WO2013150902A1 - Procédé de production d'un oligodésoxynucléotide à terminaison 5' phosphorylée par clivage d'un oligodésoxynucléotide contenant un nucléotide non naturel - Google Patents
Procédé de production d'un oligodésoxynucléotide à terminaison 5' phosphorylée par clivage d'un oligodésoxynucléotide contenant un nucléotide non naturel Download PDFInfo
- Publication number
- WO2013150902A1 WO2013150902A1 PCT/JP2013/058349 JP2013058349W WO2013150902A1 WO 2013150902 A1 WO2013150902 A1 WO 2013150902A1 JP 2013058349 W JP2013058349 W JP 2013058349W WO 2013150902 A1 WO2013150902 A1 WO 2013150902A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- dna
- reaction
- substituted
- oligodeoxynucleotide
- unsubstituted
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 *C#CC(C(N)=N1)=CN(C(C2)OC(CO)C2O)C1=O Chemical compound *C#CC(C(N)=N1)=CN(C(C2)OC(CO)C2O)C1=O 0.000 description 5
Images



























































Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/06—Pyrimidine radicals
- C07H19/073—Pyrimidine radicals with 2-deoxyribosyl as the saccharide radical
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07H—SUGARS; DERIVATIVES THEREOF; NUCLEOSIDES; NUCLEOTIDES; NUCLEIC ACIDS
- C07H19/00—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof
- C07H19/02—Compounds containing a hetero ring sharing one ring hetero atom with a saccharide radical; Nucleosides; Mononucleotides; Anhydro-derivatives thereof sharing nitrogen
- C07H19/04—Heterocyclic radicals containing only nitrogen atoms as ring hetero atom
- C07H19/16—Purine radicals
- C07H19/173—Purine radicals with 2-deoxyribosyl as the saccharide radical
Definitions
- the present invention relates to a method for producing a 5'-terminal phosphorylated oligodeoxynucleotide by cleaving an oligodeoxynucleotide containing an unnatural base.
- DNA deoxyribonucleic acid
- a method using a restriction enzyme is generally used as a technique for cleaving DNA
- a method using a DNA ligase is generally used as a technique for ligating DNA.
- a DNA fragment having a protruding end is prepared using a type II restriction enzyme (Non-Patent Documents 1 and 2), and another DNA fragment having a protruding end complementary to the protruding end is prepared.
- DNA fragments can be ligated using a DNA ligation enzyme.
- the above method has some problems.
- the first and second problems cause a decrease in the efficiency of ligation between DNA fragments by the DNA ligation enzyme, and the third problem is a new problem formed as a result of ligating DNA fragments. It becomes a big restriction of the base sequence in DNA.
- Patent Documents 1 and 2 and Non-Patent Documents 3 to 5 were amplified after the DNA was amplified by polymerase chain reaction (PCR) or the like using a DNA primer into which uridine (ribonucleotide) was introduced as an unnatural base. A method for cleaving DNA at the position of uridine (ribonucleotide) is described.
- Non-Patent Documents 6 to 12 show that after amplifying DNA by PCR or the like using a DNA primer into which 2′-deoxyuridine is introduced as an unnatural base, the amplified DNA is placed at the position of 2′-deoxyuridine. Describes how to cut.
- the method described in the above document uses an enzyme such as RNase or uracil-removing enzyme for cleaving DNA. Therefore, it is disadvantageous in terms of cost.
- an enzymatic cleavage reaction must satisfy special reaction conditions relating to pH and / or metal ion concentration.
- Non-Patent Document 13 describes a method for cleaving DNA via a DNA base elimination reaction known as depletion and the resulting basic site. It is known that depurination is easily caused under acidic conditions, but very difficult to occur under basic conditions (Non-patent Document 14).
- Non-Patent Document 15 describes a deoxynucleotide containing a non-natural base that causes a reaction similar to that of depuration under basic conditions in which the depletion is unlikely to occur with a natural base.
- a complicated operation of oxygen bubbling is required for DNA cleavage.
- non-natural base that is stable under conditions of DNA synthesis by an automatic DNA synthesizer and / or DNA replication conditions such as PCR and functions as a template in a DNA replication reaction in the same manner as natural bases. No deoxynucleotides have been reported.
- DNA oligomer When an oligodeoxynucleotide (hereinafter also referred to as “DNA oligomer” or “oligo DNA”) is artificially synthesized, an automatic DNA synthesizer is used.
- a DNA oligomer synthesized using an automatic DNA synthesizer is usually not phosphorylated at the 5 'end. For this reason, when the synthetic DNA oligomer is ligated to other DNA (DNA derivative) using a DNA ligation enzyme, it is necessary to phosphorylate the 5 'end in advance.
- Methods for phosphorylating the 5 ′ and / or 3 ′ end of DNA include a method of treating a DNA fragment with a polynucleotide kinase (in the case of 5 ′ end phosphorylation), and a method for terminal phosphorylation of synthetic DNA using phosphorylated amidite (Patent Document 3 and Non-Patent Documents 16 and 17).
- the phosphorylated amidite has a coupling efficiency not 100%, and thus phosphorylated DNA and non-phosphorylated DNA are generated.
- Patent Documents 1 and 2 and Non-Patent Documents 3 to 12 known as methods for cleaving DNA use an enzyme such as RNase or uracil-removing enzyme as described above for cleaving DNA. Therefore, it is disadvantageous in terms of cost.
- an enzymatic cleavage reaction must satisfy special reaction conditions relating to pH and / or metal ion concentration.
- the method described in Non-Patent Document 15 is advantageous in terms of cost and reaction efficiency because DNA is cleaved by a chemical reaction without using an enzyme.
- deoxynucleotides containing non-natural bases described in the literature are under conditions for DNA synthesis by an automatic DNA synthesizer compared to deoxynucleotides containing corresponding natural bases (A, T, G or C). And / or low stability under DNA replication conditions such as PCR and low fidelity when used in in vivo and in vitro reactions such as DNA replication reactions.
- a complicated operation of oxygen bubbling is required for DNA cleavage.
- Patent Document 3 and Non-Patent Documents 16 and 17 known as methods for phosphorylating the 5 ′ and / or 3 ′ end of DNA have a coupling efficiency of phosphorylated amidite not 100% as described above.
- the DNA is cleaved into two fragments by a chemical reaction, and the 5 ′ end of the resulting DNA fragment becomes the phosphate end.
- the method of introducing a deoxynucleotide containing a non-natural base into a DNA strand as a cleavage site is advantageous in that it is low-cost, has few restrictions on reaction conditions, and is easy to carry out experimental operations. .
- the present invention introduces deoxynucleotides containing unnatural bases into the DNA strand, thereby cleaving the DNA strand with a low-cost and simple experimental operation by a chemical reaction, and the resulting end is phosphorylated. It is an object of the present invention to provide an oligodeoxynucleotide.
- the present inventors have used the above object by using a deoxynucleotide or deoxynucleoside containing a non-natural base having a moiety removable by a nucleophile.
- the present invention has been completed.
- the gist of the present invention is as follows.
- Nucleophiles and formula I (A) n -BC [Where: A is an oligodeoxynucleotide, B is a deoxynucleoside containing a non-natural base having a moiety E removable by a nucleophile; C is an oligodeoxynucleotide, n is 0 or 1, When n is 1, the 3 ′ end of A and the 5 ′ position of B are linked by a phosphodiester bond, The 3 ′ position of B and the 5 ′ end of C are linked by a phosphodiester bond, The phosphodiester bond between B and C is linked to the oligodeoxynucleotide moiety phosphorylated at the 5 'end of C from the 3' position of B with the elimination of E induced by nucleophile attack.
- deoxynucleoside B containing the unnatural base is:
- R is hydrogen, substituted or unsubstituted alkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted heterocyclyl, substituted or unsubstituted aryl, substituted or unsubstituted arylalkyl, substituted or unsubstituted heteroaryl, Substituted or unsubstituted heteroarylalkyl or carbonyl;
- R ′ is hydrogen or substituted or unsubstituted alkyl, alkenyl or alkynyl; r is an integer from 0 to 4;
- R ′′ is halogen or substituted or unsubstituted alkyl, alkenyl or alkynyl;
- R ′ ′′ is —COZ, —C ⁇ CZ or —CN;
- Z is hydrogen, substituted or unsubstituted alkyl, substituted or unsubstituted cycloalkyl, substituted or unsubsti
- deoxynucleoside B containing a non-natural base having a moiety E removable by a nucleophile or a derivative thereof, and a natural base
- an oligodeoxynucleotide synthesis step of synthesizing an oligodeoxynucleotide represented by formula I using a substrate containing one or more deoxynucleosides or derivatives thereof the method of.
- Formula I (A) n -BC [Where: A is an oligodeoxynucleotide, B is a deoxynucleoside containing a non-natural base having a moiety E removable by a nucleophile; C is an oligodeoxynucleotide, n is 0 or 1, When n is 1, the 3 ′ end of A and the 5 ′ position of B are linked by a phosphodiester bond, The 3 ′ position of B and the 5 ′ end of C are linked by a phosphodiester bond, The phosphodiester bond between B and C is linked to the oligodeoxynucleotide moiety phosphorylated at the 5 'end of C from the 3' position of B with the elimination of E induced by nucleophile attack.
- the 5 ′ end of C contained in the linear double-stranded DNA fragment is phosphorylated, and at least the primer portion represented by Formula I Forming a linear double-stranded DNA fragment having a 3 ′ protruding end containing a sequence complementary to a part thereof, and a linear double-stranded DNA fragment having the 3 ′ protruding end; And a ligation step of ligating another double-stranded DNA fragment; A method for ligating double-stranded DNA.
- A is an oligodeoxynucleotide
- B is a deoxynucleoside containing a non-natural base having a moiety E removable by a nucleophile
- C is an oligodeoxynucleotide
- n is 0 or 1
- n 1
- the 3 ′ end of A and the 5 ′ position of B are linked by a phosphodiester bond
- the 3 ′ position of B and the 5 ′ end of C are linked by a phosphodiester bond
- the phosphodiester bond between B and C is linked to the oligodeoxynucleotide moiety phosphorylated at the 5 'end of C from the 3' position of B with the elimination of E induced by nucleophile attack
- R is hydrogen, substituted or unsubstituted alkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted heterocyclyl, substituted or unsubstituted aryl, substituted or unsubstituted arylalkyl, substituted or unsubstituted heteroaryl, Substituted or unsubstituted heteroarylalkyl or carbonyl;
- R ′ is methyl; r is an integer from 0 to 4;
- R ′′ is halogen or substituted or unsubstituted alkyl, alkenyl or alkynyl;
- R ′ ′′ is —COZ, —C ⁇ CZ or —CN;
- Z is hydrogen, substituted or unsubstituted alkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted heterocyclyl, substituted or unsubstituted aryl, substituted
- the present invention it is possible to cleave a DNA strand by a chemical reaction at a low cost and with a simple experimental operation, and to provide an oligodeoxynucleotide whose resulting end is phosphorylated.
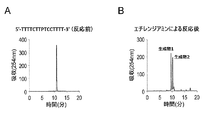
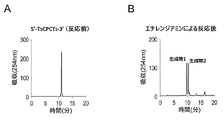
- A DNA oligomer before reaction
- B Reaction mixture after reaction with 2-aminoethanol
- C Enlarged view of the HPLC chromatogram of the reaction mixture after reaction.
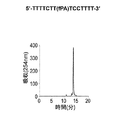
- the usage example 1-7 it is a figure which shows the reverse phase HPLC chromatogram of the DNA oligomer after cutting out and deprotecting.
- the usage example 1-8 it is a figure which shows the reverse phase HPLC chromatogram of the DNA oligomer after cutting-out and a deprotection.
- it is a figure which shows the reverse phase HPLC chromatogram of the DNA oligomer after reaction by ethylenediamine.
- the usage example 1-9 it is a figure which shows the reverse phase HPLC chromatogram of the DNA oligomer after cutting out and deprotecting.
- the usage example 1-10 it is a figure which shows the reverse phase HPLC chromatogram of the DNA oligomer after 120 degreeC processing in a 100 micrometer phosphate buffer.
- the usage example 1-11 it is a figure which shows the reverse phase HPLC chromatogram of the DNA oligomer by which the 5'-terminal after cut-out and deprotection was DMTr protected.
- the usage example 1-11 it is a figure which shows the reverse phase HPLC chromatogram of the DNA oligomer by which the 5'-terminal after the reaction by ethylenediamine was phosphorylated.
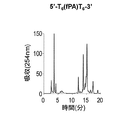
- the usage example 1-12 it is a figure which shows the reverse phase HPLC chromatogram of TT1 and TT2 after cutting and deprotection.
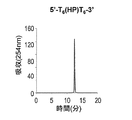
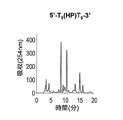
- the synthesis example 2 it is a figure which shows the reverse phase HPLC chromatogram of the synthetic
- the usage example 2-1 it is a figure which shows the reverse phase HPLC chromatogram before and behind reaction of the synthetic
- PC 5'-TTTTCTT
- PC combination DNA oligomer
- combination DNA oligomer 5'-TTTTCTT (PC) TCCTTTT-3 '.
- combination DNA oligomer (5'-TTTTTT (PC) TTTTTT-3 ').
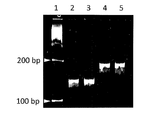
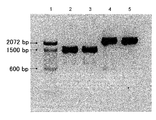
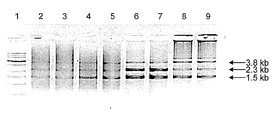
- the target DNA was amplified by polymerase chain reaction using a DNA oligomer containing an unnatural base as a primer, and the amplification product was analyzed by 10% non-denaturing polyacrylamide gel electrophoresis.
- Lane 1 100 base pair gene marker
- Lane 1 100 base pair gene marker
- the ligation product obtained by cleaving and reacting the obtained 5 ′ terminal phosphorylated DNA fragments of genes 1 and 2 with ligase; Lane 5: DNA amplification products of genes 1 and 2 (X Pa) are cleaved according to the present invention
- A Reaction mixture after reaction with methylamine
- B Reaction mixture after reaction with ethylenediamine.
- synthesis example 8-1 it is a figure which shows the reverse phase HPLC chromatogram of the synthetic
- synthesis example 8-1 it is a figure which shows the reverse phase HPLC chromatogram of the synthetic
- the usage example 8-1 it is a figure which shows the reverse phase HPLC chromatogram after reaction of the synthetic
- A Reaction mixture after reaction with methylamine
- B Reaction mixture after reaction with ethylenediamine.
- synthesis example 9-5 it is a figure which shows the reverse phase HPLC chromatogram of the synthetic
- synthesis example 9-5 it is a figure which shows the reverse phase HPLC chromatogram of the synthetic
- the usage example 9-1 it is a figure which shows the reverse phase HPLC chromatogram after reaction of the synthetic
- Lane 1 DNA marker
- Lane 2 Amplification product when LL1 and LL3 are used as primer sets
- Lane 3 Amplification product when LL2 and LL4 are used as primer sets
- Lane 4 LL5 and LL8 as primer sets Amplification product when used
- Lane 5 Amplification product when LL6 and LL7 are used as a primer set.
