WO2024252885A1 - セロビオースの製造方法 - Google Patents
セロビオースの製造方法 Download PDFInfo
- Publication number
- WO2024252885A1 WO2024252885A1 PCT/JP2024/018152 JP2024018152W WO2024252885A1 WO 2024252885 A1 WO2024252885 A1 WO 2024252885A1 JP 2024018152 W JP2024018152 W JP 2024018152W WO 2024252885 A1 WO2024252885 A1 WO 2024252885A1
- Authority
- WO
- WIPO (PCT)
- Prior art keywords
- phosphorylase
- cellobiose
- sucrose
- yeast
- seq
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Ceased
Links
Images





Classifications
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N15/00—Mutation or genetic engineering; DNA or RNA concerning genetic engineering, vectors, e.g. plasmids, or their isolation, preparation or purification; Use of hosts therefor
- C12N15/09—Recombinant DNA-technology
- C12N15/63—Introduction of foreign genetic material using vectors; Vectors; Use of hosts therefor; Regulation of expression
- C12N15/79—Vectors or expression systems specially adapted for eukaryotic hosts
- C12N15/80—Vectors or expression systems specially adapted for eukaryotic hosts for fungi
- C12N15/81—Vectors or expression systems specially adapted for eukaryotic hosts for fungi for yeasts
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/10—Transferases (2.)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/90—Isomerases (5.)
- C12N9/92—Glucose isomerase (5.3.1.5; 5.3.1.9; 5.3.1.18)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12P—FERMENTATION OR ENZYME-USING PROCESSES TO SYNTHESISE A DESIRED CHEMICAL COMPOUND OR COMPOSITION OR TO SEPARATE OPTICAL ISOMERS FROM A RACEMIC MIXTURE
- C12P19/00—Preparation of compounds containing saccharide radicals
- C12P19/12—Disaccharides
Definitions
- the present invention relates to a method for producing cellobiose from sucrose using yeast that expresses an enzyme on its surface.
- Enzymatic degradation and enzymatic synthesis methods have been known as methods for producing cellobiose.
- an enzymatic degradation method is known in which a cellulase preparation is reacted with cellulose to obtain cellobiose (Patent Document 1, Patent Document 2).
- Non-Patent Document 1 an enzymatic synthesis method is known in which starch is used as a starting material and ⁇ -glucan phosphorylase and cellobiose phosphorylase are allowed to act on the starch to obtain cellobiose
- Patent Document 3 a method is known in which sucrose is used as a starting material and sucrose phosphorylase, glucose isomerase, and cellobiose phosphorylase are allowed to act on the sucrose to obtain cellobiose.
- cellulose is used as the raw material.
- Cellulose is the main component of plant cell walls, but it usually exists as a mixture with hemicellulose, lignin, etc. Therefore, when using cellulose in such plant cell walls as a raw material, it is first necessary to remove the hemicellulose, lignin, etc. by methods such as alkali treatment, which poses the problem of high production costs for the raw material.
- the enzymatic synthesis method has low conversion efficiency from starch to glucose 1-phosphate and from sucrose to cellobiose, and requires reactions to be carried out under mild conditions (around 30°C) using enzymes, resulting in a slow reaction rate and making it unsuitable for low-cost cellobiose production.
- the object of the present invention is to provide a method for producing cellobiose from sucrose with a high conversion rate and at low cost.
- the present inventors have discovered that by allowing sucrose to coexist in a reaction solution containing yeast expressing sucrose phosphorylase (hereinafter sometimes referred to as "SP") and cellobiose phosphorylase (hereinafter sometimes referred to as “CBP”) and glucose isomerase (hereinafter sometimes referred to as "XI”), it is possible to produce cellobiose at a high conversion rate and at low cost, and have completed the present invention.
- SP sucrose phosphorylase
- CBP cellobiose phosphorylase
- XI glucose isomerase
- the present invention includes the following inventions.
- [Item 1] A method for producing cellobiose, comprising a reaction step of allowing sucrose to coexist in a reaction solution containing a recombinant yeast expressing sucrose phosphorylase and cellobiose phosphorylase and glucose isomerase.
- [Item 2] The production method according to Item 1, wherein the recombinant yeast is a yeast of the genus Saccharomyces, Pichia, or Yarrowia.
- [Item 3] The production method according to Item 1 or 2, wherein the sucrose phosphorylase is derived from a bacterium of the genus Bifidobacterium, Leuconostoc, or Streptococcus.
- sucrose phosphorylase comprises a secreted sucrose phosphorylase secreted and expressed from a recombinant yeast, and an tethered sucrose phosphorylase expressed in a state tethered to the cell wall of the recombinant yeast.
- the cellobiose phosphorylase is derived from a bacterium of the genus Clostridium, Ruminococcus, Cellulomonas, or Thermotoga.
- [Item 6] The method according to any one of Items 1 to 5, wherein the recombinant yeast is a recombinant yeast that expresses both sucrose phosphorylase and cellobiose phosphorylase in a single cell.
- the recombinant yeast is a combination of a recombinant yeast expressing sucrose phosphorylase and a recombinant yeast expressing cellobiose phosphorylase.
- the recombinant yeast is a yeast transformed with an expression vector carrying a sucrose phosphorylase gene.
- [Item 9] The production method described in Item 8, wherein the expression vector comprises a combination of a first expression vector carrying both a sucrose phosphorylase gene and a yeast cell wall domain gene, and a second expression vector carrying only the sucrose phosphorylase gene but not the yeast cell wall domain gene.
- the glucose isomerase is derived from a bacterium of the genus Streptomyces.
- the method according to any one of Items 1 to 10 further comprising treating the recombinant yeast at 45° C. to 75° C. for 15 to 150 minutes before allowing sucrose to coexist in the reaction solution.
- the present invention makes it possible to produce cellobiose from sucrose with a high conversion rate and low cost.
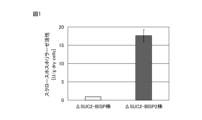
- FIG. 1 is a graph showing the results of measuring the sucrose phosphorylase activity of a recombinant yeast strain expressing sucrose phosphorylase.
- FIG. 2 is a graph showing the changes over time in the concentrations of sucrose, glucose, fructose, and cellobiose in the reaction supernatant in an experiment to produce cellobiose from sucrose using a recombinant yeast strain expressing sucrose phosphorylase in combination with a recombinant yeast strain expressing cellobiose phosphorylase.
- the graph in Figure 2(a) shows the results when a strain expressing cellobiose phosphorylase (CsCBP) derived from Clostridium stercorarium was used
- the graph in Figure 2(b) shows the results when a strain expressing cellobiose phosphorylase (CuCBP) derived from Cellulomonas uda was used
- the graph in Figure 2(c) shows the results when a strain expressing cellobiose phosphorylase (TaCBP) derived from Thermosipho africanus was used
- the graph in Figure 2(d) shows the results when a strain expressing cellobiose phosphorylase (TnCBP) derived from Thermotoga neapolitana was used.
- Figure 3 is a graph showing the time course of the concentrations of sucrose, glucose, fructose, and cellobiose in the reaction supernatant in an experiment to produce cellobiose from sucrose using a single recombinant yeast strain co-expressing sucrose phosphorylase and cellobiose phosphorylase.
- the graph in Figure 3(a) shows the results when a recombinant yeast with Saccharomyces cerevisiae as the host was used
- the graph in Figure 3(b) shows the results when a recombinant yeast with Pichia pastoris as the host was used.
- Figure 4 is a graph showing the time course of the concentrations of sucrose, glucose, fructose, and cellobiose in the reaction supernatant of an experiment to produce cellobiose from sucrose using a single recombinant yeast strain co-expressing sucrose phosphorylase and cellobiose phosphorylase with Pichia pastoris as the host.
- the graph in Figure 4(a) shows the results when the Pp-SP2CBP strain was cultured using glucose as the carbon source
- the graph in Figure 4(b) shows the results when the Pp-SP2CBP strain was cultured using glycerol as the carbon source.
- Figure 5 is a graph showing the time course of sucrose, glucose, fructose, and cellobiose concentrations in the reaction supernatant of a repeated experiment of cellobiose production from sucrose using a single recombinant yeast strain co-expressing sucrose phosphorylase and cellobiose phosphorylase.
- the graph in Figure 5(a) shows the results when recombinant yeast with Saccharomyces cerevisiae as the host was cultured using glucose as the carbon source
- the graph in Figure 5(b) shows the results when recombinant yeast with Pichia pastoris as the host was cultured using glycerol as the carbon source.
- identity of amino acid sequences means the proportion of identical amino acid residues
- similarity means the proportion of identical or similar amino acid residues.
- homology and identity of amino acid sequences can be determined, for example, by the BLAST method (NCBI PBLAST default conditions). Furthermore, for example, when it is expressed as “80% or more homology,” it clearly includes the case of "80% or more identity.”
- similar amino acid residues refers to amino acid residues having side chains with similar chemical properties (e.g., charge or hydrophobicity).
- similar amino acid residues include the following combinations: (1) Amino acid residues having an aliphatic side chain: glycine (Gly or G), alanine (Ala or A), valine (Val or V), leucine (Leu or L), and isoleucine (Ile or I) residues. (2) Amino acid residues having aliphatic hydroxyl side chains: serine (Ser or S) and threonine (Thr or T) residues.
- Amino acid residues having amide-containing side chains asparagine (Asn or N) and glutamine (Gln or Q) residues.
- Amino acid residues having aromatic side chains phenylalanine (Phe or F), tyrosine (Tyr or Y), and tryptophan (Trp or W) residues.
- Amino acid residues having basic side chains lysine (Lys or K), arginine (Arg or R), and histidine (His or H) residues.
- Amino acid residues having acidic side chains aspartic acid (Asp or D) and glutamic acid (Glu or E) residues.
- Amino acid groups having sulfur-containing side chains cysteine (Cys or C) and methionine (Met or M) residues. Furthermore, the combination of (1) and a methionine (Met or M) residue, and the combination of (4) and a histidine (His or H) residue are also treated as similar amino acid residues.
- One aspect of the present invention relates to a method for producing cellobiose.
- the method includes a step of allowing sucrose to coexist in a reaction solution containing a recombinant yeast expressing sucrose phosphorylase (SP) and cellobiose phosphorylase (CBP) and glucose isomerase (XI).
- SP sucrose phosphorylase
- CBP cellobiose phosphorylase
- XI glucose isomerase
- the yeast species is not particularly limited as long as it is capable of expressing sucrose phosphorylase (SP) and cellobiose phosphorylase (CBP).
- examples include yeasts of the genus Saccharomyces, Pichia, or Yarrowia.
- yeasts that do not have invertase activity on the cell surface such as Pichia pastoris.
- yeasts that have strong invertase activity on the cell surface such as Saccharomyces cerevisiae, it is preferable to use a strain in which invertase activity has been lost by gene knockout or the like.
- Pichia pastoris since Pichia pastoris has been classified and renamed as Komagataella pastoris and Komagataella phaffii, Pichia pastoris is a synonym for both Komagataella pastoris and Komagataella phaffii.
- sucrose phosphorylase The origin (species) of sucrose phosphorylase (SP) is not particularly limited. Examples include those derived from bacteria of the genus Bifidobacterium, Leuconostoc, or Streptococcus. Of these, those derived from bacteria of the genus Bifidobacterium are preferred, and those derived from Bifidobacterium longum are particularly preferred.
- the amino acid sequence of sucrose phosphorylase (SP) from Bifidobacterium longum is shown in SEQ ID NO: 34, and an example of a base sequence encoding this is shown in SEQ ID NO: 5.
- sucrose phosphorylase (SP) derived from Bifidobacterium longum examples include proteins consisting of an amino acid sequence having 80% or more, 85% or more, 88% or more, 90% or more, 93% or more, 95% or more, 97% or more, 98% or more, or 99% or more homology (preferably identity) with SEQ ID NO:34.
- sucrose phosphorylase When sucrose phosphorylase (SP) is expressed in recombinant yeast, it can be in the form of a secreted enzyme secreted and expressed from the recombinant yeast, or in the form of an tethered enzyme expressed in a state tethered to the cell wall of the recombinant yeast, but either form is acceptable. However, in the present invention, it is preferable to allow the secreted sucrose phosphorylase (SP) secreted and expressed from the recombinant yeast and the tethered sucrose phosphorylase (SP) expressed in a state tethered to the cell wall of the recombinant yeast to coexist in the reaction solution. This makes it possible to improve the conversion efficiency by sucrose phosphorylase (SP). The reason for this is unclear, but it is speculated to be due to the formation of a dimer between the secreted sucrose phosphorylase (SP) and the tethered sucrose phosphorylase (SP).
- CBP cellobiose phosphorylase
- SEQ ID NO: 35 amino acid sequence of cellobiose phosphorylase (CBP) from Clostridium stercorarium is shown in SEQ ID NO: 35, and an example of the base sequence encoding this is shown in SEQ ID NO: 6.
- CBP cellobiose phosphorylase
- CBP cellobiose phosphorylase
- it may be in the form of a secreted enzyme secreted and expressed from the recombinant yeast, or in the form of an tethered enzyme expressed in a state tethered to the cell wall of the recombinant yeast, either of which may be used.
- it is preferable to express it in the form of tethered cellobiose phosphorylase (CBP) expressed in a state tethered to the cell wall of the recombinant yeast.
- recombinant yeast As the recombinant yeast, a recombinant yeast that co-expresses sucrose phosphorylase (SP) and cellobiose phosphorylase (CBP) in a single cell may be used, or a recombinant yeast that expresses sucrose phosphorylase (SP) may be used in combination with a recombinant yeast that expresses cellobiose phosphorylase (CBP).
- SP sucrose phosphorylase
- CBP cellobiose phosphorylase
- sucrose phosphorylase SP
- CBP cellobiose phosphorylase
- a recombinant yeast that co-expresses the secreted enzyme and the tethered enzyme in a single cell may be used, or a recombinant yeast that expresses the secreted enzyme may be used in combination with a recombinant yeast that expresses the tethered enzyme.
- the method for preparing recombinant yeast expressing sucrose phosphorylase (SP) and/or cellobiose phosphorylase (CBP) is not particularly limited.
- one or more types of yeast are transformed with one or more expression vectors carrying the sucrose phosphorylase (SP) gene and/or the cellobiose phosphorylase (CBP) gene.
- expression vectors include plasmids, cosmids, lambda phages, etc., with plasmids being preferred.
- sucrose phosphorylase (SP) gene and/or cellobiose phosphorylase (CBP) gene are operably linked to regulatory sequences such as a promoter and a terminator, and carried in the expression vector in the form of an expression cassette that can be expressed autonomously in yeast cells.
- regulatory sequences such as a promoter and a terminator
- Such expression vectors can be constructed using standard genetic engineering techniques known to those skilled in the art, by referring appropriately to literature such as Michael R. Green et al., Molecular Cloning: A Laboratory Manual, Fourth Edition, (2012), Cold Spring Harbor Laboratory.
- the recombinant yeast is preferably pre-cultured in the presence of a carbon source prior to the reaction.
- a carbon source there are no particular limitations on the type of carbon source used when culturing the recombinant yeast cells, and it may be selected appropriately depending on the type of yeast. Examples include glucose and glycerol. Of these, in the present invention, it is preferable to use at least glycerol as a carbon source. This can improve the conversion rate of sucrose to cellobiose.
- a pretreatment prior to the reaction to stop the uptake of glucose.
- An example of such a pretreatment is a heat treatment in which the yeast is exposed to a certain temperature for a certain period of time.
- the treatment conditions are sufficient as long as they can stop the uptake of glucose by the yeast used.
- the heating temperature can be, for example, 45°C or higher, or 50°C or higher, or 55°C or higher, and can be, for example, 75°C or lower, or 70°C or lower, or 65°C or lower.
- the heating time can be, for example, 15 minutes or more, or 45 minutes or more, or 70 minutes or more, and can be, for example, 150 minutes or less, 120 minutes or less, or 100 minutes or less.
- the temperature can be about 60°C, and the time can be about 90 minutes.
- One example of such a pretreatment method is shaking a suspension of the transformed yeast under the above conditions.
- glucose isomerase (XI) is allowed to coexist in a reaction solution containing recombinant yeast expressing sucrose phosphorylase (SP) and cellobiose phosphorylase (CBP).
- the amount of recombinant yeast cells in the reaction solution is not particularly limited, but can be, for example, 25 g wet cells/L or more, 50 g wet cells/L or more, or 75 g wet cells/L or more, and can be, for example, 200 g wet cells/L or less, 150 g wet cells/L or less, or 100 g wet cells/L or less.
- Glucose isomerase also known as xylose isomerase, XI
- XI xylose isomerase
- Glucose isomerase may be expressed in yeast or other microorganisms in the same manner as other enzymes, but it is preferable to use the isolated glucose isomerase (XI) enzyme itself.
- the isolated glucose isomerase (XI) enzyme may be either a commercially available product or one obtained by culturing a microorganism that produces these enzymes. It may also be either a purified product or an unpurified product. In this case, it is preferable to add the isolated glucose isomerase (XI) enzyme to a reaction solution containing recombinant yeast that expresses sucrose phosphorylase (SP) and cellobiose phosphorylase (CBP) and allow them to coexist.
- SP sucrose phosphorylase
- CBP cellobiose phosphorylase
- the amount of glucose isomerase (XI) is not particularly limited, but is usually preferably 0.01 units or more, 0.1 units or more, or 1.0 units or more per mole of sucrose.
- 1 unit (1 U) means the amount of enzyme that breaks down 1 ⁇ mol of fructose in 1 wt/vol % sucrose at 50°C and pH 7.0 per minute.
- the reaction is carried out in the presence of sucrose in a reaction solution containing recombinant yeast expressing sucrose phosphorylase (SP) and cellobiose phosphorylase (CBP) and glucose isomerase (XI).
- sucrose may be naturally occurring or chemically synthesized.
- concentration as high as possible is preferable.
- solubility of the cellobiose produced is about 15 to 20%, if it is desired to keep it dissolved, it is preferable to use a sucrose concentration of about 10 to 20%.
- the conditions for the cellobiose synthesis reaction are not limited, but may be, for example, as follows. That is, the reaction temperature may be, for example, 35°C or higher, 45°C or higher, or 50°C or higher, and, for example, 60°C or lower, or 70°C or lower.
- the reaction time may be, for example, 60 minutes or more, 12 hours or more, or 24 hours or more, and may be, for example, 36 hours or less, or 48 hours or less.
- the pressure during the reaction is not particularly limited, and may be reduced pressure, normal pressure, or pressurized, but is usually carried out under normal pressure.
- the atmosphere during the reaction is also not particularly limited, and any atmosphere may be used as long as the enzyme activity is maintained even in an atmosphere in which the recombinant yeast used cannot grow.
- cellobiose can be obtained by separating and purifying it from the reaction solution as necessary.
- any method can be used, including, for example, filtration separation, centrifugation, membrane separation, etc. These methods allow efficient separation of the used bacteria and cellobiose.
- it may be purified to a higher degree by treatment with an ion exchange resin or recrystallization.
- the recombinant yeast may be separated and recovered from the reaction solution as necessary and reused in the cellobiose synthesis reaction. Even when the recombinant yeast of the present invention is separated and recovered from the reaction solution and reused in this way, the enzyme activity may be maintained, and high cellobiose synthesis efficiency may be repeatedly obtained. That is, according to a preferred embodiment, the cellobiose production method of the present invention further includes recovering the recombinant yeast from the reaction solution after the reaction step and subjecting it to another reaction step.
- the method for separating and recovering the recombinant yeast from the reaction solution is not limited, but examples include centrifugation.
- the number of times the recombinant yeast is separated and recovered and reused is not particularly limited, but according to one embodiment, it can be subjected to a reaction, for example, two or more times, three or more times, four or more times, or five or more times.
- the activity of the recombinant yeast when reused is not particularly limited, but according to one embodiment, it is preferable that 60% or more, 70% or more, 80% or more, or 90% or more of the activity in the previous reaction is maintained in the next reaction.
- Example Group I Cellobiose production using transformed yeast (yeast displaying sucrose phosphorylase on the surface, yeast displaying cellobiose phosphorylase on the surface, and yeast displaying sucrose phosphorylase + cellobiose phosphorylase on the surface)]
- yeast Saccharomyces cerevisiae BY4741 strain MAT ⁇ his3 leu2 met15 ura3 strain
- yeast Saccharomyces cerevisiae BY4741 ⁇ SUC2 strain MAT ⁇ his3 leu2 met15 ura3 suc2 strain
- the yeast Saccharomyces cerevisiae BY4741 strain was obtained from ATCC (American Type Culture Collection), and the yeast Saccharomyces cerevisiae BY4741 ⁇ SUC2 strain was obtained from Yeast deletion MAT-A complete set (Thermo Fisher Scientific).
- Plasmids containing the following expression cassettes X1 to X9 were prepared by the following procedure.
- vector plasmid pIEG-SSSD (a surface expression vector having an expression cassette in which the SED1 promoter derived from Saccharomyces cerevisiae, the secretory signal peptide sequence of SED1 derived from Saccharomyces cerevisiae, the coding region of the endoglucanase II (TrEGII) gene derived from Trichoderma reesei, the coding region (SED1 anchoring region) of the SED1 gene derived from Saccharomyces cerevisiae, and the terminator region of the DIT1 gene derived from Saccharomyces cerevisiae are arranged in this order: Applied Microbiology and Biotechnology, Vol.105, 2021,
- the resulting vector plasmid was amplified by PCR using primer pair SED1a-F (SEQ ID NO: 14) and SED1ss-R (SEQ ID NO: 15) with pIBISP-SSSD (SEQ ID NO: 5895-590
- the resulting plasmid containing expression cassette X1 (SED1 promoter (SEQ ID NO: 1) + SED1 secretion signal (SEQ ID NO: 2) + BlSP (SEQ ID NO: 5) + SED1 anchoring region (SEQ ID NO: 3) + DIT1 terminator (SEQ ID NO: 4)) was named pIBlSP-SSSD.
- the primer pair BlSP-F (SEQ ID NO: 18) and DIT1t-R (SEQ ID NO: 19) were used to amplify by PCR to prepare a DNA fragment containing the coding region of the sucrose phosphorylase (BlSP) gene derived from Bifidobacterium longum and the terminator region of the DIT1 gene derived from Saccharomyces cerevisiae.
- BlSP sucrose phosphorylase
- vector plasmid pIL2-BG-SSS (a surface expression vector having an expression cassette in which the SED1 promoter derived from Saccharomyces cerevisiae, the secretory signal peptide sequence of SED1 derived from Saccharomyces cerevisiae, the coding region of the ⁇ -glucosidase 1 (AaBGL1) gene derived from Aspergillus aculeatus, the coding region (SED1 anchoring region) of the SED1 gene derived from Saccharomyces cerevisiae, and the terminator region of the ⁇ -agglutinin gene derived from Saccharomyces cerevisiae are arranged in this order: Metabolic Engineering, Vol.
- pRS403-F SEQ ID NO: 20
- pRS403-S SEQ ID NO: 15
- pRS403-F SEQ ID NO: 20
- pRS403-S SEQ ID NO: 15
- pRS403-F SEQ ID NO: 20
- a DNA fragment was prepared that contained the entire length of the vector plasmid excluding the coding region of the ⁇ -glucosidase 1 (AaBGL1) gene derived from Aspergillus aculeatus, the coding region (SED1 anchoring region) of the SED1 gene derived from Saccharomyces cerevisiae, and the terminator region of the ⁇ -agglutinin gene derived from Saccharomyces cerevisiae. These fragments were ligated by the In-Fusion method.
- the resulting plasmid containing the expression cassette X2 (SED1 promoter (SEQ ID NO: 1) + SED1 secretion signal (SEQ ID NO: 2) + BlSP (SEQ ID NO: 5) + DIT1 terminator (SEQ ID NO: 4)) was named pIL2-BlSP-SSD.
- Preparation Example 1-3 Preparation of vector plasmid containing Clostridium stercorarium-derived cellobiose phosphorylase (CsCBP) surface expression cassette X3) A DNA fragment containing the coding region of the cellobiose phosphorylase (CsCBP) gene derived from Clostridium stercorarium was prepared by gene synthesis.
- CsCBP Clostridium stercorarium-derived cellobiose phosphorylase
- vector plasmid pIEG-SSSD (a surface expression vector having a cassette in which the SED1 promoter derived from Saccharomyces cerevisiae, the secretory signal peptide sequence of SED1 derived from Saccharomyces cerevisiae, the coding region of the endoglucanase II (TrEGII) gene derived from Trichoderma reesei, the coding region (SED1 anchoring region) of the SED1 gene derived from Saccharomyces cerevisiae, and the terminator region of the DIT1 gene derived from Saccharomyces cerevisiae are arranged in this order: Applied Microbiology and Biotechnology, Vol.105, 2021, Using the primer pair SED1a-F (SEQ ID NO: 14) and SED1ss-R (SEQ ID NO: 15) and PCR amplification was performed using the 5'-terminal end of the primer pair SED1a-F (SEQ ID NO: 14)
- SED1p-F SEQ ID NO: 21
- DIT1t-R SEQ ID NO: 19
- vector plasmid pIU5-CBH D (a surface expression vector having a cassette in which the Saccharomyces cerevisiae-derived SED1 promoter, the coding region of the Talaromyces emersonii-derived cellobiohydrolase (TsCBH1) gene, the coding region of the Saccharomyces cerevisiae-derived SED1 gene (SED1 anchoring region), and the terminator region of the Saccharomyces cerevisiae-derived ⁇ -agglutinin gene are arranged in this order: Biotechnology and Bioengineering, Vol.
- TsCBH1 Talaromyces emersonii-derived cellobiohydrolase
- the vector plasmid was amplified by PCR using primer pair pRS403-F (SEQ ID NO: 20) and pRS403-R (SEQ ID NO: 22) to prepare a DNA fragment containing the entire length of the vector plasmid excluding the expression cassette region. These fragments were ligated by the In-Fusion method.
- the resulting plasmid containing expression cassette X3 (SED1 promoter (SEQ ID NO: 1) + SED1 secretion signal (SEQ ID NO: 2) + CsCBP (SEQ ID NO: 6) + SED1 anchoring region (SEQ ID NO: 3) + DIT1 terminator (SEQ ID NO: 4)) was named pIU5-CsCBP-SSSD.
- the vector plasmid pIU5-CsCBP-SSSD was used as a template, and the primer pair SED1a-F (SEQ ID NO: 14) and SED1ss-R (SEQ ID NO: 15) were used to amplify by PCR to prepare a DNA fragment containing the full length of the vector plasmid excluding the coding region of the CsCBP gene. These fragments were ligated by the In-Fusion method.
- the resulting plasmid containing the expression cassette X4 (SED1 promoter (SEQ ID NO: 1) + SED1 secretion signal (SEQ ID NO: 2) + CuCBP (SEQ ID NO: 7) + SED1 anchoring region (SEQ ID NO: 3) + DIT1 terminator (SEQ ID NO: 4)) was named pIU5-CuCBP-SSSD.
- the vector plasmid pIU5-CsCBP-SSSD was used as a template, and the primer pair SED1a-F (SEQ ID NO: 14) and SED1ss-R (SEQ ID NO: 15) were used to amplify the vector plasmid by PCR, to prepare a DNA fragment containing the full length of the vector plasmid excluding the coding region of the CsCBP gene. These fragments were ligated by the In-Fusion method.
- the resulting plasmid containing the expression cassette X5 (SED1 promoter (SEQ ID NO: 1) + SED1 secretion signal (SEQ ID NO: 2) + TaCBP (SEQ ID NO: 8) + SED1 anchoring region (SEQ ID NO: 3) + DIT1 terminator (SEQ ID NO: 4)) was named pIU5-TaCBP-SSSD.
- Preparation Example 1-6 Preparation of vector plasmid containing Thermotoga neapolitana-derived cellobiose phosphorylase (TnCBP) surface expression cassette X6) A DNA fragment containing the coding region of the Thermotoga neapolitana-derived cellobiose phosphorylase (TnCBP) gene was prepared by gene synthesis.
- TnCBP Thermotoga neapolitana-derived cellobiose phosphorylase
- the vector plasmid pIU5-CsCBP-SSSD was used as a template, and the primer pair SED1a-F (SEQ ID NO: 14) and SED1ss-R (SEQ ID NO: 15) were used to amplify the vector plasmid by PCR, to prepare a DNA fragment containing the full length of the vector plasmid excluding the coding region of the CsCBP gene. These fragments were ligated by the In-Fusion method.
- the resulting plasmid containing the expression cassette X6 (SED1 promoter (SEQ ID NO: 1) + SED1 secretion signal (SEQ ID NO: 2) + TnCBP (SEQ ID NO: 9) + SED1 anchoring region (SEQ ID NO: 3) + DIT1 terminator (SEQ ID NO: 4)) was named pIU5-TnCBP-SSSD.
- Preparation Example 1-7 Preparation of vector plasmid containing Bifidobacterium longum-derived sucrose phosphorylase (BISP) surface expression cassette X7) Using the plasmid pIBlSP-SSSD as a template, amplified by PCR using the primer pair BlSP-NheI-F (SEQ ID NO: 23) and BlSP-XhoI-R (SEQ ID NO: 24), and further treated with NheI and XhoI, to prepare a DNA fragment containing the coding region of the BlSP gene.
- BlSP-NheI-F SEQ ID NO: 23
- BlSP-XhoI-R SEQ ID NO: 24
- vector plasmid pIBG-PpSSG61 (a surface expression vector having an expression cassette in which the SPI1 promoter derived from Pichia pastoris, the secretory signal peptide sequence of SPI1 derived from Pichia pastoris, the coding region of the ⁇ -glucosidase 1 (AaBGL1) gene derived from Aspergillus aculeatus, the coding region of the GCW61 gene derived from Pichia pastoris (GCW61 anchoring region), and the terminator region of the AOX1 gene derived from Pichia pastoris are arranged in this order: Biotechnology and Bioengineering, Vol.120, 2023, 1097-1107) was treated with NheI and XhoI to prepare a DNA fragment containing the entire length of the vector plasmid excluding the coding region of the AaBGL1 gene.
- plasmid pIBG-PpGMG30 (a surface expression vector having an expression cassette in which a GAPDH promoter derived from Pichia pastoris, a secretory signal peptide sequence of ⁇ -factor derived from Saccharomyces cerevisiae, a coding region of ⁇ -glucosidase 1 (AaBGL1) gene derived from Aspergillus aculeatus, a coding region of GCW30 gene derived from Pichia pastoris (GCW30 anchoring region), and a terminator region of AOX1 gene derived from Pichia pastoris are arranged in this order: Biotechnology and Bioengineering, Vol.120, 2023, 1097-1107) was treated with XhoI and Ml
- the vector plasmid was amplified by PCR using pIH-PpSS (SEQ ID NO: 28) and a DNA fragment containing the entire length of the vector plasmid excluding the coding region of the TrEGII gene and the GCW34 anchoring region was prepared. These fragments were ligated by the In-Fusion method.
- the resulting plasmid containing the expression cassette X8 (SPI1 promoter (SEQ ID NO: 10) + SPI1 secretion signal (SEQ ID NO: 11) + BlSP (SEQ ID NO: 5) + AOX1 terminator (SEQ ID NO: 13)) was named pIH-BlSP-PpSS.
- a DNA fragment containing the full length of the vector plasmid excluding the coding region of the BlSP gene was prepared by treating the vector plasmid pIBlSP-PpSSG30 with NheI and XhoI. These fragments were ligated.
- this plasmid was used as a template and amplified by PCR using the primer pair CsCBP-F (SEQ ID NO: 31) and AOX1t-R (SEQ ID NO: 32) to prepare a DNA fragment containing the coding region of the CsCBP gene, the GCW30 anchoring region, and the AOX1 terminator region.
- a vector plasmid pIZ-CBH1-PpSSG34 (a surface expression vector having an expression cassette in which the coding region of SPI1 derived from Pichia pastoris, the coding region of the cellobiohydrolase (TsCBH1) gene derived from Talaromyces emersonii, the coding region of the GCW34 gene derived from Pichia pastoris (GCW34 anchoring region), and the terminator region of the AOX1 gene derived from Pichia pastoris are arranged in this order: Biotechnology and Bioengineering, Vol.
- RNA fragment was prepared containing the entire length of the vector plasmid excluding the coding region of the TsCBH1 gene, the GCW34 anchoring region, and the AOX1 terminator region. These fragments were ligated by the In-Fusion method.
- the resulting plasmid containing the expression cassette X9 (SPI1 promoter (SEQ ID NO: 10) + SPI1 secretion signal (SEQ ID NO: 11) + CsCBP (SEQ ID NO: 6) + GCW30 anchoring region (SEQ ID NO: 12) + AOX1 terminator (SEQ ID NO: 13)) was named pIZ-CsCBP-PpSSG30.
- Preparation Example 2-1 Production of sucrose phosphorylase surface-displaying yeast
- the plasmid pIBlSP-SSSD described in Preparation Example 1-1 was treated with NdeI, and then used in yeast Saccharomyces cerevisiae BY4741 ⁇ SUC2 strain, which was transformed by the lithium acetate method.
- This transformed strain is referred to as the ⁇ SUC2-BlSP strain.
- the plasmid pIBlSP-PpSSG30 described in Preparation Example 1-7 was treated with EcoRV, and then used in yeast Pichia pastoris CBS7435 strain, which was transformed by the lithium acetate method. This transformed strain is referred to as the Pp-BlSP strain.
- the plasmid pIL2-BlSP-SSD described in Preparation Example 1-2 was treated with NdeI, and then used in the ⁇ SUC2-BlSP strain to transform it by the lithium acetate method. This transformed strain is called the ⁇ SUC2-BlSP2 strain.
- the plasmid pIH-BlSP-PpSS described in Preparation Example 1-8 was treated with BsrGI, and then used in the Pp-BlSP strain to transform it by the lithium acetate method. This transformed strain is called the Pp-BlSP2 strain.
- Preparation Example 2-2 Production of cellobiose phosphorylase surface-displaying yeast
- the plasmids pIU5-CsCBP-SSSD, pIU5-CuCBP-SSSD, pIU5-TaCBP-SSSD, and pIU5-TnCBP-SSSD described in Preparation Examples 1-3 to 1-6 were treated with SpeI, and then used in yeast Saccharomyces cerevisiae BY4741 ⁇ SUC2 strain to transform the strains by the lithium acetate method.
- These transformed strains are referred to as ⁇ SUC2-CsCBP strain, ⁇ SUC2-CuCBP strain, ⁇ SUC2-TaCBP strain, and ⁇ SUC2-TnCBP strain, respectively.
- Preparation Example 2-3 Production of yeast displaying sucrose phosphorylase + cellobiose phosphorylase on the surface
- the plasmid pIU5-CsCBP-SSSD described in Preparation Example 1-3 was treated with SpeI, and then used in the ⁇ SUC2-B1SP2 strain to transform the strain by the lithium acetate method. This transformed strain is referred to as the ⁇ SUC2-SP2CBP strain.
- the plasmid pIZ-CsCBP-PpSSG30 described in Preparation Example 1-9 was treated with NsiI, and then used in the Pp-B1SP2 strain to transform the strain by the electroporation method. This transformed strain is referred to as the Pp-SP2CBP strain.
- yeasts transformed in A the ⁇ SUC2-BlSP strain, ⁇ SUC2-BlSP2 strain, ⁇ SUC2-CsCBP strain, ⁇ SUC2-CuCBP strain, ⁇ SUC2-TaCBP strain, ⁇ SUC2-TnCBP strain, and ⁇ SUC2-SP2CBP strains using Saccharomyces cerevisiae as a host were pretreated with 5 mL of YPD medium (1% dried yeast extract (Nacalai Tesque), Bacto TM Peptone (Life Technologies, Inc.)).
- YPD medium 1% dried yeast extract (Nacalai Tesque), Bacto TM Peptone (Life Technologies, Inc.)
- the bacteria were transferred to 2% Nacalai Tesque (Nippon Biotechnology) and 2% D-glucose (Nacalai Tesque) and pre-cultured at 30°C and 200 rpm for 18 hours, and then inoculated into 50 mL of YPD medium to an OD600 of 0.05 and cultured with shaking at 30°C and 150 rpm for 48 hours.
- Pp-BlSP strain, Pp-BlSP2 strain, and Pp-SP2CBP strain which use Pichia pastoris as a host, were transplanted into 5 mL of YPG medium (1% dry yeast extract (Nacalai Tesque), 2% Bacto TM Peptone (Life Technologies), 2% glycerol (Nacalai Tesque)), pre-cultured for 18 hours at 30 ° C. and 200 rpm, and then inoculated into 50 mL of YPG medium so that the OD600 was 0.05, and cultured with shaking at 30 ° C. and 150 rpm for 48 hours.
- YPG medium 1% dry yeast extract (Nacalai Tesque), 2% Bacto TM Peptone (Life Technologies), 2% glycerol (Nacalai Tesque)
- reaction solution composition: 10 g/L sucrose (Nacalai Tesque); 80 mM sodium phosphate buffer (pH 7.0); and pretreated cells of the Pp-BlSP strain or Pp-BlSP2 strain prepared in Preparation Example 2-1 (final cell concentration 100 g wet cells/L)) was prepared and reacted at 35 rpm and 60°C for 60 minutes.
- the reaction solution was kept at 100°C for 10 minutes, then cooled on ice for 10 minutes, centrifuged to precipitate the cells, etc., and the supernatant was collected and used for analysis of fructose concentration.
- the amount of enzyme that liberates 1 ⁇ mol of fructose in 1 minute is defined as 1 IU.
- Sampling was performed twice, for 4 hours and 24 hours. To stop the reaction, the sampling liquid was placed in a sampling container and kept at 100°C for 10 minutes, then cooled on ice for 10 minutes, and centrifuged to precipitate the cells, etc., and the supernatant was collected and used for analysis.
- sampled liquid was placed in a sampling container, kept at 100° C. for 10 minutes, then cooled on ice for 10 minutes and centrifuged to precipitate the bacteria and the like, and the supernatant was collected and used for analysis.
- Figure 2 shows the time course of the sucrose, glucose, fructose, and cellobiose concentrations in the reaction supernatant of an experiment to produce cellobiose from sucrose using a combination of yeast D that displays sucrose phosphorylase and yeast D that displays cellobiose phosphorylase.
- yeast D displays sucrose phosphorylase
- yeast D displays cellobiose phosphorylase
- Figure 3 shows the time course of the concentrations of sucrose, glucose, fructose, and cellobiose in the reaction supernatant of an experiment to produce cellobiose from sucrose using a sucrose phosphorylase + cellobiose phosphorylase surface-displaying yeast ( ⁇ SUC2-SP2CBP strain) with Saccharomyces cerevisiae as the host.
- the graph in Figure 3(a) shows the results when the ⁇ SUC2-SP2CBP strain was used with Saccharomyces cerevisiae as the host, and the graph in Figure 3(b) shows the results when the Pp-SP2CBP strain was used with Pichia pastoris as the host.
- Figure 3(a) which used a single recombinant yeast strain co-expressing sucrose phosphorylase and cellobiose phosphorylase with the same Saccharomyces cerevisiae as a host, shows that sucrose was converted to cellobiose at a high conversion rate (about 70%) despite the same total amount of yeast cells used in the reaction.
- Figure 4 shows the time course of the concentrations of sucrose, glucose, fructose, and cellobiose in the reaction supernatant of an experiment to produce cellobiose from sucrose using a sucrose phosphorylase + cellobiose phosphorylase surface-displaying yeast (Pp-SP2CBP strain) with E. Pichia pastoris as the host.
- the graph in Figure 4(a) shows the results when the Pp-SP2CBP strain was cultured using glucose as the carbon source
- the graph in Figure 4(b) shows the results when the Pp-SP2CBP strain was cultured using glycerol as the carbon source.
- the conversion rate of sucrose to cellobiose in the Pp-SP2CBP strain was approximately 40% when it was cultured using glucose as the carbon source, but increased to approximately 80% when it was cultured using glycerol as the carbon source. This indicates that it is preferable to use glycerol as a carbon source when culturing a yeast that displays sucrose phosphorylase and cellobiose phosphorylase on the surface using Pichia pastoris as a host.
- Example Group II Repeated cellobiose production by reusing transformed yeast (sucrose phosphorylase + cellobiose surface display yeast)]
- ⁇ SUC2-SP2CBP strain prepared in A was transferred to 5 mL of YPD medium (1% dried yeast extract (Nacalai Tesque), 2% Bacto TM Peptone (Life Technologies), 2% D-glucose (Nacalai Tesque)) and pre-cultured at 30°C and 200 rpm for 18 hours, and then inoculated into 50 mL of YPD medium to an OD600 of 0.05, and cultured with shaking at 30°C and 150 rpm for 48 hours.
- YPD medium 1% dried yeast extract (Nacalai Tesque), 2% Bacto TM Peptone (Life Technologies), 2% D-glucose (Nacalai Tesque)
- the Pp-SP2CBP strain was transferred to 5 mL of YPG medium (1% dried yeast extract (Nacalai Tesque), 2% Bacto TM Peptone (Life Technologies), 2% glycerol (Nacalai Tesque)), pre-cultured at 30 ° C. and 200 rpm for 24 hours, and then inoculated into 50 mL of YPG medium so that the OD600 was 0.05, and cultured with shaking at 30 ° C. and 150 rpm for 48 hours.
- YPG medium 1% dried yeast extract (Nacalai Tesque), 2% Bacto TM Peptone (Life Technologies), 2% glycerol (Nacalai Tesque)
- cellobiose production reaction solution 100 g/L sucrose (Nacalai Tesque); 80 mM sodium phosphate buffer (pH 7.0); 1 wt% magnesium sulfate; 1.5 IU/mL glucose isomerase (xylose isomerase, Godo Shusei Co., Ltd., derived from Streptomyces griseofuscus); and pretreated cells of ⁇ SUC2-SP2CBP strain or Pp-SP2CBP strain prepared in G-1 (final cell concentration 100 g wet cells/L)) were prepared and reacted at 35 rpm and 60 ° C. for 24 hours (first time).
- the yeast cells were collected by centrifugation of the reacted liquid, resuspended in a new reaction liquid, and reacted at 35 rpm and 60 ° C. for 24 hours (second time).
- the third reaction was carried out in the same manner. Sampling was performed three times for each reaction, at 4 hours, 8 hours, and 24 hours. To stop the reaction, each sample was placed in a sampling container, kept at 100°C for 10 minutes, and then cooled on ice for 10 minutes and centrifuged to precipitate the bacteria and the like. The supernatant was collected and used for analysis.
- sucrose phosphorylase + cellobiose phosphorylase surface-displaying yeast with Pichia pastoris cultured using glycerol as a carbon source was used as a transformed yeast, it was confirmed that the yeast of the Pp-SP2CBP strain maintained a high cellobiose conversion rate of about 80% even when the reaction was repeated three times at 60 ° C. for 24 hours.
- the manufacturing method of the present invention can be used as a method for manufacturing cellobiose for use in food or pharmaceutical preparations.
Landscapes
- Life Sciences & Earth Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Genetics & Genomics (AREA)
- Engineering & Computer Science (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Biotechnology (AREA)
- General Engineering & Computer Science (AREA)
- Biomedical Technology (AREA)
- General Health & Medical Sciences (AREA)
- Microbiology (AREA)
- Biochemistry (AREA)
- Molecular Biology (AREA)
- Medicinal Chemistry (AREA)
- Mycology (AREA)
- Biophysics (AREA)
- Physics & Mathematics (AREA)
- Plant Pathology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
Abstract
低コストで効率の良いセロビオースの製造方法を提供する。本方法は、スクロースホスホリラーゼ及びセロビオースホスホリラーゼを表層に発現させた酵母とグルコースイソメラーゼを含有する反応液にスクロースを共存させる工程を含む。
Description
本発明は、酵素表層発現酵母によりスクロースからセロビオースを製造する方法に関する。
セロビオースの製造方法としては、従来より酵素分解法と酵素合成法とが知られている。酵素分解法としては、例えば、セルラーゼ製剤をセルロースと反応させることによりセロビオースを得る方法(特許文献1、特許文献2)が知られている。酵素合成法としては、例えば、デンプンを出発物質として、αグルカンホスホリラーゼ及びセロビオースホスホリラーゼを作用させることでセロビオースを得る方法(非特許文献1)や、スクロースを出発物質として、スクロースホスホリラーゼ、グルコースイソメラーゼ、及びセロビオースホスホリラーゼを作用させることでセロビオースを得る方法(特許文献3、特許文献4)が知られている。
酵素分解法では原料としてセルロースを使用する。セルロースは植物の細胞壁の主成分であるが、通常はヘミセルロース、リグニン等との混合物として存在する。そこで、斯かる植物細胞壁中のセルロースを原料として使用する場合には、まずアルカリ処理等の方法でヘミセルロース、リグニン等を除かねばならず、そのため原料の製造コストが高くなるという課題がある。
酵素合成法では、デンプンからグルコース1リン酸への変換効率や、スクロースからセロビオースの変換効率が低い上に、酵素を使用した温和な条件(30℃程度)で反応させる必要があるため、反応速度が遅く、低コストでのセロビオース生産には適していないという課題がある。
Suzuki et al., J. Appl. Glycosci., (2010), 57:113-119
本発明の目的は、スクロースから高い転換率及び低コストでセロビオースを製造できる方法を提供することにある。
本発明者らは、スクロースホスホリラーゼ(以下適宜「SP」と記載することがある)及びセロビオースホスホリラーゼ(以下適宜「CBP」と記載することがある)を発現させた酵母とグルコースイソメラーゼ(以下適宜「XI」と記載することがある)を含有する反応液にスクロースを共存させることにより、高い転換率で安価にセロビオースを製造可能となることを見出し、本発明を完成させた。
即ち、本発明は以下の発明を包含する。
[項1]スクロースホスホリラーゼ及びセロビオースホスホリラーゼを発現する組換え酵母とグルコースイソメラーゼとを含有する反応液中にスクロースを共存させる反応工程を含む、セロビオースの製造方法。
[項2]組換え酵母が、サッカロマイセス(Saccharomyces)属、ピキア(Pichia)属、又はヤロウィア(Yarrowia)属の酵母である、項1に記載の製造方法。
[項3]スクロースホスホリラーゼが、ビフィドバクテリウム(Bifidobacterium)属、ロイコノストック(Leuconostoc)属、又はストレプトコッカス(Streptococcus)属の細菌に由来する、項1又は2に記載の製造方法。
[項4]スクロースホスホリラーゼが、組換え酵母から分泌発現される分泌型スクロースホスホリラーゼと、組換え酵母の細胞壁に係留された状態で発現される係留型スクロースホスホリラーゼとを含む、項1~3の何れか一項に記載の製造方法。
[項5]セロビオースホスホリラーゼが、クロストリジウム(Clostridium)属、ルミノコッカス(Ruminococcus)属、セルロモナス(Cellulomonas)属、又はサーモトガ(Thermotoga)属の細菌に由来する、項1~4の何れか一項に記載の製造方法。
[項6]前記組換え酵母が、スクロースホスホリラーゼ及びセロビオースホスホリラーゼを単一の菌体で共に発現する組換え酵母である、項1~5の何れか一項に記載の製造方法。
[項7]前記組換え酵母が、スクロースホスホリラーゼを発現する組換え酵母と、セロビオースホスホリラーゼを発現する組換え酵母との組み合わせである、項1~6の何れか一項に記載の製造方法。
[項8]前記組換え酵母が、スクロースホスホリラーゼ遺伝子を担持する発現ベクターにより形質転換された酵母である、項1~7の何れか一項に記載の製造方法。
[項9]前記発現ベクターが、スクロースホスホリラーゼ遺伝子及び酵母の細胞壁ドメイン遺伝子を共に担持する第1の発現ベクターと、酵母の細胞壁ドメイン遺伝子を担持せずスクロースホスホリラーゼ遺伝子のみを担持する第2の発現ベクターとの組み合わせを含む、項8に記載の製造方法。
[項10]グルコースイソメラーゼが、ストレプトマイセス(Streptomyces)属の細菌に由来する、項1~9の何れか一項に記載の製造方法。
[項11]反応液中にスクロースを共存させる前に、組換え酵母を45℃~75℃で15分~150分処理する工程を更に含む、項1~10の何れか一項に記載の製造方法。
[項12]前記反応工程の前に、炭素源としてグリセロールを用いて酵母細胞を培養する工程を含む、請求項1~11の何れか一項に記載の製造方法。
[項13]前記反応工程の後に、製造されたセロビオースを反応液から分離する工程をさらに含む、項1~12の何れか一項に記載の製造方法。
[項14]前記反応工程の後に、前記組換え酵母を反応液から回収し、再度の反応工程に供することをさらに含む、項1~13の何れか一項に記載の製造方法。
[項1]スクロースホスホリラーゼ及びセロビオースホスホリラーゼを発現する組換え酵母とグルコースイソメラーゼとを含有する反応液中にスクロースを共存させる反応工程を含む、セロビオースの製造方法。
[項2]組換え酵母が、サッカロマイセス(Saccharomyces)属、ピキア(Pichia)属、又はヤロウィア(Yarrowia)属の酵母である、項1に記載の製造方法。
[項3]スクロースホスホリラーゼが、ビフィドバクテリウム(Bifidobacterium)属、ロイコノストック(Leuconostoc)属、又はストレプトコッカス(Streptococcus)属の細菌に由来する、項1又は2に記載の製造方法。
[項4]スクロースホスホリラーゼが、組換え酵母から分泌発現される分泌型スクロースホスホリラーゼと、組換え酵母の細胞壁に係留された状態で発現される係留型スクロースホスホリラーゼとを含む、項1~3の何れか一項に記載の製造方法。
[項5]セロビオースホスホリラーゼが、クロストリジウム(Clostridium)属、ルミノコッカス(Ruminococcus)属、セルロモナス(Cellulomonas)属、又はサーモトガ(Thermotoga)属の細菌に由来する、項1~4の何れか一項に記載の製造方法。
[項6]前記組換え酵母が、スクロースホスホリラーゼ及びセロビオースホスホリラーゼを単一の菌体で共に発現する組換え酵母である、項1~5の何れか一項に記載の製造方法。
[項7]前記組換え酵母が、スクロースホスホリラーゼを発現する組換え酵母と、セロビオースホスホリラーゼを発現する組換え酵母との組み合わせである、項1~6の何れか一項に記載の製造方法。
[項8]前記組換え酵母が、スクロースホスホリラーゼ遺伝子を担持する発現ベクターにより形質転換された酵母である、項1~7の何れか一項に記載の製造方法。
[項9]前記発現ベクターが、スクロースホスホリラーゼ遺伝子及び酵母の細胞壁ドメイン遺伝子を共に担持する第1の発現ベクターと、酵母の細胞壁ドメイン遺伝子を担持せずスクロースホスホリラーゼ遺伝子のみを担持する第2の発現ベクターとの組み合わせを含む、項8に記載の製造方法。
[項10]グルコースイソメラーゼが、ストレプトマイセス(Streptomyces)属の細菌に由来する、項1~9の何れか一項に記載の製造方法。
[項11]反応液中にスクロースを共存させる前に、組換え酵母を45℃~75℃で15分~150分処理する工程を更に含む、項1~10の何れか一項に記載の製造方法。
[項12]前記反応工程の前に、炭素源としてグリセロールを用いて酵母細胞を培養する工程を含む、請求項1~11の何れか一項に記載の製造方法。
[項13]前記反応工程の後に、製造されたセロビオースを反応液から分離する工程をさらに含む、項1~12の何れか一項に記載の製造方法。
[項14]前記反応工程の後に、前記組換え酵母を反応液から回収し、再度の反応工程に供することをさらに含む、項1~13の何れか一項に記載の製造方法。
本発明によれば、高い転換率及び低コストでスクロースからセロビオースを製造することが可能となる。
以下、本発明を具体的な実施の形態に即して詳細に説明する。但し、本発明は以下の実施の形態に束縛されるものではなく、本発明の趣旨を逸脱しない範囲において、任意の形態で実施することが可能である。なお、本明細書に記載した特許文献及び非特許文献は、その全体が援用により本願に組み込まれる。
なお、本明細書において、アミノ酸配列の「同一性」(identity)とは、一致するアミノ酸残基の割合を意味し、「相同性」(similarity)とは、一致又は類似するアミノ酸残基の割合を意味する。アミノ酸配列の相同性及び同一性は、例えばBLAST法(NCBIのPBLASTのデフォルト条件)により決定することができる。また、例えば「80%以上の相同性」と表現するときには、「80%以上の同一性」の場合を含んでいることは明らかである。
また、本明細書において、「類似するアミノ酸残基」とは、同様の化学的特質(例えば、電荷又は疎水性)を持つ側鎖を有するアミノ酸残基を意味する。類似するアミノ酸残基としては、例えば以下の組合せが挙げられる。
(1)脂肪族側鎖を有するアミノ酸残基:グリシン(Gly又はG)、アラニン(Ala又はA)、バリン(Val又はV)、ロイシン(Leu又はL)、及びイソロイシン(Ile又はI)残基。
(2)脂肪族ヒドロキシル側鎖を有するアミノ酸残基:セリン(Ser又はS)及びトレオニン(Thr又はT)残基。
(3)アミド含有側鎖を有するアミノ酸残基:アスパラギン(Asn又はN)及びグルタミン(Gln又はQ)残基。
(4)芳香族側鎖を有するアミノ酸残基:フェニルアラニン(Phe又はF)、チロシン(Tyr又はY)、及びトリプトファン(Trp又はW)残基。
(5)塩基性側鎖を有するアミノ酸残基:リジン(LysまたK)、アルギニン(Arg又はR)及びヒスチジン(His又はH)残基。
(6)酸性側鎖を有するアミノ酸残基:アスパラギン酸(Asp又はD)及びグルタミン酸(Glu又はE)残基。
(7)硫黄含有側鎖を有するアミノ酸基:システイン(Cys又はC)、及びメチオニン(Met又はM)残基。
さらに、(1)とメチオニン(Met又はM)の組み合わせ、及び、(4)とヒスチジン(His又はH)残基の組み合わせも、類似するアミノ酸残基として扱われる。
(1)脂肪族側鎖を有するアミノ酸残基:グリシン(Gly又はG)、アラニン(Ala又はA)、バリン(Val又はV)、ロイシン(Leu又はL)、及びイソロイシン(Ile又はI)残基。
(2)脂肪族ヒドロキシル側鎖を有するアミノ酸残基:セリン(Ser又はS)及びトレオニン(Thr又はT)残基。
(3)アミド含有側鎖を有するアミノ酸残基:アスパラギン(Asn又はN)及びグルタミン(Gln又はQ)残基。
(4)芳香族側鎖を有するアミノ酸残基:フェニルアラニン(Phe又はF)、チロシン(Tyr又はY)、及びトリプトファン(Trp又はW)残基。
(5)塩基性側鎖を有するアミノ酸残基:リジン(LysまたK)、アルギニン(Arg又はR)及びヒスチジン(His又はH)残基。
(6)酸性側鎖を有するアミノ酸残基:アスパラギン酸(Asp又はD)及びグルタミン酸(Glu又はE)残基。
(7)硫黄含有側鎖を有するアミノ酸基:システイン(Cys又はC)、及びメチオニン(Met又はM)残基。
さらに、(1)とメチオニン(Met又はM)の組み合わせ、及び、(4)とヒスチジン(His又はH)残基の組み合わせも、類似するアミノ酸残基として扱われる。
本発明の一側面は、セロビオースの製造方法に関する。斯かる製造方法は、スクロースホスホリラーゼ(SP)及びセロビオースホスホリラーゼ(CBP)を発現する組換え酵母と、グルコースイソメラーゼ(XI)とを含有する反応液中に、スクロースを共存させる工程を含む。
酵母の種は、スクロースホスホリラーゼ(SP)及びセロビオースホスホリラーゼ(CBP)を発現可能なものであれば、特に限定されない。例としては、サッカロマイセス(Saccharomyces)属、ピキア(Pichia)属、又はヤロウィア(Yarrowia)属の酵母が挙げられる。中でも、スクロースがインベルターゼの作用により分解されることを防ぐために、細胞表層にインベルターゼ活性を持たない酵母を用いることが好ましく、例としては、ピキア・パストリス(Pichia pastoris)が挙げられる。サッカロマイセス・セレビシエ(Saccharomyces cerevisiae)のように細胞表層に強いインベルターゼ活性を持つ酵母については、遺伝子のノックアウトなどによりインベルターゼ活性を喪失させた株を用いることが好ましい。なお、ピキア・パストリス(Pichia pastoris)は、コマガタエラ・パストリス(Komagataella pastoris)及びコマガタエラ・ファフィ(Komagataella phaffii)に分類及び改名されているため、ピキア・パストリス(Pichia pastoris)は、コマガタエラ・パストリス(Komagataella pastoris)及びコマガタエラ・ファフィ(Komagataella phaffii)の両方の同義語である。
スクロースホスホリラーゼ(SP)の由来(種)は特に限定されない。例としては、ビフィドバクテリウム(Bifidobacterium)属、ロイコノストック(Leuconostoc)属、又はストレプトコッカス(Streptococcus)属の細菌に由来するものが挙げられる。中でも、ビフィドバクテリウム(Bifidobacterium)属の細菌に由来するものが好ましく、ビフィドバクテリウム・ロングム(Bifidobacterium longum)に由来するものがとりわけ好ましい。ビフィドバクテリウム・ロングム(Bifidobacterium longum)のスクロースホスホリラーゼ(SP)のアミノ酸配列を配列番号34に示し、これをコードする塩基配列の一例を配列番号5に示す。ビフィドバクテリウム・ロングム(Bifidobacterium longum)に由来するスクロースホスホリラーゼ(SP)としては、前記の配列番号34と80%以上、又は85%以上、又は88%以上、又は90%以上、又は93%以上、又は95%以上、又は97%以上、又は98%以上、又は99%以上の相同性(好ましくは同一性)を有するアミノ酸配列からなるタンパク質が挙げられる。
スクロースホスホリラーゼ(SP)を組換え酵母に発現させる場合、組換え酵母から分泌発現される分泌型酵素の形態と、組換え酵母の細胞壁に係留された状態で発現される係留型酵素の形態とが考えられるが、どちらの形態でもよい。ただし本発明では、組換え酵母から分泌発現される分泌型スクロースホスホリラーゼ(SP)と、組換え酵母の細胞壁に係留された状態で発現される係留型スクロースホスホリラーゼ(SP)とを、反応液内に共存させることが好ましい。これにより、スクロースホスホリラーゼ(SP)による変換効率を向上させることが可能となる。その理由は定かではないが、分泌型スクロースホスホリラーゼ(SP)と係留型スクロースホスホリラーゼ(SP)が二量体を形成することによるものと推測される。
セロビオースホスホリラーゼ(CBP)の由来(種)は特に限定されないが、クロストリジウム(Clostridium)属、ルミノコッカス(Ruminococcus)属、セルロモナス(Cellulomonas)属、又はサーモトガ(Thermotoga)属の細菌に由来するものが挙げられる。中でも、クロストリジウム(Clostridium)属の細菌に由来するものが好ましく、クロストリジウム・ステルコラリウム(Clostridium stercorarium)由来のものがとりわけ好ましい。クロストリジウム・ステルコラリウム(Clostridium stercorarium)のセロビオースホスホリラーゼ(CBP)のアミノ酸配列を配列番号35に示し、これをコードする塩基配列の一例を配列番号6に示す。クロストリジウム・ステルコラリウム(Clostridium stercorarium)に由来するセロビオースホスホリラーゼ(CBP)としては、前記の配列番号35と80%以上、又は85%以上、又は88%以上、又は90%以上、又は93%以上、又は95%以上、又は97%以上、又は98%以上、又は99%以上の相同性(好ましくは同一性)を有するアミノ酸配列からなるタンパク質が挙げられる。
セロビオースホスホリラーゼ(CBP)を組換え酵母に発現させる場合、組換え酵母から分泌発現される分泌型酵素の形態と、組換え酵母の細胞壁に係留された状態で発現される係留型酵素の形態とが考えられるが、どちらの形態でもよい。ただし本発明では、組換え酵母の細胞壁に係留された状態で発現される係留型セロビオースホスホリラーゼ(CBP)の形態とすることが好ましい。
組換え酵母としては、スクロースホスホリラーゼ(SP)及びセロビオースホスホリラーゼ(CBP)を単一の菌体で共発現する組換え酵母を用いてもよく、スクロースホスホリラーゼ(SP)を発現する組換え酵母と、セロビオースホスホリラーゼ(CBP)を発現する組換え酵母とを組み合わせて用いてもよい。また、スクロースホスホリラーゼ(SP)及び/又はセロビオースホスホリラーゼ(CBP)として、分泌型酵素と係留型酵素とを併用する場合には、分泌型酵素及び係留型酵素を単一の菌体で共発現する組換え酵母を用いてもよく、分泌型酵素を発現する組換え酵母と、係留型酵素を発現する組換え酵母とを組み合わせて用いてもよい。
スクロースホスホリラーゼ(SP)及び/又はセロビオースホスホリラーゼ(CBP)を発現する組換え酵母を調製する手法は特に制限されない。例としては、スクロースホスホリラーゼ(SP)遺伝子及び/又はセロビオースホスホリラーゼ(CBP)遺伝子を担持する1種又は2種以上の発現ベクターで、1種又は2種以上の酵母を形質転換する手法を挙げることができる。発現ベクターとしては、プラスミド、コスミド、ラムダファージ等が挙げられるが、プラスミドが好ましい。また、スクロースホスホリラーゼ(SP)遺伝子及び/又はセロビオースホスホリラーゼ(CBP)遺伝子を、更にプロモーター、ターミネーター等の調節配列を作動式に連結し、酵母細胞内で自律的に発現可能な発現カセットの形態として、発現ベクターに担持させることが好ましい。なお、斯かる発現ベクターの構築は、当業者に公知の遺伝子工学の標準的な手法を用いて、例えばMichael R. Green et al, Molecular Cloning: A Laboratory Manual, Fourth Edition, (2012), Cold Spring Harbor Laboratory等の文献を適宜参照することにより実施することが可能である。
組換え酵母は、反応に先立って炭素源の存在下で前培養することが好ましい。組換え酵母の細胞を培養する際に用いる炭素源の種類は特に制限されず、酵母の種類に応じて適宜選択すればよい。例としてはグルコース、グリセロール等が挙げられる。中でも、本発明では炭素源として少なくともグリセロールを使用することが好ましい。これにより、スクロースからセロビオースへの転換率を向上できる。
組換え酵母は、反応に先立って前処理に供し、グルコースの取り込みを停止させることが好ましい。かかる前処理としては、一定の温度に一定時間に亘って晒す加熱処理が挙げられる。この場合の処理条件は、用いる酵母のグルコース取り込みを停止させることができさえすればよく、加熱温度としては例えば45℃以上、又は50℃以上、又は55℃以上とすることができ、また、例えば75℃以下、又は70℃以下、又は65℃以下とすることができる。また、加熱時間としては例えば15分以上、又は45分以上、又は70分以上とすることができ、また、例えば150分以内、又は120分間以内、又は100分以内とすることができる。好ましい一例によれば、温度は約60℃、時間は約90分間とすることができる。かかる前処理の方法の一例としては、形質転換酵母の懸濁液を前記条件で振とうすることが挙げられる。
本発明では、スクロースホスホリラーゼ(SP)及びセロビオースホスホリラーゼ(CBP)を発現する組換え酵母を含む反応液中に、グルコースイソメラーゼ(XI)を共存させる。反応液中の組換え酵母の菌体量は、特に制限されるものではないが、例えば25g湿潤細胞/L以上、又は50g湿潤細胞/L以上、又は75g湿潤細胞/L以上、また、例えば200g湿潤細胞/L以下、又は150g湿潤細胞/L以下、又は100g湿潤細胞/L以下とすることができる。
グルコースイソメラーゼ(別名キシロースイソメラーゼ、XI)としては、限定されるものではないが、例えば、ストレプトマイセス(Streptomyces)属の酵母に由来するものが挙げられる。中でも、ストレプトマイセス・グリセオフスクス(Streptomyces griseofuscus)に由来するものがとりわけ好ましい。なお、グルコースイソメラーゼとしては種々の市販品を入手できるところ、それらを用いることができる。
グルコースイソメラーゼ(XI)は、他の酵素と同様に酵母又はその他の微生物に発現させてもよいが、単離されたグルコースイソメラーゼ(XI)酵素そのものを用いることが好ましい。単離されたグルコースイソメラーゼ(XI)酵素としては、市販品あるいはこれら酵素を生産する微生物等の培養により得られたもののいずれでもよい。また精製品あるいは未精製品のいずれでもよい。この場合、スクロースホスホリラーゼ(SP)及びセロビオースホスホリラーゼ(CBP)を発現する組換え酵母を含む反応液に、単離されたグルコースイソメラーゼ(XI)酵素を加えて共存させることが好ましい。
グルコースイソメラーゼ(XI)の量は、特に制限されないが、通常はスクロース1モルに対し0.01単位以上、又は0.1単位以上、又は1.0単位以上とすることが好ましい。ここで1単位(1U)とは、50℃において1wt/vol%のスクロースに、pH7.0において1分間に1μmolのフルクトースを分解する酵素量を意味する。
本発明では、スクロースホスホリラーゼ(SP)及びセロビオースホスホリラーゼ(CBP)を発現する組換え酵母とグルコースイソメラーゼ(XI)とを含む反応液中に、スクロースを共存させて反応を実施する。スクロースは、天然に存在するものでもよく、化学合成されたものでもよい。スクロースの量は、特に制限されないが、生産性の点からはできるだけ高い濃度が好ましい。ただし、生成するセロビオースの溶解度は15から20%程度なので、溶解した状態を保ちたい場合には10から20%程度のスクロース濃度を用いるのが好ましい。
セロビオース合成反応時の条件は、制限されるものではないが、例えば以下の通りとすることができる。即ち、反応温度としては例えば35℃以上、又は45℃以上、又は50℃以上、また、例えば60℃以下、又は70℃以下とすることができる。また、反応時間としては例えば60分以上、又は12時間以上、又は24時間以上とすることができ、また、例えば36時間以内、又は48時間以内とすることができる。反応時の圧力は特に制限されず、減圧下でも常圧下でも加圧下でもよいが、通常は常圧下で行うことができる。反応時の雰囲気も特に制限されず、使用する組換え酵母が生育できない雰囲気下でも酵素の活性が保持される限り、任意の雰囲気とすることができる。
反応の終了後、必要に応じてセロビオースを反応液から分離・精製することにより、セロビオースを得ることができる。分離・精製の手法は特に制限されず、任意の手法を用いることができるが、例えばろ過分離、遠心分離、膜分離等が挙げられる。これらの方法により、用いた菌体とセロビオースを効率的に分離することができる。さらに、得られたセロビオースあるいはその溶液中の不純物を除くために、イオン交換樹脂で処理をしたり、再結晶をすることでより高度に精製してもよい。
なお、反応の終了後、必要に応じて組換え酵母を反応液から分離・回収し、セロビオース合成反応に再利用してもよい。本発明の組換え酵母は、こうして反応液から分離・回収して再利用した場合でも、酵素活性が維持され、繰り返し高いセロビオース合成効率が得られる場合がある。即ち、好ましい態様によれば、本発明のセロビオースの製造方法は、前記反応工程の後に、前記組換え酵母を反応液から回収し、再度の反応工程に供することをさらに含む。組換え酵母を反応液から分離・回収する手法は限定されないが、例えば遠心分離等が挙げられる。前記組換え酵母の分離・回収及び再利用の回数は特に限定されないが、一態様によれば、例えば2回以上、又は3回以上、又は4回以上、又は5回以上の反応に供することが出来る。前記組換え酵母の再利用時の活性は特に限定されないが、一態様によれば、前段の反応に於ける活性の60%以上、又は70%以上、又は80%以上、又は90%以上の活性が、次段の反応でも維持されることが好ましい。
以下、本発明を実施例に則して更に詳細に説明するが、これらの実施例はあくまでも説明のために便宜的に示す例に過ぎず、本発明は如何なる意味でもこれらの実施例に限定されるものではない。
なお、本実施例に示すPCR法は、何れもKOD OneTM PCR Master Mix(東洋紡)を用いて実施した。また、本実施例に示す全ての酵母への遺伝子導入は、酢酸リチウム法によって実施した。
[実施例群I:形質転換酵母(スクロースホスホリラーゼ表層提示酵母、セロビオースホスホリラーゼ表層提示酵母、及びスクロースホスホリラーゼ+セロビオースホスホリラーゼ表層提示酵母)を利用したセロビオース生産]
A.形質転換酵母の作製
