WO2009116612A1 - 親水膜 - Google Patents
親水膜 Download PDFInfo
- Publication number
- WO2009116612A1 WO2009116612A1 PCT/JP2009/055398 JP2009055398W WO2009116612A1 WO 2009116612 A1 WO2009116612 A1 WO 2009116612A1 JP 2009055398 W JP2009055398 W JP 2009055398W WO 2009116612 A1 WO2009116612 A1 WO 2009116612A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- group
- compound
- silane
- hydrophilic
- film
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C03—GLASS; MINERAL OR SLAG WOOL
- C03C—CHEMICAL COMPOSITION OF GLASSES, GLAZES OR VITREOUS ENAMELS; SURFACE TREATMENT OF GLASS; SURFACE TREATMENT OF FIBRES OR FILAMENTS MADE FROM GLASS, MINERALS OR SLAGS; JOINING GLASS TO GLASS OR OTHER MATERIALS
- C03C17/00—Surface treatment of glass, not in the form of fibres or filaments, by coating
- C03C17/28—Surface treatment of glass, not in the form of fibres or filaments, by coating with organic material
- C03C17/30—Surface treatment of glass, not in the form of fibres or filaments, by coating with organic material with silicon-containing compounds
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09D—COATING COMPOSITIONS, e.g. PAINTS, VARNISHES OR LACQUERS; FILLING PASTES; CHEMICAL PAINT OR INK REMOVERS; INKS; CORRECTING FLUIDS; WOODSTAINS; PASTES OR SOLIDS FOR COLOURING OR PRINTING; USE OF MATERIALS THEREFOR
- C09D183/00—Coating compositions based on macromolecular compounds obtained by reactions forming in the main chain of the macromolecule a linkage containing silicon, with or without sulfur, nitrogen, oxygen, or carbon only; Coating compositions based on derivatives of such polymers
- C09D183/04—Polysiloxanes
- C09D183/08—Polysiloxanes containing silicon bound to organic groups containing atoms other than carbon, hydrogen, and oxygen
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09D—COATING COMPOSITIONS, e.g. PAINTS, VARNISHES OR LACQUERS; FILLING PASTES; CHEMICAL PAINT OR INK REMOVERS; INKS; CORRECTING FLUIDS; WOODSTAINS; PASTES OR SOLIDS FOR COLOURING OR PRINTING; USE OF MATERIALS THEREFOR
- C09D4/00—Coating compositions, e.g. paints, varnishes or lacquers, based on organic non-macromolecular compounds having at least one polymerisable carbon-to-carbon unsaturated bond ; Coating compositions, based on monomers of macromolecular compounds of groups C09D183/00 - C09D183/16
- C09D4/06—Organic non-macromolecular compounds having at least one polymerisable carbon-to-carbon unsaturated bond in combination with a macromolecular compound other than an unsaturated polymer of groups C09D159/00 - C09D187/00
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09D—COATING COMPOSITIONS, e.g. PAINTS, VARNISHES OR LACQUERS; FILLING PASTES; CHEMICAL PAINT OR INK REMOVERS; INKS; CORRECTING FLUIDS; WOODSTAINS; PASTES OR SOLIDS FOR COLOURING OR PRINTING; USE OF MATERIALS THEREFOR
- C09D5/00—Coating compositions, e.g. paints, varnishes or lacquers, characterised by their physical nature or the effects produced; Filling pastes
-
- C—CHEMISTRY; METALLURGY
- C03—GLASS; MINERAL OR SLAG WOOL
- C03C—CHEMICAL COMPOSITION OF GLASSES, GLAZES OR VITREOUS ENAMELS; SURFACE TREATMENT OF GLASS; SURFACE TREATMENT OF FIBRES OR FILAMENTS MADE FROM GLASS, MINERALS OR SLAGS; JOINING GLASS TO GLASS OR OTHER MATERIALS
- C03C2217/00—Coatings on glass
- C03C2217/70—Properties of coatings
- C03C2217/75—Hydrophilic and oleophilic coatings
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G77/00—Macromolecular compounds obtained by reactions forming a linkage containing silicon with or without sulfur, nitrogen, oxygen or carbon in the main chain of the macromolecule
- C08G77/04—Polysiloxanes
- C08G77/22—Polysiloxanes containing silicon bound to organic groups containing atoms other than carbon, hydrogen and oxygen
- C08G77/28—Polysiloxanes containing silicon bound to organic groups containing atoms other than carbon, hydrogen and oxygen sulfur-containing groups
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y10—TECHNICAL SUBJECTS COVERED BY FORMER USPC
- Y10T—TECHNICAL SUBJECTS COVERED BY FORMER US CLASSIFICATION
- Y10T428/00—Stock material or miscellaneous articles
- Y10T428/31504—Composite [nonstructural laminate]
- Y10T428/31652—Of asbestos
- Y10T428/31663—As siloxane, silicone or silane
Definitions
- the present invention relates to a hydrophilic film having excellent scratch resistance and transparency, and a laminate.
- Nonpatent literature 1 Nonpatent literature 2
- Patent Document 1 The present inventors have already proposed a method for hydrophilizing the surface (Patent Document 1). According to this method, an extremely high hydrophilic film can be obtained, and it is suitably used for antifogging and antifouling applications. However, the adhesion to inorganic materials such as glass may not always be high. Further studies such as searching for primers were necessary.
- the surface of the inorganic hard coat layer is treated with a reactive silane coupling material, and then a hydrophilic monomer is added to the reactive group remaining on the surface.
- the method (patent document 2, patent document 3) which is made to graft (react) is mentioned.
- Patent Document 4 a method using a silane coupling material having a mercapto group and a hydrophilic monomer having a functional group having a carbon-carbon double bond
- Patent Document 5 A method using a silane coupling material and a hydrophilic monomer having a mercapto group
- Patent Document 5 a method using a silane coupling material having an amino group and a hydrophilic monomer having a functional group having a carbon-carbon double bond
- Patent Document 6 a method using a silane coupling material having an amino group and a hydrophilic monomer having a functional group having a carbon-carbon double bond
- Patent Literature 7 Patent Literature 8
- Patent Document 9 Patent Document 10, Patent Document 11, Patent Document 12
- Patent Document 9 Patent Document 9
- Patent Document 12 Patent Document 9
- Patent Documents 2 to 8 hydrophilization is possible, but since a trifunctional or lower silane coupling material is used, the crosslinking density is low, the hardness is insufficient, and high scratch resistance is difficult to obtain.
- the methods of Patent Documents 9 to 12 can also be hydrophilized, but it is difficult to obtain high scratch resistance, and problems such as safety due to the sulfuric acid treatment process and apparatus corrosion remain.
- WO2007 / 064003 JP-A-4-225301 JP-A-8-259270 JP 2003-5499 A Japanese Patent Laid-Open No. 2000-104046 JP 2007-313694 A Japanese Patent Laid-Open No.
- the present invention provides a hydrophilic film having high adhesion to an inorganic material, safer, excellent in transparency, and having both extremely high hydrophilicity and scratch resistance, and a laminate having the hydrophilic film. is there.
- the present inventors have formed a layer (cured product) from a mixture containing a specific silane compound having a mercapto group or the like and a specific silane compound on a substrate.
- Layer a specific hydrophilic compound is applied to the layer and allowed to react, thereby obtaining a hydrophilic film having excellent transparency with improved adhesion to the substrate and improved scratch resistance.
- the present invention has been found.
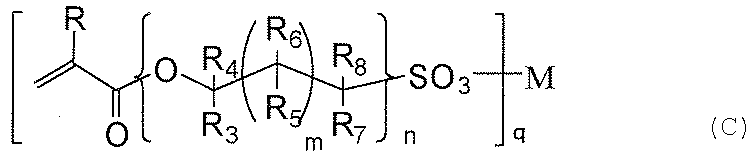
- the hydrophilic film of the present invention comprises (a) one silicon atom, a group selected from the group consisting of a mercapto group, an amino group, a (meth) acryloyl group, and a vinyl group, an alkoxy group, a halogen group, and a hydroxy group.
- a compound represented by the following general formula (c) on the surface of a layer formed from a mixture containing a silane compound having a silane compound having no reactivity with a carbon-carbon double bond Apply Selected from the group consisting of a (meth) acryloyl group contained in a compound represented by the following general formula (c) and a mercapto group, amino group, (meth) acryloyl group, and vinyl group derived from the compound (a). It is obtained by reacting at least a part with a group.
- R represents H or CH 3.
- R 3 to R 8 independently represent H, CH 3 , or OH.
- M represents an integer of 0 to 18, and n represents 1. Represents an integer of ⁇ 10, q represents 1 or 2.
- M represents H, Li, Na, K, Rb, Mg, Ca, Sr, or Ba.
- the molar ratio of the compound (a) / the compound (b) is preferably 1/2 to 1/200, and more preferably 1/3 to 1/100.
- a laminate can be obtained by forming the hydrophilic film on a substrate.
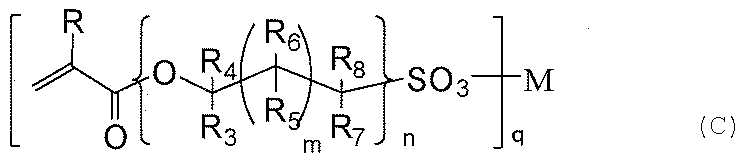
- the method for producing a laminate having a hydrophilic film comprises: (a) one silicon atom, a group selected from the group consisting of a mercapto group, an amino group, a (meth) acryloyl group, and a vinyl group; A silane compound having a group bonded to at least one silicon atom selected from the group consisting of a group, a halogen group, and a hydroxy group, and (b) selected from the group consisting of an alkoxy group, a halogen group, and a hydroxy group, Preparing a mixture containing a silane compound having a crosslinkable group bonded to a silicon atom and having no group reactive with a carbon-carbon double bond, and applying the silane mixture to a substrate; A hydrolysis reaction and a condensation reaction are allowed to proceed to form a layer on the surface of the substrate, and (c) a compound represented by the following general formula (c) is applied to the surface of the layer And selected from the group consisting of
- R represents H or CH 3.
- R 3 to R 8 independently represent H, CH 3 , or OH.
- M represents an integer of 0 to 18, and n represents 1. Represents an integer of ⁇ 10, q represents 1 or 2.
- M represents H, Li, Na, K, Rb, Mg, Ca, Sr, or Ba.
- M in the compound (c) is H, Li, Na, Rb, Mg, Ca, Sr, or Ba, and the graft reaction is performed at 130 ° C. It is preferable to proceed under the above temperature conditions.
- the present invention it is possible to provide a transparent film excellent in adhesiveness with an inorganic material and suitable for antifogging and antifouling applications and having both scratch resistance and hydrophilicity at a high level.
- hydrophilic film In order to obtain a hydrophilic film according to the present invention, it is first necessary to form a layer from a mixture containing two specific silane compounds (silane mixture).
- the mixture forming the layer includes (a) one silicon atom, a group selected from the group consisting of a mercapto group, an amino group, a (meth) acryloyl group and a vinyl group, an alkoxy group, a halogen group, and a hydroxy group. It is characterized in that a silane compound having a group bonded to at least one silicon atom selected from the group consisting of groups (hereinafter also referred to as silane compound (a)) is included.
- the alkoxy group is not particularly limited, but is usually an alkoxy group having 1 to 4 carbon atoms.
- Examples of the alkoxy group include a methoxy group, an ethoxy group, a propoxy group, and a butoxy group.
- halogen group examples include a fluoro group, a chloro group, a bromo group, and an iodo group.
- a chloro group is preferable from the viewpoint of cost and the like.
- the total number of alkoxy groups, halogen groups, and hydroxy groups bonded to one silicon atom in the silane compound (a) is not particularly limited as long as the silane compound (a) has a mercapto group, but usually 1 to 3 range.
- the total number of alkoxy groups, halogen groups and hydroxy groups bonded to silicon atoms is preferably in the range of 2 to 3, more preferably 3. preferable. This is because when the total number of these groups bonded to the silicon atom is within the above range, the crosslinking density of the resulting film tends to increase.
- the silane compound (a) may further contain other groups such as an aromatic group and an aliphatic group.
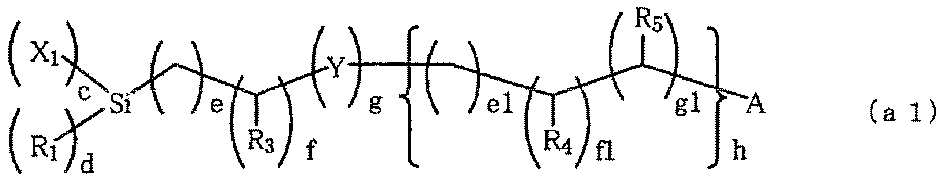
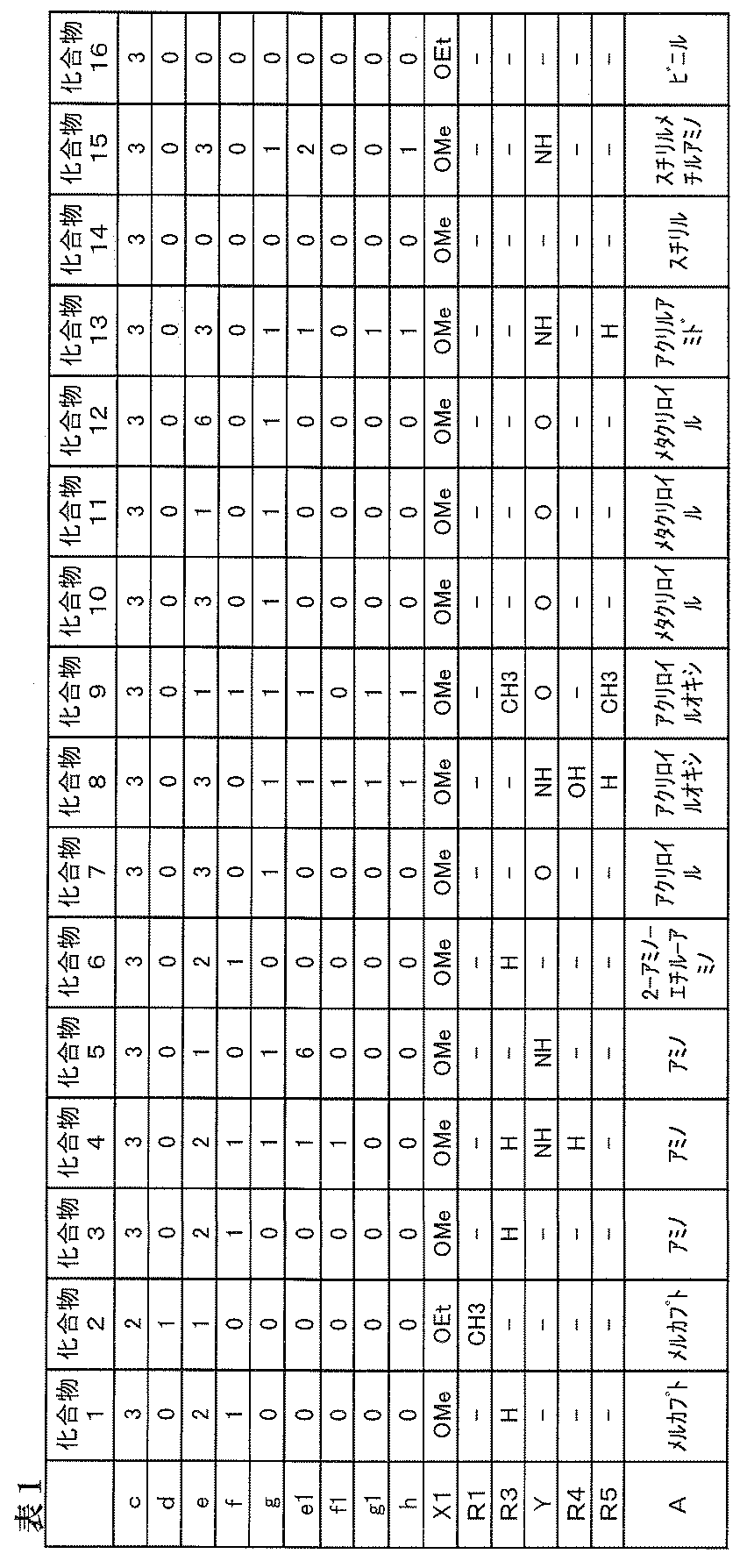
- silane compounds (a) compounds represented by the following general formula (a1) are preferable.
- R 1 represents H or an alkyl group having 1 to 4 carbon atoms.
- R 1 an alkyl group having 1 to 2 carbon atoms is relatively preferable.
- X 1 represents a halogen group, an OH group (hydroxy group), or an alkoxy group having 1 to 4 carbon atoms.
- a halogen group, an OH group (hydroxy group), and an alkoxy group having 1 to 2 carbon atoms are preferable from the viewpoint of reactivity. Further, in consideration of storage stability, an alkoxy group having 1 to 2 carbon atoms is preferable. Groups tend to be more preferred.
- R 3 to R 5 independently represent H, CH 3 , or OH.
- Y independently represents O, S, NH, or NCH3.
- O and NH are preferable, and O tends to be more preferable.
- c represents an integer of 1 to 3
- d represents an integer of 0 to 2
- c + d 3.
- e, e1, f, and f1 independently represent an integer of 0 to 10. Among these, an integer of 0 to 5 is relatively preferable, and an integer of 0 to 3 tends to be more preferable.
- g and g1 independently represent 0-2.
- h represents an integer of 0 to 3.
- an integer of 0 to 2 is preferable, and 0 to 1 tends to be more preferable.
- A represents a mercapto group, amino group, (meth) acryloyl group or vinyl group having reactivity with a carbon-carbon double bond.
- the mercapto group, amino group, (meth) acryloyl group or vinyl group is a group containing a mercapto group, an amino group, a (meth) acryloyl group or a vinyl group, respectively, by combining with other carbon groups. It may be.
- Such mercapto group-containing groups include mercapto-methyl group, 2-mercapto-ethyl group, 2-mercapto-ethyloxy group, 2-mercapto-ethylthio group, 1-mercapto-ethyl group, 3-mercapto-propyl group.
- Examples of the group containing an amino group include an amino group, an N-methylamino group, an N-ethylamino group, an N-hydroxyethyl-amino group, an aminomethyl group, a 1- and 2-aminoethyl group, 1-, 2- and 3-aminopropyl groups, 1,2-diaminoethyl groups, 1,2-, 1,3-, 2,3-diaminopropyl groups and other amino group-substituted alkyl groups, benzylamino groups, aminophenyl Group, benzamide group, acetamide group and the like.
- Examples of the group containing (meth) acryloyl group include (meth) acryloyl group, (meth) acryloyloxy group, (meth) acryloylthio, (meth) acrylamide group and the like.
- Examples of the group containing a vinyl group include an alkenyl group such as a vinyl group, an allyl group and an isopropenyl group, an alkenyl carbonate group such as an allyl carbonate group, and a vinyl aromatic group such as a styryl group and an ⁇ -methyl-styryl group. Is mentioned.
- M in the compound (a1) represents H, Li, Na, K, Rb, Mg, Ca, Sr or Ba.
- M is preferably K from the viewpoint that the efficiency of the graft reaction by heat of less than 130 degrees or ultraviolet irradiation is good, but as will be described later, the graft reaction proceeds with heat of 130 or more. In this case, even if M is H, Li, Na, Rb, Mg, Ca, Sr or Ba, high graft reaction efficiency is exhibited, so that sufficient hydrophilicity can be imparted to the hydrophilic film. .
- the silane compound (a) may be used alone or in combination of two or more.
- the mixture forming the layer has (b) a group bonded to at least four silicon atoms selected from the group consisting of an alkoxy group, a halogen group, and a hydroxy group. And a silane compound that does not have a group reactive with a carbon-carbon double bond (hereinafter also referred to as silane compound (b)).
- the alkoxy group or halogen atom bonded to the silicon atom is easily hydrolyzed by moisture and converted into a hydroxysilyl group.
- This hydroxysilyl group causes a condensation reaction with other hydroxysilyl groups, alkoxysilyl groups, or hydroxy groups (OH groups) on the substrate surface. Therefore, a mixture made of a compound having an alkoxy group, a halogen group, or a hydroxy group bonded to a silicon atom forms a siloxane bond by hydrolysis and condensation reaction, and the entire mixture is cross-linked and cured mainly by silica. A physical layer is formed.
- the silane compound (a) needs to have at least one group selected from the group consisting of a mercapto group mercapto group, an amino group, a (meth) acryloyl group and a vinyl group when viewed as a silane compound, an alkoxy group, A group selected from a halogen group and a hydroxy group can have at most 3 per silicon atom.
- the formed layer has insufficient crosslinking density, and for example, a film that can withstand severe scratch tests is formed. This is difficult and is not necessarily sufficient in terms of hardness.
- the crosslinking density of the cured product layer formed from the silane compound is not sufficient. Absent.
- silane compound (a) in addition to the silane compound (a), it is bonded to at least four silicon atoms selected from the group consisting of an alkoxy group, a halogen group, and a hydroxy group. It is important to use a silane compound (b) having the above-mentioned group and not having a group having reactivity with a carbon-carbon double bond.
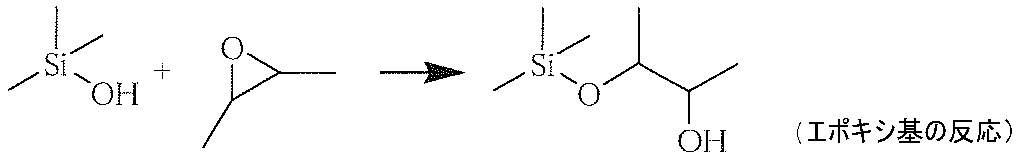
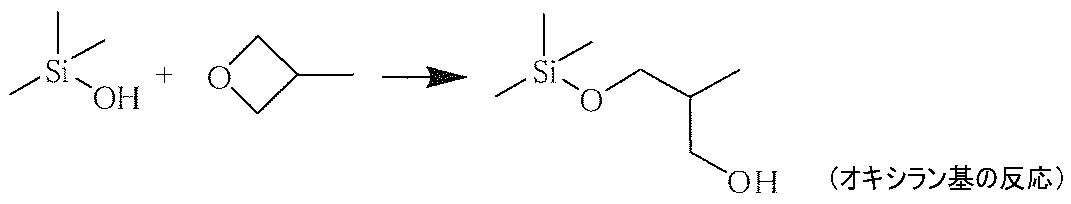
- an alkoxy group or halogen group having a 3- to 4-membered cyclic ether group (epoxy group or oxirane group) that reacts with a hydroxyl group bonded to a silicon atom formed by hydrolysis of a silane compound, and bonded to the silicon atom
- a silane compound (b) having at least two groups selected from the group consisting of hydroxy groups is effective in improving the crosslinking density.
- An example of a reaction between a hydroxyl group bonded to a silicon atom and a 3- to 4-membered cyclic ether group is described below.
- silane compound (b) together with the silane compound (a), it is possible to withstand a severe abrasion test and to form a hardened material layer having a high hardness.
- the number of groups bonded to silicon atoms selected from the group consisting of alkoxy groups, halogen groups, and hydroxy groups contained in the silane compound (b) is 4 to 4 20 is preferable, and 4 to 10 is more preferable.
- bonded with the silicon atom contained in the silane compound, the halogen group, the hydroxy group, and the silicon atom The total number of interlinkage groups is preferably more than 3 in terms of one silicon atom.
- the silicon-bonding group is a group that bonds a silicon atom to a silicon atom, and examples thereof include —O— in a siloxane bond and an alkylene group between silicon atoms.
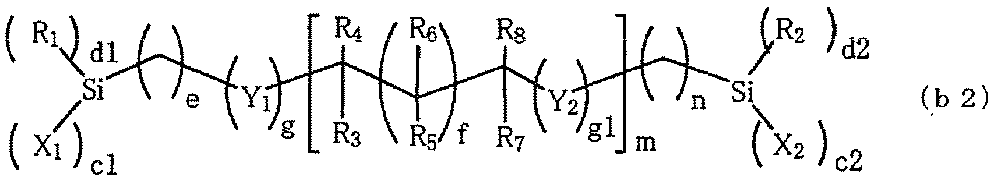
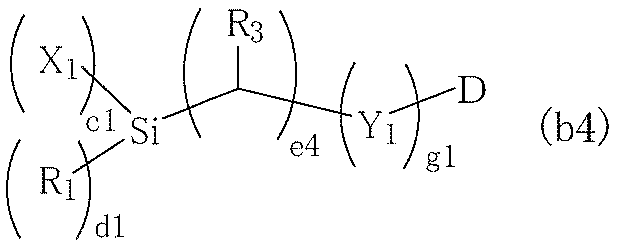
- silane compounds (b) compounds represented by the following general formulas (b1) to (b4) are preferable.
- X 1 to X 4 independently represent a halogen atom, an OH group (hydroxy group), or an alkoxy group having 1 to 4 carbon atoms.
- a halogen atom, an OH group, and an alkoxy group having 1 to 2 carbon atoms are preferable from the viewpoint of reactivity, and in consideration of storage stability, an alkoxy group having 1 to 2 carbon atoms is preferable. Tends to be more preferable.
- Examples of the compound represented by the formula (b1) include tetramethoxy-silane, tetraethoxy-silane, tetrapropoxy-silane, tetrabutoxy-silane, terafluoro-silane, tetrachloro-silane, tetrabromo-silane, and tetraiodo.
- Silane tetrahydroxy-silane, trihydroxy-methoxy-silane, dihydroxy-dimethoxy-silane, hydroxy-trimethoxy-silane, trihydroxy-ethoxy-silane, dihydroxy-diethoxy-silane, hydroxy-triethoxy-silane, trihydroxy-chloro -Silane, dihydroxy-dichloro-silane, hydroxy-trichloro-silane and the like.
- R 1 and R 2 independently represent H or an alkyl group having 1 to 4 carbon atoms. Among these R 1 and R 2 , an alkyl group having 1 to 2 carbon atoms is preferable.
- R 3 to R 8 independently represent H, CH 3 , or OH.
- X 1 and X 2 independently represent a halogen atom, an OH group (hydroxy group), or an alkoxy group having 1 to 4 carbon atoms.
- a halogen atom, an OH group, and an alkoxy group having 1 to 2 carbon atoms are preferable from the viewpoint of reactivity. Further, in consideration of storage stability, an alkoxy group having 1 to 2 carbon atoms is preferable. It tends to be more favorable.
- c1 and c2 independently represent an integer of 2 to 3
- d1 and d2 independently represent an integer of 0 to 1
- c1 + c2 + d1 + d2 6.
- e and f independently represent an integer of 0 to 10.
- an integer of 0 to 5 is preferable, and an integer of 0 to 3 is more preferable.
- g and g1 independently represent an integer of 0 to 2.
- m represents an integer of 0 to 18.
- an integer of 0 to 8 is preferable, and an integer of 0 to 2 is more preferable.
- n represents an integer of 1 to 10.
- an integer of 1 to 6 is relatively preferable, and an integer of 1 to 4 is more preferable.
- Y 1 and Y 2 independently represent O, S, NH, or NCH 3 .
- O and NH are relatively preferable, and O is more preferable.
- Examples of the compound represented by the formula (b2) include bis (trimethoxy-silyl) methane, 1,2-bis (trimethoxy-silyl) ethane, 1,6-bis (trimethoxy-silyl) hexane, bis ⁇ 3 -(Trimethoxy-silyl) propyl ⁇ sulfide, bis ⁇ 3- (trimethoxy-silyl) propyl ⁇ amine, N, N'-bis ⁇ 3- (trimethoxy-silyl) propyl ⁇ -1,2-ethylenediamine, bis (triethoxy- Silyl) methane, 1,2-bis (triethoxy-silyl) ethane, 1,8-bis (triethoxy-silyl) octane, O, O'-bis ⁇ 3- (triethoxy-silyl) propyl ⁇ -poly having a molecular weight of about 1000 (1,2-propylene oxide), 1- (triethoxy-s
- R 3 to R 8 independently represent H, CH 3 , or OH.
- R 11 to R 13 independently represent H or an alkyl group having 1 to 4 carbon atoms. Among these R 11 to R 13 , an alkyl group having 1 to 2 carbon atoms is preferable.
- X 1 to X 3 independently represent a halogen atom, an OH group (hydroxy group), or an alkoxy group having 1 to 4 carbon atoms.
- a halogen atom, an OH group, and an alkoxy group having 1 to 2 carbon atoms are preferable from the viewpoint of reactivity. Further, in consideration of storage stability, an alkoxy group having 1 to 2 carbon atoms is preferable. Tends to be more preferable.
- c1 to c3 independently represent an integer of 2 to 3
- d1 to d3 independently represent an integer of 0 to 1
- c1 + c2 + c3 + d1 + d2 + d3 9.
- n1 to n3 independently represent an integer of 1 to 10. Among these n1 to n3, an integer of 1 to 6 is relatively preferable, and an integer of 1 to 4 is more preferable.
- Examples of the compound represented by the formula (b3) include N, N ′, N ′′ -tris ⁇ (trimethoxy-silyl) methyl ⁇ isocyanurate, N, N ′, N ′′ -tris ⁇ 2- (trimethoxy- Silyl) ethyl ⁇ isocyanurate, N, N ′, N ′′ -tris ⁇ 3- (trimethoxy-silyl) propyl ⁇ isocyanurate, N, N ′, N ′′ -tris ⁇ 4- (trimethoxy-silyl) butyl ⁇ isocyanurate N, N ′, N ′′ -tris ⁇ 6- (trimethoxy-silyl) hexyl ⁇ isocyanurate, N, N ′, N ′′ -tris ⁇ 8- (trimethoxy-silyl) octyl ⁇ isocyanurate, N, N ′, N ′′ -tris ⁇ 10- (
- R 1 represents H or an alkyl group having 1 to 4 carbon atoms.
- R 1 an alkyl group having 1 to 2 carbon atoms is preferable.
- R 3 represents H, CH 3 , or OH.
- X 1 represents a halogen atom, an OH group (hydroxy group), or an alkoxy group having 1 to 4 carbon atoms.
- a halogen atom, an OH group, and an alkoxy group having 1 to 2 carbon atoms are preferable from the viewpoint of reactivity, and an alkoxy group having 1 to 2 carbon atoms is more preferable in consideration of storage stability.
- c1 represents an integer of 2 to 3
- d1 represents an integer of 0 to 1
- c1 + d1 3.
- e4 represents an integer of 0 to 20.
- an integer of 0 to 10 is preferable, and an integer of 0 to 5 is more preferable.
- g1 represents an integer of 0-2.
- Y 1 represents O, S, NH, or NCH 3 .
- O and NH are relatively preferable, and O is more preferable.
- D represents a functional group having a 3- to 4-membered cyclic ether structure.
- the functional group having such a structure include an epoxy group, a glycidyl group, a cyclohexene oxide group, a styrene oxide group, an oxetane group, an oxetanyl-methyl group, an oxetanyl-ethyl group, and a 2-methyl-2-oxetanyl-methyl group. 2-ethyl-2-oxetanyl-methyl group.
- more preferred forms include an epoxy group, a glycidyl group, a cyclohexene oxide group, an oxetane group, and a 2-ethyl-2-oxetanyl-methyl group. Further, an epoxy group, a glycidyl group, or a cyclohexene oxide group tends to be more preferable.
- Examples of the compound represented by the above formula (b4) include epoxy-trimethoxysilane, epoxy-triethoxysilane, glycidyl-trimethoxysilane, glycidyl-oxy-trimethoxysilane, 2-trimethoxysilyl-ethyl (3 , 4-cyclohexene oxide), 4- (trimethoxysilyl) -styrene oxide, glycidyloxymethyl-trimethoxysilane, glycidyloxyethyl-trimethoxysilane, glycidyloxypropyl-trimethoxysilane, glycidyloxypropyl- (methyl-dimethoxy) -Silane), glycidyloxypropyl-triethoxysilane, glycidyloxypropyl-trichlorosilane, glycidyloxypropyl-trihydroxysilane, and the like.
- the silane compound (b) may be used alone or in combination of two or more.
- the above-mentioned silane mixture may contain the silane compound (a) and the silane compound (b), respectively, or as a polycondensate of the silane compound (a) and the silane compound (b). It may be.
- a polycondensate include an oligomeric silane compound obtained by polycondensation of a silane compound (a) and a silane compound (b) (for example, X-41-X which is a polycondensation product of MPMOS and tetraethoxysilane). 1805 (product name; Shin-Etsu Chemical Co., Ltd.)).
- R represents H or CH 3 .
- R 3 to R 8 independently represent H, CH 3 , or OH.
- m represents an integer of 0 to 18, and n represents an integer of 1 to 10.
- q represents 1 or 2.
- M represents H, Li, Na, K, Rb, Mg, Ca, Sr, or Ba.
- the hydrophilic compound (c) reacts with or reacts with a group (for example, a mercapto group) present on the surface of a cured product layer obtained from a mixture containing the silane compound (a) and the silane compound (b). Excellent and hydrophilic.
- a group for example, a mercapto group
- the hydrophilic compound (c) is excellent in hydrophilicity in that the sulfonic acid group or sulfonic acid metal base (lithium sulfonate group, sodium sulfonate group, potassium sulfonate group, rubidium sulfonate group, sulfonic acid group) This is because a magnesium group, a calcium sulfonate group, a strontium sulfonate group, and a barium sulfonate group) are included.
- a hydrophilic group a sodium phosphate group, a phosphate group, a potassium carboxylate group, a sodium carboxylate group, a carboxyl group, a hydroxy group, etc. are known in addition to a sulfonic acid group or a sulfonic acid metal base. .
- the sulfonic acid group or the sulfonic acid metal base has at least a part of the carbon-carbon double bond contained in the compound (c), a mercapto group derived from the compound (a), Even after reacting with an amino group, a (meth) acryloyl group or a vinyl group, it is characterized by high hydrophilicity.
- a sulfonic acid group or a sulfonic acid metal base lithium sulfonate group, sodium sulfonate group, potassium sulfonate group, rubidium sulfonate is contained in the compound (c).
- the hydrophilic group of the hydrophilic compound (c) is not particularly limited as long as it is a sulfonic acid group or the above-described sulfonic acid metal base, but the characteristics and cost of each hydrophilic group are limited. It can be selected as appropriate in consideration.
- hydrophilic group of the hydrophilic compound (c) is a rubidium sulfonate group or the like, it tends to be undesirable from the viewpoint of cost.
- the hydrophilic compound (c) is further characterized in that an acryloyl group is contained as shown in the general formula (c).
- an acryloyl group in the hydrophilic compound (c) it exists in a cured product layer mainly composed of silica formed from a mixture containing the silane compound (a) and the silane compound (b).
- the reaction with the mercapto group, amino group, (meth) acryloyl group or vinyl group derived from the silane compound (a) becomes good.
- H and OH are preferable, and H is more preferable.
- an integer of 0 to 8 is preferable, and an integer of 0 to 2 is more preferable.
- an integer of 1 to 6 is preferable, and an integer of 1 to 4 is more preferable.
- Examples of the compound (c) include 2-sulfoethyl acrylate, 3-sulfopropyl acrylate, 2-hydroxy-3-sulfopropyl acrylate, 4-sulfobutyl acrylate, 6-sulfohexyl acrylate, 8-sulfooctyl acrylate, Examples include 9-sulfo-3,6-dioxanonyl acrylate, 10-sulfodecyl acrylate, and lithium, sodium, potassium, rubidium, magnesium, calcium, strontium, or nalium salts thereof. .
- the hydrophilic compound (c) may be used alone or in combination of two or more.
- the hydrophilic film of the present invention is produced on the substrate surface as follows.
- silane mixture a mixture containing the silane compound (a) and the silane compound (b) (hereinafter also referred to as “silane mixture”) and water are reacted on the substrate surface, an alkoxy group or a halogen atom bonded to a silicon atom. Is hydrolyzed to form a hydroxysilyl group.
- This hydroxyl group usually undergoes a condensation reaction (for example, a reaction for forming a siloxane bond) with other hydroxysilyl groups or alkoxysilyl groups by heating or the like.
- a layer mainly composed of silica also referred to as a cured product layer or a reactive silica layer
- crosslinking of the entire silane mixture is reacted on the substrate surface.
- the hydroxysilyl group formed from the silane mixture during the above reaction causes a condensation reaction or a strong interaction with a functional group such as a hydroxy group (OH group) on the substrate surface by heating or the like.
- a functional group such as a hydroxy group (OH group) on the substrate surface by heating or the like.
- the adhesion between the substrate and the cured product layer will be excellent.
- the hydrophilic film of the present invention is obtained by applying the hydrophilic compound (c) to the cured product layer obtained as described above, a group derived from the silane compound (a), and a group possessed by the hydrophilic compound (c) It is obtained by reacting.
- a film formed by curing the hydrophilic compound (c) alone tends to have insufficient hardness. Therefore, from the viewpoint of increasing the hardness of the hydrophilic film, it is also important to sufficiently increase the hardness of the cured product layer.
- it is difficult to use the silane compound (a) alone, and it is necessary to use a silane mixture containing the silane compound (a) and the silane compound (b). is there.
- the molar ratio of the compound (a) / the compound (b) contained in the silane mixture is preferably 1/2 or less, more preferably 1/3 or less, and even more preferably 1/4 or less.
- the hydrophilic compound (c) is sufficiently in close contact with the cured product layer obtained from the silane mixture from the viewpoint of improving hydrophilicity. Therefore, the acryloyl group contained in the hydrophilic compound (c) sufficiently reacts with the mercapto group, amino group, (meth) acryloyl group or vinyl group derived from the silane compound (a) contained in the cured product layer. Is desired. Therefore, the molar ratio of the compound (a) / the compound (b) contained in the silane mixture is preferably 1/200 or more, more preferably 1/100 or more, more preferably 1/50 or more, More preferably, it is 30 or more.
- the silane mixture containing the silane compound (a) and the silane compound (b) may further contain a solvent.
- the solvent include alcohols such as methanol, ethanol, IPA (isopropanol), n-butanol and methoxyethanol, aprotic polar solvents such as acetonitrile, DMF and DMSO, and ester systems such as ethyl acetate, butyl acetate and isoamyl acetate.
- examples thereof include solvents, ketone solvents such as acetone, methyl ethyl ketone, and methyl isobutyl ketone, and mixed solvents thereof.
- filler examples include wood powder, pulp, cotton chips, asbestos, glass fiber, carbon fiber, mica, walnut shell powder, rice husk powder, graphite, diatomaceous earth, white clay, silica, silica sol, nanosize silica, methyl Silica, surface-modified silica, fumed silica, precipitated silica, anhydrous silica, carbon black, calcium carbonate, magnesium carbonate, clay, talc, titanium oxide, cerium oxide, magnesium carbonate, quartz powder, aluminum fine powder, iron oxide , Flint powder, zinc powder and the like. These fillers may be used alone or in combination of two or more.
- the silane mixture may contain various additives as necessary.
- additives examples include UV absorbers, hindered amine light stabilizers (HALS), radical scavengers, antioxidants, polymerization inhibitors, anti-aging agents, ozone degradation inhibitors, metal deactivators, and storage stability.
- HALS hindered amine light stabilizers
- radical scavengers antioxidants
- polymerization inhibitors anti-aging agents
- ozone degradation inhibitors metal deactivators
- storage stability examples include improvers, pigments, dyes, binders, leveling agents, and curing aids such as alkoxy titanium.
- water is usually required for the hydrolysis reaction (hydroxysilyl group formation reaction) and the condensation reaction (siloxane bond formation reaction) to proceed. It is. Water may be derived from moisture in the air, but usually water is added to the silane mixture.
- the amount of water added is usually 0.1 to 100 equivalents, preferably 0.5 to 10 equivalents relative to the total moles of alkoxy groups or halogens bonded to the silicon atoms of the silane compound (a) and the silane compound (b). Equivalent, more preferably in the range of 1 to 5 equivalents.
- the alkoxy group or halogen atom bonded to the silicon atom is hydrolyzed to produce a hydroxysilyl group.
- the catalyst used for the hydrolysis examples include acids such as hydrochloric acid, sulfuric acid, phosphoric acid, citric acid and acetic acid, bases such as NaOH, KOH and ammonia, and inorganic salts such as KF and NaF.
- acids such as hydrochloric acid, sulfuric acid, phosphoric acid, citric acid and acetic acid
- bases such as NaOH, KOH and ammonia
- inorganic salts such as KF and NaF.
- the catalyst used for the hydrolysis also serves as a catalyst for the condensation reaction between the produced hydroxysilyl group and a reactive group on the substrate surface, such as a silanol group, and the condensation reaction between the produced hydroxy groups.
- the amount of these catalysts added is usually 0.1 to 100 wt%, preferably 0.3 to 50 wt%, more preferably 0.5 to 20 wt% with respect to the total weight of the silane compound (a) and the silane compound (b). % Range.
- the hydrolysis of the alkoxysilyl group and the halogenosilyl group usually proceeds at room temperature, but may be heated in some cases for the purpose of improving the hydrolysis rate. However, if it is heated to an excessively high temperature, for example, near 100 ° C., the heat curing reaction between the hydroxysilyl groups generated by hydrolysis proceeds, and the silane compound (a) and the silane compound (b) are mainly contained in a short time. The silane mixture becomes a gel and tends to cause a problem that it cannot be applied to the substrate.
- the hydrolysis temperature is usually in the range of 0 to 50 ° C., preferably in the range of 10 to 40 ° C., more preferably in the range of 20 to 30 ° C. It is.
- a layer (cured material layer) mainly composed of silica is formed by applying a mixture formed of the silane mixture and water to the substrate and further causing the condensation reaction to proceed.
- a mercapto group, amino group, (meth) acryloyl group or vinyl group derived from the silane compound (a) is present.
- the hydrophilic film of the present invention is obtained by applying the hydrophilic compound (c) to the cured product layer obtained as described above, and mercapto group, amino group, (meth) acryloyl group or vinyl derived from the silane compound (a). It is obtained by reacting a group with an acryloyl group possessed by the hydrophilic compound (c) by heat or radiation.
- hydrophilic compound (c) to the cured product layer may be performed by applying only the hydrophilic compound (c). However, since the hydrophilic compound (c) has a potassium sulfonate group, depending on the compound, the crystallinity is high and the operability may not be excellent.
- hydrophilic compound (c) when the hydrophilic compound (c) is applied to the cured product layer, a solution in which the hydrophilic compound is dissolved in a solvent (hereinafter also referred to as a hydrophilic compound solution (c ′)) is prepared, and this hydrophilic compound is prepared.
- the solution (c ′) may be applied to the cured product film and dried, and then reacted (grafted) by heat or radiation.
- Examples of the solvent used for the preparation of the hydrophilic compound solution (c ′) include alcohols such as methanol, ethanol, IPA (isopropanol), n-butanol and methoxyethanol; aprotic such as acetonitrile, DMF and DMSO. Polar solvents; water, and mixed solvents thereof.
- alcohols such as methanol, ethanol, IPA (isopropanol), n-butanol and methoxyethanol
- aprotic such as acetonitrile, DMF and DMSO.
- Polar solvents water, and mixed solvents thereof.
- solvents such as methanol, ethanol, IPA (isopropanol), n-butanol and methoxyethanol; water and mixed solvents thereof are preferable.
- the amount of these solvents used is appropriately determined depending on the type of solvent used.
- a catalyst, a polymerization initiator, etc. are included from the viewpoint of promoting the reactivity with the mercapto group derived from the silane compound (a) contained in the said hardened
- reaction (grafting) is performed by heating, for example, tertiary amines such as triethylamine, pyridine, N-methylmorpholine, triethylenediamine, alkali metal alkoxides such as sodium methoxide, t-butoxypotassium, sodium hydroxide, etc.
- basic catalysts such as inorganic bases such as potassium hydroxide and potassium carbonate.
- the addition amount of the catalyst and the polymerization initiator added to promote the reaction (grafting) with the reactive group derived from the silane compound (a) existing in the cured product layer is different from that of the hydrophilic compound (c). On the other hand, it is usually in the range of 0.1 to 100 wt%, preferably 0.3 to 30 wt%, more preferably 1 to 10 wt%.
- the hydrophilic compound solution (c ′) may further contain an additive.
- the above additives include polymerization accelerators, ultraviolet absorbers, infrared absorbers, hindered amine light stabilizers (HALS), radical scavengers, antioxidants, polymerization inhibitors, dyes, metal deactivators, and improved storage stability.
- the said additive can be added in the range which does not impair the reactivity (graft) with the hardened
- the hydrophilic compound (c) or the hydrophilic compound solution (c ′) By applying the hydrophilic compound (c) or the hydrophilic compound solution (c ′) to the cured product layer and carrying out the above reaction (grafting), the hydrophilic (c) compound is bonded to the cured product layer surface. Thus, the hydrophilic film of the present invention is formed.
- the hydrophilic film of the present invention is an organic substrate made of PMMA, polycarbonate (PC), PET, ABS, polyvinyl chloride, polyethylene (PE), polypropylene (PP), polylactic acid, etc., iron, stainless steel, aluminum, nickel, zinc , Various metals such as gold, silver, copper, etc., and products obtained by plating the surfaces of various substrates with those metals, and oxides of these metals, glass, ceramics, cement, slate, stone materials such as marble and granite, On the surface of an inorganic base material such as mortar (the surface in contact with the silica layer of the present invention is a base material formed of an inorganic base material typified by the above substance) By applying a solution containing the above-mentioned silane mixture, to which a hydrolysis catalyst is added, and curing by heating, the surface of the substrate is derived from the silane compound (a). That a mercapto group, an amino group, there are (meth) acryloy
- the hydrophilic compound (c) or the hydrophilic compound solution (c ′) is applied, dried, and reacted (grafted) with heat or radiation to obtain a cured product layer.
- the mercapto group, amino group, (meth) acryloyl group or vinyl group present on the surface is reacted with at least a part of the acryloyl group of the hydrophilic compound (c) to form a covalent bond.
- the surface of the reaction (graft) is washed away by washing with water to remove excess hydrophilic compound (c) and the like, and the transparent hydrophilic film of the present invention is obtained.
- a hydrophilic compound (c) or a hydrophilic compound solution (c ′) is applied on the cured layer formed mainly on silica, which is formed on the surface of the base material, and reacted (grafted).
- a laminate in which a hydrophilic film having excellent hydrophilicity derived from the component (c) is formed on the base material is obtained.
- the base material and the component derived from the hydrophilic compound (c) are firmly integrated through the cured product layer mainly composed of silica formed from the silane mixture.
- the reason for such strong integration is that the hydrosilylation and condensation of the silane mixture containing the silane compound (a) and the silane compound (b) on the base material allows the hydroxysilyl groups to adhere to each other or the surface of the base material.
- the substrate and the cured product layer are firmly integrated by covalent bond or strong interaction by reaction with a functional group that is supposed to remain in the substrate (for example, dehydration condensation crosslinking reaction by siloxane bond), and further curing It is presumed that the physical layer and the hydrophilic compound (c) are formed by a covalent bond formed by the above reaction (grafting) and firmly integrated.
- inorganic base materials are relatively preferable in terms of easy adhesion.
- an inorganic substrate whose surface is formed from glass, metal, metal plating, metal oxide, ceramics, etc. is preferable, and an inorganic substrate whose surface is formed from glass, metal, metal oxide, ceramics, etc.
- a base material is more preferable, and an inorganic base material whose surface is formed from glass is more preferable.
- the substrate may be surface-treated in order to enhance adhesion.
- the surface treatment for example, corona treatment, flame treatment, plasma treatment, ozone treatment, low temperature plasma treatment using oxygen gas or nitrogen gas, physical treatment such as glow discharge treatment, oxidation treatment using an oxidizing agent, acid or Examples thereof include chemical treatment such as etching with alkali.
- a method for easily measuring the degree of contamination on the surface of a substrate or the like is, for example, measurement of a water contact angle.
- contaminants adhering to the substrate surface include sebum, oils and fats, silicone, plasticizers that have bleed out from packaging, etc., and external air hydrophobic substances that scatter from the exhaust gas and float in the atmosphere. Substances often exhibit hydrophobic properties.
- the surface of the contaminated inorganic base material is often different from the inherent hydrophilicity of the base material.
- the “silane solution” of the present invention is applied to the contaminated inorganic substrate surface, cissing is likely to occur, and a uniform reactive silica layer (cured film layer) cannot be formed.
- the adhesiveness at the interface with the silica layer tends to decrease.
- the reactive silica layer is formed on the adhered contaminants, the adhesive force at the interface between the substrate surface and the reactive silica layer is further reduced.
- inorganic base materials are easily contaminated, and among the inorganic base materials, in particular, contaminants easily adhere to the surface of glass. For example, it is easily contaminated with silicone from a packing material and a scratch-proof sheet, and a hydrophobic substance floating in the air. Originally, the hydrophilicity of glass is very high, and it is considered that the water contact angle is 5 ° (Surface Science vol.22, pp. 55-63, 2001), but the water contact angle of the purchased glass surface is 50-70 °. Yes, it is very polluted.
- the hydrophilic film of the present invention is laminated, it is difficult to ensure sufficient adhesion when the purchased glass is used as it is, and even when ultrasonically washed in water (water contact 25 to 35). °) Sufficient adhesion tends to be difficult to obtain.
- the water contact angle on the glass surface is preferably at least 15 ° or less, more preferably 10 ° or less.
- the water contact angle of the original surface of the substrate is ⁇ 10 °.
- the range tends to be preferable, and the range of ⁇ 5 ° tends to be more preferable.
- Examples of the method for applying the solution containing the silane mixture used in the present invention to the substrate surface and the method for applying the hydrophilic compound (c) or the hydrophilic compound solution (c ′) to the cured product layer include, for example, a brush.
- Examples thereof include a coating method, a spray coating method, a wire bar method, a bar coater method, a blade method, a roll coating method, a spin coating method, a dipping method, and other known coating methods.
- the solution containing the silane mixture to be applied to the substrate is usually a hydroxyl group obtained by hydrolyzing at least part of the alkoxy group and halogen group bonded to the silicon atom derived from the silane compound (a) and the silane compound (b).
- the compound converted into is contained, and by heating this, dehydration condensation between hydroxy groups bonded to silicon atoms, dehydration of hydroxy groups on the surface of a substrate such as glass and hydroxy groups bonded to silicon atoms It is cured by a reaction such as condensation, hydrolysis of the alkoxy group bonded to the remaining silicon atoms by the generated water, or hydrolysis of the halogen group, further dehydration condensation, and the layer (both the cured product layer and the reactive silica layer). Say.) Is formed.
- the above heating may also serve as drying for the purpose of evaporating the solvent, and is usually heated in the temperature range of room temperature to 200 ° C. for 0.01 to 240 hours.
- the heating temperature is preferably higher to some extent. In this case, it is usually in the range of 1 to 480 minutes at 50 to 180 ° C., preferably in the range of 5 to 240 minutes at 80 to 150 ° C., more preferably. Is in the range of 10 to 180 minutes at 100 to 130 ° C. or in the range of 5 to 120 minutes at 140 to 180 ° C. If the heating temperature is too high, cooling takes time, and productivity may be reduced.
- the atmosphere during heating may be performed in the air or in an inert gas atmosphere such as nitrogen, but the air is simple and preferable.
- the pressure at that time may be performed under atmospheric pressure or reduced pressure, but atmospheric pressure is simple and preferable.
- the thickness of the cured product layer mainly composed of silica thus laminated on the surface of the substrate is appropriately determined according to the purpose and the substrate, but is usually in the range of 0.001 to 20 ⁇ m, preferably 0.8. It is in the range of 005 to 10 ⁇ m, more preferably in the range of 0.01 to 5 ⁇ m.
- a hydrophilic compound (c) or a hydrophilic compound solution (c ′) (graft solution) is applied onto the obtained cured product layer and reacted (graft reaction), whereby the hydrophilic film is based on the present invention. It can be formed on a material.
- This grafting reaction is usually carried out by heat or radiation, as in the reaction for forming a cured product layer.
- hydrophilic compound (c) a compound having a potassium sulfonate group (hydrophilic compound (c1)).
- hydrophilic compound (c2) a compound having a sulfonic acid metal base other than a potassium sulfonate group or a sulfonic acid (hydrophilic compound (c2)), it is preferable that the grafting proceeds in the following manner.
- the graft is heated or irradiated as described above. It is preferable to proceed by irradiation or the like, and both heating and radiation irradiation may be used in combination for proceeding with this reaction.
- reaction (grafting) by heat may also serve as evaporation of the solvent contained in the hydrophilic compound solution (c ′).
- the above heating is usually performed in the temperature range of normal temperature to 200 ° C. for 0.01 to 240 hours.
- the heating temperature is preferably higher to some extent. In this case, it is usually in the range of 1 to 480 minutes at 50 to 180 ° C., preferably in the range of 5 to 240 minutes at 80 to 150 ° C. The range is preferably 100 to 130 ° C. and 10 to 180 minutes. If the heating temperature is too high, cooling takes time, and productivity may be reduced.
- the atmosphere during heating is usually performed in a simple atmosphere. However, when it is desired to strictly prevent the reduction in the reaction rate between the mercapto group and the acrylic group of the hydrophilic compound (c) due to the oxidation of the mercapto group derived from the compound (a) (—SH ⁇ —S ⁇ S—). It is preferable to carry out in an inert gas atmosphere.
- the pressure during the reaction (grafting) may be performed under pressure, atmospheric pressure, or reduced pressure, but atmospheric pressure is simple and preferable.
- the hydrophilic compound (c) When the hydrophilic compound (c) is reacted with radiation, it can be carried out in the atmosphere or in an inert gas atmosphere such as nitrogen. If it is carried out in an inert gas atmosphere, the reaction (grafting) time can be shortened or the amount of radiation irradiation energy can be reduced, although it becomes complicated and the apparatus becomes complicated.
- Examples of the radiation include energy rays having a wavelength range of 0.0001 to 800 nm.
- ⁇ rays, ⁇ rays, ⁇ rays, X rays, electron rays, ultraviolet rays, visible light, and the like can be used.
- ultraviolet rays having a wavelength region of 200 to 450 nm are preferable, ultraviolet rays having a wavelength region of 370 to 445 nm are more preferable, ultraviolet rays having a wavelength region of 370 to 430 nm are still more preferable, and wavelength region is 370.
- Ultraviolet rays in the range of ⁇ 400 nm are particularly preferred. When ultraviolet rays having a wavelength in the above range are used, there are few problems such as thermal deformation during reaction (grafting), and the reaction (grafting) can be completed in a relatively short time even when an ultraviolet absorber is added.
- the graft reaction proceeds by heating. It is preferable. This is because when the graft reaction is carried out by radiation alone without using heat, the mercapto group, amino group, (meth) acryloyl group or vinyl group derived from the compound (a), and the hydrophilic compound (c) This is because the reaction rate with the acrylic group is low and sufficient hydrophilicity may not be imparted to the hydrophilic film.
- the temperature of the heat is preferably 130 ° C. or higher, more preferably 130 to 190 ° C., and still more preferably 140 to 180 ° C. in order to promote the reaction efficiently and reduce unreacted substances. If the heat temperature is less than 130 ° C., sufficient grafting reaction does not proceed and a hydrophilic film having sufficient hydrophilicity may not be formed.
- the heating time is usually in the range of 0.01 to 240 hours.
- the silane compound (b) is represented by the formula (b1 ) Or a silane compound represented by formula (b2), and the silane compound represented by formula (b1) is slightly more preferable than the silane compound represented by (b2).
- silane compounds represented by the above formula (b1) more preferable is a silane compound having 12 or less total carbon atoms in one molecule, such as tetramethoxy-silane, tetraethoxy-silane, Examples include hydroxy-triethoxysilane, dihydroxy-diethoxy-silane, trihydroxy-ethoxy-silane, tetrahydroxy-silane, tetrachloro-silane, tetra-n-propoxy-silane, tetra-isopropoxy-silane and the like.
- silane compounds represented by the formula (b2) more preferred are silane compounds having 1 to 20 total carbon atoms in one molecule, such as bis (trimethoxysilyl) methane, Bis (triethoxysilyl) methane, bis (trihydroxysilyl) methane, 1,2-bis (trimethoxysilyl) ethane, 1,2-bis (triethoxysilyl) ethane, 1,2-bis (tri-n- Propoxysilyl) ethane, bis (triisopropoxysilyl) ethane, 1,2-bis (trichloro) ethane, 1- (hydroxy-diethoxy-silyl) -2- (triethoxy-silyl) ethane, 1,2-bis (hydroxy) -Diethoxy-silyl) ethane, 1- (dihydroxy-ethoxy-silyl) -2- (hydroxy-diethoxy-silyl) ethane, 1,2 Bis (dihydroxy-ethoxy-silyl
- the thickness of the film formed from the component derived from the hydrophilic compound (c) is expected to be mostly monomolecularly bonded to the cured product layer on the surface of the cured product layer.
- a small number is estimated to be in the range of 0.001 to 0.5 ⁇ m because the polymer of the hydrophilic compound (c) is expected to be bonded to the cured product layer.
- the hydrophilic compound (c) is considered to have a lower scratch resistance when the chain length of the polymer derived from the hydrophilic compound (c) formed by the reaction (grafting) is longer.
- the thickness of the film formed from the components derived from is estimated to be preferably in the range of 0.001 to 0.1 ⁇ m, more preferably in the range of 0.001 to 0.01 ⁇ m.
- the laminate obtained by the present invention may be a film, a sheet, or a molded body. Since the laminate obtained by the present invention has high hydrophilicity and scratch resistance, it can be suitably used as an antifogging material, an antifouling (self-cleaning) material, an antistatic material, a quick drying material or the like. For example, it is used as a covering used for outer walls, exteriors, inner walls, interiors, floors, etc. of buildings, ships, aircrafts, and vehicles.
- the laminate obtained by the present invention includes clothing materials such as clothes, fabrics, and fibers; optical articles such as optical films, optical disks, glasses, contact lenses, and goggles; displays such as flat panels and touch panels, and the like Display material; Glass substrate of solar cell or protective transparent plate of outermost layer of solar cell; Lighting article such as lamp and light and its lighting member; Cooling fin such as heat exchanger, cosmetic container and its container material, reflecting film, reflecting plate Reflective materials such as sound insulation boards installed on expressways, window glass, mirrors, furniture, furniture materials, bathroom materials, kitchen materials, ventilation fans, piping, wiring, electrical appliances, electrical parts, etc. Used.
- the surface is subjected to corona treatment, flame treatment, plasma treatment, ozone treatment, low temperature plasma treatment using oxygen gas or nitrogen gas, physical treatment such as glow discharge treatment, oxidation, etc.
- Surface treatment by chemical treatment such as oxidation treatment with an agent, etching treatment with acid or alkali, etc., laminating inorganic materials such as ITO and silica, etc., hydrophilic compound as other component (c) or the like
- An organic material such as a resin may be laminated by further coating a “graft solution” containing
- the water contact angle was measured at room temperature (25 ° C.) using a CA-V model manufactured by Kyowa Interface Science Co., Ltd.
- Example 1 (Washing substrate) A glass plate having a thickness of 2 mm (surface water contact angle 57 to 70 °) was used as a substrate. The glass plate used as a base material was first cleaned by the following procedure.
- silane solution (1) Preparation of solution containing silane mixture (Silane solution (1))
- silane compound (a) 3-mercaptopropyl-trimethoxysilane (hereinafter abbreviated as MPMOS; molecular weight, 196.3) 3.3 g (17 mmol)
- silane compound (b) tetramethoxysilane (hereinafter referred to as TMOS).
- a uniform liquid (silane mixture) was prepared by mixing 12.7 g (83 mmol) of molecular weight 152.2) and 400 ml of ethanol (specific gravity 0.79).
- the molar ratio of MPMOS / TMOS in the silane solution (1) is 1/5
- the amount of sulfuric acid is 3 wt% (the total amount of MPMOS and TMOS)
- the amount of water is 1.8 times mol (with respect to silicon atoms).
- hydrophilic compound solution (1) As the hydrophilic compound (c), 2.5 g (10.8 mmol) of 3-sulfopropyl acrylate / potassium salt (hereinafter abbreviated as SPA-K, molecular weight 232.3), and 130 ml of 2-methoxyethanol (specific gravity 0.96). ) Were first mixed to prepare a mixed solution. 0.75 g (7.4 mmol, 69 mol% vs. SPA-K, 31 wt% vs. SPA-K) of triethylamine (hereinafter abbreviated as TEA, molecular weight 101.2) as a catalyst was added to this mixed solution and mixed and dissolved well. 128 g of a solution having a solid content concentration of 2 wt% (hydrophilic compound solution (1)) was obtained.
- TEA triethylamine
- a solution (silane solution 1) containing a silane mixture was applied to the glass plate surface (water contact angle 5 to 6 °) subjected to the above-described cleaning with a bar coater # 10, pre-dried at 50 ° C. for 5 minutes, and then 120 ° C. And then dried by heating for 1 hour.
- a cured product layer mainly composed of silica in which a mercapto group derived from the silane compound (a) was present was formed.
- the thickness of the cured product layer was about 0.5 ⁇ m, and the water contact angle on the surface of the cured product layer was 46 °.
- the hydrophilic compound solution (1) was applied to the surface of the cured product layer with a bar coater # 10, preliminarily dried at 50 ° C. for 5 minutes, and then heated and dried at 120 ° C. for 1 hour. By this operation, SPA-K reacted (grafted) to the mercapto group present in the cured product layer. Thereafter, the mixture was cooled to room temperature, and SPA-K that was not involved in the reaction (grafting) was washed away with water, and further dried using an air gun. The obtained film had excellent hydrophilicity, was transparent, had no tackiness, and was firmly adhered to the glass plate. The film evaluation results are shown in Table 2.
- Example 2 As described in Table 2, a film was prepared on a glass plate and tested in the same manner as in Example 1 except that the molar ratio of MPMOS / TMOS in the silane solution (1) was changed. The film evaluation results are shown in Table 2.
- the glass plate on which the cured product layer was laminated was slightly clouded before washing with acetone, but returned to transparency by washing with acetone, and no significant changes were observed in the appearance and antireflection properties of the glass plate.
- the thickness of the cured product layer was about 0.5 ⁇ m
- the water contact angle on the surface of the cured product layer was 58 °
- formation of the cured product layer by MPMOS was confirmed.
- the film formed on the obtained glass plate was tested in the same manner as in Example 1. As a result, no major change was observed in the appearance of the film surface, the water contact angle on the surface was 16 °, and the film surface was hydrophilic. It was confirmed that the functional antoxMS-2N was immobilized on the glass plate surface.
- the evaluation results are shown in Table 4.
- Example 8 Comparative Examples 3 to 4
- Table 5 the type of the hydrophilic compound (c) to be grafted onto the cured product layer surface was changed, and the hydrophilic compound (c) was further subjected to the same thermal grafting conditions as in Example 1 or Comparative Example 3.
- a film was formed on a glass plate in the same manner as in Example 1 except that the cured product layer surface was grafted under the UV grafting conditions.
- each of the films obtained by thermal grafting or UV grafting was tested in the same manner as in Example 1.
- the film evaluation results are shown in Table 5.
- scratchability 1, scratchability 2 and adhesion tests the same results were shown for the film obtained by thermal grafting and the film obtained by UV grafting.
- Example 9 As described in Table 6, a film was produced on a glass plate and tested in the same manner as in Example 1 except that the type of the hydrophilic compound (c) to be grafted on the cured product layer surface was changed. The results are shown in Table 6.
- Examples 12 to 19 As described in Tables 7 and 8, except that the curing conditions of the cured film layer or the thermal grafting conditions of the hydrophilic compound (c) were changed, a film was produced on the glass plate in the same manner as in Example 1, Tested. The results are shown in Tables 7 and 8.
- Example 24 (Preparation of silane solution (3)) Oligomeric silane compound obtained by reacting (condensation polymerization) MPMOS and tetraethoxysilane in a molar ratio of 1: 4.7 as a silane compound (a) and (b) in a conical flask equipped with a stir bar
- Product name X-41-1805 (Shin-Etsu Chemical Co., Ltd., mercapto equivalent 862 g / mol) 1.08 g (1.25 mmol) and silane compound (b) component TEOS (tetraethoxysilane) 2.60 g ( 12.5 mmol) was weighed and 93.5 g of ethanol was added and stirred to prepare a uniform solution. Note that the blending molar ratio of X-41-1805 and TEOS (X-41-1805 / TEOS) is 1/10.
- Example 2 2.9 g of 5 wt% sulfuric acid was added to the prepared uniform liquid and hydrolyzed by stirring at room temperature for 10 minutes to obtain a uniform silane solution (3) having a solid content of 4 wt%.
- the silane solution (3) was used to form a cured film layer in which mercapto groups derived from X-41-1805 remained on the cleaned glass plate surface.
- the hydrophilic compound solution (1) prepared in Example 1 was applied again with a bar coater # 2, similarly, preliminarily dried at 50 ° C. for 5 minutes, and then at 150 ° C. for 1 hour.
- Examples 28 to 30 As described in Table 10, a film was produced on a glass plate and tested in the same manner as in Example 24 except that the cured film layer was formed by changing the curing conditions of the cured film layer. The results are shown in Table 10.
- Example 31 A time-of-flight secondary ion mass spectrometer for the ionic strength of each surface of a cured film layer obtained in the same manner as in Example 20 and a film obtained by grafting a hydrophilic compound to the cured film layer. Using (TOF-SIMS), analysis was performed under the following conditions to investigate the functional group concentration on each surface. The results are shown in Table 10.
- Examples 34 to 37, Comparative Examples 6 to 12 As described in Table 12, a film was prepared on a glass plate and tested in the same manner as in Example 24 except that the type of the hydrophilic compound (c) to be grafted onto the cured product layer surface was changed. The results are shown in Table 12.
- silane solution (4) 11.0 g of 5 wt% aqueous ammonia was added to the prepared uniform liquid and stirred at room temperature for 10 minutes to obtain a uniform silane solution (4) having a solid content of 3 wt%.
- the silane solution (4) was applied to the surface of a glass plate (water contact angle 5-6 °) washed in the same manner as in Example 1 with a bar coater # 12, and after preliminary drying at 50 ° C. for 5 minutes, 70 Heat-drying was carried out at 2 ° C. for 2 hours to form a cured film layer in which amino groups derived from AAMOS remained on the glass surface.
- the thickness of this silica layer was about 0.4 ⁇ m.
- Example 2 On the surface of the obtained cured film layer, the hydrophilic compound solution (1) prepared in Example 1 was grafted with SPA-K to the amino group on the surface of the cured film layer under the same conditions as in Example 1. A film was formed. Table 13 shows the evaluation results of the obtained film.
- Example 39 (Preparation of silane solution (5)) A silane solution (5) having a solid content of 4 wt% was prepared in the same manner as in Example 1 except that a compound having an acryloyl group (AOMOS) was used as the silane compound (a) instead of MPMOS. (Formation of cured film layer) In the same manner as in Example 1, the silane solution (3) was applied to the cleaned glass plate surface (water contact angle 5 to 6 °) with a bar coater # 10, preliminarily dried at 50 ° C. for 5 minutes, and then 120 Heating and drying at 1 ° C.
- AOMOS acryloyl group
- hydrophilic compound solution (2) SPA-K 2.0 g (8.61 mol) in a light-shielded sample bottle, 2.0 g (100 wt% vs. SPA-K) Darocur 1173 (Ciba Specialty Chemicals) as a UV polymerization initiator, and 2-methoxy as a solvent 100 g of ethanol was added and mixed to prepare a uniform hydrophilic compound solution (2) having a hydrophilic monomer concentration of 2 wt%.
- the hydrophilic compound solution (2) is applied to the surface of the reactive silica layer obtained above with a bar coater # 2, dried in an oven at 50 ° C. for 5 minutes, and then 30 with a metal halide lamp (illuminance 130 mW / cm 2 ). After irradiating for 2 seconds (3900 mJ / cm 2 ), carrying out a grafting reaction and washing with water, SPA-K that was not involved in the reaction with acryloyl groups (grafting) was washed away, and then dried with an air gun. A film was formed. Table 14 shows the evaluation results of the obtained film.
- Examples 40 to 43, Comparative Examples 13 to 14 As described in Table 13, a film was formed on a glass plate in the same manner as in Example 38 except that the type of compound (a) and the compounding molar ratio of compound (a) component and compound (b) component were changed. Were made and tested. The results are shown in Table 14.
- Example 44 As described in Table 15, as the compound (b), instead of 2.6 g (12.5 mmol) of TEOS, TMOSI (N, N ′, N ′′ -tris (3-trimethoxysilyl-propyl) isocyanate Example 1 except that a silane solution containing 2.6 g (4.22 mol) was prepared and the compounding molar ratio of X-41-1805 and TEOS (X-41-1805 / TEOS) was 1/5 Films were prepared and tested on glass plates as in 24. The results are listed in Table 15.
- Example 45 As described in Table 16, except that the substrate was changed from a glass plate to a stainless plate etched with 2 wt% sulfuric acid (immersion for 10 minutes at room temperature) after being cleaned by the same cleaning method as in Example 1. In the same manner as in Example 24, a film was produced on a glass plate and tested. The results are listed in Table 16.
- Example 46 to 47 As described in Table 16, the thickness of the cured film layer is changed, and the base material is changed from a glass plate to a PC (polycarbonate) plate subjected to surface modification treatment (Japanese Patent No. 3557194) under the following conditions. At the same time, a membrane was prepared and tested in the same manner as in Example 24 except that the heating grafting conditions were 120 ° C. ⁇ 3 hours after preliminary drying at 50 ° C. ⁇ 5 minutes. The results are listed in Table 16.
- ⁇ Itro treatment (surface treatment) conditions The combustible gas prepared by continuously mixing the following itrogas and city gas was ignited, and the substrate surface was treated by passing the substrate twice through the generated flame at a linear velocity of 0.7 m / sec. .
- Example 48 As described in Table 17, films were prepared on glass plates in the same manner as in Examples 27 and 24. After the obtained film was treated under the following immersion conditions or weather resistance conditions, the same evaluation as in Example 24 was performed. The results are shown in Table 17. (Immersion conditions and weather resistance conditions) Water immersion condition 1: The obtained film was immersed in boiling water (100 ° C.) for 1 hour. -Water immersion condition 2: The obtained film was immersed in warm water (60 ° C) for 60 days. Chemical soaking condition 1: The obtained film was immersed in a mold killer (trade name: Johnson Co., Ltd., including hypochlorite, sodium hydroxide, surfactant (alkylamine oxide), etc.) for 7 days.
- a mold killer trade name: Johnson Co., Ltd., including hypochlorite, sodium hydroxide, surfactant (alkylamine oxide), etc.
- Chemical soaking condition 2 The obtained film was soaked for 7 days in Kitchen Hiter (trade name: Kao Corporation, including sodium hypochlorite, cleaning components, etc.). And weather conditions: The resulting film, the back panel temperature (BPT) 63, in an atmosphere of rainfall 18 minutes / 2 hours, 60 W / m 2 using a xenon arc lamp, the light of wavelength 300 ⁇ 400 nm, 1000 Time exposure.
- Kitchen Hiter trade name: Kao Corporation, including sodium hypochlorite, cleaning components, etc.
- weather conditions The resulting film, the back panel temperature (BPT) 63, in an atmosphere of rainfall 18 minutes / 2 hours, 60 W / m 2 using a xenon arc lamp, the light of wavelength 300 ⁇ 400 nm, 1000 Time exposure.
- Example 49 As described in Table 17, a film was produced on a glass plate in the same manner as in Example 24 and tested in the same manner as in Example 47. The results are shown in Table 17.
- Example 50 As described in Table 17, a film was produced on a glass plate in the same manner as in Example 24, except that zirconium octylate was added to the silane mixture so as to be 1.0% by weight as an additive, Tested as in Example 47. The results are shown in Table 17.
- Example 51 A membrane (hydrophilic compound (c): 3-sulfopropyl acrylate (SPA)) produced on a glass plate in the same manner as in Example 34 was placed in the laboratory (temperature 25 ° C. ⁇ 3 ° C., humidity 35% ⁇ 10%). Left for 10 days to 4 months. After elapse of a predetermined standing period, the membrane was washed with running water using [Bencot M-3] (Asahi Kasei) and dried using an air gun. Thereafter, the water contact angle of the film after each standing period was measured. The results are shown in Table 18.
- Example 15 In the same manner as in Example 1, the washed glass plate (the glass plate on which no film was formed) was left in the same manner as in Example 51, and then the water contact angle of the glass plate after each leaving period passed. was measured. The results are shown in Table 18.
- Example 52 (Preparation of silane solution (6))
- X-41 to 1805 shown in Example 24 as the silane compounds (a) and (b)
- silane compound (b) TEOS 13.0 g (62.5 mmol) was weighed, and 152.0 g of methanol was added and stirred to prepare a uniform solution.
- the compounding molar ratio of X-41-1805 (Shin-Etsu Chemical Co., Ltd., mercapto equivalent 862 g / mol) and TEOS (X-41-1805 / TEOS) is 1/50.
- Example 53 (Preparation of silane solution (7))
- silane compounds (a) and (b) in Example 52, X-41-1805 (1.08 g, 1.25 mmol), and silane compound (b), 13.0 g (62.5 mmol) of TEOS was weighed, and 7.0 g (29.6 mmol) of 3-glycidoxypropyltrimethoxysilane (hereinafter, GPMOS molecular weight: 236.3) was further added as the silane compound (b). Weighed, added 228.0 g of methanol and stirred to prepare a uniform solution.
- the blending molar ratio of X-41 to 1805, TEOS and GPMOS is 1/50 / 23.7.
- Example 54 (Preparation of silane solution (8)) In an Erlenmeyer flask with a tight stopper equipped with a stirrer, 3.00 g (15.3 mmol) of MPMOS shown in Example 1 as a silane compound (a) component and 9.6 g of TEOS as a silane compound (b) component (45) .8 mmol) was weighed, 441.0 g of methanol was added and stirred to prepare a uniform solution. The molar ratio of MPMOS to TEOS (MPMOS / TEOS) is 1/3.
- the thickness of the cured product layer was about 0.4 ⁇ m.
- (Grafting) A film was produced on the surface of the obtained cured film layer in the same manner as in Example 52, and then tested in the same manner as in Example 52. The evaluation results obtained are shown in Table 20.
- [Example 55] (Formation of cured product layer) As shown in Table 20, the mixing molar ratio of MPMOS and TEOS in the silane solution (8) and the mixing weight ratio of MPMOS, TEOS and silica were not changed, and the solid content concentration was changed to 10 wt%.
- a cured product layer was formed in the same manner as in Example 54 except that the silane solution was applied on a glass plate with a bar coater # 30.
- the thickness of the cured product layer was about 3 ⁇ m. (Grafting) A film was produced on the surface of the obtained cured film layer in the same manner as in Example 52, and then tested in the same manner as in Example 52. The evaluation results obtained are shown in Table 20.
Landscapes
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Organic Chemistry (AREA)
- Wood Science & Technology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Geochemistry & Mineralogy (AREA)
- Laminated Bodies (AREA)
- Coating Of Shaped Articles Made Of Macromolecular Substances (AREA)
- Paints Or Removers (AREA)
Abstract
Description
下記一般式(c)で表される化合物に含まれる(メタ)アクリロイル基と、上記化合物(a)に由来するメルカプト基、アミノ基、(メタ)アクリロイル基、およびビニル基からなる群より選ばれる基との少なくとも一部を反応させて得られることを特徴とする。
上記親水膜において、上記化合物(a)/上記化合物(b)のモル比が、1/2~1/200であることが好ましく、1/3~1/100であればより好ましい。
また、親水膜を有する積層体の製造方法において、上記化合物(c)のMが、H、Li、Na、Rb、Mg、Ca、Sr、またはBaであり、かつ、上記グラフト反応を、130℃以上の温度条件下で進行させることが好ましい。
上記層を形成する混合物には、(a)1個のシリコン原子と、メルカプト基、アミノ基、(メタ)アクリロイル基およびビニル基からなる群より選ばれる基と、アルコキシ基、ハロゲン基、およびヒドロキシ基からなる群より選ばれる少なくとも1個のシリコン原子に結合した基とを有するシラン化合物(以下、シラン化合物(a)ともいう。)が含まれる点に特徴がある。
これら基の中でも、コスト等の面からクロロ基が好ましい。
上記シラン化合物(a)は、芳香族基、脂肪族基などさらに他の基を含んでいてもよい。

e、e1、f、およびf1は、独立して、0~10の整数を表し、これらの中でも0~5の整数が比較的に好ましく、0~3の整数であればより好ましい傾向にある。
上記層を形成する混合物には、上記シラン化合物(a)に加えて、(b)アルコキシ基、ハロゲン基、およびヒドロキシ基からなる群より選ばれる、少なくとも4個のシリコン原子に結合した基を有し、かつ、炭素-炭素二重結合との反応性を有する基を有さないシラン化合物(以下、シラン化合物(b)ともいう。)がさらに含まれる点に特徴がある。
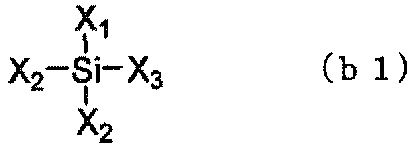

シラン化合物(a)およびシラン化合物(b)を含む混合物から得られる硬化物層の架橋密度を向上させ、より強固な膜を形成させる点からは、c1=c2=c3=3、d1=d2=d3=0が好ましい。
本発明では、シラン化合物(a)とシラン化合物(b)とを含む混合物を加水分解、縮合させて得られる、シリカを主体とする硬化物層の表面に、(c)下記一般式(c)で表される化合物(以下、親水性化合物(c)ともいう。)を塗布し、親水性化合物(c)に含まれる炭素-炭素二重結合と、上記化合物(a)に由来するメルカプト基との少なくとも一部を反応させる点に特徴がある。

本発明の親水膜は、以下のようにして、基材表面に製造される。
本発明の親水膜は、PMMA、ポリカーボネート(PC)、PET、ABS、塩ビ、ポリエチレン(PE)、ポリプロピレン(PP)、およびポリ乳酸などからなる有機基材、鉄、ステンレススチール、アルミニウム、ニッケル、亜鉛、金、銀、銅などの各種金属、及び各種基材の表面をそれらの金属でメッキ処理された物、並びにそれら金属の酸化物、ガラス、セラミックス、セメント、スレート、大理石や御影石などの石材、モルタルなどの無機基材(本発明のシリカ層と接する表面が、上記物質に代表される無機基材により形成されている基材をいう。)などの表面に、まず、必要に応じて、水、加水分解触媒を加えた、上述のシラン混合物を含む溶液を塗布し、加熱硬化することによって、基材表面上に、シラン化合物(a)に由来するメルカプト基、アミノ基、(メタ)アクリロイル基またはビニル基が存在する、硬化物層を形成する。
これら無機基材の中でも、ガラス、金属、金属メッキ、金属酸化物、セラミックスなどからその表面が形成された無機基材が好ましく、ガラス、金属、金属酸化物、セラミックスからその表面が形成された無機基材がより好ましく、ガラスからその表面が形成された無機基材がさらに好ましい。
水接触角の測定は、協和界面科学社製CA-V型を用いて、室温(25℃)にて測定した。
スチールウール#0000を用い、0.1Kgfの荷重をかけて、10往復擦った。評価は、傷の数を目視で数え、傷がなかった場合を○、傷が10本以内だった場合を△、10本を越えた場合を×とした。
スチールウール#0000を用い、2Kgfの荷重をかけて、10往復擦った。評価は、傷の数を目視で数え、傷がなかった場合を○、傷が10本以内だった場合を△、10本を越えた場合を×とした。
カッターナイフとカッターガイドとを用いて、幅3mm間隔で、碁盤目状に切り込みをいれた(合計25マス)。次に、碁盤目の上に、空気が入らないようにセロハン粘着テープ(ニチバン(株)、セロテープ(登録商標)、幅24mm)を貼り付け、手で擦って、粘着テープを完全に付着させた。最後に、接着面に対して垂直方向に、瞬間的に引き剥がした。評価は、剥れなかったマスの数を、目視で数え、100マスに換算して表記した。
(基材の洗浄)
厚さ2mmのガラス板(表面水接触角57~70°)を基材として用いた。以下の手順で、まず基材となるガラス板の洗浄を行った。
シラン化合物(a)として、3-メルカプトプロピル-トリメトキシシラン(以下、MPMOSと略す。分子量、196.3)3.3g(17mmol)、シラン化合物(b)として、テトラメトキシシラン(以下、TMOSと略す。分子量152.2)12.7g(83mmol)、およびエタノール400ml(比重0.79)を混合して均一液(シラン混合物)を作製した。この均一液に、5wt%硫酸水10.4g(硫酸5mmol、水548mmol)を加えて、室温で10分間攪拌し、固形分濃度5wt%の均一なシラン混合物を含む溶液(シラン溶液(1))を342.4g得た。ここで、シラン溶液(1)中のMPMOS/TMOSのモル比は1/5、であり、硫酸量は3wt%(対MPMOSとTMOSの合計量)、水量は1.8倍モル(対シリコン原子に結合したアルコキシ基)であった。
親水性化合物(c)として、3-スルホプロピルアクリレート・カリウム塩(以下、SPA-Kと略す。分子量232.3)2.5g(10.8mmol)、および2-メトキシエタノール130ml(比重0.96)を混合してまず混合液を調製した。この混合液に、触媒としてトリエチルアミン(以下、TEAと略す。分子量101.2)0.75g(7.4mmol、69mol%対SPA-K、31wt%対SPA-K)を加えて、よく混合溶解し、固形分濃度2wt%の溶液(親水性化合物溶液(1))を128g得た。
上述の洗浄を経たガラス板表面(水接触角5~6°)に、シラン混合物を含む溶液(シラン溶液1)をバーコーター#10で塗布し、50℃で5分間予備乾燥した後、120℃で1時間加熱乾燥を行った。この操作により、シラン化合物(a)に由来するメルカプト基が存在するシリカを主体とする硬化物層が形成された。この硬化物層の厚みは約0.5μmであり、この硬化物層表面の水接触角は46°であった。
上記硬化物層表面に、親水性化合物溶液(1)をバーコーター#10で塗布し、50℃で5分間予備乾燥した後、120℃で1時間加熱乾燥を行った。この操作により、硬化物層に存在するメルカプト基にSPA-Kが反応(グラフト)した。その後、室温まで冷却して、反応(グラフト)に関与しなかったSPA-Kを水洗により洗い流し、さらにエアーガンを用いて乾燥した。得られた膜は、親水性に優れ、透明で、タック性もなく、ガラス板に強固に密着していた。膜の評価結果を表2に示す。
表2に記載されるように、シラン溶液(1)中のMPMOS/TMOSの配合モル比を変更したこと以外は、実施例1と同様にガラス板上に膜を作製し試験した。膜の評価結果を表2に示す。
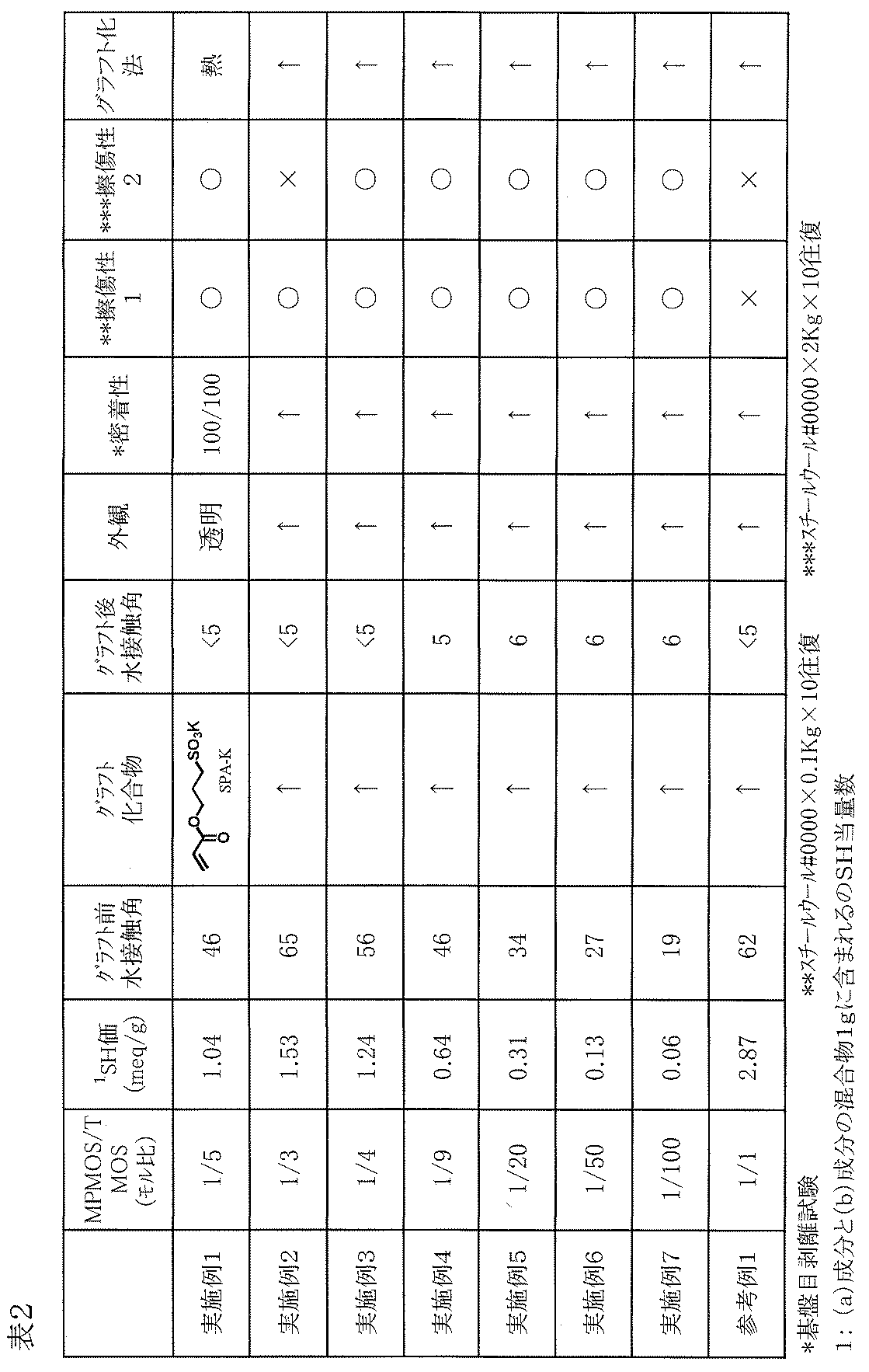
表3に記載されるように、シラン溶液(1)中のMPMOS/TMOSの配合モル比を変更し、さらに硬化物層表面にグラフトさせる親水性化合物(c)の種類を変更したこと以外は、実施例1と同様にガラス板上に膜を作製し試験した。膜の評価結果を表3に示す。

(硬化物層の形成)
実施例1と同様に、ガラス板を洗浄し、洗浄後のガラス板の水接触角を5~6°とさせた。このガラス板に、0.5wt%のMPMOS(3-メルカプトプロピルトリメトキシシラン)を含むイソプロピルアルコール溶液(シラン溶液(2))をスプレーで塗布し、50℃で5分間予備乾燥した後、120℃で1時間加熱乾燥を行った。その後、室温まで冷却して、硬化物層をアセトンにより洗浄し、エアーガンで乾燥した。なお、アセトン洗浄前には、硬化物層を積層したガラス板は若干白濁していたが、アセトン洗浄により透明に戻り、ガラス板の外観・反射防止の特性に大きな変化は見られなかった。この硬化物層の厚みは約0.5μmであり、この硬化物層の表面の水接触角は58°であり、MPMOSによる硬化物層の形成が確認できた。
メタノール250gに2-スルホエチルメタクリレート・ナトリウム塩(日本乳化剤(株):antoxMS-2N)25.0gを加えて、室温で溶解しようとしたが、溶解できなかった(antoxMS-2N濃度=9.1wt%)。
得られた親水性化合物溶液(2)中に、MPMOSにより硬化物層が形成されたガラス板を、室温で3分間浸漬し、引き上げて窒素ボックス中で3分間(室温)保持した後、メタルハライドランプ(出力120W/cm2)の紫外線光源直下20cmで40秒間の紫外線照射(大気下)を行い、メタクリル基とメルカプト基との反応を行った。紫外線照射されたガラス板を純水で洗浄し、エアーガンで乾燥した。
表4に記載されるように、シラン溶液(2)にTMOSを加え、MPMOS/TMOSの配合モル比を変更して硬化物層を形成したこと以外は、比較例2と同様にガラス板上に膜を作製し試験した。膜の評価結果を表4に示す。

表5に記載されるように、硬化物層表面にグラフトさせる親水性化合物(c)の種類を変更し、さらに親水性化合物(c)を、実施例1と同様の熱グラフト条件または比較例3のUVグラフト条件で硬化物層表面にグラフトさせたこと以外は、実施例1と同様にガラス板上に膜を作製した。
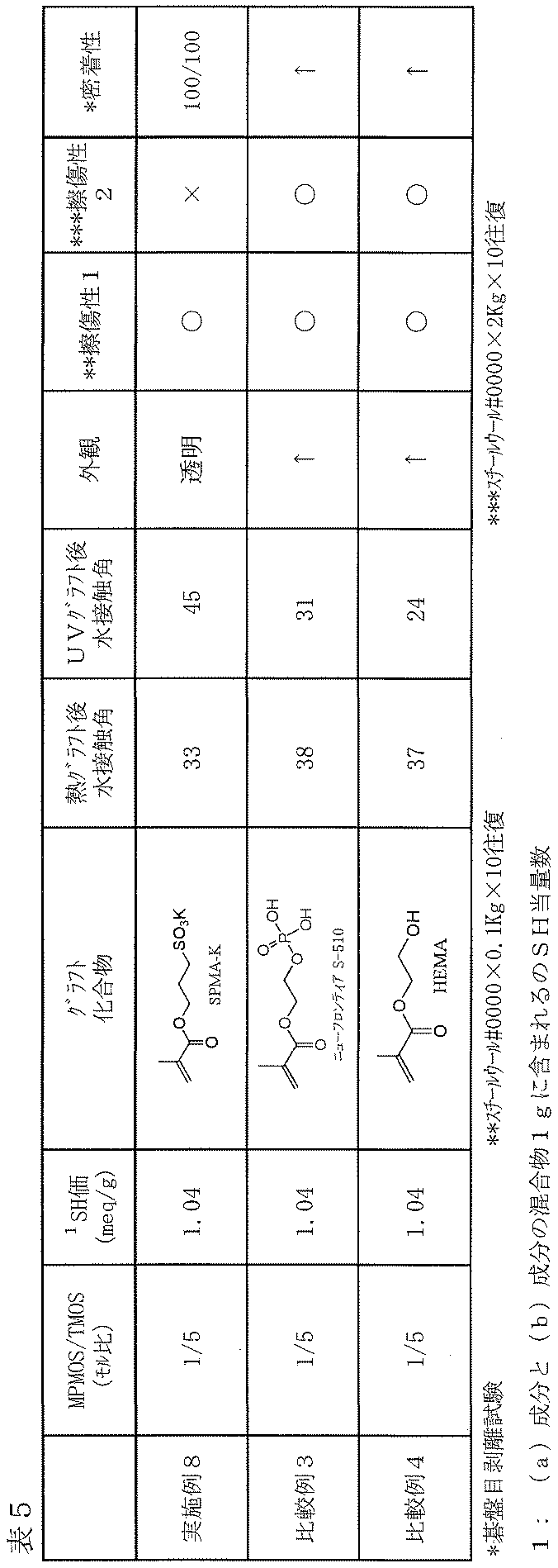
表6に記載されるように、硬化物層表面にグラフトさせる親水性化合物(c)の種類を変更したこと以外は、実施例1と同様に、ガラス板上に膜を作製し、試験した。結果を表6に示す。
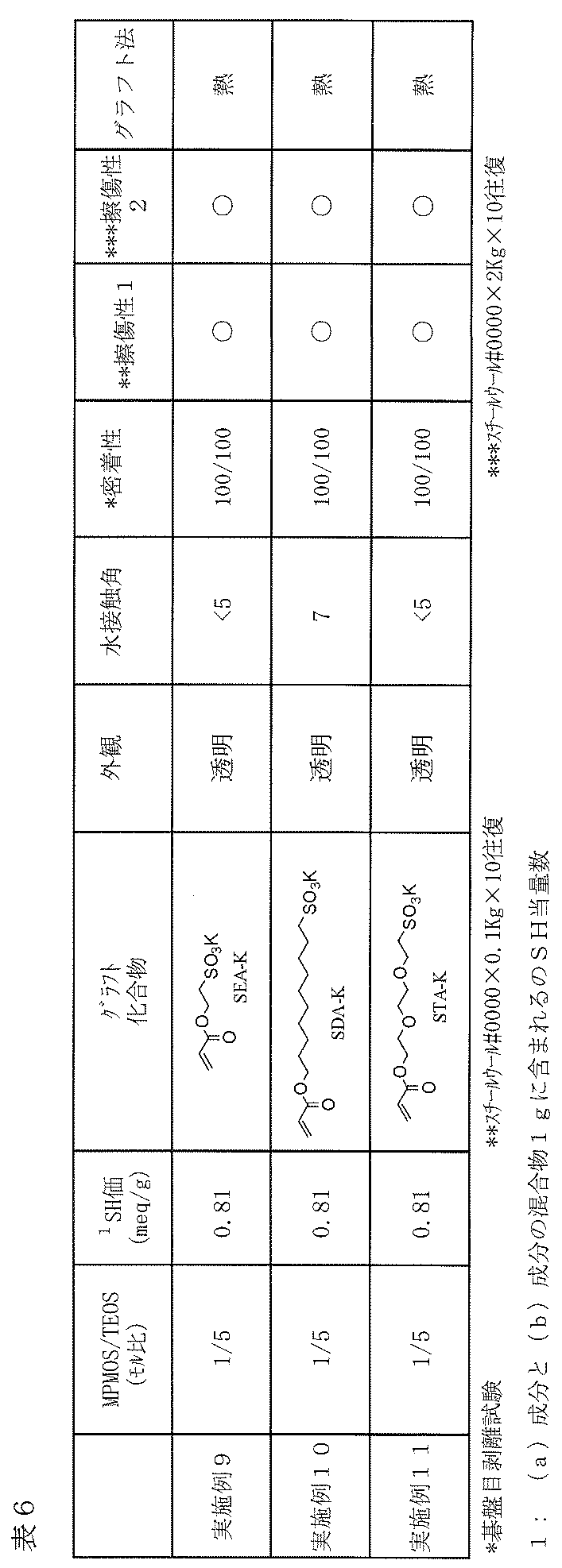
表7、8に記載されるように、硬化膜層の硬化条件または親水性化合物(c)の熱グラフト条件を変更したこと以外は、実施例1と同様にガラス板上に膜を作製し、試験した。結果を表7、8に示す。
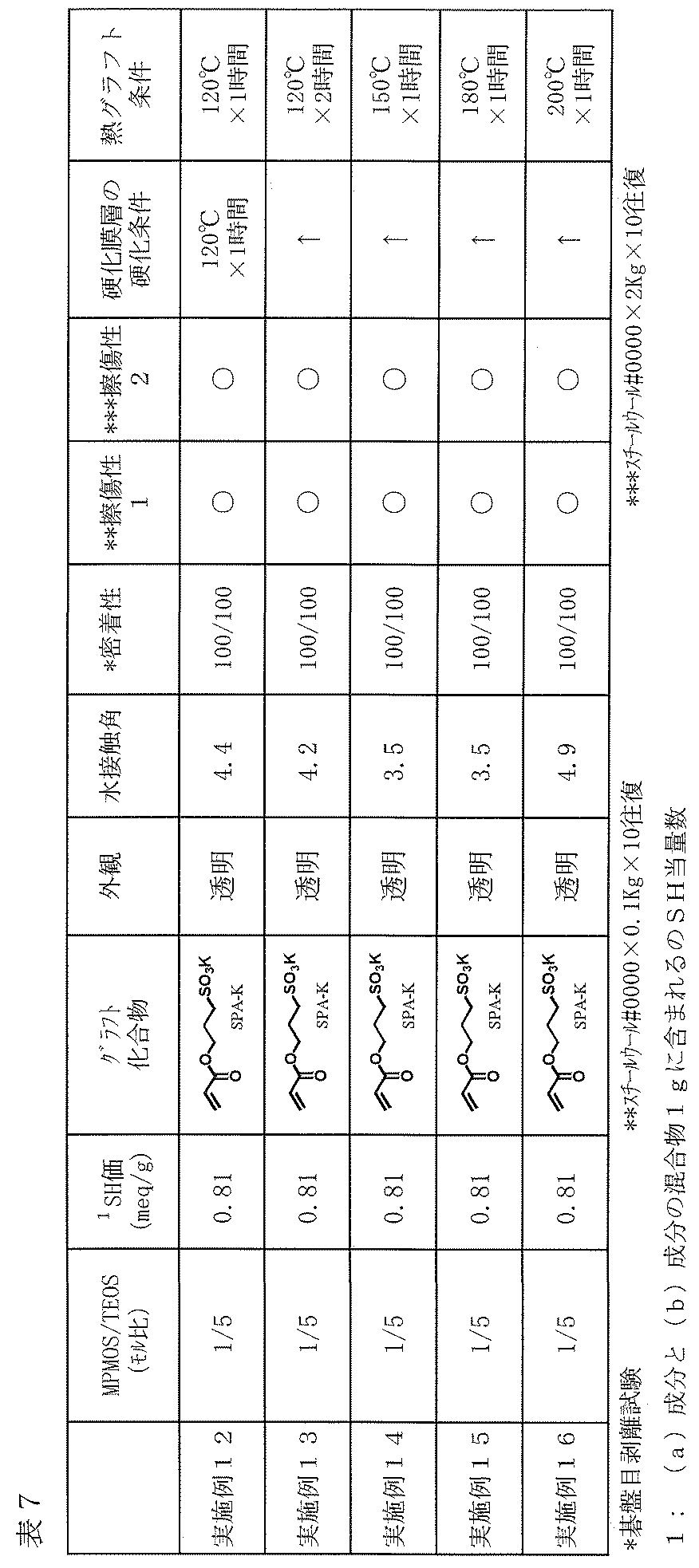

(シラン溶液(3)の調製)
攪拌子を備えた密栓付三角フラスコに、シラン化合物(a)および(b)として、MPMOSとテトラエトキシシランを凡そ1:4.7(モル比)で反応(縮重合)させたオリゴマー型シラン化合物である製品名X-41-1805(信越化学工業(株)、メルカプト当量862g/mol)1.08g(1.25mmol) とシラン化合物(b)成分であるTEOS(テトラエトキシシラン) 2.60g(12.5mmol)を秤量し、エタノール93.5gを加えて攪拌して均一液を調製した。なお、X-41-1805とTEOSの配合モル比(X-41-1805/TEOS)は1/10である。
(硬化物層の形成)
上記シラン溶液(3)を、実施例1と同様に、洗浄されたガラス板表面にX-41-1805に由来するメルカプト基が残存する硬化膜層を形成させた。
(グラフト化)
得られた硬化膜層の表面に、実施例1で調製した親水性化合物溶液(1)をバーコーター#2で再び塗布し、同様に50℃×5分間の予備乾燥後、150℃で1時間の加熱乾燥を行うことによって、硬化膜層表面のメルカプト基にSPA-Kをグラフトさせた。グラフト後、室温まで冷却し、水洗することによって、メルカプト基との反応(グラフト)に関与しなかったSPA-Kを洗い流し、エアーガンにて乾燥した。得られた膜は透明で、タック性もなく、ガラス表面に強固に密着していた。このガラス板上に形成された膜について、実施例1と同様に試験し、さらに、下記条件および評価基準に基づいて、テーバー磨耗性試験を行った。得られた評価結果を表9に示す。
中心に内径6mmの穴の開いた青板ガス(縦10cm×横10cm×厚さ0.2cm)にコーテイングされたサンプル板を、以下の条件で耐磨耗性の試験を行った。なお、表9中の試験結果は、磨耗輪が通過した箇所4点のヘーズ値を以下の条件で測定し、平均値を記載した。
(測定条件)
・試験機器: (株)東洋精機製作所 「ロータリーアブレッサー」 #2114
008-09
・磨耗輪: 大和化成工業(株) 「C180-OXF」
・荷重: 500g (250g×2)
・測定機器: 日本電色工業(株) 「ヘーズメーター NDH 2000」
[実施例20~23、25~27]
表9に記載されるように、シラン溶液(3)にTMOSを加え、X-41-1805/TMOSの配合モル比を変更して硬化物層を形成したこと以外は、実施例24と同様にガラス板上に膜を作製し、試験した。結果を表9に示す。
基材の青板ガラス(縦10cm×横10cm×厚さ0.2cm)について、実施例24と同様なテーバー磨耗試験を行った。結果を表9に示す。

表10に記載されるように、硬化膜層の硬化条件を変更して硬化膜層を形成したこと以外は、実施例24と同様にガラス板上に膜を作製し、試験した。結果を表10に示す。
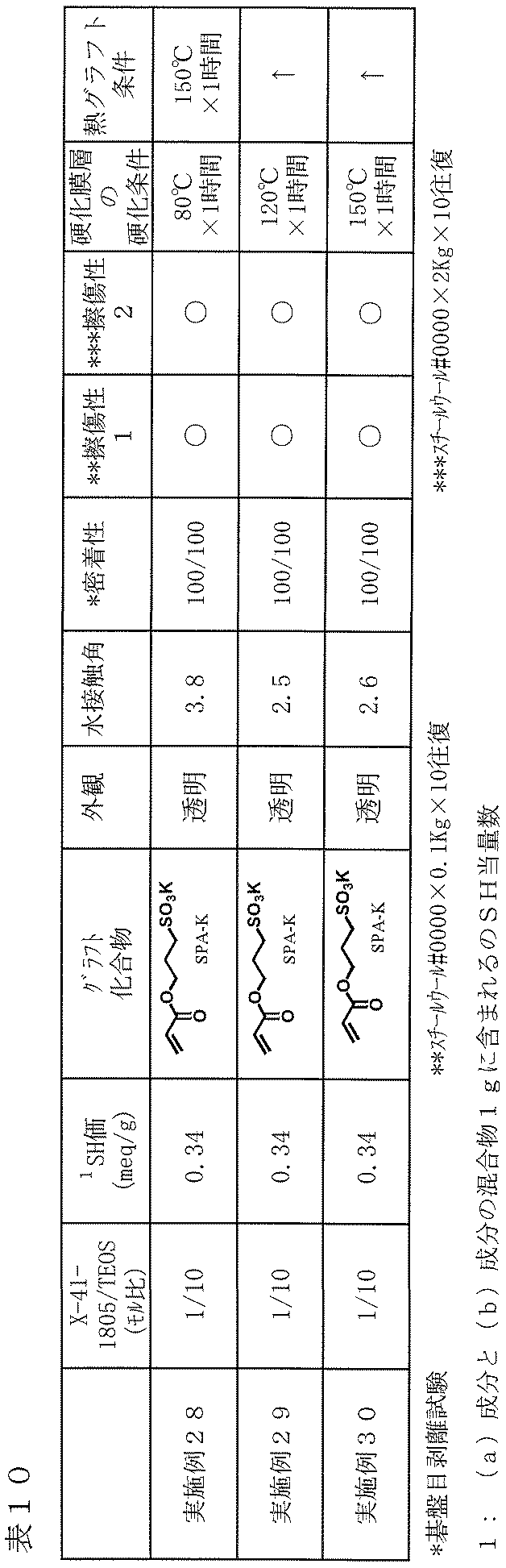
実施例20と同様にして得られた、硬化膜層および、この硬化膜層に親水性化合物をグラフトして得られた膜について、各表面のイオン強度を、飛行時間型二次イオン質量分析装置(TOF-SIMS)を用いて、以下の条件で分析して、各表面の官能基濃度を調査した。結果を表10に示す。
<イオン強度の分析条件>
・試験機器: ION・TOF社製 「TOF-SIMS」
・一次イオン: Bi3+
・加速電圧: 25kV
・分析領域: 500μm×500μm
[実施例32~33]
表11に記載されるように、実施例24、26と同様にしてガラス板上に膜を形成し、実施例31と同様にして各表面のイオン強度を分析した。結果を表11に示す。

表12に記載されるように、硬化物層表面にグラフトさせる親水性化合物(c)の種類を変更したこと以外は、実施例24と同様にガラス板上に膜を作成し、試験した。結果を表12に示す。
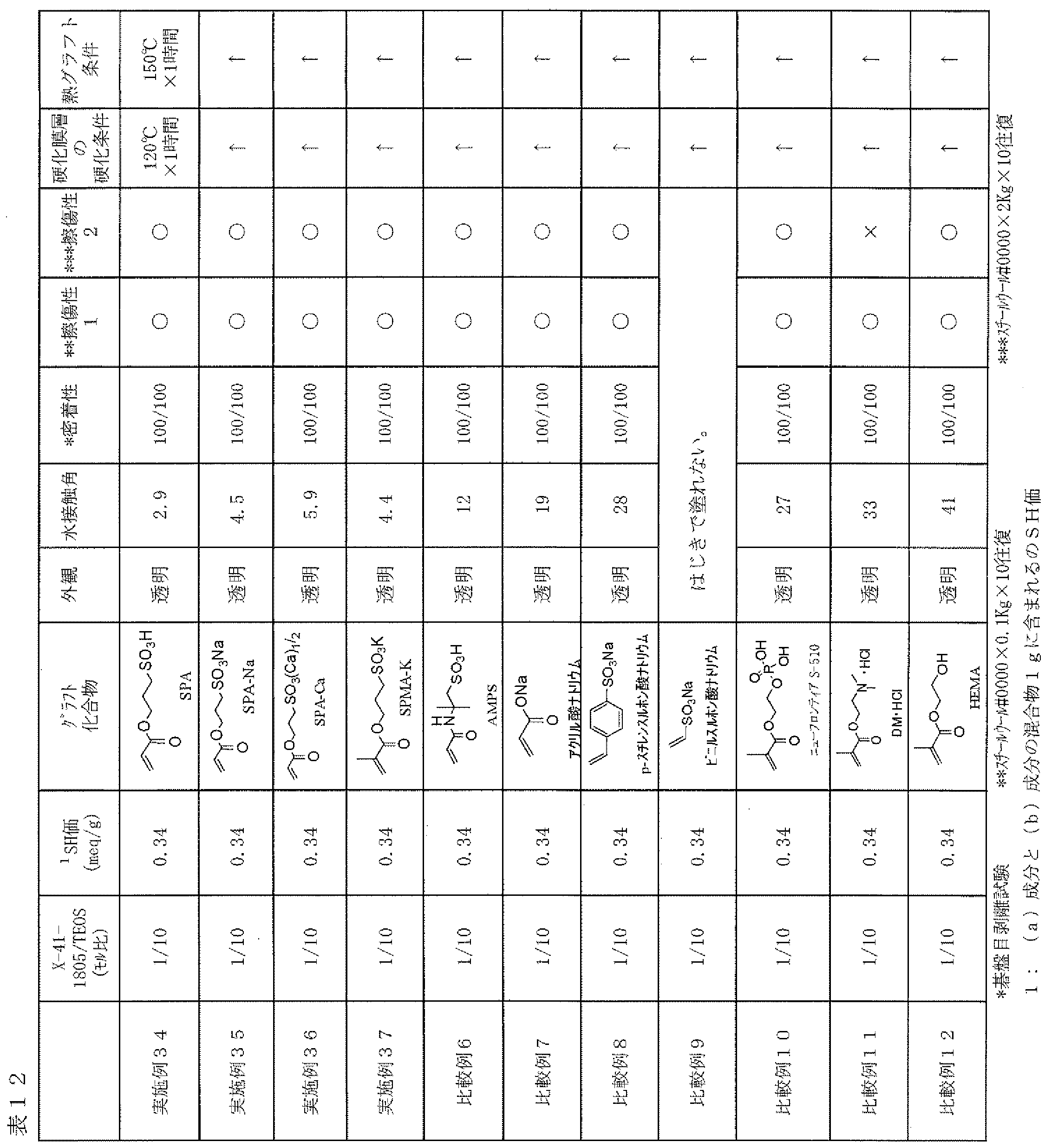
(シラン溶液(4)の調製)
攪拌子を備えた密栓付三角フラスコに、シラン化合物(a)としてN-(2-アミノエチル)-3-アミノプロピル-トリメトキシシラン(AAMOS)0.55g(2.5mmol)と(b)成分であるTEOS(テトラエトキシシラン)2.60g(12.5mmol)を秤量し、メタノール86.3gを加えて均一液を調製した。なお、シラン化合物(a)とシラン化合物(b)との配合モル比(シラン化合物(a)/シラン化合物(b))は1/5である。
(硬化膜層の形成)
上記シラン溶液(4)を、実施例1と同様に洗浄されたガラス板(水接触角5~6°)表面に、バーコーター#12で塗布し、50℃×5分間の予備乾燥後、70℃で2時間の加熱乾燥を行い、ガラス表面にAAMOSに由来するアミノ基が残存する硬化膜層を形成させた。本シリカ層の膜厚は凡そ0.4μmであった。
(グラフト化)
得られた硬化膜層の表面に、実施例1で調製した親水性化合物溶液(1)を、実施例1と同様の条件で、硬化膜層表面のアミノ基にSPA-Kをグラフトさせて、膜を形成した。得られた膜の評価結果を表13に示す。
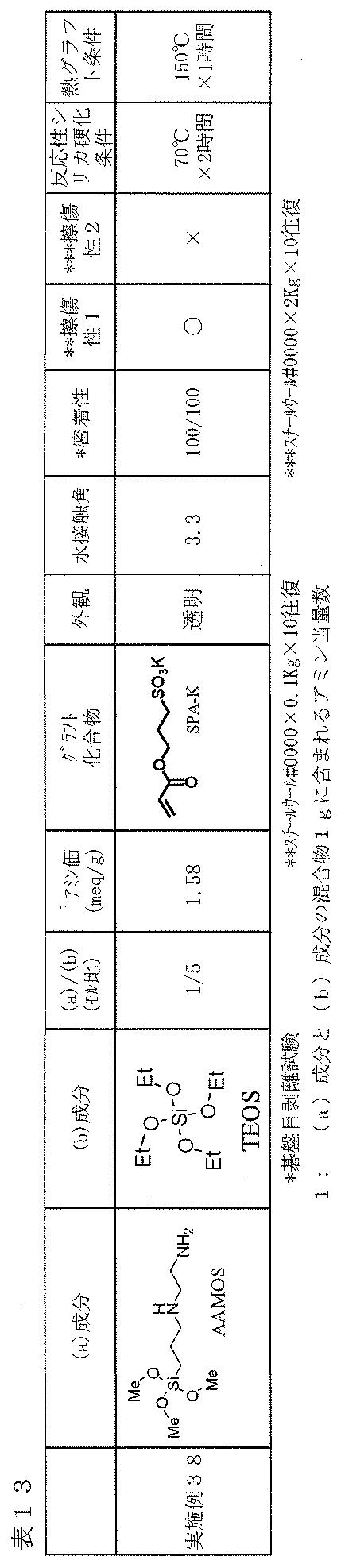
(シラン溶液(5)の調製)
シラン化合物(a)としてMPMOSの代わりにアクリロイル基を有する化合物(AOMOS)を用いたこと以外は、実施例1と同様にして、固形分4wt%のシラン溶液(5)を調製した。
(硬化膜層の形成)
上記シラン溶液(3)を、実施例1と同様に、洗浄されたガラス板表面(水接触角5~6°)にバーコーター#10で塗布し、50℃×5分間の予備乾燥後、120℃×1時間の加熱乾燥を行い、ガラス板表面の、AOMOSに由来する重合性の炭素-炭素二重結合を有する基(アクリロイル基)が残存した硬化膜層を形成した。
(親水性化合物溶液(2)の調製)
遮光サンプル瓶にSPA-K 2.0g(8.61mol)、UV重合開始剤としてダロキュアー1173(チバ・スペシャリティ・ケミカルズ(株))2.0g(100wt%対SPA-K)、溶剤として2-メトキシエタノール100gを加えて混合し、親水モノマー濃度2wt%の均一な親水性化合物溶液(2)を調製した。
(UVグラフト化)
上記で得られた反応性シリカ層表面に、上記親水性化合物溶液(2)をバーコーター#2で塗布し、50℃×5分間オーブンで乾燥後、メタルハライドランプ(照度130mW/cm2)で30秒間照射(3900mJ/cm2)して、グラフト化反応を行い、水洗することによって、アクリロイル基との反応(グラフト)に関与しなかったSPA-Kを洗い流した後に、エアーガンにて乾燥して、膜を形成した。得られた膜の評価結果を表14に示す。
表13に記載されるように、化合物(a)の種類や化合部(a)成分と化合物(b)成分の配合モル比を変更したこと以外は、実施例38と同様にガラス板上に膜を作製し、試験した。結果を表14に示す。

表15に記載されているように、化合物(b)として、TEOS 2.6g(12.5mmol)の代わりに、TMOSI(N,N´,N″-トリス(3-トリメトキシシリル-プロピル)イソシアヌレート) 2.6g(4.22mol)を含むシラン溶液を調製し、X-41-1805とTEOSとの配合モル比(X-41-1805/TEOS)を1/5としたこと以外は実施例24と同様に、ガラス板上に膜を作製し、試験した。結果を表15に記載する。
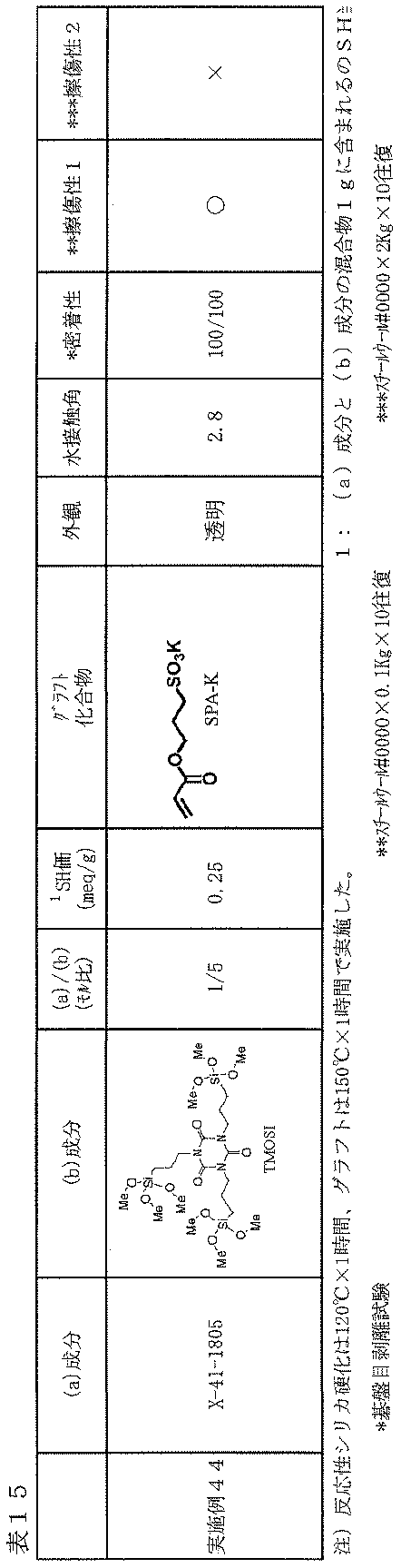
表16に記載されているように、基材をガラス板から、実施例1と同様の洗浄法で洗浄した後に2wt%硫酸でエッチング(室温で10分間浸漬)されたステンレス板に変更した以外は、実施例24と同様にガラス板上に膜を作製し、試験した。結果を表16に記載する。
表16に記載されているように、硬化膜層の厚さを変更するとともに、基材をガラス板から、下記条件で表面改質処理(特許3557194号)されたPC(ポリカーボネート)板に変更するとともに、加熱グラフト条件を50℃×5分間の予備乾燥後、120℃×3時間としたこと以外は、実施例24と同様に膜を作製し、試験した。結果を表16に記載する。
<イトロ処理(表面処理)条件>
下記イトロガスと都市ガスとを連続混合しながら調製した可燃性ガスに着火し、生成した火炎中に、基材を0.7m/秒の線速度で2回通過させて、基材表面を処理した。
イトロガス:表面改質剤としてテトラメチルシラン1.0×10-4モル%とテトラメトキシシラン1.0×10-5モル%を含む空気混合圧縮ガス
(なお、テトラメチルシランおよびテトラメトキシシランのモル%は、イトロガス全体量を100モル%としている。)

表17に記載されているように、実施例27および実施例24と同様にガラス板上に膜を作製した。得られた膜を、下記の各浸漬条件あるいは耐候条件で処理した後に、実施例24と同様な評価を行った。結果を表17に示す。
(浸漬条件および耐候条件)
・水浸漬条件1:得られた膜を、沸騰水(100℃)に1時間浸漬した。
・水浸漬条件2:得られた膜を、温水(60℃)に60日間浸漬した。
・薬品浸漬条件1:得られた膜を、カビキラー(商品名:ジョンソン株式会社、次亜塩素酸塩、水酸化ナトリウム、界面活性剤(アルキルアミンオキシド)等を含む。)に7日間浸漬した。
・薬品浸漬条件2:得られた膜を、キッチンハイター(商品名:花王株式会社、次亜塩素酸ナトリウム、洗浄成分等を含む。)に7日間浸漬した。
・耐候条件:得られた膜を、バックパネル温度(BPT)63、降雨条件18分/2時間の雰囲気下において、キセノンアーク灯を用いて60W/m2、波長300~400nmの光に、1000時間曝露した。
表17に記載されているように、実施例24と同様にガラス板上に膜を作製し、実施例47と同様に試験した。結果を表17に示す。
表17に記載されているように、添加剤としてオクチル酸ジルコニウムをシラン混合物に1.0重量%となるように添加したこと以外は、実施例24と同様にガラス板上に膜を作製し、実施例47と同様に試験した。結果を表17に示す。

実施例34と同様にしてガラス板上に作製した膜(親水性化合物(c):3-スルホプロピルアクリレート(SPA))を実験室内(気温25℃±3℃、湿度35%±10%)に10日~4ヶ月放置した。予め定めた放置期間の経過後、当該膜を[ベンコットM-3](旭化成)を用いて流水で洗い、エアーガンを用いて乾燥した。その後、各放置期間を経過した膜の水接触角を測定した。結果を表18に示す。
実施例1と同様にして、洗浄されたガラス板(膜を作製していないガラス板)についても、実施例51と同様に放置し、その後、各放置期間を経過した当該ガラス板の水接触角を測定した。結果を表18に示す。
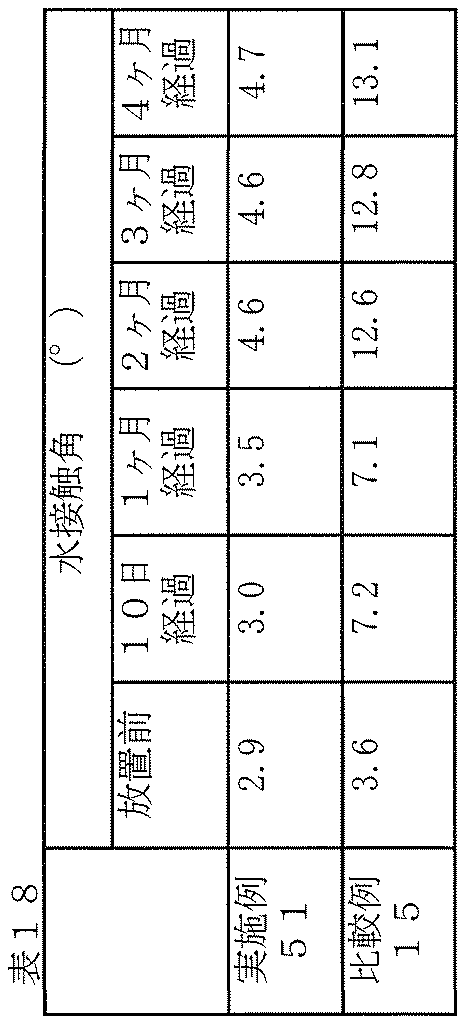
(シラン溶液(6)の調製)
攪拌子を備えた密栓付三角フラスコに、シラン化合物(a)および(b)として、実施例24に示したX-41-1805 1.08g(1.25mmol)と、シラン化合物(b)として、TEOS 13.0g(62.5mmol)を秤量し、メタノール152.0gを加えて攪拌して均一液を調製した。なお、X-41-1805(信越化学工業(株)、メルカプト当量862g/mol)とTEOSの配合モル比(X-41-1805/TEOS)は1/50である。
(硬化物層の形成)
上記シラン溶液(6)を、実施例1と同様にして洗浄したガラス板表面にバーコーター#30で塗布し、50℃で5分間予備乾燥した後、150℃で1時間加熱乾燥を行った。この硬化物層の厚みは約3μmであった。
(グラフト化)
得られた硬化膜層の表面に、実施例24と同様に膜を作製し、その後、実施例1及び実施例24と同様に試験した。得られた評価結果を表19に示す。
[実施例53]
(シラン溶液(7)の調製)
攪拌子を備えた密栓付三角フラスコに、シラン化合物(a)および(b)として、実施例52と同様にX-41-1805 1.08g(1.25mmol)と、シラン化合物(b)として、TEOS 13.0g(62.5mmol)を秤量し、更に、シラン化合物(b)として、3-グリシドキシプロピルトリメトキシシラン(以下、GPMOS 分子量:236.3)7.0g(29.6mmol)を秤量し、メタノール 228.0gを加えて攪拌して均一液を調製した。なお、X-41-1805とTEOS及びGPMOSの配合モル比(X-41-1805/TEOS/GPMOS)は1/50/23.7である。
(硬化物層の形成)
上記シラン溶液(7)を、実施例52と同様にしてガラス板上に硬化物層を形成した。この硬化物層の厚みは約3μmであった。
(グラフト化)
得られた硬化膜層の表面に、実施例52と同様にして膜を作製し、その後、実施例52と同様に試験した。得られた評価結果を表19に示す。

(シラン溶液(8)の調製)
攪拌子を備えた密栓付三角フラスコに、シラン化合物(a)成分として、実施例1で示したMPMOS 3.00g(15.3mmol)と、シラン化合物(b)成分であるTEOS 9.6g(45.8mmol)を秤量し、メタノール441.0gを加えて攪拌して均一液を調製した。なお、MPMOSとTEOSの配合モル比(MPMOS/TEOS)は1/3である。
(硬化物層の形成)
上記シラン溶液(8)を、実施例1と同様にしてガラス板上に硬化物層を形成した。この硬化物層の厚みは約0.4μmであった。
(グラフト化)
得られた硬化膜層の表面に、実施例52と同様にして膜を作製し、その後、実施例52と同様に試験した。得られた評価結果を表20に示す。
[実施例55]
(硬化物層の形成)
表20に記載されるように、シラン溶液(8)中のMPMOSとTEOSの配合モル比、及びMPMOSとTEOS及びシリカの配合重量比を変更せず、固形分濃度を10wt%に変更して調製したシラン溶液を、ガラス板上にバーコーター#30で塗布したこと以外は、実施例54と同様にして硬化物層を形成した。この硬化物層の厚みは約3μmであった。
(グラフト化)
得られた硬化膜層の表面に、実施例52と同様にして膜を作製し、その後、実施例52と同様に試験した。得られた評価結果を表20に示す。
表20に記載されるように、シラン溶液(8)中のMPMOS/TEOSの配合モル比、及びMPMOS/TEOS/シリカの配合重量比を変更したこと以外は、実施例54と同様にしてガラス板上に膜を作製し、その後、実施例52と同様に試験した。得られた評価結果を表20に示す。
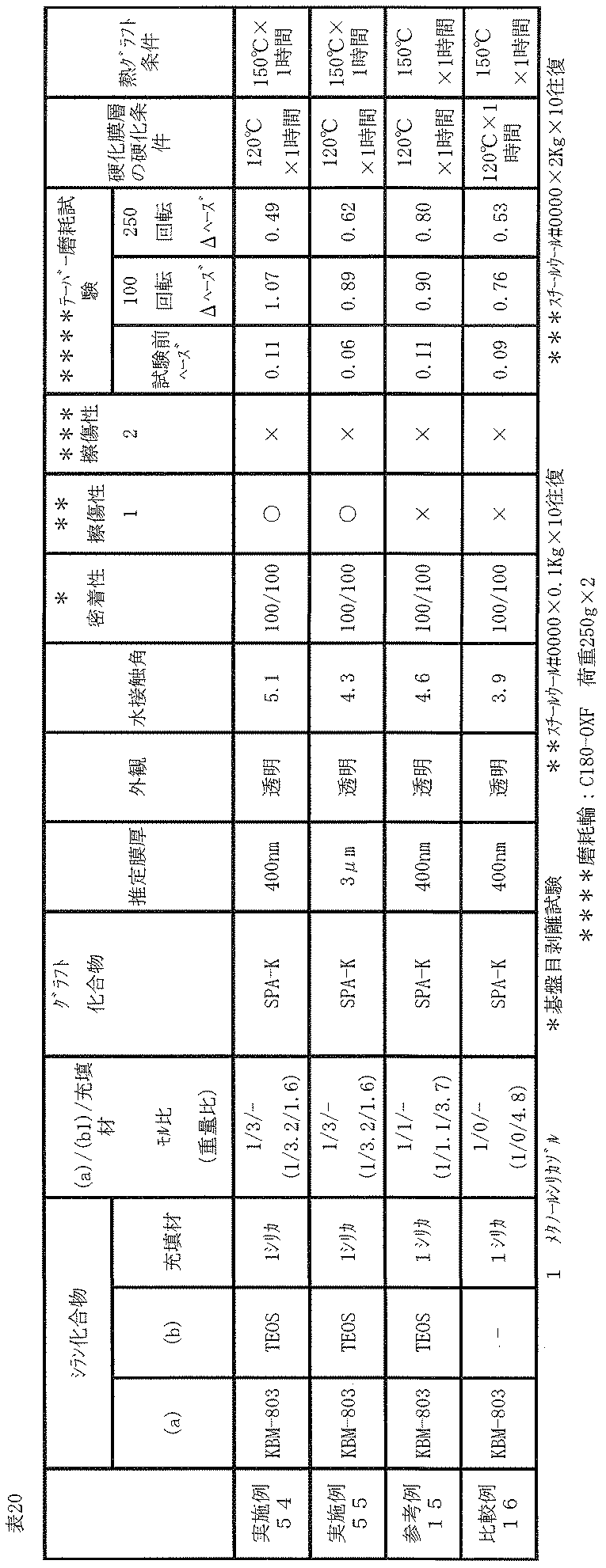
Claims (5)
- (a)1個のシリコン原子と、メルカプト基、アミノ基、(メタ)アクリロイル基、ビニル基からなる群より選ばれる基と、アルコキシ基、ハロゲン基、およびヒドロキシ基からなる群より選ばれる少なくとも1個のシリコン原子に結合した基と、を有するシラン化合物
ならびに(b)アルコキシ基、ハロゲン基およびヒドロキシ基からなる群より選ばれる、シリコン原子に結合した架橋性基を有し、かつ炭素―炭素二重結合との反応性を有する基を有さないシラン化合物を含む混合物から形成された層の表面に、(c)下記一般式(c)で表される化合物を塗布し、
下記一般式(c)で表される化合物に含まれる(メタ)アクリロイル基と、上記化合物(a)に由来するメルカプト基、アミノ基、(メタ)アクリロイル基、およびビニル基からなる群より選ばれる基との少なくとも一部を反応させて得られる親水膜。
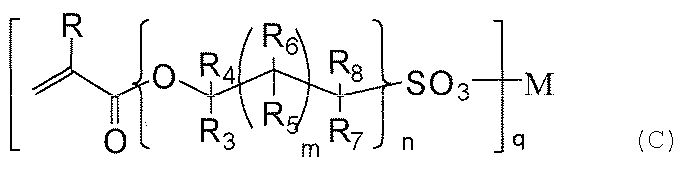
- 上記化合物(a)/上記化合物(b)のモル比が、1/2~1/200である請求項1に記載の親水膜。
- 請求項1または2に記載の親水膜を有する積層体。
- (a)1個のシリコン原子と、メルカプト基、アミノ基、(メタ)アクリロイル基およびビニル基からなる群より選ばれる基と、アルコキシ基、ハロゲン基、およびヒドロキシ基からなる群より選ばれる少なくとも1個のシリコン原子に結合した基と、を有するシラン化合物、ならびに(b)アルコキシ基、ハロゲン基およびヒドロキシ基からなる群より選ばれる、シリコン原子に結合した架橋性基を有し、かつ炭素―炭素二重結合との反応性を有する基を有さないシラン化合物を含む混合物を調製し、
当該シラン混合物を基材に塗布して、加水分解反応および縮合反応を進行させて、当該基材の表面に層を形成し、
当該層の表面に、(c)下記一般式(c)で表される化合物を塗布して、下記一般式(c)で表される化合物に含まれる(メタ)アクリロイル基と、上記化合物(a)に由来するメルカプト基、アミノ基、(メタ)アクリロイル基およびビニル基からなる群より選ばれる基との少なくとも一部をグラフト反応を進行させて、親水膜を形成する親水膜を有する積層体の製造方法。
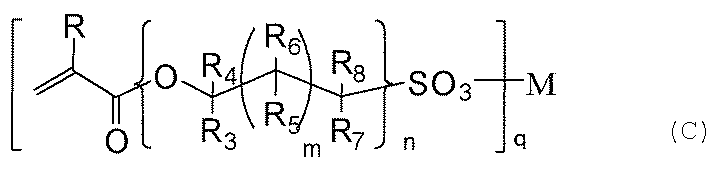
- 上記化合物(c)のMが、H、Li、Na、Rb、Mg、Ca、Sr、またはBaであり、かつ、上記グラフト反応を、130℃以上の温度条件下で進行させる請求項4に記載の親水膜を有する積層体の製造方法。
Priority Applications (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN200980109158XA CN101970553B (zh) | 2008-03-21 | 2009-03-19 | 亲水膜 |
| JP2010503919A JP5308436B2 (ja) | 2008-03-21 | 2009-03-19 | 親水膜 |
| US12/933,743 US9512034B2 (en) | 2008-03-21 | 2009-03-19 | Hydrophilic film |
| KR1020137010840A KR20130059453A (ko) | 2008-03-21 | 2009-03-19 | 친수막 |
| KR1020107020441A KR101300382B1 (ko) | 2008-03-21 | 2009-03-19 | 친수막 |
| EP09721264.1A EP2256153B1 (en) | 2008-03-21 | 2009-03-19 | Hydrophilic film |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2008-073500 | 2008-03-21 | ||
| JP2008073500 | 2008-03-21 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2009116612A1 true WO2009116612A1 (ja) | 2009-09-24 |
Family
ID=41091010
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2009/055398 Ceased WO2009116612A1 (ja) | 2008-03-21 | 2009-03-19 | 親水膜 |
Country Status (6)
| Country | Link |
|---|---|
| US (1) | US9512034B2 (ja) |
| EP (1) | EP2256153B1 (ja) |
| JP (1) | JP5308436B2 (ja) |
| KR (2) | KR20130059453A (ja) |
| CN (1) | CN101970553B (ja) |
| WO (1) | WO2009116612A1 (ja) |
Cited By (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2599799A4 (en) * | 2010-07-29 | 2014-11-05 | Mitsui Chemicals Inc | LAYERED FILM AND HYDROPHILIC MATERIAL THEREWITH |
| US8961682B2 (en) | 2012-07-26 | 2015-02-24 | Empire Technology Development Llc | Hydrophilic paints using pigments coated with anti-oxidants |
| JP2020536996A (ja) * | 2017-10-04 | 2020-12-17 | エムシーエス・インダストリーズ・インコーポレイテッド | 防曇コーティングと塗布方法 |
| JP2021011530A (ja) * | 2019-07-05 | 2021-02-04 | 岩崎電気株式会社 | 無機材料の表面改質方法 |
| WO2023062932A1 (ja) * | 2021-10-11 | 2023-04-20 | 株式会社トプコン | 手術顕微鏡用光学素子およびその製造方法、医療用光学機器 |
Families Citing this family (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP6049979B2 (ja) * | 2009-07-03 | 2016-12-21 | ソニー株式会社 | 光学素子、および表示装置 |
| US9273222B2 (en) * | 2011-10-14 | 2016-03-01 | Mitsui Chemicals, Inc. | Composition and film comprising same |
| US8912244B2 (en) * | 2012-03-09 | 2014-12-16 | Protocol Environmental Solutions Inc. | Non-film forming compositions and methods of protecting cured concrete and cementitious materials |
| JP5881828B2 (ja) * | 2012-06-12 | 2016-03-09 | 三井化学株式会社 | 親水性の変性アクリル樹脂膜 |
| BR112015025893A2 (pt) * | 2013-04-12 | 2017-07-25 | Mitsui Chemicals Inc | filme que compreende copolímero ou composição |
| EP3081578A4 (en) * | 2013-12-11 | 2017-06-21 | Mitsui Chemicals, Inc. | Composition for hydrophilic cured product |
| CN111708107B (zh) | 2014-02-12 | 2022-06-24 | 视觉缓解公司 | 易清洁涂层 |
| US10563067B2 (en) * | 2014-07-16 | 2020-02-18 | Lixil Corporation | Hydrophilic treatment coating composition and hydrophilizing treatment method |
| DE102014220798A1 (de) | 2014-10-14 | 2016-04-14 | Scheuten S.À.R.L. | Hydrophil beschichtetes Isolierglas für Gewächshäuser |
| KR102345588B1 (ko) * | 2017-03-29 | 2021-12-29 | 닛테츠 닛신 세이코 가부시키가이샤 | 도장 금속판의 제조 방법 |
| WO2018179466A1 (ja) * | 2017-03-29 | 2018-10-04 | 日新製鋼株式会社 | 塗装金属板およびその製造方法 |
| CN111669969B (zh) * | 2018-02-01 | 2022-02-25 | Agc株式会社 | 基材及共聚物 |
| DE112019001895T5 (de) * | 2018-04-10 | 2020-12-24 | AGC Inc. | Medizinische vorrichtung |
| CN110406218B (zh) * | 2019-08-27 | 2024-08-09 | 河北维嘉无纺布股份有限公司 | 一种卫生用pla复合无纺布及其制备方法 |
| JP7096517B2 (ja) * | 2020-11-25 | 2022-07-06 | ダイキン工業株式会社 | 表面処理剤 |
| CN115369544B (zh) * | 2022-08-29 | 2024-04-09 | 鹤山市旗康服装有限公司 | 一种耐磨吸湿透气性牛仔面料及其制备方法 |
| CN118724092A (zh) * | 2023-03-31 | 2024-10-01 | 宝山钢铁股份有限公司 | 一种废平整液回用的方法 |
Citations (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH04225301A (ja) | 1990-12-27 | 1992-08-14 | Seiko Epson Corp | 防曇性能を有する光学物品の製造方法 |
| JPH05341108A (ja) | 1993-01-20 | 1993-12-24 | Seiko Epson Corp | 無機コート膜の表面改質方法 |
| JPH05341107A (ja) | 1986-01-21 | 1993-12-24 | Seiko Epson Corp | 無機コート膜を有する光学材料 |
| JPH0682605A (ja) | 1993-04-08 | 1994-03-25 | Seiko Epson Corp | 無機コート膜を有する光学物品及びその表面改質法 |
| JPH0688902A (ja) | 1993-02-03 | 1994-03-29 | Seiko Epson Corp | 無機コート膜を有する光学物品及びその表面改質方法 |
| JPH08259270A (ja) | 1995-03-17 | 1996-10-08 | Seiko Epson Corp | 防曇性能を有する光学物品 |
| JPH0977891A (ja) * | 1995-01-11 | 1997-03-25 | Sekisui Chem Co Ltd | 基材密着性の良好な表面層を有する物品および該物品の製造方法 |
| JPH1045927A (ja) * | 1996-08-08 | 1998-02-17 | Nof Corp | 防曇膜及びその製造方法 |
| JP2000104046A (ja) | 1998-09-29 | 2000-04-11 | Seiko Epson Corp | 光学物品に防曇性能を付与する方法及び該方法によって得られる防曇性光学物品 |
| JP2001154503A (ja) | 1999-11-26 | 2001-06-08 | Ricoh Co Ltd | 画像形成装置 |
| JP2001154502A (ja) | 1999-11-24 | 2001-06-08 | Konica Corp | 画像形成装置 |
| JP2003005499A (ja) | 2001-06-25 | 2003-01-08 | Fuji Xerox Co Ltd | 現像装置 |
| JP3557194B2 (ja) | 2002-02-13 | 2004-08-25 | 泰浩 森 | 固体物質の表面改質方法、表面改質された固体物質および固体物質の表面改質装置 |
| WO2007064003A1 (ja) | 2005-12-02 | 2007-06-07 | Mitsui Chemicals, Inc. | 単層膜およびこれからなる親水性材料 |
| JP2007313674A (ja) | 2006-05-23 | 2007-12-06 | Fujifilm Corp | 表面親水性部材、その製造方法及び表面親水性部材を用いた平版印刷版原版 |
Family Cites Families (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2711456B2 (ja) * | 1988-08-08 | 1998-02-10 | 鐘淵化学工業株式会社 | 硬化性樹脂及びその製造法 |
| JPH0726207A (ja) * | 1993-07-07 | 1995-01-27 | Mitsui Toatsu Chem Inc | プライマー用組成物、その硬化層及びそれを用いた透明樹脂成型体 |
| JP2001194503A (ja) | 2000-01-06 | 2001-07-19 | Seiko Epson Corp | 光学物品に防曇性能を付与する方法及び防曇性光学物品 |
| JP2001194502A (ja) | 2000-01-06 | 2001-07-19 | Seiko Epson Corp | 光学物品に防曇性能を付与する方法及び防曇性光学物品 |
| JP4686839B2 (ja) * | 2000-10-23 | 2011-05-25 | 東レ株式会社 | モノマー、ポリマーおよびそれを用いた眼用レンズ |
| AR038269A1 (es) * | 2002-01-09 | 2005-01-12 | Novartis Ag | Articulos polimericos que tienen un recubrimiento lubrico, y metodo para fabricarlos |
| EP1366828A1 (en) * | 2002-05-31 | 2003-12-03 | Rohm And Haas Company | Multi-layer coating composition and method of preparation |
| CN1229173C (zh) * | 2002-08-23 | 2005-11-30 | 上海制皂有限公司 | 一种有机硅阴离子表面活性剂及其合成方法 |
| JP3912520B2 (ja) * | 2002-09-26 | 2007-05-09 | 信越化学工業株式会社 | 改質木材の製造方法 |
| DE10259241A1 (de) * | 2002-12-17 | 2004-07-01 | Röhm GmbH & Co. KG | Verfahren zur Herstellung von wasserspreitenden Kunststoffkörpern |
| DE10259240A1 (de) * | 2002-12-17 | 2004-07-08 | Röhm GmbH & Co. KG | Umformbare wasserspreitende Kunststoffkörper und Verfahren zu dessen Herstellung |
| JP2006053501A (ja) * | 2004-07-16 | 2006-02-23 | Fuji Photo Film Co Ltd | ハロゲン化銀写真感光材料 |
| EP1760084A1 (de) * | 2005-09-02 | 2007-03-07 | Sika Technology AG | Tensid-stabilisierte Organoalkoxysilanzusammensetzung |
-
2009
- 2009-03-19 KR KR1020137010840A patent/KR20130059453A/ko not_active Withdrawn
- 2009-03-19 EP EP09721264.1A patent/EP2256153B1/en not_active Not-in-force
- 2009-03-19 KR KR1020107020441A patent/KR101300382B1/ko not_active Expired - Fee Related
- 2009-03-19 JP JP2010503919A patent/JP5308436B2/ja not_active Expired - Fee Related
- 2009-03-19 US US12/933,743 patent/US9512034B2/en not_active Expired - Fee Related
- 2009-03-19 WO PCT/JP2009/055398 patent/WO2009116612A1/ja not_active Ceased
- 2009-03-19 CN CN200980109158XA patent/CN101970553B/zh not_active Expired - Fee Related
Patent Citations (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH05341107A (ja) | 1986-01-21 | 1993-12-24 | Seiko Epson Corp | 無機コート膜を有する光学材料 |
| JPH04225301A (ja) | 1990-12-27 | 1992-08-14 | Seiko Epson Corp | 防曇性能を有する光学物品の製造方法 |
| JPH05341108A (ja) | 1993-01-20 | 1993-12-24 | Seiko Epson Corp | 無機コート膜の表面改質方法 |
| JPH0688902A (ja) | 1993-02-03 | 1994-03-29 | Seiko Epson Corp | 無機コート膜を有する光学物品及びその表面改質方法 |
| JPH0682605A (ja) | 1993-04-08 | 1994-03-25 | Seiko Epson Corp | 無機コート膜を有する光学物品及びその表面改質法 |
| JPH0977891A (ja) * | 1995-01-11 | 1997-03-25 | Sekisui Chem Co Ltd | 基材密着性の良好な表面層を有する物品および該物品の製造方法 |
| JPH08259270A (ja) | 1995-03-17 | 1996-10-08 | Seiko Epson Corp | 防曇性能を有する光学物品 |
| JPH1045927A (ja) * | 1996-08-08 | 1998-02-17 | Nof Corp | 防曇膜及びその製造方法 |
| JP2000104046A (ja) | 1998-09-29 | 2000-04-11 | Seiko Epson Corp | 光学物品に防曇性能を付与する方法及び該方法によって得られる防曇性光学物品 |
| JP2001154502A (ja) | 1999-11-24 | 2001-06-08 | Konica Corp | 画像形成装置 |
| JP2001154503A (ja) | 1999-11-26 | 2001-06-08 | Ricoh Co Ltd | 画像形成装置 |
| JP2003005499A (ja) | 2001-06-25 | 2003-01-08 | Fuji Xerox Co Ltd | 現像装置 |
| JP3557194B2 (ja) | 2002-02-13 | 2004-08-25 | 泰浩 森 | 固体物質の表面改質方法、表面改質された固体物質および固体物質の表面改質装置 |
| WO2007064003A1 (ja) | 2005-12-02 | 2007-06-07 | Mitsui Chemicals, Inc. | 単層膜およびこれからなる親水性材料 |
| JP2007313674A (ja) | 2006-05-23 | 2007-12-06 | Fujifilm Corp | 表面親水性部材、その製造方法及び表面親水性部材を用いた平版印刷版原版 |
Non-Patent Citations (4)
| Title |
|---|
| HYOMEN KAGAKU, SURFACE SCIENCE, vol. 22, 2001, pages 55 - 63 |
| KOBUNSHI, POLYMER, vol. 44, no. 5, pages 307 |
| MIRAI ZAIRYO, FUTURE MATERIAL, vol. 2, no. 1, pages 36 - 41 |
| See also references of EP2256153A4 * |
Cited By (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2599799A4 (en) * | 2010-07-29 | 2014-11-05 | Mitsui Chemicals Inc | LAYERED FILM AND HYDROPHILIC MATERIAL THEREWITH |
| US9034464B2 (en) | 2010-07-29 | 2015-05-19 | Mitsui Chemicals, Inc. | Single layer film and hydrophilic material comprising the same |
| US8961682B2 (en) | 2012-07-26 | 2015-02-24 | Empire Technology Development Llc | Hydrophilic paints using pigments coated with anti-oxidants |
| JP2020536996A (ja) * | 2017-10-04 | 2020-12-17 | エムシーエス・インダストリーズ・インコーポレイテッド | 防曇コーティングと塗布方法 |
| JP2021011530A (ja) * | 2019-07-05 | 2021-02-04 | 岩崎電気株式会社 | 無機材料の表面改質方法 |
| WO2023062932A1 (ja) * | 2021-10-11 | 2023-04-20 | 株式会社トプコン | 手術顕微鏡用光学素子およびその製造方法、医療用光学機器 |
Also Published As
| Publication number | Publication date |
|---|---|
| KR20130059453A (ko) | 2013-06-05 |
| JPWO2009116612A1 (ja) | 2011-07-21 |
| KR20100113634A (ko) | 2010-10-21 |
| EP2256153A4 (en) | 2012-10-03 |
| KR101300382B1 (ko) | 2013-08-26 |
| EP2256153B1 (en) | 2014-06-25 |
| JP5308436B2 (ja) | 2013-10-09 |
| EP2256153A1 (en) | 2010-12-01 |
| US20110008630A1 (en) | 2011-01-13 |
| US9512034B2 (en) | 2016-12-06 |
| CN101970553B (zh) | 2012-11-21 |
| CN101970553A (zh) | 2011-02-09 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP5308436B2 (ja) | 親水膜 | |
| JP6625987B2 (ja) | ポリフッ素含有シロキサン被覆 | |
| CN112218728B (zh) | 隐形指纹涂料及其形成方法 | |
| CN105102498B (zh) | 共聚物及包含该共聚物的亲水性材料 | |
| JP2020044841A (ja) | 防曇膜つき透明物品 | |
| CN101675133A (zh) | 可手工涂布的溶胶凝胶膜形成用涂布液 | |
| TW201111459A (en) | Near-infrared shield coating agent which is curable with ordinary temperature and the near-infrared shield coating prepared using the same and the process for preparing the coating | |
| CN101638482A (zh) | 聚合物及其制备方法和抗污涂料组合物及抗污涂层材料 | |
| JP2010248468A (ja) | 撥水撥油樹脂組成物および塗装品 | |
| CN105073917A (zh) | 由共聚物或组合物形成的膜 | |
| JP2020164842A (ja) | 防曇剤組成物、該組成物から形成される防曇膜を有する防曇性物品 | |
| CN1860196A (zh) | 具有低折射率及斥水性的覆膜 | |
| WO2018207811A1 (ja) | 防汚性物品および防汚性物品の製造方法 | |
| CN107903734B (zh) | 具有自修复性能的耐水长效防雾抗霜的高透光率涂层的制备方法 | |
| CN107708879A (zh) | 飞行器透明体的拒水性表面处理和处理飞行器透明体的方法 | |
| JP2014234506A (ja) | 新規化合物、撥水膜形成用組成物、撥水膜付き基体および輸送機器用物品 | |
| TW202104528A (zh) | 皮膜、積層體以及積層體的製造方法 | |
| WO2018042302A1 (en) | Curable silsesquioxane polymer comprising inorganic oxide nanoparticles, articles, and methods | |
| JP2017101135A (ja) | 親水性被膜、親水性被膜形成物品、親水性被膜形成用塗布液及び親水性被膜形成物品の製造方法 | |
| JP2016050276A (ja) | 塗装鋼板用表面処理剤 | |
| TW201842098A (zh) | 親水性構造體及親水性構造體的製造方法 | |
| JP2021091859A (ja) | 親水性ポリマーを含む防曇剤 | |
| CN1298792C (zh) | 含可水解有机硅化合物的组合物和由该组合物得到的涂层 | |
| CN104926148A (zh) | 一种防雾抗划涂层及其制备方法 | |
| WO2014021135A1 (ja) | 撥水液、撥水性物品、及びそれらの製法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| WWE | Wipo information: entry into national phase |
Ref document number: 200980109158.X Country of ref document: CN |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 09721264 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2010503919 Country of ref document: JP |
|
| ENP | Entry into the national phase |
Ref document number: 20107020441 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 12933743 Country of ref document: US Ref document number: 2009721264 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |