WO2016114308A1 - 芳香族アミン化合物の製造方法 - Google Patents
芳香族アミン化合物の製造方法 Download PDFInfo
- Publication number
- WO2016114308A1 WO2016114308A1 PCT/JP2016/050849 JP2016050849W WO2016114308A1 WO 2016114308 A1 WO2016114308 A1 WO 2016114308A1 JP 2016050849 W JP2016050849 W JP 2016050849W WO 2016114308 A1 WO2016114308 A1 WO 2016114308A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- formula
- aromatic
- compound represented
- compound
- potassium hydroxide
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- IHOXZXORJFHUNH-NTEUORMPSA-N CC(C(OCCCCCCOc1ccc(/C=C/C(OCCc(ccc([N+]([O-])=O)c2)c2[N+]([O-])=O)=O)cc1)=O)=C Chemical compound CC(C(OCCCCCCOc1ccc(/C=C/C(OCCc(ccc([N+]([O-])=O)c2)c2[N+]([O-])=O)=O)cc1)=O)=C IHOXZXORJFHUNH-NTEUORMPSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C213/00—Preparation of compounds containing amino and hydroxy, amino and etherified hydroxy or amino and esterified hydroxy groups bound to the same carbon skeleton
- C07C213/02—Preparation of compounds containing amino and hydroxy, amino and etherified hydroxy or amino and esterified hydroxy groups bound to the same carbon skeleton by reactions involving the formation of amino groups from compounds containing hydroxy groups or etherified or esterified hydroxy groups
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J27/00—Catalysts comprising the elements or compounds of halogens, sulfur, selenium, tellurium, phosphorus or nitrogen; Catalysts comprising carbon compounds
- B01J27/06—Halogens; Compounds thereof
- B01J27/135—Halogens; Compounds thereof with titanium, zirconium, hafnium, germanium, tin or lead
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C213/00—Preparation of compounds containing amino and hydroxy, amino and etherified hydroxy or amino and esterified hydroxy groups bound to the same carbon skeleton
- C07C213/10—Separation; Purification; Stabilisation; Use of additives
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C219/00—Compounds containing amino and esterified hydroxy groups bound to the same carbon skeleton
- C07C219/32—Compounds containing amino and esterified hydroxy groups bound to the same carbon skeleton having amino groups bound to carbon atoms of six-membered aromatic rings and esterified hydroxy groups bound to acyclic carbon atoms or to carbon atoms of rings other than six-membered aromatic rings of the same carbon skeleton
Definitions
- the present invention relates to a method for producing an aromatic amine compound.
- Patent Document 1 and Non-Patent Document 1 describe a method of adding a sodium hydrogen carbonate aqueous solution to the reaction mixture after the reaction to neutralize it, and filtering the Sn-derived precipitate.
- Patent Document 2 describes a method of distilling off the solvent after the reaction, adding an aqueous sodium hydroxide solution to the residue to adjust the pH to about 12 to 14, and then extracting the product with ethyl acetate.
- Patent Document 3 describes a method in which sodium hydroxide is added to a reaction solution until it becomes strongly basic, and then the product is extracted with diethyl ether.
- Patent Document 4 describes a method in which a potassium hydroxide aqueous solution is added to a reaction solution that is an ethyl acetate solution, and a product is extracted with ethyl acetate, and then generated by silica gel column chromatography.
- the present invention provides a novel method for easily and efficiently removing an Sn compound produced when an aromatic nitro compound is reduced using SnCl 2 (stannous chloride) to produce an aromatic amine compound.
- the purpose is to provide.
- the present invention is as follows. 1.
- the aromatic nitro compound represented by the formula 1 is reduced using stannous chloride to produce the aromatic amine compound represented by the formula 2:
- the reaction liquid obtained by reducing the aromatic nitro compound represented by Formula 1 is separated by bringing it into contact with a mixed liquid of a compound containing a benzene ring and an aqueous potassium hydroxide solution, and the organic layer obtained is represented by Formula 2.
- a method for producing an aromatic amine compound, characterized in that the aromatic amine compound is obtained.
- Ar represents an aromatic group.
- the Sn compound in the reaction solution is simply and efficiently obtained. It can be removed and a high purity aromatic amine compound can be obtained.
- the aromatic nitro compound represented by the formula 1 is reduced using stannous chloride according to the following reaction formula (1) to produce the aromatic amine compound represented by the formula 2. is there.
- Ar represents an aromatic group, and the number of NO 2 groups is at least one, but may be two or more, preferably 1 to 4, more preferably 1 to 3, particularly 1 or 2 is preferred. It goes without saying that the valence of the aromatic group which is Ar varies depending on the number of NO 2 groups.
- the aromatic ring of the aromatic group may be substituted with an alkyl group or a halogen atom.