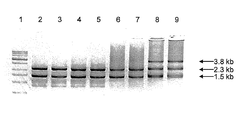
- a ligation product of a 5′-terminal phosphorylated DNA fragment obtained by cleaving a DNA amplification product containing 5-ethynyl-2′-deoxyuridine (Pa) by the cleavage reaction of the present invention It is a figure which shows the result analyzed by 1% agarose gel electrophoresis.
- Lane 1 DNA marker
- Lanes 2 and 3 Ligation reaction product when ligation reaction is performed at 10 temperature cycles
- Lanes 4 and 5 Ligation reaction product when ligation reaction is performed at 20 temperature cycles
- Lanes 6 and 7 Ligation product when ligation reaction was performed with 30 temperature cycles.
- Lanes 2, 4, and 6 Ligation product when LL1 and LL3 are used as a primer set and ligation reaction between amplification product DNA when LL6 and LL7 are used as a primer set
- Lanes 3, 5 and 7 Ligation reaction when amplification product DNA when LL2 and LL4 are used as a primer set and amplification product DNA when LL6 and LL7 are used as a primer set product.
- Lane 1 DNA marker
- Lanes 2 and 3 Cleavage product of DNA cleavage reaction after 9 hours of methylamine treatment
- Lanes 4 and 5 DNA cleavage reaction after 24 hours of methylamine treatment
- Lane 6 and 7 DNA cleavage treatment after 48 hours of methylamine treatment
- Lane 8 and 9 DNA cleavage after 48 hours of methylamine treatment
- Lanes 2, 4, 6, and 8 Cleavage of the amplification product DNA when LL1 and LL3 are used as a primer set and the amplification product DNA when LL6 and LL7 are used as a primer set Reaction product or ligation product when cleaved and ligated; Lanes 3, 5, 7, and 9: DNA of amplification product when LL2 and LL4 are used as a primer set, and LL6 and LL7 as a primer set A cleavage reaction product in the case of a cleavage reaction with the amplification product DNA or a ligation reaction product in a cleavage reaction and a ligation reaction.
- Lane 1 DNA marker
- Lanes 2, 3, 6, and 7 Amplification product DNA when LL1 and LL4 are used as a primer set and amplification product DNA when LL5 and LL8 are used as a primer set
- Lanes 4, 5, 8, and 9 DNA of amplification product when LL2 and LL3 are used as a primer set, and DNA of amplification product when LL6 and LL7 are used as a primer set Ligation reaction product when ligation reaction is performed.
- Lanes 2 to 5 Ligation reaction product when DNA is ligated using T4 DNA ligase
- Lanes 6 to 9 Ligation reaction product when DNA is ligated using Taq DNA ligase.
- Lanes 2, 4, 6, and 8 Ligation reaction product when DNA is ligated using a mixed solution of 2 ⁇ L of the amplified product DNA; Lanes 3, 5, 7, and 9: 4 ⁇ L of the amplified product DNA Ligation reaction product when DNA is ligated using a mixed solution of
- oligodeoxynucleotide means deoxyribonucleic acid (DNA), single-stranded or double-stranded oligodeoxynucleotide or polydeoxynucleotide, or a derivative thereof.
- the oligodeoxynucleotide (DNA oligomer or oligo DNA) may be natural or synthetic, and may be a polymerase chain reaction (PCR) product.
- Suitable oligodeoxynucleotide derivatives include, but are not limited to, for example, oligodeoxynucleotides in which the phosphodiester bond in the oligodeoxynucleotide is converted to a phosphorothioate bond, an N3′-P5 ′ phosphoramidite bond, or a peptide bond
- Deoxyribose in the derivative, oligodeoxynucleotide is morpholine, 2'-O-propylribose, 2'-O-methylribose, 2'-fluoro-2'-deoxyribose, treofuranose (TNA) or 2'-position
- Examples include oligodeoxynucleotide derivatives converted to ribose (locked nucleic acid (LNA) or cross-linked nucleic acid (BNA)) obtained by cross-linking O and C at the 4 'position with methylene, and peptide nucleic acids (
- the 5 'end and / or the 3' end of the oligodeoxynucleotide may be phosphorylated or not phosphorylated unless otherwise specified.
- the 5′-position hydroxyl group is a protecting group as in the case of synthesis under the condition that the 5′-position hydroxyl group protecting group of the last deoxynucleotide is not deprotected. It may be in a protected form. In either case, the oligodeoxynucleotide shall be included.
- protecting group means a group that binds to a terminal hydroxyl group to substantially suppress or prevent unwanted side reactions, and includes, but is not limited to, for example, a trityl group, a mono-substituted group, or Mention may be made of disubstituted trityl groups, o-nitrobenzyl groups and levulinyl groups. Dimethoxytrityl (DMTr) is preferred.
- the protecting group is removed by performing a predetermined deprotection treatment.
- examples of the deprotecting agent used for the deprotection treatment include acids such as trichloroacetic acid and trifluoroacetic acid.
- examples of the deprotecting agent used for performing the deprotecting treatment include hydrazine.
- examples of the deprotection treatment include UV light irradiation treatment.
- Oligodeoxynucleotide may be in a form in which the hydroxyl group at the 5 'end and / or the 3' end and / or the base is labeled with a probe molecule. In either case, the oligodeoxynucleotide shall be included.
- probe (label) molecule refers to a molecule conventionally used in the art for labeling oligodeoxynucleotides, such as biotin, polyethylene glycol (PEG), fluorescent dye, dicoxygenin, protein (eg, And a linker molecule for providing a labeling molecule). Fluorescent dyes selected from the group consisting of fluorescein derivatives and cyanine dyes are preferred.
- the probe molecule may be in its native form, with other functional groups such as active esters (eg succinimidyl esters), azide groups, amino groups, thiol groups, maleimide groups or alkynyl groups. It may be in a form in which a group capable of forming a covalent bond is introduced. Any form shall be included in the probe molecule.
- alkyl means a straight or branched chain aliphatic hydrocarbon group containing the specified number of carbon atoms.
- “1-20 alkyl” and “C 1 -C 20 alkyl” mean a straight or branched hydrocarbon chain containing at least 1 and at most 20 carbon atoms.
- Suitable alkyls include, but are not limited to, methyl, ethyl, n-propyl, isopropyl, n-butyl, sec-butyl, isobutyl, tert-butyl, n-pentyl, n-hexyl, n-heptyl and n -Octyl and the like can be mentioned.
- alkenyl means a group in which one or more C—C single bonds of the alkyl are substituted with double bonds.
- Suitable alkenyls include, but are not limited to, vinyl, 1-propenyl, allyl, 1-methylethenyl (isopropenyl), 1-butenyl, 2-butenyl, 3-butenyl, 1-methyl-2-propenyl, 2 Examples include -methyl-2-propenyl, 1-methyl-1-propenyl, 2-methyl-1-propenyl, 1-pentenyl, 1-hexenyl, n-heptenyl and 1-octenyl.
- alkynyl means a group in which one or more C—C single bonds of the alkyl are substituted with triple bonds.
- Suitable alkynyls include but are not limited to ethynyl, 1-propynyl, 2-propynyl, 1-butynyl, 2-butynyl, 3-butynyl, 1-methyl-2-propynyl, 1-pentynyl, 1-hexynyl 1-heptynyl, 1-octynyl and the like.
- cycloalkyl means an alicyclic alkyl containing the specified number of carbon atoms.
- Cycloalkyl having 3 to 20 members” and “C 3 to C 20 cycloalkyl” means a cyclic hydrocarbon group containing at least 3 and at most 20 carbon atoms. To do. Suitable cycloalkyls include, but are not limited to, cyclopropyl, cyclobutyl, cyclopentyl, cyclohexyl, and the like.
- cycloalkenyl means a group in which one or more C—C single bonds of the cycloalkyl are substituted with double bonds.
- Suitable cycloalkenyl includes, but is not limited to, cyclopropenyl, cyclobutenyl, cyclopentenyl, cyclohexenyl, and the like.
- cycloalkynyl means a group in which one or more C—C single bonds of the cycloalkyl are substituted with triple bonds.
- Suitable cycloalkynyl includes, but is not limited to, cyclobutynyl, cyclopentynyl, cyclohexynyl, and the like.
- heterocyclyl means that one or more carbon atoms of the cycloalkyl, cycloalkenyl or cycloalkynyl are each independently a nitrogen atom (N), a sulfur atom (S) and an oxygen atom (O). It means a group substituted by a selected hetero atom.
- N nitrogen atom
- S sulfur atom
- O oxygen atom
- (cyclic) 3-20 membered heterocyclyl” and “C 3 -C 20 heterocyclyl” include one or more cyclic hydrocarbon groups containing at least 3 and at most 20 carbon atoms. It means a group in which carbon atoms are independently substituted with the above heteroatoms.
- substitution with N or S includes substitution with N-oxide or S oxide or dioxide, respectively.
- Suitable heterocyclyls include, but are not limited to, pyrrolidinyl, tetrahydrofuranyl, dihydrofuranyl, tetrahydrothienyl, tetrahydropyranyl, dihydropyranyl, tetrahydrothiopyranyl, piperidinyl, morpholinyl, thiomorpholinyl, and piperazinyl. it can.
- aryl means an aromatic group having 5 to 20 carbon atoms.
- aryl of (ring) 5-20” and “C 5 -C 20 aryl” mean an aromatic group containing at least 5 and at most 20 carbon atoms.
- Suitable aryls include, but are not limited to, phenyl, naphthyl, anthryl (anthracenyl), and the like.
- arylalkyl means a group in which one of the hydrogen atoms of the alkyl is substituted with the aryl.
- Suitable arylalkyls include, but are not limited to, benzyl, 1-phenethyl, 2-phenethyl, and the like.
- arylalkenyl means a group in which one of the alkenyl hydrogen atoms is substituted with the aryl.
- Suitable arylalkenyls include, but are not limited to, styryl and the like.
- heteroaryl refers to a heteroatom in which one or more carbon atoms of the aryl are each independently selected from a nitrogen atom (N), a sulfur atom (S), and an oxygen atom (O).
- N nitrogen atom
- S sulfur atom
- O oxygen atom
- “(heterocyclic) 5-20 membered heteroaryl” and “C 5 -C 20 heteroaryl” are one or more carbons of an aromatic group containing at least 5 and at most 20 carbon atoms. It means a group in which atoms are each independently substituted with the above heteroatoms.
- substitution with N or S includes substitution with N-oxide or S oxide or dioxide, respectively.
- Suitable heteroaryl include, but are not limited to, furanyl, thienyl, pyrrolyl, imidazolyl, pyrazolyl, triazolyl, tetrazolyl, thiazolyl, oxazolyl, isoxazolyl, oxadiazolyl, thiadiazolyl, isothiazolyl, pyridyl, pyridazinyl, pyrazinyl, pyrimidinyl, quinolinyl, Examples thereof include isoquinolinyl and indolyl.
- heteroarylalkyl means a group in which one of the hydrogen atoms of the alkyl is substituted with the heteroaryl.
- carbonyl means a monovalent group having a carbonyl group. Suitable carbonyls include, but are not limited to, formyl, acetyl, benzoyl, carboxy, methoxycarbonyl, halocarbonyl, carbamoyl (aminocarbonyl), and the like.
- Each of the groups described above is independently selected from the group consisting of an oxygen atom (O), a nitrogen atom (N), a sulfur atom (S), a silicon atom (Si), and a phosphorus atom (P).
- the above heteroatoms may be included. It is preferably an oxygen atom, a nitrogen atom or a sulfur atom.
- one or more carbon atoms constituting the above group are each independently substituted with these heteroatoms.
- the groups described above are intended to include not only the form itself, but also the form in which some of the carbon atoms are replaced with heteroatoms.
- the groups described above are each independently unsubstituted or one or more halogen, hydroxyl group, substituted or unsubstituted amino group, nitro group, cyano group, mercapto group, carbonyl group, amide group Or a substituted or unsubstituted monovalent hydrocarbon group which may contain a hetero atom (eg, substituted or unsubstituted C 1 -C 20 alkyl, C 2 -C 20 alkenyl, C 2 -C 20 alkynyl, C 1 ⁇ C 20 alkoxy, C 3 ⁇ C 20 cycloalkyl, C 3 ⁇ C 20 cycloalkyloxy, C 3 ⁇ C 20 cycloalkenyl, C 3 ⁇ C 20 cycloalkyl alkenyloxy, C 4 ⁇ C 20 cycloalkynyl, C 4 ⁇ C 20 cycloalkynyloxy, C 3 -C 20 heterocyclyl, C 3 -C 20 heterocyclyl,
- halogen or “halo” means fluorine, chlorine, bromine or iodine.
- Oligodeoxynucleotide> [2-1. Oligodeoxynucleotide]
- the inventors of the present invention have studied means for performing DNA strand cleavage and phosphorylation of a cleaved end by chemical reaction at low cost and simple experimental operation. As a result, deoxynucleosides containing unnatural bases described below are obtained.
- a nucleophile such as a primary amine
- the DNA oligomer is cleaved at a deoxynucleoside site containing a non-natural base to form an oligodeoxynucleotide phosphorylated at the 5 'end.
- non-natural base exhibits substantially the same or substantially the same complementarity as the natural base under DNA replication conditions such as PCR
- DNA containing the non-natural base can be used in vivo and in vivo.
- in vitro reaction it is possible to exhibit a function as a template that is substantially the same or substantially the same as DNA comprising a natural base.
- A is an oligodeoxynucleotide
- B is a deoxynucleoside containing a non-natural base having a moiety E removable by a nucleophile
- C is an oligodeoxynucleotide
- n is 0 or 1
- n 1
- the 3 ′ end of A and the 5 ′ position of B are linked by a phosphodiester bond
- the 3 ′ position of B and the 5 ′ end of C are linked by a phosphodiester bond
- the phosphodiester bond between B and C is such that the oligodeoxynucleotide part phosphorylated at the 5 'end of C from the 3' position of B with E elimination induced by nucleophile attack. It is cut by detaching.
- oligodeoxynucleotides A and C are preferably oligodeoxynucleotides having the characteristics described above.
- the number of bases of oligodeoxynucleotides A and C is not particularly limited, but in the case of oligodeoxynucleotide A, it is usually in the range of 0 to 100,000, preferably in the range of 0 to 20,000, and in the range of 0 to 10,000. It is more preferable. In the case of oligodeoxynucleotide C, it is usually in the range of 1 to 100,000, preferably in the range of 1 to 20,000, and more preferably in the range of 1 to 10,000.
- the deoxynucleoside B containing a non-natural base having a moiety E removable by a nucleophile is preferably a deoxynucleoside containing a non-natural base having the characteristics described below.
- the hydroxyl group at the 5 ′ end of deoxynucleoside B containing an unnatural base may be in an unprotected form, and is preferably a trityl group or a mono- or di-substituted trityl group, preferably May be in a form protected with a protecting group as described above, such as a DMTr group. Any form shall be encompassed by the oligonucleotide represented by Formula I.
- the DNA strand is cleaved by a chemical reaction at a low cost and with a simple experimental operation, and the resulting end is phosphorylated at the end. Can be obtained.
- Deoxynucleoside B containing an unnatural base used in the method of the present invention has a moiety that can be eliminated by a nucleophile. Specifically, it has the following features.
- DNA synthesis conditions means general synthesis conditions such as phosphoramidite method applied when DNA synthesis is performed by an automatic DNA synthesizer usually used in the art.
- the automatic DNA synthesizer include, but are not limited to, ABI 392 DNA / RNA synthesizer (Applied Biosystems) and NTS H-6 DNA / RNA synthesizer (Nippon Techno Service).
- DNA synthesis conditions are based on a combination of deoxynucleoside B containing a non-natural base and one or more deoxynucleosides containing a natural base used as a substrate when synthesizing an oligodeoxynucleotide represented by Formula I. For example, it can be set as appropriate by referring to the instruction manual attached to the DNA automatic synthesizer.
- (I-1) Must not substantially react with iodine in the presence of iodine used for the oxidation of the phosphite part of the coupling reaction product.
- (I-3) Must not substantially react with alkali in the presence of an alkali such as aqueous ammonia used for excision and deprotection of the synthesized DNA (alkaline conditions).
- DNA replication conditions means general reaction conditions applied when DNA is replicated by a PCR reaction usually used in the art. DNA replication conditions are based on the base sequence of the oligodeoxynucleotide used as a template for replicating the oligodeoxynucleotide represented by Formula I, for example, Kramer M. F. and Coen D. M., “Enzymatic It can be set appropriately by referring to amplification of DNA by PCR: standard procedures and optimization. ”, Curr Protoc Cytom. 2006 Aug;
- Characteristic (ii) “Stablely present under DNA replication conditions such as in-vivo and / or in-vitro reaction (for example, PCR reaction)” specifically means that the following characteristics are satisfied.
- a buffer solution usually used in PCR for example, a tris buffer containing a salt such as magnesium chloride or potassium chloride and having a pH of around 8 at room temperature (optionally containing a surfactant or BSA)
- Heating at 95 ° C in the PCR cycle does not substantially decompose.
- deoxynucleoside B containing an unnatural base preferably has the following characteristics (vi) and (vii).
- the unnatural base has a structure of a purine type or pyrimidine type base substituted with a substituted or unsubstituted alkenyl or alkynyl.
- deoxynucleoside B containing an unnatural base preferably has the following characteristics (vi ′) and (vii ′).
- the unnatural base has a structure of a water adduct or a nucleophile adduct of a purine or pyrimidine base substituted with a substituted or unsubstituted alkenyl or alkynyl, or a tautomer thereof. .
- deoxynucleoside B containing a non-natural base preferably has the following characteristics (vi ′′) and (vii ′′).
- a non-natural base has a structure of a purine base substituted with a substituted or unsubstituted aryl having carbonyl, substituted or unsubstituted alkynyl or cyano.
- deoxynucleoside B containing a non-natural base means not only compound B itself but also a water adduct or a nucleophile adduct of the compound or a tautomer thereof. .
- deoxynucleoside B containing an unnatural base has the following formulas (vi) and (vii):
- R and R ′ represent the same groups as defined below.
- the following reaction proceeds when an oligodeoxynucleotide containing B is reacted in the presence of a nucleophile (basic conditions).
- the effect of this invention is not limited to the following reaction mechanism.
- DNA means oligodeoxynucleotide
- Nu means nucleophile
- deoxynucleoside B containing an unnatural base has the following characteristics (vi ′) and (vii ′):
- R means the same group as defined below, DNA means oligodeoxynucleotide, and Nu means nucleophile.
- deoxynucleoside B containing an unnatural base has the following characteristics (vi ′) and (vii ′):
- R represents the same group as defined below.
- a compound represented by the formula by reacting an oligodeoxynucleotide containing B in the presence of a nucleophile (under basic conditions), a water addition reaction or a nucleophile addition reaction of the compound can proceed. There is sex.
- R means the same group as defined below, DNA means an oligodeoxynucleotide, Nu means a nucleophile, and R Nu means a group generated upon addition of a nucleophile. .
- deoxynucleoside B containing an unnatural base has the following characteristics (vi ′′) and (vii ′′):
- r and R ′′ mean the same groups as defined below, DNA means oligodeoxynucleotide, and Nu means nucleophile.
- a DNA strand can be reacted at a low cost and with a simple experimental operation by reacting in the presence of a nucleophile (under basic conditions). As a result, it is possible to obtain an oligodeoxynucleotide whose resulting end is phosphorylated.
- the cleavage of DNA by the method of the present invention is accompanied by the elimination of E from the deoxynucleoside containing a non-natural base induced by the attack of a nucleophile, the single-base deoxynucleoside B It becomes possible to introduce a cleavage site at an arbitrary position of the DNA strand by insertion or substitution.
- the hydroxyl group at the 5 ′ end of deoxynucleoside B containing a non-natural base prepared as a monomer unit is a trityl group or mono-substituted. Alternatively, it is preferably in a form protected with a protecting group as described above, such as a disubstituted trityl group, preferably a DMTr group.
- the hydroxyl group at the 5 ′ end of deoxynucleoside B containing an unnatural base is unprotected, or is a trityl group, monosubstituted or disubstituted It is preferably in a form protected with a protecting group as described above, such as a trityl group, preferably a DMTr group.
- deoxynucleotide B containing an unnatural base means a form in which the hydroxyl group at the 5 ′ end of deoxynucleoside B containing an unnatural base is phosphorylated.
- Deoxynucleoside B containing unnatural bases is:
- R is hydrogen, substituted or unsubstituted alkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted A heterocyclyl, substituted or unsubstituted aryl, substituted or unsubstituted arylalkyl, substituted or unsubstituted heteroaryl, substituted or unsubstituted heteroarylalkyl or carbonyl.
- R is hydrogen, substituted or unsubstituted C 1 -C 10 alkyl, substituted or unsubstituted C 3 -C 20 cycloalkyl, substituted or unsubstituted C 3 -C 20 heterocyclyl, substituted or unsubstituted Substituted C 5 -C 20 aryl, substituted or unsubstituted C 6 -C 20 arylalkyl, substituted or unsubstituted C 5 -C 20 heteroaryl, substituted or unsubstituted C 5 -C 20 heteroarylalkyl or carbonyl It is preferable that Particularly suitable R can include hydrogen, methyl, ethyl, phenyl, hydroxymethyl, aminomethyl, mercaptomethyl, halomethyl, formyl, carboxyl, carbamoyl and acetyl. Particular preference is given to hydrogen, methyl, phenyl or carbamoyl.
- R ′ is hydrogen or substituted or unsubstituted alkyl, alkenyl or alkynyl. It is. Specifically, R ′ is preferably a hydrogen atom or a substituted or unsubstituted C 1 -C 10 alkyl, C 2 -C 10 alkenyl or C 2 -C 10 alkynyl, particularly methyl. preferable.
- r is an integer from 0 to 4, and R ′′ is halogen or substituted or unsubstituted alkyl, alkenyl, or alkynyl. Specifically, r is an integer from 0 to 4 and R ′′ is halogen or substituted or unsubstituted C 1 -C 10 alkyl, C 2 -C 10 alkenyl or C 2 -C 10 alkynyl. Preferably, r is particularly preferably 0.
- R ′ ′′ is —COZ, —C ⁇ C—Z, or —CN. Specifically, R ′′ ′′ is preferably —COZ.
- Z is hydrogen, substituted or unsubstituted alkyl, substituted or unsubstituted cycloalkyl, substituted or unsubstituted heterocyclyl, substituted or unsubstituted aryl, substituted or unsubstituted aryl Alkyl, substituted or unsubstituted heteroaryl, or substituted or unsubstituted heteroarylalkyl.
- Z is hydrogen, substituted or unsubstituted C 1 -C 10 alkyl, substituted or unsubstituted C 3 -C 20 cycloalkyl, substituted or unsubstituted C 3 -C 20 heterocyclyl, substituted or unsubstituted C 5 ⁇ C 20 aryl substituted, substituted or unsubstituted C 6 ⁇ C 20 arylalkyl, substituted or unsubstituted C 5 ⁇ C 20 heteroaryl, or substituted or unsubstituted of C 5 ⁇ C 20 heteroarylalkyl
- Particularly suitable R may include hydrogen, methyl, ethyl, phenyl, hydroxymethyl, aminomethyl, mercaptomethyl or halomethyl. Hydrogen, methyl or phenyl is preferable, and hydrogen is more preferable.
- a part of carbon atoms may be replaced with one or more, preferably 1 to 5, more preferably 1 to 3 heteroatoms.
- the hetero atom is preferably a hetero atom selected from the group consisting of an oxygen atom, a nitrogen atom, a sulfur atom, a silicon atom and a phosphorus atom.
- the group is one or more halogen, hydroxyl group, substituted or unsubstituted amino group, nitro group, cyano group, mercapto group, carbonyl group, amide group, or hetero atom.
- a substituted or unsubstituted monovalent hydrocarbon group eg substituted or unsubstituted C 1 -C 20 alkyl, C 1 -C 20 alkoxyl, C 3 -C 20 cycloalkyl, C 3 -C 20 cycloalkyloxy, C 3 ⁇ C 20 heterocyclyl, C 3 ⁇ C 20 heterocyclyloxy, C 5 ⁇ C 20 aryl, C 5 ⁇ C 20 aryloxy, C 6 ⁇ C 20 arylalkyl, C 5 ⁇ C 20 heteroaryl C 5 -C 20 heteroaryloxy or C 6 -C 20 heteroarylalkyl, in which some of the carbon atoms may be replaced by heteroatoms). That's right.
- Deoxynucleoside B containing a non-natural base includes not only the compound itself but also a salt thereof.
- the counter ion of the salt of deoxynucleoside B containing a non-natural base includes, but is not limited to, for example, a cation such as sodium ion, potassium ion, calcium ion or magnesium ion, or chloride ion, bromide ion, Formate, acetate, maleate, fumarate, benzoate, ascorbate, pamoate, succinate, bismethylenesalicylate, methanesulfonate, ethanedisulfonate, propionate, tartaric acid Ion, salicylate ion, citrate ion, gluconate ion, aspartate ion, stearate ion, palmitate ion, itaconic acid ion, glycolate ion, p-aminobenzoate ion, glutamate ion
- Deoxynucleoside B containing an unnatural base includes not only the compound itself but also a solvate thereof.
- Solvents that can form solvates with deoxynucleoside B containing an unnatural base include, but are not limited to, for example, methanol, ethanol, 2-propanol (isopropyl alcohol), dimethyl sulfoxide (DMSO), acetic acid, Organic solvents such as ethanolamine or ethyl acetate, or water are preferred.
- deoxynucleoside B containing an unnatural base includes not only the compound itself but also its protected form.
- protected form means a form in which a protecting group is introduced into one or more functional groups.
- a DNA strand can be reacted at a low cost and with a simple experimental operation by reacting in the presence of a nucleophile (under basic conditions). As a result, it is possible to obtain an oligodeoxynucleotide whose resulting end is phosphorylated.
- FIG. 1 is a process diagram showing one embodiment of a method for producing an oligodeoxynucleotide of the present invention.
- a preferred embodiment of the method of the present invention will be described in detail with reference to FIG.
- the method of the present invention comprises a nucleophile and a compound of formula I: (A) n -BC And an oligodeoxynucleotide phosphorylated at the 5 ′ end of C, by contacting with an oligodeoxynucleotide represented by formula II: (A) n -B ' A 5′-terminal phosphorylated oligonucleotide forming step for obtaining an oligodeoxynucleotide represented by (Step S2).
- This step cleaves the phosphodiester bond between B and C by removing the oligodeoxynucleotide moiety phosphorylated at the 5 ′ end of C from the 3 ′ position of B, thereby 'To obtain oligodeoxynucleotides phosphorylated at the ends.
- B ′ is the residue part of B that is formed with the elimination of E by the attack of the nucleophile.
- a and B ′ are linked by a phosphodiester bond. More specifically, B 'is linked to the 3' end of A by a phosphodiester bond at a position derived from the 5 'position of B.
- the 5′-position of B ′ means a position derived from the 5′-position of B in B ′.
- a nucleophile and formula I ′ BC
- the phosphodiester bond between B and C is removed from the 3 'position of B by contacting the phosphodiester bond between B and C by contacting with the oligodeoxynucleotide represented by Resulting in oligodeoxynucleotides phosphorylated at the 5 ′ end of the C.
- n 1, the phosphodiester bond between B and C is brought into contact with the 5 ′ end of C by contacting the nucleophile with the oligodeoxynucleotide represented by Formula I.
- This step does not require enzyme treatment, and the cleavage of the oligodeoxynucleotide represented by Formula I or Formula I ′ and phosphorylation of the 5 ′ end are performed by a chemical reaction (nucleophilic reaction by a nucleophile). Therefore, it is possible to produce oligodeoxynucleotides phosphorylated at the 5 'end with a low-cost and simple experimental operation as compared with the prior art method using an enzyme such as polynucleotide kinase.
- R Nu is a non-natural type obtained by adding one molecule of a nucleophile (Nu) to a degraded nucleotide. This means that the base has been removed.
- the above reaction mechanism applies not only to non-natural bases corresponding to pyrimidine-type bases such as thymine and cytosine, but also to deoxynucleoside B containing non-natural bases corresponding to purine-type bases such as adenine and guanine. Is done.
- the reaction mechanism includes not only deoxynucleoside B containing unnatural bases having characteristics (vi) and (vii), but also characteristics (vi ′) and (vii ′) or characteristics (vi ′′) and ( The same applies to deoxynucleoside B containing non-natural bases having vii ′′) in consideration of the above explanation regarding deoxynucleoside B containing these non-natural bases.
- the depurination reaction is a reaction in which adenine and / or guanine, which are purine bases contained in DNA, are eliminated from the nucleoside.
- adenine and guanine are easily released by protonation, the depurination reaction hardly occurs under basic conditions, and easily occurs under acidic conditions.
- the pyrimidine bases cytosine and thymine are less likely to desorb than purine bases, and the pyrimidine base is not likely to be desorbed from DNA not only under basic and neutral conditions but also under acidic conditions.
- Non-Patent Documents 26 to 29 a reaction in which hydrogen at the 3-position of the base moiety is eliminated.
- a reaction in which DNA is cleaved by a mechanism that is considered to be eliminated from a nucleoside at the same time that the unnatural base contained in deoxynucleoside B is cyclized has not been reported so far. Is a new reaction found.
- DNA containing an unnatural base of a thymidine derivative that acts as a thymidine in a DNA replication reaction using DNA polymerase is known (Patent Documents 4 and 5 and Non-Patent Documents 30 to 34).
- Patent Documents 4 and 5 and Non-Patent Documents 30 to 34 when reacted under basic conditions (in the presence of a nucleophile), the natural bases contained in the DNA are not damaged and are specific only at the sites of deoxynucleotides containing unnatural bases.
- a reaction in which DNA is cleaved and the 5 ′ end resulting from cleavage is phosphorylated has not been reported so far, and is a novel reaction found by the present inventors.
- the oligodeoxynucleotide represented by the formula I used in this step may be prepared by purchasing a pre-synthesized one or by performing the oligodeoxynucleotide synthesis step described below. May be. In either case, it is included in the embodiment of this step.
- the nucleophilic agent used in this step attacks the deoxynucleoside B site containing the unnatural base contained in the oligodeoxynucleotide represented by Formula I, and removes the detachable moiety E from B. And an oligodeoxynucleotide moiety phosphorylated at the 5 ′ end of C along with the elimination of E, can be cleaved by elimination from the 3 ′ position of B.
- a nucleophile is not limited, For example, a primary amine can be mentioned.
- a primary amine selected from the group consisting of methylamine, ethylenediamine, 2-aminoethanol, ethylamine, propylamine, 2-aminoethanethiol, 1,3-diaminopropane and tris (2-aminoethyl) amine is preferred.
- the phosphodiester bond between B and C contained in the oligodeoxynucleotide represented by formula I is converted to the oligodeoxynucleotide moiety phosphorylated at the 5 ′ end of C. It is possible to obtain oligodeoxynucleotide C in which the 5 ′ end is phosphorylated by cleaving it from the 3 ′ position.
- This step is usually carried out by bringing a nucleophile and an oligodeoxynucleotide represented by the formula I into contact under predetermined reaction conditions.
- the reaction conditions may be set as appropriate based on the combination of the nucleophile used in this step and deoxynucleoside B containing an unnatural base.
- the reaction temperature is preferably in the range of 0 to 90 ° C., more preferably in the range of 4 to 70 ° C.
- the reaction condition is a heating condition
- the reaction temperature is preferably in the range of 60 to 90 ° C., more preferably in the range of 70 to 80 ° C.
- reaction temperature is 90 ° C or lower, particularly in the range of 4 to 70 ° C, the lower the temperature, the more alkali denaturation of the long-chain oligonucleotide can be suppressed.
- the reaction time is preferably a time in the range of 0.1 to 168 hours, more preferably a time in the range of 0.1 to 48 hours, further preferably a time in the range of 0.1 to 24 hours, A time in the range of 12 hours is particularly preferred.
- This step is usually carried out by bringing a nucleophile and an oligodeoxynucleotide represented by the formula I into contact in a medium.
- the medium may be appropriately set based on the combination of the nucleophile used in this step and deoxynucleoside B containing an unnatural base. Specifically, water is preferable.
- This step may be carried out using a deprotected oligodeoxynucleotide represented by Formula I, for example, a 5 ′ end is a trityl group or a mono- or di-substituted trityl group, preferably a DMTr group.
- a deprotected oligodeoxynucleotide represented by Formula I for example, a 5 ′ end is a trityl group or a mono- or di-substituted trityl group, preferably a DMTr group.
- the oligodeoxynucleotide represented by the formula I protected with the protecting group described above may be used.
- the oligodeoxynucleotide represented by Formula II obtained in this step is in a form in which the 5 'end is protected.
- deprotection may be performed by performing the deprotection treatment described above in the 3′-terminal phosphorylated oligodeoxynucleotide
- the oligodeoxynucleotide C phosphorylated at the 5 ′ end obtained in this step and optionally the oligodeoxynucleotide represented by the formula II are separated from each other, the raw material and the auxiliary by a purification means usually used in the art. By being separated from the product, it can be purified and isolated. Therefore, this step may further include a purification and isolation step.
- purification means include, but are not limited to, medium pressure liquid chromatography (MPLC), high performance liquid chromatography (HPLC), vacuum concentration (evaporation), lyophilization, recrystallization, agarose gel electrophoresis, poly Mention may be made of acrylamide gel electrophoresis, reverse phase or treatment with a simple cartridge filled with an ion exchange carrier and ethanol precipitation, and combinations thereof.
- the purification and isolation step is not performed in this step, and in the 3′-terminal phosphorylated oligodeoxynucleotide forming step described below, oligodeoxynucleotide C having a phosphorylated 5 ′ end and the 3 ′ end are phosphorylated.
- the purification and isolation of the oligodeoxynucleotides may be performed together. By performing the above-described purification and isolation step, it is possible to produce oligodeoxynucleotide C having a highly purified 5 'end phosphorylated.
- the method of the present invention optionally has the formula II: (A) n -B ' Is reacted with an oligodeoxynucleotide of formula IIa: A-PO 3 H 2 A 3′-terminal phosphorylated oligodeoxynucleotide forming step for obtaining an oligodeoxynucleotide phosphorylated at the 3 ′ end represented by the formula (3) may be further included (step S3).
- This step cleaves the phosphodiester bond between B ′ and A by removing the oligodeoxynucleotide moiety phosphorylated at the 3 ′ end of A from B, which is represented by formula IIa 3 'To obtain oligodeoxynucleotides phosphorylated at the ends.
- the 3 'end of A is phosphorylated by cleaving the phosphodiester bond between B' and A contained in the oligodeoxynucleotide represented by Formula II by a chemical reaction (elimination reaction). Resulting in oligodeoxynucleotides. Therefore, it is possible to produce an oligodeoxynucleotide phosphorylated at the 3 'end with a low-cost and simple experimental operation, compared to the terminal phosphorylation method of synthetic DNA using phosphorylated amidite.
- R Nu is a non-natural type obtained by adding one molecule of a nucleophile (Nu) to a degraded nucleotide. This means that the base has been removed.
- the oligodeoxynucleotide A- (B-1 ′) represented by the formula II formed along with the elimination of E due to the attack of the nucleophile in the previous step is subjected to heat treatment in this step to obtain B-1
- the phosphodiester bond between 'and A is cleaved by elimination of the oligodeoxynucleotide moiety phosphorylated at the 3' end of A from B-1 'and phosphorylated at the 3' end of A Resulting in oligodeoxynucleotides.
- This step is usually carried out by reacting the oligodeoxynucleotide represented by formula II under predetermined reaction conditions.
- the reaction conditions may be set as appropriate based on deoxynucleosides B and B 'formed by cleavage of the unnatural base used.
- the reaction temperature is preferably in the range of 0 to 120 ° C., more preferably in the range of 4 to 70 ° C.
- the reaction temperature is preferably in the range of 90 to 120 ° C, more preferably in the range of 110 to 120 ° C.
- the reaction time is preferably a time in the range of 0.1 to 168 hours, more preferably a time in the range of 0.1 to 48 hours, further preferably a time in the range of 0.1 to 24 hours, A time in the range of 1 hour is particularly preferred.
- the pH of the reaction system is usually neutral conditions, specifically, a value in the range of 6 to 8 is preferable, and a value in the range of 6.8 to 7.2 is more preferable.
- This step is usually performed by reacting an oligodeoxynucleotide represented by formula II in a medium.
- the medium may be appropriately set based on the oligodeoxynucleotide represented by formula II used in this step. Specifically, water is preferable.
- the medium may also contain one or more buffer components in order to maintain the pH of the reaction system in the preferred pH range described above. Examples of the buffer component include, but are not limited to, phosphate and cacodylate, and combinations thereof. A phosphate is preferred.
- the concentration of the buffer component is preferably in the range of 10 to 1000 ⁇ mM, and more preferably in the range of 10 to 100 ⁇ mM.
- the oligodeoxynucleotide phosphorylated at the 3 ′ end represented by Formula IIa obtained in this step is purified by being separated from the raw materials and by-products by purification means usually used in the art. And can be isolated. Therefore, this step may further include a purification and isolation step.
- purification means include, but are not limited to, medium pressure liquid chromatography (MPLC), high performance liquid chromatography (HPLC), vacuum concentration (evaporation), lyophilization and recrystallization, agarose gel electrophoresis, poly Mention may be made of acrylamide gel electrophoresis, reverse phase or treatment with a simple cartridge filled with an ion exchange carrier and ethanol precipitation, and combinations thereof.
- This step may be performed using a deprotected oligodeoxynucleotide represented by Formula II, for example, a 5 ′ end is a trityl group or a mono- or di-substituted trityl group, preferably a DMTr group.
- a deprotected oligodeoxynucleotide represented by Formula II for example, a 5 ′ end is a trityl group or a mono- or di-substituted trityl group, preferably a DMTr group.
- the oligodeoxynucleotide represented by the formula II protected with the protecting group described above may be used.
- the oligodeoxynucleotide phosphorylated at the 3 'end represented by Formula IIa obtained in this step is in a form in which the 5' end is protected.
- deprotection can be performed by performing the deprotection treatment described above to obtain an oligodeoxynucleotide in which the 3 ′ end represented by the formula IIa in the deprotected form is phosphorylated. It becomes possible.
- step S1 Using a substrate comprising a derivative and one or more deoxynucleosides or derivatives thereof comprising a natural base, the formula I: (A) n -BC
- An oligodeoxynucleotide synthesis step for synthesizing the oligodeoxynucleotide represented by the formula (1) may be further included (step S1). The purpose of this step is to obtain an oligodeoxynucleotide represented by the formula I, which is a raw material for the 5′-terminal phosphorylated oligodeoxynucleotide forming step.
- n 0 or 1.
- deoxynucleoside B containing a non-natural base having a part E that can be eliminated by a nucleophile can be deoxynucleoside B containing a non-natural base described above.
- one or more deoxynucleosides containing a natural base are adenine, thymine, guanine and other than deoxynucleotide B containing an unnatural base among the deoxynucleotides contained in the oligodeoxynucleotide represented by Formula I.
- deoxynucleotide containing a natural base selected from the group consisting of cytosine, and a deoxynucleoside containing a natural base having a purity usually used in the art can be used.
- a derivative of deoxynucleoside B containing a non-natural base and a derivative of deoxynucleoside containing a natural base having a moiety E that can be eliminated by a nucleophile is not limited, for example, at the 3 ′ position. Mention may be made of derivatives of the deoxynucleoside having a phosphoramidite group. By using the above derivative, it is possible to obtain the oligodeoxynucleotide represented by the formula I by an automatic DNA synthesizer usually used in the art.
- the substrate used in this step is a deoxynucleoside B containing a non-natural base having a moiety E that can be eliminated by a nucleophile described above or a derivative thereof, and one or more deoxy containing a natural base.
- the substrate may optionally include other components such as deoxynucleosides or derivatives thereof containing other unnatural bases, or labeling molecules such as fluorescent dyes, biotin or dichogigenin.
- This step is carried out by contacting the substrate with a medium such as acetonitrile or N, N-dimethylformamide and an activator such as tetrazole, a tetrazole derivative or 4,5-dicyanoimidazole, if desired. can do.
- a medium such as acetonitrile or N, N-dimethylformamide
- an activator such as tetrazole, a tetrazole derivative or 4,5-dicyanoimidazole, if desired. can do.
- this step may be performed under the DNA synthesis conditions described above.
- the oligodeoxynucleotide represented by formula I can be obtained in high yield.
- a DNA strand can be cleaved by a low-cost and simple experimental operation, and the resulting oligodeoxynucleotide whose end is phosphorylated can be obtained.
- the present invention also provides formula I: (A) n -BC [Wherein A, B, C and n represent the same meaning as defined above, When n is 1, the 3 ′ end of A and the 5 ′ position of B are linked by a phosphodiester bond, The 3 ′ position of B and the 5 ′ end of C are linked by a phosphodiester bond, The phosphodiester bond between B and C is linked to the oligodeoxynucleotide moiety phosphorylated at the 5 'end of C from the 3' position of B with the elimination of E induced by nucleophile attack.
- the 5 ′ end of C contained in the linear double-stranded DNA fragment is phosphorylated, and at least the primer portion represented by Formula I Forming a linear double-stranded DNA fragment having a 3 ′ protruding end containing a sequence complementary to a part thereof, and a linear double-stranded DNA fragment having the 3 ′ protruding end; And a ligation step of ligating another double-stranded DNA fragment; And a method for ligating double-stranded DNA.
- means for amplifying a linear double-stranded DNA fragment is not limited, and examples thereof include a polymerase chain reaction (PCR).
- this step may be performed under the DNA replication conditions described above.
- the oligodeoxynucleotide represented by the formula I the oligodeoxynucleotide C may have a base sequence complementary to at least a part of the template DNA that is a target of the polymerase chain reaction.
- nucleophile used in the 3 'protruding end forming step and the specific reaction conditions of this step it is preferable to apply the conditions described above with respect to the 5' end phosphorylated oligodeoxynucleotide forming step.
- the method of the present invention can cleave DNA without considering the presence or absence of a restriction enzyme recognition sequence in the DNA fragment to be cleaved, and the length of the protruding portion at the protruding end. And it has the advantage that the arrangement can be set arbitrarily. Therefore, a linear double-stranded DNA fragment having a 3 ′ overhang produced by the method of the present invention is a DNA fragment produced using a type II restriction enzyme during ligation using a DNA ligation enzyme. Compared to the above, it is possible to exhibit high connection efficiency.
- Kit for use in the production method of oligodeoxynucleotide> By the method of the present invention, oligodeoxynucleotide C phosphorylated at the 5 ′ end and / or oligodeoxynucleotide phosphorylated at the 3 ′ end represented by Formula IIa can be obtained. Therefore, the present invention also relates to a kit for use in the above-described method for producing the oligodeoxynucleotide of the present invention or the method for ligating double-stranded DNA.
- the kit has the formula I: (A) n -BC Or a deoxynucleoside B containing a non-natural base or a derivative thereof having a moiety E removable by a nucleophile.
- n 0 or 1.
- the kit further comprises a natural base selected from the group consisting of adenine, thymine, guanine and cytosine in addition to the oligodeoxynucleotide represented by formula I or deoxynucleoside B containing a non-natural base or a derivative thereof. It may contain one or more additional elements selected from the group consisting of one or more deoxynucleosides or derivatives thereof, nucleophiles, media, catalysts, DNA-ligating enzymes (ligases) and instructions for use.
- the nucleophile, the medium, the catalyst, and the DNA ligating enzyme can be appropriately selected from those that can be used in the method of the present invention described above.
- the kit of the present invention is not necessarily provided in a form in which the above elements are integrated, and may be provided in a form in which the elements are separately packaged. Any form shall be included in the kit.
- the oligodeoxynucleotide production method of the present invention is carried out, and the oligodeoxynucleotide C and / or formula IIa phosphorylated at the 5 ′ end by a low-cost and simple experimental operation It is possible to obtain an oligodeoxynucleotide phosphorylated at the 3 ′ end represented by
- a DNA strand is cleaved by a chemical reaction at a low cost and with a simple experimental operation, and further, the terminal phosphorylation of a synthetic DNA using an enzymatic method such as polynucleotide kinase or a phosphorylated amidite. It becomes possible to perform terminal phosphorylation with higher yield compared to the oxidation method.
- the chemical shift of 1 H-NMR was described in ppm based on the chemical shift (2.49 ppm) of the residual proton in DMSO-d6.
- the HRMS spectrum was measured using SYNAPT G2 MS manufactured by Waters.
- the MALDI TOF-MS spectrum was measured using AXIMA Assurance manufactured by Shimadzu Corporation.
- the target substance was purified with a silica gel column (developing solvent was 2 to 5% MeOH, 2% Et 3 N / CH 2 Cl 2 ).
- the solvent of the objective fraction was distilled off under reduced pressure and dried with a vacuum pump. Dissolve in a small amount of methylene chloride, add about 400 mL of hexane, filter the resulting white precipitate, and dry with a vacuum pump to obtain 1.20 g (1.64 mmol, 82.0% yield) of the target substance (molecular weight 731.88 (one triethylamine molecule) was obtained as a white powder.
- Loamidite (molecular weight 236.68, specific gravity 1.061 g / mL) was added, and the mixture was stirred at room temperature for 30 minutes. 25 mL of AcOEt and 25 mL of saturated saline were added and separated, and the organic layer was further washed twice with saturated saline. The organic layer was dried over anhydrous magnesium sulfate and filtered through a filter, and the solvent of the filtrate was distilled off under reduced pressure. After azeotroping with acetonitrile three times, 10 mL of acetonitrile was added. The solution was passed through a 0.45 ⁇ m filter and a small amount of molecular sieve was added. Attached to an automatic DNA synthesizer and used for DNA synthesis.
- the ammonia was distilled off under reduced pressure with a centrifugal concentrator, the solution containing DNA was passed through a 0.45 ⁇ m filter, and then the main peak eluted at a retention time of around 10 minutes was purified by reverse phase HPLC (0 -50% CH 3 CN / 50 mM ammonium formate aqueous solution, 20 minutes, detected at 254 nm), ammonium formate and the solvent were distilled off by centrifugal concentrator and lyophilization.
- the obtained DNA oligomer was identified by MALDI TOF-MS measurement.
- the synthesized DNA oligomer was made into an aqueous solution by adding about 500 ⁇ L of distilled water.
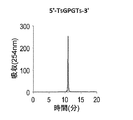
- Figures 2 to 7 show the reverse-phase HPLC chromatograms of the synthesized DNA oligomers.
- the retention time of the synthesized DNA oligomer in the reverse phase HPLC is as follows.
- 10 ⁇ L of DNA aqueous solution contains 10 ⁇ L of enzymatic degradation solution (calf intestinal alkaline phosphatase (100 U / mL) and P1 nuclease (100 U / mL) And then decomposed to deoxynucleoside by standing at room temperature for 12 hours by reverse-phase HPLC (3-20% CH 3 CN / 50 mM ammonium formate aqueous solution, 20 minutes, detected at 254 nm) Went by.
- enzymatic degradation solution calf intestinal alkaline phosphatase (100 U / mL) and P1 nuclease (100 U / mL)
- reverse-phase HPLC 3-20% CH 3 CN / 50 mM ammonium formate aqueous solution, 20 minutes, detected at 254 nm
- the molar absorption constant at 260 nm of 5′-TTTTCTTPTCCTTTT-3 ′ was calculated to be 119,000. This value was very close to the calculated value of 119,400 for the molar absorption constant at 260 nm of 5'-TTTTCTTTTCCTTTT-3 '(Predicting ultraviolet spectrum of single and double stranded deoxyribonucleic acids. Tataurov, AV, You , Y. and Owczarzy, R. Biophys. Chem. 2008, 133, 66-70), the molar extinction coefficient at 260 nm of a DNA oligomer containing one P is the molar extinction of DNA calculated by substituting P for T.
- the DNA oligomer was diluted to measure the absorbance at 260 nm, and the molar extinction coefficient at 260 nm calculated by substituting P for T (Predicting ultraviolet spectrum of single and double stranded deoxyribonucleic acids) Tataurov, AV, You, Y. and Owczarzy, R. Biophys. Chem. 2008, 133, 66-70), the concentration of the original DNA oligomer was determined.
- the molar extinction constant ⁇ of each DNA oligomer at 260 nm when P is replaced with T is as follows.
- the DNA oligomer is cleaved.
- the rate of the cleavage reaction and the one DNA fragment produced by the cleavage are different.
- the DNA fragment generated by the primary amine used is different because the amine used is added to the newly generated DNA fragment at the 3′-end. This DNA fragment may be further degraded to the 3′-phosphate end, in which case the same DNA fragment is generated regardless of the primary amine used.
- a DNA fragment that newly generates a 5′-phosphate terminus the same DNA fragment is generated regardless of the primary amine used when the source DNA oligomer is the same. Specific reaction of the primary amine is shown below.
- the molecular weight of the fractionated product was measured by MALDI TOF-MS. Further, the ratio of deoxynucleoside obtained by enzymatic decomposition was determined for a part of the product. Regarding pTCCTTTT, it was confirmed that the retention time in the reverse phase HPLC of the product treated with alkaline phosphatase coincided with TCCTTTT synthesized separately, and the molecular weight by MALDI TOF-MS measurement also coincided.
- a part of the product 2 may be further decomposed to become a 3′-phosphate end.
- Fig. 8 shows reversed-phase HPLC chromatograms of the synthesized DNA oligomer before and after the reaction.
- R Nu means a P-derived moiety that is generated in association with the cleavage reaction.
- R Nu is expected to be formed by adding one molecule of a nucleophile to the degraded nucleotide and removing the unnatural base.
- Figures 10 to 15 show the reverse-phase HPLC chromatograms before and after the reaction of each synthesized DNA oligomer.
- R Nu means a P-derived moiety that is generated in association with the cleavage reaction.
- R Nu is expected to be formed by adding one molecule of a nucleophile to the degraded nucleotide and removing the unnatural base.
- Fig. 16 shows the reversed-phase HPLC chromatograms before and after the reaction of the synthesized DNA oligomer.
- R Nu means a P-derived moiety that is generated in association with the cleavage reaction.
- R Nu is expected to be formed by adding one molecule of a nucleophile to the degraded nucleotide and removing the unnatural base.
- a DNA oligomer having the sequence 5′-PTTATTGTT-3 ′ was synthesized by the same method as described above. Cutting out from the carrier was performed with a 1: 1 mixed solution of 28% aqueous ammonia and 40% aqueous methylamine. The excised DNA oligomer solution was placed in a screw tube and deprotected by heating in a water bath at 70 ° C. for 16 hours. Ammonia and methylamine were removed under reduced pressure using a centrifugal concentrator and passed through a 0.45 ⁇ m filter.
- the fraction containing the main peak was separated by reverse phase HPLC (0-50% CH 3 CN / 50 mM ammonium formate aqueous solution, 20 minutes, detected at 254 nm), and formic acid was collected using a centrifugal concentrator and a freeze dryer. Ammonium and solvent were distilled off.
- MALDI TOF-MS measurement it was confirmed that the obtained DNA oligomer coincided with the molecular weight of 5′-TTATTGTT-3 ′ phosphorylated at the 5′-terminus.
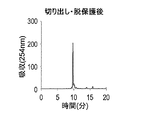
- FIG. 17 shows a reverse phase HPLC chromatogram of the DNA oligomer after cutting and deprotection.
- a DNA oligomer having the sequence 5′-PTTATTGTT-3 ′ was synthesized by the same method as described above. Cleavage from the carrier was performed with 28% aqueous ammonia, and deprotection was performed by standing at room temperature (about 25 ° C.) for 16 hours. Ammonia was distilled off under reduced pressure using a centrifugal concentrator and passed through a 0.45 ⁇ m filter. Thereafter, the fraction containing the main peak was separated by reverse phase HPLC (0-50% CH 3 CN / 50 mM ammonium formate aqueous solution, 20 minutes, detected at 254 nm), and formic acid was collected using a centrifugal concentrator and a freeze dryer.
- FIG. 18 shows a reverse phase HPLC chromatogram of the DNA oligomer after cutting and deprotection
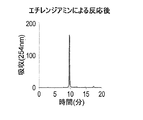
- FIG. 19 shows a reverse phase HPLC chromatogram of the DNA oligomer after the reaction with ethylenediamine.
- a DNA oligomer having the sequence 5′-TTATTGTTPT-3 ′ was synthesized by the same method as described above. Cutting out from the carrier was performed with a 1: 1 mixed solution of 28% aqueous ammonia and 40% aqueous methylamine. The excised DNA oligomer solution was placed in a screw tube and deprotected by heating in a water bath at 70 ° C. for 16 hours. Ammonia and methylamine were removed under reduced pressure using a centrifugal concentrator and passed through a 0.45 ⁇ m filter.
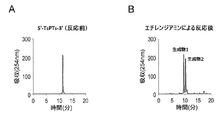
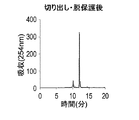
- Fig. 20 shows the reverse-phase HPLC chromatogram of the DNA oligomer after excision and deprotection
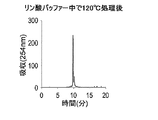
- Fig. 21 shows the reverse-phase HPLC chromatogram of the DNA oligomer after treatment at 120 ° C in 100 ⁇ mM phosphate buffer.
- R Nu is expected to be formed by adding one molecule of a nucleophile to the degraded nucleotide and removing the unnatural base.
- a DNA oligomer having the sequence 5′-TTATTGTTPT-3 ′ was synthesized by the same method as described above. Cleavage from the carrier was performed with 28% aqueous ammonia, and deprotection was performed by standing at room temperature (about 25 ° C.) for 16 hours. Ammonia was distilled off under reduced pressure using a centrifugal concentrator and passed through a 0.45 ⁇ m filter. Thereafter, the fraction containing the main peak was separated by reverse phase HPLC (0-50% CH 3 CN / 50 mM ammonium formate aqueous solution, 20 minutes, detected at 254 nm), and formic acid was collected using a centrifugal concentrator and a freeze dryer.
- Ammonium and solvent were distilled off. 30 ⁇ L of distilled water and 30 ⁇ L of ethylenediamine were added to the resulting lyophilized product and heated in a water bath at 70 ° C. for 3 hours. The reaction mixture was neutralized with acetic acid and then passed through a 0.45 ⁇ m filter. After that, the peak with a retention time of around 10 minutes was purified by reverse phase HPLC (0-50% CH 3 CN / 50 mM ammonium formate aqueous solution, 20 minutes, detected at 254 nm), using a centrifugal concentrator and a freeze dryer. Ammonium formate and the solvent were distilled off.
- Figure 22 shows the reverse-phase HPLC chromatogram of the DNA oligomer after excision and deprotection
- Figure 23 shows the reverse-phase HPLC chromatogram of the DNA oligomer after the reaction with ethylenediamine.
- the reverse phase HPLC chromatogram of the oligomer is shown in FIG. 24, respectively.
- R Nu is expected to be formed by adding one molecule of a nucleophile to the degraded nucleotide and removing the unnatural base.
- a DNA oligomer having the sequence 5′-PTTATTGTT-3 ′ was synthesized by the same method as described above. However, the DMTr group at the 5′-end of the last deoxynucleoside was not deprotected and was synthesized under the conditions of “DMTr ON”. Cleavage from the carrier was performed with 28% aqueous ammonia, and deprotection was performed by standing at room temperature (about 25 ° C.) for 16 hours. Ammonia was distilled off under reduced pressure using a centrifugal concentrator and passed through a 0.45 ⁇ m filter.
- the fraction containing the main peak was separated by reverse phase HPLC (0-50% CH 3 CN / 50 mM ammonium formate aqueous solution, 20 minutes, detected at 254 nm), and formic acid was collected using a centrifugal concentrator and a freeze dryer. Ammonium and solvent were distilled off. 100 ⁇ L of distilled water and 100 ⁇ L of ethylenediamine were added to the resulting lyophilized product and heated in a water bath at 70 ° C. for 8 hours.
- the reaction mixture was diluted to 1 mL with water and neutralized with acetic acid, and then the fraction containing the main peak was separated by reverse phase HPLC (0-50% CH 3 CN / 50 mM ammonium formate aqueous solution, 20 min, 254 (detected at nm), ammonium formate and the solvent were distilled off using a centrifugal concentrator and a freeze dryer. It was confirmed by MALDI TOF-MS that the obtained DNA oligomer had the same molecular weight as 5′-TTATTGTT-3 ′ phosphorylated at the 5′-end.
- Figure 25 shows the reverse-phase HPLC chromatogram of the DNA oligomer with DMTr protection at the 5'-end after excision and deprotection.
- Figure 25 shows the reverse-phase HPLC chromatogram of the DNA oligomer phosphorylated at the 5'-end after reaction with ethylenediamine. Are shown in FIG.
- DNA oligomers 5'-end of the post-reaction with ethylenediamine was phosphorylated main peak: 5'-pTTATTGTT-3 'molecular formula C 80 H 104 N 22 O 54 P 8 FW 2485.6 [MH] - 2484.6 Found 2484.6
- the reaction scheme is shown below.
- TS1 5'-FAM-ATAGTGAGTCGTATTAGGCCCGATCCAGGCCATCGCGCATGCTCAG-3 '(SEQ ID NO: 7)
- TS3 5'-ATAGTGAGTCGTATTAGGCCCGATCCAGGCCATCGCGCATGCTCAG-3 '(SEQ ID NO: 8)
- TF3 5'-NH 2 -TTAACTGAGCATGCGCGATGGCCTG-3 '(SEQ ID NO: 9)
- TT1 5'-pGATCGGGCCTAATACGACTCACTAT-FAM-3 '(SEQ ID NO: 10)
- TT2 5'-pATCGGGCCTAATACGACTCACTAT-FAM-3 '(SEQ ID NO: 11)
- TT1 and TT2 which are DNA oligomers phosphorylated at the 5 ′ end, were prepared by the following method.
- a DNA oligomer having sequences of 5′-PGATCGGGCCTAATACGACTCACTAT-FAM-3 ′ (SEQ ID NO: 12) and 5′-PATCGGGCCTAATACGACTCACTAT-FAM-3 ′ (SEQ ID NO: 13) The compound was synthesized in the same manner as in the above Use Example 1-7. Cutting out from the carrier was performed with a 1: 1 mixed solution of 28% aqueous ammonia and 40% aqueous methylamine. The DNA oligomer solution thus cut out was put in a screw tube and deprotected by heating in a water bath at 70 ° C. for 16 hours.
- 27A and B show reverse phase HPLC chromatograms of TT1 and TT2 after cutting and deprotection, respectively.
- TT1 Main peak: 5'-pGATCGGGCCTAATACGACTCACTAT-FAM-3 ' molecular formula C 270 H 332 N 94 O 161 P 26 FW 8275.4 [MH] - 8274.4 Found 8273.7
- TT2 Main peak: 5'-pATCGGGCCTAATACGACTCACTAT-FAM-3 ' molecular formula C 260 H 320 N 89 O 155 P 25 FW 7946.2 [MH] - 7945.2 Found 7946.1
- 5′-FAM The structure of a DNA oligomer in which the 5 ′ end is FAM-modified is shown below.
- DNA oligomer whose 5 ′ end is modified with an amino group (hereinafter also referred to as “5′-NH 2 ”) is shown below.
- FAM-3 ′ The structure of a DNA oligomer (hereinafter also referred to as “FAM-3 ′”) in which the 3 ′ end is FAM-modified is shown below.
- the fluorescent dye FAM when the polyacrylamide gel is irradiated with ultraviolet rays, the fluorescent dye FAM emits fluorescence, so that the FAM-modified DNA oligomer is detected as a white band on the gel.
- the first lane band from the left is TS1 which is a DNA oligomer consisting of 46 bases
- the second and sixth lane bands from the left are the DNA oligomers consisting of 25 bases and 24 bases, TT1 and TT2, respectively. , Respectively.
- the third to fifth lanes from the left are the DNA oligomers obtained by ligation reaction of TT1 and TF3 with TS3 as a template for 1, 2 or 3 hours on the gel of FAM-modified TT1-derived DNA oligomers. Indicates the position. Since the position of the main band detected in these lanes is close to the position of TS1 consisting of 46 bases, TT1 and TF3 were connected in the ligation reaction mixture, and a DNA oligomer consisting of 50 bases was formed. It is suggested.
- the first lane from the right shows the position on the gel of the DNA oligomer in the control group obtained by ligating TT2 and TF3 for 3 hours using TS3 as a template. Since the position of the main band detected in this lane is close to the position of TT2, which is a DNA oligomer before ligation consisting of 24 bases, TT2 annealed with the template TS3 but was not ligated with TF3. Is suggested.
- TT2 is a DNA oligomer consisting of a base sequence with one base deleted from the 5 'end of TT1, so when TT2 and TF3 are annealed using TS3 as a template, a 1 base gap is generated between TT2 and TF3. ( Figure 28A). For this reason, it is considered that the ligation reaction did not proceed in the control sample.
- Figures 29-30 show the reverse-phase HPLC chromatograms of the synthesized DNA oligomers.
- the retention time of the synthesized DNA oligomer in the reverse phase HPLC is as follows.
- FIGS. 31 and 33 The reverse phase HPLC chromatograms of the synthesized DNA oligomers before and after the reaction are shown in FIGS. 31 and 33, and the MALDI-TOF-MS spectra of the product 1 and other by-products are shown in FIGS. 32 and 34, respectively.
- MALDI TOF-MS data of product 1 in each DNA oligomer is as follows.
- R Nu is expected to be formed by adding one molecule of nucleophile to the degraded nucleotide and removing the unnatural base.
- this deoxynucleoside has an ethynyl group, it can be easily coupled with another labeled molecule (for example, a fluorescent dye) having an azide group by click chemistry.
- a labeled molecule for example, a fluorescent dye
- the above mechanism makes it possible to detect the deoxynucleoside incorporated into the DNA of the cell with a label such as a fluorescent dye.
- Raman spectroscopy the deoxynucleoside can be detected without performing the labeling reaction as described above. Therefore, the deoxynucleoside can be monitored even in living cells (Non-patent Document 64).
- Non-patent Documents 68 and 69 disclose methods for synthesizing 5-ethynyl-2'-deoxyuridine.
- a method of synthesizing a DNA oligomer containing 5-ethynyl-2'-deoxyuridine using an automatic DNA synthesizer has already been reported (Non-patent Documents 68 and 69).
- the deoxynucleoside is applied to DNA strand breaks and phosphorylated phosphorylated ends.
- the phosphoramidite which is a monomer unit of 5-ethynyl-2′-deoxyuridine, was synthesized according to the methods described in Non-Patent Documents 66 and 68.
- a DNA oligomer containing 5-ethynyl-2′-deoxyuridine was synthesized by the same method as in Synthesis Example 1-4 (Non-patent Document 69). Cleavage from the carrier and deprotection were performed by standing in 50 mM K 2 CO 3 / MeOH at room temperature (about 25 ° C.) for 16 hours.
- the DNA oligomer contains adenine and / or guanine
- 500 ⁇ L of water is added to the DNA oligomer, and then methanol is distilled off under reduced pressure using a centrifugal concentrator.
- Methanol was distilled off under reduced pressure using a centrifugal concentrator, 1 mL of distilled water was added, and then passed through a 0.45 ⁇ m filter.
- the fraction containing the main peak was separated by reverse phase HPLC (0-50% CH 3 CN / 50 mM ammonium formate aqueous solution, 20 minutes, detected at 254 nm), and formic acid was collected using a centrifugal concentrator and a freeze dryer. Ammonium and solvent were distilled off. The obtained DNA oligomer was identified by MALDI TOF-MS measurement.
- 35 and 36 show reverse phase HPLC chromatograms of the synthesized DNA oligomers.
- the retention time of the synthesized DNA oligomer in the reverse phase HPLC is as follows.
- DNA oligomer containing 5-ethynyl-2′-deoxyuridine> [Use Example 3-1: Cleavage reaction of DNA oligomer containing 5-ethynyl-2'-deoxyuridine] 30 ⁇ L of DNA oligomer aqueous solution (strand concentration 500 ⁇ M) and 30 ⁇ L of methylamine (40% aqueous solution) were added to the screw tube, and heated in a water bath at 70 ° C. for 10 minutes or 30 minutes.
- Methylamine was distilled off from the reaction mixture under reduced pressure using a centrifugal concentrator, the reaction mixture was analyzed by reverse phase HPLC, and the product was separated (0-50% CH 3 CN / 50 mM ammonium formate aqueous solution, 20 Min, detected at 254 nm). The molecular weight of the fractionated product was measured by MALDI TOF-MS (sample product after reaction for 30 minutes).
- MALDI TOF-MS data of product 1 in each DNA oligomer is as follows.
- R Nu is expected to be formed by adding one molecule of a nucleophile to the degraded nucleotide and removing the unnatural base.
- a DNA oligomer containing 5-ethynyl-2'-deoxyuridine was almost completely cleaved by heating at 70 ° C for 30 minutes. From this result, a DNA oligomer containing 5-ethynyl-2′-deoxyuridine is a DNA oligomer containing 5-phenylethynyl-2′-deoxyuridine (Use Example 1) and 5- (1-propynyl) -2′- It was revealed that the cleavage reaction proceeded very rapidly as compared with a DNA oligomer containing deoxyuridine (Use Example 2).
- products 1 and 2 were produced by the cleavage reaction.
- the fractionated products 1 and 2 were measured for molecular weight by MALDI-TOF-MS.
- MALDI-TOF-MS data for products 1 and 2 are as follows.
- R Nu is expected to be formed by adding one molecule of a nucleophile to the degraded nucleotide and removing the unnatural base.
- Product 1 was confirmed to have the same retention time by HPLC analysis by overstrike with the decomposition product of methylamine.
- the nucleophilic species of this reaction is considered to be hydroxide ions.
- the reaction of DNA oligomers containing cytosine resulted in more than two product peaks. This result suggests that deamination of cytosine occurs when heated under alkaline conditions.
- the target substance was purified with a silica gel column (developing solvent was 2 to 5% MeOH, 2% Et 3 N / CH 2 Cl 2 ).
- the solvent of the objective fraction was distilled off under reduced pressure and dried with a vacuum pump. Dissolve in a small amount of methylene chloride, add hexane, distill off the solvent under reduced pressure, and dry with a vacuum pump to obtain 1.49 g (1.94 mmol, yield 79%) of the target substance (including 1.5 molecules of molecular weight 766.43 triethylamine) Was obtained as a white powder.
- the carbomethoxyvinyl group in the base moiety is converted to a carbamoylvinyl group under deprotection conditions.
- Ammonia was distilled off under reduced pressure using a centrifugal concentrator and passed through a 0.45 ⁇ m filter. Thereafter, the fraction containing the main peak was separated by reverse phase HPLC (0-50% CH 3 CN / 50 mM ammonium formate aqueous solution, 20 minutes, detected at 254 nm), and formic acid was collected using a centrifugal concentrator and a freeze dryer. Ammonium and solvent were distilled off.
- the obtained DNA oligomer was identified by MALDI TOF-MS measurement.
- the retention time of the synthesized DNA oligomer in the reverse phase HPLC is as follows.
- reaction mixture was neutralized with acetic acid and analyzed by reverse phase HPLC to fractionate the product (0-50% CH 3 CN / 50 mM aqueous ammonium formate, 20 min, detected at 254 nm).
- the molecular weight of the fractionated product was measured by MALDI TOF-MS (sample product after reaction for 8 hours).
- FIGS. 42 and 44 show reverse phase HPLC chromatograms of the synthesized DNA oligomers before and after the reaction
- FIGS. 43 and 45 show MALDI-TOF-MS spectra of the product 1 and other by-products, respectively.
- MALDI TOF-MS data of product 1 in each DNA oligomer is as follows.
- R Nu is expected to be formed by adding one molecule of a nucleophile to the degraded nucleotide and removing the unnatural base.
- the organic layer was washed twice with 100 mL of saturated brine, and then the organic layer was dried over magnesium sulfate. The organic layer after drying was filtered, and the solvent of the filtrate was distilled off under reduced pressure, followed by drying with a vacuum pump. About 30 mL of 1,4-dioxane was added and dissolved, and then 961 ⁇ L (15.0 mmol) of propargylamine (molecular weight 55.08, specific gravity 0.86) was added and stirred at 25 ° C. for 2 hours. The solvent was distilled off under reduced pressure, 30 mL of methanol and 30 mL of 28% aqueous ammonia were added, and the mixture was stirred at 25 ° C. for 24 hours.
- the target substance was purified with a silica gel column (developing solvent was 2 to 5% MeOH, 2% Et 3 N / CH 2 Cl 2 ).
- the solvent of the objective fraction was distilled off under reduced pressure and dried with a vacuum pump. Dissolve in a small amount of methylene chloride, add hexane, distill off the solvent under reduced pressure, and dry with a vacuum pump to obtain 631 mg (0.924 mmol, 83% yield) of the target substance (molecular weight 682.85; including one triethylamine molecule) was obtained as a white powder.
- the fraction containing the main peak was separated by reverse phase HPLC (0-50% CH 3 CN / 50 mM ammonium formate aqueous solution, 20 minutes, detected at 254 nm), and formic acid was collected using a centrifugal concentrator and a freeze dryer. Ammonium and solvent were distilled off. The obtained DNA oligomer was identified by MALDI TOF-MS measurement.
- Fig. 46 shows the reverse phase HPLC chromatogram of the synthesized DNA oligomer.
- the retention time of the synthesized DNA oligomer in the reverse phase HPLC is as follows.
- reaction mixture was neutralized with acetic acid and analyzed by reverse phase HPLC to fractionate the product (0-50% CH 3 CN / 50 mM aqueous ammonium formate, 20 min, detected at 254 nm).
- the molecular weight of the fractionated product was measured by MALDI TOF-MS (sample product after 16 hours reaction).
- FIGS. 48 and 50 show MALDI-TOF-MS spectra of product 1 and other by-products, respectively.
- MALDI TOF-MS data of product 1 in each DNA oligomer is as follows.
- R Nu is expected to be formed by adding one molecule of a nucleophile to the degraded nucleotide and removing the unnatural base.
- the sequence design of the DNA oligomer containing an unnatural base is performed so that a protruding end that can be ligated by ligase is generated, Polymerase chain reaction was performed using two target DNAs as templates.
- the base sequence of the target DNA consists of gene 1 (136 base pairs) corresponding to positions 28634-28770 and gene 2 (179 base pairs) corresponding to positions 28743-28932 from ⁇ DNA.
- 5′-GTAGCGATCAAGCCATGAATGTAACG-3 ′ (SEQ ID NO: 22) as the forward primer strand of gene 1 and 5′-ACAGGCGAATCGCAAXCACTG-3 ′
- X 5-phenylethynyl-2′-deoxyuridine (P) or 5 as the reverse primer strand -Ethynyl-2′-deoxyuridine (Pa))
- SEQ ID NO: 23 as 5′-ATTGCGATTCGCCTGXCTCTGC-3 ′
- 5′-GGTTGACTTAAATCGACCAGTAACAGG-3 ′ (SEQ ID NO: 25) were designed
- the polymerase chain reaction was performed using a final concentration of 0.3 ⁇ M primer, 1 ⁇ DNA, 0.4 ⁇ mM dNTPs, 1 U KOD Fx neo, and a buffer attached to the product. -According to catalog number (TP600), annealing was performed at 94 ° C for 2 minutes, denaturation: 98 ° C for 10 seconds, elongation: 68 ° C for 10 seconds (denaturation ⁇ extension repeated 35 times). The amplified product was subjected to 10% non-denaturing polyacrylamide gel to confirm the amplified product by polymerase chain reaction. The results are shown in FIG.
- the gel after electrophoresis is infiltrated into an ethidium bromide solution at room temperature for 30 minutes, thereby performing a double-stranded DNA selective staining operation and detecting fluorescence emitted by ultraviolet irradiation to detect DNA.
- the strand is detected as a white band.
- the band in Lane 1 was a TrackIt (trademark) 100 bp DNA ladder (catalog number 48810488-058, manufactured by Invitrogen) in which a band was detected every 100 base pairs as a gene marker.
- 1 shows amplification products
- the amplification product of gene 2 when using as a primer strand is shown.
- the amplification of the target product was confirmed from the mobility of 100 and 200 base pairs derived from the marker.
- the supernatant was collected, 150 ⁇ L of PCI mixed solution was added again, and then the solution was vigorously mixed using a vortex mixer and centrifuged until the two layers were uniformly separated. This operation was repeated a total of 5 times.
- the supernatant obtained by the above operation was applied to a Bio-Spin 6 column (catalog number 732-6227 manufactured by BIO-RAD), and then purified by ethanol precipitation.
- the 5′-end phosphorylated DNA fragment was dried using a centrifugal concentrator and redissolved in 5 ⁇ L of TE buffer.
- the lane 1 band used was a TrackIt (trademark) 100 bp DNA ladder in which a band was detected every 100 base pairs as a gene marker.
- the band in lane 6 is a DNA fragment having the nucleotide sequence corresponding to the 28634-28932th position from the 5 ′ end in ⁇ DNA, that is, gene 3 (299 nucleotides) having the same nucleotide sequence as the ligation product obtained by linking genes 1 and 2.
- the amplification product of the pair) is shown.
- Synthesis Example 7 DNA oligomer containing 2′-deoxy-4-N- ⁇ N ′, N′-di (n-butyl) formamidine ⁇ -5-phenylethynylcytidine>
- Synthesis Example 7-1 Synthesis of 2′-deoxy-4-N- ⁇ N ′, N′-di (n-butyl) formamidine ⁇ -5-phenylethynylcytidine] 2′-Deoxy-5-phenylethynylcytidine was synthesized according to Non-Patent Document 41.
- the solvent of the reaction solution was distilled off under reduced pressure, and the target substance was purified with a silica gel column (developing solvent: CH 2 Cl 2 only to 5-10% MeOH / CH 2 Cl 2 ).
- the solvent of the objective fraction was distilled off under reduced pressure to obtain 700 mg (1.50 mmol, yield 69%) of the objective substance (molecular weight 466.57) as a light brown oily substance.
- the solvent of the objective fraction was distilled off under reduced pressure, and a small amount of methylene chloride and hexane were added and azeotroped. 924 mg (1.20 mmol, 80% yield) of the desired product was obtained as a white powder.
- the ammonia was distilled off under reduced pressure using a centrifugal concentrator, the solution containing DNA was passed through a 0.45 ⁇ m filter, and the main peak was purified by reverse-phase HPLC (0-50% CH 3 CN / 50 mM formic acid. Ammonium aqueous solution, 20 minutes, detected at 254 nm), ammonium formate and the solvent were distilled off by centrifugal concentrator and lyophilization. To the obtained residue, 100 ⁇ L of 80% aqueous acetic acid solution was added, mixed well, and allowed to stand at 25 ° C. for 30 minutes.
- distilled water was added to the mixture so as to be about 1 mL, passed through a 0.45 ⁇ m filter, and the main peak eluted at a retention time of 13 to 14 minutes was purified by reverse phase HPLC (8- 20% CH 3 CN / 50 mM ammonium formate aqueous solution, 20 minutes, detected at 254 nm), ammonium formate and the solvent were distilled off by centrifugal concentrator and lyophilization.
- the obtained DNA oligomer was identified by MALDI TOF-MS measurement.
- the synthesized DNA oligomer was made into an aqueous solution by adding about 500 ⁇ L of distilled water.
- the concentration of DNA was determined by calculating the molar extinction coefficient of 260 nm by approximating Q to C, and calculating the concentration from the UV absorption of 260 nm.
- the MALDI TOF-MS data of the synthesized DNA oligomer is as follows.
- methylamine was distilled off under reduced pressure with a centrifugal concentrator and analyzed by reverse phase HPLC to fractionate the product (8-20% CH 3 CN / 50 mM ammonium formate aqueous solution, 20 minutes, at 254 nm. detection). The molecular weight of the fractionated product was measured by MALDI TOF-MS.
- 55 and 56 show reverse phase HPLC chromatograms after the reaction of each synthesized DNA oligomer.
- R Nu means a Q-derived moiety that is generated in accordance with the cleavage reaction.
- R Nu is expected to be formed by adding one molecule of a nucleophile to the degraded nucleotide and removing the unnatural base.
- the reaction mixture was neutralized with acetic acid and analyzed by reverse phase HPLC (8-20% CH 3 CN / 50 mM aqueous ammonium formate, 20 min, detected at 254 nm).
- An HPLC chart of the reaction mixture of 5′-T 6 (Q) T 6 -3 ′ is shown in FIG. 57, and an HPLC chart of the reaction mixture of 5′-TTTTCTT (Q) TCCTTTT-3 ′ is shown in FIG.
- 5'-T 6 QT 6 -3 was quantified based on the peak area of 'which is the 5'-cleavage product of 5'-T 6 p-3' , the 5'-T 6 QT 6 -3 '
- the amount of cleavage product on the 5′-side was calculated to be 3.6 times higher with methylamine treatment than with ethylenediamine treatment.
- Q obtained in Synthesis Example 7 has a plurality of tautomers as shown below. Therefore, in oligodeoxynucleotides containing Q, it is considered that the elimination reaction of E and the cleavage reaction of oligodeoxynucleotide proceed when a tautomer of Q reacts with a nucleophile.
- DNA means an oligodeoxynucleotide
- Nu means a nucleophile
- Synthesis Example 8 DNA oligomer containing 5- (2-oxy-2-phenylethyl) -2′-deoxyuridine (HP)>
- Synthesis Example 8-1 Synthesis of DNA oligomer containing 5- (2-oxy-2-phenylethyl) -2′-deoxyuridine (HP)] Based on a known reaction at the nucleoside level, in the DNA oligomer containing P obtained in Synthesis Example 1, the base portion of P was cyclized with silver ions (A facile synthesis of fluorophores based on 5-phenylethynyluracils.Hudson, HER And Moszybski, JM Synlett 2006, 18, p. 2997-3000), followed by sodium hydroxide treatment to convert P to 5- (2-oxy-2-phenylethyl) -2'-deoxyuridine (Non-patent document 39).
- the obtained solution was passed through a 0.45 ⁇ m filter, and purified by reverse phase HPLC (8-20% CH 3 CN / 50 mM aqueous ammonium formate solution, 20 minutes, detected at 254 nm) and desalted.
- the product obtained as the main peak was identified by MALDI TOF-MS measurement.
- DNA concentration was determined by approximating HP with T and calculating a molar extinction coefficient of 260 nm, and calculating the concentration from 260 nm UV absorption.
- the retention time of the synthesized DNA oligomer in the reverse phase HPLC is as follows.
- the MALDI TOF-MS data of the synthesized DNA oligomer is as follows.
- 5'-TTTTTT (HP) molecular formula for TTTTTT-3 'C 137 H 174 N 26 O 90 P 12, [MH] - Calculated 3995.6, Found 3995.6 (SEQ ID NO: 28)
- 5′-TTTTCTT (HP) TCCTTTT-3 ′, the molecular formula C 154 H 197 N 33 O 101 P 14 , [MH] ⁇ calculated value 4559.0, measured value 4559.4 (SEQ ID NO: 29) ⁇ 16.
- methylamine was distilled off under reduced pressure with a centrifugal concentrator and analyzed by reverse phase HPLC to fractionate the product (8-20% CH 3 CN / 50 mM ammonium formate aqueous solution, 20 minutes, at 254 nm. detection). The molecular weight of the fractionated product was measured by MALDI TOF-MS.
- 61 and 62 show reverse phase HPLC chromatograms after the reaction of each synthesized DNA oligomer.
- R Nu means a part derived from HP that is generated in accordance with the cleavage reaction.
- R Nu is expected to be formed by adding one molecule of a nucleophile to the degraded nucleotide and removing the unnatural base.
- the reaction mixture was neutralized with acetic acid and analyzed by reverse phase HPLC (8-20% CH 3 CN / 50 mM aqueous ammonium formate, 20 min, detected at 254 nm).
- An HPLC chart of the reaction mixture of 5′-T 6 (HP) T 6 -3 ′ is shown in FIG. 63, and an HPLC chart of the reaction mixture of 5′-TTTTCTT (HP) TCCTTTT-3 ′ is shown in FIG.
- 5'-TTTTCTTp-3 ' which is a 5'-side cleavage product of 5'-TTTTCTT (HP) TCCTTTT-3'
- 5'-TTTTCTT (HP) TCCTTTT-3 The amount of cleavage product on the 5'-side of ' was calculated to be 2.1 times higher with methylamine treatment than with ethylenediamine treatment. From this result, it was suggested that in the cleavage reaction of DNA oligomer using methylamine or primary amine of ethylenediamine, the one having the highest reaction rate is the cleavage reaction using methylamine.
- the HP obtained in Synthesis Example 8 has a structure in which water is added to the triple bond of P obtained in Synthesis Example 1.
- P can be heated in the presence of an amine used as a nucleophile to form an amine adduct in which an amine is added to a triple bond, and to form HP via tautomerism and amine elimination. There is sex.
- R Nu means a group generated with the addition of a nucleophile.
- the oligodeoxynucleotide is considered to be cleaved by elimination of a portion derived from an unnatural base while causing the following intramolecular cyclization reaction.
- an oligodeoxynucleotide containing an amine adduct is considered to be cleaved after causing an intramolecular cyclization reaction as described below.
- the solvent of the reaction solution was distilled off under reduced pressure, 200 mL of methanol was added and filtered. The residue was washed with methanol. The filtrate was collected, the solvent was distilled off under reduced pressure, and the target substance was purified with a silica gel column (developing solvent was 10 to 20% MeOH / CH 2 Cl 2 ). The solvent of the objective fraction was distilled off under reduced pressure, and the precipitate formed with a small amount of methanol and acetonitrile was filtered and dried under reduced pressure.
- the solvent was distilled off under reduced pressure, and the target substance was purified with a silica gel column (developing solvent was 5 to 10% MeOH / CH 2 Cl 2 ).
- the solvent of the objective fraction was distilled off under reduced pressure, and the precipitate formed with a small amount of ethyl acetate and hexane was filtered and dried under reduced pressure. 2.10 g (4.11 mmol, 66% yield) of the target substance (molecular weight 510.59) was obtained as a pale yellow powder.
- the solvent of the objective fraction was distilled off under reduced pressure, and a small amount of methylene chloride and hexane were added and azeotroped. 2.20 g (2.71 mmol, 66% yield) of the desired product (molecular weight 812.95) was obtained as a pale yellow powder.
- N, N, N ′, N′-tetraisopropyl phosphoramidite (molecular weight 236.68, specific gravity 1.061 g / mL) was added, and the mixture was stirred at room temperature for 30 minutes. 25 mL of AcOEt and 25 mL of saturated saline were added, and the mixture was separated. The organic layer was further washed twice with saturated brine. The obtained organic layer was dried over anhydrous magnesium sulfate and filtered through a filter, and the solvent of the filtrate was distilled off under reduced pressure. After azeotroping with acetonitrile three times, 10 mL of acetonitrile was added and dissolved, and a small amount of molecular sieve was added. Attached to an automatic DNA synthesizer and used for DNA synthesis.
- the DNA was synthesized by the phosphoramidite method with DMTr ON using a DNA synthesis protocol of a normal automatic DNA synthesizer with 1 ⁇ mol scale. Cutting out from the carrier was performed with 28% aqueous ammonia, and deprotection was performed by standing at 25 ° C. for 16 hours. After deprotection, the ammonia was distilled off under reduced pressure using a centrifugal concentrator, the solution containing DNA was passed through a 0.45 ⁇ m filter, and the main peak was purified by reverse-phase HPLC (0-50% CH 3 CN / 50 mM formic acid.
- Ammonium aqueous solution 20 minutes, detected at 254 nm), ammonium formate and solvent were distilled off by centrifugal concentrator and lyophilization. To the obtained residue, 100 ⁇ L of 80% aqueous acetic acid solution was added, mixed well, and allowed to stand at 25 ° C. for 30 minutes.
- distilled water was added to the mixture to make about 1 mL, passed through a 0.45 ⁇ m filter, and the main peak eluted at a retention time of 13 to 14 minutes was purified by reverse phase HPLC (5- 20% CH 3 CN / 50 mM ammonium formate aqueous solution, 20 minutes, detected at 254 nm), ammonium formate and the solvent were distilled off by centrifugal concentrator and lyophilization.
- the obtained DNA oligomer was identified by MALDI TOF-MS measurement.
- the synthesized DNA oligomer was made into an aqueous solution by adding about 500 ⁇ L of distilled water.
- the concentration of DNA was determined by approximating fPG to C and calculating a molar extinction coefficient of 260 nm, and calculating the concentration from 260 nm UV absorption.
- 65 and 66 show reverse phase HPLC chromatograms of the synthesized DNA oligomers.
- the retention time of the synthesized DNA oligomer in the reverse phase HPLC is as follows.
- the MALDI TOF-MS data of the synthesized DNA oligomer is as follows.
- methylamine was distilled off under reduced pressure with a centrifugal concentrator and analyzed by reverse phase HPLC to fractionate the product (5-20% CH 3 CN / 50 mM ammonium formate aqueous solution, 20 minutes, at 254 nm. detection). The molecular weight of the fractionated product was measured by MALDI TOF-MS.
- 67 and 68 show reverse phase HPLC chromatograms after the reaction of each synthesized DNA oligomer.
- the MALDI TOF-MS data of products 1 and 2 in each DNA oligomer are as follows.
- R Nu means a part derived from fPG that is generated along with the cleavage reaction.
- R Nu is expected to be formed by adding one molecule of a nucleophile to the degraded nucleotide and removing the unnatural base.
- 8-Bromo-2'-deoxyadenosine was synthesized according to a known document (Japanese Patent Laid-Open No. 2011-37796). 2.20 g (6.66 mmol) 8-bromo-2'-deoxyadenosine (molecular weight 330.14), 2.00 g (13.3 mmol) 2-formylbenzeneboronic acid (molecular weight 149.94) and 150 mg (0.666 mmol) palladium acetate (molecular weight) 224.51) and 525 mg (2.00 mmol) of triphenylphosphine (molecular weight 262.29) and 1.84 g (13.3 mmol) of potassium carbonate were added to an eggplant flask, and 40 mL of acetonitrile and 20 mL of water were added thereto at 80 ° C.
- the solvent was distilled off under reduced pressure, and the target substance was purified with a silica gel column (developing solvent was 2 to 5% MeOH / CH 2 Cl 2 ).
- the solvent of the desired fraction was distilled off under reduced pressure, a small amount of methylene chloride was added, diethyl ether was added, and the precipitate formed by adding hexane was filtered and dried under reduced pressure. 1.40 g (2.83 mmol, yield 65%) of the target substance (molecular weight 494.59) was obtained as a white powder.
- the target substance was purified with a silica gel column (developing solvent was 2% Et 3 N, 2% MeOH / CH 2 Cl 2 ).
- the solvent of the objective fraction was distilled off under reduced pressure, and a small amount of methylene chloride and hexane were added and azeotroped. 1.24 g (1.56 mmol, 59% yield) of the desired product (molecular weight 796.95) was obtained as a white powder.
- the DNA was synthesized by the phosphoramidite method with DMTr ON using a DNA synthesis protocol of a normal automatic DNA synthesizer with 1 ⁇ mol scale. Cutout was performed with 28% aqueous ammonia, and deprotection was performed by standing at 25 ° C. for 16 hours. After deprotection, the ammonia was distilled off under reduced pressure using a centrifugal concentrator, the solution containing DNA was passed through a 0.45 ⁇ m filter, and the main peak was purified by reverse-phase HPLC (0-50% CH 3 CN / 50 mM formic acid.
- Ammonium aqueous solution 20 minutes, detected at 254 nm), ammonium formate and solvent were distilled off by centrifugal concentrator and lyophilization. To the obtained residue, 100 ⁇ L of 80% aqueous acetic acid solution was added, mixed well, and allowed to stand at 25 ° C. for 30 minutes.
- distilled water was added to the mixture so as to be about 1 mL, passed through a 0.45 ⁇ m filter, and then the main peak eluted at a retention time of 13-14 minutes was purified by reverse phase HPLC (5- 20% CH 3 CN / 50 mM ammonium formate aqueous solution, 20 minutes, detected at 254 nm), ammonium formate and the solvent were distilled off by centrifugal concentrator and lyophilization.
- the obtained DNA oligomer was identified by MALDI TOF-MS measurement.
- the synthesized DNA oligomer was made into an aqueous solution by adding about 500 ⁇ L of distilled water.
- DNA concentration was determined by approximating fPA to C and calculating a molar extinction coefficient of 260 nm, and calculating the concentration from 260 nm UV absorption.
- the retention time of the synthesized DNA oligomer in the reverse phase HPLC is as follows.
- the MALDI TOF-MS data of the synthesized DNA oligomer is as follows.
- 5′-TTTTCTT (fPA) TCCTTTT-3 ′ the molecular formula C 154 H 196 N 36 O 99 P 14 , calculated value of [MH] ⁇ 4568.0, measured value 4568.5 (SEQ ID NO: 33) ⁇ 20.
- methylamine was distilled off under reduced pressure with a centrifugal concentrator and analyzed by reverse phase HPLC to fractionate the product (5-20% CH 3 CN / 50 mM ammonium formate aqueous solution, 20 minutes, at 254 nm. detection). The molecular weight of the fractionated product was measured by MALDI TOF-MS.
- 71 and 72 show reverse phase HPLC chromatograms after the reaction of each synthesized DNA oligomer.
- the MALDI TOF-MS data of products 1 and 2 in each DNA oligomer are as follows.
- R Nu means a part derived from fPA that is generated along with the cleavage reaction.
- Degradation reaction of DNA oligomer by methylamine is shown below.
- the product on the left corresponds to the product 2
- the product on the right corresponds to the product 1.
- R Nu is expected to be formed by adding one molecule of a nucleophile to the degraded nucleotide and removing the unnatural base.
- the oligodeoxynucleotide containing fPG or fPA is considered to be cleaved by elimination of a portion derived from an unnatural base while causing the following intramolecular cyclization reaction.
- the following reaction formula is for oligodeoxynucleotides containing fPG, but similar reactions are thought to proceed in the case of oligodeoxynucleotides containing fPA.
- LL1 5'-AGG (Pa) GCTTTATGACTCTGCCG-3 '(SEQ ID NO: 34)
- LL2 5'-AGGTGCTTTA (Pa) GACTCTGCCG-3 '(SEQ ID NO: 35)
- LL3 5'-ACCGAAGCG (Pa) GGTGTTTACGAAGG-3 '(SEQ ID NO: 36)
- LL4 5'-AGCG (Pa) GGTGTTTACGAAGG-3 '(SEQ ID NO: 37)
- LL5 5'-ACGC (Pa) TCGGTATCGCTCTG-3 '(SEQ ID NO: 38)
- LL6 5'-ACGCTTCGG (Pa) ATCGCTCTG-3 '(SEQ ID NO: 39)
- LL7 5'-ATAAAGCACC (Pa) CGGTGGTGAAAGG-3 '(SEQ ID NO: 40)
- LL8 5'-ACC (Pa) CGGTGGTGAAAGGGC
- Strand concentration 10 ⁇ M 2 ⁇ L of primer, 1 ⁇ L of 1 ng / ⁇ L ⁇ DNA, 10 ⁇ L of 2 mM dNTPs, 1 ⁇ L (1 U) of KOD Fx neo (Toyobo KFX-201), and buffer supplied with the product ( ⁇ 2) of 25 ⁇ L was added to the polymerase chain reaction tube.
- FIG. 73 shows the gel after ethidium bromide staining. In the photo in the figure, black and white are reversed.
- the bands in lane 1 are DNA markers (100, 200, 300, 400, 500, 600, 700, 800, 900, 1000, 1100, 1200, 1300, 1400, 1500 and 2072 bp, bands that appeared strongly. Are 600, 1500 and 2072 bp DNA).
- the lane 2 band shows the amplification product DNA when LL1 and LL3 are used as the primer set, and the lane 3 band shows the amplification product DNA when LL2 and LL4 are used as the primer set.
- the lane 4 band shows the amplification product DNA when LL5 and LL8 are used as the primer set, and the lane 5 band shows the amplification product DNA when LL6 and LL7 are used as the primer set. Show.
- the band in lane 1 shows DNA markers (0.5, 1.0, 1.5, 2, 3, 4, 6, 8, 10, and 12 kb).
- the bands in lanes 2 and 3 show the DNA of the ligation product when the ligation reaction was performed in 10 temperature cycles, and the bands in lanes 4 and 5 were ligated in 20 temperature cycles. In this case, the ligation reaction product DNA is shown, and the bands in lanes 6 and 7 show the ligation reaction product DNA when the ligation reaction is performed in 30 temperature cycles.
- the bands in lanes 2, 4 and 6 were ligated with the amplification product DNA when LL1 and LL3 were used as a primer set and the amplification product DNA when LL6 and LL7 were used as a primer set.
- the ligation reaction product DNA is shown, and the bands in lanes 3, 5 and 7 are the amplification product DNA when LL2 and LL4 are used as a primer set, and the bands when LL6 and LL7 are used as a primer set.
- the DNA of the ligation product when the ligation reaction is performed with the DNA of the amplification product is shown.
- a 3.8-kb kb DNA band was confirmed by ligation reaction of 1.5-kb and 2.3-kb raw DNA. This suggests that the DNA was cleaved by methylamine treatment, and hybridization of two DNA strands having one nucleotide gap (deletion site) occurred during the heating and cooling cycles for ligation. Ligase is thought to cause a covalent bond between the two DNAs. However, it is unclear only from the results of analysis by agarose gel electrophoresis how often the two DNAs were covalently linked by ligase. When hybridized DNA is introduced into E. coli or the like, nicks (cuts) that are not linked are generally repaired.
- the band in lane 1 shows DNA markers (0.5, 1.0, 1.5, 2, 3, 4, 6, 8, 10, and 12 kb).
- the bands in lanes 2 and 3 show the DNA of the cleavage reaction product when the DNA cleavage reaction was performed with methylamine treatment for 9 hours, and the bands in lanes 4 and 5 were subjected to methylamine treatment for 24 hours.
- the DNA of the cleavage reaction product when the DNA cleavage reaction is performed is shown, and the bands in lanes 6 and 7 indicate the DNA of the cleavage reaction product when the DNA cleavage reaction is performed for 48 hours with methylamine treatment. Is shown.
- the bands in lanes 8 and 9 indicate the DNA of the ligation product when the ligation reaction was performed with 30 temperature cycles after methylamine treatment for 48 hours to cleave the DNA.
- the bands in lanes 2, 4, 6, and 8 cleave the amplification product DNA when LL1 and LL3 are used as a primer set and the amplification product DNA when LL6 and LL7 are used as a primer set.
- 5 position substituted 2'-deoxyuridine derivatives are known to be suitable as substrates or templates for DNA replication reactions, and report of DNA replication reactions using 5 position substituted 2'-deoxyuridine triphosphates.
- a DNA replication and amplification reaction using 5- (1-propynyl) -2'-deoxyuridine triphosphate having a structure similar to Pa has also been reported, and the replication reaction proceeds efficiently with a relatively high degree of loyalty. It is known (Non-patent Document 77).
- Non-patent Document 77 Until now, there has been no report of DNA replication reaction using Pa as a template. In this use example, DNA ligation was confirmed, suggesting that DNA elongation occurred using Pa as a template.
- the band in lane 1 shows DNA markers (0.5, 1.0, 1.5, 2, 3, 4, 6, 8, 10, and 12 kb).
- the bands in lanes 2, 3, 6, and 7 were obtained by ligating the amplification product DNA when LL1 and LL4 were used as a primer set with the amplification product DNA when LL5 and LL8 were used as a primer set.
- the ligation reaction product DNA is shown, and the bands in lanes 4, 5, 8 and 9 are the amplification product DNA when LL2 and LL3 are used as the primer set, and LL6 and LL7 as the primer set.
- the DNA of the ligation product in the case of ligation reaction with the DNA of the amplification product in this case is shown.
- the bands in lanes 2 to 5 show the DNA of the ligation product when the DNA is ligated using T4 DNA ligase, and the bands in lanes 6 to 9 ligate the DNA using Taq DNA ligase.
- the DNA of the ligation reaction product is shown.
- the bands in lanes 2, 4, 6, and 8 show the DNA of the ligation product when the DNA is ligated using a mixed solution of 2 ⁇ L of the amplified product DNA, and lanes 3, 5, Bands 7 and 9 indicate the DNA of the ligation product when the DNA is ligated using 4 ⁇ L of the amplification product DNA mixed solution.
- a DNA strand is cleaved by a chemical reaction at a low cost and with a simple experimental operation, and compared with an enzymatic method such as polynucleotide kinase or a terminal phosphorylation method of synthetic DNA using a phosphorylated amidite. It is possible to obtain oligodeoxynucleotides whose ends are phosphorylated with higher yield.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Biochemistry (AREA)
- Biotechnology (AREA)
- General Health & Medical Sciences (AREA)
- Genetics & Genomics (AREA)
- Molecular Biology (AREA)
- Saccharide Compounds (AREA)
- Measuring Or Testing Involving Enzymes Or Micro-Organisms (AREA)
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2012-084249 | 2012-04-02 | ||
| JP2012084249 | 2012-04-02 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2013150902A1 true WO2013150902A1 (fr) | 2013-10-10 |
Family
ID=49300395
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2013/058349 Ceased WO2013150902A1 (fr) | 2012-04-02 | 2013-03-22 | Procédé de production d'un oligodésoxynucléotide à terminaison 5' phosphorylée par clivage d'un oligodésoxynucléotide contenant un nucléotide non naturel |
Country Status (2)
| Country | Link |
|---|---|
| JP (1) | JPWO2013150902A1 (fr) |
| WO (1) | WO2013150902A1 (fr) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2016094845A2 (fr) | 2014-12-12 | 2016-06-16 | Woolf Tod M | Compositions et procédés d'édition d'acides nucléiques dans des cellules à l'aide d'oligonucléotides |
Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1999011819A1 (fr) * | 1997-08-28 | 1999-03-11 | The Perkin-Elmer Corporation | Detection amelioree de mutations dans des acides nucleiques, par clivage chimique |
| WO2003048387A2 (fr) * | 2001-12-04 | 2003-06-12 | Solexa Limited | Nucleotides marques |
| WO2004011665A2 (fr) * | 2002-05-17 | 2004-02-05 | Nugen Technologies, Inc. | Procedes de fragmentation, d'etiquetage et d'immobilisation d'acides nucleiques |
-
2013
- 2013-03-22 JP JP2014509104A patent/JPWO2013150902A1/ja active Pending
- 2013-03-22 WO PCT/JP2013/058349 patent/WO2013150902A1/fr not_active Ceased
Patent Citations (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO1999011819A1 (fr) * | 1997-08-28 | 1999-03-11 | The Perkin-Elmer Corporation | Detection amelioree de mutations dans des acides nucleiques, par clivage chimique |
| WO2003048387A2 (fr) * | 2001-12-04 | 2003-06-12 | Solexa Limited | Nucleotides marques |
| WO2004011665A2 (fr) * | 2002-05-17 | 2004-02-05 | Nugen Technologies, Inc. | Procedes de fragmentation, d'etiquetage et d'immobilisation d'acides nucleiques |
Non-Patent Citations (3)
| Title |
|---|
| ANDERSON, AARON S. ET AL.: "Independent generation and reactivity of 2'-deoxy-5- methyleneuridin-5-yl, a significant reactive intermediate produced from thymidine as a result of oxidative stress.", THE JOURNAL OF ORGANIC CHEMISTRY, vol. 65, no. 15, 2000, pages 4648 - 4654 * |
| PESNOT, THOMAS ET AL.: "Two-step synthesis of novel, bioactive derivatives of the ubiquitous cofactor nicotinamide adenine dinucleotide (NAD).", JOURNAL OF MEDICINAL CHEMISTRY, vol. 54, no. 10, 2011, pages 3492 - 3499 * |
| SCHLITT, KATHERINE M. ET AL.: "Concerning the hydrolytic stability of 8-aryl-2'- deoxyguanosine nucleoside adducts: implications for abasic site formation at physiological pH.", THE JOURNAL OF ORGANIC CHEMISTRY, vol. 74, no. 16, 2009, pages 5793 - 5802 * |
Cited By (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2016094845A2 (fr) | 2014-12-12 | 2016-06-16 | Woolf Tod M | Compositions et procédés d'édition d'acides nucléiques dans des cellules à l'aide d'oligonucléotides |
| EP4372091A2 (fr) | 2014-12-12 | 2024-05-22 | Tod M. Woolf | Compositions et procédés d'édition d'acides nucléiques dans des cellules à l'aide d'oligonucléotides |
Also Published As
| Publication number | Publication date |
|---|---|
| JPWO2013150902A1 (ja) | 2015-12-17 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CA2459347C (fr) | Compositions d'acides nucleiques bloques et leurs utilisations | |
| EP3137478B1 (fr) | Groupes protecteurs phosphoreux et leurs procédés de préparation et utilisation | |
| JP2004500330A (ja) | グアニジニウム官能化オリゴマーとその製造法 | |
| EP1483280A2 (fr) | Base azotee fluorescente et nucleosides comprenant cette base | |
| CN102443030A (zh) | 光可断裂的标记核苷酸和核苷与标记的核苷酸和核苷以及其在dna测序中的使用方法 | |
| Boháčová et al. | Protected 5-(hydroxymethyl) uracil nucleotides bearing visible-light photocleavable groups as building blocks for polymerase synthesis of photocaged DNA | |
| Sørensen et al. | Functionalized LNA (locked nucleic acid): high-affinity hybridization of oligonucleotides containing N-acylated and N-alkylated 2′-amino-LNA monomers | |
| Christensen et al. | Intercalating Nucleic Acids with Pyrene Nucleotide Analogues as Next‐Nearest Neighbors for Excimer Fluorescence Detection of Single‐Point Mutations under Nonstringent Hybridization Conditions | |
| R Kore et al. | Recent developments in the synthesis and applications of C5-substituted pyrimidine nucleosides and nucleotides | |
| Matyašovský et al. | 2-Substituted 2′-deoxyinosine 5′-triphosphates as substrates for polymerase synthesis of minor-groove-modified DNA and effects on restriction endonuclease cleavage | |
| JP4903163B2 (ja) | オリゴヌクレオチド合成のためのホスホルアミダイト活性化剤 | |
| WO2013150902A1 (fr) | Procédé de production d'un oligodésoxynucléotide à terminaison 5' phosphorylée par clivage d'un oligodésoxynucléotide contenant un nucléotide non naturel | |
| US8946397B1 (en) | Tagged nucleosides that leave no scar upon cleavage | |
| Seela et al. | Synthesis and Enzymic Hydrolysis of Oligoribonucleotides Incorporating 3‐Deazaguanosine: The Importance of the Nitrogen‐3 Atom of Single Conserved Guanosine Residues on the Catalytic Activity of the Hammerhead Ribozyme | |
| Seela et al. | 2′‐Deoxyuridine and 2′‐Deoxyisocytidine as Constituents of DNA with Parallel Chain Orientation: The Stabilization of the iCd⋅ Gd Base Pair by the 5‐Methyl Group | |
| WO2015167620A1 (fr) | Composés, compositions et procédés comprenant des fragments thermiquement labiles | |
| JP4180681B2 (ja) | アンチセンスオリゴヌクレオチド | |
| WO2022175685A1 (fr) | Guanines modifiées | |
| Jestřábová et al. | Hydrophobic alkyl-linked base-modified pyrimidine and 7-deazapurine 2′-deoxyribonucleoside phosphoramidites: synthesis and application in solid-phase synthesis of modified and hypermodified oligonucleotides | |
| Lin et al. | 7-Deaza-2, 8-diazaadenine containing oligonucleotides: synthesis, ring opening and base pairing of 7-halogenated nucleosides | |
| Babu et al. | Novel nucleic acid architectures involving locked nucleic acid (LNA) and pyrene residues: Results from an Indo-Danish collaboration | |
| Sunaga et al. | Systematic Evaluation of Hybridization Property and Photo-Crosslinking Ability of Modified Oligonucleotides Containing Benzophenone-Bearing Glycol Nucleoside Analogs | |
| CA3235799A1 (fr) | Procede de production de polynucleotides | |
| Ohkubo et al. | Non-protected Synthesis of Oligonucleotides | |
| WO2004048376A1 (fr) | Nucleosides de napthylidine bicycliques |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 13772655 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2014509104 Country of ref document: JP Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| 122 | Ep: pct application non-entry in european phase |
Ref document number: 13772655 Country of ref document: EP Kind code of ref document: A1 |