WO2013129709A2 - ポリオルガノシロキサン含有グラフト共重合体、樹脂組成物及び成形体 - Google Patents
ポリオルガノシロキサン含有グラフト共重合体、樹脂組成物及び成形体 Download PDFInfo
- Publication number
- WO2013129709A2 WO2013129709A2 PCT/JP2013/067938 JP2013067938W WO2013129709A2 WO 2013129709 A2 WO2013129709 A2 WO 2013129709A2 JP 2013067938 W JP2013067938 W JP 2013067938W WO 2013129709 A2 WO2013129709 A2 WO 2013129709A2
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- polyorganosiloxane
- mass
- graft copolymer
- resin composition
- powder
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B29—WORKING OF PLASTICS; WORKING OF SUBSTANCES IN A PLASTIC STATE IN GENERAL
- B29D—PRODUCING PARTICULAR ARTICLES FROM PLASTICS OR FROM SUBSTANCES IN A PLASTIC STATE
- B29D7/00—Producing flat articles, e.g. films or sheets
- B29D7/01—Films or sheets
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F220/00—Copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and only one being terminated by only one carboxyl radical or a salt, anhydride ester, amide, imide or nitrile thereof
- C08F220/02—Monocarboxylic acids having less than ten carbon atoms; Derivatives thereof
- C08F220/10—Esters
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F283/00—Macromolecular compounds obtained by polymerising monomers on to polymers provided for in subclass C08G
- C08F283/12—Macromolecular compounds obtained by polymerising monomers on to polymers provided for in subclass C08G on to polysiloxanes
- C08F283/124—Macromolecular compounds obtained by polymerising monomers on to polymers provided for in subclass C08G on to polysiloxanes on to polysiloxanes having carbon-to-carbon double bonds
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F285/00—Macromolecular compounds obtained by polymerising monomers on to preformed graft polymers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G77/00—Macromolecular compounds obtained by reactions forming a linkage containing silicon with or without sulfur, nitrogen, oxygen or carbon in the main chain of the macromolecule
- C08G77/42—Block-or graft-polymers containing polysiloxane sequences
- C08G77/445—Block-or graft-polymers containing polysiloxane sequences containing polyester sequences
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K3/00—Use of inorganic substances as compounding ingredients
- C08K3/01—Use of inorganic substances as compounding ingredients characterized by their specific function
- C08K3/016—Flame-proofing or flame-retarding additives
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L27/00—Compositions of homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by a halogen; Compositions of derivatives of such polymers
- C08L27/02—Compositions of homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by a halogen; Compositions of derivatives of such polymers not modified by chemical after-treatment
- C08L27/12—Compositions of homopolymers or copolymers of compounds having one or more unsaturated aliphatic radicals, each having only one carbon-to-carbon double bond, and at least one being terminated by a halogen; Compositions of derivatives of such polymers not modified by chemical after-treatment containing fluorine atoms
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L51/00—Compositions of graft polymers in which the grafted component is obtained by reactions only involving carbon-to-carbon unsaturated bonds; Compositions of derivatives of such polymers
- C08L51/08—Compositions of graft polymers in which the grafted component is obtained by reactions only involving carbon-to-carbon unsaturated bonds; Compositions of derivatives of such polymers grafted on to macromolecular compounds obtained otherwise than by reactions only involving unsaturated carbon-to-carbon bonds
- C08L51/085—Compositions of graft polymers in which the grafted component is obtained by reactions only involving carbon-to-carbon unsaturated bonds; Compositions of derivatives of such polymers grafted on to macromolecular compounds obtained otherwise than by reactions only involving unsaturated carbon-to-carbon bonds on to polysiloxanes
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L69/00—Compositions of polycarbonates; Compositions of derivatives of polycarbonates
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G2261/00—Macromolecular compounds obtained by reactions forming a carbon-to-carbon link in the main chain of the macromolecule
- C08G2261/50—Physical properties
- C08G2261/62—Mechanical aspects
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L2201/00—Properties
- C08L2201/02—Flame or fire retardant/resistant
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L2201/00—Properties
- C08L2201/08—Stabilised against heat, light or radiation or oxydation
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L2205/00—Polymer mixtures characterised by other features
- C08L2205/03—Polymer mixtures characterised by other features containing three or more polymers in a blend
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L2207/00—Properties characterising the ingredient of the composition
- C08L2207/04—Thermoplastic elastomer
Definitions
- the present invention relates to a polyorganosiloxane-containing graft copolymer that improves the impact resistance of a thermoplastic resin and exhibits high color development and flame retardancy in a molded product obtained from the thermoplastic resin.
- the present invention also relates to a powder containing a polyorganosiloxane-containing graft copolymer that suppresses generation of outgas due to thermal decomposition of a thermoplastic resin and provides a thermoplastic resin composition having both heat decomposition resistance and pigment colorability.
- Aromatic polycarbonate resin is a general-purpose engineering plastic that excels in transparency, impact resistance, heat resistance, dimensional stability, etc., and its excellent characteristics make it an automotive field, OA equipment fields such as printers, and electric / electronic fields such as mobile phones. It is widely used industrially as a material for these.
- Patent Document 1 discloses 10 to 60% by mass of a vinyl monomer containing a (meth) acrylate monomer having an aryl group or a benzyl group in the presence of 40 to 90% by mass of a polyorganosiloxane rubber. Graft copolymers obtained by polymerization have been proposed.
- Patent Document 2 discloses a composite rubber system obtained by grafting a vinyl monomer containing 80% by mass or more of an aromatic alkenyl compound as a composition to a composite rubber containing polyorganosiloxane and polyalkyl (meth) acrylate. Graft copolymers have been proposed.
- Patent Document 3 discloses a silicone / acrylic composite rubber-based graft copolymer obtained by graft-polymerizing one or more vinyl monomers to a composite rubber including a polyorganosiloxane rubber and a polyalkyl (meth) acrylate rubber.
- a silicone / acrylic composite rubber-based graft copolymer having a number average particle diameter of 300 to 2000 nm and a proportion of particles less than 300 nm in all particles being 20% by volume or less has been proposed.
- Patent Document 4 discloses a silicone / acrylic composite rubber graft copolymer obtained by graft polymerizing one or more vinyl monomers to a composite rubber containing polyorganosiloxane rubber and polyalkyl (meth) acrylate rubber.
- the polyorganosiloxane content is 15 to 70% by mass
- the composite rubber content is 75 to 90% by mass
- the amount of iron charged is 0.0001 to 2 ppm in the graft copolymer latex.
- the resulting molded product is excellent in flame retardancy but has low color developability. Further, in the method proposed in Patent Document 2, the resulting molded article is excellent in color developability, but is not satisfactory in impact resistance and flame retardancy.
- Patent Document 3 there is no description about generation of outgas at the time of high temperature molding, and there is no description about chemical resistance. Since the graft copolymer disclosed in Patent Document 4 has a small particle diameter, the pigment colorability is insufficient.
- An object of the present invention is to provide a graft copolymer capable of imparting impact resistance, flame retardancy, and color development to a thermoplastic resin (hereinafter referred to as “first problem”).
- Another object of the present invention is to provide a resin composition that suppresses generation of outgas due to thermal decomposition of a thermoplastic resin and has both heat decomposition resistance and pigment colorability (hereinafter referred to as “second problem”). .)
- the first problem is solved by the following first invention group.
- a vinyl monomer mixture containing an alkyl group or an aromatic group-containing (meth) acrylic acid ester (b 1 ) and an aromatic vinyl monomer (b 2 ) was graft-polymerized onto a polyorganosiloxane rubber.
- a polyorganosiloxane-containing graft copolymer (G1) having a volume average particle diameter of 200 to 2000 nm and a polyorganosiloxane content of 0.1 to 69% by mass .
- the resin composition containing the above four types of materials (1) to (4) is supplied to a devolatilizing extruder (PCM-30 manufactured by Ikegai Co., Ltd.) heated to a barrel temperature of 280 ° C. Kneading is performed at 150 rpm to obtain pellets.
- the pellets are supplied to a 100 t injection molding machine (SE-100DU manufactured by Sumitomo Heavy Industries, Ltd.) and molded under the conditions of a cylinder temperature of 280 ° C. and a mold temperature of 90 ° C. to obtain the following test pieces.
- SE-100DU manufactured by Sumitomo Heavy Industries, Ltd.
- the Charpy impact strength of “Test piece 1” is measured at a temperature of ⁇ 30 ° C. in accordance with JIS K 7111/1 / 1eA.
- Total light transmittance (%] Based on JIS K 7375, the total light transmittance in D65 light source of "test piece 2" is measured using Nippon Denshoku Industries Co., Ltd. HAZE Meter NDH4000.
- thermoplastic resin (A) the polyorganosiloxane-containing graft copolymer (G1) according to any one of [1] to [9], a fluororesin (C), and a flame retardant (D)
- a thermoplastic resin composition A thermoplastic resin composition.
- thermoplastic resin composition according to [10], wherein the thermoplastic resin (A) is a thermoplastic resin having at least one bond selected from a carbonate bond, an ester bond, and an amide bond.
- thermoplastic resin composition according to any one of [10] to [12], wherein the flame retardant (D) includes at least one flame retardant selected from a phosphorus flame retardant and an organometallic salt flame retardant. object.
- the flame retardant (D) includes at least one flame retardant selected from a phosphorus flame retardant and an organometallic salt flame retardant. object.
- thermoplastic resin composition obtained by molding the thermoplastic resin composition according to any one of [10] to [13].
- the second problem is solved by the following second invention group.
- the sp value calculated by the Fedors method of the graft portion of the polyorganosiloxane-containing graft copolymer (G2) is 20.15 to 21.00, The described powder.
- a powder in which the body is a monomer mixture containing an aromatic vinyl monomer, the powder has an alkali metal content of 0 to 20 ppm and an alkaline earth metal content of 0 to 150 ppm.
- the above five types of materials (1) to (5) were blended and supplied to a devolatilizing extruder (PCM-30 manufactured by Ikegai Co., Ltd.) heated to a barrel temperature of 280 ° C.
- the resin composition pellets are obtained by kneading.
- a 100t injection molding machine SE-100DU manufactured by Sumitomo Heavy Industries, Ltd.
- a mold having a hot runner test piece size: length 100 mm ⁇ width 100 mm ⁇ thickness 2 mm, pin gate
- cylinder temperature 310 ° C. runner temperature
- the pellets are molded under conditions of 310 ° C. and a mold temperature of 90 ° C. to produce a first shot injection molded body.
- the resin composition is allowed to stay in the injection molding machine for 6 minutes and then one-shot molding is performed to obtain “test piece 11”.
- ⁇ Evaluation condition 12> The number of silver streaks generated near the gate of “Test piece 11” is visually confirmed.
- ⁇ Evaluation condition 13> The resin composition pellets were molded with a 100-ton injection molding machine (SE-100DU manufactured by Sumitomo Heavy Industries, Ltd.) under the conditions of a cylinder temperature of 310 ° C. and a mold temperature of 90 ° C. to obtain “test piece 13” (length 100 mm). , Width 50 mm, thickness 2 mm).
- JIS Z 8729 display method of object color by L * a * b * color system
- measurement is performed using a spectroscopic color difference meter SE-2000 manufactured by Nippon Denshoku Industries Co., Ltd. according to JIS Z8722 as follows. Then, the object color of the “test piece 13” is measured.
- Apparatus Spectroscopic color difference meter SE-2000 (manufactured by Nippon Denshoku Industries Co., Ltd., 0-45 ° spectroscopic system) Measurement range: 380 to 780 nm, Measurement light source: C light (2 ° field of view), The L * value is calculated from the tristimulus values (XYZ) using the CIE color difference formula.
- thermoplastic resin is an aromatic polycarbonate resin.
- thermoplastic resin By blending the graft copolymer of the first invention group with a thermoplastic resin, it is possible to obtain a resin composition having high impact resistance without significantly impairing the color developability and flame retardancy of the thermoplastic resin. it can.
- thermoplastic resin When the powder of the second invention group is blended in a thermoplastic resin, generation of outgas due to thermal decomposition of the thermoplastic resin can be suppressed, and a resin composition excellent in heat decomposition resistance can be obtained. Further, a resin composition having pigment colorability can be obtained.
- (meth) acrylate means at least one of “acrylate” and “methacrylate”.
- the “(co) polymer” means at least one of “polymer” and “copolymer”.
- the polyorganosiloxane-containing graft copolymer (G1) of the first invention group (hereinafter sometimes abbreviated as “graft copolymer (G1)” or “copolymer (G1)”) is an alkyl group or This is a copolymer obtained by graft polymerization of a vinyl monomer mixture containing an aromatic group-containing (meth) acrylic acid ester (b 1 ) and an aromatic vinyl monomer (b 2 ) onto a polyorganosiloxane rubber.
- the polyorganosiloxane-containing graft copolymer (G2) of the second invention group (hereinafter sometimes abbreviated as “graft copolymer (G2)” or “copolymer (G2)”) is a polyorganosiloxane. It is a copolymer obtained by graft-polymerizing one or more vinyl monomers to a rubber.
- the powder of the polyorganosiloxane-containing graft copolymer (G2) of the second invention group is a powder of the graft copolymer (G2).
- This powder is a powder recovered from the latex of the graft copolymer by a method such as spray drying or coagulation.
- the polyorganosiloxane rubber is preferably a polyorganosiloxane rubber or a composite rubber containing polyorganosiloxane and polyalkyl (meth) acrylate.
- a polyorganosiloxane is a polymer containing an organosiloxane unit as a constituent unit.
- the polyorganosiloxane rubber can be obtained by polymerizing organosiloxane or an organosiloxane mixture containing components used as necessary. Examples of components used as necessary include a siloxane-based crosslinking agent, a siloxane-based graft crossing agent, and a siloxane oligomer having a terminal blocking group.
- the organosiloxane either a chain organosiloxane or a cyclic organosiloxane can be used. Cyclic organosiloxane is preferred because it has high polymerization stability and a high polymerization rate.
- the cyclic organosiloxane is preferably a 3- to 7-membered ring, and examples thereof include the following.
- siloxane crosslinking agent those having a siloxy group are preferable.
- siloxane-based crosslinking agent By using a siloxane-based crosslinking agent, a polyorganosiloxane having a crosslinked structure can be obtained.
- the siloxane crosslinking agent include trifunctional or tetrafunctional crosslinking agents such as trimethoxymethylsilane, triethoxyphenylsilane, tetramethoxysilane, tetraethoxysilane, tetra-n-propoxysilane, and tetrabutoxysilane. Can be mentioned. Among these, a tetrafunctional crosslinking agent is preferable, and tetraethoxysilane is more preferable.
- the content of the siloxane crosslinking agent is preferably 0.1 to 30% by mass in 100% by mass of the organosiloxane mixture.
- the siloxane-based graft crossing agent has a siloxy group and a functional group copolymerizable with a vinyl monomer.
- a siloxane-based graft crossing agent By using a siloxane-based graft crossing agent, a polyorganosiloxane having a functional group copolymerizable with a vinyl monomer can be obtained.
- an alkyl (meth) acrylate component for composite rubber described later or a vinyl monomer can be grafted to the polyorganosiloxane by radical polymerization.
- siloxane-based graft crossing agent examples include siloxane represented by the formula (1).
- R 1 represents a methyl group, an ethyl group, a propyl group, or a phenyl group.
- R 2 represents an organic group in the alkoxyl group, and examples thereof include a methyl group, an ethyl group, a propyl group, and a phenyl group.
- n represents 0, 1 or 2.
- R represents any group represented by formulas (2) to (5).
- R 3 and R 4 each represent hydrogen or a methyl group
- p represents an integer of 1 to 6.
- Examples of the functional group represented by the formula (2) include a methacryloyloxyalkyl group.
- Examples of the siloxane having this group include the following. ⁇ -methacryloyloxyethyldimethoxymethylsilane, ⁇ -methacryloyloxypropylmethoxydimethylsilane, ⁇ -methacryloyloxypropyldimethoxymethylsilane, ⁇ -methacryloyloxypropyltrimethoxysilane, ⁇ -methacryloyloxypropylethoxydiethylsilane, ⁇ -methacryloyloxypropyl Diethoxymethylsilane, ⁇ -methacryloyloxybutyldiethoxymethylsilane, etc.
- Examples of the functional group represented by the formula (3) include a vinylphenyl group.
- Examples of the siloxane having this group include vinylphenylethyldimethoxysilane.
- siloxane having a functional group represented by the formula (4) examples include vinyltrimethoxysilane and vinyltriethoxysilane.
- Examples of the functional group represented by the formula (5) include a mercaptoalkyl group.
- Examples of the siloxane having this group include the following. ⁇ -mercaptopropyldimethoxymethylsilane, ⁇ -mercaptopropylmethoxydimethylsilane, ⁇ -mercaptopropyldiethoxymethylsilane, ⁇ -mercaptopropylethoxydimethylsilane, ⁇ -mercaptopropyltrimethoxysilane and the like.
- siloxane-based graft crossing agents may be used alone or in combination of two or more.
- the content of the siloxane grafting agent is preferably 0.05 to 20% by mass in 100% by mass of the organosiloxane mixture.
- siloxane oligomer having a terminal blocking group refers to a siloxane oligomer having an alkyl group or the like at the end of an organosiloxane oligomer and stopping the polymerization of polyorganosiloxane.
- siloxane oligomer having a terminal blocking group examples include hexamethyldisiloxane, 1,3-bis (3-glycidoxypropyl) tetramethyldisiloxane, and 1,3-bis (3-aminopropyl) tetramethyldisiloxane. And methoxytrimethylsilane.
- an organosiloxane or the organosiloxane mixture is emulsified with an emulsifier and water to prepare an emulsion, and then polymerized at a high temperature using an acid catalyst. The acid is then neutralized with an alkaline substance to obtain a polyorganosiloxane rubber latex.
- an “organosiloxane mixture” is used as a raw material for rubber will be described, but the same production process can be applied to the case where “organosiloxane” is used.
- emulsion preparation methods include a method using a homomixer that makes fine particles by shearing force by high-speed rotation, a method that mixes by high-speed agitation using a homogenizer that makes fine particles by jet output from a high-pressure generator, etc. Is mentioned.
- a method using a homogenizer is a preferable method because the particle size distribution of the polyorganosiloxane rubber latex becomes narrow.
- a method for mixing the acid catalyst during the polymerization (1) a method of adding and mixing together with the organosiloxane mixture, the emulsifier and water, and (2) a solution of the acid catalyst aqueous solution in the emulsion of the organosiloxane mixture all at once.
- examples thereof include (3) a method in which an emulsion of an organosiloxane mixture is dropped into a high-temperature acid aqueous solution at a constant rate and mixed. Since it is easy to control the particle diameter of the polyorganosiloxane, a method of keeping the emulsion of the organosiloxane mixture at a high temperature and then adding the acid catalyst aqueous solution all at once is preferable.
- the polymerization temperature is preferably 50 ° C. or higher, and more preferably 70 ° C. or higher.
- the polymerization time is usually 2 hours or longer, preferably 5 hours or longer when the acid catalyst aqueous solution is added all at once to the emulsion of the organosiloxane mixture for polymerization.
- the resulting latex is polymerized at a high temperature of 50 ° C. or higher. It can also be held at the following temperature for about 5 to 100 hours.
- the polymerization reaction of the organosiloxane mixture can be terminated by neutralizing the latex to pH 6 to 8 with an alkaline substance such as sodium hydroxide, potassium hydroxide, or an aqueous ammonia solution.
- an alkaline substance such as sodium hydroxide, potassium hydroxide, or an aqueous ammonia solution.
- the emulsifier used in the above production method is not particularly limited as long as the organosiloxane mixture can be emulsified, but an anionic emulsifier or a nonionic emulsifier is preferable.
- the anionic emulsifier include sodium alkylbenzene sulfonate, sodium alkyldiphenyl ether disulfonate, sodium alkyl sulfate, sodium polyoxyethylene alkyl sulfate, and sodium polyoxyethylene nonyl phenyl ether sulfate.
- nonionic emulsifiers include the following. Polyoxyethylene alkyl ether, polyoxyethylene alkylene alkyl ether, polyoxyethylene distyrenated phenyl ether, polyoxyethylene tribenzyl phenyl ether, polyoxyethylene polyoxypropylene glycol and the like.
- emulsifiers may be used alone or in combination of two or more.
- the amount of the emulsifier used is preferably 0.05 to 10 parts by mass, more preferably 0.1 to 5 parts by mass with respect to 100 parts by mass of the organosiloxane mixture.
- the particle diameter of the polyorganosiloxane rubber latex can be adjusted to a desired value. If the usage-amount of an emulsifier is 0.05 mass part or more, the emulsification stability of the emulsion of an organosiloxane emulsion mixture will become enough.
- the amount of the emulsifier used is 10 parts by mass or less, coloring of the powder containing the graft copolymer due to the emulsifier can be suppressed, and the thermal decomposition resistance of the resin composition containing the graft copolymer and the resin is reduced. Can be suppressed.