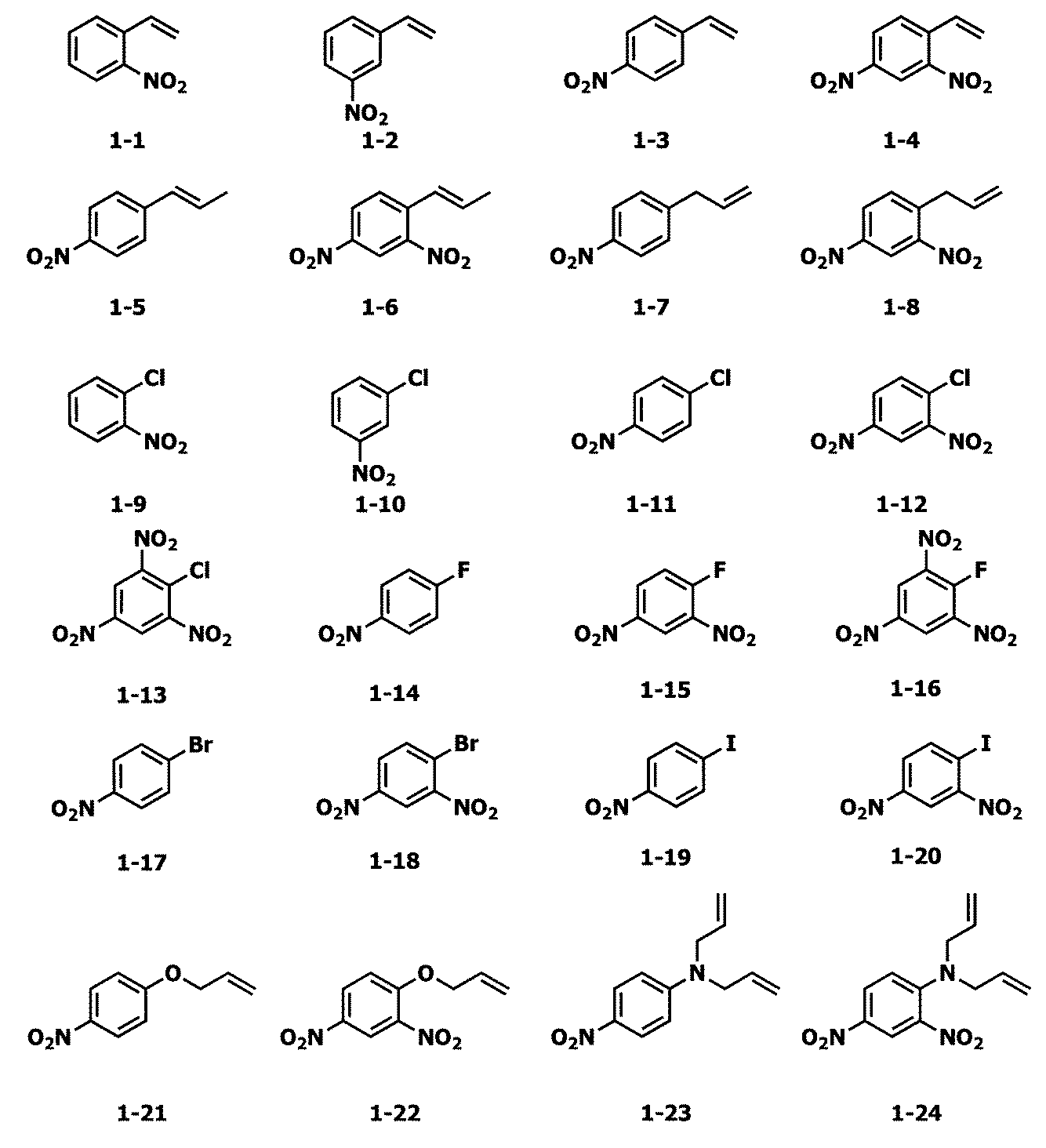
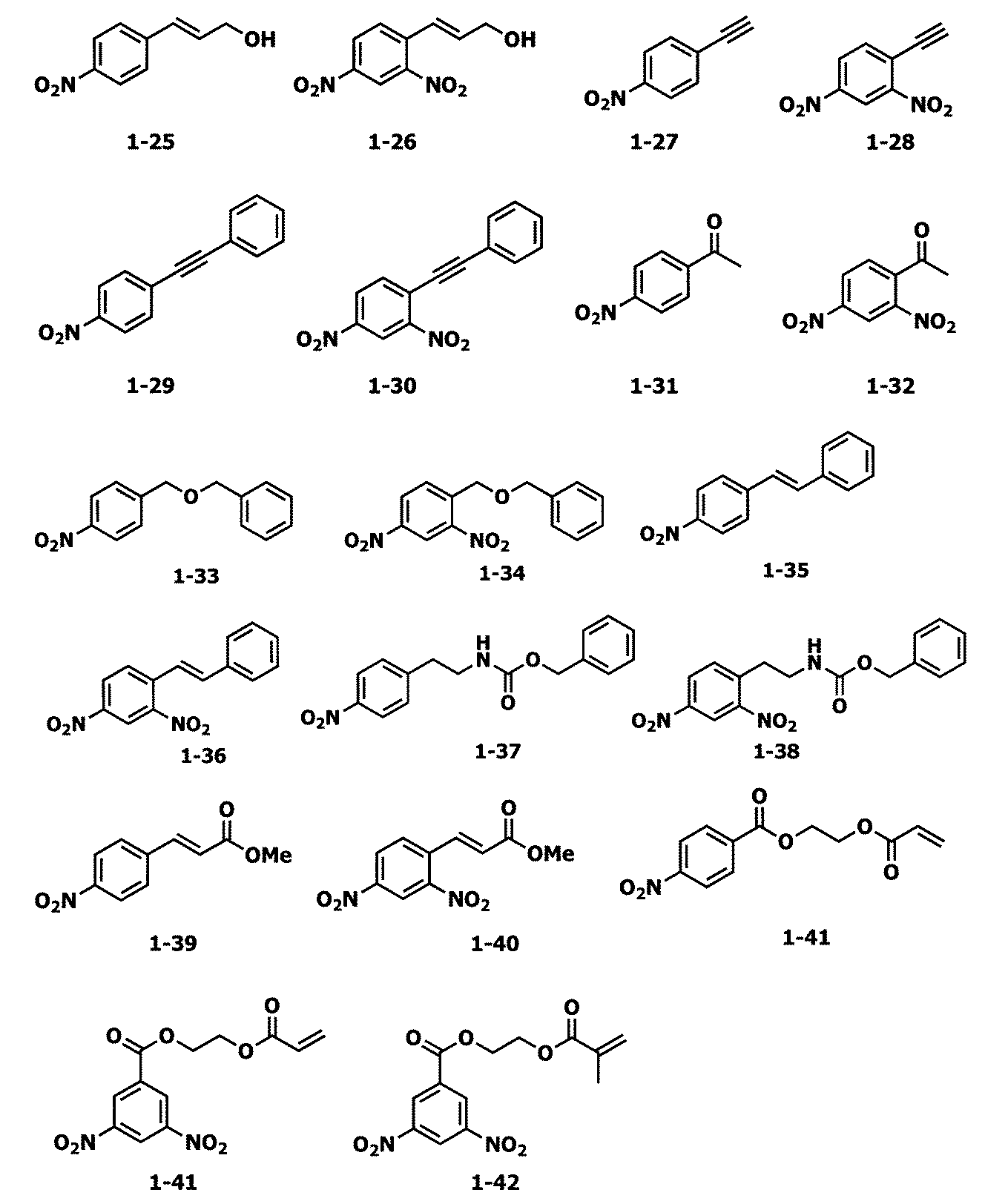
- Examples of the nitro compound represented by the formula 1 used in the present invention include the following formulas 1-1 to 1-53.
