[001] A presente invenção refere-se a partículas inaláveis com preendendo uma forma amorfa estabilizada de tiotrópio, com um agente de estabilização, e a essas partículas opcionalmente misturadas com um ou mais excipientes grossos. Ela também se refere a uma composição farmacêutica que compreende os mesmos, a um processo para a preparação dos mesmos, e o uso dos mesmos para o tratamento de asma ou doença pulmonar obstrutora crônica (COPD).
ANTECEDENTE DA TÉCNICA
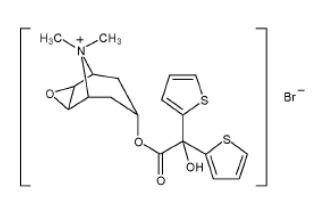
[002] O brometo de tiotrópio é um antagonista do receptor mus- carínico com um efeito anticolinérgico altamente eficaz usado como um bronco dilatador ativo longo. Ele foi primeiro descrito no Pedido de Patente Europeia EP 418716, e tem a estrutura química que se segue:
[003] O brometo de tiotrópio é usado para o tratamento de doen ças respiratórias, especificamente a doença pulmonar obstrutora crônica (COPD) e a asma.
[004] Sais diferentes desse produto (cloreto, brometo, iodeto, etc.), bem como as formas cristalinas diferentes do mesmo são conhecidos.
[005] Para o tratamento de doenças respiratórias tais como a asma ou a COPD, é útil a administração da substância ativa através de inalação. Em comparação aos outros inaladores, os inaladores de pó seco (DPI) oferecem flexibilidade em termos de faixa de dose no- minal, isto é, a quantidade de substância ativa que pode ser administrada em uma única inalação, tornando os mesmos especialmente interessantes como uma ferramenta de administração. Desse modo, o uso de pós inalantes que contenham substâncias ativas a serem administradas com um DPI é de importância específica.
[006] O composto ativo, com a finalidade de passar para dentro dos pulmões tem que ser inalante, isto é, ele deve estar presente em partículas do tamanho de cerca de 1 até 10 μm. As partículas microfi- nas desse tamanho podem ser obtidas, por exemplo, através de mi- cronização, precipitação controlada a partir de solventes adequados ou através de secagem por pulverização, se as condições do processo forem selecionadas, controladas e executadas de forma adequada.
[007] Tradicionalmente, os pós secos têm sido formulados como um fármaco micronizado com uma partícula de veículo mais grossa, tipicamente a lactose. As formulações de pó seco de tiotrópio microni- zado combinadas com uma partícula de veículo mais grossa também têm sido descrita no estado da técnica. Por exemplo, a EP 1292281 descreve um processo de preparação que inclui a micronização compreendendo diversas etapas de misturação dos excipientes. No entanto, a micronização é um método fracamente reproduzível e altamente afetado por uma pequena variabilidade no material de partida. Além disso, essas composições, incluindo o medicamento comercializado da Boehringer Ingelheim, Spiriva®, tem sido provado como exibindo um desempenho fraco em termos de capacidade de dispersão e fração de partícula fina. Além do mais o processo para a sua preparação exibe a dificuldade de serem obtidas misturas homogêneas entre o tiotrópio, os veículos finos e grossos uma vez que são necessárias múltiplas e longas etapas de misturação. Em outro exemplo, a EP 1508330 descreve uma cápsula que contém tiotrópio misturado com um excipiente aceitável, que contém de preferência mono-hidrato de brometo de tio- trópio cristalino. No entanto, o agente de estabilização é misturado de forma mecânica com partículas amorfas de tiotrópio, isso significa que não há um contato direto entre o agente de estabilização e o tiotrópio amorfo.
[008] Desse modo, existe a necessidade com relação a um aper feiçoamento das composições inaláveis de pó seco de tiotrópio, que evitem as desvantagens das formulações da técnica anterior e exibam melhoramentos com relação à homogeneidade das misturas de pó, da capacidade de fluidez do pó e/ou da dispersão do pó, e propriedades aerodinâmicas.
SUMÁRIO DA INVENÇÃO
[009] Os inventores descobriram uma forma amorfa anidra esta bilizada de tiotrópio com um agente de estabilização que dá lugar a composições de pó inaláveis que exibem um alto grau de homogeneidade, e flutuações menores nas propriedades de dispersão. Esses fatores são cruciais para assegurar que à proporção que pode ser inalada da substância ativa seja liberada de forma reproduzível em quantidades constantes e com a mais baixa possibilidade de variabilidade. As composições da invenção também tém propriedades de fluxo melhoradas, uma alta capacidade de dispersão e uma Fração de Partícula Fina (FPF) melhorada através da utilização de um inalador de pó seco, como será mostrado nos exemplos.
[0010] Além disso, ao contrário do que ocorre no caso de prepara ção de partículas inaláveis que contenham uma forma cristalina, em que condições de processo acuradas e laboriosas para ser obtida a forma desejada são necessárias, as partículas inaláveis da invenção podem ser obtidas através de um processo simples e rápido uma vez que elas estão em forma amorfa.
[0011] Por esse motivo, um primeiro aspecto da presente invenção se refere a partículas inaláveis que compreendem uma forma amorfa anidra estabilizada de tiotrópio com um agente de estabilização, especificamente um derivado de açúcar. As partículas inaláveis podem compreender uma matriz na qual o tiotrópio está intimamente disperso em um estado molecular, ou tiotrópio amorfo disperso na superfície do agente de estabilização, isto é as partículas do agente de estabilização são revestidas por uma camada fina de tiotrópio amorfo.
[0012] Em fármacos tais como o tiotrópio, que exibem especifica mente uma alta eficiência, somente pequenas quantidades da substância ativa são necessárias em cada dose única para ser conseguido o efeito terapêutico desejado. Como uma consequência, a substância ativa tem que ser diluída com uma ou mais substâncias farmacologi- camente inativas (excipientes adequados) com a finalidade de serem obtidos pós fluíveis. A diluição tem que ser tal que a quantidade aplicada a partir do inalador do pó contenha exatamente a dose desejada. Esses excipientes farmacologicamente inativos são não somente usados como diluentes, porém também com relação à capacidade dos mesmos de dar propriedades de uma boa capacidade de fluir á composição de pó, facilitando dessa forma o processo de misturação.
[0013] Uma abordagem usada para melhorar a capacidade de fluir do pó se refere ao uso de um ou mais excipientes mais grossos. Esses excipientes mais grossos usados como veículos tem que ter um tamanho de partícula que torna os mesmos não possível de inalação, uma vez que partículas microfinas tendem a mostrar uma forte adesão e coesão que por sua vez leva a propriedades de um fluxo fraco e a agregação do pó. Desse modo, é vantajosa a mistura das partículas inaláveis da invenção com um ou mais excipientes grossos que tenham um tamanho médio de partícula de 15 a 250 μm.
[0014] Por isso, outro aspecto da invenção se refere a partículas inaláveis que compreendem uma forma amorfa estabilizada de tiotrópio com um agente de estabilização, especificamente um derivado de açú- car, em que as partículas são misturadas com um ou mais excipientes grossos que tenham um tamanho médio de partícula de 15 a 250 μm. Essas partículas também serão referidas aqui, neste pedido de patente como partículas misturadas com um ou mais excipientes grossos.
[0015] Foi descoberto que a forma amorfa estabilizada do tiotrópio com um agente de estabilização da presente invenção também permite serem obtidas formulações de pó seco com uma capacidade de fluidez aceitável através da utilização somente de ingredientes finos. Como será explicado abaixo em mais detalhe, isso pode ser conseguido através da preparação de partículas inaláveis através de um método adequado de secagem se iniciando a partir de soluções de tiotrópio e de um agente de estabilização em uma diluição apropriada, Assim, em tal caso, onde não há necessidade de utilizar nenhum excipiente grosso, a etapa de misturação não é necessária.
[0016] As partículas inaláveis da presente invenção como defini das acima, incluindo as partículas inaláveis sem o excipiente grosso, e as partículas inaláveis com um ou mais excipientes grossos, podem ser administradas a um paciente que esteja sofrendo de uma doença respiratória na forma de uma composição farmacêutica apropriada. Desse modo, outro aspecto da presente invenção se refere a composições farmacêuticas que compreendem as partículas inaláveis da presente invenção como definidas acima, incluindo as partículas inaláveis sem o excipiente grosso, e as partículas inaláveis com um ou mais excipientes grossos. Essas composições serão descritas abaixo.
[0017] As partículas inaláveis da invenção podem ser preparadas de forma conveniente através de um método de secagem apropriado a partir de uma solução ou de uma suspensão de tiotrópio e do agente de estabilização. Por esse motivo, outro aspecto da invenção se refere a um processo para a preparação de partículas inaláveis que compreende as etapas que se seguem: (a) dissolvendo ou dispersando o agente de estabilização em um solvente volátil miscível em água contendo opcionalmente água, para a formação de uma solução ou uma suspensão; (b) dissolvendo um sal de tiotrópio ou um solvato do mesmo, ou qualquer forma solida do mesmo em um solvente volátil miscí- vel em água contendo opcionalmente água; (c) misturando a solução da etapa (b) e a solução ou a suspensa da etapa (a); e (d) secando por pulverização a solução/suspensão da etapa (c) para serem obtidas as partículas desejadas.
[0018] Como mencionado acima, as partículas estabilizadas de tio- trópio com o agente de estabilização podem ser misturadas com um ou mais excipientes grossos. Por esse motivo, outro aspecto da invenção se refere a um processo para a preparação das mesmas, que compreende as etapas de (a) a (d), como definidas acima, compreendendo também a etapa de misturar as partículas da etapa (d) com um ou mais excipientes grossos que tenham um tamanho médio de partícula de 15 a 250 μm.
[0019] Como descrito previamente, as composições da invenção são úteis para o tratamento de determinadas doenças respiratórias. Por essa razão, outro aspecto da presente invenção se refere às partículas inaláveis como definidas previamente, para uso no tratamento de asma ou da doença pulmonar obstrutora crônica (COPD). Desse modo, este aspecto se refere ao uso das partículas inaláveis como definidas previamente, para a fabricação de um medicamento para o tratamento da asma e da COPD, e também pode ser formulado como um método para o tratamento da asma e da COPD compreendendo a administração de uma quantidade eficaz das partículas inaláveis da invenção previamente definidas, incluindo as partículas inaláveis sem o excipiente grosso, e as partículas inaláveis com um ou mais excipien- tes grossos, para um paciente que esteja necessitando da mesma.
[0020] Esses aspectos da presente invenção serão ainda descritos na seção de descrição detalhada que se segue. A não ser que definidos de outra forma, todos os termos técnicos e científicos usados aqui, neste pedido de patente, tem o mesmo significado como comumente entendido por uma pessoa versada na técnica à qual esta invenção pertence. Os métodos e os materiais similares ou equivalentes àqueles descritos aqui, neste pedido de patente podem ser usados na pratica da presente invenção. Através de toda a descrição e das reivindicações, a palavra "compreende" e as variações da mesma não estão destinadas a excluir outras características técnicas, aditivos, componentes ou etapas. Os aspectos adicionais, vantagens e características da invenção se tornarão aparentes àquelas pessoas versadas na técnica na ocasião do exame da descrição podem ser aprendidas através da pratica da invenção.
BREVE DESCRIÇÃO DOS DESENHOS
[0021] A figura 1 mostra os padrões PXRD do brometo de tiotrópio não tradado (A) e brometo de tiotrópio seco por pulverização depois de armazenagem durante 6 meses a 25°C (B).
[0022] A figura 2 mostra os padrões PXRD do brometo de tiotrópio - pó de lactose seco por pulverização, obtido a partir de soluções de etanol e água imediatamente depois da secagem por pulverização (A) e depois da armazenagem durante 6 meses a 25°C (B).
[0023] A figura 3 mostra as imagens SEM com relação aos pós secos por pulverização obtidos a partir de tiotrópio - lactose em solução em diferentes ampliações.
[0024] A figura 4 mostra as imagens SEM com relação aos pós secos por pulverização obtidos a partir de tiotrópio - lactose em solução em diferentes ampliações.
DESCRIÇÃO DETALHADA DA INVENÇÃO
[0025] De acordo com a invenção, um primeiro aspecto se refere a partículas inaláveis que compreendem uma forma amorfa anidra estabilizada de tiotrópio com um agente de estabilização como definido acima.
[0026] O termo tiotrópio se refere a qualquer sal farmaceuticamen- te aceitável que compreenda tiotrópio como o cátion de amônio livre e um ânion como um contra íon. Os sais de tiotrópio não limitativos que podem ser usados dentro do âmbito da presente invenção são aqueles compostos que contêm, por exemplo, cloreto, brometo, iodeto, meta- nossulfonato, para-toluenossulfonato, benzenossulfonato ou sulfato de metila. De acordo com a invenção, o tiotrópio presente na forma amorfa estabilizada é anidro. Em uma modalidade de preferência, as partículas inaláveis da invenção compreendem uma forma amorfa estabilizada de um sal de tiotrópio com um agente de estabilização. Em uma modalidade de maior preferência, o sal de tiotrópio é o brometo de tio- trópio. A expressão tiotrópio base se refere ao tiotrópio como o cátions de amônio livre.
[0027] O agente de estabilização como descrito aqui, neste pedido de patente tem que ser capaz de estabilizar a substância ativa. Ele se refere a qualquer ingrediente inativo farmaceuticamente aceitável (isto é, qualquer excipiente farmacêutico) que possa ser usado nas composições de acordo com a invenção com a finalidade de impedir ou retardar a transformação física (isto é, a partir de uma forma amorfa para uma forma cristalina) e/ou a degradação química (como por exemplo, por hidrólise, oxidação ou qualquer outro mecanismo de degradação ) do tiotrópio. Isso significa que a transformação química do tiotrópio amorfo ou a degradação química do mesmo durante a armazenagem (como por exemplo, a 25°C/ 60% RH ou 40°C/ 75% RH) é mais vagarosa quando as partículas inaláveis contêm ambos um agente de estabilização e um tiotrópio amorfo na sua composição em comparação com as partículas inaláveis que contêm somente o tiotrópio amorfo.
[0028] Em uma modalidade da invenção, o agente de estabiliza ção pode ser um derivado de açúcar. Os exemplos não limitativos de açucares são os monos sacarídeos tais como a glicose ou a arabinose, dissacarídios tais como a lactose, sacarose, maltose ou trealose, oligo- e polissacarídios, tais como o dextrano e poli alcoóis tais como o sorbitol, manitol, ou glicerol. Em uma modalidade de preferência o agente de estabilização é a lactose (anidra ou o mono-hidrato).
[0029] Dentro do âmbito da invenção, o termo "amorfo" significa que a formulação em pó contém menos do que 10% de frações cristalinas.
[0030] O termo "inalável" significa que as partículas são adequa das para a administração pulmonar. As partículas inaláveis podem ser dispersas e inaladas por meio de um inalador, de tal forma que as partículas entram nos pulmões e são capazes de desenvolver uma atividade sistêmica opcionalmente através dos alvéolos. Em uma modalidade da invenção, as partículas inaláveis que compreendem a matriz amorfa de tiotrópio e o agente de estabilização tem um tamanho médio aerodinâmico de partícula de até 10 μm. Em uma modalidade de preferência, o tamanho médio aerodinâmico de partícula está na faixa de 0,5 a 6 μm.
[0031] Em uma modalidade preferida, a % em peso do brometo de tiotrópio referida ao peso do brometo de tiotrópio e do agente de estabilização é compreendida entre 0,1 - 10%, de preferência entre 4 a 8%.
[0032] Na presente invenção, as partículas inaláveis compreen dem uma forma amorfa estabilizada de tiotrópio com um agente de estabilização. A expressão "forma amorfa estabilizada de tiotrópio " como descrita na invenção se refere às composições obtidas através dos processos de produção usados no âmbito desta invenção para obter partículas inaláveis que contenham um agente de estabilização e o tiotrópio amorfo.
[0033] A expressão "forma amorfa estabilizada" inclui ambas as partículas, nas quais a forma estabilizada amorfa do tiotrópio e o agente de estabilização compreendem: (a) uma matriz na qual o tiotrópio está intimamente disperso em um estado molecular, isto é, partículas inaláveis obtidas através da secagem de uma solução que contenha ambos o agente de estabilização e o tiotrópio; e (b) tiotrópio amorfo disperso na superfície do agente de estabilização, isto é, o agente de estabilização é revestido por uma camada fina de tiotrópio amorfo, em cujo caso as partículas inaláveis são obtidas através da secagem de uma suspensão do agente de estabilização que contém o tiotrópio em solução).
[0034] Em ambos os casos, o contato direto entre o agente de es tabilização e o tiotrópio amorfo (isto é, um contato intimo entre as moléculas do agente de estabilização e o tiotrópio, ou o contato intimo entre as partículas do agente de estabilização e a camada de revestimento do tiotrópio amorfo) é necessário para ser obtido uma forma amorfa estabilizada do tiotrópio. Isso significa que uma melhor estabilidade do tiotrópio amorfo é obtida quando as partículas inaláveis são obtidas através da utilização de processos como o descrito nesta invenção em comparação aos processos de produção com base no uso de somente uma etapa de combinação, na qual o agente de estabilização é misturado mecanicamente com partículas de tiotrópio amorfo (isto é, com a utilização de um misturador apropriado para a mistura- ção de pós farmacêuticos).
[0035] Com a finalidade de serem obtidas composições para inala ção com boas propriedades de fluxo pode ser necessário a mistura das partículas inaláveis que compreendem uma forma amorfa de tiotrópio e um agente de estabilização com um ou mais excipientes grossos.
[0036] Dessa forma, dependendo da proporção de tiotrópio / agen te de estabilização, quando a forma amorfa estabilizada do tiotrópio com o agente de estabilização compreende uma matriz na qual o tio- trópio esta intimamente disperso em um estado molecular, as composições farmacêuticas da invenção podem ser preparadas tanto a partir de: (a) as partículas obtidas depois da aplicação do método de secagem adequado; ou (b) as partículas obtidas depois da aplicação do método de secagem adequado convenientemente misturadas com um ou mais excipientes grossos que tenham um tamanho médio de partícula de 15 até 250 μm.
[0037] Quando a forma amorfa estabilizada do tiotrópio com o agente de estabilização compreende o tiotrópio amorfo disperso na superfície do agente de estabilização, as composições farmacêuticas da invenção tem que ser preparadas a partir das partículas obtidas depois da aplicação do método de secagem adequado, misturadas de forma conveniente com um ou mais excipientes grossos que tenham um tamanho médio de partícula de 15 até 250 μm.
[0038] Em uma modalidade preferida da invenção, somente é usado o excipiente grosso, e ele tem de preferência um tamanho médio de partícula variando a partir de 50 até 150 μm.
[0039] O agente de estabilização e os excipientes grossos podem ser usados em substâncias quimicamente diferentes ou idênticas. Em uma modalidade de preferência específica, o agente de estabilização e o excipiente grosso compreendem o mesmo composto químico. Em uma modalidade preferida, o agente de estabilização, bem como o ex- cipiente grosso compreendem lactose, opcionalmente na forma do mono-hidrato.
[0040] As composições farmacêuticas da invenção compreendem de preferência a partir de 0,02 até 0,8% da base de tiotrópio.
[0041] A não ser que declaradas de outra forma aqui, neste pedido de patente, as percentagens dadas dentro do âmbito da presente invenção são sempre percentagens em peso.
[0042] De preferência, as composições da invenção estão na for ma de uma cápsula para inalação. A cápsula pode ser formada de vários materiais, tais como gelatina, derivados de celulose, amido, derivados de amido, quitosana e plásticos sintéticos. Em uma modalidade de preferência, a cápsula é formada de derivados de celulose, os exemplos não limitativos de tais derivados são a hidroxipropilmetilcelu- lose (HPMC), hidroxipropilcelulose (HPC), metilcelulose, hidroximetil- celulose ou hiroxipropilcelulose. Em uma modalidade de mais preferência da invenção, a cápsula de inalação compreende HPMC. Na modalidade de maior preferência, as cápsulas HPMC exibem uma umidade residual (teor de água abaixo de 4%).
[0043] As partículas inaláveis da invenção podem ser preparadas tanto se iniciando a partir de uma solução como de uma suspensão do agente de estabilização em um solvente adequado. Uma vantagem do presente método é que as composições farmacêuticas da invenção dão valores de pH neutro depois da dissolução em água. Isso é muito interessante em termos de tolerância no pulmão na medida em que os valores de pH neutro dos pulmões não devem ser modificados depois da inalação.
[0044] No caso do início a partir de uma solução do agente de es tabilização, o agente de estabilização deve ser dissolvido em um solvente miscível em água que contenha água (solução da etapa a) e essa solução é misturada com a solução obtida a partir do tiotrópio no mesmo solvente (solução da etapa b). Neste caso, como se tornará óbvio para uma pessoa versada na técnica, essas etapas podem ser realizadas em qualquer ordem com a finalidade de ser obtida uma solução que compreenda o tiotrópio e o agente de estabilização.
[0045] Depois da aplicação do método adequado, as partículas resultantes obtidas compreendem uma matriz na qual o tiotrópio está intimamente disperso em um estado molecular no agente de estabili- zação (isto é, uma matriz de lactose contendo o tiotrópio é formada) com a finalidade de ser obtido um efeito estabilizador sobre o ingrediente ativo na forma amorfa.
[0046] Como descrito anteriormente, dependendo das quantidades relativas de tiotrópio e do agente de estabilização usadas, pode não ser necessária a misturação das partículas obtidas com um ou mais excipientes grossos com a finalidade de serem obtidas composições com boas propriedades de fluxo. Dessa forma, em uma diluição apropriada, de preferência 0,02 a 0,8% do tiotrópio/ tiotrópio e agente de estabilização não há mais excipiente grosso a ser adicionado, e a etapa de misturação não é necessária, de tal modo que é conseguido uma boa homogeneidade do pó.
[0047] Quando as partículas inaláveis da invenção são preparadas a partir de uma suspensão do agente de estabilização, as etapas (a) e (b) são executadas de preferência separadamente, e de mais preferência, o agente de estabilização é primeiro disperso e homogeneizado (por exemplo, com a utilização de homogeneização de alta velocidade ou de alta pressão) com a finalidade de ser obtida uma suspensão homogênea do agente de estabilização, adicionando antes a solução do ingrediente ativo (solução da etapa (b); Nesse caso, depois da aplicação do método de secagem apropriado, as partículas resultantes compreendem o tiotró- pio amorfo disperso na superfície das partículas do agente de estabilização. Nesse caso, o agente de estabilização está de preferência na forma de partículas que tenham um tamanho médio de partícula de 1 a 9 μm.
[0048] O sistema de solvente usado para a preparação das partí culas inaláveis descritas acima compreendem um solvente volátil mis- cível com a água, contendo opcionalmente água. Como explicado acima, a presença da água dá lugar a soluções do agente de estabilização, enquanto que a ausência da água dá lugar a suspensões do referido agente. No caso em que a solução do agente de estabilização for usada, dependendo da quantidade do agente de estabilização usada, deve ser adicionado o solvente apropriado miscível em água com a finalidade de solubilizar totalmente o agente de estabilização. Por outro lado, o ingrediente ativo é sempre dissolvido. De preferência, o solvente volátil miscível em água é um (C1-C4) álcool. A expressão (C1C4) álcool se refere a uma cadeia de alquila linear ou ramificada que compreende a partir de 1 até 4 átomos de carbono e um ou mais grupos hidroxila. Este termo inclui, sem limitação, metanol, etanol, propanol, isopropanol e butanol. Em uma modalidade preferida da invenção, o solvente de álcool usado é o etanol ou o isopropanol.
[0049] O método de secagem usado é o de secagem por pulveri zação. Esse método permite contribuir para ser obtido um controle acurado do tamanho médio de partícula e da distribuição do tamanho de partícula, e também o melhoramento das propriedades do pó macroscópico tal como a capacidade de fluir e a capacidade de dispersão. Um Secador Pulverizador típico permite a recuperação de uma faixa de tamanho de partículas indo a partir de 0,5 até 30 μm. O limite mais baixo é dado através da separação do ciclone usado: as partículas menores não mais podem ser separadas e irão para o filtro.
[0050] Os processos descritos para a preparação das partículas inaláveis têm a vantagem de que evita a micronização através da utilização de um processo mais reproduzível, uma vez que a micronização é um método de micronização fraco e altamente dependente das propriedades físicas do material de partida. No caso da presente invenção, o material de partida (composto de tiotrópio) pode estar em qualquer forma física, uma vez que tenha sido primeiramente dissolvido. Desse modo, o composto de tiotrópio de partida pode ser qualquer sal de tiotrópio ou um solvato do mesmo, ou qualquer forma solida do mesmo, incluindo o racemato ou qualquer outro enantiômero do mesmo ou as misturas dos mesmos.
[0051] Como mencionado acima, as partículas inaláveis da inven ção exibem uma fração de partícula fina aperfeiçoada. A expressão "fração de partícula fina" (FPF) descreve a parte inalável do pó. A FPF é a dose (expressa em % em peso) de partículas que tem um diâmetro aerodinâmico inferior a 5 μm com relação à dose nominal (Dose de partícula fina/ dose carregada x 100). A dose de partícula fina (FPD) pode ser determinada através de métodos descritos na USP and European Pharmacopoeia, com relação à avaliação aerodinâmica de partícula fina. Em pó que é bem dispersável a FPF é de mais do que 20%, de preferência mais do que 30%. A expressão "diâmetro aerodinâmico médio de partícula" também conhecida como diâmetro aerodinâmico médio de massa (MMAD) indica o tamanho aerodinâmico da partícula no qual 50% das partículas do pó têm um diâmetro aerodinâmico menor.
EXEMPLOS
[0052] Os exemplos que se seguem são proporcionados para as finalidades de ilustração, e não estão destinados a serem limitativos da presente invenção. Cromatografia Líquida de Alto Desempenho (HPLC)
[0053] A determinação do teor de tiotrópio nas formulações secas por pulverização e as combinações finais de tiotrópio e lactose foram executadas através de HPLC
[0054] O sistema de HPLC consiste em um sistema de Cromato- grafia Liquida de Alto Desempenho Serie HP 1200, Agilent Technologies, Bélgica) equipado com uma bomba quaternária, e um amostrador automático e um conjunto de diodo detectador de UV ajustado a 238 nm. O sistema de separação foi uma coluna C18 de fase reversa de 125 mm x 4 mm de aço inoxidável (tamanho de partícula de 5 μm (Alltima, Alltech, Bélgica). Amostras de 100 μl de volume foram injetadas, A temperatura foi ajustada para 35°C e a velocidade de fluxo foi de 0,8 ml por minuto. O tempo total de operação foi de 10 minutos. - Tampão: KH2PO4 100 mM ajustado para o pH 4 com o ácido alfa fosfórico - Fase móvel: 80 vol. Tampão de fosfato: 20 vol. acetonitrila - Fase de diluição: 75 vol. de água: 25 vol. de metanol
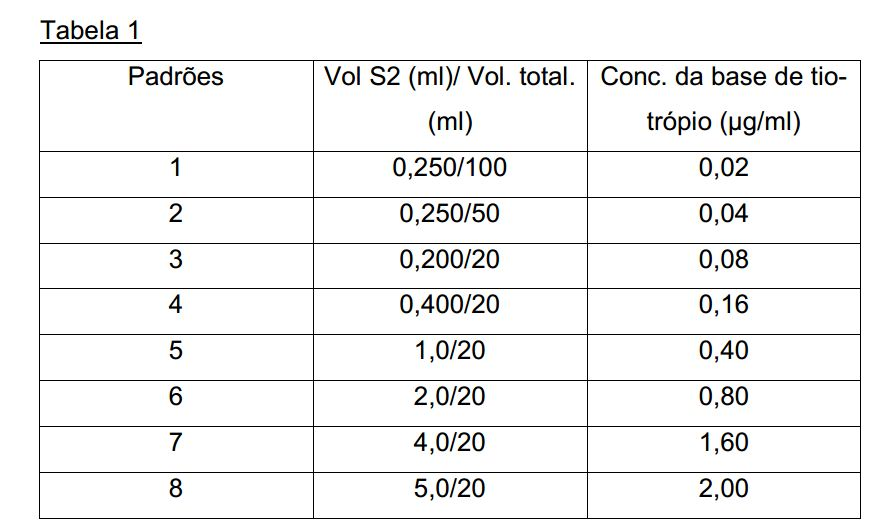
[0055] Na tabela 1 são mostradas as soluções- padrão que con têm tiotrópio. Soluções- padrão
[0056] S1: 2,5 mg de mono-hidrato de BR tiotrópio em 10 ml de metanol (= 200 μg/ ml de base de tiotrópio)
[0057] S2: 1,0 ml da solução S1 em 25 ml da fase de diluição (= 8 μg/ ml) Tabela 1
Preparação de formulações estáveis de tiotrópio por secagem por pulverização. (a) Preparação de partículas inaláveis compreendendo brometo de tiotrópio e microfinos de lactose (Lactochem, Borculo; tamanho médio de partícula de cerca de 8 μm).
[0058] As formulações foram preparadas, em escala de laborató rio, através de secagem por pulverização com a utilização de um Büchi Mini Spray Dryer B-191a (Büchi laboratory-Techniques, Suíça). Solu- ções de suspensões diferentes foram preparadas em etanol ou isopropanol que continham ou não continham quantidades diferentes de água.
[0059] Primeiramente, a lactose foi dissolvida ou dispersa no meio solvente com a finalidade de serem obtidas soluções (na presença de quantidades suficientes de água) ou suspensões (na ausência de água). Em seguida, o brometo de tiotrópio foi dissolvido no meio de solvente e as soluções ou suspensões obtidas foram misturadas. Na ausência de água, a lactose foi em primeiro lugar dispersa e homogeneizada (por exemplo com a utilização de homogeneização de alta velocidade ou de alta pressão) com a finalidade de ser obtida uma suspensão homogênea, antes da adição da solução de tiotrópio.
[0060] As preparações foram em seguida secas por pulverização com agitação constante. As condições que se seguem foram usadas durante a secagem por pulverização: fluxo de ar de pulverização, 800 l/hora; fluxo de ar de secagem, 35 m3 hora; taxa de alimentação da suspensão, 3,5 a 4,0 g/minuto; tamanho do bocal 0,5 mm. A temperatura de entrada foi estabelecida a 70°C ou 120°C (na presença de água) e, nessas condições, a temperatura de saída variou entre 44°C e 46°C ou 65°C e 68°C. O pó resultante foi insuflado através de um separador de ciclone e recolhido em um contêiner. Os pós foram armazenados em secadores em condições de temperatura diferentes (25°C, 40°C e 60°C).
[0061] Em seguida a esse processo, foram obtidos os exemplos que se seguem: Exemplo 1: Solução de tiotrópio e lactose em etanol - água Etanol 100 g Água 100 g Microfino de lactose 4,480g Mono-hidrato de brometo de tiotrópio 0,360g Exemplo 2: Tiotrópio - solução de lactose em isopropanol - Água Isopropanol Água Microfino de lactose 100g 100g 4,480g Mono-hidrato de brometo de tiotrópio 0,360g Exemplo 3: Suspensões deTiotrópio - lactose em Etanol Etanol 200g Microfino de lactose 4,480g Mono-hidrato de brometo de tiotrópio 0,360g Exemplo 4: Suspensões de tiotrópio e lactose em isopropanol Isopropanol Microfino de lactose 200g 4,480g Mono-hidrato de brometo de tiotrópio 0,360g Exemplo 5: Solução de Tiotrópio e Lactose em Etanol - Água Etanol Água Mono-hidrato de lactose malha 200 100 g 100 g 4,480g (tamanho médio de particular de cerca de 74 μm) Mono-hidrato de brometo de tiotrópio 0,360g Exemplo 6: Solução de Tiotrópio e Lactose em isopropanol - Água Isopropanol Água Mono-hidrato de Lactose malha 200 100g 100g 4,480g (tamanho médio de particular de cerca de 74 μm) Mono-hidrato de brometo de tiotrópio 0,360g Exemplo 7: Solução de Tiotrópio e Lactose em Etanol - Água Etanol Água Microfino de lactose 500 g 500 g 88,02g Mono-hidrato de brometo de tiotrópio 0,360g (b) Preparação de partículas inaláveis obtidas na etapa (1) misturadas com um excipiente grosso de mono-hidrato de lactose malha 200 (ta-manho médio de partícula de cerca de 74 μm ou lactose anidra seca com cilindro (tamanho médio de partícula de cerca de 120 μm)
[0062] Para essa finalidade foi usado um misturador em escala de laboratório de três dimensões bem- conhecido: Turbula 2C (Bachofen AG, Switzerland). Um recipiente de 50 ml de plástico de polietileno não reticulado foi enchido até 59% do seu volume interno e a mistura (cerca de 20 g) foi feita em velocidades de mistura de 46,2 rpm.
[0063] Cerca de 25% de lactose foi primeiro adicionado para forrar as paredes do vaso de mistura com a finalidade de reduzir a adição futura do fármaco. O pó seco por pulverização obtido na etapa (a) foi em seguida adicionado e misturado manualmente com a lactose, Em seguida, o resto da lactose foi adicionado e a misturação foi executada durante cerca de 30 minutos.
[0064] O exemplo 1 foi combinado com o excipiente grosso (mono- hidrato de lactose e lactose anidra) para a formação dos lotes 1A (exemplo 8) e 1B (exemplo 9) respectivamente.
[0065] O exemplo 3 foi combinado com o excipiente grosso (mono- hidrato de lactose e lactose anidra) para a formação dos lotes 3A (exemplo 10) e 3B (exemplo 11) respectivamente. Exemplo 8: Operação de combinação (Turbula)- mono-hidrato de lactose Pó seco por pulverização obtido como na etapa (a) do Exemplo 1 1,200 g Mono-hidrato de lactose (74 μm, DMV) 20,810 g Peso total 22,010 g Exemplo 9: Operação de combinação (Turbula)- lactose anidra Pó seco por pulverização obtido como na etapa (a) do Exemplo 1 1,200 g lactose anidra (120 μm, DMV) 20,810 g Peso total 22,010 g Exemplo 10: Operação de combinação (Turbula)- mono-hidrato de lactose Pó seco por pulverização obtido como na etapa (a) do Exemplo 31,200 g Mono-hidrato de lactose (74 μm, DMV) 20,810 g Peso total 22,010 g Exemplo 11: Operação de combinação (Turbula) - lactose anidra Pó seco por pulverização obtido como na etapa (a) do Exemplo 31,200 g lactose anidra (120 μm, DMV) 20,810 g Peso total 22,010 g Composição unitária final dos exemplos anteriores Tiotrópio (base) 18 μg Mono-hidrato de lactose (agente de estabilização) 0,2775 mg Mono-hidrato de lactose ou lactose anidra (excipiente grosso) 5,2025 mg
[0066] Nesses exemplos, o conteúdo final de brometo de tiotrópio foi em torno de 0,39% (em torno de 0,33% para a base de tiotrópio) em peso como referido com relação ao total da composição). Difração de RaiosX em Pó (XRPD).
[0067] Os padrões de Difração de RaiosX em Pó (XRPD) foram obtidos com a utilização de um Siemens Diffractometer D5000 (Siemens, Alemanha), com uma linha de Cu como a fonte de radiação (WL1 = 1.5406 A, WL2 = 1.54439 A), e rodadas- padrão com a utilização de uma voltagem de 40 kV, uma corrente de 40 mA e uma taxa de varredura de 0,02° por minuto sobre uma faixa de 2 θ de 2° até 70°.
[0068] Nas figuras 1A e 2A, a análise de PXRD mostrou que o processo de secagem por pulverização permitiu a transformação de ambas uma solução de brometo de tiotrópio isolada e uma solução de brometo de tiotrópio e lactose, de acordo com a composição descrita no exemplo 1, em uma forma amorfa. No entanto, como mostrado na figura 1B o brometo de tiotrópio amorfo seco por pulverização não era estável, na medida em que a forma amorfa foi transformada em tiotró- pio cristalino durante a armazenagem. Em contraste, como mostrado na figura 2B, a presença de lactose nos pós secos por pulverização permitiu a estabilização do tiotrópio como uma forma amorfa de tiotró- pio e lactose mesmo depois da armazenagem.
Análise do Tamanho de Partículas
[0069] O tamanho das partículas foi medido através de uma técni ca com base em espalhamento de luz de laser. A distribuição do volume do tamanho de partícula foi medida com um difratômetro a laser Malvern Mastersizer 2000® com a utilização de um sistema de amostragem a seco (Scirocco 2000, Malvern, Reino Unido) com um SOP (Procedimento de Operação- Padrão) adequado.
[0070] A distribuição do tamanho de partícula é caracterizada pelo diâmetro médio de massa-volume (d(0,5)), isto é, o tamanho em mí- crons no qual 50% da amostra é menor e 50% é maior, e o diâmetro médio de massa- volume (D [4,3]) Os valores apresentados são a média de pelo menos 3 determinações.
[0071] A análise do tamanho de partícula mostrou que ambas as formulações obtidas a partir da lactose em solução (Exemplo 1) e em suspensão (exemplo 3) apresentaram propriedades de tamanho de partícula apropriadas para a administração profunda ao pulmão.
[0072] De fato, os diâmetros de volume médio d(0,5) para os tipos de formulação de solução e de suspensão foram de 2,9 μm e 3,0 μm respectivamente e o diâmetros de volume médios D [4,3] com relação às mesmas formulações foram de 10,3 μm e 10,5 μm, respectivamente. Além disso mais do que 70% das partículas apresentaram um tamanho de partícula abaixo de 5,0 μm o que é em geral considerado como o tamanho limite para uma boa penetração no pulmão. Microscopia eletrônica de varredura (SEM)
[0073] A avaliação do tamanho e da morfologia das partículas foi conseguida através de Microscopia Eletrônica de Varredura (SEM) com a utilização de um microscópio JSM-610 (Jeol, Japão).
[0074] As amostras foram espalhadas em uma película fina de uma resina epóxi de dois componentes que foi em seguida revestida com uma camada de platina. A aceleração durante a observação foi de 25 kV.
[0075] A figura 3 mostra as imagens SEM com relação aos pós secos por pulverização obtidos a partir de tiotrópio e lactose em solução, de acordo com a composição descrita no exemplo 1 em amplificações diferentes.
[0076] A figura 4 mostra as imagens SEM com relação aos pós secos por pulverização obtidos a partir de tiotrópio e lactose em sus-pensão, de acordo com a composição descrita no exemplo 3 em am-plificações diferentes.
[0077] A morfologia e a estrutura da superfície das formulações analisadas por SEM mostraram que os pós secos por pulverização consistiam de aglomerados soltos. O tamanho dos aglomerados variou até cerca de 100 a 500 μm. Em ampliações maiores, foi possível observar que esses aglomerados são compostos por partículas pequenas da faixa de cerca de um micrômetro e que tem um formato mais homogêneo do tipo esférico no caso do pó obtido a partir de soluções do que a partir de suspensões. Avaliação da homogeneidade da distribuição do fármaco em pós secos por pulverização e em misturas finais de tiotrópio e de lactose
[0078] A homogeneidade de distribuição do fármaco foi avaliada a partir de HPLC, com utilização do método descrito acima.
[0079] Os resultados mostrados na tabela 2 mostraram os dados referentes às amostras pesadas (unidade de dose em cápsula), con-centração de tiotrópio em soluções injetadas a partir da análise de HPLC, quantidades de tiotrópio em formulações décadas por pulverização expressas em mg/g e em percentagem. Esses dados mostram uma distribuição do fármaco relativamente homogênea com relação a dois lotes secos por pulverização, exibindo os teores de fármaco apro-priados de cerca de 7% e 6,7% para soluções secas por pulverização contendo etanol e água e isopropanol e água. Tabela 2
[0080] A distribuição aerodinâmica do tamanho de partícula com re lação às novas formulações de Tiotrópio foi determinada com a utilização de um Multi-Stage Liquid Impinger (MsLI). Um dispositivo de inalação de pó seco (Fantasmino®) foi equipado com uma cápsula HPMC tamanho 3 (Capsugel, França). A velocidade de fluxo foi ajustada para uma queda de pressão de 4 kPa, como é típica para a inspiração por um paciente, resultando em uma velocidade de fluxo de 100 l/minuto durante 2,4 se-gundos. Cinco cápsulas carregadas com 5,5, mg de pó (18 μg de tiotró- pio) foram tomadas para cada teste, A deposição do fármaco no disposi-tivo, a garganta, as quatro etapas e o filtro (etapa 5) foram determinados através de análise por Cromatografia Liquida de Alta Pressão (HPLC). Para exatidão, cada teste foi repetido três vezes.
[0081] A distribuição aerodinâmica do tamanho de partícula com relação ao produto de referência Spiriva ® foi determinada com a utili-zação de MsLi com o dispositivo Handihaler® e contendo cápsulas de gelatina. A velocidade de fluxo foi de 10 l/ minuto durante 4 segundos.
[0082] Os resultados indicaram que a FDP, que aproximadamente corresponde a deposição do fármaco nas etapas 3, 4 e filtro (diâme- tros em corte de 5,27 μm, 2,40 μm, e 1,32 μm, respectivamente), variou no interior de uma faixa de 4,9 μg (FPF de 27%) e de 6,4 μg (FPF de 35%) com relação às formulações desenvolvidas. Esses resultados mostraram valores de FPD significativamente mais altos em comparação com o produto de referência. Por certo, o valor de FPD obtido para o Spiriva ® foi de 1,6 μg, que corresponde a um FPF de 8,9%. Tabela 3
Avaliação preliminar da estabilidade das novas formulações
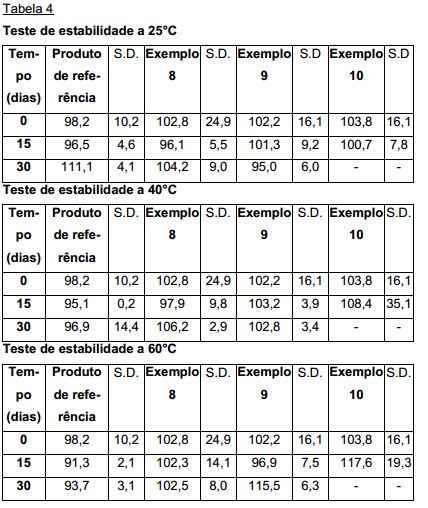
[0083] Três formulações misturadas de mono-hidrato de lactose grosso e tiotrópio que apresentavam homogeneidade, a saber, os exemplos 8, 9 e 10 foram colocados em recipientes bem- fechados e armazenados durante 15 dias e 1 mês a 25°C (temperatura ambiente), 40°C e 60°C. O teor de tiotrópio das amostras diferentes foi avaliado através de análise HPLC como descrito abaixo em tempos de armaze-nagem diferentes daquele do produto de referência Spiriva®.
[0084] Os resultados obtidos mostraram que os três lotes apresenta ram uma estabilidade aceitável em até 30 dias de tempo de armazena- gem nas 3 temperaturas de armazenagem. O teor de tiotrópio das novas formulações pode ser comparado àquele do produto de referência.
[0085] Na tabela 4, os valores de estabilidade das formulações diferentes em 30 dias e a 25°C, 40°C e 60°C estão mostrados, Especi-ficamente é dada a indicação sobre a percentagem do teor de tiotrópio determinado por HPLC bem como os valores do desvio- padrão (S.D.) Tabela 4 Teste de estabilidade a 25°C