- Examples of the acid catalyst used for polymerization of the organosiloxane mixture include sulfonic acids such as aliphatic sulfonic acid, aliphatic substituted benzenesulfonic acid, and aliphatic substituted naphthalenesulfonic acid, and mineral acids such as sulfuric acid, hydrochloric acid, and nitric acid. These acid catalysts may be used individually by 1 type, and may use 2 or more types together. Among these, the use of mineral acids such as sulfuric acid, hydrochloric acid, and nitric acid can narrow the particle size distribution of the polyorganosiloxane rubber latex, and further, molding caused by the emulsifier component in the polyorganosiloxane rubber latex. The appearance defect of the product can be reduced.
- sulfonic acids such as aliphatic sulfonic acid, aliphatic substituted benzenesulfonic acid, and aliphatic substituted naphthalenesul
- the mass average particle diameter of the polyorganosiloxane rubber latex for the first invention group is preferably 150 nm to 1000 nm.
- the volume average particle size of the graft copolymer (G1) obtained from the polyorganosiloxane rubber can be adjusted to 200 to 2000 nm.
- the mass average particle diameter of the latex of the polyorganosiloxane rubber for the second invention group is preferably 250 nm to 1000 nm.
- the particle size measured by the absorbance method of the graft copolymer obtained from the polyorganosiloxane rubber can be adjusted to 300 to 2000 nm.
- the “mass average particle diameter Dw / number average particle diameter Dn” of the latex of the polyorganosiloxane rubber is preferably 1.0 to 1.7.
- Dw / Dn By setting Dw / Dn to 1.0 to 1.7, a graft copolymer having high pigment coloring property can be obtained.
- the particle size is measured using a CHDF2000 type particle size distribution meter manufactured by MATEC USA.
- Cartridge dedicated capillary cartridge for particle separation (trade name; C-202), Carrier liquid: dedicated carrier liquid (trade name: 2XGR500), Carrier liquid: almost neutral, Carrier liquid flow rate: 1.4 ml / min, Carrier liquid pressure: about 4,000 psi (2,600 kPa), Measurement temperature: 35 ° C Sample usage: 0.1 ml.
- the standard particle size substance 12 types of particles having a particle size in the range of 40 to 800 nm and monodispersed polystyrene with a known particle size manufactured by DUKE, USA are used.
- an emulsifier may be added as necessary for the purpose of improving mechanical stability.
- the emulsifier the same anionic emulsifier and nonionic emulsifier as those exemplified above are preferable.
- composite rubber a composite rubber containing polyorganosiloxane and polyalkyl (meth) acrylate (hereinafter abbreviated as “composite rubber”) can be used as the polyorganosiloxane rubber.
- the composite rubber is a rubber containing the polyorganosiloxane and a polyalkyl (meth) acrylate having a homopolymer having a glass transition temperature Tg of 0 ° C. or less.
- the composite rubber is preferably a rubber obtained by polymerizing an alkyl (meth) acrylate in the presence of a polyorganosiloxane rubber.
- the polyalkyl (meth) acrylate (PA) constituting the composite rubber can be obtained by polymerizing an alkyl (meth) acrylate component (hereinafter abbreviated as "(meth) acrylate component for composite rubber").
- the (meth) acrylate component for composite rubber contains an alkyl (meth) acrylate having a glass transition temperature Tg represented by the following FOX formula of 0 ° C. or lower and ⁇ 100 ° C. or higher and a crosslinkable monomer. Is preferred.
- Tg Glass transition temperature (° C.) of the copolymer
- wi mass fraction of monomer i
- Tgi Glass transition temperature (° C.) of a homopolymer obtained by polymerizing monomer i.
- the value described in POLYMER HANDBOOK Volume 1 is used as the value of Tgi.
- Examples of the (meth) acrylate component for composite rubber include the following. Methyl (meth) acrylate, ethyl (meth) acrylate, n-propyl (meth) acrylate, i-propyl (meth) acrylate, n-butyl (meth) acrylate, i-butyl (meth) acrylate, t-butyl (meth) Acrylate, 2-ethylhexyl (meth) acrylate. These may be used alone or in combination of two or more.
- an alkyl (meth) acrylate having a glass transition temperature of a homopolymer of 0 ° C. or lower and ⁇ 100 ° C. or higher is used. It is preferable to use 50% by mass or more, and more preferably 80% by mass or more. However, this mass% is a value based on 100 mass% of the total amount of the (meth) acrylate component for composite rubber used for polymerization.
- alkyl (meth) acrylate having a homopolymer glass transition temperature of 0 ° C. or lower examples include ethyl acrylate, n-propyl acrylate, n-butyl acrylate, i-butyl acrylate, and 2-ethylhexyl acrylate. These may be used alone or in combination of two or more. Among these, n-butyl acrylate is particularly preferable in consideration of the impact resistance of the thermoplastic resin composition and the gloss of the molded product.
- crosslinkable monomer examples include the following polyfunctional monomers. Allyl methacrylate, triallyl cyanurate, triallyl isocyanurate, divinylbenzene, ethylene glycol diester dimethacrylate, propylene glycol diester dimethacrylate, 1,3-butylene glycol diester dimethacrylate, 1,4-butylene glycol diester dimethacrylate 1,6-hexanediol diacrylic acid, triallyl trimellitic acid and the like. These may be used alone or in combination of two or more.
- the use amount of the crosslinkable monomer in 100% by mass of the polyalkyl (meth) acrylate component is preferably 0.1 to 20.0% by mass, More preferably, the content is 0.3 to 10.0% by mass.
- the crosslinkable monomer in an amount of 0.1 to 20.0% by mass a molded article having excellent impact strength can be obtained.
- the use amount of the crosslinkable monomer in 100% by mass of the polyalkyl (meth) acrylate component is preferably 0.1 to 2.0% by mass, More preferably, it is 0.3 to 1.8% by mass.
- the content of polyorganosiloxane in 100% by mass of the composite rubber is preferably 5 to 65% by mass, and more preferably 10 to 40% by mass. If the content of polyorganosiloxane is 5% by mass or more, a resin composition having excellent impact strength and chemical resistance at low temperatures can be obtained. Moreover, if it is 65 mass% or less, the resin composition excellent in pigment coloring property can be obtained.
- examples of the method of adding the (meth) acrylate component for composite rubber to the latex of the polyorganosiloxane rubber include a method of adding the whole amount at once, a method of adding dropwise at a constant rate. It is done.
- an emulsifier When producing a composite rubber latex, an emulsifier can be added to stabilize the latex and control the particle diameter of the composite rubber.
- the emulsifier is not particularly limited, and an anionic emulsifier and a nonionic emulsifier are preferable.
- anionic emulsifiers include the following. Sodium alkylbenzene sulfonate, sodium alkyl diphenyl ether disulfonate, sodium alkyl sulfate, polyoxyethylene alkyl sodium sulfate, polyoxyethylene nonyl phenyl ether sodium sulfate, sodium sarcosine, fatty acid potassium, fatty acid sodium, alkenyl succinate dipotassium, rosin acid soap, Polyoxyethylene alkyl sodium phosphate, polyoxyethylene alkyl calcium phosphate, etc.
- nonionic emulsifiers examples include polyoxyethylene alkyl ether, polyoxyethylene distyrenated phenyl ether, and polyoxyethylene tribenzyl phenyl ether. These emulsifiers may be used individually by 1 type, and may use 2 or more types together.
- radical polymerization initiator used for the polymerization of the (meth) acrylate component for composite rubber examples include azo initiators, peroxides, and redox initiators in which peroxides and reducing agents are combined. These may be used alone or in combination of two or more. In these, an azo initiator and a redox initiator are preferable from the viewpoint of suppressing outgassing of a resin composition (particularly an aromatic polycarbonate resin composition).
- azo initiator examples include the following. Oil-soluble azo initiators such as 2,2′-azobisisobutyronitrile and dimethyl 2,2′-azobis (2-methylpropionate); 4,4′-azobis (4-cyanovaleric acid) 2,2′-azobis [N- (2-carboxymethyl) -2-methylpropionamidine] hydrate, 2,2′-azobis- (N, N′-dimethyleneisobutylamidine) dihydrochloride, , 2′-azobis [2- (2-imidazolin-2-yl) propane] dihydrochloride and other water-soluble azo initiators. These may be used alone or in combination of two or more.
- Oil-soluble azo initiators such as 2,2′-azobisisobutyronitrile and dimethyl 2,2′-azobis (2-methylpropionate); 4,4′-azobis (4-cyanovaleric acid) 2,2′-azobis [N- (2-carboxymethyl) -2-methylpropionamidine] hydrate, 2,2
- peroxides include the following. Inorganic peroxides such as hydrogen peroxide, potassium persulfate, ammonium persulfate, diisopropylbenzene hydroperoxide, p-menthane hydroperoxide, cumene hydroperoxide, t-butyl hydroperoxide, succinic acid peroxide, t- Butyl peroxyneodecanoate, t-butyl peroxyneoheptanoate, t-butyl peroxypivalate, 1,1,3,3-tetramethylbutylperoxy-2-ethylhexanoate, t-butyl Organic peroxides such as peroxy-2-ethylhexanoate. These peroxides may be used alone or in combination of two or more.
- Inorganic peroxides such as hydrogen peroxide, potassium persulfate, ammonium persulfate, diisopropylbenzene hydroperoxide, p-
- organic peroxides are preferably used from the viewpoint of suppressing outgassing of the aromatic polycarbonate resin composition.
- diisopropylbenzene hydroperoxide, p-menthane hydroperoxide, and cumene hydroperoxide are more preferable from the viewpoint of chemical resistance.
- a peroxide When a peroxide is combined with a reducing agent to form a redox initiator, the above peroxide, a reducing agent such as sodium formaldehyde sulfoxylate, L-ascorbic acid, fructose, dextrose, sorbose, inositol, and sulfuric acid It is preferable to use a combination of monoiron and ethylenediaminetetraacetic acid disodium salt.
- a reducing agent such as sodium formaldehyde sulfoxylate, L-ascorbic acid, fructose, dextrose, sorbose, inositol, and sulfuric acid It is preferable to use a combination of monoiron and ethylenediaminetetraacetic acid disodium salt.
- reducing agents may be used alone or in combination of two or more.
- sodium formaldehyde sulfoxylate it is preferable to suppress the usage-amount as much as possible from a viewpoint of suppressing the outgas of a resin composition.
- the amount of radical polymerization initiator used is preferably 0.01 to 1 part by mass with respect to 100 parts by mass of the composite rubber when an azo initiator is used.
- the amount of peroxide used is preferably 0.01 to 1 part by mass with respect to 100 parts by mass of the composite rubber.
- the amount of the reducing agent used is preferably 0.01 to 1 part by mass with respect to 100 parts by mass of the composite rubber.
- the composite rubber for the second invention group particularly when sodium formaldehyde sulfoxylate is used as the reducing agent, from the viewpoint of setting the alkaline earth metal content of the graft copolymer powder to 0 to 150 ppm. 0.01 to 0.2 parts by mass is particularly preferable.
- the polyorganosiloxane-containing graft copolymer (G1) of the first invention group comprises (meth) acrylic acid ester (b 1 ) having an alkyl group or an aromatic group with respect to a polyorganosiloxane rubber and an aromatic vinyl monomer.
- the graft copolymer (G1) can be obtained by graft polymerization of the vinyl monomer mixture in the presence of the polyorganosiloxane rubber.
- the (meth) acrylic acid ester (b 1 ) having an alkyl group or an aromatic group may be simply referred to as “(meth) acrylic acid ester (b 1 )”.
- the graft component contains a component derived from (meth) acrylic acid ester (b 1 )
- the graft copolymer (G1) is compatible and dispersible in the thermoplastic resin (A) such as polycarbonate resin. Excellent.
- the graft copolymer (G1) contains a component derived from the aromatic vinyl monomer (b 2 ), a resin composition comprising the graft copolymer (G1) and the thermoplastic resin (A) is difficult. Excellent flammability.
- examples of the (meth) acrylic acid ester (b 1 ) of the present invention include the following.
- Alkyl methacrylates such as methyl methacrylate, ethyl methacrylate, n-butyl methacrylate, i-butyl methacrylate; alkyl acrylates such as methyl acrylate; These monomers may be used individually by 1 type, and may use 2 or more types together.
- the (meth) acrylic acid ester having an aromatic group means a (meth) acrylic acid ester having an aromatic hydrocarbon group such as a phenyl group, and examples thereof include the following. Phenyl (meth) acrylate, benzyl (meth) acrylate, 4-t-butylphenyl (meth) acrylate, monobromophenyl (meth) acrylate, dibromophenyl (meth) acrylate, 2,4,6-tribromophenyl (meth) Acrylate, monochlorophenyl (meth) acrylate, dichlorophenyl (meth) acrylate, trichlorophenyl (meth) acrylate, naphthyl (meth) acrylate, and the like. These monomers may be used individually by 1 type, and may use 2 or more types together.
- aromatic vinyl monomer (b 2 ) of the present invention examples include the following. Styrene, ⁇ -methylstyrene, p-methylstyrene, p-t-butylstyrene, p-methoxystyrene, o-methoxystyrene, 2,4-dimethylstyrene, chlorostyrene, bromostyrene, vinyltoluene, vinylnaphthalene, vinylanthracene etc. These monomers may be used individually by 1 type, and may use 2 or more types together.
- the vinyl monomer mixture of the present invention may contain another copolymerizable monomer (b 3 ) as long as the object of the present invention is not impaired.
- the other monomer (b 3 ) include the following. Carboxyl group-containing monomers such as (meth) acrylic acid and carboxyethyl (meth) acrylate; vinyl cyanide monomers such as (meth) acrylonitrile; vinyl ether monomers such as vinyl methyl ether and vinyl ethyl ether; benzoic acid Vinyl carboxylate monomers such as vinyl, vinyl acetate and vinyl butyrate; (meth) acrylates having reactive functional groups such as glycidyl (meth) acrylate, allyl (meth) acrylate and 1,3-butylene dimethacrylate; ethylene, Olefins such as propylene and butylene. These monomers may be used individually by 1 type, and may use 2 or more types together.
- the amount of the aromatic vinyl monomer (b 2 ) used in 100% by mass of the vinyl monomer mixture is preferably 2 to 95% by mass, more preferably 2 to 71% by mass, and further preferably 10 to 65% by mass.
- the amount of (meth) acrylic acid ester (b 1 ) used in 100% by mass of the vinyl monomer mixture is preferably 5 to 98%, more preferably 29 to 98% by mass, and even more preferably 35 to 90% by mass. .
- the amount of the other monomer (b 3 ) used in 100% by mass of the vinyl monomer mixture is preferably 5% by mass or less.
- the amount of the aromatic vinyl monomer (b 2 ) used is 2% by mass or more, the flame retardancy of the molded article is good, and when it is 95% by mass or less, the graft copolymer weight in the thermoplastic resin (A)
- the compatibility and dispersibility of the combined body (G1) are improved. If the compatibility and dispersibility are poor, the graft copolymer (G1) may be observed as a foreign substance in the molded body, or the flame retardancy of the molded body may be deteriorated.
- the amount of (meth) acrylic acid ester (b 1 ) used in the vinyl monomer mixture is 5% by mass or more, the compatibility and dispersibility of the graft copolymer (G1) in the thermoplastic resin (A) When it is 98% by mass or less, the flame retardancy of the molded article is good.
- the content of the component derived from the aromatic vinyl monomer (b 2 ) in 100% by mass of the graft copolymer (G1) is preferably 0.1 to 20% by mass, more preferably 1 to 15% by mass, More preferably, it is 3 to 10% by mass.
- the content of “polyorganosiloxane” in 100% by mass of the graft copolymer (G1) is 0.1 to 69% by mass. This amount is preferably 5 to 60% by mass, more preferably 10 to 40% by mass, further preferably 15 to 40% by mass, and particularly preferably 25 to 40% by mass. .
- the content of the polyorganosiloxane is 0.1% by mass or more, the impact strength and flame retardancy at low temperatures of the molded product are good, and when it is 69% by mass or less, the color developability of the molded product is good.
- the content of “polyorganosiloxane rubber” in 100% by mass of the graft copolymer (G1) is preferably 10 to 99% by mass. If the content of the polyorganosiloxane rubber is 10% by mass or more, the impact strength of the molded article at a low temperature is sufficient, and if it is 99% by mass or less, the surface appearance of the resin composition is preferable. Furthermore, from the viewpoint of improving the chemical resistance of the resin composition, the content is more preferably 60 to 97% by mass, further preferably 75 to 95% by mass, and particularly preferably 80 to 95% by mass.
- the volume average particle diameter of the graft copolymer (G1) is 200 to 2000 nm.
- the particle size is preferably 250 to 1000 nm, more preferably 300 to 800 nm, still more preferably 350 to 750 nm, particularly preferably 400 to 700 nm, and 480 to 700 nm. Most preferably it is.
- the volume average particle diameter of the graft copolymer (G1) is 200 nm or more, the molded product obtained by blending the graft copolymer (G1) with the thermoplastic resin has impact resistance (particularly low temperature impact resistance). And flame retardancy is improved.
- the volume average particle diameter of the graft copolymer (G1) is 2000 nm or less
- a molded product obtained by blending the graft copolymer (G1) with a thermoplastic resin has color development and impact resistance (especially low temperature). Impact resistance is improved and the surface appearance is improved.
- the volume average particle diameter of the graft copolymer (G1) a value measured by the following method can be adopted.
- the latex of the graft copolymer is diluted with distilled water, and the median diameter in volume average is determined using a laser diffraction scattering type particle size distribution analyzer (SALD-7100, manufactured by Shimadzu Corporation).
- SALD-7100 laser diffraction scattering type particle size distribution analyzer
- the sample concentration of latex is appropriately adjusted so as to be within an appropriate range in the scattered light intensity monitor attached to the apparatus.
- As the standard particle size substance 12 types of particles having monodisperse polystyrene having a known particle size and having a particle size in the range of 20 to 800 nm are used.
- the graft copolymer (G2) of the second invention group is a copolymer obtained by graft polymerization of one or more vinyl monomers to a polyorganosiloxane rubber.
- the glass transition temperature Tg represented by the formula of FOX is preferably higher than 0 ° C, more preferably 50 ° C or higher, and 80 ° C or higher. Further preferred.
- vinyl monomer examples include the following. Aromatic vinyl monomers such as styrene, ⁇ -methylstyrene, vinyltoluene; (meth) acrylic acid such as methyl (meth) acrylate, ethyl (meth) acrylate, butyl (meth) acrylate, 2-ethylhexyl (meth) acrylate Esters; vinyl cyanide monomers such as acrylonitrile and methacrylonitrile. These may be used individually by 1 type and may use 2 or more types together.
- Aromatic vinyl monomers such as styrene, ⁇ -methylstyrene, vinyltoluene
- (meth) acrylic acid such as methyl (meth) acrylate, ethyl (meth) acrylate, butyl (meth) acrylate, 2-ethylhexyl (meth) acrylate Esters
- vinyl cyanide monomers such as acrylonitrile and methacrylonitrile
- the glass transition temperature Tg of the graft part In order for the glass transition temperature Tg of the graft part to exceed 0 ° C., it is preferable to use 50% by mass or more, more preferably 80% by mass or more of a vinyl monomer having a homopolymer Tg exceeding 0 ° C. preferable.
- this mass% is a value based on 100 mass% of the total amount of vinyl monomers to be subjected to graft polymerization.
- Examples of the vinyl monomer having a Tg of the homopolymer exceeding 0 ° C. include the following. Alkyl methacrylates such as methyl methacrylate, ethyl methacrylate, n-butyl methacrylate and i-butyl methacrylate; alkyl acrylates such as methyl acrylate; These may be used individually by 1 type and may use 2 or more types together.
- an alkyl acrylate having a Tg of 0 ° C. or less such as ethyl acrylate and n-butyl acrylate may be used.
- the content of the alkyl acrylate having a Tg of 0 ° C. or less in the homopolymer is preferably 0.05 to 20% by mass in 100% by mass of the vinyl monomer.
- the content of the polyorganosiloxane rubber in 100% by mass of the graft copolymer (G2) is preferably 10 to 99% by mass.
- the content of the polyorganosiloxane rubber is 10% by mass or more, the impact strength at low temperatures of the molded article is good. Moreover, if it is 99 mass% or less, the surface external appearance of a molded object will become favorable.
- the content of the polyorganosiloxane rubber is more preferably 60 to 97% by mass, and particularly preferably 75 to 95% by mass.
- the vinyl monomer mixture for graft polymerization preferably contains a “crosslinkable monomer” from the viewpoint of improving the chemical resistance and thermal decomposition resistance of the resin composition. .
- crosslinkable monomer examples include the following polyfunctional monomers. Allyl methacrylate, triallyl cyanurate, triallyl isocyanurate, divinylbenzene, ethylene glycol dimethacrylate, propylene glycol dimethacrylate, 1,3-butylene glycol dimethacrylate, 1,4-butylene glycol dimethacrylate, 1,6-hexanediol Diacrylate, triallyl trimellitate, etc. These may be used individually by 1 type and may use 2 or more types together.
- the content of the component derived from the crosslinkable monomer in the graft copolymer is preferably 0.5 to 2.0% by mass in 100% by mass of the graft copolymer, 0.5 to 1 More preferably, it is 0.0 mass%.