- nitro compound represented by the formula 1 used in the present invention examples include dinitro compounds described in P81 of International Patent Application Publication WO2007-071091, P82 to P88 of International Patent Application Publication WO2007-071091.
- Compounds described herein shall be read as dinitro
- nitro compounds represented by Formula 1 when the number of NO 2 groups is 2, it is represented by Formula 3 below.
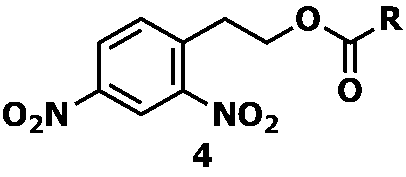
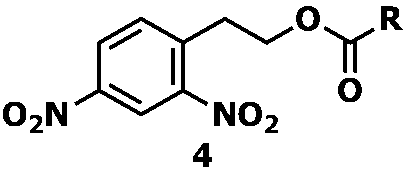
- examples of the nitro compound represented by Formula 3 include a dinitro compound represented by Formula 4 below.
- the two NO 2 groups are present at the meta position, but may be present at the ortho or para position, with the meta position being preferred.
- R represents an organic group that is inert to reduction by stannous chloride, and includes, for example, methyl, ethyl, n-propyl, i-propyl, n-butyl, i-butyl, s -Alkyl groups such as butyl, t-butyl, n-hexyl, n-octyl and n-decyl; cycloalkyl such as cyclopentyl and cyclohexyl; bicycloalkyl such as bicyclohexyl; vinyl Alkenyl such as 1-propenyl group, 2-propenyl group, isopropenyl group, 1-methyl-2-propenyl group, 1 or 2 or 3-butenyl group, 1-hexenyl group, ⁇ -styryl group, ⁇ -styryl group, etc.
- aryl group such as phenyl group, xylyl group, tolyl group, biphenyl group and naphthyl group; aralkyl group such as benzyl group, phenylethyl group and phenylcyclohexyl group Among them, a vinyl group or a ⁇ -styryl group is preferable.
- Some or all of the hydrogen atoms in these organic groups are halogen atoms, hydroxyl groups, thiol groups, phosphate ester groups, ester groups, carboxyl groups, phosphate groups, thioester groups, amide groups, organooxy groups, organo groups.
- a silyl group an organothio group, an organoamino group, a carbamate group, an acyl group, an alkyl group, a cycloalkyl group, a bicycloalkyl group, an alkenyl group, an aryl group, an aralkyl group, or the like.
- these may have a ring structure.
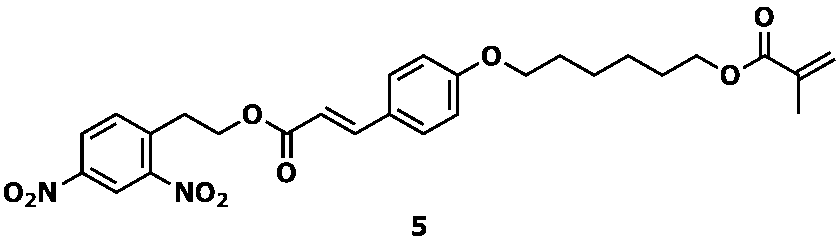
- Examples of the nitro compound represented by Formula 4 include a dinitro compound represented by Formula 5 below.
- the reduction reaction can be carried out with or without using a reaction solvent, but it is preferably carried out with a reaction solvent.
- the reaction solvent is not particularly limited as long as it is inert to the reaction.
- hydrocarbons such as hexane, cyclohexane, benzene and toluene; halogenated hydrocarbons such as carbon tetrachloride, chloroform and 1,2-dichloroethane; alcohols such as methanol, ethanol and isopropyl alcohol; diethyl ether and diisopropyl ether Ethers such as 1,4-dioxane and tetrahydrofuran; ketones such as acetone, methyl ethyl ketone and methyl isobutyl ketone; nitriles such as acetonitrile and propionitrile; carboxylic acid esters such as ethyl acetate and ethyl propionate; Nitrogen-containing aprotic
- Sulfur aprotic polar solvent pyridine, pyridine picoline, and the like; water; and the like. These solvents may be used alone or as a mixture of two or more thereof.
- a mixed solvent of water and a solvent selected from tetrahydrofuran and 1,4-dioxane is preferable, and a mixed solvent of tetrahydrofuran and water is more preferable.
- the mixing ratio of the organic solvent and water is 1:50 to 50: 1, preferably 1:10 to 10: 1 by mass ratio.
- the amount (concentration) of the reaction solvent used is not particularly limited, but 1 to 100 times the reaction solvent can be used with respect to 1 part by mass of the aromatic nitro compound represented by Formula 1.
- the amount is preferably 1.5 to 50 times by mass, more preferably 2.5 to 10 times by mass.
- the reaction temperature is not particularly limited, but is, for example, ⁇ 90 to 200 ° C., preferably 0 to 150 ° C., more preferably 10 to 120 ° C.
- the reaction time is usually 0.05 to 200 hours, preferably 0.5 to 100 hours.
- the amount of stannous chloride used in the reduction reaction is not particularly limited, but is preferably 1 to 10 mole times the amount of 1 mole of the nitro group of the aromatic nitro compound represented by the formula 1, more preferably Is 2 to 5 mole times.
- the aromatic nitro compound represented by the formula 1 has two nitro groups, it is preferably 2 to 20 mole times, more preferably 4 to 10 mole times the amount of the aromatic nitro compound. Amount.
- the specific reduction reaction is preferably carried out as follows. That is, an aromatic nitro compound as a starting material, stannous chloride, and a reaction solvent such as tetrahydrofuran and water are charged into a reactor, and stirred at 10 to 120 ° C., preferably for 1 to 20 hours. .
- the starting material is charged in a mixture of stannous chloride and the reaction solvent, or the aromatic nitro compound is added in portions, or the aromatic nitro compound is added to the reaction solvent.
- a method of dissolving and adding is preferable.
- the end point of the reaction can be confirmed by thin layer chromatography or high performance liquid chromatography.
- the aromatic nitro compound represented by the formula 1 is reduced to produce the aromatic amine represented by the formula 2, and the reaction liquid containing the aromatic amine, the unreacted raw material, and the Sn compound. Is obtained.
- a compound containing a benzene ring has a boiling point of preferably 80 to 170 ° C., more preferably 80 to 150 ° C., since it can be easily concentrated after separation.
- Examples of such a compound containing a benzene ring include benzene, toluene, m-xylene, o-xylene, p-xylene, mesitylene, anisole, chlorobenzene, o-dichlorobenzene, and the like. Toluene is particularly preferred because it is easy to dry.
- Examples of the liquid separation step for separating the Sn compound from the reaction solution to obtain the target product include the following four methods when toluene is used as the compound containing a benzene ring.
- Methodhod 1 After adding an aqueous potassium hydroxide solution to the reaction solution, toluene is added to separate the solution, and the desired product is obtained from the organic layer.
- Methodhod 2 Toluene is added to the reaction solution, washed with an aqueous potassium hydroxide solution, and separated to obtain the desired product from the organic layer.
- Methodhod 3) A reaction solution is added to a container containing toluene and an aqueous potassium hydroxide solution, and the mixture is separated to obtain a target product from the organic layer.
- Methodhod 4 Toluene and an aqueous potassium hydroxide solution are added to the reaction solution at one time, and the mixture is separated to obtain the desired product from the organic layer.
- any of these methods can be used, but when mixing with an aqueous potassium hydroxide solution, the temperature can be controlled to be constant, and no poor stirring due to precipitation of the target product or Sn compound occurs during mixing.
- the above (Method 3) is particularly preferable in that the Sn compound does not precipitate after mixing and the interface between the two layers is easy to distinguish.
- the amount of potassium hydroxide in the aqueous potassium hydroxide solution used in the liquid separation step is preferably set to an amount for converting the Sn compound in the reaction solution into potassium hexahydrostannate having high water solubility.
- the amount is preferably 3 mol times or more, more preferably 3 to 10 mol times, and particularly preferably 3 to 6 mol times with respect to 1 mol.
- the concentration of the aqueous potassium hydroxide solution used is not particularly limited. However, if the target product does not decompose, the higher the concentration, the more preferable it is that treatment can be performed with a small amount of aqueous potassium hydroxide solution. When the target product decomposes, it can be carried out by lowering the concentration of potassium hydroxide. Therefore, the concentration of the potassium hydroxide aqueous solution is preferably 1 to 50% by weight, and more preferably 10 to 50% by weight.
- the ratio of the amount of the compound containing a potassium hydroxide aqueous solution and a compound containing a benzene ring is such that the interface at the time of liquid separation is easy to separate and the volume efficiency is improved. Is preferred, 75: 1 to 1:75 is more preferred, and 50: 1 to 1:50 is particularly preferred.
- the temperature in the liquid separation is not particularly limited as long as the target product does not decompose and dissolve, and is preferably 10 to 80 ° C., and more preferably 10 to 60 ° C. in terms of easy temperature control.
- the time required for liquid separation is from 0.01 to 4 hours, preferably from 0.02 to 1 hour, from the point of view.
- the compound layer containing a benzene ring which is an organic layer obtained by the above liquid separation, contains an aromatic amine compound that is a target product.
- the organic layer may be concentrated as it is to separate and recover the target product.
- a poor solvent can be added to the dissolved organic layer to crystallize the target product, and the aromatic amine compound can be recovered by crystallization.
- hydrocarbons such as hexane, heptane and cyclohexane, alcohols such as ethanol, methanol and i-propyl alcohol are preferably used.
- purification treatment such as recrystallization may be performed.
- the aqueous layer formed by the above liquid separation contains potassium hexahydrostannate having high solubility resulting from the used stannous chloride used for the reduction reaction.
- these tin compounds can be recovered.
- the said reaction mixture was dripped at the mixed solution of toluene (75.0g) and 40 mass% potassium hydroxide aqueous solution (105g). At this time, there was no precipitate during the dropping, and the mixture could be stirred well. Thereafter, the mixture was stirred for 10 minutes, and the aqueous layer was discarded. The obtained organic layer was added to a 6% by mass aqueous potassium hydroxide solution (75 g) and stirred, and then the aqueous layer was discarded. Thereafter, the operation of adding water (75 g), stirring, and discarding the aqueous layer was repeated 5 times.
