-
GEBIET DER ERFINDUNG:
-
Die vorliegende Erfindung betrifft
ein neues substituiertes Thiazolo[3,2-a]azepinderivat oder ein pharmakologisch
annehmbares Salz davon, sowie ein Verfahren zu dessen Herstellung.
Genauer betrifft die vorliegende Erfindung ein neues substituiertes
Thiazolo[3,2-a]azepinderivat oder ein pharmakologisch annehmbares
Salz davon, das als Medikament nützlich
ist, sowie ein industriell vorteilhaftes Verfahren zur Herstellung dieses
Derivats.
-
BESCHREIBUNG DES STANDES
DER TECHNIK:
-
In den letzten Jahren wurden Inhibitoren
gegen neuturale End-Peptidase (NEP-24, 11, nachfolgend als NEP abgekürzt) und
Angiotensin I-umwandelndes Enzym (nachfolgend als ACE abgekürzt) als
neue Heilmittel gegen Herzversagen erkannt.
-
Artrialnatriuretisches Peptid (nachfolgend
als ANP abgekürzt)
ist ein Hormon, das im lebenden Körper vorkommt, das nicht nur
potente hydrouretische und natriuretische Aktivitäten und
eine vasodilierende Wirkung zeigt, sondern ferner eine inhibitorische
Wirkung gegen die Freisetzung von Norepinephrin durch Depression der
sympathetischen Nerven, eine Aktivität der Inhibierung der Sekretion
von Renin aus der Niere und eine Aktivität der Inhibierung der Sekretion
von Aldesteron aus den Adrenalindrüsen, und darüber hinaus
eine perfusionsabsenkende Aktivität durch Verstärkung der
venösen
Wasserdurchlässigkeit
usw. Die Aktivitäten
von ANP bei Patienten, die beispielsweise an kongestivem Herzversagen,
begleitet von einer verstärkten
Vorbelastung, leiden, werden als zur Behandlung von nicht nur Herzversagen,
sondern auch Hochdruck als bevorzugt angesehen.
-
Hier besteht jedoch das Problem,
dass die klinische Anwendung von ANP derzeit auf akute Stadien beschränkt ist,
da ANP ein Peptid ist und daher nicht oral verabreicht werden kann
und eine schlechte metabolische Stabilität aufweist. Ferner wurde berichtet,
dass die Aktivitäten
von ANP abnehmen, wenn es über einen
langen Zeitraum verabreicht wird. Demzufolge muss man bei seiner
Verwendung grosse Vorsicht walten lassen.
-
Nach angemessener Berücksichtigung
der obigen Eigenschaften von ANP sind diejenigen, die kürzlich als
ANP-verwandte Zubereitungen für
die orale Verabreichung erkannt wurden, die oben beschriebenen neuturalen
End-Peptidase-Inhibitoren (nachfolgend als NEP-Inhibitoren abgekürzt). Es
wurde berichtet, dass bei der Verabreichung bei einem Patienten
mit Herzversagen ein NEP-Inhibitor die ANP-Konzentration so anhebt,
dass er eine natriuretische Aktivität zeigt. Die NEP-Inhibitoren
aus dem Stand der Technik wirken jedoch nur geringfügig auf
das kardiale Blutverhalten ein, und folglich wurden Abnahmen der
Vor- und Nachbelastung nicht klar hervorgebracht.
-
Andererseits inhibieren ACE-Inhibitoren,
die als Vasodilatoren nützlich
sind, die Bildung von Angiotensin (II) (nachfolgend als AT-II abgekürzt), das
ein Herzversagen hervorrufender Faktor ist, und zeigen dadurch eine
signifikante Verbesserung der Schwere der NYHA-Erkrankung und bezüglich der
Erhöhung
der Toleranz gegenüber
Bewegung bei chronischem Herzversagen, und folglich wurde deren
Eignung, einschliesslich ihrer Wirkungen auf die Verlängerung
der Lebenserwartung, nachgewiesen. Jedoch ist das Wirksamkeitsverhältnis der
ACE-Inhibitoren aus dem Stand der Technik bei Patienten nicht immer
hoch und die Wirksamkeit jedes dieser Inhibitoren schwankt von Patient
zu Patent. Ferner wurde auf das Problem hingewiesen, dass die Inhibitoren
beispielsweise Nebeneffekte hervorrufen, wie beispielsweise Hypotension,
so dass die Verabreichung derselben bei Patienten mit renaler Hypofunktion
beschränkt
werden muss.
-
Wie oben beschrieben, werden NEP-Inhibitoren
und ACE-Inhibitoren als neue Mittel gegen Herzversagen angesehen,
jedoch haben die NEP-Inhibitoren und ACE-Inhibitoren aus dem Stand
der Technik nur einen begrenzte Anwendbarkeit. Daher wurde die Entwicklung
eines Medikaments, das sowohl Vorzüge bezüglich der NEP-inhibierenden
Wirkung als auch der ACE-inhibierenden Wirkung zeigt, mit Spannung
erwartet.
-
JP-A-6-56790 und die verwandte EP-A-0
599 444 offenbaren die folgenden Verbindungen, die NEP-inhibierende
Wirkung und ACE-inhibierende Wirkung zeigen:
worin R
1 Wasserstoff,
R
3-CO- oder R
18-S-
ist; R
2 und R
19 sind
jeweils unabhängig
voneinander Wasserstoff, Alkyl, Cycloalkyl-(CH
2)
m-, substituiertes Alkyl, Aryl-(CH
2)
m-, substituiertes
Aryl-(CH
2)
m- oder
Heteroaryl-(CH
3)
m-;
n ist 0 oder 1, mit der Massgabe, dass n 0 sein muss, wenn R
1 und R
19 beide etwas
anderes sind als Wasserstoff; m ist 0 oder eine ganze Zahl von 1–6, R
3 ist Alkyl, substituiertes Alkyl, Cycloalkyl-(CH
2)
m-, Aryl-(CH
2)
m-, substituiertes
Aryl-(CH
2)
m- oder
Heteroaryl-(CH
2)
m-;
R
18 ist Alkyl, substituiertes Alkyl, Cycloalkyl-(CH
2)
m-, Aryl-(CH
2)
m-, substituiertes
Aryl-(CH
2)
m- oder Heteroaryl-(CH
2)
m-; R
12 ist
Wasserstoff, Alkyl, substituiertes Alkyl, Aryl-(CH
2)
m-, substituiertes Aryl-(CH
2)
m-, Heteroaryl-(CH
2)
m-,
und v
und w sind jeweils 1 oder 2.
-
Diese Verbindungen sind jedoch von
den erfindungsgemässen
Verbindungen strukturell unterschiedlich, und darüber hinaus
sind die NEP-inhibierende Wirkung und die ACE-inhibierende Wirkung
zur Erfüllung der
bisher geforderten Wirksamkeiten jeweils zu schlecht, und weiterhin
sind die Verbindungen bezüglich
der Wirksamkeit bei oraler Verabreichung problematisch. Daher ist
ihre klinische Anwendung beschränkt.
Ferner offenbart WO 94/10193 ähnliche
Verbindungen wie die in JP-A-6-56790 offenbarten.
-
Unter den oben beschriebenen Umständen haben
die hiesigen Erfinder ihre Untersuchungen zur Auffinden eines Medikaments
begonnen, das exzellente inhibitorische Wirkungen gegen sowohl NEP
als auch ACE zeigen, und die eine hohe Wirksamkeit bei Verabreichung
auf beliebigem Wege zeigen können.
Als Ergebnis haben sie herausgefunden, dass das obige Ziel mit den
folgenden Verbindungen erreicht werden kann, wodurch die vorliegende
Erfindung erhalten wurde.
-
OFFENBARUNG DER ERFINDUNG:
-
Die vorliegende Erfindung betrifft
ein substituiertes Thiazolo[3,2-a]azepinderivat der allgemeinen
Formel (I) oder ein pharmakologisch annehmbares Salz davon:
worin
R
1 ein Wasserstoffatom ist oder eine Schutzgruppe
für eine
Thiolgruppe, ausgewählt
aus einer C
1-6-Alkylgruppe, Gruppen, die
von aliphatischen gesättigten
Monocarbonsäuren
abgeleitet sind, Gruppen, die von aliphatischen ungesättigten
Carbonsäuren
abgeleitet sind, Gruppen, die von carbocyclischen Carbonsäuren abgeleitet
sind, Gruppen, die von heterocyclischen Carbonsäuren abgeleitet sind, Gruppen,
die von Hydroxycarbonsäuren
und Alkoxycarbonsäuren
abgeleitet sind, Phenyl- oder Naphthylgruppen, Heteroarylgruppen, worin
die Heteroarylgruppen einen Ring aufweisen, der aus 3–8 Ringgliedern
und 1–4
Heteroatomen aufgebaut ist, Benzyl, Furoylmethyl, Thienylmethyl
und Pyridylmethyl;
R
2 ist ein Wasserstoffatom,
eine C
1-6-Alkylgruppe, eine Phenylgruppe,
die einen Substituenten aufweisen kann, eine Heteroarylgruppe, die
einen Substituenten aufweisen kann, eine C
1-6-Alkoxygruppe
oder eine C
1-6-Alkylthiogruppe, worin der
oben genannte Substituent der Phenyl- oder Heteroarylgruppe ausgewählt ist
aus C
1-6-Alkyl, Halogenatomen, C
1-6-Alkoxy, Nitro und Amino, und worin die
Heteroarylgruppe ein 3- bis 8-gliedriger Ring mit 1–4 Heteroatomen
ist;
R
3, R
4 und
R
5 können
identisch oder voneinander verschieden sein und repräsentieren
jeweils ein Wasserstoffatom, eine C
1-6-Alkylgruppe,
eine C
1-6-Alkoxylgruppe, eine C
1-6-Alkylthiogruppe,
oder alternativ dazu können R
3, R
4 oder R
5 zusammen mit dem Kohlenstoffatom, an das
sie gebunden sind, einen Ring bilden, mit der Massgabe, dass der
Fall, dass R
3, R
4 und
R
5 alle Wasserstoffatome darstellen, ausgenommen
ist;
R
6 und R
7 sind
jeweils ein Wasserstoffatom;
R
8 ist
ein Wasserstoffatom oder eine Schutzgruppe für eine Carboxylgruppe, ausgewählt aus
einer C
1-6-Alkylgruppe, Benzyl, 1-Naphthylmethyl,
2-Naphthylethyl,
2-Pyridylmethyl, 3-Pyridylpropyl und 2-Thienylethyl; und
n
und m sind jeweils unabhängig
voneinander 0, 1 oder 2.
-
Darüber hinaus betrifft die vorliegende
Erfindung eine pharmazeutische Zusammensetzung, die die obigen substituierten
Thiazolo[3,2-a]azepinderivate enthält, und deren Verwendung zur
Herstellung eines Medikaments zur Verhinderung und Behandlung von
Erkrankungen, bei denen eine NEP-inhibierende Wirkung wirksam ist,
oder Erkrankungen, bei denen eine ACE-inhibierende Wirkung wirksam
ist. Erfindungsgemäss können sie
zur Herstellung eines diuretischen Medikaments oder zur Herstellung
von Medikamenten zur Vorbeugung und Behandlung von medizinischen
Zuständen,
wie beispielsweise akutem und chronischem Herzversagen, Angina pectoris,
Bluthochdruck, Restenose, Arteriosklerose oder Nierenversagen, verwendet
werden.
-
Ferner betrifft die vorliegende Erfindung
Verbindungen der obigen Formel (I), worin R1 eine
Thiolgruppen-Schutzgruppe ist, und R2 bis
R8 und m und n sind wie oben definiert,
sowie Verbindungen der Formel (I), worin R8 eine
Carboxylgruppen-Schutzgruppe ist, und R1 bis
R7 und m und n sind wie oben definiert.
-
Schliesslich werden erfindungsgemäss verschiedene
Verfahren zur Herstellung der obigen substituierten Thiazolo[3,2-a]azepinderivate
und deren Zwischenprodukte bereitgestellt, wie in den Ansprüchen definiert.
-
In den obigen Definitionen ist die
C1-6-Alkylgruppe, die in die Definition
von R2, R3, R4 und R5 eingeschlossen
ist, eine lineare oder verzweigte Alkylgruppe. Beispiele hierfür schliessen
Methyl, Ethyl, n-Propyl, Isopropyl, n-Butyl, Isobutyl, sek-Butyl,
tert-Butyl, n-Pentyl, Isopentyl, Neopentyl, tert-Pentyl, 1-Methylbutyl, 2-Methylbutyl,
1,2-Dimethylpropyl, n-Hexyl, Isohexyl, 1-Methylpentyl, 2-Methylpentyl,
3-Methylpentyl, 1,1-Dimethylbutyl,
1,2-Dimethylbutyl, 2,2-Dimethylbutyl, 1,3-Dimethylbutyl, 2,3-Dimethylbutyl,
3,3-Dimethylbutyl, 1-Ethylbutyl, 2-Ethylbutyl, 1,1,2-Trimethylpropyl,
eine 1,2,2-Trimethylpropylgruppe, 1-Ethyl-1-methylpropyl, 1-Ethyl-2-methylpropyl
und dergleichen ein. Unter diesen bevorzugt sind eine Methylgruppe,
eine Ethylgruppe, eine n-Propylgruppe, eine Isopropylgruppe, eine
n-Butylgruppe, eine Isobutylgruppe und eine sek-Butylgruppe.
-
Die in die Definition von R2, R3, R4 und
R5 eingeschlossene C1-6-Alkoxygruppe schliesst
beispielsweise eine Methoxygruppe, eine Ethoxygruppe, eine n-Propoxygruppe
oder dergleichen ein.
-
Die in die Definitionen von R2, R3, R4 und
R5 eingeschlossene C1-6-Alkylthiogruppe ist
eine Alkylthiogruppe, die 1–6
Kohlenstoffatome aufweist, und ist beispielsweise eine Methylthiogruppe,
eine Ethylthiogruppe, eine n-Propylthiogruppe oder dergleichen.
-
In der Heteroarylgruppe, die einen
Substituenten aufweisen kann, die in die Definitionen von R2 eingeschlossen ist, ist das Heteroaryl
ein Ring, der aus 3–8
Gliedern aufgebaut ist, vorzugsweise 5 oder 6 Gliedern, und das
1–4 Heteroatome,
wie beispielsweise Stickstoff, Schwefel oder Sauerstoff, aufweist.
In der "Phenylgruppe,
die einen Substituenten aufweisen kann" und der "Heteroarylgruppe, die einen Substituenten
aufweisen kann",
die in die Definitionen von R2 eingeschlossen
sind, kann der "Substituent" C1-6-Alkylgruppen
einschliessen, wie beispielsweise Methyl, Ethyl, n-Propyl und t-Butyl;
Halogenatome, wie beispielsweise ein Fluoratom, ein Chloratom, ein
Bromatom und ein Iodatom; C1-6-Alkoxygruppen,
wie beispielsweise Methoxy, Ethoxy, n-Propoxy und t-Butoxy; eine
Nitrogruppe; eine Aminogruppe, die mono- oder disubstituiert sein
kann; oder dergleichen. Mit diesen Substituenten kann eine 1- bis
3-fache Substitution vorhanden sein.
-
Die in die Definition von R1 eingeschlossene Thiolgruppen-Schutzgruppe
schliesst beispielsweise Niederalkylgruppen ein, wie Methyl, Ethyl,
n-Propyl und t-Butyl; Acetylgruppen, die beispielsweise Gruppen
sind, die abgeleitet sind von aliphatischen gesättigten Monocarbonsäuren, wie
beispielsweise eine Acetylgruppe, eine Propionylgruppe, eine Butyrylgruppe,
eine Pivaloylgruppe, eine Palmitoylgruppe und eine Stearoylgruppe;
Gruppen, die von aliphatischen ungesättigten Carbonsäuren abgeleitet
sind, wie beispielsweise eine Acryloylgruppe, eine Propioloylgruppe,
eine Methacryloylgruppe, eine Crotonoylgruppe und eine Oleoylgruppe; Gruppen,
die von carbocyclischen Carbonsäuren
abgeleitet sind, wie beispielsweise eine Benzoylgruppe, eine Naphthoylgruppe,
eine Toluoylgruppe, eine Apotoylgruppe und eine Cinnamoylgruppe;
Gruppen, die von carbocyclischen Carbonsäuren abgeleitet sind, wie beispielsweise
eine Furoylgruppe, eine Thenoylgruppe, eine Nicotinoylgruppe und
eine Isonicotinoylgruppe; Acylgruppen, die beispielsweise Gruppen
einschliessen, die von Hydroxycarbonsäuren oder Alkoxycarbonsäuren abgeleitet
sind, wie beispielsweise eine Glykoloylgruppe, eine Lactoylgruppe,
eine Glyceroylgruppe, eine Maloylgruppe, eine Tartaroylgruppe, eine
Benziloylgruppe, eine Salicyloylgruppe, eine Anisoylgruppe, eine
Vanilloylgruppe und eine Piperonyloylgruppe; Arylgruppen, wie beispielsweise
Phenyl und Naphthyl; Heteroarylgruppen, wie beispielsweise Furoyl,
Pyridyl und Thienyl; Arylalkylgruppen, wie beispielsweise Benzyl;
Heteroarylalkylgruppen, wie beispielsweise eine Furoylmethylgruppe,
eine Thienylmethylgruppe und eine Pyridylmethylgruppe; und dergleichen.
-
Die in der Definition von R8 eingeschlossene Carboxylgruppen-Schutzgruppe
schliesst eine Niederalkylgruppe ein, wie beispielsweise Methyl,
Ethyl, n-Propyl und t-Butyl; eine Arylalkylgruppe, wie beispielsweise Benzyl,
1-Naphthylmethyl und 2-Naphthylethyl; eine Heteroarylalkylgruppe,
wie beispielsweise 2-Pyridylmethyl, 3-Pyridylpropyl und 2-Thienylethyl; oder
dergleichen. Kurz gesagt, es kann eine beliebige Gruppe sein, so
lange sie in vivo unter Erhalt einer Carboxylgruppe abgespalten
wird.
-
Unter den "zwei Substituenten, die einander benachbart
sind, und mit den Kohlenstoffatomen, an die sie gebunden sind, einen
Ring bilden können" innerhalb der Definitionen
von R3, R4 und R5, ist der gebildete Ring vorzugsweise ein
Ring, der aus 5–8
Gliedern aufgebaut ist.
-
Darüber hinaus schliessen die pharmakologisch
annehmbaren Salze nicht nur anorganische Salze ein, wie beispielsweise
ein Hydrochlorid, ein Sulfat und ein Nitrat, sondern auch organische
Salze, wie beispielsweise ein Maleat, ein Citrat und ein Acetat,
sowie Salze mit Alkalimetallen, wie beispielsweise ein Natriumsalz
und ein Kaliumsalz, und darüber
hinaus Salze mit Aminosäuren,
wie beispielsweise ein Aspartat und ein Glutamat.
-
Die erfindungsgemässen Verbindungen weisen exzellente
inhibitorische Wirkungen gegen sowohl NEP als auch ACE auf. Die
Verbindungen der folgenden allgemeinen Formel (I') sind unter den erfindungsgemässen am
meisten bevorzugt, da sie eine hohe Bioverfügbarkeit besitzen und selbst
bei oraler Verabreichung eine exzellente Wirkung zeigen:
worin
R
1 ein Wasserstoffatom oder eine Thiolgruppen-Schutzgruppe
ist, wie in den Ansprüchen
definiert; R
4 und R
5 können identisch
oder voneinander verschieden sein und sind jeweils ein Wasserstoffatom,
eine C
1-6-Alkylgruppe, eine C
1-6-Alkoxylgruppe,
eine C
1-6-Alkylthiogruppe, oder alternativ
dazu können
die beiden Substituenten R
4 und R
5, die einander benachbart sind, zusammen
mit den Kohlenstoffatomen, an die sie gebunden sind, einen Ring
bilden, mit der Massgabe, dass der Fall, dass sowohl R
4 als
auch R
5 Wasserstoffatome darstellen, ausgeschlossen
ist, und insbesondere sind diejenigen bevorzugt, worin R
4 ein Wasserstoffatom ist, und R
5 ist
eine C
1-6-Alkylgruppe, und die C
1-6-Alkylgruppe ist in diesem Fall vorzugsweise
eine Methylgruppe;
R
6 und R
7 sind jeweils ein Wasserstoffatom; und R
8 ist ein Wasserstoffatom oder eine Carboxylgruppen-Schutzgruppe
wie in den Ansprüchen
definiert.
-
Unter den erfindungsgemässen Verbindungen
(I) sind weiter bevorzugte Verbindungen diejenigen, die durch die
allgemeine Formel (IA) repräsentiert
werden:
worin
R
1 ein Wasserstoffatom oder eine Thiolgruppen-Schutzgruppe
darstellt, wie in den Ansprüchen
definiert; R
5 ist eine C
1-6-Alkylgruppe,
eine C
1-6-Alkoxygruppe oder eine C
1-6-Alkylthiogruppe,
vorzugsweise eine C
1-6-Alkylgruppe, am meisten
bevorzugt eine Methylgruppe;
R
6 und
R
7 sind jeweils ein Wasserstoffatom, und
R
8 ist ein Wasserstoffatom oder eine Carboxylgruppen-Schutzgruppe
wie in den Ansprüchen
definiert, und am meisten bevorzugt ein Wasserstoffatom. Ferner
sind die am meisten bevorzugten Verbindungen unter den erfindungsgemässen Verbindungen
(IA) diejenigen, die durch die folgende allgemeine Formel (IA') repräsentiert werden:
worin
R
1 ein Wasserstoffatom oder eine Thiolgruppen-Schutzgruppe
darstellt, wie in den Ansprüchen
definiert, vorzugsweise ein Wasserstoffatom oder eine Acetylgruppe;
R
5 ist eine C
1-6-Alkylgruppe,
eine C
1-6-Alkoxygruppe oder eine C
1-6-Alkylthiogruppe;
R
6 und
R
7 sind jeweils ein Wasserstoffatom; und
R
8 ist ein Wasserstoffatom oder eine Carboxylgruppen-Schutzgruppe,
wie in den Ansprüchen
definiert, wobei der Fall, dass es ein Wasserstoffatom darstellt,
am meisten bevorzugt ist.
-
Unter den Verbindungen der Formel
(I') sind die Verbindungen
der allgemeinen Formel (I'') am meisten bevorzugt:
worin
R
1 ein Wasserstoffatom oder eine Thiolgruppen-Schutzgruppe
darstellt, wie in den Ansprüchen
definiert; R
4 und R
5 können identisch
oder voneinander verschieden sein und sind jeweils ein Wasserstoffatom,
eine C
1-6-Alkylgruppe, eine C
1-6-Alkoxylgruppe
oder eine C
1-6-Alkylthiogruppe, oder alternativ
dazu können
R
4 und R
5 zusammen
mit den Kohlenstoffatomen, an die sie gebunden sind, einen Ring
bilden, mit der Massgabe, dass der Fall, dass R
4 und
R
5 beide Wasserstoffatome darstellen, ausgenommen
ist;
R
6 und R
7 repräsentieren
jeweils ein Wasserstoffatom; und R
8 ist
ein Wasserstoffatom oder eine Schutzgruppe für eine Carboxylgruppe wie in
den Ansprüchen
definiert.
-
Unter den erfindungsgemässen Verbindungen
(IA') sind die am
meisten bevorzugten Verbindungen diejenigen, die durch die folgenden
beiden Formeln dargestellt werden, die denjenigen entsprechen, worin
R5 in Formel (IA') eine Methylgruppe ist:
-
-
Die beiden folgenden Verbindungen,
worin R1, R5 und
R7 alle Wasserstoffatome darstellen, gehören zu den
erfindungsgemäss
am meisten bevorzugten Verbindungen.
-
Unter diesen sind die Verbindungen,
in denen R8 ein Wasserstoffatom darstellt,
wie folgt:
-
-
Die eine Gruppe bevorzugter Verbindungen,
wie oben beschrieben, sind Verbindungen, die durch Einführung einer
(2S,3S)-3-Methyl-2-thiopentanamido-Gruppe in ein Thiazolo[3,2-a]azepin-Grundgrüst in der 6-Position
erhalten wird, und Verbindungen mit einem Substituenten, wie beispielsweise
einer C1-6-Alkylgruppe am Thiazol[3,2-a]azepin-Grundgerüst in der
9-Position. Obwohl die oben genannte JP-A-6- und die EP-Schrift als
Stand der Technik Verbindungen mit einem Thiazolo[3,2-a]azepin-Grundgerüst vorschlagen,
ist in den darin offenbarten Verbindungen ein am Thiazolo[3,2-a]-azepin-Grundgerüst in der
6-Position vorhandener Substituent höchstens eine Benzylgruppe,
und sie offenbaren keine erfindungsgemässe Gruppe, die die folgende spezifische
sterische Struktur aufweist:
-
-
Die hiesigen Erfinder haben die Einführung einer
(2S,3S)-3-Methyl-2-thiopentanamido-Gruppe
mit einer spezifischen Konfiguration in einen Thiazolo[3.2-a]azepin-Ring
in der 6-Position auf Grundlage von vollständig anderen Überlegungen
eingeführt,
und haben zufällig
herausgefunden, dass eine solche Einführung eine Verbindung liefert,
die als Doppelinhibitor gegen sowohl NEP als auch ACE im Vergleich
zu den im obigen Stand der Technik beschriebenen extrem exzellent
sind. Die Erfindung wurde auf Basis dieses Befundes erhalten.
-
Ferner sind die erfindungsgemässen Verbindungen
solche, die erhalten werden durch Einführen einer C1-6-Alkylgruppe
(am meisten bevorzugt einer Methylgruppe) in die 9-Position des
Thiazolo[3,2-a]azepin-Rings.
-
Folglich wurde die vorliegende Erfindung
auf Basis eines vollständig
neuen Konzepts erhalten, wonach die erfindungsgemässen überlegenen
Verbindungen (I) jeweils einen Thiazolo[3,2-a]azepin-Ring aufweisen,
dessen 6-Position mit
mit einer spezifischen Konfiguration
substituiert ist, und deren 9-Position mit einer C
1-6-Alkylgruppe,
wie beispielsweise einer Methylgruppe, substituiert ist. Durch die
Einführung
dieses neuen Konzepts wurden die erfindungsgemässen Verbindungen erhalten,
die exzellente Doppelinhibitoren darstellen.
-
Genauer haben die überlegenen
erfindungsgemässen
Verbindungen die Eigenschaften, dass sie nicht nur eine exzellente
doppelinhibitorische Wirkung aufweisen, sondern auch eine verbesserte
Bioverfügbarkeit, und
dass sie auch bei oraler Verabreichung eine exzellente Wirkung im
Vergleich zu Verbindungen aus dem Stand der Technik zeigen.
-
Unter diesen sind die folgenden Verbindungen
am meisten bevorzugt:
-
-
Obwohl die erfindungsgemässen Verbindungen
nach einem bekannten Verfahren oder einer Kombination bekannter
Verfahren hergestellt werden können,
war dies problematisch, da die Ausgangsverbindungen teuer sind und
die Vorgehensweise kompliziert ist. Daher haben die hiesigen Erfinder
nach einem Verfahren zur industriell vorteilhaften Herstellung der
erfindungsgemässen
Verbindungen gesucht. Als Ergebnis haben sie die nachfolgend beschriebenen
Herstellungsverfahren aufgefunden.
-
-
-
-
In der Formelreihe repräsentieren
R3, R4 und R5 jeweils unabhängig voneinander ein Wasserstoffatom, eine
Niederalkylgruppe, eine Niederalkoxygruppe, eine Niederalkylthiogruppe,
eine Arylgruppe, die einen Substituenten aufweisen kann, oder eine
Heteroarylgruppe, die einen Substituenten aufweisen kann, oder alternativ
dazu können
R3, R4 oder R5 zusammen mit dem Kohlenstoffatom, an das
sie gebunden sind, einen Ring bilden, mit der Massgabe, dass der
Fall, dass R3, R4 und
R5 alle Wasserstoffatome darstellen, ausgenommen ist;
R6 und R7 repräsentieren
jeweils unabhängig
voneinander ein Wasserstoffatom, eine Niederalkylgruppe, eine Arylgruppe,
die substituiert sein kann, oder eine Aralkylgruppe, die substituiert
sein kann; R1a ist eine Acylgruppe; R8a ist eine Schutzgruppe für eine Carboxylgruppe;
R12 ist eine Gruppe, die zusammen mit dem
endocyclischen Stickstoffatom ein Aldehydäquivalent bildet; Z ist eine
Acylgruppe oder eine Carbamatgruppe; und m und n haben die gleichen
Bedeutungen wie in der allgemeinen Formel (I).
-
1. Schritt:
-
Dieser Schritt umfasst die Acylierung
eines Pipecolinsäurederivats
(18) unter Erhalt eines N-Acylpipecolinsäurederivats (19). Die Verbindung
(19) kann nach einem herkömmlichen
Verfahren erhalten werden. Die Verbindung (19) kann erhalten werden
durch beispielsweise Umsetzung der Verbindung (18) mit einem Säureanhydrid,
wie beispielsweise Acetanhydrid, bei Raumtemperatur bis 100°C, durch
Umsetzung der Verbindung (18) mit einem Säurehalogenid, wie beispielsweise
Acetylchlorid und Benzoylchlorid, in Gegenwart einer Base, wie beispielsweise
Pyridin und Dimethylaminopyridin, bei 0°C bis Raumtemperatur, oder ferner
durch die sogenannte Schotten-Baumann-Reaktion,
die die Umsetzung der Verbindung (18) mit einem Säurehalogenid
in Gegenwart einer Base, z. B. Natriumhydroxid oder Natriumhydrogencarbonat,
umfasst.
-
2. Schritt:
-
Dieser Schritt umfasst die Veresterung
der Carbonsäure
des im 1. Schritt erhaltenen N-Acylpipecolinsäurederivats (19) unter Erhalt
eines Esters (20). Die Estergruppe ist vorzugsweise eine Gruppe,
die unter solchen Bedingungen entschützt werden kann, unter denen
herkömmliche
Alkylester während
der Entschützung des
Esters nicht hydrolysiert werden, beispielsweise ein t-Butylester,
ein Benzylester, der mit einer Methoxygruppe oder dergleichen substituiert
sein kann, und ein Alkylsilylethylester. Wenn ein t-Butylester hergestellt wird,
kann dieser hergestellt werden durch Umsetzung der Verbindung (19)
mit Isobutylen in einem organischen Lösungs mittel, wie beispielsweise
Dioxan und Tetrahydrofuran, in Gegenwart eines sauren Katalysators,
wie beispielsweise Schwefelsäure
und p-Toluolsulfonsäure,
oder durch Umsetzung der Verbindung (19) mit t-Butanol in Gegenwart
eines Kondensationsmittels, wie beispielsweise N,N'-Dicyclohexylcarbodiimid (DCC)
und 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimid-hydrochlorid (DEC).
wenn andererseits ein Ester, wie beispielsweise ein Benzylester,
ein Methoxybenzylester und ein Alkylsilylethylester, hergestellt
wird, so kann die Verbindung (20) erhalten werden durch Veresterung
mit einem Veresterungsmittel, wie beispielsweise einem Benzylhalogenid,
einem Methoxybenzylhalogenid und einem Alkylsilylethylhalogenid
in Gegenwart einer Base, wie beispielsweise Kaliumcarbonat, Natriumcarbonat
und einem Alkylamin, in einem inerten organischen Lösungsmittel,
wie beispielsweise Tetrahydrofuran, Dimethylformamid und Dichlormethan.
-
3. Schritt:
-
Dieser Schritt umfasst die elektrolytische
Oxidation des Pipecolinsäurederivats
(20), das im 2. Schritt erhalten wurde, unter Erhalt eines Hemiacetals
(21). Die elektrolytische Oxidation kann unter verschiedenen Bedingungen
durchgeführt
werden. Das Hemiacetal (V) kann beispielsweise erhalten werden durch
elektrolytische Oxidation der Verbindung (20) mit Platin, Kohlenstoff,
rostfreiem Stahl, Bleioxid oder dergleichen als Elektrode unter
Verwendung eines Elektrolyten als unterstützendem Elektrolyten, der die
elektrische Leitfähigkeit
in einem wässrigen
System oder einem organischen Lösungsmittelsystem
erhöht,
wie beispielsweise Tetraalkylammoniumperchlorate, z. B. Tetraethylammoniumperchlorat
oder Tetramethylammoniumperchlorat; Alkalimetallsalzen, z. B. Natriumperchlorat
oder Lithiumperchlorat; Tetraalkylammoniumsulfonaten, z. B. Tetraethylammonium-p-toluolsulfonat;
Tetraalkylammoniumtetrafluorboraten; und Tetraalkylammoniumhexafluorphosphaten,
in einem Lösungsmittel,
wie beispielsweise einem Wasser/Acetonitril-System, einem Wasser/Alkohol-System
und einem Wasser/Essigsäure-System.
Die hindurchgeführte
Strommenge beträgt
im allgemeinen 2 F oder mehr pro Mol an verwendeter Verbindung (20).
Insbesondere wenn Platin oder Kohlenstoff als Elektrode und Tetraethylammoniumperchlorat,
Tetraethylammoniumtetrafluorborat oder Tetramethylammoniumhexafluorphosphat
als unterstützender
Elektrolyt verwendet werden, werden bessere Ergebnisse erzielt.
-
4. Schritt:
-
Dieser Schritt umfasst die Umsetzung
des im 3. Schritt erhaltenen Hemiacetals (22) mit einem Cysteinesterderivat
(23) unter Erhalt eines Thiazolidinderivats (24). In der Praxis
kann das Thiazolidinderivat (24) erhalten werden durch Zugabe des
Cysteinesterderivats (23) zu dem Reaktionssystem nach der Beendigung des
3. Schritts ohne Isolierung des Hemiacetals (22). Wenn optisch aktives
L- oder D-Cystein als das in der Reaktion einzusetzende Cystein
verwendet wird, liegt die absolute Konfiguration der Carboxylgruppe
in der 4-Position des Thiazolidinrings der Verbindung (24) in der
R- oder S-Konfiguration vor.
-
5. Schritt:
-
Dieser Schritt umfasst die selektive
Entschützung
der Schutzgruppe der Carbonsäure,
die in dem Thiazolidinderivat (24) aus dem 4. Schritt durch R9 repräsentiert
wird, wodurch ein Carbonsäurederivat
(25) erhalten wird. Das Carbonsäurederivat
(25) kann erhalten werden durch Behandlung mit einem Ent-t-butylierungsmittel,
wie beispielsweise Trifluoressigsäure, Salzsäure und Iodtrimethylsilan,
wenn die Verbindung (24) ein t-Butylester ist, oder durch Mittel,
die üblicherweise
nur die entsprechende Ester-Schutzgruppe
entschützen, beispielsweise
katalytische Hydrierung, Salzsäure,
2,3-Dichlor-5,6-dicyano-1,4-benzochinon (DDQ) oder Tetraalkylammoniumfluorid,
wenn die Verbindung (24) ein Ester ist, wie beispielsweise ein Benzylester,
ein Methoxybenzylester und ein Alkylsilylethylester.
-
6. Schritt:
-
Dieser Schritt umfasst die Cyclisierung
des im 5. Schritt erhaltenen Thiazolidincarbonsäurederivats (25) durch Kondensation,
wodurch ein Thiazoloazepinderivat (26) erhalten wird. Die Cyclisierung
kann mit einem herkömmlichen
Kondensationsmittel durchgeführt
werden. Das cyclisierte Produkt (26) kann beispielsweise erhalten
werden durch Umsetzung der Verbindung (25) mit 1-Ethoxycarbonyl-2-ethoxy-1,2-dihydrochinon
(EEDQ), DCC, DEC oder dergleichen in einem Lösungsmittel, wie beispielsweise
Ethanol, Tetrahydrofuran und Dichlormethan.
-
7. Schritt:
-
Dieser Schritt umfasst die Entschützung der
N-Acetylgruppe des im 6. Schritt erhaltenen Thiazolazepinderivats
(26) unter Erhalt eines Aminosäurederivats
(27). Obwohl verschiedene Möglichkeiten
zur Entfernung der N-Acetylgruppe bekannt sind, kann das angestrebte
Aminosäurederivat
(27) beispielsweise erhalten werden durch Erwärmen in einer alkoholischen
Lösung,
einer verdünnten
Mineralsäure,
wie beispielsweise Salzsäure
und Schwefelsäure,
durch Behandlung. in einer alkoholischen Lösung von Natriumhydroxid, Kaliumhydroxid
oder dergleichen, oder durch Umsetzung mit Phos phorpentachlorid
oder Oxalylchlorid in Pyridin, gefolgt von der Behandlung mit einem
Alkohol.
-
8. Schritt:
-
Dieser Schritt umfasst die Kondensation
des Aminosäurederivats
(27) aus dem 7. Schritt mit einem Carbonsäurederivat der allgemeinen
Formel (29) oder einem aktiven Derivat davon, wie beispielsweise
einem Säurehalogenid
davon, unter Erhalt eines Amidderivats (30). Diese Kondensation
kann nach einem herkömmlichen
Verfahren durchgeführt
werden, und ein Beispiel hierfür
schliesst die Kondensation des Aminosäurederivats (27) mit dem Carbonsäurederivat
(29) in Gegenwart eines üblichen
Kondensationsmittels, z. B. EEDQ, DCC, DEC oder Diethylcyanophosphonat,
ein. Jedes organische Lösungsmittel,
das gegenüber
der Reaktion inert ist, kann als Reaktionslösungsmittel verwendet werden,
und Beispiele hierfür
schliessen Methylenchlorid und Tetrahydrofuran ein. Wenn die Reaktion über ein
Säurehalogenid,
wie beispielsweise ein Säurechlorid
des Carbonsäurederivats
(29), durchgeführt
wird, kann die Verbindung (30) durch Umsetzung des Carbonsäurederivats
(29) mit einem üblichen
Chlorierungsmittel, z. B. Thionylchlorid oder Oxalylchlorid, in
einem geeigneten inerten Lösungsmittel
unter Bildung eines Säurechlorids
davon und Umsetzung desselben mit dem Aminosäurederivat (27) erhalten werden.
-
9. Schritt:
-
Dieser Schritt umfasst die Hydrolyse
des im B. Schritt erhaltenen α-Acylthiocarbonsäureamidderivats (30)