- the content of this component is 0.5% by mass or more, the chemical resistance and thermal decomposition resistance of the molded body are good, and when it is 2.0% by mass or less, the impact strength of the molded body is good.
- the content of the crosslinkable monomer in the monomer mixture for graft polymerization is preferably 0.5 to 10% by mass in 100% by mass of the monomer mixture for graft polymerization. More preferably, it is 5 to 5% by mass. If the content of the crosslinkable monomer is 0.5% by mass or more, the chemical resistance and thermal decomposition resistance of the molded product are good, and if it is 5% by mass or less, the impact strength of the molded product is good. .
- the sp value calculated by the Fedors method of the graft part is preferably 20.15 to 21.00. If the sp value is 20.15 or more, the chemical resistance of the molded article is good, and if it is 21.00 or less, the impact strength of the molded article is good.
- the sp value of the graft part is calculated based on the sp value calculated by the Ferors method of each vinyl monomer described in POLYMER HANDBOOK Volume 1 (WILEY-INTERSCIENCE).
- a vinyl monomer mixture consists of 3 types of vinyl monomers, it calculates using the following formula
- the sp value of the graft part can be adjusted by the composition of the vinyl monomer used. For example, methyl methacrylate (sp value: 20.15), methacrylic acid (sp value: 23.43), 2-hydroxyethyl methacrylate (sp value: 24.98), phenyl methacrylate (sp value: 20.95), etc. By using a small amount, the sp value of the graft part can be adjusted to 20.15 to 21.00.
- the vinyl monomer mixture for grafting contains an “aromatic vinyl monomer” from the viewpoint of improving the thermal decomposition resistance of the resin composition.
- aromatic vinyl monomer examples include styrene, ⁇ -methylstyrene, vinyltoluene and the like. These may be used individually by 1 type and may use 2 or more types together.
- the content of the aromatic vinyl monomer is preferably 10 to 90% by mass, more preferably 30 to 80% by mass, and further preferably 40 to 70% by mass in 100% by mass of the monomer mixture for graft polymerization. . If the content of the aromatic vinyl monomer is 10% by mass or more, the thermal decomposition resistance of the resin composition is good, and if it is 90% by mass or less, the impact resistance of the molded article is good, which is preferable.
- the particle size of the graft copolymer (G2) measured by the absorbance method is preferably 300 to 2000 nm, preferably 300 to 800 nm, and more preferably 350 to 650 nm.
- This particle size is a particle size calculated from the absorbance of latex. The measuring method will be described later.
- the particle diameter is smaller than 300 nm, the pigment colorability and chemical resistance of the resin composition are lowered, which is not preferable.
- the external appearance of a resin composition will deteriorate when a particle diameter exceeds 2000 nm, it is unpreferable.
- Examples of the polymerization method for the graft portion include a method in which a vinyl monomer for graft polymerization is added to a latex of a polyorganosiloxane rubber and the polymerization is performed in one or more stages. When the polymerization is performed in multiple stages, it is preferable to polymerize by adding the vinyl monomer for graft polymerization in a divided manner into the latex of the polyorganosiloxane rubber-based rubber and adding sequentially or continuously. Such a polymerization method has good polymerization stability and can stably obtain a latex of a graft copolymer having a desired particle size and particle size distribution.
- an emulsifier can be added as necessary.
- the emulsifier include the same emulsifiers as those used in the production of the composite rubber, and anionic emulsifiers and nonionic emulsifiers are preferable.
- Examples of the polymerization initiator used for the polymerization of the graft part include the same polymerization initiators used for producing the composite rubber, and azo initiators and redox initiators are preferable.
- a spray drying method or a coagulation method can be used.
- the spray drying method is a method in which the latex of the graft copolymer is sprayed in the form of fine droplets in a dryer and dried by applying a heating gas for drying.
- Examples of the method for generating fine droplets include a rotating disk type, a pressure nozzle type, a two-fluid nozzle type, and a pressurized two-fluid nozzle type.
- the capacity of the dryer may be a small capacity as used in a laboratory or a large capacity as used industrially.
- the temperature of the heating gas for drying is preferably 200 ° C. or less, more preferably 120 to 180 ° C. Two or more types of graft copolymer latices produced separately may be spray dried together.
- an optional component such as silica can be added to the latex of the graft copolymer and spray dried.
- the coagulation method is a method of coagulating the latex of the graft copolymer, separating the graft copolymer, collecting it, and drying it.
- a graft copolymer latex is introduced into hot water in which a coagulant is dissolved, salted out, and solidified to separate the graft copolymer.
- the separated wet graft copolymer is dehydrated to recover the graft copolymer having a reduced water content.
- the recovered graft copolymer is dried using a press dehydrator or a hot air dryer.
- the coagulant examples include inorganic salts such as aluminum chloride, aluminum sulfate, sodium sulfate, magnesium sulfate, sodium nitrate, and calcium acetate, and acids such as sulfuric acid, and calcium acetate is particularly preferable. These coagulants may be used alone or in combination of two or more. When used in combination, it is necessary to select a combination that does not form an insoluble salt in water. For example, the combined use of calcium acetate and sulfuric acid or a sodium salt thereof is not preferable because a calcium salt insoluble in water is formed.
- the above-mentioned coagulant is usually used as an aqueous solution.
- the concentration of the aqueous solution of the coagulant is preferably 0.1% by mass or more, particularly 1% by mass or more from the viewpoint of stably coagulating and recovering the graft copolymer.
- the concentration of the coagulant aqueous solution is 20% by mass or less, particularly 15% by mass or less. It is preferable.
- the quantity of the coagulant aqueous solution with respect to latex is not specifically limited, It is preferable that it is 10 to 500 mass parts with respect to 100 mass parts of latex.
- the method of bringing the latex into contact with the coagulant aqueous solution is not particularly limited, but the following methods are usually mentioned.
- (1) A method in which a latex is continuously added to the aqueous solution of the coagulant while stirring and the solution is maintained for a certain period of time. The mixture containing the coagulated polymer and water is continuously withdrawn from the container.
- the temperature at which the latex is brought into contact with the coagulant aqueous solution is not particularly limited, but is preferably 30 ° C. or higher and 100 ° C. or lower.
- the contact time is not particularly limited.
- the agglomerated graft copolymer is washed with about 1 to 100 times by weight of water, and the wet graft copolymer separated by filtration is dried using a fluidized dryer or a pressure dehydrator.
- the drying temperature and drying time may be appropriately determined depending on the glass transition temperature of the obtained graft copolymer.
- the graft copolymer may be directly sent to an extruder or a molding machine for producing the resin composition, and mixed with a thermoplastic resin to obtain a molded body. Is possible.
- the graft copolymer is preferably recovered using a coagulation method from the viewpoint of heat decomposability when it is used as a resin composition.
- the polyorganosiloxane-containing graft copolymer (G1) of the first invention group is obtained from the “test piece” measured under the following “evaluation condition 2” for the test piece produced under the following “preparation condition 1”.
- the Charpy impact strength [kJ / m 2 ], total light transmittance [%], and flame retardancy preferably satisfy the following performance conditions (1) to (3). (1) 15 ⁇ “Charpy impact strength” ⁇ 70 (2) 60 ⁇ “Charpy impact strength” + “total light transmittance” ⁇ 1.36 (3) Flame retardancy by UL94 standard vertical combustion test at 1/16 inch thickness is “V-1” or more.
- the resin composition containing the above four types of materials (1) to (4) is supplied to a devolatilizing extruder (PCM-30 manufactured by Ikegai Co., Ltd.) heated to a barrel temperature of 280 ° C. Kneading is performed at 150 rpm to obtain pellets.
- the pellets are supplied to a 100 t injection molding machine (SE-100DU manufactured by Sumitomo Heavy Industries, Ltd.) and molded under the conditions of a cylinder temperature of 280 ° C. and a mold temperature of 90 ° C. to obtain the following test pieces.
- SE-100DU manufactured by Sumitomo Heavy Industries, Ltd.
- Total light transmittance (%] Based on JIS K7375, the total light transmittance in the D65 light source of "test piece 2" is measured using HAZE Meter NDH4000 by Nippon Denshoku Industries Co., Ltd.
- each of these test pieces satisfies the following performance conditions (11), (12) and (13). (11) 25 ⁇ “Charpy impact strength” ⁇ 70 (12) 60 ⁇ “Charpy impact strength” + “total light transmittance” ⁇ 1.36 (13) The flame retardancy by the UL94 standard vertical combustion test at 1/16 inch thickness is “V-1” or more.
- each of these test pieces satisfies the following performance conditions (21), (22) and (23).
- (21) 25 ⁇ “Charpy impact strength” ⁇ 40
- (22) 60 ⁇ “Charpy impact strength” + “total light transmittance” ⁇ 1.36 ⁇ 100
- Flame retardancy in the UL94 standard vertical combustion test at 1/16 inch thickness is “V-0” or more.
- the molded product obtained by satisfying the condition (1) has a low-temperature impact strength at a practically usable level, and by satisfying the condition (2), high color developability and low-temperature impact strength. It can be said that a molded article having an excellent balance can be obtained. Furthermore, it can be said that the molded body obtained by satisfying the condition (3) has flame retardancy that can be used practically.
- the increase in Charpy impact strength can be achieved by increasing the volume average particle diameter of the graft copolymer (G1), using a component derived from (meth) acrylic acid ester (b 1 ) as the graft component, and the like. it can.
- the increase in the total light transmittance can be achieved by setting the polyorganosiloxane content of the graft copolymer (G1) to 0.1 to 69% by mass.
- the performance of flame retardancy “V-1” or higher is to increase the volume average particle diameter of the graft copolymer (G1) and to use a component derived from the aromatic vinyl monomer (b 2 ) as the graft component. Can be achieved by etc.
- the powder of the polyorganosiloxane-containing graft copolymer (G2) of the second invention group is a powder of the graft copolymer (G2).
- This powder may contain other (co) polymers other than the graft copolymer (G2).
- the other (co) polymer is, for example, a (co) polymer that is polymerized without graft bonding to a polyorganosiloxane rubber when a vinyl monomer is graft polymerized.
- the particle diameter of the graft copolymer (G2) measured by the absorbance method is 300 to 2000 nm, and the alkali metal content of the powder is 0 to 20 ppm. And the alkaline earth metal content is 0 to 150 ppm.
- the alkali metal needs to be contained in an amount of 20 ppm or less, and is preferably 1 to 15 ppm because it decomposes the aromatic polycarbonate resin catalytically.
- alkali metals it is particularly necessary to suppress the total content of potassium and sodium. A method for measuring the alkali metal content will be described later.
- Examples of the method for setting the alkali metal content of the powder to 0 to 20 ppm include the following methods. (1) A method in which an alkali metal salt type is not used as an emulsifier, a polymerization initiator or the like during graft polymerization, or a method of suppressing the amount of use thereof. (2) A powder collected by spray recovery or coagulation recovery is water or organic. A method of washing with a solvent or the like, and (3) a method of combining these two.
- the content of alkaline earth metal in the powder of the graft copolymer (G2) is 0 to 150 ppm. If the content of the alkaline earth metal exceeds 150 ppm, the thermal decomposition resistance of the resin composition obtained from the powder and the resin is lowered, which is not preferable.
- Alkaline earth metals have a weaker action than alkali metals, but in particular, catalytically decompose aromatic polycarbonate resins. Therefore, it is necessary to suppress the content to 150 ppm or less, and 10 to 140 ppm is preferable. Among alkaline earth metals, it is particularly necessary to suppress the total content of calcium and magnesium. A method for measuring the alkaline earth metal content will be described later.
- Examples of the method for setting the alkaline earth metal content of the powder to 0 to 150 ppm include the following methods. (1) A method in which an alkaline earth metal salt type is not used as an emulsifier, a polymerization initiator or the like in the graft polymerization, or a coagulant in the coagulation recovery, or a method for suppressing the use amount thereof ( 2) A method in which the powder recovered by spraying or coagulation is washed with water or an organic solvent, and (3) A method in which both are combined.
- a redox initiator using sodium formaldehyde sulfoxylate as a polymerization initiator is used as a polymerization initiator
- the coagulation recovery is performed using an alkaline earth metal salt type coagulant
- the polymerization initiator is grafted together. It remains as an alkaline earth metal salt in the powder containing the polymer. Since this alkaline earth metal salt lowers the heat decomposability of the resin composition obtained from this powder and resin, a method of not using sodium formaldehyde sulfoxylate or suppressing the amount of use is preferred.
- the sulfur content of the powder of the graft copolymer (G2) is preferably 0 to 200 ppm.
- the sulfur content is an indicator of the amount of sulfates remaining in the powder.
- Sulfates are a general term for sulfur oxo acids such as sulfuric acid, sulfonic acid, sulfinic acid, and salts thereof.
- sulfates decompose the aromatic polycarbonate resin catalytically, so the content thereof must be suppressed to 200 ppm or less as the sulfur content, and 10 to 190 ppm is preferable.
- the sulfur content exceeds 200 ppm, the heat decomposability of the resin composition is lowered, which is not preferable. A method for measuring the sulfur content will be described later.
- Examples of the method for setting the sulfur content in the powder to 0 to 200 ppm include the following methods. (1) A method that does not use sulfates as an emulsifier, a polymerization initiator, etc. during graft polymerization, or a method that suppresses the amount used thereof. (2) A spray collected or coagulated recovered powder is washed with water or an organic solvent. (3) A method of combining both of these.
- a redox initiator using sodium formaldehyde sulfoxylate as a reducing agent it remains as sulfates in the graft copolymer powder.
- a method of not using sodium formaldehyde sulfoxylate or suppressing the amount of use thereof is preferable.
- the powder of the graft copolymer (G2) is a polyorganosiloxane obtained by graft polymerizing one or more vinyl monomers to a polyorganosiloxane rubber.
- the graft copolymer powder is a monomer mixture in which the one or more vinyl monomers contain an “aromatic vinyl monomer”, and the alkali metal content of the powder is 0 It is ⁇ 20 ppm and the alkaline earth metal content is 0 to 150 ppm. Since a monomer mixture containing an aromatic vinyl monomer is used as a vinyl monomer for graft polymerization, a resin composition having excellent thermal decomposition resistance and impact resistance can be obtained.
- the number of silver generations measured in the following “evaluation condition 12” for the resin composition used in the “preparation condition 11” is 0.
- the content of the aromatic vinyl monomer is preferably 10 to 90% by mass, more preferably 30 to 80% by mass, and further preferably 40 to 70% by mass in 100% by mass of the monomer mixture for graft polymerization. .
- the content of the component derived from the crosslinkable monomer is preferably 0.5 to 2.0% by mass in 100% by mass of the graft copolymer. It is more preferably 5 to 1.0% by mass.
- the content of the crosslinkable monomer in 100% by mass of the monomer mixture for graft polymerization is preferably 0.5 to 10% by mass, and more preferably 0.5 to 5% by mass.
- the powder is particularly preferably L * measured by the following “evaluation condition 13” of the resin composition used in the “preparation condition 11” is 20 or less.
- JIS Z 8729 display method of object color by L * a * b * color system
- measurement is performed using a spectroscopic color difference meter SE-2000 manufactured by Nippon Denshoku Industries Co., Ltd. according to JIS Z8722 as follows. Then, the object color of the “test piece 13” is measured.
- Apparatus Spectroscopic color difference meter SE-2000 (manufactured by Nippon Denshoku Industries Co., Ltd., 0-45 ° spectroscopic system) Measurement range: 380 to 780 nm, Measurement light source: C light (2 ° field of view), The L * value is calculated from the tristimulus values (XYZ) using the CIE color difference formula.
- the graft copolymer (G1) of the first invention group can be mixed with the thermoplastic resin (A) and used as a thermoplastic resin composition.
- a thermoplastic resin composition the thermoplastic resin composition containing a thermoplastic resin (A), a graft copolymer (G), a fluorine resin (C), and a flame retardant (D) is preferable.
- thermoplastic resin (A) examples include the following. Olefin resins such as polypropylene (PP) and polyethylene (PE); polystyrene (PS), high impact polystyrene (HIPS), (meth) acrylate / styrene copolymer (MS), styrene / acrylonitrile copolymer (SAN), Styrene / maleic anhydride copolymer (SMA), acrylonitrile / butadiene / styrene copolymer (ABS), acrylic ester / styrene / acrylonitrile copolymer (ASA), acrylonitrile / ethylene / propylene rubber / styrene copolymer (ASA) Styrene (St) resin such as AES); acrylic (Ac) resin such as polymethyl methacrylate (PMMA); polycarbonate resin (PC resin); polyamide (PA) resin
- the thermoplastic resin (A) is a heat having at least one bond selected from a carbonate bond, an ester bond, and an amide bond from the viewpoint of improving the impact resistance and flame retardancy of the obtained molded product.
- a plastic resin is preferred.
- the thermoplastic resin having at least one bond selected from a carbonate bond, an ester bond, and an amide bond include PC-based resins, PBT, PET, PA resins, and PLA.
- a resin called an alloy blend containing these resins may be used.
- an aromatic polycarbonate resin is particularly preferable.
- the aromatic polycarbonate-based resin is a thermoplastic aromatic polycarbonate polymer which may have a branched chain, obtained by reacting an aromatic hydroxy compound or a small amount thereof with a diester of phosgene or carbonic acid. It is a copolymer.
- the production method of the aromatic polycarbonate resin is not particularly limited, and a known method, that is, a phosgene method (interfacial polymerization method), a melting method (transesterification method) or the like is employed. Since the amount of terminal OH groups tends to affect the thermal stability, hydrolysis stability, etc., the PC-based resin is manufactured by the melting method and adjusts the degree of pressure reduction during the reaction in the present invention. Accordingly, it is possible to use an aromatic polycarbonate resin in which the terminal OH group amount is adjusted.
- PC resin examples include the following.
- the amount of the graft copolymer (G1) used relative to the thermoplastic resin (A) is preferably 0.5 to 90 parts by mass of the graft copolymer (G1) with respect to 100 parts by mass of the thermoplastic resin (A). 0.5 to 20 parts by mass is more preferable, and 1 to 7 parts by mass is even more preferable. When the amount of the graft copolymer (G1) used is 0.5 to 90 parts by mass, a resin composition having excellent impact resistance and surface appearance can be obtained.
- the fluororesin (C) can be used for the purpose of preventing dripping during combustion.
- the fluororesin (C) include polytetrafluoroethylene and modified polytetrafluoroethylene.
- the modified polytetrafluoroethylene include SAN-modified polytetrafluoroethylene and acrylic-modified polytetrafluoroethylene.
- fluororesin (C) known ones can be used, those appropriately synthesized may be used, and commercially available products may be used. As a commercial item, the following are mentioned, for example. Polytetrafluoroethylene such as “Polyflon FA-500” (trade name, manufactured by Daikin Industries, Ltd.); SAN-modified polytetrafluoroethylene such as “BLENDEX B449” (trade name, manufactured by GE Specialty Chemicals); Acrylic modified polytetrafluoroethylene such as “A-3000”, “methabrene A-3750”, “methabrene A-3800” (trade name, manufactured by Mitsubishi Rayon Co., Ltd.). These fluororesins (C) may be used alone or in combination of two or more.
- SAN-modified polytetrafluoroethylene acrylic-modified polytetratetrafluoroethylene, and acrylic-modified polytetra Fluoroethylene is preferred, and acrylic-modified polytetrafluoroethylene is more preferred.
- the content of polytetrafluoroethylene in SAN-modified polytetrafluoroethylene or acrylic-modified polytetrafluoroethylene is preferably 10 to 80% by mass in 100% by mass of the fluororesin (C), and 20 More preferably, it is ⁇ 70% by mass.
- the content of polytetrafluoroethylene in SAN-modified polytetrafluoroethylene and acrylic-modified polytetrafluoroethylene is 10% by mass or more, the resulting molded article is excellent in flame retardancy.
- the content of polytetrafluoroethylene in SAN-modified polytetrafluoroethylene and acrylic-modified polytetrafluoroethylene is 80% by mass or less, the resulting molded article is excellent in appearance.
- the blending amount of the fluororesin (C) is preferably 0.01 to 10 parts by mass, more preferably 0.1 to 5 parts by mass with respect to 100 parts by mass of the thermoplastic resin (A). More preferably, it is 0.3 to 2 parts by mass.
- the blending amount of the fluororesin (C) is 0.01 parts by mass or more, the obtained molded article is excellent in flame retardancy.
- the original property of a thermoplastic resin (A) is not impaired as the compounding quantity of a fluorine resin (C) is 10 mass parts or less.
- a flame retardant (D) As a flame retardant (D), a well-known flame retardant can be used, for example, the following are mentioned.
- Halogen flame retardants comprising a combination of halogenated compounds such as halogenated bisphenol A, halogenated polycarbonate oligomers, brominated epoxy compounds, and flame retardant aids such as antimony oxide; organic salt flame retardants; phosphate ester flame retardants, Phosphorus flame retardants such as halogenated phosphoric acid ester flame retardants; sulfonic acid flame retardants such as metal salts of aromatic sulfonic acids and metal salts of perfluoroalkane sulfonic acids; branched phenyl silicone compounds and phenyl silicone resins Silicone flame retardants such as organopolysiloxanes.
