WO2014147889A1 - ハロヒダントイン化合物の製造方法 - Google Patents
ハロヒダントイン化合物の製造方法 Download PDFInfo
- Publication number
- WO2014147889A1 WO2014147889A1 PCT/JP2013/080919 JP2013080919W WO2014147889A1 WO 2014147889 A1 WO2014147889 A1 WO 2014147889A1 JP 2013080919 W JP2013080919 W JP 2013080919W WO 2014147889 A1 WO2014147889 A1 WO 2014147889A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- value
- solvent
- compound
- dimethylhydantoin
- halohydantoin
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/54—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
- C07D233/66—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D233/72—Two oxygen atoms, e.g. hydantoin
- C07D233/80—Two oxygen atoms, e.g. hydantoin with hetero atoms or acyl radicals directly attached to ring nitrogen atoms
- C07D233/82—Halogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D233/00—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings
- C07D233/54—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members
- C07D233/66—Heterocyclic compounds containing 1,3-diazole or hydrogenated 1,3-diazole rings, not condensed with other rings having two double bonds between ring members or between ring members and non-ring members with hetero atoms or with carbon atoms having three bonds to hetero atoms with at the most one bond to halogen, e.g. ester or nitrile radicals, directly attached to ring carbon atoms
- C07D233/72—Two oxygen atoms, e.g. hydantoin
Definitions
- the present invention relates to a method for producing a halohydantoin compound.
- halohydantoin compounds are widely used as photosensitizers for photographs or the like, or as halogenating agents or oxidizing agents in the production process of pharmaceuticals, agricultural chemicals, compounds and the like.
- 1,3-diiodo-5,5-dimethylhydantoin which is one of halohydantoin compounds, is a promising compound with a high iodine content and a more economical production method.
- Non-Patent Document 1 discloses that 5,5-dimethylhydantoin and iodine monochloride are reacted with an aqueous sodium hydroxide solution and carbon tetrachloride, and the resulting crystals are washed with water, and then acetic anhydride. It is described that, after washing with ethyl, it is purified by drying at 60 ° C. under reduced pressure. Further, it is described that the obtained 1,3-diiodo-5,5-dimethylhydantoin is a reagent that is stable enough to be stored in a dark place in a desiccator without recrystallization.
- Patent Document 1 5,5-dimethylhydantoin and iodine monochloride are reacted with each other in an aqueous solution of sodium hydroxide using N, N-dimethylformamide or acetic acid-n-butyl solvent. Is collected by filtration, and the resulting crystals are purified by drying under reduced pressure.
- Non-Patent Document 1 The effective iodine of 1,3-diiodo-5,5-dimethylhydantoin by the purification method described in Non-Patent Document 1 is 65%, the yield is as low as 75%, and the purity is not described.
- Patent Document 1 does not describe the purity of 1,3-diiodo-5,5-dimethylhydantoin obtained by the purification method.
- halohydantoin compounds are unstable even at room temperature and need to be refrigerated. Further, when the halohydantoin compound is suspended in water, it decomposes gradually and liberates halogen alone. Furthermore, the halohydantoin compound has a problem that it becomes more unstable when heated in the state of a wet body containing a mixed liquid component such as water and an organic solvent in a certain amount or more. Further, when the halogen halohydantoin compound is dried under reduced pressure, the halogen alone generated by thermal decomposition sublimates, so that the halogen sublimated solidifies in the vacuum line and closes the vacuum line.
- the pressure cannot be reduced, the pressure in the dryer increases, and the temperature in the dryer increases, so that the halohydantoin compound becomes more unstable. Therefore, the halohydantoin compound is decomposed and the hydantoin compound and halogen alone are liberated. For this reason, the purity of the halohydantoin compound decreases. Further, the free halogen alone causes a problem of corrosion of equipment used for coloring and refining the halohydantoin compound.
- the present invention has been made in view of the above problems, and its object is to reduce impurities from a composition containing a halohydantoin compound while suppressing decomposition of the halohydantoin compound that causes coloring of the halohydantoin compound and corrosion of equipment. It is an object to provide a method for producing a halohydantoin compound by removing.
- a production method is a method for producing a halohydantoin compound in which a composition containing an halohydantoin compound and impurities is treated, wherein the composition is added to a mixed solvent of water and an organic solvent. It is characterized by including a washing step of washing by adding an object.
- a halohydantoin compound by removing impurities from the composition containing the halohydantoin compound while suppressing decomposition of the halohydantoin compound that causes coloring of the halohydantoin compound and corrosion of equipment. There is an effect.
- the production method according to the present invention is a production method of a halohydantoin compound for treating a composition containing a halohydantoin compound and impurities, and the washing step comprises adding the above composition to a mixed solvent of water and an organic solvent for washing. Is a manufacturing method.
- the halohydantoin compound is unstable in a wet state containing some liquid components. Therefore, there is a method of drying the wet body, but when purified by heating and drying under reduced pressure, the halohydantoin compound becomes more unstable and decomposes to liberate the hydantoin compound and the halogen alone. Thereby, the purity of the halohydantoin compound decreases, and the halohydantoin compound is colored by the liberated halogen alone.
- the present inventors diligently studied to solve the above problems. As a result, the present inventors have found that the purity can be improved and the coloring can be reduced by washing the halohydantoin compound, which is reduced in purity and markedly colored, with a mixed solvent of water and an organic solvent. That is, according to this invention, the manufacturing method of a halohydantoin compound with high purity and little coloring can be provided.
- the composition to be treated contains a halohydantoin compound and impurities.
- the lower limit of the amount of impurities contained in the composition is preferably 4% by weight, more preferably 5% by weight, and even more preferably 6% by weight with respect to the entire composition.
- an upper limit is 30 weight%, More preferably, it is 25 weight%, More preferably, it is 20 weight%.
- the above composition has an L * value of 70 or less, an a * value of 5 or more, and a b * value of 14 or less in an L * a * b * color system (CIE 1976 L * a * b * color space). It is preferable that A halohydantoin compound with less coloring can be obtained by treating a highly colored composition by the production method according to the present invention.
