WO2016159652A1 - 조직투과성 펩타이드와 항-혈관 내피세포 성장인자 제제가 융합된 융합단백질을 유효성분으로 포함하는 안질환 예방 및 치료용 약학적 조성물 - Google Patents
조직투과성 펩타이드와 항-혈관 내피세포 성장인자 제제가 융합된 융합단백질을 유효성분으로 포함하는 안질환 예방 및 치료용 약학적 조성물 Download PDFInfo
- Publication number
- WO2016159652A1 WO2016159652A1 PCT/KR2016/003254 KR2016003254W WO2016159652A1 WO 2016159652 A1 WO2016159652 A1 WO 2016159652A1 KR 2016003254 W KR2016003254 W KR 2016003254W WO 2016159652 A1 WO2016159652 A1 WO 2016159652A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- fusion protein
- tissue
- vegf
- ranibizumab
- seq
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/17—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- A61K38/177—Receptors; Cell surface antigens; Cell surface determinants
- A61K38/179—Receptors; Cell surface antigens; Cell surface determinants for growth factors; for growth regulators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/17—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- A61K38/18—Growth factors; Growth regulators
- A61K38/1858—Platelet-derived growth factor [PDGF]
- A61K38/1866—Vascular endothelial growth factor [VEGF]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/0005—Vertebrate antigens
- A61K39/0011—Cancer antigens
- A61K39/00113—Growth factors
- A61K39/001135—Vascular endothelial growth factor [VEGF]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/395—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum
- A61K39/39533—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals
- A61K39/3955—Antibodies; Immunoglobulins; Immune serum, e.g. antilymphocytic serum against materials from animals against proteinaceous materials, e.g. enzymes, hormones, lymphokines
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/50—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates
- A61K47/51—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent
- A61K47/62—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient the non-active ingredient being chemically bound to the active ingredient, e.g. polymer-drug conjugates the non-active ingredient being a modifying agent the modifying agent being a protein, peptide or polyamino acid
- A61K47/64—Drug-peptide, drug-protein or drug-polyamino acid conjugates, i.e. the modifying agent being a peptide, protein or polyamino acid which is covalently bonded or complexed to a therapeutically active agent
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/475—Growth factors; Growth regulators
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N5/00—Undifferentiated human, animal or plant cells, e.g. cell lines; Tissues; Cultivation or maintenance thereof; Culture media therefor
- C12N5/10—Cells modified by introduction of foreign genetic material
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2319/00—Fusion polypeptide
- C07K2319/70—Fusion polypeptide containing domain for protein-protein interaction
- C07K2319/74—Fusion polypeptide containing domain for protein-protein interaction containing a fusion for binding to a cell surface receptor
Definitions
- composition for the prevention and treatment of eye diseases comprising a fusion protein fused with a tissue-penetrating peptide and an anti-vascular endothelial growth factor agent as an active ingredient
- the present invention relates to a pharmaceutical composition for preventing and treating ocular diseases, including a fusion protein in which a tissue-penetrating peptide and an anti-vascular endothelial growth factor agent are fused as an active ingredient, and more specifically, a tissue-penetrating peptide and an anti- Pharmaceutical composition for the prevention and treatment of ocular diseases, including a fusion protein fused with vascular endothelial growth factor (ant i-VEGF) preparation, and a tissue-penetrating peptide and anti-vascular endothelial growth factor (ant i-VEGF) ) Converting a recombinant vector comprising a nucleic acid sequence encoding the fusion protein to which the agent is fused into a host cell; Culturing the superior cells; And a method for producing an anti-vascular endothelial growth factor agent having improved resistance and efficacy, comprising recovering the fusion protein from the cell, administering an effective amount of the fusion protein according to the present invention to
- the macula lutea is the part of the eye that is densely packed with cells in the retina of the eye to receive light most clearly and accurately, and macular degenerat ion is a disease that causes degeneration of the macular degeneration due to various causes. ).
- the macular degeneration is one of the three major causes of blindness together with glaucoma and diabetic retinopathy.
- the most common cause of macular degeneration is increased age, and other causes of family history, race, and hobby are known to cause macular degeneration, and damage to the macula results in loss of the ability to recognize details such as small prints, facial features, or small objects. do.
- macular degeneration There are two types of macular degeneration, non-exudative (dry) macular degeneration and exudative (wet) macular degeneration, and 90% of people with macular degeneration have a dry form of symptoms.
- dry macular degeneration waste products form a yellow precipitate called drusen that can accumulate in the tissue under the macula. Can be.
- the presence of drusen interferes with blood flow to the retina, especially the macula, and when the blood flow decreases, it reduces the supply of nutrients to the macula, stopping or contracting the efficient action of photosensitive cells.
- wet macular degeneration new, weak blood vessels grow in or below the retina, causing liquid and blood to leak into the space below the macula.
- choroidal neovascularization where the choroid is the region of the vessels below the retina and neovascularization refers to the growth of new blood vessels in tissues, as can be inferred from the name of choroidal neovascularization. In this case, blood vessels develop from the choroid into the macula and grow.
- macular degeneration is considered to be a disease that is common to older people, it is known that the number of patients in their 4s and 50s has recently increased rapidly. The main causes of the lowered age group of macular degeneration are increased dietary intake, westernization, smoking and drinking. , Bad lifestyles such as UV exposure are pointed out.
- Diabetic Macular Edema is explained by the thickening of the retina and / or hard alum within a disc diameter of the center of the retina.
- DME and diabetic retinopathy are microvascular complications in diabetic patients, weaken vision, and eventually cause blindness. Patients with DR may progress to DME and develop DME after destruction of the blood-retinal barrier due to leakage of expanded permeable capillaries and microaneurysms.
- DME is associated with choroidal neovascularization that penetrates damaged or tissue-broken Brook membranes.
- the representative therapeutic agents used for the prevention or treatment of various ocular diseases related to neovascularization in the eye including macular degeneration, diabetic macular edema, ranibizumab, bevacizumab, beep Alf ibercept, conbercept, and the like.
- biopharmaceuticals for the treatment of major eye diseases such as macular degeneration and diabetic retinal edema, including the aforementioned drugs are mainly used for the treatment of diseases of the posterior eye including the retina in the form of intraocular injections.
- ranibizumab and aflibercept as eye drops to solve problems such as decreased patient convenience, increased side effects, and psychological fears.
- the fourth and fourth domains are Fc-linked fusion proteins that are currently being developed as eye drops, but reportedly, bioavailability is known to be less than about 5% (Wang et al., 2013. PLOS ONE).
- Ranibizumab, sold under the brand name Lucentis was noted as a breakthrough treatment by 90% of patients with macular degeneration at the time of development, but only 30% of patients with banung patients showed improvement in vision.
- resistance is caused by continuous drug administration (Syed et al., 2012. Nature Rev. Drug Discov.) To improve this, the binding force to VEGF-A is increased by 100 times or more.
- Appliverscept an Fc fusion antibody designed to inhibit VEGF-B and P1GF, was released in 2011, but has shown rapid growth, but there is no difference in clinical efficacy between the two products.
- 10% of patients with anti-VEGF agents are not expected to have a therapeutic effect, which is thought to be caused by growth factor-dependent neovascular bypass pathways other than VEGF-A.
- resistance was induced and drug reactivity decreased with increasing frequency of administration (Lux et al., 2007. Br. J. Ophthalmol.). It is known to be a result of vascular strengthening and pericyte-dependent VEGF production resulting from increased coverage of pericyte, which contributes to stabilizing endothelial cells in the rat.
- an object of the present invention is to provide a pharmaceutical composition for the prevention and treatment of eye diseases comprising a fusion protein fused to a tissue-penetrating peptide and an anti-vascular endothelial growth factor (ant i-VEGF) preparation as an active ingredient. .
- Another object of the present invention is the step of (a) converting a recombinant vector comprising a nucleic acid sequence encoding a fusion protein fused to a tissue-penetrating peptide and an anti-vascular endothelial growth factor (ant i-VEGF) preparation into a host cell ; (b) culturing the cells; And (c) recovering the fusion protein from the cell, and providing a method of producing an anti-vascular endothelial growth factor agent having improved resistance and improved efficacy.
- ant i-VEGF anti-vascular endothelial growth factor
- Another object of the present invention is to treat an ocular disease comprising administering to an individual in need thereof an effective amount of a fusion protein fused with a tissue-permeable tempide and an anti-vascular endothelial growth factor (ant i-VEGF) agent.
- a tissue-permeable tempide and an anti-vascular endothelial growth factor (ant i-VEGF) agent are fused.
- the present invention provides a pharmaceutical composition for the prevention and treatment of eye diseases comprising a fusion protein fused with a tissue-penetrating peptide and an anti-vascular blood cell growth factor (ant i-VEGF) preparation as an active ingredient.
- a pharmaceutical composition for the prevention and treatment of eye diseases comprising a fusion protein fused with a tissue-penetrating peptide and an anti-vascular blood cell growth factor (ant i-VEGF) preparation as an active ingredient.
- ant i-VEGF anti-vascular blood cell growth factor
- the present invention provides a host comprising a recombinant vector comprising a nucleic acid sequence encoding a fusion protein fused with a tissue-permeable peptide and an anti-vascular endothelial growth factor (ant i-VEGF) agent.
- the present invention comprises administering to a subject in need thereof an effective amount of a fusion protein in which a tissue permeable peptide and an anti-vascular endothelial growth factor (ant i-VEGF) agent are fused.
- a tissue permeable peptide and an anti-vascular endothelial growth factor (ant i-VEGF) agent are fused.
- the present invention provides a preparation for treating ophthalmic diseases comprising a fusion protein in which a tissue-penetrating peptide and an anti-vascular endothelial growth factor (ant i-VEGF) agent are fused as an active ingredient.
- a tissue-penetrating peptide and an anti-vascular endothelial growth factor (ant i-VEGF) agent are fused as an active ingredient.
- the present invention provides a pharmaceutical composition for the prevention and treatment of eye diseases comprising a fusion protein fused with a tissue-penetrating peptide and an anti-vascular endothelial growth factor (ant i-VEGF) agent as an active ingredient. to provide.
- vascular endothelial growth factor -A (VEGF-A) is well known to induce blood ejection (extravasat ion). This is also called the vascular permeabi li factor. This action is known to be due to binding to the vascular endothelial growth factor receptor (VEGFR2).
- VEGFR2 vascular endothelial growth factor receptor
- vascular endothelial growth factor -A mutations although not able to bind to the vascular endothelial growth factor receptor, are vascular permeable. This is an example of how this is being promoted. This suggests that there is another receptor of vascular endothelial growth factor-A (Stacker et al., 1999. J. Biol. Chem.).
- Neuropilin is a transmembrane glycoprotein and has two forms, NRP1 and NRP2.
- Neurophylline acts as a core receptor for VEGF receptors (VEGFRs) by binding VEGF family ligands.
- NRP1 functions as a co-receptor of VEGFRl, VEGFR2, VEGFR3 and binds to various VEGF ligands.
- NRP2 acts as a co-receptor of VEGFR2 and VEGFR3, contributing to lymphatic tube formation (lymphangiogenesis) and cell adhesion (adhesion).