- the solid (1.00 g) obtained above was dissolved in toluene (7.00 g) at 50 ° C., and 0.1 g of activated carbon (special white birch, manufactured by Nippon Enviro Chemical Co., Ltd.) was added and stirred for 30 minutes. Thereafter, the activated carbon was removed by filtration, and the filtrate was concentrated. As a result, the Sn content in the target product was less than 1 ppm.
- activated carbon special white birch, manufactured by Nippon Enviro Chemical Co., Ltd.
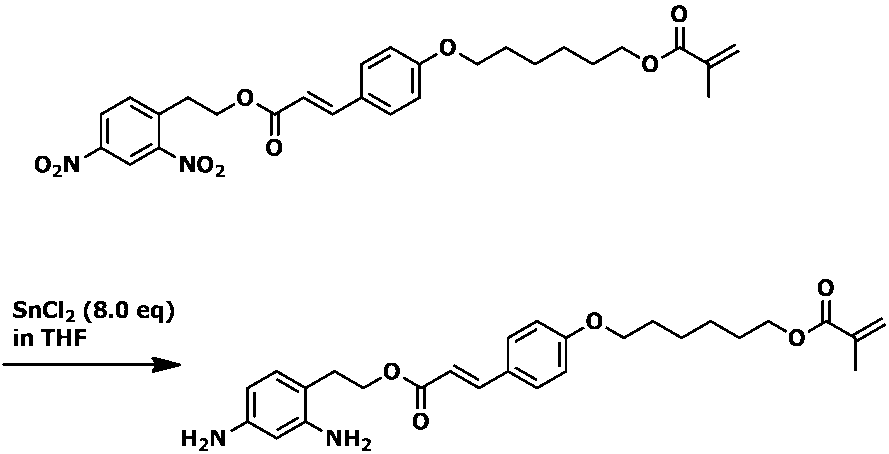
- Example 1 the amount of raw material (E) -4- (6- (methacryloyloxy) hexyloxy) cinnamic acid (2- (2,4-dinitrophenyl) ethyl) ester was 1.0 g, and other raw materials was carried out in the same manner as in Example 1 except that the ratio was changed to the same ratio as in Example 1. 5 g of ethyl acetate was added to the reaction mixture containing the desired product (E) -4- (6- (methacryloyloxy) hexyloxy) cinnamic acid (2- (2,4-diaminophenyl) ethyl) ester. Thereafter, sodium hydrogen carbonate (2.4 g) was further added. As a result, a large amount of white precipitate was formed, and stirring became impossible.
- Comparative Example 2 A reaction mixture comprising (E) -4- (6- (methacryloyloxy) hexyloxy) cinnamic acid (2- (2,4-diaminophenyl) ethyl) ester, obtained in the same manner as in Comparative Example 1. 7.2 g, which is one third of the above, was collected, to which 3.3 g of ethyl acetate was added, and a 40 mass% aqueous sodium hydroxide solution was added dropwise. When 1.3 g was added dropwise, As a result, a large amount of white precipitate was formed and stirring became impossible. Further, when 1.9 g of 40% by mass aqueous sodium hydroxide solution was added dropwise, the precipitate was dissolved, but then left to stand for 3 days.
- Comparative Example 3 The reaction was carried out in the same manner as in Comparative Example 1, and the resulting reaction mixture containing (E) -4- (6- (methacryloyloxy) hexyloxy) cinnamic acid (2- (2,4-diaminophenyl) ethyl) ester was added. On the other hand, 5.0 g of ethyl acetate was added, and a 40% by mass aqueous potassium hydroxide solution (7.0 g) was further added dropwise. During the dropwise addition, a white precipitate was formed and the stirring property was deteriorated, but after the entire amount was dropped, the precipitate was dissolved. Then, when it left still for several days, the precipitate generate
- the method of the present invention is a method for producing an aromatic amine compound by reducing an aromatic nitro compound with stannous chloride, and the Sn compound can be easily and efficiently removed, and has high purity.
- an aromatic amine compound can be obtained, which is industrially useful.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Description
特許文献2では、反応後に溶媒を留去し、残渣に水酸化ナトリウム水溶液を加え、pHを約12~14にした後、酢酸エチルで生成物を抽出する方法が記載されている。
特許文献3では、反応液に水酸化ナトリウムを強塩基性になるまで加えた後、ジエチルエーテルで生成物を抽出する方法が記載されている。
特許文献4では、酢酸エチル溶液である反応液に、水酸化カリウム水溶液を加え、酢酸エチルで生成物を抽出したあと、シリカゲルカラムクロマトグラフィーによって生成する方法が記載されている。
1.下記の反応式(1)に従って、式1で表わされる芳香族ニトロ化合物を、塩化第一スズを用いて還元して式2で表される芳香族アミン化合物を製造する方法において、
式1で表わされる芳香族ニトロ化合物を還元して得られる反応液を、ベンゼン環を含む化合物と水酸化カリウム水溶液との混合液に接触させて分液させ、得られる有機層から式2で表される芳香族アミン化合物を得ることを特徴とする芳香族アミン化合物を製造する方法。
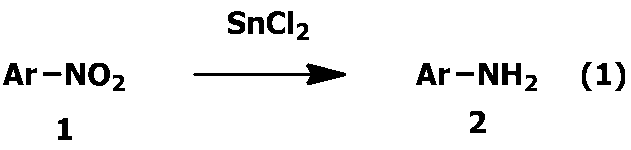
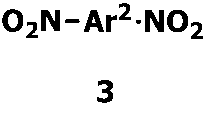

6.水酸化カリウムの量が、スズ原子1モルに対して、3モル倍量以上である、上記1~5のいずれかに記載の方法。
7.水酸化カリウム水溶液とベンゼン環を含む化合物の使用量比が、質量比で50:1~1:50である上記1~6のいずれかに記載の方法。
8.分液工程の温度が、10~80℃である上記1~7のいずれかに記載の方法。
9.水酸化カリウム水溶液とベンゼン環を含む化合物の混合液中に、芳香族ニトロ化合物及び塩化第一スズを含む反応液を加える、上記1~8のいずれかに記載の方法。
10.前記ベンゼン環を含む化合物が沸点80~170℃を有する、上記1~9のいずれかに記載の方法。
11.前記ベンゼン環を含む化合物がトルエンである、上記1~10のいずれかに記載の方法。
12.反応溶媒にテトラヒドロフランを用いる、上記1~11のいずれかに記載の方法。
13.反応溶媒に水を用いる、上記1~12のいずれかに記載の方法。





式4中、Rは塩化第一スズによる還元に不活性な有機基を表し、例えば、メチル基、エチル基、n-プロピル基、i-プロピル基、n-ブチル基、i-ブチル基、s-ブチル基、t-ブチル基、n-ヘキシル基、n-オクチル基、n-デシル基等のアルキル基;シクロペンチル基、シクロヘキシル基等のシクロアルキル基;ビシクロヘキシル基等のビシクロアルキル基;ビニル基、1-プロペニル基、2-プロペニル基、イソプロペニル基、1-メチル-2-プロペニル基、1または2または3-ブテニル基、1-ヘキセニル基、α-スチリル基、β-スチリル基等のアルケニル基;フェニル基、キシリル基、トリル基、ビフェニル基、ナフチル基等のアリール基;ベンジル基、フェニルエチル基、フェニルシクロヘキシル基等のアラルキル基、であり、なかでも、ビニル基又はβ-スチリル基が好ましい。
なお、これらの有機基の水素原子の一部または全部は、ハロゲン原子、水酸基、チオール基、リン酸エステル基、エステル基、カルボキシル基、リン酸基、チオエステル基、アミド基、オルガノオキシ基、オルガノシリル基、オルガノチオ基、オルガノアミノ基、カルバミン酸エステル基、アシル基、アルキル基、シクロアルキル基、ビシクロアルキル基、アルケニル基、アリール基、アラルキル基などで置換されていてもよい。また、これらは環状構造であってもよい。
反応溶媒としては、反応に不活性なものであれば特に限定はされない。例えば、ヘキサン、シクロヘキサン、ベンゼン、トルエン等の炭化水素類;四塩化炭素、クロロホルム、1,2-ジクロロエタン等のハロゲン系炭化水素類;メタノール、エタノール、イソプロピルアルコール等のアルコール類;ジエチルエーテル、ジイソプロピルエーテル、1,4-ジオキサン、テトラヒドロフラン等のエーテル類;アセトン、メチルエチルケトン、メチルイソブチルケトン等のケトン類;アセトニトリル、プロピオニトリル等のニトリル類;酢酸エチル、プロピオン酸エチル等のカルボン酸エステル類;N,N-ジメチルホルムアミド、N,N-ジメチルアセトアミド、N-メチル-2-ピロリドン、1,3-ジメチル-2-イミダゾリジノン等の含窒素非プロトン性極性溶媒;ジメチルスルホキシド、スルホラン等の含硫黄非プロトン性極性溶媒;ピリジン、ピコリン等のピリジン類;水;等が挙げられる。これらの溶媒は単独で用いても、これらのうちの2種類以上を混合して用いてもよい。好ましくは、テトラヒドロフラン及び1,4-ジオキサンから選ばれる溶媒と、水との混合溶媒であり、さらに好ましくは、テトラヒドロフランと水との混合溶媒である。
水と有機溶媒の混合溶媒を使用する場合、有機溶媒と水との混合比率は、質量比で1:50~50:1、好ましくは1:10~10:1である。
出発原料の仕込み順は、反応中間体の副反応を避けるため、塩化第一スズ、及び反応溶媒の混合物中に、芳香族ニトロ化合物を分割して添加するか、又は反応溶媒に芳香族ニトロ化合物を溶かして添加する方法が好ましい。反応の終点は、薄層クロマトグラフィーや高速液体クロマトグラフィーなどによって確認できる。
上記のようにして、式1で表される芳香族ニトロ化合物は還元され、式2で表される芳香族アミンが生成し、かかる芳香族アミン、未反応の原料、及びSn化合物を含む反応液が得られる。
反応液からSn化合物を分離し、目的物を得る分液工程としては、例えばベンゼン環を含む化合物としてトルエンを用いる場合には、以下の4方法が挙げられる。
(方法1)反応液に水酸化カリウム水溶液を添加した後、トルエンを加えて分液し、有機層から目的物を得る。
(方法2)反応液にトルエンを添加し、水酸化カリウム水溶液で洗浄し、分液して、有機層から目的物を得る。
(方法3)トルエンと水酸化カリウム水溶液の入った容器に、反応液を添加し、分液して、有機層から目的物を得る。
(方法4)反応液にトルエンと水酸化カリウム水溶液を一度に加えて分液し、有機層から目的物を得る。
分液工程で使用される水酸化カリウム水溶液中の水酸化カリウムの量は、反応液中のSn化合物を、水溶性の高いヘキサヒドロスズ酸カリウムにするための量にするのが好ましく、スズ原子1モルに対して3モル倍以上が好ましく、より好ましくは、3~10モル倍、特に好ましくは3~6モル倍である。
また、水酸化カリウム水溶液とベンゼン環を含む化合物の使用量の比率は、分液時の界面が分離しやすく、且つ、容積効率をよくするという点から、質量比で100:1~1:100が好ましく、75:1~1:75がよりに好ましく、50:1~1:50が特に好ましい。
HPLC分析
装置:LC-20Aシステム(島津製作所社製)
カラム:Inertsil ODS-3(4.6mmΦ×250mm、ジーエルサイエンス社製)
検出器:UV検出(波長254nm)
溶離液:アセトニトリル/0.1wt%酢酸水溶液(70/30、v/v)
さらに、上記で得られた固体(1.00g)をトルエン(7.00g)に50℃で溶解し、活性炭0.1g(特製白鷺)日本エンバイロケミカル社製)を加えて、30分間撹拌した。その後、活性炭をろ過で除き、ろ液を濃縮した結果、目的物中のSn含量は1ppm未満となった。
実施例1において、原料の(E)-4-(6―(メタクリロイルオキシ)ヘキシルオキシ)桂皮酸(2-(2,4-ジニトロフェニル)エチル)エステルの量を1.0gとし、その他の原料を実施例1と同様の比率にした以外は、実施例1と同様に実施した。目的物である(E)-4-(6―(メタクリロイルオキシ)ヘキシルオキシ)桂皮酸(2-(2,4-ジアミノフェニル)エチル)エステルを含む反応混合物に対して、酢酸エチル5gを加えた後、さらに炭酸水素ナトリウム(2.4g)を加えた結果、白色の析出物が大量に生成し、撹拌不能となった。
比較例1と同様にして実施し、得られた、(E)-4-(6―(メタクリロイルオキシ)ヘキシルオキシ)桂皮酸(2-(2,4-ジアミノフェニル)エチル)エステルを含む反応混合物の3分の1の量である7.2gを採取し、これに対して、酢酸エチル3.3gを加え、さらに40質量%水酸化ナトリウム水溶液を滴下したところ、1.3gを滴下した時点で、白色の析出物が大量に生成し、撹拌不能となった。さらに、1.9gの40質量%水酸化ナトリウム水溶液を滴下したところ、析出物は溶解したが、その後3日静置したところ、析出物が発生した。
