发明概述
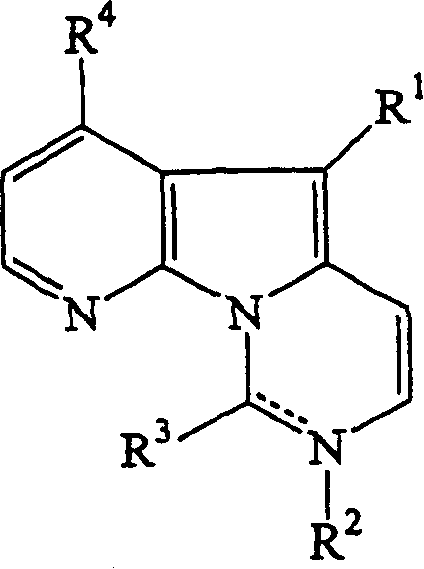
本发明的一方面涉及式(I)的化合物及其药学上可接受的盐:
其中:
R1为芳族取代基;
当虚线为不存在时,R2为氢或取代基,或当虚线表示键,使得在带有R2的氮和带有R3的碳之间为双键时,R2不存在;
当虚线不存在时,R3为氧代基=O,或当虚线表示键,使得在带有R2的氮和带有R3的碳之间为双键时,R3为取代基;
R4为氢或取代基。
基团R1一般为4至10元、更优选5或6元,最优选6元芳族环。在本发明中R1还可以是稠合环。所述环可具有一个或多个杂原子,具有1至3个选自氮、氧或硫的环杂原子是合适的,特别是2个杂原子。特别优选为氮杂原子,例如R1为嘧啶环,特别是具有下式的4-嘧啶基取代基:
所述芳族环可被取代,如被一个或多个选自以下的基团取代:烷基、烷氧基、硫代烷基、卤基、氨基、取代氨基、卤代烷基、烷氧基烷基、芳基、羟基、羧基、烷氧基羰基或其它常规的基团,包括甲磺酰基。在下文中给出的其它基团也可用作取代基。
当R2存在时,优选为氢、氮保护基或一些其它的取代基。氮保护基的例子如甲氧基甲基或甲苯磺酰基是众所周知的,无须进行详细的讨论。其它取代基的例子包括可通过其中R1为氢的化合物的反应,在该位置上进行取代的任何基团。更具体的可参考下文给出的其它基团。
R3为氧代基,或者可为通过所述氧代化合物的反应而引入的取代基,包括氨基、取代氨基(包括受保护的氨基)和硫代烷基。更具体的可参考下文给出的其它基团。
R4为氢,或取代基,如烷氧基(特别是甲氧基)、羟基、卤基(特别是氯基),或其它可通过亲核取代或通过其它衍生化作用引入的基团,包括硫代烷基或甲磺酰基。当R4为氢时,所述化合物为deoxyvariolin B衍生物。当R4为羟基时,所述化合物为variolin B衍生物。更具体的可参考下文给出的其它基团。
优选R1为在2位取代的4-嘧啶基。适合的取代基包括氨基及其衍生物,如N-酰基,特别是N-乙酰基。其它亲核取代基也包括在内,如烷氧基或烷硫基取代基,特别是甲硫基。
优选不存在R2。
优选R3为氨基及其衍生物,如N-酰基,特别是N-乙酰基。
优选R4为氢。
优选虚线表示键。
特别优选的一类化合物包括其中:
R1为在2位上被以下基团取代的4-嘧啶基:氨基、N-酰基(特别是N-乙酰基)、烷硫基(特别是甲硫基)、烷基或芳基-亚磺酰基(特别是甲亚磺酰基)、烷基或芳基磺酰基(特别是甲磺酰基);
R2不存在;
R3为任选受保护的氨基或N-酰基,特别是N-乙酰基;并且
R4为氢、羟基或甲氧基的式(I)化合物。
本发明还涉及药学上可接受的盐。
可用于本发明的取代基的例子包括OH、OR′、SH、SR′、SOR′、SO2R′、NH2、NHR′、N(R′)2、NHCOR′、N(COR′)2、NHSO2R′、C(=O)R′、CO2H、CO2R′、C1-C12烷基和C1-C12卤代烷基,所述R′基团或各个R′基团独立选自OH、C1-C12烷基、C1-C12卤代烷基、芳基(可任选被选自C1-C6烷基、C1-C6烷氧基、C1-C6烷硫基、NH2、C1-C6烷基氨基、二(C1-C6烷基)氨基、NO2、CN和卤素的基团取代)、芳烷基或芳基链烯基(其中的芳基部分可任选被选自C1-C6烷基、C1-C6烷氧基、C1-C6烷硫基、NH2、C1-C6烷基氨基、二(C1-C6烷基)氨基、NO2、CN和卤素的基团取代),并且其中R1为式N(R′)2或N(COR′)2的基团,各个R′基团可相同或不同,或两个R′基团与它们连接的氮原子一起形成一个5-12元杂环。
在本说明书的定义中,烷基可为直链或支链基团,并优选具有1至约12个碳原子,更优选1至约8个碳原子,更为优选1至约6个碳原子,最优选1、2、3或4个碳原子。在本发明的化合物中特别优选烷基为甲基、乙基和丙基,包括异丙基。除非另有声明,否则用于此处的术语“烷基”同时包括环状和非环状基团,而环状基团包括至少三个碳环成员。
卤代烷基为被一个或多个卤原子(优选氟、氯、溴或碘)取代的上述烷基(包括环烷基),并优选具有1至约12个碳原子,更优选1至约8个碳原子,更为优选1至约6个碳原子,最优选1、2、3或4个碳原子。在本发明的化合物中特别优选的卤代烷基为被1、2或3个相同或不同的卤原子取代的甲基、乙基和丙基(包括异丙基),特别是氟代甲基、氟代氯代甲基、三氟代甲基和三氯代甲基。
本发明的化合物中优选的链烯基和炔基具有一个或多个不饱和键,并具有2至约12个碳原子,更优选2至约8个碳原子,更为优选2至约6个碳原子,甚至更优选2、3或4个碳原子。此处所用术语“链烯基和炔基”同时包括环状和非环状基团,而通常更优选直链或支链非环状基团。
在本发明的化合物中优选的烷氧基包括具有一个或多个(但优选只有一个)氧键和1至约12个碳原子,更优选1至约8个碳原子,更为优选1至约6个碳原子,最优选1、2、3或4个碳原子的基团。
在本发明的化合物中优选的烷硫基具有一个或多个(但优选只有一个)硫醚键,并具有1至约12个碳原子,更优选1至约8个碳原子,更为优选1至约6个碳原子。特别优选具有1、2、3或4个碳原子的烷硫基。
在本发明的化合物中优选的烷基亚磺酰基包括那些具有一个或多个亚砜基(SO)和1至约12个碳原子,更优选1至约8个碳原子,更为优选1至约6个碳原子的基团。特别优选具有1、2、3或4个碳原子的烷基亚磺酰基。
在本发明的化合物中优选的烷基磺酰基包括那些具有一个或多个砜基(SO2)和1至约12个碳原子,更优选1至约8个碳原子,更为优选1至约6个碳原子的基团。特别优选具有1、2、3或4个碳原子的烷基磺酰基。
在本发明的化合物中优选的烷酰基包括那些具有一个或多个羰基(CO)和1至约12个碳原子,更优选1至约8个碳原子,更为优选1至约6个碳原子(包括羰基上的碳)的基团。特别优选具有1、2、3或4个碳原子的烷酰基,尤其是甲酰基、乙酰基、丙酰基、丁酰基和异丁酰基。
在本发明的化合物中优选的烷氨基具有一个或多个(但优选只有一个)NH键和1至约12个碳原子,更优选1至约8个碳原子,更为优选1至约6个碳原子。特别优选具有1、2、3或4个碳原子的烷氨基,尤其是甲氨基、乙氨基、丙氨基和丁氨基。
在本发明的化合物中优选的二烷氨基具有一个或多个(但优选只有一个)与两个烷基结合的氮原子,两个烷基的每一个可具有1至约12个碳原子,更优选1至约8个碳原子,更为优选1至约6个碳原子。所述烷基可相同或不同。特别优选其中每个烷基具有1、2、3或4个碳原子的二烷氨基,尤其是二甲氨基、二乙氨基、N-甲基乙氨基、N-乙基丙氨基、二丙氨基、二丁氨基和N-甲基丁氨基。
在本发明的化合物中优选的烷酰基氨基具有一个与烷基结合的NH-CO-键,所述烷基具有1至约12个碳原子,更优选1至约8个碳原子,更为优选1至约6个碳原子。特别优选具有1、2、3或4个碳原子的烷酰基氨基,尤其是甲酰基氨基、乙酰基氨基、丙酰基氨基和丁酰基氨基。特别优选乙酰基氨基。
在本发明的化合物中优选的二烷酰基氨基具有一个与两个上述烷酰基结合的氮原子,两个烷酰基各自可相同或不同,具有1至约12个碳原子,更优选1至约8个碳原子,更为优选1至约6个碳原子。特别优选其中每个烷酰基具有1、2、3或4个碳原子的二烷酰基氨基,尤其是二甲酰基氨基、甲酰基乙酰基氨基、二乙酰基氨基、二丙酰基氨基和二丁酰基氨基。特别优选二乙酰基氨基。
在本发明的化合物中优选的烷基磺酰基氨基具有一个与烷基结合的NH-SO2-键,所述烷基具有1至约12个碳原子,更优选1至约8个碳原子,更为优选1至约6个碳原子。特别优选具有1、2、3或4个碳原子的烷基磺酰基氨基,尤其是甲磺酰基氨基、乙磺酰基氨基、丙磺酰基氨基和丁磺酰基氨基。
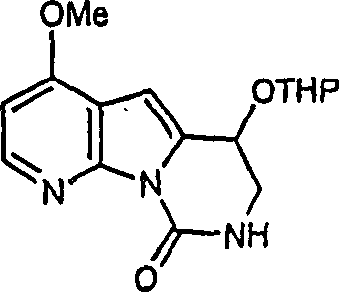
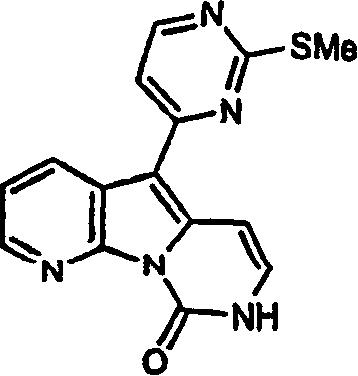
本发明化合物的具体例子包括在以下各页中的化合物(1)、(2)、(5)、(16)、(18)和(21),以及我们标记为(20a)的化合物,该化合物在方案4中为化合物(20)和(21)的中间体,具有下式的结构,其中不存在R2,R3为乙酰氨基,R4为氢和R1为2-甲硫基嘧啶-4-基:
本发明还提供包括本发明的化合物和药学上可接受的载体的药用组合物,提供本发明化合物在制备药物中的用途。还提供治疗方法。
药用组合物的实例包括含有适当组分的供口服、局部或肠胃外给药的任何固体(如片剂、丸剂、胶囊剂、颗粒剂)或液体(溶液剂、混悬剂或乳剂),且它们可包含所述纯的化合物或者与任何载体或其它药理活性的化合物相结合。当这些组合物经肠胃外给药时,须经灭菌处理。
本发明的各种化合物或组合物可通过任何合适的方法给药,如静脉内输注、口服制剂给药、腹膜内和静脉内给药。优选采用的输注时间低于24小时,更优选2-12小时,最优选2-6小时。特别理想的是短时间的输注,这样不必留在医院进行过夜治疗。但是,如果需要,输注时间可为12-24小时或者更长。可在2-4周的适当间隔内进行输注。含有本发明化合物的药用组合物可通过在缓释制剂中的脂质体或毫微球囊体、或者通过其它标准传递方式进行传递。
所述化合物的正确剂量将根据具体的剂型、施用方式、具体的部位、宿主以及所治疗的肿瘤而变化。还应考虑到其它因素如年龄、体重、性别、饮食、给药时间、排出率、宿主的症状、联合使用的药物、反应敏感性以及疾病的严重程度。可在最大的耐受剂量范围内连续或周期性给药。
本发明还提供用于联合疗法的药用组合物,所述药用组合物包括本发明的化合物和至少一种其它具有治疗活性的化合物。这些其它的化合物可具有抗肿瘤活性,或可具有某些与本发明化合物的抗肿瘤活性一起发挥作用的活性。
这些其它药物可作为同一组合物中的一部分,或者可作为独立的组合物同时或在不同时间进行给药。所述其它药物不受特别限定,适当的候选药物包括:
a)具有抗有丝分裂作用的药物,尤其是那些靶向细胞骨架成分的药物,包括微管调节剂,如紫杉烷类药物(如紫杉酚、紫杉醇、taxotere、docetaxel)、鬼臼毒素或长春花碱类(长春新碱、长春碱);
b)抗代谢药物,如5-氟尿嘧啶、阿糖胞苷、吉西他滨、嘌呤类似物(如喷司他丁、甲氨蝶呤);
c)烷基化剂,如氮芥类(如环磷酰胺或异环磷酰胺);
d)靶向DNA的药物,如蒽环类(antracycline)药物多柔比星(adriamycin)、多柔比星(doxorubicin)、pharmorubicin或表柔比星;
e)靶向拓扑异构酶的药物,如依托泊苷;
f)激素和激素激动剂或拮抗剂,如雌激素、抗雌激素(他莫昔芬及相关化合物)和雄激素、氟他胺、亮丙瑞林、戈舍瑞林、环丙孕酮(cyprotrone)或奥曲肽;
g)靶向肿瘤细胞信号传导的药物,包括抗体衍生物,如herceptin;
h)烷基化剂,如铂类药物(顺铂、卡铂(carbonplatin)、奥沙利铂、卡铂(paraplatin))或亚硝基脲;
i)有效影响肿瘤代谢的药物,如基质金属蛋白酶抑制剂;
j)基因疗法和反义剂;
k)抗体疗法;
l)其它源于海洋的生物活性化合物,特别是didemnins如aplidine,或海鞘素,如ET 743。
m)止吐药,具体如地塞米松。
方案4的化合物的细胞毒性由以下的IC50μM数据来说明:
|
化合物 |
P-388 |
A-549 |
HT-29 |
|
52120avariolinB |
0.36>3>30.85 |
0.040.160.290.17 |
0.040.160.290.09 |
本发明还涉及从7-氮杂吲哚开始或在更后的阶段开始合成所述化合物的方法。以7-氮杂吲哚为原料,在2位碳上锂化,引入C2-侧链,随后环化得到了三环吡啶并吡咯并嘧啶酮(11)。通过杂芳基偶合反应引入第四个芳环。
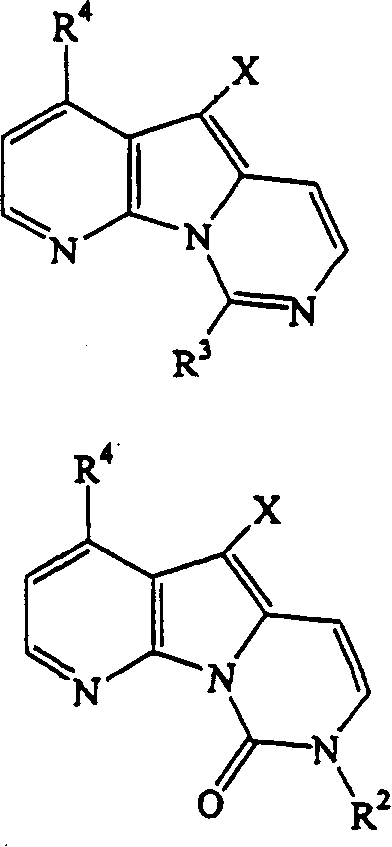
因此,本发明提供一种制备本发明化合物的方法,所述方法包括使任选取代的5-卤代吡啶并[3′,2′:4,5]吡咯并[1,2-c]嘧啶或8,9-二氢-5-卤代吡啶并[3′,2′:4,5]吡咯并[1,2-c]嘧啶-9-酮与衍生的芳族化合物,如甲锡烷基芳基化合物,特别是三甲基甲锡烷基芳基化合物,尤其是三甲基甲锡烷基嘧啶衍生物反应。可将所得的产物进一步反应以改变取代基。可将原料化合物中的氨基或其它活性取代基保护起来,并随后脱保护。
因此,本发明优选的中间体化合物具有下式的结构:
X为卤基,R2、R3和R4如上定义,其中X具体为碘基、R2为保护基团、R3为受保护的氨基和R4为氢、羟基或甲氧基。
我们所研究的合成variolin B的方法以deoxyvariolin B(5)为目标。我们的逆合成是以由7-氮杂吲哚制备的共同的吡啶并吡咯并嘧啶三环体系为基础。关键步骤是用于引入嘧啶取代基的由Pd(O)催化的杂芳基偶合。
在7-氮杂吲哚的2位上引入官能化的2碳链(参见J.Org.Chem.1965,30,2531-2533)通过2-锂-衍生物与2-邻苯二甲酰亚氨基乙醛(6)的反应来实现。2-邻苯二甲酰亚氨基乙醛(6)本身通过以下步骤获得:在140℃下,使2-氨基乙醛二甲缩醛与邻苯二甲酸酐在二氯甲烷中反应15分钟,将氨基保护;接着用10%的盐酸在回流下将所述缩醛基团水解;收率为75%。关于7-氮杂吲哚的锂化,现有技术仅描述的是对其N-苯基磺酰基衍生物的锂化,参见Tetrahedron,1997,53,3637-3648。而我们使用Katritzky描述的方法(参见J.Am.Chem.Soc.1986,108,6808-6809),该方法采用的是对原位形成的1-羧酸锂盐的2-锂化,这是因为我们发现这样的收率比采用1-苯基磺酰基-7-氮杂吲哚的高,另外避免了N-保护基团的引入和除去两步独立的步骤。由此,双锂衍生物(7)与醛(6)的反应以44%的收率得到醇(8)。醇的保护(变为四氢吡喃醚的形式)得到非对映异构体的混合物。该混合物没有被进一步分离,这是因为在合成后期两个立体异构中心(stereogeniccenters)都将不复存在。邻苯二甲酰亚胺残基的肼解定量地得到胺(9)。所得的胺(9)在二氯甲烷中,以二异丙基乙胺(DIPEA)为碱,采用三光气处理转化为四氢嘧啶酮(10),收率76%。通过酸水解除去羟基保护基团,接着通过甲磺酸盐对所述醇进行脱水得到二氢嘧啶酮(11)(方案1)。
方案1:吡啶并[3′,2′:4,5]吡咯并[1,2-c]嘧啶-1-酮(11)9的合成
i: n-BuLi、THF、-78℃至室温;
ii: CO2、-78℃;
iii:t-BuLi、THF、-78℃;
iv: 5THF、-78℃至室温(44%);
v: DHP、HCl-苯、CHCl3,(87%);
vi: NH2NH2·H2O、EtOH,(100%);
vii:(Cl3CO)2CO、DIPEA、CH2Cl2、室温(76%);
viii:4N HCl、CH2Cl2(100%);
ix: MsCl、TEA、CH2Cl2、0℃(95%)
按照我们以前将杂芳基与7-氮杂吲哚偶合(参见Synthesis 1999,615-620),以及将二氢吡咯并[1,2-c]嘧啶-1-酮锂化(参见J.Soc.Chem.Perkin Trans.I,1999,249-255)的经验,我们计划由受保护的卤代衍生物(13)来制备锡衍生物(14)。在DMF中,以氢化钠为碱,采用甲基氯甲基醚对三环嘧啶酮(11)进行保护,得到(12)。在氢氧化钾中使用N-溴代琥珀酰亚胺(NBS)或碘对(12)进行卤化,分别得到(13a)(80%)和(13b)(62%)。可通过对比1H-NMR波谱证实在C-5位上引入卤素:(12)在δ6.41ppm处出现的H-5单峰在(13a)和(13b)的波谱中没有出现。遗憾的是我们无法分离出锡衍生物(14),例如采用丁基锂处理(13a),随后用氯化三甲基锡猝灭(参见Synthesis 1999,615-620),得到一种络合物混合物,但无法将其解析出来。试图通过在二噁烷中,以Pd(PPh3)4为催化剂,用六甲基二锡处理(13b)来使碘-锡互换,得到(14)和(12)的混合物(比率为7∶3),但无法通过柱层析分离出14。
方案2
i: MOMCl,NaH,DMF,0℃(87%);
ii: NBS,CH2Cl2,0℃(80%);
ii:I2,KOH,DMF,0℃(62%)
我们现在改用三甲基甲锡烷基嘧啶(15)和碘代-7-氮杂吲哚(13b)来实施偶联反应。对嘧啶(15)的制备(参见Tetrahedron 1989,45,993-1006)的改进之处在于:使用Pd(OAc)2和PPh3的THF溶液。与现有技术描述的工作比较,减少了TBAF的量和反应时间。
将碘代衍生物(1mmol)、(15)(3mmol)、催化剂A或B、LiCL(3mmol)的二噁烷(20ml)溶液回流5小时。蒸发溶剂,将剩余物通过快速柱层析纯化。(13b)和(15)间的偶联反应在所有试验条件下都得到(12)和(16)的混合物,无法将它们分离(表1)。
表1.(13b)和(15)在二噁烷中的偶联反应
|
(15)/(13b)a |
Catal/Ligb |
LiCl/CuIc |
16/12d |
%e |
|
1.11.22 |
ABB |
3/--3/--3/0.1 |
2∶12∶13∶1 |
565253 |
a:摩尔比;
b:A=Pd(PPh3)4,0.1当量;B=Pd2(dba)3 0.1当量和PPh3 0.2当量;
c:相对于1当量的13b;
d:由1H-NMR测得的比;
e:由1H-NMR测量的由粗反应混合物计算的(16)的收率。
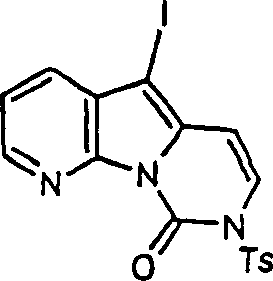
由于难以将(16)纯化,我们尝试采用新的保护基团。碘代化合物(17)(与(13b)的区别在于保护基团不同)通过(11)与甲苯磺酰氯和氢化钠的DMF溶液反应,接着用NIS碘化来合成。
(17)和三甲基甲锡烷基嘧啶(15)间的偶联反应产生四环化合物(18),但即使采用表1所示用于(13b)和(15)的偶联反应的最佳反应条件,也只能得到10%的收率。
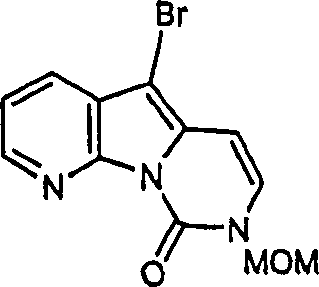
方案4.deoxyvariolin B的合成
i: TsCl,NaH,DMF(40%);
ii: NIS.CHCl3,室温(80%);
iii: 15,Pd2(dba)3,PPh3,LiCl,CuI(10%);
iv: TMSCl,HMDSA,2,6-二甲基吡啶;
v: NH3,150℃,60psi(30%两步);
vi: Ac2O,THF(75%);
vii:NIS.CHCl3(95%);
viii:iii随后为HCl-MeOH,(45%);
ix: MCPBA,CH2Cl2,0℃(90%);
x:NH4OH,二噁烷,80℃(90%)。
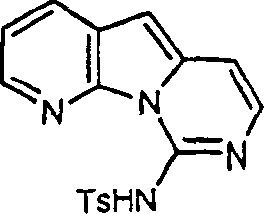
下一种近似的方法是通过将嘧啶酮(11)转化成iodoaniidopyrimidine(20)来改变C环的官能化。氨基衍生物19通过采用TMSCl和六甲基二硅氮烷(HMDSA)作为甲硅烷基化剂将(11)O-甲硅烷基化,接着用氨亲核取代制得,参见Lebgs Ann.Chem.,1975,988-1002。胺(19)的酰基化和在富含π电子的环空位上的卤代可以很好的收率进行。
采用前述相同的反应条件和催化剂,使(20)和(15)进行杂芳基偶联得到各种酰化和去保护的胺的混合物,该产物用无水HCl的甲醇溶液进行甲醇解,得到胺(21),收率45%。通过用氨基取代新产生的嘧啶环上的甲硫基制得deoxyvariolin B(5)。用间氯过苯甲酸氧化(21),接着用氢氧化铵将所得的砜取代为氨基,以极好的收率得到(5),参见Tetrahedron 1989,45,993-1006和Katrizky,A.R.;Rees,C.W.Comprehensive Heterocyclic Chemistry,Pergamon Press,Oxford,1984,第3卷,111页。
一种更通用的合成方案为:
试剂:
i: n-BuLi,THF,-78℃至室温;
ii: CO2,-78℃;
iii:t-BuLi,THF,-78℃;
iv: 6,THF,-78℃至室温;
v: DHP,HCl,苯,CHCl3,Δ;
vi: NH2MH2.H2O,EtOH,Δ;
vii:(Cl3CO)2CO,DIPEA,CH2Cl2,室温;
viii:4N HCl,CH2Cl2;
ix: MsCl,TEA,CH2Cl2,0℃;
x: MOMCl或TsCl,NaH,DMF,0℃;
xi: NBS/NIS,CH2Cl2,0℃或I2,KOH,,0℃;
xii: 15,Pd2(dba)3,PPh3,LiCl,CuI,二噁烷,Δ;
xiii: TMSCl,HMDSA,二甲基吡啶,D;
xiv: NH3,150℃,60psi;
xv: Ac2O,THF,室温;
xvi: NIS,CHCl3,0℃;
xvii: TsN=CCl2,DIPEA,CH2Cl2,室温;
xviii:4N HCl,CHCl3,室温;
xix: MsCl,TEA,CH2Cl2,室温;
xx: NIS,CHCl3,-30℃;
xxi: 无水MeOH-HCl或48%HBr,Δ。
由4-甲氧基-7-氮杂吲哚制备三环化合物11b(X=OMe),参见J.Heterocyclic Chem.1989,26,317,接着按照相同的方式制备11a。通过用三甲基甲硅烷基氯(TMSCl)和六甲基二硅氮烷(HMDSA)六甲基二硅氮烷进行O-甲硅烷基化,接着用氨进行亲核取代将11b转化为19b,收率仅为22%。将19b酰化并将所得的乙酰基衍生物碘化得到碘代乙酰胺20b。由于20b上存在H3-H4和H7-H8偶合质子,因此在1HNMR上显示为两组独立的AB芳族体系。20b和2-乙酰基氨基-4-三甲基甲锡烷基嘧啶(27)在钯催化下偶联,接着用酸处理得到O-methylvarioline B(5b)。以4-氯-2-甲磺酰基嘧啶为原料,用氨的异丙醇溶液对其甲磺酰基进行亲核取代,接着用乙酸酐酰化,随后以Pd(PPh3)4为催化剂,用六甲基二锡的二噁烷溶液进行卤素金属的互换制得甲锡烷基嘧啶(27),收率40%,参见Heterocycles,1977,8,299。
为了改进三环体系19的制备,略去嘧啶环的形成步骤。通过使9a-c与N-二氯亚甲基-4-甲基苯磺酰胺和DIPEA的二氯甲烷溶液反应,接着进行O-去保护和脱氢作用制得三环化合物22a-c,参见Chem.Ver.1966,99,1252。按照将10→11的相同转化方法,通过对22进行酸催化的O-去保护,接着脱氢制得N-甲苯磺酰基衍生物23。使用Na的氨溶液或Na的萘溶液将23的N-甲苯磺酰基保护基去除得到19,中等收率。
在前述相同条件下使23与甲锡烷基衍生物27进行杂芳基偶联反应得到25,具有非常好的收率。
可通过用甲醇的盐酸溶液进行酸催化的甲醇解来实施所述N-乙酰基的去保护。可通过用HBr处理来实现25a→26的转化。化合物26是一种仅一个氮受保护的新varioline B衍生物。
已经得到varoline B的几种衍生物,其中三种的差别仅是一个基团的变化:5a为dehydroxyvarioline B、5b为methylvarioline B和26为tosylvarioline B。
可按照用于23的相同条件除去26的甲苯磺酰基,得到variolineB。
按照类似的试验方法,由23c可制得化合物25c,由25c又可制得吡啶环的衍生物。
我们以前的试验表明杂芳族锡衍生物和13b、17和24的偶联具有很好的结果,使通式中的R1具有多样性。
我们已经研究出一种通用的合成方法,该方法不仅可用于合成这类海洋生物碱的这些基团,而且还可用于天然产物的其它衍生物。
本申请要求在先申请的优先权。为了使在先申请的所有内容均包括本说明书中,我们特别地通过引用该公开而将其结合到本文中来。
实施例
实施例1
2-(1-羟基-2-邻苯二甲酰亚氨基乙基)-7-氮杂吲哚(8a)
往经冷却(-78℃)的7-氮杂吲哚(7.6g,64mmol)的无水THF(150ml)溶液中加入正丁基锂(44ml,1.6M的己烷溶液),将所得混合液搅拌10分钟。使无水CO2通过所述混合液鼓泡40分钟。蒸发溶剂,将剩余物溶解在新鲜的无水THF(400ml)中。将溶液在-78℃下冷却,加入叔丁基锂(42ml,1.7M的己烷溶液)。搅拌所述混合液20分钟。缓慢加入邻苯二甲酰亚氨基乙醛(ftalimidoacetaldehyde)(14g,71mmol)的THF(400ml)溶液。1.5小时后,用饱和NH4Cl水溶液(100ml)猝灭所述反应液,蒸发有机溶剂。将混合物溶解在二氯甲烷中,用水洗涤。将有机溶液干燥并蒸发。混合物经快速柱层析纯化。用CH2Cl2/丙酮(95/5)洗脱,得到7-氮杂吲哚(3.8g,50%),接着用CH2Cl2/MeOH(98/2)洗脱得到8a(8.7g,44%)的白色固体。
mp 231-232℃(CH2Cl2/MeOH).
