Wirkstoffhaltige Fasernflächengebilde mit einstellbarer Wirkstofffreisetzung, ihre Anwendungen und Verfahren zur ihrer Herstellung
Die Erfindung betrifft wirkstoffhaltige Faserflächengebilde mit einstellbarem Wirkstoff- freisetzungsprofil, umfassend einen faserförmigen, polymeren löslichen und/oder abbaubaren Wirkstoffträger und wenigstens einen mit dem Träger assoziierten und von dem Faserflächengebilde freisetzbaren Wirkstoff; wirkstoffhaltige Formulierungen, umfassend solche Faserflächengebilde; die Verwendung erfindungsgemäßer wirkstoffhal- tiger Faserflächengebilde zur Herstellung wirkstoffhaltiger Formulierungen; und Verfah- ren zur Herstellung erfindungsgemäßer Faserflächengebilde.
Stand der Technik
Zur Herstellung von Nano- und Mesofasern sind dem Fachmann eine Vielzahl an Ver- fahren bekannt, von denen dem Elektrospinnverfahren („Electrospinning") derzeit die größte Bedeutung zukommt. Bei diesem Verfahren, welches beispielsweise von D.H. Reneker, H. D. Chun in Nanotechn. 7 (1996), Seite 216 f. beschrieben ist, wird üblicherweise eine Polymerschmelze oder eine Polymerlösung an einer als Elektrode dienenden Kante einem hohen elektrischen Feld ausgesetzt. Dies kann beispielsweise dadurch erreicht werden, dass die Polymerschmelze oder Polymerlösung in einem elektrischen Feld unter geringem Druck durch eine mit einem Pol einer Spannungsquelle verbundene Kanüle extrudiert wird. Aufgrund der dadurch erfolgenden elektrostatischen Aufladung der Polymerschmelze oder Polymerlösung entsteht ein auf die Gegenelektrode gerichteter Materialstrom, der sich auf dem Wege zur Gegenelektrode verfestigt. In Abhängigkeit von den Elektrodengeometrien werden mit diesem Verfahren Vliese bzw. so genannte Nonwovens oder Ensembles geordneter Fasern erhalten.
In der DE-A1 -10133393 wird ein Verfahren zur Herstellung von Hohlfasern mit einem Innendurchmesser von 1 bis 100 nm offenbart, bei dem eine Lösung eines wasser- unlöslichen Polymers - beispielsweise eine Poly-L-Iactid-Lösung in Dichlormethan oder eine Polyamid-46-Lösung in Pyridin - elektroversponnen wird. Ein ähnliches Verfahren ist auch aus der W0-A1 -01/09414 und der DE-A1 -10355665 bekannt.
Aus der DE-A1 -19600162 ist ein Verfahren zur Herstellung von Rasenmäherdraht oder textilen Flächengebilden bekannt, bei dem Polyamid, Polyester oder Polypropylen als
fadenbildendes Polymer, ein Maleinsäureanhydrid-modifizierter Polyethylen/Polypropy- len-Kautschuk sowie ein oder mehrere Alterungsstabilisatoren zusammengegeben, aufgeschmolzen und miteinander vermischt werden, bevor diese Schmelze schmelzversponnen wird.
Die DE-A1-10 2004 009 887 betrifft ein Verfahren zur Herstellung von Fasern mit einem Durchmesser von < 50 μm durch elektrostatisches Verspinnen oder Versprühen einer Schmelze von mindestens einem thermoplastischen Polymeren.
Eine ausführliche Darstellung von Elektrospinnverfahren, Eigenschaften von Fasern und ihren möglichen Anwendungen ist zu finden in A. Greiner und J. Wendorff, An- gew. Chemie Int. Ed., 2007, 119, 5770-5805.
Ein weiteres geeignetes Verfahren zur Herstellung von Faservliesen ist das sogenann- te Zentrifugalverfahren (auch Rotorspinnen genannt). In der EP 624 665B1 und der EP 1 088 918 A1 (beide BASF Anmeldungen) wird ein Verfahren zur Herstellung von Fasergebilden aus Melamin-Formaldehyd-Harz und ihrer Blends mit thermoplastischen Polymeren mittels Zentrifugalspinnverfahren auf einem Schleuderteller offenbart.
Ein Verfahren und eine Vorrichtung zur Herstellung von Fasern aus der Schmelze unterschiedlicher Polymermaterialien mit Hilfe der Zentrifugalkräfte werden auch in der DE10 2005 048 939 A1 beschrieben.
In der WO-A-2001 /54667 und der WO-A-2004/014304 werden amorphe pharmazeuti- sehe Formulierungen und Verfahren zu ihrer Herstellung offenbart. Durch Elektrospin- nen von Polymer-Pharmawirkstoff-Lösungen gelang es, stabile amorphe Formulierungen zu erzeugen. Es werden aber keine speziellen Angaben zur Wirkstoff-Freisetzung und ihrer Kontrolle gemacht.
Die WO-A-2007/082936 beschreibt die Verwendung amphiphiler, selbstassemblieren- der Proteine zur Formulierung schwer wasserlöslicher Effektstoffe durch Dispergieren der Effektstoffe in einem proteinhaltigen Schutzkolloid. Nach Mischung der schwer wasserlöslicher Effektstoffe und der amphiphilen, selbstassemblierenden Proteine in einer gemeinsamen dispersen Phase und anschließender Phasentrennung in eine pro- tein- und effektstoffreiche Phase sowie eine protein- und effektstoffarme Phase liegen
Protein-Microbeads vor, in welche die schwer wasserlöslichen Effektstoffe verkapselt sind.
Die WO-A-2007/093232 beschreibt nanopartikuläre Formulierungen von Pflanzen- schutzwirkstoffen, in welcher die Nanopartikel Kern-Schale-Strukturen mit einen mittleren Teilchendurchmesser von 0,05 bis 2,0 μm aufweisen und der Pflanzenschutzwirkstoff im Kern röntgenamorph zusammen mit einem oder mehreren Polymeren vorliegt, wobei das Polymer nicht oder nur teilweise in Wasser oder wässrigen Lösungen oder Wasser-Lösungsmittelgemischen löslich ist und die Schale aus einer stabilisierenden Hüllmatrix besteht. Herstellbar werden diese Formulierungen nach einem Verfahren, welches dadurch gekennzeichnet ist, dass man (a) eine Lösung des Pflanzenschutzwirkstoffs in einem nicht mit Wasser mischbaren organischen Lösungsmittel herstellt, (b) das Kernpolymer in einem nicht mit Wasser mischbaren organischen Lösungsmittel löst; und die aus (a) und (b) resultierende Mischung mit einer wässrigen Lösung um- fassend Komponenten der Hüllmatrix durch Einspritzen der entsprechenden Lösungen in eine Mischkammer emulgiert und das organische Lösungsmittel nach dem Emulgie- ren entfernt.
Zusammenfassung der Erfindung:
Die bisher bekannten polymerbasierten Formulierungen von Wirk- und Effektstoffen sind noch immer mit Nachteilen behaftet. Insbesondere die gezielte Einstellung der Freisetzung der formulierten Wirkstoffe, z.B. über längere Zeiträume, stellt ein bisher nicht zufriedenstellend gelöstes Problem dar.
Die Einschließung von agrochemischen, pharmazeutischen und kosmetischen Effektstoffen in synthetische Polymere oder in Polymermischungen im Temperaturbereich von 5-900C, unter Normaldruck aus einer verdünnten Polymer-Wirkstoff-Lösung wäre besonders vorteilhaft für schwer lösliche und temperaturempfindliche Effektstoffe. Von besonderem Wert wäre dabei die Anwendung von bioabbaubaren bzw. biokompatiblen Polymeren.
Es bestand daher die Aufgabe, ein Verfahren bereitzustellen, das die Formulierung solcher Wirkstoffe unter Verwendung von Polymermaterialien als Formulierungshilfs- stoff erlaubt und dabei eine bessere Einstellung der Wirkstofffreisetzung ermöglicht als dies aus dem Stand der Technik bekannt ist.
Im Folgenden werden die Begriffe Wirkstoffe und Effektstoffe synonym verwendet
Figurenbeschreibung:
In den beiliegenden Figuren zeigt:
Figur 1A REM-Aufnahmen von PVP-Polymerfasern, erhältlich durch Elektroverspinnen von PVP-Polymerlösungen mit unterschiedlichen Gehalten des Wirkstoffes Epoxicona- zol.
Figur 1 B die Wiederfindungsraten für den Wirkstoff Epoxiconazol in verschiedenen erfindungsgemäß hergestellten PVP-Matrices (Faservliese 1 und 2) im Vergleich zu entsprechenden Eichproben.
Figur 2 die Ergebnisse von WAXS-Messungen an frisch hergestellten Faserflächengebilden aus PVP-Epoxiconazol bei Gehalten von 9, 23 und 33 Gew.-% Epoxiconazol im Vergleich zu reinem PVP bzw. kristallinem Epoxiconazol.
Figur 3 die Ergebnisse von WAXS-Messungen an erfindungsgemäß hergestellten Faserflächengebilden aus PVP-Epoxiconazol, welche bei unterschiedlichen Temperaturen gelagert worden waren, im Vergleich zu reinem PVP bzw. kristallinem Epoxiconazol; jede Probe wurde dazu jeweils 24 Stunden bei +400C, -10° C und 00C und danach 72 Std. bei 200C gelagert.
Figur 4A REM-Aufnahmen erfindungsgemäß hergestellter PVP-ß-Carotinfasern mit unterschiedlichen ß-Carotin-Gehalten.
Figur 4B die Wiederfindungsraten für den Wirkstoff ß-Carotin in verschiedenen erfin- dungsgemäß hergestellten PVP-Matrices (Faservliese 1 und 2) im Vergleich zu entsprechenden Eichproben.
Figur 5 die Ergebnisse von WAXS-Messungen an erfindungsgemäß hergestellten Faserflächengebilden aus PVP-ß-Carotin. im Vergleich zu reinem PVP bzw. kristallinem ß-Carotin.
Figur 6 die Ergebnisse von WAXS-Messungen an erfindungsgemäß hergestellten Faserflächengebilden aus PVP-ß-Carotin, welche bei unterschiedlichen Temperaturen gelagert worden waren, im Vergleich zu reinem PVP bzw. kristallinem ß-Carotin; jede Probe wurde dazu jeweils 24 Stunden bei +400C, -10° C und 00C und danach 72 Std. bei 200C gelagert.
Figur 7 REM-Aufnahmen erfindungsgemäß hergestellter PMMA-Epoxiconazolfasern mit unterschiedlichen Epoxiconazol-Gehalten.
Figur 8 die Ergebnisse der WAXS-Messungen an Faserflächengebilden aus PMMA- Epoxiconazol bei verschiedenen Epoxiconazol-Gehalten.
Figur 9 die jeweiligen Freisetzungsprofile von Epoxiconazol aus dem biologisch abbaubaren Polyester Ecoflex® als Film bzw. Faserflächengebilde.
Figur 10 die unterschiedlichen Freisetzungsprofile von Epoxiconazol aus biologisch abbaubarem Polyester Exoflex®, PVP und PMMA.
Figur 1 1 die unterschiedlichen Freisetzungsprofile von Epoxiconazol aus Faserflä- chengebilden, hergestellt aus PVP, PMMA und 1 : 1- bzw. 1 :5-Blends von PVP und PMMA.
Figur 12 mikroskopische Aufnahmen von Querschnitten durch wirkstofffreie Fasern aus PMMA und PVP;
Figur 13 die unterschiedlichen Freisetzungsprofile von Epoxiconazol aus Faserflächengebilden, hergestellt aus PVP, Ecoflex® und einem 1 :1-Blend von PVP und E- coflex;
Figur 14 elektronenmikroskopische (SEM) Aufnahmen von C16-Spinnenseidenprotein- Flächengebilden (Fasern) mit eingeschlossenem Wirkstoff Clotrimazol;
Figur 15 kristallinitätsuntersuchungen (WAXS in Transmission) des Wirkstoffes Clotrimazol in den durch Elektrospinnen gewonnenen C16-Spinnenseidenprotein- Formulierungen im Vergleich zu Clotrimazol-Reinsubstanz;
Figur 16 die Freisetzung des Wirkstoffs Clotrimazol aus einer durch Elektrospinnen gewonnenen und zu Tabletten verpressten C16-Spinnenseidenprotein-Formulierung in Kaliumphosphat-Puffer (Kontrolle) sowie artifiziellen Magen- und Darmsaft. Als 100 %- Wert wurden die in der Tabelle gemäß Beispiel 10 aufgeführten Gesamtwirkstoff- Konzentrationen angesetzt.
Detaillierte Beschreibung der Erfindung:
1. Definition verwendeter Begriffe
Werden keine anderen Angaben gemacht, so gelten im Rahmen der vorliegenden Beschreibung folgende Bedeutungen technischer Begriffe:
Unter einem „Trägerpolymer" versteht man synthetische Polymere bzw. ihre Abmi- schungen, Biopolymere bzw. ihre Abmischungen, oder auch Abmischungen aus mindestens einem synthetischen und einem Biopolymer, wobei das Trägerpolymer die Fähigkeit besitzt, mit dem oder den zu formulierenden Wirkstoffen/Effektstoffen nicht- kovalente Wechselwirkungen einzugehen, bzw. partikuläre Wirkstoffe (dispergiert oder kristallin) zu umschließen.
Bei einer „nicht-kovalenten" Wechselwirkung handelt es sich um alle, dem Fachmann bekannten Typen von Bindungen, wobei keine Ausbildung von kovalenten Bindungen zwischen Wirkstoff und Trägerpolymer erfolgt. Als Beispiel können, ohne auf diese beschränkt zu sein, folgende genannt werden: Wasserstoffbrückenbildung, Komplexbildung, lonenwechselwirkung.
Unter einem „Wirkstoff" oder „Effektstoff" versteht man synthetische oder natürliche, niedermolekulare Substanzen mit hydrophilen, lipophilen oder amphiphilen Eigenschaf- ten, welche in der Agrochemie, Pharmazie, Kosmetik oder Nahrungs- und Futtermittelindustrie Anwendung finden können; ebenso wie biologische aktive Makromoleküle welche in ein erfindungsgemäßes Faserflächengebilde eingebettet oder daran adsorbiert werden können, wie z.B. Peptide (wie Oligopeptide mit 2 bis 10 Aminosäureresten und Polypeptide mit mehr als 10, wie z.B. 1 1 bis 100 Aminosäureresten) sowie Enzy- me und einzel- oder doppelsträngige Nukleinsäuremoleküle (wie Oligonukleotide mit 2 bis 50 Nukleinsäuretesten und Polynukleotide mit mehr als 50 Nukleinsäureresten).
„Niedermolekular" bedeutet dabei Molmassen von weniger als 5000, insbesondere weniger als 2000, wie beispielsweise 100 bis 1000 Gramm pro Mol.
„Hochmolekular" bedeutet dabei Molmassen von mehr als 5000, insbesondere weniger als 10.000, wie beispielsweise 10.000 bis 1.000.000 Gramm pro Mol.
Die Begriffe „Wirkstoff" und „Effektstoff' werden als Synonym verwendet.
Der Begriff „Faserflächengebilde" umfasst erfindungsgemäß sowohl einzelne Polymerfasern als auch die Zusammenlagerung einer Vielzahl solcher Fasern, beispielsweise zu Faservliesen.
Ein „Wirkstoffträger" ist faserförmig ausgebildet und trägt, vorzugsweise in adsorbierter, nicht kovalent gebundener Form an der Faseroberfläche und/oder in das Fasermaterial integriert, den oder die erfindungsgemäß zu verarbeiteten Wirkstoffe. Der Wirkstoff kann dabei über die Faser gleichmäßig oder ungleichmäßig verteilt vorliegen. Der Wirkstoff kann zudem in amorpher, teilkristalliner oder kristalliner Form am/im Wirkstoffträger reversibel adsorbiert sein.
Ein „löslicher" Wirkstoffträger ist in einem wässrigen oder organischen Lösungsmittel, vorzugsweise einem wässrigen Lösungsmittel, wie beispielsweise Wasser oder einem Wasser-basiertes Lösungsmittel, in einem pH-Bereich von pH 2 bis 13, wie z.B. 4 bis 1 1 , teilweise oder vollständig löslich. Somit kann die Löslichkeit in Wasser in einem großen Bereich variieren - d.h. von guter, d.h. rascher und vollständiger oder im Wesentlichen vollständiger Löslichkeit bis sehr langsamer und vollständiger oder unvollständiger Löslichkeit.
Als polymere Bestandteile der erfindungsgemäßen Wirkstoffzubereitungen eignen sich prinzipiell alle Polymere, die in einem Temperaturbereich zwischen 0 und 2400C, einem Druckbereich zwischen 1 und 100 bar, einem pH-Bereich von 0 bis 14 oder lo- nenstärken bis 10 mol/l in Wasser oder/und in organischen Lösungsmitteln löslich sind.
Ein „abbaubarer" Wirkstoffträger liegt dann vor, wenn die Faserstruktur durch chemi- sehe, biologische oder physikalische Prozesse, wie beispielsweise durch Einwirkung
von Licht oder andere Strahlung, Lösungsmittel, chemische oder biochemische Oxida- tion, Hydrolyse, Proteolyse teilweise oder vollständig zerstört wird.
Biochemische Prozesse können dabei von Enzymen oder Mikroorganismen, wie bei- spielsweise von Prokaryonten oder Eukaryonten, wie z.B. Bakterien, Hefen, Pilzen vermittelt werden.
Unter „Mischbarkeit" von Polymeren versteht man erfindungsgemäß, dass bei einem Gemisch aus wenigstens zwei verschiedenen Polymeren ein Polymer für das andere als Lösungsmittel fungieren kann. Dies bedeutet, dass ein einphasiges System zwischen den zwei verschiedenen Polymeren entsteht. Bei nicht-mischbaren Komponenten werden entsprechend zwei unterschiedliche Phasen vorliegen.
Unter einem „Kompositpolymer" versteht man erfindungsgemäß ein homogenes oder inhomogenes Gemisch aus wenigstens einer faserbildenden Polymerkomponente mit wenigstens einem niedermolekularen oder hochmolekularen Zusatz, wie insbesondere einen nicht einpolymerisierbaren Zusatz, wie beispielsweise einen Wirkstoff oder Effektstoff gemäß obiger Definition.
Unter einer „prozessierten Form" eines Faserflächengebildes versteht man, dass das ursprünglich bei der Herstellung des Faserflächengebildes anfallende Produkt weiterverarbeitet wird; beispielsweise, dass die Fasern komprimiert oder tablettiert werden, auf einen weiteren Träger aufgebracht werden und/oder einer Zerkleinerung zur Verkürzung der Faserlänge unterzogen werden.
Werden keine anderen Angaben gemacht, so betreffen Molekulargewichtsangaben für Polymere Mn oder Mw-Werte.
2. Bevorzugte Ausführungsformen
Ein erster Gegenstand der Erfindung betrifft wirkstoffhaltiges Faserflächengebilde, umfassend einen faserförmigen, polymeren löslichen und/ oder abbaubaren Wirkstoffträger und einen, mit dem Träger assoziierten, und von dem Faserflächengebilde freisetzbaren niedermolekularen Wirkstoff oder mehrere Wirkstoffe, wie z. B. 2, 3, 4 oder 5 Wirkstoffe, aus der gleichen oder aus verschiedenen Wirkstoffklassen oder mit gleicher oder unterschiedlicher Wirkungsweise, wobei der Träger ein Kompositpolymer ist, das
ein Gemisch aus zwei oder mehreren, wie z. B. 2, 3, 4 oder 5 Polymerkomponenten umfasst, wobei sich diese wenigstens zwei Polymerkomponenten in wenigstens einer Eigenschaft unterscheiden, die ausgewählt ist unter a) Löslichkeit in wässrigen oder nicht-wässrigen Lösungsmitteln, b) Molekulargewicht (Mn oder Mw) c) Glasübergangstemperatur (Tg)/Schmelzpunkt (Smp); und d) Abbaubarkeit, wie chemische oder insbesondere biologische Abbaubarkeit, wie z.B. durch wenigstens ein Enzym oder wenigstens einen Mikroorganismus, oxidativ oder/und hydrolytisch und/oder durch Strahlung.
Insbesondere unterscheiden sich die Polymerkomponenten bezüglich Löslichkeit und/oder Abbaubarkeit .
Weiterhin können sich die wenigstens zwei Polymerkomponenten unterscheiden durch
(i) unterschiedlichen Beladungsdichten mit dem oder den Wirkstoffen;
(ii) unterschiedliche spezifische Oberfläche der Fasern (d.h. Durchmesser);
(iii) unterschiedliche physikalische Strukturierung der Fasern, wie z.B. in Form von unterschiedlicher Porosität und/oder Oberflächenrauhigkeit (Topographie), Phasen- trennung.
Insbesondere ist dabei der wenigstens eine Wirkstoff in amorpher oder teilkristalliner Form enthalten
Beispielsweise kann in dem Faserflächengebilde der Wirkstoff in den Träger integriert (eingebettet) und/oder daran adsorbiert sein.
Insbesondere bevorzugt ist der faserförmige, wirkstoffhaltige Träger durch ein Spinnverfahren erhältlich.
Insbesondere wird der faserförmige, wirkstoffhaltige Träger durch ein Elektrospinnver- fahren mit einer elektroverspinnbaren Lösung, enthaltend, jeweils in gelöster Form, den wenigstens einen Wirkstoff und das Gemisch aus wenigstens zwei Polymerkomponenten, hergestellt.
Die in dem erfindungsgemäßen Faserflächengebilde enthaltenen Polymerkomponenten sind miteinander mischbar oder wenigstens zwei der Polymerkomponenten sind nicht miteinander mischbar.
Die erfindungsgemäß verwendeten Polymerkomponenten sind insbesondere ausgewählt unter synthetischen Polymeren und natürlichen Polymeren (Biopolymeren), wie insbesondere amphiphilen selbstassemblierenden Proteinen, wobei die Biopolymere gegebenenfalls zusätzlich chemisch und/oder enzymatisch modifiziert sind.
Die amphiphilen selbstassemblierenden Proteine sind z.B. Microbead-bildende Proteine, oder intrinsisch entfaltete Proteine. Beispielsweise ist das amphiphile selbstas- semblierende Protein ein Seidenprotein, wie insbesondere ein Spinnenseidenprotein, vorzugsweise ein C16-, R16- oder S16-Protein (vgl. SEQ ID NO: 2, 4 bzw. 6); oder ein von diesen Proteinen abgeleitetes verspinnbares Protein mit einer Sequenzidentität von wenigstens etwa 50%, wie z.B. wenigsten 60, 70, 80, 90, 95, 96, 97, 98 oder 99% Sequenzidentität.
Das synthetische Polymer ist entweder ein Homo- oder ein Copolymer.
Das Trägerpolymer ist insbesondere ausgewählt unter a) Mischungen aus mindestens 2 mischbaren synthetischen Homo- oder Copolyme- ren; b) Mischungen aus mindestens 2 nicht mischbaren synthetischen Homo- oder Copolymer c) Mischungen aus mindestens 2 mischbaren Biopolymeren; d) Mischungen aus mindestens 2 nicht mischbaren Biopolymeren; e) Mischungen aus mindestens einem synthetischen Homo- oder Copolymer und mindestens einem Biopolymer, die miteinander mischbar sind; und f) Mischungen aus mindestens einem synthetischen Homo- oder Copolymer und mindestens einem Biopolymer, die miteinander nicht mischbar sind.
Dabei weisen die Polymerkomponenten unabhängig voneinander Molmassen im Bereich von etwa 500 bis 10.000.000, wie z.B. 1.000 - 1.000.000 oder 10.000 - 500.000 oder 20.000 - 250.000 auf.
Der Durchmesser der Wirkstoffträgerfasern beträgt etwa 10 nm bis 100 μm, wie z.B.
50 nm bis 10 μm, oder 100 nm bis 2 μm.
Weiterhin kann erfindungsgemäß die Wirkstoffbeladung etwa 0.01 bis 80 Gew.-%, wie z. B. 1 bis 70 Gew.-% oder 10 bis 50 Gew.-%, bezogen auf den Feststoffgehalt des Faserflächengebildes betragen.
Insbesondere ist das erfindungsgemäße Faserflächengebilde ausgewählt unter Polymerfasern und Polymervliesstoffen.
Außerdem können die Fasern eine zusätzliche physikalische Strukturierung aufweisen, wie z.B. Porosität. Weiterhin kann zur Erhöhung der Viskosität bzw. Viskoelastizität und zur besseren Verspinnbarkeit der Lösung mindestens ein weiteres Polymer enthalten sein. Außerdem kann mindestens ein niedermolekulares Additiv, wie z. B. ein organisches oder anorganisches Salz zur Erhöhung der elektrischen Leitfähigkeit der verspinnbaren Lösung, Penetrationshilfsmittel für Wirkstoffe, Hilfsstoffe zur Erhöhung der Bioverfügbarkeit etc., enthalten sein.
In den Faserflächengebilden ist der Wirkstoff insbesondere molekular dispers (d.h. die Wirkstoffmoleküle liegen einzeln in der Polymermatrix vor, sind also darin gelöst) oder nanopartikulär dispers (d.h. die Moleküle sind zu Teilchen (Clustern) aggregiert mit Dimensionen in Bereich weniger Nanometer) in den Fasern enthalten.
Gegenstand der Erfindung sind auch wirkstoffhaltige Formulierungen, umfassend ein Faserflächengebilde nach obiger Definition in prozessierter Form, gegebenenfalls in Kombination mit wenigstens einem weiteren Formulierungshilfsmittel, welches das Faserflächengebilde in zerkleinerter oder nicht-zerkleinerter Form umfasst. Beispielsweise kann das Faserflächengebilde in kompaktierter (gepresster) Form (wie Tabletten oder Kapseln), in Pulverform, oder aufgetragen auf ein Trägersubstrat vorliegen.
Erfindungsgemäße Formulierungen sind insbesondere ausgewählt unter kosmetischen (insbesondere haut- und haarkosmetischen), human- und tierpharmazeutischen, agrochemischen (insbesondere Fungiziden, Herbiziden, Insektiziden und sonstige Pflanzenschutzformulierungen) Formulierungen, Nahrungs- und Futtermittelzusätzen (wie z.B. Nahrungs- und Futterergänzungsmitteln).
Gegenstand der Erfindung ist auch die Verwendung eines wirkstoffhaltigen Faserflächengebildes nach obiger Definition zur Herstellung einer wirkstoffhaltigen Formulierung nach obiger Definition, und insbesondere die Verwendung einer wirkstoffhaltigen Formulierung nach obiger Definition zur kontrollierten Abgabe (Freisetzung) eines darin enthaltenen Wirkstoffs.
Schließlich ist Gegenstand der Erfindung ein Verfahren zur Herstellung eines Faserflächengebildes nach obiger Definition, wobei man a) den wenigstens einen Wirkstoff zusammen mit den Trägerpolymerkomponenten in einer gemeinsamen flüssigen Phase mischt und b) anschließend die Einbettung des Wirkstoffes in eine polymere Kompositfaser durch Spinnverfahren durchführt.
Dabei mischt man den wenigstens einen Wirkstoff und die Polymerkomponenten in einer Lösungsmittelphase und verspinnt diese Mischung.
Es kann aber auch der wenigstens eine Wirkstoff und die Polymerkomponenten in einem Gemisch aus wenigstens zwei miteinander mischbaren Lösungsmitteln gemischt werden, wobei Wirkstoffe und Polymere mindestens in einem der Lösungsmittel löslich sind, und die so erhaltene Mischung versponnen wird.
Das Spinnverfahren kann ein Elektrospinnverfahren oder ein Zentrifugen(Rotor)spinn- verfahren sein. Insbesondere wird das Spinnverfahren bei einer Temperatur im Bereich von etwa 0 bis 900C durchgeführt.
Die vorliegende Erfindung betrifft außerdem die Faserflächengebilde, wobei
(i) der Durchmesser der Fasern 10 nm bis 100 μm, bevorzugt 50 nm bis 10 μm, besonders bevorzugt 100 nm bis 2 μm, beträgt (ii) die Effektstoffbeladung von 0.01 bis 80 Gew.-%, bevorzugt 1 bis 60 Gew.-%, besonders bevorzugt 5 bis 50 Gew.-%, bezogen auf den Gesamtfeststoff der Formulierung, beträgt, (iii) der Effektstoff röntgenamorph oder teilkristallin (als feinteilige Dispersion) in den
Fasern zusammen mit den Polymeren und ggf. Additiven vorliegt
Für bestimmte Anwendungen könnte es vorteilhaft sein, wenn die Polymere nach dem Entfernen vom gemeinsamen Lösungsmittel sich entmischen und zwei oder mehr Phasen bilden.
Durch Spinnprozesse können aus wässrigen Lösungen oder organischen Lösungsmitteln, in denen synthetische oder Biopolymere und Effektstoffe gelöst oder dispergiert vorliegen, Flächengebilde (Fasern, Vliesstoffe, Beschichtungen) hergestellt werden.
Diese Polymer- und wirkstoffreichen Phasen können als Beschichtungen (Lagen auf einem Substrat), als mechanisch stabile Wirkstoff-enthaltende Polymerstrukturen abgetrennt und ggf. getrocknet genutzt, sowie zu Tabletten oder Kapseln verarbeitet werden.
Gegenstand der Erfindung sind außerdem Faserflächengebilde nach obiger Definition, welche im wesentlichen frei ist von niedermolekularen Wirkstoffen und/ oder hochmolekularen Wirkstoffen.
Schließlich betrifft die Erfindung die Verwendung eines solchen wirkstofffreien Faserflächengebildes zu Herstellung einer wirkstoffhaltigen Formulierung, insbesondere wo- bei die Formulierung ausgewählt ist unter kosmetischen, human- und tierpharmazeutischen, agrochemischen Formulierungen, Nahrungs- und Futtermittelzusätzen.
Dazu kann z.B. das wirkstofffreie Faserflächengebilde im wesentlichen wie hierin beschrieben durch Verspinnen geeigneter Polymere hergestellt werden und in einem nächsten Schritt ein oder mehrere Wirkstoffe damit assoziiert, wie z.B. adsorbiert, d.h. nicht-kovalent gebunden werden.
3. Weitere Ausgestaltungen der Erfindung
(i) Formulierung von Wirkstoffen
Die erfindungsgemäßen Formulierungen von Wirkstoffen können unter Verwendung von synthetischen und/oder Biopolymeren auf verschiedene Art und Weise nach bekannten Methoden hergestellt werden. Die Wirkstoffe können z.B. durch Spinnverfahren in Faserflächengebilde verpackt oder verkapselt werden.
Die Fasern und Flächengebilde aus Polymer-Wirkstoff-Kompositionen können mit allen, dem Fachmann bekannten Spinnverfahren ausgehend von einer Lösung oder einer feinteiligen Dispersion oder einem Gel hergestellt werden. Besonders geeignet sind Spinnverfahren aus der Lösung oder einer feinteiligen Dispersion, darunter besonders bevorzugt sind Zentrifugenspinnen (Rotorspinnen) und Elektrospinnen (elektrostatisches Spinnen).
Bei der Verspinnung von Formulierungen zu Fasern sind Faserdurchmessern von 10 nm bis 100 μm, bevorzugt mit Durchmesser von 50 nm bis 10 μm, besonders bevorzugt von 100 nm bis 2 μm, grundsätzlich geeignet.
Beim Elektroverspinnen (elektrostatisches Spinnen) wird die zu formulierende Lösung oder feinteilige Dispersion in ein elektrisches Feld mit der Stärke zwischen 0,01 bis 10 kV/cm, besonders bevorzugt zwischen 1 und 6 kV/cm und ganz besonders bevorzugt zwischen 2 und 4 kV/cm, eingebracht. Sobald die elektrischen Kräfte die Oberflächenspannung der Formulierung übersteigen, erfolgt der Massentransport in Form eines Jets auf die gegenüberliegende Elektrode. Das Lösungsmittel verdampft im Zwischenelektrodenraum und der Feststoff der Formulierung liegt dann als Fasern auf der Ge- genelektrode vor. Die Spinnelektrode kann Düsen- oder Spritzen- basiert sein oder Walzengeometrie haben. Das Spinnen kann in beiden vertikalen Richtungen (von unten nach oben und von oben nach unten) und in horizontaler Richtung erfolgen.
Ein weiteres erfindungsgemäß geeignetes Verfahren ist das Zentrifugenspinnen (Ro- torspinnen). Bei diesem Verfahren wird das Ausgangsmaterial als Lösung oder feinteilige Dispersion in ein Feld mit Gravitationskräften eingebracht. Dazu wird das Faserrohmaterial in ein Behältnis gegeben und das Behältnis in Rotation versetzt, wobei das fluidisierte Faserrohmaterial durch Zentripetal- bzw. Zentrifugalkräfte aus dem Behältnis in Form von Fasern ausgetragen wird. Die Fasern können anschließend durch Gasstrom abtransportiert und zu Flächengebilden zusammengelegt werden.
Die Formulierung der Wirkstoffe kann erfindungsgemäß durch Einschließen in die durch die erfindungsgemäßen Verfahren hergestellten Faserflächengebilde erfolgen. Dieser Prozess umfasst meist zwei Schritte. Im ersten Schritt wird eine Spinnlösung aus Wirkstoff(en) und Trägerpolymer(en) durch Mischen der Komponenten in einer gemeinsamen Phase hergestellt. Dazu können Wirkstoff und Polymere direkt durch ein
Lösungsmittel oder eine Lösungsmittelmischung in Lösung gebracht werden. Alternativ können Wirkstoff und Polymere zunächst in unterschiedlichen Lösungsmitteln gelöst und die Lösungen im Anschluss miteinander vermischt werden, so dass wiederum eine gemeinsame Phase entsteht. Bei der gemeinsamen Phase kann es sich auch um eine molekular-disperse Phase oder eine kolloidal-disperse Phase handeln.
Das Lösen von Wirkstoff und Polymer in verschiedenen Lösungsmitteln und das anschließende Mischen beider Lösungen sind insbesondere dann von Vorteil, wenn sich Wirkstoff und Polymer nicht in einem gemeinsamen Lösungsmittel oder Lösungsmittel- gemisch lösen lassen. Auf diese Art und Weise lassen sich auch kolloidal-disperse Lösungen hydrophober Wirkstoffe herstellen, indem der in einem geeigneten Lösungsmittel gelöste Wirkstoff in ein anderes Lösungsmittel verdünnt wird, in dem dieser Wirkstoff unlöslich ist.
Geeignete Lösungsmittel sollten grundsätzlich die Ausbildung von Faserflächengebilden nicht behindern und den Wirkstoff nicht irreversibel inaktivieren.
Als Lösungsmittel kommen zum einen Wasser und auch Mischungen aus Wasser und wassermischbaren, organischen Lösungsmitteln in Frage. Bespiele für geeignete, was- sermischbare Lösungsmittel sind, ohne dabei einschränkend zu sein, Alkohole wie Methanol, Ethanol und Isopropanol, fluorierte Alkohole, wie Hexafluorisopropanol und Trifluorethanol, Alkanone, wie Aceton; oder auch Sulfoxide, wie z.B. Dimethylsulfoxid; oder Formamide wie Dimethylformamid; oder andere organische Lösungsmittel, wie z.B. Tetrahydrofuran und Acetonitril oder N-Methyl-2-pyrrolidon oder Formiat. Im AII- gemeinen kann mit allen Lösungsmitteln und Lösungsmittelgemischen gearbeitet werden, in denen sich die Trägerpolymere lösen lassen. Weitere Beispiele für geeignete Lösungsmittel sind ionische Flüssigkeiten wie z.B. 1-Ethyl(-3-methylimidazolin (EMIM)- Acetat, wässrige Lösungen chaotroper Salze, wie z.B. Harnstoff, Guanidiniumhydro- chlorid und Guanidiniumthiocyanat, oder organische Säuren, wie z.B. Ameisensäure, Essigsäure etc.
In weiterer Ausführungsform können Lösungsmittel oder Lösungsmittelgemische verwendet werden, die nicht mit Wasser mischbar sind. Der Begriff "nicht mit Wasser mischbares organisches Lösungsmittel" beschreibt organische Lösungsmittel, die in Wasser eine Löslichkeit von weniger als 50%, vorzugsweise weniger als 25%, beson-
ders bevorzugt weniger als 10% ganz besonders bevorzugt 40 weniger als 10% hat, in einer äussert bevorzugten Ausführungsform weniger als 5%.
Folgende Lösungsmittel seien beispielhaft genannt, ohne jedoch einschränkend zusein: Cyclohexan, Cyclopentan, Pentan, Hexan, Heptan, 2-Methylpentan, 3- Methylpentan, 2-Methylhexan, 3-Methylhexan, 2-Methylbutan, 2,3-Dimethylbutan, Me- thylcyclopentan, Methylcyclohexan, 2,3-Dimethylpentan, 2,4-Dimethylpentan, Benzol, I- Penten, 2-Penten, I-Hexen, I-Hepten, Cyclohexen, I-Butanol, Ethylvinylether, Propy- lether, Isopropylether, Butylvinylether, Butylethylether, 1 ,2-Epoxybutan, Furan, Tetra- hydropyran,l-Butanal, 2-Methylpropanal, 2-Pentanon, 3-Pentanon, Cyclohexanon, FIu- orbenzol, Hexafluorbenzol, Ethylformiat, Propylformiat, Isopropylformiat, Ethylacetat, Vinylacetat, Isopropylacetat, Ethylpropionat, Methylacrylat, Ethylacrylat, Methylmethac- rylat, Chlorethan, I-Chlorpropan, 2-Chlorpropan, 1 -Chlorbutan, 2-Chlorbutan, 1-Chlor- 2-methylpropan, 2-Chlor-2-methylpropan, l-Chlor-3-methylbutan, 3-Chlorpropen, Dich- lormethan, Trichlormethan, Tetrachlormethan, 1 -Dichlorethan, 1 ,2-Dichlorethan, 1 ,2- Dichlorpropan, 1 ,1 ,1-Trichlorethan, 1 ,2-Dichlorethylen, 1 ,2-Dichlorethylen, Trichlorethy- len, Brommethan, I -Brompropan, 2-Brompropan, I-Brombutan, 2-Brombutan, 2-Brom- 2-methylpropan, Brommethylen, lodmethan, lodethan, 2-lodpropan, Trichlorfluor- methan, Dichlorfluormethan, Dibromfluormethan, Bromchlormethan, Bromchlorfluormethan, 1 ,1 ,2-Trichlor-1 ,2,2-trifluorethan, 1 ,1 ,2,2-Tetrachlordifluorethan, 1 ,2- Dibromtetrafluorethan, 1 ,2-Dibrom-l ,I-Diflourethan,1 ,1 -Dichlor-2,2-Difluorethylen, Pro- pionitril, Acrylonitril, Methacrylonitril, Triethylamin, Schwefelkohlenstoff, I-Butanthiol, Methylsulfid, Ethylsulfid und Tetramethylsilan.
ii) Fasernflächengebilde und deren Polymerkomponenten
Die erfindungsgemäßen Fasern in Fasernflächengebilden können aus einer, zwei, drei oder mehr Phasen bestehen.
In einer weiteren Ausführungsform der vorliegenden Erfindung besteht die Faser der erfindungsgemäßen Faserflächengebilde aus mindestens drei Phasen, wobei die eine Phase aus amorphen oder teilkristallinen oder kristallinen Partikeln des Wirkstoffs besteht, die andere Phase eine molekulardisperse Verteilung des Wirkstoffs in einer Polymermatrix darstellt, und die dritte Phase eine wirkstofffreie Polymerphase darstellt.
In einer weiteren Ausführungsform der vorliegenden Erfindung besteht die Faser der erfindungsgemäßen Fasergebilde aus mindestens zwei Phasen, wobei die eine Phase aus amorphen oder teilkristallinen oder kristallinen Partikeln des Wirkstoffs besteht, die andere Phase eine molekulardisperse Verteilung des Wirkstoffs in einer Polymermatrix darstellt.
In einer weiteren Ausführungsform der vorliegenden Erfindung besteht die Faser der erfindungsgemäßen Fasernflächengebilde aus mindestens zwei Phasen, wobei die eine Phase aus amorphem oder teilkristallinem oder kristallinem Wirkstoff besteht, und die andere Phase eine wirkstofffreie Polymermatrix darstellt.
In einer weiteren, bevorzugten Ausführungsform der vorliegenden Erfindung besteht die Faser der erfindungsgemäßen Faserflächengebilde aus einer molekulardispersen Verteilung des Wirkstoffs in einer Polymermatrix.
Bei Verwendung von nicht mischbaren Polymeren A und B kann es zur Ausbildung weiterer Phasen, z.B. die aus Wirkstoff und Polymer A mit geringen Mengen an Polymer B, bzw. aus Wirkstoff und Polymer B mit geringen Mengen an Polymer A bestehen.
Als polymere Bestandteile der erfindungsgemäßen Wirkstoffzubereitungen eignen sich prinzipiell alle natürlichen und synthetischen Polymere, die in einem Temperaturbereich zwischen 0 und 2400C, einem Druckbereich zwischen 1 und 100 bar, einem pH- Bereich von 0 bis 14 oder lonenstärken bis 10 mol/l in Wasser oder/und in organischen Lösungsmitteln löslich sind.
Es können ein oder mehrere Polymere eingesetzt werden. Die Molmassen der verwendeten Polymere liegen im Bereich von 500 - 10.000.000 g/mol, bevorzugt im Bereich 1.000 - 1.000.000 g/mol. Grundsätzlich kommen alle für den Anwendungsbereich Pharmakologie, Pflanzenschutz, Kosmetik, Nahrungs- und Futtermittelherstellung geeigneten Polymere in Betracht.
Die Polymere mit hohem Molekulargewicht (ab 500.000) sind dann vorteilhaft, wenn ein schlecht löslicher Effektstoff formuliert werden sollte. Diese Polymere erfordern eine sehr geringe Konzentration in der Formulierung, um daraus Faserflächengebilde
zu erhalten. Entsprechend niedrig wird auch die Effektstoffkonzentration in der Formulierung sein.
Ist es beabsichtigt, Zubereitungen von einem amorphen Wirkstoff mit verbesserter Langzeitstabilität zu erhalten, sollen die Polymere entweder eine starke nicht-kovalente Wechselwirkung mit Wirkstoff aufweisen oder ihre Glasübergangstemperatur (Tg) vorzugsweise oberhalb der Spinntemperatur haben. In diesem Fall bleibt der Wirkstoff nach dem Entfernen des Lösungsmittels im Polymer molekulardispers oder feindispers gelöst, da im ersten Fall die Wechselwirkung mit dem Träger und im zweiten Fall die fehlende Mobilität der Polymerketten unterhalb der Glasübergangstemperatur die Bewegung der Wirkstoffmoleküle behindern. Gegebenenfalls kann noch mindestens einen Zusatzstoff enthalten sein, der die Agglomerierung des Wirkstoffes verhindert.
Geeignete synthetische Polymere sind z. B. ausgewählt aus der Gruppe bestehend aus Homo- und Copolymerisaten von aromatischen Vinylverbindungen, Homo- und Copolymerisaten von Alkylacrylaten, Homo- und Copolymerisaten von Alkylmethacryla- ten, Homo- und Copolymerisaten von α-Olefinen, Homo- und Copolymere von aliphati- schen Dienen, Homo- und Copolymerisaten von Vinylhalogeniden, Homo- und Copolymerisaten von Vinylacetaten, Homo- und Copolymerisaten von Acrylnitrilen, Homo- und Copolymerisaten von Urethanen, Homo- und Copolymerisaten von Vinylamiden und Copolymeren aufgebaut aus zwei oder mehr der die vorstehend genannten Polymere bildenden Monomereinheiten.
Als Trägerpolymere kommen insbesondere Polymere auf Basis der folgenden Mono- mere in Betracht:
Acrylamid, Adipinsäure, Allylmethacrylat, alpha-Methylstyrol, Butadien, Butandiol, Bu- tandioldimethacrylat, Butandioldivinylether, Butandioldimethacrylat, Butandiolmonoac- rylat, Butandiolmonomethacrylat, Butandiolmonovinylether, Butylacrylat, Butylmethac- rylat, Cyclohexylvinylether, Diethylenglycoldivinylether, Diethylenglycolmonovinylether, Ethylacrylat, Ethyldiglycolacrylat, Ethylen, Ethylenglycolbutylvinylether, Ethylenglycol- dimethacrylat, Ethylenglycoldivinylether, Ethylhexylacrylat, Ethylhexylmethacrylat, E- thylmethacrylat, Ethylvinylether, Glycidylmethacrylat, Hexandioldivinylether, Hexandi- olmonovinylether, Isobuten, Isobutylacrylat, Isobutylmethacrylat, Isopren, Isopropylac- rylamid, Methylacrylat, Methylenbisacrylamid, Methylmethacrylat, Methylvinylether, n- Butylvinylether, NMethyl-N-vinylacetamid, N-Vinylcaprolactam, N-Vinylimidazol, N- Vinylpiperidon, NVinylpyrrolidon, Octadecylvinylether, Phenoxyethylacrylat, Polytetra-
hydrofuran-2-divinylether, Propylen, Styrol, Terephthalsäure, tert-Butylacrylamid, tert- Butylacrylat, tert-Butylmethacrylat, Tetraethylenglycoldivinylether, Triethylenglycoldi- methylacrylat, Triethylenglycoldivinylether, Triethylenglycoldivinylmethylether, Tri- methylolpropantrimethacrylate, Trimethylolpropantrivinylether, Vinyl-(2- ethylhexyl)ether, Vinyl-4-tertbutyl-benzoat, Vinylacetat, Vinylchlorid, Vinyldodecylether, Vinylidenchlorid, Vinylisobutylether, Vinylisopropylether, Vinylpropylether und Vinyl-tert- butylether.
Der Begriff „Polymere" umfasst sowohl Homo- als auch Copolymere. Als Copolymere kommen sowohl statistische als auch alternierende Systeme, Blockcopolymere oder Pfropfcopolymere in Frage. Der Begriff Copolymere umfasst Polymere, die aus zwei oder mehr verschiedenen Monomeren aufgebaut sind, oder bei denen sich der Einbau mindestens eines Monomers in die Polymerkette auf verschiedene Art und Weise realisieren lässt, wie es z.B. bei den Stereoblockcopolymeren der Fall ist.
Es können auch Abmischungen von Homo- und Copolymeren sein. Die Homo- und Copolymere können mischbar und nicht mischbar sein.
Folgende Polymere sind vorzugsweise zu nennen: Polyvinylether wie z.B. Polybenzyloxyethylen, Polyvinylacetale, Polyvinylester wie z.B. Polyvinylacetat, Polyoxytetramethylen, Polyamide, Polycarbonate, Polyester, Polysilo- xane, Polyurethane, Polyacrylamide, wie z.B. Poly(N-isopropylacrylamid), Polymethac- rylamide, Polyhydroxybutyrate, Polyvinylalkohole, acetylierte Polyvinylalkohole, Polyvi- nylformamid, Polyvinylamine, Polycarbonsäuren (Polyacrylsäure, Polymethacrylsäure), Polyacrylamid, Polyitaconsäure, Poly(2-hydroxyethylacrylat), Poly(N-isopropylacryl- amid), Polysulfonsäure (Poly(2-acrylamido-2-methyl-1-propanesulfonsäure) oder PAMPS), Polymethacrylamid, Polyalkylenoxiden, z. B. Polyethylenoxiden; PoIy-N- vinylpyrrolidon; Maleinsäuren, Poly(ethylenimin), Polystyrolsulfonsäure, Polyacrylate, wie z.B. Polypheno xyethylacrylat, Polymethylacrylat, Polyethylacrylat, Polydodecylac- rylat, Poly(ibornylacrylat),Poly(n-butylacrylat), Poly(t-butylacrylat), Polycyclohexylacry- lat, Poly(2-ethylhexylacrylat), Polyhydroxypropylacrylat, Polymethacrylate, wie z.B. Polymethylmethacrylat, Poly(n-amylmethacrylat), Poly(n-butylmethacrylat), Polyethyl- methacrylat, Poly(hydroxypropylmethacrylat), Polycyclohexylmethacrylat, Poly(2- e- thylhexylmethacrylat), Polylaurylmethacrylat, Poly(t-butylmethacrylat), Polybenzyl- methacrylat, Poly(ibornylmethacrylat), Polyglycidylmethacrylat und Polystearylmethac- rylat, Polystyrol, sowie Copolymere auf Basis von Styrol, z.B. mit Maleinsäureanhydrid,
Styrol-Butadien-Copolymere, Methylmethacrylat-Styrol-Copolymere, N-Vinylpyrrolidon- Copolymere, Polycaprolactone, Polycaprolactame, Poly(N-vinylcaprolactam).
Ganz besonders bevorzugt sind Poly-N-vinylpyrrolidon, Polymethylmethacrylat, Acry- lat-Styrol-Copolymere, Polyvinylalkohol, Polyvinylacetat, Polyamid, Polyester
Weiter sind natürliche Polymere oder Biopolymere geeignet:
Als nicht limitierende Beispiele sind zu nennen: Cellulose, Celluloseether wie z.B. Me- thylcellulose (Substitutionsgrad 3 - 40 %), Ethylcellulose, Butylcellulose, Hydroxy- methylcellulosen; Hydroxyethylcellulosen; Hydroxypropylcellulosen, Isopropylcellulose,
Cellulose-Ester, wie z.B. Celluloseacetat, Stärken, modifizierte Stärken wie z.B Methyl- ether-Stärke, Gummi arabicum, Chitin, Schellak, Gelatine, Chitosan, Pektin, Casein,
Alginat, sowie Copolymere und Blockcopolymere aus den Monomeren der o.g. Verbin- düngen.; sowie Nukleinsäuremoleküle.
Insbesondere sind erfindungsgemäß eingesetzte Biopolymere biologisch abbaubar.
Weitere geeignete bioabbaubare Biopolymere sind amphiphile, selbstassemblierende Proteine. Amphiphile, selbstassemblierende Proteine bestehen aus Polypeptiden, die aus Aminosäuren, insbesondere aus den 20 natürlich vorkommenden Aminosäuren, aufgebaut sind. Die Aminosäuren können auch modifiziert, beispielsweise acetyliert, glycosyliert, farnesyliert, sein.
Geeignete amphiphile, selbstassemblierende Proteine sind insbesondere solche Prote- ine, welche Protein-Microbeads ausbilden können und die in der WO-A-20077082936 beschrieben sind, worauf hier ausdrücklich Bezug genommen werden soll.
Weitere geeignete Proteine für die Formulierung von Wirkstoffen mittels Spinnverfahren sind Seidenproteine. Darunter verstehen wir im Folgenden solche Proteine, die hoch repetitive Aminosäuresequenzen enthalten und im Tier in einer flüssigen Form gespeichert werden und bei deren Sekretion durch Scherung oder Verspinnen Fasern entstehen (Craig, C. L. (1997) Evolution of arthropod silks. Annu. Rev. Entomol. 42: 231-67).
Besonders geeignete Proteine für die Formulierung von Wirkstoffen mittels Spinnverfahren sind Spinnenseidenproteine, die in ihrer ursprünglichen Form aus Spinnen isoliert werden konnten.
Ganz besonders geeignete Proteine sind Seidenproteine, die aus der „Major Ampulla- te"-Drüse von Spinnen isoliert werden konnten.
Bevorzugte Seidenproteine sind ADF3 und ADF4 aus der der „Major Ampullate"-Drüse von Araneus diadematus (Guerette et al., Science 272, 5258:1 12-5 (1996)).
Ebenso geeignete Proteine für die Formulierung von Wirkstoffen mittels Spinnverfahren sind natürliche oder synthetische Proteine, die sich von natürlichen Seidenproteinen ableiten und welche unter Verwendung gentechnologischer Arbeitsmethoden hete- rolog in prokaryontischen oder eukaryontischen Expressionssystemen hergestellt wur- den. Nichtlimitierende Beispiele für prokaryontische Expressionsorganismen sind E- scherichia coli, Bacillus subtilis, Bacillus megaterium, Corynebacterium glutamicum u.a.. Nichtlimitierende Beispiele für eukaryontische Expressionsorganismen sind Hefen, wie Saccharomyces cerevisiae, Pichia pastoris u.a., filamentöse Pilze, wie Aspergillus niger, Aspergillus oryzae, Aspergillus nidulans, Trichoderma reesei, Acremonium chrysogenum u.a., Säugetierzellen, wie Heia-Zellen, COS-Zellen, CHO-Zellen u.a., Insektenzellen, wie Sf9-Zellen, MEL-Zellen u.a..
Besonders bevorzugt für die Formulierung von Wirkstoffen mittels Spinnverfahren sind synthetische Proteine, welche auf Wiederholungseinheiten von natürlichen Seidenpro- teinen basieren. Neben den synthetischen repetitiven Seidenprotein-Sequenzen können diese zusätzlich eine oder mehrere natürliche nicht-repetitve Seidenprotein- Sequenzen enthalten (Winkler und Kaplan, J Biotechnol 74:85-93 (2000)).
Unter den synthetischen Seidenproteinen bevorzugt für die Formulierung von Wirkstof- fen mittels Spinnverfahren sind synthetische Spinnenseidenproteine, welche auf Wiederholungseinheiten von natürlichen Spinnenseidenproteinen basieren. Neben den synthetischen repetitiven Spinnenseidenprotein-Sequenzen können diese zusätzlich eine oder mehrere natürliche nicht-repetitve Spinnenseidenprotein-Sequenzen enthalten.
Unter den synthetischen Spinnenseidenproteinen ist bevorzugt das sog. C16-Protein zu nennen (Huemmerich et al. Biochemistry, 43(42):13604-13612 (2004)). Dieses Protein hat die in SEQ ID NO: 2 dargestellte Polypeptidsequenz.
Neben der in SEQ ID NO:2 dargestellten Polypeptidsequenz sind auch besonders funktionale Äquivalente, funktionale Derivate und Salze dieser Sequenz bevorzugt.
Weiterhin sind für die Formulierung von Wirkstoffen mittels Spinnverfahren synthetische Proteine bevorzugt, welche auf Wiederholungseinheiten von natürlichen Seiden- proteinen kombiniert mit Sequenzen von Insektenstrukturproteinen wie dem Resilin (Elvin et al., 2005, Nature 437: 999-1002) basieren.
Unter den Kombinationsproteinen aus Seidenproteinen und Resilinen besonders bevorzugt sind die R16- und S16-Proteine. Diese Proteine haben die in SEQ I D NO: 4 und SEQ ID NO: θdargestellten Polypeptidsequenzen.
Neben der in SEQ ID NO: 4 und SEQ ID NO: 6 dargestellten Polypeptidsequenzen sind auch besonders funktionale Äquivalente, funktionale Derivate und Salze dieser Sequenzen bevorzugt.
Unter „funktionalen Äquivalenten" versteht man erfindungsgemäß insbesondere auch Mutanten, welche in wenigstens einer Sequenzposition der oben genannten Aminosäuresequenzen eine andere als die konkret genannte Aminosäure aufweisen aber trotzdem die Eigenschaft zur Verpackung von Effektstoffen besitzt. „Funktionale Äquivalen- te" u mfassen som it d ie du rch eine oder meh rere Am inosäu re-Additionen, - Substitutionen, -Deletionen und/oder -Inversionen erhältlichen Mutanten, wobei die genannten Veränderungen in jeglicher Sequenzposition auftreten können, solange sie zu einer Mutante mit dem erfindungsgemäßen Eigenschaftsprofil führen. Funktionale Äquivalenz ist insbesondere auch dann gegeben, wenn die Reaktivitätsmuster zwi- sehen Mutante und unverändertem Polypeptid qualitativ übereinstimmen.
„Funktionale Äquivalente" im obigen Sinne sind auch „Präkursoren" der beschriebenen Polypeptide sowie „funktionale Derivate" und „Salze" der Polypeptide.
„Präkursoren" sind dabei natürliche oder synthetische Vorstufen der Polypeptide mit oder ohne die gewünschte biologische Aktivität.
Beispiele für geeignete Aminosäuresubstitutionen sind folgender Tabelle zu entnehmen:
Ursprünglicher Rest Beispiele der Substitution
AIa Ser
Arg Lys
Asn GIn; His
Asp GIu
Cys Ser
GIn Asn
GIu Asp
GIy Pro
His Asn ; GIn
Ne Leu; VaI
Leu Ne; VaI
Lys Arg ; GIn ; GIu
Met Leu ; Ne
Phe Met ; Leu ; Tyr
Ser Thr
Thr Ser
Trp Tyr
Tyr Trp ; Phe
VaI Ne; Leu
Unter dem Ausdruck „Salze" versteht man sowohl Salze von Carboxylgruppen als auch Säureadditionssalze von Aminogruppen der erfindungsgemäßen Proteinmoleküle. Salze von Carboxylgruppen können in an sich bekannter Weise hergestellt werden und umfassen anorganische Salze, wie zum Beispiel Natrium-, Calcium-, Ammonium-, Eisen- und Zinksalze, sowie Salze mit organischen Basen, wie zum Beispiel Aminen, wie Triethanolamin, Arginin, Lysin, Piperidin und dergleichen. Säureadditionssalze, wie zum Beispiel Salze mit Mineralsäuren, wie Salzsäure oder Schwefelsäure und Salze mit organischen Säuren, wie Essigsäure und Oxalsäure sind ebenfalls Gegenstand der Erfindung.
„Funktionale Derivate" erfindungsgemäßer Polypeptide können an funktionellen Aminosäure-Seitengruppen oder an deren N- oder C-terminalen Ende mit Hilfe bekannter Techniken ebenfalls hergestellt werden. Derartige Derivate umfassen beispielsweise aliphatische Ester von Carbonsäuregruppen, Amide von Carbonsäuregruppen, erhältlich durch Umsetzung mit Ammoniak oder mit einem primären oder sekundären Amin; N-Acylderivate freier Aminogruppen, hergestellt durch Umsetzung mit Acylgruppen;
oder O-Acylderivate freier Hydroxygruppen, hergestellt durch Umsetzung mit Acylgrup- pen.
Erfindungsgemäß mit umfasste „funktionale Äquivalente" sind Homologe zu den hierin konkret offenbarten Proteinen/Polypeptiden. Diese besitzen wenigstens 60%, wie z.B. 70, 80 oder 85%, wie z.B. 90, 91 , 92, 93, 94, 95, 96, 97, 98 oder 99%, Identität zu einer der konkret offenbarten Aminosäuresequenzen.
Unter „Identität" zwischen zwei Sequenzen wird insbesondere die Identität der Reste über d i e jeweils gesamte Sequenzlänge verstanden, insbesondere die Identität, die durch Vergleich mit Hilfe der Vector NTI Suite 7.1 (Vector NTI Advance 10.3.0, In- vitrogen Corp.) (bzw. Software der Firma Informax (USA) unter Anwendung der Clustal
Methode (Higgins DG, Sharp PM. Fast and sensitive multiple sequence alignments on a microcomputer. Comput Appl. Biosci. 1989 Apr;5(2):151-1 ) unter Einstellung folgen- der Parameter berechnet wird:
Multiple ahgnment parameter:
Gap opening penalty 10
Gap extension penalty 0,05
Gap Separation penalty ränge 8
Gap Separation penalty off
% identity for alignment delay 40
Residue specific gaps off
Hydrophilic residue gap off
Transition weighing 0
Pairwise alignment parameter:
FAST algorithm off
K-tuple size 1
Gap penalty 3
Window size 5
Number of best diagonals 5
Von besonderem Interesse sind weiterhin synthetische, biologisch abbaubare Polyme- re.
Die Angabe "biologisch abbaubare Polymere" soll alle Polymere umfassen, die die in DIN V 54900 gegebene Definition der Bioabbaubarkeit erfüllen, insbesondere kompostierbare Polyester.
Im Allgemeinen bedeutet die biologische Abbaubarkeit, dass die Polymere, wie z.B. Polyester, in einer angemessenen und nachweisbaren Zeitspanne zerfallen. Der Abbau kann hydrolytisch und/oder oxidativ erfolgen und zum überwiegenden Teil durch die Einwirkung von Mikroorganismen, wie Bakterien, Hefen, Pilzen und Algen, bewirkt werden. Die biologische Abbaubarkeit lässt sich z.B. dadurch bestimmen, dass Polyes- ter mit Kompost gemischt und für eine bestimmte Zeit gelagert werden. Gemäß ASTM D 5338, ASTM D 6400 und DIN V 54900 wird CO2-freie Luft beispielsweise durch gereiften Kompost während des Kompostierens strömen gelassen und dieser einem definierten Temperaturprogramm unterworfen. Hierbei wird die biologische Abbaubarkeit über das Verhältnis der Netto-Cθ2-Freisetzung der Probe (nach Abzug der C02- Freisetzung durch den Kompost ohne Probe) zur maximalen CO2-Freisetzung der Probe (berechnet aus dem Kohlenstoffgehalt der Probe) als biologische Abbaubarkeit definiert. Biologisch abbaubare Polyester zeigen in der Regel schon nach wenigen Tagen der Kompostierung deutliche Abbauerscheinungen wie Pilzbewuchs, Riss- und Lochbildung. Beispiele für biologisch abbaubare Polymere sind biologisch abbaubare PoIy- ester wie zum Beispiel Polylactid, Polycaprolacton, Polyalkylenadipatterephthalate, Polyhydroxyalkonoate (Polyhydroxybutyrat) und Polylactidglycosid. Besonders bevorzugt sind biologisch abbaubare Polyalkylenadipatterephthalate, vorzugsweise Polybu- tylenadipatterephthalate. Geeignete Polyalkylenadipatterephthalate sind z.B. in der DE
4 440 858 beschrieben (und sind kommerziell erhältlich, z.B. Ecoflex von BASF).
Die Polymerstrukturen können als Wirkstoff-enthaltende Faserflächengebilde (z.B. Polymer-Fasern, Polymer-Vliesstoffe) hergestellt und während des Spinnvorganges auf Substraten wie z. B. Mikrofaser-Vliesen abgelegt werden. Anschließend können diese zu Tabletten oder Kapseln verpresst werden.
Der Spinnlösung können zusätzlich weitere Stoffe zugegeben werden, um z.B. Kristallisation des Wirkstoffes in den Fasern später zu beeinflussen (z.B. zu hemmen) oder bevorzugte Anwendungseigenschaften, wie Bioverfügbarkeit, zu erreichen. Bevorzugte Zusatzstoffe sind dabei z.B. ionische (kationisch oder anionisch) und nichtionische Tenside. Geeignete Mengen der Zusatzstoffe in der Spinnlösung sind 0,01 Gew. % bis
5 Gew.-% .
Zusätzlich können der Spinnlösung oder den daraus hergestellten Polymer-Flächengebilden Stoffe zugesetzt werden, welche eine Sprengung der Tabletten oder Kapseln und damit eine verbesserte Dispergierung der zu den Tabletten oder Kapseln verpress- ten Polymer-Flächengebilde ermöglicht.
Erfindungsgemäß wurde insbesondere beobachtet, dass durch geeignete Kombination von Polymerkomponenten für die Ausbildung des erfindungsgemäßen wirkstoffhaltigen Fasernflächengebildes dessen Wirkstofffreisetzungseigenschaften gezielt beeinfluss- bar sind. Insbesondere gelingt dies durch Kombination von mindestens zwei Polymerkomponenten, welche sich in wenigstens einer der folgenden Eigenschaften unterscheiden: a) Löslichkeit in wässrigen oder nicht-wässrigen Lösungsmitteln, b) Molekulargewicht c) Glasübergangstemperatur und/oder Schmelzpunkt c) Abbaubarkeit (insbesondere biologische oder chemische Abbaubarkeit, wobei eine biologische Abbaubarkeit insbesondere durch wenigstens ein Enzym oder einen Mikroorganismus induziert werden kann und die chemische Abbaubarkeit beispielsweise auf hydrolytischem oder oxidativem Weg erfolgen kann. Außerdem kann eine Ab- baubarkeit auch physikalisch induziert werden, wie insbesondere durch Einwirkung von Licht)
Auf diese Weise kann die vorliegende Erfindung dazu benutzt werden, um die Wirkstofffreisetzung an den jeweiligen Bedarf des Anwenders anzupassen. Das jeweils bereitzustellende Freisetzungsprofil kann aufgrund systematischer Überlegungen oder durch wenige Vorversuche empirisch ermittelt werden. Wie in den enthaltenen Ausführungsbeispielen näher erläutert, können erfindungsgemäß durch Kombination unterschiedlicher Polymerkomponenten neue Freisetzungsprofile für Wirkstoffe erzeugt werden, welche sich den Freisetzungsprofilen, die man für die Polymereinzelkomponenten beobachtet, deutlich unterscheiden. So kann beispielsweise beobachtet werden, dass eine Wirkstofffreisetzung durch das Polymerkombination im Vergleich zu Freisetzung durch die Polymereinzelkomponenten deutlich früher einsetzt, oder dass sich die Freisetzungsprofile für erfindungsgemäße Polymerkombinationen durch eine höhere oder geringere Freisetzungsrate im Vergleich zu den Einzelkomponenten über den gesam- ten oder über einen Teil des Beobachtungszeitraumes auszeichnen.
Nicht limitierende Beispiele für besonders geeignete Polymerkombinationen sind zusätzlich zu den in den Beispielen veranschaulichten Kombinationen folgende:
Polyester/Polyacrylat (nicht mischbar)
Polyester/Polystyrol bzw. Styrol-Copolymer (Acrylat o. Butadien) (nicht mischbar) Polyamid/Spinnenseideprotein (mischbar)
Styrol bzw. Styrol-Copolymer/Spinnenseideprotein
PVP/Spinnenseideprotein (mischbar)
PVA/Spinnenseideprotein
PEO/Spinnenseideprotein Polyamid/PVP (mischbar)
Polyamid/Polyacrylsäure (mischbar)
Polymilchsäure/PVP (evtl nicht mischbar)
Polymilchsäure/Polyacrylat
Polyester/Polyacrylat/PVP (nicht mischbar) Polyester/Polymilchsäure/PVP (nicht mischbar oder mischbar)
Polyester/Stärke (mischbar)
PVP/Stärke
Cellulose bzw. Derivate/Polyacrylat
Cellulose Acetat/Polyethylen-Vinylacetat (mischbar) Polyvinylalkohol/Polyvinylacetat (mischbar)
Collagen/PVA
Collagen/PEO
Chitosan/PVA (PEO)
Polyurethan/PVP (mischbar) Polyurethan/Polyester (evtl nicht mischbar)
Polyurethan/Spinnenseideprotein (mischbar)
Polycarbonat/Polycaprolactone (Mischbar)
Polycarbonat/Polyester (mischbar)
Polyacrylat/Polyvinylchlorid (mischbar)
(iii) Wirkstoffe
Im Folgenden werden die Begriffe Wirkstoffe und Effektstoffe synonym verwendet. Da- bei handelt es sich sowohl um wasserlösliche als auch schwer-wasserlösliche Effektstoffe. Die Begriffe schwer-wasserlösliche und hydrophobe Wirk- oder Effektstoffe wer-
den synonym verwendet. Als schwer wasserlöslichen Wirkstoffe werden im Folgenden solche Verbindungen bezeichnet, deren Wasserlöslichkeit bei 200C < 1 Gew.-%, bevorzugt < 0,5 Gew.-%, besonders bevorzugt < 0,25 Gew.-%, ganz besonders bevorzugt < 0,1 Gew.-% beträgt. Als wasserlösliche Wirkstoffe werden im Folgenden solche Verbindungen bezeichnet, deren Wasserlöslichkeit bei 200C > 1 Gew.-%, bevorzugt > 10 Gew.-%, besonders bevorzugt > 40 Gew.-%, ganz besonders bevorzugt > 70 Gew.- % beträgt.
Geeignete Effektstoffe sind Farbstoffe, insbesondere die in der folgenden Tabelle genannten:
Besonders vorteilhafte Farbstoffe sind die in der folgenden Liste genannten öllöslichen oder in Öl dispergierbaren Verbindungen. Die Colour Index Nummern (CIN) sind dem Rowe Colour Index, 3. Auflage, Society of Dyers and Colourists, Bradford, England, 1971 entnommen.
Weitere bevorzugte Effektstoffe sind Fettsäuren, insbesondere gesättigte Fettsäuren, die eine Alkylverzweigung tragen, besonders bevorzugt verzweigte Eicosansäuren, wie 18-Methyl-eicosansäure.
Weitere bevorzugte Effektstoffe sind Carotinoide. Unter Carotinoide sind erfindungsgemäß folgende Verbindungen sowie deren veresterte oder glykosylierte Derivate zu verstehen: ß-Carotin, Lycopin, Lutein, Astaxanthin, Zeaxanthin, Cryptoxanthin, Citra- naxanthin , Canthaxanthin , Bixin , ß-Apo-4-carotinal, ß-Apo-8-carotinal, ß-Apo-8- carotinsäureester, Neurosporen, Echinenon, Adonirubin, Violaxanthin, Torulen, Toru- larhodin, einzeln oder als Mischung. Bevorzugt verwendete Carotinoide sind ß-Carotin, Lycopin, Lutein, Astaxanthin, Zeaxanthin, Citranaxanthin und Canthaxanthin.
Weitere bevorzugte Effektstoffe sind Vitamine, insbesondere Retinoide und deren Es- ter.
Unter Retinoide sind im Rahmen der vorliegenden Erfindung Vitamin A Alkohol (Retinol) und seine Derivate wie Vitamin A Aldehyd (Retinal), Vitamin A Säure (Retinsäure) und Vitamin A Ester (z.B. Retinylacetat, Retinylpropionat und Retinylpalmitat) gemeint. Der Begriff Retinsäure umfasst dabei sowohl all-trans Retinsäure als auch 13-cis Retinsäure. Die Begriffe Retinol und Retinal umfassen bevorzugt die all-trans Verbindungen. Als bevorzugtes Retinoid verwendet man für die erfindungsgemäßen Formulierungen all-trans-Retinol, im Folgenden als Retinol bezeichnet.
