WO2005010030A2 - C5a-REZEPTOR-ANTAGONISTEN - Google Patents
C5a-REZEPTOR-ANTAGONISTEN Download PDFInfo
- Publication number
- WO2005010030A2 WO2005010030A2 PCT/EP2004/008057 EP2004008057W WO2005010030A2 WO 2005010030 A2 WO2005010030 A2 WO 2005010030A2 EP 2004008057 W EP2004008057 W EP 2004008057W WO 2005010030 A2 WO2005010030 A2 WO 2005010030A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- substituted
- group
- alkyl
- amino acid
- cycloalkyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 CCC1C=*=CC1 Chemical compound CCC1C=*=CC1 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K7/00—Peptides having 5 to 20 amino acids in a fully defined sequence; Derivatives thereof
- C07K7/50—Cyclic peptides containing at least one abnormal peptide link
- C07K7/54—Cyclic peptides containing at least one abnormal peptide link with at least one abnormal peptide link in the ring
- C07K7/56—Cyclic peptides containing at least one abnormal peptide link with at least one abnormal peptide link in the ring the cyclisation not occurring through 2,4-diamino-butanoic acid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/16—Central respiratory analeptics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/02—Drugs for dermatological disorders for treating wounds, ulcers, burns, scars, keloids, or the like
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/04—Antipruritics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P17/00—Drugs for dermatological disorders
- A61P17/06—Antipsoriatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/02—Drugs for skeletal disorders for joint disorders, e.g. arthritis, arthrosis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P19/00—Drugs for skeletal disorders
- A61P19/08—Drugs for skeletal disorders for bone diseases, e.g. rachitism, Paget's disease
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P21/00—Drugs for disorders of the muscular or neuromuscular system
- A61P21/04—Drugs for disorders of the muscular or neuromuscular system for myasthenia gravis
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/02—Drugs for disorders of the nervous system for peripheral neuropathies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/06—Antihyperlipidemics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/02—Immunomodulators
- A61P37/06—Immunosuppressants, e.g. drugs for graft rejection
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/02—Antithrombotic agents; Anticoagulants; Platelet aggregation inhibitors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/08—Vasodilators for multiple indications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/705—Receptors; Cell surface antigens; Cell surface determinants
Definitions
- the present invention relates to C5a receptor antagonists and uses thereof.
- complement system In addition to the adaptive immune system, there is a system that is clearly older in terms of the ancestry to ward off infections.
- This system is called the complement system and consists of more than 30 soluble and membrane-bound proteins.
- the complement system can be activated together with the adaptive immune response or independently, e.g. to kill parhogenic bacteria. Excessive activation or insufficient regulation of the complement system is associated with a large number of inflammatory diseases, e.g. septic shock, reperfusion damage, rheumatoid arthritis, graft rejection, acute respiratory distress syndrome (ARDS), systemic lupus erythematosus (SLE) and gomeralonephritis.
- septic shock e.g. septic shock, reperfusion damage, rheumatoid arthritis, graft rejection, acute respiratory distress syndrome (ARDS), systemic lupus erythematosus (SLE) and gomeralonephritis.
- ARDS
- the complement system can be activated in three different ways. These are called classic, alternative and mannose-binding lectin (MBL) pathways. All paths run through the sequential processing - and thus activation - of inactive professions of proteases. Since the activated protease can activate the next proform, the triggering reaction is amplified comparable to the coagulation cascade.
- MBL mannose-binding lectin
- C3b is a key component of a central protease in the complement cascade, the C5 convertase.
- C3b is part of the C5 convertase of both the classic and the alternative route.
- the MLB route also leads via the convertases of the classic route.
- the C5 convertase is responsible for the progress of the complement cascade and catalyzes the cleavage of C5.
- C3b is covalently bound to the surface of, for example, bacteria, which can thereby preferably be taken up by macrophages. The same applies to the clarification of immune complexes.
- C3a is the small fragment that is formed alongside C3b by the cleavage of C3.
- C3a is a relatively weak chemokine and belongs to the anaphylatoxins.
- C5b is formed by splitting C5.
- the fission product is the starting point for the formation of the membrane attack complex (MAC).
- the MAC forms a pore through which the plasma membrane of bacteria as well as the body's own cells can be perforated. This can lead to lysis of the perforated cells.
- C5a is the 74 amino acid, N-terminal cleavage product of the ⁇ chain of the plasma protein C5 and is released by the activity of the C5 convertase.
- C5a is bound by its receptor, which is called C5aR or CD88, with high affinity and triggers a variety of pro-inflammatory effects. It is one of the strongest chemokines and, like C3a, belongs to the anaphylatoxins.
- the C5aR can be found on a variety of cells. The receptor is found particularly on damrophils, macrophages, smooth muscle cells and endothelial cells.
- C5a The release of C5a is directly or indirectly responsible for a variety of diseases. Examples include sepsis (Huber-Lang et al. 2001 Faseb Journal 15: 568-570), multiple sclerosis (Mullerladner et al. 1996 Journal of Neurological Science 144: 135- 141), reperfusion damage (Riley et al. 2000 Journal of Thoriacic and Cardiovascular Surgery 120: 350-358), psoriasis (Bergh et al. 1993 Archives of Dermatological Research 285: 131-134), rheumatoid arthritis (Woodruff et al.
- C5aR antagonist also referred to herein as a C5aR antagonist.
- small molecules include L-156602 (Merck), RPR120033 (Rhone-Poulenc), W-54011 (Mitsubishi Pharma) and NGD 2000-1 (Neurogen). All previously known inhibitors with a molecular weight of ⁇ 500 g mol have at least one of the following disadvantages: low specificity, agonistic activity, too low affinity, poor solubility, insufficient metabolic stability or inhibition of P450 enzymes.
- C5aR antagonists Another way to develop C5aR antagonists has been through the use of recombinant proteins.
- Examples of such protein-based antagonists are CGS 32359 (Ciba-Geigy, Pellas et al. 1998 Journal of Immunology 160: 5616-5621), ⁇ prfl-A8 (Heller et al. 1999 Journal of Immunology 163: 985-994) and Antibodies, which can be of recombinant or non-recombinant origin (Huber-Lang et al. 2001 Faseb Journal 15: 568-570).
- These C5aR antagonists are proteins and therefore expensive to manufacture. They are characterized by a relatively high affinity and specificity, but they have the disadvantage of a high immunogenicity. In addition, proteins can only be obtained using complex processes such as Administer injections efficiently.
- sequence information from the C-terminal region of C5a was used to develop peptide antagonists.
- Peptides as therapeutically usable antagonists of the C5aR have the advantage of lower production costs, no immunogenicity and high plasma stability compared to protein therapeutics and are also more specific than most of the previously known small molecules.
- Peptide antagomes have been described in large numbers. A common characteristic of almost all peptide C5aR antagonists is their origin in the C-terminus of C5a.
- peptide C5aR antagonists or partial agonists are described, inter alia, in the following patent applications or patents: US 4,692,511, US 5,663,148, WO 90/09162, WO 92/11858, WO 92/12168, WO 92/21361, WO 94 / 07518, WO 94/07815, WO 95/25957, WO 96/06629, WO 99/00406 and WO 99/13899, WO 03/033528.
- De Martino et al. (1995 Journal of Biological Chemistry 270: 15966-15969) a first structural attempt is made to explain the importance of C-terminal arginine in peptide C5aR ligands.
- WO 90/09162 presents 38 peptide inhibitors with their IC50 values (Examples 2, 13, 23, 31, 91, 106, 111, 117, 131, 150, 165, 182, 188, 202, 213, 220, 229 , 245, 247, 249, 279, 282, 295, 296, 305, 316, 338, 348, 377, 402, 404, 409, 421, 424, 432, 445, 455, 460).
- 37 peptides have a C-terminal arginine and only one peptide carries another C-terminal amino acid (tyrosine, example 305).
- sequence of example 305 from WO 90/09162 is Ac-, Phe-Lys-Ala-Cha-Ala-Leu-ala-Tyr-OH and an ido value for the binding of 0.17 ⁇ M is described, which is less corresponds to one tenth of the activity of other peptides described with a C-terminal arginine (for example Ac-Phe-Lys-Ala-Cha-Ala-Leu-N-Methyl (D) ala-Arg-OH (Example 296) and (N- Ethyl) Phe-Lys-Ala-Cha-Ala-Leu-N-Methyl (D) ala-Arg-OH (Example 402) with an IC 50 value of 0.012 ⁇ M or 0.011 ⁇ M).
- a C-terminal arginine for example Ac-Phe-Lys-Ala-Cha-Ala-Leu-N-Methyl (D) al
- this tyrosine-containing compound even shows only an IC 50 value of only 1.3 ⁇ M.
- Functional assay systems usually have a higher predictive value for activity in vivo than binding assays. This makes it clear that the use of a C-terminal tyrosine did not result in a compound that can be used to develop a therapeutically useful C5aR antagonist. This may also be the reason why the authors have not described any other tyrosine-containing peptides with a value for the activity.
- WO 92/12168 describes a further 20 peptides with their IC 50 values (binding to C5aR). 19 of these have a terminal arginine, which can exist in both the D and the L form. A peptide carries a phenylbutanoyl residue at the C-terminal, which could enter into a hydrophobic interaction.
- This peptide (Example 170) has the sequence (N-methyl) Phe-Lys-Pro-cha-Phe-Phenylbutanoyl and is given an ICs ⁇ value of only 2.6 ⁇ M, which does not appear to be promising for use as a medicament
- ICs ⁇ value of only 2.6 ⁇ M, which does not appear to be promising for use as a medicament
- a direct comparison between C-terminal argininyl and phenylbutanoyl is only partially possible in this application, since the directly comparable mechanism with a C-terminal arginine was not disclosed.
- Example 105 from WO 92/12168 ((N-methyl) Phe-Lys-Procha- ⁇ ⁇ CH 2 -N (CH 2 CH 2 C 6 H 5 ) ⁇ - Arg-OH) is most likely to be compared with Example 170 is suitable.
- the IC 50 value for this hexamer is 0.36 ⁇ M.
- the substitution of Arg leads to a significant decrease in activity in these examples as well.
- IC 50 values given in the international applications mentioned WO 90/09162, WO 92/12168 and WO 94/07518 were obtained by measurements with isolated membranes of polymorphic damrophil granulocytes (PMN membranes), since at the time the experiments were carried out there were no C5aR overexpressing cells could be produced.
- the resulting values do not reflect the affinity of the connection on whole cells.
- the connections to receptors on whole cells are significantly less affine (Kawai et al. 1991 Journal of Medicmical Chemistry 34: 2068-71; Rollins et al. 1988 Journal of Biological Chemistry 263: 520-526). However, it is more meaningful to measure their biological activity instead of the binding of the antagonists to the receptors.
- Such functional assays are often used for G protein coupled receptors.
- WO 99/00406 describes a series of cyclic and linear peptide inhibitors, the common feature of which is the C-terminal arginine.
- the necessary positive charge which is realized, for example, by arginine, is explicitly dealt with (WO 99/00406 page 12, line 13ff).
- C5a-desArg carboxypeptidases
- WO 03/033528 reports individual substitutions of different amino acids in the Ac-Phe [Orn-Pro-cha-Trp-Arg] molecule (compound 1). It is stated that, for example, the replacement of arginine in compound 1 by homoargimn (compound 44), CitruUin (compound 45), lysine (compound 47) or canavanine (compound 48) leads to a deterioration in the affinity for the C5a receptor and the antagonistic properties leads.
- WO 03/033528 does state that the arginine substitution from 1 (arginine) to 45 (citruUin) gives a compound that is said to have remarkable antagonistic activity (p. 44, lines 28ff.).
- the limit of what is classified as remarkable is arbitrary and the significant decrease in antagonistic activity by a factor of 24 underlines the importance of arginine at the C-terminal position of peptide C5aR antagonists known in the art.
- the peptide 45 containing citruUin is moreover the only peptide which does not carry a net positive charge under physiological conditions and for which a value for the binding and the antagonistic activity is described in WO 03/033528.
- the object of the present invention is to provide antagonists of the C5a receptor.
- a further object on which the present invention is based is to provide medicaments which can be used in the treatment of clinical pictures in which the C5a receptor is causally, indirectly or symptomatically involved.
- the object is achieved according to the invention by a compound, preferably a C5a receptor antagonist, with the following structure:
- X2 is a radical that mimics the biological binding properties of a phenylalanine unit
- X3 and X4 are individually and independently of one another a spacer, the spacer preferably being selected from the group comprising amino acids, amino acid analogs and amino acid derivatives,
- X5 is a radical that mimics the biological binding properties of a cyclohexylalamn or homoleucine unit
- X6 is a radical that mimics the biological binding properties of a tryptophan unit
- X7 is a radical that mimics the biological binding properties of a norleucine or phenylalanine unit
- the connecting lines - in formula (I) denote chemical bonds, the chemical bond being selected individually and independently, preferably from the group comprising covalent bonds, ionic bonds and coordinative bonds, the bond preferably being a chemical bond and more preferably the chemical bond Binding is such a bond which is selected from the group comprising amide bonds, disulfide bonds, ether bonds, thioether bonds, oxime bonds and aminotriazine bonds.
- X3 and X7 are each an amino acid, an amino acid derivative or an amino acid analogue, the chemical bond between X3 and X7 being formed with the participation of at least one molecular part each of X3 and X7, and the molecular parts for X3 and X7 individually and independently are selected from one another from the group comprising the C-terminus, the N-Tenninus and the respective side chain of the amino acid.
- XI is a radical with a mass of about 1-300, the radical preferably being selected from the group consisting of R5, R5-CO-, R5-N (R6) -CO-, R5-O-CO-, R5- SO 2 -, R5-N (R6) -C (NH) -, wherein R5 and R6 are preferably selected individually and independently of one another from the group which is substituted by H, alkyl, substituted alkyl, cycloalkyl, substituted cycloalkyl, heterocyclyl Contains heterocyclyl, aryl and substituted aryl;
- X2 and X6 are each independently an aromatic amino acid, a derivative or an analog thereof;
- X5 and X7 are individually and independently of one another a hydrophobic amino acid, a derivative or an analogue thereof.
- X2, X5, X6 and X7 individually and independently of one another have the following structure:
- XC is (R4) or N, Rl is optionally present and when Rl is present, Rl is a radical selected from the group consisting of>N-R1B,> C (R1B) (R1D) and> O, where R1B and R1D are individually and independently selected are from the group comprising H, alkyl, substituted alkyl, cycloalkyl, substituted cycloalkyl, heterocyclyl, substituted heterocyclyl, aryl, substituted aryl, heteroaryl, substituted heteroaryl, arylalkyl, substituted arylalkyl, substituted arylalkyl, cycloalkylalkyl and substituted cycloalkylalkyl;
- R4 is a radical, the radical being selected from the group comprising H, F, CH 3 , CF 3 , alkyl and substituted alkyl;
- R3 is a radical, the radical containing an aromatic group and being selected from the group consisting of aryl, substituted aryl, heteroaryl, substituted heteroaryl, arylalkyl, substituted arylalkyl, heteroarylalkyl, substituted heteroarylalkyl, alkyloxy -Alkyl, substituted alkyloxy-alkyl, alkyloxy-cycloalkyl, substituted alkyloxy-cycloalkyl, alkyloxy-heterocyclyl, substituted alkyloxy-heterocyclyl, alkyloxy-aryl, substituted alkyloxy-aryl, alkyloxy-heteroaryl, substituted alkyloxy-heteroaryl, alkylthio-alkyl, substituted alkylthio -Alkyl, alkylthio-cycloalkyl and substituted alkylthio-cycloalkyl; and
- R3 is a radical, the radical containing an aliphatic or aromatic group and preferably being selected from the group consisting of alkyl, substituted alkyl, cycloalkyl, substituted cycloalkyl, heterocyclyl, substituted heterocyclyl, aryl, substituted Aryl, heteroaryl, substituted heteroaryl, arylalkyl, substituted arylalkyl, heteroarylalkyl, substituted heteroarylalkyl, cycloalkylalkyl, substituted cycloalkylalkyl, heterocyclylalkyl, substituted heterocyclylalkyl, alkyloxyalkyl, substituted alkyloxyalkyl, alkyloxycycloalkyl, substituted alkyloxy heterocyclyl, substituted alkyloxy heterocyclyl, substituted alkyloxy heterocyclyl, alkyloxy aryl, substituted alkyloxy heterocyclyl, alkyloxy aryl, substituted alkyloxy
- a ring is formed with the participation of R3 and R4.
- R3 is selected individually and independently of one another from the group consisting of phenyl, substituted phenyl, benzyl, substituted benzyl, 1,1-diphenylmethyl, substituted 1,1-diphenylmethyl, naphthylmethyl, substituted Naphthylmethyl, thienymethyl, substituted thienylmethyl, benzothienylmethyl, substituted benzothienylmethyl, imidazolylmethyl, substituted imidazolylmethyl, idolylmethyl and substituted indolylmethyl.
- R3 is selected individually and independently of one another from the group consisting of C3-C5-alkyl, substituted C3-C5-alkyl, C5-C7-cycloalkyl, substituted C5-C7-cycloalkyl, C5-C7-cycloalkylmethyl, substituted C5-C7-cycloalkylmefhyl, cycloalkylethyl, substituted cycloalkylethyl, benzyl, substituted benzyl, phenylethyl, naphthylmefhyl, thienylmethyl, propenyl, propinyl, methylfhioethyl, imidazolylmethyl, substituted imidazololylmethylmethyl.
- XI is selected from the group consisting of H, acetyl, propanoyl, butanoyl, benzoyl, fluoromethylcarbonyl, difluoromethylcarbonyl, phenyl, oxycarbonyl, methyloxycarbonyl, phenylaminocarbonyl, methylaminocarbonyl, phenylsulfonyl, 2,6-dioxo - Includes hexahydro-pyrimidine-4-carbonyl and methyl-sulfonyl.
- X2 is an amino acid derivative of an amino acid selected from the group consisting of phenylalanine, 2-fluoro-phenylalanine, 3-fluorophenylalanine, 4-fluoro-phenylalanine, 2-chlorophenylalanine, 3-chlorophenylalanine, 4-chlorophenylalanine, 1-naphthylalanine , 2- Thienylalanine, 3-thienylalanine, 3,3-diphenylalanine, tyrosine, tryptophan, histidine and respective derivatives thereof;
- X2 and XI taken together are PhCH 2 CH 2 CO- or PhCH 2 -;
- X6 is an amino acid derivative of an amino acid selected from the group consisting of tryptophan, phenylalanine, tyrosine, histidine, 1-naphthylalanine, benzothienylalanine, 2-ammoindan-2-carboxylic acid, 2-thienylalanine, 3-thienylalanine, 2-fluoro-phenylalanine , 3-fluoro-phenylalanine, 4-fluoro-phenylalanine, 2-chlorophenylalanine, 3-chlorophenylalanine, 4-chlorophenylalanine and respective derivatives thereof;
- X5 is an amino acid derivative of an amino acid selected from the group consisting of D-cyclohexylalanine, D-cyclohexylglycine, D-homo-cyclohexylalanine, D-homoleucine, D-cysteine (fBu), D-cysteine (iPr), octahydroindole-2 -carboxylic acid, 2-methyl-D-phenylalanine and respective derivatives thereof; and
- X7 is an amino acid derivative of an amino acid that is selected from the group consisting of norvaline, norleucine, homo-leucine, leucine, isoleucine, valine, cysteine, cysteine (Me), cysteine (Et), cysteine (Pr), methionine, AUylglycine, Propargylglycine, cyclohexylglycine, cyclohexylalanine, phenylalanine, tyrosine, tryptophan, histidine, 1-naphthylalanine, 2-thienylalanine, 3-thienylalanine and respective derivatives thereof.
- XI and / or X4 has one or more water solubility-improving groups, the water solubility-improving group being selected from the group consisting of hydroxy, keto, carboxamido, ether, urea, carbamate, amino, substituted amino, guanidino, Includes pyridyl and carboxyl.
- the tasks are solved according to the invention by a compound, preferably a C5a receptor antagonist, with the structure X1-X2-X3-X4-X5-X6-X7-1 CD
- X4 is a cyclic or a non-cyclic amino acid, the cyclic amino acid being selected from the group consisting of proline, pipecolic acid, azetidine-2-carboxylic acid, tetrahydroisoquinoline-3-carboxylic acid, tefrahydroisoquinoline-1-carboxylic acid, octahydroindole-2-carboxylic acid, l- Aza-bicyclo- [3.3.0] octane-2-carboxylic acid, 4-phenyl-pynolidine-2-carboxylic acid, cis-Hyp and frans-Hyp, and the non-cyclic amino acid selected from the group consisting of Ser, Gin, Asn , Cys (O 2 CH 2 CH 2 CONH 2 ), Arg, Hyp (COCH 2 OCH 2 CH 2 OCH 2 CH 2 ⁇ CH 3 ), Hyp (CONH-CH 2 CH (OH) -CH 2
- the connecting lines - in formula (I) denote chemical bonds, the chemical bond being selected individually and independently, preferably from the group comprising covalent bonds, ionic bonds and coordinative bonds, the bond preferably being a chemical bond and more preferably the chemical bond Binding is such a bond which is selected from the group comprising amide bonds, disulfide bonds, ether bonds, thioether bonds, oxime bonds and aminotriazine bonds.
- the amino acid represented by X4 is preferably selected from the group consisting of proline, pipecolic acid, azetidine-2-carboxylic acid, tetrahydroisoquinoline-3-carboxylic acid, tefrahydroisoquinoline-1-carboxylic acid, octahydroindole-2-carboxylic acid, 1-azabicyclo- [3.3.0] octane-2-carboxylic acid, 4-phenyl-pynolidine-2-carboxylic acid, Hyp, Ser, Gin, Asn, Cys (O 2 CH 2 CH 2 CONH 2 ) and Arg.
- X2 is an amino acid derivative of an amino acid which is selected from the group consisting of phenylalanine, 2-fluoro-phenylalanine, 3-fluoro-phenylalanine, 4-fluoro-phenylalanine, 2-chlorophenylalanine, 3-chlohenylalanine, 4-chlorophenylalanine, 1- Naphthylalanine, 2-thienylalanine, 3-thienylalanine, 3,3-diphenylalanine, tyrosine, tryptophan, histidine and respective derivatives thereof;
- X2 and XI taken together are PhCH 2 CH 2 CO- or PhCH 2 -;
- X6 is an amino acid derivative of an amino acid selected from the group consisting of tryptophan, phenylalanine, tyrosine, histidine, 1-naphthylalanine, benzothienylalanine, 2-aminoindan-2-carboxylic acid, 2-thienylalanine, 3-thienylalanine, 2-fluoro-phenylalanine , 3-fluorophenylalanine, 4-fluorophenylalanine, 2-chlorophenylalanine, 3-chlorophenylalanine, 4-chlorophenylalanine and respective derivatives thereof;
- X5 is an amino acid derivative of an amino acid selected from the group consisting of D-cyclohexylalanine, D-cyclohexylglycine, D-homo-cyclohexylalanine, D-homoleucine, D-cysteine (tBu), D-cysteine (iPr), octahydroindole-2 -carboxylic acid, 2-methyl-D-phenylalanine and respective derivatives thereof; and
- X7 is an amino acid derivative of an amino acid that is selected from the group consisting of norvaline, norleucine, homo-leucine, leucine, isoleucine, valine, cysteine, cysteine (Me), cysteine (Et), cysteine (Pr), methioriine, AUylglycine, Propargylglycine, cyclohexylglycine, cyclohexylalanine, phenylalanine, tyrosine, tryptophan, histidine, 1-naphthylalanine, 2-thienylalanine, 3-thienylalanine and respective derivatives thereof.
- the object is achieved according to the invention by a compound, preferably a C5a receptor antagonist, with the structure
- X3 has the following structure
- X is C (R4) or N
- Rl is optionally present and if Rl is present, Rlein is a radical selected from the group comprising> N-R1B,> C (R1B) (R1D) and> O, where R1B and R1D are selected individually and independently of one another from the group comprising H, alkyl, substituted alkyl, cycloalkyl, substituted cycloalkyl, heterocyclyl, substituted heterocyclyl, aryl, substituted aryl, heteroaryl, substituted heteroaryl, arylakyl, substituted arylalkyl, cycloalkylalkyl and substituted cycloalkylalkyl;
- R4 is a radical, the radical being selected from the group comprising H, F, CF 3 , alkyl and substituted alkyl;
- the structure (IV) is preferably bound to the molecular constituents X2 and X4 via Rl and R2;
- R3 is a radical selected from the group consisting of H, alkyl, substituted alkyl, cycloalkyl, substituted cycloalkyl, heterocyclyl, substituted heterocyclyl, aryl, substituted aryl, heteroaryl, substituted heteroaryl, cycloalkylalkyl, substituted cycloalkylalkyl, heterocyclylalkyl, substituted heterocyclylalkyl, arylalkyl, substituted arylalkyl, heteroarylalkyl and substituted heteroarylalkyl.
- YB, YBl and YB2 are individually and independently selected from the group consisting of H, alkyl, substituted alkyl, cycloalkyl, substituted cycloalkyl, heterocyclyl, substituted heterocyclyl, aryl, substituted aryl, heteroaryl, substituted heteroaryl, arylakyl, substituted arylalkyl , Cycloalkylalkyl and substituted cycloalkylalkyl.
- R3 is a radical selected from the group consisting of methyl, ethyl, propyl, butyl, benzyl and
- Y is optionally present and if Y is present, Y is a radical selected from the group comprising -N (YB) ⁇ , -O-, -S- and -SS-, and YB preferably as in claim 14 is defined.
- X2 is an amino acid derivative of an amino acid that is selected from the group consisting of phenylalanine, 2-fluoro-phenylalanine, 3-fluoro-phenylalanine, 4-fluoro-phenylalanine, 2-chlorophenylalanine, 3-chlorophenylalanine, 4-chlorophenylalanine, 1-naphthylalanine , 2-thienylalanine, 3-thienylalanine, 3,3-diphenylalanine, tyrosine, tryptophan, histidine and respective derivatives thereof;
- X2 and XI taken together are PhCH CH 2 CO- or PhCH 2 -;
- X6 is an amino acid derivative of an amino acid selected from the group consisting of tryptophan, phenylalanine, tyrosine, histidine, 1-naphthylalanine, benzothienylalanine, 2-aminoindan-2-carboxylic acid, 2-thienylalanine, 3-thienylalanine, 2-fluoro-phenylalanine , 3-fluoro-phenylalanine, 4-fluoro-phenylalanine, 2-chloro-phenylalanine, 3-chlorophenylalanine, 4-chlorophenylalanine and respective derivatives thereof;
- X5 is an amino acid derivative of an amino acid selected from the group consisting of D-cyclohexylalanine, D-cyclohexylglycine, D-homo-cyclohexylalanine, D-homoleucine, D-cysteine (tBu), D-cysteine (iPr), octahydroindole-2 -carboxylic acid, 2-methyl-D-phenylalanine and respective derivatives thereof; and
- X7 is an amino acid derivative of an amino acid selected from the group consisting of norvaline, norleucine, homo-leucine, leucine, isoleucine, valine, cysteine, cysteine (Me), cysteine (Et), cysteine (Pr), methionine, allylglycine, Propargylglycine, cyclohexylglycine, cyclohexylalamine, phenylalanine, tyrosine, tryptophan, histidine, 1-naphthylalanine, 2-thienylalanine, 3-thienylalanine, and respective derivatives thereof.
- X3 is an amino acid derivative of an amino acid, the amino acid being selected from the group consisting of alpha-amino-glycine, alpha-beta-diaminopropionic acid (Dap), alpha-gamma - Diaminobutyric acid (Dab), ormthine, lysine, homolysin, Phe (4-NH2), 2-amino-3- (4- ⁇ iperidinyl) propionic acid and 2-amino-3- (3-piperidinyl) propionic acid, and the amino acid the side chain is derivatized.
- the object is achieved according to the invention by a compound, preferably a C5a receptor antagonist, preferably according to one of the aspects one to four of the present invention, with the following structure:
- A is selected from the group comprising H, NH2, NHAlkyl, NAlkyl2, NHAcyl and OH,
- B is selected from the group comprising CH2CAryl), CH (aryl) 2, CH2 (heteroaryl), substituted CH2 (aryl), aryl, substituted aryl and heteroaryl,
- Cl and C2 are selected individually and independently from the group comprising alkyl and substituted alkyl, it being possible optionally for a bond to be formed between Cl and C2,
- D is selected from the group comprising alkyl, cycloalkyl, CH2 (cycloalkyl), CH2CH2 (cycloalkyl), CH2Ph (2-Me) and CH2-S-alkyl,
- Z3 is optionally present and if Z3 is present it is selected from the group consisting of CO and CH2.
- A is selected from the group comprising H, NH2, NHEt, NHAc, OH,
- B is selected from the group comprising CH2Ph, CH2Ph (4-F), CH (Ph) 2, CH2Thienyl, CH2Naphtyl, phenyl, Ph (4-F) and thienyl,
- Cl is selected from the group comprising H and methyl
- C2 is selected from the group comprising methyl and CH2OH, or when Cl and C2 are connected by a bond, the resulting structure is selected from the group consisting of - (CH2) 2-, - (CH2) 3-, - (CH2) 4- and -CH2CH (OH) CH2-.
- D is selected from the group consisting of CH2CH2iPr, CH2iPr, Cyclohexyl, CH2Cyclohexyl, CH2CH2Cyclohexyl, CH2Ph (2-Me), CH2-S-tBu and CH2-S-iPr,
- E is selected from the group consisting of CH2Ph, CH2Ph (2-Cl), CH2Ph (3-Cl), CH2Ph (4-Cl), CH2Ph (2-F), CH2Ph (3-F), CH2Ph (4-F ), CH2Indolyl, CH2Thienyl, CH2Benzothienyl and CH2Naphtyl,
- the object is achieved according to the invention by a compound, preferably a C5a receptor antagonist, the compound having the following structure:
- dl, d2, d3 and d4 denote the distances from A, B, C and D in at least one energetically accessible conformer of the connection and have the following values:
- a and C are individually and independently of one another a hydrophobic radical, the hydrophobic radical being selected from the group comprising alkyl, cycloalkyl, heterocyclyl, aryl and heteroaryl;
- B and D are individually and independently of one another an aromatic or heteroaromatic radical, the aromatic radical preferably being aryl, and preferably the heteroaromatic radical being heteroaryl.
- a and C are selected individually and independently of one another from the group consisting of C3-C6-alkyl, C5-C7-cycloalkyl, methylthioethyl, methylthio-tert-butyl, idolyl, phenyl, naphthyl, thienyl, propenyl , Propynyl, hydroxyphenyl, idolyl and imidazolyl;
- B is selected from the group consisting of phenyl, substituted phenyl, naphthyl, thienyl, benzothienyl, hydroxyphenyl, indolyl, and imidazolyl;
- D is selected from the group consisting of phenyl, naphthyl, thienyl, thiazolyl, furanyl, hydroxyphenyl, indolyl and imidazolyl.
- the object is achieved according to the invention by a compound, preferably a C5a receptor antagonist,
- A, B, C and D denote the C-alpha atoms in amino acids, amino acid analogs or amino acid derivatives
- amino acids the alpha atoms of which are represented by B and D, individually and independently of one another have an aromatic or heteroaromatic amino acid side chain which comprises an aryl, arylalkyl, heteroaryl or heteroarylalkyl group.
- the amino acid whose alpha atom is represented by A is selected from the group comprising C3-C6-alkyl, methylthioethyl, propenyl, propynyl, R5, methyl-R5 and ethyl-R5, where R5 is a radical selected is from the group comprising C5-C7 cycloalkyl, phenyl, substituted phenyl, hydroxyphenyl, indolyl, imidazolyl, naphthyl and thienyl;
- the amino acid whose alpha atom is represented by B is selected from the group comprising R5, methyl-R5 and ethyl-R5, where R5 is a radical selected from the group consisting of phenyl, substituted phenyl, naphthyl , Thienyl, benzothienyl, hydroxyphenyl, indolyl and imidazolyl;
- the amino acid, the alpha atom of which is represented by C is selected from the group comprising C3-C6-alkyl, R5, methyl-R5 and ethyl-R5, where R5 is a radical derived from the Group is selected which includes C5-C7 cycloalkyl, phenyl, 1-methyl-phenyl, 2-methylphenyl, 3-methyl-phenyl and S-tBu; and the amino acid whose alpha atom is represented by D is selected from the group comprising R5, methyl-R5 and ethyl R5, where R5 is a radical selected from the group consisting of phenyl, naphthyl, thienyl , Thiazolyl, furanyl, hydroxyphenyl, indolyl and imidazolyl.
- the object is achieved according to the invention by a compound, preferably a C5a receptor antagonist, with the following structure:
- X2 is a radical that mimics the biological binding properties of a phenylalanine unit
- X3 and X4 are individually and independently of one another a spacer, the spacer preferably being selected from the group comprising amino acids, amino acid analogs and amino acid derivatives,
- X5 is a radical that mimics the biological binding properties of a cyclohexylalamn or homoleucine unit
- X6 is a radical that mimics the biological binding properties of a tryptophan unit
- X7 is a radical that mimics the biological binding properties of a norleucine or phenylalanine unit
- X8 is a radical, the radical optionally being contained in structure LT and, if present, is selected from the group which is substituted by H, NH 2 , OH, NH-OH, NH-Oalkyl, amino, substituted amino, alkoxy Alkoxy, hydrazino, substituted hydrazino, aminooxy, substituted aminooxy, alkyl, substituted alkyl, cycloalkyl, substituted cycloalkyl, heterocyclyl, substituted heterocyclyl, heteroaryl, substituted heteroaryl, arylalkyl, substituted arylalkyl, substituted aryl, amino acid, amino acid analog and amino acid derivative;
- the connecting lines - in formula (LT) denote chemical bonds, the chemical bond being selected individually and independently, preferably from the group comprising covalent bonds, ionic bonds and coordinative bonds, the bond preferably being a chemical bond, and more preferably the chemical bond Binding is such a bond which is selected from the group comprising amide bonds, disulfide bonds, ether bonds, thioether bonds, oxime bonds and aminotriazine bonds.
- XI is a radical with a mass of about 1-300, the radical preferably being selected from the group consisting of R5, R5-CO-, R5-N (R6) -CO-, R5-O-CO-, R5- SO 2 -, R5-N (R6) -C (NH) -, wherein R5 and R6 are preferably selected individually and independently of one another from the group which is substituted by H, alkyl, substituted alkyl, cycloalkyl, substituted cycloalkyl, heterocyclyl Contains heterocyclyl, aryl and substituted aryl;
- X2 and X6 are each independently an aromatic amino acid, derivative or analogue thereof; X5 and X7 are individually and independently of one another a hydrophobic amino acid, a derivative or an analogue thereof.
- X2, X5, X6 and X7 individually and independently of one another have the following structure:
- X is C (R4) or N
- Rl is optionally present and when Rl is present is a radical selected from the group consisting of> N-R1B,> C (R1B) (R1D) and> O, where R1B and R1D are individually and independently selected from the group comprising H, alkyl, substituted alkyl, cycloalkyl, substituted cycloalkyl, heterocyclyl, substituted heterocyclyl, aryl, substituted aryl, heteroaryl, substituted heteroaryl, arylalkyl, substituted arylalkyl, substituted arylalkyl, cycloalkylalkyl and substituted cycloalkylalkyl;
- R4 is a radical, the radical being selected from the group comprising H, F, CH 3 , CF 3 , alkyl and substituted alkyl;
- R3 is a radical, the radical containing an aromatic group and being selected from the group consisting of aryl, substituted aryl, heteroaryl, substituted heteroaryl, arylalkyl, substituted arylalkyl, heteroarylalkyl, substituted heteroarylalkyl, alkyloxy -Alkyl, substituted alkyloxy alkyl, alkyloxy cycloalkyl, substituted alkyloxy cycloalkyl, alkyloxy heterocyclyl, substituted alkyloxy heterocyclyl, alkyloxy aryl, substituted alkyloxy aryl, substituted alkyloxy heteroaryl, substituted alkyloxy heteroaryl, alkylthioalkyl
- R3 is a radical, the radical containing an aliphatic or aromatic group and is preferably selected from the group consisting of alkyl, substituted alkyl, cycloalkyl, substituted cycloalkyl, heterocyclyl, substituted heterocyclyl, aryl, substituted Aryl, heteroaryl, substituted heteroaryl, arylalkyl, substituted arylalkyl, heteroarylalkyl, substituted heteroarylalkyl, cycloalkylalkyl, substituted cycloalkylalkyl, heterocyclylalkyl, substituted heterocyclylalkyl, alkyloxyalkyl, substituted alkyloxyalkyl, alkyloxycycloalkyl, substituted alkyloxycyclo, substituted alkyloxycyclo, substituted alkyloxyalkyl, substituted alkyloxyalkyl Alkyloxy heterocyclyl, alkyloxy aryl, substituted alkyloxy aryl, substituted al
- a ring is formed with the participation of R3 and R4.
- R3 is selected individually and independently of one another from the group which substitutes phenyl, substituted phenyl, benzyl, substituted benzyl, 1,1-diphenylmethyl, substituted 1,1-diphenylmethyl, naphthylmethyl Naphthylmethyl, thienylmethyl, substituted thienylmethyl, benzothienylmethyl, substituted berizothienymethyl, imidazolylmethyl, substituted imidazolylmethyl, indolylmethyl and substituted indolylmethyl.
- R3 is selected from the group consisting of C3-C5-alkyl, substituted C3-C5-alkyl, C5-C7-cycloalkyl, substituted C5-C7-cycloalkyl, C5-C7-cycloalkylmethyl, substituted C5-C7-cycloalkylethyl, cycloalkylethyl, substituted cycloalkylethyl, benzyl, substituted benzyl, phenylethyl, naphthylmethyl, thienylmethyl, propenyl, imethyl, methylazolio substituted imidazolylmethyl, indolylmethyl and substituted indolylmethyl.
- X8 is selected from the group comprising H, ORl and NR1R2, where R1 and R2 are individually and independently selected from the group consisting of H, Alkyl, aryl, cycloalkyl and arylalkyl.
- XI is selected from the group consisting of H, acetyl, propanoyl, butanoyl, benzoyl, fluoromethylcarbonyl, difluoromethylcarbonyl, phenyl, oxycarbonyl, methyloxycarbonyl, phenylaminocarbonyl, methylaminocarbonyl, phenyl sulfonyl, 2,6-dioxo-hexahydro-pyrimidine-4-carbonyl and methyl-sulfonyl.
- XI and / or X4 have one or more groups which improve water solubility, the group which improves water solubility being selected from the group consisting of hydroxyl, keto, carboxamido, Efher, urea, carbamate and amino , substituted amino, guanidino, pyridyl and carboxyl.
- the object is achieved according to the invention by a compound, preferably a C5a receptor antagonist, with the structure
- X4 is a cyclic or a non-cyclic amino acid, the cyclic amino acid being selected from the group consisting of proline, pipecolic acid, azetidine-2-carboxylic acid, tefrahydroisoquinoline-3-carboxylic acid, tefrahydroisoquinoline-1 carboxylic acid, octahydroindole-2- carboxylic acid, l-aza-bicyclo- [3.3.0] -octane-2-carboxylic acid, 4-phenyl-pynolidin-2-carboxylic acid, cis-Hyp and trans-Hyp and the non-cyclic amino acid selected from the group consisting of Ser, Gin, Asn, Cys (O 2 CH 2 CH 2 CONH 2 ), Arg, Hyp (COCH 2 OCH 2 CH 2 OCH 2 CH 2 OCH 3 ), Hyp (CONH-CH 2 CH (OH) -CH 2 OH) and respective derivatives thereof
- the amino acid represented by X4 is preferably selected from the group consisting of proline, pipecolic acid, azetidine-2-carboxylic acid, tefrahycfroisochmolin-3-carboxylic acid,
- Tetrahydroisoquinoline-1-carboxylic acid Tetrahydroisoquinoline-1-carboxylic acid, octahydroindole-2-carboxylic acid, l-aza-bicyclo- [3.3.0] - octane-2-carboxylic acid, 4-phenyl-pynolidine-2-carboxylic acid, Hyp, Ser, Gin, Asn, Cys ( O 2 CH 2 CH 2 CONH 2 ) and Arg.
- the object is achieved according to the invention by a compound, preferably a C5a receptor antagonist, with the structure
- XI -X2 and X4-X8 are defined according to the seventh and eighth aspects of the present invention and wherein
- X3 has the following structure: R1 - X - R2 I R3 Y (IN), wonn
- X is C (R4) or ⁇
- Rl is optionally present and when Rl is present, Rl is a radical selected from the group consisting of> ⁇ -R1B,> C (R1B) (R1D) and> O, where R1B and R1D are independently selected from the group comprising H, alkyl, substituted alkyl, cycloalkyl, substituted cycloalkyl, heterocyclyl, substituted heterocyclyl, aryl, substituted aryl, heteroaryl, substituted heteroaryl, arylakyl, substituted arylalkyl, cycloalkylalkyl and substituted cycloalkylalkyl;
- R4 is a radical, the radical being selected from the group comprising H, F, CF 3 , alkyl and substituted alkyl;
- the structure (IV) is preferably bound to the molecular constituents X2 and X4 via Rl and R2;
- R3 is a radical selected from the group consisting of H, alkyl, substituted alkyl, cycloalkyl, substituted cycloalkyl, cycloalkylalkyl, substituted cycloalkylalkyl, heterocyclyl, substituted heterocyclyl, heterocyclylalkyl, substituted heterocyclylalkyl, aryl, substituted aryl, arylalkyl, substituted arylalkyl Heteroaryl, substituted heteroaryl, heteroarylalkyl, substituted heteroarylalkyl, acyl, substituted acyl, alkoxyalkyl, substituted alkoxyalkyl, aryloxyalkyl, substituted aryloxyalkyl, sulfhydryla ⁇ kyl, substituted sulfhydrylalkyl, hydroxyalkyl, substituted hydroxyalkyl, Carboxyalkyl, substituted carboxyalkyl, carboxamidoalkyl, substituted
- R3 is a radical with the structure
- n 1, 2, 3 or 4;
- a ring is formed between each two molecular parts of the compound, the molecular parts being selected individually and independently of one another from the group comprising YBl, YB2, YB3 and YB 4.
- the ring is formed with the participation of YB2 and YB3.
- X2 is an amino acid derivative of an amino acid selected from the group consisting of phenylalanine, 2-fluoro-phenylalanine, 3-fluoro-phenylalanine, 4-fluoro-phenylalanine, 2-chlorophenylalanine, 3-chlorophenylalanine, 4-chlorophenylalanine, 1-naphthylalanine , 2-thienylalanine, 3-thienylalanine, 3,3-diphenylalanine, tyrosine, tryptophan, histidine and respective derivatives thereof;
- X2 and XI taken together are PhCH 2 CH 2 CO- or PhCH 2 -;
- X6 is an amino acid derivative of an amino acid selected from the group consisting of tryptophan, phenylalanine, tyrosine, histidine, 1-naphthylalanine, benzothienylalanine, 2-aminoindan-2-carboxylic acid, 2-thienylalanine, 3-thienylalanine, 2-fluoro-phenylalanine , 3-fluoro-phenylalanine, 4-fluoro-phenylalanine, 2-chloro-phenylalanine, 3-chloro-phenylalanine, 4-chloro-phenylalanine and respective derivatives thereof;
- X5 is an amino acid derivative of an amino acid selected from the group consisting of D-cyclohexylalanine, D-cyclohexylglycine, D-homo-cyclohexylalanine, D-homoleucine, D-cysteine (tBu), D-cysteine (iPr), octahydroindole-2 -carboxylic acid, 2-methyl-D-phenylalanine and respective derivatives thereof; and X7 is an amino acid derivative of an amino acid selected from the group consisting of norvaline, norleucine, homo-leucine, leucine, isoleucine, valine, cysteine, cysteine (Me), cysteine (Et), cysteine (Pr), mefhionine, allylglycine, Propargylglycine, cyclohexylglycine, cyclohexylalamine, phenylalanine, tyrosine, tryptophan
- X3 is an amino acid derivative of an amino acid, the amino acid being selected from the group consisting of alpha-amino-glycine, alpha-beta-diaminopropionic acid (Dap), alpha- gamma-diaminobutyric acid (Dab), ornithine, lysine, homolysin, Phe (4-NH2), 2-amino-3- (4-piperidinyl) propionic acid and 2-amino-3- (3-piperidinyl) propionic acid, and the amino acid is derivatized on the side chain.
- the amino acid is derivatized on the side chain.
- the object is achieved according to the invention by a compound, preferably a C5a receptor antagonist, preferably according to the seventh to ninth aspect of the present invention, with the following structure:
- A is selected from the group comprising H, NH2, NHAlkyl, NAlkyl2, NHAcyl, substituted NHAcyl and OH,
- B is selected from the group comprising CH2CAryl), CH (aryl) 2, CH2 (heteroaryl) and substituted CH2 (aryl), Cl and C2 are selected individually and independently from the group comprising alkyl and substituted alkyl, it being possible optionally for a bond to be formed between Cl and C2,
- D is selected from the group consisting of alkyl, cycloalkyl, CH2 (cycloalkyl), CH2CH2 (cycloalkyl), CH2Ph (2-Me) and CH2-S-alkyl,
- E is selected from the group comprising CH2 (aryl), substituted CH2 (aryl) and CH2 (heteroaryl),
- Z2 is -R3-Y-, where R3 is selected from the group comprising H, alkyl, arylalkyl and Y is optionally present, and when Y is present, Y is selected from the group consisting of H, N (YB1) (YB2), N (YB1) C (N-YB2) -N (YB3) (YB4),
- YBl, YB2, YB3 and YB4 are individually and independently selected from the group comprising H, CN and alkyl and optionally a ring is formed with the participation of at least two of YBl, YB2, YB3 and YB4, and
- G is selected from the group comprising H, ORl and NR1R2, where R1 and R2 are individually and independently selected from the group comprising H, alkyl, aryl, cycloalkyl and arylalkyl.
- A is selected from the group comprising H, NH2, NHEt, NHAc, OH
- B is selected from the group comprising CH2Ph, CH2Ph (4-F), CH (Ph) 2, CH2Thienyl and CH2Naphtyl,
- Cl is selected from the group comprising H and methyl
- C2 is selected from the group comprising methyl and CH2OH, or when Cl and C2 are connected by a bond, the resulting structure is selected from the group consisting of - CCH2) 2-, - (CH2) 3-, - (CH2) 4- and -CH2CH (OH) CH2-.
- D is selected from the group consisting of CH2CH2iPr, CffiiPr, Cyclohexyl, CH2Cyclohexyl, CH2CH2Cyclohexyl, CH2Ph (2-Me), CH2-S-tBu and CH2-S-iPr,
- E is selected from the group consisting of CH2Ph, CH2Ph (2-Cl), CH2Ph (3-Cl), CH2Ph (4-Cl), CH2Ph (2-F), CH2Ph (3-F), CH2PhC4-F), CH2Indolyl, CH2Thienyl, CH2Benzothienyl and CH2NaphtyI,
- Z2 is -R3-Y-, wherein R3 is selected from the group comprising CH2, (CH2) 2, (CH2) 3, (CH2) 4 and CH2-C6H4, and Y is selected from the group consisting of NH2, NHEt, N (Et) 2,
- G is selected from the group consisting of NH2, NHMe, OH, and H.
- X7ist and X8 is G.
- the object is achieved according to the invention by a pharmaceutical formulation comprising at least one compound according to one of the preceding claims and additionally a pharmaceutically acceptable carrier.
- the object is achieved according to the invention by using at least one compound according to the first to tenth aspect of the present invention for producing a medicament.
- the medicament is used for the prevention and / or treatment of a disease in which the complement system is activated and / or in which the inhibition of the complement system causes a relief of the symptoms.
- the medicament is used for the prevention and / or treatment of a disease in which the inhibition of the activation of the C5a receptor alone and / or in combination with other therapeutic agents causes a relief of the symptoms.
- the disease and / or the symptoms to be treated are selected from the group of autoimmune diseases, acute inflammatory diseases, trauma, local inflammation, shock, burns.
- the diseases are selected from the group comprising rheumatoid arthritis, ankylose spondylitis, sarcoidosis, systemic lupus erythematosus, multiple sclerosis, psoriasis, septic shock, hemonhagic shock, SIRS (septic inflammatory response syndrome), MOF (multiple organ failure), asthma, vasculitis, myocarditis, dermatomyositis, inflammatory Darmerl ⁇ rankept (D3D: inflammatory bowel disease), pemphigus, Myasfhenia gravis, glomerulonephritis, acute respiratory failure, stroke, myocardial infarction, reperfusion injury, neurocognitive dysfunctions, antiphospholipid syndrome, burns, inflammatory diseases of the eye, local manifestations of systemic diseases, inflammatory vascular diseases and acute injuries to the central nervous system.
- SIRS septic inflammatory response syndrome
- MOF multiple organ failure
- asthma vasculitis
- myocarditis derm
- the inflammatory disease of the eye is selected from the group consisting of uveitis, age-related macular degeneration, diabetic retinopathy, diabetic macular edema, ocular Pemphigoid, Keratoconjunctivitis, Stevens-Johnson Syndrome and Graves Ophthalmophatie includes.
- the disease is a local manifestation of systemic diseases, the systemic disease being selected from the group comprising rheumatism, SLE and type I and type II diabetes.
- the manifestations are selected from the group consisting of the manifestations on the eye, on or in the brain, on the vessels, on the heart, on the lungs, on the kidneys, on the liver, of the gasttointestinal tract , the spleen, the skin, on the bone system, on the lyphatic system and in the blood.
- the inflammatory vascular disease is selected from the group comprising vasculitis, vascular leakage and atherosclerosis.
- the object is achieved according to the invention by using at least one compound according to the first to tenth aspect of the present invention for the prevention and / or support of surgical interventions, in particular for the manufacture of a medicament suitable therefor.
- the medicament is used for the prevention and / or support of surgical interventions.
- the medicament is used to support and / or to prevent and / or aftercare of a surgical intervention, the surgical intervention being selected from the group consisting of CABG, PACT, PTA , MidCAB, OPCAB, thrombolysis, organ transplantation and vascular occlusion (clamping).
- the medicament is used for thrombolytic treatment. In one embodiment of the twelfth and thirteenth aspect of the present invention, it is provided that the medicament is used in the context of a dialysis treatment, if appropriate before, during or after.
- the medicament is used to prevent damage to an organ that has been transplanted and or to be transplanted.
- the medicament is used for the prevention or treatment of organ rejection reactions.
- the present invention relates to a method for the treatment of patients, the method comprising the administration of one or more of the compounds according to the invention.
- the treatment can be a treatment in the narrower sense, but also includes preventive treatment and follow-up treatment.
- CPB Cardiopuhnunary Bypass
- the patient to be treated is preferably a mammal, more preferably farm animals, sports and pets, and most preferably humans.
- the patient is one who needs treatment.
- the patient suffers from one of the above diseases, for the treatment and / or prevention of which the compounds according to the invention can be used.
- the present invention thus provides for the first time those antagonists of the C5a receptor which overcome the inherent pharmacological disadvantages of the prior art antagonistically active peptides provided with a positive charge.
- the present invention is based on the surprising finding that, in contrast to the teaching of the prior art, antagonists of the C5a receptor can also be obtained which do not carry a positive net charge under physiological conditions, in particular at a pH of 7.4 and / or whose C-terminal amino acid has no positive charge under physiological conditions.
- the positive charge in peptides can be very disadvantageous from a pharmacological point of view. So positive charges e.g. lead to histamine release and cause less membrane passage (cf. Example 15). It is therefore particularly desirable to develop a peptide antagonist that does not have a net positive charge (hereinafter also referred to as a compound).
- Avoiding a C-terminal positive charge can also have other positive effects.
- Receptor specificity or parameters that are important in vivo such as pharmacokinetics, plasma protein binding or mutagenicity, can be positively influenced.
- the compounds mentioned in the present invention were tested in a primary test for their IC 50 values in a functional test system.
- all compounds, peptides and peptidomimetics are regarded as within the meaning of the present invention have noteworthy inhibitory activity, which has an IC 5 o value in a functional assay system as described in Example 1, point of less than 200 nM.
- the compounds according to the invention are antagonists of the C5a receptor. Even more preferably, these are designed as peptides or peptidomimetics. Furthermore, the present invention is based on the surprising finding that the compounds to be used according to the invention as antagonists of the C5a receptor carry an uncharged C-terminal amino acid, amino acid derivative or amino acid analogue.
- Particularly preferred compounds and antagonists according to the present invention are the following cyclic compounds.
- linear, that is to say structurally flexible, peptides can be potent inhibitors as well as structurally fixed cyclic peptides. This may be due to the substitution of the C-terminally charged arginine by hydrophobic amino acids, amino acid derivatives or amino acid analogs.
- linear peptide inhibitors according to the invention are in particular the compounds listed in the following table:
- linear peptides are generally significantly worse antagonists of C5a than cyclic peptides, such as those described in WO 99/00406 (for example Ac-Phe- [Lys-Pro-cha -T ⁇ -arg], Ac-Phe- [Orn-Pro-cha-T ⁇ -arg], Ac-Phe- [Orn-Pro-cha-T ⁇ -Arg], Ac-Phe- [Lys-Pro-cha-T ⁇ Arg]).
- the most active linear peptide described in WO 99/00406 has the sequence Me-Phe-Lys-Pro-cha-T ⁇ -arg and shows an IC 50 value of 0.085 ⁇ M (measured using the cellular myeloperoxidase release assay) human PMNs).
- the comparable cyclic peptide Ac-Phe- [Lys-Pro-cha-T ⁇ -arg] shows an IC 5 o-value of 0.012 uM. ha WO 99/00406 states that less structural flexibility of the cyclic peptide leads to a reduction, ie improvement, in the IC 50 value.
- the present invention thus describes for the first time peptidic or peptidomimetic C5aR antagonists with inhibitory activities with an IC 50 ⁇ 200 nM which do not carry a positive net charge under physiological pH values (pH 7.4) and / or whose C-terminal amino acid carries no positive charge ,
- the IC value is determined using a functional test (Kohl 1997 The Anaphylatoxins. In: Dodds, AW, Sim, RB (Eds.), Complement: A Practical Approach. Oxford, pp. 135-163).
- the compounds according to the invention can thus be used as C5aR antagonists, in particular also under physiological conditions.
- Another characteristic of the compounds according to the invention is the lack of agonistic activity in a cellular assay up to a concentration of at least 1430 nM.
- Example 12 shows an example of the results of measurements of a selection of the peptides according to the invention by means of a method with which the agonism with respect to the C5aR is determined. It is obvious that the compounds of the invention up to? show no agonistic activity at the maximum concentration used.
- HOCH 2 (CHOH) -C NO-CH 2 -CO-Phe- [Orn-Pro-cha- T ⁇ -Nle], Ph-CH 2 -CH 2 -CO- [Orn-Pro-cha-T ⁇ -Nle], Ac-Phe- [Orn-Hyp-cha-T ⁇ -Phe], H-Phe- [Orn-Pro-cha-T ⁇ -Phe], Ac-Phe- [Orn-Pro-cha-T ⁇ -Phe], Ac-Lys-Phe- [Orn-Pro-cha-T ⁇ -Nle], H-Phe- [Orn-Pro-cha-T ⁇ -Nle], H-Phe- [Orn-Ser-cha-T ⁇ -Nle], Ac-Phe- [Orn-Pro-cha-T ⁇ -Eafj, Ac-Phe-Orn-Pro-cha-T ⁇ -P
- the following peptides in particular are notably inhibitory: Ac-Phe- [Orn-Pro-cha-T ⁇ -Phe], Ac-Phe- [Orn-Hyp-cha-T ⁇ -Phe], Ac-Phe- [Orn-Pro-cha -T ⁇ -PafJ, Ac-Phe- [Orn-Pro-cha-T ⁇ -Ecr], Ac-Phe- [Orn-Pro-cha-T ⁇ -Ppa], Ac-Phe- [Orn-Pro-cha-T ⁇ - Nle], Ac- Phe- [Orn-Pro-cha-T ⁇ -Met], Ac-Phe- [Orn-Pro-cha-T ⁇ -Nva], Ac-Phe- [Orn-Pro-cha-T ⁇ -HIe] , Ac-Phe- [Orn-Pro-cha-T ⁇ -Eafj, Ac-Phe- [Orn-Pro-cha-T ⁇ -Ebd], Ac-Phe- [Orn-Pro-cha-T ⁇ -Eag],
- the oral intake of peptides is influenced by various factors such as size, charge and hydrophobicity.
- Peptides are generally considered to be poorly available orally (Burton et al. 1996 Journal of Pharmaceutical Sciences 85: 1337-1340).
- a model to estimate oral Abso ⁇ tion represents the measurement of the AB permeability through a monolayer layer of intestinal epithelial cells (eg CaCo2 or TC-7) (Example 15, Lennernäs 1997 Journal of Pharmacy and Pharmacology 49: 627-38).
- the compounds according to the invention which can be used as C5aR antagonists, have a significantly improved AB permeability due to the hydrophobic substitution of the C-terminal arginine.
- the antagonist Ac-Phe- [Orn-Hyp-cha-T ⁇ -Phe] shows a surprisingly good permeability of 14.3xl0 "6 cm / s compared to the poor permeability of 0.52xl0 " 6 cm / s of the charged antagonist Ac-Phe- [Orn-Pro-cha-T ⁇ -Arg].
- the high permeability is numerically in a range that is close to the substances that are readily available for oral use.
- An example of an orally very readily available compound is propanolol, which in this Lennernäs test shows an AB permeability of 31.1 10 "6 cm / s.
- the compounds according to the invention are those which are designed in such a way that groups which improve water solubility are additionally added to them at XI and / or X4.
- groups which have strong interactions with water and are strongly solvated is particularly favorable for improving water solubility. Commonly used examples are: hydroxy, keto, carboxamido, efher, urea, carbamate, amino, substituted amino, guanidino, pyridyl, carboxyl.
- the groups described can be expressly introduced at all positions of XI and / or X4 and both one and more of the groups which improve water solubility can be inserted. Examples of the introduction of several groups are the linking of carbohydrate tests or ethylene glycols.
- the present invention therefore also includes peptic or peptidomimetic C5aR antagonists, in particular according to the present invention, the solubility of which has been improved by additional modifications.
- modifications are known to the person skilled in the art and include, for example, the introduction of the abovementioned groups which improve water solubility. The following examples demonstrate that this is an effective measure or leads to highly effective antagonists.
- Compound 1 dissolves 8% in aqueous HEPES buffer (pH 7.4) according to Example 13.
- compound 40 is 94% soluble in HEPES buffer.
- Compound 2, which has an additional OH group compared to compound 1, is 13% soluble.
- the solubility becomes increased from 22% (compound 28) to 84% (compound 4). This is the case even though connection 4 is not loaded. This ensures that the peptides and peptidomimetics according to the invention can be converted into a readily water-soluble form despite their hydrophobic character.
- the term “contains” means that the respective structural element is contained, but the structure is not restricted to this.
- substituted in preferred embodiments means that one or more hydrogen atoms of a group or compound which is substituted is replaced by another atom, a group of atoms, a molecule or a group of molecules.
- substituents or substitutions Such an atom, group of atoms, molecules , as well as molecular groups, is / are referred to as substituents or substitutions.
- the substitution assumes that the normal valence of the respective atom is not exceeded and that the substitution results in a stable compound.
- Substituents or substitutions can preferably be selected individually or in any combination from the group consisting of hydroxyl, alkoxyl, mercapto, alkyl, alkenyl, alkynyl, alkoxy, alkylsulfinyl, cycloalkyl, heterocyclyl, aryl, arylalkyl, arylalkoxy, heteroaryl, aryloxy, halogen, trifluoromethyl, Difluoromethyl, cyano, nitto, azido, amino, aminoalkyl, carboxamido, -C (O) H, acyl, oxyacyl, carboxyl, carbamate, trialkylsilyl, sulfonyl, sulfonamide and sulfuryl.
- alkyl means a saturated aliphatic radical consisting of 1-10 carbon atoms or a mono- or polyunsaturated aliphatic hydrocarbon radical which contains between 2 and 12 carbon atoms and at least one double or triple bond.
- alkyl includes both straight chain and branched alkyl radicals. Straight-chain alkyl radicals having 1 to 8 carbon atoms are preferred.
- alkyl includes any analogs that can be composed of combination names of the prefixes “alk” or “alkyl”.
- alkoxy or "alkylthio” describes an alkyl group which is bonded via an oxygen or a sulfur.
- cycloalkyl means cyclic derivatives of an alkyl group as defined above, optionally unsaturated and / or substituted. Saturated cycloalkyl groups, in particular cycloalkyl groups having 3 to 8 carbon atoms, are preferred. Cycloalkyl groups which have 3 to Contain 6 carbon atoms.
- aryl denotes an aromatic radical having 6 to 14 carbon atoms, where “substituted aryl” stands for aryl radicals which carry one or more substituents.
- halogenated derivatives that can carry one or more halogen atoms.
- the halogenated derivatives include any halogen radical as defined below.
- halogen denotes a halogen radical from the group consisting of fluorine, chlorine, bromine and iodine. Fluorine, chlorine and bromine are preferred here.
- heteroaryl denotes a 5- to 8-membered, preferably 5- to 6-membered, monocyclic or 8 to 11-membered bicyclic, aromatic, heterocyclic radical, where each heterocycle can consist of carbon atoms as well as 1-4 heteroatoms from the series N, O or S.
- the heterocycle can be connected by any atom of the cycle, so that a stable structure results.
- Preferred heteroaryl radicals in the context of this invention are, for example, furyl, thienyl, pynolyl, oxazolyl, thiazolyl, imidazolyl, pyrazolyl, isoxazolyl, oxadiazolyl, triazolyl, tetrazolyl, thiadiazolyl, pyridinyl, pyridazinyl, pyrimidinyl, pyrazinylolylolylolylolylolylolylindolylylolylolylolylindolylolylindolylolylindolylolylindolylolylindolylolylindolylolylindolylolylindolylolylindolylolylindolylolylindolylolylindolylolylindolylolylindolylolylindolylolylindolylolylindolyl
- heterocyclyl means a 5- to 8-membered, preferably 5- to 6-membered, monocyclic or 8 to 11-membered bicyclic, heterocyclic radical, which is either saturated or unsaturated, however
- Each heterocycle consists of carbon atoms as well as 1-4 heteroatoms from the series N, O or S.
- the heterocycle can be connected by any atom of the cycle, so that a stable structure results.
- Preferred heteroaryl radicals are within the scope of this invention
- Each aryl or heteroaryl compound also includes the partially or fully hydrogenated derivatives.
- quinolyl and decahydroquinolinyl include tetrahydroquinolinyl.
- Naphthyl can also include the hydrogenated derivatives such as tetrahydronaphthyl.
- nitrogen or“ N ”and“ sulfur ”or“ S ” also include any oxidized derivatives of nitrogen such as nitrones, N-oxides or of sulfur such as sulfoxides, sulfones and quaternized forms of basic nitrogen such as HC1- or TFA salts included.
- Radicals can be mono- as well as di-, tri- or tetraradicals. This means that the meaning of different terms can also change slightly.
- a diradical that is described as “propyl” automatically means “propyplene” (eg - (CH 2 ) 3 -).
- the present invention includes all isotopes of atoms in the described compounds.
- Isotopes are atoms that have the same atomic number but different mass numbers.
- the tritium and deuterium are isotopes of hydrogen
- examples of carbon isotopes are n C, 13 C and 14 C.
- the term "energetically accessible conformer” means any conformer of a compound that falls within a 20 kcal / mol window above the conformation with the lowest energy.
- a Monte Carlo or systematic conformation search using MM2, MM3 or MMFF - Force fields implemented in molecular modeling programs such as MacroModel® v 7.0, Schrödinger Inc. Portland, Oregon, USA (http://www.schrodinger.com).
- Aino acids are known to the person skilled in the art and are defined in that both an amino and a carboxyl group are present in one molecule. This can mean both natural and unnatural amino acids. Examples are a-, ß-, and -amino acids, wherein preferably ⁇ -amino acids, particularly preferably ⁇ -L-amino acids can be used. If an amino acid is not specified in more detail (eg "tryptophan"), then both the L and the D form are meant.
- a natural amino acid is an L-amino acid selected from the group consisting of glycine, leucine, isoleucine, valine, alanine, phenylalanine, tyrosine, tryptophan, aspartic acid, asparagine, glutamic acid, glutamine, cysteine, methionine, arginine, lysine, proline, serine, threonine and histidine.
- An unnatural amino acid is a non-proteinogenic amino acid that includes, but is not limited to, D-amino acids, N-alkyl amino acids, homo-amino acids, ot, - disubstituted amino acids, dehydro-amino acids.
- Amino acid derivatives are compounds that arise from amino acids by modifying them at the N and / or C terminus.
- Non-limiting examples are reactions of the carboxyl group to salts, esters, acylhydrazides, hydroxamic acids or amides and the amino group to amides, ureas, thioureas, thioamides, sulfonamides, phosphoric acid amides, boric acid amides or alkylamines.
- Parts of compounds that result from modifications of amino acids at the C and or N terminus can also be referred to as amino acid units.
- the amino acids can also be derivatized on their side chains.
- a derivatized amino acid is one in which the side chain is derivatized once or several times, this type of derivatization is usually specifically mentioned here.
- a preferred derivatization of the side chain can take place in particular where the side chain has a functional group.
- a preferred functional group is, for example, an amino group, carboxyl group, thiol group or alcohol group.
- Amino acid analogs are compounds that arise from amino acids by replacing the amino and / or carboxyl group with other groups that can mimic these.
- Non-limiting examples are the incorporation of thioamides, ureas, thioureas, acylhydrazides, esters, alkylamines, sulfonamides, phosphoric acid amides, ketones, alcohols, boronic acid amides, benzodiazepines and other aromatic or non-aromatic heterocycles (for an overview see MA Estiarte, DH Ristry in Burgers Medicinal , 6th Edition, Volume 1, Part 4, John Wiley & Sons, New York, 2002).
- Aromatic amino acids are amino acids that contain aryl or heteroaryl groups.
- Non-limiting examples are phenylalanine, 2-fluorophenylalanine, 3-fluorophenylalanine, 4-fluoro-phenylalanine, 2-chlorophenylalanine, 3-chlorophenylalanine, 4-chlorophenylalanine, tyrosine, histidine, tryptophan, homo- Phenylalanine, homo-tyrosine, homo-histidine, homo-tryptophan, 1-naphthylalanine, 2-naphthylalanine, 2-thienylalanine, 3-thienylalanine, benzothienylalanine, furylalanine, thiazolylalanine, pyridylalanine, 2-aminohydroanoic acid carboxylic acid, biphenylalanine, 3,3-diphenylalanine and corresponding D- and ß-amino acids.
- Hydrophobic amino acids are amino acids that contain hydrophobic alkyl, cycloalkyl, heterocyclyl, aryl or heteroaryl groups.
- Non-limiting examples are leucine, isoleucine, valine, phenylalanine, tyrosine, histidine, cysteine, cysteine (iPr), cysteine (tBu), methionine, Prohn, tryptophan, norleucine, norvaline, homoleucine, cyclohexylalanine, cyclopentylalanine, 1-, naphthylanine Naphthylalanine, 2-thienylalanine, 3-thienylalanine, benzothienylalanine, allylglycine, propargylglycine, 2-methylphenylalanine, 3-methylphenylalanine, 4-methylphenylalanine, homocyclohexylalanine, cyclohexylhexylglycine, N-cyclohexy
- the biological binding properties of an amino acid unit are those binding properties that the respective amino acid exhibits when interacting with a biological molecule.
- Biological molecules are in particular those that have a biological function. Examples of such biological molecules, but not limited to them, are, for example, protein or peptide-based receptors.
- Groups or units that mimic or mimic the biological binding properties of an amino acid are defined as groups that can enter into at least one or the same or similar interaction with a receptor or interaction partner, preferably a biological receptor or biological interaction partner as the amino acid itself.
- groups to consider the most widespread in terms of the most preferred interactions of the amino acid in question with biological receptors.
- the oxygen atom of the carbonyl group of an amino acid is able to act as a hydrogen bond acceptor, whereas the NH proton can interact as a hydrogen bond donor.
- amino acids can interact with receptors via their side chains. phenylalanine and tryptophan can have both hydrophobic interactions via the side chain methylene group or the aromatic groups and ⁇ -7T interactions via the aromatic groups.
- tryptophan indole group can act as a hydrogen bond donor via the NH group.
- cyclohexylalanine and norleucine can enter into hydrophobic interactions with biological receptors via their alkyl or cycloalkyl side chains. For all amino acids, not only the entire side chain, but also parts of it, can interact fundamentally.
- a group or a unit which is to mimic or mimic the biological binding property of an amino acid or is to have this property can enter into at least one of the above interactions of the respective amino acid, then it can mimic its biological binding properties.
- Spacers as used herein, in preferred embodiments, unless otherwise stated in individual cases, are organic radicals with a mass of about 1-300, which enable a covalent linkage of different chemical groups. Examples are simple groups like
- R is a residue with a mass of about 1-300.
- R is a radical selected from the group consisting of H, alkyl, substituted alkyl, cycloalkyl, substituted cycloalkyl, cycloalkylalkyl, substituted cycloalkylalkyl, heterocyclyl, substituted heterocyclyl, heterocyclylalkyl, substituted heterocyclylalkyl, aryl, substituted aryl, arylalkyl, substituted aryl , Heteroaryl, Substituted Heteroaryl, Heteroarylalkyl, Substituted Heteroarylalkyl, Acyl, Substituted Acyl, Alkoxyalkyl, Substituted Alkoxyalkyl, Aryloxyalkyl, Substituted Aryloxyalkyl, Sulfhydrylalkyl, Substituted Sulfhydrylalkylalkyl, Sulf
- Spacers are preferably selected from the following group:
- R is preferably a radical selected from the group containing H, alkyl, substituted alkyl, arylalkyl, substituted arylalkyl, heteroarylalkyl, substituted heteroarylalkyl.
- Peptides that carry a net positive charge can cause histamine release (Jasani et al. 1979 Biochemical Journal 181: 623-632).
- subcutaneous application or the setting of subcutaneous depots is not possible with such connections.
- the resorption of the pharmaceuticals is particularly important. If the boundary conditions are otherwise the same, charged molecules are generally less well absorbed than uncharged ones (Veber et al. 2002 Journal of Medicinal Chemistry 45: 2615-2623). Due to the lack of net charge of the compounds according to the invention, they are also suitable for use as an orally administrable medicament.
- the compounds according to the invention can be used for the manufacture of medicaments, in particular for the manufacture of medicaments for the prevention and / or treatment of inimuno-inflammatory diseases.
- diseases include in particular the following diseases: autoimmune diseases, acute inflammatory diseases, trauma, local inflammation, septic and hemonhagic shock.
- these diseases are selected from the group consisting of rheumatoid arthritis, systemic lupus eryfhematodes, multiple sclerosis, psoriasis, septic shock, asthma, vasculitis, dermatomyositis, inflammatory bowel disease (IBD: inflammatory bowel disease), pemphigus, myomeric genesis, myasthenia gravis acute respiratory insufficiency, stroke, heart attack, reperfusion damage, neurocognitive dysfunction, antiphospholipid syndrome, burns, inflammatory diseases of the eye such as uveitis, age-related macular degeneration (English: age-related macular degeneration), diabetic retinopathy, diseases like local rheumatism, local manifestations system SLE, diabetes in the eye, brain, vessels, heart, luge, kidney, liver, gastrointestinal tract, spleen, skin or other organ systems, inflammatory vascular diseases such as vasculitis, atherosclerosis, and acute injuries to the center ral nervous system
- the present invention also relates to formulations, in particular pharmaceutical formulations, which contain at least one of the compounds according to the invention.
- Pharmaceutical active ingredients are often mixed with other pharmaceutically acceptable ingredients in order to ensure an improved effect, such as, for example, improved transport, shelf life, temporal behavior when released and the like.
- a large number of corresponding formulations are known to the person skilled in the art.
- Inert diluents, calcium carbonate, sodium carbonate, lactose, calcium phosphate, sodium phosphate, starch, alginates, gelatin, magnesium stearate and talc are known as constituents of such formulations.
- Certain ingredients can be added to allow a time-delayed release of the active pharmaceutical ingredients.
- examples include glycerol monostearate and glycerol distearate.
- the pharmaceutically active ingredient being mixed with calcium carbonate, calcium phosphate or kaolin.
- the pharmaceutically active ingredients are mixed, for example, with oils (peanut oil, liquid paraffin, olive oil).
- the pharmaceutically active constituents can in particular be mixed with the following constituents: carboxymethyl cellulose, methyl cellulose, hydropropyl methyl cellulose, sodium algenate, polyvinylpynolidone, lecithin, polymerization products of alkylene oxides and fatty acids such as polyoxyethylene stearate, heptadecaethyleneoxycetanol,
- Polyoxyethylene sorbitol monooleate and polyoxyethylene sorbitan monooleate are referred to as polyoxyethylene sorbitol monooleate.
- Various additives can be used for preservation. Examples include ethyl or n-propyl p-hydroxybenzonate.
- Certain formulations are used to enable special forms of application.
- Examples of application forms of compounds according to the invention are oral, subcutaneous, intravenous, topical, intramuscular, rectal and inhalation applications.
- the compounds according to the invention can be present as pharmaceutically acceptable salts.
- Fig.l is a bar graph showing the influx of new trophies in the immune complex mediated peritonitis, expressed as the average number of polymo ⁇ hkernigen Cells / field when Compound 149 is administered compared to vehicle alone;
- Fig. 2 is a graph showing the C5a-induced neutropenia in rats expressed as% monrophile over time when compound 149 or vehicle was administered.
- Acetonitrile grade, J.T. Baker
- Dichloromethane for synthesis, Merck Eurolab
- Diethylefher for synthesis, Merck Eurolab
- N, N-dimethylformamide LAB, Merck Eurolab
- Dioxane for synthesis, Aldrich
- Methanol for synthesis, Merck Eurolab
- Water was demineralized using a demineralizer (Milli-Q Plus, Millipore).
- the reagents used were from Advanced ChemTech (Bamberg, Germany), Sigma-Aldrich-Fluka (Deisenhofen, Germany), Bachern (Heidelberg, Germany), JT Baker (Phillipsburg, USA), Lancaster (Mühmeim / Main, Germany), Merck Eurolab (Darmstadt, Germany), ⁇ eosystem (Strasbourg, France), ⁇ ovabiochem (Bad Soden, Germany, from 2003 Merck Biosciences, Darmstadt, Germany) and Acros (Geel, Belgium, sales company Fisher Scientific GmbH, viewing distance, Germany), Peptech (Cambridge, MA, USA), Synthetech (Albany, OR, USA), Pharmacore (High Point, NC, USA), Anaspec (San Jose, CA, USA) and used without further purification.
- Non-commercially available unnatural amino acids or carboxylic acids for N-terminal modification were produced according to standard instructions.
- Fmoc-cis-Hyp-OH was obtained by reacting H-cis-Hyp-OH with Fmoc-OSu [Paquet et al. 1982 Canadian Journal of Chemistry 60: 976-980A].
- Fmoc-Phe (4-STrt-Amidino) -OH was synthesized by a known protocol [Pearson et al. 1996 Journal of Medicinal Chemistry 39: 1372-1382].
- Side chain-modified cysteine derivatives have been prepared by alkylation of Fmoc-cysteine-OH with alkyl halides.
- concentrations of the reagents in percent are volume percent (v / v).
- Analytical chromatography was carried out using a Hewlett Packard Series 1100 system (degasser G1322A, quaternary pump G1311A, automatic sampler G1313A, thermostatted column compartment G 1316A, variable UV detector G1314A) and coupled ESI-MS (Finnigan LCQ ion trap mass spectrometer). Control software from Firnigan was used for this (Navigator Ver 1.1 spl). Helium served as the collision gas in the ion trap.
- the separation was carried out on RP-18 column material (Vydac 218 TP5215, 2.1 x 150 mm, 5 ⁇ m, C18, 300 A with guard column (Merk)) at 30 ° C and a flow of 0.3 ml / min using a linear gradient for all chromatograms (5-95% B within 25 min, where A: 0.05% TFA in water and B: 0.05% TFA in CH 3 CN).
- the dead time between injection and UV detection (HPLC) was 1.65 min, between UV detection and mass detection 0.21 min.
- the accuracy of the mass spectrometer is approximately ⁇ 0.2 amu.
- Linear peptides were synthesized according to the Fmoc-'Bu strategy in a batch process. Either manual synthesis in polypropylene syringes with frits was carried out or automatic synthesizers (Syro from Multisyntech, Witten or Sophas from Zinsser, Frankfurt) were used.
- the C-terminal amino acid was either attached to trityl chloride resin (approx. 200 mg resin; loading of reactive groups approx. 1.5 mmol / g; attachment by reaction with 0.8 eq. Fmoc -Amino acid and 3.0 eq.DIPEA in CH 2 C1 2 for 2 hours; loading of the amino acid obtained approx. 0.2-0.4 mmol / g) or there was a link to Wang resin (200-500 mg resin ; Loading of reactive groups approx. 0.6 mmol / g; connection by reaction with 4 eq. Fmoc-amino acid and 4 eq. DIC and 3 eq. NMI in DMF for 3 hours; loading of the amino acid obtained approx. 0.2-0, 6 mmol / g).
- the first amino acid was obtained by Fmoc elimination from Fmoc rinkamide resin (approx. 200 mg resin; Fmoc elimination with 20% piperidine in DMF for 20 min) and subsequent coupling of the Fmoc Amino acid (single or multiple reaction with 5 eq. Fmoc-amino acid; 5 eq. HBTU and 15 eq. DIPEA in DMF for 30-60 min) attached.
- the desired peptide was produced by a sequence of Fmoc cleavage and attachment of the required Fmoc amino acid or carboxylic acid, as required.
- the resin was reacted with 20% piperidine in DMF for 20 min. Couplings were made by single or multiple implementation with 5 eq. the amino acid, 5 eq. HBTU and 15 eq. DIPEA in DMF for 30-60 min.
- N-terminal acetyl groups the N-terminal free resin-bound peptide was reacted with 10% acetic anhydride and 20% DIPEA in DMF for 20 min.
- TFA 95% TFA, 2.5% water, 2.5% TLPS or a similar acidic solution was used to split off the completely synthesized peptide from the resin and to remove side protective groups.
- the TFA was then removed on a rotary evaporator or the peptide obtained is precipitated with methyl T-sutyl ether at 0 ° C. and isolated after centrifugation and decanting of the supernatant solution.
- the peptide was dissolved with a mixture of 2N HCl and MeCN and lyophilized.
- the linear peptide synthesized according to AAV1 with a free N-terminus was pre-cleaved from the resin with 10 eq. of the corresponding aldehyde in 5% acetic acid and 5% trimethyl orthoformate in THF. After about 4 hours, the imine obtained was 5 eq. Sodium cyanoborohydride reduced overnight.
- the linear peptide Ac-Phe-Orn-Hyp-cha-T ⁇ -Phe-OH was prepared after linear peptide synthesis according to AAV 1 and cyclized according to AAV 2. Due to the higher nucleophilicity of amines compared to alcohols, there was no product besides the desired cyclized product due to undesired esterification of the Hyp-OH group with the C-terminal carboxyl group. Purification of the crude product obtained by HPLC led to 26.9 mg of the desired white solid Ac-Phe- [Orn-Hyp-cha-T ⁇ -Phe] (2).
- the resin-bound peptide H-Orn-Pro-cha-T ⁇ -Nle-Trityl resin was prepared after linear peptide synthesis according to AAV 1 and subjected to reductive alkylation with benzaldehyde according to AAV 3. Cyclization according to AAV 2 and subsequent purification by HPLC led to 0.9 mg of the desired product 56 as a white solid.
- the linear peptide Ac-Phe-Orn-Hyp-cha-T ⁇ -Nle-OH was prepared according to AAV 1, cyclized according to AAV 2 and the cyclized peptide Ac-Phe- [Orn-Hyp-cha-T ⁇ -Nle] per HPLC cleaned. 35.4 ⁇ l (40 eq.) 2- (2- (2-methoxyethoxy) ethoxy) acetic acid were reacted for 15 min at 40 ° C. with 50.3 ⁇ l (120 eq.) Thionyl chloride.
- the linear peptide Ac-Phe-Orn-Hyp-cha-T ⁇ -Nle-OH was prepared according to AAV 1, cyclized according to AAV 2 and the cyclized peptide Ac-Phe- [Orn-Hyp-cha-T ⁇ -Nle] produced by HPLC cleaned. Then 5.0 mg of the peptide was reacted with 26.1 mg of 4-isocyanatomethyl-2,2-dimethyl- [1,3] dioxolane and 1.88 ⁇ l (2.0 eq.) DIPEA in 0.3 ml of MeCN , After 3 days of stirring at 40 ° C., the solvent was removed on a rotary evaporator and the crude product obtained was purified by HPLC. 0.22 mg of the desired white solid 6 was obtained.
- the linear peptide Ac-Phe-Orn-Pro-cha-T ⁇ -Orn-OH was prepared according to AAV 1, cyclized according to AAV 2 and the resulting cyclized peptide Ac-Phe- [Om-Pro-cha-T ⁇ -Orn] via HPLC. Then 2.6 mg of the peptide was reacted with 22.6 mg (30 eq.) 2- (methylmercapto) -2-imidazoline hydroiodide and 29.7 ⁇ l (60 eq.) DIPEA in 260 ⁇ l MeOH. After stirring for 2 days at 50 ° C., the solvent was removed on a rotary evaporator and the crude product obtained was purified by HPLC. 0.86 mg of the desired white solid 7 was obtained.
- the peptide Ph-CH 2 -CH 2 -CO-Orn-Pro-cha-T ⁇ -Nle-OH was produced after linear peptide synthesis according to AAV 1, 3-phenylproionic acid being used as the N-terminal carboxylic acid. It was cyclized according to AAV 2 and the crude product obtained was purified by HPLC. 3.13 mg of the desired white solid 41 were obtained.
- Example 11 Determination of the IC 50 value in an enzyme release assay
- Basophilic leukemia cells from rats which express the human C5aR (CD88) are in DMEM with 10% fetal calf serum, 100 U / ml penicillin, 100 ⁇ g / ml streptomycin and 2 mM glutamine (all media components Biochrome, Berlin) to grown to confluence at 37 ° C and 10% CO 2 . All of the following information relates to a culture bottle with an area of 75 cm 2 . Used medium is poured off.
- Cells are washed with 10 ml PBS (Dulbecco's PBS, Biochrome) and then overlaid with 3 ml Cell Dissociation Solution (CDS, Sigma). Cells are incubated at RT for 1 min. The CDS is then removed and the cells are incubated for a further 10-15 min at 37 ° C. for detachment. 20 ⁇ l solution of the compound to be tested is used in the assay. The assay solution must not contain more than 2.8% DMSO. Dilution series are diluted in 1/3 or 1/2 steps.