- NRPl / NRP2 acts as a co-receptor of Plexin family receptors and binds to secreted semaphorin 3-family ligands (class 3 semaphorins: Sema3A, Sema3B, Sema3C, Sema3D, Sema3E, Sema3F, and Sema3G).
- the tissue permeability is a tissue that specifically recognizes and accumulates neurophyllin overexpressed tissue, or widens the cell gap of vascular endothelial cells to promote the out-of-vascular flow of drugs, and serves as a barrier to water-soluble molecules. By means of adjusting the corneal cell gap it means having one of the properties to promote the distribution of drugs in the eye.
- the term "neuropilin (NRP)" in the present invention is a transmembrane glycoprotein, and has two forms of NRP1 and NRP2. Neurophyllin consists of five domains. From the N-terminus, al / a2 domains are classified as CUB domains, and the Ig-like C2 type portion of semaphorin binds.
- this site forms a complex with plexin and serves to increase the binding force with semaphorin-plexin.
- the bl and b2 domains of neuropilins are classified as FV / VIII domains, and the C-terminal sites of VEGF family ligands or secreted semaphorin 3 family ligands (secreted Sema3s) bind.
- the VEGF ligand and the semaphorin trifamily ligands have a recognition site (RXRR, Arg-X-Arg-Arg) of purine proteolytic enzymes, and are commonly used at the C-terminus by arginine (Furin processing). Arg) ends with amino acid residues (Adams et al., 1997.
- Anti-vascular endothelial growth factor preparations bind to molecules that interfere with the interaction between VEGF and native VEGF receptors, such as VEGF or VEGF receptors to prevent or otherwise inhibit the interaction between VEGF and VEGF receptors. It includes molecules.
- VEGF antagonists include anti-VEGF antibodies, anti-VEGF receptor antibodies and VEGF receptor-based chimeric molecules.
- fusion refers to the integration of two molecules having different or identical functions or structures, and fusion by all physical, chemical or biological methods in which tissue-permeable peptides can bind to anti-vascular endothelial growth factor preparations. Can be.
- the fusion may preferably be by a linker peptide, which may bind, for example, to the C-terminus of the Fab (antigen binding fragment) or Fc fragment of the antibody.
- the ocular disease of the present invention preferably means ocular disease caused by angiogenesis.
- the expression “ocular disease due to angiogenesis” means any disease of the eye by or related to blood vessel growth or proliferation, or by vascular leakage.
- the binding of neurophilin receptors increases the tissue permeability of the drug, and the cover age of the per icyte may be deteriorated and thus the effect may be exerted in patients who exhibit resistance to the drug.
- the dosing cycle is improved and the development of eye drops is possible.
- the variant of ranibizumab introduced a point mutation in which the cysteine (C) is substituted with serine (S) in the amino acid sequence of ranibizumab greatly increases productivity.
- the effect of increasing productivity was maintained even in the improved fusion of the tissue permeable peptide (TPP) to the ranibizumab variant (Example 1).
- the affinity to the neurophilin receptor of the fusion protein fused with a tissue-permeable peptide to the C-terminus of the ranibizumab variant was compared with the tight junction between endothelial cells and the ability.
- the fusion protein was found to bind very well to a level similar to that of neurophylline receptor NRP 1 and Sema3A ligand.
- the adhesion and resolution between endothelial cells was very excellent (Example ⁇ 1-3>).
- a fusion protein fused with a tissue-permeable peptide at the C-terminus of the ranibizumab variant may improve permeability through binding with a neurophylline receptor distributed in eye endothelial cells.
- the degree of permeability was compared over time ( ⁇ Example 4>).
- the anti-vascular endothelial growth factor preparations in which the tissue-penetrating peptide was conjugated started permeation from corneal epithelial tissue from 1 hour of experiment, and confirmed that the intraocular drug distribution was significantly higher than ranibizumab at 2 hours.
- the possibility of dose control and administration interval increase through permeability improvement was confirmed.
- the fusion protein according to the present invention rapidly penetrates the cornea, a tissue that acts as a barrier to water-soluble molecules, to reach the inside of the eyeball compared to ranibizumab.
- the formulation could be developed as an eye drop by improving the formulation.
- the effect of increasing the permeability of eye tissues by the fusion of tissue-permeable peptides was similarly observed in the whole ant ibody form having a large molecular weight and complex protein structure.
- a fusion protein fused with a tissue-permeable peptide at the FC-terminus of bevacizumab in the form of immunoglobulin G (IgG) also showed a markedly increased eye permeability compared to bevacizumab, an improved body of bevacizumab according to the present invention. It can also be seen that the therapeutic effect can be developed as a drug.
- the fusion protein fusion protein form of VEGF receptors (VEGFR1, VEGFR2) to the Fc fragment was also applied to the tissue-penetrating peptide at the FC-terminus in the same manner as bevacizumab.
- the drug may be developed as a drug having a significantly improved therapeutic effect compared to the Fc fusion protein.
- the angiogenesis inhibitory effect of the fusion protein and ranibizumab of the present invention and the neovascular inhibitory effect in the drug resistance model were evaluated using various animal disease models. Experimental results using a corneal neovascular model showed that the fusion protein of the present invention exhibited significant neovascularization effects of at least 50% compared to the control group. It was confirmed that it was possible to increase the efficacy by the tissue-permeable peptide fused at the C-terminus by showing an effect equivalent to ranibizumab (Example ⁇ 5-1>).
- the fusion protein of the present invention showed more than twice the efficacy of ranibizumab compared to ranibizumab in angiogenesis inhibitory effect in the drug resistance model, in combination with ant i-VEGF aptaraer and ant i-PDGF ant ibody preparation.
- Levels similar to those of the administered literature (Jo et al., 2006. Am. J. Pathol. 168), with about 70% of patients receiving ranibizumab who had only been maintained without vision improvement. It can be expected that there will be (Example ⁇ 5-2>).
- the superior effect of inhibiting angiogenesis in ocular disease of the fusion protein according to the present invention is a model of choroidal neovascularization (CNV) used as an actual macular degeneration efficacy model and an oxygen-induced neovascularization (0IR) model used as a retinopathy efficacy model.
- CNV choroidal neovascularization
- IR oxygen-induced neovascularization
- the same is observed in the present invention, and it can be predicted that the therapeutic effect will be remarkably enhanced in comparison with ranibizumab ( ⁇ Example 6>, ⁇ Example 7>).
- prevention means any action that inhibits or delays the progression of eye diseases by administration of a composition of the present invention.
- treatment refers to any action by which administration of a composition of the present invention improves or advantageously alters the symptoms of an ocular disease. More specifically, the term 'treatment' refers generically to ameliorating the symptoms of eye diseases, which may include treating, substantially preventing, or improving the condition of eye diseases, as long as they originate from eye diseases. It includes, but is not limited to, alleviating, restraining, or preventing branch symptoms or most symptoms.
- the term 'effective amount' refers to an amount that improves the symptoms of an ocular disease when administered to an individual, and includes an amount that heals or substantially prevents or improves an ocular disease.
- the term 'individual' may be an animal, preferably a mammal, particularly an animal including a human, or may be a cell, tissue, organ or the like derived from the animal. The subject may be a patient in need of treatment. All.
- composition according to the invention can be administered to any mammal, including humans, by any means.
- it can be administered orally or parenterally.
- Parenteral administration methods include, but are not limited to, intravenous, intramuscular, intraarterial, intramedullary, intradural, intracardiac, transdermal, subcutaneous, intraperitoneal, intranasal, intestinal, topical, sublingual, or rectal Administration.
- the immunogenic complex protein according to the present invention may be administered in a monograph or in combination with a known compound having the effect of preventing and treating a target disease.
- the dosage of the pharmaceutical composition of the present invention to the human body may vary depending on the age, weight, sex, dosage form, health condition, and degree of disease of the patient. 10 mg / day, preferably 0.1 to 2 mg / day per eye. In the case of eye drops, the dosage range is from 0.01 mg to 1 ml of eye drop, preferably 0.01 to 10 mg / day. In addition, divided doses may be administered at regular intervals according to the judgment of the doctor or pharmacist.
- the composition of the present invention may further include a pharmaceutically acceptable additive, and the pharmaceutically acceptable additive may include starch, gelatinized starch, microcrystalline cellulose, lactose, povidone, colloidal silicon dioxide and calcium hydrogen phosphate.
- the pharmaceutically acceptable additive according to the present invention is preferably included in an amount of 0.01 to 90 parts by weight based on the composition, but is not limited thereto.
- the pharmaceutical composition may be in various forms suitable for any route of administration, including but not limited to injections, eye drops, eye ointments, ocular implants, and when formulated it is commonly used layering agents, extenders, binders It may be prepared using a diluent or excipient such as a wetting agent, a disintegrant, and a surfactant.
- injectables include, but are not limited to, intraocular injection including intravitreal injection and conjunctival injection. Injectables may include conventional additives such as solubilizers, isotonic agents, suspending agents, emulsifiers, stabilizers, preservatives, and the like.
- Suitable carriers for the injection of the present invention include physiological saline, bacteriostatic water, cremofo ER EL (Basp, Pa., NJ) or Phosphate Complete Brine (PBS).
- the composition must be sterile and must be fluid enough to be easily injected.
- the composition must be stable under the conditions of manufacture and storage and must be preserved against the contaminating action of microorganisms such as bacteria and fungi.
- the carrier may be a solvent or a dispersion medium containing, for example, water, ethane, polyols (such as glycerol, propylene glycol and liquid polyethylene glycol, etc.), and appropriate mixtures thereof.
- Proper fluidity can be maintained using, for example, a coating such as lecithin, maintaining the required particle size in the case of dispersions, and using surfactants.
- Microbial action can be prevented by the use of various antibacterial and antifungal agents such as parabens, chlorobutane, phenol, ascorbic acid, chimerosal and the like.
- isotonic agents for example, sugars, polyalcohols such as manny, sorbitan, trehalose, sodium chloride in the compositional process.
- Absorption of the injectable composition can be enhanced by including in the composition an agent that delays absorption, such as aluminum monostearate, hyaluronic acid, and gelatin.
- the eye drops may be water soluble ophthalmic solutions, non-aqueous ophthalmic solutions or ophthalmic emulsions.
- conventionally known optional ingredients in eye drops for example, Layers, viscous agents, stabilizers, isotonic agents, preservatives and the like can be blended.
- buffers such as a citrate, a phosphate, an acetate, an amino acid salt, are mentioned as a buffer.
- polyvinyl alcohol, hydroxypropyl cell rose, methyl cell rose, povidone, hydroxypropyl methyl cell rose, ethyl cell rose, hydroxyethyl sal rose, carmellose, polyethylene glycol, chondroitin or salts thereof Can be mentioned.
- salts such as sodium chloride and potassium chloride
- Polyhydric alcohols such as glycerin and propylene glycol
- Sugars such as glucose, sucrose and trehalose
- Sugar alcohols such as Xilitle and Solbi
- Polyethers such as polyethylene glycol
- Amide sulfonic acids such as taurine, etc.
- the pH at room temperature is preferably 4.5 to 8.5, more preferably 5.5 to 8, particularly Preferably it is 6-8.
- the pH is measured using a pH meter at room temperature (eg, Accumet model 25 pH / Ion meter, manufactured by Fisher Scient ific).
