WO2019043778A1 - 硬化性組成物、硬化性ペースト材、硬化性シート材、硬化性型取り材、硬化方法および硬化物 - Google Patents
硬化性組成物、硬化性ペースト材、硬化性シート材、硬化性型取り材、硬化方法および硬化物 Download PDFInfo
- Publication number
- WO2019043778A1 WO2019043778A1 PCT/JP2017/030905 JP2017030905W WO2019043778A1 WO 2019043778 A1 WO2019043778 A1 WO 2019043778A1 JP 2017030905 W JP2017030905 W JP 2017030905W WO 2019043778 A1 WO2019043778 A1 WO 2019043778A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- curable
- curable composition
- compound
- group
- curing
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
- 0 C*(C)c1ccc(*)cc1 Chemical compound C*(C)c1ccc(*)cc1 0.000 description 1
Images





Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G59/00—Polycondensates containing more than one epoxy group per molecule; Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups
- C08G59/18—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing
- C08G59/20—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing characterised by the epoxy compounds used
- C08G59/22—Di-epoxy compounds
- C08G59/24—Di-epoxy compounds carbocyclic
- C08G59/245—Di-epoxy compounds carbocyclic aromatic
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G65/00—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule
- C08G65/02—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule from cyclic ethers by opening of the heterocyclic ring
- C08G65/04—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule from cyclic ethers by opening of the heterocyclic ring from cyclic ethers only
- C08G65/22—Cyclic ethers having at least one atom other than carbon and hydrogen outside the ring
- C08G65/223—Cyclic ethers having at least one atom other than carbon and hydrogen outside the ring containing halogens
- C08G65/226—Cyclic ethers having at least one atom other than carbon and hydrogen outside the ring containing halogens containing fluorine
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G59/00—Polycondensates containing more than one epoxy group per molecule; Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups
- C08G59/02—Polycondensates containing more than one epoxy group per molecule
- C08G59/022—Polycondensates containing more than one epoxy group per molecule characterised by the preparation process or apparatus used
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G59/00—Polycondensates containing more than one epoxy group per molecule; Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups
- C08G59/18—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing
- C08G59/20—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing characterised by the epoxy compounds used
- C08G59/22—Di-epoxy compounds
- C08G59/226—Mixtures of di-epoxy compounds
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G59/00—Polycondensates containing more than one epoxy group per molecule; Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups
- C08G59/18—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing
- C08G59/68—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing characterised by the catalysts used
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G59/00—Polycondensates containing more than one epoxy group per molecule; Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups
- C08G59/18—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing
- C08G59/68—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing characterised by the catalysts used
- C08G59/687—Macromolecules obtained by polymerising compounds containing more than one epoxy group per molecule using curing agents or catalysts which react with the epoxy groups ; e.g. general methods of curing characterised by the catalysts used containing sulfur
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G65/00—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule
- C08G65/02—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule from cyclic ethers by opening of the heterocyclic ring
- C08G65/04—Macromolecular compounds obtained by reactions forming an ether link in the main chain of the macromolecule from cyclic ethers by opening of the heterocyclic ring from cyclic ethers only
- C08G65/06—Cyclic ethers having no atoms other than carbon and hydrogen outside the ring
- C08G65/16—Cyclic ethers having four or more ring atoms
- C08G65/18—Oxetanes
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K5/00—Use of organic ingredients
- C08K5/36—Sulfur-, selenium-, or tellurium-containing compounds
- C08K5/37—Thiols
- C08K5/375—Thiols containing six-membered aromatic rings
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L63/00—Compositions of epoxy resins; Compositions of derivatives of epoxy resins
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K3/00—Use of inorganic substances as compounding ingredients
- C08K3/34—Silicon-containing compounds
- C08K3/36—Silica
Definitions
- the present invention relates to a curable composition, a curable paste material, a curable sheet material, a curable mold material, a curing method and a cured product.
- Gaps occur between parts due to part-to-part tolerances or structural assembly tolerances. It is practiced to insert shims into the generated gaps. The following method is taken as a method of producing a shim that reproduces a gap. (1) Gap measurement by filler gauge. (2) Application of a contact or non-contact gap measuring instrument (Gap Master etc.). (3) Mold removal by epoxy resin. Among the above-mentioned methods, when a complicated 3D shape molding or a highly accurate shim shape is required, the method (3) is applied from the viewpoint of the gap measurement accuracy.
- the allowable temperature of the seal bonding surface on which the sealing material is applied is, for example, less than 60 ° C, and the allowable temperature of the primer coated surface on which the primer is applied is, for example, less than 93 ° C. Therefore, no dramatic effect can be expected in methods for promoting resin curing such as curing acceleration by heating, and sufficient time reduction is a severe situation.
- Patent Document 1 includes an aromatic epoxy compound, an alicyclic epoxy compound or an aliphatic epoxy compound, and a polyfunctional aliphatic oxetane compound as a cationically polymerizable organic substance mixture, and the aromatic epoxy compound is a main component. Certain compositions have been proposed. Patent Document 1 also describes that this composition contains a cationic polymerization initiator. The cationic polymerization initiator is used in a proportion of 7 to 10 parts by mass with respect to 100 parts by mass of the cationically polymerizable organic substance mixture. Patent Document 1 does not describe using the composition for producing a shim. The composition described in Patent Document 1 needs to be heated at 130 to 180 ° C. when it is cured, and deviates from the above-mentioned allowable temperature.
- the present invention is a curable composition capable of securing a sufficiently long pot life, and which can be rapidly cured even at a temperature of 65 ° C. or less after the pot life has elapsed, a curable paste material using the same, and a curable sheet material It is an object of the present invention to provide a curable mold material, a curing method and a cured product.
- the present invention has the following aspects.
- a cationically polymerizable compound, a thermal polymerization initiator, and a storage stabilizer The cationically polymerizable compound comprises at least two selected from the group consisting of glycidyl ether compounds, alicyclic epoxy compounds, and oxetane compounds,
- the content of the thermal polymerization initiator is 0.3 to 3 parts by mass with respect to 100 parts by mass of the cationically polymerizable compound
- Curable composition which can be chain-hardened by the thermal energy generated by the polymerization reaction of the said cationically polymerizable compound.
- the curable composition of [1] which is chain curable at a temperature of 65 ° C. or less.
- the glycidyl ether compound is at least one selected from the group consisting of a bisphenol A type diglycidyl ether compound and a bisfer F type diglycidyl ether compound,
- the alicyclic epoxy compound is 3,4-epoxycyclohexylmethyl-3,4-epoxycyclohexane carboxylate,
- the oxetane compound is at least one member selected from the group consisting of 4,4′-bis [(3-ethyl-3-oxetanyl) methoxymethyl] biphenyl and 3-ethyl-3-hydroxymethyl oxetane [1]
- R 1 represents a hydrogen atom, a methyl group, an acetyl group or a methoxycarbonyl group
- R 2 and R 3 each independently represent a hydrogen atom, a halogen atom or an alkyl group having 1 to 4 carbon atoms
- R 4 Represents a hydrogen atom, a halogen atom, a nitro group, a methyl group or a methoxy group
- R 5 represents an alkyl group having 1 to 4 carbon atoms
- X represents SbF 6 , PF 6 , AsF 6 or BF 4 .
- R 6 represents an acetyl group or a methoxycarbonyl group
- A represents CH 3 SO 4 .
- a curable paste material comprising the curable composition of any one of [9] [1] to [8].
- a curable sheet material comprising the curable composition according to any one of [1] to [8].
- a curable mold material comprising the curable composition of any one of [1] to [8].
- the curable composition of any one of [12] [1] to [8], the curable paste material of [9], the curable sheet material of [10], or the curable mold material of [11] A curing method in which curing is performed at a temperature not higher than ° C. [13] A curable composition according to any one of [1] to [8], a curable paste material for [9], a curable sheet material for [10], or a curable mold material for [11] Hardened. [14] The cured product of [10] having a hardness of 60 Hs or more.
- the present invention it is possible to secure a sufficient pot life of a sufficient length, and a curable composition which can be rapidly cured even at a temperature of 65 ° C. or less after the pot life has elapsed, a curable paste material using the same, and curability
- a sheet material, a curable mold material, a curing method and a cured product can be provided.
- the curable composition of the present invention comprises a cationically polymerizable compound, a thermal polymerization initiator, and a storage stabilizer.
- the curable composition of the present invention can further contain a filler.
- the curable composition of the present invention may further contain other components other than the cationically polymerizable compound, the thermal polymerization initiator, the storage stabilizer, and the filler, as needed, as long as the effects of the present invention are not impaired. .
- the cationically polymerizable compound contains at least two selected from the group consisting of glycidyl ether compounds, alicyclic epoxy compounds, and oxetane compounds.
- the cationically polymerizable compound may further contain another cationically polymerizable compound other than a glycidyl ether compound, an alicyclic epoxy compound and an oxetane compound.
- the total content of the glycidyl ether compound, the alicyclic epoxy compound and the oxetane compound in the cationically polymerizable compound is preferably 50 to 100% by mass, more preferably 80 to 100% by mass, with respect to the total mass of the cationically polymerizable compound. preferable.
- “-” indicating a numerical range means that numerical values described before and after that are included as the lower limit value and the upper limit value.
- the glycidyl ether compound has one or more glycidyl ether groups.
- glycidyl ether compounds include bisphenol A type diglycidyl ether compounds, bisferr F type diglycidyl ether compounds, hydrogenated bisphenol type glycidyl ether compounds, other aromatic glycidyl ether compounds, aliphatic glycidyl ether compounds, and the like.
- One of these glycidyl ether compounds may be used alone, or two or more thereof may be used in combination.
- the glycidyl ether compound is at least selected from the group consisting of bisphenol A type diglycidyl ether compounds and bisfer F type diglycidyl ether compounds from the viewpoints of availability, heat resistance, rigidity, strength characteristics, versatility, low cost, etc. One is particularly preferred.
- the alicyclic epoxy compound has one or more alicyclic epoxy groups.
- An alicyclic epoxy group is a state in which one oxygen atom is bonded to two carbon atoms (usually, carbon atoms adjacent to each other) of cyclically bonded carbon atoms forming an aliphatic ring of an alicyclic compound.
- Epoxy group of Examples of alicyclic epoxy compounds include dicyclopentadiene dioxide, limonene dioxide, di (3,4-epoxycyclohexyl) adipate, (3,4-epoxycyclohexyl) methyl-3,4-epoxycyclohexanecarboxylate, 3, 4-epoxy-6-methylcyclohexylmethyl-3,4-epoxy-6-methylcyclohexanecarboxylate, ethylene-1,2-di (3,4-epoxycyclohexanecarboxylic acid) ester and the like.
- One of these alicyclic epoxy compounds may be used alone, or two or more thereof may be used in combination.
- alicyclic epoxy compound 3,4-epoxy-6-methylcyclohexylmethyl-3,4-epoxy from the viewpoint of high reactivity, heat resistance, rigidity, strength characteristics, versatility, availability, low cost, etc.
- -6-Methylcyclohexane carboxylate is particularly preferred.
- the oxetane compound has one or more oxetane groups.
- the oxetane compound 3-ethyl-3-hydroxymethyl oxetane (hereinafter referred to as "EHO"), bisphenol A type oxetane compound, bisphenol oxetane compound, bisphenol S type oxetane compound, xylylene type oxetane compound, phenol novolac type oxetane Compound, cresol novolac type oxetane compound, alkylphenol novolac type oxetane compound, biphenol type oxetane compound, bixylenol type oxetane compound, naphthalene type oxetane compound, dicyclopentadiene type oxetane compound, phenol and aromatic aldehyde having a phenolic hydroxyl group
- Examples include oxetanides
- the oxetane compound is selected from the group consisting of 4,4′-bis [(3-ethyl-3-oxetanyl) methoxymethyl] biphenyl (hereinafter also referred to as “OXBP”) and EHO from the viewpoint of high reactivity. At least one is preferred. OXBP is preferred in that the heat resistance of the cured product is more excellent. EHO is preferred in that it is inexpensive.
- OXBP 4,4′-bis [(3-ethyl-3-oxetanyl) methoxymethyl] biphenyl
- EHO is preferred in that it is inexpensive.
- an epoxidized olefin As another cationically polymerizable compound, an epoxidized olefin, a vinyl ether compound, etc. are mentioned, for example.
- the epoxidized olefin include epoxidized linseed oil, epoxidized castor oil, epoxidized soybean oil and the like.
- vinyl ether compounds ethylene glycol divinyl ether, diethylene glycol divinyl ether, triethylene glycol divinyl ether, propylene glycol divinyl ether, dipropylene glycol divinyl ether, butanediol divinyl ether, hexanediol divinyl ether, cyclohexane dimethanol divinyl ether, trimethylolpropane Di or trivinyl ether compounds such as trivinyl ether; ethyl vinyl ether, n-butyl vinyl ether, isobutyl vinyl ether, octadecyl vinyl ether, cyclohexyl vinyl ether, hydroxybutyl vinyl ether, 2-ethylhexyl vinyl ether, cyclohexane dimethanol monovinyl ether, n-propyl Vinyl ether, isopropyl vinyl ether, isopropenyl ether -o- propylene carbonate, do
- the cationically polymerizable compound comprises at least one glycidyl ether compound, at least one alicyclic epoxy compound, and at least one oxetane compound.
- the glycidyl ether compound has relatively low polymerization reactivity, when the cationic polymerizable compound contains the glycidyl ether compound, it is possible to suppress the curing speed from becoming too fast.
- glycidyl ether compounds are relatively inexpensive, costs can be reduced.
- the alicyclic epoxy compound has relatively high polymerization reactivity
- the calorific value per unit time increases, and chain curing tends to proceed.
- the oxetane compound has high cationic polymerization reactivity and increases the calorific value per unit time similarly to the alicyclic epoxy, the cationic polymerizable compound containing the oxetane compound can help to facilitate chain curing.
- the content of the glycidyl ether compound is preferably 10 to 80% by mass, more preferably 20 to 60% by mass, and still more preferably 30 to 50% by mass, with respect to the total mass of the cationically polymerizable compound. 35 to 45% by weight is particularly preferred. If the content of the glycidyl ether compound is equal to or more than the above lower limit value, more excellent initial reaction stability, an effect of suppressing an increase in curing rate, and physical properties can be obtained. It is also cheaper. If the content of the glycidyl ether compound is equal to or less than the above upper limit value, more excellent chain curing reactivity can be obtained.
- the content of the alicyclic epoxy compound is preferably 10 to 80% by mass, more preferably 20 to 60% by mass, still more preferably 25 to 45% by mass, with respect to the total mass of the cationically polymerizable compound. % Is particularly preferred. If the content of the alicyclic epoxy compound is not less than the above lower limit value, more excellent chain curing reactivity can be obtained. If the content of the alicyclic epoxy compound is equal to or less than the above upper limit value, more excellent initial reaction stability and an effect of suppressing an increase in curing rate can be obtained.
- the content of the oxetane compound is preferably 5 to 70% by mass, more preferably 10 to 50% by mass, still more preferably 15 to 35% by mass, particularly preferably 20 to 30% by mass, based on the total mass of the cationically polymerizable compound. preferable. If the content of the oxetane compound is equal to or more than the above lower limit value, more excellent chain curing reactivity can be obtained. If the content of the oxetane compound is less than or equal to the above upper limit value, more excellent initial reaction stability and an effect of suppressing the increase in curing rate can be obtained.
- the cationically polymerizable compound is selected from the group consisting of bisphenol A type diglycidyl ether compounds and bisphor F type diglycidyl ether compounds and at least one glycidyl ether compound And 3,4-epoxy-6-methylcyclohexylmethyl-3,4-epoxy-6-methylcyclohexanecarboxylate and at least one oxetane compound selected from the group consisting of OXBP and EHO.
- the preferable range of the content of each of the glycidyl ether compound and the oxetane compound is the same as that described above.
- the preferred range of the content of 3,4-epoxy-6-methylcyclohexylmethyl-3,4-epoxy-6-methylcyclohexanecarboxylate is the same as the preferred range of the content of the alicyclic epoxy compound.
- the content of OXBP is preferably 5 to 50% by mass, more preferably 5 to 40% by mass, still more preferably 10 to 30% by mass, with respect to the total mass of the cationically polymerizable compound. And 15 to 25% by mass are particularly preferred. If the content of OXBP is equal to or more than the above lower limit value, more excellent chain curing reactivity, heat resistance and rigidity can be obtained. If the content of OXBP is less than or equal to the above upper limit value, a more excellent initial reaction stability and an effect of suppressing an increase in curing rate can be obtained. It is also cheaper.
- the content of EHO is preferably 5 to 40% by mass, more preferably 5 to 25% by mass, still more preferably 5 to 15% by mass, based on the total mass of the cationically polymerizable compound. And 5 to 10% by mass are particularly preferable.
- the content of EHO is equal to or more than the above lower limit, more excellent chain curing reactivity can be obtained. It is also cheaper. If the content of EHO is less than or equal to the above upper limit value, more excellent initial reaction stability, an effect of suppressing an increase in curing rate, and heat resistance can be obtained.
- the cationically polymerizable compound is a bisphenol A type diglycidyl ether compound, and 3,4-epoxy-6-methylcyclohexylmethyl-3,4-epoxy-6-methyl. It contains cyclohexane carboxylate, OXBP and EHO.
- the preferable range of the content of the bisphenol A type diglycidyl ether compound is the same as the preferable range of the content of the above glycidyl ether compound.
- the preferred ranges of the contents of 3,4-epoxy-6-methylcyclohexylmethyl-3,4-epoxy-6-methylcyclohexanecarboxylate, OXBP and EHO are the same as described above.
- the thermal polymerization initiator is a compound which is decomposed by heat to generate a Lewis acid or a protic acid.
- the polymerization reaction of the cationically polymerizable compound is initiated by the action of the Lewis acid or protic acid.