unter Erhalt eines α-Mercaptocarbonsäureamidderivats
(31).
-
Es kann nach einem herkömmlichen
Hydrolyseverfahren hydrolysiert werden, d. h. in einer verdünnten wässrigen
Lösung
eines Alkalis, wie beispielsweise Natriumhydroxid und Lithiumhydroxid,
oder in einer verdünnten
wässrigen
Lösung
einer Mineralsäure.
-
10. Schritt:
-
Dieser Schritt umfasst die Acylierung
des im 9. Schritt erhaltenen α-Mercaptocarbonsäureamidderivats
(31) unter Erhalt eines α-Acylthiocarbonsäureamidderivats
(32).
-
Die Reaktion kann in herkömmlicher
Weise durchgeführt
werden. Das α-Acylcarbonsäureamidderivat (32)
kann beispielsweise erhalten werden durch Umsetzen des α-Mercaptocarbonsäureamidderivats
(31) mit einem Acylierungsmittel, wie beispielsweise einem Säureanhydrid,
z. B. Acetanhydrid, und einem Säurehalogenid
in einem nicht-wässrigen
Lösungsmittel,
wie beispielsweise Acetonitril, Tetrahydrofuran und Dichlormethan
in Gegenwart eines Katalysators, wie beispielsweise Kobaltchlorid,
oder durch Behandlung in Gegenwart einer Base, wie beispielsweise
Kaliumhydrogencarbonat, Natriumhydrogencarbonat und Triethylamin,
ebenfalls in einem wässrigen
Lösungsmittel.
-
Insbesondere können mit einem Acylierungsmittel,
das ein aktiver Ester ist, der hergestellt wird durch Umsetzung
einer Carbonsäure
mit Carbodiimidazol, bessere Ergebnisse erzielt werden.
-
In dem obigen Herstellungsverfahren
sind die Verbindungen der allgemeinen Formel (27) extrem wichtige
Zwischenprodukte zur Herstellung der erfindungsgemässen Verbindung.
-
-
-
In der Formelreihe repräsentieren
R3, R4 und R5 jeweils unabhängig voneinander ein Wasserstoffatom, eine
C1-6-Alkylgruppe, eine C1-6-Alkoxylgruppe
oder eine C1-6-Alkylthiogruppe, oder alternativ
dazu können
R3, R4 oder R5 zusammen mit dem Kohlenstoffatom, an das
sie gebunden sind, einen Ring bilden, mit der Massgabe, dass der
Fall, dass R3, R4 und
R5 alle Wasserstoffatome darstellen, ausgenommen
ist;
R6 und R7 sind
jeweils ein Wasserstoffatom; R1a ist eine
Acylgruppe; R8a ist eine Schutzgruppe für eine Carboxylgruppe;
X ist eine Abgangsgruppe, wie beispielsweise ein Halogenatom, eine
Methansulfonyloxygruppe oder eine p-Toluolsulfonyloxygruppe; und
m und n haben die gleichen Bedeutungen wie in der allgemeinen Formel
(I).
-
1. Schritt:
-
Dieser Schritt umfasst die Kondensation
des im Herstellungsverfahren A erhaltenen Aminosäurederivats mit einem Carbonsäurederivat
der allgemeinen Formel (33) oder einem aktiven Derivat davon, wie
beispielsweise einem Säurehalogenid
davon, unter Erhalt eines Amidderivats (34). Diese Kondensation
wird in der gleichen Weise durchgeführt wie im B. Schritt des Herstellungsverfahrens
A, ausser dass ein α-Hydroxycarbonsäurederivat
(33) anstelle des Carbonsäurederivats
(29) verwendet wird.
-
2. Schritt:
-
Dieser Schritt umfasst die Halogenierung
des im 1. Schritt erhaltenen Hydroxycarbonsäureamidderivats (34) unter
Erhalt eines α-Halogencarbonsäureamidderivats
(35). Zur Halogenierung der Verbindung (34) unter sterischer Inversion
der Hydroxylgruppe sind verschiedene Verfahren bekannt, beispielsweise
(i) ein Verfahren, das deren Umsetzung mit Dialkylazodicarboxylat,
Triphenylphosphin und entweder Zinkbromid oder Zinkoxid in einem
organischen Lösungsmittel,
wie beispielsweise Tetrahydrofuran, umfasst, (ii) ein Verfahren, das
deren Umsetzung mit einer Organophosphorverbindung, wie beispielsweise
Trialkylphosphin, Triphenylphosphin und Triphenylphosphit, und einer
Halogenverbindung, wie beispielsweise N-Halogensuccinimid und Brom/Iod,
in einem organischen Lösungsmittel,
wie beispielsweise Acetonitril, Dimethylformamid und Dichlormethan,
in Gegenwart oder Abwesenheit einer Base, wie beispielsweise Pyridin,
umfasst, und (iii) ein Verfahren, das deren Umsetzung mit Tosylchlorid,
Trifluormethansulfonsäureanhydrid
oder dergleichen in einem inerten Lösungsmittel, wie beispielsweise
Dichlormethan, in Gegenwart einer Base, wie beispielsweise Pyridin und
Triethylamin, unter Bildung eines Sulfonsäureesters, gefolgt von der
Umsetzung mit einem Halogenierungsmittel, wie beispielsweise Lithiumhalogenid,
um fasst. Insbesondere ist ein Verfahren, in dem Triphenylphosphin
und Brom unter den Bedingungen (ii) verwendet werden, bevorzugt.
-
3. Schritt:
-
Dieser Schritt umfasst die Einführung einer
Acylthiogruppe in das im 2. Schritt erhaltene α-Halogencarboxylamidderivat
(35) unter Erhalt eines α-Acylthiocarboxylamidderivats
(36).
-
Die Reaktion kann in herkömmlicher
Weise durchgeführt
werden. Das α-Acylthiocarboxylamidderivat (36)
kann beispielsweise erhalten werden durch Umsetzen des α-Halogencarboxylamidderivats
(35) mit einem Thiocarbonsäuresalz,
wie beispielsweise Kaliumthioacetat oder Natriumthioacetat, in einem
polaren Lösungsmittel,
wie beispielsweise Acetonitril und Aceton, oder durch Umsetzen der
Verbindung (35) mit einer Thiocarbonsäure, wie beispielsweise Thioessigsäure und
Thiobenzoesäure,
in Gegenwart eine Base, wie beispielsweise Kaliumcarbonat und Cäsiumcarbonat.
-
4. Schritt:
-
Dieser Schritt umfasst die Hydrolyse
des im 3. Schritt erhaltenen α-Acylthiocarboxylamidderivats
(36) unter Erhalt eines α-Mercaptocarboxylamidderivats
(37). Es kann gemäss
einer herkömmlichen
Hydrolyse hydrolysiert werden, d. h. in einer verdünnten wässrigen
Lösung
eines Alkalis, wie beispielsweise Natriumhydroxid und Lithiumhydroxid,
oder in einer verdünnten
wässrigen
Lösung
einer Mineralsäure.
-
5. Schritt:
-
Dieser Schritt umfasst die Acylierung
des im 4. Schritt erhaltenen α-Mercaptocarboxylamidderivats (37)
unter Erhalt eines α-Acylthiocarboxylamidderivats
(38).
-
Die Reaktion wird in herkömmlicher
Weise durchgeführt.
Das α-Acylthiocarboxylamidderivat
(38) kann beispielsweise erhalten werden durch Umsetzen des α-Mercaptocarboxylamidderivats
(37) mit einem Acylierungsmittel, wie beispielsweise einem Säureanhydrid,
z. B. Acetanhydrid, und einem Säurehalogenid
in einem nicht-wässrigen
Lösungsmittel,
wie beispielsweise Acetonitril, Tetrahydrofuran und Dichlormethan,
in Gegenwart eines Katalysators, wie beispielsweise Kobaltchlorid,
oder durch Behandlung in Gegenwart einer Base, wie beispielsweise
Kaliumhydrogencarbonat, Natriumhydrogencarbonat und Triethylamin,
ebenfalls in einem wässrigen
Lösungsmittel.
-
Insbesondere können mit einem aktiven Ester,
der durch Umsetzung einer Carbonsäure mit Carbodiimidazol hergestellt
wird, als Acylierungsmittel bessere Ergebnisse erzielt werden.
-
-
In der Formelreihe repräsentieren
R3, R4 und R5 jeweils unabhängig voneinander ein Wasserstoffatom, eine
C1-6-Alkylgruppe, eine C1-6-Alkoxylgruppe
oder eine C1-6-Alkylthiogruppe, oder alternativ
dazu können
R3, R4 oder R5 zusammen mit dem Kohlenstoffatom, an das
sie gebunden sind, einen Ring bilden, mit der Massgabe, dass der
Fall, dass R3, R4 und
R5 alle Wasserstoffatome darstellen, ausgenommen
ist;
R6 und R7 sind
jeweils ein Wasserstoffatom; R1a ist eine
Acylgruppe; R8a ist eine Schutzgruppe für eine Carboxylgruppe;
X ist eine Abgangsgruppe, wie beispielsweise ein Halogenatom, eine
Methansulfonyloxygruppe oder eine p-Toluolsulfonyloxygruppe; und
m und n haben die gleichen Bedeutungen wie in der allgemeinen Formel
(I).
-
1. Schritt:
-
Dieser Schritt umfasst die Hydrolyse
der Estergruppe des Halogenids (35), das im Herstellungsverfahren
2 erhalten wurde, unter Erhalt eines Carbonsäurederivats (39). Die Hydrolyse
kann gemäss
einer herkömmlichen
Hydrolyse durchgeführt
werden, d. h. in einer verdünnten
wässrigen
Lösung
eines Alkalis, wie beispielsweise Natriumhydroxid und Lithiumhydroxid,
oder in einer verdünnten
wässrigen
Lösung
einer Mineralsäure.
-
2. Schritt:
-
Dieser Schritt umfasst die Einführung einer
Acylthiogruppe in das im 1. Schritt erhaltene α-Halogencarboxylamidderivat
(39) unter Erhalt eines α-Acylthiocarboxylamidderivats
(38). Die Reaktion kann in herkömmlicher
Weise durchgeführt
werden. Das α-Acylthiocarboxylamidderivat
(38) kann beispielsweise erhalten werden durch Umsetzen des α-Halogencarboxylamidderivats
(38) mit einem Thiocarbonsäuresalz,
wie beispielsweise Kaliumthioacetat und Natriumthioacetat, in einem
polaren Lösungsmittel,
wie beispielsweise Acetonitril, Dimethylsulfoxid und Aceton, oder
durch Umsetzen des Derivats (38) mit einer Thiocarbonsäure, wie beispielsweise
Thioessigsäure
und Thiobenzoesäure,
in Gegenwart eine Base, wie beispielsweise Kaliumcarbonat und Cäsiumcarbonat.
-
-
In den Formeln haben R2 und
m die oben beschriebenen Bedeutungen.
-
Dieser Schritt umfasst den Austausch
der Aminogruppe einer natürlichen
oder nicht-natürlichen
Aminosäure
(40) durch eine Hydroxylgruppe unter Erhalt einer α-Hydroxycarbonsäure (33).
Der Austausch durch eine Hydroxylgruppe wird entweder durch Umsetzen
der Aminosäure
(40) mit einem Nitrierungsmittel, wie beispielsweise Natriumnitrit
in verdünnter
Schwefelsäure
oder durch Umsetzen der Aminosäure
(40) mit Natriumnitrit in Essigsäure
unter Bildung eines Acetats und nachfolgender Hydrolyse durchgeführt.
-
Für
den Fall, dass bei den unter den erfindungsgemässen Verbindungen bevorzugten
Verbindungen (I')
R4 und R5 jeweils
Wasserstoffatome darstellen, ist bereits ein Verfahren bekannt,
worin die Verbindung erhalten wird, indem die Verbindung (II) und
die Verbindung (i), (ro) oder (ha) der Amidierung unterworfen wird.
-
-
-
Man kann jedoch kaum sagen, dass
das obige Verfahren industriell vorteilhaft ist, da alle obigen
Verbindungen (i), (ro) und (ha) teuer sind und D-Allo-isoleucin,
dessen Massenproduktion sehr arbeitsaufwendig ist, als Ausgangsmaterial
verwendet wird. Die nachfolgend beschriebenen Verfahren sind industriell
vorteilhafte Verfahren, nach denen die Verbindung (I') in hoher Ausbeute
in einer vorteilhaften Vorgehensweise erhalten werden kann.
-
-
-
1. Schritt:
-
Dieser Schritt umfasst die Hydroxylierung
der Aminogruppe von L-Isoleucin (9) in einer herkömmlichen Weise
unter erhalt einer α-Hydroxycarbonsäure (10).
Obwohl die Hydroxylierung nach einem herkömmlicherweise angewandten Verfahren
durchgeführt
werden kann, wird sie vorzugsweise entweder durch Umsetzung von
L-Isoleucin (9) mit einem Nitrierungsmittel, wie beispielsweise
Natriumnitrit, in verdünnter
Schwefelsäure oder
durch Umsetzung des L-Isoleucins (9) mit Natriumnitrit in Essigsäure unter
Bildung eines Acetats und nachfolgender Hydrolyse durchgeführt.
-
2. Schritt:
-
Dieser Schritt umfasst die Kondensation
der im 1. Schritt erhaltenen α-Hydroxycarbonsäure (10)
mit einem Aminderivat (11) in herkömmlicher Weise unter Erhalt
eines Hydroxycarboxylamidderivats (12).
-
Die Reaktion kann nach einem herkömmlichen
Verfahren durchgeführt
werden. Das Amidderivat (12) kann beispielsweise erhalten werden
durch Umsetzen der α-Hydroxycarbonsäure (10)
mit dem Aminderivat (11) in Gegenwart eines üblichen Kondensationsmittels,
beispielsweise EEDQ, DCC, DEC oder Diethylcyanophosphonat, in einem
inerten Lösungsmittel,
wie beispielsweise Methylenchlorid und Tetrahydrofuran.
-
3. Schritt:
-
Dieser Schritt umfasst die Halogenierung
des Hydroxycarboxylamidderivats (12) in herkömmlicher Weise unter Erhalt
eines α-Halogencarboxylamidderivats
(13).
-
Es kann ein beliebiges übliches
Verfahren angewandt werden, so lange es ein Verfahren ist, das die Halogenierung
unter sterischer Inversion erzielt. Beispiele für solche Verfahren sind beispielsweise
(i) ein Verfahren, das deren Umsetzung mit Dialkylazodicarboxylat,
Triphenylphosphin und entweder Zinkbromid oder Zinkoxid in einem
organischen Lösungsmittel,
wie beispielsweise Tetrahydrofuran, umfasst, (ii) ein Verfahren, das
deren Umsetzung mit einer Organophosphorverbindung, wie beispielsweise
Trialkylphosphin, Triphenylphosphin und Triphenylphosphit, und einer
Halogenverbindung, wie beispielsweise N-Halogensuccinimid und Brom/Iod,
in einem organischen Lösungsmittel,
wie beispielsweise Acetonitril, Dimethylformamid und Dichlormethan,
in Gegenwart oder Abwesenheit einer Base, wie beispielsweise Pyridin,
umfasst, und (iii) ein Verfahren, das deren Umsetzung mit Tosylchlorid,
Trifluormethansulfonsäureanhydrid
oder dergleichen in einem inerten Lösungsmittel, wie beispielsweise
Dichlormethan, in Gegenwart einer Base, wie beispielsweise Pyridin und
Triethylamin, unter Bildung eines Sulfonsäureesters, gefolgt von der
Umsetzung mit einem Halogenierungsmittel, wie beispielsweise Lithiumhalogenid,
umfasst. Besonders bevorzugt ist ein Verfahren, das die Umsetzung
mit einer Organophosphorverbindung, wie beispielsweise einem Trialkylphosphin,
Triphenylphosphin und Triphenylphosphit, und einer Halogenverbindung,
wie beispielsweise N-Halogensuccinimid und Brom/Iod, in einem organischen
Lösungsmittel,
wie beispielsweise Acetonitril, Dimethylformamid und Dichlormethan,
in Gegenwart oder Abwesenheit einer Base, wie beispielsweise Pyridin,
umfasst. Das Verfahren, das die Verwendung von Triphenylphosphin
und Brom als Reagenzien umfasst, ist besonders bevorzugt.
-
4. Schritt:
-
Dieser Schritt umfasst die Einführung einer
Acylthiogruppe in das im 2. Schritt erhaltene α-Halogencarboxylamidderivat
(13) unter Erhalt eines α-Acylthiocarboxylamidderivats
(8a).
-
Die Reaktion kann in herkömmlicher
Weise durchgeführt
werden. Das α-Acylthiocarboxylamidderivat (8a)
kann beispielsweise erhalten werden durch Umsetzen des α-Halogencarboxylamidderivats
(13) mit einem Thiocarbonsäuresalz,
wie beispielsweise Kaliumthioacetat oder Natriumthioacetat, in einem
polaren Lösungsmittel,
wie beispielsweise Acetonitril und Aceton, oder durch Umsetzen des
Derivats (13) mit einer Thiocarbonsäure, wie beispielsweise Thioessigsäure und
Thiobenzoesäure,
in Gegenwart eine Base, wie beispielsweise Kaliumcarbonat und Cäsiumcarbonat.
-
5. Schritt:
-
Dieser Schritt wird durchgeführt, wenn
R1 und R8 Wasserstoffatome
sind oder wenn R1 eine Acylgruppe und R8 ein Wasserstoffatom ist. Anders gesagt
handelt es sich um einen Schritt, in dem ein (2S,3S)-3-Methyl-2-thiopentansäurederivat
(8b) erhalten wird durch Hydrolyse des im 4. Schritt erhaltenen α-Acylthiocarboxylamidderivats
(8a) in einer herkömmlichen
Wiese.
-
Die Hydrolyse kann gemäss einer
herkömmlichen
Hydrolyse durchgeführt
werden, d. h. in einer verdünnten
wässrigen
Lösung
eines Alkalis, wie beispielsweise Natriumhydroxid und Lithiumhydroxid,
oder in einer verdünnten
wässrigen
Lösung
einer Mineralsäure.
Wenn die angestrebte Verbindung eine solche ist, in der R1 eine Acylgruppe ist, wird der nachfolgende
6. Schritt unter Verwendung des erhaltenen (2S,3S)-3-Methyl-2-thio-pentansäurederivats
(8b) durchgeführt.
-
6. Schritt:
-
Dieser Schritt wird durchgeführt, wenn
die Zielverbindung eine solche ist, in der R1 eine
Acylgruppe ist. Anders ausgedrückt
handelt es sich um einen Schritt, der die Acylierung des im 5. Schritt
erhaltenen (2S,3S)-3-Methyl-2-thiopentansäurederivats
(8b) in herkömmlicher
Weise unter Erhalt eines α-Acylthiocarboxylamidderivats
(8c) umfasst.
-
Die Reaktion kann nach einem in herkömmlicher
Weise angewandten Verfahren durchgeführt werden. Das α-Acylthiocarboxylamidderivat
(8c) kann beispielsweise erhalten werden durch Umsetzen des α-Mercaptocarboxylamidderivats
(8b) mit einem Acylierungsmittel, wie beispielsweise einem Säureanhydrid,
z. B. Acetanhydrid, und einem Säurehalogenid
in einem nichtwässrigen
Lösungsmittel,
wie beispielsweise Acetonitril, Tetrahydrofuran und Dichlormethan,
oder durch dessen Behandlung in Gegenwart einer Base, wie beispielsweise
Kaliumhydrogencarbonat, Natriumhydrogencarbonat und Triethylamin,
oder Kobaltchlorid, ebenfalls in einem wässrigen Lösungsmittel.
-
Die Zielverbindung kann auch nach
dem unten beschriebenen Verfahren hergestellt werden, nach dem das α-Halogenhydroxycarboxylamidderivat
(13) durch Hydroxylierung von L-Isoleucin und Kondensation mit dem
Aminderivat (11) nach dem Herstellungsverfahren 5 erhalten wurde.
-
-
1. Schritt:
-
Dieser Schritt umfasst die Hydrolyse
des im 3. Schritt des Herstellungsverfahrens 5 erhaltenen α-Halogencarboxylamidderivats
(13) in einer herkömmlichen
Weise unter Erhalt einer Carbonsäure
(14).
-
Die Hydrolyse kann gemäss einer
herkömmlichen
Hydrolyse durchgeführt
werden, d. h. in einer verdünnten
wässrigen
Lösung
eines Alkalis, wie beispielsweise Natriumhydroxid und Lithiumhydroxid,
oder in einer verdünnten
wässrigen
Lösung
einer Mineralsäure.
-
2. Schritt:
-
Dieser Schritt umfasst die Einführung einer
Acylthiogruppe in das im 1. Schritt erhaltene α-Halogencarboxylamidderivat
(14) unter Erhalt eines α-Acylthiocarboxylamidderivats
(8c). Die Reaktion kann in herkömmlicher
Weise durchgeführt
werden. Das α-Acylthiocarboxylamidderivat
(8c) kann beispielsweise erhalten werden durch Umsetzen des α-Halogencarboxylamidderivats
(14) mit einem Thiocarbonsäuresalz,
wie beispielsweise Kaliumthioacetat und Natriumthioacetat in einem
polaren Lösungsmittel,
wie beispielsweise Acetonitril, Dimethylsulfoxid und Aceton, oder
durch Umsetzen des Derivats (14) mit einer Thiocarbonsäure, wie beispielsweise
Thioessigsäure
und Thiobenzoesäure,
in Gegenwart eine Base, wie beispielsweise Kaliumcarbonat und Cäsiumcarbonat.
-
US
4 415 496 und
US 4 617
301 offenbaren unter den Aminen der allgemeinen Formel
(II) die Amine (II'''), in denen R
3,
R
4 und R
5 alle Wasserstoffatome
darstellen. Als Verfahren zur Herstellung dieser Amine (II''') sind
bisher beispielsweise ein in
US
4 415 496 beschriebenes Verfahren bekannt, das (S)-2-Amino-6-hydroxyhexansäure als
Ausgangsmaterial verwendet, sowie ein in
US 4 617 301 und
US 5 118 810 beschriebenes Verfahren,
das ε-N-BOC-L-Lysin
als Ausgangsmaterial verwendet. Man kann jedoch kaum sagen, dass
diese Vorgehensweisen vorteilhaft sind, da die Ausgangsmaterialien
schwer verfügbar
sind, und sie erfordern zahlreiche Stufen, sowie in all diesen ein
Ionenaustauscherharz und viel Raney-Nickel. Die nachfolgend beschriebenen
Herstellungsverfahren sind solche, die nicht nur die Herstellung
der Amine (II'''), deren industrielle Herstellung in
verfahrenstechnischer wie industrieller Hinsicht beschränkt war,
sondern auch die Herstellung der Amine, in denen eines oder zwei
oder mehr von R
3, R
4 und
R
5 kein Wasserstoffatom darstell(t/en),
die nach den als Herstellungsverfahren für die Amine (II''')
offenbarten Herstellungsverfahren nicht leicht hergestellt werden
konnten, zu niedrigen Kosten in hoher Ausbeute in verfahrenstechnisch
vorteilhafter weise ermöglichen.
-
-
-
In der Formelreihe repräsentieren
R3, R4 und R5 jeweils unabhängig voneinander ein Wasserstoffatom, eine
C1-6-Alkylgruppe, eine C1-6-Alkoxylgruppe
oder eine C1-6-Alkylthiogruppe oder alternativ
dazu können
R3, R4 oder R5 zusammen mit dem Kohlenstoffatom, an das
sie gebunden sind, einen Ring bilden, mit der Massgabe, dass der
Fall, dass R3, R4 und
R5 alle Wasserstoffatome darstellen, ausgenommen
ist;
R6 und R7 repräsentieren
jeweils unabhängig
voneinander ein Wasserstoffatom; R2a ist
eine Schutzgruppe für eine
Carboxylgruppe; R12 ist eine Gruppe, die
zusammen mit dem endocyclischen Stickstoffatom ein Aldehydäquivalent
bildet; Z ist eine Acylgruppe oder eine Carbamatgruppe.
-
1. Schritt:
-
Dieser Schritt umfasst die Acylierung
eines optisch aktiven (2S)-Pipecolinsäurederivats
(15) unter Erhalt eines N-Acylpipecolinsäurederivats (16). Die Verbindung
(16) kann nach einer herkömmlichen
Acylierung erhalten werden. Die Verbindung (16) kann erhalten werden
durch beispielsweise Umsetzung der Verbindung (15) mit einem Säureanhydrid,
wie beispielsweise Acetanhydrid, bei Raumtemperatur bis 100°C, durch
Umsetzung der Verbindung (15) mit einem Säurehalogenid, wie beispielsweise
Acetylchlorid und Benzoylchlorid, in Gegenwart einer Base, wie beispielsweise
Pyridin und Dimethylaminopyridin, bei 0°C bis Raumtemperatur, oder ferner
durch die sogenannte Schotten-Baumann-Reaktion, die die Umsetzung
der Verbindung (15) mit einem Säureanhydrid
oder einem Säurehalogenid
in Gegenwart einer Base, z. B. Natriumhydroxid, Natriumcarbonat
oder Natriumhydrogencarbonat, umfasst.
-
2. Schritt:
-
Dieser Schritt umfasst die Veresterung
der Carbonsäure
des im 1. Schritt erhaltenen N-Acylpipecolinsäurederivats (16) unter Erhalt
eines Esters (2').
Die Estergruppe ist vorzugsweise eine Gruppe, die unter solchen
Bedingungen entschützt
werden kann, unter denen herkömmliche
Alkylester während
der Entschützung des
Esters nicht hydrolysiert werden, beispielsweise ein t-Butylester,
ein Benzylester, der mit einer Methoxygruppe oder dergleichen substituiert
sein kann, und ein Alkylsilylethylester. wenn ein t-Butylester hergestellt wird,
kann dieser hergestellt werden durch Umsetzung der Verbindung (16)
mit Isobutylen in einem etherischen Lösungsmittel, wie beispielsweise
Dioxan und Tetrahydrofuran, oder in einem organischen Lösungsmittel,
wie Dichlormethan, in Gegenwart eines sauren Katalysators, wie beispielsweise
Schwefelsäure
und p-Toluolsulfonsäure,
oder durch Umsetzung der Verbindung (16) mit t-Butanol in Gegenwart
eines Kondensationsmittels, wie beispielsweise Dicyclohexylazodicarboxylat
(DCC) und 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimid-hydrochlorid
(DEC). Wenn andererseits ein Ester, wie beispielsweise ein Benzylester,
ein Methoxybenzylester und ein Alkylsilylethylester, hergestellt
wird, so kann die Verbindung (2')
erhalten werden durch Veresterung mit einem Veresterungsmittel,
wie beispielsweise einem Benzylhalogenid, einem Methoxybenzylhalogenid
und einem Alkylsilylethylhalogenid, in Gegenwart einer Base, wie
beispielsweise Kaliumcarbonat, Natriumcarbonat und einem Alkylamin,
in einem inerten organischen Lösungsmittel,
wie beispielsweise Tetrahydrofuran, Dimethylformamid und Dichlormethan.
-
3. Schritt:
-
Dieser Schritt umfasst die elektrolytische
Oxidation des Pipecolinsäurederivats
(2'), das im 2.
Schritt erhalten wurde, unter Erhalt eines Hemiacetals (3'). Die elektrolytische
Oxidation kann unter verschiedenen Bedingungen durchgeführt werden.
Das Hemiacetal (3')
kann beispielsweise erhalten werden durch elektrolytische Oxidation
der Verbindung (2')
mit Platin, Kohlenstoff, rostfreiem Stahl, Bleioxid oder dergleichen
als Elektrode unter Verwendung eines Elektrolyten als unterstützendem
Elektrolyten, der die elektrische Leitfähigkeit in einem wässrigen
System oder einem organischen Lösungsmittelsystem
erhöht,
wie beispielsweise Tetraalkylammoniumperchlorat, z. B. Tetraethylammoniumperchlorat
oder Tetramethylammoniumperchlorat; Alkalimetallsalzen, z. B. Natriumperchlorat
oder Lithiumperchlorat; Tetraalkylammoniumsulfonaten, z. B. Tetraethylammonium-p-toluolsulfonat;
Tetraalkylammoniumtetrafluorboraten; und Tetraalkylammoniumhexafluorphosphaten,
in einem Lösungsmittel,
wie beispielsweise einem Wasser/Acetonitril-System, einem Wasser/Alkohol-System
und einem Wasser/Essigsäure-System. Die hindurchgeführte Strommenge
beträgt
im allgemeinen 2 F oder mehr pro Mol an verwendeter Verbindung (2'). Insbesondere wenn
Platin oder Kohlenstoff als Elektrode und Tetraethylammoniumperchlorat,
Tetraethylammoniumtetrafluorborat, Tetramethylammoniumhexafluorphosphat
oder Tetraethylammonium-p-toluolsulfonat als unterstützender
Elektrolyt verwendet werden, werden bessere Ergebnisse erzielt.
-
4. Schritt:
-
Dieser Schritt umfasst die Umsetzung
des im 3. Schritt erhaltenen Hemiacetals (3) mit einem L-Cysteinesterderivat
(4) unter Erhalt eines Thiazolidinderivats (5). Das Thiazolidinderivat
(5) kann erhalten werden durch Zugabe des L-Cysteinesterderivats
(4) zu dem Reaktionssystem nach Beendigung des 3. Schritts ohne Isolierung
des Hemiacetals (3) zur Durchführung
der Behandlung.
-
5. Schritt:
-
Dieser Schritt umfasst die selektive
Entschützung
der Schutzgruppe der Carbonsäure,
die in dem Thiazolidinderivat (5')
aus dem 4. Schritt durch R11 repräsentiert
wird, wodurch ein Carbonsäurederivat
(6') erhalten wird.
Das Carbonsäurederivat
(6') kann erhalten
werden durch Behandlung mit einem Ent-t-butylierungsmittel, wie
beispielsweise Trifluoressigsäure,
Salzsäure
und Iodtrimethylsilan, wenn die Verbindung (5') ein t-Butylester ist, oder durch Mittel,
die üblicherweise
nur die entsprechende Ester-Schutzgruppe
entschützen, beispielsweise
katalytische Hydrierung, Salzsäure,
2,3-Dichlor-5,6-dicyano-1,4-benzochinon (DDQ) oder Tetraalkylammoniumfluorid,
wenn die Verbindung (5')
ein Ester ist, wie beispielsweise ein Benzylester, ein Methoxybenzylester
und ein Alkylsilylethylester.
-
6. Schritt:
-
Dieser Schritt umfasst die Cyclisierung
des im 5. Schritt erhaltenen Thiazolidincarbonsäurederivats (6') durch Kondensation,
wodurch ein Aminosäurederivat
(7') erhalten wird.
Die Cyclisierung kann mit einem herkömmlichen Kondensationsmittel
durchgeführt
werden. Das Aminosäurederivat
(7') als cyclisiertes
Produkt kann beispielsweise erhalten werden durch Umsetzung der
Verbindung (6')
mit 2-Ethoxycarbonyl-1-ethoxy-1,2-dihydrochinon (EEDQ), DCC, DEC
oder dergleichen in einem Lösungsmittel,
wie beispielsweise Ethanol, Tetrahydrofuran und Dichlormethan.
-
7. Schritt:
-
Dieser Schritt umfasst die Entschützung der
N-Acetylgruppe in dem im 6. Schritt erhaltenen Aminosäurederivat
(7') unter Erhalt
eines Aminosäurederivats
(1'). Obwohl verschiedene
Möglichkeiten
zur Entfernung der N-Acetylgruppe bekannt sind, kann das angestrebte
Aminosäurederivat
beispielsweise erhalten werden durch Erwärmen in einer alkoholischen
Lösung,
einer verdünnten
Mineralsäure,
wie beispielsweise Salzsäure
und Schwefelsäure,
durch Behandlung in einer alkoholischen Lösung von Natriumhydroxid, Kaliumhydroxid
oder dergleichen, oder durch Umsetzung mit Phosphorpentachlorid
oder Oxalylchlorid in Pyridin, gefolgt von der Behandlung mit einem
Alkohol.
-
HERSTELLUNGSVERFAHREN
B
-
Der 1. und 2. Schritt des Herstellungsverfahrens
A können
auch nach der folgenden Vorgehensweise durchgeführt werden:
-
-
In der Formelreihe repräsentieren
R3, R4 und R5 jeweils unabhängig voneinander ein Wasserstoffatom, eine
C1-6-Alkylgruppe, eine C1-6-Alkoxylgruppe
oder eine C1-6-Alkylthiogruppe, oder alternativ
dazu können
R3, R4 und R5 zusammen mit dem Kohlenstoffatom, an das
sie gebunden sind, einen Ring bilden, mit der Massgabe, dass der
Fall, dass R3, R4 und
R5 alle Wasserstoffatome darstellen, ausgeschlossen
ist;
R11 ist eine Schutzgruppe für eine Carboxylgruppe;
und Z ist eine Acylgruppe oder eine Carbamatgruppe.
-
1. Schritt:
-
Dieser Schritt umfasst die t-Butylveresterung
eines optisch aktiven (2S)-Pipecolinsäurederivats (15) unter Erhalt
eines Esters (17). Der Ester (15) kann in der gleichen Weise erhalten
werden wie im 2. Schritt des Her stellungsverfahrens A beschrieben,
d. h. durch Umsetzung der Verbindung (2) mit Isobutylen in einem
organischen Lösungsmittel,
wie beispielsweise Dioxan und Tetrahydrofuran, in Gegenwart eines
sauren Katalysators, wie beispielsweise Schwefelsäure und
p-Toluolsulfonsäure,
oder durch Umsetzung der Verbindung (2) mit t-Butanol in Gegenwart
eines Kondensationsmittels, wie beispielsweise Dicyclohexylazodicarboxylat (DCC)
und 1-(3-Dimethylaminopropyl)-3-ethylcarbodiimid (DEC).
-
2. Schritt:
-
Dieser Schritt umfasst die Acylierung
des Stickstoffatoms des im 1. Schritt erhaltenen Esters (17) unter Erhalt
eines Acylpipecolinsäurederivats
(2). Die Verbindung (2) kann in der gleichen Weise erhalten werden wie
im 1. Schritt des Herstellungsverfahrens A beschrieben. Das heisst,
die Verbindung (2) kann erhalten werden durch Umsetzung der Verbindung
(17) mit einem Säureanhydrid,
wie beispielsweise Acetanhydrid, bei Raumtemperatur bis 100°C, oder durch
Umsetzung der Verbindung (15) mit einem Säurehalogenid, wie beispielsweise
Acetylchlorid und Benzoylchlorid, in Gegenwart einer Base, wie beispielsweise
Pyridin und Dimethylaminopyridin, bei 0°C bis Raumtemperatur, oder ferner
durch die sogenannte Schotten-Baumann-Reaktion, die die Umsetzung der Verbindung
(15) mit einem Säurehalogenid
in Gegenwart einer Base, z. B. Natriumhydroxid oder Natriumhydrogencarbonat,
umfasst.
-
HERSTELLUNGSVERFAHREN
C
-
Wenn R5 eine
verzweigte Alkylgruppe ist, kann die Herstellung auch nach dem folgenden
Verfahren erfolgen:
-
-
-
1. Schritt:
-
Dieser Schritt umfasst die elektrolytische
Oxidation des Pipecolinsäurederivats
(18'), das in herkömmlicher
Weise erhalten wurde, unter Erhalt eines Hemiacetals (19'). Die elektrolytische
Oxidation kann unter verschiedenen Bedingungen durchgeführt werden.
Das Hemiacetal (19')
kann beispielsweise erhalten werden durch elektrolytische Oxidation
der Verbindung (18')
mit Platin, Kohlenstoff, rostfreiem Stahl, Bleioxid oder dergleichen
als Elektrode unter Verwendung eines Alkalimetallsalzes, wie beispielsweise
Tetraethylammoniumperchlorat und Tetramethylammoniumperchlorat,
eines Tetraalkylammoniumhexafluorphosphats, wie beispielsweise Tetraethylammonium-p-toluolsulfonat
oder dergleichen, als unterstützendem
Elektrolyten in einem Lösungsmittel,
wie beispielsweise einem Wasser/Alkohol-System und einem Wasser/Essigsäure-System.
Die hindurchgeführte
Strommenge beträgt
im allgemeinen 2 F oder mehr pro Mol an verwendeter Verbindung (18'). Insbesondere wenn
Platin oder Kohlenstoff als Elektrode und Tetraethylammoniumtetrafluorborat
oder Tetramethylammoniumhexafluorphosphat als unterstützender
Elektrolyt verwendet werden, werden bessere Ergebnisse erzielt.
-
2. Schritt:
-
Dieser Schritt umfasst die Durchführung der
1,2-Eliminierung mit dem im 1. Schritt erhaltenen Hemiacetal (19') unter Erhalt eines
Iminoderivats (40). Die Verbindung (40) kann durch herkömmliche
Eliminierung, wie beispielsweise saure Katalyse, und eine thermische
Reaktion erhalten werden.
-
3. Schritt:
-
Dieser Schritt umfasst die Acylierung
des im 2. Schritt erhaltenen Iminoderivats (40) unter Erhalt eines Ketons
(41). Im allgemeinen können
verschiedene Acylgruppen unter Ausnutzung der elektrophilen Substitution
mit der Iminogruppe eingeführt
werden. Das Keton (41) kann beispielsweise erhalten werden durch
das Vilsmeier-Verfahren, das in einem inerten Lösungsmittel, wie beispielsweise
Dichlormethan, Chloroform und Dimethylformamid, unter Verwendung
von Phosphoroxychlorid, Thionylchlorid oder dergleichen durchgeführt wird,
durch ein Formylierungsverfahren, wie beispielsweise die Gattermann-Koch-Synthese
oder die Friedel-Crafts-Synthese, unter Verwendung von Aluminiumchlorid,
Titantetrachlorid oder dergleichen.
-
4. Schritt:
-
Dieser Schritt umfasst die Reduktion
der Carbonylgruppe des im 3. Schritt erhaltenen Ketons (41) unter
Erhalt einer Methylenverbindung (42). Die Reduktion (Agar) des Ketons
kann in herkömmlicher
Weise durchgeführt
werden. Die Methylenverbindung (42) kann beispielsweise erhalten
werden durch katalytische Hydrierung, Wolff-Kishner-Reduktion unter
Verwendung von Hydrazin oder Reduktion unter Verwendung eines Hydrosilans,
wie beispielsweise Trichlorsilan und Triethylsilan.
-
HERSTELLUNGSVERFAHREN
D
-
Wenn R5 eine
verzweigte Alkylgruppe ist, kann die Herstellung auch nach dem folgenden
Verfahren erfolgen:
-
-
1. Schritt:
-
Dieser Schritt umfasst die Umwandlung
der Carbonylgruppe der im 3. Schritt von Herstellungsverfahren C
erhaltenen Acylverbindung (43) in ein Olefin unter Erhalt einer
Olefinverbindung (44). Die Olefinverbindung (44) kann erhalten werden
durch eine Umwandlungsreaktion von Carbonyl zu Olefin, z. B. die
Wittig-Reaktion unter Verwendung von Alkylidinphosphoran und einer
starken Base, wie beispielsweise Natriumamid und N-Butyllithium;
oder nach dem Horner-Verfahren unter Verwendung eines Phosphonsäureesters.
-
2. Schritt:
-
Dieser Schritt umfasst die Reduktion
der Doppelbindung der im 1. Schritt erhaltenen Olefinverbindung (44),
wodurch ein gesättigtes
Derivat (45) erhalten wird. Das gesättigte Derivat (45) kann nach
einer herkömmlichen
Reaktion zur Reduktion einer Doppelbindung erhalten werden, beispielsweise
durch katalytische Hydrierung.
-
Wie oben beschrieben, können die
erfindungsgemässen
Verbindungen industriell vorteilhaft hergestellt werden und sind
auch in dieser Hinsicht exzellente Verbindungen.
-
Nachfolgend werden zur Illustration
der Nützlichkeit
der erfindungsgemässen
Verbindungen pharmakologische Experimentalbeispiele detailliert
beschrieben.
-
PHARMAKOLOGISCHES EXPERIMENTALBEISPIEL
1
-
Bestimmung der NEP-inhibierenden
Wirkungen von Medikamenten mit Rattennierenrinde:
-
(1) Experimentelles Verfahren:
-
Die NEP-Aktivität wurde bestimmt mit der Membranfraktion,
die aus der Nierenrinde von Ratten nach dem Verfahren von Booth
und Kenny (A Rapid Method for the Purification of Microvilli from
Rabbit Kidney, Andrew G. Booth und A. John Kenny, Biochem. J., 1974,
142, 575–581)
hergestellt wurde.
-
Die NEP-Aktivität wurde nach dem Verfahren
von Orlowsky und Wilk (Purification and Specificity of a Membrane-Bound
Metalloendpeptidase from Bovine Pituitaries, Marian Orlowsky und
Shrwin Wilk, Biochemistry, 1981, 20, 4942–4950) nach dem folgenden Verfahren
bestimmt.
-
Benzoyl-glyceryl-arginyl-arginyl-2-naphthylamid
[Benzoyl-Gly-Arg-Arg-2-naphthylamid
(Nova Biochem. Schweiz)] wurde als Substrat verwendet. Das Naphthylamin,
das in Anwesenheit der NEP-Enzymprobe freigesetzt wurde, und überschüssige Leucinaminopeptidase
(Sigma Chemical Co., USA) wurde mit First Garnet (Sigma Chemical
Co., USA) farbentwickelt und die Absorbanz bei einer Wellenlänge von
540 nm wurde bestimmt.
-
Die NEP-inhibierende Aktivität wurde
durch Zugabe der Testverbindung zu dem obigen Experimentalsystem
in Endkonzentrationen von 1, 3, 10, 30, 100, 300 und 1.000 nM bestimmt,
wodurch eine Inhibierungskurve gebildet wurde, und die Konzentration,
bei der 50% der Aktivität
inhibiert wurden, wurde als IC50 bestimmt.
[4S-(4α,7α(R*),12bβ]]-7-[(1-oxo-2(S)-thio-3-phenylpropyl)amino]-1,2,3,4,6,7,8,12b-octahydro-6-oxopyrido[2,1-a]
[2]benzazepin-4-carbonsäure
(eine in JP-A-6-56790 offenbarte Verbindung) wurde als Kontrollverbindung
verwendet.
-
(2) Experimentelle Ergebnisse:
-
Die Ergebnisse des obigen Experiments
sind in der nachstehend beschriebenen Tabelle 1 angegeben.
-
PHARMAKOLOGISCHES EXPERIMENTALBEISPIEL
2
-
Bestimmung der ACE-inhibierenden
Aktivitäten
von Medikamenten mit Rattenlunge:
-
(1) Experimentelles Verfahren:
-
Die ACE-inhibierende Aktivität wurde
mit der Membranfraktion untersucht, die nach dem Verfahren von Wu-Wong
et al. (Characterization of Endothelin Converting Enzyme in Rat
Lung, R. Junshyum, Wu-Wong, Gerald P. Budzik, Edward M. Devine und
Terry J. Opgenorth, Biochem. Biophys. Res. Commun., 1990, 171, 1291–1296) aus
Rattenlunge hergestellt wurde.
-
Die ACE-Aktivität wurde durch eine Modifikation
(worin der pH-Wert des Boratpuffers auf 8,3 verändert wurde) des Cushman-Cheung-Verfahrens
(Spectrophotometric Assay and Properties of the Angiotensin-Converting
Enzyme of Rabbit Lung, D. W. Cushman und H. S. Cheung, 1971, 20,
1637–1648)
bestimmt.
-
Das aus Hippuryl-histidyl-leucin
[Hippuryl-His-Leu (Peptide Institute Inc., Japan) in Gegenwart von ACE
freigesetzte Hippurat wurde mit Ethylacetat extrahiert und die Absorbanz
bei einer Wellenlänge
von 228 nm bestimmt.
-
Die ACE-inhibierende Aktivität wurde
durch Zugabe der Testverbindung zu dem obigen Experimentalsystem
in Endkonzentrationen von 1, 3, 10, 30, 100, 300 und 1.000 nm bestimmt,
wodurch eine Inhibierungskurve gebildet wurde, und die Konzentration,
bei der 50% der Aktivität
inhibiert wurden, wurde als IC50 bestimmt. [4S-(4α,7α(R*),12bβ]]-7-[(1-oxo-2(S)-thio-3-phenylpropyl)amino]-1,2,3,4,6,7,8,12b-octahydro-6-oxopyrido[2,1-a][2]benz azepin-4-carbonsäure (eine
in JP-A-6-56790 offenbarte Verbindung) wurde als Kontrollverbindung verwendet.
-
(2) Experimentelle Ergebnisse:
-
Die Ergebnisse des nach dem obigen
Experimentalverfahren durchgeführten
Experiments sind in Tabelle 1 angegeben.
-
NEP-
und ACE-inhibierende Aktivitäten
der Beispielsverbindungen und der Vergleichsverbindung
-
Anmerkung:
-
-
BEISPIELE
-
Zur weiteren Erleichterung des Verständnisses
der vorliegenden Erfindung werden nun Beispiele beschrieben. Es
bedarf jedoch keiner Erwähnung,
dass die vorliegende Erfindung nicht auf diese beschränkt ist. Vor
den Beispielen werden die Herstellungsbeispiele der Verbindungen,
die als Ausgangsverbindungen für
die erfindungsgemässen
Verbindungen verwendet werden, als Synthesebeispiele angegeben.
-
SYNTHESEBEISPIEL
1
Ethyl-5-methylpyridinium-2-carboxylat:
-
200 ml Ethanol und 100 ml (1,88 mol)
konzentrierte Schwefelsäure
wurden zu 55,5 g 5-Methylpyridin-2-carbonitril hinzugegeben, wodurch
eine homogene Lösung
gebildet wurde, gefolgt von 2-tägigem
Erwärmen
unter Rückfluss.
Die Reaktionsflüssigkeit
wurde langsam in eine gesättigte
wässrige
Natriumhydrogencarbonatlösung
unter Eiskühlung
zur Neutralisierung der Schwefelsäure gegossen, gefolgt von Extraktion
mit Dichlormethan. Die organische Schicht wurde mit einer gesättigten
wässrigen
Kochsalzlösung
gewaschen und über
wasserfreiem Natriumsulfat getrocknet. Nach der Filtration wurde
das Filtrat eingeengt, wodurch 78,1 g eines braunen Öls der Titelverbindung
als Rohprodukt erhalten wurden.
1H-NMR
(400 MHz, CDCl3) δ: 8,57 (1H, m), 8,03 (1H, dt,
J = 8,0, 0,5 Hz), 7,63 (1H, ddd, J = 1,0, 2,5, 8,0 Hz), 4,47 (2H,
q, J = 7,0 Hz), 2,42 (3H, s), 1,44 (3H, t, J = 7,0 Hz).
-
SYNTHESEBEISPIEL
2
2-Carboxy-5-methylpyridiniumchlorid:
-
78,1 g des in Synthesebeispiel 1
erhaltenen rohen Ethyl-5-methylpyridini-2-carboxylat-Produkts wurden
in 200 ml 6N-Salzsäure
aufgelöst
und dann für
16 Stunden unter Rückfluss
erwärmt.
Die Reaktionslösung wurde
unter reduziertem Druck eingeengt. Dann wurde Acetonitril zu dem
Rückstand
hinzugegeben und die dadurch ausgefällten weissen Kristalle durch
Filtration abgetrennt, mit Acetonitril gewaschen und bei 90°C getrocknet,
wodurch 26,3 g der Titelverbindung erhalten wurden. Ausbeute: 37%.
1H-NMR (400 MHz, CDCl3) δ: 8,51 (1H,
m), 8,37 (1H, m), 8,21 (1H, d, J = 8,0 Hz), 2,42 (3H, s).
-
SYNTHESEBEISPIEL
3
(2S*,5S*)-2-Carboxy-5-methylpiperidiniumchlorid und (2S*,5R*)-2-Carboxy-5-methylpiperidiniumchlorid:
-
26,3 g (151 mmol) des in Synthesebeispiel
2 erhaltenen 2-Carboxy-5-methylpyridiniumchlorids
wurden in 300 ml Ethanol-Wasser (1 : 1) aufgelöst. Dann wurden 2 g Platinoxid
zugegeben, gefolgt von Hydrierung bei 50°C und 16 Atm über Nacht.
Nach Entfernung des Katalysators durch Filtration wurde das Filtrat
unter reduziertem Druck eingeengt und die so erhaltenen weissen
Kristalle wurden bei 90°C
getrocknet, wodurch 27,0 g der Titelverbindung als Mischung (Diastereomerenverhältnis 3
: 1) erhalten wurden. Ausbeute: 99%.
1H-NMR
(400 MHz, D2O) δ: 4,06 (3/4H, t, J = 5,0 Hz),
3,71 (1/4H, m), 3,24 (1/4H, ddd, J = 1,5, 4,0, 13,0 Hz), 3,10 (3/4H,
dd, J = 4,5, 13,0 Hz), 2,82 ((3/4H, dd, J = 10,0, 13,0 Hz), 2,53
(1/4H, t, J = 13,0 Hz), 2,22–2,04
(1H, m), 1,90–1,52
(2H, m), 1,22–1,04
(1H, m), 0,82 (3 × 3/4H,
d, J = 7,0 Hz), 0,81 (3 × 1/4H,
d, J = 7,0 Hz).
-
SYNTHESEBEISPIEL
4
(2S*,5S*)-N-Acetyl-5-methylpiperidin-2-carbonsäure:
-
27,0 g (150 mmol) der Mischung von
(2S*,5S*)-2-Carboxy-5-methylpiperidiniumchlorid und (2S*,5S*)-2-Carboxy-5-methylpiperidiniumchlorid
aus Synthesebeispiel 3 wurden in 700 ml Dichlormethan suspendiert.
Dann wurden 21 ml (150 mmol) Triethylamin hinzugegeben, gefolgt
von 2-stündigem
Rühren
bei Raumtemperatur. Durch Kristallisation wurden weisse Kristalle
abgetrennt, mit Dichlormethan gewaschen und dann bei 50°C getrocknet,
wodurch 15,9 g (2S*,5S*)-5-Methylpiperidin-2-carbonsäure erhalten
wurden. Ausbeute: 74%.
-
15,9 g (111 mmol) der oben beschriebenen
(2S*,5S*)-5-Methylpiperidin-2-carbonsäure wurden
in 220 ml Dichlormethan/Wasser (1 : 1) aufgelöst. 93,3 g (1,11 mol) Natriumhydrogencarbonat
und 21,0 ml (222 mmol) Acetanhydrid wurden in dieser Reihenfolge
bei Raumtemperatur zugegeben, gefolgt von 3-tägigem Rühren. Die Reaktionslösung wurde
unter Eiskühlung
in 6 N-Salzsäure
gegossen, und dann wurde mit Chloroform extrahiert. Dann wurde die
organische Schicht über
wasserfreiem Natriumsulfat getrocknet. Nach Filtration wurde das
Filtrat unter reduziertem Druck eingeengt, wodurch 20,1 g der Titelverbindung
als farbloses Öl
erhalten wurden. Ausbeute: 98%.
1H-NMR
(400 MHz, CDCl3) δ: 10,17 (1H, br), 5,41 (1H,
d, J = 5,5 Hz), 4,54–4,44
(2 × 1/4H,
m), 3,62 (1H, dd, J = 4,5, 13,5 Hz), 2,90 (1H, dd, J = 12,0, 13,5
Hz), 2,39–2,26
(2 × 3/4H,
m), 2,17 (3 × 3/4H,
s), 2,13 (3 × 1/4H, s),
1,96–1,52
(2H, m), 1,15–1,03
(1H, m), 0,92 (3 × 3/4H,
d, J = 6,5 Hz), 0,90 (3 × 1/4H,
d, J = 7,0 Hz).
-
SYNTHESEBEISPIEL
5
t-Butyl-(2S*,5S*)-N-acetyl-5-methylpiperidyl-2-carboxylat:
-
16,3 g (88 mmol) der in Synthesebeispiel
4 erhaltenen (2S*,5S*)-N-Acetyl-5-methylpiperidin-2-carbonsäure wurden
in 180 ml Dichlormethan aufgelöst
und dann wurden 6,1 ml (0,11 mol) konzentrierte Schwefelsäure zugegeben.
Dann wurde Isobutylengas in das Reaktionssystem eingeführt, gefolgt
von 4-tägigem
Rühren
bei Raumtemperatur. Die Reaktionsflüssigkeit wurde unter Eiskühlung in
eine gesättigte
wässrige
Natriumcarbonatlösung
gegossen und dann wurde mit Chloroform extrahiert. Die organische
Schicht wurde mit einer gesättigten
wässrigen
Natriumhydrogencarbonatlösung
und einer gesättigten
wässrigen
Kochsalzlösung
gewaschen und dann über
wasserfreiem Natriumsulfat getrocknet. Nach Filtration wurde das
Filtrat unter reduziertem Druck eingeengt, wodurch 16,4 g der Titelverbindung
als farbloses Öl
erhalten wurden. Ausbeute: 77%.
1H-NMR
(400 MHz, CDCl3) δ: 5,26 (1H, dd, J = 1,0, 6,0
Hz), 4,50–4,32
(3/4H, m), 3,59 (1H, dd, J = 4,5, 13,0 Hz), 2,90 (1H, dd, J = 12,0,
13,0 Hz), 2,30–2,17
(5/4H, m), 2,13 (3 × 3/4H,
s), 2,07 (3 × 1/4H,
s), 1,73–1,56
(2H, m), 1,47 (9 × 1/4H,
s), 1,46 (9 × 3/4H,
s), 1,05–0,94
(1H, m), 0,91 (3 × 3/4H,
d, J = 6,5 Hz), 0,90 (3 × 1/4H,
d, J = 7,0 Hz).
-
SYNTHESEBEISPIEL
6
Methyl(2RS,4R)-2-[(1S,4S)-4-acetylamino-4-(t-butoxycarbonyl)-1-methylbutyl]thiazolidin-4-carboxylat
und
Methyl-(2RS,4R)-2-[(1R,4R)-acetylamino-4-(t-butoxycarbonyl)-2-methylbutyl]thiazolidin-4-carboxylat:
-
9,41 g (39 mmol) des in Synthesebeispiel
5 erhaltenen t-Butyl(2S*,5S*)-N-acetyl-5-methylpiperidin-2-carboxylats
wurden in 150 ml Methanol aufgelöst,
gefolgt von der Zugabe von Tetraethylammonium-p-toluolsulfonat (Et4NOTs,
1,5 g, 1 G/V%). Mit Kohlenstoffelektroden wurde unter Bedingungen
von 11,4 F/mol und einer Stromdichte von 60 mA/cm2 bei
Raumtemperatur ein konstanter Strom (480 mA) hindurchgeleitet. Nach Einengung
der Reaktionslösung
unter reduziertem Druck wurde der Rückstand in Ethylacetat aufgelöst, mit Wasser
und einer gesättigten
wässrigen
Kochsalzlösung
gewaschen, und dann wurde die organische Schicht über wasserfreiem
Natriumsulfat getrocknet. Nach Filtration wurde das Filtrat unter
reduziertem Druck eingeengt, wodurch 11,5 g (2S*,5S*)-N-Acetyl-6-methoxy-5-methylpiperidin-2-carboxylat
als Rohprodukt erhalten wurden.
-
11,5 g des obigen (2S*,5S*)-N-Acetyl-6-methoxy-5-methylpiperidin-2-carboxylats wurden
in 100 ml Essigsäure-Wasser
(1 : 1) aufgelöst,
und dann wurden 6,0 ml (55 mmol) N-Methylmorpholin und 8,7 g (51 mmol)
Methyl-L-cysteinat-hydrochlorid
hinzugegeben, gefolgt von 3-tägigem
Rühren
bei Raumtemperatur unter einer Stickstoffatmosphäre. Die Reaktionsflüssigkeit
wurde zur Entfernung der Essigsäure
eingeengt. Nach Extraktion mit Dichlormethan wurde die organische
Schicht mit Wasser und einer gesättigten
wässrigen
Kochsalzlösung
gewaschen und über
wasserfreiem Magnesiumsulfat getrocknet. Nach Filtration wurde das
Filtrat unter reduziertem Druck eingeengt, und der erhaltene Rückstand
wurde mittels Silicagel-Säulenchromatografie
(Elution mit Dichlormethan : Ethanol = 98 : 2) unter Erhalt von
9,21 g der Titelverbindung als blassgelbes Öl (Diastereomerenverhältnis 1
: 1 : 1 : 1) gereinigt. Ausbeute: 63%.
1H-NMR
(400 MHz, CDCl3) δ: 6,18–6,04 (1H, m), 4,56–4,36 (2H,
m), 4,14–4,04
(2 × 1/4H,
m), 3,82–3,68
(2 × 1/4H,
m), 3,79 (3 × 2/4H,
s), 3,77 (3 × 2/4H,
s), 3,30–3,25
(2 × 1/4H,
m), 3,20–3,16
(2 × 1/4H,
m), 3,04–2,97
(2 × 1/4H,
m), 2,80–2,70
(2 × 1/4H,
m), 2,03 (3 × 2/4H,
s), 2,02 (3 × 1/fH,
s), 2,01 (3 × 1/4H,
s), 2,00–1,50
(5H, m), 1,482 (9 × 1/fH,
s), 1,478 (9 × 1/4H,
s), 1,473 (9 × 1/4H,
s), 1,470 (9 × 1/fH,
s), 1,10 (3 × 1/4H,
d, J = 7,0 Hz), 1,04 (3 × 1/4H,
d, J = 7,0 Hz), 1,03 (3 × 1/4H,
d, J = 7,0 Hz), 0,97 (3 × 1/4H,
d, J = 7,0 Hz).
-
SYNTHESEBEISPIEL
7
Methyl[3R-(3α,6α,9β,9aβ)]-6-acetylamino-9-methyl-5-oxo-octahydrothiazolo[3,2-a]azepin-3-carboxylat
und
Methyl[3R-(3α,6α,9α,9aβ)]-6-acetylamino-9-methyl-5-oxo-octahydrothiazolo[3,2-a]azepin-3-carboxylat:
-
50 ml Trifluoressigsäure wurden
unter Eiskühlung
zu 8,30 g (22,2 mmol) der in Synthesebeispiel 6 erhaltenen Mischung
aus Methyl(2RS,4R)-2-[(1S,4S)-4-acetylamino-4-(t-butoxycarbonyl)-1-methylbutyl]-thiazolidin-4-carboxylat
und Methyl(2RS,4R)-2-[(1R,4R)-4-acetylamino-4-(t-butoxycarbonyl)-1-methylbutyl]thiazolidin-4-carboxylat
zugegeben, gefolgt von einer langsamen Anhebung der Temperatur auf
Raumtemperatur. Nach 6-stündigem
Rühren
wurde das Lösungsmittel
abdestilliert und dann wurde eine azeotrope Destillation mit Toluol
durchgeführt,
wodurch 9,84 g einer Mischung des Trifluoressigsäuresalzes von Methyl(2R,4R)-2-[(1S,4RS)-4-acetylamino-4-carboxy-1-methylbutyl]-thiazolidin-4-carboxylat
und des Trifluoressigsäuresalzes
von Methyl(2R,4R)-2-[(1R,4RS)-4-acetylamino-4-carboxy-1-methylbutyl]-thiazolidin-4-carboxylat
(Isomerenverhältnis
1,4 : 1,4 : 1 : 1) als Rohprodukt erhalten wurden. 9,84 g dieses
Rohprodukts wurden in 150 ml Tetrahydrofuran aufgelöst, gefolgt
von der Zugabe von 9,8 ml (89 mmol) N-Methylmorpholin zur Einstellung
des pH-Werts auf 7.2-Ethoxy-1-ethoxycarbonyl-1,2-dihydrochinolin (EEDQ, 6,59 g, 27 mmol)
wurde bei Raumtemperatur zugefügt,
gefolgt von Rühren
in einer Stickstoffatmosphäre
bei Raumtemperatur über Nacht.
Nach Einengung der Reaktionsflüssigkeit
unter reduziertem Druck wurden 100 ml 2N-Salzsäure zu dem Rückstand
zur Einstellung des pH-Werts auf 1 oder darunter gegeben, gefolgt
von der Extraktion mit Dichlormethan. Die organische Schicht wurde
mit einer gesättigten
wässrigen
Natriumhydrogencarbonatlösung und
einer gesättigten
wässrigen
Kochsalzlösung
gewaschen und dann über
wasserfreiem Magnesiumsulfat getrocknet. Nach der Filtration wurde
das Filtrat unter reduziertem Druck eingeengt, und der erhaltene
Rückstand
wurde mittels Silicagel- Säulenchromatografie
(Elution mit Dichlormethan : Ethanol = 98 : 2) gereinigt, und durch
Umkristallisation (Ethylacetat-Hexan) wurden 2,59 g einer Mischung
der Titelverbindungen (Isomerenverhältnis 2 : 1) in Form weisser
Kristalle erhalten. Ausbeute: 39%.
1H-NMR
(400 MHz, CDCl3) δ: 6,83–6,74 (1H, m), 5,33 (2/3H,
dd, J = 3,2, 6,4 Hz), 5,23 (1/3H, s), 4,96 (1/3H, t, J = 6,8 Hz),
4,82 (2/3H, d, J = 9,5 Hz), 4,60–4,58 (1H, m), 3,79 (3H, s),
3,32 (1/3H, dd, 6,4, 11,6 Hz), 3,22 (2/3H, dd, J = 3,2, 11,6 Hz),
3,14 (1/3H, dd, J = 6,8, 11,6 Hz), 3,10 (2/3H, dd, J = 6,8, 11,6
Hz), 2,01 (3 × 2/3H,
s), 2,00 (3 × 1/3H,
s), 2,10– 1,89
(3H, m), 1,80–1,66
(2H, m), 1,12 (3 × 1/3H,
d, J = 7,2 Hz), 1,00 (3 × 2/3H,
d, J = 6,8 Hz).
-
SYNTHESEBEISPIEL
8
2-Acetyl-decahydro-(4aR,8aR)-isochinolin-3(S)-carbonsäure:
-
Eine Mischung, die (4aR,8aS)-Isomer,
(4aR,8aR)-Isomer und trans-Isomer
von Decahydroisochinolin-3(S)-carbonsäure umfasste, wurde in 72 ml
Wasser aufgelöst,
gefolgt von der Zugabe von 60,9 g (725 mmol) Natriumhydrogencarbonat
und 72 ml Dichlormethan bei Raumtemperatur. Anschliessend wurden
tropfenweise 27,4 ml (290 mmol) Acetanhydrid zugegeben, gefolgt
von 22-stündigem
Rühren.
Unlösliche
Bestandteile wurde durch Filtration abgetrennt. Nach Zugabe von
6N-Salzsäure
zur Einstellung des pH-Werts auf 3 wurde Kochsalz bis zur Sättigung
zugegeben. Dann wurde mit Chloroform extrahiert und die organische Schicht über wasserfreiem
Natriumsulfat getrocknet. Nach Filtration wurde das Filtrat unter
reduziertem Druck eingeengt. Dann wurde Dichlormethan zugegeben,
wodurch 5,45 g Kristalle der Titelverbindung erhalten wurden. Ausbeute:
33,4% (zwei Stufen).
-
SYNTHESEBEISPIEL
9
t-Butyl-2-acetyl-decahydro-(4aR,8aR)-isochinolin-3(S)-carboxylat:
-
5,21 g der Titelverbindung wurden
in der gleichen Weise wie in Synthesebeispiel 5 unter Verwendung der
in Synthesebeispiel 8 erhaltenen Verbindung erhalten. Ausbeute:
77%.
-
SYNTHESEBEISPIEL
10
Methyl-(2RS,4R)-2-[(1R,2R)-2-[(2S)-2-acetylamino-2-(t-butoxycarbonyl)ethyl]cyclohexyl]-thiazolidin-4-carboxylat:
-
1,61 g der Titelverbindung wurden
unter Verwendung der in Synthesebeispiel 9 hergestellten Verbindung
in gleicher Weise wie in Synthesebeispiel 6 erhalten. Ausbeute:
21%.
1H-NMR (400 MHz, CDCl3) δ: 7,28 und
6,15 (gesamt 1H, jeweils brd), 4,57–3,75 (gesamt 3H, m), 3,78
und 3,76 (gesamt 3H, jeweils s), 3,30–3,20 (gesamt 1H, m), 3,04
und 2,76 (gesamt 1H, dd und t), 2,01 und 1,97 (gesamt 3H, jeweils
s), 1,50 und 1,47 (gesamt 9H, jeweils s), 2,40–1,05 (gesamt 12H, m) .
-
SYNTHESEBEISPIEL
11
Methyl-(3R,6S,7aR,11aR,11bR)-6-acetylamino-5-oxo-2,3,5,6,7,7a,11a,11b-octahydrocyclohexyl[c]thiazolo[3,2-a]azepin-3-carboxylat:
-
0,48 g der Titelverbindung wurden
unter Verwendung der in Synthesebeispiel 10 hergestellten Verbindung
in gleicher Weise wie in Synthesebeispiel 7 erhalten. Ausbeute:
36%. Die absolute Konfiguration wurde anhand des mittels NMR-Spektroskopie
ermittelten NOE bestimmt.
1H-NMR (400
MHz, CDCl3) δ: 6,76 (1H, brd, J = 6,0 Hz),
5,14 (1H, s), 4,91 (1H, t , J = 7,0 Hz), 4,56 (1H, ddd, J = 1,8,
6,0, 11,4 Hz), 3,79 (3H, s), 3,29 (1H, dd, J = 7,0, 11,6 Hz), 3,13
(1H, dd, J = 7,0, 11,6 Hz), 2,35– 2,30 (1H, m), 2,07–1,15 (11H,
m), 2,00 (3H, s).
NOE δ:
3,29 (H2β) ↓ → 1,70 (H11)
5,14
(H11b) ↓ → 1,85 (H11a),
2,33 (H7a), 4,56 (H6),
4,91 (H3) ↓ → 3,13 (H2α)
4,56 (H6) ↓ → 2,33 (H7α)
-
SYNTHESEBEISPIEL
12
Mischung von Methyl-[3R-(3α,6α,8α,9αβ)]-6-acetylamino-8-methyl-5-oxo-octahydrothiazolo[3,2-a]-azepin-3-carboxylat
und Methyl-[3R-(3α,6α,8β,9αβ)]-6-acetylamino-8-methyl-5-oxo-octahydrothiazolo[3,2-a]-azepin-3-carboxylat:
-
Eine Mischung, die die obigen Titelverbindungen
in einem Verhältnis
von etwa 1 : 1 umfasste, wurde unter Verwendung von DL-(2S*,4S*)-2-Carboxy-4-methylpiperidiniumchlorid
in der gleichen Weise wie in Synthesebeispiel A-4-7 synthetisiert.
-
t-Butyl-(S)-N-acetyl-5-formyl-1,2,3,4-tetrahydropyridin-2-carboxylat:
-
82 ml (880 mmol) Phosphoroxychlorid
wurden zu 137 ml (1,77 mol) Dimethylformamid bei 0°C hinzugegeben,
und dann wurde bei –10
bis 0°C
eine Lösung
von 39,8 g (177 mmol) des in Synthesebeispiel 13 erhaltenen t-Butyl-(S)-N-acetyl-1,2,3,4-tetrahydropyridin-2-carboxylats
in 40 ml Dimethylformamid hinzugegeben, gefolgt von der langsamen
Anhebung der Temperatur auf Raumtemperatur. Nach 1-stündigem Rühren wurde
die Reaktionsflüssigkeit
in 2,0 l einer wässrigen
Lösung
von 365 g (4,49 mol) Natriumsulfat gegossen und dann wurde mit Ethylacetat
extrahiert. Die organische Schicht wurde mit einem gesättigten
Natriumhydrogencarbonat- und einer gesättigten wässrigen Kochsalzlösung gewaschen
und dann über
Natriumsulfat getrocknet. Das Lösungsmittel
wurde abdestilliert und der Rückstand
wurde aus Isopropylether umkristallisiert, wodurch 18,1 g der Titelverbindung
erhalten wurden. Ausbeute: 40%.
1H-NMR
(400 MHz, CDCl3) δ: 9,35 (1/6H, s), 9,30 (5/6H,
s), 8,16 (1/6H, s), 7,50 (5/6H, s), 5,13 (5/6H, s), 4,62 (1/6H,
br), 2,60–2,40
(2H, m), 2,41 (3 × 5/6H,
s), 2,22 (3 × 1/6H,
s), 1,98–1,70
(2H, m), 1,45 (9H, s).
-
SYNTHESEBEISPIEL
15
t-Butyl-(2S,5S)-N-acetyl-5-methylpiperidin-2-carboxylat:
-
140 mg (0,529 mmol) des in Synthesebeispiel
14 erhaltenen t-Butyl-(S)-N-acetyl-5-formyl-1,2,3,4-tetrahydropyridin-2-carboxylats
wurden in 20 ml Ethanol aufgelöst,
gefolgt von der Zugabe von 5% Pd/C (140 ml). Zur Durchführung der
Hydrierung wurde eine Behandlung in einer Wasserstoffatmosphäre von 3
kg/cm2 unter Verwendung einer Mitteldruckausrüstung für die katalytische
Reduktion durchgeführt.
Der Katalysator wurde mittels Filtration entfernt und das Filtrat
unter Erhalt von 140 mg der Titelverbindung eingeengt. Ausbeute:
100%.
-
SYNTHESEBEISPIEL
16
t-Butyl-(S)-N-acetyl-5-vinyl-1,2,3,4-tetrahydropyridin-2-carboxylat:
-
Eine etherische Lösung (42,5 ml) von n-Butyllithium
wurde zu einer suspendierten Lösung
von 15,2 g (42,5 mmol) Methyltriphenylphosphoniumbromid in Diethylether
(80 ml) bei 30°C
oder darunter hinzugegeben. Zu dieser Lösung wurde eine THF-Lösung von
8,98 g des in Synthesebeispiel 14 erhaltenen t-Butyl-(S)-N-acetyl-5-formyl-1,2,3,4-tetrahydropyridin-2-carboxylats
bei Raumtemperatur hinzugegeben, gefolgt von Rühren über Nacht. Dann wurde Wasser
zu der Reaktionsflüssigkeit
hinzugegeben, gefolgt von der Extraktion mit Ethylacetat. Nach Waschen
mit einer gesättigten
wässrigen
Kochsalzlösung
wurde über
Natriumsulfat getrocknet. Durch Reinigung mittels Silicagel- Säulenchromatografie (Elution
mit n-Hexan : Ethylacetat = 2 : 1) wurden 4,08 g der Titelverbindung
erhalten. Ausbeute: 46%.
1H-NMR (400
MHz, CDCl3) δ: 7,35 (1/6H, s), 6,69 (5/6H,
s), 6,38 (1/6H, dd, J = 10,8, 17,6 Hz), 6,30 (5/6H, dd, J = 10,8,
17,2 Hz), 5,11 (5/6H, m), 5,05 (5/6H, d, J = 17,2 Hz), 5,03 (1/6H,
d, J = 17,6 Hz), 4,95 (5/6H, d, J = 10,8 Hz), 4,93 (1/6H, d, J =
10,8 Hz), 4,53 (1H, m), 2,52– 2,38
(1H, m), 2,34–2,24
(1H, m), 2,26 (3 × 5/6H,
s), 2,14 (3 × 1/6H,
s), 2,04–1,78
(2H, m), 1,45 (9 × 1/6H,
s), 1,44 (9 × 5/6H,
s).
-
SYNTHESEBEISPIEL
17
t-Butyl-(2S,5S)-N-acetyl-5-ethylpiperidin-2-carboxylat:
-
4,08 (16,3 mmol) des in Synthesebeispiel
16 erhaltenen t-Butyl-(S)-N-acetyl-5-vinyl-1,2,3,4-tetrahydropyridin-2-carboxylats
wurden in 150 ml Ethanol aufgelöst,
gefolgt von der Zugabe von 4,0 g 10% Pd/C. Die Hydrierung wurde
durch Behandlung in einer Wasserstoffatmosphäre von 3 kg/cm2 unter
Verwendung einer Mitteldruckausrüstung
für die
katalytische Reaktion durchgeführt.
Der Katalysator wurde durch Filtration entfernt und das Filtrat
unter Erhalt von 140 mg der Titelverbindung eingeengt. Ausbeute:
100%.
1H-NMR (400 MHz, CDCl3) δ:
5,27 (1H, d, J = 6,0 Hz), 4,52–4,35
(3/4H, m), 3,64 (1H, dd, J = 4,5, 13,0 Hz), 2,91 (1H, dd, J = 12,0,
13,0 Hz), 2,35–2,15
(5/4H, m), 2,13 (3 × 3/4H,
s), 2,07 (3 × 1/4H,
s), 1,80–1,50
(2H, m), 1,47 (9 × 1/4H,
s), 1,46 (9 × 1/4s),
1,35–1,20
(2H, m), 1,05–0,95
(1H, m), 0,93 (3 × 3/4H,
t, J = 7,6 Hz), 0,90 (3 × 1/4H,
t, J = 7,6 Hz).
-
SYNTHESEBEISPIEL
18
Methyl-(2RS,4R)-2-[(1S,4S)-4-acetylamino-4-t-butoxycarbonyl)-1-ethylbutyl]thiazolidin-4-carboxylat:
-
4,29 g (16,8 mmol) des in Synthesebeispiel
17 erhaltenen t-Butyl-(2S,5S)-N-acetyl-5-ethylpiperidin-2-carboxylats
wurden in 43 ml Methanol aufgelöst,
gefolgt von der Zugabe von 0,43 g Tetraethylammoniumtosylat. Mittels
Kohlenstoffelektroden unter Bedingungen von 5F/mol bei Raumtemperatur
wurde ein konstanter Strom (0,33 A) hindurchpassiert. Die Reaktionslösung wurde
unter reduziertem Druck eingeengt und der Rückstand in Ethylacetat aufgelöst. Dann
wurde mit Wasser und einer gesättigten
wässrigen
Kochsalzlösung
gewaschen und die organische Schicht über wasserfreiem Natriumsulfat
getrocknet. Nach Filtration wurden 5,08 g (2S,5S)-N-Acetyl-6-methoxy-5-methylpiperidin-2-carboxylat
des Filtrats als Rohprodukt erhalten. Anschliessend wurde das obige
Rohprodukt in 60 ml Essigsäure-Wasser
(1 : 1) aufgelöst,
und 2,4 ml (23,7 mmol) N-Methylmorpholin und 3,46 g (20,2 mmol)
Methyl-L-cysteinat-hydrochlorid wurden hinzugegeben, gefolgt von
3-tägigem
Rühren
bei Raumtemperatur unter einer Stickstoffatmosphäre. Die Reaktionsflüssigkeit wurde
in eine wässrige
Lösung
(120 ml) von 49 g Natriumhydrogencarbonat gegossen, gefolgt von
Extraktion mit Ethylacetat. Dann wurde mit einer gesättigten
wässrigen
Kochsalzlösung
gewaschen und über
wasserfreiem Natriumsulfat getrocknet. Nach Filtration wurde das
Filtrat eingeengt und der Rückstand
mittels Silicagel-Säulenchromatografie
(Dichlormethan : Ethanol = 100 : 1) unter Erhalt von 3,62 g der
Titelverbindung gereinigt. Ausbeute: 55%.
1H-NMR
(400 MHz, CDCl3) δ: 6,11 (2/3H, d, J = 7,6 Hz),
6,06 (1/3H, d, J = 8,0 Hz), 4,60 (1/3H, m), 4,55–4,40 (2 × 2/3H, m), 4,12 (1/3H, m),
3,78 (3 × 2/3H,
s), 3,77 (3 × 1/3H,
s), 3,85–3,70
(1H, m), 3,28 (2/3H, dd, J = 7,2, 10,4 Hz), 3,17 (1/3H, dd, J =
7,6, 10,8 Hz), 3,03 (1/3H, 1/3H, dd, J = 5,6, 10,8 Hz), 2,77 (2/3H,
dd, 10,0, 10,4 Hz), 2,02 (3H, s), 1,90–1,20 (7H, m), 1,48 (9 × 1/3H,
s), 1,47 (9 × 2/3H,
s), 0,92 (3 × 2/3H,
t, J = 7,6 Hz), 0,90 (3 × 1/3H,
t, J = 7,6 Hz), (als 1 : 2 Diastereomerenmischung).
-
SYNTHESEBEISPIEL
19
Methyl-[3R-(3α,6α,9β,9aβ)]-6-acetylamino-9-ethyl-5-oxo-octahydrothiazolo[3,2-a]azepin-3-carboxylat:
-
21,5 ml Trifluoressigsäure wurden
zu 3,62 g des in Synthesebeispiel 18 erhaltenen Methyl-(2RS,4R)-2-[(1S,4S)-4-acetylamino-4-t-butoxycarbonyl)-1-ethylbutyl]-thiazolidin-4-carboxylats
unter Eiskühlung
hinzugegeben, gefolgt von einer langsamen Anhebung der Temperatur
auf Raumtemperatur. Nach 5-stündigem
Rühren
wurde das Lösungsmittel
abdestilliert, gefolgt von einer azeotropen Destillation mit Toluol, wodurch
das Trifluoressigsäuresalz
von Methyl-(2R,4R)-2-[(1S,4RS)-4-acetylamino-4-carboxy-1-ethylbutyl]-thiazolidin-4-carboxylat
als Rohprodukt erhalten wurde. Das Rohprodukt wurde in 60 ml Tetrahydrofuran aufgelöst, gefolgt
von der Zugabe von 4,09 ml (37,2 mmol) N-Methylmorpholin zur Einstellung
des pH-Werts auf 7. 2-Ethoxy-1-ethoxycarbonyl-1,2-dihydrochinolin
(EEDQ, 2,76 g, 11,2 mmol) wurde bei Raumtemperatur zugegeben, gefolgt
von Rühren
bei Raumtemperatur unter einer Stickstoffatmosphäre über Nacht. Nach Einengung der
Reaktionsflüssigkeit
unter reduziertem Druck wurden 100 ml 2N-Salzsäure zur Einstellung des pH-Werts
auf 1 oder weniger zum Rückstand
hinzugegeben, gefolgt von Extraktion mit Dichlormethan. Die organische
Schicht wurde mit einer gesättigten
wässrigen
Natriumhydrogencarbonatlösung
und einer gesättigten
wässrigen
Kochsalzlösung
gewaschen und dann über
wasserfreiem Magnesiumsulfat getrocknet. Nach Filtration wurde das
Filtrat unter reduziertem Druck eingeengt und der erhaltene Rückstand
mittels Silicagel-Säulenchromatografie
(Elution mit Dichlormethan : Ethanol = 100 : 1-100 : 3) unter Erhalt von 1,3 g der Titelverbindung
als weisse Kristalle gereinigt. Ausbeute: 44%.
1H-NMR
(400 MHz, CDCl3) δ: 6,80 (1H, br), 5,30 (1H, dd,
J = 3,6, 6,8 Hz), 4,87 (1H, d, J = 9,2 Hz), 4,58 (1H, m), 3,79 (3H,
s), 3,22 (1H, dd, 3,6, 11,6 Hz), 3,10 (1H, dd, J = 6,8, 11,6 Hz),
2,18–2,08
(1H, m), 2,01 (3H, s), 1,84–1,56
(5H, m), 1,31 (1H, m), 0,92 (3H, t, J = 7,6 Hz).
-
BEISPIEL
1
Methyl-[3R-(3α,6α,9β,9aβ)]-6-amino-9-methyl-5-oxo-octahydrothiazolo[3,2-a]azepin-3-carboxylat
und
Methyl-[3R-(3α,6α,9α,9aβ)]-6-amino-9-methyl-5-oxo-octahydrothiazolo[3,2-a]azepin-3-carboxylat:
-
2,59 g (8,6 mmol) der Mischung (Isomerenverhältnis: 2
: 1) von Methyl-[3R-(3α,6α,9β,9aβ)]-6-amino-9-methyl-5-oxo-octahydrothiazolo[3,2-a]azepin-3-carboxylat und
Methyl-[3R-(3α,6α,9α,9aβ)]-6-amino-9-methyl-5-oxo-octahydrothiazolo[3,2-a]azepin-3-carboxylat,
wie in Synthesebeispiel 7 erhalten, wurden in einer 10%-igen Lösung von
Salzsäure
in Methanol (100 ml) aufgelöst,
gefolgt von Erwärmen
unter Rückfluss für 26 Stunden.
Nach Entfernen des Lösungsmittels
durch Destillation unter reduziertem Druck wurde 2N-Salzsäure zugegeben,
gefolgt von Waschen mit Dichlormethan. Nach Alkalisierung der wässrigen
Schicht mit wässrigem
Ammoniak wurde mit Dichlormethan extrahiert, und die organische
Schicht wurde über
wasserfreiem Kaliumcarbonat getrocknet. Nach Filtration wurde das
Filtrat unter reduziertem Druck eingeengt, wodurch 2,01 g einer
Mischung der Titelverbindungen (Isomerenverhältnis: 2 : 1) als farbloses Öl erhalten
wurden. Ausbeute: 90%.
1H-NMR (400
MHz, CDCl3) δ: 5,35 (2/3H, dd, J = 3,2, 6,8
Hz), 4,99 (1/3H, t, J = 6,8 Hz), 4,76 (2/3H, d, J = 10,0 Hz), 4,78–4,70 (1/3H,
m), 3,78 (3 × 2/3H,
s), 3,76 (3 × 1/3H,
s), 3,55 (2/3H, dd, J = 2,2, 10,6 Hz), 3,49 (1/3H, dd, J = 2,0,
10,8 Hz), 3,30 (1/3H, dd, J = 6,0, 11,6 Hz), 3,21 (2/3H, dd, J =
3,2, 12,0 Hz), 3,11 (1/3H, dd, J = 7,2, 11,6 Hz), 3,09 (2/3H, dd,
J = 6,4, 12,0 Hz), 2,13–1,50
(7H, m), 1,12 (3 × 1/3H,
d, J = 6,8 Hz), 1,00 (3 × 2/3H,
d, J = 6,8 Hz).
-
BEISPIEL
2
Methyl-[3R-(3α,6α,9β,9aβ)]-6-[[(2S,3S)-1-oxo-2-acetylthio-3-methylpentyl]amino]-9-methyl-5-oxo-octahydrothiazolo[3,2-a]azepin-3-carboxylat und
Methyl-[3R-(3α,6α,9α,9aβ)]-6-[[(2S,3S)-1-oxo-2-acetylthio-3-methylpentyl]amino]-9-methyl-5-oxo-octahydrothiazolo[3,2-a]azepin-3-carboxylat:
-
Eine Lösung von 1,78 g (9,3 mmol)
von (2S,3S)-2-Acetylthio-3-methylpentansäure in Tetrahydrofuran (100
ml) wurde zu 2,01 g (7,8 mmol) der Mischung (Isomerenverhältnis: 2
: 1) von Methyl-[3R-(3α,6α,9β,9aβ)]-6-amino-9-methyl-5-oxo-octahydrothiazolo[3,2-a]azepin-3-carboxylat
und Methyl-[3R-(3α,6α,9α,9aβ)]-6-amino-9-methyl-5-oxo-octahydrothiazolo[3,2-a]azepin-3-carboxylat
aus Beispiel 1 unter Eiskühlung
hinzugegeben. Zu dieser Lösung
wurden nacheinander 1,79 g (9,3 mmol) 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimid-hydrochlorid
(DEC·HCl),
1,03 ml (9,3 mmol) N-Methylmorpholin und 1,26 g (9,3 mmol) 1-Hydroxy-1H-benzotriazol-monohydrat
(HOBt) hinzugegeben, gefolgt von 18-stündigem Rühren bei Raumtemperatur unter
einer Stickstoffatmosphäre.
Nach der Zugabe von Wasser und Extraktion mit Ethylacetat wurde
die organische Schicht mit 1N-Salzsäure, einer gesättigten
wässrigen
Natriumhydrogencarbonatlösung
und einer gesättigten
wässrigen
Kochsalzlösung
gewaschen und über
wasserfreiem Magnesiumsulfat getrocknet. Nach Filtration wurde das
Filtrat unter reduziertem Druck eingeengt und der erhaltene Rückstand mittels
Silicagel-Säulenchromatografie
(eluiert mit Hexan : Ethylacetat = 3 : 1) gereinigt. 837 mg Methyl-[3R-(3α,6α,9β,9aβ)]-6-[[(2S,3S)-1-oxo-2-acetylthio-3-methylpentyl]amino]-9-methyl-5-oxo-octahydrothiazolo[3,2-a]azepin-3-carboxylat
als farbloses Öl
wurden als erster Effluent zurückgewonnen.
Ausbeute: 42%. Die absolute Konfiguration der Verbindung wurde mit
einem NOE-Experiment bestimmt.
1H-NMR
(400 MHz, CDCl3) δ: 7,37 (1H, d, J = 6,0 Hz),
5,36 (1H, dd, J = 3,0, 7,0 Hz), 4,80 (1H, d, J = 9,5 Hz), 4,53 (1H,
m), 3,97 (1H, d, J = 7,0 Hz), 3,79 (3H, s), 3,22 (1H, dd, J = 3,0,
11,5 Hz), 3,10 (1H, dd, J = 7,0, 11,5 Hz), 2,38 (3H, s), 2,14–1,90 (3H,
m), 1,78–1,62
(3H, m), 1,57 (1H, m), 1,16 (1H, m), 1,00 (3H, d, J = 6,5 Hz), 0,99
(3H, d, J = 7,0 Hz), 0,88 (3H, t, J = 7,5 Hz).
NOE δ: 1,00 (9-Me) ↓ → 4,80 (H9a)
3,10
(H2α) ↓ → 4,80 (H9a),
5,36 (H3)
3,22 (H2β) ↓ → 1,95 (H9),
3,79
(3-COOMe) 4,53 (H6) ↓ → 4,80 (H9a)
-
Ferner wurden 532 mg Methyl-[3R-(3α,6α,9α,9aβ)]-6-[[(2S,3S)-1-oxo-2-acetylthio-3-methylpentyl]amino]-9-methyl-5-oxo-octahydrothiazolo[3,2-a]azepin-3-carboxylat
als farbloses Öl
als zweiter Effluent zurückgewonnen.
Ausbeute: 16%. Die absolute Konfiguration der Verbindung wurde mit
einem NOE-Experiment bestimmt.
1H-NMR
(400 MHz, CDCl3) δ: 7,32 (1H, brd, J = 6,1 Hz),
5,20 (1H, s), 5,00 (1H, dd, J = 6,0, 6,4 Hz), 4,48 (1H, m), 3,96
(1H, d, J = 6,6 Hz), 3,78 (3H, s), 3,32 (1H, dd, J = 6,0, 11,7 Hz),
3,13 (1H, dd, J = 6,4, 11,7 Hz), 2,37 (3H, s), 2,20–1,50 (7H,
m), 1,15 (1H, m), 1,10 (3H, d, J = 7,4 Hz), 0,98 (3H, d, J = 6,8
Hz), 0,87 (3H, t, J = 7,2 Hz).
NOE δ: 1,10 (9-Me) ↓ → 3,32 (H2β),
3,78
(3-COOMe) 3,13 (H2α) ↓ → 5,20 (H9a),
5,00
(H3) 4,48 (H6) ↓ → 5,20 (H9a)
-
BEISPIEL
3
[3R-(3α,6α,9β,9aβ)]-6-[[(2S,3S)-1-Oxo-2-thio-3-methylpentyl]amino]-9-methyl-5-oxo-octahydrothiazolo[3,2-a]azepin-3-carbonsäure:
-
167 mg (0,39 mmol) des in Beispiel
2 erhaltenen Methyl-[3R-(3α,6α,9β,9aβ)]-6-[[(2S,3S)-1-oxo-2-acetylthio-3-methylpentyl]amino]-9-methyl-5-oxo-octahydrothiazolo[3,2-a]azepin-3-carboxylats
wurden in 5 ml entlüftetem
Ethanol aufgelöst,
und dann wurden 2,0 ml (2,0 mmol) einer wässrigen 1N-Lithiumhydroxidlösung unter
Eiskühlung
hinzugegeben, gefolgt von 1-stündigem Rühren bei
Raumtemperatur unter einer Stickstoffatmosphäre. Die Reaktionslösung wurde
durch Zugabe von 7,5 ml 2N-Salzsäure
unter Eiskühlung
angesäuert.
Nach Verdünnung
mit Wasser wurde mit Dichlormethan extrahiert. Die organische Schicht
wurde mit einer gesättigten
wässrigen
Kochsalzlösung
gewaschen und über wasserfreiem
Magnesiumsulfat getrocknet. Nach Filtration wurde das Filtrat unter
reduziertem Druck eingeengt. Der erhaltene amorphe Feststoff wurde
umkristallisiert (Dichlormethan-Hexan) und für 12 Stunden mit Warmluft bei
50°C getrocknet,
wodurch 118 mg der Titelverbindung in Form weisser Kristalle erhalten
wurden. Ausbeute: 81%).
1H-NMR (400
MHz, CDCl3) δ: 7,66 (1H, d, J = 6,5 Hz),
5,39 (1H, dd, J = 3,0, 7,0 Hz), 4,86 (1H, d, J = 9,5 Hz), 4,60 (1H,
m), 3,29 (1H, dd, J = 3,0, 12,0 Hz), 3,22 (1H, dd, J = 7,0, 9,0
Hz), 3,13 (1H, dd, J = 7,0, 12,0 Hz), 2,10–1,90 (4H, m), 1,87 (1H, d,
J = 9,0 Hz), 1,81–1,64
(2H, m), 1,61 (1H, m), 1,21 (1H, m), 1,03 (3H, d, J = 7,0 Hz), 1,00
(3H, d, J = 7,0 Hz), 0,90 (3H, t, J = 7,0 HZ).
-
BEISPIEL
4
Methyl-(3R,6S,7aR,11aR,11bR)-6-amino-5-oxo-2,3,5,6,7,7a,11a,11b-octahydrocyclohexyl[c]thiazolo[3,2-a]azepin-3-carboxylat:
-
0,23 g der Titelverbindung wurden
unter Verwendung der in Synthesebeispiel 11 erhaltenen Verbindung
in der gleichen Weise wie in Beispiel 1 erhalten. Ausbeute: 57%.
1H-NMR (400 MHz, CDCl3) δ: 5,08 (1H,
s), 4,94 (1H, t, J = 6,8 Hz), 3,78 (3H, s), 3,54–3,52 (1H, m), 3,27 (1H, dd,
J = 6,8, 11,6 Hz), 3,11 (1H, dd, J = 6,8, 11,6 Hz), 2,23–1,18 (14H,
m).
-
BEISPIEL
5
Methyl-(3R,6S,7aR,11aR,11bR)-6-[[(2S,3S)-1-oxo-2-acetylthio-3-methylpentyl]amino]-5-oxo-2,3,5,6,7,7a,11a,11b-octahydrocyclohexyl-[c]thiazolo[3,2-a]azepin-3-carboxylat:
-
0,32 g der Titelverbindung wurden
unter Verwendung der in Beispiel 4 erhaltenen Verbindung in der gleichen
Weise wie in Beispiel A erhalten. Ausbeute: 88%.
1H-NMR
(400 MHz, CDCl3) δ: 7,33 (1H, brd, J = 6,0 Hz),
5,14 (1H, s), 4,96 (1H, t, J = 6,6 Hz), 4,57–4,52 (1H, m), 3,97 (1H, d,
J = 6,8 Hz), 3,79 (3H, s), 3,30 (1H, dd, J = 6,6, 11,6 Hz), 3,14
(1H, dd, J = 6,6, 11,6 Hz), 2,38 (3H, s), 2,40–0,85 (15H, m), 0,99 (3H, d,
J = 6,8 Hz), 0,89 (3H, t, J = 7,4 Hz).
-
BEISPIEL
6
(3R,6S,7aR,11aR,11bR)-6-[[(2S,3S)-1-Oxo-2-thio-3-methylpentyl]amino]-5-oxo-2,3,5,6,7,7a,11a,11b-octahydrocyclohexyl[c]thiazolo[3,2-a]azepin-3-carbonsäure:
-
0,18 g der Titelverbindung wurden
unter Verwendung der in Beispiel 5 erhaltenen Verbindung in der gleichen
Weise wie in Beispiel 3 als weisse Kristalle erhalten. Ausbeute:
63%.
1H-NMR (400 MHz, CDCl3) δ: 7,33 (1H,
brd, J = 6,0 Hz), 5,14 (1H, s), 4,96 (1H, t, J = 6,6 Hz), 4,57–4,52 (1H, m),
3,97 (1H, d, J = 6,8 Hz), 3,79 (3H, s, 3,30 (1H, dd, J = 6,6, 11,6
Hz), 3,14 (1H, dd, J = 6,6, 11,6 Hz), 2,38 (3H, s), 2,40–0,85 (15H,
m), 0,99 (3H, d, J = 6,8 Hz, 0,89 (3H, t, J = 7,4 Hz).
-
BEISPIEL
7
Methyl-[3R-(3α,6α,8α,9aβ)]-6-amino-8-methyl-5-oxo-octahydrothiazolo[3,2-a]azepin-3-carboxylat
und
Methyl-[3R-(3α,6α,8β,9aβ)]-6-amino-8-methyl-5-oxo-octahydrothiazolo[3,2-a]azepin-3-carboxylat:
-
940 mg (3,12 mmol) der Mischung in
einem Verhältnis
von etwa 1 : 1 von Methyl-[3R-(3α,6α,8α,9aβ)]-6-acetylamino-8-methyl-5-oxo-octahydrothiazolo[3,2-a]azepin-3-carboxylat
und Methyl-[3R-(3α,6α,8β,9aβ)]-6-acetylamino-8-methyl-5-oxo-octahydrothiazolo[3,2-a]azepin-3-carboxylat,
die in Synthesebeispiel 12 erhalten wurde, wurden in 24 ml einer
10%-igen Salzsäurelösung in
Methanol aufgelöst, gefolgt
von 24-stündigem
Erwärmen
unter Rückfluss.
Nach Abdestillieren des Lösungsmittels
unter reduziertem Druck wurde Wasser zugegeben und mit Dichlormethan
gewaschen. Die erhaltene wässrige
Schicht wurde durch Zugabe einer gesättigten wässrigen Natriumhydrogencarbonatlösung basisch
gemacht und dann mit Dichlormethan extrahiert, gefolgt von Trocknen über wasserfreiem
Natriumsulfat. Der durch Einengung erhaltene Rückstand wurde mittels Silicagel-Säulenchromatografie
(Chloroform : Methanol : wässriger
Ammoniak = 98 : 2 : 0,2) gereinigt, wodurch 220 mg einer Mischung
der beiden Titelverbindungen im Verhältnis von etwa 1,4 : 1 erhalten
wurden. Ausbeute: 27%.
1H-NMR (400
MHz, CDCl3) δ: 5,29 (1H × 1,4/2,4, dd, J = 2,4, 6,4
Hz), 5,00 (1H × 1,4/2,4,
d, J = 10,4 Hz), 3,78 (3H × 1,4/2,4,
s), 3,54 (1H × 1,4/2,4,
dd, J = 1,2, 10,8 Hz), 3,26 (1H × 1,4/2,4, dd, J = 2,4, 11,6
Hz), 3,17 (1H × 1,4/2,4,
dd, J = 6,4, 11,6 Hz), 1,43–2,05
(7H × 1,4/2,4,
m), 1,00 (3H × 1,4/2,4,
d, J = 6,8 Hz) und
1H-NMR (400 MHz,
CDCl3) δ:
5,21 (1H × 1,0/2,4,
dd, J = 3,2, 6,4 Hz), 5,15 (1H × 1,0/2,4,
dd, J = 2,2, 10,2 Hz), 3,78 (3H × 1,0/2,4, s), 3,71 (1H × 1,0/2,4,
dd, J = 3,2, 10,8 Hz), 3,27 (1H × 1,0/2,4, dd, J = 3,2, 12,0
Hz), 3,18 (1H × 1,0/2,4,
dd, J = 6,4, 12,0 Hz), 1,60–2,33
(7H × 1,0/2,4,
m), 1,16 (3H × 1,0/2,4,
d, J = 7,2 Hz).
-
BEISPIEL
8
Methyl-[3R-(3α,6α,8β,9aβ)]-6-[[(2S,3S)-1-oxo-2-acetylthio-3-methylpentyl]amino]-8-methyl-5-oxo-octahydrothiazolo[3,2-a]azepin-3-carboxylat und
Methyl-[3R-(3α,6α,8α,9aβ)]-6-[[(2S,3S)-1-oxo-2-acetylthio-3-methylpentyl]amino]-8-methyl-5-oxo-octahydrothiazolo[3,2-a]azepin-3-carboxylat:
-
Eine Lösung von 214 mg (1,12 mmol)
(2S,3S)-2-Acetylthio-3-methylpentansäure in Tetrahydrofuran (17
ml) wurde zu 215 mg (0,83 mmol) der Mischung (Isomerenverhältnis: 1
: 1,4) von Methyl-[3R-(3α,6α,8β,9aβ)]-6-amino-8-methyl-5-oxo-octahydrothiazolo[3,2-a]azepin-3-carboxylat
und Methyl-[3R-(3α,6α,8α,9aβ)]-6-amino-8-methyl-5-oxo-octahydrothiazolo[3,2-a]azepin-3-carboxylat,
die in Beispiel 7 erhalten wurde, unter Eiskühlung hinzugegeben. Zu dieser
Lösung
wurden 207 mg (1,08 mmol) 1-Ethyl-3-(3-dimethylaminopropyl)-carbodiimid-hydrochlorid
(DEC·HCl),
0,12 ml (1,08 mmol) N-Methylmorpholin und 166 mg (1,08 mmol) 1-Hydroxy-1H-benzotriazol-monohydrat
(HOBt) nacheinander hinzugegeben, gefolgt von 18-stündigem Rühren bei
Raumtemperatur unter einer Stickstoffatmosphäre. Die Reaktionslösung wurde
eingeengt, mit Wasser versetzt und mit Ethylacetat extrahiert. Anschliessend
wurde die organische Schicht mit 1N Salzsäure, einer gesättigten
wässrigen
Natriumhydrogencarbonatlösung
und einer gesättigten wässrigen
Kochsalzlösung
gewaschen und über
wasserfreiem Magnesiumsulfat getrocknet. Nach Filtration wurde der
durch Einengung des Filtrats unter reduziertem Druck erhaltene Rückstand
mittels Silicagel-Säulenchromatografie
(eluiert mit Hexan : Ethylacetat = 3 : 1) gereinigt, wodurch 254
mg einer Mischung (Isomerenverhältnis:
1 : 1,3) der beiden Titelverbindungen erhalten wurden. Diese Mischung
wurde ferner zur Auftrennung und Reinigung auf eine präparative
Säule,
YMC-Pack SIL (SH-043-5) (eluiert mit Hexan : Ethylacetat = 4 : 1)
gegeben. Auf diese Weise wurden 97 mg Methyl-[3R-(3α,6α,8R,9aβ)]-6-[[(2S,3S)-1-oxo-2-acetylthio-3-methylpentyl]amino]-8-methyl-5-oxo-octahydrothiazolo[3,2-a]azepin-3-carboxylat
als farbloses Öl
aus dem ersten Effluenten erhalten. Ausbeute: 27%. Die absolute
Konfiguration der Verbindung wurde mit einem NOE-Experiment bestimmt.
1H-NMR (400 MHz, CDCl3) δ: 7,26 (1H,
brd, J = 6,1 Hz), 5,21 (1H, dd, J = 2,9, 6,6 Hz), 5,20 (1H, dd,
J = 1,5, 10,6 Hz), 4,75 (1H, ddd, J = 5,0, 6,1, 9,0 Hz), 3,95 (1H,
d, J = 7,1 Hz), 3,79 (3H, s), 3,28 (1H, dd, J = 2,9, 12,0 Hz), 3,20
(1H, dd, J = 6,6, 12,0 Hz), 2,37 (3H, s), 2,30–1,10 (8H, m), 1,26 (3H, d,
J = 7,1 Hz), 0,99 (3H, d, J = 6,6 Hz), 0,88 (3H, t, J = 7,4 Hz).
NOE δ: 1,26 (8-Me) ↓ → 4,75 (H6α), 5,20 (H9aα),
68
(H9α) 5,20
(H9aα) ↓ → 1,68 (H9α),
4,75
(H6α) 2,28
(H8β) ↓ → 1,68 (H9α)
-
136 mg Methyl-[3R-(3α,6α,8α,9aβ)]-6-[[(2S,3S)-1-oxo-2-acetylthio-3-methylpentyl]amino]-8-methyl-5-oxo-octahydrothiazolo[3,2-a]azepin-3-carboxylat wurden
als farbloses Öl
aus dem zweiten Effluenten erhalten. Ausbeute: 38%. Die absolute
Konfiguration der Verbindung wurde mit einem NOE-Experiment bestimmt.
1H-NMR (400 MHz, CDCl3) δ: 7,38 (1H,
brd, J = 6,0 Hz), 5,27 (1H, dd, J = 2,4, 6,4 Hz), 5,03 (1H, d, J
= 10,4 Hz), 4,55 (1H, dd, J = 6,4, 10,0 Hz), 3,97 (1H, d, J = 6,8
Hz), 3,79 (3H, s), 3,27 (1H, dd, J = 2,4, 11,8 Hz), 3,19 (1H, dd,
J = 5,4, 11,8 Hz), 2,38 (3H, s), 2,15–1,11 (8H, m), 0,99 (6H, d,
J = 6,8 Hz), 0,89 (3H, t, J = 7,4 Hz).
NOE δ: 2,09 (H8) ↓ → 1,88 (H9α), 1,92 (H7α), 4,55 (H6), 5,03 (H9a)
5,03
(H9a) ↓ → 1,88 (H9α), 4,55 (H6)
4,55
(H6) ↓ → 1,92 (H7α)
-
BEISPIEL
9
[3R-(3α,6α,8α,9aβ)]-6-[[(2S,3S)-1-Oxo-2-thio-3-methylpentyl]amino]-8-methyl-octahydro-5-oxothiazolo[3,2-a]azepin-3-carbonsäure:
-
130 mg (0,30 mmol) des in Beispiel
8 erhaltenen Methyl-[3R-(3α,6α,8α,9aβ)]-6-[[(2S,3S)-1-oxo-2-acetylthio-3-methylpentyl]amino]-8-methyl-octahydro-5-oxothiazolo[3,2-a]azepin-3-carboxylats
wurden in 4,3 ml entlüftetem
EtOH aufgelöst.
Dann wurden 2,1 ml einer wässrigen
1N-Lithiumhydroxidlösung unter
Eiskühlung
hinzugegeben, gefolgt von 1-stündigem
Rühren
bei Raumtemperatur unter einer Stickstoffatmosphäre. Nach Ansäuerung durch
Zugabe von 1,5 ml 2N-Salzsäure unter
Eiskühlung
wurde Wasser zugegeben und dann mit Dichlormethan extrahiert. Nach
Waschen mit einer gesättigten
wässrigen
Kochsalzlösung
wurde über
wasserfreiem Magnesiumsulfat getrocknet und unter reduziertem Druck
eingeengt. Der erhaltene amorphe Feststoff wurde umkristallisiert
(Ethylacetat : Hexan) und mit Warmluft bei 50°C für 24 Stunden getrocknet, wodurch
90 mg der Titelverbindung erhalten wurden. Ausbeute: 80%.
1H-NMR (400 MHz, CDCl3) δ: 7,61 (1H,
brd, J = 6,4 Hz), 5,29 (1H, dd, J = 2,4, 6,4 Hz), 5,07 (1H, d, J
= 10,4 Hz), 4,62 (1H, dd, J = 6,8, 11,2 Hz), 3,37 (1H, dd, J = 2,4,
12,0 Hz), 3,22 (1H, dd, J = 6,4, 88 Hz), 3,21 (1H, dd, J = 6,4,
12,0 Hz), 2,19–1,18
(8H, m), 1,87 (1H, d, J = 8,8 Hz), 1,01 (3H, d, J = 6,4 Hz), 1,00
(3H, d, J = 6,8 Hz), 0,91 (3H, t, J = 7,4 Hz).
-
BEISPIEL
10
[3R-(3α,6α,8β,9aβ)]-6-[[(2S,3S)-1-Oxo-2-thio-3-methylpentyl]amino]-8-methyl-octahydro-5-oxothiazolo[3,2-a]azepin-3-carbonsäure:
-
93 mg (0,216 mmol) des in Beispiel
8 erhaltenen Methyl-[3R-(3α,6α,8β,9aβ)]-6-[[(2S,3S)-1-oxo-2-acetylthio-3-methylpentyl]amino]-8- methyloctahydro-5-oxothiazolo[3,2-a]azepin-3-carboxylats
wurden in 3,1 ml entlüftetem
EtOH aufgelöst.
Dann wurden 1,5 ml einer wässrigen
1N-Lithiumhydroxidlösung unter
Eiskühlung
hinzugegeben, gefolgt von 1-stündigem
Rühren
bei Raumtemperatur unter einer Stickstoffatmosphäre. Nach Ansäuerung durch
Zugabe von 1,1 ml 2N-Salzsäure unter
Eiskühlung
wurde Wasser zugegeben und dann mit Dichlormethan extrahiert. Nach
Waschen mit einer gesättigten
wässrigen
Kochsalzlösung
wurde über
wasserfreiem Magnesiumsulfat getrocknet und unter reduziertem Druck
eingeengt. Der erhaltene amorphe Feststoff wurde umkristallisiert
(Dichlormethan-Hexan) und mit Warmluft bei 50°C für 24 Stunden getrocknet, wodurch
66 mg der Titelverbindung erhalten wurden. Ausbeute: 82%.
1H-NMR (400 MHz, CDCl3) δ: 7,49 (1H,
brd, J = 6,0 Hz), 5,27–5,21
(2H, m), 4,84–4,77
(1H, m), 3,37 (1H, dd, J = 2,4, 12,0 Hz), 3,22 (1H, dd, J = 7,0,
12,0 Hz), 3,19 (1H, dd, J = 7,2, 8,8 Hz), 1,88 (1H, d, J = 8,8 Hz), 2,33–1,58 (7H,
m), 1,28 (3H, d, J = 7,2 Hz), 1,30–1,18 (1H, m), 1,00 (3H, d,
J = 6,8 Hz), 0,90 (3H, t, J = 7,2 Hz).
-
BEISPIEL
11
[3R-(3α,6α,9α,9aβ)]-6-[[(2S,3S)-1-Oxo-2-thio-3-methylpentyl]amino]-9-methyl-5-oxo-octahydrothiazolo[3,2-a]azepin-3-carbonsäure:
-
1,43 g (3,33 mmol) des in Beispiel
2 erhaltenen Methyl-[3R-(3α,6α,9α,9aβ)]-6-[[(2S,3S)-1-oxo-2-acetylthio-3-methylpentyl]amino]-9-methyl-5-oxo-octahydrothiazolo[3,2-a]azepin-3-carboxylats
wurden in 30 ml entlüftetem
Ethanol aufgelöst
und dann wurden unter Eiskühlung
20 ml (20 mmol) einer wässrigen
1N-Lithiumhydroxidlösung
hinzugegeben, gefolgt von 1-stündigem Rühren bei
Raumtemperatur in einer Stickstoffatmosphäre. Die Reaktionslösung wurde
durch Zugabe von 50 ml 2N-Salzsäure
unter Eiskühlung
angesäuert
und mit Wasser verdünnt,
gefolgt von Extraktion mit Dichlormethan. Die organische Schicht
wurde mit einer gesättigten
wässrigen
Kochsalzlösung
gewaschen und über
wasserfreiem Magnesiumsulfat getrocknet. Nach Filtration wurde das
Filtrat unter reduziertem Druck eingeengt. Der erhaltene amorphe
Feststoff wurde umkristallisiert (Dichlormethan-Hexan) und mit Warmluft
bei 50°C
für 12
Stunden getrocknet, wodurch 1,10 g der Titelverbindung in Form weisser
Kristalle erhalten wurden. Ausbeute: 89%.
1H-NMR
(400 MHz, CDCl3) δ: 7,57 (1H, brd, J = 6,4 Hz),
5,25 (1H, s), 5,08 (1H, dd, J = 3,2, 6,8 Hz), 4,60 (1H, m), 3,48
(1H, dd, J = 3,2, 11,6 Hz), 3,25 (1H, dd, J = 6,4, 8,4 Hz), 3,13
(1H, dd, J = 6,8, 11,6 Hz), 2,16– 1,54 (7H, m), 1,85 (1H, d,
J = 8,4 Hz), 1,24 (1H, m), 1,03 (3H, d, J = 6,4 Hz), 1,01 (3H, d,
J = 6,4 Hz), 0,91 (3H, t, J = 7,2 Hz).
-
BEISPIEL
12
[3R-(3α,6α,9β,9aβ)]-6-[[(2S,3S)-1-Oxo-2-acetylthio-3-methylpentyl]amino]-9-methyl-5-oxo-octahydrothiazolo[3,2-a]azepin-3-carbonsäure:
-
522 mg (1,39 mmol) der in Beispiel
3 erhaltenen [3R-(3α,6α,9β,9aβ)]-6-[[(2S,3S)-1-Oxo-2-thio-3-methylpentyl]amino]-9-methyl-5-oxo-octahydrothiazolo[3,2-a]azepin-3-carbonsäure wurden
in Acetonitril (15 ml)-Tetrahydrofuran
(15 ml) aufgelöst.
54 mg (0,42 mmol) wasserfreies Kobaltchlorid und 170 ml (1,81 mmol) Acetanhydrid
wurden bei Raumtemperatur unter einer Stickstoffatmosphäre hinzugegeben,
gefolgt von 5-stündigem
Rühren.
Zu der Reaktionslösung
wurde Wasser hinzugegeben, und dann mit Ethylacetat extrahiert.
Die organische Schicht wurde mit einer gesättigten wässrigen Kochsalzlösung gewaschen
und über
wasserfreiem Magnesiumsulfat getrocknet. Nach Filtration wurde das
Filtrat unter reduziertem Druck eingeengt. Der erhaltene amorphe
Feststoff wurde umkristallisiert, (Dichlormethan-Hexan) und mit
Warmluft bei 50°C
für 18
Stunden getrocknet, wodurch 439 mg der Titelverbindung in Form weisser
Kristalle erhalten wurden. Ausbeute: 76%.
1H-NMR
(400 MHz, CDCl3) δ: 7,38 (1H, brd, J = 6,0 Hz),
5,39 (1H, dd, J = 2,8, 6,4 Hz), 4,83 (1H, d, J = 9,6 Hz), 4,56 (1H,
m), 3,96 (1H, d, J = 6,8 Hz), 3,29 (1H, dd, J = 2,8, 11,6 Hz), 3,12
(1H, dd, J = 6,4, 11,6 Hz), 2,38 (3H, s), 2,14–1,88 (4H, m), 1,77–1,64 (2H,
m), 1,58 (1H, m), 1,16 (1H, m), 1,01 (3H, d, J = 7,2 Hz), 1,00 (3H,
d, J = 6,8 Hz), 0,88 (3H, t, J = 7,2 Hz).
-
BEISPIEL
13
[3R-(3α,6α,9β,9aβ)]-6-[[(2S,3S)-1-Oxo-2-propionylthio-3-methylpentyl]amino]-9-methyl-5-oxo-octahydrothiazolo[3,2-a]azepin-3-carbonsäure:
-
30 mg der Titelverbindung wurden
in Form weisser Kristalle aus 60 mg (0,16 mmol) der in Beispiel
A-3 erhaltenen [3R-(3α,6α,9β,9aβ)]-6-[[(2S,3S)-1-Oxo-2-thio-3-methylpentyl]amino]-9-methyl-5-oxo-octahydrothiazolo[3,2-a]azepin-3-carbonsäure, 17
ml (0,19 mmol) Propionylchlorid und 6 mg (0,05 mmol) wasserfreiem Kobaltchlorid
in der gleichen Weise wie in Beispiel 12 erhalten. Ausbeute: 44%.
1H-NMR (400 MHz, CDCl3) δ: 7,40 (1H,
brd, J = 6,4 Hz), 5,39 (1H, dd, J = 2,4, 6,8 Hz), 4,83 (1H, d, J
= 9,6 Hz), 4,56 (1H, m), 3,98 (1H, d, J = 6,8 Hz), 3,29 (1H, dd,
J = 2,8, 11,6 Hz), 3,11 (1H, dd, J = 6,8, 11,6 Hz), 2,63 (2H, q,
J = 7,6 Hz), 2,16–1,88
(4H, m), 1,76–1,64
(2H, m), 1,57 (1H, m), 1,19 (3H, t, J = 7,6 Hz), 1,17 (1H, m), 1,01 (3H,
d, J = 6,8 Hz), 1,00 (3H, d, J = 6,8 Hz), 0,88 (3H, t, J = 7,2 Hz).
-
BEISPIEL
14
[3R-(3α,6α,9β,9aβ)]-6-[[(2S,3S)-1-Oxo-2-benzoylthio-3-methylpentyl]amino]-9-methyl-5-oxo-octahydrothiazolo[3,2-a]azepin-3-carbonsäure:
-
490 mg der Titelverbindung wurden
in Form weisser Kristalle aus 434 mg (1,16 mmol) der in Beispiel A-3
erhaltenen [3R-(3α,6α,9β,9aβ)]-6-[[(2S,3S)-1-Oxo-2-thio-3-methylpentyl]amino]-9-methyl-5-oxo-octahydrothiazolo[3,2-a]azepin-3-carbonsäure, 300
ml (1,33 mmol) Benzoesäureanhydrid
und 45 mg (0,35 mmol) wasserfreiem Kobaltchlorid in der gleichen
Weise wie in Beispiel 12 erhalten. Ausbeute: 88%.
1H-NMR
(400 MHz, CDCl3) δ: 8,00–7,96 (2H, m), 7,62–7,42 (4H,
m), 5,38 (1H, dd, J = 2,4, 6,4 Hz), 4,84 (1H, d, J = 9,6 Hz), 4,59
(1H, m), 4,20 (1H, d, J = 7,2 Hz), 3,27 (1H, dd, J = 2,4, 11,6 Hz),
3,10 (1H, dd, J = 6,4, 11,6 Hz), 2,22–1,60 (7H, m), 1,25 (1H, m),
1,06 (3H, d, J = 6,8 Hz), 1,00 (3H, d, J = 6,8 Hz), 0,92 (3H, t,
J = 7,2 Hz).
-
BEISPIEL
15
[3R-(3α,6α,9β,9aβ)]-6-[[(2S,3S)-1-Oxo-2-(1,1-dimethylpropionyl)thio-3-methylpentyl]amino]-9-methyl-5-oxo-octahydrothiazolo[3,2-a]azepin-3-carbonsäure:
-
58 mg der Titelverbindung wurden
in Form weisser Kristalle aus 54 mg (0,14 mmol) der in Beispiel
A-3 erhaltenen [3R-(3α,6α,9β,9aβ)]-6-[[(2S,3S)-1-Oxo-2-thio-3-methylpentyl]amino]-9-methyl-5-oxo-octahydrothiazolo[3,2-a]azepin-3-carbonsäure, 27
ml (0,22 mmol) Pivaloylchlorid und 6 mg (0,05 mmol) wasserfreiem
Kobaltchlorid in der gleichen Weise wie in Beispiel 12 erhalten.
Ausbeute: 88%.
1H-NMR (400 MHz, CDCl3) δ:
7,41 (1H, brd, J = 6,0 Hz), 5,39 (1H, dd, J = 2,4, 6,4 Hz), 4,83
(1H, d, J = 9,6 Hz), 4,56 (1H, m), 3,92 (1H, d, J = 6,8 Hz), 3,29
(1H, dd, J = 2,4, 11,6 Hz), 3,10 (1H, dd, J = 6,4, 11,6 Hz), 2,18–1,52 (7H,
m), 1,25 (9H, s), 1,20 (1H, m), 1,01 (3H, d, J = 6,8 Hz), 1,00 (3H,
d, J = 6,8 Hz), 0,87 (3H, t, J = 7,2 Hz).
-
BEISPIEL
16
[3R-(3α,6α,9β,9aβ)]-6-[[(2S,3S)-1-Oxo-2-(4-morpholinyl)acetylthio-3-methylpentyl]amino]-9-methyl-5-oxo-octahydrothiazolo[3,2-a]azepin-3-carbonsäure:
-
44 mg (0,24 mmol) 4-Morpholinylessigsäure-hydrochlorid
wurden in entlüftetem
wasserfreiem N,N-Dimethylformamid (1,2 ml) in einer Stickstoffatmosphäre aufgelöst, und
dann wurden unter Eiskühlung
27,3 mg (1,68 mmol) N,N'-Carbodiimidazol
hinzugegeben, gefolgt von 1-stündigem
Rühren
bei Raumtemperatur. Eine Lösung
von 60 mg (0,16 mmol) der in Beispiel 3 erhaltenen [3R-(3α,6α,9β,9aβ)]-6-[[(2S,3S)-1-Oxo-2-thio-3-methylpentyl]amino]-9-methyl-5-oxo-octahydrothiazolo[3,2-a]azepin-3-carbonsäure in entlüftetem trockenem
Tetrahydrofuran (1,6 ml) wurde unter Eiskühlung zugegeben, gefolgt von
weiterem 2-tägigen
Rühren
bei Raumtemperatur. Nach Einengung der Reaktionslösung wurden
Ethylacetat und eine gesättigte
wässrige
Kochsalzlösung
zugegeben, wodurch eine Flüssig-Flüssig-Auftrennung
bewirkt wurde. Die organische Schicht wurde mit einer gesättigten
wässrigen
Kochsalzlösung gewaschen
und dann über
wasserfreiem Magnesiumsulfat getrocknet. Nach Filtration wurde das
Filtrat unter reduziertem Druck eingeengt. Der erhaltene amorphe
Feststoff wurde umkristallisiert (Ethylacetat-Ether-Hexan) und mit
Warmluft bei 50°C über Nacht
getrocknet, wodurch 68 mg der Titelverbindung in Form weisser Kristalle
erhalten wurden. Ausbeute: 85%.
1H-NMR
(400 MHz, CDCl3) δ: 7,40 (1H, brd, J = 6,0 Hz),
5,35 (1H, dd, J = 2,4, 6,8 Hz), 4,82 (1H, d, J = 9,2 Hz), 4,55 (1H,
m), 3,92 (1H, d, J = 6,8 Hz), 3,77 (4H, t, J = 4,4 Hz), 3,31 (2H,
s), 3,28 (1H, dd, J = 2,4, 11,6 Hz), 3,12 (1H, dd, J = 6,8,11,6
Hz), 2,62 (4H, m), 2,14–1,52
(7H, m), 1,17 (1H, m), 1,01 (3H, d, J = 6,4 Hz), 1,00 (3H, d, J
= 6,8 Hz), 0,87 (3H, t, J = 7,6 Hz).
-
BEISPIEL
17
[3R-(3α,6α,9β,9aβ)]-6-[[(2S,3S)-1-Oxo-2-(4-thiomorpholinyl)acetylthio-3-methylpentyl]amino]-9-methyl-5-oxo-octahydrothiazolo[3,2-a]azepin-3-carbonsäure:
-
81 mg der Titelverbindung wurden
in Form weisser Kristalle aus 70 mg (0,19 mmol) der in Beispiel
3 erhaltenen [3R-(3α,6α,9β,9aβ)]-6-([(2S,3S)-1-Oxo-2-thio-3-methylpentyl]amino]-9-methyl-5-oxo- octahydrothiazolo[3,2-a]azepin-3-carbonsäure, 55
mg (0,28 mmol) 4-Thiomorpholinylessigsäure-hydrochlorid und 33 mg (0,21
mmol) N,N'-Carbodiimidazol in
gleicher Weise wie in Beispiel 16 erhalten. Ausbeute: 84%.
1H-NMR (400 MHz, CDCl3) δ: 7,40 (1H,
brd, J = 6,4 Hz), 5,33 (1H, m), 4,83 (1H, d, J = 9,6 Hz), 4,56 (1H,
m), 3,89 (1H, d, J = 7,2 Hz), 3,32–3,26 (1H, m), 3,30 (2H, s),
3,12 (1H, dd, J = 6,4, 11,2 Hz), 2,88–2,82 (2H, m), 2,76–2,70 (2H,
m), 2,14–1,90
(4H, m), 1,78–1,54
(3H, m), 1,18 (1H, m), 1,01 (3H, d, J = 6,8 Hz), 1,00 (3H, d, J
= 6,4 Hz), 090 (3H, t, J = 7,6 Hz).
-
BEISPIEL
18
3R-(3α,6α,9β,9aβ)]-6-[[(2S,3S)-1-Oxo-2-(4-dioxo-thiomorpholinyl)acetylthio-3-methylpentyl]amino]-9-methyl-5-oxo-octahydrothiazolo[3,2-a]azepin-3-carbonsäure:
-
73 mg der Titelverbindung wurden
in Form weisser Kristalle aus 70 mg (0,19 mmol) der in Beispiel
3 erhaltenen [3R-(3α,6α,9β,9aβ)]-6-([(2S,3S)-1-Oxo-2-thio-3-methylpentyl]amino]-9-methyl-5-oxo-octahydrothiazolo[3,2-a]azepin-3-carbonsäure, 54
mg (0,28 mmol) 4-Dioxothiomorpholinylessigsäure und 33 mg (0,21 mmol) N,N'-Carbodiimidazol
in gleicher Weise wie in Beispiel 16 erhalten. Ausbeute: 71%.
1H-NMR (400 MHz, CDCl3) δ: 7,50 (1H,
brd, J = 6,4 Hz), 5,27 (1H, dd, J = 3,2, 6,8 Hz), 4,81 (1H, d, J
= 9,6 Hz), 4,55 (1H, m), 3,96 (1H, d, J = 6,4 Hz), 3,46 (2H, s),
3,27 (H, dd, J = 3,2, 12,0 Hz), 3,24–3,10 (5H, m), 2,18–1,92 (4H,
m), 1,76–1,63
(2H, m), 1,55 (1H, m), 1,17 (1H, m), 1,01 (3H, d, J = 6,8 Hz), 1,00
(3H, d, J = 6,8 Hz), 0,90 (3H, t, J = 7,6 Hz).
-
BEISPIEL
19
[3R-(3α,6α,9β,9aβ)]-6-[[(2S,3S)-1-Oxo-2-(nicotinoyl)thio-3-methylpentyl]amino]-9-methyl-5-oxo-octahydrothiazolo[3,2-a]azepin-3-carbonsäure:
-
28 mg der Titelverbindung wurden
in Form weisser Kristalle aus 50 mg (0,13 mmol) der in Beispiel
3 erhaltenen [3R-(3α,6α,9β,9aβ)]-6-[[(2S,3S)-1-Oxo-2-thio-3-methylpentyl]amino]-9-methyl-5-oxo-octahydrothiazolo[3,2-a]azepin-3-carbonsäure, 18
mg (0,15 mmol) Nicotinsäure
und 23 mg (0,14 mmol) N,N'-Carbodiimidazol
in gleicher Weise wie in Beispiel 12 erhalten. Ausbeute: 44%.
1H-NMR (400 MHz, CDCl3) δ: 9,17 (1H,
br), 8,80 (1H, br), 8,22 (1H, brd, J = 8,4 Hz), 7,58 (1H, br), 7,43
(1H, m), 5,27 (1H, br), 4,82 (1H, br), 4,60 (1H, br), 4,23 (1H,
brd, J = 7,2 Hz), 3,34 (1H, br), 3,12 (1H, br), 2,24–1,92 (4H,
m), 1,80–1,58
(3H, m), 1,24 (1H, m), 1,06 (3H, d, J = 6,8 Hz), 1,00–0,84 (6,
m).
-
BEISPIEL
20
[3R-(3α,6α,9α,9aβ)]-6-[[(2S,3S)-1-Oxo-2-acetylthio-3-methylpentyl]amino]-9-methyl-5-oxo-octahydrothiazolo[3,2-a]azepin-3-carbonsäure:
-
163 mg der Titelverbindung wurden
in Form weisser Kristalle aus 212 mg (0,57 mmol) der in Beispiel 11
erhaltenen [3R-(3α,6α,9α,9aβ)]-6-[[(2S,3S)-1-Oxo-2-thio-3-methylpentyl]amino]-9-methyl-5-oxo-octahydrothiazolo[3,2-a]azepin-3-carbonsäure, 64
ml (0,68 mmol) Acetanhydrid und 22 mg (0,17 mmol) wasserfreiem Kobaltchlorid
in gleicher Weise wie in Beispiel 12 erhalten. Ausbeute: 69%.
1H-NMR (400 MHz, CDCl3) δ: 7,30–7,20 (1H,
m), 5,28 (1H, s), 5,08 (1H, dd, J = 2,4, 6,4 Hz), 4,59 (1H, m),
3,95 (1H, d, J = 7,2 Hz), 3,48 (1H, dd, J = 2,4, 11,6 Hz), 3,12
(1H, dd, J = 6,4, 11,6 Hz), 2,40 (3H, s), 2,16–1,52 (7H, m), 1,18 (1H, m),
1,01 (3H, d, J = 6,4 Hz), 1,00 (3H, d, J = 7,2 Hz), 0,91 (3H, t,
J = 7,2 Hz).
-
BEISPIEL
21
[3R-(3α,6α,9α,9aβ)]-6-[[(2S,3S)-1-Oxo-2-benzoylthio-3-methylpentyl]amino]-9-methyl-5-oxo-octahydrothiazolo[3,2-a]azepin-3-carbonsäure:
-
163 mg der Titelverbindung wurden
in Form weisser Kristalle aus 265 mg (0,71 mmol) der in Beispiel 11
erhaltenen [3R-(3α,6α,9α,9aβ)]-6-[[(2S,3S)-1-Oxo-2-thio-3-methylpentyl]amino]-9-methyl-5-oxo-octahydrothiazolo[3,2-a]azepin-3-carbonsäure, 176
mg (0,78 mmol) Benzoesäureanhydrid
und 28 mg (0,21 mmol) wasserfreiem Kobaltchlorid in gleicher Weise
wie in Beispiel 12 erhalten. Ausbeute: 48%.
1H-NMR
(400 MHz, CDCl3) δ: 7,99 (2H, m), 7,60–7,40 (4H,
m), 5,22 (1H, s), 5,03 (1H, br), 4,60 (1H, m), 4,08 (1H, br), 3,42
(1H, br), 3,03 (1H, br), 2,20–1,60
(7H, m), 1,24 (1H, m), 1,10–0,90
(9H, m).
-
BEISPIEL
22
Methyl-[3R-(3α,6α,9α,9aβ)]-6-amino-9-ethyl-5-oxo-octahydrothiazolo[3,2-a]azepin-3-carboxylat:
-
1,3 g (4,13 mmol) des in Synthesebeispiel
19 erhaltenen Methyl-[3R-(3α,6α,9α,9aβ)]-6-acetylamino-9-ethyl-5-oxo-octahydrothiazolo[3,2-a]azepin-3-carboxylats wurden
in einer 10%-igen Salzsäureösung (50 ml)
in Methanol aufgelöst,
gefolgt von 2-stündigem
Erwärmen
unter Rückfluss.
Nach Abdestillieren des Lösungsmittel
unter reduziertem Druck wurde Wasser zugegeben und dann mit Dichlormethan
gewaschen. Nachdem die erhaltene wässrige Schicht durch Zugabe
einer gesättigten
wässrigen
Natriumhydrogencarbonatlösung
basisch gemacht worden war, wurde mit Dichlormethan extrahiert und
dann über
wasserfreiem Natriumsulfat getrocknet. Nach Filtration wurde das
Filtrat unter reduziertem Druck eingeengt, wodurch 0,83 g der Titelverbindung
als farbloses Öl
erhalten wurden. Ausbeute: 74%.
1H-NMR
(400 MHz, CDCl3) δ: 5,32 (1H, dd, J = 3,5, 6,8
Hz), 4,89 (d, J = 9,2 Hz), 3,78 (3H, s), 3,55 (1H, dd, J = 2,0,
10,5 Hz), 3,21 (1H, dd, J = 3,5, 11,6 Hz), 3,09 (1H, dd, J = 6,8,
11,6 Hz), 2,20–1,40
(9,0, m), 0,92 (3H, t, J = 7,6 Hz).
-
BEISPIEL
23
Methyl-[3R-(3α,6α,9α,9aβ)]-6-[[(2S,3S)-1-oxo-2-acetylthio-3-methylpentyl]amino]-9-ethyl-5-oxo-octahydrothiazolo[3,2-a]azepin-3-carboxylat:
-
Eine Lösung von 0,70 g (3,66 mmol)
(2S,3S)-2-Acetylthio-3-methylpentansäure in Tetrahydrofuran
(50 ml) wurde zu 0,83 g (3,05 mmol) des in Beispiel 22 erhaltenen
[3R-(3α,6α,9α,9aβ)]-6-Amino]-9-ethyl-5-oxo-octahydrothiazolo[3,2-a]azepin-3-carboxylats
unter Eiskühlung
hinzugegeben. Zu dieser Lösung
wurden nacheinander 0,70 g (3,66 mmol) 1-Ethyl-3-(3-dimethylaminopropyl)carbodiimid-hydrochlorid
(DEC·HCl),
0,4 ml (3,66 mmol) N-Methylmorpholin und 0,50 g (9,3 mmol) 1-Hydroxy-1H-benzotriazolmonohydrat
(HOBt) hinzugegeben, gefolgt von 10-stündigem Rühren bei Raumtemperatur unter
einer Stickstoffatmosphäre.
Zu der Reaktionslösung
wurde Wasser hinzugegeben und dann mit Ethylacetat extrahiert. Dann
wurde die organische Schicht mit 1N-Salzsäure, einer gesättigten
wässrigen
Natriumhydrogencarbonatlösung
und einer gesättigten wässrigen
Kochsalzlösung
gewaschen und über
wasserfreiem Magnesiumsulfat getrocknet. Nach Filtration wurde das
Filtrat unter reduziertem Druck eingeengt und der erhaltene Rückstand
mittels Silicagel-Säulenchromatografie
(Hexan-Ethylacetat
= 3 : 1) gereinigt, wodurch 476 mg der Titelverbindung als farbloses Öl erhalten
wurden. Ausbeute: 35%.
1H-NMR (400
MHz, CDCl3) δ: 7,36 (1H, d, J = 6,0 Hz),
5,34 (1H, dd, J = 3,2, 6,4 Hz), 4,86 (1H, d, J = 9,6 Hz), 4,54 (1H,
m), 3,97 (1H, d, J = 6,8 Hz), 3,78 (3H, s), 3,21 (1H, dd, J = 3,2,
12,0 Hz), 3,11 (1H, dd, J = 6,4, 12,0 Hz), 2,38 (3H, s), 2,16–2,04 (2H,
m), 1,82–1,52
(6H, m), 1,31 (1H, m), 16 (1H, m), 0,99 (3H, d, J = 6,4 Hz), 0,91 (3H,
t, J = 7,2 Hz), 0,88 (3H, t, J = 7,2 Hz).
-
BEISPIEL
24
[3R-(3α,6α,9α,9aβ)]-6-[[(2S,3S)-1-Oxo-2-thio-3-methylpentyl]amino]-9-ethyl-5-oxo-octahydrothiazolo[3,2-a]azepin-3-carbonsäure:
-
476 mg (1,07 mmol) des in Beispiel
23 erhaltenen Methyl-[3R-(3α,6α,9α,9aβ)]-6-[[(2S,3S)-1-oxo-2-acetylthio-3-methylpentyl]amino]-9-ethyl-5-oxo-octahydrothiazolo[3,2-a]azepin-3-carboxylats
wurden in 10,7 ml entlüftetem
Ethanol aufgelöst,
und unter Eiskühlung
wurden 5,36 ml einer entlüfteten
wässrigen
1N-Lithiumhydroxidlösung
hinzugegeben, gefolgt von 1-stündigem
Rühren bei
Raumtemperatur unter einer Stickstoffatmosphäre. Die Reaktionslösung wurde
durch Zugabe von 2N-Salzsäure
unter Eiskühlung
angesäuert
und mit Wasser verdünnt,
gefolgt von der Extraktion mit Dichlormethan. Die organische Schicht
wurde mit einer gesättigten
wässrigen
Kochsalzlösung
gewaschen und über wasserfreiem
Magnesiumsulfat getrocknet. Nach Filtration wurde das Filtrat unter
reduziertem Druck eingeengt. Der erhaltene amorphe Feststoff wurde
mit Hexan trituriert und dann durch Filtration abgetrennt. Dieser wurde
für 12
Stunden mit Warmluft bei 50°C
getrocknet, wodurch 300 mg der Titelverbindung erhalten wurden. Ausbeute:
72%.
1H-NMR (400 MHz, CDCl3) δ: 7,66 (1H,
d, J = 6,4 Hz), 5,36 (1H, dd, J = 3,2, 6,4 Hz), 4,91 (1H, d, J =
9,2 Hz), 4,61 (1H, m), 3,28 (1H, dd, J = 3,2, 12,0 Hz), 3,22 (1H,
dd, J = 6,8, 8,8 Hz), 3,13 (1H, dd, J = 6,4, 12,0 Hz), 2,16–2,08 (2H,
m), 1,98 (1H, m), 1,87 (1H, d, J = 8,8 Hz), 1,84–1,56 (5H, m), 1,38–1,18 (2H,
m), 0,99 (3H, d, J = 6,4 Hz), 0,93 (3H, t, J = 7,2 Hz), 0,90 (3H,
t, J = 7,6 Hz).
 und v und w sind jeweils 1 oder 2.
und v und w sind jeweils 1 oder 2.
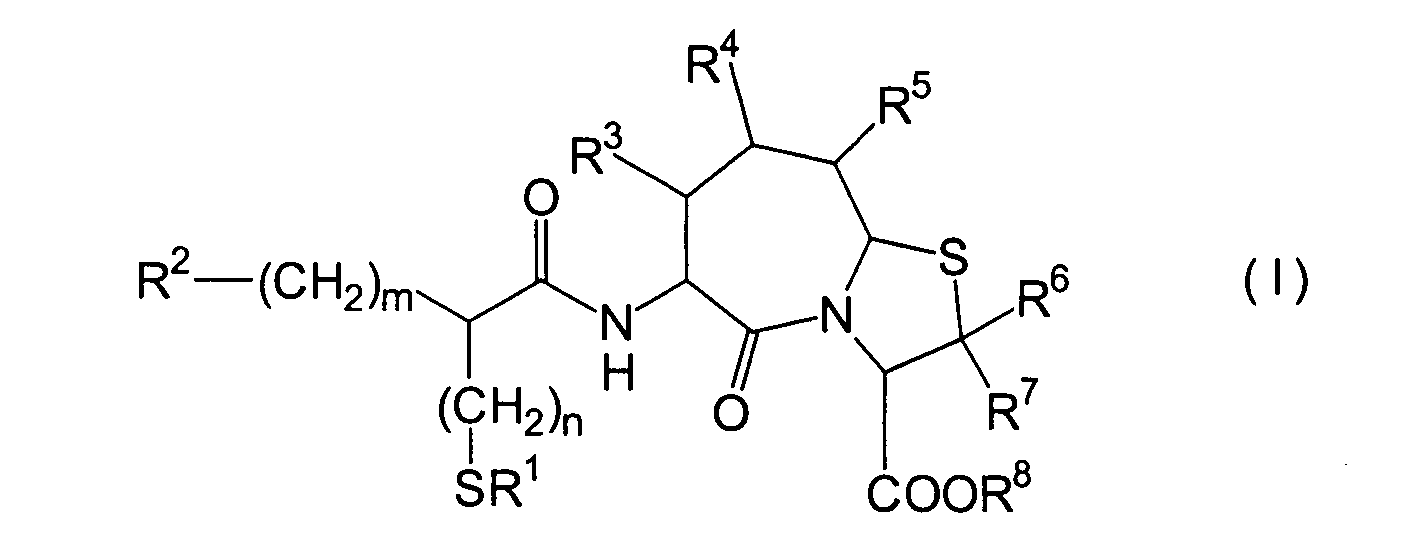
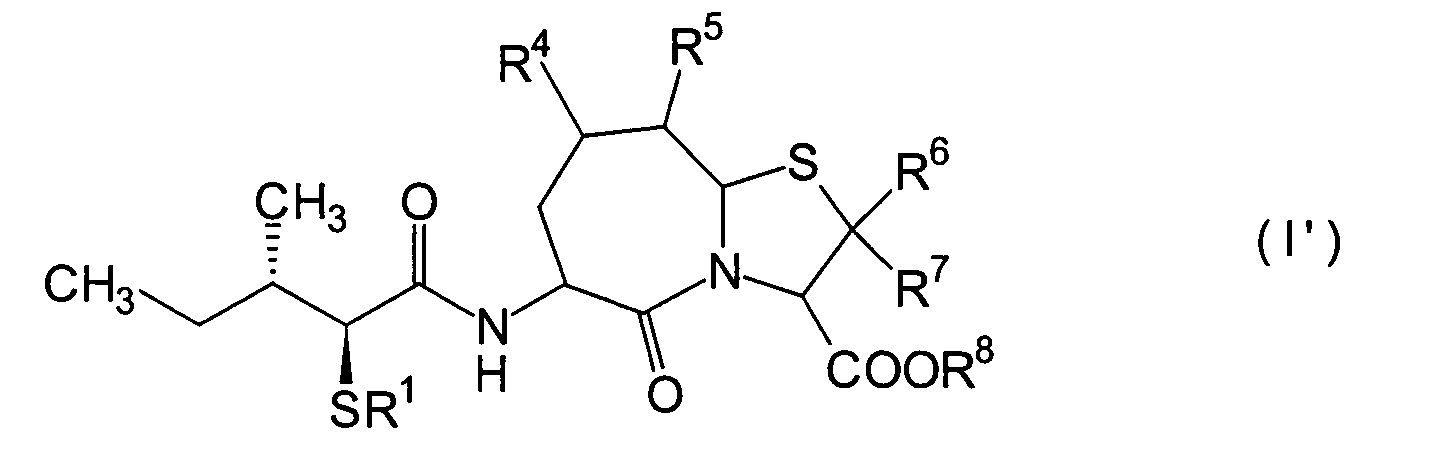

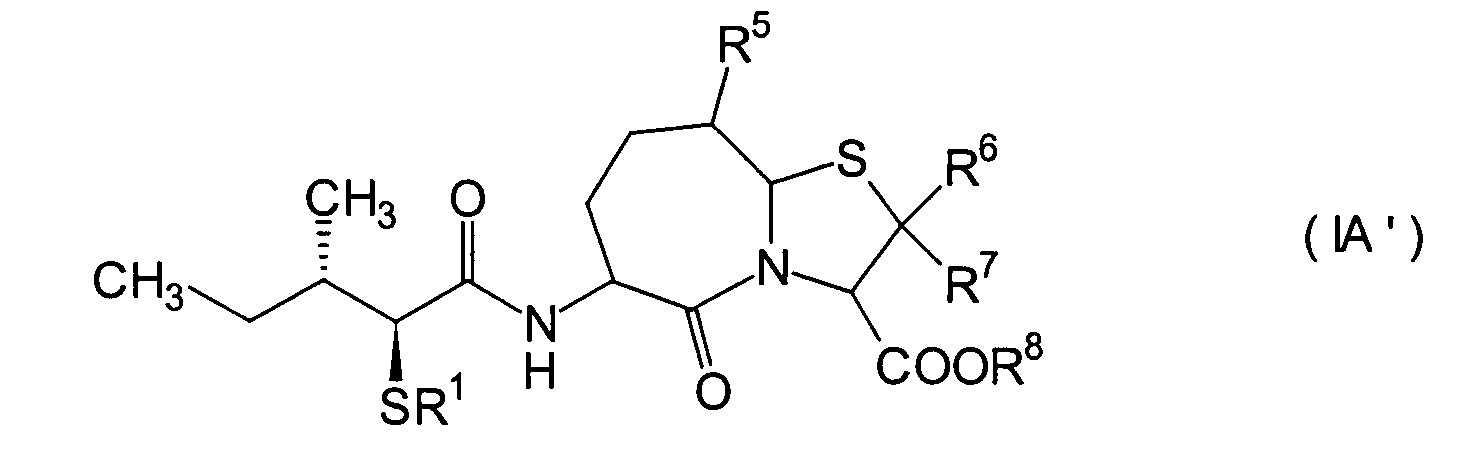



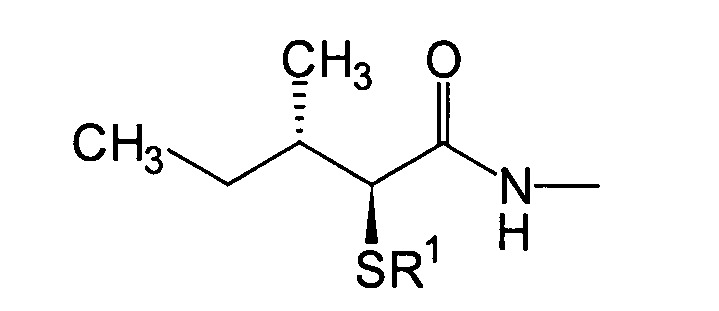





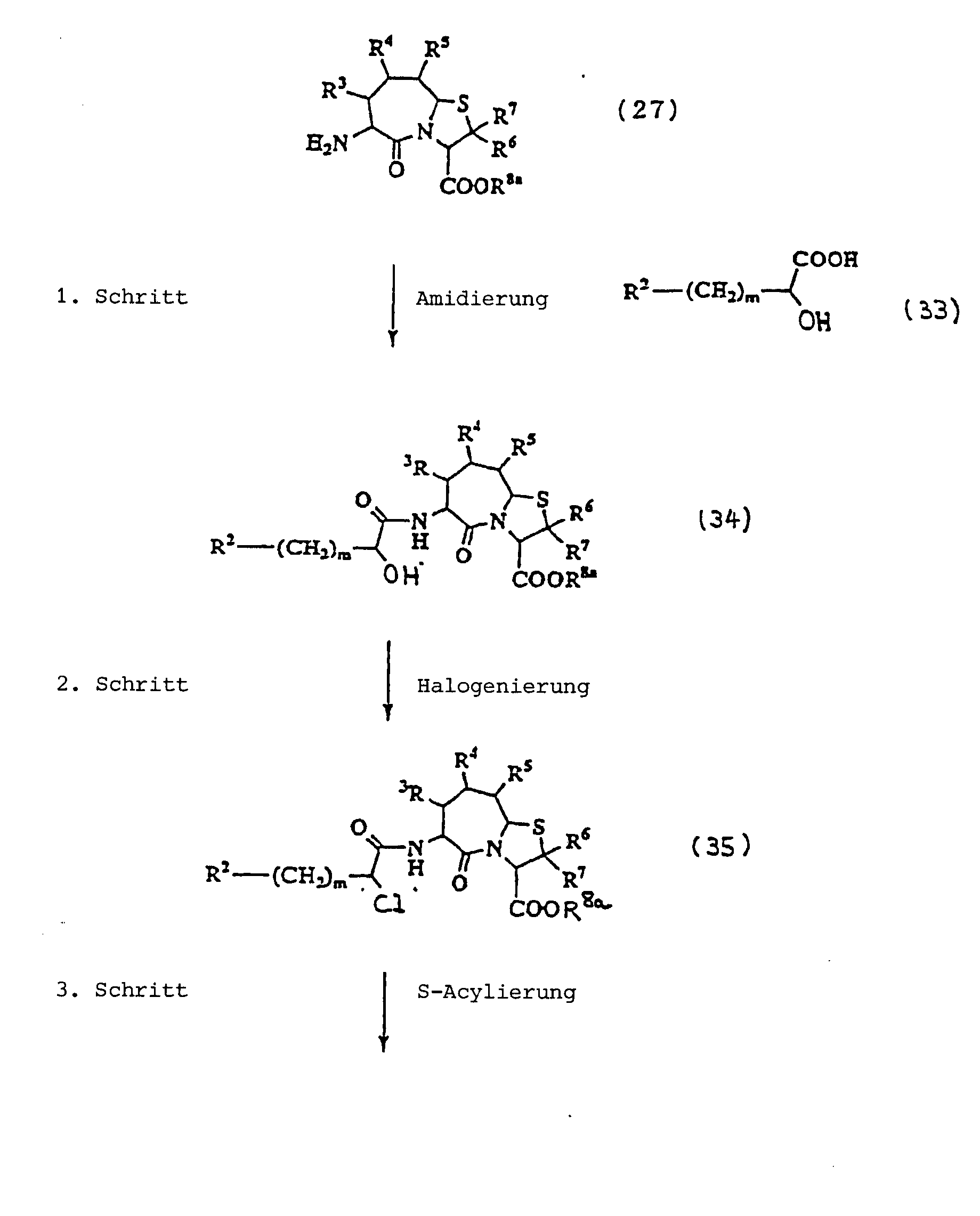
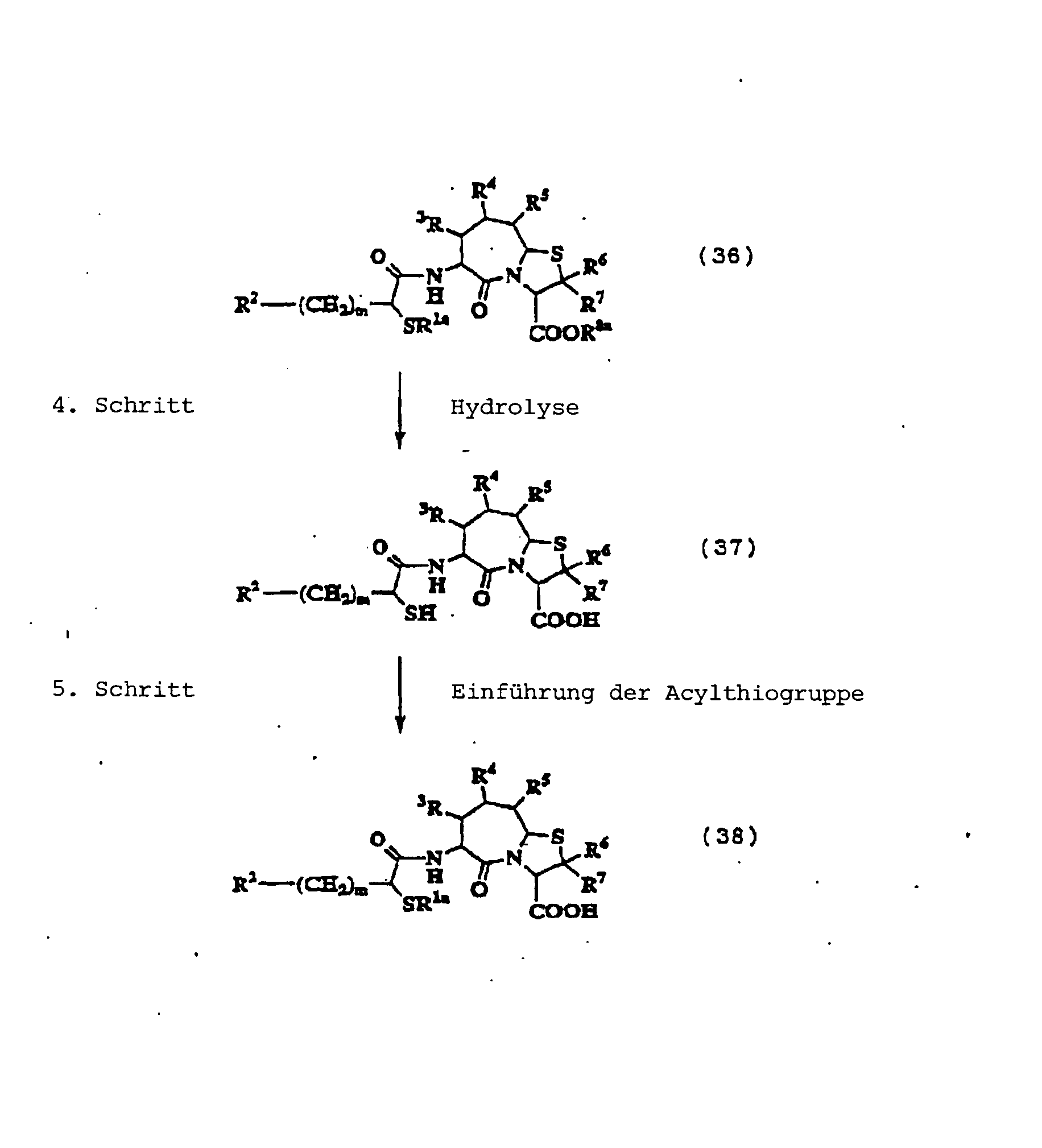
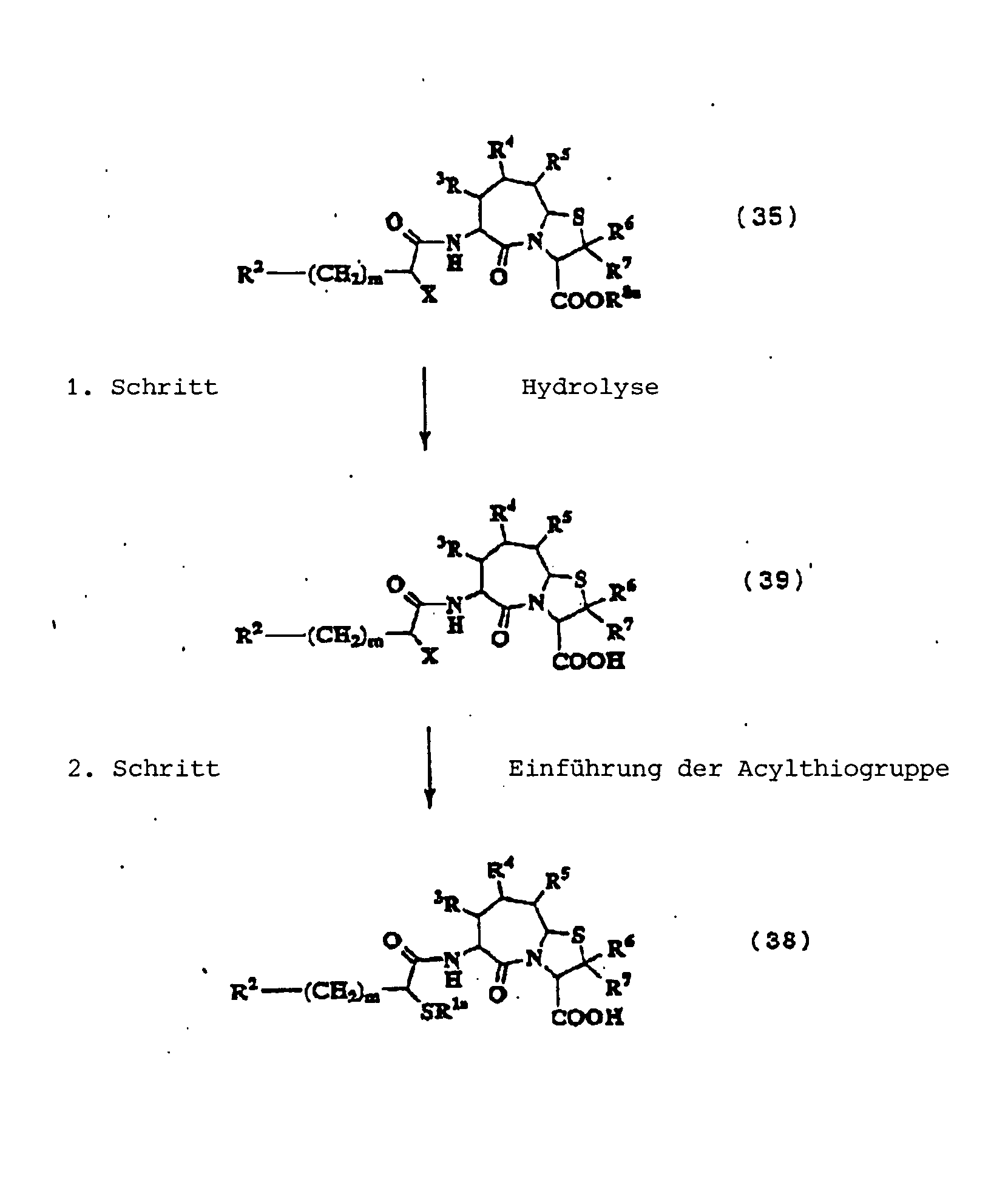
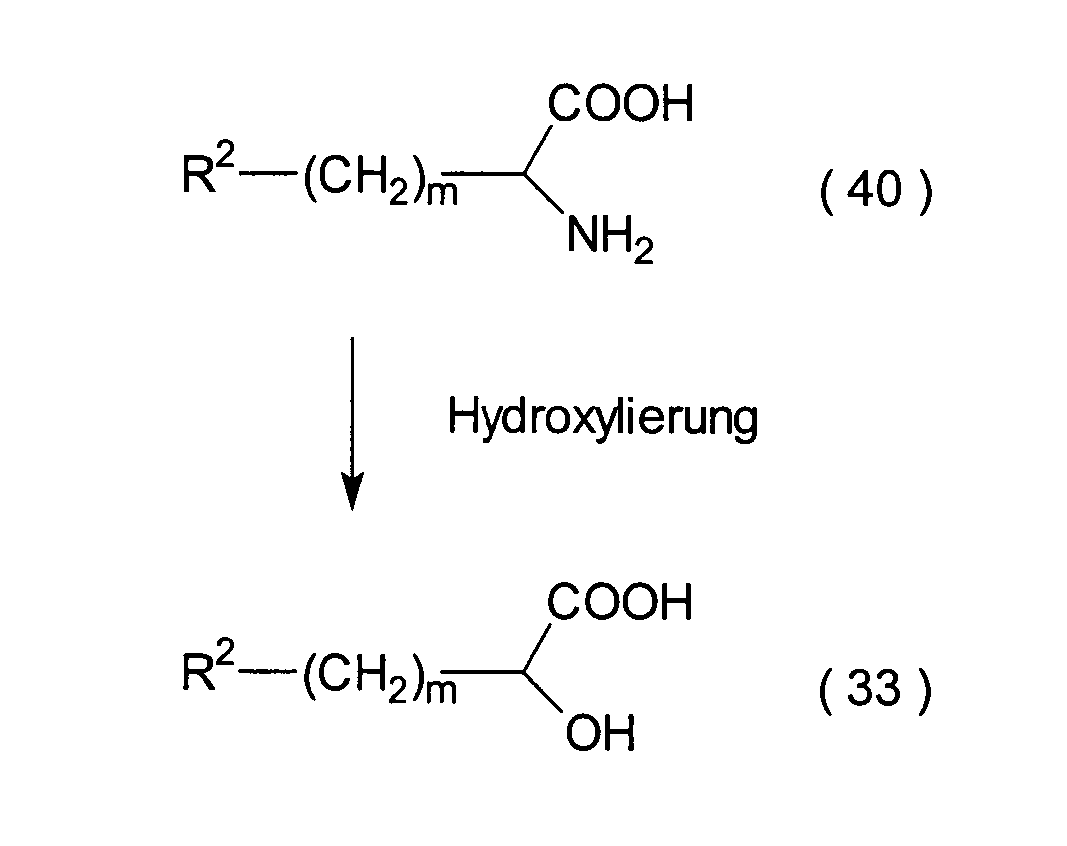





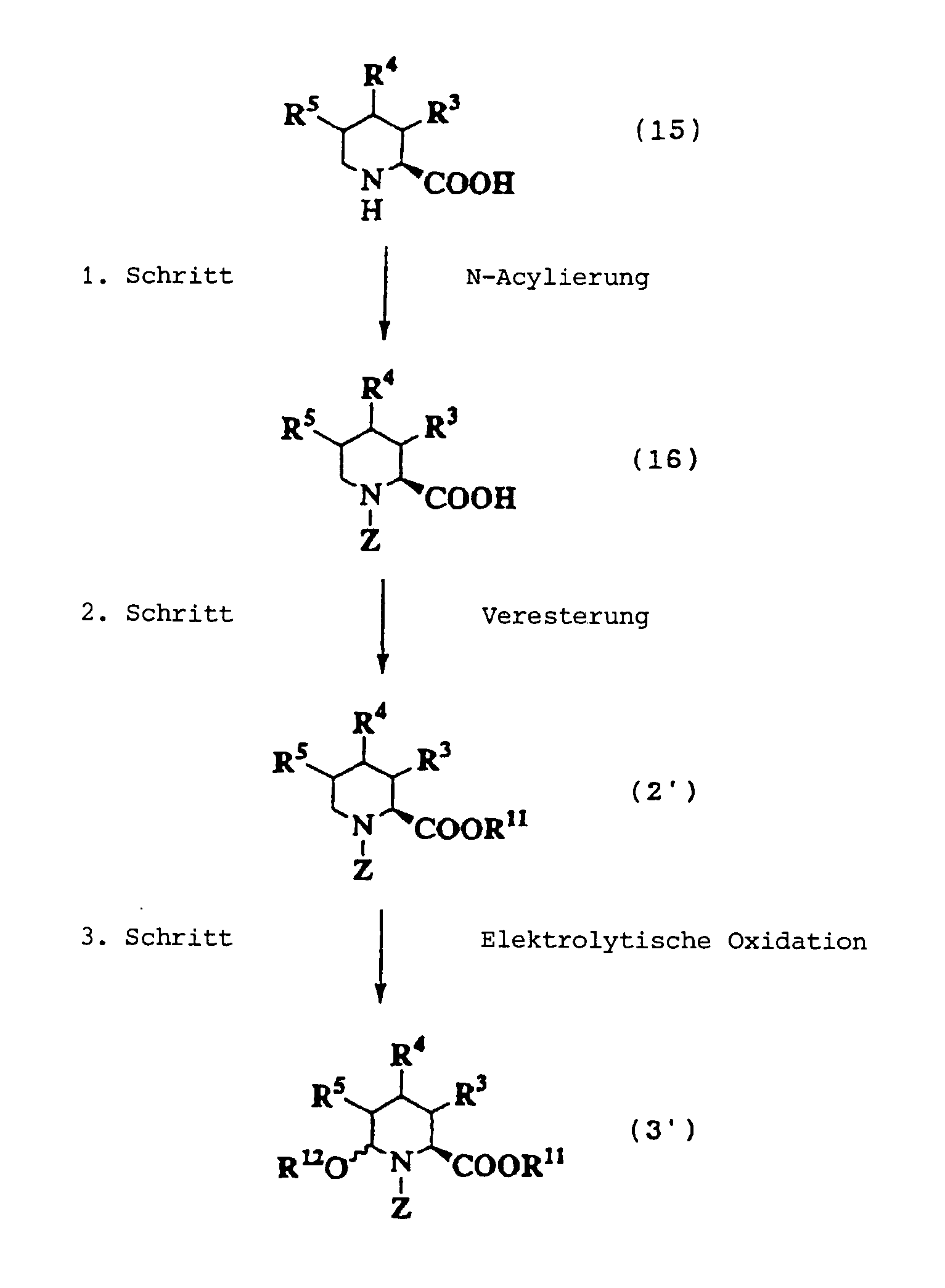
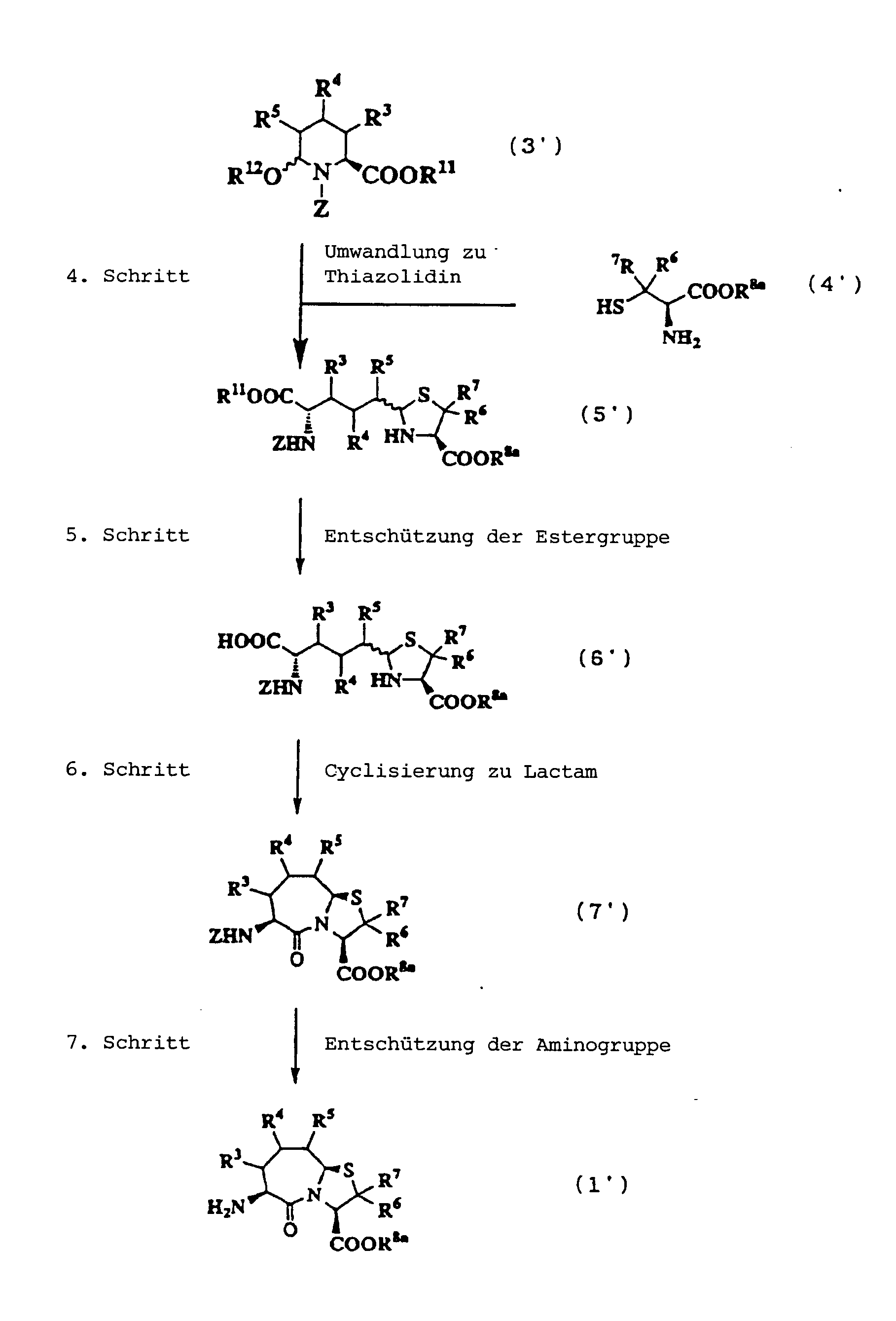
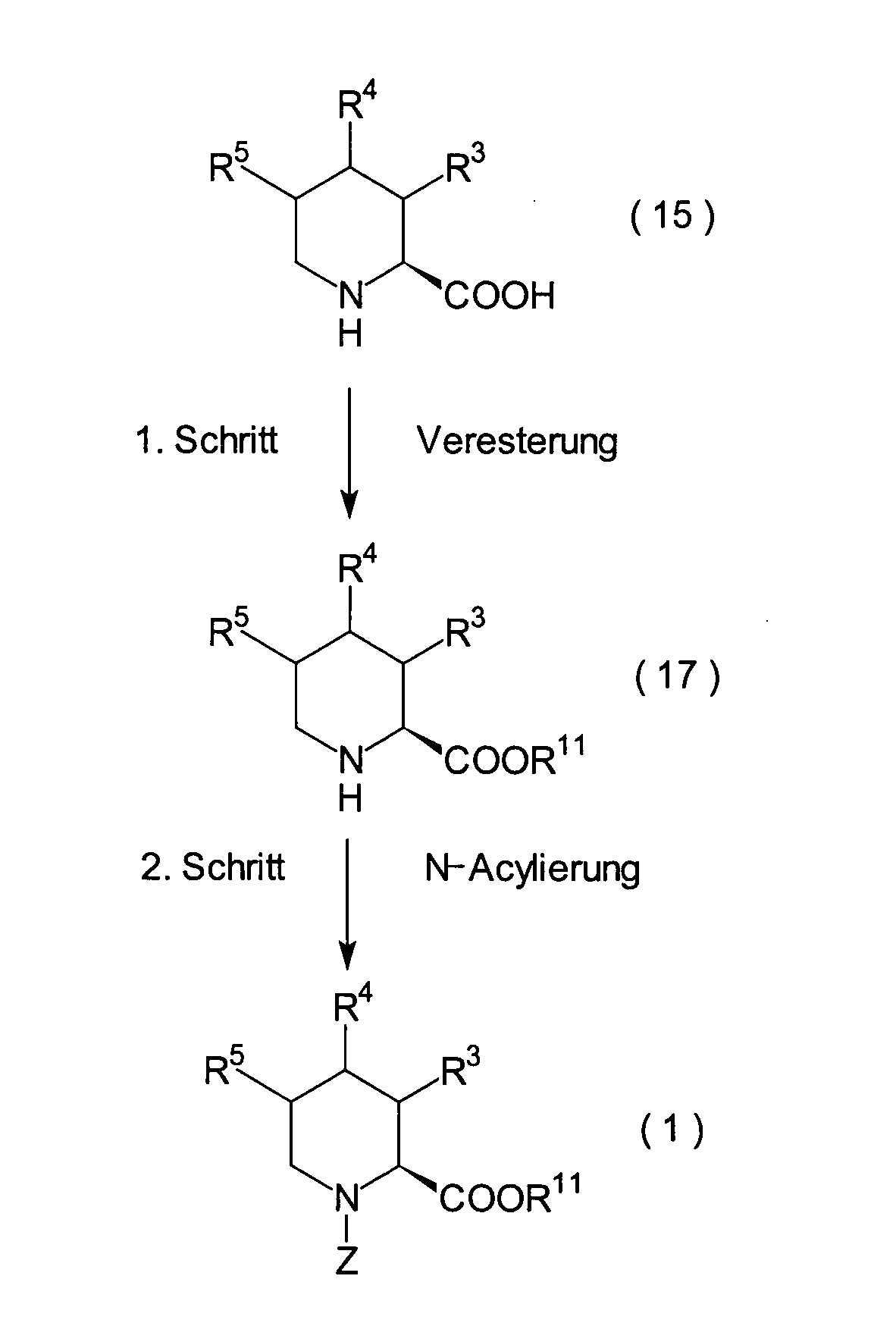


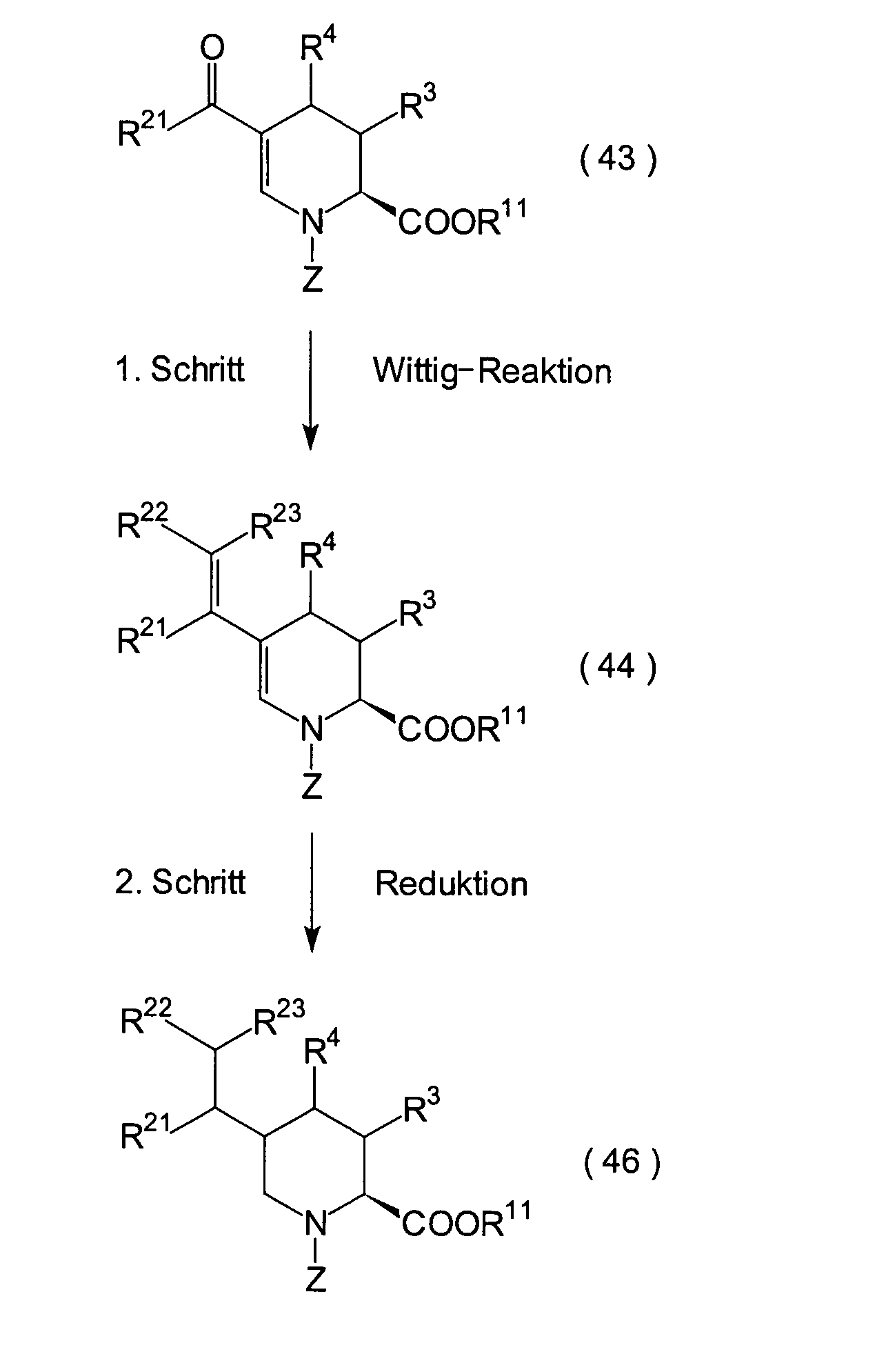


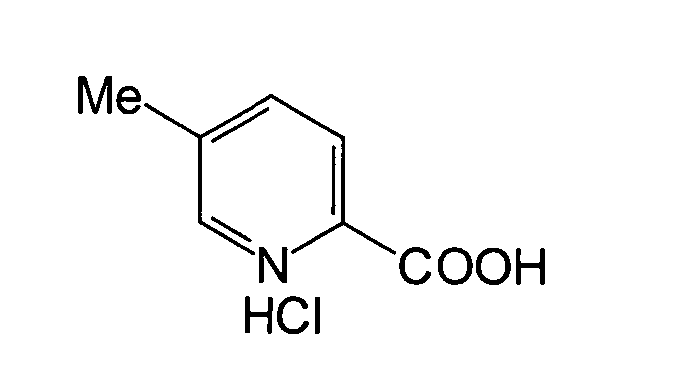
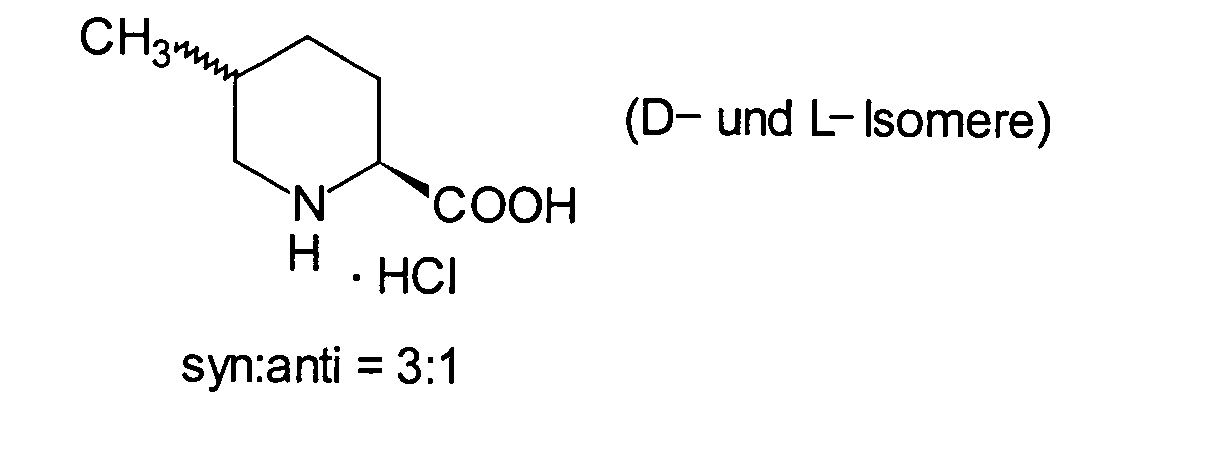



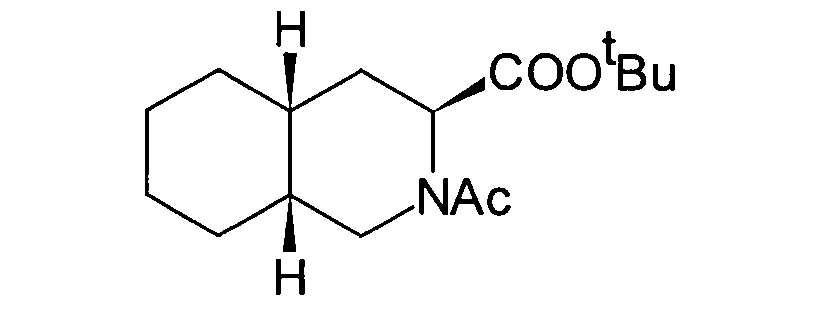

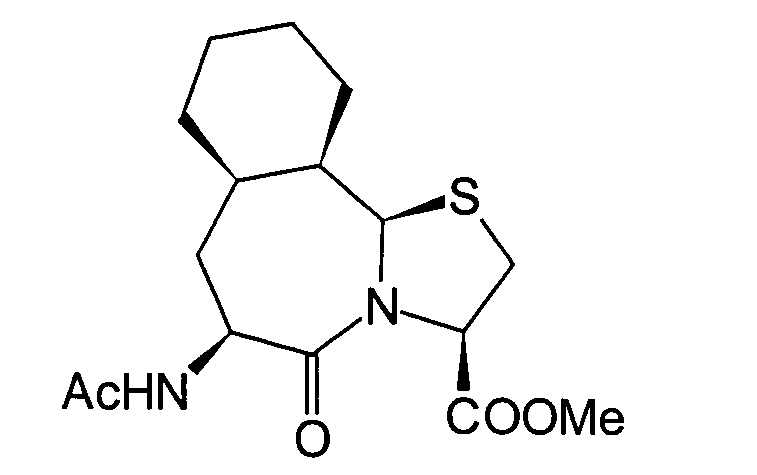



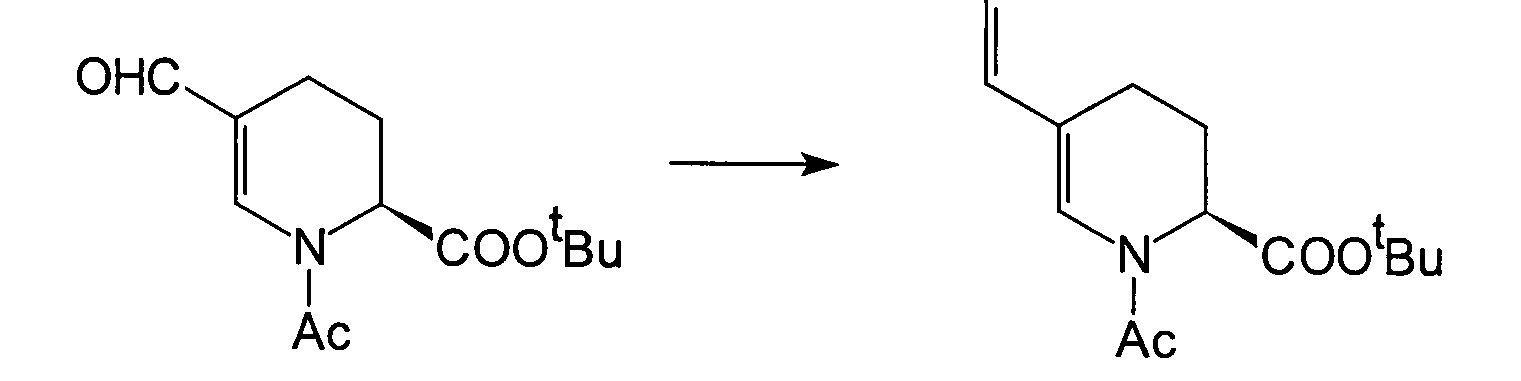
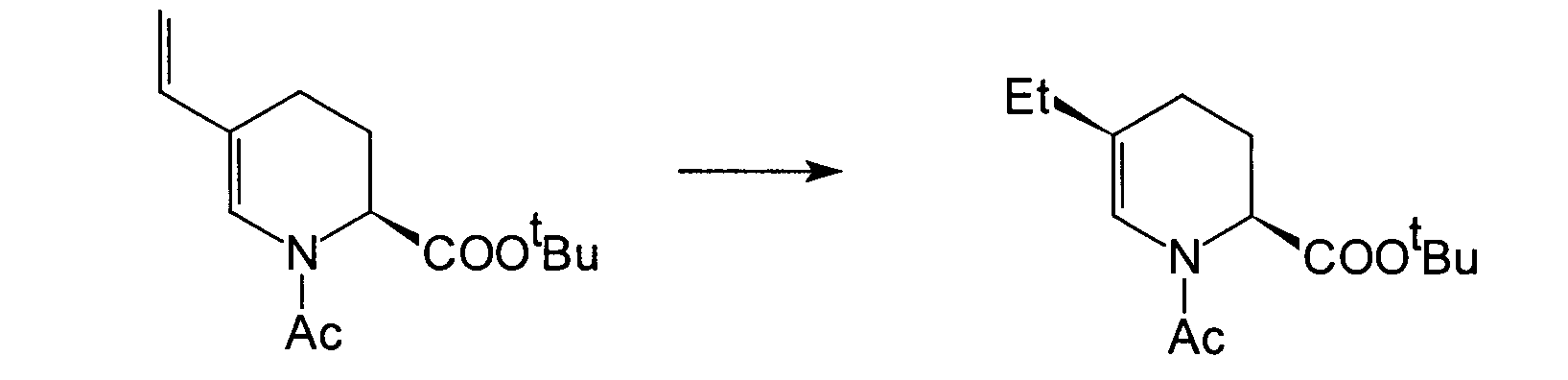

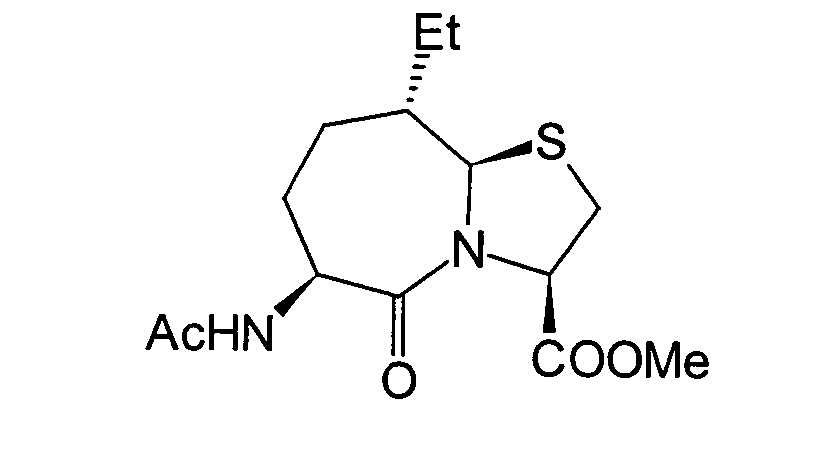

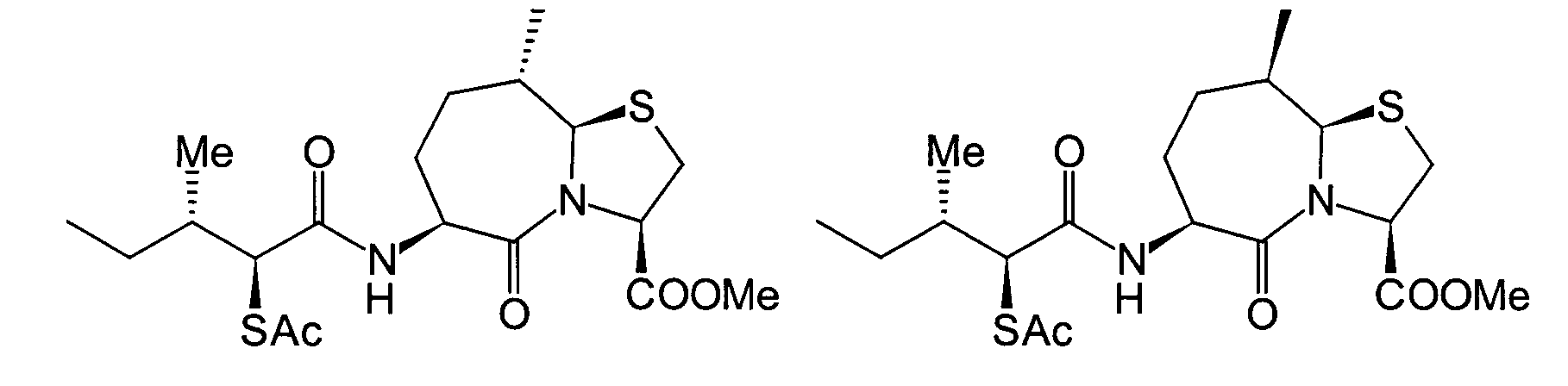




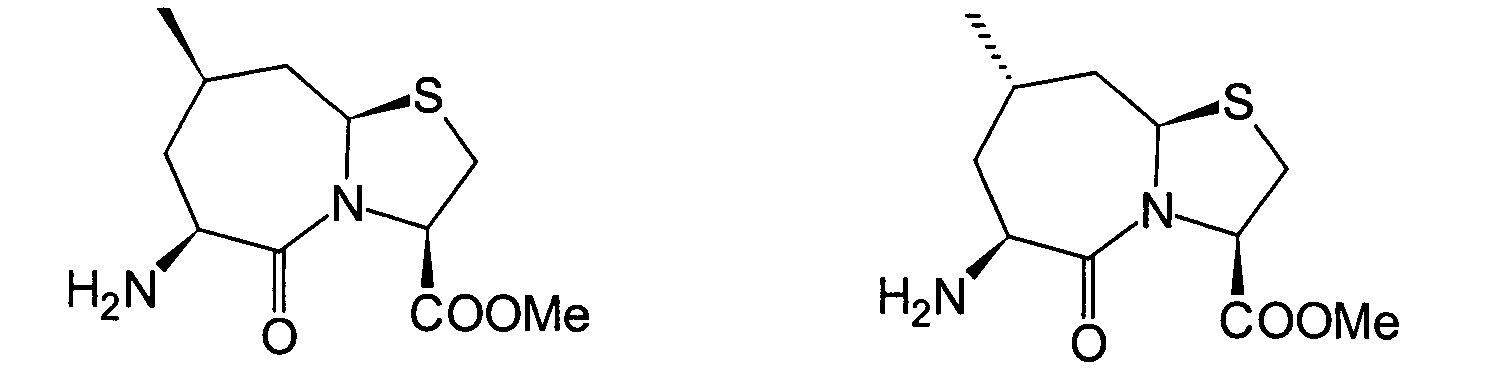
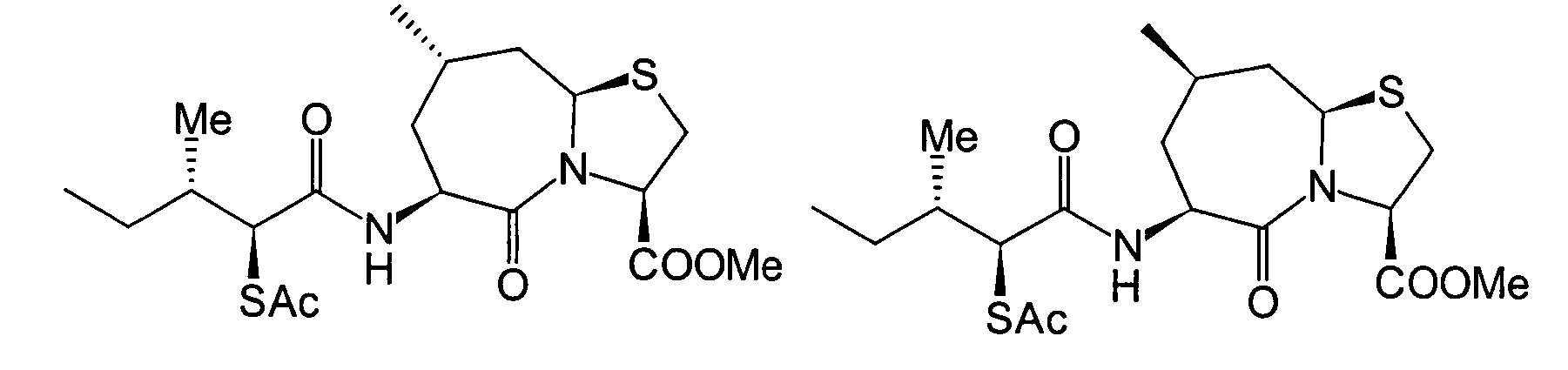
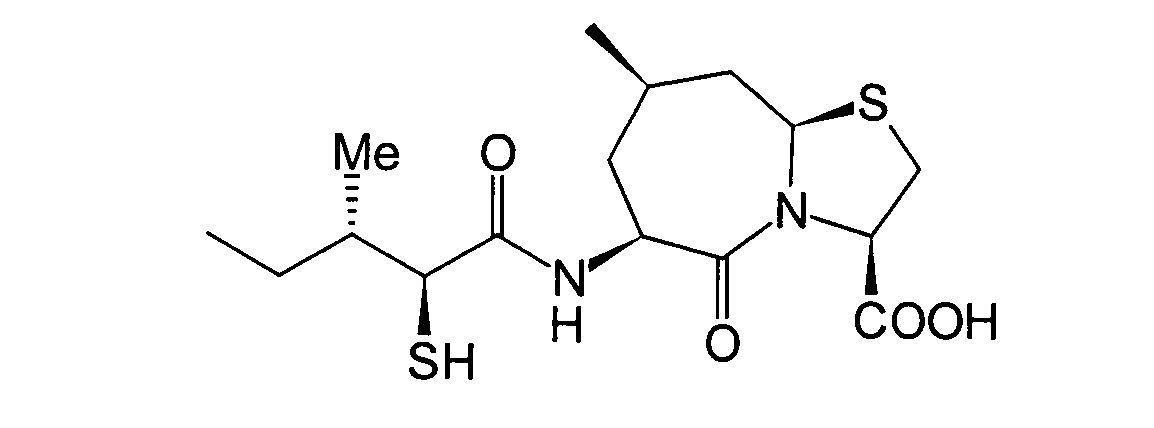
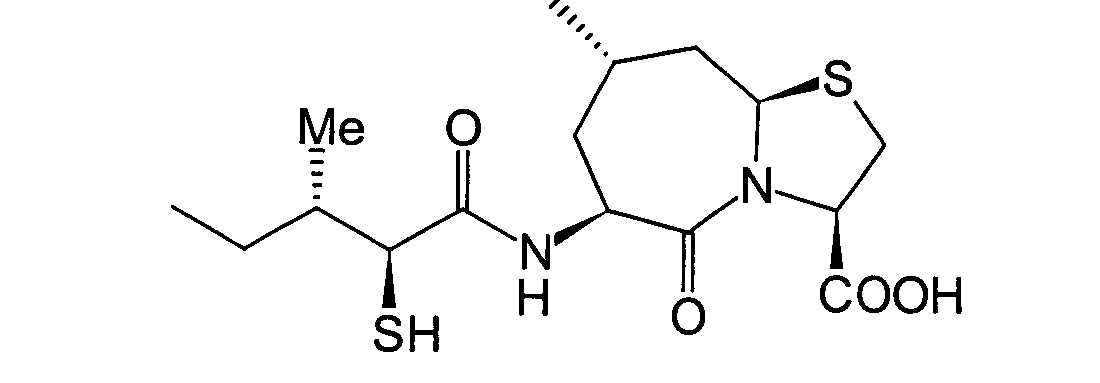