- Some ophthalmic drugs have poor permeability through the eye barrier and cannot be administered by eye drops. Thus, ointments can be used to extend contact time and increase the amount of drug absorbed.
- water-insoluble polymers include ethyl sal, ethylene vinyl acetate copolymer, polymethacrylate, ethyl methacrylate, methyl methacrylate, trimetal ammonium ethyl, copolymer, and methacrylate. Acid metal, butyl methacrylate, dimetal aminoethyl methacrylate, copolymer, etc. are mentioned.
- biodegradable polymers include polylactic acid, polylactic acid, glucolic acid co-polymer, polycyanoacrylate, polyalkylcyanoacrylate, poly? ⁇ , and caprolactone.
- water-soluble polymer cellulose derivatives such as hydroxypropane metal cellulose loose furate, carboxymethyl ethyl cellulose, hydroxypropyl cellulose, calcium alkynate, chitosan, albumin, gelatin, methacrylic acid Acid metal copolymer etc. are mentioned.
- oil-soluble component include tripalmitin, cetyl alcohol, cholesterol, various phospholipids, cetyl palmitate, and cholesterol palmitate.
- carrier components usually have a sustained release (slow release function) of the active ingredient containing an ophthalmic therapeutically active substance.
- a carrier component with a high specific gravity which can be used without participating in a specific gravity regulator
- hydroxypropyl methyl cellulose phthalate 200731 weight 1.65
- hydroxypropyl methyl salo phthalate 220824 weight 1.82
- carboxymethyl Ethyl salose specific gravity 1.59
- specific gravity regulators used for specific gravity adjustment of carrier particles include, for example, insoluble components such as titanium oxide (4.17), tripotassium phosphate (specific gravity 3.14), anhydrous potassium hydrogen phosphate (specific ratio 2.89), and phosphoric acid.
- the insert is similar to a soft contact lens placed on the cornea, except that it is usually placed in the upper appendage rather than attached to the open cornea, or less frequently in the lower conjunctival sac. Inserts are generally made of a biologically soluble material that dissolves or disintegrates in the tear fluid while releasing the drug.
- Solid preparations for oral administration include tablets, pills, powders, granules, capsules, and the like, and these solid preparations may be mixed with Tyros with at least one excipient such as starch, calcium carbonate, sucrose, lactose or gelatin. Can be formulated. In addition to simple excipients, lubricants such as magnesium styrate talc may also be used. Oral liquid preparations include suspending agents, liquid solutions, emulsions and syrups. Water and liquid paraffin, which are commonly used as simple diluents, may include various excipients, for example wetting agents, sweeteners, fragrances, and preservatives. .
- compositions of the present invention may be used in any physiologically acceptable carrier, parasitic agent or stabilizer (Remington: The Science and Pract ice of Pharmacy, 19th Edite, Al fonso, R., ed, Mack Publishing Co. (Easton, PA: 1995)).
- Acceptable carriers, excipients or stabilizers are nontoxic to recipients at the dosages and concentrations employed, and include monolayer solutions such as phosphoric acid, citric acid and other organic acids; Antioxidants including ascorbic acid; Low molecular weight (less than about 10 residues) polypeptides; Proteins such as serum albumin, gelatin or immunoglobulins; Hydrophilic polymers such as polyvinylpyrrolidone; Amino acids such as glycine, glutamine, asparagine, arginine or lysine; Monosaccharides, disaccharides and other carbohydrates including glucose, mannose, trehalose, or dextrin ol; Chelating agents such as EDTA; Sugar alcohols such as manny or sorbbi; Salt-forming counterions such as sodium; And / or nonionic surfactants such as tungsten, pluronics or polyethylene glycol (PEG).
- monolayer solutions such as phosphoric acid, citric acid and other organic acids
- the present invention provides a pharmaceutical composition characterized in that the tissue-permeable peptide comprises any one amino acid sequence selected from the group consisting of SEQ ID NO: 1 to 7.
- the tissue-permeable peptide represented by the amino acid sequence selected from the group consisting of SEQ ID NOs: 1 to 7, the amino acid sequence and the length of the binding site of the VEGF A ligand that binds to the blb2 domain of neurophylline, sema known to bind to neurophylline It is a peptide designed based on the similarity of their C-terminal sequences by analyzing the nucleotide sequences of the Purine C-terminal sequences of Forin 3A and Semaphorin 3F.
- the present invention provides the anti-vascular endothelial growth factor preparations include bevacizumab, ranibizumab, r84 (PLoS One. 2010 Aug 6; 5 (8) ), Aplibercept (af l ibercept), convergence (cdnbercept), CT0KW02005056764A2), D0M15-10-1KW02008149147A2), D0M15-26-
- EPI-0030 W02011023130A1
- a pharmaceutical composition comprising an agent used to prevent or treat an ocular disease, a biosimilar thereof, and a variant thereof, more preferably Rani. Bizumab, bevacizumab, Able Libercept, and Conceptcept may be, but are not limited to.
- the term 'biosimi lar' refers to similarity in terms of quality, efficacy and safety by imitating patent-expired original biopharmaceuticals already developed / sold using biotechnology such as genetic recombination and cell culture technology. Means a drug that has been proven to replicate.
- the variant of the present invention is characterized in that the cysteine, which is an amino acid of the heavy chain constant 1 domain and the light chain constant domain, is deleted or substituted with another amino acid residue including serine, except cysteine. It provides a pharmaceutical composition.
- ranibizumab in which the last cysteine in the nucleotide sequence of ranibizumab was changed to serine was prepared (Example ⁇ 1-1>) .
- the present invention is a ranibizumab (ranibizumab) is a variant consisting of a light chain having an amino acid sequence represented by SEQ ID NO: 8 and a heavy chain having an amino acid sequence represented by SEQ ID NO: 10 It provides a pharmaceutical composition, characterized in that.
- the tissue permeable peptide of the one aspect may further include a linker template.
- the linker peptide may consist of 1 to 50 amino acids, bar Preferably it may consist of 4-20, more preferably 4-15 amino acids.
- the linker peptide may be composed of glycine (Glycine, G), serine (Sine) or alanine (alanine, A), preferably the sequence of the linker peptide is (GA) n or (GGGGS) ' ⁇ Consisting of amino acid sequences (wherein n and m are each independently an integer from 1 to 20, and means the number of times the sequence in parentheses is repeated), more preferably GAGA or (GGGGS 3 ).
- the tissue-penetrating peptide was confirmed to produce a fusion protein fused to ranibizumab variants through the linker.
- the fusion protein is a form in which TPP # 2 is linked with a linker ((GGGGS) 3 ) to the ranibizumab variant (IDB0061) according to the present invention, and is composed of an amino acid sequence represented by SEQ ID NO: 12 and SEQ ID NO: 14 Protein (IDB0062); In the form Raney connected in non - mainstream Thank variant (IDB0061) TPP # 5, the linker ((GGGGS) 3) in accordance with the present invention, the fusion protein (IDB0064) consisting of amino acids sequence shown in SEQ ID NO: 16 and SEQ ID NO: 18; In bevacizumab, a TPP # 2 is linked to a heavy chain by a linker ((GGGGS) 3 ), and is a fusion protein (IDB0072) consisting of an amino acid sequence
- the pharmaceutical composition of the present invention can be used to treat ocular diseases according to any neovascular system.
- the expression 'ocular disease along neovascularization' means any disease of the eye by or related to blood vessel growth or proliferation or by blood vessel leakage.
- the ocular diseases according to angiogenesis include proliferative vitreoretinopathy, macular degeneration, pigment retinopathy, diabetic retinopathy, choroidal neovascularization, neovascular glaucoma, ischemic optic neuropathy, prematurity retinopathy, prematurity retinopathy, epidemic keratoconjunctivitis, neovascular iris Disease, posterior keratofibrosis, atopic keratitis, keratoconjunctivitis keratitis, psoriatic psoriatic keratitis, flicative keratoconjunctivitis, scleritis and diabetic macular edema, more preferably macular degeneration and diabetes It may be, but is not limited to, sex macular edema.
- the present invention provides a recombinant vector comprising a nucleic acid sequence encoding a fusion protein fused to a tissue-permeable peptide and an anti-vascular endothelial growth factor (ant i-VEGF) agent Converting to host cells; (b) culturing the cells; And (c) recovering the fusion protein from the cell, and providing a method for producing an anti-vascular endothelial growth factor agent having improved resistance and improved efficacy.
- the nucleic acid sequence encoding the fusion protein may be selected from the group consisting of SEQ ID NO: 13, SEQ ID NO: 15, SEQ ID NO: 17, SEQ ID NO: 19, SEQ ID NO: 21, and SEQ ID NO: 23.
- the nucleic acid encoding the light and heavy chains of the ranibizumab improver IDB0062 according to the present invention is in the nucleotide sequences of SEQ ID NO: 13 and SEQ ID NO: 15, respectively, and the nucleic acids encoding the light and heavy chains of IDB0064 are SEQ ID NO: 17 and The nucleic acids encoding the base sequence of SEQ ID NO: 19 and the light and heavy chains of the bevacizumab modifier IDB0072 are as described in nucleotide sequences of SEQ ID NO: 21 and SEQ ID NO: 23, respectively.
- the vector refers to an expression vector prepared by a person skilled in the art to express the fusion protein of the present invention by inserting the polynucleotide of the present invention into the vector according to any method known in the art using an appropriate transcription / translation control sequence.
- the polynucleotide sequence cloned according to the present invention may be operably linked to an appropriate expression control sequence, and the operably linked gene sequence and expression control sequence may be combined with a selection marker and a replication initiation point (repl icat ion or igin). It can be included in one expression vector that it contains.
- the expression control sequence refers to a DNA sequence that controls the expression of a polynucleotide sequence operably linked in a particular host cell.
- One such regulatory sequence is one selected from the group consisting of a promoter for transcription, a sequence of operators to regulate transcription, a sequence encoding a suitable mRNA liposome binding site, and a sequence that controls termination of transcription and translation. It may contain the above.
- the vector used as the mother vector of the expression vector is not particularly limited, and this invention Any plasmid, virus or other mediator commonly used for expression in microorganisms used as host cells in the art may be used.
- the plasmids include E. coli-derived plasmids (pBR322, pBR325, pUC118 and pUC119, pET-22b (+)), Bacillus subtilis-derived plasmids (pUBllO and pTP5), and yeast-derived plasmids ( ⁇ 13, ⁇ 24 and YCp50).
- the virus may be, but is not limited to, an animal virus such as a retrovirus, adenovirus or vaccinia virus, and a crypt virus such as baclovirus.
- the host cell can choose to control the expression of the inserted sequence or to produce the desired product from the gene in a particular desired manner.
- Different host cells have characteristic and specific mechanisms for the translation and post-translational processing and modification of proteins. Suitable cell lines or host systems can be chosen that provide the desired modification and processing of the heterologous protein expressed.
- Expression in yeast can produce biologically active products.
- Expression in eukaryotic cells can increase the likelihood of 'natural' folding.
- Host cells capable of stably and continuously cloning and expressing the vector of the present invention may use any host cell known in the art, for example, E. coli JM109, E. coli BL21DE, E. coli DH5, E coli RR1, E. coli LE392, E. coli B, E.