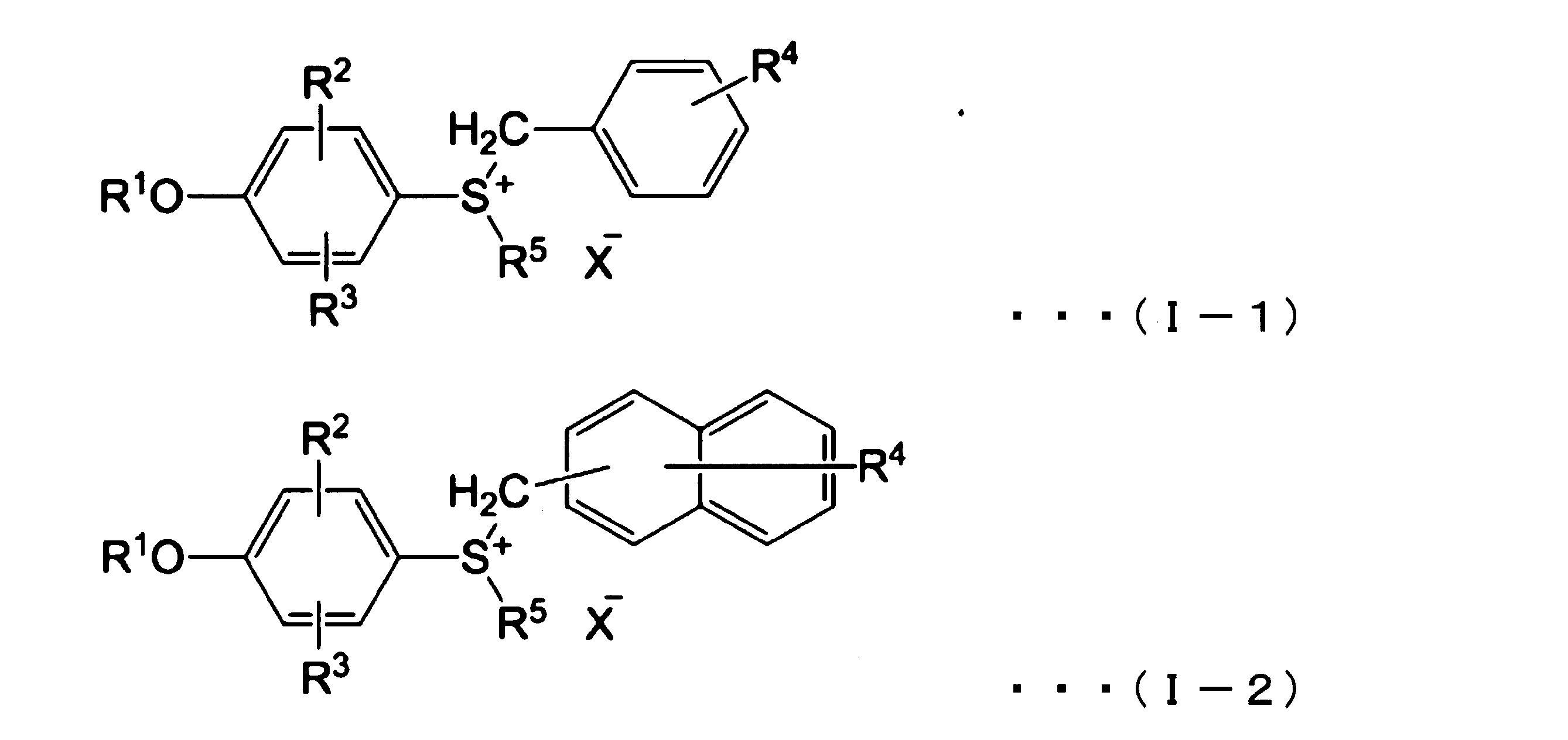
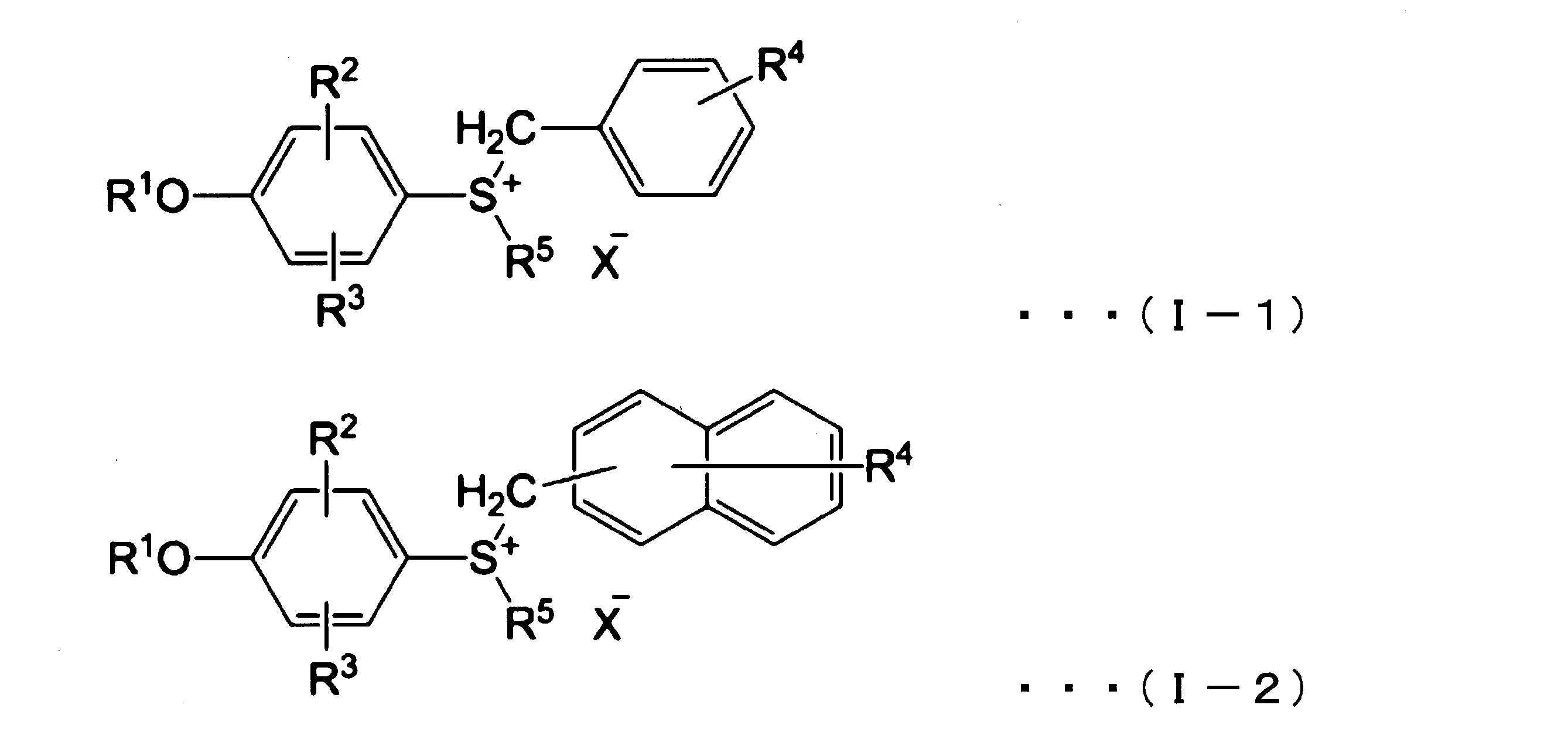
- a thermal polymerization initiator is preferably a compound capable of decomposing at a temperature of 65 ° C. or less to generate a Lewis acid or a protic acid. Examples of such a compound include a sulfonium salt represented by the following formula (I-1), a sulfonium salt represented by the following formula (I-2), and the like.
- One of these thermal polymerization initiators may be used alone, or two or more thereof may be used in combination.
- R 1 represents a hydrogen atom, a methyl group, an acetyl group (CH 3 CO) or a methoxycarbonyl group (CH 3 OCO)
- R 2 and R 3 each independently represent a hydrogen atom, a halogen atom or 1 carbon atom
- R 4 represents a hydrogen atom, a halogen atom, a nitro group, a methyl group or a methoxy group
- R 5 represents an alkyl group having 1 to 4 carbon atoms
- X represents SbF 6 , PF 6 , Show AsF 6 or BF 4 .
- the thermal polymerization initiator is preferably at least one selected from the group consisting of sulfonium salts represented by the above formulas (I-1) or (I-2) from the viewpoint of chain curing properties.
- sulfonium salt represented by the formula (I-1) or (I-2) R 1 is a hydrogen atom, an acetyl group or a methoxycarbonyl group, R 2 and R 3 are each a hydrogen atom, and R 4 Is a hydrogen atom, a halogen atom, a nitro group or a methyl group, and a sulfonium salt in which R 5 is a methyl group is preferred.
- R 1 is a methoxycarbonyl group
- R 2 and R 3 are each a hydrogen atom
- R 4 is a hydrogen atom
- R 5 is a methyl group
- X A sulfonium salt in which is SbF 6, that is, a sulfonium salt represented by the following formula (I-1-1) is preferable.
- the storage stabilizer is a compound capable of capturing the Lewis acid or protic acid generated by the decomposition of the thermal polymerization initiator.
- the curable composition contains a storage stabilizer, the polymerization of the cationically polymerizable compound is suppressed, and the pot life of the curable composition is extended.
- Examples of the storage stabilizer include sulfonium salts represented by the following formula (II), and the like. One of these storage stabilizers may be used alone, or two or more thereof may be used in combination.
- R 6 represents an acetyl group or a methoxycarbonyl group
- A represents CH 3 SO 4 .
- the storage stabilizer is preferably at least one selected from the group consisting of sulfonium salts represented by formula (II) from the viewpoint of storage stability properties.
- the filler is used to adjust the properties such as viscosity of the curable composition, to reduce the amount of heat generation during curing by reducing the proportion of the cationically polymerizable compound, to suppress curing shrinkage / improve dimensional stability, improve hardness, improve rigidity, etc. It is used for the purpose.
- the filler examples include glass, silica, ceramic, plastic, metal and the like. At least one selected from the group consisting of glass, silica and ceramic is preferable in terms of property stability, dispersibility, availability, variety of shape and performance, low cost and the like.
- the form of the filler examples include powder, fiber, and flakes, and the like, and easiness of mixing and preparation, easiness to conform to the shape (fillability in the gap portion, shape following property at the time of crushing, etc.) In terms of point, powdery form is preferable.
- the powdery filler may have a distribution in particle size. 200 micrometers or less are preferable, 50 micrometers or less are more preferable, 20 micrometers or less are more preferable, and, as for the average particle diameter of a powdery filler, 5 micrometers or less are especially preferable.
- the lower limit of the average particle size is not particularly limited, and may be, for example, less than 1 ⁇ m.
- the average particle size is measured by a particle size distribution measuring device or the like. In order to control the viscosity characteristics of the curable composition, a plurality of powdery fillers having different average particle sizes may be used in combination.
- powdery silica is preferable in that it is inexpensive and products of various particle sizes are commercially available and it is easy to select the particle size.
- Other components include, for example, a sensitizer, a curing accelerator, a pigment / dye, an antifoaming agent, a viscosity modifier and the like. These other components may be used alone or in combination of two or more.
- the content of the thermal polymerization initiator in the curable composition is 0.3 to 3 parts by mass, preferably 0.5 to 2 parts by mass, with respect to 100 parts by mass of the cationically polymerizable compound, and 0.75 to 1 .5 parts by weight are particularly preferred.
- the content of the thermal polymerization initiator is a solid content equivalent.
- the content of the storage stabilizer in the curable composition can be appropriately set in consideration of the desired pot life and curing time of the curable composition.
- the higher the content of the storage stabilizer the later the initiation of the polymerization reaction of the cationically polymerizable compound tends to be delayed, and the pot life and curing time tend to be longer.
- the content of the storage stabilizer in the curable composition is preferably 0.3 parts by mass or more with respect to 100 parts by mass of the thermal polymerization initiator.
- the content of the storage stabilizer is preferably 0.3 to 5 parts by mass, more preferably 1 to 4 parts by mass, with respect to 100 parts by mass of the thermal polymerization initiator, from the balance of pot life and curing time. Particularly preferred is 5 to 3.5 parts by mass.
- the content of the filler is preferably 50% by mass or more, more preferably 60% by mass or more, still more preferably 70% by mass or more, based on the total mass of the curable composition. Mass% or more is particularly preferable.
- the upper limit of the content of the filler which varies depending on the density of the filler, may be, for example, 80% by mass with respect to the total mass of the curable composition.
- the content of the other components in the curable composition may be, for example, 0 to 20% by mass based on the total mass of the curable composition.
- the curable composition of the present invention can be chain-cured by the thermal energy generated by the polymerization reaction (exothermic polymerization reaction) of the cationically polymerizable compound.
- "Chain curing” is a curing form that uses its own reaction heat energy as a driving force. When thermal energy (primary thermal energy) is applied to a part of the curable composition, an initial exothermic polymerization reaction occurs, and thermal energy (secondary thermal energy) is generated accordingly.
- the heat energy (secondary heat energy) generated by the first heat generation polymerization reaction (the second heat energy) continuously repeats the heat generation polymerization reaction and the accompanying generation of the second heat energy, and the curing of the curable composition proceeds in a chained manner, thereby curing The entire sex composition cures.
- the curable composition can be chain-cured can be determined by the following procedure.
- the curable composition is filled in a cylindrical mold having a length of 200 mm and a diameter of mm 10 mm to make a sample (for example, one having a stopper at one end of a resin tube / resin hose, a test tube, etc. is used as a casting mold ).
- a solder iron adjusted to an arbitrary temperature is brought into contact with the position of the upper end portion (a place where contact with the curable composition is possible) of this sample.
- the curable composition can be chain-cured, an exothermic polymerization reaction occurs after contact with a heat source, and curing proceeds in the longitudinal direction of the sample by secondary thermal energy generated by the exothermic polymerization reaction, and the entire sample is cured. .
- the cured part can be confirmed by visual observation, temperature measurement of each point in the longitudinal direction, or the like.
- the curable composition of the present invention is preferably chain curable at a temperature of 65 ° C. or less.
- the chain curable temperature is a temperature at which the first exothermic polymerization reaction occurs in chain curing, that is, the temperature at which chain curing can be initiated.
- the temperature of the curable composition may exceed the upper limit of the chain curable temperature by secondary heat energy after initiation of chain curing.
- the chain curing start temperature is more preferably 45 ° C. or less.
- the lower limit of the chain curing start temperature is not particularly limited, and may be appropriately selected in consideration of the balance with the pot life.
- the chain curing start temperature may be, for example, 0 ° C. or higher, 5 ° C. or higher, 10 ° C. or higher, 15 ° C. or higher, or 20 ° C. or higher. Good.
- the temperature at which chain curing can start is particularly preferably around room temperature (RT).
- room temperature indicates a temperature within 23 ⁇ 3 ° C.
- the chain cure start temperature can be adjusted by the type of thermal polymerization initiator.
- the curable composition of the present invention preferably has a working life of 20 minutes or more at room temperature, more preferably 30 minutes or more, still more preferably 45 minutes or more, and particularly preferably 60 minutes or more.
- the pot life indicates the time until the material can be handled in the handling environment (room temperature). While the curable composition is soft when the pot life at room temperature is equal to or more than the above lower limit value, it is possible to complete an operation such as a coating operation in molding.
- the pot life at room temperature of the curable composition is preferably 20 minutes or more, more preferably 30 minutes or more, still more preferably 45 minutes or more, and particularly preferably 60 minutes or more.
- the pot life of the curable composition can be adjusted by the content of the storage stabilizer, the type of the polymerization initiator, the type of the cationically polymerizable resin component, the amount of the filler added, and the like.
- the curing time of the curable composition of the present invention at room temperature is preferably (use time at room temperature + 6 hours) or less, and more preferably (use time at room temperature + 2 hours) or less.
- the pot life at room temperature is 60 minutes, 7 hours or less is preferable, and 3 hours or less is more preferable.
- the curing time of the curable composition of the present invention at 60 ° C. is preferably 3 hours or less (excluding pot life), and more preferably 1 hour (excluding pot life) or less.
- the curing time is from the time of applying the primary heat energy to the curable composition (from the time of exposure to room temperature environment in the case of room temperature curing) to the time of curing of the curable composition and having predetermined physical properties. Indicates the time of The point at which the curing of the curable composition is completed means the point at which the physical property change due to the curing reaction disappears. In addition, the curing time at the time of actually curing a curable composition changes with primary heat energy (temperature) provided to a curable composition, atmospheric temperature, etc.
- the viscosity at room temperature of the curable composition of the present invention is preferably 100 Pa ⁇ s or more, and more preferably 1000 Pa ⁇ s or more.
- the curable composition is deformed under pressure.
- the viscosity is at least the above lower limit, molds can be sufficiently obtained.
- the rigidity of the sandwiching structural members made of aluminum, titanium, carbon fiber reinforced plastic (CFRP), etc.
- CFRP carbon fiber reinforced plastic
- the viscosity is a value measured by a B-type viscometer. The same measurement value can be obtained by the viscoelasticity measuring device.
- the viscosity can be adjusted by the content of the filler, the cationically polymerizable resin component, and the like. For example, as the content of the filler is higher, the viscosity tends to be higher.
- the hardness when hardening the curable composition of this invention ie, the hardness of hardened
- 60 Hs or more is more preferable.
- the cured product is processed or handled (handled) and shaped.
- the upper limit of the hardness of the cured product is not particularly limited, and may be, for example, 80 Hs.
- the hardness (Shore hardness) is measured by a hardness tester. The hardness can be adjusted by the filler loading amount, the cationically polymerizable resin component, and the like. For example, the greater the filler loading, the higher the hardness tends to be.
- the curable composition can be prepared by mixing a cationically polymerizable compound, a thermal polymerization initiator, a storage stabilizer, and if necessary, a filler and other components.
- the mixing is usually performed at a temperature lower than the chain curable temperature of the curable composition.
- the mixing of the polymerization initiator is preferably performed at (chain curing start temperature -10 ° C) or less, more preferably (chain curing start temperature -15 ° C) or less, (chain cure start temperature -20) It is more preferable to carry out below (C).
- the mixing temperature of the polymerization initiator is preferably 5 to 20 ° C., and more preferably 10 to 15 ° C. It can suppress that chain curing advances at the time of mixing as mixing temperature is below the said upper limit. It is easy to mix each material as mixing temperature is more than the above-mentioned lower limit.
- the mixing method is not particularly limited, and a known mixing method can be used, but since heat is generated depending on the mixing method, it is necessary to work carefully.
- the order of mixing the respective components is not particularly limited, and all the components may be mixed at one time or may be sequentially mixed, but after mixing of the polymerization initiator, the temperature at the time of mixing the polymerization initiator (the following Should be maintained). From the above, as the order of mixing, the order of mixing described later is preferable in terms of ease of handling.
- the thermal polymerization initiator and the storage stabilizer are mixed to form a first mixture, and the cationically polymerizable compound and the filler are mixed to form a second mixture, the first mixture and the second mixture. It is preferred to mix with the mixture.
- the filler is blended, the viscosity becomes high, and the concentration of the thermal polymerization initiator or the like tends to be uneven at the time of mixing. If a portion where the amount of the storage stabilizer is low locally with respect to the thermal polymerization initiator is generated, the polymerization reaction may start at that portion.
- the curable composition prepared is typically in the form of a paste.
- the paste-like curable composition can be used as a curable paste material.
- the prepared curable composition may be formed into a sheet.
- the sheet-like curable composition can be used as a curable sheet material.
- a film material such as a resin film may be laminated on one side or both sides of the sheet-like curable composition (a layer made of the curable composition).
- a laminate in which a film material is laminated on one side or both sides of a sheet-like curable composition can also be used as a curable sheet material.
- the storage temperature in the frozen storage is preferably ⁇ 15 ° C. or less, more preferably ⁇ 40 ° C. or less, and particularly preferably ⁇ 55 ° C. or less.
- the lower limit of the storage temperature is not particularly limited, and may be, for example, ⁇ 65 ° C. or less.
- the curable composition of the present invention comprises a cationically polymerizable compound, a thermal polymerization initiator, and a storage stabilizer, and the cationically polymerizable compound is a glycidyl ether compound, an alicyclic epoxy compound, and an oxetane compound.
- the thermal polymerization initiator content is 0.3 to 3 parts by mass with respect to 100 parts by mass of the cationically polymerizable compound, and is generated by the polymerization reaction of the cationically polymerizable compound As it can be chain-hardened by thermal energy, a sufficiently long pot life can be secured. Moreover, it can be rapidly cured even at a temperature of 65 ° C. or less after the pot life time has elapsed.
- the reasons for being able to secure a sufficiently long pot life are mainly due to the following. That is, when the curable composition of the present invention is placed in a working environment or a working environment (under an ambient temperature of pot life), the thermal polymerization initiator decomposes to a slight extent, and the Lewis acid or protic acid Occur. Most of the Lewis acid or protic acid generated within the pot life is captured by the storage stabilizer, little or no exothermic polymerization reaction occurs, and chain curing does not start. In addition, even with the heat energy generated by the slightly generated exothermic polymerization reaction, further chain curing does not start because a glycidyl ether type compound or the like having high stability absorbs some of the heat energy.
- This effect is high particularly when the filler is included. Since the storage stabilizer is consumed by capture of the Lewis acid or protic acid, when the amount of the generated Lewis acid or protic acid exceeds the amount of the storage stabilizer, the chain polymerization is promoted by the exothermic polymerization reaction of the cationically polymerizable compound. Enough secondary heat energy is generated. This secondary heat energy initiates chain curing and cures the entire curable composition.
- the application of the curable composition of the present invention is not particularly limited.
- it may be used for a curable mold material, a curable sheet material, a curable paste material, a potting material, a casting material, an adhesive, a paint, a coating agent, etc. it can.
- the hardenable mold material used to make the shims placed in the gaps between the structural members such as the gap between the wing of the aircraft and the engine nacelle, has a short working time while ensuring some pot life It is desirable to do.
- it may be desired to satisfy the following conditions 1 to 7.
- the curable composition of the present invention can satisfy these conditions. Therefore, for the application of the curable composition of the present invention, a curable mold material for producing a shim disposed in a gap between structural members is suitable.
- Condition 1 Material temperature suppression by reaction heat at the time of hardening It is required that the surface temperature of the structural member does not deviate from the allowable temperature by the reaction heat at the time of hardening of the mold material.
- the acceptable temperature of the seal adhesive surface on which the seal material is applied is typically less than 60 ° C, and the acceptable temperature of the primer coated surface on which the primer is applied is typically less than 93 ° C.
- Condition 2 Influence of the hardened resin on the drilling drill Shim is often manufactured in the state of having a hole of the same size. In this case, the mold material is drilled together with the structural member when it is partially cured. It is required that the molding material does not affect the drill blade at the time of drilling and does not reduce the processing accuracy.
- Condition 3 Hardness that the cured product does not deform Since the die-casting material after curing is directly transported and shape-measured by FARO ARM, it is required to be held without deformation of the shape in each operation.
- Condition 4 Shortening of the curing time in the low temperature region As an example, there is a usable time of about 1 hour under the temperature condition satisfying the condition 1, and a degree that drilling can be performed 1 hour after the curing starts (for example, hardness 30 Hs or more) It is desirable that the resin be cured to have a hardness of 60 Hs or more after 6 hours from the start of curing.
- Condition 5 Viscosity when the structural members do not deform during molding When a gap between the structural members is molded, a certain pressure is applied to the molding material.
- the sandwiching structural member has a viscosity that does not deform. At the same time, it is also required to have sufficient viscosity to take a mold.
- Condition 6 Material cost From the production cost requirements of the shim, it is required that the material cost of the mold material be as low as possible.
- Condition 7 Influence on the human body Since the molding work is handled directly by the workers, it is required from the viewpoint of safety that it does not contain materials that affect the human body.
- the curable paste material of the present invention contains the above-mentioned curable composition of the present invention.
- the curable paste material of the present invention can be used in the same applications as the curable composition of the present invention.
- the curable sheet material of the present invention comprises the curable composition of the present invention.
- the curable sheet material include a sheet made of the curable composition of the present invention, a sheet in which a film substrate such as a resin film is laminated on one side or both sides of a layer made of the curable composition of the present invention.
- a film substrate such as a resin film
- the curable sheet material of the present invention can be used in the same applications as the curable composition of the present invention.
- the curable mold material of the present invention comprises the curable composition of the present invention.
- the form of the curable mold material of the present invention is not particularly limited, and may be, for example, a paste, a sheet, or the like.
- the curable paste material of the present invention described above can be used as a paste-like curable mold material.
- the above-mentioned curable sheet material of the present invention can be used as a sheet-like curable mold material.
- the objects to be molded by the curable mold material of the present invention include gaps between structural members, teeth, bush gaps, sealing, hole filling, and the like, and gaps between structural members are preferable.
- curable composition etc. The above-mentioned curable composition, curable paste material, curable sheet material or molding material (hereinafter collectively referred to as "curable composition etc.") can be cured to form a cured product. . If the curable composition or the like is given any shape before curing, a cured product having any shape can be obtained.
- the curable composition and the like can be cured by applying thermal energy (primary thermal energy) to a part of the curable composition and the like.
- the method for applying thermal energy is not particularly limited.
- a method of placing a curable composition or the like under temperature conditions that enables chain curing, a part of a curable composition or the like using a heating means, chain curing Methods such as heating to a possible temperature may be mentioned.
- the heating means include a dryer, a rod-like heater, a heater-blanket, an infrared heater, an oven and the like.
- Preferred chain curable temperatures are as described above. Therefore, as temperature at the time of hardening a hardenability constituent etc., 65 ° C or less is preferred, 45 ° C or less is still more preferred, and about room temperature (within 25 ⁇ 3 ° C) is especially preferred.
- the curable composition or the like before complete curing may be subjected to processing such as drilling.
- the hardness of the curable composition or the like at the time of processing is preferably 30 Hs or more, more preferably 60 Hs or more.
- the application of the cured product is not particularly limited.
- it can be used for a mold for producing a shim, a mold for producing a bush, a sealing material or a filling material, a mold for producing the same, a casting material, an adhesive application, a coating application, a coating agent application and the like.
- An example of the shape of a mold for producing a shim is a shape having a maximum length of about 2 m, a width of about 0.2 m, and a thickness of about 0.2 to 3 mm.
- Part shows “mass part”.
- the materials used in each example are shown below.
- (Material used) 828 “jER (registered trademark) 828” manufactured by Mitsubishi Chemical Corporation, a bisphenol A-type diglycidyl ether compound.
- 2021P “Celloxide (registered trademark) 2021P” manufactured by Daicel, 3,4-epoxycyclohexylmethyl-3,4-epoxycyclohexanecarboxylate.
- OXBP "ETERNACOLL (registered trademark) OXBP” manufactured by Ube Industries, Ltd., 4,4'-bis [(3-ethyl-3-oxetanyl) methoxymethyl] biphenyl.
- EHO "ETERNACOLL EHO” manufactured by Ube Industries, Ltd., 3-ethyl-3-hydroxymethyl oxetane.
- SI-60 Thermal polymerization initiator, "Sanaid SI-60” manufactured by Sanshin Chemical Industry Co., Ltd., a sulfonium salt represented by the above formula (I-2).
- SI-45 Thermal polymerization initiator, "Sanaid SI-45” manufactured by Sanshin Chemical Industry Co., Ltd., a sulfonium salt represented by the above formula (I-1-1).
- SI-B2A Thermal polymerization initiator, "Sanaid SI-B2A” manufactured by Sanshin Chemical Industry Co., Ltd.
- UVI-6974 Photopolymerization initiator, "UVI-6974” manufactured by Dow Chemical Co., diphenyl [4- (phenylthio) phenyl] sulfonium hexafluoroantimonate (V).
- Storage stabilizer "SI-auxiliary agent” manufactured by Sanshin Chemical Industry Co., Ltd., a compound represented by the above formula (II).
- Each of SI-60, SI-45, SI-B2A and storage stabilizer is used as a 50 mass% solution (solvent: ⁇ -butyrolactone), and UVI-6974 is a 50 mass% solution (solvent: propylene carbonate). Using.
- EXV-5 Brand name, sold by Laser Net, silica filler.
- URE-FIL 9 trade name, silica filler (fumed silica (nano particle)).
- FB-5D trade name, Denka company, silica filler, powdery, average particle size 5 ⁇ m.
- FB-20D trade name, Denka company, silica filler, powdery, average particle size 20 ⁇ m.
- FB-950 trade name, Denka company, silica filler.
- UVI-6974 is a 50% by weight solution of propylene carbonate from the beginning, it was used as it was when used.
- the mixture ratio of the solid contents of each polymerization initiator shown in Table 1 and the other mixture ratio (for example, each amount of 2 times each) shown in Table 1 were charged into a light shielding container to prepare a mixed solution of the polymerization initiator (mixture liquid This step is unnecessary if it is not necessary).
- Predetermined amounts of the respective main ingredient components shown in Tables 1 to 3 were mixed to prepare a main ingredient mixture.
- a mixed solution of a polymerization initiator (or a polymerization stabilizer-containing polymerization initiator solution) was metered into this main agent mixture so as to obtain predetermined amounts of each polymerization initiator and storage stabilizer shown in Tables 1 to 3.
- the mixture was rapidly stirred and mixed for about 1 minute to obtain a curable composition.
- pot life and curing completion time were measured in the following procedure. The results are shown in Tables 1 to 3.
- the “main agent” in Tables 1 to 3 indicates a cationically polymerizable compound, and the amounts (parts) of the main agent, the filler, the thermal polymerization initiator, the storage stabilizer and the like are solid amounts.
- the pot life of the curable composition was determined by leaving the produced curable composition in a room temperature environment, measuring the temperature of the composition during that time, and determining the time when the resin temperature began to rise due to chain curing.
- the curing completion time of the curable composition was determined by placing the produced curable composition in a room temperature environment, measuring the temperature of the composition during that time, and showing a peak of temperature rise when the resin temperature exceeded 45 ° C. It asked for time.
- Examples 26 to 31 Weigh each solid in a light-shielding container so that the ratio of SI-45 and SI-Auxiliaries will be the same as the mixing ratio in Table 3 (for example, 2 times each) An amount of ⁇ -butyrolactone equal to the total amount of solid content was added to dissolve the solid content, and a storage stabilizer-containing polymerization initiator solution was prepared. A predetermined amount of each of the main agent components shown in Table 3 was mixed to prepare a main agent mixture, and a predetermined amount of filler shown in Table 3 was added thereto, mixed and kneaded. Once a uniform paste mixture was obtained it was cooled to below 15 ° C.
- a storage stabilizer-containing polymerization initiator solution is weighed and added to the paste mixture which has reached 15 ° C. or less so as to have predetermined amounts of each polymerization initiator and storage stabilizer shown in Table 3, and after addition, it becomes uniform
- the mixture was kneaded to obtain a paste-like curable composition.
- About the obtained curable composition it carried out similarly to Example 1, and measured pot life and hardening completion time. The results are shown in Table 3.
- FIG. 1 shows a graph in which the elapsed time from the start of heating of the curable composition of Examples 3 to 25 is taken on the horizontal axis, and the temperature of the curable composition is taken on the vertical axis.
- FIG. 2 shows a graph in which the elapsed time from the start of heating of the curable compositions of Examples 26 to 31 is taken on the horizontal axis, and the temperature of the curable composition is taken on the vertical axis.
- the curable compositions of Examples 1-31 had a pot life of at least 20 minutes and cure was complete within 2.2 hours.
- the curable composition of Comparative Example 1 containing only an alicyclic epoxy compound as a cationic polymerizable compound and containing no storage stabilizer did not have a usable life of less than 20 minutes.
- the curable composition of Comparative Example 2 containing only a glycidyl ether compound as a cationic polymerizable compound and containing no storage stabilizer did not complete curing within 8 hours.
- Examples 32 to 50, Comparative Examples 3 to 7 Weigh each solid in a light-shielding container so that the ratio of SI-45 and SI-Auxiliaries will be the same as the mixing ratio of Table 4 (for example, in each case 2 times each), and weigh it An amount of ⁇ -butyrolactone equal to the total amount of solid content was added to dissolve the solid content, and a storage stabilizer-containing polymerization initiator solution was prepared. 10 g of SI-B2A was weighed in a separate light shielding container, and 10 g of ⁇ -butyrolactone was added thereto to dissolve the solid content to prepare a 50 mass% solution of SI-B2A.
- UVI-6974 is a 50% by weight solution of propylene carbonate from the beginning, it was used as it was when used.
- the mixture ratio of the solid contents of each polymerization initiator shown in Table 4 to each other and the same ratio (for example, each amount of 2 times each) shown in Table 4 was charged into a light shielding container to prepare a mixed solution of the polymerization initiator (mixed liquid This step is unnecessary if it is not necessary).
- a predetermined amount of each main agent component shown in Table 4 was mixed to prepare a main agent mixture, and a predetermined amount of filler shown in Table 4 was added thereto, mixed and kneaded. Once a uniform paste mixture was obtained it was cooled to below 15 ° C.
- the mixture of polymerization initiators (or a storage stabilizer-containing polymerization initiator solution) is metered and charged to the paste mixture which has reached 15 ° C. or less so as to obtain predetermined amounts of each polymerization initiator and storage stabilizer shown in Table 4. After the addition, the mixture was kneaded so as to be uniform to obtain a paste-like curable composition.
- FIG. 3 is a schematic top view explaining an evaluation sample in an application simulation test
- FIG. 4 is a side view thereof
- FIG. 5 is a schematic view for explaining the implementation situation of the application simulation test.
- the outline of the test is as follows. A plate of two different materials of CFRP plate 1 and Ti plate 3 and a heater 11 having a rod-like tip 11a which can be heated to 60 ° C. were prepared.
- the CFRP plate 1 and the Ti plate 3 each have an elliptical shape having a major axis of 700 mm and a minor axis of 300 mm.
- Five through holes 3a (diameter 15 mm) for inserting the rod-like end 11a of the heater 11 are formed in the range from the center to one end of the Ti plate 3 in the major axis direction.
- the five through holes 3a are formed at positions of four vertices and a center of a square in top view.
- the paste-like curable composition obtained above is applied and sandwiched between the two plates in a state where the resin film 5 is disposed on the contact surface with each plate, and the spacer 9 (thickness 3. It clamped and fixed, with a clearance gap maintained by 0 mm.
- the rod end 11a of the heater 11 was heated and maintained at 60 ° C., inserted into the through hole 3a of the Ti plate 3 of the evaluation sample and fixed for 1 hour, and then the heater 11 was removed.
- the test was performed in the form which removed the heater 11 from the beginning about a part of test.
- the contact point with the inserted rod-like tip 11a is a forced curing site, a point away from the heater 11 without the influence of the heater 11 (for example, the end of the right side of the ellipse in FIG.
- the following evaluation was performed by making the part side (end part of the long axis right end part) the end of the curable laminate 7) as a natural curing site. The results are shown in Table 5.
- ⁇ Attainment temperature (natural / forced)> It was judged from the peak value of the measured resin temperature according to the following criteria. ⁇ (especially good): 60 ° C. or less. ⁇ (Good): Exceeds 60 ° C. and 93 ° C. (within 30 minutes) ⁇ (defect): over 60 ° C. and 93 ° C. or less (30 minutes or more). X (not good): Exceeds 93 ° C.
- ⁇ Empty time> The temperature of the composition was measured, and the time in which the resin temperature exceeded 45 ° C. or the shorter of the peak temperature was determined as the pot life of the cured product on the basis of the following criteria. ⁇ (especially good): 45 minutes or more. ⁇ (Good): 20 minutes or more and less than 45 minutes. ⁇ (defect): 15 minutes or more and less than 20 minutes. X (not good): less than 15 minutes.
- the “main agent” in Tables 4 to 5 indicates a cationically polymerizable compound.
- the ratio of the thermal polymerization initiator to 100 parts by mass of the main agent in Table 5 and the ratio of the storage stabilizer to 100 parts by mass of the thermal polymerization initiator are respectively values in solid content.
- the curable compositions of the examples were sufficiently long in pot life and, after the pot life had elapsed, were able to be cured rapidly even at a temperature of 60 ° C. or less and natural curing (room temperature).
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Epoxy Resins (AREA)
- Polyethers (AREA)
- Compositions Of Macromolecular Compounds (AREA)
Abstract
本発明の硬化性組成物は、カチオン重合性化合物と、熱重合開始剤と、保存安定剤とを含み、前記カチオン重合性化合物が、グリシジルエーテル化合物、脂環式エポキシ化合物、およびオキセタン化合物からなる群から選ばれる少なくとも2種を含み、前記熱重合開始剤の含有量が、前記カチオン重合性化合物100質量部に対して0.3~3質量部であり、前記カチオン重合性化合物の重合反応により発生した熱エネルギーにより連鎖硬化可能である。
Description
本発明は、硬化性組成物、硬化性ペースト材、硬化性シート材、硬化性型取り材、硬化方法および硬化物に関する。
部品単品公差や構造組立公差により部品間にギャップが発生する。発生したギャップにシムを挿入することが行われている。ギャップを再現するシムを製作する手法として下記手法が取られている。
(1)フィラーゲージによるギャップ測定。
(2)接触式または非接触式のギャップ測定器(Gap Master等)の適用。
(3)エポキシ樹脂による型取り。
上述の手法のうち、複雑な3D形状の型取りや高精度なシム形状が要求される場合には、ギャップ測定精度の観点から手法(3)が適用されている。
(1)フィラーゲージによるギャップ測定。
(2)接触式または非接触式のギャップ測定器(Gap Master等)の適用。
(3)エポキシ樹脂による型取り。
上述の手法のうち、複雑な3D形状の型取りや高精度なシム形状が要求される場合には、ギャップ測定精度の観点から手法(3)が適用されている。
手法(3)では、樹脂準備、塗付作業、樹脂硬化時間、硬化樹脂測定作業等、シム製作までに多大な時間を要する。将来的な高レート生産達成には、シム製作までの時間の短縮が課題となる。しかし、樹脂の可使時間(ポットライフ)の長さと硬化時間の短さとの相反する関係により劇的な時間短縮は見込めない。すなわち、硬化時間の短い樹脂は可使時間も短く、塗付作業を行うのに充分な可使時間を確保できない。したがって、硬化時間の短い樹脂を採用できない。加えて、型取りに際しては通常、対象構造部材の表面温度が許容温度を逸脱しないことが要求される。シール材が塗布されているシール接着面の許容温度は例えば60℃未満であり、プライマーが塗布されているプライマー塗布面の許容温度は例えば93℃未満である。したがって、加温による硬化促進等、樹脂硬化促進を図る手法にも劇的な効果が見込めず、十分な時間短縮は厳しい状況である。
ところで特許文献1には、カチオン重合性有機物質混合物として芳香族エポキシ化合物と、脂環式エポキシ化合物または脂肪族エポキシ化合物と、多官能脂肪族オキセタン化合物とを含み、芳香族エポキシ化合物が主成分である組成物が提案されている。特許文献1にはまた、この組成物にカチオン重合開始剤を含有させることが記載されている。カチオン重合開始剤は、カチオン重合性有機物質混合物100質量部に対して7~10質量部の割合で用いられている。
特許文献1には、組成物をシム製作に用いることは記載されていない。特許文献1に記載の組成物は、硬化させる際に130~180℃で加熱する必要があり、上述の許容温度を逸脱する。
特許文献1には、組成物をシム製作に用いることは記載されていない。特許文献1に記載の組成物は、硬化させる際に130~180℃で加熱する必要があり、上述の許容温度を逸脱する。
本発明は、十分な長さの可使時間を確保でき、可使時間経過後に65℃以下の温度でも迅速に硬化可能な硬化性組成物、これを用いた硬化性ペースト材、硬化性シート材、硬化性型取り材、硬化方法および硬化物を提供することを目的とする。
本発明は以下の態様を有する。
〔1〕カチオン重合性化合物と、熱重合開始剤と、保存安定剤とを含み、
前記カチオン重合性化合物が、グリシジルエーテル化合物、脂環式エポキシ化合物、およびオキセタン化合物からなる群から選ばれる少なくとも2種を含み、
前記熱重合開始剤の含有量が、前記カチオン重合性化合物100質量部に対して0.3~3質量部であり、
前記カチオン重合性化合物の重合反応により発生した熱エネルギーにより連鎖硬化可能である、硬化性組成物。
〔2〕65℃以下の温度で連鎖硬化可能である、〔1〕の硬化性組成物。
〔3〕フィラーをさらに含む、〔1〕または〔2〕の硬化性組成物。
〔4〕室温における可使時間が20分間以上である、〔1〕~〔3〕のいずれかの硬化性組成物。
〔5〕前記グリシジルエーテル化合物が、ビスフェノールA型ジグリシジルエーテル化合物およびビスフェールF型ジグリシジルエーテル化合物からなる群から選ばれる少なくとも1種であり、
前記脂環式エポキシ化合物が、3,4-エポキシシクロヘキシルメチル-3,4-エポキシシクロヘキサンカルボキシレートであり、
前記オキセタン化合物が、4,4’-ビス[(3-エチル-3-オキセタニル)メトキシメチル]ビフェニルおよび3-エチル-3-ヒドロキシメチルオキセタンからなる群から選ばれる少なくとも1種である、〔1〕~〔4〕のいずれかの硬化性組成物。
〔6〕前記熱重合開始剤が、下記式(I-1)または(I-2)で表されるスルホニウム塩からなる群から選ばれる少なくとも1種である、〔1〕~〔5〕のいずれかの硬化性組成物。
〔1〕カチオン重合性化合物と、熱重合開始剤と、保存安定剤とを含み、
前記カチオン重合性化合物が、グリシジルエーテル化合物、脂環式エポキシ化合物、およびオキセタン化合物からなる群から選ばれる少なくとも2種を含み、
前記熱重合開始剤の含有量が、前記カチオン重合性化合物100質量部に対して0.3~3質量部であり、
前記カチオン重合性化合物の重合反応により発生した熱エネルギーにより連鎖硬化可能である、硬化性組成物。
〔2〕65℃以下の温度で連鎖硬化可能である、〔1〕の硬化性組成物。
〔3〕フィラーをさらに含む、〔1〕または〔2〕の硬化性組成物。
〔4〕室温における可使時間が20分間以上である、〔1〕~〔3〕のいずれかの硬化性組成物。
〔5〕前記グリシジルエーテル化合物が、ビスフェノールA型ジグリシジルエーテル化合物およびビスフェールF型ジグリシジルエーテル化合物からなる群から選ばれる少なくとも1種であり、
前記脂環式エポキシ化合物が、3,4-エポキシシクロヘキシルメチル-3,4-エポキシシクロヘキサンカルボキシレートであり、
前記オキセタン化合物が、4,4’-ビス[(3-エチル-3-オキセタニル)メトキシメチル]ビフェニルおよび3-エチル-3-ヒドロキシメチルオキセタンからなる群から選ばれる少なくとも1種である、〔1〕~〔4〕のいずれかの硬化性組成物。
〔6〕前記熱重合開始剤が、下記式(I-1)または(I-2)で表されるスルホニウム塩からなる群から選ばれる少なくとも1種である、〔1〕~〔5〕のいずれかの硬化性組成物。
ここで、R1は水素原子、メチル基、アセチル基またはメトキシカルボニル基を示し、R2およびR3はそれぞれ独立に、水素原子、ハロゲン原子または炭素数1~4のアルキル基を示し、R4は水素原子、ハロゲン原子、ニトロ基、メチル基またはメトキシ基を示し、R5は炭素数1~4のアルキル基を示し、XはSbF6、PF6、AsF6またはBF4を示す。
〔7〕前記保存安定剤が、下記式(II)で表されるスルホニウム塩からなる群から選ばれる少なくとも1種である、〔1〕~〔6〕のいずれかの硬化性組成物。
〔7〕前記保存安定剤が、下記式(II)で表されるスルホニウム塩からなる群から選ばれる少なくとも1種である、〔1〕~〔6〕のいずれかの硬化性組成物。

ここで、R6はアセチル基またはメトキシカルボニル基を示し、AはCH3SO4を示す。
〔8〕前記保存安定剤の含有量が、前記熱重合開始剤100質量部に対して0.3~5質量部である、〔1〕~〔7〕のいずれかの硬化性組成物。
〔9〕〔1〕~〔8〕のいずれかの硬化性組成物を含む、硬化性ペースト材。
〔10〕〔1〕~〔8〕のいずれかの硬化性組成物を含む、硬化性シート材。
〔11〕〔1〕~〔8〕のいずれかの硬化性組成物を含む、硬化性型取り材。
〔12〕〔1〕~〔8〕のいずれかの硬化性組成物、〔9〕の硬化性ペースト材、〔10〕の硬化性シート材、または〔11〕の硬化性型取り材を、65℃以下の温度で硬化させる、硬化方法。
〔13〕〔1〕~〔8〕のいずれかの硬化性組成物、〔9〕の硬化性ペースト材、〔10〕の硬化性シート材、または〔11〕の硬化性型取り材を硬化させた、硬化物。
〔14〕硬度が60Hs以上である、〔10〕の硬化物。
〔8〕前記保存安定剤の含有量が、前記熱重合開始剤100質量部に対して0.3~5質量部である、〔1〕~〔7〕のいずれかの硬化性組成物。
〔9〕〔1〕~〔8〕のいずれかの硬化性組成物を含む、硬化性ペースト材。
〔10〕〔1〕~〔8〕のいずれかの硬化性組成物を含む、硬化性シート材。
〔11〕〔1〕~〔8〕のいずれかの硬化性組成物を含む、硬化性型取り材。
〔12〕〔1〕~〔8〕のいずれかの硬化性組成物、〔9〕の硬化性ペースト材、〔10〕の硬化性シート材、または〔11〕の硬化性型取り材を、65℃以下の温度で硬化させる、硬化方法。
〔13〕〔1〕~〔8〕のいずれかの硬化性組成物、〔9〕の硬化性ペースト材、〔10〕の硬化性シート材、または〔11〕の硬化性型取り材を硬化させた、硬化物。
〔14〕硬度が60Hs以上である、〔10〕の硬化物。
本発明によれば、十分な長さの可使時間を確保でき、可使時間経過後に65℃以下の温度でも迅速に硬化可能な硬化性組成物、これを用いた硬化性ペースト材、硬化性シート材、硬化性型取り材、硬化方法および硬化物を提供できる。
〔硬化性組成物〕
本発明の硬化性組成物は、カチオン重合性化合物と、熱重合開始剤と、保存安定剤とを含む。
本発明の硬化性組成物は、フィラーをさらに含むことができる。
本発明の硬化性組成物は、必要に応じて、本発明の効果を損なわない範囲で、カチオン重合性化合物、熱重合開始剤、保存安定剤およびフィラー以外の他の成分をさらに含むことができる。
本発明の硬化性組成物は、カチオン重合性化合物と、熱重合開始剤と、保存安定剤とを含む。
本発明の硬化性組成物は、フィラーをさらに含むことができる。
本発明の硬化性組成物は、必要に応じて、本発明の効果を損なわない範囲で、カチオン重合性化合物、熱重合開始剤、保存安定剤およびフィラー以外の他の成分をさらに含むことができる。
(カチオン重合性化合物)
カチオン重合性化合物は、グリシジルエーテル化合物、脂環式エポキシ化合物、およびオキセタン化合物からなる群から選ばれる少なくとも2種を含む。
カチオン重合性化合物は、グリシジルエーテル化合物、脂環式エポキシ化合物およびオキセタン化合物以外の他のカチオン重合性化合物をさらに含んでいてもよい。
カチオン重合性化合物は、グリシジルエーテル化合物、脂環式エポキシ化合物、およびオキセタン化合物からなる群から選ばれる少なくとも2種を含む。
カチオン重合性化合物は、グリシジルエーテル化合物、脂環式エポキシ化合物およびオキセタン化合物以外の他のカチオン重合性化合物をさらに含んでいてもよい。
カチオン重合性化合物中のグリシジルエーテル化合物、脂環式エポキシ化合物およびオキセタン化合物の合計の含有量は、カチオン重合性化合物の総質量に対し、50~100質量%が好ましく、80~100質量%がより好ましい。
なお、本明細書および特許請求の範囲において、数値範囲を示す「~」は、その前後に記載された数値を下限値および上限値として含むことを意味する。
なお、本明細書および特許請求の範囲において、数値範囲を示す「~」は、その前後に記載された数値を下限値および上限値として含むことを意味する。
グリシジルエーテル化合物は、グリシジルエーテル基を1以上有する。
グリシジルエーテル化合物としては、ビスフェノールA型ジグリシジルエーテル化合物、ビスフェールF型ジグリシジルエーテル化合物、水添ビスフェーノール型グリシジルエーテル化合物、その他の芳香族グリシジルエーテル化合物、脂肪族グリシジルエーテル化合物等が挙げられる。これらのグリシジルエーテル化合物はいずれか1種を単独で用いてもよく2種以上を併用してもよい。
グリシジルエーテル化合物としては、ビスフェノールA型ジグリシジルエーテル化合物、ビスフェールF型ジグリシジルエーテル化合物、水添ビスフェーノール型グリシジルエーテル化合物、その他の芳香族グリシジルエーテル化合物、脂肪族グリシジルエーテル化合物等が挙げられる。これらのグリシジルエーテル化合物はいずれか1種を単独で用いてもよく2種以上を併用してもよい。
グリシジルエーテル化合物としては、入手性、耐熱性、剛性、強度特性、汎用性、低コスト等の観点から、ビスフェノールA型ジグリシジルエーテル化合物およびビスフェールF型ジグリシジルエーテル化合物からなる群から選ばれる少なくとも1種が特に好ましい。
脂環式エポキシ化合物は、脂環式エポキシ基を1以上有する。脂環式エポキシ基とは、脂環式化合物の脂肪族環を形成する環状に結合した炭素原子のうち2個の炭素原子(通常は互いに隣接する炭素原子)に酸素原子1個が結合した状態のエポキシ基をいう。
脂環式エポキシ化合物としては、ジシクロペンタジエンジオキサイド、リモネンジオキサイド、ジ(3,4-エポキシシクロヘキシル)アジペート、(3,4-エポキシシクロヘキシル)メチル-3,4-エポキシシクロヘキサンカルボキシレート、3,4-エポキシ-6-メチルシクロヘキシルメチル-3,4-エポキシ-6-メチルシクロヘキサンカルボキシレート、エチレン-1,2-ジ(3,4-エポキシシクロヘキサンカルボン酸)エステル等が挙げられる。これらの脂環式エポキシ化合物はいずれか1種を単独で用いてもよく2種以上を併用してもよい。
脂環式エポキシ化合物としては、ジシクロペンタジエンジオキサイド、リモネンジオキサイド、ジ(3,4-エポキシシクロヘキシル)アジペート、(3,4-エポキシシクロヘキシル)メチル-3,4-エポキシシクロヘキサンカルボキシレート、3,4-エポキシ-6-メチルシクロヘキシルメチル-3,4-エポキシ-6-メチルシクロヘキサンカルボキシレート、エチレン-1,2-ジ(3,4-エポキシシクロヘキサンカルボン酸)エステル等が挙げられる。これらの脂環式エポキシ化合物はいずれか1種を単独で用いてもよく2種以上を併用してもよい。
脂環式エポキシ化合物としては、高反応性、耐熱性、剛性、強度特性、汎用性、入手性、低コスト等の観点から、3,4-エポキシ-6-メチルシクロヘキシルメチル-3,4-エポキシ-6-メチルシクロヘキサンカルボキシレートが特に好ましい。
オキセタン化合物は、オキセタン基を1以上有する。
オキセタン化合物としては、3-エチル-3-ヒドロキシメチルオキセタン(以下、「EHO」とも記す。)、ビスフェノールA型オキセタン化合物、ビスフェノールオキセタン化合物、ビスフェノールS型オキセタン化合物、キシリレン型オキセタン化合物、フェノールノボラック型オキセタン化合物、クレゾールノボラック型オキセタン化合物、アルキルフェノールノボラック型オキセタン化合物、ビフェノール型オキセタン化合物、ビキシレノール型オキセタン化合物、ナフタレン型オキセタン化合物、ジシクロペンタジエン型オキセタン化合物、フェノール類とフェノール性水酸基を有する芳香族アルデヒドとの縮合物のオキセタン化物等が挙げられる。これらのオキセタン化合物はいずれか1種を単独で用いてもよく2種以上を併用してもよい。