Weitere bevorzugte Effektstoffe sind Vitamine, Provitamine und Vitaminvorstufen aus den Gruppen A, B, C, E und F, insbesondere 3,4-Didehydroretinol, ß-Carotin (Provitamin des Vitamin A), Palmitinsäureester der Ascorbinsäure, Tocopherole, insbesondere α-Tocopherol sowie seine Ester, z.B. das Acetat, das Nicotinat, das Phosphat und das Succinat; weiterhin Vitamin F, worunter essentielle Fettsäuren, besonders Linolsäure, Linolensäure und Arachidonsäure, verstanden werden.
Weitere bevorzugte Effektstoffe sind lipophile, öllösliche Antioxidantien aus der Gruppe Vitamin E, d.h. Tocopherol und dessen Derivate, Gallussäureester, Flavonoide und Carotinoide sowie Butylhydroxytoluol/anisol.
Ein weiterer bevorzugter Effektstoff ist Liponsäure und geeignete Derivate (Salze, Ester, Zucker, Nukleotide, Nukleoside, Peptide und Lipide).
Weitere bevorzugte Effektstoffe sind UV-Lichtschutzfilter. Darunter sind organische Substanzen zu verstehen, die in der Lage sind, ultraviolette Strahlen zu absorbieren
und die aufgenommene Energie in Form längerwelliger Strahlung, z.B. Wärme, wieder abzugeben.
Als öllösliche UV-B-Filter können z.B. folgende Substanzen verwendet werden:
3-Benzylidencampher und dessen Derivate, z.B. 3-(4-Methylbenzyliden)campher; 4-Aminobenzoesäu rederivate, vorzugsweise 4-(Dimethylamino)benzoesäure-2- ethylhexylester, 4-( Dimethylamino)benzoesäure-2-octylester und 4-(Dimethylamino)- benzoesäureamylester; Ester der Zimtsäure, vorzugsweise 4-Methoxyzimtsäure-2- ethylhexylester, 4 Methoxyzimtsäurepropylester, 4-Methoxyzimtsäureisoamylester, 4 Methoxyzimtsäureisopentylester, 2-Cyano-3-phenyl-zimtsäure-2-ethylhexylester (Oc- tocrylene);
Ester der Salicylsäure, vorzugsweise Salicylsäure-2-ethylhexylester, Salicylsäure-4 isopropylbenzylester, Salicylsäurehomomenthylester; Derivate des Benzophenons, vorzugsweise 2-Hydroxy-4-methoxybenzophenon, 2-Hydroxy-4-methoxy-4'-methylben- zophenon, 2,2'-Dihydroxy-4-methoxybenzophenon; Ester der Benzalmalonsäure, vorzugsweise 4-Methoxybenzmalonsäuredi-2-ethylhexylester; Triazinderivate, wie z.B. 2,4,6-Trianilino-(p-carbo-2'-ethyl-1 '-hexyloxy)-1 ,3,5-triazin (Octyltriazone) und Dioctyl Butamido Triazon (Uvasorb® HEB):
Propan-1 ,3-dione, wie z.B. 1 -(4-tert. Butylphenyl)-3-(4'-methoxyphenyl)propan-1 ,3- dion.
Besonders bevorzugt ist die Verwendung von Estern der Zimtsäure, vorzugsweise 4- Methoxyzimtsäure-2 -ethylhexylester, 4-Methoxyzimtsäureisopentylester, 2-Cyano-3- phenyl-zimtsäure-2-ethylhexylester (Octocrylene).
Des Weiteren ist die Verwendung von Derivaten des Benzophenons, insbesondere 2- Hydroxy-4-methoxybenzophenon, 2-Hydroxy-4-methoxy-4"-methylbenzophenon, 2,2'- Dihydroxy-4-methoxybenzophenon sowie der Einsatz von Propan-1 ,3-dionen, wie z.B. 1 -(4-tert. Butylphenyl)-3-(4-'methoxyphenyl)propan-1 ,3-dion bevorzugt.
Als typische UV-A-Filter kommen in Frage:
Derivate des Benzoylmethans, wie beispielsweise 1-(4'-tert.Butylphenyl)-3-(4'-methoxy- phenyl)propan-1 ,3-dion, 4-tert. -Butyl-4'-methoxydibenzoylmethan oder 1-Phenyl-3-(4'- isopropylphenyl)-propan-1 ,3-dion;
Amino-hydroxy-substituierte Derivate von Benzophenonen wie z.B. N,N-Diethylamino- hydroxybenzoyl-n-hexylbenzoat.
Die UV-A und UV-B-Filter können selbstverständlich auch in Mischungen eingesetzt werden.
Geeignete UV-Filtersubstanzen sind in der folgenden Tabelle genannt.
Neben den beiden vorgenannten Gruppen primärer Lichtschutzstoffe können auch sekundäre Lichtschutzmittel vom Typ der Antioxidantien eingesetzt werden, die die photochemische Reaktionskette unterbrechen, welche ausgelöst wird, wenn UV- Strahlung in die Haut eindringt. Typische Beispiele hierfür sind Tocopherole (Vitamin E) und öllösliche Ascorbinsäurederivate (Vitamin C).
Erfindungsgemäß können geeignete Derivate (Salze, Ester, Zucker, Nukleotide, Nukleoside, Peptide und Lipide) der genannten Verbindungen als Effektstoffe verwendet werden.
Weiter bevorzugt sind sogenannte Peroxydzersetzter, d.h. Verbindungen die in der Lage sind Peroxyde, besonders bevorzugt Lipidperoxide zu zersetzen. Darunter sind organische Substanzen zu verstehen, wie z.B. 5-Pyrimidinol- sowie 3-Pyridinolderivate und Probucol.
Weiterhin handelt es sich bei den genannten Peroxidzersetzern bevorzugt um die in den Patentanmeldungen WO-A-02/07698 und WO-A03/059312, auf deren Inhalt hiermit ausdrücklich Bezug genommen wird, beschriebenen Substanzen, bevorzugt die dort beschriebenen Bor-enthaltenden oder Stickstoff-enthaltenden Verbindungen, die Peroxide oder Hydroperoxide zu den entsprechenden Alkoholen ohne Bildung radikalischer Folgestufen reduzieren können. Ferner können für diesen Zweck sterisch gehinderte Amine eingesetzt werden.
Eine weitere Gruppe sind Antiirritantien, die eine entzündungshemmende Wirkung auf durch UV-Licht geschädigte Haut besitzen. Solche Stoffe sind beispielsweise Bisabolol, Phytol und Phytantriol.
Eine weitere Gruppe von Effektstoffen sind Wirkstoffe, die im Pflanzenschutz einge- setzt werden können, beispielsweise Herbizide, Insektizide und Fungizide.
Die folgende Liste von Insektiziden zeigt mögliche Pflanzenschutzwirkstoffe auf, soll aber nicht auf diese beschränkt sein:
A.1. Organo(thio)phosphate: azinphos-methyl, chlorpyrifos, chlorpyrifos-methyl, chlor- fenvinphos, diazinon, disulfoton, ethion, fenitrothion, fenthion, isoxathion, malathion, methidathion, methyl-parathion, oxydemeton-methyl, paraoxon, parathion, phenthoate, phosalone, phosmet, phosphamidon, phorate, phoxim, pirimiphos-methyl, profenofos, prothiofos, sulprophos, tetrachlorvinphos, terbufos, triazophos, trichlorfon;
A.2. Carbamate: alanycarb, bendiocarb, benfuracarb, carbaryl, carbofuran, carbosul- fan, fenoxycarb, furathiocarb, methiocarb, methomyl, oxamyl, pirimicarb, thiodicarb, triazamate;
A.3. Pyrethroide: allethrin, bifenthrin, cyfluthrin, cyhalothrin, cyphenothrin, cyper-5 methrin, alpha-cypermethrin, beta-cypermethrin, zeta-cypermethrin, deltamethrin, es- fenvalerate, etofenprox, fenpropathrin, fenvalerate, imiprothrin, lambda-cyhalothrin, permethrin, prallethrin, pyrethrin I and II, resmethrin, silafluofen, tau-fluvalinate, tefluthrin, tetramethrin, tralomethrin, transfluthrin;
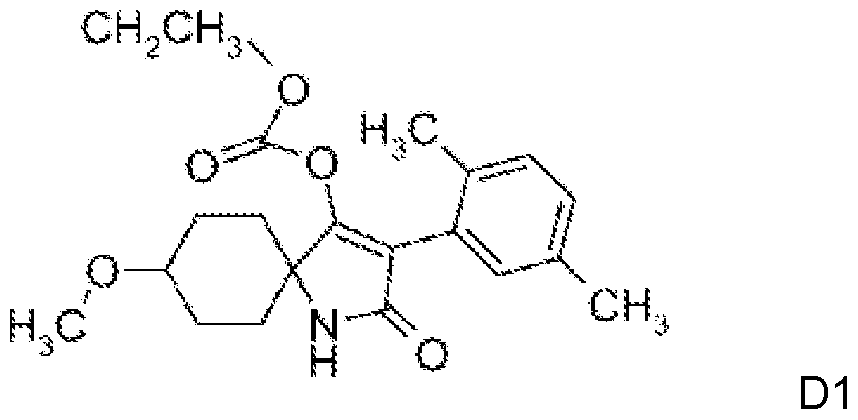
A.4. Wachstumsregulatoren: a) chitin synthesis inhibitors: benzoylureas: chlorfluazu- ron, cyramazin, diflubenzuron, flucycloxuron, flufenoxuron, hexaflumuron, lufenuron, novaluron, teflubenzuron, triflumuron; buprofezin, diofenolan, hexythiazox, etoxazole, clofentazine; b) ecdysone antagonists: halofenozide, methoxyfenozide, tebufenozide, azadirachtin; c) juvenoids: pyriproxyfen, methoprene, fenoxycarb; d) lipid biosynthesis inhibitors: spirodiclofen, spiromesifen, a tetronic acid derivative of formula D1 ,
A.5. Nicotin Rezeptor Agonisteni Antagonisten: clothianidin, dinotefuran, thiacloprid;
A.6. GABA Antagonisten: acetoprole, endosulfan, ethiprole, fipronil, vaniliprole;
A.7. Macrolid-Insektizide: abamectin, emamectin, milbemectin, lepimectin, spinosad;
A.8. MET1 I Acarizide: fenazaquin, pyridaben, tebufenpyrad, tolfenpyrad;
A.9. MET1 Il and III Verbindung: acequinocyl, fluacyprim, hydramethylnon; A.10. Entkoppler-Verbindungen: chlorfenapyr;
A.11 . Hemmer der oxidativen Phosphorylierung: cyhexatin, diafenthiuron, fenbutatin oxide, propargite;
A.12. Häutungsstörende Verbindungen: cryomazine;
A.13. Hemmer der Mixed-Function-Oxidase: piperonyl butoxide; A.14. Natriumkanalblocker: indoxacarb, metaflumizone;
A.15. Verschiedene: benclothiaz, bifenazate, flonicamid, pyridalyl, pymetrozine, sulfur, thiocyclam und aminoisothiazole Verbindungen der Formel D2,
wobei R
1 für -CH
2OCH
2CH
3 oder H und R
H für CF
2CF
2CF
3 oder CH
2CH(CH
3)
3 steht, anthranilamide Verbindungen der Formel D3:
wobei B1 für Wasserstoff oder Chlor, B2für Brom oder CF3, und RB für CH3 oder CH(CHs)2 steht, und malononitrile Verbindungen wie in JP 2002 284608, WO 02/189579, WO 02/190320, WO 02/190321 , WO 04/106677, WO 04/120399, oder der JP 2004 99597 beschrieben, N-R'-2,2-Dihalo-l-R"cyclo-propancarboxamid-2-(2,6- dichlor-α, a, a, α-trifluor-p-t o l y l ) h y d ra zo n o d e r N-R'-2,2-Di(R"')propionamid-2-(2,6- dichlor-α,α,α,α-trifluor-p-tolyl)-hydrazon, worin R' für Methyl oder Ethyl steht, HaIo für Chlor oder Brom steht, R" für Wasserstoff oder Methyl und R'" für Methyl oder Ethyl stehen;
Die folgende Liste von Fungiziden zeigt mögliche Wirkstoffe auf, soll aber nicht auf diese beschränkt sein:
1. Strobilurine
Azoxystrobin, Dimoxystrobin, Enestroburin, Fluoxastrobin, Kresoxim-methyl, Metomi- nostrobin, Picoxystrobin, Pyraclostrobin, Trifloxystrobin, Orysastrobin, (2-Chlor-5-[1-(3- methyl-benzyloxyimino)-ethyl]-benzyl)-carbaminsäuremethylester, (2-Chlor-5-[1 -(6-me- thyl-pyridin-2-ylmethoxyimino)-ethyl]-benzyl)-carbaminsäuremethylester, 2-(ortho-((2,5- Dimethylphenyl-oxymethylen)phenyl)-3-methoxy-acrylsäuremethylester;
2. Carbonsäureamide
- Carbonsäureanilide: Benalaxyl, Benodanil, Boscalid, Carboxin, Mepronil, Fenfuram, Fenhexamid, Flutolanil, Furametpyr, Metalaxyl, Ofurace, Oxadixyl, Oxycarboxin,
Penthiopyrad, Thifluzamide, Tiadinil, 4-Difluormethyl-2-methyl-thiazol-5-carbonsäure-
(4'-brom-biphenyl-2-yl)-a m i d , 4-Difluor-2-methyl-triazol-5-carbonsäure-(4'-trifluor-me- thyl-biphenyl-2-yl)-amid, 4-Difluor-2-methyl-triazol-5-carbonsäure-(4'-chlor-3'-fluor-bi- phenyl-2-yl)-amid, 3-Difluor-1-methyl-pyrazol-4-carbonsäure-(3',4'-dichlor-4-fluor-biphe- nyl-2-yl)-amid;
- Carbonsäuremorpholide: Dimethomorph, Flumorph;
- Benzoesäureamide: Flumetover, Fluopicolide (Picobenzamid), Zoxamide;
- Sonstige Carbonsäureamide: Carpropamid, Diclocymet, Mandipropamid, N-(2-(4-[3- (4-Chlor-phenyl)-prop-2-inyloxy]-3-methoxy-phenyl)-ethyl)-2-methansulfonylamino-3- methyl-butyramid, N-(2-(4-[3-(4-Chlor-phenyl)-prop-2-inyloxy]-3-methoxy-phenyl)- ethyl)-2-ethansulfonylamino-3-methyl-butyramid;
3. Azole
- Triazole: Bitertanol, Bromuconazole, Cyproconazole, Difenoconazole, Diniconazole,
Enilconazole, Epoxiconazole, Fenbuconazole, Flusilazole, Fluquinconazole, Flutria- fol,Hexaconazol, Imibenconazole, Ipconazole, Metconazole, Myclobutanil, Pencona- zole,Propiconazole, Prothioconazole, Simeconazole, Tebuconazole, Tetracona- zole,Ti"iadimenol, Triadimefon, Triticonazole;
- Imidazole: Cyazofamid, Imazalil, Pefurazoate, Prochloraz, Triflumizole;
- Benzimidazole: Benomyl, Carbendazim, Fuberidazole, Thiabendazole; - Sonstige: Ethaboxam, Etridiazole, Hymexazole;
4. Stickstoffhaltige Heterocyclylverbindungen:
- Pyridine: Fluazinam, Pyrifenox, 3-[5-(4-Chlor-phenyl)-2,3-dimethyl-isoxazolidin-3-yl]- pyridin;
- Pyrimidine: Bupirimate, Cyprodinil, Ferimzone, Fenarimol, Mepanipyrim, Nuarimol, Pyrimethanil;
- Piperazine: Triforine;
- Pyrrole: Fludioxonil, Fenpiclonil;
- Morpholine: Aldimorph, Dodemorph, Fenpropimorph, Tridemorph;
- Dicarboximide: Iprodione, Procymidone, Vinclozolin; - sonstige: Acibenzolar-S-methyl, Anilazin, Captan, Captafol, Dazomet, Diclomezine, Fenoxanil, Folpet, Fenpropidin, Famoxadone, Fenamidone, Octhilinone, Probenazole, Proquinazid, Quinoxyfen, Tricyclazole, 5-Chlor-7-(4-methyl-piperidin-1-yl)-6-(2,4,6- trifluor-phenyl)-[1 ,2,4]triazolo[1 ,5-alpyrimidin, 2-Butoxy-6-iodo-3-propyl-chromen-4-on, 3-(3-Brom-6-fluor-2-methyl-indol-1-sulfonyl)-[1 ,2,4]triazol-1-sulfonsäuredimethylamid;
5. Carbamate und Dithiocarbamate
- Carbamate: Diethofencarb, Flubenthiavalicarb, Iprovalicarb, Propamocarb, 3-(4- Chlor-phenyl)-3-(2-isopropoxycarbonylamino-3-methyl-butyrylamino)-propionsäureme- thylester, N-(1-(1-(4-cyanophenyl)ethansulfonyl)-but-2-yl) carba minsäure-(4-fluor-25 phenyl)ester;
6. Sonstige Fungizide
- Organometallverbindungen: Fentin-Salze;
- Schwefelhaltige Heterocyclylverbindungen: Isoprothiolane, Dithianon; - Organophosphorverbindungen: Edifenphos, Fosetyl, Fosetyl-aluminium, Iprobenfos, Pyrazophos, Tolclofos-methyl, Phosphorige Säure und ihre Salze;
- Organochlorverbindungen: Thiophanate Methyl, Chlorothalonil, Dichlofluanid, To- lylfluanid, Flusulfamide, Phthalide, Hexachlorbenzene, Pencycuron, Quintozene;
- Nitrophenylderivate: Binapacryl, Dinocap, Dinobuton; - Sonstige: Spiroxamine, Cyflufenamid, Cymoxanil, Metrafenone.