- yeast Sacharomycecerevisiae
- nasal cells e.g., human cells
- CH0 cell line Choinese hamster ovary
- W138, ⁇ , COS-7, 293 as host cells
- HepG2, 3 ⁇ 3, RIN and MDCK cell lines Methods for transforming host cells by delivering the vector into the host cells are known. Any method is possible and is not specifically limited.
- Escherichia coli can be transformed by heat shock or electroporat ion methods, and calcium phosphate precipitat ion, DEAE-dextran method, and electricity can be used to construct production cell lines using animal cells.
- Electroporat ion, direct microinject ion, DNA-loaded liposome method, lipofectamine-DNA complex, cell sonication traits may be altered by sonicat ion, gene bombardment using high veloci ty microprojectiles, polycat ion, and receptor-mediated transfect ion. You can switch. Some of these techniques can be refined for in vivo or ex vivo use. Cultivation of the transformed cells is carried out under appropriate conditions that allow the expression of the fusion protein, which can be carried out according to methods well known to those skilled in the art.
- the transformed cells can be cultured in large quantities by conventional culture methods.
- As the culture medium a medium consisting of a carbon source, a nitrogen source, vitamins and minerals may be used.
- a 2X YT medium may be used.
- Culture of the cells is possible under normal cell culture conditions and can be incubated for 10 hours to 40 hours, for example, in the temperature range of 15 ° C to 45 ° C.
- Centri fugat ion or filtrat ion may be used to remove the cells in the culture and recover only the culture medium, which steps may be performed by the skilled person as needed.
- the culture medium (filtrate) from which the cells have been removed can be stored and stored for a short period of time so as not to lose its activity in the usual way.
- Fusion proteins expressed in transformed cells can be purified in a conventional manner, for example salting out (eg ammonium sulfate precipitation, sodium phosphate precipitation), solvent precipitation (acetone, ethanol). Purification of the fusion protein of the present invention by applying a single or a combination of techniques such as protein fraction precipitation, dialysis, gel filtration, ion exchange, reverse phase column chromatography, affinity chromatography, and ultrafiltration.
- salting out eg ammonium sulfate precipitation, sodium phosphate precipitation
- solvent precipitation acetone, ethanol
- Purification of the fusion protein of the present invention by applying a single or a combination of techniques such as protein fraction precipitation, dialysis, gel filtration, ion exchange, reverse phase column chromatography, affinity chromatography, and ultrafiltration.
- the pharmaceutical composition comprising a fusion protein fused with a tissue-penetrating peptide of the present invention and an anti-vascular endothelial growth factor (ant i-VEGF) agent is an improved tissue permeability of the anti-vascular endothelial growth factor formulation.
- the intraocular injection improves the drug delivery to the choroidal tissues, and can be developed as an eye drop as well as the effect of treating, reducing the dose, or increasing the cycle of drug-resistant patients.
- FIG. 1A is a schematic diagram of a fusion protein fused with a tissue permeable peptide and an anti-vascular endothelial growth factor antigen binding fragment (Fab).
- Figure 1B is a schematic diagram showing the binding of the fusion protein fused with VEGF and neurophyllin receptor -1 fused to the tissue-penetrating peptad and anti-vascular endothelial growth factor.
- FIG. 1C is a schematic diagram of ameliorators in which tissue-permeable peptides are bound to various vascular endothelial growth factor inhibitor forms (Fab, whole IgG, Fc fusion protein).
- Figure 2 is a result of analyzing the main product according to the production of ranibizumab and the fusion protein of the present invention by HPLC (IDB0062: Fab fusion protein fused with ranibizumab variants and tissue-permeable peptide TPP # 2).
- AU means an arbitrary unit.
- Figure 3 shows the results of the analysis of the primary purified products of the fusion proteins in a 12% non-reducing SDS-PAGE gel (IDB0062, Fab fusion protein fused with the ranibizumab variant and tissue-permeable peptide TPP # 2; IDB0064, Fab fusion protein fused with ranibizumab variant and tissue permeable peptide TPP # 5).
- FIG. 4A shows the results of binding affinity analysis of ranibizumab, ranibizumab variants, and VEGF of the fusion proteins according to the present invention.
- Figure 4B is a result of evaluating the binding ability of the tissue-penetrating peptides and neuropillin receptor -1 of the fusion protein according to the present invention (IDB0061, ranibizumab variant; IDB0062, ranibizumab variant and tissue permeable peptide TPP # 2 fusion) Fab fusion protein; Fc-TPP # 2, Fc fusion protein in which the tissue-permeable peptide TPP # 2 is fused to the FC-terminus of IgGl.
- IDB0061 ranibizumab variant
- IDB0062 ranibizumab variant and tissue permeable peptide TPP # 2 fusion
- Fc-TPP # 2 Fc fusion protein in which the tissue-permeable peptide TPP # 2 is fused to the FC-terminus of IgGl
- Figure 5A is a result of evaluating the storage conditions of the fusion protein according to the present invention.
- 5B is a result of evaluating the stability of the fusion protein with repeated shock / thawing.
- Figure 6A is a result of evaluating the angiogenesis inhibitory effect in the corneal neovascular prevention model in the renal neovascular prevention model of ranibizumab, ranibizumab variants and fusion proteins according to the present invention.
- FIG. 6B is a graph showing the numerical results of FIG. 6A.
- 7A is a result of evaluation of angiogenesis inhibitory effect in the corneal neovascular resistance model in the neovascular resistance model of ranibizumab and the fusion protein according to the present invention.
- FIG. 7B is a graph showing the numerical results of FIG. 7A.
- 7C is a result of evaluating the pericyte coverage reduction effect of ranibizumab and the fusion protein according to the present invention.
- Figure 8A is the result of evaluation by the ocular osmolality oligoscope microscope of the fusion protein ranibizumab improved IDB0062 according to the present invention.
- 8B is a graph showing the numerical results of FIG. 8A.
- 8C is a result of analyzing the distribution of FITC conjugat ionized protein in the cross section of eye tissues reacted for 2 hours by fluorescence microscopy with DAPI staining.
- FIG. 8D is a graph showing numerical results of FIG. 8C.
- Figure 8E is a result of analyzing the fragments of the retinal tissue by confocal microscopy, the distribution according to the retinal permeability of IDB0062.
- Figure 9A is the result of evaluating the ocular performance of the fusion protein bevacizumab improved IDB0072 according to the present invention through a light microscope.
- 9B is a graph showing the numerical results of 9A.
- FIG. 10A shows the inhibition of proliferation of choroidal neovascularization following drug treatment in ranibizumab and the choroidal neovascularization (CNV) model of the fusion protein according to the present invention.
- FIG. FIG. 10B is a graph showing the results of FIG. 10A numerically.
- FIG. 11A is a result of analyzing by fluorescence microscopy that leakage of the end of blood vessels is suppressed by drug treatment in ranibizumab and the oxygen-induced retinopathy (0IR) model of the fusion protein according to the present invention.
- FIG. 11B is a graph showing the numerical results of FIG. 11A.
- 12A is a fluorescent microscope after i solect in B4 staining that the formation of neovascularization and vascular bundles (tufts) is suppressed by drug treatment in ranibizumab and oxygen-induced retinopathy (0IR) model of the fusion protein according to the present invention. This is the result of analysis.
- FIG. 12B is a graph showing numerical results of FIG. 12A.
- FIG. FIG. 13 shows the results of intraocular injection of IDB0062 and ranibizumab in rats, followed by analysis of the amount of drug present in retinal tissues by western blot assay.
- ranibizumab The low production yield of ranibizumab is due to the need for complex post-processing due to the low culture productivity of the Fab fragment and the polymorphism of the produced protein through periplasmic expression.
- the ranibizumab variant IDB0061 was produced. Through this, it was confirmed that the production yield was dramatically improved by increasing the culture productivity and simplifying the purification process.
- ranibizumab synthesizes the gene by changing the last cysteine of seranibisumab nucleotide sequence to serine. Since transformaUon to SUPEX5 (KCTC 12657BP) production cell line was then subjected to expression test.
- the construction of the light chain portion of ranibizumab variant IDB0061 is as follows. The nucleic acid sequence of ranibizumab was obtained from related patents, and codon optimization was performed. Based on this, the nucleic acid of the Fab light chain variable region (VL) containing the signal sequence was synthesized, and the CL region of the Fab was ant i-.
- a vector of albumin Fab SL335 was prepared by PCR using pr imer (SEQ ID NOs: 24, 25) as a template.
- pr imer SEQ ID NOs: 24, 25
- the construction of the heavy chain portion of ranibizumab variant IDB0061 is as follows. Retrieving a Raney non - mainstream Thank of the nucleic acid sequence from the patent and the nucleic acid of the codon optimized as a rear Based on this working signal sequence (gl II) a heavy chain variable region (VH) Fab contains, synthesized, CH1 domain of the Fab Was prepared by PCR using a pr imer (SEQ ID NO: 32, 33) as a template of the SL335 vector, an ant i- albumin Fab.
- Point mutat ions were generated in the amino acid sequence of ranibizumab using methods commonly used in the field of protein engineering. Specifically, cysteine, the 214th amino acid in the light chain amino acid sequence of Rani vizumab (serine) and cysteine, the 226th amino acid in the heavy chain amino acid sequence of ranibizumab, was substituted with serine.
- the light and heavy chain amino acid sequences of the ranibizumab variant IDB0061 in which the point mutations occurred were represented by SEQ ID NO: 8 and SEQ ID NO: 10, respectively, and the nucleic acid sequences encoding them were represented by SEQ ID NO: 9 and SEQ ID NO: 11, respectively.
- ranibizumab variant IDB0061 was cloned into pHEKA vector and trans format ion to SUPEX5 strain. Specifically, competent cel l 100 ⁇ was put in the lmra cuvette, and 3 ⁇ l of the DNA was added thereto, followed by electric shock at 1800 V to introduce a gene to complete the production cell line. Inoculate 1% ( ⁇ / ⁇ ) of ranibizumab and IDB0061-producing cell lines in 50 ml of 2x YT media (100 mM potassium phosphate buffer, H 7.2, 50 ug / ml kanamycin) for 18 hours at 220 ° C at 28 ° C. The preculture was carried out.
- 2x YT media 100 mM potassium phosphate buffer, H 7.2, 50 ug / ml kanamycin
- the present inventors can bind both neurophylline l (NRPl) and 2 to the C-terminus of ranibizumab or specifically bind to neurophylline 1 to increase the efficacy and overcome resistance of ranibizumab, an anti-VEGF agent.
- Tissue permeable peptide t issue penetrating peptide, TPP
- TPP Tissue permeable peptide
- FIG. 1 The schematic diagram of the fusion protein in which ⁇ is fused to ranibizumab is shown in FIG. 1. Specifically, various ⁇ s (peptides specifically binding to neurophylline receptors) described in Table 3 at the C-terminus of ranibizumab variant IDB0061 prepared in Example ⁇ 1-1> by April Bio A fused fusion protein and a cell line producing the same were produced.
- ⁇ # 2 having the amino acid sequence of SEQ ID NO: 2 in the ⁇ shown in Table 3 is a modification of the C-terminal sites of VEGF 165 and semaphorin 3-family ligands, which are intrinsic ligands of neurophylline
- TPP # 5 is As the blb2 domain protein of neurophylline 1 and the blb2 domain protein of neurophyllin 1 as competitors, a peptide derived from a clone that selectively binds to the bl domain of neurophyllin 1 was isolated and identified.