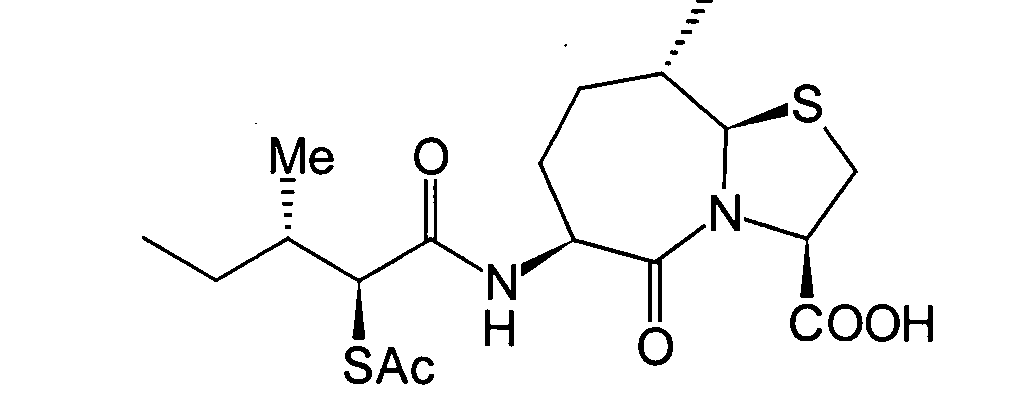

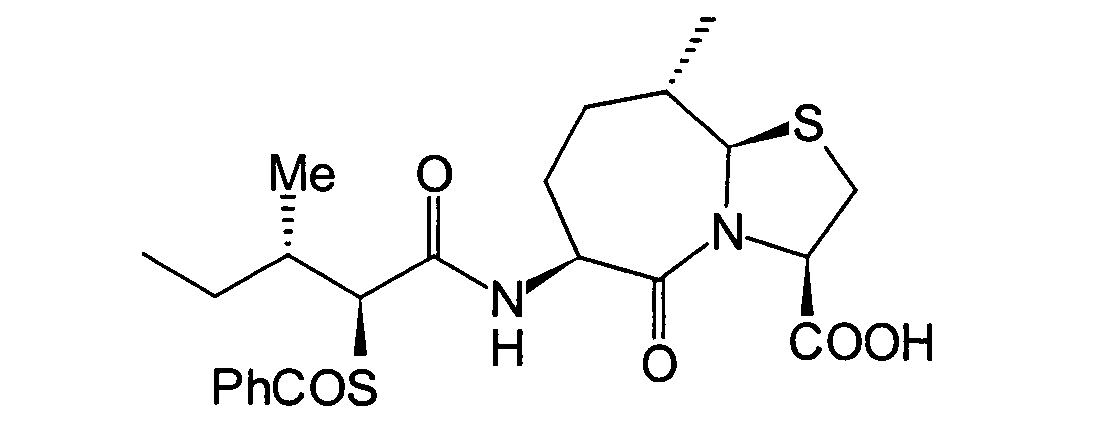
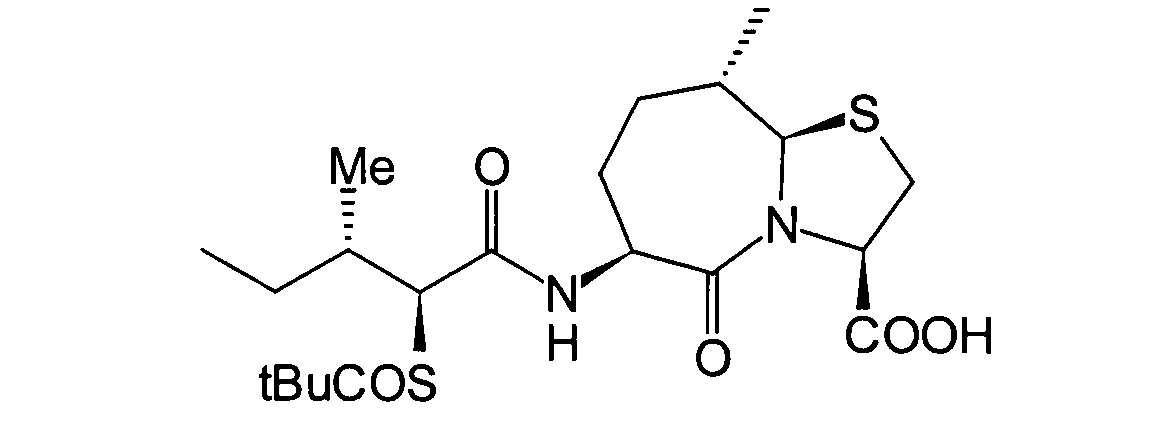
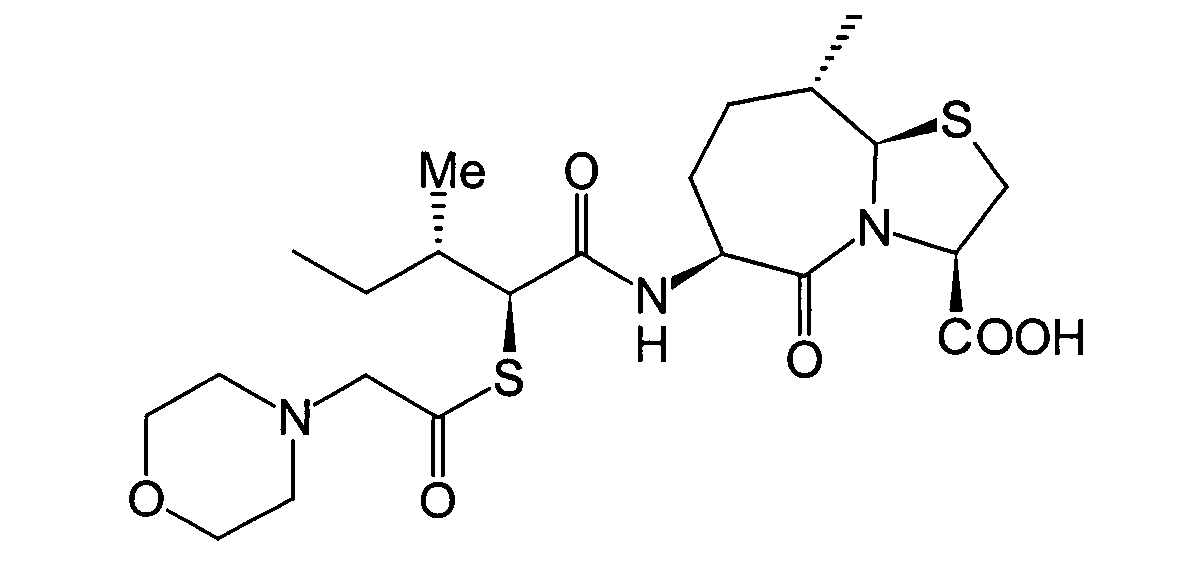

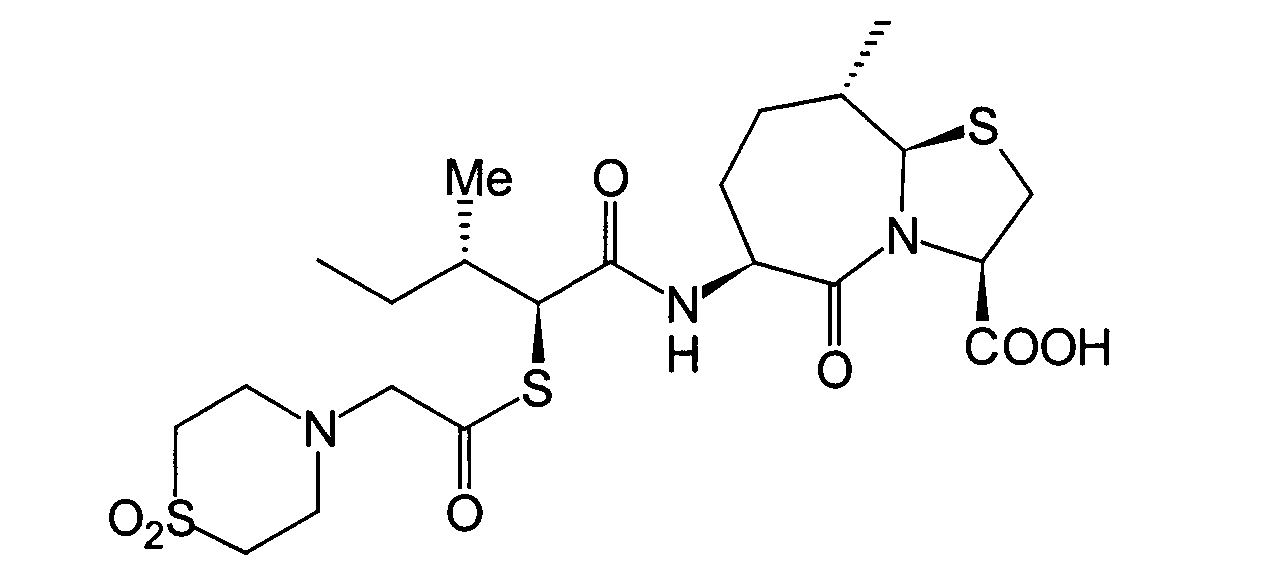

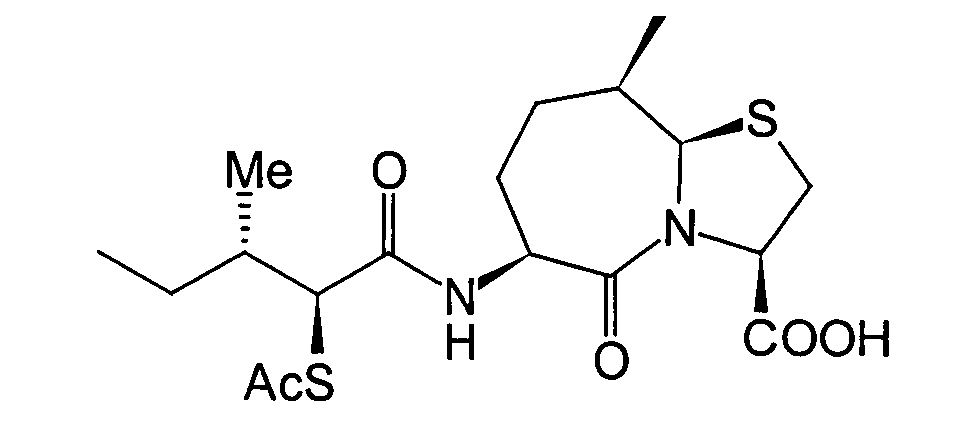
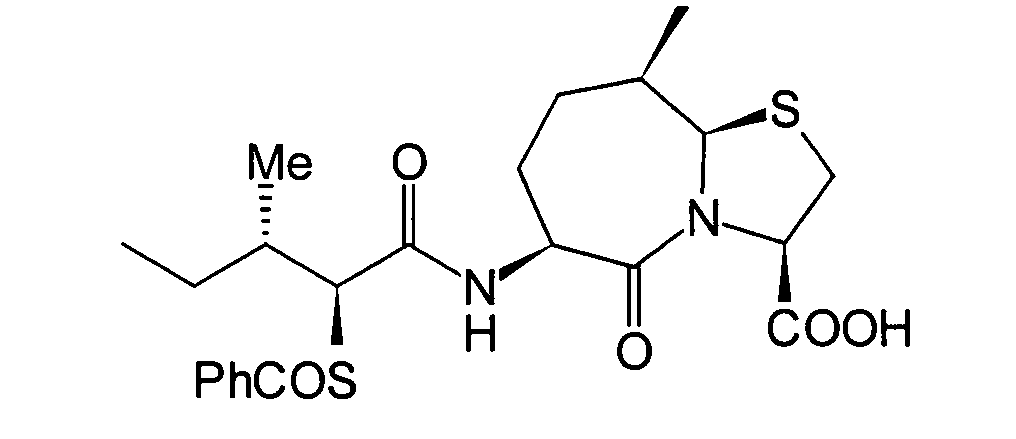

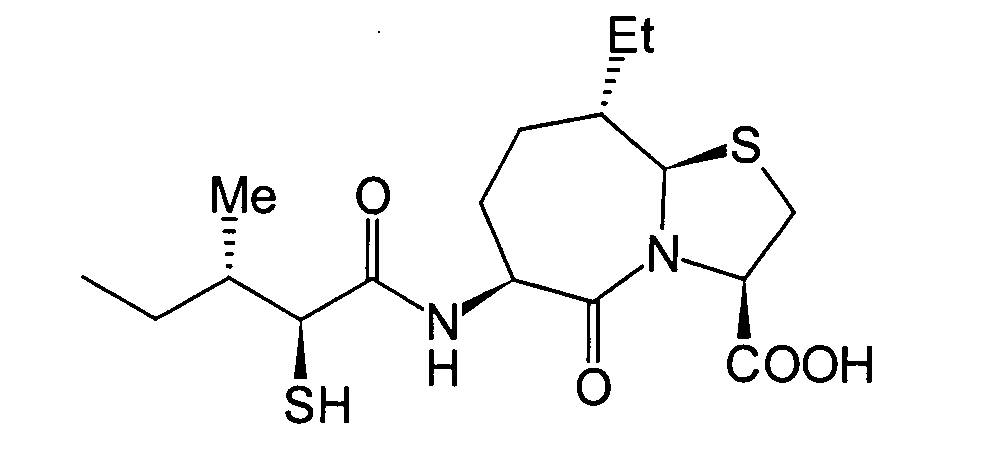
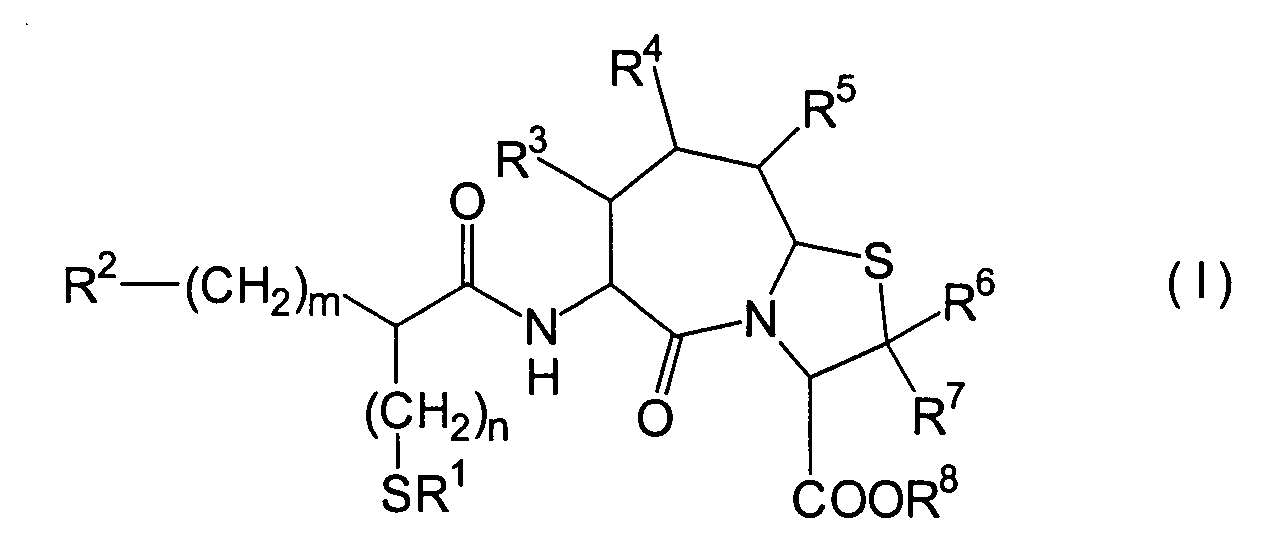
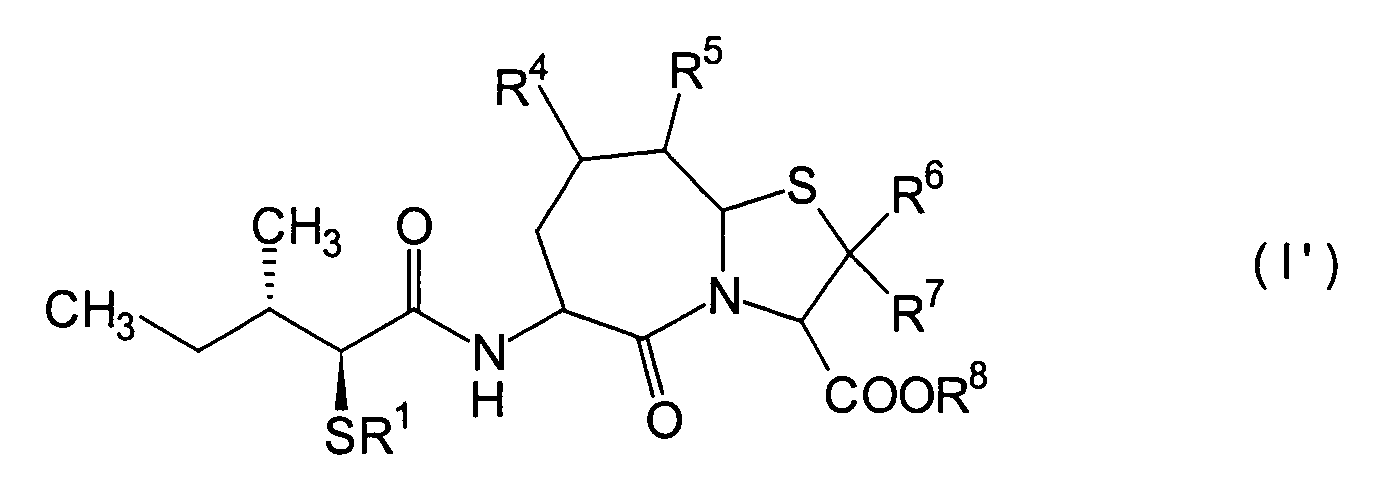



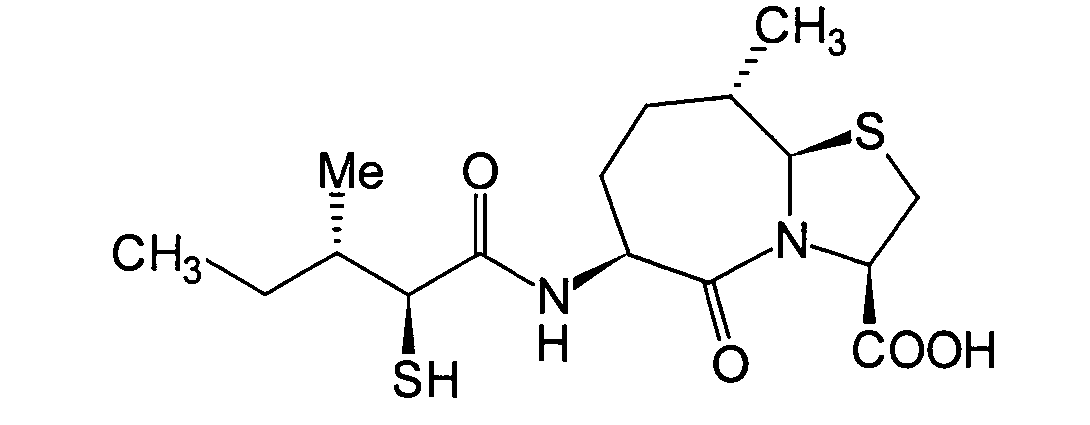



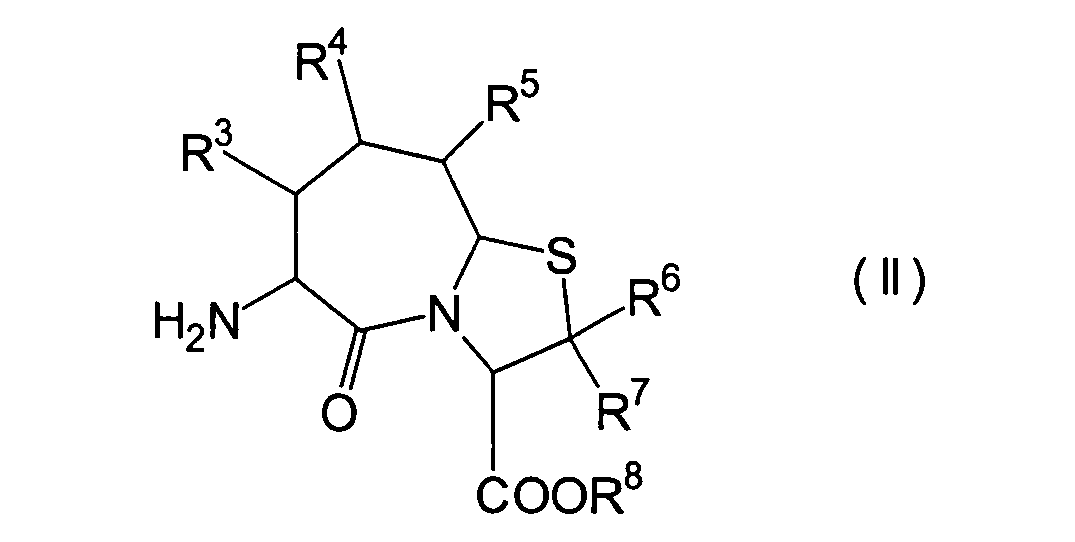
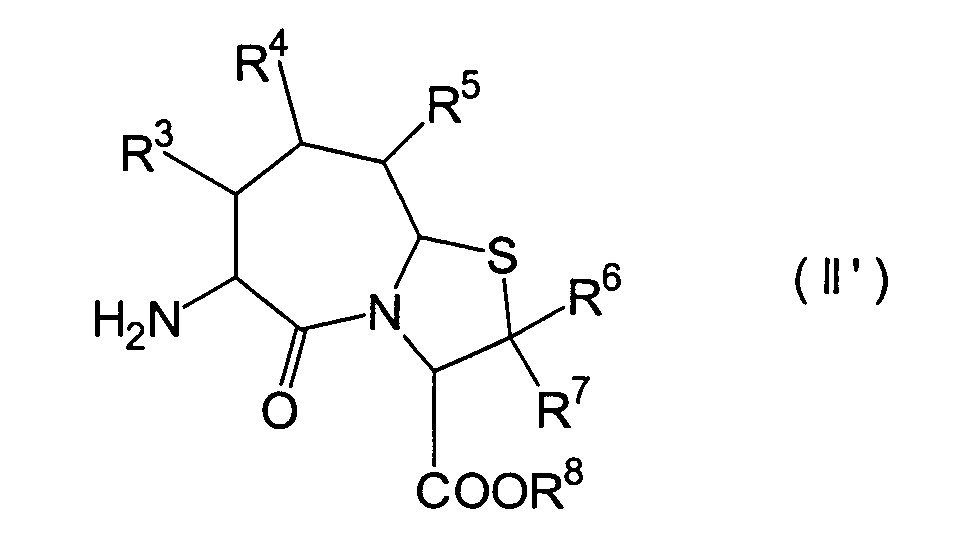

 R3, R4, R5, Z und R11 haben die Bedeutungen wie oben beschrieben und R12 ist eine Gruppe, die zusammen mit dem endocyclischen Stickstoffatom ein Aldehydäquivalent bildet.
R3, R4, R5, Z und R11 haben die Bedeutungen wie oben beschrieben und R12 ist eine Gruppe, die zusammen mit dem endocyclischen Stickstoffatom ein Aldehydäquivalent bildet.
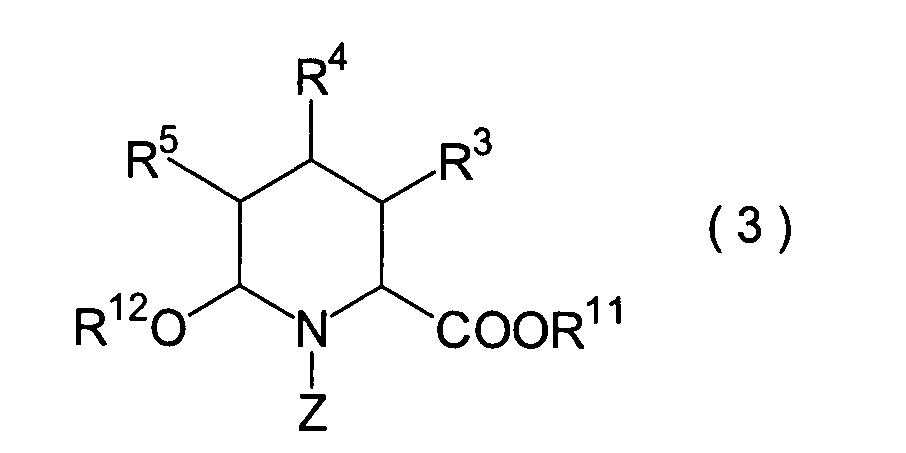 worin R2 ein Wasserstoffatom oder eine Schutzgruppe für eine Carbonsäure ist; und R6 und R7 sind jeweils ein Wasserstoffatom, wodurch ein Thiazolidinderivat (5) erhalten wird:
worin R2 ein Wasserstoffatom oder eine Schutzgruppe für eine Carbonsäure ist; und R6 und R7 sind jeweils ein Wasserstoffatom, wodurch ein Thiazolidinderivat (5) erhalten wird: worin R2 bis R7 und R11 wie oben definiert sind.
worin R2 bis R7 und R11 wie oben definiert sind.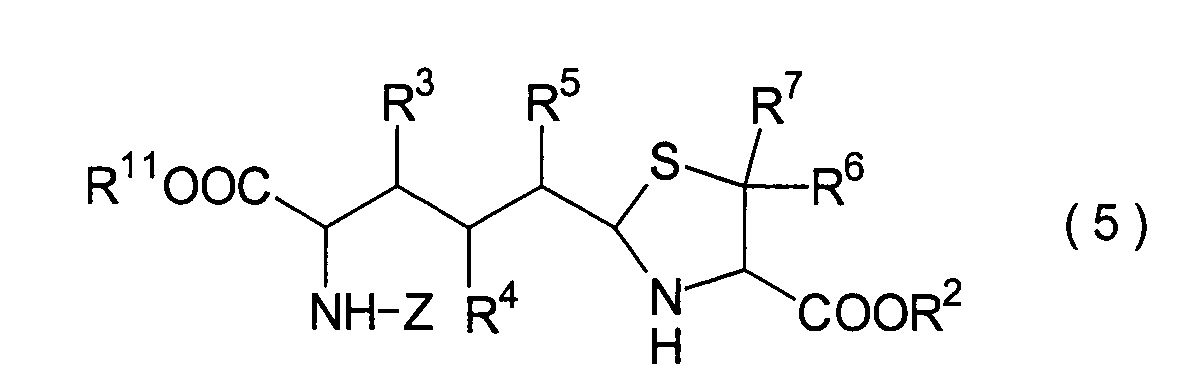
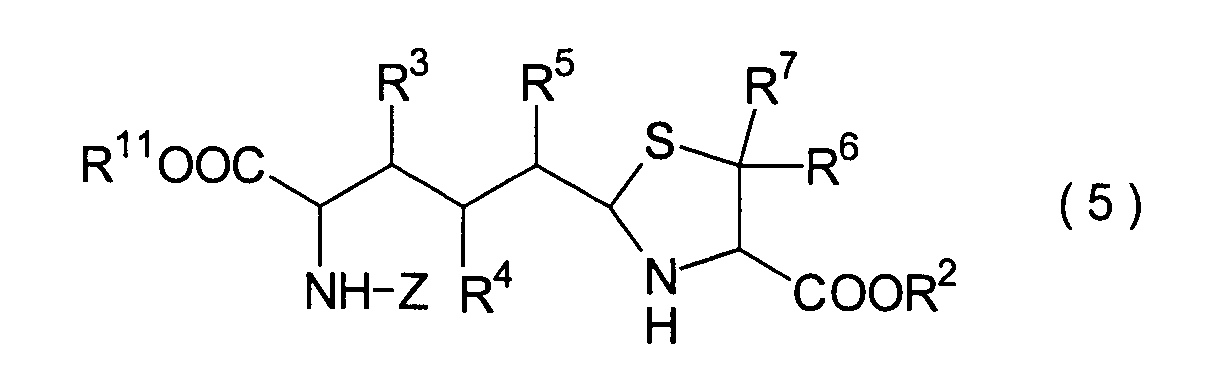 worin R2, R3, R4, R5, R6, R7 und Z die gleichen Bedeutungen haben wie oben beschrieben, Cyclisieren dieses Derivats unter Erhalt eines Aminosäurederivats der allgemeinen Formel (7):
worin R2, R3, R4, R5, R6, R7 und Z die gleichen Bedeutungen haben wie oben beschrieben, Cyclisieren dieses Derivats unter Erhalt eines Aminosäurederivats der allgemeinen Formel (7):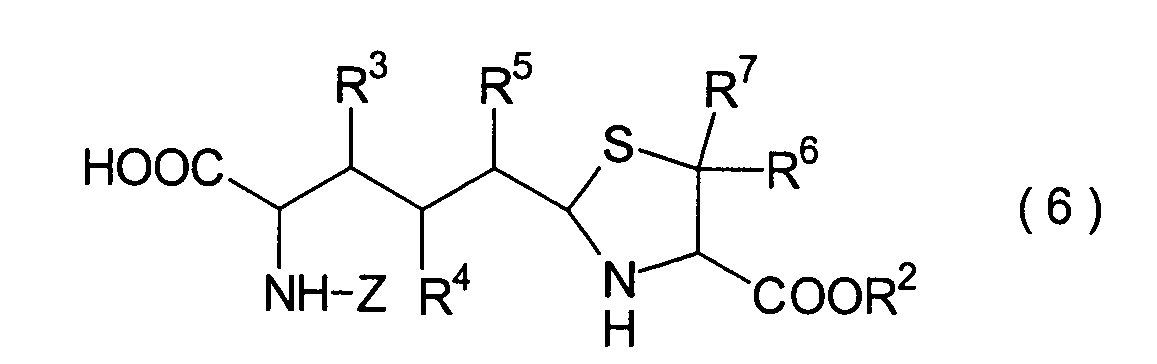 worin R2, R3, R4, R5, R6, R7 und Z die oben beschriebenen Bedeutungen aufweisen, und, sofern erforderlich, Umwandlung dieses Derivats in ein Aminosäurederivat der allgemeinen Formel (1):
worin R2, R3, R4, R5, R6, R7 und Z die oben beschriebenen Bedeutungen aufweisen, und, sofern erforderlich, Umwandlung dieses Derivats in ein Aminosäurederivat der allgemeinen Formel (1):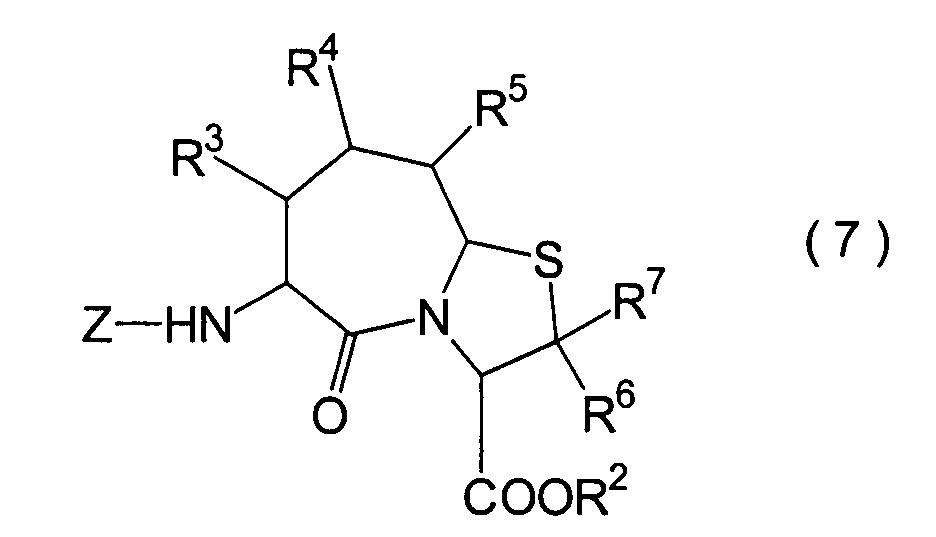 worin R2, R3, R4, R5, R6 und R7 die oben beschriebenen Bedeutungen besitzen.
worin R2, R3, R4, R5, R6 und R7 die oben beschriebenen Bedeutungen besitzen.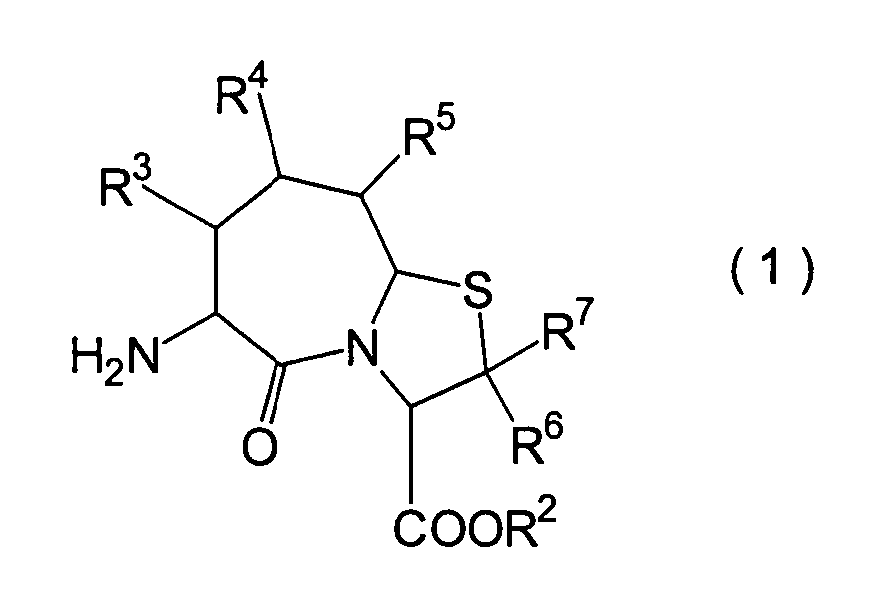
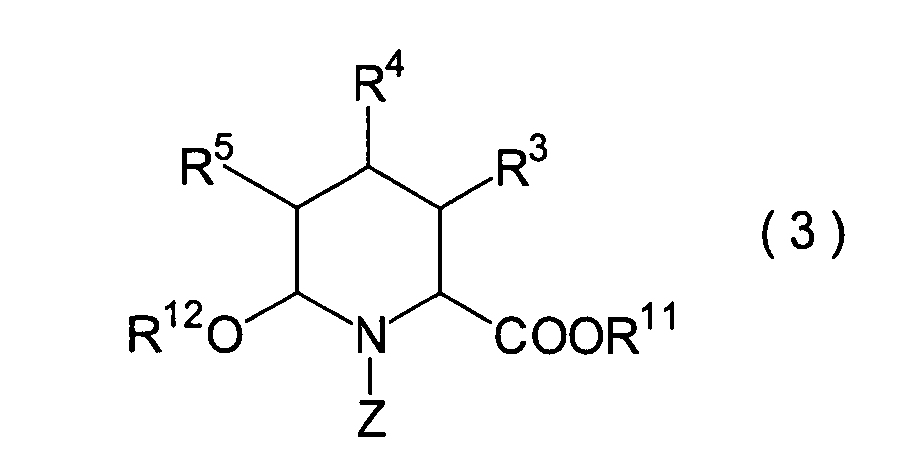 worin R2 ein Wasserstoffatom oder eine Schutzgruppe für eine Carbonsäure ist; und R6 und R7 sind jeweils ein Wasserstoffatom, wodurch ein Thiazolidinderivat der allgemeinen Formel (5) erhalten wird:
worin R2 ein Wasserstoffatom oder eine Schutzgruppe für eine Carbonsäure ist; und R6 und R7 sind jeweils ein Wasserstoffatom, wodurch ein Thiazolidinderivat der allgemeinen Formel (5) erhalten wird: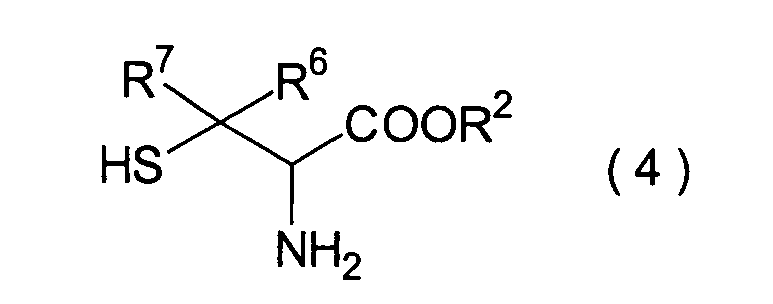 worin R2 eine Schutzgruppe für eine Carbonsäure ist; R6 und R7 sind ein Wasserstoffatom; R3, R4 und R5 repräsentieren jeweils unabhängig voneinander ein Wasserstoffatom, eine C1-6-Alkylgruppe, eine C1-6-Alkoxylgruppe oder eine C1-6-Alkylthiogruppe, mit der Massgabe, dass der Fall dass R3, R4 und R5 alle Wasserstoffatome darstellen, ausgenommen ist; R11 ist eine Schutzgruppe für eine Carbonsäure; und Z ist eine Acylgruppe oder eine Carbamatgruppe, Cyclisierung eines Thiazolidinderivats, das durch weitere Entschützung des obigen Derivats erhalten wird und das durch die allgemeine Formel (6) repräsentiert wird:
worin R2 eine Schutzgruppe für eine Carbonsäure ist; R6 und R7 sind ein Wasserstoffatom; R3, R4 und R5 repräsentieren jeweils unabhängig voneinander ein Wasserstoffatom, eine C1-6-Alkylgruppe, eine C1-6-Alkoxylgruppe oder eine C1-6-Alkylthiogruppe, mit der Massgabe, dass der Fall dass R3, R4 und R5 alle Wasserstoffatome darstellen, ausgenommen ist; R11 ist eine Schutzgruppe für eine Carbonsäure; und Z ist eine Acylgruppe oder eine Carbamatgruppe, Cyclisierung eines Thiazolidinderivats, das durch weitere Entschützung des obigen Derivats erhalten wird und das durch die allgemeine Formel (6) repräsentiert wird: worin R2, R3, R4, R5, R6, R7 und Z die gleichen Bedeutungen haben wie oben beschrieben, wodurch ein Aminosäurederivat der allgemeinen Formel (7) erhalten wird:
worin R2, R3, R4, R5, R6, R7 und Z die gleichen Bedeutungen haben wie oben beschrieben, wodurch ein Aminosäurederivat der allgemeinen Formel (7) erhalten wird: worin R2, R3, R4, R5, R6, R7 und Z die oben beschriebenen Bedeutungen aufweisen, und, sofern erforderlich, Entschützen dieses Derivats zu einem Aminosäurederivat der allgemeinen Formel (1):
worin R2, R3, R4, R5, R6, R7 und Z die oben beschriebenen Bedeutungen aufweisen, und, sofern erforderlich, Entschützen dieses Derivats zu einem Aminosäurederivat der allgemeinen Formel (1): worin R2 bis R7 die oben beschriebenen Bedeutungen aufweisen.
worin R2 bis R7 die oben beschriebenen Bedeutungen aufweisen.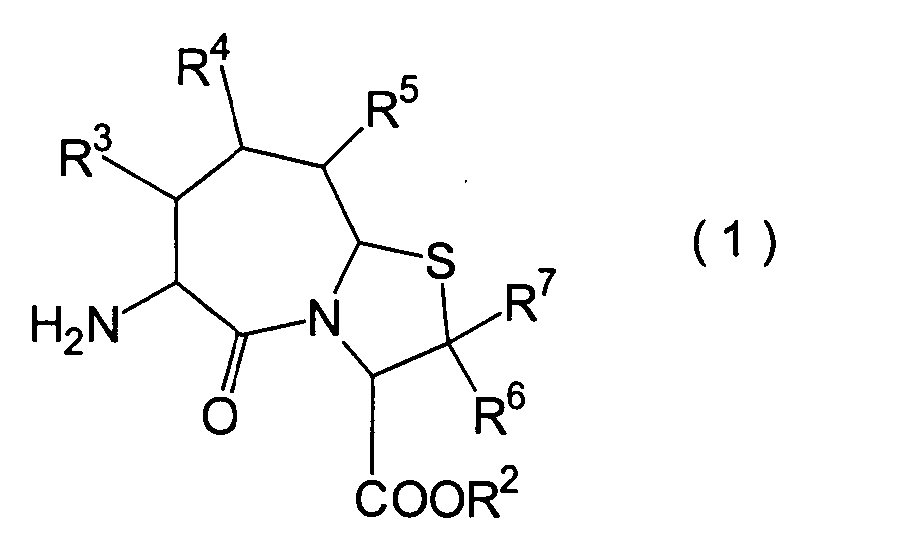
 worin R3, R4 und R5 unabhängig voneinander ein Wasserstoffatom, eine C1-6-Alkylgruppe, eine C1-6-Alkoxylgruppe oder eine C1-6-Alkylthiogruppe repräsentieren, mit der Massgabe, dass der Fall, dass R3, R4 und R5 alle Wasserstoffatome sind, ausgenommen ist; R11 ist eine Schutzgruppe für eine Carbonsäure; und R12 ist eine Gruppe, die zusammen mit dem endocyclischen Stickstoffatom ein Aldehydäquivalent bildet, Umsetzung des erhaltenen Hemiacetals (3) mit einem Cysteinderivat der allgemeinen Formel (4):
worin R3, R4 und R5 unabhängig voneinander ein Wasserstoffatom, eine C1-6-Alkylgruppe, eine C1-6-Alkoxylgruppe oder eine C1-6-Alkylthiogruppe repräsentieren, mit der Massgabe, dass der Fall, dass R3, R4 und R5 alle Wasserstoffatome sind, ausgenommen ist; R11 ist eine Schutzgruppe für eine Carbonsäure; und R12 ist eine Gruppe, die zusammen mit dem endocyclischen Stickstoffatom ein Aldehydäquivalent bildet, Umsetzung des erhaltenen Hemiacetals (3) mit einem Cysteinderivat der allgemeinen Formel (4):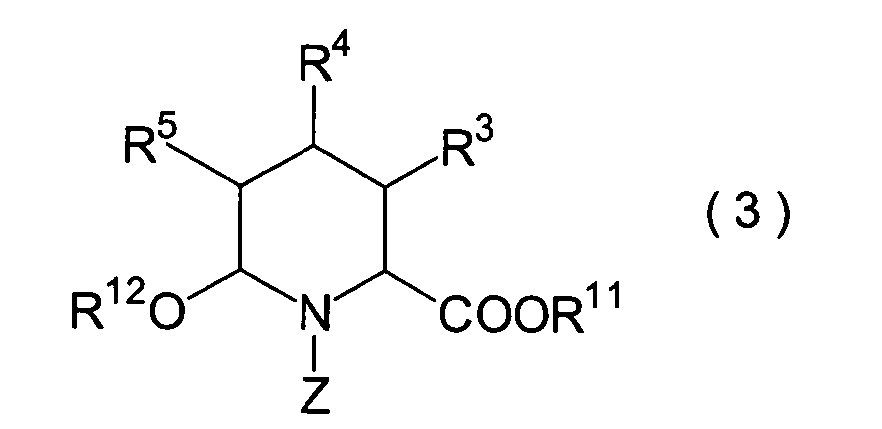 worin R2 ein Wasserstoffatom oder eine Schutzgruppe für eine Carbonsäure ist; und R6 und R7 sind jeweils ein Wasserstoffatom, wodurch ein Thiazolidinderivat der allgemeinen Formel (5) erhalten wird:
worin R2 ein Wasserstoffatom oder eine Schutzgruppe für eine Carbonsäure ist; und R6 und R7 sind jeweils ein Wasserstoffatom, wodurch ein Thiazolidinderivat der allgemeinen Formel (5) erhalten wird: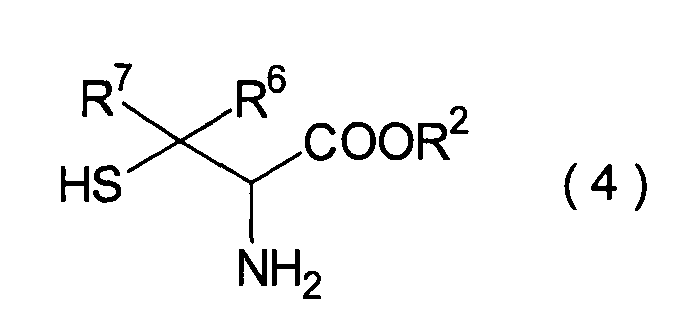 worin R2 eine Schutzgruppe für eine Carbonsäure ist; R6 und R7 sind ein Wasserstoffatom; R3, R4 und R5 repräsentieren jeweils unabhängig voneinander ein Wasserstoffatom, eine C1-6-Alkylgruppe, eine C1-6-Alkoxylgruppe oder eine C1-6-Alkylthiogruppe, mit der Massgabe, dass der Fall dass R3, R4 und R5 alle Wasserstoffatome darstellen, ausgenommen ist; R11 ist eine Schutzgruppe für eine Carbonsäure; und Z ist eine Acylgruppe oder eine Carbamatgruppe, Cyclisierung eines Thiazolidinderivats, das durch weitere Entschützung des obigen Derivats erhalten wird und das durch die allgemeine Formel (6) repräsentiert wird:
worin R2 eine Schutzgruppe für eine Carbonsäure ist; R6 und R7 sind ein Wasserstoffatom; R3, R4 und R5 repräsentieren jeweils unabhängig voneinander ein Wasserstoffatom, eine C1-6-Alkylgruppe, eine C1-6-Alkoxylgruppe oder eine C1-6-Alkylthiogruppe, mit der Massgabe, dass der Fall dass R3, R4 und R5 alle Wasserstoffatome darstellen, ausgenommen ist; R11 ist eine Schutzgruppe für eine Carbonsäure; und Z ist eine Acylgruppe oder eine Carbamatgruppe, Cyclisierung eines Thiazolidinderivats, das durch weitere Entschützung des obigen Derivats erhalten wird und das durch die allgemeine Formel (6) repräsentiert wird: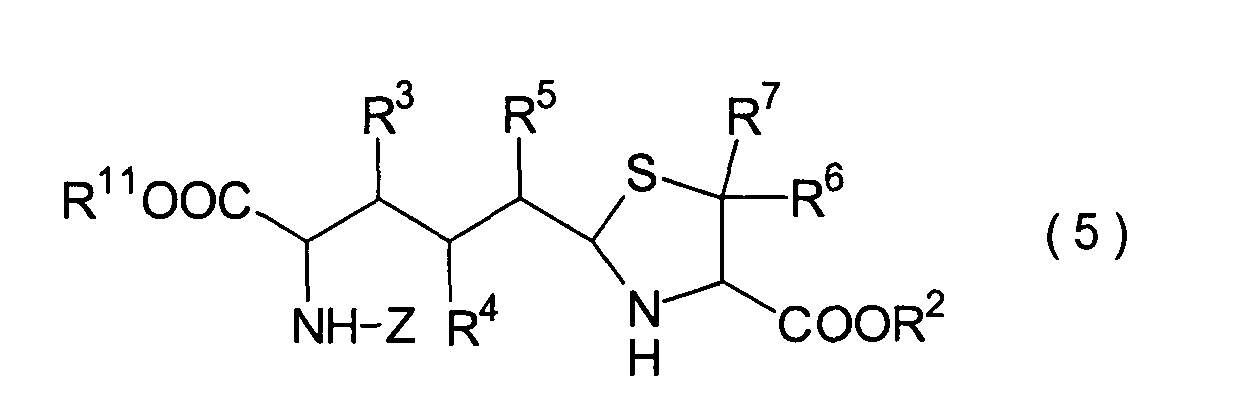 worin R2, R3, R4, R5, R6, R7 und Z die gleichen Bedeutungen haben wie oben beschrieben, wodurch ein Aminosäurederivat der allgemeinen Formel (7) erhalten wird:
worin R2, R3, R4, R5, R6, R7 und Z die gleichen Bedeutungen haben wie oben beschrieben, wodurch ein Aminosäurederivat der allgemeinen Formel (7) erhalten wird: worin R2, R3, R4, R5, R6, R7 und Z die oben beschriebenen Bedeutungen aufweisen, und, sofern erforderlich, Entschützen dieses Derivats zu einem Aminosäurederivat der allgemeinen Formel (1):
worin R2, R3, R4, R5, R6, R7 und Z die oben beschriebenen Bedeutungen aufweisen, und, sofern erforderlich, Entschützen dieses Derivats zu einem Aminosäurederivat der allgemeinen Formel (1):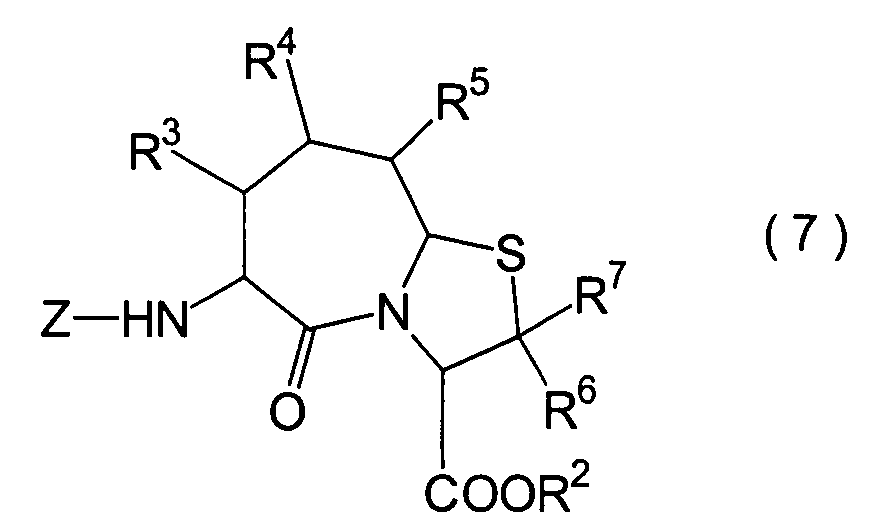 worin R2 bis R7 die oben beschriebenen Bedeutungen aufweisen.
worin R2 bis R7 die oben beschriebenen Bedeutungen aufweisen.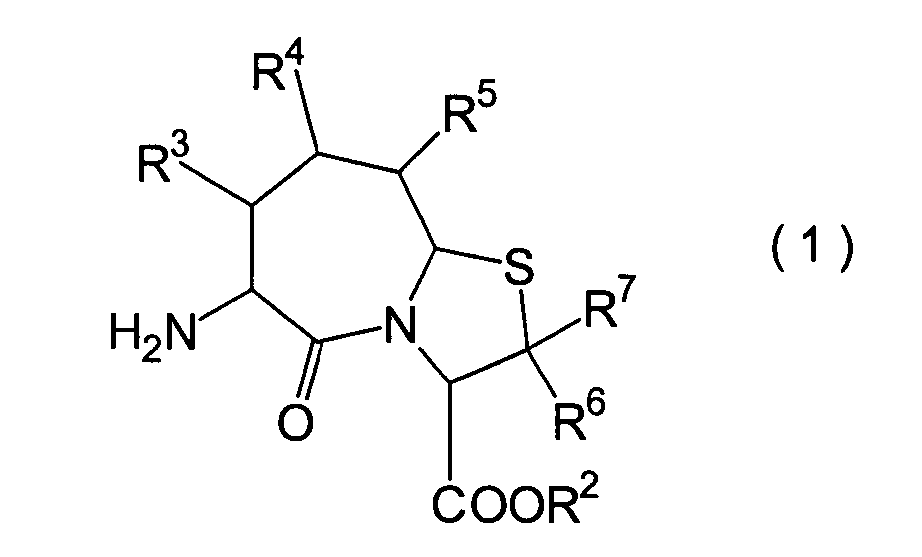
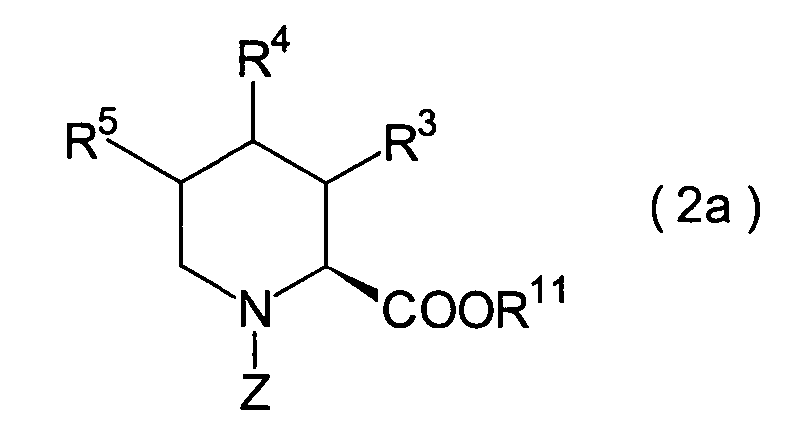 worin R3, R4 und R5 unabhängig voneinander ein Wasserstoffatom, eine C1-6-Alkylgruppe, eine C1-6-Alkoxylgruppe oder eine C1-6-Alkylthiogruppe repräsentieren, mit der Massgabe, dass der Fall, dass R3, R4 und R5 alle Wasserstoffatome sind, ausgenommen ist; R11 ist eine Schutzgruppe für eine Carbonsäure und Z ist eine Acylgruppe oder eine Carbamatgruppe; und R12 ist eine Gruppe, die zusammen mit dem endocyclischen Stickstoffatom ein Aldehydäquivalent bildet.
worin R3, R4 und R5 unabhängig voneinander ein Wasserstoffatom, eine C1-6-Alkylgruppe, eine C1-6-Alkoxylgruppe oder eine C1-6-Alkylthiogruppe repräsentieren, mit der Massgabe, dass der Fall, dass R3, R4 und R5 alle Wasserstoffatome sind, ausgenommen ist; R11 ist eine Schutzgruppe für eine Carbonsäure und Z ist eine Acylgruppe oder eine Carbamatgruppe; und R12 ist eine Gruppe, die zusammen mit dem endocyclischen Stickstoffatom ein Aldehydäquivalent bildet.
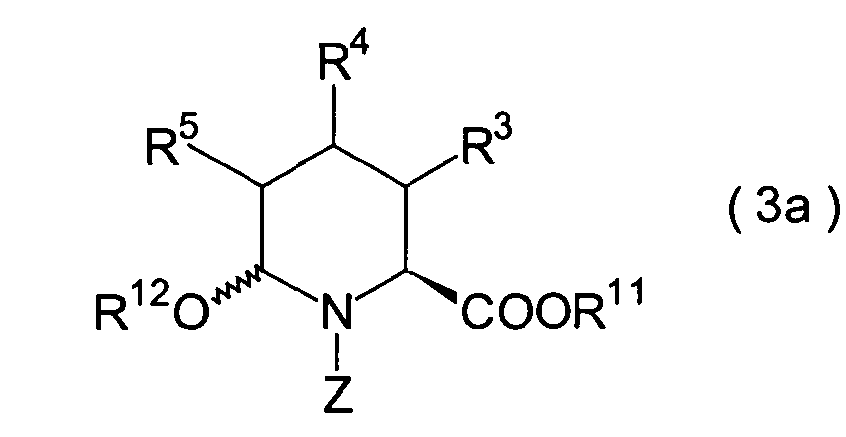 worin R8 ein Wasserstoffatom oder eine Schutzgruppe für eine Carbonsäure ist; und R6 und R7 repräsentieren ein Wasserstoffatom, wodurch ein Thiazolidinderivat (5a) erhalten wird:
worin R8 ein Wasserstoffatom oder eine Schutzgruppe für eine Carbonsäure ist; und R6 und R7 repräsentieren ein Wasserstoffatom, wodurch ein Thiazolidinderivat (5a) erhalten wird: worin R3 bis R8 und R11 wie oben definiert sind.
worin R3 bis R8 und R11 wie oben definiert sind.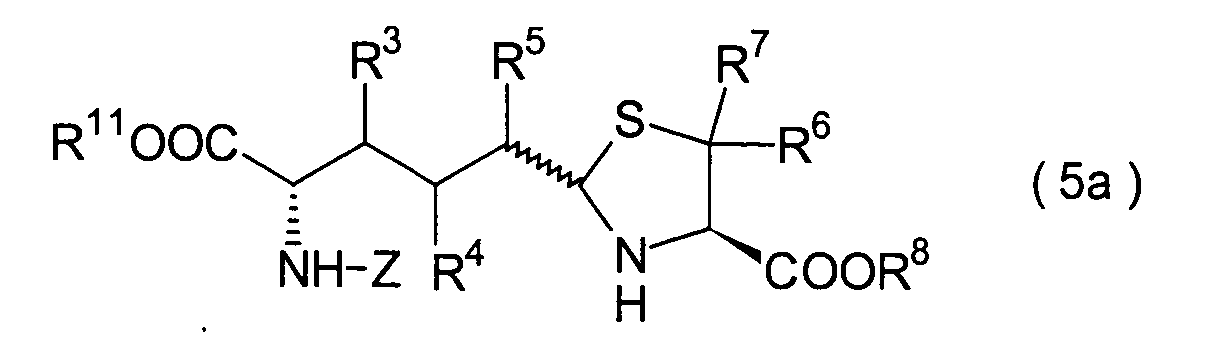
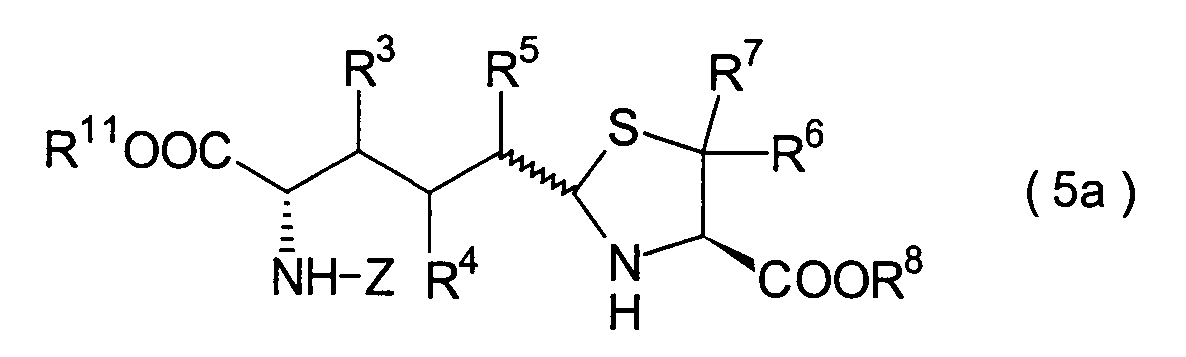 worin R3, R4, R5, R6, R7, R8 und Z die oben beschriebenen Bedeutungen aufweisen, Cyclisierung dieses Derivats unter Erhalt eines Aminosäurederivats der allgemeinen Formel (7a):
worin R3, R4, R5, R6, R7, R8 und Z die oben beschriebenen Bedeutungen aufweisen, Cyclisierung dieses Derivats unter Erhalt eines Aminosäurederivats der allgemeinen Formel (7a):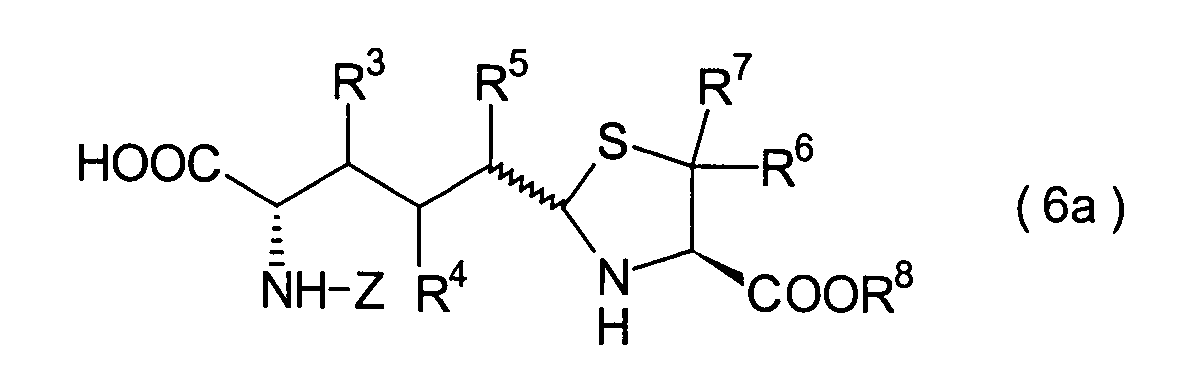 worin R3, R4, R5, R6, R7, R8 und Z die oben beschriebenen Bedeutungen aufweisen, und, sofern erforderlich, Umwandlung in ein Aminosäurederivat der allgemeinen Formel (1a):
worin R3, R4, R5, R6, R7, R8 und Z die oben beschriebenen Bedeutungen aufweisen, und, sofern erforderlich, Umwandlung in ein Aminosäurederivat der allgemeinen Formel (1a): worin R3 bis R8 wie oben definiert sind.
worin R3 bis R8 wie oben definiert sind.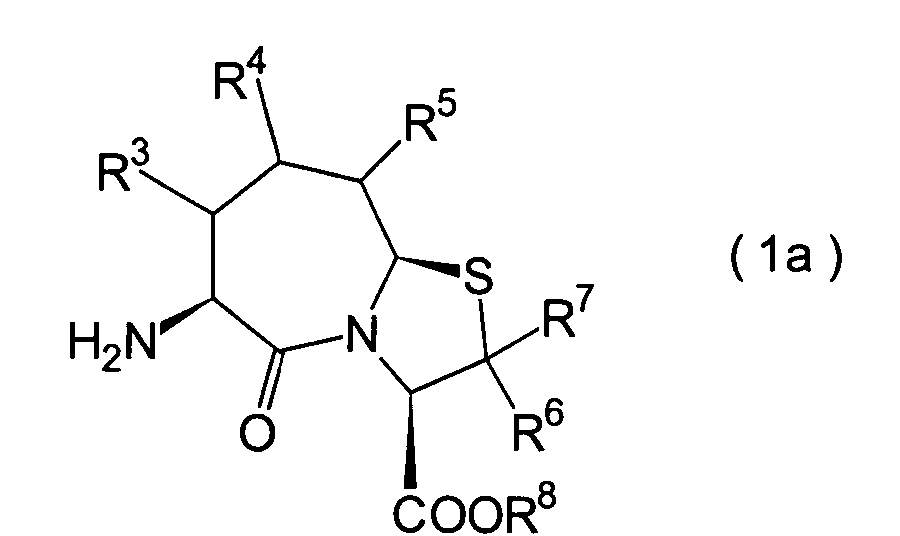
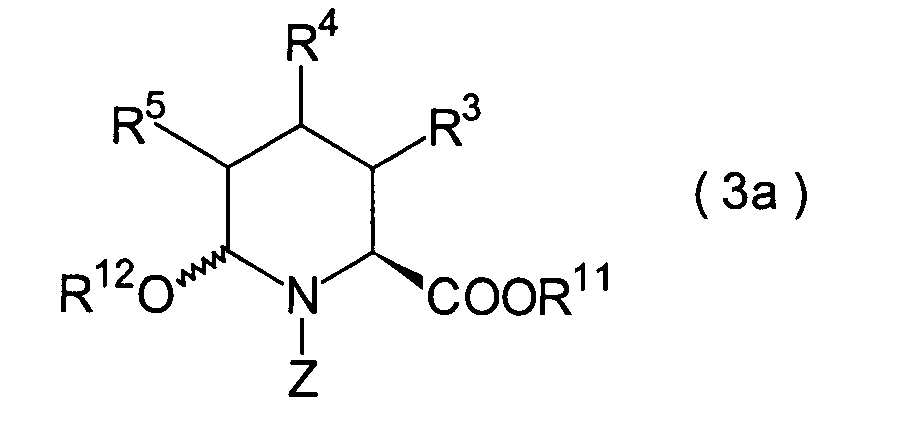 worin R8 ein Wasserstoffatom oder eine Schutzgruppe für eine Carbonsäure ist; und R6 und R7 repräsentieren ein Wasserstoffatom, wodurch ein Thiazolidinderivat der allgemeinen Formel (5a) erhalten wird:
worin R8 ein Wasserstoffatom oder eine Schutzgruppe für eine Carbonsäure ist; und R6 und R7 repräsentieren ein Wasserstoffatom, wodurch ein Thiazolidinderivat der allgemeinen Formel (5a) erhalten wird: worin R8 ein Wasserstoffatom oder eine Schutzgruppe für eine Carbonsäure ist, R6 und R7 repräsentieren ein Wasserstoffatom; R3, R4 und R5 repräsentieren jeweils unabhängig voneinander ein Wasserstoffatom, eine C1-6-Alkylgruppe, eine C1-6-Alkoxylgruppe oder eine C1-6-Alkylthiogruppe, mit der Massgabe, dass der Fall, dass R3, R4 und R5 alle Wasserstoffatome darstellen, ausgenommen ist; R11 repräsentiert eine Schutzgruppe für eine Carbonsäure; und Z ist eine Acylgruppe oder eine Carbamatgruppe, Cyclisierung eines Thiazolidinderivats, das erhalten wird durch weitere Entschützung des obigen Derivats, und das durch die allgemeine Formel (6a) repräsentiert wird:
worin R8 ein Wasserstoffatom oder eine Schutzgruppe für eine Carbonsäure ist, R6 und R7 repräsentieren ein Wasserstoffatom; R3, R4 und R5 repräsentieren jeweils unabhängig voneinander ein Wasserstoffatom, eine C1-6-Alkylgruppe, eine C1-6-Alkoxylgruppe oder eine C1-6-Alkylthiogruppe, mit der Massgabe, dass der Fall, dass R3, R4 und R5 alle Wasserstoffatome darstellen, ausgenommen ist; R11 repräsentiert eine Schutzgruppe für eine Carbonsäure; und Z ist eine Acylgruppe oder eine Carbamatgruppe, Cyclisierung eines Thiazolidinderivats, das erhalten wird durch weitere Entschützung des obigen Derivats, und das durch die allgemeine Formel (6a) repräsentiert wird: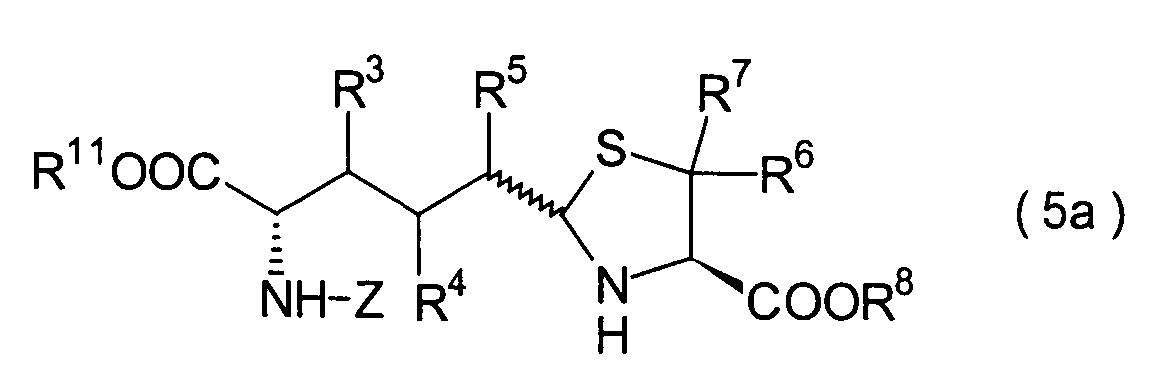 worin R3, R4, R5, R6, R7, R8 und Z die oben beschriebenen Bedeutungen aufweisen, wodurch ein Aminosäurederivat der allgemeinen Formel (7a) erhalten wird:
worin R3, R4, R5, R6, R7, R8 und Z die oben beschriebenen Bedeutungen aufweisen, wodurch ein Aminosäurederivat der allgemeinen Formel (7a) erhalten wird: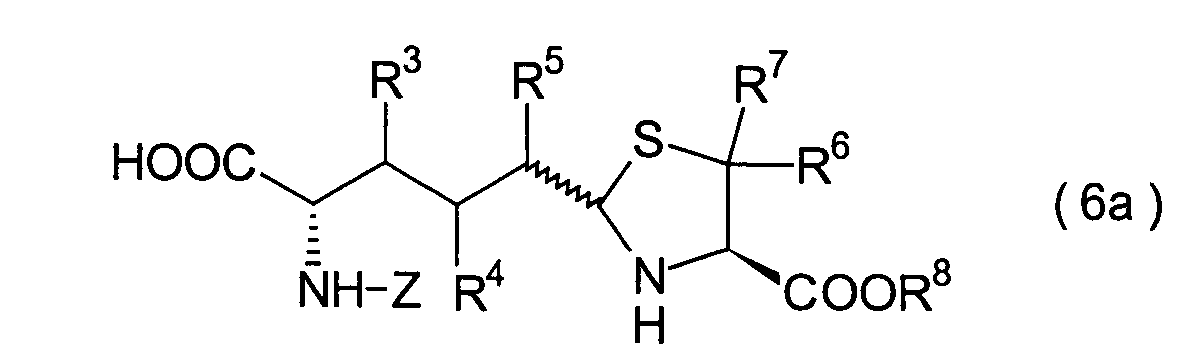 worin R3 bis R8 und Z die gleichen Bedeutungen haben wie oben definiert, und, sofern erforderlich, Entschützen dieses Derivats unter Erhalt eines Aminosäurederivats der allgemeinen Formel (1a):
worin R3 bis R8 und Z die gleichen Bedeutungen haben wie oben definiert, und, sofern erforderlich, Entschützen dieses Derivats unter Erhalt eines Aminosäurederivats der allgemeinen Formel (1a):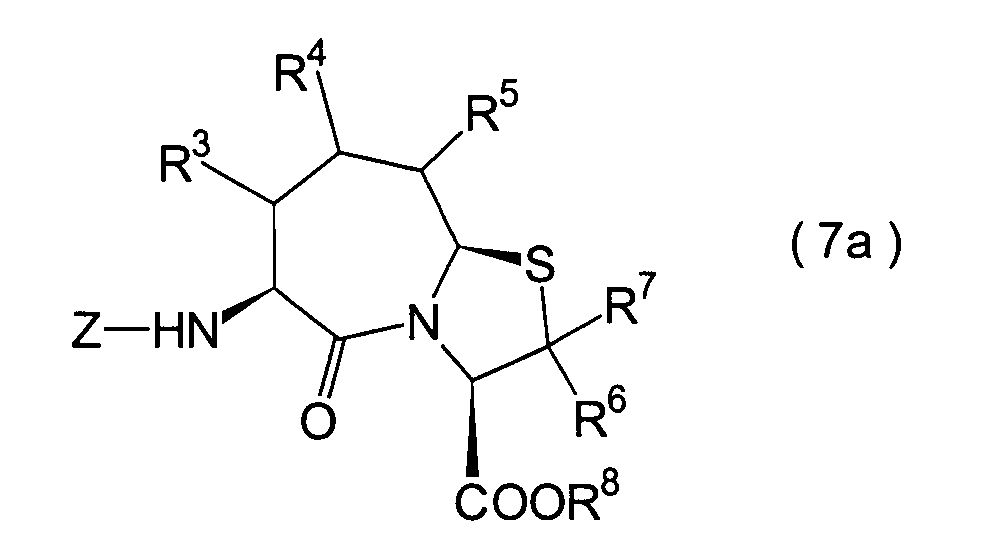 worin R3 bis R8 die gleichen Bedeutungen aufweisen wie oben definiert.
worin R3 bis R8 die gleichen Bedeutungen aufweisen wie oben definiert.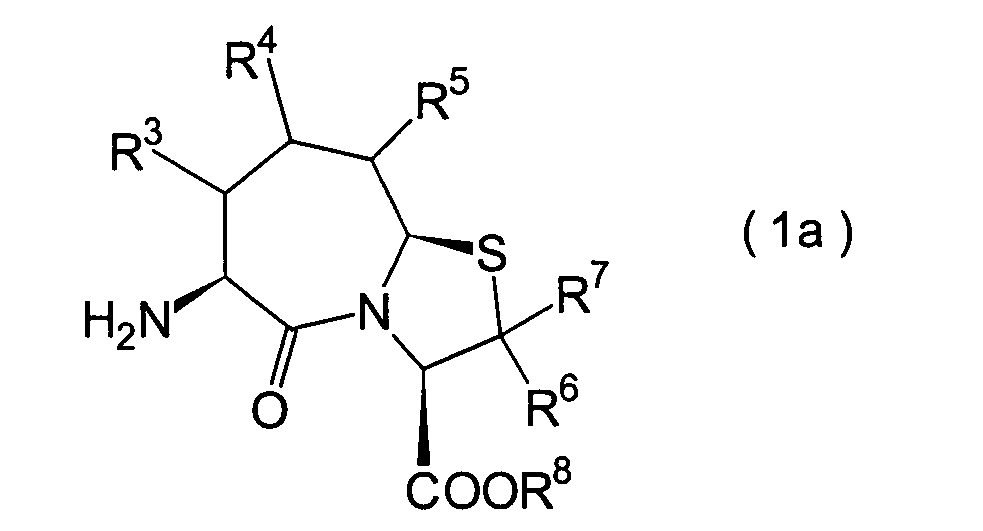
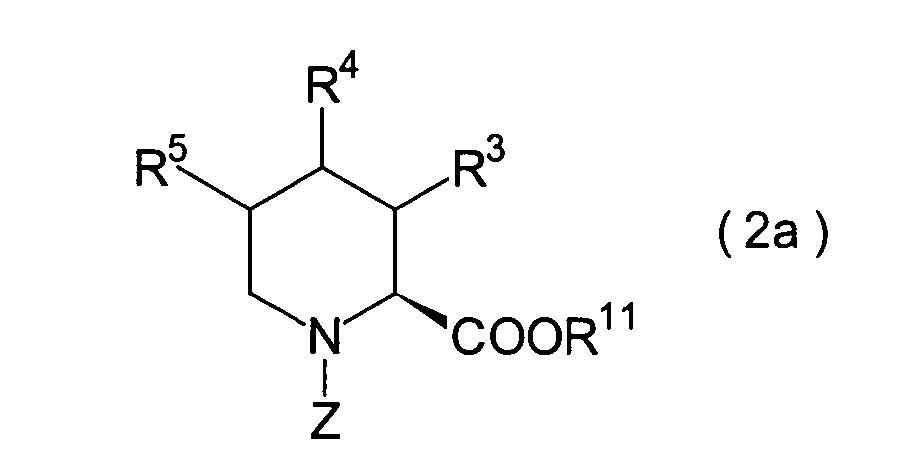 worin R3, R4 und R5 unabhängig voneinander ein Wasserstoffatom, eine C1-6-Alkylgruppe, eine C1-6-Alkoxylgruppe oder eine C1-6-Alkylthiogruppe re präsentieren, mit der Massgabe, dass der Fall, dass R3, R4 und R5 alle Wasserstoffatome sind, ausgenommen ist; R11 ist eine Schutzgruppe für eine Carbonsäure und Z repräsentiert eine Acylgruppe; und R12 ist eine Gruppe, die zusammen mit dem endocyclischen Stickstoffatom ein Aldehydäguivalent bildet, Umsetzen des erhaltenen Hemiacetals (3a) mit einem Cysteinderivat der allgemeinen Formel (4a):
worin R3, R4 und R5 unabhängig voneinander ein Wasserstoffatom, eine C1-6-Alkylgruppe, eine C1-6-Alkoxylgruppe oder eine C1-6-Alkylthiogruppe re präsentieren, mit der Massgabe, dass der Fall, dass R3, R4 und R5 alle Wasserstoffatome sind, ausgenommen ist; R11 ist eine Schutzgruppe für eine Carbonsäure und Z repräsentiert eine Acylgruppe; und R12 ist eine Gruppe, die zusammen mit dem endocyclischen Stickstoffatom ein Aldehydäguivalent bildet, Umsetzen des erhaltenen Hemiacetals (3a) mit einem Cysteinderivat der allgemeinen Formel (4a):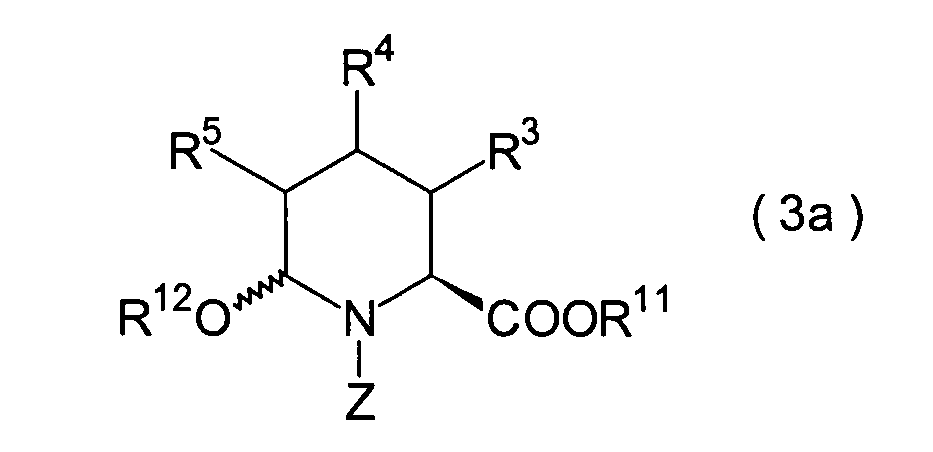 worin R8 ein Wasserstoffatom oder eine Schutzgruppe für eine Carbonsäure ist; und R6 und R7 repräsentieren ein Wasserstoffatom, wodurch ein Thiazolidinderivat der allgemeinen Formel (5a) erhalten wird:
worin R8 ein Wasserstoffatom oder eine Schutzgruppe für eine Carbonsäure ist; und R6 und R7 repräsentieren ein Wasserstoffatom, wodurch ein Thiazolidinderivat der allgemeinen Formel (5a) erhalten wird: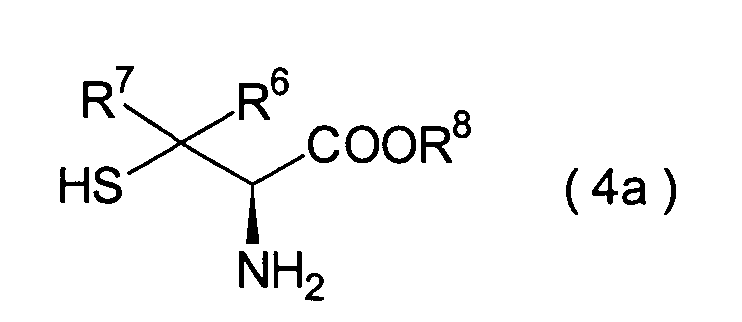 worin R8 ein Wasserstoffatom oder eine Schutzgruppe für eine Carbonsäure ist, R6 und R7 repräsentieren ein Wasserstoffatom; R3, R4 und R5 repräsentieren jeweils unabhängig voneinander ein Wasserstoffatom, eine C1-6-Alkylgruppe, eine C1-6-Alkoxylgruppe oder eine C1-6-Alkylthiogruppe, mit der Massgabe, dass der Fall, dass R3, R4 und R5 alle Wasserstoffatome darstellen, ausgenommen ist; R11 repräsentiert eine Schutzgruppe für eine Carbonsäure; und Z ist eine Acylgruppe oder eine Carbamatgruppe, Cyclisierung eines Thiazolidinderivats, das erhalten wird durch weitere Entschützung des obigen Derivats, und das durch die allgemeine Formel (6a) repräsentiert wird:
worin R8 ein Wasserstoffatom oder eine Schutzgruppe für eine Carbonsäure ist, R6 und R7 repräsentieren ein Wasserstoffatom; R3, R4 und R5 repräsentieren jeweils unabhängig voneinander ein Wasserstoffatom, eine C1-6-Alkylgruppe, eine C1-6-Alkoxylgruppe oder eine C1-6-Alkylthiogruppe, mit der Massgabe, dass der Fall, dass R3, R4 und R5 alle Wasserstoffatome darstellen, ausgenommen ist; R11 repräsentiert eine Schutzgruppe für eine Carbonsäure; und Z ist eine Acylgruppe oder eine Carbamatgruppe, Cyclisierung eines Thiazolidinderivats, das erhalten wird durch weitere Entschützung des obigen Derivats, und das durch die allgemeine Formel (6a) repräsentiert wird: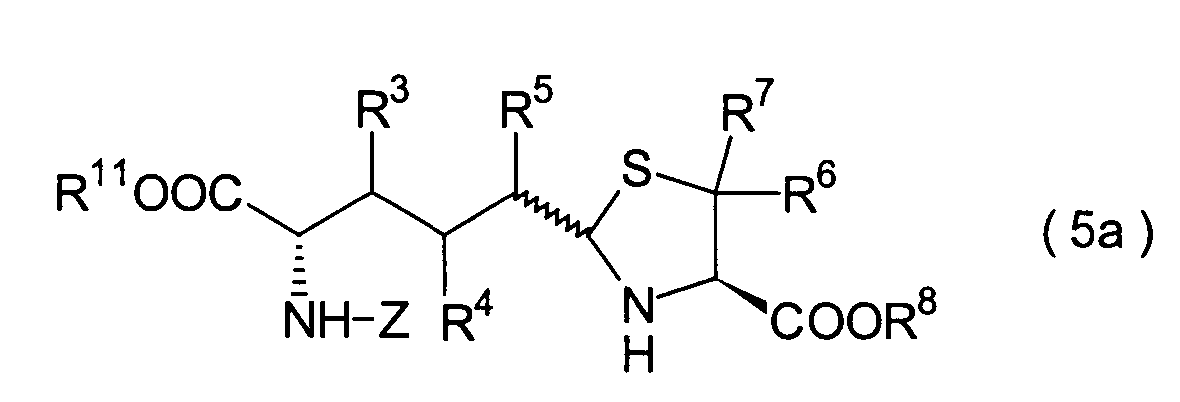 worin R3, R4, R5, R6, R7, R8 und Z die oben beschriebenen Bedeutungen aufweisen, wodurch ein Aminosäurederivat der allgemeinen Formel (7a) erhalten wird:
worin R3, R4, R5, R6, R7, R8 und Z die oben beschriebenen Bedeutungen aufweisen, wodurch ein Aminosäurederivat der allgemeinen Formel (7a) erhalten wird: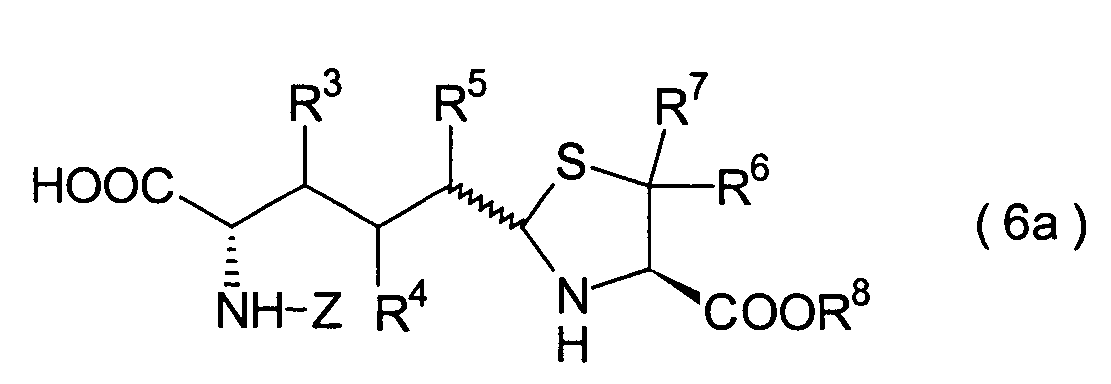 worin R3 bis R8 und Z die gleichen Bedeutungen haben wie oben definiert, und, sofern erforderlich, Entschützen dieses Derivats unter Erhalt eines Aminosäurederivats der allgemeinen Formel (1a):
worin R3 bis R8 und Z die gleichen Bedeutungen haben wie oben definiert, und, sofern erforderlich, Entschützen dieses Derivats unter Erhalt eines Aminosäurederivats der allgemeinen Formel (1a):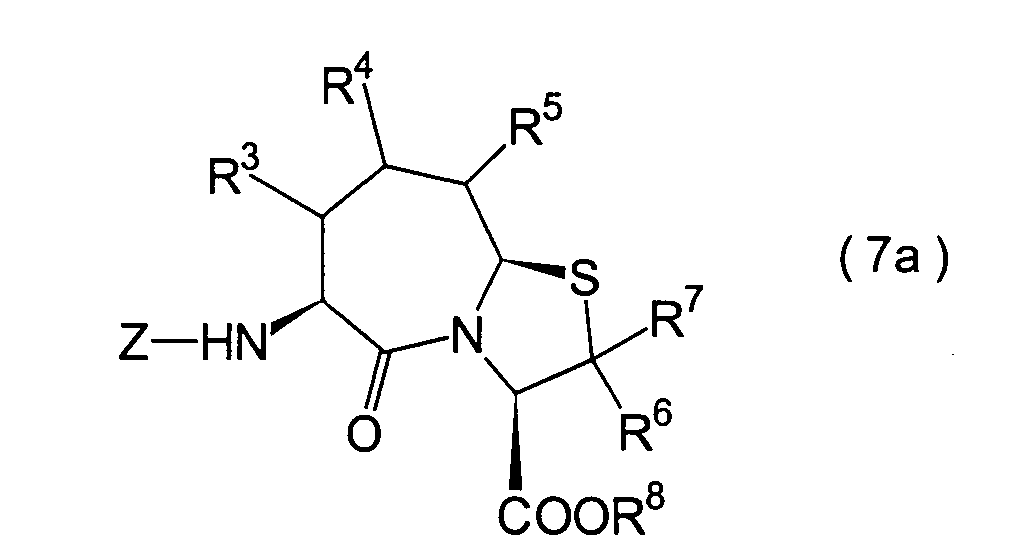 worin R3 bis R8 die gleichen Bedeutungen aufweisen wie oben definiert.
worin R3 bis R8 die gleichen Bedeutungen aufweisen wie oben definiert.
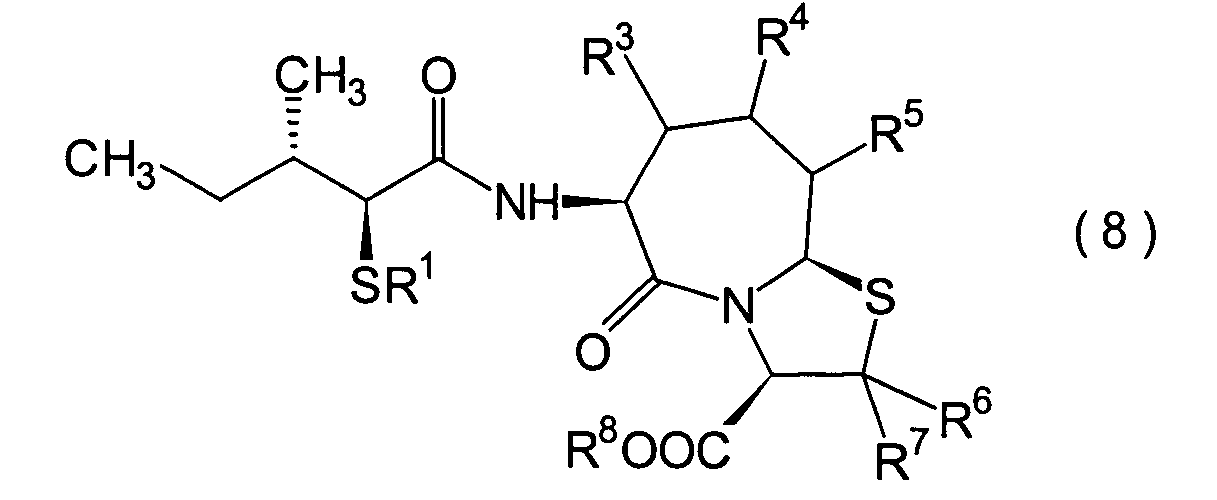 wodurch eine α-Hydroxycarbonsäure (10) erhalten wird:
wodurch eine α-Hydroxycarbonsäure (10) erhalten wird:

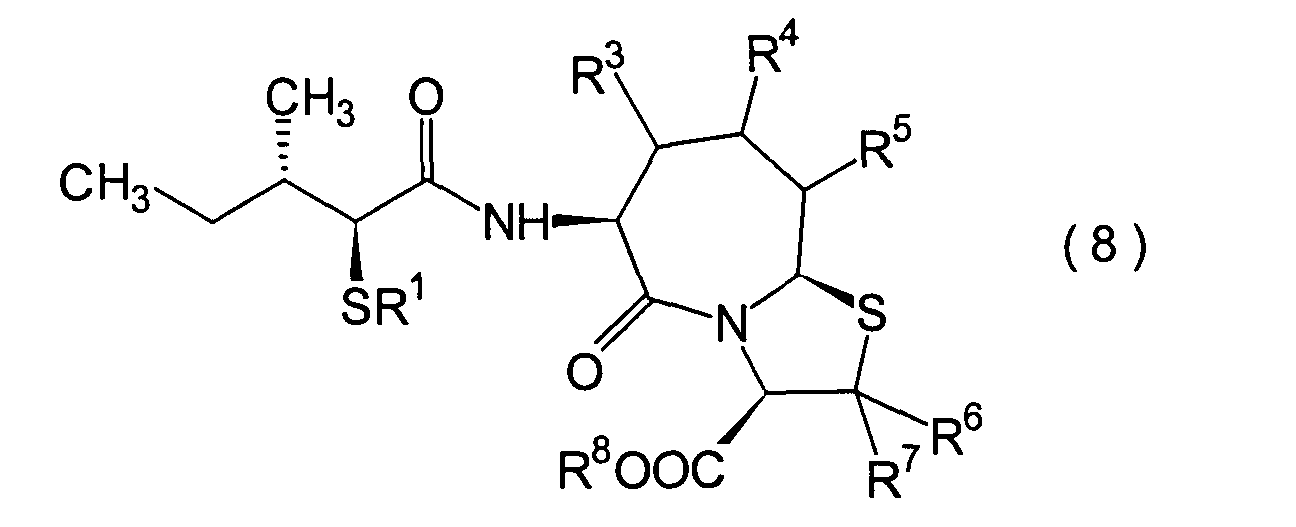 worin R2a ein Wasserstoffatom oder eine Schutzgruppe für eine Carboxylgruppe ist, durch Kupplung einer α-Hydroxycarbonsäure (10):
worin R2a ein Wasserstoffatom oder eine Schutzgruppe für eine Carboxylgruppe ist, durch Kupplung einer α-Hydroxycarbonsäure (10):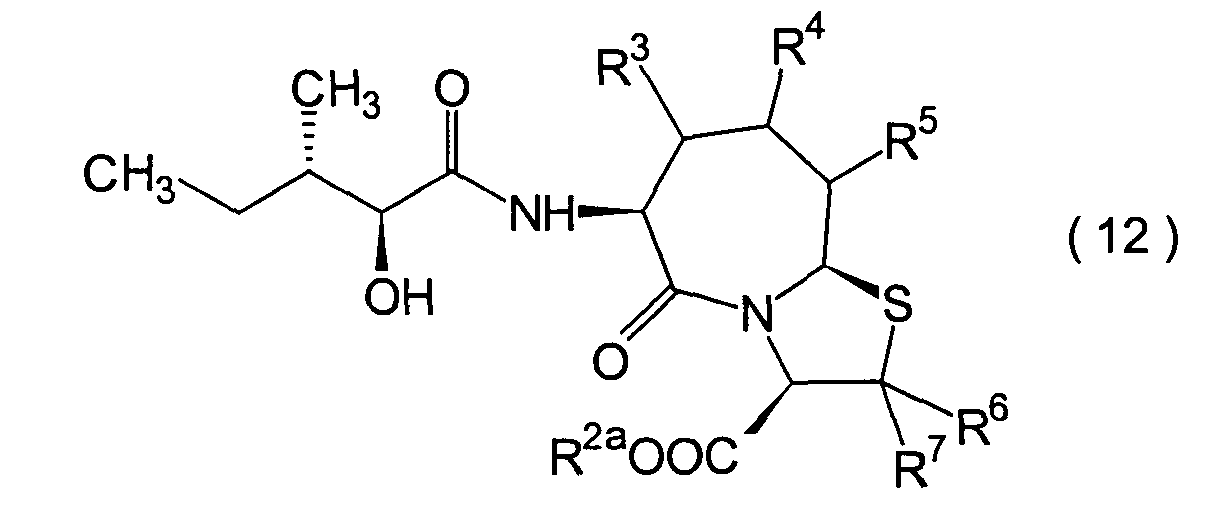 mit einem Aminderivat der allgemeinen Formel (11):
mit einem Aminderivat der allgemeinen Formel (11): worin R3, R4 und R5 jeweils unabhängig voneinander ein Wasserstoffatom, eine C1-6-Alkylgruppe, eine C1-6-Alkoxylgruppe oder eine C1-6-Alkylthiogruppe darstellen, mit der Massgabe, dass der Fall, dass R3, R4 und R5 alle Wasserstoffatome darstellen, ausgeschlossen ist; R6 und R7 repräsentieren jeweils ein Wasserstoffatom; und R8a ist eine Schutzgruppe für eine Carbonsäure.
worin R3, R4 und R5 jeweils unabhängig voneinander ein Wasserstoffatom, eine C1-6-Alkylgruppe, eine C1-6-Alkoxylgruppe oder eine C1-6-Alkylthiogruppe darstellen, mit der Massgabe, dass der Fall, dass R3, R4 und R5 alle Wasserstoffatome darstellen, ausgeschlossen ist; R6 und R7 repräsentieren jeweils ein Wasserstoffatom; und R8a ist eine Schutzgruppe für eine Carbonsäure.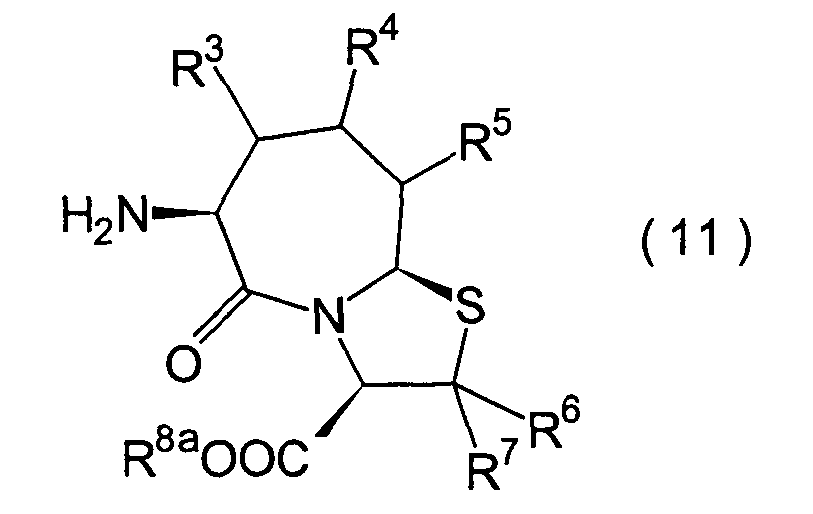
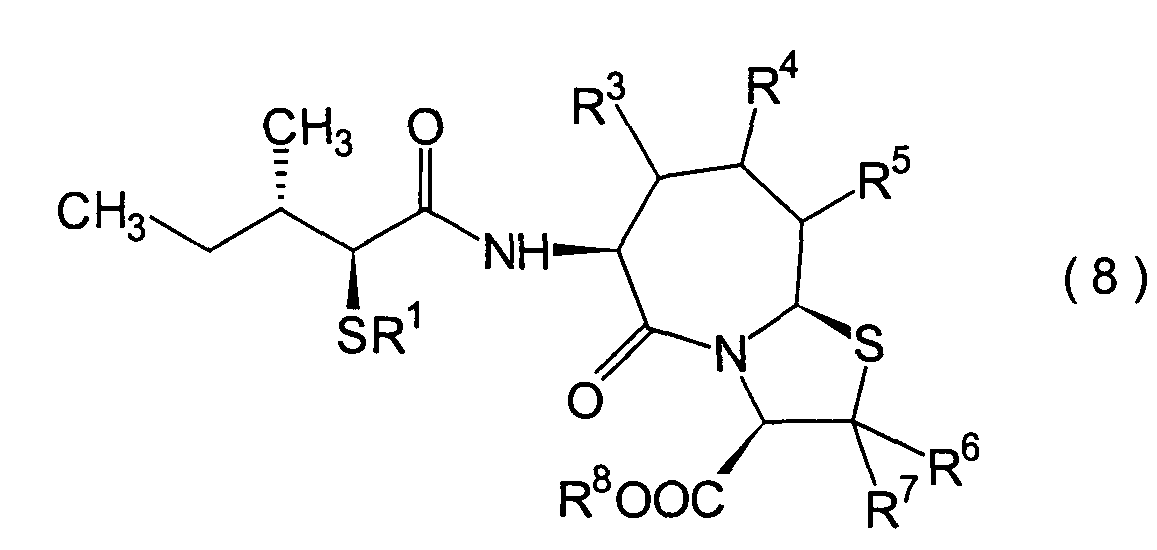 und, falls notwendig, Hydrolysieren desselben, worin R3, R4 und R5 jeweils unabhängig voneinander ein Wasserstoffatom, eine C1-6-Alkylgruppe, eine C1-6-Alkoxylgruppe oder eine C1-6-Alkylthiogruppe darstellen, mit der Massgabe, dass der Fall, dass R3, R4 und R5 alle Wasserstoffatome darstellen, ausgeschlossen ist; R6 und R7 repräsentieren jeweils ein Wasserstoffatom; und R2a ist ein Wasserstoffatom oder eine Schutzgruppe für eine Carbonsäure.
und, falls notwendig, Hydrolysieren desselben, worin R3, R4 und R5 jeweils unabhängig voneinander ein Wasserstoffatom, eine C1-6-Alkylgruppe, eine C1-6-Alkoxylgruppe oder eine C1-6-Alkylthiogruppe darstellen, mit der Massgabe, dass der Fall, dass R3, R4 und R5 alle Wasserstoffatome darstellen, ausgeschlossen ist; R6 und R7 repräsentieren jeweils ein Wasserstoffatom; und R2a ist ein Wasserstoffatom oder eine Schutzgruppe für eine Carbonsäure.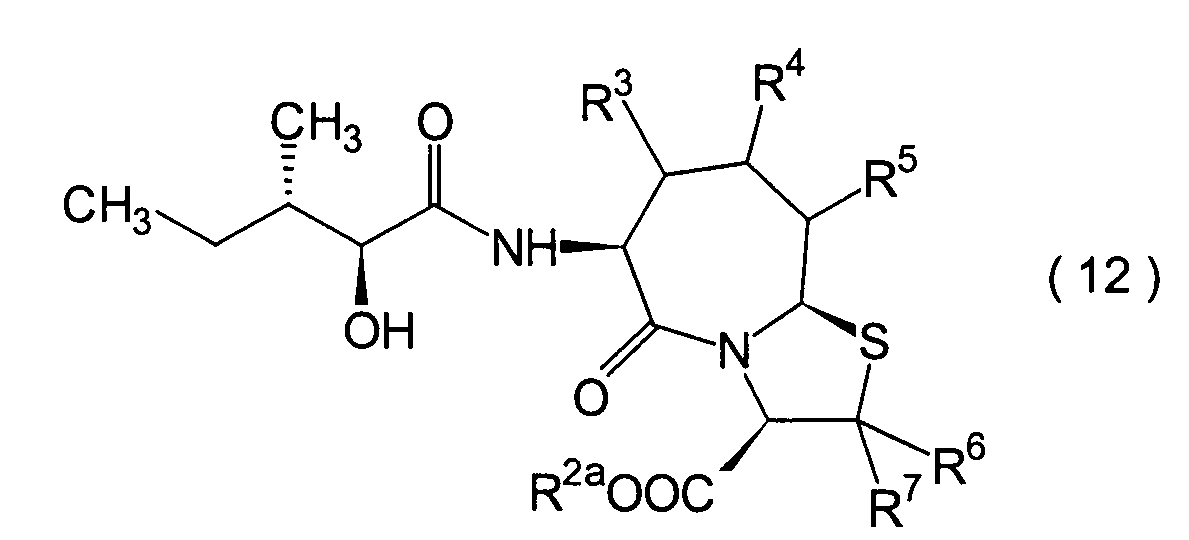
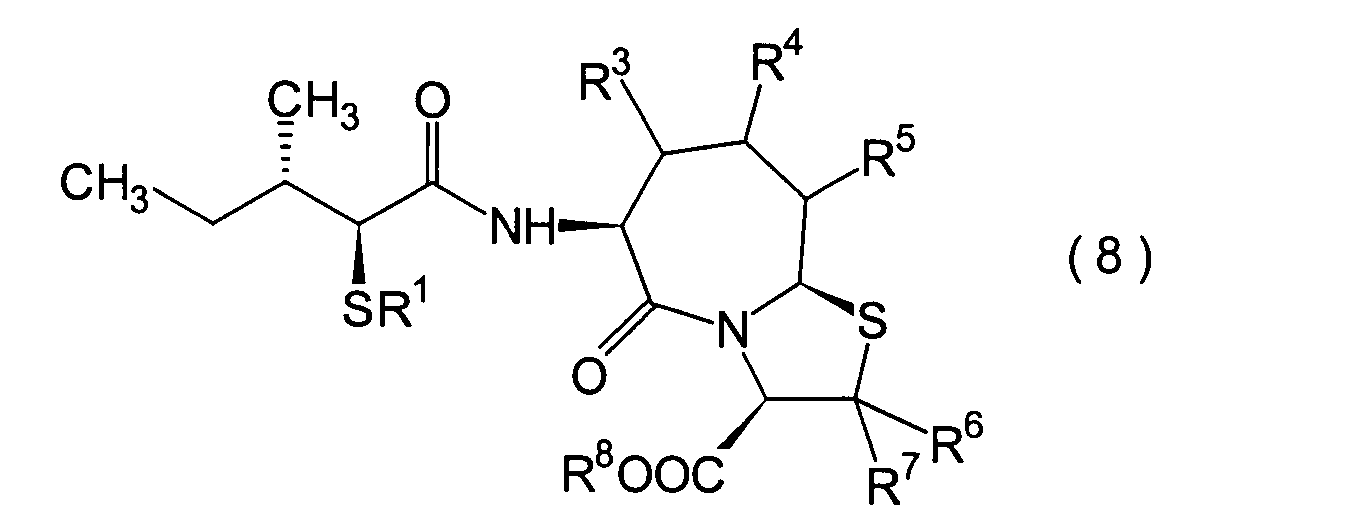 wodurch eine α-Hydroxycarbonsäure erhalten wird:
wodurch eine α-Hydroxycarbonsäure erhalten wird: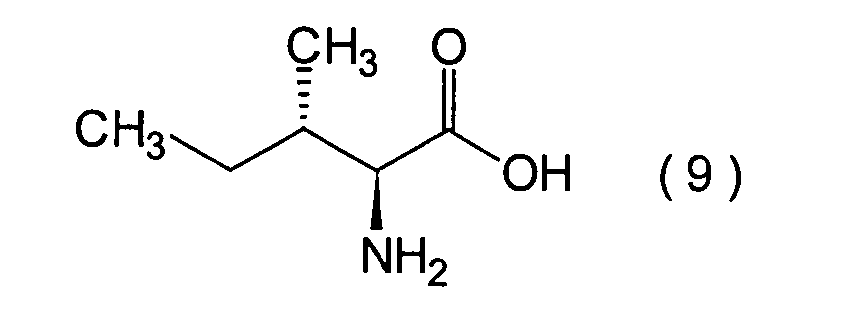 sowie die Kupplung dieser Säure mit einem Aminderivat der allgemeinen Formel (11):
sowie die Kupplung dieser Säure mit einem Aminderivat der allgemeinen Formel (11):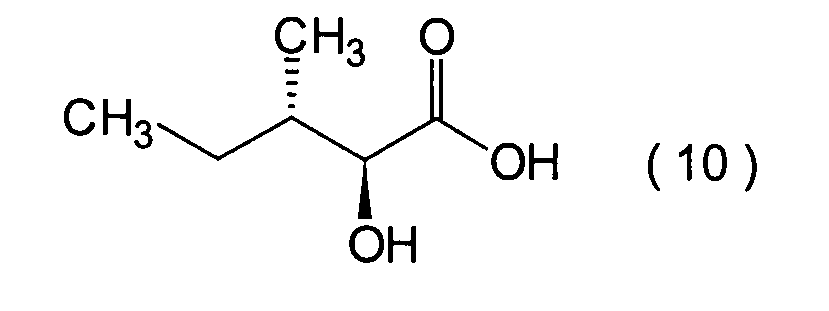 worin R3, R4 und R5 jeweils unabhängig voneinander ein Wasserstoffatom, eine C1-6-Alkylgruppe, eine C1-6-Alkoxylgruppe oder eine C1-6-Alkylthiogruppe darstellen, mit der Massgabe, dass der Fall, dass R3, R4 und R5 alle Wasserstoffatome darstellen, ausgeschlossen ist; R6 und R7 repräsentieren jeweils ein Wasserstoffatom; und R8a ist eine Schutzgruppe für eine Carbonsäure, wodurch ein α-Hydroxycarbonsäureamidderivat der allgemeinen Formel (12) erhalten wird:
worin R3, R4 und R5 jeweils unabhängig voneinander ein Wasserstoffatom, eine C1-6-Alkylgruppe, eine C1-6-Alkoxylgruppe oder eine C1-6-Alkylthiogruppe darstellen, mit der Massgabe, dass der Fall, dass R3, R4 und R5 alle Wasserstoffatome darstellen, ausgeschlossen ist; R6 und R7 repräsentieren jeweils ein Wasserstoffatom; und R8a ist eine Schutzgruppe für eine Carbonsäure, wodurch ein α-Hydroxycarbonsäureamidderivat der allgemeinen Formel (12) erhalten wird: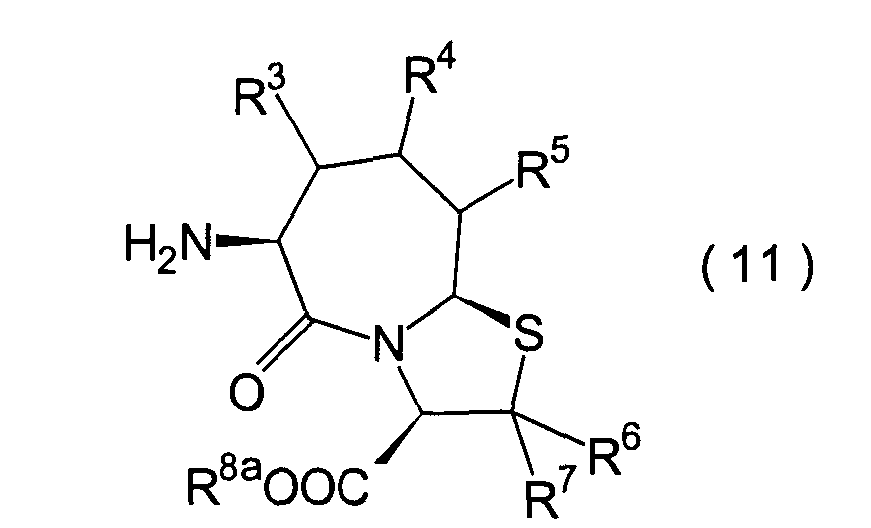 worin R2a ein Wasserstoffatom oder eine Schutzgruppe für eine Carbonsäure ist.
worin R2a ein Wasserstoffatom oder eine Schutzgruppe für eine Carbonsäure ist.
 worin R3, R4 und R5 jeweils unabhängig voneinander ein Wasserstoffatom, eine C1-6-Alkylgruppe, eine C1-6-Alkoxylgruppe oder eine C1-6-Alkylthiogruppe darstellen, mit der Massgabe, dass der Fall, dass R3, R4 und R5 alle Wasserstoffatome darstellen, ausgeschlossen ist; R6 und R7 repräsentieren jeweils ein Wasserstoffatom; und R8a ist eine Schutzgruppe für eine Carbonsäure, wodurch ein α-Hydroxycarbonsäureamidderivat der allgemeinen Formel (12) erhalten wird:
worin R3, R4 und R5 jeweils unabhängig voneinander ein Wasserstoffatom, eine C1-6-Alkylgruppe, eine C1-6-Alkoxylgruppe oder eine C1-6-Alkylthiogruppe darstellen, mit der Massgabe, dass der Fall, dass R3, R4 und R5 alle Wasserstoffatome darstellen, ausgeschlossen ist; R6 und R7 repräsentieren jeweils ein Wasserstoffatom; und R8a ist eine Schutzgruppe für eine Carbonsäure, wodurch ein α-Hydroxycarbonsäureamidderivat der allgemeinen Formel (12) erhalten wird: worin R2a ist ein Wasserstoffatom oder eine Schutzgruppe für eine Carbonsäure darstellt, sowie die Einführung einer Acylthiogruppe und, sofern erforderlich, Hydrolyse derselben, wodurch ein (2S,3S)-3-Methyl-2-thiopentansäurederivat der allgemeinen Formel (8) erhalten wird:
worin R2a ist ein Wasserstoffatom oder eine Schutzgruppe für eine Carbonsäure darstellt, sowie die Einführung einer Acylthiogruppe und, sofern erforderlich, Hydrolyse derselben, wodurch ein (2S,3S)-3-Methyl-2-thiopentansäurederivat der allgemeinen Formel (8) erhalten wird: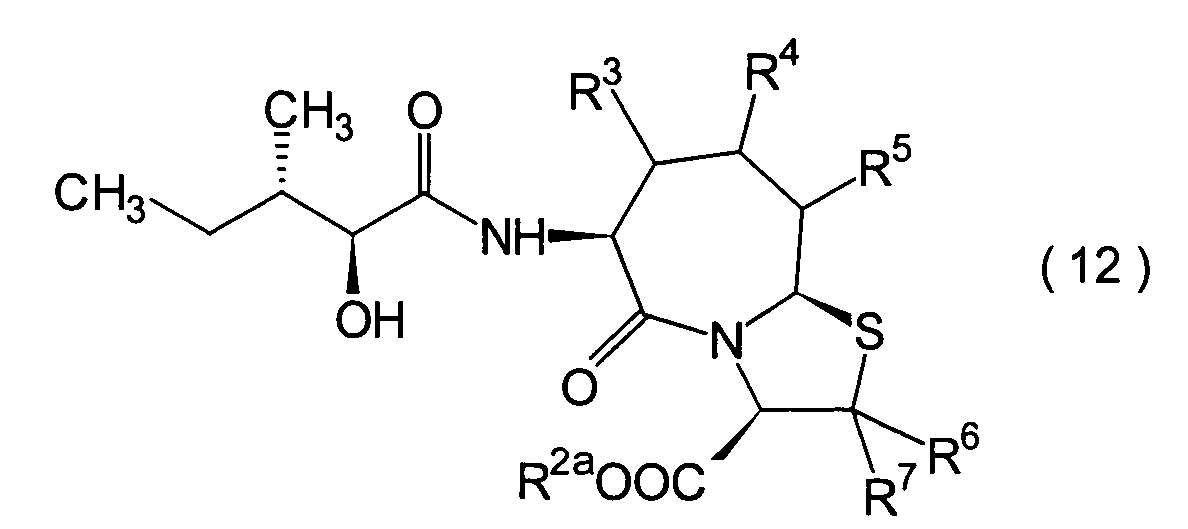 worin R1 ein Wasserstoffatom oder eine Acylgruppe ist; und R5 ist ein Wasserstoffatom oder eine Schutzgruppe für eine Carboxylgruppe.
worin R1 ein Wasserstoffatom oder eine Acylgruppe ist; und R5 ist ein Wasserstoffatom oder eine Schutzgruppe für eine Carboxylgruppe.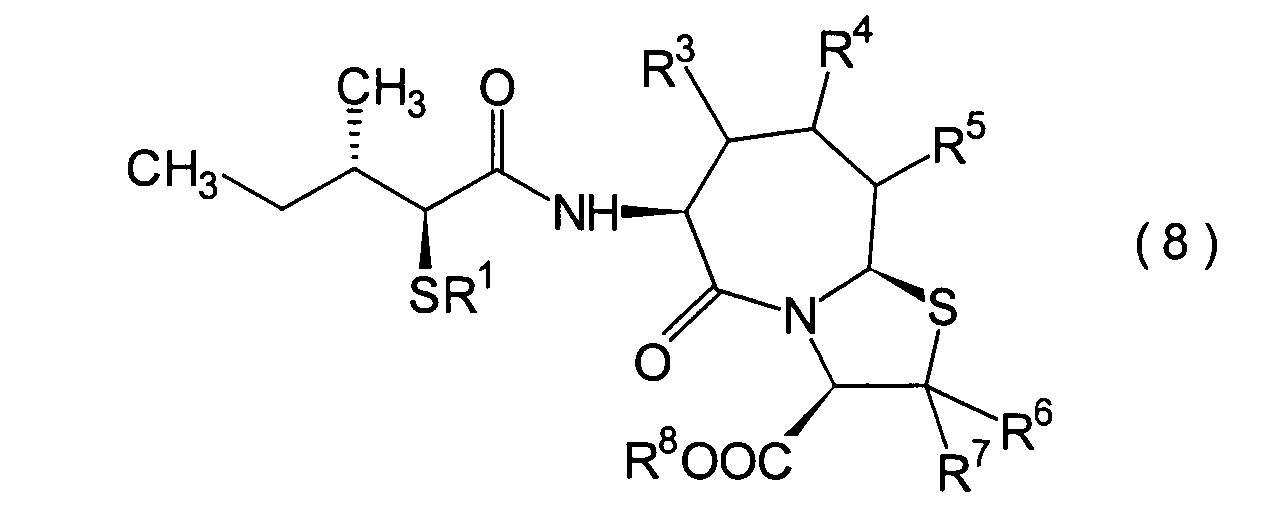
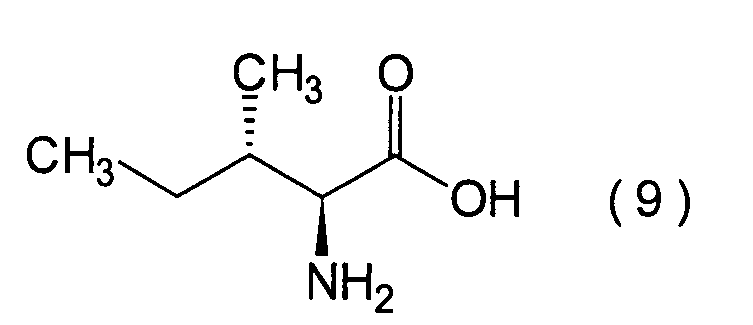 sowie die Kupplung dieser Säure mit einem Aminderivat der allgemeinen Formel (11):
sowie die Kupplung dieser Säure mit einem Aminderivat der allgemeinen Formel (11): worin R3, R4 und R5 jeweils unabhängig voneinander ein Wasserstoffatom, eine C1-6-Alkylgruppe, eine C1-6-Alkoxylgruppe oder eine C1-6-Alkylthiogruppe darstellen, mit der Massgabe, dass der Fall, dass R3, R4 und R5 alle Wasserstoffatome darstellen, ausgeschlossen ist; R6 und R7 repräsentieren jeweils ein Wasserstoffatom; und R8a ist eine Schutzgruppe für eine Carbonsäure, wodurch ein α-Hydroxycarbonsäureamidderivat der allgemeinen Formel (12) erhalten wird:
worin R3, R4 und R5 jeweils unabhängig voneinander ein Wasserstoffatom, eine C1-6-Alkylgruppe, eine C1-6-Alkoxylgruppe oder eine C1-6-Alkylthiogruppe darstellen, mit der Massgabe, dass der Fall, dass R3, R4 und R5 alle Wasserstoffatome darstellen, ausgeschlossen ist; R6 und R7 repräsentieren jeweils ein Wasserstoffatom; und R8a ist eine Schutzgruppe für eine Carbonsäure, wodurch ein α-Hydroxycarbonsäureamidderivat der allgemeinen Formel (12) erhalten wird: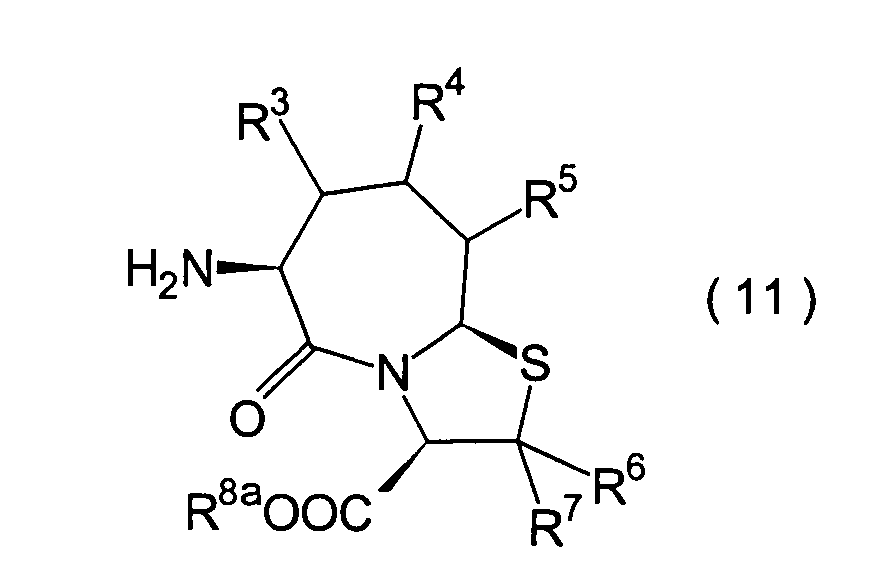 worin R2a ein Wasserstoffatom oder eine Schutzgruppe für eine Carbonsäure ist, sowie die Einführung einer Acylthiogruppe und, soweit erforderlich, Hydrolyse derselben, wodurch ein (2S,3S)-3-Methyl-2-thiopentansäurederivat der allgemeinen Formel (8) erhalten wird:
worin R2a ein Wasserstoffatom oder eine Schutzgruppe für eine Carbonsäure ist, sowie die Einführung einer Acylthiogruppe und, soweit erforderlich, Hydrolyse derselben, wodurch ein (2S,3S)-3-Methyl-2-thiopentansäurederivat der allgemeinen Formel (8) erhalten wird: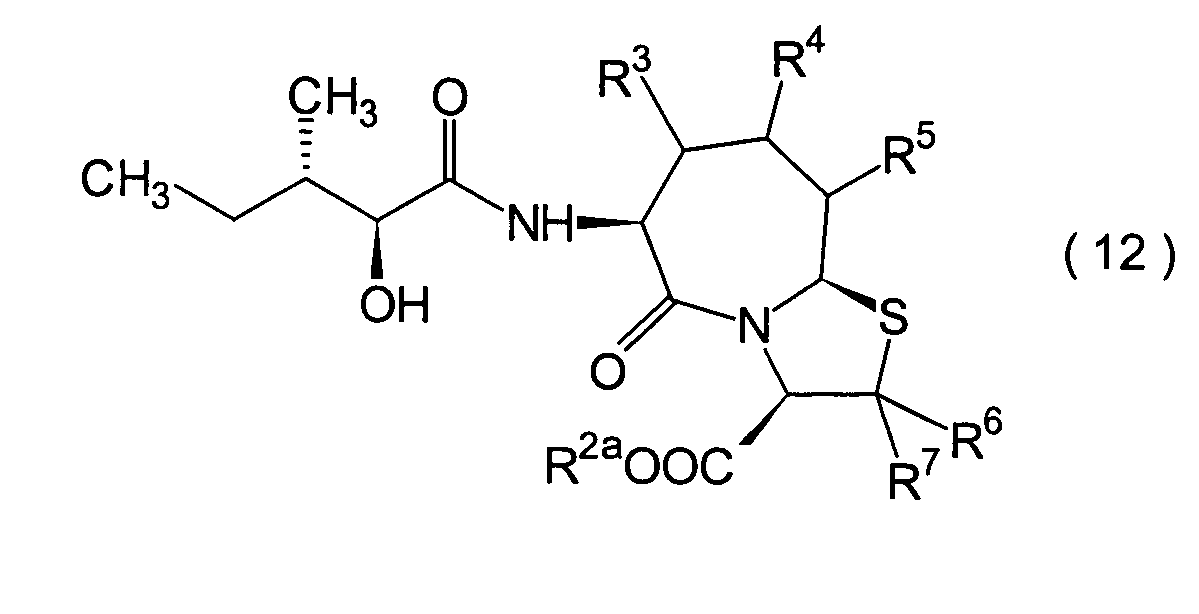 worin R1 ein Wasserstoffatom oder eine Acylgruppe ist; und R8 ist ein Wasserstoffatom oder eine Schutzgruppe für eine Carboxylgruppe.
worin R1 ein Wasserstoffatom oder eine Acylgruppe ist; und R8 ist ein Wasserstoffatom oder eine Schutzgruppe für eine Carboxylgruppe.
 das einen Schritt umfasst, worin es erhalten wird durch Halogenierung des α-Hydroxycarbonsäureamidderivats unter Erhalt eines α-Halogencarbonsäureamidderivats der allgemeinen Formel (13):
das einen Schritt umfasst, worin es erhalten wird durch Halogenierung des α-Hydroxycarbonsäureamidderivats unter Erhalt eines α-Halogencarbonsäureamidderivats der allgemeinen Formel (13): worin X ein Halogenatom ist; R3, R4 und R5 sind jeweils unabhängig voneinander ein Wasserstoffatom, eine C1-6-Alkylgruppe, eine C1-6-Alkoxylgruppe oder eine C1-6-Alkylthiogruppe, mit der Massgabe, dass der Fall, dass R3, R4 und R5 alle Wasserstoffatome darstellen, ausgeschlossen ist; R6 und R7 repräsentieren jeweils ein Wasserstoffatom; und R2a ist ein Wasserstoffatom oder eine Schutzgruppe für eine Carbonsäure, Einführen einer Acylthiogruppe und, sofern erforderlich, Hydrolyse.
worin X ein Halogenatom ist; R3, R4 und R5 sind jeweils unabhängig voneinander ein Wasserstoffatom, eine C1-6-Alkylgruppe, eine C1-6-Alkoxylgruppe oder eine C1-6-Alkylthiogruppe, mit der Massgabe, dass der Fall, dass R3, R4 und R5 alle Wasserstoffatome darstellen, ausgeschlossen ist; R6 und R7 repräsentieren jeweils ein Wasserstoffatom; und R2a ist ein Wasserstoffatom oder eine Schutzgruppe für eine Carbonsäure, Einführen einer Acylthiogruppe und, sofern erforderlich, Hydrolyse.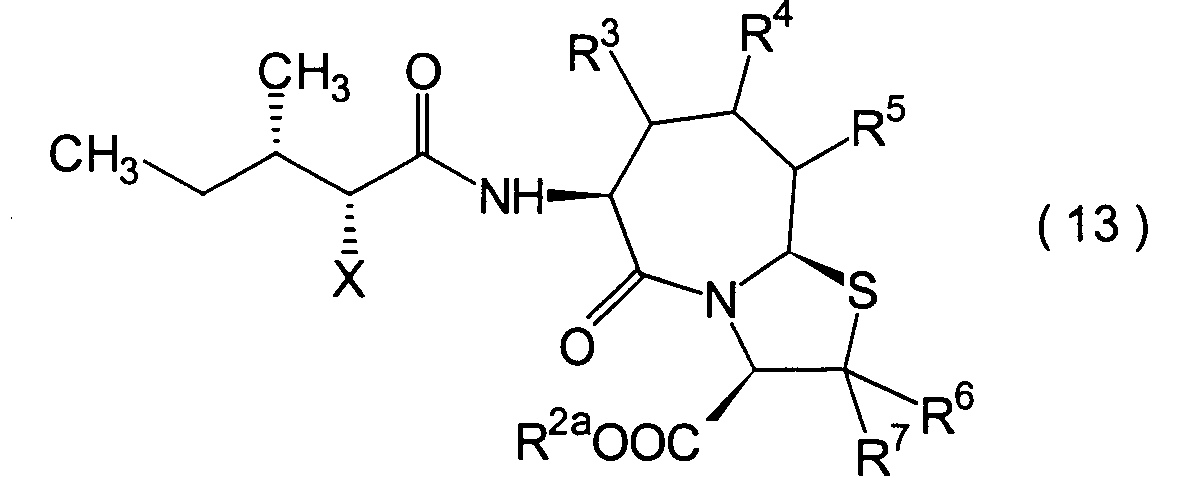
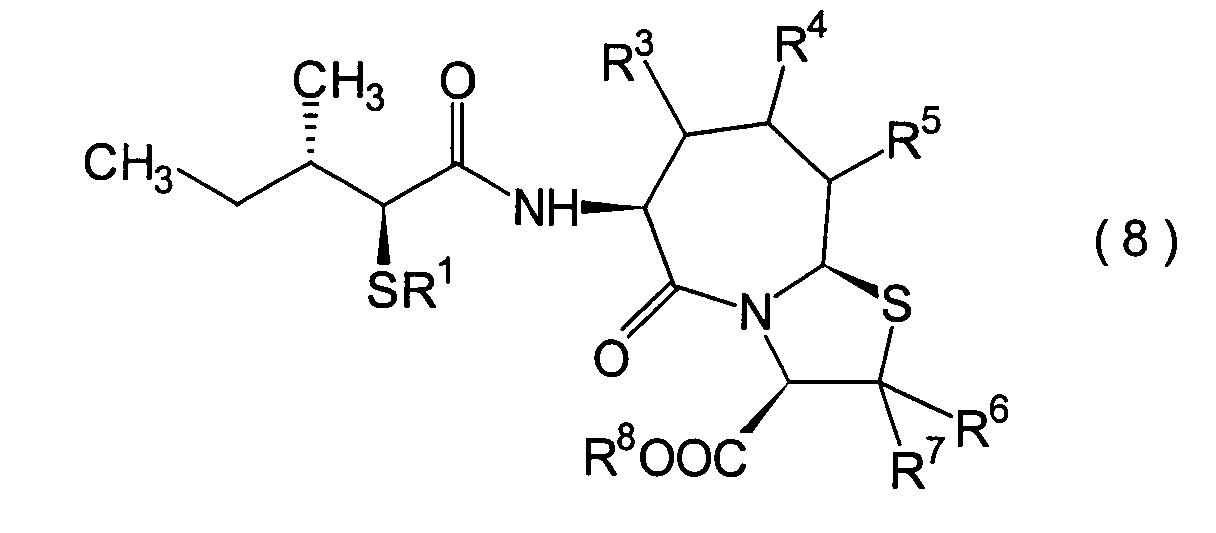 das einen Schritt umfasst, der die Halogenierung des α-Hydroxycarbonsäureamidderivats (12) umfasst, wodurch ein α-Halogencarbonsäureamidderivat der allgemeinen Formel (13) erhalten wird:
das einen Schritt umfasst, der die Halogenierung des α-Hydroxycarbonsäureamidderivats (12) umfasst, wodurch ein α-Halogencarbonsäureamidderivat der allgemeinen Formel (13) erhalten wird: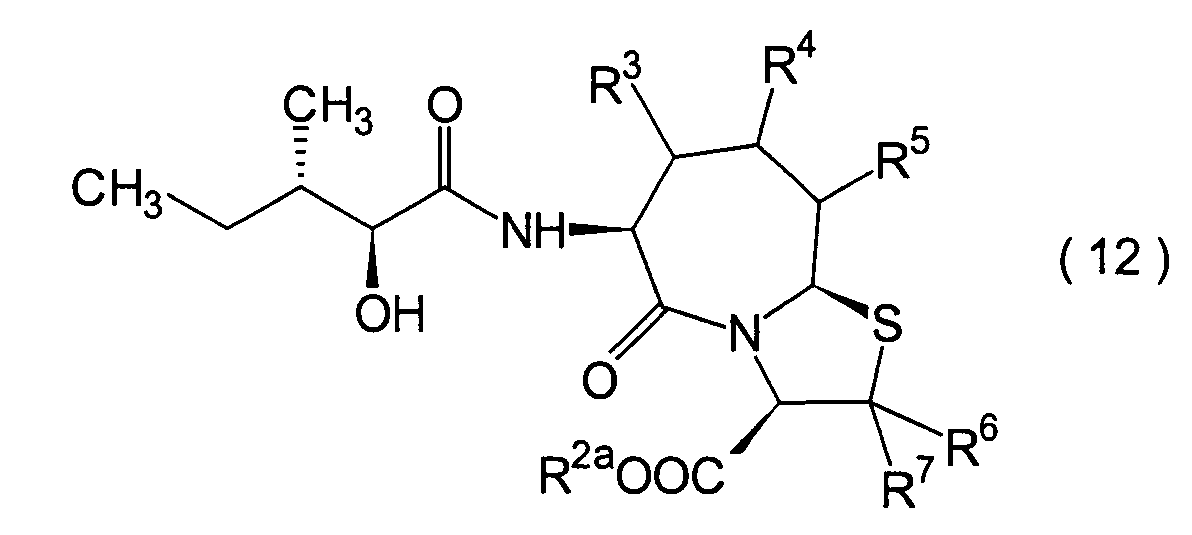 worin X ein Halogenatom ist; sowie bei Bedarf dessen Hydrolyse, wodurch ein α-Halogencarbonsäurederivat der allgemeinen Formel (14) erhalten wird:
worin X ein Halogenatom ist; sowie bei Bedarf dessen Hydrolyse, wodurch ein α-Halogencarbonsäurederivat der allgemeinen Formel (14) erhalten wird: worin R3, R4 und R5 jeweils unabhängig voneinander ein Wasserstoffatom, eine C1-6-Alkylgruppe, eine C1-6-Alkoxylgruppe oder eine C1-6-Alkylthiogruppe darstellen, mit der Massgabe, dass der Fall, dass R3, R4 und R5 alle Wasserstoffatome darstellen, ausgeschlossen ist; R6 und R7 repräsentieren jeweils ein Wasserstoffatom; und R2a ist ein Wasserstoffatom oder eine Schutzgruppe für eine Carbonsäure, und ferner Einführung einer Acylthiogruppe.
worin R3, R4 und R5 jeweils unabhängig voneinander ein Wasserstoffatom, eine C1-6-Alkylgruppe, eine C1-6-Alkoxylgruppe oder eine C1-6-Alkylthiogruppe darstellen, mit der Massgabe, dass der Fall, dass R3, R4 und R5 alle Wasserstoffatome darstellen, ausgeschlossen ist; R6 und R7 repräsentieren jeweils ein Wasserstoffatom; und R2a ist ein Wasserstoffatom oder eine Schutzgruppe für eine Carbonsäure, und ferner Einführung einer Acylthiogruppe.