CN1294591A - 嘌呤衍生物的制备方法 - Google Patents
嘌呤衍生物的制备方法 Download PDFInfo
- Publication number
- CN1294591A CN1294591A CN99804402A CN99804402A CN1294591A CN 1294591 A CN1294591 A CN 1294591A CN 99804402 A CN99804402 A CN 99804402A CN 99804402 A CN99804402 A CN 99804402A CN 1294591 A CN1294591 A CN 1294591A
- Authority
- CN
- China
- Prior art keywords
- alkyl
- general formula
- compound
- aryl
- hydrogen
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Granted
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D473/00—Heterocyclic compounds containing purine ring systems
- C07D473/40—Heterocyclic compounds containing purine ring systems with halogen atoms or perhalogeno-alkyl radicals directly attached in position 2 or 6
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D473/00—Heterocyclic compounds containing purine ring systems
- C07D473/26—Heterocyclic compounds containing purine ring systems with an oxygen, sulphur, or nitrogen atom directly attached in position 2 or 6, but not in both
- C07D473/32—Nitrogen atom
Landscapes
- Organic Chemistry (AREA)
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Medicinal Chemistry (AREA)
- General Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Animal Behavior & Ethology (AREA)
- Pharmacology & Pharmacy (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Virology (AREA)
- Communicable Diseases (AREA)
- General Chemical & Material Sciences (AREA)
- Oncology (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Catalysts (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
- Saccharide Compounds (AREA)
- Transition And Organic Metals Composition Catalysts For Addition Polymerization (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
Abstract
Description


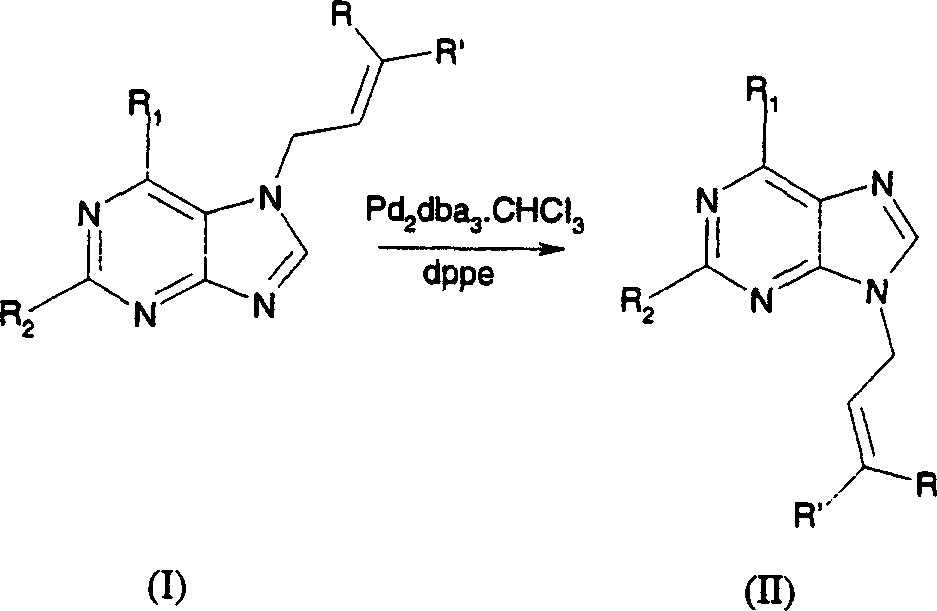



Claims (11)


Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GBGB9807116.0A GB9807116D0 (en) | 1998-04-02 | 1998-04-02 | Novel process |
| GB9807116.0 | 1998-04-02 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| CN1294591A true CN1294591A (zh) | 2001-05-09 |
| CN1125068C CN1125068C (zh) | 2003-10-22 |
Family
ID=10829765
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CN99804402A Expired - Fee Related CN1125068C (zh) | 1998-04-02 | 1999-03-30 | 嘌呤衍生物的制备方法 |
Country Status (16)
| Country | Link |
|---|---|
| US (1) | US6437125B1 (zh) |
| EP (1) | EP1068210B1 (zh) |
| JP (1) | JP4459439B2 (zh) |
| KR (1) | KR100613812B1 (zh) |
| CN (1) | CN1125068C (zh) |
| AT (1) | ATE268332T1 (zh) |
| AU (1) | AU3605199A (zh) |
| BR (1) | BR9908959A (zh) |
| CA (1) | CA2323965A1 (zh) |
| DE (1) | DE69917765T2 (zh) |
| DK (1) | DK1068210T3 (zh) |
| ES (1) | ES2222702T3 (zh) |
| GB (1) | GB9807116D0 (zh) |
| IL (1) | IL138289A0 (zh) |
| PT (1) | PT1068210E (zh) |
| WO (1) | WO1999051604A2 (zh) |
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN110386935A (zh) * | 2018-04-20 | 2019-10-29 | 重庆常捷医药有限公司 | 一种泛昔洛韦中间体的合成方法 |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE60202245T2 (de) | 2001-08-30 | 2005-09-01 | Ajinomoto Co., Inc. | Herstellungsmethode von Famciclovir und Herstellungs- bzw. Kristallisationsmethode eines entsprechenden Intermediates |
| AU2003268213A1 (en) * | 2002-08-26 | 2004-03-11 | Teva Pharmaceutical Industries Ltd. | Crystalline solid famciclovir forms i, ii, iii and preparation thereof |
| US7473780B2 (en) * | 2004-05-18 | 2009-01-06 | Teva Pharmeceutical Industries Ltd. | Drying process for preparing crystalline solid famciclovir |
| GB2426247A (en) * | 2005-05-20 | 2006-11-22 | Arrow Int Ltd | Methods of preparing purine derivatives such as famciclovir |
Family Cites Families (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| DE3485225D1 (de) * | 1983-08-18 | 1991-12-05 | Beecham Group Plc | Antivirale guanin-derivate. |
| US5684153A (en) * | 1984-08-16 | 1997-11-04 | Beecham Group Plc | Process for the preparation of purine derivatives |
| EP0182024B1 (en) * | 1984-09-20 | 1991-04-03 | Beecham Group Plc | Purine derivatives and their pharmaceutical use |
| GB8817607D0 (en) | 1988-07-23 | 1988-09-01 | Beecham Group Plc | Novel process |
| US5688948A (en) * | 1991-09-18 | 1997-11-18 | Ajinomoto Co., Inc. | Process for isomerizing acyclic nucleosides and process for separating purine nucleosides |
| GB9407698D0 (en) * | 1994-04-19 | 1994-06-15 | Smithkline Beecham Plc | Pharmaceuticals |
| EP0827960A1 (en) * | 1996-09-10 | 1998-03-11 | Ajinomoto Co., Inc. | Process for producing purine derivatives |
-
1998
- 1998-04-02 GB GBGB9807116.0A patent/GB9807116D0/en not_active Ceased
-
1999
- 1999-03-30 AT AT99917959T patent/ATE268332T1/de not_active IP Right Cessation
- 1999-03-30 JP JP2000542325A patent/JP4459439B2/ja not_active Expired - Fee Related
- 1999-03-30 AU AU36051/99A patent/AU3605199A/en not_active Abandoned
- 1999-03-30 CN CN99804402A patent/CN1125068C/zh not_active Expired - Fee Related
- 1999-03-30 US US09/623,700 patent/US6437125B1/en not_active Expired - Lifetime
- 1999-03-30 CA CA002323965A patent/CA2323965A1/en not_active Abandoned
- 1999-03-30 WO PCT/EP1999/002309 patent/WO1999051604A2/en not_active Ceased
- 1999-03-30 KR KR1020007010706A patent/KR100613812B1/ko not_active Expired - Fee Related
- 1999-03-30 IL IL13828999A patent/IL138289A0/xx not_active IP Right Cessation
- 1999-03-30 DK DK99917959T patent/DK1068210T3/da active
- 1999-03-30 ES ES99917959T patent/ES2222702T3/es not_active Expired - Lifetime
- 1999-03-30 EP EP19990917959 patent/EP1068210B1/en not_active Expired - Lifetime
- 1999-03-30 DE DE69917765T patent/DE69917765T2/de not_active Expired - Fee Related
- 1999-03-30 BR BR9908959-9A patent/BR9908959A/pt not_active Application Discontinuation
- 1999-03-30 PT PT99917959T patent/PT1068210E/pt unknown
Cited By (1)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN110386935A (zh) * | 2018-04-20 | 2019-10-29 | 重庆常捷医药有限公司 | 一种泛昔洛韦中间体的合成方法 |
Also Published As
| Publication number | Publication date |
|---|---|
| PT1068210E (pt) | 2004-10-29 |
| WO1999051604A3 (en) | 2000-02-17 |
| CN1125068C (zh) | 2003-10-22 |
| AU3605199A (en) | 1999-10-25 |
| JP2002510692A (ja) | 2002-04-09 |
| DE69917765D1 (de) | 2004-07-08 |
| HK1035896A1 (zh) | 2001-12-14 |
| ATE268332T1 (de) | 2004-06-15 |
| EP1068210A2 (en) | 2001-01-17 |
| DK1068210T3 (da) | 2004-09-20 |
| KR20010034701A (ko) | 2001-04-25 |
| KR100613812B1 (ko) | 2006-08-18 |
| ES2222702T3 (es) | 2005-02-01 |
| JP4459439B2 (ja) | 2010-04-28 |
| IL138289A0 (en) | 2001-10-31 |
| WO1999051604A2 (en) | 1999-10-14 |
| GB9807116D0 (en) | 1998-06-03 |
| CA2323965A1 (en) | 1999-10-14 |
| BR9908959A (pt) | 2000-12-05 |
| US6437125B1 (en) | 2002-08-20 |
| EP1068210B1 (en) | 2004-06-02 |
| DE69917765T2 (de) | 2005-07-14 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| KR0160144B1 (ko) | 뉴클레오시드의 부분 입체 이성체를 선택적으로 합성하는 방법 | |
| CN1290841C (zh) | 核苷类似物的非对映选择合成方法 | |
| JPH08504212A (ja) | 抗ウイルス特性を有する置換1,3‐オキサチオランを調製する方法 | |
| CN1153769C (zh) | 嘌呤衍生物及其中间体以及该衍生物的制备方法 | |
| CN1125068C (zh) | 嘌呤衍生物的制备方法 | |
| CN1120044A (zh) | N-9取代的鸟嘌呤化合物的制备 | |
| KR101157468B1 (ko) | 1,3-디옥솔란 뉴클레오시드 제조방법 | |
| JP2002510692A5 (zh) | ||
| KR100491941B1 (ko) | 푸린유도체의제조방법 | |
| CA2220995C (fr) | Nouveaux derives 7,12-dioxa-benzo¬a|anthraceniques substitues, leur procede de preparation et les compositions pharmaceutiques qui les contiennent | |
| CN1122805A (zh) | 9-(2-羟乙氧基甲基)鸟嘌呤的制备方法 | |
| HK1035896B (zh) | 制造嘌呤衍生物的方法 | |
| JP2005521660A (ja) | ジオキソランヌクレオシド類似体の製造方法 | |
| HK1034512B (zh) | 嘌呤衍生物的制造方法及其中间产物 | |
| JPH05271139A (ja) | 被保護ホルミルシクロペンタンジオール類、ヒドロキシメチルシクロペンタンジオール類、それらの製造方法及び中間体類 | |
| EP0882050B1 (en) | Process for preparing purine derivatives |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| BB1A | Publication of application | ||
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| ASS | Succession or assignment of patent right |
Owner name: BELLSOS INTELLECTUAL PROPERTY CO., LTD. Free format text: FORMER OWNER: SMITHKLINE BEECHAM PLC Effective date: 20010809 |
|
| C41 | Transfer of patent application or patent right or utility model | ||
| TA01 | Transfer of patent application right |
Effective date of registration: 20010809 Address after: Bermuda Hamilton Applicant after: Novartis Int Pharm Ltd. Address before: The British England Middlesex County Applicant before: Smithkline Beecham PLC |
|
| C14 | Grant of patent or utility model | ||
| GR01 | Patent grant | ||
| REG | Reference to a national code |
Ref country code: HK Ref legal event code: GR Ref document number: 1050419 Country of ref document: HK |
|
| C17 | Cessation of patent right | ||
| CF01 | Termination of patent right due to non-payment of annual fee |
Granted publication date: 20031022 Termination date: 20110330 |