比較例1と同様に実施し、得られた、(E)-4-(6―(メタクリロイルオキシ)ヘキシルオキシ)桂皮酸(2-(2,4-ジアミノフェニル)エチル)エステルを含む反応混合物に対して、酢酸エチル5.0gを加え、さらに40質量%水酸化カリウム水溶液(7.0g)を滴下した。滴下中、白色の析出物が生成し、撹拌性が悪化したが、全量滴下終了後は、析出物は溶解した。その後、数日静置したところ、析出物が、再び発生した。
なお、2015年1月13日に出願された日本特許出願2015-004365号の明細書、特許請求の範囲、及び要約書の全内容をここに引用し、本発明の明細書の開示として、取り入れるものである。
Claims (13)
- 下記の反応式(1)に従って、式1で表わされる芳香族ニトロ化合物を、塩化第一スズを用いて還元して式2で表される芳香族アミン化合物を製造する方法において、
式1で表わされる芳香族ニトロ化合物を還元して得られる反応液を、ベンゼン環を含む化合物と水酸化カリウム水溶液との混合液に接触させて分液させ、得られる有機層から式2で表される芳香族アミン化合物を得ることを特徴とする芳香族アミン化合物を製造する方法。

- 式1で表される化合物が、式3で表される芳香族ジニトロ化合物である請求項1に記載の方法。
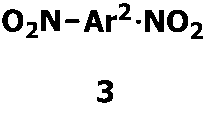
- 式1で表される化合物が、式4で表される芳香族ジニトロ化合物である請求項1に記載の方法。
- 式1で表される化合物が、式5で表される芳香族ジニトロ化合物である請求項1に記載の方法。

- 前記水酸化カリウム水溶液の濃度が、1~50重量%である請求項1~4のいずれか一項に記載の方法。
- 水酸化カリウムの量が、スズ原子1モルに対して、3モル倍量以上である請求項1~5のいずれか一項に記載の方法。
- 水酸化カリウム水溶液とベンゼン環を含む化合物の使用量比が、質量比で50:1~1:50である請求項1~6のいずれか一項に記載の方法。
- 分液工程の温度が、10~80℃である請求項1~7のいずれか一項に記載の方法。
- 水酸化カリウム水溶液とベンゼン環を含む化合物の混合液中に、前記反応液を加える請求項1~8のいずれか一項に記載の方法。
- ベンゼン環を含む化合物が沸点80~170℃を有する請求項1~10のいずれか一項に記載の方法。
- ベンゼン環を含む化合物がトルエンである請求項1~10のいずれか一項に記載の方法。
- 反応溶媒にテトラヒドロフランを用いる請求項1~11のいずれか一項に記載の方法。
- 反応溶媒に水を用いる請求項1~12のいずれか一項に記載の方法。
Priority Applications (4)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR1020177018798A KR20170102484A (ko) | 2015-01-13 | 2016-01-13 | 방향족 아민 화합물의 제조 방법 |
| JP2016569482A JP7109880B2 (ja) | 2015-01-13 | 2016-01-13 | 芳香族アミン化合物の製造方法 |
| EP16737379.4A EP3246308B1 (en) | 2015-01-13 | 2016-01-13 | Method for producing aromatic amine compound |
| CN201680004493.3A CN107108464A (zh) | 2015-01-13 | 2016-01-13 | 芳香族胺化合物的制造方法 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2015-004365 | 2015-01-13 | ||
| JP2015004365 | 2015-01-13 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2016114308A1 true WO2016114308A1 (ja) | 2016-07-21 |
Family
ID=56405849
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2016/050849 Ceased WO2016114308A1 (ja) | 2015-01-13 | 2016-01-13 | 芳香族アミン化合物の製造方法 |
Country Status (6)
| Country | Link |
|---|---|
| EP (1) | EP3246308B1 (ja) |
| JP (1) | JP7109880B2 (ja) |
| KR (1) | KR20170102484A (ja) |
| CN (1) | CN107108464A (ja) |
| TW (1) | TWI697472B (ja) |
| WO (1) | WO2016114308A1 (ja) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPWO2016114312A1 (ja) * | 2015-01-13 | 2017-10-19 | 日産化学工業株式会社 | 反応混合物中のスズ化合物の処理方法 |
Families Citing this family (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN113999136B (zh) * | 2021-12-07 | 2023-08-04 | 山东第一医科大学(山东省医学科学院) | 大黄酰胺衍生物及其制备方法和应用、recql4特异性表达的肝癌抑制剂 |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH02275857A (ja) * | 1989-01-13 | 1990-11-09 | Yoshitomi Pharmaceut Ind Ltd | ピリジン誘導体 |
| JP2002506466A (ja) * | 1997-05-20 | 2002-02-26 | エルシコン・インコーポレーテッド | フッ化アミン物質 |
| JP2010518060A (ja) * | 2007-02-06 | 2010-05-27 | シプラ・リミテッド | エチル−n−(2,3−ジクロロ−6−ニトロベンジル)グリシン塩酸塩の製造方法 |
| WO2014208609A1 (ja) * | 2013-06-25 | 2014-12-31 | 日産化学工業株式会社 | 液晶配向剤、液晶配向膜、液晶表示素子 |
Family Cites Families (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| TWI758241B (zh) | 2015-01-13 | 2022-03-21 | 日商日產化學工業股份有限公司 | 反應混合物中之錫化合物之處理方法 |
-
2016
- 2016-01-13 WO PCT/JP2016/050849 patent/WO2016114308A1/ja not_active Ceased
- 2016-01-13 CN CN201680004493.3A patent/CN107108464A/zh active Pending
- 2016-01-13 JP JP2016569482A patent/JP7109880B2/ja active Active
- 2016-01-13 TW TW105100957A patent/TWI697472B/zh active
- 2016-01-13 EP EP16737379.4A patent/EP3246308B1/en active Active
- 2016-01-13 KR KR1020177018798A patent/KR20170102484A/ko not_active Withdrawn
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH02275857A (ja) * | 1989-01-13 | 1990-11-09 | Yoshitomi Pharmaceut Ind Ltd | ピリジン誘導体 |
| JP2002506466A (ja) * | 1997-05-20 | 2002-02-26 | エルシコン・インコーポレーテッド | フッ化アミン物質 |
| JP2010518060A (ja) * | 2007-02-06 | 2010-05-27 | シプラ・リミテッド | エチル−n−(2,3−ジクロロ−6−ニトロベンジル)グリシン塩酸塩の製造方法 |
| WO2014208609A1 (ja) * | 2013-06-25 | 2014-12-31 | 日産化学工業株式会社 | 液晶配向剤、液晶配向膜、液晶表示素子 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP3246308A4 * |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPWO2016114312A1 (ja) * | 2015-01-13 | 2017-10-19 | 日産化学工業株式会社 | 反応混合物中のスズ化合物の処理方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP3246308B1 (en) | 2020-03-11 |
| TW201634439A (zh) | 2016-10-01 |
| KR20170102484A (ko) | 2017-09-11 |
| JP7109880B2 (ja) | 2022-08-01 |
| EP3246308A1 (en) | 2017-11-22 |
| JPWO2016114308A1 (ja) | 2017-10-19 |
| CN107108464A (zh) | 2017-08-29 |
| EP3246308A4 (en) | 2018-08-08 |
| TWI697472B (zh) | 2020-07-01 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20140235862A1 (en) | Method for producing 4, 4-difluoro-3,4-dihydroisoquinoline derivatives | |
| WO2016114308A1 (ja) | 芳香族アミン化合物の製造方法 | |
| US10752608B2 (en) | Method for producing crystal of uracil compound | |
| RS53735B1 (sr) | Postupak proizvodnje toluidin jedinjenja | |
| EP3088391A1 (en) | Method for producing benzyl ester 2-aminonicotinate derivative | |
| JP6822145B2 (ja) | 反応混合物中のスズ化合物の処理方法 | |
| KR20060130483A (ko) | 비피리디늄 화합물의 제조방법 | |
| WO2007145260A1 (ja) | アゼチジノンカルボン酸の改良された晶析方法 | |
| EP0894784B1 (en) | 4-fluorosalicylic acid derivatives and process for producing the same | |
| JP6263814B2 (ja) | 細胞死抑制剤及び新規化合物 | |
| EP2522651B1 (en) | Process for the purification of 2,6-diisopropyl phenol | |
| JPH05271169A (ja) | 新規な光学活性tert−ロイシン・1−(4−置換フェニル)エタンスルホン酸塩およびその製造法 | |
| WO2015159672A1 (ja) | ハロゲン化合物の製造方法、カリウム塩の製造方法、及びカリウム塩 | |
| JPH05125023A (ja) | 2−ビスアリールアミノ−9,9−ジアルキルフルオレンの単離方法 | |
| JP2689600B2 (ja) | α―イソプロピル―p―クロロフェニル酢酸の光学分割法 | |
| JP2018087178A (ja) | アジルサルタンの製造方法 | |
| JP2012184175A (ja) | 高純度3−アセチル−9−エチルカルバゾールの製造方法。 | |
| JP2020063197A (ja) | 2,6−ナフタレンジカルボン酸ジアリルの製造方法 | |
| JP2005112807A (ja) | アミノアルコキシカルボスチリル誘導体の製造方法。 | |
| JP2003277322A (ja) | アルコキシ安息香酸誘導体類の製造方法 | |
| JPS62154B2 (ja) | ||
| JP2004352676A (ja) | 新規フタルアミド酸誘導体及びその製造法 | |
| JP2011068581A (ja) | ヒドロキシビニルナフタレン化合物の製造方法 | |
| JP2008127308A (ja) | 光学活性5−オキソピロリジン−3−カルボン酸誘導体の製造法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 16737379 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2016569482 Country of ref document: JP Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 20177018798 Country of ref document: KR Kind code of ref document: A |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REEP | Request for entry into the european phase |
Ref document number: 2016737379 Country of ref document: EP |