- composition refers to those containing impurities within the above range, and those whose impurities are less than 4% by weight by the production method of the present invention may be simply referred to as halohydantoin compounds. .
- examples of the halohydantoin to be produced include compounds represented by the following chemical formula I.
- R 1 , R 2 , X 1 and X 2 may be in any combination within the above-mentioned range.
- R 1 and R 2 may be the same or different and are each independently H, a substituted or unsubstituted aliphatic hydrocarbon group having 1 to 10 carbon atoms, a substituted or unsubstituted carbon group having 3 to 10 is an alicyclic hydrocarbon group, or a substituted or unsubstituted allyl group or aralkyl group having 6 to 10 carbon atoms, more preferably H or an aliphatic hydrocarbon group having 1 to 8 carbon atoms, More preferably, it is H or a methyl group, most preferably R 1 and R 2 are both methyl groups;
- X 1 and X 2 may be the same or different and are each independently H or a halogen atom, more preferably H, Br or I, and still more preferably H or I. Most preferably, X 1 and X 2 are both I, except that X 1 and X 2 are both H.
- the method for obtaining the halohydantoin compound is not particularly limited, and may be synthesized according to a conventionally known method. An example of a method for synthesizing a halohydantoin compound will be described below.
- a hydantoin compound and a halogen compound such as iodine monochloride are reacted in an aqueous solution in the presence of a base.
- a halogen compound such as iodine monochloride
- the wet body containing a halohydantoin compound is obtained.
- the liquid component is subsequently removed by heating and drying.
- a drying method by heating a drying method generally used industrially may be applied. For example, a method of drying under reduced pressure using a conical vacuum dryer may be used.
- the halohydantoin compounds synthesized in this way can contain a large amount of impurities and can be highly colored. In the present invention, such a halohydantoin compound, that is, a composition is treated.
- the impurity may be, for example, at least one of a halogen simple substance, a hydantoin compound, and an inorganic salt.
- the halogen simple substance and the hydantoin compound may be generated by being unreacted during the above synthesis method or by being decomposed and liberated during heat drying. Examples of the halogen alone include iodine, bromine, and chlorine. Examples of the hydantoin compound include hydantoin, 1-methylhydantoin, 5-methylhydantoin, and 5,5-dimethylhydantoin.
- the inorganic salt is a by-product of a base and a halogen compound used when synthesizing the halohydantoin compound, and examples thereof include lithium chloride, sodium chloride, potassium chloride, and magnesium chloride.
- the composition to be treated is not limited to the one that has been heat-dried in the above-described synthesis method, and for example, a wet-state composition obtained in the synthesis reaction may be applied. Good.
- composition may contain components other than the impurities.
- examples of other components include water or an organic solvent.
- Such a composition is also a subject of the production method according to the present invention.
- the washing step is a step of washing by adding the above composition to a mixed solvent of water and an organic solvent.
- the composition is washed using a mixed solvent obtained by previously mixing water and an organic solvent. That is, either water or an organic solvent is not used alone or separately.
- the production method according to the present invention is a method for regenerating a halohydantoin compound that does not meet product specifications.
- the cleaning method is not particularly limited, and may be cleaned according to a conventionally known method. For example, it can be washed by adding water and an organic solvent into a reaction vessel equipped with a stirrer to make a mixed solvent, and then adding the composition and stirring.
- cleaning method is not limited to this,
- the composition may be put in Nutsche and a centrifuge, and it may wash
- the lower limit of the temperature of the mixed solvent is preferably ⁇ 10 ° C., more preferably ⁇ 5 ° C., and further preferably 0 ° C.
- an upper limit is 50 degreeC, More preferably, it is 40 degreeC, More preferably, it is 30 degreeC. If it is ⁇ 10 ° C. or higher, there is an advantage that the cleaning effect is high and the purity can be improved and coloring can be reduced, and if it is 50 ° C. or lower, the decomposition of the halohydantoin compound can be suppressed. is there.
- By washing the composition in such a low temperature range decomposition of the halohydantoin compound contained in the composition can be suppressed, and a highly pure halohydantoin compound can be obtained.
- the lower limit of the treatment time during the washing step is preferably 1 minute, more preferably 10 minutes, and even more preferably 20 minutes. Moreover, it is preferable that an upper limit is 24 hours, More preferably, it is 12 hours, More preferably, it is 6 hours.
- the lower limit of the content of the organic solvent contained in the mixed solvent is preferably 10% by weight, more preferably 12% by weight, and even more preferably 15% by weight with respect to the entire mixed solvent. Moreover, it is preferable that an upper limit is 90 weight%, More preferably, it is 85 weight%, More preferably, it is 80 weight%.
- the organic solvent is, for example, at least one organic solvent selected from an ester solvent, an alcohol solvent, an aromatic solvent, an ether solvent, and a chlorine solvent having a boiling point temperature of 30 ° C. or higher and 200 ° C. or lower.
- the present invention can be suitably applied to things.
- ester solvents include methyl acetate, ethyl acetate, propyl acetate, isopropyl acetate, butyl acetate, acetic acid-isobutyl, acetic acid-sec-butyl, acetic acid-tert-butyl, pentyl acetate, hexyl acetate, methyl propionate, Examples include ethyl propionate, propyl propionate, butyl propionate, methyl butyrate, ethyl butyrate, propyl butyrate, and butyl butyrate.
- alcohol solvents examples include methanol, ethanol, propanol, isopropanol, butanol, isobutanol, sec-butanol, and tert-butanol.
- Aromatic solvents include benzene, toluene, ethylbenzene, propylbenzene, cumene, butyl zensen, isobutylbenzene, sec-butylbenzene, tert-butylbenzene, o-xylene, m-xylene, p-xylene, mesitylene, 1, Examples include 2,3-trimethylbenzene, 1,2,4-trimethylbenzene, o-cymene, m-cymene, and p-cymene.