- phosphorus flame retardants such as phosphate ester flame retardants
- Organic metal salt flame retardants such as metal salts are preferred.
- phosphate ester flame retardant examples include the following. Trimethyl phosphate, triethyl phosphate, tributyl phosphate, trioctyl phosphate, tributoxyethyl phosphate, triphenyl phosphate, tricresyl phosphate, cresyl diphenyl phosphate, octyl diphenyl phosphate, isopropylphenyl diphosphate, tris (butoxyethyl) phosphate, trisisobutyl Phosphate, bis- (isopropylphenyl) diphenyl phosphate, tris- (isopropylphenyl) phosphate, 1,3 phenylene bis (diphenyl phosphate), 1,3 phenylene bis (di2,6 xylenyl phosphate), bisphenol A bis (Diphenyl phosphate), resorcinol bisdiphenyl phosphate, octyl dip
- Organic metal salt-based flame retardants are advantageous in that they exhibit a flame-retardant effect when added in a very small amount, and thus are difficult to reduce the heat resistance of the molded product, and can impart a considerable amount of antistatic properties to the molded product.
- the organometallic salt flame retardant most advantageously used in the present invention is a fluorine-containing organometallic salt compound.
- the fluorine-containing organometallic salt compound refers to a metal salt compound composed of an anion component composed of an organic acid having a fluorine-substituted hydrocarbon group and a cation component composed of a metal ion.
- metal salts of fluorine-substituted organic sulfonic acids metal salts of fluorine-substituted organic sulfates, and metal salts of fluorine-substituted organic phosphates are preferred.
- a fluorine-containing organometallic salt compound may be used individually by 1 type, and may use 2 or more types together.
- a metal salt of a fluorine-substituted organic sulfonic acid is preferable, and a metal salt of a sulfonic acid having a perfluoroalkyl group is particularly preferable.
- a metal salt of an organic sulfonic acid not containing a fluorine atom is suitable.
- the metal salt include aliphatic sulfonic acid metal salts and aromatic sulfonic acid metal salts. Of these, metal salts of aromatic sulfonic acids are preferred.
- Examples of the metal species constituting the metal ions of the organic metal salt flame retardant include alkali metals such as sodium and potassium, and alkaline earth metals such as calcium.
- Specific examples of the organic metal salt flame retardant include the following. 4-methyl-N- (4-methylphenyl) sulfonyl-benzenesulfonamide potassium salt, potassium diphenylsulfone-3-sulfonate, potassium diphenylsulfone-3-3′-disulfonate, sodium paratoluenesulfonate, perfluoro Butanesulfonic acid potassium salt and the like. These may be used alone or in combination of two or more.
- the blending amount of the flame retardant (D) is preferably 0.01 to 20 parts by mass with respect to 100 parts by mass of the thermoplastic resin (A).
- the blending amount of the flame retardant (D) is 0.01 parts by mass or more, the obtained molded article is excellent in flame retardancy.
- the original property of a thermoplastic resin (A) is not impaired as the compounding quantity of a flame retardant (D) is 20 mass parts or less.
- the optimum blending amount varies depending on the type of flame retardant (D).
- the blending amount of the phosphate ester flame retardant is more preferably 1 to 10 parts by mass, and the blending amount of the organometallic salt flame retardant is more preferably 0.01 to 2 parts by mass.
- the thermoplastic resin composition of the first invention group can contain an antioxidant (E) as necessary.
- the antioxidant (E) is a component not only for the purpose of suppressing the oxidative decomposition of the resin during the production of the molded body, but also for improving the flame retardancy of the molded body.
- Antioxidant (E) will not be specifically limited if it is used at the time of normal shaping
- the blending amount of the antioxidant (E) is preferably 0.05 to 2 parts, more preferably 0.05 to 0.8 parts with respect to 100 parts by mass of the thermoplastic resin (A).
- the blending amount of the antioxidant (E) is 0.05 parts or more, the obtained molded article is excellent in flame retardancy. Moreover, the fall of the impact resistance of the molded object obtained as the compounding quantity of antioxidant (E) is 2 parts or less can be suppressed.
- thermoplastic resin composition of the first invention group the following components can be further blended as necessary.
- thermoplastic resin composition of the first invention group is not particularly limited.
- a thermoplastic resin (A), a graft copolymer (G1) and, if necessary, a fluorine-based resin (C), a flame retardant (D), an antioxidant (E), various additives, V-type It can be prepared by mixing and dispersing with a blender, Henschel mixer or the like, and melt-kneading the mixture using a kneader such as an extruder, a Banbury mixer, a pressure kneader, or a roll. These components can be mixed batchwise or continuously, and the mixing order of the components is not particularly limited.
- the melt-kneaded product can be made into pellets and used for various moldings.
- the powder of the graft copolymer (G2) of the second invention group of the present invention can be mixed with a resin and used as a resin composition.
- the resin that can be used in the second invention group is not particularly limited, and examples thereof include one or more resins selected from “curable resin”, “thermoplastic resin”, and “thermoplastic elastomer”.
- the amount of the powder of the graft copolymer (G2) added to the resin is preferably 0.5 to 90% by weight and more preferably 0.5 to 20% by weight in a total of 100% by mass of the resin and the powder.
- the added amount of the powder is 0.5 to 90% by weight, a resin composition having excellent impact resistance and surface appearance can be obtained.
- the curable resin examples include an epoxy resin, a phenol resin, an unsaturated polyester resin, a melamine resin, and a urea resin.
- an epoxy resin is preferable because it has excellent electrical characteristics and is suitable for semiconductor encapsulation. These may be used individually by 1 type and may use 2 or more types together.
- the curable resin can be classified into a thermosetting resin and a photocurable resin, and any of them may be used.
- Examples of the epoxy resin include dicyclopentadiene type, cresol novolac type, phenol novolac type, bisphenol type, and biphenyl type. These may be used individually by 1 type and may use 2 or more types together.
- As an epoxy resin since the dispersibility of the powder of a graft copolymer (G2) becomes favorable, a solid thing is preferable.
- epoxy resin curing agents examples include phenolic curing agents such as phenol novolac resins and cresol novolac resins; amine curing agents; acid anhydride curing agents. These may be used individually by 1 type and may use 2 or more types together.
- the amount of the curing agent used is preferably a stoichiometric amount of epoxy group.
- the phenol resin examples include a resol type phenol resin and a novolac type phenol resin.
- the phenol resin may be modified with drying oil, xylene resin, melamine resin or the like.
- a phenol resin since the dispersibility of the powder containing a graft copolymer becomes favorable, a solid thing is preferable.
- the phenolic resin is a novolac type phenolic resin
- a polyamine such as hexamine, an epoxy resin, an isocyanate compound, a polyformaldehyde compound, a resol type phenolic resin or the like is used in combination as a curing agent.
- polyhydric alcohols eg ethylene glycol, dipropylene glycol,
- the unsaturated polyester resin may be a resin obtained by copolymerizing a monomer copolymerizable with the unsaturated dibasic acid.
- the monomer copolymerizable with the unsaturated dibasic acid include styrene, t-butylstyrene, divinylbenzene, diallyl phthalate, vinyl toluene, and (meth) acrylates.
- thermoplastic resin examples include the resins exemplified as the thermoplastic resin (A). Of these, the following resins or alloys are preferred. St resin, PC resin, PA resin, PET resin, PBT resin, (m-) PPE resin, POM resin, PU resin; Alloy of PC resin such as PC / ABS and St resin, PA such as PA / ABS Alloy of resin and St resin, alloy of PA resin and TPE, alloy of PA resin such as PA / PP and polyolefin resin, alloy of PC resin such as PC / PBT and PEs resin; PPE / PBT, Alloys between PPE resins such as PPE / PA. PC resin is more preferable.
- thermoplastic elastomer examples include the following. Styrene elastomer, olefin elastomer, vinyl chloride elastomer, urethane elastomer, polyester elastomer, polyamide elastomer, fluorine elastomer, 1,2-polybutadiene, trans 1,4-polyisoprene. Among these, urethane elastomers, polyester elastomers, and polyamide elastomers are preferable.
- curable resin composition examples include (1) a method of mixing each component in a solution state, and (2) each of the components. Examples thereof include a method in which components are melt-mixed using a mixing roll, a kneader, etc., cooled, and then pulverized or tableted.
- the curable resin composition can contain various additives as long as it does not depart from the object of the present invention.
- additives include the following.
- thermoplastic resin composition examples include the following methods. (1) A method of melt-mixing with an extruder, kneader, mixer, etc. after mixing the powder of the graft copolymer (G2) with a Henschel mixer, a tumbler, etc., and the powder or granules of a thermoplastic resin, (2) A method in which the remaining materials are sequentially mixed with a previously melted thermoplastic resin.
- the thermoplastic resin composition can contain various additives as long as it does not depart from the object of the present invention.
- additives include the following. Stabilizers such as phenol stabilizers, phosphorus stabilizers, UV absorbers, amine light stabilizers; flame retardants such as phosphorus, bromine, silicone, and organic metal salts; various physical properties such as hydrolysis resistance Modifiers for imparting; fillers such as titanium oxide and talc; dyes and pigments; plasticizers.
- thermoplastic resin a thermoplastic resin is preferable and an aromatic polycarbonate resin is particularly preferable from the viewpoint that the effect of suppressing the outgas generation can be maximized.
- aromatic polycarbonate resin used in the thermoplastic resin composition of the second invention group include the aromatic polycarbonate resins exemplified as the thermoplastic resin (A).
- the method for preparing the aromatic polycarbonate resin composition is not particularly limited, and examples thereof include the following methods.
- the powder of the graft copolymer (G2), the aromatic polycarbonate resin, and various additives used as necessary are mixed and dispersed by a V-type blender or a Henschel mixer.
- this mixture is melt-kneaded using a kneader such as an extruder or a Banbury mixer, a pressure kneader, or a roll.
- the said aromatic polycarbonate resin composition can contain the following various additives in the range which does not impair the objective of this invention.
- Flameproofing agents eg, fluorinated polyolefins, silicones and aramid fibers
- lubricants eg, talc, CaCO 3 and glass flakes
- nucleating agents eg, pentaerythritol tetrastearate
- nucleating agents eg, pentaerythritol tetrastearate
- nucleating agents eg, pentaerythritol tetrastearate
- nucleating agents eg, antistatic agents, stabilizers, fillers
- Reinforcing materials eg glass fibers, carbon fibers, mica, kaolin, talc, CaCO 3 and glass flakes
- dyes and pigments may be used individually by 1 type and may use 2 or more types together.
- thermoplastic resin composition of the first invention group and the second invention group of the present invention a molding method used for molding a normal thermoplastic resin composition, for example, an injection molding method, an extrusion molding method is used. , Blow molding method, calendar molding method and the like.
- the aromatic polycarbonate resin composition of the present invention can provide an excellent molded article having both heat resistance and impact resistance while suppressing generation of outgas, particularly when molding at a high temperature of 300 ° C. or higher.
- the molded product of the first invention group of the present invention has excellent impact resistance, flame retardancy, and color development, it can be used as various materials in the automotive field, OA equipment field, home appliance, electrical / electronic field, etc. Can be widely used.
- Examples of the molding method of the curable resin composition of the second invention group of the present invention include transfer molding, sheet compound molding, and bulk molding. Moreover, when a curable resin composition is a solution state, it can also apply
- the molded product of the second invention group of the present invention is not particularly limited in use, and can be widely used industrially as a material in the automotive field, OA equipment field, electrical / electronic field and the like.
- Examples 1 to 14 and Comparative Examples 1 to 13 relate to the first invention group, and Examples 21 to 46 and Comparative Examples 21 to 25 relate to the second invention group.
- Examples 1 to 5 and Comparative Examples 1 to 4 relate to a polyorganosiloxane-containing graft copolymer, and Examples 6 to 14 and Comparative Examples 5 to 13 relate to a polycarbonate resin composition.
- Examples 21 to 33 and Comparative Examples 21 and 22 relate to a powder containing a polyorganosiloxane-containing graft copolymer, and Examples 34 to 46 and Comparative Examples 23 to 25 relate to a polycarbonate resin composition.
- volume average particle diameter of graft copolymer This evaluation relates to the first invention group.
- the latex of the graft copolymer is diluted with distilled water, and the median diameter in volume average is determined using a laser diffraction scattering type particle size distribution analyzer (SALD-7100, manufactured by Shimadzu Corporation).
- SALD-7100 laser diffraction scattering type particle size distribution analyzer
- the sample concentration of latex is appropriately adjusted so as to be within an appropriate range in the scattered light intensity monitor attached to the apparatus.
- As the standard particle size substance 12 types of particles having monodisperse polystyrene having a known particle size and having a particle size in the range of 20 to 800 nm are used.
- Charpy impact strength Specimens (length 80.0mm, width 10.0mm, thickness 4mm, with V notch) at temperatures of 23 ° C and -30 ° C according to JIS K 7111/1 / 1eA Measure Charpy impact strength.
- Total light transmittance (color development) This evaluation relates to the first invention group. Based on JIS K 7375, the total light transmittance in a D65 light source is measured about a test piece (length 100mm, width 50mm, thickness 2mm) using Nippon Denshoku Industries Co., Ltd. HAZE Meter NDH4000.
- thermoplastic resin composition is molded into 1/16 inch combustion rods and subjected to the UL-94V test.
- the number of silver streaks of the molded product is visually confirmed in accordance with the above-mentioned “evaluation condition 12 (heat-resistant decomposition property)” and used as an index of heat-resistant decomposition property. As the outgas generation amount increases, the number of silver streaks increases, indicating that the thermal decomposition resistance is low. ++: Best (0), +: Good (1-2), -: Evil (3-10), ---: Worst (10 or more).
- DBSNa sodium dodecylbenzenesulfonate
- the emulsion was put in a separable flask having a capacity of 5 liters equipped with a cooling condenser.
- the emulsion was heated to a temperature of 80 ° C. and then a mixture of 0.20 parts sulfuric acid and 49.8 parts distilled water was continuously added over 3 minutes.
- the polymerization reaction was carried out while maintaining the temperature heated to 80 ° C. for 7 hours, and then cooled to room temperature (25 ° C.), and the resulting reaction solution was kept at room temperature for 6 hours. Thereafter, a 5% aqueous sodium hydroxide solution was added to neutralize the reaction solution to pH 7.0 to obtain a polyorganosiloxane rubber latex (S-1).
- the solid content of this latex was 29.8%. Further, the number average particle size (Dn) of the latex measured by a capillary particle size distribution meter (CHDF2000 type manufactured by MATEC, USA) was 384 nm, the mass average particle size (Dw) was 403 nm, and Dw / Dn was 1.05. .
- the emulsion was charged into a separable flask having a capacity of 5 liters equipped with a cooling condenser.
- the emulsion was heated to a temperature of 85 ° C., and this temperature was maintained for 6 hours for a polymerization reaction, then cooled to room temperature (25 ° C.), and the resulting reaction product was kept at room temperature for 12 hours. Thereafter, a 5% aqueous sodium hydroxide solution was added to neutralize the reaction solution to pH 7.0 to obtain a polyorganosiloxane rubber latex (S-2).
- This latex was 28.3%. Moreover, Dn of this latex was 86 nm, Dw was 254 nm, and Dw / Dn was 2.95.
- emulsifier As an emulsifier, 6.0 parts of dipotassium alkenyl succinate and 230 parts of distilled water were charged into a separable flask having a capacity of 2 liters equipped with a stirring blade, a condenser, a thermocouple, and a nitrogen inlet, and were stirred at room temperature under a nitrogen stream at Stir for minutes.
- the dipotassium alkenyl succinate was used in a state of being dissolved in advance in a part of the distilled water.
- the temperature of the liquid in the flask was raised to 70 ° C., and an aqueous solution in which 0.2 part of potassium persulfate was dissolved in 3 parts of distilled water was added to the flask. Further, a mixture comprising 50 parts of methyl methacrylate (MMA), 30 parts of styrene (St), 20 parts of n-butyl acrylate (n-BA) and 0.1 part of n-octyl mercaptan was dropped into the flask over 4 hours. Then, radical polymerization was performed. After completion of the dropwise addition, the liquid in the flask was stirred for 1 hour while maintaining the temperature at 70 ° C. to obtain a latex (p2-1) containing a vinyl polymer (p2). The content of the vinyl polymer (p2) in this latex was 30%.
- MMA methyl methacrylate
- St styrene
- n-BA n-butyl acrylate
- radical polymerization was
- “Fullon AD939E” (Asahi Glass Co., Ltd.), which is a latex containing 166.7 parts of the latex (p2-1) and the PTFE polymer (p1) in a 5 liter reactor equipped with a stirrer PTFE concentration 60%, PTFE mass average molecular weight of about 15 million, polyoxyalkylene alkyl ether concentration 3%) 83.3 parts were charged and stirred for 5 minutes to obtain latex (j-2).
- the latex contained 50 parts of PTFE, 2.5 parts of polyoxyalkylene alkyl ether, and 50 parts of the vinyl polymer (p2).
- Example 1 The polyorganosiloxane rubber latex (S-1) obtained in Production Example 1 was collected in a 29.5 parts, 5 liter separable flask in terms of polymer, and 100 parts of deionized water was added and mixed. Next, in this separable flask, a mixture of 58.9 parts of n-butyl acrylate (n-BA), 1.8 parts of allyl methacrylate (AMA) and 0.25 parts of tert-butyl hydroperoxide (t-BH) was added. Added.
- n-BA n-butyl acrylate
- AMA allyl methacrylate
- t-BH tert-butyl hydroperoxide
- the nitrogen atmosphere in the flask was replaced by passing a nitrogen stream through the separable flask, and the liquid temperature was raised to 50 ° C.
- the liquid temperature reached 50 ° C, 0.001 part of ferrous sulfate (Fe), 0.003 part of ethylenediaminetetraacetic acid disodium salt (EDTA), and 0.3 part of sodium formaldehyde sulfoxylate (SFS) were removed.
- An aqueous solution dissolved in 2.5 parts of ionic water was added to initiate radical polymerization.
- the liquid temperature was maintained at 65 ° C. for 1 hour to obtain a latex of a composite rubber of polyorganosiloxane and poly n-butyl acrylate.
- graft copolymer (G-1) 500 parts of an aqueous solution having a calcium acetate concentration of 1% by mass is maintained at a temperature of 30 ° C., and while stirring, 300 parts of the latex of the graft copolymer (G-1) is gradually added dropwise to the solution. did.
- the obtained graft copolymer (G-1) was filtered and dehydrated. Further, 10 times the amount of water was added to 100 parts of the graft copolymer, followed by washing for 10 minutes in a flask equipped with a stirrer, followed by filtration and dehydration. This operation was repeated twice and then dried to obtain a graft copolymer (G-1) powder.
- Table 1 shows the polymerization rate and volume average particle diameter of the graft copolymer (G-1).
- this polymerization rate is a polymerization rate of the monomer component used in all the processes from manufacture of composite rubber to graft polymerization.
- Examples 2 to 4 Comparative Examples 1 and 2
- a polyorganosiloxane-containing graft copolymer (G-2 to 4 and G ′) was prepared in the same manner as in Example 1 except that the types and amounts of the respective raw materials used in Example 1 were changed to the conditions shown in Table 1. -1 to 2) were produced, and a graft copolymer powder was obtained.
- Table 1 shows the polymerization rate and volume average particle diameter of each graft copolymer obtained.
- Example 5 The latex (S-1) obtained in Production Example 1 was sampled into a separable flask having a volume of 5 liters in terms of polymer, and 200 parts of distilled water was added and mixed. Next, a mixture of 59.1 parts of n-BA, 0.9 part of AMA, and 0.18 part of t-BH was added to the separable flask.
- the nitrogen atmosphere in the flask was replaced by passing a nitrogen stream through the separable flask, and the liquid temperature was raised to 50 ° C.
- the liquid temperature reaches 50 ° C.
- 0.18 part of sodium formaldehyde sulfoxylate (SFS) are distilled.
- An aqueous solution dissolved in 10 parts of water was added to initiate radical polymerization.
- the liquid temperature was maintained at 65 ° C. for 1 hour to obtain a latex of a composite rubber of polyorganosiloxane and poly n-butyl acrylate.
- graft copolymer (G-5) was filtered and dehydrated. Further, 10 times the amount of water was added to 100 parts of the graft copolymer, followed by washing for 10 minutes in a flask equipped with a stirrer, followed by filtration and dehydration. This operation was repeated twice, followed by drying to obtain a graft copolymer (G-5) powder.
- the nitrogen atmosphere in the flask was replaced by passing a nitrogen stream through the separable flask, and the liquid temperature was raised to 60 ° C.
- the liquid temperature reached 60 ° C.