Die folgende Liste von Herbiziden zeigt mögliche Wirkstoffe auf, soll aber nicht auf diese beschränkt sein:
Verbindungen, die die Biosynthese von Lipiden inhibieren, z.B. Chlorazifop, Clodina- fop, Clofop, Cyhalofop, Ciclofop, Fenoxaprop, Fenoxaprop-p, Fenthiaprop, Fluazifop, Fluazifop-P, Haloxyfop, Haloxyfop-P, Isoxapyrifop, Metamifop, Propaquizafop, Quizalo- fop, Quizalofop-P, Trifop, bzw. deren Esters, Butroxydim, Cycloxydim, Profoxydim, Sethoxydim, Tepraloxydim, Tralkoxydim, Butylate, Cycloat, Diallat, Dimepiperat, EPTC, Esprocarb, Ethiolate, Isopolinate, Methiobencarb, Molinate, Orbencarb, Pebulate, Pro- sulfocarb, Sulfallat, Thiobencarb, Thiocarbazil, Triallat, Vernolat, Benfuresat, Ethofu-5 mesat und Bensulid;
ALS-Inhibitoren wie Amidosulfuron, Azimsulfuron, Bensulfuron, Chlorimuron, Chlorsul- furon, Cinosulfuron, Cyclosulfamuron, Ethametsulfuron, Ethoxysulfuron, Flazasulfuron,
Flupyrsulfuron, Foramsulfuron, Halosulfuron, Imazosulfuron, lodosulfuron, Mesosulfu- ron, Metsulf u ron, Nicosulfuron, Oxasulfuron, Primisulfuron, Prosulfuron, Pyrazosulfu- ron, Rimsulfuron, Sulfometuron, Sulfosulfuron, Thifensulfuron, Triasulfuron, Tribenuron,
Trifloxysulfuron, Triflusulfuron, Tritosulfuron, Imazamethabenz, Imazamox, Imazapic, Imazapyr, Imazaquin, Imazethapyr, Cloransulam, Diclosulam, Florasulam, Flumetsu- lam, Metosulam, Penoxsulam, Bispyribac, Pyriminobac, Propoxycarbazone, Flucarba- zone, Pyribenzoxim, Pyriftalid und Pyrithiobac; sofern der pH Wert 8 < ist;
Verbindungen, die die Photosynthese inhibieren wie Atraton, Atrazine, Ametryne, A- ziprotryne, Cyanazine, Cyanatryn, Chlorazine, Cyprazine, Desmetryne, Dimethametry-
ne, Dipropetryn, Eglinazine, Ipazine, Mesoprazine, Methometon, Methoprotryne, Pro- cyazine, Proglinazine, Prometon, Prometryne, Propazine, Sebuthylazine, Secbumeton, Simazine, Simeton, Simetryne, Terbumeton, Terbuthylazine und Terbutryne;
Protoporphyrinogen-IX Oxidase-Inhibitoren wie Acifluorfen, Bifenox, Chlomethoxyfen, Chlornitrofen, Ethoxyfen, Fluorodifen, Fluoroglycofen, Fluoronitrofen, Fomesafen, Fury- loxyfen, Halosafen, Lactofen, Nitrofen, Nitrofluorfen, Oxyfluorfen, Fluazolate, Pyraflu- fen, Cinidon-ethyl, Flumiclorac, Flumioxazin, Flumipropyn, Fluthiacet, Thidiazimin, Oxadiazon, Oxadiargyl, Azafenidin, Carfentrazone, Sulfentrazone, Pentoxazone, Benz- fendizone, Butafenacil, Pyraclonil, Profluazol, Flufenpyr, Flupropacil, Nipyraclofen und Etnipromid;
Herbizide wie Metflurazon, Norflurazon, Flufenican, Diflufenican, Picolinafen, Beflubu- tamid, Fluridone, Flurochloridone, Flurtamone, Mesotrione, Sulcotrione, Isoxachlortole, Isoxaflutole, Benzofenap, Pyrazolynate, Pyrazoxyfen, Benzobicyclon, amitrole, cloma- zone, Aclonifen, 4-(3-trifluormethylphenoxy)- 2-(4-trifluoromethylphenyl)pyrimidin, und 3-heterocyclyl-substituierte Benzoylderivate der Formel (vgl. WO-A-96/26202, WO-A- 97/411 16, WO-A-97/41 117 und WO-A-97/41 1 18)
worin die Substituenten R8 bis R13 folgende Bedeutung haben:
R8, R10 Wasserstoff, Halogen, CrC5-Alkyl, CrC5-Haloalkyl, CrC5-Alkoxy, Haloalkoxy, d-Cs-Alkylthio, CrC5-Alkylsulfinyl oder CrC5-Alkylsulfonyl; R9 bedeutet ein heterocyclisches Radikal aus der Gruppe bestehend aus Thiazol-2-yl, thiazol-4-yl, Thiazol-5-yl, lsoxazol-3-yl, lsoxazol-4-yl, lsoxazol-5-yl, 4,5-dihydroisoxazol- 3-yl, 4,5-dihydroisoxazol-4-yl und 4,5-dihydroisoxazol-5-yl, worin die genannten Radikale einen oder mehrere Substituenten tragen können z.B. mono-, di-,5 tri- oder tetrasubstituiert sein können durch Halogen, d-C4-Alkyl, Ci-C4-AIkOXy, d-C4-Haloalkyl, d- C4-Haloalkoxy oder CrC4-Alkylthio;
R11 = Wasserstoff, Halogen oder CrC5-Alkyl;
R12 = d-C6-Alkyl;
R13 = Wasserstoff oder CrC6-Alkyl, sofern der pH Wert < 8 ist;
Mitose-Inhibitoren wie Benfluralin, Butralin, Dinitramine, Ethalfluralin, Fluchloralin, i- Sopropalin, Methalpropalin, Nitralin, Oryzalin, Pendimethalin, Prodiamine, Profluralin, Trifluralin, Amiprofos-methyl, Butamifos, Dithiopyr, Thiazopyr, Propyzamide, Chlorthal, Carbetamide, Chlorpropham and Propham;
VLCFA-Inhibitoren wie Acetochlor, Alachlor, Butachlor, Butenachlor, Delachlor, Dietha- tyl, Dimethachlor, Dimethenamid, Dimethenamid-P, Metazachlor, Metolachlor, SMeto- lachlor, Pretilachlor, Propisochlor, Prynachlor, Terbuchlor, Thenylchlor, Xylachlor,
CDEA, Epronaz, Diphenamid, Napropamide, Naproanilide, Pethoxamid, Flufenacet,
Mefenacet, Fentrazamide, Anilofos, Piperophos, Cafenstrole, Indanofan und Tridiphan;
Inhibitoren für die Biosynthese von Cellulose wie Dichlobenil, Chlorthiamid, Isoxaben und Flupoxam;
Herbizide wie Dinofenat, Dinoprop, Dinosam, Dinoseb, Dinoterb, DNOC, Etinofen und
Medinoterb;
außerdem: Benzoylprop, Flamprop, Flamprop-M, Bromobutide, Chlorflurenol, Cin- methylin, Methyldymron, Etobenzanid, Pyributicarb, Oxaziclomefone, Triaziflam und Methyl bromide.
Im Pflanzenschutz verwendete Wirkstoffe können auch zur Bekämpfung von Schädlingen (z.B. Schaben, Ameisen, Termiten u.a.) im urbanen Raum (z.B. Wohnsiedlungen, Haus- und Gartenbereich, Gaststätten, Parkanlagen, Industrieflächen u.a.) eingesetzt werden und sind speziell für diese Anwendungen eine weitere Gruppe von geeigneten Effektstoffen.
Auch Wirkstoffe zur Bekämpfung von Schädlingen aus dem Bereich der Wirbeltiere (z.B. Ratten, Mäuse u.a.) können mit den erfindungsgemäßen Verfahren formuliert werden und die resultierenden Wirkstoffzubereitungen zur entsprechenden Schädlingsbekämpfung im landwirtschaftlichen und urbanen Raum angewendet werden.
Des weiteren geeignet sind Wirkstoffe für die pharmazeutische Anwendung, insbesondere solche für die orale Verabreichung. Das erfindungsgemäße Verfahren ist prinzi-
piell unabhängig von der medizinischen Indikation auf eine beliebige Vielzahl von Wirkstoffen anwendbar.
Insbesondere sind wasserlösliche Wirkstoffe für die pharmazeutische Anwendung zu nennen, insbesondere solche für die orale Verabreichung. Dies betrifft sowohl verschreibungspflichtige als auch nicht verschreibungspflichtige Wirkstoffe. Die Erfindung ist prinzipiell unabhängig von der medizinischen Indikation auf eine Vielzahl von Wirkstoffen anwendbar.
Nicht limitierende Beispiele für geeignete schwer wasserlösliche pharmazeutische Wirkstoffe sind in der folgenden Tabelle genannt.
Beispiele für wasserlösliche pharmazeutische Wirkstoffe sind husten- und schleimlösende Wirkstoffe, wie insbesondere Guaiacol Glykol Ether (auch Guaifenesin genannt) und dessen Derivate.
Weitere bevorzugte pharmazeutische Wirkstoffe sind Antikörper und andere in der Pharmazie verwendete Proteine, z. B. Enzyme oder Peptide, oder Nukleinsäuren.
(iv) Wirkstoff-Freisetzung aus den Formulierungen
Die Freisetzung der Wirkstoffe aus den mit den erfindungsgemäßen Verfahren herge- stellten Formulierungen kann durch Desorption in geeignete Lösungsmittel, durch den Abbau der Faserflächengebilde durch Hydrolyse, Oxidation oder biologisch durch Enzyme, wie z.B. Proteasen, oder ganze Mikroorganismen oder durch Auflösen des Faserflächengebilde durch geeignete Lösungsmittel erfolgen und durch Diffusion des
Wirkstoffes auf die Faseroberfläche. Geeignete Lösungsmittel für die Desorption sind alle Lösungsmittel oder Lösungsmittelgemische, in denen sich der Wirkstoff lösen lässt. Lösungsmittel, die die Faserflächengebilde auflösen können, können Lösungsmittel nur geeignet für das Trägerpolymersystem sein, oder geeignet für das Trägerpolymersys- tem und den Wirkstoff sein.
Ein besonderer Vorteil dieser Erfindung ist die verzögerte Wirkstofffreisetzung, wobei chemische Faktoren, wie z.B. Zusammensetzung des Trägers mit einer definierten Ausgestaltung der Nano- und Mesofasern (kontrollierte spezifische Oberfläche) kombi- niert werden können. Dadurch kann die Freisetzung deutlich präziser kontrolliert werden.
Die Kinetik und das Profil der Freisetzung der Effektstoffmoleküle kann z.B. gesteuert werden: (i) durch die Beladungsdichte des Trägerpolymers mit Wirkstoffen; (ii) durch spezifische Oberfläche der Fasern (d.h. Durchmesser); (iii) durch das Verwenden eines Polymergemisches aus mindestens 2 Polymeren als Trägerpolymer, die nicht gleich gut in gleichem Lösungsmittel löslich sind. D.h. durch Variation des Verhältnisses des löslichen und schwer bzw. unlöslichen Po- lymers im bestimmten Lösungsmittel;
(iv) durch Verwendung eines bioabbaubaren synthetischen Polymers oder eines
Biopolymers als Träger; (v) durch Variation des Verhältnisses in der Mischung eines nicht bioabbaubaren
Polymers mit einem bioabbaubarem Polymer; (vi) durch Variation der chemischen Struktur des nicht bioabbaubaren Polymers (z.B. wasserlöslich/wasserunlöslich) in der Mischung eines nicht bioabbaubaren Polymers mit einem bioabbaubaren Polymer; (vii) durch Variation des Verhältnisses in der Mischung eines nicht bioabbaubaren
Polymers mit einem bioabbaubaren Polymer; (viii) durch das Verwenden eines Polymergemisches aus mindestens 2 nicht bioabbaubaren Polymeren als Trägerpolymer, die nicht gleich gut in dem Freisetzungsmedium löslich sind und einem bioabbaubaren Polymer; (ix) durch Verwendung von einer Mischung aus nicht mischbaren Polymeren, wobei die Fasern eine chemische Strukturierung (Phasenseparation) aufweisen; (x) Durch Verwendung von Homopolymeren, Copolymeren oder Polymer-Blends, die mindestens eine Phase mit Tg unterhalb der Anwendungstemperatur aufweisen.
(xi) durch physikalische Strukturierung in Form von Porosität und/oder Oberflächenrauhigkeit (Topographie); und (xii) Kombination obiger Maßnahmen.
Ein weiterer Gegenstand der Erfindung ist die Verwendung der unter Benutzung der beschriebenen Polymere hergestellten Fasernflächengebilde zur Speicherung, zum Transport oder zur Freisetzung von Wirkstoffen in kosmetischen Produkten, humanen und tierischen Pharmaprodukten, Pflanzenschutzprodukten, Nahrungs- und Futtermitteln. Dabei dienen die Faserflächengebilde weiterhin dem Schutz der verpackten Wirk- Stoffe vor Umwelteinflüssen, wie z.B. oxidativen Prozessen oder UV-Strahlung, oder vor Zerstörung durch Reaktion mit anderen Bestandteilen der Produkte oder vor biologischem Abbau durch Enzyme (z.B. Proteasen) oder Mikroorganismen. Der Wirkstoff kann durch Desorption, biologischen Abbau, gezielte Freisetzung oder langsame Freisetzung oder Kombination dieser Mechanismen aus den Faserflächengebilden freige- setzt werden.
Durch Variation der Aminosäuresequenz der beschriebenen amphiphilen selbstas- semblierenden Proteine bzw. Fusionierung mit zusätzlichen Protein- oder Peptidse- quenzen ist es möglich, Strukturen zu generieren, welche bestimmte Oberflächen, z.B. Haut, Haar, Blätter, Wurzeln spezifisch erkennen bzw. von diesen Oberflächen oder den enthaltenen Rezeptoren erkannt und gebunden werden.
Dadurch ist es möglich, die mit den beschriebenen amphiphilen selbstassemblierenden Proteinen formulierten Wirkstoffe effektiver an den gewünschten Wirkort zu bringen bzw. die Wirkstoff auf nähme zu verbessern.
Weiterhin ist es durch Variation der Aminosäuresequenz der für die Wirkstoffformulierung beschriebenen amphiphilen selbstassemblierenden Proteine bzw. Fusionierung mit zusätzlichen Protein- oder Peptidsequenzen möglich, Wirkstoffe gezielt an ge- wünschte Wirkorte zu lenken, um damit z.B. eine höhere Spezifität, geringeren Wirkstoffverbrauch oder Wirkstoffdosis, eine schnellere oder stärkere Wirkung zu erzielen.
EXPERIMENTELLER TEIL
Allgemeiner Teil
a) Elektrospinnverfahren
Die zur Durchführung des erfindungsgemäßen Verfahrens geeignete Vorrichtung zum Elektrospinnen umfasst eine an deren Spitze mit einer mit einem Pol einer Spannungsquelle verbundenen Kapillardüse versehene Spritze zur Aufnahme der erfindungsgemäßen Formulierung. Gegenüber dem Ausgang der Kapillardüse ist in einem Abstand von etwa 20 cm eine mit dem anderen Pol der Spannungsquelle verbundene quadratische Gegenelektrode angeordnet, die als Kollektor für die gebildeten Fasern fungiert. Während des Betriebs der Vorrichtung wird an den Elektroden eine Spannung zwischen 15 kV und 35 kV eingestellt und die Formulierung unter einem geringen Druck durch die Kapillardüse der Spritze ausgetragen. Aufgrund der durch das starke elektrische Feld von 0,9 bis 2 kV/cm erfolgenden elektrostatischen Aufladung der For- mulierung entsteht ein auf die Gegenelektrode gerichteter Materialstrom, der sich auf dem Wege zur Gegenelektrode unter Faserbildung verfestigt, infolge dessen sich auf der Gegenelektrode Fasern mit Durchmessern im Mikro- und Nanometerbereich abscheiden.
Eine weitere mögliche Vorrichtung zur Durchführung des erfindungsgemäßen Verfahrens umfasst eine Walze, die sich in einem Behälter mit Spinnlösung dreht. Die Walze kann glatt sein oder eine physikalische Strukturierung, z.B. Nadeln oder Riefen aufweisen. Die Spinnlösung gerät bei jeder Drehung der Walze in das starke elektrische Feld und mehrere Materialströme werden gebildet. Die Gegenelektrode befindet sich über der Spinnelektrode. Die Fasern werden auf einen Trägervlies, z.B. Polypropylen abgeschieden. Beispielsweise kann eine Nanospider-Apparatur der Firma Elmarco verwendet werden. Die Spannung beträgt dabei etwa 82 kV bei einem Elektrodenabstand von 18cm. Die Temperatur beträgt etwa 23 0C und die relative Luftfeuchtigkeit 35 %. Es wird eine gezackte Elektrode zum Verspinnen verwendet. Um ein möglichst dickes Protein-Flächengebilde (z.B. Protein-Filme, Protein-Fasern, Protein-Vliesstoffe) zu erreichen wird das Trägervlies im Stillstand belassen. Alternativ kann das Trägervlies aber auch u nter Vorsch u b bewegt werden , u m defin iert d ü n nere Protein- Flächengebilde-Schichten zu erzielen.
b)Probenvorbereitung für WAXS-Messung
Die Proben wurden zwischen zwei Klebebandstreifen (handelsübliches Produkt der Firma Scotch) präpariert und deren Transmission gemessen.
c) Wirkstofffreisetzungsversuche
Die Untersuchungen der Freisetzung der Wirkstoffe aus den Fasernflächengebilden erfolgte nach der Long Time Encapsulation Analysis Methode. Bei dieser Methode werden die verkapselten Wirkstoffe in definierter Konzentration unterhalb der Löslichkeitsgrenze des Wirkstoffes in vollentsalztem (VE) Wasser angesetzt. Die Proben werden über einen Zeitraum von Minuten bis zu mehreren Wochen unter Rühren gehalten. In logarithmisch abgestuften Zeitabständen wird jeweils eine Probe genommen und der darin befindliche freie Wirkstoff chromatographisch untersucht. Anhand der zuvor durchgeführten Eichung des Wirkstoffes kann somit die freigesetzte Menge bestimmt werden.
Wirkstofffreisetzungsversuche mit proteinhaltigen Formulierungen können außerdem in zwei weiteren Ansatzvarianten durchgeführt werden:
Per oral aufzunehmende Wirkstoff-Formulierungen (wie z.B. Clotrimazol - gepresst zu Tabletten) können in künstlichem Magensaft (0,1 g NaCI; 0,16 g Pepsin; 0,35 ml HCl auf 50 ml auffüllen, pH 1-2) und künstlichem Darmsaft (3,4 g KH2PO4 in 12,5 ml Wasser lösen + 3,85 ml 0,2N NaOH auf 25 ml auffüllen + 0,5 g Pankreatin auf 50 ml auffüllen, pH 6,8) analysiert werden, um die Wirkstoff-Freisetzung unter proteolytisch aktiven Bedingungen im Verdauungstrakt zu simulieren. Kontrollansätze (ohne Proteasen) erfolgten in 5 mM Kaliumphosphatpuffer (pH 8,0), wobei unter diesen Bedingungen nur eine geringe Wirkstoff-Freisetzung beobachtet werden sollte. Pro Tablette wurden 20 ml des jeweiligen Verdauungssaftes oder Puffers zugegeben und die Ansätze bei 37 0C und 80 upm leicht schüttelnd inkubiert. Zu verschiedenen Zeitpunkten werden je 500 μl Probe für eine Wirkstoff-Quantifizierung mittels HPLC oder Photometer ent- nommen. Um bei schlecht wasserlöslichen Wirkstoffen, z.B. Clotrimazol, auch nach der Freisetzung entstandene Wirkstoff-Aggregate zu erfassen, wurde die absorptionspho- tometrische Quantifizierung nach Extraktion mit THF (3 ml Überstand + 3 ml THF + Spatelspitze NaCI, kräftiges vortexen, 1 min 15.000 x g, Oberphase vermessen, ggf. verdünnen) durchgeführt.
Bei anderen Wirkstoffen (nicht per oral aufgenommene pharmazeutische oder andere Wirkstoffe), z.B. Uvinul A+ und Metazach lor, kann die Freisetzungsanalyse durch Versetzen definierter Mengen an Protein-Wirkstoff-Flächengebilden mit unspezifischer Proteinase K-Lösung erfolgen. Die Protein-Wirkstoff-Flächengebilde wurden in 0,25-0,5 % [w/v] Proteinase K (Roche, Germany, gelöst in 5 mM Kaliumphosphatpuffer) bei 120-150 upm schüttelnd inkubiert. Zu verschiedenen Zeitpunkten wurden die noch intakten Protein-Wirkstoff-Flächengebilde durch Zentrifugation abgetrennt, die Überstände mit einem 4-5-fachen Überschuss an THF versetzt und der Wirkstoffgehalt anschließend absorptionsphotometrisch bestimmt. Bei allen Ansätzen wurden die freigesetzten Wirkstoff-Mengen nach Abgleich mit einer Wirkstoff-spezifischen Eichreihe ermittelt.
Beispiel 1 - Herstellung und Eigenschaften der Kompositfasern aus PVP und Epoxiconazol
Zur Herstellung von Kompositfasern wurden Polymerlösungen aus PoIy(I -vinyl-2- pyrrolidinone) Kollidon K-90 (PVP) (Mw=1.100.000 g/mol, Tg= 1800C, BASF SE) und Fungizid Epoxiconazol (1 -{[3-(2-Chlorphenyl)-2-(4-fluorphenyl)oxiran-2-yl]methyl}-1 H- 1 ,2,4-triazol) im Ethanol/Wasser Gemisch (90:10) hergestellt und zu Fasern versponnen. Dabei wurden die Lösungen mit einer Spritzenanlage unter Spannungen zwischen 35 und 45 kV versponnen.
Die Einwaagen sind in folgender Tabelle aufgeführt.
Die Angaben zur Konzentration des Wirkstoffes Epoxiconazol beziehen sich auf den Gesamtfeststoff (PVP+Wirkstoff). Die Konzentration des Trägerpolymers bezieht sich auf die Gesamtmasse von Lösungsmittel und Polymer vor der Zugabe des Wirkstoffes.
Die Figur 1A zeigt die Fasermorphologie in Abhängigkeit vom Wirkstoffgehalt.
Um Aussagen über den Anteil von Epoxiconazol in den Fasern machen zu können, wurde zu Beginn eine Eichung durchgeführt. Dazu wurden Lösungen von Epoxiconazol mit Konzentrationen von 10 bis 40 Gew.-% (bezogen auf den Feststoffgehalt) und 5.7 Gew.-% PVP (bezogen auf das Gesamtgewicht der Formulierung vor der Zugabe des Wirkstoffes) in Ethanol/Wasser (90/10) hergestellt, auf einen Si-Wafer aufgetragen und deren Transmission wurde mittels IR-Spektroskopie untersucht. Das Verhältnis der spezifischen Banden von PVP und Epoxiconazol wurde ausgewertet und daraus eine Eichgerade erstellt.
Die hergestellten wirkstoffhaltigen Faserflächengebilde wurden ebenfalls in dem Etha- nol/Wasser-Gemisch aufgelöst und analog zu den Eichproben als Film auf Si-Wafer aufgetragen, IR-spektroskopisch vermessen und über die Eichgerade wurden die Konzentrationen von Epoxiconazol bestimmt. Die Eichwerte zusammen mit den Befunden aus Faserflächengebilden sind in der Figur 1 B dargestellt.
Die Graphik zeigt, dass nach dem Verspinnen der Fasern noch in etwa die gleiche eingesetzte Menge an Epoxiconazol vorhanden ist. Mehrere Messungen zeigen, dass das Ergebnis reproduzierbar ist.
Der Wirkstoff Epoxiconazol befindet sich im Faserflächengebilde in einem amorphen Zustand. Dies belegen die Röntgenweitwinkelstreumessungen (WAXS), die mit einem Bruker-Diffraktometer D5005 (monochromatisierte Cu-Kα-Strahlung) in Transmission durchgeführt wurden. Ergebnisse der WAXS Messungen an frisch präparierten Fasern- flächengebilden aus PVP-Epoxiconazol sind in Figur 2 dargestellt.
Die Proben wurden dabei auf bzw. zwischen zwei Klebebandstreifen eingeschlossen. Bei dem scharfen Peak bei etwa 2Θ=18° handelt es sich um eine Verunreinigung, da dieser Peak bereits im reinen PVP zu beobachten ist und nicht dem Epoxiconazol zu- geordnet werden kann.
Um die Lagerstabilität dieser Faserflächengebilde zu prüfen, wurden die Proben bei +400C, -10° C und 00C jeweils 24 Std. und 72 Std. bei 200C gelagert und anschließend mittels Weitwinkelröntgensteuerung erneut untersucht.
Die Ergebnisse der WAXS-Messungen an bei unterschiedlichen Temperaturen gelagerten Fasernflächengebilden aus PVP-Epoxiconazol sind in Figur 3 dargestellt.
Figur 3 zeigt deutlich, dass die Zubereitungen lagerstabil sind - Wirkstoff ändert seine amorphe Morphologie im Laufe der Lagerung bei unterschiedlichen Temperaturen nicht.
Beispiel 2 - Herstellung und Eigenschaften der Kompositfasern aus PVP und be- ta -Carotin.
ß-Carotin wird zur Färbung fetthaltiger Lebensmittel wie Butter, Margarine, Käse, Mayonnaise und - in wasserdispergierbarer Form - auch wasserhaltiger Lebensmittel wie z.B. Fruchtgetränke, Puddings, Zuckerwaren verwendet. ß-Carotin wird auch als Farbstoff für Kosmetika und als Futtermittelzusatzstoff eingesetzt. Zur Herstellung von Kompositfasern wurden Polymerlösungen aus PoIy(I -vinyl-2-pyrrolidinone) Kollidon K-90 (PVP) (Mw=1.100.000 g/mol, Tg= 1800C, BASF SE) und dem Farbstoff ß-Carotin im Chloroform hergestellt und zu Fasern versponnen. Dazu wurden die Lösungen mit einer Spritzenanlage unter Spannungen zwischen 40 und 45 kV versponnen. Zusätzlich wurde 0.5 Gew.-% bezogen auf die Gesamtformulierung an Benzyltributylammoni- umbromid dazugegeben, um die elektrische Leitfähigkeit der Lösung zu erhöhen. Dies wirkt positiv auf die Fasernmorphologie und Durchmesserverteilung: es bilden sich nämlich weniger Verdickungen (Beads) und die Faserndurchmesserverteilung wird enger.
Die Einwaagen sind in folgender Tabelle aufgeführt.
Die Angaben zur Konzentration des Effektstoffes ß-Carotin beziehen sich auf die Gesamtmasse von PVP und Effektstoff. Die Konzentration des Trägerpolymers bezieht sich auf die Gesamtmasse von Lösungsmittel und Polymer.
Figur 4A zeigt die Fasermorphologie in Abhängigkeit vom Effektgehalt.
Um Aussagen über den Anteil von ß-Carotin in den Fasern machen zu können, wurde zu Beginn eine Eichung durchgeführt. Dazu wurden Lösungen von ß-Carotin mit Kon- zentrationen von 10 bis 40 Gew.-% (bezogen auf den Feststoffgehalt) und 6 Gew.-% PVP (bezogen auf Gesamtgewicht der Formulierung vor der Zugabe des Wirkstoffes) in Chloroform hergestellt, auf einen Si-Wafer aufgetragen und deren Transmission wurde mittels IR-Spektroskopie untersucht. Das Verhältnis der spezifischen Banden vom PVP und ß-Carotin wurde ausgewertet und eine Eichgerade erstellt.
Die hergestellten effektstoffhaltigen Faserflächengebilde wurden im Chloroform aufgelöst und wie die Eichproben als Film auf Si-Wafer aufgetragen, IR spektroskopisch vermessen und über die Eichgerade die ß-Carotin-Konzentrationen ausgewertet. Die Eichwerte zusammen mit den Befunden aus Faserflächengebilden sind in Figur 4B dargestellt.
Das Diagramm von Figur 4B zeigt, dass nach dem Verspinnen die Fasern noch etwa die eingesetzte Menge an ß-Carotin aufweisen. Mehrere Messungen zeigen, dass das Ergebnis reproduzierbar ist.
Der Effektstoff ß-Carotin befindet sich in einem amorphen Zustand. Dies zeigen die Röntgenweitwinkelstreumessungen (WAXS), die mit einem Bruker-Diffraktometer D5005 (monochromatisierte Cu-Kα-Strahlung) in Transmission durchgeführt wurden.
Die Proben wurden dabei zwischen zwei Klebebandstreifen eingeschlossen.
Figur 5 zeigt die Ergebnisse der WAXS Messungen an frisch präparierten Fasernflä- chengebilden aus PVP-ß-Carotin
Um die Lagerstabilität dieser Faserflächengebilde zu prüfen, wurden die Proben bei +400C, -10° C und 00C jeweils 24 Std. und bei 200C mindestens 72 Std. gelagert und anschließend mittels Weitwinkelröntgensteuerung erneut untersucht.
Figur 6 zeigt Ergebnisse der WAXS Messungen an bei unterschiedlichen Temperaturen gelagerten Fasernflächengebilden aus PVP-ß-Carotin.
Figur 6 zeigt deutlich, dass die Zubereitungen lagerstabil sind. Der Wirkstoff ändert seine amorphe Morphologie im Laufe der Lagerung bei unterschiedlichen Temperaturen nicht.
Beispiel 3 - Herstellung und Eigenschaften der Kompositfasern aus PMMA und Epoxiconazol.
Um die breite Anwendbarkeit der Methodeweiter zu veranschaulichen, wurden Kompositfasern aus Poly(methylmethacrylat) und Fungizid Epoxiconazol hergestellt.
Z u r H e rstellung von Kompositfasern wurden Polymerlösungen aus Po- ly(methylmethacrylat) Plexiglas® (PMMA) (Mw=430 000 g/mol, Tg= 1 100C (ISO 11357)) und Fungizid Epoxiconazol (1-{[3-(2-Chlorphenyl)-2-(4-fluorphenyl)oxiran-2-yl]methyl}- 1 H-1 ,2,4-triazol) in einem Ethanol/Chloroform-Gemisch (6:1 1 ) hergestellt und zu Fasern versponnen. Dazu wurden die Lösungen mit einer Spritzenanlage unter Spannungen zwischen 40 und 45 kV versponnen.
Die Einwaagen sind in folgender Tabelle aufgeführt.
Die Angaben zur Konzentration des Wirkstoffes Epoxiconazol beziehen sich auf den Gesamtfeststoff (PMMA+Wirkstoff). Die Konzentration des Trägerpolymers bezieht sich auf Gesamtmasse von Lösungsmittel und Polymer vor der Eingabe des Wirkstof- fes.
Figur 7 zeigt die Fasermorphologie in Abhängigkeit vom Wirkstoffgehalt.
Der Wirkstoff Epoxiconazol befindet sich in den Faserflächengebilden im amorphen Zustand. Dies zeigen die Röntgenweitwinkelstreumessungen (WAXS), die mit einem Bruker-Diffraktometer D5005 (monochromatisierte Cu-Kα-Strahlung) in Transmission durchgeführt wurden. Die Proben wurden dabei auf bzw. zwischen Scotchband präpariert.
Figur 8 zeig die Ergebnisse der WAXS Messungen an Fasernflächengebilden aus PMMA-Epoxiconazol
Beispiel 4 - Einfluss der spezifischen Oberfläche auf die Wirkstofffreisetzung
Der weitere Vorteil der Fasern ist ihre große spezifische Oberfläche verglichen mit Filmen oder anderen Formulierungsformen. Um dies zu belegen, wurde die Freisetzung des Wirkstoffes aus Fasern und Filmen untersucht.
Die Faserflächengebilde für die Freisetzungstests wurden aus einer Lösung enthaltend 12 Gew.-% Ecoflex® (aliphatisch-aromatisches Copolyester der BASF SE auf Basis von Butandiol, Adipinsäure und Terephthalsäure, Tg=-30°C, Tm=1 15°C, Mn=35.000;
Mw = 1 18.000; vergleiche auch http://iwww.plasticsportal.com/products/ecoflex.html)
(bezogen auf die Gesamtmasse der Formulierung vor der Eingabe des Wirkstoffes),
Lösungsmittelgemisch Chloroform/i-Propanol (95:5) und 10 Gew.-% Epoxiconazol (be- zogen auf Feststoff (Polymer+Wirkstoff)) versponnen.
Als Vergleichsprobe zum Faserflächengebilde wurde auf einen Objektträger die gleiche Polymer-Wirkstoff lösung aus 12 Gew.-% Ecoflex (bezogen auf Gesamtmasse der Formulierung vor der Zugabe des Wirkstoffes) und 10 Gew.-% Epoxiconazol (bezogen auf Feststoffgehalt) aufgestrichen, das Lösemittel verdunstet und dann mit einer Ra- sierklinge der Polymer/Wirkstoff-Film vom Objektträger abgetrennt.
Die beiden Proben wurden in einer Konzentration von 7mg/l in VE-Wasser eingewogen und in einem 0,5-l-Erlenmeyerkolben bei konstanter Drehzahl auf einem Magnetrührer ununterbrochen gerührt. Die Messung erfolgte nach der oben beschriebenen Methode. Die entnommenen Proben wurden an einem HPLC- Gerät Series 1100 von Agilent bei einer Wellenlänge von 220 nm auf freien Wirkstoff analysiert.
Figur 9 zeigt die Freisetzungsprofile von Epoxiconazol aus bioabbaubarem Polyester Ecoflex als Film und als Faserflächengebilde
Aus Figur 9 geht hervor, dass die Freisetzung von der spezifischen Oberfläche des Trägers abhängig ist und auf diese Weise gesteuert werden kann.
Beispiel 5: Wirkstofffreisetzung aus Polymeren mit unterschiedlicher Löslich- keit.
Die Freisetzung kann zusätzlich über die Löslichkeit des Trägerpolymers im Lösungsmittel gesteuert werden. Als Beispiel wurden Faserflächengebilde aus Polyvinylpyrroli- don, Polymethymethacrylat und Ecoflex mit Epoxiconazol hergestellt und die Freiset- zung in VE-Wasser nach der in Beispiel 4 beschriebenen Methode gemessen. Die Proben wurden folgendermaßen hergestellt: a) 5 Gew.-% PVP, 20 Gew.-% Epoxiconazol in Ethanol-Wasser-Gemisch (9:1 ); b) 12 Gew. % Ecoflex, 20 Gew. % Epoxiconazol in Chloroform-i-Propanol-Gemisch (95:5); c) 6 Gew.-% PMMA, 20 Gew.-% Epoxiconazol in Chloroform-Ethanol-Gemisch (11 :6)).
Die Angaben zur Konzentration des Wirkstoffes Epoxiconazol beziehen sich auf den Gesamtfeststoff (PVP+Wirkstoff). Die Konzentration des Trägerpolymers bezieht sich auf Gesamtmasse von Lösungsmittel und Polymer vor der Eingabe des Wirkstoffes.
Figur 10 zeigt die Freisetzungsprofile von Epoxiconazol aus bioabbaubarem Polyester Ecoflex, PVP und PMMA.
Das wasserlösliche PVP setzt Epoxiconazol relativ schnell frei. Bereits nach 2 Min. sind etwa 40 % des Epoxiconazols aus den Fasern ausgetreten. Aus Ecoflex-Fasern wird Epoxiconazol verzögert erst nach circa 10 min langsam freigesetzt. Erst nach einem Tag sind 40 % des Wirkstoffs aus den Fasern ausgetreten. Ecoflex ist nicht wasserlöslich. Die verzögerte und langsame Freisetzung könnte demzufolge auf Diffusion des Epoxiconazols an die Oberfläche der Fasern bzw. auf einen partiellen Abbau des Polyesters zurückzuführen sein. Im Gegensatz zu PVP- und Ecoflex-Fasern wird aus PMMA-Fasern in den ersten zwei Tagen kein Epoxiconazol freigesetzt. PMMA-Fasern sind nicht wasserlöslich und scheinbar ist die Diffusion des Epoxiconazols aus den Fasern in Wasser auch sehr langsam bzw. nicht möglich.
Beispiel 6 - Freisetzung aus einem Blend von schlecht mischbaren Polymeren
Das Freisetzungsprofil kann auch über die Polymerzusammensetzung von Faseflächengebilden beeinflusst werden. So können schlecht bzw. begrenzt mischbare Trägerpolymere eingesetzt werden. Die Freisetzung von Epoxiconazol aus PVP- und PMMA-Fasern sowie Fasern aus deren Blends PVP-PMMA(1 :1 ) und PVP-PMMA(1 :5) wurde anhand folgender Proben getestet:
a) 5 Gew.-% PVP, 20 Gew.-% Epoxiconazol in Ethanol-Wasser-Gemisch (9:1 ); b) 2.5 Gew.-% PVP, 2.5 Gew.-% PMMA, 20 Gew.-%. Epoxiconazol in Chloroform- Ethanol-Gemisch (11 :6); c) 1 Gew.-% PVP, 5 Gew.-% PMMA, 20 Gew.-% Epoxiconazol in Chloroform-Ethanol (1 1 :6); d) 6 Gew.-% PMMA, 20 Gew.-% Epoxiconazol in Chloroform-Ethanol-Gemisch (11 :6).
Die Angaben zur Konzentration des Wirkstoffes Epoxiconazol beziehen sich auf den Gesamtfeststoff (Trägerpolymer+Wirkstoff). Die Konzentration des Trägerpolymers bezieht sich auf Gesamtmasse von Lösungsmittel und Polymer vor der Eingabe des Wirkstoffes.
Figur 11 zeigt die Freisetzungsprofile von Epoxiconazol aus Faserflächengebilden hergestellt aus PVP und dessen Blends mit PMMA.
Die Freisetzung aus den Polymerblends entspricht sehr gut den erwarteten Verhalten aus den Freisetzungsprofilen der Fasern aus PVP oder PMMA. So nimmt mit steigendem PMMA-Anteil die Freisetzung ab. Die schnelle anfängliche Freisetzung - der erste Messpunkt liegt bereits deutlich über 0% - ist bei den Faserflächengebilden zu beobachten. Dieses Verhalten kann dadurch erklärt werden, dass die beiden Trägerpolymere nicht mischbar sind und eine Struktur bilden, in der PVP-reiche und PVP arme Domänen vorliegen. Diese Strukturierung ist in TEM-Aufnahmen sehr gut ersichtlich. Acrylat wird dabei hell dargestellt. Figur 12 zeigt Querschnitte der Fasern aus PMMA und PVP (5:1 ).
Man sieht, dass sich die PVP-reiche Phase bevorzugt auf der Faseroberfläche befindet, während die Acrylat-Phase im Inneren dominiert. Eine schnelle Freisetzung kann mit dem Auflösen der PVP-reichen Phase erklärt werden.
Beispiel 7 - Freisetzung aus dem Blend von mischbaren Polymeren
Werden mischbare Polymere verwendet, so entstehen Fasernflächengebilden mit homogenen Komponentenverteilung über die ganzen Fasern. Für die Untersuchungen des Freisetzungsprofils wurden folgende Proben herangezogen:
a) 5 Gew.-% PVP, 20 Gew.-% Epoxiconazol in Ethanol-Wasser-Gemisch (9:1 ); b) 4 Gew.-% PVP, 4 Gew.-% Ecoflex, 20 Gew.-% Epoxiconazol in Chloroform-i- Propanol-Gemisch (95:5); c) 12 Gew.-% Ecoflex, 20 Gew.-% Epoxiconazol in Chloroform-i-Propanol-Gemisch (95:5).
Die Angaben zur Konzentration des Wirkstoffes Epoxiconazol beziehen sich auf den Gesamtfeststoff (Trägerpolymer+Wirkstoff). Die Konzentration des Trägerpolymers bezieht sich auf Gesamtmasse von Lösungsmittel und Polymer vor der Eingabe des Wirkstoffes.
Figur 13 zeigt die Freisetzungsprofile von Epoxiconazol aus Faserflächengebilden hergestellt aus PVP und seinen Blends mit Ecoflex.
Man beobachtet, dass die schnelle, für PVP typische Wirkstofffreisetzung im Blend nicht mehr vorhanden ist; das Profil entspricht dem am wenigstens löslichen Polymer Ecoflex und ist zeitlich schneller geworden.
Beispiel 8 - Herstellung des C16-Spinnenseidenproteins
Die Herstellung des C16-Spinnenseidenproteins erfolgte biotechnologisch unter Verwendung plasmidhaltiger Escherichia coli Expressionsstämme. Design und Klonierung des C16-Spinnenseidenproteins (auch ADF4 genannt) sind in Hümmerich et al. (Bio- chemistry 43, 2004, 13604-13012) beschrieben. Im Gegensatz zum dort beschriebenen Verfahren wurde C16-Spinnenseidenprotein im E. co//-Stamm BL21 Gold (DE3) (Stratagene) hergestellt. Die Anzucht erfolgt in Techfors-Fermentern (Infors HAT, Schweiz) unter Verwendung eines Minimalmediums und Fed-Batch Techniken.
Minimalmedium: 2,5 g/l Citronensäuremonohydrat 4 g/l Glycerin
12,5 g/l Kaliumdihydrogenphosphat 6,25 g/l Ammoniumsulfat 1 ,88 g/l Magnesiumsulfatheptahydrat
0,13 g/l Calciumchloriddihydrat
15.5 ml/l Spurenelementelösung (40 g/l Citronensäuremonohydrat;
1 1 g/l Zink(ll)sulfatheptahydrat; 8,5 g/l Diammoniumei- sen(ll)sulfatheptahydrat; 3 g/l Mangan(ll)sulfatmonohydrat; 0,8 g/l Kupfer(ll)sulfatpentahydrat; 0,25 g/l Kobalt(ll)sulfathepta- hydrat) 3 ml/l Vitaminlösung (6,3 mg/ml Thiaminhydrochlorid; 0,67 mg/ml
Vitamin B12) pH 6,3
Feed-Lösung: 790 g/l Glycerin
6,9 g/l Citronensäuremonohydrat
13.6 g/l Natriumsulfat
1 ,05 g/l Diammoniumeisen(ll)sulfatheptahydrat 13 mg/l Thiaminhydrochlorid
Die Zellen wurden bei 37°C bis zu einer OD6oo von 100 angezogen, danach erfolgte die Induktion der Proteinexpression mit 0, 1 mM Isopropyl ß-D-1-thiogalactopyranoside (IPTG). Am Ende der Fermentation (8 bis 12 Stunden nach Induktion) wurden die Kulturen geerntet. Der Hauptanteil des Proteins befand sich in „Inclusion Bodies".
Nach der Zellernte wurde das Pellet in 2OmM 3-(-N-Morpholino)propanesulfonic acid (MOPS) pH 7,0 resuspendiert (5L Puffer pro Kilogramm Feuchtmasse). Anschließend erfolgte der Zell-Aufschluss unter Verwendung eines Micofluidizer M-1 10EH (Microflui- dics, US) bei Drücken von 1200 bis 1300 bar. Nach Sedimentation enthielt das Pellet nach Aufschluss neben den „Inclusion Bodies" noch Zelltrümmer und Membranbestandteile, welche durch zwei Waschschritte entfernt wurden. In einem ersten Waschschritt wurde das Pellet in 2,5 Volumen Tris-Puffer (50 mM Tris/HCI, 0,1 % Triton X- 100, pH 8,0) resuspendiert und anschließend der verbleibende Feststoff durch Zentri- fugation sedimentiert. Ein zweiter Waschschritt erfolgte unter Verwendung von Tris- Puffer (50 mM Tris/HCI, 5mM EDTA, pH 8,0). Das abermals nach Sedimentation erhaltene Pellet war nahezu frei von Membran- und Zelltrümmern.
Die gereinigten „Inclusion Bodies" wurden in Guanidiniumthiocyanat (Roth, Germany) gelöst, wobei pro 1 g Pellet (Feuchtmasse) 1 ,6 g Guanidiniumthiocyanat hinzugegeben wurden. Die „Inclusion Bodies" lösten sich unter Rühren bei leichter Erwärmung (500C). Zur Abtrennung evtl. vorhandener nicht-löslicher Bestandteile wurde anschließend noch eine Zentrifugation durchgeführt. Um eine wässrige C16-Spinnenseidenproteinlö- sung zu erhalten, wurde dann eine 16-stündige Dialyse gegen 5 mM Kaliumphosphatpuffer (pH 8,0) durchgeführt (Verdünnungsfaktor der Dialyse: 200).
Verunreinigende E. co//-P roteine bildeten bei der Dialyse Aggregate, welche durch Zentrifugation abgetrennt werden konnten. Die erhaltene Proteinlösung wies eine Reinheit von -95% C16-Spinnenseidenprotein auf.
Die erhaltene wässrige Proteinlösung kann entweder direkt zum Elektroverspinnen verwendet oder zwecks besserer Lagerfähigkeit zu Protein-Microbeads weiterverarbeitet werden. Zur Herstellung von C16-Protein-Microbeads wurde die wässrige C16- Spinnenseidenproteinlösung wird mit 0,25 Volumen einer 4-molaren Ammoniumsulfatlösung versetzt. Unter Einwirkung des Ammoniumsulfats assemblieren die Proteinmo- nomere zu kugelförmigen Gebilden, welche hier als Microbeads bezeichnet werden.
Die Microbeads wurden durch Zentrifugation abgetrennt, drei Mal mit destilliertem Wasser gewaschen und anschließend gefriergetrocknet.
Beispiel 9 - Formulierung von Clotrimazol als Effektstoff mittels Elektroverspin- nen
Um die Verwendbarkeit des beschriebenen Verfahrens für die Formulierung von aktiven Substanzen insbesondere schwer wasserlöslichen zu zeigen, wurde beispielhaft der pharmazeutische Wirkstoff Clotrimazol mittels Elektroverspin nen in C 1 6- Spinnenseidenprotein-Flächengebilde verkapselt.
Für die Herstellung einer verspinnbaren Lösung wurden C16-Spinnenseidenprotein- Microbeads (14 % [w/w]) und der Wirkstoff Clotrimazol (10 % [w/w]) zusammen in A- meisensäure (98-100% p.a.) gelöst. Es wurden 200 ml Ameisensäure in einem Be- cherglas vorgelegt und anschließend sukzessive 50,4 g C16-Spinnenseidenprotein und 36 g Clotrimazol (Fa. Sigma, Deutschland) eingerührt. Nachdem die Stoffe vollständig gelöst waren wurde die Lösung mit Ameisensäure (98-100%) auf 360 g aufgefüllt.
Alternativ kann auch wasserlösliche C16-Spinnenseidenproteinlösung (siehe Beispiel 1 ) als Ausgangsstoffbasis genutzt werden. Der Wirkstoff wird dann direkt in der wässri- gen Proteinlösung gelöst bzw. bei Einsatz höherer Wirkstoffkonzentrationen in einem alternativen Lösungsmittel (z.B. Ameisensäure) vorgelöst und dann mit der Proteinlösung gemischt. Um die Viskosität der Spinnlösung zu erhöhen können dann zusätzlich wasserlösliche Polymere oder Polymerdispersionen zugemischt werden.
Die Lösung aus C16-Spinnenseidenprotein und Clotrimazol wurde drei Stunden lang in einer Nanospider-Apparatur der Firma Elmarco versponnen. Die Spannung betrug 82 kV bei einem Elektrodenabstand von 18 cm. Die Temperatur betrug 23 0C und die relative Luftfeuchtigkeit 35 %. Es wurde eine gezackte Elektrode zum Verspinnen verwen- det. Um ein möglichst dickes Protein-Flächengebilde zu erreichen wurde das Trägervlies im Stillstand belassen. Alternativ kann das Trägervlies aber auch unter Vorschub bewegt werden, um definiert dünnere Protein-Flächengebilde-Schichten zu erzielen. Die aus dem Ansatz gewonnenen Protein-Fasern wurden anschließend bei 400C und unter Vakuum über Nacht getrocknet.
D i e e l e kt ro n e n m i k ro s ko p i s c h e An a l y s e d e r s o h e rg e s te l l te n C 1 6- Spinnenseidenprotein-Flächengebilde mit eingeschlossenem Clotrimazol ergab, dass es sich hauptsächlich um Fasern mit einem Durchmesser etwa 50 nm bis zu 1 μm handelt (Figur 14).
Im Gegensatz zum reinen Clotrimazol zeigen sich in der C16-Spinnenseidenprotein / Clotrimazol Formulierung in der Röntgenbeugung keine kristallinen Peaks (Figur 15). Demnach ist davon auszugehen, dass der Wirkstoff amorph oder als feste Lösung verkapselt wurde, was seine Bioverfügbarkeit positiv beeinflussen kann.
Um die Wirkstoff-Freisetzung aus einer möglichst relevanten Applikationsform zu überprüfen, wurden aus den C16-Spinnenseidenprotein-Flächengebilden Tabletten ver- presst. Es wurden jeweils 300 mg Material unter Vakuum und 100 Bar Druck ca. 10 min lang in einer KBr-Presse (Firma: Paul-Otto-Weber, Deutschland) verpresst. Die Tabletten hatten einen Durchmesser von etwa 13 mm und eine Dicke von etwa 2 mm.
Die Freisetzung von Clotrimazol aus den Tabletten wurde in zwei verschiedenen Ansätzen getestet. Es wurden künstlicher Magensaft (0,1 g NaCI; 0,16 g Pepsin; 0,35 ml HCl auf 50 ml auffüllen, pH 1-2) und künstlicher Darmsaft (3,4 g KH2PO4 in 12,5 ml Wasser lösen + 3,85 ml 0,2N NaOH auf 25 ml auffüllen + 0,5 g Pakreatin auf 50 ml auffüllen, pH 6,8) verwendet, um die Wirkstoff-Freisetzung unter proteolytisch aktiven Bedingungen im Verdauungstrakt zu simulieren. Ein weiterer Ansatz erfolgte in 5 mM Kaliumphosphatpuffer (pH 8,0), wobei unter diesen Kotrollbedingungen nur eine geringe Wirkstoff-Freisetzung beobachtet werden sollte. Pro Tablette wurden 20 ml des je- weiligen Verdauungssaftes oder Puffers zugegeben und die Ansätze bei 37 0C und 80 upm leicht schüttelnd inkubiert. Die Quantifizierung des freigesetzten Clotrimazols erfolgte aufgrund seiner schlechten Wasserlöslichkeit (und damit Neigung zu Aggregatbildung in wässrigen Systemen) nach Extraktion des Überstandes mit THF durch ab- sorptionsphotometrische Bestimmung bei 262 nm (3 ml Überstand + 3 ml THF + Spa- telspitze NaCI, kräftiges vortexen, 1 min 15.000 x g, Oberphase vermessen, ggf. verdünnen).
Während im Kontrollansatz (Puffer ohne Proteasen) lediglich maximal 2 % der verkapselten Wirkstoffmenge freigesetzt werden, wird in Magensaft gesteuert durch die vor- handene enzymatische Aktivität (Proteasen) etwa 50 % Freisetzung innerhalb 24 h erzielt (Figur 16). Dabei wird der Wirkstoff Clotrimazol kontinuierlich freigesetzt. Im
Darmsaft hingegen werden nach 24 h nur etwa 20 % des Wirkstoffes freigesetzt (Figur 16). Die C16-Spinnenseidenprotein / Clotrimazol-Formulierung scheint unter den dabei vorherrschenden eher neutralen pH-Werte im betrachteten Zeitbereich so stabil, dass nur eine abgeschwächte Freisetzung zu beobachten ist.
Um den nach 24 h aus der Formulierung noch nicht freigesetzten Anteil an Clotrimazol zu bestimmen, wurden die Ansätze mit den nicht proteolytisch abgebauten C16- Spinnenseidenprotein-Fasern mit 3 ml Tetrahydrofuran (THF) versetzt und für max. 48 h weiter schüttelnd inkubiert. Anschließend wurde der Wirkstoffgehalt absorptionspho- tometrisch bei 262 nm quantifiziert. Aus dem Endwert und den vorher bestimmten Zwischenwerten konnte dadurch die Beladungsdichte der C16- Spinnenseidenproteinformulierung mit dem Wirkstoff Clotrimazol bestimmt werden. Die Beladungsdichte lag bei allen untersuchten Tabletten zwischen 27 % und 33 % [w/w], woraus sich eine durchschnittliche Beladungsdichte des zu Tabletten verpressten C16- Spinnenseidenprotein-Flächengebildes mit etwa 30 % [w/w] Clotrimazol ergab (vgl. Tabelle unten).
Beladungsdichten der C16-Spinnenseidenprotein-Formulierung (Tabletten) mit dem Wirkstoff Clotrimazol.
Auf die Offenbarung der hierin zitierten Druckschriften wird ausdrücklich Bezug genommen