- 75 ⁇ l of RBL cells treated as follows are added to the 20 ⁇ l solutions of the compounds: after the detachment phase, the cells are tapped violently and taken up in 10 ml of HAG-CM heated to 37 ° C. (20 mM HEPES; 125 mM NaCl, 5 mM KC1, 1mM CaCl 2 , 1mM MgCl 2 , 0.5mM glucose, 0.25% BSA.HEPES preparation :: 2.3 g / 1 HEPES salt + 2.66 g / 1 HEPES acid). Cells are counted and centrifuged (200g, 10 min).
- the cell pellet is taken up with preheated HAG-CM (ie Hepes-buffered NaCl-glucose solution with calcium and magnesium), and the cell density is adjusted to 2 ⁇ 10 6 cells / ml.
- HAG-CM Hepes-buffered NaCl-glucose solution with calcium and magnesium
- the cells are incubated at 37 ° C for 5 min.
- 27 ml of a cytochalasin B solution 100 ⁇ g / ml in DMSO, Sigma
- 75 ⁇ l of cell suspension become the 20 ⁇ l solution with the one to be tested Connection given. This results in a volume of 95 ⁇ l per well.
- the cells are incubated at 37 ° C for 10 min.
- Example 12 Determination of EC 5 0- value in an enzyme release assay
- the solubility of compounds was determined as follows: 20 ⁇ l of a 10 mM stock solution (in DMSO) of the compound are diluted in 980 ⁇ l of the solvent to be examined. After 24 h incubation at RT with shaking, the samples are centrifuged at 11,000 rpr ⁇ in an Eppendorf centrifuge. The supernatant is quantified photometrically. The optical density of the sample and a control value in 60% MeOH serves as a measure of the solubility. Substances that dissolve as well in the solvent to be examined as in the control are tested for their maximum solubility as follows. For this, 10 mg / ml of compound are dissolved in the solvents of choice. The undissolved portion is removed after 24 hours by centrifugation (see above). The supernatant is measured photometrically and related to a corresponding control target value (60% MeOH). The solubility for some of the compounds of the invention are given in Table 6.
- Example 14 Development of the pharmacophore model on which the antagonists are based
- a pharmacophore model was developed based on the structure-activity relationships of these and other peptides.
- the distance between the groups critical for the activity (2 hydrophobic and two aromatic groups) was predicted using the following method:
- the pharmacophore model was created on the basis of a 2 ns long molecular dynamics simulation (step size 2 fs) of the compound 28.
- the simulation was carried out using the AMBER94 force field as well as an explicit water model (TTP3) and periodic boundary conditions.
- TTP3 explicit water model
- the statistical analysis of the snapshots from the last nanosecond of the trajectory (1000 structures) led to the distances between the mass centers of the pharmacophoric groups given below.
- the Startstr ⁇ ktur for the molecular dynamic simulation was based on ensemble-dynamic calculations with seven to limiting each other structurally, in the lower nanomolar range (IC 5 0) active cyclic peptides.
- Example 15 Determination of AB permeability in a TC-7 based assay system
- HBSS-MES HBSS-MES
- 14 C-mannitol (approx. 4 ⁇ M) is mixed into the sample. This solution is then centrifuged and the supernatant is added to the apical side of a TC-7 cell culture (passage 15, in a 24-well transwell plate) so that a DMSO concentration of 1% is established.
- HBSS-HEPES (5mM, pH 7.4) is on the basolatheral side. The cells are then incubated at 37 ° C. for 120 min.
- the integrity of the TC-7 cell layer is checked via the added mannitol (P app ⁇ 2.5 10 "6 cm / s).
- the permeability P app [cm / s] is equal (VRxCR 120 ) / ( ⁇ txAx (C D> ⁇ md- CR, ⁇ id)) where V R is the volume of the receiver chamber, C R120 is the concentration of the test compound in the receiving chamber after 120 min, ⁇ t is the incubation time, A is the area of the TC-7 cell layer, CD, m i d is the midpoint concentration of the test substance in the donor chamber and C R , m i d is the concentration of the test compound in the receiver chamber.
- the linear peptide 52 was prepared according to AAV 1 after linear peptide synthesis and purified by HPLC. 10.5 mg of compound 52 were obtained as a white solid.
- Example 18 Synthesis of Ac-Phe-Orn-Pro-cha-Trp-NH-CH 2 -CH 2 -Ph (72) 200 mg of bromo (4-methoxyphenyl) methylpolystyrene resin is mixed with 5 ml of a 50% solution of phenylethylamine incubated in THF (v / v) at room temperature for 18 h. The resin is then washed (DMF; 3 x 5.0 ml, MeOH; 3 x 5.0 ml, DCM; 3 x 5.0 ml) and the peptide is synthesized according to AAV 1. After HPLC purification, 4.1 of compound 72 is obtained as a white solid.
- the resin-bound peptide H-Phe-Om-Pro-cha-Bta-Phe-Rink amide resin was produced according to AAV 1.
- Diphenylmethyl isocyanate (5 eq.) And DFPEA (10 eq.) In DMF were then added and agitation was carried out for 2 hours. After cleavage from the resin with a mixture of 95% TFA, 2.5% water and 2.5% TIPS was purified by HPLC. 0.92 mg of the compound was obtained as a white solid.
- the resin-bound peptide H-Gly-Phe-Orn-Pro-cha-Bta-Phe-Rink amide resin was produced according to AAV 1. Disuccinimidyl carbonate (3 eq.) And DIPEA (3 eq.) In DMF were then added and agitation was carried out for 3 hours. Then another 3 eq. DIPEA was added and agitation was carried out for an additional 5 hours at room temperature. After cleavage from the resin with a mixture of 95% TFA, 2.5% water and 2.5% TIPS was purified by HPLC. 3.8 mg of the compound were obtained as a white solid.
- the resin-bound peptide H-Phe-Orn-Pro-cha-Bta-Phe-Rink amide resin was produced according to AAV 1. Then succinic anhydride (5 eq.) And DLPEA (10 eq.) In DMF were added and agitation was carried out for 2 hours. The mixture was filtered and washed with DMF (5x), MeOH (5x) and CH 2 C1 2 (5x). The resin was then reacted with HBTU (5 eq.) And DLPEA (10 eq.) In DMF for 1 day. The peptide was cleaved from the resin with a mixture of 95% TFA, 2.5% water and 2.5% TfPS and purified by HPLC to give 0.47 mg of the compound as a white solid.
- the resin was washed (DMF; 3 x 5.0 ml, MeOH; 3 x 5.0 ml, DCM; 3 x 5.0 ml) and 4 ml of a 5 M solution of "butylamine were added for 2 ⁇ 30 min Washing the resin (DMF; 3 x 5.0 ml, MeOH; 3 x 5.0 ml, DCM; 3 x 5.0 ml) the rest of the synthesis of the peptomer was carried out according to AAV1.
- Example 28 Effectiveness of compound 149 in a model of immune complex mediated peritonitis
- the peritonitis mediated by the immune complex is part of the pathological occurrence in connection with diseases mediated by the immune complex, e.g. Vasculitis, nephritis, arthritis or farmer's lung.
- diseases mediated by the immune complex e.g. Vasculitis, nephritis, arthritis or farmer's lung.
- the corresponding model was developed by Heller et al. (1999 Journal of Immunology 163: 985-994) and is based on the proinflammatory effect of brain complex formation from i.v. given antigen and i.p. given Antikö ⁇ er.
- BALB / c mice (6-8 weeks old) were given iv with compound 149 according to the invention (1 mg / kg body weight in 200 ⁇ l vehicle) 15 min before the initiation of the reverse passive arthus Response treated.
- the Arthus reaction was triggered by the application of OSA (20 mg / kg iv in 200 ⁇ l PBS) and polyclonal anti-OVA Ab (rabbit; 800 ⁇ g / mouse ip). After 6 hours a peritoeneal lavage was carried out with 2 ml PBS 0.1% BSA.
- the PE cells obtained were stained using DIFF-Quick. At least 20 visual fields (100x magnification) were microscopically analyzed for the presence of granulocytes.
- C5a-induced neutropenia is a model for shock-like diseases (septic shock) in which, among other things, the systemic effects of C5a such as A drop in blood pressure and neutropenia plays an important role.
- the attachment of the neuttophils to the vessel wall, triggered by C5a, is the reason for the decrease in the number of neutrophils in the circulating blood (neutropenia). This process of recruiting neuttophils also plays a crucial role in many other diseases such as reperfusion damage.
- the model has been also by Short et al. (1999 British Journal of Pharmacology 125: 551-554).
- mice Female Wistar rats are i.p. anesthetized with ketamine (80 mg / kg) and xylazine (12 mg / kg). The rats are inhibited and a catheter is inserted into the jugular vein and the following treatment follows:
- the rats are pretreated with vehicle or compound 149 according to the invention by means of i.v. Infusion. A blood sample is taken one minute beforehand.
- the rats are treated with 2 ⁇ g kg hrC5a iv (2 ⁇ g / kg, 1 min). Blood samples are taken shortly before and at various times after HRC5a administration.
- Blood samples (approx. 0.2 ml) are taken from the jugular vein in lithium-heparin tubes. Differential blood images are obtained from the blood samples.
- White blood cells are determined with a hematology cell counter.
- Blood smears are made from the heparimized blood samples. Each sample is dehydrated with methanol before staining. After fixation, each slide is incubated with May Grünwald stain for 5 min. Then the slides are washed with aqua dest. rinsed. It is then stained with Giemsa stain for 2 min and the slides are washed once more in distilled water.
- the differential time number is determined as the sum of neuttophils, eosinophils, easophils, lymphocytes and monocytes from 100 cells. Then the percentage of neutrophils in the number of all white blood cells is determined.
- FIG. 2 shows that the administration of compound 149 significantly reduces the C5a-induced neutropenia and thus has the desired anti-inflammatory effect in this inflammation model.
- CitruUin although uncharged under physiological conditions, is nevertheless polar, albeit less polar than a charged guanidine. This is e.g. through the calculated logP values of various amino acid derivatives, as shown in the following overview:
- the logP value indicates the distribution ratio of a connection between a water and an octanol phase and is lower the polar the connection is.
- the logP values were predicted using the Cherndraw program (available from CambridgeSoft, Cambridge, Great Britain).
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Organic Chemistry (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Pharmacology & Pharmacy (AREA)
- Animal Behavior & Ethology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Immunology (AREA)
- Diabetes (AREA)
- Physical Education & Sports Medicine (AREA)
- Orthopedic Medicine & Surgery (AREA)
- Dermatology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Molecular Biology (AREA)
- Genetics & Genomics (AREA)
- Neurology (AREA)
- Biophysics (AREA)
- Biochemistry (AREA)
- Hematology (AREA)
- Heart & Thoracic Surgery (AREA)
- Pulmonology (AREA)
- Cardiology (AREA)
- Rheumatology (AREA)
- Gastroenterology & Hepatology (AREA)
- Zoology (AREA)
- Urology & Nephrology (AREA)
- Obesity (AREA)
- Biomedical Technology (AREA)
- Neurosurgery (AREA)
- Cell Biology (AREA)
- Toxicology (AREA)
- Transplantation (AREA)
Abstract
Description

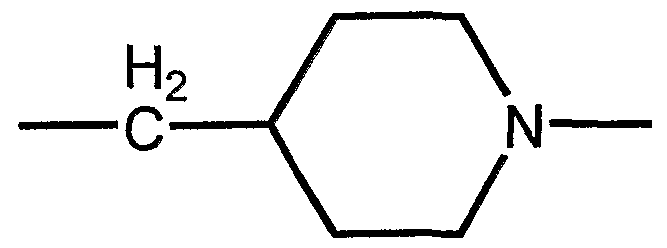





























































Claims

















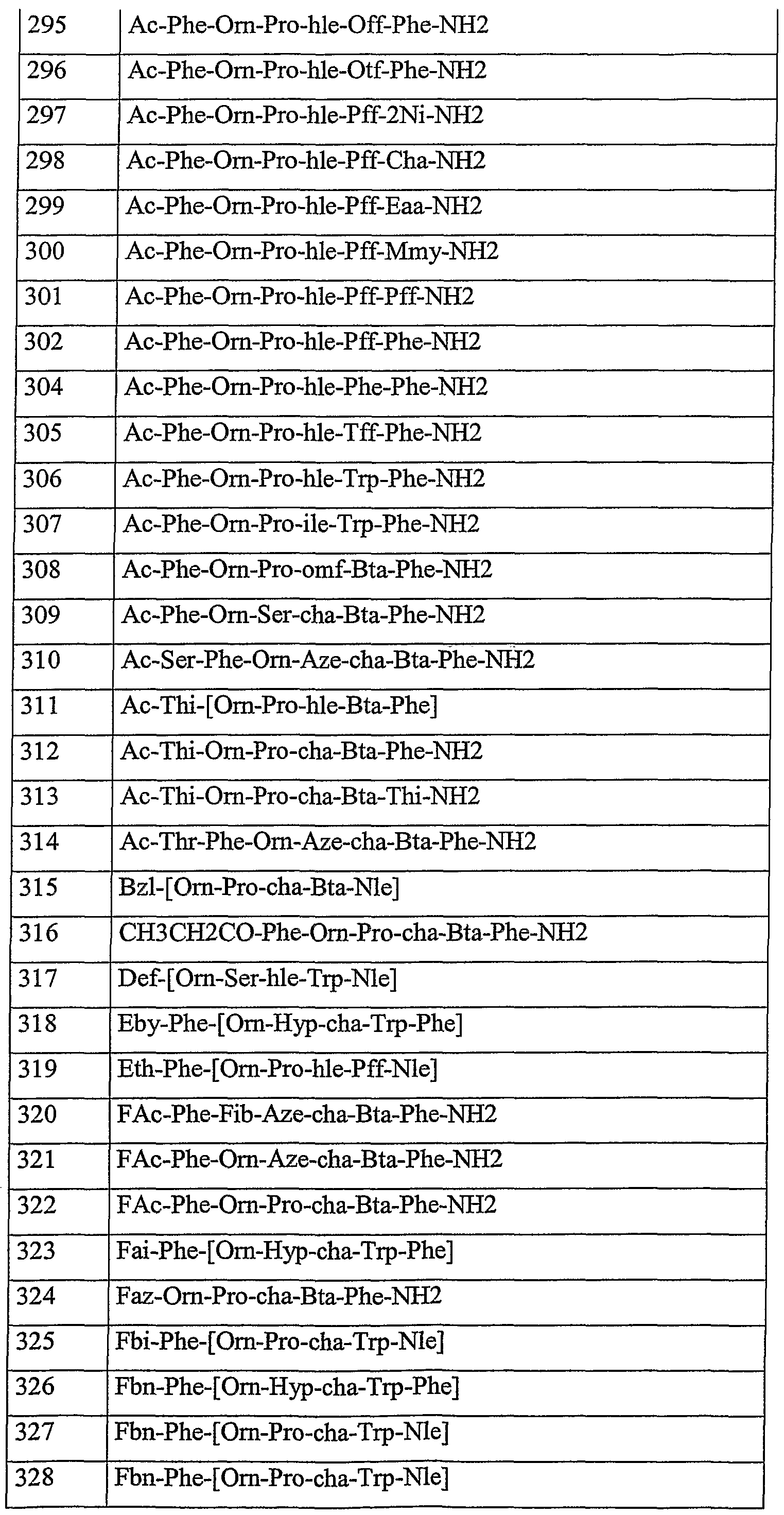


Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2006519897A JP4644194B2 (ja) | 2003-07-17 | 2004-07-19 | C5a受容体拮抗物質 |
| US10/564,788 US7727960B2 (en) | 2003-07-17 | 2004-07-19 | C5a receptor antagonists |
| AU2004259282A AU2004259282A1 (en) | 2003-07-17 | 2004-07-19 | C5a receptor antagonists |
| CA002532994A CA2532994A1 (en) | 2003-07-17 | 2004-07-19 | C5a receptor antagonists |
| EP04763337A EP1646643A2 (de) | 2003-07-17 | 2004-07-19 | C5a-REZEPTOR-ANTAGONISTEN |
| US12/748,680 US20110003756A1 (en) | 2003-07-17 | 2010-03-29 | C5a Receptor Antagonists |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP03016233A EP1498422A1 (de) | 2003-07-17 | 2003-07-17 | C5a-Rezeptor-Antagonisten |
| EP03016233.3 | 2003-07-17 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US12/748,680 Division US20110003756A1 (en) | 2003-07-17 | 2010-03-29 | C5a Receptor Antagonists |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2005010030A2 true WO2005010030A2 (de) | 2005-02-03 |
| WO2005010030A3 WO2005010030A3 (de) | 2006-06-22 |
Family
ID=33462142
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/EP2004/008057 Ceased WO2005010030A2 (de) | 2003-07-17 | 2004-07-19 | C5a-REZEPTOR-ANTAGONISTEN |
Country Status (8)
| Country | Link |
|---|---|
| US (2) | US7727960B2 (de) |
| EP (2) | EP1498422A1 (de) |
| JP (2) | JP4644194B2 (de) |
| AU (1) | AU2004259282A1 (de) |
| CA (1) | CA2532994A1 (de) |
| SG (1) | SG144934A1 (de) |
| WO (1) | WO2005010030A2 (de) |
| ZA (1) | ZA200600344B (de) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006074964A1 (en) * | 2005-01-17 | 2006-07-20 | Jerini Ag | C5a receptor antagonists |
| EP1739078A1 (de) | 2005-05-30 | 2007-01-03 | Jerini AG | C5a-Rezeptor-Antagonisten |
| JP2009544627A (ja) * | 2006-07-21 | 2009-12-17 | プロミックス・ピーティーワイ・リミテッド | 内膜過形成および関連する状態に対する治療 |
| WO2018020358A1 (en) | 2016-07-29 | 2018-02-01 | Pfizer Inc. | Cyclic peptides as c5 a receptor antagonists |
Families Citing this family (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9839667B2 (en) | 2005-10-14 | 2017-12-12 | Allergan, Inc. | Prevention and treatment of ocular side effects with a cyclosporin |
| JP2011195451A (ja) * | 2008-06-20 | 2011-10-06 | Fukuoka Univ | ペプチド |
| WO2010126833A1 (en) * | 2009-04-27 | 2010-11-04 | Jerini Ophthalmic Inc. | Sustained release formulations of peptidomimetic drugs and uses thereof |
| WO2011123795A1 (en) | 2010-04-02 | 2011-10-06 | Battelle Memorial Institute | Methods for associating or dissociating guest materials with a metal organic framework, systems for associating or dissociating guest materials within a series of metal organic frameworks, and gas separation assemblies |
| EP3524258B1 (de) | 2011-06-22 | 2025-10-01 | Apellis Pharmaceuticals, Inc. | Verfahren zur behandlung chronischer erkrankungen mit komplementinhibitoren |
| US9289467B2 (en) | 2011-08-10 | 2016-03-22 | Case Western Reserve University | Compositions and methods for treating bone conditions |
| US10220002B2 (en) | 2011-12-02 | 2019-03-05 | Board Of Regents Of The University Of Nebraska | Controlled-release peptide compositions and uses thereof |
| AU2016347456B2 (en) | 2015-10-27 | 2018-12-13 | F. Hoffmann-La Roche Ag | Peptide macrocycles against acinetobacter baumannii |
| EP3388444A1 (de) | 2017-04-10 | 2018-10-17 | F. Hoffmann-La Roche AG | Antibakterielle peptidmakrozyklen und verwendung davon |
| WO2019040973A1 (en) * | 2017-09-01 | 2019-03-07 | Alsonex Pty Ltd | METHOD FOR SOLID PHASE SYNTHESIS OF CYCLIC PENTAPEPTIDES |
| US11505573B2 (en) | 2018-03-28 | 2022-11-22 | Hoffmann-La Roche Inc. | Peptide macrocycles against Acinetobacter baumannii |
| US11819532B2 (en) | 2018-04-23 | 2023-11-21 | Hoffmann-La Roche Inc. | Peptide macrocycles against Acinetobacter baumannii |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH04503073A (ja) * | 1989-01-31 | 1992-06-04 | アボツト・ラボラトリーズ | アナフィラトキシン受容体リガンド |
| US5387671A (en) * | 1990-12-27 | 1995-02-07 | Abbott Laboratories | Hexa- and heptapeptide anaphylatoxin-receptor ligands |
| AUPO755097A0 (en) * | 1997-06-25 | 1997-07-17 | University Of Queensland, The | Receptor agonist and antagonist |
| AUPR833401A0 (en) * | 2001-10-17 | 2001-11-08 | University Of Queensland, The | G protein-coupled receptor antagonists |
| KR100853220B1 (ko) * | 2002-04-04 | 2008-08-20 | 삼성전자주식회사 | 표시 장치용 박막 트랜지스터 어레이 기판의 제조 방법 |
| AUPS160602A0 (en) | 2002-04-08 | 2002-05-16 | University Of Queensland, The | Therapeutic method |
| AU2002952086A0 (en) * | 2002-10-16 | 2002-11-07 | The University Of Queensland | Treatment of osteoarthritis |
-
2003
- 2003-07-17 EP EP03016233A patent/EP1498422A1/de not_active Withdrawn
-
2004
- 2004-07-19 SG SG200805390-2A patent/SG144934A1/en unknown
- 2004-07-19 AU AU2004259282A patent/AU2004259282A1/en not_active Abandoned
- 2004-07-19 US US10/564,788 patent/US7727960B2/en not_active Expired - Fee Related
- 2004-07-19 WO PCT/EP2004/008057 patent/WO2005010030A2/de not_active Ceased
- 2004-07-19 EP EP04763337A patent/EP1646643A2/de not_active Withdrawn
- 2004-07-19 CA CA002532994A patent/CA2532994A1/en not_active Abandoned
- 2004-07-19 JP JP2006519897A patent/JP4644194B2/ja not_active Expired - Fee Related
-
2006
- 2006-01-13 ZA ZA200600344A patent/ZA200600344B/en unknown
-
2010
- 2010-03-29 US US12/748,680 patent/US20110003756A1/en not_active Abandoned
- 2010-10-13 JP JP2010230873A patent/JP2011057681A/ja not_active Withdrawn
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2006074964A1 (en) * | 2005-01-17 | 2006-07-20 | Jerini Ag | C5a receptor antagonists |
| EP1739078A1 (de) | 2005-05-30 | 2007-01-03 | Jerini AG | C5a-Rezeptor-Antagonisten |
| JP2009544627A (ja) * | 2006-07-21 | 2009-12-17 | プロミックス・ピーティーワイ・リミテッド | 内膜過形成および関連する状態に対する治療 |
| EP2049141A4 (de) * | 2006-07-21 | 2011-10-26 | Promics Pty Ltd | Behandlung von intimahyperplasie und relevanten erkrankungen |
| WO2018020358A1 (en) | 2016-07-29 | 2018-02-01 | Pfizer Inc. | Cyclic peptides as c5 a receptor antagonists |
| CN109563136A (zh) * | 2016-07-29 | 2019-04-02 | 辉瑞公司 | 作为C5a受体拮抗剂的环肽 |
Also Published As
| Publication number | Publication date |
|---|---|
| AU2004259282A1 (en) | 2005-02-03 |
| EP1498422A1 (de) | 2005-01-19 |
| CA2532994A1 (en) | 2005-02-03 |
| WO2005010030A3 (de) | 2006-06-22 |
| JP2007534619A (ja) | 2007-11-29 |
| ZA200600344B (en) | 2007-01-31 |
| US7727960B2 (en) | 2010-06-01 |
| US20060183883A1 (en) | 2006-08-17 |
| SG144934A1 (en) | 2008-08-28 |
| US20110003756A1 (en) | 2011-01-06 |
| JP4644194B2 (ja) | 2011-03-02 |
| JP2011057681A (ja) | 2011-03-24 |
| EP1646643A2 (de) | 2006-04-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| DE60038734T2 (de) | Melanocortinrezeptor-liganden | |
| US20110003756A1 (en) | C5a Receptor Antagonists | |
| DE60119714T2 (de) | Peptidderivat | |
| DE60102899T2 (de) | Verfahren zur lösung von glucagon-ähnlichen peptid-1 (glp-1) verbindungen | |
| DE69434217T2 (de) | Conotoxine | |
| DE69838678T2 (de) | ZYKLISCHE ANTAGONISTEN DER C5a REZEPTOREN SOWIE DER G-PROTEIN GEKOPPELTEN REZEPTOREN | |
| DE60130192T2 (de) | Kahalalide f und verwandte verbindungen | |
| WO2011063367A1 (en) | Melanocortin-1 receptor-specific linear peptides | |
| US20080161232A1 (en) | C5a Receptor Antagonist | |
| DE69906108T2 (de) | Peptid-derivat | |
| US20160108090A1 (en) | Dynorphin a analogs with bradykinin receptors specificity for modulation of neuropathic pain | |
| DE102004051014A1 (de) | Chemisch modifizierte Peptidanaloga | |
| DE69701269T2 (de) | Zyklische Motilin-ähnliche Polypeptide mit die gastrointestinale Motorik stimulierender Aktivität | |
| DE69232716T2 (de) | Peptide mit thrombospondin-ähnlicher Aktivität und ihre therapeutische Verwendung | |
| WO2002055552A2 (de) | Lösliche cyclische analoga des beta amyloiden peptides | |
| EP3230300B1 (de) | Inhibitoren zur hemmung der tumormetastasierung | |
| JP4565087B2 (ja) | 新規なχ−コノトキシン・ペプチド(−I) | |
| US20230391838A1 (en) | Conformationally-Constrained Alpha-RGIA Analogues | |
| DE10315030B4 (de) | Zyklische Peptide, Verfahren zu deren Herstellung und deren medizinische Verwendung | |
| HK1107999A (en) | C5a receptor antagonists | |
| AU2024314499A1 (en) | Modified polypeptide and use thereof in field of analgesia | |
| CN117683088A (zh) | 一种具有血浆稳定性的多肽及其制备方法 | |
| Albericio et al. | Kahalalide F and related compounds | |
| DD299043A5 (de) | Verfahren zur herstellung neuartiger oral wirksamer neuropsychopharmaka | |
| DE10018335A1 (de) | Raumform von TPR-Strukturmotiv enthaltenden Polypeptiden mit Chaperon-Bindungsfunktion, deren Kristalle und Verbindungen zur Inhibierung derartiger Polypeptide |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| AK | Designated states |
Kind code of ref document: A2 Designated state(s): AE AG AL AM AT AU AZ BA BB BG BR BW BY BZ CA CH CN CO CR CU CZ DE DK DM DZ EC EE EG ES FI GB GD GE GH GM HR HU ID IL IN IS JP KE KG KP KR KZ LC LK LR LS LT LU LV MA MD MG MK MN MW MX MZ NA NI NO NZ OM PG PH PL PT RO RU SC SD SE SG SK SL SY TJ TM TN TR TT TZ UA UG US UZ VC VN YU ZA ZM ZW |
|
| AL | Designated countries for regional patents |
Kind code of ref document: A2 Designated state(s): BW GH GM KE LS MW MZ NA SD SL SZ TZ UG ZM ZW AM AZ BY KG KZ MD RU TJ TM AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IT LU MC NL PL PT RO SE SI SK TR BF BJ CF CG CI CM GA GN GQ GW ML MR NE SN TD TG |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application | ||
| DPEN | Request for preliminary examination filed prior to expiration of 19th month from priority date (pct application filed from 20040101) | ||
| WWE | Wipo information: entry into national phase |
Ref document number: 2006/00344 Country of ref document: ZA Ref document number: 200600344 Country of ref document: ZA |
|
| ENP | Entry into the national phase |
Ref document number: 2532994 Country of ref document: CA |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2004763337 Country of ref document: EP Ref document number: 2006183883 Country of ref document: US Ref document number: 2006519897 Country of ref document: JP Ref document number: 10564788 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2004259282 Country of ref document: AU |
|
| WWP | Wipo information: published in national office |
Ref document number: 2004763337 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 10564788 Country of ref document: US |