オキセタン化合物としては、3-エチル-3-ヒドロキシメチルオキセタン(以下、「EHO」とも記す。)、ビスフェノールA型オキセタン化合物、ビスフェノールオキセタン化合物、ビスフェノールS型オキセタン化合物、キシリレン型オキセタン化合物、フェノールノボラック型オキセタン化合物、クレゾールノボラック型オキセタン化合物、アルキルフェノールノボラック型オキセタン化合物、ビフェノール型オキセタン化合物、ビキシレノール型オキセタン化合物、ナフタレン型オキセタン化合物、ジシクロペンタジエン型オキセタン化合物、フェノール類とフェノール性水酸基を有する芳香族アルデヒドとの縮合物のオキセタン化物等が挙げられる。これらのオキセタン化合物はいずれか1種を単独で用いてもよく2種以上を併用してもよい。
オキセタン化合物としては、高反応性の観点から、4,4’-ビス[(3-エチル-3-オキセタニル)メトキシメチル]ビフェニル(以下、「OXBP」とも記す。)およびEHOからなる群から選ばれる少なくとも1種が好ましい。硬化物の耐熱性がより優れる点では、OXBPが好ましい。安価である点では、EHOが好ましい。
他のカチオン重合性化合物としては、例えば、エポキシ化オレフィン、ビニルエーテル化合物等が挙げられる。
エポキシ化オレフィンとしては、エポキシ化亜麻仁油、エポキシ化ひまし油、エポキシ化大豆油等が挙げられる。
ビニルエーテル化合物としては、エチレングリコールジビニルエーテル、ジエチレングリコールジビニルエーテル、トリエチレングリコールジビニルエーテル、プロピレングリコールジビニルエーテル、ジプロピレングリコールジビニルエーテル、ブタンジオールジビニルエーテル、ヘキサンジオールジビニルエーテル、シクロヘキサンジメタノールジビニルエーテル、トリメチロールプロパントリビニルエーテル等のジまたはトリビニルエーテル化合物;エチルビニルエーテル、n-ブチルビニルエーテル、イソブチルビニルエーテル、オクタデシルビニルエーテル、シクロヘキシルビニルエーテル、ヒドロキシブチルビニルエーテル、2-エチルヘキシルビニルエーテル、シクロヘキサンジメタノールモノビニルエーテル、n-プロピルビニルエーテル、イソプロピルビニルエーテル、イソプロペニルエーテル-o-プロピレンカーボネート、ドデシルビニルエーテル、ジエチレングリコールモノビニルエーテル、オクタデシルビニルエーテル等のモノビニルエーテル化合物等が挙げられる。
これら他のカチオン重合性化合物はいずれか1種を単独で用いてもよく2種以上を併用してもよい。
エポキシ化オレフィンとしては、エポキシ化亜麻仁油、エポキシ化ひまし油、エポキシ化大豆油等が挙げられる。
ビニルエーテル化合物としては、エチレングリコールジビニルエーテル、ジエチレングリコールジビニルエーテル、トリエチレングリコールジビニルエーテル、プロピレングリコールジビニルエーテル、ジプロピレングリコールジビニルエーテル、ブタンジオールジビニルエーテル、ヘキサンジオールジビニルエーテル、シクロヘキサンジメタノールジビニルエーテル、トリメチロールプロパントリビニルエーテル等のジまたはトリビニルエーテル化合物;エチルビニルエーテル、n-ブチルビニルエーテル、イソブチルビニルエーテル、オクタデシルビニルエーテル、シクロヘキシルビニルエーテル、ヒドロキシブチルビニルエーテル、2-エチルヘキシルビニルエーテル、シクロヘキサンジメタノールモノビニルエーテル、n-プロピルビニルエーテル、イソプロピルビニルエーテル、イソプロペニルエーテル-o-プロピレンカーボネート、ドデシルビニルエーテル、ジエチレングリコールモノビニルエーテル、オクタデシルビニルエーテル等のモノビニルエーテル化合物等が挙げられる。
これら他のカチオン重合性化合物はいずれか1種を単独で用いてもよく2種以上を併用してもよい。
本発明の硬化性組成物の好ましい一態様において、カチオン重合性化合物は、少なくとも1種のグリシジルエーテル化合物と、少なくとも1種の脂環式エポキシ化合物と、少なくとも1種のオキセタン化合物とを含む。
これらの化合物のうち、グリシジルエーテル化合物は、比較的重合反応性が低いため、カチオン性重合性化合物がグリシジルエーテル化合物を含むことで、硬化速度が速くなりすぎるのを抑制できる。また、グリシジルエーテル化合物は比較的安価であるため、コストを低減できる。脂環式エポキシ化合物は、比較的重合反応性が高いため、カチオン性重合性化合物が脂環式エポキシ化合物を含むことで、単位時間あたりの発熱量が増え、連鎖硬化が進みやすくなる。オキセタン化合物は、脂環エポキシと同様にカチオン重合反応性が高く、単位時間当たりの発熱量が増えるため、カチオン性重合性化合物がオキセタン化合物を含むことで、連鎖硬化を進みやすくする一助にできる。
これらの化合物のうち、グリシジルエーテル化合物は、比較的重合反応性が低いため、カチオン性重合性化合物がグリシジルエーテル化合物を含むことで、硬化速度が速くなりすぎるのを抑制できる。また、グリシジルエーテル化合物は比較的安価であるため、コストを低減できる。脂環式エポキシ化合物は、比較的重合反応性が高いため、カチオン性重合性化合物が脂環式エポキシ化合物を含むことで、単位時間あたりの発熱量が増え、連鎖硬化が進みやすくなる。オキセタン化合物は、脂環エポキシと同様にカチオン重合反応性が高く、単位時間当たりの発熱量が増えるため、カチオン性重合性化合物がオキセタン化合物を含むことで、連鎖硬化を進みやすくする一助にできる。
上記の好ましい態様において、グリシジルエーテル化合物の含有量は、カチオン重合性化合物の総質量に対し、10~80質量%が好ましく、20~60質量%がより好ましく、30~50質量%がさらに好ましく、35~45質量%が特に好ましい。グリシジルエーテル化合物の含有量が上記下限値以上であれば、より優れた初期の反応安定性、硬化速度上昇の抑制効果、および物性特性が得られる。また、より低コストである。グリシジルエーテル化合物の含有量が上記上限値以下であれば、より優れた連鎖硬化反応性が得られる。
脂環式エポキシ化合物の含有量は、カチオン重合性化合物の総質量に対し、10~80質量%が好ましく、20~60質量%がより好ましく、25~45質量%がさらに好ましく、30~40質量%が特に好ましい。脂環式エポキシ化合物の含有量が上記下限値以上であれば、より優れた連鎖硬化反応性が得られる。脂環式エポキシ化合物の含有量が上記上限値以下であれば、より優れた初期の反応安定性、および硬化速度上昇の抑制効果が得られる。
オキセタン化合物の含有量は、カチオン重合性化合物の総質量に対し、5~70質量%が好ましく、10~50質量%がより好ましく、15~35質量%がさらに好ましく、20~30質量%が特に好ましい。オキセタン化合物の含有量が上記下限値以上であれば、より優れた連鎖硬化反応性が得られる。オキセタン化合物の含有量が上記上限値以下であれば、より優れた初期の反応安定性、および硬化速度上昇の抑制効果が得られる。
本発明の硬化性組成物のより好ましい一態様において、カチオン重合性化合物は、ビスフェノールA型ジグリシジルエーテル化合物およびビスフェールF型ジグリシジルエーテル化合物からなる群から選ばれる少なくとも1種のグリシジルエーテル化合物と、3,4-エポキシ-6-メチルシクロヘキシルメチル-3,4-エポキシ-6-メチルシクロヘキサンカルボキシレートと、OXBPおよびEHOからなる群から選ばれる少なくとも1種のオキセタン化合物とを含む。
上記のより好ましい態様において、グリシジルエーテル化合物およびオキセタン化合物それぞれの含有量の好ましい範囲は、上記と同様である。3,4-エポキシ-6-メチルシクロヘキシルメチル-3,4-エポキシ-6-メチルシクロヘキサンカルボキシレートの含有量の好ましい範囲は、上記脂環式エポキシ化合物の含有量の好ましい範囲と同様である。
OXBPおよびEHOの両方を含む場合、OXBPの含有量は、カチオン重合性化合物の総質量に対し、5~50質量%が好ましく、5~40質量%がより好ましく、10~30質量%がさらに好ましく、15~25質量%が特に好ましい。OXBPの含有量が上記下限値以上であれば、より優れた連鎖硬化反応性、耐熱性、剛性が得られる。OXBPの含有量が上記上限値以下であれば、より優れた初期の反応安定性、および硬化速度上昇の抑制効果が得られる。また、より低コストである。
OXBPおよびEHOの両方を含む場合、EHOの含有量は、カチオン重合性化合物の総質量に対し、5~40質量%が好ましく、5~25質量%がより好ましく、5~15質量%がさらに好ましく、5~10質量%が特に好ましい。EHOの含有量が上記下限値以上であれば、より優れた連鎖硬化反応性が得られる。また、より低コストである。EHOの含有量が上記上限値以下であれば、より優れた初期の反応安定性、硬化速度上昇の抑制効果、および耐熱性が得られる。
本発明の硬化性組成物のさらに好ましい一態様において、カチオン重合性化合物は、ビスフェノールA型ジグリシジルエーテル化合物と、3,4-エポキシ-6-メチルシクロヘキシルメチル-3,4-エポキシ-6-メチルシクロヘキサンカルボキシレートと、OXBPと、EHOとを含む。
上記のさらに好ましい態様において、ビスフェノールA型ジグリシジルエーテル化合物の含有量の好ましい範囲は、上記グリシジルエーテル化合物の含有量の好ましい範囲と同様である。3,4-エポキシ-6-メチルシクロヘキシルメチル-3,4-エポキシ-6-メチルシクロヘキサンカルボキシレート、OXBP、EHOそれぞれの含有量の好ましい範囲は、上記と同様である。
(熱重合開始剤)
熱重合開始剤は、熱により分解し、ルイス酸またはプロトン酸を発生し得る化合物である。ルイス酸またはプロトン酸の作用によってカチオン重合性化合物の重合反応が開始される。
熱重合開始剤としては、65℃以下の温度での硬化性組成物の連鎖硬化を可能とする観点から、65℃以下の温度で分解し、ルイス酸またはプロトン酸を発生し得る化合物が好ましい。このような化合物としては、例えば下記式(I-1)で表されるスルホニウム塩、下記式(I-2)で表されるスルホニウム塩等が挙げられる。これらの熱重合開始剤はいずれか1種を単独で用いてもよく2種以上を併用してもよい。
熱重合開始剤は、熱により分解し、ルイス酸またはプロトン酸を発生し得る化合物である。ルイス酸またはプロトン酸の作用によってカチオン重合性化合物の重合反応が開始される。
熱重合開始剤としては、65℃以下の温度での硬化性組成物の連鎖硬化を可能とする観点から、65℃以下の温度で分解し、ルイス酸またはプロトン酸を発生し得る化合物が好ましい。このような化合物としては、例えば下記式(I-1)で表されるスルホニウム塩、下記式(I-2)で表されるスルホニウム塩等が挙げられる。これらの熱重合開始剤はいずれか1種を単独で用いてもよく2種以上を併用してもよい。
ここで、R1は水素原子、メチル基、アセチル基(CH3CO)またはメトキシカルボニル基(CH3OCO)を示し、R2およびR3はそれぞれ独立に、水素原子、ハロゲン原子または炭素数1~4のアルキル基を示し、R4は水素原子、ハロゲン原子、ニトロ基、メチル基またはメトキシ基を示し、R5は炭素数1~4のアルキル基を示し、XはSbF6、PF6、AsF6またはBF4を示す。
熱重合開始剤としては、連鎖硬化特性の観点から、上記式(I-1)または(I-2)で表されるスルホニウム塩からなる群から選ばれる少なくとも1種が好ましい。
式(I-1)または(I-2)で表されるスルホニウム塩としては、R1が水素原子、アセチル基またはメトキシカルボニル基であり、R2およびR3がそれぞれ水素原子であり、R4が水素原子、ハロゲン原子、ニトロ基またはメチル基であり、R5がメチル基であるスルホニウム塩が好ましい。中でも、式(I-1)で表され、R1がメトキシカルボニル基であり、R2およびR3がそれぞれ水素原子であり、R4が水素原子であり、R5がメチル基であり、XがSbF6であるスルホニウム塩、すなわち下記式(I-1-1)で表されるスルホニウム塩が好ましい。
式(I-1)または(I-2)で表されるスルホニウム塩としては、R1が水素原子、アセチル基またはメトキシカルボニル基であり、R2およびR3がそれぞれ水素原子であり、R4が水素原子、ハロゲン原子、ニトロ基またはメチル基であり、R5がメチル基であるスルホニウム塩が好ましい。中でも、式(I-1)で表され、R1がメトキシカルボニル基であり、R2およびR3がそれぞれ水素原子であり、R4が水素原子であり、R5がメチル基であり、XがSbF6であるスルホニウム塩、すなわち下記式(I-1-1)で表されるスルホニウム塩が好ましい。

(保存安定剤)
保存安定剤は、熱重合開始剤の分解により発生したルイス酸またはプロトン酸を捕捉し得る化合物である。硬化性組成物が保存安定剤を含むことで、カチオン重合性化合物の重合が抑制され、硬化性組成物の可使時間が長くなる。
保存安定剤は、熱重合開始剤の分解により発生したルイス酸またはプロトン酸を捕捉し得る化合物である。硬化性組成物が保存安定剤を含むことで、カチオン重合性化合物の重合が抑制され、硬化性組成物の可使時間が長くなる。
保存安定剤としては、例えば、下記式(II)で表されるスルホニウム塩等が挙げられる。これらの保存安定剤はいずれか1種を単独で用いてもよく2種以上を併用してもよい。

保存安定剤としては、保存安定性特性の観点から、式(II)で表されるスルホニウム塩からなる群から選ばれる少なくとも1種が好ましい。
(フィラー)
フィラーは、硬化性組成物の粘度等の性状の調整、カチオン重合性化合物の割合を低下させることによる硬化時の発熱量の低減、硬化収縮抑制/寸法安定性向上、硬度向上、剛性向上等の目的で用いられる。
フィラーは、硬化性組成物の粘度等の性状の調整、カチオン重合性化合物の割合を低下させることによる硬化時の発熱量の低減、硬化収縮抑制/寸法安定性向上、硬度向上、剛性向上等の目的で用いられる。
フィラーとしては、ガラス、シリカ、セラミック、プラスチック、金属等が挙げられる。性状安定性、分散性、入手性、形状・性能の多様性、低コスト等の点で、ガラス、シリカおよびセラミックからなる群から選ばれる少なくとも1種が好ましい。
フィラーの形態としては、粉体状、繊維状、フレーク状等が挙げられ、混合や調製の容易さ、形状への馴染み易さ(隙間部分への充填性、押し潰し時の形状追従性等)の点で、粉体状が好ましい。
フィラーの形態としては、粉体状、繊維状、フレーク状等が挙げられ、混合や調製の容易さ、形状への馴染み易さ(隙間部分への充填性、押し潰し時の形状追従性等)の点で、粉体状が好ましい。
粉体状のフィラーは、粒度に分布を有していてもよい。
粉体状のフィラーの平均粒度は、200μm以下が好ましく、50μm以下がより好ましく、20μm以下がさらに好ましく、5μm以下が特に好ましい。平均粒度が上記上限値以下であると、カチオン重合性化合物等との混和性、形状への馴染み易さ等がより優れる。平均粒度の下限は特に限定されず、例えば1μm未満であってよい。平均粒度は粒度分布測定装置等により測定される。
硬化性組成物の粘度特性を制御するために、平均粒度が異なる複数の粉体状のフィラーを併用してもよい。
粉体状のフィラーの平均粒度は、200μm以下が好ましく、50μm以下がより好ましく、20μm以下がさらに好ましく、5μm以下が特に好ましい。平均粒度が上記上限値以下であると、カチオン重合性化合物等との混和性、形状への馴染み易さ等がより優れる。平均粒度の下限は特に限定されず、例えば1μm未満であってよい。平均粒度は粒度分布測定装置等により測定される。
硬化性組成物の粘度特性を制御するために、平均粒度が異なる複数の粉体状のフィラーを併用してもよい。
フィラーとしては、安価で、多様な粒度の製品が市販されており粒度を選びやすい点で、粉体状のシリカが好ましい。
(他の成分)
他の成分としては、例えば増感剤、硬化促進剤、顔料/染料、消泡剤、粘度調整剤等が挙げられる。これらの他の成分はいずれか1種を単独で用いてもよく2種以上を併用してもよい。
他の成分としては、例えば増感剤、硬化促進剤、顔料/染料、消泡剤、粘度調整剤等が挙げられる。これらの他の成分はいずれか1種を単独で用いてもよく2種以上を併用してもよい。
(各成分の含有量)
硬化性組成物中の熱重合開始剤の含有量は、カチオン重合性化合物100質量部に対して0.3~3質量部であり、0.5~2質量部が好ましく、0.75~1.5質量部が特に好ましい。熱重合開始剤の含有量は、固形分換算量である。熱重合開始剤の含有量が上記下限値以上であると、連鎖硬化が可能であり、また、硬化開始後の硬化速度が充分に速くなる。熱重合開始剤の含有量が上記上限値を超えても、硬化速度の向上効果はほとんど見られず、コストの増大を招く。
硬化性組成物中の熱重合開始剤の含有量は、カチオン重合性化合物100質量部に対して0.3~3質量部であり、0.5~2質量部が好ましく、0.75~1.5質量部が特に好ましい。熱重合開始剤の含有量は、固形分換算量である。熱重合開始剤の含有量が上記下限値以上であると、連鎖硬化が可能であり、また、硬化開始後の硬化速度が充分に速くなる。熱重合開始剤の含有量が上記上限値を超えても、硬化速度の向上効果はほとんど見られず、コストの増大を招く。
硬化性組成物中の保存安定剤の含有量は、硬化性組成物の所望の可使時間および硬化時間を考慮して適宜設定し得る。保存安定剤の含有量が多いほど、カチオン重合性化合物の重合反応の開始が遅くなり、可使時間および硬化時間が長くなる傾向がある。
例えば室温における可使時間を20分間以上とする場合には、硬化性組成物中の保存安定剤の含有量は、熱重合開始剤100質量部に対して0.3質量部以上が好ましい。
保存安定剤の含有量としては、可使時間および硬化時間のバランスから、熱重合開始剤100質量部に対して0.3~5質量部が好ましく、1~4質量部が更に好ましく、1.5~3.5質量部が特に好ましい。
例えば室温における可使時間を20分間以上とする場合には、硬化性組成物中の保存安定剤の含有量は、熱重合開始剤100質量部に対して0.3質量部以上が好ましい。
保存安定剤の含有量としては、可使時間および硬化時間のバランスから、熱重合開始剤100質量部に対して0.3~5質量部が好ましく、1~4質量部が更に好ましく、1.5~3.5質量部が特に好ましい。
硬化性組成物がフィラーを含む場合、フィラーの含有量は、硬化性組成物の総質量に対し、50質量%以上が好ましく、60質量%以上がより好ましく、70質量%以上がさらに好ましく、75質量%以上が特に好ましい。フィラーの含有量の上限は、フィラーの密度によっても異なるが、例えば硬化性組成物の総質量に対し、80質量%であってよい。
硬化性組成物中の他の成分の含有量は、例えば、硬化性組成物の総質量に対しに対し、0~20質量%であってよい。
本発明の硬化性組成物は、カチオン重合性化合物の重合反応(発熱重合反応)により発生した熱エネルギーにより連鎖硬化可能である。
「連鎖硬化」とは、自己の反応熱エネルギーをドライブフォースとする硬化形態である。硬化性組成物の一部に熱エネルギー(一次熱エネルギー)が付与されると、最初の発熱重合反応が起こり、それに伴って熱エネルギー(二次熱エネルギー)が発生する。最初の発熱重合反応により発生した熱エネルギー(二次熱エネルギー)により連続して発熱重合反応およびそれに伴う二次熱エネルギーの発生が繰り返され、硬化性組成物の硬化が連鎖的に進行し、硬化性組成物全体が硬化する。
「連鎖硬化」とは、自己の反応熱エネルギーをドライブフォースとする硬化形態である。硬化性組成物の一部に熱エネルギー(一次熱エネルギー)が付与されると、最初の発熱重合反応が起こり、それに伴って熱エネルギー(二次熱エネルギー)が発生する。最初の発熱重合反応により発生した熱エネルギー(二次熱エネルギー)により連続して発熱重合反応およびそれに伴う二次熱エネルギーの発生が繰り返され、硬化性組成物の硬化が連鎖的に進行し、硬化性組成物全体が硬化する。
硬化性組成物が連鎖硬化可能であるか否かは、以下の手順で判定できる。
硬化性組成物を長さ200mm、Φ10mmの筒状の型に充填して試料とする(例えば樹脂チューブ・樹脂ホースの類の片端に栓をした物や、試験管等を注型型として使用する)。この試料の上端部(硬化性組成物への接触が可能な箇所)の位置に、熱源として、任意の温度(一次熱エネルギー付与温度)に調整されたハンダゴテを接触させる。硬化性組成物が連鎖硬化可能であれば、熱源の接触後、発熱重合反応が生じ、その発熱重合反応により発生した二次熱エネルギーにより試料の長手方向に硬化が進行し、試料全体が硬化する。硬化している部分は、目視、長手方向の各点の温度計測等により確認できる。
硬化性組成物を長さ200mm、Φ10mmの筒状の型に充填して試料とする(例えば樹脂チューブ・樹脂ホースの類の片端に栓をした物や、試験管等を注型型として使用する)。この試料の上端部(硬化性組成物への接触が可能な箇所)の位置に、熱源として、任意の温度(一次熱エネルギー付与温度)に調整されたハンダゴテを接触させる。硬化性組成物が連鎖硬化可能であれば、熱源の接触後、発熱重合反応が生じ、その発熱重合反応により発生した二次熱エネルギーにより試料の長手方向に硬化が進行し、試料全体が硬化する。硬化している部分は、目視、長手方向の各点の温度計測等により確認できる。
本発明の硬化性組成物は、65℃以下の温度で連鎖硬化可能であることが好ましい。連鎖硬化可能な温度が65℃以下であると、シム製作の際に要求される許容温度を逸脱することなく型取りを行うことができる。
連鎖硬化可能な温度とは、連鎖硬化における最初の発熱重合反応が起こる温度、つまり連鎖硬化開始可能温度である。連鎖硬化開始後に二次熱エネルギーによって硬化性組成物の温度が、連鎖硬化可能な温度の上限を超えてもよい。
連鎖硬化開始可能温度は、45℃以下がさらに好ましい。
連鎖硬化開始可能温度の下限は特に限定されず、可使時間とのバランスを考慮して適宜選定し得る。連鎖硬化開始可能温度が高くなると可使時間が長くなる傾向がある。したがって、可使時間を確保する観点からは、連鎖硬化開始可能温度が高い方が好ましい。連鎖硬化開始可能温度は例えば0℃以上であってもよく、5℃以上であってもよく、10℃以上であってもよく、15℃以上であってもよく、20℃以上であってもよい。
連鎖硬化開始可能温度は、室温(RT)程度が特に好ましい。ここで「室温」とは、23±3℃以内の温度を示す。
連鎖硬化開始可能温度は、熱重合開始剤の種類によって調整できる。
連鎖硬化可能な温度とは、連鎖硬化における最初の発熱重合反応が起こる温度、つまり連鎖硬化開始可能温度である。連鎖硬化開始後に二次熱エネルギーによって硬化性組成物の温度が、連鎖硬化可能な温度の上限を超えてもよい。
連鎖硬化開始可能温度は、45℃以下がさらに好ましい。
連鎖硬化開始可能温度の下限は特に限定されず、可使時間とのバランスを考慮して適宜選定し得る。連鎖硬化開始可能温度が高くなると可使時間が長くなる傾向がある。したがって、可使時間を確保する観点からは、連鎖硬化開始可能温度が高い方が好ましい。連鎖硬化開始可能温度は例えば0℃以上であってもよく、5℃以上であってもよく、10℃以上であってもよく、15℃以上であってもよく、20℃以上であってもよい。
連鎖硬化開始可能温度は、室温(RT)程度が特に好ましい。ここで「室温」とは、23±3℃以内の温度を示す。
連鎖硬化開始可能温度は、熱重合開始剤の種類によって調整できる。
本発明の硬化性組成物は、室温における可使時間が20分間以上であることが好ましく、30分間以上がより好ましく、45分間以上がさらに好ましく、60分間以上が特に好ましい。
可使時間とは、材料の取扱い環境(室温)での取扱い可能なの時点までの時間を示す。
室温における可使時間が上記下限値以上であると、硬化性組成物が柔らかい間に、型取りにおける塗付作業等の作業を完了することができる。
可使時間とは、材料の取扱い環境(室温)での取扱い可能なの時点までの時間を示す。
室温における可使時間が上記下限値以上であると、硬化性組成物が柔らかい間に、型取りにおける塗付作業等の作業を完了することができる。
硬化性組成物の室温における可使時間は、20分以上が好ましく、30分以上がより好ましく、45分以上がさらに好ましく、60分以上が特に好ましい。
硬化性組成物の可使時間は、保存安定剤の含有量、重合開始剤の種類、カチオン重合性樹脂成分の種類、フィラーの添加量等により調整できる。
硬化性組成物の可使時間は、保存安定剤の含有量、重合開始剤の種類、カチオン重合性樹脂成分の種類、フィラーの添加量等により調整できる。
本発明の硬化性組成物の室温における硬化時間は、(室温における可使時間+6時間)以下が好ましく、(室温における可使時間+2時間)以下がより好ましい。例えば室温における可使時間が60分間である場合には、7時間以下が好ましく、3時間以下がより好ましい。
本発明の硬化性組成物の60℃における硬化時間は、3時間(可使時間は除く)以下が好ましく、1時間(可使時間は除く)以下がより好ましい。
本発明の硬化性組成物の60℃における硬化時間は、3時間(可使時間は除く)以下が好ましく、1時間(可使時間は除く)以下がより好ましい。
硬化時間とは、硬化性組成物に一次熱エネルギーを付与した時点から(室温硬化の場合、室温環境下に暴露した時点から)、硬化性組成物が硬化して所定の物性を有した時点までの時間を示す。硬化性組成物が硬化完了した時点とは、所定の物性を有し、硬化反応による物性変化がなくなった時点を示す。
なお、硬化性組成物を実際に硬化させる際の硬化時間は、硬化性組成物に付与する一次熱エネルギー(温度)、雰囲気温度等によって異なる。
なお、硬化性組成物を実際に硬化させる際の硬化時間は、硬化性組成物に付与する一次熱エネルギー(温度)、雰囲気温度等によって異なる。
本発明の硬化性組成物の室温における粘度は、100Pa・s以上が好ましく、1000Pa・s以上がより好ましい。構造間のギャップを型取る際、硬化性組成物(型取り材)に圧力をかけて変形させる。粘度が上記下限値以上であると、十分に型を取れる。一方で、挟み込む構造部材(アルミニウム製、チタン製、炭素繊維強化プラスチック(CFRP)製等)の部材剛性で異なってくるが、これら挟み込み構造部材が変形しない粘度であることが好ましく、例えば、5000Pa・s以下が好ましい。
上記粘度は、B型粘度計により測定される値である。なお、粘弾性測定装置によっても同様の測定値が得られる。
粘度は、フィラーの含有量、カチオン重合性樹脂成分等により調整できる。例えばフィラーの含有量が多いほど、粘度が高くなる傾向がある。
上記粘度は、B型粘度計により測定される値である。なお、粘弾性測定装置によっても同様の測定値が得られる。
粘度は、フィラーの含有量、カチオン重合性樹脂成分等により調整できる。例えばフィラーの含有量が多いほど、粘度が高くなる傾向がある。
本発明の硬化性組成物を硬化させたときの硬度、つまり硬化物の硬度は、30Hs以上が好ましく、60Hs以上がより好ましい。型取り作業または取回し作業の際、硬化物は加工または運搬され(ハンドリング)、形状測定される。硬化物の硬度が30Hs以上であると、各作業の際のハンドリングが可能となり硬化物が変形しにくい。硬化物の硬度が60Hs以上であると、大寸法の型取り作業であっても変形しにくい。硬化物の硬度の上限は特に限定されず、例えば80Hsであってよい。
硬度(ショア硬度)は、硬度計により測定される。
硬度は、フィラー充填量、カチオン重合性樹脂成分等により調整できる。例えばフィラー充填量が多いほど、硬度が高くなる傾向がある。
硬度(ショア硬度)は、硬度計により測定される。
硬度は、フィラー充填量、カチオン重合性樹脂成分等により調整できる。例えばフィラー充填量が多いほど、硬度が高くなる傾向がある。
(硬化性組成物の調製方法)
硬化性組成物は、カチオン重合性化合物と、熱重合開始剤と、保存安定剤と、必要に応じてフィラー、他の成分を混合することにより調整できる。
混合は通常、硬化性組成物の連鎖硬化可能な温度よりも低温で行われる。重合開始剤の混合は、(連鎖硬化開始可能温度-10℃)以下で行うことが好ましく、(連鎖硬化開始可能温度-15℃)以下で行うことがより好ましく、(連鎖硬化開始可能温度-20℃)以下で行うことがさらに好ましい。従って例えば硬化性組成物が30℃で連鎖硬化開始可能である場合、重合開始剤の混合温度は、5~20℃が好ましく、10~15℃がより好ましい。混合温度が上記上限値以下であると、混合時に連鎖硬化が進むことを抑制できる。混合温度が上記下限値以上であると、各材料を混合しやすい。
混合方法は特に限定されず、公知の混合方法を用いることができるが、混合方法によっては熱の発生があるため、これに注意して作業する必要がある。
各成分の混合順序は特に限定されず、全ての成分を一括で混合してもよく、順次混合してもよいが、重合開始剤が混合された以降は、重合開始剤混合時の温度(以下)を維持するようにする必要がある。以上のことから、混合の順序としては、後述の混合順番が取扱いの容易性の点で好ましい。
硬化性組成物は、カチオン重合性化合物と、熱重合開始剤と、保存安定剤と、必要に応じてフィラー、他の成分を混合することにより調整できる。
混合は通常、硬化性組成物の連鎖硬化可能な温度よりも低温で行われる。重合開始剤の混合は、(連鎖硬化開始可能温度-10℃)以下で行うことが好ましく、(連鎖硬化開始可能温度-15℃)以下で行うことがより好ましく、(連鎖硬化開始可能温度-20℃)以下で行うことがさらに好ましい。従って例えば硬化性組成物が30℃で連鎖硬化開始可能である場合、重合開始剤の混合温度は、5~20℃が好ましく、10~15℃がより好ましい。混合温度が上記上限値以下であると、混合時に連鎖硬化が進むことを抑制できる。混合温度が上記下限値以上であると、各材料を混合しやすい。
混合方法は特に限定されず、公知の混合方法を用いることができるが、混合方法によっては熱の発生があるため、これに注意して作業する必要がある。
各成分の混合順序は特に限定されず、全ての成分を一括で混合してもよく、順次混合してもよいが、重合開始剤が混合された以降は、重合開始剤混合時の温度(以下)を維持するようにする必要がある。以上のことから、混合の順序としては、後述の混合順番が取扱いの容易性の点で好ましい。
硬化性組成物がフィラーを含む場合、熱重合開始剤と保存安定剤とを混合して第一混合物とし、カチオン重合性化合物とフィラーとを混合して第二混合物とし、第一混合物と第二混合物とを混合することが好ましい。フィラーを配合すると高粘度になり、混合時に熱重合開始剤等の濃度のムラが生じやすい。局所的に熱重合開始剤に対して保存安定剤が少ない部分が生じると、その部分で重合反応が開始するおそれがある。上記の順序で混合することで、局所的に熱重合開始剤に対して保存安定剤が少ない領域が生じることを抑制できる。
調製された硬化性組成物は、典型的には、ペースト状である。ペースト状の硬化性組成物は、硬化性ペースト材として用いることができる。
調製された硬化性組成物を、シート状に成形してもよい。シート状の硬化性組成物は、硬化性シート材として用いることができる。
シート状の硬化性組成物(硬化性組成物からなる層)の片面または両面に樹脂フィルム等のフィルム材が積層されていてもよい。シート状の硬化性組成物の片面または両面にフィルム材が積層された積層体も、硬化性シート材として用いることができる。
シート状の硬化性組成物(硬化性組成物からなる層)の片面または両面に樹脂フィルム等のフィルム材が積層されていてもよい。シート状の硬化性組成物の片面または両面にフィルム材が積層された積層体も、硬化性シート材として用いることができる。
硬化性組成物、硬化性ペースト材または硬化性シート材を製造後、硬化させる前に保管期間を設ける場合、保管方法としては冷凍保管が好ましい。
冷凍保管における保管温度は、-15℃以下が好ましく、-40℃以下がより好ましく、-55℃以下が特に好ましい。保管温度の下限は特に限定されず、例えば-65℃以下であってよい。
冷凍保管における保管温度は、-15℃以下が好ましく、-40℃以下がより好ましく、-55℃以下が特に好ましい。保管温度の下限は特に限定されず、例えば-65℃以下であってよい。
本発明の硬化性組成物にあっては、カチオン重合性化合物と、熱重合開始剤と、保存安定剤とを含み、カチオン重合性化合物が、グリシジルエーテル化合物、脂環式エポキシ化合物、およびオキセタン化合物からなる群から選ばれる少なくとも2種を含み、熱重合開始剤の含有量がカチオン重合性化合物100質量部に対して0.3~3質量部であり、カチオン重合性化合物の重合反応により発生した熱エネルギーにより連鎖硬化可能であるため、十分な長さの可使時間を確保できる。また、可使時間経過後に65℃以下の温度でも迅速に硬化可能である。
十分な長さの可使時間を確保できる理由は主に以下によるものである。すなわち、本発明の硬化性組成物が作業環境または取回し環境に置かれると(可使時間の環境温度下)、僅かではあっても熱重合開始剤が分解し、ルイス酸またはプロトン酸が発生する。可使時間内において発生したルイス酸またはプロトン酸の多くは保存安定剤により捕捉され、発熱重合反応は生じないか生じてもわずかであり、連鎖硬化は開始しない。加えて、僅かに生じた発熱重合反応による発生熱エネルギーでさえも、その何割かを安定性が高いグリシジルエーテル型化合物等が吸収することでさらに連鎖硬化は開始しない。特にフィラーを含む場合にはこの効果が高い。保存安定剤はルイス酸またはプロトン酸の捕捉により消費されるため、発生したルイス酸またはプロトン酸の量が保存安定剤の量を上回ると、カチオン重合性化合物の発熱重合反応によって、連鎖硬化を進めるのに十分な二次熱エネルギーが発生する。この二次熱エネルギーにより連鎖硬化が開始し、硬化性組成物の全体が硬化する。
本発明の硬化性組成物の用途は特に限定されず、例えば硬化性型取り材、硬化性シート材、硬化性ペースト材、ポッティング材、注型材、接着剤、塗料、コーティング剤等に用いることができる。
構造部材間のギャップ、例えば航空機の主翼とエンジンナセルとの間の隙間、に配置されるシムの作製に用いられる硬化性型取り材には、ある程度の可使時間を確保しつつ硬化時間を短くすることが望まれる。また、以下の条件1~7を満たすことが望まれることがある。本発明の硬化性組成物は、これらの条件を満たし得る。したがって、本発明の硬化性組成物の用途としては、構造部材間のギャップに配置されるシムを作製するための硬化性型取り材が好適である。
構造部材間のギャップ、例えば航空機の主翼とエンジンナセルとの間の隙間、に配置されるシムの作製に用いられる硬化性型取り材には、ある程度の可使時間を確保しつつ硬化時間を短くすることが望まれる。また、以下の条件1~7を満たすことが望まれることがある。本発明の硬化性組成物は、これらの条件を満たし得る。したがって、本発明の硬化性組成物の用途としては、構造部材間のギャップに配置されるシムを作製するための硬化性型取り材が好適である。
条件1:硬化時の反応熱による物温抑制
構造部材の表面温度が、型取り材の硬化時の反応熱によって、許容温度を逸脱しないことが求められる。シール材が塗布されているシール接着面の許容温度は典型的には60℃未満であり、プライマーが塗布されているプライマー塗布面の許容温度は典型的には93℃未満である。
条件2:硬化樹脂による穴明けドリルへの影響
シムは正寸穴を有した状態で製作されることが多い。この場合、型取り材は、ある程度硬化した段階で、構造部材と共に穴明け加工される。型取り材は、穴明け時にドリルの刃に影響を及ぼさず、加工精度を落とさないことが求められる。
条件3:硬化物が変形しない硬度
硬化後の型取り材は、直接運搬され、FARO ARMにより形状測定されるため、各作業で形状が変形することなく保持されることが求められる。
条件4:低温領域における硬化時間の短縮
一例としては、条件1を満たす温度条件下で、1時間程度の可使時間があり、硬化開始から1時間後に、穴あけが出来る程度(例えば硬度30Hs以上)に硬化し、硬化開始から6時間後に完全硬化し硬度60Hs以上になること、が望まれる。
条件5:型取り時に構造部材が変形しない粘性
構造部材間のギャップを型取る際、型取り材にある一定の圧力がかかる。その際、挟み込む構造部材が変形しない程度の粘性を有することが求められる。同時に、型を取れるだけの十分な粘性も併せ持つことが求められる。
条件6:材料コスト
シムの生産コストの要求より、型取り材の材料コストはなるべく低いことが求められる。
条件7:人体への影響
型取り作業は作業者が直接扱う為、安全上の観点から人体へ影響を及ぼす材料を含まないことが求められる。
構造部材の表面温度が、型取り材の硬化時の反応熱によって、許容温度を逸脱しないことが求められる。シール材が塗布されているシール接着面の許容温度は典型的には60℃未満であり、プライマーが塗布されているプライマー塗布面の許容温度は典型的には93℃未満である。
条件2:硬化樹脂による穴明けドリルへの影響
シムは正寸穴を有した状態で製作されることが多い。この場合、型取り材は、ある程度硬化した段階で、構造部材と共に穴明け加工される。型取り材は、穴明け時にドリルの刃に影響を及ぼさず、加工精度を落とさないことが求められる。
条件3:硬化物が変形しない硬度
硬化後の型取り材は、直接運搬され、FARO ARMにより形状測定されるため、各作業で形状が変形することなく保持されることが求められる。
条件4:低温領域における硬化時間の短縮
一例としては、条件1を満たす温度条件下で、1時間程度の可使時間があり、硬化開始から1時間後に、穴あけが出来る程度(例えば硬度30Hs以上)に硬化し、硬化開始から6時間後に完全硬化し硬度60Hs以上になること、が望まれる。
条件5:型取り時に構造部材が変形しない粘性
構造部材間のギャップを型取る際、型取り材にある一定の圧力がかかる。その際、挟み込む構造部材が変形しない程度の粘性を有することが求められる。同時に、型を取れるだけの十分な粘性も併せ持つことが求められる。
条件6:材料コスト
シムの生産コストの要求より、型取り材の材料コストはなるべく低いことが求められる。
条件7:人体への影響
型取り作業は作業者が直接扱う為、安全上の観点から人体へ影響を及ぼす材料を含まないことが求められる。
〔硬化性ペースト材〕
本発明の硬化性ペースト材は、上記本発明の硬化性組成物を含む。
本発明の硬化性ペースト材は、本発明の硬化性組成物と同様の用途に用いることができる。
本発明の硬化性ペースト材は、上記本発明の硬化性組成物を含む。
本発明の硬化性ペースト材は、本発明の硬化性組成物と同様の用途に用いることができる。
〔硬化性シート材〕
本発明の硬化性シート材は、上記本発明の硬化性組成物を含む。
硬化性シート材の例としては、本発明の硬化性組成物からなるシート、本発明の硬化性組成物からなる層の片面または両面に樹脂フィルム等のフィルム基材が積層されたシート等が挙げられる。
本発明の硬化性シート材は、本発明の硬化性組成物と同様の用途に用いることができる。
本発明の硬化性シート材は、上記本発明の硬化性組成物を含む。
硬化性シート材の例としては、本発明の硬化性組成物からなるシート、本発明の硬化性組成物からなる層の片面または両面に樹脂フィルム等のフィルム基材が積層されたシート等が挙げられる。
本発明の硬化性シート材は、本発明の硬化性組成物と同様の用途に用いることができる。
〔硬化性型取り材〕
本発明の硬化性型取り材は、上記本発明の硬化性組成物を含む。
本発明の硬化性型取り材の形態は特に限定されず、例えばペースト状、シート状等であってよい。上述の本発明の硬化性ペースト材をペースト状の硬化性型取り材として用いることができる。上述の本発明の硬化性シート材をシート状の硬化性型取り材として用いることができる。
本発明の硬化性型取り材によって型取りをする対象としては、構造部材間のギャップ、歯型、ブッシュ隙間、封孔または穴埋め等が挙げられ、構造部材間のギャップが好ましい。
本発明の硬化性型取り材は、上記本発明の硬化性組成物を含む。
本発明の硬化性型取り材の形態は特に限定されず、例えばペースト状、シート状等であってよい。上述の本発明の硬化性ペースト材をペースト状の硬化性型取り材として用いることができる。上述の本発明の硬化性シート材をシート状の硬化性型取り材として用いることができる。
本発明の硬化性型取り材によって型取りをする対象としては、構造部材間のギャップ、歯型、ブッシュ隙間、封孔または穴埋め等が挙げられ、構造部材間のギャップが好ましい。
〔硬化方法、硬化物〕
上述の硬化性組成物、硬化性ペースト材、硬化性シート材または型取り材(以下、これらをまとめて「硬化性組成物等」とも記す。)は、硬化させて硬化物とすることができる。硬化させる前に硬化性組成物等に任意の形状を付与しておくと、任意の形状を有する硬化物が得られる。
上述の硬化性組成物、硬化性ペースト材、硬化性シート材または型取り材(以下、これらをまとめて「硬化性組成物等」とも記す。)は、硬化させて硬化物とすることができる。硬化させる前に硬化性組成物等に任意の形状を付与しておくと、任意の形状を有する硬化物が得られる。
硬化性組成物等は、硬化性組成物等の一部に熱エネルギー(一次熱エネルギー)を付与することにより硬化させることができる。
熱エネルギーの付与方法としては、特に限定されず、例えば硬化性組成物等を、連鎖硬化可能な温度条件下に置く方法、硬化性組成物等の一部を、加熱手段を用いて、連鎖硬化可能な温度に加熱する方法等が挙げられる。加熱手段としては、ドライヤー、棒状ヒーター、ヒータ―ブランケット、赤外線ヒーター、オーブン等が挙げられる。
熱エネルギーの付与方法としては、特に限定されず、例えば硬化性組成物等を、連鎖硬化可能な温度条件下に置く方法、硬化性組成物等の一部を、加熱手段を用いて、連鎖硬化可能な温度に加熱する方法等が挙げられる。加熱手段としては、ドライヤー、棒状ヒーター、ヒータ―ブランケット、赤外線ヒーター、オーブン等が挙げられる。
好ましい連鎖硬化可能な温度は上記のとおりである。したがって、硬化性組成物等を硬化させる際の温度としては、65℃以下が好ましく、45℃以下がさらにが好ましく、室温程度(25±3℃以内)が特に好ましい。
連鎖硬化が開始した後、完全硬化する前の硬化性組成物等に、穴明け加工、等の加工を施してもよい。
加工を施す際の硬化性組成物等の硬度は、30Hs以上が好ましく、60Hs以上がさらに好ましい。
加工を施す際の硬化性組成物等の硬度は、30Hs以上が好ましく、60Hs以上がさらに好ましい。
硬化物の用途は特に限定されない。例えばシム作製のための型、ブッシュ作製のための型、封孔材または穴埋め材およびこれらを作製する際の型、注型材、接着剤用途、塗料用途、コーティング剤用途等に用いることができる。
シム作製のための型の形状の一例は、最大長さ約2m、幅約0.2m、板厚約0.2~3mmの形状である。
シム作製のための型の形状の一例は、最大長さ約2m、幅約0.2m、板厚約0.2~3mmの形状である。
以下、実施例を示して本発明をさらに詳しく説明する。ただし本発明はそれらの実施例によって何ら限定されるものではない。「部」は「質量部」を示す。
各例で用いた材料を以下に示す。
各例で用いた材料を以下に示す。
(使用材料)
828:三菱ケミカル社製「jER(登録商標) 828」、ビスフェノールA型ジグリシジルエーテル化合物。
2021P:ダイセル社製「セロキサイド(登録商標) 2021P」、3,4-エポキシシクロヘキシルメチル-3,4-エポキシシクロヘキサンカルボキシレート。
OXBP:宇部興産社製「ETERNACOLL(登録商標) OXBP」、4,4’-ビス[(3-エチル-3-オキセタニル)メトキシメチル]ビフェニル。
EHO:宇部興産社製「ETERNACOLL EHO」、3-エチル-3-ヒドロキシメチルオキセタン。
828:三菱ケミカル社製「jER(登録商標) 828」、ビスフェノールA型ジグリシジルエーテル化合物。
2021P:ダイセル社製「セロキサイド(登録商標) 2021P」、3,4-エポキシシクロヘキシルメチル-3,4-エポキシシクロヘキサンカルボキシレート。
OXBP:宇部興産社製「ETERNACOLL(登録商標) OXBP」、4,4’-ビス[(3-エチル-3-オキセタニル)メトキシメチル]ビフェニル。
EHO:宇部興産社製「ETERNACOLL EHO」、3-エチル-3-ヒドロキシメチルオキセタン。
SI-60:熱重合開始剤、三新化学工業社製「サンエイドSI-60」、上記式(I-2)で表されるスルホニウム塩。
SI-45:熱重合開始剤、三新化学工業社製「サンエイドSI-45」、上記式(I-1-1)で表されるスルホニウム塩。
SI-B2A:熱重合開始剤、三新化学工業社製「サンエイドSI-B2A」。
UVI-6974:光重合開始剤、ダウ・ケミカル社製「UVI-6974」、ジフェニル[4-(フェニルチオ)フェニル]スルホニウム ヘキサフルオロアンチモナート(V)。
保存安定剤:三新化学工業社製「SI-助剤」、上記式(II)で表される化合物。
SI-60、SI-45、SI-B2A、保存安定剤はそれぞれ、50質量%溶液(溶媒:γ-ブチロラクトン)にして用い、UVI-6974は50質量%溶液(溶媒:プロピレンカーボネート)のものを用いた。
SI-45:熱重合開始剤、三新化学工業社製「サンエイドSI-45」、上記式(I-1-1)で表されるスルホニウム塩。
SI-B2A:熱重合開始剤、三新化学工業社製「サンエイドSI-B2A」。
UVI-6974:光重合開始剤、ダウ・ケミカル社製「UVI-6974」、ジフェニル[4-(フェニルチオ)フェニル]スルホニウム ヘキサフルオロアンチモナート(V)。
保存安定剤:三新化学工業社製「SI-助剤」、上記式(II)で表される化合物。
SI-60、SI-45、SI-B2A、保存安定剤はそれぞれ、50質量%溶液(溶媒:γ-ブチロラクトン)にして用い、UVI-6974は50質量%溶液(溶媒:プロピレンカーボネート)のものを用いた。
EXV-5:商品名、レーザーネット社販売、シリカフィラー。
URE-FIL9:商品名、シリカフィラー(ヒュームドシリカ(ナノ粒子))。
FB-5D:商品名、デンカ社、シリカフィラー、粉体状、平均粒度5μm。
FB-20D:商品名、デンカ社、シリカフィラー、粉体状、平均粒度20μm。
FB-950:商品名、デンカ社、シリカフィラー。
URE-FIL9:商品名、シリカフィラー(ヒュームドシリカ(ナノ粒子))。
FB-5D:商品名、デンカ社、シリカフィラー、粉体状、平均粒度5μm。
FB-20D:商品名、デンカ社、シリカフィラー、粉体状、平均粒度20μm。
FB-950:商品名、デンカ社、シリカフィラー。
(実施例1~25、比較例1~2)
SI-45、SI-助剤およびSI-60を(SI-60を使用しない組成の場合には除外)、表1~3の互いの混合比率と同比率(例えば其々2倍量ずつ)となるようにそれぞれの固形分を遮光性の容器に量り取り、これに量り取った固形分の全量と同量のγ-ブチロラクトンを投入して固形分を溶かし、保存安定剤入り重合開始剤溶液を作製した。
別の遮光性容器にSI-B2Aを10g量り取り、これにγ-ブチロラクトンを10g投入して固形分を溶かし、SI-B2A の50質量%溶液を作製した。UVI-6974は最初からプロピレンカーボネートの50質量%溶液であるので使用する際はこのまま使用した。遮光性容器に表1に示す各重合開始剤の固形分の互いの混合比率と同比率(例えば其々2倍量ずつ)を投入し、重合開始剤の混合液を作製した(混合液を用いなくても良い場合はこの工程は不要)。
表1~3に示す各主剤成分を所定量混合して主剤混合物を作製した。
この主剤混合物に、表1~3に示す各重合開始剤および保存安定剤の所定量となる様に重合開始剤の混合液(または保存安定剤入り重合開始剤溶液)を計量・投入した。重合開始剤投入後、素早く1分間程度 攪拌・混合して硬化性組成物を得た。
得られた硬化性組成物について、下記の手順で、可使時間、硬化完了時間を測定した。結果を表1~3に示す。
なお、表1~3中の「主剤」はカチオン重合性化合物を示し、主剤、フィラー、熱重合開始剤、保存安定剤等の量(部)は固形分量である。
SI-45、SI-助剤およびSI-60を(SI-60を使用しない組成の場合には除外)、表1~3の互いの混合比率と同比率(例えば其々2倍量ずつ)となるようにそれぞれの固形分を遮光性の容器に量り取り、これに量り取った固形分の全量と同量のγ-ブチロラクトンを投入して固形分を溶かし、保存安定剤入り重合開始剤溶液を作製した。
別の遮光性容器にSI-B2Aを10g量り取り、これにγ-ブチロラクトンを10g投入して固形分を溶かし、SI-B2A の50質量%溶液を作製した。UVI-6974は最初からプロピレンカーボネートの50質量%溶液であるので使用する際はこのまま使用した。遮光性容器に表1に示す各重合開始剤の固形分の互いの混合比率と同比率(例えば其々2倍量ずつ)を投入し、重合開始剤の混合液を作製した(混合液を用いなくても良い場合はこの工程は不要)。
表1~3に示す各主剤成分を所定量混合して主剤混合物を作製した。
この主剤混合物に、表1~3に示す各重合開始剤および保存安定剤の所定量となる様に重合開始剤の混合液(または保存安定剤入り重合開始剤溶液)を計量・投入した。重合開始剤投入後、素早く1分間程度 攪拌・混合して硬化性組成物を得た。
得られた硬化性組成物について、下記の手順で、可使時間、硬化完了時間を測定した。結果を表1~3に示す。
なお、表1~3中の「主剤」はカチオン重合性化合物を示し、主剤、フィラー、熱重合開始剤、保存安定剤等の量(部)は固形分量である。
<可使時間>
硬化性組成物の可使時間は、作製した硬化性組成物を室温環境に静置して、その間の組成物の温度を計測し、連鎖硬化によって樹脂温度が上昇し始めた時間により求めた。
硬化性組成物の可使時間は、作製した硬化性組成物を室温環境に静置して、その間の組成物の温度を計測し、連鎖硬化によって樹脂温度が上昇し始めた時間により求めた。
<硬化完了時間>
硬化性組成物の硬化完了時間は、作製した硬化性組成物を室温環境に静置して、その間の組成物の温度を計測し、樹脂温度が45℃を超えて温度上昇のピークを示した時間として求めた。
硬化性組成物の硬化完了時間は、作製した硬化性組成物を室温環境に静置して、その間の組成物の温度を計測し、樹脂温度が45℃を超えて温度上昇のピークを示した時間として求めた。
(実施例26~31)
SI-45とSI-助剤を、表3の互いの混合比率と同比率(例えば其々2倍量ずつ)となるようにそれぞれの固形分を遮光性の容器に量り取り、これに量り取った固形分の全量と同量のγ-ブチロラクトンを投入して固形分を溶かし、保存安定剤入り重合開始剤溶液を作製した。
表3に示す各主剤成分を所定量混合して主剤混合物を作製し、これに表3に示す所定量のフィラーを投入し、混合および混練した。一様なペースト混合物となったら、これを15℃以下になるまで冷却した。
15℃以下となったペースト混合物に、表3に示す各重合開始剤および保存安定剤の所定量となる様に保存安定剤入り重合開始剤溶液を計量および投入し、投入後、一様なるように混練し、ペースト状の硬化性組成物を得た。
得られた硬化性組成物について、実施例1と同様にして、可使時間、硬化完了時間を測定した。結果を表3に示す。
SI-45とSI-助剤を、表3の互いの混合比率と同比率(例えば其々2倍量ずつ)となるようにそれぞれの固形分を遮光性の容器に量り取り、これに量り取った固形分の全量と同量のγ-ブチロラクトンを投入して固形分を溶かし、保存安定剤入り重合開始剤溶液を作製した。
表3に示す各主剤成分を所定量混合して主剤混合物を作製し、これに表3に示す所定量のフィラーを投入し、混合および混練した。一様なペースト混合物となったら、これを15℃以下になるまで冷却した。
15℃以下となったペースト混合物に、表3に示す各重合開始剤および保存安定剤の所定量となる様に保存安定剤入り重合開始剤溶液を計量および投入し、投入後、一様なるように混練し、ペースト状の硬化性組成物を得た。
得られた硬化性組成物について、実施例1と同様にして、可使時間、硬化完了時間を測定した。結果を表3に示す。
図1に、実施例3~25の硬化性組成物の加温を開始した時点からの経過時間を横軸に、硬化性組成物の温度を縦軸にとったグラフを示す。
図2に、実施例26~31の硬化性組成物の加温を開始した時点からの経過時間を横軸に、硬化性組成物の温度を縦軸にとったグラフを示す。
図2に、実施例26~31の硬化性組成物の加温を開始した時点からの経過時間を横軸に、硬化性組成物の温度を縦軸にとったグラフを示す。

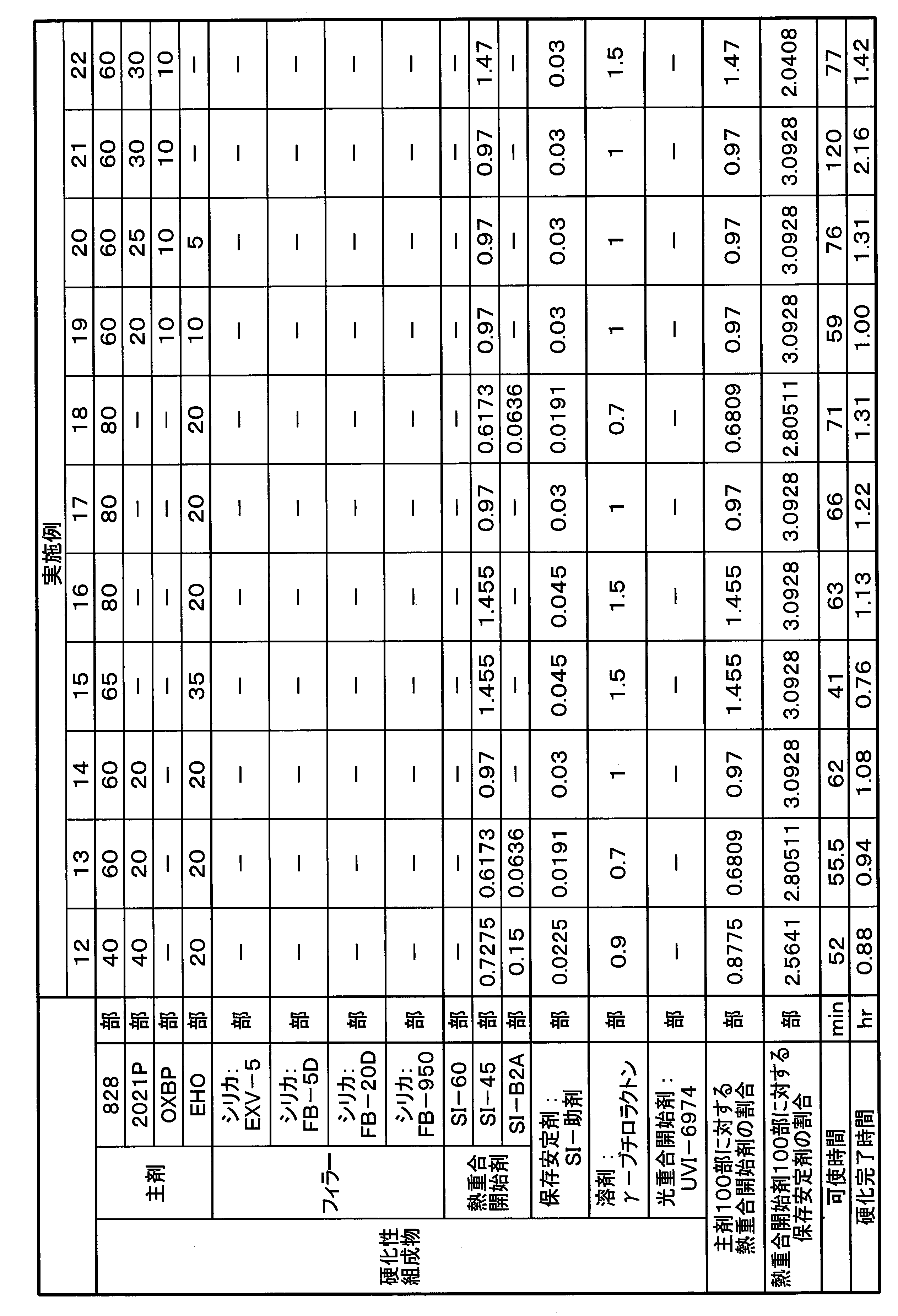
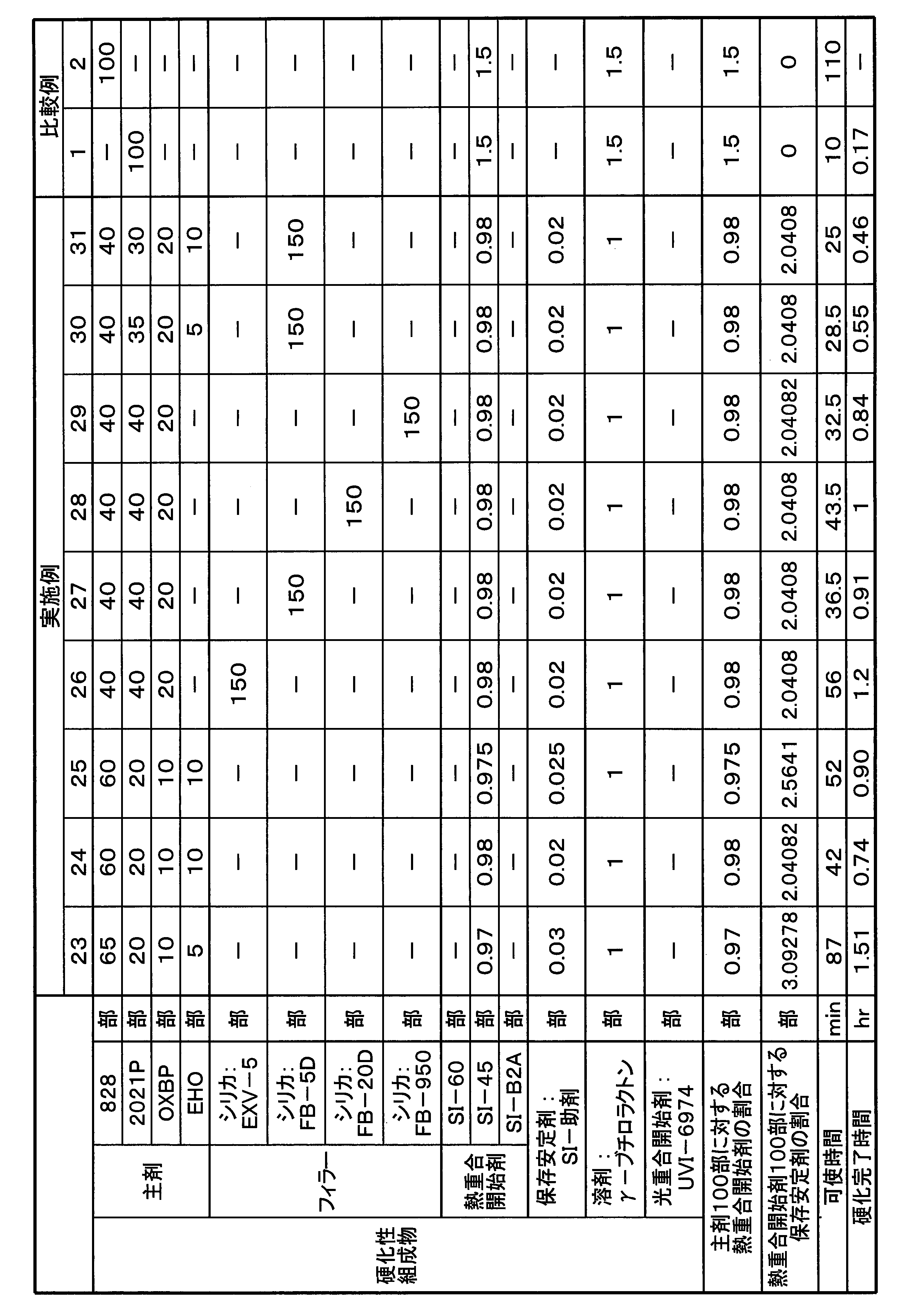
実施例1~31の硬化性組成物は、20分間以上の可使時間を有し、かつ2.2時間以内に硬化が完了した。
カチオン性重合性化合物として脂環式エポキシ化合物のみを含み、保存安定剤を含まない比較例1の硬化性組成物は、可使時間が20分間に満たなかった。
カチオン性重合性化合物としてグリシジルエーテル化合物のみを含み、保存安定剤を含まない比較例2の硬化性組成物は、8時間以内に硬化が完了しなかった。
カチオン性重合性化合物として脂環式エポキシ化合物のみを含み、保存安定剤を含まない比較例1の硬化性組成物は、可使時間が20分間に満たなかった。
カチオン性重合性化合物としてグリシジルエーテル化合物のみを含み、保存安定剤を含まない比較例2の硬化性組成物は、8時間以内に硬化が完了しなかった。
図1と図2の対比から、フィラーを含むことで、硬化性組成物の発熱を抑制できることが確認できた。
(実施例32~50、比較例3~7)
SI-45とSI-助剤を、表4の互いの混合比率と同比率(例えば其々2倍量ずつ)となるようにそれぞれの固形分を遮光性の容器に量り取り、これに量り取った固形分の全量と同量のγ-ブチロラクトンを投入して固形分を溶かし、保存安定剤入り重合開始剤溶液を作製した。
別の遮光性容器にSI-B2Aを10g量り取り、これにγ-ブチロラクトンを10g投入して固形分を溶かし、SI-B2A の50質量%溶液を作製した。UVI-6974は最初からプロピレンカーボネートの50質量%溶液であるので使用する際はこのまま使用した。遮光性容器に表4に示す各重合開始剤の固形分の互いの混合比率と同比率(例えば其々2倍量ずつ)を投入し、重合開始剤の混合液を作製した(混合液を用いなくても良い場合はこの工程は不要)。
表4に示す各主剤成分を所定量混合して主剤混合物を作製し、これに表4に示す所定量のフィラーを投入し、混合および混練した。一様なペースト混合物となったら、これを15℃以下になるまで冷却した。
15℃以下となったペースト混合物に、表4に示す各重合開始剤および保存安定剤の所定量となる様に重合開始剤の混合液(または保存安定剤入り重合開始剤溶液)を計量および投入し、投入後、一様なるように混練し、ペースト状の硬化性組成物を得た。
SI-45とSI-助剤を、表4の互いの混合比率と同比率(例えば其々2倍量ずつ)となるようにそれぞれの固形分を遮光性の容器に量り取り、これに量り取った固形分の全量と同量のγ-ブチロラクトンを投入して固形分を溶かし、保存安定剤入り重合開始剤溶液を作製した。
別の遮光性容器にSI-B2Aを10g量り取り、これにγ-ブチロラクトンを10g投入して固形分を溶かし、SI-B2A の50質量%溶液を作製した。UVI-6974は最初からプロピレンカーボネートの50質量%溶液であるので使用する際はこのまま使用した。遮光性容器に表4に示す各重合開始剤の固形分の互いの混合比率と同比率(例えば其々2倍量ずつ)を投入し、重合開始剤の混合液を作製した(混合液を用いなくても良い場合はこの工程は不要)。
表4に示す各主剤成分を所定量混合して主剤混合物を作製し、これに表4に示す所定量のフィラーを投入し、混合および混練した。一様なペースト混合物となったら、これを15℃以下になるまで冷却した。
15℃以下となったペースト混合物に、表4に示す各重合開始剤および保存安定剤の所定量となる様に重合開始剤の混合液(または保存安定剤入り重合開始剤溶液)を計量および投入し、投入後、一様なるように混練し、ペースト状の硬化性組成物を得た。
図3~5を用いて、硬化性組成物の評価に用いた適用模擬試験を説明する。図3は適用模擬試験における評価試料を説明する模式上面図であり、図4はその側面図である。図5は、適用模擬試験の実施状況を説明する模式図である。試験の概要は以下のとおりである。
CFRPプレート1およびTiプレート3の2種類の材質の異なるプレートと、60℃に加温可能な棒状先端部11aを有するヒータ11とを用意した。CFRPプレート1およびTiプレート3はそれぞれ長径700mm、短径300mmの楕円形状である。Tiプレート3の長径方向の中央から一端までの範囲内には、ヒータ11の棒状先端部11aを挿入するための貫通孔3a(直径15mm)が5つ形成されている。5つの貫通孔3aは、上面視で四角形の4つの頂点および中心の位置に形成されている。
この2枚のプレートの間に、各プレートとの接触面に樹脂フィルム5を配した状態で、上記で得られたペースト状の硬化性組成物を塗布して挟み、スペーサ9(厚さ3.0mm)によって隙間を維持したままクランプして固定した。これにより、CFRPプレート1と樹脂フィルム5と硬化性組成物の層7(厚さ3.0mm)と樹脂フィルム5とTiプレート3とがこの順に積層した評価試料を得た。スペーサ9は、Tiプレート3の長径方向の一端から中央までの範囲内に、5つの貫通孔3aを囲むように4箇所に配置した。また、各スペーサ9は、CFRPプレート1と、CFRPプレート1側の樹脂フィルム5との間に挿入した。クランプ位置は、4箇所のスペーサ9それぞれの近傍(図3中の×印の位置)とした。
クランプ固定後に、ヒータ11の棒状先端部11aを60℃に加温および保持し、評価試料のTiプレート3の貫通孔3aに挿し入れて1時間固定し、その後ヒータ11は取り外した。但し、一部の試験については最初からヒータ11を取り外した形態で試験を実施した。評価試料の硬化性組成物の層7のうち、挿入した棒状先端部11aとの接触箇所を強制硬化部位、ヒータ11の影響のないヒータ11から離れた箇所(例えば、図3の楕円右側の端部側(長軸右端部側)で硬化性積層物7の端部)を自然硬化部位として下記評価を行った。結果を表5に示す。
CFRPプレート1およびTiプレート3の2種類の材質の異なるプレートと、60℃に加温可能な棒状先端部11aを有するヒータ11とを用意した。CFRPプレート1およびTiプレート3はそれぞれ長径700mm、短径300mmの楕円形状である。Tiプレート3の長径方向の中央から一端までの範囲内には、ヒータ11の棒状先端部11aを挿入するための貫通孔3a(直径15mm)が5つ形成されている。5つの貫通孔3aは、上面視で四角形の4つの頂点および中心の位置に形成されている。
この2枚のプレートの間に、各プレートとの接触面に樹脂フィルム5を配した状態で、上記で得られたペースト状の硬化性組成物を塗布して挟み、スペーサ9(厚さ3.0mm)によって隙間を維持したままクランプして固定した。これにより、CFRPプレート1と樹脂フィルム5と硬化性組成物の層7(厚さ3.0mm)と樹脂フィルム5とTiプレート3とがこの順に積層した評価試料を得た。スペーサ9は、Tiプレート3の長径方向の一端から中央までの範囲内に、5つの貫通孔3aを囲むように4箇所に配置した。また、各スペーサ9は、CFRPプレート1と、CFRPプレート1側の樹脂フィルム5との間に挿入した。クランプ位置は、4箇所のスペーサ9それぞれの近傍(図3中の×印の位置)とした。
クランプ固定後に、ヒータ11の棒状先端部11aを60℃に加温および保持し、評価試料のTiプレート3の貫通孔3aに挿し入れて1時間固定し、その後ヒータ11は取り外した。但し、一部の試験については最初からヒータ11を取り外した形態で試験を実施した。評価試料の硬化性組成物の層7のうち、挿入した棒状先端部11aとの接触箇所を強制硬化部位、ヒータ11の影響のないヒータ11から離れた箇所(例えば、図3の楕円右側の端部側(長軸右端部側)で硬化性積層物7の端部)を自然硬化部位として下記評価を行った。結果を表5に示す。
<自然硬化>
図3~4の様にしてペースト状の硬化性組成物をプレート1,3の間に挟み、厚さ3mmの硬化性組成物の層7を形成した。これらの試料を図5の様にして上述した条件で硬化させ、ヒータ11の影響のないヒータ11から離れた箇所を自然硬化部位とした。硬化が完了するまでの間、硬化性組成物の温度および硬度(ショア硬度)を測定した。硬度(ショア硬度)は、硬度計により測定し、硬化開始から6時間後の硬度を以下の基準で判定した。
◎(特に良好):3時間以内に硬度が60Hs以上。
○(良好):硬度が60Hs以上。
△(不良):硬度が30Hs以上60Hs未満。
×(不可):硬度が30Hs未満。
図3~4の様にしてペースト状の硬化性組成物をプレート1,3の間に挟み、厚さ3mmの硬化性組成物の層7を形成した。これらの試料を図5の様にして上述した条件で硬化させ、ヒータ11の影響のないヒータ11から離れた箇所を自然硬化部位とした。硬化が完了するまでの間、硬化性組成物の温度および硬度(ショア硬度)を測定した。硬度(ショア硬度)は、硬度計により測定し、硬化開始から6時間後の硬度を以下の基準で判定した。
◎(特に良好):3時間以内に硬度が60Hs以上。
○(良好):硬度が60Hs以上。
△(不良):硬度が30Hs以上60Hs未満。
×(不可):硬度が30Hs未満。
<強制硬化>
図3~4の様にしてペースト状の硬化性組成物7をプレート1,3の間に挟み、厚さ3mmの硬化性組成物の層を形成した。これらの試料を図5の様にして上述した条件で硬化させ、ヒータ11の棒状先端部(60℃保持)が接触している箇所を強制硬化部位とした。1時間後にヒータを取り外し、強制硬化部位の硬度(ショア硬度)を硬度計により測定し、硬化開始から1時間後の硬度を以下の基準で判定した。
◎(特に良好):硬度が60Hs以上。
○(良好):硬度が30Hs以上60Hs未満。
△(不良):硬度が15Hs以上30Hs未満。
×(不可):硬度が15Hs未満。
図3~4の様にしてペースト状の硬化性組成物7をプレート1,3の間に挟み、厚さ3mmの硬化性組成物の層を形成した。これらの試料を図5の様にして上述した条件で硬化させ、ヒータ11の棒状先端部(60℃保持)が接触している箇所を強制硬化部位とした。1時間後にヒータを取り外し、強制硬化部位の硬度(ショア硬度)を硬度計により測定し、硬化開始から1時間後の硬度を以下の基準で判定した。
◎(特に良好):硬度が60Hs以上。
○(良好):硬度が30Hs以上60Hs未満。
△(不良):硬度が15Hs以上30Hs未満。
×(不可):硬度が15Hs未満。
<到達温度(自然/強制)>
計測した樹脂温度のピーク値から以下の基準で判定した。
◎(特に良好):60℃以下。
○(良好):60℃を超え93℃以下(30分以内)。
△(不良):60℃を超え93℃以下(30分以上)。
×(不可):93℃を超過。
計測した樹脂温度のピーク値から以下の基準で判定した。
◎(特に良好):60℃以下。
○(良好):60℃を超え93℃以下(30分以内)。
△(不良):60℃を超え93℃以下(30分以上)。
×(不可):93℃を超過。
<可使時間>
組成物の温度を計測し、樹脂温度が45℃を超えた時間またはピーク温度示した時間の短い方の時間を硬化物の可使時間として、以下の基準で判定した。
◎(特に良好):45分間以上。
○(良好):20分間以上45分間未満。
△(不良):15分間以上20分間未満。
×(不可):15分間未満。
組成物の温度を計測し、樹脂温度が45℃を超えた時間またはピーク温度示した時間の短い方の時間を硬化物の可使時間として、以下の基準で判定した。
◎(特に良好):45分間以上。
○(良好):20分間以上45分間未満。
△(不良):15分間以上20分間未満。
×(不可):15分間未満。
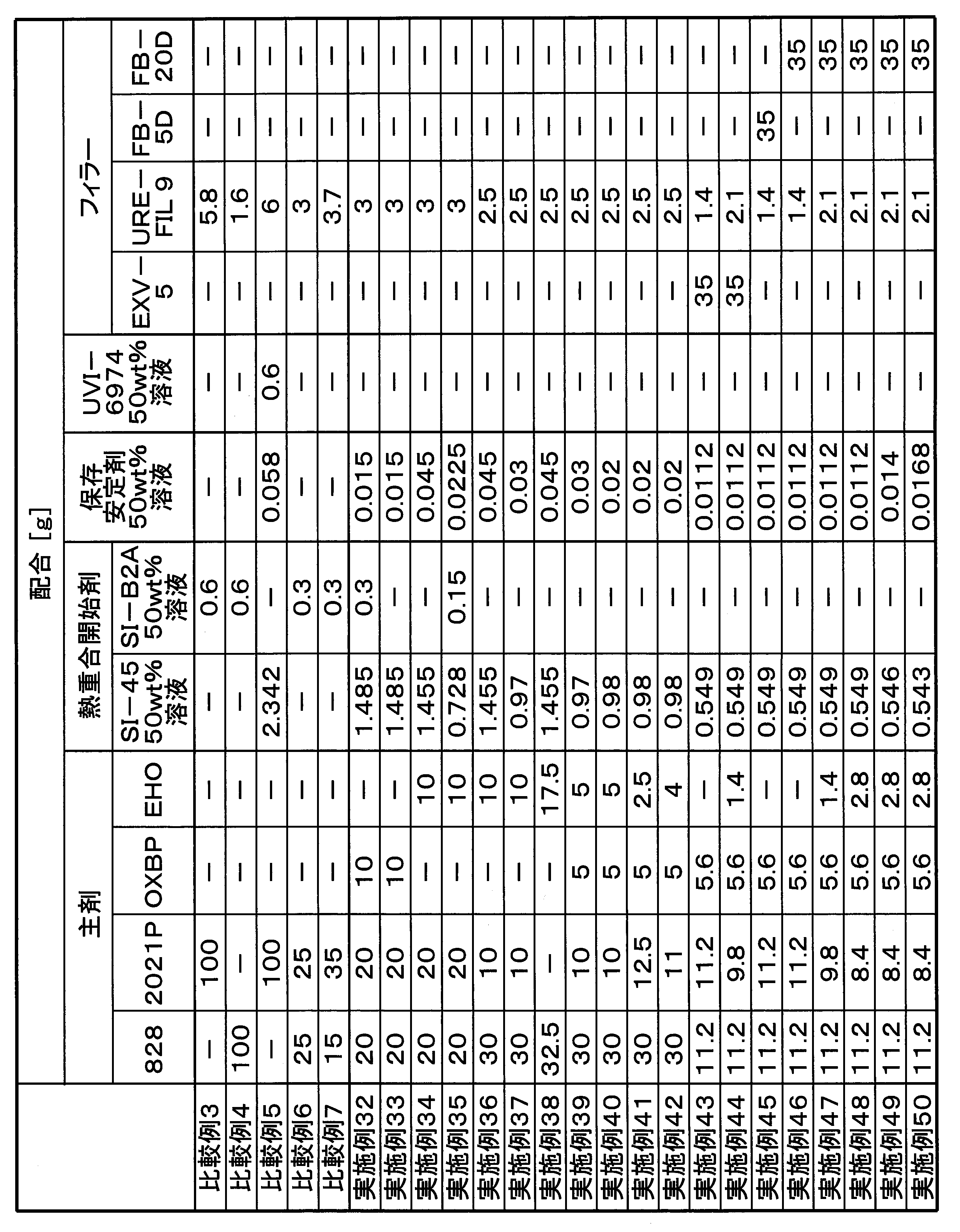

表4~5中の「主剤」はカチオン重合性化合物を示す。表5中の主剤100質量部に対する熱重合開始剤の割合、熱重合開始剤100質量部に対する保存安定剤の割合はそれぞれ固形分での値である。
実施例の硬化性組成物は、可使時間が十分に長く、かつ可使時間経過後に60℃以下の温度、並びに自然硬化(室温)でも迅速に硬化可能であった。
Claims (14)
- カチオン重合性化合物と、熱重合開始剤と、保存安定剤とを含み、
前記カチオン重合性化合物が、グリシジルエーテル化合物、脂環式エポキシ化合物、およびオキセタン化合物からなる群から選ばれる少なくとも2種を含み、
前記熱重合開始剤の含有量が、前記カチオン重合性化合物100質量部に対して0.3~3質量部であり、
前記カチオン重合性化合物の重合反応により発生した熱エネルギーにより連鎖硬化可能である、硬化性組成物。 - 65℃以下の温度で連鎖硬化可能である、請求項1に記載の硬化性組成物。
- フィラーをさらに含む、請求項1または2に記載の硬化性組成物。
- 室温における可使時間が20分間以上である、請求項1~3のいずれか一項に記載の硬化性組成物。
- 前記グリシジルエーテル化合物が、ビスフェノールA型ジグリシジルエーテル化合物およびビスフェールF型ジグリシジルエーテル化合物からなる群から選ばれる少なくとも1種であり、
前記脂環式エポキシ化合物が、3,4-エポキシシクロヘキシルメチル-3,4-エポキシシクロヘキサンカルボキシレートであり、
前記オキセタン化合物が、4,4’-ビス[(3-エチル-3-オキセタニル)メトキシメチル]ビフェニルおよび3-エチル-3-ヒドロキシメチルオキセタンからなる群から選ばれる少なくとも1種である、請求項1~4のいずれか一項に記載の硬化性組成物。 - 前記熱重合開始剤が、下記式(I-1)または(I-2)で表されるスルホニウム塩からなる群から選ばれる少なくとも1種である、請求項1~5のいずれか一項に記載の硬化性組成物。

- 前記保存安定剤が、下記式(II)で表されるスルホニウム塩からなる群から選ばれる少なくとも1種である、請求項1~6のいずれか一項に記載の硬化性組成物。
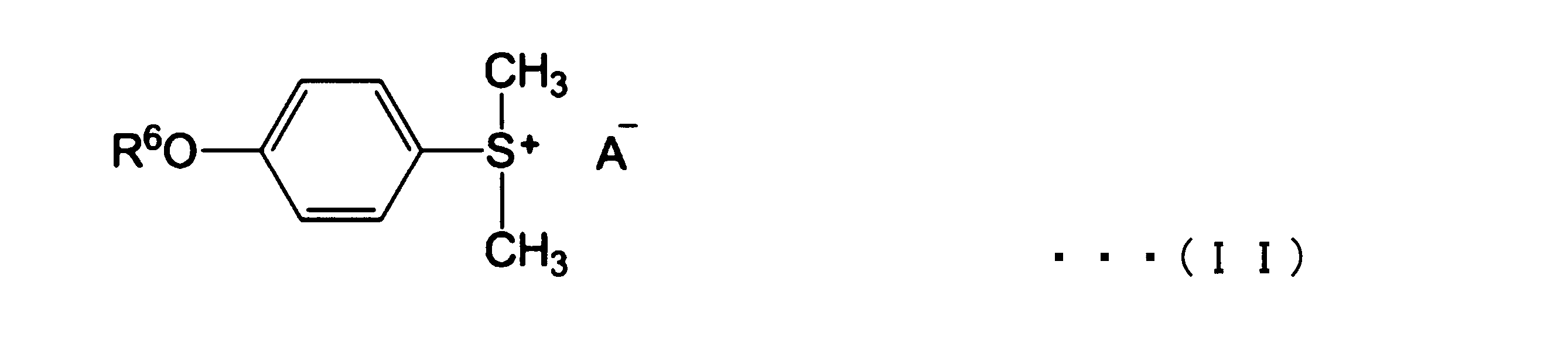
- 前記保存安定剤の含有量が、前記熱重合開始剤100質量部に対して0.3~5質量部である、請求項1~7のいずれか一項に記載の硬化性組成物。
- 請求項1~8のいずれか一項に記載の硬化性組成物を含む、硬化性ペースト材。
- 請求項1~8のいずれか一項に記載の硬化性組成物を含む、硬化性シート材。
- 請求項1~8のいずれか一項に記載の硬化性組成物を含む、硬化性型取り材。
- 請求項1~8のいずれか一項に記載の硬化性組成物、請求項9に記載の硬化性ペースト材、請求項10に記載の硬化性シート材、または請求項11に記載の硬化性型取り材を、65℃以下の温度で硬化させる、硬化方法。
- 請求項1~8のいずれか一項に記載の硬化性組成物、請求項9に記載の硬化性ペースト材、請求項10に記載の硬化性シート材、または請求項11に記載の硬化性型取り材を硬化させた、硬化物。
- 硬度が60Hs以上である、請求項13に記載の硬化物。
Priority Applications (5)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN201780093371.0A CN110945051A (zh) | 2017-08-29 | 2017-08-29 | 固化性组合物、固化性膏材料、固化性片材、固化性取模材料、固化方法以及固化物 |
| US16/636,673 US11667748B2 (en) | 2017-08-29 | 2017-08-29 | Curable composition, curable paste material, curable sheet material, curable modeling material, curing method, and cured product |
| PCT/JP2017/030905 WO2019043778A1 (ja) | 2017-08-29 | 2017-08-29 | 硬化性組成物、硬化性ペースト材、硬化性シート材、硬化性型取り材、硬化方法および硬化物 |
| EP17923577.5A EP3647337B1 (en) | 2017-08-29 | 2017-08-29 | Curable composition, curable paste material, curable sheet material, curable modeling material, curing method, and cured product |
| JP2019538782A JP6956190B2 (ja) | 2017-08-29 | 2017-08-29 | 硬化性組成物、硬化性ペースト材、硬化性シート材、硬化性型取り材、硬化方法および硬化物 |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| PCT/JP2017/030905 WO2019043778A1 (ja) | 2017-08-29 | 2017-08-29 | 硬化性組成物、硬化性ペースト材、硬化性シート材、硬化性型取り材、硬化方法および硬化物 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2019043778A1 true WO2019043778A1 (ja) | 2019-03-07 |
Family
ID=65527177
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2017/030905 Ceased WO2019043778A1 (ja) | 2017-08-29 | 2017-08-29 | 硬化性組成物、硬化性ペースト材、硬化性シート材、硬化性型取り材、硬化方法および硬化物 |
Country Status (5)
| Country | Link |
|---|---|
| US (1) | US11667748B2 (ja) |
| EP (1) | EP3647337B1 (ja) |
| JP (1) | JP6956190B2 (ja) |
| CN (1) | CN110945051A (ja) |
| WO (1) | WO2019043778A1 (ja) |
Cited By (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2021083632A1 (de) | 2019-10-31 | 2021-05-06 | Delo Industrie Klebstoffe Gmbh & Co. Kgaa | KATIONISCH FEUCHTEINDUZIERT HÄRTBARE MASSE, VERWENDUNG DER MASSE SOWIE VERFAHREN ZUM FÜGEN, VERGIEßEN UND BESCHICHTEN VON SUBSTRATEN |
| JP2021147584A (ja) * | 2020-03-23 | 2021-09-27 | ヘンケルジャパン株式会社 | デュアル硬化型接着剤組成物 |
| WO2022012996A1 (de) | 2020-07-16 | 2022-01-20 | Delo Industrie Klebstoffe Gmbh & Co. Kgaa | Wiederlösbare zusammensetzungen auf basis von polyacetalen |
| JPWO2022092080A1 (ja) * | 2020-10-30 | 2022-05-05 | ||
| JP2022544023A (ja) * | 2019-07-25 | 2022-10-17 | スリーエム イノベイティブ プロパティズ カンパニー | アセンブリにシムを入れる方法 |
| DE102021110210A1 (de) | 2021-04-22 | 2022-10-27 | Delo Industrie Klebstoffe Gmbh & Co. Kgaa | Verfahren zum Herstellen eines optischen Moduls und optisches Modul |
| JP2022175092A (ja) * | 2021-05-12 | 2022-11-25 | ヘンケルジャパン株式会社 | 可視又は赤外レーザー硬化型のカチオン重合系エポキシ樹脂組成物 |
| DE102021122835A1 (de) | 2021-09-03 | 2023-03-09 | Delo Industrie Klebstoffe Gmbh & Co. Kgaa | Warmhärtende Masse auf Basis von (Meth)Acrylaten und Peroxodicarbonaten |
| WO2023147993A1 (de) | 2022-02-04 | 2023-08-10 | Delo Industrie Klebstoffe Gmbh & Co. Kgaa | Kationisch polymerisierbare flammgeschützte massen |
| US11919204B2 (en) | 2019-01-25 | 2024-03-05 | Mitsubishi Heavy Industries, Ltd. | Mold-making method to gap, shim manufacturing method, manufacturing method of mold-making kit, and mold-making kit |
Families Citing this family (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP4050061B1 (en) * | 2021-02-26 | 2025-10-15 | Henkel AG & Co. KGaA | Near-infrared (nir) sensitized adhesive and sealant compositions |
| DE102022106647A1 (de) | 2022-03-22 | 2023-09-28 | Delo Industrie Klebstoffe Gmbh & Co. Kgaa | Niedertemperaturhärtende Massen auf Basis von Glycidylethern |
Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2012219262A (ja) * | 2011-04-14 | 2012-11-12 | Sony Chemical & Information Device Corp | 異方性導電フィルム |
| JP2013186976A (ja) * | 2012-03-06 | 2013-09-19 | Sekisui Chem Co Ltd | 有機エレクトロルミネッセンス表示素子用上下導通材料及び有機エレクトロルミネッセンス表示素子 |
| WO2015046333A1 (ja) * | 2013-09-27 | 2015-04-02 | 株式会社ダイセル | 半導体積層用接着剤組成物 |
| WO2015129670A1 (ja) * | 2014-02-27 | 2015-09-03 | 積水化学工業株式会社 | 有機エレクトロルミネッセンス表示素子封止用硬化性樹脂組成物、有機エレクトロルミネッセンス表示素子封止用硬化性樹脂シート、及び、有機エレクトロルミネッセンス表示素子 |
| JP2016069433A (ja) * | 2014-09-27 | 2016-05-09 | アイカ工業株式会社 | 熱カチオン硬化性樹脂組成物 |
| WO2016158523A1 (ja) | 2015-03-30 | 2016-10-06 | 株式会社Adeka | 組成物 |
| JP2016210849A (ja) * | 2015-04-30 | 2016-12-15 | 株式会社Adeka | 硬化物の製造方法、硬化物、硬化性組成物および接着剤 |
| JP2017008145A (ja) * | 2015-06-17 | 2017-01-12 | 株式会社ダイセル | 硬化性組成物、接着シート、積層物及び装置 |
Family Cites Families (12)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH055006A (ja) * | 1990-11-16 | 1993-01-14 | Nippon Kayaku Co Ltd | カチオン重合性有機材料組成物および当該組成物の安定化法 |
| JP3950241B2 (ja) | 1997-10-17 | 2007-07-25 | 三菱重工業株式会社 | 樹脂組成物、樹脂硬化物、及び構造物の補修方法、補強方法、補修用材料、補強用材料 |
| US6919420B2 (en) * | 2002-12-05 | 2005-07-19 | International Business Machines Corporation | Acid-cleavable acetal and ketal based epoxy oligomers |
| JP2006225545A (ja) | 2005-02-18 | 2006-08-31 | Yokohama Rubber Co Ltd:The | 硬化性樹脂組成物 |
| JP2008169327A (ja) * | 2007-01-12 | 2008-07-24 | Toray Fine Chemicals Co Ltd | 接着剤組成物 |
| JP2009132834A (ja) * | 2007-11-30 | 2009-06-18 | Nippon Shokubai Co Ltd | 硬化性樹脂組成物、光学部材用硬化性材料、及び、光学部材 |
| JP2011080017A (ja) | 2009-10-09 | 2011-04-21 | Denso Corp | カチオン重合性組成物 |
| JP5754073B2 (ja) | 2010-03-17 | 2015-07-22 | 株式会社デンソー | 硬化複合体の製造方法、および硬化複合体 |
| JP5922544B2 (ja) * | 2011-10-03 | 2016-05-24 | 積水化学工業株式会社 | 異方性導電材料及び接続構造体 |
| JP5636462B2 (ja) | 2013-04-02 | 2014-12-03 | 株式会社オートネットワーク技術研究所 | 感光性組成物の硬化方法 |
| JP6400006B2 (ja) * | 2013-07-09 | 2018-10-03 | 株式会社Adeka | カチオン重合性組成物 |
| JP6762745B2 (ja) | 2016-03-29 | 2020-09-30 | 株式会社Adeka | 硬化性組成物 |
-
2017
- 2017-08-29 CN CN201780093371.0A patent/CN110945051A/zh active Pending
- 2017-08-29 EP EP17923577.5A patent/EP3647337B1/en active Active
- 2017-08-29 US US16/636,673 patent/US11667748B2/en active Active
- 2017-08-29 WO PCT/JP2017/030905 patent/WO2019043778A1/ja not_active Ceased
- 2017-08-29 JP JP2019538782A patent/JP6956190B2/ja active Active
Patent Citations (8)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP2012219262A (ja) * | 2011-04-14 | 2012-11-12 | Sony Chemical & Information Device Corp | 異方性導電フィルム |
| JP2013186976A (ja) * | 2012-03-06 | 2013-09-19 | Sekisui Chem Co Ltd | 有機エレクトロルミネッセンス表示素子用上下導通材料及び有機エレクトロルミネッセンス表示素子 |
| WO2015046333A1 (ja) * | 2013-09-27 | 2015-04-02 | 株式会社ダイセル | 半導体積層用接着剤組成物 |
| WO2015129670A1 (ja) * | 2014-02-27 | 2015-09-03 | 積水化学工業株式会社 | 有機エレクトロルミネッセンス表示素子封止用硬化性樹脂組成物、有機エレクトロルミネッセンス表示素子封止用硬化性樹脂シート、及び、有機エレクトロルミネッセンス表示素子 |
| JP2016069433A (ja) * | 2014-09-27 | 2016-05-09 | アイカ工業株式会社 | 熱カチオン硬化性樹脂組成物 |
| WO2016158523A1 (ja) | 2015-03-30 | 2016-10-06 | 株式会社Adeka | 組成物 |
| JP2016210849A (ja) * | 2015-04-30 | 2016-12-15 | 株式会社Adeka | 硬化物の製造方法、硬化物、硬化性組成物および接着剤 |
| JP2017008145A (ja) * | 2015-06-17 | 2017-01-12 | 株式会社ダイセル | 硬化性組成物、接着シート、積層物及び装置 |
Non-Patent Citations (1)
| Title |
|---|
| See also references of EP3647337A4 |
Cited By (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US11919204B2 (en) | 2019-01-25 | 2024-03-05 | Mitsubishi Heavy Industries, Ltd. | Mold-making method to gap, shim manufacturing method, manufacturing method of mold-making kit, and mold-making kit |
| JP2022544023A (ja) * | 2019-07-25 | 2022-10-17 | スリーエム イノベイティブ プロパティズ カンパニー | アセンブリにシムを入れる方法 |
| JP7713925B2 (ja) | 2019-07-25 | 2025-07-28 | スリーエム イノベイティブ プロパティズ カンパニー | アセンブリにシムを入れる方法 |
| WO2021083632A1 (de) | 2019-10-31 | 2021-05-06 | Delo Industrie Klebstoffe Gmbh & Co. Kgaa | KATIONISCH FEUCHTEINDUZIERT HÄRTBARE MASSE, VERWENDUNG DER MASSE SOWIE VERFAHREN ZUM FÜGEN, VERGIEßEN UND BESCHICHTEN VON SUBSTRATEN |
| JP2021147584A (ja) * | 2020-03-23 | 2021-09-27 | ヘンケルジャパン株式会社 | デュアル硬化型接着剤組成物 |
| CN115315498A (zh) * | 2020-03-23 | 2022-11-08 | 汉高股份有限及两合公司 | 可双重固化粘合剂组合物 |
| JP7516081B2 (ja) | 2020-03-23 | 2024-07-16 | ヘンケルジャパン株式会社 | デュアル硬化型接着剤組成物 |
| WO2022012996A1 (de) | 2020-07-16 | 2022-01-20 | Delo Industrie Klebstoffe Gmbh & Co. Kgaa | Wiederlösbare zusammensetzungen auf basis von polyacetalen |
| DE102020118813A1 (de) | 2020-07-16 | 2022-01-20 | Delo Industrie Klebstoffe Gmbh & Co. Kgaa | Wiederlösbare Zusammensetzungen auf Basis von Polyacetalen |
| JPWO2022092080A1 (ja) * | 2020-10-30 | 2022-05-05 | ||
| WO2022092080A1 (ja) * | 2020-10-30 | 2022-05-05 | 株式会社Adeka | 重合性組成物、硬化物及び硬化物の製造方法 |
| WO2022223191A1 (de) | 2021-04-22 | 2022-10-27 | Delo Industrie Klebstoffe Gmbh & Co. Kgaa | Verfahren zum herstellen eines optischen moduls und optisches modul |
| DE102021110210A1 (de) | 2021-04-22 | 2022-10-27 | Delo Industrie Klebstoffe Gmbh & Co. Kgaa | Verfahren zum Herstellen eines optischen Moduls und optisches Modul |
| JP2022175092A (ja) * | 2021-05-12 | 2022-11-25 | ヘンケルジャパン株式会社 | 可視又は赤外レーザー硬化型のカチオン重合系エポキシ樹脂組成物 |
| WO2023030763A1 (de) | 2021-09-03 | 2023-03-09 | Delo Industrie Klebstoffe Gmbh & Co. Kgaa | Warmhärtende masse auf basis von (meth)acrylaten und peroxodicarbonaten |
| DE102021122835A1 (de) | 2021-09-03 | 2023-03-09 | Delo Industrie Klebstoffe Gmbh & Co. Kgaa | Warmhärtende Masse auf Basis von (Meth)Acrylaten und Peroxodicarbonaten |
| WO2023147993A1 (de) | 2022-02-04 | 2023-08-10 | Delo Industrie Klebstoffe Gmbh & Co. Kgaa | Kationisch polymerisierbare flammgeschützte massen |
| DE102022102650A1 (de) | 2022-02-04 | 2023-08-10 | Delo Industrie Klebstoffe Gmbh & Co. Kgaa | Kationisch polymerisierbare flammgeschützte Massen |
Also Published As
| Publication number | Publication date |
|---|---|
| US20200190251A1 (en) | 2020-06-18 |
| EP3647337A1 (en) | 2020-05-06 |
| EP3647337B1 (en) | 2021-10-27 |
| EP3647337A4 (en) | 2020-06-17 |
| US11667748B2 (en) | 2023-06-06 |
| JP6956190B2 (ja) | 2021-11-02 |
| JPWO2019043778A1 (ja) | 2020-06-18 |
| CN110945051A (zh) | 2020-03-31 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| WO2019043778A1 (ja) | 硬化性組成物、硬化性ペースト材、硬化性シート材、硬化性型取り材、硬化方法および硬化物 | |
| CN102378772B (zh) | 产生低放热量的环氧树脂基芯填料材料 | |
| JP6175515B2 (ja) | アンダーフィル組成物およびそれを使用したパッケージング工程 | |
| CN102639589B (zh) | 链式固化性树脂组合物和纤维增强复合材料 | |
| JP4925900B2 (ja) | 光学的立体造形用樹脂組成物 | |
| JP2012517507A (ja) | 2液型液体シム組成物 | |
| CN105339411A (zh) | 用作蜂窝状小室的填料的环氧树脂基组合物 | |
| CN107987474A (zh) | 一种用于快速成型的光固化树脂及制备方法 | |
| JP6013906B2 (ja) | 液状エポキシ樹脂組成物 | |
| KR20190053901A (ko) | 광-고정가능한 주조 화합물 및 상기 화합물을 이용하여 기판/부품을 선택적으로 주조하는 방법 | |
| WO2017002315A1 (ja) | 放熱材料接着用組成物、接着剤付放熱材料、インレイ基板、及びその製造方法 | |
| JP7094233B2 (ja) | ギャップの型取り方法、シムの製造方法、及び型取りキットの製造方法 | |
| JP6367668B2 (ja) | 熱カチオン硬化性樹脂組成物 | |
| JP5269734B2 (ja) | 重合硬化性組成物、その重合硬化方法、および重合硬化樹脂組成物 | |
| JP4843256B2 (ja) | 硬化促進剤 | |
| CN116496741A (zh) | 一种双重固化环氧胶黏剂及其制备方法 | |
| JP6849899B2 (ja) | エポキシ樹脂組成物 | |
| KR20240081446A (ko) | 액상 봉지제, 전자 부품과 그 제조 방법, 및 반도체 장치 | |
| TWI805680B (zh) | 固化性環氧樹脂組成物及其固化物 | |
| EP4714994A1 (en) | Two-component composition providing fire retardency | |
| JP2021031604A (ja) | 自己修復性エポキシ樹脂組成物 | |
| JP6653152B2 (ja) | エポキシ樹脂組成物 | |
| JP2011079942A (ja) | 液状エポキシ樹脂組成物 | |
| WO2023147993A1 (de) | Kationisch polymerisierbare flammgeschützte massen | |
| JPWO2020066746A1 (ja) | 繊維強化複合材料用樹脂組成物及びそれを用いた繊維強化複合材料 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 17923577 Country of ref document: EP Kind code of ref document: A1 |
|
| ENP | Entry into the national phase |
Ref document number: 2019538782 Country of ref document: JP Kind code of ref document: A |
|
| ENP | Entry into the national phase |
Ref document number: 2017923577 Country of ref document: EP Effective date: 20200128 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |