BRPI0918652B1 - Composição farmacêutica compreendendo um meio hidrofóbico e uma forma sólida que compreende polipeptídeo e sal de ácido graxo de cadeia média, processo de produção da mesma e forma de dosagem oral - Google Patents
Composição farmacêutica compreendendo um meio hidrofóbico e uma forma sólida que compreende polipeptídeo e sal de ácido graxo de cadeia média, processo de produção da mesma e forma de dosagem oral Download PDFInfo
- Publication number
- BRPI0918652B1 BRPI0918652B1 BRPI0918652-2A BRPI0918652A BRPI0918652B1 BR PI0918652 B1 BRPI0918652 B1 BR PI0918652B1 BR PI0918652 A BRPI0918652 A BR PI0918652A BR PI0918652 B1 BRPI0918652 B1 BR PI0918652B1
- Authority
- BR
- Brazil
- Prior art keywords
- formulation
- fatty acid
- medium
- glyceryl
- formulations
- Prior art date
Links
- -1 MEDIUM CHAIN FATTY ACID SALT Chemical class 0.000 title claims abstract description 132
- 230000002209 hydrophobic effect Effects 0.000 title claims abstract description 130
- 239000007787 solid Substances 0.000 title claims abstract description 104
- 239000008194 pharmaceutical composition Substances 0.000 title claims abstract description 41
- 239000006186 oral dosage form Substances 0.000 title claims abstract description 7
- 108090000765 processed proteins & peptides Proteins 0.000 title claims description 42
- 102000004196 processed proteins & peptides Human genes 0.000 title claims description 28
- 229920001184 polypeptide Polymers 0.000 title claims description 13
- 238000004519 manufacturing process Methods 0.000 title abstract description 21
- 239000000203 mixture Substances 0.000 claims abstract description 701
- 238000000034 method Methods 0.000 claims abstract description 70
- 239000002775 capsule Substances 0.000 claims abstract description 49
- 239000004359 castor oil Substances 0.000 claims abstract description 49
- 235000019438 castor oil Nutrition 0.000 claims abstract description 49
- ZEMPKEQAKRGZGQ-XOQCFJPHSA-N glycerol triricinoleate Natural products CCCCCC[C@@H](O)CC=CCCCCCCCC(=O)OC[C@@H](COC(=O)CCCCCCCC=CC[C@@H](O)CCCCCC)OC(=O)CCCCCCCC=CC[C@H](O)CCCCCC ZEMPKEQAKRGZGQ-XOQCFJPHSA-N 0.000 claims abstract description 48
- 239000000725 suspension Substances 0.000 claims abstract description 38
- VLPFTAMPNXLGLX-UHFFFAOYSA-N trioctanoin Chemical compound CCCCCCCC(=O)OCC(OC(=O)CCCCCCC)COC(=O)CCCCCCC VLPFTAMPNXLGLX-UHFFFAOYSA-N 0.000 claims abstract description 36
- 150000003839 salts Chemical class 0.000 claims abstract description 27
- 150000004667 medium chain fatty acids Chemical class 0.000 claims abstract description 22
- DEQANNDTNATYII-OULOTJBUSA-N (4r,7s,10s,13r,16s,19r)-10-(4-aminobutyl)-19-[[(2r)-2-amino-3-phenylpropanoyl]amino]-16-benzyl-n-[(2r,3r)-1,3-dihydroxybutan-2-yl]-7-[(1r)-1-hydroxyethyl]-13-(1h-indol-3-ylmethyl)-6,9,12,15,18-pentaoxo-1,2-dithia-5,8,11,14,17-pentazacycloicosane-4-carboxa Chemical group C([C@@H](N)C(=O)N[C@H]1CSSC[C@H](NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@H](CCCCN)NC(=O)[C@@H](CC=2C3=CC=CC=C3NC=2)NC(=O)[C@H](CC=2C=CC=CC=2)NC1=O)C(=O)N[C@H](CO)[C@H](O)C)C1=CC=CC=C1 DEQANNDTNATYII-OULOTJBUSA-N 0.000 claims description 114
- 108010016076 Octreotide Proteins 0.000 claims description 114
- 229960002700 octreotide Drugs 0.000 claims description 111
- 229920002307 Dextran Polymers 0.000 claims description 102
- WWZKQHOCKIZLMA-UHFFFAOYSA-N octanoic acid Chemical compound CCCCCCCC(O)=O WWZKQHOCKIZLMA-UHFFFAOYSA-N 0.000 claims description 90
- BYKRNSHANADUFY-UHFFFAOYSA-M sodium octanoate Chemical compound [Na+].CCCCCCCC([O-])=O BYKRNSHANADUFY-UHFFFAOYSA-M 0.000 claims description 79
- 239000002245 particle Substances 0.000 claims description 72
- GHBFNMLVSPCDGN-UHFFFAOYSA-N rac-1-monooctanoylglycerol Chemical group CCCCCCCC(=O)OCC(O)CO GHBFNMLVSPCDGN-UHFFFAOYSA-N 0.000 claims description 54
- 235000014113 dietary fatty acids Nutrition 0.000 claims description 47
- 239000000194 fatty acid Substances 0.000 claims description 47
- 229930195729 fatty acid Natural products 0.000 claims description 47
- UYXTWWCETRIEDR-UHFFFAOYSA-N Tributyrin Chemical compound CCCC(=O)OCC(OC(=O)CCC)COC(=O)CCC UYXTWWCETRIEDR-UHFFFAOYSA-N 0.000 claims description 44
- OBETXYAYXDNJHR-UHFFFAOYSA-N alpha-ethylcaproic acid Natural products CCCCC(CC)C(O)=O OBETXYAYXDNJHR-UHFFFAOYSA-N 0.000 claims description 43
- 239000004094 surface-active agent Substances 0.000 claims description 41
- 230000008569 process Effects 0.000 claims description 32
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 claims description 32
- 229940087068 glyceryl caprylate Drugs 0.000 claims description 26
- 235000010482 polyoxyethylene sorbitan monooleate Nutrition 0.000 claims description 26
- 229920000053 polysorbate 80 Polymers 0.000 claims description 26
- 229920000642 polymer Polymers 0.000 claims description 23
- 239000000843 powder Substances 0.000 claims description 22
- UFTFJSFQGQCHQW-UHFFFAOYSA-N triformin Chemical compound O=COCC(OC=O)COC=O UFTFJSFQGQCHQW-UHFFFAOYSA-N 0.000 claims description 22
- 239000001267 polyvinylpyrrolidone Substances 0.000 claims description 20
- 229920000036 polyvinylpyrrolidone Polymers 0.000 claims description 20
- 235000013855 polyvinylpyrrolidone Nutrition 0.000 claims description 20
- 229920001282 polysaccharide Polymers 0.000 claims description 15
- 239000005017 polysaccharide Substances 0.000 claims description 15
- IIZPXYDJLKNOIY-JXPKJXOSSA-N 1-palmitoyl-2-arachidonoyl-sn-glycero-3-phosphocholine Chemical group CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCC\C=C/C\C=C/C\C=C/C\C=C/CCCCC IIZPXYDJLKNOIY-JXPKJXOSSA-N 0.000 claims description 14
- 235000010445 lecithin Nutrition 0.000 claims description 14
- 239000000787 lecithin Substances 0.000 claims description 14
- 229940067606 lecithin Drugs 0.000 claims description 14
- 239000003921 oil Substances 0.000 claims description 14
- LDVVTQMJQSCDMK-UHFFFAOYSA-N 1,3-dihydroxypropan-2-yl formate Chemical compound OCC(CO)OC=O LDVVTQMJQSCDMK-UHFFFAOYSA-N 0.000 claims description 13
- 150000004665 fatty acids Chemical class 0.000 claims description 10
- FIWQZURFGYXCEO-UHFFFAOYSA-M sodium;decanoate Chemical compound [Na+].CCCCCCCCCC([O-])=O FIWQZURFGYXCEO-UHFFFAOYSA-M 0.000 claims description 9
- 239000002253 acid Substances 0.000 claims description 8
- 125000004432 carbon atom Chemical group C* 0.000 claims description 8
- 239000000263 2,3-dihydroxypropyl (Z)-octadec-9-enoate Substances 0.000 claims description 7
- RZRNAYUHWVFMIP-GDCKJWNLSA-N 3-oleoyl-sn-glycerol Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OC[C@H](O)CO RZRNAYUHWVFMIP-GDCKJWNLSA-N 0.000 claims description 7
- 150000002148 esters Chemical class 0.000 claims description 7
- RZRNAYUHWVFMIP-UHFFFAOYSA-N monoelaidin Natural products CCCCCCCCC=CCCCCCCCC(=O)OCC(O)CO RZRNAYUHWVFMIP-UHFFFAOYSA-N 0.000 claims description 7
- 239000000244 polyoxyethylene sorbitan monooleate Substances 0.000 claims description 7
- RTZKZFJDLAIYFH-UHFFFAOYSA-N ether Substances CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 claims description 6
- 239000011734 sodium Substances 0.000 claims description 6
- LTOCMXUTASYUOC-UHFFFAOYSA-M sodium;nonanoate Chemical compound [Na+].CCCCCCCCC([O-])=O LTOCMXUTASYUOC-UHFFFAOYSA-M 0.000 claims description 6
- 229920001214 Polysorbate 60 Polymers 0.000 claims description 5
- 239000002480 mineral oil Substances 0.000 claims description 5
- 235000010446 mineral oil Nutrition 0.000 claims description 5
- VBICKXHEKHSIBG-UHFFFAOYSA-N 1-monostearoylglycerol Chemical compound CCCCCCCCCCCCCCCCCC(=O)OCC(O)CO VBICKXHEKHSIBG-UHFFFAOYSA-N 0.000 claims description 4
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 claims description 4
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 claims description 4
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 claims description 4
- 125000001931 aliphatic group Chemical group 0.000 claims description 4
- 125000003976 glyceryl group Chemical group [H]C([*])([H])C(O[H])([H])C(O[H])([H])[H] 0.000 claims description 4
- 229910052744 lithium Inorganic materials 0.000 claims description 4
- 229920001983 poloxamer Polymers 0.000 claims description 4
- 229920002451 polyvinyl alcohol Polymers 0.000 claims description 4
- 229910052700 potassium Inorganic materials 0.000 claims description 4
- 239000011591 potassium Substances 0.000 claims description 4
- ARIWANIATODDMH-UHFFFAOYSA-N rac-1-monolauroylglycerol Chemical compound CCCCCCCCCCCC(=O)OCC(O)CO ARIWANIATODDMH-UHFFFAOYSA-N 0.000 claims description 4
- 229910052708 sodium Inorganic materials 0.000 claims description 4
- JZVZOOVZQIIUGY-UHFFFAOYSA-M sodium;tridecanoate Chemical compound [Na+].CCCCCCCCCCCCC([O-])=O JZVZOOVZQIIUGY-UHFFFAOYSA-M 0.000 claims description 4
- ZORQXIQZAOLNGE-UHFFFAOYSA-N 1,1-difluorocyclohexane Chemical compound FC1(F)CCCCC1 ZORQXIQZAOLNGE-UHFFFAOYSA-N 0.000 claims description 3
- 229920002125 Sokalan® Polymers 0.000 claims description 3
- 239000002563 ionic surfactant Substances 0.000 claims description 3
- 239000002736 nonionic surfactant Substances 0.000 claims description 3
- BTURAGWYSMTVOW-UHFFFAOYSA-M sodium dodecanoate Chemical compound [Na+].CCCCCCCCCCCC([O-])=O BTURAGWYSMTVOW-UHFFFAOYSA-M 0.000 claims description 3
- UDWXLZLRRVQONG-UHFFFAOYSA-M sodium hexanoate Chemical compound [Na+].CCCCCC([O-])=O UDWXLZLRRVQONG-UHFFFAOYSA-M 0.000 claims description 3
- NMTDPTPUELYEPL-UHFFFAOYSA-M sodium;heptanoate Chemical compound [Na+].CCCCCCC([O-])=O NMTDPTPUELYEPL-UHFFFAOYSA-M 0.000 claims description 3
- JUQGWKYSEXPRGL-UHFFFAOYSA-M sodium;tetradecanoate Chemical compound [Na+].CCCCCCCCCCCCCC([O-])=O JUQGWKYSEXPRGL-UHFFFAOYSA-M 0.000 claims description 3
- ZOOPHYLANWVUDY-UHFFFAOYSA-M sodium;undecanoate Chemical compound [Na+].CCCCCCCCCCC([O-])=O ZOOPHYLANWVUDY-UHFFFAOYSA-M 0.000 claims description 3
- 235000011069 sorbitan monooleate Nutrition 0.000 claims description 3
- 239000001593 sorbitan monooleate Substances 0.000 claims description 3
- 229940035049 sorbitan monooleate Drugs 0.000 claims description 3
- QHZLMUACJMDIAE-SFHVURJKSA-N 1-hexadecanoyl-sn-glycerol Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@@H](O)CO QHZLMUACJMDIAE-SFHVURJKSA-N 0.000 claims description 2
- XZIIFPSPUDAGJM-UHFFFAOYSA-N 6-chloro-2-n,2-n-diethylpyrimidine-2,4-diamine Chemical compound CCN(CC)C1=NC(N)=CC(Cl)=N1 XZIIFPSPUDAGJM-UHFFFAOYSA-N 0.000 claims description 2
- QHZLMUACJMDIAE-UHFFFAOYSA-N Palmitic acid monoglyceride Natural products CCCCCCCCCCCCCCCC(=O)OCC(O)CO QHZLMUACJMDIAE-UHFFFAOYSA-N 0.000 claims description 2
- RVGRUAULSDPKGF-UHFFFAOYSA-N Poloxamer Chemical compound C1CO1.CC1CO1 RVGRUAULSDPKGF-UHFFFAOYSA-N 0.000 claims description 2
- 239000002202 Polyethylene glycol Substances 0.000 claims description 2
- 235000004443 Ricinus communis Nutrition 0.000 claims description 2
- 150000003863 ammonium salts Chemical class 0.000 claims description 2
- 150000001491 aromatic compounds Chemical class 0.000 claims description 2
- 239000003833 bile salt Substances 0.000 claims description 2
- 150000001923 cyclic compounds Chemical class 0.000 claims description 2
- 239000003599 detergent Substances 0.000 claims description 2
- 150000002191 fatty alcohols Chemical class 0.000 claims description 2
- 229940068939 glyceryl monolaurate Drugs 0.000 claims description 2
- 229940075507 glyceryl monostearate Drugs 0.000 claims description 2
- 239000001788 mono and diglycerides of fatty acids Substances 0.000 claims description 2
- 230000007935 neutral effect Effects 0.000 claims description 2
- WWZKQHOCKIZLMA-UHFFFAOYSA-M octanoate Chemical compound CCCCCCCC([O-])=O WWZKQHOCKIZLMA-UHFFFAOYSA-M 0.000 claims description 2
- 239000012188 paraffin wax Substances 0.000 claims description 2
- 229960000502 poloxamer Drugs 0.000 claims description 2
- 229920001223 polyethylene glycol Polymers 0.000 claims description 2
- 235000010483 polyoxyethylene sorbitan monopalmitate Nutrition 0.000 claims description 2
- 239000000249 polyoxyethylene sorbitan monopalmitate Substances 0.000 claims description 2
- LKUNXBRZDFMZOK-UHFFFAOYSA-N rac-1-monodecanoylglycerol Chemical compound CCCCCCCCCC(=O)OCC(O)CO LKUNXBRZDFMZOK-UHFFFAOYSA-N 0.000 claims description 2
- DCBSHORRWZKAKO-UHFFFAOYSA-N rac-1-monomyristoylglycerol Chemical compound CCCCCCCCCCCCCC(=O)OCC(O)CO DCBSHORRWZKAKO-UHFFFAOYSA-N 0.000 claims description 2
- 229940035044 sorbitan monolaurate Drugs 0.000 claims description 2
- HIXDQWDOVZUNNA-UHFFFAOYSA-N 2-(3,4-dimethoxyphenyl)-5-hydroxy-7-methoxychromen-4-one Chemical compound C=1C(OC)=CC(O)=C(C(C=2)=O)C=1OC=2C1=CC=C(OC)C(OC)=C1 HIXDQWDOVZUNNA-UHFFFAOYSA-N 0.000 claims 2
- JNYAEWCLZODPBN-JGWLITMVSA-N (2r,3r,4s)-2-[(1r)-1,2-dihydroxyethyl]oxolane-3,4-diol Chemical compound OC[C@@H](O)[C@H]1OC[C@H](O)[C@H]1O JNYAEWCLZODPBN-JGWLITMVSA-N 0.000 claims 1
- CERQOIWHTDAKMF-UHFFFAOYSA-M Methacrylate Chemical compound CC(=C)C([O-])=O CERQOIWHTDAKMF-UHFFFAOYSA-M 0.000 claims 1
- 150000004676 glycans Chemical class 0.000 claims 1
- 239000003814 drug Substances 0.000 abstract description 160
- 229940124597 therapeutic agent Drugs 0.000 abstract description 143
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 abstract description 26
- 201000010099 disease Diseases 0.000 abstract description 17
- 150000001298 alcohols Chemical class 0.000 abstract description 6
- 238000009472 formulation Methods 0.000 description 496
- NOESYZHRGYRDHS-UHFFFAOYSA-N insulin Chemical compound N1C(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(NC(=O)CN)C(C)CC)CSSCC(C(NC(CO)C(=O)NC(CC(C)C)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CCC(N)=O)C(=O)NC(CC(C)C)C(=O)NC(CCC(O)=O)C(=O)NC(CC(N)=O)C(=O)NC(CC=2C=CC(O)=CC=2)C(=O)NC(CSSCC(NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2C=CC(O)=CC=2)NC(=O)C(CC(C)C)NC(=O)C(C)NC(=O)C(CCC(O)=O)NC(=O)C(C(C)C)NC(=O)C(CC(C)C)NC(=O)C(CC=2NC=NC=2)NC(=O)C(CO)NC(=O)CNC2=O)C(=O)NCC(=O)NC(CCC(O)=O)C(=O)NC(CCCNC(N)=N)C(=O)NCC(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC=CC=3)C(=O)NC(CC=3C=CC(O)=CC=3)C(=O)NC(C(C)O)C(=O)N3C(CCC3)C(=O)NC(CCCCN)C(=O)NC(C)C(O)=O)C(=O)NC(CC(N)=O)C(O)=O)=O)NC(=O)C(C(C)CC)NC(=O)C(CO)NC(=O)C(C(C)O)NC(=O)C1CSSCC2NC(=O)C(CC(C)C)NC(=O)C(NC(=O)C(CCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(N)CC=1C=CC=CC=1)C(C)C)CC1=CN=CN1 NOESYZHRGYRDHS-UHFFFAOYSA-N 0.000 description 160
- 239000002609 medium Substances 0.000 description 140
- 230000000694 effects Effects 0.000 description 117
- 241000700159 Rattus Species 0.000 description 105
- FZWBNHMXJMCXLU-BLAUPYHCSA-N isomaltotriose Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1OC[C@@H]1[C@@H](O)[C@H](O)[C@@H](O)[C@@H](OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C=O)O1 FZWBNHMXJMCXLU-BLAUPYHCSA-N 0.000 description 98
- 108090001061 Insulin Proteins 0.000 description 80
- 102000004877 Insulin Human genes 0.000 description 80
- 229940125396 insulin Drugs 0.000 description 80
- 210000001630 jejunum Anatomy 0.000 description 51
- PPXUHEORWJQRHJ-UHFFFAOYSA-N ethyl isovalerate Chemical compound CCOC(=O)CC(C)C PPXUHEORWJQRHJ-UHFFFAOYSA-N 0.000 description 42
- 238000010521 absorption reaction Methods 0.000 description 40
- 229960002446 octanoic acid Drugs 0.000 description 40
- HTQBXNHDCUEHJF-XWLPCZSASA-N Exenatide Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(N)=O)C(=O)NCC(=O)NCC(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CO)C(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CO)C(N)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@@H](NC(=O)[C@H](C)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCSC)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CO)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)CNC(=O)[C@@H](N)CC=1NC=NC=1)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=CC=C1 HTQBXNHDCUEHJF-XWLPCZSASA-N 0.000 description 39
- 108010011459 Exenatide Proteins 0.000 description 39
- 229960001519 exenatide Drugs 0.000 description 38
- 230000001965 increasing effect Effects 0.000 description 37
- 108010049264 Teriparatide Proteins 0.000 description 35
- 239000000243 solution Substances 0.000 description 33
- OGBMKVWORPGQRR-UMXFMPSGSA-N teriparatide Chemical compound C([C@H](NC(=O)[C@H](CCSC)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@@H](N)CO)C(C)C)[C@@H](C)CC)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC=1N=CNC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC=1N=CNC=1)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC=1C=CC=CC=1)C(O)=O)C1=CNC=N1 OGBMKVWORPGQRR-UMXFMPSGSA-N 0.000 description 33
- 229960005460 teriparatide Drugs 0.000 description 32
- 150000001875 compounds Chemical class 0.000 description 31
- 210000004369 blood Anatomy 0.000 description 25
- 239000008280 blood Substances 0.000 description 25
- 102000018997 Growth Hormone Human genes 0.000 description 24
- 108010051696 Growth Hormone Proteins 0.000 description 24
- 238000002474 experimental method Methods 0.000 description 24
- 108090000623 proteins and genes Proteins 0.000 description 24
- 239000000122 growth hormone Substances 0.000 description 23
- 230000035515 penetration Effects 0.000 description 22
- 108010000817 Leuprolide Proteins 0.000 description 21
- GFIJNRVAKGFPGQ-LIJARHBVSA-N leuprolide Chemical compound CCNC(=O)[C@@H]1CCCN1C(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CC(C)C)NC(=O)[C@@H](CC(C)C)NC(=O)[C@@H](NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C2=CC=CC=C2NC=1)NC(=O)[C@H](CC=1N=CNC=1)NC(=O)[C@H]1NC(=O)CC1)CC1=CC=C(O)C=C1 GFIJNRVAKGFPGQ-LIJARHBVSA-N 0.000 description 21
- 229960004338 leuprorelin Drugs 0.000 description 21
- 238000011282 treatment Methods 0.000 description 21
- MYPYJXKWCTUITO-LYRMYLQWSA-N vancomycin Chemical compound O([C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@H]1OC1=C2C=C3C=C1OC1=CC=C(C=C1Cl)[C@@H](O)[C@H](C(N[C@@H](CC(N)=O)C(=O)N[C@H]3C(=O)N[C@H]1C(=O)N[C@H](C(N[C@@H](C3=CC(O)=CC(O)=C3C=3C(O)=CC=C1C=3)C(O)=O)=O)[C@H](O)C1=CC=C(C(=C1)Cl)O2)=O)NC(=O)[C@@H](CC(C)C)NC)[C@H]1C[C@](C)(N)[C@H](O)[C@H](C)O1 MYPYJXKWCTUITO-LYRMYLQWSA-N 0.000 description 21
- 108010059993 Vancomycin Proteins 0.000 description 20
- 238000004108 freeze drying Methods 0.000 description 20
- 238000011068 loading method Methods 0.000 description 20
- 102000004169 proteins and genes Human genes 0.000 description 20
- 229960003165 vancomycin Drugs 0.000 description 20
- MYPYJXKWCTUITO-UHFFFAOYSA-N vancomycin Natural products O1C(C(=C2)Cl)=CC=C2C(O)C(C(NC(C2=CC(O)=CC(O)=C2C=2C(O)=CC=C3C=2)C(O)=O)=O)NC(=O)C3NC(=O)C2NC(=O)C(CC(N)=O)NC(=O)C(NC(=O)C(CC(C)C)NC)C(O)C(C=C3Cl)=CC=C3OC3=CC2=CC1=C3OC1OC(CO)C(O)C(O)C1OC1CC(C)(N)C(O)C(C)O1 MYPYJXKWCTUITO-UHFFFAOYSA-N 0.000 description 20
- 238000002156 mixing Methods 0.000 description 19
- 230000035699 permeability Effects 0.000 description 19
- 239000008186 active pharmaceutical agent Substances 0.000 description 18
- 238000005469 granulation Methods 0.000 description 18
- 230000003179 granulation Effects 0.000 description 18
- 239000003381 stabilizer Substances 0.000 description 18
- 239000004615 ingredient Substances 0.000 description 17
- GLZPCOQZEFWAFX-UHFFFAOYSA-N Geraniol Chemical compound CC(C)=CCCC(C)=CCO GLZPCOQZEFWAFX-UHFFFAOYSA-N 0.000 description 16
- 102000002265 Human Growth Hormone Human genes 0.000 description 16
- 108010000521 Human Growth Hormone Proteins 0.000 description 16
- 239000000854 Human Growth Hormone Substances 0.000 description 16
- IYFATESGLOUGBX-YVNJGZBMSA-N Sorbitan monopalmitate Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@@H](O)[C@H]1OC[C@H](O)[C@H]1O IYFATESGLOUGBX-YVNJGZBMSA-N 0.000 description 16
- 210000000936 intestine Anatomy 0.000 description 16
- 238000012360 testing method Methods 0.000 description 16
- CEAZRRDELHUEMR-URQXQFDESA-N Gentamicin Chemical compound O1[C@H](C(C)NC)CC[C@@H](N)[C@H]1O[C@H]1[C@H](O)[C@@H](O[C@@H]2[C@@H]([C@@H](NC)[C@@](C)(O)CO2)O)[C@H](N)C[C@@H]1N CEAZRRDELHUEMR-URQXQFDESA-N 0.000 description 15
- 229930182566 Gentamicin Natural products 0.000 description 15
- 239000003795 chemical substances by application Substances 0.000 description 15
- 239000000945 filler Substances 0.000 description 15
- 229960002518 gentamicin Drugs 0.000 description 15
- 239000003550 marker Substances 0.000 description 15
- KBPLFHHGFOOTCA-UHFFFAOYSA-N 1-Octanol Chemical compound CCCCCCCCO KBPLFHHGFOOTCA-UHFFFAOYSA-N 0.000 description 14
- 108010047761 Interferon-alpha Proteins 0.000 description 14
- 102000006992 Interferon-alpha Human genes 0.000 description 14
- TWRXJAOTZQYOKJ-UHFFFAOYSA-L Magnesium chloride Chemical compound [Mg+2].[Cl-].[Cl-] TWRXJAOTZQYOKJ-UHFFFAOYSA-L 0.000 description 14
- 108010010056 Terlipressin Proteins 0.000 description 14
- 239000011159 matrix material Substances 0.000 description 14
- 150000004804 polysaccharides Chemical class 0.000 description 14
- 150000003384 small molecules Chemical class 0.000 description 14
- BENFXAYNYRLAIU-QSVFAHTRSA-N terlipressin Chemical compound NCCCC[C@@H](C(=O)NCC(N)=O)NC(=O)[C@@H]1CCCN1C(=O)[C@H]1NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC=2C=CC=CC=2)NC(=O)[C@H](CC=2C=CC(O)=CC=2)NC(=O)[C@@H](NC(=O)CNC(=O)CNC(=O)CN)CSSC1 BENFXAYNYRLAIU-QSVFAHTRSA-N 0.000 description 14
- 229960003813 terlipressin Drugs 0.000 description 14
- 238000012384 transportation and delivery Methods 0.000 description 14
- 241000282887 Suidae Species 0.000 description 13
- 229940024606 amino acid Drugs 0.000 description 13
- 150000001413 amino acids Chemical class 0.000 description 13
- 235000019198 oils Nutrition 0.000 description 13
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 12
- 229940079593 drug Drugs 0.000 description 12
- 230000009368 gene silencing by RNA Effects 0.000 description 12
- 238000001990 intravenous administration Methods 0.000 description 12
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 11
- HTTJABKRGRZYRN-UHFFFAOYSA-N Heparin Chemical class OC1C(NC(=O)C)C(O)OC(COS(O)(=O)=O)C1OC1C(OS(O)(=O)=O)C(O)C(OC2C(C(OS(O)(=O)=O)C(OC3C(C(O)C(O)C(O3)C(O)=O)OS(O)(=O)=O)C(CO)O2)NS(O)(=O)=O)C(C(O)=O)O1 HTTJABKRGRZYRN-UHFFFAOYSA-N 0.000 description 11
- 241001465754 Metazoa Species 0.000 description 11
- 108091030071 RNAI Proteins 0.000 description 11
- 238000001035 drying Methods 0.000 description 11
- MHMNJMPURVTYEJ-UHFFFAOYSA-N fluorescein-5-isothiocyanate Chemical compound O1C(=O)C2=CC(N=C=S)=CC=C2C21C1=CC=C(O)C=C1OC1=CC(O)=CC=C21 MHMNJMPURVTYEJ-UHFFFAOYSA-N 0.000 description 11
- 239000008103 glucose Substances 0.000 description 11
- 210000004379 membrane Anatomy 0.000 description 11
- 239000012528 membrane Substances 0.000 description 11
- 102000040430 polynucleotide Human genes 0.000 description 11
- 108091033319 polynucleotide Proteins 0.000 description 11
- 239000002157 polynucleotide Substances 0.000 description 11
- 239000007864 aqueous solution Substances 0.000 description 10
- 230000000968 intestinal effect Effects 0.000 description 10
- 239000000546 pharmaceutical excipient Substances 0.000 description 10
- WBHHMMIMDMUBKC-XLNAKTSKSA-N ricinelaidic acid Chemical compound CCCCCC[C@@H](O)C\C=C\CCCCCCCC(O)=O WBHHMMIMDMUBKC-XLNAKTSKSA-N 0.000 description 10
- 229960003656 ricinoleic acid Drugs 0.000 description 10
- FEUQNCSVHBHROZ-UHFFFAOYSA-N ricinoleic acid Natural products CCCCCCC(O[Si](C)(C)C)CC=CCCCCCCCC(=O)OC FEUQNCSVHBHROZ-UHFFFAOYSA-N 0.000 description 10
- 210000002784 stomach Anatomy 0.000 description 10
- 150000003626 triacylglycerols Chemical class 0.000 description 10
- RZRNAYUHWVFMIP-KTKRTIGZSA-N 1-oleoylglycerol Chemical compound CCCCCCCC\C=C/CCCCCCCC(=O)OCC(O)CO RZRNAYUHWVFMIP-KTKRTIGZSA-N 0.000 description 9
- FBPFZTCFMRRESA-KVTDHHQDSA-N D-Mannitol Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@H](O)CO FBPFZTCFMRRESA-KVTDHHQDSA-N 0.000 description 9
- 229930195725 Mannitol Natural products 0.000 description 9
- 208000035475 disorder Diseases 0.000 description 9
- KANJSNBRCNMZMV-ABRZTLGGSA-N fondaparinux Chemical compound O[C@@H]1[C@@H](NS(O)(=O)=O)[C@@H](OC)O[C@H](COS(O)(=O)=O)[C@H]1O[C@H]1[C@H](OS(O)(=O)=O)[C@@H](O)[C@H](O[C@@H]2[C@@H]([C@@H](OS(O)(=O)=O)[C@H](O[C@H]3[C@@H]([C@@H](O)[C@H](O[C@@H]4[C@@H]([C@@H](O)[C@H](O)[C@@H](COS(O)(=O)=O)O4)NS(O)(=O)=O)[C@H](O3)C(O)=O)O)[C@@H](COS(O)(=O)=O)O2)NS(O)(=O)=O)[C@H](C(O)=O)O1 KANJSNBRCNMZMV-ABRZTLGGSA-N 0.000 description 9
- 229960001318 fondaparinux Drugs 0.000 description 9
- RZRNAYUHWVFMIP-HXUWFJFHSA-N glycerol monolinoleate Natural products CCCCCCCCC=CCCCCCCCC(=O)OC[C@H](O)CO RZRNAYUHWVFMIP-HXUWFJFHSA-N 0.000 description 9
- 230000003870 intestinal permeability Effects 0.000 description 9
- 239000000594 mannitol Substances 0.000 description 9
- 235000010355 mannitol Nutrition 0.000 description 9
- 125000003729 nucleotide group Chemical class 0.000 description 9
- 230000001225 therapeutic effect Effects 0.000 description 9
- 239000005792 Geraniol Substances 0.000 description 8
- GLZPCOQZEFWAFX-YFHOEESVSA-N Geraniol Natural products CC(C)=CCC\C(C)=C/CO GLZPCOQZEFWAFX-YFHOEESVSA-N 0.000 description 8
- 239000003240 coconut oil Substances 0.000 description 8
- 235000019864 coconut oil Nutrition 0.000 description 8
- 231100000673 dose–response relationship Toxicity 0.000 description 8
- 229940113087 geraniol Drugs 0.000 description 8
- 229920003088 hydroxypropyl methyl cellulose Polymers 0.000 description 8
- 210000004731 jugular vein Anatomy 0.000 description 8
- 238000011552 rat model Methods 0.000 description 8
- 238000007920 subcutaneous administration Methods 0.000 description 8
- 230000001839 systemic circulation Effects 0.000 description 8
- 241000282693 Cercopithecidae Species 0.000 description 7
- 101000976075 Homo sapiens Insulin Proteins 0.000 description 7
- 102000003982 Parathyroid hormone Human genes 0.000 description 7
- 108090000445 Parathyroid hormone Proteins 0.000 description 7
- 238000010171 animal model Methods 0.000 description 7
- 230000004888 barrier function Effects 0.000 description 7
- 125000004122 cyclic group Chemical group 0.000 description 7
- 230000007423 decrease Effects 0.000 description 7
- NCXUIEDQTCQZRK-UHFFFAOYSA-L disodium;decanedioate Chemical compound [Na+].[Na+].[O-]C(=O)CCCCCCCCC([O-])=O NCXUIEDQTCQZRK-UHFFFAOYSA-L 0.000 description 7
- 235000010979 hydroxypropyl methyl cellulose Nutrition 0.000 description 7
- 239000001866 hydroxypropyl methyl cellulose Substances 0.000 description 7
- UFVKGYZPFZQRLF-UHFFFAOYSA-N hydroxypropyl methyl cellulose Chemical compound OC1C(O)C(OC)OC(CO)C1OC1C(O)C(O)C(OC2C(C(O)C(OC3C(C(O)C(O)C(CO)O3)O)C(CO)O2)O)C(CO)O1 UFVKGYZPFZQRLF-UHFFFAOYSA-N 0.000 description 7
- PBGKTOXHQIOBKM-FHFVDXKLSA-N insulin (human) Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@H]1CSSC[C@H]2C(=O)N[C@H](C(=O)N[C@@H](CO)C(=O)N[C@H](C(=O)N[C@H](C(N[C@@H](CO)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC=3C=CC(O)=CC=3)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC=3C=CC(O)=CC=3)C(=O)N[C@@H](CSSC[C@H](NC(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=3C=CC(O)=CC=3)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](C)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C(C)C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=3NC=NC=3)NC(=O)[C@H](CO)NC(=O)CNC1=O)C(=O)NCC(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)NCC(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H]([C@@H](C)O)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H]([C@@H](C)O)C(O)=O)C(=O)N[C@@H](CC(N)=O)C(O)=O)=O)CSSC[C@@H](C(N2)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C(C)C)NC(=O)[C@@H](NC(=O)CN)[C@@H](C)CC)[C@@H](C)CC)[C@@H](C)O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@@H](NC(=O)[C@@H](N)CC=1C=CC=CC=1)C(C)C)C1=CN=CN1 PBGKTOXHQIOBKM-FHFVDXKLSA-N 0.000 description 7
- 210000004185 liver Anatomy 0.000 description 7
- 229940127215 low-molecular weight heparin Drugs 0.000 description 7
- 229910001629 magnesium chloride Inorganic materials 0.000 description 7
- 239000000199 parathyroid hormone Substances 0.000 description 7
- 210000002966 serum Anatomy 0.000 description 7
- 239000000126 substance Substances 0.000 description 7
- 241001631457 Cannula Species 0.000 description 6
- DHMQDGOQFOQNFH-UHFFFAOYSA-N Glycine Chemical compound NCC(O)=O DHMQDGOQFOQNFH-UHFFFAOYSA-N 0.000 description 6
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 6
- 241000282898 Sus scrofa Species 0.000 description 6
- 229920000669 heparin Polymers 0.000 description 6
- 239000002773 nucleotide Substances 0.000 description 6
- 210000000496 pancreas Anatomy 0.000 description 6
- 229960001319 parathyroid hormone Drugs 0.000 description 6
- 239000000137 peptide hydrolase inhibitor Substances 0.000 description 6
- 229920002683 Glycosaminoglycan Polymers 0.000 description 5
- 241000699666 Mus <mouse, genus> Species 0.000 description 5
- 208000008589 Obesity Diseases 0.000 description 5
- 108020004459 Small interfering RNA Proteins 0.000 description 5
- 125000003118 aryl group Chemical group 0.000 description 5
- 230000008901 benefit Effects 0.000 description 5
- 230000015556 catabolic process Effects 0.000 description 5
- HVYWMOMLDIMFJA-DPAQBDIFSA-N cholesterol Chemical compound C1C=C2C[C@@H](O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 HVYWMOMLDIMFJA-DPAQBDIFSA-N 0.000 description 5
- 238000006731 degradation reaction Methods 0.000 description 5
- 206010012601 diabetes mellitus Diseases 0.000 description 5
- 238000005516 engineering process Methods 0.000 description 5
- 229920000159 gelatin Polymers 0.000 description 5
- 235000019322 gelatine Nutrition 0.000 description 5
- 229960002897 heparin Drugs 0.000 description 5
- 201000011200 hepatorenal syndrome Diseases 0.000 description 5
- 229940088597 hormone Drugs 0.000 description 5
- 239000005556 hormone Substances 0.000 description 5
- 208000015181 infectious disease Diseases 0.000 description 5
- 235000020824 obesity Nutrition 0.000 description 5
- 208000007232 portal hypertension Diseases 0.000 description 5
- 238000002360 preparation method Methods 0.000 description 5
- 239000002994 raw material Substances 0.000 description 5
- 210000000813 small intestine Anatomy 0.000 description 5
- 206010000599 Acromegaly Diseases 0.000 description 4
- OGSPWJRAVKPPFI-UHFFFAOYSA-N Alendronic Acid Chemical compound NCCCC(O)(P(O)(O)=O)P(O)(O)=O OGSPWJRAVKPPFI-UHFFFAOYSA-N 0.000 description 4
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 4
- 206010012735 Diarrhoea Diseases 0.000 description 4
- AOJJSUZBOXZQNB-TZSSRYMLSA-N Doxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-TZSSRYMLSA-N 0.000 description 4
- 101001032756 Rattus norvegicus Granzyme-like protein 1 Proteins 0.000 description 4
- YASAKCUCGLMORW-UHFFFAOYSA-N Rosiglitazone Chemical compound C=1C=CC=NC=1N(C)CCOC(C=C1)=CC=C1CC1SC(=O)NC1=O YASAKCUCGLMORW-UHFFFAOYSA-N 0.000 description 4
- 208000009311 VIPoma Diseases 0.000 description 4
- 239000000556 agonist Substances 0.000 description 4
- 229940062527 alendronate Drugs 0.000 description 4
- 238000004458 analytical method Methods 0.000 description 4
- 239000012736 aqueous medium Substances 0.000 description 4
- OGBUMNBNEWYMNJ-UHFFFAOYSA-N batilol Chemical class CCCCCCCCCCCCCCCCCCOCC(O)CO OGBUMNBNEWYMNJ-UHFFFAOYSA-N 0.000 description 4
- 239000011230 binding agent Substances 0.000 description 4
- 150000001768 cations Chemical class 0.000 description 4
- 239000000470 constituent Substances 0.000 description 4
- 239000003623 enhancer Substances 0.000 description 4
- 235000013305 food Nutrition 0.000 description 4
- 230000002496 gastric effect Effects 0.000 description 4
- 238000003018 immunoassay Methods 0.000 description 4
- 238000001727 in vivo Methods 0.000 description 4
- 210000005027 intestinal barrier Anatomy 0.000 description 4
- 230000007358 intestinal barrier function Effects 0.000 description 4
- 239000003055 low molecular weight heparin Substances 0.000 description 4
- 229920000609 methyl cellulose Polymers 0.000 description 4
- 235000010981 methylcellulose Nutrition 0.000 description 4
- 239000001923 methylcellulose Substances 0.000 description 4
- 150000002772 monosaccharides Chemical class 0.000 description 4
- OHDXDNUPVVYWOV-UHFFFAOYSA-N n-methyl-1-(2-naphthalen-1-ylsulfanylphenyl)methanamine Chemical compound CNCC1=CC=CC=C1SC1=CC=CC2=CC=CC=C12 OHDXDNUPVVYWOV-UHFFFAOYSA-N 0.000 description 4
- 239000000816 peptidomimetic Substances 0.000 description 4
- 230000036470 plasma concentration Effects 0.000 description 4
- 230000002035 prolonged effect Effects 0.000 description 4
- 238000011321 prophylaxis Methods 0.000 description 4
- CXMXRPHRNRROMY-UHFFFAOYSA-N sebacic acid Chemical compound OC(=O)CCCCCCCCC(O)=O CXMXRPHRNRROMY-UHFFFAOYSA-N 0.000 description 4
- 159000000000 sodium salts Chemical class 0.000 description 4
- NHXLMOGPVYXJNR-ATOGVRKGSA-N somatostatin Chemical compound C([C@H]1C(=O)N[C@H](C(N[C@@H](CO)C(=O)N[C@@H](CSSC[C@@H](C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC=2C=CC=CC=2)C(=O)N[C@@H](CC=2C=CC=CC=2)C(=O)N[C@@H](CC=2C3=CC=CC=C3NC=2)C(=O)N[C@@H](CCCCN)C(=O)N[C@H](C(=O)N1)[C@@H](C)O)NC(=O)CNC(=O)[C@H](C)N)C(O)=O)=O)[C@H](O)C)C1=CC=CC=C1 NHXLMOGPVYXJNR-ATOGVRKGSA-N 0.000 description 4
- 238000001228 spectrum Methods 0.000 description 4
- PFNFFQXMRSDOHW-UHFFFAOYSA-N spermine Chemical compound NCCCNCCCCNCCCN PFNFFQXMRSDOHW-UHFFFAOYSA-N 0.000 description 4
- TYFQFVWCELRYAO-UHFFFAOYSA-N suberic acid Chemical compound OC(=O)CCCCCCC(O)=O TYFQFVWCELRYAO-UHFFFAOYSA-N 0.000 description 4
- 208000001072 type 2 diabetes mellitus Diseases 0.000 description 4
- PPXXXBJQFYWSJN-VWMHFEHESA-N (2s)-2-amino-5-(diaminomethylideneamino)pentanoic acid;octanoic acid Chemical compound CCCCCCCC(O)=O.OC(=O)[C@@H](N)CCCN=C(N)N PPXXXBJQFYWSJN-VWMHFEHESA-N 0.000 description 3
- 208000030507 AIDS Diseases 0.000 description 3
- 208000004611 Abdominal Obesity Diseases 0.000 description 3
- 208000020084 Bone disease Diseases 0.000 description 3
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 3
- 206010065941 Central obesity Diseases 0.000 description 3
- 108010000437 Deamino Arginine Vasopressin Proteins 0.000 description 3
- 102000004190 Enzymes Human genes 0.000 description 3
- 108090000790 Enzymes Proteins 0.000 description 3
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 3
- 239000001828 Gelatine Substances 0.000 description 3
- 108010072051 Glatiramer Acetate Proteins 0.000 description 3
- 239000004471 Glycine Substances 0.000 description 3
- 102000004269 Granulocyte Colony-Stimulating Factor Human genes 0.000 description 3
- 108010017080 Granulocyte Colony-Stimulating Factor Proteins 0.000 description 3
- 206010056438 Growth hormone deficiency Diseases 0.000 description 3
- 208000032843 Hemorrhage Diseases 0.000 description 3
- 241000282412 Homo Species 0.000 description 3
- 206010021518 Impaired gastric emptying Diseases 0.000 description 3
- 206010049287 Lipodystrophy acquired Diseases 0.000 description 3
- 208000001145 Metabolic Syndrome Diseases 0.000 description 3
- 239000004372 Polyvinyl alcohol Substances 0.000 description 3
- 229940124158 Protease/peptidase inhibitor Drugs 0.000 description 3
- 206010056658 Pseudocyst Diseases 0.000 description 3
- 229920002472 Starch Polymers 0.000 description 3
- 102100026383 Vasopressin-neurophysin 2-copeptin Human genes 0.000 description 3
- 201000000690 abdominal obesity-metabolic syndrome Diseases 0.000 description 3
- 230000002159 abnormal effect Effects 0.000 description 3
- 150000007513 acids Chemical class 0.000 description 3
- KBZOIRJILGZLEJ-LGYYRGKSSA-N argipressin Chemical compound C([C@H]1C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CSSC[C@@H](C(N[C@@H](CC=2C=CC(O)=CC=2)C(=O)N1)=O)N)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCN=C(N)N)C(=O)NCC(N)=O)C1=CC=CC=C1 KBZOIRJILGZLEJ-LGYYRGKSSA-N 0.000 description 3
- 230000009286 beneficial effect Effects 0.000 description 3
- 230000000740 bleeding effect Effects 0.000 description 3
- 229940126600 bulk drug product Drugs 0.000 description 3
- IFKLAQQSCNILHL-QHAWAJNXSA-N butorphanol Chemical compound N1([C@@H]2CC3=CC=C(C=C3[C@@]3([C@]2(CCCC3)O)CC1)O)CC1CCC1 IFKLAQQSCNILHL-QHAWAJNXSA-N 0.000 description 3
- 229960001113 butorphanol Drugs 0.000 description 3
- 210000001072 colon Anatomy 0.000 description 3
- 230000003247 decreasing effect Effects 0.000 description 3
- 230000007812 deficiency Effects 0.000 description 3
- 229960004281 desmopressin Drugs 0.000 description 3
- NFLWUMRGJYTJIN-NXBWRCJVSA-N desmopressin Chemical compound C([C@H]1C(=O)N[C@H](C(N[C@@H](CC(N)=O)C(=O)N[C@@H](CSSCCC(=O)N[C@@H](CC=2C=CC(O)=CC=2)C(=O)N1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)NCC(N)=O)=O)CCC(=O)N)C1=CC=CC=C1 NFLWUMRGJYTJIN-NXBWRCJVSA-N 0.000 description 3
- 229940042399 direct acting antivirals protease inhibitors Drugs 0.000 description 3
- 239000002552 dosage form Substances 0.000 description 3
- 238000012377 drug delivery Methods 0.000 description 3
- 239000013583 drug formulation Substances 0.000 description 3
- 239000000839 emulsion Substances 0.000 description 3
- 210000000981 epithelium Anatomy 0.000 description 3
- 208000001288 gastroparesis Diseases 0.000 description 3
- ZHYZQXUYZJNEHD-VQHVLOKHSA-N geranic acid Chemical class CC(C)=CCC\C(C)=C\C(O)=O ZHYZQXUYZJNEHD-VQHVLOKHSA-N 0.000 description 3
- 229930008392 geranic acid Natural products 0.000 description 3
- 230000012010 growth Effects 0.000 description 3
- 239000002628 heparin derivative Substances 0.000 description 3
- 230000006872 improvement Effects 0.000 description 3
- 238000011065 in-situ storage Methods 0.000 description 3
- 210000004347 intestinal mucosa Anatomy 0.000 description 3
- 208000006132 lipodystrophy Diseases 0.000 description 3
- BTAUEIDLAAYHSL-UHFFFAOYSA-M lithium;octanoate Chemical group [Li+].CCCCCCCC([O-])=O BTAUEIDLAAYHSL-UHFFFAOYSA-M 0.000 description 3
- 239000004570 mortar (masonry) Substances 0.000 description 3
- 239000004006 olive oil Substances 0.000 description 3
- 235000008390 olive oil Nutrition 0.000 description 3
- RLEFZEWKMQQZOA-UHFFFAOYSA-M potassium;octanoate Chemical compound [K+].CCCCCCCC([O-])=O RLEFZEWKMQQZOA-UHFFFAOYSA-M 0.000 description 3
- 238000012545 processing Methods 0.000 description 3
- 229920006395 saturated elastomer Polymers 0.000 description 3
- 229960005480 sodium caprylate Drugs 0.000 description 3
- 239000011343 solid material Substances 0.000 description 3
- 235000011071 sorbitan monopalmitate Nutrition 0.000 description 3
- 239000001570 sorbitan monopalmitate Substances 0.000 description 3
- 229940031953 sorbitan monopalmitate Drugs 0.000 description 3
- 239000008107 starch Substances 0.000 description 3
- 235000019698 starch Nutrition 0.000 description 3
- 239000000829 suppository Substances 0.000 description 3
- 230000009897 systematic effect Effects 0.000 description 3
- RCINICONZNJXQF-MZXODVADSA-N taxol Chemical class O([C@@H]1[C@@]2(C[C@@H](C(C)=C(C2(C)C)[C@H](C([C@]2(C)[C@@H](O)C[C@H]3OC[C@]3([C@H]21)OC(C)=O)=O)OC(=O)C)OC(=O)[C@H](O)[C@@H](NC(=O)C=1C=CC=CC=1)C=1C=CC=CC=1)O)C(=O)C1=CC=CC=C1 RCINICONZNJXQF-MZXODVADSA-N 0.000 description 3
- ZHYZQXUYZJNEHD-UHFFFAOYSA-N trans-geranic acid Natural products CC(C)=CCCC(C)=CC(O)=O ZHYZQXUYZJNEHD-UHFFFAOYSA-N 0.000 description 3
- 238000009849 vacuum degassing Methods 0.000 description 3
- 230000000007 visual effect Effects 0.000 description 3
- HDTRYLNUVZCQOY-UHFFFAOYSA-N α-D-glucopyranosyl-α-D-glucopyranoside Natural products OC1C(O)C(O)C(CO)OC1OC1C(O)C(O)C(O)C(CO)O1 HDTRYLNUVZCQOY-UHFFFAOYSA-N 0.000 description 2
- RXBQNMWIQKOSCS-UHFFFAOYSA-N (7,7-dimethyl-4-bicyclo[3.1.1]hept-3-enyl)methanol Chemical compound C1C2C(C)(C)C1CC=C2CO RXBQNMWIQKOSCS-UHFFFAOYSA-N 0.000 description 2
- 108091032973 (ribonucleotides)n+m Proteins 0.000 description 2
- 102000040650 (ribonucleotides)n+m Human genes 0.000 description 2
- BBMCTIGTTCKYKF-UHFFFAOYSA-N 1-heptanol Chemical compound CCCCCCCO BBMCTIGTTCKYKF-UHFFFAOYSA-N 0.000 description 2
- OWEGMIWEEQEYGQ-UHFFFAOYSA-N 100676-05-9 Natural products OC1C(O)C(O)C(CO)OC1OCC1C(O)C(O)C(O)C(OC2C(OC(O)C(O)C2O)CO)O1 OWEGMIWEEQEYGQ-UHFFFAOYSA-N 0.000 description 2
- RZVAJINKPMORJF-UHFFFAOYSA-N Acetaminophen Chemical compound CC(=O)NC1=CC=C(O)C=C1 RZVAJINKPMORJF-UHFFFAOYSA-N 0.000 description 2
- WSVLPVUVIUVCRA-KPKNDVKVSA-N Alpha-lactose monohydrate Chemical compound O.O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@H]1O[C@@H]1[C@@H](CO)O[C@H](O)[C@H](O)[C@H]1O WSVLPVUVIUVCRA-KPKNDVKVSA-N 0.000 description 2
- DKPFZGUDAPQIHT-UHFFFAOYSA-N Butyl acetate Natural products CCCCOC(C)=O DKPFZGUDAPQIHT-UHFFFAOYSA-N 0.000 description 2
- YNXLOPYTAAFMTN-SBUIBGKBSA-N C([C@H](N)C(=O)N1CCC[C@H]1C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)NCC(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CO)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](C)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(N)=O)C1=CC=C(O)C=C1 Chemical compound C([C@H](N)C(=O)N1CCC[C@H]1C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CCCCN)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](C)C(=O)N1[C@@H](CCC1)C(=O)NCC(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CO)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](C)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(N)=O)C1=CC=C(O)C=C1 YNXLOPYTAAFMTN-SBUIBGKBSA-N 0.000 description 2
- 108060001064 Calcitonin Proteins 0.000 description 2
- 102000055006 Calcitonin Human genes 0.000 description 2
- 206010007270 Carcinoid syndrome Diseases 0.000 description 2
- 206010053567 Coagulopathies Diseases 0.000 description 2
- UHDGCWIWMRVCDJ-CCXZUQQUSA-N Cytarabine Chemical compound O=C1N=C(N)C=CN1[C@H]1[C@@H](O)[C@H](O)[C@@H](CO)O1 UHDGCWIWMRVCDJ-CCXZUQQUSA-N 0.000 description 2
- 108020004414 DNA Proteins 0.000 description 2
- 239000004375 Dextrin Substances 0.000 description 2
- 229920001353 Dextrin Polymers 0.000 description 2
- 208000001976 Endocrine Gland Neoplasms Diseases 0.000 description 2
- 102000003951 Erythropoietin Human genes 0.000 description 2
- 108090000394 Erythropoietin Proteins 0.000 description 2
- 108010054218 Factor VIII Proteins 0.000 description 2
- 102000001690 Factor VIII Human genes 0.000 description 2
- 208000007984 Female Infertility Diseases 0.000 description 2
- 102000003974 Fibroblast growth factor 2 Human genes 0.000 description 2
- 108090000379 Fibroblast growth factor 2 Proteins 0.000 description 2
- 102000012673 Follicle Stimulating Hormone Human genes 0.000 description 2
- 108010079345 Follicle Stimulating Hormone Proteins 0.000 description 2
- 108700012941 GNRH1 Proteins 0.000 description 2
- 108010010803 Gelatin Proteins 0.000 description 2
- NMJREATYWWNIKX-UHFFFAOYSA-N GnRH Chemical compound C1CCC(C(=O)NCC(N)=O)N1C(=O)C(CC(C)C)NC(=O)C(CC=1C2=CC=CC=C2NC=1)NC(=O)CNC(=O)C(NC(=O)C(CO)NC(=O)C(CC=1C2=CC=CC=C2NC=1)NC(=O)C(CC=1NC=NC=1)NC(=O)C1NC(=O)CC1)CC1=CC=C(O)C=C1 NMJREATYWWNIKX-UHFFFAOYSA-N 0.000 description 2
- 239000000579 Gonadotropin-Releasing Hormone Substances 0.000 description 2
- 102000006771 Gonadotropins Human genes 0.000 description 2
- 108010086677 Gonadotropins Proteins 0.000 description 2
- 239000000095 Growth Hormone-Releasing Hormone Substances 0.000 description 2
- ZRALSGWEFCBTJO-UHFFFAOYSA-N Guanidine Chemical compound NC(N)=N ZRALSGWEFCBTJO-UHFFFAOYSA-N 0.000 description 2
- 208000005176 Hepatitis C Diseases 0.000 description 2
- 108010007267 Hirudins Proteins 0.000 description 2
- 102000007625 Hirudins Human genes 0.000 description 2
- 206010021928 Infertility female Diseases 0.000 description 2
- IMQLKJBTEOYOSI-GPIVLXJGSA-N Inositol-hexakisphosphate Chemical compound OP(O)(=O)O[C@H]1[C@H](OP(O)(O)=O)[C@@H](OP(O)(O)=O)[C@H](OP(O)(O)=O)[C@H](OP(O)(O)=O)[C@@H]1OP(O)(O)=O IMQLKJBTEOYOSI-GPIVLXJGSA-N 0.000 description 2
- 102000014150 Interferons Human genes 0.000 description 2
- 108010050904 Interferons Proteins 0.000 description 2
- 102000000588 Interleukin-2 Human genes 0.000 description 2
- 108010002350 Interleukin-2 Proteins 0.000 description 2
- 102000009151 Luteinizing Hormone Human genes 0.000 description 2
- 108010073521 Luteinizing Hormone Proteins 0.000 description 2
- GUBGYTABKSRVRQ-PICCSMPSSA-N Maltose Natural products O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@@H](CO)OC(O)[C@H](O)[C@H]1O GUBGYTABKSRVRQ-PICCSMPSSA-N 0.000 description 2
- LRHPLDYGYMQRHN-UHFFFAOYSA-N N-Butanol Chemical compound CCCCO LRHPLDYGYMQRHN-UHFFFAOYSA-N 0.000 description 2
- AMQJEAYHLZJPGS-UHFFFAOYSA-N N-Pentanol Chemical compound CCCCCO AMQJEAYHLZJPGS-UHFFFAOYSA-N 0.000 description 2
- CMWTZPSULFXXJA-UHFFFAOYSA-N Naproxen Natural products C1=C(C(C)C(O)=O)C=CC2=CC(OC)=CC=C21 CMWTZPSULFXXJA-UHFFFAOYSA-N 0.000 description 2
- 208000001132 Osteoporosis Diseases 0.000 description 2
- 229930012538 Paclitaxel Natural products 0.000 description 2
- 108091005804 Peptidases Proteins 0.000 description 2
- 108010088847 Peptide YY Proteins 0.000 description 2
- ISWSIDIOOBJBQZ-UHFFFAOYSA-N Phenol Chemical compound OC1=CC=CC=C1 ISWSIDIOOBJBQZ-UHFFFAOYSA-N 0.000 description 2
- IMQLKJBTEOYOSI-UHFFFAOYSA-N Phytic acid Natural products OP(O)(=O)OC1C(OP(O)(O)=O)C(OP(O)(O)=O)C(OP(O)(O)=O)C(OP(O)(O)=O)C1OP(O)(O)=O IMQLKJBTEOYOSI-UHFFFAOYSA-N 0.000 description 2
- 239000004365 Protease Substances 0.000 description 2
- 102100037486 Reverse transcriptase/ribonuclease H Human genes 0.000 description 2
- IWUCXVSUMQZMFG-AFCXAGJDSA-N Ribavirin Chemical compound N1=C(C(=O)N)N=CN1[C@H]1[C@H](O)[C@H](O)[C@@H](CO)O1 IWUCXVSUMQZMFG-AFCXAGJDSA-N 0.000 description 2
- 102100022831 Somatoliberin Human genes 0.000 description 2
- 101710142969 Somatoliberin Proteins 0.000 description 2
- 102000005157 Somatostatin Human genes 0.000 description 2
- 108010056088 Somatostatin Proteins 0.000 description 2
- 101000857870 Squalus acanthias Gonadoliberin Proteins 0.000 description 2
- 241000191967 Staphylococcus aureus Species 0.000 description 2
- MUMGGOZAMZWBJJ-DYKIIFRCSA-N Testostosterone Chemical compound O=C1CC[C@]2(C)[C@H]3CC[C@](C)([C@H](CC4)O)[C@@H]4[C@@H]3CCC2=C1 MUMGGOZAMZWBJJ-DYKIIFRCSA-N 0.000 description 2
- IQFYYKKMVGJFEH-XLPZGREQSA-N Thymidine Chemical compound O=C1NC(=O)C(C)=CN1[C@@H]1O[C@H](CO)[C@@H](O)C1 IQFYYKKMVGJFEH-XLPZGREQSA-N 0.000 description 2
- 102000003978 Tissue Plasminogen Activator Human genes 0.000 description 2
- 108090000373 Tissue Plasminogen Activator Proteins 0.000 description 2
- HDTRYLNUVZCQOY-WSWWMNSNSA-N Trehalose Natural products O[C@@H]1[C@@H](O)[C@@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@H](O)[C@@H](O)[C@@H](O)[C@@H](CO)O1 HDTRYLNUVZCQOY-WSWWMNSNSA-N 0.000 description 2
- ISAKRJDGNUQOIC-UHFFFAOYSA-N Uracil Chemical compound O=C1C=CNC(=O)N1 ISAKRJDGNUQOIC-UHFFFAOYSA-N 0.000 description 2
- GXBMIBRIOWHPDT-UHFFFAOYSA-N Vasopressin Natural products N1C(=O)C(CC=2C=C(O)C=CC=2)NC(=O)C(N)CSSCC(C(=O)N2C(CCC2)C(=O)NC(CCCN=C(N)N)C(=O)NCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(CCC(N)=O)NC(=O)C1CC1=CC=CC=C1 GXBMIBRIOWHPDT-UHFFFAOYSA-N 0.000 description 2
- 108010004977 Vasopressins Proteins 0.000 description 2
- 102000002852 Vasopressins Human genes 0.000 description 2
- RJURFGZVJUQBHK-UHFFFAOYSA-N actinomycin D Natural products CC1OC(=O)C(C(C)C)N(C)C(=O)CN(C)C(=O)C2CCCN2C(=O)C(C(C)C)NC(=O)C1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)NC4C(=O)NC(C(N5CCCC5C(=O)N(C)CC(=O)N(C)C(C(C)C)C(=O)OC4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-UHFFFAOYSA-N 0.000 description 2
- 230000009471 action Effects 0.000 description 2
- 239000002998 adhesive polymer Substances 0.000 description 2
- 230000002776 aggregation Effects 0.000 description 2
- 238000004220 aggregation Methods 0.000 description 2
- 235000010443 alginic acid Nutrition 0.000 description 2
- 229920000615 alginic acid Polymers 0.000 description 2
- HDTRYLNUVZCQOY-LIZSDCNHSA-N alpha,alpha-trehalose Chemical compound O[C@@H]1[C@@H](O)[C@H](O)[C@@H](CO)O[C@@H]1O[C@@H]1[C@H](O)[C@@H](O)[C@H](O)[C@@H](CO)O1 HDTRYLNUVZCQOY-LIZSDCNHSA-N 0.000 description 2
- 229940126574 aminoglycoside antibiotic Drugs 0.000 description 2
- 239000002647 aminoglycoside antibiotic agent Substances 0.000 description 2
- 208000007502 anemia Diseases 0.000 description 2
- 239000000730 antalgic agent Substances 0.000 description 2
- 239000003242 anti bacterial agent Substances 0.000 description 2
- 230000003276 anti-hypertensive effect Effects 0.000 description 2
- 239000003146 anticoagulant agent Substances 0.000 description 2
- 229940127090 anticoagulant agent Drugs 0.000 description 2
- 239000003160 antidiuretic agent Substances 0.000 description 2
- 229940124538 antidiuretic agent Drugs 0.000 description 2
- 239000002111 antiemetic agent Substances 0.000 description 2
- 229940125683 antiemetic agent Drugs 0.000 description 2
- 239000002282 antimigraine agent Substances 0.000 description 2
- 229940125684 antimigraine agent Drugs 0.000 description 2
- 229940034982 antineoplastic agent Drugs 0.000 description 2
- 239000002246 antineoplastic agent Substances 0.000 description 2
- VSRXQHXAPYXROS-UHFFFAOYSA-N azanide;cyclobutane-1,1-dicarboxylic acid;platinum(2+) Chemical compound [NH2-].[NH2-].[Pt+2].OC(=O)C1(C(O)=O)CCC1 VSRXQHXAPYXROS-UHFFFAOYSA-N 0.000 description 2
- SESFRYSPDFLNCH-UHFFFAOYSA-N benzyl benzoate Chemical compound C=1C=CC=CC=1C(=O)OCC1=CC=CC=C1 SESFRYSPDFLNCH-UHFFFAOYSA-N 0.000 description 2
- GUBGYTABKSRVRQ-QUYVBRFLSA-N beta-maltose Chemical compound OC[C@H]1O[C@H](O[C@H]2[C@H](O)[C@@H](O)[C@H](O)O[C@@H]2CO)[C@H](O)[C@@H](O)[C@@H]1O GUBGYTABKSRVRQ-QUYVBRFLSA-N 0.000 description 2
- 238000004166 bioassay Methods 0.000 description 2
- 230000015572 biosynthetic process Effects 0.000 description 2
- 230000023555 blood coagulation Effects 0.000 description 2
- 239000000872 buffer Substances 0.000 description 2
- RYYVLZVUVIJVGH-UHFFFAOYSA-N caffeine Chemical compound CN1C(=O)N(C)C(=O)C2=C1N=CN2C RYYVLZVUVIJVGH-UHFFFAOYSA-N 0.000 description 2
- BBBFJLBPOGFECG-VJVYQDLKSA-N calcitonin Chemical compound N([C@H](C(=O)N[C@@H](CC(C)C)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(=O)NCC(=O)N[C@@H](CO)C(=O)NCC(=O)N[C@@H]([C@@H](C)O)C(=O)N1[C@@H](CCC1)C(N)=O)C(C)C)C(=O)[C@@H]1CSSC[C@H](N)C(=O)N[C@@H](CO)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CO)C(=O)N[C@@H]([C@@H](C)O)C(=O)N1 BBBFJLBPOGFECG-VJVYQDLKSA-N 0.000 description 2
- 229960004015 calcitonin Drugs 0.000 description 2
- 238000004364 calculation method Methods 0.000 description 2
- 150000001720 carbohydrates Chemical class 0.000 description 2
- 229960004562 carboplatin Drugs 0.000 description 2
- 208000002458 carcinoid tumor Diseases 0.000 description 2
- 230000001413 cellular effect Effects 0.000 description 2
- 239000002738 chelating agent Substances 0.000 description 2
- PBAYDYUZOSNJGU-UHFFFAOYSA-N chelidonic acid Natural products OC(=O)C1=CC(=O)C=C(C(O)=O)O1 PBAYDYUZOSNJGU-UHFFFAOYSA-N 0.000 description 2
- 238000007385 chemical modification Methods 0.000 description 2
- 235000012000 cholesterol Nutrition 0.000 description 2
- QMVPMAAFGQKVCJ-UHFFFAOYSA-N citronellol Chemical compound OCCC(C)CCC=C(C)C QMVPMAAFGQKVCJ-UHFFFAOYSA-N 0.000 description 2
- FDJOLVPMNUYSCM-UVKKECPRSA-L cobalt(3+);[(2r,3s,4r,5s)-5-(5,6-dimethylbenzimidazol-1-yl)-4-hydroxy-2-(hydroxymethyl)oxolan-3-yl] [(2r)-1-[3-[(2r,3r,4z,7s,9z,12s,13s,14z,17s,18s,19r)-2,13,18-tris(2-amino-2-oxoethyl)-7,12,17-tris(3-amino-3-oxopropyl)-3,5,8,8,13,15,18,19-octamethyl-2,7, Chemical compound [Co+3].N#[C-].C1([C@H](CC(N)=O)[C@@]2(C)CCC(=O)NC[C@@H](C)OP([O-])(=O)O[C@H]3[C@H]([C@H](O[C@@H]3CO)N3C4=CC(C)=C(C)C=C4N=C3)O)[N-]\C2=C(C)/C([C@H](C\2(C)C)CCC(N)=O)=N/C/2=C\C([C@H]([C@@]/2(CC(N)=O)C)CCC(N)=O)=N\C\2=C(C)/C2=N[C@]1(C)[C@@](C)(CC(N)=O)[C@@H]2CCC(N)=O FDJOLVPMNUYSCM-UVKKECPRSA-L 0.000 description 2
- OROGSEYTTFOCAN-DNJOTXNNSA-N codeine Chemical compound C([C@H]1[C@H](N(CC[C@@]112)C)C3)=C[C@H](O)[C@@H]1OC1=C2C3=CC=C1OC OROGSEYTTFOCAN-DNJOTXNNSA-N 0.000 description 2
- 230000000052 comparative effect Effects 0.000 description 2
- 229940038717 copaxone Drugs 0.000 description 2
- 230000009260 cross reactivity Effects 0.000 description 2
- 229960000684 cytarabine Drugs 0.000 description 2
- OPTASPLRGRRNAP-UHFFFAOYSA-N cytosine Chemical compound NC=1C=CNC(=O)N=1 OPTASPLRGRRNAP-UHFFFAOYSA-N 0.000 description 2
- 230000006378 damage Effects 0.000 description 2
- MWKFXSUHUHTGQN-UHFFFAOYSA-N decan-1-ol Chemical compound CCCCCCCCCCO MWKFXSUHUHTGQN-UHFFFAOYSA-N 0.000 description 2
- 238000011161 development Methods 0.000 description 2
- 230000018109 developmental process Effects 0.000 description 2
- 229960002086 dextran Drugs 0.000 description 2
- 235000019425 dextrin Nutrition 0.000 description 2
- 201000010064 diabetes insipidus Diseases 0.000 description 2
- 150000002016 disaccharides Chemical class 0.000 description 2
- SDWYUQHONRZPMW-UHFFFAOYSA-L disodium;octanedioate Chemical compound [Na+].[Na+].[O-]C(=O)CCCCCCC([O-])=O SDWYUQHONRZPMW-UHFFFAOYSA-L 0.000 description 2
- LQZZUXJYWNFBMV-UHFFFAOYSA-N dodecan-1-ol Chemical compound CCCCCCCCCCCCO LQZZUXJYWNFBMV-UHFFFAOYSA-N 0.000 description 2
- POULHZVOKOAJMA-UHFFFAOYSA-N dodecanoic acid Chemical compound CCCCCCCCCCCC(O)=O POULHZVOKOAJMA-UHFFFAOYSA-N 0.000 description 2
- 229960004679 doxorubicin Drugs 0.000 description 2
- 230000002526 effect on cardiovascular system Effects 0.000 description 2
- 239000002702 enteric coating Substances 0.000 description 2
- 238000009505 enteric coating Methods 0.000 description 2
- 229940105423 erythropoietin Drugs 0.000 description 2
- 229960000301 factor viii Drugs 0.000 description 2
- 206010016165 failure to thrive Diseases 0.000 description 2
- 238000011010 flushing procedure Methods 0.000 description 2
- 229940028334 follicle stimulating hormone Drugs 0.000 description 2
- 239000012634 fragment Substances 0.000 description 2
- 230000006870 function Effects 0.000 description 2
- 230000005176 gastrointestinal motility Effects 0.000 description 2
- 239000000499 gel Substances 0.000 description 2
- 239000008273 gelatin Substances 0.000 description 2
- 239000007903 gelatin capsule Substances 0.000 description 2
- 235000011852 gelatine desserts Nutrition 0.000 description 2
- 235000021474 generally recognized As safe (food) Nutrition 0.000 description 2
- 235000021473 generally recognized as safe (food ingredients) Nutrition 0.000 description 2
- ZHYZQXUYZJNEHD-VQHVLOKHSA-M geranate Chemical compound CC(C)=CCC\C(C)=C\C([O-])=O ZHYZQXUYZJNEHD-VQHVLOKHSA-M 0.000 description 2
- 239000002622 gonadotropin Substances 0.000 description 2
- 208000037824 growth disorder Diseases 0.000 description 2
- 239000003102 growth factor Substances 0.000 description 2
- ZSIAUFGUXNUGDI-UHFFFAOYSA-N hexan-1-ol Chemical compound CCCCCCO ZSIAUFGUXNUGDI-UHFFFAOYSA-N 0.000 description 2
- FUZZWVXGSFPDMH-UHFFFAOYSA-M hexanoate Chemical compound CCCCCC([O-])=O FUZZWVXGSFPDMH-UHFFFAOYSA-M 0.000 description 2
- WQPDUTSPKFMPDP-OUMQNGNKSA-N hirudin Chemical compound C([C@@H](C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C=CC(OS(O)(=O)=O)=CC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(N)=O)C(O)=O)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC=1NC=NC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](CCCCN)NC(=O)[C@H]1N(CCC1)C(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)CNC(=O)[C@@H](NC(=O)[C@@H](NC(=O)[C@H]1NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CCC(O)=O)NC(=O)CNC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)CNC(=O)[C@H](CC(C)C)NC(=O)[C@H]([C@@H](C)CC)NC(=O)[C@@H]2CSSC[C@@H](C(=O)N[C@@H](CCC(O)=O)C(=O)NCC(=O)N[C@@H](CO)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@H](C(=O)N[C@H](C(NCC(=O)N[C@@H](CCC(N)=O)C(=O)NCC(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CCCCN)C(=O)N2)=O)CSSC1)C(C)C)NC(=O)[C@H](CC(C)C)NC(=O)[C@H]1NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)CNC(=O)[C@H](CO)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H]([C@@H](C)O)NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@@H](NC(=O)[C@H](CC=2C=CC(O)=CC=2)NC(=O)[C@@H](NC(=O)[C@@H](N)C(C)C)C(C)C)[C@@H](C)O)CSSC1)C(C)C)[C@@H](C)O)[C@@H](C)O)C1=CC=CC=C1 WQPDUTSPKFMPDP-OUMQNGNKSA-N 0.000 description 2
- 229940006607 hirudin Drugs 0.000 description 2
- MGXWVYUBJRZYPE-YUGYIWNOSA-N incretin Chemical class C([C@@H](C(=O)N[C@@H](CO)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC=1C=CC=CC=1)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CCCCN)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CCC(N)=O)C(O)=O)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@@H](N)CC=1C=CC(O)=CC=1)[C@@H](C)O)[C@@H](C)CC)C1=CC=C(O)C=C1 MGXWVYUBJRZYPE-YUGYIWNOSA-N 0.000 description 2
- 239000000859 incretin Substances 0.000 description 2
- 230000002401 inhibitory effect Effects 0.000 description 2
- 230000005764 inhibitory process Effects 0.000 description 2
- 150000002500 ions Chemical class 0.000 description 2
- 230000007794 irritation Effects 0.000 description 2
- 229960001021 lactose monohydrate Drugs 0.000 description 2
- 108010021336 lanreotide Proteins 0.000 description 2
- 230000000670 limiting effect Effects 0.000 description 2
- 150000002632 lipids Chemical class 0.000 description 2
- 238000001294 liquid chromatography-tandem mass spectrometry Methods 0.000 description 2
- 239000006194 liquid suspension Substances 0.000 description 2
- 229940040129 luteinizing hormone Drugs 0.000 description 2
- RLSSMJSEOOYNOY-UHFFFAOYSA-N m-cresol Chemical compound CC1=CC=CC(O)=C1 RLSSMJSEOOYNOY-UHFFFAOYSA-N 0.000 description 2
- 229920002521 macromolecule Polymers 0.000 description 2
- 229940057917 medium chain triglycerides Drugs 0.000 description 2
- 208000030159 metabolic disease Diseases 0.000 description 2
- XZWYZXLIPXDOLR-UHFFFAOYSA-N metformin Chemical compound CN(C)C(=N)NC(N)=N XZWYZXLIPXDOLR-UHFFFAOYSA-N 0.000 description 2
- 229960003105 metformin Drugs 0.000 description 2
- 230000004048 modification Effects 0.000 description 2
- 238000012986 modification Methods 0.000 description 2
- 239000003147 molecular marker Substances 0.000 description 2
- BQJCRHHNABKAKU-KBQPJGBKSA-N morphine Chemical compound O([C@H]1[C@H](C=C[C@H]23)O)C4=C5[C@@]12CCN(C)[C@@H]3CC5=CC=C4O BQJCRHHNABKAKU-KBQPJGBKSA-N 0.000 description 2
- 229960002009 naproxen Drugs 0.000 description 2
- CMWTZPSULFXXJA-VIFPVBQESA-N naproxen Chemical compound C1=C([C@H](C)C(O)=O)C=CC2=CC(OC)=CC=C21 CMWTZPSULFXXJA-VIFPVBQESA-N 0.000 description 2
- 239000003887 narcotic antagonist Substances 0.000 description 2
- ZWRUINPWMLAQRD-UHFFFAOYSA-N nonan-1-ol Chemical compound CCCCCCCCCO ZWRUINPWMLAQRD-UHFFFAOYSA-N 0.000 description 2
- 229960001494 octreotide acetate Drugs 0.000 description 2
- 229920001542 oligosaccharide Polymers 0.000 description 2
- 229960001592 paclitaxel Drugs 0.000 description 2
- 229960005415 pasireotide Drugs 0.000 description 2
- 108700017947 pasireotide Proteins 0.000 description 2
- VMZMNAABQBOLAK-DBILLSOUSA-N pasireotide Chemical compound C([C@H]1C(=O)N2C[C@@H](C[C@H]2C(=O)N[C@H](C(=O)N[C@H](CC=2C3=CC=CC=C3NC=2)C(=O)N[C@H](C(N[C@@H](CC=2C=CC(OCC=3C=CC=CC=3)=CC=2)C(=O)N1)=O)CCCCN)C=1C=CC=CC=1)OC(=O)NCCN)C1=CC=CC=C1 VMZMNAABQBOLAK-DBILLSOUSA-N 0.000 description 2
- 230000007170 pathology Effects 0.000 description 2
- REIUXOLGHVXAEO-UHFFFAOYSA-N pentadecan-1-ol Chemical compound CCCCCCCCCCCCCCCO REIUXOLGHVXAEO-UHFFFAOYSA-N 0.000 description 2
- 239000008177 pharmaceutical agent Substances 0.000 description 2
- 150000002989 phenols Chemical class 0.000 description 2
- 229940068041 phytic acid Drugs 0.000 description 2
- 235000002949 phytic acid Nutrition 0.000 description 2
- 239000000467 phytic acid Substances 0.000 description 2
- 210000003240 portal vein Anatomy 0.000 description 2
- OXCMYAYHXIHQOA-UHFFFAOYSA-N potassium;[2-butyl-5-chloro-3-[[4-[2-(1,2,4-triaza-3-azanidacyclopenta-1,4-dien-5-yl)phenyl]phenyl]methyl]imidazol-4-yl]methanol Chemical compound [K+].CCCCC1=NC(Cl)=C(CO)N1CC1=CC=C(C=2C(=CC=CC=2)C2=N[N-]N=N2)C=C1 OXCMYAYHXIHQOA-UHFFFAOYSA-N 0.000 description 2
- 238000001556 precipitation Methods 0.000 description 2
- CPTBDICYNRMXFX-UHFFFAOYSA-N procarbazine Chemical compound CNNCC1=CC=C(C(=O)NC(C)C)C=C1 CPTBDICYNRMXFX-UHFFFAOYSA-N 0.000 description 2
- 229960000624 procarbazine Drugs 0.000 description 2
- 230000001681 protective effect Effects 0.000 description 2
- 230000009467 reduction Effects 0.000 description 2
- 230000002829 reductive effect Effects 0.000 description 2
- 230000001105 regulatory effect Effects 0.000 description 2
- 230000004044 response Effects 0.000 description 2
- 230000002441 reversible effect Effects 0.000 description 2
- 229960000329 ribavirin Drugs 0.000 description 2
- HZCAHMRRMINHDJ-DBRKOABJSA-N ribavirin Natural products O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1N=CN=C1 HZCAHMRRMINHDJ-DBRKOABJSA-N 0.000 description 2
- 229960004586 rosiglitazone Drugs 0.000 description 2
- 230000028327 secretion Effects 0.000 description 2
- 229940125723 sedative agent Drugs 0.000 description 2
- 239000000932 sedative agent Substances 0.000 description 2
- 239000004055 small Interfering RNA Substances 0.000 description 2
- SLVJLFOSPGAWHC-UHFFFAOYSA-M sodium;8-hydroxy-8-oxooctanoate Chemical compound [Na+].OC(=O)CCCCCCC([O-])=O SLVJLFOSPGAWHC-UHFFFAOYSA-M 0.000 description 2
- 230000007928 solubilization Effects 0.000 description 2
- 238000005063 solubilization Methods 0.000 description 2
- 229960000553 somatostatin Drugs 0.000 description 2
- 229940075620 somatostatin analogue Drugs 0.000 description 2
- 229940035636 somatostatin and analogues Drugs 0.000 description 2
- 229940063675 spermine Drugs 0.000 description 2
- 150000005846 sugar alcohols Polymers 0.000 description 2
- 230000002195 synergetic effect Effects 0.000 description 2
- 230000009885 systemic effect Effects 0.000 description 2
- HLZKNKRTKFSKGZ-UHFFFAOYSA-N tetradecan-1-ol Chemical compound CCCCCCCCCCCCCCO HLZKNKRTKFSKGZ-UHFFFAOYSA-N 0.000 description 2
- 238000002560 therapeutic procedure Methods 0.000 description 2
- MGSRCZKZVOBKFT-UHFFFAOYSA-N thymol Chemical compound CC(C)C1=CC=C(C)C=C1O MGSRCZKZVOBKFT-UHFFFAOYSA-N 0.000 description 2
- 210000001519 tissue Anatomy 0.000 description 2
- 229960000187 tissue plasminogen activator Drugs 0.000 description 2
- 239000003071 vasodilator agent Substances 0.000 description 2
- 229960003726 vasopressin Drugs 0.000 description 2
- 229940045999 vitamin b 12 Drugs 0.000 description 2
- 230000003442 weekly effect Effects 0.000 description 2
- NOOLISFMXDJSKH-UTLUCORTSA-N (+)-Neomenthol Chemical compound CC(C)[C@@H]1CC[C@@H](C)C[C@@H]1O NOOLISFMXDJSKH-UTLUCORTSA-N 0.000 description 1
- CRDAMVZIKSXKFV-FBXUGWQNSA-N (2-cis,6-cis)-farnesol Chemical compound CC(C)=CCC\C(C)=C/CC\C(C)=C/CO CRDAMVZIKSXKFV-FBXUGWQNSA-N 0.000 description 1
- 239000000260 (2E,6E)-3,7,11-trimethyldodeca-2,6,10-trien-1-ol Substances 0.000 description 1
- BJFIDCADFRDPIO-DZCXQCEKSA-N (2S)-N-[(2S)-6-amino-1-[(2-amino-2-oxoethyl)amino]-1-oxohexan-2-yl]-1-[[(4R,7S,10S,13S,16S,19R)-19-amino-7-(2-amino-2-oxoethyl)-10-(3-amino-3-oxopropyl)-16-[(4-hydroxyphenyl)methyl]-6,9,12,15,18-pentaoxo-13-(phenylmethyl)-1,2-dithia-5,8,11,14,17-pentazacycloeicos-4-yl]-oxomethyl]-2-pyrrolidinecarboxamide Chemical compound NCCCC[C@@H](C(=O)NCC(N)=O)NC(=O)[C@@H]1CCCN1C(=O)[C@H]1NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CC=2C=CC=CC=2)NC(=O)[C@H](CC=2C=CC(O)=CC=2)NC(=O)[C@@H](N)CSSC1 BJFIDCADFRDPIO-DZCXQCEKSA-N 0.000 description 1
- KIUKXJAPPMFGSW-DNGZLQJQSA-N (2S,3S,4S,5R,6R)-6-[(2S,3R,4R,5S,6R)-3-Acetamido-2-[(2S,3S,4R,5R,6R)-6-[(2R,3R,4R,5S,6R)-3-acetamido-2,5-dihydroxy-6-(hydroxymethyl)oxan-4-yl]oxy-2-carboxy-4,5-dihydroxyoxan-3-yl]oxy-5-hydroxy-6-(hydroxymethyl)oxan-4-yl]oxy-3,4,5-trihydroxyoxane-2-carboxylic acid Chemical compound CC(=O)N[C@H]1[C@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@H](O[C@H]2[C@@H]([C@@H](O[C@H]3[C@@H]([C@@H](O)[C@H](O)[C@H](O3)C(O)=O)O)[C@H](O)[C@@H](CO)O2)NC(C)=O)[C@@H](C(O)=O)O1 KIUKXJAPPMFGSW-DNGZLQJQSA-N 0.000 description 1
- LNAZSHAWQACDHT-XIYTZBAFSA-N (2r,3r,4s,5r,6s)-4,5-dimethoxy-2-(methoxymethyl)-3-[(2s,3r,4s,5r,6r)-3,4,5-trimethoxy-6-(methoxymethyl)oxan-2-yl]oxy-6-[(2r,3r,4s,5r,6r)-4,5,6-trimethoxy-2-(methoxymethyl)oxan-3-yl]oxyoxane Chemical compound CO[C@@H]1[C@@H](OC)[C@H](OC)[C@@H](COC)O[C@H]1O[C@H]1[C@H](OC)[C@@H](OC)[C@H](O[C@H]2[C@@H]([C@@H](OC)[C@H](OC)O[C@@H]2COC)OC)O[C@@H]1COC LNAZSHAWQACDHT-XIYTZBAFSA-N 0.000 description 1
- GDPHPXYFLPDZGH-XBTMSFKCSA-N (2s)-2-[[(2s)-2-[[(2s)-2-[[2-[[(2r)-2-[[(2s)-2-amino-3-(4-hydroxyphenyl)propanoyl]amino]propanoyl]amino]acetyl]amino]-3-phenylpropanoyl]amino]-4-methylpentanoyl]amino]-5-(diaminomethylideneamino)pentanoic acid Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCN=C(N)N)C(O)=O)NC(=O)CNC(=O)[C@@H](C)NC(=O)[C@@H](N)CC=1C=CC(O)=CC=1)C1=CC=CC=C1 GDPHPXYFLPDZGH-XBTMSFKCSA-N 0.000 description 1
- FELGMEQIXOGIFQ-CYBMUJFWSA-N (3r)-9-methyl-3-[(2-methylimidazol-1-yl)methyl]-2,3-dihydro-1h-carbazol-4-one Chemical compound CC1=NC=CN1C[C@@H]1C(=O)C(C=2C(=CC=CC=2)N2C)=C2CC1 FELGMEQIXOGIFQ-CYBMUJFWSA-N 0.000 description 1
- PUDHBTGHUJUUFI-SCTWWAJVSA-N (4r,7s,10s,13r,16s,19r)-10-(4-aminobutyl)-n-[(2s,3r)-1-amino-3-hydroxy-1-oxobutan-2-yl]-19-[[(2r)-2-amino-3-naphthalen-2-ylpropanoyl]amino]-16-[(4-hydroxyphenyl)methyl]-13-(1h-indol-3-ylmethyl)-6,9,12,15,18-pentaoxo-7-propan-2-yl-1,2-dithia-5,8,11,14,17-p Chemical compound C([C@H]1C(=O)N[C@H](CC=2C3=CC=CC=C3NC=2)C(=O)N[C@@H](CCCCN)C(=O)N[C@H](C(N[C@@H](CSSC[C@@H](C(=O)N1)NC(=O)[C@H](N)CC=1C=C2C=CC=CC2=CC=1)C(=O)N[C@@H]([C@@H](C)O)C(N)=O)=O)C(C)C)C1=CC=C(O)C=C1 PUDHBTGHUJUUFI-SCTWWAJVSA-N 0.000 description 1
- FDKXTQMXEQVLRF-ZHACJKMWSA-N (E)-dacarbazine Chemical compound CN(C)\N=N\c1[nH]cnc1C(N)=O FDKXTQMXEQVLRF-ZHACJKMWSA-N 0.000 description 1
- QMVPMAAFGQKVCJ-SNVBAGLBSA-N (R)-(+)-citronellol Natural products OCC[C@H](C)CCC=C(C)C QMVPMAAFGQKVCJ-SNVBAGLBSA-N 0.000 description 1
- ICLYJLBTOGPLMC-KVVVOXFISA-N (z)-octadec-9-enoate;tris(2-hydroxyethyl)azanium Chemical compound OCCN(CCO)CCO.CCCCCCCC\C=C/CCCCCCCC(O)=O ICLYJLBTOGPLMC-KVVVOXFISA-N 0.000 description 1
- RCLLNBVPCJDIPX-UHFFFAOYSA-N 1-(2-chloroethyl)-3-[2-(dimethylsulfamoyl)ethyl]-1-nitrosourea Chemical compound CN(C)S(=O)(=O)CCNC(=O)N(N=O)CCCl RCLLNBVPCJDIPX-UHFFFAOYSA-N 0.000 description 1
- PWMWNFMRSKOCEY-UHFFFAOYSA-N 1-Phenyl-1,2-ethanediol Chemical compound OCC(O)C1=CC=CC=C1 PWMWNFMRSKOCEY-UHFFFAOYSA-N 0.000 description 1
- XFRVVPUIAFSTFO-UHFFFAOYSA-N 1-Tridecanol Chemical compound CCCCCCCCCCCCCO XFRVVPUIAFSTFO-UHFFFAOYSA-N 0.000 description 1
- GYSCBCSGKXNZRH-UHFFFAOYSA-N 1-benzothiophene-2-carboxamide Chemical compound C1=CC=C2SC(C(=O)N)=CC2=C1 GYSCBCSGKXNZRH-UHFFFAOYSA-N 0.000 description 1
- LEBVLXFERQHONN-UHFFFAOYSA-N 1-butyl-N-(2,6-dimethylphenyl)piperidine-2-carboxamide Chemical compound CCCCN1CCCCC1C(=O)NC1=C(C)C=CC=C1C LEBVLXFERQHONN-UHFFFAOYSA-N 0.000 description 1
- VSNHCAURESNICA-NJFSPNSNSA-N 1-oxidanylurea Chemical compound N[14C](=O)NO VSNHCAURESNICA-NJFSPNSNSA-N 0.000 description 1
- SGTNSNPWRIOYBX-UHFFFAOYSA-N 2-(3,4-dimethoxyphenyl)-5-{[2-(3,4-dimethoxyphenyl)ethyl](methyl)amino}-2-(propan-2-yl)pentanenitrile Chemical compound C1=C(OC)C(OC)=CC=C1CCN(C)CCCC(C#N)(C(C)C)C1=CC=C(OC)C(OC)=C1 SGTNSNPWRIOYBX-UHFFFAOYSA-N 0.000 description 1
- JVKUCNQGESRUCL-UHFFFAOYSA-N 2-Hydroxyethyl 12-hydroxyoctadecanoate Chemical compound CCCCCCC(O)CCCCCCCCCCC(=O)OCCO JVKUCNQGESRUCL-UHFFFAOYSA-N 0.000 description 1
- JRLVYNDEHNATSC-UHFFFAOYSA-N 2-amino-3,7-dihydropurine-6-thione hydroxyurea Chemical compound N1C(N)=NC=2N=CNC2C1=S.ONC(=O)N JRLVYNDEHNATSC-UHFFFAOYSA-N 0.000 description 1
- WADQOGCINABPRT-UHFFFAOYSA-N 3-chloro-2-methylphenol Chemical compound CC1=C(O)C=CC=C1Cl WADQOGCINABPRT-UHFFFAOYSA-N 0.000 description 1
- AOJJSUZBOXZQNB-VTZDEGQISA-N 4'-epidoxorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(=O)CO)[C@H]1C[C@H](N)[C@@H](O)[C@H](C)O1 AOJJSUZBOXZQNB-VTZDEGQISA-N 0.000 description 1
- SQDAZGGFXASXDW-UHFFFAOYSA-N 5-bromo-2-(trifluoromethoxy)pyridine Chemical compound FC(F)(F)OC1=CC=C(Br)C=N1 SQDAZGGFXASXDW-UHFFFAOYSA-N 0.000 description 1
- USSIQXCVUWKGNF-UHFFFAOYSA-N 6-(dimethylamino)-4,4-diphenylheptan-3-one Chemical compound C=1C=CC=CC=1C(CC(C)N(C)C)(C(=O)CC)C1=CC=CC=C1 USSIQXCVUWKGNF-UHFFFAOYSA-N 0.000 description 1
- SLXKOJJOQWFEFD-UHFFFAOYSA-N 6-aminohexanoic acid Chemical compound NCCCCCC(O)=O SLXKOJJOQWFEFD-UHFFFAOYSA-N 0.000 description 1
- STQGQHZAVUOBTE-UHFFFAOYSA-N 7-Cyan-hept-2t-en-4,6-diinsaeure Natural products C1=2C(O)=C3C(=O)C=4C(OC)=CC=CC=4C(=O)C3=C(O)C=2CC(O)(C(C)=O)CC1OC1CC(N)C(O)C(C)O1 STQGQHZAVUOBTE-UHFFFAOYSA-N 0.000 description 1
- 229930000680 A04AD01 - Scopolamine Natural products 0.000 description 1
- 229930024421 Adenine Natural products 0.000 description 1
- GFFGJBXGBJISGV-UHFFFAOYSA-N Adenine Chemical compound NC1=NC=NC2=C1N=CN2 GFFGJBXGBJISGV-UHFFFAOYSA-N 0.000 description 1
- 108700029992 Ala(2)-Arg(6)- enkephalin-Leu Proteins 0.000 description 1
- WKEMJKQOLOHJLZ-UHFFFAOYSA-N Almogran Chemical compound C1=C2C(CCN(C)C)=CNC2=CC=C1CS(=O)(=O)N1CCCC1 WKEMJKQOLOHJLZ-UHFFFAOYSA-N 0.000 description 1
- 208000024827 Alzheimer disease Diseases 0.000 description 1
- QGZKDVFQNNGYKY-UHFFFAOYSA-O Ammonium Chemical compound [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 1
- 206010002091 Anaesthesia Diseases 0.000 description 1
- 108020005544 Antisense RNA Proteins 0.000 description 1
- 102000007592 Apolipoproteins Human genes 0.000 description 1
- 108010071619 Apolipoproteins Proteins 0.000 description 1
- 102100033367 Appetite-regulating hormone Human genes 0.000 description 1
- 108010039627 Aprotinin Proteins 0.000 description 1
- BSYNRYMUTXBXSQ-UHFFFAOYSA-N Aspirin Chemical compound CC(=O)OC1=CC=CC=C1C(O)=O BSYNRYMUTXBXSQ-UHFFFAOYSA-N 0.000 description 1
- 201000001320 Atherosclerosis Diseases 0.000 description 1
- 241000894006 Bacteria Species 0.000 description 1
- 208000035143 Bacterial infection Diseases 0.000 description 1
- DWRXFEITVBNRMK-UHFFFAOYSA-N Beta-D-1-Arabinofuranosylthymine Natural products O=C1NC(=O)C(C)=CN1C1C(O)C(O)C(CO)O1 DWRXFEITVBNRMK-UHFFFAOYSA-N 0.000 description 1
- 229940122361 Bisphosphonate Drugs 0.000 description 1
- 108010006654 Bleomycin Proteins 0.000 description 1
- 108010039209 Blood Coagulation Factors Proteins 0.000 description 1
- 102000015081 Blood Coagulation Factors Human genes 0.000 description 1
- 241000283690 Bos taurus Species 0.000 description 1
- COVZYZSDYWQREU-UHFFFAOYSA-N Busulfan Chemical compound CS(=O)(=O)OCCCCOS(C)(=O)=O COVZYZSDYWQREU-UHFFFAOYSA-N 0.000 description 1
- FVLVBPDQNARYJU-XAHDHGMMSA-N C[C@H]1CCC(CC1)NC(=O)N(CCCl)N=O Chemical compound C[C@H]1CCC(CC1)NC(=O)N(CCCl)N=O FVLVBPDQNARYJU-XAHDHGMMSA-N 0.000 description 1
- 206010006895 Cachexia Diseases 0.000 description 1
- 241000282472 Canis lupus familiaris Species 0.000 description 1
- 208000024172 Cardiovascular disease Diseases 0.000 description 1
- 108010020326 Caspofungin Proteins 0.000 description 1
- 108090000994 Catalytic RNA Proteins 0.000 description 1
- 102000053642 Catalytic RNA Human genes 0.000 description 1
- 229940123150 Chelating agent Drugs 0.000 description 1
- BHYOQNUELFTYRT-UHFFFAOYSA-N Cholesterol sulfate Natural products C1C=C2CC(OS(O)(=O)=O)CCC2(C)C2C1C1CCC(C(C)CCCC(C)C)C1(C)CC2 BHYOQNUELFTYRT-UHFFFAOYSA-N 0.000 description 1
- 229920001287 Chondroitin sulfate Polymers 0.000 description 1
- 208000000419 Chronic Hepatitis B Diseases 0.000 description 1
- 208000006154 Chronic hepatitis C Diseases 0.000 description 1
- PTOAARAWEBMLNO-KVQBGUIXSA-N Cladribine Chemical compound C1=NC=2C(N)=NC(Cl)=NC=2N1[C@H]1C[C@H](O)[C@@H](CO)O1 PTOAARAWEBMLNO-KVQBGUIXSA-N 0.000 description 1
- GJSURZIOUXUGAL-UHFFFAOYSA-N Clonidine Chemical compound ClC1=CC=CC(Cl)=C1NC1=NCCN1 GJSURZIOUXUGAL-UHFFFAOYSA-N 0.000 description 1
- 206010009657 Clostridium difficile colitis Diseases 0.000 description 1
- 102000007644 Colony-Stimulating Factors Human genes 0.000 description 1
- 108010071942 Colony-Stimulating Factors Proteins 0.000 description 1
- CMSMOCZEIVJLDB-UHFFFAOYSA-N Cyclophosphamide Chemical compound ClCCN(CCCl)P1(=O)NCCCO1 CMSMOCZEIVJLDB-UHFFFAOYSA-N 0.000 description 1
- 102000004127 Cytokines Human genes 0.000 description 1
- 108090000695 Cytokines Proteins 0.000 description 1
- NOOLISFMXDJSKH-UHFFFAOYSA-N DL-menthol Natural products CC(C)C1CCC(C)CC1O NOOLISFMXDJSKH-UHFFFAOYSA-N 0.000 description 1
- 108010092160 Dactinomycin Proteins 0.000 description 1
- GHVNFZFCNZKVNT-UHFFFAOYSA-N Decanoic acid Natural products CCCCCCCCCC(O)=O GHVNFZFCNZKVNT-UHFFFAOYSA-N 0.000 description 1
- 206010051055 Deep vein thrombosis Diseases 0.000 description 1
- 229920000045 Dermatan sulfate Polymers 0.000 description 1
- JXNRXNCCROJZFB-UHFFFAOYSA-N Di-Me ester-(2R, 3E)-Phytochromobilin Natural products NC(N)=NCCCC(C(O)=O)NC(=O)C(N)CC1=CC=C(O)C=C1 JXNRXNCCROJZFB-UHFFFAOYSA-N 0.000 description 1
- 108010016626 Dipeptides Proteins 0.000 description 1
- 238000002965 ELISA Methods 0.000 description 1
- 206010014486 Elevated triglycerides Diseases 0.000 description 1
- 241000196324 Embryophyta Species 0.000 description 1
- 241000792859 Enema Species 0.000 description 1
- 108010092674 Enkephalins Proteins 0.000 description 1
- 208000008967 Enuresis Diseases 0.000 description 1
- HTIJFSOGRVMCQR-UHFFFAOYSA-N Epirubicin Natural products COc1cccc2C(=O)c3c(O)c4CC(O)(CC(OC5CC(N)C(=O)C(C)O5)c4c(O)c3C(=O)c12)C(=O)CO HTIJFSOGRVMCQR-UHFFFAOYSA-N 0.000 description 1
- 241000283073 Equus caballus Species 0.000 description 1
- 208000000624 Esophageal and Gastric Varices Diseases 0.000 description 1
- 239000001856 Ethyl cellulose Substances 0.000 description 1
- ZZSNKZQZMQGXPY-UHFFFAOYSA-N Ethyl cellulose Chemical compound CCOCC1OC(OC)C(OCC)C(OCC)C1OC1C(O)C(O)C(OC)C(CO)O1 ZZSNKZQZMQGXPY-UHFFFAOYSA-N 0.000 description 1
- DBVJJBKOTRCVKF-UHFFFAOYSA-N Etidronic acid Chemical compound OP(=O)(O)C(O)(C)P(O)(O)=O DBVJJBKOTRCVKF-UHFFFAOYSA-N 0.000 description 1
- 208000001362 Fetal Growth Retardation Diseases 0.000 description 1
- 208000001640 Fibromyalgia Diseases 0.000 description 1
- 241000192125 Firmicutes Species 0.000 description 1
- GHASVSINZRGABV-UHFFFAOYSA-N Fluorouracil Chemical compound FC1=CNC(=O)NC1=O GHASVSINZRGABV-UHFFFAOYSA-N 0.000 description 1
- 206010070531 Foetal growth restriction Diseases 0.000 description 1
- 101710198884 GATA-type zinc finger protein 1 Proteins 0.000 description 1
- 108060003199 Glucagon Proteins 0.000 description 1
- 102000051325 Glucagon Human genes 0.000 description 1
- DTHNMHAUYICORS-KTKZVXAJSA-N Glucagon-like peptide 1 Chemical compound C([C@@H](C(=O)N[C@@H]([C@@H](C)CC)C(=O)N[C@@H](C)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCCCN)C(=O)NCC(=O)N[C@@H](CCCNC(N)=N)C(N)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CCCCN)NC(=O)[C@H](C)NC(=O)[C@H](C)NC(=O)[C@H](CCC(N)=O)NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CO)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@@H](N)CC=1N=CNC=1)[C@@H](C)O)[C@@H](C)O)C(C)C)C1=CC=CC=C1 DTHNMHAUYICORS-KTKZVXAJSA-N 0.000 description 1
- 102100039620 Granulocyte-macrophage colony-stimulating factor Human genes 0.000 description 1
- 101710119601 Growth hormone-releasing peptides Proteins 0.000 description 1
- 206010053759 Growth retardation Diseases 0.000 description 1
- 229920002971 Heparan sulfate Polymers 0.000 description 1
- STECJAGHUSJQJN-GAUPFVANSA-N Hyoscine Natural products C1([C@H](CO)C(=O)OC2C[C@@H]3N([C@H](C2)[C@@H]2[C@H]3O2)C)=CC=CC=C1 STECJAGHUSJQJN-GAUPFVANSA-N 0.000 description 1
- 208000035150 Hypercholesterolemia Diseases 0.000 description 1
- 208000031226 Hyperlipidaemia Diseases 0.000 description 1
- 206010020772 Hypertension Diseases 0.000 description 1
- 208000001953 Hypotension Diseases 0.000 description 1
- 206010021137 Hypovolaemia Diseases 0.000 description 1
- 206010021143 Hypoxia Diseases 0.000 description 1
- MPBVHIBUJCELCL-UHFFFAOYSA-N Ibandronate Chemical compound CCCCCN(C)CCC(O)(P(O)(O)=O)P(O)(O)=O MPBVHIBUJCELCL-UHFFFAOYSA-N 0.000 description 1
- XDXDZDZNSLXDNA-TZNDIEGXSA-N Idarubicin Chemical compound C1[C@H](N)[C@H](O)[C@H](C)O[C@H]1O[C@@H]1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2C[C@@](O)(C(C)=O)C1 XDXDZDZNSLXDNA-TZNDIEGXSA-N 0.000 description 1
- XDXDZDZNSLXDNA-UHFFFAOYSA-N Idarubicin Natural products C1C(N)C(O)C(C)OC1OC1C2=C(O)C(C(=O)C3=CC=CC=C3C3=O)=C3C(O)=C2CC(O)(C(C)=O)C1 XDXDZDZNSLXDNA-UHFFFAOYSA-N 0.000 description 1
- 108010021625 Immunoglobulin Fragments Proteins 0.000 description 1
- 102000008394 Immunoglobulin Fragments Human genes 0.000 description 1
- 238000012404 In vitro experiment Methods 0.000 description 1
- UGQMRVRMYYASKQ-KQYNXXCUSA-N Inosine Chemical compound O[C@@H]1[C@H](O)[C@@H](CO)O[C@H]1N1C2=NC=NC(O)=C2N=C1 UGQMRVRMYYASKQ-KQYNXXCUSA-N 0.000 description 1
- 229930010555 Inosine Natural products 0.000 description 1
- 206010022489 Insulin Resistance Diseases 0.000 description 1
- 108090000723 Insulin-Like Growth Factor I Proteins 0.000 description 1
- 102100026720 Interferon beta Human genes 0.000 description 1
- 108090000467 Interferon-beta Proteins 0.000 description 1
- 102000015696 Interleukins Human genes 0.000 description 1
- 108010063738 Interleukins Proteins 0.000 description 1
- 208000008081 Intestinal Fistula Diseases 0.000 description 1
- 102000036770 Islet Amyloid Polypeptide Human genes 0.000 description 1
- LPHGQDQBBGAPDZ-UHFFFAOYSA-N Isocaffeine Natural products CN1C(=O)N(C)C(=O)C2=C1N(C)C=N2 LPHGQDQBBGAPDZ-UHFFFAOYSA-N 0.000 description 1
- YQEZLKZALYSWHR-UHFFFAOYSA-N Ketamine Chemical compound C=1C=CC=C(Cl)C=1C1(NC)CCCCC1=O YQEZLKZALYSWHR-UHFFFAOYSA-N 0.000 description 1
- FBOZXECLQNJBKD-ZDUSSCGKSA-N L-methotrexate Chemical compound C=1N=C2N=C(N)N=C(N)C2=NC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)C=C1 FBOZXECLQNJBKD-ZDUSSCGKSA-N 0.000 description 1
- URLZCHNOLZSCCA-VABKMULXSA-N Leu-enkephalin Chemical compound C([C@@H](C(=O)N[C@@H](CC(C)C)C(O)=O)NC(=O)CNC(=O)CNC(=O)[C@@H](N)CC=1C=CC(O)=CC=1)C1=CC=CC=C1 URLZCHNOLZSCCA-VABKMULXSA-N 0.000 description 1
- JAQUASYNZVUNQP-USXIJHARSA-N Levorphanol Chemical compound C1C2=CC=C(O)C=C2[C@]23CCN(C)[C@H]1[C@@H]2CCCC3 JAQUASYNZVUNQP-USXIJHARSA-N 0.000 description 1
- NNJVILVZKWQKPM-UHFFFAOYSA-N Lidocaine Chemical compound CCN(CC)CC(=O)NC1=C(C)C=CC=C1C NNJVILVZKWQKPM-UHFFFAOYSA-N 0.000 description 1
- 108010048179 Lypressin Proteins 0.000 description 1
- 241000282567 Macaca fascicularis Species 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- RJQXTJLFIWVMTO-TYNCELHUSA-N Methicillin Chemical compound COC1=CC=CC(OC)=C1C(=O)N[C@@H]1C(=O)N2[C@@H](C(O)=O)C(C)(C)S[C@@H]21 RJQXTJLFIWVMTO-TYNCELHUSA-N 0.000 description 1
- 229920000168 Microcrystalline cellulose Polymers 0.000 description 1
- 229930192392 Mitomycin Natural products 0.000 description 1
- 208000004221 Multiple Trauma Diseases 0.000 description 1
- 208000023637 Multiple injury Diseases 0.000 description 1
- RXBQNMWIQKOSCS-RKDXNWHRSA-N Myrtenol Natural products C1[C@H]2C(C)(C)[C@@H]1CC=C2CO RXBQNMWIQKOSCS-RKDXNWHRSA-N 0.000 description 1
- NWIBSHFKIJFRCO-WUDYKRTCSA-N Mytomycin Chemical compound C1N2C(C(C(C)=C(N)C3=O)=O)=C3[C@@H](COC(N)=O)[C@@]2(OC)[C@@H]2[C@H]1N2 NWIBSHFKIJFRCO-WUDYKRTCSA-N 0.000 description 1
- STECJAGHUSJQJN-UHFFFAOYSA-N N-Methyl-scopolamin Natural products C1C(C2C3O2)N(C)C3CC1OC(=O)C(CO)C1=CC=CC=C1 STECJAGHUSJQJN-UHFFFAOYSA-N 0.000 description 1
- UBQYURCVBFRUQT-UHFFFAOYSA-N N-benzoyl-Ferrioxamine B Chemical compound CC(=O)N(O)CCCCCNC(=O)CCC(=O)N(O)CCCCCNC(=O)CCC(=O)N(O)CCCCCN UBQYURCVBFRUQT-UHFFFAOYSA-N 0.000 description 1
- ZDZOTLJHXYCWBA-VCVYQWHSSA-N N-debenzoyl-N-(tert-butoxycarbonyl)-10-deacetyltaxol Chemical compound O([C@H]1[C@H]2[C@@](C([C@H](O)C3=C(C)[C@@H](OC(=O)[C@H](O)[C@@H](NC(=O)OC(C)(C)C)C=4C=CC=CC=4)C[C@]1(O)C3(C)C)=O)(C)[C@@H](O)C[C@H]1OC[C@]12OC(=O)C)C(=O)C1=CC=CC=C1 ZDZOTLJHXYCWBA-VCVYQWHSSA-N 0.000 description 1
- CHJJGSNFBQVOTG-UHFFFAOYSA-N N-methyl-guanidine Natural products CNC(N)=N CHJJGSNFBQVOTG-UHFFFAOYSA-N 0.000 description 1
- 108020001621 Natriuretic Peptide Proteins 0.000 description 1
- 102000004571 Natriuretic peptide Human genes 0.000 description 1
- 238000011887 Necropsy Methods 0.000 description 1
- 206010028980 Neoplasm Diseases 0.000 description 1
- 108010025020 Nerve Growth Factor Proteins 0.000 description 1
- 102000007072 Nerve Growth Factors Human genes 0.000 description 1
- SNIOPGDIGTZGOP-UHFFFAOYSA-N Nitroglycerin Chemical compound [O-][N+](=O)OCC(O[N+]([O-])=O)CO[N+]([O-])=O SNIOPGDIGTZGOP-UHFFFAOYSA-N 0.000 description 1
- 239000000006 Nitroglycerin Substances 0.000 description 1
- 206010067482 No adverse event Diseases 0.000 description 1
- 108091028043 Nucleic acid sequence Proteins 0.000 description 1
- 108091034117 Oligonucleotide Proteins 0.000 description 1
- UQCNKQCJZOAFTQ-ISWURRPUSA-N Oxymorphone Chemical compound O([C@H]1C(CC[C@]23O)=O)C4=C5[C@@]12CCN(C)[C@@H]3CC5=CC=C4O UQCNKQCJZOAFTQ-ISWURRPUSA-N 0.000 description 1
- 102400000050 Oxytocin Human genes 0.000 description 1
- 101800000989 Oxytocin Proteins 0.000 description 1
- XNOPRXBHLZRZKH-UHFFFAOYSA-N Oxytocin Natural products N1C(=O)C(N)CSSCC(C(=O)N2C(CCC2)C(=O)NC(CC(C)C)C(=O)NCC(N)=O)NC(=O)C(CC(N)=O)NC(=O)C(CCC(N)=O)NC(=O)C(C(C)CC)NC(=O)C1CC1=CC=C(O)C=C1 XNOPRXBHLZRZKH-UHFFFAOYSA-N 0.000 description 1
- 229910019142 PO4 Inorganic materials 0.000 description 1
- 206010063350 Pancreatic leak Diseases 0.000 description 1
- 206010033645 Pancreatitis Diseases 0.000 description 1
- 206010033647 Pancreatitis acute Diseases 0.000 description 1
- 235000019483 Peanut oil Nutrition 0.000 description 1
- 241001494479 Pecora Species 0.000 description 1
- 229920002845 Poly(methacrylic acid) Polymers 0.000 description 1
- 108091036414 Polyinosinic:polycytidylic acid Proteins 0.000 description 1
- 201000010769 Prader-Willi syndrome Diseases 0.000 description 1
- 102100040918 Pro-glucagon Human genes 0.000 description 1
- 206010060862 Prostate cancer Diseases 0.000 description 1
- 208000000236 Prostatic Neoplasms Diseases 0.000 description 1
- 101000781681 Protobothrops flavoviridis Disintegrin triflavin Proteins 0.000 description 1
- 208000003100 Pseudomembranous Enterocolitis Diseases 0.000 description 1
- 206010037128 Pseudomembranous colitis Diseases 0.000 description 1
- 108091008103 RNA aptamers Proteins 0.000 description 1
- 238000012228 RNA interference-mediated gene silencing Methods 0.000 description 1
- 241000700157 Rattus norvegicus Species 0.000 description 1
- 108091028664 Ribonucleotide Proteins 0.000 description 1
- IIDJRNMFWXDHID-UHFFFAOYSA-N Risedronic acid Chemical compound OP(=O)(O)C(P(O)(O)=O)(O)CC1=CC=CN=C1 IIDJRNMFWXDHID-UHFFFAOYSA-N 0.000 description 1
- 206010040047 Sepsis Diseases 0.000 description 1
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 1
- 229920001304 Solutol HS 15 Polymers 0.000 description 1
- 102000013275 Somatomedins Human genes 0.000 description 1
- FOCVUCIESVLUNU-UHFFFAOYSA-N Thiotepa Chemical compound C1CN1P(N1CC1)(=S)N1CC1 FOCVUCIESVLUNU-UHFFFAOYSA-N 0.000 description 1
- 239000005844 Thymol Substances 0.000 description 1
- DKJJVAGXPKPDRL-UHFFFAOYSA-N Tiludronic acid Chemical compound OP(O)(=O)C(P(O)(O)=O)SC1=CC=C(Cl)C=C1 DKJJVAGXPKPDRL-UHFFFAOYSA-N 0.000 description 1
- IVTVGDXNLFLDRM-HNNXBMFYSA-N Tomudex Chemical compound C=1C=C2NC(C)=NC(=O)C2=CC=1CN(C)C1=CC=C(C(=O)N[C@@H](CCC(O)=O)C(O)=O)S1 IVTVGDXNLFLDRM-HNNXBMFYSA-N 0.000 description 1
- KJADKKWYZYXHBB-XBWDGYHZSA-N Topiramic acid Chemical compound C1O[C@@]2(COS(N)(=O)=O)OC(C)(C)O[C@H]2[C@@H]2OC(C)(C)O[C@@H]21 KJADKKWYZYXHBB-XBWDGYHZSA-N 0.000 description 1
- 108060008683 Tumor Necrosis Factor Receptor Proteins 0.000 description 1
- 208000026928 Turner syndrome Diseases 0.000 description 1
- JXNRXNCCROJZFB-RYUDHWBXSA-N Tyr-Arg Chemical compound NC(=N)NCCC[C@@H](C(O)=O)NC(=O)[C@@H](N)CC1=CC=C(O)C=C1 JXNRXNCCROJZFB-RYUDHWBXSA-N 0.000 description 1
- 206010056091 Varices oesophageal Diseases 0.000 description 1
- 206010046996 Varicose vein Diseases 0.000 description 1
- 206010047249 Venous thrombosis Diseases 0.000 description 1
- JXLYSJRDGCGARV-WWYNWVTFSA-N Vinblastine Natural products O=C(O[C@H]1[C@](O)(C(=O)OC)[C@@H]2N(C)c3c(cc(c(OC)c3)[C@]3(C(=O)OC)c4[nH]c5c(c4CCN4C[C@](O)(CC)C[C@H](C3)C4)cccc5)[C@@]32[C@H]2[C@@]1(CC)C=CCN2CC3)C JXLYSJRDGCGARV-WWYNWVTFSA-N 0.000 description 1
- 229940122803 Vinca alkaloid Drugs 0.000 description 1
- 108700005077 Viral Genes Proteins 0.000 description 1
- 208000036142 Viral infection Diseases 0.000 description 1
- 208000027276 Von Willebrand disease Diseases 0.000 description 1
- JLCPHMBAVCMARE-UHFFFAOYSA-N [3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[3-[[3-[[3-[[3-[[3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-[[5-(2-amino-6-oxo-1H-purin-9-yl)-3-hydroxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxyoxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(5-methyl-2,4-dioxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(6-aminopurin-9-yl)oxolan-2-yl]methoxy-hydroxyphosphoryl]oxy-5-(4-amino-2-oxopyrimidin-1-yl)oxolan-2-yl]methyl [5-(6-aminopurin-9-yl)-2-(hydroxymethyl)oxolan-3-yl] hydrogen phosphate Polymers Cc1cn(C2CC(OP(O)(=O)OCC3OC(CC3OP(O)(=O)OCC3OC(CC3O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c3nc(N)[nH]c4=O)C(COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3COP(O)(=O)OC3CC(OC3CO)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3ccc(N)nc3=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cc(C)c(=O)[nH]c3=O)n3cc(C)c(=O)[nH]c3=O)n3ccc(N)nc3=O)n3cc(C)c(=O)[nH]c3=O)n3cnc4c3nc(N)[nH]c4=O)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)n3cnc4c(N)ncnc34)O2)c(=O)[nH]c1=O JLCPHMBAVCMARE-UHFFFAOYSA-N 0.000 description 1
- NYSJHRJTGAOUNZ-UHFFFAOYSA-L [Na+].C(CCCCCCCCCCCCC)(=O)[O-].[Na+].C(CCCCCCCCCCCCC)(=O)[O-] Chemical compound [Na+].C(CCCCCCCCCCCCC)(=O)[O-].[Na+].C(CCCCCCCCCCCCC)(=O)[O-] NYSJHRJTGAOUNZ-UHFFFAOYSA-L 0.000 description 1
- 238000012084 abdominal surgery Methods 0.000 description 1
- 238000002835 absorbance Methods 0.000 description 1
- 238000009825 accumulation Methods 0.000 description 1
- FHEAIOHRHQGZPC-KIWGSFCNSA-N acetic acid;(2s)-2-amino-3-(4-hydroxyphenyl)propanoic acid;(2s)-2-aminopentanedioic acid;(2s)-2-aminopropanoic acid;(2s)-2,6-diaminohexanoic acid Chemical compound CC(O)=O.C[C@H](N)C(O)=O.NCCCC[C@H](N)C(O)=O.OC(=O)[C@@H](N)CCC(O)=O.OC(=O)[C@@H](N)CC1=CC=C(O)C=C1 FHEAIOHRHQGZPC-KIWGSFCNSA-N 0.000 description 1
- DEXPIBGCLCPUHE-UISHROKMSA-N acetic acid;(4r,7s,10s,13r,16s,19r)-10-(4-aminobutyl)-n-[(2s,3r)-1-amino-3-hydroxy-1-oxobutan-2-yl]-19-[[(2r)-2-amino-3-naphthalen-2-ylpropanoyl]amino]-16-[(4-hydroxyphenyl)methyl]-13-(1h-indol-3-ylmethyl)-6,9,12,15,18-pentaoxo-7-propan-2-yl-1,2-dithia-5, Chemical compound CC(O)=O.C([C@H]1C(=O)N[C@H](CC=2C3=CC=CC=C3NC=2)C(=O)N[C@@H](CCCCN)C(=O)N[C@H](C(N[C@@H](CSSC[C@@H](C(=O)N1)NC(=O)[C@H](N)CC=1C=C2C=CC=CC2=CC=1)C(=O)N[C@@H]([C@@H](C)O)C(N)=O)=O)C(C)C)C1=CC=C(O)C=C1 DEXPIBGCLCPUHE-UISHROKMSA-N 0.000 description 1
- 229960001138 acetylsalicylic acid Drugs 0.000 description 1
- 150000001252 acrylic acid derivatives Chemical class 0.000 description 1
- RJURFGZVJUQBHK-IIXSONLDSA-N actinomycin D Chemical compound C[C@H]1OC(=O)[C@H](C(C)C)N(C)C(=O)CN(C)C(=O)[C@@H]2CCCN2C(=O)[C@@H](C(C)C)NC(=O)[C@H]1NC(=O)C1=C(N)C(=O)C(C)=C2OC(C(C)=CC=C3C(=O)N[C@@H]4C(=O)N[C@@H](C(N5CCC[C@H]5C(=O)N(C)CC(=O)N(C)[C@@H](C(C)C)C(=O)O[C@@H]4C)=O)C(C)C)=C3N=C21 RJURFGZVJUQBHK-IIXSONLDSA-N 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 201000003229 acute pancreatitis Diseases 0.000 description 1
- 239000000654 additive Substances 0.000 description 1
- 230000000996 additive effect Effects 0.000 description 1
- 229960000643 adenine Drugs 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 230000032683 aging Effects 0.000 description 1
- 238000013019 agitation Methods 0.000 description 1
- 239000000783 alginic acid Substances 0.000 description 1
- 229960001126 alginic acid Drugs 0.000 description 1
- 150000004781 alginic acids Chemical class 0.000 description 1
- 229960002133 almotriptan Drugs 0.000 description 1
- WUOACPNHFRMFPN-UHFFFAOYSA-N alpha-terpineol Chemical compound CC1=CCC(C(C)(C)O)CC1 WUOACPNHFRMFPN-UHFFFAOYSA-N 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 125000000539 amino acid group Chemical group 0.000 description 1
- 229960002684 aminocaproic acid Drugs 0.000 description 1
- 229940126575 aminoglycoside Drugs 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- BSJGASKRWFKGMV-UHFFFAOYSA-L ammonia dichloroplatinum(2+) Chemical compound N.N.Cl[Pt+2]Cl BSJGASKRWFKGMV-UHFFFAOYSA-L 0.000 description 1
- 230000037005 anaesthesia Effects 0.000 description 1
- 229940035676 analgesics Drugs 0.000 description 1
- 125000000129 anionic group Chemical group 0.000 description 1
- 230000000843 anti-fungal effect Effects 0.000 description 1
- 230000001775 anti-pathogenic effect Effects 0.000 description 1
- 230000000840 anti-viral effect Effects 0.000 description 1
- 229940088710 antibiotic agent Drugs 0.000 description 1
- 229940121375 antifungal agent Drugs 0.000 description 1
- 239000000427 antigen Substances 0.000 description 1
- 108091007433 antigens Proteins 0.000 description 1
- 102000036639 antigens Human genes 0.000 description 1
- 239000002220 antihypertensive agent Substances 0.000 description 1
- 239000003096 antiparasitic agent Substances 0.000 description 1
- 229940125687 antiparasitic agent Drugs 0.000 description 1
- 229960004405 aprotinin Drugs 0.000 description 1
- 238000010420 art technique Methods 0.000 description 1
- 238000003556 assay Methods 0.000 description 1
- 208000022362 bacterial infectious disease Diseases 0.000 description 1
- 210000000227 basophil cell of anterior lobe of hypophysis Anatomy 0.000 description 1
- 229960002903 benzyl benzoate Drugs 0.000 description 1
- IQFYYKKMVGJFEH-UHFFFAOYSA-N beta-L-thymidine Natural products O=C1NC(=O)C(C)=CN1C1OC(CO)C(O)C1 IQFYYKKMVGJFEH-UHFFFAOYSA-N 0.000 description 1
- JGQFVRIQXUFPAH-UHFFFAOYSA-N beta-citronellol Natural products OCCC(C)CCCC(C)=C JGQFVRIQXUFPAH-UHFFFAOYSA-N 0.000 description 1
- 229940093761 bile salts Drugs 0.000 description 1
- 230000000975 bioactive effect Effects 0.000 description 1
- 230000003115 biocidal effect Effects 0.000 description 1
- 239000003124 biologic agent Substances 0.000 description 1
- 230000031018 biological processes and functions Effects 0.000 description 1
- 229940088623 biologically active substance Drugs 0.000 description 1
- 229960000074 biopharmaceutical Drugs 0.000 description 1
- 150000004663 bisphosphonates Chemical class 0.000 description 1
- 108010055460 bivalirudin Proteins 0.000 description 1
- 229960001500 bivalirudin Drugs 0.000 description 1
- OIRCOABEOLEUMC-GEJPAHFPSA-N bivalirudin Chemical compound C([C@@H](C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H]([C@@H](C)CC)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C=CC(O)=CC=1)C(=O)N[C@@H](CC(C)C)C(O)=O)NC(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@H](CC(N)=O)NC(=O)CNC(=O)CNC(=O)CNC(=O)CNC(=O)[C@H]1N(CCC1)C(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H]1N(CCC1)C(=O)[C@H](N)CC=1C=CC=CC=1)C1=CC=CC=C1 OIRCOABEOLEUMC-GEJPAHFPSA-N 0.000 description 1
- 229960001561 bleomycin Drugs 0.000 description 1
- OYVAGSVQBOHSSS-UAPAGMARSA-O bleomycin A2 Chemical compound N([C@H](C(=O)N[C@H](C)[C@@H](O)[C@H](C)C(=O)N[C@@H]([C@H](O)C)C(=O)NCCC=1SC=C(N=1)C=1SC=C(N=1)C(=O)NCCC[S+](C)C)[C@@H](O[C@H]1[C@H]([C@@H](O)[C@H](O)[C@H](CO)O1)O[C@@H]1[C@H]([C@@H](OC(N)=O)[C@H](O)[C@@H](CO)O1)O)C=1N=CNC=1)C(=O)C1=NC([C@H](CC(N)=O)NC[C@H](N)C(N)=O)=NC(N)=C1C OYVAGSVQBOHSSS-UAPAGMARSA-O 0.000 description 1
- 230000008499 blood brain barrier function Effects 0.000 description 1
- 230000017531 blood circulation Effects 0.000 description 1
- 239000003114 blood coagulation factor Substances 0.000 description 1
- 230000036765 blood level Effects 0.000 description 1
- 210000001218 blood-brain barrier Anatomy 0.000 description 1
- 230000037396 body weight Effects 0.000 description 1
- 210000004556 brain Anatomy 0.000 description 1
- 229960003150 bupivacaine Drugs 0.000 description 1
- RMRJXGBAOAMLHD-IHFGGWKQSA-N buprenorphine Chemical compound C([C@]12[C@H]3OC=4C(O)=CC=C(C2=4)C[C@@H]2[C@]11CC[C@]3([C@H](C1)[C@](C)(O)C(C)(C)C)OC)CN2CC1CC1 RMRJXGBAOAMLHD-IHFGGWKQSA-N 0.000 description 1
- 229960001736 buprenorphine Drugs 0.000 description 1
- 229960002092 busulfan Drugs 0.000 description 1
- 229960002546 butalbital Drugs 0.000 description 1
- UZVHFVZFNXBMQJ-UHFFFAOYSA-N butalbital Chemical compound CC(C)CC1(CC=C)C(=O)NC(=O)NC1=O UZVHFVZFNXBMQJ-UHFFFAOYSA-N 0.000 description 1
- 229940084891 byetta Drugs 0.000 description 1
- 229960001948 caffeine Drugs 0.000 description 1
- VJEONQKOZGKCAK-UHFFFAOYSA-N caffeine Natural products CN1C(=O)N(C)C(=O)C2=C1C=CN2C VJEONQKOZGKCAK-UHFFFAOYSA-N 0.000 description 1
- 201000011510 cancer Diseases 0.000 description 1
- 239000000828 canola oil Substances 0.000 description 1
- 235000019519 canola oil Nutrition 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 230000002612 cardiopulmonary effect Effects 0.000 description 1
- 239000002327 cardiovascular agent Substances 0.000 description 1
- JYIKNQVWKBUSNH-WVDDFWQHSA-N caspofungin Chemical compound C1([C@H](O)[C@@H](O)[C@H]2C(=O)N[C@H](C(=O)N3CC[C@H](O)[C@H]3C(=O)N[C@H](NCCN)[C@H](O)C[C@@H](C(N[C@H](C(=O)N3C[C@H](O)C[C@H]3C(=O)N2)[C@@H](C)O)=O)NC(=O)CCCCCCCC[C@@H](C)C[C@@H](C)CC)[C@H](O)CCN)=CC=C(O)C=C1 JYIKNQVWKBUSNH-WVDDFWQHSA-N 0.000 description 1
- 229960003034 caspofungin Drugs 0.000 description 1
- 230000001925 catabolic effect Effects 0.000 description 1
- 229920002678 cellulose Polymers 0.000 description 1
- 235000010980 cellulose Nutrition 0.000 description 1
- 230000008859 change Effects 0.000 description 1
- JCKYGMPEJWAADB-UHFFFAOYSA-N chlorambucil Chemical compound OC(=O)CCCC1=CC=C(N(CCCl)CCCl)C=C1 JCKYGMPEJWAADB-UHFFFAOYSA-N 0.000 description 1
- 229960004630 chlorambucil Drugs 0.000 description 1
- BHYOQNUELFTYRT-DPAQBDIFSA-N cholesterol sulfate Chemical compound C1C=C2C[C@@H](OS(O)(=O)=O)CC[C@]2(C)[C@@H]2[C@@H]1[C@@H]1CC[C@H]([C@H](C)CCCC(C)C)[C@@]1(C)CC2 BHYOQNUELFTYRT-DPAQBDIFSA-N 0.000 description 1
- 150000001841 cholesterols Chemical class 0.000 description 1
- 229940059329 chondroitin sulfate Drugs 0.000 description 1
- 230000001684 chronic effect Effects 0.000 description 1
- 208000020832 chronic kidney disease Diseases 0.000 description 1
- 208000022831 chronic renal failure syndrome Diseases 0.000 description 1
- 230000004087 circulation Effects 0.000 description 1
- 235000000484 citronellol Nutrition 0.000 description 1
- HJKBJIYDJLVSAO-UHFFFAOYSA-L clodronic acid disodium salt Chemical compound [Na+].[Na+].OP([O-])(=O)C(Cl)(Cl)P(O)([O-])=O HJKBJIYDJLVSAO-UHFFFAOYSA-L 0.000 description 1
- 229960002896 clonidine Drugs 0.000 description 1
- 239000011248 coating agent Substances 0.000 description 1
- 238000000576 coating method Methods 0.000 description 1
- 229960004126 codeine Drugs 0.000 description 1
- 230000000112 colonic effect Effects 0.000 description 1
- 229940000425 combination drug Drugs 0.000 description 1
- 230000000295 complement effect Effects 0.000 description 1
- 239000003184 complementary RNA Substances 0.000 description 1
- 238000007596 consolidation process Methods 0.000 description 1
- 238000010276 construction Methods 0.000 description 1
- 238000013270 controlled release Methods 0.000 description 1
- 230000001276 controlling effect Effects 0.000 description 1
- 235000005687 corn oil Nutrition 0.000 description 1
- 239000002285 corn oil Substances 0.000 description 1
- 210000004087 cornea Anatomy 0.000 description 1
- 235000012343 cottonseed oil Nutrition 0.000 description 1
- 239000002385 cottonseed oil Substances 0.000 description 1
- PZAQDVNYNJBUTM-UHFFFAOYSA-L cyclohexane-1,2-diamine;7,7-dimethyloctanoate;platinum(2+) Chemical compound [Pt+2].NC1CCCCC1N.CC(C)(C)CCCCCC([O-])=O.CC(C)(C)CCCCCC([O-])=O PZAQDVNYNJBUTM-UHFFFAOYSA-L 0.000 description 1
- 229960004397 cyclophosphamide Drugs 0.000 description 1
- 229940104302 cytosine Drugs 0.000 description 1
- 229960003901 dacarbazine Drugs 0.000 description 1
- 229960000640 dactinomycin Drugs 0.000 description 1
- 229960004969 dalteparin Drugs 0.000 description 1
- STQGQHZAVUOBTE-VGBVRHCVSA-N daunorubicin Chemical compound O([C@H]1C[C@@](O)(CC=2C(O)=C3C(=O)C=4C=CC=C(C=4C(=O)C3=C(O)C=21)OC)C(C)=O)[C@H]1C[C@H](N)[C@H](O)[C@H](C)O1 STQGQHZAVUOBTE-VGBVRHCVSA-N 0.000 description 1
- 229960000975 daunorubicin Drugs 0.000 description 1
- 229960000958 deferoxamine Drugs 0.000 description 1
- SQIFACVGCPWBQZ-UHFFFAOYSA-N delta-terpineol Natural products CC(C)(O)C1CCC(=C)CC1 SQIFACVGCPWBQZ-UHFFFAOYSA-N 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- AVJBPWGFOQAPRH-FWMKGIEWSA-L dermatan sulfate Chemical compound CC(=O)N[C@H]1[C@H](O)O[C@H](CO)[C@H](OS([O-])(=O)=O)[C@@H]1O[C@H]1[C@H](O)[C@@H](O)[C@H](O)[C@H](C([O-])=O)O1 AVJBPWGFOQAPRH-FWMKGIEWSA-L 0.000 description 1
- 229940051593 dermatan sulfate Drugs 0.000 description 1
- UREBDLICKHMUKA-CXSFZGCWSA-N dexamethasone Chemical compound C1CC2=CC(=O)C=C[C@]2(C)[C@]2(F)[C@@H]1[C@@H]1C[C@@H](C)[C@@](C(=O)CO)(O)[C@@]1(C)C[C@@H]2O UREBDLICKHMUKA-CXSFZGCWSA-N 0.000 description 1
- 229960003957 dexamethasone Drugs 0.000 description 1
- 239000000032 diagnostic agent Substances 0.000 description 1
- 229940039227 diagnostic agent Drugs 0.000 description 1
- 238000010586 diagram Methods 0.000 description 1
- ATKXDQOHNICLQW-UHFFFAOYSA-N dichloralphenazone Chemical compound OC(O)C(Cl)(Cl)Cl.OC(O)C(Cl)(Cl)Cl.CN1C(C)=CC(=O)N1C1=CC=CC=C1 ATKXDQOHNICLQW-UHFFFAOYSA-N 0.000 description 1
- 229960005422 dichloralphenazone Drugs 0.000 description 1
- DCOPUUMXTXDBNB-UHFFFAOYSA-N diclofenac Chemical compound OC(=O)CC1=CC=CC=C1NC1=C(Cl)C=CC=C1Cl DCOPUUMXTXDBNB-UHFFFAOYSA-N 0.000 description 1
- 229960001259 diclofenac Drugs 0.000 description 1
- RBOXVHNMENFORY-DNJOTXNNSA-N dihydrocodeine Chemical compound C([C@H]1[C@H](N(CC[C@@]112)C)C3)C[C@H](O)[C@@H]1OC1=C2C3=CC=C1OC RBOXVHNMENFORY-DNJOTXNNSA-N 0.000 description 1
- 229960000920 dihydrocodeine Drugs 0.000 description 1
- HESHRHUZIWVEAJ-JGRZULCMSA-N dihydroergotamine Chemical compound C([C@H]1C(=O)N2CCC[C@H]2[C@]2(O)O[C@@](C(N21)=O)(C)NC(=O)[C@H]1CN([C@H]2[C@@H](C3=CC=CC4=NC=C([C]34)C2)C1)C)C1=CC=CC=C1 HESHRHUZIWVEAJ-JGRZULCMSA-N 0.000 description 1
- 229960004704 dihydroergotamine Drugs 0.000 description 1
- HSUGRBWQSSZJOP-RTWAWAEBSA-N diltiazem Chemical compound C1=CC(OC)=CC=C1[C@H]1[C@@H](OC(C)=O)C(=O)N(CCN(C)C)C2=CC=CC=C2S1 HSUGRBWQSSZJOP-RTWAWAEBSA-N 0.000 description 1
- 229960004166 diltiazem Drugs 0.000 description 1
- ORXJMBXYSGGCHG-UHFFFAOYSA-N dimethyl 2-methoxypropanedioate Chemical compound COC(=O)C(OC)C(=O)OC ORXJMBXYSGGCHG-UHFFFAOYSA-N 0.000 description 1
- SWSQBOPZIKWTGO-UHFFFAOYSA-N dimethylaminoamidine Natural products CN(C)C(N)=N SWSQBOPZIKWTGO-UHFFFAOYSA-N 0.000 description 1
- 208000016097 disease of metabolism Diseases 0.000 description 1
- YGEVMZLRIOFHSG-UHFFFAOYSA-L disodium dodecanoate Chemical compound [Na+].[Na+].CCCCCCCCCCCC([O-])=O.CCCCCCCCCCCC([O-])=O YGEVMZLRIOFHSG-UHFFFAOYSA-L 0.000 description 1
- 238000009826 distribution Methods 0.000 description 1
- 229960003668 docetaxel Drugs 0.000 description 1
- FGXWKSZFVQUSTL-UHFFFAOYSA-N domperidone Chemical compound C12=CC=CC=C2NC(=O)N1CCCN(CC1)CCC1N1C2=CC=C(Cl)C=C2NC1=O FGXWKSZFVQUSTL-UHFFFAOYSA-N 0.000 description 1
- 229960001253 domperidone Drugs 0.000 description 1
- 229940126534 drug product Drugs 0.000 description 1
- 239000012636 effector Substances 0.000 description 1
- 229960002472 eletriptan Drugs 0.000 description 1
- OTLDLQZJRFYOJR-LJQANCHMSA-N eletriptan Chemical compound CN1CCC[C@@H]1CC1=CN=C2[C]1C=C(CCS(=O)(=O)C=1C=CC=CC=1)C=C2 OTLDLQZJRFYOJR-LJQANCHMSA-N 0.000 description 1
- 210000003989 endothelium vascular Anatomy 0.000 description 1
- 239000007920 enema Substances 0.000 description 1
- 229940079360 enema for constipation Drugs 0.000 description 1
- 230000002708 enhancing effect Effects 0.000 description 1
- 229960000610 enoxaparin Drugs 0.000 description 1
- 230000007613 environmental effect Effects 0.000 description 1
- 229960001904 epirubicin Drugs 0.000 description 1
- 210000002919 epithelial cell Anatomy 0.000 description 1
- 229960003133 ergot alkaloid Drugs 0.000 description 1
- OFKDAAIKGIBASY-VFGNJEKYSA-N ergotamine Chemical compound C([C@H]1C(=O)N2CCC[C@H]2[C@]2(O)O[C@@](C(N21)=O)(C)NC(=O)[C@H]1CN([C@H]2C(C3=CC=CC4=NC=C([C]34)C2)=C1)C)C1=CC=CC=C1 OFKDAAIKGIBASY-VFGNJEKYSA-N 0.000 description 1
- 229960004943 ergotamine Drugs 0.000 description 1
- XCGSFFUVFURLIX-UHFFFAOYSA-N ergotaminine Natural products C1=C(C=2C=CC=C3NC=C(C=23)C2)C2N(C)CC1C(=O)NC(C(N12)=O)(C)OC1(O)C1CCCN1C(=O)C2CC1=CC=CC=C1 XCGSFFUVFURLIX-UHFFFAOYSA-N 0.000 description 1
- 208000024170 esophageal varices Diseases 0.000 description 1
- 201000010120 esophageal varix Diseases 0.000 description 1
- 150000002170 ethers Chemical class 0.000 description 1
- 235000019325 ethyl cellulose Nutrition 0.000 description 1
- 229920001249 ethyl cellulose Polymers 0.000 description 1
- 229940009626 etidronate Drugs 0.000 description 1
- VJJPUSNTGOMMGY-MRVIYFEKSA-N etoposide Chemical compound COC1=C(O)C(OC)=CC([C@@H]2C3=CC=4OCOC=4C=C3[C@@H](O[C@H]3[C@@H]([C@@H](O)[C@@H]4O[C@H](C)OC[C@H]4O3)O)[C@@H]3[C@@H]2C(OC3)=O)=C1 VJJPUSNTGOMMGY-MRVIYFEKSA-N 0.000 description 1
- 229960005420 etoposide Drugs 0.000 description 1
- 230000008020 evaporation Effects 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 229940043259 farnesol Drugs 0.000 description 1
- 229930002886 farnesol Natural products 0.000 description 1
- 229960002428 fentanyl Drugs 0.000 description 1
- IVLVTNPOHDFFCJ-UHFFFAOYSA-N fentanyl citrate Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O.C=1C=CC=CC=1N(C(=O)CC)C(CC1)CCN1CCC1=CC=CC=C1 IVLVTNPOHDFFCJ-UHFFFAOYSA-N 0.000 description 1
- 208000030941 fetal growth restriction Diseases 0.000 description 1
- 239000004503 fine granule Substances 0.000 description 1
- 229960000390 fludarabine Drugs 0.000 description 1
- GIUYCYHIANZCFB-FJFJXFQQSA-N fludarabine phosphate Chemical compound C1=NC=2C(N)=NC(F)=NC=2N1[C@@H]1O[C@H](COP(O)(O)=O)[C@@H](O)[C@@H]1O GIUYCYHIANZCFB-FJFJXFQQSA-N 0.000 description 1
- 229960002949 fluorouracil Drugs 0.000 description 1
- 235000012631 food intake Nutrition 0.000 description 1
- 229940085942 formulation r Drugs 0.000 description 1
- 229960002284 frovatriptan Drugs 0.000 description 1
- SIBNYOSJIXCDRI-SECBINFHSA-N frovatriptan Chemical compound C1=C(C(N)=O)[CH]C2=C(C[C@H](NC)CC3)C3=NC2=C1 SIBNYOSJIXCDRI-SECBINFHSA-N 0.000 description 1
- 210000003736 gastrointestinal content Anatomy 0.000 description 1
- 210000001035 gastrointestinal tract Anatomy 0.000 description 1
- SDUQYLNIPVEERB-QPPQHZFASA-N gemcitabine Chemical compound O=C1N=C(N)C=CN1[C@H]1C(F)(F)[C@H](O)[C@@H](CO)O1 SDUQYLNIPVEERB-QPPQHZFASA-N 0.000 description 1
- 229960005277 gemcitabine Drugs 0.000 description 1
- 235000021472 generally recognized as safe Nutrition 0.000 description 1
- 229960003776 glatiramer acetate Drugs 0.000 description 1
- MASNOZXLGMXCHN-ZLPAWPGGSA-N glucagon Chemical compound C([C@@H](C(=O)N[C@H](C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H]([C@@H](C)O)C(O)=O)C(C)C)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](C)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CO)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CO)NC(=O)[C@H](CC=1C=CC(O)=CC=1)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@H](CC=1C=CC=CC=1)NC(=O)[C@@H](NC(=O)CNC(=O)[C@H](CCC(N)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](N)CC=1NC=NC=1)[C@@H](C)O)[C@@H](C)O)C1=CC=CC=C1 MASNOZXLGMXCHN-ZLPAWPGGSA-N 0.000 description 1
- 229960004666 glucagon Drugs 0.000 description 1
- 230000002641 glycemic effect Effects 0.000 description 1
- 229960003711 glyceryl trinitrate Drugs 0.000 description 1
- PCHJSUWPFVWCPO-UHFFFAOYSA-N gold Chemical compound [Au] PCHJSUWPFVWCPO-UHFFFAOYSA-N 0.000 description 1
- 239000010931 gold Substances 0.000 description 1
- 229910052737 gold Inorganic materials 0.000 description 1
- 208000002566 gonadal dysgenesis Diseases 0.000 description 1
- 210000003714 granulocyte Anatomy 0.000 description 1
- 229960004198 guanidine Drugs 0.000 description 1
- 208000014951 hematologic disease Diseases 0.000 description 1
- 230000002489 hematologic effect Effects 0.000 description 1
- 208000031169 hemorrhagic disease Diseases 0.000 description 1
- 230000002008 hemorrhagic effect Effects 0.000 description 1
- 208000002672 hepatitis B Diseases 0.000 description 1
- 208000010710 hepatitis C virus infection Diseases 0.000 description 1
- 229940022353 herceptin Drugs 0.000 description 1
- 238000011540 hip replacement Methods 0.000 description 1
- 238000010562 histological examination Methods 0.000 description 1
- 229920002674 hyaluronan Polymers 0.000 description 1
- 229960003160 hyaluronic acid Drugs 0.000 description 1
- OROGSEYTTFOCAN-UHFFFAOYSA-N hydrocodone Natural products C1C(N(CCC234)C)C2C=CC(O)C3OC2=C4C1=CC=C2OC OROGSEYTTFOCAN-UHFFFAOYSA-N 0.000 description 1
- WVLOADHCBXTIJK-YNHQPCIGSA-N hydromorphone Chemical compound O([C@H]1C(CC[C@H]23)=O)C4=C5[C@@]12CCN(C)[C@@H]3CC5=CC=C4O WVLOADHCBXTIJK-YNHQPCIGSA-N 0.000 description 1
- 229960001410 hydromorphone Drugs 0.000 description 1
- 150000002433 hydrophilic molecules Chemical class 0.000 description 1
- 201000001421 hyperglycemia Diseases 0.000 description 1
- 230000036543 hypotension Effects 0.000 description 1
- 230000007954 hypoxia Effects 0.000 description 1
- 229960005236 ibandronic acid Drugs 0.000 description 1
- 229960000908 idarubicin Drugs 0.000 description 1
- 229960001101 ifosfamide Drugs 0.000 description 1
- HOMGKSMUEGBAAB-UHFFFAOYSA-N ifosfamide Chemical compound ClCCNP1(=O)OCCCN1CCCl HOMGKSMUEGBAAB-UHFFFAOYSA-N 0.000 description 1
- 239000012216 imaging agent Substances 0.000 description 1
- 230000001771 impaired effect Effects 0.000 description 1
- 238000000338 in vitro Methods 0.000 description 1
- 230000002779 inactivation Effects 0.000 description 1
- 238000010348 incorporation Methods 0.000 description 1
- 238000009776 industrial production Methods 0.000 description 1
- 208000027866 inflammatory disease Diseases 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- ZPNFWUPYTFPOJU-LPYSRVMUSA-N iniprol Chemical compound C([C@H]1C(=O)NCC(=O)NCC(=O)N[C@H]2CSSC[C@H]3C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@H](C(N[C@H](C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=4C=CC(O)=CC=4)C(=O)N[C@@H](CC=4C=CC=CC=4)C(=O)N[C@@H](CC=4C=CC(O)=CC=4)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C)C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CSSC[C@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CC=4C=CC=CC=4)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCCCN)NC(=O)[C@H](C)NC(=O)[C@H](CCCNC(N)=N)NC2=O)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CSSC[C@H](NC(=O)[C@H](CC=2C=CC=CC=2)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H]2N(CCC2)C(=O)[C@@H](N)CCCNC(N)=N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N2[C@@H](CCC2)C(=O)N2[C@@H](CCC2)C(=O)N[C@@H](CC=2C=CC(O)=CC=2)C(=O)N[C@@H]([C@@H](C)O)C(=O)NCC(=O)N2[C@@H](CCC2)C(=O)N3)C(=O)NCC(=O)NCC(=O)N[C@@H](C)C(O)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@H](C(=O)N[C@@H](CC=2C=CC=CC=2)C(=O)N[C@H](C(=O)N1)C(C)C)[C@@H](C)O)[C@@H](C)CC)=O)[C@@H](C)CC)C1=CC=C(O)C=C1 ZPNFWUPYTFPOJU-LPYSRVMUSA-N 0.000 description 1
- 229960003786 inosine Drugs 0.000 description 1
- 229940079322 interferon Drugs 0.000 description 1
- 229940047124 interferons Drugs 0.000 description 1
- 229940047122 interleukins Drugs 0.000 description 1
- 229940029329 intrinsic factor Drugs 0.000 description 1
- 238000011835 investigation Methods 0.000 description 1
- UWKQSNNFCGGAFS-XIFFEERXSA-N irinotecan Chemical compound C1=C2C(CC)=C3CN(C(C4=C([C@@](C(=O)OC4)(O)CC)C=4)=O)C=4C3=NC2=CC=C1OC(=O)N(CC1)CCC1N1CCCCC1 UWKQSNNFCGGAFS-XIFFEERXSA-N 0.000 description 1
- 229960004768 irinotecan Drugs 0.000 description 1
- 229960003046 isometheptene Drugs 0.000 description 1
- XVQUOJBERHHONY-UHFFFAOYSA-N isometheptene Chemical compound CNC(C)CCC=C(C)C XVQUOJBERHHONY-UHFFFAOYSA-N 0.000 description 1
- YWXYYJSYQOXTPL-SLPGGIOYSA-N isosorbide mononitrate Chemical compound [O-][N+](=O)O[C@@H]1CO[C@@H]2[C@@H](O)CO[C@@H]21 YWXYYJSYQOXTPL-SLPGGIOYSA-N 0.000 description 1
- 229960003827 isosorbide mononitrate Drugs 0.000 description 1
- 229960003299 ketamine Drugs 0.000 description 1
- OZWKMVRBQXNZKK-UHFFFAOYSA-N ketorolac Chemical compound OC(=O)C1CCN2C1=CC=C2C(=O)C1=CC=CC=C1 OZWKMVRBQXNZKK-UHFFFAOYSA-N 0.000 description 1
- 229960004752 ketorolac Drugs 0.000 description 1
- 210000003734 kidney Anatomy 0.000 description 1
- 230000003907 kidney function Effects 0.000 description 1
- 238000013150 knee replacement Methods 0.000 description 1
- 108010053037 kyotorphin Proteins 0.000 description 1
- 150000002597 lactoses Chemical class 0.000 description 1
- 229960002437 lanreotide Drugs 0.000 description 1
- 229960003406 levorphanol Drugs 0.000 description 1
- 229960004194 lidocaine Drugs 0.000 description 1
- 239000002502 liposome Substances 0.000 description 1
- 230000003908 liver function Effects 0.000 description 1
- 229960003837 lypressin Drugs 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 238000005259 measurement Methods 0.000 description 1
- HAWPXGHAZFHHAD-UHFFFAOYSA-N mechlorethamine Chemical compound ClCCN(C)CCCl HAWPXGHAZFHHAD-UHFFFAOYSA-N 0.000 description 1
- 229960004961 mechlorethamine Drugs 0.000 description 1
- 229960001924 melphalan Drugs 0.000 description 1
- SGDBTWWWUNNDEQ-LBPRGKRZSA-N melphalan Chemical compound OC(=O)[C@@H](N)CC1=CC=C(N(CCCl)CCCl)C=C1 SGDBTWWWUNNDEQ-LBPRGKRZSA-N 0.000 description 1
- 229940041616 menthol Drugs 0.000 description 1
- 108020004999 messenger RNA Proteins 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 229960001797 methadone Drugs 0.000 description 1
- 229960000485 methotrexate Drugs 0.000 description 1
- 229960003085 meticillin Drugs 0.000 description 1
- 239000004530 micro-emulsion Substances 0.000 description 1
- 235000019813 microcrystalline cellulose Nutrition 0.000 description 1
- 239000011859 microparticle Substances 0.000 description 1
- 239000004005 microsphere Substances 0.000 description 1
- 238000003801 milling Methods 0.000 description 1
- 229960004857 mitomycin Drugs 0.000 description 1
- KKZJGLLVHKMTCM-UHFFFAOYSA-N mitoxantrone Chemical compound O=C1C2=C(O)C=CC(O)=C2C(=O)C2=C1C(NCCNCCO)=CC=C2NCCNCCO KKZJGLLVHKMTCM-UHFFFAOYSA-N 0.000 description 1
- 229960001156 mitoxantrone Drugs 0.000 description 1
- 239000011259 mixed solution Substances 0.000 description 1
- 210000001616 monocyte Anatomy 0.000 description 1
- 229960005181 morphine Drugs 0.000 description 1
- 210000004877 mucosa Anatomy 0.000 description 1
- 201000006417 multiple sclerosis Diseases 0.000 description 1
- 210000003205 muscle Anatomy 0.000 description 1
- 208000010125 myocardial infarction Diseases 0.000 description 1
- UZHSEJADLWPNLE-GRGSLBFTSA-N naloxone Chemical compound O=C([C@@H]1O2)CC[C@@]3(O)[C@H]4CC5=CC=C(O)C2=C5[C@@]13CCN4CC=C UZHSEJADLWPNLE-GRGSLBFTSA-N 0.000 description 1
- 229960004127 naloxone Drugs 0.000 description 1
- DQCKKXVULJGBQN-XFWGSAIBSA-N naltrexone Chemical compound N1([C@@H]2CC3=CC=C(C=4O[C@@H]5[C@](C3=4)([C@]2(CCC5=O)O)CC1)O)CC1CC1 DQCKKXVULJGBQN-XFWGSAIBSA-N 0.000 description 1
- 229960003086 naltrexone Drugs 0.000 description 1
- 239000002105 nanoparticle Substances 0.000 description 1
- 229960005254 naratriptan Drugs 0.000 description 1
- UNHGSHHVDNGCFN-UHFFFAOYSA-N naratriptan Chemical compound C=12[CH]C(CCS(=O)(=O)NC)=CC=C2N=CC=1C1CCN(C)CC1 UNHGSHHVDNGCFN-UHFFFAOYSA-N 0.000 description 1
- 239000000692 natriuretic peptide Substances 0.000 description 1
- 230000017066 negative regulation of growth Effects 0.000 description 1
- 239000003900 neurotrophic factor Substances 0.000 description 1
- 229960001597 nifedipine Drugs 0.000 description 1
- HYIMSNHJOBLJNT-UHFFFAOYSA-N nifedipine Chemical compound COC(=O)C1=C(C)NC(C)=C(C(=O)OC)C1C1=CC=CC=C1[N+]([O-])=O HYIMSNHJOBLJNT-UHFFFAOYSA-N 0.000 description 1
- 210000002445 nipple Anatomy 0.000 description 1
- 231100000252 nontoxic Toxicity 0.000 description 1
- 231100000956 nontoxicity Toxicity 0.000 description 1
- 102000039446 nucleic acids Human genes 0.000 description 1
- 108020004707 nucleic acids Proteins 0.000 description 1
- 150000007523 nucleic acids Chemical class 0.000 description 1
- 125000005474 octanoate group Chemical group 0.000 description 1
- 125000005473 octanoic acid group Chemical group 0.000 description 1
- 239000002674 ointment Substances 0.000 description 1
- 229960005343 ondansetron Drugs 0.000 description 1
- 229940126701 oral medication Drugs 0.000 description 1
- 150000002894 organic compounds Chemical class 0.000 description 1
- 229940082615 organic nitrates used in cardiac disease Drugs 0.000 description 1
- DWAFYCQODLXJNR-BNTLRKBRSA-L oxaliplatin Chemical compound O1C(=O)C(=O)O[Pt]11N[C@@H]2CCCC[C@H]2N1 DWAFYCQODLXJNR-BNTLRKBRSA-L 0.000 description 1
- 229960001756 oxaliplatin Drugs 0.000 description 1
- 230000003647 oxidation Effects 0.000 description 1
- 238000007254 oxidation reaction Methods 0.000 description 1
- 229960005118 oxymorphone Drugs 0.000 description 1
- XNOPRXBHLZRZKH-DSZYJQQASA-N oxytocin Chemical compound C([C@H]1C(=O)N[C@H](C(N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CSSC[C@H](N)C(=O)N1)C(=O)N1[C@@H](CCC1)C(=O)N[C@@H](CC(C)C)C(=O)NCC(N)=O)=O)[C@@H](C)CC)C1=CC=C(O)C=C1 XNOPRXBHLZRZKH-DSZYJQQASA-N 0.000 description 1
- 229960001723 oxytocin Drugs 0.000 description 1
- 239000006179 pH buffering agent Substances 0.000 description 1
- 239000005022 packaging material Substances 0.000 description 1
- WRUUGTRCQOWXEG-UHFFFAOYSA-N pamidronate Chemical compound NCCC(O)(P(O)(O)=O)P(O)(O)=O WRUUGTRCQOWXEG-UHFFFAOYSA-N 0.000 description 1
- 229940046231 pamidronate Drugs 0.000 description 1
- 229960005489 paracetamol Drugs 0.000 description 1
- BUUKFBVDKSFMHN-LKMAISLMSA-N parathar acetate Chemical compound CC(O)=O.C([C@H](NC(=O)[C@H](CCSC)NC(=O)[C@H](CC(C)C)NC(=O)[C@H](CCC(N)=O)NC(=O)[C@@H](NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](CO)NC(=O)[C@@H](NC(=O)[C@@H](N)CO)C(C)C)[C@@H](C)CC)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC(C)C)C(=O)NCC(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CO)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N[C@@H](CC=1C2=CC=CC=C2NC=1)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@@H](CC(O)=O)C(=O)N[C@@H](C(C)C)C(=O)N[C@@H](CC=1NC=NC=1)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](CC=1C=CC=CC=1)C(O)=O)C1=CN=CN1 BUUKFBVDKSFMHN-LKMAISLMSA-N 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 230000037361 pathway Effects 0.000 description 1
- 239000000312 peanut oil Substances 0.000 description 1
- 230000006320 pegylation Effects 0.000 description 1
- 230000002688 persistence Effects 0.000 description 1
- 239000000825 pharmaceutical preparation Substances 0.000 description 1
- WVDDGKGOMKODPV-ZQBYOMGUSA-N phenyl(114C)methanol Chemical compound O[14CH2]C1=CC=CC=C1 WVDDGKGOMKODPV-ZQBYOMGUSA-N 0.000 description 1
- NBIIXXVUZAFLBC-UHFFFAOYSA-K phosphate Chemical compound [O-]P([O-])([O-])=O NBIIXXVUZAFLBC-UHFFFAOYSA-K 0.000 description 1
- 239000010452 phosphate Substances 0.000 description 1
- WTJKGGKOPKCXLL-RRHRGVEJSA-N phosphatidylcholine Chemical compound CCCCCCCCCCCCCCCC(=O)OC[C@H](COP([O-])(=O)OCC[N+](C)(C)C)OC(=O)CCCCCCCC=CCCCCCCCC WTJKGGKOPKCXLL-RRHRGVEJSA-N 0.000 description 1
- 238000013310 pig model Methods 0.000 description 1
- 239000013612 plasmid Substances 0.000 description 1
- 229920003023 plastic Polymers 0.000 description 1
- 239000004033 plastic Substances 0.000 description 1
- 231100000614 poison Toxicity 0.000 description 1
- 229920000058 polyacrylate Polymers 0.000 description 1
- 239000004584 polyacrylic acid Substances 0.000 description 1
- 229940115272 polyinosinic:polycytidylic acid Drugs 0.000 description 1
- 235000010989 polyoxyethylene sorbitan monostearate Nutrition 0.000 description 1
- 239000001818 polyoxyethylene sorbitan monostearate Substances 0.000 description 1
- 239000003910 polypeptide antibiotic agent Substances 0.000 description 1
- 235000019422 polyvinyl alcohol Nutrition 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 201000009104 prediabetes syndrome Diseases 0.000 description 1
- XOFYZVNMUHMLCC-ZPOLXVRWSA-N prednisone Chemical compound O=C1C=C[C@]2(C)[C@H]3C(=O)C[C@](C)([C@@](CC4)(O)C(=O)CO)[C@@H]4[C@@H]3CCC2=C1 XOFYZVNMUHMLCC-ZPOLXVRWSA-N 0.000 description 1
- 229960004618 prednisone Drugs 0.000 description 1
- 230000002265 prevention Effects 0.000 description 1
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 1
- 230000000770 proinflammatory effect Effects 0.000 description 1
- 229940124272 protein stabilizer Drugs 0.000 description 1
- 230000017854 proteolysis Effects 0.000 description 1
- 238000001959 radiotherapy Methods 0.000 description 1
- 229960004432 raltitrexed Drugs 0.000 description 1
- 239000002464 receptor antagonist Substances 0.000 description 1
- 229940044551 receptor antagonist Drugs 0.000 description 1
- 210000000664 rectum Anatomy 0.000 description 1
- 238000012429 release testing Methods 0.000 description 1
- 238000009256 replacement therapy Methods 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 210000001533 respiratory mucosa Anatomy 0.000 description 1
- 230000000717 retained effect Effects 0.000 description 1
- 239000002336 ribonucleotide Substances 0.000 description 1
- 125000002652 ribonucleotide group Chemical group 0.000 description 1
- 108091092562 ribozyme Proteins 0.000 description 1
- 229940089617 risedronate Drugs 0.000 description 1
- 229960000425 rizatriptan Drugs 0.000 description 1
- TXHZXHICDBAVJW-UHFFFAOYSA-N rizatriptan Chemical compound C=1[C]2C(CCN(C)C)=CN=C2C=CC=1CN1C=NC=N1 TXHZXHICDBAVJW-UHFFFAOYSA-N 0.000 description 1
- 238000005070 sampling Methods 0.000 description 1
- STECJAGHUSJQJN-FWXGHANASA-N scopolamine Chemical compound C1([C@@H](CO)C(=O)O[C@H]2C[C@@H]3N([C@H](C2)[C@@H]2[C@H]3O2)C)=CC=CC=C1 STECJAGHUSJQJN-FWXGHANASA-N 0.000 description 1
- 229960002646 scopolamine Drugs 0.000 description 1
- 229960003440 semustine Drugs 0.000 description 1
- 230000035945 sensitivity Effects 0.000 description 1
- 239000008159 sesame oil Substances 0.000 description 1
- 235000011803 sesame oil Nutrition 0.000 description 1
- 230000035939 shock Effects 0.000 description 1
- 239000001632 sodium acetate Substances 0.000 description 1
- 235000017281 sodium acetate Nutrition 0.000 description 1
- 239000007790 solid phase Substances 0.000 description 1
- 239000002904 solvent Substances 0.000 description 1
- 229940078986 somatuline Drugs 0.000 description 1
- 239000003549 soybean oil Substances 0.000 description 1
- 235000012424 soybean oil Nutrition 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 238000001694 spray drying Methods 0.000 description 1
- 238000007619 statistical method Methods 0.000 description 1
- 230000003637 steroidlike Effects 0.000 description 1
- 150000003431 steroids Chemical class 0.000 description 1
- 238000003756 stirring Methods 0.000 description 1
- 238000003860 storage Methods 0.000 description 1
- 238000005728 strengthening Methods 0.000 description 1
- ZSJLQEPLLKMAKR-GKHCUFPYSA-N streptozocin Chemical compound O=NN(C)C(=O)N[C@H]1[C@@H](O)O[C@H](CO)[C@@H](O)[C@@H]1O ZSJLQEPLLKMAKR-GKHCUFPYSA-N 0.000 description 1
- 229960001052 streptozocin Drugs 0.000 description 1
- 150000003441 suberic acid derivatives Chemical class 0.000 description 1
- GGCSSNBKKAUURC-UHFFFAOYSA-N sufentanil Chemical compound C1CN(CCC=2SC=CC=2)CCC1(COC)N(C(=O)CC)C1=CC=CC=C1 GGCSSNBKKAUURC-UHFFFAOYSA-N 0.000 description 1
- 229960004739 sufentanil Drugs 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- 229960003708 sumatriptan Drugs 0.000 description 1
- KQKPFRSPSRPDEB-UHFFFAOYSA-N sumatriptan Chemical compound CNS(=O)(=O)CC1=CC=C2NC=C(CCN(C)C)C2=C1 KQKPFRSPSRPDEB-UHFFFAOYSA-N 0.000 description 1
- 238000001356 surgical procedure Methods 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 238000009121 systemic therapy Methods 0.000 description 1
- 239000003826 tablet Substances 0.000 description 1
- 229950010168 tauromustine Drugs 0.000 description 1
- 229960000338 teriparatide acetate Drugs 0.000 description 1
- 150000003505 terpenes Chemical class 0.000 description 1
- 229940116411 terpineol Drugs 0.000 description 1
- 229960003604 testosterone Drugs 0.000 description 1
- 229960001196 thiotepa Drugs 0.000 description 1
- 229940104230 thymidine Drugs 0.000 description 1
- 229960000790 thymol Drugs 0.000 description 1
- 229940019375 tiludronate Drugs 0.000 description 1
- 229960005062 tinzaparin Drugs 0.000 description 1
- 229960004394 topiramate Drugs 0.000 description 1
- 239000003440 toxic substance Substances 0.000 description 1
- 231100000419 toxicity Toxicity 0.000 description 1
- 230000001988 toxicity Effects 0.000 description 1
- 231100000041 toxicology testing Toxicity 0.000 description 1
- 239000003053 toxin Substances 0.000 description 1
- 231100000765 toxin Toxicity 0.000 description 1
- CRDAMVZIKSXKFV-UHFFFAOYSA-N trans-Farnesol Natural products CC(C)=CCCC(C)=CCCC(C)=CCO CRDAMVZIKSXKFV-UHFFFAOYSA-N 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 230000032258 transport Effects 0.000 description 1
- 229940117013 triethanolamine oleate Drugs 0.000 description 1
- 102000003298 tumor necrosis factor receptor Human genes 0.000 description 1
- KJIOQYGWTQBHNH-UHFFFAOYSA-N undecanol Chemical compound CCCCCCCCCCCO KJIOQYGWTQBHNH-UHFFFAOYSA-N 0.000 description 1
- 229940035893 uracil Drugs 0.000 description 1
- 208000027185 varicose disease Diseases 0.000 description 1
- 230000000304 vasodilatating effect Effects 0.000 description 1
- 229940124549 vasodilator Drugs 0.000 description 1
- 229940035629 vasopressin and analogues Drugs 0.000 description 1
- 239000003981 vehicle Substances 0.000 description 1
- 210000003462 vein Anatomy 0.000 description 1
- 229960001722 verapamil Drugs 0.000 description 1
- HOFQVRTUGATRFI-XQKSVPLYSA-N vinblastine Chemical compound C([C@@H](C[C@]1(C(=O)OC)C=2C(=CC3=C([C@]45[C@H]([C@@]([C@H](OC(C)=O)[C@]6(CC)C=CCN([C@H]56)CC4)(O)C(=O)OC)N3C)C=2)OC)C[C@@](C2)(O)CC)N2CCC2=C1N=C1[C]2C=CC=C1 HOFQVRTUGATRFI-XQKSVPLYSA-N 0.000 description 1
- 229960003048 vinblastine Drugs 0.000 description 1
- OGWKCGZFUXNPDA-XQKSVPLYSA-N vincristine Chemical compound C([N@]1C[C@@H](C[C@]2(C(=O)OC)C=3C(=CC4=C([C@]56[C@H]([C@@]([C@H](OC(C)=O)[C@]7(CC)C=CCN([C@H]67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)C[C@@](C1)(O)CC)CC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-XQKSVPLYSA-N 0.000 description 1
- 229960004528 vincristine Drugs 0.000 description 1
- OGWKCGZFUXNPDA-UHFFFAOYSA-N vincristine Natural products C1C(CC)(O)CC(CC2(C(=O)OC)C=3C(=CC4=C(C56C(C(C(OC(C)=O)C7(CC)C=CCN(C67)CC5)(O)C(=O)OC)N4C=O)C=3)OC)CN1CCC1=C2NC2=CC=CC=C12 OGWKCGZFUXNPDA-UHFFFAOYSA-N 0.000 description 1
- FKBHRUQOROFRGD-IELIFDKJSA-N vinorelbine Chemical compound C1N(CC=2[C]3C=CC=CC3=NC=22)CC(CC)=C[C@H]1C[C@]2(C(=O)OC)C1=CC([C@]23[C@H]([C@@]([C@H](OC(C)=O)[C@]4(CC)C=CCN([C@H]34)CC2)(O)C(=O)OC)N2C)=C2C=C1OC FKBHRUQOROFRGD-IELIFDKJSA-N 0.000 description 1
- 229960002066 vinorelbine Drugs 0.000 description 1
- 230000009385 viral infection Effects 0.000 description 1
- 230000003612 virological effect Effects 0.000 description 1
- 239000004034 viscosity adjusting agent Substances 0.000 description 1
- 239000007966 viscous suspension Substances 0.000 description 1
- 108010047303 von Willebrand Factor Proteins 0.000 description 1
- 208000012137 von Willebrand disease (hereditary or acquired) Diseases 0.000 description 1
- 230000003313 weakening effect Effects 0.000 description 1
- 238000004260 weight control Methods 0.000 description 1
- 208000016261 weight loss Diseases 0.000 description 1
- 230000004580 weight loss Effects 0.000 description 1
- 230000029663 wound healing Effects 0.000 description 1
- 229960001600 xylazine Drugs 0.000 description 1
- BPICBUSOMSTKRF-UHFFFAOYSA-N xylazine Chemical compound CC1=CC=CC(C)=C1NC1=NCCCS1 BPICBUSOMSTKRF-UHFFFAOYSA-N 0.000 description 1
- XRASPMIURGNCCH-UHFFFAOYSA-N zoledronic acid Chemical compound OP(=O)(O)C(P(O)(O)=O)(O)CN1C=CN=C1 XRASPMIURGNCCH-UHFFFAOYSA-N 0.000 description 1
- 229960004276 zoledronic acid Drugs 0.000 description 1
- 229960001360 zolmitriptan Drugs 0.000 description 1
- UTAZCRNOSWWEFR-ZDUSSCGKSA-N zolmitriptan Chemical compound C=1[C]2C(CCN(C)C)=CN=C2C=CC=1C[C@H]1COC(=O)N1 UTAZCRNOSWWEFR-ZDUSSCGKSA-N 0.000 description 1
Images










Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/44—Oils, fats or waxes according to two or more groups of A61K47/02-A61K47/42; Natural or modified natural oils, fats or waxes, e.g. castor oil, polyethoxylated castor oil, montan wax, lignite, shellac, rosin, beeswax or lanolin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/08—Solutions
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7028—Compounds having saccharide radicals attached to non-saccharide compounds by glycosidic linkages
- A61K31/7034—Compounds having saccharide radicals attached to non-saccharide compounds by glycosidic linkages attached to a carbocyclic compound, e.g. phloridzin
- A61K31/7036—Compounds having saccharide radicals attached to non-saccharide compounds by glycosidic linkages attached to a carbocyclic compound, e.g. phloridzin having at least one amino group directly attached to the carbocyclic ring, e.g. streptomycin, gentamycin, amikacin, validamycin, fortimicins
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/7088—Compounds having three or more nucleosides or nucleotides
- A61K31/713—Double-stranded nucleic acids or oligonucleotides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/70—Carbohydrates; Sugars; Derivatives thereof
- A61K31/715—Polysaccharides, i.e. having more than five saccharide radicals attached to each other by glycosidic linkages; Derivatives thereof, e.g. ethers, esters
- A61K31/716—Glucans
- A61K31/721—Dextrans
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/04—Peptides having up to 20 amino acids in a fully defined sequence; Derivatives thereof
- A61K38/08—Peptides having 5 to 11 amino acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/04—Peptides having up to 20 amino acids in a fully defined sequence; Derivatives thereof
- A61K38/08—Peptides having 5 to 11 amino acids
- A61K38/09—Luteinising hormone-releasing hormone [LHRH], i.e. Gonadotropin-releasing hormone [GnRH]; Related peptides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/04—Peptides having up to 20 amino acids in a fully defined sequence; Derivatives thereof
- A61K38/12—Cyclic peptides, e.g. bacitracins; Polymyxins; Gramicidins S, C; Tyrocidins A, B or C
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/04—Peptides having up to 20 amino acids in a fully defined sequence; Derivatives thereof
- A61K38/14—Peptides containing saccharide radicals; Derivatives thereof, e.g. bleomycin, phleomycin, muramylpeptides or vancomycin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/17—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- A61K38/19—Cytokines; Lymphokines; Interferons
- A61K38/21—Interferons [IFN]
- A61K38/212—IFN-alpha
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/17—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- A61K38/22—Hormones
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/17—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- A61K38/22—Hormones
- A61K38/2214—Motilins
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/17—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- A61K38/22—Hormones
- A61K38/26—Glucagons
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/17—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- A61K38/22—Hormones
- A61K38/27—Growth hormone [GH], i.e. somatotropin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/17—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- A61K38/22—Hormones
- A61K38/28—Insulins
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/17—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- A61K38/22—Hormones
- A61K38/29—Parathyroid hormone, i.e. parathormone; Parathyroid hormone-related peptides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/17—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- A61K38/22—Hormones
- A61K38/30—Insulin-like growth factors, i.e. somatomedins, e.g. IGF-1, IGF-2
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/16—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- A61K38/17—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- A61K38/22—Hormones
- A61K38/31—Somatostatins
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/08—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing oxygen, e.g. ethers, acetals, ketones, quinones, aldehydes, peroxides
- A61K47/12—Carboxylic acids; Salts or anhydrides thereof
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/08—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing oxygen, e.g. ethers, acetals, ketones, quinones, aldehydes, peroxides
- A61K47/14—Esters of carboxylic acids, e.g. fatty acid monoglycerides, medium-chain triglycerides, parabens or PEG fatty acid esters
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/24—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing atoms other than carbon, hydrogen, oxygen, halogen, nitrogen or sulfur, e.g. cyclomethicone or phospholipids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/26—Carbohydrates, e.g. sugar alcohols, amino sugars, nucleic acids, mono-, di- or oligo-saccharides; Derivatives thereof, e.g. polysorbates, sorbitan fatty acid esters or glycyrrhizin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/30—Macromolecular organic or inorganic compounds, e.g. inorganic polyphosphates
- A61K47/32—Macromolecular compounds obtained by reactions only involving carbon-to-carbon unsaturated bonds, e.g. carbomers, poly(meth)acrylates, or polyvinyl pyrrolidone
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/30—Macromolecular organic or inorganic compounds, e.g. inorganic polyphosphates
- A61K47/34—Macromolecular compounds obtained otherwise than by reactions only involving carbon-to-carbon unsaturated bonds, e.g. polyesters, polyamino acids, polysiloxanes, polyphosphazines, copolymers of polyalkylene glycol or poloxamers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K51/00—Preparations containing radioactive substances for use in therapy or testing in vivo
- A61K51/02—Preparations containing radioactive substances for use in therapy or testing in vivo characterised by the carrier, i.e. characterised by the agent or material covalently linked or complexing the radioactive nucleus
- A61K51/04—Organic compounds
- A61K51/08—Peptides, e.g. proteins, carriers being peptides, polyamino acids, proteins
- A61K51/083—Peptides, e.g. proteins, carriers being peptides, polyamino acids, proteins the peptide being octreotide or a somatostatin-receptor-binding peptide
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K51/00—Preparations containing radioactive substances for use in therapy or testing in vivo
- A61K51/02—Preparations containing radioactive substances for use in therapy or testing in vivo characterised by the carrier, i.e. characterised by the agent or material covalently linked or complexing the radioactive nucleus
- A61K51/04—Organic compounds
- A61K51/08—Peptides, e.g. proteins, carriers being peptides, polyamino acids, proteins
- A61K51/10—Antibodies or immunoglobulins; Fragments thereof, the carrier being an antibody, an immunoglobulin or a fragment thereof, e.g. a camelised human single domain antibody or the Fc fragment of an antibody
- A61K51/1021—Antibodies or immunoglobulins; Fragments thereof, the carrier being an antibody, an immunoglobulin or a fragment thereof, e.g. a camelised human single domain antibody or the Fc fragment of an antibody against cytokines, e.g. growth factors, VEGF, TNF, lymphokines or interferons
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K51/00—Preparations containing radioactive substances for use in therapy or testing in vivo
- A61K51/02—Preparations containing radioactive substances for use in therapy or testing in vivo characterised by the carrier, i.e. characterised by the agent or material covalently linked or complexing the radioactive nucleus
- A61K51/04—Organic compounds
- A61K51/08—Peptides, e.g. proteins, carriers being peptides, polyamino acids, proteins
- A61K51/10—Antibodies or immunoglobulins; Fragments thereof, the carrier being an antibody, an immunoglobulin or a fragment thereof, e.g. a camelised human single domain antibody or the Fc fragment of an antibody
- A61K51/1024—Antibodies or immunoglobulins; Fragments thereof, the carrier being an antibody, an immunoglobulin or a fragment thereof, e.g. a camelised human single domain antibody or the Fc fragment of an antibody against hormones, hormone-releasing or hormone-inhibiting factors
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K51/00—Preparations containing radioactive substances for use in therapy or testing in vivo
- A61K51/02—Preparations containing radioactive substances for use in therapy or testing in vivo characterised by the carrier, i.e. characterised by the agent or material covalently linked or complexing the radioactive nucleus
- A61K51/04—Organic compounds
- A61K51/08—Peptides, e.g. proteins, carriers being peptides, polyamino acids, proteins
- A61K51/10—Antibodies or immunoglobulins; Fragments thereof, the carrier being an antibody, an immunoglobulin or a fragment thereof, e.g. a camelised human single domain antibody or the Fc fragment of an antibody
- A61K51/1045—Antibodies or immunoglobulins; Fragments thereof, the carrier being an antibody, an immunoglobulin or a fragment thereof, e.g. a camelised human single domain antibody or the Fc fragment of an antibody against animal or human tumor cells or tumor cell determinants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0031—Rectum, anus
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0053—Mouth and digestive tract, i.e. intraoral and peroral administration
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/10—Dispersions; Emulsions
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/4841—Filling excipients; Inactive ingredients
- A61K9/4858—Organic compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/4841—Filling excipients; Inactive ingredients
- A61K9/4866—Organic macromolecular compounds
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/48—Preparations in capsules, e.g. of gelatin, of chocolate
- A61K9/4891—Coated capsules; Multilayered drug free capsule shells
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/04—Drugs for disorders of the alimentary tract or the digestive system for ulcers, gastritis or reflux esophagitis, e.g. antacids, inhibitors of acid secretion, mucosal protectants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/10—Laxatives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/12—Antidiarrhoeals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/14—Prodigestives, e.g. acids, enzymes, appetite stimulants, antidyspeptics, tonics, antiflatulents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/16—Drugs for disorders of the alimentary tract or the digestive system for liver or gallbladder disorders, e.g. hepatoprotective agents, cholagogues, litholytics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P1/00—Drugs for disorders of the alimentary tract or the digestive system
- A61P1/18—Drugs for disorders of the alimentary tract or the digestive system for pancreatic disorders, e.g. pancreatic enzymes
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P13/00—Drugs for disorders of the urinary system
- A61P13/12—Drugs for disorders of the urinary system of the kidneys
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/04—Anorexiants; Antiobesity agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P3/00—Drugs for disorders of the metabolism
- A61P3/08—Drugs for disorders of the metabolism for glucose homeostasis
- A61P3/10—Drugs for disorders of the metabolism for glucose homeostasis for hyperglycaemia, e.g. antidiabetics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/04—Antibacterial agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/20—Antivirals for DNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/02—Drugs for disorders of the endocrine system of the hypothalamic hormones, e.g. TRH, GnRH, CRH, GRH, somatostatin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/06—Drugs for disorders of the endocrine system of the anterior pituitary hormones, e.g. TSH, ACTH, FSH, LH, PRL, GH
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P5/00—Drugs for disorders of the endocrine system
- A61P5/06—Drugs for disorders of the endocrine system of the anterior pituitary hormones, e.g. TSH, ACTH, FSH, LH, PRL, GH
- A61P5/08—Drugs for disorders of the endocrine system of the anterior pituitary hormones, e.g. TSH, ACTH, FSH, LH, PRL, GH for decreasing, blocking or antagonising the activity of the anterior pituitary hormones
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P7/00—Drugs for disorders of the blood or the extracellular fluid
- A61P7/04—Antihaemorrhagics; Procoagulants; Haemostatic agents; Antifibrinolytic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/08—Vasodilators for multiple indications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/12—Antihypertensives
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/14—Vasoprotectives; Antihaemorrhoidals; Drugs for varicose therapy; Capillary stabilisers
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
- A61K38/04—Peptides having up to 20 amino acids in a fully defined sequence; Derivatives thereof
- A61K38/08—Peptides having 5 to 11 amino acids
- A61K38/095—Oxytocins; Vasopressins; Related peptides
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/16—Agglomerates; Granulates; Microbeadlets ; Microspheres; Pellets; Solid products obtained by spray drying, spray freeze drying, spray congealing,(multiple) emulsion solvent evaporation or extraction
- A61K9/1605—Excipients; Inactive ingredients
- A61K9/1617—Organic compounds, e.g. phospholipids, fats
- A61K9/1623—Sugars or sugar alcohols, e.g. lactose; Derivatives thereof; Homeopathic globules
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Engineering & Computer Science (AREA)
- Epidemiology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Immunology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Organic Chemistry (AREA)
- Gastroenterology & Hepatology (AREA)
- Endocrinology (AREA)
- Zoology (AREA)
- Diabetes (AREA)
- Molecular Biology (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Hematology (AREA)
- Cardiology (AREA)
- Heart & Thoracic Surgery (AREA)
- Virology (AREA)
- Dispersion Chemistry (AREA)
- Nutrition Science (AREA)
- Obesity (AREA)
- Biochemistry (AREA)
- Inorganic Chemistry (AREA)
- Reproductive Health (AREA)
- Physiology (AREA)
- Optics & Photonics (AREA)
- Physics & Mathematics (AREA)
- Biophysics (AREA)
Abstract
composições farmacêuticas, seus processos de produção, bem como forma de dosagem oral. as composições farmacêuticas descritas aqui incluem uma suspensão que compreende uma mistura na forma sólida de uma quantidade terapeuticamente eficaz de um agente terapêutico e pelo menos um sal de um ácido graxo de cadeia média e um meio hidrofóbico, ex, óleo de rícino ou tricaprilato de glicerila ou uma mistura dos mesmos dos mesmos. as composições farmacêuticas descritas aqui contêm sal de ácido graxo de cadeia média e são substancialmente livre de alcoóis. as composições farmacêuticas podem ser encapsuladas em uma cápsula. os métodos de tratamento ou prevenção de doenças pela administração de tais composições para sujeitos afetados também são descritos.
Description
[001] O presente pedido reivindica o benefício de prioridade da U.S. S.N. 61/097,716, depositada em 17 de setembro de 2008, U.S.S.N. 61/141,686, depositada em 31 de dezembro de 2008 e U.S.S.N. 61/161,387, depositada em 18 de março de 2009, cada qual é incorporada neste por referência em sua totalidade.
[002] A presente invenção refere-se de forma geral às composições farmacêuticas que permitem o fornecimento aprimorado, por exemplo, o fornecimento oral e métodos de uso de tais composições.
[003] Técnicas que permitem a transferência eficiente de uma substância de interesse através de uma barreira biológica são de interesse considerável nos campos da biotecnologia e medicina. Por exemplo, tais técnicas podem ser usadas para o transporte de uma variedade de substâncias diferentes através de uma barreira biológica regulada por juntas apertadas (isto é, o epitélio mucoso, que inclui o epitélio intestinal e respiratório, e o endotélio vascular, que inclui the barreira hematoencefálica, membrana nasal, córnea e outras membranas oculares, e membranas genitourinárias). Em específico, existe um grande interesse no fornecimento oral de agentes terapêuticos para evitar o uso de mais meios invasivos de administração e, assim, melhorar a conveniência e aceitação do paciente.
[004] Vários veículos de fornecimento de fármacos já foram utilizados, entre eles lipossomas, nanopartículas lipídicas ou poliméricas, e microemulsões. Esses aprimoraram a biodisponibilidade oral de certos fármacos, principalmente o efeito de proteção que oferecem. No entanto, para os fármacos mais relevantes, a biodisponibilidade permanece muito baixa e não alcança os objetivos terapêuticos mínimos.
[005] Assim, há uma necessidade de um meio eficiente, específico, não invasivo e de baixo risco para destinar várias barreiras biológicas para o fornecimento de vários agentes terapêuticos como peptídios e polipeptídios, fármacos de macromoléculas e outros agentes terapêuticos que incluem pequenas moléculas com baixa biodisponibilidade.
[006] Os inventores da presente invenção descobriram que a absorção de certos agentes terapêuticos em um indivíduo pode ser aprimorada quando administrada em uma composição descrita neste. Por exemplo, um agente terapêutico administrado em fórmula de acordo com uma ou mais modalidades apresenta uma biodisponibilidade aprimorada (BA) com relação ao mesmo agente terapêutico administrado através de uma via similar, porém em uma composição substancialmente livre da cadeia média de componente de sal de ácido graxo descrito neste ou que possui uma quantidade menor da cadeia média de componente de sal de ácido graxo descrita neste. Tal aprimoramento na BA relativa pode ser na ordem de pelo menos cerca de 1,5, 2, 3, 5, 10, 50 ou 100 vezes. Sob alguns aspectos, uma composição descrita neste aprimora a absorção no aparelho digestivo (GI) de um agente terapêutico que é geralmente caracterizado por baixa ou nenhuma biodisponibilidade e/ou absorção oral. Tais agentes terapêuticos podem ter baixa ou nenhuma biodisponibilidade, por exemplo, em solução aquosa, e em outras fórmulas orais conhecidas na técnica. Sob pelo menos um aspecto, uma composição descrita neste aprimora a biodisponibilidade através do aprimoramento da permeabilidade da parede/barreira GI para as moléculas de fármacos. Por exemplo, uma composição descrita neste pode facilitar a absorção através da penetração da parede/barreira GI primariamente através da desselagem de juntas apertadas entre as células epiteliais GI, MS também poderá funcionar com a absorção transcelular. Os inventores presentes criaram um processo para a produção de uma composição farmacêutica (produto de fármaco em massa) que envolve o preparo de uma composição solúvel em água compreendendo uma quantia terapeuticamente eficaz de pelo menos um agente terapêutico e a cadeia média de sal de ácido graxo (e outros ingredientes - vide abaixo), secando (por exemplo, por liofilização) a composição solúvel em água para obter um pó sólido, e a suspensão do material liofilizado (o pó sólido) em um meio hidrofóbico (oleoso), preferencialmente óleo de rícino ou tricaprilato de glicerila (incluindo outros ingredientes, por exemplo, PVP e tensoativos e modificadores de viscosidade - vide abaixo), para produzir uma suspensão contendo em forma sólida o agente terapêutico e a cadeia média de sal de ácido graxo, produzindo, portanto, o produto de fármaco em massa, que deve conter pelo menos 10% em peso de cadeia média de sal de ácido graxo. A forma sólida pode compreender uma partícula (por exemplo, consiste essencialmente de partículas, ou consiste em partículas. A partícula pode ser produzida por liofilização ou por granulação. O produto de fármaco em massa pode então ser encapsulado em cápsulas que serão cobertas por uma camada sensível ao pH e pode ser usada para o fornecimento oral. Um processo típico para a produção da fórmula reivindicada é apresentada na figura 1 , em que a insulina é exemplificada como o ingrediente farmacêutico ativo (API) e a cadeia média de sal de ácido graxo é um octanoato de sódio (Na-C8), também é conhecida como caprilato de sódio.
[007] A presente invenção demonstra o fornecimento do produto ao intestino, que é um modelo para o fornecimento oral, e a partir daí, para a corrente sanguínea com alta biodisponibilidade.
[008] Assim, um aspecto da invenção apresenta uma composição. A composição inclui um agente terapêutico e uma cadeia média de sal de ácido graxo associados a um meio substancialmente hidrofóbico, preferencialmente óleo de rícino, em que o agente terapêutico e a cadeia média de sal de ácido graxo do mesmo estão em forma sólida, por exemplo, na mesma forma sólida como uma partícula, obtida como uma secagem a partir de um meio aquoso, por exemplo, por liofilização do meio aquoso, e em que a cadeia média de sal de ácido graxo é presente em 10 % em peso ou mais, preferencialmente 12 -15%, por exemplo, cerca de 12%, cerca de 13%, cerca de 14%, ou cerca de 15% ou cerca de 16%, ou cerca de 17%, e em que a composição contém outros ingredientes (como descrito neste) porém é substancialmente livre de um "agente fluidizante da membrana". Os "agentes fluidizantes das membranas" são definidos como vários alcoóis de cadeia média cíclica lineares, ramificados, aromáticos, em específico, geraniol e octanol.
[009] As presentes composições da invenção não são emulsões. Quase todas as presentes composições são suspensões oleosas e a quantidade de água em composições é muito baixo; algumas das presentes composições que não são suspensões incorporam uma grande quantidade (cerca de 78% de ácido octanoico) e são soluções.
[010] Em composições da invenção, o agente terapêutico e a cadeia média de sal de ácido graxo têm contato íntimo com o meio substancialmente hidrofóbico. Por exemplo, um pó compreendendo o agente terapêutico e a cadeia média de sal de ácido graxo é coberto, imerso ou suspenso no meio substancialmente hidrofóbico. Durante o processo de produção o meio aquoso que contém o agente terapêutico e a cadeia média de sal de ácido graxo e outros ingredientes é seco (por exemplo, por liofilização) para obter a fração hidrofílica que é um pó (por exemplo, uma forma sólida compreendendo uma pluralidade de partículas), e uma partícula em tal pó que contém todos os ingredientes em que o agente terapêutico e a cadeia média de sal de ácido graxo estejam juntos em uma única partícula. A forma sólida pode ser, por exemplo, uma partícula granulada ou uma partícula liofilizada.
[011] Em algumas modalidades, o agente terapêutico é selecionado a partir de grupo que consiste em peptídios, polissacarídeos, polinucleotídeos, e pequenas moléculas. O agente terapêutico pode ser uma proteína. Por exemplo, o agente terapêutico pode ser insulina. Em outras modalidades, o agente terapêutico é um polinucleotídeo, por exemplo, composto de DNA ou RNA. Em algumas modalidades, o agente terapêutico é uma pequena molécula, um fármaco com pouca solubilidade, ou um fármaco altamente cristalino. O agente terapêutico pode ser um hormônio de crescimento. Em pelo menos uma modalidade, o agente terapêutico é teriparatide. Em algumas modalidades, o agente terapêutico pode ser leuprolide ou alendronato ou octreotídeo.
[012] Em algumas modalidades, a composição inclui uma pluralidade de cadeias médias de sal de ácido graxos e derivados das mesmas. Por exemplo, a partícula sólida pode também incluir uma pluralidade de cadeias médias de sal de ácido graxos e derivados das mesmas.
[013] Em algumas modalidades, a cadeia média de sal de ácido graxo é selecionada a partir de grupo que consiste em hexanoato de sódio, heptanoato de sódio, octanoato de sódio, nonanoato de sódio, decanoato de sódio, undecanoato de sódio, dodecanoato de sódio, tridecanoato de sódio e tetradecanoato de sódio ou uma combinação destes. De acordo com uma ou mais modalidades, a composição é substancialmente livre de decanoato de sódio, tridecanoato de sódio e tetradecanoato de sódio. Em algumas modalidades, a cadeia média de ácidos graxos é octanoato de sódio e o octanoato de sódio é presente em uma concentração acima de 10%, por exemplo, cerca de 11% a cerca de 50% peso/peso (p/p). Em algumas modalidades, o meio substancialmente hidrofóbico compreende um triglicerídeo. Por exemplo, o triglicerídeo pode ser selecionado a partir de grupo que consiste em tributirato de gliceril, mono-oleato de glicerol, monocaprilato de glicerila e tricaprilato de glicerila.
[014] Em algumas modalidades, o meio substancialmente hidrofóbico compreende óleo mineral, óleo de rícino, azeite, óleo de milho, óleo de coco, óleo de amendoim, óleo de soja, óleo de semente de algodão, óleo de gergelim ou óleo de canola, ou combinações dos mesmos.
[015] Em algumas modalidades a composição solúvel em água contém uma cadeia média de sal de ácido graxo e o meio hidrofóbico contém a correspondente cadeia média de ácidos graxos; em algumas modalidades específicas a cadeia média de sal de ácido graxo é um sal de ácido octanoico como octanoato de sódio e a cadeia média de ácidos graxos é ácido octanoico.
[016] Em algumas modalidades, a composição solúvel em água contém a cadeia média de sal de ácido graxo e o meio hidrofóbico contém a correspondente cadeia média de monoglicerídio ou a correspondente cadeia média de triglicerídeo ou uma combinação destes;
[017] em algumas modalidades específicas, a cadeia média de sal de ácido graxo é octanoato de sódio e o monoglicerídio é monocaprilato de glicerila e o triglicerídeo é tricaprilato de glicerila.
[018] Em algumas modalidades, a composição também inclui um ou mais excipientes. Os excipientes pode ser um sal, por exemplo, MgCl2 ou uma amina contendo composto ou manitol. Em algumas modalidades, o excipiente é na mesma forma sólida que o agente terapêutico.
[019] Em algumas modalidades o excipiente é um estabilizador. Os inventores inesperadamente descobriram que apesar do polivinilpirolidina (PVP) em específico, PVP- 12 ser conhecido na técnica como um estabilizador, em fórmulas da invenção age para aumentar o efeito do potenciador de permeabilidade na absorção do agente terapêutico.
[020] Em algumas modalidades, a composição também inclui um ou mais tensoativos. Por exemplo, o tensoativo pode ser selecionado a partir de grupo que consiste em monopalmitato do sorbitano (Span- 40®), mono-oleato de polioxietilenossorbitano (Tween80), lecitina, e mono-oleato de glicerol (GMO). Em uma ou mais modalidades, o tensoativo compreende a partir de cerca de 0,1% a cerca de 6% em peso de a composição.
[021] Em modalidades preferenciais, a composição é em forma de dose oral. Por exemplo, a composição pode ser preenchida em cápsula sólida ou maleável. Em algumas modalidades, a composição é em forma de um supositório. De acordo com uma ou mais modalidades, a composição pode ser em forma de um conjunto de enemas.
[022] Em algumas modalidades, a biodisponibilidade do agente terapêutico, quando administrada a um indivíduo, é de pelo menos 1,5-2% em relação a administração parenteral (subcutânea ou intravenosa). Em algumas modalidades, a composição, quando administrada a um indivíduo, fornece cerca de 2%, cerca de 3%, cerca de 5%, cerca de 10%, ou cerca de 20% ou cerca de 30% absorção do agente terapêutico através de uma barreira biológica. Os níveis de absorção alcançaram a produção dos níveis terapêuticos necessários para a indicação interessada.
[023] Sob um aspecto, a invenção apresenta um método de tratamento de um distúrbio em um indivíduo. O método inclui a administração ao indivíduo de qualquer uma das composições descritas aqui.
[024] Em algumas modalidades, a composição é administrada oralmente. Em outras modalidades, a composição é administrada retalmente, sublingualmente ou através de administração bucal.
[025] Em algumas modalidades, a doença pode ser anemia. De acordo com uma ou mais modalidades, a doença é osteoporose. A doença pode ser infertilidade feminina. Em outras modalidades, a doença é falha de crescimento ou deficiência de hormônio de crescimento. Em pelo menos uma modalidade, a doença é perda de peso, ou enfraquecimento, acromegalia ou diabetes relacionados a HIV.
[026] Em algumas modalidades o agente terapêutico é octreotídeo e a doença é acromegalia, mobilidade de GI anormal, gastroparesia, diarreia ou hipertensão portal.
[027] Em algumas modalidades, o método pode incluir o encapsulamento da suspensão para formar uma cápsula. O método pode também incluir a cobertura da cápsula.
[028] Em algumas modalidades, o método pode incluir o fornecimento de instruções para administrar a cápsula a um indivíduo. As instruções podem se referir à administração da cápsula a um indivíduo para qualquer indicação descrita neste. Sob um aspecto, a invenção apresenta uma cápsula fornecida com instruções relacionadas à administração da cápsula a um indivíduo para qualquer indicação descrita neste.
[029] Ainda outros aspectos, modalidades e vantagens de tais aspectos e modalidades exemplares, são discutidas em detalhes abaixo. Ademais, deve ser compreendido que ambas as informações acima e as descrições que seguem são meramente exemplos ilustrativos de vários aspectos e modalidades, e têm a intenção de fornecer uma visão geral ou base para a compreensão da natureza e caráter dos aspectos e modalidades reivindicados. Os desenhos anexados são incluidos para fornecer ilustrações e uma maior compreensão dos vários aspectos e modalidades, e são incorporados em e constituem parte deste relatório descritivo. Os desenhos, juntamente com o resto deste relatório descritivo, servem para explicar os princípios e operações dos aspectos e modalidades descritos e reivindicados.
[030] Ao longo deste depósito, várias publicações, incluindo patentes dos Estados Unidos, são referenciados por autor e ano, e patentes e aplicações por número. As descrições dessas publicações e patentes e pedidos de patentes em suas totalidades são incorporadas por referência a este pedido, a fim de descrever mais completamente o estado da técnica à qual pertence esta invenção.
[031] Vários aspectos de pelo menos uma modalidade são discutidos abaixo com referência às figuras anexadas. Nas figuras, que não são intecionadas como escalas, cada componente idêntico ou quase idêntico que é ilustrado em várias figuras é representado por um número semelhante. Para maior clareza, nem todo componente pode ser identificado em cada desenho. As figuras são fornecidas para propósitos de ilustração e explicação e não têm a intenção de ser uma definição dos limites da invenção. Nas figuras:
[032] A figura 1 apresenta um processo para a produção de uma fórmula de insulinaa de uma composição de acordo com uma ou mais modalidades como referenciado nos Exemplos anexados;
[033] As figuras 2-5 apresentam dados referenciados nos Exemplos 3 até 6; A figura 6 apresenta dados referenciados no Exemplo 8; A figura 7 apresenta dados da permeabilidade de marcador de peso molecular referenciados no Exemplo 33; A figura 8 apresenta dados da permeabilidade ao longo do tempo referenciados no Exemplo 34; e
[034] As figuras 9 e 10 apresentam dados relacionados a administração de doctereotídeo a macacos referenciados no Exemplo 35.
[035] As composições descritas aqui podem ser administradas a um indivíduo para fornecer a biodisponibilidade aprimorada de um agente terapêutico. Composições farmacêuticas:
[036] As composições farmacêuticas descritas aqui incluem um agente terapêutico e uma cadeia média de sal de ácido graxo em contato íntimo ou associação com um meio substancialmente hidrofóbico. Por exemplo, o agente terapêutico e a cadeia média de ácidos graxos ou derivados do mesmo podem ser cobertos, suspensos, borrifados por ou imerso em um meio substancialmente hidrofóbico formando uma suspensão. As composições da invenção não são emulsões. Quase todas as composições são suspensões oleosas e a quantidade de água em composições é muito baixa; algumas das presentes composições que não são suspensões incorporam uma grande quantidade (cerca de 78% de ácido octanoico) e são soluções por análise visual. A suspensão pode ser uma suspensão líquida incorporando material sólido, ou uma suspensão semissólida incorporando material sólido (uma pomada).
[037] Muitas das composições descritas aqui compreendem uma suspensão que compreende uma mistura de um meio hidrofóbico e a forma sólida em que a forma sólida compreende uma quantia terapeuticamente eficaz de um agente terapêutico e pelo menos um sal de cadeia média de ácidos graxos, e em que a cadeia média de sal de ácido graxo apresenta uma composição em uma quantidade de 10% ou mais por peso. A forma sólida pode compreender uma partícula (por exemplo, consistir essencialmente em partículas, ou consistir em partículas). A partícula pode ser produzida por liofilização ou por granulação. Em algumas modalidades, preferencialmente após a fresagem, 90% (v/v) das partículas estão abaixo de 130 mícrons, e 50% (v/v) das partículas estão abaixo de 45 mícrons.
[038] Um composto de carga é um agente terapêutico (por exemplo, insulina) ou um composto teste (por exemplo, dextrano de grande peso molecular) que é formulado como descrito neste com composições da invenção.
[039] Os inventores foram específicos em incluir em muitas das composições da invenção apenas excipientes que são geralmente reconhecidos como seguros, com base nos dados disponíveis no uso humano, segurança de animais e regras regulatórias (por exemplo, excipientes GRAS). Algumas composições da invenção podem ter outros tipos de excipientes (por exemplo, não GRAS). Em algumas modalidades, as composições da invenção possuem quantidades de excipientes que estão dentro das doses diárias máximas como observado em tais dados disponíveis para cada excipiente específico.
[040] A cadeia média de sal de ácido graxo pode geralmente facilitar ou aumentar a permeabilidade e/ou absorção do agente terapêutico. Em algumas modalidades, a cadeia média de sal de ácido graxos inclui derivados de cadeia média de sal de ácido graxos. O agente terapêutico e a cadeia média de sal de ácido graxo são em forma sólida, por exemplo, a partícula sólida como uma partícula liofilizada, partícula granulada, comprimido ou microesfera. Em modalidades preferenciais, o agente terapêutico e a cadeia média de sal de ácido graxo são ambos na mesma forma sólida, por exemplo, ambos na mesma partícula. Em outras modalidades, o agente terapêutico e a cadeia média de sal de ácido graxo podem cada um ser em uma diferente forma sólida, por exemplo, cada um em uma partícula diferente. As composições descritas aqui são substancialmente livres de qualquer "agente fluidizante das membranas" definido como alcoóis de cadeia média cíclica lineares, ramificadas, aromáticas, em específico, geraniol e octanol. Por exemplo, as composições preferencialmente incluem o agente fluidizante das membranas, porém, certas modalidades podem incluir, por exemplo, menos de 1% ou menos de 0,5% ou menos de 0,1% em peso de agente fluidizante das membranas. Diferente de emulsões, onde a água é um constituinte essencial da fórmula, as composições descritas aqui fornecem uma forma sólida como uma partícula contendo o agente terapêutico, que é então associada com o meio hidrofóbico (oleoso). A quantidade de água em composições é geralmente menos de 3% em peso, geralmente menos de cerca de 2% ou cerca de 1% ou menos por peso.
[041] As composições descritas aqui são suspensões que compreendem uma mistura de um meio hidrofóbico e uma forma sólida em que a forma sólida compreende uma quantia terapeuticamente eficaz de um agente terapêutico e pelo menos um sal de uma cadeia média de ácidos graxos. A forma sólida pode ser uma partícula (por exemplo, consistir essencialmente em partículas, ou consistir em partículas). A partícula pode ser produzida por liofilização ou por granulação. A cadeia média de sal de ácido graxo geralmente é apresentada em composições descritas aqui em uma quantidade de 10% ou mais por peso. Em certas modalidades, a cadeia média de sal de ácido graxo é apresentada em uma composição em uma quantidade de 10%-50%, preferencialmente 11%-18% ou cerca de 11%-17% ou 12%-16% ou 12%-15% ou 13%-16% ou 13%-15% ou 14%-16% ou 14%-15% ou 15%-16% ou mais preferencialmente 15% ou 16% em peso, e a cadeia média de ácidos graxos tem um comprimento de cadeia de cerca de 6 a cerca de 14 átomos de carbono preferencialmente 8, 9 ou 10 átomos de carbono. Em algumas modalidades, em composições descritas sobre, a forma sólida incluindo o agente terapêutico também inclui um estabilizador (por exemplo, um estabilizador da estrutura de proteína). Estabilizadores de estrutura de proteína são compostos que estabilizam a estrutura de proteína sob condições aquosas ou não aquosas ou podem reduzir ou prevenir a agregação do agente terapêutico, por exemplo, durante um processo de secagem como liofilização ou outro passo de processamento. Estabilizadores de estrutura podem ser moléculas polianiônicas, como ácido fítico, íons polivalentes como Ca, Zn ou Mg, sacarídeos como um dissacarídeo (por exemplo, trehalose, maltose) ou um oligo ou polissacarídeo como dextrina ou dextrano, ou a poliálcool como manitol, ou um aminoácido como glicina, ou moléculas policatiônicas, como espermina, ou tensoativos como mono-oleato sorbitano de polioxietileno(Tween 80) ou ácido plurônico. Polímeros descarregados, como manitol, metil celulose e álcool polivinílico, são também estabilizadores adequados.
[042] Apesar de polivinilpirrolidona (PVP) ser conhecido na técnica como um estabilizador, os inventores inesperadamente descobriram que, em composições da invenção descritas aqui, PVP, em específico, PVP- 12, age para aumentar o efeito do potenciador de permeabilidade em uma forma sinergética; ademais, o aumento do nível de PVP- 12 to 10% aumentou a absorção do agente terapêutico no sangue devido a atividade aprimorada das fórmulas. Os inventores demonstraram que o dextrano possui um efeito similar (mas menor) como o PVP. Outra matriz formando polímeros possui um efeito similar.
[043] Em algumas modalidades, como quando o agente terapêutico é a pequena molécula, um agente de crescimento pode ser adicionado, por exemplo, manitol ou glicina.
[044] Em certas modalidades das composições descritas aqui, o agente terapêutico é uma proteína, um polipeptídio, um peptídio, um glicosaminoglicano, uma pequena molécula, um polissacarídeo ou um polinucleotídeo inter alia, como octreotídeo, hormônio de crescimento, hormônio de paratireoide, aminoácidos de hormônio de paratireoide 134 [PTH(l-34) chamado de tereparatídeo], uma heparina de baixo peso molecular ou fondaparinux inter alia. Heparinas de baixo peso molecular são definidas como sais de heparina contendo um peso molecular médio de menos de 8000 Da e para o qual pelo menos 60% de todas as correntes contem um peso molecular menor que 8000 Da.
[045] Em uma modalidade específica das composições descritas aqui, o sal de ácido graxo é octanoato de sódio e o meio hidrofóbico é óleo de rícino; em outra modalidade específica uma composição também compreende mono-oleato de glicerol e monopalmitato do sorbitano ou monocaprilato de glicerila e tricaprilato de glicerila e mono- oleato de polioxietilenossorbitano; em outra modalidade específica a composição também compreende tributirato de glicerila, lecitina, etilisovalerato e pelo menos um estabilizador. Em modalidades específicas, o agente terapêutico é octreotídeo, hormônio de crescimento, hormônio de paratireoide, teriparatide, interferon-alfa (IFN- α), uma heparina de baixo peso molecular, fondaparinux, siRNA, somatostatina e análogos (agonistas) do mesmo, incluindo peptidomiméticos, exenatida, vancomicina ou gentamicina inter alia. Agentes terapêuticos:
[046] As composições farmacêuticas descritas aqui podem ser usadas com uma variedade de agentes terapêuticos (também chamado de ingrediente farmacêutico ativo =API). Em algumas modalidades, a composição farmacêutica inclui uma pluralidade de agentes terapêuticos (causadores). Os agentes terapêuticos podem ser na mesma forma sólida (por exemplo, na mesma partícula), ou os agentes terapêuticos podem cada um ser em uma forma sólida independente (por exemplo, cada um em diferentes partículas. Em algumas modalidades, o agente terapêutico é em forma de uma partícula, por exemplo, uma partícula sólida ou granulada. A partícula é associada com ou está em contato íntimo com um meio substancialmente hidrofóbico, por exemplo, um meio hidrofóbico descrito aqui.
[047] Agentes terapêuticos que podem ser usados em composições descritas aqui incluem qualquer molécula ou composto servindo como, por exemplo, um agente biológico, terapêutico, farmacêutico ou de diagnóstico incluindo um agente de imagem. Os agentes terapêuticos incluem fármacos e outros agentes incluindo, porém não limitado a, aos listados na Farmacopeia dos Estados Unidos e em outras farmacopeias conhecidas. Agentes terapêuticos são incorporados nas fórmulas da invenção sem qualquer modificação química. Agentes terapêuticos incluem proteínas, polipeptídios, peptídios, polinucleotídeos, polissacarídeos e pequenas moléculas.
[048] O termo "pequena molécula" deve ser compreendido como referência a um composto orgânico de pequeno peso molecular que pode ser sinteticamente produzido ou obtido de fontes naturais e tipicamente possui um peso molecular de menos de 2000 Da, ou menos de 1000 Da ou até menos de 600 Da, por exemplo, menos de ou cerca de 550 Da ou menos de ou cerca de 500 Da ou menos de ou cerca de 400 Da; ou cerca de 400 Da a cerca de 2000 Da; ou cerca de 400 Da a cerca de 1700 Da. Exemplos de pequenas moléculas são ergotamina (peso molecular =582 Da), fondaparinux (peso molecular = 1727 Da), leuprolide (peso molecular = 1209 Da), vancomicina (peso molecular = 1449 Da), gentamicina (peso molecular = 478 Da) e doxorrubicina (peso molecular =544). O termo "polinucleotídeo"refere-se a qualquer molécula composta de nucleotídeos de DNA, nucleotídeos de RNA ou uma combinação de ambos os tipos que compreendem dois ou mais de bases guanidina, citosina, timidina, adenina, uracil ou inosina, inter alia. Um polinucleotídeo pode incluir nucleotídeos naturais, nucleotídeos quimicamente modificados e nucleotídeos sintéticos, ou análogos químicos dos mesmos e pode ser de um filamento ou filamento duplo. O termo inclui "oligonucleotídeos" e engloba "ácidos nucleicos".
[049] Por "RNA interferente pequeno"(siRNA) deve ser compreendido uma molécula de RNA (ribonucleotídeo) que diminui ou silencia (previne) a expressão de um gene/ mRNA de sua contraparte endógena ou celular. O termo é entendido como compreendendo "Interferência de RNA" (RNAi), e "RNA de cadeia dupla " (dsRNA).
[050] Por "polipeptídio" deve ser compreendida uma molécula composta de aminoácidos covalentemente ligados e o termo inclui peptídios, polipeptídios, proteínas e peptidomiméticos. Um peptidomimético é um composto contendo elementos estruturais não peptídicos que são capazes de imitar ações biológicas de um paptídeo de origem natural. Algumas das características clássicas dos peptídios como ligações peptídicas enzimaticamente separáveis geralmente não são apresentadas em peptidomiméticos.
[051] O termo "aminoácido"refere-se a uma molécula que consiste em qualquer um dos 10 aminoácidos que ocorrem naturalmente, aminoácidos que tenham sido quimicamente modificados ou aminoácidos sintéticos.
[052] Por "polissacarídeo" deve ser compreendido um polímero linear ou ramificado composto de monossacarídeos covalentemente ligados; a glicose é o monossacarídeo mais comum e existem pelo menos oito unidades de monossacarídeos em um polissacarídeo e geralmente muitos mais. Polissacarídeos possuem uma fórmula geral de Cx(H2O)y em que x é geralmente um número alto entre 200 e 2500. Considerando que as unidades repetidas na base do polímero geralmente dão monossacarídeos de seis carbonos, a fórmula geral também pode ser representada como (C6H10O5)n em que 40<n<3000, isto é, são normalmente entre 40 e 3000 unidades de monossacarídeos em um polissacarídeo.
[053] Um "glicosaminoglicano" é um polissacarídeo que contém amino contendo açúcares.
[054] Agentes terapêuticos aniônicos exemplares incluem polinucleotídeos de várias origens, e, particularmente, de origem humana, viral, animal, eucariótica ou procariótica, vegetal, ou sintética etc., incluindo sistemas para fornecimento de genes terapêuticos. Um polinucleotídeo de interesse pode ser de uma variedade de tamanhos, variando de, por exemplo, um nucleotídeo de único traço para um fragmento de gente ou todo o gene. Pode ser um gene viral ou um plasmídeos. Polinucleotídeos exemplares servindo como agentes terapêuticos incluem sequências de DNA específicas (por exemplo, codificação de genes), sequências de RNA específicas (por exemplo, aptâmeros de RNA, RNA antissenso, RNA interferente curto (siRNA) ou um RNA de inibição específico (RNAi)), poli CPG, ou poli I:C polímeros de polinucleotídeos sintéticos.
[055] Com alternativa, o agente terapêutico pode ser uma proteína, como, por exemplo, uma enzima, um hormônio, uma incretina, um proteoglicano, uma ribozima, uma citocina, um peptídio, uma apolipoproteína, um fator de crescimento, uma molécula bioativa, um antígeno, ou um anticorpo ou fragmentos do mesmo etc. O peptídio pode ser um peptídio pequeno, por exemplo, a partir de cerca de 2 a cerca de 40 aminoácidos, examplos incluem antagonistas receptores de fibriogen (RGD contendo peptídios que são tetrapeptídios contendo um peso molecular médio de cerca de 600. Peptídios exemplares são somatostatina e análogos do mesmo, por exemplo, octreotídeo e lanreotide (Somatuline) que são ambos octapeptídios cíclicos e pasireotide (SOM-230) que é um hexapeptídio cíclico (Weckbecker et al, 2002, Endocrinology 143(10) 4123-4130; Schmid, 2007, Molecular e Cellular Endocrinology 286, 69-74). Outros peptídios exemplares são acetato glatiramer (Copaxone®) que é um tetrapeptídio, terlipressin que é um 12 análogo de aminoácido (agonista) de lisina vasopressin (ADH) e exenatida, um aminoácido peptídio 39 que é um agente mimético de incretina, e outros análogos de peptídio como glucagon- l(GLP-l). (Byetta® é a marca registrada para exenatida (Eli Lilly e Company / Amylin Pharmaceuticals, Inc.). Outros peptídios incluem dalargin que é um hexapeptídio, e quiotorfina que é um dipeptídio. Peptídios incluem hormônio de crescimento liberando peptídios que são peptídios de cerca de 12 aminoácidos ou menos; vide por exemplo peptídios divulagados nas patentes US números 4411890 ( Momany) e 4839344 (Bowers et al)
[056] Exemplos de outros peptídios que podem ser usados na prática desta invenção são descritos na Patente US No. 4589881 (30 ou mais resíduos de aminoácidos) de Pierschbacher et al; Patente US No. 4544500 (20-30 resíduos) de Bittle et al; e EP0204480 ( >34 resíduos) de Dimarchi et al e teriparatide. Em algumas modalidades, o agente terapêutico pode incluir um polissacarídeo, como um glicosaminoglicano. Glicosaminoglicanos exemplares incluem heparina, derivados de heparina, sulfato de heparan, sulfato de condroitina, sulfato de dermatano, e ácido hialurônico. Exemplos de derivados de heparina incluem, porém não são limitados a, heparinas de baixo peso molecular como enoxaparina, dalteparina e tinzaparina. Um agente terapêutico com um efeito similar a heparina é fondaparinux.
[057] Outros exemplos de agentes terapêuticos incluem, porém não são limitados a hormônios como insulina, eritropoietina (EPO), peptídio como glucacon 1 (GLP-I), hormônio simulando melanocito (alfa-MSH), hormônio de paratireoide (PTH), teriparatide, hormônio de crescimento (GH), leuprolida, acetato de leuprolida, fator VIII, hormônio de crescimento liberando hormônio (GHRH), aminoácidos de peptídios YY 3-36 (PYY(3_36)), calcitonina, somatotropina, somatostatina, somatomedina, interleucinas como interleucina-2 (IL-2), alfa-1- antiripsina, granulócito/fator estimulador de colônia de monócito (GM- CSF), fator estimulador de colônia de granulócitos (G-CSF), T20, testosterona, interferons como interferon- alfa (IFN-α) IFN-β e IFN-Y, hormônio luteinizador (LH), hormônio estimulador de foliculos (FSH), gonadotropina crônica humana (hCG), encefalina, dalargina, quiotorfina, fator de crescimento de fibroblasto básico (bFGF), hirudina, hirulógo, hormônio luteinizador liberando hormônio (LHRH), análogo de gonadotropina liberando hormônio (GnRH), peptídio natriurético derivado do cérebro (BNP), ativador de tecido plasminogene (TPA), oxitocina, e análogos e combinações do mesmo. Outros exemplos de agentes terapêuticos incluem, porém não são limitados a agentes analgésicos, agentes antienxaqueca, agentes anticoagulantes, agente antieméticos, cardiovasculares, anti-hipertensivos e agentes vasodilatores, sedativos, antagonistas narcóticos, agentes de quelação, agentes antidiuréticos e agentes antineoplásticos.
[058] Analgésicos incluem, porém não são limitados a, fentanila, sufentanila, butorfanol, buprenorfina, levorfanol, morfina, hidromorfona, hidrocodeína, oximorfona, a metadona, a lidocaína, a bupivacaína, naproxeno, diclofenaco, paverin, e análogos do mesmo. Agentes antienxaqueca incluem, porém não são limitados a, naratriptano, naproxeno, almotriptano, butalbital, frovatriptano, sumatriptano, rizatriptano, acetaminofeno, isometepteno, butorfanol, dicloralfenazona, alcaloides da cravagem como dihidroergotamina e ergotamina, fármacos não esteroides anti-inflamatórias como cetoprofeno (AINEs) e cetorolaco, eletriptano, butorfanol, topiramato, zolmitriptano, aspirina, cafeína e codeína, e análogos e combinações do mesmo. Agentes anticoagulantes incluem, porém não são limitados a heparina, hirudina, heparinas de baixo peso molecular e análogos do mesmo e fondaparinux. Agentes antieméticos incluem porém não são limitados a escopolamina, ondansetron domperidona, etoclopramida, e análogos do mesmo. Agentes cardiovasculares, anti-hipertensivos e vasodilatadores incluem, porém não são limitados a, diltiazem, clonidina, nifedipina, verapamil, isossorbida-5-mononitrato, nitratos orgânicos, nitroglicerina e análogos do mesmo. Sedativos incluem, porém não são limitados a, benzodiazeínas, fenotiozinas e análogos do mesmo. Antagonistas narcóticos incluem, porém não são limitados a, naltrexona, naloxona e análogos do mesmo. Agentes de quelação incluem, porém não são limitados a deferoxamina e análogos do mesmo. Agentes antidiuréticos incluem, porém não são limitados a, desmopressina, vasopressina e análogos (agonistas) do mesmo como terlipressina; o nome comercial da terlipressina é glypressin ®. Agentes antineoplásicos incluem, porém não são limitados a, 5- fluorouracila, bleomicina, vincristina temezolamida, procarbazina, 6-tioguanina hidroxiureia, citarabina, ciclofosfamida, doxorrubicina, alcaloides da vinca, epirrubicina, etoposide, ifosfamida, carboplatina e outros fármacos antineoplásicos baseados em platina (aomo carboplatina (Paraplatin ®, tetraplatin oxaliplatina, aroplatin e transplatin), vimblastina, vinorelbina, clorambucila, bussulfan, mecloretamina, mitomicina, dacarbazina, tiotepa, daunorrubicina, idarubicina, procarbazina mitoxantrona, Al esperamicin, dactinomicina, plicamicina, carmustina, lomustina (CCNU), tauromustina, estreptozocina, melfalano dactinomicina, prednisona, dexametasona 2-clorodeoxiadenosina, citarabina, docetaxel, fludarabina, gemcitabina, herceptin, hidroxiureia, irinotecan, metotrexato, rituxin, tomudex, semustina e topotecano, taxol e comospostos como taxol e análogos e combinações do mesmo.
[059] Exemplos adicionais de agentes terapêuticos incluem, porém não são limitados a fatores de coagulação e fatores neurotróficos, fragmentos anti-TNF de anticorpos e de receptores de TNF.
[060] Agentes terapêuticos também incluem agentes farmaceuticamente ativos selecionados a partir de grupo que consiste em vitamina B 12, um bisfosfonato (por exemplo, pamidronato, fosfato, alendronato, etidronato, tiludronato, ácido zoledrônico risedronato, clodronato de sódio, ou ácido ibandrónico), taxol, caspofungina, ou um antibiótico aminoglicosídeo. Agentes terapêuticos adicionais incluem uma toxina, ou um agente antipatogênico, como um antibiótico (por exemplo, vancomicina), um antiviral, antifúngico, ou um agente antiparasitário. O agente terapêutico pode por si só ser diretamente ativo ou ativado in situ por uma composição, por uma substância distinta, ou pelas condições ambientais.
[061] Em algumas modalidades, a composição pode incluir uma pluralidade de agentes terapêuticos (fármacos de combinação). Por exemplo, a composição pode incluir um fator VIII e vWF, GLP-I e PYY, IFN-α e análogos de nucleotídeos (isto é ribavirina), e alendronato ou insulina e GLP- 1.
[062] Em algumas modalidades, a composição pode incluir a pequena molécula e um peptídio ou proteína. Combinações exemplares incluem uma combinação de IFN-α e análogos de nucleotídeos (isto é ribavirina) para o tratamento da hepatite C, teriparatida e alendronato para o tratamento de doenças ósseas, uma combinação de GH mais os medicamentos para a terapia de HIV (por exemplo, HAART) para simultaneamente tratar uma infecção viral e a lipodistrofia que acompanha a AIDS ou os efeitos colaterais de enfraquecimento da AIDS. Combinações de duas pequenas moléculas podem ser usadas quando uma delas geralmente tem baixa biodisponibilidade ou absorção mesmo se a outra geralmente possuir biodisponibilidade ou absorção eficaz, como alguns antibióticos (por exemplo, uma combinação de vancomicina e gentamicina como um aminoglicosídeo. Combinações exemplares para a prevenção e tratamento de distúrbios metabólicos como obesidade e diabetes incluem também uma combinação de insulina e metformina, rosiglitazona e insulina, GLP-I (ou exenatida) e metformina, e GLP-I (ou exenatida) e rosiglitazona.
[063] Indicações e condições que podem ser tratadas por fondaparinux formulado como descrito neste inclui trombose venosa profunda, substituição de quadril ou joelho e pacientes presos em camas.
[064] Em algumas modalidades das composições descritas aqui, a composição inclui uma combinação de uma proteína ou peptídio com pequenas moléculas que, ou tem ou não tem boa absorção ou biodisponibilidade. Por exemplo, uma composição pode incluir pelo menos um agente terapêutico que pode geralmente ser caracterizado como pouco absorvível ou de pouca biodisponibilidade. Uma composição pode também ser usada para a administração de agentes terapêuticos que são absorvidos no estômago e/ou intestino, porém causam irritação no estômago e/ou intestino e, portanto são difíceis de tolerar. Em tal situação, um indivíduo pode se beneficiar caso a biodisponibilidade do agente terapêutico tenha aumentado e se mais do agente terapêutico foi absorvido diretamente na corrente sanguínea; caso menos do agente terapêutico for administrado haverá claramente mais chances de chance de causar irritação ao estômago e/ou intestino. Assim, composições da invenção são visadas para compreender, portanto dois ou mais agentes terapêuticos.
[065] No geral, a composição pode incluir a partir de cerca de 0,01% a cerca de 50% em peso de o agente terapêutico , por exemplo, cerca de 0,01, 0,02 0,05, 1, 2, 3, 4, 5, 6, 7, 8, 9, 10, 15, 20, 25, 30, 35, 40, 45, ou 50% em peso. O máximo incluído em uma composição é geralmente na faixa de cerca de 6%-33% em peso do agente terapêutico. Em algumas modalidades das composições descritas aqui, a forma sólida incluindo o agente terapêutico também inclui um estabilizador (por exemplo, um estabilizador de estrutura de proteína). Estabilizadores de estrutura de proteína são compostos que estabilizam a estrutura de proteína sob condições aquosas ou não aquosas ou podem reduzir ou prevenir a agregação do agente terapêutico, por exemplo, durante um processo de secagem como liofilização ou outra etapa de processamento. Estabilizadores de estrutura podem ser moléculas polianiônicas, como ácido fítico, íons polivalentes como Ca, Zn ou Mg, sacarídeos como a dissacarídeo (por exemplo, trealose, maltose) ou um oligo ou polissacarídeo como dextrina ou dextrano, ou a poliálcool como manitol, ou um aminoácido como glicina, ou moléculas policatiônicas, como espermina, ou tensoativos como Tween 80 ou Span 40 ou ácido plurônico. Polímeros descarregados, como metil celulose e álcool polivinílico, são também estabilizadores adequados. Cadeia média de sal de ácido graxo:
[066] As composições descritas aqui incluem o sal de uma cadeia média de ácidos graxos ou derivados do mesmo em forma sólida. Por exemplo, o sal de uma cadeia média de ácidos graxos é em forma de uma partícula como uma partícula sólida. Em algumas modalidades, a partícula pode ser caracterizada com uma partícula granulada. Em pelo menos algumas modalidades, a forma sólida pode geralmente resultar de uma secagem por spray ou processo de evaporação. Em modalidades preferenciais, o sal de uma cadeia média de ácidos graxos é na mesma partícula que o agente terapêutico. Por exemplo, o agente terapêutico e o sal de uma cadeia média de ácidos graxos podem ser preparados juntamente através de um primeiro preparo de uma solução como uma solução aquosa compreendendo o agente terapêutico e o sal de uma cadeia média de ácidos graxos e colifilizando a solução para fornecer a forma sólida ou partícula que compreende o agente terapêutico e o sal de uma cadeia média de ácidos graxos (e outros ingredientes). Como descrito acima, as partículas sólidas resultantes estão associadas com um meio hidrofóbico. Por exemplo, as partículas sólidas podem ser suspensas ou imersas em um meio hidrofóbico. Em diferentes modalidades das composições descritas aqui a cadeia média de sal de ácido graxo pode ser na mesma partícula ou em uma partícula diferente da API. Foi descoberto que biodisponibilidade de um composto de carga seria menor se a cadeia média de ácidos graxos estivesse em uma diferente partícula da que o agente terapêutico, isto é haveria biodisponibilidade aprimorada se a cadeia média de sal de ácido graxo e o composto de carga fossem secos após a solubilização conjunta na fração hidrofílica. Acredita-se que se a cadeia média de sal de ácido graxo e o composto de carga forem secos após solubilização conjunta na fração hidrofílica estarão então na mesma partícula no pó final.
[067] A cadeia média de sal de ácido graxo inclui aqueles contendo uma cadeia de carbono com o comprimento a partir de cerca de 6 a cerca de 14 átomos de carbono. Exemplos de sais de ácidos graxos são hexanoato de sódio, heptanoato de sódio, octanoato de sódio (também chamado de caprilato de sódio), nonanoato de sódio, decanoato de sódio, undecanoato de sódio, decanoato de sódio, tridecanoato de sódio, e tetradecanoato de sódio. Em algumas modalidades, a cadeia média de sal de ácido graxo contém um cátion selecionado a partir de grupo que consiste em potássio, lítio, amônia e outros cátions monovalentes, por exemplo, a cadeia média de sal de ácido graxo é selecionada a partir de octanoato de lítio ou octanoato de potássio ou octanoato de arginina ou outros sais monovalentes de cadeia média de ácidos graxos. Os inventores descobriram que o aumento da quantidade de cadeia média de sal de ácido graxo aumentou a biodisponibilidade da fórmula resultante. Em específico, o aumento da quantidade de cadeia média de sal de ácido graxo, em específico, octanoato de sódio, cerca de 10% a uma faixa de cerca de 12% to 15 % aumentou a biodisponibilidade dos agentes terapêuticos nas composições farmacêuticas descritas aqui.
[068] No geral, a quantidade de cadeia média de sal de ácido graxo em composições descritas aqui pode ser de 10% mais a cerca de 50% em peso da composição farmacêutica bruta. Por exemplo, a cadeia média de sal de ácido graxo pode ser apresentada em uma quantidade de cerca de 10% -50%, preferencialmente cerca de 11%-40% mais preferencialmente cerca de 11%-28% em peso por exemplo em cerca de 12%-13%, 13%-14%, 14%-15% , 15%-16%, 16%- 17%, 17%-18%, 18%-19%, 19%-20%, 20%-21%, 21%-22%, 22%-23%, 23%-24%,2 4%- 25%, 25%-26%, 26%-27%, ou 27%-28% em peso da composição farmacêutica bruta. Em outras modalidades, a cadeia média de sal de ácido graxo pode estar presente em uma quantidade de pelo menos cerca de 11%, pelo menos cerca de 11%, pelo menos cerca de 13%, pelo menos cerca de14%, pelo menos cerca de 15% pelo menos cerca de 16%,pelo menos cerca de 17%, pelo menos cerca de 18%, pelo menos cerca de 19%, pelo menos cerca de 20%, pelo menos cerca de 21%, pelo menos cerca de 22%, pelo menos cerca de 23%, pelo menos cerca de 24%, pelo menos cerca de 25%, pelo menos cerca de 26%, pelo menos cerca de 27% ou pelo menos cerca de 28% em peso da composição farmacêutica bruta. Em modalidades específicas a cadeia média de sal de ácido graxo (sódio, potássio, lítio ou sal de amônia ou uma mistura dos mesmos) é presente em cerca de 12% -21% em peso da composição farmacêutica bruta preferencialmente 11%-18% ou cerca de 11%-17% ou 12%-16% ou 12%-15% ou 13%-16% ou 13%- 15% ou 14%-16% ou 14%-15% ou 15%-16% ou mais preferencialmente 15% ou 16%. Em modalidades específicas a cadeia média de sal de ácido graxo (contendo uma cadeia de carbono com o comprimento de a partir de cerca de 6 a cerca de 14 átomos de carbono especificamente 8, 9 ou 10 átomos de carbono) é presente em cerca de 12% -21% em peso de da composição farmacêutica bruta preferencialmente 11 % - 18 % cerca de 11 % - 17 % ou 12%-16% ou 12%-15% ou 13%-16% ou 13%-15% ou 14%-16% ou 14%-15% ou 15%-16% ou mais preferencialmente 15% ou 16%. Em modalidades específicas a cadeia média de sal de ácido graxo (por exemplo sais de ácido octanoico, sais de ácido subérico, sais de ácido gerânico) é presente em cerca de 12% -21% em peso da composição farmacêutica bruta preferencialmente 11%-18% cerca de 11%-17% ou 12%-16% ou 12%-15% ou 13%-16% ou 13%-15% ou 14%-16% ou 14%-15% ou 15%-16% ou mais preferencialmente 15% ou 16%. Em certas modalidades, a cadeia média de sal de ácido graxo é apresentada em um pó sólido em uma quantidade de 50% a 90%, preferencialmente em uma quantidade de 70% a 80%.
[069] Uma modalidade da invenção compreende uma composição compreendendo a suspensão que consiste essencialmente em uma admistura de um meio hidrofóbico e a forma sólida em que a forma sólida compreende uma quantia terapeuticamente eficaz de um agente terapêutico e pelo menos um sal da cadeia média de ácidos graxos, e em que a cadeia média de sal de ácido graxo não é um sal de sódio. O sal pode ser o sal de um outro cátion, por exemplo, lítio, potássio ou amônio; um sal de amônia é preferido.
[070] Em certas modalidades a composição da invenção compreende uma suspensão que compreende uma admistura de um meio hidrofóbico e a forma sólida em que a forma sólida compreende uma quantia terapeuticamente eficaz de um agente terapêutico, pelo menos um sal de a cadeia média de ácidos graxos e uma matriz formando polímero, e em que a matriz formando polímero é apresentam em uma composição em uma quantidade de 3% ou mais por peso. Em certas modalidades a composição compreende uma suspensão que consiste essencialmente em uma admistura de um meio hidrofóbico e a forma sólida em que a forma sólida compreende uma quantia terapeuticamente eficaz de um agente terapêutico, pelo menos um sal de a cadeia média de ácidos graxos e uma matriz formando polímero, e em que a matriz formando polímero é apresentada em uma composição em uma quantidade de 3% ou mais por peso. Em modalidades específicas, a matriz formando polímero é dextrano ou polivinilpirrolidona (PVP). Em modalidades específicas, a polivinilpirrolidona é apresentada em uma composição em uma quantidade de cerca de 2% a cerca de 20% em peso, preferencialmente em uma quantidade de cerca de 3% a cerca de 18 % em peso, mais preferencialmente em uma quantidade de cerca de 5% a cerca de 15 % em peso, mais preferencialmente em uma quantidade de cerca de 10 % em peso. Em certas modalidades específicas, a polivinilpirrolidona é PVP- 12 e/ou possui peso molecular de cerca de 3000. Outra matriz formando polímeros possui um efeito similar em composições da invenção; como matrizes formando polímeros incluem polissacarídeos iônicos (por exemplo, ácido algínico e alginates) ou polissacarídeos neutros (por exemplo dextrano e HPMC), ácido poliacrílico e derivados de ácido polimetacrílico e alcoóis orgânicos de alto peso molecular (por exemplo álcool polivinílico).
[071] É geralmente aceito na técnica de fornecimento de proteínas, polipeptídios e peptídios que inibidores de protease normalmente devem ser adicionados à fórmula para prevenir a degradação de API. No entanto, nas fórmulas da invenção instantânea, não é necessário inibidores de protease. As fórmulas da invenção parecem garantir estabilidade ao agente terapêutico para a degradação de protease dentro do limite de tempo da atividade, isto é, as fórmulas da invenção são aparentemente inibidoras de ambiente para atividade de enzima. Adicionalmente, os inventores executaram um experimento em que o inibidor de protease aprotinina foi adicionado foi adicionado em uma fórmula, o que não promoveu nenhum efeito beneficial na atividade. Um experimento similar foi executado em que o inibidor de protease ácido ε-aminocaproico foi adicionado em uma fórmula, o que também não promoveu nenhum efeito beneficial na atividade. Portanto, em algumas modalidades, a composição farmacêutica descrita neste é substancialmente livre de um inibidor de protease.
[072] Em modalidades da invenção, os compostos gerais, incluindo o agente terapêutico e a cadeia média de sal de ácido graxo são solubilizados em meio aquoso e então secos para produzir um pó. O processo de secagem pode ser alcançado, por exemplo, por liofilização ou granulação. O pó obtido é chamado de "fração hidrofílica". Na fração hidrofílica a água é geralmente apresentada em uma quantidade de menos de 6%. A liofilização pode ser conduzida como apresentado nos Exemplos deste e por métodos conhecidos na técnica, por exemplo, como descrito em Lyophilisation: Introduction e Basic Principles , Thomas Jennings, publicado por Interpharm/CRC Press Ltd (1999, 2002) O liofilizado pode, por exemplo, ser moído (por exemplo, menos de 150 mícrons) ou cimentado em argamassa. Durante a produção industrial, o liofilizado é preferencialmente moído antes da mistura da fração hidrofílica e o meio hidrofóbico de forma a produzir reprodutibilidade de lote para lote. A granulação pode ser conduzida como apresentado nos Exemplos deste e por métodos conhecidos na técnica , por exemplo, como descrito em Granulation, Salman et al , eds, Elsevier (2006) e em Handbook of Pharmaceutical Granulation Technology, 2a edição, Dilip M. Parikh, ed., (2005).
[073] Vários aglutinantes podem ser utilizados no processo de granulação, como celuloses (incluindo celuloses microcristalinas), lactoses (por exemplo, monohidrato de lactose), dextroses, amido e manitol e outros ligantes como descritos nas duas referências anteriores. Meio hidrofóbico: Óleo:
[074] Como descrito acima, em composições da invenção descritas aqui, o agente terapêutico e a cadeia média de sal de ácido graxo estão em contato íntimo ou associação com um meio hidrofóbico. Por exemplo, um ou ambos podem ser cobertos, suspensos, imersos ou de qualquer forma associados a um meio hidrofóbico. Meios hidrofóbicos adequados podem conter, por exemplo, moléculas alifáticas, cíclicas ou aromáticas. Exemplos de um meio hidrofóbico alifático adequado incluem, porém não são limitados a, óleo mineral, monoglicerídios de ácido graxo, diglicerídeos, triglicerídeos, éteres, ésteres, e combinações do mesmo. Exemplos de ácidos graxos adequados são ácido octanoico, ácido decanoico e ácido dodecanoico, também ácidos graxos C7 e C9 e ácidos di-ácidicos como ácido sebácico e ácido subérico, e derivados dos mesmos. Exemplos de triglicerídeos incluem, porém não são limitados a, cadeias lonfas de triglicerídeos, cadeias médias de triglicerídeos, e cadeias curtas de triglicerídeos. Por exemplo, a cadeia longa de triglicerídeo pode ser óleo de rícino ou óleo de coco ou azeite, e a cadeia curta de triglicerídeo pode ser tributirato de gliceril e a cadeia média triglicerídeo pode ser tricaprilato de glicerila. Monoglicerídios são considerados como tensoativos e são descritos abaixo. Ésteres exemplares incluem isovalerato de etil e acetato de butila. Exemplos de um meio hidrofóbico cíclico adequado incluem, porém não são limitados a, terpenoides, colesterol, derivados de colesterol (por exemplo, sulfato de colesterol), e ésteres de colesterol de ácidos graxos. Um exemplo não limitante de um meio hidrofóbico aromático inclui benzoato de benzila. Em algumas modalidades das composições descritas aqui, é desejável que o meio hidrofóbico inclua uma pluralidade de moléculas hidrofóbicas. Em algumas modalidades das composições descritas aqui, o meio hidrofóbico também inclui um ou mais tensoativos (vide abaixo).
[075] Em algumas modalidades das composições descritas aqui, o meio hidrofóbico também inclui um ou mais polímeros adesivos como metilcelulose, etilcelulose, hidroxipropilmetilcelulose (HPMC), ou derivados de poli(acrilato) Carbopol®934P (C934P). Tais polímeros adesivos podem auxiliar na consolidação da fórmula e/ou auxilia a sua aderência em superfícies da mucosa.
[076] As composições desta invenção descritas aqui podem também incluir um agente ativo de superfície. Por exemplo, o agente ativo de superfície pode ser um componente do meio hidrofóbico como descrito acima, e/ou o agente ativo de superfície pode ser um componente de uma forma sólida como descrito acima, por exemplo em uma forma sólida ou partícula que inclui o agente terapêutico.
[077] Agentes ativos de superfície adequados incluem tensoativos iônicos e não iônicos. Exemplos de tensoativos iônicos são lecitina (fosfatidil colina), sais de bile e detergentes. Exemplos de tensoativos não iônicos incluem monoglicerídios, cremofora, um éter de álcool graxo polietileno glicol, a éster de ácido graxo sorbitano, um éster de ácido graxo sorbitano polioxietileno, Solutol HS 15, ou um poloxamer ou uma combinação destes. Exemplos de monoglicerídios são monocaprilato de glicerila (também chamado de mono-octanoato de glicerila), monodecanoato de glixerila, monolaurato de glicerila, monomiristato de glicerila, monoestearato de glicerila, monopalmitato de glicerila, e mono- oleato de glicerol. Exemplos de ésteres de ácido graxo sorbitano incluem monolaurato de sorbitano, mono-oleato de sorbitano, e monopalmitato de sorbitano (Span 40), ou uma combinação destes. Exemplos de ésteres de ácido graxo sorbitano polioxietileno incluem mono-oleato de sorbitano de polioxietileno (Tween 80), monoestearato de sorbitano polioxietileno, monopalmitato de sorbitano polioxietileno ou uma combinação destes. Os preparos comerciais de monoglicerídios usados também contêm várias quantidades de diglicerídeos e triglicerídeos.
[078] Composições descritas aqui incluindo um agente ativo de superfície geralmente incluem menos de cerca de 12% em peso de total de agente ativo de superfície (por exemplo, menos de cerca de 10%, menos de cerca de 8%, menos de cerca de 6%, menos de cerca de 4%, menos de cerca de 2%, ou menos de cerca de 1%). Em modalidades específicas, da invenção a soma total de todos os tensoativos é de cerca de 6%. Métodos de produção das composições farmacêuticas e as composições produzidas:
[079] Também incluídos na invenção estão os métodos de produção das composições descritas aqui. Assim, uma modalidade da invenção é um processo para a produção de composição farmacêutica que compreende o preparo de uma composição solúvel em água compreendendo uma quantia terapeuticamente eficaz de pelo menos um agente terapêutico e uma cadeia média de sal de ácido graxo (como descrito acima), secando a composição solúvel em água para obter um pó sólido, e suspendendo o pó sólido em um meio hidrofóbico para produzir uma suspensão contendo em forma sólida o agente terapêutico e a cadeia média de sal de ácido graxo, produzindo, portanto uma composição farmacêutica, em que a composição farmacêutica contém 10% ou mais por peso de cadeia média de sal de ácido graxo.
[080] Uma modalidade é um processo para a produção de composição farmacêutica que compreende o fornecimento de um pó sólido de uma quantia terapeuticamente eficaz de pelo menos um agente terapêutico e um pó sólido compreendendo a cadeia média de sal de ácido graxo, e a suspensão do pó sólido em um meio hidrofóbico para produzir uma suspensão contendo em forma sólida o agente terapêutico e a cadeia média de sal de ácido graxo, produzindo, portanto uma composição farmacêutica em que a composição farmacêutica contém 10% ou mais por peso de cadeia média de sal de ácido graxo.
[081] Em uma modalidade dos processos e composições descritos aqui, a composição solúvel em água é uma solução aquosa. Em certas modalidades a secagem da composição solúvel em água é alcançada por liofilização ou por granulação. No processo de granulação um ligante pode ser adicionado a uma composição solúvel em água antes da secagem. Em certas modalidades a etapa de secagem remove água suficiente para o conteúdo da água em uma composição farmacêutica ser menor que cerca de 6% em peso, cerca de 5% em peso, cerca de 4% em peso, cerca de 3% ou cerca de 2 % ou cerca de 1% em peso. Em certas modalidades dos processos e composições descritos aqui a etapa de secagem remove água suficiente para o conteúdo da água no pó sólido ser menor que 6% ou 5% ou 4% ou 3% ou preferencialmente menor que 2% em peso. O conteúdo de água é normalmente baixo e a água pode ser adsorvida para a fase sólida durante a liofilização isto é a água pode ser retida por ligações intermoleculares. Em certas modalidades, a composição solúvel em água adicionalmente compreende um estabilizador, por exemplo, metil celulose. Em modalidades preferenciais dos processos e composições descritos aqui, o meio hidrofóbico é óleo de rícino ou tricaprilato de glicerila ou tributirato de gliceril ou uma combinação destes e pode adicionalmente conter ácido octanoico; em certas modalidades o meio hidrofóbico compreende um composto alifático, olefínico, cíclico ou aromático, um óleo mineral, uma parafina, um ácido graxo como ácido octanoico, um monoglicerídio, um diglicerídio, um triglicerídeo, um éter ou um éster, ou uma combinação destes. Em certas modalidades dos processos e composições descritos aqui, o triglicerídeo é uma cadeia londa de triglicerídeo, uma cadeia média de triglicerídeo, preferencialmente tricaprilato de glicerila, ou uma cadeia curta de triglicerídeo, preferencialmente tributirato de glicerila, e a cadeia longa de triglicerídeo é óleo de rícino ou óleo de coco ou uma combinação destes. Em certas modalidades dos processos e composições descritos aqui, o meio hidrofóbico compreende óleo de rícino ou tricaprilato de glicerila ou tributirato de glicerila ou uma combinação ou mistura dos mesmos, e pode adicionalmente compreender ácido octanoico. Em certas modalidades dos processos e composições descritos aqui o meio hidrofóbico compreende tricaprilato de glicerila ou um éster de baixo peso molecular, por exemplo, isovalerato de etila ou acetato de butila. Em certas modalidades dos processos e composições descritos aqui, o componente principal por peso do meio hidrofóbico é óleo de rícino e pode adicionalmente compreender tricaprilato de glicerila. Em certas modalidades dos processos e composições descritos aqui, o componente básico por peso do meio hidrofóbico é tricaprilato de glicerila e pode adicionalmente compreenser óleo de rícino.
[082] Uma fórmula básica é fornecida como modalidade em que o meio hidrofóbico consiste essencialmente em óleo de rícino, mono- oleato de glicerol e tributirato de glicerila; em também uma modalidade da fórmula básica, a fração hidrofílica consiste essencialmente em agente terapêutico, PVP- 12 e octanoato de sódio.
[083] Uma fórmula específica é fornecida como modalidade em que o meio hidrofóbico consiste essencialmente de tricaprilato de glicerila, óleo de rícino, monocaprilato de glicerila, e Tween 80, e a fração hidrofílica consiste essencialmente em agente terapêutico (por exemplo, octreotídeo), PVP- 12 e octanoato de sódio. Outra fórmula específica é fornecida como modalidade em que o meio hidrofóbico compreende tricaprilato de glicerila, óleo de rícino, monocaprilato de glicerila, e Tween 80, e a fração hidrofílica compreende um agente terapêutico (por exemplo, octreotídeo), PVP-12 e octanoato de sódio. Em certas modalidades o meio hidrofóbico consiste essencialmente em tricaprilato de glicerila e em certas modalidades adicionalmente contém óleo de rícino e /ou monocaprilato de glicerila.
[084] Em certas modalidades a composição compreende uma suspensão, que consiste essencialmente em uma mistura de um meio hidrofóbico e uma forma sólida, onde a forma sólida compreende uma quantidade terapeuticamente eficaz de um agente terapêutico e pelo menos um sal de um ácido graxo de cadeia média, e em que a de cadeia média sal de ácido graxo presente na composição de um montante de 10% ou mais de peso. Em certas modalidades do meio hidrofóbico constituído essencialmente de tricaprilato de glicerila e monocaprilato de glicerila; ou os meios hidrofobicos consistem essencialmente em óleo de rícino, tricaprilato de glicerila e monocaprilato de glicerila. Em certas modalidades do meio hidrofóbico compreende um triglicerídeo e um monoglicerídeo e, em certas modalidades específicas o monoglicerídeo tem o mesmo radical de ácidos graxos como as triglicérides. Em algumas destas modalidades triglicerídeos é tricaprilato de glicerila e o monoglicerídeo é monocaprilato de glicerila. Em certas modalidades o meio da cadeia de ácidos graxos de sal na composição solúvel em água tem o mesmo radical de ácidos graxos como o monoglicerídeo de cadeia média ou como os triglicerídeos de cadeia média ou uma combinação dos dois. Em algumas destas modalidades o meio da cadeia de sal de ácido graxo é caprilato de sódio (octanoato de sódio) e monoglicerídeo é monocaprilato de glicerila e os triglicerídeos é tricaprilato de glicerila.
[085] Muitas das composições descritas aqui documento compreendem uma suspensão que compreende uma mistura de um meio hidrofóbico e uma forma sólida, onde a forma sólida compreende uma quantidade terapeuticamente eficaz de um agente terapêutico e pelo menos um sal de ácido graxo de cadeia média, e no qual o sal de ácido graxo de cadeia média presente na composição de um montante de 10% ou mais de peso. O sólido pode ser uma partícula (por exemplo, consistem essencialmente em partículas, ou consiste em partículas). A partícula pode ser produzida por liofilização ou por granulação.
[086] Em uma modalidade particular, a formulação é constituída essencialmente de uma suspensão que compreende uma mistura de um meio hidrofóbico e uma forma sólida, onde a forma sólida compreende uma quantidade terapeuticamente eficaz de um agente terapêutico e cerca de 10-20% de preferência 15% de sal de ácido graxo de cadeia média preferencialmente octanoato de sódio, e cerca de 510% de preferência de 10% PVP-12, e onde o meio hidrofóbico compreende cerca de 20-80%, de preferência 30-70% de triglicerídeos de preferência tricaprilato de glicerila ou tributirato de glicerila ou óleo de rícino ou uma mistura destes, cerca de 3-10% tensoativos, de preferência cerca de 6%, de preferência monocaprilato de glicerila e Tween 80 e cerca de 1% de água, em particular modalidades do agente terapêutico está presente em um montante inferior a 33%, ou menos de 25%, ou menos de 10%, ou menos de 1% ou menos de 0,1%. O sólido pode ser uma partícula (por exemplo, consistem essencialmente de partículas, ou consiste em partículas). A partícula pode ser produzida por liofilização ou por granulação. Em uma modalidade particular o estado sólido pode ser uma partícula e pode ser produzido por liofilização ou por granulação.
[087] Em uma modalidade adicional da formulação consiste essencialmente de uma suspensão que compreende uma mistura de um meio hidrofóbico e uma forma sólida, onde a forma sólida compreende uma quantidade terapeuticamente eficaz de um agente terapêutico e cerca de 10-20% de preferência 15% sal de ácido graxo de cadeia média e de preferência octanoato de sódio cerca de 5 - 10% de preferência de 10% PVP-12, e onde o meio hidrofóbico compreende cerca de 20-80%, de preferência 30-70% de triglicerídeos de cadeia média ou curta, de preferência tricaprilato de glicerila ou tributirato de glicerila, cerca de 0-50% de preferência 0-30% de óleo de rícino, cerca de 3-10% tensoativos, de preferência cerca de 6%, de preferência monocaprilato de glicerila e Tween 80, e cerca de 1% de água, em modalidades especificas do agente terapêutico está presente em um montante inferior a 33%, ou menos de 25%, ou menos de 10%, ou menos de 1% ou menos de 0,1%.
[088] Em uma modalidade particular, a formulação é constituída essencialmente de uma suspensão que compreende uma mistura de um meio hidrofóbico e uma forma sólida, onde a forma sólida compreende uma quantidade terapeuticamente eficaz de um agente terapêutico e octanoato de sódio de cerca de 15% e cerca de 10% PVP- 12; e onde o meio hidrofóbico reúne cerca de 41% tricaprilato de glicerila, cerca de 27% óleo de rícino, cerca de 4% monocaprilato de glicerila, cerca de 2% Tween 80, cerca de 1% de água e 1% ou menos de agente terapêutico; quando o agente terapêutico for octreotida e estiver presente em cerca de 0,058%.
[089] Em outra modalidade específica a formulação consiste essencialmente em uma suspensão que compreende uma mistura de um meio hidrofóbico e um sólido no qual a forma sólida compreende uma quantidade terapeuticamente eficaz de um agente terapêutico e octanoato de sódio cerca de 15% e cerca de 10% PVP-12 e onde o meio hidrofóbico compreende cerca de 68% de tricaprilato de glicerila, cerca de 4% monocaprilato de glicerila, cerca de 2% Tween 80, cerca de 15% octanoato de sódio, cerca de 10% PVP- 12, cerca de 1% de água e menos de 1% de agente terapêutico; quando o agente terapêutico for octreotida e estiver presente em cerca de 0,058%.
[090] Uma modalidade é uma composição que compreende uma suspensão que compreende uma mistura de um meio hidrofóbico e um sólido no qual a forma sólida compreende uma quantidade terapeuticamente eficaz de octreotida e pelo menos um sal de um ácido graxo de cadeia média; em uma modalidade adicional o sal de ácido graxo de cadeia média está presente na composição de um montante de 10% ou mais, em peso, de preferência 15%, em peso, em uma modalidade adicional a forma sólida adicionalmente compreende uma matriz de polímero de formação. Em uma modalidade adicional a matriz polimérica formando é dextran ou polivinilpirrolidona (PVP). Em uma modalidade específica a matriz polimérica formando é polivinilpirrolidona e polivinilpirrolidona está presente na composição de uma quantidade de cerca de 2% a cerca de 20% em peso, de preferência cerca de 10% em peso. Em uma modalidade específica a polivinilpirrolidona é PVP-12 e / ou a polivinilpirrolidona tem um peso molecular de aproximadamente 3000. Em modalidades específicas o meio hidrofóbico é constituído essencialmente por tricaprilato de glicerila e a forma sólida consiste adicionalmente em PVP-12 e octanoato de sódio. Em mais modalidades específicas o meio hidrofóbico adicionalmente consiste em óleo de rícino ou monocaprilato de glicerila ou uma combinação do mesmo e um tensoativo. Em outras modalidades específicas o meio hidrofóbico consiste em tricaprilato de glicerila, monocaprilato de glicerila, e mono-oleato de polioxietileno sorbitano (Tween 80). Em uma modalidade outras o forma sólida consiste essencialmente de octreotida, PVP-12 e octanoato de sódio. Em uma modalidade específica o composição contém cerca de 41 % de tricaprilato de glicerila, cerca de 27% óleo de rícino, cerca de 4% monocaprilato de glicerila, cerca de 2% Tween 80, cerca de 15% octanoato de sódio, cerca de 10% PVP-12, a água cerca de 1% e cerca de 0,058% octreotida. Em outra modalidade específica a composição contém cerca de 68% de tricaprilato de glicerila, cerca de 4% monocaprilato de glicerila, cerca de 2% Tween 80, cerca de 15% octanoato de sódio, cerca de 10% PVP-12, cerca de 1% de água e cerca de 0,058% de octreotida.
[091] Em todas as formulações acima, as percentagens são recitadas peso / peso e a forma sólida pode ser uma partícula (por exemplo, consistem essencialmente em partículas, ou consiste em partículas). As partículas podem ser produzidas por liofilização ou por granulação.
[092] Sob condições normais de armazenamento, a agente terapêutico dentro das formulações da invenção é estável durante um período prolongado de tempo. O estado químico e físico da formulação é estável. Uma vez administrado o agente a intestino terapêutica é protegida contra danos por meio GI desde as formulações são à base de óleo e, portanto, um ambiente separado local é criado no intestino, onde o agente terapêutico é contido em gotículas de óleo, que confere estabilidade in vivo.
[093] Em certas modalidades o processo produz uma composição que consiste essencialmente em um agente terapêutico e um sal de ácido graxo de cadeia média e um meio hidrofóbico. Em modalidades da invenção o pó sólido (forma sólida) é constituído essencialmente de um agente terapêutico e um sal de ácido graxo de cadeia média. Outras modalidades da invenção são composições farmacêuticas produzidas pelo processo de descrever aqui. Em certas composições farmacêuticas que o agente terapêutico é uma proteína, um polipeptídio, um peptídio, um glicosaminoglicano, um polissacarídeo, uma molécula pequena ou um polinucleotídeo e modalidades específica o agente terapêutico é a insulina, hormônio do crescimento, hormônio da paratireoide, a teriparatida, o interferão alfa-(IFN-α), uma heparina de baixo peso molecular, leuprolide, fondaparinux, octreotida, a exenatida, terlipressina, vancomicina ou gentamicina. Modalidades específicas da invenção compreendem uma forma de dosagem oral compreendendo a composição farmacêutica, em especial, uma forma de dosagem oral que é revestimento entérico. Outras modalidades da invenção compreendem uma cápsula contendo as composições da invenção, e em várias modalidades a cápsula é um gel rígido ou uma cápsula de gel macio e, geralmente, a cápsula é de revestimento entérico. Outras concretizações da invenção compreendem uma forma de dosagem retal que compreende a composição farmacêutica, em específica um supositório, ou uma forma de dosagem oral. Um kit com instruções e da forma farmacêutica também está prevista.
[094] O agente terapêutico ou sal de ácido graxo de cadeia média, ou qualquer combinação de agente terapêutico e outros componentes, tais como estabilizadores de proteínas, podem ser preparados em uma solução de mistura (por exemplo, formando uma solução aquosa ou mistura) que pode ser liofilizados juntos e, em seguida, suspenso em um meio hidrofóbico. Outros componentes da composição opcionalmente também podem ser liofilizados ou adicionados durante a reconstituição dos materiais sólidos.
[095] Em algumas modalidades, o agente terapêutico é solubilizado em uma mistura, por exemplo, incluindo um ou mais componentes adicionais tais como sal de ácido graxo de cadeia média, um estabilizador e / ou um agente ativo de superfície, e que o solvente é removido para fornecer um pó sólido resultante (forma sólida), que está suspenso em um meio hidrofóbico. Em algumas modalidades, o agente terapêutico e / ou a sal de ácido graxo de cadeia média podem ser formados em uma partícula granulado que é, então, associada ao meio hidrofóbico (por exemplo suspensos no meio hidrofóbico ou revestidos com a meio hidrofóbico). Em geral, a composições aqui descritos são substancialmente livre de "agentes de fluidização da membrana", tais como alcoóis de cadeia média.
[096] "Os agentes de fluidização da membrana” são definidos como álcoois de cadeia média, que tem um comprimento de cadeia de carbono de 4-15 átomos de carbono (por exemplo, incluindo 5-15, 5-12, 6, 7, 8, 9, 10 ou 11 átomos de carbono ). Por exemplo, um agente de fluidização da membrana pode ser um álcool (por exemplo, saturados ou insaturados) linear, ramificado (por exemplo, saturados ou insaturados), cíclico (por exemplo, saturados ou insaturados), ou aromático. Exemplos de álcoois lineares adequadas incluem, mas não estão limitados a, butanol, pentanol, hexanol, heptanol, octanol, nonanol, decanol, undecanol, dodecanol, tridecanol, tetradecanol, e pentadecanol. Exemplos de álcoois ramificados incluem, mas não estão limitados a, geraniol, farnesol, rhodinol, citronelol. Um exemplo de um álcool cíclico inclui, mas não está limitado a, mentol, terpineol, perílico, mirtenol e álcool. Exemplos de álcoois aromáticos apropriados incluem, mas não estão limitados a, álcool benzílico, ácido 4-hidroxicinâmicos, timol, glicol de estireno, e compostos fenólicos. Exemplos de compostos fenólicos incluem, mas não estão limitados a, fenol, m-cresol e m- clorocresol.
[097] Se desejar, a composição farmacêutica pode também conter pequenas quantidades de substâncias tóxicas não-auxiliares como agentes tamponantes do pH, e outras substâncias como, por exemplo, acetato de sódio e oleato de trietanolamina.
[098] Em pelo menos uma modalidade, um agente terapêutico, tal como uma proteína, pode ser modificado quimicamente para aumentar a sua meia-vida na circulação. Por exemplo, a agente terapêutico pode sofrer um processo como peguilação.
[099] Em algumas modalidades o processo para a produção de uma composição farmacêutica compreende preparar uma composição solúvel em água compreendendo uma quantidade terapeuticamente eficaz de pelo menos um agente terapêutico e um sal de ácido graxo de cadeia média, secagem na composição solúvel em água para obter um pó sólido, e dissolver a pó sólido em uma solução que consiste essencialmente em ácido octanoico, produzindo, assim, a composição farmacêutica, que é uma solução. Em algumas modalidades, a forma sólida pode ser uma partícula (por exemplo, consistem essencialmente em partículas, ou consiste em partículas). Em algumas modalidades, a partícula pode ser produzida por liofilização ou por granulação. Em algumas modalidades deste processo, o ácido octanoico está presente na composição a um nível de cerca de 60% para cerca de 90% ou a um nível de cerca de 70 a cerca de 85% de preferência cerca de 78%. Em algumas modalidades deste processo, o sal de sódio de ácidos graxos é octanoato, em outras modalidades deste processo, o sal de ácido graxo de cadeia média está presente na composição de uma quantidade de cerca de 11% para cerca de 40% em peso ou por uma quantia de cerca de 11% para cerca de 28% em peso ou por uma quantia de cerca de 15% em peso. Em algumas modalidades desse processo a composição adicionalmente compreende uma matriz polimérica formando e modalidades específica deste processo, a matriz polimérica formando é dextran ou polivinilpirrolidona (PVP), em outras modalidades deste processo, a polivinilpirrolidona está presente na composição em uma quantidade de cerca de 2% para cerca de 20% em peso ou por uma quantia de cerca de 5% para cerca de 15%, em peso, de preferência em um montante de cerca de 10% em peso. Em certas modalidades deste processo é a polivinilpirrolidona PVP-12 e / ou tem um peso molecular de aproximadamente 3000. A composição pode incluir tensoativos como descrito acima. Os produtos farmacêuticos desses processos outras são encarnações da invenção, por exemplo, uma composição contendo ácido octanoico em um nível de cerca de 60% para cerca de 90% ou a um nível de cerca de 70 a cerca de 85% de preferência cerca de 78%;. sal de ácido graxo, preferencialmente octanoato de sódio, presente na composição de uma quantidade de cerca de 11% para cerca de 40% em peso ou por uma quantia de cerca de 11% para cerca de 28% em peso ou por uma quantia de cerca de 15% em peso; polímero formando matriz, por exemplo polivinilpirrolidona, de preferência PVP-12, presente na composição de uma quantidade de cerca de 2% a cerca de 20% em peso ou, preferencialmente, um montante de cerca de 5% para cerca de 15%, em peso, de preferência em um montante de cerca de 10% em peso; e tensoativos, como descrito acima. Também pode haver pequenas quantidades de outros componentes hidrofóbicos, como descrito acima.
[0100] Cápsulas: Composições farmacêuticas preferidas são as formas farmacêuticas orais ou supositórios. Exemplos de formas farmacêuticas incluem cápsulas gelatinosas ou vegetarianas, como as cápsulas de amido hidroxilpropil-metilcelulose ("HPMC), revestidos entéricos, que contém o produto de fármaco a granel. As cápsulas que podem ser utilizadas para encapsular as composições desta invenção são conhecidas na técnica e são descritas, por exemplo, em Pharmaceutical Capsules editado por Podczech e Jones, Pharmaceutical Press (2004) e em Hard gelatin capsules today - e tomorrow, 2a edição, Steggeman ed publicado por Capsugel Library (2002).
[0101] Formulações complementares: As composições da invenção podem ser formuladas utilizando outros métodos conhecidos na técnica, por exemplo, conforme descrito nas seguintes publicações: Pharmaceutical Dosage Forms Vols 1-3 ed. Lieberman, Lachman e Schwartz, publicado por Marcel Dekker Inc, New York(1989); Water-insoluble Drug Formulation 2a edição, Liu, editor, publicado por CRC Press, Taylor e Francis Group (2008); Therapeutic Peptides e Proteins: Formulation, Processing e Delivery Systems, 2a edição por Aj ay K. Banga (autor) publicado por CRC Press , Taylor e Francis Group (2006); Protein Formulation e Delivery, 2a edição, McNally e Hasted eds, publicado por Informa Healthcare USA Inc(2008); e Advanced Drug Formulation to Optimize Therapeutic Outcomes, Williams et al eds, publicado por Informa Healthcare USA (2008).
[0102] As composições da invenção podem ser formulados com micropartículas de tecnologia, por exemplo, conforme descrito no Microparticulate Oral Drug Delivery, Gerbre- Selassie ed., publicado por Marcel Dekker Inc (1994) e em Dey et al, Multiparticulate Drug Delivery Systems for Controlled Release, Tropical Journal of Pharmaceutical Research, September 2008; 7 (3): 1067-1075.
[0103] Métodos de tratamento: As composições aqui descritas apresentam entrega por via enteral eficaz de uma substância biologicamente ativa inalterada (ou seja, um agente terapêutico) e, portanto, têm muitos usos. Por exemplo, as composições aqui descritas podem ser usadas no tratamento da diabetes.
[0104] Em especial, insulina para tratar e prevenir os indivíduos (pacientes) que sofrem de diabetes tipo II (profilaxia da diabetes), e para tratar pacientes que sofrem de disglicemia, pré-diabetes e síndrome metabólica e outras condições, pode ser administrada de acordo com um ou mais modalidades da invenção. A síndrome metabólica é uma combinação de distúrbios médicos que aumentam o risco de desenvolver doença cardiovascular e diabetes. A síndrome metabólica é um conjunto de sintomas diferentes: (1) hiperglicemia (resistência à insulina, diabetes tipo II, etc.); (2) diminuição do colesterol HDL, (3) triglicerídeos elevados; (4) a pressão arterial elevada; (5) a obesidade central; e estado pró-inflamatório (6).
[0105] Uma modalidade da invenção é um método de tratamento ou prevenção de um sofrimento indivíduo às condições acima, onde a quantidade de insulina suficiente para tratar a doença é uma baixa dose de insulina formulada dentro das composições da invenção. Baixa dose de insulina é fornecida por menos de 300 ou inferior a 200 unidades por exemplo cápsula 40-200 Unidades por cápsula.
[0106] Terlipressina (ou análogos de vasopressina outros) para o tratamento de indivíduos (pacientes) que sofrem de síndrome hepato-renal (HRS), incluindo HRS I e II, sangramento de varizes esofágicas, hipertensão portal e demais condições pode ser administrada de acordo com uma ou mais modalidades de invenção. Tais formulações terlipressina também podem ser usadas para a profilaxia primária e secundária de hemorragia por varizes. Uma composição da invenção compreende uma suspensão que compreende uma mistura de um meio hidrofóbico e uma forma sólida onde a forma sólida compreende uma quantidade terapeuticamente eficaz de terlipressina (ou análogos de vasopressina outros) e pelo menos um sal de um ácido graxo de cadeia média.
[0107] A exenatida para melhorar o controle glicêmico em indivíduos que sofrem de diabetes tipo II e para tratar outras condições, como a obesidade e para o uso no controle do peso pode ser administrado em conformidade com uma ou mais modalidades da invenção.
[0108] Interferon-alfa para o tratamento de indivíduos portadores de hepatite C crônica e hepatite B crônica e para tratar outras condições, incluindo o câncer pode ser administrado de acordo com uma ou mais modalidades da invenção.
[0109] Copaxone para o tratamento de indivíduos que sofrem de esclerose múltipla e para tratar outras condições, incluindo doenças inflamatórias pode ser administrado de acordo com uma ou mais modalidades da invenção.
[0110] A desmopressina para o tratamento de indivíduos que sofrem de enurese noturna primária, o diabetes insípido (DI) ou distúrbios hemorrágicos (Doença de von Willebrand e Hemopilia A) pode ser administrada de acordo com uma ou mais modalidades da invenção. Preparações desmopressina oral conhecidos na técnica sofrem extremamente baixa biodisponibilidade oral.
[0111] A octreotida foi sintetizada pela primeira vez em 1979, e é uma octapeptídio que imita somatostatina natural farmacologicamente, embora seja um inibidor mais potente do hormônio do crescimento, glucagon e insulina do que a hormônio natural. Octreotida ou outros análogos da somatostatina podem ser administrados de acordo com uma ou mais modalidades da invenção para uso no tratamento ou prevenção de uma doença ou distúrbio em um sofrimento de objeto de uma doença como acromegalia, motilidade gastrointestinal anormal, rubor episódios associados à síndrome carcinoide, Hipertensão portal, uma tumor da glândula endócrina (como carcinoides, vipoma), gastroparesia, diarreia, vazamento de pâncreas ou de uma pseudo-cisto no pâncreas. A diarreia pode resultar de radioterapia ou pode ocorrer, por exemplo, em indivíduos com peptídio intestinal vasoativo tumores secretores (VIPomas). Além disso, os pacientes que se submetem à cirurgia de pâncreas podem sofrer de secreção do pâncreas extrínseca e são vulneráveis ao desenvolvimento de vazamento pancreático ou pseudo-cistos, que podem ser tratados por produtos octreotida da invenção. Algumas modalidades preferidas são direcionados a um método de tratar um objeto com uma doença como acromegalia, mobilidade gastrointestinal anormal, rubor episódios associados à síndrome carcinoide, hipertensão portal, um tumor da glândula endócrina (como carcinoides, vipoma), gastroparesia, diarreia, vazamento pancreático ou um pseudo-cisto no pâncreas, o qual compreende administrar ao indivíduo uma composição da invenção, em que o agente terapêutico é octreotida, em uma quantidade suficiente para tratar o distúrbio. Formulações de octreotida da invenção podem também ser utilizadas para a profilaxia primária e secundária do sangramento de varizes, que pode ser causada por hipertensão portal, as varizes podem ser gástricas ou esofágicas. Outros usos de formulações de octreotida da invenção estão no tratamento de choque hipovolêmico (por exemplo, hemorrágico) ou vasodilatadores (ex. sépticas) de origem, a síndrome hepatorrenal (HRS) de reanimação, cardiopulmonar e hipotensão induzida por anestesia. Outros análogos da somatostatina podem ser utilizados nos métodos e composições em que octreotida é usada.
[0112] Vancomicina (peso molecular 1449 Da) é um antibiótico glicopeptídio utilizado na profilaxia e tratamento de infecções causadas por bactérias Gram-positivas. A indicação inicial para a vancomicina foi para o tratamento de Staphylococcus aureus resistentes à meticilina (MRSA). Vancomicina nunca se tornou um tratamento de primeira linha para Staphylococcus aureus, sendo uma das razões pela qual a vancomicina deve ser administrada por via intravenosa. Os preparos da técnica de vancomicina devem ser administrados por via intravenosa para a terapia sistêmica, desde que a vancomicina não atravesse a mucosa intestinal. É uma grande molécula hidrofílica que fraciona insatisfatoriamente através da mucosa gastrointestinal. A única indicação para a terapia de vancomicina oral é para o tratamento da colite pseudomembranosa onde deve ser administrado por via oral para chegar ao local de infecção no cólon. A Vancomicina para uso no tratamento ou prevenção da infecção em um indivíduo pode ser administrada oralmente para o objeto de acordo com uma ou mais modalidades da invenção. Algumas modalidades preferidas da invenção são dirigidas a um método para tratar ou prevenir uma infecção em um tema que compreende administrar ao indivíduo uma composição da invenção, em que o agente terapêutico é a vancomicina, em quantidade suficiente para tratar ou prevenir a infecção.
[0113] Gentamicina (peso molecular = 478) é um antibiótico aminoglicosídeousado para tratar vários tipos de infecções bacterianas, particularmente aquelas causadas por bactérias gram-negativas. Quando a gentamicina é administrada por via oral nas formulações do estado da técnica, não é sistemicamente ativa. Isto é porque ela não é absorvida de forma apreciável no intestino delgado.
[0114] Além disso, as composições da invenção também podem ser usadas para tratar doenças decorrentes de aterosclerose e a formação de trombos e êmbolos, tais como infarto do miocárdio e acidentes vasculares cerebrais. Especificamente, as composições podem ser usadas para entregar heparina ou heparina de baixo peso molecular ou fondaparinux em todo a epitélio da mucosa.
[0115] As composições desta invenção também podem ser usadas para tratar doenças hematológicas e estados de deficiência, tais como anemia e hipóxia, que são favoráveis à administração de fatores de crescimento hematológicos. As composições da invenção podem ser usadas para fornecer vitamina B 12 em um objeto de alta biodisponibilidade em que o epitélio da mucosa do objeto carece de fator intrínseco suficiente. G-CSF também pode ser administrada de acordo com várias modalidades. Além disso, as composições da presente invenção podem ser usadas para tratar a osteoporose, por exemplo, através da administração por via enteral de teriparatida PTH, calcitonina ou uma ou duas vezes ou mais por dia.
[0116] Hormônio do crescimento humano (hGH) para tratar a deficiência de hormônio de crescimento, em especial nas crianças pode ser administrada de acordo com uma ou mais modalidades. Em algumas modalidades preferidas, uma composição aqui descrita compreendendo o hormônio do crescimento pode ser administrada a um assunto para tratar ou prevenir distúrbios metabólicos e lipídicos, relacionados a, por exemplo, obesidade, obesidade abdominal, hiperlipidemia ou hipercolesterolemia. Por exemplo, uma composição da invenção compreendendo hormônio do crescimento pode ser administrada oralmente, a um indivíduo, assim, tratar a obesidade (por exemplo, a obesidade abdominal). Em algumas modalidades preferidas, uma composição aqui descrita compreendendo hormônio de crescimento é administrada a um indivíduo para tratar ou prevenir lipodistrofia pelo HIV (caquexia pela AIDS), ou para tratar a síndrome de Prader-Willi, a perturbação do crescimento devido a secreção insuficiente de hormônio de crescimento (por exemplo, associada à disgenesia gonadal ou síndrome de Turner), distúrbios do crescimento em crianças pré-púberes com insuficiência renal crônica, e como terapia de substituição em adultos com pronunciada deficiência em hormônio do crescimento. As composições da invenção compreendendo hormônio de crescimento podem ser administradas oralmente, a um indivíduo para promover a cicatrização da ferida e atenuar as respostas catabólicos em queimaduras graves, sepse, politrauma, grandes operações, a pancreatite aguda e fístula intestinal. Muitas outras condições além do crescimento GH causam deficiências pobres, mas os benefícios do crescimento (ganho de altura) são muitas vezes mais pobres do que quando a deficiência do GH é tratada. Exemplos de outras causas de falta que podem ser tratadas com composições da invenção compreendendo o hormônio do crescimento são retardo no crescimento intrauterino, e estatura curta idiopática grave. Outros usos potenciais das composições da invenção compreendendo hormônio de crescimento incluem o tratamento para reverter ou prevenir os efeitos do envelhecimento em idosos, auxiliar de fortalecimento muscular e no tratamento da fibromialgia.
[0117] Algumas modalidades preferidas são direcionadas a um método de tratar uma doença, como obesidade, lipodistrofia do HIV, doença metabólica, ou déficit de crescimento em um assunto que compreende a administração à composição um assunto da invenção em que o agente terapêutico (efetor) é o hormônio do crescimento, em uma quantidade suficiente para tratar o distúrbio.
[0118] Algumas modalidades preferidas são direcionadas a um método de tratamento de um distúrbio ósseo em um tema que compreende administrar ao indivíduo uma composição da invenção, em que o agente terapêutico é o hormônio da paratireoide teriparatida ou, em uma quantidade suficiente para tratar o distúrbio ósseo.
[0119] Algumas modalidades preferidas são direcionadas a um método de tratamento ou prevenção de um distúrbio de coagulação do sangue em um tema que compreende administrar ao indivíduo uma composição da invenção em que o agente terapêutico é a heparina ou derivados de heparina ou fondaparinux, em uma quantidade suficiente para tratar ou evitar o transtorno de coagulação do sangue.
[0120] Leuprolide (agonista de GnRH), formulado em uma modalidade da invenção pode ser entregue para o tratamento da infertilidade feminina (por exemplo, uma ou duas doses diárias), câncer de próstata e doença de Alzheimer.
[0121] Uma modalidade da invenção refere-se a um método de tratar um objeto que sofrem de uma doença ou distúrbio que compreende administrar ao indivíduo uma composição da invenção em uma quantidade suficiente para tratar a doença. Outra modalidade da invenção refere-se a composições da invenção para uso no tratamento de uma doença ou distúrbio em um indivíduo. Outra modalidade da invenção diz respeito à utilização de um agente terapêutico para o fabrico de um medicamento pelo processo da invenção para o tratamento de um distúrbio.
[0122] O regime de dosagem utilizando o composto é selecionado de acordo com uma variedade de fatores, incluindo o tipo, espécie, idade, peso, sexo e condição clínica do paciente, a severidade da condição a ser tratada, a via de administração, a insuficiência renal e hepática função do doente, e os compostos particulares ou sal do mesmo empregado. Um médico ou veterinário normalmente qualificado pode determinar facilmente e prescrever o montante efetivo do fármaco necessário para prevenir, combater ou deter o progresso da doença. Doses orais da presente invenção, quando utilizadas para os efeitos indicados, podem ser fornecidas sob a forma de cápsulas contendo 0.001,0.0025, 0.005, 0.01,0.025, 0.05, 0.1,0.25, 0.5, 1.0, 2.5, 5.0, 10.0, 15.0, 25.0, 50.0 ou 100, 200, 300, 400, 500, 600, 700, 800 ou 1000 mg do agente terapêutico.
[0123] Compostos da presente invenção podem ser administrados em dose única diária, ou a dosagem diária total pode ser administrada em doses divididas por dois, três, quatro, cinco ou seis vezes ao dia. Em algumas modalidades, a composição é administrada em uma dose diária de cerca de 0,01 a cerca de 5000 mg / dia, por exemplo, administrado uma vez ao dia (por exemplo, de manhã ou antes de dormir) ou duas vezes ou mais por dia (por exemplo, de manhã e antes de dormir).
[0124] Um produto representante da invenção é uma formulação com base em API administradas por via oral, cápsulas de revestimento entérico: cada cápsula contém co-API liofilizado com PVP-12 e octanoato de sódio e suspensas em uma hidrofóbica (lipofílica) meio contendo: tricaprilato de glicerila, monocaprilato de glicerila, e Tween 80; em outro produto representante da invenção Óleo de rícino é adicionalmente presente. As composições aqui descritas podem ser administradas a um objeto, ou seja, um animal humano ou um, a fim de tratar o assunto com uma quantidade farmacologicamente ou terapeuticamente eficaz de um agente terapêutico aqui descrito. O animal pode ser um mamífero, por exemplo, um camundongo, rato, porco, cavalo, vaca ou carneiro. Conforme utilizado aqui o termo "quantidade farmacologicamente ou terapeuticamente eficaz" significa que a quantidade de um fármaco ou agente farmacêutico (o agente terapêutico) que irá desencadear a resposta biológica ou médica de um tecido, sistema de animal ou ser humano que está sendo procurado por um pesquisador ou médico.
[0125] As formulações da invenção permitem a incorporação do agente terapêutico para a formulação sem qualquer modificação química do agente terapêutico. Além disso, como mostrado acima, diversos agentes terapêuticos têm sido formulados com sucesso dentro das formulações da invenção, incluindo os polipéptidos, nucleotídeos, pequenas moléculas e proteínas mesmo tamanho médio. Além disso, as formulações da invenção permitem alta flexibilidade no carregamento do agente terapêutico. Capacidade de carga é dependente do agente terapêutico. Até a data, os limites de capacidade de carga não foram alcançados, porém de carga de até 1,5% peso / peso (polipeptídios) e 6% em peso / peso (pequenas moléculas) foi alcançada e maior carga de até 33% está prevista. Finalmente, as formulações da invenção protegem os compostos da carga de inativação no ambiente GI devido, por exemplo, a degradação proteolítica e oxidação.
[0126] A função e as vantagens destes e de outras modalidades vão ser mais bem compreendidas a partir dos exemplos a seguir. Estes exemplos pretendem ser ilustrativo da natureza e não devem ser considerados como a limitação do âmbito dos sistemas e métodos discutidos neste documento.
[0127] Tabela 1A apresenta um exemplo de uma composição, de acordo com uma ou mais encarnações. Mais especificamente, esta composição é uma formulação de insulina. A insulina foi obtida a partir de Diosynth Biotechnology; octanoato de sódio e NaOH da Merck; MgCl2, MC400, Span40, lecitina e óleo de rícino da Spectrum; PVP- 12 da BASF; isovalerato de etilaisovalerato de etila da Merck/Sigma; tributirato de glicerila da Acros/Penta; e mono-oleato glicerol da Abitec Corp. Tabela 1ª de uma composição para uma API (Ingrediente Farmacêutico Ativo), em conformidade com uma ou mais modalidades. Mais especificamente, esta composição é uma formulação de leuprolide. Tabela 1B
de uma composição para uma API (Ingrediente Farmacêutico Ativo), em conformidade com uma ou mais modalidades. Mais especificamente, esta composição é uma formulação de leuprolide. Tabela 1B Continuação...
Continuação...
[0128] Tabela 1C apresenta um exemplo de uma composição para uma API de acordo com uma ou mais encarnações. Mais especificamente, esta composição é uma formulação para produção de dextrana (FD4). O FD4 é marcadas com FITC dextran com peso molecular de 4.4kDa (Sigma, FD4) e esta é a dextrana, que foi utilizada em todo o contexto, salvo indicação em contrário. Esta formulação especial contém óleo de coco (Sigma) em vez de GTB. Tabela 1C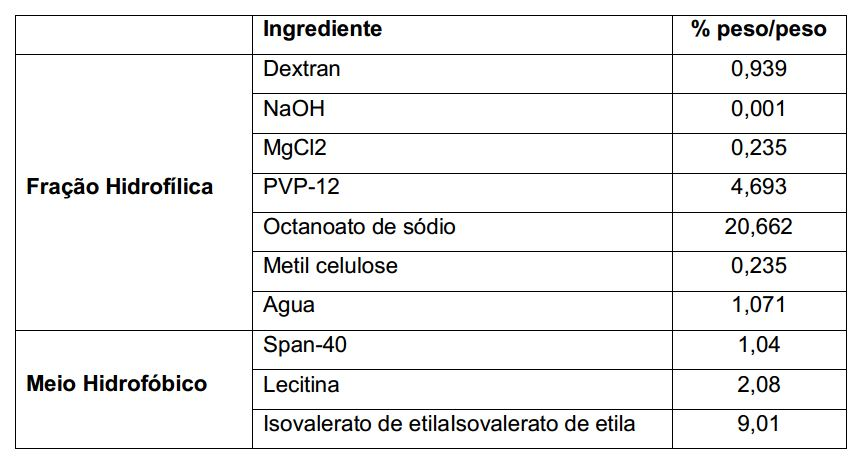 Continuação...
Continuação...
[0129] As formulações acima são utilizadas para uma ampla variedade de agentes terapêuticos e dar boa biodisponibilidade para o complexo de carga em modelos animais descritos abaixo. Note-se que a quantia líquida de agente terapêutico pode variar conforme o caso, em qualquer das formulações, podendo haver pequenas variações nas formulações, por exemplo, NaOH nem sempre é utilizado, óleo de coco pode ser usado em vez de tributirato de glicerila; MgCh nem sempre é utilizado (por exemplo, com o hGH não é utilizado); todos os ingredientes podem ser substituídos conforme acima descrito na especificação.
[0130] figura 1 ilustra um método de produzir uma composição em conformidade com uma ou mais modalidades. Por exemplo, este método pode ser aplicado para produzir as composições apresentadas acima no Exemplo 1.
[0131] figura 2 apresenta os dados relativos aos níveis séricos de insulina após a administração retal de ratos. Os ratos foram anestesiados e foram administrados 100 L da formulação de medicamentos a granel que contenham uma dose de insulina de 328 μg/rato (9 IU/rato). Amostras de sangue foram coletadas às 0, 3, 6, 10, 15, 25, 30, 40, 60 e 90 minutos após a administração de soro e foi preparado para determinação de insulina humana por um kit de imunoensaio sem reatividade cruzada entre ratos e insulina humana.
[0132] Os dados são apresentados como MEIO±DP, n = 5. O painel esquerdo da figura 2 refere-se à administração de insulina humana com octanoato de sódio (Na-C8) ou fração hidrofílica sólidos em suspensão na água (partículas sólidas na água). O painel da direita da figura 2 refere-se à administração da formulação de insulina total (partículas sólidas em meio hidrofóbico). Tabela 2 abaixo apresenta um resumo das AUC valores calculados a partir das curvas de concentração versus tempo. Tabela 2 Dados são MEIO±DP
Dados são MEIO±DP
[0133] A exposição média (expressa por valores AUC) à insulina após a administração retal de insulina SCD-se cerca de 50 vezes maior do que a exposição após a administração sem um meio hidrofóbico. Exposição mínima foi detectada em ratos de insulina administrada com octanoato de sódio sozinho ou como parte das partículas sólidas da fração hidrofílica (conforme listado no Exemplo 1) em suspensão na água. Estes dados demonstram a sinergia entre octanoato de sódio sólido e um meio hidrofóbico.
[0134] figura 3 apresenta os dados relativos aos níveis séricos de insulina e os níveis de glicose no sangue após a administração retal de solução de insulina e insulina na formulação de ratos. Os ratos foram anestesiados e 100 L de artigo de teste foi administrado (na formulação de insulina ou insulina em PBS) contendo uma dose de insulina de 328 μg/rato (9 IU/rato).Amostras de sangue foram coletadas em 0, 3, 6, 10, 15, 25, 30, 40, 60 e 90 minutos pós-administração. Nível de glicose foi imediatamente determinada com um glicosímetro e soro foi preparado para determinação de insulina humana por um kit de imunoensaio sem reatividade cruzada entre ratos e insulina humana.
[0135] Os níveis de glicose são apresentados como a percentagem de níveis de forma basal medida antes da administração (tempo 0). Os dados da figura 3 são apresentados como MEIO±DP, n=5.
[0136] Os níveis de insulina (painel esquerdo na figura 3) e glicose (painel da direita da figura 3) após a administração retal de insulina humana solubilizada em PBS (solução de insulina) ou incorporada na formulação são apresentados. Os níveis de insulina aumentaram rapidamente no soro de ratos após administração retal de insulina na formulação. Os níveis máximos foram medidos após a administração dentro de 6 minutos e uma queda gradual detectados até atingir níveis basais em aproximadamente 90 minutos após a administração. Este aumento acentuado e significativo de insulina foi acompanhado por uma queda significativa nos níveis de glicose atingindo uma média de 20% dos níveis iniciais já aos 30 min após a administração. Em contrapartida, a administração retal de insulina em PBS causada apenas por uma muito ligeira redução da glicose, que é idêntica à observada após o tratamento com o controle PBS sozinho.
[0137] figura 4 apresenta dados relativos a alterações na glicemia e concentrações séricas de insulina após a administração subcutânea (subcutânea), de solução de insulina (20 μg/rato) e administração retal na formulação de insulina (em 328 μg/rato). Amostras de sangue foram coletadas em 0, 3, 6, 10, 15, 25, 30, 40, 60 e 90 minutos após a administração retal e de 0, 15, 30, 45, 60, 90 min, 2, 3 e 4 horas após a administração de SC. A glicose foi determinada imediatamente com um glicosímetro e um kit de insulina por imunoensaio. Os níveis de glicose são apresentados como a percentagem de níveis de forma basal medida antes da administração (tempo 0). Os dados da HG. 4 é apresentado como MEIO±DP, n=5.
[0138] Os níveis de absorção da insulina de cólon em ratos após administração de insulina na formulação foram comparados com os níveis de insulina absorvida após administração SC. Exposição à insulina foi calculada a partir da área sob a curva de concentração plasmática versus tempo (AUC) e a atividade calculada como a biodisponibilidade relativa (RBA), de acordo com a seguinte equação:
[0139] rBA = (AUC(O-*) retal/AUC(O-~) SC) * (dose SC / dose retal)
[0140] Penetração de insulina na corrente sanguínea ocorre durante uma estreita janela de tempo, geralmente dentro de cerca de 10 minutos de insulina retal na administração da formulação. O aumento dos níveis séricos de insulina é comparada a uma queda nos níveis de glicose no sangue.
[0141] A fim de obter informações sobre a biodisponibilidade de insulina quando a insulina formulada é apresentado no cólon, AUQ(O- *) foi determinada para a administração retal e SC e o valor RBA de insulina humana foi de 29,4 3,4% com coeficiente de variação (CV) = 11,4% .
[0142] A administração retal de várias formulações contendo insulina foi realizada em centenas de animais. O ensaio foi desenvolvido e qualificado como um bioensaio para apoiar o desenvolvimento da plataforma e os testes de liberação do lote com uma escala linear de 10 -200 μg/rato, repetibilidade de 39% e a precisão intermediária de 33%.
[0143] A formulação de insulina aqui descrita foi testada em quatro estudos diferentes, utilizando um total de 25 ratos. A rBA foi de 3411 ± 12,6% com CV de 28,9%.
[0144] O local de destino de absorção da plataforma de administração oral da invenção é geralmente o intestino delgado. Para testar a atividade de formulação de insulina em ratos intestino, dois grandes obstáculos foram abordados: 1. Cápsulas de revestimento entérico para os ratos não estão disponíveis e, portanto, desvio no estômago que permite a administração intrajejunal direta é necessária. 2. A insulina é extensivamente metabolizada pelo fígado, em seres humanos 50-80% de insulina endógena, secretada pelas β- células do pâncreas, é sequestrada pelo fígado e, portanto, não pode ser detectado na circulação sistêmica. A insulina administrada por via intestinal (por meio de formulação de insulina) imita o percurso endógeno de insulina, o fluxo sanguíneo intestinal é drenado para a veia portal que leva diretamente para o fígado. Portanto, para determinar a absorbância de insulina, as amostras de sangue devem ser retiradas da veia portal (circulação portal, antes do fígado), bem como a veia jugular (circulação sistêmica, após o fígado).
[0145] Um modelo do rato especializado, no qual três cânulas diferentes são implantadas cirurgicamente em ratos anestesiados foi desenvolvido: 1. Cânula Jejunal - desvio do estômago, permite a administração formulação de insulina, 2. Portal da cânula da veia - coleta de sangue antes do fígado, a insulina determina que atravesse a parede GI no sangue, e 3. Cânula jugular - para determinar os níveis de insulina sistêmica. Usando esse modelo, a biodisponibilidade da insulina na formulação (rBA) foi determinada.
[0146] A figura 5 apresenta dados de um estudo representativo relativos aos níveis de insulina na circulação porta e sistêmica após a administração intrajejunal de controlar a insulina e formulação de insulina em ratos. Ratos (8 ratos por grupo) foram anestesiados e seu jejuno exposto por cirurgia abdominal. O jejuno contendo alça intestinal foi colocado em gaze e mantido úmido e totalmente intacto ao longo de todo o estudo. A cânula temporária foi inserida no jejuno e da insulina formulada foi administrada. O sangue foi coletado a partir de ambas as veias jugulares portal e nos pontos ao mesmo tempo, com cerca de 4 pontos por tempo de rato. O valor de MEIO±DP de cada momento foi usado para criar uma curva de concentração plasmática vs tempo. AUC foi determinado e o rBA foi calculado.
[0147] Os níveis de insulina na circulação portal e sistêmica subiram dramaticamente depois da intra-administração no jejuno de insulina na formulação. Isto está em contraste com a absorção de insulina mínima detectada quando o controle foi administrado por insulina. A janela de absorção foi curta e o pico de níveis de insulina de 6 minutos. Este perfil é semelhante ao observado após a administração retal de insulina formulada (veja acima). Níveis aumentados de insulina foram detectados no portal em comparação com a circulação sistêmica, com a RBA de 10,1% em comparação com 5,6%, respectivamente.
[0148] Tabela 3 detalha os componentes de um conjunto de formulações dextrano que foram preparados como descrito nos exemplos a seguir. O caprato de sódio foi obtido a partir Fluka/Sigma, o azeite de oliva da Fluka, o ácido octanoico da Sigma e o óleo mineral da Acros. Tabela 3ª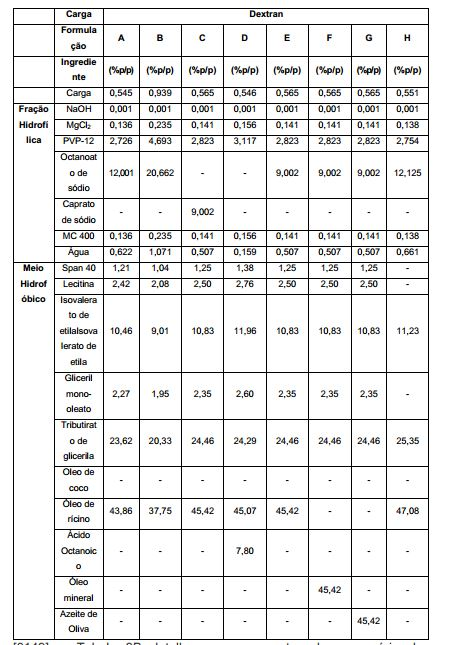
[0149] Tabela 3B detalha os componentes de uma série de formulações de acetato de teriparatida e leuprolide que foram preparados como descrito nos exemplos a seguir. A teriparatida foi obtida de Novetide, e o leuprolide foi obtido de Bambio. Tabela 3B
[0150] Tabela 3C detalha os componentes de formulações hGH que foram preparados como descrito e os exemplos a seguir. O hormônio do crescimento foi obtido a partir de PLR, Israel (GHP-24). Tabela 3C
[0151] O processo de produção de todas estas formulações acima é, essencialmente, como descrito na figura 1 e no Exemplo 11.
[0152] O efeito do aumento da quantidade de octanoato de sódio (Na-C8) na formulação sobre a atividade da formulação foi testado em formulações contendo dextrano (MW médio = 4,4 kDa, FITC) como composto de carga e de diferentes doses de Na-C8 a saber , Uma formulação na Tabela 3A (que contém octanoato de sódio 12% em peso) e dextran semelhantes formulações contendo diferentes doses Na C8: 9%, 6% e 3%, respectivamente.
[0153] Para testar a atividade dessas formulações no jejuno de ratos não anestesiados, um modelo do rato foi estabelecida, na qual duas cânulas diferentes são implantados cirurgicamente em ratos machos Sprague-Dowley 1 - Cânula jejunal para desvio do estômago e permitir a administração formulação direta para o jejuno. 2 - Cânula jugular, para determinação dos níveis sistemáticos da dextrana administrada após a administração jejunal. Ratos são autorizados a recuperação por 4 dias antes do estudo e são privados de alimentos por 18 horas antes do início do estudo.
[0154] A figura 6 apresenta dados de um estudo que determina dextran marcado com FITC (4,4 kDa) biodisponibilidade em ratos não anestesiados após a administração intrajejunal de formulações contendo diferentes quantidades de Na-C8 ou dextran marcado com FITC solubilizado com o C-Na 8 em solução solução salina (controle).
[0155] A biodisponibilidade das formulações diferentes dextran e o controle foi avaliada pela administração das diferentes formulações diretamente para o jejuno de ratos não anestesiados e os valores de dextrana de medição de plasma, 3, 6, 10, 25, 60 e 90 minutos após a administração. Níveis de dextran plasma após a administração de dextrana na formulação ou em solução solução salina foram comparados com os níveis de dextran plasma após a administração intravenosa. Valores de exposição, a AUC (0-90), foi determinada para a administração intravenosa e jejunal e a biodisponibilidade absoluta (ABA) foi calculado de acordo com a seguinte equação: aBA = (AUC jejunal (0-90)) / (iv AUC (0 - 90)) * (iv dose / dose jejunal). Os dados são apresentados como MEIO ± DP (n > 5 animais por grupo).
[0156] Os resultados mostram que o aumento da quantidade de Na- C 8 incorporados na formulação melhora a biodisponibilidade da dextrana de maneira dose-resposta, atingindo quase 30% no ABA 12% (p / p) a dose. Dextran administrado com Na-C8 em doses similares e suspensas em uma solução solução salina (ou seja, não formulou) apresentaram biodisponibilidade muito baixa (-6% ABA). Mais resultados de dose-responsiva são mostradas no Exemplo 26.
[0157] O efeito sobre a atividade de formulação de mudar a proporção (peso/peso) entre a fração hidrofílica e hidrofóbica do meio foi testado em formulações contendo dextrano (MW médio = 4,4 kDa, FITC) como carga (formulações A e B na Tabela 3A). O modelo em rato vivo não anestesiado descrito no Exemplo 8 foi utilizado para comparar a atividade das formulações descritas.
[0158] Tabela 4 apresenta os dados de biodisponibilidade, após administração intrajejunal de formulações compreendendo uma relação diferente da fração hidrofílica para hidrofóbica média. Tabela 4
[0159] As formulações A e B oram administradas d iretamente para o jejuno de ratos não anestesiados e os valores de dextrana plasma foram medidos em 3, 6, 10, 25, 60 e 90 minutos pós-administração de formulação. Os níveis de absorção de dextran jejuno de ratos após a administração de dextrana na formulação foram comparados com os níveis de dextran absorvida após a administração intravenosa. Valores de exposição, a AUC (0-90), foram determinados para a administração intravenosa e jejunal e a biodisponibilidade absoluta (ABA) foi determinado segundo a seguinte equação: ABA = Gejunal AUC (0-90)) / (iv AUC (0-90) ) * (iv dose / dose jejunal). Os dados são apresentados como Mean + SD (n > 5 ratos por grupo). Os resultados mostram que a mudança da relação entre a fração hidrofílica e hidrofóbica no meio dessas formulações, com uma % em peso baixo de agente terapêutico não teve efeito significativo sobre a biodisponibilidade da carga, o que dá uma flexibilidade na elaboração de formulações de carga adicional.
[0160] A fim de testar a capacidade da plataforma de formulação, a atividade de formulações contendo três compostos diferentes de carga (APIs), foi testado em três diferentes modelos animais: a administração jejunal em ratos não-anestesiados, a administração retal de ratos anestesiados e administração jejunal a porcos não-anestesiados.
[0161] A Tabela 5 resume os resultados de experimentos representantes testando a biodisponibilidade de formulações contendo diferentes APIs nos três diferentes modelos animais descritos acima. Tabela 5 * BA Absoluto (comparado a IV) ** BA Relativo (comparado a SC) A. Absorção de Leuprolide após a administração jejunal de leuprolide na formulação de ratos.
* BA Absoluto (comparado a IV) ** BA Relativo (comparado a SC) A. Absorção de Leuprolide após a administração jejunal de leuprolide na formulação de ratos.
[0162] Tabela 5-III apresenta os dados de um estudo representativo relacionados a leuprolide % aBA após a administração IV (intravenosa) solução de leuprolide (em 75 μg/kg) e administração jejunal de leuprolide na formulação (a 450 μg/kg; formulação K, tabela 3B) de ratos não anestesiados, como descrito anteriormente no Exemplo 8.
[0163] Amostras de sangue foram retiradas da veia jugular, 3, 6, 10, 15, 25, 40, 60 e 90 minutos após a administração jejunal e aos 3, 10, 25, 40, 90 min, 2 horas, 3,3 e 5 após a administração IV , O plasma foi preparado e os níveis de leuprolide foram determinados em cada amostra. Níveis de Leuprolide na circulação sistêmica aumentou drasticamente após a administração jejunal de leuprolide na formulação. Pico de níveis sanguíneos de Leuprolide de 3 minutos após a administração. A ABA média obtida após administração jejunal de leuprolide na formulação foi calculado como descrito nos exemplos acima, foi de 10,1%. Em um experimento de controle, administração jejunal de leuprolide em PBS demonstrado penetração insignificante para a corrente sanguínea.
[0164] Uma formulação leuprolide similares contendo de 12% octanoato de sódio conforme descrito na Tabela 1B foi preparado, foi testado no modelo acima referido e que apresentou biodisponibilidade do seguinte modo:
[0165] rBA (em comparação com SC) = 21,1 ± 12,0% (CV = 57%).
[0166] Tabela 5-1 apresenta os dados de um estudo representativo relativos aos perfis de plasma teriparatida concentração-tempo após a administração subcutânea de solução de teriparatida (menos 85 μg/formulação e administração jejunal de teriparatida (teriparatida) na formulação (a 550 μg/kg; formulação I, Tabela 3B) em ratos não anestesiados, como descritos anteriormente no Exemplo 8. Amostras de sangue foram retiradas da veia jugular, 3, 6, 10, 25, 60 e 90 minutos após a administração jejunal e aos 3, 10, 30, 60, 90 min, 2 e 3 horas após a administração SC, o plasma foi preparado e os níveis de teriparatida foram determinados em cada amostra. Níveis de teriparatídeo na circulação sistêmica aumentou drasticamente após a administração jejunal de teriparatida em formulação. Pico de níveis de teriparatídeo de 3 minutos após a administração. A média RBA obtida após administração jejunal de teriparatida na formulação foi calculada conforme descrita nos exemplos acima, e foi de 14,0%. Em um experimento de controle, administração jejunal de teriparatida em solução solução salina não demonstrou a penetração na corrente sanguínea.
[0167] Tabela 5-11 apresenta os dados de um estudo representativo relativos aos perfis de plasma teriparatida concentração- tempo após a administração subcutânea de solução de teriparatida (a 10,65 μg/kg) e administração jejunal de teriparatida na formulação (a 100 μg/kg; formulação I, Tabela 3B ) para suínos não anestesiados. Um modelo de suínos foi estabelecido em que duas cânulas de diferentes foram cirurgicamente implantadas permanentemente na fêmea suínos domésticos: 1 - cânula jejunal para ignorar o estômago e permitir a administração formulação direta para o jejuno. 2 - cateterização da veia jugular para determinar os níveis sistemáticos da carga administrada após a administração jejunal.
[0168] Os porcos foram autorizados a recuperação durante 7 dias antes do experimento e privados de alimentos 18-20 horas antes do início do experimento.
[0169] Amostras de sangue foram retiradas da veia jugular em 0, 3, 6, 10, 15, 25, 40, 60, 90 minutos, 2, 2,5 e 3 horas após a administração jejunal e em 0, 3, 6, 10, 15, 20, 30, 45, 60, 90 min, 2, 2,5, 3 e 4 horas após a administração SC, o plasma foi preparado e os níveis de teriparatida foram determinados em cada amostra. Níveis de teriparatídeo na circulação sistêmica aumentaram drasticamente após a administração jejunal de teriparatida em formulação. Pico de níveis de teriparatídeo em 10 minutos após a administração. O rBA médio obtido após administração jejunal de teriparatida na formulação foi calculado conforme descrito nos exemplos acima, e foi de 15.0%.
[0170] Um experimento com porcos semelhante foi realizado usando dextran (FD4, formulação A na Tabela 3A) e determinou-se que a biodisponibilidade média de dextrana foi de 20% nos suínos em relação ao IV.
[0171] D. Absorção hGH após a administração retal de hGH na formulação de ratos
[0172] Tabela 5 - IV apresenta os dados de um estudo representativo relativos aos perfis de plasma hGH concentração-tempo após a administração subcutânea de solução de hGH (menos 81 μg/kg) e administração retal de hGH na formulação (a 800 μg/kg; formulação P, a Tabela 3C), em ratos anestesiados
[0173] Ratos machos Sprague-Dowley foram privados de alimento por 18 horas antes do início do experimento. Os ratos foram anestesiados com uma solução de cetamina: xilazina. A formulação (100 μL/rato) foi administrado por via retal com um Venflon 14G. Amostras de sangue foram retiradas da veia jugular, 3, 6, 10, 15 minutos, 40, 60 e 90 após a administração retal e em 15, 30, 45, 60, 90 min, 2, 3 e 4 horas após a administração SC, o plasma foi preparado e os níveis de hGH foram determinados em cada amostra. Os níveis de hGH na circulação sistêmica aumentaram dramaticamente após a administração retal de hGH na formulação. Pico dos níveis de hGH em 15 minutos. O RBA média obtida após administração retal de hGH na formulação foi calculado como descrito nos exemplos acima, foi de 17,9%. Em uma experiência separada hGH foi administrada ao jejuno e da ABA foi menor. Em um experimento de controle, a administração retal de hGH em PBS não demonstrou a penetração na corrente sanguínea.
[0174] Assim, os resultados apresentados na Tabela 5 demonstram que a exposição substancial foi obtida para todas as cargas compostos testados em todos os modelos testados em animais.
[0175] Os resultados acima demonstram que as formulações aqui descritas permitem a entrega de uma ampla gama de diferentes macromoléculas através do epitélio intestinal em diferentes modelos animais.
[0176] Produção da fração hidrofílica: Para 200 mL de água os seguintes ingredientes foram adicionados lentamente, um por um (com 2-3 minutos entre cada ingrediente de mistura): 172 mg de teriparatida, 200 mg de MgCh, 4.0 g de PVP-12, 17.52 g de octanoato de sódio e 10.0 g de solução aquosa a 2% MC-400, preparada de acordo com o seguinte: 1 g de MC-400 em pó foi adicionado a 50 mL de água À 60+2 °C enquanto mistura. Após 5 minutos de mistura, o béquer foi transferido para o gelo até que uma solução clara fosse obtida.
[0177] Após a adição do CM-400 da solução, a solução foi misturada por mais 5 minutos e em seguida, liofilizado para cerca de 24 h. Este procedimento produziu cerca de 22g da fração hidrofílica.
[0178] Produção do meio hidrofóbico: 2 g de Span 40, 4 g de lecitina e 3,8 g de OGM foram dissolvidos em 17,3 g de isovalerato de etilaisovalerato de etila enquanto mistura. Para esta solução foram adicionados 39,1 g de GTB e 72,6 g de óleo de rícino. Este procedimento produziu cerca de 136-138 g de meio hidrofóbico.
[0179] Produção do medicamento a granel: mistura da fração hidrofílica e hidrofóbica do meio foi realizada em 20 ± 2 ° C.
[0180] 15,7 g da fração hidrofílica foram adicionados lentamente durante a mistura de 84,3 g de meio hidrofóbico a 50 a 600 RPM. Após a adição de todas as frações hidrofílicas, a velocidade de mistura foi aumentada para 2000 ± 200 RPM por 20-10 min, seguida por 4-8 ciclos de 15 minutos misturando a 600 ± 50 RPM e 2 min de mistura 2000 ± 200 RPM.
[0181] Desgaseificação a vácuo foi aplicada da seguinte forma: 5 minutos a 600 mbar, 5 min a 500 mbar e 30 - 120 min a 400 milibares. A suspensão resultante foi colocada em um frasco escuro de 100 ml e armazenada a 2-8 ° C. Esta é a formulação teriparatida designada no "I" descrito na Tabela 3B
[0182] Todas as outras formulações descritas a seguir foram produzidas por este método, variando os ingredientes e quantidades de acordo com as indicações dadas nas tabelas em questão (ver, por exemplo, Exemplo 29). Um diagrama desse método (com insulina como carga) é mostrado na figura 1.
[0183] O efeito do tipo de óleo incorporado na formulação (em meio hidrofóbico) sobre a atividade de formulação foi testado. Formulações contendo dextrano (MW médios = 4,4 kDa, FITC) como composto de carga e de diferentes tipos de óleos no meio hidrofóbico (formulações E, F e G da Tabela 3A) foram testadas em ratos.
[0184] Para testar a atividade dessas formulações no jejuno de ratos não anestesiados, foi estabelecido um modelo em rato no qual duas cânulas diferentes são implantados cirurgicamente em ratos machos Sprague-Dowley: 1 - Cânula jejunal para desviar do estômago e permitir a administração da formulação direta para o jejuno. 2 - Cânula na veia jugular para determinação os níveis sistemáticos do dextrano administrado após a administração jejunal. Os ratos se recuperam por 4 dias antes do estudo e são privados de alimentos por 18 horas antes do início do estudo.
[0185] A Tabela 6 apresenta dados de um estudo em ratos não anestesiados após a administração intrajejunal de formulações contendo diferentes óleos no meio hidrofóbico. Tabela 6
[0186] Formulações contendo diferentes óleos foram administradas diretamente ao jejuno de ratos não anestesiados e os níveis de dextrano no plasma foram medidos em 3, 6, 10, 25, 60 e 90 minutos após a administração da formulação. Os níveis de absorção de dextrano a partir do jejuno de ratos após a administração de dextrano na formulação foram comparados com os níveis de dextrano absorvido após a administração intravenosa. Valores de exposição, AUC (0-90), foram determinados para a administração intravenosa e jejunal e a Biodisponibilidade absoluta (aBA) foi determinada de acordo com a seguinte equação: aBA = (AUC jejunal (0-90))/(AUC iv (0 - 90))*(dose iv/dose jejunal). Os dados são apresentados como Média + DP (n > 5 ratos por grupo).
[0187] Biodisponibilidade semelhante foi obtida quando o dextrano foi incorporado em formulações contendo óleo de rícino ou óleo de coco. Uma boa biodisponibilidade também foi obtida no jejuno de ratos quando teriparatídeo foi usado como composto de carga utilizando as formulações I e J; essas formulações contêm óleo de rícino e GTB, óleo de rícino e óleo de coco, respectivamente.
[0188] Os resultados mostraram que as formulações contendo diferentes tipos de óleos em seu meio hidrofóbico são ativas, permitindo a penetração da carga (dextrano, a teriparatídeo) carreada pela formulação. Assim, os dados demonstraram que todos os óleos testados permitem a biodisponibilidade da carga transportada pela formulação. Óleo de rícino e óleo de coco podem ser superiores a outros óleos testados.
[0189] Produção da fração hidrofílica:Em uma bolsa plástica, os seguintes ingredientes foram adicionados: 1,00 g de PVP-30, 6,70 g de octanoato de sódio e 13,00 g de lactose mono-hidratada como ligante. Após 5 minutos de mistura, todo o pó foi transferido para um almofariz e pistilo.
[0190] Uma solução aquosa de dextrano FD4 foi preparada como a seguir: 0,42 g de dextrano foram dissolvidas em 1,2 g de WFI. Toda a solução de dextrano foi então adicionada lentamente ao pó ao mesmo tempo em que se utiliza uma baixa agitação de cisalhamento em um almofariz e pistilo; a agitação levou cerca de 45 min.
[0191] A mistura foi então transferida para uma bandeja de liofilização e foi seca em estufa por cerca de 20 h à 500C. Este procedimento produziu cerca de 20g de fração hidrofílica, que era um granulado fino.
[0192] Produção do meio hidrofóbico:2 g de Span 40, 4 g de lecitina e 3,8 g de GMO foram dissolvidos em 17,3 g de isovalerato de etilaisovalerato de etila durante a mistura. Para esta solução foram adicionados 39,1 g de GTB e 72,6 g de óleo de rícino. Este procedimento produziu cerca de 136-138 g de meio hidrofóbico.
[0193] Produção do produto de matéria-prima: A mistura da fração hidrofílica e do meio hidrofóbico foi realizada a 20+20C. 19,00 g (29,58% do BDP final) da fração hidrofílica foram adicionados lentamente durante a mistura de 45,23 g (70,42% do BDP final) do meio hidrofóbico a 600+50 RPM. Após a adição de toda a fração hidrofílica, a velocidade de mistura foi aumentada para 2000+200 RPM por 2-10 min, seguidos por 4-8 ciclos de 15 min de mistura a 600+50 RPM e 2 min de mistura a 2000+200 RPM. A desgaseificação a vácuo foi aplicada em seguida como se segue: 5 min a 600 mbar, 5 minutos a 500 mbar e 30-120 min a 400 mbar. A suspensão resultante foi colocada em uma garrafa escura de 100 mL e armazenada a 2-8°C.
[0194] Estudo em ratos:A suspensão acima foi administrada por via retal em ratos, conforme descrito acima nos Exemplos e os resultados foram os seguintes: 35% de BA, 12,9% de SD. Outro lote de suspensão preparada por granulação, como descrita acima, foi preparado e foi administrado ao jejuno de ratos, como descrito acima, nos Exemplos e os resultados foram os seguintes: 21,8% de BA, 4,0% de SD. Um intervalo de formulações é preparado de uma forma similar, usando de granulação e incorporando uma seleção de agentes terapêuticos e variando a quantidade de octanoato de sódio.
[0195] Experimentos in vitro foram realizados utilizando separadamente três tipos de soluções: o meio hidrofóbico, como descrito nos Exemplos acima, isovalerato de etilaisovalerato de etila sozinho e isovalerato de etilaisovalerato de etila contendo 5% de cada um dos seguintes tensoativos: lecitina, span 40 e mono-oleato de glicerila. Três tipos de cápsulas não seladas, gelatinosas, de amido e de HPMC, foram, cada um, preenchidos com cada uma destas soluções. As cápsulas preenchidas foram então mantidas in vitro por 29 dias a 22±2°C, 30-50% de umidade relativa. Cápsulas gelatinosas e de HPMC renderam os melhores resultados, ou seja, sem deformação da cápsula.
[0196] Experimentos semelhantes foram realizados usando as mesmas três soluções e cápsulas gelatinosas e de HPMC. As cápsulas foram preenchidas com as soluções, seladas (ligadas) e, em seguida, foram mantidas por 8 dias a 22±2°C, 30-50% de umidade relativa. Ambos os tipos de cápsulas apresentaram estabilidade para as soluções testadas, isto é, não houve vazamento nem deformação das cápsulas.
[0197] Formulações foram preparadas com dextrano (FD4) semelhante à Formulação A da Tabela 3A exceto pelo octanoato de sódio 12% (0,722M) ter sido substituído por uma molaridade igual de octanoato de lítio ou octanoato de potássio ou octanoato de arginina (o último como um modelo para um sal de amônio). Estas formulações são mostradas abaixo na Tabela 7A. Tabela 7ª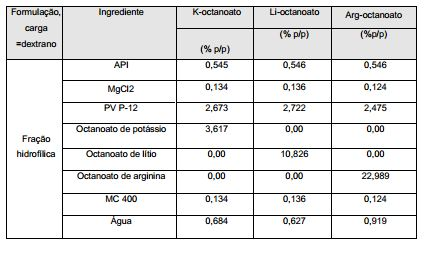
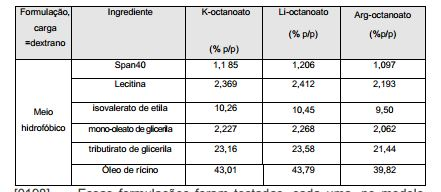
[0198] Essas formulações foram testadas, cada uma, no modelo jejunal de rato descrito no Exemplo 8. Os resultados foram obtidos e a biodisponibilidade foi calculada. Os resultados são apresentados na Tabela 7B abaixo. Tabela 7B
[0199] A formulação A utilizada no experimento acima era de um lote diferente daquele utilizado no Exemplo 8, e assim os resultados de BA dados aqui para a formulação A diferem ligeiramente daqueles mencionados na Tabela 4.
[0200] Os resultados acima mostram que, quando octanoato de sódio 12% foi substituído na formulação por uma molaridade equivalente de octanoato de lítio ou octanoato de potássio, a formulação ainda teve biodisponibilidade, mas em um nível inferior. A formulação de octanoato de arginina teve atividade semelhante à formulação octanoato de sódio 12%.
[0201] Formulações contendo geraniol (BASF) e octanol (Spectrum/MP) foram preparadas como descrito acima, usando os ingredientes mostrados abaixo na Tabela 8. O dodecanoato de sódio foi obtido a partir de Spectrum/Acros).
[0202] Formulação Q- baixa% de sal de ácido graxo de cadeia média: Uma formulação de dextrano (FD4) foi elaborada essencialmente como descrito no Exemplo 11, contendo um total de 2,9% de sal de ácido graxo de cadeia média - (1,042% de octanoato de sódio + 1,869% de dodecanoato de sódio) - e também contendo geraniol e octanol no meio hidrofóbico, todos, como mostrado na Tabela 8 abaixo.
[0203] Formulação R- mais de 10% de sal de ácido graxo de cadeia média: Uma formulação de dextrano foi elaborada essencialmente como descrito para a formulação A, exceto que geraniol e octanol foram adicionados ao meio hidrofóbico, todos como mostrados na Tabela 8. Tabela 8

[0204] A formulação Q (baixa% de sal AGCM) foi testada no modelo do rato intrajejunal acima descrito e a biodisponibilidade foi calculada: aBA = 4,4%, DP = 3,8 (n = 12). A formulação R (mais de 10% de sal AGCM) foi testada no modelo de rato intrajejunal acima descrito e a biodisponibilidade foi calculada: aBA = 22,7%. DP = 1,6 (n = 6). A BA dessas formulações não difere significativamente de formulações semelhantes, descritas nos Exemplos acima, que não contêm geraniol.
[0205] Foram preparadas formulações para gentamicina e para RNA, essencialmente como descrito no Exemplo 11, com os ingredientes do produto de matéria-prima como mostrado abaixo nax Tabela 9. A gentamicina foi obtida na Applichem e o RNA era sal de sódio do ácido poli-inosínico-policitidílico (Sigma). Tabela 9ª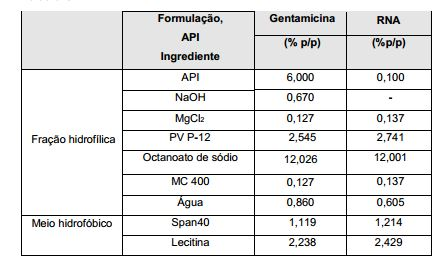

[0206] A formulação de gentamicina foi testada no modelo jejunal em ratos descrito acima e no modelo retal em ratos descrito acima (por exemplo, Exemplos 4 e 5). A gentamicina foi testada usando um imunoensaio (ELISA). Os resultados são mostrados na tabela 9B abaixo; % de BA é calculada comparada em relação à administração IV. Mostrou-se que as formulações fornecem biodisponibilidade à gentamicina. Tabela 9B
[0207] Da mesma forma, a formulação de RNA da Tabela 9A é testada no modelo jejunal em ratos e no modelo retal em ratos descritos acima. O RNA é analisado e espera-se que a formulação forneça biodisponibilidade ao RNA.
[0208] O efeito sobre a atividade da formulação da retirada de tensoativos do meio hidrofóbico foi testado em formulações contendo dextrano (PM médio = 4,4 kDa, marcado com FITC) como carga (formulações A e H na Tabela 3A).
[0209] A Tabela 10 apresenta dados de um estudo em ratos não anestesiados após a administração intrajejunal de formulações com ou sem tensoativos (por exemplo, Span40, lecitina, mono-oleato de glicerila) no meio hidrofóbico. Tabela 10
[0210] Formulações com ou sem tensoativos no meio hidrofóbico foram administradas diretamente ao jejuno de ratos não anestesiados e os níveis de dextrano no plasma foram medidos em 3, 6, 10, 25, 60 e 90 minutos após a administração da formulação. Os níveis de absorção de dextrano a partir do jejuno de ratos após a administração de dextrano na formulação foram comparados com os níveis de dextrano absorvido após a administração intravenosa.
[0211] Valores de exposição, AUC (0-90), foram determinados para a administração intravenosa e jejunal e a biodisponibilidade absoluta (aBA) foi determinada de acordo com a seguinte equação: aBA = AUC jejunal (0-90)/(AUC iv (0-90))*(dose iv/dose jejunal). Os dados são apresentados como aBA média + DP.
[0212] A menor biodisponibilidade foi alcançada quando o dextrano foi incorporado em uma formulação que não continha tensoativos no meio hidrofóbico (formulação H) em comparação com uma formulação contendo tensoativos no meio hidrofóbico (formulação A). Os resultados demonstram que a retirada de tensoativos do meio hidrofóbico afeta adversamente a atividade da formulação.
[0213] O efeito sobre a atividade da formulação da retirada de ácidos graxos de cadeia média (AGCM) da fração hidrofílica foi testado em formulações contendo dextrano (PM médio = 4,4 kDa, marcado com FITC) como carga.
[0214] A Tabela 11 apresenta dados de um estudo em ratos não anestesiados após a administração intrajejunal de formulações com ou sem octanoato de sódio na fração hidrofílica (formulações A e D na Tabela 3A, respectivamente). Tabela 11
[0215] As formulações descritas acima foram administradas diretamente ao jejuno de ratos não anestesiados e os níveis de dextrano no plasma foram medidos em 3, 6, 10, 25, 60 e 90 minutos após a administração da formulação. Os níveis de absorção de dextrano a partir do jejuno de ratos após a administração de dextrano na formulação foram comparados com os níveis de dextrano absorvido após a administração intravenosa. Valores de exposição, AUC (0-90), foram determinados para a administração intravenosa e jejunal e a biodisponibilidade absoluta (aBA) foi determinada de acordo com a seguinte equação: aBA = AUC jejunal (0-90)/(AUC iv (0-90))*(dose IV/dose jejunal). Os dados são apresentados como aBA média + DP.
[0216] Insignificante penetração de dextrano foi alcançada quando o dextrano foi incorporada em uma formulação que não tinha ácidos graxos de cadeia média na fração hidrofílica (formulação D, % de aBA = 0,6 + 1,0) em comparação com uma formulação contendo octanoato de sódio a 12% p/p na fração hidrofílica (formulação A, % de aBA = 28,0% + 6,8).
[0217] Os resultados demonstram que uma formulação sem ácidos graxos de cadeia média na fração hidrofílica não é ativa.
[0218] Um experimento semelhante foi realizado utilizando octreotídeo como carga na formulação melhorada (ver abaixo). O rBA foi de 0,11% (CV = 158%)
[0219] O efeito sobre a atividade de formulação da simplificação da formulação foi testado em formulações contendo dextrano (PM médio = 4,4 kDa, marcad com FITC) ou octreotídeo (Novetide) como carga. A formulação básica descrita nos Exemplos acima (por exemplo, formulações designadas A, I e P) foi simplificada pela não adição de MgCl2 e MC 400 na fração hidrofílica e pela não adição de span40, lecitina e iso-valerato de etila no meio hidrofóbico. Há um aumento concomitante nas quantidades de mono-oleato de glicerila (tensoativo) e tributirato de glicerila adicionadas ao meio hidrofóbico. Tais formulações são mostradas abaixo na Tabela 12A. Estas formulações simplificadas visualmente, não mostram precipitação, embora as partículas sejam visíveis ao microscópio, ou seja, são suspensões estáveis. Tabela 12ª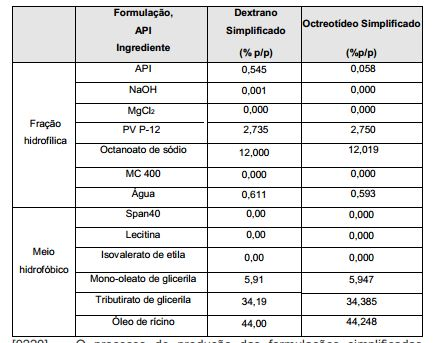
[0220] O processo de produção das formulações simplificadas acima é essencialmente como descrito na figura 1 e no Exemplo 11 para as formulações básicas. A formulação de octreotídeo básico é mostrada na Tabela 12B abaixo. Tabela 12B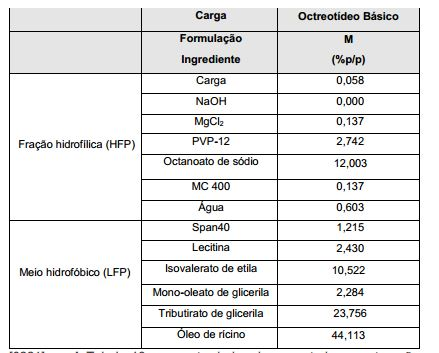
[0221] A Tabela 13 apresenta dados de um estudo em ratos não anestesiados após a administração intrajejunal de duas formulações de dextrano diferentes - formulação A da Tabela 3A e formulação simplificada mostrada na Tabela 12A. Tabela 13
[0222] Os resultados acima mosl ram que valores de AUC semelhantes foram obtidos quando o dextrano foi incorporado em uma formulação contendo a formulação básica (formulação A) em comparação com uma formulação simplificada.
[0223] A tabela 14 abaixo apresenta dados de um estudo em ratos não anestesiados após a administração intrajejunal de duas formulações de octreotídeo diferentes - a formulação básica mostrada na Tabela 12B e a formulação simplificada mostrada na Tabela 12A. Os níveis de absorção de octreotídeo a partir do jejuno de ratos após a administração de octreotídeo na formulação básica e formulação simplificada foram obtidos. valores de exposição, AUC (0-25), foram determinados. Tabela 14
[0224] Os resultados acima na Tabe a 14 mostram que os valores de AUC foram um pouco menos quando o octreotídeo estava incorporado em uma formulação simplificada em comparação com a formulação integral.
[0225] O efeito sobre a atividade da formulação da substituição do óleo de rícino (e tributirato de glicerila e iso-valerato de etila) por ácido octanoico (Aldritch) foi testada utilizando uma formulação contendo dextrano como carga. Isto foi feito para manter o motivo C8 na formulação, ou seja, considerou-se que poderia ser vantajoso ter ácido C8 no meio hidrofóbico, além do sal C8 na fração hidrofílica.
[0226] O efeito da adição de ácido ricinoleico (Spectrum) também foi testado pela feitura de uma formulação de dextrano contendo ácido octanoico/ácido ricinoleico. O ácido ricinoleico foi escolhido já que o principal componente triglicerídico no óleo de rícino é formado a partir do ácido ricinoleico. Três formulações de dextrano foram preparadas como mostrado na Tabela 15A abaixo. A formulação de dextrano básica foi preparada essencialmente como descrito nos exemplos acima. A formulação de dextrano octanoico foi preparada essencialmente como descrito nos Exemplos acima, mas em que o óleo de rícino, tributirato de glicerila e iso-valerato de etila foram substituídos por ácido octanoico. Descobriu-se que esta formulação era uma solução por meio de análise visual, mas não foi executada análise de solubilidade verdadeira. Parece que o ácido octanoico em concentração elevada (cerca de 78% desta formulação) dissolve a fração hidrofílica sólida, com o PVP e o octanoato de sódio sendo solúveis em ácido octanoico em alta concentração. A formulação de ácido octanoico/ricinoleico e dextrano foi preparada essencialmente como descrito nos Exemplos acima, mas em que o óleo de rícino, tributirato de glicerila e iso-valerato de etila foram substituídos por uma mistura de ácido octanoico e ácido ricinoleico. Esta formulação era uma suspensão, como é usual para a maioria das formulações desta invenção. Tabela 15ª

[0227] As formulações descritas acima na Tabela 15A foram administradas diretamente ao jejuno de ratos não anestesiados e os níveis de dextrano no plasma foram medidos após a administração da formulação. Valores de exposição, AUC, foram determinados para as diferentes formulações. Esses resultados são apresentados na Tabela 15B abaixo. Tabela 15B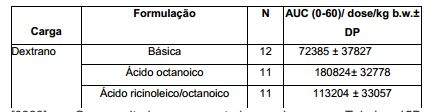
[0228] Os resultados apresentados acima na Tabela 15B demonstram que a absorção do dextrano foi muito melhorada (mais de duas vezes) na formulação contendo ácido octanoico. Além disso, a forma do gráfico foi alterada mostrando liberação mais lenta, porém mais longa. Isso pode ser vantajoso, já que permite que a API tenha ação prolongada no organismo. Os resultados de dextrano ricinoleico/ octanoico mostraram menos atividade do que a formulação de ácido octanoico, mas ainda eram melhores que a formulação básica.
[0229] Já que as formulações de ácido octanoico e ácido ricinoleico/ácido octanoico mostraram atividade elevada, formulações semelhantes foram preparadas com exenatídeo como carga. Três formulações de exenatídeo foram produzidas como mostrado na Tabela 16A abaixo. A formulação de exenatídeo básica foi preparada essencialmente como descrito nos exemplos acima. A formulação de exenatídeo/octanoico foi preparada essencialmente como descrito nos Exemplos acima
[0230] Exemplos, mas onde o óleo de rícino, tributirato de glicerila e iso-valerato de etila foram substituídos por ácido octanoico. Descobriu-se que esta formulação contendo aproximadamente 78% de ácido octanoico era uma solução por meio de análise visual, como era a formulação de dextrano semelhante acima. A formulação de ácido octanoico/ricinoleico e exenatídeo foi preparada essencialmente como descrito nos Exemplos acima, mas em que o óleo de rícino, tributirato de glicerila e iso-valerato de etila foram substituídos por uma mistura de ácido octanoico e ácido ricinoleico. Tabela 16ª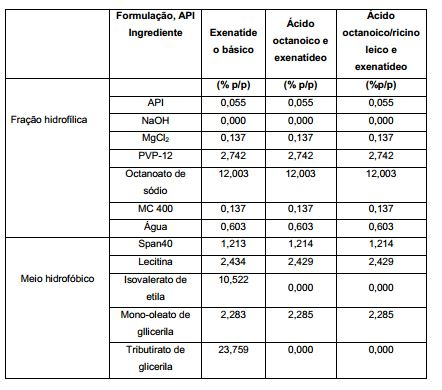

[0231] As formulações descritas acima na Tabela 16A foram administradas diretamente ao jejuno de ratos não anestesiados e os níveis de exenatídeo no plasma foram medidos após a administração da formulação. Valores de exposição, AUC, foram determinados para as diferentes formulações. Esses resultados são apresentados na Tabela 16B abaixo. Tabela 16B
[0232] Os resultados apresentados acima na Tabela 16B demonstram que a formulação de exenatídeo contendo ácido octanoico apresentou biodisponibilidade, mas a absorção de exenatídeo estava diminuída em comparação com a formulação básica. A forma do gráfico foi alterada, mostrando liberação mais lenta, porém mais longa como no caso da formulação de ácido octanoico e dextrano acima; este perfil de PK prolongado pode ser vantajoso. Observe que no caso da formulação de ácido octanoico, AUC 0-180 min foi utilizada para os cálculos de BA devido ao perfil de PK prolongado. A formulação de exenatídeo e ácido ricinoleico/octanoico teve biodisponibilidade ainda menor que a formulação de ácido octanoico.
[0233] A. Formulações de octreotídeo: O efeito sobre a atividade de formulação da variação da quantidade de ácido octanoico foi testado em formulações contendo octreotídeo como carga. Quatro formulações de octreotídeos foram preparadas com 0%, 5%, 10% ou 15% de ácido octanoico, como mostrado na Tabela 17 abaixo. As formulações são formulações de octreotídeo básicas preparadas, essencialmente, como descrito acima, em que a quantidade de ácido octanoico varia conforme descrito e a quantidade de outros ingredientes no meio hidrofóbico (isovalerato de etila e tributirato de glicerila) foram reduzidas concomitantemente. (Nestas formulações a fração hidrofílica foi simplificada para omitir MgCl2 e MC400.) Tabela 17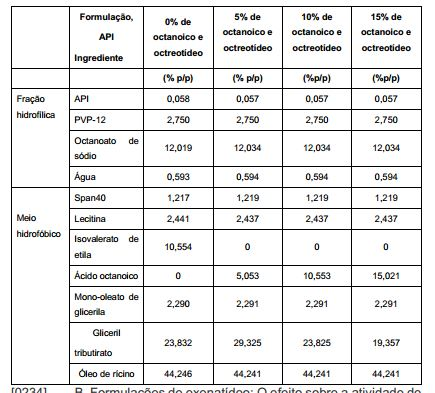
[0234] B. Formulações de exenatídeo: O efeito sobre a atividade de formulação da variação da quantidade de ácido octanoico foi testado em formulações contendo exenatídeo como carga. Cinco formulações de exenatídeo foram preparadas com 0%, 10%, 15%, 20% ou 35% de ácido octanoico, como mostrado na Tabela 18 abaixo. As formulações são formulações de exenatídeo básicas preparadas, essencialmente, como descrito acima, em que a quantidade de ácido octanoico varia conforme descrito e a quantidade de outros ingredientes no meio hidrofóbico (isovalerato de etila e tributirato de glicerila) foi reduzida concomitantemente. Tabela 18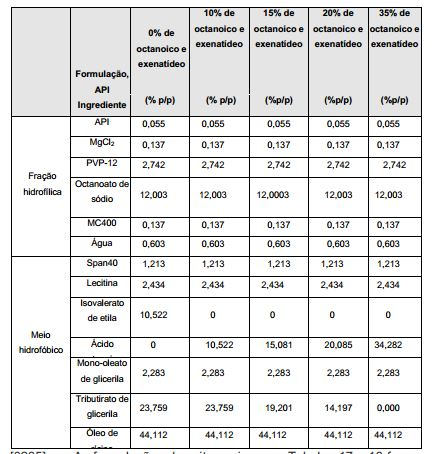
[0235] As formulações descritas acima nas Tabelas 17 e 18 foram administradas diretamente ao jejuno de ratos não anestesiados e os níveis de octreotídeo ou exenatídeo no plasma foram medidos após a administração da formulação. Valores de exposição, AUC, foram determinados para as diferentes formulações. Esses resultados são apresentados na Tabela 19 abaixo. Tabela 19
[0236] Os resultados apresentados acima na Tabela 19 mostramque a formulação de octreotídeo mostra aumento de atividade em comparação com a formulação básica já que a quantidade de ácido octanoico é aumentada para 15% (a quantidade máxima testada). Além disso, os resultados apresentados acima na Tabela 19 demonstram que a formulação de exenatídeo mostra aumento de atividade em comparação com a formulação básica já que a quantidade de ácido octanoico é aumentada para 15% e a atividade diminui em níveis mais elevados de ácido octanoico.
[0237] A. Sebacato de sódio (sal dissódico do ácido decanodioico): O efeito sobre a atividade da formulação da substituição de octanoato de sódio por sebacato de sódio (sal de ClO dissódico) em uma formulação de dextrano foi testado. O sebacato de sódio foi preparado in situ a partir de ácido sebácico (Aldrich) e hidróxido de sódio. A formulação produzida é descrito na Tabela 20 abaixo. A formulação foi preparada essencialmente como descrito acima, mas 12% de octanoato de sódio foram substituídos por sebacato de sódio, na mesma concentração molar que o octanoato de sódio, ou seja, uma quantidade equimolar de sebacato de sódio foi utilizads (ou seja, 0,72 M). Tabela 20
[0238] A formulação descrita acima na Tabela 20 foi administradadiretamente ao jejuno de ratos não anestesiados e os níveis de dextrano no plasma foram medidos após a administração da formulação. Valor de exposição, AUC, foi determinado para a formulação e esta é comparada com uma formulação semelhante preparada com octanoato de sódio. Esses resultados são apresentados na Tabela 21 abaixo. Tabela 21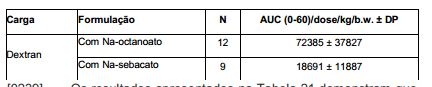
[0239] Os resultados apresenl ados na Tabela 21 demonstram que a formulação de dextrano contendo sebacato de sódio apresentou atividade, mas a absorção de dextrano estava diminuída em relação à formulação contendo uma quantidade equimolar de octanoato de sódio.
[0240] Foram preparadas formulações contendo octreotídeos em que 12% do octanoato de sódio foram substituídos por uma quantidade equimolar (0,72 M) de suberato monossódico ou de suberato dissódico, que são sais C8. Estes sais de sódio foram preparados in situ a partir de ácido subérico (Tokyo Chemical Industry Co.) e hidróxido de sódio. Tabela 22ª
[0241] As formulações descritas acima na Tabela 22 são administradas diretamente ao jejuno de ratos não anestesiados e os níveis de octreotídeo no plasma são medidos após a administração da formulação. Valores de exposição, AUC, são determinados para as formulações e estas são comparadas com uma formulação semelhante preparada com octanoato de sódio.
[0242] Duas formulações contendo octreotídeo foram preparadas essencialmente como descrito acima, em que 12% do octanoato de sódio foram substituídos por 18% de sal de sódio do ácido gerânico (0,95M) e 14,6% (0,77m) de sal de sódio de ácido gerânico, que é ácido 3,7-dimetil-2,6-octadienoico (obtido de SAFC). As formulações produzidas são descritas na Tabela 22B abaixo. Tabela 22B
[0243] As formulações descritas acima na Tabela 22B foram administradas diretamente ao jejuno de ratos não anestesiados e os níveis de octreotídeo no plasma foram medidos após a administração da formulação. Valores de exposição, AUC, foram determinados para as formulações e estas foram comparadas com uma formulação semelhante preparada com octanoato de sódio. Os resultados são apresentados abaixo na Tabela 22C e demonstram que a formulação com 18% de geranato de sódio tiveram atividade semelhante à da formulação com 12% de octanoato de sódio e a formulação com 14,6% de geranato de sódio teve aumento da atividade. Tabela 22C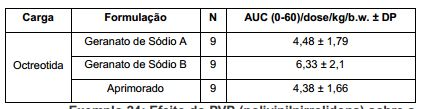
[0244] O efeito sobre a atividade de formulação da substituição de PVP-12 por manitol (Sigma) foi testado em formulações contendo exenatídeo como carga. Entendia-se na técnica que o PVP-12 é um estabilizante e poderia ser substituído na formulação por outro estabilizante como o manitol. A formulação mostrada na Tabela 23 abaixo foi preparada. Esta formulação é
[0245] uma formulação básica de exenatídeo preparada essencialmente como descrito acima, mas onde PVP-12 é substituída por manitol. Tabela 23
 Continuação...
Continuação...
[0246] A formulação descrita acima na Tabela 23 foi administrada diretamente ao jejuno de ratos não anestesiados e os níveis de exenatídeo no plasma foram medidos após a administração da formulação. Valores de exposição, AUC, foram determinados para a formulação em comparação com a formulação básica. Esses resultados são apresentados na Tabela 24 abaixo. Tabela 24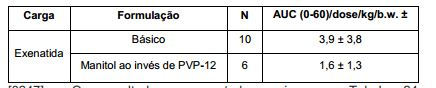
[0247] Os resultados apresenl ados acima na Tabela 24 demonstram o resultado surpreendente e inesperado de que a formulação de exenatídeo sem PVP-12 teve significativa diminuição da atividade em comparação com a formulação básica. Foi, assim, decidido investigar adicionalmente o efeito da PVP sobre a biodisponibilidade.
[0248] O efeito sobre a atividade de formulação da variação do peso molecular da PVP foi testado em formulações contendo exenatídeo como carga. Três formulações de exenatídeo foram preparadas usando PVP-12, PVP-17 ou PVP-25 (todos obtidos da BASF). PVP-12, PVP-17 e PVP-25 são todos polímeros de polivinilpirrolidona; os pesos moleculares médios são cerca de 2500-3000, 10000 e 30000, respectivamente. As formulações são formulações de exenatídeo básicas preparadas essencialmente como descrito acima, em que a PVP varia conforme descrito e em que a fração hidrofílica foi simplificada para omitir MgCl2 e MC400. Tabela 25
[0249] As três formulações descritas acima na Tabela 25 foram administradas diretamente ao jejuno de ratos não anestesiados e os níveis de exenatídeo no plasma foram medidos após a administração da formulação. Valores de exposição, AUC, foram determinados para as formulações. Os resultados são apresentados na Tabela 26 abaixo. Tabela 26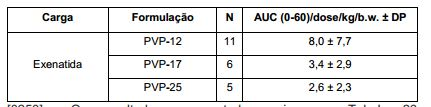
[0250] Os resultados apresentados acima na Tabela 26 demonstram que as formulações de exenatídeo contendo PVP-12 apresentaram atividade muito maior que as formulações de exenatídeo contendo PVP-17 e PVP-25. Assim, o efeito da PVP-12 só foi investigado adicionalmente e foi decidido realizar um estudo de dose- resposta usando PVP-12. O efeito do aumento da quantidade de PVP- 12 na formulação sobre a atividade da formulação foi testado em formulações contendo octreotídeo como composto de carga e diferentes doses de PVP-12 como mostrado na Tabela 27 abaixo. As doses de PVP-12 testadas foram 2,75% (a dose padrão utilizada nas formulações acima) e 5,0%, 7,5% e 10,0% de PVP-12; a fração hidrofílica foi simplificada para omitir MgCh e MC400. A formulação contendo 10% de PVP era semissólida, ou seja, aparentemente era uma suspensão semissólida. Tabela 27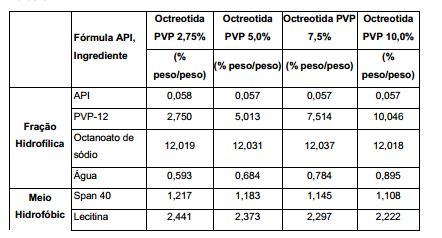

[0251] As formulações descritas acima na Tabela 27 foram administradas diretamente ao jejuno de ratos não anestesiados e os níveis de octreotídeo no plasma foram medidos após a administração da formulação. Valores de exposição, AUC, foram determinados para as quatro diferentes formulações. Esses resultados são apresentados na Tabela 28A abaixo. Tabela 28ª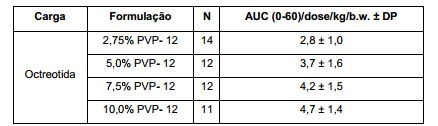
[0252] Os resultados apresentados anteriormente na Tabela 28A demonstram que a absorção de octreotídeo aumentou dramaticamente conforme a quantidade de PVP na formulação aumentava. A formulação contendo 10% de PVP-12 teve a absorção de octreotídeo cerca de 1,7 vezes maior que a formulação contendo 2,75% de PVP-12. Uma formulação octreotídeo melhorada em que havia 10% de PVP-12, mas nenhum octanoato de sódio apresentou praticamente nenhuma atividade. O rBA foi de 0,11% (CV = 158%) n=5.
[0253] Parece que o sal de ácido graxo de cadeia média atua como um intensificador de permeabilidade (facilitando ou aumentando a permeabilidade e/ou a absorção do agente terapêutico) e que a PVP serve para aumentar o efeito do intensificador de permeabilidade, de forma sinérgica já que o PVP sozinho tem praticamente nenhum efeito. Veja também o Exemplo 31.
[0254] Um experimento adicional foi realizado para investigar se os 10% de PVP-12 poderiam ser substituídos por dextrano e ainda manter a atividade da formulação. O dextrano foi fabricado pela Fluka; o peso molecular médio é ~6000. As formulações foram preparadas essencialmente como descrito acima, em que a PVP e o dextrano variam conforme descrito e em que a fração hidrofílica foi simplificada para omitir MgCl2 e MC400 e onde o octanoato de sódio foi elevado a 15%; veja o Exemplo 26. Tabela 28B
[0255] As três formulações descritas acima na Tabela 28B foram administradas diretamente ao jejuno de ratos não anestesiados e os níveis de octreotídeo no plasma foram medidos após a administração da formulação. Valores de exposição, AUC, foram determinados para as formulações. Os resultados são apresentados na Tabela 28C abaixo. Tabela 28C
[0256] Os resultados apresentados acima na Tabela 28C demonstram que a absorção de octreotídeo diminuiu quando a PVP na formulação foi substituída por dextrano, mas a atividade ainda era significativa. A formulação contendo dextrano a 10% teve absorção de octreotídeo de cerca de 75% da formulação contendo 10% de PVP e a formulação contendo dextrano a 5% teve absorção de octreotídeo de cerca de 73% da formulação contendo 10% de PVP.
[0257] O efeito sobre a atividade de formulação da substituição de octanoato de sódio por outros sais de sódio de ácido graxo de cadeia média foi testado em formulações contendo octreotida como carga. Três formulações de octreotídeo foram preparadas, como mostrado na Tabela 29 abaixo. Estas são todas formulações básicas preparadas essencialmente como descrito acima, onde a fração hidrofílica foi simplificada para omitir MgCh e MC400 e em que o sal de ácido graxo de cadeia média é uma quantidade equimolar de octanoato de sódio, nonanoato de sódio ou decanoato de sódio. Tabela 29
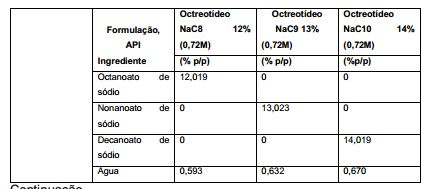 Continuação...
Continuação...
[0258] As formulações descritas acima na Tabela 29 foram administradas diretamente ao jejuno de ratos não anestesiados e os níveis de octreotídeo no plasma foram medidos após a administração da formulação. Valores de exposição, AUC, foram determinados para as formulações. Os resultados são apresentados na Tabela 30 abaixo. Tabela 30
[0259] Os resultados apresentados acima na Tabela 30 demonstram que, quando o octanoato de sódio na formulação é substituído por nonanoato de sódio ou por decanoato de sódio há uma atividade similar. Com base na análise estatística, não há diferença na atividade entre as três formulações.
[0260] A dose-resposta de octanoato de sódio a 12%, 15% e 18% foi testada pela feitura das formulações apresentadas na Tabela 31. Estas são todas formulações básicas preparadas essencialmente como descrito acima, onde a fração hidrofílica foi simplificada para omitir MgCl2 e MC400 e o composto de carga foi octreotídeo. Além disso, a formulação foi corrigida por viscosidade, isto é, a mesma viscosidade ou similar foi mantida para todas as três formulações; isso foi conseguido através da variação das quantidades de óleo de rícino e tributirato de glicerila. Tabela 31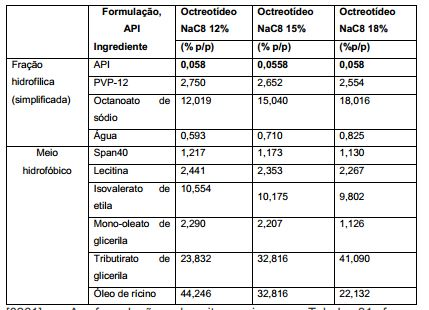
[0261] As formulações descritas acima na Tabela 31 foram administradas diretamente ao jejuno de ratos não anestesiados e os níveis de octreotídeo no plasma foram medidos após a administração da formulação. Valores de exposição, AUC, foram determinados para as formulações. Os resultados são apresentados na Tabela 32 abaixo. Tabela 32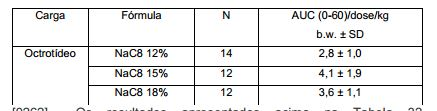
[0262] Os resultados apresentados acima na Tabela 32 demonstram que, quando octanoato de sódio na formulação é aumentado de 12% para 15%, há um aumento na atividade, mas um aumento adicional de octanoato de sódio para 18% não leva a uma maior atividade que aquela obtida em 15%. Assim, cerca de 15% de octanoato de sódio parece ser a quantidade preferida.
[0263] A Tabela 33 abaixo descreve várias formulações de octreotídeo. A primeira coluna, formulação (a), é a formulação básica preparada essencialmente como descrito acima, onde a fração hidrofílica foi simplificada para omitir MgCl2 e MC400 e o composto de carga é octreotídeo. Os tensoativos são Span 40, lecitina e mono-oleato de glicerila e pelo cálculo o HLB é aproximadamente 5-6. Em outras formulações (formulações b, c e d) o HLB foi alterado como indicado (para 3,5, 6,7 e 14), substituindo Span 40 e lecitina por diferentes quantidades de Tween 80 e variando a quantidade de mono-oleato de glicerila. Tabela 33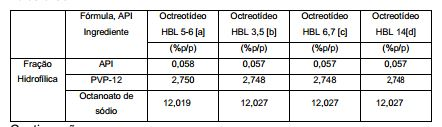 Continuação...
Continuação...
[0264] As formulações descritas acima na Tabela 33 foram administradas diretamente ao jejuno de ratos não anestesiados e os níveis de octreotídeo no plasma foram medidos após a administração da formulação. Valores de exposição, AUC, foram determinados para as formulações. Os resultados são apresentados na Tabela 34 abaixo. Tabela 34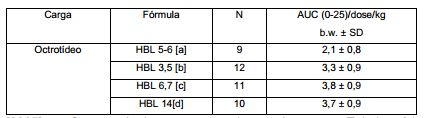
[0265] Os resultados apresentados acima na Tabela 34 demonstram que todas as três novas formulações que substituem Span 40 e lecitina por Tween 80 [b, c e d] tiveram atividade muito melhor do que a formulação básica [a], mesmo apesar do HLB em [b] ser menor, em [c] foi um pouco maior e em [d] foi muito maior do que o HLB dos tensoativos em (a). Além disso, as atividades de todas as novas formulações [(b, c e d] foram estatisticamente muito semelhantes. Assim, o HLB isolado dos tensoativos não parece afetar a atividade, mas as características dos tensoativos parecem desempenhar um papel importante. Em particular, a substituição de Span 40 e lecitina por Tween 80 é vantajosa para a atividade destas formulações de octreotídeo.
[0266] Com base na acumulação de resultados descritos acima, incluindo os resultados de dose-resposta para PVP-12, os resultados de dose-resposta para octanoato de sódio e os resultados para tensoativo nomeadamente, uma série de formulações de octreotídeo foi preparada com 10% de PVP-12 e 15% de octanoato de sódio e variando a proporção de tricaprilato de glicerila para de óleo de rícino. Além disso, mono-oleato de glicerila e tributirato de glicerila foram substituídos (se usados) por monocaprilato de glicerila e tricaprilato de glicerila (ambos fornecidos pela Abitec). Isto é para manter o motivo C8 na formulação. Assim, a fração hidrofílica contém um sal de um ácido C8 (octanoato) e o meio hidrofóbico contém monoglicerídeos e triglicerídeos que incorporam o mesmo ácido C8. Os inventores acreditam que o uso de compostos C-8, tanto na fração hidrofílica como no meio hidrofóbico pode ser vantajoso para a biodisponibilidade. As quantidades de Tween 80 e monocaprilato de glicerila também foram variadas nas formulações. As formulações foram preparadas como mostrado na Tabela 35A abaixo. As formulações I, II, V e VI eram semissólidas (aparentemente suspensões) e as formulações III e IV eram as suspensões líquidas usuais. Tabela 35ª Continuação...
Continuação...
[0267] As formulações descritas acima na Tabela 35A foram administradas diretamente ao jejuno de ratos não anestesiados e os níveis de octreotídeo no plasma foram medidos após a administração da formulação. Valores de exposição, AUC, foram determinados para as formulações. Os resultados são apresentados na Tabela 35B abaixo. Tabela 35B
[0268] Os resultados apresentados acima na Tabela 35B demonstram que as formulações I e IV têm a maior atividade. Como o óleo de rícino está ausente da formulação IV, isso demonstra que o óleo de rícino não é essencial para atividade. Parece que uma alta proporção de GTC: óleo de rícino, por exemplo, 6:4 é benéfica para atividade. Além disso, como a formulação V (que tem baixa atividade) tem a mesma proporção GTC: óleo de rícino como a formulação I, parece que adicionalmente GMC (ou outro monoglicerídeo) é desejável para a atividade. Além disso, uma formulação semelhante à formulação I da Tabela 36 foi elaborada, mas o octanoato de sódio foi omitido. Esta formulação não apresentou virtualmente qualquer atividade, rBA = 0,1%.
[0269] O produto de matéria-prima da formulação IV (melhorada, sem óleo de rícino) foi moída com uma tela de 150 mícrons, e depois o tamanho de partículas foi determinado utilizando a tecnologia de difração de laser Malvern. Resultados preliminares indicaram que 90% (v/v) das partículas estavam abaixo de 130 mícrons e 50% (v / v) das partículas estavam abaixo de 45 microns.
[0270] Experimentos preliminares usando formulações semelhantes para a formulação I, mas com diferentes quantidades aumentadas de octreotídeo, deram todos BA semelhante, isto é, havia aproximadamente exposição linear independente da carga de API. Um experimento preliminar, utilizando uma formulação semelhante à formulação IV com carga de octreotídeo ainda maior - 1,5% (p/p) - também deu BA semelhante.
[0271] Uma formulação semelhante melhorada para a formulação I acima foi preparada usando FD4 como carga, em vez de octreotídeo, e foi comparada a uma formulação básica. Estas formulações são descritas na Tabela 36A abaixo. Tabela 36 A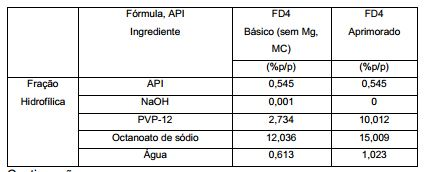 Continuação...
Continuação...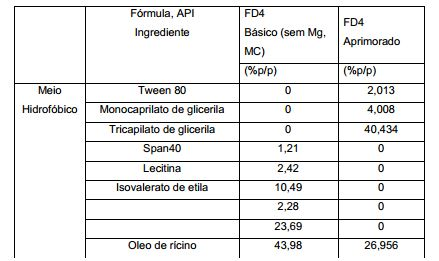
[0272] As formulações descritas acima na Tabela 36A foram administradas diretamente ao jejuno de ratos não anestesiados e os níveis de FD4 no plasma foram medidos após a administração da formulação. Valores de exposição, AUC, foram determinados para as formulações. Os resultados são apresentados na Tabela 36B abaixo Tabela 36B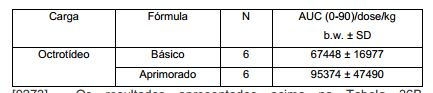
[0273] Os resultados apresentados acima na Tabela 36B demonstram que a formulação melhorada tem atividade muito maior que a formulação básica.
[0274] A formulação de octreotídeo no Exemplo 28 (Tabela 6, na primeira coluna) foi elaborada essencialmente como descrito nos Exemplos acima. Abaixo segue o processo de produção detalhado para esta formulação.
[0275] Para 150 mL de água os seguintes ingredientes foram lentamente adicionados e misturados: 24,05 g de octanoato de sódio, 16,04 g de PVP-12 e 92,4 g de 10 mg/mL de solução aquosa de octreotídeo. A solução resultante foi liofilizada.
[0276] 3,25 g de Tween 80, 6,47 g de monocaprilato de glicerila, 65,25 g de tricaprilato de glicerila e 43,50 g de óleo de rícino foram misturados. Produção do produto de matéria-prima:
[0277] 26,08 g da fração hidrofílica foram adicionados lentamente a 73,92 g do meio hidrofóbico a 20 ± 2 0C durante a mistura. Após a adição da fração hidrofílica inteira, a velocidade de mistura foi aumentada. A desgaseificação a vácuo foi em seguida aplicada e a suspensão resultante foi armazenada a 2-8°C.
[0278] Para permitir que maiores quantidades de octreotídeo fossem dissolvidas o seguinte método foi criado: 1. A quantidade de água da preparação da fração hidrofílica foi a mesma que o volume calculado do produto de matéria-prima final.
[0279] 2. A PVP-12 foi dissolvida em metade da quantidade acima da água.
[0280] 3. O octanoato de sódio foi dissolvido na segunda metade da quantidade de água.
[0281] 4. O octreotídeo foi dissolvido na solução de PVP-12 (do parágrafo 2). 5. A solução de octanoato de sódio foi adicionada à solução de octreotídeo e PVP-12.
[0282] Nesta fase houve alguma precipitação, mas ela tornou-se solúvel após a mistura.
[0283] Para testar a atividade das formulações da invenção quando administradas em cápsulas, um modelo animal que permite a administração de cápsula a porcos (porcos domésticos) foi estabelecido. A fim de desviar do estômago e permitir a administração direta de cápsulas ao intestino delgado do porco, um modelo bem estabelecido em cães ("Nipple Valve model"; Wilsson-Rahmberg &O. Jonsson, Laboratory Animals (1997), 31, 231 -240) foi adaptado para o porco comercial.
[0284] As duas formulações de octreotídeo mostradas abaixo na Tabela 37 foram preparadas. A formulação de octreotídeo (x) foi preparada essencialmente como descrito acima para a formulação básica, onde a fração hidrofílica foi simplificada para omitir MgCh e MC400. A formulação de octreotídeo (y) foi preparada essencialmente como descrito acima para a formulação de octreotídeo melhorada. As formulações preencheram cápsulas de gelatina (da Capsugel), formulação básica (x) em 0,42 mL/cápsula e a formulação melhorada (y) em 0,44mL/cápsula, resultando em 5mg de peso líquido de octreotídeo em ambos os tipos de cápsulas preenchidas. As cápsulas não receberam revestimento entérico, isto é, eram não revestidas. Tabela 37

[0285] As formulações acima descritas na Tabela 37 foram administradas diretamente ao intestino delgado dos porcos não anestesiados através do desvio gástrico descrito acima, e os níveis de octreotídeo no plasma foram medidos após a administração. Valores de exposição, AUC, foram determinados para as formulações. A % de BA foi calculada em relação à exposição a octreotídeo após administração subcutânea. Os resultados obtidos são apresentados na Tabela 38 abaixo. Tabela 38
[0286] Os resultados acima na Tabela 38 mostram que havia biodisponibilidade no modelo suíno para formulações encapsuladas, para ambas as formulações, básica e melhorada. A biodisponibilidade de Octreotídeo da formulação melhorada foi cerca de três vezes o nível de biodisponibilidade da formulação básica.
[0287] Os resultados aqui apresentados para biodisponibilidade estão subestimados porque o tempo de amostragem não foi suficiente para os níveis de octreotídeo voltarem à linha de base (0 ng/mL). Isto foi devido ao tempo de exposição inesperadamente mais longo em porcos em relação ao que tinha sido previamente medido em ratos. A forma do gráfico foi alterada em comparação com os resultados de ratos mostrando mais tempo para atingir os níveis de pico máximo e tempo prolongado em que o octreotídeo é residente no sangue. Isso pode ser vantajoso, já que permite que o octreotídeo tenha ação prolongada no organismo. Assim, a biodisponibilidade real em porcos deve ser maior que os números apresentados.
[0288] Com base nos resultados em ratos, o nível de biodisponibilidade em porcos de octreotídeo administrado em solução aquosa é extrapolado para ser cerca de 0,1%. Este nível de biodisponibilidade está abaixo do nível de sensibilidade do bioensaio utilizado para porcos.
[0289] Além dos resultados para PVP no Exemplo 24, o efeito sobre a atividade do aumento da quantidade de PVP-12 na formulação melhorada foi estudado. As formulações melhoradas, feitas essencialmente como descrito acima, continham octreotídeo como composto de carga e diferentes doses de PVP-12 como mostrado na Tabela 39 abaixo. As doses de PVP-12 testadas foram de 7,5%, 10,0% e 15,0% de PVP-12. As formulações contendo 10% e 15,0% de PVP eram semissólidas, isto é, eram aparentemente suspensões semissólidas - e a formulação contendo 7,5% de PVP era uma suspensão viscosa. Tabela 39

[0290] As formulações descritas acima na Tabela 39 foram administradas diretamente ao jejuno de ratos não anestesiados e os níveis de octreotídeo no plasma foram medidos após a administração da formulação. Valores de exposição, AUC, foram determinados para as três formulações. Esses resultados são apresentados na Tabela 40 abaixo. Tabela 40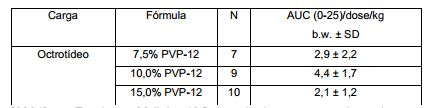
[0291] Ergui pag 88 linha 13Os resull tados apresentados acima na Tabela 40 demonstram que a absorção de octreotídeo foi a maior quando a PVP na formulação era 10% e aumentando a quantidade para 15% resulta em diminuição significativa da atividade. Isto confirma a escolha de 10% de PVP na formulação melhorada.
[0292] Três diferentes formulações básicas de três compostos de carga diferentes foram preparadas (dextrano, gentamicina e exenatídeo), essencialmente como descrito acima (em que a formulação básica é a fração hidrofílica não simplificada básica). Cada uma dessas três formulações foi administrada diretamente ao jejuno de ratos não anestesiados e os níveis de carga no plasma foram medidos após a administração da formulação. Valores de exposição, AUC, foram determinados para as formulações. Além disso, uma formulação semelhante foi preparada com um composto de carga não relevante (uma formulação simulada). Separadamente, a formulação simulada foi administrada concomitantemente com dextrano, gentamicina ou exenatídeo em solução aquosa e valores de exposição, AUC, foram determinados. A administração concomitante foi atingida pela administração da carga em solução aquosa seguida imediatamente pela administração da formulação simulada através de uma cânula-jejunal implantada (desvio gástrico). Para cada composto, a exposição após a administração da carga formulada foi comparada com a exposição após a administração da carga não formulada (concomitante). Os resultados comparativos são apresentados na Tabela 41. Os resultados mostram que há maior atividade (biodisponibilidade) quando a carga é formulada em relação à não formulada (concomitante) em todos os três casos, e que o exenatídeo mostrou, decididamente, o maior aumento na atividade devido à formulação. Nota-se que dextrano e gentamicina são compostos que não são sensíveis à degradação por protease, enquanto o exenatídeo sendo um peptídio está sujeito à degradação por enzimas intestinais. A grande diferença na atividade entre o exenatídeo formulado em comparação com o exenatídeo não formulado pode ser devido ao efeito protetor da formulação contra a degradação. Tabela 41
[0293] A. Limitação de tamanho: A tecnologia e as formulações descritas acima têm como objetivo melhorar a permeabilidade do intestino, permitindo a distribuição específica de proteínas, peptídios e outras moléculas de outro modo impermeáveis em toda esta barreira. Um certo grau de penetração não específica do conteúdo intestinal pode resultar como um efeito colateral deste aumento da permeabilidade específica. O tamanho das moléculas que poderiam penetrar no intestino de uma forma não específica foi avaliado utilizando-se diferentes marcadores de tamanho molecular.
[0294] Para avaliar o limite de tamanho molecular da permeabilidade GI aumentada, cinco diferentes dextranos marcados com FITC de diferentes pesos moleculares foram escolhidos para servir como marcadores moleculares para testar o aumento da permeabilidade intestinal; o peso molecular médio dos cinco dextranos foi de 4,4, 10, 20, 40 e 70 kDa, equivalente a um raio de 14, 23, 33, 45 e 60 A, respectivamente. Esses marcadores de tamanho diferentes foram administrados diretamente ao jejuno de ratos não anestesiados, através de uma cânula implantada no intestino e mostraram virtualmente nenhuma penetração do intestino basal quando testados isoladamente. Cada um desses marcadores foi então administrado diretamente ao jejuno de ratos não anestesiados, juntamente com 300μL de formulação básica, e o seu grau de penetração foi avaliado pelo teste de níveis de dextrano no sangue.
[0295] Os níveis de dextrano no plasma foram medidos na pré- dosagem e aos 3 ', 6', 10 ', 25', 60 ', 90' minutos após a administração da formulação. valores de exposição, AUC (0-90), foram determinados e os resultados são mostrados na figura 7. Os dados são apresentados como MÉDIA ± DP, n>4.
[0296] Os resultados mostram que enquanto o menor marcador molecular testado (dextrano de PM médio = 4,4 kDa), penetra no intestino, quando administrado concomitantemente com uma formulação, conforme o tamanho da molécula aumenta, diminui o grau de penetração: uma molécula marcadora de 10 kDa penetra em uma menor medida e um marcador de 20 kDa de uma forma ainda menor. Uma molécula marcadora de 40 kDa mostra penetração mínima, enquanto uma molécula marcadora de 70 kDa não mostra penetração alguma (penetração basal). Estes resultados indicam que 40-70 kDa é um tamanho de corte para aumento de permeabilidade não específica por formulações da invenção. Assim, a administração de um grande volume de formulação (300μL) ao jejuno de ratos resultou em aumento da permeabilidade da barreira intestinal, e esta maior permeabilidade está restrita pelo tamanho molecular, apresentando um tamanho de corte de 40-70kDa e penetração mínima em 40kDa.
[0297] Valores publicados do tamanho de moléculas de risco (peso molecular e raio), que poderia estar presente no intestino são mostrados abaixo na Tabela 42. Tabela 42
[0298] A Tabela 42 demonstra que as moléculas potencialmente perigosas presentes no intestino estão acima do tamanho de corte de aumento de permeabilidade pelas formulações testadas, como mostrado acima. Assim, estes resultados sugerem que as formulações testadas não facilitarão a penetração de moléculas perigosas através da barreira intestinal e estas formulações podem, portanto, ser consideradas seguras. Outras formulações da invenção obtêm resultados semelhantes.
[0299] B. Formulação de dosagem repetida: A fim de investigar se a dosagem repetida de formulação afeta a permeabilidade intestinal, a formulação melhorada de octreotídeo (12% de octanoato de sódio com óleo de rícino) foi administrada a ratos durante 14 dias sequenciais utilizando o modelo in vivo acima (rato implantado com duas cânulas no jejuno) . Nos dias 1, 7 e 14 de administração, um marcador de permeabilidade de dextrano (FITC-dextrano de 4,4 kDa de PM; FD4), foi administrado 60 minutos após a administração da formulação. Isto foi para avaliar a permeabilidade do intestino pela penetração do FD4 do intestino para o sangue. Nenhuma diferença significativa na exposição a FD4 após 14 dias da dosagem de formulação repetida foi encontrada. Estes resultados sugerem que não há aumento da permeabilidade intestinal após este período de dosagem repetida de formulação e que a maior permeabilidade intestinal continua a ser um processo reversível durante este período.
[0300] Os resultados sugerem que a formulação não causa dano ao tecido intestinal, mas age especificamente abrindo a barreira intestinal, não mostrando efeito de aumento de permeabilidade aditivo.
[0301] Além do estudo no Exemplo acima, um estudo foi desenvolvido a fim de definir a evolução ao longo do tempo de aumento da permeabilidade intestinal devido às formulações da invenção, e a reversibilidade deste processo, utilizando dextrano como marcador de permeabilidade.
[0302] Para definir a janela de tempo de aumento da permeabilidade intestinal, um modelo in vivo foi desenvolvido em ratos, em que uma ou duas cânulas são implantadas no jejuno dos ratos. Dextrano marcado com FITC (peso molecular médio de 4,4kDa, FD4), que não tem virtualmente nenhuma penetração no intestino basal, serviu como um marcador molecular para testar a permeabilidade intestinal. Foi delineado um experimento em que o marcador de dextrano foi administrado concomitante à formulação (por uma cânula implantada no jejuno), ou em diferentes intervalos de tempo a partir da administração da formulação (por uma segunda cânula implantada no jejuno em separado). A permeabilidade intestinal foi avaliada pelo teste de penetração de FD4 no sangue. Aos ratos foi administrada uma formulação básica concomitantemente com o marcador de dextrano, ou a formulação básica e, em seguida, o marcador de dextrano em diferentes intervalos de tempo (10, 30 e 60 minutos). Amostras de sangue foram analisadas para a pré-administração da concentração de dextrano e aos 3, 6, 10, 25, 60 e 90 min após a administração de dextrano. Os resultados são mostrados na figura 8. Os dados são apresentados como Média ± DP, n>=5.
[0303] A figura 8 demonstra que o marcador de dextrano penetra no intestino na maior magnitude, quando administrado em conjunto com a formulação. Um intervalo de 10 minutos entre a administração da formulação e a administração do marcador de dextrano resulta em quantidade significativamente menor de penetração do marcador, e aumentando adicionalmente o intervalo resulta em redução exponencial da penetração do marcador.
[0304] Estes resultados mostram que, embora haja algum grau de intensificação de permeabilidade não específica pela formulação, ela está restrita a um período curto de tempo após a administração da formulação. A permeabilidade do intestino diminui drasticamente com o tempo e 60 minutos a partir da administração da formulação não há mais penetração do marcador. Assim, a administração da formulação ao intestino de ratos resulta em um período muito curto de hiperpermeabilidade da barreira intestinal. Outras formulações da invenção obtêm resultados semelhantes.
[0305] A fim de testar a farmacocinética de octreotídeo após a administração oral de octreotídeo formulado a macacos, cinco macacos cinomolgos receberam doses por via oral com cápsulas contendo uma formulação de óleo de rícino melhorada de octreotídeo (similar à formulação I da Tabela 35 - mas com maior carga de octreotídeo) . As cápsulas utilizadas foram cápsulas gelatinosas de tamanho 1 revestidas com 6,7% do revestimento entérico Acryl-EZE®; este revestimento impede a desintegração da cápsula no estômago e permite a abertura das cápsulas no intestino delgado dos animais tratados. A dose de octreotídeo utilizada foi de 5mg/cápsula.
[0306] Macacos foram mantidos em jejum durante a noite anterior à administração da cápsula. Após a administração oral, amostras de sangue foram retiradas por um período de 9,75 horas, processadas para plasma e analisadas quanto ao teor de octreotídeo pelo método LC/MS/MS: veja a figura 9.
[0307] Experimentos semelhantes foram realizados com a formulação de nenhum óleo de rícino/GTC melhorada (semelhante à formulação IV da Tabela 35, mas com maior carga da API) e resultados similares foram obtidos. Experiências semelhantes também foram realizadas com diferentes revestimentos entéricos e resultados semelhantes foram obtidos. A fim de comparar a farmacocinética de octreotídeo após a administração da formulação de octreotídeo melhorada, com a farmacocinética de octreotídeo injetado, solução de acetato de octreotídeo (0,1 mg/macaco) foi administrada por via subcutânea a dois macacos do grupo acima para servir como referência. Amostras de sangue foram retiradas por um período de quatro horas, processadas para plasma e analisadas quanto ao teor de octreotídeo pelo método LC/MS/MS.
[0308] As farmacocinéticas de octreotídeo posterior ao octreotídeo oral e solução de octreotídeo injetada por via subcutânea foram comparadas (veja as figuras 9 e 10). Os resultados da formulação oral apresentaram absorção ao longo de um período de poucas horas. A forma do gráfico foi modificada em relação ao subcutâneo, apresentando liberação mais lenta, porém mais longa de octreotídeo no sangue. Isto pode ser vantajoso já que isso permite a persistência de octreotídeo por mais tempo no sangue potencialmente prolongando a janela de atividade.
[0309] Uma dose aprovada para o acetato de octreotídeo injetado em seres humanos é de 0,1mg/paciente. Os resultados acima nos macacos sugerem que a formulação melhorada contendo cerca de 10 mg de octreotídeo por dose irá gerar exposição terapêutica em seres humanos.
[0310] Formulações de octreotídeo básicas e melhoradas da invenção foram mantidas tanto em 40C como em 250C e foram testadas pelo conteúdo de octreotídeo periodicamente. Descobriu-se que ambas as formulações eram estáveis.
[0311] A. Vancomicina: A Tabela 43 abaixo descreve uma formulação melhorada de vancomicina, contendo 10% de PVP e 15% de octanoato de sódio na fração hidrofílica, e contendo tricaprilato de glicerila como o principal constituinte do meio hidrofóbico. A vancomicina foi obtida a partir de Gold Biotechnology. Tabela 43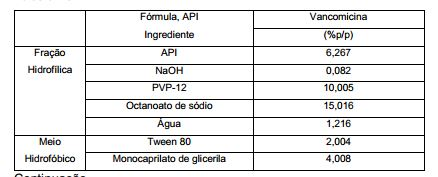 Continuação...
Continuação...
[0312] Em um experimento preliminar, a formulação descrita acima na Tabela 43 foi administrada diretamente ao jejuno de ratos não anestesiados e os níveis de vancomicina no plasma foram medidos após a administração da formulação. Valores de exposição, AUC, foram determinados para a formulação. Os resultados são aqueles em que a BA absoluta é de cerca de 5% (comparada a IV, n = 6). Quando a vancomicina em solução solução salina foi administrada ao jejuno de ratos não anestesiados, nenhum BA foi detectado.
[0313] Interferon-alfa: A Tabela 44 abaixo descreve uma formulação melhorada de interferon-alfa, contendo 10% de PVP e 15% de octanoato de sódio na fração hidrofílica, e contendo tricaprilato de glicerila como o principal constituinte do meio hidrofóbico. O interferon- alfa é fornecido em um tampão (da Intas Biopharmaceuticals) e os ingredientes do tampão de interferon-alfa na formulação são marcados com um asterisco (*). Tabela 44 Continuação...
Continuação...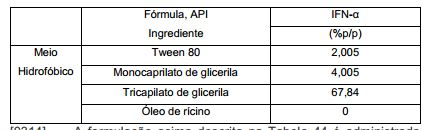
[0314] A formulação acima descrita na Tabela 44 é administrada diretamente ao jejuno de ratos não anestesiados. Os níveis plasmáticos de interferon-alfa são medidos após a administração da formulação.
[0315] C. Terlipressina: A Tabela 45 abaixo descreve uma formulação básica de terlipressina e uma formulação melhorada de terlipressina contendo 10% de PVP e 15% de octanoato de sódio na fração hidrofílica, e contendo tricaprilato de glicerila como o principal constituinte do meio hidrofóbico. A terlipressina foi obtida a partir de Bambio. A formulação básica foi elaborada essencialmente como descrito acima e a formulação melhorada também é elaborada essencialmente como descrito acima. Tabela 45
 Continuação...
Continuação...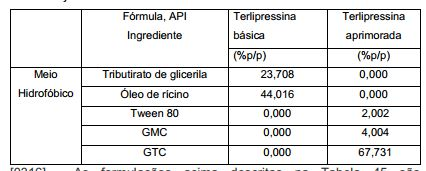
[0316] As formulações acima descritas na Tabela 45 são administradas diretamente ao jejuno de ratos não anestesiados. Os níveis plasmáticos de terlipressina são medidos após a administração da formulação.
[0317] Um dos melhores efeitos caracterizados de octreotídeo é a inibição da liberação de hormônio do crescimento. A fim de testar a eficácia de uma formulação de octreotídeo da invenção sobre a inibição do hormônio do crescimento, um modelo em ratos foi usado em que os níveis do hormônio do crescimento endógeno de rato (rGH) foram monitorados após a administração da formulação de octreotídeo ao jejuno do modelo de rato não anestesiado (descrito acima). Demonstrou-se que a administração de uma formulação básica de octreotídeo (contendo 12% de octanoato de sódio) ao jejuno de ratos reduz níveis de rGH em 87,4% em relação à administração de um controle de solução salina.
[0318] Este resultado demonstra que as formulações de octreotídeo aqui descritas permitem a distribuição de octreotídeo em sua forma ativa a partir do lúmen intestinal para a corrente sanguínea.
[0319] Um estudo de administração de toxicidade de 28 dias do controle de formulação (excipientes apenas, sem carga) foi realizado em ratos Wistar. Os animais do grupo teste foram administrados diariamente por via retal com a dose máxima viável de formulação (100 μL/animal/dia) durante 28 dias consecutivos. O grupo teste foi comparado a dois grupos controle: um grupo naive (sem tratamento) e um grupo ao qual foi administrado solução salina, (n= 15 / grupo).
[0320] Observações clínicas gerais foram feitas duas vezes por dia, e detalhadas observações clínicas foram realizadas semanalmente. Peso corporal e consumo de alimentos foram medidos semanalmente. Patologia clínica e patologia bruta foram realizadas um dia após o último tratamento. O exame histológico foi realizado no reto, cólon, fígado e rins, e não foram detectados efeitos tóxicos. Houve histopatologia limpa com nenhum GI local ou descobertas sistêmicas, nenhuma descoberta clínica relacionada à formulação, nenhuma alteração nos parâmetros químicos sanguíneos e hematológicos, nenhuma descoberta macroscópica na necropsia e nenhuma mortalidade. Em conclusão, este experimento demonstrou que não houve toxicidade observada durante uma dosagem diária por via retal de formulação de ratos durante 28 dias consecutivos.
[0321] Tendo assim descrito diversos aspectos de pelo menos uma modalidade, é para ser apreciado que várias alterações, modificações e melhorias ocorrerão prontamente para aqueles versados na técnica. Tais alterações, modificações e melhorias são destinadas a fazer parte dessa divulgação e destinam-se a estar dentro do escopo da invenção.
[0322] Assim, a descrição e desenhos precedentes são a título de exemplo apenas, e o escopo da invenção deve ser determinado a partir da construção adequada das reivindicações anexadas e seus equivalentes.
Claims (18)
1. Composição farmacêutica, caracterizada pelo fato de que compreende uma suspensão compreendendo uma mistura de um meio hidrofóbico e uma forma sólida, sendo que a forma sólida compreende uma quantidade terapeuticamente eficaz de um polipeptídeo, e pelo menos um sal de um ácido graxo de cadeia média, sendo que o sal de ácido graxo de cadeia média está presente na composição em uma quantidade de 10% a 50% em peso, 11% a 40% em peso, ou 12% a 18% em peso, ou em que o sal de ácido graxo de cadeia média está presente na forma sólida em uma quantidade de 50% a 90% em peso ou 70% a 80% em peso.
2. Composição de acordo com a reivindicação 1, caracterizada pelo fato de que o polipeptídeo é octreotídeo.
3. Composição de acordo com a reivindicação 1 ou 2, caracterizada pelo fato de que a forma sólida compreende uma partícula e/ou um pó.
4. Composição de acordo com qualquer uma das reivindicações 1 a 3, caracterizada pelo fato de que o teor de água na composição farmacêutica é inferior a 6% em peso ou inferior a 2% em peso, ou o teor de água na forma sólida é inferior a 6% em peso ou inferior a 2% em peso.
5. Composição de acordo com qualquer uma das reivindicações 1 a 4, caracterizada pelo fato de que o sal de ácido graxo de cadeia média apresenta um comprimento de cadeia de cerca de 6 a cerca de 14 átomos de carbono, ou o sal de ácido graxo de cadeia média é hexanoato de sódio, heptanoato de sódio, octanoato de sódio, nonanoato de sódio, decanoato de sódio, undecanoato de sódio, dodecanoato de sódio, tridecanoato de sódio ou tetradecanoato de sódio, ou um sal de potássio ou lítio ou amônio correspondente ou uma combinação dos mesmos.
6. Composição de acordo com qualquer uma das reivindicações 1 a 5, caracterizada pelo fato de que compreende ainda um polímero formador de matriz, presente na composição em uma quantidade de 3% ou mais em peso.
7. Composição de acordo com a reivindicação 6, caracterizada pelo fato de que o polímero formador de matriz é selecionado do grupo que consiste em: dextrano, polivinilpirrolidona (PVP), polissacarídeos iônicos ou neutros, poli(ácido acrílico), derivados de ácido poli metacrilato ou poli(vinil álcool).
8. Composição de acordo com a reivindicação 7, caracterizada pelo fato de que a polivinilpirrolidona está presente na composição em uma quantidade de 2% a 20% em peso, 5% a 15% em peso ou 10 % em peso.
9. Composição de acordo com a reivindicação 7 ou 8, caracterizada pelo fato de que a polivinilpirrolidona é PVP-12 e/ou a polivinilpirrolidona tem um peso molecular de cerca de 3000.
10. Composição de acordo com qualquer uma das reivindicações 1 a 9, caracterizada pelo fato de que o meio hidrofóbico compreende óleo de rícino ou tricaprilato de glicerila ou tributirato de glicerila ou uma combinação dos mesmos.
11. Composição de acordo com qualquer uma das reivindicações 1 a 10, caracterizada pelo fato de que o componente principal em peso do meio hidrofóbico é tricaprilato de glicerila, ou o meio hidrofóbico consiste essencialmente em tricaprilato de glicerila.
12. Composição de acordo com qualquer uma das reivindicações 1 a 11, caracterizada pelo fato de que o meio hidrofóbico compreende um composto alifático, olefínico, cíclico ou aromático, ou sendo que o meio hidrofóbico compreende um óleo mineral, uma parafina, um ácido graxo como o ácido octanoico, um monoglicerídeo, um diglicerídeo, um triglicerídeo, um éter ou um éster, ou uma combinação dos mesmos, sendo que, preferencialmente, o meio hidrofóbico compreende um triglicerídeo que é um triglicerídeo de cadeia longa, um triglicerídeo de cadeia média, um triglicerídeo de cadeia curta ou uma mistura dos mesmos.
13. Composição de acordo com qualquer uma das reivindicações 1 a 12, caracterizada pelo fato de que o meio hidrofóbico compreende ainda um tensoativo iônico ou um tensoativo não-iônico.
14. Composição de acordo com a reivindicação 13, caracterizada pelo fato de que o tensoativo é lecitina ou um sal de bile ou um detergente, ou o tensoativo é um monoglicerídeo, um cremóforo, um éter de álcool graxo de polietileno glicol, um éster de ácido graxo de sorbitano, um éster de ácido graxo de polioxietileno sorbitano, ésteres de polioxietileno de ácido 12-hidroxiesteárico, ou um poloxâmero ou uma combinação dos mesmos, sendo que, preferencialmente, o monoglicerídeo é monocaprilato de glicerila, mono-octanoato de glicerila, monodecanoato de glicerila, monolaurato de glicerila, monomiristato de glicerila, monopalmitato de glicerila ou mono-oleato de glicerila ou monoestearato de glicerila ou uma combinação dos mesmos, ou o éster de ácido graxo sorbitano compreende monolaurato de sorbitano, mono-oleato de sorbitano ou monopalmitato de sorbitano ou uma combinação dos mesmos, ou o éster de ácido graxo sorbitano de polioxietileno compreende mono-oleato de polioxietileno sorbitano, monoestearato de polioxietileno sorbitano ou monopalmitato de polioxietileno sorbitano ou uma combinação dos mesmos.
15. Composição de acordo com a reivindicação 11, caracterizada pelo fato de que o meio hidrofóbico contém ainda óleo de rícino e/ou monocaprilato de glicerila.
16. Processo para produzir uma composição farmacêutica como definida em qualquer uma das reivindicações 1 a 15, caracterizado pelo fato de que compreende prover uma forma sólida de uma quantidade terapeuticamente eficaz de um polipéptídeo e uma forma sólida que compreende um sal de ácido graxo de cadeia média, e suspender as formas sólidas em um meio hidrofóbico, para produzir uma suspensão que contém na forma sólida o octreotídeo e o sal de ácido graxo de cadeia média, produzindo assim a composição farmacêutica, sendo que a composição farmacêutica contém 10% ou mais em peso do sal de ácido graxo de cadeia média.
17. Composição, de acordo com qualquer uma das reivindicações 1 a 15, caracterizada pelo fato de que compreende 10% a 20%, preferencialmente 12% a 21%, preferencialmente 15%, de sal de ácido graxo de cadeia média, preferencialmente octanoato de sódio, 5% a 10%, preferencialmente 10%, de PVP-12, em que o meio hidrofóbico compreende 20% a 80%, preferencialmente 30% a 70%, de triglicerídeo, preferencialmente tricaprilato de glicerila ou tributirato de glicerila ou óleo de rícino ou uma mistura dos mesmos, 3% a 10% de tensoativos, preferencialmente 6%, de monocaprilato de glicerila e mono-oleato de polioxietileno sorbitano e 1% de água, sendo que o octreotídeo está presente em uma quantidade de menos de 33%, ou menos de 25%, ou menos de 10%, ou menos de 1% ou menos de 0,1%.
18. Forma de dosagem oral, caracterizada pelo fato de que contém a composição farmacêutica como definida em qualquer uma das reivindicações 1 a 15 ou 17, que pode ser opcionalmente uma cápsula e pode ser um gel rígido ou gel macio e pode opcionalmente ser de revestimento entérico.
Applications Claiming Priority (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US9771608P | 2008-09-17 | 2008-09-17 | |
| US61/097,716 | 2008-09-17 | ||
| US14168608P | 2008-12-31 | 2008-12-31 | |
| US61/141,686 | 2008-12-31 | ||
| US16138709P | 2009-03-18 | 2009-03-18 | |
| US61/161,387 | 2009-03-18 | ||
| PCT/IB2009/007155 WO2010032140A2 (en) | 2008-09-17 | 2009-09-17 | Pharmaceutical compositions and related methods of delivery |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| BRPI0918652A2 BRPI0918652A2 (pt) | 2015-08-25 |
| BRPI0918652B1 true BRPI0918652B1 (pt) | 2021-10-19 |
Family
ID=41277888
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| BRPI0918652-2A BRPI0918652B1 (pt) | 2008-09-17 | 2009-09-17 | Composição farmacêutica compreendendo um meio hidrofóbico e uma forma sólida que compreende polipeptídeo e sal de ácido graxo de cadeia média, processo de produção da mesma e forma de dosagem oral |
Country Status (25)
| Country | Link |
|---|---|
| US (12) | US20100105627A1 (pt) |
| EP (3) | EP3861986B1 (pt) |
| JP (7) | JP5623406B2 (pt) |
| KR (2) | KR101814186B1 (pt) |
| CN (2) | CN102176900B (pt) |
| AU (1) | AU2009294320B2 (pt) |
| BR (1) | BRPI0918652B1 (pt) |
| CA (3) | CA3080587C (pt) |
| CY (1) | CY1119148T1 (pt) |
| DK (1) | DK2343982T3 (pt) |
| ES (2) | ES2629131T3 (pt) |
| GB (2) | GB2463978B8 (pt) |
| HR (1) | HRP20170929T1 (pt) |
| HU (1) | HUE033611T2 (pt) |
| IL (3) | IL211799A (pt) |
| LT (1) | LT2343982T (pt) |
| MX (3) | MX395136B (pt) |
| NZ (1) | NZ591810A (pt) |
| PL (1) | PL2343982T3 (pt) |
| PT (1) | PT2343982T (pt) |
| RU (3) | RU2678833C2 (pt) |
| SI (1) | SI2343982T1 (pt) |
| SM (1) | SMT201700304T1 (pt) |
| WO (1) | WO2010032140A2 (pt) |
| ZA (1) | ZA201101657B (pt) |
Families Citing this family (75)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US8119159B2 (en) * | 1999-02-22 | 2012-02-21 | Merrion Research Iii Limited | Solid oral dosage form containing an enhancer |
| US7658938B2 (en) | 1999-02-22 | 2010-02-09 | Merrion Reasearch III Limited | Solid oral dosage form containing an enhancer |
| US20070148228A1 (en) * | 1999-02-22 | 2007-06-28 | Merrion Research I Limited | Solid oral dosage form containing an enhancer |
| US7731947B2 (en) | 2003-11-17 | 2010-06-08 | Intarcia Therapeutics, Inc. | Composition and dosage form comprising an interferon particle formulation and suspending vehicle |
| CA2563533C (en) | 2004-04-15 | 2013-10-01 | Shmuel A. Ben-Sasson | Compositions capable of facilitating penetration across a biological barrier |
| WO2006083761A2 (en) | 2005-02-03 | 2006-08-10 | Alza Corporation | Solvent/polymer solutions as suspension vehicles |
| US11246913B2 (en) | 2005-02-03 | 2022-02-15 | Intarcia Therapeutics, Inc. | Suspension formulation comprising an insulinotropic peptide |
| BRPI0710503A2 (pt) | 2006-04-07 | 2011-08-16 | Merrion Res Iii Ltd | uso de uma composição farmacêutica, composição farmacêutica, e, forma de dosagem oral |
| KR101106510B1 (ko) | 2006-05-30 | 2012-01-20 | 인타르시아 세라퓨틱스 인코포레이티드 | 투피스, 내부채널 삼투압 전달 시스템 유동 조절기 |
| EP2359808B1 (en) | 2006-08-09 | 2013-05-22 | Intarcia Therapeutics, Inc | Osmotic delivery systems and piston assemblies |
| WO2008133908A2 (en) | 2007-04-23 | 2008-11-06 | Intarcia Therapeutics, Inc. | Suspension formulations of insulinotropic peptides and uses thereof |
| CA2726861C (en) | 2008-02-13 | 2014-05-27 | Intarcia Therapeutics, Inc. | Devices, formulations, and methods for delivery of multiple beneficial agents |
| RU2517135C2 (ru) * | 2008-05-07 | 2014-05-27 | Меррион Рисерч Iii Лимитед | Композиции пептидов и способы их получения |
| KR101814186B1 (ko) | 2008-09-17 | 2018-03-14 | 키아스마 인코포레이티드 | 약제학적 조성물 및 연관된 투여방법 |
| CA2751854A1 (en) * | 2009-02-25 | 2010-09-02 | Merrion Research Iii Limited | Composition and drug delivery of bisphosphonates |
| RU2547990C2 (ru) | 2009-09-28 | 2015-04-10 | Интарсия Терапьютикс, Инк. | Быстрое достижение и/или прекращение существенной стабильной доставки лекарственного средства |
| US8759284B2 (en) | 2009-12-24 | 2014-06-24 | Rani Therapeutics, Llc | Therapeutic agent preparations for delivery into a lumen of the intestinal tract using a swallowable drug delivery device |
| US20110182985A1 (en) * | 2010-01-28 | 2011-07-28 | Coughlan David C | Solid Pharmaceutical Composition with Enhancers and Methods of Preparing thereof |
| US20110311621A1 (en) * | 2010-03-16 | 2011-12-22 | Paul Salama | Pharmaceutical compositions and methods of delvery |
| US9089484B2 (en) * | 2010-03-26 | 2015-07-28 | Merrion Research Iii Limited | Pharmaceutical compositions of selective factor Xa inhibitors for oral administration |
| CA2808978A1 (en) | 2010-08-23 | 2012-03-01 | Shmuel Ben-Sasson | Compositions for gastric delivery of active agents |
| KR20140046395A (ko) * | 2010-12-15 | 2014-04-18 | 메리온 리서치 Ⅲ 리미티드 | 경구 투여를 위한 선택적 인자 xa 억제제의 제약 조성물 |
| US9284367B2 (en) | 2010-12-23 | 2016-03-15 | Rani Therapeutics, Llc | Therapeutic agent preparations for delivery into a lumen of the intestinal tract using a swallowable drug delivery device |
| US8734429B2 (en) | 2010-12-23 | 2014-05-27 | Rani Therapeutics, Llc | Device, system and methods for the oral delivery of therapeutic compounds |
| US9629799B2 (en) | 2010-12-23 | 2017-04-25 | Rani Therapeutics, Llc | Therapeutic agent preparations for delivery into a lumen of the intestinal tract using a swallowable drug delivery device |
| US8980822B2 (en) | 2010-12-23 | 2015-03-17 | Rani Therapeutics, Llc | Therapeutic agent preparations comprising pramlintide for delivery into a lumen of the intestinal tract using a swallowable drug delivery device |
| US9415004B2 (en) | 2010-12-23 | 2016-08-16 | Rani Therapeutics, Llc | Therapeutic agent preparations for delivery into a lumen of the intestinal tract using a swallowable drug delivery device |
| US8809269B2 (en) | 2010-12-23 | 2014-08-19 | Rani Therapeutics, Llc | Therapeutic agent preparations comprising insulin for delivery into a lumen of the intestinal tract using a swallowable drug delivery device |
| US8969293B2 (en) | 2010-12-23 | 2015-03-03 | Rani Therapeutics, Llc | Therapeutic agent preparations comprising exenatide for delivery into a lumen of the intestinal tract using a swallowable drug delivery device |
| US9402807B2 (en) | 2010-12-23 | 2016-08-02 | Rani Therapeutics, Llc | Therapeutic agent preparations for delivery into a lumen of the intestinal tract using a swallowable drug delivery device |
| US10639272B2 (en) | 2010-12-23 | 2020-05-05 | Rani Therapeutics, Llc | Methods for delivering etanercept preparations into a lumen of the intestinal tract using a swallowable drug delivery device |
| US9402806B2 (en) | 2010-12-23 | 2016-08-02 | Rani Therapeutics, Llc | Therapeutic agent preparations for delivery into a lumen of the intestinal tract using a swallowable drug delivery device |
| US9861683B2 (en) | 2010-12-23 | 2018-01-09 | Rani Therapeutics, Llc | Therapeutic agent preparations for delivery into a lumen of the intestinal tract using a swallowable drug delivery device |
| US9259386B2 (en) | 2010-12-23 | 2016-02-16 | Rani Therapeutics, Llc | Therapeutic preparation comprising somatostatin or somatostatin analogoue for delivery into a lumen of the intestinal tract using a swallowable drug delivery device |
| US9283179B2 (en) | 2010-12-23 | 2016-03-15 | Rani Therapeutics, Llc | GnRH preparations for delivery into a lumen of the intestinal tract using a swallowable drug delivery device |
| KR20140026354A (ko) | 2011-01-07 | 2014-03-05 | 메리온 리서치 Ⅲ 리미티드 | 경구 투여용 철의 제약 조성물 |
| WO2012097155A1 (en) * | 2011-01-14 | 2012-07-19 | Chiasma Inc. | Improved pharmaceutical compositions for delivery of ferric iron compounds, and methods of use thereof |
| CN102145172B (zh) * | 2011-01-28 | 2012-09-26 | 瑞普(天津)生物药业有限公司 | 一种提高氨基糖苷类药物吸收度的组合物 |
| US20120208755A1 (en) | 2011-02-16 | 2012-08-16 | Intarcia Therapeutics, Inc. | Compositions, Devices and Methods of Use Thereof for the Treatment of Cancers |
| EP2526971A1 (en) * | 2011-05-25 | 2012-11-28 | ArisGen SA | Mucosal delivery of drugs |
| RU2639483C2 (ru) | 2012-03-19 | 2017-12-21 | Сидара Терапьютикс, Инк. | Режимы дозирования для соединений класса эхинокандинов |
| WO2014165607A2 (en) * | 2013-04-02 | 2014-10-09 | Stealth Peptides International, Inc. | Aromatic-cationic peptide formulations, compositions and methods of use |
| ES2502691B1 (es) * | 2013-09-25 | 2015-07-07 | Sani-Red, S.L. | Método de conservación y estabilización de proteínas, aplicable para desarrollos industriales de formulaciones de productos sanitarios, farmacéuticos y cosméticos |
| UY35790A (es) * | 2013-10-21 | 2015-05-29 | Teva Pharma | Marcadores genéticos que predicen la respuesta al acetato de glatiramer |
| US10493122B2 (en) | 2014-03-17 | 2019-12-03 | Mapi Pharma Ltd. | Sublingual delivery of glatiramer acetate |
| US10384959B2 (en) * | 2014-07-03 | 2019-08-20 | Shlomo Nir | Method of making and using granulated micelle-clay complexes for removal of pollutants from water |
| US9889085B1 (en) | 2014-09-30 | 2018-02-13 | Intarcia Therapeutics, Inc. | Therapeutic methods for the treatment of diabetes and related conditions for patients with high baseline HbA1c |
| AU2015360485B2 (en) * | 2014-12-10 | 2020-06-25 | Amryt Endo, Inc. | Oral octreotide administered in combination with other therapeutic agents |
| JP7211704B2 (ja) | 2015-01-29 | 2023-01-24 | ノヴォ ノルディスク アー/エス | Glp-1アゴニスト及び腸溶コーティングを含む錠剤 |
| WO2016126830A1 (en) | 2015-02-03 | 2016-08-11 | Chiasma Inc. | Method of treating diseases |
| RU2730996C2 (ru) | 2015-06-03 | 2020-08-26 | Интарсия Терапьютикс, Инк. | Системы установки и извлечения имплантата |
| GB201515387D0 (en) * | 2015-08-28 | 2015-10-14 | Amazentis Sa | Compositions |
| EP3405211A4 (en) * | 2016-01-21 | 2019-10-09 | Chiasma Inc. | ORAL OCTREOTIDE FOR THE TREATMENT OF DISEASES |
| WO2017134655A1 (en) * | 2016-02-01 | 2017-08-10 | Yissum Research Development Company Of The Hebrew University Of Jerusalem Ltd | Improved oral absorption of octreotide and salts thereof |
| KR102574993B1 (ko) | 2016-05-16 | 2023-09-06 | 인타르시아 세라퓨틱스 인코포레이티드 | 글루카곤-수용체 선택적 폴리펩티드 및 이들의 이용 방법 |
| USD860451S1 (en) | 2016-06-02 | 2019-09-17 | Intarcia Therapeutics, Inc. | Implant removal tool |
| USD840030S1 (en) | 2016-06-02 | 2019-02-05 | Intarcia Therapeutics, Inc. | Implant placement guide |
| IL313083A (en) * | 2016-10-21 | 2024-07-01 | Amryt Endo Inc | Terlipressin compositions and their methods of use |
| EP3538132B1 (en) | 2016-11-14 | 2020-12-30 | University of Copenhagen | Rectal insulin for treatment of inflammatory bowel diseases |
| JP7286542B2 (ja) | 2017-01-03 | 2023-06-05 | インターシア セラピューティクス,インコーポレイティド | Glp-1受容体アゴニストの持続的投与及び薬物の同時投与を含む方法 |
| IL324591A (en) | 2017-08-31 | 2026-01-01 | Daiichi Sankyo Co Ltd | Improved antibody-drug conjugate production methods |
| WO2019157453A1 (en) * | 2018-02-12 | 2019-08-15 | Trilogy Therapeutics, Inc. | Caspofungin compositions for inhalation |
| AU2019239073B2 (en) | 2018-03-19 | 2024-12-05 | Epicyt Pharma Ab | A composition for use in the treatment of conditions caused by calcium deficiency |
| BR112021004199A2 (pt) | 2018-09-07 | 2021-05-25 | R.P. Scherer Technologies, Llc | estabilização de formas de dosagem à base de lipídio sólido ou semissólido através da cura e da adição de tensoativo(s) de baixo teor de hlb |
| GB201904771D0 (en) | 2019-04-04 | 2019-05-22 | Orexo Ab | New pharmaceutical compositions |
| GB201904767D0 (en) | 2019-04-04 | 2019-05-22 | Orexo Ab | New pharmaceutical compositions |
| RU2717101C1 (ru) * | 2019-06-03 | 2020-03-18 | Андрей Александрович Иващенко | Анелированные 9-гидрокси-1,8-диоксо-1,3,4,8-тетрагидро-2Н-пиридо[1,2-a]пиразин-7-карбоксамиды - ингибиторы интегразы ВИЧ, способы их получения и применения |
| CN110720641A (zh) * | 2019-10-18 | 2020-01-24 | 广东海洋大学 | 一种具有减脂增肌功效的蛋白冻干块及其制备方法 |
| GB202004115D0 (en) * | 2020-03-20 | 2020-05-06 | Invex Therapeutics Ltd | Modified release formulations and dosage regimens |
| US20220202911A1 (en) * | 2020-12-28 | 2022-06-30 | Amryt Endo, Inc. | Oral octreotide therapy in combination with digoxin or lisinopril |
| US11141457B1 (en) | 2020-12-28 | 2021-10-12 | Amryt Endo, Inc. | Oral octreotide therapy and contraceptive methods |
| EP4373293A1 (en) * | 2021-07-22 | 2024-05-29 | Nicoventures Trading Limited | Compositions comprising constituents, derivatives or extracts of cannabis |
| US20260000620A1 (en) * | 2022-01-13 | 2026-01-01 | Amryt Endo, Inc. | Oral dosage forms |
| CN120936346A (zh) * | 2023-03-17 | 2025-11-11 | 百时美施贵宝公司 | 用于改善口服生物利用度的药物组合物 |
| WO2025247506A1 (en) * | 2024-05-31 | 2025-12-04 | Laxxon Medical Ag | Paste with an api and a permeation enhancer for 3d-printing |
Family Cites Families (202)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CH455823A (de) | 1964-08-01 | 1968-05-15 | Ceskoslovenska Akademie Ved | Verfahren zur Herstellung von Peptiden |
| US4411890A (en) | 1981-04-14 | 1983-10-25 | Beckman Instruments, Inc. | Synthetic peptides having pituitary growth hormone releasing activity |
| SE376722B (pt) * | 1971-03-30 | 1975-06-09 | S E Friberg | |
| BE795516A (fr) * | 1972-02-17 | 1973-08-16 | Ciba Geigy | Preparations de peptides huileuses et injectables et procede pour leur preparation |
| US4489097A (en) * | 1976-07-28 | 1984-12-18 | The Procter & Gamble Company | Intravenous solutions with antimicrobial agent |
| JPS53107408A (en) * | 1977-02-28 | 1978-09-19 | Yamanouchi Pharmaceut Co Ltd | Micellar preparation for rectal infusion |
| GB2051574B (en) * | 1979-05-10 | 1984-01-18 | Kyoto Pharma Ind | Adjuvant for promoting absorption of pharmacologically active substances through the rectum |
| US4544500A (en) | 1982-04-14 | 1985-10-01 | Scripps Clinic And Research Foundation | Synthetic foot and mouth disease antigen |
| US4589881A (en) | 1982-08-04 | 1986-05-20 | La Jolla Cancer Research Foundation | Polypeptide |
| US4508828A (en) | 1983-03-21 | 1985-04-02 | Immuno Nuclear Corporation | Bioassay of parathyroid hormone |
| US4572915A (en) * | 1984-05-01 | 1986-02-25 | Bioglan Laboratories | Clear micellized solutions of fat soluble essential nutrients |
| US4985404A (en) * | 1984-10-04 | 1991-01-15 | Monsanto Company | Prolonged release of biologically active polypeptides |
| US4650665A (en) * | 1985-02-08 | 1987-03-17 | Ethicon, Inc. | Controlled release of pharmacologically active agents from an absorbable biologically compatible putty-like composition |
| US4650787A (en) | 1985-04-25 | 1987-03-17 | Schally Andrew Victor | Biologically active octapeptides |
| US4732971A (en) | 1985-06-03 | 1988-03-22 | Eli Lilly And Company | Synthetic vaccines for foot and mouth disease |
| HU198626B (en) * | 1986-05-27 | 1989-11-28 | Sandoz Ag | Process for producing pharmaceutical compositions comprising somatostatin analogues as active ingredient |
| DE3709861A1 (de) * | 1987-03-25 | 1988-10-06 | Henkel Kgaa | Emulgierende suppositoriengrundmassen und daraus hergestellte zaepfchen |
| US4839344A (en) | 1987-06-12 | 1989-06-13 | Eastman Kodak Company | Polypeptide compounds having growth hormone releasing activity |
| DE3738236A1 (de) * | 1987-11-11 | 1989-05-24 | Euro Celtique Sa | Beisskapsel |
| FR2627696B1 (fr) * | 1988-02-26 | 1991-09-13 | Fournier Innovation Synergie | Nouvelle forme galenique du fenofibrate |
| NO179479C (no) * | 1988-03-11 | 1996-10-16 | Teikoku Seiyaku Kk | Fremgangsmåte for fremstilling av et intravaginalt farmasöytisk preparat |
| GB8822857D0 (en) | 1988-09-29 | 1988-11-02 | Patralan Ltd | Pharmaceutical formulations |
| ZA898331B (en) | 1988-11-22 | 1990-07-25 | Hoffmann La Roche | Pharmaceutical compositions |
| NZ233403A (en) * | 1989-04-28 | 1992-09-25 | Mcneil Ppc Inc | Simulated capsule-like medicament |
| FI920646A0 (fi) * | 1989-08-17 | 1992-02-14 | Cortecs Ltd | Farmaceutiska preparat. |
| US5665384A (en) * | 1990-04-06 | 1997-09-09 | Rhone-Poulenc Rorer S.A. | Oily capsules of ketoprofen |
| GB9013448D0 (en) | 1990-06-15 | 1990-08-08 | Sandoz Ltd | Pharmaceutical resorption-improved somatostatin compositions,their preparation and use |
| SE9003100D0 (sv) | 1990-09-28 | 1990-09-28 | Kabivitrum Ab | Lipid formulation system |
| IT1243435B (it) | 1990-10-05 | 1994-06-10 | Altergon Sa | Composizioni farmaceutiche per uso topico comprendenti acido ialuronico sale sodico e sostanze disinfettanti |
| US5254331A (en) * | 1991-09-12 | 1993-10-19 | Chanel, Inc. | Skin cream composition |
| EP0661052A1 (en) * | 1991-11-08 | 1995-07-05 | Kyoto Pharmaceutical Industries, Ltd. | Pharmaceutical preparation containing prostaglandin compound for rectal or vaginal administration |
| US5206219A (en) * | 1991-11-25 | 1993-04-27 | Applied Analytical Industries, Inc. | Oral compositions of proteinaceous medicaments |
| US5478577A (en) | 1993-11-23 | 1995-12-26 | Euroceltique, S.A. | Method of treating pain by administering 24 hour oral opioid formulations exhibiting rapid rate of initial rise of plasma drug level |
| US5246716A (en) * | 1992-01-10 | 1993-09-21 | W. Neudorff Gmbh Kg | Fatty acid-based antifungal composition having residual activity |
| DE4203170A1 (de) | 1992-02-05 | 1993-08-12 | Basf Ag | Verfahren zur herstellung von e-oximethern von phenylglyoxylsaeureestern |
| GB9203769D0 (en) | 1992-02-21 | 1992-04-08 | Sandoz Ltd | Improvements in or relating to organic compounds |
| IL103224A (en) * | 1992-09-18 | 1998-08-16 | Teva Pharma | Stabilized pharmaceutical compositions containing derivatives of vitamins d2 and d3 |
| US5288492A (en) * | 1992-11-13 | 1994-02-22 | Morris Michael A | Decongestant composition containing aloe vera |
| DK17093D0 (da) * | 1993-02-15 | 1993-02-15 | Lyfjathroun H F | Farmaceutisk praeparat til topisk administrering af antigener og/eller vacciner til pattedyr via slimhinder |
| IT1263840B (it) * | 1993-03-30 | 1996-09-04 | Giuseppe Furiosi | Formulazioni orali di ubidecarenone in forma di capsule |
| US5318781A (en) * | 1993-04-06 | 1994-06-07 | Hoffmann-La Roche Inc. | Absorption enhancement of antibiotics |
| SE9302135D0 (sv) * | 1993-06-18 | 1993-06-18 | Kabi Pharmacia Ab | New pharmaceutical composition |
| US5506203C1 (en) * | 1993-06-24 | 2001-02-06 | Astra Ab | Systemic administration of a therapeutic preparation |
| TW402506B (en) * | 1993-06-24 | 2000-08-21 | Astra Ab | Therapeutic preparation for inhalation |
| US5462726A (en) * | 1993-12-17 | 1995-10-31 | Bristol-Myers Squibb Company | Method of inhibiting side effects of solvents containing ricinoleic acid or castor oil or derivatives thereof employing a thromboxane A2 receptor antagonist and pharmaceutical compositions containing such solvents |
| GB9405304D0 (en) * | 1994-03-16 | 1994-04-27 | Scherer Ltd R P | Delivery systems for hydrophobic drugs |
| US5561115A (en) * | 1994-08-10 | 1996-10-01 | Bayer Corporation | Low temperature albumin fractionation using sodium caprylate as a partitioning agent |
| GB9417524D0 (en) * | 1994-08-31 | 1994-10-19 | Cortecs Ltd | Pharmaceutical compositions |
| KR980700872A (ko) * | 1994-12-28 | 1998-04-30 | 야마구찌 다까시 | 경점막 투여용 제제(transmucosal preparation) |
| US5708017A (en) | 1995-04-04 | 1998-01-13 | Merck & Co., Inc. | Stable, ready-to-use pharmaceutical paste composition containing proton pump inhibitors |
| US5827534A (en) * | 1995-05-24 | 1998-10-27 | University Of Maryland At Baltimore | Oral dosage composition comprising zonnula occludens toxin and a therapeutic agent for intestinal delivery |
| US6696413B2 (en) * | 1995-06-16 | 2004-02-24 | Hexal Ag | Pharmaceutical preparation with cyclosporin A |
| GB9516268D0 (en) | 1995-08-08 | 1995-10-11 | Danbiosyst Uk | Compositiion for enhanced uptake of polar drugs from the colon |
| US5686488A (en) * | 1995-08-25 | 1997-11-11 | Alcon Laboratories, Inc. | Polyethoxylated castor oil products as anti-inflammatory agents |
| EP0760237A1 (en) * | 1995-08-30 | 1997-03-05 | Cipla Limited | Oil-in-water microemulsions |
| US5665711A (en) * | 1995-12-12 | 1997-09-09 | Yoshitomi Pharmaceutical Industries, Ltd. | Antitumor composition for oral administration |
| US5858401A (en) * | 1996-01-22 | 1999-01-12 | Sidmak Laboratories, Inc. | Pharmaceutical composition for cyclosporines |
| US6280745B1 (en) * | 1997-12-23 | 2001-08-28 | Alliance Pharmaceutical Corp. | Methods and compositions for the delivery of pharmaceutical agents and/or the prevention of adhesions |
| US6190702B1 (en) | 1996-03-28 | 2001-02-20 | Takeda Chemical Industries, Ltd. | Sustained-released material prepared by dispersing a lyophilized polypeptide in an oil phase |
| US6214792B1 (en) | 1996-04-12 | 2001-04-10 | David Lew Simon | Method for treating acute and severe diarrhea |
| US5726154A (en) * | 1996-06-28 | 1998-03-10 | University Of Utah Research Foundation | Stabilization and oral delivery of calcitonin |
| WO1998002186A1 (en) * | 1996-07-11 | 1998-01-22 | Farmarc Nederland B.V. | Inclusion complex containing indole selective serotonin agonist |
| US6512010B1 (en) * | 1996-07-15 | 2003-01-28 | Alza Corporation | Formulations for the administration of fluoxetine |
| US5760096A (en) * | 1996-10-18 | 1998-06-02 | Thornfeldt; Carl R. | Potent penetration enhancers |
| US7091183B1 (en) | 1996-12-03 | 2006-08-15 | Boston Medical Center Corporation | Specific antagonists for glucose-dependent insulinotropic polypeptide (GIP) |
| JP3502762B2 (ja) * | 1997-02-25 | 2004-03-02 | 日清オイリオ株式会社 | 油性組成物及びその製造方法 |
| US6193986B1 (en) | 1997-02-25 | 2001-02-27 | The Nisshin Oil Mills, Ltd. | Oily composition with increased stability and process for producing the same |
| JPH10265380A (ja) * | 1997-03-17 | 1998-10-06 | Bristol Myers Squibb Co | 抗ガン剤 |
| WO1998042311A1 (en) * | 1997-03-25 | 1998-10-01 | Takeda Chemical Industries, Ltd. | Gastrointestinal mucosa-adherent pharmaceutical composition |
| DE69807281T2 (de) * | 1997-05-14 | 2003-04-17 | Mitsubishi Chemical Corp., Tokio/Tokyo | Difluprednat enthaltende Zusammensetzungen |
| IT1296914B1 (it) * | 1997-12-01 | 1999-08-03 | Maria Rosa Gasco | Composizione farmaceutica comprendente microparticelle atte al passaggio transmucosale ed al superamento della barriera |
| IL123143A (en) * | 1998-02-02 | 2001-08-26 | Agis Ind 1983 Ltd | Pharmaceutical compositions containing mupirocin |
| JPH11246439A (ja) * | 1998-03-02 | 1999-09-14 | Hisamitsu Pharmaceut Co Inc | 経粘膜用吸収促進剤 |
| US6326360B1 (en) * | 1998-03-11 | 2001-12-04 | Grelan Pharmaceuticals Co., Ltd. | Bubbling enteric coated preparations |
| US6322550B2 (en) * | 1998-04-14 | 2001-11-27 | Hisamitsu Pharmaceutical Co., Inc. | Method for transdermal administration of GP IIb/IIIa antagonist |
| CA2331388A1 (en) * | 1998-06-26 | 2000-01-06 | Michael Thomas Clark | Improved process for preparing schiff base adducts of amines with o-hydroxy aldehydes and compositions of matter based thereon |
| US6150333A (en) | 1998-07-30 | 2000-11-21 | Biomeasure, Inc. | Methods of using a somatostatin analogue |
| US6284223B1 (en) * | 1998-10-15 | 2001-09-04 | Fluoroprobe, Inc. | Method for viewing tumor tissue located within a body cavity |
| IT1302682B1 (it) * | 1998-10-16 | 2000-09-29 | Formenti Farmaceutici Spa | Composizioni farmaceutiche orali contenenti buprenorfina |
| GB9823246D0 (en) * | 1998-10-24 | 1998-12-16 | Danbiosyst Uk | A nasal drug delivery composition |
| US6838091B2 (en) * | 1998-12-18 | 2005-01-04 | Abbott Laboratories | Formulations comprising lipid-regulating agents |
| US6368622B2 (en) * | 1999-01-29 | 2002-04-09 | Abbott Laboratories | Process for preparing solid formulations of lipid regulating agents with enhanced dissolution and absorption |
| WO2000047203A1 (en) | 1999-02-12 | 2000-08-17 | Mqs, Inc. | Formulation and system for intra-oral delivery of pharmaceutical agents |
| US20070148228A1 (en) | 1999-02-22 | 2007-06-28 | Merrion Research I Limited | Solid oral dosage form containing an enhancer |
| US8119159B2 (en) | 1999-02-22 | 2012-02-21 | Merrion Research Iii Limited | Solid oral dosage form containing an enhancer |
| US7658938B2 (en) * | 1999-02-22 | 2010-02-09 | Merrion Reasearch III Limited | Solid oral dosage form containing an enhancer |
| DE60038097T2 (de) | 1999-02-22 | 2009-02-12 | Merrion Research I Ltd. | Feste orale dosierungsform enthaltend einen resorptionsverstärker |
| US6267985B1 (en) | 1999-06-30 | 2001-07-31 | Lipocine Inc. | Clear oil-containing pharmaceutical compositions |
| US6383527B1 (en) * | 1999-03-04 | 2002-05-07 | Nps Pharmaceuticals, Inc. | Compositions comprising valerian extracts, isovaleric acid or derivatives thereof with a NSAID |
| US6632443B2 (en) * | 2000-02-23 | 2003-10-14 | National Research Council Of Canada | Water-soluble compositions of bioactive lipophilic compounds |
| US6485706B1 (en) * | 1999-06-04 | 2002-11-26 | Delrx Pharmaceutical Corp. | Formulations comprising dehydrated particles of pharma-ceutical agents and process for preparing the same |
| US20030235595A1 (en) | 1999-06-30 | 2003-12-25 | Feng-Jing Chen | Oil-containing, orally administrable pharmaceutical composition for improved delivery of a therapeutic agent |
| US6159935A (en) | 1999-11-29 | 2000-12-12 | Pharmacia & Upjohn Co. | Method for preventing diarrhea |
| FR2803202B1 (fr) * | 2000-01-03 | 2004-04-16 | Capsulis | Compositions pharmaceutiques destinees a une adminstration par voie orale |
| GB0018891D0 (en) | 2000-08-01 | 2000-09-20 | Novartis Ag | Organic compounds |
| CN1141974C (zh) * | 2000-06-07 | 2004-03-17 | 张昊 | 结肠定位释放的口服生物制剂 |
| US6664234B1 (en) * | 2000-06-30 | 2003-12-16 | Monsanto Technology Llc | Non-aqueous injectable formulation preparation with pH adjusted for extended release of somatotropin |
| FR2811571B1 (fr) | 2000-07-11 | 2002-10-11 | Flamel Tech Sa | Composition pharmaceutique orale, permettant la liberation controlee et l'absorption prolongee d'un principe actif |
| EP1188443A1 (en) | 2000-09-15 | 2002-03-20 | Ferring BV | Improved protocol for paracentesis |
| US20020091623A1 (en) * | 2000-12-04 | 2002-07-11 | Daniels Alan F. | System and methods for an electronic real estate trading environment |
| JP5026655B2 (ja) * | 2001-04-18 | 2012-09-12 | プロメティック、バイオサイエンシーズ、インコーポレーテッド | 好中球の生存及び活性化因子としての中鎖脂肪酸、グリセリド及び類似体 |
| EP1392272B1 (en) * | 2001-05-11 | 2014-08-06 | Merrion Research III Limited | Permeation enhancers |
| US6528667B2 (en) * | 2001-05-14 | 2003-03-04 | Phoenix Research Corporation | Phosphated castor oil and derivatives |
| US20030070584A1 (en) | 2001-05-15 | 2003-04-17 | Cynthia Gulian | Dip coating compositions containing cellulose ethers |
| WO2003004001A1 (en) | 2001-07-06 | 2003-01-16 | Lifecycle Pharma A/S | Controlled agglomeration |
| US6720002B2 (en) * | 2001-07-20 | 2004-04-13 | R.P. Scherer Technologies, Inc. | Antihistamine formulations for soft capsule dosage forms |
| CN1335182A (zh) | 2001-08-08 | 2002-02-13 | 华中科技大学 | 胰岛素口腔喷剂及其制备工艺 |
| KR100425755B1 (ko) * | 2001-08-27 | 2004-04-03 | 주식회사 원진신약 | 이트라코나졸을 함유하는 조성물 및 그 제조방법 |
| US20030095928A1 (en) * | 2001-09-19 | 2003-05-22 | Elan Pharma International Limited | Nanoparticulate insulin |
| US20060003012A9 (en) | 2001-09-26 | 2006-01-05 | Sean Brynjelsen | Preparation of submicron solid particle suspensions by sonication of multiphase systems |
| US20060052404A1 (en) * | 2001-10-26 | 2006-03-09 | Rudolph Alfred R | Pharmaceutical formulations comprising substituted xanthine compounds |
| US6710195B2 (en) * | 2001-11-26 | 2004-03-23 | Supergen, Inc. | Method for preparing and using polyoxyethylated castor oil in pharmaceutical compositions |
| AU2002353118A1 (en) * | 2001-12-11 | 2003-07-24 | Dor Biopharma, Inc. | Lipid particles and suspensions and uses thereof |
| AU2002364586A1 (en) | 2001-12-21 | 2003-07-30 | Delta Biotechnology Limited | Albumin fusion proteins |
| US6890961B2 (en) * | 2002-02-01 | 2005-05-10 | Micelle Products, Inc. | Clear micellized formulations of β-carotene and method of treating leukoplakia |
| US20030162695A1 (en) | 2002-02-27 | 2003-08-28 | Schatzberg Alan F. | Glucocorticoid blocking agents for increasing blood-brain barrier permeability |
| WO2003079991A2 (en) | 2002-03-20 | 2003-10-02 | Advanced Inhalation Research, Inc. | Method for administration of growth hormone via pulmonary delivery |
| US20050287203A1 (en) * | 2002-05-08 | 2005-12-29 | Nijs De H | Formulation comprising testosteron undecanoate and castor oil |
| CZ294371B6 (cs) * | 2002-06-10 | 2004-12-15 | Pliva - Lachema, A. S. | Stabilizovaná farmaceutická kompozice na bázi polyoxyethylovaného ricinového oleje a způsob její přípravy |
| FR2842736B1 (fr) * | 2002-07-26 | 2005-07-22 | Flamel Tech Sa | Formulation pharmaceutique orale sous forme d'une pluralite de microcapsules permettant la liberation prolongee de principe(s) actif(s) peu soluble(s) |
| JP4771697B2 (ja) * | 2002-09-24 | 2011-09-14 | サントリーホールディングス株式会社 | 認知能力の正常応答の低下予防、改善又は向上作用を有する組成物 |
| WO2004035624A2 (en) | 2002-10-14 | 2004-04-29 | Novo Nordisk A/S | Glucagon - like peptide - 2 variants |
| US20040097419A1 (en) * | 2002-11-19 | 2004-05-20 | Holger Petersen | Organic compounds |
| CA2509365C (en) * | 2002-12-09 | 2012-08-07 | American Bioscience, Inc. | Compositions and methods of delivery of pharmacological agents |
| ATE419846T1 (de) * | 2003-02-07 | 2009-01-15 | Prometic Biosciences Inc | Fettsäuren mittlerer kettenlänge, glyceride und analoga als stimulatoren der erythropoiese |
| US7803781B2 (en) | 2003-02-28 | 2010-09-28 | Isis Pharmaceuticals, Inc. | Modulation of growth hormone receptor expression and insulin-like growth factor expression |
| US20040185170A1 (en) | 2003-03-21 | 2004-09-23 | Shubha Chungi | Method for coating drug-containing particles and formulations and dosage units formed therefrom |
| GB0307082D0 (en) | 2003-03-27 | 2003-04-30 | Gyne Ideas Ltd | Drug delivery device and method |
| DE10328905B4 (de) | 2003-06-26 | 2015-07-09 | Hochland Natec Gmbh | Form- und Kühlvorrichtung |
| EP1648472A2 (en) | 2003-07-01 | 2006-04-26 | Ranbaxy Laboratories, Ltd. | Stable oral compositions of azithromycin monohydrate |
| WO2005021013A1 (ja) * | 2003-09-01 | 2005-03-10 | Earthus, Inc. | β−ヒドロキシ短〜中鎖脂肪酸重合体 |
| US20070172517A1 (en) | 2003-09-17 | 2007-07-26 | Ben-Sasson Shmuel A | Compositions capable of facilitation penetration across a biological barrier |
| WO2005034913A1 (ja) * | 2003-10-10 | 2005-04-21 | Eisai Co., Ltd. | 新規液剤組成物 |
| WO2005041901A2 (en) | 2003-11-03 | 2005-05-12 | Elixir Pharmaceuticals, Inc. | Therapeutics using somatostatin agonists |
| PL1682091T3 (pl) | 2003-11-07 | 2017-09-29 | Camurus Ab | Kompozycje lipidów i kationowych peptydów |
| AR046773A1 (es) | 2003-12-23 | 2005-12-21 | Novartis Ag | Formulaciones farmaceuticas de bisfosfonatos |
| ATE429900T1 (de) * | 2004-03-10 | 2009-05-15 | Bayer Schering Pharma Ag | Zusammensetzungen aus drospirenon in molekularer dispersion |
| US20050209441A1 (en) * | 2004-03-22 | 2005-09-22 | Lile Jackson D | Process for promoting proper folding of human serum albumin using a human serum albumin ligand |
| US20070219131A1 (en) * | 2004-04-15 | 2007-09-20 | Ben-Sasson Shmuel A | Compositions capable of facilitating penetration across a biological barrier |
| CA2563533C (en) | 2004-04-15 | 2013-10-01 | Shmuel A. Ben-Sasson | Compositions capable of facilitating penetration across a biological barrier |
| US20050256097A1 (en) * | 2004-05-11 | 2005-11-17 | Kosan Biosciences, Inc. | Pharmaceutical solution formulations containing 17-AAG |
| US20050266087A1 (en) * | 2004-05-25 | 2005-12-01 | Gunjan Junnarkar | Formulations having increased stability during transition from hydrophobic vehicle to hydrophilic medium |
| CN1960746A (zh) | 2004-05-25 | 2007-05-09 | 阿尔扎公司 | 稳定性提高的含生物分子配方 |
| US20060014712A1 (en) * | 2004-05-30 | 2006-01-19 | Cemines, Inc. | Controlled delivery of therapeutic compounds |
| US20060002989A1 (en) * | 2004-06-10 | 2006-01-05 | Ahmed Salah U | Formulations of sumatriptan for absorption across biological membranes, and methods of making and using the same |
| PL1778187T3 (pl) | 2004-08-04 | 2012-09-28 | Camurus Ab | Kompozycje tworzące nielamelarne dyspersje |
| JO2937B1 (en) | 2004-08-11 | 2016-03-15 | فيرينغ.بي.في | Tight muscles of the peptide vascular tensioner receptor |
| JP4993852B2 (ja) | 2004-09-17 | 2012-08-08 | サントリーホールディングス株式会社 | ストレスに起因する行動異常を伴う症状あるいは疾患の予防又は改善作用を有する組成物 |
| AU2005286640A1 (en) * | 2004-09-21 | 2006-03-30 | Anesiva, Inc. | Delivery of polynucleotides |
| JP2008531591A (ja) * | 2005-02-24 | 2008-08-14 | エラン・ファルマ・インターナショナル・リミテッド | ドセタキセルおよびそれらの類似体のナノ粒子製剤 |
| WO2006123360A2 (en) * | 2005-03-01 | 2006-11-23 | Sun Pharmaceutical Industries Limited | Microspheres containing octreotide acetate |
| US7759312B2 (en) * | 2005-03-11 | 2010-07-20 | Endo Pharmaceuticals Solutions Inc. | Delivery of dry formulations of octreotide |
| CN101193626A (zh) | 2005-03-11 | 2008-06-04 | 益德威士医药股份有限公司 | 奥曲肽的受控释放制剂 |
| US8232241B2 (en) | 2005-05-23 | 2012-07-31 | Mayo Foundation For Medical Education And Research | Treating liver diseases |
| US7288520B2 (en) | 2005-07-13 | 2007-10-30 | Allergan, Inc. | Cyclosporin compositions |
| US20070021325A1 (en) | 2005-07-21 | 2007-01-25 | Mediplex Corporation | Drug formulation containing a solubilizer for enhancing solubility, absorption, and permeability |
| US20070066512A1 (en) | 2005-09-12 | 2007-03-22 | Dominique Verhelle | Methods and compositions using immunomodulatory compounds for the treatment of disorders associated with low plasma leptin levels |
| FR2891459B1 (fr) | 2005-09-30 | 2007-12-28 | Flamel Technologies Sa | Microparticules a liberation modifiee d'au moins un principe actif et forme galenique orale en comprenant |
| US20070104741A1 (en) | 2005-11-07 | 2007-05-10 | Murty Pharmaceuticals, Inc. | Delivery of tetrahydrocannabinol |
| NO20220050A1 (no) | 2005-11-21 | 2008-08-12 | Novartis Ag | Neuroendokrin tumorbehandling |
| ES2755032T3 (es) * | 2005-12-22 | 2020-04-21 | Novartis Ag | Formulación de liberación sostenida que comprende octreótido y dos o más polímeros de poliláctido-co-glicólido |
| CL2007000345A1 (es) | 2006-02-09 | 2008-01-11 | Univ Maryland | Composicion farmaceutica que comprende una cantidad terapeuticamente efectiva de uno o mas agentes terapeuticos y una cantidad mejoradora de la absorcion intestinal de uno o mas agonistas de receptores de zonulina/zot; y uso de la composicion para tratar la diabetes. |
| KR101194586B1 (ko) | 2006-02-10 | 2012-10-25 | 훼링 비.브이. | 신규 화합물 |
| EP1988886A1 (en) | 2006-02-13 | 2008-11-12 | Intelgenx Corporation | Delayed release pharmaceutical oral dosage form and method of making same |
| US20070224142A1 (en) | 2006-03-22 | 2007-09-27 | Swaile David F | Hydrogenated castor oil based compositions as a replacement for petrolatum |
| WO2007117556A2 (en) | 2006-04-06 | 2007-10-18 | Activbiotics, Incorporated | Pharmaceutical compositions and uses thereof |
| BRPI0710503A2 (pt) * | 2006-04-07 | 2011-08-16 | Merrion Res Iii Ltd | uso de uma composição farmacêutica, composição farmacêutica, e, forma de dosagem oral |
| US8071136B2 (en) | 2006-04-21 | 2011-12-06 | Bioactives, Inc. | Water-soluble pharmaceutical compositions of hops resins |
| GB2438834A (en) | 2006-06-08 | 2007-12-12 | Optinose As | Intranasal protein administration |
| JP2009539862A (ja) | 2006-06-09 | 2009-11-19 | メリオン リサーチ Iii リミテッド | 強化剤を含む固体経口投与剤形 |
| US7998927B2 (en) | 2006-06-23 | 2011-08-16 | Aegis Therapeutics, Llc | Stabilizing alkylglycoside compositions and methods thereof |
| KR100816065B1 (ko) * | 2006-11-27 | 2008-03-24 | 동국제약 주식회사 | 초기 방출억제 특성이 우수한 서방출성 마이크로캡슐의제조방법 및 이에 의해 제조되는 마이크로캡슐 |
| US9918934B2 (en) * | 2006-12-12 | 2018-03-20 | Edgar Joel Acosta-Zara | Linker-based lecithin microemulsion delivery vehicles |
| WO2008092084A2 (en) * | 2007-01-26 | 2008-07-31 | Centocor, Inc. | Injectable non-aqueous suspension with high concentration of therapeutic agent |
| AU2008215659B2 (en) * | 2007-02-16 | 2012-11-01 | Aska Pharmaceutical Co., Ltd. | Pharmaceutical composition comprising microparticle oily suspension |
| US7960336B2 (en) | 2007-08-03 | 2011-06-14 | Pharmain Corporation | Composition for long-acting peptide analogs |
| RU2453332C2 (ru) | 2007-10-16 | 2012-06-20 | Байокон Лимитид | Твердая фармацевтическая композиция (варианты) и способ контроля концентрации глюкозы с ее помощью, способ получения твердой фармацевтической композиции (варианты), таблетка (варианты) и способ получения амфорных частиц |
| GB2454672A (en) | 2007-11-13 | 2009-05-20 | Mologic Ltd | Chromogenic protease substrates |
| EP2249823A4 (en) * | 2008-02-15 | 2011-05-18 | Us Gov Health & Human Serv | OCTANIC ACID FORMULATIONS AND TREATMENT PROCESSES THEREWITH |
| RU2517135C2 (ru) | 2008-05-07 | 2014-05-27 | Меррион Рисерч Iii Лимитед | Композиции пептидов и способы их получения |
| JP5740689B2 (ja) | 2008-08-18 | 2015-06-24 | エンテラ バイオ エルティーディー. | タンパク質の経口投与用の方法および組成物 |
| KR101814186B1 (ko) | 2008-09-17 | 2018-03-14 | 키아스마 인코포레이티드 | 약제학적 조성물 및 연관된 투여방법 |
| US20100151033A1 (en) | 2008-12-15 | 2010-06-17 | Novartis Ag | Octreotide depot formulation with constantly high exposure levels |
| EP2477566B1 (en) | 2009-09-14 | 2017-03-22 | Synthes GmbH | Variable angle compression plate |
| JP2011113487A (ja) | 2009-11-30 | 2011-06-09 | Hochiki Corp | 警報器の設置方法、警報器および支持部材 |
| WO2011112576A1 (en) | 2010-03-10 | 2011-09-15 | Ambrilia Biopharma Inc. | Microspheres for sustained release of octreotide acetate |
| US20110311621A1 (en) | 2010-03-16 | 2011-12-22 | Paul Salama | Pharmaceutical compositions and methods of delvery |
| TWI704919B (zh) | 2012-05-31 | 2020-09-21 | 日商大塚製藥股份有限公司 | 用於預防及/或治療多囊性腎臟病之藥物 |
| CN104379150A (zh) | 2012-06-21 | 2015-02-25 | 安吉奥金尼药品有限公司 | 缓解肿瘤症状的方法和组合物 |
| WO2014049515A1 (en) | 2012-09-25 | 2014-04-03 | Piramal Enterprises Limited | Pyrrolidine substituted flavones for treatment of renal cystic diseases |
| WO2014063080A1 (en) | 2012-10-19 | 2014-04-24 | Synta Pharmaceuticals Corp. | Treating polycystic kidney disease with hsp90 inhibitory compounds |
| WO2014165607A2 (en) | 2013-04-02 | 2014-10-09 | Stealth Peptides International, Inc. | Aromatic-cationic peptide formulations, compositions and methods of use |
| JO3371B1 (ar) | 2013-07-26 | 2019-03-13 | Ferring Bv | ناهضات مستقبل فاسوبريسين 2 |
| WO2016048984A1 (en) | 2014-09-25 | 2016-03-31 | Cortendo Ab (Publ) | Methods and compositions for the treatment of cushing's syndrome using 2s, 4r ketoconazole |
| US10335452B2 (en) | 2014-10-24 | 2019-07-02 | Mallinckrodt Hospital Products IP Limited | Method of treating patients with hepatorenal syndrome type 1 |
| AU2015360485B2 (en) | 2014-12-10 | 2020-06-25 | Amryt Endo, Inc. | Oral octreotide administered in combination with other therapeutic agents |
| WO2016126830A1 (en) | 2015-02-03 | 2016-08-11 | Chiasma Inc. | Method of treating diseases |
| CN107635574B (zh) | 2015-03-30 | 2021-12-28 | 桑尼布鲁克研究院 | 治疗癌症的方法 |
| JP2017014206A (ja) | 2015-06-30 | 2017-01-19 | ナノアンティバイオティクス,インコーポレイテッド | 腹水の治療 |
| EP3405211A4 (en) | 2016-01-21 | 2019-10-09 | Chiasma Inc. | ORAL OCTREOTIDE FOR THE TREATMENT OF DISEASES |
| IL313083A (en) | 2016-10-21 | 2024-07-01 | Amryt Endo Inc | Terlipressin compositions and their methods of use |
| US11141457B1 (en) | 2020-12-28 | 2021-10-12 | Amryt Endo, Inc. | Oral octreotide therapy and contraceptive methods |
-
2009
- 2009-09-17 KR KR1020167033506A patent/KR101814186B1/ko active Active
- 2009-09-17 CA CA3080587A patent/CA3080587C/en active Active
- 2009-09-17 RU RU2015112963A patent/RU2678833C2/ru active
- 2009-09-17 KR KR1020117008503A patent/KR101683314B1/ko active Active
- 2009-09-17 BR BRPI0918652-2A patent/BRPI0918652B1/pt active IP Right Grant
- 2009-09-17 CA CA2737456A patent/CA2737456C/en active Active
- 2009-09-17 ES ES09814164.1T patent/ES2629131T3/es active Active
- 2009-09-17 PT PT98141641T patent/PT2343982T/pt unknown
- 2009-09-17 MX MX2017008558A patent/MX395136B/es unknown
- 2009-09-17 EP EP20207996.8A patent/EP3861986B1/en active Active
- 2009-09-17 CA CA2963659A patent/CA2963659C/en active Active
- 2009-09-17 NZ NZ591810A patent/NZ591810A/xx unknown
- 2009-09-17 PL PL09814164T patent/PL2343982T3/pl unknown
- 2009-09-17 EP EP17161908.3A patent/EP3210474B1/en active Active
- 2009-09-17 HU HUE09814164A patent/HUE033611T2/en unknown
- 2009-09-17 US US12/561,738 patent/US20100105627A1/en not_active Abandoned
- 2009-09-17 EP EP09814164.1A patent/EP2343982B1/en active Active
- 2009-09-17 MX MX2011002836A patent/MX2011002836A/es active IP Right Grant
- 2009-09-17 JP JP2011527428A patent/JP5623406B2/ja active Active
- 2009-09-17 CN CN200980140593.9A patent/CN102176900B/zh active Active
- 2009-09-17 ES ES20207996T patent/ES3058497T3/es active Active
- 2009-09-17 LT LTEP09814164.1T patent/LT2343982T/lt unknown
- 2009-09-17 DK DK09814164.1T patent/DK2343982T3/en active
- 2009-09-17 GB GB0916349A patent/GB2463978B8/en active Active
- 2009-09-17 HR HRP20170929TT patent/HRP20170929T1/hr unknown
- 2009-09-17 SM SM20170304T patent/SMT201700304T1/it unknown
- 2009-09-17 CN CN201710231884.5A patent/CN106974888B/zh active Active
- 2009-09-17 MX MX2015012756A patent/MX348705B/es unknown
- 2009-09-17 AU AU2009294320A patent/AU2009294320B2/en active Active
- 2009-09-17 RU RU2011115060/15A patent/RU2552324C2/ru active
- 2009-09-17 SI SI200931682T patent/SI2343982T1/sl unknown
- 2009-09-17 GB GB1119686.2A patent/GB2482810A/en not_active Withdrawn
- 2009-09-17 WO PCT/IB2009/007155 patent/WO2010032140A2/en not_active Ceased
-
2010
- 2010-12-29 US US12/981,036 patent/US20110257095A1/en not_active Abandoned
-
2011
- 2011-03-03 ZA ZA2011/01657A patent/ZA201101657B/en unknown
- 2011-03-17 IL IL211799A patent/IL211799A/en active IP Right Grant
- 2011-09-15 US US13/233,824 patent/US8535695B2/en active Active
- 2011-10-25 US US13/281,129 patent/US8329198B2/en active Active
-
2014
- 2014-01-27 JP JP2014012140A patent/JP2014074069A/ja not_active Withdrawn
- 2014-02-24 US US14/188,139 patent/US9265812B2/en active Active
-
2016
- 2016-01-25 JP JP2016011278A patent/JP6258368B2/ja active Active
- 2016-02-16 US US15/044,949 patent/US9566246B2/en active Active
- 2016-04-17 IL IL245172A patent/IL245172B/en active IP Right Grant
-
2017
- 2017-01-03 US US15/397,177 patent/US20170112938A1/en not_active Abandoned
- 2017-02-22 JP JP2017030930A patent/JP2017088625A/ja not_active Withdrawn
- 2017-06-22 CY CY20171100657T patent/CY1119148T1/el unknown
-
2018
- 2018-05-16 US US15/981,423 patent/US20190038758A1/en not_active Abandoned
- 2018-11-26 JP JP2018220221A patent/JP6771528B2/ja active Active
- 2018-11-26 JP JP2018220222A patent/JP2019048872A/ja not_active Withdrawn
-
2019
- 2019-01-28 RU RU2019102221A patent/RU2019102221A/ru not_active Application Discontinuation
-
2020
- 2020-07-20 IL IL276157A patent/IL276157A/en unknown
- 2020-08-11 US US16/990,071 patent/US11400159B2/en active Active
- 2020-12-28 JP JP2020218478A patent/JP7252934B2/ja active Active
-
2022
- 2022-08-02 US US17/879,441 patent/US11986529B2/en active Active
- 2022-08-02 US US17/879,430 patent/US11969471B2/en active Active
-
2025
- 2025-01-13 US US19/019,327 patent/US20250345438A1/en active Pending
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US11986529B2 (en) | Pharmaceutical compositions and related methods of delivery | |
| AU2020281053B2 (en) | Pharmaceutical compositions and related methods of delivery | |
| HK40057062A (en) | Pharmaceutical compositions comprising polypeptides and related methods of delivery | |
| HK1243597B (en) | Pharmaceutical compositions comprising polypeptides and related methods of delivery |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| B06F | Objections, documents and/or translations needed after an examination request according [chapter 6.6 patent gazette] | ||
| B06T | Formal requirements before examination [chapter 6.20 patent gazette] | ||
| B07D | Technical examination (opinion) related to article 229 of industrial property law [chapter 7.4 patent gazette] |
Free format text: DE ACORDO COM O ARTIGO 229-C DA LEI NO 10196/2001, QUE MODIFICOU A LEI NO 9279/96, A CONCESSAO DA PATENTE ESTA CONDICIONADA A ANUENCIA PREVIA DA ANVISA. CONSIDERANDO A APROVACAO DOS TERMOS DO PARECER NO 337/PGF/EA/2010, BEM COMO A PORTARIA INTERMINISTERIAL NO 1065 DE 24/05/2012, ENCAMINHA-SE O PRESENTE PEDIDO PARA AS PROVIDENCIAS CABIVEIS. |
|
| B07E | Notification of approval relating to section 229 industrial property law [chapter 7.5 patent gazette] | ||
| B06A | Patent application procedure suspended [chapter 6.1 patent gazette] | ||
| B09A | Decision: intention to grant [chapter 9.1 patent gazette] | ||
| B16A | Patent or certificate of addition of invention granted [chapter 16.1 patent gazette] |
Free format text: PRAZO DE VALIDADE: 20 (VINTE) ANOS CONTADOS A PARTIR DE 17/09/2009, OBSERVADAS AS CONDICOES LEGAIS. PATENTE CONCEDIDA CONFORME ADI 5.529/DF, QUE DETERMINA A ALTERACAO DO PRAZO DE CONCESSAO. |
|
| B25A | Requested transfer of rights approved |
Owner name: AMRYT ENDO, INC. (US) |