- fusion of Avast in to act as a bivalent to the neurophylline receptor is designed to have tissue permeability with similar affinity to VEGF and Sema3A ligand.
- the production of the light chain portion of ⁇ fused ranibizumab variant IDB0062 was obtained by obtaining a nucleic acid sequence of ranibizumab from the relevant patent, and after the codon optimization process, based on the light chain variable region (which contains a signal sequence) VL) nucleic acid was synthesized, and the light chain constant region (CL) C-terminal cysteine was synthesized with a nucleic acid (CL'-l inker-TPP # 2) including a linker and TPP # 2 sequence in some of the serine-substituted sequences. It was.
- the CL region of the Fab was prepared by PCR using primers (SEQ ID NOs: 24, 25) using SL335, which is an anti-albumin Fab, as a template, and primers (SEQ ID NO: 24) using the synthesized CL'-linker-TPP # 2 nucleic acid template. No. 26, 27) to prepare a CL'-linker-TPP # 2 by PCR. These two products were linked by linking PCR to prepare CL-1 inker -TPP # 2 oligonucleotide.
- VL was prepared using a primer (SEQ ID NOs: 28, 29), and then an assembly PCR using a primer (SEQ ID NOs: 30, 31, BamHI and Xhol sequences) with CL-linker-TPP # 2.
- SEQ ID NOs: 28, 29 a primer for PCR amplification of a VL sequence.
- the production of the heavy chain portion of the ranivarizumab variant IDB0062 fused to ⁇ # 2 was obtained from the related patents of the ranibizumab mutant from a related patent, and after the codon optimization process, the heavy chain containing the signal sequence (glll) was included.
- Nucleic acid of the variable region (VH) was synthesized, and the nucleic acid (CH1'-1 inker-TPP # 2) including the linker and TPP # 2 sequences in some sequences in which the cysteine of the heavy chain constant region (CHI) was substituted with serine.
- the CH1 region of the Fab consists of a primer of the SL335 anti- albumin Fab as a template.
- VH was prepared using a primer (SEQ ID NO: 36, 37) as a template of the synthesized VH nucleic acid, and then subjected to assembly PCR using a primer (SEQ ID NO: 38, 39, EcoRI and Hindlll sequences) together with CL-linker-TPP # 2. The final VH-CH1-1 inker-TPP # 2 sequence was completed.
- the light chain was digested with BamHI and X) I, the heavy chain with EcoRI and Hindlll, and cloned into the pHEKA vector digested with the same restriction enzyme.
- PHEKA vector containing the IDB0062 sequence completed by the above method was trans format ionized to SUPEX5 strain. Specifically, the competent cell ⁇ in a 1 ⁇ cuvette, 3 ⁇ 1 of the DNA was added thereto, followed by electric shock at 1800V to introduce a gene to complete the production cell line.
- the light chain and heavy chain amino acid sequences of IDB0062 thus prepared were represented by SEQ ID NO: 12 and SEQ ID NO: 14, respectively, and the nucleic acid sequences encoding them were represented by SEQ ID NO: 13 and SEQ ID NO: 15, respectively.
- Example ⁇ 1-2> affinity for the neurophyllin receptor and the tight junction and the ability between endothelial cells were compared.
- affinity for neurophylline receptors was performed for NRP Kneuropilin 1).
- SPR surface plasmon resonance
- Biacore2000 GE Healthcare
- each of the neuropilrin 1 domains was immobilized in 10 mM Na-acetate complete solution (pH 4.0) and immobilized on a CM5 sensor chip (GE Healthcare, USA) at about 1,000 response units (RU).
- HBS-EP buffer (lOmM HEPES, 2 mM ethylenediaminetetraacetic acid, and 0.005% surfactant P20, pH 7.4, GE Healthcare) was analyzed at a flow rate of 30 ⁇ 1 / min, and VEGF165 was 5nM at 80nM and semaphorin 3A at 62. 5 ⁇ , TPP was analyzed at a concentration of 25625M to 1.5625uM. Regeneration of CM5 chip after binding / dissociation analysis was performed by flowing buffer (20mM NaOH, 1M NaCl, pH 10.0) at 30 ⁇ 1 / min for 1 minute. It became.
- the sensorgrams obtained by 3 min of binding and 3 min of dissociation were normalized and subtracted from the blank cel l to calculate affinity.
- the tight junction lysis between the endothelial cells was evaluated by measuring the degree of inhibition of VE-cadherin and E-cadherin of the proteins. Inhibition of the expression of VE-cadherin and E-cadherin in endothelial cells is known to impair the tight junct ion between the endothelial cells, and through the intraocular administration of the drug to the intravenous Or tissue penetration).
- As an experimental method for indirectly confirming vascular permeability enhancement of TPP changes of VE-cadherin through Western blot were confirmed.
- vascular permeability enhancement seeded HUVEC cells at 3 ⁇ 10 5 per well in 6-wel l plates. After incubation for 24 hours, Western blot was performed by treating TPP with luM for 10 minutes. The gels subjected to SDS_PAGE were transferred to PVDF membranes and examined using a primary antibody (SantaCruz) that recognizes VE-cadherin and ⁇ -act in and a secondary antibody (SantaCurz) bound to HRP. The assay was performed using ImageQuant LAS4000 mini ( GE Healthcare).
- TPP # 2-fused Fc (Fc-TPP # 2) binds to the neurophylline receptor (NRP 1) at a high level (similar to that of Sema3A igand), as shown in Table 4 below. It has been shown to significantly inhibit VE-cadherin. In the case of Fc (Fc-TPP # 5) fused with TPP # 5, the binding force with NRP 1 was higher than that of Sema3A, and the effect of VE-cadherin was more excellent. IDB0062 was also confirmed to bind to neurophyllin receptor (NRP 1) at high levels, and the degree of VE-cadherin inhibition was also inhibited similarly to Fc-TPP # 2.
- the productivity of IDB0062 was confirmed in the same manner as in Example ⁇ 1-1>. As a result, as shown in Table 1, it was confirmed that IDB0062 fused with TPP # 2 to IDB0061 also showed about 5 times higher productivity than ranibizumab.
- the purity of the first purified product using the adsorption column was analyzed by HPLC. HPLC analysis experiment was carried out in the following manner. The purified product was first purified using an adsorption column, concentrated using amicon (Mi l ipore, 10K), and exchanged with formulat ion buf fer (10 mM hist idine, 0.1% tween20, 10% trehalose) to give a final concentration of 0.5 mg / Dilute to tnl.
- TPP # 5 in which the VE-cadherin inhibitory effect was confirmed in Example ⁇ 1-3> was linked by a linker to produce a fused ranibizumab modifier IDB0064 and confirmed productivity. .
- IDB0064 Specific production process of IDB0064 is as follows. Gene cloning was performed to bind IDB0061 to a new ⁇ , ⁇ # 5. A specific primer (SEQ ID NOs: 40, 41, 42) was used to replace C-terminal ⁇ from ⁇ # 2 to ⁇ # 5 with IDB0062 as a template, and the first sequence shared by TPP # 2 and ⁇ # 5 was used. The primer was prepared by holding the anneal ing region and then extending the TPP # 5 sequence.
- PCR was carried out 30 cycles in the order of denaturat ion (95 ° C, 40sec), anneal ing (65 ° C, 40sec), extent ion (72 ° C, lmin) and then Fab light chain-TPP # 5 Of and Fab
- the gene of heavy chain-TPP # 5 was obtained.
- the light chain portion of the PCR product was treated with BamHI and Xhol, and the heavy chain portion was treated with EcoRI and Hindl ll and then inserted into the pHEKA vector by l igat ion and trans-format ion into the production cell line SUPEX5.
- IDB0064 The light and heavy chain amino acid sequences of IDB0064 prepared as described above were represented by SEQ ID NO: 16 and SEQ ID NO: 18, respectively, and the nucleic acid sequences encoding them were represented by SEQ ID NO: 17 and SEQ ID NO: 19, respectively.
- IDB0064 production sachets were inoculated with 1% ( ⁇ / ⁇ ) in 50 ml of 2 X YT media ClOOmM potassium phosphate buffer, H 7.2, 50 ug / ml kanamycin) and incubated for 18 hours at 28 ° C at 220 rpm for 18 h.
- the binding capacity of IDB0062 to the neurophilin 1 receptor was confirmed to maintain similar binding activity as the control drug (Fc-TPP # 2). This means that the ⁇ # 2 peptide fused to the C-terminus of ranibizumab binds well to neurophyllin 1 receptor.
- the plate was washed three times, and then treated with Goat ant i-human kappa light chain Ab-HRP (Sigma Aldrich, A7l64) in a buffer of 5,000 times, treated with ⁇ per well and reacted at 37 ° C for 1 hour. .
- Goat ant i-human kappa light chain Ab-HRP Sigma Aldrich, A7l64
- TMB substrateCBethyl, E102 was treated with ⁇ per well for 2 ⁇ 3 minutes and the stop solutionClN HC1) was treated with ⁇ per well after the reaction was finished, and the value was measured at 450nm using an ELISA plate reader.
- VEGF vascular endothelial growth factor
- carbonate coating buffer 0.1M NaHC0 3 , pH 9.6
- SPL Immunoplate Maxi binding
- the plate is washed three times, and the Goat anti-human kappa light chain Ab-HRP (Sigraa Aldrich, A7164) is added to the blocking buffer. Diluted 5,000 times, treated with ⁇ per well and reacted at 37 ° C for 30 minutes. After washing the plate 7 times, TMB substrate (Bethyl, E102) was treated with ⁇ ⁇ . Per well, and after 2-3 minutes reaction, stop solut ionClN HC1) was treated with ⁇ ⁇ . Per well to stop reaction and 450nm using ELISA plate reader. The value was measured at. As shown in FIG.
- the ELISA assay of IDB0062 confirmed that the VEGF-A binding ability was somewhat inferior to ranibizumab, and that VEGF-A was changed as the tertiary structure of IDB0062 was changed by a sequence change of a specific amino acid. It was expected that the binding CDRCComp ary-Det erm ini ng region) was partially changed. However, since the binding capacity of ranibizumab is considerably higher than that of a general antibody, it is believed that this weakening of binding does not result in a substantial decrease in clinical efficacy.
- the first purified tablets were changed to formulat ion buffer, concentrated to the concentration used in animal experiments (5mg / ml), and then subjected to stability experiments. Stability experiments by storage conditions were divided by ⁇ ⁇ ⁇ and stored at 4 ° C and -80 ° C for 5 weeks, thawed in ice for 1 week, and analyzed by VEGF binidng ELISA assay. ⁇ / thawing stability was evaluated by VEGF binding ELISA assay by taking some samples while repeating the sample was frozen at -80 ° C and thawed on ice five times.
- IDB0062 was stable at VEGF-A binding capacity up to 5 weeks storage at 4 ° C and -80 ° C, even if the physical shock through 5 times repeated cold / thaw As it did not affect, it was judged that there would be no stability deterioration due to the amino acid sequence change.
