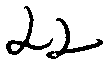CAMPO DA INVENÇÃO
A presente invenção refere-se a novos derivados de purina 2,6,9-substituídos e suas aplicações biológicas. Particularmente, a invenção refere-se a derivados de purina tendo propriedades antiproliferativas, que são úteis no tratamento de distúrbios proliferativos como câncer, leucemia, psoríase e semelhantes.
ANTECEDENTES
A iniciação, progressão, e conclusão do ciclo celular de mamíferos são reguladas por vários complexos de quinase dependentes de ciclina (CDK), que são críticos para o crescimento celular. Estes complexos compreendem pelo menos uma subunidade catalítica (o próprio CDK) e uma regulatória (ciclina). Alguns dos complexos mais importantes para a regulação do ciclo celular incluem ciclina A (CDK1 - também conhecido como cdc2, e CDK2), ciclina B1-B3 (CDK1), ciclina D1-D3 (CDK2, CDK4, CDK5, CDK6), ciclina E (CDK2). Cada um destes complexos está envolvido em uma fase particular do ciclo celular. Nem todos os membros da família CDK estão envolvidos exclusivamente no controle do ciclo celular, no entanto. Assim, CDKs 7, 8 e 9 estão implicados na regulação da transcrição, e CDK5 desempenha um papel em função da célula secretória e neuronal.
A atividade de CDKs é regulada pós-translacionalmente, por associações transitórias com outras proteínas, e por alterações de sua localização intracelular. O desenvolvimento de tumor está intimamente associado com alteração genética e desregulação de CDKs e seus reguladores, sugerindo que inibidores de CDKs podem ser terapêuticas anti-câncer úteis. De fato, resultados anteriores sugerem que células transformadas e normais diferem em sua exigência para, por exemplo, ciclina A/CDK2, e que pode ser possível desenvolver novos agentes antineoplásicos isento da toxicidade para o hospedeiro geral observada com drogas citostáticas e citotóxicas convencionais. Apesar da inibição de CDKs relacionados com o ciclo celular ser claramente relevante em, por exemplo, aplicações de oncologia, isto
Petição 870180152822, de 19/11/2018, pág. 6/18 pode não ser o caso para a inibição de CDKs regulando a RNA polimerase. Por outro lado, a inibição da função T de CDK9/ciclina foi recentemente ligada à prevenção de replicação de HIV e a descoberta de nova biologia de CDK, assim, continua a abrir novas indicações terapêuticas para os inibidores de CDK. (Sausville, E.A. Trends Molec. Med. 2002, 8,S32-S37).
A função de CDKs é para fosforilar e, assim, ativar ou desativar algumas proteínas, incluindo, por exemplo, proteínas de retinoblastoma, lamina, histona H1, e componentes do eixo celular mitótico. A etapa catalítica mediada por CDKs envolve uma reação de fosfo-transferência de ATP para o substrato de enzima macromolecular. Vários grupos de compostos (estudados em por exemplo Fischer, P.M. Curr. Opin. Drug Discovery Dev. 2001, 4, 623-634) foram verificados como possuindo propriedades antiproliferativas devido ao antagonismo ATP específico para CDK.
WO 98/05335 (CV Therapeutics Inc) descreve derivados de purina 2,6,9-trissubstituídos que são inibidores seletivos de quinases do ciclo celular. Tais compostos são úteis no tratamento de distúrbios auto-imunes, por exemplo, artrite reumatóide, lupus, diabetes tipo I, esclerose múltipla, tratamento de câncer, doença cardiovascular, tal como restenose, doença de hospedeiro v transplante, gota, doença de rim policístico, e outras doenças proliferativas cuja patogenese envolve proliferação anormal de células.
WO 99/07705 (The Regents of the University of Califórnia) descreve análogos de purina que inibem inter alia proteína quinases, proteínas G e polimerases. Mais especificamente, a invenção refere-se a métodos de usar estes análogos de purina para tratar distúrbios proliferativos celulares e doenças neurodegenerativas.
WO 97/20842 (CNRS) também descreve derivados de purina mostrando propriedades antiproliferativas que são úteis no tratamento de câncer, psoríase, e distúrbios neurodegenerativos.
A presente invenção procura prover novos derivados de purina 2,6,9-substituídos, particularmente aqueles tendo propriedades antiproliferativas.
DESCRIÇÃO DA INVENÇÃO
Um primeiro aspecto da invenção refere-se a um composto de fórmula I
.A ou um sal farmaceuticamente aceitável do mesmo, em que um dentre R1 e R2 é metila, etila ou isopropila, e o outro é H;
R3 e R4 são, cada um independentemente, H, Ci-C6 alquila ramificada ou não-ramificada, ou arila, e em que pelo menos um dentre R3 e R4 é diferente de H;
R5 é um grupo C1-C5 alquila ramificada ou não-ramificada, ou um grupo C-i-C6 cicloalquila, cada um podendo ser opcionalmente substituído com um ou mais grupos OH;
R6, R7, R8 e R9 são, cada um independentemente, H, halogênio, NO2, OH, OMe, CN, NH2, COOH, CONH2, ou SO2NH2.
Um segundo aspecto da invenção refere-se a uma composição farmacêutica compreendendo um composto de fórmula 1 e um excipiente, diluente ou veículo farmaceuticamente aceitável.
Um terceiro aspecto da invenção refere-se ao uso de um composto de fórmula 1 na preparação de um medicamento para o tratamento de um ou mais dos seguintes distúrbios:
um distúrbio proliferativo;
um distúrbio viral;
um derrame;
alopécia;
um distúrbio do sistema nervoso central;
um distúrbio neurodegenerativo; e diabetes.
Um quarto aspecto da invenção refere-se ao uso de um composto de fórmula 1 como um agente antimitótico.
Um quinto aspecto da invenção refere-se ao uso de um composto de fórmula 1 para inibir uma proteína quinase.
Um sexto aspecto da invenção refere-se a um método de tratamento de uma doença proliferativa, referido método compreendendo a administração a um mamífero de uma quantidade terapeuticamente eficaz de
um composto de fórmula 1.
Um sétimo aspecto da invenção refere-se ao uso de um composto da invenção em um teste para identificar outros compostos candidatos que influenciam a atividade de uma ou mais enzimas CDK.
DESCRIÇÃO DETALHADA
Como mencionado acima, um primeiro aspecto da invenção refere-se a um composto de fórmula 1 como definido acima.
Sabe-se, na técnica que a via de desativação metabólica in vivo principal do agente inibidor de CDK antiproliferativo experimental roscovitina (remeter à publicação do pedido de patente internacional PCT WO 20 97/20842; Wang, S., McCIue, S. J., Ferguson, J. R., Hull, J. D., Stokes, S.,
Parsons, S., Westwood, R., e Fischer, P. M. Tetrahedron: Asymmetry 2001, 12, 2891-2894) compreende a oxidação do grupo carbinol em um grupo carboxila e subseqüente excreção deste metabólito [Nutley, B. P., Raynaud, F.
I., Wilson, S. C., Fischer, P., McCIue, S., Goddard, P. M., Jarman, M., Lane,
D., e Workman, P. Clin. Câncer Res. 2000, 6 Suppl. (Proc. 11° AACR-NCIEORTC Intl. Conf. N° 318)]. O material sintético autêntico, idêntico com este metabólito, mostra reduzida atividade biológica in vitro. Assim, roscovitina e o derivado carboxila inibem a atividade de CDK2/ciclina E com valores IC50 de 0,08 e 0,24 μΜ, respectivamente. Similarmente, os valores IC5o anti30 proliferativos médios em um painel representativo de linhagens de células de tumor transformadas humanas, para roscovitina e o derivado carboxila foram de cerca de 10 e > 50 μΜ, respectivamente.
Metabólito carboxila
Assim, em uma modalidade preferida, a invenção procura prover novos derivados de purina que demonstram melhorada resistência à desativação metabólica.
Em uma modalidade preferida da invenção, um dentre R1 e R2 é 5 etila ou isopropila, e o outro é H.
> Em outra modalidade preferida da invenção, R5 é isopropila ou ciclopentila.
Λ
Em uma modalidade preferida, R6, R7, R8 e R9são todos H. Em uma modalidade preferida, R1 ou R2 é etila e o outro é H.
Em uma modalidade preferida, R3 e R4 são cada independente-
mente, H, metila, etila, propila, butila ou fenila.
Assim, em uma modalidade preferida, R3 e R4 são, cada um independentemente, H, metila, etila, n-propila, isopropila, n-butila, s-butila, t butila ou fenila.
Em uma modalidade mais preferida, R3 e R4, são cada um independentemente, H, metila, etila, propila ou butila.
Assim, em uma modalidade preferida, R3 e R4 são, cada um independentemente, H, metila, etila, n-propila, isopropila, n-butila, s-butila ou tbutila.
Em uma modalidade ainda mais preferida, R3 e R4 são, cada um independentemente, H, metila, etila, isopropila ou t-butila.
Em uma modalidade especialmente preferida, referido composto de fórmula 1 é selecionado dentre os seguintes:
(2S3F?)-3-{9-lsopropil-6-[(piridin-3-ilmetil)-amino]-9H-purin-2-ilamino}-pentan2-ol;
(2/?3S)-3-{9-lsopropil-6-[(piridin-3-ilmetil)-amino]-9H-purin-2-ilamino}-pentan2-ol;
(3/?S,4R)-4-{9-lsopropil-6-[(piridin-3-ilmetil)-amino]-9H-purin-2-ilamino}-hexan-3-ol;
(3/?S,4S)-4-{9-lsopropil-6-[(piridin-3-ilmetil)-amino]-9H-purin-2-ilamino}-hexan-3-ol;
(3RS,4/?)-4-{9-lsopropil-6-[(piridin-3-ilmetil)-amino]-9H-purin-2-ilamino}-2metil-hexan-3-ol;
(3/?S,4S)-4-{9-lsopropil-6-[(piridin-3-ilmetil)-amino]-9H-purin-2-ilamino}-2metil-hexan-3-ol;
(3F?S,4/?)-4-{9-lsopropil-6-[(piridin-3-ilmetil)-amino]-9H-purin-2-ilamino}-2,2dimetil-hexan-3-ol;
(3/?S,4S)-4-{9-lsopropil-6-[(piridin-3-ilmetil)-amino]-9H-purin-2-ilamino}-2,2dimetil-hexan-3-ol;
(3/?)-3-{9-lsopropil-6-[(piridin-3-ilmetil)-amino]-9H-purin-2-ilamino}-2-metilpentan-2-ol;
(3S)-3-{9-lsopropil-6-[(piridin-3-ilmetil)-amino]-9H-purin-2-ilamino}-2-metilpentan-2-ol;
(3S)-3-{9-lsopropil-6-[(piridin-3-ilmetil)-amino]-9H-purin-2-ilamino}-2-metilpentan-2-ol; e (3R)-3-{9-lsopropil-6-[(piridin-3-ilmetil)-amino]-9H-purin-2-ilamino}-2-metilpentan-2-ol.
Ainda mais preferivelmente, referido composto de fórmula 1 é selecionado dentre os seguintes:
(2S3/?)-3-{9-lsopropil-6-[(piridin-3-ilmetil)-amino]-9H-purin-2-ilamino}-pentan2-ol;
(2R3S)-3-{9-lsopropil-6-[(piridin-3-ilmetil)-amino]-9H-purin-2-ilamino}-pentan2-ol;
(3/?S,4/?)-4-{9-lsopropil-6-[(piridin-3-ilmetil)-amino]-9H-purin-2-ilamino}hexan-3-ol;
999 9 9
9
9 999 (3RS,4S)-4-{9-lsopropil-6-[(piridin-3-ilmetil)-amino]-9H-purin-2-ilamino}hexan-3-ol;
(3RS,4S)-4-{9-lsopropil-6-[(piridin-3-ilmetil)-amino]-9H-purin-2-ilamino}-2,2dimetil-hexan-3-ol;
(3/?)-3-{9-lsopropil-6-[(piridin-3-ilmetil)-amino]-9H-purin-2-ilamino}-2-metilpentan-2-ol; e (3S)-3-{9-lsopropil-6-[(piridin-3-ilmetil)-amino]-9H-purin-3-ilamino}-2-metilpentan-2-ol.
Ainda mais preferivelmente, referido composto de fórmula 1 é selecionado dentre os seguintes:
(3R)-3-{9-isopropil-6-[(piridin-3-ilmetil)-amino]-9H-purin-2ilamino}-2-metil-pentan-2-ol;
(3S)-3-{9-isopropil-6-[(piridin-3-ilmetil)-amino]-9H-purin-2ilamino}-2-metil-pentan-2-ol;
(2S3R)-3-{9-isopropil-6-[(piridin-3-ilmetil)-amino]-9H-purin-2ilamino}-pentan-2-ol;
(2R3S)-3-{9-isopropil-6-[(piridin-3-ilmetil)-amino]-9H-purin-2ilamino}-pentan-2-ol; e qualquer isômero ótico de 3-{9-isopropil-6-[(piridin-3-ilmetil)-amino]-9H-purin-2-ilamino}-pentan-2-ol.
Composições farmacêuticas
Um segundo aspecto da invenção refere-se a uma composição farmacêutica compreendendo um composto de fórmula 1 misturado com um excipiente, diluente ou veículo farmaceuticamente aceitável, ou uma mistura dos mesmos. Apesar dos compostos da presente invenção (incluindo seus sais farmaceuticamente aceitáveis, ésteres e solvatos farmaceuticamente aceitáveis) poderem ser administrados sozinhos, eles serão geralmente ad20 ministrados em mistura com um veículo farmacêutico, excipiente ou diluente, particularmente para terapia humana. A composição farmacêutica pode ser para uso humano ou animal, em medicina humana e veterinária.
Os exemplos destes excipientes apropriados para as várias formas diferentes de composição farmacêutica descritas aqui podem ser en• · · ·»· · ··· · »·· ··· • · · · ··· ·· · · • 9 · · ··<··· · ···· · · ♦ · · · • ····· · · · ±γ contrados em Handbook of Pharmaceutical Excipients, 2a ed., (1994), Editado por A Wade e PJ Weller.
Os veículos ou diluentes aceitáveis para uso terapêutico são bem conhecidos na técnica farmacêutica e são descritos, por exemplo, em 5 Remington's Pharmaceutical Sciences, Mack Publishing Co. (A. R. Gennaro edit. 1985).
Os exemplos de veículos apropriados incluem lactose, amido, glicose, metil celulose, estearato de magnésio, manitol, sorbitol, e semelhantes. Os exemplos de diluentes apropriados incluem etanol, glicerol e
A escolha de veículo, excipiente ou diluente farmacêutico pode ser selecionada com relação à via pretendida de administração e prática farmacêutica padrão. As composições farmacêuticas podem compreender como, ou em adição a, o veículo, excipiente ou diluente, qualquer aglutinan15 te(s) apropriado(s), lubrificante(s), agente(s) de suspensão, agente(s) de revestimento, agente(s) solubilizante(s).
Os exemplos de aglutinantes apropriados incluem amido, gelatina, açúcares naturais, tais como glicose, lactose anidra, lactose de fluxo livre, beta-lactose, adoçantes de milho, gomas naturais e sintéticas, tais como acácia, tragacanto ou alginato de sódio, carboximetil celulose e polietileno
glicol.
Os exemplos de lubrificantes apropriados incluem oleato de sódio, estearato de sódio, estearato de magnésio, benzoato de sódio, acetato de sódio, cloreto de sódio e semelhantes.
Os conservantes, estabilizadores, corantes e mesmo agentes flavorizantes podem ser providos na composição farmacêutica. Os exemplos de conservantes incluem benzoato de sódio, ácido sórbico e ésteres de ácido p-hidroxibenzóico. Os antioxidantes e agentes de suspensão podem ser também usados.
Sais/Ésteres
Os compostos da presente invenção podem estar presentes como sais ou ésteres, em particular sais ou ésteres farmaceuticamente • ··· · ··· · ··· ··· ·· · ·· ·· · · ··· · ···· · • · · · ····· · ······· ·· · • ····· ·· · aceitáveis.
Os sais farmaceuticamente aceitáveis dos compostos da invenção incluem os sais de base ou de adição de ácido apropriados dos mesmos. Um estudo de sais farmacêuticos apropriados pode ser encontrado em 5 Berge et al, J Pharm Sei, 66, 1-19 (1977). Os sais são formados, por exemplo, com ácidos inorgânicos fortes, tais como ácidos minerais, por exemplo, ácido sulfúrico, ácido fosfórico ou ácidos hidrohálicos; com ácidos carboxílicos orgânicos fortes, como ácido alcano carboxílicos de 1 a 4 átomos de carbono, que são não-substituídos ou substituídos (por exemplo, por halo-

gênio), tal como ácido acético; com ácidos dicarboxílicos saturados ou insaturados, por exemplo, oxálico, malônico, succínico, maléico, fumárico, ftálico ou tetraftálico; com ácidos hidroxicarboxílicos, por exemplo, ácido ascórbico, glicólico, láctico, málico, tartárico ou cítrico; com aminoácidos, por exemplo, ácido aspártico ou glutâmico; com ácido benzóico; ou com ácidos sulfônicos 15 orgânicos, tais como ácidos (C1-C4) alquil ou aril sulfônicos, que são não, substituídos ou substituídos (por exemplo, por um halogênio), tais como ácido metano- ou p-toluenossulfônico.
Os ésteres são formados ou usando ácidos orgânicos ou álcoois/hidróxidos, dependendo do grupo funcional sendo esterificado. Os ácidos orgânicos incluem ácidos carboxílicos, como ácidos alcanocarboxílicos de 1

a 12 átomos de carbono, que são não-substituídos ou substituídos (por exemplo, por halogênio), tal como ácido acético; com ácido dicarboxílico saturado ou insaturado, por exemplo, oxálico, malônico, succínico, maléico, fumárico, ftálico ou tetraftálico; com ácidos hidroxicarboxílicos, por exemplo, 25 ácido ascórbico, glicólico, láctico, málico, tartárico ou cítrico; com aminoácidos, por exemplo, ácido aspártico ou glutâmico; com ácido benzóico; ou com ácidos sulfônicos orgânicos, tais como ácidos (C1-C4) alquil ou aril sulfônicos, que são não-substituídos ou substituídos (por exemplo, por um halogênio), tais como ácido metano- ou p-toluenossulfônico. Os hidróxidos apropri30 ados incluem hidróxidos inorgânicos, tais como hidróxido de sódio, hidróxido de potássio, hidróxido de cálcio, hidróxido de alumínio. Os álcoois incluem alcanoálcoois de 1-12 átomos de carbono que podem ser não-substituídos
ou substituídos (por exemplo, por um halogênio). ENANTIÔMEROS/TAUTÔMEROS
Em todos os aspectos da presente invenção previamente discutidos, a invenção inclui, onde apropriado, todos os enantiômeros e tautômeros de compostos de fórmula I. O versado na técnica irá reconhecer compostos que possuem propriedades óticas (um ou mais átomos de carbono quirais), ou características tautoméricas. Os enantiômeros e/ou tautômeros podem ser isolados/preparados por métodos conhecidos na técnica. ESTEREOISÔMEROS E ISÔMEROS GEOMÉTRICOS
Alguns dos compostos da invenção podem existir como estereoisômeros e/ou isômeros geométricos - por exemplo eles podem possuir um ou mais centros assimétricos e/ou geométricos e, assim, podem existir em duas ou mais formas estereoisoméricas e/ou geométricas. A presente invenção contempla o uso de todos os estereoisomeros e/ou isômeros geométricos destes agentes inibidores, e misturas dos mesmos. Os termos usados nas reivindicações englobam estas formas, desde que as referidas formas retenham a atividade funcional apropriada (apesar de não necessariamente no mesmo grau).
A presente invenção também inclui todas as variações isotópicas apropriadas do agente ou um sal farmaceuticamente aceitável do mesmo. Uma variação isotópica de um agente da presente invenção, ou um sal farmaceuticamente aceitável do mesmo, é definida como uma na qual pelo menos um átomo é substituído por um átomo tendo o mesmo número atômico, mas uma massa atômica diferente da massa atômica geralmente encontrada na natureza. Os exemplos de isótopos que podem ser incorporados no agente e sais farmaceuticamente aceitáveis dos mesmos incluem isótopos de hidrogênio, carbono, nitrogênio, oxigênio, fósforo, enxofre, flúor e cloro, tais como 2H, 3H, 13C, 14C, 15N, 17O, 18O, 31P, 32P, 35S, 18F e 36CI, respectivamente. Certas variações isotópicas do agente e sais farmaceuticamente aceitáveis dos mesmos, por exemplo, aquelas nas quais um isótopo radioativo, tal como 3H ou 14C, é incorporado, são utilizáveis em estudos de distribuição de tecido de substrato e/ou droga. Os isótopos tritiados, isto é, 3H, e
2θ carbono-14, isto é, 14C, são particularmente preferidos por sua facilidade de preparação e capacidade de detecção. Além disso, a substituição com isótopos, tal como deutério, isto é, 2H, podem proporcionar certas vantagens terapêuticas resultantes da maior estabilidade metabólica, por exemplo, au5 mentada meia-vida in vivo, ou reduzidas exigências de dosagem e, portanto, podem ser preferidas em algumas circunstâncias. As variações isotópicas do agente da presente invenção e sais farmacêutica mente aceitáveis do mesmo desta invenção podem geralmente ser preparadas por procedimentos convencionais, usando variações isotópicas apropriadas de reagen-
tes apropriados.
SOLVATOS
A presente invenção também inclui formas de solvato dos compostos da presente invenção. Os termos usados nas reivindicações englobam estas formas.
\ 15 POLIMORFOS
A invenção, além disso, refere-se a compostos da presente invenção em suas várias formas cristalinas, formas polimórficas e formas anidras. É bem estabelecido na indústria farmacêutica que os compostos químicos podem ser isolados em qualquer uma destas formas por variação leve 20 do método de purificação e ou forma de isolamento dos solventes usados na
preparação sintética destes compostos. PRÓ-DROGAS
A invenção ainda inclui compostos da presente invenção em forma de pró-droga. Estas pró-drogas são geralmente compostos de fórmula 25 I em que um ou mais grupos apropriados foram modificados, de modo que a modificação pode ser revertida quando da administração a um indivíduo humano ou mamífero. Tal reversão é geralmente realizada por uma enzima naturalmente presente em tal indivíduo, apesar de ser possível para um segundo agente ser administrado junto com esta pró-droga, a fim de realizar a 30 reversão in vivo. Os exemplos destas modificações incluem éster (por exemplo, qualquer um dos descritos acima), em que a reversão pode ser realizada por uma esterase, etc. Outros tais sistemas serão bem conhecidos dos versados na técnica.
ADMINISTRAÇÃO
As composições farmacêuticas da presente invenção podem ser adaptadas para vias de administração oral, retal, vaginal, parenteral, intra5 muscular, intraperitoneal, intra-arterial, intratecal, intrabrônquica, subcutânea, intradérmica, intravenosa, nasal, bucal, ou sublingual.
Para administração oral, uso particular é feito de comprimidos, pílulas, tabletes, cápsulas de gelatina, pastilhas, e cápsulas. Preferivelmente, estas composições contém de 1 a 250 mg e mais preferivelmente de ΙΟ-
Ι 00 mg de ingrediente ativo por dose.
Outras formas de administração compreendem soluções ou emulsões que podem ser injetadas de modo intravenoso, intra-arterial, intratecal, subcutâneo, intradérmico, intraperitoneal ou intramuscular, e que são preparadas a partir de soluções estéreis ou esterilizáveis. As composi15 ções farmacêuticas da presente invenção também podem estar na forma de supositórios, supositório vaginal, suspensões, emulsões, loções, ungüentos, cremes, géis, pulverizações, soluções ou pós de polvilhamento.
Um meio alternativo de administração transdérmica é por uso de um adesivo na pele. Por exemplo, o ingrediente ativo pode ser incorporado em um creme consistindo de uma emulsão aquosa de polietileno glicol ou
parafina líquida. O ingrediente ativo também pode ser incorporado, em uma concentração de entre 1 e 10% em peso, em um ungüento consistindo de uma cera branca ou base de parafina flexível branca, junto com aqueles estabilizadores e conservantes, como pode ser requerido.
As formas injetáveis podem conter entre 10- 1000 mg, preferivelmente entre 10- 250 mg, de ingrediente ativo por dose.
As composições podem ser formuladas em forma de dosagem unitária, isto é, na forma de porções discretas contendo uma dose unitária, ou múltipla, ou uma subunidade de uma dose unitária.
DOSAGEM
Um versado na técnica pode facilmente determinar uma dose apropriada de uma das composições instantâneas para administrar a um
indivíduo sem indevida experimentação. Tipicamente, um médico irá determinar a dosagem real que será a mais apropriada para um paciente individual, e irá depender de vários fatores, incluindo a atividade do composto específico empregado, a estabilidade metabólica e a extensão de ação deste 5 composto, a idade, peso corporal, saúde geral, sexo, dieta, modo e tempo de administração, velocidade de excreção, combinação de drogas, a severidade da condição particular, e o indivíduo submetido à terapia. As dosagens descritas aqui são exemplares do caso médio. Pode-se ter, certamente, casos individuais onde faixas de dose maiores ou menores são necessárias, e
estas estão no escopo desta invenção.
Dependendo da necessidade, o agente pode ser administrado em uma dose de 0,01 a 30 mg/kg peso corporal, tal como 0,1 a 10 mg/kg, mais preferivelmente de 0,1 a 1 mg/kg peso corporal.
Em uma modalidade exemplar, uma ou mais doses de 10 a 150 15 mg/dia serão administradas ao paciente para o tratamento da malignidade.
USO TERAPÊUTICO
Os compostos da presente invenção foram verificados como possuindo atividade antiproliferativa e acredita-se serem assim de uso no tratamento de distúrbios proliferativos, como cânceres, leucemias, ou outros distúrbios associados com proliferação celular descontrolada, tais como pso-
ríase e restenose.
Como definido aqui, um efeito antiproliferativo no escopo da presente invenção pode ser demonstrado pela capacidade de inibir a proliferação celular em um teste de célula completa in vitro”, por exemplo, usando qualquer uma das linhagens de células A549, HeLa, HT-29, MCF7, Saos-2,
CCRF-CEM, HL-60 e K-562, ou mostrando inibição de quinase em um teste apropriado. Estes testes, incluindo métodos para seu desempenho, são descritos em maiores detalhes nos exemplos anexos. Usando estes testes, pode ser determinado se um composto é antiproliferativo no contexto da presente invenção.
Uma modalidade preferida da presente invenção assim refere-se ao uso de um ou mais compostos da invenção na preparação de um medi14
camento para o tratamento de um distúrbio proliferativo.
Como usado aqui, preparação de um medicamento inclui o uso de um composto da invenção diretamente como o medicamento, além de seu uso em um programa de triagem para outros agentes terapêuticos, ou 5 em qualquer estágio da fabricação deste medicamento.
O termo distúrbio proliferativo é usado aqui em um sentido amplo para incluir qualquer distúrbio que requer controle do ciclo celular, por exemplo, distúrbios cardiovasculares, tais como restenose e cardiomiopatia, distúrbios auto-imunes, tais como glomerulonefrite e artrite reumatóide, dis-
túrbios dermatológicos, tal como psoríase, distúrbios anti-inflamatórios, antifúngicos e anti-parasíticos, tais como malária, enfizema e alopécia. Nestes distúrbios, os compostos da presente invenção podem induzir a apoptose ou manter estase dentro das células desejadas, como requerido. Preferivelmente, o distúrbio proliferativo é um câncer ou leucemia.
Em outra modalidade preferida, o distúrbio proliferativo é psoríase.
Os compostos da invenção podem inibir qualquer uma das etapas ou estágios no ciclo celular, por exemplo, formação de envelope nuclear, saída a partir da fase quiescente do ciclo celular (GO), progressão de G1, 20 descondensação de cromossomos, ruptura de envelope nuclear, START,

iniciação de replicação de DNA, progressão de replicação de DNA, terminação de replicação de DNA, duplicação de centrossoma, progressão G2, ativação de funções mitóticas ou meióticas, condensação de cromossomos, separação de centrossomas, nucleação de microtúbulos, formação de eixo celular e função, interações com proteínas motoras de microtúbulos, separação e segregação de cromatídeos, inativação de funções mitóticas, formação de anel contrátil, e funções de citocinese. Particularmente, os compostos da invenção podem influenciar algumas funções de genes, tais como ligação de cromatina, formação de complexos de replicação, licenciamento de repli30 cação, fosforilação ou outra atividade de modificação secundária, degradação proteolítica, ligação de microtúbulos, ligação de actina, ligação de septina, atividade de nucleação de centro de organização de microtúbulos, e liga15 ção para componentes de vias de sinalização de ciclo celular.
Um outro aspecto da invenção refere-se a um método de tratamento de uma doença proliferativa, referido método compreendendo a administração a um mamífero de uma quantidade terapeuticamente eficaz de 5 um composto de fórmula 1.
Em uma modalidade preferida deste aspecto, o distúrbio proliferativo é câncer ou leucemia.
Em uma modalidade ainda mais preferida deste aspecto, o composto é administrado em uma quantidade suficiente para inibir pelo menos
uma enzima CDK.
Preferivelmente, o composto da invenção é administrado em uma quantidade suficiente para inibir pelo menos um dentre CDK1, CDK2,
CDK3, CDK4, CDK6, CDK7, CDK8 e/ou CDK9.
Mais preferivelmente, o composto da invenção é administrado em uma quantidade suficiente para inibir pelo menos um dentre CDK2 e/ou CDK4.
Ainda mais preferivelmente, a enzima CDK é CDK2.
Em uma modalidade preferida deste aspecto, o composto é administrado oralmente.
Outro aspecto da invenção refere-se ao uso de um composto de fórmula 1 como um agente antimitótico.
Ainda outro aspecto da invenção refere-se ao uso de um composto de fórmula 1 para o tratamento de um distúrbio neurodegenerativo.
Preferivelmente, o distúrbio neurodegenerativo é apoptose neu25 ronal.
Outro aspecto da invenção refere-se ao uso de um composto de fórmula 1 como um agente antiviral.
Assim, outro aspecto da invenção refere-se ao uso de um composto da invenção na preparação de um medicamento para o tratamento de 30 um distúrbio viral, tais como citomegalovírus humano (HCMV), vírus de herpes simples tipo 1 (HSV-1), vírus de imunodeficiência humana tipo 1 (HIV-1), e vírus varicela zóster (VZV).
Em uma modalidade mais preferida da invenção, o composto da invenção é administrado em uma quantidade suficiente para inibir um ou mais CDKs da célula hospedeira envolvida em replicação viral, isto é, CDK2, CDK7, CDK8, e CDK9 [Wang D, De la Fuente C, Deng L, Wang L, Zilberman 5 I, Eadie C, Healey M, Stein D, Denny T, Harrison LE, Meijer L, Kashanchi F.
Inhibition of human immunodeficiency virus type 1 transcription by chemical ciclin-dependent kinase inhibitors. J. Virol. 2001; 75: 7266-7279].
Como definido aqui, um efeito antiviral dentro do escopo da presente invenção pode ser demonstrado pela capacidade de inibir CDK2,
CDK7, CDK8 ou CDK9.
Em uma modalidade particularmente preferida, a invenção refere-se o uso de um ou mais compostos da invenção no tratamento de distúrbio viral que é dependente ou sensível a CDK. Os distúrbios dependentes de CDK são associados com um nível acima do normal de atividade de uma ou 15 mais enzimas CDK. Tais distúrbios preferivelmente associados com um nível anormal de atividade de CDK2, CDK7, CDK8 e/ou CDK9. Um distúrbio sensível a CDK é um distúrbio em que uma aberração no nível de CDK não é a causa primária, mas está a jusante da aberração metabólica primária. Em tais cenários, CDK2, CDK7, CDK8 e/ou CDK9 podem ser referidos como 20 parte da via metabólica sensível e os inibidores de CDK podem ser, então,
ativos no tratamento destes distúrbios.
Outro aspecto da invenção refere-se ao uso de compostos da invenção, ou sais farmaceuticamente dos mesmos, na preparação de um medicamento para o tratamento de diabetes.
Em uma modalidade particularmente preferida, o diabetes é diabetes tipo II.
GSK3 é uma das várias proteínas quinases que fosforilam a glicogene sintase (GS). A estimulação de síntese de glicogene por insulina em músculo esquelético resulta da desfosforilação e ativação de GS. A ação de 30 GSK3 em GS assim resulta na desativação do último e, assim, supressão de conversão da glicose em glicogene em músculos.
Diabetes tipo II (diabetes melito não-dependente de insulina) é uma doença multi-fatorial. Hiperglicemia é devido a resistência à insulina no fígado, músculos, e outros tecidos, junto com a secreção prejudicada de insulina. O músculo esquelético é o local principal para absorção de glicose estimulada por insulina, ali ela ou é removida da circulação ou convertida em 5 glicogene. A deposição de glicogene no músculo é o determinante principal em homeostase de glicose e diabéticos do tipo II tem armazenamento de glicogene no músculo defeituoso. Há evidência de que um aumento na atividade de GSK3 é importante em diabetes tipo II [Chen, Y.H.; Hansen, L.; Chen, M.X.; Bjorbaek, C.; Vestergaard, H.; Hansen, T.; Cohen, P.T.; Peder-

sen, O. Diabetes, 1994, 43, 1234]. Além disso, foi demonstrado que GSK3 é super-expressado em células musculares de diabéticos tipo II e que uma correlação inversa existe entre atividade de GSK3 de músculo esquelético e ação da insulina. [Nikoulina, S.E.; Ciaraldi, T.P.; Mudaliar, S.; Mohideen, P.;
Carter, L.; Henry, R.R. Diabetes, 2000, 49, 263].
A inibição de GSK3 é, então, de significado terapêutico no tratamento de diabetes, particularmente tipo II, e neuropatia diabética.
É notável que GSK3 é conhecido para fosforilar muitos substratos diferentes de GS, e está assim envolvido na regulação de vias bioquímicas múltiplas. Por exemplo, GSK é altamente expressado nos sistemas ner20 vosos central e periférico.
Outro aspecto da invenção, então, refere-se ao uso de compostos da invenção, ou sais farmaceuticamente aceitáveis dos mesmos, na preparação de um medicamento para o tratamento de distúrbios do sistema nervoso central, por exemplo, distúrbios neurodegenerativos.
Preferivelmente, o distúrbio do sistema nervoso central é doença de Alzheimer.
Tau é um substrato de GSK-3 que foi implicado na etiologia de doença de Alzheimer. Em células nervosas saudáveis, Tau co-reune-se com tubulina dentro dos microtúbulos. No entanto, na doença de Alzheimer, tau 30 forma grandes emaranhados de filamentos, que rompem as estruturas de microtúbulos na célula do nervo, assim prejudicando o transporte de nutrientes, bem como a transmissão de mensagens neuronais.
Sem desejar se limitar por teorias, acredita-se que os inibidores de GSK3 podem ser capazes de evitar e/ou reverter a hiperfosforilação anormal da proteína associada com microtúbulos tau, que é uma característica invariante de doença de Alzheimer e várias outras doenças neurodege5 nerativas, como palsia supranuclear progressiva, degeneração corticobasal e doença de Pick. Mutações no gene tau provocam formas herdadas de demência fronto-temporal, ainda enfatizando a relevância de disfunção de proteína tau para o processo neurodegenerativo [Goedert, M. Curr. Opin. Gen. Dev., 2001, 11, 343].
Outro aspecto da invenção refere-se ao uso de compostos da invenção, ou sais farmaceuticamente aceitáveis dos mesmos, na preparação de um medicamento para o tratamento de distúrbio bipolar.
Ainda outro aspecto da invenção refere-se ao uso de compostos da invenção, ou sais farmaceuticamente aceitáveis dos mesmos, na prepa15 ração de um medicamento para o tratamento de um derrame cerebral.
A redução de uma apoptose neuronal é um objetivo terapêutico importante no contexto de trauma na cabeça, derrame cerebral, epilepsia, e doença neuronal motora [Mattson, M.P. Nat. Rev. Mol. Cell. Biol., 2000, 1, 120]. Então, GSK3 como um fator pró-apoptótico em células neuronais torna esta proteína quinase um alvo terapêutico atraente para o projeto de drogas
inibidoras para tratar estas doenças.
Ainda outro aspecto da invenção refere-se ao uso de compostos da invenção, ou sais farmaceuticamente dos mesmos, na preparação de um medicamento para o tratamento de alopécia.
O crescimento de cabelo é controlado pela via de sinalização
Wnt, particularmente Wnt-3. Em sistemas de modelo de cultura de tecido da pele, a expressão de mutantes não-degradáveis de β-catenina leva a um aumento dramático na população de células -tronco putativas, que tem maior potencial proliferativo [Zhu, A.J.; Watt, F.M. Development, 1999, 126, 30 2285]. Esta população de células -tronco expressa um maior nível de βcatenina associada com não-caderina. [DasGupta, R.; Fuchs, E. Development, 1999, 126, 4557], que pode contribuir para seu potencial proliferativo elevado. Além disso, os camundongos transgênicos super-expressando uma β-catenina truncada na pele sofrem de morfogenese de folículo capilar de Novo, que normalmente é apenas estabelecida durante a embriogenese. A aplicação ectópica de inibidores de GSK3 pode assim ser terapeuticamente 5 útil no tratamento de calvície e na restauração de crescimento capilar após alopécia induzida por quimioterapia. Um outro aspecto da invenção refere-se a um método de tratamento de distúrbio dependente de GSK3, o referido método compreendendo a administração a um indivíduo, em necessidade do mesmo, de um composto de acordo com a invenção, ou um sal farmaceuti-

camente aceitável do mesmo, como definido acima, em uma quantidade suficiente para inibir GSK3.
Preferivelmente, o composto da invenção, ou sal farmaceuticamente aceitável do mesmo, é administrado em uma quantidade suficiente para inibir GSK3 β.
Em uma modalidade da invenção, o composto da invenção é administrado em uma quantidade suficiente para inibir pelo menos uma enzima PLK.
As quinases semelhantes a pólo (PLKs) constituem uma família de serina/treonina proteína quinases. Os mutantes mitóticos de Drosophila melanogaster no pólo local demonstram anormalidades no eixo celular
[Sunkel et al., J. Cell Sei., 1988, 89, 25] e o pólo foi verificado como codificando quinase mitótica [Llamazares et al., Genes Dev., 1991, 5, 2153]. Em seres humanos, existem três PLKS intimamente relacionados [Glover et al.,
Genes Dev., 1998, 12, 3777]. Eles contêm um domínio de quinase catalítica amino-terminal altamente homólogo e seus términos carboxila contêm duas ou três regiões conservadas, as caixas de pólo. A função das caixas de pólo permanece entendida de modo incompleto, porque elas estão implicadas na marcação de PLKs em compartimentos subcelulares [Lee et al., Proc. Natl. Acad. Sei. USA, 1998, 95, 9301; Leung et al., Nat. Struct. Biol., 2002, 9, 30 719], mediação de interações com outras proteínas [Kauselmann et al., EMBO J., 1999, 18, 5528], ou podem constituir parte de domínio autoregulatório [Nigg, Curr. Opin. Cell Biol., 1998, 10, 776]. Além disso, a atividade de PLK1
|
* · |
e · · λ · · |
99 9 |
• |
99 9
9 |
999
9 |
| |
• |
• 9 9 · |
9 |
♦ · 9 |
9 |
9 |
| |
|
|
|
|
|
9 |
| |
• |
« 9 99· |
9 |
9 |
9 |
• |
dependente da caixa de pólo é requerida para a transição apropriada de metafase/anafase e citocinese [Yuan et al., Câncer Res., 2002, 62, 4186; Seong et al., J. Biol. Chem., 2002, 277, 32282].
Estudos têm mostrado que PLKs humanos regulam alguns as5 pectos fundamentais de mitose [Lane et al., J. Cell. Biol., 1996, 135, 1701;
Cogswell et al., Cell Growth Differ., 2000, 11, 615]. Em particular, a atividade de PLK1 é acreditada como sendo necessária para a maturação funcional de centrossomas em profase prematura/tardia G2 e subseqüente estabeleci-
mento de um eixo celular bipolar. A depleção de PLK1 celular através da técnica de RNA de interferência pequena (sirna) também confirmou que esta proteína é requerida para processos mitóticos múltiplos e complementação de citocinese [Liu et al., Proc. Natl. Acad. Sei. USA, 2002, 99, 8672].
Em uma modalidade mais preferida da invenção, o composto da invenção é administrado em uma quantidade suficiente para inibir PLK1.
Dentre os três PLKs humanos, PLK1 é o melhor caracterizado;
ele regula vários efeitos do ciclo de divisão celular, incluindo o início de mitose [Toyoshima-Morimoto et al., Nature, 2001, 410, 215; Roshak et al., Cell.
Signalling, 2000, 12, 405], ativação do ponto de verificação do dano ao DNA [Smits et al., Nat. Cell Biol., 2000, 2, 672; van Vugt et al., J. Biol. Chem., 20 2001, 276, 41656], regulação do complexo promotor da anafase [Sumara et
al., Mol. Cell, 2002, 9, 515; Golan et al., J. Biol. Chem., 2002, 277, 15552;
Kotani et al., Mol. Cell, 1998, 1, 371], fosforilação da proteasoma [Feng et al., Cell Growth Differ., 2001, 12, 29], e duplicação e maturação de centrossoma [Dai et al., Oncogene, 2002, 21, 6195].
Especificamente, a iniciação de mitose requer a ativação de fator de promoção da fase-M (MPF), o complexo entre a quinase dependente de ciclina CDK1 e as ciclinas de tipo B [Nurse, Nature, 1990, 344, 503]. A última acumula durante as fases S e G2 do ciclo celular e promove a fosforilação inibidora de complexo MPF por WEE1, MIK1, e MYT1 quinases. No final da 30 fase G2, a desfosforilação correspondente pela fosfatase de duplaespecificidade, CDC25C, inicia a ativação de MPF [Nigg, Nat. Rev. Mol. Cell
Biol., 2001, 2, 21]. Na interfase, a ciclina B localiza para o citoplasma [Ha21
|
• · |
• · ··· · |
• · · |
• |
• · ·
• |
···
• |
| |
• e · · · |
• |
·· · |
• |
• |
| |
|
|
|
|
• |
| |
• · · ··· |
• |
• |
• |
• |
gting et al., EMBO J., 1998, 17, 4127], ela em seguida se torna fosforilada durante a profase e este evento causa translocação nuclear [Hagting et al.,
Curr. Biol., 1999, 9, 680; Yang et al., J. Biol. Chem., 2001, 276, 3604], O acúmulo nuclear de MPF ativo durante a profase é importante, como se pen5 sa, para iniciar os eventos da fase M [Takizawa et al., Curr. Opin. Cell Biol.,
2000, 12, 658]. No entanto, MPF nuclear é mantido inativo por WEE1, salvo se contra atuado por CDC25C. A própria fosfatase CDC25C, localizada no citoplasma durante a interfase, se acumula no núcleo na profase [Seki et al.,
Mol. Biol. Cell, 1992, 3, 1373; Heald et al., Cell, 1993, 74, 463; Dalal et al.,
Mol. Cell. Biol., 1999, 19, 4465]. A entrada nuclear tanto de ciclina B [Toyoshima-Morimoto et al., Nature, 2001, 410, 215], como de CDC25C [Toyoshima-Morimoto et al., EMBO Rep., 2002, 3, 341], é promovida através da fosforilação por PLK1 [Roshak et al., Cell. Signalling, 2000, 12, 405], Esta quinase é um regulador importante de iniciação da fase M.
. 15 Em uma modalidade particularmente preferida, os compostos da invenção são inibidores antagonísticos de ATP de PLK1.
No presente contexto, o antagonismo de ATP refere-se à capacidade de um composto inibidor de diminuir ou evitar a atividade catalítica de
PLK, isto é, fosfotransferir a partir de ATP para um substrato PLK macromo20 lecular, devido à ligação reversível ou irreversível no sítio ativo da enzima,
em tal modo como para afetar ou abolir a ligação de ATP.
Em outra modalidade preferida, o composto da invenção é administrado em uma quantidade suficiente para inibir PLK2 e/ou PLK3,
PLK2 de mamíferos (também conhecido como SNK) e PLK3 (também conhecido como PRK e FNK) foram originalmente mostrados como sendo produtos de gene prematuro imediato. A atividade de PLK3 quinase parece ter pico durante a fase G2 e S tardia. Isto também é ativado durante a ativação do ponto de verificação do dano ao DNA e severa tensão oxidativa. PLK3 também desempenha um papel importante na regulação de dinâ30 mica de microtúbulo e função de centrossoma na célula, e expressão de
PLK3 desregulada resulta em parada do ciclo celular e apoptose [Wang et al., Mol. Cell. Biol., 2002, 22, 3450]. PLK2 é o homólogo menos bem enten22 d ido dos três PLKs. Tanto PLK2 como PLK3 podem ter funções pósmitóticas adicionais importantes [Kauselmann et al., EMBO J., 1999, 18, 5528].
Outro aspecto da invenção refere-se ao uso de um composto de 5 fórmula 1 para inibir uma proteína quinase.
Em uma modalidade preferida deste aspecto, a proteína quinase é uma quinase dependente de ciclina. Preferivelmente, a proteína quinase é CDK1, CDK2, CDK3, CDK4, CDK6, CDK7, CDK8 ou CDK9, mais preferivelmente CDK2.
Outro aspecto da invenção refere-se a um método para inibir uma proteína quinase, referido método compreendendo contatar a referida proteína quinase com um composto de fórmula 1.
Em uma modalidade preferida deste aspecto, a proteína quinase é uma quinase dependente de ciclina, ainda mais preferivelmente CDK2.
TESTES
Outro aspecto da invenção refere-se ao uso de um composto, como definido acima, em um teste para identificar outros compostos candidatos que influenciam a atividade de uma ou mais enzimas CDK.
Preferivelmente, o teste é capaz de identificar compostos candi20 datos que são capazes de inibir uma ou mais enzimas CDK.
Mais preferivelmente, o teste é um teste de ligação competitivo.
Preferivelmente, o composto candidato é gerado por modificação de SAR convencional de um composto da invenção.
Como usado aqui, o termo modificação de SAR convencional refere-se a métodos padrão conhecidos na técnica para variar um dado composto por meio de derivatização química.
Assim, em um aspecto, o composto identificado pode atuar como um modelo (por exemplo, um padrão) para o desenvolvimento de outros compostos. Os compostos empregados em tal teste podem estar livres em solução, fixados a um suporte sólido, nascidos em uma superfície celular, ou localizados intracelularmente. A abolição de atividade ou a formação de complexos de ligação entre o composto e o agente sendo testado podem
ser medidas.
O teste da presente invenção pode ser uma triagem, através do qual vários agentes são testados. Em um aspecto, o método de teste da presente invenção é uma triagem de alta produção.
Esta invenção também contempla o uso de testes de triagem de drogas competitivas, nos quais os anticorpos neutralizantes capazes de ligar a um composto especificamente competem com um composto de teste para ligar a um composto.
Outra técnica para triar provê uma triagem de alta produção
(HTS) de agentes tendo uma afinidade de ligação apropriada para as substâncias e é baseado no método descrito em detalhes em WO 84/03564.
Espera-se que os métodos de teste da presente invenção sejam apropriados para tanto triagem em pequena como larga escala dos compostos de teste assim como em testes quantitativos.
- 15 Preferivelmente, o teste de ligação competitiva compreende contatar um composto de fórmula 1 com uma enzima CDK, na presença de um substrato conhecido da referida enzima CDK, e detectar qualquer mudança na interação entre a referida enzima CDK e o referido substrato conhecido.
Um sexto aspecto da invenção provê um método para detectar a ligação de um ligando a uma enzima CDK, o referido método compreenden-
do as etapas de:
(i) contatar um ligando com uma enzima CDK, na presença de um substrato conhecido de referida enzima CDK;
(ii) detectar qualquer mudança na interação entre a referida en- zima CDK e o referido substrato conhecido, e em que referido ligando é um composto de fórmula 1.
Um aspecto da invenção refere-se a um método compreendendo as etapas de:
(a) realizar um método de teste como descrito acima, (b) identificar um ou mais ligandos capazes de ligar a um domínio de ligação de ligando, e (c) preparar uma quantidade dos referidos um ou mais ligandos.
• · · · · ·
Outro aspecto da invenção provê um processo compreendendo as etapas de:
(a) realizar um método de teste como descrito acima, (b) identificar um ou mais ligandos capazes de ligar a um domí- nio de ligação de ligando, e (c) preparar uma composição farmacêutica compreendendo os referidos um ou mais ligandos.
Outro aspecto da invenção provê um processo compreendendo as etapas de:
(a) realizar um método de teste como descrito acima, (b) identificar um ou mais ligandos capazes de ligar a um domí- nio de ligação de ligando, e (c) modificar os referidos um ou mais ligandos capazes de ligar a um domínio de ligação de ligando;
(d) realizar o método de teste descrito acima, (e) opcionalmente preparar uma composição farmacêutica compreendendo os referidos um ou mais ligandos.
A invenção também refere-se a um ligando identificado pelo método descrito acima.
Ainda outro aspecto da invenção refere-se a uma composição farmacêutica compreendendo um ligando identificado pelo método descrito acima.
Outro aspecto da invenção refere-se ao uso de um ligando, identificado pelo método como descrito acima na preparação de uma com25 posição farmacêutica para uso no tratamento de distúrbios proliferativos.
Os métodos acima podem ser usados para triagem de um ligando utilizável como um inibidor de uma ou mais enzimas CDK.
PROCESSO
Um outro aspecto da invenção refere-se a um processo para a preparação de um composto de fórmula I como definido acima, referido processo compreendendo reagir um composto de fórmula V com um composto de fórmula VI
• · · · · ·
em que R1 * * *'9são como definidos na reivindicação 1 e X é Cl ou F.
Preferivelmente, o referido composto de fórmula V é preparado pelas seguintes etapas:
(i) reagir um composto de fórmula II com um composto de fórmula III para formar um composto de fórmula IV;
(ii) alquilar o referido composto de fórmula IV com um haloge- neto de alquila, R5 *-X’, para formar um composto de fórmula V.
Preferivelmente, o referido composto de fórmula VI é preparado pelas seguintes etapas:
R1 R2
XII
XIII
VI (i) oxidar um composto de fórmula VIII, em que PG é um grupo de proteção, para formar um composto de fórmula IX;
(ii) alquilar o referido composto de fórmula IX para formar um composto de fórmula X;
(iii) remover o grupo de proteção PG do referido composto de fórmula X para formar um composto de fórmula IX, que é equivalente à formula VI (onde um dentre R3 ou R4 é H).
Alternativamente, o referido composto de fórmula VI é preparado pelas seguintes etapas:
(i) oxidar um composto de fórmula VIII, em que PG é um grupo de proteção, para formar um composto de fórmula IX;
(ii) alquilar o referido composto de fórmula IX para formar um composto de fórmula X;
(iii) oxidar o referido composto de fórmula X para formar um 15 composto de fórmula XI;
(iv) alquilar o referido composto de fórmula XI para formar um composto de fórmula XII;
(v) remover o grupo de proteção PG do referido composto de fórmula XIII para formar um composto de fórmula VI.
Mais preferivelmente, a oxidação nas etapas (i) e (iii) dos pro27
cessos acima são obtidas por meio de oxidação de Swern.
Preferivelmente, a reação de alquilação das etapas (ii) e (iv) dos processos acima são obtidas por tratamento de um composto com um reagente de alquillítio, na presença de um catalisador complexo de brometo de 5 cobre/sulfeto de dimetila.
Em uma modalidade preferida alternativa, R3 = R4 e o referido composto de fórmula VI é preparado por um processo que compreende as etapas de:
XVI
XVII
(i) converter um composto de fórmula XVI, onde PG’ é um grupo 10 de proteção e R é um grupo alquila, em um composto de fórmula XVII;
(ii) remover os grupos de proteção PG’ do referido composto de fórmula XVII para formar um composto de fórmula VI.
Preferivelmente, o referido composto de fórmula XVI é converti do em um composto de fórmula XVII via uma reação de Grignard dupla.
Mais preferivelmente, em relação a esta modalidade, o referido composto de fórmula VI é preparado pelas seguintes etapas:
R1 R2
HO
nh2 o
XIV
o
XV
R1 R2
MeO nh2.hci
R1 R2
MeO ,PG'
N
O PG'
R1 R2
HO. ,PG' R3 \4 PG’
XVIa
XVII (i) reagir um composto de fórmula XIV com SOCI2 e MeOH para formar um composto de fórmula XV;
VI (ii) proteger o grupo amino do referido composto de fórmula XV para formar um composto de fórmula XVIa;
(iii) reagir o referido composto de fórmula XVIa com um reagente de Grignard R3X, onde R3 é como definido na reivindicação 1 e X é um halogeneto, para formar um composto de fórmula XVII;
(iv) remover os grupos de proteção PG’ do referido composto de fórmula XVII para formar o referido composto de fórmula VI.
Os grupos de proteção apropriados PG e PG’ serão familiares para os versados na técnica relevante. A título de exemplo, preferivelmente
o grupo de proteção PG’ é um grupo benzila, Bn e grupo de proteção PG é um grupo tritila.
Outros detalhes da preparação de compostos da presente invenção são descritos nos exemplos anexos sob o cabeçalho Síntese.
A presente invenção é ainda descrita a título dos seguintes exemplos.
EXEMPLOS
Em contraste com roscovitina, os compostos da presente invenção contém substituintes C-2 de purina modificados. Particularmente, os compostos da invenção contém substituintes C-2 tendo um grupo álcool se20 cundário ou terciário, em vez de um grupo álcool primário. Sem desejar se
limitar por teoria, acredita-se que a presença destes substituintes C-2 modificados leva a uma redução na conversão metabólica álcool- carboxila.
A fim de se desviar da redução na solubilidade aquosa esperada como um resultado da incorporação de substituintes alquila adicionais no substituinte C-2, o grupo benzilamina C-6 da roscovitina foi substituído com um grupo (piridin-3-il)-metilamino. Os exemplos anexos demonstram que esta modificação é tolerada em termos de atividade biológica (CDK2/ciclina
E ou A, inibição de CDK1/ciclina B e efeito antiproliferativo em linhagem de células de tumor humano).
Assim, a presente invenção demonstra que a modificação de substituintes C-2 e C-6 da purina de roscovitina proporciona novos compostos com melhorada utilidade terapêutica. De fato, foi demonstrado que a co29
|
• · • · |
• · • · · |
• |
• |
··»
• |
• • |
• · ·
• |
| |
• |
• · |
• |
• |
• |
·· · |
• |
| |
• |
• |
* |
• · · |
• |
• |
• |
··· locação de um ou dois substituintes de alquila inferior no carbinol C do substituinte C-2 da purina presente em roscovitina é não apenas tolerado em termos de reter a desejada atividade biológica (potência e seletividade de inibição de proteína quinase; citotoxicidade), mas em alguns casos provê compostos mais potentes. Além disso, a inclusão de um grupo (piridin-3-il)metilamino, em vez de grupo benzilamino, assegura melhorada hidrofilicidade e perfis de solubilidade aquosa para os compostos desta invenção, comparados com roscovitina (coeficientes de divisão de n-octanol/água calculados: : 2,5 < ClogP < 3,8 comparado a ClogP = 3,7 para roscovitina). Além
disso, os compostos selecionados exemplificados aqui foram mostrados como possuindo melhorada resistência a degradação metabólica, usando um sistema de modelo in vitro”apropriado.
Síntese
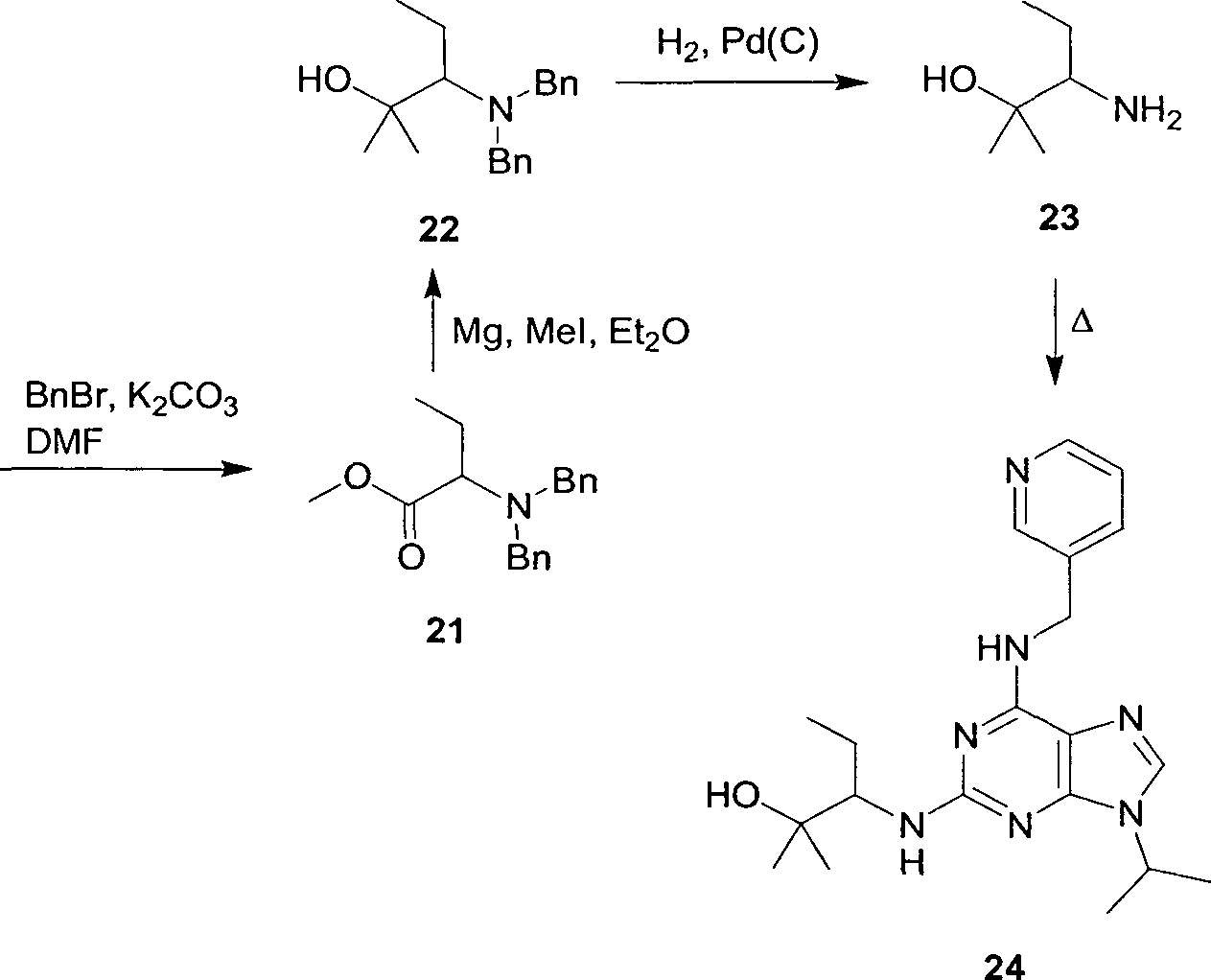
Os compostos da estrutura geral 1 podem ser preparados por métodos conhecidos na técnica (revisto em Fischer, P. M., e Lane, D. P.
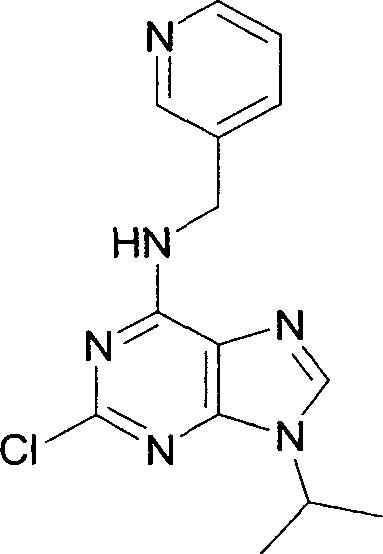
Curr. Med. Chem. 2001, 7, 1213-1245). Uma via sintética conveniente é mostrada abaixo no Esquema 1 e começa com 2,6-dicloropurina (2, X = Cl), ou 2-amino-6-cloropurina (2, X = NH2), comercialmente disponíveis. No último caso, o grupo amino é transformado para prover o material de partida particularmente apropriado 6 -cloro-2-flúor-purina (2, X = F; Gray, N. S.,
Kwon, S., e Schultz, P. G. Tetrahedron Lett. 1997, 38, 1161-1164.). A aminação seletiva na posição C-6 mais reativa com a piridilmetilamina apropriada 3, então, proporciona o intermediário 4. Este é alquilado na posição N-9, por exemplo, por substituição nucleófila usando o halogeneto de alquila 25 apropriado R5-X. O produto 5 é finalmente aminado com uma hidroxietilamina 6, em temperatura elevada.
Esquema 1
Os amino álcoois substituídos 6 (R1 ou R2 <> H) podem ser sintetizados a partir de α-amino álcoois 7 (R1 ou R2 <> H), como mostrado abaixo no Esquema 2. Muitos dos últimos são comercialmente disponíveis; alternativamente, eles podem ser preparados prontamente por redução de a- aminoácidos correspondentes. A reação inicial na metodologia sintética adotada foi a proteção tritila da função amino para proporcionar o intermediário 8 (R1 ou R2 <> H; Evans, P. A., Holmes, A. B., e Russell, K. J. Chem. Soc., Perkin Trans. 1, 1994, 3397-3409). Este foi submetido à oxidação de Swern para o aldeído correspondente 9 (R1 ou R2 <> H; Takayama, H., Ichikawa, T., Kuwajima, T., Kitajima, M., Seki, H., Aimi, N., e Nonato, M. G. J. Am. Chem. Soc. 2000, 122, 8635-8639). A introdução do substituinte R3 (se R2 <> H), ou R4 (se R1 <> H), foi obtida via alquilação controlada por quelação (Reetz, Μ. T., Roelfing, K., e Griebenow, N. Tetrahedron Lett. 1994, 35, 1969-1972) usando o reagente de alquillítio apropriado e um catalisador complexo de brometo de cobre/sulfeto de dimetila em éter dietílico. Dependendo do substituinte a ser introduzido, este procedimento deu intermediários 10 em excesso diastereomérico (de) de 50 - 80%. Alternativamente, métodos aquirais podem ser usados, opcionalmente seguidos por separação/re15 • · · · · ·

solução dos isômeros óticos. Para a produção de amino-álcoois, onde ambos R3 e R4 são diferentes de Η, o intermediário 10 foi submetido a outra reação de oxidaçâo de Swern para a cetona respectiva 12, seguido por introdução do segundo substituinte através de alquilação. A etapa final na 5 síntese para todos os amino-álcoois foi a remoção do grupo tritila, usando ácido trifluoroacético para proporcionar 6 ou 11.
R1 R2
R1 R2 RJ>r2
HO^Xn_C(C6H5)3 ---O<yXNX(C6H5)3
H ’H H
Esquema 2
Nestes casos onde amino-álcoois contém dois substituintes
idênticos no carbinol C (6, R1 ou R2 <> H; R3 = R4, não H), estes podem ser obtidos diretamente do éster de α-aminoácido correspondente, por exemplo, por dupla alquilação de Grignard (Guenther, B. R., e Kirmse, W. Liebigs Ann. Chem. 1980, 518-532).
Testes de quinase
Os compostos dos exemplos abaixo foram investigados para sua atividade inibidora de CDK2/ciclina E, CDK1/ciclina B, CDK4/ciclina D1 e
CDK7/ciclina H, ERK-2, e PKA. As quinases dependentes de ciclina, humanas, recombinantes, unidas com His6 CDK1/ciclina B1, CDK2/ciclina E,
CDK4 e CDK7/ciclina H, foram expressadas em células sf9 usando um sistema de expressão de baculovírus. A ciclina D1 recombinante foi expressa20 das em E. coli. Proteínas foram purificadas por cromatografia de afinidade
Mi por quelato metálico, em uma homogeneidade maior do que 90%. Os testes de quinase foram realizados em placas de 96 cavidades, usando CDK/ciclinas recombinantes, ERK-2 ativo recombinante (Upstate Biotechnology), ou subunidade catalítica de quinase dependente de AMP(PKA) 5 (Calbiochem Cat. 539487). Testes foram realizados em tampão de teste (βglicerofosfato 25 mM, MOPS 20 mM, EGTA 5 mM, DTT 1 mM, Na3VO3 1 mM, pH 7,4), em que foram adicionados 2-4 μg de enzima ativa com substratos apropriados (histona purificada H1 para CDK2, proteína GSTretinoblastoma recombinante (resíduos 773-928) para CDK4, peptídeo bioti-
nil-Ahx-(Tyr-Ser-Pro-Thr-Ser-Pro-Ser)4 para CDK7, proteína básica mielina para ERK-2, ou peptídeo Kemptide (Fluka Biochemika Cat. 60645) para
PKA). A reação foi iniciada por adição de mistura Mg/ATP (MgCI2 15 mM + 100 μΜ ATP com 30-50 kBq por reservatório de (γ-32Ρ]-ΑΤΡ) e as misturas incubadas durante 10 min (CDK2/ciclina E, ERK-2, PKA), ou 45 min (CDK4/ciclina D1, CDK7/ciclina H) como requerido, a 30°C. As reações foram interrompidas por gelo, seguido por filtração através de placas de filtro p81 ou placas de filtro GF/C (para CDK4) (Whatman Polyfiltronics, Kent, UK), exceto para cDK7 onde, após interromper a reação por gelo, 10 μΙ_ de mg/mL de avidina foram adicionados a cada reservatório e adicional mente incubados durante 10 min, seguido por filtração como para o teste

CDK2. Após lavar 3 vezes com ácido ortofosfórico aquoso 75 mM, as placas foram secadas, cintilante adicionado e a radioatividade incorporada medida em um contador de cintilação (TopCount, Packard Instruments, Pangbourne, Berks, UK). Os compostos para o teste de quinase foram feitos como solu25 ções estoque A 10 mM em DMSO e diluídos em 10% de DMSO em tampão de teste. Os dados foram analisados usando software de ajuste de curva (GraphPad Prism versão 3,00 para Windows, GraphPad Software, San Diego Califórnia USA), para determinar os valores de IC50 (concentração de composto de teste que inibe A atividade de quinase A 50%.). Estes valores para os compostos da presente invenção são mostrados na Tabela 1.
Teste de citotoxicidade MTT
Os compostos dos exemplos abaixo foram submetidos a um teste de proliferação celular padrão usando as seguintes linhagens de células de tumor humano: A549, HeLa, HT-29, MCF7, Saos-2, CCRF-CEM, HL• · · · · ·
60, e K-562, As linhagens de células foram obtidas a partir da ATCC (American Type Culture Collection, 10801 University Boulevard, Manessas, VA 5 20110-2209, USA). Os testes de 72-h MTT padrão (azul tiazolila, brometo de
3-[4,5-dimetiltiazol-2-il]-2,5-difeniltetrazólio) foram realizados (Haselsberger,
K.; Peterson, D. C.; Thomas, D. G.; Darling, J. L. Anti Câncer Drugs 1996, 7, 331-8; Loveland, B. E.; Johns, T. G.; Mackay, I. R.; Vaillant, F.; Wang, Z. X.;
Hertzog, P. J. Biochemistry International 1992, 27, 501-10). Em resumo: as
células foram semeadas em placas de 96 cavidades de acordo com o tempo de duplicação e incubadas durante a noite a 37°C. Os compostos de teste foram feitos em DMSO e uma série de diluição a 1/3 preparada em meio de células de 100 μΙ_, adicionados a células (em triplicatas) e incubados durante 72 horas, a 37°C. Um MTT foi feito como uma solução estoque de 5 mg/mL em meio de célula e esterilizado em filtro. Os meios foram removidos das células, seguido por uma lavagem com 200 μΙ_ de PBS. A solução de MTT foi então adicionada a 20 μΙ_ por cavidade e incubada no escuro, a 37°C durante 4 h. A solução de MTT foi removida e as células novamente lavadas com 200 μΙ_ de PBS. Um corante MTT foi solubilizado com 200 μΙ_, por cavi20 dade, de DMSO, com agitação. A absorvância foi lida a 540 nm e os dados
analisados usando software de ajuste de curva (GraphPad Prism versão
3,00 para Windows, GraphPad Software, San Diego Califórnia USA), para determinar os valores de IC50 (concentração de composto de teste que inibe a atividade de quinase a 50%.). Estes valores para os compostos da pre25 sente invenção são mostrados na Tabela 2.
Citotoxicidade seletiva
Os compostos representativos desta invenção foram examinados para seu efeito antiproliferativo contra linhagens de células de tumor humano e linhagens de células humanas não-transformadas imortalizadas (linhagens de células normais). Os resultados são resumidos na tabela 3.
Pode-se notar que os compostos examinados são agentes antiproliferativos várias vezes mais potentes, comparados com roscovitina. Além disso, seu
efeito citotóxico é significativamente mais pronunciado contra linhagens de células transformadas versus não-transformadas.
Teste comparativo de metabolismo in vitro”
Incubações microssõmicas e preparação de amostras para análise
Os microssomas foram obtidos de Totem Biologicals, Northampton, Inglaterra. A proteína microssômica (0,2 mg) e roscovitina ou composto de teste desta invenção (concentração final 10 μΜ) foram misturados em solução salina tamponada com fosfato (100 μΙ_), contendo NADPH (20 mM), MgCI2 (10 mM), e EDTA (1,5 mM). As amostras foram incubadas
durante 30 min e a reação interrompida pela adição de metanol resfriado com gelo (300 μΙ_), contendo olomoucina (Vesely, J., Havlicek, L., Strnad,
M., Blow, J.J., Donella-Deana, A., Pinna, L., Letham, D.S., Kato, J., Detivaud, L., Leclerc, S., Meijer, L. Eur. J. Biochem. 1994, 224, 771-786) como um padrão interno. As curvas de calibração foram preparadas a 0, 1 e 10 μΜ . 15 em microssomas pré-incubados durante 30 min, e estes foram também tratados com metanol contendo olomoucina. Todas as amostras foram então centrifugadas e os sobrenadantes analisados por espectrometria de massa cromatografia líquida.
Cromatografia líquida - Espectrometria de massa
A coluna de cromatografia foi uma coluna zwiteriônica Supelco
LC-ABZ, 50 X 4,6 mm, 5 μιτι (Supelco Inc., Supelco Park, Bellefonte, PA, USA). Os eluentes gradientes consistiam em metanol (A) e 0,1% de ácido fórmico em água (B). O gradiente começou com 10:90 (A:B v/v), que foi mantido isocraticamente durante 0,5 min, seguido por um aumento linear a
90:10 (A:B v/v) durante 6 min, que foi então mantido nestas condições durante mais 4 min. A taxa de fluxo foi de 1 mL/min por todo o tempo. Para LCUV-EM, as amostras foram introduzidas usando um auto-amostrador Gilson
215 (Anachem Ltd., Bedfordshire, UK), fixado a uma bomba quaternária
Thermoseparations P4000, coluna (como descrita acima), e conjunto detec30 tor Thermoseparations UV1000 a 254 nm (Thermoquest Ltd., Hemel
Hempstead, Hertfordshire, UK). O eluente do detector passou, sem separação, em um espectrômetro de massa com coletor de íons Thermoquest
LCQ, equipado com uma fonte de eletropulverização operada em modo positivo. As condições do espectrômetro de massa foram gás de preenchimento 80, gás auxiliar 20 (ambas unidades arbitrárias), voltagem capilar 4 a
4,5 kV e temperatura capilar aquecida 250 a 280°C. A faixa de massa foi de 5 50-750. O tempo de varredura foi controlado por coletor de íons que foi fixa- do para um tempo de injeção de íons máximo de 200 EM, ou o tempo requerido para injetar 2 χ 108 íons; para cada varredura, o sistema automaticamente usado, em qualquer tempo, foi alcançado primeiro.
ANÁLISE DE DADOS
Para analisar os resultados, traços de íons selecionados de íons
MH+ do composto de teste e do padrão interno foram extraídos e a área dos picos relevantes obtida. A razão da área de pico (composto de teste/padrão interno) da incubação de teste foi então comparada com as relações da área de pico obtidas da curva de calibração do composto de teste. A partir destes . 15 valores, a concentração de composto de teste remanescente após 30 min de incubação de proteína microssômica foi determinada. Os resultados para compostos representativos da presente invenção são resumidos na tabela 3, onde a estabilidade metabólica do composto é também comparada com a de roscovitina em termos de metabolismo (coluna A), inibição de CDK2 in vitro (coluna B) e citotoxicidade in vitro em linhagens de células de tumor (colu-

na C). A eficácia in vitro comparativa (coluna A x C) e a exposição celular (coluna A x C) são também mostradas. Estes resultados sugerem que os compostos da presente invenção terão uma melhorada eficácia in vivo, comparada com roscovitina. Os coeficientes de divisão n-octanol/água cal25 culados (ClogP) são também incluídos na tabela 3. Pode-se notar que aqueles compostos com melhorada atividade celular e estabilidade metabólica também possuem menor ClogP do que roscovitina, sugerindo melhorada solubilidade aquosa e, assim, facilidade de formulação para administração de drogas in vivo.
• · · · · ·
Ácido (2R)-2-(6-Benzilamino-9-isopropil-9H-purin-2-ilamino)-butírico
Benzil-(2-flúor-9-isopropil-9/7-purin-6-il)-amina (151 mg, 0,5 mmol) foi dissolvida em NMP (5 mL) e DBU (1,5 mL, 10 mmoles). Ácido (R)~ (-)-2-Aminobutírico (99% ee/GLC; 1,03 g, 10 mmoles) foi então adicionado e 5 a mistura foi agitada sob N2> a 160°C, durante 1 h. Após resfriar, a mistura foi diluída com ácido cítrico (10% de solução aquosa) e CH2CI2 (25 mL cada). As fases foram separadas e a fração orgânica foi extraída com salmoura (2 χ 10 mL), secada sobre MgSO4, filtrada, e evaporada. O resíduo foi redissolvido em MeCN e foi fracionado por RP-HPLC preparativa (Vydac 218TP1022,
9 mL/min, 22,5 - 32,5% MeCN em H2O, contendo 0,1% de CF3COOH, durante 40 min). Frações apropriadas foram reunidas e liofilizadas para dar o composto do título puro (137 mg, 74,4%), como um sólido de branco indefinido amorfo. Anal. RP-HPLC (Vydac 218TP54, 1 mL/min): tR = 16,04 min (0
- 60% de MeCN), 15,95 min (22,5 - 32,5% de MeCN em H2O, contendo 15 0,1% de CF3COOH, acima de 20 min), pureza: > 98% (λ = 214 nm). 1H-RMN (de-DMSO, 300 MHz) <5: 0,95 (t, J = 7,3 Hz, 3H, CH2CH3); 1,51 (d, J = 6,7 Hz,
6H, CH(CH3)2); 1,78 (m, J = 7,3 Hz, 2H, CH2CH3); 4,27 (m, 1H, CHCH2);
4,64 (hept., J = 6,7 Hz, 1H, CH(CH3)2); 4,69 (m, 2H, CH2Ph); 7,25 - 7,41 (m,
6H, ArH). DE-MALDI-TOF EM (matriz de ácido α -ciano-4-hidroxicinâmico): 20 [M + H]+ = 369,41, FAB-EM: [M + H]+ = 369,2033 (Ci9H25N6O2 requer
369,2039).
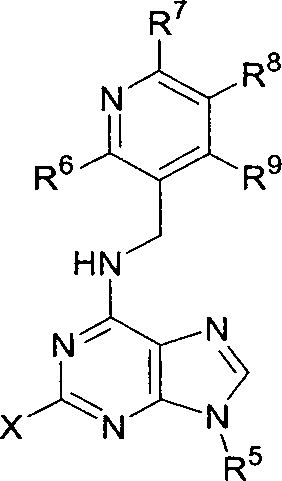
(R)-2-(Tritil-amino)-butan-1-o/
A uma solução agitada de (R)-(-)-2-aminobutan-1-ol (10 g, 1 eq,
112,18 mmoles) em DCM (500 mL), sob uma atmosfera de argônio, em temperatura ambiente, foi adicionado DIEA (30 mL, 1,54 eq, 172,22 mmoles)
seguido por cloreto de tritila (35,4 mL, 1,13 eq, 126,98 mmoles). A mistura de reação foi agitada em temperatura ambiente durante 48 h, quando TLC (hexano :éter:MeOH; 55:40:5) indicou que a reação tinha ocorrido até completar. O solvente foi evaporado em vácuo e o resíduo precipitou a partir de acetona (50 mL) com hexano (900 mL), com agitação, o precipitado foi re« 10 movido por filtração e o filtrado foi evaporado em vácuo. O resíduo foi dissolvido em hexano (1 L), filtrado, e o filtrado foi evaporado em vácuo para dar o composto do título como um óleo amarelo-claro. Rendimento: 32 g (86%). 1H-RMN (de-DMSO, 250 MHz): δ 0,56 (t, 3H, J = 7,41 Hz, NHCH(CH?CH3)CH?OH). 1,10 (m, 2H, -NHCH(ÇH2CH3)CH2OH), 2,22 (m,
1H, -NHCH(CH2CH3)CH2OH), 2,38 (m, 1H, -NHÇH(CH2CH3)CH2OH), 2,72 +
3,00 (2 x m, 2H, -NHCH(CH2CH3)ÇH2OH), 4,28 (t, 1H, J = 5,26 Hz, NHCH(CH2CH3) CH2OH), 7,14-7,49 (m, 15H, 3 x Ph).
(S)-2-(Tritil-amino)-butan-1-ol
A uma solução agitada de (S)-(+)-2-aminobutan-1-ol (10 g, 1 eq,
112,18 mmoles) em DCM (500 mL), sob uma atmosfera de argônio, em temperatura ambiente, foi adicionado DIEA (30 mL, 1,54 eq, 172,22 mmoles) seguido por cloreto de tritila (35,4 mL, 1,13 eq, 126,98 mmoles). A mistura φ φ φ φ
φ φ
φ φ φ φ φ φ φ φ · φφ φ • Φ φ • φ φ φ de reação foi agitada nesta temperatura durante 48 h, quando TLC (hexano :éter:MeOH; 55:40:5) indicou que a reação tinha ocorrido até completar. O solvente foi evaporado em vácuo e o resíduo precipitou a partir de acetona (50 mL) com hexano (900 mL) com agitação, o precipitado foi removido por 5 filtração e o filtrado foi evaporado em vácuo. O resíduo foi dissolvido em hexano (1 L), filtrado, e o filtrado foi evaporado em vácuo para dar o composto do título como um óleo amarelo-claro. Rendimento: 33 g (89%). 1H-RMN (d6DMSO, 250 MHz): 5 0,58 (t, 3H, J = 7,26 Hz, -NHCH(CH2ÇH3)CH2OH), 1,10 (m, 2H,-NHCH(ÇH2CH3)CH2OH), 2,24 (m, 1H, -NHCH(CH2CH3)CH2OH), 10 2,39 (m, 1H, -NHÇH(CH2CH3)CH2OH), 2,76 & 3,03 (2 x m, 2H, NHCH(CH2CH3)ÇH2OH), 4,32 (t, 1H, J = 4,97 Hz, -NHCH (CH2CH3) CH2OH), 7,15-7,52 (m, 15H, 3 x Ph).
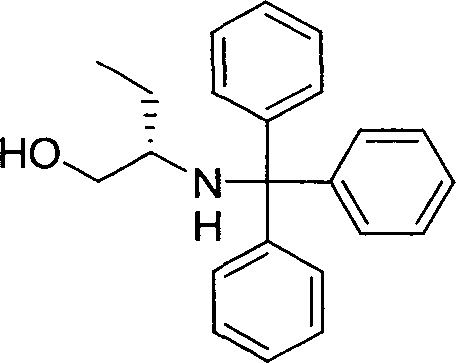
(R)-2-(Tritil-amino)-butiraldeído
A uma solução agitada de DMSO (3,0 mL, 2,8 eq, 42,28 mmo15 les) em DCM (30 mL), sob uma atmosfera de argônio a -45°C, foi adiciona-
do cloreto de oxalila (2 M em DCM, 10,56 mL, 1,40 eq, 21,12 mmoles), gota a gota. A mistura de reação foi agitada a -45°C, durante 1 h, após o que uma solução de (R)-2-(tritil-amino)-butan-1-ol (5 g, 1 eq, 15,08 mmoles) em
DCM (30 mL) foi adicionada, gota a gota, com agitação. A mistura de reação foi agitada nesta temperatura, durante 3 h, quando TLC (hexano :éter; 80:20) indicou que a reação tinha ocorrido até completar. À mistura de reação foi adicionada uma solução de TEA (10,5 mL, 5 eq, 75,33 mmoles) em DCM (30 mL), e a solução deixada aquecer para temperatura ambiente, durante 16 h.
A mistura de reação foi diluída com mais DCM (200 mL) e lavada com água (250 mL). A fase aquosa foi extraída com DCM (3 χ 50 mL), e a fase orgânica combinada lavada com salmoura (50 mL), secada (MgSO4) e evaporada em vácuo. O resíduo foi dissolvido em éter (30 mL), o precipitado sólido re··· ··· ··♦ movido porfiltração e o filtrado foi evaporado em vácuo. O resíduo foi dissolvido em hexano (50 mL), o precipitado sólido removido por filtração e o filtrado foi evaporado em vácuo, para dar o composto do título como um óleo amarelo-claro. Rendimento: 2,59 g (52%). 1H-RMN (d6-DMSO, 250 MHz): <5 5 0,77 (t, 3H, J = 7,42 Hz, -NHCH(CH2ÇH3)CHO), 1,34-1,61 (m, 2H, NHCH(ÇH2CH3)CHO), 2,92 (m, 1H, -NHÇH(CH2CH3)CHO), 3,62 (d, 1H, J = 8,21 Hz, -NHCH(CH2CH3)CHO), 7,16-7,46 (m, 15H, 3 χ Ph), 8,77 (d, 1H, J = 3,00 Hz, -NHCH(CH2CH3)ÇHO).
(S)-2-(Tritil-amino)-butiraldeído
«10 A uma solução agitada de DMSO (2,4 mL, 2,8 eq, 33,82 mmoles) em DCM (30 mL) sob uma atmosfera de argônio a -45°C, foi adicionado cloreto de oxalila (2 M em DCM, 8,45 mL, 1,40 eq, 16,9 mmoles), gota a gota. A mistura de reação foi agitada a -45°C, durante 1 h, após o que uma solução de (S)-2-(tritil-amino)-butan-1-ol (4 g, 1 eq, 12,07 mmoles) em DCM (30 mL) foi adicionada, gota a gota, com agitação. A mistura de reação foi
agitada nesta temperatura durante 3 h, quando TLC (hexano :éter; 80:20) indicou que a reação tinha ocorrido até completar. À mistura de reação foi adicionada uma solução de TEA (8,4 mL, 5 eq, 60,27 mmoles) em DCM (30 mL), e a solução deixada aquecer, em temperatura ambiente, durante 16 h.
A mistura de reação foi diluída com mais DCM (100 mL) e lavada com água (250 mL). A fase aquosa foi extraída com DCM (3 χ 50 mL), e a fase orgânica combinada lavada com salmoura (50 mL), secada (MgSO4) e evaporada em vácuo. O resíduo foi dissolvido em éter (30 mL), o precipitado sólido removido por filtração e o filtrado foi evaporado em vácuo. O resíduo foi dissol vido em hexano (50 mL), o precipitado sólido removido por filtração e o filtrado foi evaporado em vácuo para dar o composto do título como um óleo amarelo-claro. Rendimento: 3,64 g (91%). 1H-RMN (d6-DMSO, 250 MHz): δ
tf
0,77 (t, 3H, J = 7,42 Hz, -NHCH(CH2ÇH3)CHO), 1,37-1,59 (m, 2H, NHCH(ÇH2CH3)CHO), 2,93 (m, 1H, -NHÇH(CH2CH3)CHO), 3,62 (d, 1H, J = 5,84 Hz, -NHCH(CH2CH3)CHO), 7,16-7,46 (m, 15H, 3 χ Ph), 8,77 (d, 1H, J = 3,00 Hz, -NHCH(CH2CH3)ÇHO).
(2S,3R)-3-(Tritil-amino)-pentan-2-ol

A uma suspensão agitada de CuBr.SMe2 (2,74 g, 2,2 eq, 13,33 mmoles) em Et2O (100 mL), sob uma atmosfera de argônio, a -70°C, foi adicionado metil-lítio (1,6 M em Et2O, 16,6 mL, 4,4 eq, 26,56 mmoles), gota a gota, e a solução deixada aquecer para a temperatura ambiente. A mistura foi reesfriada a -70°C, à qual foi adicionada uma solução de (R)-2-(tritilamino)-butiraldeído (2 g, 1 eq, 6,05 mmoles) em Et2O (25 mL), gota a gota, com agitação. A mistura de reação foi agitada nesta temperatura durante 2 h, quando TLC (hexano :éter; 80:20) indicou que a reação tinha ocorrido até completar. À mistura de reação foi adicionada uma solução aquosa saturada de NH4CI (100 mL) e deixada aquecer para a temperatura ambiente durante 16 h. A mistura de reação foi extraída com éter (2 χ 200 mL), e a fase orgânica combinada lavada com salmoura (50 mL), secada (MgSO4) e evaporada em vácuo. O resíduo foi purificado por cromatografia em coluna de sílicagel, eluído com hexano :éter (80:20), para dar o composto do título como um óleo amarelo-claro. Rendimento: 1,91 g (91%). (80% de 2S,3R: 20% de 2R,3R). 1H-RMN (d6-DMSO, 250 MHz): δ 0,47 & 0,55 (2 x t, J = 7,43 & 7,27 Hz, NHCH(CH2ÇH3)CH(CH3)OH), 0,99-1,12 (m, 5H, -NHCH(CH2CH3)CH(CH3)OH). 2,03 (m, 1H, -NHÇH(CH2CH3)CH(CH3)OH), 3,32-3,51 (m, 1H, -NHCH(CH2CH3)
ÇH(CH3)OH), 4,40 (d, 1H, J = 3,79 Hz, -NHCH(CH2CH3)CH(CH3)OH), 7,14-7,51 (m, 25 15H, 3 χ Ph).
(2R,3S)-3-(Tritil-amino)-pentan-2-ol
A uma suspensão agitada de CuBr.SMe2 (2,74 g, 2,2 eq, 13,33 mmoles) em éter (100 mL), sob uma atmosfera de argônio, a -70 °C, foi adicionado metil lítio(1,6 M em éter, 15,13 mL, 4,0 eq, 24,21 mmoles) gota a
gota, e a solução deixada aquecer para a temperatura ambiente. A mistura foi reesfriada para -70°C, à qual foi adicionada uma solução de (S)-2-(tritilamino)-butiraldeído (2 g, 1 eq, 6,05 mmoles) em Et2O (25 mL), gota a gota com agitação. A mistura de reação foi agitada nesta temperatura durante 2 h, e então a -55°C durante 4 h, quando TLC (hexano :Et2O; 80:20) indicou 10 que a reação tinha ocorrido até completar. À mistura de reação foi adicionada uma solução aquosa saturada de NH4CI (100 mL) e deixada aquecer »
para a temperatura ambiente, durante 16 h. A mistura de reação foi extraída com Et2O (2 x 200 mL), e a fase orgânica combinada lavada com salmoura (50 mL), secada (MgSO4) e evaporada em vácuo. O resíduo foi purificado por cromatografia em coluna de sílica-gel, eluído com hexano :Et2O (80:20),
para dar o composto do título como um óleo amarelo-claro. Rendimento:
1,37 g (66%). (80% de 2R,3S: 20% de 2S,3S). 1H-RMN (d6-DMSO, 250
MHz): Õ 0, 0,47 & 0,55 (2 x t, J = 7,50 & 7,26 Hz -NHCH(CH2CH3)CH(CH^OH). 0,99-1,12 (m, 5H, -NHCH(CH2CH^CH(CH3)OH). 2,01 (m, 1H, 20 NHÇH(CH2CH3)CH(CH3)OH), 3,22-3,43 (m, 1H, -NHCH(CH2CH3) CH(CH3)OH). 4,41 (d, 1H, J = 3,31 Hz, -NHCH(CH2CH3)CH(CH3)OH), 7,14-7,56 (m, 15H, 3 x Ph). (3RS,4R)-4-(Tritil-amino)-hexan-3-ol
HO.
N H
• · · · · ·
A uma solução agitada de (R)-2-(tritil-amino)-butiraldeído (1,5 g, eq, 4,53 mmoles) em Et2O (150 mL), sob uma atmosfera de argônio a 78°C, foi adicionado brometo de etil-magnésio (3 M em Et2O, 1,51 mL, 1 eq, . 4,53 mmoles), gota a gota. A solução foi agitada a -78°C durante 2 h, então deixada aquecer para a temperatura ambiente durante 16 h. A mistura foi reesfriada para 0°C, H2O (150 mL) adicionada, e a fase orgânica separada.
A fase aquosa foi extraída com mais Et2O (2 χ 50 mL), e a fase orgânica combinada lavada com salmoura (50 mL), secada (MgSCL) e evaporada em vácuo. O resíduo foi purificado por cromatografia em coluna de sílica-gel, 10 eluído com hexano :éter (90:10), para dar o composto do título como um óleo amarelo-claro. Rendimento: 1,13 g (69%). (57% de 3S,4R: 43% de 3R,4R). 1H-RMN (d6-DMSO, 250 MHz): δ 0,45 & 0,69 (t & m, 6H, J = 7,43 Hz, -NHCH(CH2CH3)CH(CH2CH3)QH), 1,12-1,29 (m, 4H, -NHCH (ÇH2CH3)CH (ÇH2CH3)OH), 2,16 (m, 1H, -NHCH(CH2CH3)CH(CH2CH3)OH), . 15 2,54 (m, 1H, -NHÇH(CH2CH3)CH(CH2CH3)OH), 3,21-3,40 (m, 1H, NHCH(CH2CH3) ÇH(CH2 CH3)OH), 4,29+4,39 (2 χ d, 1H, J = 4,42 & 5,37 Hz, -NHCH(CH2CH3)CH (CH2CH3)OH), 7,15-7,52 (m, 15H, 3 χ Ph). (3RS,4S)-4-(Tritil-amino)-hexan-3-ol
A uma solução agitada de (R)-2-(tritil-amino)-butiraldeído (1,5 g,
1 eq, 4,53 mmoles) em Et2O (150 mL), sob uma atmosfera de argônio, a 78°C, foi adicionado brometo de etil-magnésio (3 M em Et2O, 1,51 mL, 1 eq, 4,53 mmoles), gota a gota. A solução foi agitada a -78°C durante 2h, então deixada aquecer para a temperatura ambiente durante 16 h. A mistura foi reesfriada para 0°C, H2O (150 mL) adicionada, e a fase orgânica separada.
A fase aquosa foi extraída com mais Et2O (2 χ 50 mL), e a fase orgânica combinada lavada com salmoura (50 mL), secada (MgSO4) e evaporada em vácuo. O resíduo foi purificado por cromatografia em coluna de sílica-gel,
eluído com hexano:éter (90:10), para dar o composto do título como um óleo amarelo-claro. Rendimento: 1,19 g (73%). (65% de 3R,4S: 35% de 3S,4S). 1H-RMN (de-DMSO, 250 MHz): δ 0,46+0,69 (t & m, 6H, J = 7,34 Hz, NHCH(CH?CH3)CH(CH?CH3)OH), 1,13-1,29 (m, 4H, -NHCHfCHgCHs)
CH(ÇH2_CH3)OH), 2,17 (m, 1H, -NHCH(CH2CH3)CH(CH2CH3)OH), 2,55 (m, 1H, -NHÇH___(CH2CH3)CH(CH2CH3)OH), 3,20-3,39 (m, 1H, NHCH(CH2CH3)ÇHCH2 CH3) OH), 4,29 & 4,39 (2 χ d, 1H, J = 4,74 & 5,53 Hz, -NHCH(CH2CH3)CH (CH2CH3)OH), 7,15-7,52 (m, 15H, 3 χ Ph). (3RS,4R)-2-Metil-4-(tritil-amino)-hexan-3-ol
» 10 A uma suspensão agitada de CuBr.SMe2 (1,37 g, 2,2 eq, 6,66 mmoles) em Et2O (100 mL), sob uma atmosfera de argônio, a -78°C, foi adicionado isopropil lítio (0,7 M em pentano, 17,29 mL, 4 eq, 12,1 mmoles), gota a gota, e a solução deixada aquecer para a temperatura ambiente. A mistura foi reesfriada para -70°C, à qual foi adicionada uma solução de (R)15 2-(tritil-amino)-butiraldeído (1 g, 1 eq, 3,03 mmoles) em Et2O (25 mL), gota a

gota, com agitação. A mistura de reação foi agitada nesta temperatura durante 1 h, então deixada aquecer para -55°C e agitada nesta temperatura durante 3 h. À mistura de reação foi adicionada uma solução aquosa saturada de NH4CI (100 mL) e deixada aquecer para a temperatura ambiente du20 rante 16 h. A mistura de reação foi extraída com Et2O (2 χ 200 mL), e a fase orgânica combinada lavada com salmoura (50 mL), secada (MgSO4) e eva porada em vácuo. O resíduo foi purificado por cromatografia em coluna de gradiente de sílica-gel, eluído com hexano :éter (100:0 -> 90:10), para dar o composto do título como um óleo incolor. Rendimento: 0,53 g (47%). (50% de 3S,4R: 50% de 3R,4R) 1H-RMN (d6-DMSO, 250 MHz): <5 0,44 (t, 3H, J =
7,03 Hz, -NHCH(CH?CH3)CH(CH(CH3),)OH). 0,52 & 0,77 (2 χ d, 6H, J = 6,48 Hz, NHCH (CH?CH3)CH(CH(CH3)2)OH), 0,79-1,13 (m, 2H, -NHCH(ÇH2CH3)CH
(CH(CH3)2)OH), 1,72 (m, 1H, -NHCH(CH2CH3)CH(CH (CH3)2)OH), 2,11 (m, 1H, NHÇH(CH2CH3)CH(CH(CH3)2)OH), 2,77 (m, 1H, -NH CH(CH2CH3)CH (ÇH(CH3)2)OH), 2,99 (m, 1H, -NHCH(CH2CH3)ÇH(CH(CH3)2) OH), 4,55 (d, 1H, J =
5,21 Hz, -NHCH(CH2CH3)CH(CH(CH3)2)OH), 7,15-7,46 (m, 15H, 3 x Ph).
(3RS,4S)-2-Metil-4-(tritil-amino)-hexan-3-ol


A uma suspensão agitada de CuBr.SMe2 (1,37 g, 2,2 eq, 6,66 mmoles) em Et2O (100 mL), sob uma atmosfera de argônio, a -78°C, foi adicionado isopropil-lítio (0,7 M em pentano, 17,29 mL, 4 eq, 12,1 mmoles), gota a gota e a solução deixada aquecer para a temperatura ambiente. A . 10 mistura foi reesfriada para -70°C, à qual foi adicionada uma solução de (S)2-(tritil-amino)-butiraldeído (1 g, 1 eq, 3,03 mmoles) em EtsO (25 mL), gota a gota, com agitação. A mistura de reação foi agitada nesta temperatura durante 1 h, então deixada aquecer para -55°C e agitada nesta temperatura durante 3 h. À mistura de reação foi adicionada uma solução aquosa satura15 da de NH4CI (100 mL) e deixada aquecer para a temperatura ambiente durante 16 h. A mistura de reação foi extraída com Et2O (2 χ 200 mL), e a fase orgânica combinada lavada com salmoura (50 mL), secada (MgSO4) e evaporada em vácuo. O resíduo foi purificado por cromatografia em coluna de sílica-gel, eluído com hexano :éter (100:0 -> 90:10), para dar o composto do 20 título como um óleo incolor; Rendimento: 0,36 g (32%). (50% de 3R,4S: 50% de 3S,4S). 1H-RMN (d6-DMSO, 250 MHz): <5 0,44 (t, 3H, J = 6,79 Hz, NHCH(CH2ÇH3)CH(CH(CH3)2)OH), 0,52 & 0,76 (2 χ d, 6H, J = 6,63 Hz, NHCH (CH2CH3)CH(CH(ÇH3)2)OH), 0,80-1,15 (m, 2H, -NHCH(CH2CH3)CH (ΟΗ(ΟΗ3)2)ΟΗ), 1,70 (m, 1H, -NHCH(CH2CH3)CH(CH (CH3)2)OH), 2,10 (m, 1H, 25 -ΝΗΟΗ(ΟΗ2ΟΗ3)ΟΗ(ΟΗ(ΟΗ3)2)ΟΗ), 2,76 (m, 1H, -NHCH(CH2CH3)CH (ÇH(CH3)2)OH), 2,99 (m, 1H, -NHCH(CH2CH3)ÇH(CH(CH3)2) OH), 4,55 (d, 1H, J = 5,84 Hz, -NHCH(CH2CH3)CH(CH(CH3)2)OH), 7,17-7,46 (m, 15H, 3 χ Ph).
(3RS,4R)-2,2-Dimetil-4-(tritil-amino)-hexan-3-ol
A uma suspensão agitada de CuBr.SMe2 (1,37 g, 2,2 eq, 6,66 mmoles) em Et2O (100 mL), sob uma atmosfera de argônio, a -78°C, foi adicionado tercbutillítioíf ,5 M em pentano, 8,0 mL, 4 eq, 12,0 mmoles), gota a

gota, e a solução deixada aquecer para a temperatura ambiente. A mistura foi reesfriada para -55°C, à qual foi adicionada uma solução de (R)-2-(tritilamino)-butiraldeído (1 g, 1 eq, 3,03 mmoles) em Et2O (25 mL) gota a gota, com agitação, e agitada nesta temperatura durante 3 h. À mistura de reação foi adicionada uma solução aquosa saturada de NH4CI (100 mL) e deixada . 10 aquecer para a temperatura ambiente durante 16 h. A mistura de reação foi extraída com Et2O (2 χ 200 mL), e a fase orgânica combinada lavada com salmoura (50 mL), secada (MgSO4) e evaporada em vácuo. O resíduo foi purificado por cromatografia em coluna de gradiente de sílica-gel, eluído com hexano :éter (100:0 -> 90:10), para dar o composto do título como um óleo amarelo-claro. Rendimento: 0,57 g (49%). (55% de 3S,4R: 45% de 3R,4R).
1H-RMN (d6-DMSO, 250 MHz): δ 0,36 & 0,86 (2 χ t, 3H, J = 7,42 Hz, -NH
CH(CH2ÇH3)CH(C(CH3)3)OH), 0,57 & 0,71 (2 χ s, 9H, -NHCH(CH2CH3)CH (C(CH3)3)OH), 1,38-1,52 (m, 2H, -NHCH (ÇH2CH3)CH(C(CH3)3)OH), 1,99 (m,
1H, -NHCH(CH2CH3)CH(C(CH3)3)OH), 2,27 (m, 1H, -NHÇH(CH2CH3)CH (C(CH3)3)OH), 2,95 (m, 1H, -NHCH(CH2CH3)ÇH(C (CH3)3)OH), 4,22 & 4,77 (2 x d, 1H, J = 4,42 5,21 Hz, -NHCH(CH2CH3)CH(C (CH3)3)OH), 7,14-7,52 (m, 15H, 3 χ Ph).
(3RS,4S)-2,2-Dimetil-4-(tritil-amino)-hexan-3-ol
A uma suspensão agitada de CuBr.SMe2 (1,37 g, 2,2 eq, 6,66 mmoles) em EÍ2O (100 mL), sob uma atmosfera de argônio, a -78°C, foi adicionado terc-butil-lítio (1,5 M em pentano, 8,0 mL, 4 eq, 12,0 mmoles), gota a
gota, e a solução deixada aquecer para a temperatura ambiente. A mistura foi resfriada para -55°C, à qual foi adicionada uma solução de (S)-2-(tritilamino)-butiraldeído (1 g, 1 eq, 3,03 mmoles) em Et2O (25 mL), gota a gota, com agitação, e agitada nesta temperatura durante 3 h. À mistura de reação foi adicionada uma solução aquosa saturada de NH4CI (100 mL) e deixada . 10 aquecer para a temperatura ambiente durante 16 h. A mistura de reação foi extraída com Et2O (2 χ 200 mL), e a fase orgânica combinada lavada com salmoura (50 mL), secada (MgSO4) e evaporada em vácuo. O resíduo foi purificado por cromatografia em coluna de sílica-gel, eluído com hexano :
Et2O (100:0 -> 90:10), para dar o composto do título como um óleo amarelo15 claro. Rendimento: 0,47 g (40%). (53% de 3R,4S: 47% de 3S,4S). 1H-RMN (de-DMSO, 250 MHz): δ 0,37 & 0,87 (2 χ t, 3H, J = 7,46 Hz, -NHCH (CH?CH3)CH(C(CH3)3)OH). 0,58 & 0,71 (2 χ s, 9H, -NHCH(CH2CH3)CH (C(CH3)3)OH), 1,38-1,52 (m, 2H, -NHCH(CH2CH3) CH(C(CH3)3)OH), 2,00 (m, 1H, -NHCH(CH2CH3)CH(C(CH3)3)OH), 2,28 (m, 1H, -NHÇH(CH2CH3)CH 20 (C(CH3)3)OH), 2,95 (m, 1H, -NHCH(CH2CH3)ÇH(C(CH3)3)OH), 4,24 & 4,79 (2 χ d, 1H, J = 5,21 & 6,16 Hz, -NHCH(CH2CH3)CH(C(CH3)3)OH), 7,15-7,53 (m, 15H, 3 χ Ph).
(3R)-3-(Tritil-amino)-pentan-2-ona
9 · ·
9
9
A uma solução agitada de DMSO (2,19 mL, 2,8 eq, 30,86 mmo-
les) em DCM (30 mL), sob uma atmosfera de argônio, a -45°C, foi adicionado cloreto de oxalila (2 M em DCM, 7,69 mL, 1,4 eq, 15,38 mmoles), gota a gota. A mistura de reação foi agitada a -45°C durante 1 h, após o que solu5 ção (2S,3/?)-3-(tritil-amino)-pentan-2-ol (3,81 g, 1 eq, 11,04 mmoles) em
DCM (20 mL) foi adicionada, gota a gota, com agitação. A mistura de reação foi agitada nesta temperatura durante 4 h, quando TLC (hexano :éter; 80:20) indicou que a reação tinha ocorrido até completar. À mistura de reação foi adicionada N-etilpiperidina (7,54 mL, 5 eq, 54,88 mmoles), e a solução dei10 xada aquecer para a temperatura ambiente durante 16 h. A mistura de reação foi diluída com mais DCM (50 mL) e lavada com água (200 mL). A fase aquosa foi extraída com DCM (2 χ 50 mL), e a fase orgânica combinada lavada com salmoura (50 mL), secada (MgSO4) e evaporada em vácuo. O resíduo foi dissolvido em Et2O (100 mL), o precipitado sólido removido por fil15 tração e o filtrado foi evaporado em vácuo. O resíduo foi dissolvido em he-
xano (50 mL), o precipitado sólido removido por filtração e o filtrado foi evaporado em vácuo, para dar o composto do título como um óleo amareloclaro. Rendimento: 3,78 g (100%). 1H-RMN (d6-DMSO, 250 MHz): 5 0,73 (t,
3H, J = 7,35 Hz, -NHCH(CH?CH3)C(CH3)O). 1,47-1,60 (m, 5H, 20 NHCH(CH2CH-0C(CH3)Q), 3,12 (d, 1H, J = 8,38 Hz, -NHCH(CH2CH3)C(CH3)O),
3,32 (m, 1H, -NHÇH(CH2CH3) C(CH3)O), 7,16-7,49 (m, 15H, 3 χ Ph). (3S)-3-(Tritil-amino)-pentan-2-ona
A uma solução agitada de DMSO (1,95 mL, 2,8 eq, 27,48 mmo48
|
• · |
• |
• · - · |
• |
• · · |
• |
• · · |
• · · |
|
• · |
• |
• · · |
|
• |
• |
• |
• |
| |
• |
• * · |
• |
• |
·· · |
• |
• |
| |
|
|
|
|
|
|
• |
| |
• |
• · |
• · · |
• |
• |
• |
• |
les) em DCM (30 mL), sob uma atmosfera de argônio, a -45°C, foi adicionado cloreto de oxalila (2 M em DCM, 6,85 mL, 1,4 eq, 13,70 mmoles), gota a gota. A mistura de reação foi agitada a -45°C durante 1 h, após o que solução de (2R,3S)-3-(tritil-amino)-pentan-2-ol (3,39 g, 1 eq, 9,83 mmoles) em 5 DCM (20 mL) foi adicionada, gota a gota, com agitação. A mistura de reação foi agitada nesta temperatura durante 4 h, quando TLC (hexano :éter; 80:20) indicou que a reação tinha ocorrido até completar. À mistura de reação foi adicionada /V-etilpiperidina (6,71 mL, 5 eq, 48,84 mmoles), e a solução deixada aquecer para a temperatura ambiente durante 16 h. A mistura de rea-

ção foi diluída com mais DCM (50 mL) e lavada com água (200 mL). A fase aquosa foi extraída com DCM (2 χ 50 mL), e a fase orgânica combinada lavada com salmoura (50 mL), secada (MgSCU) e evaporada em vácuo. O resíduo foi dissolvido em Et2O (100 mL), o precipitado sólido removido por filtração e o filtrado foi evaporado em vácuo. O resíduo foi dissolvido em he15 xano (50 mL), o precipitado sólido removido por filtração e o filtrado foi evaporado em vácuo, para dar o composto do título como um óleo amareloclaro. Rendimento: 3,15 g (93%). 1H-RMN (d6-DMSO, 250 MHz): δ 0,73 (t,
3H, J = 7,50 Hz, -NHCH(CH2ÇH3)C(CH3)O), 1,45-1,62 (m, 5H, NHCH(CH2CH.)C(CH3)O)· 3,12 (d, 1H, J = 8,53 Hz, -NHCH(CH2CH3)C(CH3)O), 3,31 (m, 1H, -NHÇH(CH2CH3)C(CH3)O), 7,13-7,45 (m, 15H, 3 x Ph).
(3R)-2-Metil-3-(tritil-amino)-pentan-2-ol
A uma solução agitada de (3F?)-3-(tritil-amino)-pentan-2-ona (0,87 g, 1 eq, 2,54 mmoles) em Et2O (100 mL), sob uma atmosfera de argônio, em temperatura ambiente, foi adicionado iodeto de metil-magnésio (3 M 25 em éter, 2,54 mL, 3 eq, 7,62 mmoles), gota a gota. A solução foi colocada em um banho de óleo preaquecido a 45°C e refluxada nesta temperatura durante 16 h. A mistura foi resfriada para 0°C, H2O (100 mL) adicionada, a
• ·4· · ··· · ··· ··· ·· · ·· ·· · · • · · · ····· · ···· · ♦ · · · · • ····· · · · solução filtrada através de Celite, e o Celite lavado com mais Et2O (50 mL). A fase orgânica combinada foi separada, a fase aquosa foi extraída com
Et2O (2 χ 50 mL), e a fase orgânica combinada lavada com salmoura (50 mL), secada (MgSO4) e evaporada em vácuo. O resíduo foi purificado por 5 cromatografia em coluna de sílica-gel, eluído com hexano :éter (100:0 ->
90:10) para dar o composto do título como um óleo amarelo-claro. Rendimento: 0,21 g (23%). 1H-RMN (d6-DMSO, 250 MHz): <5 0,26 (t, J = 7,42 Hz, NHCH(CH2ÇH3)CH(CH3)OH), 1,00 & 1,25 (2 χ s, 6H, -NHCH(CH2CH3)
C(CH3)2 OH). 0,72-1,43 (m, 2H, -NHCH(ÇH2CH3)C(CH3)2OH), 1,84 (m, 1H, 10 NHCH(CH2 CH3)C(CH3)2OH), 2,90 (m, 1H, -NHÇH(CH2CH3)C(CH3)2OH),
4,32 (s, 1H, -NHCH (CH2CH3)C(CH3)2OH), 7,17-7,46 (m, 15H, 3 χ Ph). (3S)-2-Metil-3-(tritil-amino)-pentan-2-ol
A uma solução agitada de (3S)-3-(tritil-amino)-pentan-2-ona (0,59 g, 1 eq, 1,72 mmol) em Et2O (100 mL), sob uma atmosfera de argônio, em temperatura ambiente, foi adicionado iodeto de metil magnésio (3 M em

Et2O, 1,72 mL, 3 eq, 5,16 mmoles), gota a gota. A solução foi colocada em um banho de óleo préaquecido a 45°C e refluxada nesta temperatura durante 16 h. A mistura foi resfriada para 0 °C, H2O (100 mL) adicionada, a solução filtrada através de Celite, e Celite lavado com mais Et2O (50 mL). A fase orgânica combinada foi separada, a fase aquosa foi extraída com Et2O (2 χ 50 mL), e a fase orgânica combinada lavada com salmoura (50 mL), secada (MgSCU) e evaporada em vácuo. O resíduo foi purificado por cromatografia em coluna de sílica-gel, eluído com hexano : Et2O (100:0 -> 90:10), para dar o composto do título como um óleo amarelo-claro. Rendi25 mento: 0,10 g (16%). 1H-RMN (d6-DMSO, 250 MHz): <5 0,27 (t, J = 7,10 Hz, NHCH(CH2 ÇH3)CH(CH3)OH), 0,99 & 1,25 (2 χ s, 6H, -NHCH(CH2CH3) C(CH3)2OH), 0,75-1,42 (m, 2H, -NHCH(ÇH2CH3)C(CH3)2OH), 1,88 (m, 1H, 50
NHCH(CH2CH3)C(CH3)2QH)) 2,92 (m, 1H, -NHÇH(CH2CH3)C(CH3)2OH),
4,32 (s, 1H, -NHCH(CH2CH3) C(CH3)2OH), 7,18-7,46 (m, 15H, 3 χ Ph). (2S,3R)-3-Amino-pentan-2-ol
A uma solução agitada de (2S,3/?)-3-(tritil-amino)-pentan-2-ol (1,32 g, 1 eq, 3,83 mmoles) em DCM (50 mL) sob uma atmosfera de argônio, em temperatura ambiente, foi adicionado CF3COOH (10 mL), gota a
gota, e a solução foi agitada nesta temperatura durante 1h. O solvente foi evaporado em vácuo e o resíduo foi precipitado de Et2O (15 mL) com hexano (300 mL), com agitação, para dar um óleo amarelo. O solvente foi de10 cantado do óleo, e o óleo foi lavado com hexano (30 mL) e secado em vácuo para dar o composto do título como um óleo amarelo-claro. Rendimento:
0,30 g (99%). (80% de 2S,3R: 20% de 2R,3R). 1H-RMN (d6-DMSO, 250 MHz): δ 0,915 & 0,924 (2 χ t, 3H, J = 7,50 & 7,58 Hz,
NH2CH(CH2ÇH3)CH(CH3)OH), 1,06 & 1,13 (2 χ d, J = 6,48 & 6,32 Hz), 15 NH2CH (CH2CH3)CH(ÇH3)OH), 1,41-1,59 (m, 2H, NHzCHíCHgCHa)
CH(CH3)OH), 2,77 & 2,93 (2 x m, 1H, NH2CH(CH2CH3)CH(CH3)OH), 3,62-3,72 & 3,80-3,90 (2 χ m, 1H, NH2CH(CH2CH3)ÇH(CH3)OH), 7,75 (bs, 2H, NH?).
(2R,3S)-3-Amino-pentan-2-ol
A uma solução agitada de (2R,3S)-3-(tritil-amino)-pentan-2-ol (1,64 g, 1 eq, 4,75 mmoles) em DCM (50 mL), sob uma atmosfera de argônio, em temperatura ambiente, foi adicionado CF3COOH (10 mL), gota a gota, e a solução foi agitada nesta temperatura durante 1 h. O solvente foi evaporado em vácuo e o resíduo foi precipitado de Et2O (15 mL) com hexano (300 mL), com agitação, para dar um óleo amarelo. O solvente foi de25 cantado do óleo, e o óleo foi lavado com hexano (30 mL) e secado em vácuo para dar o composto do título como um óleo amarelo-claro. Rendimento:
• · · • · ·
0,30 g (98%). (80% de 2R,3S: 20% de 2S,3S). 1H-RMN (d6-DMSO, 250 MHz): δ 0,913 & 0,923 (2 x t, 3H, J = 7,50 & 7,50 Hz,
NH2CH(CH2ÇH3)CH(CH3)OH), 1,11 & 1,18 (2 x d, J = 6,48 & 6,48 Hz),
NH2CH(CH2CH3)CH(ÇH3)OH), 1,41-1,65 (m, 2H, NH2CH(ÇH2CH3)
CH(CH3)OH), 2,76 + 2,93 (2 x m, 1H, NH2CH(CH2CH3)CH(CH3)OH). 3,61-3,69 & 3,80-3,90 (2 x m, 1H, NH2CH(CH2CH3)ÇH(CH3)OH), 7,73 (bs, 2H, NH?). (3RS,4R)-4-Amino-hexan-3-ol
A uma solução agitada de (3RS,4R)-4-(tritil-amino)-hexan-3-ol (1,13 g, 1 eq, 3,14 mmoles) em DCM (15 mL), sob uma atmosfera de argô nio, em temperatura ambiente, foi adicionado CF3COOH (7 mL), gota a gota, e a solução foi agitada nesta temperatura durante 4 h. O solvente foi evaporado em vácuo, EtOH (20 mL) adicionado, e removido em vácuo, e este processo repetido duas vezes. O resíduo foi precipitado de Et2O (5 mL) com hexano (40 mL), com agitação, para dar um óleo amarelo. O solvente foi de15 cantado do óleo, e o óleo foi lavado com hexano (30 mL) e secado em vácuo, para dar o composto do título como um óleo amarelo-claro. Rendimento: 0,37 g (100%). (57% de 3S,4R: 43% de 3R,4R). 1H-RMN (d6-DMSO, 250
MHz): 50,79 & 0,92 (t & m, 6H, J = 7,42 Hz, NH?CH(CH?CH3)CH(CH2CH3)OH). 1,30-1,67 (m, 4H, NH2CH(ÇH2CH3)CH (CH2 CH^OH), 2,70 (m, 1H, 20 NH2CH(CH2CH3)CH(CH2CH3)OH), 2,84 & 2,96 (2 x m, 1H,
NH2ÇH(CH2CH3)CH(CH2CH3)OH), 3,41 & 3,56 (2 x m, 1H, NH2CH (CH2CH3)ÇH(CH2CH3)OH), 7,71 (bs, 2H, NH2CH(CH2CH3)CH(CH2CH3)OH). (3RS,4S)-4-Amino-hexan-3-ol
A uma solução agitada de (3RS,4S)-4-(tritil-amino)-hexan-3-ol (1,19 g, 1 eq, 3,31 mmoles) em DCM (15 mL), sob uma atmosfera de argônio, em temperatura ambiente, foi adicionado CF3COOH (7 mL), gota a gota,
e a solução foi agitada nesta temperatura durante 4 h. O solvente foi evaporado em vácuo, EtOH (20 mL) adicionado, e removido em vácuo, e este processo repetido duas vezes. O resíduo foi precipitado de Et2O (5 mL) com hexano (40 mL), com agitação, para dar um óleo amarelo. O solvente foi de5 cantado do óleo, e o óleo foi lavado com hexano (30 mL) e secado em vácuo para dar o composto do título. Rendimento: 0,39 g (99%). (65% de 3R,4S:
35% de 3S,4S). 1H-RMN (d6-DMSO, 250 MHz): 5 0,79 & 0,92 (t & m, 6H, J =
7,50 Hz, NH2CH(CH2ÇH3)CH(CH2ÇH3)OH), 1,22-1,68 (m, 4H, NH?CH(CH2CH3) CH(ÇH2_CH3)OH), 2,71 (m, 1H, NH?CH(CH?CH^CH(CH?CH3)OH). 2,83 & 2,95 (2 x m, 1H, NH?CH(CH?CH3)CH(CH?CH3)OH). 3,39 & 3,54 (2 x m, 1H, NH2CH (CH?CH.3)CH (CH?CH3)OH). 7,77 (bs, 2H, NH2CH(CH?CH.^CH(CH2CH^QH). (3RS,4R)-4-Amino-2-metil-hexan-3-ol
A uma solução agitada de (3RS,4R)-2-metil-4-(tritil-amino)-hexan-3-ol (0,53 g, 1 eq, 1,41 mmol) em DCM (20 mL), sob uma atmosfera de argônio, em temperatura ambiente, foi adicionado CF3COOH (5 mL), gota a gota, e a solução foi agitada nesta temperatura durante 1 h. O solvente foi
evaporado em vácuo, o resíduo foi precipitado de Et2O (10 mL) com hexano (90 mL) com agitação para dar um óleo amarelo. O solvente foi decantado do óleo, e o óleo foi lavado com hexano (20 mL) e secado em vácuo para dar o composto do título como um óleo amarelo-claro. Rendimento: 0,18 g (100%). (50% de 3S,4R: 50% de 3R,4R). 1H-RMN (d6-DMSO, 250 MHz): 5
0,85-0,99 (m, 9H, NH2CH (CHzÇth) CHÍCHÍCH^OH). 1,42-1,79 (m, 2H, NH2CH(ÇH2CH3)CH (CH(CH3)2)OH), 2,95 (m, 1H,
NH?CH(CH?CH3)CH(CH(CH3)?)OH). 3,18 (m, 1H, NH2CH(CH2CH3)CH(ÇH 25 (CH3)2)OH), 3,37 (m, 1H, NH2CH(CH2CH3)ÇH(CH(CH3)2)OH), 7,58 (bs, 2H,
NH2CH(CH2CH3)CH(CH(CH3)2)OH).
(3RS,4S)-4-Amino-2-metil-hexan-3-ol
A uma solução agitada de (3F?S,4S)-2-metil-4-(tritil-amino)-hexan-3-ol (0,36 g, 1 eq, 0,97 mmol) em DCM (20 mL), sob uma atmosfera de argônio, em temperatura ambiente, foi adicionado CF3COOH (5 mL), gota a 5 gota, e a solução foi agitada nesta temperatura durante 1 h. O solvente foi evaporado em vácuo, o resíduo foi precipitado de Et2O (10 mL) com hexano (90 mL), com agitação, para dar um óleo amarelo. O solvente foi decantado do óleo, e o óleo foi lavado com hexano (20 mL) e secado em vácuo para dar o composto do título como um óleo amarelo-claro. Rendimento: 0,13 g (100%). (50% de 3R,4S: 50% de 3S,4S) 1H-RMN (d6-DMSO, 250 MHz): δ
0,85-1,01 (m, 9H, NH2CH (CH7CHg)CH(CH(CH3)2)OH). 1,44-1,76 (m, 2H,
NH2CH(ÇH2CH3)CH (CH(CH3)2) OH), 2,94 (m, 1H, NH2CH(CH2CH3)CH(CH(CH3)?)OH).
3,17 (m, 1H, NH2CH(CH2CH3)CH(ÇH(CH3)2)OH), 3,40 (m, 1H, NH2CH(CH2CH3)ÇH (CH(CH3)2) OH), 7,54 (bs, 2H, NH2CH(CH2CH3)CH(CH(CH3)2)OH).
(3RS,4R)-4-Amino-2,2-dimetil-hexan-3-ol
A uma solução agitada de (3F?S,4F?)-2,2-dimetil-4-(tritil-amino)hexan-3-ol (0,57 g, 1 eq, 1,47 mmol) em DCM (10 mL), sob uma atmosfera de argônio, em temperatura ambiente, foi adicionado CF3COOH (5 mL), gota a gota, e a solução foi agitada nesta temperatura durante 1 h. O solvente foi 20 evaporado em vácuo, o resíduo foi precipitado de Et2O (3 mL) com hexano (20 mL), com agitação, para dar um óleo amarelo. O solvente foi decantado do óleo, e o óleo foi lavado com hexano (20 mL) e secado em vácuo para dar o composto do título como um óleo amarelo-claro. Rendimento: 0,21 g (100%). (55% de 3S,4R: 45% de 3R,4R). 1H-RMN (d6-DMSO, 250 MHz): δ
0,84-0,99 (m, 3H, NHzCHÍCHzÇHgJCHÍCÍCHs^OH), 1,25-1,29 (m, 9H,
NH2CH(CH2CH3) CH(C (CH3)g)OH), 1,20-1,72 (m, 2H, NH2CH(ÇH2CH3)CH(C (CH3)3)OH), 3,14 (m, 1H, NH2ÇH(CH2CH3)CH(C(CH3)3)OH), 3,39 (m, 1H,
NH2CH(CH2CH3) CH(C(CH3)3) OH), 3,65 (m, 1H, NH2CH(CH2CH3)ÇH(C (CH3)3)OH), 7,43, 7,77 & 8,54 (3 χ bs, 2H, NH2CH(CH2CH3)CH(CH(CH3)2)OH).
(3RS,4S)-4-Amino-2,2-dimetil-hexan-3-ol
A uma solução agitada de (3RS,4S)-2,2-dimetil-4-(tritil-amino)hexan-3-ol (0,47 g, 1 eq, 1,21 mmol) em DCM (10 mL), sob uma atmosfera de argônio, em temperatura ambiente, foi adicionado CF3COOH (5 mL), gota 10 a gota, e a solução foi agitada nesta temperatura durante 1 h. O solvente foi evaporado em vácuo, o resíduo foi precipitado de Et2O (3 mL) com hexano (20 mL), com agitação, para dar um óleo amarelo. O solvente foi decantado do óleo, e o óleo foi lavado com hexano (20 mL) e secado em vácuo para dar o composto do título como um óleo amarelo-claro. Rendimento: 0,18 g 15 (99%). (53% de 3R,4S: 47% de 3S,4S). 1H-RMN (d6-DMSO, 250 MHz): δ
0,86-0,99 (m, 3H, NH2CH(CH2ÇH3)CH(C(CH3)3)OH), 1,25-1,30 (m, 9H, NH2CH(CH2CH3)CH(C(ÇH3)3) OH), 1,20-1,67 (m, 2H, NH2CH(ÇH2CH3)CH (C(CH3)3)OH), 3,14 (m, 1H, NH2ÇH_(CH2CH3)CH(C(CH3)3)OH), 3,38 (m, 1H,
NH2CH(CH2CH3) CH(C(CH3)3)OH), 3,64 (m, 1H, NH2CH(CH2CH3)ÇH(C 20 (CH3)3)OH), 7,41,7,73 & 8,44 (3 χ bs, 2H, NH2CH(CH2CH3)CH(CH(CH3)2)OH).
(3R)-3-Amino-2-metil-pentan-2-ol
A uma solução agitada de (3R)-2-metil-3-(tritil-amino)-pentan-2ol (0,21 g, 1 eq, 0,60 mmol) em DCM (5 mL), sob uma atmosfera de argônio, em temperatura ambiente, foi adicionado CF3COOH (2,5 mL), gota a gota, e a solução foi agitada nesta temperatura durante 1 h. O solvente foi evaporado em vácuo e o resíduo foi precipitado de Et2O (15 mL) com hexano (300 mL), com agitação, para dar um óleo amarelo. O solvente foi decantado do óleo, e o óleo foi lavado com hexano (30 mL) e secado em vácuo para dar o composto do título como um óleo amarelo-claro; Rendimento: 0,07 g (100%). 1H-RMN (d6-DMSO, 250 MHz): δ 0,97 (t, 3H, J = 7,42 Hz, NH2CH (CH2ÇH3)C(CH3)2OH), 1,06 & 1,19 (2 x s, 6H, NH2CH(CH2CH3)-C(CH3) 2OH), 1,28-1,71 (m, 2H, NH2CH (CH2CH3) C(CH3)2OH), 2,72 (m, 1H, NH2ÇH(CH2CH3)C(CH3)2OH), 5,21 (s, 1H, NH2CH (CH2CH3)C(CH3)2OH), 10 7,63 (bs, 2H, NH2CH(CH2CH3)C(CH3)2OH).
(3S)-3-Amino-2-metil-pentan-2-ol
A uma solução agitada de (3S)-2-metil-3-(tritil-amino)-pentan-2ol (0,38 g, 1 eq, 1,06 mmol) em DCM (5 mL), sob uma atmosfera de argônio, em temperatura ambiente, foi adicionado CF3COOH (2,5 mL), gota a gota, e 15 a solução foi agitada nesta temperatura durante 1 h. O solvente foi evaporado em vácuo e o resíduo foi precipitado de Et2O (15 mL) com hexano (300 mL), com agitação, para dar um óleo amarelo. O solvente foi decantado do óleo, e o óleo foi lavado com hexano (30 mL) e secado em vácuo para dar o composto do título como um óleo amarelo-claro. Rendimento: 0,12 g (99%).
1H-RMN (de-DMSO, 250 MHz): δ 0,97 (t, 3H, J = 7,42 Hz, NH2CH(CH2 ÇH3)C(CH3)2OH), 1,07 & 1,19 (2 x s, 6H, NH2CH(CH2CH3)CÍÇH3)2OH), 1,281,61 (m, 2H, NH2CH (ÇH2CH3)C(CH3)2OH), 2,72 (m, 1H, NH2CH(CH?CHÁ)C (CH3)2OH), 5,21 (s, 1H, NH2CH(CH2CH3)C (CH3)?OH). 7,63 (bs, 2H, NH?CH (CH2CH3)C(CH3)2OH).
6-Cloro-2-flúor-9H-purina
Este composto foi preparado por uma modificação de procedimentos na literatura (Gray, N.S.; Kwon, S.; Schultz, P.G. Tetrahedron Lett. 1997, 38(7), 1161-1164,)
6-Cloro-9/-/-purin-2-ilamina (75,0 g, 0,44 mol) foi colocada em suspensão em HBF4 aquoso (1,5 L de solução a 48% em peso/peso em H2O). Esta mistura foi resfriada a -15°C e foi agitada com vigor. NaNO2 (2,5
L de uma solução aquosa 0,3 M) foi então adicionado lentamente, durante 5 75 min, com agitação e controle de temperatura cuidadoso (< 10°C). Após
completa adição, a solução amarelo pálido foi ainda agitada em temperatura ambiente, durante 30 min. Ela foi então resfriada para -15°C e foi neutralizada com cuidado para pH = 6,2 com NaOH (50% em peso/volume de solução aquosa). Esta solução foi evaporada por rotação até semi-secura. A torta resultante foi dividida com uma espátula e secada sob vácuo elevado, durante a noite. O pó amarelo resultante foi carregado seco em uma coluna de cromatografia flash (24 X 15 cm, leito de SiO2), que foi eluído com CH2CI2/MeOH, 9:1, As frações apropriadas foram coletadas, reunidas e evaporadas. Após secar em vácuo, o composto do título (34,8 g, 48%) foi obtido 15 como um pó incolor. TLC: Rf = 0,25 (CH2CI2/MeOH, 9:1), material de partida
Rf = 0,16, m/z 173 (MH+, 100), 175 (MH+2, 33).
(2-flúor-9H-purin-6-il)-piridin-3-ilmetil-amina
A uma solução agitada de 6-cloro-2-fluoropurina (0,9 g, 1 eq,
5,22 mmoles) em n-BuOH (60 mL), sob uma atmosfera de argônio, resfriada a -20°C, foi adicionado DIEA (2,5 mL, 2,75 eq, 14,35 mmoles) seguido por
C-piridin-3-il-metilamina (0,58 mL, 1,1 eq, 5,69 mmoles). A mistura de reação • · · · · ·
foi agitada a -20°C, durante 3 h, e então deixada retornar à temperatura ambiente depois de 2 h, quando TLC (CHCh: MeOH; 90:10) indicou que a reação tinha ocorrido até completar. O solvente foi evaporado em vácuo e o resíduo foi purificado por cromatografia de coluna de gradiente em sílica-gel, 5 eluído com CHCh:MeOH (97:3 -> 90:10), para dar o composto do título como um sólido branco. Rendimento: 1,08 g (85%). Ponto de fusão 218-220 °C. 1H-RMN (de-DMSO, 250 MHz): 5 4,65 (d, 2H, J = 5,37 Hz, -HNCH?-Pir), 7,34, 8,43, 8,57 (3 x m, 4H, Pir), 8,10 (s, 1H, -N=CH-NH-), 8,83 (bs, 1H, HNCH?-Pir), 13,07 (bs, 1H, -N=CH-NH-). FABMS m/z (intensidade relativa):
245 ([M+H]+, 40), 176 (27), 154 (100), 136 (74). Massa exata (M+H): Real:
245,0951, Medida: 245,0942, Microanálise (Esperada: Medida) CnHgNeF.0,2 H2O: C; 53,31: 54,03, H; 3,82: 3,83, N; 33,91: 32,74, (2-flúor-9-isopropil-9H-purin-6-il)-piridin-3-ilmetil-amina
A uma solução agitada de (2-flúor-9/7-purin-6-il)-piridin-3-ilmetil amina (1,00 g, 1 eq, 4,10 mmoles) em DMA (12 mL), sob uma atmosfera de argônio, em temperatura ambiente, foi adicionado K2CO3 (em pó, anidro,
2,75 g, 4,85 eq, 19,90 mmol) seguido por 2-bromopropano (3,75 mL, 9,75 eq, 39,93 mmoles). A mistura de reação foi agitada em temperatura ambiente durante 48 h, quando TLC (CHCI3:MeOH; 90:10) indicou que a reação tinha ocorrido até completar. O solvente foi evaporado em vácuo e o resíduo dividido entre água (200 mL) e EtOAc (100mL), a fase aquosa foi separada e extraída com mais EtOAc (2 χ 50 mL). A fase orgânica em bruto foi lavada com salmoura (50 mL), secada (MgSO4) e evaporada em vácuo, e o resíduo foi purificado por cromatografia de coluna de gradiente em sílica-gel, eluído
com CHCI3:MeOH (100:0 -> 95:5), para dar o composto do título como um sólido branco. Rendimento: 0,73 g (62%). Ponto de fusão 131-133 °C. 1HRMN (de-DMSO, 250 MHz): δ 1,49 (2 χ s, 6H, -CH(CH3)?). 4,63 (m, 3H, ÇH(CH3)2 & -HNCHg-Pir), 7,34, 7,24, 8,44, 8,58 (4 χ m, 4H, Pir), 8,26 (s, 1H,
-N=CH-N-), 8,95 (bs, 1H, -HNCH?-Pir). FABMS m/z (intensidade relativa):
287 ([M+H]+, 100), 245 (8), 154 (23), 136 (18). Massa exata (M+H): Real:
287,1420, Medida: 287,1412, Microanálise (Esperada: Medida) Ci4Hi5N6F: C; 58,73: 58,57, H; 5,28: 5,21, N; 29,35: 29,27, (2S3R)-3-{9-lsopropil-6-í(piridin-3-ilmetil)-aminol-9H-purin-2-ilamino}-pentan10 2-ol
A uma solução agitada de (2-flúor-9-isopropil-9H-purin-6-il)-piridin-3-ilmetil-amina (30 mg, 1 eq, 0,10 mmol) em n-BuOH/DMSO (2,5 mL, 4:1), em temperatura ambiente, sob uma atmosfera de argônio, foi adiciona-
do DIEA (0,2 mL, 10,96 eq, 1,14 mmol), seguido por (2S,3R)-3-amino-pentan-2-ol (60 mg, 5,5 eq, 0,58 mmol). A mistura de reação foi colocada em um banho de óleo pré-aquecido a 160°C e agitada nesta temperatura, durante
h. A mistura de reação foi deixada resfriar para a temperatura ambiente e o solvente foi evaporado em vácuo. O resíduo foi dividido entre EtOAc (50 mL) e água (50 mL), a fase aquosa foi extraída com mais EtOAc (2 χ 25 mL), e a fase orgânica combinada foi lavada com salmoura (50 mL), secada (MgSO4) e evaporada em vácuo. O resíduo foi purificado por cromatografia de coluna de gradiente em sílica-gel eluído com CHCI3:MeOH (100:0 ->
98:2), para dar o composto do título como um sólido branco. Rendimento: 22 mg (57%). (80% de 3R,2S: 20% de 3R,2R). 1H-RMN (d-CDCI3, 250 MHz): δ
0,90, 1,05 (2 χ t, 3H, J = 6,95 & 7,42 Hz, -NHCH (CH?CH3)CH(CH3)OH). 1,17 &
1,25 (2 χ d, 3H, J = 6,32 & 6,16 Hz, -NHCH (CH2CH3)CH(ÇH3)OH) 1,57 (d, 6H, J = 6,79 Hz, -CH(CH3)?), 1,77-2,09 (m, 2H, -NHCH(ÇH2CH3)CH(CH3)OH), 3,914,03 (m, 2H, -NHÇH(CH2CH3) ÇH(CH3)OH), 4,58-4,69 (m, 1H, -ÇH(CH3)2), 4,83-5,00 (m, 3H, -HNCH2-Pir & OH), 6,16 (m, 1H, 5 NHCH(CH2CH3)CH(CH3)OH), 7,24-7,33 (m, 3H, 2 x Pir-H & -N=ÇH-N-), 7,55 (m, 1H, Pir-H), 7,74 (d, 1H, J = 7,42 Hz, Pir-H), 8,67 (m, 1H, HNCH2-Pir). FABMS m/z (intensidade relativa): 370 ([M+H]+, 100), 324 (35), 289 (37), 243 (65), 199 (85). Massa exata (M+H): Real: 370,2355, Medida: 370,2347.
(2R3S)-3-{9-lsopropil-6-í(piridin-3-ilmetil)-aminol-9H-purin-2-ilamino}-pentan-
A uma solução agitada de (2-flúor-9-isopropil-9/7-purin-6-il)-piridin-3-ilmetil-amina (30 mg, 1 eq, 0,10 mmol) em n-BuOH/DMSO (2,5 mL, 4:1), em temperatura ambiente, sob uma atmosfera de argônio, foi adiciona-
do DIEA (0,2 mL, 10,96 eq, 1,14 mmol), seguido por (2F?,3S)-3-aminopentan-2-ol (60 mg, 5,5 eq, 0,58 mmol). A mistura de reação foi colocada em um banho de óleo pré-aquecido a 160°C e agitada nesta temperatura, durante 72 h. A mistura de reação foi deixada resfriar a temperatura ambiente e o solvente foi evaporado em vácuo. O resíduo foi dividido entre EtOAc (50 mL) e água (50 mL), a fase aquosa foi extraída com mais EtOAc (2 χ 25 mL), e a fase orgânica combinada foi lavada com salmoura (50 mL), secada (MgSO4) e evaporada em vácuo. O resíduo foi purificado por cromatografia de coluna de gradiente em sílica-gel eluído com CHCEMeOH (100:0 ->
98:2), para dar o composto do título como um sólido branco. Rendimento: 25 mg (65%). (80% de 3S,2R: 20% de 3S,2S). 1H-RMN (d-CDCI3, 250 MHz): δ
0,90 & 1,05 (2 χ t, 3H, J = 6,48 & 7,42 Hz, -NHCHÍCHyCHg) CH(CH3)OH), 1,16 & 1,24 (2 χ d, 3H, J = 6,31 & 6,16 Hz, -NHCH(CH2CH3) CH(CH3)QH) 1,57 (d, 6H, J = 6,79 Hz, -CH(CH3)?). 1,77-2,09 (m, 2H, -NHCH(ÇH2CH3)CH(CH3)OH), 3,91-4,04 (m, 2H, -NHÇH(CH2CH3)ÇH(CH3)OH), 4,58-4,69 (m, 1H, -ÇH(CH3)2),
4,83 (m, 3H, -HNCH2-Pir & OH), 6,11-6,23 (m, 1H, 5 NHCH(CH2CH3)CH(CH3)OH), 7,24-7,32 (m, 3H, 2 χ Pir-H + -N=ÇH-N-), 7,537,57 (m, 1H, Pir-H), 7,74 (d, 1H, J = 7,58 Hz, Pir-H), 8,67 (m, 1H, HNCH?-Pir). FABMS m/z (intensidade relativa): 370 ([M+H]+, 100), 324 (40), 289 (15), 243 (10), 233 (13), 199 (10). Massa exata (M+H): Real: 370,2355, Medida: 370,2347.
(3RS,4R)-4-{9-lsopropil-6-[(piridin-3-ilmetil)-amino1-9H-purin-2-ilamino}hexan-3-ol
A uma solução agitada de (2-flúor-9-isopropil-9/7-purin-6-il)piridin-3-ilmetil-amina (20 mg, 1 eq, 0,07 mmol) em n-BuOH/DMSO (3,75
mL, 4:1), em temperatura ambiente sob uma atmosfera de argônio, foi adicionado DIEA (0,18 mL, 15 eq, 1,03 mmol), seguido por (3F?S,4/?)-4-aminohexan-3-ol (110 mg, 13 eq, 0,94 mmol). A mistura de reação foi colocada em um banho de óleo pré-aquecido a 140°C e agitada nesta temperatura, durante 72 h. A mistura de reação foi deixada resfriar a temperatura ambiente e o solvente foi evaporado em vácuo. O resíduo foi dividido entre EtOAc (50 20 mL) e salmoura/água (1:1, 100 mL), a fase aquosa foi extraída com mais
EtOAc (2 χ 25 mL), e a fase orgânica combinada foi lavada com salmoura (50 mL), secada (MgSO4) e evaporada em vácuo. O resíduo foi purificado por cromatografia de coluna de gradiente em sílica-gel eluído com
CHCI3:MeOH (100:0 -> 98:2), para dar o composto do título como um sólido branco. Rendimento: 23 mg (75%). (57% de 4R.3S: 43% de 4R,3R). 1H61
|
·· |
• · |
• ·· |
• |
··· |
• |
• · · |
• · · |
|
• · |
• · · |
• |
·· |
• |
• |
• |
• |
| |
• · · |
• |
• |
• |
·· · |
• |
• |
| |
|
|
|
|
|
|
• |
| |
• · |
• |
• · · |
• |
• |
• |
• |
RMN (d-CDCI3, 250 MHz): δ 0,87-1,07 (m, 6H, -NHCH(CH?CH3)
CH(CH2ÇH3)OH), 1,56 (d, 6H, J = 6,63 Hz, & -CH(CH3)2), 1,43-1,63 (m, 4H, NHCH(CH2CH3)CH(CH2CH3)OH). 3,44 (d, 1H, J = 6,31 Hz, OH), 3,58-3,71 (m, 1H, -NHCH(CH2CH3)ÇH(CH2CH3)OH), 3,89-4,01 (m, 1H, 5 NHCH(CH2CH3)ÇH(CH2 CH3)OH), 4,56-4,70 (m, 1H, -ÇH(CH3)2), 4,76-4,95 (m, 2H, -HNCH?-Pir), 5,20-5,29 & 6,17-6,34 (m, 1H, -NHCH(CH2CH3) CH(CH2CH3)OH), 7,19-7,38 (m, 3H, 2 χ Pir-H & -N=CH-N-), 7,48-7,60 (m,
1H, Pir-H), 7,72 (d, 1H, J = 7,74 Hz, Pir-H), 8,67 (m, 1H, HNCH2-Pir). FABMS m/z (intensidade relativa): 384 ([M+H]+, 100), 324 (50), 297 (30). Massa exata (M+H): Real: 384,2512, Medida: 384,2500.
(3RS,4S)-4-{9-lsopropil-6-[(piridin-3-ilmetil)-aminol-9H-purin-2-ilamino}hexan-3-ol
A uma solução agitada de (2-flúor-9-isopropil-9/-/-purin-6-il)-piri-
din-3-ilmetil-amina (20 mg, 1 eq, 0,07 mmol) em n-BuOH/DMSO (3,75 mL,
4:1), em temperatura ambiente, sob uma atmosfera de argônio, foi adicionado DIEA (0,18 mL, 15 eq, 1,03 mmol), seguido por (3RS,4S)-4-amino-hexan
3-ol (110 mg, 13 eq, 0,94 mmol). A mistura de reação foi colocada em um banho de óleo pré-aquecido a 140°C e agitada nesta temperatura, durante
h. A mistura de reação foi deixada resfriar a temperatura ambiente e o solvente foi evaporado em vácuo. O resíduo foi dividido entre EtOAc (50 mL) e salmoura/água (1:1, 100 mL), a fase aquosa foi extraída com mais EtOAc (2 χ 25 mL), e a fase orgânica combinada foi lavada com salmoura (50 mL), secada (MgSO4) e evaporada em vácuo. O resíduo foi purificado por cromatografia de coluna de gradiente em sílica-gel eluído com CHCI3:MeOH (100:0 -> 98:2), para dar o composto do título como um sólido branco. Ren• · · ·
dimento: 15,5 mg (58%). (57% de 4S.3R: 43% de 4S,3S). 1H-RMN (d-CDCI3,
250 MHz): 5 0,84-1,09 (m, 6H, -NHCH(CH?CH3) CH(CH?CH3)OH), 1,57 (d, 6H, J = 6,48 Hz, & -CH(CHj)?), 1,42-1,64 (m, 4H, -NHCH(CH2CH3)
CH(CH2CH3)OH), 3,45 (d, 1H, J = 6,31 Hz, OH), 3,55-3,71 (m, 1H, 5 NHCH(CH2CH3)ÇH(CH2CH3)OH), 3,88-4,03 (m, 1H, -NHCH(CH2CH3)ÇH (CH2CH3)OH), 4,56-4,74 (m, 1H, -ÇH(CH3)2), 4,78-4,99 (m, 2H, -HNCH?-Pir), 5,20-5,29 & 6,20-6,39 (m, 1H, -NHCH(CH2CH3)CH(CH2CH3)OH), 7,20-7,36 (m, 3H, 2 χ Pir-H + -N=CH-N-), 7,49-7,60 (m, 1H, Pir-H), 7,72 (d, 1H, J =
7,82 Hz, Pir-H), 8,71 (m, 1H, HNCH2-Pir). FABMS m/z (intensidade relativa):
384 ([M+H]+, 100), 324 (35), 297 (15). Massa exata (M+H): Real: 384,2512,
Medida: 384,2500. (3RS,4R)-4-{9-lsopropil-6-[(piridin-3-ilmetil)-amino1-9H-purin-2-ilamino}-2metil-hexan-3-ol
A uma solução agitada de (2-flúor-9-isopropil-9H-purin-6-il)-piridin-3-ilmetil-amina (30 mg, 1 eq, 0,10 mmol) em n-BuOH/DMSO (2,5 mL,
4:1), em temperatura ambiente, sob uma atmosfera de argônio, foi adicionado DIEA (0,10 mL, 5,5 eq, 0,57 mmol), seguido por (3RS,4F?)-4-amino-2metil-hexan-3-ol (42 mg, 3,0 eq, 0,32 mmol). A mistura de reação foi colocada em um banho de óleo pré-aquecido a 140°C e agitada nesta temperatura, 20 durante 72 h. A mistura de reação foi deixada resfriar a temperatura ambiente e o solvente foi evaporado em vácuo. O resíduo foi dividido entre EtOAc (50 mL) e salmoura/água (1:1, 100 mL), a fase aquosa foi extraída com mais EtOAc (2 χ 25 mL), e a fase orgânica combinada foi lavada com salmoura (50 mL), secada (MgSO4) e evaporada em vácuo. O resíduo foi purificado por cromatografia de coluna de gradiente em sílica-gel eluído com • · · · • · ·

CHCI3:MeOH (100:0 -> 98:2), para dar o composto do título como um sólido branco. Rendimento: 6,3 mg (15%). 50% de 4R,3S: 50% de 4R,3R). 1H-RMN (d-CDCI3, 250 MHz): δ 0,93-1,04 (m, 9H, -NHCH(CH2ÇH3) CH(CH(CH3)2) OH), 1,57 (d, 6H, J = 6,95 Hz, -CH(CH3)?), 1,65-1,89 (m, 4H, -NHCHÍCH?
CH3)CH(ÇH(CH3)2)OH), 3,22-3,32 (m, 1H, -NHÇH(CH2CH3)CH (CH(CH3)2) OH), 3,82-3,97 (m, 1H, -NHCH(CH2CH3) ÇH(CH(CH3)2)OH), 4,54-4,70 (m, 1H, -ÇH(CH3)2), 4,78-4,88 (m, 2H, -HNÇH2-Pir), 5,03-5,14 & 6,08-6,20 (m, 1H, -NHCH(CH2CH3)CH(CH2CH3)OH), 7,22-7,36 (m, 3H, 2 χ Pir-H & -N=CHN-), 7,49-7,58 (m, 1H, Pir-H), 7,71 (d, 1H, J = 7,90 Hz, Pir-H), 8,47-8,73 (m, 10 1H, HNCH?-Pir). FABMS m/z (intensidade relativa): 398 ([M+H]+, 100), 324 (50). Massa exata (M+H): Real: 398,2668, Medida: 398,2654.
(3RS,4S)-4-{9-lsopropil-6-[(piridin-3-ilmetil)-amino1-9H-purin-2-ilamino}-2metil-hexan-3-ol
A uma solução agitada de (2-flúor-9-isopropil-9/-/-purin-6-il)-piridin-3-ilmetil-amina (30 mg, 1 eq, 0,10 mmol) em n-BuOH/DMSO (2,5 mL,
4:1), em temperatura ambiente, sob uma atmosfera de argônio, foi adicionado DIEA (0,20 mL, 11 eq, 1,14 mmol), seguido por (3RS,4S)-4-amino-2metil-hexan-3-ol (28 mg, 2,0 eq, 0,21 mmol). A mistura de reação foi colocada em um banho de óleo pré-aquecido a 160°C e agitada nesta temperatura, durante 72 h. A mistura de reação foi deixada resfriar a temperatura ambiente e o solvente foi evaporado em vácuo. O resíduo foi dividido entre EtOAc (50 mL) e salmoura/água (1:1, 100 mL), a fase aquosa foi extraída com mais EtOAc (2 χ 25 mL), e a fase orgânica combinada foi lavada com salmoura (50 mL), secada (MgSO4) e evaporada em vácuo. O resíduo foi purificado por cromatografia de coluna de gradiente em sílica-gel eluído com • · · · · ·
CHCI3:MeOH (100:0 —> 98:2), para dar o composto do título como um sólido branco. Rendimento: 4,3 mg (10%). 50% de 4S,3R: 50% de 4S,3S). 1H-RMN (d-CDCIs, 250 MHz): δ 0,91-1,05 (m, 9H, -NHCH(CH?CH3) CH(CH(CH3)2)
OH), 1,57 (d, 6H, J = 6,95 Hz, -CH(CH3)2), 1,60-1,95 (m, 4H, -NHCH(CH2 5 CH3)CH(CH(CH3)?)OH), 3,23-3,33 (m, 1H, -NHÇH(CH2CH3)CH(CH (CH3)2)
OH), 3,84-4,04 (m, 1H, -NHCH(CH2CH3) ÇH(CH(CH3)2)OH), 4,56-4,70 (m, 1H, -ÇH(CH3)2), 4,77-4,95 (m, 2H, -HNÇH2-Pir), 5,26-5,43+6,27-6,50 (m, 1H, -NHCH(CH2CH3)CH(CH2CH3)OH), 7,23-7,37 (m, 3H, 2 x Pir-H & -N=CH-N-), 7,51-7,63 (m, 1H, Pir-H), 7,73 (d, 1H, J = 7,74 Hz, Pir-H), 8,48-8,76 (m, 1H, 10 HNCH?-Pir). FABMS m/z (intensidade relativa): 398 ([M+H]+, 100), 324 (60).
Massa exata (M+H): Real: 398,2668, Medida: 398,2654.
(3RS,4R)-4-{9-lsopropil-6-r(piridin-3-ilmetil)-amino1-9H-purin-2-ilamino)-2,2dimetil-hexan-3-ol
A uma solução agitada de (2-flúor-9-isopropil-9/-/-purin-6-il)-piri din-3-ilmetil-amina (30 mg, 1 eq, 0,10 mmol) em n-BuOH/DMSO (5 mL, 4:1), em temperatura ambiente, sob uma atmosfera de argônio, foi adicionado
DIEA (0,10 mL, 5,5 eq, 0,57 mmol), seguido por (3RS,4R)-4-amino-2,2dimetil-hexan-3-ol (52 mg, 3,41 eq, 0,36 mmol). A mistura de reação foi colocada em um banho de óleo pré-aquecido a 140°C e agitada nesta tempera20 tura, durante 72 h. A mistura de reação foi deixada resfriar a temperatura ambiente e o solvente foi evaporado em vácuo. O resíduo foi dividido entre
EtOAc (50 mL) e salmoura/água (1:1, 100 mL), a fase aquosa foi extraída com mais EtOAc (2 χ 25 mL), e a fase orgânica combinada foi lavada com salmoura (50 mL), secada (MgSO4) e evaporada em vácuo. O resíduo foi • · · • · · · · ·
purificado por cromatografia de coluna de gradiente em sílica-gel eluído com CHCI3:MeOH (100:0 -> 98:2), para dar o composto do título como um sólido branco. Rendimento: 7,3 mg (17%). (55% de 4R.3S: 45% de 4R,3R). 1HRMN (d-CDCI3, 250 MHz): δ 0,99-1,03 (m, 12H, -NHCH(CH9CH3) CH(C 5 (CH3)3)OH), 1,56 & 1,58 (2 χ d, 6H, J = 6,63 & 6,79 Hz, -CH(CH3)2), 1,691,91 (m, 2H, -NHCH(CH?CH3)CH(C(CH3)3)OH), 3,55 (d, 1H, J = 1,74 Hz, NHCH(CH2CH3)ÇH(C(CH3)3)OH), 3,76-3,87 (m, 1H, -NHÇH(CH2CH3)CH(C (CH3)3)OH), 4,57-4,70 (m, 1H, -ÇH(CH3)2), 4,80-4,89 (m, 2H, -HNCH2-Pir), 5,22-5,35 & 6,07-6,23 (m, 1H, -NHCH(CH2CH3)CH(C(CH3)3)OH), 7,24-7,36 10 (m, 3H, 2 χ Pir-H & -N=CH-N-), 7,55 (s, 1H, Pir-H), 7,74 (d, 1H, J = 7,58 Hz,
Pir-H), 8,50-8,74 (m, 1H, HNCH?-Pir). FABMS m/z (intensidade relativa): 412 ([M+H]+, 100), 324 (80). Massa exata (M+H): Real: 412,2825, Medida: 412,2835.
(3RS,4S)-4-{9-lsopropil-6-[(piridin-3-ilmetil)-aminol-9H-purin-2-ilamino}-2,215 dimetil-hexan-3-ol
A uma solução agitada de (3RS,4R)-4-{9-lsopropil-6-[(piridin-3ilmetil)-amino]-9H-purin-2-ilamino}-2,2-dimetil-hexan-3-ol (30 mg, 1 eq, 0,10 mmol) em n-BuOH/DMSO (5 mL, 4:1), em temperatura ambiente, sob uma atmosfera de argônio, foi adicionado DIEA (0,10 mL, 5,5 eq, 0,57 mmol), se20 guido por (3/?S,4S)-4-amino-2,2-dimetil-hexan-3-ol (43 mg, 2,81 eq, 0,29 mmol). A mistura de reação foi colocada em um banho de óleo pré-aquecido a 140°C e agitada nesta temperatura, durante 72 h. A mistura de reação foi deixada resfriar a temperatura ambiente e o solvente foi evaporado em vácuo. O resíduo foi dividido entre EtOAc (50 mL) e salmoura/água (1:1, 100 25 mL), A fase aquosa foi extraída com mais EtOAc (2 χ 25 mL), e a fase orgâ• · · • ·· ·· ·

nica combinada foi lavada com salmoura (50 mL), secada (MgSO4) e evaporada em vácuo. O resíduo foi purificado por cromatografia de coluna de gradiente em sílica-gel eluído com CHCI3:MeOH (100:0 -» 98:2), para dar o composto do título como um sólido branco. Rendimento: 5,4 mg (13%).(53% 5 de 4S,3R: 47% de 4S,3S). 1H-RMN (d-CDCI3, 250 MHz): <5 0,99-1,03 (m,
12H, -NHCH(CH?CH3)CH(C(CH3)3)OH), 1,57 & 1,59 (2 χ d, 6H, J = 6,79 &
6,79 Hz, -CH(CH3)2), 1,70-1,93 (m, 2H, -NHCH(CH2CH3) CH(C(CH3)3)OH),
3,55 (d, 1H, J = 2,05 Hz, -NHCH(CH2CH3)ÇH(C(CH3)3)OH), 3,77-3,90 (m,
1H, -NHÇH(CH2CH3)CH(C(CH3)3)OH), 4,58-4,70 (m, 1H, -ÇH(CH3)2), 4,8210 4,94 (m, 2H, -HNCH2-Pir), 5,31-5,45 & 6,12-6,34 (m, 1H, -NHCH (CH2CH3)CH (C(CH3)3)OH), 7,26-7,40 (m, 3H, 2 χ Pir-H & -N=CH-N-). 7,55 (s, 1H, Pir-H), 7,75 (d, 1H, J = 7,42 Hz, Pir-H), 8,46-8,81 (m, 1H, HNCH?-Pir). FABMS m/z (intensidade relativa): 412 ([M+H]+, 100), 324 (70). Massa exata (M+H): Real: 412,2825, Medida: 412,2835.
(3R)-3-{9-lsopropil-6-í(piridin-3-ilmetil)-amino1-9H-purin-2-ilamino}-2-metilpentan-2-ol
A uma solução agitada de (2-flúor-9-isopropil-9/7-purin-6-il)-piridin-3-ilmetil-amina (20 mg, 1 eq, 0,07 mmol) em n-BuOH/DMSO (1,25 mL, 4:1), em temperatura ambiente, sob uma atmosfera de argônio, foi adiciona20 do DIEA (0,25 mL, 20,5 eq, 1,44 mmol), seguido por (3R)-3-amino-2-metilpentan-2-ol (22 mg, 2,7 eq, 0,19 mmol). A mistura de reação foi colocada em um banho de óleo pré-aquecido a 140°C e agitada nesta temperatura, durante 72 h. A mistura de reação foi deixada resfriar a temperatura ambiente e o solvente foi evaporado em vácuo. O resíduo foi dividido entre EtOAc (50 25 mL) e salmoura/água (1:1, 100 mL), a fase aquosa foi extraída com mais
EtOAc (2 χ 25 mL), e a fase orgânica combinada foi lavada com salmoura (50 mL), secada (MgSO4) e evaporada em vácuo. O resíduo foi purificado por cromatografia de coluna de gradiente em sílica-gel eluído com
CHCI3:MeOH (100:0 -> 98:2), para dar o composto do título como um sólido 5 branco. Rendimento: 3,7 mg (14%). 1H-RMN (d-CDCI3, 250 MHz): <5 1,00 (t,
3H, J = 7,27 Hz, -NHCH(CH9CH3)C(CH3)9OH), 1,21 & 1,31 (2 χ s, 6H, NHCH(CH2CH3)C(ÇH3)2OH), 1,57 (d, 6H, J = 6,63 Hz, -CH(CH3)?), 1,69-1,89 (m, 2H, -NHCH(CH2CH3)C(CH3)?QH), 3,66-3,83 (m, 1H, -NHÇH(CH2CH3)
C(CH3)2OH), 4,59-4,73 (m, 1H, -ÇH(CH3)2), 4,77-5,00 (m, 2H, -HNCH?-Pir). 10 7,26-7,46 (m, 3H, 2 χ Pir-H & -N=ÇH-N-), 7,49-7,69 (m, 1H, Pir-H), 7,78 (d,
1H, J = 7,10 Hz, Pir-H). FABMS m/z (intensidade relativa): 384 ([M+H]+, 65), 366 (23), 324 (100), 217 (35), 192 (55). Massa exata (M+H): Real: 384,2512, Medida: 384,2494.
(3S)-3-{9-lsopropil-6-F(piridin-2-ilmetil)-amino]-9H-purin-3-ilamino}-2-metil15 pentan-2-ol
A uma solução agitada de (2-flúor-9-isopropil-9H-purin-6-il)-piridin-3-ilmetil-amina (30 mg, 1 eq, 0,10 mmol) em n-BuOH/DMSO (1,25 mL, 4:1), em temperatura ambiente, sob uma atmosfera de argônio, foi adicionado DIEA (0,25 mL, 14,4 eq, 1,44 mmol), seguido por (3S)-3-amino-2-metil20 pentan-2-ol (40 mg, 3,2 eq, 34 mmoles). A mistura de reação foi colocada em um banho de óleo pré-aquecido a 140°C e agitada nesta temperatura, durante 72 h. A mistura de reação foi deixada resfriar a temperatura ambiente e o solvente foi evaporado em vácuo. O resíduo foi dividido entre EtOAc (50 mL) e salmoura/água (1:1, 100 mL), a fase aquosa foi extraída com mais 25 EtOAc (2 χ 25 mL), e a fase orgânica combinada foi lavada com salmoura

(50 mL), secada (MgSO4) e evaporada em vácuo. O resíduo foi purificado por cromatografia de coluna de gradiente em sílica-gel eluído com CHCI3:MeOH (100:0 -> 98:2), para dar o composto do título como um sólido branco. Rendimento: 6,5 mg (16%). 1H-RMN (d-CDCI3, 250 MHz): δ 1,00 (t,
3H, J = 7,42 Hz, -NHCH(CH2CH3)C(CH3)7OH), 1,22 & 1,31 (2 χ s, 6H, NHCH(CH2CH3)C(CH3)2OH), 1,57 (d, 6H, J = 6,79 Hz, -CH(CH3)2), 1,70-1,90 (m, 2H, -NHCH(ÇH2CH3)C(CH3)2OH), 3,66-3,81 (m, 1H, -NHÇH(CH2CH3)
C(CH3)2OH), 4,56-4,73 (m, 1H, -ÇH(CH3)2), 4,78-5,01 (m, 2H, -HNCH2-Pir), 7,20-7,45 (m, 3H, 2 χ Pir-H & -N=CH-N-). 7,48-7,69 (m, 1H, Pir-H), 7,77 (d, 10 1H, J = 7,74 Hz, Pir-H). FABMS m/z (intensidade relativa): 384 ([M+H]+,
100), 366 (30), 324 (85), 286 (40), 242 (65), 192 (63), 176 (70). Massa exata (M+H): Real: 384,2512, Medida: 384,2494.
Para a preparação em larga escala dos enantiômeros individuais de 3-{9-isopropil-6-[(piridin-3-ilmetil)-amino]-9/-/-purin-2-ilamino}-2-metil-pen15 tan-2-ol 24 (esquema 3 abaixo), um procedimento diferente foi adotado. Dicloropurina 14 foi aminada a C-6 com piridilmetilamina 15 para proporcionar o intermediário 16, que foi então alquilado a N-9 com 2-bromopropano 17, A cloropurina resultante 18 foi aminada a C-2 com o enantiômero apropriado de 3-amino-2-metil-pentan-2-ol 23.
• · · · · ·
Esquema 3
Foi previamente demonstrado que o F?-3-amino-2-metil-pentan2-ol 23 oticamente puro pode ser obtido diretamente por conversão de ácido
R-2-amino-butírico 19 para o éster metílico 20, seguido por dupla metilação de Grignard (Guenther, B.R.; Kirmse, W. Liebigs Ann. Chem., 1980, 518).
Este método não foi apropriado para trabalho em escala, em termos de rendimento e pureza do produto. A proteção do grupo amino como o derivado dibenzila (21, Bn = benzila) foi assim adotada. A metilação do éster amino 21 forneceu o álcool 22 com elevado rendimento e pureza. Os grupos ben zila são então removidos hidrogenoliticamente para proporcionar os enan10 tiômeros puros de 3-amino-2-metil-pentan-2-ol 23, (2-Cloro-9H-purin-6-il)-piridin-3-ilmetil-amina
2,6-Dicloro-9H-purina (157,2 g, 0,83 mol), butan-1-ol (1,2 L), e
Et3N (290 mL, 2,08 moles) foram agitados e aquecidos a 50°C. 3-C-Piridin-3il-metilamina (94 mL, 0,76 mol) foi adicionada e a reação aquecida a 120°C durante 1 h. Aquecimento foi cessado e o frasco deixado resfriar para a temperatura ambiente, em seguida para 0°C, usando um banho de água gelada.
O precipitado de cor de tom pêssego foi coletado por filtração, lavado com H2O (1 L), hexano (2 x 0,5 L) e secado, para dar um sólido de cor pêssego. Uma segunda coleta foi obtida por evaporação do filtrado combinado e lava10 gens, trituração com H2O (100 mL) e CHCI3 (100 ml), filtração, lavagem com hexano e secagem. As coletas foram combinadas para dar o composto do título (196 g, 91%). 1H-RMN (DMSO-d6): δ 4,67-4,69 (2H, d, -CH2-NH-), 5,20
(1H, br. s, NH), 7,34-7,39 (dd, ArH), 7,76-7,79 (1H, d, ArH), 8,17 (1H, s, ArH -N=CH-N-), 8,46-8,48 (1H, d, ArH), 8,61 (1H, s, ArH), 8,80 (1H, br. s, NH). (2-Cloro-9-isopropil-9H-purin-6-il)-piridin-3-ilmetil-amina
(2-Cloro-9H-purin-6-il)-piridin-3-ilmetil-amina (110,3 g, 0,37 mol) foi dissolvida em DMF seco (1,5 L) e aquecida a 70°C, com agitação. K2CO3 to

(230 g, 1,67 mol) e 2-bromopropano (300 mL, 3,20 moles) foram adicionados e a reação foi agitada a 70°C, durante a noite. A mistura de reação foi deixada resfriar para a temperatura ambiente e K2CO3 foi removido por filtração e lavagem com EtOAc. O filtrado combinado e lavagens foram concentrados em um evaporador giratório e, em seguida, sob vácuo elevado, para remover a maior parte do DMF. O resíduo resultante foi agitado com vigor em H2O (1 L) e CH2CI2 (1 L) até a dissolução estar completa. A camada aquosa foi extraída com mais CH2CI2 (2 X 200 mL), os extratos orgânicos foram combinados, secados sobre MgSO4, e evaporados para dar o composto do título (87,2 g, 78%), como um pó bege. 1H-RMN (DMSO-d6): <5 1,50-1,53 (6H, d, CH(CH3)2), 4,67-4,72 (3H, m, -CH2-NH- & □CH(CH3)2), 5,20 (1H, br. s, NH), 7,34-7,39 (dd, ArHX 7,76-7,79 (1H, d, ArHl 8,33 (1H, s, ArH, -N=CH-N-). 8,46-8,48 (1H, d, ArHX 8,61 (1H, s, ArHl 8,92 (1H, br. s, NH).
Cloridrato de éster metílico de ácido R-2-Amino-butírico
o
Ácido (D)-(-)-2-Aminobutírico (50,0 g, 0,48 mol) foi colocado em suspensão em MeOH (270 mL) e resfriado para -10°C (banho de gelo-sal), com agitação, sob N2. SOCI2 (64 mL, 0,864 mol) foi adicionado, gota a gota, durante 90 min. O frasco foi equipado com um condensador de refluxo e aquecido para 70°C, durante 1 h, então resfriado e evaporado (co-evaporando com MeOH). O resíduo foi separado e secado sob vácuo elevado, para dar o composto do título como um pó branco (75 g, 100%). 1H-RMN (CDCI3): δ 0,88-0,95 (3H, t, CH3CH2-), 1,48 (2H, br. s, NH2), 1,50-1,80 (2H, m, CH3CH2-), 3,35-3,40 (1H, t, CHNH2), 3,69 (3H, s, OCH3).
Éster metílico de ácido R-2-Dibenzilamino-butírico

A cloridrato de éster metílico de ácido R-2-amino-butírico (74,5 g, 0,485 mol) em DMF seco (500 mL), sob N2, em temperatura ambiente, foi adicionado K2CC>3 (147 g, 1,065 mol), seguido por adição, gota a gota, de brometo de benzila (127 mL, 1,065 mol.). Após 2 h de agitação, K2CO3 foi filtrado e lavado (EtOAc, 200 mL). O filtrado combinado e lavagens foram evaporados em vácuo. O resíduo oleoso foi dividido entre EtOAc (1,5 L) e H2O (1,5 L). A camada orgânica foi separada e a camada aquosa foi extraída com mais EtOAc (1 L). As frações orgânicas combinadas foram concentradas. O resíduo foi purificado por cromatografia flash em sílica-gel seca (3:7 CH2CI2/hexano). As frações foram combinadas, concentradas em vácuo, e secadas para dar o composto do título (136,1 g, 94%). 1H-RMN (CDCI3): □ 0,90-0,96 (3H, t, CH3CH2-), 1,75-1,81 (2H, m, CH3-CH2-), 3,23-3,29 (1H, t, CH2-CH-), 3,50-3,57 (2H, d, -NCH2Ph), 3,77 (3H, s, -OCH3), 3,93-3,99 (2H, d, -NCH2Ph), 7,22-7,41 (10H, m, ArH).
R-3-Dibenzilamino-2-metil-pentan-2-ol

A um frasco de 3 gargalos de 5L, seco, equipado com um condensador e borbulhador de N2, foram carregadas aparas de Mg (53,7 g, 2,22 moles) e Et2O seco (2 L). A mistura foi agitada e resfriada para 0°C, usando um banho de gelo. Mel (138,2 mL, 2,22 moles) foi então adicionado, gota a gota, durante 1 h. Éster metílico de ácido F?-2-Dibenzilamino-butírico em Et2O seco (1 L) foi então adicionado, lentamente (durante 10 min), e a reação foi deixada aquecer para a temperatura ambiente, se tornando muito espessa. A reação foi deixada em agitação durante 72 h, resfriada para 0°C e extinta por adição lenta de solução de NH4CI aquosa saturada (350 g em 1 L). Após 30 min de agitação, a mistura de reação foi filtrada através de uma almofada de Celite e lavada com Et2O (3 X 0,5 L). A camada etérea foi separada e a camada aquosa extraída com mais Et2O (1 L). As frações orgânicas foram combinadas, secadas sobre MgSO4 e concentradas. O resíduo foi purificado por cromatografia flash em sílica-gel (2% EtOAc em hexano), para • · · • · · · • · · · • · · • · ·
|
• ··
• |
• ·· |
···
• |
|
• |
• |
• |
|
• |
• |
• |
|
• · |
• |
• · |
|
• |
·· · |
• |
|
• |
··· |
··· |
|
• |
• |
• |
|
• · |
• |
• |
|
·· · |
• |
• |
|
• |
• |
• |
|
• |
• |
• |
dar o composto do título (88,4 g, 82%) como um óleo incolor. 1H-RMN (CDCh): δ 1,06 (3H, s, -CCH3), 1,15-1,26 (6H, m (3H s & 3H m, -CCH3 & CH2CH3), 1,50-1,70 (1H, m, -CHHCH3), 1,70-1,90 (1H, m, -CHHCH3), 2,592,64 (1H, dd, -CHN-), 3,61-3,67 (2H, d, -NCH2Ph), 3,97-4,02 (2H, d, 5 NCH2Ph), 4,25 (1H, br. s, -OH), 7,24-7,37 (10 H, m, ArH).
R-3-Amino-2-metil-pentan-2-ol
R-3-Dibenzilamino-2-metil-pentan-2-ol (21,0 g, 70,9 mmol) e
Pd(C) (2,1 g, 20% de Pd em peso seco, 50% de H2O) em MeOH (100 mL) foram agitados em um hidrogenador Parr, a 4,5 kg/cm2 (65 psi) durante a noite. O catalisador foi removido por filtração através de uma almofada de
Celite, o filtrado concentrado e secado sob vácuo elevado, para dar o composto do título como um óleo amarelo-claro (7,585 g, 91%). 1H-RMN (CDCI3): δ 0,90-1,05 (6H, m (3H s & 3H m), -CCH3 & -CH2CH3), 1,18 (3H, s, CCH3), 1,55-1,62 (2H, m, -CH2CH3), 1,88-2,25 (2H, br. s, -NH2), 2,35-2,40 (1H, dd,-CHN-).
(3R)-3-{9-lsopropil-6-r(piridin-3-ilmetil)-amino1-9H-purin-2-ilamino}-2-metilpentan-2-ol
(2-Cloro-9-isopropil-9/7-purin-6-il)-piridin-3-ilmetil-amina (24,4 g,
82,4 mmoles), R-3-amino-2-metil-pentan-2-ol (24,4 g, 209 mmoles), e K2CO3 (24,4 g, 177 mmoles) foram aquecidos a 150°C, durante 72 h, sob N2. A mistura foi resfriada, diluída com CH2CI2, e K2CO3 removido por filtração. O filtrado foi evaporado, em seguida destilado (Kugelrohr) sob vácuo elevado,

| |
para remover R-3-amino-2-metil-pentan-2-ol não-reagido. O resíduo foi submetido a dois ciclos de cromatografia flash em sílica-gel (primeira coluna eluindo com 5 a 10% de gradiente linear de MeOH em CH2CI2; segunda co- |
| |
luna eluindo com 2,5% de MeOH em CH2CI2). O resíduo foi dissolvido em |
|
5 |
EtOAc quente (150 mL), a solução concentrada a um volume de aproximadamente 100 mL e deixada resfriar. Et2O (10 mL) foi adicionado para induzir a cristalização e a solução foi resfriada para 0°C. Os cristais foram coletados por filtração, lavados com um pouco de Et2O e secados para proporcionar 0 composto do título puro (5,70 g, 18%) como um sólido de branco indefinido. |
|
• 10 |
O filtrado combinado e as lavagens foram concentradas, 0 resíduo foi redissolvido em um volume mínimo de Me2CO quente e deixado resfriar. O produto sólido foi filtrado e redissolvido em um volume mínimo de EtOAc quente e deixado resfriar. Os sólidos formados foram removidos por filtração e 0 filtrado foi novamente concentrado até secura, redissolvido em uma mistura |
|
. 15 |
de Me2CO quente e EtOAc, resfriado, filtrado e secado, para dar uma segunda coleta de composto do título puro (2,5 g, 5,6 mmoles, 7%) como um sólido de branco indefinido. Ponto de fusão 138-145 °C. [a]D 20 +32,54 (c 0,48, MeOH). LC-EM: 97,86% puro, m/z 384 [M + H]+, 406 [M + Na]+, C20H29N7O = 383,49, Microanálise (% encontrado (teoria)): C, 62,41 (62,64); |
|
20
• |
H, 7,56 (7,62); N, 25,76 (25,57). 1H-RMN (CDCI3): δ 0,93-0,99 (3H, t, CH2CH3), 1,17 & 1,27 (6H, 2s, -C(CH3)2OH), 1,51-1,54 (6 H, d, -NCH(CH3)2),
I, 65-1,80 (2H, m, -CH2CH3), 2,15 (1H, br. s, NH ou OH), 3,60-3,67 (m, 1H, NHCH(CH2CH3)-), 4,57-4,62 (1H, m, -NCH(CH3)2), 4,77-4,80 (2H, m, HNCH2-Pir), 5,15 (br. s, 1H, NH ou OH), 6,14 (1H, br. s, NH ou OH), 7,21- |
|
25 |
7,26 (1H, m, Pry-H), 7,48 (1H, s, -N=CH-N-), 7,68-7,71 (app. d, 1H, Pir-H), 8,51-8,52 (app. d, 1H, Pir-H), 8,64 (1H, app. s, Pir-H). 13C-RMN (CDCI3): consistente com estrutura contendo 20 átomos C; 10 C alifático a δ 10,21, 20,88, 20,92, 21,90, 23,09, 26,64, 40,29 (fraco), 44,72, 61,70 & 72,45 ppm; e 10 C aromático a 112,96, 121,73, 132,97, 133,07, 133,65, 146,98, 147,67, |
|
30 |
148,89 (fraco), 152,97 & 158,77 ppm. |

(3S)-3-(9-lsopropil-6-Kpiridin-3-ilmetil)-amino]-9H-purin-2-ilamino}-2-metil pentan-2-ol

Este composto foi obtido usando procedimentos análogos aos descritos acima para o enantiômero (/?), mas partindo com ácido (L)-(+)-2aminobutírico. Ponto de fusão: 140,5-146 °C. [a]D 20 -29,00 (c 0,46, MeOH). LC-EM: 97,85% puro, m/z 384 [M + H]+, C20H29N7O = 383,49, Microanálise (% encontrado (teoria)): C, 62,51 (62,64); H, 7,61 (7,62); N, 25,70 (25,57). 1H-RMN: 5 0,93-0,99 (3H, t, -CH2CH3), 1,17 & 1,27 (6H, 2s, -C(CH3)2OH), 1,51-1,54 (6 H, d, -NCH(CH3)2), 1,65-1,80 (2H, m, -CH2CH3), 2,15 (1H, br. s, NH ou OH), 3,60-3,67 (m, 1H, -NHCH(CH2CH3)-), 4,57-4,62 (1H, m, NCH(CH3)2), 4,77-4,80 (2H, m, -HNCH2-Pir), 5,15 (br. s, 1H, NH ou OH), 6,14 (1H, br. s, NH ou OH), 7,21-7,26 (1H, m, Pry-H), 7,48 (1H, s, -N=CH-N), 7,68-7,71 (app. d, 1H, Pir-H), 8,51-8,52 (app. d, 1H, Pir-H), 8,64 (1H, app. s, Pir-H). 13C-RMN (CDCI3): consistente com estrutura contendo 20 átomos C, 10 C alifático a 5 10,21, 20,88, 20,92, 21,90, 23,09, 26,64, 40,27 (fraco), 44,71, 61,70 & 72,45 ppm; e 10 C aromático a 112,95, 121,72, 132,96, 133,07, 133,65, 146,85, 146,98, 148,86 (fraco), 152,97 & 158,77 ppm.
Várias modificações e variações da invenção serão evidentes para os versados na técnica sem sair do escopo e espírito da invenção. Apesar da invenção ter sido descrita em conexão com modalidades preferidas específicas, deve-se entender que a invenção, como reivindicada, não deve ser indevidamente limitada a estas modalidades específicas. De fato, várias modificações dos modos descritos da realização da invenção, que são óbvias para os versados nos campos relevantes, são planejadas para serem cobertas pela presente invenção.
Tabela 1
• ·· ·· ·
|
Nome |
Inibição de quinase (μΜ) |
|
CDK2/ciclina E |
CDK 1/ciclina B |
CDK4/ciclin a D1 |
CDK7/ciclina H |
PKA |
ERK2 |
|
IC50 |
SD |
Ιθ50 |
SD |
IC50 |
SD |
IC50 |
SD |
IC50 |
IC5o |
SD |
|
Roscovitina |
0,10 |
0,10 |
2,7 |
2,5 |
14 |
4 |
0,49 |
0,26 |
>50 |
1,2 |
1,3 |
|
(2S3R)-3-{9lsopropil-6[(piridin-3ilmetil)amino]-9Hpurin-2ilamino}pentan-2-ol |
0,09 |
0,02 |
4,9 |
0,3 |
14 |
4 |
0,50 |
0,40 |
>200 |
23 |
12 |
|
(2R3S)-3-{9lsopropil-6[(piridin-3ilmetil)amino]-9Hpurin-2ilamino}pentan-2-ol |
0,03 |
0,01 |
2,2 |
1,2 |
6,8 |
3,2 |
1,3 |
1,0 |
>200 |
20 |
6 |
|
(3RS,4R)-4{9-lsopropil-6[(piridin-3ilmetil)amino]-9Hpurin-2ilamino}hexan-3-ol |
0,22 |
0,10 |
12 |
1 |
25 |
0 |
1,1 |
0,3 |
>200 |
37 |
6 |
|
(3RS,4S)-4{9-lsopropil-6[(piridin-3ilmetil)amino]-9Hpurin-2ilamino}hexan-3-ol |
0,11 |
0,04 |
13 |
4 |
11 |
1 |
1,5 |
0,6 |
>200 |
17 |
8 |



|
Nome |
Inibição de quinase (μΜ) |
|
CDK2/ciclin aE |
CDK 1/ciclina B |
CDK4/ciclin a D1 |
CDK7/ciclina H |
PKA |
ERK2 |
|
IC50 |
SD |
IC50 |
SD |
IC50 |
SD |
IC50 |
SD |
IC5O |
IC50 |
SD |
|
(3RS,4R)-4-{9lsopropil-6[(piridin-3ilmetil)-amino]9H-purin-2ilamino}-2-metilhexan-3-ol |
0,69 |
0,30 |
20 |
8 |
21 |
15 |
2,2 |
1,4 |
200 |
111 |
11 |
|
(3F?S,4S)-4-{9lsopropil-6[(piridin-3ilmetil)-amino]9H-purin-2ilamino}-2-metilhexan-3-ol |
1,1 |
0,6 |
22 |
12 |
31 |
9 |
3,5 |
0,4 |
>200 |
67 |
6 |
|
(3RS,4R)-4-{9lsopropil-6[(piridin-3ilmetil)-amino]9H-purin-2ilamino}-2,2dimetil-hexan-3ol |
1,0 |
0,0 |
23 |
8 |
24 |
3 |
1,6 |
0,1 |
>200 |
51 |
8 |
|
(3RS,4S)-4-{9lsopropil-6[(piridin-3ilmetil)-amino]9H-purin-2ilamino}-2,2dimetil-hexan-3ol |
0,39 |
0,27 |
21 |
2 |
13 |
0 |
3,5 |
0,6 |
>200 |
23 |
4 |
|
(3R)-3-{9lsopropil-6[(piridin-3ilmetil)-amino]9H-purin-2ilamino}-2-metilpentan-2-ol |
0,20 |
0,08 |
6,2 |
1,1 |
7,5 |
5 |
3,1 |
0,4 |
>200 |
91 |
21 |
|
(3S)-3-{9lsopropil-6[(piridin-3ilmetil)-amino]9H-purin-2ilamino}-2-metilpentan-2-ol |
0,07 |
0,01 |
4,4 |
1,9 |
12 |
1 |
2,5 |
1,5 |
>200 |
35 |
10 |


|
78 : |
• · ·
• · · ·
• · · ·
• · ·
• · ·
• · |
··· · · · · · • ·· · ·
• · · · ·
• · · ·· ·
• · · · ♦ · • · · · · · |
|
Tabela 2 |
|
|
|
Nome |
Atividade In vitro antiproliferativa (72-h MTT IC50, μΜ) |
| |
IC50 a |
Desvio padrão |
|
Roscovitina |
12 |
3 |
|
(2S3R)-3-{9-lsopropil-6-[(piridin-3-ilmetil)-amino]-9H-
purin-2-ilamino}-pentan-2-ol |
1,5 |
0,4 |
|
(2R3S)-3-(9-lsopropil-6-[(piridin-3-ilmetil)-amino]-9H-
purin-2-ilamino}-pentan-2-ol |
0,8 |
0,3 |
|
(3RS,4R)-4-{9-lsopropil-6-[(piridin-3-ilmetil)-amino]-9H-
purin-2-ilamino}-hexan-3-ol |
10 |
1 |
|
(3RS,4S)-4-{9-lsopropil-6-[(piridin-3-ilmetil)-amino]-9H-
purin-2-ilamino}-hexan-3-ol |
5,5 |
2,8 |
|
(3RS,4/?)-4-{9-lsopropil-6-[(piridin-3-ilmetil)-amino]-9H-
purin-2-ilamino}-2-metil-hexan-3-ol |
28 |
10 |
|
(3/?S,4S)-4-{9-lsopropil-6-[(piridin-3-ilmetil)-amino]-9H-
purin-2-ilamino}-2-metil-hexan-3-ol |
19 |
2 |
|
(3RS,4R)-4-{9-lsopropil-6-[(piridin-3-ilmetil)-amino]-9Hpurin-2-ilamino}-2,2-dimetil-hexan-3-ol |
19 |
2 |
|
(3/?S,4S)-4-{9-lsopropil-6-[(piridin-3-ilmetil)-amino]-9Hpurin-2-ilamino}-2,2-dimetil-hexan-3-ol |
15 |
14 |
|
(3R)-3-{9-lsopropil-6-[(piridin-3-ilmetil)-amino]-9H-purin-
2-ilamino)-2-metil-pentan-2-ol |
5,6 |
2,1 |
|
(3S)-3-{9-lsopropil-6-[(piridin-3-ilmetil)-amino]-9H-purin-
3-ilamino}-2-metil-pentan-2-ol |
4,0 |
0,5 |
a Linhagens de células de tumor humano: A549, HT29, Saos-2
• · · · · ·
Tabela 3
|
Linhagem de células |
72-h MTT IC50 (μΜ) |
|
Código |
Tipo |
Fonte |
Roscovitina |
(2S3R)-3{9-lsopropil6-[(piridin-3ilmetil)amino]-9Hpurin-2ilamino}pentan-2-ol |
(2R3S)-3{9-lsopropil6-[(piridin-3ilmetil)amino]-9Hpurin-2ilamino}pentan-2-ol |
(3S)-3-{9lsopropil-6[(piridin-3ilmetil)-amino]9H-purin-3ilamino}-2metil-pentan-2ol |
|
A549 |
Aderente |
Carcinoma de pulmão |
9,7 ± 1,1 |
2,1 ±0,4 |
0,9 ±0,1 |
3,6 ±0,2 |
|
HeLa |
Aderente |
Adenocarcinoma cervical |
16,5 ±
1,8 |
2,9 ±0,2 |
1,4 ± 0,2 |
4,7 ±0,5 |
|
HT-29 |
Aderente |
adenocarcinoma coloretal |
10,2 ±
2,7 |
2,3 ±0,6 |
1,1 ±0,2 |
2,3 ±0,9 |
|
MCF7 |
Aderente |
Adenocarcinoma mamário |
8,7 ± 0,5 |
1,6 ±0,2 |
0,7 ±0,1 |
2,3 ±0,3 |
|
Saos-2 |
Aderente |
Osteosarcoma |
15,3 ±
4,2 |
2,8 ±0,1 |
1,2 ±0,2 |
3,9 ±0,8 |
|
CCRFCEM |
Suspensão |
Leucemia linfoblástica aguda |
16,4 ±
0,8 |
n.d. |
n.d. |
3,7 ±0,3 |
|
HL-60 |
Suspensão |
Leucemia promielocítica aguda |
13,3 ±
1,1 |
n.d. |
n.d. |
3,0 ±0,2 |
|
K-562 |
Suspensão |
Leucemia mielogenosa crônica |
40,4 ±
4,4 |
n.d. |
n.d. |
8,4 ± 1,2 |
|
Média (linhagens de células de câncer |
16,3 ±
10,2 |
2,3 ±0,5 |
1,1 ±0,3 |
4,0 ±2,0 |
|
Hs27 |
Normal |
Fibroblasto de prepúcio |
>20 |
10,2 ±0,6 |
5,9 ± 0,3 |
11,3 ±0,7 |
|
IMR90 |
Normal |
Fibroblasto de pulmão de embrião |
>20 |
>20 |
>20 |
> 20 |
|
WI38 |
Normal |
Fibroblasto de pulmão fetal |
>20 |
11,2 ± 1,2 |
6,1 ±0,3 |
> 20 |
|
Média (linhagens de células normais) |
>20 |
> 10 |
> 10 |
> 10 |


• · · • · · · · ·
Tabela 4
|
Nome |
ClogP |
% Droga após 30min. incubação microssômica |
A:% composto resta nte/% roscovitina restante |
B: IC50
CDK2 roscovitina/ICso CDK2 composto |
C: IC50 citotox. roscovitina/ICso citotox. composto |
Αχ B |
AxC |
|
Roscovitina |
3,7 |
33 |
1,0 |
1,0 |
1,0 |
1,0 |
1,0 |
|
(2S3R)-3-{9-lsopropil-
6-[(piridin-3-ilmetil)amino]-9H-purin-2ilamino}-pentan-2-ol |
2,5 |
90 |
2,7 |
1,1 |
8,0 |
3,1 |
22 |
|
(2R3S)-3-{9-lsopropil-
6-[(piridin-3-ilmetil)amino]-9H-purin-2ilamino}-pentan-2-ol |
2,5 |
67 |
2,0 |
3,7 |
14 |
7,5 |
29 |
|
(3RS,4R)-4-{9lsopropil-6-[(piridin-3ilmetil)-amino]-9Hpurin-2-ilamino}hexan-3-ol |
3,0 |
70 |
2,1 |
0,5 |
1,1 |
1,0 |
2,4 |
|
(3RS,4S)-4-{9lsopropil-6-[(piridin-3ilmetil)-amino]-9Hpurin-2-ilamino}hexan-3-ol |
3,0 |
73 |
2,2 |
1,0 |
2,1 |
2,1 |
4,7 |
|
(3RS,4S)-4-{9lsopropil-6-[(piridin-3ilmetil)-amino]-9Hpu rin-2-i Iam i no}-2,2dimetil-hexan-3-ol |
3,8 |
50 |
1,5 |
0,3 |
0,8 |
0,4 |
1,2 |
|
(3R)-3-{9-lsopropil-6[(piridin-3-ilmetil)amino]-9H-purin-2ilamino}-2-metilpentan-2-ol |
2,9 |
84 |
2,6 |
0,5 |
2,1 |
1,3 |
5,3 |
|
(3S)-3-{9-lsopropil-6[(piridin-3-ilmetil)amino]-9H-purin-3ilamino}-2-metilpentan-2-ol |
2,9 |
78 |
2,4 |
1,4 |
2,9 |
3,2 |
6,9 |
REIVINDICAÇÕES
1. Composto, caracterizado pelo fato de que apresenta a fórmula