WO2024200927A1 - Procédé de préparation d'organopolysiloxanes à fonctions (méth)acrylates - Google Patents
Procédé de préparation d'organopolysiloxanes à fonctions (méth)acrylates Download PDFInfo
- Publication number
- WO2024200927A1 WO2024200927A1 PCT/FR2024/000042 FR2024000042W WO2024200927A1 WO 2024200927 A1 WO2024200927 A1 WO 2024200927A1 FR 2024000042 W FR2024000042 W FR 2024000042W WO 2024200927 A1 WO2024200927 A1 WO 2024200927A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- organopolysiloxane
- group
- composition
- iii
- chromium
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G77/00—Macromolecular compounds obtained by reactions forming a linkage containing silicon with or without sulfur, nitrogen, oxygen or carbon in the main chain of the macromolecule
- C08G77/04—Polysiloxanes
- C08G77/38—Polysiloxanes modified by chemical after-treatment
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G77/00—Macromolecular compounds obtained by reactions forming a linkage containing silicon with or without sulfur, nitrogen, oxygen or carbon in the main chain of the macromolecule
- C08G77/04—Polysiloxanes
- C08G77/14—Polysiloxanes containing silicon bound to oxygen-containing groups
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08G—MACROMOLECULAR COMPOUNDS OBTAINED OTHERWISE THAN BY REACTIONS ONLY INVOLVING UNSATURATED CARBON-TO-CARBON BONDS
- C08G77/00—Macromolecular compounds obtained by reactions forming a linkage containing silicon with or without sulfur, nitrogen, oxygen or carbon in the main chain of the macromolecule
- C08G77/04—Polysiloxanes
- C08G77/20—Polysiloxanes containing silicon bound to unsaturated aliphatic groups
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K5/00—Use of organic ingredients
- C08K5/04—Oxygen-containing compounds
- C08K5/09—Carboxylic acids; Metal salts thereof; Anhydrides thereof
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08K—Use of inorganic or non-macromolecular organic substances as compounding ingredients
- C08K5/00—Use of organic ingredients
- C08K5/56—Organo-metallic compounds, i.e. organic compounds containing a metal-to-carbon bond
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08L—COMPOSITIONS OF MACROMOLECULAR COMPOUNDS
- C08L83/00—Compositions of macromolecular compounds obtained by reactions forming in the main chain of the macromolecule a linkage containing silicon with or without sulfur, nitrogen, oxygen or carbon only; Compositions of derivatives of such polymers
- C08L83/04—Polysiloxanes
- C08L83/06—Polysiloxanes containing silicon bound to oxygen-containing groups
-
- C—CHEMISTRY; METALLURGY
- C09—DYES; PAINTS; POLISHES; NATURAL RESINS; ADHESIVES; COMPOSITIONS NOT OTHERWISE PROVIDED FOR; APPLICATIONS OF MATERIALS NOT OTHERWISE PROVIDED FOR
- C09D—COATING COMPOSITIONS, e.g. PAINTS, VARNISHES OR LACQUERS; FILLING PASTES; CHEMICAL PAINT OR INK REMOVERS; INKS; CORRECTING FLUIDS; WOODSTAINS; PASTES OR SOLIDS FOR COLOURING OR PRINTING; USE OF MATERIALS THEREFOR
- C09D183/00—Coating compositions based on macromolecular compounds obtained by reactions forming in the main chain of the macromolecule a linkage containing silicon, with or without sulfur, nitrogen, oxygen, or carbon only; Coating compositions based on derivatives of such polymers
- C09D183/10—Block or graft copolymers containing polysiloxane sequences
Definitions
- the present invention relates to a process for preparing organopolysiloxanes with (meth)acrylate functions, preferably acrylates.
- the (meth)acrylate groups comprise acrylate groups, methacrylate groups or a mixture of the two.
- These (meth)acrylate functionalities are present only in organic groups linked to the polysiloxane chain by an Si-C bond which can be present in the chain, at the end of the chain or both.
- These (meth)acrylic acid ester functions are capable of reacting by radical means under thermal activation and/or by radiation according to a polyaddition polymerization mechanism.
- (meth)acrylate-functional organopolysiloxanes are widely used in radically crosslinkable silicone compositions which can optionally be coated in a thin layer on a flexible support which is made of textile, paper, polyvinyl chloride (PVC), polyester, polypropylene, polyamide, polyethylene, polyethylene terephthalate (PET), polyurethane or non-woven glass fibers, to produce an elastomer.
- a flexible support which is made of textile, paper, polyvinyl chloride (PVC), polyester, polypropylene, polyamide, polyethylene, polyethylene terephthalate (PET), polyurethane or non-woven glass fibers, to produce an elastomer.
- silicone formulations on flexible supports targets many applications. For example, when the flexible support is a textile, water-repellent properties are targeted or when the support is a paper or a polymer such as PVC or PET, non-stick properties are most often sought.
- Release coatings are useful for many applications where it is necessary to render a surface or material that would normally adhere to them non-adherent to other materials.
- silicone compositions are used as coatings for release papers and can thus be combined with adhesive elements that can be easily released without losing their adhesive properties, such elements being pressure-sensitive adhesives for labels, decorative laminates, transfer tape, etc.
- Silicone-based release coatings applied to paper, polyethylene, polypropylene, polyester and other such substrates are also useful as release surfaces for products for food use and in particular in the industrial packaging sector. There has therefore been a long-standing interest in these (meth)acrylate-functional organopolysiloxanes and their preparation can be envisaged in various ways described in the prior art.
- silicone elastomer articles have good mechanical properties.
- organopolysiloxanes with (meth)acrylate function are obtained industrially by reaction between an organopolysiloxane functionalized by epoxy groups and (meth)acrylic acid in the presence of a chromium-based catalyst.
- Patent EP1276825-B 1 teaches us in particular the preparation of organopolysiloxanes with a (meth)acrylate function starting from organopolysiloxanes having epoxy functionalities and acrylic acid in the presence of chromium (III) acetate and two solvents including an alcohol.
- these solvents are necessary to solubilize the chromium (III) acetate catalyst.
- the yield and selectivity of such a process can be improved.
- the compositions comprising the organopolysiloxanes with an acrylate function obtained according to the teaching of this title have an unpleasant odor that is annoying for users of these compositions.
- patent application WO2017187030 teaches the preparation of organopolysiloxanes with (meth)acrylate function from organopolysiloxanes having epoxy functionalities, acrylic acid, chromium (III) acetate, solvent but in the absence of alcoholic solvent.
- This patent application WO2017187030 has advantageously shown that it makes it possible to obtain an improved yield of organopolysiloxanes with (meth)acrylate function.
- this process makes it possible to obtain oils without unpleasant odors in durations compatible with industrial production.
- organopolysiloxane oils with epoxy functionalities comprising a low epoxy functionality level are present, the implementation of this process can become complex. A decrease in the yield and selectivity for such compounds is then observed. This drop in reactivity is explained in particular by the low solubility of the chromium acetate catalyst under such conditions.
- the epoxy functionality rate is measured in moles of epoxy functions per kilogram of polyorganosiloxane oils.
- the reaction medium can be preheated to promote the solubilization of the catalyst or the reagents can be introduced at different stages of the process.
- these adjustments to the process can lead to a drop in kinetics and thus an increase in the reaction time to form the organopolysiloxanes with (meth)acrylate function.
- one of the essential objectives of the present invention is to develop a process for preparing organopolysiloxanes with (meth)acrylate function with satisfactory kinetics and selectivity while avoiding the use of alcoholic solvent.
- Another essential objective of the present invention is to provide a process for preparing robust (meth)acrylate functional organopolysiloxanes from organopolysiloxane oils having different chain lengths and/or different levels of acrylate functionality.
- Another essential objective of the present invention is to provide a catalytic system suitable for the process of the present invention.
- Another essential objective of the present invention is to provide a process for preparing organopolysiloxanes with robust (meth)acrylate function by a so-called “one-pot” reaction.
- one-pot means the introduction of all the reactants into the reactor at the initial time of said reaction, also called to.
- the term “mass percentage of a compound A” means the mass percentage of this chemical species A relative to the total mass of the mixture.
- the Applicant has developed a process which relates to a process for the preparation of a composition X comprising at least one organopolysiloxane A comprising at least one (meth)acrylate group, said process comprising the following steps: a) At least one organopolysiloxane E comprising at least one epoxy group is reacted at a temperature of 50 to 130°C, preferably 70 to 130°C and even more preferably 90 to 125°C, with acrylic acid or methacrylic acid or a mixture of the following: two, and in the presence of:
- step a) an inhibitor of polymerization of acrylic acid or methacrylic acid
- step b) The reaction medium obtained at the end of step a) is devolatilized
- step c) Said composition X is obtained comprising at least one organopolysiloxane A.
- Step a) of the process for preparing composition X according to the invention is preferably carried out in the presence of a solvent S which is not an alcohol.
- the invention is carried out in the absence of alcohol and in particular in the absence of butanol.
- the term solvent means a non-reactive solvent. Therefore, the solvent S is different from organopolysiloxane E, acrylic acid and methacrylic acid.
- composition X has the advantages of obtaining a satisfactory yield and selectivity for organopolysiloxane A. It is to the credit of the inventors to have identified that implementing the process in the presence of chromium (III) P-diketonate complexes, such as chromium (III) acetylacetonate, makes it possible to obtain better selectivity for organopolysiloxane A.
- this process has the advantage of having a process that can be adapted to the industrial scale for organopolysiloxanes E of different degrees of polymerization and/or different levels of epoxy functionality without having to adapt the implementation of the process.
- the process according to the invention can be implemented “one-pot” from composition X prepared upstream as well as premixes of composition X prepared for example the day before. This significant advantage makes it easier to implement said process on an industrial scale.
- step a) is between one and five hours when the temperature of step a) is between 90 and 125°C.
- the organopolysiloxane A obtained by the process of the invention comprises siloxyl units (I), (II), and optionally (III) of the following formulae: in which:
- Y identical or different, represent an organic group comprising an epoxy group and optionally further comprising one or more heteroatoms such as an oxygen atom, said organic group Y preferably having from 2 to 20 carbon atoms inclusive, and, more preferably still, Y is chosen from the group consisting of an alkylglycidyl ether, a linear, branched or cyclic epoxyalkyl, a linear, branched or cyclic epoxyalkenyl and a carboxylic acid glycidyl ester,
- the symbols Z 1 and Z 2 which may be identical or different, represent a monovalent organic group having from 1 to 30 carbon atoms and preferably chosen from the group consisting of alkyl groups having from 1 to 8 carbon atoms and aryl groups having from 6 to 12 carbon atoms, and even more preferably chosen from the group consisting of a methyl, ethyl, propyl, 3,3,3-trifluoropropyl, xylyl, toluyl and phenyl group, - the symbols V, which may be identical or different, represent an organic group comprising a (meth)acrylate group, said organic group V preferably having from 5 to 23 carbon atoms inclusive, and,
- organopolysiloxane A comprises, per molecule, at least two silicon atoms and at least one siloxyl unit (III).
- Organopolysiloxane E comprises siloxyl units (I) and (II) of the following formulas: in which:
- Y identical or different, represent an organic group comprising an epoxy group and optionally further comprising one or more heteroatoms such as an oxygen atom, said organic group Y preferably having from 2 to 20 carbon atoms inclusive, and, more preferably still, Y is chosen from the group consisting of an alkyldiglycidyl ether, a linear, branched or cyclic epoxyalkyl, a linear, branched or cyclic epoxyalkenyl and a carboxylic acid glycidyl ester;
- Z 1 and Z 2 which may be identical or different, represent a monovalent organic group having from 1 to 30 carbon atoms and preferably chosen from the group consisting of alkyl groups having from 1 to 8 carbon atoms and aryl groups having from 6 to 12 carbon atoms, and even more preferably chosen from the group consisting of a methyl, ethyl, propyl, 3,3,3 trifluoropropyl, xylyl, toluyl and phenyl group, and
- said organopolysiloxane E comprises, per molecule, at least two silicon atoms and at least one siloxyl unit (I)
- the symbol Y is chosen from the group consisting of the organic groups (IV) to (VIII) of the following formulas:
- Y is the organic group (VII) of the following formula:
- organopolysiloxanes E can have a linear, branched or cyclic structure and their degree of polymerization is between 2 and 5000, preferably between 2 and 1000 and even more preferably between 2 and 500.
- the organopolysiloxane E has a linear structure and comprises siloxyl units (I) and (II) of the following formulas: in which:
- Y identical or different, represent an organic group comprising an epoxy group and optionally further comprising one or more heteroatoms such as an oxygen atom, said organic group Y preferably having from 2 to 20 carbon atoms inclusive, and, more preferably still Y is chosen from the group consisting of an alkylglycidyl ether, a linear, branched or cyclic epoxyalkyl, a linear, branched or cyclic epoxyalkenyl and a glycidyl ester of carboxylic acids;
- the symbols Z 1 and Z 2 represent a monovalent organic group having from 1 to 30 carbon atoms and preferably chosen from the group consisting of alkyl groups having from 1 to 8 carbon atoms and aryl groups having from 6 to 12 carbon atoms, and even more preferably chosen from the group consisting of a methyl, ethyl, propyl, 3,3,3-trifluoropropyl, xylyl, touyl and phenyl group, and
- organopolysiloxane E comprising, per molecule, at least two silicon atoms and at least one siloxyl unit (I).
- the organopolysiloxane E consists essentially of siloxyl units "D" selected from the group consisting of the siloxyl units Y 2 SiO 2 / 2 , YZ ⁇ iO ⁇ and Z 2 2 SiO 2 / 2 and of siloxyl units "M" selected from the group consisting of the siloxyl units YsSiOia, YZ' 2 SiOi/ 2 , Y 2 Z 1 SiOi/ 2 and Z 2 3SiOi/ 2 .
- the symbols Y, Z 1 and Z 2 are as described above.
- the organopolysiloxane E is essentially composed of siloxyl units "D" chosen from the group consisting of the siloxyl units YZ 1 SiO2/2 and Z 2 2SiO2/2 and of siloxyl units "M” chosen from the group consisting of the siloxyl units YZ ⁇ SiOia, and Z 2 3SiOi/2.
- the symbols Y, Z 1 and Z 2 are as described above.
- Organopolysiloxane E has a dynamic viscosity at 25°C of between 1 and 100,000 mPa.s, preferably between 10 and 50,000 mPa.s and even more preferably between 10 and 10,000 mPa.s, even more preferably between 10 and 5000 mPa.s.
- the organopolysiloxane E contains per molecule from 1 to 300 siloxyl units (I) bearing at least one organic group comprising an epoxy group. According to a preferred embodiment of the invention, the organopolysiloxane E contains per molecule from 2 to 250 siloxyl units (I) and more preferably the organopolysiloxane E contains per molecule from 20 to 300 siloxyl units (I).
- the organopolysiloxane E contains from 0.5 to 30% by mass of organic groups Y comprising an epoxy group, preferably from 1 to 20% by mass and even more preferably from 1 to 10% by mass relative to the total mass of the organopolysiloxane E.
- the content of organic groups Y comprising an epoxy group is from 0.05 to 3 mol per kilogram of organopolysiloxanes E, preferably from 0.1 to 1.5 mol, preferentially from 0.1 to 1 mol per kilogram of organopolysiloxane E.
- the organopolysiloxane E is chosen from the compounds of formulae (EI) to (E-IV) below: (EI) in which R is an alkyl group comprising from 2 to 5 carbon atoms, a is between 2 and 50 and preferably between 2 and 15 and b is between 20 and 400.
- organopolysiloxane A a conversion rate of the epoxy functions of organopolysiloxane E that is total or partial may be sought.
- An organopolysiloxane A comprising epoxy functions and (meth)acrylate functions may thus be obtained, which may be used, for example, as an adhesion modulator or as an adhesion promoter, or an organopolysiloxane A comprising only or essentially (meth)acrylate functions, used, for example, as an essential constituent of radically crosslinkable silicone compositions to produce an elastomer.
- the molar ratio R between the (meth)acrylic acid and the epoxy functions of organopolysiloxane E used in the process will be adapted accordingly.
- step a) the molar ratio R between the (meth)acrylic acid and the epoxy group(s) carried by the organopolysiloxane E is greater than 1.05 and preferably ranges from 1.05 to 15, and even more preferably ranges from 1.05 to 10.
- (meth)acrylic acid includes acrylic acid and methacrylic acid.
- the molar ratio R between the acrylic acid and the epoxy group(s) carried by the organopolysiloxane E is greater than 1.05 and preferably is between 1.05 and 15, and even more preferably is between 1.05 and 10.
- Catalyst C is a chromium complex in oxidation state (III).
- Catalyst C is a chromium complex of the following formula (1):
- catalyst C is a chromium complex of the following formula (1):
- Ri and R3 identical or different, represent a C1 to C30 hydrocarbon radical, a C6 to C30 hydrocarbon radical comprising an aromatic ring, or Ri and R3 together form, with the atoms to which they are linked, a monocycle consisting of 6 to 10 carbon atoms, and
- R2 is hydrogen or a C1 to C30 hydrocarbon radical, or R1 and R2 together with the atoms to which they are bonded form a monocycle of 5 to 10 atoms.
- the identical or different ligands L1 represent a P-diketonate anion represented by the following formula (2): where R1 and R3, independently of each other, represent a C1 to C30 hydrocarbon radical, or a C6 to C30 hydrocarbon radical comprising an aromatic ring, and R2 is hydrogen or a C1 to C30 hydrocarbon radical.
- catalyst C is a chromium (III) complex of the following formula (1):
- the catalyst C is a chromium complex of the following formula (1): [CrCL s] (1) in which the symbols L 1 are identical or different ligands represent a P-diketonate anion represented by the following formula (3): where R, identical or different, represent a C1 to C30 hydrocarbon radical, or a C6 to C30 hydrocarbon radical comprising an aromatic ring
- the catalyst C is a chromium P-diketonate with an oxidation state (III) chosen from the group consisting of chromium (III) acetylacetonate (2,4-pentanedionate), chromium (III) hexafluoroacetylacetonate (1,1,1,5,5,5-hexafluoro-2,4-pentanedionate (F-acac)) (III), chromium 2,2,6,6-tetramethyl-
- Chromium(III)-3,5-heptanedionate Chromium(III)-3,5-heptanedionate, chromium(III)-3,5-heptanedionate, 2,2,7-trimethyl-
- Chromium (III) 3.5-octanedionate Chromium (III) 3.5-octanedionate.
- chromium (III) P-diketonate can be generated in-situ.
- the concentration of catalyst C expressed in mol % relative to the epoxy groups of the organopolysiloxane E is between 0.05 and 1%, preferably between 0.05 and 0.5%, more preferably between 0.05 and 0.3% and even more preferably between 0.05 and 0.25%.
- Another advantage of the process according to the invention is that it is possible to use a small amount of catalyst.
- said method is carried out without solvent S.
- the process is characterized in that in step a), the composition X further comprises at least one solvent S chosen from the group consisting of methyl isobutyl ketone, methyl ethyl ketone, toluene, xylene, chlorobenzene and their mixtures.
- solvent S chosen from the group consisting of methyl isobutyl ketone, methyl ethyl ketone, toluene, xylene, chlorobenzene and their mixtures.
- the content of solvent S used in step a) of the process as described above is from 0 to 15% by mass, preferably from 2% to 15% by mass, more preferably from 4 to 12% by mass, relative to the total mass of the reaction medium used in step a) of said process.
- step a) of the process according to the invention if the amount of solvent S is greater than 15% by mass relative to the total mass of the reaction medium used in step a) of the process according to the invention, the reaction time required to achieve a conversion rate of the epoxy groups greater than 98% increases. Thus, it is important to find the right compromise between solubilization of the system and kinetics of the reaction.
- the catalyst C, the (meth)acrylic acid polymerization inhibitor, the solvent S and part or all of the (meth)acrylic acid may be pre-mixed before the addition of the organopolysiloxane E.
- the catalyst C, the (meth)acrylic acid polymerization inhibitor, the solvent S, all of the (meth)acrylic acid and the organopolysiloxane E may be pre-mixed to form a premix before carrying out the process of the invention.
- this premix may be prepared the day before being used. in the process of the invention.
- This embodiment is particularly advantageous because it makes the process of the invention flexible and particularly suitable for industrial scale.
- a polymerization inhibitor of (meth)acrylic acid is introduced.
- Polymerization inhibitors of (meth)acrylic acid are widely known and, as examples, we can cite phenolic compounds such as 4-methoxyphenol, hydroquinone and methylhydroquinone or alkyldiphenylamines such as phenothiazine.
- phenolic compounds such as 4-methoxyphenol, hydroquinone and methylhydroquinone or alkyldiphenylamines such as phenothiazine.
- the methyl ether of hydroquinone (MEHQ) or 4-methoxyphenol is used as polymerization inhibitor of (meth)acrylic acid.
- the amount of polymerization inhibitor used is between 0.01 and 1% by mass relative to the mass of the organopolysiloxane E and even more preferably between 0.01 and 0.5% relative to the mass of the organopolysiloxane E.
- step b) of the process according to the invention the reaction medium obtained at the end of step a) is devolatilized.
- the reaction medium from step a) is heated to a temperature between 80 and 130°C under reduced pressure.
- This devolatilization step makes it possible to evaporate the solvent S and the excess (meth)acrylic acid.
- the solvent S used in the process according to the invention can be recycled.
- a filtration step can further be carried out before or after step b).
- a filtration step is added to the method according to the invention, it takes place after step b).
- the process according to the invention consists of the following steps: a) at least one organopolysiloxane E comprising at least one epoxy group is reacted at a temperature between 70 and 130°C, preferably between 90 and 130°C and even more preferably between 100 and 125°C, with acrylic acid or methacrylic acid or a mixture of the two, in the absence of alcohol and in the presence of:
- step a) an inhibitor of polymerization of acrylic acid or methacrylic acid
- step b) the reaction medium obtained at the end of step a) is devolatilized
- steps a) and b) of the method When implementing the method, it is possible but not necessary to carry out steps a) and b) under an inert atmosphere. According to a preferred embodiment, when a phenolic compound is used as an inhibitor of (meth)acrylic acid, steps a) and b) of the method will be carried out under air and even more preferably under bubbling of dry air.
- composition X capable of being obtained by the process described above.
- Another subject of the invention relates to a method for producing a coating on a substrate comprising the following steps: a) a composition X is prepared according to the method as described above, b) a radically crosslinkable silicone composition W is prepared comprising: i. said composition X ii. a photoinitiator, and iii. Optionally at least one additive, c) said composition W is applied to a substrate, and d) said composition W is crosslinked by exposure to radiation.
- the substrate is a flexible support made of textile, paper, polyvinyl chloride, polyester, polypropylene, polyamide, polyethylene, polyethylene terephthalate, polyurethane or non-woven glass fibers.
- the radiation is ultraviolet light with a wavelength of less than 420 nm.
- a source of radiation mention may in particular be made of light sources such as light-emitting diodes, better known by the acronym “LED” (Light-Emitting Diodes) which deliver point UV or visible light.
- the irradiation source is a block of light-emitting diodes (LEDs), preferably a block of light-emitting diodes (LEDs) having a wavelength of 355, 365, 385 or 405 nm.
- the power of the irradiation source may be at least 1, 10 or 50 mW/cm 2 . It may be between 1 and 1,000 mW/cm 2 , preferably between 1 and 200 mW/cm 2 , preferentially between 150 mW/cm 2 , and more preferentially between 1 and 20 mW/cm 2 .
- the irradiation time can be short and is generally less than 1 second and is of the order of a few hundredths of a second for low coating thicknesses. The crosslinking obtained is excellent even in the absence of any heating.
- crosslinking step d) takes place at a temperature between 40 and 100°C.
- the curing time can be adjusted, in particular, by the number of lamps used, by the duration of exposure to UV, by the wavelength of said lamps and by the distance between the composition and the lamp.
- the quantity of composition W deposited on the substrate is variable and most often ranges between 0.1 and 5g/m 2 of treated surface. This quantity depends on the nature of the support and the non-stick properties sought. It is most often between 0.5 and 1.5g/m 2 .
- This process is particularly suitable for preparing a non-stick silicone coating on a substrate which is a flexible support made of textile, paper, polyvinyl chloride, polyester, polypropylene, polyamide, polyethylene, polyethylene terephthalate, polyurethane or non-woven glass fibers. These coatings are particularly suitable for their use in the field of non-stick.
- radical photoinitiators include: ⁇ -hydroxyketones, benzoin ethers and ⁇ -amino aromatic ketones.
- radical photoinitiators include the following products: isopropylthioxanthone, benzophenone, camphorquinone, 9-xanthenone, anthraquinone, 1-4 dihydroxyanthraquinone, 2-methylanthraquinone, 2,2-bis(3-hydroxy-1,4-naphthoquinone), 2-6-dihydroxyanthraquinone, 1-hydroxycyclohexylphenylketone, 1,5-dihydroxyanthraquinone, 1,3-diphenyl-1,3-propanedione, 5,7-dihydroxyflavone, dibenzoylperoxide, 2-benzoylbenzoic acid, 2-hydroxy-2-methylpropiophenone, 2-phenylacetophenone, 2,4,6-trimethylbenzenoyldiphenoxyphosphine oxide and its derivatives, anthrone, bis(2,6-trimethylbenzenoyldiphenoxyphosphine oxide) and its derivatives
- dimethylbenzoyl)-2,4,4-trimethylpentylphosphine 4,4'- dimethoxybenzoin, phenantrenequinone, 2-ethylanthraquinone, 2-methylanthraquinone, 1,8- dihydroxyanthraquinone, dibenzoylperoxide, 2,2-dimethoxy-2-phenylacetophenone, benzoin, 2-hydroxy-2-methylpropiophenone, benzaldehyde, 4-(2-hydroxyethoxy)phenyl-(2-hydroxy-2-methylpropyl) ketone, benzoylacetone, ethyl(2,4,6- trimethylbenzoyl)phenylphosphinate and mixtures thereof.
- Examples of commercial radical photoinitiator products include the products marketed by the company CIBA-GEIGY: Irgacure ® 369, Irgacure ® 651, Irgacure ® 907, Darocure ® 1173, etc.
- the amount of photoinitiator in composition W is generally between 0.001 and 5%, most often between 0.005 and 3% by mass relative to the total mass of composition W.
- At least one additive for regulating the release force of a silicone/adhesive interface may be included in the composition, which is chosen from:
- Suitable organic (meth)acrylate derivatives include, in particular, epoxidized (meth)acrylate compounds, (meth)acryloglyceropolyesters, (meth)acrylouretanes, (meth)acrylopoly ethers, (meth)acrylopolyesters, (meth)acrylo-acrylics.
- trimethylolpropane triacrylate tripropylene glycol diacrylate and pentaerythritol tetraacrylate.
- the additive used is a silicone with (meth)acrylate function(s).
- (meth)acrylate functions carried by the silicone and particularly suitable for the invention mention may more particularly be made of acrylate derivatives, methacrylates, (meth)acrylate ethers and meth(acrylate) esters linked to the polysiloxane chain by a Si-C bond.
- acrylate derivatives are described in particular in patents EP 281718, FR2632960 and EP940458.
- composition W can be added to accelerate the crosslinking of composition W.
- additives such as thiols or aromatic amines can be added to accelerate the crosslinking of composition W.
- Another object of the invention relates to a substrate comprising at least one coating capable of being obtained according to the method as described above.
- Another subject of the invention relates to a method for producing an elastomer article by additive manufacturing comprising the following steps: a) a composition X is prepared according to the method as described above, b) an irradiation source and a photocrosslinkable silicone composition W2 are used comprising: i. said composition X ii. a photoinitiator, and iii. optionally at least one additive, c) selectively irradiating at least a portion of the photocrosslinkable silicone composition W2 using the irradiation source to form a portion of the silicone elastomer article; and d) repeating step ii) a sufficient number of times to produce the silicone elastomer article.
- the additive manufacturing method is an additive manufacturing method by vat photopolymerization, in particular, by laser stereolithography (SLA) printing, by digital light processing (DLP), or by continuous liquid interface production (CLIP).
- SLA laser stereolithography
- DLP digital light processing
- CLIP continuous liquid interface production
- the irradiation source is an LED lamp, preferably an LED lamp having a wavelength of 355, 365, 385 or 405 nm.
- the nature of the photoinitiators and any additives used in the photocrosslinkable composition W2 are identical to those previously set out for the composition W.
- the photocrosslinkable silicone composition W2 may comprise a filler D.
- the filler D makes it possible to improve the mechanical properties of the silicone elastomer article obtained at the end of the method, while retaining good elastomeric properties.
- the filler D makes it possible to improve the modulus at break of the silicone elastomer article obtained, while retaining a high elongation at break.
- a final object of the invention concerns the silicone elastomer article obtained according to said additive manufacturing process.
- Organopolysiloxanes E used in the examples Organopolysiloxane El:
- Organopolysiloxane E3 Organopolysiloxane E4:
- Example 1 Preparation of compositions comprising polyorganosiloxanes containing acrylate groups
- the acrylate yield is calculated as the ratio between the number of moles of acrylate dosed in the composition and the number of moles of theoretical acrylate moles calculated multiplied by 100.
- the duration for the 96% acrylate yield is the reaction time necessary to reach such a yield, noted t96% acrylate in Table 2 below.
- the comparative catalyst is Cr(OAc)3 introduced at 0.5mol% relative to the epoxide concentration for comparative tests 3 and 4.
- the comparative catalyst concentration is equal to 0.25mol% relative to the epoxide concentration for comparative tests 1 and 2.
- the acrylate selectivity is equal to the acrylate yield on the conversion rate of the epoxy groups.
- the objective is to obtain the highest possible selectivity.
- the results are presented in the following table. *For comparative test 3, the conditions had to be drastically adapted in order to guarantee the solubility of the catalyst. Thus, on the one hand the catalyst content had to be reduced to guarantee its solubility and a premix containing acrylic acid, the solvent, the catalyst and the polymerization inhibitor had to be heated for 2 hours at 45 °C. Heating the reaction medium to 45 °C is necessary to guarantee the homogeneity of the reaction medium after the addition of one third of the total mass of epoxy oil 2. Then, the reaction medium is heated to 120 °C and the oil is added by pouring over a period of one hour. The subsequent steps of the process are identical to those of the process of the present invention.
- the comparative test in the presence of chromium acetate and n-butanol shows an acrylate selectivity of less than 95% (comparative test 1). Furthermore, as previously mentioned in the present application, the presence of n-butanol leads to the production of a product with an unpleasant odor.
- the process according to the present invention makes it possible to obtain very good yields and selectivities in acrylate from all types of organopolysiloxane E oils under conditions which are suitable and adaptable to the industrial scale.
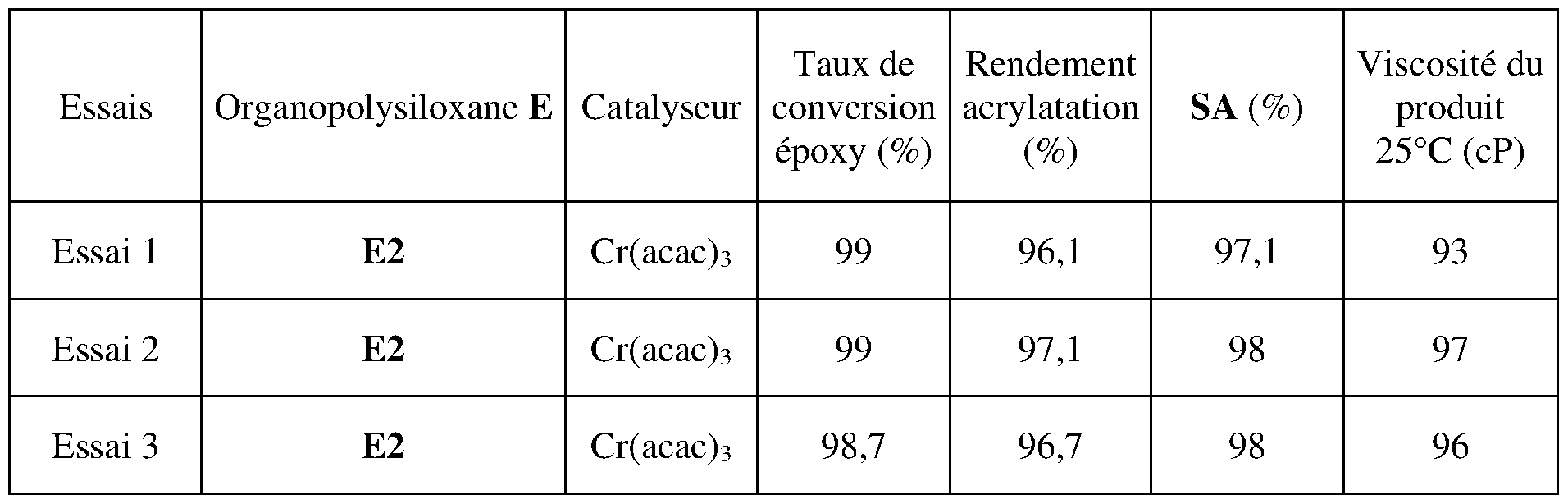
- the table below implements the method of the present invention according to three different routes.
- test 1 the organopolysiloxane oil E2 was introduced by pouring into the reaction medium containing the other constituents at a temperature of 60°C and then the reactor was heated to 120°C under conditions similar to those described previously.
- test 2 the organopolysiloxane oil E2 was introduced into the reactor heated to 45°C, then the other constituents were added to the reactor which was then heated to 120°C under conditions similar to those previously described.
- a premix made the day before is made up of: - the dissolution of the catalyst Cr(acac)3 and the inhibitor 4-methoxyphenol (MeHQ) in acrylic acid in the presence of MIBK solvent at room temperature (21°C) for a period of 5 min.
- the satisfactory miscibility of the catalytic system used in the context of the present invention allows the production of homogeneous premixes at room temperature which are directly introduced in a "one-pot" into the reactor to implement the process of the present invention.
- This flexibility makes it possible to facilitate the implementation of such a process on an industrial scale by avoiding the need for pouring of reactants, fractional heating or other obstacles to the industrialization of such a process.
Landscapes
- Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Life Sciences & Earth Sciences (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
- Wood Science & Technology (AREA)
- General Chemical & Material Sciences (AREA)
- Silicon Polymers (AREA)
Abstract
Description














Claims


Priority Applications (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| CN202480023265.5A CN120981509A (zh) | 2023-03-31 | 2024-03-29 | 具有(甲基)丙烯酸酯官能团的有机聚硅氧烷的制备方法 |
| EP24723583.1A EP4688920A1 (fr) | 2023-03-31 | 2024-03-29 | Procédé de préparation d'organopolysiloxanes à fonctions (méth)acrylates |
| KR1020257035708A KR20250166282A (ko) | 2023-03-31 | 2024-03-29 | (메트)아크릴레이트 관능기를 갖는 오르가노폴리실록산의 제조 방법 |
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| FRFR2303171 | 2023-03-31 | ||
| FR2303171 | 2023-03-31 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2024200927A1 true WO2024200927A1 (fr) | 2024-10-03 |
Family
ID=87554771
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/FR2024/000042 Ceased WO2024200927A1 (fr) | 2023-03-31 | 2024-03-29 | Procédé de préparation d'organopolysiloxanes à fonctions (méth)acrylates |
Country Status (4)
| Country | Link |
|---|---|
| EP (1) | EP4688920A1 (fr) |
| KR (1) | KR20250166282A (fr) |
| CN (1) | CN120981509A (fr) |
| WO (1) | WO2024200927A1 (fr) |
Citations (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0281718A2 (fr) | 1987-02-24 | 1988-09-14 | Rhone-Poulenc Chimie | Procédé de préparation d'un organopolysiloxane à fonction acrylate et/ou méthacrylate |
| FR2632960A1 (fr) | 1988-06-15 | 1989-12-22 | Goldschmidt Ag Th | Polysiloxanes comportant des groupes (meth)acrylates lies par l'intermediaire de groupes sic, et leur utilisation en tant que produits de revetement, durcissables sous l'effet d'un rayonnement, pour supports bidimensionnels |
| US5236637A (en) | 1984-08-08 | 1993-08-17 | 3D Systems, Inc. | Method of and apparatus for production of three dimensional objects by stereolithography |
| EP0940458A1 (fr) | 1998-03-03 | 1999-09-08 | Th. Goldschmidt AG | Utilisation d'ester de (meth)acrylate de polyols d'organosiloxane comme additif pour revêtements durcissables par rayonnement |
| EP1276825B1 (fr) | 2000-04-11 | 2004-09-15 | Rhodia, Inc. | Compositions de liberation durcissables par rayonnement, utilisation et substrats de liberation enduits |
| CN101555386A (zh) * | 2009-05-07 | 2009-10-14 | 中国科学院广州化学研究所 | 一种(甲基)丙烯酸改性环氧化有机硅紫外光固化涂料及其制备方法 |
| WO2014126837A2 (fr) | 2013-02-12 | 2014-08-21 | Eipi Systems, Inc. | Impression à interface liquide continue |
| WO2015197495A1 (fr) | 2014-06-27 | 2015-12-30 | Koninklijke Philips N.V. | Dispositif et procédé d'impression pour l'impression 3d |
| WO2016181149A1 (fr) | 2015-05-13 | 2016-11-17 | Photocentric Limited | Procédé de fabrication d'un objet |
| WO2017187030A1 (fr) | 2016-04-29 | 2017-11-02 | Bluestar Silicones France Sas | Procede de preparation d'organopolysiloxanes avec des fonctions (meth)acrylates |
| CN113831536A (zh) * | 2021-10-15 | 2021-12-24 | 江南大学 | 一种酯环环氧丙烯酸酯杂化硅树脂、制备方法及应用 |
-
2024
- 2024-03-29 CN CN202480023265.5A patent/CN120981509A/zh active Pending
- 2024-03-29 KR KR1020257035708A patent/KR20250166282A/ko active Pending
- 2024-03-29 WO PCT/FR2024/000042 patent/WO2024200927A1/fr not_active Ceased
- 2024-03-29 EP EP24723583.1A patent/EP4688920A1/fr active Pending
Patent Citations (11)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5236637A (en) | 1984-08-08 | 1993-08-17 | 3D Systems, Inc. | Method of and apparatus for production of three dimensional objects by stereolithography |
| EP0281718A2 (fr) | 1987-02-24 | 1988-09-14 | Rhone-Poulenc Chimie | Procédé de préparation d'un organopolysiloxane à fonction acrylate et/ou méthacrylate |
| FR2632960A1 (fr) | 1988-06-15 | 1989-12-22 | Goldschmidt Ag Th | Polysiloxanes comportant des groupes (meth)acrylates lies par l'intermediaire de groupes sic, et leur utilisation en tant que produits de revetement, durcissables sous l'effet d'un rayonnement, pour supports bidimensionnels |
| EP0940458A1 (fr) | 1998-03-03 | 1999-09-08 | Th. Goldschmidt AG | Utilisation d'ester de (meth)acrylate de polyols d'organosiloxane comme additif pour revêtements durcissables par rayonnement |
| EP1276825B1 (fr) | 2000-04-11 | 2004-09-15 | Rhodia, Inc. | Compositions de liberation durcissables par rayonnement, utilisation et substrats de liberation enduits |
| CN101555386A (zh) * | 2009-05-07 | 2009-10-14 | 中国科学院广州化学研究所 | 一种(甲基)丙烯酸改性环氧化有机硅紫外光固化涂料及其制备方法 |
| WO2014126837A2 (fr) | 2013-02-12 | 2014-08-21 | Eipi Systems, Inc. | Impression à interface liquide continue |
| WO2015197495A1 (fr) | 2014-06-27 | 2015-12-30 | Koninklijke Philips N.V. | Dispositif et procédé d'impression pour l'impression 3d |
| WO2016181149A1 (fr) | 2015-05-13 | 2016-11-17 | Photocentric Limited | Procédé de fabrication d'un objet |
| WO2017187030A1 (fr) | 2016-04-29 | 2017-11-02 | Bluestar Silicones France Sas | Procede de preparation d'organopolysiloxanes avec des fonctions (meth)acrylates |
| CN113831536A (zh) * | 2021-10-15 | 2021-12-24 | 江南大学 | 一种酯环环氧丙烯酸酯杂化硅树脂、制备方法及应用 |
Also Published As
| Publication number | Publication date |
|---|---|
| EP4688920A1 (fr) | 2026-02-11 |
| KR20250166282A (ko) | 2025-11-27 |
| CN120981509A (zh) | 2025-11-18 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| EP3353252B1 (fr) | Composition silicone reticulable pour la realisation de revetements anti-adherents pour supports souples et additif promoteur d'accrochage contenu dans cette composition | |
| EP2563870B1 (fr) | Composition silicone reticulable pour la realisation de revetements anti-adherents pour supports souples et additif promoteur d'accrochage contenu dans cette composition | |
| EP3215557B1 (fr) | Nouveaux catalyseurs de reticulation de compositions silicones | |
| FR2712297A1 (fr) | Compositions à base d'époxysilicone, durcissables par les rayons UV, antiadhésives. | |
| CA3022629C (fr) | Procede de preparation d'organopolysiloxanes avec des fonctions (meth)acrylates | |
| EP2024438B1 (fr) | Composition silicone reticulable pour la realisation de revetements anti-adherents pour films polymeres | |
| FR2806930A1 (fr) | Utilisation d'un derive de bore a titre de catalyseur thermoactivable pour la polymerisation et/ou reticulation de silicone par deshydrogenocondensation | |
| EP0562922A1 (fr) | Compositions à base de polyorganosiloxanes à groupements fonctionnels réticulables et leur utilisation pour la réalisation de revêtements antiadhésifs | |
| EP3083855B1 (fr) | Nouveau systeme d'inhibition d'hydrosilylation photoactivable | |
| EP1242553A1 (fr) | Complexe silicone reticulable thermiquement/adhesif dont l'interface possede une force de decollement modulable | |
| EP2655517A1 (fr) | Inhibiteurs de réaction d'hydrosilylation et leur application pour la préparation de compositions silicones durcissables stables | |
| EP3320051B1 (fr) | Procede de preparation d'organopolysiloxanes avec des fonctions (meth)acrylates | |
| FR2824835A1 (fr) | Composition silicone polymerisable reticulable par voie cationique, sous activation thermique et au moyen d'un amorceur de type adduit acide/base de lewis | |
| EP0304381B1 (fr) | Article stratifié souple pour adhésif de transfert | |
| WO2001085864A1 (fr) | Complexe silicone/adhesif dont l'interface possede une force de decollement modulable par irradiation par faisceau d'electrons | |
| FR2717482A1 (fr) | Complexes organoplatiniques et systèmes catalytiques photoactivables d'hydrosilylation en contenant. | |
| EP0750624A1 (fr) | Complexes du platine et catalyseurs d'hydrosilylation photoactivables les contenant | |
| EP4688920A1 (fr) | Procédé de préparation d'organopolysiloxanes à fonctions (méth)acrylates | |
| WO2012084992A1 (fr) | Composition silicone reticulable par hydrosilylation et procede de revetement ou de fabrication d'objets a partir de cette composition | |
| FR2707655A1 (fr) | Nouveaux polymères siliconés à fonctions oléfiniques, leur procédé de préparation et compositions durcissables comprenant lesdits polymères. | |
| EP0402274A1 (fr) | Composition durcissable, sous un rayonnement, à base de polyorganosiloxane à fonction propanaldéhyde |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 24723583 Country of ref document: EP Kind code of ref document: A1 |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 202547092011 Country of ref document: IN |
|
| ENP | Entry into the national phase |
Ref document number: 2025557104 Country of ref document: JP Kind code of ref document: A |
|
| WWE | Wipo information: entry into national phase |
Ref document number: CN2024800232655 Country of ref document: CN Ref document number: 2025557104 Country of ref document: JP |
|
| ENP | Entry into the national phase |
Ref document number: 1020257035708 Country of ref document: KR Free format text: ST27 STATUS EVENT CODE: A-0-1-A10-A15-NAP-PA0105 (AS PROVIDED BY THE NATIONAL OFFICE) |
|
| WWE | Wipo information: entry into national phase |
Ref document number: KR1020257035708 Country of ref document: KR |
|
| WWE | Wipo information: entry into national phase |
Ref document number: 2024723583 Country of ref document: EP |
|
| WWP | Wipo information: published in national office |
Ref document number: 202547092011 Country of ref document: IN |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |
|
| ENP | Entry into the national phase |
Ref document number: 2024723583 Country of ref document: EP Effective date: 20251031 |
|
| ENP | Entry into the national phase |
Ref document number: 2024723583 Country of ref document: EP Effective date: 20251031 |
|
| ENP | Entry into the national phase |
Ref document number: 2024723583 Country of ref document: EP Effective date: 20251031 |
|
| ENP | Entry into the national phase |
Ref document number: 2024723583 Country of ref document: EP Effective date: 20251031 |
|
| ENP | Entry into the national phase |
Ref document number: 2024723583 Country of ref document: EP Effective date: 20251031 |
|
| ENP | Entry into the national phase |
Ref document number: 2024723583 Country of ref document: EP Effective date: 20251031 |
|
| ENP | Entry into the national phase |
Ref document number: 2024723583 Country of ref document: EP Effective date: 20251031 |
|
| ENP | Entry into the national phase |
Ref document number: 2024723583 Country of ref document: EP Effective date: 20251031 |
|
| WWP | Wipo information: published in national office |
Ref document number: 2024723583 Country of ref document: EP |