CN1170832C - 取代噻唑并[3,2-α]氮杂�衍生物 - Google Patents
取代噻唑并[3,2-α]氮杂�衍生物 Download PDFInfo
- Publication number
- CN1170832C CN1170832C CNB991236092A CN99123609A CN1170832C CN 1170832 C CN1170832 C CN 1170832C CN B991236092 A CNB991236092 A CN B991236092A CN 99123609 A CN99123609 A CN 99123609A CN 1170832 C CN1170832 C CN 1170832C
- Authority
- CN
- China
- Prior art keywords
- hydrogen atom
- methyl
- general formula
- formula
- obtains
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D513/00—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00
- C07D513/02—Heterocyclic compounds containing in the condensed system at least one hetero ring having nitrogen and sulfur atoms as the only ring hetero atoms, not provided for in groups C07D463/00, C07D477/00 or C07D499/00 - C07D507/00 in which the condensed system contains two hetero rings
- C07D513/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K38/00—Medicinal preparations containing peptides
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K5/00—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof
- C07K5/04—Peptides containing up to four amino acids in a fully defined sequence; Derivatives thereof containing only normal peptide links
- C07K5/06—Dipeptides
- C07K5/06139—Dipeptides with the first amino acid being heterocyclic
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/55—Design of synthesis routes, e.g. reducing the use of auxiliary or protecting groups
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Molecular Biology (AREA)
- Genetics & Genomics (AREA)
- Biophysics (AREA)
- Biochemistry (AREA)
- Immunology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Animal Behavior & Ethology (AREA)
- Epidemiology (AREA)
- Pharmacology & Pharmacy (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Thiazole And Isothizaole Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
由通式(II)表示的取代噻唑并〔3,2-α〕氮杂䓬衍生物及其药理学上允许的盐,式中,R<sup>3</sup>、R<sup>4</sup>以及R<sup>5</sup>可以相同,也可以不同,表示氢原子、低级烷氧基、低级烷基、低级烷硫基、可以带有取代基的芳基、可以带有取代基的杂环基;另外,R<sup>3</sup>、R<sup>4</sup>或R<sup>5</sup>中相邻的2个也可以同与它们相结合的碳原子一起形成环,但是,R<sup>3</sup>、R<sup>4</sup>以及R<sup>5</sup>均为氢原子的情况除外,R<sup>6</sup>以及R<sup>7</sup>可以相同也可以不同,表示氢原子、低级烷基;R<sup>8</sup>表示氢原子或羧基的保护基。
Description
本发明涉及新的取代噻唑并〔3,2-α〕氮杂衍生物及其药理学上容许的盐及其制造方法。更详细地是涉及作为医药有用的新的取代噻唑并〔3,2-α〕氮杂衍生物及其药理学上允许的盐及该衍生物在工业上有利的制造方法。
近年来,作为新的心脏功能障碍治疗药受到重视的有心房性钠利尿肽分解酶(Neutural Endpeptidase:NEP-24,11以下简称NEP)阻碍剂及血管紧张素I转换酶(以下简称ACE)阻碍剂。
心房性钠利尿肽(Artrial Natruiuretic Peptide:以下简称为ANP)是存在于人体内的激素,除具有较强的水、钠利尿作用及血管扩张作用等之外,还具有由交感神经抑制引起的去甲肾上腺素游离抑制作用、从肾分泌肾素的抑制作用、从肾上腺分泌醛固酮的抑制作用、及通过使静脉中的水透过性元进而引起的灌流降低作用等。例如,在伴随前负荷的上升的缺血性心脏功能障碍的患者中ANP的作用不只限于心脏功能障碍,对于高血压病的治疗也优选使用。
但是因为ANP为肽类,不可能经口给药,代谢稳定性差,目前仅限于急性期的临床使用。另外,还有有关由于长期给药而引起作用减弱的报告,在使用上要注意。
因此,根据ANP的上述特征,作为经口给药型的ANP的有关制剂受到注目的是上述的ANP分解酶阻碍剂(以下简称NEP阻碍剂)。NEP阻碍剂,对心脏功能障碍患者给药后可使血中ANP浓度升高,显示出钠利尿作用。但现有的NEP阻碍剂对心血管行动状态的作用轻微,前负荷及后负荷的减轻不明确。
一方面,作为血管扩张药之一的ACE阻碍剂,通过抑制作为心脏功能障碍的恶化因素的血管紧张素II(以下简称为AT-II)的生成,而显示出对慢性心脏功能障碍、NYHA重症程度的有效改善及运动耐容能力的提高,证明了包括延长生命效果的有效性。但现有的ACE阻碍剂对于患者的有效率不高,根据患者的不同,其效果的偏差很大。而且具有引起低血压症等副作用,如肾机能低下等,在给药方面受到限制。
如上所述,NEP阻碍剂及ACE阻碍剂作为新的心脏功能障碍阻碍药而受到注目,便现有的NEP阻碍剂及ACE阻碍剂在有用性方面受到限制。因此急待开发一种兼有NEP阻碍作用及ACE阻碍作用二者的长处的药物。
在特开平6-56790号中记载了如下所示的兼有NEP阻碍作用及ACE阻碍作用的化合物组。

(式中,R1为氢原子、R3-CO或R18-S-;R2及R19分别独立表示氢原子、烷基、环烷基-(CH2)m-、取代烷基、芳基-(CH2)m-、取代芳基-(CH2)m-、或杂环基-(CH2)m-;n为0或1,但当R2及R19均不为0氢原子时,n必须为0;m为0或1-6的整数;R3为烷基、取代烷基、环烷基-(CH2)m-、芳基-(CH2)m-、取代芳基-(CH2)m-、或杂环基-(CH2)m-,R18为烷基、取代烷基、环烷基-CH2)m-、芳基-(CH2)m-、取代芳基-(CH2)m-或杂环基-(CH2)m-。R12为氢原子、烷基、取代烷基、芳基-(CH2)m-、取代芳基-(CH2)m-、杂环基-(CH2)m-、
或
。v及w为1或2)。
但该化合物组与本发明化合物的构造不同,而且并不是同时具有能够满足先前所期待的NEP;阻碍作用及ACE阻碍作用的物质,在口服有效性方面也有问题,只限于治疗现场使用。在WO94/10193号公报及特开平6-56790号中均记载有同样的化合物。
鉴于以上情况,本发明人等对具有优良的NEP阻碍作用及ACE阻碍作用的、不论给药途径如何均具有优良效果的药物进行了深入的研究。结果发现了如下所示的化合物组,达到了所希望的目的,完成了本发明。
即,本发明涉及以通式(I)表示的取代噻唑并〔3,2-α〕氮杂衍生物及其药理学上允许的盐,

(式中,R1表示氢原子或硫醇基的保护基,R2表示氢原子、低级烷基、可以带有取代基的芳基、可以带有取代基的杂环基、低级烷氧基、低级烷硫基,R3、R4及R5相同或不同,表示氢原子、低级烷基、低级烷氧基、低级烷硫基、可以带有取代基的芳基、可以带有取代基的杂环基,另外,R3、R4或R5中相邻的2个也可与它们相结合的碳原子一起形成环。但R3、R4及R5均为氢原子的情况除外。
R6及R7相同或不同,表示氢原子、低级烷基,R8表示氢原子或羧基的保护基。
n、m独立地表示0或1、2)
上述定义中,R2、R3、R4、R5、R6及R7的定义中的低级烷基是指碳原子数1-6的直链或支链的烷基,如甲基、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基、叔丁基、正戊基、异戊基、新戊基、叔戊基、1-甲基丁基、2-甲基丁基、1,2-二甲基丙基、正己基、异己基、1-甲基戊基、2-甲基戊基、3-甲基戊基、1,1-二甲基丁基、1,2-二甲基丁基、2,2-二甲基丁基、1,3-二甲基丁基、2,3-二甲基丁基、3,3-二甲基丁基、1-乙基丁基、2-乙基丁基、1,1,2-三甲基丙基、1,2,2-三甲基丙基、1-乙基-1-甲基丙基、1-乙基-2-甲基丙基等。其中优选的是甲基、乙基、正丙基、异丙基、正丁基、异丁基、仲丁基。
在R2、R3、R4及R5的定义中的低级烷氧基是指碳原子数1-6的烷氧基,如甲氧基、乙氧基、正丙氧基等。
在R2、R3、R4及R5的定义中的低级烷硫基是指碳原子数1-6的烷硫基,如甲硫基、乙硫基、正丙硫基等。
在R2、R3、R4及R5的定义中的可以带有取代基的芳基中的芳基是指苯基、1-萘基、2-萘基、蒽基等。
在R2、R3、R4及R5的定义中的可以带有取代基的杂环基中的杂环基是指含有1-4个氮原子、硫原子或氧原子等杂原子的3-8员环优选5-6员环。
另外,在R2、R3、R4及R5的定义中的“可以带有取代基的芳基”、“可以带有取代基的杂环基”中,取代基是指甲基、乙基、正丙基、叔丁基等低级烷基、氟原子、氯原子、溴原子、碘原子等卤素原子、甲氧基、乙氧基、正丙氧基、叔丁氧基等低级烷氧基、硝基、单或二取代氨基等。这些取代基为1~3个。
在R1的定义中的硫醇基的保护基是指,如甲基、乙基、正丙基、叔丁基等低级烷基、乙酰基、丙酰基、丁酰基、三甲基乙酰基、棕榈酰基、硬脂酰基等由脂肪饱和单羧酸衍生的基团;丙烯酰基、丙炔酰基、异丁烯酰基、巴豆酰基、油酰基等由脂肪族不饱和羧酸衍生的基团;苯甲酰基、萘酰基、甲苯酰基、apotoyl基、肉桂酰基等由碳环式羧酸衍生的基团;呋喃甲酰基、噻吩甲酰基、烟酰基、异菸酰基等由碳环式羧酸衍生的基团;乙醇酰基、丙醇酰基、甘油酰基、马来酸基、酒石酰基、二苯乙醇酰基、水杨酰基、茴香酰基、香草酰基、胡椒酰基等由羟基羧酸或烷氧基羧酸衍生的基团等酰基、苯基、萘基等芳基、呋喃甲酰基、吡啶基、噻嗯基等杂环基、苄基等芳烷基、呋喃甲酰基甲基、噻嗯基甲基、吡啶基甲基等杂环芳烷基等。
在R8的定义中的羧基的保护基是指甲基、乙基、正丙基、叔丁基等低级烷基、苄基、1-萘甲基、2-萘乙基等芳烷基、2-吡啶基甲基、3-吡啶基丙基、2-噻嗯基乙基等杂环烷基等。总之,只要在体内脱离释放出羧基,则可以是任何物质。
在R3、R4及R5的定义中的“相邻的2个取代基与与其结合的碳原子一起形成环”优选5-8员环。
另外,药理学上允许的盐是指盐酸盐、硫酸盐、硝酸盐等无机盐,马来酸盐、柠檬酸盐、醋酸盐等有机酸盐,钠盐、钾盐等碱金属盐,天冬氨酸盐、谷氨酸盐等氨基酸盐等。
本发明的化合物组,同时具有优良的NEP阻碍作用及ACE阻碍作用,以下述通式(I’)表示的化合物其生物利用度好,口服效果优良,是本发明的化合物组中最优选的化合物组。
(式中,R1表示氢原子或硫醇基的保护基,R4及R5相同或不同,表示氢原子、低级烷基、低级烷氧基、低级烷硫基、带有取代基的芳基、带有取代基的杂环基。另外,R4及R5中相邻的2个取代基也可与与它们相结合的碳原子一起形成环。但R4及R5均为氢原子的情况除外。特别优选R4为氢原了、R5为低级烷基的物质。此时低级烷基优选甲基。
R6及R7相同或者不同表示氢原子或低级烷基,最优选均为氢原子的情况。R8表示氢原子或羧基的保护基。)
本发明化合物(I)中最优选的化合物以下述通式(A)表示,

(式中,R1表示氢原子或硫醇基的保护基,R5表示低级烷基、低级烷氧基、低级烷硫基、带有取代基的芳基、带有取代基的杂环基,优选低级烷基,最优选甲基。
R6及R7相同或不同,表示氢原子或低级烷基,最优选均为氢原子的情况。
R8表示氢原子或羧基的保护基,最优选氢原子。)
本发明化合物(A)中,最优选的化合物组以下述通式(A’)表示。

(式中,R1表示氢原子或硫醇基的保护基,优选氢原子或乙酰基。R5表示低级烷基、低级烷氧基、低级烷硫基、也可带有取代基的芳基、也可以带有取代基的杂环基。
R6及R7相同或不同,表示氢原子或低级烷基,最优选均为氢原子的情况。
R8表示氢原子或羧基的保护基,最优选为氢原子。)
本发明化合物(A’)中,最优选的化合物为式(A’)中R5为甲基的以下述2个通式表示的化合物。
在该2个通式中,R1、R6及R7均为氢原子的以下2个化合物为本发明最优选的化合物。
其中,R8为氢原子的化合物如下所示。
上述一组优选的化合物组为在噻唑并〔3,2-α〕氮杂骨架的6位上导入了(2S,3S)-3-甲基-2-硫代戊酰胺基的化合物,且在噻唑并〔3,2-α〕氮杂骨架的9位上具有低级烷基等取代基的化合物。在现有技术特开平6-号及EP中已经提出了具有噻唑并〔3,2-α〕氮杂骨架的化合物,但所记载的化合物在噻唑并〔3,2-α〕氮杂骨架的6位上的取代基几平均为苄基,但没有记载如本发明的具有特定立体构造的基团,即
本发明人等从完全不同的思路出发,通过在噻唑并〔3,2-α〕氮杂环的6位上导入具有特定的立体构造的(2S,3S)-3-甲基-2-硫代戊酰胺基,偶然发现了与上述现有技术中记载的化合物相比,作为NEP及ACE二者的双重抑制剂极优良的化合物,完成了本发明。
本发明为在噻唑并〔3,2-α〕氮杂环的9位上导入低级烷基等最优选为甲基的化合物。
因此,本发明的优良的化合物组(I)为具有噻唑并〔3,2-α〕氮杂环的,在6位上被具有特定的立体构造的

所取代,并且在9位上被甲基等低级烷基取代的全新的化合物,由于引入了这样的新概念,从而成功地得到了具有优良的双重抑制作用的本发明化合物。
即,本发明化合物的优良的化合物组与现有技术中记载的化合物相比,不只具有优良的双重阻碍活性,而且生物利用度高,口服效果优良。
其中,如下所示的化合物为最优选的化合物。
本发明的化合物组可以通过已知的方法或已知的方法组合制造,但存在原料化合物价格高、操作性不好等问题。因此对有利于工业制造的本发明的化合物组的制造方法进行了研究,结果发现了如下所示的制造方法。
制造方法1:

(上述式中,R3、R4、R5分别独立表示氢原子、低级烷基、低级烷氧基、低级烷硫基、可以带有取代基的芳基、可以带有取代基的杂环基,R3、R4或R5也可与与它们结合的碳原子一起形成环,但R3、R4及R5均为氢原子的情况除外。R6、R7分别独立表示氢原子、低级烷基、也可以取代的芳基或也可以取代的芳烷基,R1a表示酰基,R8a表示羧基的保护基,R12表示与环内的氮原子一起形成醛等价体的基团,Z表示酰基、氨甲酰基,几及m与通式(I)中含义相同。)
(工序1)
本工序是将哌啶酸衍生物(18)酰基化而制得N-酰基哌啶酸衍生物(19)的工序。如将化合物(18)与乙酸酐等酸酐在室温-100℃下反应,或将化合物(18)与乙酰氯、苯甲酰氯等酰基卤在吡啶、二甲胺基吡啶等碱的存在下,在室温下反应,或是将化合物(18)与酰基卤在氢氧化钠或碳酸氢钠等碱的存在下反应,即通过所说的肖-鲍氏反应制得化合物(19)。
(工序2)
本工序是将由工序1得到的N-酰基哌啶酸衍生物(19)的羧酸酯化,制得酯(20)的工序。作为酯基,优选可以以叔丁基酯、甲氧基等取代的苄基酯、烷基甲硅烷基乙酯等通常的烷基酯在脱酯化时在不水解的条件下可以脱酯化的基团。当为叔丁基酯时,可通过将化合物(19)在二噁烷、四氢呋喃等有机溶剂中,在硫酸、对甲苯磺酸等酸催化剂存在下与异丁烯反应,或将化合物(19)在N,N’-二环己基碳化二亚胺(DCC)、1-(3-二甲胺基丙基)-3-乙基碳化二亚胺(DEC)等缩合剂的存在下与叔丁醇反应而合成。当为苄基酯、甲氧基苄基酯、烷基甲硅烷基乙基酯等酯对,可以使用苄基氯、甲氧基苄基氯、烷基甲硅烷基乙基氯等酯化剂,在碳酸钾、碳酸钠、烷基胺等碱的存在下,在四氢呋喃、二甲基甲酰胺、乙氯甲烷等惰性有机溶剂中进行酯化,可以制得化合物(20)。
(工序3)
本工序是将由工序2制得的哌啶酸衍生物(20)电解氧化,制得半缩醛(21)的工序。电解氧化可以在各种条件下进行,例如可以使用铂、碳精、不锈钢、氧化铅等作为电极,作为支持电解质,可以使用四乙基铵高氯酸盐、四甲基铵高氯酸盐等四烷基铵高氯酸盐、氯化钠、氯化锂等碱金属盐、四乙基铵对甲苯磺酸盐等四烷基铵磺酸盐、四烷基铵四氟硼酸盐、四烷基铵六氟磷酸盐等,使用在水或有机溶剂中容易使电流通过的电解质,在水-乙腈、水-乙醇、水-醋酸等溶剂中将化合物(20)电解氧化,可以制得半缩醛(V)。通常通电的电流量为使用的化合物(20)的2法拉第/摩尔以上。特别是以铂或碳精作为电极,以四乙基铵高氯酸盐、四乙基铵四氟硼酸盐、四甲基铵六氟磷酸盐作为支持电解质使用时,可以得到较好的结果。
(工序4)
本工序是将由工序3得到的半缩醛(22)与半胱氨酸的酯衍生物(23)反应,制得噻唑烷(24)的工序。实际上,不必分离半缩醛(22),在第3工序结束后,在反应系中加入半胱氯酯的酸衍生物(23),处理后得到噻唑烷(24)。另外,在本反应中使用的半胱氨酸,在使用光学活性L-半胱氨酸或D-半胱氨酸的情况下,化合物(24)中的噻唑烷环的4位的羧基的绝对构型;分别为R或S。
(工序5)
本工序是将由工序4得到的噻唑烷衍生物(24)的以R9所示的羧酸的保护基有选择地脱保护,制得羧酸衍生物(25)的工序。当化合物(24)为叔丁基酯时,通过以三氟乙酸、盐酸、碘代三甲基硅烷等脱叔丁基剂处理,当为苄基酯、甲氧基苄基酯、烷基甲硅烷基乙基酯等酯类时,通过接触加氢、盐酸、2,3-二氯-5,6-二氰基-1,4-对苯醌(DDQ),四烷基氟化铵等通常的只将对应的酯保护基脱保护的方法,制得羧酸衍生物(25)。
(工序6)
本工序是将由工序5得到的噻唑烷羧酸衍生物(25)进行缩合环化,制得噻唑并氨杂衍生物(26)的工序。可利用常用的缩合剂进行环化,例如通过将1-乙氧羰基-2-乙氧基-1,2-二氢喹啉(EEDQ)、DCC、DEC等在乙醇、四氢呋喃、二氯甲烷等溶剂中,与化合物(25)反应、可制得环化体(26)。
(工序7)
本工序是将由工序6得到的噻唑并氮杂衍生物(26)的N-乙酰基脱保护,制得氨基酸衍生物(27)的工序。已知有多种N-乙酰基的脱乙酰基化,例如通过在盐酸或硫酸等稀矿酸的醇溶液中加热,以氢氧化钠、氢氧化钾等醇溶液处理,或使五氯化磷或草酰氯在吡啶中反应,然后用乙醇处理,可以得到目的产物氨基酸衍生物(27)。
(工序8)
本工序是将由工序7得到的氨基酸衍生物(27)与通式(29)所示的羧酸衍生物或酰基卤等活性衍生物进行缩合,制得酰胺衍生物(30)的工序。该缩合反应通过常法进行,例如将氨基酸衍生物(27)及羧酸衍生物(29)在EEDQ、DCC、DEC或二乙基氰基膦酸盐等,在常用的缩合试剂的存在下进行缩合反应。反应溶剂可以使用所有与反应无关的有机溶剂,如二氯甲烷、四氢呋喃等。当为羧酸衍生物(29)的酰氯等酰基卤时,将羧酸衍生物(29)在适当的惰性溶剂中,使用氯化亚硫酰、草酰氯等常用的氯化剂作为酰氯,与氨基酸衍生物(27)反应,制得化合物(30)。
(工序9)
本工序是将由工序8得到的α-酰基硫代羧酸酰胺衍生物(30)水解,制得α-巯基羧酸酰胺衍生物(31)。
利用通常的水解,即可以在氢氧化钠、氢氧化锂等稀碱水溶液或稀无机酸水溶剂中进行水解。
(工序10)
本工序是通过将由工序9得到的α-巯基羧酸酰胺衍生物(31)酰基化,制得α-酰基硫代羧酸酰胺衍生物(32)的工序。
反应可利用常法进行,例如将醋酸酐等酸酐、酰基卤等酰基化剂与α-巯基羧酸酰胺衍生物(31),在乙腈、四氢呋喃、二氯甲烷等非水溶剂中,在氯化钴等催化剂存在下进行反应,或是在水系溶剂中,在碳酸氢钾、碳酸氢钠、三乙胺等碱的存在下处理,可以制得α-酰基硫代羧酸酰胺衍生物(32)。
作为酰基化剂,使羧酸与碳化二咪唑作用得到的活性酯可以得到比较好的结果。
在本制造方法中,以通式(27)所示的化合物是制造本发明化合物组时非常重要的中间体。
制造方法2:
(上述式中,R3、R4、R5分别独立表示氢原子、低级烷基、低级烷氧基、低级烷硫基、可以带有取代基的芳基、可以带有取代基的杂环基、R3、R4或R5也可与与它们结合的碳原子一起形成环。但R3、R4及R5均为氢原子的情况除外。R6、R7分别独立表示氢原子、低级烷基、可以带有取代基的芳基或可以带有取代基的芳烷基。R1a表示酰基,R8a表示羧基的保护基,X表示卤原子、甲磺酰氧基、对甲苯磺酰氧基等脱离基。m及n与通式(I)中含义相同。)
(工序1)
本工序是将由制法A得到的氨基酸衍生物(27)及以通式(33)表示的羧酸衍生物或其酰其卤等活性衍生物进行缩合,制得酰胺衍生物(34)的工序。该缩合反应可通过与制法A的工序8相同的操作,使用α-羟基羧酸衍生物(33)代替羧酸衍生物(29)。
(工序2)
本工序是将由工序1得到的羟基羧酸酰胺衍生物(34)进行卤化,制得α-卤代羧酸酰胺衍生物(35)的工序。作为伴随化合物(34)的羧基的立体反转进行卤化的方法,例如有(i)在四氢呋喃等有机溶剂中,与偶氮二羧酸二烷基酯、三苯基膦及溴化锌或碘化锌反应的方法、(ii)在乙腈、二甲基甲酰胺、二氯甲烷等有机溶剂中,在吡啶等碱的存在或非存在条件下,使三烷基膦、三苯基膦、亚磷酸三苯基等有机膦化合物与N-氯代琥珀酰亚胺、溴碘等卤素化合物反应的方法、(iii)在二氯甲烷等惰性溶剂中,在吡啶、三乙胺等碱的存在下,使与对甲苯碘酰氯或三氟甲苯磺酸酐等反应,生成磺酸酯后,与氯化锂等卤化试剂反应的方法等多种方法,在(ii)的条件下,特别优选使用三苯基膦、溴的方法。
(工序3)
在工序是将由工序2得到的α-卤代羧酸酰胺衍生物(35)酰基硫化,制得α-酰基硫代羧酸酰胺衍生物(36)的工序。
反应可通过常法进行,例如将α-卤代羧酸酰胺衍生物(35)在乙腈、丙酮等极性溶剂中与硫代醋酸钾、硫代醋酸钠等硫代羧酸盐反应,或在碳酸钾、碳酸铯等碱存在下与硫代醋酸、硫代苯甲酸等硫代羧酸反应,可以得到α-酰基硫代羧酸酰胺衍生物(36)。
(工序4)
本工序是将由工序3得到的α-酰基硫代羧酸酰胺衍生物(36)进行水解,制得α-巯基羧酸酰胺衍生物(37)的工序。可利用通常的水解,即在氢氧化钠、氢氧化锂等稀碱水溶液或稀无机酸水溶液中进行水解。
(工序5)
本工序是将由工序4得到的α-巯基羧酸酰胺衍生物(37)酰基化,制得α酰基硫代羧酸酰胺衍生物(38)的工序。
反应可利用常法进行,例如将乙酸酐等酸酐、酰基卤等酰基化剂与α-巯基羧酸酰胺衍生物(37),在乙腈、四氢呋喃、二氯甲烷等非水溶剂中,在钴等催化剂的存在下进行反应,或是在水系溶剂中,在碳酸氢钾、碳酸氢钠、三乙胺等碱的存在下进行反应,制得α-酰基硫代羧酸酰胺衍生物(38)。
作为酰化剂,使羧酸与碳化二咪唑反应得到的活性酯可以得到特别优良的结果。
制造方法3

(上述式中,R3、R4、R5分别独立表示氢原子、代级烷基、低级烷氧基、低级烷硫基、可以带有取代基的芳基、可以带有取代基的杂环基,另外,R3、R4或R5也可与与它们结合的碳原子一起形成环。但R3、R4及R5均为氢原子的情况除外。R6、R7分别独立表示氢原子、低级烷基、可以被取代的芳基或也可以被取代的芳烷基。R1a表示酰基。R8a表示羧基的保护基。X为卤素原子、甲磺酰氧基、对甲苯磺酰氧基等脱离基,m及n与通式(I)中含义相同)。
(工序1)
本工序是将由制造方法2得到的卤化物(35)的酯基加水分解制得羧酸衍生物(39)的工序。利用通常的加水分解,即可在氢氧化钠、氢氧化锂等稀碱水溶液或稀无机酸水溶液中进行水解。
(工序2)
本工序是将由工序1得到的α-卤代羧酸酰胺衍生物(39)进行酰基硫化,制得α-酰基硫代羧酸酰胺衍生物(38)的工序。反应可依常法进行,例如将α卤代羧酸酰胺衍生物(38)在乙腈、二甲亚砜、丙酮等极性溶剂中,与硫代醋酸钾、硫代醋酸钠等硫代羧酸盐反应,或在碳酸钾、、碳酸铯等碱存在下与硫代苯甲酸、硫代醋酸等硫代羧酸反应,制得α-酰基硫代羧酸酰胺衍生物(38)。
制造方法4:
(上述式中,R2、m与上述含义相同)
本工序是将天然型氨基酸或非天然型氨基酸(40)的氨基羟基化,制得α-羟基羧酸(33)的工序。羟基化是将氨基酸(40)在稀硫酸中与亚硝酸钠等亚硝酸剂反应,或在醋酸中与亚硝酸钠反应成为醋酸酯后,再进行水解。
本发明的化合物中,对于优选的化合物组(I’),当R4及R5均为氢原子时,已知有将化合物(II)及化合物(一)、(二)或(三)

(二)(Y表示卤原子等脱离基)

进行酰胺化反应的方法。但,上述化合物(イ)、(ロ)及(ハ)均价格昂贵,大量合成时要以D-别异白氨酸作为起始原料,而D-别异白氨酸很难得到,因此在工业上很难说有利。以下所示方法可以高收率、操作性良好地制造化合物(I’),于工业生产有利。
制造方法5:
(工序1)
本工序是将L-异亮氨酸(9)的氨基通过常法进行羟基化,制得α-羟基羧酸(10)的工序。羟基化可利用常法进行,但优选将L-异亮氨酸(9)在稀硫酸中与亚硝酸钠等亚硝酸剂反应,或在醋酸中与亚硝酸钠反应成为醋酸酯后,再进行水解。
(工序2)
本工序是将由工序1得到的α-羟基羧酸(10)与胺衍生物(11)通过常法缩合,制得羟基羧酸酰胺衍生物(12)的工序。
反应可通过常法进行,例如将α-羟基羧酸(10)及胺衍生物(11)在EEDQ、DCC、DEC或二乙基氰基膦酸盐等常用的缩合试剂的存在下,在二氯甲烷或四氢呋喃等惰性溶剂中反应,制得酰胺衍生物(12)。
(工序3)
本工序是利用常法将羟基羧酸酰胺衍生物(12)卤化,制得α-卤代羧酸酰胺衍生物(13)的工序。
如果是利用立体反转卤化的方法,可使用常用的任何方法。例如有(i)在四氢呋喃等有机溶剂中,与偶氮二羧酸二烷基酯、三苯基膦及溴化锌或碘化锌反应的方法,(ii)在乙腈、二甲基甲酰胺、二氯甲烷等有机溶剂中,在吡啶等碱的存在或不存在下,与三烷基膦、三苯基膦、亚磷酸三苯基等有机膦化合物及N-卤代琥珀酰亚胺、溴碘等卤素化合物反应的方法,(iii)在二氯甲烷等惰性溶剂中,在吡啶、三乙胺等碱的存在下,与对甲苯磺酰氯或三氟甲磺酸酐等反应成为磺酸酯后,与卤化锂等卤化试剂反应的方法等,特别优选的是,在乙腈、二甲基甲酰胺、二氯甲烷等有机溶剂中,在吡啶等碱的存在或不存在下,与三烷基膦、三苯基膦、亚磷酸三苯基等有机膦化合物及N-卤代琥珀酰亚胺、溴、碘等卤素化合物反应的方法,此处作为试剂优选使用三苯基膦及溴的方法。
(工序4)
本工序是将由工序3得到的α-卤代羧酸酰胺衍生物(13)进行酰基硫化,制得α-酰基硫代羧酸酰胺衍生物(8a)的工序。
反应可利用常法进行,例如将α-卤代羧酸酰胺衍生物(13)在乙腈、丙酮等极性溶剂中,与硫代醋酸钾、硫代醋酸钠等硫代羧酸盐反应,或在碳酸钾、碳酸铯等碱存在下与硫代醋酸、硫代苯甲酸等硫代羧酸反应,制得α-酰基硫代羧酸酰胺衍生物(8a)。
(工序5)
本工序是目的化合物的R1及R8为氢原子时或R1为酰基、R8为氢原子时进行的工序。即,将由工序4得到的α-酰基硫代羧酸酰胺衍生物(8a)利用常法水解,制得(2S,3S)-3-甲基-2-硫代戊酸衍生物(8b)的工序。
通常的水解即在氢氧化钠、氢氧化锂等稀碱水溶液或稀无机酸水溶液中进行水解。当目的化合物的R1为酰基的情况下,将得到的(2S,3S)-3-甲基-2-硫代戊酸衍生物(8b)用于以下工序6。
(工序6)
本工序是目的化合物的R1为酰基时进行的工序。即将由工序5得到的(2S,3S)-3-甲基-2-硫代戊酸衍生物(8b)利用常法进行酰基化,制得α-酰基硫代羧酸酰胺衍生物(8c)的工序。
反应可利用常法进行,例如将醋酸酐等酸酐、酰基卤等酰基化剂及α-巯基羧酸酰胺衍生物(8b),在乙腈、四氢呋喃、二氯甲烷等非水溶剂中进行反应,或者在水系溶剂中,在碳酸氢钾、碳酸氢钠、三乙胺等碱或氯化钴的存在下进行反应,制得α-酰基硫代羧酸酰胺衍生物(8c)。
在制造方法5中,将L-异亮氨酸羟基化,将胺衍生物(11)缩合,制得α-卤代羟基羧酸酰胺衍生物(13)之后,按以下方法得到目的化合物。
制造方法6:


(工序1)
本工序是将由制造方法5的工序3得到的α-卤代羧酸酰胺衍生物(13)用常法水解,制得羧酸(14)的工序。
利用通常的水解,即在氢氧化钠、氢氧化锂等稀碱水溶液或稀无机酸水溶液中进行水解。
(工序2)
本工序是将由工序1得到的α-卤代羧酸酰胺衍生物(14)进行酰基硫化制得α-酰基硫代羧酸酰胺衍生物(8c)的工序。反应可利用常法进行,例如将α-卤代羧酸酰胺衍生物(14)在乙腈、二甲亚砜、丙酮等极性溶剂中,与硫代醋酸钾、硫代醋酸钠等硫代羧酸盐反应,或在碳酸钾、碳酸铯等碱的存在下,与硫代醋酸、硫代苯甲酸等硫代羧酸反应,制得α-酰基硫代羧酸酰胺衍生物(8c)。
以通式(II)表示的胺,其中R3、R4及R5均为氢原子的胺(II)在US4415496及US4617301中有记载。制得该胺(II)的方法,现在已知的例如有US4415496号中记载的以(S)-2-氨基-6-羟基己酸作为起始原料的方法及在US4617301号及US5118810号中记载的以ε-N-BOC-L-赖氨酸作为起始原料的方法,每个起始原料都很难得到,且步骤多,需要大量的拉萘镍及离子交换树脂,操作性差。采用以下所示的制造方法,对于目前在工业制造的操作性及工业方面受到显著制约的胺(II)、及现有技术中记载的制造有困难的R3、R4、R5中的任一个或二个以上为氢原子以外的胺,可操作性良好且低成本高收率地制得。
制造方法A
(上述式中,R3、R4、R5分别独立表示氢原子、低级烷基、低级烷氧基、低级烷硫基、可以带有取代基的芳基、可以带有取代基的杂环基。另外,R3、R4、及R5也可与与它们结合的碳原子一起形成环。但R3、R4、R5均为氢原子的情况除外。R6、R7分别独立表示氢原子、低级烷基、可以被取代的芳基或可以被取代的芳烷基。R2a表示羧基的保护基。R12表示与环内的氮原子一起形成醛基等价体的基团。Z表示酰基、氨基甲酸酯基。)
(工序1)
本工序是将哌啶酸衍生物的2S光学活性体(15)酰基化,制得N-酰基哌啶酸衍生物(16)的工序。可利用常用的酰基化方法制得化合物(16),例如将化合物(15)与醋酸酐等酸酐在室温-100℃下反应,或将化合物(15)与乙酰氯、苯甲酰氯等酰基卤在吡啶、二甲基氨基吡啶等碱的存在下,在0℃-室温下反应,或将化合物(15)与酸酐或酰基卤在碳酸钠或碳酸氢钠等碱存在下反应,即通过肖-鲍氏反应制得化合物(16)。
(工序2)
本工序是将由工序1得到的N-酰基哌啶酸衍生物(16)的羧酸酯化,制得酯(2’)的工序。作为酯基例如有叔丁基酯、甲氧基等可以被取代的苄基酯、烷基甲硅烷基乙酯等,通常的烷基酯优选地进行脱酯化时可以在不水解的条件下进行脱酯化的基团。在为叔丁基酯的情况下,将化合物(16)在二噁烷、四氢呋喃等醚类溶剂或二氯甲烷等有机溶剂中,在硫酸、对甲苯磺酸等酸催化剂的存在下与异丁烯反应,另外,可将化合物(16)在二环己基偶氮二羧酸酯(DCC)、1-(3-二甲胺基丙基)-3-乙基碳化二亚胺(DEC)等缩合剂的存在下与叔丁醇反应而合成。另外,当为苄基酯、甲氧基苄基酯、烷基甲硅烷基乙酯等酯类时,使用苄基卤化物,甲氧苄基卤化物,烷基甲硅烷基乙基卤化物等酯化剂,在碳酯钾、碳酸钠、烷基胺等碱的存在下,在四氢呋喃、二甲基甲酰胺、二氯甲烷等惰性溶剂中进行酯化,制得化合物(2’)。
(工序3)
本工序是将由工序2得到的哌啶酸衍生物(2’)进行电解氧化,制得半缩醛(3’)的工序。
电解氧化可在各种条件下进行,例如使用铂、碳精、不锈钢、氧化铅等作为电极,使用在水系或有机溶剂系中电流容易流动的电解质,如四乙基铵高氯酸盐、四甲基铵高氯酸盐等四烷基铵高氯酸盐、氯化钠、氯化锂等碱金属盐、四乙基铵对甲苯磺酸盐等四烷基铵磺酸盐、四烷基铵四氟硼酸盐、四烷基铵六氟磷酸盐等,在水-乙腈、水-乙醇、水-醋酸等溶剂中将化合物(2’)电解氧化,制得半缩醛(3’)。通电的电流量 一般为相对于使用的化合物(2’)为3法拉第/摩尔以上。特别是,当使用铂或碳精作为电极、使用四乙基铵高氯酸盐、四乙基铵四氟硼酯盐、四甲基铵六氟磷酸盐或四乙基铵对甲苯磺酸盐作为支持电解质时可以得到理想的结果。
(工序4)
本工序是将由工序3得到的半缩醛(3)与L-半胱氨酸的酯衍生物(4)反应,制得噻唑烷(5)的工序。
(工序5)
本工序是将由工序4得到的噻唑烷衍生物(5’)的以R11所示的羧酸的保护基有选择地脱保护,制得羧酸衍生物(6’)的工序。在化合物(5’)为叔丁酯的情况下,以三氟乙酸、盐酸、碘代三甲基硅烷等脱叔丁基剂处理,当为苄基酯、甲氧基苄基卤化物、烷基甲硅烷乙基酯等酯的情况下,利用接触加氢、盐酸、2,3-二氯-5,6-二氰基-1,4-对苯醌(DDQ)、四烷基铵氟化物等通常的能够只将对应的酯保护基脱保护的方法可以制得羧酸衍生物(6’)。
(工序6)
本工序是将由工序5得到的噻唑烷羧酸衍生物(6’)缩合环化,制得氨基酸衍生物(7’)的工序。可利用常用的缩合剂环化,例如将2-乙氧基-1-乙氧基-1,2-二氢喹啉(EEOQ)、DCC、EDC等在乙醇、四氢呋喃、二氯甲烷等溶剂中与化合物(6’)反应,制得环化氨基酸衍生物(7’)。
(工序7)
本工序是将由工序6得到的氨基酸衍生物(7’)的N-乙酰基脱保护,制得氨基酸衍生物(1’)的工序。已知有各种N-乙酰基的脱乙酰基化的方法,例如可以通过在盐酸或硫酸等稀无机酸的醇溶液中加热,以氢氧化钠、氢氧化钾等醇溶液处理,或是将五氯化磷或草酰氯在吡啶中的反应然后用乙醇处理,可以制得作为目的产物的氨基酸衍生物。
制造方法B
从制造方法A的工序1到工序2的工序,可以按以下方法进行。

(上述式中,R3、R4、R5分别独立表示氢原子、低级烷基、低级烷氧基、低级烷硫基、可以带有取代基的芳基、可以带有取代基的杂环基。另外,R3、R4及R5也可与与它们结合的碳原子一起形成环。但R3、R4及R5均为氢原子的情况除外。R11表示羧基的保护基,Z表示酰基或氨基甲酸酯基。)
(工序1)
本工序是将哌淀酸衍生物的2S光学活性体(15)进行叔丁基酯化,制得(17)的工序。与制造方法A的工序2所示方法相同,即将化合物(2)在二噁烷、四氢呋喃等有机溶剂中,在硫酸、对甲苯磺酸等酸催化剂存在下与异丁烯反应,或将化合物(2)在二环己基偶氮二羧酸酯(DCC)、1-(3-二甲胺基丙基)-3-乙基碳化二亚胺(DEC)等缩合剂存在下,与叔丁醇反应,可以制得酯(15)。
(工序2)
本工序是将由工序1得到的酯(17)的氮原子酰基化,制得酰基哌啶酸衍生物(2)的工序。可以利用与制造方法A的工序1所示相同的方法制得化合物(2)。即,将化合物(17)与醋酸酐等酸酐在室温-100℃条件下反应,或将化合物(15)与乙酰氯、苯甲酰氯等酰基卤在吡啶、二甲胺基吡啶等碱的存在下在0℃-室温条件下反应,另外,也可将化合物(15)与酰基卤在氢氧化钠或碳酸氢钠等碱的存在下反应,即通过肖-鲍氏反应制得化合物(2)。
制造方法C
R5为支链烷基时可以利用以下方法制得。

(工序1)
本工序是将由常法得到的哌啶酸衍生物(18’)电解氧化,制得半缩醛(19’)的工序。电解氧化可以在各种条件下进行,例如使用铂、碳精、不锈钢、氧化铅等作为电极,使用四乙基铵高氯酸盐、四甲基铵高氯酸盐等四烷基铵高氯酸盐、四乙基铵对甲苯磺酸盐等四烷基铵六氟磷酸盐等作为支持电解质,在水-乙醇、水-乙酸等溶剂中,将化合物(18’)电解氧化制得半缩醛(19’)。通电的电流量一般为相对于使用的化合物(18’)的2法拉第/摩尔以上。特别是在使用铂或碳作为电极,以四乙基铵四氟硼酸盐、四甲基铵六氟磷酸盐作为支持电解质的情况下可以得到理想的结果。
(工序2)
本工序是将由工序1得到的半缩醛(19’)进行1,2-脱离反应,制得亚胺(40)的工序。可通过酸催化剂、热反应等通常的脱离反应制得化合物(40)。
(工序3)
本工序是将由工序2得到的亚胺(40)酰基化,制得酮(41)的工序。通常可以利用向亚胺基的亲电子取代反应导入各种酰基。例如有在二氯甲烷、氯仿、二甲基甲酰胺等惰性溶剂中,通过使用氯氧化磷、亚硫酰氯等的Volsmeier法或Gattermann-koc法进行甲酰基化的反应,使用氯化铝、四氯化钛等进行的弗里德尔-克拉夫茨反应等,制得酮(41)。
(工序4)
本工序是将由工序3得到的酮(41)的羰基还原,制得甲撑体(42)的工序。可利用常法进行,例如通过接触加氢、使用肼的Wolff-Kishner还原、使用三氯硅烷或三乙基硅烷等氢化硅烷类的还原方法等,可以制得甲撑体(42)。
制造方法D
当R5为支链烷基时可利用以下方法制得。
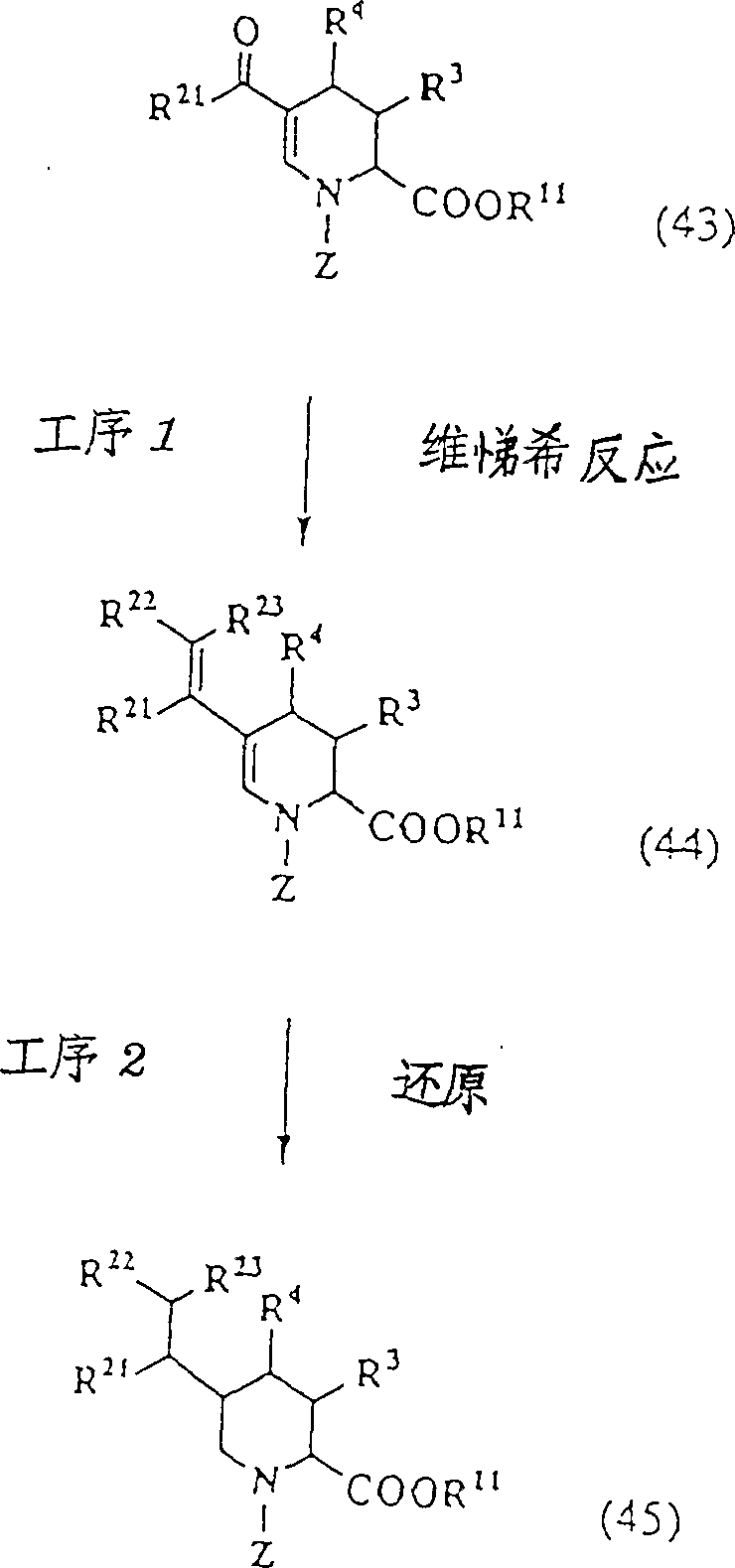
(工序1)
本工序是将由制造方法C的工序3制得的酰基体(43)的羰基烯烃化,制得烯烃(44)的工序。例如使用亚烷基·正膦与胺基钠或正丁基锂等强碱的维悌希反应,使用膦酸酯的Horner法等羰基烯烃化反应,可以制得烯烃(44)。
(工序2)
本工序是将由工序1得到的烯烃(44)的双键还原,制得饱和物(45)的工序。利用接触加氢等常用的双键还原反应,可以制得饱和物(45)。
如上所述,本发明化合物组利 于工业上制造,可以说是优良的化合物组。
下面用药理实验详细说明本发明化合物的有效性。
药理实验例1
测定使用大鼠的肾皮质的药物的NEP阻碍活性
1.实验方法
使用由大鼠的肾皮质按照Booth and Kenny方法(A RapidMethod for the purification of Microvilli from Rabbit Kidney.,Andraw G.Booth and A.John Kenny,Biochem j.,1974,142,575-581)调制的膜部分(fraction),测定NEP活性。
NEP的活性可按照Orlowsky and Wilk的方法(Purificationand specificity of a Membrane-Bound Metalloendpeptidasefrom Bovine Pituitaries.,Marian Orlowsky and Shrwin Wilk,Biochemistry,1981,20,4942-4950.),利用以下方法测定。
使用苯甲酰-甘氨酰-精氨酰-2-萘基酰胺(苯甲酰-Gly-Arg-Arg-2-萘基酰胺(Nova Biochem,Switzerland)),在NEP酶标准品及过剩的白氨酸氨基肽酶(leucine aminopeptidase(sig-ma chemical co.,U.S.A.))存在下,将游离的萘胺(Naphthylamine)用first garnet(Sigma chemical Co.,U.S.A.))使发色,测定540nm波长的吸光度。
NEP的阻碍活性是在上述实验系统中,添加被检验化合物使最终浓度分别为1、3、10、30、100、300、及1000nM,求出阻碍曲线,将显示出50%阻碍活性的浓度作为IC50。另外,使用〔4S-〔4α,7α(R*),12bβ〕〕-7-〔(1-氧代-2(S)-硫代-3-苯基丙基)氨基〕-1,2,3,4,6,7,8,(2b-八氢-6-羟基吡啶并〔2,1-α〕〔2〕苯并氮杂-4-羧酸(特开平6-56790号中记载的化合物)作为对照化合物。
2.实验结果
上述实验结果如下表1所示。
药理实验例2
测定使用大鼠肺的药物的ACE阻碍活性
1.实验方法
使用由大鼠的肺按照Wu-Wong等的方法(Characterizationof Endthalin Converting Enzyme in Rat Lung.,Junshyum R.Wu-Wong,G erald P.Budzik,Edward M.De Vine and TerryJ.Opgenorth,Biochem.Biophys.Res.Commun.,1990,171,1291-1296.)调制的膜成分,测定ACE阻碍活性。
ACE活性利用Cushman and Cheung(spectro photometric,Assay and Properties of the Angiotensin-Converting Enzyme ofRabbit Lung.,Cushman D.W.and Cheung H.S.,1971,20,1637-1648.)的改进方法(硼酸盐缓冲液(borate buffer)pH8.3)进行测定。
在ACE存在下,将从Hippuryl-His-Leu(peptidelnstituteInc.,Japan)游离的Hippurate用乙酸乙酯萃取后,测定在228nm波长处的吸光度。
ACE的阻碍活性是在上述实验系统中,添加被检验化合物使最终浓度分别为1、3,10、30、100、300、及1000nM,求出阻碍曲线,将显示50%阻碍活性的浓度作为IC50。另外,使用〔4S-〔4α,7α(R*),12bβ〕〕-7-〔(1-氧代-2(S)-硫代-3-苯基丙基)氨基〕-1,2,3,4,6,7,8,12b-八氢-6-羟基吡啶并〔2,1-α〕〔2〕苯并氮杂-4-羧酸(特开平6-56790号中记载的化合物)作为对照化合物。
2.实验结果
按上述实验方法进行的实验结果如下表1所示。
实施例化合物及比较化合物的NEP阻碍活性及ACE阻碍活性
| NEP阻碍活性IC50(nM) | ACE阻碍活性IC50(nM) | |
| 实施例3 | 4.3 | 2.5 |
| 实施例11 | 6.7 | 2.2 |
| 实施例24 | 1.5 | 2.5 |
| 实施例9 | 13 | 5.1 |
| 实施例10 | 12 | 4.3 |
| 比较化合物*1 | 27 | 9 |
*1为比较化合物:〔4S-〔4α,7α(R*),12bβ〕)-7-〔(1-氧代-2(S)-硫代-3-苯基丙基)氨基〕-1,2,3,4,6,7,8,12b-八氢-6-羟基吡啶并〔2,1-α〕〔2〕苯并氮杂-4-羧酸(化合物名MDL-100,173)
实施例
以下通过实施例详细说明本发明,但本发明并不只限定于这些实施例。首先说明作为本发明化合物的原料化合物的制造合成例。
合成例1
5-甲苯吡啶-2-羧酸乙酯

在5-甲基吡啶-2-腈55.5g中加入乙醇200ml、浓硫酸100ml(1.88mol)形成均匀溶液后加热回流2天。将反应液在冰冷下缓慢注入饱和碳酸氢钠水溶液中,将硫酸中和后,用二氯甲烷萃取,有机层用饱和食盐水洗净,用无水硫酸钠干燥。过滤后将滤液减压浓缩,得到78.1g茶褐色油状标题化合物的粗生成物。
1H-NMR(400MHz,CDCl3)δ;8.57(1H,m),8.03(1H,dt,J=8.0,0.5Hz),
7.63(1H,ddd,J=1.0,2.5,8.0Hz),4.47(2H,q,J=7.0Hz),
2.42(3H,s),1.44(3H,t,J=7.0Hz).
合成例2
2-羧基-5-甲基氯化吡啶

将由合成例1得到的5-甲基吡啶-2-羧酸乙酯的粗生成物78.1g溶于200ml 6N盐酸中,加热回流16小时。将反应溶液减压浓缩后,在残渣中加入乙腈、滤取析出的白色结晶,用乙腈洗,在90℃下干燥,得到26.3g标题化合物。收率37%。
1H-NMR(400MHz,CDCl3)δ;8.51(1H,m),8.37(1H,m),8.21(1H, d,J=8.0Hz),2.42(3H,s).
合成例3(2S*,5S*)-2-羧基-5-甲基氯化哌啶及(2S*,5R*)-2-羧基-5-甲基氯化哌啶
将由合成例2得到的2-羧基-5-甲基氯化吡啶26.3g(151mmol)溶于乙醇-水(1∶1)300ml中,加入氧化铂2g,在50℃、16个大气压下加氢反应一夜。过滤除去催化剂后,将滤液减压浓缩,将得到的白色结晶在90℃下干燥,得到27.0g标题化合物的混合物(非对映立体异构体之比3∶1)。收率99%。
1H-NMR(400MHz,D2O)δ;4.06(3/4H,t,J=5.0Hz),3.71(1/4H,m),3.24(1/4H,ddd,J=1.5,4.0,13.0Hz),3.10(3/4H,dd,J=4.5,13.0Hz),2.82(3/4H,dd,J=10.0,13.0Hz)2.53(1/4H,t,J=13.0Hz),2.22-2.04(1H,m),1.90-1.52(2H,m),1.22-1.04(1H,m),0.82(3×3/4H,d,J=7.0Hz),0.81(3×1/4H,d,J=7.0Hz).
合成例4
(2S*,5S*)-N-乙酰基-5-甲基哌啶-2-羧酸

将由合成例3得到的(2S*,5S*)-2-羧基-5-甲基氯化哌啶及(2S*,5S*)-2-羧基-5-甲基氯化哌啶混合物27.0g(150mmol)悬浮于700ml二氯甲烷中,加入21ml(150mmol)三乙胺,在室温下搅拌2小时。滤取白色结晶,用二氯甲烷洗,在50℃下干燥,得到(2S*,5S*)-5-甲基哌啶-2-羧酸15.9g。收率74%。
将上述的(2S*,5S*)-5-甲基哌啶-2-羧酸15.9g(111mmol)溶于220ml二氯甲烷-水(1∶1),在室温下依次加入碳酸氢钠93.3g(1.11mol)、醋酸酐21.0ml(222mmol),搅拌3天。将反应溶液在冰冷下注入6N盐酸中,用氯仿萃取后,将有机层用无水硫酸钠干燥。过滤后将滤液减压浓缩,得到无色油状的标题化合物20.1g。收率98%。
1H-NMR(400MHz,CDCl3)δ;10.17(1H,br),5.41(1H,d,J=5.5Hz),4.54-4.44(2×1/4H,m),3.62(1H,dd,J=4.5,13.5Hz),2.90(1H,dd,J=12.0,13.5Hz),2.39-2.26(2×3/4H,m),2.17(3×3/4H,s),2.13(3×1/4H,s),1.96-1.52(2H,m),1.15-1.03(1H,m),0.92(3×3/4H,d,J=6.5Hz),0.90(3×1/4H,d,J=7.0Hz).
(2S*,5S*)-N-乙酰基-5-甲基哌啶-2-羧酸叔丁酯
将由合成例4得到的(2S*,5S*)-N-乙酰基-5-甲基哌啶-2-羧酸16.3g(88mmol)溶于180ml二氯甲烷中,加入浓硫酸6.1ml(0.11mol),然后在反应系统内充填异丁烯气体,在室温下搅拌4天。将反应液在冰冷下注入饱和碳酸钠水溶液中,用氯仿萃取。将有机层用饱和碳酸氢钠水溶液、饱和食盐水洗净后,用无水硫酸钠干燥。过滤后将滤液减压浓缩,得到16.4g无色油状标题化合物。收率77%。1H-NMR(400MHz,CDCl3)δ;5.26(1H,dd, J=1.0,6.0Hz),
4.50-4.32(3/4H,m),3.59(1H,dd,J=4.5,13.0Hz),
2.90(1H,dd,J=12.0,13.0Hz),2.30-2.17(5/4H,m),
2.13(3×3/4H,s),2.07(3×1/4H,s),1.73-1.56(2H,m),
1.47(9×1/4H,s),1.46(9×3/4H,s),1.05-0.94(1H,m),
0.91(3×3/4H,d,J=6.5Hz),0.90(3×1/4H,d,J=7.0Hz)
合成例6
(2RS,4R)-2-〔(1S,4S)-4-乙酰胺基-4-(叔丁氧羰基)-1-甲基丁基〕噻唑烷-4-羧酸甲酯及(2RS,4R)-2-〔(1R,4R)-4-乙酰胺基-4-(叔丁氧羰基)-1-甲基丁基〕噻唑烷-4-羧酸甲酯
将由合成例5得到的(2S*,5S*)-N-乙酰基-5-甲基哌啶-2-羧酸叔丁酯9.41g(39mmol)溶于150ml甲醇中,加入四乙基铵对甲苯磺酸盐(Et4NOTs,1.5g,1w/v%),使用碳精电极,在室温下通过一定电流(480mA)11.4F/mol,电流密度为60mA/cm2。将反应溶液减压浓缩后,将残渣溶于乙酸乙酯,用水及饱和食盐水洗,将有机层用无水硫酸钠干燥。过滤后将滤液减压浓缩,得到11.5g(2S*,5S*)-N-乙酰基-6-甲氧基-5-甲基哌啶-2-羧酸酯的粗生成物。
将上述的(2S*,5S*)-N-乙酰基-6-甲氧基-5-甲基哌啶-2-羧酸酯11.5g溶于醋酸-水(1∶1)100ml中,加入N-甲基吗啉6.0ml(55mmol)、L-半胱氨酸甲酯·盐酸盐8.7g(51m-mol),在氮气环境下室温下搅拌3天。将反应液浓缩除去醋酸,用二氯甲烷萃取,将有机层用水、饱和食盐水洗,用无水硫酸镁干燥。过滤后将滤液减压浓缩,得到的残渣用硅胶柱色谱法(二氯甲烷∶乙醇=98∶2)精制,得到淡黄色油状标题化合物9.21g(非对称立体异构体之比为1∶1∶1∶1)。收率63%。
1H-NMR(400MHz,CDCl3)δ;6.18-6.04(1H,m),4.56-4.36(2H,m),4.14-4.04(2×1/4H,m),3.82-3.68(2×1/4H,m),3.79(3×2/4H,s),3.77(3×2/4H,s),3.30-3.25(2×1/4H,m),3.20-3.16(2×1/4H,m),3.04-2.97(2×1/4H,m),2.80-2.70(2×1/4H,m),2.03(3×2/4H,s),2.02(3×1/4H,s),2.01(3×1/4H,s),2.00-1.50(5H,m),1.482(9×1/4H,s),1.478(9×1/4H,s),1.473(9×1/4H,s),1.470(9×1/4H,s),1.10(3×1/4H,d,J=7.0Hz),1.04(3×1/4H,d,J=7.0Hz),1.03(3×1/4H,d,J=7.0Hz),0.97(3×1/4H,d,J=7.0Hz).
[0001]
合成例7
〔3R-(3α,6α,9β,9αβ)〕-6-乙酰胺基-9-甲基-5-氧代-八氢噻唑并〔3,2-α〕氮杂-3-羧酸甲酯及〔3R-(3α,6α,9α,9αβ)〕-6-乙酰胺基-9-甲基-5-氧代-八氢噻唑并〔3,2-α〕氮杂-3-羧酸甲酯

在由合成例6得到的(2RS,4R)-2-〔(1S,4S)-4-乙酰胺基-4-(叔丁氧羰基)-1-甲基丁基〕噻唑烷-4-羧酸甲酯及(2RS,4R)-2-〔(1R,4R)-4-乙酰胺基-4-(叔丁氧羰基)-1-甲基丁基〕噻唑烷-4-羧酸甲酯混合物8.30g(22.2mmol)中在冰冷下加入三氟醋酸50ml,缓慢升温至室温。搅拌6小时后,蒸馏除去溶剂用甲苯共沸,得到(2R,4R)-2-〔(1S,4RS)-4-乙酰胺基-4-羧基-1-甲基丁基〕噻唑烷-4-羧酸甲酯三氟醋酸盐及(2R,4R)-2-〔(1R,4RS)-4-乙酰胺基-4-羧基-1-甲基丁基〕噻唑烷-4-羧酸甲酯三氟醋酸盐混合物9.84g(异构体比1.4∶1.4∶1∶1),为粗生成物。将该9.84g粗生成物溶于150ml四氢呋喃中,加入N-甲基吗啉9.8ml(89mmol)使pH为7。室温下加入2-乙氧基-1-乙氧羰基-1,2-二氢喹啉(EEDQ,6.59g,27mmol),氮气环境下室温下搅拌一夜。将反应液减压浓缩后在残渣中加入2N盐酸100ml,使pH为1以下,用二氯甲烷萃取,有机层用饱和碳酸氢钠水溶液、饱和食盐水洗,用无水硫酸镁干燥。过滤后将滤液减压浓缩,得到的残渣用硅胶柱色谱法(乙氯甲烷∶乙醇=98∶2)精制,重结晶(乙酸乙酯-己烷)得到标题化合物混合物(异构体之比为2∶1)2∶59g,为白色结晶。收率为39%。
1H-NMR(400MHz,CDCl3)d;6.83-6.74(1H,m),5.33(2/3H,dd,J=3.2,6.4Hz),5.23(1/3H,s),4.96(1/3H,t,J=6.8Hz),4.82(2/3H,d,J=9.5Hz),4.60-4.58(1H,m),3.79(3H,s),3.32(1/3H,dd,6.4,11.6Hz),3.22(2/3H,dd,J=3.2,11.6Hz),3.14(1/3H,dd,J=6.8,11.6Hz),3.10(2/3H,dd,J=6.8,11.6Hz),2.01(3×2/3H,s),2.00(3×1/3H,s),2.10-1.89(3H,m),1.80-1.66(2H,m),1.12(3×1/3H,d,J=7.2Hz),1.00(3×2/3H,d,J=6.8Hz)
合成例8
2-乙酰基-十氢(4aR,8aR)-异喹啉-3(S)-羧酸
将由4aS,8aS异构体及4aR,8aR异构体以及反式异构体组成的十氢异喹啉-3(S)羧酸的混合物溶于72ml水,在室温下加入碳酸氢钠60.9g(725mmol)、二氯甲烷72ml,然后缓慢滴加醋酸酐27.4ml(290mmol),搅拌22小时。滤去不溶物,注入6N盐酸使pH为3,然后加入氯化钠使饱和,用氯仿萃取,有机层用无水硫酸钠干燥。过滤后将滤液减压浓缩,加入二氯甲烷得到5.45g标题化合物的结晶。收率33.4%(2步)。
合成例9
2-乙酰基-十氢-(4aR,4aR)-异喹啉-3(S)-羧酸叔丁酯
使用由合成例8得到的化合物,与合成例5同样制得5.21g标题化合物。收率77%。
合成例10
(2RS,4R)-2〔(1R,2R)-2-〔(2S)-2-乙酰胺基-2-(叔丁氧羰基)乙基〕环己基〕噻唑烷-4-羧酸甲酯
使用由合成例9得到的化合物,与合成例6同样制得1.61g标题化合物。收率21%。
1H-NMR(400MHz,CDCl3)δ;7.28 and 6.15(total 1H,each brd),4.57-3.75(total 3H,m),3.78 and 3.76(total3H,each s),3.30-3.20(total 1H,m),3.04 and 2.76(total 1H,dd and t),2.01 and 1.97(total 3H,each s),1.50 and 1.47(total 9H,each s),2.40-1.05(total 12H,m)
合成例11(3R,6S,7aR,11aR,11bR)-6-乙酰胺基-5-氧代-2,3,5,6,7,7a,11a,11b,-八氢环己基〔C〕噻唑并〔3,2-α〕氮杂-3-羧酸甲酯

使用由合成例10得到的化合物,与合成例7同样制得0.48g标题化合物。收率36%。绝对构型通过NMR的NOE确定。
1H-NMR(400MHz,CDCl3)δ;6.76(1H,brd,J=6.0Hz),5.14(1H,s),4.91(1H,t,J=7.0Hz),4.56(1H,ddd,J=1.8,6.0,11.4Hz),3.79(3H,s),3.29(1H,dd,J=7.0,11.6Hz),3.13(1H,dd,J=7.0,11.6Hz),2.35-2.30(1H,m),2.07-1.15(11H,m),2.00(3H,s)
NOEδ;3.29(H2β)↓→1.70(H11)5.14(H11b)↓→1.85(H11a),2.33(H7a),4.56(H6)
4.91(H3)↓→3.13(H2α)
4.56(H6)↓→2.33(H7α)
合成例12
〔3R-(3α,6α,8α,9αβ)〕-6-乙酰胺基-8-甲基-5-氧代-八氢噻唑并〔3,2-α〕氮杂-3-羧酸甲酯及〔3R-(3α,6α,8α,9αβ)〕-6-乙酰胺基-8-甲基-5-氧代-八氢噻唑并〔3,2-α〕氮杂-3-羧酸甲酯的混合物

使用DL-(2S*,4S*)-2-羧基-4-甲基氯化哌啶,与合成例A-4-7同样合成上述标题化合物的约1∶1的混合物。
合成例14
(S)-N-乙酰基-5-甲酰基-1,2,3,4-四氢吡啶-2-羧酸叔丁酯

在二甲基甲酰胺137ml(1.77mol)中在0℃下加入氯氧化磷82ml(880mmol),然后在-10-0℃下加入由合成例13得到的(S)-N-乙酰基-1,2,3,4-四氢吡啶-2-羧酸叔丁酯39.8g(177mol)的二甲基甲酰胺40ml溶液,缓慢升温至室温。搅拌1小时后,将反应液注入硫酸钠365g(4.49mol)的水溶液2.0L中,用乙酸乙酯萃取。将有机层用饱和碳酸氢钠、饱和食盐水洗,用硫酸钠干燥。蒸馏除去溶剂,将残渣用异丙醚重结晶,得到标题化合物18.1g。收率40%。
1H-NMR(400MHz,CDCl3)d;9.35(1/6H,s),9.30(5/6H,s),8.16(1/6H,s),7.50(5/6H,s),5.13(5/6H,s),4.62(1/6H,br),2.60-2.40(2H,m),2.41(3×5/6H,s),2.22(3×1/6H,s),1.98-1.70(2H,m),1.45(9H,s)
合成例15
(2S,5S)-N-乙酰基-5-甲基哌啶-2-羧酸叔丁酯

将由合成例14得到的(S)-N-乙酰基-5-甲酰基-1,2,3,4-四氢吡啶-2-羧酸叔丁酯140mg(0.529mmol)溶于20ml乙醇中,加入5%Pd/C(140mg)。使用中压接触还原装置,在3kg/cm2氢气中进行加氢。滤去催化剂,将滤液浓缩得到140mg标题化合物。收率100%。
合成例16
(S)-N-乙酰基-5-乙烯基-1,2,3,4-四氢吡啶-2-羧酸叔丁酯
在甲基三苯基膦溴化物15.2g(42.5mmol)的二乙醚(80ml)的悬浊液中,在30℃以下加入正丁基锂的醚溶液(42.5ml)。在该溶液中,室温下加入由合成例1 4得到的(S)-N-乙酰基-5-甲酰基-1,2,3,4-四氢吡啶-2-羧酸叔丁酯8.98g(35.5mmol)的THF溶液,搅拌1夜。在反应液中加入水,用乙酸乙酯萃取。用饱和食盐水洗,然后用硫酸钠干燥。用硅胶柱色谱法(正己烷∶乙酸乙酯=2∶1)精制,得到4.08g标题化合物。收率46%。
1H-NMR(400MHz,CDCl3)d;7.35(1/6H,s),6.69(5/6H,s),6.38(1/6H,dd,J=10.8,17.6Hz),6.30(5/6H,dd,J=10.8,17.2Hz),5.11(5/6H,m),5.05(5/6H,d,J=17.2Hz),5.03(1/6H,d,J=17.6Hz),4.95(5/6H,d,J=10.8Hz),4.93(1/6H,d,J==10.8Hz),4.53(1H,m),2.52-2.38(1H,m),2.34-2.24(1H,m),2.26(3×5/6H,s),2.14(3×1/6H,s),2.04-1.78(2H,m),1.45(9×1/6H,s),1.44(9×5/6H,s)
合成例17
(2S,3S)-N-乙酰基-5-乙基哌啶-2-羧酸叔丁酯
将由合成例16得到的(S)-N-乙酰基-5-乙烯基-1,2,3,4-四氢吡啶-2-羧酸叔丁酯4.08g(16.3mmol)溶于150ml乙醇中,加入10%Pd/c4.0g。使用中压接触还原装置,在3kg/cm2氢气中进行加氢。滤去催化剂,将滤液浓缩得到140mg标题化合物。收率100%。
1H-NMR(400MHz,CDCl3)d;5.27(1H,d,J=6.0Hz),4.52-4.35(3/4H,m),3.64(1H,dd,J=4.5,13.0Hz),2.91(1H,dd,J=12.0,13.0Hz),2.35-2.15(5/4H,m),2.13(3×3/4H,s),2.07(3×1/4H,s),1.80-1.50(2H,m),1.47(9×1/4H,s),1.46(9×1/4H,s),1.35-1.20(2H,m),1.05-0.95(1H,m),0.93(3×3/4H,t,J=7.6Hz),0.90(3×1/4H,t,J=7.6Hz)
合成例18
(2RS,4R)-2-〔(1S,4S)-4-乙酰胺基-4-(叔丁氧羰基)-1-乙基丁基〕噻唑烷-4-羧酸甲酯
将由合成例17得到的(2S,5S)-N-乙酰基-5-乙基哌啶-2-羧酸叔丁酯4.29g(16.8mmol)溶于43ml甲醇中,加入四乙铵对甲苯磺酸盐。使用碳精电极,在室温下以一定电流(0.33A)按5F/mol通电。反应溶液减压浓缩,残渣溶于乙酸乙酯。用水及饱和食盐水洗,用无水硫酸钠干燥有机层。过滤后得到(2S,5S)-N-乙酰基-6-甲氧基-5-甲基哌啶-2-羧酸酯的粗生成物5.08g。
将该粗生成物溶于60ml醋酸-水(1∶1)中,加入N-甲基吗啉2.4ml(23.7mmol)、L-半胱氨酸甲基酯,盐酸盐3.46g(20.2mmol),在室温下氮气中搅拌3天。用饱和食盐水洗,以无水硫酸钠干燥。过滤后将滤液浓缩,残渣用硅胶柱色谱法(二氯甲烷∶乙醇=100∶1)精制,得到标题化合物3.62g。收率55%。
1H-NMR(400MHz,CDCl3)d;6.11(2/3H,d,J=7.6Hz),6.06(1/3H,d,J=8.0Hz),4.60(1/3H,m),4.55-4.40(2×2/3H,m),4.12(1/3H,m),3.78(3×2/3H,s),3.77(3×1/3H,s),3.85-3.70(1H,m),3.28(2/3H,dd,J=7.2,10.4Hz),3.17(1/3H,dd,J=7.6,10.8Hz),3.03(1/3H,1/3Hdd,J=5.6,10.8Hz),2.77(2/3H,dd,10.0,10.4Hz),2.02(3H,s),1.90-1.20(7H,m),1.48(9×1/3H,s),1.47(9×2/3H,s),0.92(3×2/3H,t,J=7.6Hz),0.90(3×1/3H,t,J=7.6Hz),
(但为1∶2的非对映立体异构体的混合物)
合成例19
〔3R-(3α,6α,9β,9αβ)〕-6-乙酰胺基-9-乙基-5-氧代-八氢噻唑并〔3,2-α〕氮杂-3-羧酸甲脂

在由合成例18得到的(2RS,4R)-2-〔(1S,4S)-4-乙酰胺基-4-叔丁氧羰基)-1-乙基丁基〕噻唑烷-4-羧酸甲酯3.62g(9.31mmol)中在冰冷下加入三氟醋酸21.5ml,缓慢升温至室温。搅拌5小时,蒸馏除去溶剂,用甲苯共沸,得到(2R,4R)-2-〔(1S,4RS)-4-乙酰胺基-4-羧基-1-乙基丁基〕噻唑烷-4-羧酸甲酯三氟醋酸盐的粗生成物。将该粗生成物溶于60ml四氢呋喃中,加入N-甲基吗啉4.09ml(37.2mmol)使pH为7。室温下加入2-乙氧基-1-乙氧羰基-1,2-二氢喹啉-(EEDQ,2,76g,11.2mmol),氮气环境下室温下搅拌一夜。将反应液减压浓缩后,在残渣中加入2N盐酸100ml,使pH为1以下,用二氯甲烷萃取。将有机层用饱和碳酸氢钠水溶液、饱和食盐水洗,用无水硫酸镁干燥。过滤后减压浓缩滤液,得到的残渣用硅胶柱色谱法(二氯甲烷∶乙醇=100∶1-100∶3)精制,得到白色结晶的标题化合物1.3g。收率44%。
1H-NMR(400MHz,CDCl3)d;6.80(1H,br),5.30(1H,dd,J=3.6,6.8Hz),4.87(1H,d,J=9.2Hz),4.58(1H,m),3.79(3H,s),3.22(1H,dd,3.6,11.6Hz),3.10(1H,dd,J=6.8,11.6Hz),2.18-2.08(1H,m),2.01(3H,s),1.84-1.56(5H,m),1.31(1H,m),0.92(3H,t,J=7.6Hz)
实施例1
〔3R-(3α,6α,9β,9αα)〕-6-氨基-9-甲基-5-氧代-八氢噻唑并〔3,2-α〕氮杂-3-羧酸甲酯及〔3R-(3α,6α,9β,9αβ)〕-6-氨基-9-甲基-5-氧代-八氢噻唑并〔3,2-α〕氮杂-3-羧酸甲酯

将由合成例7得到的〔3R-(3α,6α,9β,9αβ)〕-6-乙酰胺基-9-甲基-5-氧代-八氢噻唑并〔3,2-α〕氮杂-3-羧酸甲酯及〔3R-(3α,6α,9β,9αβ)〕-6-乙酰胺基-9-甲基-5-氧代-八氢噻唑并〔3,2-α〕氮杂-3-羧酸甲酯混合物(异构体之比为2∶1)2.59g溶于10%盐酸甲醇溶液(100ml)中,加热回流26小时。减压下蒸馏除去溶剂后加入2N盐酸,用二氯甲烷洗。在水层中加入氨水使成碱性后,用二氯甲烷萃取,有机层用无水碳酸钾干燥。过滤后将滤液减压浓缩,得到无色油状的标题化合物混合物(异构体之比为2∶1)2.01g。收率为90%。
1H-NMR(400MHz,CDCl3)d;5.35(2/3H,dd,J=3.2,6.8Hz),4.99(1/3H,t,J=6.8Hz),4.76(2/3H,d,J=10.0Hz),4.78-4.70(1/3H,m),3.78(3×2/3H,s),3.76(3×1/3H,s),3.55(2/3H,dd,J=2.2,10.6Hz),3.49(1/3H,dd,J=2.0,10.8Hz),3.30(1/3H,dd,J=6.0,11.6Hz),3.21(2/3H,dd,J=3.2,12.0Hz),3.11(1/3H,dd,J=7.2,11.6Hz),3.09(2/3H,dd,J=6.4,12.0Hz),2.12-1.50(7H,m),1.12(3×1/3H,d,J=6.8Hz),1.00(3×2/3H,d,J=6.8Hz).
实施例2
〔3R-(3α,6α,9β,9αβ)〕-6-〔〔(2S,3S)-1-氧代-2-乙酰硫基-3-甲基戊基〕氨基〕-9-甲基-5-氧代-八氢噻唑并〔3,2-α〕氮杂-3-羧酸甲酯及〔3R-(3α,6α,9α,9αβ)〕-6-〔〔(2S,3S)-1-氧代-2-乙酰硫基-3-甲基戊基〕氨基〕-9-甲基-5-氧代-八氢噻唑并〔3,2-α〕氮杂-3-羧酸甲酯
在由实施例1得到的〔3R-(3α,6α,9β,9αβ)〕-6-氨基-9-甲基-5-氧代-八氢噻唑并〔3,2-α〕氮杂-3-羧酸甲酯及〔3R-(3α,6α,9α,9αβ)〕-6-氨基-9-甲基-5-氧代-八氢噻唑并〔3,2-α〕氮杂-3-羧酸甲酯混合物(异构体之比2∶1)2.01g(7.8mmol)中,在冰冷下加入(2S,3S)-2-乙酰硫基-3-甲基戊酸1.78g(9.3mmol)的四氢呋喃(100)溶液。在该溶液中依次加入1-乙基-3-(3-二甲胺基丙基)碳化二亚胺盐酸盐(DEC·HCl)1.79g(9.3mmol)、N-甲基吗啉1.03ml(9.3mmol)、1-羟基-1H-苯并三唑1水和物(HOBT)1.26g(9.3mmol)后,在氮气环境中室温下搅拌18小时。在反应溶液中加入水,用乙酸乙酯萃取后,将有机层用1NHCl、饱和碳酸氢钠水溶液、饱和食盐水洗,用无水硫酸镁干燥。过滤后,将滤液减压浓缩,得到的残渣用硅胶柱色谱法(己烷∶乙酸乙酯=3∶1)精制,得到第一洗脱物为无色油状的〔3R-(3α,6α,9β,9αβ)〕-6-〔〔(2S,3S)-1-氧代-2-乙酰硫基-3-甲基戊基〕氨基〕-9-甲基-5-氧代-八氢噻唑并〔3,2-α〕氮杂-3-羧酸甲酯837mg。收率42%。化合物的绝对构型由NOE实验确定。
1H-NMR(400MHz,CDCl3)d;7.37(1H,d,J=6.0Hz),5.36(1H,dd,J=3.0,7.0Hz),4.80(1H,d,J=9.5Hz),4.53(1H,m),3.97(1H,d,J=7.0Hz),3.79(3H,s),3.22(1H,dd,J=3.0,11.5Hz),3.10(1H,dd,J=7.0,11.5Hz),2.38(3H,s),2.14-1.90(3H,m),1.78-1.62(3H,m),1.57(1H,m),1.16(1H,m),1.00(3H,d,J=6.5Hz),0.99(3H,d,J=7.0Hz),0.88(3H,t,J=7.5Hz).
NOEδ;1.00(9-Me)↓→4.80(H9a)
3.10(H2α)↓→4.80(H9a),5.36(H3)
3.22(H2β)↓→1.95(H9),3.79(3-COOMe)
4.53(H6)↓→4.80(H9a)
得到第二溶出物为无色油状的〔3R-(3α,6α,9α,9αβ)〕-6-〔〔(2S,3S)-1-氧代-2-乙酰硫基-3-甲基戊基〕氨基〕-9-甲基-5-氧代-八氢噻唑并〔3,2-α〕氮杂-3-羧酸甲酯532mg。收率16%。化合物的绝对构型由NOE的实验确定。
1H-NMR(400MHz,CDCl3)d;7.32(1H,brd,J=6.1Hz),5.20(1H,s),5.00(1H,dd,J=6.0,6.4Hz),4.48(1H,m),3.96(1H,d,J=6.6Hz),3.78(3H,s),3.32(1H,dd,J=6.0,11.7Hz),3.13(1H,dd,J=6.4,11.7Hz),2.37(3H,s),2.20-1.50(7H,m),1.15(1H,m),1.10(3H,d,J=7.4Hz),0.98(3H,d,J=6.8Hz),0.87(3H,t,J=7.2Hz).
NOEδ;1.10(9-Me)↓→3.32(H2β),3.78(3-COOMe)
3.13(H2α)↓→5.20(H9a),5.00(H3)
4.48(H6)↓→5.20(H9a)
实验例3
〔3R-((3α,6α,9α,9αβ)〕-6-〔〔(2S,3S)-1-氧代-2-硫代-3-甲基戊基〕氨基〕-9-甲基-5-氧代-八氢噻唑并〔3,2-α〕氮杂-3-羧酸
将由实施例2得到的〔3R-(3α,6α,9β,9αβ)〕-6-〔〔(2S,3S)-1-氧代-2-乙酰硫基-3-甲基戊基〕氨基〕-9-甲基-5-氧代-八氢噻唑并〔3,2-α〕氮杂-3-羧酸甲酯167mg(0.39mmol)溶于5ml脱气乙醇中,在冰冷下加入1N氢氧化锂水溶液2.0ml(2.0mmol),氮气环境中室温下搅拌1小时。在反应溶液中冰冷下加入2N盐酸7.5ml使成酸性,用水稀释后以二氯甲烷萃取。将有机层用饱和食盐水洗,以无水硫酸镁干燥。过滤后,将滤液减压浓缩,得到的无定形物进行重结晶(二氯甲烷-己烷),50℃下热风干燥12小时,得到白色结晶标题化合物118mg。收率81%。
1H-NMR(400MHz,CDCl3)d;7.66(1H,d,J=6.5Hz),5.39(1H,dd,J=3.0,7.0Hz),4.86(1H,d,J=9.5Hz),4.60(1H,m),3.29(1H,dd,J=3.0,12.0Hz),3.22(1H,dd,J=7.0,9.0Hz),3.13(1H,dd,J=7.0,12.0Hz),2.10-1.90(4H,m),1.87(1H,d,J=9.0Hz),1.81-1.64(2H,m),1.61(1H,m),1.21(1H,m),1.03(3H,d,J=7.0Hz),1.00(3H,d,J=7.0Hz),0.90(3H,t,J=7.0Hz).
实施例4(3R,6S,7aR,11aR,11bR)-6-氨基-5-氧代-2,3,5,6,7,7α,11a,11b-八氢环己基〔C〕噻唑并〔3,2-α〕氮杂-3-羧酸甲酯
使用由合成例11得到的化合物,与实施例1同样制得标题化合物0.23g。收率57%。
1H-NMR(400MHz,CDCl3)δ;5.08(1H,s),4.94(1H,t,J=6.8Hz),3.78(3H,s),3.54-3.52(1H,m),3.27(1H,dd,J=6.8,11.6Hz),3.11(1H,dd,J=6.8,11.6Hz)2.23-1.18(14H,m)
实施例5
(3R,6S,7aR,11aR,11bR)-6-〔〔(2S,3S)-1-氧代-2-乙酰硫基-3-甲基戊基〕氨基〕-5-氧代-2,3,5,6,7,7a,11a,11b-八氢环己基〔C〕噻唑并〔3,2-α〕氮杂-3-羧酸甲酯

使用由实施例4得到的化合物,与实施例A同样制得标题化合物0.32g。收率88%。
1H-NMR(400MHz,CDCl3)δ;7.33(1H,brd,J=6.0Hz),5.14(1H,s),4.96(1H,t,J=6.6Hz),4.57-4.52(1H,m),3.97(1H,d,J=6.8Hz),3.79(3H,s),3.30(1H,dd,J=6,6,11.6Hz),3.14(1H,dd,J=6,6,11.6Hz2.38(3H,s),2.40-0.85(15H,m),0.99(3H,d,J=6.8Hz)0.89(3H,t,J=7.4Hz)
实施例6
(3R,6S,7aR,11aR,11bR)-6-〔〔(2S,3S)-1-氧代-2-乙酰硫基-3-甲基戊基〕氨基〕-5-氧代-2,3,5,6,7,7a,11a,11b-八氢环己基〔C〕噻唑并〔3,2-α〕氮杂-3-羧酸

使用由实施例5得到的化合物,与实施例3同样制得白色结晶的标题化合物0.18g。收率63%。
1H-NMR(400MHz,CDCl3)δ;7.33(1H,brd,J=6.0Hz),5.14(1H,s),4.96(1H,t,J=6.6Hz),4.57-4.52(1H,m),3.97(1H,d,J=6.8Hz),3.79(3H,s),3.30(1H,dd,J=6.6,11.6Hz),3.14(1H,dd,J=6.6,11.6Hz)2.38(3H,s),2.40-0.85(15H,m),0.99(3H,d,J=6.8Hz),0.89(3H,t,J=7.4Hz)
实施例7
〔3R-(3α,6α,8α,9αβ〕-6-氨基-8-甲基-5-氧代-八氢噻唑并〔3,2-α〕氮杂-3-羧酸甲酯及〔3R-(3α,6α,8α,9αβ〕-6-氨基-8-甲基-5-氧代-八氢噻唑并〔3,2-α〕氮杂-3-羧酸甲酯混合物

将由合成例12得到的〔3R-(3α,6α,8α,9αβ〕-6-乙酰胺基-8-甲基-5-氧代-八氢噻唑并〔3,2-α〕氮杂-3-羧酸甲酯及〔3R-(3α,6α,8β,9αβ〕-6-乙酰胺基-8-甲基-5-氧代-八氢噻唑并〔3,2-α〕氮杂-3-羧酸甲酯的约1∶1的混合物940mg(3.12mmol)溶于10%盐酸甲醇溶液24ml中,加热回流24小时。减压下蒸馏除去溶剂,加入水后以二氯甲烷洗,在得到的水层中加入饱和碳酸氢钠水溶液使成碱性后用二氯甲烷萃取,用无水硫酸钠干燥。减压浓缩后得到的残渣用硅胶柱色谱法(氯仿∶甲醇∶氨水=98∶2∶0.2)精制,得到标题化合物的约1.4∶1的混合物220mg。收率27%。
1H-NMR(400MHz,CDCl3)δ;5.29(1H×1.4/2.4,dd,J=2.4,6.4Hz),5.00(1H×1.4/2.4,d,J=10.4Hz),3.78(3H×1.4/2.4,s),3.54(1H×1.4/2.4,dd,J=1.2,10.8Hz),3.26(1H×1.4/2.4,dd,J=2.4,11.6Hz),3.17(1H×1.4/2.4,dd,J=6.4,11.6Hz),1.43-2.05(7H×1.4/2.4,m),1.00(3H×1.4/2.4,d,J=6.8Hz)
および
1H-NMR(400MHz,CDCl3)δ;5.21(1H×1.0/2.4,dd,J=3.2,6.4Hz),5.15(1H×1.0/2.4,dd,J=2.2,10.2Hz),3.78(3H×1.0/2.4,s),3.71(1H×1.0/2.4,dd,J=3.2,10.8Hz),3.27(1H×1.0/2.4,dd,J=3.2,12.0Hz),3.18(1H×1.0/2.4,dd,J=6.4,12.0Hz),1.60-2.33(7H×1.0/2.4,m),1.16(3H×1.0/2.4,d,J=7.2Hz)
实施例8
〔3R-(3α,6α,8β,9αβ〕-6-〔〔(2S,3S)-1-氧代-2-乙酰硫基-3-甲基戊基〕氨基〕-8-甲基-5-氧代-八氢噻唑并〔3,2-α〕氮杂-3-羧酸甲酯及〔3R-(3α,6α,8α,9αβ〕-6-〔〔(2S,3S)-1-氧代-2-乙酰硫基-3-甲基戊基〕氨基〕-8-甲基-5-氧代-八氢噻唑并〔3,2-α〕氮杂-3-羧酸甲酯
在由实施例7得到的〔3R-(3α,6α,8β,9αβ〕-6-氨基-8-甲基-5-氧代-八氢噻唑并〔3,2-α〕氮杂-3-羧酸甲酯及〔3R-(3α,6α,8α,9αβ〕-6-氨基-8-甲基-5-氧代-八氢噻唑并〔3,2-α〕氮杂-3-羧酸甲酯的混合物(异构体之比为1∶1.4)215mg(0.83mmol)中,在冰冷下加入(2S,3S)-乙酰硫基-3-甲基戊酸214mg(1.12mmol)的四氢呋喃(17ml)溶液。在该溶液中依次加入1-乙基-3-(3-二甲胺基丙基)碳化二亚胺盐酸盐(DEC、HCl)207mg(1.08mmol)、N-甲基吗啉0.12ml(1.08mmol)后,在氮气环境下室温下搅拌18小时。浓缩反应溶液后加入水,用乙酸乙酯萃取,将有机层用1N盐酸,饱和碳酸氢钠水溶液、饱和食盐水洗,用无水硫酸镁干燥。过滤后,减压浓缩滤液,得到的残渣用硅胶柱色谱法(己烷∶乙酸乙酯=3∶1)精制,得到二种标题化合物的混合物(异构体之比为1∶1.3)254mg。再将该混合物用分离柱YMC-PackSIL(SH-043-5)(己烷∶乙酸乙酯=4∶1)分离精制,由第一洗脱液得到无色油状的〔3R-(3α,6α,8β,9αβ〕-6-〔〔(2S,3S)-1-氧代-2-乙酰硫基-3-甲基戊基〕氨基〕-8-甲基-5-氧代-八氢噻唑并〔3,2-α〕氮杂-3-羧酸甲酯97mg。收率27%。化合物的绝对构型由NOE实验确定。
1H-NMR(400MHz,CDCl3)δ;7.26(1H,brd,J=6.1Hz),5.21(1H,dd,J=2.9,6.6Hz),5.20(1H,dd,J=1.5,10.6Hz),4.75(1H,ddd,J=5.0,6.1,9.0Hz),3.95(1H,d,J=7.1Hz),3.79(3H,s),3.28(1H,dd,J=2.9,12.0Hz),3.20(1H,dd,J=6.6,12.0Hz),2.37(3H,s),2.30-1.10(8H,m),1.26(3H,d,J=7.1Hz),0.99(3H,d,J=6.6Hz),0.88(3H,t,J=7.4Hz)
NOEδ;1.26(8-Me)↓→4.75(H6α),5.20(H9aα),1.68(H9α)
5.20(H9aα)↓→1.68(H9α),4.75(H6α)
2.28(H8β)↓→1.68(H9α)
由第2溶出液得到无色结晶的〔3R-(3α,6α,8α,9αβ〕-6-〔〔(2S,3S)-1-氧代-2-乙酰硫基-3-甲基戊基〕氨基〕-8-甲基-5-氧代-八氢噻唑并〔3,2-α〕氮杂-3-羧酸甲酯136mg。收率38%。化合物的绝对构型由NOE实验确定。
1H-NMR(400MHz,CDCl3)δ;7.38(1H,brd,J=6.0Hz),5.27(1H,dd,J=2.4,6.4Hz),5.03(1H,d,J=10.4Hz),4.55(1H,dd,J=6.4,10.0Hz),3.97(1H,d,J=6.8Hz),3.79(3H,s),3.27(1H,dd,J=2.4,11.8Hz),3.19(1H,dd,J=6.4,11.8Hz),2.38(3H,s),2.15-1.11(8H,m),0.99(6H,d,J=6.8Hz),0.89(3H,t,J=7.4Hz)
NOEδ;2.09(H8)↓→1.88(H9α),1.92(H7α),4.55(H6),5.03(H9a)
5.03(H9a)↓→1.88(H9α),4.55(H6)
4.55(H6)↓→1.92(H7α)
实施例9
〔3R-(3α,6α,8α,9αβ〕-6-〔〔(2S,3S)-1-氧代-2-硫代-3-甲基戊基〕氨基〕-8-甲基-八氢-5-氧代噻唑并〔3,2-α〕氮杂-3-羧酸

将由实施例8得到的〔3R-(3α,6α,8α,9αβ〕-6-〔〔(2S,3S)-1-氧代-2-乙酰硫基-3-甲基戊基〕氨基〕-8-甲基-八氢-5-氧代噻唑并〔3,2-α〕氮杂-3-羧酸甲酯130mg(0.30mmol)溶于4.3ml脱气处理过的乙醇中。在冰冷下加入2.1ml1N氢氧化锂水溶液,氮气中室温下搅拌1小时。向其中在冰冷下加入1.5ml2N盐酸使成为酸性后加入水,用二氯甲烷萃取,用饱和食盐水洗净后用无水硫酸镁干燥,减压浓缩。将得到的无定形物重结晶(乙酸乙酯-己烷)、50℃下热风干燥24小时,得到标题化合物90mg。收率80%。
1H-NMR(400MHz,CDCl3)δ;7.61(1H,brd,J=6.4Hz),5.29(1H,dd,J=2.4,6.4Hz),5.07(1H,d,J=10.4Hz),4.62(1H,dd,J=6.8,11.2Hz),3.37(1H,dd,J=2.4,12.0Hz),3.22(1H,dd,J=6.4,8.8Hz),3.21(1H,dd,J=6.4,12.0Hz),2.19-1.18(8H,m),1.87(1H,d,J=8.8Hz),1.01(3H,d,J=6.4Hz),1.00(3H,d,J=6.8Hz),0.91(3H,t,J=7.4Hz)
实施例10
〔3R-(3α,6α,8β,9αβ〕-6-〔〔(2S,3S)-1-氧代-2-硫代-3-甲基戊基〕氨基〕-8-甲基-八氢-5-氧代-噻唑并〔3,2-α〕氮杂-3-羧酸
将由实施例8得到的〔3R-(3α,6α,8β,9αβ〕-6-〔〔(2S,3S)-1-氧代-2-乙酰硫基-3-甲基戊基〕氨基〕-8-甲基-八氢-5-氧代噻唑并〔3,2-α〕氮杂-3-羧酸甲脂93mg(0.216mmol)溶于3.1ml脱气乙醇中。然后在冰冷下加入1.5ml1N氢氧化锂水溶液,在氮气环境中室温下搅拌1小时。在冰冷下向其中加入2N盐酸1.1ml成为酸性后加入水然后以二氯甲烷萃取,用饱和食盐水洗,用无水硫酸镁干燥后减压浓缩。将得到的无定形物重结晶(二氯甲烷-己烷),在50℃下热风干燥2 4小时,得到标题化合物66mg。收率82%。
1h-NMR(400MHz,CDCl3)δ;7.49(1H,brd,J=6.0Hz),5.27-5.21(2H,m),4.84-4.77(1H,m),3.37(1H,dd,J=2.4,12.0Hz),3.22(1H,dd,J=7.0,12.0Hz),3.19(1H,dd,J=7.2,8.8Hz),1.88(1H,d,J=8.8Hz),2.33-1.58(7H,m),1.28(3H,d,J=7.2Hz),1.30-1.18(1H,m),1.00(3H,d,J=6.8Hz),0.90(3H,t,J=7.2Hz)
实施例11
〔3R-(3α,6α,9α,9αβ〕-6-〔〔(2S,3S)-1-氧代-2-硫代-3-甲基戊基〕氨基〕-9-甲基-5-氧代-八氢噻唑并〔3,2-α〕氮杂-3-羧酸
将由实施例2得到的〔3R-(3α,6α,9α,9αβ〕-6-〔〔(2S,3S)-1-氧代-2-乙酰酰硫基-3-甲基戊基〕氨基〕-9-甲基-5-氧代-八氢噻唑并〔3,2-α〕氮杂-3-羧酸甲酯1.43g(3.33mmol)溶于脱气的30ml乙醇中,在冰冷下加入1N氢氧化锂水溶液20ml(20mmol),氮气环境下,室温下搅拌1小时。反应溶液中在冰冷下加入2N盐酸50ml,成为酸性后用水稀释,用二氯甲烷萃取。将有机层用饱和食盐水洗,用无水硫酸镁干燥。过滤后将滤液减压浓缩,将得到的无定形物重结晶(二氯甲烷-己烷),在50℃下热风干燥12小时,得到白色结晶的标题化合物1.10g。收率89%。
1H-NMR(400MHz,CDCl3)d;7.57(1H,brd,J=6.4Hz),5.25(1H,s),5.08(1H,dd,J=3.2,6.8Hz),4.60(1H,m),3.48(1H,dd,J=3.2,11.6Hz),3.25(1H,dd,J=6.4,8.4Hz),3.13(1H,dd,J=6.8,11.6Hz),2.16-1.54(7H,m),1.85(1H,d,J=8.4Hz),1.24(1H,m),1.03(3H,d,J=6.4Hz),1.01(3H,d,J=6.4Hz),0.91(3H,t,J=7.2Hz).
实施例12
〔3R-(3α,6α,9β,9αβ〕-6-〔〔(2S,3S)-1-氧代-2-乙酰硫基-3-甲基戊基〕氨基〕-9-甲基-5-氧代-八氢噻唑并〔3,2-α〕氮杂-3-羧酸

将由实施例3得到的〔3R-(3α,6α,9β,9αβ〕-6-〔〔(2S,3S)-1-氧代-2-硫代-3-甲基戊基〕氨基〕-9-甲基-5-氧代-八氢噻唑并〔3,2-α〕氮杂-3-羧酸522mg(1.39mmol)溶于乙腈(15ml)-四氢呋喃(15ml)中,在氮气环境下,室温下,加入无水氯化钴54mg(0.42mmol)、醋酸酐170ml(1.81mmol),搅拌5小时。在反应溶液中,加入水后用乙酸乙酯萃取,将有机层用饱和食盐水行,用无水硫酸镁干燥。过滤后将滤液减压浓缩,将得到的无定形物重结晶(乙酸乙酯-己烷),在50℃下热风干燥18小时,得到白色结晶的标题化合物439mg。收率76%。
1H-NMR(400MHz,CDCl3)d;7.38(1H,brd,J=6.0Hz),5.39(1H,dd,J=2.8,6.4Hz),4.83(1H,d,J=9.6Hz),4.56(1H,m),3.96(1H,d,J=6.8Hz),3.29(1H,dd,J=2.8,11.6Hz),3.12(1H,dd,J=6.4,11.6Hz),2.38(3H,s),2.14-1.88(4H,m),1.77-1.64(2H,m),1.58(1H,m),1.16(1H,m),1.01(3H,d,J=7.2Hz),1.00(3H,d,J=6.8Hz),0.88(3H,t,J=7.2Hz).
实施例13
〔3R-(3α,6α,9β,9αβ〕-6-〔〔(2S,3S)-1-氧代-2-丙酰硫基-3-甲基戊基〕氨基〕-9-甲基-5-氧代-八氢噻唑并〔3,2-α〕氮杂-3-羧酸

与实施例12的方法相同,利用实施例A-3得到的〔3R-(3α,6α,9β,9αβ〕-6-〔〔(2S,3S)-1-氧代-2-硫代-3-甲基戊基〕氨基〕-9-甲基-5-氧代-八氢噻唑并〔3,2-α〕氮杂-3-羧酸60mg(0.16mmol)丙酰氯17ml(0.19mmol)及无水氯化钴6mg(0.05mmol),得到白色结晶的标题化合物30mg。收率44%。
1H-NMR(400MHz,CDCl3)d;7.40(1H,brd,J=6.4Hz),5.39(1H,dd,J=2.4,6.8Hz),4.83(1H,d,J=9.6Hz),4.56(1H,m),3.98(1H,d,J=6.8Hz),3.29(1H,dd,J=2.8,11.6Hz),3.11(1H,dd,J=6.8,11.6Hz),2.63(2H,q,J=7.6Hz),2.16-1.88(4H,m),1.76-1.64(2H,m),1.57(1H,m),1.19(3H,t,J=7.6Hz),1.17(1H,m),1.01(3H,d,J=6.8Hz),1.00(3H,d,J=6.8Hz),0.88(3H,t,J=7.2Hz).
实施例14
〔3R-(3α,6α,9β,9αβ〕-6-〔〔(2S,3S)-1-氧代-2-苯甲酰硫基-3-甲基戊基〕氨基〕-9-甲基-5-氧代-八氢噻唑并〔3,2-α〕氮杂-3-羧酸
与实施例12的方法相同,利用由实施例A-3得到的〔3R-(3α,6α,9β,9αβ〕-6-〔〔(2S,3S)-1-氧代-2-硫代-3-甲基戊基〕氨基〕-9-甲基-5-氧代-八氢噻唑并〔3,2-α〕氮杂-3-羧酸434mg(1.16mmol)苯甲酸酐300(1.33mmol)及无水氯化钴45mg(0.35mmol),得到白色结晶的标题化合物490mg。收率88%。
1H-NMR(400MHz,CDCl3)d;8.00-7.96(2H,m),7.62-7.42(4H,m),5.38(1H,dd,J=2.4,6.4Hz),4.84(1H,d,J=9.6Hz),4.59(1H,m),4.20(1H,d,J=7.2Hz),3.27(1H,dd,J=2.4,11.6Hz),3.10(1H,dd,J=6.4,11.6Hz),2.22-1.60(7H,m),1.25(1H,m),1.06(3H,d,J=6.8Hz),1.00(3H,d,J=6.8Hz),0.92(3H,t,J=7.2Hz).
实施例15
〔3R-(3α,6α,9β,9αβ〕-6-〔〔(2S,3S)-1-氧代-2-(1,1-二甲基丙酰基)硫代-3-甲基戊基〕氨基〕-9-甲基-5-氧代-八氢噻唑并〔3,2-α〕氮杂-3-羧酸

与实施例12的方法相同,由实施例A-3得到的〔3R-(3α,6α,9β,9αβ〕-6-〔〔(2S,3S)-1-氧代-2-硫代-3-甲基戊基〕氨基〕-9-甲基-5-氧代-八氢噻唑并〔3,2-α〕氮杂-3-羧酸54mg(0.14mmol)、三甲基乙酰氯、27ml(0.22mmol)、无水氯化钴6mg(0.05mmol),得到白色结晶的标题化合物58mg。收率88%。
1H-NMR(400MHz,CDCl3)d;7.41(1H,brd,J=6.0Hz),5.39(1H,dd,J=2.4,6.4Hz),4.83(1H,d,J=9.6Hz),4.56(1H,m),3.92(1H,d,J=6.8Hz),3.29(1H,dd,J=2.4,11.6Hz),3.10(1H,dd,J=6.4,11.6Hz),2.18-1.52(7H,m),1.25(9H,s),1.20(1H,m),1.01(3H,d,J=6.8Hz),1.00(3H,d,J=6.8Hz),0.87(3H,t,J=7.2Hz).
实施例16
〔3R-(3α,6α,9β,9αβ〕-6-〔〔(2S,3S)-1-氧代-2-(4-吗啉基)乙酰硫基-3-甲基戊基〕氨基〕-9-甲基-5-氧代-八氢噻唑并〔3,2-α〕氮杂-3-羧酸

在氮气环境下,将44mg(0.24mmol)4-吗啉基醋酸盐酸盐溶于脱气的无水N,N-二甲基甲酰胺(1.2ml),冰冷下加入N,N’-碳化二咪唑27.3mg(1.68mmol),室温上搅拌1小时。冰冷下加入由实施例3得到的〔3R-(3α,6α,9β,9αβ〕-6-〔〔(2S,3S)-1-氧代-2-硫代-3-甲基戊基〕氨基〕-9-甲基-5-氧代-八氢噻唑并〔3,2-α〕氮杂-3-羧酸60mg(0.16mmol)的脱气无水四氢呋喃(1.6ml)溶液,室温搅拌2天。将反应溶液浓缩后,加入乙酸乙酯及饱和食盐水分液,有机层用饱和食盐水洗,用无水硫酸镁干燥。过滤后减压浓缩滤液,将得到的无定形物重结晶(乙酸乙酯-乙醚-己烷),50℃下热风干燥一夜,得到白色结晶的标题化合物68mg。收率85%
1H-NMR(400MHz,CDCl3)d;7.40(1H,brd,J=6.0Hz),5.35(1H,dd,J=2.4,6.8Hz),4.82(1H,d,J=9.2Hz),4.55(1H,m),3.92(1H,d,J=6.8Hz),3.77(4H,t,J=4.4Hz),3.31(2H,s),3.28(1H,dd,J=2.4,11.6Hz),3.12(1H,dd,J=6.8,11.6Hz),2.62(4H,m),2.14-1.52(7H,m),1.17(1H,m),1.01(3H,d,J=6.4Hz),1.00(3H,d,J=6.8Hz),0.87(3H,t,J=7.6Hz).
实施例17
〔3R-(3α,6α,9β,9αβ〕-6-〔〔(2S.3S)-1-氧代-2-(4-硫代吗啉基)乙酰硫基-3-甲基戊基〕氮基〕-9-甲基-5-氧代-八氢噻唑并〔3,2-α〕氮杂-3-羧酸
与实施例16的方法相同,通过由实施例3得到的〔3R-(3α,6α,9β,9αβ〕-6-〔〔(2S,3S)-1-氧代-2-硫代-3-甲基戊基〕氨基〕-9-甲基-5-氧代-八氢噻唑并〔3,2-α〕氮杂-3-羧酸70mg(0.19mmol)、4-硫代吗啉基醋酸盐酸盐55mg(0.28mmol)、N-N’-碳化二咪唑33mg(0.21mmol),得到白色结晶的标题化合物81mg。收率84%。
1H-NMR(400MHz,CDCl3)d;7.40(1H,brd,J=6.4Hz),5.33(1H,m),4.83(1H,d,J=9.6Hz),4.56(1H,m),3.89(1H,d,J=7.2Hz),3.32-3.26(1H,m),3.30(2H,s),3.12(1H,dd,J=6.4,11.2Hz),2.88-2.82(2H,m),2.76-2.70(2H,m),2.14-1.90(4H,m),1.78-1.54(3H,m),1.18(1H,m),1.01(3H,d,J=6.8Hz),1.00(3H,d,J=6.4Hz),0.90(3H,t,J=7.6Hz).
实施例18
〔3R-(3α,6α,9αβ〕-6-〔〔(2S,3S)-1-氧代-2-(4-二氧硫代吗啉基)乙酰硫基-3-甲基戊基〕氨基〕-9-甲基-5-氧代-八氢噻唑并〔3,2-α〕氮杂-3-羧酸

与实施例16的方法相同,通过由实施例3得到的〔3R-(3α,6α,9β,9αβ〕-6-〔〔(2S,3S)-1-氧代-2-硫代-3-甲基戊基〕氨基〕-9-甲基-5-氧代-八氢噻唑并〔3,2-α〕氮杂-3-羧酸70mg(0.19mmol)、4-二氧硫代吗啉基醋酸54mg(0.28mmol)、N-N’-碳化二咪唑33mg(0.21mmol),得到白色结晶的标题化合物73mg。收率71%。
1H-NMR(400MHz,CDCl3)d;7.50(1H,brd,J=6.4Hz),5.27(1H,dd,J=3.2,6.8Hz),4.81(1H,d,J=9.6Hz),4.55(1H,m),3.96(1H,d,J=6.4Hz),3.46(2H,s),3.27(1H,dd,J=3.2,12.0Hz),3.24-3.10(5H,m),2.18-1.92(4H,m),1.76-1.63(2H,m),1.55(1H,m),1.17(1H,m),1.01(3H,d,J=6.8Hz),1.00(3H,d,J=6.8Hz),0.90(3H,t,J=7.6Hz).
实施例19
〔3R-(3α,6α,9β,9αβ〕-6-〔〔(2S,3S)-1-氧代-2-烟酰硫基-3-甲基戊基〕氨基〕-9-甲基-5-氧代-八氢噻唑并〔3,2-α〕氮杂-3-羧酸

与实施例12的方法相同,通过由实施例3得到的〔3R-(3α,6α,9β,9αβ〕-6-〔〔(2S,3S)-1-氧代-2-硫代-3-甲基戊基〕氨基〕-9-甲基-5-氧代-八氢噻唑并〔3,2-α〕氮杂-3-羧酸50mg(0.13mmol)、烟酸18mg(0.15mmol)、N,N’-碳化二咪唑23mg(0.14mmol),得到白色结晶的标题化合物28mg。收率44%。
1H-NMR(400MHz,CDCl3)d;9.17(1H,br),8.80(1H,br),8.22(1H,brd,J=8.4Hz),7.58(1H,br),7.43(1H,m),5.27(1H,br),4.82(1H,br),4.60(1H,br),4.23(1H,brd,J=7.2Hz),3.34(1H,br),3.12(1H,br),2.24-1.92(4H,m),1.80-1.58(3H,m),1.24(1H,m),1.06(3H,d,J=6.8Hz),1.00-0.84(6H,m)
实施例20
〔3R-(3α,6α,9α,9αβ〕-6-〔〔(2S,3S)-1-氧代-2-乙酰硫基-3-甲基戊基〕氨基〕-9-甲基-5-氧代-八氢噻唑并〔3,2-α〕氮杂-3-羧酸

与实施例12的方法相同,通过由实施例11得到的〔3R-(3α,6α,9α,9αβ〕-6-〔〔(2S,3S)-1-氧代-2-硫代-3-甲基戊基〕氨基〕-9-甲基-5-氧代-八氢噻唑并〔3,2-α〕氮杂-3-羧酸212mg(0.57mmol)、醋酸酐64ml(0.68mmol)、无水氯化钴22mg(0.17mmol),得到白色结晶的标题化合物163mg。收率69%。
1H-NMR(400MHz,CDCl3)d;7.30-7.20(1H,m),5.28(1H,s),5.08(1H,dd,J=2.4,6.4Hz),4.59(1H,m),3.95(1H,d,J=7.2Hz),3.48(1H,dd,J=2.4,11.6Hz),3.12(1H,dd,J=6.4,11.6Hz),2.40(3H,s),2.16-1.52(7H,m),1.18(1H,m),1.01(3H,d,J=6.4Hz),1.00(3H,d,J=7.2Hz),0.91(3H,t,J=7.2Hz).
实施例21
〔3R-(3α,6α,9α,9αβ〕-6-〔〔(2S,3S)-1-氧代-2-
苯甲酰硫基-3-甲基戊基〕氨基〕-9-甲基-5-氧代-八氢噻唑并〔3,2-α〕氮杂-3-羧酸
与实施例12的方法相同,通过由实施例11得到的〔3R-(3α,6α,9α,9αβ〕-6-〔〔(2S,3S)-1-氧代-2-硫代-3-甲基戊基〕氨基〕-9-甲基-5-氧代-八氢噻唑并〔3,2-α〕氮杂-3-羧酸265mg(0.71mmol)、苯甲酸酐176mg(0.78mmol)、无水氯化钴28mg(0.21mmol),得到白色结晶的标题化合物163mg。收率48%。
1H-NMR(400MHz,CDCl3)d;7.99(2H,m),7.60-7.40(4H,m),5.22(1H,s),5.03(1H,br),4.60(1H,m),4.08(1H,br),3.42(1H,br),3.03(1H,br),2.20-1.60(7H,m),1.24(1H,m),1.10-0.90(9H,m).
实施例22
〔3R-(3α,6α,9β,9αβ〕-6-氨基-9-乙基-5-氧代-八氢噻唑并〔3,2-α〕氮杂-3-羧酸甲酯
将由合成例19得到的〔3R-(3α,6α,9β,9αβ〕-6-乙酰胺基-9-乙基-5-氧代-八氢噻唑并〔3,2-α〕氮杂-3-羧酸甲酯1.3g(4.13mmol)溶于10%盐酸甲醇溶液(50ml),加热回流2天。减压下蒸馏除去溶剂后加入水,用二氯甲烷洗。在得到的水层中加入饱和碳酸氢钠水溶液成为碱性后,用二氯甲烷萃取,以无水硫酸钠干燥。过滤后减压浓缩滤液,得到无色油状的标题化合物0.83g。收率74%。
1H-NMR(400MHz,CDCl3)d;5.32(1H,dd,J=3.5,6.8Hz),4.89(1H,d,J=9.2Hz),3.78(3H,s),3.55(1H,dd,J=2.0,10.5Hz),3.21(1H,dd,J=3.5,11.6Hz),3.09(1H,dd,J=6.8,11.6Hz),2.20-1.40(9H,m),0.92(3H,t,J=7.6Hz)
实施例23
〔3R-(3α,6α,9β,9αβ〕-6-〔〔(2S,3S)-1-氧代-2-乙酰硫基-3-甲基戊基〕氨基〕-9-乙基-5-氧代-八氢噻唑并〔3,2-α〕氮杂-3-羧酸甲酯
在由实施例22得到的〔3R-(3α,6α,9β,9αβ〕-6-氨基-9-乙基-5-氧代-八氢噻唑并〔3,2-α〕氮杂-3-羧酸甲酯0.83g(3.05mmol)中在冰冷下加入(2S,3S)-2-乙酰硫基-3-甲基戊酸0.70g(3.66mmol)的四氢呋喃溶液(50ml)。在该溶液中依次加入1-乙基-3-(3-二甲胺基丙基)碳化二亚胺盐酸盐(DEC·HCl)0.70g(3.66mmol)、N-甲基吗啉0.4ml(3.66mmol)、1-羟基-1H-苯并三唑1水和物(HOBT)0.50g(9.3mmol),然后在氮气环境下室温下搅拌18小时。在反应溶液中加入水,用乙酸乙酯萃取,将有机层用1N盐酸、饱和碳酸氢钠水溶液、饱和食盐水洗,用无水硫酸镁干燥。过滤后,减压浓缩滤液,得到的残渣用硅胶柱色谱法(己烷∶乙酸乙酯=3∶1)精制,得到无色油状的标题化合物476mg。收率35%。
1H-NMR(400MHz,CDCl3)d;7.36(1H,d,J=6.0Hz),5.34(1H,dd,J=3.2,6.4Hz),4.86(1H,d,J=9.6Hz),4.54(1H,m),3.97(1H,d,J=6.8Hz),3.78(3H,s),3.21(1H,dd,J=3.2,12.0Hz),3.11(1H,dd,J=6.4,12.0Hz),2.38(3H,s),2.16-2.04(2H,m),1.82-1.52(6H,m),1.31(1H,m),1.16(1H,m),0.99(3H,d,J=6.4Hz),0.91(3H,t,J=7.2Hz),0.88(3H,t,J=7.2Hz)
实施例24
〔3R-(3α,6α,9β,9αβ〕-6-〔〔(2S,3S)-1-氧代-2-硫代-3-甲基戊基〕氨基〕-9-乙基-5-氧代-八氢噻唑并〔3,2-α〕氮杂-3-羧酸

将由实施例23得到的〔3R-(3α,6α,9β,9αβ〕-6-〔〔(2S,3S)-1-氧代-2-乙酰硫基-3-甲基戊基〕氨基〕-9-乙基-5-氧代-八氢噻唑并〔3,2-α〕氮杂-3-羧酸甲酯476mg(1.07mmol)溶于10.7ml脱气的乙醇,在冰冷下加入5.36ml脱气的1N氢氧化锂水溶液,在氮气环境下室温下搅拌1小时。在反应溶液中冰冷下加入2N盐酸成为酸性,用水稀释后用二氯甲烷萃取。将有机层用饱和食盐水洗,用无水硫酸镁干燥。过滤后减压浓缩滤液,将得到的无定形物用己烷精制,滤取。将得到的物质在50℃下热风干燥12小时,得到标题化合物300mg。收率72%。
1H-NMR(400MHz,CDCl3)d;7.66(1H,d,J=6.4Hz),5.36(1H,dd,J=3.2,6.4Hz),4.91(1H,d,J=9.2Hz),4.61(1H,m),3.28(1H,dd,J=3.2,12.0Hz),3.22(1H,dd,J=6.8,8.8Hz),3.13(1H,dd,J=6.4,12.0Hz),2.16-2.08(2H,m),1.98(1H,m),1.87(1H,d,J=8.8Hz),1.84-1.56(5H,m),1.38-1.18(2H,m),0.99(3H,d,J=6.4Hz),0.93(3H,t,J=7.2Hz),0.90(3H,t,J=7.6Hz)
Claims (3)
1.氨基酸衍生物的制造方法,其特征在于,将由通式(5a)

式中,R8表示氢原子或甲基;R6、R7分别独立表示氢原子、低级烷基、或可以带有取代基的芳烷基;R3、R4、R5分别独立表示氢原子、羟基、低级烷基、低级烷氧基、低级烷硫基、可以带有取代基的芳基或可以带有取代基的杂环基;R11表示羧基的保护基;Z表示酰基或氨基甲酸酯基,
所示的噻唑烷衍生物脱保护,得到由通式(6a)
式中,R3、R4、R5、R6、R7、R8、Z与上述意义相同,
所示的噻唑烷衍生物,将其环化得到由通式(7a)
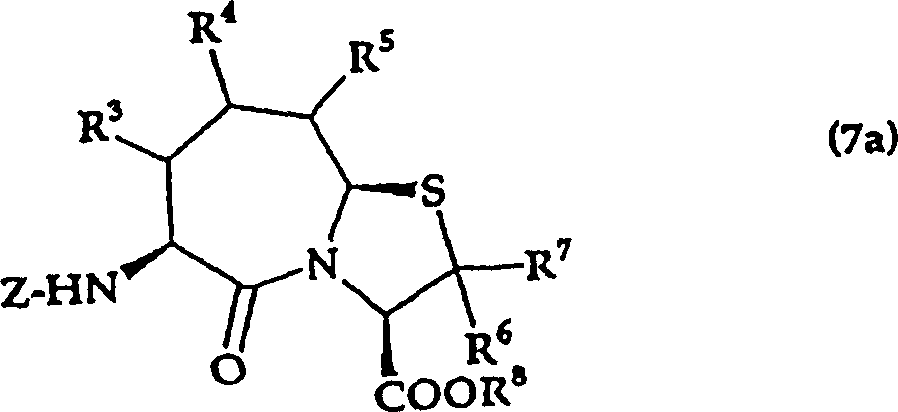
式中,R3、R4、R5、R6、R7、R8、Z与上述意义相同,
所示的氨基酸衍生物,根据需要得到由通式(1a)

所示的氨基酸衍生物。
2.权利要求1记载的氨基酸衍生物的制造方法,其特征在于,将由通式(3a)

式中,R3、R4、R5分别独立表示氢原子、羟基、低级烷基、低级烷氧基、低级烷硫基、可以带有取代基的芳基或可以带有取代基的杂环基;R11表示羧基的保护基,式中R12表示与环内的氮原子一起形成醛等价体的基团,
所示的半缩醛与由通式(4a)
式中,R8表示氢原子或甲基;R6以及R7分别独立表示氢原子、低级烷基或可以带有取代基的芳烷基,
所示的半胱氨酸衍生物反应,得到由通式(5a)

式中,R8表示氢原子或甲基;R6、R7分别独立表示氢原子、低级烷基、或可以带有取代基的芳烷基;R3、R4、R5分别独立表示氢原子、羟基、低级烷基、低级烷氧基、低级烷硫基、可以带有取代基的芳基或可以带有取代基的杂环基;R11表示羧基的保护基,Z表示酰基或氨基甲酸酯基,
所示的噻唑烷衍生物,再将该衍生物脱保护,得到由通式(6a)
式中,R3、R4、R5、R6、R7、R8、Z与上述意义相同,
所示的噻唑烷衍生物,将其环化得到由通式(7a)
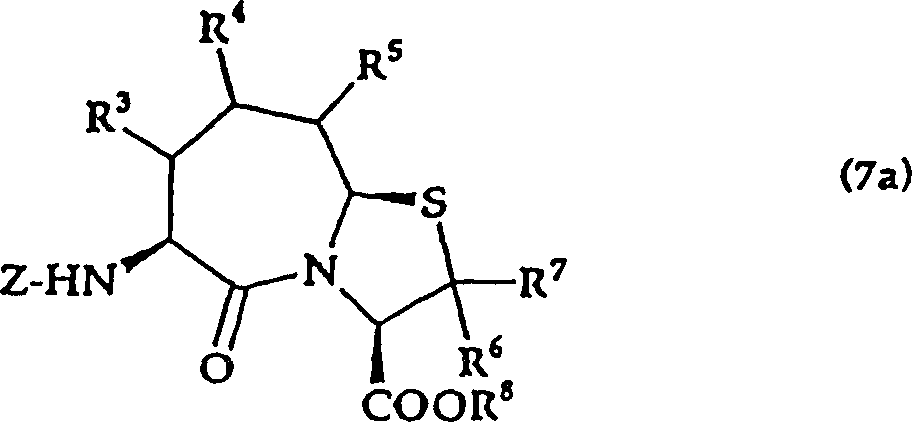
式中,R3、R4、R5、R6、R7、R8、Z与上述意义相同,
所示的氨基酸衍生物,根据需要进行脱保护,得到由通式(1a)

所示的氨基酸衍生物。
3.权利要求1记载的氨基酸衍生物的制造方法,其特征在于,将由通式(2a)

所示的哌啶酸衍生物电解氧化,得到由通式(3a)
R3、R4、R5分别独立表示氢原子、羟基、低级烷基、低级烷氧基、低级烷硫基、可以带有取代基的芳基或可以带有取代基的杂环基;R11表示羧基的保护基;式中R12表示与环内的氮原子一起形成醛等价体的基团,
所示的半缩醛,将得到的半缩醛(3a)与由通式(4a)

式中,R8表示氢原子或甲基;R6以及R7分别独立表示氢原子、低级烷基或可以带有取代基的芳烷基,
所示的半胱氨酸衍生物反应,得到由通式(5a)

式中,R8表示氢原子或甲基;R6、R7分别独立表示氢原子、低级烷基、或可以带有取代基的芳烷基;R3、R4、R5分别独立表示氢原子、羟基、低级烷基、低级烷氧基、低级烷硫基、可以带有取代基的芳基或可以带有取代基的杂环基;R11表示羧基的保护基;Z表示酰基或氨基甲酸酯基,所示的噻唑烷衍生物,再将该衍生物脱保护,得到由通式(6a)

式中,R3、R4、R5、R6、R7、R8、Z与上述意义相同,
所示的噻唑烷衍生物,将其环化得到由通式(7a)
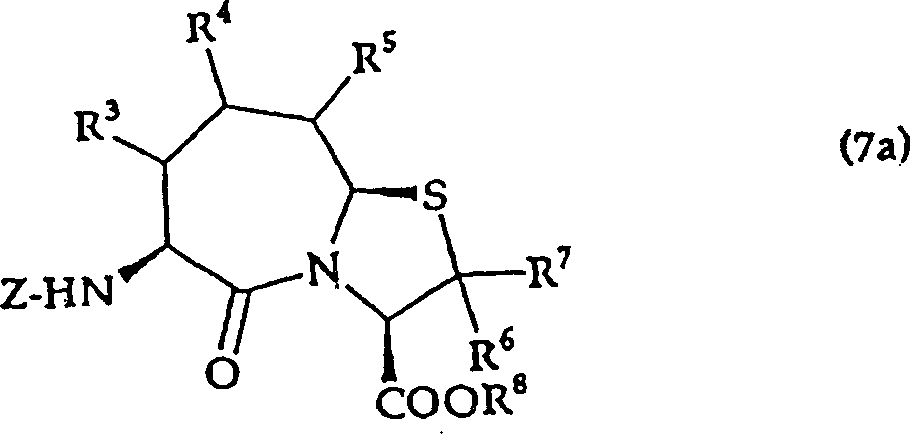
所示的氨基酸衍生物,根据需要进行脱保护,得到由通式(1a)

所示的氨基酸衍生物。
Applications Claiming Priority (6)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| JP165481/1994 | 1994-07-18 | ||
| JP6165481A JPH0827156A (ja) | 1994-07-18 | 1994-07-18 | アミノ酸誘導体の製造方法 |
| JP199180/1994 | 1994-08-24 | ||
| JP19918094A JP3444666B2 (ja) | 1994-08-24 | 1994-08-24 | (2s, 3s)−3− メチル−2− チオペンタン酸誘導体の製造方法 |
| JP6306468A JPH08165293A (ja) | 1994-12-09 | 1994-12-09 | 置換チアゾロ[3,2−a]アゼピン誘導体 |
| JP306468/1994 | 1994-12-09 |
Related Parent Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN95190779A Division CN1053908C (zh) | 1994-07-18 | 1995-06-07 | 取代噻唑并[3,2-α]氮杂䓬衍生物 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1262275A CN1262275A (zh) | 2000-08-09 |
| CN1170832C true CN1170832C (zh) | 2004-10-13 |
Family
ID=27322514
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN95190779A Expired - Fee Related CN1053908C (zh) | 1994-07-18 | 1995-06-07 | 取代噻唑并[3,2-α]氮杂䓬衍生物 |
| CNB991236092A Expired - Fee Related CN1170832C (zh) | 1994-07-18 | 1999-10-28 | 取代噻唑并[3,2-α]氮杂�衍生物 |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN95190779A Expired - Fee Related CN1053908C (zh) | 1994-07-18 | 1995-06-07 | 取代噻唑并[3,2-α]氮杂䓬衍生物 |
Country Status (17)
| Country | Link |
|---|---|
| US (2) | US5789403A (zh) |
| EP (1) | EP0719779B1 (zh) |
| KR (1) | KR100284247B1 (zh) |
| CN (2) | CN1053908C (zh) |
| AT (1) | ATE254622T1 (zh) |
| AU (1) | AU694233B2 (zh) |
| CA (1) | CA2171334C (zh) |
| DE (1) | DE69532155T2 (zh) |
| DK (1) | DK0719779T3 (zh) |
| ES (1) | ES2210292T3 (zh) |
| FI (2) | FI110001B (zh) |
| HU (1) | HU225916B1 (zh) |
| MX (1) | MX9601017A (zh) |
| NO (1) | NO308077B1 (zh) |
| NZ (1) | NZ287557A (zh) |
| PT (1) | PT719779E (zh) |
| WO (1) | WO1996002549A1 (zh) |
Families Citing this family (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US5650408A (en) * | 1995-06-07 | 1997-07-22 | Karanewsky; Donald S. | Thiazolo benzazepine containing dual action inhibitors |
| CA2251155A1 (en) * | 1997-02-05 | 1998-08-13 | Suntory Limited | Medicinal compositions for treating cardiac diseases caused by cardiac hypertrophy |
| US9823092B2 (en) | 2014-10-31 | 2017-11-21 | Allegro Microsystems, Llc | Magnetic field sensor providing a movement detector |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| ZA821904B (en) * | 1981-03-23 | 1983-01-26 | Merck & Co Inc | Bicyclic lactams as anthihypertensives |
| US4415496A (en) * | 1981-03-23 | 1983-11-15 | Merck & Co., Inc. | Bicyclic lactams |
| US4617301A (en) * | 1983-06-22 | 1986-10-14 | Merck & Co., Inc. | Sulfoxide and sulfone derivatives of bicyclic lactams as antihypertensives |
| JPS6056790A (ja) * | 1983-09-03 | 1985-04-02 | 山陽科学株式会社 | 荷役用マット |
| RU2124503C1 (ru) * | 1992-05-18 | 1999-01-10 | И.Р.Сквибб энд Санз, Инк. | Гетероциклические азотсодержащие производные карбоновой кислоты, способ их получения и фармацевтическая композиция |
| DE69329701T2 (de) * | 1992-10-30 | 2001-05-10 | Merrell Pharmaceuticals Inc., Cincinnati | Mercaptoacetylamid substituiertes bizyclisches laktam zur verwendung als enkephalinase und ace-hemmer |
| US5650408A (en) * | 1995-06-07 | 1997-07-22 | Karanewsky; Donald S. | Thiazolo benzazepine containing dual action inhibitors |
-
1995
- 1995-06-07 ES ES95921127T patent/ES2210292T3/es not_active Expired - Lifetime
- 1995-06-07 EP EP95921127A patent/EP0719779B1/en not_active Expired - Lifetime
- 1995-06-07 AT AT95921127T patent/ATE254622T1/de not_active IP Right Cessation
- 1995-06-07 KR KR1019960701358A patent/KR100284247B1/ko not_active Expired - Fee Related
- 1995-06-07 CN CN95190779A patent/CN1053908C/zh not_active Expired - Fee Related
- 1995-06-07 MX MX9601017A patent/MX9601017A/es not_active IP Right Cessation
- 1995-06-07 DE DE69532155T patent/DE69532155T2/de not_active Expired - Fee Related
- 1995-06-07 WO PCT/JP1995/001139 patent/WO1996002549A1/ja not_active Ceased
- 1995-06-07 PT PT95921127T patent/PT719779E/pt unknown
- 1995-06-07 HU HU9600679A patent/HU225916B1/hu not_active IP Right Cessation
- 1995-06-07 AU AU26301/95A patent/AU694233B2/en not_active Ceased
- 1995-06-07 DK DK95921127T patent/DK0719779T3/da active
- 1995-06-07 NZ NZ287557A patent/NZ287557A/en not_active IP Right Cessation
- 1995-06-07 CA CA002171334A patent/CA2171334C/en not_active Expired - Fee Related
- 1995-06-07 US US08/612,864 patent/US5789403A/en not_active Expired - Lifetime
-
1996
- 1996-03-14 NO NO19961051A patent/NO308077B1/no not_active IP Right Cessation
- 1996-03-14 FI FI961199A patent/FI110001B/fi active
-
1998
- 1998-05-27 US US09/085,729 patent/US6051705A/en not_active Expired - Fee Related
-
1999
- 1999-10-28 CN CNB991236092A patent/CN1170832C/zh not_active Expired - Fee Related
-
2001
- 2001-06-01 FI FI20011154A patent/FI113656B/fi not_active IP Right Cessation
Also Published As
| Publication number | Publication date |
|---|---|
| CN1134152A (zh) | 1996-10-23 |
| WO1996002549A1 (en) | 1996-02-01 |
| FI113656B (fi) | 2004-05-31 |
| DK0719779T3 (da) | 2004-03-22 |
| EP0719779A4 (en) | 1996-12-18 |
| PT719779E (pt) | 2004-04-30 |
| CN1053908C (zh) | 2000-06-28 |
| FI961199A0 (fi) | 1996-03-14 |
| HU225916B1 (en) | 2007-12-28 |
| CA2171334C (en) | 2008-10-21 |
| DE69532155T2 (de) | 2004-08-19 |
| US6051705A (en) | 2000-04-18 |
| NZ287557A (en) | 1997-04-24 |
| AU2630195A (en) | 1996-02-16 |
| FI20011154L (fi) | 2001-06-01 |
| NO961051D0 (no) | 1996-03-14 |
| AU694233B2 (en) | 1998-07-16 |
| FI110001B (fi) | 2002-11-15 |
| KR100284247B1 (ko) | 2001-10-24 |
| EP0719779A1 (en) | 1996-07-03 |
| CN1262275A (zh) | 2000-08-09 |
| EP0719779B1 (en) | 2003-11-19 |
| KR960704898A (ko) | 1996-10-09 |
| MX9601017A (es) | 1997-10-31 |
| HUT76477A (en) | 1997-09-29 |
| ES2210292T3 (es) | 2004-07-01 |
| US5789403A (en) | 1998-08-04 |
| FI961199A7 (fi) | 1996-05-13 |
| DE69532155D1 (de) | 2003-12-24 |
| NO308077B1 (no) | 2000-07-17 |
| HU9600679D0 (en) | 1996-05-28 |
| CA2171334A1 (en) | 1996-02-01 |
| NO961051L (no) | 1996-05-10 |
| ATE254622T1 (de) | 2003-12-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1192019C (zh) | 环胺衍生物 | |
| CN1054380C (zh) | 稠环化合物及其用途 | |
| CN1038839C (zh) | 一类环胺化合物的制备方法 | |
| CN1030251C (zh) | 取代的4-(喹啉-2-基-甲氧基)苯乙酸衍生物的制备方法 | |
| CN1030252C (zh) | 四氢苯并咪唑衍生物的制备方法 | |
| CN1196682C (zh) | 4-吡啶基-n-酰基-l-苯丙氨酸 | |
| CN1173497A (zh) | 含杂环碳酸衍生物 | |
| CN1088207A (zh) | 嘧啶化合物 | |
| CN1079224A (zh) | 抗菌素化合物 | |
| CN1064683A (zh) | 新的β-氨基-α羟基羧酸及其用途 | |
| CN1863797A (zh) | 具有苯甲酰胺取代基的环胺bace-1抑制剂 | |
| CN1099392A (zh) | 含有稠合双环的化合物及其制备方法 | |
| CN1049219C (zh) | 具有内皮素拮抗活性的芳香杂环并环戊烯衍生物 | |
| CN1164530A (zh) | 氨基醇衍生物和制备它们的方法 | |
| CN1124734A (zh) | 噻嗪-或硫氮杂䓬衍生物 | |
| CN1065237C (zh) | 环戊烷-和环戊烯-β-氨基酸 | |
| CN1105799A (zh) | 作为基质金属蛋白酶抑制剂的喹诺酮衍生物 | |
| CN1203058C (zh) | 哌啶衍生物及含这些衍生物作为有效成分的药物 | |
| CN1819990A (zh) | 具有抗hcv作用的化合物及其制法 | |
| CN1170832C (zh) | 取代噻唑并[3,2-α]氮杂�衍生物 | |
| CN1036758A (zh) | 双环胺化合物及其制备方法 | |
| CN1033991A (zh) | 取代羟胺类 | |
| CN1086825A (zh) | 三环和四环化合物 | |
| CN1649847A (zh) | 制备旋光亚砜衍生物的方法 | |
| CN1058773A (zh) | (二苯甲基乙氧基哌啶基)脂族酸衍生物及其在治疗过敏和气喘中的用途 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| C06 | Publication | ||
| PB01 | Publication | ||
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| C19 | Lapse of patent right due to non-payment of the annual fee | ||
| CF01 | Termination of patent right due to non-payment of annual fee |