WO2012147865A1 - オルトフタルアルデヒドの結晶性水和物、それを含有する消毒薬及び殺生物剤、並びにオルトフタルアルデヒドの製造方法 - Google Patents
オルトフタルアルデヒドの結晶性水和物、それを含有する消毒薬及び殺生物剤、並びにオルトフタルアルデヒドの製造方法 Download PDFInfo
- Publication number
- WO2012147865A1 WO2012147865A1 PCT/JP2012/061242 JP2012061242W WO2012147865A1 WO 2012147865 A1 WO2012147865 A1 WO 2012147865A1 JP 2012061242 W JP2012061242 W JP 2012061242W WO 2012147865 A1 WO2012147865 A1 WO 2012147865A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- opa
- opach
- water
- solution
- crystalline hydrate
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 CCC(C)(C1OC(CC*C(C)(*)C2OC(*)c3ccccc23)c2ccccc12)OC(*(C)C(C(N)(N)N)=O)=O Chemical compound CCC(C)(C1OC(CC*C(C)(*)C2OC(*)c3ccccc23)c2ccccc12)OC(*(C)C(C(N)(N)N)=O)=O 0.000 description 1
Images







Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D307/00—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom
- C07D307/77—Heterocyclic compounds containing five-membered rings having one oxygen atom as the only ring hetero atom ortho- or peri-condensed with carbocyclic rings or ring systems
- C07D307/87—Benzo [c] furans; Hydrogenated benzo [c] furans
- C07D307/89—Benzo [c] furans; Hydrogenated benzo [c] furans with two oxygen atoms directly attached in positions 1 and 3
-
- A—HUMAN NECESSITIES
- A01—AGRICULTURE; FORESTRY; ANIMAL HUSBANDRY; HUNTING; TRAPPING; FISHING
- A01N—PRESERVATION OF BODIES OF HUMANS OR ANIMALS OR PLANTS OR PARTS THEREOF; BIOCIDES, e.g. AS DISINFECTANTS, AS PESTICIDES OR AS HERBICIDES; PEST REPELLANTS OR ATTRACTANTS; PLANT GROWTH REGULATORS
- A01N25/00—Biocides, pest repellants or attractants, or plant growth regulators, characterised by their forms, or by their non-active ingredients or by their methods of application, e.g. seed treatment or sequential application; Substances for reducing the noxious effect of the active ingredients to organisms other than pests
- A01N25/12—Powders or granules
Definitions
- the present invention relates to a crystalline hydrate of orthophthalaldehyde represented by the following formula (1).
- Orthophthalaldehyde (hereinafter also referred to as OPA) is a product of glutaraldehyde because the product formulated at a low concentration of 0.55% by mass has an excellent bactericidal effect against microorganisms such as bacteria and has low odor.
- OPA is used as a high-level disinfectant for sterilizing and disinfecting medical instruments such as endoscopes (Patent Document 1).
- OPA is also used as a biocide for the purpose of sterilization and slime control of white water systems in paper mills, air conditioning cooling water systems and circulating water systems in various factories and facilities (Patent Documents 2 and 3).
- the aforementioned disinfectant and biocide products are usually provided in the form of easy-to-handle OPA solutions, but their production requires the use of crystalline OPA, which is difficult to handle.
- OPA is commercially available in crystalline powder or flake form, and its melting point is as low as about 56 ° C., so that it has a problem that it tends to solidify during storage at room temperature.
- OPA is an unstable substance having a dialdehyde structure, so there is a problem that the purity tends to decrease if air and moisture are not sufficiently blocked during storage.
- OPA crystals and vapors are in the skin and mucous membranes.
- OPA is not usually a solid product form, but a solution containing about 10 to 40% by mass of OPA except for a low-concentration preparation such as the above-mentioned high-level disinfectant.
- the product form is distributed.
- the solubility of OPA in water is as low as about 5.9% by mass at room temperature, it is an expensive aprotic organic solvent that is a good solvent for OPA in order to provide such a highly concentrated OPA solution as a product.
- VOC volatile organic compound
- organic solvents are used as a carbon source to assimilate microorganisms, so it is desirable to avoid mixing organic solvents in biocides as much as possible. It is rare.
- an aqueous suspension such as a flowable agent that does not use an organic solvent as much as possible as a product formulated with OPA.
- the melting point of OPA is as low as about 56 ° C., and solidification is likely to occur. Because it was not successful.
- OPA is non-oxidizing and has the advantage of low corrosiveness to metals such as iron, stainless steel, and copper, it has a low melting point and a relatively high dissolution rate in water. It was not processed into tablets and provided as a disinfectant and biocide. As described above, there was a problem in working environment in handling OPA crystals in an open system, which is also why the tableting was difficult.
- solid substances such as bromochlorodimethylhydantoin (hereinafter also referred to as BCDMH) that generates hypochlorous acid and hypobromite in water, chlorinated isocyanuric acid, and chlorite that generates chlorine dioxide are contained in tablets. Processed products have already been put on the market, but since they are all oxidative, they have a high metal corrosiveness against iron, stainless steel, copper and the like, which has been a problem.
- BCDMH bromochlorodimethylhydantoin
- an active substance having the same physiological activity as OPA that is, a biological effect for sterilization, disinfection, slime control, etc. that can be used as a disinfectant or biocide, and having less problems as described above has been desired.
- OPA a biological effect for sterilization, disinfection, slime control, etc.
- it does not have a low melting point such as OPA, is hard to be consolidated, has a strong odor like OPA, does not irritate the skin / mucous membrane, and is easy to handle
- an oxidizing substance such as BCDMH
- product forms such as aqueous suspensions and tablets that were difficult to prepare or apply with OPA It is required that it can be used after being processed, or can be used as a crystalline powder.
- ⁇ , ⁇ , ⁇ ′, ⁇ ′-tetrahalogeno-o-xylene is hydrolyzed under normal pressure or under pressure (Patent Document 4 and Patent Document 5), or naphthalene is used.
- a method of performing ozonolysis under pressure (Patent Document 6) is known. Then, the obtained OPA reaction liquid is extracted with an organic solvent to take out crude OPA once, and purification operations such as crystallization and distillation are added thereto to commercialize OPA.
- Patent Document 7 has a drawback that the purification process is very long and complicated. That is, in Patent Document 7, ⁇ , ⁇ , ⁇ ′, ⁇ ′-tetrachloro-o-xylene is hydrolyzed to give an OPA reaction solution, OPA is extracted with methyl tertiary butyl ether (MTBE), and MTBE is removed by distillation. Then, methanol was added to the evaporation residue of the obtained crude OPA to acetal, this was distilled under reduced pressure to distill OPA dimethoxyacetal, and this purified OPA dimethoxyacetal was heated in water under a sulfuric acid catalyst to form OPA.
- MTBE methyl tertiary butyl ether
- the OPA is extracted from the resulting OPA aqueous solution with diisopropyl ether (DIPE), and this DIPE solution is cooled and crystallized to crystallize OPA, which is filtered and dried to obtain a crystal product of OPA.
- DIPE diisopropyl ether
- acetalization of OPA was incorporated, and the production of high-purity OPA was a very complicated operation.
- diisopropyl ether which is also described in Patent Document 7, or an organic solvent having relatively high toxicity and high flammability, such as toluene, is usually used.
- DIPE diisopropyl ether
- organic solvent having relatively high toxicity and high flammability such as toluene
- OPACH crystalline hydrate of OPA of the present invention
- Patent Document 8 Non-Patent Document
- Document 1 Non-Patent Document 2
- Non-Patent Document 5 OPA is self-polymerized into an OPA polymer in the presence of an initiator and a catalyst at a low temperature of usually ⁇ 50 ° C. or lower. At that time, cyclic hemiacetal is connected by an ether bond and polymerization proceeds, and the hydroxyl groups at both ends of the polymer are bonded to alcohol, amine, acyl compound, etc.
- Non-Patent Document 2 the OPA polymer described in Non-Patent Document 2 is represented by the formula (2) using alcohol as an initiator and trichloroacetyl isocyanate as a stopper.
- OPA polymer has a ceiling temperature of about ⁇ 43 ° C., and it is known that the OPA polymerization does not occur at a relatively high temperature such as room temperature. There is no literature report that an oligomer was obtained.
- the crystalline hydrate of OPA of the present invention (OPACH) is a compound that can be said to be an OPA dimer.
- OPA dimer the OPA is first converted to phthalide by sunlight, which is condensed with another molecule of OPA.
- Another compound that has been produced has already been reported (for example, Non-Patent Document 3), and in this application, “OPA dimer” is not used as the name of the novel compound of the present invention.
- Non-Patent Document 4 OPA has been reported to exist in an equilibrium state represented by the following three different chemical formulas in an aqueous solution.
- Non-Patent Document 4 a part of dialdehyde (3a) normally expressed as OPA is hydrated with one molecule of water per OPA molecule in an aqueous solution to become an acyclic hydrate (3b). It is further stated that this closes to a cyclic hemiacetal (3c). The abundance ratio in a 25 ° C. aqueous solution of a certain concentration is reported that (3a) is about 20%, (3b) is about 8%, and (3c) is about 72%, and the temperature of the OPA aqueous solution is high. It is also stated that the proportion of dialdehyde (3a) increases.
- a crystalline solid which is a novel compound, is preferentially deposited over OPA in OPA in an aqueous solution or a water-miscible organic solvent containing water (hereinafter, both are collectively referred to as “in the aqueous solution”). It has been reported that the precipitates are present in a stable dispersion in the solution, and the crystalline solid obtained by solid-liquid separation and drying is also stable in air at normal temperature and pressure. It has not been.
- JP-A-63-3313705 Japanese Patent Application Laid-Open No. 06-26497 Japanese Patent Laid-Open No. 06-23368 JP 2000-26359 A Japanese Patent Laid-Open No. 09-31009 JP-A-10-182542 JP 2007-8932 A JP 59-216142 A
- the subject of the present invention is further a method for producing OPA having a higher purity than the original OPA by a simple operation of passing the crystalline hydrate as an intermediate from OPA, in particular, isolation and purification thereof. To provide a law.
- OPACH obtained by solid-liquid separation and drying is stably present in air at room temperature and normal pressure, and this OPACH has useful properties for handling and formulation as compared with conventional crystalline OPA.
- This OPACH has a physiological activity as a disinfectant and biocide similar to OPA because it is easily decomposed by heating and / or dissolution in water or an organic solvent and returns to two molecules of OPA and one molecule of water.
- OPA having higher purity than that of the original OPA can be obtained by returning OPACH to OPA.
- the present invention has been made based on this finding.
- a crystalline hydrate of orthophthalaldehyde represented by the following formula (1) A disinfectant comprising the orthophthalaldehyde crystalline hydrate according to (1).
- a method for producing orthophthalaldehyde characterized in that the crystalline hydrate of orthophthalaldehyde according to (1) is heated and / or dissolved in a solvent.
- the orthophthalaldehyde crystalline hydrate (OPACH) represented by the formula (1) of the present invention becomes orthophthalaldehyde (OPA) at the time of use, and therefore has a physiological activity as a disinfectant and biocide of OPA. It is easier to handle than OPA and can be used in the production of high-purity OPA. Accordingly, the orthophthalaldehyde crystalline hydrate (OPACH) of the present invention can be used as OPACH, or it can be decomposed into OPA and used as OPA.
- FIG. 2 is a DSC chart of OPACH manufactured in Example 1.
- FIG. 2 is an FT-IR spectrum of OPACH produced in Example 1.
- 2 is a UV spectrum of OPACH produced in Example 1. It is a UV spectrum of OPA for comparative reference.
- 1 is a 1 H-NMR spectrum (D 2 O solution) of OPA for comparison.
- 1 is a 1 H-NMR spectrum (DMSO-d 6 solution) of OPACH produced in Example 1.
- 3 is a 13 C-NMR spectrum (DMSO-d 6 solution) of OPACH produced in Example 1.
- the present invention will be specifically described below.
- First Embodiment Crystalline Hydrate of Orthophthalaldehyde The first embodiment of the present invention is formed from two molecules of orthophthalaldehyde and one molecule of water, and is stable as a crystalline solid in air at normal temperature and pressure. It is a crystalline hydrate (OPACH) of orthophthalaldehyde represented by the following formula (1).
- the crystalline hydrate of OPA of the present invention is a hydration reaction from two molecules of OPA and one molecule of water according to the following formula (4) including the above (3a), (3b) and (3c). It is a novel compound that is thought to be produced by
- the OPA crystalline hydrate (OPACH) of the present invention can be produced by performing a hydration reaction of OPA in an aqueous solution or in a water-miscible organic solvent containing water, that is, in the aqueous solution.
- OPACH produced by OPA hydration is preferentially precipitated because its solubility in the aqueous solution is lower than that of OPA.
- OPACH will precipitate if OPA is dissolved at a concentration exceeding the saturation solubility, for example, in a supersaturated state.
- OPA precipitates preferentially, and this becomes a normal OPA crystallization operation by lowering the temperature of the solution.
- an organic solvent immiscible with water for example, toluene
- OPA is precipitated instead of OPACH by cooling.
- the solvent used may be water alone, but in that case, the OPACH production efficiency decreases because the solubility of OPA as a raw material in water is as low as about 5.9% by mass. Therefore, in order to increase the production rate and yield of OPACH, an organic solvent that is a good solvent for OPA and is miscible with water is preferably used in combination with the excess amount of water. However, use of lower alcohols such as methanol, ethanol, and 2-propanol is avoided because OPA acetalization easily occurs even in the presence of water, and the production of OPACH is suppressed.
- Suitable organic solvents miscible with water include acetone, 1,2-dimethoxyethane (DME), N, N-dimethylformamide (DMF), N-methyl-2-pyrrolidone (NMP), propylene carbonate, etc. Is mentioned.