酵母サッカロマイセス・セレビシエ(Saccharomyces cerevisiae)BY4741ΔSUC2株に、調製したプラスミドを使用して酢酸リチウム法によって、スクロースホスホリラーゼ(SP)及びセロビオースホスホリラーゼ(CBP)の2種の酵素を発現する種々の組換え酵母株を構築した。以下具体的に記載する。
酵母サッカロマイセス・セレビシエ(Saccharomyces cerevisiae)BY4741ΔSUC2株に、調製したプラスミドを使用して酢酸リチウム法によって、スクロースホスホリラーゼ(SP)及びセロビオースホスホリラーゼ(CBP)の2種の酵素を発現する種々の組換え酵母株を構築した。以下具体的に記載する。
・菌株及び培地
菌株としては、酵母サッカロマイセス・セレビシエ(Saccharomyces cerevisiae)BY4741株(MATα his3 leu2 met15 ura3株)及び酵母サッカロマイセス・セレビシエ(Saccharomyces cerevisiae)BY4741ΔSUC2株(MATα his3 leu2 met15 ura3 suc2株)を用いた。酵母サッカロマイセス・セレビシエ(Saccharomyces cerevisiae)BY4741株をATCC(アメリカンタイプカルチャーコレクション)から、酵母サッカロマイセス・セレビシエ(Saccharomyces cerevisiae)BY4741ΔSUC2株をYeast deletion MAT-A complete set(Thermo Fisher Scientific)から、それぞれ入手した。
菌株としては、酵母サッカロマイセス・セレビシエ(Saccharomyces cerevisiae)BY4741株(MATα his3 leu2 met15 ura3株)及び酵母サッカロマイセス・セレビシエ(Saccharomyces cerevisiae)BY4741ΔSUC2株(MATα his3 leu2 met15 ura3 suc2株)を用いた。酵母サッカロマイセス・セレビシエ(Saccharomyces cerevisiae)BY4741株をATCC(アメリカンタイプカルチャーコレクション)から、酵母サッカロマイセス・セレビシエ(Saccharomyces cerevisiae)BY4741ΔSUC2株をYeast deletion MAT-A complete set(Thermo Fisher Scientific)から、それぞれ入手した。
・発現カセットを含むプラスミドの構築
下記発現カセットX1~X9をそれぞれ含むプラスミドを、以下の手順により調製した。
・X1:SED1プロモーター(配列番号1)+SED1分泌シグナル(配列番号2)+BlSP(配列番号5)+SED1アンカーリング領域(配列番号3)+DIT1ターミネーター(配列番号4)
・X2:SED1プロモーター(配列番号1)+SED1分泌シグナル(配列番号2)+BlSP(配列番号5)+DIT1ターミネーター(配列番号4)
・X3:SED1プロモーター(配列番号1)+SED1分泌シグナル(配列番号2)+CsCBP(配列番号6)+SED1アンカーリング領域(配列番号3)+DIT1ターミネーター(配列番号4)
・X4:SED1プロモーター(配列番号1)+SED1分泌シグナル(配列番号2)+CuCBP(配列番号7)+SED1アンカーリング領域(配列番号3)+DIT1ターミネーター(配列番号4)
・X5:SED1プロモーター(配列番号1)+SED1分泌シグナル(配列番号2)+TaCBP(配列番号8)+SED1アンカーリング領域(配列番号3)+DIT1ターミネーター(配列番号4)
・X6:SED1プロモーター(配列番号1)+SED1分泌シグナル(配列番号2)+TnCBP(配列番号9)+SED1アンカーリング領域(配列番号3)+DIT1ターミネーター(配列番号4)
・X7:SPI1プロモーター(配列番号10)+SPI1分泌シグナル(配列番号11)+BlSP(配列番号5)+GCW30アンカーリング領域(配列番号12)+AOX1ターミネーター(配列番号13)
・X8:SPI1プロモーター(配列番号10)+SPI1分泌シグナル(配列番号11)+BlSP(配列番号5)+AOX1ターミネーター(配列番号13)
・X9:SPI1プロモーター(配列番号10)+SPI1分泌シグナル(配列番号11)+CsCBP(配列番号6)+GCW30アンカーリング領域(配列番号12)+AOX1ターミネーター(配列番号13)
下記発現カセットX1~X9をそれぞれ含むプラスミドを、以下の手順により調製した。
・X1:SED1プロモーター(配列番号1)+SED1分泌シグナル(配列番号2)+BlSP(配列番号5)+SED1アンカーリング領域(配列番号3)+DIT1ターミネーター(配列番号4)
・X2:SED1プロモーター(配列番号1)+SED1分泌シグナル(配列番号2)+BlSP(配列番号5)+DIT1ターミネーター(配列番号4)
・X3:SED1プロモーター(配列番号1)+SED1分泌シグナル(配列番号2)+CsCBP(配列番号6)+SED1アンカーリング領域(配列番号3)+DIT1ターミネーター(配列番号4)
・X4:SED1プロモーター(配列番号1)+SED1分泌シグナル(配列番号2)+CuCBP(配列番号7)+SED1アンカーリング領域(配列番号3)+DIT1ターミネーター(配列番号4)
・X5:SED1プロモーター(配列番号1)+SED1分泌シグナル(配列番号2)+TaCBP(配列番号8)+SED1アンカーリング領域(配列番号3)+DIT1ターミネーター(配列番号4)
・X6:SED1プロモーター(配列番号1)+SED1分泌シグナル(配列番号2)+TnCBP(配列番号9)+SED1アンカーリング領域(配列番号3)+DIT1ターミネーター(配列番号4)
・X7:SPI1プロモーター(配列番号10)+SPI1分泌シグナル(配列番号11)+BlSP(配列番号5)+GCW30アンカーリング領域(配列番号12)+AOX1ターミネーター(配列番号13)
・X8:SPI1プロモーター(配列番号10)+SPI1分泌シグナル(配列番号11)+BlSP(配列番号5)+AOX1ターミネーター(配列番号13)
・X9:SPI1プロモーター(配列番号10)+SPI1分泌シグナル(配列番号11)+CsCBP(配列番号6)+GCW30アンカーリング領域(配列番号12)+AOX1ターミネーター(配列番号13)
(調製例1-1:ビフィドバクテリウム・ロングム(Bifidobacterium longum)由来スクロースホスホリラーゼ(BlSP)表層発現カセットX1を含有するベクタープラスミドの調製)
ビフィドバクテリウム・ロングム(Bifidobacterium longum)由来スクロースホスホリラーゼ(BlSP)遺伝子のコーディング領域を含むDNA断片を遺伝子合成により調製した。次に、ベクタープラスミドpIEG-SSSD(サッカロマイセス・セレビシエ(Saccharomyces cerevisiae)由来SED1プロモーター、サッカロマイセス・セレビシエ(Saccharomyces cerevisiae)由来SED1の分泌シグナルペプチド配列、トリコデルマ・リーセイ由来エンドグルカナーゼII(TrEGII)遺伝子のコーディング領域、サッカロマイセス・セレビシエ(Saccharomyces cerevisiae)由来SED1遺伝子のコーディング領域(SED1アンカーリング領域)、及びサッカロマイセス・セレビシエ(Saccharomyces cerevisiae)由来DIT1遺伝子のターミネーター領域がこの順に配置されている発現カセットを有する表層発現用ベクター:Applied Microbiology and Biotechnology, Vol.105, 2021, 5895-5904)を鋳型とし、プライマー対SED1a-F(配列番号14)及びSED1ss-R(配列番号15)を用いてPCR法により増幅し、TrEGII遺伝子のコーディング領域を除いたベクタープラスミド全長を含むDNA断片を調製した。これらの断片をIn-Fusion法により連結した。得られた発現カセットX1(SED1プロモーター(配列番号1)+SED1分泌シグナル(配列番号2)+BlSP(配列番号5)+SED1アンカーリング領域(配列番号3)+DIT1ターミネーター(配列番号4))を含むプラスミドをpIBlSP-SSSDと命名した。
ビフィドバクテリウム・ロングム(Bifidobacterium longum)由来スクロースホスホリラーゼ(BlSP)遺伝子のコーディング領域を含むDNA断片を遺伝子合成により調製した。次に、ベクタープラスミドpIEG-SSSD(サッカロマイセス・セレビシエ(Saccharomyces cerevisiae)由来SED1プロモーター、サッカロマイセス・セレビシエ(Saccharomyces cerevisiae)由来SED1の分泌シグナルペプチド配列、トリコデルマ・リーセイ由来エンドグルカナーゼII(TrEGII)遺伝子のコーディング領域、サッカロマイセス・セレビシエ(Saccharomyces cerevisiae)由来SED1遺伝子のコーディング領域(SED1アンカーリング領域)、及びサッカロマイセス・セレビシエ(Saccharomyces cerevisiae)由来DIT1遺伝子のターミネーター領域がこの順に配置されている発現カセットを有する表層発現用ベクター:Applied Microbiology and Biotechnology, Vol.105, 2021, 5895-5904)を鋳型とし、プライマー対SED1a-F(配列番号14)及びSED1ss-R(配列番号15)を用いてPCR法により増幅し、TrEGII遺伝子のコーディング領域を除いたベクタープラスミド全長を含むDNA断片を調製した。これらの断片をIn-Fusion法により連結した。得られた発現カセットX1(SED1プロモーター(配列番号1)+SED1分泌シグナル(配列番号2)+BlSP(配列番号5)+SED1アンカーリング領域(配列番号3)+DIT1ターミネーター(配列番号4))を含むプラスミドをpIBlSP-SSSDと命名した。
(調製例1-2:ビフィドバクテリウム・ロングム(Bifidobacterium longum)由来スクロースホスホリラーゼ(BlSP)分泌発現カセットX2を含有するベクタープラスミドの調製)
プラスミドpIBlSP-SSSDを鋳型とし、プライマー対DIT1t-F(配列番号16)及びBlSP-R(配列番号17)を用いてPCR法により増幅し、SED1アンカーリング領域を除いたプラスミド全長を含むDNA断片を調製した。この断片の両端をIn-Fusion法により連結し、閉環させた。次に、このプラスミドを鋳型とし、プライマー対BlSP-F(配列番号18)及びDIT1t-R(配列番号19)を用いてPCR法により増幅し、ビフィドバクテリウム・ロングム(Bifidobacterium longum)由来スクロースホスホリラーゼ(BlSP)遺伝子のコーディング領域及びサッカロマイセス・セレビシエ(Saccharomyces cerevisiae)由来DIT1遺伝子のターミネーター領域を含むDNA断片を調製した。また、ベクタープラスミドpIL2-BG-SSS(サッカロマイセス・セレビシエ(Saccharomyces cerevisiae)由来SED1プロモーター、サッカロマイセス・セレビシエ(Saccharomyces cerevisiae)由来SED1の分泌シグナルペプチド配列、アスペルギルス・アクレアタス由来β-グルコシダーゼ1(AaBGL1)遺伝子のコーディング領域、サッカロマイセス・セレビシエ(Saccharomyces cerevisiae)由来SED1遺伝子のコーディング領域(SED1アンカーリング領域)、及びサッカロマイセス・セレビシエ(Saccharomyces cerevisiae)由来α-アグルチニン遺伝子のターミネーター領域がこの順に配置されている発現カセットを有する表層発現用ベクター:Metabolic Engineering, Vol.57, 2020, 110-117)を鋳型とし、プライマー対pRS403-F(配列番号20)及びSED1ss-R(配列番号15)を用いてPCR法により増幅し、アスペルギルス・アクレアタス由来β-グルコシダーゼ1(AaBGL1)遺伝子のコーディング領域、サッカロマイセス・セレビシエ(Saccharomyces cerevisiae)由来SED1遺伝子のコーディング領域(SED1アンカーリング領域)、及びサッカロマイセス・セレビシエ(Saccharomyces cerevisiae)由来α-アグルチニン遺伝子のターミネーター領域を除いたベクタープラスミド全長を含むDNA断片を調製した。これらの断片をIn-Fusion法により連結した。得られた発現カセットX2(SED1プロモーター(配列番号1)+SED1分泌シグナル(配列番号2)+BlSP(配列番号5)+DIT1ターミネーター(配列番号4))を含むプラスミドをpIL2-BlSP-SSDと命名した。
プラスミドpIBlSP-SSSDを鋳型とし、プライマー対DIT1t-F(配列番号16)及びBlSP-R(配列番号17)を用いてPCR法により増幅し、SED1アンカーリング領域を除いたプラスミド全長を含むDNA断片を調製した。この断片の両端をIn-Fusion法により連結し、閉環させた。次に、このプラスミドを鋳型とし、プライマー対BlSP-F(配列番号18)及びDIT1t-R(配列番号19)を用いてPCR法により増幅し、ビフィドバクテリウム・ロングム(Bifidobacterium longum)由来スクロースホスホリラーゼ(BlSP)遺伝子のコーディング領域及びサッカロマイセス・セレビシエ(Saccharomyces cerevisiae)由来DIT1遺伝子のターミネーター領域を含むDNA断片を調製した。また、ベクタープラスミドpIL2-BG-SSS(サッカロマイセス・セレビシエ(Saccharomyces cerevisiae)由来SED1プロモーター、サッカロマイセス・セレビシエ(Saccharomyces cerevisiae)由来SED1の分泌シグナルペプチド配列、アスペルギルス・アクレアタス由来β-グルコシダーゼ1(AaBGL1)遺伝子のコーディング領域、サッカロマイセス・セレビシエ(Saccharomyces cerevisiae)由来SED1遺伝子のコーディング領域(SED1アンカーリング領域)、及びサッカロマイセス・セレビシエ(Saccharomyces cerevisiae)由来α-アグルチニン遺伝子のターミネーター領域がこの順に配置されている発現カセットを有する表層発現用ベクター:Metabolic Engineering, Vol.57, 2020, 110-117)を鋳型とし、プライマー対pRS403-F(配列番号20)及びSED1ss-R(配列番号15)を用いてPCR法により増幅し、アスペルギルス・アクレアタス由来β-グルコシダーゼ1(AaBGL1)遺伝子のコーディング領域、サッカロマイセス・セレビシエ(Saccharomyces cerevisiae)由来SED1遺伝子のコーディング領域(SED1アンカーリング領域)、及びサッカロマイセス・セレビシエ(Saccharomyces cerevisiae)由来α-アグルチニン遺伝子のターミネーター領域を除いたベクタープラスミド全長を含むDNA断片を調製した。これらの断片をIn-Fusion法により連結した。得られた発現カセットX2(SED1プロモーター(配列番号1)+SED1分泌シグナル(配列番号2)+BlSP(配列番号5)+DIT1ターミネーター(配列番号4))を含むプラスミドをpIL2-BlSP-SSDと命名した。
(調製例1-3:クロストリジウム・ステルコラリウム(Clostridium stercorarium)由来セロビオースホスホリラーゼ(CsCBP)表層発現カセットX3を含有するベクタープラスミドの調製)
クロストリジウム・ステルコラリウム(Clostridium stercorarium)由来セロビオースホスホリラーゼ(CsCBP)遺伝子のコーディング領域を含むDNA断片を遺伝子合成により調製した。次に、ベクタープラスミドpIEG-SSSD(サッカロマイセス・セレビシエ(Saccharomyces cerevisiae)由来SED1プロモーター、サッカロマイセス・セレビシエ(Saccharomyces cerevisiae)由来SED1の分泌シグナルペプチド配列、トリコデルマ・リーセイ由来エンドグルカナーゼII(TrEGII)遺伝子のコーディング領域、サッカロマイセス・セレビシエ(Saccharomyces cerevisiae)由来SED1遺伝子のコーディング領域(SED1アンカーリング領域)、及びサッカロマイセス・セレビシエ(Saccharomyces cerevisiae)由来DIT1遺伝子のターミネーター領域がこの順に配置されているカセットを有する表層発現用ベクター:Applied Microbiology and Biotechnology, Vol.105, 2021, 5895-5904)を鋳型とし、プライマー対SED1a-F(配列番号14)及びSED1ss-R(配列番号15)を用いてPCR法により増幅し、TrEGII遺伝子のコーディング領域を除いたベクタープラスミド全長を含むDNA断片を調製した。これらの断片をIn-Fusion法により連結した。次に、このプラスミドを鋳型とし、プライマー対SED1p-F(配列番号21)及びDIT1t-R(配列番号19)を用いてPCR法により増幅し、サッカロマイセス・セレビシエ(Saccharomyces cerevisiae)由来SED1プロモーター、サッカロマイセス・セレビシエ(Saccharomyces cerevisiae)由来SED1の分泌シグナルペプチド配列、クロストリジウム・ステルコラリウム(Clostridium stercorarium)由来セロビオースホスホリラーゼ(CsCBP)のコーディング領域、サッカロマイセス・セレビシエ(Saccharomyces cerevisiae)由来SED1遺伝子のコーディング領域(SED1アンカーリング領域)、及びサッカロマイセス・セレビシエ(Saccharomyces cerevisiae)由来DIT1遺伝子のターミネーター領域を含むDNA断片を調製した。また、ベクタープラスミドpIU5-CBHD(サッカロマイセス・セレビシエ(Saccharomyces cerevisiae)由来SED1プロモーター、タラロマイセス・エメルソニ(Talaromyces emersonii)由来セロビオヒドロラーゼ(TsCBH1)遺伝子のコーディング領域、サッカロマイセス・セレビシエ(Saccharomyces cerevisiae)由来SED1遺伝子のコーディング領域(SED1アンカーリング領域)、及びサッカロマイセス・セレビシエ(Saccharomyces cerevisiae)由来α-アグルチニン遺伝子のターミネーター領域がこの順に配置されているカセットを有する表層発現用ベクター:Biotechnology and Bioengineering, Vol.114, 2017, 1201-1207)を鋳型とし、プライマー対pRS403-F(配列番号20)及びpRS403-R(配列番号22)を用いてPCR法により増幅し、発現カセット領域を除いたベクタープラスミド全長を含むDNA断片を調製した。これらの断片をIn-Fusion法により連結した。得られた発現カセットX3(SED1プロモーター(配列番号1)+SED1分泌シグナル(配列番号2)+CsCBP(配列番号6)+SED1アンカーリング領域(配列番号3)+DIT1ターミネーター(配列番号4))を含むプラスミドをpIU5-CsCBP-SSSDと命名した。
クロストリジウム・ステルコラリウム(Clostridium stercorarium)由来セロビオースホスホリラーゼ(CsCBP)遺伝子のコーディング領域を含むDNA断片を遺伝子合成により調製した。次に、ベクタープラスミドpIEG-SSSD(サッカロマイセス・セレビシエ(Saccharomyces cerevisiae)由来SED1プロモーター、サッカロマイセス・セレビシエ(Saccharomyces cerevisiae)由来SED1の分泌シグナルペプチド配列、トリコデルマ・リーセイ由来エンドグルカナーゼII(TrEGII)遺伝子のコーディング領域、サッカロマイセス・セレビシエ(Saccharomyces cerevisiae)由来SED1遺伝子のコーディング領域(SED1アンカーリング領域)、及びサッカロマイセス・セレビシエ(Saccharomyces cerevisiae)由来DIT1遺伝子のターミネーター領域がこの順に配置されているカセットを有する表層発現用ベクター:Applied Microbiology and Biotechnology, Vol.105, 2021, 5895-5904)を鋳型とし、プライマー対SED1a-F(配列番号14)及びSED1ss-R(配列番号15)を用いてPCR法により増幅し、TrEGII遺伝子のコーディング領域を除いたベクタープラスミド全長を含むDNA断片を調製した。これらの断片をIn-Fusion法により連結した。次に、このプラスミドを鋳型とし、プライマー対SED1p-F(配列番号21)及びDIT1t-R(配列番号19)を用いてPCR法により増幅し、サッカロマイセス・セレビシエ(Saccharomyces cerevisiae)由来SED1プロモーター、サッカロマイセス・セレビシエ(Saccharomyces cerevisiae)由来SED1の分泌シグナルペプチド配列、クロストリジウム・ステルコラリウム(Clostridium stercorarium)由来セロビオースホスホリラーゼ(CsCBP)のコーディング領域、サッカロマイセス・セレビシエ(Saccharomyces cerevisiae)由来SED1遺伝子のコーディング領域(SED1アンカーリング領域)、及びサッカロマイセス・セレビシエ(Saccharomyces cerevisiae)由来DIT1遺伝子のターミネーター領域を含むDNA断片を調製した。また、ベクタープラスミドpIU5-CBHD(サッカロマイセス・セレビシエ(Saccharomyces cerevisiae)由来SED1プロモーター、タラロマイセス・エメルソニ(Talaromyces emersonii)由来セロビオヒドロラーゼ(TsCBH1)遺伝子のコーディング領域、サッカロマイセス・セレビシエ(Saccharomyces cerevisiae)由来SED1遺伝子のコーディング領域(SED1アンカーリング領域)、及びサッカロマイセス・セレビシエ(Saccharomyces cerevisiae)由来α-アグルチニン遺伝子のターミネーター領域がこの順に配置されているカセットを有する表層発現用ベクター:Biotechnology and Bioengineering, Vol.114, 2017, 1201-1207)を鋳型とし、プライマー対pRS403-F(配列番号20)及びpRS403-R(配列番号22)を用いてPCR法により増幅し、発現カセット領域を除いたベクタープラスミド全長を含むDNA断片を調製した。これらの断片をIn-Fusion法により連結した。得られた発現カセットX3(SED1プロモーター(配列番号1)+SED1分泌シグナル(配列番号2)+CsCBP(配列番号6)+SED1アンカーリング領域(配列番号3)+DIT1ターミネーター(配列番号4))を含むプラスミドをpIU5-CsCBP-SSSDと命名した。
(調製例1-4:セルロモナス・ウダ(Cellulomonas uda)由来セロビオースホスホリラーゼ(CuCBP)表層発現カセットX4を含有するベクタープラスミドの調製)
セルロモナス・ウダ(Cellulomonas uda)由来セロビオースホスホリラーゼ(CuCBP)遺伝子のコーディング領域を含むDNA断片を遺伝子合成により調製した。次に、ベクタープラスミドpIU5-CsCBP-SSSDを鋳型とし、プライマー対プライマー対SED1a-F(配列番号14)及びSED1ss-R(配列番号15)を用いてPCR法により増幅し、CsCBP遺伝子のコーディング領域を除いたベクタープラスミド全長を含むDNA断片を調製した。これらの断片をIn-Fusion法により連結した。得られた発現カセットX4(SED1プロモーター(配列番号1)+SED1分泌シグナル(配列番号2)+CuCBP(配列番号7)+SED1アンカーリング領域(配列番号3)+DIT1ターミネーター(配列番号4))を含むプラスミドをpIU5-CuCBP-SSSDと命名した。
セルロモナス・ウダ(Cellulomonas uda)由来セロビオースホスホリラーゼ(CuCBP)遺伝子のコーディング領域を含むDNA断片を遺伝子合成により調製した。次に、ベクタープラスミドpIU5-CsCBP-SSSDを鋳型とし、プライマー対プライマー対SED1a-F(配列番号14)及びSED1ss-R(配列番号15)を用いてPCR法により増幅し、CsCBP遺伝子のコーディング領域を除いたベクタープラスミド全長を含むDNA断片を調製した。これらの断片をIn-Fusion法により連結した。得られた発現カセットX4(SED1プロモーター(配列番号1)+SED1分泌シグナル(配列番号2)+CuCBP(配列番号7)+SED1アンカーリング領域(配列番号3)+DIT1ターミネーター(配列番号4))を含むプラスミドをpIU5-CuCBP-SSSDと命名した。
(調製例1-5:サーモシフォ・アフリカヌス(Thermosipho africanus)由来セロビオースホスホリラーゼ(TaCBP)表層発現カセットX5を含有するベクタープラスミドの調製)
サーモシフォ・アフリカヌス(Thermosipho africanus)由来セロビオースホスホリラーゼ(TaCBP)遺伝子のコーディング領域を含むDNA断片を遺伝子合成により調製した。次に、ベクタープラスミドpIU5-CsCBP-SSSDを鋳型とし、プライマー対SED1a-F(配列番号14)及びSED1ss-R(配列番号15)を用いてPCR法により増幅し、CsCBP遺伝子のコーディング領域を除いたベクタープラスミド全長を含むDNA断片を調製した。これらの断片をIn-Fusion法により連結した。得られた発現カセットX5(SED1プロモーター(配列番号1)+SED1分泌シグナル(配列番号2)+TaCBP(配列番号8)+SED1アンカーリング領域(配列番号3)+DIT1ターミネーター(配列番号4))を含むプラスミドをpIU5-TaCBP-SSSDと命名した。
サーモシフォ・アフリカヌス(Thermosipho africanus)由来セロビオースホスホリラーゼ(TaCBP)遺伝子のコーディング領域を含むDNA断片を遺伝子合成により調製した。次に、ベクタープラスミドpIU5-CsCBP-SSSDを鋳型とし、プライマー対SED1a-F(配列番号14)及びSED1ss-R(配列番号15)を用いてPCR法により増幅し、CsCBP遺伝子のコーディング領域を除いたベクタープラスミド全長を含むDNA断片を調製した。これらの断片をIn-Fusion法により連結した。得られた発現カセットX5(SED1プロモーター(配列番号1)+SED1分泌シグナル(配列番号2)+TaCBP(配列番号8)+SED1アンカーリング領域(配列番号3)+DIT1ターミネーター(配列番号4))を含むプラスミドをpIU5-TaCBP-SSSDと命名した。
(調製例1-6:サーモトガ・ネアポリタナ(Thermotoga neapolitana)由来セロビオースホスホリラーゼ(TnCBP)表層発現カセットX6を含有するベクタープラスミドの調製)
サーモトガ・ネアポリタナ(Thermotoga neapolitana)由来セロビオースホスホリラーゼ(TnCBP)遺伝子のコーディング領域を含むDNA断片を遺伝子合成により調製した。次に、ベクタープラスミドpIU5-CsCBP-SSSDを鋳型とし、プライマー対SED1a-F(配列番号14)及びSED1ss-R(配列番号15)を用いてPCR法により増幅し、CsCBP遺伝子のコーディング領域を除いたベクタープラスミド全長を含むDNA断片を調製した。これらの断片をIn-Fusion法により連結した。得られた発現カセットX6(SED1プロモーター(配列番号1)+SED1分泌シグナル(配列番号2)+TnCBP(配列番号9)+SED1アンカーリング領域(配列番号3)+DIT1ターミネーター(配列番号4))を含むプラスミドをpIU5-TnCBP-SSSDと命名した。
サーモトガ・ネアポリタナ(Thermotoga neapolitana)由来セロビオースホスホリラーゼ(TnCBP)遺伝子のコーディング領域を含むDNA断片を遺伝子合成により調製した。次に、ベクタープラスミドpIU5-CsCBP-SSSDを鋳型とし、プライマー対SED1a-F(配列番号14)及びSED1ss-R(配列番号15)を用いてPCR法により増幅し、CsCBP遺伝子のコーディング領域を除いたベクタープラスミド全長を含むDNA断片を調製した。これらの断片をIn-Fusion法により連結した。得られた発現カセットX6(SED1プロモーター(配列番号1)+SED1分泌シグナル(配列番号2)+TnCBP(配列番号9)+SED1アンカーリング領域(配列番号3)+DIT1ターミネーター(配列番号4))を含むプラスミドをpIU5-TnCBP-SSSDと命名した。
(調製例1-7:ビフィドバクテリウム・ロングム(Bifidobacterium longum)由来スクロースホスホリラーゼ(BlSP)表層発現カセットX7を含有するベクタープラスミドの調製)
プラスミドpIBlSP-SSSDを鋳型とし、プライマー対BlSP-NheI-F(配列番号23)及びBlSP-XhoI-R(配列番号24)を用いてPCR法により増幅し、さらにNheI及びXhoIで処理することで、BlSP遺伝子のコーディング領域を含むDNA断片を調製した。また、ベクタープラスミドpIBG-PpSSG61(ピキア・パストリス(Pichia pastoris)由来SPI1プロモーター、ピキア・パストリス(Pichia pastoris)由来SPI1の分泌シグナルペプチド配列、アスペルギルス・アクレアタス由来β-グルコシダーゼ1(AaBGL1)遺伝子のコーディング領域、ピキア・パストリス(Pichia pastoris)由来GCW61遺伝子のコーディング領域(GCW61アンカーリング領域)、及びピキア・パストリス(Pichia pastoris)由来AOX1遺伝子のターミネーター領域がこの順に配置されている発現カセットを有する表層発現用ベクター:Biotechnology and Bioengineering, Vol.120, 2023, 1097-1107)をNheI及びXhoIで処理し、AaBGL1遺伝子のコーディング領域を除いたベクタープラスミド全長を含むDNA断片を調製した。これらの断片をライゲーション法により連結した。次に、このプラスミドを鋳型とし、XhoI及びMluIで処理し、GCW61アンカーリング領域を除いたベクタープラスミド全長を含むDNA断片を調製した。さらに、プラスミドpIBG-PpGMG30(ピキア・パストリス(Pichia pastoris)由来GAPDHプロモーター、サッカロマイセス・セレビシエ(Saccharomyces cerevisiae)由来α-ファクターの分泌シグナルペプチド配列、アスペルギルス・アクレアタス由来β-グルコシダーゼ1(AaBGL1)遺伝子のコーディング領域、ピキア・パストリス(Pichia pastoris)由来GCW30遺伝子のコーディング領域(GCW30アンカーリング領域)、及びピキア・パストリス(Pichia pastoris)由来AOX1遺伝子のターミネーター領域がこの順に配置されている発現カセットを有する表層発現用ベクター:Biotechnology and Bioengineering, Vol.120, 2023, 1097-1107)をXhoI及びMluIで処理し、GCW30アンカーリング領域を含むDNA断片を調製した。これらの断片をライゲーション法により連結した。得られた発現カセットX7(SPI1プロモーター(配列番号10)+SPI1分泌シグナル(配列番号11)+BlSP(配列番号5)+GCW30アンカーリング領域(配列番号12)+AOX1ターミネーター(配列番号13))を含むプラスミドをpIBlSP-PpSSG30と命名した。
プラスミドpIBlSP-SSSDを鋳型とし、プライマー対BlSP-NheI-F(配列番号23)及びBlSP-XhoI-R(配列番号24)を用いてPCR法により増幅し、さらにNheI及びXhoIで処理することで、BlSP遺伝子のコーディング領域を含むDNA断片を調製した。また、ベクタープラスミドpIBG-PpSSG61(ピキア・パストリス(Pichia pastoris)由来SPI1プロモーター、ピキア・パストリス(Pichia pastoris)由来SPI1の分泌シグナルペプチド配列、アスペルギルス・アクレアタス由来β-グルコシダーゼ1(AaBGL1)遺伝子のコーディング領域、ピキア・パストリス(Pichia pastoris)由来GCW61遺伝子のコーディング領域(GCW61アンカーリング領域)、及びピキア・パストリス(Pichia pastoris)由来AOX1遺伝子のターミネーター領域がこの順に配置されている発現カセットを有する表層発現用ベクター:Biotechnology and Bioengineering, Vol.120, 2023, 1097-1107)をNheI及びXhoIで処理し、AaBGL1遺伝子のコーディング領域を除いたベクタープラスミド全長を含むDNA断片を調製した。これらの断片をライゲーション法により連結した。次に、このプラスミドを鋳型とし、XhoI及びMluIで処理し、GCW61アンカーリング領域を除いたベクタープラスミド全長を含むDNA断片を調製した。さらに、プラスミドpIBG-PpGMG30(ピキア・パストリス(Pichia pastoris)由来GAPDHプロモーター、サッカロマイセス・セレビシエ(Saccharomyces cerevisiae)由来α-ファクターの分泌シグナルペプチド配列、アスペルギルス・アクレアタス由来β-グルコシダーゼ1(AaBGL1)遺伝子のコーディング領域、ピキア・パストリス(Pichia pastoris)由来GCW30遺伝子のコーディング領域(GCW30アンカーリング領域)、及びピキア・パストリス(Pichia pastoris)由来AOX1遺伝子のターミネーター領域がこの順に配置されている発現カセットを有する表層発現用ベクター:Biotechnology and Bioengineering, Vol.120, 2023, 1097-1107)をXhoI及びMluIで処理し、GCW30アンカーリング領域を含むDNA断片を調製した。これらの断片をライゲーション法により連結した。得られた発現カセットX7(SPI1プロモーター(配列番号10)+SPI1分泌シグナル(配列番号11)+BlSP(配列番号5)+GCW30アンカーリング領域(配列番号12)+AOX1ターミネーター(配列番号13))を含むプラスミドをpIBlSP-PpSSG30と命名した。
(調製例1-8:ビフィドバクテリウム・ロングム(Bifidobacterium longum)由来スクロースホスホリラーゼ(BlSP)分泌発現カセットX8を含有するベクタープラスミドの調製)
プラスミドpIBlSP-PpSSG30を鋳型とし、プライマー対BlSP-F2(配列番号25)及びBlSP-R2(配列番号26)を用いてPCR法により増幅し、BlSP遺伝子のコーディング領域を含むDNA断片を調製した。また、ベクタープラスミドpIH-EG-SSG34(ピキア・パストリス(Pichia pastoris)由来SPI1プロモーター、ピキア・パストリス(Pichia pastoris)由来SPI1の分泌シグナルペプチド配列、トリコデルマ・リーセイ由来エンドグルカナーゼII(TrEGII)遺伝子のコーディング領域、ピキア・パストリス(Pichia pastoris)由来GCW34遺伝子のコーディング領域(GCW34アンカーリング領域)、及びピキア・パストリス(Pichia pastoris)由来AOX1遺伝子のターミネーター領域がこの順に配置されている発現カセットを有する表層発現用ベクター:Biotechnology and Bioengineering, Vol.120, 2023, 1097-1107)を鋳型とし、プライマー対AOX1t-F(配列番号27)及びSPI1s s-R(配列番号28)を用いてPCR法により増幅し、TrEGII遺伝子のコーディング領域及びGCW34アンカーリング領域を除いたベクタープラスミド全長を含むDNA断片を調製した。これらの断片をIn-Fusion法により連結した。得られた発現カセットX8(SPI1プロモーター(配列番号10)+SPI1分泌シグナル(配列番号11)+BlSP(配列番号5)+AOX1ターミネーター(配列番号13))を含むプラスミドをpIH-BlSP-PpSSと命名した。
プラスミドpIBlSP-PpSSG30を鋳型とし、プライマー対BlSP-F2(配列番号25)及びBlSP-R2(配列番号26)を用いてPCR法により増幅し、BlSP遺伝子のコーディング領域を含むDNA断片を調製した。また、ベクタープラスミドpIH-EG-SSG34(ピキア・パストリス(Pichia pastoris)由来SPI1プロモーター、ピキア・パストリス(Pichia pastoris)由来SPI1の分泌シグナルペプチド配列、トリコデルマ・リーセイ由来エンドグルカナーゼII(TrEGII)遺伝子のコーディング領域、ピキア・パストリス(Pichia pastoris)由来GCW34遺伝子のコーディング領域(GCW34アンカーリング領域)、及びピキア・パストリス(Pichia pastoris)由来AOX1遺伝子のターミネーター領域がこの順に配置されている発現カセットを有する表層発現用ベクター:Biotechnology and Bioengineering, Vol.120, 2023, 1097-1107)を鋳型とし、プライマー対AOX1t-F(配列番号27)及びSPI1s s-R(配列番号28)を用いてPCR法により増幅し、TrEGII遺伝子のコーディング領域及びGCW34アンカーリング領域を除いたベクタープラスミド全長を含むDNA断片を調製した。これらの断片をIn-Fusion法により連結した。得られた発現カセットX8(SPI1プロモーター(配列番号10)+SPI1分泌シグナル(配列番号11)+BlSP(配列番号5)+AOX1ターミネーター(配列番号13))を含むプラスミドをpIH-BlSP-PpSSと命名した。
(調製例1-9:クロストリジウム・ステルコラリウム(Clostridium stercorarium)由来セロビオースホスホリラーゼ(CsCBP)表層発現カセットX9を含有するベクタープラスミドの調製)
プラスミドpIU5-CsCBP-SSSDを鋳型とし、プライマー対CsCBP-NheI-F(配列番号29)及びCsCBP-XhoI-R(配列番号30)を用いてPCR法により増幅し、さらにNheI及びXhoIで処理することで、CsCBP遺伝子のコーディング領域を含むDNA断片を調製した。また、ベクタープラスミドpIBlSP-PpSSG30をNheI及びXhoIで処理することで、BlSP遺伝子のコーディング領域を除いたベクタープラスミド全長を含むDNA断片を調製した。これらの断片をライゲーション法により連結した。次に、このプラスミドを鋳型とし、プライマー対CsCBP-F(配列番号31)及びAOX1t-R(配列番号32)を用いてPCR法により増幅し、CsCBP遺伝子のコーディング領域、GCW30アンカーリング領域、及びAOX1ターミネーター領域を含むDNA断片を調製した。また、ベクタープラスミドpIZ-CBH1-PpSSG34(ピキア・パストリス(Pichia pastoris)由来SPI1、タラロマイセス・エメルソニ(Talaromyces emersonii)由来セロビオヒドロラーゼ(TsCBH1)遺伝子のコーディング領域、ピキア・パストリス(Pichia pastoris)由来GCW34遺伝子のコーディング領域(GCW34アンカーリング領域)、及びピキア・パストリス(Pichia pastoris)由来AOX1遺伝子のターミネーター領域がこの順に配置されている発現カセットを有する表層発現用ベクター:Biotechnology and Bioengineering, Vol.120, 2023, 1097-1107)を鋳型とし、プライマー対AOX1t-F2(配列番号33)及びSPI1s s-R(配列番号28)を用いてPCR法により増幅し、TsCBH1遺伝子のコーディング領域、GCW34アンカーリング領域、及びAOX1ターミネーター領域を除いたベクタープラスミド全長を含むDNA断片を調製した。これらの断片をIn-Fusion法により連結した。得られた発現カセットX9(SPI1プロモーター(配列番号10)+SPI1分泌シグナル(配列番号11)+CsCBP(配列番号6)+GCW30アンカーリング領域(配列番号12)+AOX1ターミネーター(配列番号13))を含むプラスミドをpIZ-CsCBP-PpSSG30と命名した。
プラスミドpIU5-CsCBP-SSSDを鋳型とし、プライマー対CsCBP-NheI-F(配列番号29)及びCsCBP-XhoI-R(配列番号30)を用いてPCR法により増幅し、さらにNheI及びXhoIで処理することで、CsCBP遺伝子のコーディング領域を含むDNA断片を調製した。また、ベクタープラスミドpIBlSP-PpSSG30をNheI及びXhoIで処理することで、BlSP遺伝子のコーディング領域を除いたベクタープラスミド全長を含むDNA断片を調製した。これらの断片をライゲーション法により連結した。次に、このプラスミドを鋳型とし、プライマー対CsCBP-F(配列番号31)及びAOX1t-R(配列番号32)を用いてPCR法により増幅し、CsCBP遺伝子のコーディング領域、GCW30アンカーリング領域、及びAOX1ターミネーター領域を含むDNA断片を調製した。また、ベクタープラスミドpIZ-CBH1-PpSSG34(ピキア・パストリス(Pichia pastoris)由来SPI1、タラロマイセス・エメルソニ(Talaromyces emersonii)由来セロビオヒドロラーゼ(TsCBH1)遺伝子のコーディング領域、ピキア・パストリス(Pichia pastoris)由来GCW34遺伝子のコーディング領域(GCW34アンカーリング領域)、及びピキア・パストリス(Pichia pastoris)由来AOX1遺伝子のターミネーター領域がこの順に配置されている発現カセットを有する表層発現用ベクター:Biotechnology and Bioengineering, Vol.120, 2023, 1097-1107)を鋳型とし、プライマー対AOX1t-F2(配列番号33)及びSPI1s s-R(配列番号28)を用いてPCR法により増幅し、TsCBH1遺伝子のコーディング領域、GCW34アンカーリング領域、及びAOX1ターミネーター領域を除いたベクタープラスミド全長を含むDNA断片を調製した。これらの断片をIn-Fusion法により連結した。得られた発現カセットX9(SPI1プロモーター(配列番号10)+SPI1分泌シグナル(配列番号11)+CsCBP(配列番号6)+GCW30アンカーリング領域(配列番号12)+AOX1ターミネーター(配列番号13))を含むプラスミドをpIZ-CsCBP-PpSSG30と命名した。
・形質転換酵母の作製
(調製例2-1:スクロースホスホリラーゼ表層提示酵母の製造)
調製例1-1に記載のプラスミドpIBlSP-SSSDをNdeIで処理し、酵母サッカロマイセス・セレビシエ(Saccharomyces cerevisiae)BY4741ΔSUC2株に供し、酢酸リチウム法により形質転換した。この形質転換株をΔSUC2-BlSP株と称する。同様に、調製例1-7に記載のプラスミドpIBlSP-PpSSG30をEcoRVで処理し、酵母ピキア・パストリス(Pichia pastoris)CBS7435株に供し、酢酸リチウム法により形質転換した。この形質転換株をPp-BlSP株と称する。
(調製例2-1:スクロースホスホリラーゼ表層提示酵母の製造)
調製例1-1に記載のプラスミドpIBlSP-SSSDをNdeIで処理し、酵母サッカロマイセス・セレビシエ(Saccharomyces cerevisiae)BY4741ΔSUC2株に供し、酢酸リチウム法により形質転換した。この形質転換株をΔSUC2-BlSP株と称する。同様に、調製例1-7に記載のプラスミドpIBlSP-PpSSG30をEcoRVで処理し、酵母ピキア・パストリス(Pichia pastoris)CBS7435株に供し、酢酸リチウム法により形質転換した。この形質転換株をPp-BlSP株と称する。
また、調製例1-2に記載のプラスミドpIL2-BlSP-SSDをNdeIで処理し、ΔSUC2-BlSP株に供し、酢酸リチウム法により形質転換した。この形質転換株をΔSUC2-BlSP2株と称する。同様に、調製例1-8に記載のプラスミドpIH-BlSP-PpSSをBsrGIで処理し、Pp-BlSP株に供し、酢酸リチウム法により形質転換した。この形質転換株をPp-BlSP2株と称する。
(調製例2-2:セロビオースホスホリラーゼ表層提示酵母の製造)
調製例1-3~1-6に記載のプラスミドpIU5-CsCBP-SSSD、pIU5-CuCBP-SSSD、pIU5-TaCBP-SSSD、又はpIU5-TnCBP-SSSDをSpeIで処理し、酵母サッカロマイセス・セレビシエ(Saccharomyces cerevisiae)BY4741ΔSUC2株に供し、酢酸リチウム法によりそれぞれ形質転換した。これらの形質転換株をそれぞれΔSUC2-CsCBP株、ΔSUC2-CuCBP株、ΔSUC2-TaCBP株、及びΔSUC2-TnCBP株と称する。
調製例1-3~1-6に記載のプラスミドpIU5-CsCBP-SSSD、pIU5-CuCBP-SSSD、pIU5-TaCBP-SSSD、又はpIU5-TnCBP-SSSDをSpeIで処理し、酵母サッカロマイセス・セレビシエ(Saccharomyces cerevisiae)BY4741ΔSUC2株に供し、酢酸リチウム法によりそれぞれ形質転換した。これらの形質転換株をそれぞれΔSUC2-CsCBP株、ΔSUC2-CuCBP株、ΔSUC2-TaCBP株、及びΔSUC2-TnCBP株と称する。
(調製例2-3:スクロースホスホリラーゼ+セロビオースホスホリラーゼ表層提示酵母の製造)
調製例1-3に記載のプラスミドpIU5-CsCBP-SSSDをSpeIで処理し、ΔSUC2-BlSP2株に供し、酢酸リチウム法により形質転換した。この形質転換株をΔSUC2-SP2CBP株と称する。同様に、調製例1-9に記載のプラスミドpIZ-CsCBP-PpSSG30をNsiIで処理し、Pp-BlSP2株に供し、エレクトロポレーション法により形質転換した。この形質転換株をPp-SP2CBP株と称する。
調製例1-3に記載のプラスミドpIU5-CsCBP-SSSDをSpeIで処理し、ΔSUC2-BlSP2株に供し、酢酸リチウム法により形質転換した。この形質転換株をΔSUC2-SP2CBP株と称する。同様に、調製例1-9に記載のプラスミドpIZ-CsCBP-PpSSG30をNsiIで処理し、Pp-BlSP2株に供し、エレクトロポレーション法により形質転換した。この形質転換株をPp-SP2CBP株と称する。
B.形質転換酵母の前処理
Aで形質転換した酵母(菌株)のうち、サッカロマイセス・セレビシエ(Saccharomyces cerevisiae)を宿主とするΔSUC2-BlSP株、ΔSUC2-BlSP2株、ΔSUC2-CsCBP株、ΔSUC2-CuCBP株、ΔSUC2-TaCBP株、ΔSUC2-TnCBP株、及びΔSUC2-SP2CBP株について、5mLのYPD培地(乾燥酵母エキス(ナカライテスク)1%、BactoTM Peptone(Life Technologies)2%、D-グルコース(ナカライテスク)2%)に移植し、30℃、200rpmにて18時間前培養したのち、50mLのYPD培地にOD600が0.05となるように植菌し、30℃、150rpmにて48時間振とう培養した。また、ピキア・パストリス(Pichia pastoris)を宿主とするPp-BlSP株、Pp-BlSP2株、及びPp-SP2CBP株について、5mLのYPG培地(乾燥酵母エキス(ナカライテスク)1%、BactoTM Peptone(Life Technologies)2%、グリセロール(ナカライテスク)2%)に移植し、30℃、200rpmにて18時間前培養したのち、50mLのYPG培地にOD600が0.05となるように植菌し、30℃、150rpmにて48時間振とう培養した。これらの培養液を遠心分離して菌体を沈降させ、純水で2回洗浄し、できる限り上澄みを除いて固体の湿潤状態の菌体(wet cell)の重量を測定し、これと同量の純水を加え菌体懸濁液を得た(約500g湿潤細胞/L)。菌体懸濁液は60℃で90分間保持し、前処理菌体とした。
Aで形質転換した酵母(菌株)のうち、サッカロマイセス・セレビシエ(Saccharomyces cerevisiae)を宿主とするΔSUC2-BlSP株、ΔSUC2-BlSP2株、ΔSUC2-CsCBP株、ΔSUC2-CuCBP株、ΔSUC2-TaCBP株、ΔSUC2-TnCBP株、及びΔSUC2-SP2CBP株について、5mLのYPD培地(乾燥酵母エキス(ナカライテスク)1%、BactoTM Peptone(Life Technologies)2%、D-グルコース(ナカライテスク)2%)に移植し、30℃、200rpmにて18時間前培養したのち、50mLのYPD培地にOD600が0.05となるように植菌し、30℃、150rpmにて48時間振とう培養した。また、ピキア・パストリス(Pichia pastoris)を宿主とするPp-BlSP株、Pp-BlSP2株、及びPp-SP2CBP株について、5mLのYPG培地(乾燥酵母エキス(ナカライテスク)1%、BactoTM Peptone(Life Technologies)2%、グリセロール(ナカライテスク)2%)に移植し、30℃、200rpmにて18時間前培養したのち、50mLのYPG培地にOD600が0.05となるように植菌し、30℃、150rpmにて48時間振とう培養した。これらの培養液を遠心分離して菌体を沈降させ、純水で2回洗浄し、できる限り上澄みを除いて固体の湿潤状態の菌体(wet cell)の重量を測定し、これと同量の純水を加え菌体懸濁液を得た(約500g湿潤細胞/L)。菌体懸濁液は60℃で90分間保持し、前処理菌体とした。
C.酵母細胞表層のスクロースホスホリラーゼ活性測定
反応液5mL(組成:10g/Lスクロース(ナカライテスク);80mMリン酸ナトリウム緩衝液(pH7.0);及び調製例2-1で調製したPp-BlSP株またはPp-BlSP2株の前処理菌体(最終菌体濃度100g湿潤細胞/L))を調製し、35rpm、60℃にて60分間反応させた。反応を停止させるために、反応液を100℃、10分間保持し、その後10分間氷冷した後に遠心して菌体などを沈殿させ、上清を集めてフルクトース濃度の分析に用いた。1分間で1μmolのフルクトースを遊離する酵素量を1IUとする。
反応液5mL(組成:10g/Lスクロース(ナカライテスク);80mMリン酸ナトリウム緩衝液(pH7.0);及び調製例2-1で調製したPp-BlSP株またはPp-BlSP2株の前処理菌体(最終菌体濃度100g湿潤細胞/L))を調製し、35rpm、60℃にて60分間反応させた。反応を停止させるために、反応液を100℃、10分間保持し、その後10分間氷冷した後に遠心して菌体などを沈殿させ、上清を集めてフルクトース濃度の分析に用いた。1分間で1μmolのフルクトースを遊離する酵素量を1IUとする。
D.スクロースホスホリラーゼ表層提示酵母とセロビオースホスホリラーゼ表層提示酵母を併用したスクロースからのセロビオース生産
反応液5mL(組成:100g/Lスクロース(ナカライテスク);80mMリン酸ナトリウム緩衝液(pH7.0);1重量%硫酸マグネシウム;1.5IU/mLグルコースイソメラーゼ(キシロースイソメラーゼ、合同酒精(株)製、ストレプトマイセス・グリセオフスクス(Streptomyces griseofuscus)由来);調製例2-1で調製したΔSUC2-BlSP2株の前処理菌体(最終菌体濃度50g湿潤細胞/L);及び調製例2-2で調製したΔSUC2-CsCBP株、ΔSUC2-CuCBP株、ΔSUC2-TaCBP株、またはΔSUC2-TnCBP株の前処理菌体(最終菌体濃度50g湿潤細胞/L))を調製し、35rpm、60℃にて24時間反応させた。サンプリングは4時間及び24時間の2回行った。反応を停止させるために、サンプリング液はサンプリング容器にとって100℃、10分間保持し、その後10分間氷冷した後に遠心して菌体などを沈殿させ、上清を集めて分析に用いた。
反応液5mL(組成:100g/Lスクロース(ナカライテスク);80mMリン酸ナトリウム緩衝液(pH7.0);1重量%硫酸マグネシウム;1.5IU/mLグルコースイソメラーゼ(キシロースイソメラーゼ、合同酒精(株)製、ストレプトマイセス・グリセオフスクス(Streptomyces griseofuscus)由来);調製例2-1で調製したΔSUC2-BlSP2株の前処理菌体(最終菌体濃度50g湿潤細胞/L);及び調製例2-2で調製したΔSUC2-CsCBP株、ΔSUC2-CuCBP株、ΔSUC2-TaCBP株、またはΔSUC2-TnCBP株の前処理菌体(最終菌体濃度50g湿潤細胞/L))を調製し、35rpm、60℃にて24時間反応させた。サンプリングは4時間及び24時間の2回行った。反応を停止させるために、サンプリング液はサンプリング容器にとって100℃、10分間保持し、その後10分間氷冷した後に遠心して菌体などを沈殿させ、上清を集めて分析に用いた。
E.スクロースホスホリラーゼ+セロビオースホスホリラーゼ表層提示酵母を用いたスクロースからのセロビオース生産
反応液5mL(組成:100g/Lスクロース(ナカライテスク);80mMリン酸ナトリウム緩衝液(pH7.0);1重量%硫酸マグネシウム;1.5IU/mLグルコースイソメラーゼ(キシロースイソメラーゼ、合同酒精(株)製、ストレプトマイセス・グリセオフスクス(Streptomyces griseofuscus)由来);及び調製例2-3で調製したΔSUC2-SP2CBP株またはPp-BlSP2株の前処理菌体(最終菌体濃度100g湿潤細胞/L))を調製し、35rpm、60℃にて24時間反応させた。サンプリングは4時間、8時間、及び24時間の3回行った。反応を停止させるために、サンプリング液はサンプリング容器にとって100℃、10分間保持し、その後10分間氷冷した後に遠心して菌体などを沈殿させ、上清を集めて分析に用いた。
反応液5mL(組成:100g/Lスクロース(ナカライテスク);80mMリン酸ナトリウム緩衝液(pH7.0);1重量%硫酸マグネシウム;1.5IU/mLグルコースイソメラーゼ(キシロースイソメラーゼ、合同酒精(株)製、ストレプトマイセス・グリセオフスクス(Streptomyces griseofuscus)由来);及び調製例2-3で調製したΔSUC2-SP2CBP株またはPp-BlSP2株の前処理菌体(最終菌体濃度100g湿潤細胞/L))を調製し、35rpm、60℃にて24時間反応させた。サンプリングは4時間、8時間、及び24時間の3回行った。反応を停止させるために、サンプリング液はサンプリング容器にとって100℃、10分間保持し、その後10分間氷冷した後に遠心して菌体などを沈殿させ、上清を集めて分析に用いた。
F.分析
C、D、及びEの上清の分析にはHPLCを用いた。分析条件は下記とした。
・装置:ポンプユニット(Shimadzu LC-20AB)、オートサンプラー(Shimadzu SIL-20AC)、カラムオーブン(Shimadzu CTO-20AC)
・使用カラム:Sugar-D(COSMOSIL、Code:05397-51、ID4.6×250mm)
・ガードカラム:Sugar-D guard column(COSMOSIL、Code:05397-81)
・移動相:アセトニトリル/水=70/30、流量1.0mL/分
・カラムオーブン温度:30℃
・検出器:示差屈折率検出器(RID)(Shimadzu RID-10A)
・分析時間:15分
・検量線:糖混合液(スクロース、グルコース、フルクトース、及びセロビオース)を使用し、1、5、10g/Lの3点で校正した。
C、D、及びEの上清の分析にはHPLCを用いた。分析条件は下記とした。
・装置:ポンプユニット(Shimadzu LC-20AB)、オートサンプラー(Shimadzu SIL-20AC)、カラムオーブン(Shimadzu CTO-20AC)
・使用カラム:Sugar-D(COSMOSIL、Code:05397-51、ID4.6×250mm)
・ガードカラム:Sugar-D guard column(COSMOSIL、Code:05397-81)
・移動相:アセトニトリル/水=70/30、流量1.0mL/分
・カラムオーブン温度:30℃
・検出器:示差屈折率検出器(RID)(Shimadzu RID-10A)
・分析時間:15分
・検量線:糖混合液(スクロース、グルコース、フルクトース、及びセロビオース)を使用し、1、5、10g/Lの3点で校正した。
G.結果
Cのスクロースホスホリラーゼ活性測定の結果を図1に示す。スクロースホスホリラーゼを細胞壁に係留された状態で発現させるための発現カセットX1のみを導入されたΔSUC2-BlSP株と比較して、発現カセットX1に加えてスクロースホスホリラーゼを分泌発現させるための発現カセットX2を導入されたΔSUC2-BlSP2株は、著しく高いスクロースホスホリラーゼ活性を示すことが分かった。このことにより、スクロースホスホリラーゼについては、係留型と分泌型を併用することが好ましいことがわかった。
Cのスクロースホスホリラーゼ活性測定の結果を図1に示す。スクロースホスホリラーゼを細胞壁に係留された状態で発現させるための発現カセットX1のみを導入されたΔSUC2-BlSP株と比較して、発現カセットX1に加えてスクロースホスホリラーゼを分泌発現させるための発現カセットX2を導入されたΔSUC2-BlSP2株は、著しく高いスクロースホスホリラーゼ活性を示すことが分かった。このことにより、スクロースホスホリラーゼについては、係留型と分泌型を併用することが好ましいことがわかった。
Dのスクロースホスホリラーゼ表層提示酵母とセロビオースホスホリラーゼ表層提示酵母を併用したスクロースからのセロビオース生産実験の反応液上清中のスクロース、グルコース、フルクトース、及びセロビオース濃度の経時変化を図2に示す。セロビオースホスホリラーゼ表層提示酵母としてΔSUC-CsCBP株を用いた場合には、スクロースのセロビオースへの転換率は反応開始から24時間で約30%であった(図2(a))。一方で、セロビオースホスホリラーゼ表層提示酵母としてΔSUC-CuCBP株(図2(b))、ΔSUC-TaCBP株(図2(c))又はΔSUC-TnCBP株(図2(d))を用いた場合には、転換率は反応開始から24時間でも10%以下であった。この結果により、CBP遺伝子としてはクロストリジウム・ステルコラリウム(Clostridium stercorarium)由来のものが好ましいことがわかった。
Eのサッカロマイセス・セレビシエ(Saccharomyces cerevisiae)を宿主としたスクロースホスホリラーゼ+セロビオースホスホリラーゼ表層提示酵母(ΔSUC2-SP2CBP株)を用いたスクロースからのセロビオース生産実験の反応液上清中のスクロース、グルコース、フルクトース、及びセロビオース濃度の経時変化を図3に示す。図3(a)のグラフはサッカロマイセス・セレビシエ(Saccharomyces cerevisiae)を宿主とするΔSUC2-SP2CBP株を用いた場合の結果を示し、図3(b)のグラフはピキア・パストリス(Pichia pastoris)を宿主とするPp-SP2CBP株を用いた場合の結果を示す。サッカロマイセス・セレビシエ(Saccharomyces cerevisiae)を宿主としてスクロースホスホリラーゼのみを発現する組換え酵母株とセロビオースホスホリラーゼのみを発現する組換え酵母株を併用した図2(a)の実験結果と比較して、同じサッカロマイセス・セレビシエ(Saccharomyces cerevisiae)を宿主としてスクロースホスホリラーゼとセロビオースホスホリラーゼを共発現する単一の組換え酵母株を用いた図3(a)では、反応に使用した酵母菌体の総量は同じであるにも関わらず、スクロースがセロビオースにより高い転換率(約70%)で転換されていることがわかる。このことにより、スクロースホスホリラーゼとセロビオースホスホリラーゼについては単一の組換え酵母株に共発現させることが好ましいことがわかった。また、ピキア・パストリス(Pichia pastoris)を宿主としてスクロースホスホリラーゼとセロビオースホスホリラーゼを共発現する単一の組換え酵母株を用いた図3(b)では、スクロースのセロビオースへの転換率は約80%にまで向上した。
Eのピキア・パストリス(Pichia pastoris)を宿主としたスクロースホスホリラーゼ+セロビオースホスホリラーゼ表層提示酵母(Pp-SP2CBP株)を用いたスクロースからのセロビオース生産実験の反応液上清中のスクロース、グルコース、フルクトース、及びセロビオース濃度の経時変化を図4に示す。図4(a)のグラフはグルコースを炭素源として培養したPp-SP2CBP株を用いた場合の結果を示し、図4(b)のグラフはグリセロールを炭素源として培養したPp-SP2CBP株を用いた場合の結果を示す。Pp-SP2CBP株のスクロースのセロビオースへの転換率は、グルコースを炭素源として培養した場合には約40%であったが、グリセロールを炭素源として培養した場合には、約80%にまで向上した。このことにより、ピキア・パストリス(Pichia pastoris)を宿主としたスクロースホスホリラーゼ+セロビオースホスホリラーゼ表層提示酵母を培養する際には、グリセロールを炭素源に用いることが好ましいことがわかった。
[実施例群II:形質転換酵母(スクロースホスホリラーゼ+セロビオース表層提示酵母)を再利用した繰り返しセロビオース生産]
A.形質転換酵母の作製
実施例群IのAで形質転換した酵母(菌株)のうち、サッカロマイセス・セレビシエ(Saccharomyces cerevisiae)を宿主とするΔSUC2-SP2CBP株、ピキア・パストリス(Pichia pastoris)を宿主とするPp-SP2CBP株を用いた。
A.形質転換酵母の作製
実施例群IのAで形質転換した酵母(菌株)のうち、サッカロマイセス・セレビシエ(Saccharomyces cerevisiae)を宿主とするΔSUC2-SP2CBP株、ピキア・パストリス(Pichia pastoris)を宿主とするPp-SP2CBP株を用いた。
B.形質転換酵母の前処理
Aで用意したΔSUC2-SP2CBP株について、5mLのYPD培地(乾燥酵母エキス(ナカライテスク)1%、BactoTM Peptone(Life Technologies)2%、D-グルコース(ナカライテスク)2%)に移植し、30℃、200rpmにて18時間前培養したのち、50mLのYPD培地にOD600が0.05となるように植菌し、30℃、150rpmにて48時間振とう培養した。また、Pp-SP2CBP株について、5mLのYPG培地(乾燥酵母エキス(ナカライテスク)1%、BactoTM Peptone(Life Technologies)2%、グリセロール(ナカライテスク)2%)に移植し、30℃、200rpmにて24時間前培養したのち、50mLのYPG培地に、OD600が0.05となるように植菌し、30℃、150rpmにて48時間振とう培養した。これらの培養液を遠心分離して菌体を沈降させ、純水で2回洗浄し、できる限り上澄みを除いて固体の湿潤状態の菌体(wet cell)の重量を測定し、これと同量の純水を加え菌体懸濁液を得た(約500g湿潤細胞/L)。菌体懸濁液は60℃で90分間保持し、前処理菌体とした。
Aで用意したΔSUC2-SP2CBP株について、5mLのYPD培地(乾燥酵母エキス(ナカライテスク)1%、BactoTM Peptone(Life Technologies)2%、D-グルコース(ナカライテスク)2%)に移植し、30℃、200rpmにて18時間前培養したのち、50mLのYPD培地にOD600が0.05となるように植菌し、30℃、150rpmにて48時間振とう培養した。また、Pp-SP2CBP株について、5mLのYPG培地(乾燥酵母エキス(ナカライテスク)1%、BactoTM Peptone(Life Technologies)2%、グリセロール(ナカライテスク)2%)に移植し、30℃、200rpmにて24時間前培養したのち、50mLのYPG培地に、OD600が0.05となるように植菌し、30℃、150rpmにて48時間振とう培養した。これらの培養液を遠心分離して菌体を沈降させ、純水で2回洗浄し、できる限り上澄みを除いて固体の湿潤状態の菌体(wet cell)の重量を測定し、これと同量の純水を加え菌体懸濁液を得た(約500g湿潤細胞/L)。菌体懸濁液は60℃で90分間保持し、前処理菌体とした。
C.形質転換酵母を再利用した繰り返しセロビオース生産
反応液5mL(組成:100g/Lスクロース(ナカライテスク);80mMリン酸ナトリウム緩衝液(pH7.0);1重量%硫酸マグネシウム;1.5IU/mLグルコースイソメラーゼ(キシロースイソメラーゼ、合同酒精(株)製、ストレプトマイセス・グリセオフスクス(Streptomyces griseofuscus)由来);及びG-1で調製したΔSUC2-SP2CBP株またはPp-SP2CBP株の前処理菌体(最終菌体濃度100g湿潤細胞/L))を調製し、35rpm、60℃にて24時間反応させた(1回目)。反応させた液の遠心分離で酵母細胞を回収し、新しい反応液に再懸濁し、35rpm、60℃にて24時間反応させた(2回目)。同様に3回目の反応を行った。サンプリングは、各回の反応において4時間、8時間、及び24時間の3回行った。反応を停止させるために、各サンプリング液はサンプリング容器にとって100℃、10分間保持し、その後10分間氷冷した後に遠心して菌体などを沈殿させ、上清を集めて分析に用いた。
反応液5mL(組成:100g/Lスクロース(ナカライテスク);80mMリン酸ナトリウム緩衝液(pH7.0);1重量%硫酸マグネシウム;1.5IU/mLグルコースイソメラーゼ(キシロースイソメラーゼ、合同酒精(株)製、ストレプトマイセス・グリセオフスクス(Streptomyces griseofuscus)由来);及びG-1で調製したΔSUC2-SP2CBP株またはPp-SP2CBP株の前処理菌体(最終菌体濃度100g湿潤細胞/L))を調製し、35rpm、60℃にて24時間反応させた(1回目)。反応させた液の遠心分離で酵母細胞を回収し、新しい反応液に再懸濁し、35rpm、60℃にて24時間反応させた(2回目)。同様に3回目の反応を行った。サンプリングは、各回の反応において4時間、8時間、及び24時間の3回行った。反応を停止させるために、各サンプリング液はサンプリング容器にとって100℃、10分間保持し、その後10分間氷冷した後に遠心して菌体などを沈殿させ、上清を集めて分析に用いた。
D.分析
上記Dで得られた上清につき、実施例群IのFに記載の方法で分析した。
上記Dで得られた上清につき、実施例群IのFに記載の方法で分析した。
E.結果
上記Dで得られた上清中のスクロース、グルコース、フルクトース、及びセロビオース濃度の経時変化を、図5(a)(ΔSUC2-SP2CBP株)及び図5(b)(Pp-SP2CBP株)に示す。この結果により、本発明の、スクロースホスホリラーゼ及びセロビオースホスホリラーゼを発現する組換え酵母とグルコースイソメラーゼとを含有する反応液中にスクロースを共存させる工程を含む、セロビオースの製造方法は、当該工程を繰り返しても高いセロビオースへの転換率が維持されることがわかった。また、グリセロールを炭素源として培養したピキア・パストリスを宿主としたスクロースホスホリラーゼ+セロビオースホスホリラーゼ表層提示酵母を形質転換酵母として用いた場合には、Pp-SP2CBP株の酵母は、60℃、24時間の反応を3回繰り返しても、約80%の高いセロビオース転換率が維持されることが確認された。
上記Dで得られた上清中のスクロース、グルコース、フルクトース、及びセロビオース濃度の経時変化を、図5(a)(ΔSUC2-SP2CBP株)及び図5(b)(Pp-SP2CBP株)に示す。この結果により、本発明の、スクロースホスホリラーゼ及びセロビオースホスホリラーゼを発現する組換え酵母とグルコースイソメラーゼとを含有する反応液中にスクロースを共存させる工程を含む、セロビオースの製造方法は、当該工程を繰り返しても高いセロビオースへの転換率が維持されることがわかった。また、グリセロールを炭素源として培養したピキア・パストリスを宿主としたスクロースホスホリラーゼ+セロビオースホスホリラーゼ表層提示酵母を形質転換酵母として用いた場合には、Pp-SP2CBP株の酵母は、60℃、24時間の反応を3回繰り返しても、約80%の高いセロビオース転換率が維持されることが確認された。
本発明の製造方法は、食品又は医薬品製剤に使用するセロビオースの製造方法として利用することができる。
Claims (14)
- スクロースホスホリラーゼ及びセロビオースホスホリラーゼを発現する組換え酵母とグルコースイソメラーゼとを含有する反応液中にスクロースを共存させる反応工程を含む、セロビオースの製造方法。
- 組換え酵母が、サッカロマイセス(Saccharomyces)属、ピキア(Pichia)属、又はヤロウィア(Yarrowia)属の酵母である、請求項1に記載の製造方法。
- スクロースホスホリラーゼが、ビフィドバクテリウム(Bifidobacterium)属、ロイコノストック(Leuconostoc)属、又はストレプトコッカス(Streptococcus)属の細菌に由来する、請求項1又は2に記載の製造方法。
- スクロースホスホリラーゼが、組換え酵母から分泌発現される分泌型スクロースホスホリラーゼと、組換え酵母の細胞壁に係留された状態で発現される係留型スクロースホスホリラーゼとを含む、請求項1~3の何れか一項に記載の製造方法。
- セロビオースホスホリラーゼが、クロストリジウム(Clostridium)属、ルミノコッカス(Ruminococcus)属、セルロモナス(Cellulomonas)属、又はサーモトガ(Thermotoga)属の細菌に由来する、請求項1~4の何れか一項に記載の製造方法。
- 前記組換え酵母が、スクロースホスホリラーゼ及びセロビオースホスホリラーゼを単一の菌体で共に発現する組換え酵母である、請求項1~5の何れか一項に記載の製造方法。
- 前記組換え酵母が、スクロースホスホリラーゼを発現する組換え酵母と、セロビオースホスホリラーゼを発現する組換え酵母との組み合わせである、請求項1~6の何れか一項に記載の製造方法。
- 前記組換え酵母が、スクロースホスホリラーゼ遺伝子を担持する発現ベクターにより形質転換された酵母である、請求項1~7の何れか一項に記載の製造方法。
- 前記発現ベクターが、スクロースホスホリラーゼ遺伝子及び酵母の細胞壁ドメイン遺伝子を共に担持する第1の発現ベクターと、酵母の細胞壁ドメイン遺伝子を担持せずスクロースホスホリラーゼ遺伝子のみを担持する第2の発現ベクターとの組み合わせを含む、請求項8に記載の製造方法。
- グルコースイソメラーゼが、ストレプトマイセス(Streptomyces)属の細菌に由来する、請求項1~9の何れか一項に記載の製造方法。
- 反応液中にスクロースを共存させる前に、組換え酵母を45℃~75℃で15分~150分処理する工程を更に含む、請求項1~10の何れか一項に記載の製造方法。
- 前記反応工程の前に、炭素源としてグリセロールを用いて酵母細胞を培養する工程を含む、請求項1~11の何れか一項に記載の製造方法。
- 前記反応工程の後に、製造されたセロビオースを反応液から分離する工程をさらに含む、請求項1~12の何れか一項に記載の製造方法。
- 前記反応工程の後に、前記組換え酵母を反応液から回収し、再度の反応工程に供することをさらに含む、請求項1~13の何れか一項に記載の製造方法。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP2023-094225 | 2023-06-07 | ||
| JP2023094225 | 2023-06-07 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| WO2024252885A1 true WO2024252885A1 (ja) | 2024-12-12 |
Family
ID=93795374
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| PCT/JP2024/018152 Ceased WO2024252885A1 (ja) | 2023-06-07 | 2024-05-16 | セロビオースの製造方法 |
Country Status (2)
| Country | Link |
|---|---|
| TW (1) | TW202511494A (ja) |
| WO (1) | WO2024252885A1 (ja) |
Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH01137979A (ja) * | 1987-11-24 | 1989-05-30 | Natl Food Res Inst | グルコースイソメラーゼ遺伝子、該遺伝子を有する組換え体および該組換え体を有する微生物 |
| JPH03130086A (ja) * | 1989-10-17 | 1991-06-03 | Natl Food Res Inst | セロビオースの製造方法 |
| JP2004222506A (ja) * | 2003-01-17 | 2004-08-12 | Nikken Kasei Kk | α−グルカンからのセロビオ−スの製造法 |
| CN108611386A (zh) * | 2016-12-12 | 2018-10-02 | 中国科学院天津工业生物技术研究所 | 多酶催化制备纤维二糖的方法 |
-
2024
- 2024-05-16 WO PCT/JP2024/018152 patent/WO2024252885A1/ja not_active Ceased
- 2024-05-29 TW TW113119839A patent/TW202511494A/zh unknown
Patent Citations (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JPH01137979A (ja) * | 1987-11-24 | 1989-05-30 | Natl Food Res Inst | グルコースイソメラーゼ遺伝子、該遺伝子を有する組換え体および該組換え体を有する微生物 |
| JPH03130086A (ja) * | 1989-10-17 | 1991-06-03 | Natl Food Res Inst | セロビオースの製造方法 |
| JP2004222506A (ja) * | 2003-01-17 | 2004-08-12 | Nikken Kasei Kk | α−グルカンからのセロビオ−スの製造法 |
| CN108611386A (zh) * | 2016-12-12 | 2018-10-02 | 中国科学院天津工业生物技术研究所 | 多酶催化制备纤维二糖的方法 |
Non-Patent Citations (5)
| Title |
|---|
| "18.3 Cellobiose Production from Sucrose", BIOCATALYSIS AND BIOTECHNOLOGY FOR FUNCTIONAL FOODS AND INDUSTRIAL PRODUCTS: INTERNATIONAL SYMPOSIUM ON BIOCATALYSIS AND BIOTECHNOLOGY ; (TAICHUNG, TAIWAN) : 2005.10.19-21, CRC PRESS, 1 January 2007 (2007-01-01) - 21 October 2005 (2005-10-21), pages 326 - 328, XP009560695, ISBN: 0-8493-9282-9 * |
| B. KULLIN ; V. R. ABRATT ; S. J. REID: "A functional analysis of the Bifidobacterium longum cscA and scrP genes in sucrose utilization", APPLIED MICROBIOLOGY AND BIOTECHNOLOGY, SPRINGER, BERLIN, DE, vol. 72, no. 5, 8 March 2006 (2006-03-08), Berlin, DE , pages 975 - 981, XP019441648, ISSN: 1432-0614, DOI: 10.1007/s00253-006-0358-x * |
| KITAOKA M. ET AL.: "Conversion of sucrose into cellbiose using sucrose phosphorylase, xylose isomerase and cellobiose phosphorylase", JAPANESE SOCIETY OF STARCH SCIENCE. JOURNAL / DENPUN KAGAKU., JAPANESE SOCIETY OF STARCH SCIENCE, TSUKUBA., JP, vol. 39, no. 4, 1 January 1992 (1992-01-01), JP , pages 281 - 283, XP002983778, ISSN: 0021-5406 * |
| KITAOKA, MOTOMITSU: "Producing cellobiose from sucrose", KAGAKU TO SEIBUTSU - CHEMISTRY AND BIOLOGY, GAKKAI SHUPPAN SENTA / JAPAN SCIENTIFIC SOCIETIES PRESS, JP, vol. 40, no. 8, 1 January 2002 (2002-01-01), JP , pages 498 - 500, XP009560076, ISSN: 0453-073X * |
| ZHONG CHAO; WEI PING; ZHANG YI-HENG PERCIVAL: "A kinetic model of one-pot rapid biotransformation of cellobiose from sucrose catalyzed by three thermophilic enzymes", CHEMICAL ENGINEERING SCIENCE, OXFORD, GB, vol. 161, 1 December 2016 (2016-12-01), GB , pages 159 - 166, XP029889317, ISSN: 0009-2509, DOI: 10.1016/j.ces.2016.11.047 * |
Also Published As
| Publication number | Publication date |
|---|---|
| TW202511494A (zh) | 2025-03-16 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP3086249B2 (ja) | 熱、酸及び/またはアルカリに高い安定性を有する細菌由来突然変異体α―アミラーゼ | |
| JP6537076B2 (ja) | 分泌シグナルペプチドならびにそれを利用したタンパク質の分泌および細胞表層提示 | |
| AU2015223025B2 (en) | Enzymatic hydrolysis of disaccharides and oligosaccharides using alpha-glucosidase enzymes | |
| CN103555690B (zh) | 一种新型果糖苷酶及其编码基因和应用 | |
| US20150167030A1 (en) | Recombinant cellulosome complex and uses thereof | |
| JP2026053673A (ja) | タガトースを産生する枯草菌の遺伝子工学菌及びタガトースの調製方法 | |
| Rolain et al. | O-glycosylation as a novel control mechanism of peptidoglycan hydrolase activity | |
| CN112063666A (zh) | 一种重组蔗糖异构酶在转化蔗糖制备异麦芽酮糖中的应用 | |
| CN110234751A (zh) | 用于改善酒精发酵的异源蛋白酶表达 | |
| KR102007890B1 (ko) | 변형된 당 대사 경로를 갖는 재조합 균주를 이용한 당이성화 효소의 스크리닝 방법 | |
| CN106459942A (zh) | 具有增强活性的外切葡聚糖酶变体及其用途 | |
| WO2024252885A1 (ja) | セロビオースの製造方法 | |
| CN103031290B (zh) | 一种耐高糖的β-葡萄糖苷酶Bgl4及其表达基因和应用 | |
| CN101165190B (zh) | 固定化双酶法制备n-乙酰神经氨酸 | |
| JP6954644B2 (ja) | アルギン酸リアーゼ及び当該酵素を用いる不飽和ウロン酸単糖の製造方法 | |
| CN112867790A (zh) | 提高苹果酸产量的新突变蛋白 | |
| CN104673814A (zh) | 一种来自于阴沟肠杆菌的l-苏氨酸醛缩酶及其应用 | |
| JP5951085B2 (ja) | 改良型β−フルクトフラノシダーゼ | |
| EP3071692B1 (fr) | Variants d'endoglucanases a activite amelioree et leurs utilisations | |
| JP2014504877A (ja) | セロデキストリンおよびβ‐D‐グルコースの高度発酵 | |
| US9382555B2 (en) | Polypetide with enhanced beta-glucosidase activity at low temperature | |
| KR102867250B1 (ko) | 진세노사이드 전환용 조성물 및 이를 이용한 진세노사이드의 전환 방법 | |
| JP7449558B2 (ja) | 新規キシログルカナーゼ及びキシログルカンオリゴ糖の製造方法 | |
| JP7319652B2 (ja) | タンパク質細胞表層発現酵母 | |
| JP2012039966A (ja) | セルロース結合ドメインが不活性化されたセロビオヒドロラーゼ活性を有するタンパク質 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 121 | Ep: the epo has been informed by wipo that ep was designated in this application |
Ref document number: 24819118 Country of ref document: EP Kind code of ref document: A1 |
|
| NENP | Non-entry into the national phase |
Ref country code: DE |