- ether solvents include diethyl ether, dipropyl ether, isopropyl ether, methyl-tert-butyl ether, methylcyclopentyl ether, dibutyl ether, anisole, ethylphenyl ether, tetrahydrofuran, tetrahydropyran, and 1,4-dioxane.
- Chlorine solvents include chloropropane, chlorobutane, chloropentane, chlorohexane, chloroheptane, chlorooctane, dichloromethane, 1,2-dichloroethane, 1,2-dichloropropane, 1,3-dichloropropane, 2,2-dichloropropane 1,2-dichlorobutane, 1,3-dichlorobutane, 1,4-dichlorobutane, chloroform, 1,1,1-trichloroethane, 1,1,2-trichloroethane, 1,1,1,2-tetrachloroethane 1,1,2,2-tetrachloroethane, 1,2,3-trichloropropane, carbon tetrachloride.
- a mixed solvent prepared by mixing water and an organic solvent in advance may be charged into a reaction vessel for washing the composition, or water and an organic solvent may be contained in the reaction vessel. You may prepare by adding a solvent, respectively.
- the impurity content is less than 4% by weight
- the L * value is more than 70
- the a * value is A halohydantoin compound having a b * value of less than 5 and a b * value exceeding 14 can be obtained.
- the content of the halogen simple substance, the hydantoin compound and the inorganic salt contained in the halohydantoin compound can be reduced to 1% by weight or less, respectively. Therefore, it is possible to produce a halohydantoin compound with high purity and little coloration.
- the composition containing the impurities in the range of 4 wt% to 30 wt% in the washing step.
- the production method according to the present invention can be suitably applied to the impurity that is at least one of a halogen simple substance, a hydantoin compound, and an inorganic salt.
- the L * value in the L * a * b * color system (CIE 1976 L * a * b * color space) defined by the International Lighting Commission is 70 or less, More preferably, the composition having an a * value of 5 or more and a b * value of 14 or less is washed.
- the organic solvent is at least one of an ester solvent, an alcohol solvent, an aromatic solvent, an ether solvent, and a chlorine solvent having a boiling point temperature of 30 ° C. or more and 200 ° C. or less. It can be suitably applied to those which are one kind of organic solvent.
- the temperature of the mixed solvent in the washing step is ⁇ 10 ° C. or higher and 50 ° C. or lower.
- the halohydantoin compound is more preferably a compound represented by the above chemical formula I.
- a 200 L glass-lined reaction kettle is charged with ion-exchanged water (74.4 kg), 12 wt% NaOH aqueous solution (17.36 kg, 52 mol), and then 5,5-dimethylhydantoin (6.7 kg, 52 mol) as a hydantoin compound. After that, it was cooled to 5 ° C. While maintaining the temperature of the contents in the reaction kettle at 0 to 5 ° C., 43 wt% of a solution of iodine monochloride in butyl acetate (19.1 kg, 50.7 mol) was added dropwise over 60 minutes.
- Part of the wet body was dried under reduced pressure using an evaporator (4 kPa, 80 ° C., 30 minutes) to obtain a dried product of 1,3-diiodo-5,5-dimethylhydantoin.
- the content of 1,3-diiodo-5,5-dimethylhydantoin in the dried product was 98.0% by weight.
- the color tone the L * value was 88.33, the a * value was 1.70, and the b * value was 16.59.
- Example 1 In Example 1, the dried product containing the halohydantoin compound obtained in Production Example 1 was treated with a mixed solvent of an organic solvent and water to produce a halohydantoin compound.
- the contents of the reaction vessel were supplied for 10 seconds under reduced pressure on a Nutsche with an inner diameter of 55 mm, in which a qualitative filter paper (No. 2) manufactured by ADVANTEC was attached to a suction bottle, and then suction was continued for 5 minutes. The filterability at this time was good.
- the crystals on the filter paper were washed with water (30 g), and then suction was continued for 20 minutes. The decompression was released, and 30.0 g of wet material on the filter paper was taken out.
- the obtained wet body was dried under reduced pressure using an evaporator (4 kPa, 80 ° C., 30 minutes) to obtain 22.8 g of a recycled product.
- the obtained recycled product was light yellow, and the color tone L * value was 86.26, the a * value was 0.44, and the b * value was 19.53.
- the content of 1,3-diiodo-5,5-dimethylhydantoin was 98.0% by weight, and the recovery rate was 88.9%.
- 5,5-dimethylhydantoin and iodine alone were not mixed.
- Example 2 In Example 2, it carried out by the same method as Example 1 except having charged 27.2g of ion-exchange water and 142.8g of butyl acetate as a mixed solvent, and obtained 22.0g of regenerated goods.
- the obtained recycled product was light yellow, and the color tone L * value was 86.46, the a * value was 1.19, and the b * value was 16.50.
- the content of 1,3-diiodo-5,5-dimethylhydantoin was 98.0% by weight, and the recovery rate was 85.8%. Contamination of 5,5-dimethylhydantoin and iodine alone was not observed.
- Example 3 In Example 3, the same reaction vessel as in Example 1 was used, the inside of the reaction vessel was sufficiently replaced with nitrogen, ion-exchanged water (153.0 g), and then methanol (17.0 g) from the dropping funnel. Was charged. After cooling the temperature in the reaction vessel to 2 ° C., the brown color obtained by the same method as in Production Example 1 (color tone L * value 55.14, a * value 5.47, b * value 8.73) ) 30 g (1,3-diiodo-5,5-dimethylhydantoin content of 91.0% by weight) is charged for 45 minutes while maintaining the temperature in the reaction vessel at 1 to 2 ° C. Stir. At this time, the pH of the contents in the reaction vessel was 2.3.
- the contents of the reaction vessel were supplied for 10 seconds under reduced pressure on a Nutsche with an inner diameter of 55 mm, in which a qualitative filter paper (No. 2) manufactured by ADVANTEC was attached to a suction bottle, and then suction was continued for 5 minutes. The filterability at this time was good.
