CN1037100C - 喹啉类抗生素合成中中间体的制备 - Google Patents
喹啉类抗生素合成中中间体的制备 Download PDFInfo
- Publication number
- CN1037100C CN1037100C CN93102103A CN93102103A CN1037100C CN 1037100 C CN1037100 C CN 1037100C CN 93102103 A CN93102103 A CN 93102103A CN 93102103 A CN93102103 A CN 93102103A CN 1037100 C CN1037100 C CN 1037100C
- Authority
- CN
- China
- Prior art keywords
- benzyl
- formula
- compound
- alkali
- alkyl
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/52—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring condensed with a ring other than six-membered
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Plural Heterocyclic Compounds (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
- Indole Compounds (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
Abstract
本发明涉及制备下式化合物的新方法,其中的R和X如上所限定。式Ⅶ的化合物是合成具有抗菌活性的氮杂双环喹啉甲酸中的有用的中间体。本发明也涉及合成这类抗生素的某些新的中间体。
Description
本发明涉及在合成喹啉类抗生素7-(1a,5a,6a)-(6-氨基-3-氮杂双环[3.1.0]己-3-基)-1-(2,4-二氟苯基)-6-氟-1,4-二氢-4-氧代-1,8-二氮萘-3-甲酸及相关的抗生素化合物中制备中间体的新方法。喹啉类抗生素7-(1a,5a,6a)-(6-氨基-3-氮杂双环[3.1.0]己-3-基-1-(2,4-二氟苯基)-6-氟-1,4-二氢-4-氧代-1,8-二氮萘-3-甲酸具有下列化学结构式 1990年7月11日提交的美国专利申请07/551,212号和1989年8月16日提交、1991年3月7日公布的世界专利申请WO91/02526号均提到了该化合物及相关的氮杂双环喹啉甲酸具有抗菌活性。前述两项申请作为参考文献全部并入本申请中。
1990年7月11日提交的美国专利申请07/551,212号和1989年8月16日提交、1991年3月7日公布的世界专利申请WO91/02526号均提到了该化合物及相关的氮杂双环喹啉甲酸具有抗菌活性。前述两项申请作为参考文献全部并入本申请中。
本发明的新方法可用于制备下式化合物该化合物为合成式I所示喹啉类抗生素及上述氮杂双环喹啉甲酸抗生素的中间体。美国专利申请07/551,212号和世界专利申请WO91/02526号中详述了可将式VII所示的化合物转换为这些抗生素化合物的方法。
本发明涉及制备下式化合物的方法式中R为(C1-C6)烷基,(C3-C6)环烷基或苄基,其中所说的苄基中的苯基部分可任意地被一个或多个取代基所取代,所述取代基分别选自卤素(如氯,氟,溴或磺),硝基,(C1-C6)烷基,(C1-C6)烷氧基,氨基和三氟甲基;该方法包括在碱存在下将下式化合物与卤代硝基甲烷反应其中R如上所限定。
在本发明的一个优选实例中,按上述方法制备的式III化合物是其中R为(C1-C6)烷基或苄基的化合物。在更优选实例中,R是苄基。
本文所用“卤素”一词指的是氯、氟、溴和碘。
本发明还涉及上述的方法,该方法还包括使所生成的式III化合物与一种还原剂反应,生成下式的化合物 其中R如上所限定。
其中R如上所限定。
本发明还涉及具有下式的化合物其中R如上所限定。
下述反应路线阐释了本发明的方法和本发明中化合物的制备。除非另有说明,反应路线及其后的讨论中的式I、式II、式III、式IV和取代基R和X均如上所限定。
反应路线
上面的反应路线阐释了式VII化合物的制备,这些化合物在上面提到的喹啉类抗生素的合成中是很有用的中间体。
上述反应路线中,在有碱存在的情况下,式II的化合物与卤代硝基甲烷、最好是氯代硝基甲烷(ClCH2NO2)或溴代硝基甲烷(BrCH2NO2))反应,生成相应的式III化合物。该反应一般是在惰性的、极性的和质子惰性的溶剂[如二甲基甲酰胺(DMF)、二甲基亚砜(DMSO)或二甲基乙酰胺(DMAC)],或惰性的醚溶剂[如乙醚、甘醇二甲醚或四氢呋喃(THF)],或其它的惰性溶剂[如苯、甲苯或氯代苯或甲苯)中进行的。优先选用甲苯。适用的反应温度范围为-78℃至80℃,0℃为优选。最好最后加碱。适用的碱包括碳酸盐碱如碳酸钾或碳酸钠,磷因酰碱如2-叔丁基亚氨基-2-二乙氨基-1,3-二甲基全氢-1,3,2-二氮杂-磷因,胺类碱如三乙胺,胍,二异丙基乙胺,四甲基胍,1,8-二氮杂双环-[5.4.0]十一碳-7-烯(DBU)和1,5-二氮杂双环-[5.4.0]壬-5-烯(DBN)。使用胺类碱是有利的,最优先选用DBU。
上述生成的式III化合物的还原生成相应的式IV化合物。适用的还原剂包括甲硼烷/甲硫醚,甲硼烷/四氢呋喃,硼氢化钠和三氟化硼合乙醚混合物,优选的还原剂为甲硼烷/四氢呋喃。还原反应典型地是在45℃-90℃的温度下进行,用惰性的醚溶剂如甘醇二甲醚,二甘醇二甲醚,乙醚,二异丙基醚或四氢呋喃。优选的反应条件是66℃和四氢呋喃。
用一种金属和无机酸处理,可将得到的式IV化合物转变成相应的式V化合物胺。锌为优选金属。适用的无机酸有盐酸,硫酸。优选盐酸。该反应一般是在低级的醇溶剂如乙醇、甲醇、1-丙醇或2-丙醇(乙醇为优选)中,在0-80℃(优选25℃)的温度下进行的。
将一种合适的氮保护基加到式V化合物的未取代的氨基氮上,则生成相应的式VI化合物,其中X为一氮保护基。许多众所周知的氮保护基可采用,包括C2-C6烷氧羰基,任意取代的苄氧羰基,芳氧羰基,甲硅烷基,三苯甲基,乙烯氧羰基,邻-硝基苯碘酰基,二苯基膦基,对甲苯磺酰基和苄基。用二碳酸二叔丁基酯或2-叔-丁氧羰基氧基亚氨基-2-苯基乙腈是很有利的。加氮保护基通常是在如下条件中进行:溶剂为一种氯化的烃溶剂如二氯甲烷或1,2-二氯乙烷,或醚溶剂如甘醇二甲醚,二甘醇二甲醚或四氢呋喃;加或不加催化量的胺碱如三乙胺,二异丙基乙胺或吡啶(优选三乙胺);反应温度为0-50℃(优选25℃)。
为R为苄基时,前述一步中通过氢解除去式VI化合物中的R基团则生成所希望的式VIII化合物。这一反应通常是使式VI化合物(其中的R为苄基)与氢气(气压为0-2000磅/平方英寸,优选50磅/平方英寸)在有稀有金属催化剂如钯、铂或铑存在的情况下进行反应。优选钯-碳或氢氧化钯-炭。反应温度范围20℃-80℃,优选25℃。溶剂通常为低级醇,优选甲醇。
当R为C1-C6烷基或C3-C6环烷基时,可通过与氯甲酸α-氯乙基酯(ACE-Cl)反应除去R基团(见Olefson et al.,J.Org.Chem.,49,2081-2(1984)和Olefson et al.,Pure & Appl.Chem.,60(11),1715-24(1988))。
用式VII化合物制备具有式I结构的喹啉抗生素和相关的氮杂环喹啉甲酸抗生素的方法在美国专利申请07/551,212号(1990年7月11日提交)和世界专利申请WO 91/02526号(1989年8月16日提交,1991年3月7日公布)中均有叙述,该两项申请作为参考文献全部并入本文。
能用本发明的方法和化合物合成的具有式I结构的抗菌化合物及其相关的氮杂环喹啉甲酸抗生素可用于治疗动物(包括人)的细菌感染。它们具有广谱抗菌作用,特别适用于治疗革兰氏阳性菌感染。
美国专利申请07/551,212号和世界专利申请WO91/05526号详述了这类抗生素化合物的适宜剂量范围和投药方法,也给出了检测这类化合物抗菌活性的方法。
下述实施例说明本发明的方法和化合物。但需指出,本发明并不局限于这些实施例的具体范围。
实施例1
1a,5a,6a-3-苄基-6-硝基-2,4-二氧代-3-氮杂双环[3.1.0]己烷
将250ml甲苯加入到N-苄基马来酰亚胺(24.3g,130mmol)和溴代硝基甲烷(18.2ml,260mmol)混合物甲,冷却到0℃。用搅拌器强烈搅拌下,在30分钟内将用200ml甲苯稀释的58ml DBU(1,8-二氮杂双环[5.4.0]十一碳-7-烯)(390mmol)加入到混合物中。室温下再将反应物搅拌2小时。将甲苯层倒出并用0.1M HCl(2×100ml)洗涤,通过硫酸镁(MgSO4)干燥。将溶剂蒸发得5.4g产物,即收率为17%。熔点=114-115.5℃。1H NMR(CDCl3):7.31(m,5H,芳族的)4.54(s,2H,苄基的),4.47(t,1H,α-硝基),3.35(d,2H,3-环)。
实施例2
1a,5a,6a-3-苄基-6-硝基-3-氮杂双环[3.1.0]己烷
将硼甲烷·四氢呋喃(THF)配合物(32.4ml,1M THF溶液,32.4mmol)加到实施例1所得的1a,5a,6a-3-苄基-6-硝基-2,4-二氧-3-氮杂双环[3.1.0]己烷中(2g,8.1mmol,溶于20mlTHF中),将混合物回流加热3小时。将反应物冷却到室温,小心地加入10ml甲醇。再回流加热15分钟。然后使溶剂蒸发,将残油溶于200ml二氯甲烷中并用水(3×100ml)洗涤。将有机物层用MgSO4干燥,蒸发后得到1.5g产物(淡色油状物),收率为90%。1H NMR(CDCl3):7.35-7.19(m,5H,芳族的)4.63(t,1H,α-硝基),3.59(s,2H,苄基的),3.14(m,2H,5-环),2.49(m,2H,5-环),2.51(m,2H,3-环)。
实施例3
1a,5a,6a-3-苄基-6-氨基-3-氮杂二环[3.1.0]己烷
在实施例2所生成的1a,5a,6a-3-苄基-6-硝基-3-氮杂二环[3.1.0]己烷(6g,27.5mmol)的50ml乙醇液中加入18.0g锌粉(275mmol)。再加入150ml 1M HCl,加入速率以使反应物温度不超过40℃(1小时)为限。将反应物在室温下搅拌3小时,然后通过硅藻土(CeliteR)过滤。将溶剂蒸发,用500ml 1M NaOH溶液将粘稠的白色残余物消化3小时。将混合物用二氯甲烷(2×300ml)提取,两次提取的有机物层合并后用盐水(3×100)洗涤,通过MgSO4干燥。将溶剂蒸发得4.06g产物,收率为79%。1H NMR(CDCl3):7.35-7.20(m,5H,芳族的)4.62(宽单线,1H,α-硝基),3.60(s,2H,苄基的),3.14(m,2H,5-环),2.52(m,2H,5-环和m,2H,环丙基)。
实施例4
1a,5a,6a-3-苄基-6-[(叔丁基甲酰)氨基]-3-氮杂双环[3.1.0]己烷
在实施例3所生成的1a,5a,6a-3-苄基-6-氨基-3-氮杂双环[3.1.0]己烷(3.75g,19.9mmol)的50ml THF液加入二碳酸二-叔-丁酯(4.78g,21.9mmol)和三乙胺(0.28ml,1.99mmol),将混合物搅拌4小时,然后蒸发掉溶剂,加入75ml二氯甲烷。将混合物用20ml水洗涤并通过MgSO4干燥。将溶剂蒸发,再溶于100mg己烷中。将混合物加热直到所有的固体物质溶解,加入2.5g活性炭,继续加热5分钟。将炭滤去。将反应混合物冷却后生成的固体物质过滤并在空气中干燥,得到5.1g产物,收率为89%。溶点=131-132℃(白色针状结晶)。1H NMR(CDCl3):7.24(m,5H,芳族的)3.54(s,2H,苄基的),3.06(m,2H,5-环),2.91(宽,1H,α-酰胺),2.43(m,2H,5-环),1.52(m,2H,3-环)。
实施例5
1a,5a,6a-[(叔丁基甲酰)氨基]-3-氮杂双环[3.1.0]己烷
在实施例4生成的1a,5a,6a-3-苄基-6-[(叔丁基甲酰)氨基]-3-氮杂双环[3.1.0]己烷(2.0g,6.94mmol)的50ml甲醇液中加入氢氧化钯/炭(Pd(OH)2/C)(50%湿)(1.0g,50%重量)。给混合物加氢(50磅/平方英寸)6小时,通过硅藻土过滤,将溶剂蒸发,得1.36g产物,收率为99%。1H NMR(CDCl3):3.22-2.95(m,4H,5-环),2.61(宽,1H,-酰胺),2.32(m,1H,α-酰胺),1.63(m,2H,3-环),1.45s,9H,丁基)。
Claims (11)
1.制备下式化合物的方法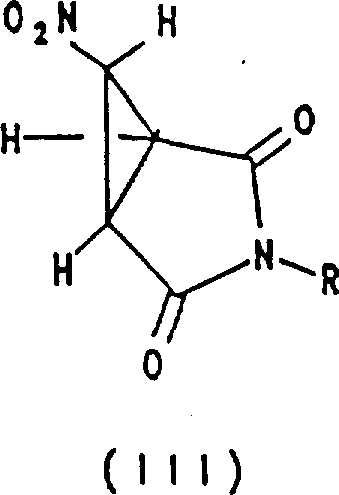 式中R为C1-C6烷基,C3-C8环烷或苄基,其中所说的苄基中的苯基部分可以独立地被一个或多个下述取代基所取代:卤素,硝基,C1-C6烷基,C1-C6烷氧基,氨基和三氟甲基;所述方法是将下式的化合物在碱存在下与卤代硝基甲烷反应
式中R为C1-C6烷基,C3-C8环烷或苄基,其中所说的苄基中的苯基部分可以独立地被一个或多个下述取代基所取代:卤素,硝基,C1-C6烷基,C1-C6烷氧基,氨基和三氟甲基;所述方法是将下式的化合物在碱存在下与卤代硝基甲烷反应 式中的R如上述所限定。
式中的R如上述所限定。
2.按权利要求1的方法,其中所生成的式III化合物是R为C1-C6烷基或苄基的化合物。
3.按权利要求2的方法,其中R为苄基。
4.按权利要求1的方法,其中所说的卤代硝基甲烷为溴代硝基甲烷或氯代硝基甲烷。
5.按权利要求1的方法,其中所述的方法是在-78℃至80℃的温度范围内实施。
6.按权利要求1的方法,其中实施所述的方法的溶剂选自:苯,甲苯,二甲基甲酰胺或四氢呋喃。
7.按权利要求6的方法,其中所述溶剂为甲苯。
8.按权利要求1的方法,其中所说的碱为碳酸盐碱,胺类碱和磷因酰胺碱。
9.按权利要求8的方法,其中所说的碱选自:碳酸钠,碳酸氢钠,碳酸钾,碳酸氢钾,2-叔丁亚氨基-2-二乙氨基-1,3-二甲基全氢化-1,3,2-二氮杂-磷因,三乙胺,胍,二异丙基乙胺,四甲基胍,1,8-二氮杂双环[5.4.0]十一碳-7-烯和1,5-二氮杂双环[4.3.0]壬-5-烯。
10.按权利要求9的方法,其中的碱为1.8-二氮杂双环[5.4.0]十一碳-7-烯。
11.具有下式的化合物式中R为C1-C6烷基或苄基,而所说的苄基中的苯基部分可任意被一个或多个选自下述的基团所取代:卤素,硝基,C1-C6烷基,C1-C6烷氧基,氨基和三氟甲基。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US844,367 | 1992-03-02 | ||
| US07/844,367 US5256791A (en) | 1992-03-02 | 1992-03-02 | Preparation of intermediates in the synthesis of quinoline antibiotics |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1076440A CN1076440A (zh) | 1993-09-22 |
| CN1037100C true CN1037100C (zh) | 1998-01-21 |
Family
ID=25292537
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN93102103A Expired - Fee Related CN1037100C (zh) | 1992-03-02 | 1993-03-01 | 喹啉类抗生素合成中中间体的制备 |
Country Status (22)
| Country | Link |
|---|---|
| US (2) | US5256791A (zh) |
| EP (1) | EP0629189B1 (zh) |
| JP (1) | JP2564247B2 (zh) |
| KR (2) | KR950700251A (zh) |
| CN (1) | CN1037100C (zh) |
| AT (1) | ATE156480T1 (zh) |
| AU (1) | AU667872B2 (zh) |
| CA (1) | CA2131160C (zh) |
| DE (1) | DE69312913T2 (zh) |
| DK (1) | DK0629189T3 (zh) |
| ES (1) | ES2105217T3 (zh) |
| FI (1) | FI106022B (zh) |
| GR (1) | GR3024789T3 (zh) |
| HU (1) | HU215837B (zh) |
| IL (1) | IL104818A (zh) |
| MX (1) | MX9301138A (zh) |
| MY (1) | MY107723A (zh) |
| NO (1) | NO300681B1 (zh) |
| NZ (1) | NZ246768A (zh) |
| TW (1) | TW211002B (zh) |
| WO (1) | WO1993018001A1 (zh) |
| ZA (1) | ZA931428B (zh) |
Families Citing this family (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CZ286896B6 (en) | 1994-01-18 | 2000-07-12 | Pfizer | Process for preparing pharmaceutically acceptable salts of naphthyridone carboxylic acid and intermediates for this preparation |
| US5475116A (en) * | 1994-04-29 | 1995-12-12 | Pfizer Inc. | Aza bicyclo[3,1,0]hexane intermediates useful in the synthesis of quinolones |
| US5929240A (en) * | 1994-12-12 | 1999-07-27 | Pfizer Inc. | Process and intermediates for preparing naphthyridonecarboxylic acid salts |
| JPH0912547A (ja) * | 1995-06-23 | 1997-01-14 | Chisso Corp | ニューキノロン系化合物中間体の製造方法 |
| GB9524466D0 (en) * | 1995-11-30 | 1996-01-31 | Pfizer Ltd | Process |
| US6057455A (en) * | 1996-07-09 | 2000-05-02 | Pfizer, Inc. | Preparation of intermediates useful in the synthesis of quinoline antibiotics |
| JPH1087617A (ja) * | 1996-07-09 | 1998-04-07 | Pfizer Inc | キノリン系抗生物質の合成に有用な中間体の製法 |
| GB9614422D0 (en) * | 1996-07-09 | 1996-09-04 | Pfizer Ltd | Novel process |
| HN1998000106A (es) | 1997-08-01 | 1999-01-08 | Pfizer Prod Inc | Composiciones parenterales de alatroflaxacino |
| DE19733439A1 (de) * | 1997-08-02 | 1999-02-04 | Bayer Ag | Neue 6-endo-Amino-3-azabicyclo(3.1.0)hexande, Verfahren zu deren Herstellung und deren Verwendung zur Herstellung von Chinolon- und Naphthyridincarbonsäure-Derviaten mit verbesserten Eigenschaften |
| US6184380B1 (en) * | 1999-01-25 | 2001-02-06 | Pfizer Inc. | Process for preparing naphthyridones and intermediates |
| US7019142B2 (en) | 1998-01-16 | 2006-03-28 | Pfizer Inc. | Process for preparing naphthyridones and intermediates |
| PA8464701A1 (es) * | 1998-01-16 | 2000-09-29 | Pfizer Prod Inc | Procedimiento e intermedios para preparar naftiridonas |
| WO2008010061A2 (en) * | 2006-07-17 | 2008-01-24 | Glenmark Pharmaceuticals S.A. | 3-azabicyclo [3.1.0] hexane vanilloid receptor ligands, pharmaceutical compositions containing them, and processes for their preparation |
| CA3142351A1 (en) | 2019-05-31 | 2020-12-03 | Ikena Oncology, Inc. | Tead inhibitors and uses thereof |
| CA3141826A1 (en) | 2019-05-31 | 2020-12-03 | Ikena Oncology, Inc. | Tead inhibitors and uses thereof |
| WO2023151188A1 (zh) * | 2022-02-08 | 2023-08-17 | 上海皓元医药股份有限公司 | 一种抗病毒药物中间体的绿色合成方法 |
Family Cites Families (6)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US4183857A (en) * | 1978-07-06 | 1980-01-15 | Shell Oil Company | 3-Benzyl-3-azabicyclo(3.1.0)hexane-2,4-dione |
| DE2962910D1 (en) * | 1978-10-27 | 1982-07-08 | Shell Int Research | A process for the preparation of 3-azabicyclo(3.1.0)hexane derivatives and modifications thereof |
| JPH0676400B2 (ja) * | 1987-08-25 | 1994-09-28 | 大日本製薬株式会社 | 新規ピリドンカルボン酸誘導体、そのエステルおよびその塩 |
| US5196548A (en) * | 1989-05-11 | 1993-03-23 | Pfizer Inc. | Preparation of diazabicyclic Intermediates |
| WO1991002526A1 (en) * | 1989-08-16 | 1991-03-07 | Pfizer Inc. | Azabicyclo quinolone carboxylic acids |
| US5200527A (en) * | 1991-04-08 | 1993-04-06 | Lonza Ltd. | Process for the production of 2-azabicyclo [2.2.1] hept-5-en-3-one |
-
1992
- 1992-03-02 US US07/844,367 patent/US5256791A/en not_active Expired - Fee Related
-
1993
- 1993-01-07 KR KR1019940703051A patent/KR950700251A/ko active Granted
- 1993-01-07 JP JP5515648A patent/JP2564247B2/ja not_active Expired - Lifetime
- 1993-01-07 NZ NZ246768A patent/NZ246768A/en unknown
- 1993-01-07 EP EP93902893A patent/EP0629189B1/en not_active Expired - Lifetime
- 1993-01-07 KR KR1019940703051A patent/KR0135626B1/ko not_active Expired - Fee Related
- 1993-01-07 AT AT93902893T patent/ATE156480T1/de active
- 1993-01-07 DK DK93902893.2T patent/DK0629189T3/da active
- 1993-01-07 DE DE69312913T patent/DE69312913T2/de not_active Expired - Fee Related
- 1993-01-07 ES ES93902893T patent/ES2105217T3/es not_active Expired - Lifetime
- 1993-01-07 WO PCT/US1993/000008 patent/WO1993018001A1/en not_active Ceased
- 1993-01-07 AU AU34300/93A patent/AU667872B2/en not_active Ceased
- 1993-01-07 CA CA002131160A patent/CA2131160C/en not_active Expired - Fee Related
- 1993-01-07 HU HU9402530A patent/HU215837B/hu not_active IP Right Cessation
- 1993-01-11 TW TW082100123A patent/TW211002B/zh active
- 1993-02-22 IL IL104818A patent/IL104818A/xx not_active IP Right Cessation
- 1993-03-01 ZA ZA931428A patent/ZA931428B/xx unknown
- 1993-03-01 CN CN93102103A patent/CN1037100C/zh not_active Expired - Fee Related
- 1993-03-01 MX MX9301138A patent/MX9301138A/es not_active IP Right Cessation
- 1993-03-01 MY MYPI93000368A patent/MY107723A/en unknown
- 1993-08-04 US US08/101,879 patent/US5298629A/en not_active Expired - Fee Related
-
1994
- 1994-09-01 NO NO943243A patent/NO300681B1/no not_active IP Right Cessation
- 1994-09-01 FI FI944013A patent/FI106022B/fi active
-
1997
- 1997-09-17 GR GR970402413T patent/GR3024789T3/el unknown
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1037100C (zh) | 喹啉类抗生素合成中中间体的制备 | |
| KR900001202B1 (ko) | 1,4-벤조디아제핀 유도체의 제조방법 | |
| CN101346355B (zh) | 制备4-(苯并咪唑基甲基氨基)-苯甲脒的盐的改良方法 | |
| US9315457B2 (en) | Crystalline polymorphic forms of an antidiabetic compound | |
| CN106458892A (zh) | 用于制备倍癌霉素前药的改进方法 | |
| SI9300286A (sl) | Ciklopentan- in -penten-beta-amino kisline | |
| CN101516872A (zh) | (2s,5r)-5-乙炔基-1-{n-(4-甲基-1-(4-羧基-吡啶-2-基)哌啶-4-基)甘氨酰}吡咯烷-2-甲腈的合成 | |
| US5475116A (en) | Aza bicyclo[3,1,0]hexane intermediates useful in the synthesis of quinolones | |
| CZ286896B6 (en) | Process for preparing pharmaceutically acceptable salts of naphthyridone carboxylic acid and intermediates for this preparation | |
| CA2068853A1 (fr) | Derives de pyridone amino acide azetidinyl substitues, leur preparation et leur application en tant que medicaments | |
| CN101679301A (zh) | 2-(2-吡啶基甲基)亚磺酰基-1h-苯并咪唑类的制备方法及其中使用的中间体化合物 | |
| KR0147888B1 (ko) | 3-(1-아미노시클로알킬)피롤리딘 유도체 및 이의 제조방법 | |
| US5929240A (en) | Process and intermediates for preparing naphthyridonecarboxylic acid salts | |
| CN111484507B (zh) | 一种氧氮杂螺环类化合物的制备方法 | |
| CN118271321A (zh) | 一种吲哚啉类化合物的合成方法 | |
| JPWO2005063678A1 (ja) | フェニル酢酸誘導体の製造方法 | |
| KR100199101B1 (ko) | 새로운 아민화합물 및 그 산부가염 | |
| CN111362852A (zh) | 一种药物中间体1-叔丁氧羰基-3-氮杂环丁酮的制备方法 | |
| Chittimalla et al. | CODEN: HLEEAI | |
| JP2002053536A (ja) | スピロアミノピロリジン誘導体およびその製造法 | |
| JPH03184975A (ja) | Nf―1616―904物質の製造法 | |
| HK1025318A (zh) | 制备托诺氟沙星酸式盐的方法 | |
| JPH0616666A (ja) | 双環状アミン誘導体 | |
| JPS62201866A (ja) | インド−ル酢酸誘導体の製造方法 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C06 | Publication | ||
| PB01 | Publication | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| C19 | Lapse of patent right due to non-payment of the annual fee | ||
| CF01 | Termination of patent right due to non-payment of annual fee |