PROCESSO PARA PREPARAR NEBIVOLOL
Descrição
[001] A presente invenção refere-se a um processo para preparar Nebivolol e, mais particularmente, a um método aperfeiçoado de desbenzilação de um composto da fórmula
útil para preparar nebivolol dotado de elevado grau de pureza.
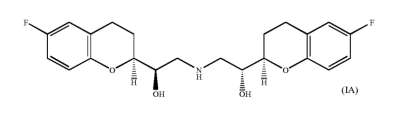
[002] Nebivolol (doravante, NBV), é uma mistura de quantidades iguais de [2S[2R*[R[R*]]]]α,α'-[imino- bis(metileno)]bis[6-fluoro-croman-2-metanol] (doravante d-NBV) de fórmula (IA)
e seu enântiomero [2R[2S*[S[S*]]]] (doravante, /-NBV) da fórmula (IB)
[003] Nebivolol é caracterizado por suas propriedades β-bloqueadoras adrenérgicas e é útil no tratamento da hipertensão essencial. Tem propriedades básicas e podem ser convertidos em seus sais de adição através do tratamento com ácidos adequados. O sal de adição de ácido clorídrico é o produto comercializado.
[004] Sabe-se na técnica que a síntese de estruturas moleculares α,α'-[imino- bis(metileno)]bis[croman-2-metanol] é um desafio para a pessoa versada, devido aos quatro átomos de carbono assimétrico produzindo uma mistura de 16 estereoisômeros (no caso de substituições assimétricas) ou uma mistura de 10 estereoisômeros (no caso de substituições simétricas). Como aparente a partir da presença de simetria na estrutura de nebivolol, um total de 10 estereoisômeros podem ser gerados.
[005] A literatura relata vários processos para a preparação de nebivolol.
[006] Patente EP 145067 (Janssen Pharmaceutica NV) descreve um método de preparar NBV, o qual compreende sintetizar misturas diastereoisoméricas de derivados de epóxido croman.
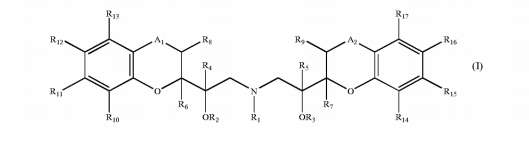
[007] Referidos derivados de epóxido representam os intermediários principais do processo, os quais são devidamente combinados para fornecer um composto de fórmula
em que R1 é hidrogênio, C1-6 alquil, aril, aril-C1-6-alquil, C1-12 alquilcarbonil ou arilcarbonil.
[008] A descrição da patente relata que o composto de fórmula I, em que R1 é um radical fenilmetil pode ser convertido em um composto de fórmula I, em que R1 é hidrogênio de acordo com procedimentos de hidrogenólise conhecidos na técnica. Em particular, no exemplo 23, uma mistura de três partes de composto (A+A-) α,α'- [[(fenilmetil)imino]-bis(metileno)]bis[3,4-deidro-2H-1-benzopiran-2-metanol] e cento e vinte partes de metanol são hidrogenadas à pressão atmosférica e à temperatura ambiente, com duas partes de Paládio sobre carbono (10%).
[009] Patente EP 334429 (Janssen Pharmaceutica NV) descreve substancialmente o mesmo processo de síntese relatado na patente anterior, e é, particularmente, direcionado para a preparação de um único isômero ótico (R,S,S,S) e (S,R,R,R) de NBV.
[0010] Neste caso, a desproteção do grupo amina é descrita como atuável através dos procedimentos de hidrogenação catalítica, tais como Paládio ou Platina suportada em carbono em um solvente adequado. No exemplo 3, uma mistura com três partes e metade de benzil derivado e duzentos e cinquenta partes de 2-metóxi etanol é hidrogenada à pressão atmosférica e à temperatura ambiente, com duas partes de Paládio sobre carbono (10%).
[0011] Pedidos de patentes internacionais WO 2008/010022 (Cimex Pharma AG e University of Zurich), WO 2006/025070 (Torrent Pharmaceutical Ltd), WO 2006/016376 (Hetero Drugs Ltd.) e WO 2004/041805 (Egis Gyogyszergyar RT) descrevem processos alternativos para preparar NBV na forma racêmica e/ou seus enântiomero puros, em que estão fornecidos processos de desbenzilação através de hidrogenação catalítica de acordo com o estado da técnica. Basicamente, o grupo benzil é removido por uma hidrogenação clássica na presença de catalisador (Pd/C).
[0012] O pedido de patente internacional co-pendente WO 2008/064827, em nome do mesmo requerente, descreve um processo para preparar nebivolol e, em particular, um processo para preparar d-nebivolol e seu enântiomero 1-nebivolol ou seus sais partindo de 2,2-dimetil-1,3-dioxolan-4-carbaldeído e um reagente de Grignard. Etapas j/u descrevem a desproteção de um derivado N-benzil de nebivolol (fórmula Xa ou Xb) de acordo com técnicas conhecidas, de preferência, através da hidrogenação catalítica. O pedido de patente ainda proporciona que o hidrogênio molecular pode ser gerado in situ por usar fontes alternativas, como o ácido fórmico, formato de amônio, ácido fosfórico, ciclohexeno e ciclohexadieno, sob condições catalíticas de redução de transferência de hidrogênio. Exemplo 10 do mesmo pedido de patente descreve a preparação de [2S,R,2' R, α'R]-α,α'-[imino bis-metileno]bis[6-fluoro-3,4-deidro-2H-1-benzopiran-2-metanol] na forma de sal de formato do N-benzil derivado correspondente pelo tratamento com formato de amônio na presença de Pd/C (10% em peso) e metanol.
[0013] Portanto, é conhecida na técnica que grupos amino protegidos podem ser desbenzilado pelo uso de hidrogênio molecular na presença de um catalisador a base de metal de transição. No entanto, uma desvantagem relevante associável à utilização de tais métodos é que com alguns substratos, sujeitos às condições de hidrogenação, podem ocorrer reações indesejadas que levam à formação de subprodutos de reação e, como consequência, reduz a pureza e o rendimento do produto final. Por exemplo, sabe-se que o processo de hidrogenólise dos grupos de proteção de benzil, na presença de grupos aromáticos de halogênio, não é quimiosseletivo, resultando na obtenção de compostos desalogenados em quantidades inaceitáveis do ponto de vista industrial (>1%).
[0014] Um método alternativo útil para a hidrogenação de compostos orgânicos é a hidrogenação catalítica conhecida por transferência de hidrogênio ou CTH (hidrogenação por transferência catalítica), o qual difere dos métodos clássicos mencionados acima, em que os átomos de hidrogênio derivam de compostos identificados como doadores de hidrogênio. Referidas CTH podem ser realizadas em condições moderadas e, acima de tudo, elas provaram ser seletivas na desbenzilação de substratos protegidos contendo, em adição, grupos de halogênio aromáticos.
[0015] No entanto, um aspecto associável aos métodos CTH é o fato de que enquanto eles são quimiosseletivos, eles apresentam a desvantagem de serem lentos e de não alcançarem à completa conversão e, desta forma, são geralmente, pouco compatíveis, ou pelo menos pouco produtivos a nível industrial. Uma das causas possíveis é representada pelo envenenamento progressivo do catalisador pelas aminas que geram como produtos da reação de N-desbenzilação.
[0016] Portanto, seria desejável estudar métodos alternativos de desbenzilação, os quais permitam superar as desvantagens dos processos descritos pelo estado da técnica.
[0017] Verificamos, surpreendentemente, um método simples e eficiente de hidrogenação de intermediários úteis para preparar NBV, o qual prevê um CTH através do uso de ácido fórmico como fonte de hidrogênio in situ.
[0018] Portanto, um primeiro objetivo da presente invenção é um processo para a desbenzilação de um composto de fórmula
que compreende a reação do referido composto com ácido fórmico na presença de um catalisador a base de paládio.
[0019] Na presente invenção, resíduo Bn significa um grupo benzil (fenilmetil) como é conhecido na técnica.
[0020] O composto de fórmula II pode ser preparado de acordo com técnicas conhecidas, por exemplo, de acordo com os processos descritos nos pedidos de patentes internacionais e patentes EP 0145067, EP 0334429, WO 2006/016376, WO 2008/064827 e WO 2008/064826.
[0021] De preferência, o objetivo do processo da presente invenção é aplicado ao composto de fórmula II, na forma de um único enântiomero RSSS (benzil derivado 1-NBV) ou SRRR (benzil derivado d-NBV), bem como, ainda mais preferencialmente, para a mistura racêmica (±)[R*,S*,S*,S*] do mesmo.
[0022] A desproteção dos grupos benzil através da hidrogenação por transferência de hidrogênio é conhecida na técnica.
[0023] Geralmente, a reação de um composto de fórmula II com ácido fórmico, na presença de um catalisador a base de paládio, é realizada na presença de um ou mais solventes orgânicos, opcionalmente em mistura.
[0024] Solventes adequados para o objetivo de desbenzilação da invenção são solventes orgânicos inertes, tais como álcoois alifáticos, éteres ou ésteres.
[0025] De preferência, o solvente da reação é selecionado a partir do metanol, etanol, isopropanol, sec-butanol, acetato de etil ou tetraidrofurano.
[0026] Ainda mais preferencialmente, sec-butanol é usado como solvente da reação.
[0027] A desbenzilação da invenção também pode ser realizada em massa.
[0028] Catalisadores a base de Pd úteis para o objetivo de desbenzilação da invenção são Pd sobre carbono (Pd/C), tanto seco quanto úmido, de preferência, até cerca de 50% p/p de água. Preferencialmente, 5% ou 10% de Pd/C é usado.
[0029] Geralmente, no objetivo de redução da invenção, uma quantidade de catalisador em torno de 2-10% p/p em relação ao substrato é utilizada.
[0030] A desbenzilação presente de um composto de fórmula II com ácido fórmico, na presença de um catalisador a base de paládio é ainda mais, preferencialmente, realizada com 5% de Pd/C úmido (cerca de 50% p/p de água) em uma quantidade de cerca de 10% p/p em relação ao substrato (cerca de 5% p/p calculado sobre o catalisador seco).
[0031] Geralmente, a reação de um composto de fórmula II com ácido fórmico, na presença de um catalisador a base de paládio, é realizada a uma temperatura compreendida entre 25°C e 100°C. Preferencialmente, a reação é realizada em cerca de 70°C.
[0032] Geralmente, no objetivo de redução da invenção, as razões estequiométricas de pelo menos 2 moles de ácido/mol de substrato são usadas. De preferência, 3 moles de ácido/mol de substrato são usadas.
[0033] Em um aspecto da invenção, a reação de um composto da fórmula II com ácido fórmico, na presença de um catalisador a base de paládio é realizada por calor (70°C±2°C) adicionando ácido fórmico a uma mistura contendo substrato e 5% de Pd/C úmido em sec-butanol. Após ainda agitar a quente por algumas horas, a mistura de reação é trabalhada com uma solução aquosa básica (por exemplo NaOH). O catalisador é então separado da mistura de reação através, por exemplo, da filtração sobre Celite e o produto é recuperado de acordo com técnicas conhecidas.
[0034] O objetivo do processo da presente invenção leva à formação de um sal de adição de nebivolol, ou seja, sal de formato de nebivolol, o qual é neutralizado em um ambiente básico, opcionalmente, in situ, para nebivolol livre de base.
[0035] Referido NBV livre de base obtido a partir do objetivo do processo da presente invenção é particularmente adequado, em termos de pureza, para as etapas subsequentes da síntese do produto final.
[0036] Portanto, um objetivo maior da presente invenção é um processo para sintetizar nebivolol ou um sal adicional deste, o qual compreende a desbenzilação de um composto da fórmula II com ácido fórmico na presença de um catalisador a base de paládio de acordo com o relatado acima.
[0037] Para os propósitos da presente invenção, é claro que é preferível ter como um substrato uma mistura racêmica dos compostos da fórmula II (mistura 1:1 de isômeros 1 e benzil derivado de d NBV) os quais devidamente tratados levam à preparação de NBV (mistura 1:1 de isômeros 1 e d), por sua vez, convertida em produto final.
[0038] Assim, por exemplo, a mistura de d-NBV e 1-NBV, obtida a partir da mistura correspondente de benzil derivados, de acordo com a invenção, é tratada com ácido clorídrico, na presença de um solvente orgânico para fornecer o sal de cloridrato de NBV respectivo.
[0039] Referido sal pode ser ainda purificado através de métodos conhecidos na técnica, como por exemplo, cristalização.
[0040] Por isso, é facilmente perceptível como o objetivo do método de redução da invenção constitui uma alternativa sintética eficiente e econômica na preparação de cloridrato de NBV do ingrediente ativo.
[0041] Referido método, in primis, prova ser quimiosseletivo na presença de compostos halogenados no anel aromático, permitindo limitar a formação de subprodutos indesejáveis, tais como, por exemplo, a impureza identificada por um ensaio de HPLC-MS possuindo a estrutura de nebivolol mono desfluorado. Essa impureza, designada aqui como nebivolol "des-F", tem a fórmula geral abaixo
[0042] Como mencionado acima, processo de hidrogenação convencional pode levar à obtenção de compostos desalogenados em grandes quantidades (>1%) e as purificações subsequentes necessárias por recristalização do produto final, além de ser caro em termos de custo, tempo e materiais de consumo, falha em limitar as impurezas de desfluorado abaixo de 0,1% exigido pelos padrões farmacêuticos.
[0043] Como se sabe, é muito importante obter um produto dotado de um grau de pureza suficiente para cumprir referidas normas. Impurezas em nebivolol, como em geral em qualquer outro princípio ativo farmacêutico, são absolutamente indesejáveis e em casos extremos, elas podem até ser prejudiciais aos pacientes tratados com formas de dosagem contendo o princípio ativo.
[0044] Portanto, um aspecto importante do objetivo do processo da invenção é a capacidade de fornecer um produto final de alta pureza em que a titulação de cada impureza é inferior a 0,1% e a soma de todas as impurezas é muito inferior a 1%, fazendo etapas adicionais de purificação caras, por exemplo, recristalização, desnecessárias.
[0045] Portanto, um objetivo maior da presente invenção é nebivolol ou um sal de adição deste, com uma pureza de pelo menos 99,9% em peso.
[0046] Um objetivo maior da presente invenção é nebivolol ou um sal de adição deste, o qual compreende menos de 0,1% em peso de nebivolol "des-F".
[0047] Um objeto ainda da presente invenção é nebivolol ou um sal de adição deste, o qual compreende menos de 0,05% em peso de nebivolol "des-F".
[0048] Portanto, um objetivo ainda da presente invenção é um processo para a síntese de nebivolol ou um sal de adição deste, com uma pureza de pelo menos 99,9% em peso, o qual inclui uma desbenzilação de acordo com o descrito acima.
[0049] Portanto, um objetivo ainda da presente invenção é um processo para a síntese de nebivolol ou um sal de adição deste, com menos de 0,1% em peso de nebivolol "des-F", o qual compreende uma desbenzilação de acordo com o descrito acima.
[0050] Portanto, um objetivo ainda da presente invenção é um processo para a síntese de nebivolol ou um sal de adição deste, com menos de 0,05% em peso de nebivolol "des-F", o qual compreende uma desbenzilação de acordo com o descrito acima.
[0051] Como mencionado acima, a principal desvantagem de CTH reside na necessidade de tempos prolongados de reação e, algumas vezes, na dificuldade em completar a reação.
[0052] O uso de ácido fórmico de acordo com a invenção, se comparado ao método de CTH na presença de formato de amônio descrito no pedido de patente internacional co-pendente WO 2008/064827, permite acelerar a reação cinética tornando-a seletiva e, nesse ínterim, rápida. Provavelmente, o mecanismo de reação que permite tal aceleração pode estar no fato de que, ao contrário de formato de amônio, ácido fórmico permite a precipitação do sal do formato de nebivolol durante a reação através de um processo de cristalização induzida.
[0053] A presença de ácido fórmico, diferente do formato de amônio, ainda, inibe o processo de envenenamento do catalisador por aminas (produto de nebivolol mesmo após desbenzilação).
[0054] Assim, do ponto de vista operacional, pode ser notado que o processo desenvolvido em comparação com o descrito em WO2008/064827, em que formato de amônio é uma fonte de hidrogênio:
- - envolve a utilização de um número menor de equivalentes de fonte de hidrogênio;
- - envolve o uso de uma quantidade menor de catalisador;
- - envolve conversões quase quantitativas, alta produtividade e altas recuperações do produto desbenzilado.
[0055] Além disso, o sal de formato de nebivolol, assim diretamente obtido, um intermediário essencial para a obtenção de nebivolol com elevada pureza, não requer purificações adicionais, tais como cromatografia preparativa em meio de ácido fórmico realizada no pedido co-pendente WO 2008/064827.
[0056] Vantagens associadas ao objetivo do método da invenção comparadas ao estado da técnica são, assim, claras.
[0057] Uma modalidade prática do objetivo do processo da presente invenção, compreende a desbenzilação de um composto da fórmula II para fornecer nebivolol livre de base, através de uma hidrogenação catalítica por transferência de hidrogênio com ácido fórmico como uma fonte de hidrogênio, e na presença de catalisador a base de Paládio.
[0058] Uma modalidade preferida prática do objetivo do processo da presente invenção compreende reagir um composto racêmico de fórmula II com ácido fórmico, na presença de catalisador Pd/C e, opcionalmente, na presença de um solvente alcoólico para fornecer sal de formato de nebivolol; o qual é neutralizado para ser livre de base através de uma reação com uma base dentre as quais um hidróxido alcalino é preferido.
[0059] Para melhor ilustrar a invenção os exemplos a seguir são fornecidos.
Exemplo 1
Síntese de [2S, αR,2'R,α'R]-α,α'-[imino- bis(metileno)]bis[6-fluoro-3,4-deidro-2H-1-benzopiran-2-metanol].
[0060] Cloridrato de [2S, αR,2' R,α'R]-α,α'- [[(fenilmetil)imino]-bis-metileno)]bis[6-fluoro-3,4-deidro-2H-1-benzopiran-2-metanol] (5,3g, 94% p/p, 9,37mmoles) foi suspenso em água (20,4g) e sec-butanol (40g), e a mistura heterogênea foi agitada sob atmosfera de nitrogênio em 25°C. 30% de hidróxido de sódio (1,5g, 11,25mmoles) foi adicionado à mistura, e a mistura foi agitada até a completa dissolução do sólido. A fase aquosa foi então separada e a fase alcoólica orgânica foi recuperada através de lavagem com solvente para fornecer mais um uma solução livre de base de [2S,αR,2'R,α'R]-α,α'-[[(fenilmetil)imino]-bis(metileno)]bis[6-fluoro-3,4-deidro-2H-1-benzopiran-2-metanol] em sec-butanol (57,1g; 8,557% p/p).
[0061] Uma parte dessa solução (52,5g; 8,557% p/p; 9,09mmoles) foi concentrada por destilação azeotrópica (Text = 95°C) à pressão atmosférica. A solução foi então diluída com sec-butanol (17,3g) e concentrada por destilação azeotrópica sob vácuo fraco. Tal sequência de operações foi repetida mais duas vezes obtendo uma solução concentrada (24,4g) que foi então levada a um volume com mais sec-butanol (20,6g). A solução foi aquecida a 70±2°C, e Paládio/carbono (0,526g, Pd/C a 5% úmido em 57%) foi adicionado à mistura durante o aquecimento. Uma vez que a temperatura foi atingida, 98% de ácido fórmico (1,279g, 27,23mmoles) foi adicionado à mistura em uma hora por uma bomba de seringa. A mistura heterogênea foi agitada a 70±2°C por 3 horas adicionais, desde o fim da adição e, em seguida, diluída em água (19,3g) e acrescida com 30% de hidróxido de sódio (1,1 g). A mistura foi mantida a 70±2°C por 15 minutos e, em seguida, filtrada a quente sob vácuo em um painel Celite, lavando com sec-butanol (8,2g) pré-aquecido a 70±2°C. O filtrado foi mantido a 60°C, em seguida, a fase aquosa foi separada enquanto que a fase orgânica foi lavada com água (2 x 19 g) a 60°C. A fase orgânica foi então concentrada por destilação sob vácuo até um volume residual de cerca de 40 ml. A mistura foi então diluída com sec-butanol (35,3g) e concentrada por destilação a vácuo até um volume residual de cerca de 45 ml. A solução orgânica foi levada a 90°C, então, foi resfriada (em 4 horas) até 25°C e mantida nesta temperatura por cerca de 16 horas.
[0062] A suspensão assim obtida foi diluída com uma mistura de sec-butanol/água (92/8 p/p) (19,9 g), aquecida a 80°C e resfriada (em 3 horas) a 25°C. Depois de mais uma hora a 25°C, o precipitado foi filtrado sob vácuo e o painel foi lavado com uma mistura sec-butanol/água (95/5 p/p) (5,4 g). O precipitado foi seco sob vácuo a 25°C para fornecer o produto desejado (S,R,R,R)-Nebivolol) como sólido branco (2,70 g, 71% de rendimento molar, titulação p/p HPLC = 97,0%, pureza HPLC = 99,7% Área).
Exemplo 2
Síntese de [2R,αS,2'S,α'S]-α,α'-[imino- bis(metileno)]bis [6-fluoro-3,4-deidro-2H-1-benzopiran-2- metanol].
[0063] Cloridrato de [2R,αS,2'S,α'S]-α,α'- [[(fenilmetil)imino]bis-metileno)]bis[6-fluoro-3,4-deidro-2H-1-benzopiran-2-metanol] (3,0g, 5,64 mmoles) foi suspenso em água (12,2g) e sec-butanol (24,1g), e a mistura heterogênea foi agitada mecanicamente sob atmosfera de nitrogênio a 25°C. O hidróxido de sódio (0,96 g de 30% p/p de solução aquosa) foi adicionado à mistura, e a mistura foi agitada até a completa dissolução do sólido. A fase aquosa foi então separada e a fase alcoólica orgânica foi diluída com álcool sec-butanol (15,7 g) e então sujeita a destilação azeotrópica sob vácuo (4 KPa). A destilação foi interrompida e a mistura foi levada a um volume com sec-butanol (23,4g). A destilação foi retomada e, no final da operação, a mistura foi diluída com sec-butanol (13,8g) para fornecer cerca de 9% em solução (p/p) de base de [2R,αS,2'S,α'S]-α,α'-[[(fenilmetil)imino]bis-metileno)] bis[6-fluoro-3,4-deidro-2H-1-benzopiran-2-metanol].
[0064] A solução foi aquecida a 70±2°C, e Paládio/Carbono (0,33 g, Pd/C a 5% úmido em 57%) foi adicionado à mistura durante o aquecimento. Uma vez que a temperatura foi atingida, uma solução de ácido fórmico (0,79g, 16,91mmoles) e sec-butanol (0,79g) foram adicionados à mistura em uma hora por uma bomba de seringa. A mistura heterogênea foi então agitada mecanicamente, por 3 horas adicionais, desde o fim da adição e, então diluída com água demi (11,5g), hidróxido de sódio (0,88 g de 30% p/p de solução aquosa) e, finalmente, sec-butanol (7,8 g). A mistura foi filtrada a quente sob vácuo em um painel de Celite (1,6 g), e o painel foi lavado com sec-butanol (12,2 g) pré-aquecido a 70±2°C. A fase aquosa foi separada e armazenada enquanto a fase orgânica foi lavada com uma solução aquosa saturada com bicarbonato de sódio (15,95g) a 70±2°C e com água demi (15,2g) a 70±2°C. A fase orgânica e a fase aquosa inicial a 60°C foram então combinadas e a mistura bifásica foi diluída com água demi (15,2g). A lavagem aquosa foi descarregada e a fase orgânica foi submetida à destilação (P = 3 KPa; Tint = 27°C). O volume foi reduzido em 25% e destilação foi interrompida. A temperatura foi ajustada para 70±2°C e, em seguida, levada a 0°C em 6 horas. Após 10 horas adicionais em 0°C, o precipitado foi filtrado sob vácuo e o painel foi lavado com sec-butanol (8,0g). O precipitado foi seco sob vácuo a 60°C para fornecer o produto desejado (R,S,S,S-Nebivolol) como sólido branco (1,63 g, rendimento molar de 67%, titulação p/p HPLC = 94,3%; pureza HPLC = 99,6% Área).
Exemplo 3
Síntese de (±) [R*,S*,S*,S*]-α,α'-[imino- bis(metileno)]bis[6-fluoro-3,4-deidro-2H-1-benzopiran-2- metanol].
[0065] (±)[R*,S*,S*,S*]-α,α'- [[(fenilmetil)imino]bis-metileno)]bis[6-fluoro-3,4-deidro-2H-1-benzopiran-2-metanol] (530 g, 1,07 moles) e Pd/C a 5% úmido em 50% (52,3 g) em sec-butanol (4970 g) foram carregados em um reator. Uma solução de ácido fórmico (98%) (150,7 g, 3,21 moles) em sec-butanol (151 g) foi adicionada à mistura aquecida a 70±2°C em cerca de uma hora. A mistura reacional foi mantida sob agitação a 70±2°C por cerca de duas horas, no final de uma solução composta de NaOH a 30% (225g) em água (1900g) foi adicionada e foi mantida sob agitação em 70±2°C até a dissolução da suspensão. A mistura foi filtrada a quente em um painel Celite lavando com sec-butanol (726g), e tolueno (530g) foi adicionado. A mistura bifásica foi mantida a 70±2°C, em seguida, a fase aquosa foi separada e a fase orgânica resultante foi lavada com uma solução aquosa de bicarbonato (180g dissolvido em 2400g de água) e então com água (2280g). A solução orgânica foi destilada sob vácuo, várias vezes, reintegrando a fase concentrada com sec-butanol novo. A fase final orgânica (cerca de 8000 ml) foi aquecida a 85-90°C até solubilização completa e, então gradualmente, resfriada a 20°C obtendo a precipitação do produto.
[0066] O sólido foi isolado por filtração e seco em estufa sob vácuo, a 50°C para fornecer o produto desejado como sólido branco (380g, o rendimento molar de 87,6%; titulação p/p HPLC ≥ 99%; pureza de HPLC ≥ 99% Área).
Exemplo 4
Síntese de Cloridrato de (±)[R*,S*,S*,S*]-α,α'-[imino-bis(metileno)]bis[6-fluoro-3,4-deidro-2H-1-benzopiran-2-metanol].
[0067] (±)[R*,S*,S*,S*]-α,α'-[imino]bis- metileno)]bis[6-fluoro-3,4-deidro-2H-1-benzopiran-2-metanol] (380g; 0,937 moles), sec-butanol (4195 g) e água (306g) foram carregados em um reator. HCl a 31% (134,4g, 1,14 moles) foi adicionado à mistura, sob agitação e aquecido a 70±2°C. A mistura foi aquecida a 70±2°C por 2 horas, resfriada a 20±2°C e mantida a essa temperatura durante pelo menos 3 horas, obtendo a precipitação do produto. O sólido foi isolado por filtração lavando com sec-butanol (422g) e seco em estufa sob vácuo a 60°C para fornecer o produto desejado como sólido branco (400,5g; rendimento molar de 96,7%; titulação p/p HPLC ≥ 99% (produto seco), HPLC de pureza ≥ 99% Área); perfil de HPLC de pureza típica: soma das impurezas = 0,06% p/p; nebivolol "des-F" = 0,04% p/p.
[0068] 1H-RMN (400 MHz; MeOD) δ(ppm): 6,85-6,77 (m, 6H), 4,15-4,11 (m, 1H), 4,07-4,01 (m, 2H), 3, 97-3, 92 (m, 1H), 3,56-3,25 (m, 4H), 2,99-2,80 (m, 4H), 2,30-2,24 (m, 1H), 2,07-1,92 (m, 2H), 1,86-1,76 (m, 1H).
MS (ESI): m/z ([M+H]+) = 406,2
PF = 225,6-226,8°C.
Exemplo 5
Comparação com a técnica anterior: N-desbenzilação por hidrogenação catalítica convencional. Síntese de (±)[R*,S*,S*,S*]-α,α'-[imino-bis(metileno)]bis[6-fluoro- 3,4-deidro-2H-1-benzopiran-2-metanol].
[0069] (±)[R*,S*,S*,S*]-α,α'-[(fenilmetil)imino-bis-metileno)]bis[6-fluoro-3,4-deidro-2H-1-benzopiran-2-metanol] (18,82 g; 0,038 moles), sec-butanol (220 ml) e Pd/C a 5% úmido em 50% (3 g) foram carregados em autoclave. A mistura foi aquecida a 80°C e autoclave foi pressurizada com hidrogênio (P = 400 kPa). A mistura foi mantida sob agitação nas condições acima mencionadas por 17 horas, e foi então filtrada a quente em um painel Celite lavando com butanol-sec a quente (150 ml) (usando Buchner alinhado com Talinhamento = 85°C). A solução foi levada à temperatura ambiente para provocar a precipitação, a mistura então obtida foi mantida sob agitação a 15°C por cerca de 2 horas para completar a precipitação do produto.
[0070] O sólido foi isolado por filtração, lavagem com água fria sec-butanol (50 ml) e seco em estufa sob vácuo a 35°C para fornecer o produto desejado como sólido branco (12,9 g; rendimento molar 83,7%); perfil de pureza HPLC: nebivolol "des-F" = 2,09% p/p.
[0071] Assim, isto resulta prontamente aparente como o uso de um método convencional de hidrogenação catalítica a fim de realizar uma desbenzilação de acordo com a invenção, consiste na formação de uma percentagem elevada (>2%) do subproduto desfluorado, nebivolol "des-F", e as desvantagens associadas às purificações subsequentes do produto descritas acima.
Exemplo 6
[0072] Comparação com a técnica anterior: N- desbenzilação por CTH de acordo com o pedido de patente international WO 2008/064827, Exemplo 10; isolamento de Nebivolol livre de base e formação de cloridrato de Nebivolol foi realizada seguindo os métodos descritos nos exemplos 3 (parte) e 4 acima.
Parte A: (±)[R*,S*,S*,S*]-α,α'-[imino- bis(metileno)]bis[6-fluoro-3,4-deidro-2H-1-benzopiran-2- metanol].
[0073] (±)[R*,S*,S*,S*]-α,α'- [(fenilmetil)imino]bis-metileno) ]bis[6-fluoro-3,4-deidro-2H-1-benzopiran-2-metanol] (5,0g, 0,0101mol) foi dissolvido em metanol (343,9g). Formato de amônio (4,8g, 0,0761mol) foi adicionado à mistura reacional, seguido por catalisador Paládio sobre carbono a 5% em peso úmido (contém ~ 50% de água) (0,5g). A mistura reacional foi aquecida em refluxo (cerca de 65°C) sob agitação por cerca de 11 horas, então, resfriada a 45°C, filtrada em um bloco celite, lavando com metanol e, finalmente concentrada sob vácuo. O resíduo bruto (6,2 g) foi diluído com 2-butanol (65,2g), então uma solução aquosa de hidróxido de sódio a 30% (2,0g) e água (29,8 g) foi adicionada, sob agitação. A suspensão resultante foi aquecida a 70±2°C até a completa dissolução obtendo uma mistura de duas fases claras. A camada aquosa foi separada e a fase orgânica resultante foi lavada com hidrogenocarbonato de sódio aquoso (1,7g dissolvidos em 22,6g de água) e depois com água (2x 21,5 g). Tolueno (5,0 g) também foi adicionado para melhorar a separação aquosa-orgânica. A fase orgânica foi concentrada duas vezes, sob pressão reduzida, acrescentando novo 2-butanol (total 32,9g) para os resíduos resultantes. A solução final orgânica (cerca de 60 ml) foi diluída com 2-butanol (9,5 g), aquecida a 85-90°C até a completa dissolução e então, gradualmente, resfriada a 20°C obtendo a precipitação do produto. O sólido foi isolado por filtração, lavando com 2-butanol (3x 2,7g) e seco sob vácuo a 60°C para fornecer o composto rotulado como um sólido branco (3,5g, ensaio (HPLC) 96,6% p/p; 82,6% de rendimento molar) Perfil de pureza (HPLC). Soma das impurezas = 0,284% p/p; Impureza Máxima Única (Nebivolol "des-F") = 0,196% p/p.
Parte B: Cloridrato de (±)[R*,S*,S*,S*]-α,α'-[imino-bis(metileno)]bis[6-fluoro-3,4-deidro-2H-1-benzopiran-2- metanol].
[0074] (±)[R*,S*,S*,S*]-α,α'-[imino-bis (metileno)]bis[6-fluoro-3,4-deidro-2H-1-benzopiran-2-metanol] (3,106 g; 96,6% p/p; 0,00740 mol) foi dissolvido em uma mistura de 2-butanol (33,1g) e água (2,0g). Ácido clorídrico aquoso concentrado (cerca de 31%) (1,1g; 0,00903 mol) foi adicionado à solução, sob agitação a 70±2°C. A mistura resultante foi aquecida a 70±2°C por 2 horas, então resfriada a 20±2°C durante 2 horas, e mantida nesta temperatura por mais 2 horas obtendo a precipitação do produto. O sólido foi isolado por filtração, lavando com 2-butanol (2 x 2g) , e seco sob vácuo a 60°C para fornecer o composto como um sólido branco (3,1g, 98,3% p/p de ensaio (HPLC); 93,2% de rendimento molar). Perfil de pureza (HPLC): Soma das impurezas = 0,224% p/p; Impureza Única Máxima (nebivolol "des-F") = 0,178% p/p.
[0075] Assim, isto resulta prontamente aparente como o processo de N-desbenzilação por CTH descrito no pedido de patente internacional WO 2008/064827, Exemplo 10, além de ser muito lento (11 horas à temperatura de refluxo), utilizando uma maior quantidade de catalisador e fonte de hidrogênio equivalente e não levando a conversão quantitativa, consiste na formação de subprodutos indesejáveis, nebivolol "des-F" (>0,1%), provando ser suficientemente quimiosseletivo.
[0076] Ao contrário, partindo do mesmo intermediário, (±)[R*,S*,S*,S*]-α,α'- [(fenilmetil)imino]bis-metileno) ]bis[6-fluoro-3,4-deidro-2H-1-benzopiran-2-metanol], usado na Parte A acima (lote EP109) e seguindo o procedimento descrito nos Exemplos 3 e 4 da presente invenção (N-desbenzilação, isolamento de NBV livre de base e formação de cloridrato), obtivemos cloridrato de NBV altamente puro (Titulação p/p HPLC = 100,0%; Perfil de pureza (HPLC): Soma das impurezas = 0,0272% p/p; Impureza Única Máxima (Nebivolol "des-F") = 0,0207% p/p).