- the crystals on the filter paper were washed with 30 g of water, and then suction was continued for 45 minutes.
- the vacuum was released and 27.5 g of wet material on the filter paper was taken out.
- the obtained wet body was dried under reduced pressure using an evaporator (4 kPa, 80 ° C., 30 minutes) to obtain 24.6 g of a recycled product.
- the obtained recycled product was light yellow, and the color tone L * value was 86.75, the a * value was 1.36, and the b * value was 15.82.
- the content of 1,3-diiodo-5,5-dimethylhydantoin was 98.4% by weight, and the recovery rate was 88.6%. Contamination of 5,5-dimethylhydantoin and iodine alone was not observed in the recycled product.
- Example 4 In Example 4, it carried out using the same reaction container as Example 3 except having charged 136.0g of ion-exchange water and 34.0g of methanol as a mixed solvent, and obtained 23.9g of regenerated products.
- the obtained recycled product was light yellow, and the color tone L * value was 87.22, the a * value was 1.15, and the b * value was 16.24.
- the content of 1,3-diiodo-5,5-dimethylhydantoin was 98.5% by weight, and the recovery rate was 86.2%. Contamination of 5,5-dimethylhydantoin and iodine alone was not observed.
- Example 5 the dried product containing the halohydantoin compound obtained in Production Example 2 was treated with a mixed solvent of an organic solvent and water to produce a halohydantoin compound.
- the contents of the reaction vessel were supplied for 10 seconds under reduced pressure on a Nutsche with an inner diameter of 55 mm, in which a qualitative filter paper (No. 2) manufactured by ADVANTEC was attached to a suction bottle, and then suction was continued for 5 minutes. The filterability at this time was good.
- the crystals on the filter paper were washed with water (30 g), and then suction was continued for 20 minutes. The decompression was released, and 30.3 g of wet material on the filter paper was taken out.
- the obtained wet body was dried under reduced pressure using an evaporator (4 kPa, 80 ° C., 30 minutes) to obtain 25.5 g of a recycled product.
- the obtained recycled product was light yellow, and the color tone L * value was 87.19, the a * value was 1.14, and the b * value was 16.64.
- the content of 1,3-diiodo-5,5-dimethylhydantoin was 98.0% by weight, and the recovery rate was 88.4%.
- 5,5-dimethylhydantoin and iodine alone were not mixed.
- Comparative Example 1 was carried out using the same reaction vessel as in Example 1. After charging butyl acetate (150.1 g) from the dropping funnel and cooling the temperature in the reaction vessel to 2 ° C., brown color obtained by the same method as in Production Example 1 (color tone L * value is 55.14, a * 30.2 g of dried product (value of 5.47, b * value of 8.73) was charged (the content of 1,3-diiodo-5,5-dimethylhydantoin was 91.0% by weight), and the reaction While maintaining the temperature in the container at 2 to 5 ° C., the mixture was stirred for 30 minutes.
- the contents of the reaction vessel were fed for 5 minutes under reduced pressure on a Nutsche with an inner diameter of 55 mm, in which a qualitative filter paper (No. 2) made by ADVANTEC was attached to the suction bottle, and then suction was continued for 5 minutes. The filterability at this time was good.
- the crystals on the filter paper were washed with 31.2 g of water, and then suction was continued for 30 minutes. The vacuum was released and 25.7 g of wet material on the filter paper was taken out.
- the obtained wet body was dried under reduced pressure using an evaporator (4 kPa, 80 ° C., 30 minutes) to obtain 22.8 g of a recycled product.
- the content of 1,3-diiodo-5,5-dimethylhydantoin in the regenerated product was 98.0% by weight, but the regenerated product was brown and the color tone L * value was 57.82, a * The value was 9.03 and the b * value was 13.30, and no decolorization was observed.
- Comparative Example 2 was carried out using the same reaction vessel as in Example 1. After charging ion-exchanged water (150.0 g) from the dropping funnel and cooling the temperature in the reaction vessel to 2 ° C., the brown color obtained by the same method as in Production Example 1 (color tone L * value is 55.14, a * 30.2 g of dried product colored in a value of 5.47, b * value of 8.73) (content of 1,3-diiodo-5,5-dimethylhydantoin is 91.0% by weight) The mixture was stirred for 30 minutes while maintaining the temperature in the reaction vessel at 2 to 5 ° C.
- the contents of the reaction vessel were supplied for 5 minutes under reduced pressure on a Nutsche with an inner diameter of 55 mm, in which a qualitative filter paper (No. 2) manufactured by ADVANTEC was attached to a suction bottle, and then suction was continued for 5 minutes. The filterability at this time was good.
- the crystals on the filter paper were washed with 31.2 g of water, and then suction was continued for 30 minutes. The vacuum was released and 25.7 g of wet material on the filter paper was taken out.
- the obtained wet body was dried under reduced pressure using an evaporator (4 kPa, 80 ° C., 30 minutes) to obtain 22.8 g of a recycled product.
- the content of 1,3-diiodo-5,5-dimethylhydantoin in the regenerated product was 98.0% by weight, but the regenerated product was brown and had a color tone L * value of 55.14, a The * value was 5.47, the b * value was 8.73, and no color was removed.
- the present invention can be suitably used for a method for producing a halohydantoin compound used as a photosensitizer for photographs, as a halogenating agent or an oxidizing agent in the production process of pharmaceuticals, agricultural chemicals, compounds, etc., or as an iodination reagent. .
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Plural Heterocyclic Compounds (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Indole Compounds (AREA)
Abstract
Description
本発明に係る製造方法において、処理対象となる組成物は、ハロヒダントイン化合物及び不純物を含む。組成物に含まれる不純物の量は、組成物全体に対して下限値が4重量%であることが好ましく、より好ましくは5重量%であり、さらに好ましくは6重量%である。また、上限値は30重量%であることが好ましく、より好ましくは25重量%であり、さらに好ましくは20重量%である。この範囲内で不純物を含む組成物を本発明に係る製造方法によって処理することにより、不純物の含有量を低下させて、純度の高いハロヒダントイン化合物を得ることができる。
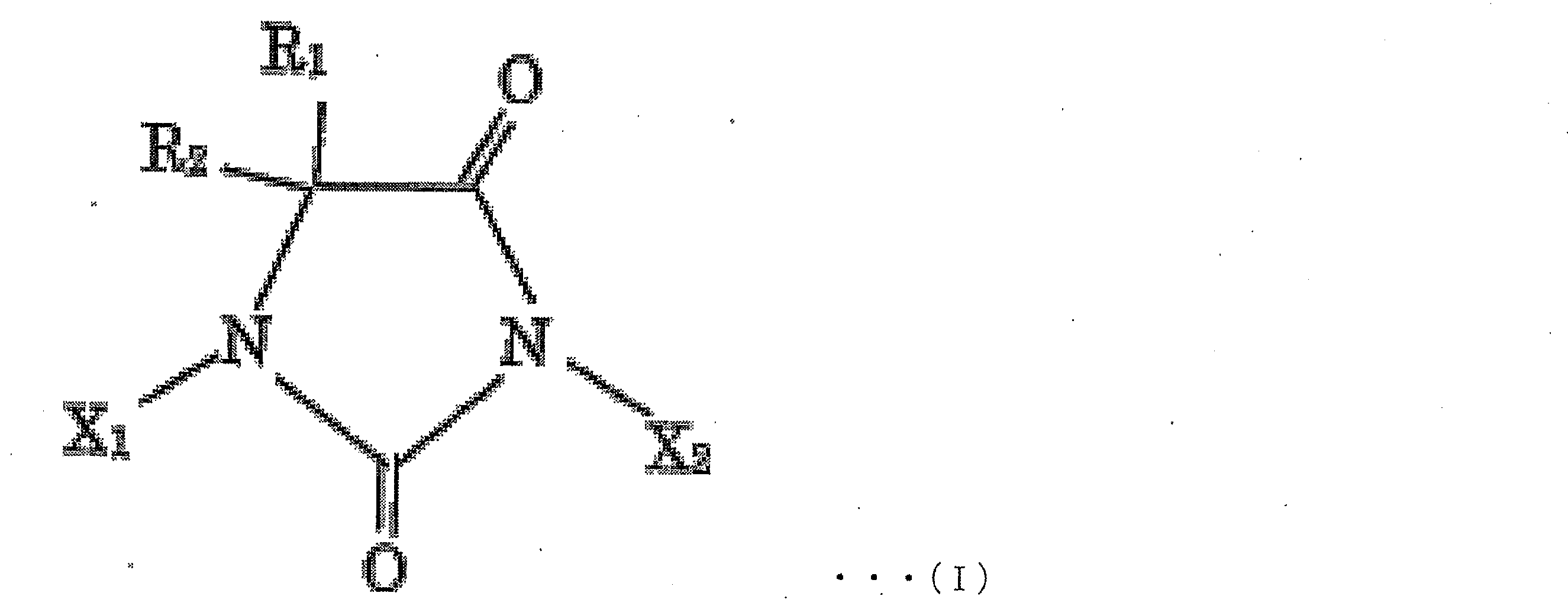
R1及びR2は、同一であっても異なっていてもよく、それぞれ独立に、H、置換もしくは非置換の炭素数1~10の脂肪族炭化水素基、置換もしくは非置換の炭素数3~10の脂環式炭化水素基、又は置換もしくは非置換の炭素数6~10のアリル基もしくはアラルキル基であり、より好ましくは、H、又は炭素数1~8の脂肪族炭化水素基であり、さらに好ましくは、H又はメチル基であり、最も好ましくはR1及びR2が共にメチル基であり;
X1及びX2は、同一であっても異なっていてもよく、それぞれ独立に、H又はハロゲン原子であり、より好ましくは、H、Br又はIであり、さらに好ましくは、H又はIであり、最も好ましくはX1及びX2が共にIであり、ただし、X1及びX2が共にHである形態は含まない)。
洗浄工程とは、水と有機溶媒との混合溶媒に、上記組成物を加えて洗浄する工程である。上述したように、本発明では、予め水と有機溶媒とを混合した混合溶媒を用いて組成物を洗浄する。すなわち、水又は有機溶媒のいずれかを単独で使用したり、それぞれ別々に使用したりするものではない。本発明によれば、混合溶媒を用いて組成物を洗浄するため、純度を向上させることのみならず、着色の少ないハロヒダントイン化合物を得ることができる。言い換えると、本発明に係る製造方法は、製品規格に満たないハロヒダントイン化合物を再生させる方法であるともいえる。
まず、以下の手順によって1,3-ジヨード-5,5-ジメチルヒダントイン(ハロヒダントイン化合物)を含む組成物を製造した。
製造例1と同様の方法で1,3-ジヨード-5,5-ジメチルヒダントインの湿体を得た。湿体の一部をサンプリングし、エバポレーターを用いて減圧乾燥(4kPa、80℃、30分間)して乾燥品を得た。1,3-ジヨード-5,5-ジメチルヒダントインの含有量は96.4重量%であり、外観は淡黄色で、色調はL*値が88.80、a*値が1.54、b*値が16.01であった。
実施例1では、上記製造例1において得られたハロヒダントイン化合物を含む乾燥品を、有機溶媒と水との混合溶媒で処理して、ハロヒダントイン化合物を製造した。
実施例2では、混合溶媒としてイオン交換水27.2g及び酢酸ブチル142.8gを仕込んだ以外は実施例1と同様の方法で実施して、再生品22.0gを得た。得られた再生品は淡黄色であり、色調L*値が86.46、a*値が1.19、b*値が16.50であった。1,3-ジヨード-5,5-ジメチルヒダントインの含有量は98.0重量%、回収率は85.8%であり、5,5-ジメチルヒダントイン、及びヨウ素単体の混入は認められなかった。
実施例3では、実施例1と同様の反応容器内を使用し、当該反応容器内を窒素で十分に置換して、滴下ロートよりイオン交換水(153.0g)、次いでメタノール(17.0g)を仕込んだ。反応容器内の温度を2℃まで冷却した後、製造例1と同様の方法で得られた褐色(色調L*値が55.14、a*値が5.47、b*値が8.73)に着色した乾燥品30g(1,3-ジヨード-5,5-ジメチルヒダントインの含有量は91.0重量%)を投入し、反応容器内の温度を1~2℃に保ちながら、45分間攪拌した。このときの反応容器内の内容物のpHは2.3であった。吸引ビンにADVANTEC社製の定性濾紙(No.2)を装着した内径55mmのヌッチェ上に、減圧下、反応容器の内容物を10秒で給液した後、5分間吸引を続けた。この際の濾過性は良好であった。水30gを用いて濾紙上の結晶を水洗し、その後、45分間吸引を続けた。減圧を解除し、濾紙上の湿体27.5gを取り出した。得られた湿体はエバポレーターを用いて減圧乾燥(4kPa、80℃、30分間)して、再生品24.6gを得た。得られた再生品は淡黄色であり、色調L*値が86.75、a*値が1.36、b*値が15.82であった。1,3-ジヨード-5,5-ジメチルヒダントインの含有量は98.4重量%、回収率は88.6%であった。再生品中に5,5-ジメチルヒダントイン、およびヨウ素単体の混入は認められなかった。
実施例4では、混合溶媒としてイオン交換水136.0g及びメタノール34.0gを仕込んだ以外は、実施例3と同様の反応容器を用いて実施して、再生品23.9gを得た。得られた再生品は淡黄色であり、色調L*値が87.22、a*値が1.15、b*値が16.24であった。1,3-ジヨード-5,5-ジメチルヒダントインの含有量は98.5重量%、回収率は86.2%であり、5,5-ジメチルヒダントイン、及びヨウ素単体の混入は認められなかった。
実施例5では、上記製造例2において得られたハロヒダントイン化合物を含む乾燥品を、有機溶媒と水との混合溶媒で処理して、ハロヒダントイン化合物を製造した。
比較例1は、実施例1と同様の反応容器を用いて実施した。滴下ロートから酢酸ブチル(150.1g)を仕込み、反応容器内の温度を2℃まで冷却した後、製造例1と同様の方法で得られた褐色(色調L*値が55.14、a*値が5.47、b*値が8.73)に着色した乾燥品30.2g(1,3-ジヨード-5,5-ジメチルヒダントインの含有量は91.0重量%)を投入し、反応容器内の温度を2~5℃に保ちながら、30分間攪拌した。吸引ビンにADVANTEC製の定性濾紙(No.2)を装着した内径55mmのヌッチェ上に、減圧下、反応容器の内容物を5分間で給液した後、5分間吸引を続けた。この際の濾過性は良好であった。水31.2gを用いて濾紙上の結晶を水洗し、その後、30分間吸引を続けた。減圧を解除し、濾紙上の湿体25.7gを取り出した。得られた湿体はエバポレーターを用いて減圧乾燥(4kPa、80℃、30分間)して、再生品22.8gを得た。得られた再生品中の1,3-ジヨード-5,5-ジメチルヒダントインの含有量は98.0重量%であったが、再生品は褐色であり、色調L*値が57.82、a*値が9.03、b*値が13.30であり、全く脱色されていなかった。
比較例2は、実施例1と同様の反応容器を用いて実施した。滴下ロートからイオン交換水(150.0g)を仕込み、反応容器内の温度を2℃まで冷却した後、製造例1と同様の方法で得られた褐色(色調L*値が55.14、a*値が5.47、b*値が8.73)に着色した乾燥品30.2g(1,3-ジヨード-5,5-ジメチルヒダントインの含有量は91.0重量%)を投入し、反応容器内の温度を2~5℃に保ちながら、30分間攪拌した。吸引ビンにADVANTEC社製の定性濾紙(No.2)を装着した内径55mmのヌッチェ上に、減圧下、反応容器の内容物を5分間で給液した後、5分間吸引を続けた。この際の濾過性は良好であった。水31.2gを用いて濾紙上の結晶を水洗し、その後、30分間吸引を続けた。減圧を解除し、濾紙上の湿体25.7gを取り出した。得られた湿体はエバポレーターを用いて減圧乾燥(4kPa、80℃、30分間)して、再生品22.8gを得た。得られた再生品中の1,3-ジヨード-5,5-ジメチルヒダントインの含有量は98.0重量%であったが、再生品は褐色であり、色調L*値が55.14、a*値が5.47、b*値が8.73であり、全く脱色されていなかった。
Claims (7)
- ハロヒダントイン化合物及び不純物を含む組成物を処理するハロヒダントイン化合物の製造方法であって、
水と有機溶媒との混合溶媒に、上記組成物を加えて洗浄する洗浄工程を含む、製造方法。 - 上記洗浄工程では、上記不純物が4重量%以上、30重量%以下の範囲で含まれている上記組成物を洗浄する、請求項1に記載の製造方法。
- 上記不純物が、ハロゲン単体、ヒダントイン化合物及び無機塩のうち少なくとも1種である、請求項1又は2に記載の製造方法。
- 上記洗浄工程では、L*a*b*表色系(CIE1976L*a*b*色空間)でL*値が70以下、a*値が5以上、b*値が14以下である上記組成物を洗浄する、請求項1~3のいずれか1項に記載の製造方法。
- 上記有機溶媒が、沸点温度30℃以上200℃以下である、エステル系溶媒、アルコール系溶媒、芳香族系溶媒、エーテル系溶媒及び塩素系溶媒のうち少なくともいずれか1種の有機溶媒である、請求項1~4のいずれか1項に記載の製造方法。
- 上記洗浄工程のときにおける、上記混合溶媒の温度が-10℃以上、50℃以下である、請求項1~5のいずれか1項に記載の製造方法。
- 上記ハロヒダントイン化合物が、下記化学式I
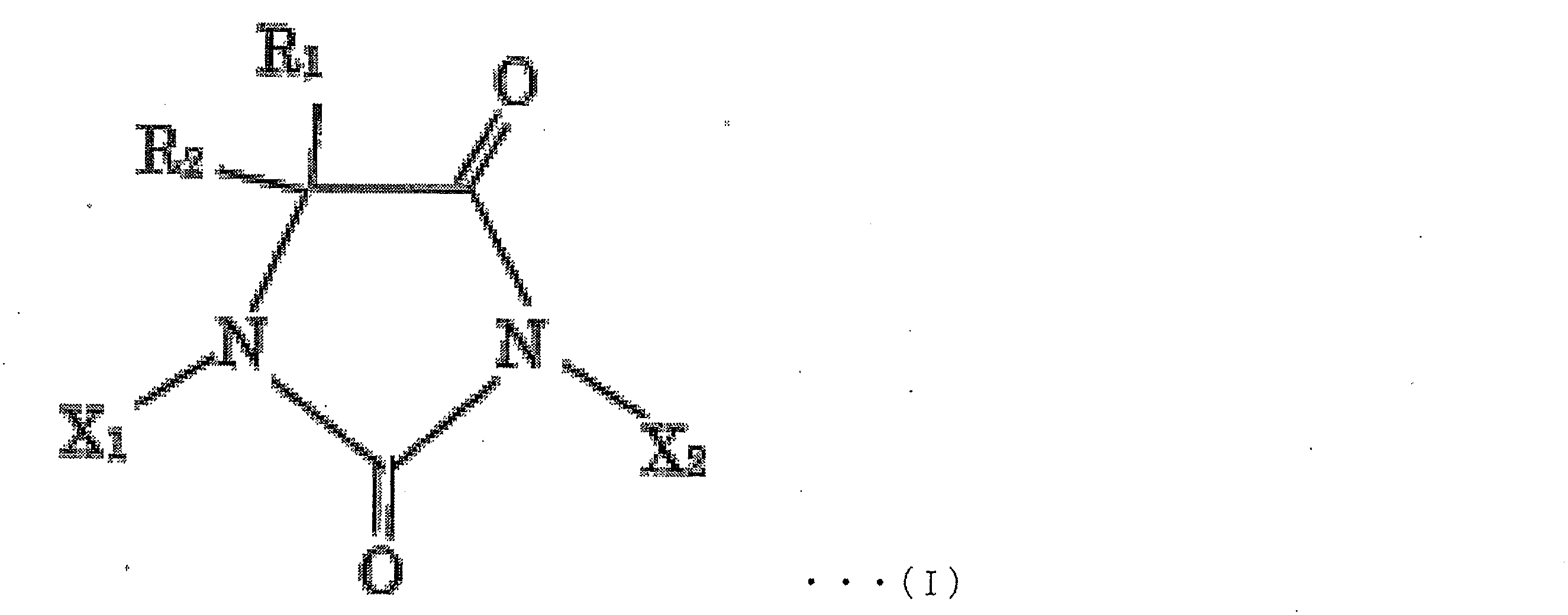
X1及びX2は、同一であっても異なっていてもよく、それぞれ独立に、H又はハロゲン原子であり、より好ましくは、H、Br又はIであり、さらに好ましくは、H又はIであり、最も好ましくはX1及びX2が共にIであり、ただし、X1及びX2が共に水素原子である形態は含まない)
で示される化合物である、請求項1~6のいずれか1項に記載の製造方法。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP13878784.1A EP2977370B1 (en) | 2013-03-19 | 2013-11-15 | Method for producing halohydantoin compound |
| US14/350,096 US9656967B2 (en) | 2013-03-19 | 2013-11-15 | Method for producing halohydantoin compound |
| JP2015506547A JP6280101B2 (ja) | 2013-03-19 | 2013-11-15 | ハロヒダントイン化合物の製造方法 |
| CN201380069768.8A CN104903298B (zh) | 2013-03-19 | 2013-11-15 | 生产卤乙内酰脲化合物的方法 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2013-056989 | 2013-03-19 | ||
| JP2013056989 | 2013-03-19 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2014147889A1 true WO2014147889A1 (ja) | 2014-09-25 |
Family
ID=51579601
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2013/080919 Ceased WO2014147889A1 (ja) | 2013-03-19 | 2013-11-15 | ハロヒダントイン化合物の製造方法 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US9656967B2 (ja) |
| EP (1) | EP2977370B1 (ja) |
| JP (1) | JP6280101B2 (ja) |
| CN (1) | CN104903298B (ja) |
| WO (1) | WO2014147889A1 (ja) |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH09316057A (ja) * | 1995-12-25 | 1997-12-09 | Bromine Compounds Ltd | ジアルキルヒダントインハロゲン化物の安定化方法 |
| JP2001334098A (ja) * | 2000-05-26 | 2001-12-04 | Toshiba Corp | 洗濯機 |
| JP2002030072A (ja) | 2000-05-10 | 2002-01-29 | Nippon Nohyaku Co Ltd | 1,3−ジヨードヒダントイン類の製造方法 |
| WO2007026766A1 (ja) * | 2005-09-02 | 2007-03-08 | Nippoh Chemicals Co., Ltd. | 1,3-ジヨードヒダントイン化合物およびその製造方法 |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4621096A (en) * | 1982-09-30 | 1986-11-04 | Great Lakes Chemical Corporation | Densified halogenated dimethylhydantoins |
| WO1997043264A1 (en) * | 1996-05-14 | 1997-11-20 | Great Lakes Chemical Corporation | Compositions and methods utilizing 1,3-bromochloro-5-methyl-5-propylhydantoin |
| US6809205B1 (en) | 2000-01-18 | 2004-10-26 | Albemarle Corporation | Process for producing N-halogenated organic compounds |
| US6508954B1 (en) * | 2000-01-18 | 2003-01-21 | Albemarle Corporation | 1,3-dibromo-5,5-dimethylhydantoin of enhanced properties |
| US6463766B2 (en) | 2000-01-28 | 2002-10-15 | Kabushiki Kaisha Toshiba | Washing machine with means for preventing propagation of microorganism |
| WO2010054009A1 (en) * | 2008-11-07 | 2010-05-14 | Novabay Pharmaceuticals, Inc. | Antimicrobial oxazolidinone, hydantoin and imidazolidinone compositions |
| JP5734124B2 (ja) * | 2011-07-21 | 2015-06-10 | 株式会社 東邦アーステック | 1,3−ジヨードヒダントイン類の製造方法 |
-
2013
- 2013-11-15 CN CN201380069768.8A patent/CN104903298B/zh not_active Expired - Fee Related
- 2013-11-15 US US14/350,096 patent/US9656967B2/en not_active Expired - Fee Related
- 2013-11-15 EP EP13878784.1A patent/EP2977370B1/en not_active Not-in-force
- 2013-11-15 WO PCT/JP2013/080919 patent/WO2014147889A1/ja not_active Ceased
- 2013-11-15 JP JP2015506547A patent/JP6280101B2/ja not_active Expired - Fee Related
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH09316057A (ja) * | 1995-12-25 | 1997-12-09 | Bromine Compounds Ltd | ジアルキルヒダントインハロゲン化物の安定化方法 |
| JP2002030072A (ja) | 2000-05-10 | 2002-01-29 | Nippon Nohyaku Co Ltd | 1,3−ジヨードヒダントイン類の製造方法 |
| JP2001334098A (ja) * | 2000-05-26 | 2001-12-04 | Toshiba Corp | 洗濯機 |
| WO2007026766A1 (ja) * | 2005-09-02 | 2007-03-08 | Nippoh Chemicals Co., Ltd. | 1,3-ジヨードヒダントイン化合物およびその製造方法 |
Non-Patent Citations (3)
| Title |
|---|
| JUJI YOSHIMURA, KAGAKU JITEN, 15 November 2012 (2012-11-15), pages 1469 - 1470, XP008180860 * |
| ORFEO O. ORAZI ET AL.: "N-Iodohydantoins. II. Iodinations with 1,3-Diiodo-5,5- dimethylhydantoin", THE JOURNAL OF ORGANIC CHEMISTRY, vol. 30, no. 4, 1965, pages 1101 - 1104, XP055257864 * |
| ORFEO O. ORAZI. ET AL.: "N-Iodohydantoins. II. Iodinations with 1,3-Diiodo-5,5-dimethylhydantoin", J. ORG. CHEM., vol. 30, 1965, pages 1101 - 1104, XP055257864, DOI: doi:10.1021/jo01015a036 |
Also Published As
| Publication number | Publication date |
|---|---|
| JPWO2014147889A1 (ja) | 2017-02-16 |
| US9656967B2 (en) | 2017-05-23 |
| EP2977370A1 (en) | 2016-01-27 |
| EP2977370A4 (en) | 2016-08-31 |
| JP6280101B2 (ja) | 2018-02-14 |
| US20150376137A1 (en) | 2015-12-31 |
| EP2977370B1 (en) | 2019-03-20 |
| CN104903298A (zh) | 2015-09-09 |
| CN104903298B (zh) | 2018-05-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP0126569A1 (en) | Bromination process for preparing decabromodiphenyl ether from diphenyl ether | |
| EP1760057A1 (en) | Method for producing polyhalogenated diamantane and derivative thereof | |
| US4214103A (en) | Purification of brominated organic products | |
| JP6280101B2 (ja) | ハロヒダントイン化合物の製造方法 | |
| GB2205830A (en) | Preparation of 1,2,5,6,9,10-hexabromocyclododecane | |
| WO2022255500A1 (ja) | エーテル化合物の製造方法 | |
| US10092853B2 (en) | Apparatus for producing halohydantoin compound | |
| JP3575839B2 (ja) | 5−アセトアセチルアミノ−2−ベンズイミダゾロンの製造方法 | |
| JP5276241B2 (ja) | L−アスコルビン酸の調製方法 | |
| RU2164233C1 (ru) | Способ получения фталоцианина хлоралюминия | |
| Markish et al. | New Aspects on the Preparation of 1, 3-Dibromo-5, 5-dimethylhydantoin | |
| JP2000095728A (ja) | ソルビン酸の製造法 | |
| BE1000126A4 (fr) | Composants de catalyseurs pour catalyseurs de polymerisation d'alpha-olefines, et procede pour les fabriquer. | |
| JPWO2014097787A1 (ja) | ハロヒダントイン化合物の製造方法及びハロヒダントイン化合物 | |
| CA1338353C (fr) | Procede de preparation d'imides halogenes | |
| US6787666B2 (en) | Process for the preparation of isolated 3,4-diaminobenzenesulphonic acid | |
| Fuchs et al. | Novel bromination reagents. Electrophilic aromatic bromination by hexabromocyclopentadiene | |
| HU185787B (en) | Process for preparing benzoxazolone | |
| Moosath et al. | Studies on hexachloroceric acid: I. Isolation of hexachloroceric acid | |
| CN101200430A (zh) | 一种溴硝醇的改进合成方法 | |
| CN109721464A (zh) | 一种2,6-二氯氯苄的合成方法 | |
| JP2023027630A (ja) | エーテル化合物の製造方法 | |
| CN106632001A (zh) | 一种4‑(溴乙酰基)吡啶氢溴酸盐的制备方法 | |
| JPS59193864A (ja) | 1−ナフト−ル−4−スルホン酸の製法 | |
| JPS63115839A (ja) | 多クロルアルキルフエノン類の製法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 13878784 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2015506547 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 14350096 Country of ref document: US |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2013878784 Country of ref document: EP |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |