidazol. Der Rest Q kann über jedes beliebige Ring-Kohlenstoffatom mit der (NH)- Gruppe der Kette verknüpft sein. Bevorzugt sind für den Chinazolinring, den Chinoxalinring, den Cinnolin- und den Phthalazinring die 5- und 8-Position, für den Naphthyridinring die 3- und 5-Position und für den Dihydroindolon-, Dihydroisoindolon-, Benzimidazol- Indazol-, Indol- und den Benzothiazolring die 7- und die 4-Position. Der Ausdruck, dass Q „über jedes beliebige Ring-Kohlenstoffatom oder jede beliebige Position verknüpft sein kann" bedeutet im Sinne der Erfindung jede
chemisch mögliche Verknüpfung zwischen einem der Kohlenstoff-Atome des Heterozyklus Q und der NH-Gruppe der Verbindung der Formel I.
Q kann substituiert sein durch einen oder mehrere Reste aus der Gruppe d-C5- Alkyl, d-C5-Alkoxy, d-C5-Alkylthio, d-C5-Perfluoralkyl, Halogen, Hydroxy, Cyano, Nitro, oder NR4R5, wobei R4 und R5 unabhängig voneinander Wasserstoff, d-Cs-Alky! oder (CO)-d-C5-Alkyl sein können.
Bevorzugt sind die d-C3-Alkylgruppe, die Cι-C5-Alkoxygruppe, die Hydroxy- gruppe, die d-Cs-Perfluoralkylgruppe und Halogenatome. Besonders bevorzugt sind die d-C3-Alkylgruppe, die Hydroxygruppe und Halogenatome.
Ein weiterer Gegenstand der vorliegenden Erfindung ergibt sich aus den in den Beispielen offenbarten Bedeutungen für A, R1, R2, R3, R4, R5, B und Q und allen daraus möglichen Kombinationen.
Die erfindungsgemäßen Verbindungen der allgemeinen Formel I können durch das Vorhandensein von Asymmetriezentren als unterschiedliche Stereoisomere vorliegen. Sowohl die Racemate als auch die getrennt vorliegenden Stereoisomere gehören zum Gegenstand der vorliegenden Erfindung.
Ein besonderer Gegenstand der vorliegenden Erfindung im Hinblick auf ihre Wirkstärke sind die getrennt vorliegenden Stereoisomere, d.h. (+)-Enantiomere und (-)-Enantiomere.
Im Falle, dass die Verbindungen der allgemeinen Formel I als Salze vorliegen, kann dies beispielsweise in der Form des Hydrochlorids, Sulfats, Nitrats, Phosphats, Pivalats, Maleats, Fumarats, Tartrats, Benzoats, Mesylats, Citrats oder Succinats sein, das nach dem Fachmann bekannten Methoden erhalten werden kann.
Die Verfahren zur Herstellung der Verbindungen aus WO98/54159, WO00/32584 und WO02/10143 können auch für die Herstellung der erfindungsgemäßen Verbindungen verwendet werden. Für die Anknüpfung der für die erfindungsgemäßen Verbindungen charakteristischen Benzothiazol- Chinazolin-, Chinoxalin-, Cinnolin-, Indazol-, Phthalazin-, 1 ,7- und 1 ,8-
Naphthyridin-, Dihydroindolon-, Dihydroisoindolon-, Benzimidazol- oder Indol- gruppe können folgende Verfahrensschritte durchgeführt werden:
A1) für B = CO
Eine D-Ketosäure der allgemeinen Formel (II), worin A, R1 und R2 die für Formel (I) angegebenen Bedeutungen haben, wird mit einem Aminobenzothiazol,
Aminochinazolin, Aminochinoxalin, Aminocinnolin oder Aminophthalazin Derivat (Q-NH2) ins D-Ketoamid (III), wobei A, R1 und R2die oben angegebene Bedeutung zukommt, in der dem Fachmann bekannten Weise übergeführt. Beispielsweise wird unter Verwendung von dehydratisierenden Kupplungsreagenzien, wie sie aus der Peptidchemie bekannt sind, z.B.
Dicyclohexylcarbodiimid oder 1 -(3-Dimethylaminopropyl)-3-ethylcarbodiimide, oder durch vorgeschaltete Umwandlung der Säure in ein Säurechlorid, z.B. mit Thionylchlorid oder POCl3 und nachfolgende Umsetzung mit Q-NH2 das D-Ketoamid (III) erhalten.
Verbindung (III) wird entweder mit einer Alkylmetallverbindung R3-M, worin R3 die oben angegebenen Bedeutungen hat und M für ein Alkalimetall (Lithium, Natrium oder Kalium) oder MgX oder ZnX mit X = Halogen (Chlor, Brom, lod), oder durch Reaktion mit Verbindung (IV),
(R6)3Si-R3
(IV)
wobei R3 die oben angegebene Bedeutung hat und R6 eine CrC5-Alkylgruppe bezeichnet und die drei R6-Gruppen nicht gleich sein müssen, in Gegenwart eines Katalysators, z.B. Fluorid-Salzen oder Basen, wie etwa Alkalicarbonaten (J. Am. Chem. Soc. 1989, 111, 393), zur Titelverbindung (I) umgesetzt.
A2) für B = CO
(H) 3. verseifen (Via) (I)
Alternativ können auch D-Ketosäuren (II) zu Verbindungen (V),
R Rx (V)
worin A, R1 und R2 wie oben beschrieben definiert sind und R7 d-C -Alkyl ist, nach den üblichen Methoden, z.B. mit Thionylchlorid in Methanol oder Ethanol oder mit Methyliodid und Alkalicarbonat, verestert werden und in Analogie zur Reaktionsfolge A1) von (IM) in (I) mit Alkylmetallverbindungen der Formel R3-M worin R3 die oben angegebenen Bedeutungen hat und M für ein Alkalimetall (Lithium, Natrium oder Kalium) oder MgX oder ZnX mit X = Halogen (Chlor, Brom, lod) steht, oder mit (R6)3Si-R3 zur Verbindung (VI) umgesetzt werden.
(VI)
Der Ester (VI) wird unter Standardbedingungen, etwa wäßriger Alkalihydroxidlösung, zur Säure (Via; R
7 = H) verseift.
Die Säure (Via) wird zur Kupplung mit einem Aminochinazolin, Aminochinoxalin, Aminocinnolin, Aminoindazol, Aminophthalazin, Aminonaphthyridin, Aminobenzothiazol Aminodihydroindolon-, Amoinodihydroisoindolon-, Aminobenzimidazol- oder Aminoindol unter Verwendung eines gängigen Aktivierungsreagenzes, z.B. Thionylchlorid, gegebenenfalls in der Gegenwart eines Katalysators wie Dimethylaminopyridin, zur Titelverbindung (I) umgesetzt.
B) für B = eine gegebenenfalls durch Methyl oder Ethyl substituierte Methylenqruppe
c) Rl R
2 R
3 ΝH
2 A H ΝH-Q
Λ AX
a)
Eine Verbindung der allgemeinen Formel (VII) oder (VIII),
worin A, B und R1, R2 und R3 die oben angegebene Bedeutung haben und LG eine beliebige Fluchtgruppe wie Halogenid oder Sulfonat bedeutet, wird mit einer Verbindung der allgemeinen Formel (IX) oder (X)
Q-NH-R9 Q-N=C=O (IX) (X)
worin R9 ein Wasserstoffatom, eine d-C5-Acylgruppe oder Alkoxy- oder
Aryloxycarbonylgruppe bedeutet und Q die oben angegebene Bedeutung hat, umgesetzt, wobei der Rest R9 abgespalten oder ein intermediär gebildetes Oxazolidinon (vgl. z.B. S.J. Brickner, D.K. Hutchinson, M.R. Barbachyn, P.R. Maπninen, D.A. Ulanowicz, S.A. Garmon, K.C. Grega, S.K. Hendges, D.S. Toops, C.W. Ford, G.E. Zurenko J. Med. Chem. 1996, 39, 673) beispielsweise mit wäßrigen Alkalihydroxiden gespalten wird, um zur Titelverbindung (I) zu gelangen.
b) Ein anderer Weg besteht darin, Verbindungen der Formel (VII) oder (VIII) mit Stickstoffnucleophilen, beispielsweise Azid-Salzen oder Ammoniak umzusetzen, wobei sich im ersten Falle eine Reduktion in der dem Fachmann bekannten Weise, z.B. mit komplexen Hydrid reagenzien, wie Lithiumaluminiumhydrid, oder durch eine Übergangsmetall-katalysierte Hydrogenolyse anschließt, um zu Verbindungen der Formel (XI) zu gelangen.
Den Resten R1-R3, A und B kommt die gleiche Bedeutung wie oben angegeben zu.
C)
Verbindung (XI) kann unter Basenkatalyse, z.B. in Gegenwart tertiärer Aminbasen oder Alkalicarbonaten oder -hydroxiden, oder unter Übergangsmetallkatalyse, z.B. Palladiumkatalyse (J.P. Wolfe, S. Wagaw, J.-F. Marcoux, S.L. Buchwald Acc. Chem. Res. 1998, 31, 805; J.F. Hartwig Acc. Chem. Res. 1998, 31, 852), mit einem halogenierten Chinazolin, Chinoxalin,
Cinnolin, Indazol, Phthalazin, Naphthyridin, Benzothiazol Dihydroindolon, Dihydroisoindolon-, Benzimidazol- oder Indol in die Titelverbindung (I) übergeführt werden.
d)
Schließlich lässt sich die Titelverbindung (I) auch durch reduktive Aminierung einer Verbindung der Formel (XII), die nach dem Fachmann bekannten Methoden aus Verbindung (VI) mittels Reduktion oder Alkylierung erhalten werden kann, mit Q-NH2 synthetisieren, wobei z.B Natriumcyanoborhydrid, Natriumtriacetoxyborhydrid oder Wasserstoff unter Palladium Katalyse als Reduktionsmittel in Betracht kommen.
R8 bedeutet Wasserstoff, Methyl oder Ethyl gemäß der für die Methylengruppe in B definierten Substituenten.
Im Falle, dass die Verbindungen der allgemeinen Formel I als Salze vorliegen, kann dies beispielsweise in der Form des Hydrochlorids, Sulfats, Nitrats, Phosphats, Pivalats, Maleats, Fumarats, Tartrats, Benzoats, Mesylats, Citrats oder Succinats sein.
Wenn die erfindungsgemäßen Verbindungen als racemische Gemische vorliegen, können sie nach dem Fachmann geläufigen Methoden der Racemattrennung in die reinen, optisch aktiven Formen aufgetrennt werden. Beispielsweise lassen sich die racemischen Gemische durch Chromatographie an einem selbst optisch aktiven Trägermaterial (CHIRALPAK AD®) in die reinen Isomere trennen. Es ist auch möglich, die freie Hydroxygruppe in einer racemischen Verbindung der allgemeinen Formel I mit einer optisch aktiven Säure zu verestem. Die erhaltenen diastereoisomeren Ester können durch fraktionierte Kristallisation oder chromatographisch getrennt werden. Die
getrennten Ester werden dann jeweils zu den optisch reinen Isomeren verseift. Als optisch aktive Säure kann beispielsweise Mandelsäure, Camphersulfonsäure oder Weinsäure verwendet werden.
Die Bindung der Substanzen an den Glucocorticoid-Rezeptor (GR) und weitere Steroidhormon-Rezeptoren (Mineralcorticoid-Rezeptor (MR), Progesteron- Rezeptor (PR) und Androgen-Rezeptor (AR)) wird mit Hilfe rekombinant hergestellter Rezeptoren überprüft. Cytosolpräparationen von Sf9 Zellen, die mit rekombinanten Baculoviren, die für den GR kodieren, infiziert worden waren, werden für die Bindungsuntersuchungen eingesetzt. Im Vergleich zur
Bezugssubstanz [3H]-Dexamethason zeigen die Substanzen eine hohe bis sehr hohe Affinität zum GR.
Darüber hinaus zeigen die hier beschriebenen durch Chinazoline, Chinoxaline, Cinnoline, Indazole, Phthalazine, Naphthyridine, Benzothiazole, Dihydroindolone, Dihydroisoindolone, Benzimidazole und Indole substituierten Verbindungen der Formel (I) eine hohe Selektivität für den Glucocorticoid- Rezeptor. So zeigt Beispiel 2 z.B. folgendes Profil: IC50(GR) = 1.8 nM; IC50(MR), lC5o(PR), IC50(AR) > 1 DM und die Verbindung aus Beispiel 52: IC50(GR) = 10 nM; ICso(MR), IC50(PR), IC50(AR) > 1 DM.
Als wesentlicher, molekularer Mechanismus für die antiinflammatorische Wirkung von Glucocorticoiden wird die durch den GR vermittelte Hemmung der Transkription von Cytokinen, Adhäsionsmolekülen, Enzymen und anderer pro - inflammatorischen Faktoren angesehen. Diese Hemmung wird durch eine Interaktion des GR mit anderen Trankriptionsfaktoren, z.B. AP-1 und NF-kappa- B, bewirkt (zur Übersicht siehe Cato, ACB and Wade E, BioEssays 18, 371 -378 1996).
Die erfindungsgemäßen Verbindungen der allgemeinen Formel I hemmen die durch Lipopolysacchard (LPS) ausgelöste Sekretion des Cytokins IL-8 in der menschlichen Monozytenzellline THP-1. Die Konzentration der Cytokine wurde im Überstand mittels kommerziell erhältlicher ELISA-Kits bestimmt.
Die antiinflammatorische Wirkung der Verbindungen der allgemeinen Formel I wurden im Tierexperiment durch Testen in der Crotonöl - induzierten Entzündung in der Ratte und der Maus getestet (J. Exp. Med. (1995), 182, 99- 108). Hierzu wurde den Tieren Crotonöl in ethanolischer Lösung topisch auf die Ohren appliziert. Die Testsubstanzen wurden gleichzeitig oder zwei Stunden vor dem Crotonöl ebenfalls topisch oder systemisch appliziert. Nach 16-24 Stunden wurden das Ohrgewicht als Maß für das entzündliche Ödem, die Peroxidaseaktivität als Maß für die Einwanderungen von Granulozyten und die Elastaseaktivität als Maß für die Einwanderung von neutrophilen Granulozyten gemessen. Die Verbindungen der allgemeinen Formel I hemmen in diesem Test sowohl nach topischer, als auch nach systemischer Applikation die drei oben genannten Entzündungsparameter.
Eine der häufigsten unerwünschten Wirkungen einer Glucocorticoid - Therapie ist der sogenannte "Steroiddiabetes" [vgl. Hatz, HJ, Glucocorticoide: Immunologische Grundlagen, Pharmakologie und Therapierichtlinien, Wissenschafliche Verlagsgesellschaft mbH, Stuttgart, 1998]. Ursache hierfür ist die Stimulation der Gluconeogenese in der Leber durch Induktion der hierfür verantwortlichen Enzyme und durch freie Aminosäuren, die aus dem Abbau von Proteinen (katabole Wirkung der Glucocorticoide) entstehen. Ein Schlüsselenzym des katabolen Stoffwechsels in der Leber ist die Tyrosin- aminotranferase (TAT). Die Aktivität dieses Enzyms kann photometrisch aus Leberhomogenaten bestimmt werden und stellt ein gutes Maß für die unerwünschten metabolischen Wirkungen der Glucocorticoide dar. Zur Messung der TAT - Induktion werden die Tiere 8 Stunden nach Gabe der Testsubstanzen getötet, die Leber entnommen und die TAT - Aktivität im Homogenat gemessen. Die Verbindungen der allgemeinen Formel I induzieren in diesem Test in Dosen, in denen sie antiinflammatorisch wirksam sind, nicht oder nur in geringem Maße die Tyrosinaminotransferase.
Aufgrund ihrer antiinflammatorischen und zusätzlichen anti-allergischen, immun- suppressiven und anti-proliferativen Wirkung können die erfindungsgemäßen Verbindungen der allgemeinen Formel I als Medikamente zur Behandlung oder
Prophylaxe folgender Krankheitszustände bei Säugetieren und Menschen verwendet werden: Dabei steht der Begriff „ERKRANKUNG" für die folgenden Indikationen:
(i) Lungenerkrankungen, die mit entzündlichen, allergischen und / oder proliferativen Prozessen einhergehen: - Chronisch obstruktive Lungenerkrankungen jeglicher Genese, vor allem Asthma bronchiale - Bronchitis unterschiedlicher Genese - Alle Formen der restriktiven Lungenerkrankungen, vor allem allergische Alveolitis, - Alle Formen des Lungenödems, vor allem toxisches Lungenödem - Sarkoidosen und Granulomatosen, insbesondere Morbus Boeck (ii) Rheumatische Erkrankungen / Autoimmunerkrankungen / Gelenkerkrankungen, die mit entzündlichen, allergischen und / oder proliferativen Prozessen einhergehen: - Alle Formen rheumatischer Erkrankungen, insbesondere rheumatoide Arthritis, akutes rheumatisches Fieber, Polymyalgia rheumatica - Reaktive Arthritis - Entzündliche Weichteilerkrankungen sonstiger Genese - Arthritische Symtome bei degenerativen Gelenkerkrankungen (Arthrosen) - Traumatische Arthritiden - Kollagenosen jeglicher Genese, z.B. systemischer Lupus erythematodes, Sklerodermie, Polymyositis, Dermatomyositis- Sjögren-Syndrom, Still— Syndrom, Felty-Syndrom (iii) Allergien, die mit entzündlichen, und / oder proliferativen Prozessen einhergehen: - Alle Formen allergischer Reaktionen, z.B. Quincke Ödem, Heuschnupfen, Insektenstich, allergische Reaktionen auf Arzneimittel, Blutderivate, Kontrastmittel etc., Anaphylaktischer Schock, Urtikaria, Kontakdermatitis (iv) Gefäßentzündungen (Vaskulitiden) - Panarteriitis nodosa, Arteriitis temporalis, Erythema nodosum
(v) Dermatologische Erkrankungen, die mit entzündlichen, allergischen und / oder proliferativen Prozessen einhergehen: - Atopische Dermatitis (vor allem bei Kindern) - Psoriasis - Pityriasis rubra pilaris - Erythematöse Erkrankungen, ausgelöst durch unterschiedlichen Noxen, z.B. Strahlen, Chemikalien, Verbrennungen etc. - Bullöse Dermatosen - Erkrankungen des lichenoiden Formenkreises, - Pruritus (z. B. allergischer Genese) - Seborrhoisches Ekzem - Rosacea - Pemphigus vulgaris - Erythema exsudativum multiforme - Balanitis - Vulvitis - Haarausfall wie Alopecia areata - Cutane T - Zeil - Lymphome
(vi) Nierenerkrankungen, die mit entzündlichen, allergischen und / oder proliferativen Prozessen einhergehen: - Nephrotisches Syndrom - Alle Nephritiden
(vii) Lebererkrankungen, die mit entzündlichen, allergischen und / oder proliferativen Prozessen einhergehen: - akuter Leberzellzerfall - akute Hepatitis unterschiedlicher Genese, z.B. viral, toxisch, arzneimittelinduziert - chronisch aggressive und / oder chronisch intermittierende Hepatitis (viii) Gastrointestinale Erkrankungen, die mit entzündlichen, allergischen und / oder proliferativen Prozessen einhergehen: - regionale Enteritis (Morbus Crohn) - Colitis U Icerosa - Gastritis
- Refluxoesophagitis - Gastroenteritiden anderer Genese, z.B. einheimische Sprue (ix) Proktologische Erkrankungen, die mit entzündlichen, allergischen und / oder proliferativen Prozessen einhergehen: - Analekzem - Fissuren - Hämorrhoiden - idiopathische Proktitis (x) Augenerkrankungen, die mit entzündlichen, allergischen und / oder proliferativen Prozessen einhergehen: - allergische Keratitis, Uveitis, Iritis, - Konjunktivitis - Blepharitis - Neuritis nervi optici - Chorioditis - Ophtalmia sympathica (xi) Erkrankungen des Hals-Nasen-Ohren-Bereiches, die mit entzündlichen, allergischen und / oder proliferativen Prozessen einhergehen:
- allergische Rhinitis, Heuschnupfen - Otitis externa, z.B. bedingt durch Kontaktexem, Infektion etc.
- Otitis media (xii) Neurologische Erkrankungen, die mit entzündlichen, allergischen und / oder proliferativen Prozessen einhergehen: - Hirnödem, vor allem Tumor-bedingtes Hirnödem - Multiple Sklerose - akute Encephalomyelitis - Meningitis - verschieden Formen von Krampfanfällen, z.B. BNS-Krämpfe (xiii) Bluterkrankungen, die mit entzündlichen, allergischen und / oder proliferativen Prozessen einhergehen: - Erworbene hämolytische Anämie - Idopathische Thrombocytopenia
005/003098 20
(xiv) Tumorerkrankungen, die mit entzündlichen, allergischen und / oder proliferativen Prozessen einhergehen: - Akute lymphatische Leukämie - Maligne Lymphome - Lymphogranulomatosen - Lymphosarkome - Ausgedehnte Metastasierungen, vor allem bei Mamma- Bronchial- und Prostatakarzinom (xv) Endokrine Erkrankungen, die mit entzündlichen, allergischen und / oder proliferativen Prozessen einhergehen: - Endokrine Orbitopathie - Thyreotoxische Krise - Thyreoiditis de Quervain - Hashimoto Thyreoiditis - Morbus Basedow
(xvi) Organ- und Gewebstransplantationen , Graft-versus-host-disease(xvii) Schwere Schockzustände, z.B anaphylaktischer Schock , systemic inflammatory response syndrome (SIRS) (xviii) Substitutionstherapie bei: - angeborene primäre Nebenniereninsuffizienz, z.B. kongenitales adrenogenitales Syndrom - erworbene primäre Nebenniereninsuffizienz, z.B. Morbus Addison, autoimmune Adrenalitits, postinfektiös, Tumoren, Metastasen etc. - angeboren sekundäre Nebenieren Insuffizienz, z.B. kongenitaler Hypopitutitarismus - erworbene sekundäre Nebenniereninsuffizienz, z.B. postinfektiös, Tumoren etc.
(xix) Emesis, die mit entzündlichen, allergischen und / oder proliferativen Prozessen einhergehen: * - z.B. in Kombination mit einem 5-HT3-Antagonisten bei Zytostika - bedingten Erbrechen, (xx) Schmerzen bei entzündlicher Genese, z.B. Lumbago
Darüber hinaus können die erfindungsgemäßen Verbindungen der allgemeinen Formel I zur Therapie und Prophylaxe weiterer oben nicht genannter Krankheitszustände eingesetzt werden, für die heute synthetische Glucocorticoide verwendet werden (siehe dazu Hatz, HJ, Glucocorticoide: Immunologische Grundlagen, Pharmakologie und Therapierichtlinien, Wissenschafliche Verlagsgesellschaft mbH, Stuttgart, 1998).
Alle zuvor genannten Indikationen (i) bis (xx) sind ausführlich beschrieben in Hatz, HJ, Glucocorticoide: Immunologische Grundlagen, Pharmakologie und Therapierichtlinien, Wissenschafliche Verlagsgesellschaft mbH, Stuttgart, 1998.
Für die therapeutischen Wirkungen bei den oben genannten Krankheitszuständen ist die geeignete Dosis unterschiedlich und hängt beispielsweise von der Wirkstärke der Verbindung der allgemeinen Formel I, dem Wirt, der Art der Verabreichung und der Art und der Schwere der zu behandelnden Zustände, sowie der Verwendung als Prophylaktikum oder Therapeutikum ab.
Die Erfindung liefert weiterhin (i) die Verwendung eines der erfindungsgemäßen Verbindung gemäß Formel I oder deren Gemisch zur Herstellung eines Medikaments zur Behandlung von einer ERKRANKUNG; (ii) ein Verfahren zur Behandlung von einer ERKRANKUNG, welches Verfahren eine Verabreichung einer Verbindungsmenge gemäß der Erfindung umfasst, wobei die Menge die Krankheit unterdrückt, und wobei die Verbindungsmenge einem Patienten gegeben wird, der ein solches Medikament benötigt; (iii) eine pharmazeutische Zusammensetzung zur Behandlung von einer ERKRANKUNG, welche Behandlung eines der erfindungsgemäßen Verbindungen oder deren Gemisch und wenigstens einen pharmazeutischen Hilfs- und/oder Trägerstoff umfasst.
Im allgemeinen sind bei Tieren zufriedenstellende Resultate zu erwarten, wenn die täglichen Dosen einen Bereich von 1 μg bis 100.000 μg der erfindungsgemäßen Verbindung pro kg Körpergewicht umfassen. Bei größeren Säugetieren, beispielsweise dem Menschen, liegt eine empfohlene tägliche Dosis im Bereich von 1 μg bis 100.000 μg pro kg Körpergewicht. Bevorzugt ist eine Dosis von 10 bis 30.000 μg pro kg Körpergewicht, mehr bevorzugt eine Dosis von 10 bis 10.000 μg pro kg Körpergewicht. Zum Beispiel wird diese Dosis zweckmäßigerweise mehrmals täglich verabreicht. Zur Behandlung eines akuten Schocks (z.B. anaphylaktischer Schock) können Einzeldosen gegeben werden, die deutlich über den oben genannten Dosen liegen.
Die Formulierung der pharmazeutischen Präparate auf Basis der neuen Verbindungen erfolgt in an sich bekannter Weise, indem man den Wirkstoff mit den in der Galenik gebräuchlichen Trägersubstanzen, Füllstoffen, Zerfallsbeeinflussern, Bindemitteln, Feuchthaltemitteln, Gleitmitteln,
Absorptionsmitteln, Verdünnungsmitteln, Geschmackskorrigentien, Färbemitteln usw., verarbeitet und in die gewünschte Applikationsform überführt. Dabei ist auf Remington's Pharmaceutical Science, 15th ed. Mack Publishing Company, East Pennsylvania (1980) hinzuweisen.
Für die orale Applikation kommen insbesondere Tabletten, Dragees, Kapseln, Pillen, Pulver, Granulate, Pastillen, Suspensionen, Emulsionen oder Lösungen in Frage.
Für die parenterale Applikation sind Injektion- und Infusionszubereitungen möglich.
Für die intraartikulären Injektion können entsprechend zubereitet Kristallsuspensionen verwendet werden.
Für die intramuskuläre Injektion können wässrige und ölige Injektionslösungen oder Suspensionen und entprechende Depotpräparationen verwendet werden.
Für die rektale Applikation können die neuen Verbindungen in Form von Suppositorien, Kapseln, Lösungen (z.B. in Form von Klysmen) und Salben sowohl zur systemischen, als auch zur lokalen Therapie verwendet werden.
Zur pulmonalen Applikation der neuen Verbindungen können diese in Form von Aerosolen und Inhalaten verwendet werden.
Für die lokale Anwendung an Augen, äußerem Gehörgang, Mittelohr, Nasenhöhle und Nasennebenhöhlen können die neuen Verbindungen als Tropfen, Salben und Tinkturen in entsprechenden pharmazeutischen Zubereitungen verwendet werden.
Für die topische Auftrag ung sind Formulierungen in Gelen, Salben, Fettsalben, Cremes, Pasten, Puder, Milch und Tinkturen möglich. Die Dosierung der Verbindungen der allgemeinen Formel I sollte in diesen Zubereitungen 0.01 % - 20% betragen, um eine ausreichende pharmakologische Wirkung zu erzielen.
Die Erfindung umfasst ebenfalls die erfindungsgemäßen Verbindungen der allgemeinen Formel I als therapeutischen Wirkstoff. Weiterhin gehören zur Erfindung die Verbindungen der allgemeinen Formel I als therapeutischen Wirkstoff zusammen mit pharmazeutisch verträglichen und annehmbaren Hilfsstoffen und Trägerstoffen. Ebenfalls umfasst die Erfindung eine pharmazeutische Zusammensetzung, die eine oder mehrere der pharmazeutisch aktiven, erfindungsgemäßen Verbindungen oder deren Gemisch oder deren pharmazeutisch verträgliches Salz oder pharmazeutisch verträgliche Hilfsstoffe und Trägerstoffe enthält.
Die nachstehenden Beispiele dienen der näheren Erläuterung der Erfindung ohne sie darauf beschränken zu wollen. Die Synthesen von wichtigen Vorstufen, die im Rahmen des experimentellen Teils nicht offenbart sind, sind bereits Stand der Technik, und können zum Beispiel aus der WO 98/54159 , (WO 00/32584) oder WO 02/10143 entnommen werden.
Experimenteller Teil Beispiel 1
4-(5-Fluor-2-methoxyphenyl)-4-methyl-1-(phthalazin-5-ylamino)-2- (trifluormethyl)-pentan-2-ol
4-(5-Fluor-2-methoxy-phenyl)-2-hydroxy-4-methyl-2-trifluoromethyl-pentanal 0.81 ml (8.67 mmol) Oxalylclorid werden in 15 ml Dichlormethan auf -60°C gekühlt und mit 1.6 ml (22.6 mmol) Dimethylsulfoxid in 10 ml Dichlormethan versetzt. Nach 15 min werden 1.0 g (3.22 mmol) 4-(5-Fluor-2-methoxy-phenyl)- 4-methyl-2-trifluoromethyl-pentan-1 ,2-diol (WO 00/32584) in 10 ml Dichlormethan zugeben und die Mischung wird für eine Stunde bei -60°C gerührt. Es werden 4.1 ml (29 mmol) Triethylamin zugegeben und man läßt die Mischung über 30 min auf RT erwärmen. Man gießt in 50 ml Wasser und extrahiert mit CH2CI2. Die vereinigten organischen Extrakte werden mit gesättigter NaCI-Lösung gewaschen, getrocknet (Na2SO ) und im Vakuum eingeengt. Nach Chromatographie an Kieselgel mit Hexan-Essigester (0-30 %) werden 600 mg des Produkts erhalten. 'H-NMR (CDCI3); δ = 1.38 (s, 3H), .47 (s, 3H), 2.23 (d, 1 H), 3.36 (d, 1 H), 3.86 (s, 3H), 6.77 (dd, 1H), 6.87 (dd, 1H), 6.91 (ddd, 1H), 9.05 (s, 1H).
Zu 70 mg (0.48 mmol) 5-Aminophthalazin (I.A. Shaikh, F. Johnson, A.P. Grollman J. Med. Chem.1986, 26, 1329-1340) in 2 ml Essigsäure werden 200 mg (0.65 mmol) 4-(5-Fluor-2-methoxy-phenyI)-2-hydroxy-4-methyl-2- trifluoromethyl-pentanal in 5 ml Toluol gegeben. Die Reaktionslösung wird 6 Stunden zum Rückfluss unter Wasserabscheid ung erhitzt und weitere 4 Stunden über Molsieb (4 A) refluxiert. Das Lösungsmittel wird im Vakuum entfernt und Reste Essigsäure werden durch azeotrope Codestillation mit Toluen beseitigt. Nach Chromatographie an Kieselgel mit Hexan-Essigester (0- 70 %) werden 40 mg
4-(5-Fluor-2-methoxyphenyl)-4-methyl-1-(phthalazin-5-ylimino)-2-(trifluormethyl)- pentan-2-ol erhalten. Zu 10 mg Imin in 10 ml Ethylacetat und 1 ml Triethylamin werden 20 mg Palladium auf Kohle gegeben und man schüttelt 2 h unter einer Wasserstoffatmosphäre bei Normaldruck. Die Lösung wird mittels Filtration vom Katalysator befreit und eingedampft. Nach Chromatographie an Kieselgel mit Hexan-Essigester (0-70 %) werden 4 mg des gewünschten Produktes erhalten Η-NMR (CDCI3); δ = 1.44 (s, 3H), 1.66 (s, 3H), 2.07 (d, 1 H), 3.07 (d, 1 H), 3.16 (d, 1 H), 3.24 (d, 1 H), 3.85 (s, 3H), 6.42 (d, 1 H), 6.76 (m, 2H), 7.11 (dd, 1 H), 7.30 (d, 1 H), 7.66 (dd, 1 H), 9.38 (s, 1 H), 9.48 (s, 1 H).
Beispiel 2
4-(5-Fluor-2-methoxyphenyl)-4-methyl-1-(2-methychinazolin-5-ylamino)-2- (trifluormethyl)-pentan-2-ol
5-Amino-2-methychinazoIin
12,7g (mmol) 2-Methyl-5-πitro-3H-chinazolin-4-on (M.T. Bogert, V.J. Chambers J. Org Chem. 1905, 649-658) und 37,5g Phosphorpentachlorid werden in 75 ml Phosphorylchlorid über 20 Stunden unter Rückfluss erhitzt. Nach dem Abkühlen gießt man in ges. NaHCO3 Lösung und extrahiert mit Ethylacetat. Die organische Phase wird getrocknet und das Lösungsmittel entfernt. Man erhält 14g 4-Chlor-2-methyl-5-nitrochinazolin, von denen 4.5 g (20.2 mmol) in 225 ml Ethylacetat und 22.5 ml Triethylamin gelöst werden. Man gibt 2 g Palladium auf Kohle zu und rührt bei Eiskühlung 4 Stunden unter einer Wasserstoff- atmosphäre bei Normaldruck. Die Lösung wird mittels Filtration über Celite vom Katalysator befreit, wobei mit 200 ml Ethanol nachgewaschen wird, und eingedampft. Nach Chromatographie an Kieselgel mit Essigester-Ethanol (0-10%) werden 530 mg des Produkts erhalten.'H-NMR (CDCI3); δ = 2.87 (s, 3H), 4.52 (br., 2H), 6.77 (d, 1 H), 7.33 (d, 1 H), 7.65 (t, 1 H), 9.40 (s, 1 H).
180 mg (0.48 mmol) 4-(5-Fluor-2-methoxyphenyl)-2-hydroxy-4-methyl-2-(trifluor- methyl)pentanal und 50 mg 5-Amino-2-methychinazolin werden in 20 ml Dichlorethan und 2 ml Essigsäure unter kontinuierlichem langsamen Entfernen des Lösungsmittels über 5 Stunden auf 5 ml aufkonzentriert. Das restliche Lösungsmittel wird im Vakuum entfernt und Reste Essigsäure werden durch azeotrope Codestillation mit Toluen beseitigt. Nach Chromatographie an Kieselgel mit Hexan-Essigester (0-70 %) werden 58 mg 4-(5-Fluor-2- methoxyphenyl)-4-methyl-1-(2-methychinazolin-5-ylimino)-2-(trifluormethyl)- pentan-2-ol erhalten. Zum Imin in 10 ml Ethylacetat und 1 ml Triethylamin wer- den 20 mg Palladium auf Kohle gegeben und man schüttelt 2 h unter einer
Wasserstoffatmosphäre bei Normaldruck. Die Lösung wird mittels Filtration vom Katalysator befreit und eingedampft. Man nimmt in 5 ml Chloroform auf und gibt 200 mg aktiviertes Mangandioxid zu und rührt 30 min. Es wird über Celite filtriert und im Vakuum eingeengt. Nach Chromatographie an Kieselgel mit Hexan- Essigester (0-70 %) werden 22 mg des Produkts erhalten. Η-NMR (CDCI3); δ = 1.47 (s, 3H), 1.56 (s, 3H), 2.38 (d, 1 H), 2.77 (d, 1 H), 2,83 (s, 3H), 3.16 (dd, 1 H), 3.33 (dd, 1 H), 3.85 (s, 3H), 4.70 (br., 1 H), 6.05 (d, 1 H), 6.77 (dd, 1 H), 6.88 (ddd, 1 H), 7.09 (dd, 1 H), 7.24 (d, 1 H), 7.56 (t, 1H), 9.16 (s, 1 H).
Beispiel 3
4-(5-Fluor-2-hvdroχyphenyl)-4-methyl-1-(2-methvchinazolin-5-ylamino)-2- (trifluormethyl)-pentan-2-ol 103 mg (0.23 mmol) 4-(5-Fluor-2-methoxyphenyl)-4-methyl-1 -(2- methychinazoIin-5-ylamino)-2-(trifluormethyl)-pentan-2-ol in 10 ml CH2CI2 werden bei 0 °C mit 5 ml 1 M Bortribromid-CH2Cl2-Lösung versetzt. Nach 10 h werden weitere 5 ml 1M Bortribromid-CH2CI2-Lösung zugegeben und bei Raumtemperatur wird der Ansatz nach 72 h in gesättigte NaHCO3 gegossen, 20 Minuten gerührt und mit CH2CI2 extrahiert. Die vereinigten organischen Extrakte
werden mit Wasser gewaschen, getrocknet (Na2SO4) und im Vakuum eingeengt. Chromatographie mit Hexan-2-Propanol (0-20 %) an Kieselgel liefert 80 mg des Produkts.
Η-NMR (CDCI3); δ = 1.51 (s, 3H), 1.58 (s, 3H), 2.37 (d, 1 H), 2.81 (s, 3H), 2.91 (d, 1 H), 3.25 (dd, 1 H), 3.43 (dd, 1 H), 5.05 (br., 1 H), 6.20 (d, 1 H), 6.54 (dd, 1H), 6.69 (m, 1H), 7.05 (dd, 1H), 7.23 (d, 1 H), 7.59 (d, 1 H), 7.58 (d, 1 H), 8.32 (d, 1 H), 8.68 (d, 1 H).
Beispiel 4
4-(2.5-Difluorphenyl)-4-methyl-1-(2-methylchinazolin-5-ylamino)-2-
(trifluormethyl)-pentan-2-ol
4-(2,5-Difluoφhenyl)-2-hydroxy-4-methyl-2-trifIuormethyl-pentanal
5.4 g (15.5 mmol) 4-(2,5-Difluorphenyl)-2-hydroxy-4-methyl-2-trifluoromethyl- valeriansäureethylester (WO 02/10143) werden bei 0°C in Diethylether gelöst und innerhalb von 20 min mit 1.76 g (46.5 mmol) Lithiumaluminiumhydrid versetzt. Man lässt bei RT 4 h lang rühren, gibt vorsichtig soviel gesättigte NaHCO3-Lösung zu, bis keine Gasentwicklung mehr beobachtet wird. Die Mischung wird mit Essigester verdünnt, noch 15 min gerührt und dann der gebildete Niederschlag abfiltriert. Man engt ein und chromatographiert an Kieselgel mit Hexan/Ethylacetat (50 %). Man erhält 2.45 g 2,5-Difluorphenyl)-4- methyl-2-trifluormethyl-pentan-1 ,2-diol als schwach gelbliches kristallisierendes Öl. 800 mg (2.8 mmol) 4-(2,5-Difluorphenyl)-4-methyl-2-trifluormethyl-pentan- 1 ,2-diol werden in 20 ml Dichlormethan vorgelegt und bei 0°C 9.5 ml DMSO und 1.95 ml Triethylamin zugegeben. Die Lösung wird langsam mit 1.34 g (8.4 mmol) SO3-Pyridin-Komplex versetzt und 2 h bei 0°C gerührt. Die Mischung wird zwischen ges. Ammoniumchloridlösung und MTBE verteilt, die Phasen getrennt und die wässrige Phase mit MTBE extrahiert. Die vereinigten organischen Phasen werden mit Wasser und ges. NaCI-Lösung gewaschen und mit NaSO4 getrocknet. Man engt ein und chromatographiert an Kieselgel mit
Hexan/Essigester (30%). Man erhält 710 mg des gewünschten Produkts. 1H- NMR (CDCI3): δ = 1.41 (s, 3H), 1.48 (s, 3H), 2.39 (d, 2H), 3.02 (d, 1 H), 3.61 (s, 1 H), 6.84-7.18 (m, 3H), 9.23 (s, 1 H).
240 mg (0.84 mmol) 4-(2,5-Difluor-phenyl)-2-hydroxy-4-methyl-2-trifluormethyl- pentanal und 200 mg (1.26 mmol) 5-Amino-2-methyl-chinazolin werden zunächst analog Beispiel 2 umgesetzt. Nach Chromatographie an Kieselgel mit Hexan-Essigester (0-70 %) erhält man 80 mg 4-(2,5-Difluorphenyl)-4-methyl-1- (2-methylchinazolin-5-ylimino)-2-(trifIuormethyl)-pentan-2-ol, die in Essigester / Ethanol 1:1 wieder aufgenommen und mit 10 mg Palladium-Katalysator (10% auf Aktivkohle) unter Wasserstoffatmosphäre (1 atm) hydriert werden. Nach 5 Stunden bei RT wird der Katalysator abgesaugt und das Filtrat eingeengt. Man nimmt den Rückstand in Chloroform wieder auf und setzt analog Beispiel 2 mit Braunstein um. Nach chromatographischer Reinigung erhält man 15 mg des gewünschten Produkts als rotbraunen Film. MS (ESI): 440 (M+H); 1H-NMR (CDCI3): δ = 1.48 (s, 3H), 1.62 (s, 3H), 2.29 (d, 1 H), 2.61 (d, 1 H), 2.79 (s, 3H), 3.19-3.35 (m, 2H), 3.61 (s, 1 H), 4.69-4.73 (m, 1 H), 6.00 (d, 1 H), 6.83-6.91 (m, 2H), 7.08-7.14 (m, 1H), 7.23 (d, 1 H), 7.52 (dd, 1 H), 9.14 (d, 1 H).
Beispiel 5
4-(5-Fluor-2-methoxyphenyl)-4-methyl-1-(2-methylbenzothiazol-7-ylamino)-2-
(trifluormethyl)-pentan-2-o!
200 mg (0,65mmol) 4-(5-Fluor-2-methoxy-phenyl)-2-hydroxy-4-methyI-2- trifluoromethyl-pentanal und 126 mg (0,77 mmol) 7-Amino-2-methylbenzothiazol (Libeer et al. Bull.Soc.Chim.Belg.; 1971 ; 80; 43-47) werden in 8 ml Essigsäure über 5 h auf 125°C erhitzt. Nach dem Abkühlen auf Raumtemperatur versetzt man mit 214 mg (1.01 mmol) Natriumtriacetoxyborhydrid und lässt 16 h rühren. Nach der Zugabe von weiteren 100 mg (0.47 mmol) Natriumtriacetoxyborhydrid und 2 Stunden Rühren gibt man Toiuol zu und engt im Vakuum ein. Der
Rückstand wird in Ethylacetat aufgenommen, die organische Phase mit ges. Natriumhydrogencarbonat und ges. Natriumchlorid Lösung gewaschen und über Natriumsulfat getrocknet. Nach Chromatographie an Kieselgel mit Hexan- Essigester (0-50 %) werden 221 mg des Produkts erhalten. Η-NMR (CDCI3); δ = 1.45 (s, 3H), 1.58 (s, 3H), 2.25 (d, 1H), 2.78 (d, 1 H), 2.82 (s, 3H), 3.14 (s, 1 H), 3.16 (dd, 1 H), 3.28 (dd, 1 H), 3.48 (dd, 1 H), 3.84 (s, 3H), 4.23 (d, 1 H), 5.97 (d, 1 H), 6.82 (dd, 1 H), 6.96 (ddd, 1 H), 7.15 (dd, 1 H), 7.21 (t, 1 H), 7.42 (d, 1 H).
Beispiel 6
4-(5-Fluor-2-hvdroxyphenyl)-4-methyl-1-(2-methylbenzothiazol-7-ylamino)-2- (trifluormethyl)pentan-2-ol
Analog zu Beispiel 3 werden 150 mg (0.13 mmol) 4-(5-Fluor-2-methoxyphenyl)- 4-methyl-1-(2-methylbenzothiazol-7-ylamino)-2-(trifluormethyl)-pentan-2-ol in 15 ml CH2CI2 mit 6.8 ml 1 M Bortribromid-CH2Cl2-Lösung umgesetzt. Nach Chromatographie an Kieselgel mit Hexan-Essigester (0-70 %) werden 102 mg des Produkts erhalten. 1H-NMR (CDCI3); δ = 1.50 (s, 3H), 1.59 (s, 3H), 2.31 (d, 1 H), 2.79 (d, 1H), 2.80 (s, 3H), 3.27 (m, 2H), 3.40 (dd, 1 H), 3.54 (dd, 1H), 6.02 (d, 1 H), 6.11 (br., 1H), 6.65 (dd, 1 H), 6.82 (ddd, 1 H), 7.12 (dd, 1 H), 7.18 (t, 1 H), 7.40 (d, 1 H).
1-(Chinoxalin-5-ylamino)-4-(5-fluor-2-methoxyphenyl)-4-methyl-2- (trifluormethyl)-pentan-2-ol
Zu 80 mg (0.55 mmol) 5-Aminochinoxalin (J. Salon, V. Milata, N. Pronayova, J. Lesko Monatsh. Chem. 2000, 131, 293-299)in 2 ml Essigsäure werden 140 mg (0.46 mmol) 4-(5-Fluor-2-methoxy-phenyl)-2-hydroxy-4-methyl-2-trifluoromethyl- pentanal, gelöst in 5 ml Dichlorethan gegeben. Die Reaktionslösung wird 5 Stunden über Molsieb (4 A) refluxiert. Die Mischung wird zwischen Wasser und Dichlormethan verteilt und extrahiert (CH
2CI
2). Die vereinigten organischen Phasen werden gewaschen (ges. NaCl-Lösung), getrocknet (Na
2SO
4) und eingeengt. Nach chromatographischer Reinigung an Kieselgel mit Hexan/Ethylacetat (0-50%) erhält man 82 mg 1-(Chinoxalin-5-ylimino)-4-(5-fluor- 2-methoxyphenyl)-4-methyl-2-(trifluormethyl)-pentan-2-ol, die in 3 ml Methanol aufgenommen und mit 100 μl Essigsäure und 10 mg (0.26 mmol) NaBH versetzt werden. Die Reaktionsmischung wird 2 Tage bei RT gerührt, dabei werden zwei mal jeweils weitere 10 mg NaBH
4 zugesetzt. Die Mischung wird zwischen Wasser und Dichlormethan verteilt und extrahiert (CH
2CI
2). Die vereinigten organischen Phasen werden gewaschen (ges. NaCl-Lösung), getrocknet (Na
2SO
4) und eingeengt. Das Rohprodukt wird durch Chromatographie an Kieselgel mit Hexan/Essigester (10-50%) gereinigt. Man erhält 40 mg des gewünschten Produkts, das aus Hexan/Diethylether rekristallisiert werden kann. MS (ESI): 438 (M+H);
1H-NMR (CDCI
3): δ = 1.46 (s, 3H), 1.61 (s, 3H), 2.26 (d, 1 H), 2.80 (d, 1 H), 2.99 (s, 1 H), 3.22-3.49 (m, 3H), 3.85 (s, 3H), 6.07 (d, 1 H), 6.81 (dd, 1 H), 6.91-6.99 (m, 1 H), 7.19 (dd, 1H), 7.36 (dd, 1 H), 7.46 (d, 1 H), 8.61 (d, 1 H), 8.80 (d, 1 H).
Beispiel 8
1-(Chinoxalin-5-ylamino)-4-(5-fluor-2-hvdroxyphenyl)-4-methyl-2-(trifluormethyl)- pentan-2-ol
Analog zu Beispiel 3 werden 30 mg (68 μmol) 1-(Chinoxalin-5-ylamino)-4-(5- fluor-2-methoxyphenyl)-4-methyl-2-(trifluormethyl)-pentan-2-ol, gelöst in 3 ml
Dichlormethan mit 3 ml Bortribromid-Lösung (1 M in CH2CI2) umgesetzt und bei RT 24 h gerührt. Die Mischung wird zwischen Essigester und ges. NaHCO - Lösung verteilt und mit Essigester extrahiert. Die vereinigten organischen Phasen werden gewaschen (ges. NaCl-Lösung), getrocknet (Na2SO4) und eingeengt. Das Rohprodukt wird durch Chromatographie an Kieselgel mit Hexan/Essigester (20%) gereinigt. Man erhält 15 mg des gewünschten Produkts. MS (ESI): 424 (M+H); 1H-NMR (CDCI3): δ = 1.46 (s, 3H), 1.53 (s, 3H), 2.28 (d, 1 H), 2.58 (d, 1H), 2.97 (br, 1H), 3.30-3.56 (m, 4H), 6.18 (d, 1H), 6.56 (dd, 1 H), 6.76-6.83 (m, 1 H), 7.15 (dd, 1 H), 7.36 (d, 1H), 7.46 (d, 1 H), 8.65 (d, 1 H), 8.83 (d, 1H).
Beispiel 9
D-r(Chinoxalin-5-ylamino)methyll-1-(2-chlor-5-fluorphenyl)-D-(trifluormethyl) cvclobutanethanol
1-(2-Chlor-5-fluorphenyl)-Ω-hydroxy-π-(thfluormethyl)cyclobutanpropanal 3.1 g (8.7 mmol) 1-(2-Chlor-5-fluorphenyl)-D-hydroxy-D-(trifluormethyl) cyclobutanpropionsäureethylester (WO 02/10143) werden analog Beispiel 4 mit 990 mg (26.1 mmol) Lithiumaluminiumhydrid umgesetzt. Man erhält 1.80 g 1-(2- Chlor-5-fluorphenyl)-D-(hydroxy)-D-(trifluormethyl)cyclobutanpropanol als schwach gelbliches Öl. In 20 ml Dichlormethan werden 493 μl (2.56 mmol) Oxalylchlorid vorgelegt. Bei -75°C werden 802 μl (11.3 mmol) DMSO und nach 15 min rühren eine Lösung von 800 mg (2.56 mmol) des 1-[(Chlor-5- fluorphenyl)-D-(hydroxy)-D-(trifluormethyl)cyclobutanpropanol in 10 ml Dichlormethan zugetropft. Nach weiteren 15 min werden 2.20 ml (15.8 mmol) Triethylamin zugetropft und weitere 30 min bei -60°C und 30 min bei 0°C gerührt. Die Reaktion wird durch Zugabe von Wasser beendet, die Phasen getrennt und mit Dichlormethan extrahiert. Die vereinigten organischen Phasen werden mit Wasser und ges. NaCl-Lösung gewaschen und mit NaSO getrocknet. Man engt ein und chromatographiert an Kieselgel mit
Hexan/Essigester (30%). Man erhält 810 mg des gewünschten Produkts. MS (Cl): 342 (M+NH4); 1H-NMR (CDCI3): δ = 1.74-1.92 (m, 1 H), 2.00-2.70 (m, 5H), 2.86 (d, 1 H), 3.19 (d, 1 H), 3.52 (s, 1 H), 6.79-6.93 (m, 1 H), 7.10-7.24 (m, 2H), 8.94 (s, 1 H).
Zu 325 mg (0.96 mmol) 5-Aminochinoxalin (J. Salon, V. Milata, N. Pronayova, J. Lesko Monatsh. Chem. 2000, 131, 293-299) in 3 ml Essigsäure werden 200 mg (0.64 mmol) 1-[2-Chlor-5-fluorphenyl)-G-hydroxy-D- (trifluormethyl)cyclobutanpro-panal in 2 ml Toluol gegeben und 24 h bei RT gerührt. Die Lösung wird zwischen Toluol und Wasser verteilt, die wässrige Phase mit Toluol extrahiert, die vereinigten organischen Phasen mit ges. NaCl- Lösung gewaschen, getrocknet (Na2SO ) und das Lösemittel entfernt. Das rohe D-[(Chinoxalin-5-ylimino)methyl]-1-(2-chlor-5-fluorphenyl)-D-(trifluor- methyl)cyclobutanethanol wird in Methanol/Essigsäure 1 :1 aufgenommen und mit 100 mg (2.66 mmol) NaBH versetzt. Nach 6 Stunden rühren bei RT wird die Reaktion durch Zugabe von ges. NH4CI-Lösung abgebrochen und die Mischung mit Dichlormethan verdünnt. Nach Extraktion mit Dichlormethan werden die vereinigten organischen Phasen gewaschen (ges. NaCl-Lösung), getrocknet (Na2SO ) und die Lösemittel entfernt. Man erhält 280 mg Produkt als dunkelrotes Harz, das aus Hexan/Diethylether kristallisiert werden kann. MS (ESI): 454 (M+H).
Beispiel 10
4-(5-Fluor-2-methoxyphenyl)-4-methyl-1-(2-methyl-1.8-naphthyridin-5-ylamino)- 2-(trifluormethvP-pentan-2-ol 1-Amino-4-(5-fluor-2-methoxyphenyl)-4-methyl-2-(trifluormethyl)propan-2-ol 1.0 g (3.4 mmol) 2-[2-(5-Fluor-2-methoxyphenyl-2-methylpropyl]-2- (trifluormethyl)oxiran (WO 00/32584) in 68 ml THF werden mit 1.1 g Natriumazid und 180 mg Ammoniumchlorid in 14 ml Wasser und 26 ml Ethanol 6 h unter
Rückfluß erhitzt. Der Ansatz wird eingeengt, mit Ether verdünnt, mit Wasser gewaschen, getrocknet (Na
2SO
4) und eingeengt. Chromatographie an Kieselgel mit Hexan-Essigester (0-15 %) liefert 950 mg 1-Azido-4-(5-fluor-2- methoxyphenyl)-4-methyl-2-(trifluormethyl)propan-2-ol. Diese werden in 29 ml THF gelöst und bei 0 °C portionsweise mit 270 mg Lithiumaluminiumhydrid versetzt. Nach 1 h wird der Ansatz mit Essigester und Wasser behandelt und über Celite filtriert. Die Essigesterphase wird getrocknet (Na
2SO ) und im Vakuum eingeengt. 920 mg Amin werden erhalten.
1H-NMR (CDCI
3): δ = 1.4 (s, 3H), 1.5 (s, 3H), 2.15 (d, 1 H), 2.45 (d, 1 H), 2.55 (d, 1 H), 2.75 (d, 1 H), 2.80 (m), 3.8 (s, 3H), 6.8 (dd, 1 H), 6.9 (td, 1 H), 7.05 (dd, 1 H)
202 mg (1 ,13 mmol) 5-Chlor-2-methyl-1 ,8-naphthyridin (E.V. Brown, J. Org. Chem 1965, 1607-1609) werden zu 350 mg (1 ,13 mmol) 1-Amino-4-(5-fluor-2- methoxyphenyl)-4-methyl-2-(trifluormethyl)-pentan-2-ol und 128 mg (1 ,13 mmol) DABCO gegeben. Man erhitzt die für 1 ,5 h auf 150°C. Nach Chromatographie der erkalteten Schmelze an Kieselgel mit Dichlormethan/Methanol (0-10%) erhält man 385 mg gewünschtes Produkt. Η-NMR (CDCI3); δ = 1.46 (s, 3H), 1.58 (s, 3H), 2.45 (d, 1H), 2.68 (s, 3H), 2.72 (d, 1 H), 3.20 (d, 1 H), 3.38 (d, 1H), 3.83 (s, 3H), 5.86 (d, 1H), 6.77 (dd, 1 H), 6.92 (ddd, 1H), 7.08 (dd, 1 H), 7.11 (d, 1 H), 7.71 (d, 1 H), 8.50 (d, 1H).
Beispiel 11
4-(5-Fluor-2-hvdroxyphenyl)-4-methyl-1-(2-methyl-1 ,8-naphthyridin-5-ylamino)- 2-(trifluormethyl)-pentan-2-ol
Analog zu Beispiel 3 werden mg (0.13 mmol) 4-(5-Fluor-2-methoxyphenyl)-4- methyl-1-(2-methyl-1 ,8-naphthyridin-5-ylamino)-2-(trifluormethyl)-pentan-2-ol in 15 ml CH2CI2 mit ml 1M Bortribromid-CH2Cl2-Lösung umgesetzt. Nach Chromatographie an Kieselgel mit Hexan-Essigester (0-70 %) werden 102 mg des Produkts erhalten. 1H-NMR (CDCI3); δ = 1.50 (s, 3H), 1.59 (s, 3H), 2.31 (d,
1 H), 2.79 (d, 1 H), 2.80 (s, 3H), 3.27 (m, 2H), 3.40 (dd, 1 H), 3.54 (dd, 1H), 6.02 (d, 1 H), 6.11 (br., 1 H), 6.65 (dd, 1 H), 6.82 (ddd, 1H), 7.12 (dd, 1H), 7.18 (t, 1 H), 7. (d, 1 H).
Beispiel 12
1-(Cinnolin-5-ylamino)-4-(5-fluor-2-hvdroxyphenyl)-4-methyl-2-(trifluormethyl)- pentan-2-ol
240 mg (0.78 mmol) 4-(5-Fluor-2-methoxy-phenyl)-2-hydroxy-4-methyl-2- trifluoromethyl-pentanal und 170 mg (1.17 mmol) 5-Aminocinnolin (J.R. Elkins, EN. Brown J. Heterocycl. Chem. 1968, 639-646) werden in 10 ml Dichlorethan gelöst. Man setzt 1 ml Essigsäure und 30 mg gepulvertes Molsieb (4 A) zu, und refluxiert 6 h über Molsieb (4 A). Die Reaktionsmischung wird zwischen Dichlormethan und Wasser verteilt, die Phasen getrennt, die wässrige Phase mit CH2CI2 extrahiert, die vereinigten organischen Phasen mit ges. ΝaHCO3-Lösung und ges. NaCl-Lösung gewaschen und mit Na2SO getrocknet. Man engt ein und chromatographiert an Kieselgel mit Hexan/Essigester (20 bis 50%). Man erhält 70 mg 1-(Cinnolin-5-ylimino)-4-(5-fluor-2-hydroxyphenyl)-4-methyl-2- (trifluormethyl)-pentan-2-ol, von dem 30 mg in THF aufgenommen und mit 10 mg ( 0.16 mmol) Natriumcyanoborhydrid und 100 μl Essigsäure versetzt werden. Nach 6 h rühren bei RT verteilt man zwischen Wasser und CH2CI2 und trennt die Phasen. Die wässrige Phase wird mit CH2CI2 extrahiert, die vereinigten organischen Phasen mit ges. NaCl-Lösung gewaschen und mit Na2SO getrocknet. Man engt ein, nimmt den Rückstand in Chloroform wieder auf, setzt eine Spatelspitze aktiviertes Mangandioxid hinzu und rührt 2 h bei RT. Dann wird der Braunstein abfiltriert und das Filtrat eingeengt. Das Rohprodukt wird an Kieselgel mit Hexan/Essigester (20 bis 50%) chromatographiert. Man erhält 3.3 mg des gewünschten Produkts als roten Film. MS (ESI): 438 (M+H); 1H-NMR (CDCI3): δ = 1.48 (s, 3H), 1.53 (s, 3H), 2.59 (dd, 2H), 3.13 (s, 1H), 3.24
(dd, 1H), 3.37 (dd, 1 H), 3.89 (s, 3H), 4.80-4.84 (m, 1 H), 6.47 (d, 1H), 6.83 (dd, 1 H), 6.91-6.99 (m, 1 H), 7.05 (dd, 1 H), 7.76-7.87 (m, 2H), 8.11 (d, 1 H), 9.08 (d, 1 H).

4-(5-Fluor-2-methoxyphenyl)-4-methyl-1-(8-fluor-2-methylchinazolin-5-ylamino)- 2-(trifluormethyl)-pentan-2-o! 5-Amino-8-fluor-2-methylchinazolin Zu einer Lösung von 3.35 g (20.25 mmol) Chloralhydrat und 21.27 g (149.7 mmol) Natriumsulfat in 72 ml Wasser wird eine 50°C warme Lösung von 2.4 g (18.6 mmol) 2,5-Difluoranilin in 11 ml Wasser und 1.6 ml konz. Salzsäure (37%) gegeben, die bei dieser Temperatur vorher 1 h gerührt wurde. Man rührt weitere 30 min bei RT und erhitzt nach der Zugabe von 4.09 g (58.9 mmol) Hydroxylammoniumchlorid in 19 ml Wasser über 45 min auf 125°C und hält diese Temperatur für 5 min. Nach dem Abkühlen und einer weiteren Stunde wird der ausgefallene hellbraune Niederschlag abfiltriert, mit Wasser gewaschen und getrocknet. Man erhält 3.0g (15.0 mmol) des Hydroxylimins als Zwischenprodukt, die portionsweise in 15 ml konz. Schwefelsäure bei 60°C gelöst werden. Nach vollständiger Zugabe erhitzt man 2 Stunden auf 80°C und 4h auf 90°C. Man läßt abkühlen und gießt die Lösung auf 100g Eis. Man extrahiert mit Ethylacetat, wäscht die organische Phase mit Wasser, trocknet über Natriumsulfat und engt ein. Nach Chromatographie an Kieselgel mit Hexan-Essigester (0-45 %) werden 1 ,2 g (7.1 mmol) des 4,7-Difluorisatins erhalten. Zum Isatin in 30 ml einer 1 molaren Natronlauge werden über 10 min 1.8 ml einer 30%igen Wasserstoffperoxid Lösung getropft. Nach 2 Stunden Rühren bei RT wird auf 0°C gekühlt und es werden 5 ml einer 4 molaren Salzsäure zugegeben und mit 50 ml Wasser verdünnt. Man extrahiert mit Ethylacetat, trocknet über Natriumsulfat, engt ein und erhält so quantitativ 1.27 g der 3,6-Difluoranthranilsäure, die ohne weitere Aufreinigung umgesetzt wird.
Die 3,6-Difluoranthranilsäure wird in 8 ml Essigsäureanhydrid 45 min lang auf 100°C erhitzt. Nach dem Abkühlen wird die entstandene Essigsäure und überschüssiges Essigsäureanhydrid azeotrop mit Toluol im Vakuum entfernt. Der Rückstand wird unter Eiskühlung mit 40 ml einer 25%igen Ammoniaklösung verstzt und 72 Stunden gerührt. Man verdünnt mit Wasser und säuert mit
Essigsäure an. Man extrahiert mit Ethylacetat, wäscht die organische Phase mit Wasser, trocknet über Natriumsulfat und engt ein. Die so erhaltenen 1 ,03 g (5.25 mmol) 5,8-Difluor-2-methyl-3H-chinazolin-4-on und 6 g Phosphorpenta- chlorid werden in 20 ml Phosphorylchlorid über 12 h auf 125°C erhitzt. Nach dem Abkühlen gießt man in ges. NaHCO3 Lösung und extrahiert mit Ethylacetat. Die organische Phase wird getrocknet und das Lösungsmittel entfernt. Man erhält quantitativ 1.7g 4-Chlor-5,8-difluor-2-methylchinazolin, die in 60 ml Ethylacetat und 5 ml Triethylamin gelöst werden. Man gibt 600 mg Palladium auf Kohle zu und schüttelt 2 h (480 ml Wasserstoffaufnahme) unter einer Wasserstoffatmosphäre bei Normaldruck. Die Lösung wird mittels Filtration über Celite vom Katalysator befreit, wobei mit 100 ml Ethanol nachgewaschen wurde, und eingedampft. Nach Chromatographie an Kieselgel mit Hexan-Essigester- Ethanol (0-40 %) werden 550 mg 5,8-Difluor-2-methyIchinazolin erhalten. Zu 240 mg (1.3 mmol) 5,8-Difluor-2-methylchinazolin, 300 mg (1.13 mmol) 18- Krone-6 in 10 ml DMF werden 890 mg (13.7 mmol) Natriumazid gegeben und man erhitzt die Mischung über 8 h auf 125°C. Das Lösungsmittel wird im Vakuum entfernt und man chromatographiert an Kieselgel mit Ethylacetat und erhält 52 mg Produkt. H-NMR (CDCI3); δ = 2.92 (s, 3H), 4.31 (br., 2H), 6.67 (dd, 1 H), 7.38 (dd, 1 H), 9.37 (s, 1 H).
Zu 200 mg (0.48 mmol) 4-(5-Fluor-2-methoxyphenyl)-2-hydroxy-4-methyl-2- (trifluormethyl)pentanal und 40 mg (0.23 mmol) 5-Amino-8-fluor-2- methylchinazolin werden in Dichlorethan 50 mg Natriumacetat, 0,05 ml Trifluoressigsäure und 0,1 ml Essigsäure gegeben. Man erhitzt unter Rückfluss und entfernt nach 4 Stunden das Lösungsmittel im Vakuum unter Zusatz von Toluol. Nach Chromatographie an Kieselgel mit Hexan-Essigester (0-70 %) werden 58 mg 4-(5-Fluor-2-methoxyphenyl)-4-methyl-1-(8-fluor-2- methylchinazolin-5-ylimino)-2-(trifluormethyl)-pentan-2-ol erhalten. Zum Imin in
10 ml Ethylacetat und 1 ml Triethylamin werden 20 mg Palladium auf Kohle gegeben und man schüttelt 1 h unter einer Wassestoffatmosphäre bei Normaldruck. Die Lösung wird mittels Filtration vom Katalysator befreit und eingedampft. Man nimmt in 5 ml Chloroform auf und gibt 200 mg aktiviertes Mangandioxid zu und rührt 30 min. Es wird über Celite filtriert und im Vakuum eingeengt. Nach Chromatographie an Kieselgel mit Hexan-Essigester (0-70 %) werden 12 mg des Produkts erhalten. Η-NMR (CDCI3); δ = 1.46 (s, 3H), 1.55 (s, 3H), 2.37 (d, 1 H), 2.76 (d, 1 H), 2,90 (s, 3H), 3.13 (dd, 1 H), 3.27 (dd, 1 H), 3.85 (s, 3H), 4.50 (br., 1 H), 5.94 (dd, 1 H), 6.77 (dd, 1 H), 6.91 (ddd, 1 H), 7.08 (dd, 1 H), 7.30 (dd, 1 H), 9.16 (s, 1 H).
Beispiel 14
4-(5-Fluor-2-hvdroxyoxyphenyl)-4-methyl-1-(8-fluor-2-methylchinazolin-5- ylamino)-2-(trifluormethyl)-pentan-2-ol
Analog zu Beispiel 3 werden 20 mg (43 μmol) 4-(5-Fluor-2-methoxyphenyl)-4- methyl-1-(8-fluor-2-methylchinazolin-5-ylamino)-2-(trifluormethyl)-pentan-2-ol in 4 ml CH2CI2 mit 2 ml 1M Bortribromid-CH2Cl2-Lösung umgesetzt. Nach Chromatographie an Kieselgel mit Hexan/2-Propanol (10 %) werden 17 mg des Produkts erhalten. Η-NMR (CDCI3); δ = 1.50 (s, 3H), 1.57 (s, 3H), 2.35 (d, 1 H), 2.86 (s, 3H), 2,90 (d, 1 H), 3.21 (dd, 1 H), 3.36 (dd, 1 H), 4.72 (br., 1 H), 6.08 (dd, 1 H), 6.54 (dd, 1 H), 6.68 (ddd, 1 H), 7.03 (dd, 1 H), 7.33 (dd, 1 H), 9.19 (s, 1 H).
Beispiel 15
N-(2-Methylchinazolin-5-yl)-4-(5-fluor-2-methoxyphenyl)-2-hvdroxy-4-methyl-2- (trifluormethyl)pentansäureamid
104 mg (0.41 mmol) 4-(5-Fluor-2-methoxyphenyl)-4-methyl-2-oxopentansäure (WO 00/32584) und 100 mg (0.63 mmol) 5-Amino-2-methylchinazolin in 2 ml DMF werden bei RT unter Argon mit 102 mg (4.49 mmol) Dicyclohexylcarbo- diimid versetzt. Man lässt 3 Sunden bei Raumtemperatur rühren, gießt die Reaktionsmischung in Wasser, extrahiert mit Ethylacetat, wäscht die organische Phase mit Wasser und trocknet (Na
2SO
4). Nach Chromatographie an Kieselgel mit Hexan-Essigester (0-70%) werden 64.9 mg N-(2-MethylchinazoIin-5-yl)-4-(5- fluor-2-methoxyphenyI)-4-methyl-2-oxopentansäureamid erhalten, die in 2.2 ml DMF gelöst und auf 0°C gekühlt werden. Die Lösung wird mit 0.18 ml (Trifluormethyl)trimethylsilan und 243 mg Cäsiumcarbonat versetzt und 6 Stunden bei Raumtemperatur gerührt. Man gibt Wasser hinzu, extrahiert mit Ethylacetat, wäscht die organische Phase mit Wasser und trocknet über Natriumsulfat. Das eingeengte Zwischenprodukt wird in 2 ml THF aufgenommen und 100 μl einer 1 M Lösung von Tetrabutylammoniumfluorid werden zugegeben. Man rührt 30 Minuten, gibt Wasser zu, extrahiert mit Ethylacetat, wäscht die organische Phase mit Wasser und trocknet über Natriumsulfat. Nach Chromatographie an Kieselgel mit Hexan-Essigester (0-65%) werden 14.7 mg Produkt erhalten. Η-NMR (CDCI
3); δ = 1.44 (s, 3H), 1.46 (s, 3H), 2.85 (d, 1H), 2.91 (s, 1H), 3.04 (d, 1H), 3.89 (s, 3H), 4.18 (s, 1H), 6.77 (m, 2H), 6.94 (dd, 1H), 7.79 (d, 1H), 7.86 (t, 1 H), 8.05 (d, 1H), 9.08 (s, 1 H), 9.12 (s, 1H).
Beispiel 16
4-(5-Fluor-2-methoxyphenyl)-1-(1H-indazol-4-ylamino)-4-methyl-2-(trifluor- methvQpentan-2-ol 154 mg 4-(5-Fluor-2-methoxyphenyl)-2-hydroxy-4-methyl-2- (trifluormethyl)pentanal und 80 mg 1Η-lndazol-4-ylamin (v. Auwers Chem.Ber., 1920, 53, 1213) werden in 10 ml Toluol und 1,5 ml Essigsäure gelöst und 16 Stunden bei Raumtemperatur gerührt. Es wird mit Ethylacetat und Natriumhydrogencarbonatlösung versetzt, die Ethylacetatphase zweimal mit
Natriumhydrogencarbonatlösung gewaschen, über Natriumsulfat getrocknet und eingeengt. Nach Chromatographie an Kieselgel mit Hexan/Ethylacetat(1.5+1) werden 172 mg 4-(5-Fluor-2-methoxyphenyl)-1-(1H-indazol-4-yIimino)-4-methyl- 2-(trifluormethyl)pentan-2-ol erhalten. MS(EI
+): 423/424. 148 mg Imin werden in 5 ml Methanol und 0.5 ml Essigsäure gelöst, mit 60 mg Natriumcyanoborhydrid vereinigt, 2 Stunden bei 0°C und 6 Stunden bei Raumtemperatur gerührt. Es wird mit Ethylacetat und Natriumhydrogencarbonatlösung versetzt, die Ethylacetatphase zweimal mit Natriumhydrogencarbonatlösung gewaschen, getrocknet und eingeengt Nach Chromatographie an Kieselgel mit Hexan/Ethylacetat(1.5+1) werden 130 mg 4- (5-Fluor-2-methoxyphenyl)-1-(1H-indazoI-4-ylamino)- 4-methyl-2-(trifluor- methyl)pentan-2-ol erhalten. MS(EI
+): 425/426, Η-NMR (CDCI
3); δ = 1.45 (s, 3H), 1.58 (s, 3H), 2.27 (d, 1 H), 2.78 (d, 1 H), 3.18 (d, 1 H), 3.35 (d, 1H), ), 3,85 (s, 3H), 5.67 (d, 1 H), 6.83 (dd, 1 H), 6.85 (d, 1H), 6.95 (ddd, 1H), 7.12 (dd, 1 H), 7.15 (dd, 1 H), 7.86 (br, 1 H).
Beispiel 17
4-(5-Fluor-2-hvdroxyphenyl)-1-(1Hindazol-4-ylamino)-4-methyl-2- (trifluormethyl)pentan-2-ol
Analog zu Beispiel 3 werden 127 mg 4-(5-Fluor-2-methoxyphenyl)-1-(1H- indazol-4-ylamino)-4-methyl-2-(trifluormethyl)pentan-2-ol mit 10 ml 1M Bortribromid-CH2Cl2-Lösung umgesetzt. Nach Chromatographie an Kieselgel mit Hexan/Ethylacetat (40%) werden 60 mg 4-(5-FIuor-2-hydroxyphenyI)-1-(1H- indazol-4-ylamino)- 4-methyl -2-(trifluormethyI)pentan-2-ol erhalten. Fp.: 164- 165°C, MS(EI+): 411/412 Η-NMR (D6-DMSO); δ = 1.37 (s, 3H), 1.55 (s, 3H), 1.92 (d, 1 H), 2.92 (dd, 1 H), 3.03-3.18 (2H), 5.16 (t(br), 1 H), 5.58 (d, 1 H), 5.82 (s, 1 H), 6.65 (d, 1 H), 6.81 (dd, 1 H), 6.85 (ddd, 1H), 6.95 (dd, 1 H), 7.00 (dd, 1H), 7.97 (s, 1H), 9.75 (s, 1 H), 12.7 (s, 1 H)
Beispiel 18
4-(5-Fluor-2-methoxyphenyl)-4-methyl-1-(1-methyl-1Hindazol-4-ylamino)-2- (trifluormethyl)pentan-2-ol
154 mg 4-(5-Fluor-2-methoxy-phenyl)-2-hydroxy-4-methyl-2- (trifluormethyl)pentanal und 91 mg 1-Methyl-1 H-indazoI-4-ylamin (Sureau Chimia, 1961, 15, 195) werden wie im Beispiel 15 beschrieben zu 4-(5-Fluor-2- methoxyphenyl)-4-methyl-1-(1-methyl-1H-indazol-4-yIimino)-2- (trifluormethyl)pentan-2-ol umgesetzt MS(EI+): 437/438 und weiter mit Natriumcyanoborhydrid zu 4-(5-Fluor-2-methoxyphenyl)- 4-methyl-1-(1-methyl- 1 Hindazol-4-ylamino)-2-(trifluormethyl)pentan-2-ol reduziert. MS(EI+): 439/440, Η-NMR (CDCI3); δ = 1.46 (s, 3Η), 1.59 (s, 3H), 2.27 (d, 1 H), 2.77 (d, 1H), 3.05- 3.20 (3H), 3.38 (d, 1 H), 3.82 (s, 3H), 4.00 (s, 3H), 5.60 (d, 1 H), 6.75 (d, 1H), 6.84 (dd, 1 H), 6.95 (ddd, 1 H), 7.12 (dd, 1 H), 7.16 (dd, 1 H), 7.75 (s, 1 H).
Beispiel 19
4-(5-Fluor-2-hydroxyphenyl)-4-methyl-1-(1-methyl-1/-/-indazol-4-ylamino)-2- (trif!uormethy!)pentan-2-ol Analog Beispiel 3 werden aus 84 mg 4-(5-Fluor-2-methoxyphenyl)-4-methyl-1- (1-methyl-1H-indazol-4-ylamino)-2-(trifluormethyl)pentan-2-ol 22 mg 4-(5-Fluor- 2-hydroxyphenyl)-4-methyl-1-(1-methyl-1H-indazol-4-ylamino)-2- (trif!uormethyl)pentan-2-ol erhalten Fp.: 193-194°C, MS(El
+): 425/426, Η-NMR (D6-DMSO); δ = 1.40 (s, 3H), 1.53 (s, 3H), 1.91 (d, 1 H), 2.95 (dd, 1 H), 3.09-3.20 (2H), 3.90 (s, 3H), 5.26 (t(br), 1H),), 5.62 (d, 1 H), 5.83 (s, 1 H), 6.73 (d, 1 H), 6.80 (dd, 1 H), 6.85 (ddd, 1 H), 6.99-7.05 (2H), 7.93 (s, 1 H), 9.75 (s, 1 H)
Beispiel 20
4-(5-Fluor-2-methoxyphenyl)-4-methyl-1-(2-methyl-2Hindazol-4-ylamino)-2- (trifluormethyl)pentan-2-ol 154 mg 4-(5-Fluor-2-methoxy-phenyl)-2-hydroxy-4-methyl-2-
(trifluormethyl)pentanal und 91 mg 2-Methyl-2H-indazol-4-ylamin (Sureau Chimia, 1961 , 15, 195) werden wie im Beispiel 15 beschrieben zu 4-(5-Fluor-2- methoxyphenyl)- 4-methyl-1-(2-methyl-2/V-indazol-4-ylimino)-4-methyl-2- (trifluormethyl)pentan-2-ol umgesetzt [ Fp.: 92-94 , MS(EI+): 437/438] und weiter mit Natriumcyanoborhydrid zu 4-(5-Fluor-2-methoxyphenyl)-4~methyl-1- (2-methyl-2W-indazol-4-ylamino)-2-(trifluormethyl)pentan-2-ol reduziert. MS(EI+): 439/440, Η-NMR (CDCI3); δ = 1.47 (s, 3H), 1.56 (s, 3H), 2.30 (d, 1 H), 2.75 (d, 1 H), 3.14 (d, 1H), 3.29 (d(br), 1H), 3.33 (s(br), 1H), 3.75 (s(br), 1H), 3.85 (s, 3H), 4.15 (s, 3H), 5.55 (d, 1 H), 6.86 (d, 1 H), 6.95-7 07 (2H), 7.10 (dd, 1 H), 7.15 (dd, 1H), 7.66 (s, 1 H)
Beispiel 21
4-(5-Fluor-2-hydroχyphenyl)-4-methyl-1-(2-methyl-2/-/-indazol-4-ylamino)-2- (trifluormethyl)pentan-2-ol
Analog Beispiel 3 werden aus 132 mg 4-(5-Fluor-2-methoxyphenyl)-4-methyl-1- (2-methyl-2Hindazol-4-ylamino)-2-(trifluormethyl)pentan-2-ol 100 mg 4-(5- Fluor-2-hydroxyphenyl)-4-methyl-1-(2-methyl-2Η-indazol-4-ylamino)-2- (trifluormethyl)pentan-2-ol erhalten Fp.: 182°C, MS(EI+): 425/426, Η-NMR (D6- DMSO); δ = 1.40 (s, 3Η), 1.55 (s, 3H), 1 .93 (d, 1H), 2.89 (dd, 1H), 3.05-3.17
(2H), 4.07 (s, 3H), 5.00 (t(br), 1H), 5.38 (d, 1H), 5.85 (s, 1H), 6.72-6.90 (4H), 6.99 (dd, 1H), 8.16 (s, 1H), 9.73 (s, 1H)
4-(2.5-Difluorphenyl)-1-(1/-/-indazol-4-ylamino)-4-methyl-2-(trifluormethyl)pentan- 2-ol Analog Beispiel 3 werden 207 mg (0.73 mmol) 4-(2,5-Difluorphenyl)-2-hydroxy- 4-methyl-2-trifluormethyl-pentanal mit 150 mg (1.10 mmol) 1 H-lndazol-4-ylamin (v. Auwers Chem.Ber., 1920, 53, 1213) umgesetzt. Man erhält 110 mg 4-(2,5- Difluorphenyl)-1-(1H-indazol-4-ylimino)-4-methyl-2-(trifluormethyl)pentan-2-ol. 50 mg (0.12 mmol) davon werden analog Beispiel 7 mit 27 mg (0.72 mmol) NaBH reduziert. Nach Chromatographie an Kieselgel mit Hexan/Essigester (20- 30%) erhält man 18 mg des gewünschten Produkts. 1H-NMR (300 MHz, CDCI3): D = 1.48 (s, 3H), 1.62 (s, 3H), 2.27 (d, 1H), 2.52 (d, 1H), 3.23-3.38 (m, 2H), 5.67 (d, 1H), 6.83-7.01 (m, 3H), 7.08-7.18 ( , 2H), 7.89 (s, 1H)
4-(4-Brom-2-methoxyphenyl)-2-hvdroxy- N-(7/y-indazol-4-yQ-4-methyl-2-(trifluor- methvQpentansäureamid 122 mg 4-Dimethylaminopyridin werden in der Wärme in 3 ml Sulfolan
® gelöst, auf Raumtemperatur abgekühlt und mit 0.0525 ml Thionylchlorid vereinigt. Nach 45 Minuten bei Raumtemperatur wird mit 192 mg 4-(4-Brom-2-methoxyphenyl)- 2-hydroxy-4-methyl-2-(trifluormethyl)pentansäure (WO 98/54159) versetzt und
erneut 45 Minuten bei Raumtemperatur gerührt. Es wird mit 90 mg 7H-lndazol- 4-ylamin (v. Auwers Chem.Ber., 1920, 53, 1213) versetzt , 1 Stunde auf 80°C erwärmt und mit Natriumhydrogencarbonatlösung und Ethylacetat vereinigt Die Ethylacetatphase wird viermal mit Wasser gewaschen, getrocknet und eingengt. Nach Chromatographie an Kieselgel mit Hexan/Ethylacetat (50%) werden 150 mg 4-(4-Brom-2-methoxyphenyl)-2-hydroxy-N-(7H-indazol-4-yl)- 4-methyl -2- (trifluormethyl)pentansäureamid erhalten. MS(EI
+): 499/501 Η-NMR (D6- DMSO); δ = 1.38 (s, 3H), 1.50 (s, 3H), 2.17 (d, 1 H), 3.10 (d, 1 H), 3.83 (s, 3H), 6.65 (dd, 1H), 6.97 (d, 1 H), 7.03 (d, 1H), 7.08 (s, 1 H), 7.10 (d, 1H), 7.27 (dd, 1 H), 7.31 (d, 1H), 7.92 (s, 1H), 9.45 (s, 1 H), 13.1 (s, 1H)
Beispiel 24
4-(4-Brom-2-hvdroxyphenyl)-2-hydroxy-N-(7 -/-indazol-4-yl)-4-methyl-2-(trifluor- methvDpentansäureamid
Analog Beispiel 3 werden aus 100 mg 4-(4-Brom-2-methoxyphenyI)-2-hydroxy- N-( /-/-indazol-4-yl)- 4-methyl -2-(trifluormethyI)pentansäureamid 55 mg 4-(4- Brom-2-hydroxyphenyl)-2-hydroxy-N-(7H-lndazol-4-yl)- 4-methyl -2-(trifluor- methyl)pentansäureamid erhalten. MS(EI+): 485/487, 1H-NMR (D6-DMSO); δ = 1.42 (s, 3H), 1.48 (s, 3H), 2.23 (d, 1 H), 3.15 (d, 1 H), 6.54 (dd, 1 H), 6.83 (d, 1 H), 6.95 (d, 1 H), 7.00 (s, 1H), 7.10 (d, 1 H), 7.25 (dd, 1 H), 7.30 (d, 1 H), 7.95 (s, 1 H), 9.61 (s, 1 H), 9.95 (s, 1 H), 13.12 (s, 1 H)
Beispiel 25
4-(5-Fluor-2-methoxyoxyphenyl)-4-methyl-1-(7-fluor-2-methylchinazolin-5- ylamino)-2-(trifluormethyl)-pentan-2-ol
5-Amino-7-fluoro-2-methychinazolin
17 g (70,5 mmol) 3,6-Difluor-2-N-pivaloylaminobenzaldehyd (L. Florvall, I Fagervall, L.-G- Larsson, S.B. Ross, Eur.J. Med. Chem. 34 (1999) 137-151) , 9,2 g Acetamidin Hydrochlorid, 13,4g Kaliumcarbonat und 10,4 g Molekularsieb (4A) werden in 70 ml Butyronitril zusammengegeben. Man erhitzt unter heftigem Rühren 17 Stunden auf 145°C und entfernt das Lösungsmittel im Vakuum. Nach Chromatographie des Rückstands an Kieselgel mit Hexan/Ethylacetat (0-70%) erhält man 4,5 g 7-FIuoro-5-N-pivaloylamino-2-methychinazolin. 1g (3,82 mmol) 7-Fluoro-5-N-pivaloylamino-2-methychinazolin werden in 74 ml Toluen gelöst und auf -70°C gekühlt. Über 30 min werden 9,5 ml (11 ,4 mmol) einer 1 ,2 M Disobutylaluminiumhydrid Lösung in Toluol zugetropft. Man lässt die Reaktionsmischung auf -40°C erwärmen und rührt 4 Stunden bei -40°C. Es wird langsam Wasser zugegeben und 30 Minuten bei Raumtemperatur gerührt bis sich ein Niederschlag bildet, der mittels Filtration durch Cellite entfernt wird. Man trennt die Phasen, wäscht mit gesättigter Natriumchlorid Lösung und trocknet über Natriumsulfat. Nach Chromatographie an Kieselgel mit Hexan- Essigester (0-100%) werden 64 mg des Produkts erhalten. 1H-NMR (CDCI3); δ = 2.83 (s, 3H), 4.67 (br., 2H), 6.50 (dd, 1 H), 6.93 (dd, 1 H), 9.23 (s, 1 H). Zu 22 mg (0.07 mmol) 4-(5-Fluor~2-methoxyphenyI)-2-hydroxy-4-methyl-2- (trifluormethyl)-pentanal und 11 mg (0.06 mmol) 5-Amino-7-fluor-2- methylchinazolin in 4 ml Toluen werden 0.1 ml Titantetraethylat gegeben und die Mischung wird über 2,5 Stunden auf 100°C erhitzt. Nach dem Abkühlen gießt man auf Wasser und rührt heftig nach. Die Suspension wird durch Cellite filtriert wobei gründlich mit Ethylacetat nachgewaschen wird. Die Phasen des Filtrats werden getrennt und es wird nochmals mit Ethylacetat extrahiert. Man trocknet über Natriumsulfat und entfernt das Lösungsmittel im Vakuum. Das so roh erhaltene 4-(5-Fluor-2-methoxyphenyl)-1 -(7-fluor-2-methylchinazolin-5- ylimino)-4-methyl-2-(trifluormethyl)-pentan-2-ol wird in 8 ml Ethylacetat in 5 ml Ethylacetat und 0.5 ml Triethylamin werden 20 mg Palladium auf Kohle gegeben und man schüttelt 1 h unter einer Wasserstoffatmosphäre bei Normaldruck. Die Lösung wird mittels Filtration vom Katalysator befreit und eingedampft. Man nimmt in 5 ml Chloroform auf, gibt 50 mg aktiviertes Mangandioxid zu und rührt 20 min. Es wird über Cellite filtriert und im Vakuum eingeengt. Nach
Chromatographie an Kieselgel mit Hexan-Essigester (0-70 %) werden 18 mg des Produkts erhalten. 1H-NMR (CDCI3); δ = 1.47 (s, 3H), 1.54 (s, 3H), 2.44 (d, 1 H), 2.70 (d, 1 H), 2.81 (s, 3H), 3.15 (dd, 1 H), 3.30 (dd, 1 H), 3.86 (s, 3H), 4.97 (br., 1 H), 5.83 (dd, 1 H), 6.79 (dd, 1 H), 6.85 (dd, 1 H), 6.92 (ddd, 1 H), 7.06 (dd, 1 H), 8.98 (s, 1 H).
Beispiel 26
4-(5-Fluor-2-methoxyphenyl)-1-(6-methyl-1Hindazol-4-ylamino)-4-methyl-2- (trifluormethyl)pentan-2-o!
Analog Beispiel 16 wird aus 4-(5-Fluor-2-methoxyphenyl)-2-hydroxy-4-methyl-2- (trifluormethyl)pentanal und 6-Methyl-1 H-indazol-4-ylamin (v. Auwers Chem.Ber., 1920, 53, 1213) das gewünschte Produkt erhalten. 1H-NMR (CD3OD); δ = 1.45 (s, 3H), 1.64 (s, 3H), 2.01 (d, 1H), 2.23 (s, 3H), 2.78 (d, 1 H), 3.01 (d, 1 H), 3.21 (d, 1 H), 3.86 (s, 3H), 5.96 (s, 1 H), 6.93 (m, 2H), 7.14 (dd, 1 H), 7.21 (s, 1 H), 7.73 (s, 1 H).
4-(3-Fluor-2-methoxyphenyl)-1-(2-methylchinazolin-5-ylamino)-4-methyl-2- (trifluormetyl)pentan-2-ol
2, 6-Difluoranisol
20 g (153,74 mmol) 2,6-Difluorphenol werden in 200 ml Aceton gelöst und unter Stickstoff mit 42,5 g (307,48 mmol) Kaliumcarbonat versetzt. Nach Zugabe von 19,1 ml Methyliodid (2 Equivalente) wird dreineinhalb Stunden am Rückfluss gekocht. Nach dem Abkühlen wird das Reaktionsgemisch filtriert, der Filterrückstand mit Aceton gewaschen und das Filtrat bis zur trockene einrotiert.
Der Rückstand wird an Kieselgel (Laufmittel Ethylacetat Hexan) chromatographiert. Es werden 17,27 g (77,9 %) des gewünschten Produkts erhalten. Es ist zu beachten, dass das Produkt leicht flüchtig ist. Die Badtemperatur sollte 30° C nicht überschreiten und das Vakuum des Rotationsverdampfers ist anzupassen.
2-(3-Fluor-2-methoxyphenyl)-2-methylpropannitril
10 g (69,39 mol) 2,6-Difluoranisol werden in 200 ml Toluol gelöst und bei
Raumtemperatur mit 5,75 g (83,27 mmol) Isobuttersäurenitril versetzt. Innerhalb von 35 Minuten werden 166,5 ml einer 0,5 molaren Lösung von
Kaliumhexamethyldisilazid in Toluol zugetropft. Dabei erfolgt ein leichter Temperaturanstieg auf 27,5° C. Nach 16 Stunden Rühren bei Raumtemperatur wird das Reaktionsgemisch mit 200 ml Wasser und 400 ml Ethylacetat versetzt und mit 10%iger Schwefelsäure bis pH 4 angesäuert. Die organische Phase wird abgetrennt und die wässrige Phase einmal mit Ethylacetat geschüttelt (200 ml). Die vereinigten organischen Extrakte werden mit Wasser und Sole geschüttelt. Nach dem Trocknen, Filtrieren und dem Abrotieren des Lösungsmittels wird der Rückstand an Kieselgel (Laufmittel Ethylacetat/Hexan) chromatographiert. Isoliert werden 7,66 g (57,1 %) der gewünschten Verbindung.
2-(3-Fluor-2-methoxyphenyl)-2-methylpropanal
7,66 g (39,64 mmol) des oben beschriebenen Nitrils werden in 158 ml Toluol gelöst. Bei -65 bis -60°C werden innerhalb von 40 Minuten 49,5 ml einer 1,2 molaren Lösung von DIBAH in Touluol zugetropft. Nach einstündigem Rühren bei dieser Temperatur wird begonnen, 493 ml einer 10%igen L-(+)~
Weinsäurelösung zuzutropfen. Nach 100 Millilitern ist die Temperatur auf -10° C gestiegen. Der Rest der Weinsäurelösung wird schnell zugegeben und der Ansatz zwei Stunden kräftig bei Raumtemperatur gerührt. Das Reaktionsgemisch wird zweimal mit je 400 ml Diethylether geschüttelt. Die vereinigten organischen Extrakte werden mit Wasser und Sole geschüttelt, getrocknet und das Lösungsmittel abrotiert. Der erhaltene Rückstand (7,8 g = 102 %) wird roh in die nächste Stufe eingesetzt.
(E/Z)-4-(3-Fluor-2-methoxyphenyl)-4-methylpent-2-ensäureethylester Zu einer Lösung von 9,87 g (39,75 mmol) 2-Ethoxy- phosphonoessigsäuretriethylester in 40 ml absolutem THF werden bei 0° C 21 ,3 ml einer 2 molaren LDA-Iösung in THF zugetropft. Nach 30minütigem Rühren bei 0° C werden 7,8 g (39,75 mmol) 2-(3-Fluor-2-methoxyphenyl)-2- methylpropanal, gelöst in 26 ml THF, zügig bei 0° C zugetropft. Das Kältebad wird entfernt und der Ansatz 16 Stunden bei Raumtemperatur gerührt. Da Reaktinsgemsich wird auf Wasser gegossen und zweimal mit Ethylacetat extrahiert. Die vereinigten organischen Extrakte werden mit Wasser und Sole gewaschen, getrocknet und das Lösungsmittel nach dem Abfiltrieren des Trocknungsmittels abrotiert. Der Rückstand wird an Kieselgel (Laufmittel Ethylacetat/Hexan) chromatographiert. Isoloiert werden 8,39 g (68,2 %) der gewünschten Verbindung.
(E/Z)-4-(3-Fluor-2-methoxyphenyl)-4-methylpent-2-ensäure
8,39 g (27,03 mmol) (E/Z)-4-(3-Fluor-2-methoxyphenyl)-4-methylpent-2- ensäureethylester werden mit 270 ml einer 1 N NaOH in Ethanol/Wasser (2:1) versetzt und zwei Tage bei Raumtemperatur gerührt. Das Ethanol wird am Rotationsverdampfer abgezogen und der Rückstand zweimal mit je 150 ml Diethylether extrahiert. Die vereinigten organischen Extrakte werden mit Wasser gewaschen und nach DC Kontrolle verworfen. Die Wasserphasen werden mit conz. Salzsäure bis pH 3 angesäuert und zweimal mit je 300 ml Diethylether extrahiert. Die Etherextrakte werden mit Wasser und Sole gewaschen, getrocknet, das Lösungsmittel abrotiert und der Rückstand (5,89 g = 77,2 %) roh in die nächste Stufe eingesetzt.
4-(3-Fluor-2-methoxyphenyl)-4-methyl-2-oxo-pentansäure 5,89 g (20,86 mmol) (E/Z)-4-(3-Fluor-2-methoxyphenyl)-4-methylpent-2-ensäure werden bei Raumtemperatur mit 126 ml einer 1 molaren Schwefelsäure versetzt und nach Zugabe von 21 ml Eisessig 15 Stunden bei 90° C Badtemperatur gerührt. Das Reaktionsgemisch wird unter Eisbadkühlung vorsichtig (schäumt stark) mit festem Kaliumcarbonat bis pH 9 versetzt. Es wird zweimal mit Diethylether extrahiert. Die vereinigten organischen Extrakte werden mit Wasser
gewaschen und nach DC verworfen. Die vereinigten Wasserphasen werden mit conc. Salzsäure bis pH 4 angesäuert und zweimal mit je 300 ml Diethylether extrahiert. Die Etherextrakte werden mit Wasser und Sole gewaschen, getrocknet und das Lösungsmittel abrotiert. Da der Rückstand noch Essigsäure enthält, wird er zweimal mit je 100 ml Toluol abrotiert. Der verbleibende Rückstand (4,14 g = 78,1 %) wird roh in die nächste Stufe eingesetzt.
4-(3-Fluor-2-methoxyphenyl)-4-methyl-2-oxo-pentansäurethylester
4,14 g (16,28 mmol) 4-(3-Fluor-2-methoxyphenyl)-4-methyl-2-oxo-pentansäure werden in 97 ml Ethanol gelöst, mit 1,79 ml Schwefelsäure versetzt und vier Stunden am Rückfluß gekocht. Das Ethanol wird am Rotationsverdampfer abgezogen und der Rückstand vorsichtig mitgesättigter Natriumhydrogencarbonatlösung bis pH 9 versetzt. Es wird zweimal mit je 100 ml Ethylacetat extrahiert und die vereinigten organischen Extrakte mit Wasser und anschließend mit Sole gewaschen. Nach Trocken, Abfiltrieren des Trocknungsmittels und Einrotieren des Lösemittels wird der Rückstand an Kieselgel (Laufmittel Ethylacetat/Hexan) chromatographiert. Isoliert werden 4,16 g (90,6 %) der gewünschten Verbindung.
4-(3-F!uor-2-methoxyphenyl)-4-methyl-2-trifluormethyl-2-trimethylsilyloxy- pentansäureethylester
4,16 g (14,74 mmol) 4-(3-Fluor-2-methoxyphenyI)-4-methyl-2-oxo- pentansäureethylester werden in 24 ml THF gelöst und bei 0° C mit 2,51 g (17,68 mmol) (Trifluormethyl)-trimethylsilan und 36,1 mg Tetrabutylammoniumfluorid versetzt. Nach zweieinhalb Rühren zwischen 0 und 5° C wird der Ansatz auf 50 ml Eiswassergegossen. Es wird zweimal mit je 150 ml Diethylether extrahiert und die vereinigten organischen Extrakte wie üblich aufgearbeitet. Nach Chromatographie an Kieselgel (Laufmittel Ethylacetat/Hexan) werden 5,24 g (83,8 %) der gewünschten Verbindung erhalten.
4-(3-Fluor-2-methoxyphenyl)-4-methyl-2-trifluormethyl-2-trimethylsilyloxy- pentan-1-ol
5,24 g (12,34 mmol) 4-(3-Fluor-2-methoxyphenyl)-4-methyl-2-trifluormethyl-2- trimethylsilyloxy-pentansäurethylester werden in 45 ml Diethylether gelöst und bei 0 bis 5° C portionsweise mit 936,9 mg (24,69 mmol) LiAIH4 versetzt. Nach viereinhalbstündigem Rühren bei Raumtemperatur wird das Reaktionsgemisch unter Eisbadkühlung vorsichtig mit gesättigter NaHCO3 versetzt, eine Stunde in der Kälte und über Nacht bei Raumtemperatur gerührt. Nach dem üblichen Aufarbeiten werden 4,11 g (87,1 %) eines Gemisches aus der gewünschten Verbindung und der Verbindung erhalten, bei der der Silylether gewandert ist. Das Gemisch wird roh in die nächste Stufe eingesetzt werden.
4-(3-Fluor-2-methoxyphenyl)-4-methyl-2-trifluormethyl-pentan-1,2-diol 4,11 g (10,75 mmol) 4-(3-Fluor-2-methoxyphenyl)-4-methyl-2-trifluormethyl-2- trimethylsilyloxy-pentan-1-oI werden in 61 ml THF gelöst, mit 3,39 g (10,746 mmol) Bu4NF trihydrat versetzt und eine Stunde bei Raumtemperatur gerührt. Die Reaktionsmischung wird auf Wasser gegossen und zweimal mit Diethylether extrahiert. Die organischen Phasen werden wie üblich mit Wasser und Sole gewaschen. Nach dem Trocknen, Abfiltrieren des Trocknungsmittels und Einrotieren des Lösungsmittels wird der verbleibende Rückstand an Kieselgel (Laufmittel Ethylacetat/Hexan) chromatographiert. Isoliert werden 2,71 g (81 ,4 %) der gewünschten Verbindung.
4-(3-Fluor-2-methoxyphenyl)-2-hydroxy-4-methyl-2-triflυormethyl-pentanal In einen ausgeheizten Kolben werden 765 mg (6,03 mmol) Oxalylchlorid in 13 ml Dichlormethan vorgelegt. Bei -78° C werden 0,855 ml DMSO, gelöst in 2,5 ml Dichlormethan, zugetropft und der Ansatz fünf Minuten nachgerührt.
Anschließend werden 1 ,7g (5,48 mmol) 4-(3-FIuor-2-methoxyphenyl)-4-methyl- 2-trifluormethyl-pentan-1 ,2-diol, gelöst in 5 ml Dichlormethan zugetropft. Nach 15minütigem Rühren wird der Ansatz vorsichtig mit 3,79 ml (27,40 mmol) Triethylamin versetzt, fünf Minuten bei -78° C gerührt und anschließend langsam auf Raumtemperatur kommen lassen. Es werden 20 ml Wasser zugegeben und der Ansatz eine weiter Stunde bei Raumtemperatur gerührt. Nach Phasentrennung wird die wässrige Phase einmal mit 100 ml Dichlormethan geschüttelt. Die vereinigten organischen Extrakte werden mit
1%iger Schwefelsäure, 5%iger Natriumhydrogencarbonatlösung und Sole gewaschen. Nach dem üblichen Prozedere werden 1 ,617 g (96,2 %) des Aldehyds erhalten, der roh in die nächste Stufe eingesetzt wird. Analog Beispiel 25 wird aus 4-(3-Fluor-2-methoxyphenyI)-2-hydroxy-4-methyl-2- (trifluormethyl)pentanal und 5-Amino-2-methylchinazolin das gewünschte Produkt erhalten. 1H-NMR (300 MHz, CDCI3): D = 1.45 (3H), 1.57 (3H), 2.35 (1 H), 2.75 (1 H), 2.82 ( 3H), 3.00-3.40 (3H), 4.00 (3H), 4,75 (1H), 6.10 (1H), 6.90-7.02 (2H), 7.05-7.18 (1H), 7.25 (1H), 7.55 (1H), 9.10 (1H).
Beispiel 28
4-(3-Fluor-2-methoxyphenyl)-1-(1f/-indazol-4-ylamino)-4-methyl-2-(trifluor- methyl)pentan-2-ol Analog Beispiel 16 wird aus 4-(3-Fluor-2-methoxyphenyl)-2-hydroxy-4-methyl-2- (trifluormethyl)pentanal und 1 H-lndazol-4-ylamin das gewünschte Produkt erhalten.
1H-NMR (300 MHz, CDCI3): D = 1.49 (3H), 1.60 (3H), 2.30 (1H), 2.71 (1H), 3.18 (1 H), 3.30 (1H), 4.05 (3H), 5,75 (1H), 6.85 (1 H), 6.90-7.20 (4H), 7.80 (1H).
Beispiel 29
4-(3-Fluor-2-hvdroxyphenyl)-1-(1 -/-indazol-4-ylamino)-4-methyl-2- (trifluormethyl)pentan-2-ol
Analog Beispiel 3 erhält man aus 4-(3-Fluor-2-methoxyphenyl)-1-(1f7-indazol-4- ylamino)-4-methyl-2-(trifluormethyl)pentan-2-ol das gewünschte Produkt.1H- NMR (300 MHz, DMSO-d6): D = 1.42 (3H), 1.59 (3H), 1.95 (1H), 2.85-2.95 (1H), 3.03-3.18 (2H), 5.15 (1H), 5.55 (1H), 5.80 (1H), 6,65 (1H), 6.70-6.80 (1H), 6.90 (1H), 6.90-7.13 (2H), 7.95 (1H), 9.80 (1H), 12.70 (1H).
2-f(5-Chlor-1H-indazol-4-ylamino)-methvn-1 ,1 ,1-trifluor-4-(3-fluor-2- methoxyphenyl)-4-methylpentan-2-ol 5-Chlor-4-nitro-1 H-indazol
In 100 mL Essigsäure werden 2.24 g (12 mmol) 4-Chlor-2-methyl-3- nitrophenylamin, hergestellt nach Literatur (Mori et al., Chem. Pharm. Bull. 1986, 34, 4859ff. sowie Brand und Zöller, Chem. Ber. 1907, 3324ff.), gelöst. Bei 10°C werden 6.0 mL einer 2 molaren wässrigen Natriumnitritlösung zugetropft. Man gibt anschließend die Suspension innerhalb von 15 Minuten zur siedenden Essigsäure (150 mL) zu und lässt das Reaktionsgemisch 4 Stunden refluxieren. Nach Entfernen der Essigsäure im Vakuum nimmt man den Rückstand in Ethylacetat und gesättigter Natriumhydrogencarbonat Lösung auf. Die organische Phase wird mit gesättigter Natriumchloridlösung gewaschen und über Natriumsulfat getrocknet. Nach Entfernen des Lösungsmittels im Vakuum wird das Rohprodukt (1.81 g, 76%) weiter umgesetzt.
1H-NMR(300 MHz, DMSO-d6): δ = 7.65 (d, 1H), 7.97 (d, 1 H), 8.32 (s, 1 H), 13.97 (s, 1 H). 5-Chlor- 1 H-indazol-4-ylamin Eine Lösung aus 5-Chlor-4-nitro-1 H-indazol (872 mg, 4.41 mmol) wird mit 150 mg Palladium auf Kohle (10%ig) versetzt und unter Wasserstoffatmosphäre bei Raumtemperatur gerührt. Nach 45 Minuten wird der Katalysator über eine Fritte abgesaugt und mit Methanol gewaschen. Das Filtrat wird eingeengt und der Rückstand in 200 mL Ethylacetat aufgenommen und erwärmt. Nach erneutem Absaugen und Einengen des Filtrats erfolgt die Reinigung an Kieselgel mit Hexan/Ethylacetat (100-33% Hexan). Man erhält 296 mg (40% der Theorie) des Produkts. 1H-NMR(300 MHz, DMSO-d6): δ = 5.97 (s, 2H), 6.66 (d, 1 H), 7.05 (d, 1 H), 8.19 (s, 1 H), 12.83 (s, 1 H).
2-[(5-Chlor-1 H-indazol-4-ylimino)-methyl]-1 , 1 , 1-trifluor-4-(3-fluor-2- methoxyphenyl)-4-methyl-pentan-2-ol
Eine Lösung aus 4-(3-Fluor-2-methoxyphenyl)-2-hydroxy-4-methyl-2-trifluor- methylpentanal (278 mg, 0.9 mmol) und 5-Chlor-1 H-indazol-4-ylamin (121 mg, 0.72 mmol) in 20 mL Xylol wird mit Titan(IV)ethylat (0.42 mL, 2.0 mmol) versetzt und für 10 Stunden unter Rückfluss gekocht. Nach Abkühlen auf Raumtemperatur wird Xylol abdestilliert und der Rückstand an Kieselgel mit Hexan/Ethylacetat (30-100% Ethylacetat) gereinigt. Man erhält 123 mg (37% der Theorie) des Produkts.
1 H-NMR(400 MHz, CDCI
3): δ = 1.43 (s, 3H), 1.57 (s, 3H), 2.38 (d, 1 H), 3.22 (d, 1 H), 3.94 (d, 3H), 4.91 (s, 1H), 6.41-6.52 (m, 2H), 6.90 (d, 1 H), 7.28 (d, 1H), 7.38 (d, 1 H), 7.56 (s, 1 H), 7.72 (s, 1 H), 10.26 (br, 1 H). 2-[(5-Chlor-1H-indazol-4-ylamino)-methyl]-1, 1, 1-trifluor-4-(3-fluor-2' methoxyphenyl)-4-methylpentan-2-ol Eine Lösung aus 2-[(5-Chlor-1H-indazol-4-ylimino)-methyl]-1 ,1 ,1-trifluor-4-(3- fluor-2-methoxyphenyl)-4-methylpentan-2-ol (49 mg, 0.11 mmol) in 5.0 mL Methanol wird mit 15 mg Natriumborhydrid versetzt und bei Raumtemperatur gerührt. Anschließend wurden innerhalb 4 Tagen insgesamt 300 mg Natriumborhydrid portionsweise zugegeben. Das Reaktionsgemisch wird mit 10%iger Essigsäure neutralisiert. Nach Entfernen des Lösungsmittels wird der Rückstand in gesättigter Natriumhydrogencarbonatlösung und Ethylacetat aufgenommen. Man extrahiert mit Ethylacetat, wäscht die vereinigten organischen Phasen mit gesättigter Natriumchloridlösung und trocknet sie über Natriumsulfat. Nach Entfernen des Lösungsmittels im Vakuum sowie Reinigung des Rückstands mittels präparativer Dünnschichtchromatographie an Kieselgel mit Hexan/Ethylacetat (50% Ethylacetat) erhält man 24 mg (47%) des Produkts.
1H-NMR(300 MHz, CDCI
3): δ = 1.48 (s, 3H), 1.59 (s, 3H), 2.39 (d, 1H), 2.72 (d, 1H), 3.55-3.59 (m, 2H), 4.01 (d, 3H), 4.80-4.84 (m, 1 H), 6.80 (d, 1H), 6.86-6.98 (m, 2H), 7.11 (d, 1 H), 7.22 (d, 1 H), 7.70 (s, 1 H).
1.1 ,1 -Trifluor-4-(3-fluor-2-methoxyphenyl)-4-methyl-2-f (5-methyl-1 H-indazol-4- ylamino)-methyll-pentan-2-ol 5-Methyl-1 H-indazol-4-ylamin
Zu einer Lösung aus 2,4-DimethylaniIin (12.4 mL, 100 mmol) in 80 mL konzentrierter Schwefelsäure wird bei 0°C mit 5.0 mL rauchender Salpetersäure versetzt und 20 Minuten bei 4°C, anschließend 30 Minuten bei Raumtemperatur gerührt. Das Reaktionsgemisch wird auf 600 mL Eiswasser gegossen, mit 5N Natronlauge auf pH 10 eingestellt. Der Niederschlag wird abgesaugt, mit
Wasser gewaschen und getrocknet. Man erhält 15.72 g (95% der Theorie) 2,4- Dimethylnitrophenylamin als Gemisch der Regioisomere. In Analogie zur Darstellung von 5-Chlor-4-nitro-1 /-/-indazol wurden bei der Umsetzung von 2,4-Dimethylnitrophenylamin (2.0 g, 12 mmol) mit 6.0 mL einer 2 molaren wässrigen Natriumnitritlösung in Essigsäure (250 mL) 1.14 g (57% der Theorie) des Produkts als Gemisch der beiden Regioisomere erhalten. MS (ES+, Acetonitril/Wasser 1:1 + 0.01 % Ameisensäure): m/z(%) 178 (M+1 , 100). In Analogie zur Darstellung von 5-Chlor-1tV-indazol-4-ylamin wird das Regioisomeren-gemisch der vorherigen Reaktion (1.0 g, 5.64 mmol) mit 100 mg Palladium auf Kohle in Methanol unter Wasserstoffatmosphäre 16 Stunden bei Raumtemperatur umgesetzt. Nach Reinigung an Kieselgel mit Hexan /Ethylacetat (33% Hexan, dann 100% Ethylacetat) erhält man 53 mg (6% der Theorie) 5-Methyl-1Hindazol-4-ylamin. 1H-NMR(300 MHz, DMSO-d6): δ = 2.12 (s, 3H), 5.41 (s, 2H), 6.57 (d, 1 H), 6.90 (d, 1 H), 8.10 (s, 1 H), 12.5 (s, 1 H). 4-(3-Fluor-2-methoxyphenyl)-2-hydroxy-4-methyl-2-trifluor-methylpentanal (308 mg, 1.0 mmol) und 5-Methyl-1H-indazol-4-ylamin (148 mg, 1.0 mmol) werden in 15.0 mL Xylol vorgelegt und mit Titan(IV)ethyIat (0.42 mL, 2.0 mmol) versetzt. Nach 3 Stunden unter Rückfluss lässt man die Reaktionsmischung auf Raumtemperatur abkühlen. Nach Zugabe von Ethylacetat und gesättigter
Natriumchloridlösung wird 30 Minuten bei Raumtemperatur heftig gerührt. Man saugt den ausgefallenen Niederschlag ab, trennt die wässrige Phase ab und trocknet die organische Phase über Natriumsulfat. Die Reinigung erfolgt mittels Chromatographie an Kieselgel mit Hexan/Ethylacetat (30-40% Ethylacetat). Man erhält 345 mg (79% der Theorie) des 1 ,1 ,1-Trifluor-4-(3-fluor-2-methoxyphenyl)- 4-methyl-2-[(5-methyl-1H-indazol-4-ylimino)methyl]-pentan-2-ols. Eine Lösung aus 1 ,1 ,1-Trifluor-4-(3-fluor-2-methoxyphenyl)-4-methyl-2-[(5- methyl-1/- -indazol-4-ylimino)-methyl]-pentan-2-ol (151 mg, 0.34 mmol) in 20 mL Methanol gelöst, mit 30 mg Palladium auf Kohle (10%ig) versetzt und bei Raumtemperatur unter Wasserstoffatmosphäre gerührt. Nach 20 Stunden fügt man 30 mg Palladium auf Kohle hinzu und lässt die Reaktionsmischung für weitere 28 Stunden bei Raumtemperatur rühren. Der Katalysator wird über Celite abfiltriert und mit Methanol gewaschen. Nach Einengen des Filtrats sowie Reinigung des Rückstands mittels präparativer Dünnschichtchromatographie an Kieselgel mit Hexan/Ethylacetat (50% Ethylacetat) erhält man 28 mg (19% der Theorie) des Produkts. H-NMR(400 MHz, CDCI3): δ = 1.47 (s, 3H), 1.60 (s, 3H), 2.18 (s, 3H), 2.37 (d, 1 H), 2.70 (d, 1 H), 3.48 (s, 2H), 4.00 (d, 3H), 6.88-6.97 (m, ' 3H), 7.07 (d, 1H), 7.11 (d, 1H), 7.67 (s, 1 H). Beispiel 32
4-(Benzri ,31dioxol-4-yl)-4-methyl-1-(2-methvchinazolin-5-ylamino)-2- (trifluormethyl)-pentan-2-ol 1-(Benz[1,3]dioxol-4-yl)-1-methylethanol 25,5 g 4-Acetylbenzo[1 ,3]dioxol werden mit 57,2 ml Methylmagnesiumchlorid Lösung (3M in THF) in 375 ml THF bei RT unter Argon versetzt. Es wird 16 h bei RT gerührt und auf Eis / 2H Salzsäure gegeben. Es wird mit Essigester extrahiert und die organische Phase mit Wasser und Sole gewaschen und es wird getrocknet (Na
2SO
4). Man erhält 27,89 g 1-[Benz(1 ,3)dioxol-4-yI]-1- methylethanol als braunes Öl.
1H-NMR (CDCI
3), D (ppm) = 1.6 (s, 6H), 5.95 (s, 2H), 6.76 (dd, 1 H), 6.82 (t, 1 H), 6.91 (dd, 1 H)
4-(Benz[1,3Jdioxol-4-yl)-4-methyl-2-oxo-pentansäureethylester 5.0 g 1-(Benz[1 ,3]dioxol-4-yl)-1-methylethanol und 7.8 g 2-Trimethylsilyloxy- acrylsäure-ethylester werden in 100 ml Dichlormethan bei -70 °C mit 2.4 ml Zinn(IV)chlorid versetzt. Nach 5 Minuten wird die Lösung auf Kaliumcarbonatlösung gegeben und stark gerührt. Es wird durch Kieselgur filtriert, die Phasen getrennt und die wässrige Phase mit Dichlormethan extrahiert. Die organische Phase wird mit Wasser und Sole gewaschen, getrocknet (Na2SO4) und eingedampft. Nach Chromatographie an Kieselgel (Hexan / Essigester 0 -> 10%) erhält man 3.4 g der Titelverbindung als farbloses Öl. 1H-NMR (CDCI3), D (ppm) = 1.25 (t, 3H), 1.44 (s, 6H), 3.31 (s, 2H), 4.12 (q, 2H), 5.92 (s, 2H), 6.7 - 6.82 (m, 3H)
4-(Benz[1,3]dioxol-4-yl)-2-hydroxy-4-methyl-2-trifluormethyl- pentansäureethylester
4.42 g 4-(Benz[1 ,3]dioxol-4-yl)-4-methyl-2-oxo-pentansäureethylester und 6.9 ml
Trifluormethyl-trimethylsilan in 95 ml THF werden bei -70°C langsam mit 3.2 ml TBAF-Lösung (1H in THF) versetzt.Es wird 1 h bei -70°C und 2 h bei Raumtemperatur gerührt und eine Spatelspitze festes
Tetrabutylammoniumfluorif (TBAF) zugegeben. Nach 1 h Rühren wird auf 0.1 N Salzsäure gegeben und mit Essigester extrahiert. Die organische Phase wird mit Wasser und Sole gewaschen, getrocknet (Na2SO4) und eingedampft. Nach Chromatographie an Kieselgel (Hexan / Essigester 0 -> 10%) erhält man 4.55 g der Titelverbindung als gelbes Öl. 1H-NMR (CDCI3), D (ppm) = 1.19 (t, 3H), 1.39 (s, 3H), 1.46 (s, 3H), 2.29 (d, 1 H), 2.74 (d, 1 H), 3.59 (dq, 1 H), 4.05 (dq, 1 H), 5.92 (s, 1 H), 5.98 (s, 1 H), 6.68 - 6.85 (m, 3H) 4-(Benz[1,3]dioxol-4-yl)-2-hydroxy-4-methyl-2-thfluormethyl-pentanol 2.92 g 4-(Benz[1 ,3]dioxol-4-yl)-2-hydroxy-4-methyl-2-trifluormethyl- pentansäureethylester in 100 ml Diethylether werden portionsweise bei 0°C mit 478 mg Lithiumaluminiumhydrid versetzt. Nach Rühren für 10 h wird auf
gesättigte Bicarbonat Lösung gegeben und durch Kieselgur filtriert. Die Phasen werden getrennt und die Wasserphase mit Essigester extrahiert. Die organische Phase wird mit Wasser und Sole gewaschen, getrocknet (Na2SO ) und eingedampft. Nach Chromatographie an Kieselgel (Hexan / Essigester 0 -> 10%) erhält man 2.44 g der Titelverbindung als gelbes Öl.
1H-NMR (CDCI3), D (ppm) = 1.42 (s, 3H), 1.51 (s, 3H), 2.22 (d, 1 H), 2.36 (d, 1 H), 2.9 (bs, 1 H), 3.41 (d, 1 H), 3.51 (d, 1 H), 5.92 (s, 1 H), 5.95 (s, 1 H), 6.69 - 6.85 (m, 3H)
4-(Benz[1,3]dioxol-4-yl)-2-hydroxy-4-methyl-2-tήfluormethyl-pentanal
0,650 ml Oxalylchlorid in 16.0 ml Dichlormethan werden bei -78°C mit 1.2 ml DMSO in 3.0 ml Dichlormethan versetzt. Nach 5 min. werden 2 g 4- (Benz[1 ,3]dioxol-4-yI)-2-hydroxy-4-methyl-2-trifIuormethyl-pentanol in 7.0 ml Dichlormethan bei -78°C zugetropft. Nach 15 min. wird mit 4.6 ml Triethylamin versetzt und langsam auf RT erwärmt. Es wird mit Wasser und Sole gewaschen, mit Natriumsulfat getrocknet und im Vakuum eingeengt. Nach Chromatographie an Kieselgel (Hexan / Essigester 0 -> 5%) erhält man 1.64 g der Titelverbindung als gelbes Öl. 1H-NMR (CDCI3), D (ppm) = 1.40 (s, 3H), 1.44 (s, 3H), 2.24 (d, 1 H), 3.1 (d, 1 H), 3.64 (bs, 1 H), 5.94 (s, 1 H), 5.99 (s, 1 H), 6.67 - 6.9 (m, 3H), 9.05 (s, 1 H)
In Analogie zur Herstellung von Beispiel 2 wird aus 125 mg 5-Amino-2- methychinazolin und 237 mg 4-(Benz[1 ,3]dioxol-4-yl)-2-hydroxy-4-methyl-2- trifluormethyl-pentanal das entsprechende Imin erhalten und mit Palladium auf Aktivkohle reduziert. Die Reoxidation wird durch Erhitzen des Produktes in Xylol in Anwesenheit von Palladium auf Aktivkohle an der Luft erreicht. Man erhält 61 mg der Titelverbindung.
1 H-NMR (CDCI
3), D (ppm) = 1.48 (s, 3H), 1.56 (s, 3H), 2.35 (d, 1 H), 2.53 (d, 1 H), 2.85 (s, 3H), 3.23 (dd, 1 H), 3.41 (dd, 1 H), 4.74 (bt, 1H), 5.90 (s, 1 H), 5.93 (s, 1H), 6.17 (d, H), 6.78 (dd, 1 H), 6.84-6.93 (m, 2H), 7.28 (d, 1 H), 7.58 (t, 1 H), 9.21 (s, 1 H)
4-(Benzπ ,31dioxol-4-vπ-4-methyl-1-(8-fluor-2-methylchinazolin-5-ylamino)-2- (trifluormethyl)-pentan-2-ol Analog Beispiel 25 werden 90 mg 4-(Benz[1 ,3]dioxol-4-yl)-2-hydroxy-4-methyl-2- (trifluormethyl)pentanal und 50 mg 5-Amino-8-fluor-2-methychinazolin in 10 ml Toluen umgesetzt . Nach Chromatographie an Kieselgel mit Hexan-Essigester (0-70 %) werden 58 mg 4-(Benz[1,3]dioxol-4-yl)-4-methyl-1-(2-methychinazolin- 5-ylimino)-2-(trifluormethyl)-pentan-2-ol erhalten. Zum Imin in 10 ml Ethylacetat und 1 ml Triethylamin werden 20 mg Palladium auf Kohle gegeben und man schüttelt 2 h unter einer Wasserstoffatmosphäre bei Normaldruck. Die Lösung wird mittels Filtration vom Katalysator befreit und eingedampft. Man nimmt in 5 ml Chloroform auf und gibt 200 mg aktiviertes Mangandioxid zu und rührt 10 min. Es wird über Celite filtriert und im Vakuum eingeengt. Nach Chromatographie an Kieselgel mit Hexan-Essigester (0-70 %) werden 15 mg des Produkts erhalten. 1H-NMR (CDCI3); δ = 1.26 (s, 3H), 1.48 (s, 3H), 2.34 (d, 1H), 2.54 (d, 1 H), 2.91 (s, 3H), 3.19 (dd, 1H), 3.34 (dd, 1H), 4.58 (br., 1 H), 5.89 (d, 1 H), 5.94 (d, 1 H), 6.05 (dd, 1 H), 6.71-6.85 (m, 3H), 7.32 (dd, 1 H), 9.27 (s, 1 H).
Beispiel 34
4-(Benzπ ,3ldioxol-4-vπ-4-methyl-1-(1 - -indazol-4-ylamino)-2-(trifluormethyl)- pentan-2-ol
In Analogie zur Herstellung von Beispiel 30 wird das entsprechende Imin aus 168 mg 4-Amino-1 H-indazol und 383 mg 4-(Benz[1 ,3]dioxol-4-yl)-2-hydroxy-4- methyl-2-trifluormethyl-pentanal erhalten und weiter mit 488 mg
Natriumcyanoborhydrid zu 240 mg der Titelverbindung reduziert. 1H-NMR (CDCI3), D (ppm) = 1.44 (s, 3H), 1.58 (s, 3H), 2.27 (d, 1 H), 2.54 (d, 1 H), 3.22 (dd, 1H), 3.41 (dd, 1 H), 4.01-4.17 (m, 1 H), 5.81 (d, 1H), 5.92 s, 2H), 6.73-6.99 (m, 4H), 7.12 (t, 1 H), 7.88 (s, 1 H)
Beispiel 35 und 36
(-)-4-(Benzf1 ,31dioxol-4-vP-4-methyl-1 -(1 H-indazol-4-ylamino)-2-(trifluormethv0- pentan-2-ol und (+)-4-(BenzH ,3ldioxol-4-yl)-4-methyl-1-(1H-indazo!-4-v!amino)- 2-(trifluormethyl)-pentan-2-ol Trennung von (+/-)-4-(Benz[1 ,3]dioxol-4-yl)-4-methyl-1-(1H-indazol-4-ylamino)- 2-(trifluormethyI)-pentan-2-ol: Das Enantiomerengemisch wird durch Chromatographie an chiralem Trägermaterial (CHIRALPAK AD®, Fa DAICEL) mit Hexan / Ethanol (90 : 10, vvv) getrennt Man erhält so das (-)-Enantiomer: MS (esi): M++1 = 422, [D]p -50.9° °(c = 1.0, CHCI3) und das (+)-Enantiomer: MS (esi): M++1 = 422, [D]D +54.3° °(c = 1.0, CHCI3)
4-(Benz[1 ,31dioxol-4-yl)-1-(6-methyl-1H-indazol-4-ylamino)-4-methyl-2-(trifluor- methyl)pentan-2-ol Analog Beispiel 16 wird aus 4-(Benz[1 ,3]dioxol-4-yl)-2-hydroxy-4-methyl-2- (trifluormethyl)pentanal und 6-Methyl-1 H-lndazol-4-ylamin (v. Auwers Chem.Ber., 1920, 53, 1213) das gewünschte Produkt erhalten.
1H-NMR (CD3OD); δ = 1.44 (s, 3H), 1.65 (s, 3H), 2.06 (d, 1 H), 2.23 (s, 3H), 2.8 (d, 2H), 3.09 (d, 1 H), 5.89 (s, 1 H), 5.91 (s, 1 H), 6.05 (s, 1 H), 6.75 (dd, 1 H), 6.86 (t, 1 H), 6.94 (dd, 1 H), 7.22 (s, 1 H), 7.73 (s, 1 H).
Beispiel 38
4-(Benz[1,31dioxol-4-yl)-4-methyl-1-(2-methyl-benzothiazol-7-ylamino)-2- (trifluormethyl)-pentan-2-ol
In Analogie zur Herstellung von Beispiel 30 wird das entsprechende Imin aus 100 mg 7-Amino-2-methylbenzothiazol (Libeer et al. Bull.Soc.Chim.Belg:, 1971; 80; 43-47) und 154 mg 4-(Benz[1,3]dioxol-4-yl)-2-hydroxy-4-methyl-2- trifiuormethyl-pentanal erhalten und weiter mit 63 mg Natriumcyanoborhydrid zu 8 mg der Titelverbindung reduziert. H-NMR (CDCI3), D (ppm) = 1.42 (s, 3H), 1.53 (s, 3H), 2.01 (d, 1H), 2.10 (d, 1H), 2.78 (s, 3H), 5.29 (d, 1H), 5.67 (d, 1H), 6.00 (s, 2H), 6.13 (s, 1H), 6.63-6.82 (m, 3H), 7.18-7.30 (m, 2H)
Beispiel 39
4-(Benz[1,31dioxol-4-yl)-4-methyl-1-(2-methyl-1,8-naphthyridin-5-ylamino)-2- (trif I uormethvP-penta n-2-ol
Analog Beispiel 10 wird 1-Amino-4-(benz[1 ,3]dioxol-4-yl)-4-methyl-2- (trifluormethyl)propan-2-ol mit 5-Chlor-2-methyl-1,8-naphthyridin zum gewünschten Produkt umgesetzt. 1NMR (CDCI3) D (ppm) = 1.49 (s, 3H), 1.57 (s, 3H), 2.41 (d, 1H), 2.49 (d, 1H), 2.74 (s, 3H), 3.32 (d, 1H), 3.50 (d, 1 H), 5.89 (s, 1 H), 5.93 (s, 1H), 6.06 (d, 1H), 6.76-6.92 (m, 3H), 7.23 (d, 1 H), 8.05 (d, 1H), 8.61 (d, 1H)
Beispiel 40
4-(2,3-Dihvdrobenzofuranyl)-4-methyl-1-(1H-indazol-4-ylamino)-2- (trifluormethyl)-pentan-2-ol
4-(2!3-Dihydrobenzofuranyl)-2-hydroxy-4-methyl-2-(trifluormethyl)pentanal In Analogie zum beschriebenen Syntheseweg von 4-(Benz[1 ,3]dioxoI-4-yl)-2- hydroxy-4-methyl-2-trifluormethyl-pentanol (Beispiel 32) erhält man ausgehend von 513 mg 4-(2,3-Dihydrobenzofuranyl)-4-methyl-2-oxo-pentansäureethylester (WO 00/32584) 142 mg 4-(2,3-Dihydrobenzofuranyl)-2-hydroxy-4-methyl-2- (trifluormethyl)pentanol als Fraktion 2 und 84 mg der Titelverbindung als Fraktion 1 als farbloses Öl.
Fraktion 1: 1H-NMR (CDCI3), D (ppm) =1.39 (s, 3H), 1.45 (s, 3H), 2.18 (d, 1 H), 3.19 (t, 2H), 3.34 (d, 1 H), 3.62 (bs, 1H), 4.51 - 4.63 (m, 2H), 6.8 (t 1H), 6.91 (d , 1 H), 7.11 (d, 1 H), 8.96 (s, 1 H)
Fraktion 2: 1H-NMR (CDCI3), D (ppm) = 1.41 (s, 3H), 1.48 (s, 3H), 2.25 (d, 1H),
2.48 (d, 1 H), 3.2 (t, 2H), 3.42 (bs, 1H), 4.56 (t, 2H), 6.85 (t, 1 H), 7.06 - 7.15 (m,
2H)
In Analogie zur Herstellung von Beispiel 30 wird das entsprechende Imin aus 32 mg 4-Amino-1 H-indazol und 75 mg 4-(2,3-Dihydrobenzofuranyl)-2-hydroxy-4- methyl-2-(trifluormethyl)pentanal erhalten und weiter mit 134 mg Natriumcyanoborhydrid zu 64 mg der Titelverbindung reduziert. 1H-NMR (CDCI3), D (ppm) = 1.45 (s, 3H), 1.57 (s, 3H), 2.31 (d, 1 H), 2.64 (d, 1 H), 3.05- 3.19 (m, 2H), 3.21 (d, 1H), 3.39 (d, 1 H), 4.48-4.63 (m, 2H), 5.76 (d, 1H), 6.85 (d, 1 H), 6.92 (t, 1 H), 7.05-7.23 (m, 3H), 7.85 (s, 1 H)
Beispiel 41
4-(3-Chlor-2-methoxyphenyl)-1-(1H-indazol-4-ylamino)-4-methyl-2-(trifluor- methyl)pentan-2-ol
3-Chlor-2-methoxybenzylcyanid
Zu 31 ,6 g (201 ,7mmol) 3-Chlor-2-methoxytoluen in 500 ml CCI4 werden 39,4 g (221 ,3 mmol) NBS und 100 mg Benzoylperoxid gegeben. Man erhitzt über 16 Stunden unter Rückfluß, lässt abkühlen und filtriert. Das Filtrat wird vom Lösungsmittel befreit und und in214 ml Dimethylformamid und 142 ml Wasser gelöst. Man gibt bei 0°C 20,9g (322,1 mmol) Kaliumcyanid zu und rührt über 16 Stunden. Die Reaktionsmischung wird mit Wasser verdünnt und mehrfach mit erf-Butyl-methylether extrahiert. Man wäscht die organische Phase mehrfach mit gesättigter Natriumchlorid Lösung und trocknet über Natriumsulfat. Das Lösungsmittel wird im Vakuum entfernt und nach chromatographischer Reinigung an Kieselgel (Hexan / Ethylacetat 20%) erhält man 29,7g Produkt. 1H-NMR (CDCI3): δ = 3.76 (s, 2H), 3.95 (s, 3H), 7.08 (t, 1 H), 7.31 (d, 1 H), 7.37 (d, 1 H)
4-(3-Chlor-2-methoxy-phenyl)-4-methyl-2-trifluormethyl-pentan-1,2-diol 29,7 g (163,7 mmol) 4-Chlor-2-methoxybenzylcyanid und 46.5 g (327,4 mmol) Methyliodid in 260 ml DMF werden bei 0°C portionsweise mit 13.2 g (327.4 mmol) Natriumhydrid (60%ig in Öl) versetzt. Es wird über Nacht gerührt und dann mit Wasser und Essigester versetzt. Die Phasen werden getrennt und die wässrige Phase mehrfach mit Essigester extrahiert. Es wird mit Wasser und gesättigter Natriumchlorid Lösung gewaschen, mit Natriumsulfat getrocknet und im Vakuum eingeengt. Nach Chromatographie an Kieselgel (Hexan / Essigester 95:5) erhält man 32.4 g 2-(4-Chlor-2-methoxy-phenyl)-2-methylpropionitril als farbloses Öl. 7 g (33.4 mmol) des Nitrils werden in Toluol bei - 78°C langsam mit 41.6 ml (50.1 mmol) Diisobutylaluminiumhydrid Lösung (20% in Toluol) versetzt und nach 3 h bei -78°C werden.5.55 ml Isopropanol zugetropft. Man lässt auf - 5CC erwärmen und gibt 380 ml einer 10%igen wässrigen Weinsäure Lösung zu. Nach Verdünnen mit Ether wird kräftig gerührt, die organische Phase wird abge- trennt und die wässrige Phase mehrfach mit Ether extrahiert. Es wird mit Sole gewaschen, mit Natriumsulfat getrocknet und im Vakuum eingeengt. Nach Chromatographie an Kieselgel (Hexan / Essigester 95:5) erhält man 7.1 g 2-(4- Chlor-methoxy-phenyl)-2-methylpropanal als farbloses Öl. Eine Lösung von
8.95 g (33.4mmol) 2-Diethylphosphono-2-ethoxyessigsäure-ethylester in 30 ml Tetrahydrofuran wird unter Eiskühlung innerhalb von 20 Minuten mit 19 ml (38 mmol) einer 2 M Lösung von Lithiumdiisopropylamid in Tetrahydrofuran-Heptan- Toluol versetzt und 15 Minuten bei 0 °C gerührt. Innerhalb von 30 Minuten wird eine Lösung von 7.1 g (33.4 mmol) 2-(3-Chlor-2-methoxyphenyl)-2- methylpropanal in 27 ml Tetrahydrofuran bei 0°C zugetropft. Nach 20 Stunden bei RT wird Wasser zugegeben und mehrfach mit Ether und Ethylacetat extrahiert. Man wäscht mit gesättigter Ammoniumchlorid Lösung, trocknet (Na2SO ) und engt ein. Das Rohprodukt wird säulenchromatographisch an Kieselgel (Hexan / Ethylacetat 10%) gereinigt und man erhält 8.5 g 4-(3-Chlor-2- methoxy-phenyl)-4-methyl-3-ethoxy-2-en-vaIeriansäureethylester. Das Zwischenprodukt wird mit 80 ml 3 M Natronlauge / 160 ml Ethanol verseift. Man erhält 5.3 g Säure, die mit 80 ml 2 N Schwefelsäure bei 90°C über 16 Stunden gerührt werden. Nach dem Abkühlen stellt man mit Kaliumcarbonat basisch, wäscht mit Ether und säuert mit Salzsäure an. Nach Extraktion mit Ethylacetat, Waschen mit gesättigter Natriumchlorid Lösung und Entfernen des Lösungsmittels werden 4.0 g 4-(3-Chlor-2-methoxyphenyl)-4-methyl-2-oxo- valeriansäure erhalten. 6.6 g (24.3 mmol) 4-(3-Chlor-2-methoxy-phenyl)-4- methyl-2-oxo-valeriansäure und 2.74 ml (51.4 mmol) Schwefelsäure (96%ig) werden in 150 ml Ethanol 5 Stunden unter Rückfluss erhitzt. Der Ansatz wird im Vakuum eingeengt und der Rückstand in gesättigter Natriumhydrogencarbonat Lösung aufgenommen. Es wird mehrfach mit Essigester extrahiert, mit gesättigter Natriumhydrogencarbonat Lösung gewaschen, getrocknet (Natriumsulfat) und im Vakuum eingeengt. Nach chromatographischer Reinigung an Kieselgel (Hexan / Ethylacetat 10%) erhält man 5.9 g 4-(3-Chlor-2- methoxy-phenyl)-4-methyl-2-oxo-valeriansäure-ethylester. Dieser Ester und 3.4 g (23.8 mmol) (Trifluormethyl)-trimethylsilan in 34 ml THF werden mit 49 mg Tetrabutylammoniumfluorid bei 0°C versetzt. Es wird 16 h bei RT gerührt und anschließend wird die Reaktionsmischung auf Wasser gegeben. Es wird mehrfach mit Essigester extrahiert, mit gesättigter Natriumchlorid Lösung gewaschen, mit Natriumsulfat getrocknet und im Vakuum eingeengt. Man erhält 2.96 g 4-(3-Chlor-2-methoxy-phenyl)-2-hydroxy-4-methyl-2-trifluormethyl- valeriansäure-ethylester als gelbes Öl. Dieses Öl wird in 24 ml Diethylether bei
0°C mit 510 mg Lithiumaluminiumhydrid versetzt und noch 4 Stunden bei RT gerührt. Zum Ansatz werden bei 0°C vorsichtig 20 ml gesättigte Natriumhydrogencarbonat Lösung gegeben und es wird 1 Stunde kräftig nachgerührt. Man extrahiert mehrfach mittert-Butylmethylether, wäscht mit Wasser und gesättigter Natriumchlorid Lösung, trocknet mit Natriumsulfat und engt im Vakuum ein. Das Rohprodukt wird in 33 ml THF mit 1.83 (5.79 mmol) Tetrabutylammoniumfluorid Trihydrat versetzt und 16 Stunden gerührt. Man gießt auf Eiswasser, extrahiert mehrfach mit tert-Butylmethylether, wäscht mit gesättigter Natriumchlorid Lösung, trocknet mit Natriumsulfat und engt im Vakuum ein. Nach chromatographischer Reinigung an Kieselgel (Hexan / Ethylacetat 25%) erhält man 1.81g 4-(3-Chlor-2-methoxy-phenyl)-4-methyI-2-trifluormethyi-pentan-1 ,2- diol. 1H-NMR (CDCI3), D (ppm) = 1.47 (s, 3H), 1.56 (s, 3H), 2.21 (d, 1 H), 2.54 (d, 1 H), 2.91 (s, 1H), 3.31 (dd, 1 H), 3.42 (d, 1 H), 4.01 (s, 3H), 7.00 (t, 1 H), 7.20-7.35 (m, 2H) 4-(3-Chlor-2-methoxy-phenyl)-2-hydroxy-4-methyl-2-trifluoromethyl-pentanal Zu 1.2 g (3.7 mmol) Diol in 24 ml Dichlormethan und 6.4 ml DMSO gibt man 1.87g (18.5 mmol) Triethylamin und portionsweise über 10 min 1.17 g (7.4 mmol) Pyridin SO3 Komplex. Man rührt über 5 Stunden und gibt 30 ml ges. Ammoniumchlorid Lösung zu. Die Mischung wird weitere 15 min gerührt, die Phasen getrennt und es wird mit tert.-Butylethylether extrahiert. Man wäscht mit Wasser und trocknet über Natriumsulfat. Das Lösungsmittel wird im Vakuum entfernt und nach chromatographischer Reinigung an Kieselgel (Hexan / Ethylacetat, 0-50%) erhält man 0.98 g Produkt. 1H-NMR (CDCI3): δ = 1.44 (s, 3H), 1.50 (s, 3H), 2.29 (d, 2H), 3.28 (d, 1 H), 3.55 (s, 1 H), 4.01 (s, 3H) 6.95 (t, 1 H), 7.07 (dd, H), 7.30 (dd, 1 H), 8.90 (s, 1 H).
Analog Beispiel 16 wird aus 4-(3-ChIor-2-methoxyphenyl)-2-hydroxy-4-methyl-2- (trifluormethyl)pentanal und 1 H-lndazol-4-ylamin das gewünschte Produkt erhalten. 1H-NMR (300 MHz, CD3OD): D = 1.49 (3H), 1.68 (3H), 2.03 (1 H), 3.00 (1 H), 3.25 (1 H), 3.35 (1 H), 3.95 (3H), 5.60 (1H), 6.73-6.83 (2H), 7.03 (1 H), 7.18 (1 H), 7.30 (1 H), 7.95 (1 H).
4-(3-Chlor-2-hvdroxyphenyl)-1-(1H-indazol-4-vIamino)-4-methyl-2-(trifluor- methyl)pentan-2-ol
Analog Beispiel 3 erhält man aus 4-(3-Chlor-2-methoxyphenyl)-1-(1H-indazol-4- yIamino)-4-methyl-2-(trifluormethyl)pentan-2-ol das gewünschte Produkt. 1H- NMR (300 MHz, CD3OD): D = 1.50 (3H), 1.70 (3H), 2.03 (1H), 3.00 (1 H), 3.25 (1 H), 3.35 (1 H), 5.60 (1 H), 6.73-6.83 (2H), 7.03 (1 H), 7.18 (1 H), 7.30 (1 H), 7.95 (1 H).
Beispiel 43
4-(4-Fluor-2-methoxyphenyl)-4-methyl-1-(1H-indazol-4-ylamino)-2- (trifluormethyl)-pentan-2-ol 4-(4-Fluor-2-methoxyphenyl)-2-hydroxy-4-methyl-2-(trifIuormethyl)pentanol und 4-(2-Fluor-4-methoxyphenyl)-2-hydroxy-4-methyl-2-(trifluormethyl)pentanol Beide Produkte werden analog zum Syntheseweg von 4-(4-Chlor-2- methoxyphenyl)-2-hydroxy-4-methyl-2-(trifluormethyl)pentanol und 4-(2-Chlor-4- methoxyphenyl)-2-hydroxy-4-methyl-2-(trifluormethyl)pentanol (Beispiel 45) aus 3-Fluoranisol und 2-Hydroxy-4-methylen-2- (trifluormethyl)valeriansäureethylester und der anschließenden Reaktion mit Lithiumaluminiumhydrid erhalten. Die Trennung erfolgt wurch Chromatographie an Kieselgel. 1. Fraktion: 1H-NMR (CDCI3), D (ppm) = 1.40 (s, 3H), 1.52 (s, 3H), 2.25 (d, 1 H), 2.50 (d, 1 H), 2.83 (bs, 1 H), 3.36 (d, 1 H), 3.46 (d, 1 H), 3.85 (s, 3H), 6.54 - 6.69 (m, 2H), 7.19 - 7.30 (m, 1 H) 2. Fraktion: 1H-NMR (CDCI3), D (ppm) = 1.41 (s, 3H), 1.53 (s, 3H), 2.17 (d, 1 H), 2.3 (d, 1 H), 2.89 (bs, 1 H), 3.34 (bd, 1H), 3.53 (d, 1 H), 3.78 (s, 3H), 6.58 (dd, 1 H), 6.63 (dd, 1H), 7.22 (t, 1H)
4-(4-Fluor-2-methoxyphenyl)-2-hydroxy-4-methyl-2-(trifluormethyl)pentanal Analog zur Synthese von 4-(4-Chlor-2-methoxyphenyl)-2-hydroxy-4-methyl-2- (trifluormethyl)pentanal erhält man ausgehend von 3 g 4-(4-Fluor-2- methoxyphenyl)-2-hydroxy-4-methyl-2-(trifluormethyl)pentanol 1.81 g der Titelverbindung als trübes Öl.
1H-NMR (CDCI3), D (ppm) = 1.38 (s, 3H), 1.46 (s, 3H), 2.19 (d, 1H), 3.37 (d, 1 H), 3.58 (s, 1 H), 3.87 (s, 3H), 6.55 - 6.64 (m, 2H), 7.06 (dd, 1 H), 8.97 (s, 1H) In Analogie zur Herstellung von Beispiel 30 wird das entsprechende Imin aus 64 mg 4-Amino-1 H-indazol und 150 mg 4-(4-Fluor-2-methoxyphenyl)-2-hydroxy-4- methyl-2-(trifluormethyl)pentanal erhalten und weiter mit 141 mg
Natriumcyanoborhydrid zu 53 mg der Titelverbindung reduziert. 1H-NMR (CDCI3), □ (ppm) = 1.44 (s, 3H), 1.59 (s, 3H), 2.29 (d, 1H), 2.70 (d, 1H), 3.18 (d, 1 H), 3.33 (d, 1H), 3.84 (s, 3H), 5.64 (d, 1 H), 6.65 (dd, 1 H), 6.71 (dt, 1H), 6.88 (d, 1 H), 7.12 (t, 1 H), 7.37 (dd, 1 H), 7.90 (s, 1 H)
Beispiel 44
4-(4-Fluor-2-hvdroχyphenyl)-4-methyl-1-(1H-indazol-4-ylamino)-2- (trif!uormethv!)-pentan-2-ol In Analogie zur Herstellung von Beispiel 3 werden aus der Reaktion von 45 mg 4-(4-Fluor-2-methoxyphenyl)-4-methyl-1-(1H- indazol-4-ylamino)-2-(trifluormethyl)-pentan-2-ol und 1.65 ml Bortribromid (1 M in Dichlormethan) 4 mg der Titelverbindung erhalten.
1H-NMR (CDCI
3), D (ppm) = 1.48 (s, 3H), 1.52 (s, 3H), 2.26 (d, 1 H), 2.84 (d, 1H), 3.25 (d, 1 H), 3.41 (d, 1H), 4.23 (bs, 1 H), 5.72 (d, 1 H), 6.44 (dd, 1 H), 6.64 (dt, 1 H), 6.83 (d, 1 H), 7.12 (t, 1 H), 7.33 (dd, 1 H), 7.96 (s, 1 H)
Beispiel 45
4-(4-Chlor-2-methoxyphenyl)-4-methyl-1-(1H-indazol-4-ylamino)-2- (trifluormethyl)-pentan-2-ol
4-(4-Chlor-2-methoxyphenyl)-2-hydroxy-4-methyl-2-(trifluormethyl)pentanol und 4-(2-Chlor-4-methoxyphenyl)-2-hydroxy-4-methyl-2-(tήfluormethyl)pentanol Eine Lösung von 3 g 2-Hydroxy-4-methylen-2- (trifluormethyl)valeriansäureethylester in 22 ml 3-Chloranisol werden bei RT portionsweise mit Aluminiumtrichlorid versetzt. Es wird 48 h gerührt und dann mit 2 N Salzsäure und Hexan versetzt und 1 h gerührt. Es wird mit 2 N Salzsäure und Wasser gewaschen und überschüssiges 3-Chloranisol im Vakuum abdestilliert. Der verbleibende Rückstand wird durch Chromatographie an Kieselgel (Hexan / Essigester 0-10%) gereinigt. Man erhält 2.85 g eines Gemisches von 4-(4-Chlor-2-methoxyphenyl)-2-hydroxy-4-methy-2-(trifluor- methyl)valeriansäureethylester und 4-(2-Chlor-4-methoxyphenyl)-2-hydroxy-4- methy-2-(trifluormethyl)valeriansäureethylester als gelbes Öl. Dieses Substanzgemisch wird in 90 ml Ether bei 0°C mit 445 mg Lithiumaluminiumhydrid versetzt und 12 h gerührt. Der Ansatz wird auf gesättigte Natriumhydrogencarbonat-Lösung gegeben, durch Kieselgur filtriert, die Phasen werden getrennt und die wässrige Phase mit Essigester extrahiert. Es wird mit Wasser und Sole gewaschen, mit Natriumsulfat getrocknet und im Vakuum eingeengt. Nach Chromatographie an Kieselgel (Hexan / Essigester 95:5) erhält man als 1. Fraktion 1.87 mg 4-(4-Chlor-2-methoxyphenyl)-2- hydroxy-4-methyl-2-(trifluormethyl)pentan-1-ol und als 2. Fraktion 160 mg 4-(2- Chlor-4-methoxyphenyl)-2-hydroxy-4-methyl-2-(trifluormethyl)pentan-1-ol als Farblose Öle.
1. Fraktion: 1H-NMR (CDCI3), D (ppm) = 1.41 (s, 3H), 1.51 (s, 3H), 2.24 (d, 1 H), 2.51 (d, 1 H), 2.84 (bs, 1H), 3.36 (d, 1 H), 3.48 (d, 1 H), 3.85 (s, 3H), 6.88 (d, 1 H), 6.92 (dd, 1 H), 7.24 (d, 1 H)
2. Fraktion: 1H-NMR (CDCI3), D (ppm) = 1.52 (s, 3H), 1.62 (s, 3H), 2.18 (d, 1H), 2.76 (d, 1 H), 2.93 (bs, 1H), 3.33 (d, 1H), 3.55 (d, 1 H), 3.80 (s, 3H), 6.78 (dd, 1H), 6.90 (d, 1 H), 7.38 (d, 1 H) 4-(4-Chlor-2-methoxyphenyl)-2-hydroxy-4-methyl-2-(trifluormethyl)pentanal 0,425 ml Oxalylchlorid in 10 ml Dichlormethan werden bei -78°C mit 0.77 ml DMSO in 2.0 ml Dichlormethan versetzt. Nach 5 min. werden 1.38 g 4-(4-Chlor- 2-methoxyphenyl)-2-hydroxy-4-methyl-2-(trifluormethyl)pentan-1-ol in 6.0 ml Dichlormethan bei -78°C zugetropft. Nach 15 min. wird mit 2.9 ml Triethylamin versetzt und langsam auf RT erwärmt. Es wird mit Wasser und Sole gewaschen, mit Natriumsulfat getrocknet und im Vakuum eingeengt. Nach Chromatographie an Kieselgel (Hexan / Essigester 98:2) erhält man 1.16 g 4-(4-Chlor-2- methoxyphenyl)-2-hydroxy-4-methyl-2-(trifluormethyl)-pentanal als farbloses Öl. H-NMR (CDCI3), D (ppm) =1.38 (s, 3H), 1.44 (s, 3H), 2.21 (d, 1 H), 3.34 (d, 1 H), 3.57 (bs, 1 H), 3.89 (s, 3H), 6.84 (d, 1 H), 6.87 (d, 1 H), 7.04 (d, 1 H), 9.02 (s, 1 H) In Analogie zur Herstellung von 4-(5-Fluor-2-methoxyphenyl)-4-methyl-1-(2- methylphthalazin-1 -on-5-ylamino)-2-(trifluormethyl)-pentan-2-ol (Beispiel 27) wird das entsprechende Imin aus 168 mg 4-Amino-1 H-indazol und 410 mg 4-(4- Chlor-2-methoxyphenyl)-2-hydroxy-4-methyl-2-(trifluormethyl)pentanal erhalten und weiter mit 580 mg Natriumcyanoborhydrid zu 303 mg der Titelverbindung reduziert. 1H-NMR (CDCI3), D (ppm) = 1.44 (s, 3H), 1.59 (s, 3H), 2.26 (d, 1 H), 2.70 (d, 1 H), 3.17 (dd, 1H), 3.31 (dd, 1 H), 3.85 (s, 3H), 4.08 (bs, 1 H), 5.53 (d, 1 H), 6.86 (d, 1 H), 6.89 (d, 1 H), 6.99 (dd, 1 H), 7.14 (t, 1 H), 7.34 (d, 1 H), 7.86 (s, 1H)
Beispiel 46 und 47
(-)-4-(4-Chlor-2-methoxyphenyl)-4-methyl-1-(1H-indazol-4-ylamino)-2- (trifluormethyl)-pentan-2-ol und (+)-4-(4-Chlor-2-methoxyphenyl)-4-methyl-1- (1 H-indazol-4-ylamino)-2-(trifluormethv0-pentan-2-ol Trennung von (+/-)-4-(4- Chlor-2-methoxyphenyl)-4-methyl-1 -(1 H-indazol-4-ylamino)-2-(trifluormethyI)- pentan-2-ol:
Das Enantiomerengemisch wird durch Chromatographie an chiralem
Trägermaterial (CHIRALPAK AD®, Fa DAICEL) mit Hexan / Ethanol (80 : 20, vvv) getrennt. Man erhält so das
(-)-Enantiomer: MS (esi): M++1 = 442/444, [D]D -60.8° °(c = 1.0, CHCI3) und das (+)-Enantiomer: MS (esi): M++1 = 442/444, [D]Q +43.0° °(c = 1.0, CHCI3)
Beispiel 48 und 49
(-)-4-(4-Chlor-2-hvdroxyphenyl)-4-methyl-1-(1H-indazol-4-ylamino)-2- (trifluormethyl)-pentan-2-ol In Analogie zur Herstellung von Beispiel 3 werden aus der Reaktion von 100 mg (-)-4-(4-Chlor-2-methoxyphenyl)-4-methyl-1-(1H- indazol-4-ylamino)-2-(trifluormethyl)-pentan-2-ol und 3.6 ml Bortribromid (1M in Dichlormethan) 11 mg der Titelverbindung erhalten. H-NMR (CDCI3), D (ppm) = 1.49 (s, 3H), 1.62 (s, 3H), 2.25 (d, 1 H), 2.82 (d, 1H), 3.25 (d, 1 H), 3.40 (d, 1H), 5.63 (d, 1 H), 6.72 (d, 1H), 6.84 (d, 1H), 6.93 (d, 1H), 7.14 (t, 1 H), 7.31 (d, 1 H), 7.95 (s, 1 H)
(+)-4-(4-Chlor-2-hvdroχyphenyl)-4-methyl-1-(1H-indazol-4-ylamino)-2-
(trif!uormethv!)-pentan-2-o! In Analogie zur Herstellung von Beispiel 3 werden aus der Reaktion von 5.3 g (+)-4-(4-Chlor-2-methoxyphenyl)-4-methyl-1-(1H- indazol-4-ylamino)-2-(trifluormethyl)-pentan-2-ol und 190 ml Bortribromid (1 M in Dichlormethan) 2.17 g der Titelverbindung erhalten. 1H-NMR (CDCI3), D (ppm) = 1.49 (s, 3H), 1.62 (s, 3H), 2.25 (d, 1 H), 2.82 (d, 1 H), 3.25 (d, 1 H), 3.40 (d, 1 H), 5.63 (d, 1 H), 6.72 (d, 1 H), 6.84 (d, 1 H), 6.93 (d, 1 H), 7.14 (t, 1 H), 7.31 (d, 1 H), 7.95 (s, 1 H)
Beispiel 50
4-(4-Chlor-2-methoχyphenyl)-4-methyl-1-(2-methvchinazolin-5-ylamino)-2- (trifluormethyl)-pentan-2-ol
In Analogie zur Herstellung von Beispiel 2 wird aus 395 mg 5-Amino-2- methychinazolin und 299 mg 4-(4-Chlor-2-methoxyphenyl)-2-hydroxy-4-methyl- 2-(trifluormethyl)pentanal das entsprechende Imin erhalten und mit Palladium auf Aktivkohle reduziert. Die Reoxidation wird durch Erhitzen des Produktes in Xylol in Anwesenheit von Palladium auf Aktivkohle an der Luft erreicht. Man erhält 11 mg der Titelverbindung. 1H-NMR (CDCI3), D (ppm) = 1.50 (s, 3H), 1.59 (s, 3H), 2.45 (d, 1 H), 2.68 (d, 1 H), 2.86 (s, 3H), 3.18 (dd; 1 H), 3.36 (dd, 1 H), 3.89 (s, 3H), 6.08 (d, 1 H), 6.92 (d, 1 H), 6.98 (t, 1 H), 7.36 (d, 1H), 7.54 (d, 1H), 9.11 (s, 1 H)
Beispiel 51
4-(4-Chlor-2-methoxyphenyl)-1-(8-fluor-2-methvchinazolin-5-ylamino)-4-methyl- 2-(trif!uormethyl)-pentan-2-o! 1.72 g (5.30 mmol) 4-(4-Chlor-2-methoxyphenyI)-2-hydroxy-4-methyl-2- trifluormethyl-pentanai und 0.85 g (4.80 mmol) 5-Amino-8-fluor-2-methyl- chinazolin werden zunächst analog Beispiel 2 in 64 ml Dichlorethan und 13 ml Essigsäure umgesetzt. Man erhält 600 mg gereinigtes Zwischenprodukt. 200 mg (0.42 mmol) dieses Imins werden 7 ml THF gelöst und bei 0°C 60 mg (0.84 mmol) Natriumcyanoborhydrid zugesetzt. Nach 1 h werden einige Tropfen Methanol /Essigsäure (1 :1) sowie weitere 15 mg (0.21 mmol) Natriumcyanoborhydrid zugegeben und weitere 2 h gerührt. Die Reaktion wird
durch Zugabe von ges. Ammoniumchloridlösung abgebrochen und analog Beispiel 26 aufgearbeitet. Das Rohprodukt wird an Kieselgel chromatographiert (Eluent Hexan/Ethylacetat 20%). Dieses Produkt wird in wenig Chloroform aufgenommen, mit einer Spatelspitze Mangandioxid versetzt und 1 h gerührt. Der Braunstein wird abfiltriert, das Filtrat wird eingeengt und über wenig Kieselgel mit Hexan/Ethylacetat 20-50% chromatographiert. Man erhält 5 mg des gewünschten Produkts. 1H-NMR (CDCI3), D (ppm) = 1.49 (s, 3H), 1.56 (s, 3H), 2.45 (d, 1H), 2.68 (d, 1 H), 2.88 (s, 3H), 3.16 (dd, 1 H), 3.30 (dd, 1H), 3.89 (s, 3H), 5.92 (dd, 1 H), 6.85 (d, 1 H), 6.92 (dd, 1 H), 7.30 (dd, 1 H), 7.36 (d, 1H), 9.19 (s, 1 H); MS (Cl): 486/488 (M+H).
Beispiel 52
4-(4-Chlor-2-methoxyphenyl)-4-methyl-1-(2-methyl-1 ,8-naphthyridin-5-ylamino)- 2-(trifluormethyl)pentan-2-ol
Analog Beispiel 10 wird 1-Amino-4-(4-chlor-2-methoxypheπyl)-4-methyl-2- (trifluormethyl)propan-2-ol mit 5-Chlor-2-methyl-1 ,8-naphthyridin zum gewünschten Produkt umgesetzt1 H-NMR (CDCI3); δ = 1.46 (s, 3H), 1.56 (s, 3H), 2.46 (d, 1H), 2.62 (d, 1 H), 2.70 (s, 3H), 3.22 (d, 1H), 3.38 (dd, 1H), 3.84 (s, 3H), 5.89 (d, 1 H), 6.87 (d, 1 H), 6.94 (dd, 1 H), 7.14 (d, 1 H), 7.27 (d, 1 H), 7.82 (d, 1 H), 8.58 (s, 1 H).
Beispiel 53
4-(4-Chlor-2-hvdroxyphenyl)-4-methyl-1-(2-methyl-1 ,8-naphthyridin-5-ylamino)- 2-(trifluormethyl)-pentan-2-ol
Analog Beispiel 3 wird 4-(4-Chlor-2-methoxyphenyl)-4-methyl-1-(2-methyl-1 ,8- naphthyridin-5-ylamino)-2-(trifluormethyl)pentan-2-ol zum gewünschten Produkt umgesetzt.
1H-NMR (CD
3OD); δ = 1.47 (s, 3H), 1.66 (s, 3H), 2.02 (d, 1 H), 2.18 (d, 1 H), 2.72 (s, 3H), 3.14 (d, 1H), 3.24 (dd, 1 H), 5.94 (d, 1 H), 6.65 (d, 1H), 6.76 (dd, 1 H), 7.31 (d, 1 H), 7.40 (d, 1 H), 8.26 (d, 1 H), 8.35 (s, 1 H).
Beispiel 54
4-(2-Fluor-4-methoxyphenyl)-4-methyl-1-(1H-indazol-4-ylamino)-2- (trifluormethyl)-pentan-2-ol 4-(2-Fluor-4-methoxyphenyl)-2-hydroxy-4-methyl-2-(trifluormethyl)pentanal Analog zur Synthese von 4-(4-Chlor-2-methoxyphenyI)-2-hydroxy-4-methyl-2- (trifluormethyl)pentanal (Beispiel 45) erhält man ausgehend von 3 g 4-(2-Fluor- 4-methoxyphenyl)-2-hydroxy-4-methyl-2-(trifluormethyl)pentanol (Beispiel 47) 1.73 g der Titelverbindung als farbloses Öl. 1H-NMR (CDCI3), D (ppm) = 1.39 (s, 3H), 1.46 (s, 3H), 2.26 (d, 1 H), 3.09 (d, 1 H), 3.63 (s, 1 H), 3.78 (s, 3H), 6.52 - 6.65 (m, 2H), 7.03 (t, 1 H), 9.04 (s, 1H) In Analogie zur Herstellung von Beispiel 30 wird das entsprechende Imin aus 82 mg 4-Amino-1 H-indazol und 192 mg 4-(2-Fluor-4-methoxyphenyl)-2-hydroxy-4- methyl-2-(trifluormethyl)pentanal erhalten und weiter mit 281 mg Natriumcyanoborhydrid zu 152 mg der Titelverbindung reduziert.1 H-NMR (CDCI3), D (ppm) = 1.47 (s, 3H), 1.61 (s, 3H), 2.23 (d, 1 H), 2.50 (d, 1 H), 3.24 (dd, 1 H), 3.37 (dd, 1 H), 3.78 (s, 3H), 4.00-4.11 (m, 1 H), 5.70 (d, 1 H), 6.61 (dd, 1 H), 6.72 (dd, 1 H), 6.87 (d, 1H), 7.08 (t, 1 H), 7.33 (t, 1 H), 7.90 (s, 1 H)
Beispiel 55
4-(2-Fluor-4-hvdroxyphenyl)-4-methyl-1-(1H-indazol-4-ylamino)-2- (trifluormethyl)-pentan-2-ol
In Analogie zur Herstellung von Beispiel 3 werden aus der Reaktion von 80 mg 4-(2-Fluor-4-methoxyphenyl)-4-methyl-1-(1H-indazol-4-ylamino)-2- (trifluormethyI)-pentan-2-ol und 3 ml Bortribromid (1M in Dichlormethan) 76 mg der Titelverbindung erhalten. 1H-NMR (CDCI3), D (ppm) = 1.44 (s, 3H), 1.60 (s, 3H), 2.22 (d, 1 H), 2.49 (d, 1 H), 3.28 (d, 1 H), 3.37 (d, 1 H), 5.83 (d, 1 H), 6.58 (dd, 1 H), 6.66 (dd, 1 H), 6.85 (d, 1H), 7.16 (t, 1 H), 7.27 (d, 1 H), 7.88 (s, 1H)
Beispiel 56
4-(2-Chlor-4-methoxyphenyl)-4-methyl-1-(1H-indazol-4-ylamino)-2- (trifluormethyl)-pentan-2-ol 4-(2-Chlor-4-methoxyphenyl)-2-hydroxy-4-methyl-2-(trifluormethyl)pentanal Analog zur Synthese von 4-(4-Chlor-2-methoxyphenyl)-2-hydroxy-4-methyl-2- (trifluormethyl)pentanal (Beispiel 45) erhält man ausgehend von 150 mg 4-(2- Chlor-4-methoxyphenyl)-2-hydroxy-4-methyl-2-(trifluormethyl)pentanol 101 mg der Titelverbindung als gelbes Öl. 1H-NMR (CDCI3), D (ppm) = 1.52 (s, 3H), 1.54 (s, 3H), 2.24 (d, 1 H), 3.60 (d, 1 H), 3.65 (s, 1 H), 3.79 (s, 3H), 6.72 (dd, 1 H), 6.88 (d, 1H), 7.19 (d, 1 H), 9.11 (s, 1H)
In Analogie zur Herstellung von Beispiel 30 wird das entsprechende Imin aus 36 mg 4-Amino-1 H-indazol und 90 mg 4-(2-Chlor-4-methoxyphenyl)-2-hydroxy-4- methyl-2-(trifluormethyl)pentanal erhalten und weiter mit 114 mg
Natriumcyanoborhydrid zu 54 mg der Titelverbindung reduziert. 1H-NMR (CDCIs), D (ppm) = 1.56 (s, 1 H), 1.71 (s, 3H), 2.19 (d, 1 H), 3.01 (d, 1 H), 3.28 (d, 1 H), 3.37 (d, 1 H), 3.79 (s, 3H), 5.56 (d, 1 H), 6.83 (dd, 1 H), 6.86 (d, 1 H), 7.06 (t, 1 H), 7.49 (d, 1 H), 7.90 (s, 1 H)
Beispiel 57
4-(2-Chlor-4-hvdroxyphenyl)-4-methyl-1-(1H-indazol-4-ylamino)-2- (trifluormethv!)-pentan-2-o!
In Analogie zur Herstellung von Beispiel 3 werden aus der Reaktion von 27 mg 4-(2-Chlor-4-methoxyphenyl)-4-methyl-1-(1H-indazol-4-ylamino)-2- (trifluormethyl)-pentan-2-ol und 0.93 ml Bortribromid (1M in Dichlormethan) 11 mg der Titelverbindung erhalten. 1H-NMR (CDCI3), D (ppm) = 1.54 (s, 3H), 1.69 (s, 3H), 2.19 (d, 1 H), 3.00 (d, 1 H), 3.25-3.46 (m, 2H), 4.01 (bs, 1 H), 5.75 (d, 1 H), 6.77 (dd, 1 H), 6.82 (d, 1 H), 6.92 (d, 1 H), 7.11 (t, 1 H), 7.41 (d, 1 H), 7.86 (s, 1 H)
Beispiel 58
4-(4-Brom-2-methoxyphenvP-4-methyl-1-(1H-indazol-4-ylamino)-2- (trifluormethvP-pentan-2-ol
4-(4-Brom-2-methoxyphenyl)-4-methyl-2-(trifluormethyl)-pentan-1,2-diol 2,55 g (6,17 mmol) 4-(4-Brom-2-methoxyphenyl)-2-hydroxy-4-methyl-2- (trifluormethyl)-pentansäureethylester (synthetisiert in zwei Stufen ausgehend von 4-(4-Brom-2-methoxyphenyl)-2-oxopentansäure (WO 98/54159) werden in 102 ml Diethylether gelöst, bei 0 bis -5°C portionsweise mit 351,3 mg (9,256 mmol) Lithiumaluminiumhydrid versetzt und dreieinhalb Stunden bei Raumtemperatur gerührt. Das Reaktionsgemisch wird bei Eisbadkühlung
tropfenweise mit gesättigter Natriumhydrogencarbonatlösung versetzt, 15 Minuten bei 5°C und anschließend eine Stunde bei Raumtemperatur gerührt. Der ausgefallene Niederschlag wird abgesaugt, mit Diethylether nachgewaschen und das Filtrat am Rotationsverdampfer eingeengt. Der Rückstand wird an Kieselgel (Laufmittel Ethylacetat/Hexan) chromatographiert. Neben 308 mg des Aldehyds (siehe nächste Stufe) werden 2,025 g (88,4%) des Diols erhalten.
4-(4-Brom-2-methoxyphenyl)-2-hydroxy-4-methyl-2-(trifluormethyl)~pentanal 2,03 g (5,442 mmol) 4-(4-Brom-2-methoxyphenyl)-4-methyl-2-(trifluormethyl)- pentan-1 ,2-diol werden nach Swern wie in Beispiel 49 beschrieben zum
Aldehyd oxydiert. Isoliert werden 1 ,839 g (91 ,4%) der gewünschten Verbindung. 1H-NMR (300 MHz, CDCI3): D = 1.39 (3H), 1.45 (3H), 2.23 (1H), 3.35 (1 H), 3.58 (1 H), 3.90 (3H), 6.93-7.09 (3H) 9.03 (1 H). In Analogie zur Herstellung von Beispiel 30 wird das entsprechende Imin aus 181 mg 4-Amino-1 H-indazol und 500 mg 4-(4-Brom-2-methoxyphenyl)-2- hydroxy-4-methyl-2-(trifluormethyl)pentanal erhalten und weiter mit 334 mg Natriumcyanoborhydrid zu 190 mg der Titelverbindung reduziert. 1H-NMR (CDCI3), D (ppm) = 1.44 (s, 3H), 1.59 (s, 3H), 2.24 (d, 1 H), 2.71 (d, 1H), 3.17 (dd, 1 H), 3.31 (dd, 1 H), 3.87 (s, 3H), 4.01-4.10 (m, 1 H), 5.52 (d, 1 H), 6.68 (d, 1 H), 7.05 (d, 1 H), 7.15 (dd, 1 H), 7.15 (t, 1 H), 7.27 (d, 1 H), 7.88 (s, 1 H)
Beispiel 59
4-(4-Brom-2-hvdroxyphenvP-4-methyl-1-(1H-indazol-4-ylamino)-2- (trifluormethyl)-pentan-2-ol In Analogie zur Herstellung von Beispiel 3 werden aus der Reaktion von 90 mg 4-(4-Brom-2-methoxyphenyl)-4-methyl-1-(1H-indazol-4-ylamino)-2- (trifluormethyl)-pentan-2-ol und 3.8 ml Bortribromid (1M in Dichlormethan) 14 mg der Titelverbindung erhalten.
1H-NMR (CDCI3), D (ppm) = 1.48 (s, 3H), 1.60 (s, 3H), 2.22 (d, 1 H), 2.86 (d, 1 H), 3.23 (d, 1 H), 3.39 (d, 1 H), 4.22 (bs, 1 H), 5.65 (d,
1H), 6.83 (d, 1H), 6.85 (d, 1 H), 7.04 (dd, 1 H), 7.15 (t, 1 H), 7.23 (d, 1 H), 7.95 (s,
1 H)
Beispiel 60
4-(5-Chlor-2-methoxyphenyl)-1-(1H-indazol-4-ylamino)-4-methyl-2-(trifluor- methyl)pentan-2-ol
4-(5-Chlor-2-methoxyphenyl)-2-hydroxy-4-methyl-2-(trifluormethyl)pentanal 2 g (6,12 mmol) 4-(5-Chlor-2-methoxyphenyl)-4-methyl-2-trifluormethyl-pentan- 1 ,2-diol werden mit 854,6 mg (6,733 mmol) Oxalylchlorid und1 ,05 ml (14,812 mmol) DMSO wie im Beispiel 49 beschrieben nach Swern oxydiert Nach dem Aufarbeiten werden 1 ,95 g (98,4%) des gewünschten Aldehyds erhalten, der roh in die nächste Stufe eingesetzt wird. 1H-NMR (300 MHz, CDCI3): D = 1.39 (3H), 1.49 (3H), 2.27 (1 H), 3.32 (1 H), 3.59 (1H), 3.88 (3H), 6.78 (1 H), 7.10 (1 H), 7.20 (1 H), 9.09 (1 H). Analog Beispiel 16 wird aus 4-(5-Chlor-2-methoxyphenyl)-2-hydroxy-4-methyl-2- (trifluormethyl)pentanal und 1 H-lndazol-4-ylamin das gewünschte Produkt erhalten.
1H-NMR (300 MHz, CDCI3): D = 1.45 (3H), 1.60 (3H), 2.25 (1 H), 2.78 (1 H), 3.13 (1H), 3.35 (1H), 3.83 (3H), 5,60 (1 H), 6.82 (1 H), 6.87 (2H), 7.15 (1H), 7.25 (1H), 7.40 (1 H), 7.86 (1 H).
4-(6-Fluor-2-methoxyoxyphenyl)-4-methyl-1-(7-fluor-2-methylchinazolin-5- ylamino)-2-(trifluormethvP-pentan-2-ol
Analog Beispiel 25 wird 4-(6-Fluor-2-methoxyphenyl)-2-hydroxy-4-methyl-2- trifluoromethyl-pentanal mit 5-Amino-7-fluor-2-methylchinazolin zum
gewünschten Produkt umgesetzt, 1 H-NMR (CDCI3); δ = 1.59 (d, 3H), 1.69 (d, 3H), 2.35 (d, 1 H), 2.65 (d, 1 H), 2.80 (s, 3H), 3.21 (dd, 1H), 3.41 (dd, 1H), 3.86 (s, 3H), 5.02 (br., 1 H), 5.89 (dd, 1 H), 6.62 (dd, 1 H), 6.67 (d, 1 H), 6.83 (dd, 1 H), 7.14 (ddd, 1H), 8.98 (s, 1 H).
Beispiel 62
4-(6-Fluor-2-hvdroxyoχyphenyl)-1-(7-fluor-2-methylchinazolin-5-ylamino)-4- methyl-2-(trifluormethyl)pentan-2-ol
Analog Beispiel 3 wird 4-(6-Fluor-2-methoxyphenyl)-4-methyl-1-(8-fluor-2- methylchinazolin-5-ylamino)-2-(trifluormethyl)-pentan-2-ol zum gewünschten Produkt umgesetzt. 1H-NMR (CDCI3); δ = 1.64 (d, 3H), 1.73 (d, 3H), 2.40 (d, 1 H), 2.73 (d, 1 H), 2.78 (s, 3H), 3.32 (dd, 1H), 3.49 (dd, 1 H), 3.84 (s, 1 H), 5.13 (br., 1H), 5.98 (dd, 1H), 6.48 (d, 1H), 6.54 (dd, 1H), 6.80 (dd, 1H), 6.93 (ddd, 1 H), 8.97 (s, 1 H).
Beispiel 63
4-(6-FIuor-2-methoxyphenvP-4-methyl-1-(8-fluor-2-methylchinazolin-5-ylamino)- 2-(trif!uormethyl)-pentan-2-ol
Analog Beispiel 25 wird 4-(6-Fluor-2-methoxyphenyl)-2-hydroxy-4-methyl-2- trifluoromethyl-pentanal mit 5-Amino-8-fluor-2-methylchinazolin zum gewünschten Produkt umgesetzt. 1H-NMR (CDCI3); δ = 1.58 (d, 3H), 1.68 (d, 3H), 2.35 (d, 1 H), 2.69 (d, 1H), 2.90 (s, 3H), 3.04 (s, 1H), 3.23 (dd, 1 H), 3.40 (dd, 1H), 3.85 (s, 3H), 4.56 (br., 1 H), 6.05 (dd, 1 H), 6.58-6.68 (m, 2H), 7.14 (ddd, 1 H), 7.30 (dd, 1 H), 9.17 (s, 1 H).
Beispiel 64
4-(6-Fluor-2-hvdroxyphenvP-4-methyl-1-(8-fluor-2-methylchinazolin-5-ylamino)- 2-(trifluormethvP-pentan-2-ol
Analog Beispiel 3 wird 4-(6-Fluor-2~methoxyphenyl)-4-methyl-1-(8-fluor-2- methylchinazoIin-5-ylamino)-2-(trifluormethyl)-pentan-2-ol zum gewünschten Produkt umgesetzt 1H-NMR (CDCI3); δ = 1.62 (d, 3H), 1.74 (d, 3H), 2.26 (d, 1 H), 2.85 (d, 1 H), 2.63 (s, 1 H), 2.89 (s, 3H), 3.32 (dd, 1H), 3.44 (dd, 1H), 4.64 (br., 1 H), 6.10 (dd, 1 H), 6.46-6.58 (m, 2H), 6.93 (ddd, 1 H), 7.31 (dd, 1H), 9.22 (s, 1 H).
Beispiel 65
4-(3,5-Difluor-2-methoxyphenyl)-4-methyl-1-(1H-indazol-4-ylamino)-2- (trifluormethyl)-pentan-2-ol 4-(3,5-Difluor-2-methoxyphenyl)-2-hydroxy-4-methyl-2-(trifluormethyl)pentanal In Analogie zum beschriebenen Syntheseweg von 4-(Benz[1 ,3]dioxol-4-yl)-2- hydroxy-4-methyl-2-trifluormethyI-pentanal erhält man 90 mg der Titelverbindung als farbloses Öl.
1H-NMR (CDCI
3), D (ppm) = 1.39 (s, 3H), 1.45 (s, 3H), 2.33 (d, 1 H), 3.18 (d, 1 H), 3.59 (s, 1 H), 3.98 (d, 3H), 6.15 (dm, 1H), 6.72 - 6.82 (m, 1 H), 9.24 (s, 1 H) In Analogie zur Herstellung von Beispiel 30 wird das entsprechende Imin aus 35 mg 4-Amino-1 H-indazol und 85 mg 4-(3,5-Difluor-2-methoxyphenyl)-2-hydroxy- 4-methyl-2-(trifluormethyl)pentanal erhalten und weiter mit 99 mg Natriumcyanoborhydrid zu 43 mg der Titelverbindung reduziert.
1H-NMR
(CDCI
3), D (ppm) = 1.45 (s, 1 H), 1.59 (s, 3H), 2.26 (d, 1 H), 2.71 (d, 1 H), 3.22 (d, 1H), 3.34 (d, 1 H), 4.01 (d, 3H), 5.81 (d, 1 H), 6.78 (ddd, 1H), 6.88 (d, 1H), 6.93 (ddd, 1H), 7.15 (t, 1H), 7.90 (s, 1 H)
Beispiel 66
4-(3,5-Difluor-2-hvdroxyphenvP-4-methyl-1-(1H-indazol-4-ylamino)-2- (trifluormethv!)-pentan-2-ol In Analogie zur Herstellung von Beispiel 3 werden aus der Reaktion von 37 mg 4-(3,5-Difluor-2-methoxyphenyl)-4-methyI-1-(1H-indazol-4-ylamino)-2- (trifluormethyl)-pentan-2-ol und 1.3 ml Bortribromid (1M in Dichlormethan) 14 mg der Titelverbindung erhalten.
1H-NMR (CDCI
3), D (ppm) = 1.50 (s, 3H), 1.62 (s, 3H), 2.30 (d, 1H), 2.81 (d, 1 H), 3.27 (d, 1 H), 3.39 (d, 1H), 5.73 (d, 1 H), 6.78 (dt, 1 H), 6.87 (d, 1H), 6.94 (dt, 1 H), 7.12 (t, 1 H), 7.89 (s, 1 H)
Beispiel 67
4-(2,3-DihydrobenzofuranvP-4-methyl-1-(1H-indazol-4-ylamino)-2- (trifluormethvD-pentan-2-o!
In Analogie zur Herstellung von Beispiel 30 wird das entsprechende Imin aus 32 mg 4-Amino-1 H-indazol und 75 mg 4-(2,3-Dihydrobenzofuranyl)-2-hydroxy-4- methyl-2-(trifluormethyl)pentanal erhalten und weiter mit 134 mg Natriumcyanoborhydrid zu 64 mg der Titelverbindung reduziert. 1H-NMR
(CDCI3), D (ppm) = 1.45 (s, 3H), 1.57 (s, 3H), 2.31 (d, 1 H), 2.64 (d, 1 H), 3.05- 3.19 (m, 2H), 3.21 (d, 1 H), 3.39 (d, 1 H), 4.48-4.63 (m, 2H), 5.76 (d, 1 H), 6.85 (d, 1 H), 6.92 (t, 1 H), 7.05-7.23 (m, 3H), 7.85 (s, 1H)
Beispiel 68
4-(4,5-Difluor-2-methoxyphenvP-4-methyl-1-(1H-indazol-4-ylamino)-2- (trifluormethyl)-pentan-2-ol
4-(4,5-Diflυor-2-methoxyphenyl)-2-hydroxy-4-methyl-2-(trifluormethyl)pentanal In Analogie zum beschriebenen Syntheseweg von 4-(Beπz[1,3]dioxol-4-yl)-2- hydroxy-4-methyl-2-trifluormethyl-pentanal erhält man 695 mg der Titelverbindung als farbloses Öl. 1H-NMR (CDCI3), D (ppm) = 1.38 (s, 3H), 1.44 (s, 3H), 2.23 (d, 1H), 3.29 (d, 1H), 3.56 (s, 1H), 3.83 (s, 3H), 6.76 (dd, 1H), 6.96 (dd, 1H), 9.08 (s, 1H)
In Analogie zur Herstellung von Beispiel 30 wird das entsprechende Imin aus 82 mg 4-Amino-1 H-indazol und 200 mg 4-(4,5-Difluor-2-methoxyphenyl)-2-hydroxy- 4-methyl-2-(trifluormethyl)pentanal erhalten und weiter mit 319 mg Natriumcyanoborhydrid zu 155 mg der Titelverbindung reduziert. H-NMR (CDCI3), D (ppm) = 1.44 (s, 3H), 1.58 (s, 3H), 2.24 (d, 1H), 2.71 (d, 1H), 3.19 (d, 1H), 3.32 (bd, 1H), 3.82 (s, 3H), 5.63 (d, 1H), 6.70 (dd, 1H), 6.87 (d, 1H), 7.13 (t, 1H), 7.24 (dd, 1H), 7.87 (s, 1 H)
Beispiel 69
4-(4-Chlor-5-fluor-2-methoxyphenyl)-4-methyl-1-(1Hindazol-4-ylamino)-2- (trifluormethvP-pentan-2-ol 4-(4-Chlor-5-flυor-2-methoxyphenyl)-2-hydroxy-4-methyl-2- (trifluormethyl)pentanal
In Analogie zum beschriebenen Syntheseweg von 4-(Benz[1 ,3]dioxol-4-yl)-2- hydroxy-4-methyl-2-trifluormethyl-pentanoI erhält man 1.55 g 4-(4-Chlor-5-fluor- 2-methoxyphenyl)-2-hydroxy-4-methyl-2-(trifluormethyl)pentanal als Fraktion 1 und 1.32 g 4-(4-Chlor-5-fluor-2-methoxyphenyl)-2-hydroxy-4-methyl-2- (trifluormethyl)pentanol als Fraktion 2. Fraktion 1 : 1H-NMR (CDCI3), D (ppm) = 1.36 (s, 3H), 1.43 (s, 3H), 2.28 (d, 1 H), 3.27 (d, 1 H), 3.57 (s, 1 H), 3.85 (s, 3H), 6.84 (d, 1 H), 6.93 (d, 1 H), 9.11 (s, 1 H) Fraktion 2: 1H-NMR (CDCI3), D (ppm) = 1.4 (s, 3H), 1.48 (s, 3H), 2.23 (d, 1 H), 2.47 (d, 1 H), 2.91 (s, 1H), 3.35 (dd, 1H), 3.5 dd, 1 H), 3.83 (s, 3H), 6.87 (d, 1H), 7.1 (d, 1H) In Analogie zur Herstellung von Beispiel 30 wird das entsprechende Imin aus 78 mg 4-Amino-1 H-indazol und 200 mg 4-(4-Chlor-5-fluor-2-methoxyphenyl)-2- hydroxy-4-methyl-2-(trifluormethyl)pentanal erhalten und weiter mit 280 mg Natriumcyanoborhydrid zu 116 mg der Titelverbindung reduziert. H-NMR (CDCI3), D (ppm) = 1.43 (s, 3H), 1.58 (s, 3H), 2.23 (d, 1 H), 2.71 (d, 1 H), 3.19 (d, 1 H), 3.31 (bd, 1H), 3.83 (s, 3H), 5.55 (d, 1 H), 6.85 (s, 1 H), 6.87 (d, 1 H), 7.14 (t, 1 H), 7.21 (d, 1H), 7.88 (s, 1H)
4-(4-Chlor-5-fluor-2-hvdroxyphenyl)-4-methyl-1-(1 H-indazol-4-ylamino)-2- (trifluormethyl)-pentan-2-ol In Analogie zur Herstellung von Beispiel 3 werden aus der Reaktion von 100 mg 4-(4-Chlor-5-fluor-2-methoxyphenyl)-4-methyl-1- (1H-indazol-4-ylamino)-2-(trifluormethyl)-pentan-2-oI und 3.3 ml Bortribromid (1 M in Dichlormethan) 30 mg der Titelverbindung erhalten. Fp. = 171 - 173°C
4-(4-Chlor-5-fluor-2-methoxyoxyphenvP-4-methyl-1-(8-fluor-2-methylchinazolin- 5-ylamino)-2-(trifluormethyP-pentan-2-ol Analog Beispiel 25 wird 4-(4-Chlor-5-fluor-2methoxyphenyl)-2-hydroxy-4-methyl- 2-trifluoromethyI-pentanal mit 5-Amino-8-fluor-2-methyIchinazolin zum gewünschten Produkt umgesetzt.
1H-NMR (CDCI
3); δ = 1.45 (s, 3H), 1.56 (s, 3H), 2.30 (d, 1 H), 2.73 (d, 1H), 2.90 (s, 3H), 2.99 (s, 1 H), 3.14 (dd, 1 H), 3.22 (dd, 1 H), 3.83 (s, 3H), 4.48 (br., 1 H), 5.88 (dd, 1 H), 6.79 (d, 1 H), 7.17 (d, 1 H), 7.31 (dd, 1 H), 9.20 (s, 1 H).
Beispiel 72
4-Methyl-1-(2-methvchinazolin-5-ylamino)- 4-phenyl-2-(trifluormethvP-pentan-2- pj 2-Hydroxy-4-methyl-4-phenyl-2-trifluoromethyl-pentanal 10.4 g 4-Methyl-2-oxo-4-phenylpentansäure (WO98/54159) in 250 ml Dimethylformamid werden bei -5 °C mit 4.1 ml Thionylchlorid und nach 15 min mit 4 ml Methanol versetzt. Nach 15 h bei Raumtemperatur wird der Ansatz mit Wasser verdünnt und mit Essigester extrahiert. Die organischen Extrakte werden mit Wasser gewaschen, getrocknet (Na
2SO
4) und eingeengt, wobei 9.3 g 4-Methyl-2-oxo-4-phenylpentansäure-methylester erhalten werden. Diese werden in 558 ml DMF bei -5°C mit 15.5 ml (104.63 mmol) (Trifluormethyl)trimethylsilan und 20.5 g (63.28 mmol) Caesiumcarbonat versetzt und 16 h bei Raumtemperatur gerührt. Man gibt Wasser hinzu, extrahiert mit Ethylacetat, wäscht die organische Phase mit Wasser und trocknet (Na
2SO
4). Das eingeengte Zwischenprodukt wird in 200 ml THF aufgenommen und 50 ml
einer 1 M Lösung von Tetrabutylammoniumfluorid in THF werden zugegeben. Man rührt 2 Stunden, gibt Wasser zu, extrahiert mit Ethylacetat, wäscht die organische Phase mit Wasser und trocknet (Na
2SO ). Nach Chromatographie an Kieselgel mit Hexan-Essigester (0-30%) werden 8.35 g 2-Hydroxy-4-methyl- 4-phenyl-2-(trifluormetyl)pentansäure-methylester erhalten. Der Ester (8.3 g, 28.59 mmol) wird in 180 ml THF gelöst, und über einen Zeitraum von 2,5 Stunden werden 1.52 g (36.20 mmol) Lithiumaluminiumhydrid in kleinen Portionen zugegeben. Nach vollständigem Umsatz werden 5 ml Ethylacetat zugetropft und nach weiteren 10 min werden vorsichtig 10 ml Wasser zugegeben. Man filtriert vom gebildeten Niederschlag ab und wäscht sorgfältig mit Ethylacetat. Nach Chromatographie an Kieselgel mit Hexan-Essigester (0- 35%) werden 5.40 g 4-Methyl-4-phenyl-2-(trifluormetyl)pentan-1 ,2-diol erhalten. Zu 2.5g (9.53 mmol) Diol in 75 ml Dichlormethan und 28 ml DMSO gibt man 5.7 ml (40.3 mmol) Triethylamin und portionsweise über 20 min 5 g Pyridin SO
3 Komplex. Man rührt über 2 Stunden und gibt 40 ml ges. Ammoniumchlorid
Lösung zu. Die Mischung wird weitere 15 min gerührt, die Phasen getrennt und es wird mit Dichlormethan extrahiert. Man wäscht mit Wasser und trocknet über Natriumsulfat Das Lösungsmittel wird im Vakuum entfernt und man erhält 3 g Produkt. 1H-NMR (CDCI3): δ = 1.34 (s, 3H), 1.44 (s, 3H), 2.34 (d, 2H), 2.66 (d, 1 H), 3.64 (s, 1 H), 7.03-7.41 (m, 4H), 8.90 (s, 1 H).
Analog Beispiel 25 wird 2-Hydroxy-4-methyl-4-phenyl-2-trifluoromethyl-pentanal mit 5-Amino-2-methyIchinazolin zum gewünschten Produkt umgesetzt. 1H-NMR (CDCI3); δ = 1.46 (s, 3H), 1.59 (s, 3H), 2.30 (d, 1 H), 2.37 (d, 1 H), 2,84 (s, 3H), 3.10 (dd, 1H), 3.27 (dd, 1H), 4.70 (br., 1H), 6.12 (d, 1 H), 7.26 (d, 1 H), 7.28 (t, 1 H), 7.39 (t, 2H), 7.48 (d, 2H), 7.58 (t, 1 H), 9.22 (s, 1 H).
Beispiel 73
4-(2,5-DiflυorphenvP-4-methyl-1-(2-methyl-benzothiazol-7-ylamino)-2-
(trifluormethyl)-pentan-2-ol
110 mg (0.66 mmol) 2-Methyl-benzothiazol-7-ylamin in 1 ml Essigsäure werden mit 150 mg (0.53 mmol) 4-(2,5-Difluor-phenyl)-2-hydroxy-4-methyl-2- trifluormethyl-pentanal, gelöst in 10 ml Dichlorethan, versetzt, 4 h über Molsieb (4 A) refluxiert und weitere 16 h bei RT gerührt. Die Mischung wurde zwischen Wasser und Dichlormethan verteilt, mit Dichlormethan extrahiert und die vereinigten organischen Phasen gewaschen (ges. NaCI-lösung), getrocknet (Na2SO4) und eingeengt Das nach Chromatographie an Kieselgel mit Hexan/Essigester (20%) erhaltene Zwischenprodukt (97 mg) wird in Essigsäure aufgenommen, mit 10 mg NaBH4 versetzt und die Mischung 4 h bei RT gerührt. Man verteilt zwischen Wasser und Dichlormethan, extrahiert, wäscht die vereinigten organischen Phasen (ges. NaCI-lösung), trocknet (Na2SO4) und engt ein. Man erhält 90 mg Produkt, das aus Hexan/Diethylether kristallisiert werden kann. MS (Cl): 445 (M+H); 1H-NMR (CDCI3): δ = 1.61 (s, br, 6H), 2.26 (d, 1 H), 2.50 (d, 1 H), 2.83 (s, 3H), 3.15 (s, 1 H), 3.27 (d, br, 2H), 3.49 (m, 1 H), 6.02 (d, 1 H), 6.82-7.02 (m, 2H), 7.10-7.25 (m, 2H), 7.45 (dd, 1H).
Beispiel 74
4-(2-Chlorphenyl)-4-methyl-1-(2-methyl-benzothiazol-7-ylamino)-2- (trifluormethyl)-pentan-2-ol In Analogie zu Beispiel 5 wird aus 7-Amino-2-methylbenzothiazol (Libeer et al. Bull.Soc.Chim.Belg.; 1971 ; 80; 43-47) und 4-(2-Chlorphenyl)-2-hydroxy-4- methyl-2-trifluormethyl-pentanal das gewünschte Produkt erhalten. 1H-NMR (300 MHz, CDCI3): D = 1.60 (3H), 1.73 (3H), 2.20 (1 H), 2.80 (3H), 3.09 (1 H), 3.18 (1 H), 3.23 (4H), 5,78 (1 H), 7.13 (1 H), 7.20-7.35 (2H), 7.37-7.45 (2H), 7.60 (1 H).
Beispiel 75
4-(2-Methoxyphenyl)-4-methyl-1-(1H-indazol-4-ylamino)-2-(trifluormethyl)- pentan-2-ol
4-(2-Methoxyphenyl)-2-hydroxy-4-methyl-2-(trifluormethyl)pentanal In Analogie zum beschriebenen Syntheseweg von 4-(Benz[1 ,3]dioxol-4-yl)-2- hydroxy-4-methyl-2-trifluormethyl-pentanal (Beispiel 32) erhält man ausgehend von 28.6 g 4-(2-Methoxyphenyl)-4-methyl-2-oxo-pentansäureethylester (WO 98/54159) 7.9 mg der Titelverbindung als farbloses Öl. 1H-NMR (CDCI3), D
(ppm) = 1.40 (s, 3H), 1.47 (s, 3H), 2.2 (d, 1 H), 3.46 (d, 1H), 3.60 (s, 1 H), 3.88 (s, 3H), 6.83 - 6.94 (m, 2H), 7.13 (dd, 1 H), 7.24 (dt, 1 H), 8.94 (s, 1 H) In Analogie zur Herstellung von Beispiel 30 wird das entsprechende Imin aus 139 mg 4-Amino-1 H-indazol und 300 mg 4-(2-Methoxyphenyl)-2-hydroxy-4- methyl-2-(trifluormethyl)pentanal erhalten und weiter mit 627 mg Natriumcyanoborhydrid zu 310 mg der Titelverbindung reduziert. 1H-NMR (CDCI3), D (ppm) = 1.48 (s, 3H), 1.60 (s, 3H), 2.33 (d, 1H), 2.77 (d, 1 H), 3.17 (dd, 1 H), 3.36 (dd, 1 H), 3.88 (s, 3H), 3.98-4.08 (m, 1 H), 5.66 (d, 1 H), 6.83 (d, 1 H), 6.94 (dd, 1 H), 7.04 (dt, 1 H), 7.09 (t, 1 H), 7.32 (dt, 1 H), 7.43 (dd, 1 H), 7.86 (s, 1 H)
Beispiel 76
4-(2-Hvdroxyphenyl)-4-methyl-1-(1H-indazol-4-ylamino)-2-(trifluormethyl)- pentan-2-ol In Analogie zur Herstellung von Beispiel 3 werden aus der Reaktion von 20 mg
4-(2-Methoxyphenyl)-4-methyl-1-(1H-indazoI-4-ylamino)-2-(trifluormethyl)-
005/003098 85
pentan-2-ol und 0.75 ml Bortribromid (1M in Dichlormethan) 15 mg der Titelverbindung erhalten.
1H-NMR (CDCI3), D (ppm) = 1.52 (s, 3H), 1.62 (s, 3H), 2.32 (d, 1 H), 2.90 (d, 1H), 3.23 (d, 1 H), 3.43 (dd, 1H), 4.22 (bs, 1 H), 5.75 (d, 1 H), 6.72 (d, 1H), 6.79 (d, 1H), 6.97 (t, 1 H), 7.04-7.16 (m, 2H), 7.39 (d, 1H), 7.93 (s, 1H)
Beispiel 77 und 78
(-)-4-(2-Hvdroxyphenyl)-4-methyl-1-(1H-indazol-4-ylamino)-2-(trifluormethyl)- pentan-2-ol und (+)-4-(2-HydroxyphenyP-4-methyl-1-(1H-indazol-4-ylamino)-2- (trifluormethyl)-pentan-2-ol
Trennung von (+/-)-4-(2-Hydroxyphenyl)-4-methyl-1-(1H-indazol-4-ylamino)-2- (trifluormethyl)-pentan-2-ol Das Enantiomerengemisch wird durch Chromatographie an chiralem Trägermaterial (CHIRALPAK AD®, Fa DAICEL) mit Hexan / Ethanol (70 : 30, vvv) getrennt. Man erhält so das Enantiomer A: MS (ei) M+ = 392 und das Enantiomer B: MS (ei) M+ = 392
Beispiel 79

4-(4-Chlorphenyl)-4-methyl-1-(1H-indazol-4-ylamino)-2-(trifluormethyl)-pentan-2- ol 2-(4~Chloφhenyl)-2-methylpropanal 10 g 4-Chlorbenzylcyanid und 14.3 ml Methyliodid in 140 ml DMF werden bei 0°C potionsweise mit Natriumhydrid (60%ig in Öl) versetzt. Es wird über Nacht gerührt und dann mit Wasser und Essigester versetzt. Die Phasen werden getrennt und die wässrige Phase mit Essigester extrahiert. Es wird gründlich mit Wasser extrahiert, mit Sole gewaschen, mit Natriumsulfat getrocknet und im Vakuum eingeengt. Nach Chromatographie an Kieselgel
(Hexan / Essigester 95:5) erhält man 11.73 g 2-(4-Chlorphenyl)-2- methylpropionitril als farbloses Öl. Dieser wird in Toluol bei - 78°C langsam mit 55.4 ml Diisobutylaluminiumhydrid Lösung (20% in Toluol) versetzt und nach 4 h bei -78°C wurden 50 ml Essigester zugetropft. Er wird unter Erwärmung auf RT über Nacht gerührt und Wasser zugegeben. Nach filtrieren durch Kieselgur werden die Phasen getrennt und die wässrige Phase mit Essigester extrahiert. Es wird mit Wasser und Sole gewaschen, mit Natriumsulfat getrocknet und im Vakuum eingeengt. Nach Chromatographie an Kieselgel (Hexan / Essigester 95:5) erhält man 10.2 g 2-(4-Chlorphenyl)-2-methylpropanal als farbloses Öl. H- NMR (CDCI
3), D (ppm) = 1.46 (s, 6H), 7.20 (d, 1 H), 7.29-7.43 (m, 3H), 9.48 (s, 1H) 4-(4-Chlorphenyl)-4-methyl-2-oxo-valeriansäure
Eine Lösung von 15.04 g 2-Diethylphosphono-2-ethoxyessigsäure-ethylester in 50 ml Tetrahydrofuran wird unter Eiskühlung innerhalb von 20 Minuten mit 30 ml einer 2 M Lösung von Lithiumdiisopropylamid in Tetrahydrofuran-Heptan-Toluol versetzt und 15 Minuten bei 0 °C gerührt. Innerhalb von 30 Minuten wird eine Lösung von 10.2 g 2-(4-Chlorphenyl)-2-methylpropanal in 50 ml Tetrahydrofuran bei 0°C dazugetropft. Nach 20 Stunden bei RT wird 2 N Schwefelsäure zugegeben, mit Ethylacetat extrahiert, getrocknet (Na2SO4) und eingeengt. Das Rohprodukt wird mit 200 ml 2 M Natronlauge / 400 ml Ethanol verseift. Man erhält 13.8 g Säure, die mit 300 ml 2 N Schwefelsäure und 100 ml Eisessig unter starkem Rühren unter Rückfluss 3 Stunden erhitzt wird. Nach Extraktion mit Ethylacetat und Waschen mit Wasser werden 10.9 g 4-(4-Chlorphenyl)-4- methyl-2-oxo-valeriansäure als rotes Öl erhalten. 1H-NMR (CDCI3), D (ppm) = 1.47 (s, 6H), 3.28 (s, 2H), 7.28 (m, 4H), 7.73 (bs, 1 H)
4-(4-Chlorphenyl)-2-hydroxy-4-methyl-2-(trifluormethyl)-pentan-1-al In Analogie zur Synthese von 4-(3-Chlor-2-methoxy-phenyl)-2-hydroxy-4-methyl- 2-trifluoromethyl-pentanal (Beispiel 45) werden durch Veresterung von 10.9 g 4- (4-Chlorphenyl)-4-methyl-2-oxo-va!eriansäure in Ethanol / Schwefelsäure, Umsetzung der Produktes mit (Trifluormethyl)trimethylsilan und Tetrabutylammoniumfluorid und Reduktion des gebildeten Hydroxyesters mit
Lithiumaluminiumhydrid 4.22 g 4-(4-Chlorphenyl)-2-hydroxy-4-methyl-2- (trifluormethyl)pentan-l-ol als farbloses Öl erhalten.
1H-NMR (CDCI3), □ (ppm) = 1.39 (s, 3H), 1.49 (s, 3H), 2.07 (d, 1 H), 2.19 (d, 1 H), 2.83 (bs, 1 H), 3.27 (d, 1 H), 3.41 (d, 1 H), 7.26-7.38 (m, 4H). Zu 2 g (6.7 mmol) Diol in 50 ml Dichlormethan und 22 ml DMSO gibt man 6.8 ml (33.3 mmol) Triethylamin und portionsweise über 20 min 1.5 g Pyridin SO3 Komplex. Man rührt über 5 Stunden und gibt 40 ml ges. Ammoniumchlorid Lösung zu. Die Mischung wird weitere 15 min gerührt, die Phasen getrennt und es wird mit Dichlormethan extrahiert. Man wäscht mit Wasser und trocknet über Natriumsulfat Das Lösungsmittel wird im Vakuum entfernt und man erhält nach Chromatographie an Kieselgel (Hexan / Ethylacetat 0-30%) 1.27 g Produkt. 1H- NMR (CDCI3): δ = 1.34 (s, 3H), 1.44 (s, 3H), 2.34 (d, 2H), 2.66 (d, 1 H), 3.64 (s, 1 H), 7.23-7.31 (m, 4H), 8.90 (s, 1H).ln Analogie zur Herstellung von Beispiel 30 wird das entsprechende Imin aus 158 mg 4-Amino-1 H-indazol und 350 mg 4-(4- Chlorphenyl)-2-hydroxy-4-methyl-2-(trifluormethyl)pentanal erhalten und 100 mg des Imins wurden weiter mit 216 mg Natriumcyanoborhydrid zu 68 mg der Titelverbindung reduziert. 1H-NMR (CDCI3), D (ppm) = 1.42 (s, 3H), 1.59 (s, 3H), 2.19 (d, 1 H), 2.31 (d, 1 H), 3.11 (d, 1 H), 3.22 (d, 1 H), 5.67 (d, 1H), 6.90 (d, 1 H), 7.18 (t, 1 H), 7.35 (d, 2H), 7.42 (d, 2H), 7.89 (s, 1 H)
Beispiel 80
4-(4-Chlorphenyl)-4-methyl-1-(8-fluor-2-methylchinazolin-5-ylamino)-2- (trifluormethyl)-pentan-2-o! Analog Beispiel 25 wird 4-(4-Chlorphenyl)-2-hydroxy-4-methyl-2-trifluoromethyl- pentanal mit 5-Amino-8-fluor-2-methylchinazolin zum gewünschten Produkt umgesetzt. 1H-NMR (CDCI3); δ = 1.45 (s, 3H), 1.58 (s, 3H), 2.29 (d, 1 H), 2.36 (d, 1 H), 2.79 (br., 1 H), 2,90 (s, 3H), 3.05 (dd, 1H), 3.20 (dd, 1 H), 4.37 (br., 1H), 5.98 (dd, 1 H), 7.29 (dd, 1H), 7.38 (t, 2H), 7.49 (d, 2H), 9.21 (s, 1H).
Beispiel 81
4-ir4-(5-Fluor-2-methoxyphenyl)-2-hvdroxy-4-methyl-2- (trifluormethvPpentyllamino)-1,3-dihvdroindol-2-on Dimethyl-2-(2,6-dinitrophenyl)-malonat
42,95 g (311,03 mmol) Dimethylmalonat werden in 300 ml N,N- Dimethylformamid gelöst und portionsweise mit 35,15 g (296,22 mmol) Kalium- teil, butylat versetzt. Nachdem das entstandene tert. Butanol abdestilliert worden ist, wird der Reaktionsmischung auf 20° C abgekühlt. Zur Mischung werden zügig 30 g (148,11 mmol) 2,6-Dichlorbenzol portionsweise zugegeben. Nach dreistündigem Rühren bei 90° C wird über Nacht bei Raumtemperatur gerührt. Die Reaktionsmischung wird auf 800 ml 1%ige NaOH-lösung (mit Eis gekühlt) gegeben und dreimal mit Methyl-tert. butylether extrahiert. Die vereinigten Etherphasen werden nach DC Kontrolle verworfen. Die wässrige Phase wird unter Eisbadkühlung mit conc. Salpetersäure (w = 65 %) vorsichtig angesäuert. Sechsmaliges Extrahieren mit Methyl-tert. butylether, übliches Aufarbeiten der vereinigten organischen Extrakte (Wasser, Sole, Trocknen, Abfiltrieren und Abrotieren des Lösungsmittels) liefert einen Rückstand, der an Kieselgel (Laufmittel Ethylacetat/Hexan) chromatographiert wird. Isoliert werden 12,09 g (27,09 %) der gewünschten Verbindung. Methyl-(2,6-Dinitrophenyl)-acetat 10,08 g (33,8 mmol) Dimethyl-2-(2,6-dinitrophenyl)-malonat werden in 54 ml Eisessig mit 2,7 ml Perchlorsäure versetzt und bei 125° C am Rückfluß erhitzt. Dabei wurde der entstehende Etylacetat abdestilliert. Nach 90 Minuten wird die Reaktion abgebrochen, da laut DC Kontrolle kein Ausgangsmaterial mehr vorhanden ist. Die Reaktionsmischung wird auf Eiswasser gegossen und dreimal mit Ethylacetat extrahiert. Die vereinigten organischen Extrakte werden mit 5%iger Natriumhydrogencarbonatlösung, mit Wasser und mit Sole
geschüttelt. Nach dem Trocknen der organischen Phase, dem Abfiltrieren des Trocknungsmittels und dem Abrotieren des Lösungsmittels verbleibt ein Rückstand, der an Kieselgel (Laufmittel Ethylacetat/Hexan) chromatographiert wird. Isoliert werden 4,69 g der (2,6-Dinitrophenyl)-essigsäure, die anschließend 5 mit Methanol (16 ml) und conc. Schwefelsäure (0,4 ml) verestert wird. Dazu werden die Säure und die Reagentien sieben Stunden am Rückfluß gekocht Das Methanol wird abrotiert und der Rückstand in üblicher weise aufgearbeitet Nach Chromatographie an Kieselgel (Laufmittel Ethylacetat/Hexan) werden 4,43 g (89 %) des gewünschten Esters erhalten. l o 4-Amino-1, 3-dihydroindol-2-orι 4,43 g (18,45 mmol) Methyl-(2,6-Dinitrophenyl)-acetat werden in 38,8 ml Eisessig und 11 ml Wasser gegeben und mit 3,75 g Eisenpulver versetzt und vier Stunden nachgerührt. Dabei tritt eine Erwärmung auf 40 bis 60° C ein. Die Reaktionsmischung wird auf Eiswasser gegeben, mit Ethylacetat versetzt und
15 zehn Minuten kräftig gerührt. Das Gemisch wird über Glasfaserfilter filtriert, die organische Phase abgetrennt und die wässrige Phase noch zweimal mit Ethylacetat extrahiert. Die vereinigten organischen Extrakte werden mit Sole gewaschen, getrocknet und das Lösungsmittel nach dem Abfiltrieren des Trockenmittels abrotiert. Der Rückstand wird an Kieselgel (Laufmittel
20 Methanol/Dichlormethan) chromatographiert. Isoliert werden 2,38 g des 4-Nitro- indol-2-ons. Die Nitroverbindung wird abermals in Eisessig/Wasser mit 2,7 g Eisenpulver versetzt und der oben beschriebene Zyklus ein weiteres Mal durchlaufen. Isoliert werden nun 1 ,63 g des gewünschten Amins. Analog Beispiel 27 wird 4-(5-Fluor-2-methoxyphenyl)-2-hydroxy-2-trifluormethyl-
25 pentanal mit 4-Amino-1 ,3-dihydroindol-1-on zum gewünschten Produkt umgesetzt. 1H-NMR (CDCI3): δ = 1.44 (s, 3H), 1.58 (s, 3H), 2.23 (d, 1H), 2.70 (d, 1H), 3.04 (s, 1H), 3.09 (d, 1 H), 3.19 (s, 2H), 3.21 (d, 1 H), 3.85 (s, 3H), 5.71 (d, 1H), 6.32 (d, 1H), 6.84 (dd, 1H), 6.92-7.01 (m, 2H), 7.14 (dd, 1 H), 7.78 (s, br, 1 H)
4-{f4-(5-Chlor-2-methoxyphenyl)-2-hydroxy-4-methyl-2-
(trifluormethyl)pentvπamino)-1 ,3-dihv-6-fluordroindol-2-on
180 mg (0.60 mmol) 4-(4-Chlor-2-methoxyphenyl)-2-hydroxy-4-methyl-2- trifluormethyl-pentanal und 100 mg (0.60 mmol) 4-Amino-6-fluorindol-2-on werden in 15 ml Dichlorethan und 3 ml Essigsäure vorgelegt, ca. 100 mg gepulvertes Molsieb (4 A) zugegeben und die Mischung 6 h zum Rückfluss erhitzt. Nach dem Abkühlen wird abfiltriert, das Filtrat zwischen Dichlormethan und ges. NaHCO3-Lösung verteilt und die Phasen getrennt. Die wäßrige Phase wird mehrfach mit Dichlormethan extrahiert, die vereinigten organischen Phasen werden mit Wasser und ges. NaCl-Lösung gewaschen und mit Na2SO getrocknet Die Lösemittel werden entfernt und das Rohprodukt an Kieselgel chromatographiert (Eluent Hexan/Ethylacetat 20-100%). Man erhält 40 mg Imin, das in 5 ml Methanol aufgenommen wird, mit einigen Tropfen Essigsäure und ca. 20 mg Natriumcyanoborhydid versetzt und 2 h bei RT gerührt wird. Die Mischung wird durch Zugabe von ges. NaHCO3-Lösung abgebrochen. Man extrahiert mit Dichlormethan, wäscht mit ges. NaCl-Lösung und trocknet mit Na2SO4. Chromatographische Reinigung des Rohprodukts (Kieselgel, Eluent Hexan/Ethylacetat 20-100%) liefert 4 mg des gewünschten Produkts. MS (ESI): 475/477 (M+H); 1H-NMR (CDCI3): δ = 1.46 (s, 3H), 1.57 (s, 3H), 2.29 (d, 1H), 2.60 (d, 1 H), 2.78 (s, 1 H), 2.99-3.21 (m, 4H), 3.47 (t, br, 1H), 3.88 (s, 3H), 5.52 (dd, 1 H), 6.08 (dd, 1H), 6.89 (d, 1 H), 7.00 (dd, 1 H), 7.32 (dd, 1 H), 7.68 (s, br, 1H).
Beispiele 83 - 92
Allgemeine Arbeitsvorschrift zur Herstellung von N-Methylbenzimidazol- Derivaten:
0.70 mmol des substituierten Pentanais und 1.05 mmol der Aminkomponente (4-Methyl-1H-benzimidazol-4-ylamin, vgl. z.B. V. Milata, D. Ilavsky, J. Saloό,
Bull. Soc. Chim. Belg. 1997, 106, 731-732; 3-Methyi-3H-benzimidazol-4-ylamin, vgl. z.B. M. Kamel, M.l. Ali, M.M. Kamel, Tetrahedron 1966, 22, 3351-3354 ) werden mit ca. 80 mg Molsieb (4 A, gepulvert) in 10 ml Dichlorethan und 1 ml Essigsäure vorgelegt und ca. 7 h zum Rückfluss erhitzt. Nach dem abkühlen wird die Mischung mit Phosphatpufferlösung (1 M, pH 7) und Dichlormethan versetzt, abfiltriert und die Phasen getrennt. Die wäßrige Phase wird mehrfach mit Dichlormethan extrahiert, die vereinigten organischen Phasen werden mit Wasser und ges. NaCl-Lösung gewaschen und mit Na2SO4 getrocknet. Die Lösemittel werden am Rotationsverdampfer entfernt und das Zwischenprodukt durch Chromatographie an Kieselgel gereinigt (Eluent Hexan/Ethylacetat). Das gebildete Imin wird in wenig Methanol wieder aufgenommen, einige Tropfen Essigsäure zugesetzt und NaCNBH3 (2-3 Äquivalente) zugegeben. Die Lösung wird 3 h bei RT gerührt, die Reaktion durch Zugabe von Phosphatpufferlösung (1 M, pH 7) beendet und mit Dichlormethan extrahiert. Die vereinigten organischen Phasen werden mit Wasser und ges. NaCl-Lösung gewaschen und mit Na2SO getrocknet. Das Rohprodukt wird über Kieselgel mit Hexan/Ethylacetat filtriert.
Allgemeine Arbeitsvorschrift zur Methyletherspaltung bei N-Methylbenzimidazol- Derivaten:
Ca. 0.1 mmol des Methylethers werden in ca. 3 ml Dichlormethan vorgelegt und ca. 1 ml einer BBr3-Lösung (1 M in Dichlormethan) zugesetzt. Man rührt 2 h bei RT, verdünnt mit Ethylacetat, gibt die Lösung in ges. NaHCO3-Lösung, trennt die Phasen und extrahiert mit Ethylacetat. Die vereinigten organischen Phasen werden mit Wasser und ges. NaCl-Lösung gewaschen und mit Na2SO4 getrocknet. Das Rohprodukt wird aus Diethylether/Hexan kristallisiert. Man erhält Ausbeuten von ca. 50%.
Name Struktur MS 1H-NMR (300 MHz, CDCI3)
1 ,1 ,1 -Trifluor-4- 442/44 1.32 (s, 3H), 1.64 (s, 3H),
(4-chlor-2- 4 2.02 (m, 2H), 3.00-3.16 (m, hydroxy-phenyl)-
(M+H, 2H), 4.04 (s, 3H), 5.98 (d,
4-methyl-2-(4- ESI) 1H), 6.68-6.74 (m, 1H), methyl-1H- 7.04 (d, 1 H), 7.23-7.33 (m, benzimidazol-4- 3H), 8.98 (s, 1H). (CD3OD) ylamin)
1 ,1,1 -Trif luor-4- 442/44
(4-chlor-2- 4
hydroxy-phenyl)- (M+H,
4-methyl-2-(3- ESI) methyl-3H- benzimidazol-4- ylamin)
1 ,1 ,1-Trifluor-4- 426 1.47 (s, 3H), 1.64 (s, 3H),
(5-fluor-2- (M+H, 1.97-2.03 (m, 2H), 3.06 (d, hydroxy-phenyl)- ESI) 1H), 3.27 (d, 1H), 3.85 (s,
4-methyl-2-(4-
3H), 5.86 (d, 1 H), 6.57- methyl-1H- 6.82 (m, 3H), 6.98-7.06 (m, enzimidazol-4- 2H), 8.01 (s, 1H). (CD3OD) ylamin)
1 ,1,1-Trifluor-4- 426 1.47 (s, 3H), 1.63 (s, 3H),
(5-fluor-2- (M+H, 1.92-2.04 (m, 1 H), 2.87 (d, hydroxy-phenyl)- ESI) 1 H), 3.09-3.18 (m, 1H),
4-methyl-2-(3- 4.15 (s, 3H), 5.84 (d, 1H), methyl-3H-
6.40 (dd, 1H), 6.50-6.57 enzimidazol-4- (m, 1H), 6.92-7.06 (m, 3H), ylamin) 8.09 (s, 1H). (CP3OD)
Beispiel 93
4-f[4-(5-Fluor-2-methoxyphenyl)-2-hvdroxy-4-methyl-2- (trifluormethvPpentyllamino)-2,3-dihydroisoindolon-1(2H)-on 2-Methyl-3-nitrobenzoesäuremethylester
30 g (165,6 mmol) 2-Methyl-3-nitrobenzoesäure werden in 150 ml Methanol gegeben und nach Zugabe von 2,9 ml konzentrierter Schwefelsäure zwei Tage am Rückfluß gekocht. Nach dem Abkühlen wird das Kristallisat (25,55 g = 79 %) abgesaugt und so in die nächste Stufe eingesetzt. 2-(Brommethyl)-3-nitrobenzoesäuremethylester
25,55 g (130,9 mmol) 2-Methyl-3-nitrobenzoesäuremethylester werden in 300 ml Tetrachlorkohlenstoff gegeben, mit 25,6 Gramm (141 ,7 mmol) N- Bromsuccinimid und 62,8 mg Benzoylperoxid versetzt. Nach sieben Tagen Kochen am Rückfluß wird nach dem Abkühlen das Succinimid abgesaugt und anschließend das Filtrat zur Trockene einrotiert. Zurück bleibt die gewünschte Verbindung, die roh in die nächste Stufe eingesetzt wird. 2-(Azidomethyl)-3-nitrobenzoesäuremethylester
10 g (36,5 mmol) 2-(Brommethyl)-3-nitrobenzoesäuremethylester werde mit 36 ml N,N-Dimethylformamid und 24 ml Wasser versetzt. Nach Zugabe von 3,54 g Natriumazid wird der Ansatz über Nacht gerührt. Die Reaktionsmischung wird mit Methyl-tert. Butylether verdünnt, zweimal mit Wasser und einmal mit Sole gewaschen. Nach dem Trocknen über Natriumsulfat wird filtriert und das Lösungsmittel abrotiert. Das gewünschte Azid wird in einer Ausbeute von 89,6 % (7,72 g) erhalten und roh weiter eingesetzt. 4-Amino-2, 3-dihydroisoindol-1-on 1 g (4,2 mmol) 2-(Azidomethyl)-3-nitrobenzoesäuremethylester werden in 10 ml Ethanol und 2 ml Eisessig gegeben und mit 148,5 mg Pd/C versetzt. Nach dem Rühren über Nacht bei Raumtemperatur unter einer Wasserstoffatmosphäre wird der Katalysator über ein Glasfaserfilter abgesaugt und das Filtrat bis zur Trockene eingeengt. Der Rückstand wird an Kieselgel (Ethylacetat) chromatographiert. Isoliert werden 391 ,5 mg (62,4 %) der gewünschten Verbindung.
Analog Beispiel 30 wird 4-(5-Fluor-2-methoxyphenyl)-2-hydroxy-2-trifluormethyl- pentanal mit 4-Amino-2,3-dihydroisoindol-1-on zum gewünschten Produkt umgesetzt. 1H-NMR (DMSOD6): δ = 1.37 (s, 3H), 1.54 (s, 3H), 2.03 (d, 1H), 2.81 (d, 1 H), 2.81 (d, 1 H), 2.90 (dd, 1 H), 3.02 (dd, 1 H), 3.33 (s, 1 H), 3.81 (s, 3H), 4.01-4.14
(m, 2H), 4.77 (br., 1H), 5.76 (s, 1H), 6.17 (d, 1H), 6.88 (d, 1H), 7.01-7.16 (m , 4H), 8.35 (s, 1H)
Beispiel 94
4-(5-Fluor-2-hvdroxyoxyphenvP-1-(1H-indol-4-ylamino)-4-methyl-2-(trifluor- methvDpentan-2-ol
Analog Beispiel 3 wird 4-(5-Fluor-2-methoxyphenyl)-4-methyl-1-(1H-indol-5- ylamino)-2-(trifluormethyl)-pentan-2-ol zum gewünschten Produkt umgesetzt. 1H-NMR (CDCI3); δ = 1.49 (s, 3H), 1.61 (s, 3H), 2.31 (d, 1H), 2.75 (d, 1H), 3.26 (d, 1H), 3.43 (d, 1H), 3.60 (s, 1H), 5.67 (d, 1H), 6.33 (m, 1H), 6.63 (dd, 1H), 6.86-6.97 (m, 3H), 7.10 (dd, 1H), 7.15 (dd, 1H), 8.14 (br, 1H)
Beispiel 95
1,1,1 -Trifluor-2-f(8-fluor-2-methyl-chinazolin-5-ylamino)-methyl]-4-(3-methoxy- phenyl)-4-methyl-pentan-2-ol Die Verbindung wurde, ausgehend von den entsprechenden Vorstufen, analog wie in Beispiel 13 beschrieben, synthetisiert. Isoliert wurden im letzten Schritt
26,9 mg (17,8%) der gewünschten Verbindung. 1H-NMR (300 MHz, CD3OD): D = 1,49 (3H), 1,69 (3H), 2,19 (1H), 2,56 (1H), 2,85 (3H), 2,93 (1H), 3,12 (1H), 3,64 (3H), 5,90 (1H), 6,58 (1H), 7,00 (1H), 7,03- 7,20 (2H), 7,38(1 H), 9,38(1 H).
Beispiel 96
96
3-(4,4,4-Trifluor-3-[(8-fluor-2-methyl-chinazolin-5-ylamino)-methyl-3-hydroxy-1,1- dimethyl-butvD-phenol Die Verbindung wurde, ausgehend vom Ether, beschrieben in Beispiel 95, durch
Etherspaltung mit BBr3 gewonnen. Isoliert wurden im letzten Schritt 6 mg
(29,5%) der gewünschten Verbindung.
1H-NMR (300 MHz, CD3OD): D = 1 ,49 (3H), 1 ,65 (3H), 2,15 (1 H), 2,50 (1H),
2,82 (3H), 2,95 (1 H), 3,17 (1 H), 6,02 (1 H), 6,53 (1 H), 6,90-7,13 (3H), 7,39 (1 H), 9,38 (1H).
Beispiel 97
(+)-1 ,1 ,1-Trifluor-2-f(8-fluor-2-methyl-cinazolin-5-ylamino)-methyll-4-(3-methoxy- phenyl)-4-methyl-pentan-2-ol
Die nach Beispiel 95 hergestellte Verbindung wird an einer chiralen Säule (Chiralpak AD 20D, Laufmittel Hexan/Isopropanol) in ihre Enantiomeren getrennt.
(+)-Enantiomer: Beispiel 97; (-)-Enantiomer: Beispiel 98.
Beispiel 98
(-)-1 ,1 ,1-Trifluor-2-[(8-fluor-2-methyl-cinazolin-5-ylamino)-methylH-(3-methoxy- phenyl)-4-methyl-pentan-2-ol
97
Bedingungen für die Racematspaltung siehe Beispiel 97.
Beispiel 99
3-ri-(3-Chlor-2-methoxy-phenvP-cvclohexyπ-1 , 1.1-trifluor-2-ff8-fluor-2-methyl- chinazolin-5-v!amino)-methyl1-propan-2-ol
Die Verbindung wurde, ausgehend von den entsprechenden Vorstufen, analog wie in den obigen Beispielen beschrieben, synthetisiert. Isoliert wurden im letzten Schritt 32 mg (31 ,9%) der gewünschten Verbindung.
1H-NMR (CDCI3); δ = 1 ,00-2,50 (12H), 2,60-3,30 (6H), 3,79 (3H), 4,15-4,55 (1 H),
5,60-5,85 (1H), 6,82 (1H), 6,95 (1 H), 7,10-7,50 (2H), 9,10 (1H).
Beispiel 100
(-)-4-[4-(4-Chlor-2-methoxy-phenvP-2-hydroxy-4-methyl-2-trifluormethyl- pentylamino1-1 ,3-dihydro-indol-2-on
Die Verbindung wurde, ausgehend von den entsprechenden Vorstufen, analog wie in den obigen Beispielen beschrieben, synthetisiert. Isoliert wurden im letzten Schritt 117,1 mg (97,7%) der gewünschten Verbindung als Racemat. 1H-NMR (300 MHz, CDCI3): D = 1 ,44 (3H), 1 ,58 (3H), 2,09 (1 H), 2,20 (1 H), 2,65 (1H), 2,95-3,27 (3H), 3,88 (3H), 5,99 (1H), 6,34 (1 H), 6,85-7,08 (3H), 7,32 (1H), 7, 99 (1 H).
Anschließend wurde eine Racematspaltung durchgeführt. Nach Chromatographie an einer chiralen Säule (Chiralpak AD 20 D, Laufmittel
Hexan/Ethanol) wurden die beiden Enantiomeren erhalten. (-)-Enaπtiomer: Beispiel 100; (+)-Enantiomer: Beispiel 101.
Beispiel 101
(+)-4-[4-(4-Chlor-2-methoxy-phenvP-2-hvdroxy-4-methyl-2-trifluormethyl- pentylamino1-1 ,3-dihvdro-indol-2-on
Bedingungen für die Racematspaltung siehe Beispiel 101.
Beispiel 102
(-)-4-f4-(4-Chlor-2-hvdroxy-phenyP-2-hvdroxy-4-methyl-2-trifluormethyl- pentylaminol-1 ,3-dihvdro-indol-2-on
Die Verbindung wurde durch Etherspaltung der im Beispiel 100 beschriebenen
Verbindung synthetisiert.
1H-NMR (300 MHz, CD3OD): D = 1 ,32 (3H), 1 ,49 (3H), 2,43 (1 H), 2,59 (1 H),
3,10 (1 H), 3,21-3,40 (3H), 5,87 (1 H), 6,24 (1 H), 6,58 (1 H), 6,69 (1 H), 6,95 (1 H), 7,18 (1 H).
Beispiel 103
99
(+)-4-r4-(4-Chlor-2-hvdroxy-phenyl)-2-hydroxy-4-methyl-2-trifluormethyl- pentylamino1-1 ,3-dihvdro-indol-2-on Die Verbindung wurde durch Etherspaltung der im Beispiel 101 beschriebenen Verbindung synthetisiert. Für die 1NMR-Daten siehe Beispiel 102.
Beispiel 104
6-Fluor-4-f4-(2-fluor-6-methoxy-phenyl)-2-hvdroxy-4-methyl-2-trifluormethyl- pentylamino1-2,3-dihydro-isoindol-1-on
Die Verbindung wurde, ausgehend von den entsprechenden Vorstufen, analog wie in den obigen Beispielen beschrieben, synthetisiert. Isoliert wurden im letzten Schritt 62,5 mg (77,8%) der gewünschten Verbindung. 1H-NMR (300 MHz, CD3OD): D = 1,53 (3H), 1 ,79 (3H), 1 ,98 (1H), 2,95-3,12
(2H), 3,25 (1H), 3,90 (3H), 4,09-4,28 (2H), 6,00 (1H), 6,62 (1H), 6,71 (1H), 6,83 (1H), 7,19 (1H).
6-Fluor-4-f4-(2-fluor-6-hvdroxy-phenyl)-2-hydroxy-4-methyl-2-trifluormethyl- pentylaminol-2.3-dihvdro-isoindol-1-on
Die Verbindung wurde durch Etherspaltung der im vorigen Beispiel beschriebenen Verbindung mit BBr3 erhalten. Isoliert wurden 37,5 mg (69,7%) der gewünschten Verbindung.
1H-NMR (300 MHz, CD3OD): D = 1 ,54 (3H), 1 ,82 (3H), 1 ,89 (1 H), 3,05 (1 H),
3,20-3,40 (2H), 4,10-4,28 (2H), 6,05 (1 H), 6,47 (1 H), 6,58 (1 H), 6,70 (1H), 6,98
(1H).
Beispiel 106
1 ,1,1-Trifluor-4-(2-fluor-6-methoxy-phenyl)-2-[(1 H-indazol-4-ylamino)-methyI]-4- methyl-pentan-2-ol
Die Verbindung wurde, ausgehend von den entsprechenden Vorstufen, analog wie in den obigen Beispielen beschrieben, synthetisiert. Isoliert wurden im letzten Schritt 51 ,2 mg (72,8%) der gewünschten Verbindung. 1H-NMR (300 MHz, CD3OD): D = 1,54 (3H), 1 ,80 (3H), 2,03 (1 H), 3,00-3,19 (2H), 3,35 (1H), 3,85 (3H), 5,65 (1 H), 6,63 (1H), 6,70-6,84 (2H), 7,08 (1H), 7,18 (1H), 7,93 (1 H).
Beispiel 107
1 ,1 ,1-Trifluor-4-(2-fluor-6-hvdroxy-phenyl)-2-r(1 H-indazol-4-ylamino)-methyl
'|-4- methyl-pentan-2-ol
Die Verbindung wurde durch Etherspaltung der im Beispiel 106 beschriebenen Verbindung mit BBr
3 erhalten. Isoliert wurden 20,8 mg (54,2%) der gewünschten Verbindung.
1H-NMR (300 MHz, CD3OD): D = 1,57 (3H), 1,80 (3H), 1,92 (1H), 3,15 (1H), 3,20-3,50 (2H), 5,70 (1H), 6,40-6,60 (2H), 6,75 (1H), 6,88-7,10 (2H), 7,90 (1 H).