- EDTA ethylenediaminetetraacetic acid disodium salt
- SFS sodium formaldehyde sulfoxylate
- Examples 6 to 11, Comparative Examples 5 to 10 As the polyorganosiloxane-containing graft copolymer, the powder of the polyorganosiloxane-containing graft copolymer (G-1 to 5, G′-1 to 4) obtained in Examples 1 to 5 or Comparative Examples 1 to 4 was used. In accordance with the above-mentioned “Production Condition 1”, “Test piece 1” for impact resistance evaluation, “Test piece 2” for evaluation of total light transmittance, and “Test piece 3” for evaluation of flame retardancy are prepared, respectively. did. At that time, the pellets were dried at 80 ° C. for 12 hours and then supplied to an injection molding machine. Subsequently, each evaluation was performed according to the above-mentioned “evaluation condition 2”, and the evaluation results shown in Table 2 were obtained.
- Example 12 to 14 Comparative Examples 11 to 13
- Table 3 shows phenolic antioxidants (trade name: Irganox 245, manufactured by Ciba Japan Co., Ltd.) and phosphorus antioxidants (trade name: ADK STAB PEP36, manufactured by ADEKA Corporation). The amount was blended. Except for these, a polycarbonate resin composition and test pieces were obtained in the same manner as in Example 6. The evaluation results are shown in Table 3.
- the resin composition of Comparative Example 8 uses a graft copolymer (G′-3) having a polyorganosiloxane content of more than 69% by mass and containing no aromatic vinyl monomer (b 2 ). Yes. For this reason, since the graft copolymer has a low refractive index and a refractive index difference from the matrix resin becomes large, the resin composition has a low total light transmittance and a low color developability. Since the resin composition of Comparative Example 10 did not contain the graft copolymer (B), the impact resistance was low, and the flame retardancy was relatively low.
- G′-3 graft copolymer having a polyorganosiloxane content of more than 69% by mass and containing no aromatic vinyl monomer (b 2 ).
- the resin compositions of Examples 12 and 13 are superior in balance of flame retardancy, impact resistance, and total light transmittance as compared with the resin composition of Comparative Example 11. I understood that. Moreover, it turned out that the resin composition of Example 14 is excellent in the balance of a flame retardance, impact resistance, and a total light transmittance compared with the resin composition of Comparative Examples 12 and 13.
- Example 21 33.56 parts of the polyorganosiloxane rubber latex (S-1) obtained in Production Example 1 (10.0 parts in terms of polymer) was collected in a separable flask having a capacity of 5 liters, and 200 parts of distilled water was added. And mixed. Next, in this separable flask, a mixture of 59.1 parts of n-butyl acrylate (n-BA), 0.9 part of allyl methacrylate (AMA) and 0.24 part of t-butyl hydroperoxide (t-BH) was added. Added.
- n-BA n-butyl acrylate
- AMA allyl methacrylate
- t-BH t-butyl hydroperoxide
- the nitrogen atmosphere in the flask was replaced by passing a nitrogen stream through the separable flask, and the liquid temperature was raised to 50 ° C.
- the liquid temperature reaches 50 ° C.
- 0.18 part of sodium formaldehyde sulfoxylate (SFS) are distilled.
- An aqueous solution dissolved in 10 parts of water was added to initiate radical polymerization.
- the liquid temperature dropped to 65 ° C., in order to complete the polymerization of the acrylate component, the liquid temperature was maintained at 65 ° C. for 1 hour to obtain a latex of a composite rubber of polyorganosiloxane and poly n-butyl acrylate.
- Example 21 the polyorganosiloxane-containing graft copolymer (G-22 to 26, G--) was used in the same manner as in Example 21 except that the type and amount of each raw material used were changed to the conditions shown in Table 4. 33, G′-21, G′-22) were produced. Each evaluation result is shown in Table 5.
- Example 27 33.56 parts of latex (S-1) obtained in Production Example 1 (10.0 parts in terms of polymer) were collected in a separable flask having a volume of 5 liters, and 200 parts of distilled water was added and mixed. A mixture of 59.1 parts n-BA and 0.9 parts AMA was then added to the separable flask.
- Example 28 to 32 A powder of polyorganosiloxane-containing graft copolymer (G-28 to 32) was prepared in the same manner as in Example 27 except that the type and amount of each raw material used were changed to the conditions shown in Table 4. The body was manufactured. Each evaluation result is shown in Table 5.
- the graft copolymer powder of Comparative Example 21 has an alkaline earth metal content when solidified with calcium acetate. More than 150 ppm.
- the powder of the graft copolymer of Comparative Example 22 had a particle diameter of 300 nm or less because the particle diameter of the polyorganosiloxane rubber was small.
- Examples 34 to 46, Comparative Examples 23 to 25 As the polyorganosiloxane-containing graft copolymer, powders of graft copolymers (G-21 to 33, G′-21, G′-22) were used. In accordance with the above-mentioned “Resin composition and test piece preparation condition 11”, “Evaluation condition 12 (heat decomposability)” and “Evaluation condition 13 (color developability)”, respectively, evaluated. At that time, the pellets were dried at 80 ° C. for 12 hours and then supplied to an injection molding machine.
- the pellets dried at 80 ° C. for 12 hours as described above are supplied to a 100 t injection molding machine (trade name: SE-100DU, manufactured by Sumitomo Heavy Industries, Ltd.) at a cylinder temperature of 310 ° C. and a mold temperature of 90 ° C. Injection molding was performed.
- a test piece for Charpy impact strength (length 80.0 mm, width 10.0 mm, thickness 4 mm, with V notch)
- test piece for thermal decomposition evaluation (length 100 mm, Test pieces (length 100 mm, width 50 mm, thickness 2 mm) for chemical resistance evaluation were obtained.
- the evaluation results are shown in Table 6.
- the polycarbonate resin composition of Comparative Example 23 was significantly outgassed compared to the resin compositions of Examples 34 to 46 because the amount of alkaline earth metal contained in the graft copolymer powder was 150 ppm or more. Generation was confirmed, and the thermal decomposition resistance was low.
- the peroxide used for the polymerization of the alkyl (meth) acrylate component during the production of the composite rubber is cumene hydroperoxide and diisopropylbenzene hydroperoxide, so that the chemical resistance is high. It was excellent.
- polycarbonate resin compositions of Examples 35, 37, 39, 41, 42, and 43 are more excellent in chemical resistance because the content of the composite rubber in the graft copolymer is 75% by mass or more. It was.
- the polycarbonate resin composition of Example 44 had good chemical resistance because the sp value of the graft portion of the graft copolymer was high.
- the grafting monomer used in the production of the graft copolymer contains a crosslinkable monomer, and the chemical resistance of the graft copolymer itself is high. The property was good.
- the graft portion of the graft copolymer contained a styrene unit, and the thermal decomposition resistance was good.
- the polycarbonate resin composition of Comparative Example 24 has a high content of alkaline earth metal and sulfur in the graft copolymer powder, and the particle diameter of the graft copolymer is smaller than 300 nm. The color developability was low.
- the resin compositions (particularly polycarbonate resin compositions) of the first invention group and the second invention group of the present invention are both as materials in the automotive field, OA equipment fields such as printers, and electric / electronic fields such as mobile phones. Useful.
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Mechanical Engineering (AREA)
- Graft Or Block Polymers (AREA)
- Compositions Of Macromolecular Compounds (AREA)
- Macromonomer-Based Addition Polymer (AREA)
- Silicon Polymers (AREA)
Description
(1)15≦「シャルピー衝撃強度」≦70
(2)60≦「シャルピー衝撃強度」+「全光線透過率」×1.36
(3)1/16インチ厚さにおけるUL94規格垂直燃焼試験による難燃性が「V-1」以上。
(1)粘度平均分子量22,000の芳香族ポリカーボネート樹脂(三菱エンジニアリングプラスチック(株)製ユーピロンS-2000F):100質量部、
(2)ポリオルガノシロキサン含有グラフト共重合体(G1):5.5質量部、
(3)アクリル変性ポリテトラフルオロエチレン(三菱レイヨン(株)製メタブレンA-3800):0.5質量部、
(4)芳香族リン酸エステル系難燃剤(大八化学工業(株)製PX-200):5.5質量部。
(i)シャルピーノッチ付き衝撃強さ評価用「試験片1」(長さ80.0mm、幅10.0mm、厚み4mm、Vノッチ付き)。
(ii)全光線透過率評価用「試験片2」(長さ100mm、幅50mm、厚み2mm)。
(iii)1/16インチ厚さの難燃性評価用「試験片3」(長さ127mm、幅12.7mm、厚み1.6mm)。
〔シャルピー衝撃強度[kJ/m2]〕
JIS K 7111‐1/1eAに準拠して温度-30℃にて、「試験片1」のシャルピー衝撃強度を測定する。
JIS K 7375に準拠して日本電色工業(株)製HAZE Meter NDH4000を用いて、「試験片2」のD65光源における全光線透過率を測定する。
UL94V試験に準拠した垂直燃焼試験法にて、5つの「試験片3」について総燃焼時間を測定する。
(1)ポリオルガノシロキサン含有グラフト共重合体(G2)の紛体:4質量部、
(2)粘度平均分子量22,000の芳香族ポリカーボネート樹脂(三菱エンジニアリングプラスチック(株)製ユーピロンS-2000F):96質量部、
(3)Irganox1076(BASF製):0.1質量部、
(4)アデカスタブ2112((株)ADEKA製):0.1質量部、
(5)カーボンブラック#960(三菱化学(株)製):0.1質量部。
「試験片11」のゲート付近に発生するシルバーストリークの本数を目視で確認する。
前記樹脂組成物のペレットを100t射出成形機(住友重機(株)製SE-100DU)にて、シリンダー温度310℃、金型温度90℃の条件で成形して「試験片13」(長さ100mm、幅50mm、厚み2mm)を得る。
装置:分光式色差計SE-2000(日本電色工業株式会社製、0-45°後分光方式)
測定範囲:380~780nm、
測定光源:C光(2°視野)、
三刺激値(XYZ)からCIE色差式を用いてL*値を算出する。
第1の発明群のポリオルガノシロキサン含有グラフト共重合体(G1)(以下、「グラフト共重合体(G1)」または「共重合体(G1)」と略す場合がある。)は、アルキル基又は芳香族基を有する(メタ)アクリル酸エステル(b1)と芳香族ビニル単量体(b2)とを含むビニル単量体混合物をポリオルガノシロキサン系ゴムにグラフト重合した共重合体である。
ポリオルガノシロキサン系ゴムとしては、ポリオルガノシロキサンゴム、又はポリオルガノシロキサン及びポリアルキル(メタ)アクリレートを含有する複合ゴムであることが好ましい。
ポリオルガノシロキサンは、オルガノシロキサン単位を構成単位として含有する重合体である。ポリオルガノシロキサンゴムは、オルガノシロキサン、または、必要に応じて使用される成分を含むオルガノシロキサン混合物を重合することにより得ることができる。必要に応じて使用される成分としては、シロキサン系架橋剤、シロキサン系グラフト交叉剤、及び末端封鎖基を有するシロキサンオリゴマー等が挙げられる。

ポリオルガノシロキサンゴムの製造方法としては特に制限はなく、例えば、以下の製造方法を採用できる。
カートリッジ:専用の粒子分離用キャピラリー式カートリッジ(商品名;C-202)、
キャリア液:専用キャリア液(商品名;2XGR500)、
キャリア液の液性:ほぼ中性、
キャリア液の流速:1.4ml/分、
キャリア液の圧力:約4,000psi(2,600kPa)、
測定温度:35℃、
試料使用量:0.1ml。
本発明において、ポリオルガノシロキサン系ゴムとして、ポリオルガノシロキサン及びポリアルキル(メタ)アクリレートを含有する複合ゴム(以下、「複合ゴム」と略す。)を用いることができる。複合ゴムは、前記ポリオルガノシロキサンと、単独重合体のガラス転移温度Tgが0℃以下のポリアルキル(メタ)アクリレートを含有するゴムである。複合ゴムは、ポリオルガノシロキサンゴムの存在下にアルキル(メタ)アクリレートを重合して得られるゴムであることが好ましい。
Tg:共重合体のガラス転移温度(℃)、
wi:単量体iの質量分率、
Tgi:単量体iを重合して得られる単独重合体のガラス転移温度(℃)。
なお、Tgiの値としては、POLYMER HANDBOOK Volume 1(WILEY-INTERSCIENCE)に記載の値が用いられる。
複合ゴムの製造方法としては、特に制限はなく、例えば、乳化重合法、懸濁重合法、微細懸濁重合法により製造することができるが、乳化重合法を用いることが好ましい。中でも、ポリオルガノシロキサンゴムのラテックスの存在下に、複合ゴム用(メタ)アクリレート成分を乳化重合して、複合ゴムのラテックスを得る方法が特に好ましい。
第1の発明群のポリオルガノシロキサン含有グラフト共重合体(G1)は、ポリオルガノシロキサン系ゴムに対してアルキル基又は芳香族基を有する(メタ)アクリル酸エステル(b1)と芳香族ビニル単量体(b2)とを含むビニル単量体混合物をグラフトしたグラフト共重合体である。上記ポリオルガノシロキサン系ゴムの存在下でビニル単量体混合物をグラフト重合することによって、グラフト共重合体(G1)を得ることができる。以下、アルキル基又は芳香族基を有する(メタ)アクリル酸エステル(b1)を、単に「(メタ)アクリル酸エステル(b1)」という場合がある。
第2の発明群のグラフト共重合体(G2)はポリオルガノシロキサン系ゴムに1種以上のビニル単量体をグラフト重合して得られる共重合体である。グラフト共重合体(G2)のグラフト部は、前記FOXの式で表されるガラス転移温度Tgが0℃を超えることが好ましく、50℃以上であることがより好ましく、80℃以上であることがさらに好ましい。
Asp:ビニル単量体Aのsp値
Bsp:ビニル単量体Bのsp値
Csp:ビニル単量体Cのsp値
Amo:ビニル単量体Aのモル分率
Bmo:ビニル単量体Bのモル分率
Cmo:ビニル単量体Cのモル分率
Amo+Bmo+Cmo=1 。
グラフト部の重合の方法としては、例えば、ポリオルガノシロキサン系ゴムのラテックス中にグラフト重合用のビニル単量体を添加し、1段又は多段で重合する方法が挙げられる。多段で重合する場合は、ポリオルガノシロキサンゴム系ゴムのラテックス中に、グラフト重合用のビニル単量体を分割して逐次添加し又は連続的に添加して、重合することが好ましい。このような重合方法は重合安定性が良好であり、且つ所望の粒子径及び粒子径分布を有するグラフト共重合体のラテックスを安定に得ることができる。
第1の発明群のポリオルガノシロキサン含有グラフト共重合体(G1)は、下記の「作製条件1」で作製された試験片について、下記の「評価条件2」で測定される「試験片」のシャルピー衝撃強度[kJ/m2]、全光線透過率[%]及び難燃性が、以下の性能条件(1)~(3)を満たすことが好ましい。
(1)15≦「シャルピー衝撃強度」≦70
(2)60≦「シャルピー衝撃強度」+「全光線透過率」×1.36
(3)1/16インチ厚さにおけるUL94規格垂直燃焼試験による難燃性が「V-1」以上。
(1)粘度平均分子量22,000の芳香族ポリカーボネート樹脂(三菱エンジニアリングプラスチック(株)製ユーピロンS-2000F):100質量部、
(2)ポリオルガノシロキサン含有グラフト共重合体(G1):5.5質量部、
(3)アクリル変性ポリテトラフルオロエチレン(三菱レイヨン(株)製メタブレンA-3800):0.5質量部、
(4)芳香族リン酸エステル系難燃剤(大八化学工業(株)製PX-200):5.5質量部。
(i)シャルピーノッチ付き衝撃強さ評価用「試験片1」(長さ80.0mm、幅10.0mm、厚み4mm、Vノッチ付き)。
(ii)全光線透過率評価用「試験片2」(長さ100mm、幅50mm、厚み2mm)。
(iii)厚み1/16インチの難燃性評価用「試験片3」。(長さ127mm、幅12.7mm、厚み1.6mm)
<評価条件2>:
〔シャルピー衝撃強度[kJ/m2]〕
JIS K 7111‐1/1eAに準拠して温度-30℃にて、「試験片1」のシャルピー衝撃強度を測定する。
JIS K 7375に準拠して日本電色工業(株)製HAZE Meter NDH4000を用いて、「試験片2」のD65光源における全光線透過率測定する。
UL94V試験に準拠した垂直燃焼試験法にて、5つの「試験片3」について総燃焼時間を測定する。
(11)25≦「シャルピー衝撃強度」≦70
(12)60≦「シャルピー衝撃強度」+「全光線透過率」×1.36
(13)1/16インチ厚さにおけるUL94規格垂直燃焼試験による難燃性が「V-1」以上。
(21)25≦「シャルピー衝撃強度」≦40
(22)60≦「シャルピー衝撃強度」+「全光線透過率」×1.36≦100
(23)1/16インチ厚さにおけるUL94規格垂直燃焼試験による難燃性が「V-0」以上。
(1)ポリオルガノシロキサン含有グラフト共重合体(G2)の粉体:4質量部、
(2)粘度平均分子量22,000の芳香族ポリカーボネート樹脂(三菱エンジニアリングプラスチック(株)製ユーピロンS-2000F):96質量部、
(3)Irganox1076(BASF製):0.1質量部、
(4)アデカスタブ2112((株)ADEKA製):0.1質量部、
(5)カーボンブラック#960(三菱化学(株)製):0.1質量部。
「試験片11」のゲート付近に発生するシルバーストリークの本数を目視で確認する。
前記樹脂組成物のペレットを100t射出成形機(住友重機(株)製SE-100DU)に供給して、シリンダー温度310℃、金型温度90℃の条件で成形して「試験片13」(長さ100mm、幅50mm、厚み2mm)を得る。
装置:分光式色差計SE-2000(日本電色工業株式会社製、0-45°後分光方式)
測定範囲:380~780nm、
測定光源:C光(2°視野)、
三刺激値(XYZ)からCIE色差式を用いてL*値を算出する。
第1の発明群のグラフト共重合体(G1)は、熱可塑性樹脂(A)と混合して熱可塑性樹脂組成物として使用することができる。熱可塑性樹脂組成物としては、熱可塑性樹脂(A)、グラフト共重合体(G)、フッ素系樹脂(C)及び難燃剤(D)を含有する熱可塑性樹脂組成物が好ましい。
熱可塑性樹脂(A)としては、例えば、以下のものが挙げられる。ポリプロピレン(PP)、ポリエチレン(PE)等のオレフィン系樹脂;ポリスチレン(PS)、ハイインパクトポリスチレン(HIPS)、(メタ)アクリレート・スチレン共重合体(MS)、スチレン・アクリロニトリル共重合体(SAN)、スチレン・無水マレイン酸共重合体(SMA)、アクリロニトリル・ブタジエン・スチレン共重合体(ABS)、アクリル酸エステル・スチレン・アクリロニトリル共重合体(ASA)、アクリロニトリル・エチレン・プロピレンゴム・スチレン共重合体(AES)等のスチレン(St)系樹脂;ポリメチルメタクリレート(PMMA)等のアクリル(Ac)系樹脂;ポリカーボネート系樹脂(PC系樹脂);ポリアミド(PA)樹脂;ポリエチレンテレフタレート(PET)、ポリブチレンテレフタレート(PBT)、ポリ乳酸(PLA)等のPEs樹脂;(変性)ポリフェニレンエーテル((m-)PPE)樹脂、ポリオキシメチレン(POM)樹脂、ポリスルフォン(PSO)樹脂、ポリアリレート(PAr)樹脂、ポリフェニレン(PPS)樹脂等のエンジニアリングプラスチックス;熱可塑性ポリウレタン(PU)樹脂;PC/ABS等のPC樹脂とSt系樹脂とのアロイ、PVC/ABS等のPVC系樹脂とSt系樹脂とのアロイ、PA/ABS等のPA樹脂とSt系樹脂とのアロイ、PA樹脂とTPEとのアロイ、PA/PP等のPA樹脂とポリオレフィン系樹脂とのアロイ、PC/PBT等のPC樹脂とPEs樹脂とのアロイ、ポリオレフィン系樹脂/TPEとPP/PE等とのオレフィン系樹脂同士のアロイ、PPE/HIPSとPPE/PBTとPPE/PA等とのPPE系樹脂同士のアロイ、PVC/PMMA等のPVC系樹脂とAc系樹脂とのアロイ等のポリマーアロイ;硬質塩化ビニル樹脂、半硬質塩化ビニル樹脂、軟質塩化ビニル樹脂等のPVC系樹脂。
フッ素系樹脂(C)は、燃焼時の滴下防止を目的として用いることができる。フッ素系樹脂(C)としては、ポリテトラフルオロエチレン、変性ポリテトラフルオロエチレン等が挙げられる。変性ポリテトラフルオロエチレンとしては、SAN変性ポリテトラフルオロエチレン、アクリル変性ポリテトラフルオロエチレン等が挙げられる。
難燃剤(D)としては、公知の難燃剤を用いることができ、例えば以下のものが挙げられる。ハロゲン化ビスフェノールA、ハロゲン化ポリカーボネートオリゴマー、臭素化エポキシ化合物等のハロゲン系化合物と酸化アンチモン等の難燃助剤の組合せからなるハロゲン系難燃剤;有機塩系難燃剤;リン酸エステル系難燃剤、ハロゲン化リン酸エステル型難燃剤等のリン系難燃剤;芳香族スルホン酸の金属塩、パーフルオロアルカンスルホン酸の金属塩等のスルホン酸系難燃剤;分岐型のフェニルシリコーン化合物、フェニルシリコーン系樹脂等のオルガノポリシロキサン等のシリコーン系難燃剤。
第1の発明群の熱可塑性樹脂組成物中には必要に応じて、酸化防止剤(E)を含有させることができる。酸化防止剤(E)は、成形体の製造時の樹脂の酸化分解を抑制することを目的とするだけでなく、成形体の難燃性を向上させることも目的とする成分である。酸化防止剤(E)は、通常の成形時に使用されるものであれば、特に限定されない。具体例としては、例えば以下のものが挙げられる。トリス[N-(3,5-ジ-t-ブチル-4-ヒドロキシベンジル)]イソシアヌレート((株)ADEKA製、アデカスタブAO-20など)、テトラキス[3-(3,5-ジ-t-ブチル-4-ヒドロキシフェニル)プロピオニルオキシメチル]メタン(BASF社製、イルガノックス1010など)、ビス(3-tert-ブチル-4-ヒドロキシ-5-メチルベンゼンプロパン酸)エチレンビス(オキシエチレン)(BASF社製、イルガノックス245など)、3,5-ジ-tert-ブチル-4-ヒドロキシベンゼンプロピオン酸オクタデシル(BASF社製、イルガノックス1076など)、ブチリデン-1,1-ビス-(2-メチル-4-ヒドロキシ-5-t-ブチル-フェニル((株)ADEKA製、アデカスタブAO-40など)、1,1,3-トリス(2-メチル-4-ヒドロキシ-5-t-ブチルフェニル)ブタン(吉冨ファインケミカル(株)製、ヨシノックス930など)などのフェノール系酸化防止剤;ビス(2,6,ジ-t-ブチル-4-メチルフェニル)ペンタエリスリトールホスファイト((株)ADEKA製、アデカスタブPEP-36など)、トリス(2,4-ジ-t-ブチルフェニル)ホスファイト((株)ADEKA製、アデカスタブ2112など)、2,2-メチレンビス(4,6-ジ-t-ブチルフェニル)オクチルホスファイト((株)ADEKA製、アデカスタブHP-10など)などのリン系酸化防止剤;ジラウリル3,3’-チオ-ジプロピオネート(吉冨ファインケミカル(株)製、ヨシノックスDLTP)、ジミリスチル3,3’-チオ-ジプロピオネート(吉冨ファインケミカル(株)製、ヨシノックスDMTP)などのイオウ系酸化防止剤等。
第1の発明群の熱可塑性樹脂組成物中には、更に必要に応じて、以下の成分を配合することができる。可塑剤、滑剤;離型剤(例えば、ペンタエリトリトールテトラステアレート);成核剤、帯電防止剤、安定剤、充填材;強化材(ガラス繊維、炭素繊維、マイカ、カオリン、タルク、CaCO3およびガラスフレーク);色素および顔料。これらは1種を単独で用いてもよく、2種以上を併用してもよい。
第1の発明群の熱可塑性樹脂組成物の調製方法は特に限定されない。例えば、熱可塑性樹脂(A)、グラフト共重合体(G1)と、必要に応じてフッ素系樹脂(C)、難燃剤(D)、酸化防止剤(E)、各種添加剤とを、V型ブレンダーやヘンシェルミキサー等により混合分散させ、この混合物を押出機またはバンバリーミキサー、加圧ニーダー、ロール等の混練機等を用いて溶融混練することにより調製できる。これらの各成分の混合はバッチ的又は連続的に実施することができ、各成分の混合順序は特に限定されない。溶融混練物はペレットにして、各種の成形に用いることができる。
本発明の第2の発明群のグラフト共重合体(G2)の粉体は、樹脂と混合して樹脂組成物として使用することができる。第2の発明群において使用できる樹脂としては、特に制限はないが、例えば「硬化性樹脂」、「熱可塑性樹脂」及び「熱可塑性エラストマー」から選ばれる1種以上の樹脂が挙げられる。
硬化性樹脂としては、例えば、エポキシ樹脂、フェノール樹脂、不飽和ポリエステル樹脂、メラミン樹脂、尿素樹脂が挙げられる。これらの中では、電気的特性に優れ、半導体封止に適していることから、エポキシ樹脂が好ましい。これらは1種を単独で用いてもよく、二種以上を併用してもよい。硬化性樹脂は、熱硬化性樹脂と光硬化性樹脂とに分類できるが、そのいずれであってもよい。
熱可塑性樹脂としては、前記熱可塑性樹脂(A)として例示した樹脂が挙げられる。それらの中でも以下の樹脂またはアロイが好ましい。St系樹脂、PC樹脂、PA樹脂、PET樹脂、PBT樹脂、(m-)PPE樹脂、POM樹脂、PU樹脂;PC/ABS等のPC樹脂とSt系樹脂とのアロイ、PA/ABS等のPA樹脂とSt系樹脂とのアロイ、PA樹脂とTPEとのアロイ、PA/PP等のPA樹脂とポリオレフィン系樹脂とのアロイ、PC/PBT等のPC樹脂とPEs樹脂とのアロイ;PPE/PBT、PPE/PA等のPPE系樹脂同士のアロイ等。またPC樹脂がより好ましい。
熱可塑性エラストマーとしては、例えば以下のものが挙げられる。スチレン系エラストマー、オレフィン系エラストマー、塩化ビニル系エラストマー、ウレタン系エラストマー、ポリエステル系エラストマー、ポリアミド系エラストマー、フッ素系エラストマー、1,2-ポリブタジエン、トランス1,4-ポリイソプレン。これらの中でも、ウレタン系エラストマー、ポリエステル系エラストマー、ポリアミド系エラストマーが好ましい。
樹脂が硬化性樹脂である場合の樹脂組成物(以下、「硬化性樹脂組成物」という。)の調製方法としては、例えば、(1)各成分を溶液状態で混合する方法、(2)各成分をミキシングロールやニーダー等を用いて溶融混合し、冷却した後、粉砕もしくは打錠する方法が挙げられる。
(1)ヘンシェルミキサー、タンブラー等でグラフト共重合体(G2)の粉体、及び熱可塑性樹脂の粉体もしくは粒状物を混合した後に、押出機、ニーダー、ミキサー等で溶融混合する方法、
(2)予め溶融させた熱可塑性樹脂に、残りの材料を逐次混合していく方法。
本発明の第1の発明群及び第2の発明群の熱可塑性樹脂組成物の成形方法としては、通常の熱可塑性樹脂組成物の成形に用いられる成形法、例えば、射出成形法、押出成形法、ブロー成形法、カレンダー成形法等が挙げられる。
(1)ラテックスの固形分
質量w1のラテックスを180℃の熱風乾燥機中で30分間乾燥し、乾燥後の残渣の質量w2を測定し、下記式により固形分を算出する。
固形分[%]=w2/w1×100 ・・・(1)
重合体を製造する際に仕込んだ全単量体の質量w3と重合後に得られた固形分の質量w4から、下記式により重合率を算出する。
重合率[%]=w4/w3×100 ・・・(2)
この評価は第1の発明群に関するものである。グラフト共重合体のラテックスを蒸留水で希釈し、レーザー回折散乱式粒度分布計(島津製 SALD-7100)を用い、体積平均におけるメジアン径を求める。ラテックスの試料濃度は、装置に付属の散乱光強度モニターにおいて適正範囲となるよう適宜調整する。標準粒子径物質としては、粒子径既知の単分散ポリスチレンであって、粒子径が20~800nmの範囲内の12種類の粒子が用いられる。
この評価は第2の発明群に関するものである。グラフト共重合体のラテックスの濃度(固形分)を、脱イオン水を用いて0.5g/Lに希釈した液について、紫外可視分光光度計(島津製作所製 UV-mini1240)を用いて、波長700nmにおける吸光度DAを測定する。下記式により粒子径を算出する。
この評価は第2の発明群に関するものである。グラフト共重合体を含む粉体0.25gを分解容器内に量り取り、硝酸8mlを加えマイクロウエーブ(湿式分解)にて分解させる。冷却後、フッ化水素酸2 ml入れ再度マイクロウエーブで処理した後、蒸留水で50mlにメスアップし検液とする。この検液についてICP発光分析装置(IRIS Interpid II XSP:Thermo社製)を用いて、カリウム、ナトリウム、カルシウム、マグネシウム、硫黄の含有量を定量する。
JIS K 7111‐1/1eAに準拠して、温度23℃、及び-30℃にて、試験片(長さ80.0mm、幅10.0mm、厚み4mm、Vノッチ付き)のシャルピー衝撃強度を測定する。
この評価は第1の発明群に関するものである。JIS K 7375に準拠して、日本電色工業(株)製HAZE Meter NDH4000を用いて、試験片(長さ100mm、幅50mm、厚み2mm)について、D65光源における全光線透過率を測定する。
この評価は第1の発明群に関するものである。熱可塑性樹脂組成物を1/16インチの燃焼棒に成形し、UL-94V試験を行なう。
この評価は第1の発明群に関するものである。試験片(長さ80.0mm、幅10.0mm、厚み4mm、Vノッチ付き)を温度120℃のオーブン中にて12時間、熱処理する。オーブンから試験片を取り出し、温度23℃、相対湿度50%の雰囲気で24時間以上放置した後、温度23℃において、シャルピー衝撃強度を測定し、耐熱老化性の指標とする。
この評価は第2の発明群に関するものである。先ず、前述の「作成条件11」に従って、ペレットを製造する。次いで、このペレットを住友SE100DU射出成形機(住友重機械(株)製)とホットランナーを有する金型(試験片サイズ:縦100mm×横100mm×厚み2mm、ピンゲート)に供給して、シリンダー温度310℃、ランナー温度310℃、金型温度90℃の条件で射出成形する。1ショット成形後、2分間ずつ滞留させながら連続して3ショット成形して、滞留なし(0分)の試験片、2分間滞留後の試験片、4分間滞留後の試験片、及び6分間滞留後の試験片を得る。
++ : 最良 (0本)、
+ : 良 (1~2本)、
- : 悪 (3~10本)、
-- : 最悪 (10本以上)。
この評価は第2の発明群に関するものである。100t射出成形機(住友重機械(株)製SE-100DU)を用い、シリンダー温度310℃、金型温度90℃の条件で樹脂組成物を射出成形して作成した長さ100mm、幅50mm、厚さ2mmの試験片を得る。この試験片を溶剤(アセトン13質量%、トルエン20質量%、4-ヒドロキシ-4-メチル-2-ペンタノン16質量%、イソブチルアルコール16質量%、メチルエチルケトン20質量%、キシレン5質量%、シクロヘキサノン10質量%の混合物)中に室温(25℃)で2分間浸漬後、温度70℃にて30分間乾燥する。次いで、この試験片を温度23℃、相対湿度50%の空気雰囲気中に4時間静置後、デュポン面衝撃試験機(荷重:1kg、高さ:10cm、n=10)を用いて面衝撃試験を行う。以下の計算式を用いて脆性破壊率を算出し、耐薬品性の指標とする。脆性破壊率が低いほど耐薬品性が良好であることを示す。
この評価は第2の発明群に関するものである。前述の「評価条件13(発色性)」に従って、試験片のL*を測定する。
[製造例1]
テトラエトキシシラン(TEOS)2部、γ-メタクリロイロキシプロピルジメトキシメチルシラン(DSMA)2部及び、オクタメチルシクロテトラシロキサン(モメンティブ・パフォーマンス・マテリアルズ・ジャパン(株)製、製品名:TSF404)96部を混合してオルガノシロキサン混合物100部を得た。脱イオン水150部中にドデシルベンゼンスルホン酸ナトリウム(DBSNa)1部を溶解した水溶液を、前記混合物中に添加し、ホモミキサーにて10,000rpmで5分間攪拌した後、ホモジナイザーに20MPaの圧力で2回通し、安定な予備混合エマルションを得た。
環状オルガノシロキサン混合物(信越シリコーン(株)製、製品名:DMC)を97.5部、TEOSを2部及びDSMAを0.5部、混合してオルガノシロキサン混合物100部を得た。DBSNaを0.68部、ドデシルベンゼンスルホン酸(DBSH)を0.68部、脱イオン水200部中に溶解した水溶液を、前記混合物中に添加し、ホモミキサーにて10,000rpmで2分間攪拌した後、ホモジナイザーに20MPaの圧力で2回通し、安定な予備混合エマルションを得た。
これはポリテトラフルオロエチレン含有粉体(J-2)の製造例である。以下の説明において、「PTFE」は、ポリテトラフルオロエチレンを意味する。
製造例1において得たポリオルガノシロキサンゴムのラテックス(S-1)をポリマー換算で29.5部、容量5リットルのセパラブルフラスコ内に採取し、脱イオン水100部を添加混合した。次いでこのセパラブルフラスコ内に、n-ブチルアクリレート(n-BA)58.9部、アリルメタクリレート(AMA)1.8部、tert-ブチルハイドロパーオキサイド(t-BH)0.25部の混合物を添加した。
実施例1において用いた各原料の種類及び量を表1に示す条件に変更したこと以外は実施例1と同様にして、ポリオルガノシロキサン含有グラフト共重合体(G-2~4、及びG’-1~2)を製造し、更にグラフト共重合体の粉体を得た。得られた各グラフト共重合体の重合率、体積平均粒子径を表1に示す。
製造例1において得たラテックス(S-1)をポリマー換算で10.0部、容量5リットルのセパラブルフラスコ内に採取し、蒸留水200部を添加混合した。次いでこのセパラブルフラスコ内に、n-BAを59.1部、AMAを0.9部、t-BHを0.18部の混合物を添加した。
製造例1において得たラテックス(S-1)をポリマー換算で80.0部、容量5リットルのセパラブルフラスコ内に採取し、蒸留水46部を添加混合した。次いでこのセパラブルフラスコ内に、AMAを5.0部、クメンハイドロパーオキサイド(CHP)を0.11部の混合物を添加した。
製造例2によって得たラテックス(S-2)をポリマー換算で20.0部、容量5リットルのセパラブルフラスコ内に採取し、蒸留水140部を添加混合した。次いでこのセパラブルフラスコ内に、n-BAを49.0部、AMAを1.0部、t-BHを0.20部の混合物を添加した。その後は実施例1と同様にして複合ゴムのラテックスを得た。
ポリオルガノシロキサン含有グラフト共重合体として、実施例1~5または比較例1~4で得たポリオルガノシロキサン含有グラフト共重合体(G-1~5、G’-1~4)の粉体を用いて、前述の「作製条件1」に従って、それぞれ、耐衝撃性評価用「試験片1」、全光線透過率評価用「試験片2」、及び難燃性評価用「試験片3」を作製した。尚、その際、ペレットは80℃で12時間乾燥した後、射出成形機に供給した。次いで、前述の「評価条件2」に従って、各評価を実施し、表2に示す評価結果を得た。
実施例1または比較例4で得たポリオルガノシロキサン含有グラフト共重合体(G-1、G’-4)の粉体、有機スルホン酸金属塩(DIC(株)製、商品名:メガファックF-114)、滴下防止剤として製造例3で得たポリテトラフルオロエチレン含有粉体J-2、及びポリカーボネート樹脂(三菱エンジニアリングプラスチックス(株)製、商品名;ユーピロンS-2000F、粘度平均分子量22,000)、さらにフェノール系酸化防止剤(チバ・ジャパン(株)製、商品名:イルガノックス245)、リン系酸化防止剤((株)ADEKA製、商品名:アデカスタブPEP36)を表3に記載の量で配合した。これら以外は実施例6と同様にしてポリカーボネート系樹脂組成物及び各試験片を得た。評価結果を表3に示す。
ポリオルガノシロキサン含有グラフト共重合体を3.3部配合した実施例6の樹脂組成物は、比較例5の樹脂組成物に比べ、難燃性、耐衝撃性及び全光線透過率のバランスに優れることが分かった。また、ポリオルガノシロキサン含有グラフト共重合体を5.5部配合した実施例7~11の樹脂組成物は、比較例6~9の樹脂組成物に比べ、難燃性、耐衝撃性及び全光線透過率のバランスに優れることが分かった。


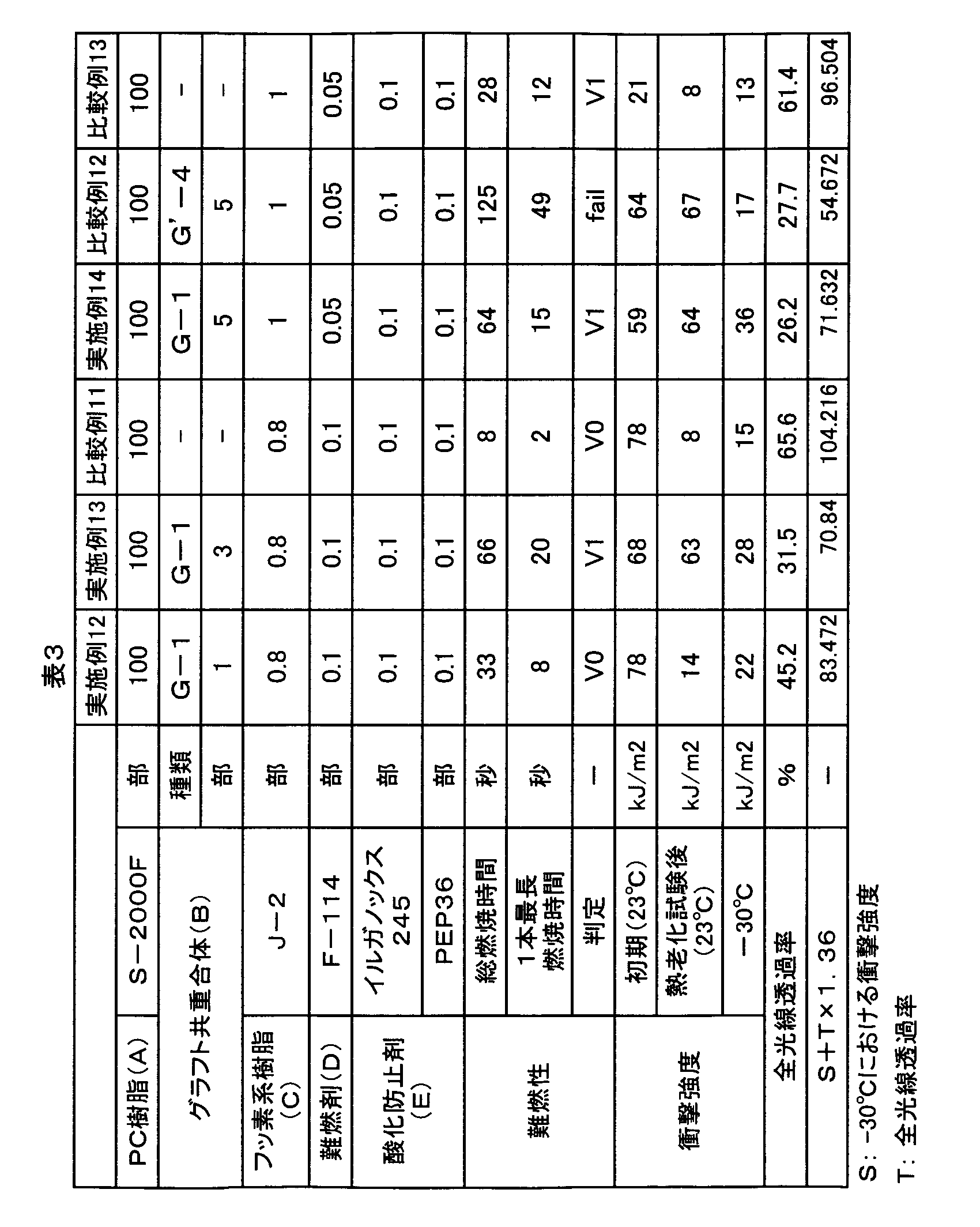
製造例1において得たポリオルガノシロキサンゴムのラテックス(S-1)33.56部(ポリマー換算で10.0部)を、容量5リットルのセパラブルフラスコ内に採取し、蒸留水200部を添加し混合した。次いでこのセパラブルフラスコ内に、n-ブチルアクリレート(n-BA)59.1部、アリルメタクリレート(AMA)0.9部、t-ブチルハイドロパーオキサイド(t-BH)0.24部の混合物を添加した。
実施例21において、用いた各原料の種類及び量を表4に示す条件に変更したこと以外は実施例21と同様にして、ポリオルガノシロキサン含有グラフト共重合体(G-22~26、G-33、G’-21、G’-22)の粉体を製造した。各評価結果を表5に示す。
製造例1において得たラテックス(S-1)33.56部(ポリマー換算で10.0部)を容量5リットルのセパラブルフラスコ内に採取し、蒸留水200部を添加混合した。次いでこのセパラブルフラスコ内に、n-BAを59.1部、及びAMAを0.9部の混合物を添加した。
実施例27において、用いた各原料の種類及び量を表4に示す条件に変更したこと以外は実施例27と同様にして、ポリオルガノシロキサン含有グラフト共重合体(G-28~32)の粉体を製造した。各評価結果を表5に示す。
実施例21~33のグラフト共重合体の粉体は、複合ゴムの製造時に重合開始剤として用いたSFSの量が0.2部より少ないため、酢酸カルシウムで凝固した場合であってもアルカリ土類金属含有量が150ppm以下であった。
ポリオルガノシロキサン含有グラフト共重合体として、グラフト共重合体(G-21~33、G’-21、G’-22)の粉体を用いた。前述の「樹脂組成物及び試験片の作製条件11」、「評価条件12(耐熱分解性)」及び「評価条件13(発色性)」に従って、それぞれ、「耐熱分解性」及び「発色性」を評価した。尚、その際、ペレットは80℃で12時間乾燥した後、射出成形機に供給した。
実施例34~46から明らかなように、アルカリ金属量が20ppm以下かつアルカリ土類金属量が150ppm以下であるグラフト共重合体の粉体を用いて得られるポリカーボネート樹脂組成物は、高温成形時のアウトガスの発生が抑制されており、耐熱分解性に優れていた。
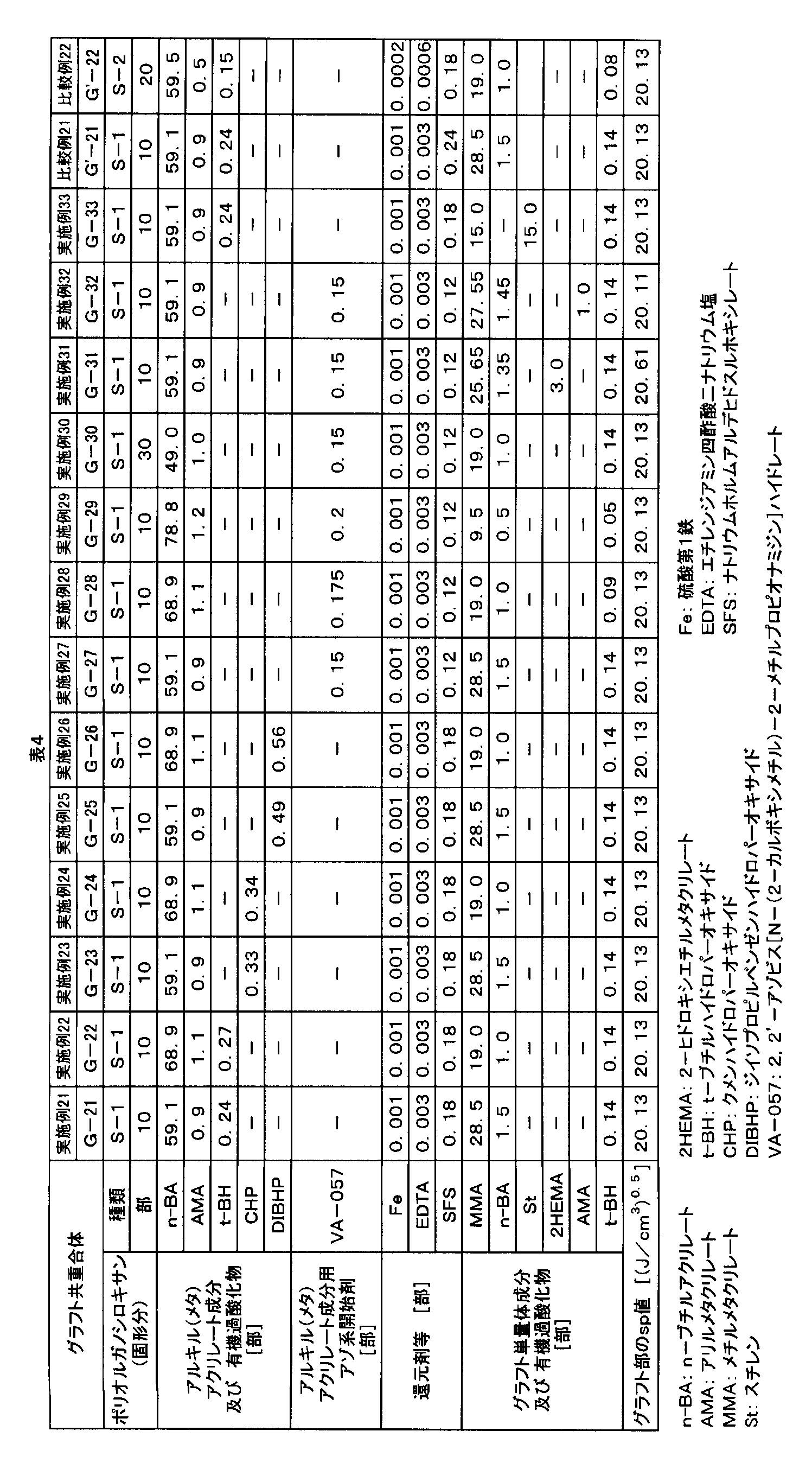
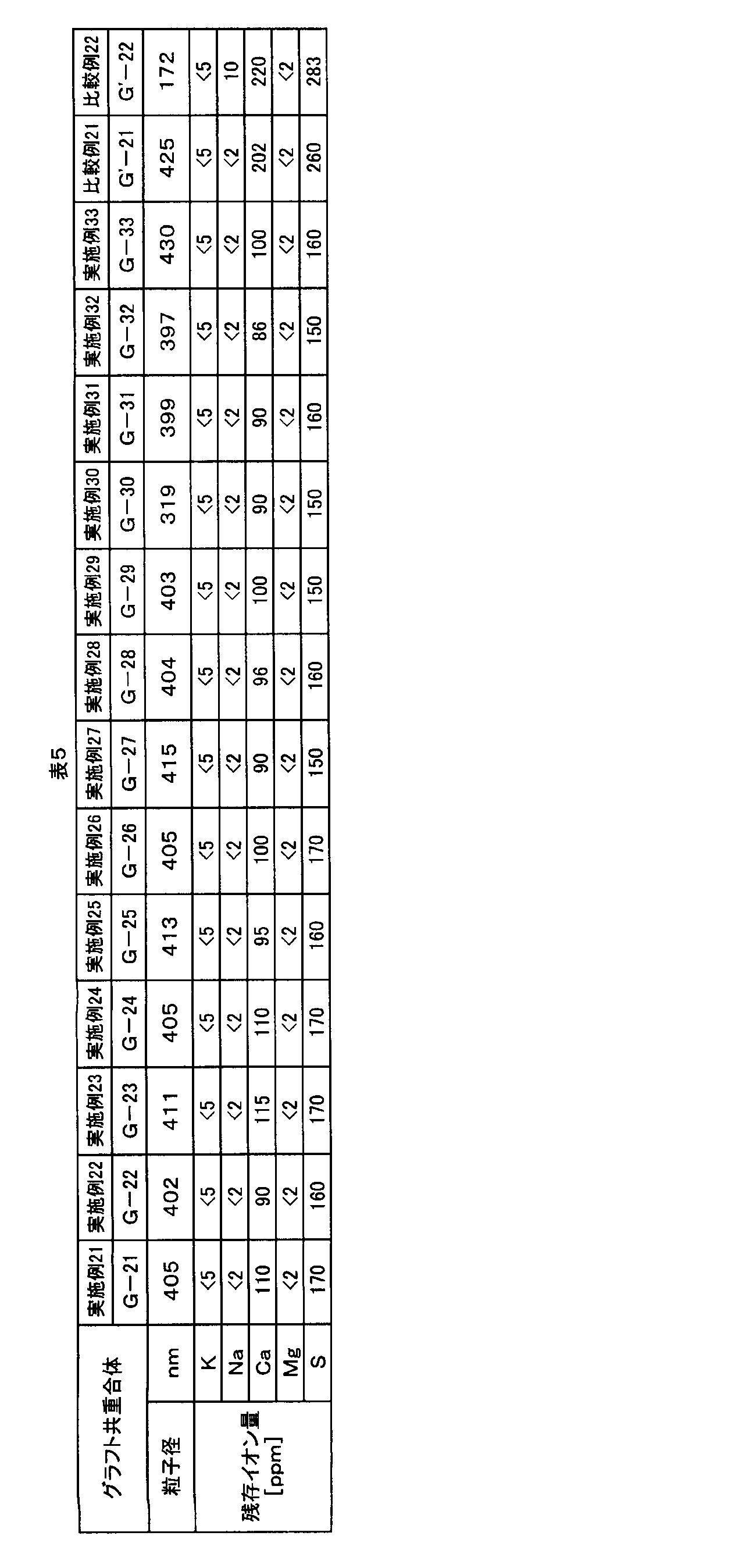

Claims (27)
- アルキル基又は芳香族基を有する(メタ)アクリル酸エステル(b1)と芳香族ビニル単量体(b2)とを含むビニル単量体混合物をポリオルガノシロキサン系ゴムにグラフト重合したポリオルガノシロキサン含有グラフト共重合体(G1)であって、体積平均粒子径が200~2000nmであり、ポリオルガノシロキサンの含有量が0.1~69質量%であるポリオルガノシロキサン含有グラフト共重合体。
- 前記ビニル単量体混合物中に前記芳香族ビニル単量体(b2)が2~95質量%含まれる請求項1に記載のポリオルガノシロキサン含有グラフト共重合体。
- 前記ポリオルガノシロキサン系ゴムが、ポリオルガノシロキサン及びポリアルキル(メタ)アクリレート(PA)を含む複合ゴムである、請求項1に記載のポリオルガノシロキサン含有グラフト共重合体。
- 下記の「作製条件1」で作製された「試験片」について、下記の「評価条件2」で測定されるシャルピー衝撃強度[kJ/m2]、全光線透過率[%]及び難燃性が、以下の性能条件(1)~(3)を満たす、請求項1に記載のポリオルガノシロキサン含有グラフト共重合体:
[ (1)15≦「シャルピー衝撃強度」≦70
(2)60≦「シャルピー衝撃強度」+「全光線透過率」×1.36
(3)1/16インチ厚さにおけるUL94規格垂直燃焼試験による難燃性が「V-1」以上。
<試験片の作製条件1>:
(1)粘度平均分子量22,000の芳香族ポリカーボネート樹脂(三菱エンジニアリングプラスチック(株)製ユーピロンS-2000F):100質量部、
(2)ポリオルガノシロキサン含有グラフト共重合体(G1):5.5質量部、
(3)アクリル変性ポリテトラフルオロエチレン(三菱レイヨン(株)製メタブレンA-3800):0.5質量部、
(4)芳香族リン酸エステル系難燃剤(大八化学工業(株)製PX-200):5.5質量部。
上記の4種類の材料(1)~(4)を配合した樹脂組成物を、バレル温度280℃に加熱した脱揮式押出機((株)池貝製PCM-30)に供給してスクリュー回転数150rpmの条件で混練してペレットを得る。このペレットを100t射出成形機(住友重機(株)製SE-100DU)に供給して、シリンダー温度280℃、金型温度90℃の条件で成形して以下の各試験片を得る。
(i)シャルピーノッチ付き衝撃強さ評価用「試験片1」(長さ80.0mm、幅10.0mm、厚み4mm、Vノッチ付き)。
(ii)全光線透過率評価用「試験片2」(長さ100mm、幅50mm、厚み2mm)。
(iii)1/16インチ厚さの難燃性評価用「試験片3」(長さ127mm、幅12.7mm、厚み1.6mm)。
<評価条件2>:
〔シャルピー衝撃強度[kJ/m2]〕
JIS K 7111‐1/1eAに準拠して温度-30℃にて、「試験片1」のシャルピー衝撃強度を測定する。
〔全光線透過率[%]〕
JIS K 7375に準拠して日本電色工業(株)製HAZE Meter NDH4000を用いて、「試験片2」のD65光源における全光線透過率を測定する。
〔難燃性〕
UL94V試験に準拠した垂直燃焼試験法にて、5つの「試験片3」の総燃焼時間を測定する。]。 - 熱可塑性樹脂(A)、請求項1~4から選ばれる一項に記載のポリオルガノシロキサン含有グラフト共重合体(G1)、フッ素系樹脂(C)及び難燃剤(D)を含有する熱可塑性樹脂組成物。
- 前記熱可塑性樹脂(A)がカーボネート結合、エステル結合、及びアミド結合から選ばれる少なくとも一つの結合を有する熱可塑性樹脂である、請求項5に記載の熱可塑性樹脂組成物。
- 前記熱可塑性樹脂(A)がポリカーボネート系樹脂である、請求項5に記載の熱可塑性樹脂組成物。
- 前記難燃剤(D)がリン系難燃剤及び有機金属塩系難燃剤から選ばれる少なくとも一種の難燃剤を含む、請求項5~7から選ばれる一項に記載の熱可塑性樹脂組成物。
- 請求項5~7から選ばれる一項に記載の熱可塑性樹脂組成物を成形して得られる成形体。
- 請求項8に記載の熱可塑性樹脂組成物を成形して得られる成形体。
- ポリオルガノシロキサン系ゴムに1種以上のビニル単量体をグラフト重合して得られるポリオルガノシロキサン含有グラフト共重合体(G2)の粉体であって、該ポリオルガノシロキサン含有グラフト共重合体(G2)の吸光度法で測定される粒子径が300~2000nmであり、該粉体のアルカリ金属含有量が0~20ppmであり、かつアルカリ土類金属含有量が0~150ppmである粉体。
- 前記粉体の硫黄含有量が0~200ppmである請求項11に記載の粉体。
- 前記ポリオルガノシロキサン系ゴムが、ポリオルガノシロキサン及びポリアルキル(メタ)アクリレート(PA)を含む複合ゴムである請求項11に記載の粉体。
- 前記1種以上のビニル単量体が、芳香族ビニル単量体を含有する単量体混合物である請求項11~13から選ばれる一項に記載の粉体。
- 前記ポリオルガノシロキサン含有グラフト共重合体(G2)のグラフト部のFedors法にて算出したsp値が、20.15~21.00である請求項11~13から選ばれる一項に記載の粉体。
- 前記1種以上のビニル単量体が、架橋性単量体を含有する単量体混合物である請求項11~13から選ばれる一項に記載の粉体。
- ポリオルガノシロキサン系ゴムに1種以上のビニル単量体をグラフト重合して得られるポリオルガノシロキサン含有グラフト共重合体(G2)の粉体であって、該1種以上のビニル単量体が、芳香族ビニル単量体を含有する単量体混合物であり、該粉体のアルカリ金属含有量が0~20ppmであり、かつアルカリ土類金属含有量が0~150ppmである粉体。
- 前記粉体の硫黄含有量が0~200ppmである請求項17に記載の粉体。
- 下記の「作製条件11」で使用される樹脂組成物について、下記の「評価条件12」で測定されるシルバー発生本数が0本である、請求項11または17に記載の粉体:
[ <樹脂組成物及び試験片の作製条件11>;
(1)ポリオルガノシロキサン含有グラフト共重合体(G2)の紛体:4質量部、
(2)粘度平均分子量22,000の芳香族ポリカーボネート樹脂(三菱エンジニアリングプラスチック(株)製ユーピロンS-2000F):96質量部、
(3)Irganox1076(BASF製):0.1質量部、
(4)アデカスタブ2112((株)ADEKA製):0.1質量部、
(5)カーボンブラック#960(三菱化学(株)製):0.1質量部
上記の5種類の材料(1)~(5)を配合し、バレル温度280℃に加熱した脱揮式押出機((株)池貝製PCM-30)に供給してスクリュー回転数150rpmの条件で混練して樹脂組成物のペレットを得る。次いで、100t射出成形機(住友重機械(株)製SE-100DU)とホットランナーを有する金型(試験片サイズ:縦100mm×横100mm×厚み2mm、ピンゲート)を用い、シリンダー温度310℃、ランナー温度310℃、金型温度が90℃の条件で前記ペレットを成形して1ショット目の射出成形体を作製する。前記射出成形機による1ショット成形後、樹脂組成物を射出成形機内に6分間滞留させた後に更に1ショット成形して、「試験片11」を得る。
<評価条件12>:
「試験片11」のゲート付近に発生するシルバーストリークの本数を目視で確認する。]。 - 前記作製条件11で使用される樹脂組成物について、下記の「評価条件13」で測定されるL*が20以下である、請求項19に記載の粉体:
[ <評価条件13>:
前記樹脂組成物のペレットを100t射出成形機(住友重機(株)製SE-100DU)にて、シリンダー温度310℃、金型温度90℃の条件で成形して「試験片13」(長さ100mm、幅50mm、厚み2mm)を得る。
JIS Z 8729(L*a*b* 表色系による物体色の表示方法)により、測定は下記のようにJISZ8722に準じて、日本電色工業(株)製分光式色差計SE-2000を用いて、「試験片13」の物体色を測定する。
装置:分光式色差計SE-2000(日本電色工業株式会社製、0-45°後分光方式)、
測定範囲:380~780nm、
測定光源:C光(2°視野)、
三刺激値(XYZ)からCIE色差式を用いてL*値を算出する。]。 - 請求項11~13、17及び18から選ばれる一項に記載の粉体及び樹脂を含む樹脂組成物。
- 前記樹脂組成物100質量%中、前記ポリオルガノシロキサン含有グラフト共重合体(G2)の紛体の含有量が0.5~90質量%である、請求項21に記載の樹脂組成物。
- 前記樹脂が熱可塑性樹脂である請求項21または22に記載の樹脂組成物。
- 前記熱可塑性樹脂が芳香族ポリカーボネート樹脂である請求項23に記載の樹脂組成物。
- 請求項21に記載の樹脂組成物を成形して得られる成形体。
- 請求項22または24に記載の樹脂組成物を成形して得られる成形体。
- 請求項23に記載の樹脂組成物を成形して得られる成形体。
Priority Applications (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR1020157011297A KR101768349B1 (ko) | 2012-10-31 | 2013-06-28 | 폴리오르가노실록산 함유 그래프트 공중합체, 수지 조성물 및 성형체 |
| EP13755215.4A EP2915828B1 (en) | 2012-10-31 | 2013-06-28 | Polyorganosiloxane-containing graft copolymer, resin composition, and molded article |
| JP2013530479A JP6191458B2 (ja) | 2012-10-31 | 2013-06-28 | ポリオルガノシロキサン含有グラフト共重合体、樹脂組成物及び成形体 |
| CN201380057340.1A CN104755516B (zh) | 2012-10-31 | 2013-06-28 | 含聚有机硅氧烷的接枝共聚物、树脂组合物以及成形体 |
| US14/439,745 US9527997B2 (en) | 2012-10-31 | 2013-06-28 | Polyorganosiloxane-containing graft copolymer, resin composition, and molded article |
| KR1020167030877A KR101824832B1 (ko) | 2012-10-31 | 2013-06-28 | 폴리오르가노실록산 함유 그래프트 공중합체, 수지 조성물 및 성형체 |
| US15/220,808 US9803082B2 (en) | 2012-10-31 | 2016-07-27 | Polyorganosiloxane-containing graft copolymer, resin composition, and molded article |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012240403 | 2012-10-31 | ||
| JP2012-240403 | 2012-10-31 |
Related Child Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US14/439,745 A-371-Of-International US9527997B2 (en) | 2012-10-31 | 2013-06-28 | Polyorganosiloxane-containing graft copolymer, resin composition, and molded article |
| US15/220,808 Division US9803082B2 (en) | 2012-10-31 | 2016-07-27 | Polyorganosiloxane-containing graft copolymer, resin composition, and molded article |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| WO2013129709A2 true WO2013129709A2 (ja) | 2013-09-06 |
| WO2013129709A3 WO2013129709A3 (ja) | 2013-11-14 |
Family
ID=49083409
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2013/067938 Ceased WO2013129709A2 (ja) | 2012-10-31 | 2013-06-28 | ポリオルガノシロキサン含有グラフト共重合体、樹脂組成物及び成形体 |
Country Status (7)
| Country | Link |
|---|---|
| US (2) | US9527997B2 (ja) |
| EP (2) | EP2915828B1 (ja) |
| JP (2) | JP6191458B2 (ja) |
| KR (2) | KR101768349B1 (ja) |
| CN (2) | CN104755516B (ja) |
| TW (1) | TWI598367B (ja) |
| WO (1) | WO2013129709A2 (ja) |
Cited By (14)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2016035021A (ja) * | 2014-08-04 | 2016-03-17 | 三菱エンジニアリングプラスチックス株式会社 | ポリカーボネート樹脂組成物および成形品 |
| JP2017088738A (ja) * | 2015-11-10 | 2017-05-25 | 三菱レイヨン株式会社 | ポリカーボネート樹脂組成物、成形体 |
| JP2017088737A (ja) * | 2015-11-10 | 2017-05-25 | 三菱レイヨン株式会社 | ポリカーボネート樹脂組成物、成形体 |
| WO2017086210A1 (ja) * | 2015-11-20 | 2017-05-26 | 出光興産株式会社 | ポリカーボネート樹脂組成物及びその成形品 |
| CN107474540A (zh) * | 2017-08-17 | 2017-12-15 | 浙江久运汽车零部件有限公司 | 一种改性耐高温空滤器硅胶进气软管 |
| JP2018002978A (ja) * | 2016-07-08 | 2018-01-11 | 三菱エンジニアリングプラスチックス株式会社 | ポリカーボネート樹脂組成物 |
| WO2018040297A1 (zh) * | 2016-08-31 | 2018-03-08 | 广州熵能创新材料股份有限公司 | 乙烯基系接枝共聚物和含有其的树脂组合物、及乙烯基系接枝共聚物的制备方法 |
| KR20180084052A (ko) * | 2015-11-20 | 2018-07-24 | 이데미쓰 고산 가부시키가이샤 | 폴리카보네이트 수지 펠릿의 제조 방법 |
| WO2018212062A1 (ja) | 2017-05-17 | 2018-11-22 | 三菱ケミカル株式会社 | ポリオルガノシロキサン含有ゴムラテックス及びその製造方法、ポリオルガノシロキサン含有ゴムグラフト重合体、粉体、樹脂組成物並びに成形体 |
| CN109897141A (zh) * | 2019-04-08 | 2019-06-18 | 淮安市博彦土木工程科学研究院有限公司 | 一种建材用高强度asa树脂的制备方法 |
| JP2019189697A (ja) * | 2018-04-20 | 2019-10-31 | 三菱エンジニアリングプラスチックス株式会社 | ポリカーボネート樹脂組成物および成形品 |
| WO2022210483A1 (ja) * | 2021-03-30 | 2022-10-06 | 三菱ケミカル株式会社 | 樹脂組成物、及び成形体 |
| CN115867611A (zh) * | 2020-06-12 | 2023-03-28 | 三菱化学株式会社 | 含聚有机硅氧烷的聚合物粒子群、组合物、树脂组合物及成形体 |
| CN120699402A (zh) * | 2025-08-18 | 2025-09-26 | 湖南省长城铭泰新材料科技有限公司 | 一种高阻隔抑菌包装材料及其制备方法和包装袋 |
Families Citing this family (25)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2716692A1 (de) * | 2012-10-08 | 2014-04-09 | WKP Products SA | Verbundwerkstoffe zur Nutzung in Spritzguss-Verfahren |
| CN110709437B (zh) | 2017-06-06 | 2022-07-05 | 三菱化学株式会社 | 含聚有机硅氧烷的接枝共聚物、热塑性树脂组合物及成形体 |
| EP4234630A3 (en) * | 2017-10-04 | 2023-10-04 | Mitsubishi Chemical Corporation | Rubber-containing graft polymer, resin composition containing rubber-containing graft polymer, and shaped article thereof |
| US10738213B2 (en) * | 2017-10-17 | 2020-08-11 | Ppg Industries Ohio, Inc. | Modified silicone coating composition |
| JP7044115B2 (ja) * | 2017-11-27 | 2022-03-30 | 三菱ケミカル株式会社 | ゴム含有グラフト重合体組成物を含む熱可塑性樹脂の樹脂改質用組成物、ゴム含有グラフト重合体含有樹脂組成物およびその成形体 |
| CN108194158B (zh) * | 2017-12-19 | 2024-02-09 | 玉环捷宇机械制造有限公司 | 一种凸轮组件及加工工艺以及应用该凸轮组件的配气机构 |
| WO2020020869A1 (en) * | 2018-07-24 | 2020-01-30 | Ineos Styrolution Group Gmbh | Process for preparing graft rubber compositions with improved dewatering |
| CN113227249B (zh) | 2018-12-27 | 2024-03-15 | 株式会社钟化 | 树脂组合物及其应用 |
| CN114341233B (zh) * | 2019-09-27 | 2025-02-07 | 株式会社钟化 | 粉粒体及粉粒体的制造方法 |
| EP4502080A3 (en) * | 2020-10-23 | 2025-04-30 | Mitsubishi Chemical Corporation | Resin composition and molded article thereof |
| WO2022186248A1 (ja) * | 2021-03-05 | 2022-09-09 | 株式会社カネカ | 樹脂組成物、重合体微粒子の製造方法および樹脂組成物の製造方法 |
| CN115043991B (zh) * | 2021-03-08 | 2023-07-25 | 中国石油天然气股份有限公司 | 一种乳液聚合法制备含氟热塑性弹性体的方法及制得的热塑性弹性体 |
| CN115043993B (zh) * | 2021-03-08 | 2023-06-30 | 中国石油天然气股份有限公司 | 一种乳液聚合法制备含羧基热塑性树脂的方法及制得的含羧基热塑性树脂 |
| CN115043997B (zh) * | 2021-03-08 | 2023-07-25 | 中国石油天然气股份有限公司 | 一种乳液聚合法制备含羧基热塑性弹性体的方法及制得的热塑性弹性体 |
| CN115043992B (zh) * | 2021-03-08 | 2023-06-30 | 中国石油天然气股份有限公司 | 一种提高了硬度的热塑性弹性体及其制备方法 |
| CN115043994B (zh) * | 2021-03-08 | 2023-07-25 | 中国石油天然气股份有限公司 | 一种提高耐热性的热塑性树脂的乳液聚合制备方法及得到的热塑性树脂 |
| CN115043995B (zh) * | 2021-03-08 | 2023-06-30 | 中国石油天然气股份有限公司 | 一种乳液聚合法制备热塑性树脂的方法及制得的热塑性树脂 |
| CN115043996B (zh) * | 2021-03-08 | 2023-07-25 | 中国石油天然气股份有限公司 | 一种乳液聚合法制备高耐候性热塑性树脂的方法及制得的含硅热塑性树脂 |
| CN115043980B (zh) * | 2021-03-08 | 2023-06-30 | 中国石油天然气股份有限公司 | 一种乳液聚合法制备耐寒性热塑性弹性体的方法及制得的含硅热塑性弹性体 |
| CN115028782B (zh) * | 2021-03-08 | 2023-07-25 | 中国石油天然气股份有限公司 | 一种乳液聚合法制备含氟高耐候性热塑性树脂的方法及制得的热塑性树脂 |
| WO2022210417A1 (ja) * | 2021-03-31 | 2022-10-06 | テクノUmg株式会社 | 熱可塑性樹脂組成物、成形品 |
| CN113278115B (zh) * | 2021-05-27 | 2022-10-04 | 英德侗富贵科技材料有限公司 | 一种核壳聚合物及其制备方法和应用 |
| CN117050316A (zh) * | 2023-08-02 | 2023-11-14 | 合肥博发新材料科技有限公司 | 含磷阻燃抑烟硅油及制备与其在阻燃抑烟技术中的应用 |
| CN121895731A (zh) * | 2024-10-21 | 2026-04-21 | 柯尼卡美能达株式会社 | 再生树脂组合物、树脂成型品、树脂壳体及电子设备 |
| CN121061992B (zh) * | 2025-11-05 | 2026-02-03 | 吉林省林业科学研究院(吉林省林业生物防治中心站) | 一种阻燃高强度纤维板及其制备方法 |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2003113299A (ja) | 2001-10-02 | 2003-04-18 | Mitsubishi Rayon Co Ltd | 熱可塑性樹脂組成物 |
| JP2004331726A (ja) | 2003-05-01 | 2004-11-25 | Mitsubishi Rayon Co Ltd | グラフト共重合体およびこれを用いた熱可塑性樹脂組成物 |
| JP2004346271A (ja) | 2003-05-26 | 2004-12-09 | Mitsubishi Rayon Co Ltd | シリコーン/アクリル複合ゴム系グラフト共重合体および熱可塑性樹脂組成物 |
| JP2011063706A (ja) | 2009-09-17 | 2011-03-31 | Mitsubishi Rayon Co Ltd | グラフト共重合体及びその製造方法、難燃剤、熱可塑性樹脂組成物、並びに成形体 |
Family Cites Families (26)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5087662A (en) * | 1988-11-14 | 1992-02-11 | General Electric Company | Polyester, polycarbonate and/or polyphenylene ether with polyorganosiloxane/polyvinyl-based graft (meth) acrylate polymers |
| EP0433906B1 (en) * | 1989-12-18 | 1995-07-26 | Toshiba Silicone Co., Ltd. | Thermoplastic resin and process for producing the same |
| US6545089B1 (en) * | 1997-09-04 | 2003-04-08 | General Electric Company | Impact modified carbonnate polymer composition having improved resistance to degradation and improved thermal stability |
| JP2000044669A (ja) * | 1998-08-04 | 2000-02-15 | Teijin Ltd | 芳香族ポリカーボネートの製造方法および真空捕集系 |
| JP4702998B2 (ja) * | 2000-12-05 | 2011-06-15 | 株式会社カネカ | ゴム変性樹脂およびそれを含有する熱可塑性樹脂組成物 |
| JP4587606B2 (ja) * | 2001-06-27 | 2010-11-24 | 株式会社カネカ | ポリカーボネート系難燃性樹脂組成物 |
| KR100851266B1 (ko) * | 2001-07-05 | 2008-08-08 | 카네카 코포레이션 | 난연성 열가소성 수지 조성물 |
| JP2003089749A (ja) * | 2001-09-18 | 2003-03-28 | Kanegafuchi Chem Ind Co Ltd | 難燃性ポリカーボネート樹脂組成物 |
| JP3865295B2 (ja) * | 2001-10-11 | 2007-01-10 | 旭化成ケミカルズ株式会社 | 難燃性樹脂組成物 |
| TWI317749B (en) * | 2002-02-15 | 2009-12-01 | Kaneka Corp | Graft copolymers and impact-resistant flame-retardant resin compositions containing the same |
| JP3884661B2 (ja) * | 2002-02-15 | 2007-02-21 | 株式会社カネカ | グラフト共重合体及びそれを含有する難燃性樹脂組成物 |
| JP2003238793A (ja) * | 2002-02-19 | 2003-08-27 | Mitsubishi Rayon Co Ltd | 熱可塑性樹脂組成物 |
| JP4015439B2 (ja) | 2002-02-25 | 2007-11-28 | 三菱レイヨン株式会社 | 熱可塑性樹脂組成物 |
| EP1699874B1 (en) * | 2003-11-26 | 2010-03-10 | Dow Corning Corporation | Silicone polymer and organic polymer containing alloy and/or hybrid emulsion compositions |
| JP2008189860A (ja) | 2007-02-07 | 2008-08-21 | Gantsu Kasei Kk | シリコーンゴムグラフト共重合体およびその製造法 |
| JP5248029B2 (ja) * | 2007-03-27 | 2013-07-31 | 三菱レイヨン株式会社 | グラフト共重合体及び樹脂組成物 |
| JP5306929B2 (ja) * | 2008-07-23 | 2013-10-02 | 三菱エンジニアリングプラスチックス株式会社 | 難燃性熱可塑性ポリエステル樹脂組成物 |
| EP2338929B8 (en) * | 2008-08-29 | 2015-01-07 | Mitsubishi Rayon Co., Ltd. | Silicone-polymer-containing vinyl polymer powder and manufacturing method thereof, resin composition, and compact |
| JP5000613B2 (ja) * | 2008-09-30 | 2012-08-15 | 株式会社日本触媒 | 有機粒子含有組成物 |
| KR101065337B1 (ko) * | 2008-12-19 | 2011-09-16 | 제일모직주식회사 | 내광성 및 난연성이 우수한 폴리카보네이트 수지 조성물 |
| JP5634031B2 (ja) * | 2009-03-27 | 2014-12-03 | 株式会社日本触媒 | 重合体粒子およびそれを用いた重合体粒子含有組成物 |
| EP2256146A1 (de) * | 2009-05-30 | 2010-12-01 | Bayer MaterialScience AG | Polycarbonate mit extrem hoher Reinheit und guter Eigenfarbe und thermischer Beständigkeit sowie eine Vorrichtung und ein Verfahren zu ihrer Herstellung |
| JP5450316B2 (ja) * | 2010-08-24 | 2014-03-26 | 株式会社日本触媒 | 重合体粒子およびそれを用いた重合体粒子含有組成物 |
| JP4930629B2 (ja) * | 2010-08-27 | 2012-05-16 | 横浜ゴム株式会社 | 熱可塑性樹脂組成物 |
| JP2012087296A (ja) * | 2010-09-22 | 2012-05-10 | Toray Ind Inc | 熱可塑性樹脂組成物およびその成形品 |
| JP5848699B2 (ja) * | 2011-02-09 | 2016-01-27 | 三菱レイヨン株式会社 | ポリオルガノシロキサンラテックス、それを用いたグラフト共重合体、熱可塑性樹脂組成物、及び成形体 |
-
2013
- 2013-06-28 WO PCT/JP2013/067938 patent/WO2013129709A2/ja not_active Ceased
- 2013-06-28 JP JP2013530479A patent/JP6191458B2/ja active Active
- 2013-06-28 KR KR1020157011297A patent/KR101768349B1/ko active Active
- 2013-06-28 US US14/439,745 patent/US9527997B2/en active Active
- 2013-06-28 EP EP13755215.4A patent/EP2915828B1/en active Active
- 2013-06-28 EP EP16172835.7A patent/EP3085715A1/en not_active Withdrawn
- 2013-06-28 CN CN201380057340.1A patent/CN104755516B/zh active Active
- 2013-06-28 KR KR1020167030877A patent/KR101824832B1/ko active Active
- 2013-06-28 CN CN201710562109.8A patent/CN107286300B/zh active Active
- 2013-07-04 TW TW102124058A patent/TWI598367B/zh active
-
2016
- 2016-06-16 JP JP2016119796A patent/JP6191735B2/ja active Active
- 2016-07-27 US US15/220,808 patent/US9803082B2/en active Active
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2003113299A (ja) | 2001-10-02 | 2003-04-18 | Mitsubishi Rayon Co Ltd | 熱可塑性樹脂組成物 |
| JP2004331726A (ja) | 2003-05-01 | 2004-11-25 | Mitsubishi Rayon Co Ltd | グラフト共重合体およびこれを用いた熱可塑性樹脂組成物 |
| JP2004346271A (ja) | 2003-05-26 | 2004-12-09 | Mitsubishi Rayon Co Ltd | シリコーン/アクリル複合ゴム系グラフト共重合体および熱可塑性樹脂組成物 |
| JP2011063706A (ja) | 2009-09-17 | 2011-03-31 | Mitsubishi Rayon Co Ltd | グラフト共重合体及びその製造方法、難燃剤、熱可塑性樹脂組成物、並びに成形体 |
Non-Patent Citations (2)
| Title |
|---|
| "Polymer Handbook", vol. 1, WILEY-INTERSCIENCE |
| See also references of EP2915828A4 |
Cited By (20)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2016035021A (ja) * | 2014-08-04 | 2016-03-17 | 三菱エンジニアリングプラスチックス株式会社 | ポリカーボネート樹脂組成物および成形品 |
| JP2017088738A (ja) * | 2015-11-10 | 2017-05-25 | 三菱レイヨン株式会社 | ポリカーボネート樹脂組成物、成形体 |
| JP2017088737A (ja) * | 2015-11-10 | 2017-05-25 | 三菱レイヨン株式会社 | ポリカーボネート樹脂組成物、成形体 |
| US11208552B2 (en) | 2015-11-20 | 2021-12-28 | Idemitsu Kosan Co., Ltd. | Method of manufacturing polycarbonate resin pellets |
| WO2017086210A1 (ja) * | 2015-11-20 | 2017-05-26 | 出光興産株式会社 | ポリカーボネート樹脂組成物及びその成形品 |
| KR102888537B1 (ko) | 2015-11-20 | 2025-11-19 | 이데미쓰 고산 가부시키가이샤 | 폴리카보네이트 수지 펠릿의 제조 방법 |
| KR20180084052A (ko) * | 2015-11-20 | 2018-07-24 | 이데미쓰 고산 가부시키가이샤 | 폴리카보네이트 수지 펠릿의 제조 방법 |
| JPWO2017086209A1 (ja) * | 2015-11-20 | 2018-09-06 | 出光興産株式会社 | ポリカーボネート樹脂ペレットの製造方法 |
| JPWO2017086210A1 (ja) * | 2015-11-20 | 2018-09-06 | 出光興産株式会社 | ポリカーボネート樹脂組成物及びその成形品 |
| JP2018002978A (ja) * | 2016-07-08 | 2018-01-11 | 三菱エンジニアリングプラスチックス株式会社 | ポリカーボネート樹脂組成物 |
| WO2018040297A1 (zh) * | 2016-08-31 | 2018-03-08 | 广州熵能创新材料股份有限公司 | 乙烯基系接枝共聚物和含有其的树脂组合物、及乙烯基系接枝共聚物的制备方法 |
| WO2018212062A1 (ja) | 2017-05-17 | 2018-11-22 | 三菱ケミカル株式会社 | ポリオルガノシロキサン含有ゴムラテックス及びその製造方法、ポリオルガノシロキサン含有ゴムグラフト重合体、粉体、樹脂組成物並びに成形体 |
| CN107474540A (zh) * | 2017-08-17 | 2017-12-15 | 浙江久运汽车零部件有限公司 | 一种改性耐高温空滤器硅胶进气软管 |
| JP2019189697A (ja) * | 2018-04-20 | 2019-10-31 | 三菱エンジニアリングプラスチックス株式会社 | ポリカーボネート樹脂組成物および成形品 |
| CN109897141B (zh) * | 2019-04-08 | 2021-03-02 | 淮安市博彦土木工程科学研究院有限公司 | 一种建材用高强度asa树脂的制备方法 |
| CN109897141A (zh) * | 2019-04-08 | 2019-06-18 | 淮安市博彦土木工程科学研究院有限公司 | 一种建材用高强度asa树脂的制备方法 |
| CN115867611A (zh) * | 2020-06-12 | 2023-03-28 | 三菱化学株式会社 | 含聚有机硅氧烷的聚合物粒子群、组合物、树脂组合物及成形体 |
| WO2022210483A1 (ja) * | 2021-03-30 | 2022-10-06 | 三菱ケミカル株式会社 | 樹脂組成物、及び成形体 |
| JPWO2022210483A1 (ja) * | 2021-03-30 | 2022-10-06 | ||
| CN120699402A (zh) * | 2025-08-18 | 2025-09-26 | 湖南省长城铭泰新材料科技有限公司 | 一种高阻隔抑菌包装材料及其制备方法和包装袋 |
Also Published As
| Publication number | Publication date |
|---|---|
| US9803082B2 (en) | 2017-10-31 |
| US20160333130A1 (en) | 2016-11-17 |
| JP6191735B2 (ja) | 2017-09-06 |
| EP2915828A2 (en) | 2015-09-09 |
| KR20160130533A (ko) | 2016-11-11 |
| JPWO2013129709A1 (ja) | 2015-12-10 |
| CN104755516A (zh) | 2015-07-01 |
| CN104755516B (zh) | 2020-01-24 |
| KR20150067248A (ko) | 2015-06-17 |
| TWI598367B (zh) | 2017-09-11 |
| KR101768349B1 (ko) | 2017-08-14 |
| EP2915828A4 (en) | 2015-11-04 |
| CN107286300B (zh) | 2021-09-28 |
| CN107286300A (zh) | 2017-10-24 |
| EP2915828B1 (en) | 2024-10-09 |
| TW201408707A (zh) | 2014-03-01 |
| US20150267048A1 (en) | 2015-09-24 |
| EP3085715A1 (en) | 2016-10-26 |
| JP6191458B2 (ja) | 2017-09-06 |
| WO2013129709A3 (ja) | 2013-11-14 |
| US9527997B2 (en) | 2016-12-27 |
| JP2016164283A (ja) | 2016-09-08 |
| KR101824832B1 (ko) | 2018-02-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP6191735B2 (ja) | 粉体、樹脂組成物及び成形体 | |
| JP6221744B2 (ja) | ポリオルガノシロキサン含有グラフト共重合体、樹脂組成物、成形体、摺動性改良剤及び摺動部材 | |
| JP6769547B2 (ja) | ポリオルガノシロキサン含有ゴムグラフト重合体 | |
| JP5765076B2 (ja) | 熱可塑性樹脂組成物 | |
| JP2011063706A (ja) | グラフト共重合体及びその製造方法、難燃剤、熱可塑性樹脂組成物、並びに成形体 | |
| JP5473496B2 (ja) | グラフト共重合体及びその製造方法、熱可塑性樹脂組成物、並びに成形品 | |
| JP6465079B2 (ja) | 摺動部材用熱可塑性樹脂組成物及び摺動部材 | |
| JP5379667B2 (ja) | 熱可塑性樹脂組成物及び成形体 | |
| JP6467909B2 (ja) | ポリカーボネート樹脂組成物及び成形体 | |
| JP5896246B2 (ja) | グラフト共重合体及びその製造方法、難燃剤、熱可塑性樹脂組成物、並びに成形体 | |
| JP2011012241A (ja) | グラフト共重合体及びその製造方法、熱可塑性樹脂組成物、並びに成形物 | |
| JP6365116B2 (ja) | 熱可塑性樹脂組成物及びその成形体。 | |
| TW201506048A (zh) | 含有聚有機矽氧烷的接枝共聚合物、樹脂組成物、成形體、滑動性改良劑及滑動構件 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| ENP | Entry into the national phase |
Ref document number: 2013530479 Country of ref document: JP Kind code of ref document: A |
|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 13755215 Country of ref document: EP Kind code of ref document: A2 |
|
| ENP | Entry into the national phase |
Ref document number: 20157011297 Country of ref document: KR Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14439745 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2013755215 Country of ref document: EP |