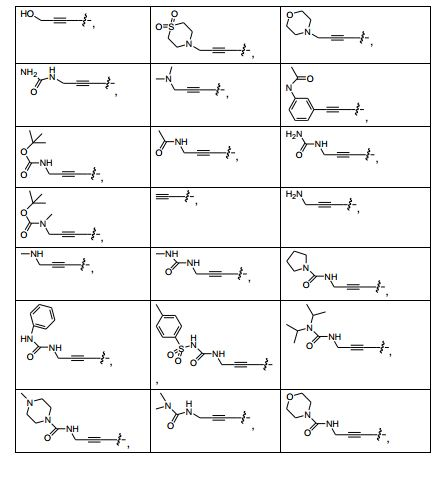
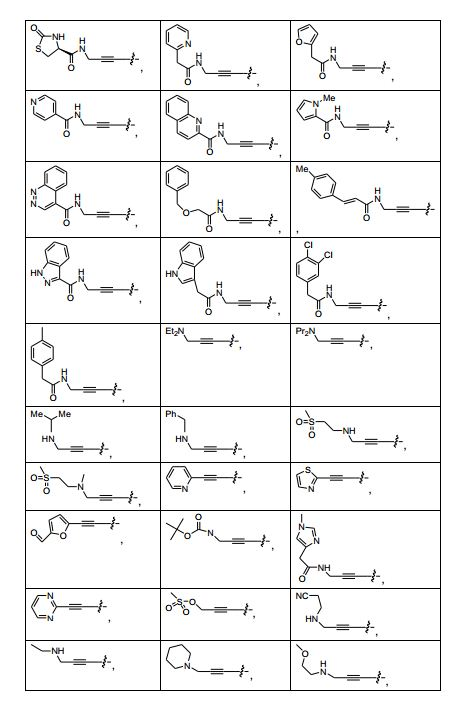
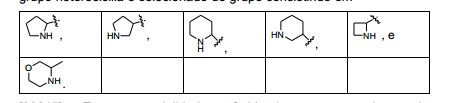
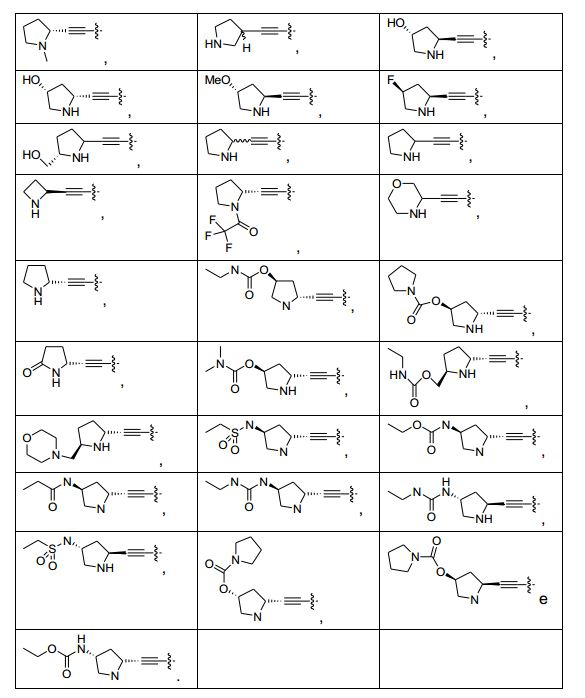
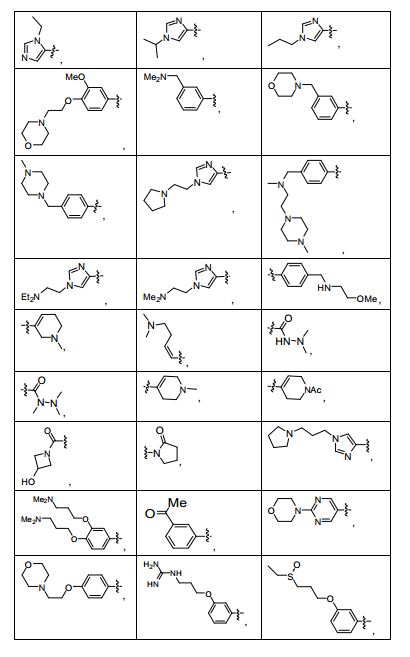
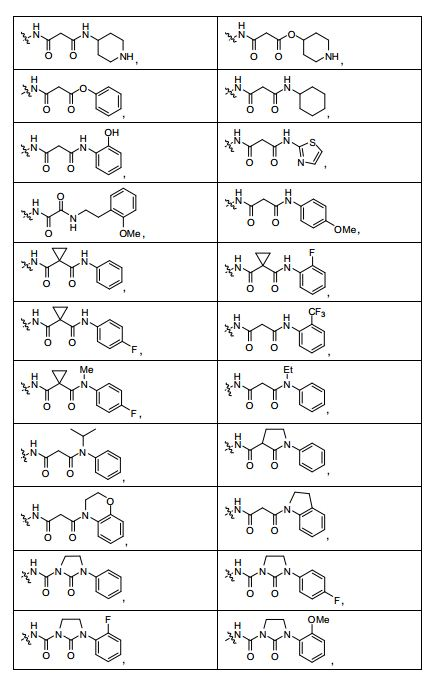
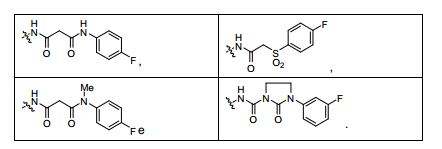
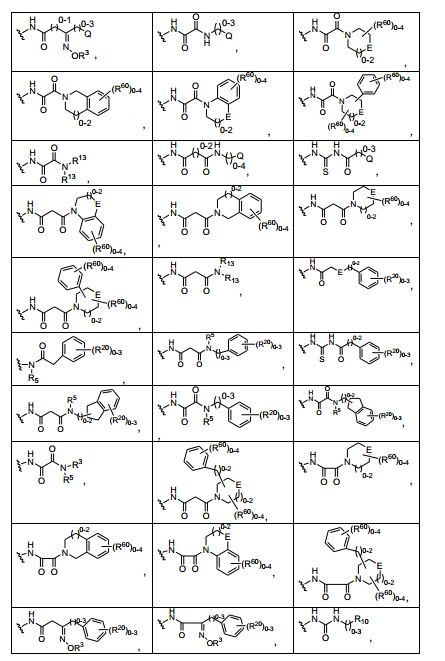
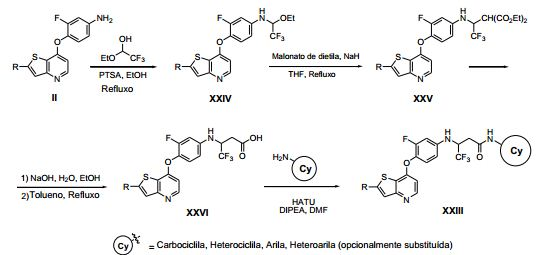
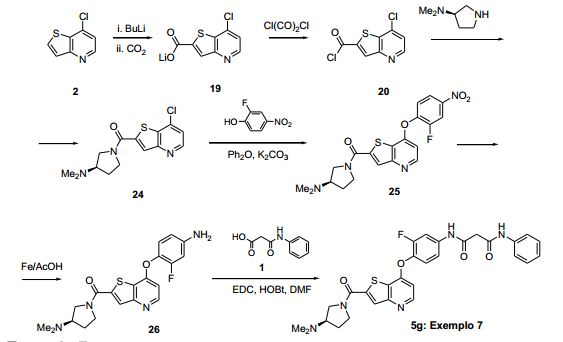
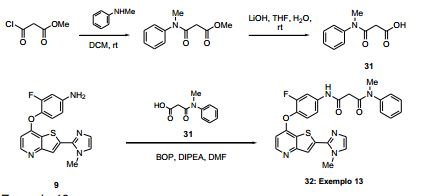
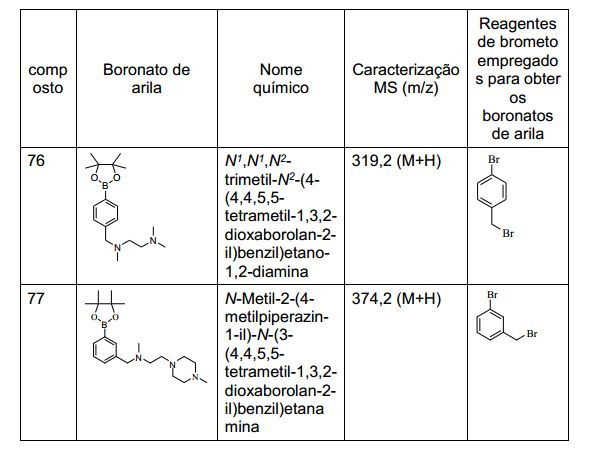
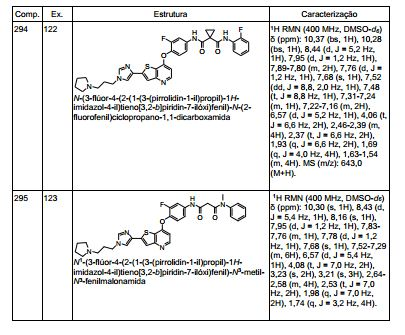
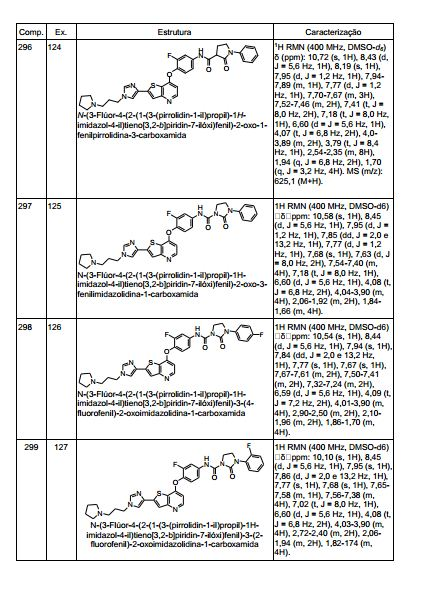
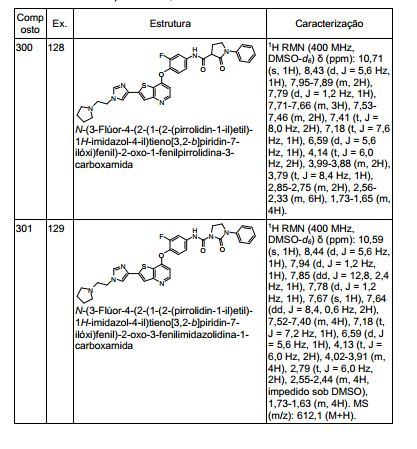
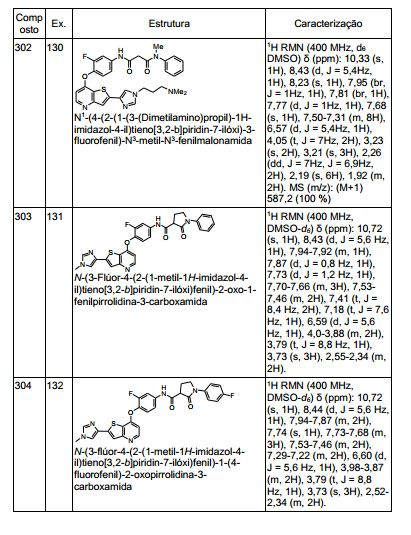
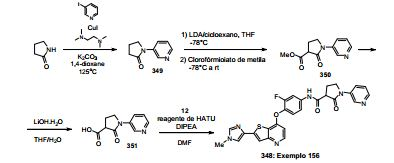
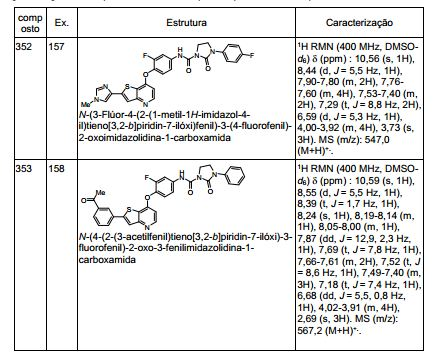
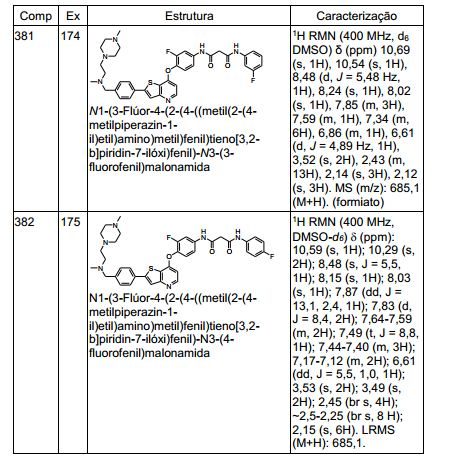
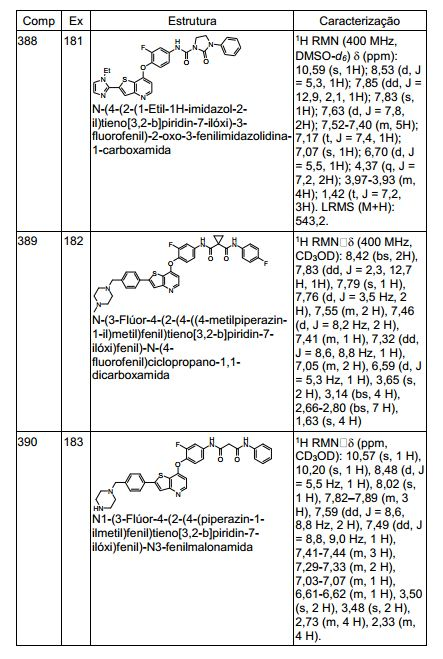
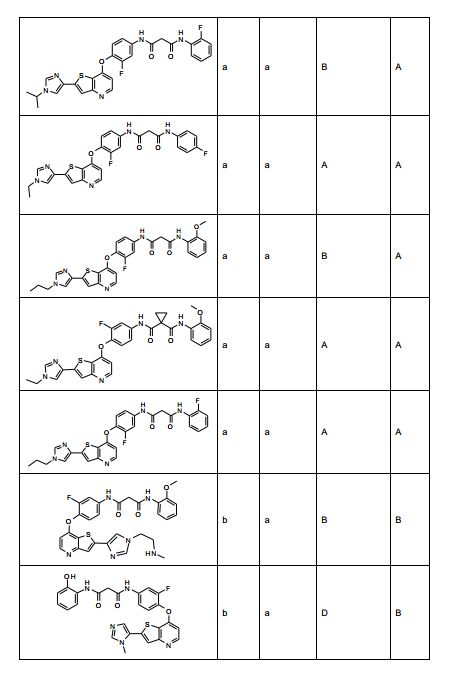
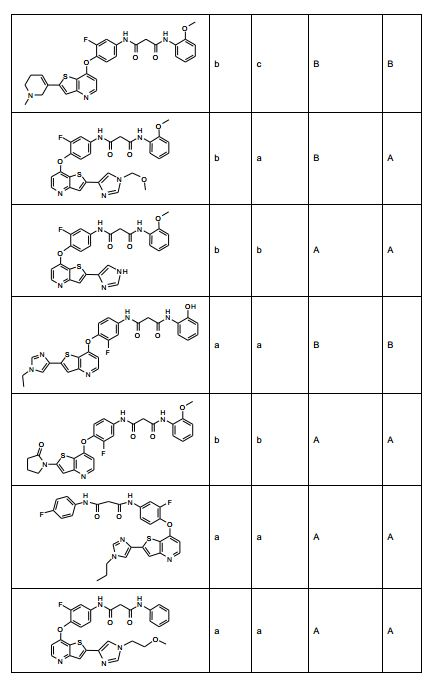
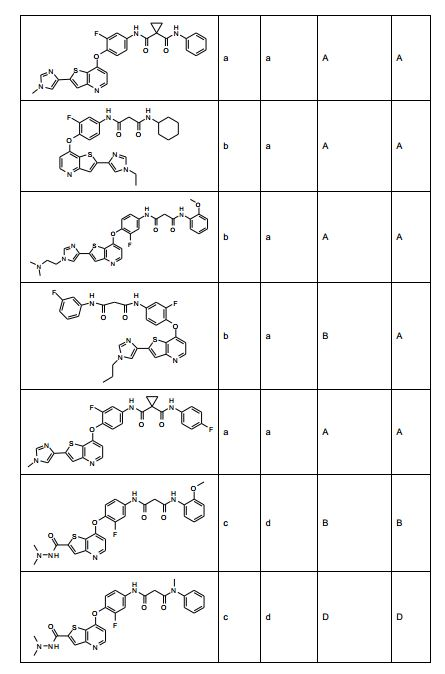
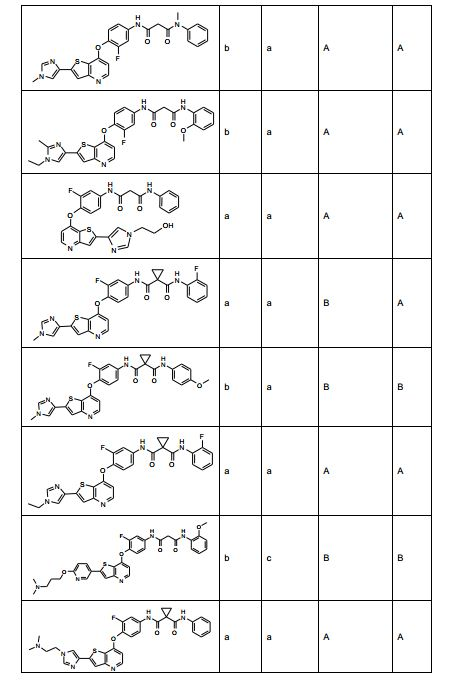
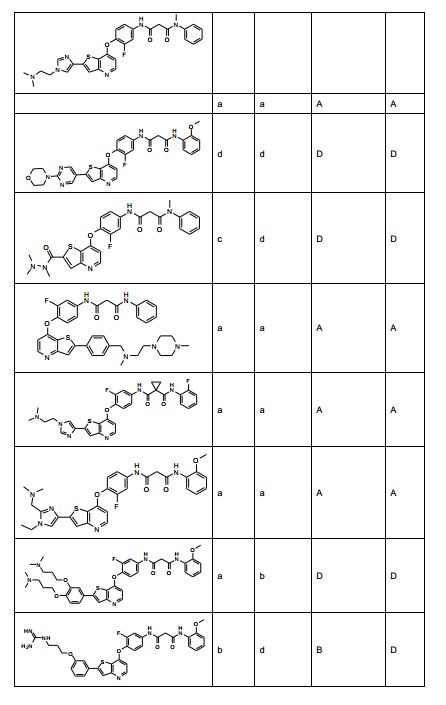
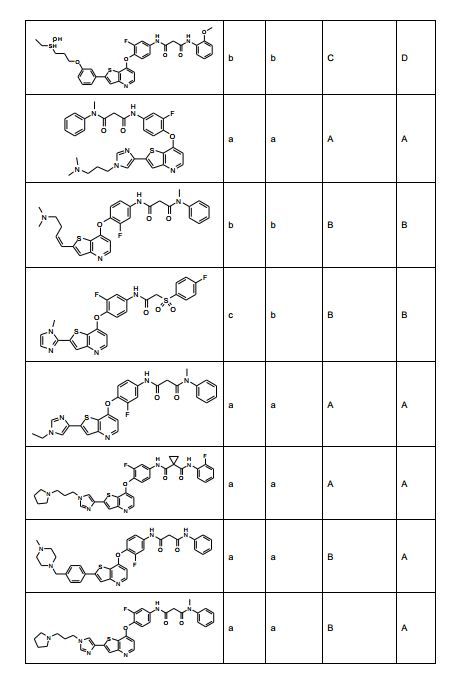
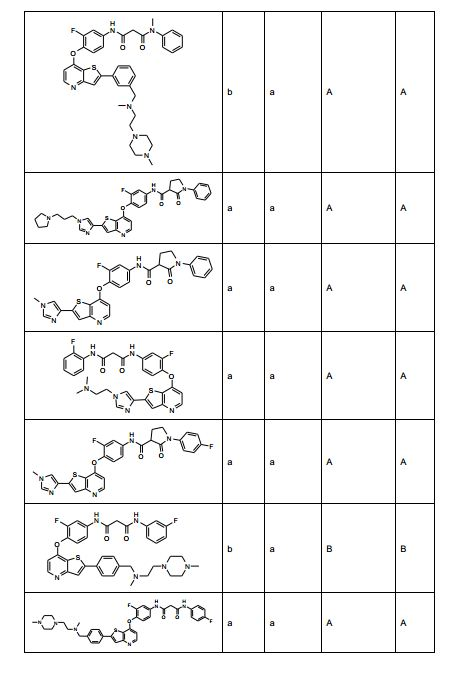
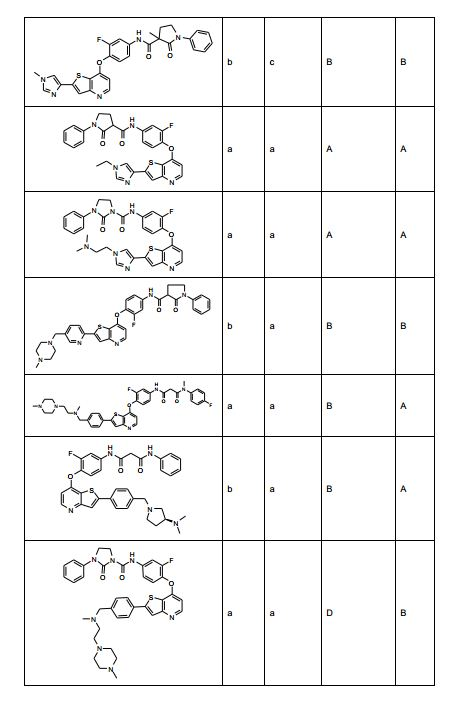
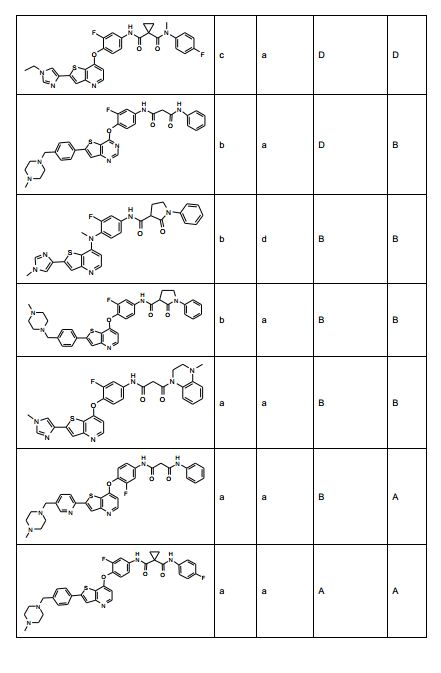
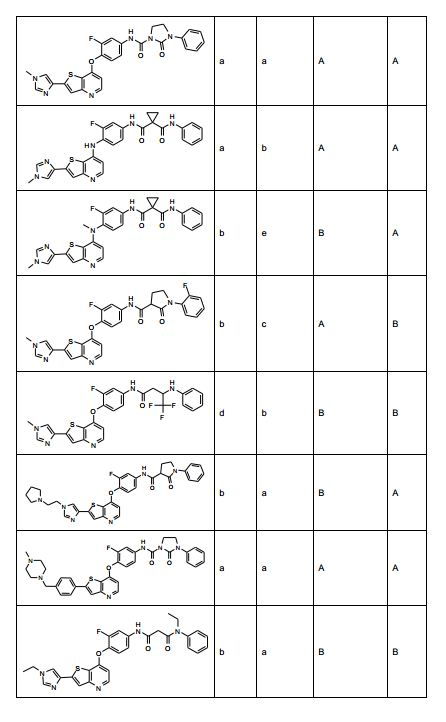
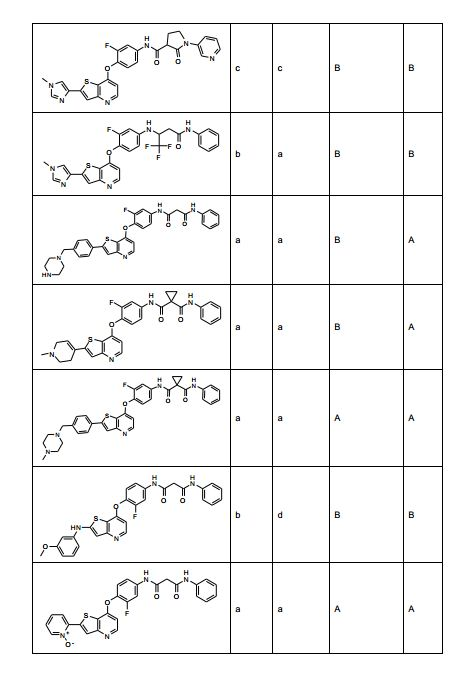
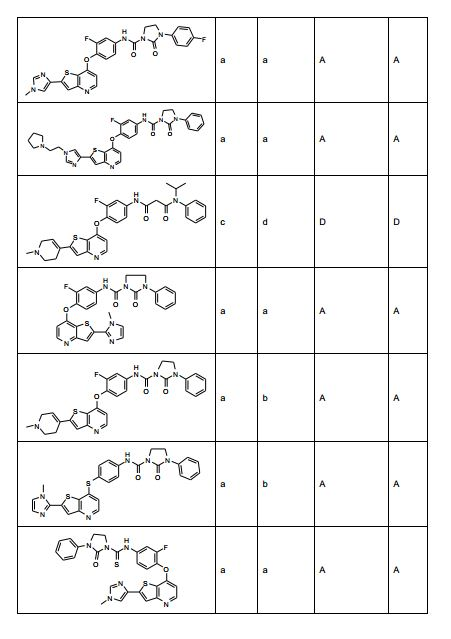
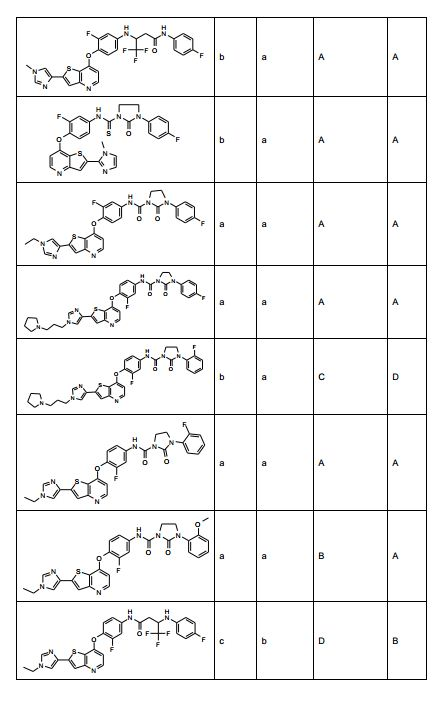
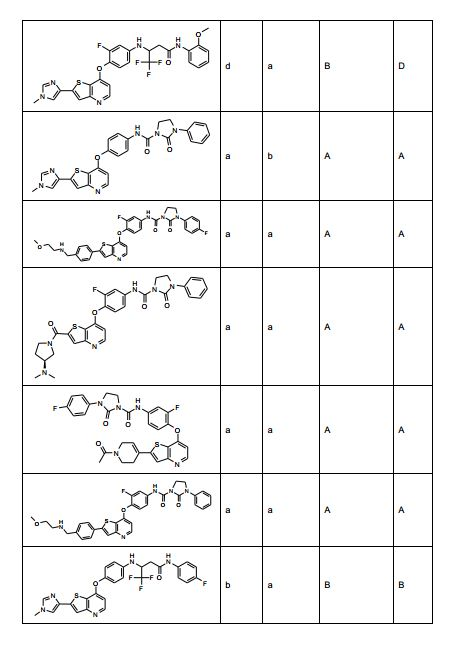
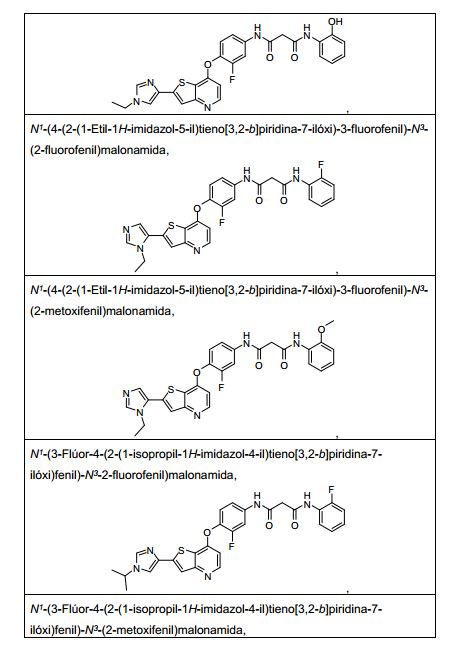
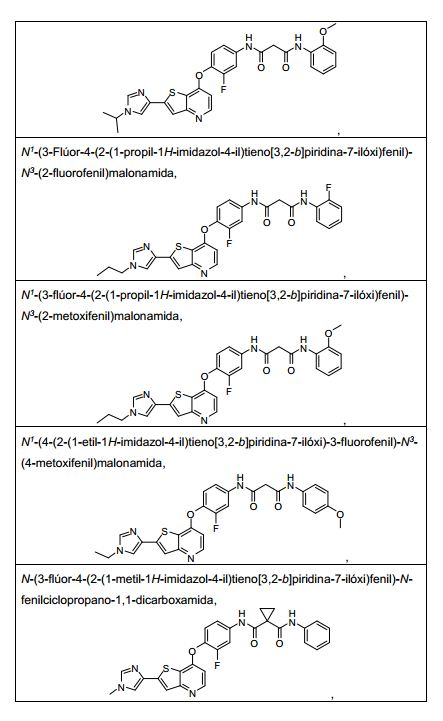
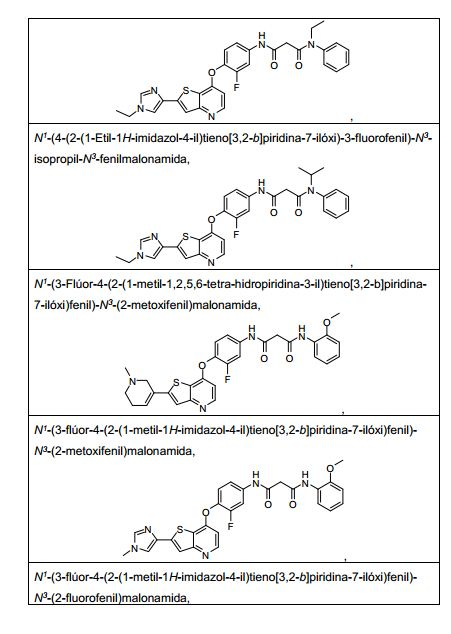
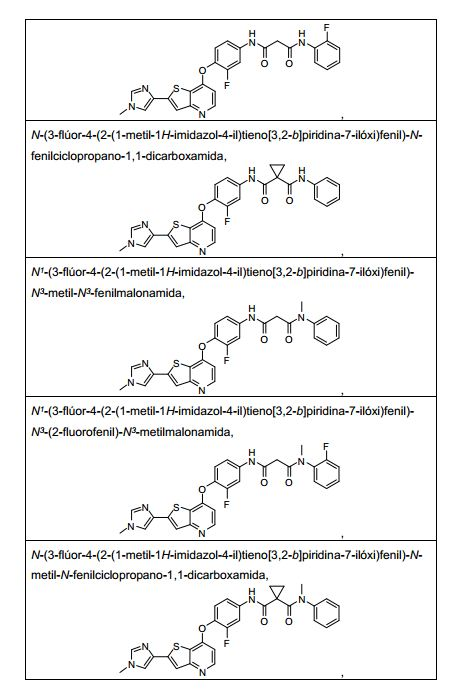
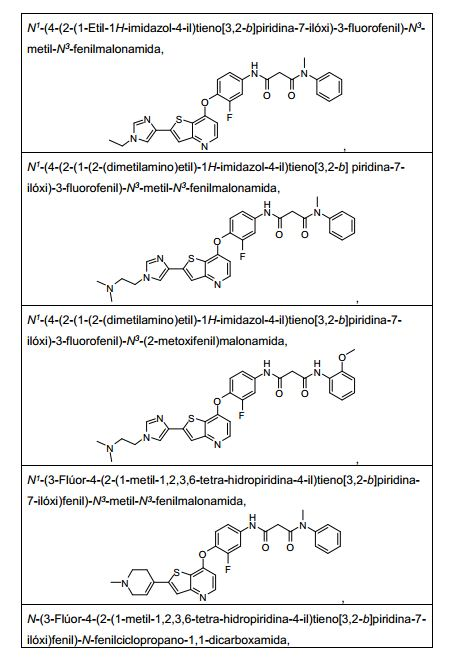
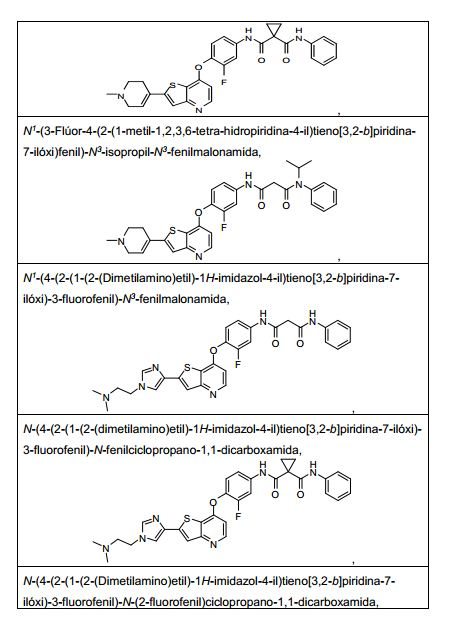
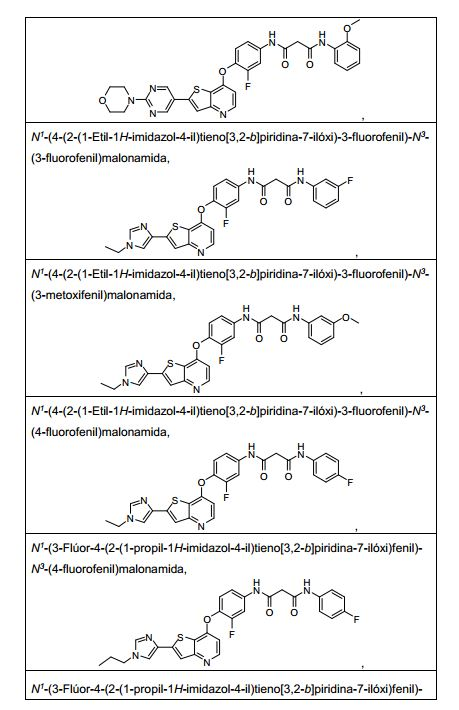
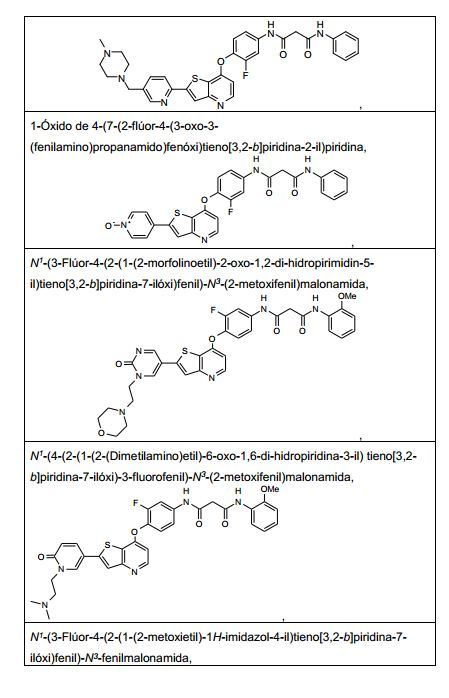
ANTECEDENTES DA PRESENTE INVENÇÃO
Campo da presente invenção
[001] A presente invenção refere-se à inibição de sinalização de receptor de VEGF e sinalização de receptor de HGF. Mais particularmente, a presente invenção refere-se a compostos e métodos para a inibição de sinalização de receptor de VEGF e sinalização de receptor de HGF.
Sumário da Técnica Relacionada
[002] Angiogênese é um componente importante de certos processos fisiológicos normais tais como embriogênese e cicatrização de ferimento, porém angiogênese aberrante contribui para alguns distúrbios patológicos e em particular para crescimento de tumor, 1,2 VEGF-A (fator de crescimento endotelial vascular A) é um fator-chave que promove neovascularização (angiogênese) de tumores, 3-7 VEGF induz migração e proliferação celular endotelial por sinalização através de dois receptores de afinidade elevada, o receptor de tirosina cinase similar à fms, Flt-1, e o receptor contendo o domínio de inserção de cinase, KDR,8,9,10. Estas respostas de sinalização são crucialmente dependentes da ativação e dimerização de receptor da atividade de tirosina cinase de receptor intrínseco (RTK). A ligação de VEGF como um homodímero ligado à dissulfeto estimula a ativação e dimerização de receptor do domínio11 RTK. A atividade de cinase autofosforila resíduos de tirosina de receptor citoplásmico, que então servem como sítios de ligação para moléculas envolvidas na propagação de uma cascata de sinalização. Embora múltiplas trilhas devam provavelmente ser elucidadas para ambos os receptores, a sinalização de KDR é mais extensivamente estudada, com uma resposta mitogênica sugerida envolver cinases12 de proteína ativada por mitógeno ERK-1 e ERK-2.
[003] Rompimento de sinalização de receptor de VEGF é um alvo terapêutico altamente atrativo em câncer, como angiogênese é um pré- requisito para todo crescimento de tumor sólido, e que o endotélio maduro permanece relativamente quiescente (com a exceção do sistema reprodutor feminino e cicatrização de ferimento). Vários métodos experimentais para inibir sinalização de VEGF foram examinados, incluindo uso de anticorpos 13,14,15 de neutralização, antagonistas 16 de receptor, receptores 17 solúveis, construções 18 antissenso e estratégias 19 negativas dominantes.
[004] A despeito da atratividade da terapia antiangiogênica por inibição de VEGF somente, várias questões podem limitar este método. Níveis de expressão de VEGF podem por si próprios ser elevados por numerosos estímulos variados e talvez mais importante, o estado hipóxico de tumores resultante da inibição de VEGFr, pode levar à indução de fatores que por si próprios promovem metástase e invasão de tumor desse modo, potencialmente solapar o impacto de inibidores de VEGF como produtos terapêuticos 20 de câncer.
[005] O receptor de HGF (fator de crescimento de hepatócito) e de HGF, c-met, estão envolvidos com a capacidade de células de tumor solaparem a atividade de inibição 20 de VEGF. HGF derivado de células de tumor circunjacentes a fibroblastos estromais ou expresso do tumor propriamente dito foi sugerido desempenhar um papel crucial em metástase, invasão e angiogênese de tumor 21,22. Por exemplo, crescimento invasivo de certas células de câncer é drasticamente realçado por interações estromais de tumor envolvendo a trilha 23,24,25 de HGF/c-Met (receptor de HGF). HGF, que foi originalmente identificado como um potente mitógeno para hepatócitos 26,27 é primariamente secretado de células estromais, e o HGF secretado pode promover a motilidade e invasão de várias células de câncer que expressam c-Met de uma maneira parácrina 28,29,30. Ligação de HGF a c-Met leva à ativação e fosforilação de receptor de trilha de sinalização de proteína cinase ativada por Ras/mitógeno (MAPK), desse modo realçando comportamentos malignos de células de câncer 30,31. Além disso, a estimulação da trilha de HGF/c-met propriamente dita pode levar à indução da expressão de VEGF, por si só contribuindo diretamente para a atividade angiogênica 32.
[006] Desse modo, métodos ou estratégias antiangiogênicas antitumor que têm como alvo tanto a sinalização de VEGF/VEGFr quanto a sinalização de HGF/c-met podem envolver a capacidade de células de tumor superarem a inibição de VEGF somente e podem representar produtos terapêuticos de câncer melhorados.
[007] Aqui descrevemos pequenas moléculas que são potentes inibidores tanto de KDR de receptor de VEGF quanto de c-met de receptor de HGF.
BREVE SUMÁRIO DA PRESENTE INVENÇÃO
[008] A presente invenção fornece novos compostos e métodos para tratamento de doenças proliferativas celulares. Os compostos da presente invenção são inibidores de função dual, capazes de inibir tanto a sinalização de receptor de VEGF quanto de HGF. Consequentemente, a presente invenção fornece novos inibidores de sinalização de receptor de VEGF e sinalização de receptor de HGF, incluindo o KDR de receptor de VEGF e o c-met de receptor de HGF.
[009] Em um primeiro aspecto, a presente invenção fornece compostos de fórmula A que são úteis como inibidores de sinalização de receptor de VEGF e sinalização de receptor de HGF e, portanto, são ferramentas de pesquisa úteis para o estudo do papel de VEGF e HGF tanto em estados normais quanto em estados de doença.
[0010] Em um segundo aspecto, a presente invenção fornece compostos de fórmula B que são úteis como inibidores de sinalização de receptor de VEFG e sinalização de receptor de HGF e, portanto, são ferramentas de pesquisa úteis para o estudo do papel de VEGF e HGF tanto em estados normais quanto em estados de doença.
[0011] Em um terceiro aspecto, a presente invenção fornece composições compreendendo um composto que é um inibidor de sinalização de receptor de VEGF e sinalização de receptor de HGF, ou um sal farmaceuticamente aceitável deste, e um veículo, excipiente, ou diluente farmaceuticamente aceitável.
[0012] O quarto aspecto da presente invenção fornece um método de inibir a sinalização de receptor de VEGF e sinalização de receptor de HGF, o método compreendendo contatar o receptor com um composto de acordo com a presente invenção, ou com uma composição de acordo com a presente invenção. Inibição da atividade de VEGF e HGF pode ser em uma célula ou um organismo multicelular. Se em um organismo multicelular, o método de acordo com este aspecto da presente invenção compreender administrar ao organismo um composto de acordo com a presente invenção, ou uma composição de acordo com a presente invenção. Preferivelmente o organismo é um mamífero, mais preferivelmente um ser humano.
[0013] Os anteriores simplesmente sumariam certos aspectos da presente invenção e não se pretende que sejam limitantes por natureza. Estes aspectos et al. aspectos e modalidades são descritos mais completamente abaixo.
DESCRIÇÃO DETALHADA DAS MODALIDADES PREFERIDAS
[0014] A presente invenção fornece compostos e métodos para inibição dos KDR de receptor de VEGF e c-met de receptor de HGF. A presente invenção também fornece composições e métodos para tratamento de condições e doenças proliferativas celulares. A patente e literatura científica referidas aqui estabelecem o conhecimento que é disponibilizado por aqueles com experiência na técnica. As patentes, pedidos, e referências publicadas que são citadas aqui são por meio deste incorporadas por referência à mesma extensão como se cada fosse específica e individualmente indicada ser incorporada por referência. No caso de incongruências, a presente descrição prevalecerá.
[0015] Para propósitos da presente invenção, as seguintes definições serão empregadas (a menos que expressamente estabelecido de outra maneira):
[0016] os termos "inibidor de sinalização de receptor de VEGF" e "inibidor de sinalização de receptor de HGF" são usados para identificar um composto tendo a estrutura como definida aqui, que é capaz de interagir com um receptor de HGF e um receptor de VEGF e inibir a atividade de HGF e VEGF. Em algumas modalidades preferidas, tal redução da atividade é pelo menos cerca de 50%, mais preferivelmente pelo menos cerca de 75%, e ainda mais preferivelmente pelo menos cerca de 90%.
[0017] Para simplicidade, porções químicas são definidas e referidas do princípio ao fim primariamente como porções químicas univalentes (por exemplo, alquila, arila, etc.). No entanto, tais termos são também empregados para transmitir porções multivalentes correspondentes sob as circunstâncias estruturais apropriadas claras para aqueles versados na técnica. Por exemplo, ao mesmo tempo que uma porção de "alquila" geralmente refere-se a um radical monovalente (por exemplo, CH3-CH2-), em certas circunstâncias uma porção de ligação bivalente pode ser "alquila," em cujo caso aqueles versados na técnica entenderão a alquila ser um radical divalente (por exemplo, -CH2-CH2-), que é equivalente ao termo "alquileno." (Similarmente, em circunstâncias nas quais uma porção divalente é requerida e é relatada como sendo "arila," aqueles versados na técnica entenderão que o termo "arila" refere-se à porção divalente correspondente, arileno). Todos os átomos são entendidos ter seu número normal de valências para formação de ligação (isto é, 4 para carbono, 3 para N, 2 para O, e 2, 4, ou 6 para S, dependendo do estado de oxidação do S). Ocasionalmente uma porção pode ser definida, por exemplo, como (A)a-B-, em que a é 0 ou 1. Em tais exemplos, quando a é 0 a porção é B- e quando a é 1 a porção é A-B-. Da mesma forma, várias porções descritas aqui existem em formas tautoméricas múltiplas, todas das quais pretende-se que sejam abrangidas por qualquer estrutura tautomérica dada.
[0018] O termo "hidrocarbila" como empregado aqui refere-se a uma alquila, alquenila, ou alquinila linear, ramificada, ou cíclica, cada como definido aqui. Uma "C0" hidrocarbila é usada para referir-se a uma ligação covalente. Desse modo, "C0-C3-hidrocarbila" inclui uma ligação covalente, metila, etila, etenila, etinila, propila, propenila, propinila e ciclopropila.
[0019] O termo "alquila" como empregado aqui refere-se a grupos alifáticos de cadeia linear e ramificada tendo de 1 a 12 átomos de carbono, preferivelmente 1-8 átomos de carbono, e mais preferivelmente 1-6 átomos de carbono, que são opcionalmente substituídos com um, dois ou três substituintes. Grupos alquila preferidos incluem, sem limitação, metila, etila, propila, isopropila, butila, isobutila, sec-butila, terc-butila, pentila e hexila. Uma "C0" alquila (como em "C0-C3-alquila") é uma ligação covalente (similar à "C0" hidrocarbila).
[0020] O termo "alquenila" como empregado aqui significa um grupo alifático de cadeia linear ou ramificada insaturado com uma ou mais ligações duplas carbono-carbono, tendo de 2 a 12 átomos de carbono, preferivelmente 2-8 átomos de carbono, e mais preferivelmente 2-6 átomos de carbono, que são opcionalmente substituídos com um, dois ou três substituintes. Grupos alquenila preferidos incluem, sem limitação, etenila, propenila, butenila, pentenila e hexenila.
[0021] O termo "alquinila" como empregado aqui significa um grupo alifático de cadeia linear ou ramificada insaturado com uma ou mais ligações triplas carbono-carbono, tendo de 2 a 12 átomos de carbono, preferivelmente 2-8 átomos de carbono, e mais preferivelmente 2-6 átomos de carbono, que são opcionalmente substituídos com um, dois ou três substituintes. Grupos alquinila preferidos incluem, sem limitação, etinila, propinila, butinila, pentinila e hexinila.
[0022] Um grupo "alquileno," "alquenileno," ou "alquinileno" é um grupo alquila, alquenila, ou alquinila, como definido aqui acima, que está posicionado entre e serve para unir dois outros grupos químicos. Grupos alquileno preferidos incluem, sem limitação, metileno, etileno, propileno e butileno. Grupos alquenileno preferidos incluem, sem limitação, etenileno, propenileno e butenileno. Grupos alquinileno preferidos incluem, sem limitação, etinileno, propinileno e butinileno.
[0023] O termo "carbociclo" como empregado aqui é pretendido significar uma porção de cicloalquila ou arila opcionalmente substituída. O termo "carbociclo" também inclui uma porção de cicloalquenila tendo pelo menos uma ligação dupla carbono-carbono.
[0024] O termo "cicloalquila" como empregado aqui inclui grupos hidrocarboneto cíclicos saturados e parcialmente insaturados tendo 3 a 12 carbonos, preferivelmente 3 a 8 carbonos, e mais preferivelmente 3 a 6 carbonos, em que o grupo cicloalquila adicionalmente é opcionalmente substituído. Grupos cicloalquila preferidos incluem, sem limitação, ciclopropila, ciclobutila, ciclopentila, ciclopentenila, ciclo- hexila, cicloexenila, ciclo-heptila, e ciclo-octila.
[0025] O termo "heteroalquila" como empregado aqui refere-se a um grupo alquila, como definido aqui acima, em que um ou mais átomos de carbono na cadeia são substituídos por um heteroátomo selecionado do grupo consistindo em O, S, NH, N-alquila, SO, SO2, SO2NH, ou NHSO2.
[0026] Um grupo "arila" é uma porção C6-C14 aromática compreendendo um a três anéis aromáticos, que são opcionalmente substituídos. Preferivelmente, o grupo arila é um grupo C6-C10 arila. Grupos arila preferidos incluem, sem limitação, fenila, naftila, antracenila, e fluorenila. Um grupo "aralquila" ou "arilalquila" compreende um grupo arila covalentemente ligado a um grupo alquila, o qual pode independentemente ser opcionalmente substituído ou não- substituído. Preferivelmente, o grupo aralquila é (C1-C6)alqu(C6- C10)arila, incluindo, sem limitação, benzila, fenetila, e naftilmetila. Uma "arilalquila inferior" refere-se a uma arilalquila onde a porção de "alquila" do grupo tem um a seis carbonos
[0027] Um grupo "heterociclila" ou "heterocíclico" é uma estrutura de anel tendo de cerca de 3 a cerca de 12 átomos, em que um ou mais átomos são selecionados do grupo consistindo em N, O, S, SO e SO2. O grupo heterocíclico é opcionalmente substituído sobre o carbono em uma ou mais posições. O grupo heterocíclico é também independente e opcionalmente substituído sobre o nitrogênio com alquila, arila, aralquila, alquilcarbonila, alquilsulfonila, arilcarbonila, arilsulfonila, alcoxicarbonila ou aralcoxicarbonila. Grupos heterocíclicos preferidos incluem, sem limitação, epóxi, aziridinila, tetra-hidrofuranila, pirrolidinila, piperidinila, piperazinila, tiazolidinila, oxazolidinila, oxazolidinonila e morfolino. Em certas modalidades preferidas, o grupo heterocíclico é fundido a um grupo arila, heteroarila ou cicloalquila. Exemplos de tais heterociclos fundidos incluem, sem limitação, tetra- hidroquinolina e di-hidrobenzofurano. Especificamente exclusos do escopo deste termo são compostos onde um átomo de O ou S anular é adjacente a outro átomo de O ou S.
[0028] Como empregado aqui, o termo "heteroarila" refere-se a grupos tendo 5 a 14 átomos de anel, preferivelmente 5, 6, 9, ou 10 átomos de anel; tendo 6, 10, ou 14 K-elétrons compartilhados em uma disposição cíclica; e tendo, além dos átomos de carbono, de um a três heteroátomos por anel selecionados do grupo consistindo em N, O, e S. O termo "heteroarila" é também pretendido abranger grupos monocíclicos e bicíclicos. Por exemplo, um grupo heteroarila pode ser pirimidinila, piridinila, benzimidazolila, tienila, benzotiazolila, benzofuranila e indolinila. Um grupo "heteroaralquila" ou "heteroarilalquila" compreende um grupo heteroarila covalentemente ligado a um grupo alquila, o qual é independente e opcionalmente substituído ou não-substituído. Grupos heteroalquila preferidos compreendem um grupo C1-C6 alquila e um grupo heteroarila tendo 5, 6, 9, ou 10 átomos de anel. Especificamente exclusos do escopo deste termo são compostos tendo átomos de O e/ou S anulares adjacentes. Exemplos de grupos heteroaralquila preferidos incluem piridilmetila, piridiletila, pirrolilmetila, pirroliletila, imidazolilmetila, imidazoliletila, tiazolilmetila e tiazoliletila. Especificamente exclusos do escopo deste termo são compostos tendo átomos de O e/ou S anulares adjacentes.
[0029] Para simplicidade, a referência a uma "Cn-Cm" heterociclila ou heteroarila significa uma heterociclila ou heteroarila tendo de "n" a "m" átomos anulares, onde "n" e "m" são números inteiros. Desse modo, por exemplo, uma C5-C6-heterociclila é um anel de 5 ou 6 membros tendo pelo menos um heteroátomo, e inclui pirrolidinila (C5) e piperidinila (C6); C6-hetoarila inclui, por exemplo, piridila e pirimidila.
[0030] Um grupo "arileno," "heteroarileno," ou "heterociclileno" é um grupo arila, heteroarila, ou heterociclila, como definido aqui acima, que está posicionado entre e serve para unir dois outros grupos químicos.
[0031] O termo "azolila" como empregado aqui é pretendido significar um grupo heterocíclico saturado ou insaturado de cinco membros contendo dois ou mais heteroátomos, como átomos de anel, selecionados do grupo consistindo em nitrogênio, enxofre e oxigênio, em que pelo menos um dos heteroátomos é um átomo de nitrogênio. Um grupo azolila como empregado na presente invenção pode ser opcionalmente substituído. Grupos azolila preferidos incluem, porém não são limitados a imidazolila, oxazolila, tiazolila, pirazolila, isoxazolila, isotiazolila, 1,3,4-tiadiazolila, 1,2,4-tiadiazolila, 1,2,4- oxadiazolila, e 1,3,4-oxadiazolila opcionalmente substituídos.
[0032] Um grupo heteroalicíclico refere-se especificamente a um radical de heterociclila não-aromática. Um heteroalicíclico pode conter insaturação, porém não é aromático.
[0033] Um grupo heterociclilalquila refere-se a um resíduo em que uma heterociclila é ligada a uma estrutura origem por meio de um de um radical de alquileno, alquilideno ou alquilidina. Exemplos incluem (4-metilpiperazin-1-il)metila, (morfolin-4-il)metila, (piridina-4-il)metila, 2- (oxazolin-2-il)etila, 4-(4-metilpiperazin-1-il)-2-butenila, e similares. Tanto a porção de radical de heterociclila quanto de alquileno, alquilideno, ou alquilidina correspondente de um grupo heterociclilalquila podem ser opcionalmente substituídas. Uma "heterociclilalquila inferior" refere-se a uma heterociclilalquila onde a porção de "alquila" do grupo tem um a seis carbonos.
[0034] Um grupo heteroaliciclilalquila refere-se especificamente a uma heterociclilalquila onde a porção de heterociclila do grupo é não- aromática.
[0035] Heterociclilas e heteroarilas preferidas incluem, porém não são limitadas a, acridinila, azocinila, benzimidazolila, benzofuranila, benzotiofenila, benzoxazolila, benztiazolila, benztriazolila, piridotriazolila, benzisoxazolila, benzisotiazolila, benzimidazolinila, carbazolila, 4aH-carbazolila, carbolinila, cromanila, cromenila, cinolinila, deca-hidroquinolinila, 2H,6H-1,5,2-ditiazinila, di-hidrofuro[2,3- b]tetra-hidrofurano, furanila, furazanila, imidazolidinila, imidazolinila, imidazolila, 1H-indazolila, indolenila, indolinila, indolizinila, indolila, 3H- indolila, isobenzofuranila, isocromanila, isoindazolila, isoindolinila, isoindolila, isoquinolinila, isotiazolila, isoxazolila, metilenodioxifenila, morfolinila, naftiridinila, octra-hidroisoquinolinila, oxadiazolila, 1,2,3- oxadiazolila, 1,2,4-oxadiazolila, 1,2,5-oxadiazolila, 1,3,4-oxadiazolila, oxazolidinila, oxazolila, oxazolidinila, pirimidinila, fenantridinila, fenantrolinila, fenazinila, fenotiazinila, fenoxatiinila, fenoxazinila, ftalazinila, piperazinila, piperidinila, piperidonila, 4-piperidonila, piperonila, pteridinila, purinila, piranila, pirazinila, pirazolidinila, pirazolinila, pirazolila, piridazinila, pirido-oxazol, piridoimidazol, piridotiazol, piridinila, piridila, pirimidinila, pirrolidinila, pirrolinila, 2H- pirrolila, pirrolila, quinazolinila, quinolinila, 4H-quinolizinila, quinoxalinila, quinuclidinila, tetra-hidrofuranila, tetra-hidroisoquinolinila, tetra-hidroquinolinila, tetrazolila, 6H-1,2,5-tiadiazinila, 1,2,3-tiadiazolila, 1,2,4-tiadiazolila, 1,2,5-tiadiazolila, 1,3,4-tiadiazolila, tiantrenila, tiazolila, tienila, tienotiazolila, tienooxazolila, tienoimidazolila, tiofenila, triazinila, 1,2,3-triazolila, 1,2,4-triazolila, 1,3,4-triazolila, e xantenila.
[0036] Como empregado aqui, quando a porção (por exemplo, cicloalquila, hidrocarbila, arila, heteroarila, heterocíclica, ureia, etc.) é descrita como "opcionalmente substituído" entende-se que o grupo opcionalmente tem de um a quatro, preferivelmente de um a três, mais preferivelmente um ou dois, substituintes de não-hidrogênio. Substituintes adequados incluem, sem limitação, halo, hidróxi, oxo (por exemplo, um -CH- anular substituído por oxo é grupo -C(O)-) nitro, arenossulfocarbila, hidrocarbila, arila, aralquila, alcóxi, arilóxi, amino, acilamino, alquilcarbamoíla, arilcarbamoíla, aminoalquila, acila, carbóxi, hidroxialquila, alcanossulfonila, arenesulfonila, alcanossulfonamido, arenesulfonamido, aralquilsulfonamido, alquilcarbonila, acilóxi, ciano, e ureído. Os substituintes preferidos, que são em si também não-substituídos (a menos que de outro modo expressamene estabelecido) são: (a) halo, hidróxi, ciano, oxo, carbóxi, formila, nitro, amino, amidino, guanidino, (b) C1-C5 alquila ou alquenila ou arilalquila imino, carbamoíla, azido, carboxamido, mercapto, hidróxi, hidroxialquila, alquilarila, arilalquila, C1-C8 alquila, C1-C8 alquenila, C1-C8 alcóxi, C1-C8 alcoxicarbonila, ariloxicarbonila, C2-C8 acila, C2-C8 acilamino, C1-C8 alquiltio, arilalquiltio, ariltio, C1-C8 alquilsulfinila, arilalquilsulfinila, arilsulfinila, C1-C8 alquilsulfonila, arilalquilsulfonila, arilsulfonila, C0-C6 N-alquila carbamoíla, C2-C15 N,N-dialquilcarbamoíla, C3-C7 cicloalquila, aroíla, arilóxi, éter de arilalquila, arila, arila fundido a um anel cicloalquila ou heterociclo ou outro arila, C3-C7 heterociclo, C5-C14 heteroarila, ou qualquer destes anéis fundido ou espiro-fundido a uma cicloalquila, heterociclila, ou arila, em que cada um dos anteriores é também opcionalmente substituído por uma ou mais porções listadas em (a), acima; e (c) -(CH2)s-NR31R32, em que s é de 0 (em cujo caso o nitrogênio é diretamente ligado à porção que é substituída) a 6, e R31 e R32 são cada qual independentemente hidrogênio, ciano, oxo, carboxamido, amidino, C1-C8 hidroxialquila, C1-C3 alquilarila, aril-C1-C3 alquila, C1-C8 alquila, C1-C8 alquenila, C1-C8 alcóxi, C1-C8 alcoxicarbonila, ariloxicarbonila, aril-C1-C3 alcoxicarbonila, C2-C8 acila, C1-C8 alquilsulfonila, arilalquilsulfonila, arilsulfonila, aroíla, arila, cicloalquila, heterociclila, ou heteroarila, em que cada um dos anteriores é também opcionalmente substituído por uma ou mais porções listadas em (a), acima; ou
[0037] R30 e R31 tomados juntamente com o N ao qual eles são ligados formam uma heterociclila ou heteroarila, cada uma das quais é opcionalmente substituída com de 1 a 3 substituintes de (a), acima.
[0038] Os substituintes especialmente preferidos nos grupos alquila incluem halogênio e hidróxi.
[0039] Os substituintes especialmente preferidos os grupos de anel, tal como arila, heteroarila, cicloalquila e heterociclila, incluem halogênio, alcóxi e alquila.
[0040] A "arenossulfocarbila" como empregado aqui é uma porção hidrocarbila, em que de um a todos os hidrogênios foram substituídos por um ou mais halo.
[0041] O termo "halogênio" ou "halo" como empregado aqui refere- se a cloro, bromo, flúor ou iodo. Como aqui empregado, o termo "acila" refere-se a um substituinte alquilcarbonila ou arilcarbonila. O termo "acilamino" refere-se a um grupo amida ligado a um átomo de nitrogênio (isto é, R-CO-NH-). O termo "carbamoíla" refere-se a um grupo amida ligado no átomo de carbono de carbonila (isto é, NH2-CO- ). O átomo de nitrogênio de um substituinte de acilamino ou carbamoíla é adicionalmente substituído. O termo "sulfonamido" refere-se a um substituinte de sulfonamida ligado pelo átomo de enxofre ou nitrogênio. O termo "amino" é entendido incluir grupos NH2, alquilamino, arilamino, e amino cíclico. O termo "ureído" como empregado aqui refere-se a uma porção de ureia substituída ou não- substituída.
[0042] O termo "radical" como empregado aqui significa uma porção química compreendendo um ou mais elétrons não-pareados.
[0043] A porção que é substituída é aquela em que um ou mais hidrogênios foram independentemente substituídos por outro substituinte químico. Como um exemplo não-limitante, fenilas substituídas incluem 2-flurofenila, 3,4-diclorofenila, 3-cloro-4-flúor- fenila, 2-flúor-3-propilfenila. Como outro exemplo não-limitante, n- octilas substituídas incluem 2,4-dimetil-5-etil-octila e 3-ciclopentil-octila. Estão incluídos nesta definição os metilenos (-CH2-) substituídos por oxigênio para formar carbonila -CO-).
[0044] Uma porção "não-substituída" como acima definido (por exemplo, cicloalquila não-substituída, heteroarila não-substituída, etc.) significa que a porção como acima definida que não tem qualquer um dos substituintes opcionais para os quais a definição da porção (acima) de outro modo aplica-se. Desse modo, por exemplo, enquanto uma "arila" inclui fenila e fenila substituída com um halo, "arila não- substituída" não inclui fenila substituída por um halo.
[0045] Um anel carboxíclico de três a oito membros saturado ou insaturado é preferivelmente um anel carbocíclico saturado ou insaturado, de quatro a sete membros, mais preferivelmente cinco a seis membros. Exemplos de anéis carbocíclicos de três a oito membros saturado ou insaturado incluem fenila, ciclopropila, ciclobutila, ciclopentila, ciclo-hexila e ciclo-heptila.
[0046] Um anel heterocíclico de três a oito membros saturado ou insaturado contém pelo menos um heteroátomo selecionado de átomos de oxigênio, nitrogênio, e enxofre. O anel heterocíclico de três a oito membros saturado ou insaturado preferivelmente contém um ou dois heteroátomos com os átomos constituintes de anel restantes sendo átomos de carbono. O anel heterocíclico de três a oito membros saturado ou insaturado é preferivelmente um anel heterocíclico de quatro a sete membros saturado ou insaturado, mais preferivelmente anel heterocíclico de cinco ou seis membros saturado ou insaturado. Exemplos de grupos heterocíclicos de três a oito membros saturado ou insaturado incluem tienila, piridila, 1,2,3-triazolila, imidazolila, isoxazolila, pirazolila, piperazinila, piperazino, piperidila, piperidino, morfolinila, morfolino, homopiperazinila, homopiperazino, tiomorfolinila, tiomorfolino, tetra-hidropirrolila e azepanila.
[0047] Um grupo carboxílico e heterocíclico saturado ou insaturado pode condensar com outro grupo saturado ou heterocíclico para formar um grupo bicíclico, preferivelmente um grupo carbocíclico ou heterocíclico bicíclico de nove a doze membros saturado ou insaturado. Os grupos bicíclico incluem naftila, quinolila, 1,2,3,4-tetra- hidroquinolila, 1,4-benzoxanila, indanila, indolila, e 1,2,3,4-tetra- hidronaftila.
[0048] Quando a carbocíclico ou heterocíclico grupo é substituído por dois grupos C1-6 alquila, os dois grupos alquila podem combinar-se entre si para formar uma cadeia de alquileno, preferivelmente uma cadeia C1-3 alquileno. Os grupos carbocíclico ou heterocíclico tendo esta estrutura reticulada incluem biciclo[2,2,2]octanila e norbornanila.
[0049] O termo "quantidade terapeuticamente eficaz" como empregado aqui é uma quantidade de um composto da presente invenção, que quando administrado a um paciente, melhora um sintoma de uma doença. A quantidade de um composto da presente invenção que constitui uma "quantidade terapeuticamente eficaz" variará dependendo do composto, o estado de doença e sua gravidade, a idade do paciente a ser tratado, e similares. A quantidade terapeuticamente eficaz pode ser determinada rotineiramente por alguém versado na técnica.
[0050] O termo "paciente" como empregado aqui para os propósitos da presente invenção inclui humanos et al. animais, particularmente mamíferos, et al. organismos. Desse modo, os compostos, composições e métodos da presente invenção são aplicáveis tanto à terapia humana quanto aplicações veterinárias. Em uma modalidade preferida o paciente é um mamífero, e em uma modalidade mais preferida o paciente é humano.
[0051] Os termos "tratando" ou "tratamento" como empregado aqui abrange o tratamento de um estado de doença em um mamífero, cujo estado de doença é caracterizado por proliferação celular anormal, e invasão e inclui pelo menos um de: (i) prevenir o estado de doença de ocorrer em um mamífero, em particular, quando um tal mamífero está predisposto ao estado de doença, porém ainda não foi diagnosticado como tendo-a; (ii) inibir o estado de doença, isto é, deter seu desenvolvimento; e (iii) aliviar o estado de doença, isto é, causar a regressão do estado de doença. Em uma modalidade preferida da presente invenção o mamífero é um ser humano. Como é conhecido na técnica, ajustes para liberação sistêmica versus localizada, idade, peso corporal, saúde geral, sexo, dieta, tempo de administração, interação de fármaco e a gravidade da condição podem ser necessários, e serão verificáveis com experimentação de via por alguém versado na técnica.
[0052] Em toda a especificação, modalidades preferidas de um ou mais substituintes químicos são identificadas. São também preferidas combinações de modalidades preferidas. Por exemplo, o parágrafo [00133]053 descreve modalidades preferidas de R7 nos compostos da presente invenção e o parágrafo 0114 descreve modalidades preferidas de G nos compostos da presente invenção. Desse modo, também contemplados como incluídos no escopo da presente invenção estão os compostos em que R7 é como descrito no parágrafo 0053 e G é como descrito no parágrafo 0114. Além disso, compostos excluídos de qualquer um dos gêneros particulares de compostos (por exemplo, através de uma cláusula de condição) destinam-se a serem excluídos do escopo da presente invenção totalmente, incluindo det al. gêneros descritos, a menos que expressamente estabelecido ao contrário.
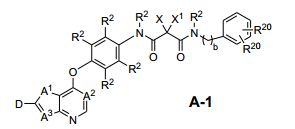
Compostos
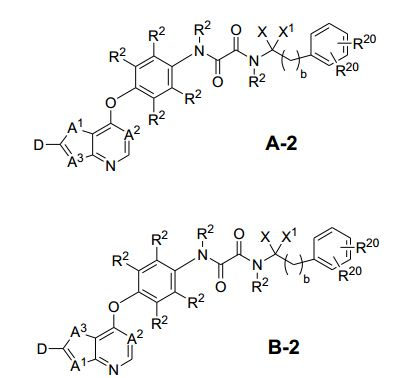
[0053] No primeiro e segundo aspectos, a presente invenção compreende compostos de fórmula (A) e formula (B), que são inibidores de sinalização de receptor de VEGF e sinalização de receptor de HGF:
[0054] e sais farmaceuticamente aceitáveis e complexos destes, em que,
[0055] D é selecionado do grupo consistindo em R7, R1 e R21, em que
[0056] R7 é selecionado do grupo consistindo em -H, halogênio, nitro, azido, C1-C6 alquila, C3-C10 cicloalquila, -C(O)NR42R43, -Y- NR42R43, -NR42C(= O)R43, -SO2R42, -SO2NR42R43, -NR37SO2R42, - NR37SO2NR42R43, -C(= N-OR42)R43, -C(= NR42)R43, -NR37C(= NR42)R43, -C(= NR42)NR37R43, -NR37C(= NR42)NR37R43, -C(O)R42, -CO2R42, - C(O)(heterociclila), -C(O)(C6-C10 arila), -C(O)(heteroarila), -Y-(C6-C10 arila), -Y-(heteroarila), -Y-(heterociclila de 5 a 10 membros), -NR6aR6b, -NR6aSO2R6b, -NR6aC(O)R6b, -OC(O)R6b, -NR6aC(O)OR6b, - OC(O)NR6aR6b,-OR6a, -SR6a, -S(O)R6a, -SO2R6a, -SO3R6a, - SO2NR6aR6b, -SO2NR42R43, -COR6a, -CO2R6a, -CONR6aR6b, -(C1- C4)fluoroalquila, -(C1-C4)fluoroalcóxi, -(CZ3Z4)aCN, em que n é um número inteiro variando de 0 a 6, e os grupos R7 anteriormente mencionados diferentes de -H e halogênio são opcionalmente substituídos por 1 a 5 R38, ou R7 é uma porção selecionada do grupo consistindo em -(CZ3Z4)a-arila, -(CZ3Z4)a-heterociclo, (C2-C6)alquinila, - (CZ3Z4)a-(C3-C6)cicloalquila, -(CZ3Z4)a-(C5-C6)cicloalquenila, (C2-C6) alquenila e (C1-C6)alquila, em que a referida porção é opcionalmente substituída com 1 a 3 grupos Y2 independentemente selecionados, onde a é 0,1, 2, ou 3, e em que quando a é 2 ou 3, as unidades CZ3Z4 podem ser iguais ou diferentes; em que
[0057] cada R6a e R6b é independentemente selecionado do grupo consistindo em hidrogênio e uma porção selecionada do grupo consistindo em -(CZ5Z6)u-(C3-C6)cicloalquila, -(CZ5Z6)u-(C5- C6)cicloalquenila, -(CZ5Z6)u-arila, -(CZ5Z6)u-heterociclo, (C2- C6)alquenila, e (C1-C6)alquila, em que a referida porção é opcionalmente substituída com 1 a 3 grupos Y3 independentemente selecionados, onde u é 0,1, 2, ou 3, e em que quando u é 2 ou 3, as unidades CZ5Z6 podem ser iguais ou diferentes, ou
[0058] R6a e R6b tomados juntamente com os átomos adjacentes formam um heterociclo;
[0059] cada Z3, Z4, Z5 e Z6 é independentemente selecionado do grupo consistindo em H, F e (C1-C6)alquila, ou
[0060] cada Z3 e Z4, ou Z5 e Z6 são selecionados juntos para formar um carbociclo, ou
[0061] dois grupos Z3 sobre átomos de carbono adjacentes são selecionados juntos para opcionalmente formar um carbociclo;
[0062] cada Y2 e Y3 é independentemente selecionado do grupo consistindo em halogênio, ciano, nitro, tetrazolila, guanidino, amidino, metilguanidino, azido, -C(O)Z7, -OC(O)NH2, -OC(O) NHZ7, - OC(O)NZ7Z8, -NHC(O)Z7, -NHC(O)NH2, -NHC(O)NHZ7, - NHC(O)NZ7Z8, -C(O)OH, -C(O)OZ7, -C(O)NH2, -C(O)NHZ7,- C(O)NZ7Z8, -P(O)3H2, -P(O)3 (Z7)2, -S(O)3H, -S(O)Z7, -S(O)2Z7, - S(O)3Z7, -Z7, -OZ7, -OH, -NH2, -NHZ7, -NZ7Z8, -C(= NH)NH2,-C(= NOH)NH2, -N-morfolino, (C2-C6)alquenila, (C2-C6)alquinila, (C1- C6)haloalquila, (C2-C6)haloalquenila, (C2-C6)haloalquinila, (C1- C6)haloalcóxi, -(CZ9Z10)rNH2, -(CZ9Z10)rNHZ3, -(CZ9Z10)rNZ7Z8, -X6 (CZ9Z10)r-(C3-C8)cicloalquila, -X6 (CZ9Z10)r-(C5-C8)cicloalquenila, -X6 (CZ9Z10)r-arila e -X6 (CZ9Z10)r-heterociclo, em que
[0063] r é 1, 2, 3 ou 4;
[0064] X6 é selecionado do grupo consistindo em O, S, NH, -C(O)-, -C(O)NH-, -C(O)O-, -S(O)-, -S(O)2- e -S(O)3-;
[0065] Z7 e Z8 são independentemente selecionados do grupo consistindo em uma alquila de 1 a 12 átomos de carbono, uma alquenila de 2 a 12 átomos de carbono, uma alquinila de 2 a 12 átomos de carbono, uma cicloalquila de 3 a 8 átomos de carbono, uma cicloalquenila de 5 a 8 átomos de carbono, uma arila de 6 a 14 átomos de carbono, um heterociclo de 5 a 14 átomos de anel, uma aralquila de 7 a 15 átomos de carbono, e uma heteroaralquila de 5 a 14 átomos de anel, ou
[0066] Z7 e Z8 juntos podem opcionalmente formar um heterociclo;
[0067] Z9 e Z10 são independentemente selecionados do grupo consistindo em H, F, uma (C1-C12)alquila, uma (C6-C14)arila, uma (C5- C14)heteroarila, uma (C7-C15)aralquila e uma (C5-C14)heteroaralquila, ou
[0068] Z9 e Z10 são tomados juntos para formar um carbociclo, ou
[0069] dois grupos Z9 sobre átomos de carbono adjacentes são tomados juntos para formar um carbociclo; ou
[0070] quaisquer dois grupos Y2 ou Y3 ligados aos átomos de carbono adjacentes podem ser tomados juntos para serem - O[C(Z9)(Z10)]rO ou -O[C(Z9)(Z10)]r+1, ou
[0071] quaisquer dois grupos Y2 ou Y3 ligados aos mesmos ou adjacentes átomos de carbono podem ser selecionados juntos para formar um carbociclo ou heterociclo; e em que
[0072] qualquer um dos substituintes acima mencionados compreendendo um grupo CH3 (metila), CH2 (metileno), ou CH (metina) que não é ligado a um grupo halogênio, SO ou SO2 ou a um átomo de N, O ou S opcionalmente transporta sobre o referido grupo um substituinte selecionado de hidróxi, halogênio, (C1-C4)alquila, (C1- C4)alcóxi e uma -N[(C1-C4)alquila][(C1-C4)alquila];
[0073] R1 é -CHCH ou -C=C-(CR45R45)n-R46;
[0074] cada R45 é independentemente selecionado do grupo consistindo em H, uma (C1-C6)alquila e uma (C3-C8)cicloalquila;
[0075] R46 é selecionado do grupo consistindo em heterociclila, - N(R47)-C(O)-N(R47)(R48), -N(R47)-C(S)-N(R47)(R48), -N(R47)-C(O)-OR48, -N(R47)-C(O)-(CH2)n-R48, -N(R47)-SO2R47, -(CH2)nNR47R48, -(CH2)nOR48, -(CH2)nSR49, -(CH2)nS(O)R49, -(CH2)nS(O)2R49, -OC(O)R49, - OC(O)OR49, -C(O)NR47R48, heteroarila opcionalmente substituída com um ou mais substituintes selecionados do grupo consistindo em halo, - CF3, (C1-C6)alcóxi, -NO2, (C1-C6)alquila, -CN, -SO2R50 e - (CH2)nNR50R51, e arila opcionalmente substituída com um ou mais substituintes selecionados do grupo consistindo em halo, -CF3, (C1- C6)alcóxi, -NO2, (C1-C6)alquila, -CN, -SO2R50 e -(CH2)nNR50R51;
[0076] R47 e R48 são independentemente selecionados do grupo consistindo em H, (C1-C6)alquila, (C3-C8)cicloalquila, heterociclila, - (CH2)nNR50R51, -(CH2)nOR50, -(CH2)nC(O)R49, -C(O)2R49, -(CH2)nSR49, - (CH2)nS(O)R49, -(CH2)nS(O)2R49, -(CH2)nR49, -(CH2)nCN, arila opcionalmente substituída com um ou mais substituintes selecionados do grupo consistindo em halo, -CF3, (C1-C6)alcóxi, -NO2, (C1-C6)alquila, -CN, -(CH2)nOR49, -(CH2)nheterociclila, -(CH2)nheteroarila, -SO2R50 e - (CH2)nNR50R51, e heteroarila opcionalmente substituída com um ou mais substituintes selecionados do grupo consistindo em halo, -CF3, (C1-C6)alcóxi, -NO2, (C1-C6)alquila, -CN, -(CH2)nOR49, - (CH2)nheterociclila, -(CH2)nheteroarila, -SO2R50 e -(CH2)nNR50R51, ou
[0077] R47 e R48, juntamente com o átomo ao qual eles são ligados, formam um anel carbo- ou heterocíclico de 3 a 8 membros ;
[0078] R49 é selecionado do grupo consistindo em (C1-C6)alquila, (C3-C8)cicloalquila, heterociclil(C1-C6)alquileno, aril(C1-C6)alquileno em que a arila é opcionalmente substituída com um ou mais substituintes selecionados do grupo consistindo em halo, -CF3, (C1-C6)alcóxi, -NO2, (C1-C6)alquila, -CN, -SO2R50 e -(CH2)nNR50R51, heteroaril(C1- C6)alquileno em que a heteroarila é opcionalmente substituída com um ou mais substituintes selecionados do grupo consistindo em halo, - CF3, (C1-C6)alcóxi, -NO2, (C1-C6)alquila, -CN, -SO2R50 e - (CH2)nNR50R51, arila opcionalmente substituída com um ou mais substituintes selecionados do grupo consistindo em halo, -CF3, (C1- C6)alcóxi, -NO2, (C1-C6)alquila, -CN, -SO2R50 e -(CH2)nNR50R51, e heteroarila opcionalmente substituída com um ou mais substituintes selecionados do grupo consistindo em halo, -CF3, (C1-C6)alcóxi, -NO2, (C1-C6)alquila, -CN, -SO2R50 e -(CH2)nNR50R51;
[0079] R50 e R51 são independentemente selecionados do grupo consistindo em H, (C1-C6)alquila, (C3-C8)cicloalquila e -C(O)R45, ou
[0080] R50 e R51, juntamente com o átomo ao qual eles são ligados, formam um anel carbo- ou heterocíclico de 3 a 8 membros; e
[0081] R21 é o grupo definido por -(Z11)-(Z12)m-(Z13)m1, em que
[0082] Z11 é heterociclila, quando m e m1 são 0, ou heterociclileno, quando ou m ou m1 é 1,
[0083] Z12 é selecionado do grupo consistindo em OC(O), OC(S) e C(O);
[0084] Z13 é selecionado do grupo consistindo em heterociclila, aralquila, N(H)R52, (C1-C3)alquila, -OR52, halo, S(O)2R56, (C1- C3)hidroxialquila e (C1-C3)haloalquila;
[0085] m é 0 ou 1;
[0086] m1 é 0 ou 1;
[0087] R52 é selecionado do grupo consistindo em H, - (CH2)qS(O)2R54, -(C1-C6) alquil- NR53R53 (C1-C3)alquila, -(CH2)qOR53, - C(O)R54 e -C(O)OR53;
[0088] q é 0, 1, 2, 3 ou 4;
[0089] cada R53 é independentemente (C1-C3)alquila;
[0090] R54 é (C1-C3)alquila ou N(H)R53;
[0091] R56 é selecionado do grupo consistindo em NH2, (C1- C3)alquila e OR52;
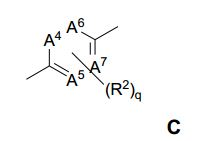
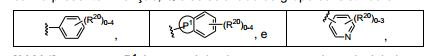
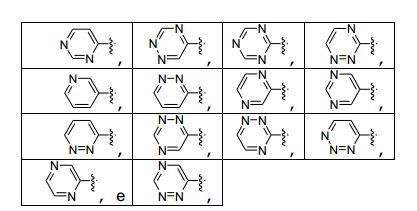
[0092] A1 é selecionado do grupo consistindo em -CH2-, -O-, -S-, - N(H)-, -N(CI-C6 alquil)-, -N-(Y-aril)-, -N-OMe, -NCH2OMe e N-Bn;
[0093] Y é uma ligação ou -(C(R11)(H))t-, em que t é um número inteiro de 1 a 6; e
[0094] R11 em cada ocorrência é independentemente selecionado do grupo consistindo em H e C1-C6 alquila, em que a C1-C6 alquila é opcionalmente substituída ;
[0095] A2 é selecionado do grupo consistindo em N e CR, em que R é selecionado do grupo consistindo em -H, halogênio, -CN, C1-C6 alquila, C2-C6 alquenila, e C2-C6 alquinila, em que a C1-C6 alquila, C2- C6 alquenila, e C2-C6 alquinila são opcionalmente substituídas;
[0096] A3 é selecionado do grupo consistindo em C-D e N;
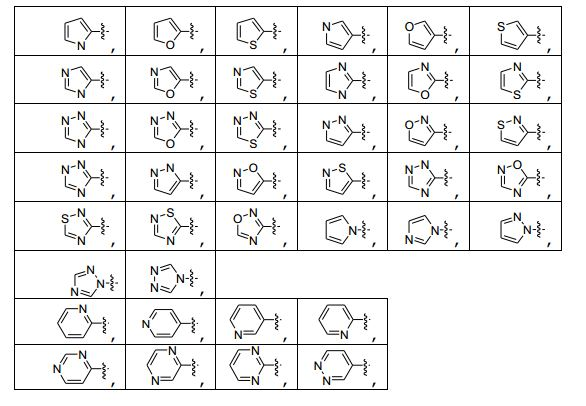
[0097] Ar é um grupo da fórmula C,
[0098] em que,
[0099] A4, A5, A6 e A7 são independentemente selecionados do grupo consistindo em N e -CH2-, com a condição de que não mais do que dois de A4, A5, A6 e A7 possam ser N;
[00100] R2 em cada ocorrência é independentemente selecionado do grupo consistindo em -H, halogênio, trihalometila, -CN, -NO2, -NH2, -OR3, -NR3R4, -S(O)0-2R3, -S(O)2NR3R3, -C(O)OR3, -C(O)NR3R3, - N(R3)SO2R3, -N(R3)C(O)R3, -N(R3)CO2R3, -C(O)R3, C1-C4 alcóxi, C1-C4 alquiltio, -O(CH2)narila, -O(CH2)nheteroarila, -(CH2)0-5 (arila), -(CH2)0-5 (heteroarila), C1-C6 alquila, C2-C6 alquenila, C2-C6 alquinila, -CH2 (CH2)0-4-T2, em que T2 é selecionado do grupo consistindo em -OH, - OMe, -OEt, -NH2, -NHMe, -NMe2, -NHEt e -NEt2, e em que a arila, heteroarila, C1-C6 alquila, C2-C6 alquenila, e C2-C6 alquinila são opcionalmente substituídas; e
[00101] q é um número inteiro de 0 a 4;
[00102] G é um grupo B-L-T, em que
[00103] B é selecionado do grupo consistindo em ausente, -N(R13)-, -N(SO2R13)-, -O-, -S(O)0-2 e -C(= O)-;
[00104] L é selecionado do grupo consistindo em ausente, -C(= S)N(R13)-, -C(= NR14)N(R13)-, -SO2N(R13)-, -SO2-, -C(= O)N(R13)-, - N(R13)-, -C(= O)C1-2alquil-N(R13)-, -N(R13)C1-2alquil-C(= O)-, -C(= O)C0- 1alquil-C(= O)N(R13)-, -C0-4alquileno, -C(= O)C0-1alquil-C(= O)OR3-, - C(= NR14)-C0-1alquil-C(= O)-, -C(= O)-, -C(= O)C0-1alquil-C(= O)- e uma heterociclila de quatro a seis membros opcionalmente substituída contendo entre um e três heteroátomos anulares incluindo pelo menos um nitrogênio; e
[00105] T é selecionado do grupo consistindo em -H, -R13, -Co— 4alquila, -C0-4alquil-Q, -O-C0-4alquil-Q, -C0-4alquil-O-Q, -N(R13)C0-4alquil- Q, -SO2C0-4alquil-Q, -C(= O)C0-4alquil-Q, -C0-4alquil-N(R13)Q e -C(= O)N(R13)-C0-4alquil-Q, em que cada C0-4alquila é opcionalmente substituída ;
[00106] R13 é selecionado do grupo consistindo em -H, -CN, -NO2, - NH2, -OR3, -NR3R4, -S(O)0-2R3, -S(O)2NR3R3, -C(O)OR3, -C(O)NR3R3, - N(R3)SO2R3, -N(R3)C(O)R3, -N(R3)CO2R3, -C(O)R3, -C(O)SR3, C1-C4 alcóxi, C1-C4 alquiltio, -O(CH2)narila, -O(CH2)nheteroarila, -(CH2)0-5 (arila), -(CH2)0-5 (heteroarila), C1-C6 alquila, C2-C6 alquenila, C2-C6 alquinila, -CH2 (CH2)0-4-T2, uma C1-4 alquilcarbonila opcionalmente substituída, e um grupo carboxíclico ou heterocíclico de três a sete membros saturado ou insaturado, em que T2 é selecionado do grupo consistindo em -OH, -OMe, -OEt, -NH2, -NHMe, -NMe2, -NHEt e -NEt2, e em que a arila, heteroarila, C1-C6 alquila, C2-C6 alquenila, e C2-C6 alquinila são opcionalmente substituídas;
[00107] dois R13, juntamente com o átomo ou átomos aos quais eles são ligados, podem combinar-se para formar um heteroalicíclico opcionalmente substituído com entre um e quatro de R60, em que o heteroalicíclico pode ter até quatro heteroátomos anulares, e o heteroalicíclico pode ter uma arila ou heteroarila fundida a ele, em cujo caso a arila ou heteroarila é opcionalmente substituída com um adicional de um a quatro de R60;
[00108] R14 é selecionado do grupo -H, -NO2, -NH2, -N(R3)R4, -CN, - OR3, uma (C1-C6)alquila opcionalmente substituída, uma heteroaliciclilalquila opcionalmente substituída, uma arila opcionalmente substituída, uma arilalquila opcionalmente substituída e um heteroalicíclico opcionalmente substituído,
[00109] cada R3 é independentemente selecionado do grupo consistindo em -H e R4;
[00110] R4 é selecionado do grupo consistindo em uma (C1- C6)alquila, uma arila, uma arilalquila inferior, uma heterociclila e uma heterociclilalquila inferior, cada uma das quais é opcionalmente substituída, ou
[00111] R3 e R4, tomados juntamente com um nitrogênio comum ao qual eles são ligados, formam uma heterociclila de cinco a sete membros opcionalmente substituída, a heterociclila de cinco a sete membros opcionalmente substituída opcionalmente contendo pelo menos um heteroátomo anular adicional selecionado do grupo consistindo em N, O, S e P;
[00112] R60 é selecionado do grupo consistindo em -H, halogênio, trihalometila, -CN, -NO2, -NH2, -OR3, -NR3R4, -S(O)0-2R3, -SO2NR3R3, - CO2R3, -C(O)NR3R3, -N(R3)SO2R3, -N(R3)C(O)R3, -N(R3)CO2R3, - C(O)R3, uma (C1-C6)alquila opcionalmente substituída, uma arila opcionalmente substituída, uma heteroarilalquila opcionalmente substituída e uma arilalquila opcionalmente substituída;
[00113] dois R60, quando ligados a um carbono não-aromático, podem ser oxo;
[00114] Q é um sistema de anel de cinco a dez membros, opcionalmente substituído por entre zero e quatro de R20;
[00115] R20 é selecionado do grupo consistindo em -H, halogênio, trihalometila, -CN, -NO2, -NH2, -OR3, -OCF3, -NR3R4, -S(O)0-2R3, - S(O)2NR3R3, -C(O)OR3, -C(O)NR3R3, -N(R3)SO2R3, -N(R3)C(O)R3, - N(R3)C(O)OR3, -C(O)R3, -C(O)SR3, C1-C4 alcóxi, C1-C4 alquiltio, - O(CH2)narila, -O(CH2)nheteroarila, -(CH2)0-5 (arila), -(CH2)0-5 (heteroarila), C1-C6 alquila, C2-C6 alquenila, C2-C6 alquinila, -CH2 (CH2)0-4-T2, uma C1-4 alquilcarbonila opcionalmente substituída, C1-4 alcóxi, um amino opcionalmente substituído por C1-4 alquila em que C1C4 alquila pode opcionalmente substituída por C1-4 alcóxi e um grupo carboxíclico ou heterocíclico de três a sete membros saturado ou insaturado, em que T2 é selecionado do grupo consistindo em -OH, - OMe, -OEt, -NH2, -NHMe, -NMe2, -NHEt e -NEt2, e em que a arila, heteroarila, C1-C6 alquila, C2-C6 alquenila, e C2-C6 alquinila são opcionalmente substituídas;
[00116] cada R38 é independentemente selecionado de halo, ciano, nitro, trifluorometóxi, trifluorometila, azido, -C(O)R40, -C(O)OR40, - OC(O)R40, -OC(O)OR40, -NR36C(O)R39, -C(O)NR36R39, -NR36R39, - OR37, -SO2NR36R39, C1-C6 alquila, -(CH2)jO(CH2)iNR36R39, - (CH2)nO(CH2)iOR37, -(CH2)nOR37, -S(O)j(C1-C6 alquila), -(CH2)n(C6-C10 arila), -(CH2)n(heterociclila de 5 a 10 membros); -C(O)(CH2)n(C6-C10 arila), -(CH2)nO(CH2)j(C6-C10 arila), -(CH2)nO(CH2)i(heterociclila de 5 a 10 membros), -C(O)(CH2)n(heterociclila de 5 a 10 membros), - (CH2)jNR39 (CH2)iNR36R39, -(CH2)jNR39CH2C(O)NR36R39, -(CH2)jNR39 (CH2)iNR37C(O)R40, -(CH2)jNR39 (CH2)nO(CH2)iOR37, -(CH2)jNR39 (CH2)iS(O)j(C1-C6 alquila), -(CH2)jNR39 (CH2)nR36, -SO2 (CH2)n(C6-C10 arila), -SO2 (CH2)n(heterociclila de 5 a 10 membros), -(CH2)nNR36R39, - NR37SO2NR36R39, SO2R36, C2-C6 alquenila, C3-C10 cicloalquila e C1-C6 alquilamino, em que j é um número inteiro variando de 0 a 2, n é um número inteiro variando de 0 a 6, i é um número inteiro variando de 2 a 6, as porções -(CH2)i- e -(CH2)n- dos grupos R38 anteriores opcionalmente incluem uma ligação dupla ou tripla de carbono- carbono, onde n é um número inteiro entre 2 e 6, e as porções de alquila, arila e heterociclila dos grupos R38 anteriores são opcionalmente substituídas por um ou mais substituintes independentemente selecionados de halo, ciano, nitro, trifluorometila, azido, -OH, -C(O)R40, -C(O)OR40, -OC(O)R40, -OC(O)OR40, - NR36C(O)R39, -C(O)NR36R39, -(CH2)nNR36R39, C1-C6 alquila, C3-C10 cicloalquila, -(CH2)n(C6-C10 arila), -(CH2)n(heterociclila de 5 a 10 membros), -(CH2)nO(CH2)iOR37, e -(CH2)nOR37, em que n é um número inteiro variando de 0 a 6 e i é um número inteiro variando de 2 a 6;
[00117] cada R36 e R39 é independentemente selecionado do grupo consistindo em H, -OH, C1-C6 alquila, C3-C10 cicloalquila, -(CH2)n(C6- C10 arila), -(CH2)n(heterociclila de 5 a 10 membros), - (CH2)nO(CH2)iOR37, -(CH2)nCN(CH2)nOR37, -(CH2)nCN(CH2)nR37, e - (CH2)nOR37, em que n é um número inteiro variando de 0 a 6 e i é um número inteiro variando de 2 a 6, e as porções de alquila, arila e heterociclila dos grupos R36 e R39 anteriores são opcionalmente substituídas por um ou mais substituintes independentemente selecionados de -OH, halo, ciano, nitro, trifluorometila, azido, -C(O)R40, -C(O)OR40, -CO(O)R40, -OC(O)OR40, -NR37C(O)R41, -C(O)NR37R41, - NR37R41, -C1-C6 alquila, -(CH2)n(C6-C10 arila), -(CH2)n(heterociclila de 5 a 10 membros), -(CH2)nO(CH2)iOR37, e -(CH2)nOR37, em que n é um número inteiro variando de 0 a 6 e i é um número inteiro variando de 2 a 6, com a condição de que quando R36 e R39 forem ambos ligados ao mesmo nitrogênio, então R36 e R39 não sejam ambos ligados ao nitrogênio diretamente através de um oxigênio;
[00118] cada R40 é independentemente selecionado de H, C1-C10 alquila, -(CH2)n(C6-C10 arila), C3-C10 cicloalquila, e -(CH2)n(heterociclila de 5 a 10 membros), em que n é um número inteiro variando de 0 a 6;
[00119] cada R37 e R41 é independentemente selecionado de H, ou OR36, C1-C6 alquila e C3-C10 cicloalquila;
[00120] cada R42 e R43 é independentemente selecionado do grupo consistindo em H, C1-C6 alquila, -Y-(C3-C10 cicloalquila), -Y-(C6-C10 arila), -Y-(C6-C10 heteroarila), -Y-(heterociclila de 5 a 10 membros), -Y- O-Y1-OR37, -Y1-CO2-R37, e -Y-OR37, em que, Y é uma ligação ou é - (C(R37)(H))n, em que n é um número inteiro variando de 1 a 6, Y1 é - (C(R37)(H))n, e e as porções de alquila, cicloalquila, arila, heteroarila e heterociclila dos grupos R42 e R43 anteriores são opcionalmente substituídas por 1 ou mais substituintes independentemente selecionados de R44; ou
[00121] R42 e R43 tomados juntamene com o nitrogênio ao qual eles são ligados formam um anel de C5-C9 azabicíclico, aziridinila, azetidinila, pirrolidinila, piperidinila, piperazinila, morfolinila, tiomorfolinila, isoquinolinila, ou di-hidroisoquinolinila, em que o referido anel de C5-C9 azabicíclico, aziridinila, azetidinila, pirrolidinila, piperidinila, piperazinila, morfolinila, tiomorfolinila, isoquinolinila, ou di- hidroisoquinolinila é opcionalmente substituído por 1 a 5 substituintes de R44, com a condição de que R42 e R43 não sejam ambos ligados ao nitrogênio diretamente através de um oxigênio;
[00122] cada R44 é independentemente selecionado do grupo consistindo em halo, ciano, nitro, trifluorometóxi, trifluorometila, azido, - C(O)R40, -C(O)OR40, -OC(O)R40, -OC(O)OR40, -NR36C(O)R39, - C(O)NR36R39, -NR36R39, -OR37, -SO2NR36R39, -SO2R36, -NR36SO2R39, - NR36SO2NR37R41, C1-C6 alquila, C2-C6 alquenila, C2-C6 alquinila, C3C10 cicloalquila, -C1-C6 alquilamino, -(CH2)jO(CH2)iNR36R39, - (CH2)nO(CH2)iOR37, -(CH2)nOR37, -S(O)j(C1-C6 alquila), -(CH2)n(C6-C10 arila), -(CH2)n(heterociclila de 5 a 10 membros), -C(O)(CH2)n(C6-C10 arila), -(CH2)nO(CH2)j(C6-C10 arila), -(CH2)nO(CH2)i(heterociclila de 5 a 10 membros), -C(O)(CH2)n(heterociclila de 5 a 10 membros), - (CH2)jNR39 (CH2)iNR36R39, -(CH2)jNR39CH2C(O)NR36R39, -(CH2)jNR39 (CH2)iNR37C(O)R40, -(CH2)jNR39 (CH2)nO(CH2)iOR37, -(CH2)jNR39 (CH2)iS(O)j(C1-C6 alquila), -(CH2)jNR39 (CH2)nR36, -SO2 (CH2)n(C6-C10 arila), e -SO2 (CH2)n(heterociclila de 5 a 10 membros) em que, j é um número inteiro de 0 a 2, n é um número inteiro de 0 a 6 e i é um número inteiro variando de 2 a 6, as porções -(CH2)i- e -(CH2)n1- dos grupos R44 anteriores opcionalmente incluem uma ligação dupla ou tripla de carbono-carbono em que n é um número inteiro de 2 a 6, e as porções alquila, arila e heterociclila dos grupos R44 anteriores são opcionalmente substituídos por 1 ou mais substituintes independentemente selecionados do grupo consistindo em halo, ciano, nitro, trifluorometila, azido, -OH, -C(O)R40, -C(O)OR40, -OC(O)R40, - OC(O)OR40,-NR36C(O)R39, -C(O)NR36R39, -(CH2)nNR36R39, -SO2R36, - SO2NR36R39, C1-C6 alquila, C3-C10 cicloalquila, -(CH2)n(C6-C10 arila), - (CH2)n(heterociclila de 5 a 10 membros), -(CH2)nO(CH2)iOR37 e - (CH2)nOR37, em que n é um número inteiro de 0 a 6 e i é um número inteiro de 2 a 6; e
[00123] Z é selecionado do grupo consistindo em -O-, -S- e -NR5-, em que R5 é selecionado do grupo consistindo em H, uma opcionalmente substituída (C1-C5)acila e C1-C6 alquil-O-C(O), em que C1-C6 alquila é opcionalmente substituída ;
[00124] com a condição de que
[00125] quando G for NR13 (C= Zp)NR13C(O)(C(X)(X1))-Q, em que Zp é O, S ou NH, X e X1 independentemente representem H, C1-C6 alquila, halo, ciano ou nitro, em que a C1-C6 alquila é opcionalmente substituída, ou X e X1 tomados juntamente com o átomo ao qual eles são ligados, formam a C3-C7 cilcoalquila, Q é selecionado do grupo consistindo em cicloalquila, heterociclila, arila e heteroarila, em que cada uma das referidas cicloalquila, heterociclila, arila e heteroarila é opcionalmente substituída com 1 a 3 R20, Ar é fenila opcionalmente substituída com 1 a 4 porções independentemente selecionadas do grupo consistindo em hidrogênio, halo, trihalometila, -CN, -NO2, -NH2, - OR3, -NR3R4, -C(O)OR3, -C(O)R3, C1-C4 alcóxi, C1-C6 alquiltio, C1-C6 alquila, C2-C6 alquenila ou C2-C6 alquinila, em que C1-C6 alquila, C2-C6 alquenila e C2-C6 alquinila são opcionalmente substituídas, e Z é O, S ou NH, então D não é R7, R1 ou R21;
[00126] quando D é selecionado do grupo consistindo em -H, halogênio, nitro, azido, -NR6aR6b, -NR6aSO2R6b, -NR6aC(O)R6b, - OC(O)R6b, -NR6aC(O)OR6b, -OC(O)NR6aR6b,-OR6a, -SR6a, -S(O)R6a, - SO2R6a, -SO3R6a, -SO2NR6aR6b, -COR6a, -CO2R6a, -CONR6aR6b, -(C1- C4)fluoroalquila, -(C1-C4)fluoroalcóxi, -(CZ3Z4)aCN, e uma porção selecionada do grupo consistindo em -(CZ3Z4)a-arila, -(CZ3Z4)a- heterociclo, (C2-C6)alquinila, -(CZ3Z4)a-(C3-C6)cicloalquila, -(CZ3Z4)a- (C5-C6)cicloalquenila, (C2-C6) alquenila e (C1-C6)alquila, em que as referidas porções opcionalmente substituídas por 1 a 3 grupos Y2 independentemente selecionados, onde a é 0,1, 2, ou 3, e em que quando a é 2 ou 3, as unidades CZ3Z4 podem ser iguais ou diferentes, A1 é -S-, A2 é -N- ou -CR-, em que R é H, F, Cl, CF3, CH3, OCH3 ou OCF3, A3 é -CH-, Z é -O- ou -S-, A6 e A7 são -CH2-, então B-L-T não é - X3-C(O)-NH-R33, em que X3 é O ou CR2aR2b, cada de R2a e R2b é independentemente selecionado do grupo consistindo em H, halogênio, ou uma porção, opcionalmente substituída com 1 a 3 grupos X4a independentemente selecionados, em que X4a é independentemente selecionado do grupo consistindo em (C1- C6)alcóxi, (C1-C6)alquilamina e (C1-C6)alquila, em que qualquer número dos átomos de hidrogênio nos grupos de (C1-C6)alcóxi e (C1- C6)alquila podem ser opcionalmente substituídos com F, ou R2a e R2b juntos podem ser oxo ou uma porção, opcionalmente substituída com 1 a 3 grupos X4 independentemente selecionados, selecionado do grupo consistindo em (C3-C6)cicloalquila, heterocicloalquila de 3 a 6 membros e = CH-(C1-C5)alquila, R33 é H ou uma porção, opcionalmente substituída com 1 a 3 grupos Y2 independentemente selecionados, selecionado do grupo consistindo em -(CZ1Z2)sCN, - (CZ1Z2)s-(C3-C8)cicloalquila, -(CZ1Z2)s-(C5-C8)cicloalquenila, (C2- C6)alquenila, (C2-C6)alquinila, -(CZ1Z2)s-arila, -(CZ1Z2)s-heterociclo e (C1-C8)alquila, onde s é 0, 1, 2 ou 3, e em que quando s é 2 ou 3, as unidades CZ1Z2 podem ser iguais ou diferentes, e em que Z1 e Z2 são cada qual independentemente selecionado do grupo consistindo em H, F, e (C1-C6)alquila ou cada Z1 e Z2 são selecionados juntos para formar um carbociclo ou dois grupos Z1 ou Z2 sobre átomos de carbono adjacentes são selecionados juntos para opcionalmente formar um carbociclo;
[00127] quando D é selecionado do grupo consistindo em H, C1-C6 alquila, C3-C10 cicloalquila, -C(O)NR42R43, -C(O)(C6-C10 arila), - (CH2)n(C6-C10 arila), -(CH2)n(heterociclila de 5 a 10 membros), - (CH2)nNR42R43, -SO2NR42R43 e -CO2R42, em que n é um número inteiro de 0 a 6, e em que as referidas porções C1-C6 alquila, -C(O)(C6-C10 arila), -(CH2)n(C6-C10 arila), -(CH2)n(heterociclila de 5 a 10 membros) são não-substituídas ou substituídas por um ou mais substituintes selecionados de R38, com a condição de que R38 não seja -(CH2)3- 6NR36R39, -NR37SO2NR36R39, SO2R36, ou C2-C6 alquenila, A1 é -S-, A2 é -N- ou -CH-, A3 é -CH-, e Z é -O-, -S- ou -NH-, então Ar-G não é grupo C6 arila não-substituída ou heterociclila de 6 membros ou grupo C6 arila ou heterociclila de 6 membros substituído por 1 a 5 substituintes selecionados de R38, com a condição de que R38 não seja -(CH2)3- 6NR36R39, -NR37SO2NR36R39, SO2R36, ou C2-C6 alquenila;
[00128] quando D é selecionado do grupo consistindo em imidazolila, oxazolila, oxadiazolila, isoxazolila, tiazolila e tiadiazolila, em que as referidas imidazolila, oxazolila, oxadiazolila, isoxazolila, tiazolila e tiadiazolila são opcionalmente substituídas por 1 a 5 substituintes selecionados de R38, com a condição de que R38 não seja nitro, azido, -C(O)OR40, -OC(O)R40, -OC(O)OR40, -C(O)(CH2)n(C6-C10 arila), -(CH2)nO(CH2)j(C6-C10 arila), C3-C10 cicloalquila ou C1-C6 alquilamino, R36 e R39 não sejam -OH, C3-C5 cicloalquila, - (CH2)nCN(CH2)nOR37 ou -(CH2)nCN(CH2)R37, R37 e R41 não sejam - OR36 ou C3-C10 cicloalquila, e R40 não é C3-C10 cicloalquila, A1 é -S-, A2 é N, CH ou C-CN, A3 é -CH-, e Z é -NH-, então Ar-G não é heterociclila de 6 membros não-substituída ou heterociclila de 6 membros opcionalmente substituída por 1 a 5 substituintes selecionados de R38, com a condição de que R38 não seja nitro, azido, -C(O)OR40, - OC(O)R40, -OC(O)OR40, -C(O)(CH2)n(C6-C10 arila), -(CH2)nO(CH2)j(C6- C10 arila), C3-C10 cicloalquila ou C1-C6 alquilamino, R36 e R39 não sejam -OH, C3-C5 cicloalquila, -(CH2)nCN(CH2)nOR37 ou -(CH2)nCN(CH2)R37, R37 e R41 não sejam -OR36 ou C3-C10 cicloalquila, e R40 não é C3-C10 cicloalquila;
[00129] quando D é selecionado do grupo consistindo em H, C1-C6 alquila, -C(O)NR36R39, -C(O)(C6-C10 arila), -(CH2)n(C6-C10 arila), e - (CH2)n(heterociclila de 5 a 10 membros), em que os referidos grupos, diferentes de H, são não-substituídos ou substituídos por um a cinco substituintes selecionados de R38, com a condição de que R38 não seja -(CH2)3-6NR36R39, -NR37SO2NR36R39, SO2R36, C2-C6 alquenila, C3-C10 cicloalquila ou C1-C6 alquilamino, R36 e R39 não sejam -OH, C3-C10 cicloalquila, -(CH2)nCN(CH2)nOR37 ou -(CH2)nCN(CH2)nR37, R37 e R41 não sejam OR36 ou C3-C10 cicloalquila e R40 não é C3-C10 cicloalquila, A1 é -S- quando A3 é -CH- ou A1 é -CH- quando A3 é -S-, A2 é -N- ou - CH-, e Z é -NH- ou N-(C1-C6 alquila), então Ar-G não é C6 arila não- substituída ou heterociclila de 6 membros ou C6 arila ou heterociclila de 6 membros com 1 a 5 substituintes selecionados do grupo R38, com a condição de que R38 não seja -(CH2)3-6NR36R39, -NR37SO2NR36R39, SO2R36, C2-C6 alquenila, C3-C10 cicloalquila ou C1-C6 alquilamino, R36 e R39 não sejam -OH, C3-C10 cicloalquila, -(CH2)nCN(CH2)nOR37 ou - (CH2)nCN(CH2)nR37, R37 e R41 não sejam OR36 ou C3-C10 cicloalquila e R40 não é C3-C10 cicloalquila;
[00130] quando D é selecionado do grupo consistindo em - C(O)NR42R43, -(CH2)nNR42R43, -NR42C(= O)R43, -SO2R42, -SO2NR42R43, -NR37SO2R42, -NR37SO2NR42R43, -C(= N-OR42)R43, -C(= NR42)R43, - NR37C(= NR42)R43, -C(= NR42)NR37R43, -NR37C(= NR42)NR37R43, - C(O)R42 e -CO2R42, em que as porções alquila, arila e heterociclila dos grupos R42 e R43 anteriores são opcionalmente substituídas com 1 a 3 substituintes independentemente selecionados do grupo R44, com a com a condição de que R44 não seja nitro, azido, -C(O)OR40, - OC(O)R40, OC(O)OR40, C3-C10 cicloalquila ou C1-C6 alquilamina, A1 é - S-, A2 é N, CH ou C(CN), A3 é CH, e Z é NH ou N-(C1-C6)alquila, então Ar-G não seja heterociclila de 6 membros, em que a heterociclila de 6 membros é opcionalmente substituída por 1 a 5 substituintes do grupo R44, com a condição de que R44 não seja nitro, azido, -C(O)OR40, - OC(O)R40, OC(O)OR40, C3-C10 cicloalquila ou C1-C6 alquilamina;
[00131] quando D é -C^CH ou -C=C-(CR45R45)n-R46, A1 é -S- e A3 é -CH- ou A1 é -CH- e A3 é -S-, A2 é N, e Z é NH ou N-(C1-C6)alquila, então Ar-G não é selecionado do grupo consistindo em arila opcionalmente substituída com um ou mais substituintes selecionados do grupo consistindo em halo, alquinila, -CF3, -(CH2)nOR57, - (CH2)nSR57, -NO2, C1-C6 alquila, -CN, -SO2R50, -(CH2)narila e - (CH2)nNR50R51, e heteroarila opcionalmente substituída com um ou mais substituintes selecionados do grupo consistindo em halo, alquinila, -CF3, -(CH2)nOR57, -(CH2)nSR57, -NO2, C1-C6alquila, -CN, - SO2R50, -(CH2)narila e -(CH2)nNR50R51, em que R57 é selecionado do grupo consistindo em H, C1-C6 alquila, -(CH2)nNR50R51, - (CH2)nheterociclila, -(CH2)narila em cuja arila é opcionalmente substituída com um ou mais substitutentes selecionados do grupo consistindo em halo, -CF3, C1-C6alcóxi, -NO2, C1-C6alquila, -CN, - SO2R50, e -(CH2)nNR50R51, arilC1-C6alquenileno em cuja arila é opcionalmente substituída por um ou mais substituintes selecionados do grupo consistindo em halo, -CF3, C1-C6alcóxi, -NO2, C1-C6alquila, - CN, -SO2R50, e -(CH2)nNR50R51, heteroarilC1-C6alquenileno em cuja heteroarila é opcionalmente substituída por um ou mais substituintes selecionados do grupo consistindo em halo, -CF3, C1-C6 alcóxi, -NO2, C1-C6 alquila, -CN, -SO2R50, e -(CH2)nNR50R51, e -(CH2)nheteroarila em cuja heteroarila é opcionalmente substituído por um ou mais substituintes selecionados do grupo consistindo em halo, -CF3, C1- C6alcóxi, -NO2, C1-C6alquila, -CN, -SO2R50, e -(CH2)nNR50R51; e
[00132] quando D é o grupo definido por -(Z11)-(Z12)m-(Z13)m1, A1 é - S- e A3 é CH, ou A1 é CH e A3 é S, A2 é N, e Z é NH, N-(C1-C3alquila) ou N-C(O)R53, então Ar-G não é o grupo definido por -(Q1)-(Q2)0-1- (Q3)0-1, em que Q1 é arileno, heteroarileno, arila ou aralquila, Q2 é O, S(O)2, ou S, e Q3 é aralquila, heteroarila, ou arila.
[00133] Em uma modalidade preferida dos compostos de acordo com a presente invenção, D é definido pelo grupo R7, em que R7 é selecionado do grupo consistindo em -H, halogênio, C1-C6 alquila, C3C10 cicloalquila, -C(O)NR42R43, -C(O)(C6-C10 arila), -C(O)(heterociclila), -C(O)(heteroarila), -Y-(C6-C10 arila), -Y-(heterociclila de 5 a 10 membros), -Y-(heteroarila), -S-arila, -S-C1-C6 alquila, -SO-C1-C6 alquila, -SO2-C1-C6 alquila, -Y-NR42R43, -SO2NR42R43 e -C(O)OR6a, em que os grupos R7 anteriormente mencionados diferentes de -H e halogênio são opcionalmente substituídos por 1 a 5 R38.
[00134] Em uma modalidade preferida dos compostos de acordo com a presente invenção, D é definido pelo grupo R7, em que R7 é selecionado do grupo consistindo em -H, -C(O)NR42R43, -Y- (heterociclila de 5 a 10 membros), -Y-(C6-C10 arila), -Y-(heteroarila), -Y- NR42R43, SO2NR42R43 e C(O)OR42, em que os grupos R7 anteriormente mencionados diferentes de -H são opcionalmente substituídos por 1 a 5 R38.
[00135] Em uma modalidade preferida dos compostos de acordo com a presente invenção, R7 é selecionado do grupo consistindo em - (CH2)n(heterociclila de 5 a 10 membros), -C(O)NR42R43, -SO2NR42R43 e -CO2R42, em que o referido grupo R7 -(CH2)n(heterociclila de 5 a 10 membros) é não-substituído ou substituído por um ou mais grupos R38.
[00136] Em uma modalidade preferida dos compostos de acordo com a presente invenção, R7 é selecionado do grupo consistindo em - (CH2)n(heterociclila de 5 a 10 membros), e -C(O)NR42R43.
[00137] Em uma modalidade preferida dos compostos de acordo com a presente invenção, R7 é -C(O)NR42R43, em que R42 e R43 são independentemente selecionados de H, (C1-C6)alquila, (C3- C10)cicloalquila, -(CH2)n(C3-C10 cicloalquila), -(CH2)n(C6-C10 arila), - (CH2)n(heterociclila de 5 a 10 membros), -(CH2)nO(CH2)iOR37, - (CH2)nOR37, em que n é um número inteiro de 0 a 6, i é um número inteiro de 2 a 6, e as porções alquila, arila e heterociclila dos referidos R42 e R43 são não-substituídos ou substituídos com um ou mais substituintes independentemente selecionados de R38, ou R42 e R43 são tomados junto com o nitrogênio ao qual eles são ligados para formar um anel de C5-C9 azabicíclico, aziridinila, azetidinila, pirrolidinila, piperidinila, piperazinila, morfolinila, tiomorfolinila, isoquinolinila, ou di-hidroisoquinolinila, em que os referidos aneis de C5-C9 azabicíclico, aziridinila, azetidinila, pirrolidinila, piperidinila, piperazinila, morfolinila, tiomorfolinila, isoquinolinila, ou di- hidroisoquinolinila são não-substituídos ou substituídos com 1 a 5 substituintes de R38, onde R42 e R43 não sejam ambos ligados ao nitrogênio diretamente através de um oxigênio.
[00138] Em uma modalidade preferida dos compostos de acordo com a presente invenção, R7 é -C(O)NR42R43, em que R42 e R43 são tomados junto com o nitrogênio ao qual eles são ligados para formar um anel de C5-C9 azabicíclico, aziridinila, azetidinila, pirrolidinila, piperidinila, piperazinila, morfolinila, tiomorfolinila, isoquinolinila, ou di- hidroisoquinolinila, em que o referido anel de C5-C9 azabicíclico, aziridinila, azetidinila, pirrolidinila, piperidinila, piperazinila, morfolinila, tiomorfolinila, isoquinolinila, ou di-hidroisoquinolinila são não- substituídos ou substituídos com 1 a 5 substituintes de R38.
[00139] Em uma modalidade preferida dos compostos de acordo com a presente invenção, R7 é -C(O)NR42R43, em que R42 e R43 são tomados junto com o nitrogênio ao qual eles são ligados para formar um anel de pirrolidinila, piperidinila, piperazinila, morfolinila, tiomorfolinila, isoquinolinila, ou di-hidroisoquinolinila, em que o referido anel de pirrolidinila, piperidinila, piperazinila, morfolinila, tiomorfolinila, isoquinolinila, ou di-hidroisoquinolinila é não-substituído ou substituído com 1 a 5 substituintes de R38.
[00140] Em uma modalidade preferida dos compostos de acordo com a presente invenção, R7 é -C(O)NR42R43, em que R42 e R43 são tomados junto com o nitrogênio ao qual eles são ligados para formar um anel pirrolidinila, piperidinila, piperazinila, morfolinila, ou tiomorfolinila, em que os referidos anéis de pirrolidinila, piperidinila, piperazinila, morfolinila, ou tiomorfolinila são não-substituídos ou substituídos com 1 a 5 substituintes de R38.
[00141] Em uma modalidade preferida dos compostos de acordo com a presente invenção, R7 é -C(O)NR42R43, em que R42 e R43 são tomados junto com o nitrogênio ao qual eles são ligados para formar um anel de pirrolidinila ou piperidinila, em que o referido anel de pirrolidinila ou piperidinila é não-substituído ou substituído com 1 a 5 substituintes de R38.
[00142] Em uma modalidade preferida dos compostos de acordo com a presente invenção, R7 é -C(O)NR42R43, em que R42 e R43 são tomados junto com o nitrogênio ao qual eles são ligados para formar um anel de pirrolidinila, em que a referida pirrolidinila é não-substituída ou substituído com 1 a 5 substituintes de R38.
[00143] Em uma modalidade preferida dos compostos de acordo com a presente invenção, R7 é -C(O)NR42R43, em que R42 e R43 são tomados junto com o nitrogênio ao qual eles são ligados para formar um anel de pirrolidin-1-ila, em que a referida pirrolidin-1-ila é não- substituída ou substituída por 1 a 5 substituintes de R38.
[00144] Em uma modalidade preferida dos compostos de acordo com a presente invenção, R7 é -(CH2)n(heterociclila de 5 a 10 membros) grupo, em que o referida grupo -(CH2)n(heterociclila de 5 a 10 membros) é não-substituído ou substituído por 1 a 5 grupos R38.
[00145] Em uma modalidade preferida dos compostos de acordo com a presente invenção, R7 é grupo -(CH2)n(heterociclila de 5-8 membros), o referido grupo -(CH2)n(heterociclila de 5-8 membros) grupo é não-substituído ou substituído por 1 a 5 grupos R38.
[00146] Em uma modalidade preferida dos compostos de acordo com a presente invenção, R7 é grupo -(CH2)n(heterociclila de 5 ou 6 membros), o referido grupo -(CH2)n(heterociclila de 5 ou 6 membros) é não-substituído ou substituído por 1 a 5 grupos R38.
[00147] Em uma modalidade preferida dos compostos de acordo com a presente invenção, R7 é grupo -(CH2)n(heterociclila de 5 membros), o referido grupo -(CH2)n(heterociclila de 5 membros) é não- substituído ou substituído por 1 a 5 grupos R38.
[00148] Em uma modalidade preferida dos compostos de acordo com a presente invenção, R7 é -(CH2)ntiazolila, em que n é um número inteiro de 0 a 6, a referida -(CH2)ntiazolila é não-substituída ou substituída por 1 a 5 grupos R38.
[00149] Em uma modalidade preferida dos compostos de acordo com a presente invenção, R7 é a tiazolila, a referida tiazolila é não- substituída ou substituída por 1 a 5 grupos R38.
[00150] Em uma modalidade preferida dos compostos de acordo com a presente invenção, R7 é uma imidazolila, a referida imidazolila é não-substituída ou substituída por 1 a 5 grupos R38.
[00151] Em uma modalidade preferida dos compostos de acordo com a presente invenção, R7 é selecionado do grupo consistindo em imidazolila, oxazolila, oxadiazolila, isoxazolila, tiazolila e tiadiazolila, em que a imidazolila, oxazolila, oxadiazolila, isoxazolila, tiazolila e tiadiazolila, cada uma das quais é opcionalmente substituída por 1 a 5 grupos R38.
[00152] Em uma modalidade preferida dos compostos de acordo com a presente invenção, R7 é selecionado do grupo consistindo em halo, -CO2H, -CONH2 e -CSNH2.
[00153] Em uma modalidade preferida dos compostos de acordo com a presente invenção, R7 é um grupo heteroarila opcionalmente substituída por uma ou mais porção selecionada do grupo consistindo em halo, ciano, nitro, trifluorometóxi, trofluorometila, azido, -C(O)R40, - C(O)OR40, -OC(O)R40, -OC(O)OR40, -NR36C(O)R39, -C(O)NR36R39, - NR36R37, -OR37, -SO2NR36R39, (C1-C6)alquila, (C3-C10)cicloalquila, - (CH2)jO(CH2)iNR36R39, -(CH2)nO(CH2)iOR37, -(CH2)nOR37, -S(O)j(C1-C6 alquila), -(CH2)n(C6-C10 arila), -(CH2)n(heterociclila de 5 a 10 membros), -C(O)(CH2)n(C6-C10 arila), -(CH2)nO(CH2)j(C6-C10 arila), - (CH2)nO(CH2)i(heterociclila de 5 a 10 membros), - C(O)(CH2)n(heterociclila de 5 a 10 membros),- (CH2)jNR39 (CH2)iNR36R39, -(CH2)jNR39CH2C(O)NR36R39,-(CH2)jNR39 (CH2)iNR37C(O)R40, (CH2)jNR39 (CH2)nO(CH2)iOR37, -(CH2)jNR39 (CH2)iS(O)j(C1-C6 alquila), -(CH2)jNR39, -(CH2)nR36, -SO2 (CH2)n(C6-C10 arila), e -SO2 (CH2)n(heterociclila de 5 a 10 membros), em que j é um número inteiro de 0 a 2, n é um número inteiro de 0 a 6, i é um número inteiro de 2 a 6, as porções -(CH2)i- e -(CH2)n- dos referidos grupos substituintes opcionalmente incluem uma ligação dupla ou tripla de carbono-carbono, onde n é um número inteiro entre 2 e 6, e as porções alquila, arila e heterociclila dos grupos substituintes são não- substituídas ou substituídas com um ou mais substituintes independentemente selecionados de halo, ciano, nitro, trifluorometila, azido, -OH, -C(O)R40, -C(O)OR40, -OC(O)R40, -OC(O)OR40, - NR36C(O)R39, -C(O)NR36R39, -(CH2)nNR36R39, (C1-C6)alquila, (C3- C10)cicloalquila, -(CH2)n(C6-C10 arila), -(CH2)n(heterociclila de 5 a 10 membros), -(CH2)nO(CH2)iOR37, e -(CH2)nOR37, em que n é um número inteiro de 0 a 6 e i é um número inteiro de 2 a 6, e em que R36 e R39 são independentemente selecionados do grupo consistindo em H, - OH, (C1-C6)alquila, (C3-C10)cicloalquila, -(CH2)n(C6-C10 arila), - (CH2)n(heterociclila de 5 a 10 membros), -(CH2)nO(CH2)iOR37 e - (CH2)nOR37, em que n é um número inteiro de 0 a 6 e i é um número inteiro de 2 a 6, e as porções alquila, arila e heterociclila dos grupos R36 e R39 são não-substituídas ou substituídas com um ou mais substituintes independentemente selecionados de hidróxi, halo, ciano, nitro, trifluorometila, azido, -C(O)R40, -C(O)OR40, -CO(O)R40, - OC(O)OR40, -NR37C(O)R41, -C(O)NR37R41, -NR37R41, (C1-C6)alquila, - (CH2)n(C6-C10 arila), -(CH2)n(heterociclila de 5 a 10 membros), - (CH2)nO(CH2)iOR37, e -(CH2)nOR37, em que n é um número inteiro de 0 a 6 e i é um número inteiro de 2 a 6, onde quando R36 e R39 são ambos ligados ao mesmo nitrogênio, então R36 e R39 não são ambos ligados ao nitrogênio diretamente através de um oxigênio.
[00154] Em uma modalidade preferida dos compostos de acordo com a presente invenção, R7 é selecionado do grupo consistindo em H, -(C1-C6)alquila, -C(O)NR36R37, -C(O)(C6-C10 arila), -(CH2)n(C6-C10 aril) e -(CH2)n(heterociclila de 5 a 10 membros), em que os grupos R7 exceto H são opcionalmente substituídos por 1 a 5 grupos R38. Preferivelmente R7 é -(CH2)n(C6-C10 aril) e -(CH2)n(heterociclila de 5 a 10 membros), opcionalmente substituída por 1 a 5 grupos de R38, mais preferivelmente fenila ou piridila, opcionalmente substituída por 1 a 5 grupos R38.
[00155] Em uma modalidade preferida dos compostos de acordo com a presente invenção, R7 é selecionado do grupo consistindo em H, -(C1-C6)alquila, -C(O)NR36R37, -C(O)(C6-C10 arila), -(CH2)n(C6-C10 aril) e -(CH2)n(heterociclila de 5 a 10 membros), em que os grupos R7 exceto H são opcionalmente substituídos por terc-butil-dimetil-silanila e 1 a 3 grupos R38.
[00156] Em uma modalidade preferida dos compostos de acordo com a presente invenção, R7 é selecionado do grupo consistindo em - C(O)NR42R43, -(CH2)nNR42R43, -NR42C(= O)R43, -SO2R42, -SO2NR42R43, -NR37SO2R42, -NR37SO2NR42R43, -C(= N-OR42)R43, -C(= NR42)R43, - NR37C(= NR42)R43, -C(= NR42)NR37R43, -NR37C(= NR42)NR37R43, - C(O)R42, -CO2R42, em que cada R42 e R43 é independentemente selecionado do grupo consistindo em H, (C1-C6)alquila, -(CH2)n(C3- C10)cicloalquila), -(CH2)n(C6-C10 arila), -(CH2)n(heterociclila de 5 a 10 membros), -(CH2)nO(CH2)iOR37, -(CH2)nOR37, em que n é um número inteiro de 0 a 6 e i é um número inteiro de 2 a 6, e as porções alquila, arila e heterociclila dos anteriores grupos R42 e R43 são opcionalmente substituídos por 1 a 3 substituintes independentemente de R38, ou R42 e R43 são tomados junto com o nitrogênio ao qual eles são ligados para formar um anel de C5-C9 azabicíclico, aziridinila, azetidinila, pirrolidinila, piperidinila, piperazinila, morfolinila, tiomorfolinila, isoquinolinila, ou di-hidroisoquinolinila, em que o referido anel de C5-C9 azabicíclico, aziridinila, azetidinila, pirrolidinila, piperidinila, piperazinila, morfolinila, tiomorfolinila, isoquinolinila, ou di-hidroisoquinolinila é não- substituído ou substituído com 1 a 5 substituintes de R38, com a condição de que R42 e R43 não sejam ambos ligados ao nitrogênio diretamente através de um oxigênio.
[00157] Em uma modalidade preferida dos compostos de acordo com a presente invenção, R7 é selecionado do grupo consistindo em - C(O)NR42R43, -SO2R42, -SO2NR42R43, -C(= N-OR42)R43 e -C(= NR42)R43.
[00158] Em uma modalidade preferida dos compostos de acordo com a presente invenção, R7 é -C(O)NR42R43, em que cada R42 e R43 é independentemente selecionado do grupo consistindo em H, (C1- C6)alquila, -(CH2)nOR37, em que n é um número inteiro de 0 a 6 e a porção alquila dos grupos anteriores R42 e R43 são opcionalmente substituídos por 1 a 3 substituintes independentemente de halo, ciano, trifluorometila, -C(O)R40, -NR37C(O)R41, -C(O)NR37R41, -NR37R41, (C1- C6)alquila, -(CH2)n(C6-C10 arila), -(CH2)n(heterociclila de 5 a 10 membros), -(CH2)nO(CH2)iOR37 e -(CH2)nOR37, em que n é um número inteiro de 0 a 6 e i é um número inteiro de 2 a 6, ou R42 e R43 são tomados junto com o nitrogênio ao qual eles são ligados para formar um anel de C5-C9 azabicíclico, aziridinila, azetidinila, pirrolidinila, piperidinila, piperazinila, morfolinila, tiomorfolinila, isoquinolinila, ou di- hidroisoquinolinila, em que o referido anel de C5-C9 azabicíclico, aziridinila, azetidinila, pirrolidinila, piperidinila, piperazinila, morfolinila, tiomorfolinila, isoquinolinila, ou di-hidroisoquinolinila é não-substituído ou substituído com 1 a 5 substituintes de R38, com a condição de que R42 e R43 não sejam ambos ligados ao nitrogênio diretamente através de um oxigênio.
[00159] Em uma modalidade preferida dos compostos de acordo com a presente invenção, R7 é -C(O)NR42R43, em que R42 e R43 são tomados junto com o nitrogênio ao qual eles são ligados para formar um anel C5-C9 azabicíclica, aziridinila, azetidinila, pirrolidinila, piperidinila, piperazinila ou morfolinila, em que o referido anel C5-C9 azabicíclica, aziridinila, azetidinila, pirrolidinila, piperidinila, piperazinila ou morfolinila é não-substituído ou substituído com 1 a 5 substituintes de R38.
[00160] Em uma modalidade preferida dos compostos de acordo com a presente invenção, R7 é -C(O)NR42R43, em que R42 e R43 são tomados junto com o nitrogênio ao qual eles são ligados para formar um anel C5-C9 azabicíclica, aziridinila, azetidinila ou pirrolidinila, em que o referido anel C5-C9 azabicíclica, aziridinila, azetidinila ou pirrolidinila é não-substituído ou substituído com 1 a 5 substituintes de R38.
[00161] Em uma modalidade preferida dos compostos de acordo com a presente invenção, R7 é -C(O)NR42R43, em que R42 e R43 são tomados junto com o nitrogênio ao qual eles são ligados para formar um anel C5-C9 azabicíclico, azetidinila ou pirrolidinila, em que o referido anel de C5-C9 azabicíclico, azetidinila ou pirrolidinila é não- substituído ou substituído com 1 a 5 substituintes de R38.
[00162] Em uma modalidade preferida dos compostos de acordo com a presente invenção, R7 é -C(O)NR42R43, em que R42 e R43 são tomados junto com o nitrogênio ao qual eles são ligados para formar um anel C5-C9 azabicíclico, em que o referido anel C5-C9 azabicíclico é não-substituído ou substituído com 1 a 5 substituintes de R38.
[00163] Em uma modalidade preferida dos compostos de acordo com a presente invenção, R7 é -C(O)NR42R43, em que R42 e R43 são tomados junto com o nitrogênio ao qual eles são ligados para formar um anel azetidinila, em que o referido anel azetidinila é não-substituído ou substituído com 1 a 5 substituintes de R38.
[00164] Em uma modalidade preferida dos compostos de acordo com a presente invenção, R7 é -C(O)NR42R43, em que R42 e R43 são tomados junto com o nitrogênio ao qual eles são ligados para formar um anel de pirrolidinila, em que o referido anel pirrolidinila é não- substituído ou substituído com 1 a 5 substituintes de R38.
[00165] Em uma modalidade preferida dos compostos de acordo com a presente invenção, R7 é selecionado do grupo consistindo em - H, halogênio, nitro, azido, -NR6aR6b, -NR6aSO2R6b, -NR6aC(O)R6b, - OC(O)R6b, -NR6aC(O)OR6b, -OC(O)NR6aR6b,-OR6a, -SR6a, -S(O)R6a, - SO2R6a, -SO3R6a, -SO2NR6aR6b, -COR6a, -CO2R6a, -CONR6aR6b, -(C1- C4)fluoroalquila, -(C1-C4)fluoroalcóxi, -(CZ3Z4)aCN, e uma porção selecionada do grupo consistindo em -(CZ3Z4)a-arila, -(CZ3Z4)a- heterociclo, (C2-C6)alquinila, -(CZ3Z4)a-(C3-C6)cicloalquila, -(CZ3Z4)a- (C5-C6)cicloalquenila, (C2-C6) alquenila e (C1-C6)alquila, em que a referida porção é opcionalmente substituída com 1 a 3 grupos Y2 independentemente selecionados, onde a é 0,1, 2, ou 3, e em que quando a é 2 ou 3, as unidades CZ3Z4 podem ser iguais ou diferentes; em que
[00166] cada R6a e R6b é independentemente selecionado do grupo consistindo em hidrogênio e uma porção selecionada do grupo consistindo em -(CZ5Z6)u-(C3-C6)cicloalquila, -(CZ5Z6)u-(C5- C6)cicloalquenila, -(CZ5Z6)u-arila, -(CZ5Z6)u-heterociclo, (C2- C6)alquenila, e (C1-C6)alquila, em que a referida porção é opcionalmente substituída com 1 a 3 grupos Y3 independentemente selecionados, onde u é 0,1, 2, ou 3, e em que quando u é 2 ou 3, as unidades CZ5Z6 podem ser iguais ou diferentes, ou
[00167] R6a e R6b tomados junto com átomos adjacentes formam um heterociclo;
[00168] cada Z3, Z4, Z5 e Z6 é independentemente selecionado do grupo consistindo em H, F e (C1-C6)alquila, ou
[00169] cada Z3 e Z4, ou Z5 e Z6 são selecionados juntos para formar um carbociclo, ou
[00170] dois grupos Z3 sobre átomos de carbono adjacentes são selecionados juntos para opcionalmente formar um carbociclo;
[00171] cada Y2 e Y3 é independentemente selecionado do grupo consistindo em halogênio, ciano, nitro, tetrazolila, guanidino, amidino, metilguanidino, azido, -C(O)Z7, -OC(O)NH2, -OC(O) NHZ7, - OC(O)NZ7Z8, -NHC(O)Z7, -NHC(O)NH2, -NHC(O)NHZ7, - NHC(O)NZ7Z8, -C(O)OH, -C(O)OZ7, -C(O)NH2, -C(O)NHZ7,- C(O)NZ7Z8, -P(O)3H2, -P(O)3 (Z7)2, -S(O)3H, -S(O)Z7, -S(O)2Z7, - S(O)3Z7, -Z7, -OZ7, -OH, -NH2, -NHZ7, -NZ7Z8, -C(= NH)NH2,-C(= NOH)NH2, -N-morfolino, (C2-C6)alquenila, (C2-C6)alquinila, (C1- C6)haloalquila, (C2-C6)haloalquenila, (C2-C6)haloalquinila, (C1- C6)haloalcóxi, -(CZ9Z10)rNH2, -(CZ9Z10)rNHZ3, -(CZ9Z10)rNZ7Z8, -X6 (CZ9Z10)r-(C3-C8)cicloalquila, -X6 (CZ9Z10)r-(C5-C8)cicloalquenila, -X6 (CZ9Z10)r-arila e -X6 (CZ9Z10)r-heterociclo, em que
[00172] r é 1, 2, 3 ou 4;
[00173] X6 é selecionado do grupo consistindo em O, S, NH, -C(O)-, -C(O)NH-, -C(O)O-, -S(O)-, -S(O)2- e -S(O)3-;
[00174] Z7 e Z8 são independentemente selecionados do grupo consistindo em uma alquila de 1 a 12 átomos de carbono, uma alquenila de 2 a 12 átomos de carbono, uma alquinila de 2 a 12 átomos de carbono, uma cicloalquila de 3 a 8 átomos de carbono, uma cicloalquenila de 5 a 8 átomos de carbono, uma arila de 6 a 14 átomos de carbono, um heterociclo de 5 a 14 átomos de anel, uma aralquila de 7 a 15 átomos de carbono, e uma heteroaralquila de 5 a 14 átomos de anel, ou
[00175] Z7 e Z8 juntos podem opcionalmente formar um heterociclo;
[00176] Z9 e Z10 são independentemente selecionados do grupo consistindo em H, F, uma (C1-C12)alquila, uma (C6-C14)arila, uma (C5- C14)heteroarila, uma (C7-C15)aralquila e uma (C5-C14)heteroaralquila, ou
[00177] Z9 e Z10 são tomados juntos para formar um carbociclo, ou
[00178] dois grupos Z9 sobre átomos de carbono adjacentes são tomados juntos para formar um carbociclo; ou
[00179] quaisquer dois grupos Y2 ou Y3 ligados aos átomos de carbono adjacentes podem ser tomados juntos para serem - O[C(Z9)(Z10)]rO ou -O[C(Z9)(Z10)]r+1, ou
[00180] quaisquer dois grupos Y2 ou Y3 ligados aos mesmos ou adjacentes átomos de carbono podem ser selecionados juntos para formar um carbociclo ou heterociclo; e em que
[00181] qualquer um dos substituintes acima mencionados compreendendo um grupo CH3 (metila), CH2 (metileno), ou CH (metina) que não é ligado a um grupo halogênio, SO ou SO2 ou a um átomo de N, O ou S opcionalmente transporta sobre o referido grupo um substituinte selecionado de hidróxi, halogênio, (C1-C4)alquila, (C1- C4)alcóxi e an -N[(C1-C4)alquila][(C1-C4)alquila].
[00182] Em uma modalidade preferida dos compostos de acordo com a presente invenção R7 é selecionado do grupo consistindo em - H, -Y-(arila), -Y-(heteroarila) e C(O)-heterociclila, cada um dos quais, exceto para -H, é opcionalmente substituída com 1 a 5 R38.
[00183] Em uma modalidade preferida dos compostos de acordo com a presente invenção, R7 é selecionado do grupo consistindo em
[00184] em que os membros do referido grupo são opcionalmente substituídos por 1 a 3 R38.
[00185] Em uma modalidade preferida dos compostos de acordo com a presente invenção, R7 é selecionado do grupo consistindo em
[00186] em que os membros do referido grupo são opcionalmente substituídas com 1 a 3 R38.
[00187] Em uma modalidade preferida dos compostos de acordo com a presente invenção, R7 é selecionado do grupo consistindo em
[00188] Em uma modalidade preferida dos compostos de acordo com a presente invenção, R7 é selecionado do grupo consistindo em fenila e piridila, que são opcionalmente substituídas por 1 a 5 R38.
[00189] Em uma modalidade preferida dos compostos de acordo com a presente invenção, D é definido pelo grupo R1, em que R1 é - CHCH ou -C=C-(CR45R45)n-R46; em que
[00190] cada R45 é independentemente selecionado do grupo consistindo em H, uma (C1-C6)alquila e uma (C3-C8)cicloalquila;
[00191] R46 é selecionado do grupo consistindo em heterociclila, - N(R47)-C(O)-N(R47)(R48), -N(R47)-C(S)-N(R47)(R48), -N(R47)-C(O)-OR48, -N(R47)-C(O)-(CH2)n-R48, -N(R47)-SO2R47, -(CH2)nNR47R48, -(CH2)nOR48, -(CH2)nSR49, -(CH2)nS(O)R49, -(CH2)nS(O)2R49, -OC(O)R49, - OC(O)OR49, -C(O)NR47R48, heteroarila opcionalmente substituída com um ou mais substituintes selecionados do grupo consistindo em halo, - CF3, (C1-C6)alcóxi, -NO2, (C1-C6)alquila, -CN, -SO2R50 e - (CH2)nNR50R51, e arila opcionalmente substituída com um ou mais substituintes selecionados do grupo consistindo em halo, -CF3, (C1- C6)alcóxi, -NO2, (C1-C6)alquila, -CN, -SO2R50 e -(CH2)nNR50R51;
[00192] R47 e R48 são independentemente selecionados do grupo consistindo em H, (C1-C6)alquila, (C3-C8)cicloalquila, heterociclila, - (CH2)nNR50R51, -(CH2)nOR50, -(CH2)nC(O)R49, -C(O)2R49, -(CH2)nSR49, - (CH2)nS(O)R49, -(CH2)nS(O)2R49, -(CH2)nR49, -(CH2)nCN, arila opcionalmente substituída com um ou mais substituintes selecionados do grupo consistindo em halo, -CF3, (C1-C6)alcóxi, -NO2, (C1-C6)alquila, -CN, -(CH2)nOR49, -(CH2)nheterociclila, -(CH2)nheteroarila, -SO2R50 e - (CH2)nNR50R51, e heteroarila opcionalmente substituída com um ou mais substituintes selecionados do grupo consistindo em halo, -CF3, (C1-C6)alcóxi, -NO2, (C1-C6)alquila, -CN, -(CH2)nOR49, - (CH2)nheterociclila, -(CH2)nheteroarila, -SO2R50 e -(CH2)nNR50R51, ou
[00193] R47 e R48, juntamente com o átomo ao qual eles são ligados, formam um anel carbo- ou heterocíclico de 3 a 8 membros;
[00194] R49 é selecionado do grupo consistindo em (C1-C6)alquila, (C3-C8)cicloalquila, heterociclil(C1-C6)alquileno, aril(C1-C6)alquileno em que a arila é opcionalmente substituída com um ou mais substituintes selecionados do grupo consistindo em halo, -CF3, (C1-C6)alcóxi, -NO2, (C1-C6)alquila, -CN, -SO2R50 e -(CH2)nNR50R51, heteroaril(C1- C6)alquileno em que a heteroarila é opcionalmente substituída com um ou mais substituintes selecionados do grupo consistindo em halo, - CF3, (C1-C6)alcóxi, -NO2, (C1-C6)alquila, -CN, -SO2R50 e - (CH2)nNR50R51, arila opcionalmente substituída com um ou mais substituintes selecionados do grupo consistindo em halo, -CF3, (C1- C6)alcóxi, -NO2, (C1-C6)alquila, -CN, -SO2R50 e -(CH2)nNR50R51, e heteroarila opcionalmente substituída com um ou mais substituintes selecionados do grupo consistindo em halo, -CF3, (C1-C6)alcóxi, -NO2, (C1-C6)alquila, -CN, -SO2R50 e -(CH2)nNR50R51;
[00195] R50 e R51 são independentemente selecionados do grupo consistindo em H, (C1-C6)alquila, (C3-C8)cicloalquila e -C(O)R45, ou
[00196] R50 e R51, juntamente com o átomo ao qual eles são ligados, formam um anel carbo- ou heterocíclico de 3 a 8 membros.
[00197] Em uma modalidade preferida dos compostos de acordo com a presente invenção,
[00198] R46 é selecionado do grupo consistindo em -N(R47)-C(O)- N(R47)(R48), -N(R47)-C(O)-(CH2)n-R48 e -(CH2)nNR47R48; em que
[00199] R47 e R48 são independentemente selecionados do grupo consistindo em H, (C1-C6)alquila, (C3-C8)cicloalquila, heterociclila, - (CH2)nNR50R51, -(CH2)nOR50, -(CH2)nS(O)2R49 e -(CH2)nCN, ou R47 e R48, juntamente com o átomo ao qual eles são ligados, formam um anel carbo- ou heterocíclico de 3 a 8 membros; e
[00200] R50 e R51 são independentemente selecionados do grupo consistindo em H e (C1-C6)alquila, ou R50 e R51, juntamente com o átomo ao qual eles são ligados, formam um anel carbo- ou heterocíclico de 3 a 8 membros.
[00201] Em uma modalidade preferida dos compostos de acordo com a presente invenção, R1 é selecionado do grupo consistindo em
[00202] Em uma modalidade preferida dos compostos de acordo com a presente invenção, D é definido pelo grupo R21, em que R21 é definido por -(Z11)-(Z12)m-(Z13)m1, em que
[00203] Z11 é heterociclila, quando m e m1 são 0, ou heterociclileno, quando ou m ou m1 é 1;
[00204] Z12 é selecionado do grupo consistindo em OC(O), OC(S) e C(O);
[00205] Z13 é selecionado do grupo consistindo em heterociclila, aralquila, N(H)R52, (C1-C3)alquila, -OR52, halo, S(O)2R56, (C1- C3)hidroxialquila e (C1-C3)haloalquila;
[00206] m é 0 ou 1;
[00207] m1 é 0 ou 1;
[00208] R52 é selecionado do grupo consistindo em H, - (CH2)qS(O)2R54, -(C1-C6) alquil- NR53R53, (C1-C3)alquila, -(CH2)qOR53, - C(O)R54 e -C(O)OR53;
[00209] q é 0, 1, 2, 3 ou 4;
[00210] cada R53 é independentemente (C1-C3)alquila;
[00211] R54 é (C1-C3)alquila ou N(H)R53; e
[00212] R56 é selecionado do grupo consistindo em NH2, (C1- C3)alquila e OR52.
[00213] Em uma modalidade preferida dos compostos de acordo com a presente invenção, Z11 é a heterociclila e m e m1 são cada qual 0.
[00214] Em uma modalidade preferida dos compostos de acordo com a presente invenção, Z11 é a heterociclila e m é 0 e n é 0, onde o grupo heterociclila é selecionado do grupo consistindo em
[00215] Em uma modalidade preferida dos compostos de acordo com a presente invenção, Z11 é heterociclileno, Z12 é OC(O), m é 1, m1 é 1 e Z13 é heterociclila.
[00216] Em uma modalidade preferida dos compostos de acordo com a presente invenção, Z11 é
[00217] Z12 é OC(O), e
[00219] Z13 é N(H)R52, em que R52 é (C1-C3)alquila.
[00220] Em uma modalidade preferida dos compostos de acordo com a presente invenção Z11 é heterociclileno, Z12 é C(O) e m é 1, m1 é 1 e Z13 é (C1-C3)haloalquila.
[00221] Em uma modalidade preferida dos compostos de acordo com a presente invenção, Z11 é
[00222] Z12 é C(O), e
[00223] Z13 é (C1-C3)haloalquila, preferivelmente -CF3.
[00224] Em uma modalidade preferida dos compostos de acordo com a presente invenção, Z11 é heterociclileno, m é 0, m1 é 1 e Z13 é heterociclila.
[00225] Em uma modalidade preferida dos compostos de acordo com a presente invenção, Z11 é
[00226] m é 0, e
[00228] Z13 é (C1-C3)alquila, ou
[00229] Z13 é -OH, ou
[00230] Z13 é -OR52, em que R52 é (C1-C3)alquila, preferivelmente - CH3 ou
[00231] Z13 é halo, preferivelmente -F, ou
[00232] Z13 é (C1-C3)hidroxialquila, preferivelmente -CH3OH.
[00233] Em uma modalidade preferida dos compostos de acordo com a presente invenção, R21 é selecionado do grupo consistindo em
[00234] Em uma modalidade preferida dos compostos de acordo com a presente invenção em que D é definido pelo grupo R21, o grupo heterocíclico ou heterociclila é opcionalmente substituído com um substituinte selecionado do grupo consistindo em (C1-C6)alquila, (C1- C6)alcóxi, (C1-C6)alquilsufanila, (C1-C6)alquilsulfenila, (C1- C6)alquilsulfonila, oxo, hidroxila, mercapto, amino opcionalmente substituída por alquila, carbóxi, carbamoíla opcionalmente substituída por alquila, alquilcarboxiamida, carboxiamida, aminossulfonila opcionalmente substituída por alquila, ureído, arilureia, ariltioureia, alquilureia, cicloalquilureia, sulfonilureia, nitro, ciano, halo, arila, aralquila, heteroarila e (C1-C6)perfluoroalquila. Um tal anel pode ser opcionalmente fundido a um ou mais outros anéis "heterocíclico" ou anéis cicloalquila. Preferidos exemplos de porções "heterocíclicas" incluem, porém não são limitados a tetra-hidrofuranila, piranila, 1,4- dioxanoíla, 1,3-dioxanila, piperidinila, piperazinila, 2,4- piperazinadionila, pirrolidinila, pirrolidinon-2-ila, pirrolidinon-3-ila, pirrolidinon-4-ila, pirrolidinon-5-ila, imidazolidinila, pirazolidinila, morfolinila, tiomorfolinila, tetra-hidrotiopiranila, tetra-hidrotiofenila, e similares
[00235] Em uma modalidade preferida dos compostos de acordo com a presente invenção em que D é definido pelo grupo R21, o grupo heterociclileno é opcionalmente substituído com substituintes selecionados do grupo consistindo em (C1-C6)alquila, (C1-C6)alcóxi, (C1-C6)alquilsufanila, (C1-C6)alquilsulfenila, (C1-C6)alquilsulfonila, oxo, hidroxila, mercapto, amino opcionalmente substituída por alquila, carbóxi, carbamoíla opcionalmente substituída por alquila, alquilcarboxiamida, carboxiamida, aminossulfonila opcionalmente substituída por alquila, ureído, arilureia, ariltioureia, alquilureia, cicloalquilureia, sulfonilureia, nitro, ciano, halo e (C1- C6)perfluoroalquila, múltiplos graus de substituição sendo permitidos. Um tal anel pode ser opcionalmente fundido a uma ou mais anéis de benzeno ou a um ou mais det al. anéis "heterocíclicos" ou anéis cicloalquila. Os exemplos preferidos de "heterociclileno" incluem porém não são limitados a tetra-hidrofuran-2,5-di-ila, morfolina-2,3-di morfolina-2,4-di-Di-ila, e similares.
[00236] Em uma modalidade preferida da presente invenção, D é selecionado do grupo consistindo em
[00237] Em uma modalidade preferida dos compostos de acordo com a presente invenção, Z é selecionado do grupo consistindo em - O-, -S-, -S(O)0-2 e -NR5-, em que R5 é selecionado do grupo consistindo em H, uma (C1-C5)acila opcionalmente substituída e C1-C6 alquil-O-C(O), em que C1-C6 alquila é opcionalmente substituída.
[00238] Em uma modalidade preferida dos compostos de acordo com a presente invenção, Z é -O-.
[00239] Em uma modalidade preferida dos compostos de acordo com a presente invenção, Ar é um grupo da fórmula C,
[00240] em que,
[00241] A4, A5, A6 e A7 são independentemente selecionados do grupo consistindo em N e -CH2-, com a condição de que não mais do que dois de A4, A5, A6 e A7 possam ser N;
[00242] R2 em cada ocorrência é independentemente selecionada do grupo consistindo em -H, halogênio, trihalometila, -CN, -NO2, -NH2, -OR3, -NR3R4, -S(O)0-2R3, -S(O)2NR3R3, -C(O)OR3, -C(O)NR3R3, - N(R3)SO2R3, -N(R3)C(O)R3, -N(R3)CO2R3, -C(O)R3, C1-C4 alcóxi, C1-C4 alquiltio, -O(CH2)narila, -O(CH2)nheteroarila, -(CH2)0-5 (arila), -(CH2)0-5 (heteroarila), C1-C6 alquila, C2-C6 alquenila, C2-C6 alquinila, -CH2 (CH2)0-4-T2, em que T2 é selecionado do grupo consistindo em -OH, - OMe, -OEt, -NH2, -NHMe, -NMe2, -NHEt e -NEt2, e em que a arila, heteroarila, C1-C6 alquila, C2-C6 alquenila, e C2-C6 alquinila são opcionalmente substituídas; e
[00243] R3 selecionado do grupo consistindo em -H e R4;
[00244] R4 é selecionado do grupo consistindo em uma (C1- C6)alquila, uma arila, uma arilalquila inferior, uma heterociclila e uma heterociclilalquila inferior, cada uma das quais é opcionalmente substituída, ou
[00245] R3 e R4, tomados juntamente com um nitrogênio comum ao qual eles são ligados, formam uma heterociclila de cinco a sete membros opcionalmente substituída, que opcionalmente contém pelo menos um heteroátomo anular adicional selecionado do grupo consistindo em N, O, S e P; e
[00246] q é um número inteiro de 0 a 4.
[00247] Em uma modalidade preferida dos compostos de acordo com a presente invenção, Ar é selecionado do grupo consistindo em fenila, pirazina, piridazina, pirimidina e piridina, em que cada uma das referidas fenila, pirazina, piridazina, pirimidina e piridina são opcionalmente substituídas com entre zero e quatro R2.
[00248] Em uma modalidade preferida dos compostos de acordo com a presente invenção, Ar é fenila, opcionalmente substituída por entre zero e quatro R2.
[00249] Em uma modalidade preferida dos compostos de acordo com a presente invenção, Ar é fenila, substituída com entre zero e quatro halo.
[00250] Em uma modalidade preferida dos compostos de acordo com a presente invenção, G é um grupo B-L-T, em que
[00251] B é selecionado do grupo consistindo em ausente, -N(R13)-, -N(SO2R13)-, -O-, -S(O)0-2 e -C(= O)-;
[00252] L é selecionado do grupo consistindo em ausente, -C(= S)N(R13)-, -C(= NR14)N(R13)-, -SO2N(R13)-, -SO2-, -C(= O)N(R13)-, - N(R13)-, -C(= O)C1-2alquil-N(R13)-, -N(R13)C1-2alquil-C(= O)-, -C(= O)C0- 1alquil-C(= O)N(R13)-, -C0-4alquileno, -C(= O)C0-1alquil-C(= O)OR3-, - C(= NR14)-C0-1alquil-C(= O)-, -C(= O)-, -C(= O)C0-1alquil-C(= O)- e uma heterociclila de quatro a seis membros opcionalmente substituída contendo entre um e três heteroátomos anulares incluindo pelo menos um nitrogênio, em que uma alquila dos grupos L anteriormente mencionados é opcionalmente independentemente substituída com um ou dois de H, (C1-C6)alquila, halo, ciano ou nitro, em que a (C1- C6)alquila é opcionalmente substituída ; e
[00253] T é selecionado do grupo consistindo em -H, -R13, -C0- 4alquila, -C0-4alquil-Q, -O-C0-4alquil-Q, -C0-4alquil-O-Q, -N(R13)C0-4alquil- Q, -SO2C0-4alquil-Q, -C(= O)C0-4alquil-Q, -C0-4alquil-N(R13)Q e -C(= O)N(R13)-C0-4alquil-Q, em que cada C0-4alquila é opcionalmente substituída;
[00254] R13 é selecionado do grupo consistindo em -H,-CN, -NO2, - NH2, -OR3, -NR3R4, -S(O)0-2R3, -S(O)2NR3R3, -C(O)OR3, -C(O)NR3R3, - N(R3)SO2R3, -N(R3)C(O)R3, -N(R3)CO2R3, -C(O)R3, -C(O)SR3, C1-C4 alcóxi, C1-C4 alquiltio, -O(CH2)narila, -O(CH2)nheteroarila, -(CH2)0-5 (arila), -(CH2)0-5 (heteroarila), C1-C6 alquila, C2-C6 alquenila, C2-C6 alquinila, -CH2 (CH2)0-4-T2, uma C1-4 alquilcarbonila opcionalmente substituída, e um grupo carboxíclico ou heterocíclico de três a sete membros saturado ou insaturado, em que T2 é selecionado do grupo consistindo em -OH, -OMe, -OEt, -NH2, -NHMe, -NMe2, -NHEt e -NEt2, e em que a arila, heteroarila, C1-C6 alquila, C2-C6 alquenila, e C2-C6 alquinila são opcionalmente substituídas;
[00255] dois R13, juntamente com o átomo ou átomos aos quais eles são ligados, podem combinar para formar um heteroalicíclico opcionalmente substituído por entre um e quatro de R60, em que o heteroalicíclico pode ter até quatro heteroátomos anulares, e o heteroalicíclico pode ter uma arila ou heteroarila fundida a ele, em cujo caso a arila ou heteroarila é opcionalmente substituída com um adicional de um a quatro de R60;
[00256] R14 é selecionado do grupo -H, -NO2, -NH2, -N(R3)R4, -CN, - OR3, uma (C1-C6)alquila opcionalmente substituída, uma heteroaliciclilalquila opcionalmente substituída, uma arila opcionalmente substituída, uma arilalquila opcionalmente substituída e um heteroalicíclico opcionalmente substituído,
[00257] cada R3 é independentemente selecionado do grupo consistindo em -H e R4;
[00258] R4 é selecionado do grupo consistindo em uma (C1- C6)alquila, uma arila, uma arilalquila inferior, uma heterociclila e uma heterociclilalquila inferior, cada uma das quais é opcionalmente substituída, ou
[00259] R3 e R4, tomados juntamente com um nitrogênio comum ao qual eles são ligados, formam uma heterociclila de cinco a sete membros opcionalmente substituída, a heterociclila de cinco a sete membros opcionalmente substituída opcionalmente contendo pelo menos um heteroátomo anular adicional selecionado do grupo consistindo em N, O, S e P;
[00260] R60 é selecionado do grupo consistindo em -H, halogênio, trihalometila, -CN, -NO2, -NH2, -OR3, -NR3R4, -S(O)0-2R3, -SO2NR3R3, - CO2R3, -C(O)NR3R3, -N(R3)SO2R3, -N(R3)C(O)R3, -N(R3)CO2R3, - C(O)R3, uma (C1-C6)alquila opcionalmente substituída, uma arila opcionalmente substituída, uma heteroarilalquila opcionalmente substituída e uma arilalquila opcionalmente substituída;
[00261] dois R60, quando ligados a um carbono não-aromático, podem ser oxo;
[00262] Q é um sistema de anel de cinco a dez membros, opcionalmente substituído por entre zero e quatro de R20; e
[00263] R20 é selecionado do grupo consistindo em -H, halogênio, trihalometila, -CN, -NO2, -NH2, -OR3, -OCF3, -NR3R4, -S(O)0-2R3, - S(O)2NR3R3, -C(O)OR3, -C(O)NR3R3, -N(R3)SO2R3, -N(R3)C(O)R3, - N(R3)C(O)OR3, -C(O)R3, -C(O)SR3, C1-C4 alcóxi, C1-C4 alquiltio, - O(CH2)narila, -O(CH2)nheteroarila, -(CH2)0-5 (arila), -(CH2)0-5 (heteroarila), C1-C6 alquila, C2-C6 alquenila, C2-C6 alquinila, -CH2 (CH2)0-4-T2, uma C1-4 alquilcarbonila opcionalmente substituída, C1-4 alcóxi, um amino opcionalmente substituído por C1-4 alquila em que o C1-C4 alquila pode ser opcionalmente substituída por C1-4 alcóxi e um grupo carboxíclico ou heterocíclico de três a sete membros saturado ou insaturado, em que T2 é selecionado do grupo consistindo em -OH, -OMe, -OEt, -NH2, -NHMe, -NMe2, -NHEt e -NEt2, e em que a arila, heteroarila, C1-C6 alquila, C2-C6 alquenila, e C2-C6 alquinila são opcionalmente substituídas.
[00264] Em uma modalidade preferida dos compostos de acordo com a presente invenção, G é selecionado do grupo consistindo em
[00265] em que R13, R14, Q e R3 são como acima definido;
[00266] qualquer grupo metileno é independente e opcionalmente substituído por R25, em que
[00267] R25 é selecionado do grupo consistindo em halogênio, trihalometila, -CN, -NO2, -NH2, -OR3, -NR3,R4, -S(O)0-2R3, -SO2NR3R3, - CO2R3, -C(O)NR3R3, -N(R3)SO2R3, -N(R3)C(O)R3, -N(R3)CO2R3, - C(O)R3, uma arila opcionalmente substituída, uma arila equila opcionalmente substituída, uma heteroarilalquila opcionalmente substituída, e uma (C1-C6)alquila opcionalmente substituída,
[00268] dois R25, juntamente com o carbono ou carbonos aos quais eles estão ligados, podem combinar para formar um alicíclico ou heteroalicíclico de três a sete membros, e
[00269] dois R25, em um carbono único podem ser oxo;
[00270] R9 é selecionado do grupo consistindo em a C1-6 alquila na qual um ou mais átomos de hidrogênio são opcionalmente substituídos por -R24, -T1-R15, ou -NR16R17, uma porção de -N(R18)(R19) e um grupo carbocíclico ou heterocíclico de três a oito membros saturado ou insaturado que é opcionalmente substituído por uma C1-6 alquila, um C1-6 alcóxi, um átomo de halogênio, nitro, uma trifluorometila, uma C1-6 alcoxicarbonila, ciano, uma ciano C1-6 alquila, um C1-6 alquiltio, um fenóxi, uma acetila, ou um anel heterociclila de cinco ou seis membros saturado ou insaturado em que, quando o grupo carbocíclico ou heterocíclico de três a oito membros é substituído por dois C1-6 alquila, os grupos alquila podem combinar-se um com o outro para formar uma cadeia alquileno, ou o grupo carbocíclico ou heterocíclico de três a oito membros pode ser um grupo bicíclico condensado com outro grupo carbocíclico ou heterocíclico de três a oito membros saturado ou insaturado,
[00271] em que
[00272] T1 é selecionado do grupo consistindo em -O-, -S- e -NH-;
[00273] R21 representa um grupo carbocíclico ou heterocíclico de três a oito membros saturado ou insaturado ;
[00274] R15, R16, e R17, que podem ser iguais ou diferentes, representam uma C1-6 alquila ou um grupo carbocíclico ou heterocíclico de três a oito membros saturado ou insaturado; em que grupo carbocíclico ou heterocíclico três a oito membros representado por R21, R15, R16, e R17 é opcionalmente substituído por uma C1-6 alquila, uma C1-6 alcóxi, um átomo de halogênio, nitro, uma trifluorometila, um C1-6 alcoxicarbonila, uma ciano, uma ciano C1-6 alquila, uma C1-6 alquiltio, uma fenóxi, uma acetila, ou um anel heterociclila de cinco ou seis membros saturado ou insaturado; e em que quando o grupo carbocíclico ou heterocíclico de três a oito membros é substituído por dois grupos C1-6 alquila, os dois grupos alquila podem combinar-se um com o outro para formar uma cadeia de alquileno; e em que o grupo carbocíclico ou heterocíclico três a oito membros pode ser um grupo bicíclico condensado com outro grupo carbocíclico ou heterocíclico de três a oito membros saturado ou insaturado; e
[00275] R18 e R19, que podem ser iguais ou diferentes, representam (1) um átomo de hidrogênio, (2) uma C1-6 alquila que é opcionalmente substituída por um C1-6 alcóxi, uma C1-6 alquiltio, ou grupo carbocíclico ou heterocíclico de três a oito membros saturado ou insaturado em que o grupo carbocíclico ou heterocíclico de três a oito membros é opcionalmente substituído uma C1-6 alquila, um C1-6 alcóxi, um átomo de halogênio, nitro, uma trifluorometila, uma C1-6 alcóxi carbonila, ciano, uma ciano C1-6 alquila, um C1-6 alquiltio, um fenóxi, uma acetila, ou um anel heterociclila de cinco ou seis membros saturado ou insaturado e em que quando o grupo carbocíclico ou heterocíclico de três a oito membros é substituído por dois grupos C1-6 alquila, os dois grupos alquila podem combinar-se um com o outro para formar uma cadeia de alquileno, ou o grupo carbocíclico ou heterocíclico de três a oito membros pode ser um grupo bicíclico condensado com outro grupo carbocíclico ou heterocíclico de três a oito membros, saturado ou insaturado, ou (3) a grupo carbocíclico ou heterocíclico de três a oito membros, saturado ou insaturado que é opcionalmente substituído por uma C1-6 alquila, um C1-6 alcóxi, um átomo de halogênio, nitro, uma trifluorometila, uma C1-6 alcóxi carbonila, ciano, uma ciano C1-6 alquila, um C1-6 alquiltio, um fenóxi, uma acetila, ou um anel heterociclila de cinco ou seis membros saturado ou insaturado e em que, quando o grupo carbocíclico ou heterocíclico de três a oito membros é substituído por dois grupos C1-6 alquila, os dois grupos alquila podem combinar-se um com o outro para formar uma cadeia alquileno, ou o grupo carbocíclico ou heterocíclico de três a oito membros pode ser um grupo bicíclico condensado com outro grupo carbocíclico ou heterocíclico de três a oito membros, saturado ou insaturado;
[00276] X e X1 são cada qual independentemente selecionado do grupo consistindo em -H, halogênio, ciano, nitro, C1-C6 alquila, ou
[00277] X e X1 juntamente com o átomo ao qual eles são ligados formam uma C3-C4 cicloalquila;
[00278] E é selecionado do grupo consistindo em -O-, -N(R13)-, - CH2- e -S(O)0-2-;
[00279] M é selecionado do grupo consistindo em -O-, -N(R13)-, - CH2- e -C(= O)N(R13);
[00280] M1 representa -C(R26)(R27)-, em que
[00281] R26 e R27 são independentemente selecionados do grupo consistindo em um átomo de hidrogênio, uma C1-4 alquila, uma C1-4 alcóxi e -N(R12), em que
[00282] R12 é um átomo de hidrogênio ou uma C1-4 alquila; e
[00283] cada V é independentemente selecionado do grupo consistindo em = N- e = C(H)-.
[00284] Em uma modalidade preferida dos compostos de acordo com a presente invenção, G é selecionado do grupo consistindo em
[00285] Em uma modalidade preferida dos compostos de acordo com a presente invenção, o grupo alquila opcionalmente substituído representado por R9 preferivelmente representa -(CH2)p-R24, -(CH2)p- T-R15, ou -(CH2)p-NR16R17 em que p é um número inteiro de 1 a 6 e R24, R15, R16, e R17 são como acima definido.
[00286] Em uma modalidade preferida dos compostos de acordo com a presente invenção em -N(R18)(R19) representado por R9, preferivelmente, R18 representa um átomo de hidrogênio ou C1-6 alquila, e R19 representa C1-6 alquila que é opcionalmente substituída por um grupo carbocíclico ou heterocíclico de cinco ou seis membros saturado ou insaturado opcionalmente substituído; ou um grupo carbocíclico ou heterocíclico de cinco ou seis membros saturado ou insaturado opcionalmente substituído.
[00287] Em uma modalidade preferida dos compostos de acordo com a presente invenção, exemplos preferidos de R9 incluem, porém não são limitados a, benzila, fluorobenzila, difluorobenzila, clorobenzila, metilbenzila, metoxibenzila, anilina, fluoroanilino, difluoroanilino, cloroanilino, metilanilino, metoxianilino, naftila, tienil-2-il- metila e tienil-3-il-metila.
[00288] Em uma modalidade preferida dos compostos de acordo com a presente invenção, exemplos de R19 incluem fenila, fluorofenila, difluorofenila, clorofenila, metilfenila, metoxifenila, piridila, isoxazolila e quinolila.
[00289] Em uma modalidade preferida dos compostos de acordo com a presente invenção, G é selecionado do grupo consistindo em :
[00290] em que cada metileno em qualquer uma das fórmulas acima, exceto aquelas em um anel representado, é independente e opcionalmente substituído por R25;
[00291] R25 é selecionado do grupo consistindo em halogênio, trihalometila, -CN, -NO2, -NH2, -OR3, -NR3R4, -S(O)0-2R3, -SO2NR3R3, - CO2R3, -C(O)NR3R3, -N(R3)SO2R3, -N(R3)C(O)R3, -N(R3)CO2R3, - C(O)R3, uma arila opcionalmente substituída, uma arilaequila opcionalmente substituída, uma heteroarilalquila opcionalmente substituída, e uma (C1-C6)alquila opcionalmente substituída,
[00292] dois R25, juntamente com o carbono ou carbonos aos quais eles estão ligados, podem combinar-se para formar um alicíclico ou heteroalicíclico de três a sete membros;
[00293] R5 é -H ou uma (C1-C6)alquila opcionalmente substituída;
[00294] R10 é uma azolila, em que uma ou mais átomos de hidrogênio são opcionalmente substituídos por uma porção selecionada do grupo consistindo em um halogênio, C1-4 alquila, C1-4 alcóxi, C1-4 alquiltio, trihalometila, nitro, amino opcionalmente independentemente substituídos por um ou dois de C1-4 alquila, uma C1-4 alcoxicarbonil C1-4 alquila, uma C1-4 alquilcarbonila e uma C3-5 cíclico alquila;
[00295] X e X1 são independentemente selecionados do grupo consistindo em -H, halogênio, ciano, nitro, C1-C6 alquila, ou
[00296] X e X1 tomados juntamente com o átomo ao qual eles são ligados, formam a C3-C7 cicloalquila;
[00297] E é selecionado do grupo consistindo em -O-, -N(R13)-, - CH2- e -S(O)0-2-.
[00298] Em uma modalidade preferida dos compostos de acordo com a presente invenção, um grupo metileno entre dois grupos carbonila é mono- ou dissubstituído com ou uma (C1-C6)alquila opcionalmente substituída ou um espirociclo opcionalmente substituído.
[00299] Em uma modalidade preferida dos compostos de acordo com a presente invenção, R10 é selecionado do grupo consistindo em
[00300] em que A8 é selecionado do grupo consistindo em -O-, -Se -NH-; e
[00301] R22 e R23 são independentemente selecionados do grupo consistindo em -H, halogênio, C1-4 alquila, C1-4 alcóxi, C1-4 alquiltio, trihalometila, nitro, amino opcionalmente independentemente substituídos por um ou dois de C1-4 alquila, a C1-4 alcoxicarbonil C1-4 alquila, a C1-4 alquilcarbonila e a C3-5 alquila cíclica.
[00302] Em uma modalidade preferida dos compostos de acordo com a presente invenção, R10 é uma azolila opcionalmente substituída selecionada do grupo consistindo em imidazolila, oxazolila, tiazolila, pirazolila, isoxazolila, isotiazolila, 1,3,4-tiadiazolila, 1,2,4-tiadiazolila, 1,2,4-oxadiazolila, e 1,3,4-oxadiazolila.
[00303] Em uma modalidade preferida dos compostos de acordo com a presente invenção, Q é selecionado do grupo consistindo em
[00304] em que P1 é um anel de cinco a sete membros, incluindo os dois átomos de carbono compartilhados do anel aromático ao qual P1 é fundido, e em que P1 opcionalmente contém entre um a três heteroátomos.
[00305] Em uma modalidade preferida dos compostos de acordo com a presente invenção, Q é selecionado do grupo consistindo em fenila, naftila, 1,2, 3,4-tetra-hidronaftila, indanila, benzodioxanila, benzofuranila, fenazinila, fenotiazinila, fenoxazinila, tetra- hidroisoquinolila, pirrolila, pirazolila, pirazolidinila, imidazolila, imidazolinila, imidazolidinila, tetra-hidropiridinila, piridinila, pirazinila, pirimidinila, piridazinila, oxazolila, oxazolinila, oxazolidinila, triazolila, isoxazolila, isoxazolidinila, tiazolila, tiazolinila, tiazolidinila, isotiazolila, isotiazolidinila, indolila, isoindolila, indolinila, isoindolinila, octra- hidroindolila, octra-hidroisoindolila, quinolila, isoquinolila, benzimidazolila, tiadiazolila, benzopiranila, benzotiazolila, benzoxazolila, furila, tienila, benzotienila, e oxadiazolila; cada opcionalmente substituído por entre um a quatro de R20, em que
[00306] cada R20 é selecionado do grupo consistindo em -H, halogênio, trihalometila, -CN, -NO2, -NH2, -OR3, -OCF3, -NR3R4, -S(O)0- 2R3, -S(O)2NR3R3, -C(O)OR3, -C(O)NR3R3, -N(R3)SO2R3, - N(R3)C(O)R3, -N(R3)C(O)OR3, -C(O)R3, -C(O)SR3, C1-C4 alcóxi, C1-C4 alquiltio, -O(CH2)narila, -O(CH2)nheteroarila, -(CH2)0-5 (arila), -(CH2)0-5 (heteroarila), C1-C6 alquila, C2-C6 alquenila, C2-C6 alquinila, -CH2 (CH2)0-4-T2, uma C1-4 alquilcarbonila opcionalmente substituída, C1-4 alcóxi, um amino opcionalmente substituído por C1-4 alquila, em que C1-C4 alquila pode ser opcionalmente substituída por C1-4 alcóxi e um grupo carboxíclico ou heterocíclico de três a sete membros saturado ou insaturado, em que T2 é selecionado do grupo consistindo em -OH, -OMe, -OEt, -NH2, -NHMe, -NMe2, -NHEt e -NEt2, e em que a arila, heteroarila, C1-C6 alquila, C2-C6 alquenila, e C2-C6 alquinila são opcionalmente substituídas.
[00307] Em uma modalidade preferida dos compostos de acordo com a presente invenção, os compostos são representados pelas fórmulas A-1 e B-1:
[00308] e sais farmaceuticamente aceitáveis e complexos destes, em que
[00309] D é selecionado do grupo consistindo em R7, R1 e R21, em que
[00310] R7 é selecionado do grupo consistindo em -H, halogênio, nitro, azido, C1-C6 alquila, C3-C10 cicloalquila, -C(O)NR42R43, -Y- NR42R43, -NR42C(= O)R43, -SO2R42, -SO2NR42R43, -NR37SO2R42, - NR37SO2NR42R43, -C(= N-OR42)R43, -C(= NR42)R43, -NR37C(= NR42)R43, -C(= NR42)NR37R43, -NR37C(= NR42)NR37R43, -C(O)R42, -CO2R42, - C(O)(heterociclila), -C(O)(C6-C10 arila), -C(O)(heteroarila), -Y-(C6-C10 arila), -Y-(heteroarila), -Y-(heterociclila de 5 a 10 membros), -NR6aR6b, -NR6aSO2R6b, -NR6aC(O)R6b, -OC(O)R6b, -NR6aC(O)OR6b, - OC(O)NR6aR6b,-OR6a, -SR6a, -S(O)R6a, -SO2R6a, -SO3R6a, - SO2NR6aR6b, -SO2NR42R43, -COR6a, -CO2R6a, -CONR6aR6b, -(C1- C4)fluoroalquila, -(C1-C4)fluoroalcóxi, -(CZ3Z4)aCN, em que n é um número inteiro variando de 0 a 6, e os grupos R7 anteriormente mencionados diferente de -H e halogênio são opcionalmente substituídos por 1 a 5 R38, ou R7 é uma porção selecionada do grupo consistindo em -(CZ3Z4)a-arila, -(CZ3Z4)a-heterociclo, (C2-C6)alquinila, - (CZ3Z4)a-(C3-C6)cicloalquila, -(CZ3Z4)a-(C5-C6)cicloalquenila, (C2-C6) alquenila e (C1-C6)alquila, em que a referida porção é opcionalmente substituída com 1 a 3 grupos Y2 independentemente selecionados, onde a é 0,1, 2, ou 3, e em que quando a é 2 ou 3, as unidades CZ3Z4 podem ser iguais ou diferentes; em que
[00311] cada R6a e R6b é independentemente selecionado do grupo consistindo em hidrogênio e uma porção selecionada do grupo consistindo em -(CZ5Z6)u-(C3-C6)cicloalquila, -(CZ5Z6)u-(C5- C6)cicloalquenila, -(CZ5Z6)u-arila, -(CZ5Z6)u-heterociclo, (C2- C6)alquenila, e (C1-C6)alquila, em que a referida porção é opcionalmente substituída com 1 a 3 grupos Y3 independentemente selecionados, onde u é 0,1, 2, ou 3, e em que quando u é 2 ou 3, as unidades CZ5Z6 podem ser iguais ou diferentes, ou
[00312] R6a e R6b tomados juntamente com os átomos adjacentes formam um heterociclo;
[00313] cada Z3, Z4, Z5 e Z6 é independentemente selecionado do grupo consistindo em H, F e (C1-C6)alquila, ou
[00314] cada Z3 e Z4, ou Z5 e Z6 são selecionados juntos para formar um carbociclo, ou
[00315] dois grupos Z3 sobre átomos de carbono adjacentes são selecionados juntos para opcionalmente formar um carbociclo;
[00316] cada Y2 e Y3 é independentemente selecionado do grupo consistindo em halogênio, ciano, nitro, tetrazolila, guanidino, amidino, metilguanidino, azido, -C(O)Z7, -OC(O)NH2, -OC(O) NHZ7, - OC(O)NZ7Z8, -NHC(O)Z7, -NHC(O)NH2, -NHC(O)NHZ7, - NHC(O)NZ7Z8, -C(O)OH, -C(O)OZ7, -C(O)NH2, -C(O)NHZ7,- C(O)NZ7Z8, -P(O)3H2, -P(O)3 (Z7)2, -S(O)3H, -S(O)Z7, -S(O)2Z7, - S(O)3Z7, -Z7, -OZ7, -OH, -NH2, -NHZ7, -NZ7Z8, -C(= NH)NH2,-C(= NOH)NH2, -N-morfolino, (C2-C6)alquenila, (C2-C6)alquinila, (C1- C6)haloalquila, (C2-C6)haloalquenila, (C2-C6)haloalquinila, (C1- C6)haloalcóxi, -(CZ9Z10)rNH2, -(CZ9Z10)rNHZ3, -(CZ9Z10)rNZ7Z8, -X6 (CZ9Z10)r-(C3-C8)cicloalquila, -X6 (CZ9Z10)r-(C5-C8)cicloalquenila, -X6 (CZ9Z10)r-arila e -X6 (CZ9Z10)r-heterociclo, em que
[00317] r é 1, 2, 3 ou 4;
[00318] X6 é selecionado do grupo consistindo em O, S, NH, -C(O)-, -C(O)NH-, -C(O)O-, -S(O)-, -S(O)2- e -S(O)3-;
[00319] Z7 e Z8 são independentemente selecionados do grupo consistindo em uma alquila de 1 a 12 átomos de carbono, uma alquenila de 2 a 12 átomos de carbono, uma alquinila de 2 a 12 átomos de carbono, uma cicloalquila de 3 a 8 átomos de carbono, uma cicloalquenila de 5 a 8 átomos de carbono, uma arila de 6 a 14 átomos de carbono, um heterociclo de 5 a 14 átomos de anel, uma aralquila de 7 a 15 átomos de carbono, e uma heteroaralquila de 5 a 14 átomos de anel, ou
[00320] Z7 e Z8 juntos podem opcionalmente formar um heterociclo;
[00321] Z9 e Z10 são independentemente selecionados do grupo consistindo em H, F, a (C1-C12)alquila, a (C6-C14)arila, a (C5- C14)heteroarila, a (C7-C15)aralquila e a (C5-C14)heteroaralquila, ou
[00322] Z9 e Z10 são tomados juntos para formar um carbociclo, ou
[00323] dois grupos Z9 sobre átomos de carbono adjacentes são tomados juntos para formar um carbociclo; ou
[00324] quaisquer dois grupos Y2 ou Y3 ligados aos átomos de carbono adjacentes podem ser tomados juntos para serem - O[C(Z9)(Z10)]rO ou -O[C(Z9)(Z10)]r+1, ou
[00325] quaisquer dois grupos Y2 ou Y3 ligados aos mesmos ou adjacentes átomos de carbono podem ser selecionados juntos para formar um carbociclo ou heterociclo; e em que
[00326] qualquer um dos substituintes acima mencionados compreendendo um grupo CH3 (metila), CH2 (metileno), ou CH (metina) que não é ligado a um grupo halogênio, SO ou SO2 ou a um átomo de N, O ou S opcionalmente transporta sobre o referido grupo um substituinte selecionado de hidróxi, halogênio, (C1-C4)alquila, (C1- C4)alcóxi e uma -N[(C1-C4)alquila][(C1-C4)alquila];
[00327] R1 é -CHCH ou -C=C-(CR45R45)n-R46;
[00328] cada R45 é independentemente selecionado do grupo consistindo em H, a (C1-C6)alquila e a (C3-C8)cicloalquila;
[00329] R46 é selecionado do grupo consistindo em heterociclila, - N(R47)-C(O)-N(R47)(R48), -N(R47)-C(S)-N(R47)(R48), -N(R47)-C(O)-OR48, -N(R47)-C(O)-(CH2)n-R48, -N(R47)-SO2R47, -(CH2)nNR47R48, -(CH2)nOR48, -(CH2)nSR49, -(CH2)nS(O)R49, -(CH2)nS(O)2R49, -OC(O)R49, - OC(O)OR49, -C(O)NR47R48, heteroarila opcionalmente substituída com um ou mais substituintes selecionados do grupo consistindo em halo, - CF3, (C1-C6)alcóxi, -NO2, (C1-C6)alquila, -CN, -SO2R50 e - (CH2)nNR50R51, e arila opcionalmente substituída com um ou mais substituintes selecionados do grupo consistindo em halo, -CF3, (C1- C6)alcóxi, -NO2, (C1-C6)alquila, -CN, -SO2R50 e -(CH2)nNR50R51;
[00330] R47 e R48 são independentemente selecionados do grupo consistindo em H, (C1-C6)alquila, (C3-C8)cicloalquila, heterociclila, - (CH2)nNR50R51, -(CH2)nOR50, -(CH2)nC(O)R49, -C(O)2R49, -(CH2)nSR49, - (CH2)nS(O)R49, -(CH2)nS(O)2R49, -(CH2)nR49, -(CH2)nCN, arila opcionalmente substituída com um ou mais substituintes selecionados do grupo consistindo em halo, -CF3, (C1-C6)alcóxi, -NO2, (C1-C6)alquila, -CN, -(CH2)nOR49, -(CH2)nheterociclila, -(CH2)nheteroarila, -SO2R50 e - (CH2)nNR50R51, e heteroarila opcionalmente substituída com um ou mais substituintes selecionados do grupo consistindo em halo, -CF3, (C1-C6)alcóxi, -NO2, (C1-C6)alquila, -CN, -(CH2)nOR49, - (CH2)nheterociclila, -(CH2)nheteroarila, -SO2R50 e -(CH2)nNR50R51, ou
[00331] R47 e R48, juntamente com o átomo ao qual eles são ligados, formam um anel carbo- ou heterocíclico de 3 a 8 membros;
[00332] R49 é selecionado do grupo consistindo em (C1-C6)alquila, (C3-C8)cicloalquila, heterociclil(C1-C6)alquileno, aril(C1-C6)alquileno em que a arila é opcionalmente substituída com um ou mais substituintes selecionados do grupo consistindo em halo, -CF3, (C1-C6)alcóxi, -NO2, (C1-C6)alquila, -CN, -SO2R50 e -(CH2)nNR50R51, heteroaril(C1- C6)alquileno em que a heteroarila é opcionalmente substituída com um ou mais substituintes selecionados do grupo consistindo em halo, - CF3, (C1-C6)alcóxi, -NO2, (C1-C6)alquila, -CN, -SO2R50 e - (CH2)nNR50R51, arila opcionalmente substituída com um ou mais substituintes selecionados do grupo consistindo em halo, -CF3, (C1- C6)alcóxi, -NO2, (C1-C6)alquila, -CN, -SO2R50 e -(CH2)nNR50R51, e heteroarila opcionalmente substituída com um ou mais substituintes selecionados do grupo consistindo em halo, -CF3, (C1-C6)alcóxi, -NO2, (C1-C6)alquila, -CN, -SO2R50 e -(CH2)nNR50R51;
[00333] R50 e R51 são independentemente selecionados do grupo consistindo em H, (C1-C6)alquila, (C3-C8)cicloalquila e -C(O)R45, ou
[00334] R50 e R51, juntamente com o átomo ao qual eles são ligados, formam um anel carbo- ou heterocíclico de 3 a 8 membros; e
[00335] R21 é o grupo definido por -(Z11)-(Z12)m-(Z13)m1, em que
[00336] Z11 é heterociclila, quando m e m1 são 0, ou heterociclileno, quando ou m ou m1 é 1;
[00337] Z12 é selecionado do grupo consistindo em OC(O), OC(S) e C(O);
[00338] Z13 é selecionado do grupo consistindo em heterociclila, aralquila, N(H)R52, (C1-C3)alquila, -OR52, halo, S(O)2R56, (C1- C3)hidroxialquila e (C1-C3)haloalquila;
[00339] m é 0 ou 1;
[00340] m1 é 0 ou 1;
[00341] R52 é selecionado do grupo consistindo em H, - (CH2)qS(O)2R54, -(C1-C6) alquil- NR53R53 (C1-C3)alquila, -(CH2)qOR53, - C(O)R54 e -C(O)OR53;
[00342] q é 0, 1, 2, 3 ou 4;
[00343] cada R53 é independentemente (C1-C3)alquila;
[00344] R54 é (C1-C3)alquila ou N(H)R53;
[00345] R56 é selecionado do grupo consistindo em NH2, (C1- C3)alquila e OR52;
[00346] A1 é selecionado do grupo consistindo em -CH2-, -O-, -S-, - N(H)-, -N(CI-C6 alquil)-, -N-(Y-aril)-, -N-OMe, -NCH2OMe e N-Bn;
[00347] Y é uma ligação ou -(C(R11)(H))t-, em que t é um número inteiro de 1 a 6; e
[00348] R11 em cada ocorrência é independentemente selecionado do grupo consistindo em H e C1-C6 alquila, em que a C1-C6 alquila é opcionalmente substituída;
[00349] A2 é selecionado do grupo consistindo em N e CR, em que R é selecionado do grupo consistindo em -H, halogênio, -CN, C1-C6 alquila, C2-C6 alquenila, e C2-C6 alquinila, em que a C1-C6 alquila, C2C6 alquenila, e C2-C6 alquinila são opcionalmente substituídas;
[00350] A3 é selecionado do grupo consistindo em C-D e N;
[00351] R2 em cada ocorrência é independentemente selecionado do grupo consistindo em -H, halogênio, trihalometila, -CN, -NO2, -NH2, -OR3, -NR3R4, -S(O)0-2R3, -S(O)2NR3R3, -C(O)OR3, -C(O)NR3R3, - N(R3)SO2R3, -N(R3)C(O)R3, -N(R3)CO2R3, -C(O)R3, C1-C4 alcóxi, C1-C4 alquiltio, -O(CH2)narila, -O(CH2)nheteroarila, -(CH2)0-5 (arila), -(CH2)0-5 (heteroarila), C1-C6 alquila, C2-C6 alquenila, C2-C6 alquinila, -CH2 (CH2)0-4-T2, em que T2 é selecionado do grupo consistindo em -OH, - OMe, -OEt, -NH2, -NHMe, -NMe2, -NHEt e -NEt2, e em que a arila, heteroarila, C1-C6 alquila, C2-C6 alquenila, e C2-C6 alquinila são opcionalmente substituídas; e
[00352] cada R3 é independentemente selecionado do grupo consistindo em -H e R4;
[00353] R4 é selecionado do grupo consistindo em uma (C1- C6)alquila, uma arila, uma arilalquila inferior, uma heterociclila e uma heterociclilalquila inferior, cada uma das quais é opcionalmente substituída, ou
[00354] R3 e R4, tomados juntamente com um nitrogênio comum ao qual eles são ligados, formam uma heterociclila de cinco a sete membros opcionalmente substituída, que opcionalmente contém pelo menos um heteroátomo anular adicional selecionado do grupo consistindo em N, O, S e P;
[00355] X e X1 são cada qual independentemente selecionado do grupo consistindo em -H, halogênio, ciano, nitro, C1-C6 alquila, ou
[00356] X e X1 tomados juntamente com o átomo ao qual eles são ligados, formam a C3-C7 cicloalquila;
[00357] b é 0, 1, 2, 3 ou 4;
[00358] R20 é selecionado do grupo consistindo em -H, halogênio, trihalometila, -CN, -NO2, -NH2, -OR3, -OCF3, -NR3R4, -S(O)0-2R3, - S(O)2NR3R3, -C(O)OR3, -C(O)NR3R3, -N(R3)SO2R3, -N(R3)C(O)R3, - N(R3)C(O)OR3, -C(O)R3, -C(O)SR3, C1-C4 alcóxi, C1-C4 alquiltio, - O(CH2)narila, -O(CH2)nheteroarila, -(CH2)0-5 (arila), -(CH2)0-5 (heteroarila), C1-C6 alquila, C2-C6 alquenila, C2-C6 alquinila, -CH2 (CH2)0-4-T2, uma C1-4 alquilcarbonila opcionalmente substituída, C1-4 alcóxi, um amino opcionalmente substituído por C1-4 alquila em que o C1-C4 alquila pode ser opcionalmente substituída por C1-4 alcóxi e um grupo carboxíclico ou heterocíclico de três a sete membros saturado ou insaturado, em que T2 é selecionado do grupo consistindo em -OH, -OMe, -OEt, -NH2, -NHMe, -NMe2, -NHEt e -NEt2, e em que a arila, heteroarila, C1-C6 alquila, C2-C6 alquenila, e C2-C6 alquinila são opcionalmente substituídas;
[00359] Em outra modalidade preferida dos compostos de acordo com a presente invenção, D é definido pelo grupo R7, em que R7 é selecionado do grupo consistindo em -H, halogênio, C1-C6 alquila, - C(O)NR42R43, -C(O)(C6-C10 arila), -C(O)(heterociclila), - C(O)(heteroarila), -Y-(C6-C10 arila), -Y-(heterociclila de 5 a 10 membros), -Y-(heteroarila), -S-arila, -S-C1-C6 alquila, -SO-C1-C6 alquila, -SO2-C1-C6 alquila, -Y-NR42R43, -SO2NR42R43 e -CO2R6a, em que os grupos R7 anteriormente mencionados diferentes de -H e halogênio são opcionalmente substituídos por 1 a 5 R38.
[00360] Em outra modalidade preferida dos compostos de acordo com a presente invenção, D é definido pelo grupo R7, em que R7 é selecionado do grupo consistindo em -H, -C(O)NR42R43, -Y-(C6-C10 arila), -Y-(heteroarila), -C(O)(heterociclila) e -Y-NR42R43, em que os grupos R7 anteriormente mencionados diferentes de -H são opcionalmente substituídos por 1 a 5 R38.
[00361] Em outra modalidade preferida dos compostos de acordo com a presente invenção, D é definido pelo grupo R7, em que R7 é selecionado do grupo consistindo em -H, -C(O)NR42R43, -Y-(C6-C10 arila), -Y-(heteroarila), -C(O)(heterociclila) e -Y-NR42R43, em que os grupos R7 anteriormente mencionados diferentes de -H são opcionalmente substituídos por 1 a 5 R38.
[00362] Em outra modalidade preferida dos compostos de acordo com a presente invenção, D é definido pelo grupo R7, em que R7 é selecionado do grupo consistindo em -H, -Y-(C6-C10 arila), -Y- (heteroarila) e -C(O)(heterociclila), em que os grupos R7 anteriormente mencionados diferentes de -H são opcionalmente substituídos por 1 a 5 R38.
[00363] Compostos preferidos de acordo com as fórmulas A-1 e B-1 têm grupos como definido nas modalidades preferidas da presente invenção.
[00364] Em modalidades preferidas dos compostos de acordo com a presente invenção, os compostos são representados pelas fórmulas A-2 e B-2:
[00365] e sais farmaceuticamente aceitáveis e complexos destes, em que
[00366] D é selecionado do grupo consistindo em R7, R1 e R21, em que
[00367] R7 é selecionado do grupo consistindo em -H, halogênio, nitro, azido, C1-C6 alquila, C3-C10 cicloalquila, -C(O)NR42R43, -Y- NR42R43, -NR42C(= O)R43, -SO2R42, -SO2NR42R43, -NR37SO2R42, - NR37SO2NR42R43, -C(= N-OR42)R43, -C(= NR42)R43, -NR37C(= NR42)R43, -C(= NR42)NR37R43, -NR37C(= NR42)NR37R43, -C(O)R42, -CO2R42, - C(O)(heterociclila), -C(O)(C6-C10 arila), -C(O)(heteroarila), -Y-(C6-C10 arila), -Y-(heteroarila), -Y-(heterociclila de 5 a 10 membros), -NR6aR6b, -NR6aSO2R6b, -NR6aC(O)R6b, -OC(O)R6b, -NR6aC(O)OR6b, - OC(O)NR6aR6b,-OR6a, -SR6a, -S(O)R6a, -SO2R6a, -SO3R6a, - SO2NR6aR6b, -SO2NR42R43, -COR6a, -CO2R6a, -CONR6aR6b, -(C1- C4)fluoroalquila, -(C1-C4)fluoroalcóxi, -(CZ3Z4)aCN, em que n é um número inteiro variando de 0 a 6, e os grupos R7 anteriormente mencionados diferente de -H e halogênio são opcionalmente substituídos por 1 a 5 R38, ou R7 é uma porção selecionada do grupo consistindo em -(CZ3Z4)a-arila, -(CZ3Z4)a-heterociclo, (C2-C6)alquinila, - (CZ3Z4)a-(C3-C6)cicloalquila, -(CZ3Z4)a-(C5-C6)cicloalquenila, (C2-C6) alquenila e (C1-C6)alquila, em que a referida porção é opcionalmente substituída com 1 a 3 grupos Y2 independentemente selecionados, onde a é 0,1, 2, ou 3, e em que quando a é 2 ou 3, as unidades CZ3Z4 podem ser iguais ou diferentes; em que
[00368] cada R6a e R6b é independentemente selecionado do grupo consistindo em hidrogênio e a porção selecionada do grupo consistindo em -(CZ5Z6)u-(C3-C6)cicloalquila, -(CZ5Z6)u-(C5- C6)cicloalquenila, -(CZ5Z6)u-arila, -(CZ5Z6)u-heterociclo, (C2- C6)alquenila, e (C1-C6)alquila, em que a referida porção é opcionalmente substituída com 1 a 3 grupos Y3 independentemente selecionados, onde u é 0,1, 2, ou 3, e em que quando u é 2 ou 3, as unidades CZ5Z6 podem ser iguais ou diferentes, ou
[00369] R6a e R6b tomados juntamente com os átomos adjacentes formam um heterociclo;
[00370] cada Z3, Z4, Z5 e Z6 é independentemente selecionado do grupo consistindo em H, F e (C1-C6)alquila, ou
[00371] cada Z3 e Z4, ou Z5 e Z6 são selecionados juntos para formar um carbociclo, ou
[00372] dois grupos Z3 sobre átomos de carbono adjacentes são selecionados juntos para opcionalmente formar um carbociclo;
[00373] cada Y2 e Y3 é independentemente selecionado do grupo consistindo em halogênio, ciano, nitro, tetrazolila, guanidino, amidino, metilguanidino, azido, -C(O)Z7, -OC(O)NH2, -OC(O) NHZ7, - OC(O)NZ7Z8, -NHC(O)Z7, -NHC(O)NH2, -NHC(O)NHZ7, - NHC(O)NZ7Z8, -C(O)OH, -C(O)OZ7, -C(O)NH2, -C(O)NHZ7,- C(O)NZ7Z8, -P(O)3H2, -P(O)3 (Z7)2, -S(O)3H, -S(O)Z7, -S(O)2Z7, - S(O)3Z7, -Z7, -OZ7, -OH, -NH2, -NHZ7, -NZ7Z8, -C(= NH)NH2,-C(= NOH)NH2, -N-morfolino, (C2-C6)alquenila, (C2-C6)alquinila, (C1- C6)haloalquila, (C2-C6)haloalquenila, (C2-C6)haloalquinila, (C1- C6)haloalcóxi, -(CZ9Z10)rNH2, -(CZ9Z10)rNHZ3, -(CZ9Z10)rNZ7Z8, -X6 (CZ9Z10)r-(C3-C8)cicloalquila, -X6 (CZ9Z10)r-(C5-C8)cicloalquenila, -X6 (CZ9Z10)r-arila e -X6 (CZ9Z10)r-heterociclo, em que
[00374] r é 1, 2, 3 ou 4;
[00375] X6 é selecionado do grupo consistindo em O, S, NH, -C(O)-, -C(O)NH-, -C(O)O-, -S(O)-, -S(O)2- e -S(O)3-;
[00376] Z7 e Z8 são independentemente selecionados do grupo consistindo em uma alquila de 1 a 12 átomos de carbono, uma alquenila de 2 a 12 átomos de carbono, uma alquinila de 2 a 12 átomos de carbono, uma cicloalquila de 3 a 8 átomos de carbono, uma cicloalquenila de 5 a 8 átomos de carbono, uma arila de 6 a 14 átomos de carbono, um heterociclo de 5 a 14 átomos de anel, uma aralquila de 7 a 15 átomos de carbono, e uma heteroaralquila de 5 a 14 átomos de anel, ou
[00377] Z7 e Z8 juntos podem opcionalmente formar um heterociclo;
[00378] Z9 e Z10 são independentemente selecionados do grupo consistindo em H, F, a (C1-C12)alquila, a (C6-C14)arila, a (C5- C14)heteroarila, a (C7-C15)aralquila e a (C5-C14)heteroaralquila, ou
[00379] Z9 e Z10 são tomados juntos para formar um carbociclo, ou
[00380] dois grupos Z9 sobre átomos de carbono adjacentes são tomados juntos para formar um carbociclo; ou
[00381] quaisquer dois grupos Y2 ou Y3 ligados aos átomos de carbono adjacentes podem ser tomados juntos para serem - O[C(Z9)(Z10)]rO ou -O[C(Z9)(Z10)]r+1, ou
[00382] quaisquer dois grupos Y2 ou Y3 ligados aos mesmos ou adjacentes átomos de carbono podem ser selecionados juntos para formar um carbociclo ou heterociclo; e em que
[00383] qualquer um dos substituintes acima mencionados compreendendo um grupo CH3 (metila), CH2 (metileno), ou CH (metina) que não é ligado a um grupo halogênio, SO ou SO2 ou a um átomo de N, O ou S opcionalmente transporta sobre o referido grupo um substituinte selecionado de hidróxi, halogênio, (C1-C4)alquila, (C1- C4)alcóxi e an -N[(C1-C4)alquila][(C1-C4)alquila];
[00384] R1 é -CHCH ou -C=C-(CR45R45)n-R46;
[00385] cada R45 é independentemente selecionado do grupo consistindo em H, a (C1-C6)alquila e a (C3-C8)cicloalquila;
[00386] R46 é selecionado do grupo consistindo em heterociclila, - N(R47)-C(O)-N(R47)(R48), -N(R47)-C(S)-N(R47)(R48), -N(R47)-C(O)-OR48, -N(R47)-C(O)-(CH2)n-R48, -N(R47)-SO2R47, -(CH2)nNR47R48, -(CH2)nOR48, -(CH2)nSR49, -(CH2)nS(O)R49, -(CH2)nS(O)2R49, -OC(O)R49, - OC(O)OR49, -C(O)NR47R48, heteroarila opcionalmente substituída com um ou mais substituintes selecionados do grupo consistindo em halo, - CF3, (C1-C6)alcóxi, -NO2, (C1-C6)alquila, -CN, -SO2R50 e - (CH2)nNR50R51, e arila opcionalmente substituída com um ou mais substituintes selecionados do grupo consistindo em halo, -CF3, (C1- C6)alcóxi, -NO2, (C1-C6)alquila, -CN, -SO2R50 e -(CH2)nNR50R51;
[00387] R47 e R48 são independentemente selecionados do grupo consistindo em H, (C1-C6)alquila, (C3-C8)cicloalquila, heterociclila, - (CH2)nNR50R51, -(CH2)nOR50, -(CH2)nC(O)R49, -C(O)2R49, -(CH2)nSR49, - (CH2)nS(O)R49, -(CH2)nS(O)2R49, -(CH2)nR49, -(CH2)nCN, arila opcionalmente substituída com um ou mais substituintes selecionados do grupo consistindo em halo, -CF3, (C1-C6)alcóxi, -NO2, (C1-C6)alquila, -CN, -(CH2)nOR49, -(CH2)nheterociclila, -(CH2)nheteroarila, -SO2R50 e - (CH2)nNR50R51, e heteroarila opcionalmente substituída com um ou mais substituintes selecionados do grupo consistindo em halo, -CF3, (C1-C6)alcóxi, -NO2, (C1-C6)alquila, -CN, -(CH2)nOR49, - (CH2)nheterociclila, -(CH2)nheteroarila, -SO2R50 e -(CH2)nNR50R51, ou
[00388] R47 e R48, juntamente com o átomo ao qual eles são ligados, formam um anel carbo- ou heterocíclico de 3 a 8 membros;
[00389] R49 é selecionado do grupo consistindo em (C1-C6)alquila, (C3-C8)cicloalquila, heterociclil(C1-C6)alquileno, aril(C1-C6)alquileno em que a arila é opcionalmente substituída com um ou mais substituintes selecionados do grupo consistindo em halo, -CF3, (C1-C6)alcóxi, -NO2, (C1-C6)alquila, -CN, -SO2R50 e -(CH2)nNR50R51, heteroaril(C1- C6)alquileno em que a heteroarila é opcionalmente substituída com um ou mais substituintes selecionados do grupo consistindo em halo, - CF3, (C1-C6)alcóxi, -NO2, (C1-C6)alquila, -CN, -SO2R50 e - (CH2)nNR50R51, arila opcionalmente substituída com um ou mais substituintes selecionados do grupo consistindo em halo, -CF3, (C1- C6)alcóxi, -NO2, (C1-C6)alquila, -CN, -SO2R50 e -(CH2)nNR50R51, e heteroarila opcionalmente substituída com um ou mais substituintes selecionados do grupo consistindo em halo, -CF3, (C1-C6)alcóxi, -NO2, (C1-C6)alquila, -CN, -SO2R50 e -(CH2)nNR50R51;
[00390] R50 e R51 são independentemente selecionados do grupo consistindo em H, (C1-C6)alquila, (C3-C8)cicloalquila e -C(O)R45, ou
[00391] R50 e R51, juntamente com o átomo ao qual eles são ligados, formam um anel carbo- ou heterocíclico de 3 a 8 membros; e
[00392] R21 é o grupo definido por -(Z11)-(Z12)m-(Z13)m1, em que
[00393] Z11 é heterociclila, quando m e m1 são 0, ou heterociclileno, quando ou m ou m1 é 1;
[00394] Z12 é selecionado do grupo consistindo em OC(O), OC(S) e C(O);
[00395] Z13 é selecionado do grupo consistindo em heterociclila, aralquila, N(H)R52, (C1-C3)alquila, -OR52, halo, S(O)2R56, (C1- C3)hidroxialquila e (C1-C3)haloalquila;
[00396] m é 0 ou 1;
[00397] m1 é 0 ou 1;
[00398] R52 é selecionado do grupo consistindo em H, - (CH2)qS(O)2R54, -(C1-C6) alquil- NR53R53 (C1-C3)alquila, -(CH2)qOR53, - C(O)R54 e -C(O)OR53;
[00399] q é 0, 1, 2, 3 ou 4;
[00400] cada R53 é independentemente (C1-C3)alquila;
[00401] R54 é (C1-C3)alquila ou N(H)R53;
[00402] R56 é selecionado do grupo consistindo em NH2, (C1- C3)alquila e OR52;
[00403] A1 é selecionado do grupo consistindo em -CH2-, -O-, -S-, - N(H)-, -N(CI-C6 alquil)-, -N-(Y-aril)-, -N-OMe, -NCH2OMe e N-Bn;
[00404] Y é uma ligação ou -(C(R11)(H))t-, em que t é um número inteiro de 1 a 6; e
[00405] R11 em cada ocorrência é independentemente selecionado do grupo consistindo em H e C1-C6 alquila, em que a C1-C6 alquila é opcionalmente substituída;
[00406] A2 é selecionado do grupo consistindo em N e CR, em que R é selecionado do grupo consistindo em -H, halogênio, -CN, C1-C6 alquila, C2-C6 alquenila, e C2-C6 alquinila, em que a C1-C6 alquila, C2C6 alquenila, e C2-C6 alquinila são opcionalmente substituídas;
[00407] A3 é selecionado do grupo consistindo em C-D e N;
[00408] R2 em cada ocorrência é independentemente selecionado do grupo consistindo em -H, halogênio, trihalometila, -CN, -NO2, -NH2, -OR3, -NR3R4, -S(O)0-2R3, -S(O)2NR3R3, -C(O)OR3, -C(O)NR3R3, - N(R3)SO2R3, -N(R3)C(O)R3, -N(R3)CO2R3, -C(O)R3, C1-C4 alcóxi, C1-C4 alquiltio, -O(CH2)narila, -O(CH2)nheteroarila, -(CH2)0-5 (arila), -(CH2)0-5 (heteroarila), C1-C6 alquila, C2-C6 alquenila, C2-C6 alquinila, -CH2 (CH2)0-4-T2, em que T2 é selecionado do grupo consistindo em -OH, - OMe, -OEt, -NH2, -NHMe, -NMe2, -NHEt e -NEt2, e em que a arila, heteroarila, C1-C6 alquila, C2-C6 alquenila, e C2-C6 alquinila são opcionalmente substituídas; e
[00409] R3 selecionado do grupo consistindo em -H e R4;
[00410] R4 é selecionado do grupo consistindo em uma (C1- C6)alquila, uma arila, uma arilalquila inferior, uma heterociclila e uma heterociclilalquila inferior, cada uma das quais é opcionalmente substituída, ou
[00411] R3 e R4, tomados juntamente com um nitrogênio comum ao qual eles são ligados, formam uma heterociclila de cinco a sete membros opcionalmente substituída, que opcionalmente contém pelo menos um heteroátomo anular adicional selecionado do grupo consistindo em N, O, S e P;
[00412] X e X1 são cada qual independentemente selecionado do grupo consistindo em -H, halogênio, ciano, nitro, C1-C6 alquila, ou
[00413] X e X1 tomados juntamente com o átomo ao qual eles são ligados formam uma C3-C7 cicloalquila;
[00414] R20 é selecionado do grupo consistindo em -H, halogênio, trihalometila, -CN, -NO2, -NH2, -OR3, -OCF3, -NR3R4, -S(O)0-2R3, - S(O)2NR3R3, -C(O)OR3, -C(O)NR3R3, -N(R3)SO2R3, -N(R3)C(O)R3, - N(R3)C(O)OR3, -C(O)R3, -C(O)SR3, C1-C4 alcóxi, C1-C4 alquiltio, - O(CH2)narila, -O(CH2)nheteroarila, -(CH2)0-5 (arila), -(CH2)0-5 (heteroarila), C1-C6 alquila, C2-C6 alquenila, C2-C6 alquinila, -CH2 (CH2)0-4-T2, uma C1-4 alquilcarbonila opcionalmente substituída, C1-4 alcóxi, um amino opcionalmente substituído por C1-4 alquila em que o C1-4 alquila pode ser opcionalmente substituída por C1-4 alcóxi e um grupo carboxíclico ou heterocíclico de três a sete membros saturado ou insaturado, em que T2 é selecionado do grupo consistindo em -OH, -OMe, -OEt, -NH2, -NHMe, -NMe2, -NHEt e -NEt2, e em que a arila, heteroarila, C1-C6 alquila, C2-C6 alquenila, e C2-C6 alquinila são opcionalmente substituídas.
[00415] Em outra modalidade preferida dos compostos de acordo com a presente invenção, D é definido pelo grupo R7, em que R7 é selecionado do grupo consistindo em -H, -C(O)NR42R43, -Y-(C6-C10 arila), -Y-(heteroarila) e -Y-NR42R43, em que os grupos R7 anteriormente mencionados diferentes de -H são opcionalmente substituídos por 1 a 5 R38.
[00416] Compostos preferidos de acordo com as fórmulas A-2 e B-2 têm grupos como definido nas modalidades preferidas da presente invenção.
[00417] Em outra modalidade preferida dos compostos de acordo com a presente invenção, os compostos são representados pelas fórmulas A-3 e B-3:
[00418] e sais farmaceuticamente aceitáveis e complexos destes, em que
[00419] D é selecionado do grupo consistindo em R7, R1 e R21, em que
[00420] R7 é selecionado do grupo consistindo em -H, halogênio, nitro, azido, C1-C6 alquila, C3-C10 cicloalquila, -C(O)NR42R43, -Y- NR42R43, -NR42C(= O)R43, -SO2R42, -SO2NR42R43, -NR37SO2R42, - NR37SO2NR42R43, -C(= N-OR42)R43, -C(= NR42)R43, -NR37C(= NR42)R43, -C(= NR42)NR37R43, -NR37C(= NR42)NR37R43, -C(O)R42, -CO2R42, - C(O)(heterociclila), -C(O)(C6-C10 arila), -C(O)(heteroarila), -Y-(C6-C10 arila), -Y-(heteroarila), -Y-(heterociclila de 5 a 10 membros), -NR6aR6b, -NR6aSO2R6b, -NR6aC(O)R6b, -OC(O)R6b, -NR6aC(O)OR6b, - OC(O)NR6aR6b,-OR6a, -SR6a, -S(O)R6a, -SO2R6a, -SO3R6a, - SO2NR6aR6b, -SO2NR42R43, -COR6a, -CO2R6a, -CONR6aR6b, -(C1- C4)fluoroalquila, -(C1-C4)fluoroalcóxi, -(CZ3Z4)aCN, em que n é um número inteiro variando de 0 a 6, e os grupos R7 anteriormente mencionados diferente de -H e halogênio são opcionalmente substituídos por 1 a 5 R38, ou R7 é porção selecionada do grupo consistindo em -(CZ3Z4)a-arila, -(CZ3Z4)a-heterociclo, (C2-C6)alquinila, - (CZ3Z4)a-(C3-C6)cicloalquila, -(CZ3Z4)a-(C5-C6)cicloalquenila, (C2-C6) alquenila e (C1-C6)alquila, em que a referida porção é opcionalmente substituída com 1 a 3 grupos Y2 independentemente selecionados, onde a é 0,1, 2, ou 3, e em que quando a é 2 ou 3, as unidades CZ3Z4 podem ser iguais ou diferentes; em que
[00421] cada R6a e R6b é independentemente selecionado do grupo consistindo em hidrogênio e uma porção selecionada do grupo consistindo em -(CZ5Z6)u-(C3-C6)cicloalquila, -(CZ5Z6)u-(C5- C6)cicloalquenila, -(CZ5Z6)u-arila, -(CZ5Z6)u-heterociclo, (C2- C6)alquenila, e (C1-C6)alquila, em que a referida porção é opcionalmente substituída com 1 a 3 grupos Y3 independentemente selecionados, onde u é 0,1, 2, ou 3, e em que quando u é 2 ou 3, as unidades CZ5Z6 podem ser iguais ou diferentes, ou
[00422] R6a e R6b tomados juntamente com os átomos adjacentes formam um heterociclo;
[00423] cada Z3, Z4, Z5 e Z6 é independentemente selecionado do grupo consistindo em H, F e (C1-C6)alquila, ou
[00424] cada Z3 e Z4, ou Z5 e Z6 são selecionados juntos para formar um carbociclo, ou
[00425] dois grupos Z3 sobre átomos de carbono adjacentes são selecionados juntos para opcionalmente formar um carbociclo;
[00426] cada Y2 e Y3 é independentemente selecionado do grupo consistindo em halogênio, ciano, nitro, tetrazolila, guanidino, amidino, metilguanidino, azido, -C(O)Z7, -OC(O)NH2, -OC(O) NHZ7, - OC(O)NZ7Z8, -NHC(O)Z7, -NHC(O)NH2, -NHC(O)NHZ7, - NHC(O)NZ7Z8, -C(O)OH, -C(O)OZ7, -C(O)NH2, -C(O)NHZ7,- C(O)NZ7Z8, -P(O)3H2, -P(O)3 (Z7)2, -S(O)3H, -S(O)Z7, -S(O)2Z7, - S(O)3Z7, -Z7, -OZ7, -OH, -NH2, -NHZ7, -NZ7Z8, -C(= NH)NH2,-C(= NOH)NH2, -N-morfolino, (C2-C6)alquenila, (C2-C6)alquinila, (C1- C6)haloalquila, (C2-C6)haloalquenila, (C2-C6)haloalquinila, (C1- C6)haloalcóxi, -(CZ9Z10)rNH2, -(CZ9Z10)rNHZ3, -(CZ9Z10)rNZ7Z8, -X6 (CZ9Z10)r-(C3-C8)cicloalquila, -X6 (CZ9Z10)r-(C5-C8)cicloalquenila, -X6 (CZ9Z10)r-arila e -X6 (CZ9Z10)r-heterociclo, em que
[00427] r é 1, 2, 3 ou 4;
[00428] X6 é selecionado do grupo consistindo em O, S, NH, -C(O)-, -C(O)NH-, -C(O)O-, -S(O)-, -S(O)2- e -S(O)3-;
[00429] Z7 e Z8 são independentemente selecionados do grupo consistindo em uma alquila de 1 a 12 átomos de carbono, uma alquenila de 2 a 12 átomos de carbono, uma alquinila de 2 a 12 átomos de carbono, uma cicloalquila de 3 a 8 átomos de carbono, uma cicloalquenila de 5 a 8 átomos de carbono, uma arila de 6 a 14 átomos de carbono, um heterociclo de 5 a 14 átomos de anel, uma aralquila de 7 a 15 átomos de carbono, e uma heteroaralquila de 5 a 14 átomos de anel, ou
[00430] Z7 e Z8 juntos podem opcionalmente formar um heterociclo;
[00431] Z9 e Z10 são independentemente selecionados do grupo consistindo em H, F, a (C1-C12)alquila, a (C6-C14)arila, a (C5- C14)heteroarila, a (C7-C15)aralquila e a (C5-C14)heteroaralquila, ou
[00432] Z9 e Z10 são tomados juntos para formar um carbociclo, ou
[00433] dois grupos Z9 sobre átomos de carbono adjacentes são tomados juntos para formar um carbociclo; ou
[00434] quaisquer dois grupos Y2 ou Y3 ligados aos átomos de carbono adjacentes podem ser tomados juntos para serem - O[C(Z9)(Z10)]rO ou -O[C(Z9)(Z10)]r+1, ou
[00435] quaisquer dois grupos Y2 ou Y3 ligados aos mesmos ou adjacentes átomos de carbono podem ser selecionados juntos para formar um carbociclo ou heterociclo; e em que
[00436] qualquer um dos substituintes acima mencionados compreendendo um grupo CH3 (metila), CH2 (metileno), ou CH (metina) que não é ligado a um grupo halogênio, SO ou SO2 ou a um átomo de N, O ou S opcionalmente transporta sobre o referido grupo um substituinte selecionado de hidróxi, halogênio, (C1-C4)alquila, (C1- C4)alcóxi e an -N[(C1-C4)alquila][(C1-C4)alquila];
[00437] R1 é -CHCH ou -C=C-(CR45R45)n-R46;
[00438] cada R45 é independentemente selecionado do grupo consistindo em H, a (C1-C6)alquila e a (C3-C8)cicloalquila;
[00439] R46 é selecionado do grupo consistindo em heterociclila, - N(R47)-C(O)-N(R47)(R48), -N(R47)-C(S)-N(R47)(R48), -N(R47)-C(O)-OR48, -N(R47)-C(O)-(CH2)n-R48, -N(R47)-SO2R47, -(CH2)nNR47R48, -(CH2)nOR48, -(CH2)nSR49, -(CH2)nS(O)R49, -(CH2)nS(O)2R49, -OC(O)R49, - OC(O)OR49, -C(O)NR47R48, heteroarila opcionalmente substituída com um ou mais substituintes selecionados do grupo consistindo em halo, - CF3, (C1-C6)alcóxi, -NO2, (C1-C6)alquila, -CN, -SO2R50 e - (CH2)nNR50R51, e arila opcionalmente substituída com um ou mais substituintes selecionados do grupo consistindo em halo, -CF3, (C1- C6)alcóxi, -NO2, (C1-C6)alquila, -CN, -SO2R50 e -(CH2)nNR50R51;
[00440] R47 e R48 são independentemente selecionados do grupo consistindo em H, (C1-C6)alquila, (C3-C8)cicloalquila, heterociclila, - (CH2)nNR50R51, -(CH2)nOR50, -(CH2)nC(O)R49, -C(O)2R49, -(CH2)nSR49, - (CH2)nS(O)R49, -(CH2)nS(O)2R49, -(CH2)nR49, -(CH2)nCN, arila opcionalmente substituída com um ou mais substituintes selecionados do grupo consistindo em halo, -CF3, (C1-C6)alcóxi, -NO2, (C1-C6)alquila, -CN, -(CH2)nOR49, -(CH2)nheterociclila, -(CH2)nheteroarila, -SO2R50 e - (CH2)nNR50R51, e heteroarila opcionalmente substituída com um ou mais substituintes selecionados do grupo consistindo em halo, -CF3, (C1-C6)alcóxi, -NO2, (C1-C6)alquila, -CN, -(CH2)nOR49, - (CH2)nheterociclila, -(CH2)nheteroarila, -SO2R50 e -(CH2)nNR50R51, ou
[00441] R47 e R48, juntamente com o átomo ao qual eles são ligados, formam um anel carbo- ou heterocíclico de 3 a 8 membros;
[00442] R49 é selecionado do grupo consistindo em (C1-C6)alquila, (C3-C8)cicloalquila, heterociclil(C1-C6)alquileno, aril(C1-C6)alquileno em que a arila é opcionalmente substituída com um ou mais substituintes selecionados do grupo consistindo em halo, -CF3, (C1-C6)alcóxi, -NO2, (C1-C6)alquila, -CN, -SO2R50 e -(CH2)nNR50R51, heteroaril(C1- C6)alquileno em que a heteroarila é opcionalmente substituída com um ou mais substituintes selecionados do grupo consistindo em halo, - CF3, (C1-C6)alcóxi, -NO2, (C1-C6)alquila, -CN, -SO2R50 e - (CH2)nNR50R51, arila opcionalmente substituída com um ou mais substituintes selecionados do grupo consistindo em halo, -CF3, (C1- C6)alcóxi, -NO2, (C1-C6)alquila, -CN, -SO2R50 e -(CH2)nNR50R51, e heteroarila opcionalmente substituída com um ou mais substituintes selecionados do grupo consistindo em halo, -CF3, (C1-C6)alcóxi, -NO2, (C1-C6)alquila, -CN, -SO2R50 e -(CH2)nNR50R51;
[00443] R50 e R51 são independentemente selecionados do grupo consistindo em H, (C1-C6)alquila, (C3-C8)cicloalquila e -C(O)R45, ou
[00444] R50 e R51, juntamente com o átomo ao qual eles são ligados, formam um anel carbo- ou heterocíclico de 3 a 8 membros; e
[00445] R21 é o grupo definido por -(Z11)-(Z12)m-(Z13)m1, em que
[00446] Z11 é heterociclila, quando m e m1 são 0, ou heterociclileno, quando ou m ou m1 é 1;
[00447] Z12 é selecionado do grupo consistindo em OC(O), OC(S) e C(O);
[00448] Z13 é selecionado do grupo consistindo em heterociclila, aralquila, N(H)R52, (C1-C3)alquila, -OR52, halo, S(O)2R56, (C1- C3)hidroxialquila e (C1-C3)haloalquila;
[00449] m é 0 ou 1;
[00450] m1 é 0 ou 1;
[00451] R52 é selecionado do grupo consistindo em H, - (CH2)qS(O)2R54, -(C1-C6) alquil- NR53R53 (C1-C3)alquila, -(CH2)qOR53, - C(O)R54 e -C(O)OR53;
[00452] q é 0, 1, 2, 3 ou 4;
[00453] cada R53 é independentemente (C1-C3)alquila;
[00454] R54 é (C1-C3)alquila ou N(H)R53;
[00455] R56 é selecionado do grupo consistindo em NH2, (C1- C3)alquila e OR52;
[00456] A1 é selecionado do grupo consistindo em -CH2-, -O-, -S-, - N(H)-, -N(CI-C6 alquil)-, -N-(Y-aril)-, -N-OMe, -NCH2OMe e N-Bn;
[00457] Y é uma ligação ou -(C(R11)(H))t-, em que t é um número inteiro de 1 a 6; e
[00458] R11 em cada ocorrência é independentemente selecionado do grupo consistindo em H e C1-C6 alquila, em que a C1-C6 alquila é opcionalmente substituída ;
[00459] A2 é selecionado do grupo consistindo em N e CR, em que R é selecionado do grupo consistindo em -H, halogênio, -CN, C1-C6 alquila, C2-C6 alquenila, e C2-C6 alquinila, em que a C1-C6 alquila, C2C6 alquenila, e C2-C6 alquinila são opcionalmente substituídas;
[00460] A3 é selecionado do grupo consistindo em C-D e N;
[00461] R2 em cada ocorrência é independentemente selecionado do grupo consistindo em -H, halogênio, trihalometila, -CN, -NO2, -NH2, -OR3, -NR3R4, -S(O)0-2R3, -S(O)2NR3R3, -C(O)OR3, -C(O)NR3R3, - N(R3)SO2R3, -N(R3)C(O)R3, -N(R3)CO2R3, -C(O)R3, C1-C4 alcóxi, C1-C4 alquiltio, -O(CH2)narila, -O(CH2)nheteroarila, -(CH2)0-5 (arila), -(CH2)0-5 (heteroarila), C1-C6 alquila, C2-C6 alquenila, C2-C6 alquinila, -CH2 (CH2)0-4-T2, em que T2 é selecionado do grupo consistindo em -OH, - OMe, -OEt, -NH2, -NHMe, -NMe2, -NHEt e -NEt2, e em que a arila, heteroarila, C1-C6 alquila, C2-C6 alquenila, e C2-C6 alquinila são opcionalmente substituídas; e
[00462] R3 selecionado do grupo consistindo em -H e R4;
[00463] R4 é selecionado do grupo consistindo em uma (C1- C6)alquila, uma arila, uma arilalquila inferior, uma heterociclila e uma heterociclilalquila inferior, cada uma das quais é opcionalmente substituída, ou
[00464] R3 e R4, tomados juntamente com um nitrogênio comum ao qual eles são ligados, formam uma heterociclila de cinco a sete membros opcionalmente substituída, que opcionalmente contém pelo menos um heteroátomo anular adicional selecionado do grupo consistindo em N, O, S e P;
[00465] X e X1 são cada qual independentemente selecionado do grupo consistindo em -H, halogênio, ciano, nitro, C1-C6 alquila, ou
[00466] X e X1 tomados juntamente com o átomo ao qual eles são ligados, formam a C3-C7 cicloalquila;
[00467] d é 0, 1, 2 ou 3;
[00468] e é 0, 1, 2 ou 3;
[00469] f é 0 ou 1;
[00470] R20 é selecionado do grupo consistindo em -H, halogênio, trihalometila, -CN, -NO2, -NH2, -OR3, -OCF3, -NR3R4, -S(O)0-2R3, - S(O)2NR3R3, -C(O)OR3, -C(O)NR3R3, -N(R3)SO2R3, -N(R3)C(O)R3, - N(R3)C(O)OR3, -C(O)R3, -C(O)SR3, C1-C4 alcóxi, C1-C4 alquiltio, - O(CH2)narila, -O(CH2)nheteroarila, -(CH2)0-5 (arila), -(CH2)0-5 (heteroarila), C1-C6 alquila, C2-C6 alquenila, C2-C6 alquinila, -CH2 (CH2)0-4-T2, uma C1-4 alquilcarbonila opcionalmente substituída, C1-4 alcóxi, um amino opcionalmente substituído por C1-4 alquila em que o C1-4 alquila pode ser opcionalmente substituída por C1-4 alcóxi e um grupo carboxíclico ou heterocíclico de três a sete membros saturado ou insaturado, em que T2 é selecionado do grupo consistindo em -OH, -OMe, -OEt, -NH2, -NHMe, -NMe2, -NHEt e -NEt2, e em que a arila, heteroarila, C1-C6 alquila, C2-C6 alquenila, e C2-C6 alquinila são opcionalmente substituídas;
[00471] Em outra modalidade preferida dos compostos de acordo com a presente invenção, D é definido pelo grupo R7, em que R7 é selecionado do grupo consistindo em -H, halogênio, C1-C6 alquila, - C(O)NR42R43, -C(O)(C6-C10 arila), -C(O)(heterociclila), - C(O)(heteroarila), -Y-(C6-C10 arila), -Y-(heterociclila de 5 a 10 membros), -Y-(heteroarila), -S-arila, -S-C1-C6 alquila, -SO-C1-C6 alquila, -SO2-C1-C6 alquila, -Y-NR42R43, -SO2NR42R43 e -CO2R6a, em que os grupos R7 anteriormente mencionados diferentes de -H e halogênio são opcionalmente substituídos por 1 a 5 R38.
[00472] Compostos preferidos de acordo com as fórmulas A-3 e B-3 têm grupos como definido nas modalidades preferidas da presente invenção.
[00473] No terceiro aspecto, a presente invenção fornece a composição compreendendo um composto de acordo com a presente invenção juntamente com um excipiente farmaceuticamente aceitável. Em uma modalidade preferida deste aspecto, a composição também compreende um agente terapêutico adicional.
[00474] O quarto aspecto da presente invenção fornece método de inibir sinalização de receptor de VEGF e sinalização de receptor de HGF, o método compreendendo contatar o receptor com um composto de acordo com a presente invenção, ou com a composição de acordo com a presente invenção. Inibição de atividade de VEGF e HGF pode ser em uma célula ou um organismo multicelular. Se em um organismo multicelular, o método de acordo com este aspecto da presente invenção compreende administrar ao organismo um composto de acordo com a presente invenção, ou uma composição de acordo com a presente invenção. Preferivelmente o organismo é um mamífero, mais preferivelmente um ser humano.
[00475] Exemplos de cinases que são inibidas pelos compostos e composições descritos aqui e contra as quais os métodos descritos aqui são úteis incluem, porém não são limitados a c-Met e KDR.
[00476] Dependendo da condição, ou doença particular, a ser tratada, agentes terapêuticos adicionais, que podem ser normalmente administrados para tratar aquela condição, podem também estar presente nas composições desta presente invenção. Em outras palavras, compostos desta presente invenção podem ser administrados como o agente farmacêutico sozinho ou em combinação com uma ou mais outros agentes terapêuticos adicionais (farmacêuticos) onde a combinação não causa nenhum efeito adverso inaceitável. Isto pode ser de particular relevância para o tratamento de doenças hiperproliferativas tais como câncer. Neste caso, o composto desta presente invenção pode ser combinado com agentes citotóxicos conhecidos, inibidores de transdução de sinal, ou com outros agentes anticâncer, bem como com misturas e combinações destes. Como empregado aqui, agentes terapêuticos adicionais que são normalmente administrados para tratar uma doença ou condição particular, são conhecidos como "apropriado para a doença, ou condição que está sendo tratada". Como empregado aqui, "agentes terapêuticos adicionais" destina-se a incluir agentes quimioterapêuticos et al. agentes antiproliferativos.
[00477] Por exemplo, agentes quimioterapêuticos ou outros agentes antiproliferativos podem ser combinados com os compostos desta presente invenção para tratar doença proliferativa ou câncer. Exemplos de agentes quimioterapêuticos ou outros agentes antiproliferativos incluem inibidores de HDAC incluindo, porém não são limitados a SAHA, MS-275, MG0103, e aqueles descritos em WO 2006/010264, WO 03/024448, WO 2004/069823, US 2006/0058298, US 2005/0288282, WO 00/71703, WO 01/38322, WO 01/70675, WO 03/006652, WO 2004/035525, WO 2005/030705, WO 2005/092899, e agentes de desmetilação incluindo, porém não limitados a, 5-aza-dC, Vidaza e Decitabine e aqueles descritos em US 6.268137. US 5.578.716. US 5.919.772. US 6.054.439. US 6.184.211. US 6.020.318. US 6.066.625. US 6.506.735. US 6.221.849. US 6.953.783. US 11/393.380 e PCT/US2006/001791.
[00478] Em outra modalidade da presente invenção, por exemplo, agentes quimioterapêuticos ou outros agentes antiproliferativos podem ser combinados com os compostos desta presente invenção para tratar doenças proliferativas e câncer. Exemplos de agentes quimioterapêuticos conhecidos incluem, porém não são limitados a, por exemplo, outras terapias ou agentes anticâncer que podem ser usados em combinação com os agentes inventivos anticâncer da presente invenção e incluem cirurgia, radioterapia (em apenas alguns exemplos, radiação gama, radioterapia de raio de nêutron, radioterapia de raio de elétron, terapia de próton, braquiterapia, e isótopos radioativos sistêmicos, para nomear alguns), terapia endócrina, taxanos (taxol, taxotere etc), derivados de platina, modificadores de resposta biológica (interferons, interleucinas, e fator de necrose de tumor (TNF), agentes de alvejamento de receptor TRAILA, para nomear alguns), hipertermia e crioterapia, agentes para atenuar quaisquer efeitos adversos (por exemplo, antieméticos), et al. fármacos quimioterapêuticos aprovados, incluindo, porém não limitados a, fármacos de alquilação (mecloretamina, clorambucila, Ciclofosfamida, Melfalan, Ifosfamida), antimetabólitos (Metotrexato, Pemetrexed etc), antagonistas de purina e antagonistas de pirimidina (6-Mercaptopurina, 5-Fluorouracila, Citarabil, Gencitabina), venenos de fuso (Vinblastina, Vincristina, Vinorelbina, Paclitaxel), podofilotoxinas (Etoposida, Irinotecan, Topotecan), antibióticos (Doxorubicina, Bleomicina, Mitomicina), nitrosoureias (Carmustina, Lomustina), íons inorgânicos (Cisplatina, Carboplatina), Inibidores de ciclo celular (inibidores de cinesina mitótica KSP, inibidores de CENP-E e CDK), enzimas (Asparaginase), e hormônios (Tamoxifen, Leuprolida, Flutamida, e Megestrol), Gleevec (TM), adriamicina, dexametasona, e ciclofosfamida. Agentes antiangiogênicos (Avastina e outros). Inibidores de cinase (Imatinib (Gleevec), Sutent, Nexavar, Erbitux, Herceptin, Tarceva, Iressa e outros). Agentes de inibição ou ativação das trilhas de câncer tais como as trilhas de mTOR, HIF (fator induzido por hipoxia) e outros. Para uma descrição mais compreensina de terapias de câncer atualizadas veja, http://www.nci.nih.gov/, uma lista dos fármacos de oncologia aprovados pelo FDA em http://www.fda.gov/cder/cancer/druglistframe.htm, e The Merck Manual, eécima oitava edição, 2006, os conteúdos inteiros dos quais são pelo presente incorporados por referência.
[00479] Em outra modalidade, os compostos da presente invenção podem ser combinados com agentes anticâncer citotóxicos. Exemplos de tais agentes podem ser encontrados na 13a edição do Merck Index (2001). Estes agentes incluem, por nenhum meio de limitação, asparaginase, bleomicina, carboplatina, carmustina, clorambucila, cisplatina, colaspase, ciclofosfamida, citarabina, dacarbazina, dactinomicina, daunorubicina, doxorubicina (adriamicina), epirubicina, etoposida, 5-fluorouracila, hexametilmelamina, hidroxiureia, ifosfamida, irinotecan, leucovorina, lomustina, mecloretamina, 6-mercaptopurina, mesna, metotrexato, mitomicina C, mitoxantrona, prednisolona, prednisona, procarbazina, raloxifen, estreptozocina, tamoxifen, tioguanina, topotecan, vinblastina, vincristina, e vindesina.
[00480] Outros fármacos citotósicos adequados para uso com os compostos da presente invenção incluem, porém não são limitados àqueles compostos reconhecidos para serem usados no tratamento de doenças neoplásicas, tais como aqueles, por exemplo, em Goodman e Gilman's The Farmacological Basis of Therapeutics (nona edição, 1996, McGraw-Hill). Estes agentes incluem, por nenhum meio de limitação, aminoglutetimida, L-asparaginase, azatioprina, 5-azacitidina cladribina, busulfan, dietilestilbestrol, 2', 2'-difluorodeoxicitidina, docetaxel, eritro-hidroxinoniladenina, etinila estradiol, 5- fluorodeoxiuridina, monofosfato de 5-fluorodeoxiuridina, fosfato de fludarabina, fluoximesterona, flutamida, caproato de hidroxiprogesterona, idarubicina, interferon, acetato medroxiprogesterona, acetato de megestrol, melfalan, mitotano, paclitaxel, pentostatina, N-fosfonoacetil-L-aspartato (PALA), plicamicina, semustina, teniposida, propionato de testosterona, tiotepa, trimetilmelamina, uridina, e vinorelbina.
[00481] Outros agentes anticâncer citotóxicos adequados para uso em combinação com os compostos da presente invenção também incluem princípios citotóxicos recentemente descobertos tais como oxaliplatina, gemcitabina, capecitabina, epotilona e seus derivados naturais ou sintéticos, temozolomida (Quinn et al., J. Clin. Oncology 2003, 21 (4), 646-651), tositumomab (Bexxar), trabedectina (Vidal et al., Proceedings of the American Society for Clinical Oncology" 2004, 23, resumo 3181), e os inibidores da proteína de fuso de cinesina Eg5 (Wood et al., Curr. Opin. Farmacol. 2001, 1, 370-377).
[00482] Em outra modalidade, os compostos da presente invenção podem ser combinados com outros inibidores de transdução de sinal. De particular interesse são inibidores de transdução de sinal que alvejam a família de EGFR, tal como EGFR, HER-2, e HER-4 (Raymond et al., Drugs 2000, 60 (Supl,1), 15-23; Harari et al., Oncogene 2000, 19 (53), 6102-6114), e seus respectivos ligandos. Exemplos de tais agentes incluem, por nenhum meio de limitação, terapias de anticorpo tal como Herceptina (trastuzumab), Erbitux (cetuximab), e pertuzumab. Exemplos de tais terapias também incluem, por nenhum meio de limitação, inibidores de cinase de molécula pequena tais como ZD-1839/Iressa (Baselga et al., Drugs 2000, 60 (Supl. 1), 33-40), OSI-774/Tarceva (Pollack et al., J. Farm. Exp. Ther. 1999, 291 (2), 739-748), CI-1033 (Bridges, Curr. Med. Chem. 1999, 6, 825-843), GW-2016 (Lackey et al., 92nd AACR Meeting, New Orleans, Mar. 24-28, 2001, resumo 4582), CP-724,714 (Jani et al., Proceedings of the American Society for Clinical Oncology 2004, 23, resumo 3122), HKI-272 (Rabindran et al., câncer Res. 2004, 64, 3958-3965), e EKB-569 (Greenberger et al., 11° NCI-EORTC- AACR Symposium on New Drugs in câncer Therapy, Amsterdam, 7 - 10 de novembro de 2000, resumo 388).
[00483] Em outra modalidade, os compostos da presente invenção podem ser combinados com outros inibidores de transdução de sinal alvejando cinases receptoras das famílias de domínio de cinase de divisão (VEGFR, FGFR, PDGFR, flt-3, c-kit, c-fms, e similares), e seus respectivos ligandos. Estes agentes incluem, por nenhum meio de limitação, anticorpos tais como Avastina (bevacizumab). Estes agentes também incluem, por nenhum meio de limitação, inibidores de molécula pequena tais como STI-571/Gleevec (Zvelebila, Curr. Opin. Oncol., Endocr. Metab. Invest. Drugs 2000, 2 (1), 74-82), PTK-787 (Wood et al., câncer Res. 2000, 60 (8), 2178-2189), SU-11248 (Demetri et al., Proceedings of the American Society for Clinical Oncology 2004, 23, resumo 3001), ZD-6474 (Hennequin et al., 92° AACR Meeting, New Orleans, 24 - 28 de março de 2001, resumo 3152), AG-13736 (Herbst et al., Clin. câncer Res. 2003, 9, 16 (supl 1), resumo C253), KRN-951 (Taguchi et al., 95° AACR Meeting, Orlando, Fla, 2004, resumo 2575), CP-547.632 (Beebe et al., câncer Res. 2003, 63, 7301-7309), CP-673.451 (Roberts et al., Proceedings of the American Association of câncer Research 2004, 45, resumo 3989), CHIR-258 (Lee et al., Proceedings of the American Association of câncer Research 2004, 45, resumo 2130), MLN-518 (Shen et al., Blood 2003, 102, 11, resumo 476), e AZD-2171 (Hennequin et al., Proceedings of the American Association of câncer Research 2004, 45, resumo 4539).
[00484] Em outra modalidade, os compostos da presente invenção podem ser combinados com inibidores da trilha de transdução de Raf/MEK/ERK (Avruch et al., Recent Prog. Horm. Res. 2001, 56, 127155), ou uma trilha de PKB (akt) (Lawlor et al., J. Cell Sci. 2001, 114, 2903-2910). Estes incluem, por nenhum meio de limitação, PD-325901 (Sebolt-Leopold et al., Proceedings of the American Association of Cancer Research 2004, 45, resumo 4003), e ARRY-142886 (Wallace et al., Proceedings of the American Association of Cancer Research 2004, 45, resumo 3891).
[00485] Em outra modalidade, os compostos da presente invenção podem ser combinados com inibidores de histona desacetilase. Exemplos de tais agentes incluem, por nenhum meio de limitação, ácido hidroxâmico suberoilanilida (SAHA), LAQ-824 (Ottmann et al., Proceedings of the American Society for Clinical Oncology 2004, 23, resumo 3024), LBH-589 (Beck et al., Proceedings of the American Society for Clinical Oncology 2004, 23, resumo 3025), MS-275 (Ryan et al., Proceedings of the American Association of Cancer Research 2004, 45, resumo 2452), FR-901228 (Piekarz et al., Proceedings of the American Society for Clinical Oncology 2004, 23, resumo 3028) e MGCD0103 (US 6.897.220).
[00486] Em outra modalidade, os compostos da presente invenção podem ser combinados com outros agentes anti-cancer tais como inibidores de proteassoma, e inibidores de m-TOR. Estes incluem, por nenhum meio de limitação, bortezomib (Mackay et al., Proceedings of the American Society for Clinical Oncology 2004, 23, resumo 3109), e CCI-779 (Wu et al., Proceedings of the American Association of Cancer Research 2004, 45, resumo 3849). Os compostos da presente invenção podem ser combinados com outros agentes anticâncer tais como inibidores de topoisomerase, incluindo, porém não limitado à camptotecina.
[00487] Aqueles agentes adicionais podem ser administrados separadamente da composição contendo o composto, como parte de um regime de dosagem múltipla. Alternativamente, aqueles agentes podem ser parte de uma forma de dosagem única, misturados junto com o composto desta presente invenção em uma composição única. Se administrados como parte de um regime de dosagem múltipla, os dois agentes ativos podem ser submetidos simultânea, sequencialmente ou dentro de um período de tempo de um com o outro que resulta na atividade desejada dos agentes.
[00488] A quantidade tanto do composto quanto do agente terapêutico adicional (naquelas composições que compreendem um agente terapêutico adicional como descrito acima) que pode ser combinada com os materiais veículos para produzir uma forma de dosagem única variará dependendo do hospedeiro tratado e do modo particular de administração.
[00489] Naquelas composições que compreendem um agente terapêutico adicional, aquele agente terapêutico adicional e o composto desta presente invenção podem agir sinergicamente.
[00490] Os dados apresentados aqui demonstram os efeitos inibidores dos inibidores de VEGF e HGF da presente invenção. Estes dados induzem alguém a razoavelmente esperar que os compostos da presente invenção sejam úteis não apenas para inibição de sinalização de receptor de VEGF e sinalização de receptor de HGF, porém também como agentes terapêuticos para o tratamento de doenças proliferativas, incluindo câncer e crescimento de tumor.
[00491] Os compostos preferidos de acordo com a presente invenção incluem aqueles descritos nos exemplos abaixo. Os compostos foram denominados usando Chemdraw Ultra versão 6,0,2 ou versão 8,0,3, que é disponibilizado pela Cambridgesoft.com, 100 Cambridge Park Drive, Cambridge, MA 02140, Namepro versão 5,09, que é disponibilizado por ACD labs, 90 Adelaide Street West, Toronto, Ontario, M5H, 3V9, Canadá, ou foi derivado dele.
Esquemas Sintéticos e Procedimentos Experimentais
[00492] Os compostos da presente invenção podem ser preparados de acordo com os esquemas de reação ou os exemplos ilustrados abaixo utilizando métodos conhecidos por alguém versado na técnica. Estes esquemas servem para exemplificar alguns procedimentos que podem ser usados para preparar os compostos da presente invenção. Alguém versado na técnica reconhecerá quet al. procedimentos sintéticos gerais podem ser usados. Os compostos da presente invenção podem ser preparados de componentes de partida que são comercialmente disponíveis. Quaisquer tipos de substituições podem ser feitos aos componentes de partida para obter os compostos da presente invenção de acordo com os procedimentos que são bem conhecidos por aqueles versados na técnica.
[00493] Os compostos com base em tieno[3,2-b]piridina de fórmula A-1 podem ser preparados de acordo com um procedimento geral mostrado no Esquema A, por meio de uma reação de acoplamento de amida entre ácidos N-aril(heteroarila)-malonâmicos [ácidos 3-oxo-3- (arilamino)- ou 3-oxo-3-(heteroarilamino)-propanóicos] (I) e derivados de tieno[3,2-b]piridina transportando um grupo amino (II). Esquema A
(Cy) = carbociclila, heterociclila, arila, heteroarila (opcionalmente substituída)
[00494] Ácidos I tipicamente podem ser preparados de acordo com o Esquema B reagindo as aminas IV ou com 3-cloro-3-oxopropanoato (V) por meio dos ésteres de amino intermediários VI que devem ser hidrolisados (procedimento de duas etapas), ou com 2,2-dimetil- [1,3]dioxano-4,6-diona (ácido de Meldrum) (VII) na presença de TMSCl, para formar o ácido desejado I em uma etapa. Esquema B
C^^' = Carbociclila, Heterociclila, Arila, Heteroarila (opcionalmente substituída)
[00495] Derivados de tieno[3,2-b ]piridina transportando um grupo amino (II) podem ser preparados de diferentes maneiras dependendo da natureza do substituinte R no anel de tiofeno do sistema de anel bicíclico de tienopiridina. Por exemplo, quando R é uma sequência sintética de porção de amida mostrada no Esquema C, pode ser empregado.
(Cy) = Carbociclila, Heterociclila, Arila, Heteroarila (opcionalmente substituída)
[00496] Desse modo, tieno[3,2-b]piridina-7-ol (VIII) reagindo com POCl3 é convertido em cloreto XI. Tratamento deste material com uma base forte tal como n-BuLi seguido pela adição de dióxido de carbono fornece o carboxilato X que é usado sem puificação na etapa seguinte, fornecendo o cloreto de acila XI em sua reação com cloreto de oxalila. O cloreto de acila XI é usado para a etapa seguinte sem outra purificação também: em sua reação com diferentes aminas primárias ou secundárias o composto XI é convertido em uma variedade de amidas primárias e secundárias XII que podem ser derivatizadas por meio de uma substituição do átomo de cloro do anel de piridina. Desse modo, XII reagindo com 2-flúor-4-nitrofenol em um solvente de alto ponto de ebulição, tal como éter de difenila na presença de uma base tal como carbonato de potássio, produziu os derivados de nitro XIII que são então reduzidos nas aminas II em tratamento com uma mistura de NiCl2/NaBH4. As aminas II (podem ser usadas para a etapa seguinte sem outra purificação) em tratamento com ácidos N-aril(heteroarila)- malonâmicos (I) fornecem malonamidas III transportando os substituintes de amido no anel de tiofeno tal como aqueles mostrados no Esquema C. Esquema D

C^^~ c Carbociclila, Heterociclila, Arila, Heteroarila (opcionalmente substituída)
[00497] Malonamidas de fórmula A-1 com base em tieno[3,2- b]piridina transportando substituintes de heteroarila em vez de porções de amido podem ser preparadas empregando procedimentos ilustrados no Esquema D. Desse modo, o tratamento do cloreto IX com uma base forte tal como n-BuLi seguido por uma adição de cloreto de tributilestanho fornece o derivado de tributilestanila XIV. Este material reagindo com diferentes brometos de heteroarila na presença de um catalisador de Pd (condições de acoplamento Stille) produz tienopiridinas substituídas por heteroarila XII (R = heteroarila) que podem também ser derivatizadas por meio de uma substituição do átomo de cloro do anel de piridina do sistema de anel de tienopiridina. Esquema E
(Cy) = Carbociclila, Heterociclila, Arila, Heteroarila (opcionalmente substituída)
[00498] Desse modo, XIV reagindo com 2-flúor-4-nitrofenol em um solvente de alto ponto de ebulição, tais como éter de difenila na presença de uma base tal como carbonato de potássio, produziu os derivados de nitro XIII que são então reduzidos nas aminas II em tratamento com ferro em um meio acídico. As aminas II (podem ser usadas para a etapa seguinte sem outra purificação) em tratamento com ácidos N-aril(heteroarila)-malonâmicos (I) fornecem malonamidas III transportando substituintes de heteroarila no anel de tiofeno tais como aqueles mostrados no Esquema D.
[00499] Malonamidas com base em tieno[3,2-b]piridina de fórmula A-1 transportando substituintes de arila no anel e tiofeno, particularmente substituintes de arila com porções básicas, podem ser preparados usando procedimentos ilustrados no Esquema E. Desse modo, tratamento do cloreto IX com uma base forte tal como n-BuLi seguido por brominação (por exemplo, com bromo elementar) fornece o brometo XV. Este material reagindo com 2-flúor-4-nitrofenol em um solvente de ponto de ebulição elevado, tal como éter de difenila na presença de uma base tal como carbonato de potássio, produziu o derivado de nitro XVI que sofreu uma reação com ácido 4- (hidroximetil)fenilborônico na presença de uma base e um catalisador Pd (reação de acoplamento Suzuki) para fornecer derivado substituído por arila XVII com um grupo hidroxila livre. O grupo hidroxila foi substituído por um halogênio (por exemplo, cloreto usando o cloreto de tionila) para formar o composto XVIII que em tratamento com aminas secundárias e primárias foi convertido em uma variedade de compostos substituídos por arila XII (R = arila substituída). O grupo nitro destas entidades básicas foi reduzido com NaBH4/NiCl2 para formar as aminas II. Estes intermediários (podem ser usados para a etapa seguinte sem outra purificação) em tratamento com ácidos N- aril(heteroarila)-malonâmicos (I) fornecem malonamidas III transportando substituintes de arila com porções básicas, ligadas ao anel de tiofeno tal como aqueles mostrados no Esquema E.
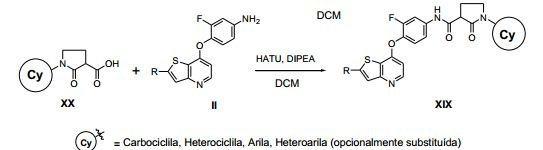
ÍCyJ = Carbociclila, Heterociclila, Arila, Heteroarila (opcionalmente substituída)
[00500] Compostos com base em tieno[3,2-b]piridina de fórmula XIX podem ser preparados de acordo com um procedimento geral mostrado no Esquema F, por meio de uma reação de acoplamento de amida entre ácidos 2-oxo-1-aril(heteroarila)pirrolidina-3-carboxílicos (XX) e derivados de tieno[3,2-b]piridina transportando um grupo amino (II). Ácidos XX podem ser preparados seguindo o procedimento de literatura [S. Danishefsky, R.K.Singh. JACS, 1975, 97, 3239-3241] ou adquiridos se comercialmente disponíveis. Esquema G
vCy) = Carbociclila, Heterociclila, Arila, Heteroarila (opcionalmente substituída)
[00501] Compostos com base em tieno[3,2-b]piridina de fórmula XXI podem ser preparados por meio de uma reação de condensação (Esquema G) entre as aminas II e cloretos de 2-oxo-3-aril(heteroarila)- 1-carbonila de uma fórmula geral XXII. Cloretos de acila XXII podem ser preparados seguindo o procedimento de literatura (Chem. Abstr.; 88; 6873 e P. Mayer, et al.; J. Med. Chem.; 2000, 43, 3653-3664) enquanto a reação pode ser realizada em solventes apróticos tais como DCM, CHCl3, tolueno, éter de dimetila de etileno glicol, MeCN, DMF, DMSO, THF, dioxano e similares, na presença de bases orgânicas tais como DIPEA, Et3N, DBU, DMAP, N-metilmorfolina, N- metilpiperidina, e similares. Esquema H
(cyj = Carbociclila, Heterociclila, Arila, Heteroarila (opcionalmente substituída)
[00502] 4,4,4-T riflúor- N -aril(heteroarila)-3-(amino)butanamidas da fórmula geral XXIII podem ser obtidas por meio de uma sequência de reação curta partindo das aminas II. As aminas II em tratamento com trifluoroacetaldeído de etila hemiacetalsob condições acídicas (por exemplo, na presença de ácido 4-toluenossulfônico) em solventes polares tal como etanol são transformados em N-(1-etóxi-2,2,2- trifluoroetil)aminas da estrutura geral XXIV. Os Compostos XXIV reagindo com malonatos sob forma de condições básicas 2-(2,2,2- triflúor-1-(amino)etil)malonatos tal como XXV. Os diésteres de amino XXV sofrem hidrólise alcalina para formar os ácidos malônicos intermediários (não mostrados no Esquema A), que são também descarboxilados, para fornecer ácidos 4,4,4-triflúor-3- (amino)butanóicos XXVI. Ácidos XXVI são acoplados com diferentes aminas primárias ou secundárias usando técnicas padrão, para produzir o composto do título XXIII.

(Cy) = Carbociclila, Heterociclila, Arila, Heteroarila (opcionalmente substituída)
[00503] 4,4,4-Triflúor- N -3-(ciclilamino)butanamidas da fórmula geral XXVII pode ser obtido por meio de uma sequência de reação curta similar como no Esquema I usando os mesmos grupos de derivados de tienopiridina II e aminas Ar-NH2. As aminas Ar-NH2 em tratamento com trifluoroacetaldeído de etila hemiacetalsob condições acídicas (por exemplo, na presença de ácido 4-toluenossulfônico) em solventes polares tal como etanol são transformados em N-(1-etóxi-2,2,2- trifluoroetil)arilaminas da estrutura geral XXVIII. Os compostos XXVIII reagindo com malonatos sob forma de condições básicas 2-(2,2,2- triflúor-1-(ciclilamino)etil)malonatos de dietila tais como XXIX. Os diésteres de amino XXIX sofrem hidrólise alcalina para formar os ácidos malônicos intermediários (não mostrados no Esquema I), que são também descarboxilados, para fornecer ácidos 4,4,4-triflúor-3- (ciclilamino)butanóicos XXX. Ácidos XXX são acoplados a várias aminas da estrutura geral II, usando técnicas padrão, para produzir o composto do título XXVII. Exemplos Particulares Esquema 1

Exemplo 1 N 1-(3-Flúor-4-(tieno[3,2-b ]piridin-7-ilóxi)fenil)-N 3-fenilmalonamida (5a) Etapa 1: Ácido 3-oxo-3-(fenilamino)propanóico (1) (procedimento de duas etapas)
[00504] A uma solução de 3-cloro-3-oxopropanoato de metila (2,0 g, 14,7 mmols) em DCM seco (100 ml) foi adicionado anilina (2,68 g, 3,6 mmol) e a mistura de reação foi agitada a 0°C durante 1 hora, evaporada, em seguida dissolvida em EtOAc, lavada com HCl diluído, NaHCO3, e salmoura. A fase orgânica foi coletada, secada sobre sulfato de sódio, filtrada, e o solvente foi evaporado para fornecer 3- oxo-3-(fenilamino)propanoato de metila como um sólido marrom que foi usado sem outra purificação (2,8g, 100%, bruto). A uma solução deste material (2,8 g, 14,5 mmol) em THF (40 ml) e água (20 ml) foi adicionado NaOH (1,16 g, 29 mmol) e a mistura de reação foi agitada durante a noite, evaporada (para remover o THF) e em seguida extraída com Et2O. A fase aquosa foi acidificada para pH 1 e extraída com EtOAc. O extrato foi seco sobre Na2SO4, filtrado e evaporado para fornecer o composto do título 1 como um sólido marrom, que foi usado sem outra purificação (2,0 g, 77% de rendimento). MS (m/z): 18,0 (100%) (M+H), 202,0 (44%) (M+Na). Etapa 2: 7-Clorotieno[3,2-b ]piridina (2)
[00505] Uma suspensão agitada de tieno[3,2-b]piridin-7-ol (5,0 g, 33,1 mmols) em POCl3 (15 mL) foi aquecida a 105°C em um banho de óleo durante 4 horas. A solução resultante foi resfriada à temperatura ambiente e o POCl3 foi removido sob pressão reduzida. O resíduo foi resfriado em um banho de gelo/água e água fria foi adicionada. A mistura foi tornada básica com solução de NH4OH concentrada e extraída com EtOAc. O extrato orgânico foi seco sobre sulfato de sódio anidro e concentrado para produzir óleo, que foi purificado por cromatografia de coluna (eluente EtOAc-hexano, 1:4) para fornecer o composto do título como um sólido marrom (4,5 g, 72% de rendimento). 1H RMN (400 MHz, CDCl3) δ (ppm): 8,60 (d, J = 4,9 Hz, 1H), 7,80 (d, J = 5,5 Hz, 1H), 7,60 (d, J = 5,5 Hz, 1H), 7,30 (d, J = 4,9 Hz, 1H). Etapa 3: 7-(2-Flúor-4-nitrofenóxi)tieno[3,2-b ]piridina (3)
[00506] Uma mistura de 2 (500 mg, 2,95 mmols), carbonato de potássio (1,62g, 11,8 mmols) e 2-flúor-4-nitrofenol (603 mg, 3,85 mmols) foi aquecida para 170°C em éter de difenila (10 ml) durante 5 horas. A mistura foi resfriada para temperatura ambiente, diluída com EtOAc e lavada com água. A fase orgânica foi coletada, secada sobre sulfato de sódio anidro e os solventes foram removidos sob pressão reduzida. O resíduo foi purificado por cromatografia de coluna, gradiente eluente EtOAc-hexano (9:1) para EtOAc, para fornecer o composto do título 3 (660 mg, 76% de rendimento). 1H RMN (400 MHz, CDCI3) δ (ppm) 8,60 (d, J = 5,54 Hz, 1H), 8,14 (m, 3H), 7,80 (d, J = 5,47 Hz, 1H), 7,61 (d, J = 5,48 Hz, 1H), 7,36 (t, J = 7,63 Hz), 6,65 (m, 1H). Etapa 4:N 1-(3-Flúor-4-(tieno[3,2-b ]piridin-7-ilóxi)fenil)-N3- fenilmalonamida (5a)
[00507] A uma solução de 3 (660 mg, 2,27 mmols) em MeOH (10 ml) foi adicionado HCl concentrado (1 ml) e Fe (1,91g, 34,8 mmols), e a mistura de reação foi agitada a 0°C durante 3 horas, neutralizada com solução de NaHCO3 aquosa e extraída com EtOAc, produzindo 3-flúor-4-(tieno[3,2-b]piridin-7-ilóxi)-fenilamina (4) como um óleo escuro (500 mg, 83%), que foi usado diretamente na etapa seguinte. A uma solução da amina 4 (200 mg, 0,69 mmol) em DMF (10 ml) foi adicionado ácido 3-oxo-3-(fenilamino)propanóico (1, 155 mg, 0,89 mmol), HOBT (121 mg, 0,89 mmol) e EDC (198 mg, 1,035 mmol). A mistura de reação foi agitada em temperatura ambiente durante a noite, evaporada até a secura e o resíduo foi dissolvido em EtOAc e lavado com água. A fase orgânica foi seca sobre sulfato de sódio, filtrada e evaporada sob pressão reduzida. O resíduo foi purificado por cromatografia de coluna (eluente EtOAc : hexano 3:1) para fornecer o composto do título 5a (30 mg, 10% de rendimento) como um sólido branco (após trituração com Et2O). 1H RMN (400 MHz, DMSO-d6) δ (ppm) :10,5 (s, 1H), 10,2 (s, 1H), 8,49 (d, J = 5,48 Hz, 1H), 8,14 (d, J = 5,48 Hz, 1H), 7,85 (dd, J = 2,35 e 13,01 Hz, 1H), 7,59 (m, 3H), 7,42 (m, 2H), 7,30 (dt, J = 1,96 e 7,43 Hz, 2H), 7,05 (t, J = 7,24 Hz, 1H), 6,65 (d, J = 5,28 Hz, 1H), 3,51 (s, 2H). Esquema 2
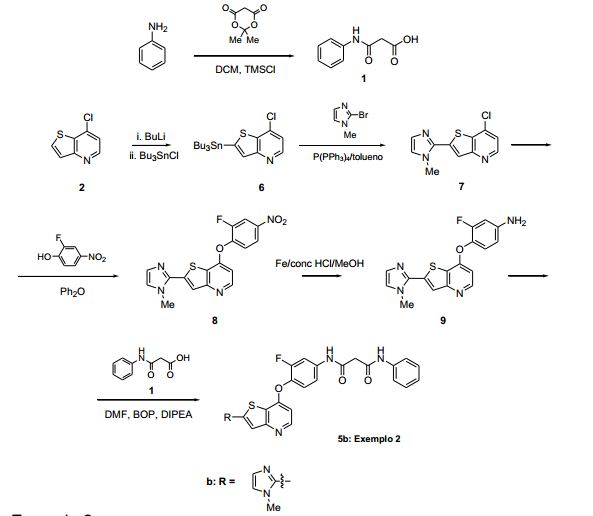
Exemplo 2 N1 -(3-Flúor-4-(2-(1-metil- 1H-imidazol-2-il)tieno[3,2-b ]piridin-7- ilóxi)fenil)-N3-fenilmalonamida (5b) Etapa 1: Ácido 3-oxo-3-(fenilamino)propanóico (1) (procedimento de uma etapa)
[00508] A uma solução de anilina (13 mL, 143 mmols) em DCM (290 mL) foi adicionado TMSCl (18,2 mL, 143 mmols) em temperatura ambiente e a mistura de reação foi agitada durante 1 hora. [Rigo, B.; Fasseur, D.; Cauliez, P. e Couturier, D. Tetrahedron Lett.; 30; 23; 1989; 3073-3076.]. 2,2-Dimetil-1,3-dioxano-4,6-diona (20,6 g, 143 mmols) foi adicionada e a mistura de reação combinada foi agitada durante a noite em temperatura ambiente, despejada em solução de NaHCO3 saturada e extraída com EtOAc. A fase aquosa foi coletada, acidificada com HCl concentrado para pH ~3 pela adição de HCl a 2N e extraída com DCM. A fase orgânica foi seca sobre Na2SO4 anidro, filtrada e concentrada sob pressão reduzida fornecendo ácido 3-oxo-3- (fenilamino)propanóico (1, 16,82 g, 65% de rendimento) como um sólido branco. MS (m/z): 180,0 (25%) (M+H), 202,0 (100%) (M+Na). Etapa 2: 7-Cloro-2-(tributilstannil)tieno[3,2-b ]piridina (6)
[00509] A uma solução de 7-clorotieno[3,2-b]piridina 2 (9,82 g, 57,89 mmols) em THF (290 mL) BuLi (2,5 N, 25 mL) foi adicionado a - 78°C e a mistura agitada durante 20 minutos na mesma temperatura. Cloreto de tributilestanho (63,68 mmols, 17,3 mL) foi adicionado em gotas e a mistura agitada durante 2 h a -78°C. A mistura homogênea desse modo obtida foi derramada em água (200 mL) e extraída com EtOAc (2x200 mL). A fase orgânica foi coletada, secada sobre Na2SO4 anidro e concentrada sob pressão reduzida. O resíduo foi purificado por cromatografia instantânea (eluente EtOAc/Hex 1:4) para fornecer 7-cloro-2-(tributilstannil)tieno[3,2-b]piridina (6, 24,79 g, 93% de rendimento) como um xarope. MS (m/z): grupo de sinais centrados por volta de 460,1 (M+1). Etapa 3: 7-Cloro-2-(1-metil- 1H-imidazol-2-il)tieno[3,2-b ]piridina (7)
[00510] A uma solução do derivado de estanho 6 (24,79 g, 54,05 mmols) e 2-bromo-1-metil-1H-imidazol (10,4 g, 64,86 mmols) [McCallum, P. W.; Weavers, R. T.; Grimmet, M. R.; Blackman, A. G.; Aust. J. Chem.; 52; 3 ;1999; 159-166.] em tolueno (180 mL) PdFPPhsh (5 g, 4,32 mmols) foi adicionado e a mistura foi refluxada sob nitrogênio durante 2 dias, resfriada para temperatura ambiente. Um precipitado foi formado o qual foi coletado por filtragem, lavado com Et2O e secado, para fornecer o composto do título 7 como um sólido cinzento (12,72 g, 94% de rendimento). MS (m/z): 250,0 (M+H). Etapa 4: 7-(2-Flúor-4-nitrofenóxi)-2-(1-metil- 1H-imidazol-2-il)tieno[3,2- b ]piridina (8)
[00511] Uma mistura de 7-cloro-2-(1-metil-1H-imidazol-2- il)tieno[3,2-b]piridina (7) (1,0 g, 4 mmols), 2-flúor-4-nitrofenol (0,942 g, 6 mmols) e K2CO3 (1,1 g, 8 mmols) em Ph2O (20 mL) foi agitada a 200°C durante 24 horas. A mistura de reação resfriada foi diluída com DCM (200 mL) e extraída com HCl a 1M (200 mL). A fase aquosa foi filtrada através de filtro de papel, basificada (pH~10) com NH4OH concentrado para produzir uma mistura turva que foi extraída com DCM. A fase orgânica foi seca sobre Na2SO4 anidro e concentrada sob pressão para fornecer o composto do título 8 como um sólido alaranjado (0,667 g, 45% de rendimento). MS (m/z): 371,0 (M+H). Etapa 5: 3-Flúor-4-(2-(1-metil- 1H-imidazol-2-il)tieno[3,2-b ]piridin-7-ilóxi) benzenamina (9)
[00512] A uma solução do composto nitro 8 (1,50 g, 4,05 mmols) e NiCh6H2O (2,02 g, 8,52 mmols) em MeOH/THF (45 mL/68 mL) a 0°C foi adicionado NaBH4 (0,618 g, 16,3 mmols) em porções (1,50 g, 4,05 mmols) com agitação vigorosa. A mistura de reação foi agitada durante 30 minutos a 0°C e concentrada sob pressão reduzida. O resíduo escuro resultante foi suspenso em HCl a 1M (10 mL) e a mistura foi basificada (pH~11) com NH4OH concentrado. A suspensão turva foi filtrada; o resíduo sólido foi separado, lavado com água e secado sob pressão reduzida para fornecer o composto do título 9 como um sólido marrom (0,73g, 52% de rendimento), o qual foi empregado na etapa seguinte sem outra purificação. MS (m/z): 341,1 (M+H). Etapa 6: N1 -(3-Flúor-4-(2-(1 -metil-1 H-imidazol-2-il)tieno[3,2-b ]piridin-7- ilóxi)fenil)-N3-fenilmalonamida (5b)
[00513] A uma solução da amina 9 (80 mg, 0,235 mmol), ácido 3- oxo-3-(fenilamino)propanóico (1) (42 mg, 0,235 mmol) e BOP (114,6 mg, 0,259 mmol) em DMF (2,4 mL), DIPEA (0,164 mL, 0,941 mmol) foi adicionado e a mistura foi agitada em temperatura ambiente durante 2 horas. A mistura de reação foi dividida entre EtOAc e água. A fase orgânica foi coletada, lavada com H2O e salmoura, secada sobre Na2SO4 anidro e concentrada. O resíduo foi purificado por cromatografia instantânea (EtOAc/MeOH 95:5) seguido por cristalização (MeOH) para fornecer o composto do título 5b (11 mg, 9% de rendimento). 1H RMN (DMSO-d6) δ (ppm): 10,55 (s, 1H), 10,19 (s, 1H), 8,5 (d, J = 5,4 Hz, 1H), 7,86 (m, 2H), 7,59 (m, 2H), 7,49 (t, J = 8,8 Hz, 1H), 7,41 (m, 2H), 7,30 (m, 2H), 7,05 (m, 2H), 6,69 (d, J = 5,4 Hz, 1H), 3,99 (s, 3H), 3,52 (s, 2H). MS (m/z): 502,2 (M+H). Esquema 3

Exemplo 3 N1 -{3-Flúor-4-[2-(1 -metil-1 H-imidazol-4-il)tieno[3,2-b ]piridin-7-ilóxi]- fenil}-N3-fenilmalonamida (5c) Etapa 1: 7-Cloro-2-(1-metil- 1H-imidazol-4-il)tieno[3,2-b ]piridina (10)
[00514] Seguindo-se o procedimento descrito para o composto 7 (Exemplo 2, Etapa 3) porém substituindo 2-bromo-1-metil-1H-imidazol por 4-bromo-1-metil-1 H-imidazol [a) Begtrup, M.; Larsen, P.; Acta Chem. Scand. 44, 10; 1990; 1050-1057. b) Begtrup, M.; Bull. Soc. Chim. Belg.; 97; 8-9; 1988; 573-598. c) Begtrup, M.; Larsen, P.; Chem. Pharm. Bull. 42, 9; 1994; 1784-1790.], o composto do título 10 foi obtido como um sólido esbranquiçado (29% de rendimento). MS (m/z): 250,1 (M+H). Etapa 2: 7-(2-Flúor-4-nitrofenóxi)-2-(1 -metil-1 H-imidazol-4-il)tieno[3,2- b ]piridina (11)
[00515] Seguindo-se o procedimento descrito para o composto nitro 8 (Exemplo 2, Etapa 4) porém substituindo o composto 7 pelo composto 10, o composto do título 11 foi obtido como um sólido vermelho (46% de rendimento). MS (m/z): 371,1 (M+H). Etapa 3: 3-Flúor-4-(2-(1-metil- 1H-imidazol-4-il)tieno[3,2-b ]piridin-7-ilóxi) benzenamina (12)
[00516] Seguindo-se o procedimento descrito para a amina 9 (Exemplo 2, Etapa 5) porém substituindo composto nitro 8 pelo composto 11, o composto do título 12 foi obtido como um sólido vermelho (82% de rendimento). MS (m/z): 341,1 (M+H). Etapa 4: N1 -(3-flúor-4-(2-(1-metil- 1H-imidazol-4-il)tieno[3,2-b]piridin-7- ilóxi)fenil)-N3-fenilmalonamida (5c)
[00517] Seguindo-se o procedimento descrito para o composto 5a (Exemplo 1, Etapa 5) porém substituindo amina 9 pelo composto 12, o composto do título 5c foi obtido como um sólido branco (78% de rendimento). MS (m/z): 502,0 (M+H). Esquema 4
Exemplo 4 N 1-(4-(2-(1 -Etil-1 H-imidazol-4-il)tieno[3,2-b ]piridin-7-ilóxi)-3-fluorofenil)- N 3-fenilmalonamida (5d) Etapa 1: 7-Cloro-2-(1-etil- 1H-imidazol-4-il)tieno[3,2-b ]piridina (13)
[00518] Seguindo-se o procedimento descrito para o composto 7 (Exemplo 2, Etapa 3) porém substituindo 2-bromo-1-metil-1H-imidazol por 4-bromo-1-etil-1 H-imodazol [a) Begtrup, M.; Larsen, P.; Acta Chem. Scand. 44, 10; 1990; 1050-1057. b) Begtrup, M.; Bull. Soc. Chim. Belg.; 97; 8-9; 1988; 573-598. c) Begtrup, M.; Larsen, P.; Chem. Pharm. Bull. 42, 9; 1994; 1784-1790.], o composto do título 13 foi obtido como um sólido amarelo (30% de rendimento). MS (m/z): 263,9 (M+H). Etapa 2: 2-(1-Etil- 1H-imidazol-4-il)-7-(2-flúor-4-nitrofenóxi)tieno[3,2- b ]piridina (14)
[00519] Seguindo-se o procedimento descrito para 8 (Exemplo 2, Etapa 4) porém substituindo o composto 7 pelo composto 10, o composto do título 14 foi obtido como um sólido amarelo (76% de rendimento). MS (m/z): 384,9 (M+H). Etapa 3: 4-(2-(1-Etil- 1H-imidazol-4-il)tieno[3,2-b ]piridin-7-ilóxi)-3- fluorobenzenamina (15)
[00520] Seguindo-se o procedimento descrito para o composto 9 (Exemplo 2, Etapa 5) porém substituindo composto nitro 8 pelo composto 14, o composto do título 15 foi obtido como um sólido amarelo (86% de rendimento). MS (m/z): 402,1 (M+H).
[00521] Seguindo-se o procedimento descrito para o composto 5a (Exemplo 1, Etapa 5) porém substituindo a amina 9 pelo composto 15, o composto do título 5d foi obtido em 57% de rendimento. MS (m/z): 516,0 (M+H). Esquema 5
Etapa 4: N 1-(4-(2-(1 -etil-1 H-imidazol-4-il)tieno[3,2-b ]piridin-7-ilóxi)-3- fluorofenil)-N 3-fenilmalonamida (5d) Exemplo 5 N 1-(3-Flúor-4-(2-(1 -metil-1 H-imidazol-5-il)tieno[3,2-b ]piridin-7- ilóxi)fenil)-N 3-fenilmalonamida (5e) Etapa 1: 7-Cloro-2-(1-metil-1 H-imidazol-5-il)tieno[3,2-b ]piridina (16)
[00522] Nitrogênio foi borbulhado através de uma mistura do derivado de estanho 6 (7,19 g, 15,7 mmols) e 5-bromo-1-metil-1H- imidazol (2,02 g, 12,5 mmols) [a) Begtrup, M.; Larsen, P.; Acta Chem. Scand. 44, 10; 1990; 1050-1057. b) Begtrup, M.; Bull. Soc. Chim. Belg.; 97; 8-9; 1988; 573-598. c) Begtrup, M.; Larsen, P.; Chem. Pharm. Bull. 42, 9; 1994; 1784-1790.] em tolueno (20 mL) durante 5 minutos. Pd(PPh3)4 (1,50 g, 1,30 mmol) foi adicionado e nitrogênio foi borbulhado por mais 5 minutos. Finalmente, a mistura foi refluxada sob nitrogênio durante a noite, a suspensão amarela resultante foi concentrada sob pressão reduzida e então purificada com cromatografia instantânea (eluente EtOAc/MeOH 90:10), para fornecer o composto do título 16 como um sólido amarelo (2,48 g, 79% de rendimento). MS (m/z): 250,0 (M+H). Etapa 2: 7-(2-Flúor-4-nitrofenóxi)-2-(1 -metil-1 H-imidazol-5-il)tieno[3,2- b ]piridina (17)
[00523] Uma mistura do composto 16 (1,52 g, 6,09 mmols), 2-flúor- 4-nitrofenol (3,87 g, 24,6 mmols) e K2CO3 (4,31 g, 31,2 mmols) em Ph2O (20 mL) foi aquecida a 190°C durante a noite. DCM (250 mL) foi adicionado à mistura marrom-escura resultante, que foi então extraída com HCl a 1M aquoso. A camada aquosa foi coletada, lavada com DCM e basificada com NH4OH. A mistura turva resultante foi extraída com DCM. A fase orgânica foi combinada, filtrada; o filtrado foi lavado com água, secado sobre Na2SO4 anidro e concentrado sob pressão reduzida. O sólido amarelo resultante foi purificado por cromatografia instantânea, eluente EtOAc/MeOH (80:20) para fornecer o composto do título 17 como um sólido amarelo (0,96 g, 43% de rendimento). MS (m/z): 371,0 (M+H). Etapa 3: 3-Flúor-4-(2-(1 -metil-1 H-imidazol-5-il)tieno[3,2-b ]piridin-7- ilóxi)benzenamina (18)
[00524] A uma mistura agitada do composto nitro 17 (0,96 g, 2,59 mmols) e NiChβ^O (1,24 g, 5,22 mmols) em MeOH/THF (26 mL/39 mL) a 0°C, NaBH4 (0,341 g, 9,01 mmols) foi adicionado em porções. A mistura de reação foi agitada durante 15 minutos, extinguida com HCl a 1M (10 mL) e concentrada sob pressão reduzida para formar um resíduo verde, que foi então dissolvido em HCl a 1M (250 mL) e basificado com NH4OH. A suspensão turva foi extraída com EtOAc, a camada orgânica foi coletada, filtrada, lavada com água e então secada sobre Na2SO4 anidro. A evaporação desta solução forneceu o composto do título 18 como um sólido marrom (0,46 g, 52% de rendimento), o qual foi empregado na etapa seguinte sem purificação adicional. MS (m/z): 341,1 (M+H). Etapa 4: N 1-(3-Flúor-4-(2-(1 -metil-1 H-imidazol-5-il)tieno[3,2-b ]piridin-7- ilóxi)fenil)-N 3-fenilmalonamida (5e)
[00525] Seguindo-se o procedimento descrito para o composto 5b (Exemplo 2, Etapa 6) porém substituindo amina 9 pelo composto 18, o composto do título 5e foi obtido em 5% de rendimento. MS (m/z): 502,0 (M+H). Esquema 6
Exemplo 6 N 1-(3-Flúor-4-(2-(pirrolidina-1-carbonil)tieno[3,2-b ]piridin-7-ilóxi)fenil)- N 3-fenilmalonamida (5f) Etapas 1-3: (7-Clorotieno[3,2-b ]piridin-2-il)(pirrolidin-1-il)metanona (21)
[00526] A uma solução agitada de 2 (11,91 g, 70,22 mmols) em THF seco (200 mL) a -78°C foi adicionado n-BuLi (33,70 mL, 84,26 mmols, solução a 2,5 M em hexanosa) e a suspensão resultante foi agitada durante 1 hora. Dióxido de carbono sólido (excesso) foi adicionado e a mistura foi deixada aquecer à temperatura ambiente durante um período de 1 hora. O solvente foi removido sob pressão reduzida e o carboxilato de lítio resultante 19 foi empregado sem outra purificação (16,43 g, quantitativo). A uma suspensão agitada de 19 (15,41 g, 70,22 mmols) em DCM seco (150 mL) foi adicionado (COCl)2 (12,25 mL, 140,44 mmols) e DMF seca (5 gotas). A mistura de reação foi aquecida ao refluxo durante 3 horas. Os solventes foram evaporados para produzir 20, que foi usado diretamente na etapa seguinte. Cloreto de acila 20 (8,14 g, 35,11 mmols) foi suspenso em DCM seco (300 mL) a 0°C, pirrolidina (3,22 mL, 38,62 mmols) foi adicionado e a mistura de reação foi agitada durante a noite. O solvente foi removido sob pressão reduzida e o resíduo foi dissolvido em EtOAc e lavado com água. A fase orgânica foi coletada e secada sobre sulfato de sódio anidro, então filtrada e concentrada sob pressão reduzida para produzir um resíduo, que foi purificado por cromatografia de coluna (eluente MeOH-CH2Cl2, 2:98, 5:95) para fornecer o composto do título 21 como um sólido marrom (16,07 g, 86% de rendimento). MS (m/z): 267,1 (M+H). Etapa 4: (7-(2-Flúor-4-nitrofenóxi)tieno[3,2-b]piridin-2-il)(pirrolidin-1-il) metanona (22)
[00527] Uma mistura de 21 (2,4 g, 8,89 mmols), K2CO3 (4,97 g, 35,96 mmols) e 2-flúor-4-nitrofenol (1,55 g, 9,89 mmols) foi aquecida a 150°C em éter de difenila (40 mL) durante 2 dias. A mistura foi purificada por cromatografia de coluna (eluentes EtOAc-hexano 5:95, 2:8, então MeOH-EtOAc 2:98, 5:95) para fornecer o composto do título 22 como um sólido amarelo (3,23 g, 93% de rendimento). MS (m/z): 388,2 (M+H). Etapa 4: (7-(4-Amino-2-fluorofenóxi)tieno[3,2-b ]piridin-2-il)(pirrolidin-1- il)metanona (23)
[00528] A uma solução de 22 (3,23 g, 8,33 mmols) e NiCl2,6H2O (3,96 g, 16,66 mmols) em MeOH/THF (100/100 mL) foi adicionado NaBH4 (1,24 g, 33,32 mmols). A mistura de reação foi agitada durante 1 hora, concentrada à secura e o sólido resultante foi dissolvido em 10% de HCl. A solução aquosa foi então tornada básica com solução de NH4OH concentrada e extraída com EtOAc. A fase orgânica foi coletada, secada sobre sulfato de sódio anidro e concentrada sob pressão reduzida para fornecer 23 como um sólido escuro (2,72 g, 91% de rendimento). MS (m/z): 358,2 (M+H). Etapa 5: N 1-(3-Flúor-4-(2-(pirrolidina-1-carbonil)tieno[3,2-b ]piridin-7- ilóxi)fenil)-N 3-fenilmalonamida (5f)
[00529] A uma solução de 23 (100 mg, 0,28 mmol), HOBT (49 mg, 0,36 mmol) e EDC (80 mg, 0,41 mmol) em DMF (5 mL) foi adicionado ácido 3-oxo-3-(fenilamino)propanóico (1) (65 mg, 0,36 mmol). A mistura de reação foi agitada durante 1 dia e foi diluída com EtOAc. A solução resultante foi lavada com água e salmoura, secada sobre sulfato de sódio anidro e concentrada para fornecer um resíduo, que foi purificado por cromatografia de coluna (eluentes MeOH-CH2Cl2, 2:98, 5:95) para produzir um material sólido, que após trituração com EtOAc/hexano forneceu 5f como um sólido branco (74 mg, 51% de rendimento). MS (m/z): 519,2 (M+H). Esquema 7
(R)-N 1-(4-(2-(3-(Dimetilamino)pirrolidina-1-carbonil)tieno[3,2-b ]piridin-7- ilóxi)-3-fluorofenil)-N 3-fenilmalonamida (5g) Etapas 1 -3: (R)-(7-Clorotieno[3,2-b ]piridin-2-il)(3-(dimetilamino) pirrolidin-1-il)metanona (24)
[00530] Seguindo-se os procedimentos descritos acima para o composto 21 (Exemplo 5f, etapas 1-3) porém substituindo pirrolidina na Etapa 3 para (R)-N,N-dimetilpirrolidin-3-amina, o composto do título 24 foi obtido como um sólido branco (58% de rendimento). MS (m/z): 310,0 (M+H). Etapa 4: (R)-(3-(Dimetilamino)pirrolidin-1-il)(7-(2-flúor-4-nitrofenóxi) tieno[3,2-b ]piridin-2-il)metanona (25)
[00531] Uma mistura da amida 24 (1,44 g, 4,65 mmols), 2-flúor-4- nitrofenol (2,21 g, 14,1 mmols) e K2CO3 (2,56 g, 18,5 mmols) em Ph2O (5,0 mL) foi aquecida a 190°C durante 3 horas. CHCI3 (100 mL) foi adicionado à mistura marrom-escuro resultante e então a mistura foi extraída com HCl a 1M. A fase aquosa foi lavada com CHCl3 e basificada com NH4OH (pH 11). A mistura turva resultante foi extraída com CHCl3 e a fase orgânica foi coletada, lavada com água, secada sobre Na2SO4 anidro então concentrada sob pressão reduzida. O material sólido amarelo remanescente foi purificado por cromatografia instantânea, eluente CHCl3/MeOH (95:5, então 80:20) para fornecer o composto do título 25 como um sólido branco (1,41 g, 3,28 mmols, 71% de rendimento). MS (m/z): 431,0 (M+H). Etapa 5: (R)-(7-(4-Amino-2-fluorofenóxi)tieno[3,2-b ]piridin-2-il)(3- (dimetilamino) pirrolidin-1-il)metanona (26)
[00532] A uma solução do composto nitro 25 (708 mg, 1,64 mmols) em AcOH (16 mL) a 90°C, foi adicionado pó de ferro (928 mg, 16,6 mmol) e a mistura de reação foi vigorosamente agitada durante 20 minutos. A suspensão cinzenta foi dissolvida em HCl a 1N (50 mL) e lavada com CHCl3 (50 mL). A fase aquosa foi basificada com NH4OH para pH ~10, extraída com CHCl3. A fase orgânica foi coletada, secada sobre Na2SO4 anidro e concentrada sob pressão reduzida para fornecer o composto do título 26 (506 mg, 77%) como um sólido branco. MS (m/z): 401,1 (M+H). Etapa 6: (R)-N 1-(4-(2-(3-(Dimetilamino)pirrolidina-1-carbonil)tieno[3,2- b ]piridin-7-ilóxi)-3-fluorofenil)-N 3-fenilmalonamida (5g)
[00533] Seguindo o procedimento descrito acima para o composto 5a (Etapa 5, Exemplo 1) porém substituindo a amina 9 pelo composto 26, o composto do título 5g foi obtido em 38% de rendimento. MS (m/z): 562,0 (M+H). Tabela 1
Esquema 8
Exemplo 8 N1 -(3-Flúor-4-(2-(1 -metil-1 H-imidazol-2-il)tieno[3,2-b ]piridin-7- ilóxi)fenil)-N3 -(2-metoxifenil)malonamida (28a) Etapa 1: ácido 3-(2-Metoxifenilamino)-3-oxopropanoico (27)
[00534] A uma solução de 2-metoxibenzenoamina (1,1 g, 8,93 mmols) em DCM (90 mL) foi adicionado TMSCl (1,1mL, 8,93mmol) em temperatura ambiente [Rigo, B.; Fasseur, D.; Cauliez, P. e Couturier, D.Tetrahedron Lett.; 30; 23; 1989; 3073-3076.]. A mistura de reação foi agitada durante 30 minutos antes da adição de 2,2-dimetil-1,3-dioxano- 4,6-diona (1,29g, 8,93mmol) e então a agitação para mistura foi continuada durante mais 2 horas. Água (1 mL) foi adicionado, e a mistura de reação foi concentrada sob pressão reduzida. O resíduo foi despejado em solução de NaHCO3 saturada, e extraído com EtOAc. A fase aquosa foi coletada, acidificada com HCl concentrado para pH ~4, extraída com EtOAc, o extrato foi seco sobre Na2SO4 anidro e concentrado. O resíduo foi purificado por cromatografia instantânea (eluente CHCl3/MeOH/AcOH 9:1:0,1) para fornecer o composto 27 (0,56 g, 30% de rendimento) como um sólido branco. 1H RMN (DMSO- d6) δ (ppm): 12,62 (bs, 1H), 9,52 (s, 1H), 8,02 (dd, 1H), 7,03 (m, 2H), 6,88 (m, 1H), 3,82 (s, 3H), 3,46 (s, 2H). MS (m/z): 210,1 (M+H). Etapa 2: N1 -(3-Flúor-4-(2-(1 -metil-1 H-imidazol-2-il)tieno[3,2-b ]piridin-7- ilóxi)fenil)-N3 -(2-metoxifenil)malonamida (28a)
[00535] A uma solução do ácido (27) (75 mg, 0,36 mmol) e HOBt (54 mg, 0,40 mmol) em DMF (6 mL), foi adicionada amina 9 (135 mg, 0,40 mmol). Após agitação durante 5 minutos, EDC (84 mg, 0,42 mmol) foi adicionado e a mistura de reação foi agitada durante mais 4 horas em temperatura ambiente. A mistura de reação foi derramada em solução de NaHCO3, extraída com EtOAc; a fase orgânica foi coletada, secada sobre Na2SO4 anidro e concentrada. O resíduo foi purificado por HPLC preparativa (coluna Aquasil C-18, gradiente MeOH/água de 60:40 para 95:5) para fornecer 28a (105 mg, 55% de rendimento) como um sólido branco. 1H RMN (DMSO-d6) δ (ppm): 10,80 (s, 1H), 9,62 (s, 1H), 8,66 (d. 1H), 8,33 (s, 1H), 8,04 (d, 1H), 7,90 (d, 1H), 7,82 (s, 1H), 7,68 (s, 1H), 7,54-7,49 (m, 2H), 7,06 (m, 2H), 6,89 (m, 2H), 4,03 (s, 3H), 3,84 (s, 3H), 3,66 (s, 2H). MS (m/z): 532,0 (M+H). Exemplo 9 N 1-(4-(2-( 1 -Etil-1 H-imidazol-4-il)tieno[3,2-b ]piridin-7-ilóxi)-3-fluorofenil)- N 3-(2-metoxifenil)malonamida (28b)
[00536] Seguindo o procedimento descrito acima para o composto 28a (Exemplo 8, Etapa 2) porém substituindo a amina 9 pela amina 15, o composto do título 28b foi obtido em 69% de rendimento. 1H RMN (DMSO-d6) δ (ppm): 10,78 (s, 1H), 9,62 (s, 1H), 8,66 (d. 1H), 8,37 (amplo, s, 1H), 8,33 (s, 1H), 8,05 (dd, 1H), 7,94 (s, 1H), 7,92 (dd, 1H), 7,55 (t, 1H), 7,47 (dd, 1H), 7,07 (m, 2H), 6,96 (d, 1H), 6,89 (m, 1H), 4,13 (q, 2H), 3,84 (s, 2H), 1,42 (t, 3H). MS (m/z): 546,0 (M+H). Exemplo 10 N 1-(3-Flúor-4-(2-(1 -metil-1 H-imidazol-5-il)tieno[3,2-b ]piridin-7- ilóxi)fenil)-N 3-(2-metoxifenil)malonamida (28c)
[00537] Seguindo o procedimento descrito acima para o composto 28a (Exemplo 8, Etapa 2) porém substituindo a amina 9 pela amina 18, o composto do título 28c foi obtido em 24% de rendimento. 1H RMN (DMSO-d6) δ (ppm): 10,58 (s, 1H), 9,62 (s, 1H), 8,49 (d, 1H), 8,05 (dd, 1H), 7,85 (m, 2H), 7,77 (s, 1H), 7,48 (t, 1H), 7,41 (m, 2H), 7,05 (m, 2H), 6,90 (m, 2H) 6,63 (dd, 1H), 3,88 (s, 3H), 3,84 (s, 3H), 3,63 (s, 2H). MS (m/z): 531,8 (M+H). Esquema 9
Exemplo 11 N1 -(3-Flúor-4-(2-(1 -metil-1 H-imidazol-2-il)tieno[3,2-b ]piridin-7- ilóxi)fenil)-N3 -(2-fluorofenil)malonamida (30a) Etapa 1: ácido 3-(2-Fluorofenilamino)-3-oxopropanoico (29)
[00538] A uma suspensão de 2-fluorobenzenoamina (1 mL, 7,80 mmols) em DCM (78 mL) foi adicionado TMSCl (0,99 mL, 7,80 mmols) em temperatura ambiente [Rigo, B.; Fasseur, D.; Cauliez, P. e Couturier, D.Tetrahedron Lett.; 30; 23; 1989; 3073-3076.]. A mistura de reação foi agitada durante 30 minutos então 2,2-dimetil-1,3-dioxano- 4,6-diona (1,12 g, 7,80 mmols) foi adicionado e a mistura combinada foi agitada durante a noite. O solvente foi removido sob pressão reduzida e o resíduo foi dissolvido em solução de NaHCO3, que foi lavada com EtOAc. A fase orgânica foi descartada e a fase aquosa foi acidificada com HCl concentrado para pH ~3, extraída com EtOAc; o extrato foi seco (Na2SO4 anidro) e concentrada. O resíduo foi purificado por cromatografia instantânea (eluente CHCl3/MeOH/AcOH 8:1:0,1) para fornecer o ácido 29 (0,4 g, 26% de rendimento) como um sólido branco. 1H RMN (DMSO-d6) δ (ppm): 12,67 (s, amplo, 1H), 9,95 (s, 1H), 7,95 (m, 1H), 7,25 (m, 1H), 7,12 (m, 1H), 3,43 (s, 2H). MS (m/z): 198,0 (M+H). Etapa 2: N1 -(3-Flúor-4-(2-(1 -metil-1 H-imidazol-2-il)tieno[3,2-b ]piridin-7- ilóxi)fenil)-N3 -(2-fluorofenil)malonamida (30a)
[00539] Partindo da amina 9 e seguindo o procedimento descrito acima para o composto 28a (Exemplo 8, Etapa 2) porém substituindo o ácido 27 pelo ácido 29, o composto do título 30a foi obtido como um sólido branco em 5% de rendimento. 1H RMN (DMSO-d6) δ (ppm): 10,56 (s, 1H), 10,04 (s, 1H), 8,50 (d. 1H), 7099-7,95 (m, 1H), 7,88 (s, 1H), 7,86 (dd, 1H), 7,50 (t, 2H) 7,42 (dd, 1H), 7,40 (s, 1H), 7,29-7,24 (m, 1H), 7,17-7,14 (m, 1H), 7,03 (s, 1H), 6,69 (d. 1H), 4,00 (s, 3H), 3,64 (s, 2H). MS (m/z): 520,1 (M+H). Exemplo 12 N1 -(4-(2-(1 -Etil-1 H-imidazol-4-il)tieno[3,2-b ]piridin-7-ilóxi)-3-fluorofenil)- N3-(2-fluorofenil)-malonamida (30b)
[00540] Seguindo o procedimento descrito acima para o composto 30a (Exemplo 12, Etapa 2) porém substituindo a amina 9 pela amina 15, o composto do título 30b foi obtido em 42% de rendimento. 1H RMN (DMSO-d6) δ (ppm): 10,55 (s, 1H), 10,05 (s, 1H), 8,41 (d. 1H), 7,97 (m, 1H), 7,95 (s, 1H), 7,85 (dd, 1H), 7,77 (d, 1H), 7,65 (s, 1H), 7,45 (dd, 1H), 7,41 (dd, 1H), 7,26 (m, 1H), 7,16 (m, 1H), 6,57 (d, 1H), 4,03 (q, 2H), 3,60 (s, 2H), 1,38 (t, 3H). MS (m/z): 534,0 (M+H). Esquema 10
Exemplo 13 N1 -(3-Flúor-4-(2-(1 -metil-1 H-imidazol-2-il)tieno[3,2-b ]piridin-7- ilóxi)fenil)-N3 -metil-N3-fenilmalonamida (32)
Etapa 1: ácido 3-(Metil(fenil)amino)-3-oxopropanoico (31)
[00541] Uma solução de 3-cloro-3-oxopropanoato de metila (1 mL, 9,32 mmols) e N-metil anilina (1,01 mL, 9,32 mmols) em DCM (18,6 mL) foi agitada durante a noite em temperatura ambiente. O solvente foi removido sob pressão reduzida e o resíduo (éster de metila de ácido N-metil-N-fenil-malonâmico, 2,55 g) foi dissolvido em THF (9 mL). Uma solução de LiOH x H2O (0,782 g, 18,64 mmols) em água (9 mL) foi adicionada e a mistura agitada durante 1 hora em temperatura ambiente. O THF foi removido sob pressão reduzida e a solução aquosa remanescente foi acidificada com HCl a 1N (até o pH~3) então extraída com EtOAc. A fase orgânica foi concentrada sob pressão reduzida para fornecer o ácido 31 (1,67 g, 93% de rendimento) como uma espuma marrom. MS (m/z): 194,0 (M+H). Etapa 2: N1 -(3-Flúor-4-(2-(1 -metil-1 H-imidazol-2-il)tieno[3,2-b ]piridin-7- ilóxi)fenil)-N3 -metil-N3-fenilmalonamida (32)
[00542] A uma solução da amina 9 (80 mg, 0,235 mmol), ácido 31 (47 mg, 0,235 mmmol), e BOP (114,6 mg, 0,254 mmol) em DMF (2,4 mL), DIPEA (0,164 mL, 0,941 mmol) foi adicionado e a mistura foi agitada durante 2 horas em temperatura ambiente. A mistura de reação foi diluída com EtOAc (100 mL), água (50 mL) foi adicionada e a mistura filtrada através de um filtro de papel. A fase orgânica foi separada, lavada com água (50 mL), salmoura (20 mL), secada (Na2SO4 anidro) e concentrada sob pressão reduzida. O resíduo foi purificado por cromatografia instantânea (eluente EtOAc/MeOH 10:1) para fornecer o composto do título 32 (36,2 mg, 30% de rendimento) como um sólido cremoso. 1H RMN (DMSO-d6) δ (ppm): 10,29 (s, 1H), 8,49 (d, J = 5,5 Hz, 1H), 7,87 (s, 1H), 7,77 (d, J = 12,7 Hz, 1H), 7,467,30 (m, 8H), 7,03 (s, 1H), 6,66 (d, J = 5,5 Hz, 1H), 3,98 (s, 3H), 3,22 (s, 2H), 2,52 (3H, s). MS (m/z): 516,3 (M+H). Esquema 11

N1 -{3-Flúor-4-[2-(1-metil-1 H-imidazol-2-il)-tieno[3,2-b ]piridin-7-ilóxi]- fenil}-N 3-piridin-4-il-malonamida (34) Etapa 1: 3-oxo-3-(piridin-4-ilamino)propanoato de lítio (33)
[00543] Uma mistura de 3-cloro-3-oxopropanoato (1 mL, 9,32 mmols), piridin-4-amina (0,88 g, 9,32 mmols), e Et3N (1,3 mL, 9,32 mmols) em DCM (18,6 mL) foi agitada durante 2 horas em temperatura ambiente. A mistura foi concentrada e o resíduo foi purificado por cromatografia instantânea (eluente EtOAc/MeOH, 19/1) para produzir 3-oxo-3-(piridin-4-ilamino)propanoato de metila (507,7 mg). Parte deste material (207 mg) foi redissolvido em THF (1 mL); tratado com LiOH x H2O (90 mg, 2,14 mmols) em H2O (1 mL) durante 1 hora em temperatura ambiente. O THF foi removido sob pressão reduzida e o resíduo foi liofilizado para fornecer o composto do título 33 (contendo 1 equivalente LiCl) (243,1 mg, 100% de rendimento) como um sólido branco, o qual foi empregado na etapa seguinte sem outra purificação. MS (m/z): 181,0 (M+H). Etapa 2: N1 -(3-Flúor-4-(2-(1 -metil-1 H-imidazol-2-il)tieno[3,2-b ]piridin-7- ilóxi)fenil)-N3 -(piridin-4-il)malonamida (34)
[00544] O composto do título foi obtido partindo da amina 9 de acordo com o procedimento descrito para 5b (Exemplo 2, Etapa 6) porém substituindo o ácido 27 pelo sal 33 (57 mg, 0,251 mmol). Após a purificação (HPLC preparativa, coluna C-18, MeOH 60% para MeOH 95% em água) o composto do título 34 foi obtido como um sólido macio branco em 23% de rendimento. 1H RMN (DMSO-d6) δ (ppm): 10,68 (s, 2H), 8,45 (d, J = 5,5 Hz, 1H), 8,42 (d, J = 5,9 Hz, 1H), 8,31 (bs, 1H), 7,86 (m, 2H), 7,56 (m, 2H), 7,49 (t, J = 8,8 Hz, 1H), 7,42 (m, 2H), 7,03 (d, J = 1 Hz, 1H), 6,69 (d, J = 5,5 Hz, 1H), 3,99 (s, 3H), 3,57 (s, 2H), 2,52 (2H, s). MS (m/z): 503,3 (M+H). Esquema 12
Exemplo 15 N1 -(3-Flúor-4-(2-(1 -metil-1 H-imidazol-2-il)tieno[3,2-b ]piridin-7- ilóxi)fenil)-N3 -(pirrolidin-3-il)malonamida (36) Etapa 1: 3-(3-cloro-3-oxopropanamido)pirrolidina-1-carboxilato de terc - Butila (35)
[00545] Uma mistura de 3-cloro-3-oxopropanoato (0,235 mL, 2,2 mmols), 3-aminopirrolidina-1-carboxilato de terc-butila (410 mg, 2,2 mmols), DCM (4,4 mL) e DIPEA (0,766 mL, 4,4 mmols) foi agitada durante 2 horas em temperatura ambiente. A mistura de reação foi concentrada sob pressão reduzida e o resíduo foi redissolvido em THF (2,2 mL), tratado com LiOH x H2O (0,185 g, 4,4 mmols) em água (2,2 mL) durante 2 horas em temperatura ambiente e concentrado sob pressão reduzida para remover o THF. A solução aquosa remanescente foi extraída com EtOAc, acidificada com HCl a 2N (até pH~5), extraída novamente com EtOAc. Os extratos orgânicos combinados foram secos sobre Na2SO4 anidro, filtrado e concentrado sob pressão reduzida para fornecer o composto do título 35 (286 mg, 48% de rendimento) como uma espuma marrom-claro o qual foi empregado na etapa seguinte sem outra purificação. MS (m/z): 273,1 (M+H). Etapa 2: N1 -(3-Flúor-4-(2-(1 -metil-1 H-imidazol-2-il)tieno[3,2-b ]piridin-7- ilóxi)fenil)-N3 -(pirrolidin-3-il)malonamida (36)
[00546] A uma solução da amina 9 (80 mg, 0,235 mmol), ácido 35 (64 mg, 0,235 mmol) e BOP (114,6 mg, 0,254 mmol) em DMF (2,4 mL) foi adicionado DIPEA (0,164 mL, 0,941 mmol) e a mistura agitada 2 horas em temperatura ambiente. EtOAc (100 mL) e água (50 mL) foram adicionados e a mistura foi filtrada através de um filtro de papel. A fase orgânica foi coletada, lavada com água (50 mL), salmoura (20 mL), secada sobre Na2SO4 anidro, filtrada e concentrada sob pressão reduzida. O resíduo foi purificado por cromatografia instantânea (eluente EtOAc/MeOH 7:1) fornecendo 3-(3-(3-flúor-4-(2-(1-metil-1H- imidazol-2-il)tieno[3,2-b]piridin-7-ilóxi)fenilamino)-3- oxopropanamido)pirrolidina-1-carboxilato de terc-butila (35a, 29,7 mg, 21% de rendimento) como um sólido transparente. MS (m/z): 595,1 (M+H). Este material foi dissolvido em uma solução de TFA (0,5 mL) e DCM (0,5 mL) e agitado durante 1 hora em temperatura ambiente. A mistura de reação foi concentrada sob pressão reduzida, água foi adicionada ao resíduo e a mistura foi liofilizada. O sólido resultante foi purificado por HPLC preparativa (coluna Aquasil C-18, gradiente: MeOH 60% para MeOH 95% em água) para fornecer o composto do título 36 como sal de tris-trifluoroacetato (13,9 mg, 35%), sólido macio cremoso. 1H RMN (DMSO-d6) δ (ppm): 10,51 (s, 1H), 8,81 (bs, 1H), 8,72 (bs, 1H), 8,57 (d, J = 5,4 Hz, 1H), 8,47 (d, J = 6,3 Hz, 1H), 7,86 (m, 1H), 7,58 (s, 1H), 7,50 (t, J = 9,0 Hz, 1H), 7,40 (m, 1H), 7,31 (s, 1H), 6,76 (d, J = 5,4 Hz, 1H), 4,29 (m, 1H), 3,82 (s, 3H), 3,40 (m, 1H), 3,36-3,20 (m, 4H), 2,99 (m, 1H), 2,16 (m, 1H), 1,85 (m, 1H). MS (m/z): 495,2 (M+H). Esquema 13

Exemplo 16 N1 -(3-Flúor-4-(2-(1 -metil-1 H-imidazol-2-il)tieno[3,2-b ]piridin-7- ilóxi)fenil)-2-metil-N3-fenilmalonamida (38) Etapa 1: 2-Metil-ácido 3-oxo-3-(fenilamino)propanóico (37)
[00547] Seguindo o procedimento descrito acima para o composto 1 (Exemplo 2, Etapa 1, Esquema 2) o composto do título 37 foi obtido como um sólido branco (58% de rendimento). 1H RMN (DMSO-d6) δ (ppm): 12,61 (s, 1H), 10,12 (s, 1H), 7,56 (m, 2H), 7,29 (m, 2H), 7,02 (m, 1H), 4,47 (q, 1H), 1,31 (d, 3H). MS (m/z): 194,1 (M+H). Etapa 2: N1 -(3-Flúor-4-(2-(1 -metil-1 H-imidazol-2-il)tieno[3,2-b ]piridin-7- ilóxi)fenil)-2-metil-N3-fenilmalonamida (38)
[00548] Partindo da amina 9 e seguindo o procedimento descrito acima para o composto 28a (Exemplo 8, Etapa 2) porém substituindo ácido 27 pelo ácido 37, o composto do título 38 foi obtido como um sólido branco (20% de rendimento). 1H RMN (d6-DMSO) δ (ppm): 10,99 (s, 1H), 10,51 (s, 1H), 8,64 (d. 1H), 8,25 (s, 1H), 7,94 (s, 1H), 7,80 (s, 1H), 7,65 (d, 2H) 7,57-7,48 (m, 2H), 7,28 (t, 2H), 7,03 (t, 1H), 6,88 (d, 1H), 4,08 (s, 3H), 1,18 (s, 3H). MS (m/z): 516,1 (M+H). Esquema 14
Exemplo 17 N1 -(3-Flúor-4-(2-(1 -metil-1 H-imidazol-2-il)tieno[3,2-b ]piridin-7- ilóxi)fenil)-2-metil-N3-(piridina-3-il)malonamida (40) Etapa 1: ácido 3-Oxo-3-(piridina-3-ilamino)propanóico (39)
[00549] A uma suspensão de piridina-3-amina (1 g, 10,6 mmols) em DCM (100 mL) foi adicionado TMSCl (1,3 mL, 10,6 mmols) em temperatura ambiente. A mistura de reação foi agitada durante 30 minutos 2,2-Dimetil-1,3-dioxano-4,6-diona (1,44 g, 10,6 mmols) foi adicionado e a mistura combinada agitada durante a noite e concentrada sob pressão reduzida. O resíduo foi dissolvido em solução diluída de NaHCO3 e lavado com EtOAc. A fase aquosa foi coletada e acidificada com HCl concentrado para pH ~3 e extraída com EtOAc. O extrato foi seco sobre Na2SO4 anidro, filtrado e concentrado. O sólido remanescente foi purificado por cromatografia instantânea (eluente CHCl3/MeOH/AcOH 8:1:0,1) para fornecer o composto do título 39 como um sólido branco (0,5 g, 26% de rendimento). 1H RMN (DMSO-d6) δ (ppm): 12,04 (bs, 1H), 9,67 (s, 1H), 8,18 (d, 1H), 8,01 (d, 1H), 7,29 (m, 1H), 2,97 (s, 2H). MS (m/z): 181,1 (M+H). Etapa 2: N1 -(3-Flúor-4-(2-(1 -metil-1 H-imidazol-2-il)tieno[3,2-b ]piridin-7- ilóxi)fenil)-N3 -(piridina-3-il)malonamida (40)
[00550] A uma solução de ácido 39 (58 mg, 0,32 mmol) e HOBt (47 mg, 0,35 mmol) em DMF (5 mL), foi adicionado amina 9 (108 mg, 0,32 mmol). Após agitação durante 5 minutos, EDC (75 mg, 0,39 mmol) foi adicionado e a mistura de reação foi agitada durante a noite em temperatura ambiente então derramada em solução de NaHCO3 diluído e extraída com EtOAc. O extrato foi seco sobre Na2SO4 anidro, filtrado e concentrado. O resíduo foi purificado por HPLC preparativa (Aquasil C-18, gradiente: MeOH em água 60% para 95%) para fornecer o composto do título 40 como um sólido branco. (20 mg, 12%) 1H RMN (DMSO-d6) δ (ppm): 10,65 (s, 1H), 10,50 (s, 1H), 8,73 (d, 1H), 8,50 (d, 1H), 8,26 (dd, 1H), 8,05 (m, 1H), 7,88 (s, 1H), 7,87 (dd, 1H), 7,48 (t, 1H), 7,41 (m, 2H), 7,35 (m, 1H), 6,69 (d, 1H), 3,99 (s, 3H), 3,56 (s, 2H). MS (m/z): 503,1 (M+H). Exemplo 18 N-{3-Flúor-4-[2-(3-metil-3 H-imidazol-4-il)-tieno[3,2-b ]piridin-7-ilóxi]- fenil}-N '-piridin-3-il-malonamida (41)
[00551] Seguindo o procedimento descrito acima para o composto 40 (Exemplo 17, Etapa 2, Esquema 14) porém substituindo a amina 9 pela amina 12, o composto do título 41 foi obtido em 33% de rendimento (Esquema 14) 1H RMN (DMSO-d6) δ (ppm): 10,58 (s, 1H), 10,44 (s, 1H), 8,74 (d, 1H), 8,49 (d, 1H), 8,27 (dd, 1H), 8,05 (m, 1H), 7,86 (m, 2H), 7,77 (s, 1H), 7,48 (t, 1H), 7,42 (m, 2H), 7,35 (q, 1H), 6,63 (d, 1H), 3,88 (s, 3H), 3,54 (s, 2H). MS (m/z): 502,7 (M+H). Esquema 15
Exemplo 19 N1 -(3-Flúor-4-(2-(1 -metil-1 H-imidazol-2-il)tieno[3,2-b ]piridin-7- ilóxi)fenil)-N3 -(piperidin-4-il)malonamida (43) Etapa 1: ácido 3-(1-(terc - Butoxicarbonil)piperidin-4-ilamino)-3- oxopropanoico (42)
[00552] Uma mistura de cloridrato de 4-aminopiperidina-1- carboxilato de terc-butila (512 mg, 2,16 mmols), 3-cloro-3- oxopropanoato de metila (232 μL, 2,16 mmols), e DIPEA (828 μL, 4,75 mmols) em DCM (20 mL) foi agitada em temperatura ambiente durante a noite. O solvente foi removido sob pressão reduzida e o resíduo foi purificado por cromatografia instantânea (eluente EtOAc) para fornecer 4-(3-metóxi-3-oxopropanamido)piperidina-1-carboxilato de terc-butila como um sólido branco (500 mg, 78%). MS (m/z): 301,1 (M+H). Este material (500 mg, 1,67 mmol) foi dissolvido em MeOH/THF/H2O (2 mL/2 mL/1 mL), e LiOH x H2O (280 mg, 6,75 mmols) foi adicionado à solução. A mistura de reação foi agitada durante 2 horas, o solvente foi removido sob pressão reduzida e o resíduo foi purificado por cromatografia instantânea (eluente CH3Cl/MeOH/AcOH) para fornecer o composto do título 42 (300 mg, 63% de rendimento) como xarope amarelado. MS (m/z): 287,1 (M+H). Etapa 2: N1 -(3-Flúor-4-(2-(1 -metil-1 H-imidazol-2-il)tieno[3,2-b ]piridin-7- ilóxi)fenil)-N3 -(piperidin-4-il)malonamida (43)
[00553] A uma solução de ácido 42 (76 mg, 0,266 mmol) e HOBt (40 mg, 0,30 mmol) foi adicionado amina 9 (100 mg, 0,29 mmol) e a mistura foi agitada em temperatura ambiente durante 5 minutos. EDC (62 mg, 0,32 mmol) foi adicionado e a mistura combinada foi agitada durante mais 48 horas. Mais 42 (38 mg, 0,133 mmol) e EDC (31mg, 0,16 mmol) foram adicionados e a mistura de reação foi agitada durante outras 24 horas. Novamente mais 42 (38 mg, 0,133 mmol) e EDC (31 mg, 0,16 mmol) foram adicionados e a mistura de reação foi agitada durante mais 24 horas então dividida entre EtOAc e solução de NaHCO3. A fase orgânica foi lavada com salmoura, secada sobre Na2SO4 anidro e então concentrada. O resíduo foi purificado por HPLC preparativa para fornecer 4-(3-(3-flúor-4-(2-(1-metil-1H-imidazol- 2-il)tieno[3,2-b]piridin-7-ilóxi)fenilamino)-3-oxopropanamido)piperidina- 1-carboxilato de terc-butila 42a (20 mg, 13% de rendimento) como um sólido branco. Este material foi dissolvido em uma solução de TFA/DCM (1 mL/2 mL), agitado durante 20 minutos e concentrado. O resíduo foi purificado por HPLC preparativa (coluna Aquasil C-18, gradiente, MeOH 60% para MeOH 95% em água) para fornecer o composto do título 43 como um sal de tris-trifluoroacetato (31 mg, 99%), sólido branco. 1H RMN (DMSO-d6) δ (ppm): 10,49 (s, 1H), 8,55 (d, 1H), 8,49 (bs, 1H), 8,27 (d, 1H), 8,00 (s, 1H), 7,86 (dd, 1H), 7,55 (s, 1H), 7,49 (t, 1H), 7,39 (dd, 2H), 7,26 (s, 1H), 6,74 (d, 2H), 4,00 (s, 2H), 3,83 (m, 1H), 3,24 (m, 2H), 3,00 (m, 2H), 1,92 (m, 2H), 1,56 (m, 2H). MS (m/z): 509,1 (M+H). Esquema 16

Éster de piperidin-4-il de ácido N-{3-flúor-4-[2-(1-metil-1H-imidazol-2- il)-tieno[3,2-b]piridin-7-ilóxi]-fenil}-malonâmico (45) Etapa 1: ácido 3-(1-(terc - Butoxicarboni)piperidin-4-ilóxi-3- oxopropanoico (44)
[00554] Uma mistura de 4-hidroxipiperidina-1-carboxilato de terc- butila (1 g, 4,97 mmols) e 2,2-dimetil-1,3-dioxano-4,6-diona (0,72 g, 4,97 mmols) em tolueno foi refluxada durante a noite [Ryu, Y.; Scott, A. I.; Tetrahedron Lett.; 44; 40; 2003; 7494-7502]. O solvente foi removido sob pressão reduzida e o resíduo foi dividido entre EtOAc e solução de NaHCO3. A fase aquosa foi coletada, acidificada com HCl a 2N e extraída com EtOAc. O extrato foi concentrado para fornecer o composto do título 44 (0,816 g, 57% de rendimento) como xarope incolor. MS (m/z): 288,1 (M+H). Etapa 2: Éster de piperidin-4-il de ácido N-{3-flúor-4-[2-(1-metil-1H- imidazol-2-il)-tieno[3,2-b]piridin-7-ilóxi]-fenil}-malonâmico (45)
[00555] A uma solução de amina 9 (355 mg, 1,05 mmol), ácido 44 (300 mg, 1,05 mmol) e BOP (464 mg, 1,05 mmol) em DMF (10 mL), DIPEA (0,22 mL, 1,26 mmol) foi adicionado e a mistura agitada em temperatura ambiente durante 2 horas, dividida entre EtOAc e água. A fase orgânica foi coletada, lavada com salmoura; secada sobre Na2SO4 anidro, filtrada e concentrada. O resíduo foi purificado por cromatografia instantânea (eluente EtOAc/MeOH) seguido por cristalização de MeOH, para produzir um material sólido o qual foi dissolvido em DCM (1 mL) e TFA (1 mL), agitado durante 30 minutos e concentrado para fornecer o composto-alvo 45 como um sal de tris- trifluoroacetato (30 mg, 5% de rendimento). 1H RMN (DMSO-d6) δ (ppm): 10,61 (s, 1H), 8,57 (d. 1H), 8,47 (bs, 1H), 8,42 (bs, 1H), 8,02 (s, 1H), 8,02 (m, 3H), 7,83 (dd, 1H), 7,57 (s, 2H), 7,51 (t, 1H), 7,38 (dd, 2H), 7,29 (s, 1H), 6,75 (d, 1H), 5,0 (m, 4H), 4,02 (s, 1H), 3,56 (s, 2H), 3,15 (m, 3H), 1,98 (m, 2H), 1,80 (m, 2H). MS (m/z): 510,0 (M+H). Esquema 17

Exemplo 21 Éster de fenila de ácido N-{3-flúor-4-[2-(1-metil-1H-imidazol-2-il)- tieno[3,2-b]piridin-7-ilóxi]-fenil}-malonâmico (47) Etapa 1: ácido 3-oxo-3-(fenilamino)propanóico (46)
[00556] Uma solução de 2,2-dimetil-1,3-dioxano-4,6-diona (5 g, 34,69 mmols) e fenol (3,26 g, 34,69 mmols) em tolueno (69 mL) [Ryu, Y.; Scott, A. I.; Tetrahedron Lett.; 44; 40; 2003; 7494-7502], foi refluxada durante 5 horas, resfriada à temperatura ambiente, e extraída com solução de NaHCO3 saturada, o extrato aquoso foi lavado com tolueno e acidificado com HCl concentrado (pH ~3). A solução acídica foi extraída com DCM e a fase orgânica foi coletada, secada sobre Na2SO4 anidro, filtrada e concentrada sob pressão reduzida para fornecer o composto do título 46 (3,88 g, 21,55 mmols, 62%) como um sólido branco. MS: 181,1 (M+H). Etapa 2: Éster de fenila de ácido N-{3-flúor-4-[2-(1-metil-1H-imidazol-2- il)-tieno[3,2-b]piridin-7-ilóxi]-fenil}-malonâmico (47)
[00557] A uma solução de ácido 46 (63,5 mg, 0,353mmol) em DCM (3,5 mL) contendo uma gota de DMF foi adicionado cloreto de oxalila (0,032 mL, 0,37 mmol) a 0°C. A mistura foi agitada 1 hora em temperatura ambiente e concentrada. O resíduo foi dissolvido em DCM (0,7 mL) e adicionado a uma solução da amina 9 (100 mg, 0,294 mmol) em uma mistura de DMF (0,5 mL) e DCM (2,5 mL). A mistura de reação foi agitada durante 2 horas em temperatura ambiente e o DCM foi removido sob pressão reduzida. O resíduo foi diluído com EtOAc e lavado com água. A fase orgânica foi seca sobre Na2SO4 anidro, filtrada e concentrada sob pressão reduzida. O resíduo foi purificado por HPLC preparativa (coluna Aquasil C-18, gradiente, MeOH 60% para MeOH 95% em água) para fornecer 47 (4,8 mg, 3,2% de rendimento) como um sólido macio branco. 1H RMN (DMSO-d6) δ (ppm): 10,70 (s, 2H), 8,5 (d, J = 5,4 Hz, 1H), 7,86 (m, 2H), 7,5 (t, J = 8,8 Hz, 1H), 7,46-7,41 (m, 4H), 7,28 (m, 2H), 7,16 (m, 2H), 7,04 (s, 1H), 6,69 (d, J = 5,4 Hz, 1H), 3,99 (s, 3H), 3,81 (s, 2H). MS (m/z): 503,3 (M+H). Esquema 18
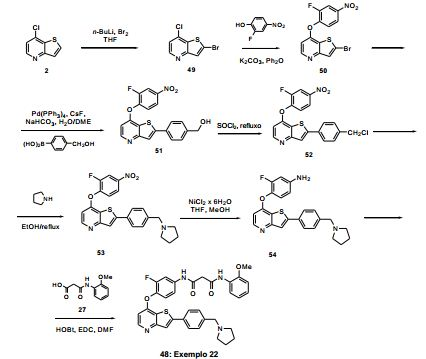
Exemplo 22 N1 -(3-Flúor-4-(2-(4-(pirrolidin-1-ilmetil)fenil)-tieno[3,2-b ]piridin-7- ilóxi)fenil)-N3 -(2-metoxifenil)malonamida (48) Etapa 1: 2-Bromo-7-cloro-tieno[3,2-b ]piridina (49)
[00558] A uma solução agitada de 2 (10,12 g, 5,59 mmols) em THF seco (200 ml) a -78°C foi adicionado n-BuLi (24 ml, 76,7 mmols, solução em hexanosa 2,5 M) e a suspensão resultante foi agitada durante 15 minutos. Bromo (18,9 g, 120 mmols) foi vagarosamente adicionado e a mistura de reação foi agitada durante mais 30 minutos, extinguida com água e diluída com EtOAc. A fase orgânica foi separada e secada sobre sulfato de sódio anidro, filtrada e concentrada sob pressão reduzida. A purificação por cromatografia de coluna (eluente EtOAc/Hexano 9:1) forneceu o composto do título 49 (10,5 g, 71% de rendimento) como um sólido amarelo. 1H RMN (CDCI3) δ (ppm): 8,62 (d, J = 5,09 Hz, 1H), 7,92 (s, 1H), 7,59 (d, J = 5,09 Hz, 1H). Etapa 2: 2-Bromo-7-(2-flúor-4-nitro-fenóxi)-tieno[3,2-b ]piridina (50)
[00559] Uma mistura de 49 (5,1 g, 20,5 mmols), carbonato de potássio (5,65 g, 4 mmols) e 2-flúor-4-nitrofenol (4,82 g, 30,7 mmols) foi aquecida a 190°C em Ph2O (25 ml) durante 3 horas. Após resfriar à temperatura ambiente ela foi diluída com DCM e filtrada. O filtrado foi concentrado e o resíduo foi purificado por cromatografia de coluna (eluente etil acetato/hexano 3:1) para fornecer o composto do título 50 como um sólido amarelo (5,4 g, 71% de rendimento). 1H RMN (DMSO-d6) δ (ppm): 8,55 (d, J = 5,28 Hz, 1H), 8,46 (dd, J = 2,5 e 10,4 Hz, 1H), 8,19 (d, J = 8,8 Hz, 1H), 7,87 (s, 1H), 7,72 (t, J = 8,4 Hz), 6,99 (d, J = 5,47 Hz, 1H). Etapa 3: (4-(7-(2-FIúor-4-nitrofenóxi)tieno[3,2-b ]piridin-2-iI)feniI)metanoI (51
[00560] A uma solução de 50 (1,0 g, 2,71 mmols) em DME seco (20 ml) foi adicionado ácido 4-(hidroximetil)fenilborônico (823 mg, 5,4 mmols), NaHCO3 (682 mg, 8,13 mmols), CsF (820 mg, 5,4 mmols) e água (10 ml), e a mistura de reação foi refluxada sob nitrogênio durante 2 horas. Após resfriar à temperatura ambiente o solvente foi removido sob pressão reduzida, o resíduo foi dissolvido em EtOAc e a solução resultante foi lavada com água, secada sobre sulfato de sódio anidro e filtrada. O filtrado foi evaporado e o sólido resultante foi triturado com Et2O para fornecer o composto do título 51 como um sólido branco (880 mg, 82% de rendimento). MS (m/z): 397,1 (M+H). Etapa 4: 2-(4-(CIorometiI)feniI)-7-(2-fIúor-4-nitrofenóxi)tieno[3,2-b ]piridina (52)
[00561] O álcool 51 (880 mg, 2,22 mmols) foi suspenso em SOCl2 (10 ml) e a mistura de reação foi refluxada durante 1 hora, resfriada e cuidadosamente derramada em uma mistura de gelo/água. Um precipitado formou-se, o qual foi coletado por filtragem e lavado com mais água fria. O material foi seco sob pressão reduzida e empregado diretamente na próxima etapa (produto bruto, >100% de rendimento). MS (m/z): 415,1 (M+H) Etapa 5: 7-(2-Flúor-4-nitrofenóxi)-2-(4-(pirrolidin-1-ilmetil)fenil)tieno[3,2- bIpiridina (53)
[00562] A uma suspensão de 52 (444 mg, 0,98 mmol, material bruto da etapa anterior) em iPrOH (10 ml) foi adicionado pirrolidina (210 mg, 2,96 mmols) e a mistura de reação foi refluxada durante 4 horas. O solvente foi removido sob pressão reduzida e o resíduo foi dissolvido em EtOAc. A solução foi lavada com água e a camada orgânica foi coletada, secada sobre sulfato de sódio anidro e filtrada. O filtrado foi evaporado e o óleo resultante foi purificado por cromatografia de coluna (eluentes EtOAc para 30% de MeOH em EtOAc), para fornecer o composto do título 53 (200 mg, 45% de rendimento) como um sólido alaranjado. MS (m/z): 450,2 (M+H). Etapa 6: 3-Flúor-4-[2-(4-pirrolidin-1-ilmetil-fenil)-tieno[3,2-b ]piridin-7- ilóxi]-fenilamina (54)
[00563] A uma solução do composto nitro 53 (256 mg, 0,57 mmol) em uma mistura de MeOH (12 mL) e THF (4 mL) a 0°C foi adicionado NiCl2,6H2O (279 mg, 1,14 mmol), seguido por adição em porções de NaBH4 (87 mg, 2,27 mmols). A mistura de reação tornou-se escura após 15 minutos, e foi filtrada através de uma almofada de Celita; o filtrado concentrado sob pressão reduzida. O resíduo foi suspenso em HCl a 2N e os sólidos foram removidos por filtragem. O filtrado foi basificado com NH4OH para pH ~10 então extraída com EtOAc. A fase orgânica foi concentrada para fornecer o composto do título 54 (260 mg, bruto, >100% de rendimento, HPLC puro) como uma espuma amarelada. MS (m/z): 420,1 (M+H). Etapa 7: N1 -(3-Flúor-4-(2-(4-(pirrolidin-1-ilmetil)fenil)-tieno[3,2-b ]piridin- 7-ilóxi)fenil)-N3-(2-metoxifenil)malonamida (48)
[00564] A uma solução de amina 54 (260 mg, 0,57 mmol, bruto) em DMF (6 mL), foram adicionados ácido 27 (108 mg, 0,52 mmol) e HOBt (77 mg, 0,57 mmol). Após agitação durante 5 minutos, EDC (120 mg, 0,63 mmol) foi adicionado, e a mistura de reação foi agitada em temperatura ambiente durante mais 4 horas e concentrada sob pressão reduzida. O resíduo foi purificado por HPLC preparativa (coluna Aquasil C-18, gradiente, MeOH 60% para MeOH 95% em água) para fornecer o composto do título 48 (160 mg, 46% de rendimento) como um sólido branco. 1H RMN (DMSO-d6) δ (ppm): 10,87 (bs, 1H), 10,75 (s, 1H), 9,62 (s, 1H), 8,60 (d. 1H), 8,17 (s, 1H), 8,02 (m, 3H), 7,89 (d, 1H), 7,74 (d, 2H), 7,51 (m, 1H), 7,05 (m, 2H), 6,90 (t, 1H), 6,79 (d, 1H), 4,39 (d, 1H), 6,57 (d, 1H), 3,84 (s, 2H), 3,69 (s, 2H), 3,35 (m, 2H), 3,06 (m, 3H), 2,00 (m, 2H), 1,87 (m, 2H). MS (m/z): 610,0 (M+H). Exemplo 23 N1 -(4-(2-(1-Etil-5-metil-1 H-imidazol-4-il)tieno[3,2-b ]piridin-7-ilóxi)-3- fluorofenil)-N3-fenilmalonamida (55) Esquema 19

[00565] A uma solução da amina 56 (WO 2006010264) (189 mg, 0,513 mmol) em DMF (10 ml) foi adicionado o ácido 1 (184 mg, 2 eq, 1,03 mmol), HOBT (139 mg, 2 eq, 1,03 mmol) e EDC (197 mg, 2 eq, 1,03 mmol). A mistura de reação foi agitada em temperatura ambiente durante a noite, concentrada à secura e dividida entre EtOAc/MeOH e água. A fase orgânica foi coletada, secada sobre sulfato de sódio e filtrada. O solvente foi removido sob pressão reduzida e o produto bruto foi purificado por cromatografia de coluna (MeOH a 10% em Et2O para MeOH a 20% em Et2O) para fornecer o composto do título 55 (80 mg, 29% de rendimento) como um sólido macio amarelo. 1H RMN (400 MHz, d6 DMSO) δ (ppm) 10,55 (s, 1H), 10,20 (s, 1H), 8,41 (d, J = 5,48 Hz, 1H), 7,85 (dd, J = 2,44 e 13,11 Hz, 1H), 7,72 (s, 1H), 7,59 (d, J = 8,61 Hz, 2H), 7,51 (s, 1H), 7,42 (m, 4H), 7,30 (dt, J = 2,15 e 6,65 Hz, 1H), 7,07 (dt, J = 1,17 e 2,35 Hz, 1H), 6,57 (dd, J = 0,78 e 5,52 Hz, 1H), 4,02 (q, J = 7,24 Hz, 2H), 3,50 (s, 2H), 3,31 (s, 3H), 1,31 (t, J = 7,24 Hz, 3H). MS (m/z): 530,0 (M+H). Exemplo 24 N1 -Cicloexil-N3 -(4-(2-(1 -etil-1 H-imidazol-4-il)tieno[3,2-b ]piridin-7-ilóxi)- 3-fluorofenil)malonamida (57) Esquema 20

Etapa 1: ácido 3-(Cicloexilamino)-3-oxopropanoico (58)
[00566] A uma solução de 3-cloro-3-oxopropanoato de metila (2,2 g, 16,1 mmols) em DCM seco (30 ml) a 0°C foi adicionado ciclo- hexilamina (4,0 g, 40,3 mmols). A mistura de reação foi agitada durante 1 hora em temperatura ambiente. A mistura de reação foi lavada com HCl diluído, NaHCO3 saturado em seguida bastante salmoura. A fase orgânica foi coletada, secada sobre sulfato de sódio anidro em seguida filtrada e concentrada. A amida bruta resultante foi empregada diretamente na próxima etapa sem nenhuma purificação adicional (3,2 g, 100%). A uma solução deste material (500 mg, 2,51 mmols) em THF/água (1:1, 20 ml) foi adicionado NaOH (200 mg, 5,02 mmols) e a mistura foi agitada durante 3 horas em temperatura ambiente. A mistura foi extraída com Et2O em seguida a fase aquosa foi acidificada para pH 1 e extraída com EtOAc. A fase orgânica foi coletada, secada sobre sulfato de sódio anidro em seguida filtrada e concentrada para fornecer o composto do título 58 como um sólido bege (450 mg, 97% de rendimento). MS (m/z): 186,2 (M+H). Etapa 2: N 1-Cicloexil-N 3-(4-(2-(1 -etil-1 H-imidazol-4-il)tieno[3,2-b ]piridin- 7-ilóxi)-3-fluorofenil)malonamida (57)
[00567] O composto do título foi obtido partindo da amina 15 (117 mg, 0,33 mmol) de acordo com o procedimento descrito para 55 porém substituindo o ácido 1 pelo ácido 58 (122 mg, 0,66 mmol). Após a purificação por cromatografia de coluna (EtOAc para MeOH a 10% em EtOAc) o composto do título 57 foi obtido como um sólido rosado (7 mg, 5% de rendimento). 1H RMN (DMSO-d6) δ (ppm): 10,45 (s, 1H), 8,40 (d, J = 5,48 Hz, 1H), 8,00 (d, J = 7,63 Hz, 1H), 7,94 (s, 1H), 7,84 (dd, J = 2,35 e 13,11 Hz, 1H), 7,71 (s, 1H), 7,65 (s, 1H), 7,39 (m, 2H), 6,54 (dd, J = 0,78 e 5,48 Hz, 1H), 4,01 (q, J = 7,24 Hz, 2H), 3,52 (m, 1H), 3,23 (s, 2H), 1,65 (m, 4H), 1,51 (m, 1H), 1,39 (t, J = 7,24 Hz, 3H), 1,16 (m, 4H). MS (m/z): 522,1 (M+H). Esquema 21
Exemplo 25 (S)-N1 -(4-(2-(4-((3-(Dimetilamino)pirrolidin-1-il)metil)fenil)tieno[3,2- b]piridin-7-ilóxi)-3-fluorofenil)-N3-fenilmalonamida (59) Etapa 1: (S)-1-(4-(7-(2-Flúor-4-nitrofenóxi)tieno[3,2-b ]piridin-2-il) benzil)-N, N-dimetilpirrolidin-3-amina (60)
[00568] A uma suspensão de 52 (1,0 g, 2,41 mmols) em DME (~ 20 ml) foi adicionado (S)-N,N-dimetilpirrolidin-3-amina (413 mg, 3,62 mmols) e TEA (486 mg, 2 eq, 4,82 mmols) e a mistura de reação foi aquecida ao refluxo durante uma hora. A mistura de reação foi resfriada à temperatura ambiente, filtrada em seguida concentrada. A mistura foi dividida entre EtOAc/H2O e o sistema bifásico foi filtrado e o sólido amarelo resultante foi coletado por filtragem para fornecer o composto do título 60 (450 mg, 38% de rendimento). A fase orgânica foi separada do sistema bifásico; ela foi então secada sobre Na2SO4, filtrada e concentrada. O resíduo foi purificado por cromatografia de coluna (9:1 EtOAc:MeOH + 1% de solução de NH4OH concentrado) para fornecer uma quantidade adicional de 60 (82 mg, 7% de rendimento). MS (m/z): 493 (M+H). Etapa 2: (S)-N 1-(4-(2-(4-((3-(Dimetilamino)pirrolidin-1-il)metil) fenil) tieno[3,2-b ]piridin-7-ilóxi)-3-fluorofenil)-N3-fenilmalonamida (59)
[00569] A uma suspensão do 60 (200 mg, 0,41 mmol) em MeOH (~ 5 ml) e água (~2 ml) foi adicionado cloreto de amônio (18 mg, 0,84 eq, 0,34 mmol) e Fe (207 mg, 9 eq, 3,69 mmols) e a mistura de reação foi aquecida ao refluxo durante 2 horas. A mistura de reação foi resfriada à temperatura ambiente, filtrada através de celite em seguida concentrada. A mistura foi dividida entre DCM/H2O e o DCM foi coletado, secado sobre Na2SO4, filtrado e concentrado para fornecer a amina 61, que foi usada diretamente na etapa seguinte (189 mg, 100%).
[00570] A uma solução do ácido 1 (148 mg, 2 eq, 0,826 mmol) em DCM seco (7 ml), a 0°C, foi adicionado, BOPCl (210 mg, 2 eq, 0,826 mmol) e a mistura de reação foi agitada durante 10 minutos. Uma solução da amina 61 (189 mg, 0,409 mmol) e iPr2NEt (316 mg, 6 eq, 2,45 mmols) em DCM seco (~7 ml) foi então adicionada e a mistura de reação foi agitada em temperatura ambiente durante 2 horas. A mistura de reação foi concentrada à secura e dividida entre EtOAc e solução de NaHCO3 saturada, a fase orgânica foi lavada duas vezes com solução saturada de NaHCO3 em seguida coletada, secada sobre sulfato de sódio e filtrada. O solvente foi removido sob pressão reduzida e o bruto foi purificado por cromatografia de coluna (2:8 MeOH/EtOAc +1% de solução de NH4OH) para fornecer o produto desejado 59 como um sólido esbranquiçado (54 mg, 21% de rendimento), 1H RMN (DMSO-d6) δ (ppm): 10,73 (s, 1H), (s, 1H), 10,33 (s, 1H), 8,48 (d, J = 5,48 Hz, 1H), 8,20 (s, 1H), 8,02 (s, 1H), 7,87 (dd, J = 2,15 e 13,1 Hz, 1H), 7,82 (d, J = 8,21 Hz, 2H), 7,50 (m, 4H), 7,30 (t, J = 8,41 Hz, 2H), 7,03 (t, J = 7,43 Hz, 1H), 6,61 (dd, J = 0,78 e 5,48 Hz, 1H), 3,60 (m, 10H), 2,64 (m, 2H), 2,51 (m, 2H), 2,42 (m, 2H), 2,32 (m, 1H), 1,87 (m, 1H), 1,61 (m, 1H). MS (m/z): 624,0 (M+H). (formiato) Esquema 22
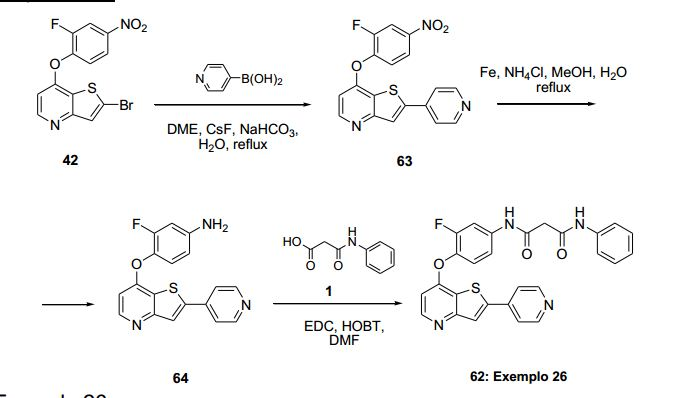
N1 -(3-flúor-4-(2-(piridin-4-il)tieno[3,2-b ]piridin-7-ilóxi)fenil)-N3 - fenilmalonamida (62) Etapa 1: 7-(2-Flúor-4-nitrofenóxi)-2-(piridin-4-il)tieno[3,2-b ]piridina (63)
[00571] Uma mistura do composto nitro 50 (890 mg, 2,41 mmols), ácido piridin-4-ilborônico (593 mg, 2 eq, 4,82 mmols), CsF (1,1 g, 3 eq, 7,23 mmols) e Pd(PPh3)4 (278 mg, 0,1 eq, 0,241 mmols) foram suspensos em DME (30 ml) e NaHCO3 (607 mg, 3 eq, 7,23 mmols), dissolvido em uma quantidade mínima de água, foi adicionada. A mistura foi desaerada por N2 borbulhante através da solução durante 10 minutos, aquecida ao refluxo durante 4 horas e concentrada à secura. O resíduo formado foi dissolvido em DCM e lavado com água. O DCM foi coletado, secado sobre sulfato de sódio, filtrado e o DCM foi removido por evaporação. O sólido resultante foi triturado com Et2O para fornecer o composto do título 63 (660 mg, 75% de rendimento), o qual foi empregado sem outra purificação. MS (m/z): 368,0 (M+H). Etapa 2: N1 -(3-Flúor-4-(2-(piridin-4-il)tieno[3,2-b ]piridin-7-ilóxi)fenil)-N3- fenilmalonamida (64)
[00572] A uma suspensão do 63 (660 mg, 1,80 mmol) em MeOH (15 ml) e água (3 ml) foi adicionado cloreto de amônio (81 mg, 0,84 eq, 1,51 mmol) e Fe (907 mg, 9 eq, 16,2 mmols) e a mistura de reação foi aquecida ao refluxo durante 2 horas. A mistura de reação foi resfriada à temperatura ambiente, filtrada através de celite em seguida concentrada. A mistura foi dividida entre DCM/H2O e o DCM foi coletado, secada sobre Na2SO4, filtrada e concentrada para produzir a amina 64 que foi usada diretamente na etapa seguinte (607 mg, 100%). A uma solução da amina 64 (607 mg, 1,80 mmol) em DMF seca (~ 7 ml) foi adicionado o ácido 1 (644 mg, 2 eq, 3,6 mmols), HOBT (365 mg, 1,5 eq, 2,7 mmols) e EDC (690 mg, 2 eq, 3,6 mmols) e a mistura de reação foi agitada em temperatura ambiente durante a noite. A mistura de reação foi concentrada à secura e dividida entre EtOAc e água, a fase orgânica foi coletada, secada sobre sulfato de sódio e filtrada. O solvente foi removido sob pressão reduzida e o bruto foi purificado por cromatografia de coluna (EtOAc) para fornecer o composto do título 62 como um sólido branco (150 mg, 17% de rendimento). 1H RMN (DMSO-d6) δ (ppm): 10,57 (s, 1H), 10,20 (s, 1H), 8,70 (m, 2H), 8,55 (d, J = 5,28 Hz, 1H), 8,37 (s, 1H), 7,89 (m, 3H), 7,60 (m, 2H), 7,58 (m, 2H), 7,30 (dt, J = 1,96 e 7,43 Hz, 2H), 7,07 (m, 1H), 6,70 (dd, J = 0,78 e 5,28 Hz, 1H), 3,51 (s, 2H). MS (m/z): 499,1 (M+H). Esquema 23
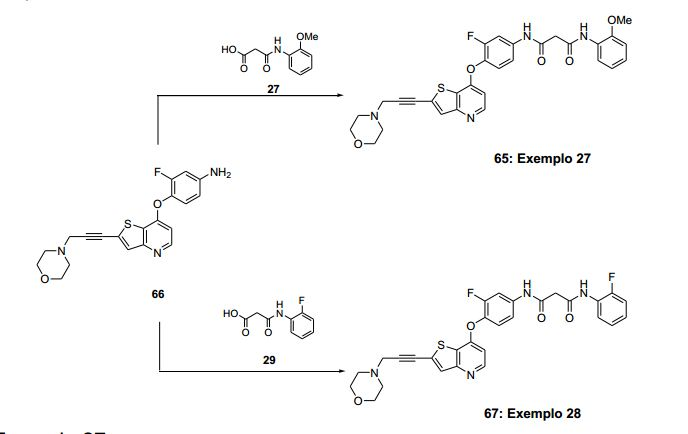
Exemplo 27 N1 -(3-Flúor-4-(2-(3-morfolinoprop-1-inil)tieno[3,2-b ]piridin-7-ilóxi)fenil)- N3-(2-metoxifenil)malonamida (65)
[00573] A uma solução da amina 66 (WO 2006010264) (1,39 g, 3,62 mmols) em DMF (15 ml) foi adicionado o ácido 27 (1,52 g, 2 eq, 7,25 mmols), HOBT (587 mg, 1,2 eq, 4,34 mmols) e EDC (691 mg, 2 eq, 7,25 mmols). A mistura de reação foi agitada em temperatura ambiente durante a noite. A mistura de reação foi concentrada à secura e dividida entre EtOAc e água, a fase orgânica foi coletada, secada sobre sulfato de sódio e filtrada. O solvente foi removido sob pressão reduzida e o bruto foi purificado por cromatografia de coluna (EtOAc para MeOH a 10% em EtOAc) para fornecer 65 (700 mg, 34% de rendimento) como um sólido branco. 1H RMN (DMSO-d6) δ (ppm): 10,58 (s, 1H), 9,62 (s, 1H), 8,53 (d, J = 5,48 Hz, 1H), 8,04 (dd, J = 1,17 e 8,80 Hz, 1H), 7,85 (dd, J = 2,35 e 12,91 Hz, 1H), 7,78 (s, 1H), 7,42 (m, 2H), 7,06 (m, 2H), 6,89 (m, 1H), 6,72 (d, J = 5,28 Hz, 1H), 3,83 (s, 3H), 2,60 (m, 8H), 2,48 (m, 4H). MS (m/z): 575,1 (M+H). Exemplo 28 N1 -(3-Flúor-4-(2-(3-morfolinoprop-1-inil)tieno[3,2-b ]piridin-7-ilóxi)fenil)- N3-(2-fluorofenil)malonamida (67)
[00574] A uma solução da amina 66 (WO 2006010264) (928 mg, 2,42 mmols) em DMF (10 mL) foi adicionado o ácido 29 (953 mg, 2 eq, 4,84 mmols), HOBT (360 mg, 1,1 eq, 2,66 mmols) e EDC (924 mg, 2 eq, 4,84 mmols). A mistura de reação foi agitada durante 72 horas. A mistura de reação foi concentrada à secura e dividida entre EtOAc e água, a fase orgânica foi coletada, secada sobre sulfato de sódio e filtrada. O solvente foi removido sob pressão reduzida e o bruto foi purificado por cromatografia de coluna (EtOAc para 10% de MeOH em EtOAc) para fornecer 67 (600 mg, 44% de rendimento) como um sólido amarelo. 1H RMN (DMSO-d6) δ (ppm): 10,58 (s, 1H), 9,62 (s, 1H), 8,53 (d, J = 5,48 Hz, 1H), 8,04 (dd, J = 1,17 e 8,80 Hz, 1H), 7,85 (dd, J = 2,35 e 12,91 Hz, 1H), 7,78 (s, 1H), 7,42 (m, 2H), 7,06 (m, 2H), 6,89 (m, 1H), 6,72 (d, J = 5,28 Hz, 1H), 3,83 (s, 3H), 2,60 (m, 8H), 2,48 (m, 4H). MS (m/z): 575,1 (M+H). Esquema 24

Exemplo 29 N1 -(4-(2-(3-(dimetilamino)prop-1-inil)tieno[3,2-b ]piridin-7-ilóxi)-3- fluorofenil)-N3-(2-fluorofenil)malonamida (68) Etapa 1: 3-(7-(2-flúor-4-nitrofenóxi)tieno[3,2-b ]piridin-2-il)-N, N- dimetilprop-2-in-1-amina (69)
[00575] A uma solução de brometo 50 (3,0 g, 8,13 mmols) em THF (40 ml) foi adicionado 4-(prop-2-inil) dimetilamina (2,70 g,32,5 mmols) [H-W. Tsou, et al. J. Med. Chem., 2001, 44, 2719-2734], trietilamina (2,05 g, 2,5 eq, 20,3 mmols), CuI (154 mg, 0,1 eq, 0,813 mmol) e Pd(PPh3)2Cl2 (319 mg, 0,056 eq, 0,046 mmol). A mistura de reação foi desgaseificada com nitrogênio e refluxada durante 2 horas, resfriada à temperatura ambiente e absorvida em sílica. A purificação por cromatografia de coluna (eluente EtOAc para 20% de MeOH em EtOAc) forneceu 69 como um sólido amarelo (2,5 g, 83% de rendimento). 1H RMN (DMSO-d6) δ (ppm): 8,65 (m, 1H), 8,48 (dd, J = 2,74 e 10,56 Hz, 1H), 8,22 (m, 1H), 7,85 (s, 1H), 7,75 (m, 1H), 6,98 (d, J = 5,28 Hz, 1H), 3,59 (s, 2H), 2,26 (s, 6H). Etapa 2: N1 -(4-(2-(3-(dimetilamino)prop-1-inil)tieno[3,2-b ]piridin-7-ilóxi)- 3-fluorofenil)-N3-(2-fluorofenil)malonamida (68)
[00576] A uma suspensão de 69 (1,0 g, 2,93 mmols) em EtOH (15 ml) e água (3 ml) foi adicionado cloreto de amônio (132 mg, 0,84 eq, 2,46 mmols) e Fe (1,48 g, 9 eq, 26,4 mmols) e a mistura de reação foi aquecida ao refluxo durante 2 horas. A mistura de reação foi resfriada à temperatura ambiente, filtrada através de celite em seguida concentrada. A mistura foi dividida entre DCM/H2O e o DCM foi coletado, secada sobre Na2SO4, filtrada e concentrada para fornecer amina 70 que foi usado diretamente na etapa seguinte (900 mg, 90% de rendimento). A uma solução da amina 70 (900 mg, 2,63 mmols) foi adicionado o ácido 27 (1,10 g, 2 eq, 5,27 mmols), HOBT (533 mg, 1,5 eq, 3,95 mmols) e EDC (1,01 g, 2 eq, 5,27 mmols). A mistura de reação foi agitada em temperatura ambiente durante a noite. A DMF foi removido por evaporação e a mistura foi dividida entre água e EtOAc. A fase orgânica foi coletada, secada sobre sulfato de sódio anidro e concentrada. A purificação por cromatografia de coluna (EtOAc para 20% de MeOH em EtOAc) forneceu o produto desejado 68 como um sólido branco (734 mg, 52% de rendimento). 1H RMN (DMSO-d6) δ (ppm): 10,58 (s, 1H), 9,62 (s, 1H), 8,53 (d, J = 5,48 Hz, 1H), 8,04 (dd, J = 1,37 e 8,90 Hz, 1H), 7,85 (dd, J = 2,35 e 12,91 Hz, 1H), 7,77 (m, 1H), 7,42 (m, 2H), 7,05 (m, 2H), 6,91 (m, 1H), 6,70 (d, J = 5,48 Hz, 1H), 3,84 (s, 3H), 3,62 (s, 2H), 3,57 (s, 2H), 2,24 (s, 6H). MS (m/z): 521,1 (M+H). Exemplo 30 N1 -(4-(2-(3-(dimetilamino)prop-1-inil)tieno[3,2-b ]piridin-7-ilóxi)-3- fluorofenil)-N3-(2-fluorofenil)malonamida (71)
[00577] O composto do título foi obtido partindo da amina 70 (1,19 g, 3,49 mmols) de acordo com o procedimento descrito para 68 porém substituindo ácido 27 pelo ácido 29 (1,37 g, 2 eq, 6,97 mmols). Após a purificação por cromatografia de coluna (EtOAc para 20% de MeOH em EtOAc) o composto do título 71 (760 mg, 42% de rendimento) foi obtido como um sólido branco. 1H RMN (DMSO-d6) δ (ppm) 10,57 (s, 1H), 10,04 (s, 1H), 8,53 (d, J = 5,28 Hz, 1H), 7,97 (m, 1H), 7,85 (dd, J = 2,35 e 12,91 Hz, 1H), 7,78 (m, 1H), 7,25 (m, 1H), 7,15 (m, 2H), 6,70 (d, J = 5,28 Hz, 1H), 3,60 (s, 2H), 3,57 (s, 2H), 2,48 (s, 6H). MS (m/z): 521,1 (M+H). Esquema 25
N-Metil-2-(4-metilpiperazin-1-il)-N-(4-(4,4,5,5-tetrametil-1,3,2- dioxaborolan-2-il)benzil)etanamina (75) Etapa 1: 2-((4-Bromobenzil)(metil)amino)etanol (72)
[00578] A uma solução de 1-bromo-4-(bromometil)benzeno (5,0 g, 20,0 mmols) em DME (30 ml) foi adicionado o álcool (3,76 g, 2,5 eq, 50,0 mmols) e a mistura de reação foi aquecida a 40°C durante uma hora. A mistura de reação foi resfriada à temperatura ambiente e concentrada. O bruto foi dissolvido em EtOAc, lavado com água e a fase orgânica foi coletada, secada sobre Na2SO4, filtrada e concentrada. A purificação por cromatografia de coluna (EtOAc) forneceu o 72 como um óleo amarelo (4,88 g, 100% de rendimento). MS (m/z): 245/247 (M+H). Etapa 2: N-(4-Bromobenzil)-2-cloro-N-metiletanamina (73)
[00579] A uma solução de 72 (2,9 g, 11,9 mmols) em tolueno (20 ml) em temperatura ambiente foi adicionado SOCl2 (2,83 g, 2 eq, 23,8 mmols) e a mistura foi aquecida a 60°C durante 3 horas. O solvente foi removido e o bruto foi dissolvido em EtOAc em seguida dividido entre água e solução de NaHCO3 saturada. A fase orgânica foi coletada, secada sobre Na2SO4, filtrada e concentrada. O bruto foi purificado por cromatografia de coluna (EtOAc a 20% em hexano) para fornecer 73 como um sólido marrom (2,9 g, 93% de rendimento). 1H RMN (DMSO- d6) δ (ppm): 7,45 (m, 2H), 7,21 (m, 2H), 3,57 (t, J = 6,85 Hz, 2H), 3,52 (s, 2H), 2,74 (t, J = 6,85 Hz, 2H), 2,26 (s, 3H). Etapa 3: N-(4-Bromobenzil)-N-metil-2-(4-metilpiperazin-1-il)etanamina (74)
[00580] A uma solução de 73 (3,5 g, 13,3 mmols) em DME (20 ml) foi adicionado N-metilpiperazina (3,33 g, 2,5 eq, 33,3 mmols) e brometo de tetrabutilamônio (cat). A mistura de reação foi aquecida ao refluxo durante 3 horas, em seguida resfriada à temperatura ambiente e concentrada. O bruto foi dissolvido em EtOAc, lavado com bastante água e a fase orgânica foi coletada, secada sobre Na2SO4 e filtrada então concentrada. A purificação por cromatografia de coluna (20% de MeOH em EtOAc + TEA a 1%) forneceu 74 como um óleo incolor (3,0 g, 69% de rendimento). MS (m/z): 326,0/328,0 (M+H). Etapa 4: N-Metil-2-(4-metilpiperazin-1-il)-N-(4-(4,4,5,5-tetrametil-1,3,2- dioxaborolan-2-il)benzil)etanamina (75)
[00581] A uma solução de 74 (600 mg, 1,84 mmol) em tolueno (20 ml) foi adicionado reagente de boro (700 mg, 1,5 eq, 2,76 mmols), Pd(PPh3)4 (214 mg, 0,1 eq, 0,184 mmol) e KOAc (541 mg, 3 eq, 5,52 mmols). A mistura de reação foi desgaseificada com N2 e aquecida ao refluxo durante 3 horas sob N2. A mistura foi resfriada à temperatura ambiente, diluída com EtOAc e água, em seguida a fase orgânica foi coletada, secada sobre Na2SO4, filtrada e concentrada para fornecer o composto do título 75 como um óleo escuro que foi empregado bruto na etapa seguinte (686 mg, 100% de rendimento). MS (m/z): 374,2 (M+H). Tabela 2 Boronatos de arila 76-77 preparados de acordo com o Esquema 25
Esquema 26
Exemplo 31 N 1-(3-Flúor-4-(2-(4-((metil(2-(4-metilpiperazin-1-il)etil)amino) metil)fenil)tieno[3,2-b ]piridin-7-ilóxi)fenil)-N3-fenilmalonamida (78) Etapa 1: N-(4-(7-(2-flúor-4-nitrofenóxi)tieno[3,2-b]piridin-2-il)benzil)-N- metil-2-(4-metilpiperazin-1-il)etanamina (79)
[00582] A uma mistura do composto nitro 50 (440 mg, 1,19 mmol) em DME (20 ml) foi adicionado o boronato 75 (667 mg, 1,5 eq, 1,79 mmol), CsF (542 mg, 3 eq, 3,57 mmols) e Pd(PPh3)4 (139 mg, 0,1 eq, 0,19 mmol) foram suspensos em DME (30 ml) e NaHCO3 (100 mg, 3 eq, 3,57 mmols), dissolvidos em uma quantidade mínima de água, foi adicionada. A mistura foi desaerada por N2 borbulhante através da solução durante 10 minutos, aquecida ao refluxo durante 4 horas. A mistura foi resfriada à temperatura ambiente, diluída com EtOAc e água em seguida a fase orgânica foi coletada, secada sobre sulfato de sódio e filtrada. O solvente foi removido e o resíduo foi purificado por cromatografia de coluna (MeOH a 20% em EtOAc + TEA a 1%) para fornecer o composto do título 79 como óleo marrom (248 mg, 39% de rendimento). MS (m/z): 536,1 (M+H). Etapas 2-3: N1 -(3-Flúor-4-(2-(4-((metil(2-(4-metilpiperazin-1 -il)etil)ami- no)metil)fenil)tieno[3,2-b ]piridin-7-ilóxi)fenil)-N3-fenilmalonamida (78)
[00583] A uma solução de 79 (200 mg, 0,37 mmol) em MeOH (10 mL) a 0°C foi adicionado NiCl2 x 6H2O (176 mg, 2 eq, 0,74 mmol) e NaBH4 (55 mg, 4 eq, 1,48 mmol). A mistura de reação foi deixada agitar durante 1 hora, concentrada à secura e o sólido resultante foi dissolvido em HCl a 2 M. Esta solução foi em seguida tornada básica com hidróxido de amônio aquoso concentrado e extraída com DCM. O extrato de DCM foi seco sobre sulfato de sódio anidro, filtrado e evaporado para fornecer a amina 80 (188 mg, 100% de rendimento), o qual foi empregado sem caracterização e outra purificação.
[00584] A uma solução do ácido 1 (165 mg, 2 eq, 0,92 mmol) em DCM seco (~5 ml), a 0°C, foi adicionado, BOPCl (234 mg, 2 eq, 0,92 mmol) e a mistura de reação foi agitada durante 10 minutos. Uma solução de 80 (232 mg, 0,46 mmol) e iPr2NEt (356 mg, 6 eq, 2,76 mmols) em DCM seco (~7 ml) foi em seguida adicionada e a mistura de reação foi agitada em temperatura ambiente durante 2 horas. A mistura de reação foi concentrada à secura e dividida entre EtOAc e solução de NaHCO3 saturada, a fase orgânica foi lavada duas vezes com NaHCO3 saturado em seguida coletada, secada sobre sulfato de sódio e filtrada. O solvente foi removido sob pressão reduzida e o bruto foi purificado por cromatografia de coluna (1:1 MeOH/EtOAc +TEA a 1%) para fornecer o composto do título 78 como sólido bege (36 mg, 12% de rendimento). 1H RMN (DMSO-d6) δ(ppm): 10,59 (s, 1H), 10,23 (s, 1H), 8,50 (d, J = 5,5 Hz, 1H), 8,04 (s, 1H), 7,89 (dd, J = 2,4 e 13,0 Hz, 1H), 7,85 (d, J = 8,4 Hz, 2H), 7,61 (dd, J = 1,1 e 8,7 Hz, 2H), 7,51 (t, J = 8,9 Hz, 1H), 7,47-7,43 (m, 3H), 7,35-7,31 (m, 2H), 7,07 (tt, J = 1,2 e 7,3 Hz, 1H), 6,64 (dd, J = 1,0 e 5,5 Hz, 1H), 3,53 (s, 2H), 3,52 (s, 2H), 2,46-2,29 (m, 12H), 2,16 (s, 3H), 2,13 (s, 3H). Exemplo 32 N1 -(3-Flúor-4-(2-(4-((metil(2-(4-metilpiperazin-1-il)etil)amino)metil) fenil)tieno[3,2-b ]piridin-7-ilóxi)fenil)-N3-metil-N3-fenilmalonamida (81)
[00585] O composto do título foi obtido partindo da amina 80 (232 mg, 0,46 mmol) de acordo com o procedimento descrito para 78 (Exemplo 31) porém substituindo o ácido 1 pelo ácido 31 (178 mg, 2 eq, 0,92 mmol). Após a purificação por cromatografia de coluna (MeOH a 40% em EtOAc + 1% de solução de NH4OH) o composto do título 81 (26 mg, 8% de rendimento) foi obtido como um sólido branco
[00586] Os compostos 82 (Exemplo 33) e 83 (Exemplo 34) foram sintetizados similarmente ao composto 78 de acordo ao Esquema 26, partindo de boronatos 76 e 77, respectivamente). Tabela 3
Esquema 27
Exemplo 35 Cloreto de 2-((4-(7-(2-flúor-4-(3-oxo-3-(fenilamino)propanamido) fenóxi)tieno[3,2-b]piridin-2-il)benzil)(metil)amino)-N,N,N- trimetiletanamínio (84) Etapa 1: 2-((4-(7-(2-Flúor-4-nitrofenóxi)tieno[3,2-b ]piridin-2-il)benzil) (metil)amino)etanol (85)
[00587] A uma suspensão de 52 (1,2 g, 2,89 mmols) em DME (30 ml) foi adicionado 2-(metilamino)etanol (2,17 g, 10 eq, 28,9 mmols) e a mistura de reação foi agitada em temperatura ambiente durante 2 horas. A mistura de reação foi concentrada então dividida entre EtOAc/H2O e o EtOAc foi coletado, secado sobre Na2SO4, filtrado e concentrado. A mistura bruta foi purificada por cromatografia de coluna (8:2 EtOAc:MeOH) para fornecer o composto do título 85 (813 mg, 45% de rendimento). 1H RMN (DMSO-d6) δ (ppm): 8,57 (d, J = 5,48 Hz, 1H), 8,48 (m, 1H), 8,21 (m, 1H), 8,08 (s, 1H), 7,83 (d, J = 8,02 Hz, 1H), 7,71 (t, J = 8,61 Hz, 2H), 7,43 (d, J = 8,22 Hz, 2H), 6,91 (d, J = 5,48 Hz, 1H), 4,41 (t, J = 5,48 Hz, 1H), 3,53 (m, 4H), 2,42 (t, J = 6,46 Hz, 2H), 2,15 (s, 3H). Etapa 2: 2-Cloro-N-(4-(7-(2-flúor-4-nitrofenóxi)tieno[3,2-b ]piridin-2- il)benzil)-N-metiletanamina (86)
[00588] A uma solução de 89 (500 mg, 1,1 mmol) em tolueno/diclorometano (1:1, 30 ml) foi adicionado SOCl2 (262 mg, 2 eq, 2,29 mmols). A mistura de reação foi aquecida a 70°C durante 2 horas em seguida concentrada à secura e o composto do título 86 foi empregado diretamente na etapa seguinte sem nenhuma purificação adicional (519 mg, 100% de rendimento). 1H RMN (DMSO-d6) δ (ppm): 8,57 (d, J = 5,48 Hz, 1H), 8,47 (m, 1H), 8,21 (m, 1H), 8,06 (s, 1H), 7,84 (d, J = 8,22 Hz, 1H), 7,71 (m, 1H), 7,43 (d, J = 8,02 Hz, 2H), 6,91 (d, J = 5,48 Hz, 1H), 3,70 (t, J = 6,65 Hz, 2H), 3,58 (s, 1H), 2,69 (m, 2H), 2,19 (s, 3H). Etapas 3-4: N1 -(4-(2-(4-(((2-Cloroetil)(metil)amino)metil)fenil)tieno[3,2- b ]piridin-7-ilóxi)-3-fluorofenil)-N3-fenilmalonamida (88)
[00589] A uma solução de 86 (375 mg, 0,79 mmol) em MeOH (15 ml) foi adicionado SnCl2 x 2H2O (891 mg, 5, eq, 3,95 mmols) e a mistura de reação foi aquecida ao refluxo durante 3 horas. A mistura foi resfriada em temperatura ambiente e derramada em gelo/água em seguida basificada para pH 9. A mistura foi filtrada e a solução aquosa foi extraída com EtOAc e o EtOAc foi lavado com solução de salmoura. A fase orgânica foi coletada, secada sobre Na2SO4, filtrada e concentrada e o 4-(2-(4-(((2-cloroetil)(metil)amino)metil)fenil) tieno[3,2-b]piridin-7-ilóxi)-3-fluorobenzenamina (87) (348 mg, 100% de rendimento) foi empregado imediatamente na etapa seguinte sem nenhuma purificação adicional.
[00590] A uma solução do ácido 1 (142 mg, 2 eq, 1,58 mmol) em DCM seco (10 ml), a 0°C, foi adicionado, BOPCl (502 mg, 2 eq, 0,826 mmol) e a mistura de reação foi agitada durante 10 minutos. Uma solução da amina 87 (348 mg, 0,79 mmol) e iPr2NEt (610 mg, 6 eq, 4,72 mmols) em DCM seco (7 ml) foi em seguida adicionado e a mistura de reação foi agitada em temperatura ambiente durante a noite. A mistura de reação foi concentrada à secura e dividida entre EtOAc e solução de NaHCO3 saturada, a fase orgânica foi lavada duas vezes com NaHCO3 saturado em seguida coletada, secada sobre sulfato de sódio e filtrada. O solvente foi removido sob pressão reduzida e o bruto foi purificado por cromatografia de coluna (EtOAc) para fornecer o produto desejado 88 como um sólido esbranquiçado (102 mg, 21% de rendimento). 1H RMN (DMSO-d6) δ (ppm): 10,58 (s, 1H), 10,22 (s, 1H), 8,48 (d, J = 5,28 Hz, 1H), 8,03 (s, 1H), 7,85 (m, 3H), 7,59 (m, 2H), 7,43 (m, 4H), 7,32 (m, 2H), 7,05 (t, J = 7,43 Hz, 1H), 6,61 (m, 1H), 3,70 (t, J = 6,65 Hz, 2H), 3,59 (s, 2H), 3,50 (s, 2H), 2,69 (m, 2H), 2,20 (s, 3H). Etapa 5: Cloreto de 2-((4-(7-(2-flúor-4-(3-oxo-3-(fenilamino) propanamido)fenóxi)tieno[3,2-b]piridin-2-il)benzil)(metil)amino)-N,N,N- trimetiletanamínio (84)
[00591] A uma solução de 88 (80 mg, 0,133 mmol) em DME (1 mL) foi adicionado NMe3 (15,7 mg, 2 eq, 0,27 mmol) e a mistura de reação foi aquecida a 100°C em um tubo selado durante uma hora. A mistura foi resfriada à temperatura ambiente e concentrada. A purificação (Gilson, 45 minutos, MeOH a 40% em água para MeOH a 80% em água) forneceu o composto do título 84 (20 mg, 9% de rendimento), 1H RMN (DMSO-d6) δ (ppm): 10,85 (s, 1H), 10,41 (s, 1H), 8,47 (m, 2H), 8,05 (s, 1H), 7,85 (m, 3H), 7,59 (m, 2H), 7,42 (m, 5H), 7,32 (m, 2H), 7,05 (t, J = 7,43 Hz, 1H), 6,63 (d, J = 5,67, 1H), 3,60 (s, 2H), 3,51 (m, 2H), 3,1 (s, 9H), 2,81 (m, 2H), 2,20 (s, 3H). MS (m/z) 626,1 (M+H). Esquema 28

Exemplo 35a N1 -(3-Flúor-4-(2-(1 -(2-metoxietil)-1 H-imidazol-4-il)tieno[3,2-b ]piridin-7- ilóxi)fenil)-N3-fenilmalonamida (91) e Exemplo 36 N1 -(3-Flúor-4-(2-(1 -(2-hidroxietil)-1 H-imidazol-4-il)tieno[3,2-b ]piridin-7- ilóxi)fenil)-N3-fenilmalonamida (89) Etapa 1: N1 -(3-Flúor-4-(2-(1 -(2-metoxietil)-1 H-imidazol-4-il)tieno[3,2- b ]piridin-7-ilóxi)fenil)-N3-fenilmalonamida (91)
[00592] A uma solução de 3-flúor-4-(2-(1-(2-metoxietil)-1H-imidazol- 4-il)tieno[3,2-b]piridin-7-ilóxi)benzenamina (90) (WO 2006010264) (135 mg, 0,35 mmol) em DMF seca (7 ml) foi adicionado o ácido (192 mg, 4,5 eq, 1,56 mmol), HOBT (72 mg, 1,5 eq, 0,63 mmol) e EDC (306 mg, 4,5 eq, 1,56 mmol) e a mistura de reação foi agitada em temperatura ambiente durante a noite. A mistura de reação foi concentrada à secura e dividida entre EtOAc e solução de NaHCO3 saturado, a fase orgânica foi coletada, secada sobre sulfato de sódio anidro e filtrada. O solvente foi removido sob pressão reduzida e o bruto foi purificado por cromatografia de coluna (EtOAc para MeOH a 15% em EtOAc) para fornecer o composto do título 91 (150 mg, 58% de rendimento). MS (m/z): 546,1 (M+H). Etapa 2: N1 -(3-Flúor-4-(2-(1 -(2-hidroxietil)-1 H-imidazol-4-il)tieno[3,2- b ]piridin-7-ilóxi)fenil)-N3-fenilmalonamida (89)
[00593] A uma solução de 91 (85 mg, 0,156 mmol) em DCM seco (2 mL) foi adicionado, a -78°C, BBr3 (0,6 mL, 4 eq, 0,62 mmol, solução em DCM a 1M) e a mistura de reação foi vagarosamente aquecida a 0°C e deixada agitar a 0°C durante 30 minutos, a mistura de reação foi extinguida com MeOH e os solventes foram removidos sob pressão reduzida. A purificação por cromatografia de coluna (Gilson, MeOH a 25% em água para MeOH a 75% em água) forneceu o composto do título 89 (26 mg, 27% de rendimento). MS (m/z): 532,1 (M+H). Esquema 29
Exemplo 37 N1 -(3-Flúor-4-(2-(1-(metoximetil)-1 H-imidazol-4-il)tieno[3,2-b ]piridin-7- ilóxi)fenil)-N3 -(2-metoxifenil)malonamida (92) Etapa 1: 7-(2-Flúor-4-nitrofenóxi)-2-(1-(metoximetil)-1 H-imidazol-4- il)tieno[3,2-b]piridina (94)
[00594] A uma solução de 7-(2-flúor-4-nitrofenóxi)-2-(1H-imidazol-4- il)tieno[3,2-b]piridina (93) (WO 2006010264) (300 mg, 0,84 mmol) em DMF seca (3 ml) a 0°C foi adicionado NaH (40 mg, 60 % de dispersão em óleo, 1,0 mmol). A mistura foi deixada aquecer à temperatura ambiente durante 0,5 hora então resfriada novamente a 0°C. MOMCl (74 mg, 1,1 eq, 0,92 mmol) foi adicionado e a mistura foi deixada aquecer à temperatura ambiente durante 20 horas, concentrada e dividida entre EtOAc e água. A fase de EtOAc foi seca sobre sulfato de sódio anidro, filtrada, concentrada e purificada por cromatografia de coluna (100 % de hexano para 100% de acetona) para fornecer o composto do título 94 (126 mg, 36% de rendimento). MS (m/z): 401,0 (M+H). Etapa 2: 3-flúor-4-(2-(1 -(metoximetil)-l H-imidazol-4-il)tieno[3,2-b ] piridin-7-ilóxi)anilina (95)
[00595] A uma suspensão de 94 (132 mg, 0,33 mmol) em EtOH (7 ml) e água (3 ml) foi adicionado cloreto de amônio (16 mg, 0,9 eq, 0,3 mmol) e Fe (157 mg, 8,5 eq, 0,28 mmol) e a mistura de reação foi aquecida ao refluxo durante 2 horas. A mistura de reação foi resfriada à temperatura ambiente, filtrada através de celite em seguida concentrada. A mistura foi dividida entre DCM/água e o DCM foi coletado, secado sobre sulfato de sódio anidro, filtrado e concentrado. O bruto 95 foi empregado diretamente na etapa seguinte (122 mg, 100% de rendimento). MS (m/z): 371,1 (M+H). Etapa 3: N1 -(3-Flúor-4-(2-(1 -(metoximetil)-l H-imidazol-4-il)tieno[3,2- b ]piridin-7-ilóxi)fenil)-N3 -(2-metoxifenil)malonamida (92)
[00596] A uma solução de 95 (136 mg, 0,37 mmol) em DMF seca (7 ml) foi adicionado o ácido 27 (300 mg, 3,0 eq, 1,1 mmol), HOBT (74 mg, 1,5 eq, 0,55 mmol) e EDC (210 mg, 3,0 eq, 1,1 mmol) e a mistura de reação foi agitada em temperatura ambiente durante a noite. A mistura de reação foi concentrada à secura e dividida entre EtOAc e solução de NaHCO3 saturada, a fase orgânica foi coletada, secada sobre sulfato de sódio anidro e filtrada. O solvente foi removido sob pressão reduzida e o bruto foi triturado com Et2O para fornecer o composto do título 92 (150 mg, 72% de rendimento). 1H RMN (DMSO- d6) δ (ppm): 10,57 (s, 1H), 9,62 (s, 1H), 8,43 (d, J = 5,48 Hz, 1H), 8,06 (m, 2H), 7,93 (m, 1H), 7,85 (m, 1H), 7,76 (s, 1H), 7,45 (m, 2H), 7,05 (m, 2H), 6,90 (m, 1H), 6,58 (m, 1H), 5,35 (s, 2H), 3,84 (m, 3H), 3,63 (s, 2H), 3,25 (s, 3H). MS (m/z): 562,1 (M+H). Esquema 30
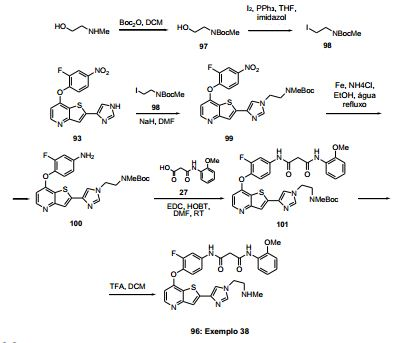
Exemplo 38 N1 -(3-Flúor-4-(2-(1 -(2-(metilamino)etil)-1 H-imidazol-4-il)tieno[3,2- b ]piridin-7-ilóxi)fenil)-N3 -(2-metoxifenil)malonamida (96) Etapa 1: 2-hidroxietil(metil)carbamato de terc-Butila (97) (J. Med. Chem., 1999, 42, 11, 2008)
[00597] A uma solução de 2-(metilamino)etanol (5,0 g, 67 mmols) em THF (50 ml) em temperatura ambiente foi adicionado Boc2O (15,7 g, 72 mmols) e a mistura de reação foi agitada em temperatura ambiente durante 4 horas. A mistura de reação foi concentrada à secura e o composto do título 97 foi empregado diretamente na etapa seguinte sem nenhuma purificação adicional (11,74 g, 100% de rendimento). MS (m/z): 176,2 (M+H). Etapa 2: 2-iodoetil(metil)carbamato de terc-Butila (98) (J. Med. Chem., 1999, 42, 11, 2008)
[00598] A uma solução de 97 (520 mg, 3,0 mmols) em THF (50 mL) foi adicionado PPh3 (1,25 g, 1,6 eq, 4,75 mmols), imidazol (306 mg, 1,5 eq, 4,5 mmols) e iodo (571 mg, 1,5 eq, 4,5 mmols). A mistura de reação foi agitada em temperatura ambiente durante uma hora e o solvente foi removido. O bruto foi dissolvido em EtOAc, lavado com solução de NaHCO3 saturada e a fase orgânica foi coletada, secada sobre sulfato de sódio anidro e filtrada. A purificação por cromatografia de coluna (hexano para EtOAc a 20% em hexano) forneceu o composto do título 98 (500 mg, 39% de rendimento). MS (m/z): 308,1 (M+Na). Etapa 3: 2-(4-(7-(2-flúor-4-nitrofenóxi)tieno[3,2-b ]piridin-2-il)-1 H- imidazol-1-il)etil(metil)carbamato de terc - Butila (99)
[00599] A uma solução de 7-(2-flúor-4-nitrofenóxi)-2-(1H-imidazol-4- il)tieno[3,2-b]piridina (93) (WO 2006010264) (150 mg, 0,42 mmol) em DMF seca (2 ml) a 0°C foi adicionado NaH (34 mg, 60 % de dispersão em óleo, 2 eq, 0,84 mmol). A mistura foi deixada aquecer à temperatura ambiente durante 0,5 hora então resfriada novamente a 0°C. O composto 98 (132 mg, 1,1 eq, 0,46 mmol) foi adicionado e a mistura foi deixada agitar em temperatura ambiente durante a noite, em seguida concentrada e dividida entre EtOAc e água. A fase de EtOAc foi seca sobre sulfato de sódio anidro, filtrada, concentrada e purificada por cromatografia de coluna (100 % de hexano para 100% de acetona) para fornecer o composto do título 99 (30 mg, 14% de rendimento). MS (m/z): 514,0 (M+H). Etapa 4: 2-(4-(7-(4-amino-2-fluorofenóxi)tieno[3,2-b ]piridin-2-il)-1 H- imidazol-1-il)etil(metil)carbamato de terc - Butila (100)
[00600] A uma suspensão de 99 (30 mg, 0,058 mmol) em EtOH (1,6 ml) e água (0,6 ml) foi adicionado cloreto de amônio (3 mg, 0,9 eq, 0,053 mmol) e Fe (28 mg, 8,5 eq, 0,49 mmol) e a mistura de reação foi aquecida ao refluxo durante 45 minutos. A mistura de reação foi resfriada à temperatura ambiente, filtrada através de celite então concentrada para fornecer o composto do título 100 (30 mg, 100% de rendimento). MS (m/z): 484,1 (M+H). Etapa 5: 2-(4-(7-(2-flúor-4-(3-(2-metoxifenilamino)-3-oxopropanamido) fenoxi)tieno[3,2-b ]piridin-2-il)-1 H-imidazol-1-il)etil(metil)carbamato de terc - Butila (101)
[00601] A uma solução de 100 (145 mg, 0,30 mmol) em DMF seca (7 ml) foi adicionado o ácido (125 mg, 2,0 eq, 0,6 mmol), HOBT (45 mg, 1,5 eq, 0,33 mmol) e EDC (115 mg, 2,0 eq, 0,6 mmol) e a mistura de reação foi agitada em temperatura ambiente durante a noite. A mistura de reação foi concentrada à secura e dividida entre EtOAc e solução de NaHCO3 saturada, a fase orgânica foi coletada, secada sobre sulfato de sódio e filtrada. O solvente foi removido sob pressão reduzida e o bruto foi purificado por cromatografia de coluna (acetona a 70% em hexano) forneceu o composto do título 101 (48 mg, 24% de rendimento). MS (m/z): 675,1 (M+H). Etapa 6: N1-(3-Flúor-4-(2-(1 -(2-(metilamino)etil)-1 H-imidazol-4-il)tieno [3,2-b ]piridin-7-ilóxi)fenil)-N3 -(2-metoxifenil)malonamida (96)
[00602] A uma solução de 101 (31 mg, 0,046 mmol) em tolueno (1 ml) foi adicionado TFA (excesso) e a mistura de reação foi agitada em temperatura ambiente durante uma hora. Os solventes foram removidos e o composto do título 96 (47 mg, 100% de rendimento) foi obtido após a trituração do sólido resultante. MS (m/z): 575,1 (M+H). Esquema 31
Exemplo 39 N1 -(4-(2-(1 H-Imidazol-4-il)tieno[3,2-b ]piridin-7-ilóxi)-3-fluorofenil)-N3 -(2- metoxifenil)malonamida (102) Etapas 1 -2: N1 -(3-Flúor-4-(2-(1 -((2-(trimetilsilil)etóxi)metil)-1 H-imidazol- 4-il)tieno[3,2-b ]piridin-7-ilóxi)fenil)-N3-(2-metoxifenil)malonamida (105)
[00603] A uma solução de 7-(2-flúor-4-nitrofenóxi)-2-(1-((2- (trimetilsilil)etóxi)metil)-1H-imidazol-4-il)tieno[3,2-b]piridina (103) (WO 2006010264) (400 mg, 0,82 mmol) em MeOH (10 mL) a 0°C foi adicionado NiCl2 x 6H2O (650 mg, 2,5 eq, 2,73 mmols) e NaBH4 (165 mg, 4 eq, 4,4 mmols). A mistura de reação foi deixada agitar durante 1 hora, concentrada à secura e o sólido resultante foi dissolvido em HCl a 2 M. Esta solução foi então tornada básica com hidróxido de amônio aquoso concentrado e extraída com DCM. O extrato de DCM foi seco sobre sulfato de sódio anidro, filtrado e evaporado para fornecer 3- flúor-4-(2-(1-((2-(trimetilsilil)etóxi)metil)-1H-imidazol-4-il)tieno[3,2- b]piridin-7-ilóxi)benzenamina (104) (350 mg, 100% de rendimento).
[00604] A uma solução de o ácido 27 (59 mg, 2 eq, 0,28 mmol) e HOBT (19 mg, 1 eq, 0,14 mmol) em DMF (3 ml) foi adicionado amina 104 (64 mg, 0,14 mmol) e a mistura de reação foi agitada durante 10 minutos. EDC (54 mg, 2 eq, 0,28 mmol) foi adicionado e a mistura de reação foi agitada em temperatura ambiente durante a noite. Os solventes foram removidos e o resíduo foi dividido entre EtOAc e água. A fase orgânica foi coletada, secada sobre sulfato de sódio anidro, filtrada e concentrada. A purificação por cromatografia de coluna (20% de acetona em hexano para 100% de acetona) forneceu o composto do título 105 (53 mg, 58% de rendimento). MS (m/z): 648,2 (M+H). Etapa 3: N 1-(4-(2-(1 H-Imidazol-4-il)tieno[3,2-b ]piridin-7-ilóxi)-3- fluorofenil)-N3-(2-metoxifenil)malonamida (102)
[00605] Uma solução de 105 (42 mg, 0,0648 mmol) em TFA (1 ml) foi agitada em temperatura ambiente durante 30 minutos, o TFA foi removido por evaporação e então HCl a 4N em dioxano (0,5 ml) foi adicionado e a mistura foi concentrada à secura. O sólido residual foi triturado com éter de dietila para fornecer o composto do título 102 (38 mg, 100% de rendimento). MS (m/z): 518,1 (M+H). Esquema 32
Exemplo 40 N 1-(4-(2-(1-Etil-2-metil-1 H-imidazol-4-il)tieno[3,2-b ]piridin-7-ilóxi)-3- fluorofenil)-N 3-(2-metoxifenil)malonamida (106) Etapa 1: 7-Cloro-2-(1 -etil-2-metil-1 H-imidazol-4-il)tieno[3,2-b ]piridina (1071
[00606] n-Butillítio foi adicionado a uma solução a -78°C de 7-cloro- 2-(1-etil-1H-imidazol-4-il)tieno[3,2-b]piridina (13, Esquema 4) em tetra- hidrofurano e agitado durante cerca de 15 minutos. Iodeto de metila foi vagarosamente adicionado e a mistura de reação foi agitada a -78°C até a conclusão da reação. A mistura foi extinguida com água então deixada aquecer à temperatura ambiente. A solução aquosa foi extraída três vezes com acetato de etila. Os extratos orgânicos combinados foram lavados com água e salmoura, em seguida secados sobre MgSO4 anidro, filtrados e evaporados. O produto bruto foi purificado por cromatografia instantânea (eluente: 100% de DCM para 2% de MeOH/98% de DCM) para fornecer o composto do título 107 (como uma mistura com cerca de 15% do material de partida 13) como um sólido amarelo (190 mg, 80% de rendimento). MS (m/z): 278,0 (M+H). Etapa 2: 2-(1-Etil-2-metil-1 H-imidazol-4-il)-7-(2-flúor-4-nitrofenóxi) tieno[3,2-b ]piridina (108)
[00607] Partindo do cloreto 107 e 2-flúor-4-nitrofenol e seguindo-se o mesmo procedimento como descrito acima para a síntese do composto 14 (Esquema 4) o composto do título 108 foi obtido em 31% de rendimento. MS (m/z): 399,0 (M+H). Etapa 3: 4-(2-(1 -Etil-2-metil-1 H-imidazol-4-il)tieno[3,2-b]piridin-7-ilóxi)- 3-fluoroanilina (109)
[00608] Partindo do composto nitro 108 e seguindo-se o mesmo procedimento como descrito acima para a síntese do composto 23 (Esquema 6), o composto do título 109 foi obtido em 16% de rendimento MS (m/z): 369,0 (M+H). Etapa 4: N 1-(4-(2-(1 -Etil-2-metil-1 H-imidazol-4-il)tieno[3,2-b]piridin-7- ilóxi)-3-fluorofenil)-N 3-(2-metoxifenil)malonamida (106)
[00609] Partindo da amina 109 e do ácido 27 e seguindo-se o mesmo procedimento como descrito para a síntese do composto 5d (Esquema 4) o composto do título 106 foi obtido em 9% de rendimento. 1H RMN (DMSO-d6). δ (ppm): 10,60 (s, 1H), 9,64 (s, 1H), 8,42 (d, J = 5,5 Hz, 1H), 8,07 (dd, J = 1,2 e 8,0 Hz, 1H), 7,87 (dd, J = 2,3 e 13,1 Hz, 1H), 7,84 (s, 1H), 7,62 (s, 1H), 7,50-7,41 (m, 2H), 7,097,05 (m, 2H), 6,94-6,90 (m, 1H), 6,57 (d, J = 5,1 Hz, 1H), 3,96 (q, J = 7,2 Hz, 2H), 3,86 (s, 3H), 3,64 (s, 2H), 2,35 (s, 3H), 1,34 (t, J = 7,2 Hz, 3H). MS (m/z): 560,0 (M+H). Esquema 33
Exemplo 41 N 1-(4-(2-(2-((Dimetilamino)metil)-1-etil-1 H-imidazol-4-il)tieno[3,2- b ]piridin-7-ilóxi)-3-fluorofenil)-N 3-(2-metoxifenil)malonamida (110) Etapa 1: 4-(7-Clorotieno[3,2-b ]piridin-2-il)-1-etil-1 H-imidazol-2- carbaldeído (111)
[00610] A uma solução de 7-cloro-2-(1-etil-1H-imidazol-4- il)tieno[3,2-b]piridina (13, Esquema 4) (1,50g, 5,69 mmols) em tetra- hidrofurano a -78°C (1,3 mL) foi adicionado n-Butillítio (3,4 mL, 8,53 mmols) e a mistura de reação foi agitada durante cerca de 15 minutos. Dimetilformamida (0,66 mL, 8,53 mmols) foi vagarosamente adicionado e a mistura de reação foi agitada a -78°C até a conclusão da reação. A mistura foi aquecida à temperatura ambiente e extinguida com água. O precipitado foi removido por filtragem, lavado com água e bem secado. O produto bruto foi purificado por trituração com diclorometano, filtrado, lavado com mais diclorometano e secado para fornecer o composto do título 111 como um sólido amarelo (682 mg, 41% de rendimento). MS (m/z): 291,9 (M+H). Etapa 2: 1 -(4-(7-Clorotieno[3,2-b ]piridin-2-il)-1-etil-1 H-imidazol-2-il)- N, N-dimetilmetanamina (112)
[00611] Uma mistura do aldeído 111 (344 mg, 1,18 mmol) e dimetilamina (0,7 mL, 1,41 mmol) em metanol (24 mL) e ácido acético (poucas gotas) foi agitada em temperatura ambiente durante a noite. Cianoboroidreto de sódio (237 mg, 3,77 mmols) foi adicionado e a mistura de reação foi deixada agitar em temperatura ambiente até a conclusão da reação. Os solventes foram evaporados e água foi adicionada ao resíduo e a mistura foi extraída com acetato de etila. As camadas orgânicas combinadas foram lavadas com salmoura, secadas sobre MgSO4 anidro, filtradas e evaporadas. O produto bruto foi purificado por cromatografia instantânea (eluente: 2,5% de MeOH/97,5% de DCM para 10% de MeOH/90% de DCM) para fornecer o composto do título 112 como um sólido amarelo (129 mg, 34% de rendimento). MS (m/z): 321,1 (M+H). Etapa 3: 1-(1 -Etil-4-(7-(2-flúor-4-nitrofenóxi)tieno[3,2-b]piridin-2-il)-1 H- imidazol-2-il)-N, N-dimetilmetanamina (113)
[00612] Partindo do cloreto 112 e 2-flúor-4-nitrofenol e seguindo o mesmo procedimento como descrito acima para a síntese do composto 14 (Esquema 4), o composto do título 113 foi obtido como sólido amarelo em 48% de rendimento. MS (m/z): 442,1 (M+H). Etapa 4: 4-(2-(2-((Dimetilamino)metil)-1-etil-1 H-imidazol-4-il)tieno[3,2- b ]piridin-7-ilóxi)-3-fluoroanilina (114)
[00613] Partindo do composto nitro 113 e seguindo-se o mesmo procedimento como descrito acima para a síntese de composto 23 (Esquema 6) o composto do título 114 foi obtido como um sólido amarelo-escuro em 78% de rendimento. MS (m/z): 412,2 (M+H). Etapa 5: N 1-(4-(2-(2-((Dimetilamino)metil)-1 -etil-1 H-imidazol-4-il)tieno[3,2- b ]piridin-7-ilóxi)-3-fluorofenil)-N3-(2-metoxifenil)malonamida (110)
[00614] Partindo da amina 114 e do ácido 27 e seguindo-se o mesmo procedimento como descrito para a síntese do composto 5d (Esquema 4) o composto do título 110 foi obtido como um sólido branco em 22% de rendimento. 1H RMN (DMSO-d6). δ (ppm): 10,59 (s, 1H), 9,64 (s, 1H), 8,43 (d, J = 5,5 Hz, 1H), 8,07 (dd, J = 1,4 e 7,4 Hz, 1H), 7,95 (s, 1H), 7,87 (dd, J = 2,2 e 12,9 Hz, 1H), 7,65 (s, 1H), 7,507,41 (m, 2H), 7,11-7,05 (m, 2H), 6,94-6,90 (m 1H), 6,57 (d, J = 4,7 Hz, 1H), 4,08 (q, J = 7,2 Hz, 2H), 3,86 (s, 3H), 3,64 (s, 2H), 3,50 (s, 2H), 2,17 (s, 6H), 1,38 (t, 3H). MS (m/z): 603,2 (M+H). Esquema 34
Exemplo 42 N 1-(3-Flúor-4-(2-(1 -metil-1 H-imidazol-5-il)tieno[3,2-b ]piridin-7- ilóxi)fenil)-N 3-(2-hidroxifenil)malonamida (115)
[00615] A uma solução do composto 28c (Exemplo 10, Esquema 8) (115 mg, 0,21 mmol) em DCM (10 mL) a -40°C foi adicionado BBr3 (1,0M, 0,86 mL, 0,86 mmol). A mistura de reação foi deixada agitar durante 1 hora em seguida diluída com MeOH (2mL) e água (1mL) e concentrada. O resíduo foi purificado por HPLC preparativa (coluna Thermo C-18, gradiente MeOH/água de 95:5 para 60:40) para fornecer o composto do título 115 (30 mg, 27% de rendimento) como um sólido branco. 1H RMN (DMSO-d6). δ (ppm): 10,57 (s, 1H), 9,88 (s, 1H), 9,59 (s, 1H), 8,49 (d, J = 5,4 Hz,1H), 7,92 (dd, J = 8,0 Hz, J = 1,5 Hz, 1H), 7,86 (dd, J = 12,7 Hz, J = 2,1Hz, 1H), 7,85 (s, 1H), 7,76 (s, 1H), 7,48 (t, J = 8,8 Hz, 1H), 7,42 (dd, J = 9,0 Hz, J = 1,7 Hz, 1H), 7,40 (s, 1H), 6,93-6,84 (m, 2H), 6,75 (t, J = 8,0 Hz, 1H), 6,63 (d, J = 5,5 Hz, 1H), 3,88 (s, 3H), 3,61 (s, 2H). MS (m/z): 518,1 (M+H). Exemplo 43 N 1-(4-(2-(1 -Etil-1 H-imidazol-4-il)tieno[3,2-b]piridin-7-ilóxi)-3-fluorofenil)- N 3-(2-hidroxifenil)malonamida (116)
[00616] Partindo do composto 28c (Exemplo 10, Esquema 8) e seguindo-se o mesmo procedimento como descrito acima para a síntese do composto 115, o composto do título 116 foi obtido como um sólido bege-claro em 23% de rendimento. 1H RMN (DMSO-d6). δ (ppm): 10,59 (s, 1H), 9,90 (s, 1H), 9,62 (s, 1H), 8,43 (d, J = 5,6 Hz, 1H), 7,96 (d, J = 1,2 Hz, 1H), 7,94 (dd, J = 1,6 e 8,0 Hz, 1H), 7,87 (dd, J = 2,0 e 12,8 Hz, 1H), 7,79 (d, J = 1,2 Hz, 1H), 7,67 (s, 1H), 7,49 (t, J = 8,8 Hz, 1H), 7,45-7,40 (m, 1H), 7,96-7,90 (m, 1H), 6,87 (dd, J = 1,2 e 8,0 Hz, 1H), 6,80-6,75 (m, 1H), 6,58 (d, J = 5,6 Hz, 1H), 4,06 (q, J = 7,2 Hz, 2H), 3,63 (s, 2H), 1,40 (t, J = 7,2 Hz, 3H). MS (m/z): 532,1 (M+H). Esquema 35
Exemplo 44 N 1-(4-(2-(1 -Etil-1 H-imidazol-5-il)tieno[3,2-b ]piridin-7-ilóxi)-3-fluorofenil)- N 3-(2-fluorofenil)malonamida (117) Etapa 1: 7-Cloro-2-(1-etil-1 H-imidazol-5-il)tieno[3,2-b ]piridina (118)
[00617] A uma solução do cloreto 2 (Esquema 1) (4,77 g, 28,12 mmols) em THF (120 mL) a -78°C foi vagarosamente adicionado n- BuLi (hexano a 2,5M, 14,06 mL, 35,15 mmols). A mistura de reação foi agitada durante uma hora a -78°C seguido pela adição vagarosa de ZnCl2 (THF a 0,5M, 70,3 mL, 35,15 mmols). Após poucos minutos, a mistura de reação foi deixada aquecer à temperatura ambiente e agitada durante uma hora.
[00618] A uma solução de 1-etil-5-iodo-1H-imidazol (2,04 g, 8,65 mmols) (Tet. Lett. 2004, 45, 5529) em THF (5 mL) foi adicionado Pd(PPh3)4 (0,81 g, 0,70 mmol) e a mistura de reação foi aquecida ao refluxo durante 1,5 hora, resfriada à temperatura ambiente em seguida diluída com hidróxido de amônio aquoso. A solução foi extraída com EtOAc e o extrato foi lavado com água e salmoura em seguida secado sobre sulfato de magnésio anidro, filtrado e evaporado sob pressão reduzida. O resíduo foi purificado por cromatografia instantânea (eluentes DCM, então DCM-MeOH, 97:3, 95:5, 9:1) para fornecer o composto do título 118 (4 g, 54% de rendimento) como um sólido amarelo. MS (m/z): 264,1 (M+H). Etapa 2: 2-(1 -Etil-1 H-imidazol-5-il)-7-(2-flúor-4-nitrofenóxi)tieno[3,2- b]piridina (119)
[00619] A uma solução de 118 (4 g, 15,16 mmols) em Ph2O (60 ml) foi adicionado 2-flúor-4-nitrofenol (4,76 g, 30,33 mmols) e carbonato de potássio (8,38 g, 60,66 mmols). A mistura de reação foi aquecida a 195°C durante 18 horas e resfriada à temperatura ambiente. O resíduo foi purificado por cromatografia de coluna (eluentes EtOAc/Hex (9/1 para 5/5) em seguida MeOH/CH2Cl2 (98/2)), para fornecer o composto do título 119 (3,05 g, 52% de rendimento) como um sólido amarelo. MS (m/z): 385,0 (M+H). Etapa 3: 4-(2-(1-Etil-1 H-imidazol-5-il)tieno[3,2-b ]piridin-7-ilóxi)-3- fluoroanilina (120)
[00620] A uma solução do composto nitro 119 (3,05 g, 7,93 mmols) em MeOH/THF (50 ml/50 mL) foi adicionado NiCl2 x 6H2O (3,77 g, 15,86 mmols) e NaBH4 (1,18 g, 31,73 mmols). A mistura de reação foi deixada agitar durante 1 hora, concentrada à secura e o sólido resultante foi dissolvido em HCl a 2 M. A solução acídica foi em seguida tornada básica com solução de hidróxido de amônio aquoso e extraída com EtOAc. O extrato orgânico foi seco sobre sulfato de sódio anidro, filtrado e evaporado. O resíduo foi purificado por cromatografia instantânea (eluentes DCM-MeOH, 98:2, 95:5, 9:1) para fornecer o composto do título 120 (2,00g, 71% de rendimento) como um sólido amarelo. MS (m/z): 355,1 (M+H). Etapa 4: N 1-(4-(2-(1-Etil-1 H-imidazol-5-il)tieno[3,2-b]piridin-7-ilóxi)-3- fluorofenil)-N 3-(2-fluorofenil)malonamida (117)
[00621] A uma solução do composto de amino 120 (400 mg, 1,08 mmol) em DMF (10 mL), ácido 3-(2-fluorofenilamino)-3-oxopropanoico (444 mg, 2,25 mmols), EDC (519 mg, 2,70 mmols) foi adicionado HOBT (364 mg, 2,70 mmols). A mistura de reação foi deixada agitar durante 1 hora. A solução foi extraída com EtOAc e o extrato foi lavado com água, cloreto de amônio aquoso e salmoura em seguida secada sobre sulfato de magnésio anidro, filtrada e evaporada sob pressão reduzida. O resíduo foi purificado por cromatografia instantânea (eluentes DCM-MeOH, 98:2, 95:5) para fornecer o composto do título 117 (348 mg, 58% de rendimento) como um sólido branco. 1H RMN (DMSO-d6). δ ppm: 10,58 (s, 1H), 10,06 (s,1H), 8,52 (d, J = 5,6 Hz, 1H), 8,10-7,76 (m, 1H), 7,94 (d, J = 1,2 Hz, 1H), 7,88 (dd, J = 2,4 e 13,2 Hz, 1H), 7,74 (s, 1H)), 7,51 (t, J = 8,8 Hz, 1H), 7,43 (dd, J = 1,6 e 8,8 Hz, 1H), 7,39 (d, J = 1,2 Hz, 1H), 7,32-7,25 (m, 1H), 7,21-7,14 (m, 2H), 6,66 (d, J = 5,6 Hz, 1H), 4,29 (q, J = 7,2 Hz, 2H), 3,62 (s, 2H), 1,34 (t, J = 7,2 Hz, 3H). MS (m/z): 534,1 (M+H). Exemplo 45 N1 -(4-(2-(1 -Etil-1 H-imidazol-5-il)tieno[3,2-b ]piridin-7-ilóxi)-3-fluorofenil)- N3-(2-metoxifenil)malonamida (121)
[00622] O composto do título 121 (Esquema 35) foi obtido similarmente ao composto 117 (Exemplo 44, Esquema 35) partindo da amina 120 e substituindo o ácido 29 pelo ácido 27. 1H RMN (DMSO- de). δα (ppm): 10,59 (s, 1H), 9,63 (s, 1H), 8,52 (d, J = 5,6 Hz, 1H), 8,07 (d, J = 7,6 Hz, 1H), 7,94 (d, J = 1,2 Hz, 1H), 7,88 (dd, J = 2,4 e 13,2 Hz, 1H), 7,74 (s, 1H), 7,51 (t, J = 8,8 Hz, 1H), 7,43 (dd, J = 1,6 e 8,8 Hz, 1H), 7,40 (d, J = 1,2 Hz, 1H), 7,12-7,4 (m, 2H), 6,96-6,88 (m, 1H), 6,66 (d, J = 5,6 Hz, 1H), 4,29 (q, J = 7,2 Hz, 2H), 3,86 (s, 3H), 3,64 (s, 2H), 1,34 (t, J = 7,2 Hz, 3H). Esquema 36
Exemplo 46 N1 -(3-Flúor-4-(2-(1 -isopropil-1 H-imidazol-4-il)tieno[3,2-b ]piridin-7- ilóxi)fenil)-N3 -2-fluorofenil)malonamida (122) Etapa 1: 7-Cloro-2-(1 -isopropil-1 H-imidazol-4-il)tieno[3,2-b ]piridina (1231
[00623] A uma solução de cloreto 2 (Esquema 1) (2,93 g, 17,31 mmols) em THF (120 mL) a -78°C foi lentamente adicionado n-BuLi (hexano a 2,5M, 8,66 mL, 21,64 mmols). A mistura de reação foi agitada durante uma hora a -78°C seguido pela lenta adição de ZnCl2 (THF a 1M, 21,6 mL, 21,64 mmols). Após alguns minutos a mistura de reação foi deixada aquecer até a temperatura ambiente e agitada durante uma hora.
[00624] A uma solução de 4-iodo-1-isopropil-1H-imidazol (2,04 g, 8,65 mmols) [Tet. Lett. 2004, 45, 5529] em THF (5 mL) foi adicionado Pd(PP3)4 (0,500 g, 0,43 mmol) e a mistura de reação que foi aquecida até o refluxo durante 5 horas, em seguida resfriada até a temperatura ambiente, diluída com hidróxido de amônio aquoso. A solução foi extraída com EtOAc e o extrato foi lavado com água e salmoura, secado sobre sulfato de magnésio anidro, filtrado e evaporado sob pressão reduzida. O resíduo foi purificado por cromatografia instantânea (eluentes DCM, em seguida DCM-MeOH, 97:3, 95:5, 9:1) para fornecer o composto do título 123 (1,16 g, 24% de rendimento) como um sólido amarelo. MS (m/z): 278,0 (M+H). Etapa 2: 7-(2-Flúor-4-nitrofenóxi)-2-(1 -isopropil-1 H-imidazol-4-il)tieno [3,2-b ]piridina (124)
[00625] A uma solução de 123 (1,16 g, 4,18 mmols) em Ph2O (20 mL) foi adicionado 2-flúor-4-nitrofenol (1,31 g, 8,37 mmols) e carbonato de potássio (2,31 g, 16,72 mmols). A mistura de reação foi aquecida para 195°C durante 18 horas, em seguida resfriada até a temperatura ambiente. O resíduo foi purificado por cromatografia de coluna, eluentes EtOAc/Hex (9/1 a 5/5), em seguida MeOH/CH2Cl2 (98/2), para fornecer o composto do título 124 (1,47 g, 88% de rendimento) como um sólido amarelo. MS (m/z): 399,0 (M+H). Etapa 3: 3-Flúor-4-(2-(1 -isopropil-1 H-imidazol-4-il)tieno[3,2-b ]piridin-7- ilóxi)anilina (125)
[00626] A uma solução do composto nitro 124 (1,47 g, 3,68 mmols) em MeOH/THF (50 mL/50 mL) foi adicionado NiCl2 x 6H2O (1,75 g, 7,37 mmols) e NaBH4 (550 mg, 14,75 mmols). A mistura de reação foi deixada agitar durante 1 hora, em seguida concentrada até a secura e o sólido resultante foi dissolvido em HCl a 2 M. A solução acídica foi então tornada básica com solução de hidróxido de amônio aquoso e extraída com EtOAc. O extrato orgânico foi seco sobre sulfato de sódio anidro, filtrado e evaporado. O resíduo foi purificado por cromatografia instantânea (eluentes DCM-MeOH, 98:2, 95:5, 9:1) para fornecer o composto do título 125 (3,31 g, 88% de rendimento) como um sólido rosa. MS (m/z): 368,1 (M+H). Etapa 4: N 1-(3-Flúor-4-(2-(1 -isopropil-1 H-imidazol-4-il)tieno[3,2- b ]piridin-7-ilóxi)fenil)-N 3-2-fluorofenil)malonamida (122)
[00627] A uma solução do composto de amino 125 (400 mg, 1,08 mmol) em DMF (20 mL), ácido 3-(2-fluorofenilamino)-3-oxopropanoico (427 mg, 2,17 mmols), EDC (352 mg, 2,60 mmols) foi adicionado HOBT (499 mg, 2,60 mmols). A mistura de reação foi deixada agitar durante 1 hora. A solução foi extraída com EtOAc e o extrato foi lavado com água, cloreto de amônio aquoso e salmoura, em seguida secado sobre sulfato de magnésio anidro, filtrado e evaporado sob pressão reduzida. O resíduo foi purificado por cromatografia instantânea (eluentes DCM-MeOH, 98:2, 95:5) para fornecer o composto do título 122 (384 mg, 65% de rendimento) como um sólido amarelo. 1H RMN (DMSO-d6). δ ppm: 10,57 (s, 1H), 10,06 (s, 1H), 8,43 (d, J = 5,6 Hz, 1H), 8,04 (dd, J = 1,6 e 7,2 Hz, 1H), 8,04-7,96 (m, 1H), 7,87 (dd, J = 2,4 e 12,8 Hz, 1H), 7,84 (d, J = 1,2 Hz, 1H), 7,67 (s, 1H), 7,49 (t, J = 8,8 Hz, 1H), 7,42 (dd, J = 1,6 e 8,8 Hz, 1H), 7,32-7,25 (m, 1H), 7,22-7,13 (m, 2H), 6,58 (d, J = 5,6 Hz, 1H), 4,48 (quin, J = 6,4 Hz, 1H), 3,63 (s, 2H),1,46 (d, J = 6,8 Hz, 6H). 3,62 (s, 2H), 1,34 (t, J = 7,2 Hz, 3H). MS (m/z): 548,1 (M+1). Exemplo 47 N 1-(3-Flúor-4-(2-(1 -isopropil-1 H-imidazol-4-il)tieno[3,2-b]piridin-7- ilóxi)fenil)-N 3-(2-metoxifenil)malonamida (126)
[00628] O composto do título 126 (Esquema 36) foi obtido similarmente ao composto 122 (Exemplo 45, Esquema 36) partindo da amina 125 e substituindo o ácido 29 pelo o ácido 27. 1H RMN (DMSO- d6). δ (ppm): 10,58 (s, 1H), 9,64 (s, 1H), 8,43 (d, J = 5,6 Hz, 1H), 8,07 (dd, J = 1,6 e 7,2 Hz, 1H), 8,05 (d, J = 1,2 Hz, 1H), 7,87 (dd, J = 2,4 e 12,8 Hz, 1H), 7,84 (d, J = 1,2 Hz, 1H), 7,67 (s, 1H), 7,49 (t, J = 8,8 Hz, 1H), 7,43 (dd, J = 1,6 e 8,8 Hz, 1H), 7,12-7,04 (m, 2H), 6,92 (ddd, J = 2,4, 7,2 e 8,8 Hz, 1H), 6,58 (d, J = 5,6 Hz, 1H), 4,48 (quin, J = 6,4 Hz, 1H), 3,86 (s, 3H), 3,64 (s, 2H),1,46 (d, J = 6,8 Hz, 6H). MS (m/z): 559,2 (M+H). Esquema 37
N 1-(3-Flúor-4-(2-(1 -isopropil-1 H-imidazol-4-il)tieno[3,2-b]piridin-7- ilóxi)fenil)-N 3-(tiazol-2-il)malonamida (127) Etapa 1: Ácido 3-oxo-3-(tiazol-2-ilamino)propanóico (129)
[00629] A uma solução de 2-aminotiazol (2,0 g, 19,97 mmols) em DCM seco (30 mL) a 0°C foi adicionado TEA (4,03 g, 2 eq, 39,94 mmols) e 3-cloro-3-oxopropanoato de metila (3,0 g, 1,1 eq, 21,97 mmols). A mistura de reação foi agitada durante 1 hora em temperatura ambiente. A mistura de reação foi concentrada até a secura, dissolvida em EtOAc e lavada bem com água. A fase orgânica foi coletada, secada sobre sulfato de sódio anidro, em seguida filtrada e concentrada. O sólido resultante foi triturado com Et2O e usado diretamente na etapa seguinte sem nenhuma purificação adicional (1,1 g, 30% de rendimento). A uma solução do éster 128 (500 mg, 2,49 mmols) em THF/água (1:1, 20 mL) foi adicionado LiOH x H2O (209 mg, 2 eq, 4,98 mmols) e a mistura agitada durante 2 horas em temperatura ambiente. A mistura foi neutralizada com solução de HCl a 1M e adsorvida sobre sílica-gel. Purificação por cromatografia de coluna (60% EtOAc em hexanos) forneceu o ácido 129 como um sólido branco (320 mg, 69% de rendimento). MS (m/z): 187,2 (M+H). Etapa 2: N 1-(3-Flúor-4-(2-(1-isopropil-1 H-imidazol-4-il)tieno[3,2- b ]piridin-7-ilóxi)fenil)-N 3-(tiazol-2-il)malonamida (127)
[00630] O composto do título foi obtido partindo da amina 125 (100 mg, 0,27 mmol) (Esquema 36) de acordo com o procedimento descrito para 55 (Exemplo 23, Esquema 19), porém substituindo o ácido 1 pelo ácido 129 (101 mg, 2 eq, 0,53 mmols). Após purificação por cromatografia de coluna (10% MeOH/EtOAc) o composto do título 127 foi obtido como um sólido branco (7 mg, 5% de rendimento). 1H RMN (DMSO-d6) δ (ppm): 12,32 (s, 1H), 10,60 (s, 1H), 8,41 (d, J = 5,48 Hz, 1H), 8,03 (s, 1H), 7,82 (m, 2H), 7,65 (s, 1H), 7,44 (m, 3H), 7,22 (d, J = 3,52 Hz, 1H), 6,55 (d, J = 5,48 Hz, 1H), 4,46 (m, 1H), 3,63 (s, 2H), 1,45 (d, J = 6,65 Hz, 6H). MS (m/z): 536,9 (M+H). Esquema 38
Exemplo 49 N 1-(3-Flúor-4-(2-(1 -propil-1 H-imidazol-4-il)tieno[3,2-b ]piridin-7- ilóxi)fenil)-N 3-(2-fluorofenil)malonamida (130) Etapa 1: 7-Cloro-2-(1-propil-1 H-imidazol-4-il)tieno[3,2-b ]piridina (131)
[00631] A uma solução de cloreto 2 (Esquema 1) (4,38 g, 25,84 mmols) em THF (120 mL) a -78°C foi lentamente adicionado n-BuLi (hexano a 2,5M, 12,9 mL, 32,31 mmols). A mistura de reação foi agitada durante uma hora a -78°C seguido por lenta adição de ZnCl2 (THF a 0,5M, 64,6 mL, 32,31 mmols). Após alguns minutos a mistura de reação foi deixada aquecer até a temperatura ambiente e agitada durante uma hora.
[00632] A uma solução de 4-iodo-1-propil-1H-imidazol (3,05 g, 12,92 mmols) [Tet. Lett. 2004, 45, 5529] em THF (5 mL) foi adicionado Pd(PPh3)4 (0,74 g, 0,64 mmol) e a mistura de reação que foi aquecida até o refluxo durante 2,5 horas, resfriada até a temperatura ambiente, em seguida diluída com hidróxido de amônio aquoso. A solução foi extraída com EtOAc, o extrato foi lavado com água e salmoura, em seguida secado sobre sulfato de magnésio anidro, filtrado e evaporado sob pressão reduzida. O resíduo foi purificado por cromatografia instantânea (eluentes DCM, em seguida DCM-MeOH, 97:3, 95:5) para fornecer o composto do título 131 (3,37 g, 47% de rendimento) como um sólido amarelo. MS (m/z): 278,0 (M+H). Etapa 2: 7-(2-Flúor-4-nitrofenóxi)-2-(1-propil-1 H-imidazol-4-il)tieno[3,2- b ]piridina (132)
[00633] A uma solução de 131 (3,37 g, 12,13 mmols) em Ph2O (40 mL) foi adicionado 2-flúor-4-nitrofenol (3,81 g, 24,26 mmols) e carbonato de potássio (6,70 g, 48,52 mmols). A mistura de reação foi aquecido para 195°C durante 20 horas, em seguida resfriada até a temperatura ambiente. O resíduo foi purificado por cromatografia de coluna, eluentes EtOAc/Hex (9/1 a 5/5), em seguida MeOH/DCM (98/2), para fornecer o composto do título 132 (4,13 g, 85% de rendimento) como um sólido amarelo. MS (m/z): 399,0 (M+H). Etapa 3: 3-Flúor-4-(2-(1 -propil-1 H-imidazol-4-il)tieno[3,2-b ]piridin-7- ilóxi)anilina (133)
[00634] A uma solução do composto nitro 132 (4,13 g, 10,36 mmols) em MeOH/THF (100 mL/100 mL) foi adicionado NiCl2 x 6H2O (4,92 g, 20,73 mmols) e NaBH4 (1,54 mg, 41,44 mmols). A mistura de reação foi deixada agitar durante 1 hora, concentrada até a secura e o sólido resultante foi dissolvido em HCl a 2 M. A solução acídica foi então tornada básica com solução de hidróxido de amônio aquoso e extraída com EtOAc. O extrato orgânico foi seco sobre sulfato de sódio anidro, filtrado e evaporado. O resíduo foi purificado por cromatografia instantânea (eluentes DCM-MeOH, 98:2, 95:5) para fornecer o composto do título 133 (3,31 g, 86% de rendimento) como um sólido rosa. MS (m/z): 368,1 (M+H). Etapa 4: N 1-(3-Flúor-4-(2-(1 -propil-1 H-imidazol-4-il)tieno[3,2-b ]piridin-7- ilóxi) fenil)-N 3-(2-fluorofenil)malonamida (130)
[00635] A uma solução do composto de amino 133 (400 mg, 1,08 mmol) em DMF (20 mL), ácido 3-(2-fluorofenilamino)-3-oxopropanoico (29) (427 mg, 2,17 mmols), EDC (352 mg, 2,60 mmols) foi adicionado HOBT (499 mg, 2,60 mmols). A mistura de reação foi deixada agitar durante 1 hora. A solução foi extraída com EtOAc e o extrato foi lavado com água, cloreto de amônio aquoso e salmoura, em seguida secado sobre sulfato de magnésio anidro, filtrado e evaporado sob pressão reduzida. O resíduo foi purificado por cromatografia instantânea (eluentes DCM-MeOH, 98:2, 95:5) para fornecer o composto do título 130 (215 mg, 36% de rendimento) como um sólido amarelo. 1H RMN (DMSO-d6). δ (ppm): 10,56 (s, 1H), 10,06 (s, 1H), 8,43 (d, J = 5,6 Hz, 1H), 8,10-7,96 (m, 1H), 7,94 (d, J = 1,2 Hz, 1H), 7,87 (dd, J = 1,6 e 12,8 Hz, 1H), 7,77 (d, J = 1,2 Hz, 1H), 7,68 (s, 1H), 7,49 (t, J = 8,8 Hz, 1H), 7,42 (dd, J = 2,0 e 8,8 Hz, 1H), 7,32-7,25 (m, 1H), 7,22-7,14 (m, 2H), 6,58 (d, J = 5,6 Hz, 1H), 3,98 (t, J = 7,2 Hz, 2H), 3,62 (s, 2H), 1,78 (sex, J = 7,2 Hz, 2H), 0,87 (t, J = 7,2 Hz, 3H). MS (m/z): 548,1 (M + H). Exemplo 50 N 1-(3-flúor-4-(2-(1 -propil-1 H-imidazol-4-il)tieno[3,2-b]piridin-7- ilóxi)fenil)-N 3-(2-metoxifenil)malonamida (134)
[00636] O composto do título 134 (Esquema 38) foi obtido similarmente ao composto 130 (Exemplo 49, Esquema 38) partindo da amina 133 e substituindo o ácido 29 pelo ácido 27. 1H RMN (DMSO- de). δ (ppm): 10,58 (s, 1H), 9,64 (s, 1H), 8,43 (d, J = 5,6 Hz, 1H), 8,07 (dd, J = 1,2 e 8,8 Hz, 1H), 7,94 (d, J = 1,2 Hz, 1H), 7,87 (dd, J = 1,6 e 12,8 Hz, 1H), 7,77 (d, J = 1,2 Hz, 1H), 7,68 (s, 1H), 7,49 (t, J = 8,8 Hz, 1H), 7,43 (dd, J = 2,0 e 8,8 Hz, 1H), 7,11-7,04 (m, 2H), 6,92 (ddd,J = 2,4, 6,4 e 8,0 Hz, 1H), 6,58 (d, J = 5,6 Hz, 1H), 3,98 (t, J = 7,2 Hz, 2H), 3,86 (s, 3H), 3,64 (s, 2H), 1,78 (sex, J = 7,2 Hz, 2H), 0,87 (t, J = 7,2 Hz, 3H). MS (m/z): 559,2 (M+H). Esquema 39
Exemplo 51 N 1-(3-Flúor-4-(2-(pirrolidina-1-carbonil)tieno[3,2-b ]piridin-7-ilóxi)fenil)- N 3-(2-metoxifenil)malonamida (135)
[00637] Partindo de (7-(4-amino-2-fluorofenóxi)tieno[3,2-b]piridin-2- il)(pirrolidin-1-il)metanona (23, Esquema 6) e o ácido 27, e seguindo o mesmo procedimento como descrito acima para a síntese de composto 130 (Esquema 38, Exemplo 49) o composto do título 135 foi obtido como sólido branco em 51% de rendimento. 1H RMN (DMSO- d6). δ (ppm): 10,65 (s, 1H), 10,25 (s, 1H), 8,61 (d, J = 5,6 Hz, 1H), 8,03 (s, 1H), 7,92-7,86 (m, J = 13,2 Hz, 1H), 7,60 (d, J = 8,4 Hz, 2H), 7,50 (t, J = 8,4 Hz, 1H), 7,47-7,20 (m, 1H), 7,30 (t, J = 8,0 Hz, 2H), 7,04 (t, J = 7,2 Hz, 1H), 6,83 (d, J = 5,6 Hz, 1H), 3,54 (t, J = 6,4 Hz, 2H), 3,53 (s, 2H), 1,97 (quin, J = 6,4 Hz, 2H), 1,89 (quin, J = 6,4 Hz, 2H). MS (m/z): 549,2 (M+H). Esquema 40
Exemplo 52 N 1-(3-Flúor-4-(2-(3-metóxi-4-(2-morfolinoetóxi)fenil)tieno[3,2-b ]piridin-7- ilóxi)fenil)-N2-(2-metoxifenetil)oxalamida (136) Etapa 1 4-(7-(2-Flúor-4-nitrofenóxi)tieno[3,2-b ]piridin-2-il)-2- metoxifenol (137)
[00638] Partindo do composto nitro 50 e 2-metóxi-4-(4,4,5,5- tetrametil-1,3,2-dioxaborolan-2-il)fenol, e seguindo o mesmo procedimento como descrito para a síntese do composto 63 (Esquema 22) o composto do título 137 foi obtido como um sólido marrom-claro em 72% de rendimento. MS (m/z): 413,1 (M+H). Etapa 2: 4-(2-(4-(7-(2-Flúor-4-nitrofenóxi)tieno[3,2-b ]piridin-2-il)-2- metoxifenóxi)etil) morfolina (138)
[00639] DEAD (0,96 mL, 6,07 mmols) foi adicionado a uma solução do composto 137 (1,78 g, 4,33 mmols), 2-morfolinoetanol (0,74 mL, 6,07 mmols) e trifenilfosfina (1,59 g, 6,07 mmols) em THF (43 mL). A mistura de reação foi agitada em temperatura ambiente até a conclusão da reação. A mistura foi extinguida com solução aquosa saturada de cloreto de amônio, em seguida extraída três vezes com acetato de etila e 3 vezes com diclorometano (com um pouquinho de metanol dentro). As camadas orgânicas combinadas foram lavadas com água e salmoura, secadas sobre MgSO4 anidro, filtradas e evaporadas. O resíduo foi triturado com éter de dietila, filtrado e secado em uma bomba a vácuo para fornecer o composto do título 138 como um sólido marrom amarelado (1,80g, 79% de rendimento). MS (m/z): 526,2 (M+H). Etapa 3: 3-Flúor-4-(2-(3-metóxi-4-(2-morfolinoetóxi)fenil)tieno[3,2- b ]piridin-7-ilóxi)anilina (139)
[00640] Partindo do composto nitro 138 e seguindo o mesmo procedimento como descrito para a síntese da amina 23 (Esquema 6) o composto do título 139 foi obtido como sólido amarelo, em 99% de rendimento (produto bruto). MS (m/z): 496,3 (M+H). Etapa 4: 2-(3-Flúor-4-(2-(3-metóxi-4-(2-morfolinoetóxi)fenil)tieno[3,2- b ]piridin-7-ilóxi)fenilamino)-2-oxoacetato de etila (140)
[00641] Clorooxoacetato de etila (0,11 mL, 0,95 mmol) foi adicionado a uma solução da amina 139 (313 mg, 0,63 mmol) e trietilamina (0,18 mL, 1,26 mmol) em diclorometano (16 mL). A mistura de reação foi agitada em temperatura ambiente até a conclusão da reação. A mistura foi extinguida com uma solução aquosa saturada de cloreto de amônio e extraída três vezes com acetato de etila. As camadas orgânicas combinadas foram lavadas com salmoura, secadas sobre MgSO4 anidro, filtradas e evaporadas. O produto bruto foi purificado por cromatografia instantânea (eluente: 5% MeOH/95% DCM a 7% MeOH/93% DCM) para fornecer o composto do título 140 como um sólido amarelo (178 mg, 47% de rendimento). MS (m/z): 596,1 (M+H). Etapa 5: N 1-(3-Flúor-4-(2-(3-metóxi-4-(2-morfolinoetóxi)fenil)tieno[3,2- b ]piridin-7-ilóxi)fenil)-N2-(2-metoxifenetil)oxalamida (136)
[00642] Éster de amino 140 (80 mg, 0,13 mmol) e 2-(2- metoxifenil)etanamina (0,2 mL, 1,34 mmol) foram misturados entre si e agitados em temperatura ambiente até a conclusão da reação. A mistura extinguida com uma solução aquosa saturada de cloreto de amônio e extraída três vezes com diclorometano. As camadas orgânicas combinadas foram lavadas com salmoura, secadas sobre MgSO4 anidro, filtradas e evaporadas. O produto bruto foi purificado por cromatografia instantânea (eluente: 3% MeOH/97% DCM a 5% MeOH/95% DCM). Este produto sólido foi então triturado com acetato de etila para fornecer o composto do título puro 136 como um sólido branco (52 mg, 57% de rendimento). 1H RMN (DMSO-d6) δ (ppm): 11,03 (s, 1H), 9,04 (t, J = 5,9 Hz, 1H), 8,48 (d, J = 5,5 Hz, 1H), 8,058,01 (m, 2H), 7,82 (d, J = 9,0 Hz, 1H), 7,53 (t, J = 9,1 Hz, 1H), 7,47 (d, J = 2,2 Hz, 1H), 7,38 (dd, J = 2,2 e 8,2 Hz, 1H), 7,21 (td, J = 1,8 e 7,8 Hz, 1H), 7,14 (dd, J = 1,8 e 7,4 Hz, 1H), 7,11 (d, J = 8,6 Hz, 1H), 6,97 (d, J = 7,4 Hz, 1H), 6,88 (td, J = 1,0 e 7,3 Hz, 1H), 6,60 (d, J = 5,5 Hz, 1H), 4,14 (t, J = 6,0 Hz, 2H), 3,89 (s, 3H), 3,80 (s, 3H), 3,59-3,57 (m, 4H), 3,46-3,41 (m, 2H), 2,83 (t, J = 7,0 Hz, 2H), 2,73-2,70 (m, 2H), um pico (4H) não mostrou-se, ele foi provavelmente sobre H2O ou DMSO. MS (m/z): 701,1 (M+H). Esquema 41

Exemplo 53 N 1-(4-(2-(1 -Etil-1 H-imidazol-4-il)tieno[3,2-b ]piridin-7-ilóxi)-3-fluorofenil)- N2 -(2-metoxifenetil)oxalamida (141) Etapa 1: 2-(3-flúor-4-(2-(1 -etil-1 H-imidazol-4-il)tieno[3,2-b ]piridin-7- ilóxi)fenilamino)-2-oxoacetato de etila (142)
[00643] Partindo da amina 15 (Esquema 4) e seguindo o mesmo procedimento como descrito para a síntese do composto 140 (Esquema 40), o composto do título 142 foi obtido como sólido branco em 14% de rendimento. MS (m/z): 455,1 (M+H). Etapa 2: N 1-(4-(2-(1-Etil-1 H-imidazol-4-il)tieno[3,2-b]piridin-7-ilóxi)-3- fluorofenil)-N 2-(2-metoxifenetil)oxalamida (141)
[00644] Partindo do éster de amino 142 e seguindo o mesmo procedimento como descrito para a síntese do composto 136 (Exemplo 52, Esquema 40), o composto do título 141 foi obtido como sólido branco em 90% de rendimento. 1H RMN (DMSO-d6) δ (ppm): 11,04 (s, 1H), 9,06 (t, J = 6,4 Hz, 1H), 8,43 (d, J = 5,6 Hz, 1H), 8,02 (dd, J = 2,4 e 12,8 Hz, 1H), 7,97 (d, J = 1,2 Hz, 1H), 7,84-7,78 (m, 1H), 7,79 (d, J = 1,2 Hz, 1H), 7,67 (s, 1H), 7,52 (t, J = 5,2 Hz, 1H), 7,20 (td, J = 1,6 e 8,0 Hz, 1H), 7,14 (dd, J = 1,6 e 7,2 Hz, 1H), 6,96 (d, J = 8,0 Hz, 1H), 6,87 (t, J = 7,2 Hz, 1H),6,58 (d, J = 5,6 Hz, 1H), 4,06 (q, J = 7,2 Hz, 2H), 3,79 (s, 3H), 3,42 (q, J = 7,2 Hz, 2H), 2,82 (t, J = 7,2 Hz, 2H), 1,40 (7,2 Hz, 3H). MS (m/z): 560,2 (M+H). Exemplo 54 N 1-(4-(2-(1 -Etil-1 H-imidazol-4-il)tieno[3,2-b ]piridin-7-ilóxi)-3-fluorofenil)- N 3-(2-fluorofenetil)oxalamida (143)
[00645] O composto do título 143 foi obtido similarmente ao composto 141 (Exemplo 53) de acordo com o Esquema 41. 1H RMN (DMSO-d6) δ (ppm): 11,04 (s, 1H), 9,16 (t, J = 6,0 Hz, 1H), 8,43 (d, J = 5,6 Hz, 1H), 8,02 (dd, J = 2,4 e 12,8 Hz, 1H), 7,96 (d, J = 1,2 Hz, 1H), 7,84-7,79 (m, 1H), 7,79 (d, J = 1,2 Hz, 1H), 7,68 (s, 1H), 7,52 (t, J = 8,8 Hz, 1H), 7,34-7,24 (m, 2H), 7,19-7,11 (m, 2H), 6,58 (d, J = 5,6 Hz, 1H), 4,06 (q, J = 7,2 Hz, 2H), 3,46 (q, J = 7,2 Hz, 2H), 2,88 (t, J = 7,2 Hz, 2H), 1,40 (t, J = 7,2 Hz, 3H). Esquema 42
Exemplo 55 N 1-(3-Flúor-4-(2-(1 -metil-1 H-imidazol-4-il)tieno[3,2-b ]piridin-7- ilóxi)fenil)-N2-(2-metoxifenetil)oxalamida (144) Etapa 1: 2-(3-flúor-4-(2-(1 -metil-1 H-imidazol-4-il)tieno[3,2-b]piridin-7- ilóxi)fenilamino)-2-oxoacetato de etila (145)
[00646] Partindo da amina 12 (Esquema 3) e seguindo o mesmo procedimento como descrito para a síntese do composto 140 (Esquema 40), o composto do título 145 foi obtido como um sólido branco em 14% de rendimento. MS (m/z): 441,1 (M+H). Etapa 2: N 1-(3-Flúor-4-(2-(3-metóxi-4-(2-morfolinoetóxi)fenil)tieno[3,2- b ]piridin-7-ilóxi)fenil)-N2-(2-metoxifenetil)oxalamida (144)
[00647] Partindo do éster de amino 145 e seguindo o mesmo procedimento como descrito para a síntese do composto 136 (Exemplo 52, Esquema 40), o composto do título 144 foi obtido como sólido branco em 33% de rendimento. 1H RMN (DMSO-d6) δ (ppm): 11,03 (s, 1H), 9,04 (t, J = 5,6 Hz, 1H), 8,43 (d, J = 5,6 Hz, 1H), 8,02 (dd, J = 2,4 e 12,8 Hz, 1H), 7,86 (s, 1H), 7,84-7,78 (m, 1H), 7,72 (s, 1H), 7,68 (s, 1H), 7,51 (t, J = 8,8 Hz, 1H), 7,20 (td, J = 1,6 e 7,6 Hz, 1H), 7,14 (dd, J = 1,6 e 7,6 Hz, 1H), 6,96 (d, J = 7,6 Hz, 1H), 6,87 (t, J = 7,6 Hz, 1H), 6,58 (d, J = 5,6 Hz, 1H), 3,799s, 3H), 3,72 (s, 3H), 3,43 (q, J = 6,8 Hz, 2H), 2,82 (t, J = 6,8 Hz, 2H). MS (m/z): 546,2 (M+H). Esquema 43
Exemplo 56 N 1-(4-(2-(3-((Dimetilamino)metil)fenil)tieno[3,2-b ]piridin-7-ilóxi)-3- fluorofenil)-N 3-(2-metoxifenil)malonamida (146) Etapa 1: (3-(7-(2-Flúor-4-nitrofenóxi)tieno[3,2-b ]piridin-2-il)fenil)metanol (147)
[00648] Partindo do composto nitro 50 e ácido 3- (hidroximetil)fenilborônico, e seguindo o mesmo procedimento como descrito para a síntese do composto 63 (Esquema 22) o composto do título 147 foi obtido como um sólido marrom-bege em 71% de rendimento. MS (m/z): 397,0 (M+H). Etapa 2: 2-(3-(Clorometil)fenil)-7-(2-flúor-4-nitrofenóxi)tieno[3,2- b ]piridina (148)
[00649] O composto de hidróxi 147 (685 mg, 1,73 mmol) foi suspenso em cloreto de tionila (8,6 mL) e a mistura de reação foi refluxada durante cerca de uma hora. A mistura foi resfriada para a TA, em seguida despejada em uma mistura de gelo/água. O sólido resultante foi coletado por filtração, lavado com água e bem secado para fornecer o composto do título 148 como um sólido amarelo (730 mg, 93% de rendimento). MS (m/z): 415,0 (M+H). Etapa 3: 1 -(3-(7-(2-Flúor-4-nitrofenóxi)tieno[3,2-b ]piridin-2-il)fenil)-N, N- dimetilmethanamina (149)
[00650] A uma suspensão do cloreto 148 (3,8 g, 8,42 mmols) em dimetilformamida (42 mL) foi adicionado dimetilamina (8,4 mL, 16,84 mmols) e a mistura de reação foi aquecida para 60°C. Após poucas horas a reação foi completada e a dimetilformamida foi evaporada. O resíduo foi triturado com acetato de etila, coletado por filtração, lavado com acetato de etila e secado para fornecer o composto do título 149 como um sólido amarelo (1,71g, 48% de rendimento). MS (m/z): 424,0 (M+H). Etapa 4: 4-(2-(3-((Dimetilamino)metil)fenil)tieno[3,2-b]piridin-7-ilóxi)-3- fluoroanilina (150)
[00651] Partindo do composto 149 e seguindo o mesmo procedimento como descrito acima para a síntese de composto 23 (Esquema 6), o composto do título 150 foi obtido em 44% de rendimento MS (m/z): 394,0 (M+H). Etapa 5: N 1-(4-(2-(3-((Dimetilamino)metil)fenil)tieno[3,2-b ]piridin-7- ilóxi)-3-fluorofenil)-N 3-(2-metoxifenil)malonamida (146)
[00652] A uma solução da amina 150 (334 mg, 0,85 mmol), ácido 3- (2-metoxifenilamino)-3-oxopropanoico (27) (355 mg, 1,70 mmol), 1- hidroxibenzotriazol (275 mg, 2,04 mmols) em dimetilformamida (8,5 mL) foi adicionado cloridrato de N-(3-dimetilaminopropil)-N- etilcarbodiimida (391 mg, 2,04 mmols) e a mistura de reação foi agitada em temperatura ambiente até a conclusão da reação. A dimetilformamida foi evaporada e o resíduo foi extinguido com uma solução saturada de bicarbonato de sódio. A camada aquosa foi extraída três vezes com acetato de etila. As camadas orgânicas combinadas foram lavadas com água e salmoura, secadas sobre MgSO4 anidro, filtradas e evaporadas para fornecer o composto do título 146 como um sólido branco (180 mg, 36% de rendimento). 1H RMN (DMSO-d6) δ (ppm): 10,59 (s, 1H), 9,64 (s, 1H), 8,52-8,50 (m, 1H), 8,08-8,06 (m, 2H), 7,88 (d, J = 12,9 Hz, 1H), 7,78 (m, 2H), 7,51-7,38 (m, 4H), 7,07 (s, 2H), 6,92 (m, 1H), 6,65 (m, 1H), 3,86 (s, 3H), 3,65 (s, 2H), 3,48 (s, 2H), 2,18 (s, 6H). MS (m/z): 585,2 (M+H).
[00653] Compostos 151 (Exemplo 57), 152 (Exemplo 58), 153 (Exemplo 59), 154 (Exemplo 60) e 155 (Exemplo 61), preparados de acordo com o Esquema 43. Tabela 4
Esquema 44
Exemplo 62 N1 -(4-(2-(1 -etil-1H-imidazol-4-il)tieno[3,2-b ]piridin-7-ilóxi)-3-fluorofenil)- N3-(4-metoxifenil)malonamida (156) Etapa 1: Ácido 3-(4-Metoxifenilamino)-3-oxopropanoico (157)
[00654] Partindo de 4-metoxianilina e seguindo o procedimento descrito acima para a síntese de composto 27 (Exemplo 8, Esquema 8), o composto do título 157 foi obtido em 56% de rendimento. MS (m/z): 210,0 (M+H). Etapa 2: N1 -(4-(2-(1-Etil-1H-imidazol-4-il)tieno[3,2-b]piridin-7-ilóxi)-3- flúor fenil)-N3 -(4-metoxifenil)malonamida (156)
[00655] Partindo da amina 15 (Esquema 4) e seguindo o procedimento descrito acima para a síntese de composto 28a (Exemplo 8, Etapa 2, Esquema 8) o composto do título 156 foi obtido em 42% de rendimento. 1H RMN (DMSO-d6) δ (ppm): 10,54 (s, 1H), 10,07 (s, 1H), 8,40 (d. J = 5,3 Hz, 1H), 7,94 (d, J = 1,2Hz, 1H), 7,64 (dd, J= 2,4 e 13,1 Hz, 1H), 7,77 (d, J = 1,2 Hz, 1H), 7,65 (s, 1H), 7,517,49 (m, 2H), 7,46 (d, J = 8,8 Hz, 1H), 7,41 (dd, J = 1,0 e 9,0 Hz, 1H), 6,89-6,87 (m, 2H), 6,56 (d, J = 5,5 Hz, 1H), 4,05 (q, 2H), 3,70 (s, 3H), 3,45 (s, 2H), 1,42 (t, 3H). MS (m/z): 546,0 (M+H). Esquema 45
Exemplo 63 N-(4-(2-(1 -Etil-1 H-imidazol-4-il)tieno[3,2-b ]piridin-7-ilóxi)-3-fluorofenil)- N-fenilciclopropano-1,1 -dicarboxamida (162a) Etapa 1: Ácido 1-(fenilcarbamoil)ciclopropanocarboxílico (161)
[00656] A uma solução de diácido 159 (2,5g, 19,2 mmols) em THF (500 mL), foi adicionado Et3N (1,40 mL, 19,2 mmols) em gotas sob nitrogênio e a mistura foi agitada durante 30 minutos a 0°C antes da adição de cloreto de tionila (2,68 mL, 19,2 mmols). A mistura de reação foi agitada durante mais 30 minutos a 0°C [para gerar in situ o cloreto de acila 160], seguido pela adição de uma solução de anilina (2,22 mL, 21,2 mmols) em THF (25 mL). A mistura de reação foi agitada durante 4 horas a 0°C, em seguida diluída com EtOAc e extraída três vezes com solução de NaOH a 2N. A fase aquosa foi titulada com solução de HCl a 2N para pH ~1-2, e em seguida extraída com EtOAc. A fase orgânica foi seca com Na2SO4 e concentrada sob vácuo para fornecer o composto do título (161) (2,86 g, 72% de rendimento) como um sólido branco. MS (m/z): 206,0 (M+H). Etapa 2: N-(4-(2-(1-Etil-1 H-imidazol-4-il)tieno[3,2-b ]piridin-7-ilóxi)-3- fluorofenil)-N-fenilciclopropano-1,1 -dicarboxamida (158)
[00657] Partindo da amina 15 e o ácido 161, e seguindo o procedimento descrito acima para a síntese do composto 28a (Exemplo 8, Etapa 2, Esquema 8) o composto do título 158 foi obtido em 65% de rendimento. 1H RMN (DMSO-d6) δ (ppm): 10,35 (s, 1H), 9,99 (s, 1H), 8,42 (d, J = 5,5 Hz, 1H), 7,94 (d, J = 1,2 Hz, 1H), 7,88 (dd, J= 2,1 e 14,1 Hz, 1H), 7,77 (d, J = 1,2 Hz, 1H), 7,66 (s, 1H), 7,61 (dd, J = 2,2 e 8,8 Hz, 2H), 7,49 (dd, J = 1,9 e 8,8 Hz, 2H), 7,45 (t, 1H, J = 8,8 Hz), 7,31-7,26 (m, 2H), 7,05 (t, 1H, J = 6,1 Hz), 6,52 (d, J = 5,4 Hz, 1H), 4,04 (q, 2H), 1,40 (s, 4H), 1,38 (t, 3H). MS (m/z): 542,1 (M+H). Esquema 46
Exemplo 64 N1 -(3-Flúor-4-(2-(1-(2-(pirrolidin-1-il)etil)-1 H-imidazol-4-il)tieno[3,2- b ]piridin-7-ilóxi)fenil)-N3 -metil-N3-fenilmalonamida (162) Etapa 1: 1-(2-Iodoetil)pirrolidina x HI (164)
[00658] Uma suspensão de trifenilfosfina (4,46 g, 17,7 mmols), imidazol (1,2 g, 17,7 mmols) e iodo (4,50 g, 17,7 mmols) em THF (90 mL) foi agitada durante 5 minutos antes de 2-(pirrolidin-1-il)etanol (163) (2 mL, 17,7 mmols) ser adicionado em gotas. A mistura de reação foi agitada durante 4 horas. O precipitado resultante foi coletado por filtração, lavado diversas vezes com EtOAc, e secado durante a noite para produzir o composto do título 164 (4,85 g, 76%) como um sólido branco. MS (m/z): 226,0 (M+H). Etapa 2: 2,4,5-Triiodo-1-(2-(pirrolidin-1-il)etil)-1 H-imidazol (166)
[00659] A uma solução de 2,4,5-tri-iodo-1H-imidazol (165) (4,08 g, 9,1 mmols) em DMF (28 mL) a 0°C, foi adicionado NaH (1,46 g, 36,4 mmols) em porções durante 30 minutos, então 1-(2-iodoetil)pirrolidina x HI 164 (4,85 g, 13,65 mmols) foi adicionado a 0°C, e a mistura de reação foi deixada aquecer até a temperatura ambiente durante um período de 4 horas. EtOAc (50 mL) foi adicionado e a mistura foi lavada com solução aquosa de NaHCO3. A fase orgânica foi separada e extraída com solução de ácido cítrico (3%), o extrato aquoso acídico foi basificado com solução de NaOH a 2N para pH ~10, o sólido resultante foi coletado por filtração para fornecer o composto do título 166 (1,75 g, 35%) como um sólido marrom-claro. MS (m/z): 543,5 (M+H). Etapa 3: 4-Iodo-1-(2-(pirrolidin-1-il)etil)-1 H-imidazol (167)
[00660] A uma solução de 2,4,5-tri-iodo-1-(2-(pirrolidin-1-il)etil)-1H- imidazol (166) (1,75 g, 3,22 mmols) em THF (32 mL) a -78°C foi adicionado tBuLi (7,57 mL, 12,88 mmols) em gotas durante um período de uma hora. A mistura de reação foi despejada em água, extraída com EtOAc, e o extrato orgânico foi separado, secado sobre Na2SO4, e concentrado a vácuo para fornecer o composto do título 167 (0,9 g, 96% de rendimento) como um xarope amarelo-escuro. 1H RMN (DMSO-d6) δD(ppm): 7,59 (s, 1H), 7,36 (s, 1H), 4,02 (m, 2H), 2,78 (m, 2H), 2,42 (m, 4H), 1,63 (m, 4H). MS (m/z): 292,1 (M+H). Etapa 4: 7-Cloro-2-(1-(2-(pirrolidin-1-il)etil)-1 H-imidazol-4-il)tieno[3,2- b]piridina (168)
[00661] Seguindo o procedimento descrito acima para a síntese de composto 123 (Esquema 36) porém substituindo 4-iodo-1-isopropil-1H- imidazol com o composto 167 o composto do título 168 foi obtido em 47% de rendimento como um sólido amarelo. MS (m/z): 333,0 (M+H). Etapa 5: 7-(2-Flúor-4-nitrofenóxi)-2-(1-(2-(pirrolidin-1-il)etil)-1 H- imidazol-4-il)tieno[3,2-b ]piridina (169)
[00662] Partindo do composto 168 e seguindo o procedimento descrito acima para a síntese de composto 124 (Esquema 36), o composto do título 169 foi obtido em 61% de rendimento como um sólido amarelo. MS (m/z): 454,0 (M+H). Etapa 6: 3-Flúor-4-(2-(1-(2-(pirrolidin-1-il)etil)-1 H-imidazol-4-il)tieno[3,2- b ]piridin-7-ilóxi)anilina (170)
[00663] A uma solução de composto nitro 169 (400 mg, 0,88 mmol) em EtOH/H2O (8 mL/4 mL) a 100°C (banho) foi adicionado pó de ferro (420 mg, 7,48 mmols) e NH4Cl (41 mg, 0,76 mmol) e a mistura de reação foi agitada vigorosamente em temperatura de refluxo durante uma hora. A mistura foi resfriada para temperatura ambiente e filtrada através de uma almofada de Celita. O filtrado foi coletado e concentrado para fornecer o composto do título 170 (4,20 mg, 88% de pureza) como um sólido amarelo. MS (m/z): 424,0 (M+H). Etapa 7: N 1-(3-flúor-4-(2-(1-(2-(pirrolidin-1-il)etil)-1 H-imidazol-4- il)tieno[3,2-b ]piridin-7-ilóxi)fenil)-N 3-metil-N3-fenilmalonamida (162).
[00664] Seguindo o procedimento descrito acima para o composto 28a (Exemplo 8, Etapa 2), porém substituindo a amina 9 pela amina 170, o composto do título 162 foi obtido em 47% de rendimento. 1H RMN (DMSO-d6) δ (ppm): 10,29 (s, 1H), 8,42 (d, J = 5,5 Hz, 1H), 7,94 (s,1H), 7,77 (m, 2H), 7,66 (s, 1H), 7,46 (m, 3H), 7,39 (m, 3H), 7,30 (m, 1H), 6,56 (d, J = 5,5 Hz, 1H), 4,11 (t, 2H), 3,19 (m, 4H), 3,14 (s, 3H), 2,77 (t, 2H), 1,65 (s, 4H). MS (m/z): 599,0 (M+H). Esquema 47
Exemplo 65 N-(4-(2-(1 -Etil-1 H-imidazol-4-il)tieno[3,2-b ]piridin-7-ilóxi)-3-fluorofenil)- N-(2-metoxifenil)ciclopropano-1,1 -dicarboxamida (171) Etapa 1: Ácido 1-(2-Metoxifenilcarbamoil)ciclopropanocarboxílico (172)
[00665] Seguindo o procedimento descrito acima para a síntese de composto 161 (Esquema 45), porém substituindo anilina por 2- metoxianilina, o composto do título 172 foi obtido em 44% de rendimento. M/S (m/z): 236,0 (M+H). Etapa 2: N-(4-(2-(1-etil-1 H-imidazol-4-il)tieno[3,2-b ]piridin-7-ilóxi)-3- fluorofenil)-N-(2-metoxifenil)ciclopropano-1,1 -dicarboxamida (171)
[00666] Partindo da amina 15 e seguindo o procedimento descrito acima para a síntese de composto 28a (Exemplo 8, Etapa 2, Esquema 8), porém substituindo o ácido 27 pelo ácido 172, o composto do título 171 foi obtido em 33% de rendimento. 1H RMN (DMSO-d6) δ (ppm): 10,29 (s, 1H), 10,11 (s, 1H), 8,42 (d, J = 5,5 Hz, 1H), 8,04 (d, 1H, J = 7,2 Hz), 7,94 (s,1H), 7,82 (d, J = 12,5 Hz, 1H), 7,77 (s, 1H), 7,66 (s, 1H), 7,50 (m, 2H), 7,05 (m, 2H), 6,90 (t, 1H), 6,56 (d, J = 5,5 Hz, 1H), 4,04 (q, 2H), 3,81 (s, 3H), 1,59 (s, 2H), 1,55 (s, 2H), 1,38 (t, 3H). MS (m/z): 572,0 (M+H). Exemplo 66 N-(3-Flúor-4-(2-(1 -(2-(pirrolidin-1-il)etil)-1 H-imidazol-4-il)tieno[3,2- b ]piridin-7-ilóxi)fenil)-N-(2-metoxifenil)ciclopropano-1,1 -dicarboxamida (173)
[00667] Seguindo o procedimento descrito acima para o composto 28a (Exemplo 8, Etapa 2, Esquema 8), porém substituindo a amina 9 pela amina 65 e ácido 27 pelo ácido 172, o composto do título 173 foi obtido em 11% de rendimento. 1H RMN (DMSO-d6) δα (ppm): 10,29 (s, 1H), 10,12 (s, 1H), 8,42 (d, J = 5,5 Hz, 1H), 8,12 (s, 1H), 8,02 (d, J = 7,6 Hz,1H), 7,92 (s, 1H), 7,82 (d, J = 13,7 Hz, 1H), 7,77 (s, 1H), 7,66 (s, 1H), 7,50 (m, 2H), 7,05 (m, 2H), 6,90 (m, 1H), 6,56 (d, J = 5,5 Hz, 1H), 4,13 (t, 2H), 3,81 (s, 3H), 2,84 (s, 2H), 2,53 (s, 2H), 2,47 (s, 2H), 1,67 (m, 4H), 1,59 (m, 2H), 1,55 (m, 2H). MS (m/z): 641,0 (M+H). Esquema 48
Exemplo 67 N-(3-Flúor-4-(2-(1 -metil-1 H-imidazol-4-il)tieno[3,2-b ]piridin-7-ilóxi)fenil)- N-(4-metoxifenil)ciclopropano-1,1 -dicarboxamida (174) Etapa 1: Ácido 1-(2-metoxifenilcarbamoil)ciclopropanocarboxílico (175)
[00668] Seguindo o procedimento descrito acima para o composto 161 (Esquema 45), porém substituindo anilina por 4-metoxianilina, o composto do título 175 foi obtido em 68% de rendimento. M/S (m/z): 236,0 (M+H). Etapa 2: N-(3-Flúor-4-(2-(1 -metil-1 H-imidazol-4-il)tieno[3,2-b]piridin-7- ilóxi) fenil)-N-(4-metoxifenil)ciclopropano-1,1-dicarboxamida (174)
[00669] Seguindo o procedimento descrito acima para o composto 5c (Esquema 3), porém substituindo o ácido de amina 1 pelo ácido 175, o composto do título 174 foi obtido em 44% de rendimento. 1H RMN (DMSO-d6) δ(ppm): 10,44 (s, 1H), 9,78 (s, 1H), 8,42 (d, J = 5,5 Hz, 1H), 7,87 (dd, J = 1,2 e 7,4 Hz, 1H), 7,84 (s,1H), 7,70 (s, 1H), 7,66 (s, 1H), 7,49 (m, 3H), 7,42 (t, J = 9,0 Hz, 1H), 6,87 (m, 2H), 6,53 (d, J = 5,5 Hz, 1H), 3,71 (s, 3H), 1,44 (t, 3H). MS (m/z): 558,0 (M+H). Esquema 49
Exemplo 68 N-(3-Flúor-4-(2-(1 -metil-1 H-imidazol-4-il)tieno[3,2-b ]piridin-7-ilóxi)fenil)- N-(2-fluorofenil)ciclopropano-1,1 -dicarboxamida (176) Etapa 1: Ácido 1-(2-fluorofenilcarbamoil)ciclopropanocarboxílico (68)
[00670] Seguindo o procedimento descrito acima para o composto 161 (Esquema 45), porém substituindo anilina por 2-flouroanilina, o composto do título 177 foi obtido em 57% de rendimento. M/S (m/z): 224,0 (M+H). Etapa 2: N-(3-Flúor-4-(2-(1 -metil-1 H-imidazol-4-il)tieno[3,2-b]piridin-7- ilóxi)fenil)-N-(4-metoxifenil)ciclopropano-1,1 -dicarboxamida (176)
[00671] Partindo da amina 12, seguindo o procedimento descrito acima para a síntese de composto 5c (Esquema 3), porém substituindo o ácido 1 pelo ácido 177, o composto do título 176 foi obtido em 30% de rendimento. 1H RMN (DMSO-d6) δ(ppm): 10,34 (s, 1H), 10,26 (s, 1H), 8,42 (d, J = 5,5 Hz, 1H), 7,86-7,82 (m, 3H), 7,70 (s, 1H), 7,66 (s, 1H), 7,51-7,43 (m, 2H), 7,28-7,23 (m, 1H), 7,18-7,15 (m, 2H), 6,55 (d, J = 5,5 Hz, 1H), 3,71 (s, 3H), 1,60-1,53 (m, 4H). MS (m/z): 546,1 (M+H). Exemplo 69 N-(4-(2-(1 -etil-1 H-imidazol-4-il)tieno[3,2-b ]piridin-7-ilóxi)-3-fluorofenil)- N-(2-fluorofenil)ciclopropano-1,1 -dicarboxamida (178)
[00672] Partindo da amina 15, seguindo o procedimento descrito acima para a síntese de composto 5d (Esquema 4) porém substituindo o ácido 1 pelo ácido 177, o composto do título 178 foi obtido em 13% de rendimento. 1H RMN (DMSO-d6) δα(ppm): 10,34 (s, 1H), 10,26 (s, 1H), 8,42 (d, J = 5,5 Hz, 1H), 7,95 (s, 1H), 7,86-7,83 (m, 2H), 7,77 (d, 1H, J = 1,2 Hz), 7,66 (s, 1H), 7,51-7,43 (m, 2H), 7,28-7,23 (m, 1H), 7,17-7,15 (m, 2H), 6,54 (d, J = 5,5 Hz, 1H), 4,04 (q, 2H), 1,60-1,53 (m, 4H), 1,38 (t, 3H). MS (m/z): 560,2 (M+H). Exemplo 70 N-(3-Flúor-4-(2-(1-(2-(pirrolidin-1-il)etil)-1 H-imidazol-4-il)tieno[3,2- b ]piridin-7-ilóxi)fenil)-N-(2-fluorofenil)ciclopropano-1,1 -dicarboxamida (179)
[00673] Partindo da amina 170 (Esquema 46), seguindo o procedimento descrito acima para o composto 28a (Exemplo 8, Etapa 2), porém substituindo o ácido 27 pelo ácido 177, o composto do título 179 foi obtido em 11% de rendimento. 1H RMN (DMSO-d6) δα (ppm): 10,28 (s, 1H), 10,26 (s, 1H), 8,42 (d, J = 5,5 Hz, 1H), 7,92 (d, J = 1,1 Hz, 1H), 7,86 (d, J = 1,9Hz, 1H), 7,83-7,79 (m, 1H), 7,76 (d, 1H, J = 1,4 Hz), 7,66 (s, 1H), 7,49 (dd, J = 2,1 e 9,0 Hz, 1H),7,45 (t, J = 8,6 Hz, 1H), 7,28-7,23 (m, 1H), 7,17-7,14 (m, 2H), 6,54 (d, 1H, J = 5,5 Hz), 4,12 (q, 2H), 2,76 (t, 2H), 2,45 (m, 2H), 1,67-1,64 (m, 4H), 1,60-1,53 (m, 4H). MS (m/z): 629,1 (M+H). Esquema 50
Exemplo 71 N-(3-Flúor-4-(2-(1 -metil-1 H-imidazol-4-il)tieno[3,2-b ]piridin-7-ilóxi)fenil)- N-(4-fluorofenil)ciclopropano-1,1 -dicarboxamida (180) Etapa 1: Ácido 1-(4-fluorofenilcarbamoil)ciclopropanocarboxílico (181)
[00674] Seguindo o procedimento descrito acima para o composto 161 (Esquema 45), porém substituindo anilina por 4-fluoroanilina, o composto do título 181 foi obtido em 57% de rendimento. MS (m/z): 224,0 (M+H). Etapa 2: N-(3-Flúor-4-(2-(1 -metil-1 H-imidazol-4-il)tieno[3,2-b]piridin-7- ilóxi) fenil)-N-(4-fluorofenil)ciclopropano-1,1 -dicarboxamida (180) 3
[00675] Partindo da amina 12 (Esquema 3) e seguindo o procedimento descrito acima para o composto 5c (Exemplo 3, Esquema 3), porém substituindo o ácido 1 pelo ácido 181, o composto do título 180 foi obtido em 24% de rendimento. 1H RMN (DMSO-d6) δ (ppm): 10,39 (s, 1H), 10,01 (s, 1H), 8,42 (d, J = 5,5 Hz, 1H), 7,87 (dd, J = 14,2 e 2,4 Hz, 1H), 7,85 (d, 1H, J = 1,2 Hz), 7,70 (d, 1H, J = 1,1 Hz), 7,66 (s, 1H), 7,64-7,61 (m, 2H), 7,50 (dd, J = 2,0 e 8,8 Hz, 1H), 7,43 (t, 1H, J = 8,8 Hz), 7,16-7,11 (m, 2H), 6,55 (d, 1H, J = 5,5 Hz), 3,71 (s, 3H), 1,44 (m, 4H). MS (m/z): 546,0 (M+H). Esquema 51
N-(3-Flúor-4-(2-(4-((metil(2-(4-metilpiperazin-1-il)etil)amino)metil) fenil)tieno[3,2-b ]piridin-7-ilóxi)fenil)-N-(4-fluorofenil)ciclopropano-1,1- dicarboxamida (182)
[00676] A uma solução do composto de amina 80 (189 mg, 0,374 mmol) (Esquema 26) em DMF (~7 mL) foram adicionados o ácido 181 (125 mg, 1,5 eq, 5,61 mmols), iPr2NEt (16 mg, 3,5 eq, 1,31 mmol) e HATU (426 mg, 3 eq, 1,12 mmol). A mistura de reação foi agitada a TA durante 24 horas. A mistura de reação foi concentrada até a secura e dividida entre solução de NaHCO3 saturada e EtOAc. O EtOAc foi lavado duas vezes com solução de NaHCO3 saturada antes de ser secado sobre Na2SO4 anidro e filtrado. O solvente foi removido sob pressão reduzida e a mistura bruta foi purificada inicialmente por cromatografia de coluna (MeOH a 25%/EtOAc + 1% de solução de NH4OH) e então usando o Gilson (30% de MeOH/H2O a 80 % de MeOH/H2O durante 45 minutos) para fornecer o composto do título 182 como um sólido branco (64 mg, 24% de rendimento). 1H RMN (DMSO-d6) δ (ppm): 8,48 (d, J = 5,48 Hz, 1H), 8,02 (s, 1H), 7,90 (dd, J = 1,77 e 12,91 Hz, 1H), 7,82 (d, J = 8,22 Hz, 2H), 7,62 (m, 2H), 7,42 (m, 4H), 7,13 (t, J = 8,99 Hz, 2H), 6,58 (d, J = 5,48 Hz, 1H), 3,51 (s, 1H), 2,40 (m, 12H), 2,14 (s, 3H), 2,10 (s, 3H), 1,44 (m, 4H). MS (m/z): 711,1 (M+H). (sal de formiato) Esquema 52

Exemplo 73 N1 -(4-(2-(1 -(2-(Dietilamino)etil)-1 H-imidazol-4-il)tieno[3,2-b ]piridin-7- ilóxi)-3-fluorofenil)-N3 -metil-N3-fenilmalonamida (183) Etapal: N, N-Dietil-2-iodoetanamina (184)
[00677] Seguindo o procedimento descrito acima para a síntese de composto 164 (Esquema 46), o composto do título 184 foi obtido em 42% de rendimento. MS (m/z): 228,0 (M+H). Etapa 2: N, N-Dietil-2-(2,4,5-tri-iodo-1 H-imidazol-1-il)etanamina (185)
[00678] Seguindo o procedimento descrito acima para a síntese de composto 166 (Esquema 46), o composto do título 185 foi obtido em 62% de rendimento. MS (m/z): 546,0 (M+H). Etapa 3: N, N-Dietil-2-(4-iodo-1 H-imidazol-1-il)etanamina (186)
[00679] Seguindo o procedimento descrito acima para a síntese de composto 167 (Esquema 46), o composto do título 186 foi obtido em 97% de rendimento. MS (m/z): 294,0 (M+H). Etapa 4: 2-(4-(7-Clorotieno[3,2-b ]piridin-2-il)-1 H-imidazol-1-il)-N, N- dietiletanamina (187)
[00680] Seguindo o procedimento descrito acima para a síntese de composto 168 (Esquema 46), o composto do título 187 foi obtido em 78% de rendimento. MS (m/z): 335,0 (M+H). Etapa 5: N, N-Dietil-2-(4-(7-(2-flúor-4-nitrofenóxi)tieno[3,2-b ]piridin-2-il)- 1H-imidazol-1-il)etanamina (188)
[00681] Seguindo o procedimento descrito acima para a síntese de composto 169 (Esquema 46), o composto do título 188 foi obtido em 10% de rendimento. MS (m/z): 456,0 (M+H). Etapa 6: 4-(2-(1 -(2-(Dietilamino)etil)-1 H-imidazol-4-il)tieno[3,2-b ]piridin- 7-ilóxi)-3-fluoroanilina (189)
[00682] Seguindo o procedimento descrito acima para a síntese de composto 170 (Esquema 46), o composto do título 189 foi obtido em 83% de rendimento. MS (m/z): 426,0 (M+H). Etapa 7: N1 -(4-(2-(1-(2-(dietilamino)etil)-1 H-imidazol-4-il)tieno[3,2- b ]piridin-7-ilóxi)-3-fluorofenil)-N3-metil-N3-fenilmalonamida (183)
[00683] Seguindo o procedimento descrito acima para a síntese de composto 163 (Esquema 46), o composto do título 183 foi obtido em 38% de rendimento. 1H RMN (DMSO-d6) δ (ppm): 10,34 (s, 1H), 8,40 (d, J = 5,3 Hz, 2H), 7,91 (s, 1H), 7,79 (m, 2H), 7,63 (s, 1H), 7,46-7,28 (m, 7H), 6,54 (d, J = 5,5 Hz, 1H), 4,03 (t, 2H), 3,20-3,19 (m, 5H), 2,70 (t, 2H), 2,44 (q, 4H), 0,89 (t, 6H), MS (m/z): 601,0 (M+H). Esquema 53
Exemplo 74 N1 -(4-(2-(1 -Etil-1 H-imidazol-4-il)tieno[3,2-b ]piridin-7-ilóxi)-3-fluorofenil)- N3-(2-(trifluorometil)fenil)malonamida (190) Etapa 1: Ácido 3-oxo-3-(2-(trifluorometil)fenilamino)propanóico (191)
[00684] Seguindo o procedimento descrito acima para a síntese de composto 27 (Esquema 8) porém substituindo 2-metoxibenzenamina com 2-(trifluorometil)benzenamina o composto do título 191 foi obtido em 6 % de rendimento. MS (m/z): 248,0 (M+H). Etapa 2: N1 -(4-(2-(1-Etil-1 H-imidazol-4-il)tieno[3,2-b]piridin-7-ilóxi)-3- flúor fenil)-N3 -(2-(trifluorometil)fenil)malonamida (190)
[00685] Partindo da amina 15 e seguindo o procedimento descrito acima para a síntese de composto 28a (Exemplo 8, Etapa 2, Esquema 8) porém substituindo o ácido 27 com o ácido 191, o composto do título 190 foi obtido em 27% de rendimento. 1H RMN (DMSO-d6) δ (ppm): 10,57 (s, 1H), 10,56 (s, 1H), 8,40 (d, J = 5,3 Hz, 1H), 8,10 (s, 1H), 7,94 (d, J = 1,2 Hz, 1H), 7,85 (dd, J= 2,2 e 13,0 Hz, 1H), 7,77 (s, 1H), 7,76 (d, J = 8,6 Hz, 1H), 7,65 (s, 1H), 7,56 (t, J = 7,8 Hz, 1H), 7,44 (t, 1H, J = 8,6 Hz), 7,41-7,40 (m, 2H), 6,56 (d, 1H), 4,03 (q, 2H), 3,54 (s, 1H), 1,33 (t, 3H). MS (m/z): 583,1,0 (M+H). Esquema 54
N1 -(4-(2-(1 -(2-(Dimetilamino)etil)-1 H-imidazol-4-il)tieno[3,2-b ]piridin-7- ilóxi)-3-fluorofenil)-N3 -(2-fluorofenil)malonamida (191) Etapa 1: 2-Iodo-N, N-dimetiletanamina (192)
[00686] Seguindo o procedimento descrito acima para a síntese de composto 164 (Esquema 46), o composto do título 192 foi obtido em 61% de rendimento. MS (m/z): 200,0 (M+H). Etapa 2: N, N-Dimetil-2-(2,4,5-tri-iodo-1 H-imidazol-1-il)etanamina (193)
[00687] Seguindo o procedimento descrito acima para a síntese de composto 166 (Esquema 46), o composto do título 193 foi obtido em 27% de rendimento. MS (m/z): 518,0 (M+H). Etapa 3: 2-(4-iodo-1H-imidazol-1-il)-N, N-dimetiletanamina (194)
[00688] Seguindo o procedimento descrito acima para o composto 167 (Esquema 46), o composto do título 194 foi obtido em 97% de rendimento. MS (m/z): 266,0 (M+H). Etapa 4: 2-(4-(7-Clorotieno[3,2-b ]piridin-2-il)-1 H-imidazol-1-il)-N, N- dimetiletanamina (195)
[00689] Seguindo o procedimento descrito acima para o composto 168 (Esquema 46), o composto do título 195 foi obtido em 41% de rendimento. MS (m/z): 307,0 (M+H). Etapa 5: 2-(4-(7-(2-Flúor-4-nitrofenóxi)tieno[3,2-b ]piridin-2-il)-1 H- imidazol-1-il)-N, N-dimetiletanamina (196)
[00690] Seguindo o procedimento descrito acima para o composto 168 (Esquema 46), o composto do título 196 foi obtido em 36% de rendimento. MS (m/z): 428,0 (M+H). Etapa 6: 4-(2-(1 -(2-(Dimetilamino)etil)-1 H-imidazol-4-il)tieno[3,2- b ]piridin-7-ilóxi)-3-fluoroanilina (197)
[00691] Seguindo o procedimento descrito acima para o composto 170 (Esquema 46), o composto do título 197 foi obtido em 93% de rendimento. MS (m/z): 398,0 (M+H). Etapa 7: N1 -(4-(2-(1-(2-(Dimetilamino)etil)-1 H-imidazol-4-il)tieno[3,2- b ]piridin-7-ilóxi)-3-fluorofenil)-N3-(2-fluorofenil)malonamida (191)
[00692] Seguindo o procedimento descrito acima para o composto 163 (Esquema 46), o composto do título 191 foi obtido em 55% de rendimento. 1H RMN (DMSO-d6) δ (ppm): 10,61 (s, 1H), 10,07 (s, 1H), 8,40 (d, J = 5,5Hz, 1H), 8,38 (s, 1H), 7,99-7,93 (m, 1H), 7,90 (d, J = 1,0 Hz, 1H), 7,85 (dd, J= 2,1 e 13,1 Hz, 1H), 7,75 (d, J = 1,0 Hz, 1H), 7,64 (s, 1H), 7,47 (t, 1H, J = 8,8 Hz), 7,40 (dd, J = 2,1 e 8,8 Hz, 1H), 7,297,23 (m,1H), 7,18-7,12 (m, 2H), 6,55 (d, J = 5,5 Hz, 1H), 4,09 (t, J = 6,2 Hz, 2H), 3,61 (s, 2H), 2,59 (t, J = 6,2 Hz, 2H), 2,17 (s, 6H). MS (m/z): 577,1 (M+H). Esquema 55
Exemplo 76 N-(4-(2-(1 -Etil-1 H-imidazol-4-il)tieno[3,2-b ]piridin-7-ilóxi)-3-fluorofenil)- N-(2-fluorofenil)-N-metilciclopropano-1,1-dicarboxamida (198) Etapa 1: Ácido 1-((4-fluorofenil)(metil)carbamoil) ciclopropanocarboxílico (199)
[00693] Seguindo o procedimento descrito acima para a síntese de composto 161 (Esquema 45), porém substituindo anilina por 4-flúor-N- metilanilina, o composto do título 199 foi obtido em 66% de rendimento. M/S (m/z): 238,0 (M+H). Etapa 2: N-(4-(2-(1-etil-1 H-imidazol-4-il)tieno[3,2-b ]piridin-7-ilóxi)-3- fluorofenil)-N-(2-fluorofenil)-N-metilciclopropano-1,1-dicarboxamida £1981
[00694] Seguindo o procedimento descrito acima para a síntese de composto 5d (Exemplo 4, Esquema 4) o composto do título 198 foi obtido em 15% de rendimento. 1HNMR (CD3OD) δ (ppm): 8,40 (d, J = 8,6Hz, 1H), 7,79 (dd, 2H), 7,65 (s, 1H), 7,46-7,38 (m, 1H), 7,31-7,27 (m, 3H), 7,20-7,16 (m, 1H), 7,06 (t, J = 8,6 Hz, 1H), 6,55 (d, J = 5,7 Hz, 1H), 4,12 (q, 2H), 3,32 (s, 3H, N-Me), 1,50 (m, 5H), 1,33 (s, 2H). MS (m/z): 574,1 (M+H). Esquema 56
Exemplo 77 N1 -Etil-N3-(4-(2-(1-etil-1 H-imidazol-4-il)tieno[3,2-b ]piridin-7-ilóxi)-3- fluorofenil)-N1 -fenilmalonamida (200) Etapa 1: Ácido 3-(etil(fenil)amino)-3-oxopropanoico (201)
[00695] Seguindo o procedimento descrito acima para o composto 31 (Esquema 10), o composto do título 201 foi obtido em 81% de rendimento. MS (m/z): 208,0 (M+H). Etapa 2: N1 -Etil-N3-(4-(2-(1-etil-1 H-imidazol-4-il)tieno[3,2-b ]piridin-7- ilóxi)-3-fluorofenil)-N1 -fenilmalonamida (200)
[00696] Seguindo o procedimento descrito acima para o composto 5d (Exemplo 4, Esquema 4), o composto do título 200 foi obtido em 49% de rendimento. 1H RMN (DMSO-d6) δ (ppm): 10,24 (s, 1H), 8,40 (d, J = 5,5 Hz, 1H), 7,94 (d, J = 1,2 Hz, 1H), 7,77 (d, J = 1,2 Hz, 1H), 7,76 (dd, J= 2,3 e 2,9 Hz, 1H), 7,65 (s, 1H), 7,49-7,27 (m, 8H), 6,54 (d, J = 5,3 Hz, 1H), 4,04 (q, 2H), 3,67 (q, 2H), 3,14 (s, 2H), 1,38 (t, 3H), 1,01 (t, 3H) MS (m/z): 544,1 (M+H). Esquema 57
Exemplo 78 N1 -(4-(2-(1 -Etil-1 H-imidazol-4-il)tieno[3,2-b ]piridin-7-ilóxi)-3-fluorofenil)- N3-isopropil-N3-fenilmalonamida (202) Etapa 1: Ácido 3-(isopropil(fenil)amino)-3-oxopropanoico (203)
[00697] Seguindo o procedimento descrito acima para o composto 31 (Esquema 10), o composto do título 203 foi obtido em 49% de rendimento. MS (m/z): 222,0 (M+H). Etapa 2: N1 -(4-(2-(1-Etil-1 H-imidazol-4-il)tieno[3,2-b]piridin-7-ilóxi)-3- fluorofenil)-N3-isopropil-N3-fenilmalonamida (202)
[00698] Seguindo o procedimento descrito acima para o composto 5d (Exemplo 4, Esquema 4), o composto do título 202 foi obtido em 28% de rendimento. 1HNMR (DMSO-d6) δ (ppm): 10,19 (s, 1H), 8,40 (d, J = 5,5 Hz, 1H), 7,94 (d, J = 1,3 Hz, 1H), 7,77-7,73 (m, 2H), 7,65 (s, 1H), 7,49-7,39 (m, 4H), 7,28-7,24 (m, 3H), 6,54 (dd, J = 0,6 e 5,3 Hz, 1H), 4,82 (m, 1H), 4,04 (q, 1H), 3,02 (s, 2H), 1,38 (t, 3H), 0,99 (s, 3H), 0,98 (s, 3H). MS (m/z): 558,0 (M+H). Esquema 58
Exemplo 79 N-(4-(2-(1 -Etil-1 H-imidazol-4-il)tieno[3,2-b ]piridin-7-ilóxi)-3-fluorofenil)- 2-oxo-1-fenilpirrolidina-3-carboxamida (204)
[00699] Seguindo o procedimento descrito acima para a síntese de composto 5d (Esquema 4), porém substituindo o ácido 1 com o ácido 205, o composto do título 204 foi obtido em 40% de rendimento. 1HNMR (DMSO-d6) δ (ppm): 10,72 (s, 1H), 8,49 (d, J = 5,7 Hz, 1H), 8,06 (d, J = 1,0 Hz, 1H), 7,96-7,90 (m, 2H), 7,72 (s, 1H), 7,67-7,65 (m, 2H), 7,53-7,49 (m, 2H), 7,41-7,37 (m, 2H), 7,16 (t, J = 7,2 Hz, 1H), 6,70 (d, J = 5,5 Hz, 1H), 4,06 (q, 2H), 3,93 (m, 2H), 3,77 (t, J = 8,2 Hz, 1H), 4,42 (m, 2H), 1,40 (t, 3H). MS (m/z): 542,0 (M+H). Exemplo 80 N-(4-(2-(1 -Etil-1 H-imidazol-4-il)tieno[3,2-b ]piridin-7-ilóxi)-3-fluorofenil)- 3-(indolin-1-il)-3-oxopropanamida (206) Etapa 1: Ácido 3-(indolin-1-il)-3-oxopropanoico (207)
[00700] Seguindo o procedimento descrito acima para o composto 31 (Esquema 10), o composto do título 207 foi obtido em 75% de rendimento. MS (m/z): 206,0 (M+H). Etapa 2: N-(4-(2-(1-Etil-1 H-imidazol-4-il)tieno[3,2-b ]piridin-7-ilóxi)-3- fluorofenil)-3-(indolin-1-il)-3-oxopropanamida (206)
[00701] Seguindo o procedimento descrito acima para o composto 5d (Esquema 4), o composto do título 206 foi obtido em 40% de rendimento. 1H RMN (DMSO-d6) δ (ppm): 10,56 (s, 1H), 8,42 (d, J = 5,5 Hz, 1H), 8,06 (d, J = 8,0Hz, 1H), 7,95 (s, J = 1,2 Hz, 1H), 7,87 (dd, J= 2,2 e 13,1 Hz, 1H), 7,78 (d, J = 1,2 Hz, 1H), 7,67 (s, 1H), 7,47 (t, J = 8,8 Hz, 1H), 7,40 (dd, J = 1,5 e 8,0 Hz, 1H), 7,25 (d, J = 7,7 Hz, 1H), 7,16 (t, J = 7,5 Hz, 1H), 7,02 (t, J = 7,3 Hz, 1H), 6,57 (d, J = 5,3 Hz, 1H), 4,16 (s, 2H), 4,06 (q, 2H), 3,69 (s, 2H), 3,32-3,15 (m, 2H), 1,39 (t, 3H). MS (m/z): 542,1 (M+H). Exemplo 81 3-(2 H-Benzo[ b ][1,4]oxazin-4 (3 H)-il)-N-(4-(2-(1-etil-1 H-imidazol-4- il)tieno[3,2-b ]piridin-7-ilóxi)-3-fluorofenil)-3-oxopropanamida (208) Etapa 1: Ácido 3-(2H-benzo[b][1,4]oxazin-4 (3H)-il)-3-oxopropanoico (209)
[00702] Seguindo o procedimento descrito acima para o composto 31 (Esquema 10), o composto do título 209 foi obtido em 75% de rendimento. MS (m/z): 222,0 (M+H). Etapa 2: 3-(2 H-Benzo[ b ][1,4]oxazin-4 (3 H)-il)-N-(4-(2-(1-etil-1 H- imidazol-4-il)tieno[3,2-b ]piridin-7-ilóxi)-3-fluorofenil)-3-oxopropanamida (208)
[00703] Seguindo o procedimento descrito acima para o composto 5d (Esquema 4), o composto do título 208 foi obtido em 40% de rendimento. 1H RMN (DMSO-d6) δ (ppm): 10,55 (s, 1H), 8,44 (d, J = 5,5 Hz, 1H), 7,98 (s, 1H), 7,83 (s, 1H), 7,82 (bs, 1H), 7,68 (s, 1H), 7,46 (t, 1H), 7,38 (bs, 1H), 7,06 (bs, 1H), 6,89 (m, 2H), 6,60 (d, J = 5,5 Hz, 1H), 4,32 (t, 2H), 4,05 (q, 2H), 3,91 (t, 2H), 3,81 (s, 2H), 1,39 (t, 3H). MS (m/z): 558,1 (M+H). Esquema 59
N-(4-(2-(1 -Etil-1 H-imidazol-4-il)tieno[3,2-b ]piridin-7-ilóxi)-3-fluorofenil)- 2-oxo-3-fenilimidazolidina-1-carboxamida(210)
[00704] A uma solução de 1-fenilimidazolidin-2-ona (211) (100 mg, 0,62 mmol) [P. Mayer, P. Brunel, C. Chaplain, C. Piedecoq, F. Calmel, P. Schambel, P. Chopin, T. Wurch, P. J. Pauwels, M. Marien, J.-L. Vidaluc, T. Imbert J. Med. Chem. 2000, 43, 3653-3664; W. Su, Y. Zhang J. Chem. Res. Synop. 2000, 9, 440-441] em THF (6 mL) foi adicionado trifosgene (189 mg, 0,62 mmol) e a solução foi agitada durante 3 horas a 60°C. A mistura de reação foi resfriada até a T.A. antes da adição de anilina 15 (229 mg, 0,65 mmol) e DIPEA (648 μL, 3,72 mmols) e agitação foi continuada durante uma hora. A mistura de reação foi concentrada e dividida entre EtOAc e água. Um precipitado foi formado que foi coletado por filtração. A camada orgânica foi separada, secada e concentrada. O resíduo foi combinado com o precipitado coletado, carregado seco para uma coluna e eluído com EtOAc/MeOH (9:1), para produzir o composto do título 210 (150 mg, 43% de rendimento) como um sólido esbranquiçado. 1H RMN (DMSO- de) δ (ppm): 10,57 (s, 1H), 8,42 (d, J = 5,5 Hz, 1H), 7,95 (d, J = 1,1 Hz, 1H), 7,83 (dd, J = 2,5 e 13,3 Hz, 2H), 7,77 (d, J = 1,2 Hz, 1H), 7,66 (s, 1H), 7,61 (dd, J = 1,0 e 8,8 Hz, 1H), 7,49-7,41 (m, 4H), 7,16 (t, J = 7,2 Hz, 1H), 6,57 (d, J = 5,5 Hz, 1H), 4,03 (q, 2H), 4,04-3,92 (m, 4H), 1,38 (t, 3H). MS (m/z): 543,0 (M+H). Tabela 5 Compostos 211-219 (Exemplos 83-91) preparados de acordo com o Esquema 59


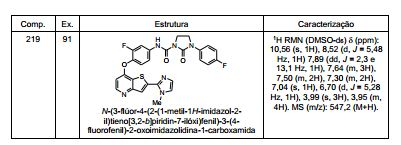
[00705] Compostos 211-214 foram sintetizados partindo da amina 15 (Esquema 4), compostos 215-216 foram preparados partindo da amina 197 (Esquema 54), composto 217 foi obtido partindo da amina 80 (Esquema 26), enquanto compostos 218-219 foram derivados partindo da amina 9 (Esquema 2). Esquema 60
Exemplo 92 N-(3-Flúor-4-(2-(4-((2-metoxietilamino)metil)fenil)tieno[3,2-b ]piridin-7- ilóxi)fenil)-3-(4-fluorofenil)-2-oxoimidazolidina-1-carboxamida (220) Etapa 1: 4-(7-(2-Flúor-4-(3-(4-fluorofenil)-2-oxoimidazolidina-1- fenóxi)tieno[3,2-b ]piridin-2-il)benzil(2-metoxietil) carbamato de terc-butila (223)
[00706] Partindo da amina 221 [preparada de acordo com o Esquema 26, usando arilboronato 76 como um intermediário (Tabela 2)] e cloreto de 3-(4-fluorofenil)-2-oxoimidazolidina-1-carbonila (222), e seguindo o procedimento descrito acima para a síntese de composto 210 (Exemplo 82, Esquema 59), o composto do título 223 foi obtido em 40% de rendimento. MS (m/z): 730,3 (M+H). Etapa 2: N -(3-Flúor-4-(2-(4-((2-metoxietilamino)metil)fenil)tieno[3,2- b ]piridin-7-ilóxi)fenil)-3-(4-fluorofenil)-2-oxoimidazolidina-1- carboxamida (220)
[00707] Uma solução de 223 (60 mg, 0,082 mmol) e TFA (1 mL) em tolueno (2 mL) foi agitada 30 minutos em temperatura ambiente. A mistura de reação foi concentrada sob pressão reduzida. O resíduo foi triturado com éter de dietila, o sólido foi coletado e secado para fornecer o composto do título 220 (70 mg, 99,5%) como um sólido esbranquiçado. 1HNMR (DMSO-d6) δα (ppm): 10,55 (s, 1H), 8,95 (s, amplo, 2H), 8,52 (d, J = 5,5 Hz, 1H), 8,13 (s, 1H), 7,97 (d, J = 8,4 Hz, 2H), 7,84 (dd, J= 2,5 e 13,1 Hz, 1H), 7,65-7,61 (m, 4H), 7,49 (t, J = 8,8 Hz, 1H), 7,44 (dd, J = 2,4 e 8,8 Hz, 1H), 7,30-7,25 (m, 2H), 6,65 (d, J = 5,5 Hz, 1H), 4,21 (t, 1H), 3,93 (m, 4H), 3,57 (t, 2H), 3,30 (s, 3H), 3,11 (m, 2H). MS (m/z): 630,3 (M+H). Esquema 61
Exemplo 93 N1-(3-Flúor-4-(2-(1 -metil-1,2,5,6-tetra-hidropiridin-3-il)tieno[3,2- b]piridin-7-ilóxi)fenil)-N3-(2-metoxifenil)malonamida (224) Etapa 1: 4-(2-Bromotieno[3,2-b]piridin-7-ilóxi)-3-fluorobenzenamina £225)
[00708] A uma mistura de composto 50 (1,0 g, 2,96 mmols) e NH4Cl (46 mg, 0,86 mmol) em EtoH (29 mL)/água (15 mL) a 100°C, Fe (1,4 g, 25,15 mmols) foi adicionado em uma porção e a mistura foi refluxada com vigorosa agitação durante 40 minutos. A mistura foi filtrada através de celite®, o Celite® lavado com EtoH e o filtrado combinado concentrado sob pressão reduzida. o resíduo foi suspenso em EtoAc, lavado com água; a fase orgânica foi seca sobre Na2So4 anidro e evaporada fornecendo o composto do título 225 (916,15 mg, 91% de rendimento). MS (m/z): 338,9 (96 %), 340,9 (100%). Etapa 2: 4-(2-Bromotieno[3,2-b]piridin-7-ilóxi)-3-fluorofenilcarbamato de terc - butila (226)
[00709] Uma solução da anilina 225 (300 mg, 0,887 mmol) e BocON (194 mg, 0,887 mmol) em MeCN (1,8 mL) foi agitada durante a noite em temperatura ambiente. A mistura bruta foi concentrada sob pressão reduzida fornecendo o composto do título 226 (380 mg, 98% de rendimento) que foi usado na próxima Etapa sem outra purificação. MS (m/z): 439,1 (96 %), 441,1 (100%). Etapa 3: 3-Flúor-4-(2-(1-metil-1,2,5,6-tetra-hidropiridin-3-il)tieno[3,2- b]piridin-7-ilóxi)fenilcarbamato de terc-butila (229) (procedimento de duas etapas)
[00710] A uma mistura de 5-bromo-1-metil-1,2,3,6-tetra-hidropiridina 227 (Drinkuth, S; Gruetsch, S; Peter, K; Christl, M. Eur. J. Org. Chem; 14; 2001; 2665-2670) (1,0 g, 5,71 mmols), bis(pinacolato)diboro (8,66 mmols, 2,18 g) e AcOK (1,7 g, 17,3 mmols) em tolueno (11,4 mL), Pd(PPh3)4 (0,171 mmol, 198 mg) foi adicionado em uma porção e a mistura foi aquecida para refluxo sob N2 durante 2 horas. A suspensão foi concentrada sob pressão reduzida, fornecendo 228 bruto que foi redissolvido em DME (29 mL) e mantido sob nitrogênio. 4-(2- Bromotieno[3,2-b]piridin-7-ilóxi)-3-fluorofenilcarbamato de terc-butila 226 (380 mg, 0,87 mmol), CsF (2,64 mmols, 401 mg), NaHCO3 (2,64 mmols, 222 mg), e água (1 mL) foram adicionados a uma alíquota da solução de DME de 228 (4 mL, 1,22 mmol) e a mistura refluxada durante a noite sob nitrogênio. O bruto foi diluído com EtOAc e extraído com HCl a 1N. A fase aquosa foi extraída com DCM, basificada para pH ~11 pela adição de solução de NaOH a 1N, extraída com EtOAc. O extrato foi seco sobre Na2SO4 anidro e concentrado sob pressão reduzida fornecendo 229 bruto (105 mg, 26% de rendimento) que foi usado na próxima Etapa sem outra purificação. MS (m/z): 456,1 (100%). Etapa 4: 3-Flúor-4-(2-(1-metil-1,2,3,6-tetra-hidropiridin-4-il)tieno[3,2- b]piridin-7-ilóxi)anilina (230)
[00711] TFA (1 mL) foi adicionado a 3-flúor-4-(2-(1-metil-1,2,3,6- tetra-hidropiridin-3-il)tieno[3,2-b]piridin-7-ilóxi)fenilcarbamato de terc- butila (229) (105 mg, 0,23 mmol) e a mistura foi agitada durante 1 hora em temperatura ambiente. A solução foi concentrada sob pressão reduzida, o resíduo co-destilado com MeCN, redissolvido em MeOH e purificado por HPLC preparativa (gradiente 40% a 95 % de MeOH em água, 45 minutos) fornecendo 230 (50 mg, 0,1 mmol, 48% de rendimento) como um sólido branco. MS (m/z): (M+1) 356,1 (100%). Etapa 5:N1-(3-Flúor-4-(2-(1-metil-1,2,3,6-tetra-hidropiridin-4- il)tieno[3,2-b]piridin-7-ilóxi)fenil)-N3-(2-metoxifenil)malonamida (224)
[00712] Uma solução de 3-flúor-4-(2-(1-metil-1,2,3,6-tetra- hidropiridin-4-il)tieno[3,2-b]piridin-7-ilóxi)anilina (230) (50 mg, 0,110 mmol), ácido 3-(2-metoxifenilamino)-3-oxopropanoico 27 (34 mg, 0,165 mmol) (Esquema 8), EDC (25 mg, 0,165 mmol) e HOBt (22 mg, 0,165 mmol) em DMF (1,1 mL) foi agitada durante a noite em temperatura ambiente. Mais 27 (34 mg, 0,165 mmol) e EDC (25 mg, 0,165 mmol) foram adicionados e a mistura agitada durante mais 6 horas. A mistura foi diluída com EtOAc, extraída com água, secada sobre Na2SO4 anidro, e concentrada sob pressão reduzida. O resíduo foi redissolvido em MeOH e purificado por HPLC preparativa (gradiente 40% a 95 % de MeOH em água, 45 minutos) seguido por cromatografia instantânea (MeOH/CHCl3/NH4Cl 1:9:0,1) fornecendo o composto do título 224 (19 mg, 0,056 mmol, 51% de rendimento) como um sólido branco. 1H RMN (400 MHz, DMSO-d6) δ (ppm): 10,61 (s, 1H), 9,64 (s, 1H), 8,45 (d, J = 5,5Hz, 1H), 8,07 (dd, J = 8,8Hz, J = 1,4Hz, 1H), 7,86 (dd, J = 2,3Hz, J = 13,1Hz, 1H), 7,49-7,41 (m, 3H), 7,11-7,05 (m,2H), 6,92 (m,1H), 6,61 (dd, J = 0,8Hz, J = 5,5Hz, 1H), 6,43 (m, 1H), 3,86 (s, 3H), 3,64 (s, 2H), 3,33 (m, 2H), 2,52-2,50 (m, 2H), 2,49 (m, 2H), 2,36-2,33 (m, 5H). MS (m/z): 547,42 (100% de rendimento). Esquema 62

(Z)-N1-(4-(2-(4-(Dimetilamino)but-1-enil)tieno[3,2-b]piridin-7-ilóxi)-3- fluorofenil)-N3-metil-N3-fenilmalonamida (231) Etapa 1: 7-Clorotieno[3,2-b]piridina-2-carbaldeído (232)
[00713] A uma solução de 7-clorotieno[3,2-b]piridina (2) (2 g, 11,83 mmols) em THF (40 mL) n-BuLi (hexano a 2,5Ms, 5,7 mL, 14,2 mmols) foi adicionado em gotas a -78°C e a mistura de reação agitada 1 hora. DMF (2,7 mL, 35,5 mmols) foi adicionada e agitação continuada mais 1 hora. A mistura de reação foi despejada em água, extraída com EtOAc, os orgânicos combinados secados sobre Na2SO4 anidro, e concentrados sob pressão reduzida. O resíduo foi triturado com éter e filtrado fornecendo o composto do título 232 (2 g, 10,12 mmols, 86% de rendimento). MS (m/z): 197,9 (36%), (M + MeOH +1) 230,0 (100%). Etapa 2: 7-(2-Flúor-4-nitrofenóxi)tieno[3,2-b]piridina-2-carbaldeído (233)
[00714] Uma mistura de aldeído 232 (500 mg, 2,53 mmols), 2-flúor- 4-nitrofenol (595 mg, 3,79 mmols), K2CO3 (700 mg, 5,06 mmols) e Ph2O (3,4 mL) foi agitada em um tubo selado durante 12 horas a 170°C. A mistura foi suspensa em uma mistura de água/EtOAc, sonicada alguns minutos e filtrada, o resíduo sólido foi lavado sucessivamente com água, EtOAc e éter, fornecendo 233 bruto (500 mg, 1,17 mmols, 46% de rendimento) que foi usado na próxima Etapa sem outra purificação. MS (m/z): (M+1) 319,0 (14%), (M + MeOH + 1) 351,0 (100%). Etapa 3: 4-(7-(2-Flúor-4-nitrofenóxi)tieno[3,2-b]piridin-2-il)-N,N- dimetilbut-3-en-1-amina (234)
[00715] A uma solução de brometo de (2- (dimetilamino)etil)trifenilfosfônio (680 mg, 1,54 mmol) em THF (7,7 mL) foi adicionado n-BuLi (hexano a 2,5Ms, 0,65 mL, 1,62 mmol) em gotas a 0°C, a mistura resultante foi aquecida até a temperatura ambiente e agitada durante 30 minutos. 7-(2-flúor-4-nitrofenóxi)tieno[3,2-b]piridina- 2-carbaldeído 233 (500 mg, 1,17 mmol) foi adicionado em uma porção e a mistura foi agitada durante 2 horas. A mistura foi despejada em água, extraída com EtOAc; a fase orgânica foi extraída com 3% de ácido cítrico, a fase aquosa combinada foi basificada para pH ~11 pela adição de NaOH a 1N. Ela foi então extraída com EtOAc, secada sobre Na2SO4 anidro e concentrada sob pressão reduzida fornecendo o composto do título 234 (360 mg, 0,929 mmol, 78% de rendimento) como uma mistura bruta que foi usada na próxima Etapa sem outra purificação. MS (m/z): (M+1) 388,1 (100%). Etapa 4: (Z)-4-(2-(4-(Dimetilamino)but-1-enil)tieno[3,2-b]piridin-7-ilóxi)- 3-fluoroanilina (235)
[00716] A uma mistura de 4-(7-(2-flúor-4-nitrofenóxi)tieno[3,2- b]piridin-2-il)-N,N-dimetilbut-3-en-1-amina (234) (171 mg, 0,44 mmol) e NH4CI (20 mg, 0,37 mmol) em EtOH (4,4 mL)/água (2,2 mL) a 100°C, Fe (209 mg, 3,75 mmols) foi adicionado em uma porção e a mistura foi aquecida para refluxo com vigorosa agitação durante 40 minutos. A mistura foi filtrada através de celite®, o Celite® lavado com EtOH e as soluções orgânicas combinadas concentradas sob pressão reduzida. O resíduo foi dissolvido em MeOH e purificado por HPLC preparativa (gradiente 40% a 95 % de MeOH em água, 45 minutos) fornecendo o composto do título 235 como um sólido amarelo (122,8 mg, 0,3 mmol, 69% de rendimento). MS (m/z): (M+1) 358,1 (100%). Etapa 5: (Z)-N1-(4-(2-(4-(Dimetilamino)but-1-enil)tieno[3,2-b]piridin-7- ilóxi)-3-fluorofenil)-N3-metil-N3-fenilmalonamida (231)
[00717] Uma solução de (Z)-4-(2-(4-(dimetilamino)but-1- enil)tieno[3,2-b]piridin-7-ilóxi)-3-fluoroanilina (235) (69,1 mg, 0,193 mmol), ácido 3-(metil(fenil)amino)-3-oxopropanoico 31 (46 mg, 0,26 mmol) (Esquema 10), e EDC (45 mg, 0,24 mmol) em DMF (2,8 mL) foi agitada durante a noite em temperatura ambiente. Mais 31 (45 mg, 0,26 mmol) e EDC (45 mg, 0,24 mmol) foram adicionados e a mistura agitada mais 6 horas. A mistura foi diluída com EtOAc, lavada com água, secada sobre Na2SO4 anidro, e concentrada sob pressão reduzida. O resíduo foi redissolvido em MeOH e purificado duas vezes por HPLC preparativa (gradiente 40% a 95 % de MeOH em água, 45 minutos) fornecendo o composto do título 231 (35 mg, 0,066 mmol, 34% de rendimento) como um sólido branco. 1H RMN (400 MHz, DMSO-d6) δ (ppm): 10,32 (s, 1H), 8,48 (d, J = 5,5Hz, 1H), 7,80 (d, J = 12,7Hz, 1H), 7,54 (s, 1H), 7,5-7,3 (m, 7H), 6,78 (d, J = 12,2Hz, 1H), 6,62 (d, J = 5,5Hz, 1H), 5,92 (tt, J = 7,2Hz, J = 4,5Hz, 11,7Hz, 1H), 3,21 (s, 5H), 2,59 (m, 2H), 2,42 (dd, 6,8Hz, 7,4Hz, 2H), 2,16 (s, 6H). MS (m/z): (M+1) 533,1 (100 %). Esquema 63

Exemplo 95 N1-(4-(2-(2,2-Dimetil-hidrazinacarbonil)tieno[3,2-b]piridin-7-ilóxi)-3- fluorofenil)-N3-(2-metoxifenil)malonamida (236) Etapa 1: 7-Cloro-N',N'-dimetiltieno[3,2-b]piridina-2-carboidrazida (237)
[00718] A uma suspensão de cloreto de 7-clorotieno[3,2-b]piridina- 2-carbonila 20 (1g, 4,33 mmols) (Esquema 6) em DCM (22 mL), 1,1- dimetiliidrazina (0,33 mL, 4,33 mmols) foi adicionado em uma porção e a mistura foi agitada durante 5 horas. Mais 1,1-dimetilidrazina (0,33 mL, 4,33 mmols) foi adicionado e a mistura foi agitada durante a noite em temperatura ambiente. O bruto foi diluído com EtOAc, extraído com HCl a 1N, a solução aquosa basificada para pH ~11 pela adição de NaOH a 1N, extraída com EtOAc, a fase orgânica secada sobre Na2SO4 anidro e concentrada sob pressão reduzida fornecendo 237 (798 mg, 3,13 mmols, 72%) como uma mistura bruta que foi usada na próxima Etapa sem outra purificação. MS (m/z): (M+1) 255,9 (100%), 257,9 (39%). Etapa 2: 7-(2-Flúor-4-nitrofenóxi)-^,,^'-dimetiltieno[3,2-b]piridina-2- carboidrazida (238)
[00719] Uma mistura de 7-cloro-N',N'-dimetiltieno[3,2-b]piridina-2- carboidrazida (237) (798 mg, 3,13 mmols), 2-flúor-4-nitrofenol (740 mg, 4,70 mmols), K2CO3 (830 mg, 6,26 mmols) e Ph2O (4,2 mL) foi agitada em um tubo selado durante 12 horas a 170°C. A mistura foi diluída com DCM, extraída com água, a fase orgânica secada com Na2SO4 anidro e concentrada. O resíduo foi purificado por cromatografia instantânea (EtOAc/hexanos 1:1) fornecendo 238 (772,5 mg, 2,05 mmols, 66% de rendimento) como uma espuma marrom. MS (m/z): (M+1) 377,0 (100%). Etapa 3: 7-(4-Amino-2-fluorofenóxi)-N',N'-dimetiltieno[3,2-b]piridina-2- carboidrazida (239)
[00720] A uma mistura de 7-(2-flúor-4-nitrofenóxi)-N',N'- dimetiltieno[3,2-b]piridina-2-carboidrazida 238 (772 mg, 2,05 mmols) e NH4Cl (93 mg, 1,74 mmol) em EtOH (20,5 mL)/água (10,3 mL) a 100°C, Fe (973 mg, 17,73 mmols) foi adicionado em uma porção e a mistura foi aquecida para refluxo com vigorosa agitação 40 minutos. A mistura foi filtrada através de celite®, o Celite® lavada com EtOH e as soluções orgânicas combinadas concentradas sob pressão reduzida. O resíduo foi purificado por cromatografia instantânea (MeOH/DCM 1:9) fornecendo 239 (605 mg, 1,75 mmol, 85%). MS (m/z): (M+1) 347,0 (100%). Etapa 4: N 1-(4-(2-(2,2-Dimetil-hidrazinacarbonil)tieno[3,2-b]piridin-7- ilóxi)-3-fluorofenil)-N3-(2-metoxifenil)malonamida (236)
[00721] Uma solução de 7-(4-amino-2-fluorofenóxi)-N',N'- dimetiltieno[3,2-b]piridina-2-carboidrazida 239 (200 mg, 0,578 mmol), ácido 3-(2-metoxifenilamino)-3-oxopropanoico 27 (195 mg, 0,693 mmol), e EDC (230 mg, 0,693 mmol) em DMF (8,3 mL) foi agitada durante a noite em temperatura ambiente. Mais 27 (195 mg, 0,693 mmol) e EDC (230 mg, 0,693 mmol) foram adicionados e a mistura agitada mais 6 horas. O bruto foi diluído com EtOAc, extraído com água, com Na2SO4 anidro, e concentrado sob pressão reduzida. O resíduo foi redissolvido em MeOH e purificado duas vezes por cromatografia instantânea (DCM/MeOH 9:1) fornecendo 236 (207 mg, 0,385 mmol, 67% de rendimento) como um sólido branco. 1H RMN (400 MHz, DMSO-d6) δ (ppm): 10,60 (s, 1H), 9,94 (s, 0,5H), 9,63 (s, 1H), 9,49 (s, 0,5H), 8,57 (dd, J = 5,5Hz, J = 7,2Hz, 1H), 8,26 (m, 1H), 8,07 (d, J = 7,4Hz,1H), 7,88 (d, J = 12,9Hz, 1H), 7,54-7,43 (m, 2H), 7,07 (m, 2H), 6,91 (m, 1H), 6,73 (m, 1H), 3,86 (s, 3H), 3,65 (s, 2H), 2,63 (s, 3H), 2,61 (s, 3H). MS (m/z): (M+1) 538,0 (100 %). Exemplo 96 N1-(4-(2-(2,2-Dimetil-hidrazinacarbonil)tieno[3,2-b]piridin-7-ilóxi)-3- fluorofenil)-N3-metil-N3-fenilmalonamida (240)
[00722] Seguindo o procedimento descrito acima para o composto 236 (Exemplo 95, Esquema 63) porém substituindo o ácido 27 com ácido 3-(metil(fenil)amino)-3-oxopropanoico (31), o composto do título 240 foi obtido em 44% de rendimento. 1H RMN (400 MHz, DMSO-d6) δ (ppm): 10,36 (s, 1H), 10,08 (br, 0,3H), 9,52 (s, 0,7H), 8,58 (dd, J = 5,5Hz, J = 8,4Hz, 1H), 8,20 (m,1H), 7,79 (m, 1H), 7,49-7,30 (m, 7H), 6,75 (d, J = 5,5Hz, 1H), 3,21 (s, 2H), 3,2 (s, 3H), 2,64 (s, 3H) 2,59 (s, 3H). MS (m/z): (M+1) 522,1 (100 %). Esquema 64
N1-(3-Flúor-4-(2-(1,2,2-trimetilidrazinacarbonil)tieno[3,2-b]piridin-7- ilóxi)fenil)-N3-(2-metoxifenil)malonamida (241) Etapa 1: 7-Cloro-N,N',N'-trimetiltieno[3,2-b]piridina-2-carboidrazida (242)
[00723] A uma solução de 7-cloro-N',N'-dimetiltieno[3,2-b]piridina-2- carboidrazida 237 (Esquema 63) (357,9 mg, 1,4 mmol) em THF (14 mL)/DMF (9 mL), a 0°C, NaH (60% em óleo mineral, 112 mg, 2,8 mmols) foi adicionado em uma porção e a mistura foi agitada durante 1 hora. MeI (0,118 mL, 2,8 mmols) foi adicionado e a mistura foi aquecida até a temperatura ambiente e agitada durante 1 hora. A suspensão foi despejada em água e extraída com EtOAc. A fase orgânica foi extraída com HCl a 0,1N, A fase aquosa basificada para pH ~ 11 pela adição de NaOH a 1N e extraída com EtOAc. A fase orgânica foi seca sobre Na2SO4 anidro e concentrada sob pressão reduzida fornecendo 242 (111,2 mg, 0,41 mmol, 29%) como um sólido amarelo. MS (m/z): (M+1) 269,9 (100%), 271,0 (38%). Etapa 2: 7-(2-Flúor-4-nitrofenóxi)-N,N',N'-trimetiltieno[3,2-b]piridina-2- carboidrazida (243)
[00724] Seguindo o procedimento descrito acima para o composto 238 (Etapa 2, Exemplo 95, Esquema 63) porém substituindo cloreto 237 com composto 242, o composto do título 243 foi obtido em 66% de rendimento. MS (m/z): (M+1) 391,1 (100%). Etapa 3: 7-(4-Amino-2-fluorofenóxi)-N,N',N'-trimetiltieno[3,2-b]piridina- 2-carboidrazida (244)
[00725] Seguindo o procedimento descrito acima para o composto 239 (Etapa 3, Exemplo 95, Esquema 63) porém substituindo 238 com composto 243, o composto do título 244 foi obtido em 33% de rendimento. MS (m/z): (M+1) 361,1 (100%). Etapa 4: N1-(3-flúor-4-(2-(1,2,2-trimetilidrazinacarbonil)tieno[3,2- b]piridin-7-ilóxi)fenil)-N3-(2-metoxifenil)malonamida (241)
[00726] Seguindo o procedimento descrito acima para o composto 236 (Etapa 4, Exemplo 95, Esquema 63) porém substituindo a amina 239 com 7-(4-amino-2-fluorofenóxi)-N,N',N'-trimetiltieno[3,2-b]piridina- 2-carboidrazida (244), o composto do título 241 foi obtido em 43% de rendimento. 1H RMN (400 MHz, DMSO-d6) δ (ppm): 10,45 (s, 1H), 8,45 (d, J = 5,4Hz, 1H), 7,79 (d, J = 13,3Hz, 1H), 7,67 (s, 1H), 7,62-7,31 (m, 7H), 6,62 (d, J = 5,4Hz, 1H), 3,40 (s, 9H), 3,23 (s, 2H), 3,21 (s, 3H). MS (m/z): (M+1) 536,2 (100 %). Esquema 65
Ácido 3-((2-fluorofenil)(metil)amino)-3-oxopropanoico (245)
[00727] A uma solução de 3-cloro-3-oxopropanoato de metila (1,75 mL, 15,99 mmols) em DCM seco (32 mL) foi adicionado 2-flúor-N- metilanilina (2 g, 15,99 mmols) e a mistura de reação foi agitada a 0°C durante 1 hora, evaporada, então redissolvida em EtOAc, lavada com NaHCO3 diluído, e salmoura. A fase orgânica foi seca sobre sulfato de sódio, e concentrada sob pressão reduzida para fornecer 3-((2- fluorofenil)(metil)amino)-3-oxopropanoato de metila como óleo amarelo que foi usado sem outra purificação (3,3 g, 16 mmols, 97%, bruto). A uma solução deste material (3,3 g, 16 mmols) em THF (16 mL) e água (16 mL) foi adicionado LiOH.H2O (1,35g, 31,5 mmols) e a mistura de reação foi agitada durante a noite, evaporada (para remover o THF) e em seguida extraída com EtOAc. A fase aquosa foi acidificada para pH ~1 pela adição de HCl a 1N e extraída com EtOAc. A solução foi seca sobre Na2SO4, filtrada e concentrada sob pressão reduzida para fornecer o composto do título 245 como um sólido marrom, que foi usado sem outra purificação (2,75 g, 13,03 mmols, 81% de rendimento). MS (m/z): (M+1) 218,0 (88%), (2M+Li) 429,0 (100%). Ácido 1-(metil(fenil)carbamoil)ciclopropanocarboxílico (246)
[00728] A uma solução de ácido ciclopropano-1,1-dicarboxílico (1,5 g, 11,53 mmols) em THF (24 mL), TEA (1,6 mL, 11,53 mmols) foi adicionado em gotas e sob agitação cloreto de tionila (0,83 mL, 11,53 mmols) e a mistura agitada 30 minutos em temperatura ambiente. Uma solução de N-metilanilina (1,3 mL, 11,53 mmols) em THF (14 mL) foi adicionada em gotas a 0 °C e a mistura foi agitada durante 2 horas. A mistura foi diluída com EtOAc, extraída com NaOH a 2N, acidificada para pH ~2 pela adição de HCl a 2N e extraída com EtOAc, secada sobre Na2SO4 anidro e concentrada sob pressão reduzida fornecendo 246 (1,24 g, 5,66 mmols, 49%) como um sólido branco. MS (m/z): (M+1) 220,0 (100%). Tabela 6
[00729] Compostos 247-252 (Exemplos 98-103) preparados partindo da amina 12 e ácidos 27, 29, 161, 31, 245 e 246, de acordo com o Esquema 3
Tabela 7
[00730] Compostos 253-255 (Exemplos 104-106) preparados partindo das aminas 15 e 197 (Esquema 54) e ácidos 31 e 27 de acordo com o Esquemas 4 e 54
Esquema 66
Exemplo 107 N1-(3-Flúor-4-(2-(1 -metil-1,2,3,6-tetra-hidropiridin-4-il)tieno[3,2- b]piridin-7-ilóxi)fenil)-N3-metil-N3-fenilmalonamida (256) Etapa 1: 1-Metil-1,2,3,6-tetra-hidropiridin-4-il trifluorometanossulfonato Í25ZI
[00731] LDA (1,5 N em THF, 8,1 mL, 12,17 mmols) foi adicionado a uma solução de 1-metilpiperidin-4-ona (1,4 mL, 12,17 mmols) em THF (16 mL) a -Z8°C, a mistura foi deixada aquecer até a temperatura ambiente e agitada durante 30 minutos. A solução foi resfriada mais uma vez até -Z8°C e PhNTf2 (5 g, 18,12 mmols) foi adicionado em uma porção, a solução aquecida até a temperatura ambiente e agitada durante 3 horas. A mistura de reação foi despejada em água, extraída com éter, a fase orgânica secada sobre Na2SO4 anidro e concentrada sob pressão reduzida. O resíduo foi purificado por cromatografia instantânea (EtOAc/Hexanos 1:5) fornecendo 257 (2,38 g, 9,7 mmols, 80%) como óleo laranja. MS (m/z): (M+1) 245,9 (100% de rendimento). Etapa 2: 7-(2-Flúor-4-nitrofenóxi)-2-(1-metil-1,2,3,6-tetra-hidropiridin-4- il)tieno[3,2-b]piridina (259)
[00732] A uma mistura de trifluorometanossulfonato de 1-metil- 1,2,3,6-tetra-hidropiridin-4-ila 257 (450 mg, 1,84 mmol), bis(pinacolato)diboro (477 mg, 2,02 mmols) e K2CO3 (541 mg, 5,52 mmols) em DME (3,7 mg), Pd(PPh3)4 (106 mg, 0,092 mmol) foi adicionado em uma porção e o sistema foi aquecido até o refluxo durante 2 horas sob N2. A mistura de reação foi resfriada e filtrada. Ao filtrado contendo o intermediário 258 foram adicionados brometo 50 (435 mg, 1,84 mmol), CsF (838 mg, 5,52 mmols), NaHCO3 (463 mg, 5,52 mmols) e água (0,8 mL), e a mistura foi aquecida até o refluxo durante mais 2 horas. A mistura de reação foi diluída com água e extraída com DCM; A fase orgânica foi extraída com HCl a 1N, A fase aquosa basificada para pH ~11 pela adição de NaOH a 2N aquoso, extraída com DCM, secada sobre Na2SO4 anidro e concentrada sob pressão reduzida fornecendo 259 (279 mg, 0,72 mmol, 39% de rendimento) como um sólido marrom. MS (m/z): (M+1) 385,9 (100%). Etapa 3: 3-Flúor-4-(2-(1-metil-1,2,3,6-tetra-hidropiridin-4-il)tieno[3,2- b]piridin-7-ilóxi)anilina (260)
[00733] A uma mistura de 4-(7-(2-flúor-4-nitrofenóxi)tieno[3,2- b]piridina-2-il)-1-metil-1,2,3,6-tetra-hidropiridina 259 (279 mg, 0,72 mmol) e NH4Cl (33 mg, 0,612 mmol) em EtOH (7,2 mL)/água (3,6 mL) a 100°C, Fe (342 mg, 6,2 mmols) foi adicionado em uma porção e a mistura aquecida até o refluxo com vigorosa agitação durante 40 minutos. A mistura foi filtrada através de celite®, o Celite® lavado com EtOH e as soluções orgânicas combinadas concentradas sob pressão reduzida. O resíduo foi dissolvido em MeOH e purificado por cromatografia instantânea (DCM/MeOH 5:1) fornecendo 260 (171,3 mg, 0,48 mmol, 67% de rendimento) como um sólido amarelo. MS (m/z): (M+1) 356,0 (100%). Etapa 4:N 1-(3-Flúor-4-(2-(1-metil-1,2,3,6-tetra-hidropiridin-4- il)tieno[3,2-b]piridin-7-ilóxi)fenil)-N3-metil-N3-fenilmalonamida (256)
[00734] Seguindo o procedimento descrito acima para o composto 5c (Esquema 3) porém substituindo o ácido 1 com o ácido 31 e amina 12 com amina 260; o composto do título 256 foi obtido em 44% de rendimento. 1H RMN (400 MHz, DMSO-d6) δ (ppm): 10,31 (s, 1H), 8,44 (d, J = 5,48Hz, 1H), 7,79 (d, 12,72Hz, 1H), 7,5-7,3 (m, 8H), 6,59 (d, J = 5,48Hz, 1H), 6,38 (t, J = 3,52Hz, 1H), 3,22 (s, 2H), 3,21 (s, 3H), 3,08 (m,2H), 2,61 (m, 4H), 2,30 (s, 3H). MS (m/z): (M+1) 531,0 (100 %). Tabela 8
[00735] Compostos 261-263 (Exemplos 108-110) preparados partindo da amina 260 (Esquema 66) e ácidos 161, 203 e 212 de acordo com o Esquemas 45, 57 e 59
Esquema 67
Exemplo 111 N-(3-Flúor-4-(2-(1 -metil-1,2,3,6-tetra-hidropiridin-4-il)tieno[3,2-b]piridin- 7-ilóxi)fenil)-3-(4-fluorofenil)-2-oxoimidazolidina-1-carboxamida (264) Etapa 1: 4-(Trifluorometilsulfonilóxi)-5,6-di-hidropiridina-1 (2 H) - carboxilato de terc - butila (265)
[00736] LDA (1,5 N em THF, 7,2 mL, 10,68 mmols) foi adicionado a uma solução de 4-oxopiperidina-1-carboxilato de terc-butila (1,94 mL, 9,74 mmols) em THF (13 mL) a -78°C, a mistura foi aquecida até a temperatura ambiente e agitada durante 30 minutos. A solução foi resfriada para -78°C e 1,1,1-triflúor-N-(piridin-2-il)-N- (trifluorometilssulfonil) metanossulfonamida (4,0 g, 11,2 mmols) foi adicionado em uma porção, a solução foi aquecida até a temperatura ambiente e agitada durante 1 hora. A mistura foi diluída com EtOAc, lavada com HCl a 1N, água, salmoura e concentrada sob pressão reduzida para um volume mínimo, e filtrada. O filtrado foi coletado, novamente concentrado e o resíduo foi destilado sob pressão reduzida fornecendo 265 (2,80 g, 8,17 mmols, 86% de rendimento) como líquido marrom. MS (m/z): (M - Boc +1) 232,1 (26%). Etapa 2: 4-(7-(2-Flúor-4-nitrofenóxi)tieno[3,2-b]piridin-2-il)-5,6-di- hidropiridina-1 (2H)-carboxilato de terc - butila (267)
[00737] Seguindo o procedimento descrito acima para o composto 259 (Esquema 66) porém substituindo composto 257 com 4- (trifluorometilsulfonilóxi)-5,6-di-hidropiridina-1 (2H)-carboxilato de terc- butila (265); o composto do título 267 foi obtido em 56% de rendimento. MS (m/z): (M+1) 472,5 (25%). Etapa 3: 7-(2-Flúor-4-nitrofenóxi)-2-(1,2,3,6-tetra-hidropiridin-4- il)tieno[3,2-b]piridina (268)
[00738] A uma solução de 4-(7-(2-flúor-4-nitrofenóxi)tieno[3,2- b]piridin-2-il)-5,6-di-hidropiridina-1 (2H)-carboxilato de terc-butila 267 (1 g, 2,13 mmols) em DCM (4,3 mL), TFA (4,3 mL) foi adicionado e a mistura de reação foi agitada durante 2 horas em temperatura ambiente. O solvente foi removido sob pressão reduzida, o resíduo foi suspenso em bicarbonato de sódio aquoso, a mistura extraída com DCM, EtOAc e DCM; as fases orgânicas combinadas foram filtradas e os sólidos recuperados secos. A fase orgânica foi seca sobre Na2SO4 anidro, concentrada sob pressão reduzida e o resíduo foi combinado com o sólido material obtido anteriormente para fornecer 268 (806 mg, 2,12 mmols, 100% de rendimento) como um sólido amarelo. MS (m/z): (M+1) 372,1 (100%). Etapa 4: 1 -(4-(7-(2-Flúor-4-nitrofenóxi)tieno[3,2-b]piridina-2-il)-5,6-di- hidropiridin-1 (2H)-il)etanona (269)
[00739] A uma suspensão de 4-(7-(2-flúor-4-nitrofenóxi)tieno[3,2- b]piridina-2-il)-1,2,3,6-tetra-hidropiridina 268 (400 mg, 1,08 mmol) e DIPEA (0,207 mL, 1,19 mmol) em DCM foi adicionado cloreto de acetila (0,15 mL, 2,16 mmols) em temperatura ambiente e a mistura (que se tornou homogênea logo depois) foi agitada durante 1 hora. A mistura bruta foi concentrada sob pressão reduzida e o resíduo purificado por cromatografia instantânea (5% de MeOH a 10% de MeOH em DCM) fornecendo 269 (358,2 mg, 0,87 mmol, 80% de rendimento) como um sólido branco. MS (m/z): (M+1) 414,4 (100%). Etapa 5: 1 -(4-(7-(4-Amino-2-fluorofenóxi)tieno[3,2-b]piridin-2-il)-5,6-di- hidropiridin-1 (2H)-il)etanona (270)
[00740] A uma mistura de 1-(4-(7-(2-flúor-4-nitrofenóxi)tieno[3,2- b]piridin-2-il)-5,6-di-hidropiridin-1 (2H)-il)etanona (269) (358,2 mg, 0,87 mmol) e NH4Cl (39,4 mg, 0,74 mmol) em EtOH (8,7 mL)/água (4,3 mL) a 100°C foi adicionado Fe (411,3 mg, 7,36 mmols) em uma porção e a mistura aquecida até o refluxo com vigorosa agitação durante 40 minutos. A mistura foi filtrada através de celite®, o Celite® lavado com EtOH e as soluções orgânicas combinadas concentradas sob pressão reduzida. O resíduo foi dissolvido em DCM, extraído com água, secado sobre Na2SO4 anidro e concentrado sob pressão reduzida fornecendo 270 (256,5 mg, 0,67 mmol, 77% de rendimento) como um sólido branco. MS (m/z): (M+1) 384,2 (100%). Etapa 6: N-(3-Flúor-4-(2-(1-metil-1,2,3,6-tetra-hidropiridin-4-il)tieno[3,2- b]piridin-7-ilóxi)fenil)-3-(4-fluorofenil)-2-oxoimidazolidina-1- carboxamida (264)
[00741] Trifosgene (209,2, 0,47 mmol) foi adicionado a uma solução de 1-(4-fluorofenil)imidazolidin-2-ona (209,2 mg, 0,71 mmol) em THF (4,7 mL) e a mistura foi aquecida até o refluxo durante 6 horas. 1-(4-(7- (4-Amino-2-fluorofenóxi)tieno[3,2-b]piridin-2-il)-5,6-di-hidropiridin-1 (2H)-il)etanona 270 (120,0 mg, 0,31 mmol) e DIPEA (0,1 mL, 0,5 mmol) foram adicionados e a mistura foi agitada durante 1 hora em temperatura ambiente. A mistura de reação foi transferida para uma coluna de cromatografia instantânea de sílica-gel e eluída com 3% de MeOH em DCM fornecendo 264 (97,6 mg, 0,16 mmol, 52% de rendimento) como um sólido branco. 1H RMN (400 MHz, DMSO-d6) δ (ppm):10,55 (s, 1H), 8,47 (d, 5,5Hz, 1H), 7,84 (dd, J = 2,4Hz, 13,1Hz, 1H), 7,71-7,63 (m, 2H), 7,54 (m, 1H), 7,48-7,42 (m, 2H), 7,31-7,27 (m, 2H), 6,63 (m,1H), 6,42 (m, 1H), 4,21 (br, 1H), 4,15 (br, 1H), 3,96-3,93 (m, 4H), 3,71-3,65 (m, 2H), 2,70 (br, 1H), 2,59 (br, 1H), 2,09 (s, 1,5H), 2,05 (s, 1,5H). MS (m/z): (M+1) 590,2 (100 %). Esquema 68

N1-(3-Flúor-4-(2-(3-hidroxiazetidina-1-carbonil)tieno[3,2-b]piridin-7- ilóxi)fenil)-N3-(2-metoxifenil)malonamida (271) Etapa 1(3-(terc - Butildimetilsililóxi)azetidin-1-il)(7-(2-flúor-4- nitrofenóxi)tieno [3,2-b]piridin-2-il)metanona (273)
[00742] Uma mistura de (3-(terc-butildimetilsililóxi)azetidin-1-il)(7- clorotieno[3,2-b]piridin-2-il)metanona 272 (749,7 mg, 1,96 mmol) (WO 2006/010264), 2-flúor-4-nitrofenol (461 mg, 2,94 mmols), K2CO3 (518 mg, 3,92 mmols) e Ph2O (2,6 mL) foi agitada em um tubo selado durante 1,5 hora a 170°C. A mistura foi diluída com DCM, extraída com água, a fase orgânica secada sobre Na2SO4 anidro e concentrada. O resíduo foi purificado por cromatografia instantânea (eluída sucessivamente com: EtOAc/hexanos 1:1, EtOAc, EtOAc/MeOH 4:1) fornecendo 273 (336 mg, 0,67 mmol, 34% de rendimento). MS (m/z): (M+1) 504,1 (100%). Etapa 2: (7-(4-amino-2-fluorofenóxi)tieno[3,2-b]piridin-2-il)(3-(terc- butildimetilsililóxi)azetidin-1-il)metanona (274)
[00743] A uma solução de (3-(terc-butildimetilsililóxi)azetidin-1-il)(7- (2-flúor-4-nitrofenóxi)tieno[3,2-b]piridin-2-il)metanona 273 (336 mg, 0,67 mmol) e NiCl2,6H2O (318 g, 1,34 mmol) em MeOH/THF (4,7 mL/4,7 mL) foi adicionado NaBH4 (101 g, 2,68 mmols) a 0°C e a mistura agitada durante 1 hora. A mistura de reação foi adicionada a uma solução de EDTA,4Na (1,1g/100 mL) e extraída com EtOAc. A camada aquosa foi filtrada e extraída novamente com EtOAc. A fase orgânica combinada foi seca sobre Na2SO4 anidro e concentrada sob pressão reduzida. O resíduo foi purificado por cromatografia instantânea (EtOAc puro) fornecendo 274 como espuma cremosa (145,2 mg, 46% de rendimento). MS (m/z): (M+1) 474,1 (100%). Etapa 3:N 1-(4-(2-(3-(terc - Butildimetilsililóxi)azetidina-1- carbonil)tieno[3,2-b]piridin-7-ilóxi)-3-fluorofenil)-N3-(2- metoxifenil)malonamida (275)
[00744] Uma mistura de (7-(4-amino-2-fluorofenóxi)tieno[3,2- b]piridin-2-il)(3-(terc-butildimetilsililóxi)azetidin-1-il)metanona 274 (145,2 mg, 0,31 mmol), 27 (77 mg, 0,367 mmol), e EDC (70,3 mg, 0,367 mmol) em DMF (4,4 mL) foi agitada durante a noite em temperatura ambiente. A mistura bruta foi diluída com EtOAc, extraída com água, secada sobre Na2SO4 anidro e concentrada sob pressão reduzida fornecendo 275 bruto (178,4 mg, 0,269 mmol, 87% de rendimento) que foi usado na próxima Etapa sem outra purificação. MS (m/z): (M+1) 665,1 (100%). Etapa 4: N 1-(3-Flúor-4-(2-(3-hidroxiazetidina-1-carbonil)tieno[3,2- b]piridin-7-ilóxi)fenil)-N3-(2-metoxifenil)malonamida x HCl (271)
[00745] A uma suspensão de N1-(4-(2-(3-(terc- butildimetilsililóxi)azetidina-1-carbonil)tieno [3,2-b]piridin-7-ilóxi)-3- fluorofenil)-N3-(2-metoxifenil) malonamida 275 (179 mg, 0,268 mmol) em MeOH (18 mL), HCl (1N em dioxano, 0,54 mL, 0,54 mmol) foi adicionado em gotas e a mistura foi agitada durante 15 minutos em temperatura ambiente. O solvente foi removido sob pressão reduzida, MeOH foi adicionado ao resíduo e concentrado, o resíduo foi suspenso em água e liofilizado fornecendo 271 (104,6 mg, 0,18 mmol, 67% de rendimento) como um sólido branco. 1H RMN (400 MHz, DMSO-d6) δ (ppm): 10,68 (s, 1H), 9,64 (s, 1H), 8,64 (d, J = 5,5Hz, 1H), 8,06 (dd, J = 1,9Hz, J = 9Hz, 1H), 7,94 (s, 1H), 7,89 (dd, J = 12,9Hz, J = 2,4Hz, 1H), 7,51 (t, J = 8,8Hz, 1H), 7,46 (dd, J = 1,6Hz, J = 8,8Hz, 1H), 7,11-7,04 (m, 2H), 6,92 (m, 1H), 6,84 (dd, J = 5,5Hz, J = 1HZ, 1H), 6,90 (m, 1H), 4,59 (m, 1H), 4,36-4,31 (m, 2H), 3,88-3,84 (m, 4H), 3,67 (s, 2H). MS (m/z): (M+1) 551,0 (100%). Esquema 69

Exemplo 113 N-(3-Flúor-4-(2-(1 -metil-1 H-imidazol-4-il)tieno[3,2-b]piridin-7-ilóxi)fenil)- 2-oxo-3-(piperidin-1-il)imidazolidina-1-carboxamida (276) Etapa 1: 1-(Piperidin-1-il)imidazolidin-2-ona (278)
[00746] A uma solução de piperidin-1-amina (5 mL, 46,32 mmols) em THF (46 mL) 1-cloro-2-isocianatoetano (4,35 mL, 50,95 mmols) foi adicionado em gotas a 0°C; após a adição ser completada, a mistura foi agitada durante 1 hora a 0°C. A reação foi extinguida com água, a mistura foi extraída com DCM e a fase orgânica foi concentrada sob pressão reduzida para produzir o cloreto 277 (não isolado). Este material foi dissolvido em THF (93 mL), NaH (60% em óleo mineral, 3,7 g, 93 mmols) foi adicionado a 0°C e a mistura agitada durante a noite. A solução extinguida com água, extraída com DCM, secada sobre Na2SO4 anidro e concentrada sob pressão reduzida. O resíduo foi triturado com hexanos e filtrado fornecendo 278 (7,2 g, 43 mmols, 92% de rendimento) como um sólido branco. MS (m/z): (M+1) 191,9 (100%). Etapa 2: N-(3-Flúor-4-(2-(1 -metil-1 H-imidazol-4-il)tieno[3,2-b]piridin-7- ilóxi) fenil)-2-oxo-3-(piperidin-1-il)imidazolidina-1-carboxamida (276)
[00747] Trifosgene (107 mg, 0,36 mmol) foi adicionado a uma solução de 1-(piperidin-1-il)imidazolidin-2-ona (278) (120 mg, 0,71 mmol) em THF (7,1 mL) e a mistura foi refluxada 6 horas. A solução foi resfriada. 3-Flúor-4-(2-(1-metil-1H-imidazol-4-il)tieno[3,2-b]piridin-7- ilóxi)anilina (12) (150,0 mg, 0,44 mmol) e DIPEA (0,186 mL, 1,07 mmol) foram adicionados e a mistura agitada 1 hora em temperatura ambiente. A mistura de reação foi filtrada, a solução transferida para uma coluna de cromatografia instantânea e eluída com 2% a 5% de MeOH em DCM fornecendo o composto do título 276 (176,4 mg, 0,33 mmol, 75% de rendimento) como um sólido cremoso. 1H RMN (400 MHz, DMSO-d6) δ (ppm): 10,57 (s, 1H), 8,4 (d, J = 5,4Hz, 1H), 7,86 (s, 1H), 7,79 (dd, J = 2Hz, J = 12,9Hz, 1H), 7,72 (s, 1H), 7,68 (s, 1H), 7,46 (t, J = 8,9Hz, 1H), 7,36 (m, 1H), 6,58 (d, 5,4Hz, 1H), 3,76 (dd, J = 7,7Hz, J = 8,2Hz, 2H), 3,73 (s, 3H), 3,54 (dd, J = 8,2Hz, J = 7,7Hz, 2H), 2,9-2,87 (m, 4H), 1,59 (m, 4H), 1,35 (m, 2H). MS (m/z): (M+1) 536,2 (100 %) Exemplo 114 3-Cicloexil-N-(3-flúor-4-(2-(1 -metil-1 H-imidazol-4-il)tieno[3,2-b]piridin-7- ilóxi)fenil)-2-oxoimidazolidina-1-carboxamida (279) Etapa 1: 1-(Piperidin-1-il)imidazolidin-2-ona (281)
[00748] Seguindo o procedimento descrito acima para o composto 278 (Esquema 69) porém substituindo piperidin-1-amina com ciclo- hexilamina, o composto do título 281 foi obtido em 23% de rendimento (via o intermediário 280). MS (m/z): (M+1) 169,2 (56%), (2M+23) 359,3 (100%). Etapa 2: 3-Cicloexil-N-(3-flúor-4-(2-(1-metil-1H-imidazol-4-il)tieno[3,2- b]piridin-7-ilóxi)fenil)-2-oxoimidazolidina-1-carboxamida (279)
[00749] Seguindo o procedimento descrito acima para o composto 276 (Exemplo 113) porém substituindo 1-(piperidin-1-il)imidazolidin-2- ona (278) por 1-ciclo-hexilimidazolidin-2-ona (281), o composto do título 279 foi obtido em 68 % de rendimento. 1H RMN (400 MHz, DMSO-d6) δ (ppm): 10,69 (s, 1H), 8,44 (dd, J = 5,5Hz, 1H), 7,86 (s, 1H), 7,79 (dd, J = 2,5Hz, J = 13Hz, 1H), 7,72 (s, 1H), 7,68 (s, 1H), 7,46 (t, J = 8,9Hz, 1H), 7,35 (d, J = 5,5Hz, 1H), 6,58 (d, J = 5,5Hz, 1H), 3,8 (dd, J = 7,6Hz, J = 8,0Hz, 2H), 3,73 (s, 3H), 3,7-3,6 (m, 1H), 3,44 (dd, J = 7,2Hz, J = 8,0Hz, 2H), 1,79-1,60 (m, 5H), 1,47-1,41 (m, 2H), 1,361,29 (m, 2H), 1,7 (m, 1H). MS (m/z): (M+1) 535,2. Esquema 70
Exemplo 115 N-(3-Flúor-4-(2-(1 -metil-1H-imidazol-4-il)tieno[3,2-b]piridin-7-ilóxi)fenil)- 2-oxo-3-fenilimidazolidina-1-carbotioamida (282)
[00750] Tiofosgene (0,052 mL, 0,68 mmol) foi adicionado a uma solução de 1-fenilimidazolidin-2-ona (100 mg, 0,62 mmol) em THF (6,2 mL) e a mistura foi aquecida a 50 °C durante a noite. A amina 12 (150,0 mg, 0,44 mmol) e DIPEA (0,115 mL, 0,66 mmol) foram adicionados e a mistura foi agitada durante 1 hora em temperatura ambiente. A mistura foi diluída com DCM, a solução foi extraída com 3% de solução de ácido cítrico, secada (Na2SO4) e concentrada sob pressão reduzida. O resíduo foi purificado por cromatografia instantânea (MeOH/DCM) 1:9 fornecendo 282 (143 mg, 0,269 mmol, 61% de rendimento) como um sólido marrom. 1H RMN (400 MHz, DMSO-d6) δ (ppm): 12,37 (s, 1H), 8,47 (d, J = 5,4Hz, 1H), 8,06 (dd, J = 2Hz, J = 11,3Hz, 1H), 7,87 (d, J = 1Hz, 1H), 7,71 (m, 2H), 7,63 (m, 2H), 7,54 (m, 2H), 7,45 (m, 2H),7,21 (dd, J = 7,2Hz, J = 7,4Hz, 1H), 6,61 (d, 5,4Hz, 1H), 4,24 (dd, J = 7,9Hz, J = 6,5Hz, 2H) 3,98 (dd, J = 7,9Hz, J = 6,5Hz, 2H), 3,72 (s, 3H). MS (m/z): (M+1) 545,1 (100 %). Exemplo 116
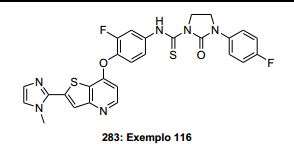
N-(3-Flúor-4-(2-(1 -metil-1 H-imidazol-2-il)tieno[3,2-b]piridin-7-ilóxi)fenil)- 3-(4-fluorofenil)-2-oxoimidazolidina-1-carboxamida (283)
[00751] O composto do título 283 foi obtido similarmente ao composto 282 (Exemplo 115) de acordo com o Esquema 69, partindo de 1-(4-fluorofenil)imidazolidin-2-ona e amina 9 (Esquema 2). 1H RMN (400 MHz, DMSO-d6) δ (ppm) 12,34 (s, 1H), 8,55 (d, J = 5,48 Hz, 1H), 8,05 (d, J = 13,1 Hz, 1H), 7,91 (s, 1H), 7,64 (m, 2H), 7,54 (m, 2H), 7,42 (s, 1H), 7,30 (t, J = 8,80 Hz, 2H), 7,05 (s, 1H), 6,71 (d, J = 5,28 Hz, 1H), 4,23 (t, J = 7,43 Hz, 2H), 3,98 (m, 5H). MS (m/z): 563,2 (M+H). Esquema 71
Exemplo 117 N 1-(3-Flúor-4-(2-(2-oxopirrolidin-1-il)tieno[3,2-b]piridin-7-ilóxi)fenil)-N3- (2-metoxifenil)malonamida (284) Etapa 1: 1-(7-(4-Amino-2-fluorofenóxi)tieno[3,2-b]piridin-2-il)pirrolidin-2- ona (285)
[00752] Uma mistura da amina 225 (200 mg, 0,59 mmol) (Esquema 61), trans-N1, N2-dimetilciclo-hexano-1,2-diamina (17 mg, 0,118 mmol), pirrolidin-2-ona (0,054 mL, 0,71 mmol), CuI (22 mg, 0,118 mmol) e K3PO4 (250 mg, 1,18 mmol) em dioxano (0,6 mL) foi agitada sob nitrogênio a 70°C durante a noite. A mistura bruta foi purificada por cromatografia instantânea (MeOH/DCM 1:19) fornecendo o composto do título 285 (83,7 mg, 0,243 mmol, 41% de rendimento) como um sólido laranja. MS (m/z): (M+1) 344,0 (100%). Etapa 2: N 1-(3-Flúor-4-(2-(2-oxopirrolidin-1-il)tieno[3,2-b]piridin-7- ilóxi)fenil)-N3-(2-metoxifenil)malonamida (284)
[00753] Uma solução da amino lactama 285 (80,7 mg, 0,243 mmol), ácido 3-(2-metoxifenilamino)-3-oxopropanoico 27 (76,1 mg, 0,365 mmol), EDC (70 mg, 0,365 mmol) e HOBt (56 mg, 0,365 mmol) em DMF (2,4 mL) foi agitada durante a noite em temperatura ambiente. Mais 27 (76,1 mg, 0,365 mmol) e EDC (70 mg, 0,365 mmol) foram adicionados e a mistura foi agitada mais 6 horas. A mistura foi diluída com EtOAc, extraída com água, secada sobre Na2SO4 anidro, e concentrada sob pressão reduzida. O resíduo foi cristalizado de MeCN fornecendo o composto do título 284 (24 mg, 0,044 mmol, 18% de rendimento). 1H RMN (400 MHz, DMSO-d6) δ (ppm): 10,73 (s, 1H), 9,63 (s, 1H), 8,64 (d, J = 6,5Hz, 1H), 8,06 (dd, J = 10Hz, J = 1,5Hz, 1H), 7,92 (dd, J = 13,1Hz, J = 2,3Hz, 1H), 7,59-7,48 (m, 2H), 7,12-7,05 (m, 3H), 6,94-6,90 (m, 2H), 4,07 (dd, J = 7,0Hz, J = 7,4Hz, 2H), 3,86 (s, 3H), 3,67 (s, 2H), 2,69 (dd, J = 7,8Hz, J = 8,2Hz, 2H), 2,24 (m, 2H). MS (m/z): (M+1) 535,1 (100 %). Esquema 72
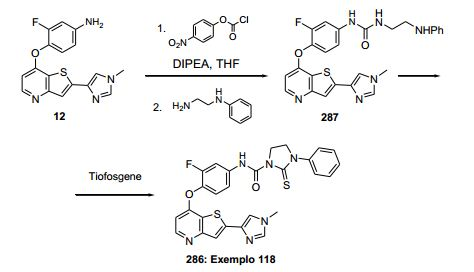
Exemplo 118 N-(3-Flúor-4-(2-(1 -metil-1 H-imidazol-4-il)tieno[3,2-b]piridin-7-ilóxi)fenil)- 3-fenil-2-tioxoimidazolidina-1-carboxamida (286)
[00754] A uma mistura de 12 (150 mg, 0,44 mmol) e DIPEA (0,084 mL, 0,48 mmol) em THF (4,5 mL), carbamato de 4-nitrofenila (97 mg, 0,48 mmol) foi adicionado e a mistura agitada 30 minutos em temperatura ambiente. Em seguida N1-feniletano-1,2-diamina (0,086 mL, 0,66 mmol) foi adicionado e a mistura foi agitada durante mais 1 hora, para formar o intermediário 287 (não isolado). Tiofosgene (0,05 mL, 0,66 mmol) e DIPEA (0,232 mL, 1,38 mmol) foram adicionados e a mistura foi agitada durante a noite em temperatura ambiente. Mais tiofosgene (0,05 mL, 0,66 mmol) foi adicionado e a mistura foi aquecida até o refluxo durante mais 3 horas. A mistura foi diluída com DCM, extraída com água, secada sobre Na2SO4 anidro e concentrada sob pressão reduzida. O resíduo foi purificado por cromatografia instantânea (5% de MeOH em DCM) seguido por trituração do sólido resultante com MeOH fornecendo o composto do título 286 (61 mg, 0,11 mmol, 25%) como um sólido cremoso. 1H RMN (400 MHz, DMSO-d6) δ (ppm): 12,56 (s, 1H), 8,45 (d, J = 5,5Hz, 1H), 7,87 (d, J = 1,2Hz, 1H), 7,82 (dd, J = 2,5Hz, J = 12,7Hz, 1H), 7,73 (d, J = 0,8Hz, 1H), 7,67 (s, 1H), 7,52-7,48 (m, 5H), 7,42-7,35 (m, 2H), 6,31 (dd, J = 5,5Hz, J = 0,8Hz, 1H), 4,27-4,22 (m, 2H), 4,13-4,09 (m, 2H), 3,73 (s, 3H). MS (m/z): (M+1) 545,2 (100 %). Tabela 9
[00755] Compostos 288-290 (Exemplos 119-121) preparados partindo da amina 197 (Esquema 54) e ácidos 1, 161 e 177
Esquema 73
4-Iodo-1-(3-(pirrolidin-1-il)propil)-1 H-imidazol (293) Etapa 1. 1-(3-Cloropropil)-4-iodo-1 H-imidazol (292)
[00756] A uma solução agitada de 4-iodo-1H-imidazol (291, 2 g, 10,3 mmols) [a) Y. He e outro., Tet. Lett. 45, 2004, 5529-5532. b) Panosyan, F.B., Still, I.W.J., Can. J. Chem. 79, 2001, 1110-1114] em tetra-hidrofurano seco (40 mL) a 0°C sob nitrogênio foi adicionado hidreto de sódio (60% em óleo, 0,91 g, 22,7 mmols). A mistura foi agitada durante 20 minutos a 0°C, 1-bromo-3-cloropropano (1,2 mL, 12,4 mmols) foi adicionado e agitação foi continuada durante 24 horas em temperatura ambiente. Água foi adicionada e a solução aquosa foi extraída duas vezes com acetato de etila. As camadas orgânicas combinadas foram secas sobre sulfato de sódio anidro e o solvente foi removido sob pressão reduzida. O resíduo foi purificado por cromatografia de coluna em sílica-gel (eluente acetato de etila- diclorometano, 4:96) para fornecer 292 (1,35 g, 5,0 mmols, 49% de rendimento) como um óleo amarelo claro. 1H RMN (400 MHz, DMSO- d6) δ ppm: 7,63 (d, J = 1,2 Hz, 1H), 7,42 (d, J = 1,2 Hz, 1H), 4,08 (t, J = 6,8 Hz, 2H), 3,55 (d, J = 6,8 Hz, 2H), 2,16 (q, J = 6,8 Hz, 2H). MS (m/z): 271,0 (M+H, 100%), 273,0 (M+H, 32%). Etapa 2: 4-Iodo-1-(3-(pirrolidin-1-il)propil)-1 H-imidazol (293)
[00757] Uma solução do cloreto 292 (1,2 g, 4,44 mmols) e pirrolidina (1,1 mL, 13,3 mmols) em DMSO seco (2 mL) foi aquecida a 60°C sob nitrogênio durante 3 horas. A mistura de reação foi resfriada, diluída com água e a fase aquosa foi extraída duas vezes com acetato de etila. As camadas orgânicas combinadas foram secas sobre sulfato de sódio anidro e o solvente foi removido sob pressão reduzida. O resíduo foi purificado por cromatografia de coluna em sílica-gel (eluente metanol-diclorometano, 5:95 a 20:80) para fornecer 293 (1,06 g, 3,47 mmols, 78% de rendimento) como um óleo incolor. 1H RMN (400 MHz, DMSO-d6) δ ppm: 7,59 (d, J = 1,2 Hz, 1H), 7,39 (d, J = 1,2 Hz, 1H), 3,98 (t, J = 7,2 Hz, 2H), 2,45-2,33 (m, 4H), 2,29 (t, J = 7,2 Hz, 2H), 1,84 (q, J = 7,2 Hz, 2H), 1,72-1,62 (m, 4H). MS (m/z): 305,9 (M+H).
[00758] Imidazol 293 foi usado para a síntese de compostos 294299 (Exemplos 122-127), Tabela 10. Tabela 10
[00759] Compostos 294-299 (Exemplos 122-127) preparados de acordo com os Esquemas 46, 58 e 59
Tabela 11
[00760] Compostos 300-309 (Exemplos 128-137) preparados de acordo com os Esquemas 46, 58 e 59
Esquema 74
1) LDA, -78°C, THF 2) Mel, -78°Caté a t.a. 205 Ácido 1-(4-metil-1,2,3,4-tetra-hidroquinoxalina-1-carbonil) ciclopropanocarboxílico (311)
[00761] A uma solução de ácido ciclopropano-1,1-dicarboxílico (1 g, 7,69 mmol) em tetra-hidrofurano seco (20 mL) a 0°C sob nitrogênio, foi adicionado trietilamina (1,07 mL, 7,69 mmols) e a mistura foi agitada durante 30 minutos. Cloreto de tionila (0,56 mL, 7,69 mmols) foi adicionado, agitação foi continuada a 0°C durante 30 minutos, 1-metil- 1,2,3,4-tetra-hidroquinoxalina (310) (1,2 g, 8,46 mmols) [Smith R.F. et al., J. Org. Chem., 24, 1959, 205 ] foi adicionado e a mistura de reação foi deixada aquecer até a temperatura ambiente. Após agitação durante 1 hora, acetato de etila foi adicionado e a mistura resultante foi extraída duas vezes com uma solução de NaOH a 1N. As camadas aquosas combinadas foram acidificadas para pH 4-5 pela adição de uma solução de HCl a 3N e extraídas 4 vezes com acetato de etila. As camadas orgânicas combinadas foram secas sobre sulfato de sódio anidro e o solvente foi removido sob pressão reduzida. O resíduo foi purificado por Biotage (Si 12M, gradiente: MeOH em diclorometano 0% a 10%) para fornecer o composto do título 311 (638 mg, 32% de rendimento). MS (m/z): 259,0 (M-H). Ácido 3-(4-metil-3,4-di-hidroquinoxalin-1 (2 H-il)-3-oxopropanoico (312)
[00762] A uma solução de 3-cloro-3-oxopropanoato de metila (1 g, 7,32 mmols) e 1-metil-1,2,3,4-tetra-hidroquinoxalina (310) (1,09 g g, 7,32 mmols) [Smith R.F. et al., J. Org. Chem., 24, 1959, 205] em diclorometano seco (40 mL) a 0°C sob nitrogênio foi lentamente adicionado N,N-diisopropiletilamina (2,55 mL, 14,6 mmols). A mistura de reação foi lentamente aquecida até a temperatura ambiente (durante 45 minutos). Uma solução aquosa saturada de bicarbonato de sódio foi adicionada e a solução aquosa foi extraída com diclorometano. A camada orgânica foi seca sobre sulfato de sódio anidro e o solvente foi removido sob pressão reduzida. O resíduo foi purificado por cromatografia de coluna em sílica-gel (eluente acetato de etila-hexano 1:1) para fornecer um sólido esbranquiçado (1,82 g). Este sólido foi dissolvido em tetra-hidrofurano (40 mL) e água (20 mL), monoidrato de hidróxido de lítio (615 mg, 14,7 mmols) e a mistura de reação foi agitada 16 horas em temperatura ambiente. A solução foi acidificada para pH 4 com uma solução de HCl a 1H e extraída 4 vezes com acetato de etila. As camadas orgânicas combinadas foram secas sobre sulfato de sódio anidro e o solvente foi removido sob pressão reduzida. O resíduo foi purificado por Biotage (Si 25M, gradiente: MeOH em diclorometano 0% a 10%) e trituração em uma mistura de éter de etila -hexano para fornecer o composto do título 312 (1,27 g, 74%) como um sólido branco. MS (m/z): 235,1 (M+H). Ácido 3-metil-2-oxo-1-fenilpirrolidina-3-carboxílico (313)
[00763] A uma solução agitada de ácido 1-fenil-2-oxo-3- pirrolidinacarboxílico (205) (200 mg, 0,975 mmol) em tetra-hidrofurano seco (5 mL) a -78°C foi adicionado LDA (1,5M de solução em cicloexano, 1,63 mL, 2,44 mmols) e a mistura foi agitada durante 40 minutos. Iodometano (152 μL, 2,44 mmols) foi adicionado e a mistura de reação foi deixada aquecer até a temperatura ambiente e agitação foi continuada durante 16 horas. Água e acetato de etila foram adicionados. A camada aquosa foi coletada, acidificada para pH 4 com uma solução de HCl a 1N e extraída duas vezes com acetato de etila. As camadas orgânicas combinadas foram secas sobre sulfato de sódio anidro e o solvente foi removido sob pressão reduzida. O resíduo foi purificado por cromatografia de coluna em sílica-gel (eluente metanol- diclorometano 2:98) para fornecer o composto do título 313 (170 mg, 80% de rendimento) como um sólido amarelo. 1H RMN (400 MHz, DMSO-d6) δ (ppm): 7,67 (dd, J = 8,8, 1,2 Hz, 2H), 7,39 (dd, J = 8,8, 7,2 Hz, 2H), 7,16 (tt, J = 7,2, 1,2 Hz, 1H), 3,92-3,80 (m, 2H), 2,54-2,46 (m, 1H), 2,08-1,98 (m, 1H), 1,34 (s, 3H). Tabela 12
[00764] Compostos 314-316 (Exemplos 138-140) preparados de acordo com o Esquema 3 partindo da amina 12 e ácidos 311-313
Esquema 75
Exemplo 141 4,4,4-T riflúor-3-(3-flúor-4-(2-(1 -metil-1 H-imidazol-4-il)tieno[3,2-b ]piridin- 7-ilóxi)fenilamino)-N-fenilbutanamida (317) Etapa 1: N-(1-Etóxi-2,2,2-trifluoroetil)-3-flúor-4-(2-(1-metil-1 H-imidazol- 4-il)tieno[3,2-b ]piridin-7-ilóxi)benzenamina (318)
[00765] Uma mistura de 12 (Esquema 3) (500 mg, 1,47 mmol), trifluoroacetaldeído de etila hemiacetal(0,35 mL, 2,94 mmols) e monoidrato de ácido 4-toluenossulfônico (280 mg, 1,47 mmol) em etanol (25 mL) foi aquecida até o refluxo durante 48 horas. A mistura de reação foi concentrada e o resíduo foi purificado por cromatografia de coluna em sílica-gel (eluente metanol-diclorometano 5:95 a 8:92) para fornecer o composto do título 318 (470 mg, 1,01 mmol, 68% de rendimento). 1H RMN (400 MHz, DMSO-d6) δ ppm: 8,42 (d, J = 5,5 Hz, 1H), 7,85 (d, J = 1,2 Hz, 1H), 7,72 (d, J = 0,8 Hz, 1H), 7,67 (s, 1H), 7,29 (t, J = 9,2 Hz, 1H), 7,08-7,02 (m, 2H), 3,86 (dd, J = 9,2, 2,0 Hz, 1H), 6,52 (d, J = 5,5 Hz, 1H), 5,68 (qd, J = 10,4, 5,2 Hz, 1H), 3,72 (s, 3H), 3,76-3,59 (m, 2H), 1,15 (t, J = 7,0 Hz, 3H). LRMS (M+1) 467,0 (100%). Etapa 2: 2-(2,2,2-Trif1úor-1-(3-flúor-4-(2-(1 -metil-1 H-imidazol-4- il)tieno[3,2-b ]piridin-7-ilóxi)fenilamino)etil)malonato de dietila (319)
[00766] A uma solução de 318 (470 mg, 1,01 mmol) e malonato de dietila (0,17 mL, 1,11 mmol) em tetra-hidrofurano anidro (10 mL) sob nitrogênio foi adicionado hidreto de sódio (60% em óleo, 89 mg, 2,22 mmols). A mistura foi aquecida para refluxo durante 2 horas, resfriada, diluída com EtOAc e água e acidificada para pH 3 usando uma solução de HCl a 1N. A camada orgânica foi separada e a camada aquosa extraída duas vezes com EtOAc. Os extratos foram combinados, secados sobre sulfato de sódio e os solventes foram removidos sob pressão reduzida. O resíduo foi purificado por cromatografia de coluna em sílica-gel (eluente MeOH-diclorometano, gradiente 0:100 a 20:80) para fornecer 319 (490 mg, 0,84 mmol, 84% de rendimento). 1H RMN (400 MHz, DMSO-d6) δ ppm: 8,42 (d, J = 5,2 Hz, 1H), 7,85 (s, 1H), 7,72 (s, 1H), 7,67 (s, 1H), 7,24 (t, J = 9,2 Hz, 1H), 6,98 (dd, J = 13,6, 2,8 Hz, 1H), 6,72 (dd, J = 9,2, 2,8 Hz, 1H), 6,62 (d, J = 10,0 Hz, 1H), 6,46 (d, J = 5,2 Hz, 1H), 5,05-4,95 (m, 1H), 4,234,07 (m, 4H), 3,91 (d, J = 9,2 Hz, 1H), 3,72 (s, 3H), 1,18 (t, J = 7,0 Hz, 3H), 1,11 (t, J = 7,0 Hz, 3H). LRMS (M+1) 581,0 (100%). Etapa 3: Ácido 4,4,4-triflúor-3-(3-flúor-4-(2-(1-metil-1 H-imidazol-4- il)tieno[3,2-b ]piridin-7-ilóxi)fenilamino)butanóico (320)
[00767] Uma solução de 319 (490 mg, 0,84 mmol) e hidróxido de sódio (338 mg, 8,44 mmols) em água (0,7 mL) e etanol (3,4 mL) foi agitada em temperatura ambiente durante 48 horas. Os solventes foram removidos sob pressão reduzida e o resíduo foi dissolvido em água (20 mL). A solução foi neutralizada para pH 4 com uma solução de HCl a 3N e o sólido desse modo formado foi filtrado, enxaguado com água e secado. O sólido foi suspenso em tolueno seco (20 mL), aquecido até o refluxo durante 1 hora sob agitação contínua, e o solvente foi removido sob pressão reduzida. O resíduo foi purificado por cromatografia de coluna em sílica-gel (eluente MeOH- diclorometano, gradiente 10:90 a 50:50) e o sólido resultante foi triturado em uma mistura de diclorometano, acetato de etila e hexano, isolado por filtração, e secado sob vácuo elevado para fornecer o composto do título 320 (150 mg, 0,31 mmol, 37% de rendimento). LRMS (M+1) 480,9 (100%). Etapa 4 4,4,4-T riflúor-3-(3-flúor-4-(2-(1 -metil-1 H-imidazol-4- il)tieno[3,2-b ]piridin-7-ilóxi)fenilamino)-N-fenilbutanamida (317)
[00768] A uma solução agitada de 320 (150 mg, 0,31 mmol), anilina (43 μL, 0,47 mmol) e N, N-diisopropiletilamina (0,19 mL, 1,09 mmol) em N,N-dimetilformamida seca (4 mL) em temperatura ambiente foi adicionado reagente de HATU (356 mg, 0,94 mmol). A mistura foi agitada em temperatura ambiente durante 16 horas. Uma solução aquosa saturada de bicarbonato de sódio foi adicionada e a solução aquosa foi extraída duas vezes com EtOAc, secada sobre sulfato de sódio anidro e o solvente foi removido sob pressão reduzida. O resíduo foi purificado por cromatografia de coluna em sílica-gel (eluente MeOH-diclorometano, gradiente 3:97 a 8:92) para fornecer o composto do título 317 (111 mg, 0,20 mmol, 64% de rendimento) como um sólido branco. 1H RMN (400 MHz, DMSO-d6) δ ppm: 10,11 (s, 1H), 8,39 (d, J = 5,6 Hz, 1H), 7,85 (d, J = 1,2 Hz, 1H), 7,72 (d, J = 0,8 Hz, 1H), 7,66 (s, 1H), 7,56 (d, J = 7,2 Hz, 2H), 7,30 (t, J = 8,0 Hz, 2H), 7,21 (t, J = 8,8 Hz, 1H), 7,05 (t, J = 7,2 Hz, 1H), 6,86 (dd, J = 13,6, 2,4 Hz, 1H), 6,67 (dd, J = 8,8, 2,0 Hz, 1H), 6,57 (d, J = 8,8 Hz, 1H), 6,44 (d, J = 5,6 Hz, 1H), 4,86-4,53 (m, 1H), 3,72 (s, 3H), 2,92 (dd, J = 15,6, 3,2 Hz, 1H), 2,76 (dd, J = 15,6, 9,6 Hz, 1H). LRMS (M+1) 556,0 (100%). Tabela 13
[00769] Compostos 321-323 (Exemplos 142-144) preparados de acordo com o Esquema 75
Esquema 76
Exemplo 145 4,4,4-T riflúor- N -(3-flúor-4-(2-(1 -metil-1 H-imidazol-4-il)tieno[3,2-b ]piridin- 7-ilóxi)fenil)-3-(fenilamino)butanamida (324) Etapa 1: N-(1-Etóxi-2,2,2-trifluoroetil)benzenamina (325)
[00770] Uma solução de anilina (2 mL, 21,9 mmols), trifluoroacetaldeído de etila hemiacetal(2,6 mL, 21,9 mmols) e monoidrato de ácido p-toluenossulfônico (220 mg, 1,16 mmol) em etanol (25 mL) foi aquecida até o refluxo durante 3 horas sob agitação contínua. A mistura de reação foi resfriada, o solvente foi removido sob pressão reduzida e o resíduo foi dissolvido em EtOAc. A camada orgânica foi lavada com uma solução aquosa saturada de bicarbonato de sódio, secada sobre sulfato de sódio anidro e o solvente foi removido sob pressão reduzida para fornecer o composto do título 325 (4,16 g, bruto) como um óleo amarelo que foi usado diretamente para a próxima etapa. Etapa 2: 2-(2,2,2-Triflúor-1-(fenilamino)etil)malonato de dietila (326)
[00771] Uma solução de malonato de dietila (1,98 mL, 13,0 mmols) em tetra-hidrofurano anidro (10 mL) foi adicionada em gotas, durante 20 minutos, em uma dispersão de hidreto de sódio (60% em óleo, 0,52 g, 13,0 mmols) em tetra-hidrofurano seco (30 mL) a 0°C, após o que o composto 325 (2,6 g, 11,9 mmols) foi adicionado e a mistura foi agitada vigorosamente a refluxo durante 16 horas. A mistura de reação foi resfriada, acidificada para pH 3 usando uma solução de HCl a 1N. A camada aquosa foi extraída duas vezes com EtOAc. Os extratos foram combinados, secados sobre sulfato de sódio e os solventes foram removidos sob pressão reduzida. O resíduo foi purificado por cromatografia de coluna em sílica-gel (eluente diclorometano-hexano, 0:100 a 60:40) para fornecer o composto do título 326 (2,16 g, 6,48 mmols, 54% de rendimento) como um óleo incolor. 1H RMN (400 MHz, CDCl3) δ ppm: 7,21-7,16 (m, 2H), 6,81-6,76 (m, 1H), 6,75-6,71 (m, 2H), 5,06 (d, J = 10,4 Hz, 1H), 4,83-4,73 (m, 1H), 4,25 (q, J = 7,2 Hz, 2H), 4,18-4,05 (m, 2H), 3,85 (d, J = 4,4 Hz, 1H), 1,28 (t, J = 7,2 Hz, 3H), 1,13 (t, J = 7,2 Hz, 3H). LRMS (M+1) 334,1 (100%). Etapa 3: Ácido 4,4,4-triflúor-3-(fenilamino)butanóico (327)
[00772] Uma solução de composto 326 (2,16 g, 6,48 mmols) e hidróxido de sódio (2,60 g, 64,8 mmols) em água (5,2 mL) e etanol (26 mL) foi agitada em temperatura ambiente durante 24 horas. Os solventes foram removidos sob pressão reduzida deixando um sólido branco que foi triturado em éter, isolado por filtração, enxaguado com éter e secado sob vácuo elevado. O sólido branco foi dissolvido em água (12 mL), e a solução foi neutralizada para pH 4 com uma solução de HCl a 3N, extraída duas vezes com EtOAc, as camadas orgânicas combinadas foram secas sobre sulfato de sódio e o solvente removido sob pressão reduzida. O sólido foi dissolvido em tolueno seco (20 mL), aquecido até o refluxo durante 1 hora com agitação contínua, e o solvente foi removido sob pressão reduzida. O resíduo foi purificado por cromatografia de coluna em sílica-gel (eluente EtOAc-hexano, gradiente 0:100 a 40:60) para fornecer o composto do título 327 (204 mg, 0,87 mmol, 13% de rendimento) como um sólido bege. 1H RMN (400 MHz, CDCl3) δ ppm: 7,24-7,18 (m, 2H), 6,82 (tt, J = 7,2, 1,0 Hz, 1H), 6,75-6,71 (m, 2H), 4,55-4,45 (m, 1H), 2,89 (dd, J = 16,0, 4,4 Hz, 1H), 2,67 (dd, J = 16,0, 8,8 Hz, 1H). LRMS (M-1) 231,9 (100%). Etapa 4: 4,4,4-Triflúor- N-(3-flúor-4-(2-(1-metil-1 H-imidazol-4- il)tieno[3,2-b]piridin-7-ilóxi)fenil)-3-(fenilamino)butanamida (324)
[00773] A uma solução agitada de composto 12 (Esquema 3) (100 mg, 0,29 mmol), composto 327 (103 mg, 0,44 mmol) e N,N- diisopropiletilamina (0,18 mL, 1,03 mmol) em N,N-dimetilformamida seca (3 mL) a 0°C foi adicionado reagente de HATU (335 mg, 0,88 mmol). A mistura foi agitada em temperatura ambiente durante 16 horas. Uma solução aquosa saturada de bicarbonato de sódio foi adicionada e a solução aquosa foi extraída duas vezes com EtOAc, secada sobre sulfato de sódio anidro e o solvente foi removido sob pressão reduzida. O resíduo foi purificado por cromatografia de coluna em sílica-gel (eluente MeOH-diclorometano, gradiente 0:100 a 15:85). O sólido resultante foi triturado em metanol, filtrado e secado sob pressão reduzida para fornecer o composto do título 324 (81 mg, 0,15 mmol, 50% de rendimento) como um sólido branco. 1H RMN (400 MHz, DMSO-d6) δ ppm: 10,50 (s, 1H), 8,41 (d, J = 5,6 Hz, 1H), 7,86 (d, J = 1,2 Hz, 1H), 7,81 (dd, J = 13,2, 2,0 Hz, 1H), 7,72 (s, 1H), 7,67 (s, 1H), 7,45 (t, J = 8,8 Hz, 1H), 7,37 (dd, J = 8,8, 1,6 Hz, 1H), 7,10 (dd, J = 8,4, 7,2 Hz, 2H), 6,75 (d, J = 8,0 Hz, 2H), 6,61 (t, J = 7,2 Hz, 1H), 6,55 (d, J = 5,6 Hz, 1H), 6,13 (d, J = 9,2 Hz, 1H), 4,75-4,65 (m, 1H), 3,72 (s, 3H), 2,94 (dd, J = 15,6, 3,6 Hz, 1H), 2,78 (dd, J = 15,9, 9,6 Hz, 1H). LRMS (M+1) 556,0 (100%). Tabela 14
[00774] Compostos 328-329 (Exemplos 146-147) preparados de acordo com o Esquema 76
Esquema 77
Exemplo 148 N 1-(4-(2-(3,4-Bis(3-(dimetilamino)propóxi)fenil)tieno[3,2-b ]piridin-7- ilóxi)-3-fluorofenil)-N 3-(2-metoxifenil)malonamida (330) Etapa 1: 2-(3,4-Dimetoxifenil)-7-(2-flúor-4-nitrofenóxi)tieno[3,2- b]piridina (331)
[00775] A uma solução agitada de 50 (400 mg, 1,08 mmol) em DME (20 mL) foram adicionados ácido 3,4-dimetoxifenilborônico (394 mg, 2,17 mmols), NaHCO3 (273 mg, 3,25 mmols), CsF (494 mg, 3,25 mmols), Pd(PPh3)4 (125 mg, 0,11 mmol) e água (10 mL). A mistura de reação foi desgaseificada durante 15 minutos com uma corrente de nitrogênio, e foi aquecida até o refluxo durante 2,5 horas sob nitrogênio. Após resfriar até a temperatura ambiente a mistura de reação foi diluída com AcOEt e sucessivamente lavada com água, NH4Cl sat., salmoura, secada sobre MgSO4 anidro, filtrada, e concentrada. O produto bruto foi purificado por cromatografia de coluna instantânea (eluentes AcOEt/CH2Cl2: 5/95 a 30/70) e triturado em AcOEt/hexanos para fornecer o composto do título 331 (273 mg, 59% de rendimento) como um sólido amarelo. MS (m/z): 427,1 (M+H)+. Etapa 2: 4-(7-(2-Flúor-4-nitrofenóxi)tieno[3,2-b ]piridin-2-il)benzeno-1,2- diol (332)
[00776] A uma solução agitada de 331 (172 mg, 0,40 mmol) em diclorometamo anidro (20 mL) a -78°C foi lentamente adicionado BBrs (~ 4 mL, 1,0 M em CH2Cl2). A temperatura foi deixada aquecer até a temperatura ambiente durante 1,5 horas, e a mistura de reação foi agitada durante a noite. Após resfriar até 0°C, MeOH e NaOH a 1N (poucos mL) foram adicionados, respectivamente. A mistura de reação foi agitada durante 1 hora, concentrada, diluída com MeOH e água, agitada durante 30 minutos, isolada por filtração, e enxaguada com MeOH. O líquido-mãe foi concentrado, dissolvido em um mínimo de MeOH, e diluído com uma quantidade pequena de água. O pH da solução foi ajustado para 4 com NaOH a 1N a fim de obter uma suspensão marrom-pálida. Após agitar durante 15 minutos, a suspensão foi filtrada, enxaguada com água, secada por ar, e secada sob vácuo elevado para fornecer o composto do título 332 (154 mg, 96% de rendimento) como um sólido verde-amarelado. MS (m/z): 399,0 (M+H)+. Etapa 3: 2-(3,4-Bis(3-cloropropóxi)fenil)-7-(2-flúor-4-nitrofenóxi) tieno[3,2-b]piridina (333)
[00777] A uma solução agitada de 332 (140 mg, 0,35 mmol) em DMF anidra (5 mL) foram adicionados 1-bromo-3-cloropropano (553 mg, 3,51 mmols) e carbonato de césio (573 mg, 1,76 mmol), respectivamente. A mistura de reação foi agitada durante 2 horas em temperatura ambiente, diluída com AcOEt, e sucessivamente lavada com água, cloreto de amônio saturado, água, e concentrada. O material bruto foi adsorvido em sílica-gel e purificado por cromatografia de coluna instantânea (eluentes AcOEt/CH2Cl2: 10/90 a 20/80) para fornecer o composto do título 333 (120 mg, 62% de rendimento) como um sólido amarelo. MS (m/z): 551,0 e 553,0 (M+H)+.. Etapa 4: 4-(2-(3,4-Bis(3-cloropropóxi)fenil)tieno[3,2-b]piridin-7-ilóxi)-3- fluoroanilina (334)
[00778] A uma suspensão agitada de composto nitro 333 (32 mg, 0,57 mmol) em uma mistura de MeOH (2 mL) e água (1 mL) foram adicionados pó de ferro (16 mg, 0,29 mmol) e NH4Cl (2,8 mg, 0,05 mmol). A mistura de reação foi aquecida até o refluxo durante 2,5 horas, resfriada até a temperatura ambiente, diluída com acetato de etila, filtrada, e enxaguada com AcOEt. O filtrado foi sucessivamente lavado com cloreto de amônio saturado, NaHCO3 sat., água, secado sobre MgSO4 anidro, filtrado, e concentrado para fornecer o composto do título 334 (25 mg, 83% de rendimento) como um sólido amarelo- pálido. MS (m/z): 521,0 e 523,0 (M+H)+. Etapa 5: N 1-(4-(2-(3,4-Bis(3-cloropropóxi)fenil)tieno[3,2-b ]piridin-7- ilóxi)-3-fluorofenil)-N 3-(2-metoxifenil)malonamida (335)
[00779] O composto do título 335 foi obtido de 334 como um sólido esbranquiçado seguindo o mesmo procedimento como em Exemplo 22, Etapa 7 (Esquema 18). Etapa 6. N 1-(4-(2-(3,4-Bis(3-(dimetilamino)propóxi)fenil)tieno[3,2- b ]piridin-7-ilóxi)-3-fluorofenil)-N 3-(2-metoxifenil)malonamida (330)
[00780] Uma solução agitada de 335 (material bruto) e um grande excesso de dimetilamina (2,3 mL, 2M em THF) em DMSO anidro (2 mL) foi agitada a 60°C durante 5 horas. A mistura foi resfriada para temperatura ambiente, e então concentrada e diretamente purificada duas vezes por HPLC preparativa (Thermo, Aquasil C18, 250x21,2 mm, 5 μm; eluente MeOH/H2O [ambos contendo 0,05% de HCO2H], gradiente linear 40/60^80/20 durante 30 minutos), para fornecer o composto do título 330 (14,4 mg, 41% de rendimento durante 2 etapas) como uma película pegajosa amarelo-pálido. 1H RMN (400 MHz, MeOH-d4) δ (ppm) : 9,00-8,20 (m, 2H), 8,11 (dd, J = 8,0, 1,6 Hz, 1H), 7,87 (dd, J = 12,5, 2,3 Hz, 1H), 7,70 (bs ,1H), 7,45-7,38 (m, 3H), 7,35 (t, J = 8,7 Hz, 1H), 7,15-7,06 (m, 2H), 7,03 (dd, J = 8,2, 1,2 Hz, 1H), 6,93 (td, J = 7,7, 1,2 Hz, 1H), 6,61 (bd, J = 4,5 Hz, 1H), 4,23 (t, J = 5,9 Hz, 2H), 4,18 (t, J = 5,8 Hz, 2H), 3,91 (s, 3H), 3,32-3,20 (m, 4H), 2,86 e 2,85 (2 s, 2x6H), 2,31-2,20 (m, 4H), um CH2 esta ausente. MS Esquema 78
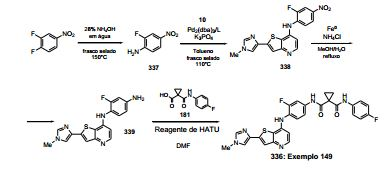
Exemplo 149 N-(3-Flúor-4-(2-(1 -metil-1 H-imidazol-4-il)tieno[3,2-b ]piridin-7- ilamino)fenil)-N -(4-fluorofenil)ciclopropano-1,1 -dicarboxamida (336) Etapa 1: 2-Flúor-4-nitroanilina (337)
[00781] Uma solução agitada de 3,4-difluoronitrobenzeno (2,00 g, 12,57 mmols) em hidróxido de amônio (20 mL, 28% em água) foi aquecida a 150°C em um frasco selado durante 3,5 horas. A mistura foi resfriada para temperatura ambiente, e a suspensão resultante foi diluída em água, agitada durante 15 minutos, o sólido foi isolado por filtração, enxaguado com água, secado por ar, e secado sob vácuo elevado para fornecer o composto do título 337 (1,76 g, 90% de rendimento) como um sólido cristalino amarelo. MS (m/z): 157,0 (M+H)+.e 179,0 (M+Na)+.. Etapa 2: N-(2-Flúor-4-nitrofenil)-2-(1 -metil-1 H-imidazol-4-il)tieno[3,2- b ]piridin-7-amina (338)
[00782] Uma suspensão agitada de 10 (500 mg, 2,00 mmols), 337 (406 mg, 2,60 mmols), Pd2 (dba)3 (73 mg, 0,08 mmol), (2-bifenil)diciclo- hexilfosfina (56 mg, 0,16 mmol), e K3PO4 (638 mg, 3,00 mmols) em tolueno (20 mL) foi desgaseificada durante 15 minutos com nitrogênio em temperatura ambiente, e então aquecida em um frasco selado a 110°C durante 22 horas (J. P. Wolfe, H. Tomori, J. P. Sadighi, J. Yin, S. L. Buchwald J. Org. Chem. 2000, 65, 1158-1174). Após resfriar até a temperatura ambiente a mistura de reação foi filtrada, enxaguada com tolueno, concentrada e adsorvida em sílica-gel. O produto bruto foi purificado por cromatografia de coluna instantânea (eluentes MeOH/CH2Cl2: 2/98 a 10/90) e precipitado em AcOEt/hexanos para fornecer o composto do título 338 (370 mg, 50% de rendimento) como um sólido laranja amarelado. 1H RMN (400 MHz, DMSO-d6) δ (ppm) : 9,57 (bs, 1H), 8,45 (d, J = 5,1 Hz, 1H), 8,22 (dd, J = 11,3, 2,5 Hz, 1H), 8,06 (dd, J = 8,9, 2,3 Hz, 1H), 7,83 (d, J = 1,2 Hz, 1H), 7,70 (d, J = 1,2 Hz, 1H), 7,63 (s ,1H), 7,24 (t, J = 8,7 Hz, 1H), 6,94 (bd, J = 4,7 Hz, 1H), 3,71 (s, 3H). MS (m/z): 370,0 (M+H)+. Etapa 3: 2-Flúor- N1 -(2-(1 -metil-1 H-imidazol-4-il)tieno[3,2-b ]piridin-7- il)benzeno-1,4-diamina (339)
[00783] A uma suspensão agitada de composto nitro 338 (370 mg, 1,00 mmol) em uma mistura de MeOH (20 mL) e água (10 mL) foram adicionados pó de ferro (280 mg, 5,01 mmols) e NH4Cl (107 mg, 2,00 mmols). A mistura de reação aquecida até o refluxo durante 2 horas, resfriada para temperatura ambiente, filtrada através de celite, e enxaguada com metanol. O filtrado foi concentrado, diluído um pouco com metanol, precipitado com AcOEt/hexanos para fornecer o composto do título 339 (463 mg, produção quantitativa, sal de amônio) como um sólido amarelo. MS (m/z): 340,0 (M+H)+. Etapa 4: N-(3-Flúor-4-(2-(1 -metil-1 H-imidazol-4-il)tieno[3,2-b]piridin-7- ilamino) fenil)-N-(4-fluorofenil)ciclopropano-1,1-dicarboxamida (336)
[00784] A uma solução agitada de 339 (80 mg, 0,24 mmol) e ácido 1-(4-fluorofenilcarbamoil) ciclopropanocarboxílico (181, 111 mg, 0,50 mmol) em DMF anidra foram adicionados DIPEA (123 μL, 0,71 mmol) e reagente de HATU (256 mg, 0,67 mmol). A mistura de reação foi agitada em temperatura ambiente durante a noite sob nitrogênio, diluída com AcOEt, e sucessivamente lavada com NaHCO3 sat., água, NH4Cl sat., água e salmoura, e concentrada. O material bruto foi primeiro purificado por cromatografia de coluna instantânea (eluentes 2% de NH4OH em metanol/CH2Cl2: 10/90) e precipitado em AcOEt (com traços de acetona)/hexanos para fornecer o composto do título 336 (75 mg, 58% de rendimento) como um sólido branco. 1H RMN (400 MHz, DMSO-d6) δ (ppm) : 10,32 (s, 1H), 10,04 (s, 1H), 8,65 (bs, 1H), 8,18 (d, J = 5,5 Hz, 1H), 7,78 (dd, J = 12,9, 2,2 Hz, 1H), 7,77 (d, J = 1,8 Hz, 1H), 7,68 (d, J = 1,2 Hz, 1H), 7,68-7,60 (m, 2H), 7,50 (s, 1H), 7,44 (dd, J = 8,4, 1,8 Hz, 1H), 7,30 (t, J = 8,9 Hz, 1H), 7,15 (t, J = 9,0 Hz, 2H), 6,36 (dd, J = 5,5, 1,6 Hz, 1H), 3,70 (s, 3H), 1,47 (s, 4H). MS (m/z): 545,0 (M+H)+. Tabela 15
[00785] Compostos 340-342 (Exemplos 150-152) preparados de acordo com o Esquema 78
Esquema 79
Exemplo 153 N-(3-Flúor-4-(metil(2-(1 -metil-1 H-imidazol-4-il)tieno[3,2-b ]piridin-7- il)amino)fenil)-N-fenilciclopropano-1,1-dicarboxamida (343) Etapa 1: N-Metil-2-(1 -metil-1 H-imidazol-4-il)tieno[3,2-b ]piridin-7-amina (344)
[00786] Uma suspensão agitada de 10 (500 mg, 2 mmol) e cloridrato de metilamina (15 g, 222 mmols) em isopropanol (50 mL) foi aquecida a 130°C em um frasco selado durante 4 dias, então resfriada até a temperatura ambiente. A mistura de reação foi despejada em água, e o pH foi ajustado para ~ 10 com NaOH a 1N. Após extração da fase aquosa com AcOEt, a camada orgânica combinada foi concentrada e diretamente purificada por cromatografia de coluna instantânea (eluentes 2% de NH4OH em metanol/CH2Cl2: 10/90 a 40/60) para fornecer o composto do título 344 (487 mg, 99% de rendimento, forma de hidrato) como um sólido bege. MS (m/z): 245,0 (M+H)+.. Etapa 2: N-(2-Flúor-4-nitrofenil)-N-metil-2-(1-metil-1 H-imidazol-4- il)tieno[3,2-b ]piridin-7-amina (345)
[00787] Uma suspensão agitada de 344 (500 mg, ~2 mmols), 3,4- difluoronitrobenzeno (795 mg, 5 mmols) e carbonato de césio (1,63 g, 5 mmols) em DMF anidra (50 mL) foi aquecida a 85°C sob nitrogênio durante 7 horas. A temperatura foi deixada resfriar até a temperatura ambiente. A mistura de reação foi despejada em água, e extraída com diclorometano. A fase orgânica combinada foi concentrada e diretamente purificada duas vezes por cromatografia de coluna instantânea (eluentes 2% de NH4OH em metanol/CH2Cl2: 5/95 a 10/90) para fornecer o composto do título 345 (144 mg, 19% de rendimento) como um sólido amarelo pegajoso. MS (m/z): 384,0 (M+H)+. Etapa 3: N-(3-Flúor-4-(metil(2-(1 -metil-1 H-imidazol-4-il)tieno[3,2- b ]piridin-7-il)amino)fenil)-N-fenilciclopropano-1,1 -dicarboxamida (343)
[00788] O composto do título 343 (sal de formiato) foi obtido em duas etapas de 345 como um sólido esbranquiçado seguindo o mesmo procedimento como em Exemplo 336, Etapa 3 e 4 (Esquema 78), porém usando na última Etapa ácido 161 em vez de 181. 1H RMN (400 MHz, DMSO-d6) δ (ppm): 10,36 (s, 1H), 10,02 (s, 1H), 8,44-8,14 (m, 2H), 7,78 (dd, J = 13,2, 2,1 Hz, 1H), 7,66 (d, J = 1,2 Hz, 1H), 7,63 (d, J = 7,6 Hz, 2H), 7,58 (d, J = 0,8, 1H), 7,47 (dd, J = 8,7, 2,1 Hz, 1H), 7,43 (s, 1H), 7,35 (t, J = 8,9 Hz, 1H), 7,31 (t, J = 8,0 Hz, 2H), 7,07 (t, J = 7,3 Hz, 1H), 6,77 (bd, J = 5,3 Hz, 1H), 3,65 (s, 3H), 3,33 (s, 3H), 1,541,44 (m, 4H). MS (m/z): 541,0 (M+H)+.. Tabela 16
[00789] Compostos 344-347 (Exemplos 154-155 preparados de acordo com o Esquema 79
Esquema 80
Exemplo 156 N-(3-Flúor-4-(2-(1 -metil-1 H-imidazol-4-il)tieno[3,2-b ]piridin-7-ilóxi)fenil)- 2-oxo-1-(piridin-3-il)pirrolidina-3-carboxamida (348) Etapa 1: 1-(Piridin-3-il)pirrolidin-2-ona (349)
[00790] Em um frasco selado, uma mistura agitada de CuI (37 mg, ~ 0,2 mmol) e carbonato de potássio (2,70 g, 19,5 mmols) foi desgaseificada durante 15 minutos. N,N’-dimetiletilenodiamina (146 μL, ~1,0 mmol), 3-iodopiridina (2,00 g, 9,8 mmols), 2-pirrolidinona (~ 1 g, 11,7 mmols) e 1,4-dioxano (10 mL) foram adicionados, respectivamente (A. Klapars, J. C. Antilla, X. Huang, S. L. Buchwald J. Am. Chem. Soc. 2001, 123, 7727-7729). O fluxo de nitrogênio foi removido, e a mistura de reação foi então aquecida a 125°C durante 18 horas. Após resfriar até a temperatura ambiente, a mistura de reação foi filtrada, enxaguada com acetato de etila, e concentrada. O produto bruto foi purificado por cromatografia de coluna instantânea (eluentes MeOH/CH2Cl2: 2/98 a 5/95) para fornecer o composto do título 349 (1,49 g, 94% de rendimento) como um líquido oleoso amarelo. MS (m/z): 163,1 (M+H)+.. Etapa 2: 2-Oxo-1-(piridin-3-il)pirrolidina-3-carboxilato de metila (350)
[00791] A uma solução agitada de 349 (1,48 g, 9,1 mmols) em THF anidro (25 mL) a -78°C sob nitrogênio foi lentamente adicionada uma solução de LDA (THF mono, 13,4 mL, 20,1 mmols, 1,5 M em cicloexano). Após 45 minutos, cloroformiato de metila (776 μL, 10,0 mmols) foi adicionado. A mistura foi aquecida até a temperatura ambiente durante 2 horas, e então agitada durante a noite. Em seguida, a mistura de reação foi extinguida pela adição de cloreto de amônio saturado e extraída com AcOEt. Após separação, a camada orgânica foi sucessivamente lavada com NH4Cl sat., água, e salmoura. A fase aquosa foi extraída duas vezes com AcOEt. As camadas orgânicas combinadas foram secas sobre sulfato de magnésio anidro, filtradas e concentradas. O produto bruto foi purificado por cromatografia de coluna instantânea (eluentes MeOH/CH2Cl2: 2/98 a 5/95) para fornecer o composto do título 350 (520 mg, 26% de rendimento) como um sólido pegajoso amarelo-pálido. 1H RMN (400 MHz, DMSO-d6) δ (ppm): 8,86 (d, J = 2,5 Hz, 1H), 8,38 (dd, J = 4,7, 1,4 Hz, 1H), 8,12-8,06 (m, 1H), 7,44 (dd, J = 8,4, 4,7 Hz, 1H), 3,99-3,84 (m, 2H), 3,81 (t, J = 8,8 Hz, 1H), 3,70 (s, 3H), 2,48-2,29 (m, 2H). MS (m/z): 221,0 (M+H)+. Etapa 3: Ácido 2-oxo-1-(piridin-3-il)pirrolidina-3-carboxílico (351)
[00792] A uma solução agitada de 350 (514 mg, 2,33 mmols) em THF (20 mL) sob nitrogênio foi adicionada uma solução de LiOH.H2O (147 mg, 3,50 mmols) em água (5 mL). A mistura de reação foi agitada durante a noite, concentrada, diluída com uma quantidade pequena de água, filtrada, neutralizada com HCl a 1N (pH ~ 5-6), e extraída duas vezes com diclorometano. A fase aquosa foi concentrada, e o resíduo foi triturado em metanol (com traços de acetona). Após filtração, o líquido-mãe foi concentrado e secado sob vácuo elevado para fornecer o composto do título 351 (493 mg, produção quantitativa, contaminado com sais) como um sólido pegajoso amarelo-pálido. MS (m/z): 207,1 (M+H)+. Etapa 4: N-(3-Flúor-4-(2-(1 -metil-1 H-imidazol-4-il)tieno[3,2-b]piridin-7- ilóxi) fenil)-2-oxo-1-(piridin-3-il)pirrolidina-3-carboxamida (348)
[00793] O composto do título 348 (Exemplo 31) foi obtido por meio de reação de acoplamento do ácido 351 e a amina 12 como um sólido bege, seguindo o mesmo procedimento como descrito acima para a síntese de composto 336 (Exemplo 149, Esquema 78); purificado usando Biotage System (Si 12M, gradiente MeOH/diclorometano: 0/100 a 20/80) seguido por trituração com diclorometano. 1H RMN (400 MHz, DMSO-d6) δ (ppm) : 10,73 (s, 1H), 8,91 (d, J = 2,8 Hz, 1H), 8,43 (d, J = 5,6 Hz, 1H), 8,39 (dd, J = 4,4, 1,6 Hz, 1H), 8,13 (qd, J = 8,4, 1,6 Hz, 1H), 7,94-7,88 (m, 1H), 7,87 (d, J = 1,0 Hz, 1H), 7,72 (d, J = 1,0 Hz, 1H), 7,69 (s, 1H), 7,53-7,43 (m, 3H), 6,60 (d, J = 5,6 Hz, 1H), 4,043,92 (m, 2H), 3,82 (t, J = 8,8 Hz, 1H), 3,73 (s, 3H), 2,55-2,36 (m, 2H). MS (m/z): 529,0 (M+H)+. Tabela 17
[00794] Compostos 352-360 (Exemplos 157-165)
Esquema 81
Exemplo 166 N 1-(3-Flúor-4-(2-(3-(3-guanidinopropóxi)fenil)tieno[3,2-b]piridin-7- ilóxi)fenil)-N3-(2-metoxifenil)malonamida (361) Etapa 1 2-(3-(3-Cloropropóxi)fenil)-4,4,5,5-tetrametil-1,3,2- dioxaborolano (363)
[00795] A 3-bromofenol (2,23 g, 12,9 mmols) em DMF (100 mL) foi adicionado, em pequenas porções, hidreto de sódio (60 % de dispersão, 0,54 g, 13 mmols) durante 30 minutos. 1-Bromo-3- cloropropano (2,1 g, 13 mmols) foi então adicionado em gotas e a mistura foi agitada a t.a. durante 24 horas. Ela foi então dividida entre éter e água, a fase orgânica foi lavada com água e salmoura, secada (MgSO4 anidro) e concentrada para fornecer 1-bromo-3-(3- cloropropóxi)benzeno (362, 2,95 g, 91 % de rendimento) (usado com está sem nenhuma purificação adicional).
[00796] Éter 362 (2,95 g, 11,8 mmols), bis(pinacolato)diboro (4,00 g, 15,8 mmols), acetato de potássio (1,20 g, 12,2 mmols) e tetracis(trifenilfosfina)paládio (0,37 g, 0,32 mmol) foram suspensos em tolueno (100 mL) e aquecidos sob refluxo durante 10 horas. A mistura foi então resfriada e o tolueno foi removido sob pressão reduzida. O resíduo foi dividido entre água e diclorometano, a fase orgânica foi seca (Na2SO4), filtrada, concentrada e purificada por cromatografia instantânea (eluente 25 % de diclorometano/hexanos) para fornecer o composto do título 363 (1,00 g, 29 % de rendimento). 1H RMN (400 MHz, DMSO-d6) δ (ppm): 7,29 (t, J = 7,8, 1H); 7,23 (dt, J = 7,2, 1,0, 1H); 7,15 (d, J = 2,5, 1H); 7,06 (ddd, J = 8,0, 2,7, 1,4, 1H); 4,06 (t, J = 6,1, 2H); 3,78 (t, J = 6,7, 2H), 2,14 (quint, J = 6,3, 2H); 1,27 (s, 12H). LRMS (M+H): 297,1. Etapa 2: 2-(3-(3-Cloropropóxi)fenil)-7-(2-flúor-4-nitrofenóxi)tieno[3,2- b]piridina (364)
[00797] Bromotienopiridina 50 (0,88 g, 2,38 mmols), boronato 363 (1,00 g, 3,40 mmols), e tetracis(trifenilfosfina)paládio (0,10 g, 0,086 mmol) foram dissolvidos em DME seco (100 mL). Fluoreto de césio (1,26 g, 8,3 mmols) e bicarbonato de sódio (0,70 g, 8,3 mmols) foram dissolvidos em água (5 mL cada) e adicionados a uma mistura de reação, que foi então aquecida até o refluxo durante 4 horas, resfriada, e concentrada. O resíduo foi dividido entre acetato de etila e água, lavado com salmoura, secado (MgSO4 anidro), filtrado, e concentrado. O resíduo foi purificado por cromatografia instantânea (eluente 75% de diclorometano/hexanos) para fornecer 364 (0,85 g, 78 % de rendimento). 1H RMN (400 MHz, CDCI3) δ (ppm): 8,60 (d, J = 6,3, 1H); 8,27-8,24 (m, 2H); 8,18 (s, 1H); 7,58-7,54 (m, 1H); 7,45-7,36 (m, 2H); 7,31 (t, J = 2,0, 1H); 7,07-7,04 (m, 1H); 6,78 (d, J = 6,1, 1H); 4,21 (t, J = 5,9, 2H); 3,79 (t, J = 6,3, 2H); 2,29 (quint, J = 6,1, 2H). LRMS (M+H): 459,1. Etapa 3: 4-(2-(3-(3-Cloropropóxi)fenil)tieno[3,2-b]piridin-7-ilóxi)-3- fluorobenzenamina (365)
[00798] Ao composto nitro 364 (0,84 g, 1,8 mmol) e hexahidrato de cloreto de níquel (0,87 g, 3,7 mmols) em 9:1 MeOH/THF foi adicionado boroidreto de sódio (0,30 g, 7,9 mmols) em pequenas porções. A mistura resultante foi agitada a t.a. durante 1 hora, então filtrada através de celite e concentrada. O resíduo foi dividido entre água e diclorometano, a fase orgânica foi coletada, lavada com salmoura, secada (MgSO4 anidro), filtrada, e concentrada. O resíduo foi purificado por cromatografia instantânea (eluente 90 % de acetato de etila/hexanos) para fornecer 365 (0,43 g, 54 % de rendimento). LRMS (M+H): 429,1. Etapa 4: N 1-(4-(2-(3-(3-Cloropropóxi)fenil)tieno[3,2-b]piridin-7-ilóxi)-3- fluorofenil)-N 3-(2-metoxifenil)malonamida (366)
[00799] A uma solução de anilina 365 (0,42 g, 0,98 mmol) em DMF (20 mL) foram adicionados ácido 103 (0,42 g, 2,0 mmols), HOBt (0,050 g, 0,38 mmol), e EDC x HCl (0,54 g, 2,8 mmols) e a mistura foi agitada a t.a. durante 24 horas. Ela foi então dividida entre acetato de etila e água. A fase orgânica foi coletada, lavada com água, NaHCO3 (aq), salmoura, secada (MgSO4 anidro), filtrada, e concentrada. O resíduo foi triturado com éter para fornecer 366 (0,60 g, 98 % de rendimento). 1H RMN (400 MHz, DMSO-d6) δ (ppm): 10,58 (s, 1H); 9,62 (s, 1H); 8,50 (d, J = 5,3, 1H); 8,11 (s, 1H); 8,06 (d, J = 8,8, 1H); 7,86 (dd, J = 12,9, 2,4, 1H); 7,49-7,39 (m, 5H), 7,09-7,03 (m, 3H); 6,92-6,88 (m, 1H); 6,63 (d, J = 5,3, 1H); 4,20 (t, J = 6,1, 2H); 3,85 (s, 3H); 3,82 (t, J = 6,7, 2H); 2,20 (quint, J = 6,3, 2H). LRMS (M+H): 620,1. Etapa 5: N1-(4-(2-(3-(3-Aminopropóxi)fenil)tieno[3,2-b]piridin-7-ilóxi)-3- fluorofenil)-N3-(2-metoxifenil)malonamida (367)
[00800] A uma solução de 366 (0,19 g, 0,31 mmol) em DMF (5 mL) foi adicionada azida de sódio (0,050 g, 0,77 mmol) e a mistura de reação foi aquecida para 100°C durante 1 hora. A mistura foi então resfriada, dividida entre acetato de etila e água, a fase orgânica foi coletada, lavada com água, salmoura, secada (MgSO4 anidro), filtrada, e concentrada. O resíduo foi filtrado através de um tampão curto de sílica, eluindo com acetato de etila, e o eluato foi concentrado. O resíduo foi dissolvido em mistura de 1:1 acetato de etila/metanol (30 mL); a está solução foi adicionado paládio (10 % sobre carbono), e a suspensão foi agitada sob uma atmosfera de hidrogênio durante 3 horas. Ela foi então filtrada através de celite e concentrada. O resíduo foi purificado por cromatografia instantânea (eluente 95:3:2 clorofórmio/metanol/NH4OH) para fornecer 367 (0,097 g, 52 % de rendimento): LRMS (M+H): 601,2. Etapa 6: N1 -(3-Flúor-4-(2-(3-(3-guanidinopropóxi)fenil)tieno[3,2- b]piridin-7-ilóxi)fenil)-N 3-(2-metoxifenil)malonamida (361)
[00801] Amina 367 (0,095 g, 0,16 mmol), pirazol-1-carboxamidina (60 mg, 0,41 mmol), e base Hunigs (0,07 g, 0,5 mmol) foram agitados em DMF seca (10 mL) durante 48 horas a t.a. A mistura foi então dividida entre acetato de etila e água. A fase aquosa foi coletada e tratada com salmoura; um precipitado foi formado, o qual foi isolado por filtração por sucção. O sólido resultante foi redissolvido em 1:1 diclorometano/metanol, filtrado, e o filtrado foi concentrado para fornecer 361 como um sólido (90 mg, 87 % de rendimento). 1H RMN (400 MHz, DMSO-d6) δ (ppm): 10,71 (br s, 1H); 9,63 (br s, 1H); 8,49 (d, J = 5,5, 1H); 8,10 (s, 1H); 8,05 (d, J = 8,4, 1H); 7,93-7,70 (m, 2H); 7,51-7,40 (m, 5H); 7,08-7,02 (m, 3H); 6,92-6,88 (m, 1H); 6,63 (d, J = 5,5, 1H); 4,13 (t, J = 5,9, 2H); 3,84 (s, 3H); 3,64 (s, 2H); 1,95 (quint, J = 6,3, 2H). [Um tripleto correspondendo a 3H é impedido pelo pico de DMSO residual]. LRMS (M+H): 643,0. Esquema 82
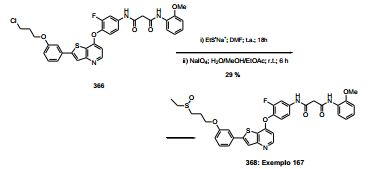
Exemplo 167 N 1-(4-(2-(3-(3-(Etilsulfinil)propóxi)fenil)tieno[3,2-b]piridin-7-ilóxi)-3- fluorofenil)-N 3-(2-metoxifenil)malonamida (368)
[00802] Ao cloreto 366 (0,048 g, 0,077 mmol) em DMF seca (10 mL) foi adicionado etanotiolato de sódio (100 mg, 1,19 mmol) e a mistura foi aquecida para 50°C, durante 18 horas. Ela foi resfriada, dividida entre acetato de etila e água, lavada com salmoura, secada (MgSO4), filtrada, passada através de um tampão curto de sílica-gel, e concentrada. O resíduo foi suspenso em 1:1 acetato de etila/metanol (50 mL) e periodato de sódio (0,060 g, 0,28 mmol) em água (5 mL) foi adicionado. A mistura de reação foi agitada durante 6 horas, concentrada e o resíduo foi dividido entre acetato de etila e água. A fase orgânica foi coletada, lavada com salmoura, secada (MgSO4 anidro), filtrada, e concentrada. HPLC de fase reversa (coluna Aquasil C-18, 60 - 95 % de MeOH/H2O + HCO2H, 30 minutos de eluição gradiente linear) do resíduo seguida por liofilização forneceu 368 (0,015 g, 29 % de rendimento). 1H RMN (400 MHz, DMSO-d6) δ (ppm): 10,71 (s, 1H); 9,66 (s, 1H); 8,51 (d, J = 5,5, 1H); 8,12 (s, 1H); 8,07 (d, J = 8,8, 1H); 7,89 (dd, J = 12,9, 2,2, 1H); 7,53-7,41 (m, 5H); 7,09-7,03 (m, 3H); 6,94-6,90 (m, 1H); 6,64 (dd, J = 5,5, 0,5, 1H); 4,22 (t, J = 6,3, 2H); 3,86 (s, 3H); 3,65 (s, 2H); 2,93-2,65 (m, 4H); 2,13 (quint, J = 8,0, 2H); 1,29 (t, J = 7,4, 3H). LCMS: (M+H) 662,0. Esquema 83

Exemplo 168 N-(3-Flúor-4-(2-(1 -metil-1 H-imidazol-2-il)tieno[3,2-b]piridin-7-ilóxi)fenil)- 2-(4-fluorofeniltio)acetamida
[00803] À anilina 9 (0,10 g, 0,30 mmol) em DMF seca (20 mL) foram adicionados ácido (p-fluorofeniltio)acético (0,11 g, 0,56 mmol), e EDC x HCl (0,13 g, 0,68 mmol) e a mistura foi agitada a t.a. durante 24 horas. Ela foi então dividida entre acetato de etila e água. A fase orgânica foi coletada, lavada com água, NaHCO3 (aq), salmoura, secada (MgSO4 anidro), filtrada e concentrada. Cromatografia de sílica-gel (eluente acetato de etila -> 5% de metanol/acetato de etila) do resíduo forneceu 369 (0,070 g, 46 % de rendimento). 1H RMN (400 MHz, DMSO-d6) δ (ppm): 10,55 (s, 1H); 8,50 (d, J = 5,5, 1H); 7,88 (s, 1H); 7,80 (dd, J = 13,1, 2,5, 1H); 7,50-7,45 (m, 3H); 7,41-7,38 (m, 2H); 7,22-7,17 (m, 2H); 7,03 (d, J = 1,2, 1H); 6,67 (d, J = 4,7, 1H); 3,98 (s, 3H); 3,84 (s, 2H). LCMS: (M+H) 508,9. Exemplo 169 N-(3-Flúor-4-(2-(1 -metil-1 H-imidazol-2-il)tieno[3,2-b]piridin-7-ilóxi)fenil)- 2-(4-fluorofenilsulfonil)acetamida (370)
[00804] A uma solução de amida 369 (0,067 g, 0,13 mmol) em diclorometano (50 mL) a 0°C foi adicionado m-CPBA (0,040 g, 0,24 mmol) e a mistura foi deixada a -10°C durante 24 horas. Ela foi então lavada com água, NaHCO3 (aq), salmoura, secada (MgSO4 anidro), filtrada, e concentrada. Cromatografia de sílica-gel (eluente 5 % de metanol/acetato de etila) do resíduo forneceu 370 (0,012 g, 18 % de rendimento). 1H RMN (400 MHz, CD3OD) δ (ppm): 8,52 (d, J = 5,7, 1H); 8,04-8,00 (m, 2H); 7,86 (s, 1H); 7,77 (dd, J = 12,5, 2,4, 1H); 7,427,32 (m, 5H); 7,20 (d, J = 1,4, 1H); 6,70 (dd, J = 5,7, 1,2, 1H); 4,35 (s, 2H); 4,04 (s, 3H). LCMS: (M+H) 541,1. Esquema 84
Exemplo 171 Ácido 2-(4-(7-(2-flúor-4-(3-oxo-3-(fenilamino)propanamido)fenóxi) tieno[3,2-b]piridin-2-il)-1 H-imidazol-1-il)acético (371) Etapa 1: 2-(4-Iodo-1 H-imidazol-1-il)acetato de etila (372)
[00805] A uma solução de 4-iodoimidazol (1,93 g, 9,95 mmols) em THF seco (50 mL) a 0°C foi adicionado hidreto de sódio (60 % de dispersão, 0,43 g, 10,8 mmols) e a mistura foi agitada durante 20 minutos. Bromoacetato de etila (1,1 mL, 1,7 g, 10 mmols) foi adicionado por seringa e a mistura turva foi agitada durante 15 minutos. Ela foi então dividida entre acetato de etila e água. A fase orgânica foi coletada, lavada com salmoura, secada (MgSO4 anidro), filtrada, e concentrada. Cromatografia de sílica-gel (50% -> 75% de acetato de etila/hexanos) do resíduo forneceu 372 (1,88 g, 67 % de rendimento). 1H RMN (400 MHz, DMSO-d6) δ (ppm): 7,57 (d, J = 1,4, 1H); 7,32 (d, J = 1,4, 1H); 4,93 (s, 2H); 4,13 (q, J = 8,0, 2H); 1,20 (t, J = 7,2, 3H). LRMS (M+H): 281,0. Etapa 2: 2-(4-(7-Clorotieno[3,2-b]piridin-2-il)-1 H-imidazol-1-il)acetato de etila (373)
[00806] A uma solução de 7-clorotienopiridina (2,23 g, 13,1 mmols) em THF seco (50 mL) a -78°C sob N2 foi adicionado n-butil-lítio (2,5 M em hexanos, 5,6 mL, 14 mmols), em gotas, com agitação. A suspensão resultante foi agitada durante 30 minutos a -78°C, então ZnCl2 (THF a 0,5M, 30 mL, 15 mmols) foi adicionado e a mistura foi deixada aquecer para 0°C. Imidazol 372 (3,20 g, 11,4 mmols) e tetracis(trifenilfosfina)paládio (0,40 g, 0,35 mmol) em THF (50 mL) foram então adicionados à suspensão de aril-lítio, e a mistura foi aquecida sob refluxo durante 2 horas, então resfriada e concentrada. O resíduo foi dividido entre acetato de etila e água. A fase orgânica foi coletada, filtrada, secada (MgSO4 anidro), filtrada novamente e concentrada. O sólido resultante foi triturado (1:1 acetato de etila/hexanos) para fornecer 373 (1,62 g, 44 % de rendimento). 1H RMN (400 MHz, DMSO-d6) δ (ppm): 8,55 (d, J = 5,1, 1H); 7,91 (d, J = 1,2, 1H); 7,78 (d, J = 1,2, 1H); 7,77 (s, 1H); 7,45 (d, J = 5,2, 1H); 5,05 (s, 2H); 4,17 (q, J = 7,2, 2H); 1,23 (t, J = 7,0, 3H). LRMS (M+H): 322,0 Etapa 3: 2-(4-(7-(2-Flúor-4-nitrofenóxi)tieno[3,2-b]piridin-2-il)-1H- imidazol-1-il)acetato de etila (374)
[00807] Uma suspensão de 373 (1,05 g, 3,26 mmols), 2-flúor-4- nitrofenol (1,10 g, 7,00 mmols), e K2CO3 (2,0 g, 15 mmols) em éter de difenila (10 mL) foi aquecida com agitação a 185°C durante 3 horas. A mistura foi resfriada, diluída com diclorometano, filtrada, e o filtrado foi concentrado. Cromatografia de sílica-gel (75% de acetato de etila/hexanos) do resíduo forneceu 374 (0,98 g, 68 % de rendimento). 1H RMN (400 MHz, CD3OD) δ (ppm): 8,52 (d, J = 5,3, 1H); 8,46 (dd, J = 10,4, 2,5, 1H); 8,20-8,17 (m, 1H); 7,89 (d, J = 1,2, 1H), 7,77-7,74 (m, 1H); 7,75 (s, 1H); 7,69 (t, J = 8,2, 1H); 6,86 (d, J = 5,3, 1H); 5,04 (s, 2H); 4,17 (q, J = 7,2, 2H); 1,22 (t, J = 7,2, 3H). LRMS (M+H): 442,9. Etapa 4: 2-(4-(7-(4-Amino-2-fluorofenóxi)tieno[3,2-b]piridin-2-il)-1H- imidazol-1-il)acetato de etila (375)
[00808] A uma solução de tienopiridina 374 (0,25 g, 0,56 mmol) e hexahidrato de cloreto de níquel (0,26 g, 1,1 mmol) em EtOH absoluto (50 mL) foi adicionado boroidreto de sódio (0,085 g, 2,2 mmols) em pequenas porções. A mistura resultante foi agitada a t.a. durante 1 hora, então filtrada através de celite, passada através de um tampão curto de sílica, eluída com 1:1 acetato de etila/etanol, e concentrada, fornecendo o composto do título 375 (0,21 g, 91 % de rendimento). LRMS (M+H): 413,1. Etapas 5-6: Ácido 2-(4-(7-(2-flúor-4-(3-oxo-3-(fenilamino)propanamido) fenóxi)tieno[3,2-b]piridin-2-il)-1 H-imidazol-1-il)acético (371)
[00809] A uma solução de ácido 1 (0,080 g, 0,44 mmol) em diclorometano (10 mL) foi adicionado BOP-Cl (0,10 g, 0,39 g) em diclorometano (10 mL) e a mistura foi agitada a t.a. durante 30 minutos. Então anilina 375 (0,11 g, 0,27 mmol) e DIPEA (0,20 mL, 0,15 g, 1,1 mmol) em diclorometano (10 mL) foram adicionados, e a mistura foi agitada a t.a. durante 72 horas. Ela foi então lavada com água, NaHCO3 a 1 M (aq), salmoura, secada (MgSO4 anidro), filtrada, e concentrada. Cromatografia de sílica-gel (5 % de metanol/acetato de etila) do resíduo forneceu o composto do título 376 (0,065 g, 43 %), contaminado com alguma anilina de partida 375. À amida impura 376 (0,050 g, 0,87 mmol) em 40 % de metanol aquoso (25 mL) foi adicionado NaOH (3M de aquosos, 1 mL, 3 mmols) e a mistura foi agitada a t.a. durante 1 hora. Ela foi então parcialmente concentrada, e o resíduo purificado por HPLC de fase reversa (coluna Aquasil C-18, 40 - 95 % de MeOH/H2O + HCO2H, 30 minutos. eluição gradiente linear) e liofilização. Trituração do sólido resultante (acetato de etila) forneceu ácido 371 (0,021 g, 44 % de rendimento). 1H RMN (400 MHz, DMSO-d6) δ (ppm): 11,00 (s, 1H); 10,61 (s, 1H); 8,33-8,31 (m, 3H); 7,87 (d, J = 14,5, 1H); 7,78 (d, J = 1,0, 1H); 7,63-7,60 (m, 4H); 7,32 7,27 (m, 4H); 7,04 (t, J = 7,2, 1H); 6,40 (d, J = 5,1, 1H); 4,38 (s, 2H); 3,51 (s, 2H, sobrepondo o pico de água). LRMS (M+H): 545,9. Esquema 85
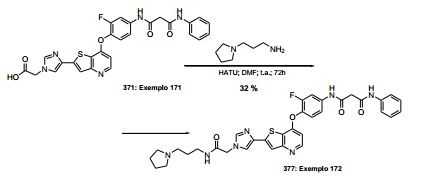
Exemplo 172 N 1-(3-Flúor-4-(2-(1-(2-oxo-2-(3-(pirrolidin-1-il)propilamino)etil)-1 H- imidazol-4-il)tieno[3,2-b]piridin-7-ilóxi)fenil)-N 3-fenilmalonamida
[00810] A uma solução de ácido 371 (0,016 g, 0,029 mmol) em DMF seca (5 mL) foi adicionado HATU (0,040 g, 0,11 mmol) e a mistura foi agitada a t.a. durante 10 minutos. 1-(3- Aminopropil)pirrolidina (0,2 mL, 0,2 g, 2 mmols) foi adicionado e a mistura resultante foi agitada a t.a. durante 72 horas e purificada por HPLC de fase reversa (coluna Aquasil C-18, 45 - 90 % de MeOH/H2O + HCO2H, 30 minutos. Eluição gradiente linear) e liofilização. Trituração do sólido resultante com éter de dietila forneceu o composto do título 372 (0,006 g, 32 % de rendimento). 1H RMN (400 MHz, CD3OD) δ (ppm): 8,40 (d, J = 5,7, 1H); 7,87 (dd, J = 12,5, 2,4, 1H); 7,80 (s, 1H); 7,75 (s, 1H); 7,67 (s, 1H); 7,60-7,56 (m, 2H); 7,43-7,30 (m, 4H); 7,14-7,09 (m, 1H); 6,59 (d, J = 5,5, 1H); 4,85 (s, 2H); 3,59 (s, 0,5H [permutando com D?]), 3,35-3,30 (m, 2H); 3,08 (br s, 4H); 2,97 (t, J = 8,0, 2H); 1,98 (br s, 4H); 1,89 (quint, J = 7,8, 2H). LRMS: (M+H) 656,0. Esquema 86
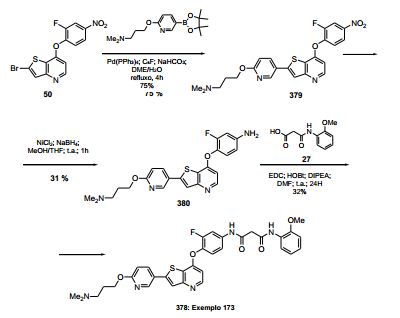
Exemplo 173 N 1-(4-(2-(6-(3-(Dimetilamino)propóxi)piridin-3-il)tieno[3,2-b]piridin-7- ilóxi)-3-fluorofenil)-N3-(2-metoxifenil)malonamida (378) Etapa 1: 3-(5-(7-(2-Flúor-4-nitrofenóxi)tieno[3,2-b]piridin-2-il)piridin-2- ilóxi)-N,N-dimetilpropan-1-amina (379)
[00811] Bromotienopiridina 50 (1,22 g, 3,30 mmols), N,N-dimetil-3- (5-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)piridin-2-ilóxi)propan-1- amina (1,15 g, 3,76 mmols), e tetracis(trifenilfosfina)paládio (0,14 g, 0,12 mmol) foram dissolvidos em DME seco (100 mL). Fluoreto de césio (1,51 g, 10,0 mmols) e bicarbonato de sódio (0,81 g, 9,6 mmols) foram dissolvidos em água (5 mL cada) e adicionados a uma mistura de reação, que foi desgaseificada com uma corrente de N2, então aquecida até o refluxo durante 4 horas, resfriada e concentrada. O resíduo foi dividido entre acetato de etila e água. A fase orgânica foi coletada, lavada com salmoura, secada (MgSO4 anidro), filtrada, e concentrada. O sólido laranja resultante foi triturado com éter para fornecer 379 (1,12 g, 75 % de rendimento). LRMS (M+H): 469,2. Etapa 2: 4-(2-(6-(3-(Dimetilamino)propóxi)piridin-3-il)tieno[3,2-b]piridin- 7-ilóxi)-3-fluorobenzenamina (380)
[00812] À tienopiridina 379 (1,11 g, 2,37 mmols) e hexahidrato de cloreto de níquel (1,11 g, 4,68 mmols) em 9:1 de MeOH/THF (50 mL) foi adicionado boroidreto de sódio (0,45 g, 11,9 mmols) em pequenas porções. A mistura resultante foi agitada a t.a. durante 1 hora, então filtrada através de celite e concentrada. O resíduo foi dividido entre água e diclorometano, a fase orgânica foi lavada com salmoura, secada (MgSO4 anidro), filtrada, e concentrada. Cromatografia instantânea (90:9:1 de clorofórmio/metanol/NH4OH) forneceu 380 (0,32 g, 31 % de rendimento). 1H RMN (400 MHz, CD3OD) δ (ppm): 8,43 (d, J = 2,5, 1H); 8,33 (d, J = 5,7, 1H); 7,93 (dd, J = 8,8, 2,5, 1H); 7,59 (s, 1H); 7,00 (t, J = 8,8, 1H); 6,77 (d, J = 8,6, 1H); 6,57 (dd, J = 12,5, 2,6, 1H); 6,53-6,49 (m, 2H); 4,29 (t, J = 6,3, 2H); 2,48-2,43 (m, 2H); 2,24 (s, 6H); 1,95-1,90 (m, 2H). LRMS (M+H): 439,1. Etapa 3: N 1-(4-(2-(6-(3-(Dimetilamino)propóxi)piridin-3-il)tieno[3,2- b]piridin-7-ilóxi)-3-fluorofenil)-N 3-(2-metoxifenil)malonamida (378)
[00813] A uma solução de anilina 380 (0,31 g, 0,71 mmol) e DIPEA (0,7 mL, 0,4 g, 3 mmols) em DMF seca (5 mL) foram adicionados ácido 27 (0,30 g, 1,4 mmol), HOBt (0,040 g, 0,30 mmol), e EDC x HCl (0,40 g, 2,1 mmols) e a mistura foi agitada a t.a. durante 18 horas. EDC x HCl adicional (0,050 g, 0,26 mmol) foi adicionado, e a mistura foi agitada durante mais 6 horas. Ela foi então dividida entre acetato de etila e água. A fase orgânica foi coletada, lavada com água, secada (MgSO4 anidro), filtrada, e concentrada. Cromatografia instantânea (clorofórmio/NH4OH) do resíduo seguida por recristalização (90% de acetato de etila/metanol) forneceu 378 (0,15 g, 32 % de rendimento) como um sólido incolor. 1H RMN (400 MHz, DMSO-d6) δ (ppm): 10,58 (br s, 1H); 9,63 (br s, 1H); 8,68 (dd, J = 2,7, 0,8, 1H); 8,48 (d, J = 5,5, 1H); 8,20 (dd, J = 8,6, 2,7, 1H); 8,05 (d, J = 7,6, 1H); 8,03 (s, 1H); 7,86 (dd, J = 13,1, 2,3, 1H); 7,49 (t, J = 8,8, 1H); 7,42 (dd, J = 8,8, 1,4, 1H); 7,09-7,03 (m, 2H); 6,94 (dd, J = 8,8, 0,7, 1H); 6,92-6,88 (m, 1H); 6,61 (dd, J = 5,5, 1,0, 1H); 4,33 (t, J = 6,7, 2H); 3,85 (s, 3H); 3,63 (s, 2H); 2,33 (t, J = 7,2, 2H); 2,13 (s, 6H); 1,84 (quint, J = 7,2, 2H). LRMS (M+H): 630,2. Tabela 18
[00814] Compostos 381-395 (Exemplos 174-188)
Esquema 87
3-Flúor-4-(2-(5-((4-metilpiperazin-1-il)metil)piridin-2-il)tieno[3,2- b]piridin-7-ilóxi)anilina (396) e 4-(2-(5-((Dimetilamino)metil)piridin-2-il)tieno[3,2-b]piridin-7-ilóxi)-3- fluoroanilina (397) Etapa 1: 7-(2-Flúor-4-nitrofenóxi)-2-iodotieno[3,2-b]piridina (399)
[00815] Uma mistura do cloreto 398 (Ragan J. A. et al., Organic Process Research e Development 2003, 7, 676-683) (7,0 g, 23,7 mmols), 2-flúor-4-nitrofenol (11,15 g, 3 eq, 71,1 mmols), K2CO3 (13,08 g, 4 eq, 94,8 mmols) em Ph2O (30 mL) foi aquecida para 200°C durante 3 horas. A mistura de reação foi resfriada até a temperatura ambiente e diluída com DCM, filtrada e concentrada. O sólido resultante foi triturado com éter de dietila, para fornecer 399 (7,3 g, 74% de rendimento), que foi usado diretamente na etapa seguinte sem nenhuma purificação adicional. MS (m/z): 417,0 (M+H). Etapa 2 6-(7-(2-Flúor-4-nitrofenóxi)tieno[3,2-b]piridin-2- il)nicotinaldeído (400)
[00816] Uma solução de 399 (1 g, 2,40 mmols) e 6- bromonicotinaldeído (450 mg, 2,40 mmols) em 1,4-dioxano (10 mL) foi tratada sequencialmente com bistrimetilestanho (500 μL, 787 mg, 2,40 mmols) e Pd(PPh3)4 (270 mg, 0,24 mmol). A mistura de reação foi então aquecida até o refluxo sob nitrogênio durante a noite, resfriada e concentrada. O produto bruto foi purificado por cromatografia instantânea usando o gradiente 5%-10% de MeOH em DCM e subsequente trituração por MeOH, fornecendo 400 puro (494 mg, 52% de rendimento). 1H RMN (400 MHz, DMSO-d6) δ (ppm): 10,11 (s, 1H), 9,10 (m, 1H), 8,65 (d, 1H, J = 5,2 Hz), 8,62 (s, 1H), 8,51 (d, 1H, J = 8,8 Hz), 8,48 (dd, 1H, J = 2,8 Hz, J = 10,4 Hz), 8,38 (dd, 1H, J = 2,1 Hz, J = 8,2 Hz), 8,20 (m, 1H), 7,73 (t, 1H, J = 9,0 Hz), 7,01 (d, 1H, J = 5,5 Hz). MS (m/z): (M+1) 395,9. Etapa 3j 7-(2-Flúor-4-nitrofenóxi)-2-(5-((4-metilpiperazin-1- il)metil)piridin-2-il)tieno[3,2-b]piridina (401)
[00817] Uma mistura de 400 (451 mg, 1,14 mmol) e 1- metilpiperazina (152 μL, 137 mg, 1,37 mmol) em DCM (7 mL) foi agitada em temperatura ambiente durante 10 minutos. Ela foi então tratada com NaBH(OAc)3 (340 mg, 1,60 mmol) e agitada em temperatura ambiente durante a noite. A mistura de reação foi diluída com DCM (20 mL) e lavada com solução de NaHCO3 saturada (20 mL). A fase orgânica foi coletada, secada sobre Na2SO4 anidro, filtrada e concentrada. O resíduo foi purificado por cromatografia instantânea usando o gradiente 30-50% de MeOH (com 2% de Et3N) em EtOAc para fornecer 401 (308 mg, 52% de rendimento). MS (m/z): (M+1) 480,0 (100%). Etapa 3: 3-Flúor-4-(2-(5-((4-metilpiperazin-1-il)metil)piridin-2- il)tieno[3,2-b]piridin-7-ilóxi)anilina (396)
[00818] A uma solução de 401 (306 mg, 0,64 mmol) e NH4Cl (30 mg, 0,54 mmol) em 2:1 de EtOH/água (10,5 mL) foi adicionado pó de ferro (304 mg, 5,43 mmols) e a suspensão foi aquecida até o refluxo durante 1 hora. A mistura de reação foi filtrada através de celite e concentrada para fornecer o composto do título 396, que foi usado sem outra purificação (343 mg, 100% de rendimento). MS (m/z): (M+1) 450,0 (100%). Etapa 4: 1 -(6-(7-(2-Flúor-4-nitrofenóxi)tieno[3,2-b]piridin-2-il)piridin-3- il)-N,N-dimetilmetanamina (402)
[00819] Uma mistura de 400 (360 mg, 0,91 mmol) e dimetilamina (2M de solução de THF, 550 nL, 1,09 mmol) em DCM (10 mL) foi agitada em temperatura ambiente durante 10 minutos. Ela foi então tratada com NaBH(OAc)3 (270 mg, 1,27 mmol) e agitada em temperatura ambiente durante a noite. A mistura de reação foi diluída com DCM (30 mL) e lavada com solução de NaHCO3 saturada (30 mL), secada sobre Na2SO4 anidro, filtrada e concentrada. O resíduo foi purificado por cromatografia instantânea usando o gradiente 1030% de MeOH em DCM como um eluente, para fornecer o composto do título 402 (321 mg, 83% de rendimento). MS (m/z): (M+1) 425,1 (100%). Etapa 5: 4-(2-(5-((Dimetilamino)metil)piridin-2-il)tieno[3,2-b]piridin-7- ilóxi)-3-fluoroanilina (397)
[00820] A uma solução de 402 (308 mg, 0,72 mmol) e NH4Cl (33 mg, 061 mmols) em 2:1 de EtOH/água (9 mL) foi adicionado pó de ferro (345 mg, 6,17 mmols) e a suspensão foi aquecida até o refluxo durante 1 hora. A mistura de reação foi filtrada através de celite e concentrada para fornecer o composto do título 397 (350 mg, produção quantitativa), que foi usado sem outra purificação. MS (m/z): (M+1) 395,1 (100%). Esquema 88
7-(4-Amino-2-fluorofenóxi)-N-(3-metoxifenil)tieno[3,2-b]piridin-2-amina (404) Etapa 1: 7-(2-Flúor-4-nitrofenóxi)-N-(3-metoxifenil)tieno[3,2-b]piridin-2- amina (403)
[00821] Uma solução de 399 (Esquema 87) (700 mg, 1,68 mmol), CS2CO3 (1,12 g, 3,43 mmols), 3-metoxianilina (190 DL, 206 mg, 1,68 mmol), Pd(OAc)2 (70 mg, 0,17 mmol) e Xantphos (1,43 g, 2,52 mmols) ( J. Org. Chem., 1999, 64, 6019-6022) em dioxano (15 mL) foi aquecida até o refluxo durante 5 horas. A mistura de reação foi resfriada, concentrada e o resíduo foi purificado por cromatografia instantânea usando 80% de EtOAc em hexanos como o eluente, para fornecer 403 (408 mg, 59% de rendimento). MS (m/z): (M+1) 412,0 (100%). Etapa 2: 7-(4-Amino-2-fluorofenóxi)-N-(3-metoxifenil)tieno[3,2-b]piridin- 2-amina (404)
[00822] A uma solução de 403 (408 mg, 0,99 mmol) e NH4Cl (45 mg, 084 mmol) em 2:1 de EtOH/água (15 mL) foi adicionado pó de ferro (472 mg, 8,43 mmols) e a suspensão foi aquecida até o refluxo durante 1 hora. A mistura de reação foi filtrada através de celite e concentrada para fornecer 404 (278 mg, 74% de rendimento), que foi usado sem outra purificação. MS (m/z): (M+1) 382,0 (100%). Tabela 19
[00823] Compostos 405-410 (Exemplos 189-192) preparados das aminas 396, 397 e 404
Esquema 89
Exemplo 193 1 -Óxido de 2-(7-(2-flúor-4-(3-oxo-3-(fenilamino)propanamido)fenóxi) tieno[3,2-b]piridin-2-il)piridina (409) Etapas1: 3-Flúor-4-(2-iodotieno[3,2-b]piridin-7-ilóxi)anilina (410)
[00824] A uma solução de 399 (2 g, 4,81 mmols) e NH4Cl (220 mg, 4,08 mmols) em 2:1 de EtOH/água (75 mL) foi adicionado pó de ferro (2,28 g, 40,8 mmols) e a suspensão foi aquecida até o refluxo durante 1 hora. A mistura de reação foi filtrada através de celite e concentrada para fornecer 410 (1,85 g, 100% de rendimento), que foi usado sem outra purificação MS. (m/z): (M+1) 386,8 (100%). Etapas 2: N 1-(3-Flúor-4-(2-iodotieno[3,2-b]piridin-7-ilóxi)fenil)-N3- fenilmalonamida (411)
[00825] Uma solução de ácido 1 (465 mg, 2,59 mmols) e BOPCl (666 mg, 2,59 mmols) foi misturada em DCM (5 mL) a 0°C e agitada na mesma temperatura durante 15 minutos. A mistura de reação foi então tratada com uma solução de 410 (500 mg, 1,29 mmol) e iPr2NEt (1,3 mL, 1 g, 7,79 mmols) em DCM (5 mL) a 0°C e deixada agitar em temperatura ambiente durante a noite. A mistura foi diluída em DCM (30 mL), lavada com solução de NaHCO3 saturada (30 mL), secada sobre Na2SO4 anidro, filtrada e concentrada. O resíduo foi purificado por cromatografia instantânea usando o gradiente 75-100% de EtOAc em hexanos como um eluente, para fornecer 411 (382 mg, 54% de rendimento). 1H RMND δ (400 MHz, CD3OD): 8,33 (d, 5,7 Hz), 7,79 (dd, 1H, J = 2,4 Hz, J = 12,3 Hz), 7,54 (m, 5H), 7,2-7,4 (m, 4H), 7,09 (t, 1H, J = 7,2 Hz), 6,50 (d, 1H, J = 5,5 Hz), 3,51 (s, 2H), 7,75 (s, 1H). MS (m/z): (M+1) 547,9 (100%). Etapas 3: 1-Óxido de 2-(7-(2-flúor-4-(3-oxo-3-(fenilamino) propanamido)fenoxi)tieno[3,2-b]piridin-2-il)piridina (409)
[00826] Uma solução de 411 (45 mg, 0,08 mmol), N-óxido de 4- bromopiridina (250 mg, 1,43 mmol), bistrimetilestanho (26 μL, 40 mg, 0,12 mmol) e Pd(PPh3)4 (10 mg, 0,01 mmol) em dioxano (1 mL) foi aquecida até o refluxo durante 4 horas. A mistura de reação foi resfriada, concentrada e o resíduo foi purificado por cromatografia instantânea usando o gradiente 5-10% de MeOH em DCM como um eluente, para fornecer 409 (11 mg, 27% de rendimento). 1H RMN δ (400 MHz, CD3OD): 8,71 (d, 1H, J = 5,4 Hz), 8,6 (m, 3H), 8,04 (dd, 1H, J = 2,5 Hz, J = 12,3 Hz), 7,85 (t, 1H, J = 8,4 Hz), 7,78 (m, 2H), 7,65 (m, 1H), 7,62 (m, 1H), 7,53 (m, 3H), 7,32 (t, 1H, J = 7,4 Hz), 6,81 (d, 1H, J = 5,5 Hz), 3,75 (s, 2H). MS (m/z): (M+1) 515,0 (100%). Esquema 90
Exemplo 194 N 1-(3-Flúor-4-(2-(1 -(2-morfolinoetil)-2-oxo-1,2-di-hidropirimidin-5- il)tieno[3,2-b]piridin-7-ilóxi)fenil)-N 3-(2-metoxifenil)malonamida (412) Etapa 1: 5-Bromo-1-(2-morfolinoetil)pirimidin-2 (1H)-ona (413)
[00827] A uma solução de 5-bromo-2-hidroxipirimidina (1,00 g, 5,75 mmols) em DMF (19 mL) foram adicionados iodeto de sódio (1,29 g, 8,6 mmols), carbonato de césio (4,68 g, 14,4 mmols), cloridrato de 4- (2-cloroetil)morfolina (1,18 g, 6,32 mmols) e a mistura de reação foi agitada a 60 °C durante 2,5 horas. A solução foi filtrada, e o filtrado foi concentrado. O resíduo foi purificado por cromatografia instantânea, eluindo com gradiente de metanol (5-15%) em diclorometano para fornecer o composto do título 413 (1,29 g, 78% de rendimento). MS (m/z): 288,1 (50%) (M+H), 290,0 (50%) (M+H). 1H RMN (DMSO-d6) δ (ppm): 8,61 (d, J = 3,3Hz, 1H), 8,46 (d, J = 3,3Hz, 1H), 3,93 (t, J = 5,9Hz, 2H), 3,51 (t, J = 4,5Hz, 4H), 2,57 (t, J = 6,1Hz, 2H), 2,45-2,39 (m, 4H). Etapa 2: 5-(7-Clorotieno[3,2-b]piridin-2-il)-1-(2-morfolinoetil)pirimidin-2 (1H)-ona (414)
[00828] A uma solução do derivado de tributilestanho 6 (1,88 g, 4,1 mmols) (Esquema 2) em tolueno (39 mL) foram adicionados 5-bromo- 1-(2-morfolinoetil)pirimidin-2-(1H)-ona (413, 1,29 g, 4,5 mmols) e Pd(PPh3)4 (474 mg, 0,41 mmol). A mistura de reação foi agitada a 100 °C durante 16 horas. A mistura foi filtrada e o solvente foi evaporado. O resíduo foi triturado em hexanos, então triturado em EtOAc para produzir o composto do título 414 (236 mg, 15% de rendimento). MS (m/z): 377,0 (M+H). 1H RMN (DMSO-d6) δ (ppm): 9,18 (d, J = 3,5Hz, 1H), 8,76 (d, J = 3,3Hz, 1H), 8,64 (d, J = 5,1Hz, 1H), 8,05 (s, 1H), 7,57 (d, J = 5,1Hz, 1H), 4,06 (t, J = 5,9Hz, 2H), 3,54 (t, J = 4,5Hz, 4H), 2,64 (t, J = 5,9Hz, 2H), 2,49-2,43 (m, 4H). Etapa 3: 5-(7-(2-Flúor-4-nitrofenóxi)tieno[3,2-b]piridin-2-il)-1-(2- morfolinoetil)pirimidin-2 (1H)-ona (415).
[00829] A uma solução da piridona 414 (236 mg, 0,63 mmol) em Ph2O (1,3 mL) foram adicionados 2-flúor-4-nitrofenol (197 mg, 1,26 mmol) e K2CO3 (173 mg, 1,26 mmol). A mistura foi agitada a 100 °C durante 1 hora, em seguida a 180 °C durante 2 horas, resfriada e evaporada sob pressão reduzida. O resíduo foi purificado por cromatografia instantânea com um gradiente de metanol (2-10%) em diclorometano para fornecer o composto do título 415 (81 mg, 26% de rendimento). MS (m/z): 498,0 (M+H). 1H RMN (DMSO-d6) δ (ppm): 9,16 (d, J = 3,3Hz, 1H), 8,66 (d, J = 3,3Hz, 1H), 8,60 (d, J = 5,3Hz, 1H), 8,47 (dd, J = 10,2,5Hz, 1H), 8,19 (d, J = 8,8Hz, 1H), 8,02 (s, 1H), 7,69 (t, J = 8,6Hz, 1H), 6,98 (d, J = 5,3Hz, 1H), 4,02 (t, J = 5,5Hz, 2H), 3,58-3,43 (m, 4H), 2,59 (t, J = 5,5Hz, 2H), 2,50-2,41 (m, 4H). Etapa 4: 5-(7-(4-Amino-2-fluorofenóxi)tieno[3,2-b]piridin-2-il)-1-(2- morfolinoetil)pirimidin-2 (1H)-ona (416)
[00830] A uma solução do composto 415 (81 mg, 0,16 mmol) em etanol (1 mL) e água (0,5 mL) foram adicionados NH4Cl (9 mg, 0,16 mmol) e pó de ferro (73 mg, 1,30 mmol). A mistura foi agitada a 80°C durante 45 minutos, filtrada através de celite, e o solvente foi evaporado para fornecer o composto do título (416). MS (m/z): 468,1 (M+H). Etapa 5 N 1-(3-Flúor-4-(2-(1-(2-morfolinoetil)-2-oxo-1,2-di- hidropirimidin-5-il)tieno[3,2-b]piridin-7-ilóxi)fenil)-N 3-(2- metoxifenil)malonamida (412)
[00831] A uma solução da amina 416 (0,16 mmol) em DMF (1 mL) foram adicionados ácido 27 (38 mg, 0,18 mmol), HOBT (24 mg, 0,18 mmol) e EDC x HCl (46 mg, 0,24 mmol). A mistura foi agitada em temperatura ambiente durante 16 horas. O solvente foi evaporado, em seguida o resíduo foi purificado por cromatografia instantânea com gradiente de metanol (0-5%) em diclorometano, seguido por purificação por HPLC preparativa com gradiente de metanol (20-100%) em água para fornecer o composto do título 412 (14 mg, 13% de rendimento). MS (m/z): 659,1 (M+H). 1H RMN (DMSO-d6) δ (ppm): 10,59 (s, 1H), 9,64 (s, 1H), 9,16 (d, J = 3,3Hz, 1H), 8,69 (d, J = 3,3Hz, 1H), 8,51 (d, J = 5,5Hz, 1H), 8,07 (d, J = 7,4Hz, 1H), 7,98 (s, 1H), 7,88 (dd, J = 13,2,3Hz, 1H), 7,51 (t, J = 9,0Hz, 1H), 7,43 (dd, J = 8,8,1,6Hz, 1H), 7,11-7,05 (m, 2H), 6,94-6,90 (m, 1H), 6,67 (d, J = 4,7Hz, 1H), 4,05 (t, J = 6,5Hz, 2H), 3,86 (s, 3H), 3,64 (s, 2H), 3,55-3,50 (m, 4H), 2,63 (t, J = 5,5Hz, 2H), 2,46-2,42 (m, 4H). Esquema 91

Exemplo 195 N 1-(4-(2-(1 -(2-(Dimetilamino)etil)-6-oxo-1,6-di-hidropiridin-3-il)tieno[3,2- b ]piridin-7-ilóxi)-3-fluorofenil)-N 3-(2-metoxifenil)malonamida (417) Etapa 1: 7-Cloro-2-(6-metoxipiridin-3-il)tieno[3,2-b]piridina (418)
[00832] A uma solução de derivado de tributilestanho 6 (2,00g, 4,36 mmols) (Esquema 91) em tolueno (10 mL) foi adicionado Ph(PPh3)4 (0,503 g, 0,436 mmol) seguido por 5-bromo-2-metoxipiridina (0,62 mL, 4,8 mmols). Gás de nitrogênio foi borbulhado diretamente dentro de uma mistura de reação durante 30 minutos antes dela ser aquecida até o refluxo durante 16 horas. A mistura de reação foi então resfriada até a temperatura ambiente e o precipitado amarelo foi coletado por filtração e lavado com hexanos. O material foi então novamente purificado por cromatografia instantânea (eluente 20 a 50% de acetato de etila em hexanos) para fornecer o composto do título 418 como um sólido branco macio (0,752g, 62% de rendimento). MS (m/z): 277,0 (M+H). Etapa 2: 7-(2-Flúor-4-nitrofenóxi)-2-(6-metoxipiridin-3-il)tieno[3,2- b]piridina (419)
[00833] Uma suspensão de 418 (0,638 g, 2,31 mmols), 2-flúor-4- nitrofenol (0,725 g, 4,61 mmols) e carbonato de potássio (0,638 g, 4,61 mmols) em éter de difenila (6 mL) foi aquecida para 170 °C durante 8 horas em um tubo de pressão. A solução escura resultante foi resfriada até a temperatura ambiente, diluída com CH2Cl2 e então filtrada. O filtrado foi concentrado e o resíduo foi purificado por cromatografia instantânea (eluente 100% de hexanos a 50/50 de acetato de etila/hexanos) para produzir o composto do título 419 como um sólido amarelo (0,395 g, 43% de rendimento). MS (m/z): 398,0 (M+H). Etapa 3: 5-(7-(2-Flúor-4-nitrofenóxi)tieno[3,2-b ]piridin-2-il)piridin-2 (1H - ona (420)
[00834] A um frasco contendo uma suspensão de 419 (0,299 g, 0,753 mmol) em acetonitrila (8 mL) foi adicionado clorotrimetilsilano (0,095 mL, 0,748 mmol) seguido por iodeto de sódio (0,112 g, 0,748 mmol). A mistura foi então aquecida para entre 70 a 100°C durante 24 horas. Durante este tempo mais quatro porções de clorotrimetilsilano (0,095 mL cada porção) e iodeto de sódio (0,112 g cada porção) foram adicionadas. A mistura de reação foi então resfriada até a temperatura ambiente e o precipitado formado foi coletado por filtração, lavado com NH4OH e água, e secado para fornecer o composto do título 420 como um sólido marrom (0,283g, 98%). MS (m/z): 384,0 (M+H), 406,0 (M+Na). Etapa 4: 1 -(2-(Dimetilamino)etil)-5-(7-(2-flúor-4-nitrofenóxi)tieno[3,2- b ]piridin-2-il)piridin-2 (1H)-ona (421)
[00835] A piridinona 420 (0,283g, 0,739 mmol), 2-cloro-N,N- dimetiletanamina (0,128 g, 0,887 mmol), carbonato de césio (0,547 g, 1,68 mmol) e iodeto de sódio (0,132 g, 0,88 mmol) foram aquecidos em DMF (10 mL) a 70 °C durante 3 dias. Uma porção adicional de 2- cloro-N,N-dimetiletanamina (64 mg, 0,44 mmol) e carbonato de césio (0,289 g, 0,887 mmol) foram adicionados. A mistura de reação foi aquecida por mais 8 horas, resfriada até a temperatura ambiente e evaporada sob pressão reduzida. O resíduo foi dividido entre água (50 mL) e 5% de etanol/diclorometano. A fase orgânica foi coletada, secada sobre Na2SO4 anidro, filtrada e concentrada. O resíduo foi então purificado por cromatografia de sílica-gel usando uma coluna Biotage de 25M e um gradiente de 3 a 20% de metanol em diclorometano com 1% de ácido acético, para fornecer o composto do título 421 como um material sólido (0,108 g, 32% de rendimento). MS (m/z): 228,0 (/2), 455,0 (M+H), 477,0 (M+Na). Etapa 5: 5-(7-(4-Amino-2-fluorofenóxi)tieno[3,2-b ]piridin-2-il)-1-(2- (dimetilamino)etil)piridin-2 (1H)-ona (422)
[00836] A uma solução do composto nitro 421 (0,103 g, 0,227 mmol) em metanol (2 mL) e THF (2 mL) a 0°C foi adicionado hexahidrato de cloreto de níquel (0,162 g, 0,681 mmol) seguido por boroidreto de sódio (28 mg, 0,749 mmol). Após uma hora de agitação a 0°C outra porção de hexahidrato de cloreto de níquel (0,108 g, 0,458 mmol) e boroidreto de sódio (19 mg, 0,50 mmol) foram adicionados. A mistura de reação foi então agitada em temperatura ambiente durante 5 horas antes de ela ser concentrada e tratada com HCl(aq) a 1N e diclorometano. A mistura foi agitada durante 10 minutos e em seguida basificada para pH 10 pela adição de NH4OH. A solução basificada foi então extraída com diclorometano. O extrato foi seco sobre Na2SO4 anidro, filtrado e concentrado. O resíduo foi purificado por cromatografia instantânea (eluir um gradiente de 10 a 100% de metanol em acetato de etila) para fornecer o composto do título 422 como sólido amarelo (26 mg, 27% de rendimento). MS (m/z): 425,0 (M+H). Etapa 6: N 1-(4-(2-(1-(2-(Dimetilamino)etil)-6-oxo-1,6-di-hidropiridin-3- il)tieno[3,2-b ]piridin-7-ilóxi)-3-fluorofenil)-N 3-(2-metoxifenil)malonamida Í4171
[00837] A uma solução de composto 422 (25 mg, 0,059 mmol) em DMF (0,5 mL) foi adicionado o ácido 27 (25 mg, 0,12 mmol), hidroxibenzotriazol (11 mg, 0,071 mmol), então EDC (23 mg, 0,12 mmol). A mistura de reação foi agitada em temperatura ambiente durante 6 horas antes da adição de NaHCO3 saturado (8 mL). A mistura foi então extraída com diclorometano; O extrato foi seco sobre Na2SO4 anidro, filtrado e concentrado. O resíduo foi então purificado por cromatografia instantânea (eluir um gradiente de 0 a 20% metanol em diclorometano) para fornecer o composto do título 417 como um sólido branco (19 mg, 53% de rendimento). 1H RMN: (DMSO-d6) δ(ppm): 10,58 (s, 1H), 9,62 (s, 1H), 8,45 (d, J = 5,6Hz, 1H), 8,28 (d, J = 2,8Hz, 1H), 8,05 (d, J = 8,4Hz, 1H), 7,95 (dd, J = 2,8, 9,6Hz, 1H), 7,887,84 (m, 2H), 7,45 (t, J = 8,8Hz, 1H), 7,43-7,40 (m, 1H), 7,07-7,03 (m, 2H), 6,92-6,88 (m, 1H), 6,60 (d, J = 5,6Hz, 1H), 6,51 (d, J = 9,6Hz 1H), 4,05 (t, J = 6,0Hz, 2H), 3,84 (s, 3H), 3,62 (s, 2H), 2,53 (t, J = 6,4Hz, 2H), 2,47 (s, 6H). MS (m/z): 308,5 (/2), 616,1 (M+H).
[00838] Os seguintes compostos et al. compostos descritos aqui, e os compostos descritos nos Exemplos de ensaio abaixo, são preparados essencialmente de acordo com os procedimentos delineados nos Esquemas, diagramas, exemplos e preparações mencionados aqui. Tabela 20
[00839] Em um terceiro aspecto, a presente invenção fornece farmaceutical composições farmacêuticas compreendendo um inibidor de sinalização de receptor de VEGF e sinalização de receptor de HGF de acordo com a presente invenção e um veículo, excipiente, ou diluente farmaceuticamente aceitável. Composições da presente invenção podem ser formuladas por qualquer método bem conhecido na técnica e podem ser preparadas para administração por qualquer via, incluindo, sem limitação, parenteral, oral, sublingual, transdérmica, tópica, intranasal, intratraqueal ou intrarretal. Em certas modalidades preferidas, composições da presente invenção são administradas intravenosamente em uma instalação de hospital. Em certas outras modalidades preferidas, a administração pode preferivelmente ser por via oral.
[00840] As características do veículo dependerão da via de administração. Como empregado aqui, o termo "farmaceuticamente aceitável" significa um material não tóxico que é compatível com um sistema biológico tal como uma célula, cultura celular, tecido, ou organismo, e que não interfere com a eficácia da atividade biológica do(s) ingrediente(s) ativos. Desse modo, as composições de acordo com a presente invenção podem conter, em adição, inibidor, diluentes, cargas, sais, tampões, estabilizantes, solubilizantes, et al. materiais bem conhecidos na técnica. A preparação de formulações farmaceuticamente aceitáveis é descrita, por exemplo, em Remington's Farmaceutical Sciences, 18a Edition, ed. A. Gennaro, Mack Publishing Co., Easton, Pa., 1990.
[00841] Como empregado aqui, o termo sais farmaceuticamente aceitáveis refere-se a sais que retêm a atividade biológica desejada dos compostos acima identificados e exibem mínimos ou nenhum efeito toxicológico indesejado. Exemplos de tais sais incluem, porém não são limitados a, sais formados com ácidos inorgânicos (por exemplo, ácido clorídrico, ácido hidrobrômico, ácido sulfúrico, ácido fosfórico, ácido nítrico, e similares), e sais formados com ácidos orgânicos tais como ácido acético, ácido oxálido, ácido tartárico, ácido succínico, ácido málico, ácido ascórbico, ácido benzóico, ácido tânico, ácido palmóico, ácido algínico, ácido poliglutâmico, ácido naftalenossulfônico, ácido naftalenodissulfônico, ácido metanossulfônico, ácido p-toluenossulfônico e ácido poligalacturônico. Os compostos podem também ser administrados como sais quaternários farmaceuticamente aceitáveis conhecidos por aqueles versados na técnica, que especificamente incluem o sal de amônio quaternário da fórmula --NR+Z--, em que R é hidrogênio, alquila, ou benzila, e Z é um contra-íon, incluindo cloreto, brometo, iodeto, --O- alquila, toluenossulfonato, metilsulfonato, sulfonato, fosfato, ou carboxilato (tal como benzoato, succinato, acetato, glicolato, maleato, malato, citrato, tartarato, ascorbato, benzoato, cinamoato, mandeloato, benziloato, e difenilacetato).
[00842] O composto ativo é incluído no veículo ou diluente farmaceuticamente aceitável em uma quantidade suficiente para liberar para um paciente uma quantidade terapeuticamente eficaz sem causar sérios efeitos tóxicos no paciente tratado. A faixa de dosagem eficas dos derivados farmaceuticamente aceitáveis podem ser calculadas com base no peso do composto origem a ser liberado. Se o derivado exibe atividade por si mesmo, a dosagem eficaz pode ser estimada como acima, usando o peso do derivado, ou por outro método conhecido por aqueles versados na técnica. Inibição de sinalização de receptor de VEGF e sinalização de receptor de HGF
[00843] Em um quarto aspecto, a presente invenção fornece método de inibir a sinalização de receptor de VEGF e sinalização de receptor de HGF em uma célula, compreendendo contatar uma célula em que a inibição de sinalização de receptor de VEGF e sinalização de receptor de HGF é desejada com um inibidor de sinalização de receptor de VEGF e sinalização de receptor de HGF de acordo com a presente invenção. Por que os compostos da presente invenção inibem a sinalização de receptor de VEGF e sinalização de receptor de HGF, eles são instrumentos de pesquisa úteis para estudo in vitro do papel de sinalização de receptor de VEGF e sinalização de receptor de HGF em processos biológicos.
[00844] Preferivelmente, o método de acordo com o quarto aspecto da presente invenção causa uma inibição de proliferação celular das células contatadas. A frase "inibição de proliferação celular" é usada para denotar uma capacidade de um inibidor de sinalização de receptor de VEGF e sinalização de receptor de HGF para retardar o crescimento de células contatadas com o inibidor quando comparado com as células não contatadas. Uma avaliação da proliferação celular pode ser feita por contagem das células contatadas e não-contatadas empregando-se uma Contadora de Células Coulter (Coulter, Miami, Fla.) ou um hemacitômetro. Onde as células estão em um crescimento sólido (por exemplo, um tumor sólido ou órgão), uma tal avaliação da proliferação celular pode ser feita avaliando-se o crescimento com compasso de calibre e comparando-se o tamanho do crescimento de células contatadas com as células não-contatadas.
[00845] Preferivelmente, o crescimento de células contatadas com o inibidor é retardado em pelo menos 50% quando comparado ao crescimento de células não-contatadas. Mais preferivelmente, a proliferação celular é inibida em 100% (isto é, as células contatadas não aumentam em número). Mais preferivelmente, a frase "inibir a proliferação celular " inclui uma redução no número ou tamanho de células contatadas, quando comparadas às células não-contatadas. Desse modo, um inibidor de sinalização de receptor de VEGF e sinalização de receptor de HGF de acordo com a presente invenção que inibe a proliferação celular em uma célula contatada pode induzir a célula contatada a sofrer retardo de crescimento, a sofrer interrupção do crescimento, a sofrer morte celular programada (isto é, a apoptose), ou a sofrer morte celular necrótica.
[00846] Em algumas modalidades preferidas, a célula contatada é uma célula neoplásica. O termo "célula neoplásica" é usada para denotar uma célula que mostra crescimento celular aberrante. Preferivelmente, o crescimento celular aberrante de uma célula neoplásica é crescimento celular aumentado. Uma célula neoplásica pode ser uma célula hiperplásica, uma célula que mostra uma falta de inibição de contato de crescimento in vitro, uma célula de tumor benigno que é incapaz de metástase in vivo, ou uma célula de câncer que é capaz de metástase in vivo e que pode recorrer após remoção experimentada. O termo "tumorigênese" é usado para denotar a indução de proliferação celular que induz ao desenvolvimento de um crescimento neoplásico.
[00847] Em algumas modalidades preferidas, a célula contatada é em um animal. Desse modo, a presente invenção fornece a método para tratamento de uma doença proliferativa ou condição em um animal, compreendendo administrar a um animal em necessidade de tal tratamento uma quantidade terapeuticamente eficaz de inibidor de sinalização de receptor de VEGF e sinalização de receptor de HGF da presente invenção. Preferivelmente, o animal é um mamífero, mais preferivelmente um mamífero domesticado. Mais preferivelmente, o animal é um ser humano.
[00848] O termo "doença proliferativa ou condição celular " é entendida referir-se a qualquer condição caracterizada por crescimento celular aberrante, preferivelmente proliferação celular anormalmente aumentada. Exemplos de tais doenças proliferativas ou condições celulares tratáveis por inibição e tratamento, incluem, porém não são limitados a câncer. Exemplos de tipos particulares de câncer incluem, porém não são limitados a câncer de mama, câncer de pulmão, câncer de cólon, câncer retal, câncer de bexiga, leucemia e câncer renal. Em modalidades particularmente preferidas, a presente invenção fornece um método para inibir a proliferação de célula neoplásica em um animal compreendendo administrar a um animal tendo pelo menos uma célula neoplásica presente em seu corpo uma quantidade terapeuticamente eficaz de um inibidor de sinalização de receptor de VEGF e sinalização de receptor de HGF da presente invenção.
EXEMPLOS DE ENSAIO
Exemplo de Ensaio 1
Inibição de Atividade de c-met e VEGF
[00849] Os seguintes protocolos foram usados para ensaiar os compostos da presente invenção. Ensaios de Tirosina cinase In Vitro Receptora (KDR de receptor de c- Met/HGF e receptor de VEGF)
[00850] Estes testes avaliam a capacidade de compostos para inibir a atividade enzimática de receptor de c-Met/HGF humano recombinante e atividade enzimática de receptor de VEGF.
[00851] Um cDNA de 1,3-kb correspondendo ao domínio intracelular de c-Met ou c-Met IC (Número de acesso Genbank NP000236-1 aminoácido 1078 a 1337) foi clonado nos sítios BamHI/XhoI do vetor pBlueBacHis2A (Invitrogen) para a produção de uma versão rotulada por histidina daquela enzima. Este constructo foi usado para gerar baculovírus recombinante usando o sistema Bac-N- Blue® de acordo com as instruções do fabricante (Invitrogen).
[00852] A proteína c-Met IC foi expressa em células Hi-5 (Trichoplusia Ni) em infecção com constructo de baculovírus recombinante. Em síntese, as células Hi-5 desenvolvidas em suspensão e mantidas em meio livre de soro (Sf900 II suplementado com gentamicina) em uma densidade celular de cerca de 2 X 106 células/mL foram infectadas com as viroses acima mencionadas em uma multiplicidade de infecção (MOI) de 0,2 durante 72 horas em 27°C com agitação a 120 rpm em uma agitadora giratória. As células infectadas foram coletadas por centrifugação a 398 g durante 15 minutos. Os péletes celulares foram congelados a -80°C até a purificação ser realizada.
[00853] Todas as etapas descritas em extração celular e purificação foram realizadas a 4°C. Os péletes celulares Hi-5 congelados infectados com o baculovírus recombinante de C-Met IC foram descongelados e suavemente ressuspensos em Tampão A (20 mM de Tris pH 8,0, glicerol a 10%, 1 μg/ mL de pepstatina, 2 μg/mL de Aprotinina e leupeptina, 50 μg/mL de PMSF, 50 μg/ mL de TLCK e E64 a 10 μM, DTT a 0,5 mM e Levamisol a 1 mM) usando 3 mL de tampão por grama de células. A suspensão foi homogeneizada por Dounce, após o que ela foi centrifugada a 22500 g, 30 minutos, 4°C. O sobrenadante (extrato celular) foi usado como material de partida para purificação de c-Met IC.
[00854] O sobrenadante foi carregado sobre uma coluna QsefaroseFF (Amersham Biosciences) equilibrada com Tampão B (Tris a 20 mM pH 8,0, de glicerol a 10%) suplementado com NaCl a 0,05 M. Seguindo um volume de coluna dez (CV) lavar com tampão de equilíbrio, proteínas de ligação foram eluídas com um gradiente linear de sal 5 CV abrangendo de 0,05 a NaCl a 1M em Tampão B. Tipicamente, a condutividade de frações selecionadas classificou-se entre 6,5 e 37 mS/cm. Este eluato de Qsefarose teve uma concentração de NaCl estimada de 0,33M e foi suplementado com uma solução de NaCl a 5M a fim de aumentar a concentração de NaCl a 0,5M e também com uma solução de Imidazol a 5M (pH 8,0) para obter uma concentração de imidazol final de 15mM. Este material foi carregado sobre uma coluna de afinidade HisTrap (GE Healthcare) equilibrada com Tampão C (50 mM a NaPO4 pH 8,0, NaCl a 0,5M, glicerol a 10%) suplementada com 15 mM de imidazol. Após uma lavagem de 10 CV com tampão de equilíbrio e uma lavagem de 8 CV com tampão C + 40 mM de imidazol, proteínas ligadas foram eluídas com um gradiente linear de 8 CV (15 a 500 mM) de imidazol em tampão C. Frações enriquecidas com C-Met IC desta etapa de cromatografia foram agrupadas com base em análise de SDS-PAGE. Este lago de enzima sofreu permuta de tampão usando coluna PD-10 (GE Healthcare) contra o tampão D (HEPES a 25 mM pH 7,5, NaCl a 0,1M, glicerol a 10% e β-mercaptoetanol a 2 mM). As concentrações de preparações de proteína C-Met IC finais foram de cerca de 0,5 mg/mL com pureza de aproximadamente 80%. Os estoques de proteína c-Met IC purificadas foram suplementados com BSA a 1 mg/mL, aliquotados e congelados a -80°C antes do uso em ensaio enzimático.
[00855] No caso de KDR de receptor de VEGF um cDNA de 1,6-kb correspondendo ao domínio catalítico de VEGFR2 ou KDR (número de acesso Genbank AF035121 amino ácido 806 a 1356) foi clonado no sítio Pst I do vetor pDEST20 Gateway (Invitrogen) para a produção de uma versão rotulada por GST daquela enzima. Este constructo foi usada para gerar baculovírus recombinante usando o sistema Bac-to- Bac® de acordo com as instruções do fabricante (Invitrogen).
[00856] A proteína GST-VEGFR2806-1356 foi expressa em células Sf9 (Spodoptera frugiperda) em infecção com constructo de baculovírus recombinante. Em síntese, células Sf9 desenvolvidas em suspensão e mantidas em meio livre de soro (Sf900 II suplementado com gentamicina) em uma densiade celular de cerca de 2 X 106 células/mL foram infectadas com as virose acima mencionada em uma multiplicidade de infecção (MOI) de 0,1 durante 72 horas a 27°C com agitação a 120 rpm em uma agitadora giratória. As células infectadas foram coletadas por centrifugação a 398 g durante 15 minutos. Os péletes celulares foram congelados a -80°C até a purificação ser realizada.
[00857] Todas as etapas descritas em extração e purificação celular foram realizadas a 4°C. Os péletes de célula Sf9 congelados infectados com o baculovírus recombinante GST-VEGFR2806-1356 foram descongelados e suavemente ressuspensas em Tampão A (PBS pH 7,3 suplementado com 1 μg/ mL de pepstatina, 2μg/mL de aprotinina e leupeptina, 50 μg/mL de PMSF, 50 μg/mL de TLCK e 10 μM de E64 e 0,5 mM de DTT) usando 3 mL de tampão por grama de células. A suspensão foi homogeneizada por Dounce e 1% de Triton X-100 foi adicionado ao homogeneizado, após o que ele foi centrifugado a 22500 g, 30 minutos, 4°C. O sobrenadante (extrato celular) foi usado como material de partida para purificação de GST-VEGFR2806-1356.
[00858] O sobrenadante foi carregado sobre uma coluna de GST- agarose (Sigma) equilibrada com PBS pH 7,3. Seguindo um volume de coluna quatro (CV) lavar com PBS pH 7,3 + 1% de Triton X-100 e 4 CV de lavagem com tampão B (Tris a 50 mM pH 8,0, 20% de glicerol e NaCl a 100 mM), as proteínas ligadas foram eluídos na etapa com 5 CV de tampão B suplementado com DTT a 5 mM e glutation a 15 mM de glutação. As frações enriquecidas com GST-VEGFR2806-1356 desta etapa de cromatografia foram agrupadas com base em traço de U.V., sito é, frações com O.D,280 elevado. As concentrações de preparações de proteína GST-VEGFR2806-1356 final foram de cerca de 0,7 mg/mL com pureza de aproximadamente 70%. Os estoques de proteína GST- VEGFR2806-1356 purificada foram aliquotados e congelados a -80°C antes do uso em ensaio enzimático.
[00859] Inibição de receptor de c-Met/HGF e VEGFR/KDR foi avaliada em um ensaio DELFIA® (Perkin Elmer). O substrato poli(Glu4,Tyr) foi imobilizado sobre placas de 96 cavidades de poliestireno de ligação elevada pretas. As placas revestidas foram lavadas e armazenadas a 4 °C. Durante o ensaio, enzimas foram pré- incubadas com inhibitor e Mg-ATP sobre gelo em placas de 96 cavidades de polipropileno durante 4 minutos, e em seguida transferidas para as placas revestidas. A reação de cinase subsequente ocorre a 30 °C durante 10 a 30 minutos. As concentrações de ATP no ensaio foram de 10 uM para C-Met (5X o Km) e 0,6 uM para VEGFR/KDR (2X o Km). A concentração de enzima foi de 25 nM (C-Met) ou 5 nM (VEGFR/KDR). Após incubação, as reações de cinase foram extinguidas com EDTA e as placas foram lavadas. O produto fosforilado foi detectado por incubação com MoAb antifosfotirosina rotulado por Európio. Após lavar as placas, MoAb ligado foi detectado por fluorescência resolvida com o tempo em uma leitora Gemini SpectraMax (Molecular Devices). Os compostos foram avaliados em uma faixa de concentrações e IC50’s (concentração de compostos fornecendo 50% de inibição de atividade enzimática) foram determinados. Ensaio com base em fosforilação de C-Met
[00860] Este teste avalia a capacidade de compostos inibirem a autofosforilação estimulada por HGF do próprio receptor de c-Met/HGF em um sistema de célula total.
[00861] A linhagem celular MNNGHOS expressando a proteína de fusão TPR-MET foi adquirida de ATCC. O TPR-MET é o produto de uma translocação cromossômica colocando o locus de TPR sobre o cromossoma 1 a montante do gene MET no cromossoma 7 codificando para seu domínio catalítico de região citoplásmica. A dimerização da oncoproteína TRP-Met de Mr 65.000 através de um dispositivo de fecho de leucina codificado pela porção de TPR induz à ativação constitutiva da met cinase. A autofosforilação constitutiva ocorre em resíduos Tyr361/365/366 de TPR-Met. Estes resíduos são homólogos a Tyr1230/1234/1235 de MET que torna-se fosforilado em dimerização do receptor em ligação de HGF.
[00862] Inibidor de c-Met formulado como estoques de 30 mM em DMSO. Para tratamentos de MNNGHOS, células, compostos foram adicionados aos meios de cultura de tecido em doses indicadas durante 3 horas antes da lise celular. As células foram lisadas em tampão de lise gelado contendo HEPES a 50 mM (pH 7,5), NaCl a 150 mM, MgCl2 a 1,5 mM, 10 % de glicerol, 1 % Triton X-100, cloridrato de fluoreto de 4-(2-aminoetil)benzenossulfonila a 1 mM, ortovanadato de sódio a 200 μM, fluoreto de sódio a 1 mM, 10 μg/mL de leupeptina, 10 μg/mL de aprotinina/mL, 1 ug/mL de pepstatina e 50 ug/mL cloridrato Na-p-Tosil-L-lisina clorometil cetona.
[00863] Os lisados foram separados em 5-20% de PAGE-SDS e imunomanchas foram realizadas usando membranas de difluoreto de polivinilideno Immobilon P (Amersham) de acordo com as instruções do fabricante para manipulação. As manchas foram lavadas em solução salina tamponada por Tris com 0,1% de detergente Tween 20 (TBST). Tyr361/365/366 de TPR-Met foram detectados com anticorpos de coelho policlonais contra Met fosforilado por tirosina (Biosource International) e peroxidase de rábano silvestre anticoelho de anticorpos secundarios (Sigma) por ensaios de quimioluminescência (Amersham, ECL) foram realizados de acordo com as instruções do fabricante e seguidos por exposição de película. O sinal foi quantificado por densitometria em Alfa-Imager. Os valores de IC50 foram definidos como a dose requerida para obter 50% de inibição dos níveis máximos de c-Met fosforilado estimulado por HGF. Tabela 21 Perfil Biológico de compostos selecionados
[00864] Este teste avalia a capacidade dos compostos de inibir o crescimento de tumor sólido.
[00865] Os xenoenxertos de tumor são estabelecidos no flanco de camundongos CD1 atímicos fêmeas (Charles River Inc.), por injeção subcutânea de 1X106 U87, A431 ou SKLMS células/camundongo. Uma vez estabelecidos, os tumores são então serialmente passados s.c. em hóspedes de camundongos nus. Fragmentos de tumor destes animais hóspedeis são usados em experimentos de avaliação de composto subsequente. Para experimentos de avaliação de composto camundongos nus fêmea pesando aproximadamente 20 g são implantados s.c. por implante cirúrgico com fragmentos de tumor de ~30 mg de tumores de doador. Quando os tumores são aproximadamente de 100 mm3 de tamanho (~ 7 - 10 dias após o implante), os animais são aleatorizados e separados em grupos de tratamento e controle. Cada grupo contém 6 a 8 camundongos transportando tumor, cada um dos quais é rotulado na orelha e seguido individualmente durante todo o experimento.
[00866] Os camundongos são pesados e as medições de tumor são feitas por compassos de calibre três vezes por semana, começando no Dia 1. Estas medições de tumor são convertidas para o volume de tumor pela fórmula bem conhecida (L+W/4)3 4/3π. O experimento é terminado quando os tumores de controle atingiram um tamanho de aproximadamente 1500 mm3. Neste modelo, a mudança em volume de tumor médio para um grupo tratado por composto/a mudança em volume de tumor médio do grupo de controle (não-tratado ou tratado com veículo) x 100 (ΔT/ΔC) é subtraído de 100 para fornecer o percentual de inibição de tamanho de tumor (%TGI) para cada composto de teste. Em adição aos volumes de tumor volumes, o peso corporal dos animais é monitorado duas vezes semanalmente durante até 3 semanas.
[00867] As atividades de diversos compostos de acordo com a presente invenção avaliadas por vários ensaios são mostradas na Tabela 21 e Tabela 22. Nestas tabelas, "a" indica atividade inibidora em uma concentração menor do que 50 nanomolares; "b" indica atividade inibidora em uma concentração > 50 porém < 250 nanomolares, "c" indica atividade inibidora a > 250 porém < 500 e "d" indica atividade inibidora em uma concentração de > 500 nanomolares; e "e" indica nenhuma atividade quando avaliada por aquele ensaio.
[00868] HGF tem atividade bem conhecida em termos de induzir a dispersão e migração (cicatrização de ferimento) (Wells et al., Cell Motila Cytoskeleton. Nov 2005 62 (3):180-94; Miura et al., Urology 2001 Dec 58 (6):1064-9; Nishimura et al., Int J Urol. 5 de maio de 1998 (3):276-81; Wang et al., Mol câncer Ther. 2003 Nov;2 (11):1085-92; e Christensen et al., câncer Res. 2003 Nov 1;63 (21):7345-55). Ensaios para avaliar a capacidade inibidora para bloquear estas atividades dependentes de HGF têm sido empregados e seguem os métodos empregados em Christensen et al. Para a Tabela 22, para colunas direcionadas à inibição de cicatrização de ferimento A549 e inibição de dispersão de DU145, os valores de IC50 são em mM, com "A" indicando IC50 menor do que 1 mM, "B" indicando IC50 de > 1 mM porém < 5 mM, "C" indicando IC50 de > 5 mM porém < 10 mM, e "D" indicando IC50 de > 10 mM.
[00869] Na Tabela 23 "a" indica % de TGI na faixa de 75-100; "b" indica % de TGI na faixa de 50-74; "c" indica % de TGI na faixa de 2549, e "d" indica % de TGI na faixa de 0-24. Regime de administração foi de uma só vez.
Tabela 23 Inibição de Crescimento de Tumor por compostos selecionados