IR(KBr)ν3200(m,NH),1760(s,C=O),1704(s,NCO),1427(m,C-N),1395(m,C-O).
1H-NMR(DMSO-d6,200MHz)δ3.88(dd,J 13.6和6.0,1H,H2’),4.00(dd,J 13.6和7.8,1H,H2’),5.06(ddd,J 7.8,6.0和5.2,1H,H1’),5.83(d,J 5.2,1H,OH),6.34(d,J 1.8,1H,H3),6.99(dd,J 8.0和4.8,1H,H5),7.81-7.88(m,4H,Phth),7.89(dd,J 8.0和1.4,1H,H4),8.14(dd,J 4.8和1.4,1H,H6),11.75(brs,1H,NH).
13C-NMR(DMSO-d6,75MHz)δ43.6(t,C2’),64.4(d,C1’),96.8(d,C3),115.4(d,C5),119.8(s,C3a),123.0(d,Phth-β),127.6(d,C4),131.6(s,Phth-ipso),134.3(d,Phth-α),140.8(s,C2),142.1(d,C6),148.6(s,C7a),167.7(s,Phth-CO).
MS(EI)m/z 308(M+1,6),307(M+,25),244(8),160(43),147(邻苯二甲酰亚胺,100),119(氮杂吲哚,52)。
对C17H13N3O3的分析计算值:C(66.44),H(4.26),N(13.67);实测值:C(65.11),H(4.26),N(13.37)。
实施例2
2-(1-羟基-2-邻苯二甲酰亚氨基乙基)-4-甲氧基-7-氮杂吲哚(8b)
按照前述方法,由4-甲氧基-7-氮杂吲哚(3.55g,24mmol)的THF(75ml)溶液、n-BuLi(16.5ml,1.6M的己烷溶液),t-BuLi(16ml,1.7M的己烷溶液)和邻苯二甲酰亚氨基乙醛(5g,26mmol)的THF(100ml)溶液得到粗制混合物,将该混合物经快速柱层析纯化。用CH2Cl2/丙酮(95/5)洗脱得到4-甲氧基氮杂吲哚(2.06g,58%),接着用CH2Cl2/MeOH(98/2)洗脱得到8b(3.68g,43%)的白色固体。
mp 225-226℃(CH2Cl2/MeOH).
IR(KBr)ν3500(s,NH/OH),1702(s,C=O),1594(m),1395(m).
1H-NMR(DMSO-d6,300MHz)δ3.88(s,3H,Me),3.86(dd,J 13.8和6.0,1H,H2’),3.95(dd,J 13.8和7.8,1H,H2’),5.00(ddd,J 7.8,6.0和5.1,1H,H1’),5.73(d,J 5.1,1H,OH),6.30(d,J 1.8,1H,H3),6.58(d,J 5.4,1H,H5),7.83(m,4H,Phth),8.02(d,J 5.4,1H,H6),11.65(br,1H,NH).
13C-NMR(DMSO-d6,75MHz)δ43.6(t,C2’),55.3(q,Me),64.2(d,C1’),94.0(d,C5),97.8(d,C3),109.5(s,C3a),123.0(d,Phth-β),131.6(s,Phth-ipso),134.3(d,Phth-α),138.1(s,C2),144.2(d,C6),150.3(s,C7a*),158.5(s,C4*),167.7(s,Phth-CO).
MS(EI)m/z 338(M+1,4),337(M+,20),319(M-H2O,44),177(100).
对C18H15N3O4·1/4H2O的分析计算值:C(63.25),H(4.57),N(12.29);实测值:C(63.32),H(4.54),N(12.07)。
实施例3
4-氯-2-(1-羟基-2-邻苯二甲酰亚氨基乙基)-7-氮杂吲哚(8c)
按照前述方法,由4-氯-7-氮杂吲哚(5g,33mmol)的THF(100ml)溶液、n-BuLi(20ml,1.6M的己烷溶液),t-BuLi(20ml,1.7M的己烷溶液)和邻苯二甲酰亚氨基乙醛(7.5g,39mmol)的THF(140ml)溶液得到粗制混合物,将该混合物经快速柱层析纯化。用CH2Cl2/丙酮(95/5)洗脱得到4-氯氮杂吲哚(4g,80%),接着用CH2Cl2/MeOH(98/2)洗脱得到8c(1.5g,12%)的白色固体。
1H-NMR(DMSO-d6,200MHz)δ3.86(m,1H,H2’),3.95(m,1H,H2’),5.01(m,1H,H1’),5.92(d,J 5.2,1H,OH),6.41(s,1H,H3),7.14(d,J 5.6,1H,H5),7.84(m,4H,Phth),8.11(d,J 5.6,1H,H6),11.75(br,1H,NH).
13C-NMR(DMSO-d6,75MHz)δ43.6(t,C2’),64.3(d,C1’),94.9(d,C3),115.3(d,C5),118.7(s,C3a),123.0(d,Phth-β),131.6(s,Phth-ipso),133.3(s,C2),134.3(d,Phth-α),142.2(s,C4),142.8(d,C6),49.2(s,C7a),167.7(s,Phth-CO).
MS(EI)m/z 342(M+1,4),341(M+,20),323(M-H2O,44),177(100).
实施例4
2-[2-邻苯二甲酰亚氨基-1-(2,3,5,6-四氢吡喃-2-基)氧乙基]-7-氮杂吲哚
往8a(10.2g,33mmol)的CHCl3(1升)溶液中加入6N HCl的无水苯(180ml)溶液。往所得混合液中加入2,3-二氢吡喃(46ml,330mmol)。将反应液回流7小时。将所述混合液冷却后,用饱和NaHCO3水溶液洗涤,干燥并蒸发。所得混合物经快速柱层析纯化。用CH2Cl2/MeOH(97/3)洗脱得到2-[2-邻苯二甲酰亚氨基-1-(2,3,5,6-四氢吡喃-2-基)氧乙基]-7-氮杂吲哚(10.8g,87%)的非对映异构体混合物(1∶1),为白色固体。
IR(薄膜法)ν1717(s,C=O),1390(m,C-O),1026(m,C-O).
1H-NMR(CDCl3,200MHz)δ1.30-1.80(m,6H,H3”,H4”和H5”),3.25-3.45(m,2H,H2’),3.80,3.98,4.22和4.38(m,dd,J 14.0和4.4,dd,J 14.8和2.0和dd,J 14.0和9.4,2H,H6”),4.58和4.72(dd,J 3.2和2.8和dd,J 3.4和3.0,1H,H2”),5.30和5.39(dd,J 8.4和5.2和dd,J 9.2和4.0,1H,H1’),6.47和6.51(d,J 1.8和d,J 1.8,1H,H3),7.08和7.15(dd,J 8.2和4.8和dd,J 8.2和5.2,1H,H5),7.69(m,2H,Phth-β),7.86(m,2H,Phth-α),7.86(m,1H,H4),8.43和8.63(dd,J 4.8和1.6和dd,J5.0和1.7,1H,H7),10.8和12.5(br,1H,NH).
13C-NMR(CDCl3,50MHz)δ18.8和19.9(t,C3”*),25.0和25.2(t,C4”*),30.2和30.8(t,C5”*),41.6和42.8(t,C6”),61.8和63.6(t,C2’),69.3和71.7(d,C1’),95.5和97.9(d,C2”),100.3和100.8(d,C3),115.9(d,C5),120.6和120.7(s,C3a),123.2和123.4(d,Phth-β),128.7和128.8(d,C4),131.8和132.0(s,Phth-ipso),133.9和134.0(d,Phth-α),136.5和137.8(s,C2),143.1(d,C6),148.7和149.2(s,C7a),168.1(s,Phth-CO).
MS(EI)m/z 391(M+,1),307(M-THP,9),147(38),85(100).
对C22H21N3O4·1/2H2O的分析计算值:C(65.99),H(5.54),N(10.49);实测值:C(66.02),H(5.80),N(10.28)。
实施例5
4-甲氧基-2-[2-邻苯二甲酰亚氨基-1-(2,3,5,6-四氢吡喃-2-基)氧乙基]-7-氮杂吲哚
按照制备2-[2-邻苯二甲酰亚氨基-1-(2,3,5,6-四氢吡喃-2-基)氧乙基]-7-氮杂吲哚的相同方法,由8b(3.8g,11mmol)的CHCl3(350ml)溶液、6N HCl的苯(35ml)溶液和2,3-二氢吡喃(10ml,110mmol)得到4-甲氧基-2-[2-邻苯二甲酰亚氨基-1-(2,3,5,6-四氢吡喃-2-基)氧乙基]-7-氮杂吲哚(3.07g,65%)的非对映异构体混合物(1∶1)。
IR(薄膜法)ν1714(s,C=O),1392(m,C-O),1026(m,C-O).
1H-NMR(CDCl3,300MHz)δ1.30-1.80(m,6H,H3”,H4”和H5”),3.25-3.45(m,2H,H2’),3.99和4.01(s,3H,OMe),3.96,4.08,4.28和4.39(dd,J 13.5和3.9,dd,J 13.8和4.6,dd,J 13.8和8.5和dd,J 13.5和9.6,2H,H6”),4.57和4.73(brt,J 3.2和brt,J 3.4,1H,H2”),5.25和5.33(dd,J6.9和2.4和dd,J 9.9和4.1,1H,H1’),6.54和6.58(brs和brs,1H,H3),6.56和6.62(d,J 5.7和d,J 5.7,1H,H5),7.68(m,2H,Phth-β),7.83(m,2H,Phth-α),8.38和8.57(d,J 5.7和d,J 5.7,1H,H7),11.7(br,1H,NH).
13C-NMR(CDCl3,50MHz)δ18.9和19.5(t,C3”*),25.0和25.3(t,C4”*),30.2和30.7(t,C5”*),42.0和43.0(t,C6”),55.4(q,MeO),61.7和62.9(t,C2’),69.4和71.5(d,C1’),94.4和95.3(d,C2”),97.7(d,C5),97.8和100.1(d,C3),110.5(s,C3a),123.1和123.2(d,Phth-β),132.1和132.1(s,Phth-ipso),133.8和133.9(d,Phth-α),134.0和135.2(s,C2),144.9和145.1(d,C6),150.6和151.1(s,C7a*),159.8(s,C4*),168.1(s,Phth-CO).
MS(EI)m/z 422(M+1,2),421(M+,4),337(M-THP,15),177(100).
HRMS m/z对C23H23N3O5的计算值:421.1637;实测值:421.1625。
实施例6
4-氯-2-[2-邻苯二甲酰亚氨基-1-(2,3,5,6-四氢吡喃-2-基)氧乙基]-7-氮杂吲哚
按照制备2-[2-邻苯二甲酰亚氨基-1-(2,3,5,6-四氢吡喃-2-基)氧乙基]-7-氮杂吲哚的相同方法,由8c(1.3g,3.8mmol)的CHCl3(50ml)溶液,6N HCl的苯(5ml)溶液和2,3-二氢吡喃(1.7ml,19mmol)得到4-氯-2-[2-邻苯二甲酰亚氨基-1-(2,3,5,6-四氢吡喃-2-基)氧乙基]-7-氮杂吲哚(1.0g,63%)的非对映异构体混合物(1∶1)。
1H-NMR(CDCl3,200MHz)δ1.30-1.80(m,6H,H3”,H4”和H5”),3.25-3.45(m,2H,H2’),3.96-4.39(m,2H,H6”),4.58和4.72(m,1H,H2”),5.30和5.40(dd,J 8.4和4.4和dd,J 9.2和4.0,1H,H1’),6.58和6.63(d,J 2.2和d,J 2.2,1H,H3),7.13和7.20(d,J 5.2和d,J 5.2,1H,H5),7.70(m,2H,Phth-β),7.86(m,2H,Phth-α),8.35和8.55(d,J 5.2和d,J 5.2,1H,H7),11.7(br,1H,NH).
13C-NMR(CDCl3,50MHz)δ18.9和19.5(t,C3”*),25.0和25.3(t,C4”*),30.2和30.8(t,C5”*),41.8和42.9(t,C6”),61.9和63.5(t,C2’),69.2和71.6(d,C1’),95.5和96.4(d,C2”),98.8和100.7(d,C3),116.1(d,C5),116.3(s,C3a),123.1和123.3(d,Phth-β),131.7和132.0(s,Phth-ipso),133.8和133.9(d,Phth-α),134.0(s,C2),143.2和143.3(d,C6).
MS(EI)m/z 426(M+1,2),425(M+,5),341(M-THP,14),177(100).
实施例7
2-[2-氨基-1-(2,3,5,6-四氢吡喃-2-基)氧乙基]-7-氮杂吲哚(9a)
往2-[2-邻苯二甲酰亚氨基-1-(2,3,5,6-四氢吡喃-2-基)氧乙基]-7-氮杂吲哚(10.2g,26mmol)的EtOH(630ml)溶液中加入NH2NH2·H2O(1.53ml,31mmol)。将所得混合液回流3小时。蒸发溶剂,将剩余物溶解在CH2Cl2中,用饱和NaHCO3水溶液洗涤。水层用CH2Cl2萃取3次。将有机溶液一起蒸发得到9a(6.72g,100%)的非对映异构体混合物(1∶1),为淡橙色固体。
IR(薄膜法)ν3200(m,NH),1421(m,C-N),1022(m,C-O).
1H-NMR(CDCl3,200MHz)δ1.40-1.90(m,6H,H3”,H4”和H5”),3.20(m,2H,H2’),3.48和3.90(m,1H,H6”),4.60和4.85(brt,J 3.5和m,1H,H2”)4.85和4.97(m和brt,J 5.7,1H,H1’),6.32和6.43(s,1H,H3),7.03和7.07(dd,J 6.6和4.8和dd,J 6.6和5.0,1H,H5),7.85和7.90(dd,J 6.6和1.4,1H,H4),8.29和8.36(dd,J 4.8和1.4和dd,J 5.0和1.4,1H,H7),10.9和12.5(br,NH).
13C-NMR(CDCl3,75MHz)δ19.7和20.0(t,C3”*),25.1和25.3(t,C4”*),30.6和30.9(t,C5”*),45.3和47.3(t,C6”),62.9和63.5(t,C2’),73.4和75.5(d,C1’),96.1和97.3(d,C2”),99.8和99.9(d,C3),115.6(d,C5),120.7(s,C3a),128.4和128.5(d,C4),138.3和139.0(s,C2),142.2和142.3(d,C6),148.5和149.0(s,C7a).
MS(CI,CH4)m/z 263(M+1,15),262(M+,100).
HRMS m/z对C14H19N3O2.H的计算值:262.1555;实测值:262.1557。
实施例8
2-[2-氨基-1-(2,3,5,6-四氢吡喃-2-基)氧乙基]-4-甲氧基-7-氮杂吲哚(9b)
按照制备9a的相同方法,以4-甲氧基-2-[2-邻苯二甲酰亚氨基-1-(2,3,5,6-四氢吡喃-2-基)氧乙基]-7-氮杂吲哚(2.9g,10mmol)的EtOH(100ml)溶液和NH2NH2·H2O(420μl,15mmol)为原料,经过3小时的反应得到9b(1.9g,95%)的非对映异构体混合物(1∶1)。
IR(薄膜法)ν3150(m,NH),1590(m,C=C),1329(m,C-N),1114(m,C-O).
1H-NMR(CDCl3,200MHz)δ1.40-1.90(m,6H,H3”-H5”),3.19(m,2H,H2’),3.48和3.90(m,1H,H6”),3.99和4.00(s,3H,MeO),4.60和4.85(m,1H,H2”)4.85和4.97(m和brt,J 5.7,1H,H1’),6.42和6.51(s,1H,H3),6.51和6.55(d,J 5.4,1H,H5),8.23和8.30(d,J 5.4,1H,H7).
13C-NMR(CDCl3,75MHz)δ20.0(t,C3”*),25.2和25.4(t,C4”*),30.7和30.9(t,C5”*),45.4和47.4(t,C6”),55.4和55.5(q,MeO),62.9和63.4(t,C2’),73.5和75.5(d,C1’),94.7和96.1(d,C2”*),97.1和99.6(d,C3),97.6(d,C5),110.5(s,C3a),135.8和136.5(s,C2),144.3和144.4(d,C6),150.3和151.3(s,C4*),159.4和159.5(s,C7a*).
MS(CI,CH4)m/z 291(M+,2),262(M-CH2NH3,12),190(M-THPO,8),177(100).
实施例9
2-[2-氨基-1-(2,3,5,6-四氢吡喃-2-基)氧乙基]-4-氯-7-氮杂吲哚(9c)
按照制备9a的相同方法,以4-氯-2-[2-邻苯二甲酰亚氨基-1-(2,3,5,6-四氢吡喃-2-基)氧乙基]-7-氮杂吲哚(950mg,2.2mmol)的EtOH(50ml)溶液和NH2NH2·H2O(250μl,4.4mmol)为原料,经过3小时的反应得到9c(640mg,97%)的非对映异构体混合物(1∶1)。
1H-NMR(CDCl3,200MHz)δ1.40-1.90(m,6H,H3”-H5”),3.19(m,2H,H2’),3.50和3.95(m,1H,H6”),4.60和4.85(m,1H,H2”)4.85和4.97(brt,1H,H1’),6.46和6.54(s,1H,H3),7.07和7.10(d,J 5.4,1H,H5),8.19和8.25(d,J 5.4,1H,H7).
13C-NMR(CDCl3,75MHz)δ19.8和20.1(t,C3”*),25.1和25.3(t,C4”*),30.6和30.9(t,C5”*),45.2和47.1(t,C6”),63.1和63.7(t,C2’),73.1和75.1(d,C1’),96.0和96.4(d,C2”*),98.3和100.2(d,C3),115.8(d,C5),120.1和120.4(s,C4),135.6和135.8(s,C4),139.1和139.8(s,C3a),142.4和142.5(d,C6),148.9和149.5(s,C7a).
MS(CI,CH4)m/z 295(M+,2),266(M-CH2NH3,12),194(M-THPO,8),177(100).
实施例10
6,7,8,9-四氢-6-(2,3,5,6-四氢吡喃-2-基)氧基吡啶并[3′,2′:4,5]吡咯并[1,2-c]嘧啶-9-酮(10a)
将9a(7.4g,28mmol)和DIPEA(5ml,28mmol)的CH2Cl2(300ml)溶液缓慢加入到三光气(2.82g,10mmol)的CH2Cl2(740ml)溶液中,室温下将所得混合液搅拌30分钟。用饱和NH4Cl水溶液和水洗涤有机混合液。将有机溶液干燥并蒸发得到10a(6.06g,76%)的非对映异构体混合物(1∶1)。
IR(KBr)ν3252(m,NH),1716(s,C=O),1407(m,C-N),1302(m,C-O).
1H-NMR(CDCl3,200MHz)δ1.40-1.80(m,6H,H3’,H4’和H5’),3.45-4.00(m,2H,H7和H6’),3.90(m,2H,H7和H6’),4.71和4.94(m,1H,H2’),5.04和5.10(dd,J 3.2和3.0和t,J 4.4,1H,H6),6.56和6.59(s,1H,H5),6.79和7.00(br,1H,NH),7.20和7.22(dd,J 7.6和4.8和dd,J 8.0和4.8,1H,H3),7.89和7.93(dd,J 7.6和1.8和dd,J 8.0和1.8,1H,H4),8.54和8.57(dd,J 4.8和1.8和dd,J 4.8和1.4,1H,H2).
13C-NMR(CDCl3,50MHz)δ18.9和19.3(t,C4’),25.3和25.4(t,C5’),30.1和30.4(t,C3’),43.5和45.3(t,C6’),62.2和62.6(t,C7),63.2和64.3(d,C6),95.7和96.7(d,C2’),102.3和103.7(d,C5),118.6(d,C3),121.3和121.7(s,C4a),129.1和129.2(d,C4),133.5和136.1(s,C5a),145.2和145.6(d,C2),148.0和148.1(s,C10a),149.8和150.2(s,C9).
MS(CI,CH4)m/z 289(M+1,6),288(M+,25),204(M-THP,23),85(THP,100).
HRMS m/z对C15H17N3O3.H的计算值:288.1348;实测值:288.1352。
实施例11
6,7,8,9-四氢-6-(2,3,5,6-四氢吡喃-2-基)氧基-4-甲氧基-吡啶并[3′,2′:4,5]吡咯并[1,2-c]嘧啶-9-酮(10b)
按照制备10a的相同方法,以三光气(20mg,0.07mmol)的CH2Cl2(3ml)溶液、9b(58mg,0.20mmol)和DIPEA(34μl,0.20mmol)的CH2Cl2(3ml)溶液为原料,在室温下反应30分钟,粗混合物经快速柱层析纯化。用CH2Cl2/MeOH(98/2)洗脱得到10b(40g,63%)的非对映异构体混合物(1∶1)。
IR(KBr)ν3258(m,NH),1714(s,C=O),1566(m,C=N),1290(m,C-O).
1H-NMR(CDCl3,200MHz)δ1.40-1.80(m,6H,H3’,H4’和H5’),3.45和3.95(m,2H,H6’),3.65和3.75(m,2H,H7),4.00(s,3H,MeO),4.67和4.94(m,1H,H2’),4.99和5.07(m,1H,H6),6.20和6.30(br,1H,NH),6.65和6.66(s,1H,H5),6.69(d,J 5.9,1H,H3),8.44和8.46(d,J 5.9,1H,H2).
13C-NMR(CDCl3,50MHz)δ18.7和19.4(t,C4’),25.3和25.4(t,C5’),30.1和30.3(t,C3’),43.5和45.3(t,C6’),61.9和62.6(t,C7),62.9和63.9(d,C6),95.5和96.2(d,C2’),99.6,100.6和101.1(d,C3和C5),130.9(s,C5a),147.3和147.7(d,C2),149.9和150.3(s,C10a or C4),159.7(s,C9).
MS(EI)m/z 318(M+1,2),317(M+,28),233(22),217(66),216(65),177(100),85(THP,100).
HRMS m/z对C16H19N3O4的计算值:317.1376;实测值:317.1383。
实施例12
6,7,8,9-四氢-6-羟基吡啶并[3′,2′:4,5]吡咯并[1,2-c]嘧啶-9-酮
往10a(6g,21mmol)的二氯甲烷(400ml)溶液中加入4N的HCl水溶液(400ml)。在室温下搅拌45分钟后,两层分离。有机溶液用4N的HCl水溶液萃取。将水溶液过滤,蒸发得到6,7,8,9-四氢-6-羟基吡啶并[3′,2′:4,5]吡咯并[1,2-c]嘧啶-9-酮(5g,100%)的盐酸盐,为淡橙色固体。
6,7,8,9-四氢-6-羟基吡啶并[3′,2′:4,3]吡咯并[1,2-c]嘧啶-9-酮盐酸盐
IR(KBr)ν3500(s,OH),1721(s,C=O),1638(m,C=C),1503(m,C=N).
1H-NMR(CD3OD,300MHz)δ3.61(dd,J 13.0和5.0,1H,H7),3.77(dd,J 13.0和4.0,1H,H7),5.23(dd,J 5.0和4.0,1H,H6),7.05(s,1H,H5),7.86(dd,J 8.0和6.0,1H,H3),8.56(dd,J 6.0和1.2,1H,H2),8.86(dd,J 8.0和1.2,1H,H4).
13C-NMR(CD3OD,75MHz)δ45.8(t,C7),59.6(d,C6),101.9(d,C5),119.0(d,C3),127.1(s,C4a),124.3(d,C4),137.4(s,C5a),139.4(d,C2),141.9(s,C10a),149.0(s,C9).
MS(CI,NH3)m/z 205(M+1,3),204(M+,4),180(100),163(50),130(90).
HRMS m/z对C10H10N3O3的计算值:204.0773;实测值:204.0772。
连续用二氯甲烷萃取6,7,8,9-四氢-6-羟基吡啶并[3′,2′:4,5]吡咯并[1,2-c]嘧啶-9-酮盐酸盐的饱和Na2CO3水溶液得到共轭碱。
6,7,8,9-四氢-6-羟基吡啶并[3′,2′:4,5]吡咯并[1,2-c]嘧啶-9-酮
IR(KBr)ν3400(m,NH/OH),1707(s,C=O),1468(m,C-N),1408(m,C-N),1297(m,C-O).
1H-NMR(DMSO-d6,300MHz)δ3.27(m,1H,H7),3.42(m,1H,H7),4.90(dd,J 9.3和5.1,1H,H6),5.88(d,J 5.1,1H,OH),6.54(s,1H,H5),7.21(dd,J 7.4和4.2,1H,H3),7.88(br,1H,NH),7.99(brd,J 7.4,1H,H2),8.30(brd,J 4.2,1H,H4).
13C-NMR(DMSO-d6,75MHz)δ41.0(t,C7),55.7(d,C6),95.4(d,C5),113.8(d,C3),116.7(s,C4a),124.2(d,C4),135.4(s,C5a),139.3(d,C2),142.9(s,C10a*),143.8(s,C9*).
实施例13
6,7,8,9-四氢-6-羟基-4-甲氧基吡啶并[3′,2′:4,5]吡咯并[1,2-c]嘧啶-9-酮
往10b(25mg,0.08mmol)的二氯甲烷(5ml)溶液中加入4N的HCl水溶液(5ml),室温下将所得混合液搅拌45分钟。分离出有机溶液,用4N的HCl水溶液萃取。过滤水溶液并蒸发得到6,7,8,9-四氢-6-羟基-4-甲氧基吡啶并[3′,2′:4,5]吡咯并[1,2-c]嘧啶-9-酮(20mg,95%)的盐酸盐,为淡橙色固体。
IR(薄膜法)ν3244(m,NH),1718(s,C=O),1627(s,NCO),1505(m,C=N),1298(m,C-O).
1H-NMR(CD3OD,200MHz)δ3.56(dd,J 13.6和4.8,1H,H7),3.71(dd,J 13.6和3.6,1H,H7),4.29(s,3H,MeO),5.11(dd,J 4.8和3.6,1H,H6),6.91(s,1H,H5),7.40(d,J 6.9,1H,H3),8.42(d,J 6.9,1H,H2).
13C-NMR(CD3OD,75MHz)δ48.9(t,C7),60.5(q,Me),62.3(d,C6),101.7(d,C5),105.5(d,C3),117.4(s,C4a),137.0(s,C5a),140.6(d,C2),141.4(s,C10a),151.9(s,C9),169.0(s,C4).
MS(EI)m/z 234(M+1,12),233(M+,77),215(M-H2O,17),55(100).
HRMS m/z对C11H11N3O3的计算值:233.0800;实测值:233.0813。
实施例14
8,9-二氢吡啶并[3′,2′:4,5]吡咯并[1,2-c]嘧啶-9-酮(11a)
往经冷却(0℃)的6,7,8,9-四氢-6-羟基吡啶并[3′,2′:4,5]吡咯并[1,2-c]嘧啶-9-酮(1g,4.2mmol)和TEA(1.74ml,13mmol)的二氯甲烷(200ml)溶液中滴加MsCl(320μl,4.2mmol)。在相同温度下将所述反应混合液搅拌30分钟,将有机溶液用饱和NH4Cl水溶液和水洗涤。将有机溶液干燥并蒸发得到11a(730mg,95%)的白色固体,产物没有进一步纯化。
mp 265-266℃(MeOH).
IR(KBr)ν3424(m,NH),1721(s,C=O),1691(m,NCO),1633(m,C=C),1408(m,C=N),1380(m),1303(m).
1H-NMR(DMSO-d6,300MHz)δ6.50(d,J 7.4,1H,H6),6.60(s,1H,H5),6.97(dd,J 7.4和5.3,1H,H7),7.37(dd,J 8.0和4.7,1H,H3),8.08(dd,J 8.0和1.7,1H,H4),8.39(dd,J 4.7和1.7,1H,H2),10.81(brd,J 5.3,1H,NH).
13C-NMR(DMSO-d6,75MHz)δ94.9(d,C5),98.0(d,C6),119.8(d,C3),123.1(s,C4a),127.5(d,C4),128.0(d,C7),137.0(s,C5a),142.5(d,C2),145.6(s,C10a*),146.7(s,C9*).
MS(EI)m/z 186(M+1,18),185(M+,15),157(M-CO,10)
(CI,NH3)m/z 204(M+18,12),187(M+2,14),186(M+1,100),109(48).
HRMS m/z对C10H7N3O的计算值:185.0589,实测值:185.0593。
实施例15
8,9-二氢-4-甲氧基吡啶并[3′,2′:4,5]吡咯并[1,2-c]嘧啶-9-酮(11b)
按照制备11a的相同方法,以6,7,8,9-四氢-6-羟基-4-甲氧基吡啶并[3′,2′:4,5]吡咯并[1,2-c]嘧啶-9-酮(113mg,0.42mmol)、TEA(195μl,1.25mmol)和MsCl(32μl,0.42mmol)的二氯甲烷(20ml)溶液为原料,反应30分钟得到11b(74mg,85%)的白色固体,产物没有进一步纯化。
IR(KBr)ν3380(m,NH),1721(s,C=O),1693(m,NCO),1633(m,C=C),1500(m,C=N),1294(m,C-O).
1H-NMR(DMSO-d6,500MHz)δ3.98(s,3H,Me),6.44(d,J 7.5,1H,H6),6.54(s,1H,H5),6.89(dd,J 7.5和2.0,1H,H7),6.96(d,J 5.5,1H,H3),8.26(d,J 5.5,1H,H2).
13C-NMR(DMSO-d6,75MHz)δ55.5(q,Me),92.6(d,C5),98.9(d,C6),101.0(d,C3),124.7(d,C7),114.0(s,C4a),134.1(s,C5a),144.9(d,C2),146.5(s,C9),147.7(s,C10a),159.1(s,C4).
MS(EI)m/z 216(M+1,17),215(M+,100),214(M-1,11),200(M-Me,59),172(48).
HRMS m/z对C11H9N3O2的计算值:215.0694;实测值:215.0690。
实施例16
8,9-二氢-8-甲氧基甲基吡啶并[3′,2′:4,5]吡咯并[1,2-c]嘧啶-9-酮(12)
往经冷却(0℃)的11a(250mg,1.4mmol)的DMF(10ml)溶液中加入NaH(65mg,1.6mmol)。将所得混合液搅拌10分钟,滴加入MOMCl(103μl,1.4mmol)。在0℃下搅拌所述混合液1小时,用水(1ml)猝灭。蒸发出溶剂,将剩余物溶解在二氯甲烷中。有机溶液用Na2CO3水溶液洗涤,蒸发和经快速柱层析纯化。用CH2Cl2/MeOH(95/5)洗脱得到12(267mg,87%)的白色固体。
IR(薄膜法)ν1711(s,C=O),1642(m,NCO),1393(m,C-O),1175(m,C-O).
1H-NMR(CDCl3,300MHz)δ3.41(s,3H,Me),5.29(s,2H,CH2),6.37(d,J 7.5,1H,H6),6.41(s,1H,H5),7.81(d,J 7.5,1H,H7),7.26(dd,J 7.8和4.8,1H,H3),7.90(dd,J 7.8和1.2,1H,H4),8.52(dd,J 4.8和1.2,1H,H2).
13C-NMR(CDCl3,50MHz)δ56.9(q,Me),78.0(t,CH2),95.9(d,C5),99.6(d,C6),119.5(d,C3),123.3(s,C4a),127.9(d,C4),128.1(d,C7),135.1(s,C5a),143.7(d,C2),146.2(s,C9),147.1(s,C10a).
MS(EI)m/z 230(M+1,30),229(M+,100).
HRMS m/z对C12H11N3O2的计算值:229.0851;实测值:229.0850。
实施例17
8,9-二氢-8-(4-甲基苯基磺酰基)吡啶并[3′,2′:4,5]吡咯并[1,2-c]嘧啶-9-酮
往经冷却(0℃)的11a(500mg,2.7mmol)的DMF(30ml)溶液中加入NaH(130mg,3.2mmol)。将所述混合液搅拌10分钟,随后加入对甲苯磺酰氯(570mg,3.0mmol)的DMF(10ml)溶液。室温下将所述混合液搅拌1小时。用水(1ml)猝灭。蒸发出溶剂,将剩余物溶解在乙酸乙酯中。有机溶液用Na2CO3水溶液洗涤,蒸发并经快速柱层析纯化。用己烷/二氯甲烷(1/1)洗脱得到8,9-二氢-8-(4-甲基苯基磺酰基)吡啶并[3′,2′:4,5]-吡咯并[1,2-c]嘧啶-9-酮(360mg,40%)的白色固体。
IR(薄膜法)ν1737(s,C=O),1642(m,NCO),1393(s,SO2),1175(m).
1H-NMR(CDCl3,200MHz)δ2.42(s,3H,Me),6.45(d,J 8.0,1H,H6),6.48(s,1H,H5),7.27(dd,J 8.2和4.7,1H,H3),7.34(d,J 8.4,2H,H3’和H5’),7.65(d,J 8.0,1H,H7),7.90(dd,J 8.2和1.4,1H,H4),8.09(d,J 8.4,2H,H2’和H6’),8.53(dd,J 4.7和1.4,1H,H2).
13C-NMR(CDCl3,50MHz)δ21.8(q,Me),98.3(d,C5),100.2(d,C6),118.5(s,C4’),120.1(d,C3),123.6(s,C4a),123.7(d,C4),128.6.(d,C7),129.6(d,C3’和C5’),129.8(d,C2’和C6’),133.8(s,C5a),141.8(s,C1’),144.6(d,C2),146.0(s,C9).
MS(EI)m/z 340(M+1,7),339(M+,32),184(M-Ts,100).
HRMS m/z对C17H13N3O3S的计算值:339.0677;实测值:339.0682。
实施例18
8,9-二氢-5-溴-8-甲氧基甲基吡啶并[3′,2′:4,5]吡咯并[1,2-c]嘧啶-9-酮(13a)
将NBS(78mg,0.44mmol)分批加入到0℃下的12a(100mg,0.44mmol)的二氯甲烷(30ml)溶液中,将混合液搅拌10分钟。将所述混合液用二氯甲烷(50ml)稀释,并用饱和NaHCO3水溶液洗涤三次。将有机层干燥,蒸发得到13a(107mg,80%)的淡黄色固体,产物没有进一步纯化。
IR(薄膜法)ν1715(s,C=O),1640(m,C=C),1092(m,C-O).
1H-NMR(CDCl3,300MHz)δ3.39(s,3H,Me),5.29(s,2H,CH2),6.43(d,J 7.8,1H,H6),6.90(d,J 7.8,1H,H7),7.33(dd,J 8.1和4.8,1H,H3),7.86(dd,J 8.1和1.5,1H,H4),8.54(dd,J 4.8和1.5,1H,H2).
13C-NMR(CDCl3,50MHz)δ57.0(q,Me),77.1(s,C5),78.3(t,CH2),97.7(d,C6),120.1(d,C3),122.7(s,C4a),126.7(d,C4),129.2(d,C7),132.8(s,C5a),144.8(d,C2),146.2(s,C9),146.5(s,C10a).
MS(EI)m/z 310(81BrM+1,5),309(81BrM+,50),308(79BrM+1,5),307(79BrM+,50),45(MOM,100).
HRMS m/z对C12H10 79BrN3O2的计算值:306.9957;实测值:306.9956。
实施例19
8,9-二氢-5-碘-8-甲氧基甲基吡啶并[3′,2′:4,5]吡咯并[1,2-c]嘧啶-9-酮(13b)
往经冷却(0℃)的11a(100mg,0.44mmol)的DMF(4ml)溶液中同时加入碘(220mg,0.86mmol)和KOH(94mg,1.66mmol)。在0℃下搅拌混合液30分钟。用0.5%NH3和0.1%Na2S2O5的水溶液(25ml)猝灭所述混合液。将溶液用乙酸乙酯萃取,有机溶液用饱和NaHCO3水溶液洗涤两次。将有机层干燥,蒸发得到13b(96mg,62%)的淡黄色固体,产物没有进一步纯化。
IR(薄膜法)ν1714(s,C=O),1637(m,C=C),1089(m,C-O).
1H-NMR(CDCl3,300MHz)δ3.45(s,3H,Me),5.36(s,2H,CH2),6.46(d,J 8.1,1H,H6),6.99(d,J 8.1,1H,H7),7.40(dd,J 8.1和4.8,1H,H3),7.81(dd,J 8.1和1.8,1H,H4),8.59(dd,J 4.8和1.8,1H,H2).
13C-NMR(CDCl3,50MHz)δ54.5(s,C5),57.1(q,Me),78.3(t,CH2),99.6(d,C6),120.3(d,C3),125.5(s,C4a),128.6(d,C4),129.8(d,C7),136.5(s,C5a),144.9(d,C2),146.0(s,C9),146.3(s,C10a).
MS(CI,NH3)m/z 357(M+2,5),356(M+1,30),230(100).
HRMS m/z对C12H10IN3O2的计算值:354.9820;实测值:354.9823。
实施例20
2-甲硫基-4-三甲基甲锡烷基嘧啶(15)
将TBAF(5ml,1M的THF溶液)滴加到4-碘-2-甲亚磺酰基嘧啶(800mg,3.2mmol)、六甲基二锡(1ml,4.8mmol)、Pd(OAc)2(45mg,0.31mmol)和PPh3(90mg,0.62mmol)的THF(10ml)溶液中。室温下将所得混合液搅拌1.5小时。真空除去溶剂,剩余物经中性氧化铝柱层析纯化。用己烷/乙酸乙酯(99/1)洗脱得到15(585mg,65%)的无色油。
IR(薄膜法)ν1539(m,C=N),1402(m),1306(m),1196(m).
1H-NMR(CDCl3,300MHz)δ0.34(s,9H,3Me),2.54(s,3H,Me),7.08(d,J 4.5,1H,H5),8.26(d,J 4.5,1H,H6).
13C-NMR(CDCl3,75MHz)δ-9.5(q,Me),14.0(q,Me),124.5(d,C5),153.6(d,C6),171.4(s,C2).
MS(EI)m/z 291(120SnM+,50),276(120SnM-Me,100).
HRMS m/z对C8H14N2S120Sn的计算值:290.9977;实测值:290.9973。
实施例21
8,9-二氢-5-碘-8-(4-甲基苯基磺酰基)吡啶并[3′,2′:4,5]吡咯并[1,2-c]嘧啶-9-酮(17)
将NIS(133mg,0.59mmol)分批加入到经冷却(0℃)的11a(200mg,0.59mmol)的二氯甲烷(20ml)溶液中,在该温度下将所得混合液搅拌30分钟。将所述混合液用二氯甲烷(50ml)稀释,用水洗涤两次。干燥有机层,蒸发得到17(218mg,80%)的淡黄色固体,产物没有进一步纯化。
IR(薄膜法)ν1735(s,C=O),1636(s,NCO),1395(m,C-N),1368(s,SO2),1175(m),1075(m).
1H-NMR(CDCl3,300MHz)δ2.38(s,3H,Me),6.43(d,J 8.3,1H,H6),7.30(m,3H,H3,H3’和H5’),7.68(dd,J 8.1和1.5,1H,H4),7.72(d,J 8.3,1H,H7),8.04(d,J 8.4,2H,H2’和H6’),8.49(dd,J 4.8和1.5,1H,H2).
13C-NMR(CDCl3,75MHz)δ21.7(q,Me),56.9(s,C5),100.0(d,C6),120.7(d,C3),123.8(s,C4’),125.3(d,C4),125.7(s,C4a),129.0(d,C7),129.5(s,C1’),129.6(d,C3’和C5’),129.7(d,C2’和C6’),133.3(s,C5a),135.0(s,C10a),145.5(d,C2),146.1(s,C9).
MS(EI)m/z 466(M+1,8),465(M+,36),310(M-Ts,100).
HRMS m/z对C17H12IN3O3S的计算值:464.9646;实测值:464.9649。
实施例22
8,9-二氢-5-(2-甲硫基嘧啶-4-基)吡啶并[3′,2′:4,5]吡咯并[1,2-c]-嘧啶-9-酮(18)
将17(50mg,0.11mmol)、2-甲硫基-4-三甲基甲锡烷基嘧啶(93mg,0.32mmol)、Pd2(dba)3(11mg,0.011mmol)、PPh3(6mg,0.022mmol)、LiCl(14mg,0.32mmol)和CuI(4mg,0.011mmol)的二噁烷(2ml)溶液回流5小时。蒸发出溶剂,粗产物经快速柱层析纯化。用CH2Cl2/MeOH/NH3水溶液(4/4/2)洗脱得到18(4mg,12%)。
1H-NMR(DMSO-d6,300MHz)δ2.60(3H,s,MeS),7.20(1H,d,J 6.3,H7),7.42(1H,dd,J7.5和4.2,H3),7.45(1H,d,J 5.7,H5’),7.70(1H,d,J 6.3,H6),8.22(1H,d,J 4.2,H2),8.39(1H,d,J 5.7,H6′),8.73(1H,d,J 7.5,H4).
MS(APCI)AP-m/z 309(M+,20),308(M-1,90),307(M-2,100);AP+m/z 331(M+Na,10),310(M+1,20),309(M+,25).
实施例23
9-氨基吡啶并[3′,2′:4,5]吡咯并[1,2-c]嘧啶(19a)
由11a:
将TMSCl(400μl,2.70mmol)加入到11a(500mg,2.70mmol)在2,6-二甲基吡啶(40ml)和HMDSA(60ml)中的溶液中,将所得混合液回流15小时。在0℃下,加入TMSTf(100μl,0.27mmol),并将NH3通过所述混合液中鼓泡15分钟。将所述混合液密封在钢制反应釜中,在150℃下加热8小时(60psi)。蒸发出溶剂,粗产物经快速柱层析纯化。用CH2Cl2/MeOH(98/2)洗脱得到19a(150mg,30%)的淡黄色固体。
由23a:
往-78℃的23a(20mg,0.06mmol)的液氨(8ml)溶液中加入小部分的Na,直到溶液保持蓝色10分钟。分批加入NH4Cl,直到蓝色消失,室温下将反应液搅拌至溶剂蒸发。将固体剩余物溶解在水中,用CH2Cl2萃取。干燥有机溶液,蒸发得到19a(4mg,38%)的黄色固体。
mp 214-215℃(摄氏度),(CH2Cl2/己烷)
IR(KBr)ν3452(m,NH),3304(m,NH),1654(m,C=C),1618(m,C=C),1570(m,C=N),1403(m,C-N).
1H-NMR(DMSO-d6,300MHz)δ6.49(s,1H,H5),6.72(d,J 6.6,1H,H6),6.80(br,1H,NH),7.30(d,J 6.6,1H,H7),7.42(dd,J 7.8和4.6,1H,H3),8.14(dd,J 7.8和1.5,1H,H4),8.33(dd,J 4.6和1.5,1H,H2),8.60(br,1H,NH).
13C-NMR(DMSO-d6,75MHz)δ88.8(d,C5),101.0(d,C6),119.6(d,C3),122.9(s,C4a),127.5(d,C4),136.8(s,C5a),138.9(d,C2),139.4(d,C7),141.8(s,C10a),148.8(s,C9).
MS(EI)m/z 185(M+1,15),184(M+,100),183(M-1,7).
HRMS m/z对C10H8N4的计算值:184.0749;实测值:184.0747。
实施例24
9-氨基-4-甲氧基吡啶并[3′,2′:4,5]吡咯并[1,2-c]嘧啶(19b)
由11b:
按照制备19a的相同方法,以11b(60mg,0.27mmol)、HMDSA(30ml)、2,6-二甲基吡啶(15ml)和TMSCl(36μl,0.27mmol)为原料。反应时间为15小时。0℃下,加入TMSTf(12μl,0.06mmol),将NH3通过所述混合液中鼓泡15分钟。在150℃(60psi)下加热的钢制反应釜中的反应时间为8小时。蒸发出溶剂,粗产物经快速柱层析纯化。用CH2Cl2/MeOH(99/1)洗脱得到19b(13mg,22%),接着用CH2Cl2/MeOH(95/5)洗脱得到11b(9mg,15%)。
由23b:
0℃下,往23b(200mg,0.54mmol)的THF(25ml)溶液中滴加前面制备的Na/萘的THF(1.43ml,0.54mmol)绿色溶液。20分钟后,在室温下搅拌所得溶液,滴加入Na/萘的THF(7.2ml,2.70mmol)溶液,将所得混合液搅拌15分钟。除去溶剂,将粗产物溶解在AcOEt中。用4N HCl萃取有机溶液3次。水层一起用NaHCO3碱化,用CH2Cl2萃取。将有机溶液干燥并蒸发,剩余物经快速柱层析纯化。用CH2Cl2/MeOH(95/5)洗脱得到19b(27mg,23%)的淡黄色固体。
1H-NMR(CDCl3+CD3OD,300MHz)δ4.01(s,3H,Me),6.45(s,1H,H5),6.61(d,J 6.6,1H,H6),6.75(d,J 5.6,1H,H3),7.14(d,J 6.6,1H,H7),8.18(d,J 5.6,1H,H2).
13C-RMN(CDCl3+CD3OD,75MHz)δ55.6(q,Me),87.5(d,C5),100.6(d,C6),102.2(d,C3),114.3(s,C4a),134.4(s,C5a),136.0(d,C2),141.7(d,C7),142.8(s,C10a),149.0(s,C9),158.8(s,C4).
MS(EI)m/z 215(M+1,7),214(M+,36),213(M-1,6),199(M-Me,36),57(100).
(ES+)m/z 216(M+2,20),215(M+1,100).
实施例25
9-乙酰基氨基吡啶并[3′,2′:4,5]吡咯并[1,2-c]嘧啶
往19a(250mg,1.36mmol)的THF(20ml)溶液中加入乙酸酐(200μl,2.04mmol),室温下将混合液搅拌20小时。除去溶剂,接着溶解在CH2Cl2中。用饱和的NaHCO3水溶液洗涤溶液。将有机层干燥并蒸发。粗产物经快速柱层析纯化。用CH2Cl2/MeOH(99/1)洗脱得到9-乙酰基氨基吡啶并[3′,2′:4,5]吡咯并[1,2-c]嘧啶(225mg,75%)的嫩黄色固体。
mp 160-161℃,(CH2Cl2/己烷)。
IR(KBr)ν3150(m,NH),1708(s,C=O),1626(m,NCO),1574(m,C=N),1372(m,C-N),1269(m).
1H-NMR(CDCl3,300MHz)δ2.63(s,3H,Me),6.53(s,1H,H5),6.97(d,J 6.6,1H,H6),7.41(dd,J 8.0和4.8,1H,H3),7.51(d,J 6.6,1H,H7),8.10(dd,J 8.0和1.5,1H,H4),8.41(dd,J 4.8和1.5,1H,H2).
13C-NMR(CDCl3,75MHz)δ26.2(q,Me),90.5(d,C5),107.2(d,C6),119.6(d,C3),122.8(s,C4a),128.4(d,C4),136.0(s,C5a),136.5(d,C2),139.6(d,C7),141.3(s,C10a),142.3(s,C9),170.0(s,CO).
MS(EI)m/z 227(M+1,3),226(M+,18),184(M-Ac,100).
HRMS m/z对C12H10N4O的计算值:226.0855;实测值:226.0852。
对C12H10N4O的分析计算值:C(63.71),H(4.46),N(24.77);实测值:C(63.65),H(4.59),N(24.80)。
实施例26
9-乙酰基氨基-4-甲氧基吡啶并[3′,2′:4,5]吡咯并[1,2-c]嘧啶
往19b(16mg,0.08mmol)的THF(2ml)溶液中加入乙酸酐(10μl,0.05mmol),室温下将所述混合液搅拌20小时。除去溶剂,接着溶解在CH2Cl2中。用饱和的NaHCO3水溶液洗涤溶液。将有机层干燥并蒸发。得到9-乙酰基氨基-4-甲氧基吡啶并[3′,2′:4,5]吡咯并[1,2-c]嘧啶(17mg,95%),产物没有进一步纯化。
1H-NMR(CDCl3,300MHz)δ2.60(s,3H,Me),4.07(s,3H,Me),6.60(s,1H,H5),6.82(d,J 5.7,1H,H3),6.96(d,J 6.6,1H,H6),7.47(d,J 6.6,1H,H7),8.29(d,J 5.7,1H,H2).
13C-NMR(CDCl3,75MHz)δ26.3(q,Me),55.8(q,Mc),88.1(d,C5),100.7(d,C6),107.7(d,C3),114.1(s,C4a),134.5(s,C5a),135.9(d,C2),142.1(d,C7),159.5(s,C4),170.1(s,CO).
MS(ES+)m/z 258(M+2,30),257(M+1,100).
实施例27
9-乙酰基氨基-5-碘吡啶并[3′,2′:4,5]吡咯并[1,2-c]嘧啶(20a)
将NIS(100mg,0.44mmol)分批加入到经冷却(0℃)的9-乙酰基氨基吡啶并[3′,2′:4,5]吡咯并[1,2-c]嘧啶(100mg,0.44mmol)的二氯甲烷(20ml)溶液中。将所得混合液搅拌15分钟。将所述溶液用二氯甲烷(50ml)稀释,用水洗涤两次。干燥有机层,蒸发得到20a(142mg,93%)的嫩黄色固体。
mp 163-164℃(摄氏度),(CH2Cl2/己烷)。
IR(KBr)ν3050(m,NH),1694(m,C=O),1573(m,C=N),1372(m,C-N),1307(m).
1H-NMR(CDCl3,200MHz)δ2.63(s,3H,Me),6.94(d,J 6.4,1H,H6),7.47(dd,J 8.2和4.8,1H,H3),7.61(d,J 6.4,1H,H7),7.93(dd,J 8.2和1.6,1H,H4),8.40(dd,J 4.8和1.6,1H,H2).
13C-NMR(CDCl3,75MHz)δ26.4(q,Me),46.7(s,C5),107.0(d,C6),120.6(d,C3),125.2(s,C4a),128.8(d,C4),136.9(s,C5a),138.6(d,C7),140.9(d,C2),141.5(s,C10a),142.7(s,C9),170.2(s,CO).
MS(EI)m/z 353(M+1,3),352(M+,22),310(M-Ac,100).
HRMS m/z对C12H9IN4O的计算值:351.9821;实测值:351.9821。
对C12H9IN4O的分析计算值:C(40.93),H(2.58),N(15.91);实测值:C(40.91),H(2.64),N(15.79)。
实施例28
9-乙酰基氨基-5-碘-4-甲氧基吡啶并[3′,2′:4,5]吡咯并[1,2-c]嘧啶(20b)
将NIS(18mg,0.078mmol)分批加入到经冷却(0℃)的9-乙酰基氨基-4-甲氧基吡啶并[3′,2′:4,5]吡咯并[1,2-c]嘧啶(20mg,0.078mmol)的二氯甲烷(15ml)溶液中。将所得混合液搅拌15分钟。将所述溶液用二氯甲烷(50ml)稀释,用水洗涤两次。干燥有机层,蒸发得到20b(27mg,93%)。
1H-NMR(CDCl3,200MHz)δ2.61(s,3H,Me),4.08(s,3H,Me),6.84(d,J 5.7,1H,H3),6.97(d,J 6.6,1H,H6),7.57(d,J 6.6,1H,H7),8.31(d,J 5.7,1H,H2).
13C-NMR(CDCl3,75MHz)δ26.3(q,Me),55.8(q,Me),101.2(d,C6),107.7(d,C3),114.1(s,C4a),133.5(s,C5a),137.7(d,C2),142.5(d,C7),142.6(s,C10a),151.8(s,C9),170.4(s,C4),176.8(s,CO).
MS(ES+)m/z 384(M+2,15),383(M+1,100),192(M+22+,50),191(M+12+,22).
实施例29
9-氨基-5-(2-甲硫基嘧啶-4-基)吡啶并[3′,2′:4,5]吡咯并[1,2-c]嘧啶(21)和9-乙酰基氨基-5-(2-甲硫基嘧啶-4-基)吡啶并[3′,2′:4,5]-吡咯并[1,2-c]嘧啶
将20a(130mg,0.37mmol)、2-甲亚磺酰基-4-三甲基甲锡烷基嘧啶(93mg,1.10mmol)、Pd2(dba)3(76mg,0.07mmol)、PPh3(39mg,0.15mmol)、LiCl(47mg,1.10mmol)和CuI(14mg,0.07mmol)在二噁烷(10ml)中的溶液回流1.5小时。除去有机溶剂,将油状物溶解在CH2Cl2中。用4N HCl萃取有机溶液4次,用固体碳酸钠碱化水溶液。接着用CH2Cl2萃取水溶液。蒸发有机层,并经快速柱层析纯化。用二氯甲烷/MeOH(99/1)洗脱得到9-乙酰基氨基-5-(2-甲亚磺酰基嘧啶-4-基)吡啶并[3′,2′:4,5]吡咯并[1,2-c]嘧啶(21mg,16%)的黄色固体,接着用CH2Cl2/MeOH(95/5)洗脱得到21(30mg,26%)的黄色固体。
将9-乙酰基氨基-5-(2-甲亚磺酰基嘧啶-4-基)吡啶并[3′,2′:4,5]吡咯并[1,2-c]嘧啶(15mg,0.043mmol)在5N HCl/MeOH(5ml)中的溶液回流1小时。除去溶剂,将剩余物溶解在饱和Na2CO3水溶液中。用二氯甲烷萃取所述溶液。蒸发出有机溶剂,得到21(12mg,90%)。
重复反应,并在临快速柱层析纯化前用HCl/MeOH处理,得到唯一的产物21(45%)。
9-氨基-5-(2-甲亚磺酰基嘧啶-4-基)吡啶并[3′,2′:4,5]吡咯并[1,2-c]嘧啶(21)
mp 223-224℃(CH2Cl2/己烷)。
IR(KBr)ν3390(m,NH),1632(m,C=C),1557(m,C=N),1517(m),1464(m,C-N),1265(m).
1H-NMR(CDCl3,300MHz)δ2.68(s,3H,Me),7.33(d,J 5.4,1H,H5’),7.49(dd,J 8.4和4.8,1H,H3),7.58(d,J 6.6,1H,H7),7.68(d,J 6.6,1H,H6),8.40(dd,J 4.8和1.6,1H,H2),8.48(d,J 5.4,1H,H6’),8.73(dd,J 8.4和1.6,1H,H4).
13C-NMR(CDCl3,50MHz)δ14.4(q,MeS),100.3(s,C5),102.1(d,C6),112.6(d,C5’),120.7(d,C3),122.0(s,C4a),128.6(d,C4),138.7(s,C5a),140.3(d,C2),142.1(s,C10a),143.4(d,C7),149.8(s,C9),156.5(d,C6’),161.2(s,C4’),172.3(s,C2’).
MS(EI)m/z 309(M+1,7),308(M+,33).
HRMS m/z对C15H12N6S的计算值:308.0844;实测值:308.0839。
UV(MeOH)λ217(16,324),252(21,415),400(11,692)。
9-乙酰基氨基-5-(2-甲亚磺酰基嘧啶-4-基)吡啶并[3′,2′:4,5]吡咯并[1,2-c]嘧啶
mp 160-162℃(CH2Cl2/己烷)。
IR(KBr)ν1704(s,C=O),1620(m,C=C),1556(m),1536(m,C=N),1517(m),1502(m),1476(m,C-N),1265(m).
1H-NMR(CDCl3,200MHz)δ2.67(s,3H,Me),2.69(s,3H,MeS),7.36(d,J 5.2,1H,H5’),7.58(dd,J 8.2和4.8,1H,H3),7.83(d,J 6.6,1H,H7),7.93(d,J 6.6,1H,H6),8.52(dd,J4.8和1.4,1H,H2),8.54(d,J 5.2,1H,H6’),8.77(dd,J 8.2和1.4,1H,H4).
13C-NMR(CDCl3,75MHz)δ14.4(q,MeS),26.4(q,Me),104.5(s,C5),107.4(d,C6),113.0(d,C5’),121.1(d,C3),121.6(s,C4a),129.5(d,C4),137.7(s,C5a),141.0(d,C2),141.1(d,C7),142.5(s,C10a),143.3(s,C9),156.8(d,C6’),160.7(s,C4’),170.2(s,C2’),172.7(s,CO).
MS(EI)m/z 351(M+1,3),350(M+,33),308(M-Ac,100).
HRMS m/z对C17H14N6OS的计算值:350.0949;实测值:350.0940。
UV(MeOH)λ255(25,480),400(17,710)。
实施例30
9-氨基-5-(2-甲亚磺酰基嘧啶-4-基)吡啶并[3′,2′:4,5]吡咯并[1,2-c]嘧啶
往经冷却(0℃)的21(20mg,0.07mmol)的二氯甲烷(5ml)溶液中加入mCPBA(32mg,0.13mmol)。将所得混合液搅拌30分钟。加入饱和Na2S2O3水溶液(1ml),并用饱和Na2CO3水溶液碱化。分离出有机层,用二氯甲烷萃取水层。干燥有机溶液,一起蒸发得到9-氨基-5-(2-甲亚磺酰基嘧啶-4-基)吡啶并[3′,2′:4,5]吡咯并[1,2-c]嘧啶(20mg,90%)。
IR(薄膜法)ν3388(m,NH),1635(m,C=C),1569(m),1519(m,C=N),1467(m),1267(s,S=O).
1H-NMR(CDCl3,200MHz)δ3.03(s,3H,Me),7.53(dd,J 8.2和4.8,1H,H3),7.63-7.78(m,3H,H7,H6和H5’),8.42(dd,J 4.8和1.4,1H,H2),8.73(d,J 5.4,1H,H6’),8.84(dd,J 8.2和1.4,1H,H4).
13C-NMR(CDCl3,50MHz)δ40.3(q,Me),99.5(s,C5),102.2(d,C6),116.5(d,C5’),121.3(d,C3),121.9(s,C4a),129.1(d,C4),140.0(s,C5a),140.8(d,C2),143.8(s,C10a),144.6(d,C7),150.0(s,C9),157.3(d,C6’),162.6(s,C4’),163.5(s,C2’).
MS(EI)m/z 324(M+,47),261(M-SOMe,100);(ES+)m/z 326(M+2,20),325(M+1,100).
实施例31
9-氨基-5-(2-甲磺酰基嘧啶-4-基)吡啶并[3′,2′:4,5]吡咯并[1,2-c]嘧啶
方法A
室温下,往9-氨基-5-(2-甲亚磺酰基嘧啶-4-基)-吡啶并[3′,2′:4,5]-吡咯并[1,2-c]嘧啶(50mg,0.16mmol)的二氯甲烷(15ml)溶液中加入mCPBA(88mg,0.36mmol)。将所得混合液搅拌2小时。加入饱和Na2S2O3水溶液(1ml),并用饱和Na2CO3水溶液碱化。分离出有机层,用二氯甲烷萃取水层。将有机溶液一起蒸发得到9-氨基-5-(2-甲磺酰基嘧啶-4-基)吡啶并[3′,2′:4,5]-吡咯并[1,2-c]嘧啶(50mg,91%)的淡橙色固体。
方法B
往21(200mg,0.65mmol)的二氯甲烷(50ml)溶液中加入mCPBA(320mg,1.30mmol)。室温下将所得混合液搅拌2小时。加入饱和Na2S2O3水溶液(5ml),并用饱和Na2CO3水溶液碱化。分离出有机层,用二氯甲烷萃取水层。将有机溶液干燥并一起蒸发得到9-氨基-5-(2-甲磺酰基嘧啶-4-基)吡啶并[3′,2′:4,5]吡咯并[1,2-c]嘧啶(201mg,91%)。
mp 118-189℃(CH2Cl2/己烷)。
IR(薄膜法)ν3340(m,NH),1569(m,C=C),1517(m),1462(m),1262(s,SO2).
1H-NMR(CDCl3,300MHz)δ3.41(s,3H,Me),7.56(dd,J 8.1和4.8,1H,H3),7.69(d,J6.6,1H,H7),7.78(d,J 5.7,1H,H5’),7.80(d,J 6.6,1H,H6),8.44(dd,J 4.8和1.5,1H,H2),8.75(d,J 5.7,1H,H6’),8.84(dd,J 8.1和1.5,1H,H4).
13C-NMR(DMSO-d6,50MHz)δ39.8(q,Me),97.4(s,C5),101.0(d,C6),118.4(d,C5’),121.0(s,C4a),121.2(d,C3),128.6(d,C4),130.3(s,C5a),140.5(d,C2),143.3(s,C10a),146.8(d,C7),149.9(s,C9),157.5(d,C6’),161.5(s,C4’),165.2(s,C2’).
MS(CI,NH3)m/z 342(M+2,6),341(M+1,20),340(M+,100),309(M-O2,5),263(M-SO2Me,25).
HRMS m/z对C15H12N3O2S的计算值:340.0742;实测值:340.0740。
实施例32
9-氨基-5-(2-氨基嘧啶-4-基)吡啶并[3′,2′:4,5]吡咯并[1,2-c]-嘧啶(5a)
方法A
将在密封管中的9-氨基-5-(2-甲磺酰基嘧啶-4-基)吡啶并[3′,2′:4,5]吡咯并[1,2-c]嘧啶(25mg,0.04mmol)在二噁烷(3ml)和23%NH3水溶液(5ml)中的溶液在80℃下加热6小时。随后冷却混合液,并除去溶剂。将剩余物溶解在饱和Na2CO3水溶液中,用二氯甲烷萃取数次。干燥有机层并蒸发。粗产物经快速柱层析纯化。用CH2Cl2/MeOH(97/3)洗脱得到5a(18mg,90%)的黄色固体。
方法B
将20a(120mg,0.36mmol)、27(200mg,0.72mmol)、Pd2(dba)3(75mg,0.06mmol)、PPh3(35mg,0.14mmol)、LiCl(42mg,1.10mmol)和CuI(12mg,0.06mmol)在二噁烷(4ml)中的溶液回流1.5小时。除去有机溶剂,将油状物溶解在HCl/MeOH中并回流1小时。蒸发出溶剂,随后溶解在二氯甲烷中。用4N HCl萃取有机溶液4次,用固体Na2CO3碱化所述水溶液。用CH2Cl2萃取水溶液。蒸发出溶剂后,所得混合物经快速柱层析纯化。用CH2Cl2/MeOH(95/5)洗脱得到5a(50mg,54%)的黄色固体。
mp 160-162℃(CH2Cl2/己烷)。
IR(薄膜法)ν3332(m,NH),1632(m,C=C),1574(m,C=N),1454(m,C-N),1262(m).
1H-NMR(DMSO-d6,500MHz)δ6.63(br,2H,2’NH2),7.12(d,J 5.5,1H,H5’),7.64(d,J8.0和4.4,1H,H3),7.70(d,J 6.6,1H,H7),7.76(d,J 6.6,1H,H6),8.29(d,J 5.5,1H,H6’),8.51(dd,J 4.4和1.4,1H,H2),8.52(br,1H,9NH),8.99(dd,J 8.0和1.4,1H,H4),9.35(br,1H,9NH).
13C-NMR(DMSO-d6,50MHz)δ99.5(s,C5),101.7(d,C6),106.8(d,C5’),120.7(d,C3),121.6(s,C4a),129.1(d,C4),138.2(s,C5a),140.1(d,C2),142.8(s,C10a),143.8(d,C7),149.7(s,C9),158.0(d,C6’),161.4(s,C4’),163.5(s,C2’).
MS(ES+)m/z 279(M+2,20),278(M+1,100),277(M+,10).
HRMS m/z对C15H11N7的计算值:277.1075;实测值:277.1071。
UV(MeOH)225(36,010),250(34,126),350(20,942),400(26,481)。
实施例33
9-氨基-5-(2-氨基嘧啶-4-基)吡啶并[3′,2′:4,5]吡咯并[1,2-c]嘧啶(5b)
按照制备5a(方法B)的相同方法,以20b(26mg,0.07mmol)、27(32mg,0.11mmol)、Pd2(dba)3(15mg,0.014mmol)、PPh3(7mg,0.028mmol)、LiCl(9mg,0.21mmol)和CuI(3mg,0.014mmol)的二噁烷(3ml)溶液为原料,回流1小时,经快速柱层析纯化后得到5b(8mg,38%)。
1H-NMR(CDCl3,200MHz)δ4.03(s,3H,Me),6.90(d,J 5.4,1H,H3),7.08(d,J 5.4,1H,H6),7.50(d,J 5.4,1H,H7),7.54(d,J 5.0,1H,H5’),8.28(d,J 5.4,1H,H2),8.47(d,J 5.0,1H,H6’)
MS(ES+)m/z 308(M+1,100),307(M+,20).
实施例34
9-甲苯磺酰氨基-6-四氢吡喃氧基吡啶并[3′,2′:4,5]吡咯并[1,2-c]嘧啶(22a)
将9a(235mg,0.88mmol)和DIPEA(505μl,1.98mmol)的CH2Cl2(20ml)溶液缓慢加入到TsNCCl2(250mg,0.99mmol)的二氯甲烷(20ml)溶液中。将所得溶液搅拌30分钟,用水洗涤。干燥有机溶液,过滤并蒸发得到粗产物。所述粗产物经快速柱层析纯化。用DCM/MeOH(99/1)洗脱得到22a(235mg,60%)的淡橙色固体。
IR(薄膜法)ν3312(m,NH),1639(s,N=C),1472(m,C-N),1277(m,C-O).
1H-NMR(CDCl3,200MHz)δ1.40-1.80(m,6H,H3’,H4’和H5’),2.38(s,3H,Me),3.45-4.00(m,4H,H7和H6’),4.65和4.95(bt,1H,H2’),5.02和5.05(bt,1H,H6),6.59和6.61(s,1H,H5),7.20和7.21(dd,J 7.8和4.8,1H,H3),7.27(d,J 8.4,2H,Ts),7.84和7.85(dd,J 7.8和1.5,1H,H4),8.12(d,J 8.4,2H,Ts),8.40(br,1H,NH),8.50和8.52(dd,J 4.8和1.5,1H,H2).
13C-NMR(CDCl3,50MHz)δ18.7和19.1(t,C4’),21.5(q,Ts),25.2和25.3(t,C5’),29.9和30.3(t,C3’),43.1和44.7(t,C6’),62.2和62.5(t,C7),62.6和63.8(d,C6),95.7和96.8(d,C2’),104.1和105.5(d,C5),119.2(d,C3),121.5(s,C4a),126.4(d,Ts),129.2(d,Ts),129.2(d,C4),131.9和134.7(s,C5a),140.0和142.5(s,C10a),145.6和145.9(d,C2),148.1(s,C9).
MS(EI)m/z 440(M+,1),376(M-SO2,19).
实施例35
4-甲氧基-9-甲苯磺酰氨基-6-四氢吡喃氧基吡啶并[3′,2′:4,5]吡咯并[1,2-c]嘧啶(22b)
按照制备22a的方法,以9b(1g,3.44mmol)、DIPEA(1.9ml,7.58mmol)和TsNCCl2(952mg,3.78mmol)为原料得到化合物22b(1.05g,65%)的淡橙色固体。
1H-NMR(CDCl3,200MHz)δ1.40-1.80(m,6H,H3’,H4’和H5’),2.38(s,3H,Me),3.45-3.95(m,4H,H7和H6’),3.97(s,3H,Me),4.62和4.95(bt,1H,H2’),5.01(m,1H,H6),6.69(m,1H,H3和H5),7.26(d,J 8.4,2H,Ts),8.10(d,J 8.4,2H,Ts),8.41和8.42(d,J5.8,1H,H2).
13C-NMR(CDCl3,50MHz)δ18.5和19.3(t,C4’),21.6(q,Ts),25.2和25.3(t,C5’),30.0和30.2(t,C3’),43.3和44.9(t,C6’),55.6(q,Me),61.9和62.3(t,C7),62.7和63.5(d,C6),95.5和96.4(d,C2’),101.3(d,C3),101.5和102.9(d,C5),126.2(s,C4a),126.5(d,Ts),129.2(d,Ts),129.3和129.5(s,C5a),132.2(s,Ts),142.5(s,C10a),147.6和147.9(d,C2),159.7(s,C9).
实施例36
4-氯-9-甲苯磺酰氨基-6-四氢吡喃氧基吡啶并[3′,2′:4,5]吡咯并[1,2-c]嘧啶(22c)
按照制备22a的方法,以9c(175mg,0.59mmol)、DIPEA(225μl,1.30mmol)和TsNCCl2(164mg,0.65mmol)为原料得到化合物22c(165mg,60%)。
IR(薄膜法)ν3309(m,NH),1634(s,N=C),1471(m,C-N),1358(m,SO2),1280(m,C-O).
1H-NMR(CDCl3,200MHz)δ1.40-1.80(m,6H,H3’,H4’和H5’),2.38(s,3H,Me),3.45-3.95(m,4H,H7和H6’),4.65和4.95(m,1H,H2’),5.01(m,1H,H6),6.71和6.72(s,1H,H5),7.24和7.25(d,J 5.4,1H,H3),7.27(d,J 8.0,2H,Ts),8.10(d,J 8.4,2H,Ts),8.39和8.41(d,J 4.8,1H,H2)8.48(br,1H,NH).
13C-NMR(CDCl3,50MHz)δ18.8和19.1(t,C4’),21.6(q,Ts),25.2和25.3(t,C5’),30.0和30.3(t,C3’),43.1和44.7(t,C6’),62.5和62.6(t,C7),63.3和63.6(d,C6),95.8和96.8(d,C2’),101.9和103.2(d,C5),119.3(d,C3),127.3(s,C4a),126.4(d,Ts),129.2(d,Ts),132.2(s,Ts),135.5和136.4(s,C5a),139.8(s,Ts),142.6(s,C10a),145.8和146.2(d,C2),147.9和148.1(s,C9).
MS(EI)m/z 475(M+,1),410(M-SO2,20).
实施例37
9-甲苯磺酰氨基-6-羟基吡啶并[3′,2′:4,5]吡咯并[1,2-c]嘧啶
将22a(210mg,0.48mmol)溶解在二氯甲烷(25mL)中,加入4NHCl水溶液(25mL),将所得混合液剧烈搅拌20分钟。将所述溶液碱化并分离出有机溶液。水层用CH2Cl2萃取两次。将有机溶液一起蒸发,并经快速柱层析纯化。用CH2Cl2/MeOH(95/5)洗脱得到9-甲苯磺酰氨基-6-羟基吡啶并[3′,2′:4,5]吡咯并[1,2-c]嘧啶(160mg,95%)的浅黄色固体。
IR(薄膜法)ν3315(m,NH),1624(s,C=N),1473(s,N-C),1276(m,SO2),1135(m,C-O).
1H-NMR(CDCl3,300MHz)δ2.35(s,3H,Me),3.50(m,1H,H7),3.75(m,1H,H7),5.04(br,1H,H6),6.55(s,1H,H5),7.00(dd,J 7.8和4.8,1H,H3),7.24(d,J 8.4,2H,Ts),7.71(dd,J 7.8和1.5,1H,H4),8.03(d,J 8.4,2H,Ts),8.27(dd,J 4.8和1.5,1H,H2),8.42(br,1H,NH).
13C-NMR(CDCl3,75MHz)δ21.5(q,Me),45.8(t,C7),60.4(d,C6),101.7(d,C5),103.7(d,C3),119.1(d,C4),122.2(s,C4a),126.5(d,Ts),129.2(d,Ts),129.4(d,C4),136.7(s,C5a),139.6(s,Ts),142.7(s,C10a),144.9(d,C2),147.5(s,Ts),148.5(s,C9).
MS(EI)m/z 357(M+1,1),356(M+,3),338(M-H2O,2),292(M-SO2,18).
实施例38
4-甲氧基-9-甲苯磺酰氨基-6-羟基吡啶并[3′,2′:4,5]吡咯并[1,2-c]嘧啶
按照制备前述化合物的方法,由22b(1.05g,2.23mmol)得到化合物4-甲氧基-9-甲苯磺酰氨基-6-羟基吡啶并[3′,2′:4,5]吡咯并[1,2-c]嘧啶(819mg,95%)的淡黄色固体。
IR(薄膜法)ν3317(m,NH),1627(s,C=N),1475(s,N-C),1293(m,SO2),1139(m,C-O).
1H-NMR(CDCl3,200MHz)δ2.35(s,3H,Me),3.50(m,1H,H7),3.75(m,1H,H7),3.85(s,3H,Me),5.02(br,1H,H6),6.43(d,J 5.4,1H,H3),6.52(s,1H,H5),7.24(d,J 8.0,2H,Ts),8.04(d,J 8.0,2H,Ts),8.09(d,J 5.4,1H,H2),8.41(br,1H,NH).
13C-NMR(CDCl3,50MHz)δ21.5(q,Me),45.9(t,C7),55.6(q,Me),60.1(d,C6),100.8(d,C3),101.4(d,C5),112.1(s,C4a),126.5(d,Ts),129.3(d,Ts),134.3(s,C5a),139.6(s,Ts),142.7(s,C10a),146.7(d,C2),148.6(s,C9),159.6(s,C4).
实施例39
4-氯-9-甲苯磺酰氨基-6-羟基吡啶并[3′,2′:4,5]吡咯并[1,2-c]嘧啶
按照制备前述化合物的方法,由22c(50mg,0.11mmol)得到化合物4-氯-9-甲苯磺酰氨基-6-羟基吡啶并[3′,2′:4,5]吡咯并[1,2-c]嘧啶(37mg,90%)。
IR(薄膜法)ν3316(m,NH),1632(s,C=N),1472(s,N-C),1277(m,SO2),1082(m,C-O).
1H-NMR(CDCl3,200MHz)δ2.35(s,3H,Me),3.56(brd,J 13.6,1H,H7),3.80(dt,J 13.6和2.6,1H,H7),4.80(br,1H,OH),5.10(br,1H,H6),6.61(s,1H,H5),7.04(d,J 5.0,1H,H3),7.24(d,J 8.2,2H,Ts),8.03(d,J 8.2,2H,Ts),8.16(d,J 5.0,1H,H2),8.46(br,1H,NH).
13C-NMR(CDCl3,50MHz)δ21.6(q,Me),45.7(t,C7),60.4(d,C6),101.5(d,C5),119.2(d,C3),121.4(s,C4a),126.5(d,Ts),129.3(d,Ts),131.0(s,Ts),136.5(s,C5a),137.4(s,Ts),142.8(s,C10a),145.2(d,C2),148.3(s,C9).
MS(EI)m/z 392(37ClM+,2),390(35ClM+,6),328(37ClM-SO2,10),326(35ClM-SO2,31).
HRMS m/z对C17H15ClN4O5S的计算值:390.0553;实测值:390.0548。
实施例40
9-甲苯磺酰氨基吡啶并[3′,2′:4,5]吡咯并[1,2-c]嘧啶(23a)
将9-甲苯磺酰氨基-6-羟基吡啶并[3′,2′:4,5]吡咯并[1,2-c]嘧啶(170mg,0.48mmol)和TEA(133μl,0.96mmol)溶解在二氯甲烷(25ml)中,滴加入甲磺酰氯(37μl,0.48mmol),将所得混合液搅拌20分钟。用水洗涤所述溶液,将有机溶液干燥并蒸发。粗混合物经快速柱层析纯化。用CH2Cl2/MeOH(98/2)洗脱得到23a(124mg,78%)的黄色固体。
IR(KBr)ν3266(m,NH),1604(s,C=N),1399,1281(m,SO2).
1H-NMR(CD3OD,300MHz)δ2.35(s,3H,Me),6.69(s,1H,H5),6.73(d,J 7.8,1H,H6),7.04(d,J 7.8,1H,H7),7.31(d,J 8.4,2H,Ts),7.44(dd,J 8.1和5.1,1H,H3),7.99(d,J8.4,2H,Ts),8.15(d,J 8.1,1H,H4),8.46(d,J 5.1,1H,H2).
MS(EI)m/z 339(M+1,4),338(M+,19),273(M-SO2,100).
HRMS m/z对C17H14N4O2S的计算值:338.0837;实测值:338.0841。
实施例41
4-甲氧基-9-甲苯磺酰氨基吡啶并[3′,2′:4,5]吡咯并[1,2-c]嘧啶(23b)
按照制备23a的方法,由4-甲氧基-9-甲苯磺酰氨基-6-羟基吡啶并[3′,2′:4,5]吡咯并[1,2-c]嘧啶(170mg,0.48mmol)得到化合物23b(124mg,78%)的淡黄色固体。
IR(KBr)ν3266(m,NH),1604(s,C=N),1399,1293(m,SO2),1141.
1H-NMR(CD3OD,300MHz)δ2.35(s,3H,Me),4.01(s,3H,Me),6.57(s,1H,H5),6.64(d,J 7.5,1H,H6),6.93(d,J 6.0,1H,H3),6.98(d,J 7.5,1H,H7),7.30(d,J 8.1,2H,Ts),8.00(d,J 8.1,2H,Ts),8.30(d,J 6.0,1H,H2).
13C-NMR(DMSO-d6,50MHz)δ21.0(q,Me),55.9(q,Me),93.0(d,C5),101.5(d,C6),102.1(d,C3),113.9(s,C4a),125.9(d,Ts),129.3(d,C2和Ts),132.6(s,C5a),142.3(s,C10a),143.9(s,C9),145.1(d,C7),158.4(s,C4).
HRMS m/z对C18H16N4O5S的计算值:368.0943;实测值:368.0941。
实施例41
4-氯-9-甲苯磺酰氨基吡啶并[3′,2′:4,5]吡咯并[1,2-c]嘧啶(23c)
按照制备23a的方法,由4-氯-9-甲苯磺酰氨基-6羟基吡啶并[3′,2′:4,5]吡咯并[1,2-c]嘧啶(165mg,0.42mmol)得到化合物23c(104mg,66%)。
IR(KBr)ν3265(m,NH),1602(s,C=N),1396,1281(m,SO2),1141.
1H-NMR(DMSO-d6,300MHz)δ2.34(s,3H,Me),6.80(s,1H,H5),6.84(d,J 7.8,1H,H6),7.17(d,J 7.8,1H,H7),7.37(d,J 7.8,2H,Ts),7.58(d,J 4.8,1H,H3),8.03(d,J 7.8,2H,Ts),8.45(d,J 4.8,1H,H2),11.20(br,1H,NH).
13C-NMR(DMSO-d6,50MHz)δ21.0(q,Me),93.7(d,C5),101.3(d,C6),119.9(d,C3),122.5(s,C4a),125.9(d,Ts),126.3(d,C2),129.4(d,Ts),133.5(s,C5a),135.6(s,Ts),142.5(s,C10a),143.5(d,C7),143.8(s,C9),145.3(s,C4).
MS(EI)m/z 374(37ClM+,5),372(35ClM+,12),309(37ClM-SO2,24),307(35ClM-SO2,63).
HRMS m/z对C17H13ClN4O2S的计算值:372.0448;实测值:372.0444。
实施例43
5-碘-9-甲苯磺酰氨基吡啶并[3′,2′:4,5]吡咯并[1,2-c]嘧啶(24a)
向-30℃的23a(10mg,0.03mmol)的二氯甲烷(5ml)溶液中分批加入N-碘代琥珀酰亚胺(7mg mg,0.03mmol),将所述混合液搅拌15分钟。用水洗涤所述溶液,干燥有机溶液,过滤并蒸发得到24a(14mg,95%)的黄色固体。
IR(KBr)ν3264(m,NH),1599(m,N=C),1393(m,C-N),1140.
1H-NMR(CD3OD,300MHz)δ2.37(s,3H,Me),6.61(d,J 7.5,1H,H6),7.15(d,J 7.5,1H,H7),7.34(d,J 8.4,2H,Ts),7.46(dd,J 7.5和4.8,1H,H3),7.81(dd,J 7.5和1.5,1H,H4),8.07(d,J 8.4,2H,Ts),8.46(dd,J 4.8和1.5,1H,H2).
MS(EI)m/z 464(M+,13),399(M-SO2,32),338(M-I,1),309(M-Ts,5),273(M-SO2-I,33),182(M-Ts-I,58).
HRMS m/z对C17H13IN4O2S的计算值:463.9804;实测值:493.9799。
实施例44
5-碘-4-甲氧基-9-甲苯磺酰氨基吡啶并[3′,2′:4,5]吡咯并[1,2-c]嘧啶(24b)
按照制备24a的方法,由23b(250mg,0.68mmol)得到化合物24b(295mg,95%)的淡黄色固体。
IR(KBr)ν3259(m,NH),1592(s,C=N),1293(m,SO2),1142.
1H-NMR(DMSO-d6,200MHz)δ2.33(s,3H,Me),3.97(s,3H,Me),6.62(d,J 7.7,1H,H6),7.06(d,J 5.6,1H,H3),7.18(d,J 7.7,1H,H7),7.35(d,J 8.2,2H,Ts),8.02(d,J 8.2,2H,Ts),8.40(d,J 5.6,1H,H2),11.15(br,1H,NH).
13C-NMR(DMSO-d6,50MHz)δ21.0(q,Me),56.2(q,Me),102.1(d,C3),113.9(s,C4a),126.1(d,Ts),129.4(d,C2和Ts),133.9(s,C5a),142.6(s,C10a),143.4(s,C9),145.1(d,C7),159.0(s,C4).
MS(EI)m/z 494(M+,31),429(M-SO2,65),368(M-I,12),339(M-Ts,15),303(M-SO2-I,58),212(M-Ts-I,100).
HRMS m/z对C18H15IN4O5S的计算值:493.9909;实测值:493.9891。
实施例45
4-甲氧基-9-甲苯磺酰氨基-5-(2-乙酰基氨基嘧啶-4-基)吡啶并[3′,2′:4,5]吡咯并[1,2-c]嘧啶(25)
将24b(295mg,0.60mmol)、27(258mg,0.9mmol)、Pd2(dba)3.CHCl3(120mg,0.12mmol)、PPh3(60mg,0.24mmol)、LiCl(74mg,1.8mmol)和CuI(23mg,0.12mmol)的二噁烷(25mL)溶液回流1小时。蒸发出溶剂,将粗产物溶解在CH2Cl2中。用4N HCl溶液萃取有机溶液3次。将水溶液一起用固体碳酸氢钠碱化,并用二氯甲烷萃取。蒸发有机溶液,将混合物经快速柱层析纯化。用CH2Cl2/MeOH(99/1)洗脱得到23b(66mg,28%),用CH2Cl2/MeOH(9/1)洗脱得到25(209mg,71%)的黄色固体。
IR(KBr)ν3379(m,NH),1592(s,C=N),1474,1449,1422,1293(SO2),1143.
1H-NMR(CDCl3,200MHz)δ2.41(s,3H,Me),2.47(s,3H,Me),4.05(s,3H,Me),6.92(br,1H,H3),7.31(d,J 8.4,2H,Ts),7.46(brd,1H,H6),7.60(d,J 6.6,1H,H7),7.98(br,1H,H5’),8.12(d,J 8.4,2H,Ts),8.38(br,1H,H2),8.50(br,1H,H6’).
MS(ES+)m/z 505(M+2,30),504(M+1,100).
实施例46
4-羟基-9-甲苯磺酰氨基-5-(2-氨基嘧啶-4-基)吡啶并[3′,2′:4,5]吡咯并[1,2-c]嘧啶(26)
将25(10mg,0.02mmol)的HBr(48%)溶液回流10分钟。将所述溶液用NaHCO3碱化,用CH2Cl2萃取。干燥有机溶液,过滤并蒸发得到26(7mg,78%)的黄色固体。
1H-NMR(CDCl3+CD3OD,300MHz)δ2.34(s,3H,Me),6.76(d,J 6.6,1H,H6),7.03(d,J6.6,1H,H5’),7.06(d,J 6.6,1H,H5’),7.23(d,J 8.1,2H,Ts),7.65(d,J 6.6,1H,H7),8.00(d,J 8.1,2H,Ts),8.01(d,J 6.6,1H,H6’),8.23(brd,1H,H2).
MS(ES+)m/z 449(M+1,10),448(M+,100).
实施例47
4-氯-2-甲磺酰基嘧啶
参见Heterocycles,1977,8,299。室温下,往4-氯-2-甲亚磺酰基嘧啶(5g,31mmol)的二氯甲烷(100ml)溶液中加入mCPBA(16.12g,94mmol),并搅拌2小时。用饱和Na2S2O3水溶液洗涤所得混合液,并将水溶液用饱和Na2CO3水溶液碱化。分离出有机层,用CH2Cl2萃取水溶液。将有机层一起蒸发得到4-氯-2-甲磺酰基嘧啶(5.5g,92%)的白色固体。
1H-NMR(CDCl3,200MHz)δ3.39(s,3H,Me),7.61(d,J 5.4,1H,H5),8.83(d,J 5.4,1H,H6).
MS(CI,CH4)m/z 192(M35Cl+,1),157(M-Cl,1),97(100).
实施例48
2-氨基-4-氯代嘧啶
室温下,往4-氯-2-甲磺酰基嘧啶(5g,26mmol)的异丙醇(20ml)溶液中加入20%的NH3水溶液(20ml),并将所述混合液搅拌20分钟。用二氯甲烷萃取所述混合液4次,真空除去有机溶剂,得到2-氨基-4-氯代嘧啶(3.3g,100%)的白色固体。
1H-NMR(CDCl3,200MHz)δ5.26(br,2H,NH2),6.67(d,J 5.2,1H,H5),8.17(d,J 5.2,1H,H6).
MS(CI,CH4)m/z 132(37ClM+1,33),131(37ClM+,2),130(35ClM+1,87),129(35ClM,8),97(100),94(M-Cl,53).
实施例49
2-乙酰基氨基-4-氯代嘧啶和4-氯-2-二乙酰基氨基嘧啶
将2-氨基-4-氯代嘧啶(500mg,3.9mmol)的乙酸酐(20ml)溶液回流30分钟。真空除去溶剂,将剩余的油状物溶解在饱和Na2CO3水溶液中。用CH2Cl2萃取水溶液。干燥有机溶液,蒸发得到油状物,所述油状物经快速柱层析纯化。用CH2Cl2/己烷(2/1)洗脱得到4-氯-2-二乙酰基氨基嘧啶(122mg,16%)的白色固体,用二氯甲烷洗脱得到2-乙酰基氨基-4-氯代嘧啶(270mg,45%)的白色固体。
2-乙酰基氨基-4-氯代嘧啶
1H-NMR(CDCl3,200MHz)δ2.51(s,3H,Me),7.04(d,J 5.3,1H,H5),8.17(d,J 5.3,1H,H6).
13C-NMR(CDCl3,50MHz)δ25.3(q,Me),116.0(d,C5),157.5(s,C4),159.2(d,C6),161.8(s,C2).
MS(EI)m/z 173(37ClM,5),171(35ClM,16),131(37ClM-Ac,32),129(35ClM-Ac,100).
4-氯-2-二乙酰基氨基嘧啶
1H-NMR(CDCl3,200MHz)δ3.32(s,6H,2Me),7.45(d,J 5.2,1H,H5),8.76(d,J 5.2,1H,H6).
13C-NMR(CDCl3,50MHz)δ26.3(q,Me),121.2(d,C5),160.0(d,C6),163.1(s,C4),171.6(s,C2).
MS(CI,CH4)m/z 215(37ClM,1),214(37ClM-1,3),213(35ClM,1),212(35ClM-1,5),174(37ClM-Ac,32),172(35ClM-Ac,100).
实施例50
2-乙酰基氨基-4-三甲基甲锡烷基嘧啶(27)
将2-乙酰基氨基-4-氯代嘧啶(170mg,1.0mmol)、六甲基二锡(400μl,1.8mmol)和Pd(PPh3)4(40mg,0.03mmol)的二噁烷(6ml)溶液回流1小时。真空除去溶剂,剩余物经中性氧化铝柱层析纯化。用己烷/AcOEt(7/3)洗脱,得到27(240mg,80%)的白色固体。
1H-NMR(CDCl3,300MHz)δ0.36(s,9H,3Me),2.53(s,3H,Me),7.13(d,J 4.8,1H,H5),7.98(br,1H,NH),8.35(d,J 4.8,1H,H6).
13C-NMR(CDCl3,75MHz)δ-9.5(q,3Me),25.3(q,Me),124.4(d,C5),154.7(d,C6).
MS(EI)m/z 300(120SnM+,1),285(120SnM-Me,34)270(120SnM-2Me,1),255(120SnM-3Me,6),244(120SnM-3Me-Ac,30),136(M-SnMe3,100).