- ex-vivo ocular penetrat ion model was constructed to evaluate the efficacy of ranibizumab improved ocular permeability.
- TPP fused at the C-terminus of IDB0062 could actually improve tissue permeability through binding to neurophylline receptors that are highly distributed in ocular endothelial cells
- the extracted eye was subjected to FITC conjugat ion ranibizumab. And the degree of permeability was compared after soaking in IDB0062 solution.
- the protein sample was exchanged with lOOmM sodium carbonate buffer (H 9.0), the concentration was corrected to lmg / ml, and then mixed with FITC (lmg / ml in DMS0) for 2 hours at room temperature. .
- the reaction was carried out by applying different reaction numbers of FITC per protein. After removing the FITC that was not conjugat ion using PD-10 desalting column, the buffer was changed to PBS and the purified product was quantitatively used as a sample for evaluation of efficacy. Conjugat ion results showed that F.P rat io of ranibizumab and IDB0062 were 1.127 and 1.133, indicating that almost the same number of moles of FTTC were bound per molecule of protein.
- IDB0062 began to penetrate into the eye rapidly through the cornea and the posterior eye from 1 hour of experiment and reached a 2 hour distribution in ocular tissue and vitreous compared to the control drug ranibizumab. 4 times significantly increased. DAPI staining can be used to clearly distinguish and analyze ocular tissues. Unlike ranibizumab, IDB0062 shows an excessive distribution of protein inside the cornea (FIG. 8C, white arrow). It can be seen that about 10-fold increase compared to bizumab (FIG. 8D). FIG.
- FIG. 8E shows the cross-section of ocular retinal tissue, while the control drug ranibizumab is mostly near the sclera, while IDB0062 passes through the ret ina pigment epithel ial cel l (RPE) layer to the retina.
- RPE pigment epithel ial cel l
- the whole antibody (bevacizumab) protein such as bevacizumab (Whole ant ibody) protein has a high molecular weight and complex protein tertiary structure, which is inferior to tissue penetration. Nevertheless, the same experiment was conducted to overcome these physical limitations by fusing tissue permeable peptides at the C-terminus of bevacizumab and to improve the permeability compared to the control drug bevacizumab.
- the manufacturing process of the bevacizumab improved body IDB0072 is as follows.
- IDB0072 is a form of antibody fusion protein in which TPP is fused by a linker to the C-terminus of bevacizumab, and the amino acid sequence of TPP is shown in Table 3 above.
- TPP having the amino acid sequence of SEQ ID NO: 1 to 4 can bind to neurophylline 1 and 2
- TPP having the amino acid sequence of SEQ ID NO: 5 to 7 is specific to neurophylline 1 only By combining It is possible.
- TPP # 2 in TPP shown in Table 3 is a modification of the C-terminal region of VEGF 165 and semaphorin 3-family ligands, which are intr insic ligands of neurophylline
- TPP # 5 is a blb2 domain of neurophylline 1 Isolation and identification of an artificial peptide derived from a selectively binding clone, of which TPP # 2 is fused with bevacizumab using a linker to act as a bivalent to the neurophylline receptor, resulting in similar affinity to VEGF and Sema3A ligands.
- IDB0072 fused to TPP # 2 having the amino acid sequence of SEQ ID NO: 2 at the C-terminus of bevacizumab was prepared.
- IDB0072 was cloned into the p C DNA3.4 vector.
- the light and heavy chain amino acid sequences of IDB0072 are set forth in SEQ ID NO: 20 and SEQ ID NO: 22, respectively, and the nucleic acid sequences encoding them are as described in SEQ ID NO: 21 and SEQ ID NO: 23, respectively.
- Plasmids encoding the light chain protein were transfected using Neon eletroporesis, and then inoculated at 3> ⁇ 1 () 6 cel ls in T25 f lask and cultured at 37 ° C. After securing stable cel l using a selection marker, the bioreactor was incubated for 7 days in a suspended state using serum-free SFM4CH0 (Hyclone) at lOOrpm, 37 ° C, pH 7.2, and 50% D0 2 . The supernatant was separated from the cells by centrifugation and sterilized using a 0.22 filter.
- SFM4CH0 Hyclone
- the protein fused with the purified antibody heavy chain constant region and the peptide specifically binding to the selected NRP1 was quantified using the absorbance and the extinction coefficient at the corrected 280nm wavelength.
- bevacizumab and IDB0072 were exchanged with 100 mM sodium carbonate buffer (pH 9.0) and the concentration corrected to 3 mg / ml.
- the mixture was mixed with FITC (lmg / ml in DMSO) and reacted at room temperature for 2 hours.
- IDB0072 reacted by applying different moles of FITC per protein from the control drug bevacizumab to enjoy the difference in conjugat ions due to TPP # 2 fusion.
- Example ⁇ 4-1> In order to evaluate the activity of the produced bevacizumab improved IDB0072 was confirmed by binding all SPR assay for neurophyllin 1 receptor. As shown in Table 5, the binding capacity of IDB0062 to the neuropilin 1 receptor showed similar binding activity to the control drug (Fc-TPP # 2) and ranibizumab monomer IDB0062. This means that the TPP # 2 peptide fused at the C-terminus of bevacizumab also binds to the neurophyllin 1 receptor as IDB0062.
- IDB0072 also improved eye permeability compared to the control drug bevacizumab. At 1 and 2 hours IDB0072 can be found to increase the intraocular distribution significantly about 1.5 times compared to bevacizumab (Fig. 9B). In addition, despite having a large molecular weight and structure, IDB0072 was found to be distributed to the cornea through the conjunctiva compared to bevacizumab. As a result, even in antibodies having a large molecular weight and complex structure, tissue permeability can be significantly increased by connecting tissue-penetrating peptides to the Fc C-terminus. These results demonstrate the possibility of improving eye tissue permeability by linking tissue-permeable peptides to various Fc fusion proteins, including antibody proteins.
- a corneal neovascular model was constructed and divided into preventive and resistant models to compare the effects of angiogenesis inhibition and angiogenesis reduction with Rani bizumab.
- a corneal neovascular model induced by alkaline burns was constructed as follows. Salose filter paper was cut into 2 ⁇ diameter and prepared and soaked in 1M NaOH solution. The mice were 6-week-old female C57BL / 6. After anesthesia (Zolet il 40mg / kg + Rompun 5mg / kg, IP), NaOH filter paper was placed on the left cornea for 30 seconds to induce alkaline burns, followed by layer washing with 40ml PBS. Corneal neovascular animal model was constructed.
- drugs were treated from the day of induction of alkali burn, and each drug was administered 4 times a day by 5 ⁇ 1 at a concentration of 5 mg / ml for 5 days.
- the eye was extracted and soaked in 4% paraformaldehyde solution for one hour and fixed.
- the cornea was separated using an anatomical microscope.
- the separated corneas were further fixed in 4% paraformaldehyde solution for 12 hours.
- the fixed cornea was washed with PBS and reacted with blocking buf fer (PBS, 0.3% BSA, 0.1% Tr i ton X100) at room temperature for 2 hours.
- the primary antibody (BD pharmingen) specific to vascular endothelial cell marker PECAM-KCD31) and the primary antibody (Mi 1 1 ipore) specific to NG-2 (per i cyte marker) were reacted overnight in the gut and secondary fluorescent antibody.
- Alexa Fluor 488, Alexa Fluor 594, Li fe Technologies reacted for 4 hours at room temperature to stain the tissue.
- the corneas were transferred to slides and mounted using an anatomical microscope with incisions in four directions toward the center of the cornea.
- the corneal slides with completed mounting were checked for vascular and peri cyte patterns using a fluorescence microscope / confocal microscope. As shown in FIG.
- IDB0062 inhibited neovascularization by about 25-30% more than ranibizumab, resulting in a significant preventive effect of at least 50% compared to vehicle. And IDB0061 also showed the same efficacy as ranibizumab, indicating that the decrease in VEGF binding ability due to the change of ranibizumab structure does not affect the actual neovascular inhibition effect, but is enhanced by -TPP fused at the C-terminus. Confirmed that it is possible.
- IDB0062 showed more than 30% of the neovascular reduction effect compared to vehicle and increased more than 2 times the efficacy of ranibizumab which showed no significant effect (FIG. 7B). These results were compared with ant i-VEGF aptamer. It is similar to the results of the literature in combination with ant i-PDGF ant ibody preparation (Jo et al., 2006. Am. J. Pathol.). Therefore, IDB0062 of the present invention was predicted to have improved vision in about 70% of ranibizumab-administered patients who only maintained without improvement of vision. In addition, as shown in FIG.
- the IDB0062 treatment group was found to have a significantly reduced per i cyte coverage of nearly 40% compared to the vehi cle treatment group, which is intended to treat patients who exhibit resistance to existing drugs.
- the results are similar to those of PDGF inhibitors being developed as concomitant medications. IDB0062 is therefore expected to be used as a monotherapy for resistance therapy, which is known to occur in 45% of patients receiving ant i-VEGF inhibitors.
- Choroidal neovascular i zat ion (CNV), a model of age-related macular degenerat ion, is laser-induced choroidal Induced by neovascularization method and confirmed the therapeutic effect of ranibizumab improved IDB0062.
- rats were randomly divided into 5 groups of 10 animals per group as shown in Table 6.
- the drug was administered intraocularly using a hamilton syringeChamilton, USA).
- the same amount of vehicle was administered to the CNV group and 100 ⁇ g / eye of ranibizumab as a control drug.
- Subjects who had surgical injuries due to drug administration or subjects who had turbidity of the lens were excluded from the experiment.
- the ranibizumab 100 ug administered group and the IDB0062 50 ug administered group suppressed angiogenesis by about 31% compared to the vehicle group, and the angiogenesis was suppressed about 36% in the IDB0062 100 ug administered group. It was confirmed. As a result, it was confirmed that IDB0062 shows the same neovascular inhibitory effect as ranibizumab at half the dose of ranibizumab.
- Oxygen-induced retinopathy as a model of ret inopathy of prematurity
- mice used mice born from the mating of 7-8 weeks old C57BL / 6 purchased from Coretech. At 7 days of age, mice (postnatal day 7, P7) were placed in an oxygen chamber and adjusted to maintain 75% (hyperoxia) oxygen concentration in the chamber (P7-P11) for 5 days. Illumination in the laboratory was allowed to equalize at 12 hour intervals, the temperature was maintained at 24 ⁇ 2 ° C, and the feed and drinking water were fed freely. After 5 days, ret inal angiogenesis was induced by exposure in the indoor atmosphere (normoxia) outside the chamber for 5 days ( ⁇ 12_ ⁇ 7). Immediately after exposure to normoxia on day P12 for drug administration randomly divided into 5 groups of 10 animals per group as described in Table 7.
- the drug is hami lton syringe (Hami l ton, USA) Intraocular administration was performed for each group. The same dose of vehicle was administered to the 0IR group and 10 ug / eye of ranibizumab as a control drug. Subjects who had surgical injuries due to the drug administration or subjects who had turbidity of the lens were excluded from the experiment.
- Oxygen-Induced Retinopathy (OIR) ⁇ Experiment 3 ⁇
- IDB0062 15 U g administration group has a dense and stable structure of blood vessels, and shows no visible leakage. see.
- streptavidin TRITC was diluted 1: 500 and reacted for 4 hours at 37 ° C, followed by 30 minutes of washing with PBS (BX51, Olympus, Japan). Observed under.
- neovascularization When P7 mice are in hyperoxia for 5 days and then transferred to normoxia, they are placed in relative ischemia. At this time, the retina grows in excess of neovascularization.
- the characteristics of neovascularization formed at this time are the fact that blood vessels grow from the retina to the vitreous, the avascular area, and form a special structure called tufts, an irregular vascular form that is easy to leak. to be.
- the formation of these tufts can be used as a quantitative measure of neovescularization.
- the salt-colored blood vessels in the vehicle group is characterized by the normal retinal vessels of the blood vessels are clearly separated and relatively thin and stable structure, the irregular and thick blood vessels together form the tufts Many new blood vessels were observed.
- Ranibizumab is identified as one band at about 48 kDa in non-reducing SDS-PAGE analysis because the heavy and light chains are linked by disulfide bonds.
- SDS-PAGE analysis shows two bands (heavy and light) at 28kDa.
- the amount of drug present in the retina was about 3.6 times higher than that of ranibizumab. This results in the fact that TPP specifically binds to NRP1 expressing tissues, showing the possibility of targeting the retinal tissue, which is a disease region, for various diseases due to neovascularization of the retina.
- Intraocular injections may increase the chance of eliminating VEGF by binding to the tissue rather than by spreading within the vi treous, resulting in the same effect at lower doses.
- IDB0062's superior efficacy is the result of efficacy evaluation using hyperretinal model.
- a pharmaceutical composition comprising a fusion protein fused with a tissue-penetrating peptide of the present invention and an anti-vascular endothelial growth factor (ant i-VEGF) agent as an active ingredient is compared with conventional anti-vascular endothelial growth factor preparations. It not only inhibits various growth factors related to neovascularization other than endothelial growth factor but also reduces the peri cyte coverage. It is thought to be able to treat augmented and drug resistant patients. In addition, since the drug delivery to the choroidal tissue is improved during intraocular injection, it is highly applicable to the industry because it is highly likely to be developed as an eye drop by reducing the dose or increasing the administration cycle and improving the eye permeability.
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Medicinal Chemistry (AREA)
- Bioinformatics & Cheminformatics (AREA)
- General Health & Medical Sciences (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Public Health (AREA)
- Epidemiology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Immunology (AREA)
- Organic Chemistry (AREA)
- Zoology (AREA)
- Genetics & Genomics (AREA)
- Gastroenterology & Hepatology (AREA)
- Microbiology (AREA)
- Mycology (AREA)
- Molecular Biology (AREA)
- Vascular Medicine (AREA)
- Biomedical Technology (AREA)
- Wood Science & Technology (AREA)
- Biotechnology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Biochemistry (AREA)
- General Engineering & Computer Science (AREA)
- Cell Biology (AREA)
- Oncology (AREA)
- Biophysics (AREA)
- Ophthalmology & Optometry (AREA)
- Physics & Mathematics (AREA)
- Toxicology (AREA)
- Plant Pathology (AREA)
- Endocrinology (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Medicinal Preparation (AREA)
Abstract
Description




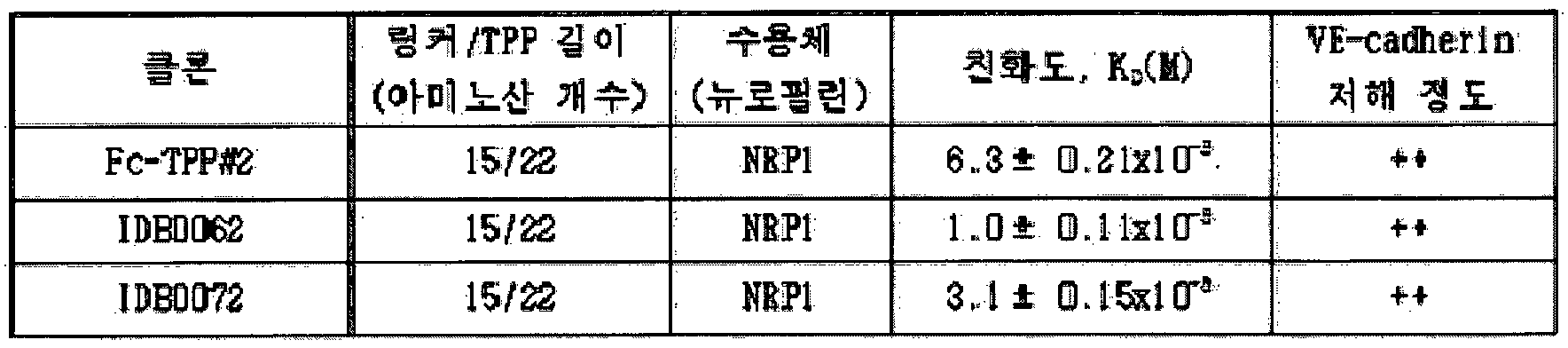


Claims
Priority Applications (11)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| AU2016241499A AU2016241499B2 (en) | 2015-03-31 | 2016-03-30 | Pharmaceutical composition for preventing and treating eye diseases, containing, as active ingredient, fusion protein in which tissue-penetrating peptide and anti-vascular endothelial growth factor preparation are fused |
| JP2017550872A JP6836512B2 (ja) | 2015-03-31 | 2016-03-30 | 組織透過性ペプチドと抗血管内皮細胞成長因子製剤が融合された融合タンパク質を有効成分として含む眼疾患の予防及び治療用薬学的組成物 |
| CA2981145A CA2981145C (en) | 2015-03-31 | 2016-03-30 | Pharmaceutical composition for preventing and treating eye diseases, containing, as active ingredient, fusion protein in which tissue-penetrating peptide and anti-vascular endothelial growth factor preparation are fused |
| MX2017012506A MX383971B (es) | 2015-03-31 | 2016-03-30 | Composición farmacéutica para la prevención y tratamiento de enfermedades del ojo que contiene, como ingrediente activo, una proteína de fusión en la cual el péptido que penetra el tejido y la preparación del factor de crecimiento endotelial antivascular se fusionan. |
| CN201680031204.9A CN108348572B (zh) | 2015-03-31 | 2016-03-30 | 含有融合组织穿透肽和抗-vegf制剂的融合蛋白的预防和治疗眼疾的药物组合物 |
| EP21195152.0A EP3973977A1 (en) | 2015-03-31 | 2016-03-30 | Pharmaceutical composition for preventing and treating eye diseases, containing, as active ingredient, fusion protein in which tissue-penetrating peptide and anti-vascular endothelial growth factor preparation are fused |
| EP16773433.4A EP3278810A4 (en) | 2015-03-31 | 2016-03-30 | Pharmaceutical composition for preventing and treating eye diseases, containing, as active ingredient, fusion protein in which tissue-penetrating peptide and anti-vascular endothelial growth factor preparation are fused |
| BR112017020948-9A BR112017020948A2 (pt) | 2015-03-31 | 2016-03-30 | composição farmacêutica, método para preparar um agente, método para tratar uma doença ocular e uso de uma proteína de fusão |
| RU2017137735A RU2712623C2 (ru) | 2015-03-31 | 2016-03-30 | Фармацевтическая композиция для предотвращения и лечения заболеваний глаз, содержащая в качестве активного ингредиента белок слияния, в котором слиты проникающий в ткань пептид и препарат против фактора роста эндотелия сосудов |
| US15/720,016 US11052130B2 (en) | 2015-03-31 | 2017-09-29 | Method for treating eye diseases with a fusion protein of a tissue-penetrating peptide and anti-vascular endothelial growth factor |
| US17/204,311 US11738064B2 (en) | 2015-03-31 | 2021-03-17 | Pharmaceutical composition for preventing and treating eye diseases, containing as active ingredient, fusion protein in which tissue-penetrating peptide and anti-vascular endothelial growth factor preparation are fused |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| KR10-2015-0045684 | 2015-03-31 | ||
| KR20150045684 | 2015-03-31 |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| US15/720,016 Continuation US11052130B2 (en) | 2015-03-31 | 2017-09-29 | Method for treating eye diseases with a fusion protein of a tissue-penetrating peptide and anti-vascular endothelial growth factor |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2016159652A1 true WO2016159652A1 (ko) | 2016-10-06 |
Family
ID=57004803
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/KR2016/003254 Ceased WO2016159652A1 (ko) | 2015-03-31 | 2016-03-30 | 조직투과성 펩타이드와 항-혈관 내피세포 성장인자 제제가 융합된 융합단백질을 유효성분으로 포함하는 안질환 예방 및 치료용 약학적 조성물 |
Country Status (11)
| Country | Link |
|---|---|
| US (2) | US11052130B2 (ko) |
| EP (2) | EP3973977A1 (ko) |
| JP (1) | JP6836512B2 (ko) |
| KR (1) | KR101846306B1 (ko) |
| CN (1) | CN108348572B (ko) |
| AU (1) | AU2016241499B2 (ko) |
| BR (1) | BR112017020948A2 (ko) |
| CA (1) | CA2981145C (ko) |
| MX (1) | MX383971B (ko) |
| RU (1) | RU2712623C2 (ko) |
| WO (1) | WO2016159652A1 (ko) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2017130194A1 (en) * | 2016-01-25 | 2017-08-03 | Rappaport Family Institute For Research In The Medical Sciences | Semaphorin 3c variants, compositions comprising said variants and methods of use thereof in treating eye diseases |
| US10561707B2 (en) | 2013-09-08 | 2020-02-18 | Technion Research And Development Foundation Ltd. | Semaphorin 3C variants, compositions comprising said variants and methods of use thereof in treating eye diseases |
| CN112368377A (zh) * | 2018-06-14 | 2021-02-12 | 爱维斯健有限公司 | 用于治疗重症联合免疫缺陷的包含细胞穿透肽和腺苷脱氨酶的融合蛋白的药物组合物 |
| US11738064B2 (en) | 2015-03-31 | 2023-08-29 | Ildong Pharm Co., Ltd. | Pharmaceutical composition for preventing and treating eye diseases, containing as active ingredient, fusion protein in which tissue-penetrating peptide and anti-vascular endothelial growth factor preparation are fused |
Families Citing this family (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US9840553B2 (en) | 2014-06-28 | 2017-12-12 | Kodiak Sciences Inc. | Dual PDGF/VEGF antagonists |
| KR20250057128A (ko) | 2015-12-30 | 2025-04-28 | 코디악 사이언시스 인코포레이티드 | 항체 및 이의 접합체 |
| EP3758737A4 (en) | 2018-03-02 | 2022-10-12 | Kodiak Sciences Inc. | IL-6 ANTIBODIES AND FUSION CONSTRUCTS AND CONJUGATES THEREOF |
| DK4364724T3 (da) * | 2018-05-10 | 2025-12-22 | Regeneron Pharma | Formuleringer med høj koncentration af VEGF-receptorfusionsprotein |
| EP3886946A1 (en) | 2019-06-05 | 2021-10-06 | Regeneron Pharmaceuticals, Inc. | Devices and methods for precision dose delivery |
| WO2021046538A1 (en) * | 2019-09-06 | 2021-03-11 | United States Government As Represented By The Department Of Veterans Affairs | Methods and systems for self-administered measurement of critical flicker frequency (cff) |
| CA3157509A1 (en) | 2019-10-10 | 2021-04-15 | Kodiak Sciences Inc. | Methods of treating an eye disorder |
| CN113384715B (zh) * | 2021-06-03 | 2023-08-04 | 上海市第十人民医院 | 一种含有细胞穿透肽融合蛋白的抗vegf药物及其制备方法和应用 |
| EP4368203A4 (en) * | 2021-07-05 | 2025-09-24 | Wuhan Neurophth Biotechnology Ltd Company | CONSTRUCTION AND USE OF ANTI-VEGF ANTIBODIES IN AN IN VIVO EXPRESSION SYSTEM |
| USD1120314S1 (en) | 2022-11-30 | 2026-03-24 | Regeneron Pharmaceuticals, Inc. | Dose delivery device |
| WO2024144099A1 (ko) * | 2022-12-27 | 2024-07-04 | 주식회사 넥스세라 | 안구 질환 치료를 위한 효율적인 약물 전달 플랫폼 |
| CN116785440A (zh) * | 2023-08-09 | 2023-09-22 | 天津医科大学总医院 | Sema4D抑制剂在制备预防和/或治疗脉络膜新生血管药物中的应用 |
| CN118108862B (zh) * | 2024-04-29 | 2024-08-09 | 上海鼎新基因科技有限公司 | 抗血管新生的融合蛋白及其应用 |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20070071756A1 (en) * | 2005-09-26 | 2007-03-29 | Peyman Gholam A | Delivery of an agent to ameliorate inflammation |
| WO2009136007A1 (en) * | 2008-05-09 | 2009-11-12 | Burnham Institute For Medical Research | Peptide homing to brain tumors |
| US8772449B2 (en) * | 2009-05-20 | 2014-07-08 | Toray Industries, Inc. | Cell-penetrating peptides |
| KR20140138539A (ko) * | 2013-05-23 | 2014-12-04 | 아주대학교산학협력단 | 뉴로필린에 특이적인 종양 침투성 펩타이드 및 이 펩타이드가 융합된 융합 단백질 |
Family Cites Families (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1711196A4 (en) | 2003-12-05 | 2011-09-14 | Bristol Myers Squibb Co | INHIBITORS OF TYPE-2 VASCULAR ENDOTHELIAL GROWTH FACTOR RECEPTORS |
| JP2010528647A (ja) | 2007-06-06 | 2010-08-26 | ドマンティス リミテッド | ポリペプチド、抗体可変ドメインおよびアンタゴニスト |
| GB0724331D0 (en) | 2007-12-13 | 2008-01-23 | Domantis Ltd | Compositions for pulmonary delivery |
| CN101951925A (zh) * | 2008-02-20 | 2011-01-19 | 建新公司 | 血管发生抑制 |
| US8821870B2 (en) | 2008-07-18 | 2014-09-02 | Allergan, Inc. | Method for treating atrophic age related macular degeneration |
| WO2011005540A1 (en) * | 2009-06-22 | 2011-01-13 | Burnham Institute For Medical Research | Methods and compositions using peptides and proteins with c-terminal elements |
| CN102002104A (zh) | 2009-08-28 | 2011-04-06 | 江苏先声药物研究有限公司 | 一种抗vegf的单克隆抗体及含有该抗体的药物组合物 |
| MX349622B (es) | 2010-09-08 | 2017-08-07 | Halozyme Inc | Metodos para evaluar e identificar o evolucionar proteinas terapeuticas condicionalmente activas. |
| ES2753135T3 (es) | 2012-05-07 | 2020-04-07 | Allergan Inc | Método de tratamiento de DMAE en pacientes resistentes a terapia anti-VEGF |
| JO3405B1 (ar) * | 2013-01-09 | 2019-10-20 | Regeneron Pharma | الأجسام المضادة لمضاد مستقبل عامل النمو المشتق من الصفائح الدموية - بيتا واستخداماتها |
| JP6138973B2 (ja) | 2013-09-19 | 2017-05-31 | フィリップス ライティング ホールディング ビー ヴィ | 連続出力調整範囲を有するコンパクト電力変換装置 |
| KR102157618B1 (ko) | 2013-10-21 | 2020-09-18 | 주식회사 아이티엘 | 서빙셀별로 듀플렉스 방식을 달리하는 무선 통신 시스템에서 통신을 수행하는 장치 및 그 방법 |
| CN108348572B (zh) | 2015-03-31 | 2021-06-29 | 日东制药株式会社 | 含有融合组织穿透肽和抗-vegf制剂的融合蛋白的预防和治疗眼疾的药物组合物 |
-
2016
- 2016-03-30 CN CN201680031204.9A patent/CN108348572B/zh not_active Expired - Fee Related
- 2016-03-30 BR BR112017020948-9A patent/BR112017020948A2/pt not_active Application Discontinuation
- 2016-03-30 CA CA2981145A patent/CA2981145C/en active Active
- 2016-03-30 EP EP21195152.0A patent/EP3973977A1/en not_active Withdrawn
- 2016-03-30 KR KR1020160038327A patent/KR101846306B1/ko not_active Expired - Fee Related
- 2016-03-30 WO PCT/KR2016/003254 patent/WO2016159652A1/ko not_active Ceased
- 2016-03-30 AU AU2016241499A patent/AU2016241499B2/en not_active Ceased
- 2016-03-30 MX MX2017012506A patent/MX383971B/es unknown
- 2016-03-30 JP JP2017550872A patent/JP6836512B2/ja not_active Expired - Fee Related
- 2016-03-30 EP EP16773433.4A patent/EP3278810A4/en not_active Withdrawn
- 2016-03-30 RU RU2017137735A patent/RU2712623C2/ru active
-
2017
- 2017-09-29 US US15/720,016 patent/US11052130B2/en not_active Expired - Fee Related
-
2021
- 2021-03-17 US US17/204,311 patent/US11738064B2/en active Active
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US20070071756A1 (en) * | 2005-09-26 | 2007-03-29 | Peyman Gholam A | Delivery of an agent to ameliorate inflammation |
| WO2009136007A1 (en) * | 2008-05-09 | 2009-11-12 | Burnham Institute For Medical Research | Peptide homing to brain tumors |
| US8772449B2 (en) * | 2009-05-20 | 2014-07-08 | Toray Industries, Inc. | Cell-penetrating peptides |
| KR20140138539A (ko) * | 2013-05-23 | 2014-12-04 | 아주대학교산학협력단 | 뉴로필린에 특이적인 종양 침투성 펩타이드 및 이 펩타이드가 융합된 융합 단백질 |
Non-Patent Citations (1)
| Title |
|---|
| SHA ET AL.: "Tumor-penetrating Peptide Fused EGFR Single-domain Antibody enhances Cancer Drug Penetration into 3D Multicellular Spheroids and Facilitates Effective Gastric Cancer Therapy", JOURNAL OF CONTROLLED RELEASE, vol. 200, 30 December 2014 (2014-12-30), pages 188 - 200, XP029222013 * |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US10561707B2 (en) | 2013-09-08 | 2020-02-18 | Technion Research And Development Foundation Ltd. | Semaphorin 3C variants, compositions comprising said variants and methods of use thereof in treating eye diseases |
| US11738064B2 (en) | 2015-03-31 | 2023-08-29 | Ildong Pharm Co., Ltd. | Pharmaceutical composition for preventing and treating eye diseases, containing as active ingredient, fusion protein in which tissue-penetrating peptide and anti-vascular endothelial growth factor preparation are fused |
| WO2017130194A1 (en) * | 2016-01-25 | 2017-08-03 | Rappaport Family Institute For Research In The Medical Sciences | Semaphorin 3c variants, compositions comprising said variants and methods of use thereof in treating eye diseases |
| CN112368377A (zh) * | 2018-06-14 | 2021-02-12 | 爱维斯健有限公司 | 用于治疗重症联合免疫缺陷的包含细胞穿透肽和腺苷脱氨酶的融合蛋白的药物组合物 |
Also Published As
| Publication number | Publication date |
|---|---|
| AU2016241499B2 (en) | 2020-12-17 |
| CN108348572A (zh) | 2018-07-31 |
| US11738064B2 (en) | 2023-08-29 |
| MX2017012506A (es) | 2018-06-27 |
| RU2017137735A3 (ko) | 2019-09-11 |
| CA2981145A1 (en) | 2016-10-06 |
| RU2712623C2 (ru) | 2020-01-30 |
| JP2018515435A (ja) | 2018-06-14 |
| US20210220436A1 (en) | 2021-07-22 |
| US11052130B2 (en) | 2021-07-06 |
| AU2016241499A1 (en) | 2017-11-09 |
| BR112017020948A2 (pt) | 2018-07-10 |
| RU2017137735A (ru) | 2019-04-30 |
| CA2981145C (en) | 2021-05-11 |
| KR20160117329A (ko) | 2016-10-10 |
| MX383971B (es) | 2025-03-14 |
| KR101846306B1 (ko) | 2018-05-18 |
| EP3278810A1 (en) | 2018-02-07 |
| EP3973977A1 (en) | 2022-03-30 |
| CN108348572B (zh) | 2021-06-29 |
| US20180133288A1 (en) | 2018-05-17 |
| JP6836512B2 (ja) | 2021-03-03 |
| EP3278810A4 (en) | 2018-11-21 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2016159652A1 (ko) | 조직투과성 펩타이드와 항-혈관 내피세포 성장인자 제제가 융합된 융합단백질을 유효성분으로 포함하는 안질환 예방 및 치료용 약학적 조성물 | |
| US20210069293A1 (en) | Modified binding proteins inhibiting the vegf-a receptor interaction | |
| AU2009319204B2 (en) | Binding proteins inhibiting the VEGF-A receptor interaction | |
| TW201350128A (zh) | 在抗vegf療法難治之患者中治療amd之方法 | |
| WO2019229116A1 (en) | Intravitreal delivery of a decorin polypeptide for the treatment of choroidal neovascularisation | |
| JP2022527276A (ja) | 眼疾患を治療するための組成物及び方法 | |
| JP7830567B2 (ja) | ペプチドおよびその医学的使用 | |
| JP2024543158A (ja) | 多重特異性リガンド結合分子及びその用途 | |
| JP7560185B2 (ja) | 補体経路阻害剤及び血管新生阻害剤を含む融合タンパク質並びにその使用 | |
| HK1179876B (en) | Modified binding proteins inhibiting the vegf-a receptor interaction | |
| HK1179876A (en) | Modified binding proteins inhibiting the vegf-a receptor interaction |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 16773433 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2981145 Country of ref document: CA |
|
| ENP | Entry into the national phase |
Ref document number: 2017550872 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: MX/A/2017/012506 Country of ref document: MX |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| REG | Reference to national code |
Ref country code: BR Ref legal event code: B01A Ref document number: 112017020948 Country of ref document: BR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2017137735 Country of ref document: RU |
|
| ENP | Entry into the national phase |
Ref document number: 2016241499 Country of ref document: AU Date of ref document: 20160330 Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 112017020948 Country of ref document: BR Kind code of ref document: A2 Effective date: 20170929 |
|
| WWW | Wipo information: withdrawn in national office |
Ref document number: 2016773433 Country of ref document: EP |