- DME 1,2-dimethoxyethane
- DMF N-dimethylformamide
- NMP N-methyl-2-pyrrolidone
- propylene carbonate etc. Is mentioned.
- it is a solvent which is not mixed with water at an arbitrary ratio such as propylene carbonate, it can be used in combination with another organic solvent miscible with water.
- organic solvent that is a good solvent for OPA and suppresses the production of OPACH even if it is miscible with water.
- organic solvent include, in addition to the above-mentioned lower alcohols, glycols such as ethylene glycol, diethylene glycol, propylene glycol, polyethylene glycol # 200, acetonitrile, tetrahydrofuran, diglyme, N, N-dimethylacetamide, etc., and their use should be avoided. preferable.
- glycols such as ethylene glycol, diethylene glycol, propylene glycol, polyethylene glycol # 200, acetonitrile, tetrahydrofuran, diglyme, N, N-dimethylacetamide, etc.
- acetonitrile acetonitrile
- tetrahydrofuran diglyme
- N N-dimethylacetamide
- suitable organic solvent etc.
- the OPA concentration and hydration reaction conditions for increasing the production rate and yield of OPACH are as follows. That is, as a measure of the OPA concentration, the aqueous solution of OPA is prepared at a concentration lower than the saturation solubility of OPA at room temperature but close thereto. At that time, the OPA concentration may be set to about 5% by mass to 40% by mass by adjusting the types of water and organic solvents and the mixing ratio thereof. When this aqueous solution is stirred at room temperature for several hours to 24 hours to carry out a hydration reaction, white OPACH crystals precipitate slowly. As described above, when the OPA concentration is set exceeding the saturation solubility in the aqueous solution, not only OPACH but also OPA yellow crystals may be precipitated, so it must be avoided.
- OPACH is produced by combining the dialdehyde (3a) and cyclic hemiacetal (3c) one molecule at a time by weak intermolecular force. It is estimated that. Therefore, it is considered that the production of OPACH is promoted when the concentrations of dialdehyde (3a) and cyclic hemiacetal (3c) are increased in the aqueous solution.
- the production of OPACH is basically subject to kinetic control, and the decomposition of OPACH which is a reverse reaction is subject to thermodynamic control.
- the production of OPACH is promoted at a temperature lower than room temperature, and the decomposition of OPACH is promoted at a high temperature. Therefore, in order to increase the production rate and yield of OPACH, it is also recommended to cool the OPACH when it starts to precipitate from the aqueous solution to lower the solution temperature to around 0 ° C. Since the OPACH crystal is white and the OPA crystal is yellow, whether or not OPACH is precipitated can be determined from the appearance of the crystal. In order to promote precipitation of OPACH, the final solution temperature may be lowered to ⁇ 10 ° C. or lower. However, energy and time are required for cooling, and the viscosity of the solution is increased and solid-liquid separation is performed.
- the merit also decreases due to the reason that workability is reduced.
- the hydration reaction from OPA to OPACH is considered to antagonize the kinetic control and the thermodynamic control as described above.
- the temperature of the aqueous solution exceeds about 30 ° C., hydration of OPA is suppressed, and it does not occur at higher temperatures.
- the precipitated OPACH may be dissolved by heating and return to OPA.
- the OPACH precipitated by the hydration reaction of OPA can be isolated as a white crystalline powder by solid-liquid separation by a method such as centrifugal filtration, suction filtration or decantation, washing with water and / or an organic solvent, and drying.
- OPACH OPACH
- the OPACH thus obtained has a melting point of about 121 ° C. as an endothermic peak in DSC analysis as shown in FIG. 1. Unlike OPA, it is almost odorless and non-irritating. Is a white powder that does not color and is harder to set than OPA at room temperature.
- OPACH can be added to an aqueous system as a powder because of such characteristics, and can be further processed into a formulation such as an aqueous suspension or a tablet, which was difficult with OPA.
- OPACH crystalline hydrate of OPA of the present invention
- two molecules of OPA and one molecule of water hydrate by a weak intermolecular force. It is a precursor compound of OPA because it is relatively stable under normal temperature and pressure, but easily returns to OPA and water by heating and dissolution. Therefore, the OPACH of the present invention can be added to an aqueous system contaminated with microorganisms and used as a disinfectant or biocide without changing to OPA.
- OPACH can be used in the form of crystals in an aqueous system, or a suspension (so-called flowable agent, preferably having a concentration of 5 to 40% by mass) dispersed in water and / or an organic solvent, or a tablet It can also be used after being processed. At that time, a part of OPACH may be changed to OPA and exist as OPA. Further, OPACH may be used as a solution by dissolving it in water and / or an organic solvent, but in that case, it may be practically used as an OPA solution.
- the organic solvent that can be used here is generally combined with water, but acetone, 1,2-dimethoxyethane (DME), ⁇ -butyrolactone, propylene carbonate, ethylene glycol, diethylene glycol, polyethylene glycol # 200 and the like.
- the thickener normally used can be added suitably.
- the thickener include xanthan gum, sodium carboxymethyl cellulose, sodium alginate and the like.
- OPACH Using OPACH as crystals, and processing into suspensions and tablets dispersed in water or organic solvents and using them as disinfectants and biocides, as previously stated, is difficult with OPA, This is a new point made possible by the present invention.
- these disinfectants and biocides using OPACH are all referred to as disinfectants and biocides containing OPACH, including the case where they are used in the form of crystals and the case where they are changed to OPA.
- the following known biocides may be used in combination. Orthophthalaldehyde, glutaraldehyde, peracetic acid, hydrogen peroxide, hypochlorous acid, ammonia monochloramine, bromochlorodimethylhydantoin (BCDMH), trichloroisocyanuric acid, sodium dichloroisocyanurate, 2,2-dibromo-3-nitrilopropion Amide, 4,5-dichloro-1,2-dithiol-3-one, methylenebisthiocyanate, 3,3,4,4-tetrachlorotetrahydrothiophene-1,1-dioxide, 1,4-bis (bromoacetoxy) -2-butene, 1,2-bis (bromoacetoxy) ethane, dichloroglyoxime, ⁇ -chlorobenzaldoxime, ⁇ -chlorobenzaldoxime a
- ⁇ Third Embodiment> Method for Producing High-Purity OPA Using OPACH As a result of intensive studies on the production mechanism of OPACH and the decomposition mechanism of OPACH into water, OPACH is heated and / or water or It was clarified that OPA having a higher degree of purification than the original OPA can be obtained by dissolving in an organic solvent and returning to OPA. This was confirmed by analyzing the purity of OPA before and after passing OPACH as an intermediate, as described in the Examples.
- the method for producing OPA of the present invention is suitable for producing OPA having a purity of 98 to 100% by mass.
- the organic solvents that can be used here are the same as those mentioned in the first embodiment. In the case of heating, 50 to 120 ° C is preferable, and 60 to 100 ° C is more preferable.
- Non-patent document 5 it is known that an OPA polymer undergoes a so-called unzipping reaction in the presence of an acid catalyst to return to OPA, and due to such characteristics, the OPA polymer is used as a resist material ( Non-patent document 5).
- the OPACH of the present invention is similar to the partial structure of the OPA polymer, but basically differs from the polymerization of OPA in that water is essential to generate OPACH by a hydration reaction as described above.
- the hydration reaction is possible near room temperature without requiring a low temperature condition of ⁇ 50 ° C. or lower like the polymerization of OPA, and both terminal groups (initiator and terminator) are required as a polymer stabilizer.
- OPACH differs from the polymerization of OPA.
- the fact that the temperature condition for producing OPACH may be around room temperature is advantageous for industrial implementation.
- OPA polymerization reaction and the hydration reaction are common in that two adjacent aldehyde groups are bonded on the benzene ring. Therefore, the molecule not having the adjacent dialdehyde structure does not participate in the reaction and remains dissolved in the aqueous solution. Since OPACH is preferably less soluble than OPA and other impurities, it can exist stably at room temperature and normal pressure as crystals dispersed in the aqueous solution or as solid-liquid separated crystals.
- OPA crystalline hydrate (OPACH) purified by removing impurities by solid-liquid separation is also a raw material of high-purity OPA, and this is organized by the following formula (5) as the reverse reaction of the hydration reaction.
- two OPA molecules and one water molecule are quantitatively obtained from one OPCHA molecule.
- OPA
- the solid material obtained by hydration of OPA is a crystalline hydrate of OPA (OPACH) and a precursor compound of OPA having a high purity. did it.
- FIG. 2 shows the FT-IR spectrum of the obtained OPACH.
- UV Ultraviolet absorption spectrum
- FIG. 5 shows the 1 H-NMR spectrum of the resulting OPA.
- OPA of D 2 O solution chart considered a mixture of dialdehyde (3a) with the cyclic hemiacetal (3c) was obtained. The abundance ratio was considered to be about 1:14 to 15 from the proton ratio of both benzene rings.
- OPACH was dissolved in DMSO-d 6 at room temperature, and a 1 H-NMR spectrum was immediately measured. The obtained 1 H-NMR spectrum is shown in FIG.
- the OPACH solution in DMSO-d 6 contained no dialdehyde (3a) and cyclic hemiacetal (3c), and a chart supporting the presence as OPACH was obtained.
- OPACH is dispersed in ion-exchanged water so that the concentration is sufficiently lower than the saturated solubility of OPA in water (about 5.9% by mass) to prepare a 1.0% by mass aqueous dispersion of OPACH.
- the time until the crystals were completely dissolved was observed while stirring at this temperature.
- the dissolution time of OPACH was 30 ° C. for 5 hours, 40 ° C. for 3 hours, and 50 ° C. for 1 hour. From the above, it was found that the dissolution time of OPACH in water depends on the temperature and can be shortened if the temperature is high, and there is no practical problem as a disinfectant and biocide.
- the typical use concentration of OPA as a high-level disinfectant is 0.55% by mass
- the concentration used by adding to a water system as a biocide is usually several mg / L to Since it is several hundred mg / L, the above-mentioned OPACH having solubility in water dissolves as OPA even when added as it is to an aqueous system contaminated with microorganisms, and thus exhibits physiological activity as OPA. It was found that the concentration was reached.
- GPC chromatogram OPACH was dissolved in DMF to make a 1.0 mass% solution.
- OPA was also dissolved in DMF to prepare a 1.0 mass% solution.
- the crystals were easily dissolved by shaking for 1 to 2 minutes at room temperature, and did not become a high-viscosity solution characteristic when the polymer was dissolved.
- GPC gel permeation chromatography
- Example 2 (Production in acetone aqueous solution) The same operation as in Example 1 was performed except that acetone was used instead of 1,2-dimethoxyethane in Example 1. As a result, OPA crystalline hydrate (OPACH) was obtained as white crystals in a yield of 47.6% based on OPA. However, the yield does not correct the mass of water added by the hydration reaction (6.29%).
- OPACH OPA crystalline hydrate
- Example 3 (Production in DMF aqueous solution) The same operation as in Example 1 was conducted except that N, N-dimethylformamide (DMF) was used instead of 1,2-dimethoxyethane in Example 1.
- DMF N, N-dimethylformamide
- OPACH OPA crystalline hydrate
- Example 4 (Production effect in aqueous solution and seed crystal addition effect) OPA (manufactured by Kay Kasei Co., Ltd., purity 99.5%) and ion-exchanged water were added to a beaker to prepare an OPA 5.0 mass% aqueous solution.
- a predetermined amount of OPA crystalline hydrate (OPACH, prepared in Example 1) was added as a seed crystal to 50 g of this aqueous solution ((A) in Table 1 below), followed by stirring at room temperature for 24 hours.
- OPA which is a yellow crystal did not precipitate in any case
- a crystalline hydrate (OPACH) of OPA which was a white crystal was precipitated as shown in Table 1 below.
- the crystals were filtered and dried and subjected to gravimetric analysis ((B) in Table 1 below), and the OPA concentration in the filtrate was determined by gas chromatography ((C) in Table 1 below).
- Example 5 (Behavior in aqueous solution) 5 g of OPACH obtained in Example 1 was added and dispersed in 50 g of ion-exchanged water and stirred at room temperature for 24 hours. Since some of the crystals were gradually dissolved, the crystals were removed through a filter and the solution portion was sampled and analyzed by gas chromatography. As a result, only one peak was observed at the same elution position as OPA in the solution part, and it was found that 2.6% by mass of OPA was contained.
- Example 6 (bactericidal effect) With respect to the aqueous solution of OPACH obtained in Example 5 (containing 2.6% by mass as OPA), the bactericidal effect for a contact time of 30 minutes against bacteria was examined. For reference, the bactericidal effect was also examined for a solution in which 0.1 mass% of OPACH was dissolved in DMSO. Moreover, in order to compare the bactericidal effect with OPA, the aqueous solution of OPA and DMSO solution were also prepared. The concentration of OPACH as OPA was calculated and matched to the concentration of OPA added. The results are shown in Table 2 below. The test conditions for the bactericidal effect are as follows.
- Test system pH 6 buffer system and pH 8 buffer system (phosphate-citrate buffer)
- Target bacteria The following three types of bacteria are mixed: Enterobacter aerogenes NBRC 13534 Staphylococcus aureus NBRC 12732 Pseudomonas aeruginosa NBRC 13275 Contact time: 30 minutes
- Example 7 Purification effect of OPA obtained via OPACH
- the OPACH was isolated by suction filtration and drying of white crystals produced by hydration in the same manner as in Example 1 except that the OPA in Example 1 was changed to 98.8% purity (manufactured by Ihara Nikkei Chemical Co., Ltd.). did.
- the OPA purity by gas chromatography was improved to 99.5%, phthalide was greatly reduced, and chlorine-containing impurities ⁇ , ⁇ , ⁇ ′, ⁇ ′-tetrachloro-o-xylene (TECOX) was not detected.
- Example 8 (Crystal solidification) A transparent hard glass cylinder having an inner diameter of 50 mm, a height of 70 mm, and a thickness of 5 mm was prepared, and the open part of the cylinder was placed up and down on a flat glass. After adding 40 g of crystal samples of OPA (recrystallized from diisopropyl ether) and OPACH (manufactured in Example 1) from the open part, a flat glass was placed on the crystal and capped. A weight of 620 g of stainless steel weight was placed on the crystal. These were allowed to stand in a constant temperature bath at 40 ° C. for 1 week, and the presence or absence of consolidation was observed.
- Example 9 Preparation of aqueous suspension and solubility in water
- 80 parts by mass of ion-exchanged water and 0.8 parts by mass of a thickener xanthan gum (trade name: Rhode Poul 23, manufactured by Rhone-Poulenc) were added to a glass bottle and dissolved by stirring.
- 20 parts by mass of OPACH was added and stirred and mixed to obtain a uniform white slurry.
- the aqueous suspension was sealed and allowed to stand at room temperature for 3 months. The crystals slightly settled (the volume of the supernatant was less than 10% of the total), but it was easily stirred into a uniform slurry (floorable agent). I'm back.
- Example 10 Manufacture of tablets and solubility in water
- the OPACH powder was sieved to collect powder having a particle size of 1.0 to 2.0 mm. This was mixed at a ratio of 85% by weight, magnesium stearate 10% by weight, and talc 5% by weight, and 10 g each was weighed and tableted by changing the tableting pressure to obtain an off-white tablet (Table 4 below).
- 1 L of clean water was weighed into a 1 L beaker, and tablets were added one by one and stirred for 7 days at room temperature using a magnetic stirrer. At that time, the stirrer was prevented from coming into direct contact with the tablet.
- Reference Example 1 (Effect of low OPA concentration) It stirred at room temperature for 24 hours like Example 1 except having reduced the density
- Reference example 2 (effect when OPA concentration is high) It stirred at room temperature for 24 hours like Example 1 except having increased the density
Landscapes
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Organic Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Health & Medical Sciences (AREA)
- Pest Control & Pesticides (AREA)
- Toxicology (AREA)
- Engineering & Computer Science (AREA)
- Dentistry (AREA)
- Wood Science & Technology (AREA)
- Zoology (AREA)
- Environmental Sciences (AREA)
- Plant Pathology (AREA)
- Agronomy & Crop Science (AREA)
- Agricultural Chemicals And Associated Chemicals (AREA)
- Furan Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
下記式(1)で表されるオルトフタルアルデヒドの結晶性水和物。
Description
本発明は、下記式(1)で表されるオルトフタルアルデヒドの結晶性水和物に関する。

オルトフタルアルデヒド(以下OPAともいう)は、0.55質量%という低濃度に製剤化された製品が細菌などの微生物に対して優れた殺菌効果を有し、低臭性でもあるためグルタルアルデヒドの代替品として内視鏡などの医療器具を殺菌・消毒するための高水準消毒薬として用いられている(特許文献1)。また、OPAは製紙工場における白水系や各種工場・施設における空調冷却水系及び循環水系などの殺菌やスライムコントロールを目的とした殺生物剤としても用いられている(特許文献2及び特許文献3)。
前述の消毒薬や殺生物剤の製品は、通常は取扱いやすいOPA溶液の形態で提供されているが、それらの製造では取扱いが厄介な結晶性のOPAを原料として用いなければならない。取扱いが厄介である理由は、OPAは結晶性粉末やフレーク状のものが商業的に提供され、その融点が約56℃と低いため、常温で保管中に固結しやすいという問題があることによる。また、OPAはジアルデヒド構造をもつ不安定な物質のため、保管中に空気や水分を十分に遮断しないと純度が低下しやすいという問題があり、さらに、OPAの結晶や蒸気は皮膚・粘膜に対して強い刺激性・腐食性をもつという問題もある。固結したOPAを使用するためには物理的に解砕する必要があって作業性が劣るという問題があり、また、OPAの結晶の取扱いでは保護具の着用、十分な局所排気や換気が必須になる。また、OPAの結晶や溶液を皮膚に付着させると皮膚が黒褐色に着色するという問題もあり、それは皮膚の新陳代謝が進むまで長期間続くことも取扱い上嫌われる理由になっている。
そのような取扱い上の問題もあり、OPAは通常では固体の製品形態でなく、前記の高水準消毒薬のような低濃度の製剤を除けば、OPAが10~40質量%程度含有される溶液の製品形態のものが流通されている。しかしながら、OPAの水への溶解度が室温で約5.9質量%と低いことから、そのようなOPAの高濃度溶液を製品として提供するにはOPAの良溶媒である高価な非プロトン性有機溶媒などを多量に使用しなければならず、その結果、有機溶媒による製品コストの上昇に加えて排水負荷やVOC(揮発性有機化合物)の原因になるといった問題があった。特に空調冷却水系における殺菌やスライムコントロールを目的とした殺生物剤の用途では、有機溶媒が炭素源となって微生物を資化するため、殺生物剤中の有機溶媒の配合を極力避けることが望まれている。
また、OPAを製剤化した製品として、有機溶媒を極力使用しないフロアブル剤のような水性懸濁液の提供が望まれていたが、OPAの融点が約56℃と低く固結が起こりやすいことなどのため、それは成功していなかった。
さらに、OPAは非酸化性のため鉄、ステンレス綱、銅などの金属に対する腐食性が低いという長所があるにもかかわらず、低融点であり水への溶解速度も比較的速すぎるため、OPAが錠剤に加工されて消毒薬及び殺生物剤として提供されることはなかった。前述のように、OPAの結晶を開放系で取り扱うこと自体に作業環境上の問題があったことも、その錠剤化が難しかった理由に挙げられる。一方、水中で次亜塩素酸や次亜臭素酸を発生するブロモクロロジメチルヒダントイン(以下BCDMHともいう)や塩素化イソシアヌル酸、並びに二酸化塩素を発生する亜塩素酸塩などの固体物質は、錠剤に加工されたものが既に上市されているが、それらはいずれも酸化性のため鉄、ステンレス鋼、銅などに対する金属腐食性が高く課題となっていた。
また、OPAを製剤化した製品として、有機溶媒を極力使用しないフロアブル剤のような水性懸濁液の提供が望まれていたが、OPAの融点が約56℃と低く固結が起こりやすいことなどのため、それは成功していなかった。
さらに、OPAは非酸化性のため鉄、ステンレス綱、銅などの金属に対する腐食性が低いという長所があるにもかかわらず、低融点であり水への溶解速度も比較的速すぎるため、OPAが錠剤に加工されて消毒薬及び殺生物剤として提供されることはなかった。前述のように、OPAの結晶を開放系で取り扱うこと自体に作業環境上の問題があったことも、その錠剤化が難しかった理由に挙げられる。一方、水中で次亜塩素酸や次亜臭素酸を発生するブロモクロロジメチルヒダントイン(以下BCDMHともいう)や塩素化イソシアヌル酸、並びに二酸化塩素を発生する亜塩素酸塩などの固体物質は、錠剤に加工されたものが既に上市されているが、それらはいずれも酸化性のため鉄、ステンレス鋼、銅などに対する金属腐食性が高く課題となっていた。
従って、OPAと同等の生理活性、すなわち消毒薬や殺生物剤に使用できる殺菌・消毒・スライムコントロールなどのための生物効果を有し、上記のような問題がより少ない活性物質が望まれていた。すなわち、OPAのような低融点でなく固結が起こりにくいものであること、OPAのような強い臭気や皮膚・粘膜への刺激性がなく取扱いが容易であること、BCDMHなどの酸化性物質とは異なり非酸化性物質で金属腐食性が低いこと、一般的に提供されている溶液の製品形態のほかにOPAでは調製又は適用が困難であった水性懸濁液や錠剤などの製品形態にも加工して使用でき、また結晶性粉末のままでも使用できることなどが要求される。
また、OPAの製造法については、α,α,α',α'-テトラハロゲノ-o-キシレンを常圧下又は加圧下で加水分解する方法(特許文献4及び特許文献5)、あるいは、ナフタレンを加圧下で加オゾン分解する方法(特許文献6)などが知られている。そして、得られたOPA反応液を有機溶媒で抽出して粗製OPAを一旦取り出し、これに晶析や蒸留などの精製操作を加えてOPAが製品化されている。
上記の2種類の反応方法のうち、特殊な設備を必要とせず安全に実施できることから広く採用されているのは、α,α,α',α'-テトラハロゲノ-o-キシレンを加水分解する方法である。この加水分解によるOPAの製造法では、OPAから副生するフタリド、フタル酸、2-カルボキシベンズアルデヒドなどの不純物のほかに未反応原料などのハロゲン含有不純物が反応液に含まれやすいことから、高純度のハロゲン非含有オルトフタルアルデヒドを製造するための方法が開示されている(特許文献7)。特に医療器具に用いられる高水準消毒薬では高純度のハロゲン非含有OPAが原料として望まれることが精製操作が不可欠となっていることの背景にある。
しかしながら、特許文献7の方法は精製工程が非常に長く煩雑となる欠点がある。すなわち、特許文献7ではα,α,α',α'-テトラクロロ-o-キシレンを加水分解してOPA反応液とし、OPAをメチルターシャリーブチルエーテル(MTBE)で抽出後、MTBEを蒸留により除き、得られた粗製OPAの蒸発残留物にメタノールを加えてアセタール化し、これを減圧蒸留してOPAジメトキシアセタールを留出させ、この精製されたOPAジメトキシアセタールを水中で硫酸触媒下に加熱してOPAに分解し、得られたOPA水溶液からOPAをジイソプロピルエーテル(DIPE)で抽出し、このDIPE溶液を冷却晶析してOPAを結晶化させ、それをろ過・乾燥してOPAの結晶製品としている。
このように、高純度でハロゲン非含有のOPAを得るためにOPAのアセタール化も組み込まれて、高純度OPAの製造は大変煩雑な操作となっていた。
このように、高純度でハロゲン非含有のOPAを得るためにOPAのアセタール化も組み込まれて、高純度OPAの製造は大変煩雑な操作となっていた。
また、OPAの晶析においては、通常、特許文献7でも述べられるジイソプロピルエーテル(DIPE)、あるいはトルエンなどの比較的毒性が強く引火性の高い有機溶媒が用いられているが、結晶化させたOPAをろ過して乾燥する際にそれらの溶媒が十分除去できず結晶中に残留しやすいという問題がある。さらにそのような再結晶の操作によって得られる結晶性のOPAは前述のように保管中に固結しやすいという問題があった。
また、OPAを直接真空蒸留して精製する方法もあるが、それにはOPAの分解を抑えて歩留まりを上げるために高真空で高理論段数の精留塔を有する蒸留設備が必要となり、設備コストが高額になるという問題があった。そのうえ、留出する溶融OPAをさらに固化してフレークやペレットなどの取り扱いやすい形状に加工することが望まれることから、蒸留によるOPAの製造法も課題があった。
ここで、本発明のOPAの結晶性水和物(以下OPACHともいう。)が新規化合物であることについて述べる。OPACHとはOPA Crystalline Hydrateの略称である。
OPAの消毒薬や殺生物剤としての用途が開示される以前から現在まで、OPAを重合してポリマーにし、これをレジスト材料として利用するための研究が行われてきた(特許文献8、非特許文献1、非特許文献2及び非特許文献5)。
OPAは、通常-50℃以下の低温において開始剤及び触媒の存在下、自己重合してOPAポリマーとなる。その際、環状のヘミアセタールがエーテル結合によりつながって重合が進み、ポリマーの両末端の水酸基には開始剤及び停止剤としてアルコール、アミン、アシル化合物などと結合させることにより室温以上の温度でも安定に存在するポリマーになる。例えば、非特許文献2に述べられるOPAポリマーは、アルコールを開始剤としトリクロロアセチルイソシアネートを停止剤とした式(2)で表される。
OPAは、通常-50℃以下の低温において開始剤及び触媒の存在下、自己重合してOPAポリマーとなる。その際、環状のヘミアセタールがエーテル結合によりつながって重合が進み、ポリマーの両末端の水酸基には開始剤及び停止剤としてアルコール、アミン、アシル化合物などと結合させることにより室温以上の温度でも安定に存在するポリマーになる。例えば、非特許文献2に述べられるOPAポリマーは、アルコールを開始剤としトリクロロアセチルイソシアネートを停止剤とした式(2)で表される。

OPAポリマーの天井温度は約-43℃と報告されているように、室温のような比較的高い温度ではOPAの重合は起きないことが知られており、また、OPAのダイマーやトリマーのようなオリゴマーが得られたとの文献報告もない。
本発明のOPAの結晶性水和物(OPACH)は、いわばOPAダイマーともいえる化合物であるが、OPAダイマーについては、OPAが先ず日光によりフタリドに変化し、これがもう1分子のOPAと縮合して生成するという別の化合物が既に報告されており(例えば、非特許文献3)、本願では本発明の新規化合物の呼称として「OPAダイマー」を用いない。
本発明のOPAの結晶性水和物(OPACH)は、いわばOPAダイマーともいえる化合物であるが、OPAダイマーについては、OPAが先ず日光によりフタリドに変化し、これがもう1分子のOPAと縮合して生成するという別の化合物が既に報告されており(例えば、非特許文献3)、本願では本発明の新規化合物の呼称として「OPAダイマー」を用いない。
一方、OPAは水溶液中では以下のような3つの異なった化学式で表される平衡状態で存在することが報告されている(非特許文献4)。

非特許文献4では、OPAとして通常に表記されるジアルデヒド(3a)は、水溶液中ではその一部がOPA1分子につき1分子の水と水和して非環状の水和物(3b)となり、さらにこれが閉環して環状のヘミアセタール(3c)になると述べられている。ある濃度の25℃水溶液中での存在比は、(3a)が約20%、(3b)が約8%、(3c)が約72%であると報告され、また、OPA水溶液の温度が高くなるほどジアルデヒド(3a)の割合が多くなることも述べられている。しかしながら、OPAが水溶液中または水を含有する水混和性の有機溶媒中(以下、両者を一括して「該水溶液中」ともいう)で新規化合物である結晶性固体をOPAの析出より優先的に生成・析出させ、その析出物が溶液中で安定に分散して存在し、さらに、それを固液分離後乾燥して得られた結晶性固体が常温常圧下空気中でも安定に存在することは報告されていない。
Chuji, Aso.:Pure Appl.Chem., 1970, Vol.23,No.2-3, pp287-304.
Oliver Coulembier et al.:Macromolecules, 2010, Vol.43, pp572-574.
Alexander Schonberg et al.:J.Am.Chem. Soc., 1955, Vol.77,pp5755-5756.
Petr Zuman et al.:Electroanalysis, 2009, Vol.21, No.3-5, pp645-649.
Minoru Tsuda et al.:J.Polymer Sci. Part A,1997, Vol.35, No.1, pp77-89.
本発明の課題は、新規化合物でありオルトフタルアルデヒド(OPA)の前駆体的化合物でもあるオルトフタルアルデヒドの結晶性水和物を提供することにある。
本発明の課題はまた、該結晶性水和物を含有する取り扱いやすい消毒薬及び殺生物剤を提供することにある。それは、OPAでは困難であった製剤の提供も含まれる。
本発明の課題はさらに、OPAから該結晶性水和物を中間体として経由させるという簡単な操作によって、元のOPAと比較して高純度となるOPAを製造する方法、特にその単離・精製法を提供することにある。
本発明の課題はまた、該結晶性水和物を含有する取り扱いやすい消毒薬及び殺生物剤を提供することにある。それは、OPAでは困難であった製剤の提供も含まれる。
本発明の課題はさらに、OPAから該結晶性水和物を中間体として経由させるという簡単な操作によって、元のOPAと比較して高純度となるOPAを製造する方法、特にその単離・精製法を提供することにある。
本発明者らは、上記課題に鑑み鋭意研究した結果、以下の知見を見出した。OPAが水溶液中または水を含有する水混和性の有機溶媒中、すなわち該水溶液中で水と結合して2分子のOPAと1分子の水からなる新規なOPAの結晶性水和物(OPACH)を優先的に生成する。これを固液分離後乾燥して得られるOPACHは常温常圧下空気中で安定に存在し、このOPACHは従来の結晶性のOPAと比較して取扱い及び製剤化のために有用な特性を有する。このOPACHは加熱及び/又は水や有機溶媒への溶解により容易に分解して2分子のOPAと1分子の水に戻ることからOPAと同様な消毒薬及び殺生物剤としての生理活性を有し、そして、このOPAからOPACHへの生成機構及びOPACHからOPAへの分解機構を鑑みてOPACHをOPAに戻すことにより元のOPAより精製度の高まったOPAが得られる。本発明はこの知見に基づきなされるに至った。
すなわち、上記課題は以下の手段により解決された。
(1)下記式(1)で表されるオルトフタルアルデヒドの結晶性水和物。

(2)(1)に記載のオルトフタルアルデヒドの結晶性水和物を含有することを特徴とする消毒薬。
(3)(1)に記載のオルトフタルアルデヒドの結晶性水和物を含有することを特徴とする殺生物剤。
(4)(1)に記載のオルトフタルアルデヒドの結晶性水和物を加熱及び/又は溶媒に溶解させることを特徴とするオルトフタルアルデヒドの製造法。
(1)下記式(1)で表されるオルトフタルアルデヒドの結晶性水和物。
(3)(1)に記載のオルトフタルアルデヒドの結晶性水和物を含有することを特徴とする殺生物剤。
(4)(1)に記載のオルトフタルアルデヒドの結晶性水和物を加熱及び/又は溶媒に溶解させることを特徴とするオルトフタルアルデヒドの製造法。
本発明の式(1)で表されるオルトフタルアルデヒドの結晶性水和物(OPACH)は、使用時にオルトフタルアルデヒド(OPA)になることからOPAの消毒薬及び殺生物剤としての生理活性を有しており、OPAより取扱いやすく、高純度OPAの製造法にも利用できる。
従って、本発明のオルトフタルアルデヒドの結晶性水和物(OPACH)は、OPACHとしても利用できるし、OPAに分解してOPAとしても利用できる。
従って、本発明のオルトフタルアルデヒドの結晶性水和物(OPACH)は、OPACHとしても利用できるし、OPAに分解してOPAとしても利用できる。
本発明の上記及び他の特徴及び利点は、適宜添付の図面を参照して、下記の記載からより明らかになるであろう。
以下、本発明を具体的に説明する。
<第一の実施形態> オルトフタルアルデヒドの結晶性水和物
本発明の第一の実施形態は、オルトフタルアルデヒド2分子と水1分子から生成し、常温常圧下空気中で結晶性固体として安定に存在し、下式(1)で表されるオルトフタルアルデヒドの結晶性水和物(OPACH)である。
<第一の実施形態> オルトフタルアルデヒドの結晶性水和物
本発明の第一の実施形態は、オルトフタルアルデヒド2分子と水1分子から生成し、常温常圧下空気中で結晶性固体として安定に存在し、下式(1)で表されるオルトフタルアルデヒドの結晶性水和物(OPACH)である。

本発明のOPAの結晶性水和物(OPACH)は、2分子のOPAと1分子の水から前記の(3a)、(3b)及び(3c)を包含する下式(4)に従って水和反応により生成すると考えられる新規化合物である。

(OPACHの製造方法)
本発明のOPAの結晶水和物(OPACH)の製造は、水溶液中または水を含有する水混和性の有機溶媒中、すなわち該水溶液中でOPAの水和反応を行うことにより可能となる。OPAの水和により生成したOPACHは、該水溶液中の溶解度がOPAより低いため優先的に析出する。その際、該水溶液中のOPA1モルに対して最低でも水0.5モルを共存させるのが必須であり、好ましくは水和反応の速度を高めるためにOPA1モルに対して10~40モル程度の過剰量の水を共存させるのがよい。
本発明のOPAの結晶水和物(OPACH)の製造は、水溶液中または水を含有する水混和性の有機溶媒中、すなわち該水溶液中でOPAの水和反応を行うことにより可能となる。OPAの水和により生成したOPACHは、該水溶液中の溶解度がOPAより低いため優先的に析出する。その際、該水溶液中のOPA1モルに対して最低でも水0.5モルを共存させるのが必須であり、好ましくは水和反応の速度を高めるためにOPA1モルに対して10~40モル程度の過剰量の水を共存させるのがよい。
水を共存させずに水混和性の有機溶媒(例えば、特許文献7におけるジイソプロピルエーテル)のみを用いると、OPAが飽和溶解度を超える濃度、例えば過飽和の状態で溶解していれば、OPACHは析出せずOPAが優先的に析出し、これは溶液の温度を下げることにより通常のOPAの晶析操作になる。一方、水と混和しない有機溶媒(例えば、トルエン)を用いると、水を併用しても分液によりOPAの大部分がトルエン相に分配してOPACHは析出しない。その際、OPAが飽和溶解度を超える濃度であれば、冷却によりやはりOPACHでなくOPAが析出することになる。
使用する溶媒は水単独でも構わないが、その場合は原料となるOPAの水への溶解度が約5.9質量%と低いことからOPACHの製造効率が低下する。従って、OPACHの生成速度及び収率を高めるため、OPAの良溶媒であり水と混和性のある有機溶媒を前記の過剰量の水と併用するのがよい。ただし、メタノール、エタノール、2-プロパノールなどの低級アルコールは、水が共存してもOPAのアセタール化が起こりやすくOPACHの生成が抑制されるため使用を避ける。水と混和性がある有機溶媒として好適なものは、アセトン、1,2-ジメトキシエタン(DME)、N,N-ジメチルホルムアミド(DMF)、N-メチル-2-ピロリドン(NMP)、炭酸プロピレンなどが挙げられる。なお、炭酸プロピレンのように水と任意の割合には混ざらない溶媒でも、水と混和性がある他の有機溶媒と併用すれば使用できる。
OPAの良溶媒であり水と混和性があってもOPACHの生成を抑制する有機溶媒がある。それらは上記の低級アルコールのほか、エチレングリコール、ジエチレングリコール、プロピレングリコール、ポリエチレングリコール#200などのグリコール類、アセトニトリル、テトラヒドロフラン、ジグライム、N,N-ジメチルアセトアミドなどが挙げられ、これらの使用は避けることが好ましい。ただし、これらの溶媒も種類によるが、その使用量が0.1%~10%程度と少なければOPACHの結晶化を完全に妨げるわけではなく、前記の好適な有機溶媒との併用により使用できることがある。
OPACHの生成速度及び収率を高めるためのOPA濃度及び水和反応条件は次のようになる。すなわち、OPA濃度の目安としては、室温においてOPAの飽和溶解度より低いがそれに近い濃度でOPAの該水溶液を調製する。その際、水及び有機溶媒の種類、それらの混合比を調節してOPA濃度が5質量%~40質量%程度になるよう設定するとよい。この該水溶液を室温において数時間から24時間程度撹拌して水和反応を行うとOPACHの白色結晶がゆっくり析出する。
前述のように、その該水溶液中での飽和溶解度を超えてOPA濃度を設定すると、OPACHだけでなくOPAの黄色結晶も析出することがあるので避ける必要がある。
前述のように、その該水溶液中での飽和溶解度を超えてOPA濃度を設定すると、OPACHだけでなくOPAの黄色結晶も析出することがあるので避ける必要がある。
現在のところ、OPACHの生成メカニズムが完全に明らかになった訳ではないが、OPACHは前記のジアルデヒド(3a)と環状のヘミアセタール(3c)が弱い分子間力によって1分子ずつ結合して生成すると推定される。従って、該水溶液中でジアルデヒド(3a)及び環状のヘミアセタール(3c)の濃度が高くなるとOPACHの生成が促進されると考えられる。また、OPACHの生成は基本的に速度論的支配を受け、逆反応となるOPACHの分解は熱力学的支配を受けるものと考えられる。すなわち、室温より低温域ではOPACHの生成が促進され、高温域ではOPACHの分解が促進される。従って、OPACHの生成速度及び収率を高めるために、該水溶液からOPACHが析出開始したら冷却を行って溶液温度を0℃付近まで低下させることも推奨される。OPACHの結晶は白色であり、OPAの結晶は黄色のため、OPACHが析出しているか否かは結晶の外観でも判別できる。なお、OPACHの析出を促進するために最終の溶液温度を-10℃以下に下げてもよいが、冷却のためにエネルギー及び時間が必要となり、溶液の粘度も上昇して固液分離する際に作業性が低下するなどの理由によりメリットも低下する。
OPAからOPACHへの水和反応は、上記のような速度論的支配と熱力学的支配が拮抗すると考えられる。該水溶液の温度がおよそ30℃を超えるとOPAの水和が抑制され、さらに高温になれば起こらなくなる。析出したOPACHは加熱により溶解してOPAに戻ることもある。
なお、OPAの水和反応を促進するため、あらかじめ該水溶液のpHを中性付近に設定することが好ましく、強い酸性や強いアルカリ性では水和反応が抑制される。また、実施例に述べるように、OPACHの析出を促進するため、該水溶液にOPACHを種晶として添加することも有効である。
OPAからOPACHへの水和反応は、上記のような速度論的支配と熱力学的支配が拮抗すると考えられる。該水溶液の温度がおよそ30℃を超えるとOPAの水和が抑制され、さらに高温になれば起こらなくなる。析出したOPACHは加熱により溶解してOPAに戻ることもある。
なお、OPAの水和反応を促進するため、あらかじめ該水溶液のpHを中性付近に設定することが好ましく、強い酸性や強いアルカリ性では水和反応が抑制される。また、実施例に述べるように、OPACHの析出を促進するため、該水溶液にOPACHを種晶として添加することも有効である。
OPAの水和反応により析出したOPACHは、遠心ろ過、吸引ろ過、デカンテーションなどの方法により固液分離して水及び/又は有機溶媒により洗浄後乾燥することにより白色結晶性粉末として単離できる。
(OPACH)
このようにして得られたOPACHは、図1に示すようにDSC分析における吸熱ピークとしての融点が約121℃であり、OPAとは異なってほぼ無臭で刺激性もなく、皮膚に触れても皮膚を着色せず、室温ではOPAより固結しにくい白色粉末である。OPACHはこのような特性のため粉末のまま水系に添加でき、さらにOPAでは難しかった水性懸濁液や錠剤などの製剤に加工して提供することが可能である。
なお、OPACHが皮膚、すなわちタンパク質に触れても着色しない理由は、OPACHがベンゼン環上にアルデヒド基をもたないことから、タンパク質と結合してシッフ塩基を形成しないためと考えられる。一方のOPAは、ベンゼン環上の2つのアルデヒド基がタンパク質と結合して2つのシッフ塩基を形成して長い共鳴構造をもつ発色団をつくり着色すると考えられる。このOPAのアミノ酸との反応による発色メカニズムについてはPeter C.Zhu et al.: Chemical Detoxifying Neutralization of ortho-Phthalaldehyde:Seeking the “Greenest”. in Pesticide Decontamination and Detoxification, 2003, Chapter 7, pp85-97, ACS Publications.(アメリカ化学会出版局 2003年10月刊)において解説されている。
このようにして得られたOPACHは、図1に示すようにDSC分析における吸熱ピークとしての融点が約121℃であり、OPAとは異なってほぼ無臭で刺激性もなく、皮膚に触れても皮膚を着色せず、室温ではOPAより固結しにくい白色粉末である。OPACHはこのような特性のため粉末のまま水系に添加でき、さらにOPAでは難しかった水性懸濁液や錠剤などの製剤に加工して提供することが可能である。
なお、OPACHが皮膚、すなわちタンパク質に触れても着色しない理由は、OPACHがベンゼン環上にアルデヒド基をもたないことから、タンパク質と結合してシッフ塩基を形成しないためと考えられる。一方のOPAは、ベンゼン環上の2つのアルデヒド基がタンパク質と結合して2つのシッフ塩基を形成して長い共鳴構造をもつ発色団をつくり着色すると考えられる。このOPAのアミノ酸との反応による発色メカニズムについてはPeter C.Zhu et al.: Chemical Detoxifying Neutralization of ortho-Phthalaldehyde:Seeking the “Greenest”. in Pesticide Decontamination and Detoxification, 2003, Chapter 7, pp85-97, ACS Publications.(アメリカ化学会出版局 2003年10月刊)において解説されている。
<第二の実施形態> OPACHを含有する消毒薬及び殺生物剤
本発明のOPAの結晶性水和物(OPACH)は、弱い分子間力により2分子のOPAと1分子の水が水和反応により結合したものであり、常温常圧下では比較的安定に存在するが、加熱や溶解などによってOPAと水へ容易に戻るためOPAの前駆体的化合物である。従って、本発明のOPACHをOPAに変えることなく、微生物で汚染された水系に添加して消毒薬や殺生物剤として用いることも可能である。この場合、OPACHを結晶のまま水系に投入して用いることもできるし、水及び/又は有機溶媒に分散させた懸濁液(いわゆるフロアブル剤、濃度は5~40質量%が好ましい)、あるいは錠剤などに加工して用いることもできる。その際、OPACHの一部がOPAに変化しOPAとして存在しても構わない。また、OPACHを水及び/又は有機溶媒に溶解させて溶液として用いてもよいが、その場合は事実上OPA溶液としての使用になることがある。
なお、ここで用いることのできる有機溶媒としては、水との併用が一般的になるが、アセトン、1,2-ジメトキシエタン(DME)、γ-ブチロラクトン、炭酸プロピレン、エチレングリコール、ジエチレングリコール、ポリエチレングリコール#200などがあげられる。
また、懸濁液とするときは、適宜、通常用いられる増粘剤を添加することができる。増粘剤としては例えばキサンタンガム、カルボキシメチルセルロースナトリウム、アルギン酸ナトリウムなどがあげられる。
本発明のOPAの結晶性水和物(OPACH)は、弱い分子間力により2分子のOPAと1分子の水が水和反応により結合したものであり、常温常圧下では比較的安定に存在するが、加熱や溶解などによってOPAと水へ容易に戻るためOPAの前駆体的化合物である。従って、本発明のOPACHをOPAに変えることなく、微生物で汚染された水系に添加して消毒薬や殺生物剤として用いることも可能である。この場合、OPACHを結晶のまま水系に投入して用いることもできるし、水及び/又は有機溶媒に分散させた懸濁液(いわゆるフロアブル剤、濃度は5~40質量%が好ましい)、あるいは錠剤などに加工して用いることもできる。その際、OPACHの一部がOPAに変化しOPAとして存在しても構わない。また、OPACHを水及び/又は有機溶媒に溶解させて溶液として用いてもよいが、その場合は事実上OPA溶液としての使用になることがある。
なお、ここで用いることのできる有機溶媒としては、水との併用が一般的になるが、アセトン、1,2-ジメトキシエタン(DME)、γ-ブチロラクトン、炭酸プロピレン、エチレングリコール、ジエチレングリコール、ポリエチレングリコール#200などがあげられる。
また、懸濁液とするときは、適宜、通常用いられる増粘剤を添加することができる。増粘剤としては例えばキサンタンガム、カルボキシメチルセルロースナトリウム、アルギン酸ナトリウムなどがあげられる。
OPACHを結晶のまま使用すること、並びに水や有機溶媒に分散させた懸濁液及び錠剤に加工して消毒薬や殺生物剤として使用することは、先に述べたようにOPAでは従来難しく、本発明により新たに可能になる点である。本発明ではこれらOPACHを用いた消毒薬、殺生物剤を、結晶のままで使用する場合や、OPAに変化して存在する場合も含め、全て、OPACHを含有する消毒薬、殺生物剤という。
本発明のOPACHを消毒薬や殺生物剤として使用する際、以下のような公知の殺生物剤を併用してもよい。
オルトフタルアルデヒド、グルタルアルデヒド、過酢酸、過酸化水素、次亜塩素酸、アンモニアモノクロラミン、ブロモクロロジメチルヒダントイン(BCDMH)、トリクロロイソシアヌル酸、ジクロロイソシアヌル酸ナトリウム、2,2-ジブロモ-3-ニトリロプロピオンアミド、4,5-ジクロロ-1,2-ジチオール-3-オン、メチレンビスチオシアネート、3,3,4,4-テトラクロロテトラヒドロチオフェン-1,1-ジオキシド、1,4-ビス(ブロモアセトキシ)-2-ブテン、1,2-ビス(ブロモアセトキシ)エタン、ジクロログリオキシム、α-クロロベンズアルドキシム、α-クロロベンズアルドキシムアセテート、α-クロロ-4-クロロベンズアルドキシム、2-(p-ヒドロキシフェニル)-グリオキシロヒドロキシモイルクロリド、2-ブロモ-2-ニトロプロパン-1,3-ジオール、2,2-ジブロモ-2-ニトロエタノール、1,3-ジアセトキシ-2-ブロモ-2-ニトロプロパン、5-クロロ-2-メチル-4-イソチアゾリン-3-オン、2-メチル-4-イソチアゾリン-3-オン、2-n-オクチル-4-イソチアゾリン-3-オン、4,5-ジクロロ-2-n-オクチル-4-イソチアゾリン-3-オン、1,2-ベンズイソチアゾリン-3-オン、N-n-ブチル-1,2-ベンズイソチアゾリン-3-オン、1,2-ジブロモ-2,4-ジシアノブタン、N,N’-メチレンビスモルホリン、ジメチロールジメチルヒダントイン、モノメチロールジメチルヒダントイン、ヘキサヒドロ-1,3,5-トリス-2-ヒドロキシエチル-s-トリアジン、ヘキサヒドロ-1,3,5-トリエチル-s-トリアジン、4,4-ジメチル-1,3-オキサゾリジン、2,4,5,6-テトラクロロイソフタロニトリル、5-クロロ-2,4,6-トリフルオロイソフタロニトリル、α-ブロモシンナムアルデヒド、3-ヨード-2-プロピニルブチルカーバメート、p-クロロフェニル-3-ヨードプロパギルホルマール、1-{(ジヨードメチル)スルホニル}-4-メチルベンゼン、ポリビニルピロリドンヨード、カルベンダジム、チアベンダゾール、2-(チオシアノメチルチオ)ベンゾチアゾール、2,3,5,6-テトラクロロ-4-(メチルスルホニル)ピリジン、シプロコナゾール、プロピコナゾール、テブコナゾール、メチルパラベン、エチルパラベン、プロピルパラベン、オルトフェニルフェノール、オルトフェニルフェノールナトリウム、2-イソプロピル-5-メチルフェノール、3-メチル-4-イソプロピルフェノール、ジクロロフェン、トリクロサン、トリクロロカルバニリド、ヒノキチオール、亜鉛ピリチオン、銅ピリチオン、ナトリウムピリチオン、イルガロール-1051、N-アルキル(C12-C18)ジメチルアンモニウムクロリド、ベンジルジメチルドデシルアンモニウムクロリド、ジデシルジメチルアンモニウムクロリド、セチルトリメチルアンモニウムクロリド、セチルピリジニウムクロリド、ベンゾトニウムクロリドなどの第四級アンモニウム塩、1,4-ビス(3,3’-(1-デシルピリジニウム)メチルオキシ)ブタンジブロミド、N,N’-ヘキサメチレンビス(4-カルバモイル-1-デシルピリジニウムブロミドなどのビス型第四級アンモニウム塩、テトラキスヒドロキシメチルホスホニウムサルフェート、トリブチルテトラデシルホスホニウムクロリド、トリブチルヘキサデシルホスホニウムクロリド、トリブチルドデシルホスホニウムブロミド、ポリヘキサメチレンビグアニド塩酸塩、クロルヘキシジン塩酸塩、グルコン酸クロルヘキシジン。
オルトフタルアルデヒド、グルタルアルデヒド、過酢酸、過酸化水素、次亜塩素酸、アンモニアモノクロラミン、ブロモクロロジメチルヒダントイン(BCDMH)、トリクロロイソシアヌル酸、ジクロロイソシアヌル酸ナトリウム、2,2-ジブロモ-3-ニトリロプロピオンアミド、4,5-ジクロロ-1,2-ジチオール-3-オン、メチレンビスチオシアネート、3,3,4,4-テトラクロロテトラヒドロチオフェン-1,1-ジオキシド、1,4-ビス(ブロモアセトキシ)-2-ブテン、1,2-ビス(ブロモアセトキシ)エタン、ジクロログリオキシム、α-クロロベンズアルドキシム、α-クロロベンズアルドキシムアセテート、α-クロロ-4-クロロベンズアルドキシム、2-(p-ヒドロキシフェニル)-グリオキシロヒドロキシモイルクロリド、2-ブロモ-2-ニトロプロパン-1,3-ジオール、2,2-ジブロモ-2-ニトロエタノール、1,3-ジアセトキシ-2-ブロモ-2-ニトロプロパン、5-クロロ-2-メチル-4-イソチアゾリン-3-オン、2-メチル-4-イソチアゾリン-3-オン、2-n-オクチル-4-イソチアゾリン-3-オン、4,5-ジクロロ-2-n-オクチル-4-イソチアゾリン-3-オン、1,2-ベンズイソチアゾリン-3-オン、N-n-ブチル-1,2-ベンズイソチアゾリン-3-オン、1,2-ジブロモ-2,4-ジシアノブタン、N,N’-メチレンビスモルホリン、ジメチロールジメチルヒダントイン、モノメチロールジメチルヒダントイン、ヘキサヒドロ-1,3,5-トリス-2-ヒドロキシエチル-s-トリアジン、ヘキサヒドロ-1,3,5-トリエチル-s-トリアジン、4,4-ジメチル-1,3-オキサゾリジン、2,4,5,6-テトラクロロイソフタロニトリル、5-クロロ-2,4,6-トリフルオロイソフタロニトリル、α-ブロモシンナムアルデヒド、3-ヨード-2-プロピニルブチルカーバメート、p-クロロフェニル-3-ヨードプロパギルホルマール、1-{(ジヨードメチル)スルホニル}-4-メチルベンゼン、ポリビニルピロリドンヨード、カルベンダジム、チアベンダゾール、2-(チオシアノメチルチオ)ベンゾチアゾール、2,3,5,6-テトラクロロ-4-(メチルスルホニル)ピリジン、シプロコナゾール、プロピコナゾール、テブコナゾール、メチルパラベン、エチルパラベン、プロピルパラベン、オルトフェニルフェノール、オルトフェニルフェノールナトリウム、2-イソプロピル-5-メチルフェノール、3-メチル-4-イソプロピルフェノール、ジクロロフェン、トリクロサン、トリクロロカルバニリド、ヒノキチオール、亜鉛ピリチオン、銅ピリチオン、ナトリウムピリチオン、イルガロール-1051、N-アルキル(C12-C18)ジメチルアンモニウムクロリド、ベンジルジメチルドデシルアンモニウムクロリド、ジデシルジメチルアンモニウムクロリド、セチルトリメチルアンモニウムクロリド、セチルピリジニウムクロリド、ベンゾトニウムクロリドなどの第四級アンモニウム塩、1,4-ビス(3,3’-(1-デシルピリジニウム)メチルオキシ)ブタンジブロミド、N,N’-ヘキサメチレンビス(4-カルバモイル-1-デシルピリジニウムブロミドなどのビス型第四級アンモニウム塩、テトラキスヒドロキシメチルホスホニウムサルフェート、トリブチルテトラデシルホスホニウムクロリド、トリブチルヘキサデシルホスホニウムクロリド、トリブチルドデシルホスホニウムブロミド、ポリヘキサメチレンビグアニド塩酸塩、クロルヘキシジン塩酸塩、グルコン酸クロルヘキシジン。
<第三の実施形態> OPACHを用いた高純度OPAの製造方法
本発明者らは、OPACHの生成機構及びそのOPAと水への分解機構を鋭意検討した結果、OPACHを加熱及び/又は水や有機溶媒へ溶解してOPAに戻すことにより元のOPAより精製度の高まったOPAが得られることを明らかにした。これは、実施例に述べるように、OPACHを中間体として経由させた前後のOPAの純度を分析することにより確認された。本発明のOPAの製造方法は、純度98~100質量%のOPAを製造するのに好適である。ここで用いることのできる有機溶媒は、第一の実施形態で挙げたものと同様のものである。加熱する場合は50~120℃が好ましく、60~100℃がさらに好ましい。
本発明者らは、OPACHの生成機構及びそのOPAと水への分解機構を鋭意検討した結果、OPACHを加熱及び/又は水や有機溶媒へ溶解してOPAに戻すことにより元のOPAより精製度の高まったOPAが得られることを明らかにした。これは、実施例に述べるように、OPACHを中間体として経由させた前後のOPAの純度を分析することにより確認された。本発明のOPAの製造方法は、純度98~100質量%のOPAを製造するのに好適である。ここで用いることのできる有機溶媒は、第一の実施形態で挙げたものと同様のものである。加熱する場合は50~120℃が好ましく、60~100℃がさらに好ましい。
ところで、OPAポリマーは、酸触媒などの存在下、いわゆるアンジッピング反応(unzipping reaction)という分解反応が起きてOPAに戻ることが知られており、このような特性によりOPAポリマーはレジスト材料として用いられる(非特許文献5)。
本発明のOPACHはOPAポリマーの部分構造と似ているが、前述したようにOPACHを水和反応により生成させるため水が必須となる点がOPAの重合とは基本的に異なる。また、OPAの重合のような-50℃以下の低温条件を必要とせず室温付近で水和反応が可能なこと、そして、ポリマー安定剤としての両末端基(開始剤及び停止剤)を必要としない点でもOPACHはOPAの重合とは異なる。特にOPACHを製造するための温度条件が室温付近でよいことは工業的実施に有利である。
OPAの重合反応と水和反応では共にベンゼン環上に隣接する二つのアルデヒド基が結合する点は共通する。従って、この隣接したジアルデヒド構造を持たない分子は反応に関与せず、該水溶液中に溶解したまま残ることになる。好適にもOPACHはOPA及び他の不純物より溶解度が低いため、該水溶液中に分散した結晶としても固液分離された結晶としても常温常圧下で安定に存在できる。
OPACHの取扱いはOPAより容易であり、加熱や溶媒への溶解を行えばOPAに戻ることは前述した通りである。従って、固液分離により不純物を除去して精製されたOPAの結晶性水和物(OPACH)は高純度のOPAの原料でもあり、これは水和反応の逆反応として下式(5)で整理されるように、OPCHA1分子からOPA2分子と水1分子が定量的に得られることになる。

以下、本発明を実施例及び比較例により具体的に説明するが、本発明はこれらに限定されるものではない。
実施例1(ジメトキシエタン水溶液中での製造)
フラスコにOPA(ケイ・アイ化成社製、純度99.5%)、1,2-ジメトキシエタン及び水道水を1,2-ジメトキシエタン/水=1/5(重量比)となるように加えてOPAの14.3重量%溶液を調製した。室温で24時間撹拌して水和反応を行うと白色結晶が析出してスラリーとなった。これを吸引ろ過し少量の上記混合溶媒で洗浄後乾燥すると、白色結晶がOPAに対して69.7%の収率で得られた。ただし、収率は水和反応で付加された水の質量(6.29%)を補正していない。前記のように、この物質は赤外吸収スペクトル(KBr錠剤法)において、アルデヒド基に特有な1690cm-1の吸収がなくOPAの結晶性水和物(OPACH)であった(図2参照)。
フラスコにOPA(ケイ・アイ化成社製、純度99.5%)、1,2-ジメトキシエタン及び水道水を1,2-ジメトキシエタン/水=1/5(重量比)となるように加えてOPAの14.3重量%溶液を調製した。室温で24時間撹拌して水和反応を行うと白色結晶が析出してスラリーとなった。これを吸引ろ過し少量の上記混合溶媒で洗浄後乾燥すると、白色結晶がOPAに対して69.7%の収率で得られた。ただし、収率は水和反応で付加された水の質量(6.29%)を補正していない。前記のように、この物質は赤外吸収スペクトル(KBr錠剤法)において、アルデヒド基に特有な1690cm-1の吸収がなくOPAの結晶性水和物(OPACH)であった(図2参照)。
このOPAの水和によって得られた固体物質がOPAの結晶性水和物(OPACH)であること、さらに高純度のOPAの前駆体的化合物であることは、次の分析結果及び試験結果から確認できた。
1)フーリエ変換赤外吸収スペクトル(FT-IR)
OPACHをKBr錠剤法により測定した。得られたOPACHのFT-IRスペクトルを図2に示す。
OPACHはOPAのC=O伸縮振動の吸収波数として特徴的な1690cm-1がなく、3350cm-1に鋭い吸収が観察された。この3350cm-1の吸収は分子内水素結合のOH基に帰属すると考えられた。
OPACHをKBr錠剤法により測定した。得られたOPACHのFT-IRスペクトルを図2に示す。
OPACHはOPAのC=O伸縮振動の吸収波数として特徴的な1690cm-1がなく、3350cm-1に鋭い吸収が観察された。この3350cm-1の吸収は分子内水素結合のOH基に帰属すると考えられた。
2)紫外吸収スペクトル(UV)
OPACH及びOPAをそれぞれアセトニトリルに溶解して10.00mg/L溶液を調製しUVスペクトルを測定した。得られたOPACH及びOPAのUVスペクトルを図3及び図4に示す。
両者のUVスペクトルは一致したが、220nmを吸収極大とするピークの面積比(16.5144/17.6153)から、OPACHはOPAに対して93.7%の吸収強度を示した。
OPACH及びOPAをそれぞれアセトニトリルに溶解して10.00mg/L溶液を調製しUVスペクトルを測定した。得られたOPACH及びOPAのUVスペクトルを図3及び図4に示す。
両者のUVスペクトルは一致したが、220nmを吸収極大とするピークの面積比(16.5144/17.6153)から、OPACHはOPAに対して93.7%の吸収強度を示した。
3)1H-NMRスペクトル
先ず参考のため、OPAをD2Oに溶解して1H-NMRスペクトルを測定した。その際、試料は室温で約4時間かけて完全に溶解してから測定を行った。得られたOPAの1H-NMRスペクトルを図5に示す。OPAのD2O溶液はジアルデヒド(3a)と環状ヘミアセタール(3c)の混合物と考えられるチャートが得られた。その存在比は両者のベンゼン環のプロトン比から約1:14~15と考えられた。(Robert S. McDonald et al.:Can.J.Chem., 1979, Vol.57,pp506-516.を参照。)
次に、OPACHを室温でDMSO-d6に溶解して直ちに1H-NMRスペクトルを測定した。得られた1H-NMRスペクトルを図6に示す。OPACHのDMSO-d6溶液はジアルデヒド(3a)及び環状ヘミアセタール(3c)は含まれず、OPACHとしての存在が支持されるチャートが得られた。化学シフト(δ,ppm):6.30(2H、d)、6.42(2H、s)、7.23(2H、d)、7.41(8H、m)。
先ず参考のため、OPAをD2Oに溶解して1H-NMRスペクトルを測定した。その際、試料は室温で約4時間かけて完全に溶解してから測定を行った。得られたOPAの1H-NMRスペクトルを図5に示す。OPAのD2O溶液はジアルデヒド(3a)と環状ヘミアセタール(3c)の混合物と考えられるチャートが得られた。その存在比は両者のベンゼン環のプロトン比から約1:14~15と考えられた。(Robert S. McDonald et al.:Can.J.Chem., 1979, Vol.57,pp506-516.を参照。)
次に、OPACHを室温でDMSO-d6に溶解して直ちに1H-NMRスペクトルを測定した。得られた1H-NMRスペクトルを図6に示す。OPACHのDMSO-d6溶液はジアルデヒド(3a)及び環状ヘミアセタール(3c)は含まれず、OPACHとしての存在が支持されるチャートが得られた。化学シフト(δ,ppm):6.30(2H、d)、6.42(2H、s)、7.23(2H、d)、7.41(8H、m)。
4)水への溶解度
OPACH5gをイオン交換水50gに加えて分散させ室温下で24時間撹拌した。結晶の一部は徐々に溶解したので、フィルターを通して結晶を取り除き溶液部をサンプリングし、ガスクロマトグラフィーにより分析した。その結果、溶液部はOPAを2.6質量%含有していた。このことから、OPACHは室温では水に徐々に溶解しOPAとなって水溶液中に存在し、その24時間後のOPA濃度は2.6質量%となることが分かった。
次に、OPAの水に対する飽和溶解度(約5.9質量%)より十分低濃度となるようOPACHをイオン交換水に分散させてOPACHの1.0質量%水分散液を調製し、これを所定の温度に保持して撹拌を行い結晶が完全に溶解するまでの時間を観察した。その結果、OPACHの溶解時間は30℃で5時間、40℃で3時間、50℃で1時間であった。以上のことから、OPACHの水への溶解時間は温度に依存し、温度が高ければ短縮することができ、消毒薬及び殺生物剤としての実用上問題ないことが分かった。
冒頭に述べたように、OPAの高水準消毒薬としての代表的な使用濃度は0.55質量%であり、また殺生物剤として水系に添加して使用される濃度は通常数mg/L~数百mg/Lであることから、上記の水への溶解性を有するOPACHは、微生物で汚染された水系にそのまま添加されてもOPAとして溶解していくことから、OPAとしての生理活性を示す濃度に到達することが分かった。
OPACH5gをイオン交換水50gに加えて分散させ室温下で24時間撹拌した。結晶の一部は徐々に溶解したので、フィルターを通して結晶を取り除き溶液部をサンプリングし、ガスクロマトグラフィーにより分析した。その結果、溶液部はOPAを2.6質量%含有していた。このことから、OPACHは室温では水に徐々に溶解しOPAとなって水溶液中に存在し、その24時間後のOPA濃度は2.6質量%となることが分かった。
次に、OPAの水に対する飽和溶解度(約5.9質量%)より十分低濃度となるようOPACHをイオン交換水に分散させてOPACHの1.0質量%水分散液を調製し、これを所定の温度に保持して撹拌を行い結晶が完全に溶解するまでの時間を観察した。その結果、OPACHの溶解時間は30℃で5時間、40℃で3時間、50℃で1時間であった。以上のことから、OPACHの水への溶解時間は温度に依存し、温度が高ければ短縮することができ、消毒薬及び殺生物剤としての実用上問題ないことが分かった。
冒頭に述べたように、OPAの高水準消毒薬としての代表的な使用濃度は0.55質量%であり、また殺生物剤として水系に添加して使用される濃度は通常数mg/L~数百mg/Lであることから、上記の水への溶解性を有するOPACHは、微生物で汚染された水系にそのまま添加されてもOPAとして溶解していくことから、OPAとしての生理活性を示す濃度に到達することが分かった。
5)GPCクロマトグラム
OPACHをDMFに溶解して1.0質量%溶液とした。比較のためOPAもDMFに溶解して1.0質量%溶液を調製した。いずれの場合も室温で1~2分間振り混ぜることにより結晶は容易に溶解し、ポリマーを溶解した際に特有となる高粘度溶液にはならなかった。これらの溶液を下記条件のゲルパーミエーションクロマトグラフィー(GPC)で測定してGPCクロマトグラムをとり、ピーク溶出時間及びピーク面積を測定した。また、ポリマー又はオリゴマーが溶出された場合を考慮して、それらの分子量をキャリブレーションするために標準ポリスチレン混合物(重量平均分子量Mw:5.3×102 ~ 9.89×104)をDMF/THF混合溶媒(50/50重量比)に溶解した標準溶液についてもGPCクロマトグラムをとった。
OPACHをDMFに溶解して1.0質量%溶液とした。比較のためOPAもDMFに溶解して1.0質量%溶液を調製した。いずれの場合も室温で1~2分間振り混ぜることにより結晶は容易に溶解し、ポリマーを溶解した際に特有となる高粘度溶液にはならなかった。これらの溶液を下記条件のゲルパーミエーションクロマトグラフィー(GPC)で測定してGPCクロマトグラムをとり、ピーク溶出時間及びピーク面積を測定した。また、ポリマー又はオリゴマーが溶出された場合を考慮して、それらの分子量をキャリブレーションするために標準ポリスチレン混合物(重量平均分子量Mw:5.3×102 ~ 9.89×104)をDMF/THF混合溶媒(50/50重量比)に溶解した標準溶液についてもGPCクロマトグラムをとった。
GPC条件:
装置 :島津製作所製 LC Solution
カラム:昭和電工製 Shodex OHpak SB-803 HQ 8.0×300mm(2本)及びガードカラムとして、Shodex OHpak SB-G 6.0×50mm(1本)
カラム温度:40℃
移動相:DMF 0.6ml/min
検出器:フォトダイオードアレイ検出器(UV波長 275nm)
注入量:10マイクロリットル
装置 :島津製作所製 LC Solution
カラム:昭和電工製 Shodex OHpak SB-803 HQ 8.0×300mm(2本)及びガードカラムとして、Shodex OHpak SB-G 6.0×50mm(1本)
カラム温度:40℃
移動相:DMF 0.6ml/min
検出器:フォトダイオードアレイ検出器(UV波長 275nm)
注入量:10マイクロリットル
GPC分析の結果、OPACH及びOPAは、いずれも溶出時間33.0分の1ピークのみが溶出された。また、OPAのピーク面積に対するOPACHのピーク面積は、前記UVスペクトルにおけるOPAのピーク面積に対するOPACHのピーク面積にほぼ一致して93%となった。これらの結果からOPACHにはポリマーは含まれず、1.0質量%のOPACHは、0.93質量%のOPAとして分離され検出されたことになる。なお、標準ポリスチレン混合物のGPCクロマトグラムは、各ピークがきれいに分離され、それらの溶出時間は17.4分から29.4分の範囲にあった。OPAの主要な不純物であるフタリドの溶出時間は、OPAの33.0分よりわずかに速い32.2分であった。
6)熱分解によるOPA及び水の回収
攪拌装置、温度計、コンデンサー、受器を備えた丸底四つ口フラスコにOPACH100g及びトルエン200gを加えた。これを撹拌しながらオイルバスを用いて90℃で12時間加熱した。OPACHの結晶は徐々に溶解して黄色透明のトルエン溶液となり、水とトルエンが共沸脱水されてコンデンサーから受器に留出した。冷却後、フラスコ内のトルエン溶液をガスクロマトグラフィーにより分析した結果、トルエン溶液にはOPAが93.5g溶解していた。留出液を分液したところ水の質量は6.2gであった。以上のことから、OPACHの100質量部は加熱によりOPA93.5質量部及び水6.2質量部を生成したことになり、実験誤差も考慮するとOPACHは定量的にOPAと水になることの裏付けが得られた。
攪拌装置、温度計、コンデンサー、受器を備えた丸底四つ口フラスコにOPACH100g及びトルエン200gを加えた。これを撹拌しながらオイルバスを用いて90℃で12時間加熱した。OPACHの結晶は徐々に溶解して黄色透明のトルエン溶液となり、水とトルエンが共沸脱水されてコンデンサーから受器に留出した。冷却後、フラスコ内のトルエン溶液をガスクロマトグラフィーにより分析した結果、トルエン溶液にはOPAが93.5g溶解していた。留出液を分液したところ水の質量は6.2gであった。以上のことから、OPACHの100質量部は加熱によりOPA93.5質量部及び水6.2質量部を生成したことになり、実験誤差も考慮するとOPACHは定量的にOPAと水になることの裏付けが得られた。
7)殺菌効果
前記4)の水への溶解度測定において得られたOPACHの2.6質量%水溶液(=実際にはOPAとして溶解)について、細菌に対する殺菌効果を調べた。OPAとの効果を比較するためOPA水溶液も調製してOPAとしての添加濃度を合わせた。その結果、OPACH水溶液とOPA水溶液は殺菌効果が一致した。詳細は実施例6を参照。
前記4)の水への溶解度測定において得られたOPACHの2.6質量%水溶液(=実際にはOPAとして溶解)について、細菌に対する殺菌効果を調べた。OPAとの効果を比較するためOPA水溶液も調製してOPAとしての添加濃度を合わせた。その結果、OPACH水溶液とOPA水溶液は殺菌効果が一致した。詳細は実施例6を参照。
8)元素分析及びX線構造解析
OPACHの元素分析を行った結果、OPACHの元素比はC:H:O=67.13:4.90:27.97であった。これよりOPACHの分子式はC16H14O5となることが裏付けられた。C16H14O5としての計算値はCが67.13%、Hが4.93%、Oが27.94%である。また、OPACHのX線構造解析を行った結果、これは前記の式(1)で表される物質と確定された。
これらの結果から、OPACHの分子量は286.28となり、OPACHの1分子はOPAとして93.71%、H2Oとして6.29%を含むことが分かる。
本発明のOPACHの化学名は、3,3’-オキシビス(1,3-ジヒドロイソベンゾフラン-1-オール)と命名される。
OPACHの元素分析を行った結果、OPACHの元素比はC:H:O=67.13:4.90:27.97であった。これよりOPACHの分子式はC16H14O5となることが裏付けられた。C16H14O5としての計算値はCが67.13%、Hが4.93%、Oが27.94%である。また、OPACHのX線構造解析を行った結果、これは前記の式(1)で表される物質と確定された。
これらの結果から、OPACHの分子量は286.28となり、OPACHの1分子はOPAとして93.71%、H2Oとして6.29%を含むことが分かる。
本発明のOPACHの化学名は、3,3’-オキシビス(1,3-ジヒドロイソベンゾフラン-1-オール)と命名される。
9)13C-NMRスペクトル
OPACHを室温でDMSO-d6に溶解して直ちに13C-NMRスペクトルを測定した。結果を図7に示す。化学シフト(δ,ppm):99.65、100.94、122.71、122.95、128.76、129.16、138.48、140.76。
OPACHを室温でDMSO-d6に溶解して直ちに13C-NMRスペクトルを測定した。結果を図7に示す。化学シフト(δ,ppm):99.65、100.94、122.71、122.95、128.76、129.16、138.48、140.76。
実施例2(アセトン水溶液中での製造)
実施例1における1,2-ジメトキシエタンに変えてアセトンを用いたことを除き、実施例1と同様に操作した。その結果、OPAの結晶性水和物(OPACH)が白色結晶としてOPAに対して47.6%の収率で得られた。ただし、収率は水和反応で付加された水の質量(6.29%)を補正していない。
実施例1における1,2-ジメトキシエタンに変えてアセトンを用いたことを除き、実施例1と同様に操作した。その結果、OPAの結晶性水和物(OPACH)が白色結晶としてOPAに対して47.6%の収率で得られた。ただし、収率は水和反応で付加された水の質量(6.29%)を補正していない。
実施例3(DMF水溶液中での製造)
実施例1における1,2-ジメトキシエタンに変えてN,N-ジメチルホルムアミド(DMF)を用いたことを除き、実施例1と同様に操作した。その結果、OPAの結晶性水和物(OPACH)が白色結晶としてOPAに対して48.2%の収率で得られた。ただし、収率は水和反応で付加された水の質量(6.29%)を補正していない。
実施例1における1,2-ジメトキシエタンに変えてN,N-ジメチルホルムアミド(DMF)を用いたことを除き、実施例1と同様に操作した。その結果、OPAの結晶性水和物(OPACH)が白色結晶としてOPAに対して48.2%の収率で得られた。ただし、収率は水和反応で付加された水の質量(6.29%)を補正していない。
実施例4(水溶液中での製造及び種晶の添加効果)
ビーカーにOPA(ケイ・アイ化成社製、純度99.5%)及びイオン交換水を加えてOPA5.0質量%水溶液を調製した。この水溶液50gに種晶としてOPAの結晶性水和物(OPACH、実施例1で調製したもの)を所定量添加した後(下表1中の(A))、室温で24時間撹拌した。その結果、黄色結晶であるOPAはいずれの場合も析出せず白色結晶であるOPAの結晶性水和物(OPACH)が下表1の通り析出した。結晶をろ過乾燥して重量分析し(下表1中の(B))、ろ液中のOPA濃度をガスクロマトグラフィーにより求めた(下表1中の(C))。
ビーカーにOPA(ケイ・アイ化成社製、純度99.5%)及びイオン交換水を加えてOPA5.0質量%水溶液を調製した。この水溶液50gに種晶としてOPAの結晶性水和物(OPACH、実施例1で調製したもの)を所定量添加した後(下表1中の(A))、室温で24時間撹拌した。その結果、黄色結晶であるOPAはいずれの場合も析出せず白色結晶であるOPAの結晶性水和物(OPACH)が下表1の通り析出した。結晶をろ過乾燥して重量分析し(下表1中の(B))、ろ液中のOPA濃度をガスクロマトグラフィーにより求めた(下表1中の(C))。
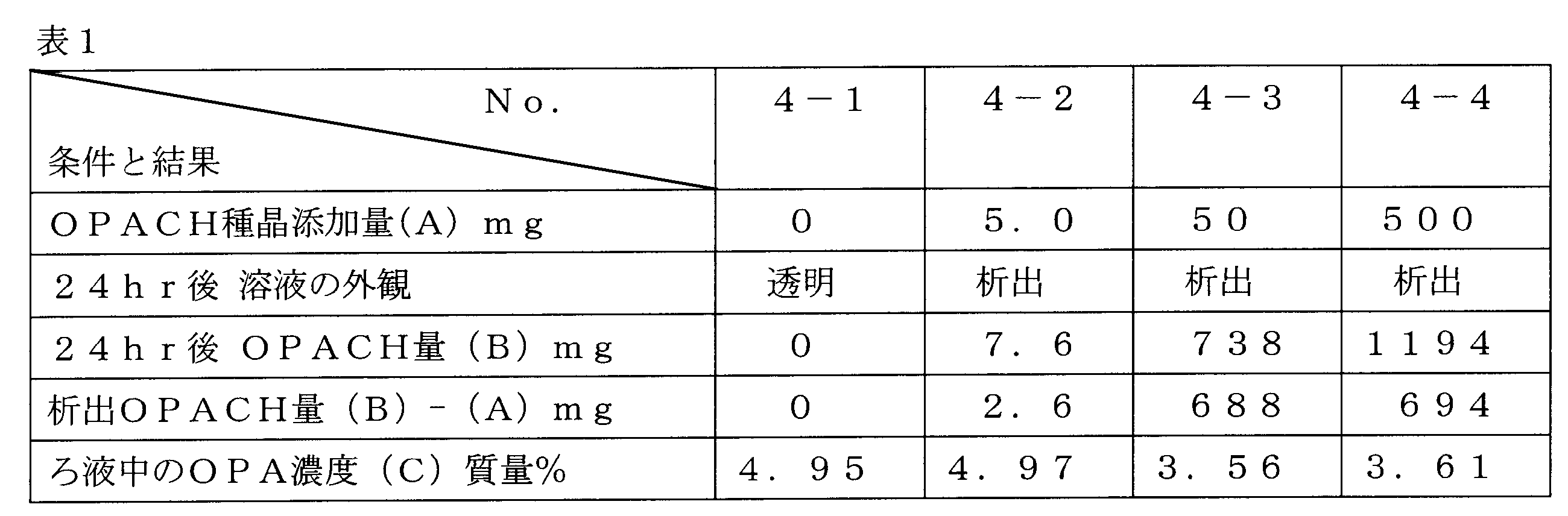
表1の結果からOPAの5.0質量%水溶液からOPACHが生成し、その際に種晶としてOPACHを一定量添加すると(No.4-3及びNo.4-4)、OPACHの無添加(No.4-1)及び少量添加(No.4-2)に比べてOPACHの生成が促進されることが分かった。
実施例5(水溶液中の挙動)
実施例1で得られたOPACH5gをイオン交換水50gに加えて分散させ室温下で24時間撹拌した。結晶の一部は徐々に溶解したので、フィルターを通して結晶を除いて溶液部をサンプリングしてガスクロマトグラフィーにより分析した。その結果、溶液部にはOPAと同じ溶出位置に1ピークのみが認められ、OPAとして2.6質量%含有されることが分かった。
実施例1で得られたOPACH5gをイオン交換水50gに加えて分散させ室温下で24時間撹拌した。結晶の一部は徐々に溶解したので、フィルターを通して結晶を除いて溶液部をサンプリングしてガスクロマトグラフィーにより分析した。その結果、溶液部にはOPAと同じ溶出位置に1ピークのみが認められ、OPAとして2.6質量%含有されることが分かった。
実施例6(殺菌効果)
実施例5で得られたOPACHの水溶液(OPAとして2.6質量%含有)について、細菌に対する接触時間30分の殺菌効果を調べた。参考のためOPACHをDMSOに0.1質量%溶解した溶液についても同様に殺菌効果を調べた。また、OPAとの殺菌効果を比較するため、OPAの水溶液及びDMSO溶液も調製した。OPACHはOPAとしての濃度を算出してOPAの添加濃度に合わせた。結果を下表2に示す。
殺菌効果の試験条件は次の通りである。
試験系:pH6緩衝液系及びpH8緩衝液系(リン酸-クエン酸緩衝液)
対象菌:以下の3種の細菌を混合
Enterobacter aerogenes NBRC 13534
Staphylococcus aureus NBRC 12732
Pseudomonas aeruginosa NBRC 13275
接触時間:30分
実施例5で得られたOPACHの水溶液(OPAとして2.6質量%含有)について、細菌に対する接触時間30分の殺菌効果を調べた。参考のためOPACHをDMSOに0.1質量%溶解した溶液についても同様に殺菌効果を調べた。また、OPAとの殺菌効果を比較するため、OPAの水溶液及びDMSO溶液も調製した。OPACHはOPAとしての濃度を算出してOPAの添加濃度に合わせた。結果を下表2に示す。
殺菌効果の試験条件は次の通りである。
試験系:pH6緩衝液系及びpH8緩衝液系(リン酸-クエン酸緩衝液)
対象菌:以下の3種の細菌を混合
Enterobacter aerogenes NBRC 13534
Staphylococcus aureus NBRC 12732
Pseudomonas aeruginosa NBRC 13275
接触時間:30分

表2の結果からOPACHの水溶液及びDMSO溶液は、OPAの水溶液及びDMSO溶液と同等の殺菌効果を示すことが分かった。従って、殺菌効果の結果からもOPACHは溶液中又はその添加された水系においてはOPAとして存在することが裏付けられた。
実施例7(OPACHを経由して得られたOPAの精製効果)
実施例1におけるOPAを純度98.8%のもの(イハラニッケイ化学工業社製)に変えて実施例1と同様に水和反応を行い生成した白色結晶を吸引ろ過・乾燥してOPACHを単離した。これをトルエン中に分散させて加熱し熱分解させたところ、下表3のようにガスクロマトグラフィーによるOPA純度が99.5%に向上し、フタリドが大幅に減少するとともに塩素含有不純物であるα,α,α',α'-テトラクロロ-o-キシレン(TECOX)は検出されなかった。
実施例1におけるOPAを純度98.8%のもの(イハラニッケイ化学工業社製)に変えて実施例1と同様に水和反応を行い生成した白色結晶を吸引ろ過・乾燥してOPACHを単離した。これをトルエン中に分散させて加熱し熱分解させたところ、下表3のようにガスクロマトグラフィーによるOPA純度が99.5%に向上し、フタリドが大幅に減少するとともに塩素含有不純物であるα,α,α',α'-テトラクロロ-o-キシレン(TECOX)は検出されなかった。

表3の結果からOPAはOPACHを経由させるという簡単な操作により精製できることが分かった。
実施例8(結晶の固結性)
内径50mm、高さ70mm、厚さ5mmの透明な硬質ガラス製の円筒を用意し、その円筒の開放部を上下にして平板ガラス上に置いた。これにOPA(ジイソプロピルエーテルから再結晶したもの)及びOPACH(実施例1で製造したもの)の結晶サンプル40gをそれぞれ開放部から加えた後、結晶の上に平板ガラスを置いて蓋をし、さらに質量620gのステンレス製オモリを載せて結晶に加重をかけた。これらを40℃の恒温槽内に1週間静置して固結の有無を観察した。
内径50mm、高さ70mm、厚さ5mmの透明な硬質ガラス製の円筒を用意し、その円筒の開放部を上下にして平板ガラス上に置いた。これにOPA(ジイソプロピルエーテルから再結晶したもの)及びOPACH(実施例1で製造したもの)の結晶サンプル40gをそれぞれ開放部から加えた後、結晶の上に平板ガラスを置いて蓋をし、さらに質量620gのステンレス製オモリを載せて結晶に加重をかけた。これらを40℃の恒温槽内に1週間静置して固結の有無を観察した。
その結果、OPAは硬い固結が認められたが、OPACHは全く固結せず、さらさらとした流動性のある粉末のままであった。
実施例9(水性懸濁液の製造及び水への溶解性)
ガラス瓶にイオン交換水80質量部及び増粘剤のキサンタンガム(ローヌ・プーラン社製商品名:ロードポール23)0.8質量部を加えて撹拌溶解した。これにOPACH20質量部を加えて撹拌混合して均一な白色スラリー液とした。この水性懸濁液を密栓して室温で3ケ月間放置したところ結晶はわずかに沈降したが(上澄み部の容積は全体の10%未満)、撹拌により容易に均一なスラリー液(フロアブル剤)に戻った。
このOPACHの20%水性懸濁液3質量部を蒸留水97質量部で希釈して3%分散液とし、室温(20℃)及び40℃に維持して撹拌を行い、OPACHの結晶が完全に溶解するまでの時間を調べた。その結果、OPACHは室温(20℃)では約5時間、40℃では約2時間で完全に溶解し透明溶液となった。この溶液に含まれるOPAの濃度は高水準消毒薬で一般的な0.56%である(≒3×0.20×0.937)。
ガラス瓶にイオン交換水80質量部及び増粘剤のキサンタンガム(ローヌ・プーラン社製商品名:ロードポール23)0.8質量部を加えて撹拌溶解した。これにOPACH20質量部を加えて撹拌混合して均一な白色スラリー液とした。この水性懸濁液を密栓して室温で3ケ月間放置したところ結晶はわずかに沈降したが(上澄み部の容積は全体の10%未満)、撹拌により容易に均一なスラリー液(フロアブル剤)に戻った。
このOPACHの20%水性懸濁液3質量部を蒸留水97質量部で希釈して3%分散液とし、室温(20℃)及び40℃に維持して撹拌を行い、OPACHの結晶が完全に溶解するまでの時間を調べた。その結果、OPACHは室温(20℃)では約5時間、40℃では約2時間で完全に溶解し透明溶液となった。この溶液に含まれるOPAの濃度は高水準消毒薬で一般的な0.56%である(≒3×0.20×0.937)。
実施例10(錠剤の製造及び水への溶解性)
OPACHの粉末をフルイに掛けて1.0~2.0mmの粒径の粉末を集めた。これを85重量%、ステアリン酸マグネシウム10重量%、タルク5重量%の割合で混合した後、10gずつ秤りとって打錠圧を変えて打錠し灰白色の錠剤を得た(下表4)。
1L容ビーカーに上水1Lを計り入れ、錠剤を1個ずつ投入してマグネチックスターラーを用いて室温で7日間撹拌した。その際、撹拌子が錠剤に直接接触しないようにした。その結果、いずれの打錠圧の錠剤も水中で膨潤、軟化、クラック発生などが起こらず、表面から徐々に溶解することが観察された。特に打錠圧の最も低い60kgf/cm2の錠剤は、錠剤表面が少し黄変しながら溶解した。これは、その水溶液を一部サンプリングしてアセトニトリルで希釈後、ガスクロマトグラフィーによりOPA濃度を分析した結果、水溶液中には7日間でOPAが3650mg/L溶解していたことにより確認された。
OPACHの粉末をフルイに掛けて1.0~2.0mmの粒径の粉末を集めた。これを85重量%、ステアリン酸マグネシウム10重量%、タルク5重量%の割合で混合した後、10gずつ秤りとって打錠圧を変えて打錠し灰白色の錠剤を得た(下表4)。
1L容ビーカーに上水1Lを計り入れ、錠剤を1個ずつ投入してマグネチックスターラーを用いて室温で7日間撹拌した。その際、撹拌子が錠剤に直接接触しないようにした。その結果、いずれの打錠圧の錠剤も水中で膨潤、軟化、クラック発生などが起こらず、表面から徐々に溶解することが観察された。特に打錠圧の最も低い60kgf/cm2の錠剤は、錠剤表面が少し黄変しながら溶解した。これは、その水溶液を一部サンプリングしてアセトニトリルで希釈後、ガスクロマトグラフィーによりOPA濃度を分析した結果、水溶液中には7日間でOPAが3650mg/L溶解していたことにより確認された。

参考例1(OPA濃度が低い場合の影響)
実施例1におけるOPA溶液の濃度14.3質量%を5.0質量%に減量したことを除き、実施例1と同様に室温で24時間撹拌した。しかし、結晶は析出せず溶液のままであったため、さらに延長して通算5日間まで撹拌したが結晶は全く析出しなかった。よって、その溶媒系においてOPAの濃度を低く設定するとOPACHは生成し難くなり、適切なOPA濃度にする必要があることが分かる。
実施例1におけるOPA溶液の濃度14.3質量%を5.0質量%に減量したことを除き、実施例1と同様に室温で24時間撹拌した。しかし、結晶は析出せず溶液のままであったため、さらに延長して通算5日間まで撹拌したが結晶は全く析出しなかった。よって、その溶媒系においてOPAの濃度を低く設定するとOPACHは生成し難くなり、適切なOPA濃度にする必要があることが分かる。
参考例2(OPA濃度が高い場合の影響)
実施例1におけるOPA溶液の濃度14.3質量%を20.0質量%に増量したことを除き、実施例1と同様に室温で24時間撹拌した。その結果OPAの黄色結晶のみが析出した。さらに延長して通算5日間まで撹拌したが、OPACHの白色結晶は析出せずOPAが析出したままであった。よって、その溶媒系においてOPAの溶解度を超えて高く濃度設定するとOPAが析出してOPACHは生成し難くなるため、適切なOPA濃度にする必要があることが分かる。
実施例1におけるOPA溶液の濃度14.3質量%を20.0質量%に増量したことを除き、実施例1と同様に室温で24時間撹拌した。その結果OPAの黄色結晶のみが析出した。さらに延長して通算5日間まで撹拌したが、OPACHの白色結晶は析出せずOPAが析出したままであった。よって、その溶媒系においてOPAの溶解度を超えて高く濃度設定するとOPAが析出してOPACHは生成し難くなるため、適切なOPA濃度にする必要があることが分かる。
本発明をその実施態様とともに説明したが、我々は特に指定しない限り我々の発明を説明のどの細部においても限定しようとするものではなく、添付の請求の範囲に示した発明の精神と範囲に反することなく幅広く解釈されるべきであると考える。
本願は、2011年4月27日に日本国で特許出願された特願2011-99736に基づく優先権を主張するものであり、これはここに参照してその内容を本明細書の記載の一部として取り込む。
Claims (4)
- 下記式(1)で表されるオルトフタルアルデヒドの結晶性水和物。

- 請求項1に記載のオルトフタルアルデヒドの結晶性水和物を含有することを特徴とする消毒薬。
- 請求項1に記載のオルトフタルアルデヒドの結晶性水和物を含有することを特徴とする殺生物剤。
- 請求項1に記載のオルトフタルアルデヒドの結晶性水和物を加熱及び/又は溶媒に溶解させることを特徴とするオルトフタルアルデヒドの製造法。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2011099736A JP5806848B2 (ja) | 2011-04-27 | 2011-04-27 | 3,3’−オキシビス(1,3−ジヒドロイソベンゾフラン−1−オール)、それを含有する消毒薬及び殺生物剤、並びにオルトフタルアルデヒドの製造方法 |
| JP2011-099736 | 2011-04-27 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2012147865A1 true WO2012147865A1 (ja) | 2012-11-01 |
Family
ID=47072380
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2012/061242 Ceased WO2012147865A1 (ja) | 2011-04-27 | 2012-04-26 | オルトフタルアルデヒドの結晶性水和物、それを含有する消毒薬及び殺生物剤、並びにオルトフタルアルデヒドの製造方法 |
Country Status (2)
| Country | Link |
|---|---|
| JP (1) | JP5806848B2 (ja) |
| WO (1) | WO2012147865A1 (ja) |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5810567A (ja) * | 1981-06-15 | 1983-01-21 | イ−ストマン・コダツク・カンパニ− | フタルアルデヒド付加物及びこれを含有する画像形成組成物 |
| JPH11140010A (ja) * | 1997-11-11 | 1999-05-25 | Ki Kasei Kk | オルトフタルアルデヒド安定化組成物 |
| JP2000178222A (ja) * | 1998-12-16 | 2000-06-27 | Ki Kasei Kk | オルトフタルアルデヒド安定化組成物及びその製造方法 |
-
2011
- 2011-04-27 JP JP2011099736A patent/JP5806848B2/ja active Active
-
2012
- 2012-04-26 WO PCT/JP2012/061242 patent/WO2012147865A1/ja not_active Ceased
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPS5810567A (ja) * | 1981-06-15 | 1983-01-21 | イ−ストマン・コダツク・カンパニ− | フタルアルデヒド付加物及びこれを含有する画像形成組成物 |
| JPH11140010A (ja) * | 1997-11-11 | 1999-05-25 | Ki Kasei Kk | オルトフタルアルデヒド安定化組成物 |
| JP2000178222A (ja) * | 1998-12-16 | 2000-06-27 | Ki Kasei Kk | オルトフタルアルデヒド安定化組成物及びその製造方法 |
Non-Patent Citations (3)
| Title |
|---|
| RAISTRICK,H. ET AL.: "Biochemistry of microorganisms. XCVII. Flavipin, a crystalline metabolite of Aspergillus flavipes and Aspergillus terreus", BIOCHEMICAL JOURNAL, vol. 63, 1956, pages 395 - 406 * |
| SCHONBERG,A. ET AL.: "Photochemical reactions in sunlight. XVIII. Dimerization of o- phthalaldehyde", JOURNAL OF THE AMERICAN CHEMICAL SOCIETY, vol. 77, 1955, pages 5755 - 5756 * |
| WATANABE,M. ET AL.: "A facile synthesis of optically active 3-ethyl- and 3-butylphthalides via catalytic enantioselective addition of dialkylzinc reagents to o-phthalaldehyde", JOURNAL OF ORGANIC CHEMISTRY, vol. 57, no. 2, 1992, pages 742 - 744 * |
Also Published As
| Publication number | Publication date |
|---|---|
| JP2012229187A (ja) | 2012-11-22 |
| JP5806848B2 (ja) | 2015-11-10 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| Sun et al. | A new cyclic N-halamine biocidal polymer | |
| DE69429829T2 (de) | Biozide polymere zyklische n-halamine | |
| US5670646A (en) | Monomeric and polymeric cyclic amine and-halamine compounds | |
| EP0239896B1 (en) | N,n'-dihalo-2-imidazolidinones | |
| RU2670439C2 (ru) | Производное замещенного пиразолилпиразола и его применение в качестве гербицида | |
| HU189592B (en) | Herbicidal compositions containing 2-/2-fluor-4-halogen-5-substituted-phanyl/-hydantoines and process for the production of the active substance | |
| Farah et al. | Antimicrobial N-brominated hydantoin and uracil grafted polystyrene beads | |
| JP5806848B2 (ja) | 3,3’−オキシビス(1,3−ジヒドロイソベンゾフラン−1−オール)、それを含有する消毒薬及び殺生物剤、並びにオルトフタルアルデヒドの製造方法 | |
| JPH02193948A (ja) | 抗微生物性を有する化合物及びその製造法 | |
| CN104230838B (zh) | 农用杀虫剂茚虫威高纯度关键中间体的制备方法 | |
| KR101569556B1 (ko) | 살미생물제 | |
| US4996220A (en) | Method of producing a clathrate compound | |
| JPH07101890A (ja) | 包接化合物 | |
| DE102009060249A1 (de) | Das Verfahren der Desinfektionsmittelgewinnung durch die Vermischung von der Polyhexamethylenguanidinbase mit ihren Derivaten, die Phosphat-Anionen und Jod enthalten | |
| EP3237490B1 (fr) | Nouveaux polymeres contenant des fonctions sulfonates metalliques, leurs procedes de preparation et leurs utilisations comme antibacteriens, fongicides, antimicrobiens | |
| CN106632032B (zh) | 地喹氯铵及其制备方法 | |
| Limban et al. | Some new 2-(4-ethyl-phenoxymethyl) benzoic acid thioureides: Synthesis and spectral characterization | |
| JP6275596B2 (ja) | テルミサルタンのアンモニウム塩の製造方法 | |
| KR100369843B1 (ko) | 3-이소티아졸론 용액 조성물 | |
| CN103333165A (zh) | 2-苯基-5-(3,4-二甲基)吡咯基-1,3,4-噁二唑及制备方法 | |
| KR100433207B1 (ko) | 3-이소티아졸론 용액 조성물 | |
| JPH0733755A (ja) | 新規な分子間化合物及びその製造方法 | |
| JP4774192B2 (ja) | 1,2−ジクロロエタンを含まないゾニサミドの結晶の製造方法およびゾニサミドの高純度結晶 | |
| US6624316B2 (en) | Method for obtaining 2-bromo-5-(2-bromo-2-nitrovinyl)-furan | |
| KR100433205B1 (ko) | 3-이소티아졸론 용액 조성물 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 12776931 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 12776931 Country of ref document: EP Kind code of ref document: A1 |