DE60009030T2 - Katalytisches crack-verfahren mit mcm-68-katalysator - Google Patents
Katalytisches crack-verfahren mit mcm-68-katalysator Download PDFInfo
- Publication number
- DE60009030T2 DE60009030T2 DE60009030T DE60009030T DE60009030T2 DE 60009030 T2 DE60009030 T2 DE 60009030T2 DE 60009030 T DE60009030 T DE 60009030T DE 60009030 T DE60009030 T DE 60009030T DE 60009030 T2 DE60009030 T2 DE 60009030T2
- Authority
- DE
- Germany
- Prior art keywords
- catalyst
- mcm
- crystalline material
- membered rings
- atoms
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
- 239000003054 catalyst Substances 0.000 title claims description 91
- 238000000034 method Methods 0.000 title claims description 40
- 238000004523 catalytic cracking Methods 0.000 title claims description 13
- 239000010457 zeolite Substances 0.000 claims description 29
- 229910021536 Zeolite Inorganic materials 0.000 claims description 26
- HNPSIPDUKPIQMN-UHFFFAOYSA-N dioxosilane;oxo(oxoalumanyloxy)alumane Chemical compound O=[Si]=O.O=[Al]O[Al]=O HNPSIPDUKPIQMN-UHFFFAOYSA-N 0.000 claims description 26
- 239000000203 mixture Substances 0.000 claims description 25
- 150000002430 hydrocarbons Chemical class 0.000 claims description 22
- 230000008569 process Effects 0.000 claims description 20
- 229930195733 hydrocarbon Natural products 0.000 claims description 19
- 239000011148 porous material Substances 0.000 claims description 18
- 239000004215 Carbon black (E152) Substances 0.000 claims description 17
- 125000004429 atom Chemical group 0.000 claims description 17
- 239000002178 crystalline material Substances 0.000 claims description 13
- 239000002808 molecular sieve Substances 0.000 claims description 13
- URGAHOPLAPQHLN-UHFFFAOYSA-N sodium aluminosilicate Chemical compound [Na+].[Al+3].[O-][Si]([O-])=O.[O-][Si]([O-])=O URGAHOPLAPQHLN-UHFFFAOYSA-N 0.000 claims description 13
- 239000010703 silicon Substances 0.000 claims description 6
- 229910052710 silicon Inorganic materials 0.000 claims description 6
- XUIMIQQOPSSXEZ-UHFFFAOYSA-N Silicon Chemical compound [Si] XUIMIQQOPSSXEZ-UHFFFAOYSA-N 0.000 claims description 5
- 229910052782 aluminium Inorganic materials 0.000 claims description 5
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 claims description 5
- XEEYBQQBJWHFJM-UHFFFAOYSA-N Iron Chemical compound [Fe] XEEYBQQBJWHFJM-UHFFFAOYSA-N 0.000 claims description 4
- 238000002441 X-ray diffraction Methods 0.000 claims description 4
- ZOXJGFHDIHLPTG-UHFFFAOYSA-N Boron Chemical compound [B] ZOXJGFHDIHLPTG-UHFFFAOYSA-N 0.000 claims description 3
- 229910052796 boron Inorganic materials 0.000 claims description 3
- 229910052732 germanium Inorganic materials 0.000 claims description 3
- GNPVGFCGXDBREM-UHFFFAOYSA-N germanium atom Chemical compound [Ge] GNPVGFCGXDBREM-UHFFFAOYSA-N 0.000 claims description 3
- 125000004430 oxygen atom Chemical group O* 0.000 claims description 3
- GYHNNYVSQQEPJS-UHFFFAOYSA-N Gallium Chemical compound [Ga] GYHNNYVSQQEPJS-UHFFFAOYSA-N 0.000 claims description 2
- ATJFFYVFTNAWJD-UHFFFAOYSA-N Tin Chemical compound [Sn] ATJFFYVFTNAWJD-UHFFFAOYSA-N 0.000 claims description 2
- RTAQQCXQSZGOHL-UHFFFAOYSA-N Titanium Chemical compound [Ti] RTAQQCXQSZGOHL-UHFFFAOYSA-N 0.000 claims description 2
- 229910052733 gallium Inorganic materials 0.000 claims description 2
- 229910052738 indium Inorganic materials 0.000 claims description 2
- APFVFJFRJDLVQX-UHFFFAOYSA-N indium atom Chemical compound [In] APFVFJFRJDLVQX-UHFFFAOYSA-N 0.000 claims description 2
- 229910052742 iron Inorganic materials 0.000 claims description 2
- 229910052718 tin Inorganic materials 0.000 claims description 2
- 239000011135 tin Substances 0.000 claims description 2
- 239000010936 titanium Substances 0.000 claims description 2
- 229910052719 titanium Inorganic materials 0.000 claims description 2
- 229910052757 nitrogen Inorganic materials 0.000 description 41
- KWYUFKZDYYNOTN-UHFFFAOYSA-M Potassium hydroxide Chemical compound [OH-].[K+] KWYUFKZDYYNOTN-UHFFFAOYSA-M 0.000 description 30
- 238000005336 cracking Methods 0.000 description 27
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical group O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 24
- 239000007787 solid Substances 0.000 description 24
- -1 C 5 olefins Chemical class 0.000 description 23
- 239000000047 product Substances 0.000 description 22
- 238000006243 chemical reaction Methods 0.000 description 21
- 239000000463 material Substances 0.000 description 20
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 19
- 239000000654 additive Substances 0.000 description 17
- 239000003921 oil Substances 0.000 description 17
- 235000019198 oils Nutrition 0.000 description 17
- 230000000996 additive effect Effects 0.000 description 16
- 238000004231 fluid catalytic cracking Methods 0.000 description 14
- 239000003502 gasoline Substances 0.000 description 14
- 230000015572 biosynthetic process Effects 0.000 description 12
- 239000013078 crystal Substances 0.000 description 10
- 230000000694 effects Effects 0.000 description 9
- 239000007789 gas Substances 0.000 description 9
- 238000003786 synthesis reaction Methods 0.000 description 9
- 239000004927 clay Substances 0.000 description 8
- 230000009849 deactivation Effects 0.000 description 8
- 239000000377 silicon dioxide Substances 0.000 description 8
- 239000011159 matrix material Substances 0.000 description 7
- 238000003756 stirring Methods 0.000 description 7
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 6
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 6
- 238000002425 crystallisation Methods 0.000 description 6
- NNPPMTNAJDCUHE-UHFFFAOYSA-N isobutane Chemical compound CC(C)C NNPPMTNAJDCUHE-UHFFFAOYSA-N 0.000 description 6
- 238000004519 manufacturing process Methods 0.000 description 6
- YKTSYUJCYHOUJP-UHFFFAOYSA-N [O--].[Al+3].[Al+3].[O-][Si]([O-])([O-])[O-] Chemical compound [O--].[Al+3].[Al+3].[O-][Si]([O-])([O-])[O-] YKTSYUJCYHOUJP-UHFFFAOYSA-N 0.000 description 5
- WNROFYMDJYEPJX-UHFFFAOYSA-K aluminium hydroxide Chemical compound [OH-].[OH-].[OH-].[Al+3] WNROFYMDJYEPJX-UHFFFAOYSA-K 0.000 description 5
- 229940075614 colloidal silicon dioxide Drugs 0.000 description 5
- 230000008025 crystallization Effects 0.000 description 5
- RAABOESOVLLHRU-UHFFFAOYSA-N diazene Chemical compound N=N RAABOESOVLLHRU-UHFFFAOYSA-N 0.000 description 5
- 229910000071 diazene Inorganic materials 0.000 description 5
- 239000012153 distilled water Substances 0.000 description 5
- 239000002245 particle Substances 0.000 description 5
- 239000011541 reaction mixture Substances 0.000 description 5
- 238000010992 reflux Methods 0.000 description 5
- CSCPPACGZOOCGX-UHFFFAOYSA-N Acetone Chemical compound CC(C)=O CSCPPACGZOOCGX-UHFFFAOYSA-N 0.000 description 4
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 4
- QUSNBJAOOMFDIB-UHFFFAOYSA-N Ethylamine Chemical compound CCN QUSNBJAOOMFDIB-UHFFFAOYSA-N 0.000 description 4
- 238000005481 NMR spectroscopy Methods 0.000 description 4
- 229910052784 alkaline earth metal Inorganic materials 0.000 description 4
- 239000002585 base Substances 0.000 description 4
- 238000009835 boiling Methods 0.000 description 4
- 210000000078 claw Anatomy 0.000 description 4
- 150000004985 diamines Chemical class 0.000 description 4
- NLYAJNPCOHFWQQ-UHFFFAOYSA-N kaolin Chemical compound O.O.O=[Al]O[Si](=O)O[Si](=O)O[Al]=O NLYAJNPCOHFWQQ-UHFFFAOYSA-N 0.000 description 4
- 239000002904 solvent Substances 0.000 description 4
- 229910004298 SiO 2 Inorganic materials 0.000 description 3
- MCMNRKCIXSYSNV-UHFFFAOYSA-N ZrO2 Inorganic materials O=[Zr]=O MCMNRKCIXSYSNV-UHFFFAOYSA-N 0.000 description 3
- 238000005299 abrasion Methods 0.000 description 3
- GTDPSWPPOUPBNX-UHFFFAOYSA-N ac1mqpva Chemical compound CC12C(=O)OC(=O)C1(C)C1(C)C2(C)C(=O)OC1=O GTDPSWPPOUPBNX-UHFFFAOYSA-N 0.000 description 3
- 150000001336 alkenes Chemical class 0.000 description 3
- 125000000217 alkyl group Chemical group 0.000 description 3
- 235000012211 aluminium silicate Nutrition 0.000 description 3
- 125000004432 carbon atom Chemical group C* 0.000 description 3
- 230000003197 catalytic effect Effects 0.000 description 3
- 239000000571 coke Substances 0.000 description 3
- 238000009838 combustion analysis Methods 0.000 description 3
- 238000001816 cooling Methods 0.000 description 3
- 239000000499 gel Substances 0.000 description 3
- 125000004435 hydrogen atom Chemical class [H]* 0.000 description 3
- 239000001282 iso-butane Substances 0.000 description 3
- 238000002844 melting Methods 0.000 description 3
- 230000008018 melting Effects 0.000 description 3
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 3
- 239000000843 powder Substances 0.000 description 3
- 239000011734 sodium Substances 0.000 description 3
- 238000010025 steaming Methods 0.000 description 3
- 229910018072 Al 2 O 3 Inorganic materials 0.000 description 2
- 239000005995 Aluminium silicate Substances 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-N Ammonia Chemical compound N QGZKDVFQNNGYKY-UHFFFAOYSA-N 0.000 description 2
- RTZKZFJDLAIYFH-UHFFFAOYSA-N Diethyl ether Chemical compound CCOCC RTZKZFJDLAIYFH-UHFFFAOYSA-N 0.000 description 2
- OAICVXFJPJFONN-UHFFFAOYSA-N Phosphorus Chemical compound [P] OAICVXFJPJFONN-UHFFFAOYSA-N 0.000 description 2
- ATUOYWHBWRKTHZ-UHFFFAOYSA-N Propane Chemical compound CCC ATUOYWHBWRKTHZ-UHFFFAOYSA-N 0.000 description 2
- GWEVSGVZZGPLCZ-UHFFFAOYSA-N Titan oxide Chemical compound O=[Ti]=O GWEVSGVZZGPLCZ-UHFFFAOYSA-N 0.000 description 2
- 239000003513 alkali Substances 0.000 description 2
- PNEYBMLMFCGWSK-UHFFFAOYSA-N aluminium oxide Inorganic materials [O-2].[O-2].[O-2].[Al+3].[Al+3] PNEYBMLMFCGWSK-UHFFFAOYSA-N 0.000 description 2
- ILRRQNADMUWWFW-UHFFFAOYSA-K aluminium phosphate Chemical class O1[Al]2OP1(=O)O2 ILRRQNADMUWWFW-UHFFFAOYSA-K 0.000 description 2
- 150000001412 amines Chemical class 0.000 description 2
- QGZKDVFQNNGYKY-UHFFFAOYSA-O ammonium group Chemical group [NH4+] QGZKDVFQNNGYKY-UHFFFAOYSA-O 0.000 description 2
- 239000011324 bead Substances 0.000 description 2
- 238000001354 calcination Methods 0.000 description 2
- 238000006555 catalytic reaction Methods 0.000 description 2
- 230000008859 change Effects 0.000 description 2
- 239000007795 chemical reaction product Substances 0.000 description 2
- 239000003795 chemical substances by application Substances 0.000 description 2
- 239000010771 distillate fuel oil Substances 0.000 description 2
- 239000012467 final product Substances 0.000 description 2
- 229910052739 hydrogen Inorganic materials 0.000 description 2
- HVTICUPFWKNHNG-UHFFFAOYSA-N iodoethane Chemical compound CCI HVTICUPFWKNHNG-UHFFFAOYSA-N 0.000 description 2
- 239000000395 magnesium oxide Substances 0.000 description 2
- CPLXHLVBOLITMK-UHFFFAOYSA-N magnesium oxide Inorganic materials [Mg]=O CPLXHLVBOLITMK-UHFFFAOYSA-N 0.000 description 2
- 229910052751 metal Inorganic materials 0.000 description 2
- 239000002184 metal Substances 0.000 description 2
- 229910044991 metal oxide Inorganic materials 0.000 description 2
- 150000004706 metal oxides Chemical class 0.000 description 2
- TVMXDCGIABBOFY-UHFFFAOYSA-N octane Chemical compound CCCCCCCC TVMXDCGIABBOFY-UHFFFAOYSA-N 0.000 description 2
- 239000012071 phase Substances 0.000 description 2
- 229910052698 phosphorus Inorganic materials 0.000 description 2
- 239000011574 phosphorus Substances 0.000 description 2
- 238000000634 powder X-ray diffraction Methods 0.000 description 2
- 229910052761 rare earth metal Inorganic materials 0.000 description 2
- 230000008929 regeneration Effects 0.000 description 2
- 238000011069 regeneration method Methods 0.000 description 2
- 125000001424 substituent group Chemical group 0.000 description 2
- 238000012360 testing method Methods 0.000 description 2
- 238000007669 thermal treatment Methods 0.000 description 2
- 238000011282 treatment Methods 0.000 description 2
- 238000007740 vapor deposition Methods 0.000 description 2
- AMGNHZVUZWILSB-UHFFFAOYSA-N 1,2-bis(2-chloroethylsulfanyl)ethane Chemical compound ClCCSCCSCCCl AMGNHZVUZWILSB-UHFFFAOYSA-N 0.000 description 1
- PAWQVTBBRAZDMG-UHFFFAOYSA-N 2-(3-bromo-2-fluorophenyl)acetic acid Chemical compound OC(=O)CC1=CC=CC(Br)=C1F PAWQVTBBRAZDMG-UHFFFAOYSA-N 0.000 description 1
- RYGMFSIKBFXOCR-UHFFFAOYSA-N Copper Chemical compound [Cu] RYGMFSIKBFXOCR-UHFFFAOYSA-N 0.000 description 1
- 239000005630 Diquat Substances 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 229910010082 LiAlH Inorganic materials 0.000 description 1
- 235000019502 Orange oil Nutrition 0.000 description 1
- 239000004743 Polypropylene Substances 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- 239000004809 Teflon Substances 0.000 description 1
- 229920006362 Teflon® Polymers 0.000 description 1
- 239000002253 acid Substances 0.000 description 1
- 238000010306 acid treatment Methods 0.000 description 1
- 150000001342 alkaline earth metals Chemical class 0.000 description 1
- PHKCHPXVCZIZPB-UHFFFAOYSA-N aluminum dioxosilane oxygen(2-) zirconium(4+) Chemical compound [O-2].[O-2].[Zr+4].[Si](=O)=O.[O-2].[Al+3] PHKCHPXVCZIZPB-UHFFFAOYSA-N 0.000 description 1
- GHTGICGKYCGOSY-UHFFFAOYSA-K aluminum silicon(4+) phosphate Chemical compound [Al+3].P(=O)([O-])([O-])[O-].[Si+4] GHTGICGKYCGOSY-UHFFFAOYSA-K 0.000 description 1
- HPTYUNKZVDYXLP-UHFFFAOYSA-N aluminum;trihydroxy(trihydroxysilyloxy)silane;hydrate Chemical compound O.[Al].[Al].O[Si](O)(O)O[Si](O)(O)O HPTYUNKZVDYXLP-UHFFFAOYSA-N 0.000 description 1
- 229910021529 ammonia Inorganic materials 0.000 description 1
- 239000000538 analytical sample Substances 0.000 description 1
- 239000012223 aqueous fraction Substances 0.000 description 1
- 244000052616 bacterial pathogen Species 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- LTPBRCUWZOMYOC-UHFFFAOYSA-N beryllium oxide Inorganic materials O=[Be] LTPBRCUWZOMYOC-UHFFFAOYSA-N 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 235000012970 cakes Nutrition 0.000 description 1
- 238000004364 calculation method Methods 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 150000001768 cations Chemical class 0.000 description 1
- 238000007385 chemical modification Methods 0.000 description 1
- 239000003245 coal Substances 0.000 description 1
- 239000008119 colloidal silica Substances 0.000 description 1
- 239000002131 composite material Substances 0.000 description 1
- 239000000356 contaminant Substances 0.000 description 1
- 229910052802 copper Inorganic materials 0.000 description 1
- 239000010949 copper Substances 0.000 description 1
- 238000002447 crystallographic data Methods 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- NLUNLVTVUDIHFE-UHFFFAOYSA-N cyclooctylcyclooctane Chemical compound C1CCCCCCC1C1CCCCCCC1 NLUNLVTVUDIHFE-UHFFFAOYSA-N 0.000 description 1
- 238000007405 data analysis Methods 0.000 description 1
- 238000000354 decomposition reaction Methods 0.000 description 1
- 230000001066 destructive effect Effects 0.000 description 1
- 230000001627 detrimental effect Effects 0.000 description 1
- GUJOJGAPFQRJSV-UHFFFAOYSA-N dialuminum;dioxosilane;oxygen(2-);hydrate Chemical compound O.[O-2].[O-2].[O-2].[Al+3].[Al+3].O=[Si]=O.O=[Si]=O.O=[Si]=O.O=[Si]=O GUJOJGAPFQRJSV-UHFFFAOYSA-N 0.000 description 1
- 229910001649 dickite Inorganic materials 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- TVUBDAUPRIFHFN-UHFFFAOYSA-N dioxosilane;oxygen(2-);titanium(4+) Chemical compound [O-2].[O-2].[Ti+4].O=[Si]=O TVUBDAUPRIFHFN-UHFFFAOYSA-N 0.000 description 1
- SYJFEGQWDCRVNX-UHFFFAOYSA-N diquat Chemical compound C1=CC=[N+]2CC[N+]3=CC=CC=C3C2=C1 SYJFEGQWDCRVNX-UHFFFAOYSA-N 0.000 description 1
- 238000004821 distillation Methods 0.000 description 1
- 235000021463 dry cake Nutrition 0.000 description 1
- 150000002170 ethers Chemical class 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 238000001704 evaporation Methods 0.000 description 1
- 239000000284 extract Substances 0.000 description 1
- 238000000605 extraction Methods 0.000 description 1
- 239000012013 faujasite Substances 0.000 description 1
- 238000011049 filling Methods 0.000 description 1
- 239000000706 filtrate Substances 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 239000010419 fine particle Substances 0.000 description 1
- ZZUFCTLCJUWOSV-UHFFFAOYSA-N furosemide Chemical compound C1=C(Cl)C(S(=O)(=O)N)=CC(C(O)=O)=C1NCC1=CC=CO1 ZZUFCTLCJUWOSV-UHFFFAOYSA-N 0.000 description 1
- 239000011521 glass Substances 0.000 description 1
- 150000004820 halides Chemical class 0.000 description 1
- 229910052621 halloysite Inorganic materials 0.000 description 1
- 229910052736 halogen Inorganic materials 0.000 description 1
- 150000002367 halogens Chemical class 0.000 description 1
- 239000001257 hydrogen Substances 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- 238000005984 hydrogenation reaction Methods 0.000 description 1
- 150000004679 hydroxides Chemical class 0.000 description 1
- 238000005470 impregnation Methods 0.000 description 1
- 230000006872 improvement Effects 0.000 description 1
- 239000012535 impurity Substances 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 229910010272 inorganic material Inorganic materials 0.000 description 1
- 239000011147 inorganic material Substances 0.000 description 1
- 229910052809 inorganic oxide Inorganic materials 0.000 description 1
- 229910052622 kaolinite Inorganic materials 0.000 description 1
- 239000010410 layer Substances 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 239000012280 lithium aluminium hydride Substances 0.000 description 1
- AXZKOIWUVFPNLO-UHFFFAOYSA-N magnesium;oxygen(2-) Chemical compound [O-2].[Mg+2] AXZKOIWUVFPNLO-UHFFFAOYSA-N 0.000 description 1
- 230000014759 maintenance of location Effects 0.000 description 1
- 239000011738 major mineral Substances 0.000 description 1
- 235000011963 major mineral Nutrition 0.000 description 1
- 150000002739 metals Chemical class 0.000 description 1
- 229910052901 montmorillonite Inorganic materials 0.000 description 1
- 229910052680 mordenite Inorganic materials 0.000 description 1
- 239000004570 mortar (masonry) Substances 0.000 description 1
- VLKZOEOYAKHREP-UHFFFAOYSA-N n-Hexane Chemical compound CCCCCC VLKZOEOYAKHREP-UHFFFAOYSA-N 0.000 description 1
- JRZJOMJEPLMPRA-UHFFFAOYSA-N olefin Natural products CCCCCCCC=C JRZJOMJEPLMPRA-UHFFFAOYSA-N 0.000 description 1
- 239000010502 orange oil Substances 0.000 description 1
- 239000012044 organic layer Substances 0.000 description 1
- TWNQGVIAIRXVLR-UHFFFAOYSA-N oxo(oxoalumanyloxy)alumane Chemical compound O=[Al]O[Al]=O TWNQGVIAIRXVLR-UHFFFAOYSA-N 0.000 description 1
- RVTZCBVAJQQJTK-UHFFFAOYSA-N oxygen(2-);zirconium(4+) Chemical compound [O-2].[O-2].[Zr+4] RVTZCBVAJQQJTK-UHFFFAOYSA-N 0.000 description 1
- 239000008188 pellet Substances 0.000 description 1
- 239000003208 petroleum Substances 0.000 description 1
- 230000010287 polarization Effects 0.000 description 1
- 229920001155 polypropylene Polymers 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 238000001556 precipitation Methods 0.000 description 1
- 238000002360 preparation method Methods 0.000 description 1
- 238000004321 preservation Methods 0.000 description 1
- 238000011027 product recovery Methods 0.000 description 1
- QQONPFPTGQHPMA-UHFFFAOYSA-N propylene Natural products CC=C QQONPFPTGQHPMA-UHFFFAOYSA-N 0.000 description 1
- 125000004805 propylene group Chemical group [H]C([H])([H])C([H])([*:1])C([H])([H])[*:2] 0.000 description 1
- 238000000197 pyrolysis Methods 0.000 description 1
- 230000005855 radiation Effects 0.000 description 1
- 150000002910 rare earth metals Chemical group 0.000 description 1
- 230000000717 retained effect Effects 0.000 description 1
- 238000007363 ring formation reaction Methods 0.000 description 1
- 150000003839 salts Chemical class 0.000 description 1
- 239000000523 sample Substances 0.000 description 1
- 239000003079 shale oil Substances 0.000 description 1
- 150000004760 silicates Chemical class 0.000 description 1
- 235000012239 silicon dioxide Nutrition 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000007790 solid phase Substances 0.000 description 1
- 241000894007 species Species 0.000 description 1
- 229910001220 stainless steel Inorganic materials 0.000 description 1
- 239000010935 stainless steel Substances 0.000 description 1
- 230000003068 static effect Effects 0.000 description 1
- 239000000725 suspension Substances 0.000 description 1
- 239000011269 tar Substances 0.000 description 1
- 239000011275 tar sand Substances 0.000 description 1
- 125000000383 tetramethylene group Chemical group [H]C([H])([*:1])C([H])([H])C([H])([H])C([H])([H])[*:2] 0.000 description 1
- 238000004227 thermal cracking Methods 0.000 description 1
- 239000004408 titanium dioxide Substances 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 238000005292 vacuum distillation Methods 0.000 description 1
- 238000010792 warming Methods 0.000 description 1
- 238000005406 washing Methods 0.000 description 1
- 229910052727 yttrium Inorganic materials 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C30—CRYSTAL GROWTH
- C30B—SINGLE-CRYSTAL GROWTH; UNIDIRECTIONAL SOLIDIFICATION OF EUTECTIC MATERIAL OR UNIDIRECTIONAL DEMIXING OF EUTECTOID MATERIAL; REFINING BY ZONE-MELTING OF MATERIAL; PRODUCTION OF A HOMOGENEOUS POLYCRYSTALLINE MATERIAL WITH DEFINED STRUCTURE; SINGLE CRYSTALS OR HOMOGENEOUS POLYCRYSTALLINE MATERIAL WITH DEFINED STRUCTURE; AFTER-TREATMENT OF SINGLE CRYSTALS OR A HOMOGENEOUS POLYCRYSTALLINE MATERIAL WITH DEFINED STRUCTURE; APPARATUS THEREFOR
- C30B28/00—Production of homogeneous polycrystalline material with defined structure
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C6/00—Preparation of hydrocarbons from hydrocarbons containing a different number of carbon atoms by redistribution reactions
- C07C6/08—Preparation of hydrocarbons from hydrocarbons containing a different number of carbon atoms by redistribution reactions by conversion at a saturated carbon-to-carbon bond
- C07C6/12—Preparation of hydrocarbons from hydrocarbons containing a different number of carbon atoms by redistribution reactions by conversion at a saturated carbon-to-carbon bond of exclusively hydrocarbons containing a six-membered aromatic ring
- C07C6/126—Preparation of hydrocarbons from hydrocarbons containing a different number of carbon atoms by redistribution reactions by conversion at a saturated carbon-to-carbon bond of exclusively hydrocarbons containing a six-membered aromatic ring of more than one hydrocarbon
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J29/00—Catalysts comprising molecular sieves
- B01J29/04—Catalysts comprising molecular sieves having base-exchange properties, e.g. crystalline zeolites
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01J—CHEMICAL OR PHYSICAL PROCESSES, e.g. CATALYSIS OR COLLOID CHEMISTRY; THEIR RELEVANT APPARATUS
- B01J29/00—Catalysts comprising molecular sieves
- B01J29/04—Catalysts comprising molecular sieves having base-exchange properties, e.g. crystalline zeolites
- B01J29/06—Crystalline aluminosilicate zeolites; Isomorphous compounds thereof
- B01J29/70—Crystalline aluminosilicate zeolites; Isomorphous compounds thereof of types characterised by their specific structure not provided for in groups B01J29/08 - B01J29/65
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B39/00—Compounds having molecular sieve and base-exchange properties, e.g. crystalline zeolites; Their preparation; After-treatment, e.g. ion-exchange or dealumination
- C01B39/02—Crystalline aluminosilicate zeolites; Isomorphous compounds thereof; Direct preparation thereof; Preparation thereof starting from a reaction mixture containing a crystalline zeolite of another type, or from preformed reactants; After-treatment thereof
- C01B39/06—Preparation of isomorphous zeolites characterised by measures to replace the aluminium or silicon atoms in the lattice framework by atoms of other elements, i.e. by direct or secondary synthesis
- C01B39/08—Preparation of isomorphous zeolites characterised by measures to replace the aluminium or silicon atoms in the lattice framework by atoms of other elements, i.e. by direct or secondary synthesis the aluminium atoms being wholly replaced
- C01B39/085—Group IVB- metallosilicates
-
- C—CHEMISTRY; METALLURGY
- C01—INORGANIC CHEMISTRY
- C01B—NON-METALLIC ELEMENTS; COMPOUNDS THEREOF; METALLOIDS OR COMPOUNDS THEREOF NOT COVERED BY SUBCLASS C01C
- C01B39/00—Compounds having molecular sieve and base-exchange properties, e.g. crystalline zeolites; Their preparation; After-treatment, e.g. ion-exchange or dealumination
- C01B39/02—Crystalline aluminosilicate zeolites; Isomorphous compounds thereof; Direct preparation thereof; Preparation thereof starting from a reaction mixture containing a crystalline zeolite of another type, or from preformed reactants; After-treatment thereof
- C01B39/46—Other types characterised by their X-ray diffraction pattern and their defined composition
- C01B39/48—Other types characterised by their X-ray diffraction pattern and their defined composition using at least one organic template directing agent
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2/00—Preparation of hydrocarbons from hydrocarbons containing a smaller number of carbon atoms
- C07C2/54—Preparation of hydrocarbons from hydrocarbons containing a smaller number of carbon atoms by addition of unsaturated hydrocarbons to saturated hydrocarbons or to hydrocarbons containing a six-membered aromatic ring with no unsaturation outside the aromatic ring
- C07C2/64—Addition to a carbon atom of a six-membered aromatic ring
- C07C2/66—Catalytic processes
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C4/00—Preparation of hydrocarbons from hydrocarbons containing a larger number of carbon atoms
- C07C4/08—Preparation of hydrocarbons from hydrocarbons containing a larger number of carbon atoms by splitting-off an aliphatic or cycloaliphatic part from the molecule
- C07C4/12—Preparation of hydrocarbons from hydrocarbons containing a larger number of carbon atoms by splitting-off an aliphatic or cycloaliphatic part from the molecule from hydrocarbons containing a six-membered aromatic ring, e.g. propyltoluene to vinyltoluene
- C07C4/14—Preparation of hydrocarbons from hydrocarbons containing a larger number of carbon atoms by splitting-off an aliphatic or cycloaliphatic part from the molecule from hydrocarbons containing a six-membered aromatic ring, e.g. propyltoluene to vinyltoluene splitting taking place at an aromatic-aliphatic bond
- C07C4/18—Catalytic processes
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C5/00—Preparation of hydrocarbons from hydrocarbons containing the same number of carbon atoms
- C07C5/22—Preparation of hydrocarbons from hydrocarbons containing the same number of carbon atoms by isomerisation
- C07C5/27—Rearrangement of carbon atoms in the hydrocarbon skeleton
- C07C5/2702—Catalytic processes not covered by C07C5/2732 - C07C5/31; Catalytic processes covered by both C07C5/2732 and C07C5/277 simultaneously
- C07C5/2708—Catalytic processes not covered by C07C5/2732 - C07C5/31; Catalytic processes covered by both C07C5/2732 and C07C5/277 simultaneously with crystalline alumino-silicates, e.g. molecular sieves
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C6/00—Preparation of hydrocarbons from hydrocarbons containing a different number of carbon atoms by redistribution reactions
- C07C6/08—Preparation of hydrocarbons from hydrocarbons containing a different number of carbon atoms by redistribution reactions by conversion at a saturated carbon-to-carbon bond
- C07C6/12—Preparation of hydrocarbons from hydrocarbons containing a different number of carbon atoms by redistribution reactions by conversion at a saturated carbon-to-carbon bond of exclusively hydrocarbons containing a six-membered aromatic ring
- C07C6/123—Preparation of hydrocarbons from hydrocarbons containing a different number of carbon atoms by redistribution reactions by conversion at a saturated carbon-to-carbon bond of exclusively hydrocarbons containing a six-membered aromatic ring of only one hydrocarbon
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07C—ACYCLIC OR CARBOCYCLIC COMPOUNDS
- C07C2529/00—Catalysts comprising molecular sieves
- C07C2529/04—Catalysts comprising molecular sieves having base-exchange properties, e.g. crystalline zeolites, pillared clays
- C07C2529/06—Crystalline aluminosilicate zeolites; Isomorphous compounds thereof
- C07C2529/70—Crystalline aluminosilicate zeolites; Isomorphous compounds thereof of types characterised by their specific structure not provided for in groups C07C2529/08 - C07C2529/65
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Engineering & Computer Science (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Life Sciences & Earth Sciences (AREA)
- Geology (AREA)
- Materials Engineering (AREA)
- Crystallography & Structural Chemistry (AREA)
- Inorganic Chemistry (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- General Chemical & Material Sciences (AREA)
- Metallurgy (AREA)
- Organic Low-Molecular-Weight Compounds And Preparation Thereof (AREA)
- Catalysts (AREA)
- Silicates, Zeolites, And Molecular Sieves (AREA)
- Production Of Liquid Hydrocarbon Mixture For Refining Petroleum (AREA)
- Low-Molecular Organic Synthesis Reactions Using Catalysts (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Paper (AREA)
- Heterocyclic Carbon Compounds Containing A Hetero Ring Having Oxygen Or Sulfur (AREA)
Description
- Diese Erfindung betrifft ein Verfahren zum katalytischen Cracken von Kohlenwasserstoffeinsatzmaterialien und insbesondere ein Verfahren zum katalytischen Cracken von Kohlenwasserstoffeinsatzmaterialien, das einen Katalysator verwendet, der MCM-68 umfasst.
- Katalytisches Cracken und insbesondere fluidkatalytisches Cracken (FCC) wird routinemäßig verwendet, um schwere Kohlenwasserstoffeinsatzmaterialien in leichtere Produkte, wie Benzin und Destillatbereichsfraktionen, umzuwandeln. Es besteht jedoch ein steigender Bedarf, die Ausbeute an leichten Olefinen, insbesondere C3- bis C5-Olefinen, im Produktbereich von katalytischen Crackverfahren zu erhöhen. Zum Beispiel sind C3- bis C5-Olefine bei der Herstellung von Ethern und Alkylat brauchbar, an denen hoher Bedarf als octansteigernde Additive für Benzin besteht.
- Konventionelle Verfahren zum katalytischen Cracken von schweren Kohlenwasserstoffeinsatzmaterialien zu Benzin und Destillatfraktionen verwenden typischerweise ein großporiges Molekularsieb, wie Zeolith Y, als primäre Crackkomponente. Es ist außerdem wohlbekannt, einen Zeolithen mittlerer Porengröße, wie ZSM-5, zu der Crackkatalysatorzusammensetzung zuzugeben, um die Octanzahl der Benzinfraktion zu steigern (siehe z.B. US-Patent Nr. 4,828,679).
- US-Patent Nr. 5,472,594 offenbart, dass die Ausbeute an C4- und C5-Olefinen beim katalytischen Cracken gesteigert werden kann, indem ein Phosphor enthaltender Zeolith mittlerer Porengröße, wie ZSM-5, zu einem konventionellen Zeolith Y Crackkatalysator gegeben wird, so dass das Gewichtsverhältnis von Phosphor enthaltendem, Zeolithen mittlerer Porengröße zu Zeolith Y im Bereich 0,005 bis 0,010 liegt.
- Es ist außerdem, z.B. aus US-Patent Nr. 4,740,292, bekannt, dass Zeolith ß zu einem konventionellen Zeolith Y Crackkatalysator gegeben werden kann, um die Ausbeute an C4-Olefinen zu erhö hen. Allerdings ist die kommerzielle Verwendbarkeit dieses Verfahrens bis heute durch die hydrothermale Stabilität der existierenden Formen von Zeolith β beschränkt.
- Gemäß der vorliegenden Erfindung ist nun gefunden worden, dass der neue Zeolith, MCM-68, Aktivität für katalytisches Cracken sowohl als primärer Crackkatalysator als auch als additiver Katalysator in Zusammenhang mit einem konventionellen Crackkatalysator aufweist. MCM-68 zeigt, insbesondere wenn er als additiver Katalysator verwendet wird, verbesserte Selektivität hinsichtlich Butylenen und Isobutan, sowie eine Verbesserung der Octanzahl der Benzinfraktion. MCM-68 zeigt außerdem ausgezeichnete hydrothermale Stabilität.
- Daher betrifft die vorliegende Erfindung ein Verfahren zum katalytischen Cracken eines Kohlenwasserstoffeinsatzmaterials, bei dem das Einsatzmaterial mit einer Katalysatorzusammensetzung kontaktiert wird, die ein poröses kristallines Material, MCM-68, umfasst, das mindestens ein Kanalsystem, in dem jeder Kanal durch einen 12-gliedrigen Ring von vierflächig koordinierten Atomen definiert ist, und mindestens zwei weitere, unabhängige Kanalsysteme enthält, in denen jeweils jeder Kanal durch einen 10-gliedrigen Ring von vierflächig koordinierten Atomen definiert ist, wobei die Anzahl an einzelnen Kanälen mit 10-gliedrigen Ringen doppelt so groß wie die Zahl der Kanäle mit 12-gliedrigen Ringen ist.
- Bevorzugt umfasst das poröse kristalline Material ein Gerüst aus vierflächigen Atomen, die durch Sauerstoffatome verbrückt sind, wobei das Gerüst der vierflächigen Atome durch eine Einheitszelle mit Atomkoordinaten in Nanometern definiert ist, die in Tabelle 1 gezeigt sind, und wobei jede Koordinatenposition innerhalb von ±0,05 nm variieren kann.
- Bevorzugt umfasst die Katalysatorzusammensetzung außerdem ein großporiges Molekularsieb mit einer Porengröße von mehr als etwa 7 x 10–10 m (Å).
-
1 ist eine schematische dreidimensionale Darstellung einer Einheitszelle von MCM-68, die nur die vierflächigen Atome und die Verbindung zwischen den vierflächigen Atomen zeigt, und -
2 ist eine schematische dreidimensionale Darstellung ähnlich1 , aber von einer Mehrzahl von Einheitszellen. - Die vorliegende Erfindung liefert ein Verfahren zur Umwandlung von Einsatzmaterialkohlenwasserstoffverbindungen zu Produktkohlenwasserstoffverbindungen mit niedrigerem Molekulargewicht als dem der Einsatzmaterialkohlenwasserstoffverbindungen. Insbesondere liefert die vorliegende Erfindung ein Verfahren zum katalytischen Cracken eines Kohlenwasserstoffeinsatzmaterials in Gegenwart eines Crackkatalysators unter katalytischen Crackbedingungen, zu einer Mischung von Produkten, die Benzin, Alkylat und C3- bis C5-Olefine und -Paraffine umfassen. Katalytische Crackeinheiten, die für das erfindungsgemäße Verfahren zugänglich sind, arbeiten bei Temperaturen von 200°C bis 870°C und unter verringertem, atmosphärischem oder Über-Druck. Das katalytische Verfahren kann entweder ein Festbett-, Bewegtbett- oder Wirbelbettverfahren sein und der Kohlenwasserstofffluss kann entweder im Gleichstrom oder im Gegenstrom zum Katalysatorfluss erfolgen. Das erfindungsgemäße Verfahren ist insbesondere auf die fluidkatalytischen Crack- (FCC) oder thermophoren katalytischen Crack- (TCC) Verfahren anwendbar.
- Das TCC-Verfahren ist ein Bewegtbettverfahren und der Katalysator liegt in Form von Pellets oder Kügelchen mit einer durchschnittlichen Teilchengröße von etwa 1/64stel bis 1/4 Zoll vor. Aktive, heiße Katalysatorkügelchen bewegen sich im Gleichstrom mit einem Kohlenwasserstoff-Einsatzprodukt abwärts durch eine Crackreaktionszone. Die Kohlenwasserstoffprodukte werden von dem verkokten Katalysator getrennt und gewonnen, und der Katalysator wird an dem unteren Ende der Zone wiedergewonnen und regeneriert.
- Typischerweise schließen TCC-Umwandlungsbedingungen eine durchschnittliche Reaktortemperatur von 450°C bis 510°C, ein Katalysator/Öl-Volumenverhältnis von 2 bis 7, eine Reaktorraumgeschwindigkeit von 1 bis 2,5 Volumen/Stunde/Volumen und ein Verhältnis von Rückführungs- zu frischem Einsatzmaterial von 0 bis 0,5 (Volumen) ein.
- Das erfindungsgemäße Verfahren ist insbesondere auf fluidkatalytisches Cracken (FCC) anwendbar, bei dem der Crackkatalysator typischerweise ein feines Pulver mit einer Teilchengröße von 10 bis 200 μm ist. Dieses Pulver wird im Allgemeinen in dem Einsatzmaterial suspendiert und in einer Reaktionszone aufwärts getrieben. Ein relativ schweres Kohlenwasserstoffeinsatzmaterial, z.B. ein Gasöl, wird mit dem Crackkatalysator gemischt, um eine fluidisierte Suspension zu liefern, und in einem verlängerten Reaktor, oder Steigrohr, bei erhöhten Temperaturen gecrackt, um eine Mischung von leichteren Kohlenwasserstoffprodukten zu liefern. Die gasförmigen Reaktionsprodukte und der erschöpfte Katalysator werden aus dem Steigrohr in einen Separator, z.B. eine Zykloneinheit, entfernt, die sich in dem oberen Abschnitt eines eingeschlossenen Strippbehälters, oder Strippers, befindet, wobei die Reaktionsprodukte in eine Produktgewinnungszone überführt werden und der erschöpfte Katalysator in ein dichtes Katalysatorbett innerhalb des unteren Abschnitts des Strippers eingeführt wird. Um mitgeführte Kohlenwasserstoffe aus dem erschöpften Katalysator zu entfernen, bevor letzterer zu einer Katalysatorregenerierungseinheit befördert wird, wird ein inertes Strippgas, z.B. Dampf, durch das Katalysatorbett geleitet, wo es solche Kohlenwasserstoffe desorbiert und sie in die Produktgewinnungszone befördert. Der fluidisierbare Katalysator wird kontinuierlich zwischen dem Steigrohr und dem Regenerator im Kreis geführt und dient dazu, Hitze von Letztgenanntem zu Zuvorgenanntem zu bringen und dadurch die thermischen Bedürfnisse der Crackreaktion, die endotherm ist, zu befriedigen.
- Typischerweise schließen FCC-Umwandlungsbedingungen eine Steigrohr-Toptemperatur von 500°C bis 595°C, bevorzugt von 520°C bis 565°C und am bevorzugtesten von 530°C bis 550°C, ein Katalysator/Öl-Gewichtsverhältnis von 3 bis 12, bevorzugt 4 bis 11 und am bevorzugtesten von 5 bis 10 und eine Katalysatorverweilzeit von 0,5 bis 15 Sekunden, bevorzugt von 1 bis 10 Sekunden ein.
- Das zu spaltende Kohlenwasserstoffeinsatzmaterial kann insgesamt oder zum Teil ein Gasöl (z.B. leichtes, mittleres oder schweres Gasöl) mit einem anfänglichen Siedepunkt oberhalb 204°C, einem 50%-Punkt von mindestens 260°C und einem Endpunkt von mindestens 315°C einschließen. Das Einsatzmaterial kann außerdem Vakuumgasöle, thermische Öle, Rückstandsöle, Cyclierungsprodukte, Gesamttoprohöle, Teersandöle, Schieferöle, synthetische Öle, schwere Kohlenwasserstofffraktionen aus der zerstörenden Hydrierung von Kohle, Teer, Tonen, Asphalten, Hydro-behandelten Einsatzmaterialien, die aus irgendeinem der vorherigen erhalten wurden, und dergleichen einschließen. Es sei darauf hingewiesen, dass die Destillation von höhersiedenden Erdölfraktionen oberhalb 400°C unter Vakuum ausgeführt werden muss, um thermisches Cracken zu vermeiden. Die hierin verwendeten Siedetemperaturen sind zur Vereinfachung als auf atmosphärischen Druck korrigierter Siedepunkt ausgedrückt. Rückstandsoder tiefergeschnittene Gasöle mit hohen Gehalten an Metallen können ebenfalls unter Verwendung des erfindungsgemäßen Verfahrens gecracked werden.
- Die in dem erfindungsgemäßen Verfahren verwendete Katalysatorzusammensetzung umfasst das neue poröse kristalline Material, MCM-68, entweder als den primären Crackbestandteil oder als einen additiven Bestandteil in Verbindung mit einem konventionellen Crackkatalysator. MCM-68 ist ein einphasiges kristallines Material, das ein einzigartiges dreidimensionales Kanalsystem hat, das mindestens ein Kanalsystem, in dem jeder Kanal durch einen 12-gliedrigen Ring von vierflächig koordinierten Atomen definiert ist, und mindestens zwei weitere unabhängige Kanalsysteme umfasst, in denen jeder Kanal durch einen 10-gliedrigen Ring von vierflächig koordinierten Atomen definiert ist, wobei die Anzahl an einzelnen Kanälen mit 10-gliedrigen Ringen doppelt so groß wie die Zahl der Kanäle mit 12-gliedrigen Ringen ist. Die normale kristalline Form von MCM-68 enthält ein Kanalsystem aus 12-gliedrigen Ringen und zwei Kanalsysteme aus 10-gliedrigen Ringen, wobei sich die Kanäle von jedem System senkrecht zu den Kanälen der anderen Systeme ausdehnen und die Kanäle der 12-gliedrigen Ringe im Allgemeinen gerade und die Kanäle der 10-gliedrigen Ringe gewunden (sinusförmig) sind.
- Die Struktur von MCM-68 kann durch seine Einheitszelle definiert werden, die die kleinste strukturelle Einheit ist, die alle strukturellen Elemente des Materials enthält. Tabelle 1 führt die Positionen von jedem vierflächigen Atom in der Einheitszelle in Nanometern auf, wobei jedes vierflächige Atom an ein Sauerstoffatom gebunden ist, das auch an ein benachbartes vierflächiges Atom gebunden ist. Die Struktur, die durch Tabelle 1 repräsentiert wird, ist in
1 (die nur die vierflächigen Atome zeigt) gezeigt. Ausgedehntere Versionen der Struktur werden einfach erzeugt, indem identische Einheitszellen in irgendeiner der x-, y- oder z-Richtungen angehängt werden. Eine ausgedehntere die Poren veranschaulichende Struktur ist in2 gezeigt . Da sich die vierflächigen Atome infolge von anderen kristallinen Kräften (z.B. Anwesenheit von anorganischen oder organischen Spezies) bewegen können, ist ein Bereich von ±0,05 nm für jede Koordinatenposition enthalten. TABELLE 1TABELLE 1 (Fortsetzung) TABELLE 1 (Fortsetzung)
TABELLE 1 (Fortsetzung)

- MCM-68 kann in nahezu reiner Form mit kleinen oder nicht-detektierbaren Verunreinigungskristallphasen hergestellt werden und hat in seiner calcinierten Form ein Röntgenbeugungsmuster, das sich von den Mustern anderer bekannter wie-synthetisiert oder thermisch behandelter kristalliner Materialien durch die in Tabelle 2 aufgeführten Linien unterscheidet. In seiner Form wie-synthetisierter hat das erfindungsgemäße kristalline MCM-68 Material ein Röntgenbeugungsmuster, das sich von anderen bekannten wie-synthetisierten oder thermisch behandelten kristallinen Materialien durch die in Tabelle 3 aufgeführten Linien unterscheidet. TABELLE 2
d (10–10m) (d(Å)) Relative Intensität [100 × I/I(o)] 13,60+/–0,39 S 13,00+/–0,37 VS 10,92+/–0,31 M 10,10+/–0,29 M 9,18+/–0,26 VS 8,21+/–0,23 W 4,58+/–0,13 W 4,54+/–0,13 W 4,45+/–0,13 VW-W 4,32+/–0,12 VW 4,22+/–0,12 VW 4,10+/–0,12 VS 4,05+/–0,11 M 3,94+/–0,11 M 3,85+/–0,11 M 3,80+/–0,11 VW 3,40+/–0,10 W 3,24+/–0,09 W 2,90+/–0,08 VW d (10–10m) (d(Å)) Relative Intensität [100 × I/I(o)] 13,56+/–0,39 VW 12,93+/–0,37 M-S 10,92+/–0,31 W 10,16+/–0,29 VW-W 9,15+/–0,26 VW-W 8,19+/–0,23 VW 4,58+/–0,13 W 4,54+/–0,13 W 4,44+/–0,12 W 4,32+/–0,12 VW 4,23+/–0,12 VW 4,10+/–0,12 VS 4,06+/–0,12 M 3,98+/–0,11 M 3,88+/–0,11 M 3,80+/–0,11 VW 3,40+/–0,10 VW 3,24+/–0,09 W 2,90+/–0,08 VW - Diese Röntgenbeugungsdaten wurden mit einem Scintag Beugungssysstem, das mit einem Germaniumfestphasendetektor ausgestattet ist, unter Verwendung von Kupfer K-α-Strahlung gesammelt. Die Beugungsdaten wurden durch stufenweises Scannen in 0,02 Graden von 2 θ aufgenommen, wobei θ der Bragg-Winkel ist und eine Zählzeit von 10 Sekunden für jede Stufe verwendet wurde. Die interplanaren Abstände, d's, wurden in Å-Einheiten berechnet, und die relativen Intensitäten der Linien, wobei I/I0 1/100stel der Intensität der stärksten Linie über dem Hintergrund ist, wurden unter Verwendung einer Profilanpassungsroutine (oder einem zweite-Ableitungs-Algorithmus) ermittelt. Die Intensitäten sind in Bezug auf Lorentz- und Polarisationseffekte unkorrigiert. Die relativen Intensitäten sind in Form der Symbole vs = sehr stark (80–100), s = stark (60–80), m = mittel (40–60), w = schwach (20–40) und vw = sehr schwach (0–20) angegeben. Es sei darauf hingewiesen, dass die für diese Probe als Einzellinien angegebenen Daten aus einer Mehrzahl an überlappenden Linien bestehen können, die unter bestimmten Bedingungen, wie Unterschieden in kristallographischen Wechseln, als aufgelöste oder teilweise aufgelöste Linien erscheinen können. Typischerweise können kristallographische Wechsel kleine Wechsel in den Einheitszellparametern und/oder einen Wechsel in der Kristallsymmetrie ohne einen Wechsel in der Struktur einschließen. Diese kleinen Einflüsse, die Wechsel in relativen Intensitäten einschließen, können auch als Ergebnis von Unterschieden im Kationengehalt, in der Gerüstzusammensetzung, in der Art und dem Grad der Porenfüllung, in der Kristallgröße und -form, in der bevorzugten Orientierung und in der thermischen und/oder hydrothermischen Vorgeschichte auftreten.
- MCM-68 hat eine Zusammensetzung, die das molare Verhältnis:
- MCM-68 kann aus einer Reaktionsmischung hergestellt werden, die Quellen an Alkali- oder Erdalkalimetallkation (M), z.B. Natrium und/oder Kalium, ein Oxid eines dreiwertigen Elements X, z.B. Aluminium und/oder Bor, ein Oxid eines vierwertigen Elements Y, z.B. Silicium, Dirigiermittel (Q) (directing agent) und Wasser enthält, wobei diese Reaktionsmischung eine Zusammensetzung ausgedrückt in Molverhältnissen von Oxiden innerhalb der folgenden Bereiche hat:
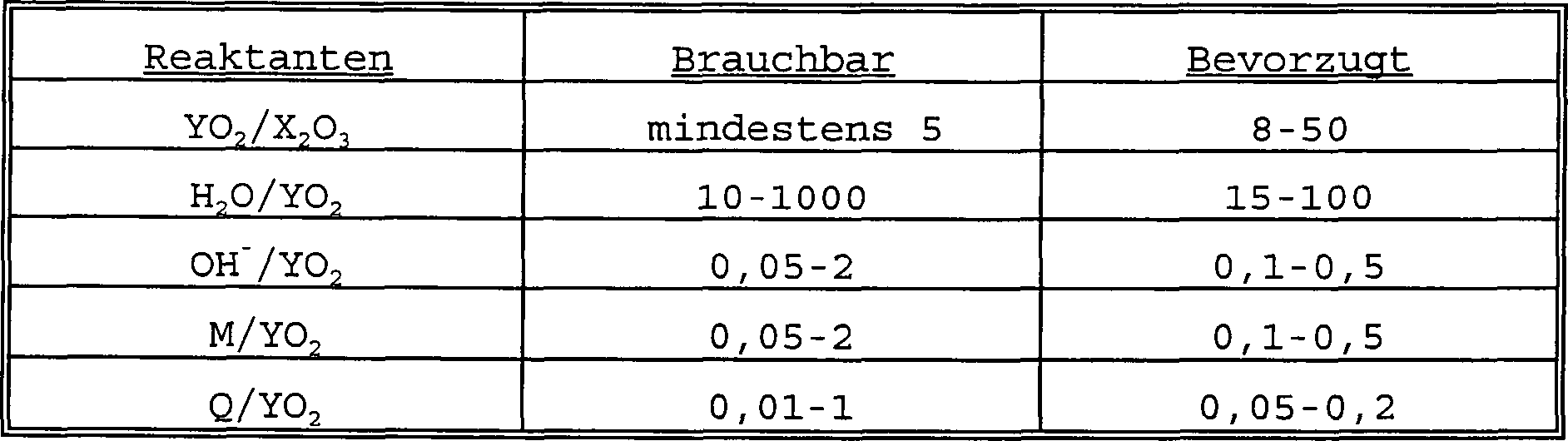
- Das organische Dirigiermittel Q, das hierin verwendet wird, ist ausgewählt aus den neuen Dikationen N,N,N',N'-Tetraalkylbicyclo[2.2.2]oct-7-en-2,3:5,6-dipyrrolidinium-Dikation und N,N,N',N'-Tetraalkylbicyclo[2.2.2]octan-2,3:5,6-dipyrrolidinium-Dikation, die durch die folgenden Formeln repräsentiert werden können:N,N,N',N'-Tetraalkylbicyclo[2.2.2]oct-7-en-2,3:5,6-dipyrrolidinium
 N,N,N',N'-Tetraalkylbicyclo[2.2.2]octan-2,3:5,6-dipyrrolidinium,
N,N,N',N'-Tetraalkylbicyclo[2.2.2]octan-2,3:5,6-dipyrrolidinium,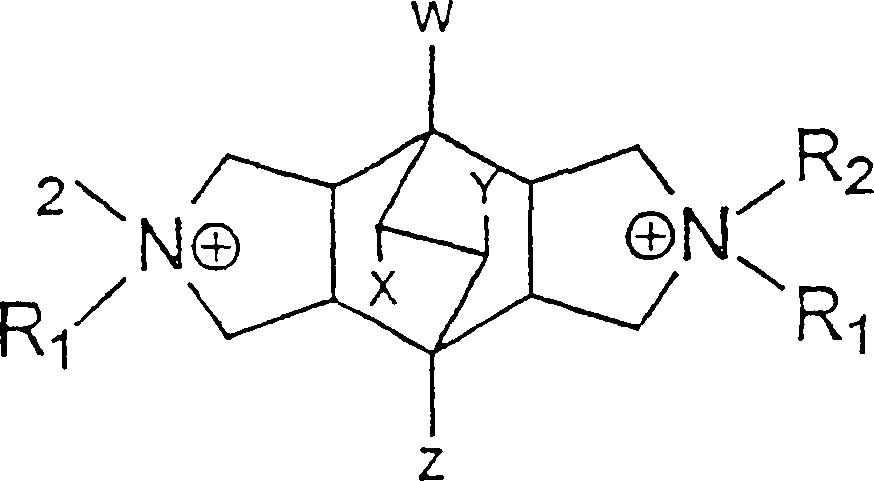
wobei R1, R2 dieselben oder verschiedene Substituenten ausgewählt aus Alkylgruppen mit 1 bis 6 Kohlenstoffatomen, Phenyl- und Benzylgruppen sein können, oder R1 und R2 als eine cyclische Gruppe mit 3 bis 6 Kohlenstoffatomen verknüpft sein können, und W, X, Y, Z dieselben oder verschiedene Substituenten ausgewählt aus Wasserstoff, Alkylgruppen mit 1 bis 6 Kohlenstoffatomen, Phenylgruppen und Halogenen sein können. - In einem bevorzugten Beispiel ist das organische Dirigiermittel das N,N,N',N'-Tetraethyl-exo,exo-bicyclo[2.2.2]oct-7-en-2,3:5,6-dipyrrolidinium (Bicyclodiquat-Et4)-Dikation, das die Formal C20H36N2 ++ hat, die wie folgt dargestellt werden kann:

- Die Quelle an organischem Dikation kann jedes Salz sein, das nicht nachteilig für die Bildung des erfindungsgemäßen kristallinen Materials ist, z.B. das Halogenid-, z.B. Iodid-, oder Hydroxidsalz.
- Die neuen organischen Dikationen, die verwendet werden, um das erfindungsgemäße MCM-68 zu synthetisieren, können z.B. aus exo,exo-Bicyclo[2.2.2]oct-7-en-2,3:5,6-tetracarbonsäuredianhydrid hergestellt werden, das ein kommerziell verfügbares Material ist. Das Dianhydrid wird zunächst mit Ammoniak oder einem Amin zur Reaktion gebracht, um ein Diimid zu ergeben, das dann mit LiAlH4 reduziert wird, um das Diamin zu ergeben. Das Diamin kann dann mit einem Alkyl-, Phenyl- oder Benzylhalogenid alkyliert werden, um das quaternäre Dikation zu ergeben. Entsprechend kann das Bicyclooctandiquat aus dem Dianhydrid hergestellt werden, was in der Literatur bekannt ist, oder es kann durch Hydrierung des Bicyclooctendianhydrids hergestellt werden.
- Die Kristallisation von MCM-68 kann entweder bei statischen oder gerührten Bedingungen in einem geeigneten Reaktorgefäß, wie z.B. Polypropylengefäßen oder Teflon beschichteten oder Edelstahlautoklaven, bei einer Temperatur von 80°C bis 250°C für einen Zeitraum durchgeführt werden, der dafür ausreicht, dass Kristallisation bei der verwendeten Temperatur stattfindet, z.B. von 12 Stunden bis 100 Tagen. Danach werden die Kristalle von der Flüssigkeit getrennt und gewonnen.
- Es sei darauf hingewiesen, dass die Reaktionsmischungsbestandteile von mehr als einer Quelle geliefert werden können. Die Reaktionsmischung kann entweder absatzweise oder kontinuierlich hergestellt werden. Die Kristallgröße und Kristallisationszeit wird mit der Art der verwendeten Reaktionsmischung und den Kristallisationsbedingungen variieren.
- Die Synthese von MCM-68 kann durch die Anwesenheit von mindestens 0,01%, bevorzugt 0,10% und noch bevorzugter 1 Kristallkeimen (bezogen auf das Gesamtgewicht) an kristallinem Produkt vereinfacht werden.
- Vor seiner Verwendung in dem erfindungsgemäßen Verfahren wird das MCM-68 wie synthetisiert einer Behandlung zur Entfernung eines Teils oder aller organischen Bestandteile unterzogen. Dies wird einfach erreicht, indem für mindestens 1 Minute und im Allgemeinen nicht länger als 20 Stunden auf eine Temperatur von mindestens 370°C erhitzt wird. Während unteratmosphärischer Druck für die thermische Behandlung verwendet werden kann, ist atmosphärischer Druck aus Gründen der Einfachheit erwünscht. Die thermische Behandlung kann bei einer Temperatur bis zu 925°C durchgeführt werden. Das thermisch behandelte Produkt kann dann in seine aktive Wasserstoffform umgewandelt werden, typischerweise durch konventionelle Stufen von wiederholtem Ammoniumaustausch gefolgt von Calcinierung.
- In seiner Wasserstoffform zeigt MCM-68 typischerweise eine hohe Säureaktivität, mit einem α-Wert von 900 bis 1000. Der α-Wert ist eine ungefähre Anzeige der katalytischen Crackaktivität des Katalysators im Vergleich zu einem Standardkatalysator, und er gibt die relative Geschwindigkeitskonstante (Geschwindigkeit der normalen Hexanumwandlung pro Volumen an Katalysator pro Zeiteinheit) an. Er basiert auf der Aktivität von Siliciumdioxid-Aluminiumoxid-Crackkatalysator, der als ein a von 1 genommen wird (Geschwindigkeitskonstante = 0,016 sek–1). Der α-Test ist in US-Patent Nr. 3,354,078, im Journal of Catalysis, 4, 527 (1965), 6, 278 (1966) und 61, 395 (1980) beschrieben. Die experimentellen Bedingungen des hierin verwendeten Tests schließen ein konstante Temperatur von 538°C und eine variable Flussrate, wie sie im Detail im Journal of Catalysis 61, 395 (1980) beschrieben ist, ein.
- Beim erfindungsgemäßen Verfahren kann das MCM-68 als der primäre Crackkatalysator entweder allein oder in Verbindung mit einer Additivkomponente, wie ZSM-5 oder Zeolith β, verwendet werden. Alternativ kann das MCM-68 als eine Additivkomponente in Verbindung mit einem konventionellen Crackkatalysator, bevorzugt einem großporigen Molekularsieb mit einer Porengröße von mehr als 7 × 10–10 m (Å), verwendet werden. Typischerweise beträgt das Gewichtsverhältnis des MCM-68 zu dem großporigen Molekularsieb 0,005 bis 0,50, bevorzugt 0,01 bis 0,25.
- Die primäre Crackkomponente kann jedes konventionelle großporige Molekularsieb mit Crackaktivität sein, einschließlich Zeolith X (US-Patent 2,882,442), REX, Zeolith Y (US-Patent 3,130,007), ultrastabilem Y-Zeolith (USY) (US-Patent 3,449,070), Seltene Erden ausgetauschtem Y (REY) (US-Patent 4,415,438), Seltene Erden ausgetauschtem USY (REUSY), dealuminiertem Y (DeAl Y) (US-Patent 3,442,792, US-Patent 4,331,694), ultrahydrophobem Y (UHPY) (US-Patent 4,401,556), und/oder dealuminierten Silikonangereicherten Zeolithen, z.B. LZ-210 (US-Patent 4,678,765). Bevorzugt sind höhere Siliciumdioxidformen von Zeolith Y, Zeolith ZK-5 (US-Patent 3,247,195, Zeolith ZK-4 (US-Patent 3,314,752), ZSM-20 (US-Patent 3,972,983), Zeolith β (US-Patent 3,308,069) und Zeolith L (US-Patente Nrn. 3,216,789 und 4,701,315). Natürlich vorkommende Zeolithe wie Faujasit, Mordenit und andere können auch verwendet werden. Diese Materialien können konventionellen Behandlungen, wie Imprägnierung oder Innenaustausch mit Seltenen Erdenelementen, unterzogen werden, um die Stabilität zu erhöhen. Das bevorzugte großporige Molekularsieb der oben angegebenen ist Zeolith Y, bevorzugter ein REY, USY oder REUSY.
- Andere geeignete großporige kristalline Molekularsiebe schließen säulenförmige Silikate und/oder Tone, Aluminiumphosphate, z.B. ein ALPO4-5, ALPO4-8, VPI-5, Siliumaluminiumphosphate, z.B. SAPO-5, SAPO-37, SAPO-31, SAPO-40 und andere Metallaluminiumphosphate ein. Diese sind vielfältig in den US-Patenten Nrn. 4,310,440, 4,440,871, 4,554,143, 4,567,029, 4,666,875, 4,742,033, 4,880,611, 4,859,314 und 4,791,083 beschrieben.
- Der Crackkatalysator wird normalerweise außerdem ein oder mehrere Matrix- oder Bindematerialien enthalten, die den Temperaturen und anderen Bedingungen, z.B. mechanischem Abrieb, die während dem Cracken auftreten, widerstehen. Es ist im Allgemeinen notwendig, dass die Katalysatoren mechanischem Abrieb, d.h. der Bildung von Feingut, also kleinen Teilchen von z.B. weniger als 20 μm ist, widerstehen. Die Zyklen von Cracken und Regenerierung bei hohen Flussraten und Temperaturen, wie in einem FCC-Verfahren, haben eine Tendenz, den Katalysator in Feingut, im Vergleich zu einem durchschnittlichen Durchmesser der Katalysatorteilchen von 60 bis 90 μm, aufzubrechen. In einem FCC-Verfahren reichen die Katalysatorteilchen von 10 bis 200 μm, bevorzugt von 20 bis 120 um. Exzessive Bildung von Katalysatorfeingut steigert die Katalysator-Kosten des Raffineriebetreibers.
- Die Matrix kann sowohl physikalische als auch katalytische Funktionen erfüllen. Matrixmaterialien schließen aktive oder inaktive anorganische Materialien, wie Tone, und/oder Metalloxide, wie Aluminiumoxid oder Siliciumdioxid, Titandioxid, Zirkoniumdioxid oder Magnesiumoxid, ein. Das Metalloxid kann in der Form eines Sols oder einer gelförmigen Ausfällung oder Gels vorliegen.
- Die Verwendung eines aktiven Matrixmaterials in Verbindung mit dem Molekularsiebbestandteil, der damit kombiniert wird, kann die Umwandlung und/oder Selektivität der Gesamtkatalysatorzusammensetzung in bestimmten Kohlenwasserstoffumwandlungsverfahren erhöhen. Inaktive Materialien können als Verdünnungsmittel dienen, um die Menge an Umwandlung in einem gegebenen Verfahren zu kontrollieren, so dass die Produkte ökonomisch und in einer ordentlichen Weise ohne Anwendung anderer Mittel zur Kontrolle der Reaktionsgeschwindigkeit erhalten werden können. Diese Materialien können wie natürlich vorkommende Tone eingeschlossen werden, um die Abriebswiderstandsfähigkeit des Katalysators unter kommerziellen Betriebsbedingungen zu verbessern.
- Natürlich vorkommende Tone, die mit dem Katalysator vermischt werden können, schließen die Montmorillonit- und Kaolinfamilien ein, die die Subbentonite und die Kaoline, die allgemein als Dixie-, McNamee-, Gerogia- oder Florida-Tone bekannt sind, und andere einschließen, in denen der Hauptmineralbestandteil Halloysite, Kaolinit, Dickit, Nacrit oder Anauxit ist. Solche Tone können im Rohzustand, wie ursprünglich abgebaut, verwendet oder zunächst einer Calcinierung, Säurebehandlung oder chemischen Modifizierung unterzogen werden.
- Zusätzlich zu den oben genannten Materialien können Katalysatoren mit einem porösen Matrixmaterial, wie Siliciumdioxid-Aluminiumoxid, Siliciumdioxid-Magnesiumoxid, Siliciumdioxid-Zirkoniumdioxid,Siliciumdioxid-Thoroxid,Siliciumdioxid-Berylliumoxid, Siliciumdioxid-Titandioxid, sowie ternären Materialien, wie Siliciumdioxid-Aluminiumoxid-Thoroxid,Siliciumdioxid-Aluminiumoxid-Zirkoniumdioxid, Siliciumdioxid-Aluminiumoxid-Magnesiumoxid, Siliciumdioxid-Magnesiumoxid-Zirkoniumdioxid vermischt sein. Die Matrix kann in Form eines Co-Gels vorliegen. Eine Mischung dieser Komponenten kann ebenso verwendet werden.
- Im Allgemeinen können die relativen Verhältnisse von feinverteiltem kristallinen Molekularsiebbestandteil und anorganischer Oxidmatrix weit variieren, wobei der Molekularsiebbestandteil 1 bis 90 Gew.-% und üblichererweise 2 bis 80 Gew.-% des Komposits ausmacht.
- Die Erfindung wird nun in Bezug auf die folgenden Beispiele genauer beschrieben:
- Beispiel 1
- Synthese von N,N'-Diethyl-exo,exo-bicyclo[2.2.2]oct-7-en-2,3:5,6-tetracarbonsäurediimid
- An einem mit einem magnetischen Rührstab ausgestatteten 2000 ml-Dreihalsrundbodenkolben wurden ein Rückflusskühler und ein Thermometer angebracht. Der Kolben wurde dann mit 70 Gew.-% Ethylamin in Wasser (515,25 g, 8 mol) portionsweise gefolgt von exo,exo-Bicyclo[2.2.2]oct-7-en-2,3:5,6-tetracarbonsäuredianhydrid (99,28 g, 0,4 mol) unter heftigem Rühren beschickt. Nach 2 Stunden Rühren bei Raumtemperatur wurde Wasser (300 ml) zugegeben. Die Mischung wurde dann 48 Stunden lang bei 70°C und dann 18 Stunden lang bei 100°C gerührt, um das überschüssige Amin zu vertreiben. Die Reaktion wurde dann auf Raumtemperatur gekühlt und das verbleibende Ethylamin tropfenweise mit konzentrierter HCl gequenscht. Der Feststoff wurde dann abgenutscht, mit Wasser (400 ml) gewaschen und in einem Vakuumexsikkator über Trockenerde getrocknet, um 120,90 g (100%) Diimid als weiße Kristalle zu ergeben.
Schmelzpunkt: 265–266°C
NMR: Lösungsmittel = CDCl3
13C(δ/ppm): 12,846; 33,411; 33,776, 42,763; 130,685; 176,438.
1H(δ/ppm): 1,07 (6H, t), 2,97 (4H, s), 3,47 (4H, q4), 3,78 (2H, br, s), 6,10 (2H, t). Verbrennungsanalyse für C16H18N2O4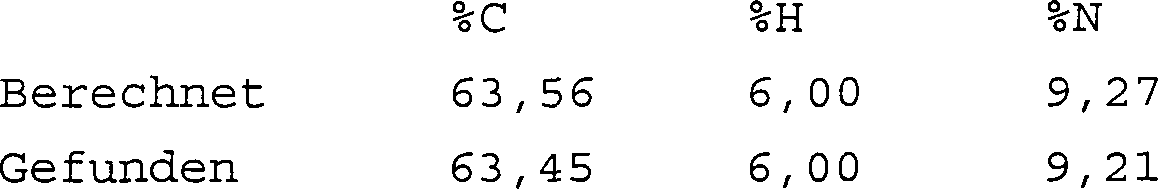
- Beispiel 2
- Synthese von N,N'-Diethyl-exo,exo-bicyclo[2.2.2]oct-7-en-2,3:5,6-dipyrrolidin
- Die gesamte Glasausrüstung dieses Verfahrens wurde für mindestens 12 Stunden in einem Ofen bei 150°C getrocknet. Ein mit einem magnetischen Rührstab, einem Thermometer und einer graduierten druckausgleichenden und mit einer Septumkappe versiegelten Zugabeeinheit ausgestatteter 2000 ml-Dreihalsrundbodenkolben wurde ausgiebig mit N2 gespült. An diesen waren ein Soxhletextraktor mit einer N,N'-Diethyl-exo,exo-bicyclo[2.2.2]oct-7-en-2,3:5,6-tetracarbonsäurediimid (33,26 g, 110 mmol) enthaltenden Hülse auf dem sich ein Rückflusskühler befand und ein Inline-Gasblubber angeschlossen. Das System wurde dann mit Lithiumaluminiumhydridpulver (12,52 g, 330 mmol) und wasserfreiem THF (1650 ml) über einen Zugabeschacht beschickt. Nach 24 Stunden Rückfluss, um das Diimid vollständig zu extrahieren und bereitzustellen, wurde die Reaktion auf 5 °C gekühlt. Dann wurde die Reaktion mit Wasser (12,5 ml), 15% NaOH-Lösung (12,5 ml) und Wasser (37,6 ml) gequenscht, wobei die Temperatur unter 10°C gehalten wurde. Nach Erwärmen auf Raumtemperatur und Abnutschen der Feststoffe gefolgt von Waschen mit Dichlormethan (660 ml), wurde Wasser (220 ml) zu den kombinierten Filtraten gegeben, die dann unter Verwendung von konzentrierter HCl auf pH = 1–2 angesäuert wurden. Die organische Schicht wurde dann abgetrennt, Wasser (220 ml) zugegeben und der pH mit konzentrierter HCl auf 1–2 eingestellt. Diese wässrige Schicht wurde abgetrennt und mit den der vorherigen wässrigen Fraktion kombiniert, mit 50% NaOH-Lösung auf pH = 11–12 basisch gemacht und mit Dichlormethan (5 × 275 ml) extrahiert. Diese vereinigten organischen Fraktionen wurden über Na2SO4 getrocknet, filtriert und im Vakuum verdampft, um ein Gelb/Orangeöl zu ergeben, das unter Kühlung verfestigten kann (22,56 g, 83%). Das Öl wurde mit Ether (2 × 150 ml) extrahiert, die Fraktionen wurden filtriert, vereinigt, über Na2SO4 getrocknet, refiltriert und das Lösungsmittel unter Vakuum verdampft, um ein goldenes Öl zu ergeben, das sich unter Kühlung (20,15 g, 74%) verfestigt. 1H und 13C-NMR-Analyse des gelben Rohfeststoffs zeigten keine sichtbaren Verunreinigungen und das Diamin wurde in dieser Form in der folgenden Diiodidherstellung verwendet. Allerdings wurde eine analytische Probe des Diamins durch Vakuumdestillation des gelben Feststoffs ((0,333 Pa) (10 mTorr), 106–110 °C) hergestellt, um ein klares Öl (52% Effizienz) zu ergeben, das beim Kühlen als ein weißer Feststoff kristallisierte.
Schmelzpunkt: 57–58°C
NMR: Lösungsmittel = CDCl3
13C(δ/ppm): 13,837; 35,491; 44,210; 49,831, 58,423; 135,294.
1H(δ/ppm): 1,05 (6H, t), 1,85 (4H, t), 2,37 (4H, q4), 2,49 (6H, br, d), 3,04 (4H, t), 6,07 (2H, t). Verbrennungsanalyse für C16H26N2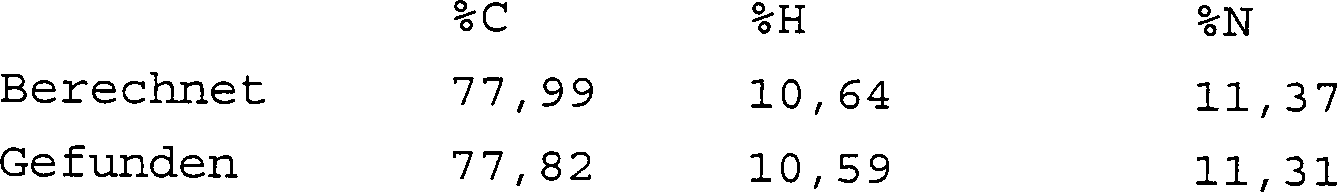
- Beispiel 3
- Synthese von N,N,N',N'-Tetraethyl-exo,exo-bicyclo[2.2.2]oct-7-en-2,3:5,6-dipyrrolidiniumdiiodid (Bycyclodiquat-Et4 2I)
- An einen mit einem magnetischen Rührstab ausgestatteten 1000 ml-Dreihalsrundbodenkolben wurden ein Rückflusskühler, ein. Thermometer und ein druckausgleichender Zugabeschacht, der eine Lösung von Iodethan (67,37 g, 432 mmol) in Ethanol (216 ml) enthielt, angeschlossen. Der Kolben wurde dann mit N,N'-Diethylexo,exo-bicyclo[2.2.2]oct-7-en-2,3:5,6-dipyrrolidin (35,48 g, 144 mmol) und Ethanol (144 ml) beschickt. Nach Rühren bis alle Feststoffe gelöst waren, wurde die Iodethanlösung langsam zugegeben und die Mischung über Nacht unter Rückfluss erhitzt. Nach anschließendem Kühlen auf 10 °C wurden die Feststoffe abgenutscht und mit Aceton (144 ml) gewaschen. Der resultierende schmutzigweiße Feststoff wurde dann in Aceton (500 ml) 15 Minuten lang unter Rückfluss erhitzt, abgenutscht und in einem Vakuumexsikkator über Trockenerde getrocknet, um einen braunen Feststoff, 70,78 g (88%) zu ergeben.
Schmelzpunkt: > 270°C (Zersetzung)
NMR: Lösungsmittel = D2O
13C(6/ppm): 10,115, 10,932, 35,721; 42,597; 55,604; 58,370; 67,030; 130,870.
1H(δ/ppm): 1,28 (12H, t), 2,85 (8H, br, s), 2,92 (2H, br, s), 3,32 (8H, q6), 3,81 (4H, d) 6,45 (2H, t). Verbrennungsanalyse für C20H36N2I2
- Beispiel 4
- Synthese von Aluminumsilikat MCM-68
- 14 g kolloidales Siliciumdioxidsol (30 Gew.% SiO2: Aldrich Ludox SM-30) und 22,096 g destilliertes Wasser wurden mit 0,6056 g Al(OH)3 (Aluminiumhydroxid, fest) gemischt. Zu dieser Reaktionsmischung wurden 7,354 g KOH (88,8% Reinheit) (Kaliumhydroxid, 20 Gew.-% Lösung) und dann 3,912 g Bicyclodiquat-Et4 2I– (N,N,N',N'-Tetraethyl-exo,exo-bicyclo[2.2.2]oct-7-en-2,3:5,6-dipyrrolidiniumdiiodid, fest) gegeben. Die Reaktion kann durch die folgenden Molverhältnisse angegeben werden:
Si/Al2 18 H2O/Si 30 OH/Si 0,375 K+/Si 0,375 Bicyclodiquat-Et42I/Si 0,10 - Die kombinierte Mischung wurde in einen Autoklauen gegeben und ohne zu rühren 300 Stunden lang auf 160°C erhitzt. Das Produkt wurde dann filtriert und mit Wasser gewaschen und über Nacht unter einer IR-Lampe getrocknet. Der Feststoff wurde anschließend in Luft bei einer Temperatur von 540°C 8 Stunden lang calciniert, um das als MCM-68 bezeichnete Material zu ergeben.
- Beispiel 5
- Ammoniumaustausch und Herstellung von H-MCM-68
- Das calcinierte MCM-68 Material aus Beispiel 4 wurde viermal mit einer 1M Ammoniumnitratlösung bei 80°C innenausgetauscht, dann filtriert, gewaschen und unter einer IR-Lampe getrocknet. Anschließend wurde es bei 540°C für 8 Stunden in Luft calciniert. Das erhaltene H-MCM-68 hatte einen α-Wert von 1000.
- Beispiel 6
- Synthese von Aluminiumsilikat MCM-68
- 7 g kolloidales Siliciumdioxid (30 Gew.-%), Al(OH)3 (Aluminiumhydroxid, fest), KOH (Kaliumhydroxid, 20 Gew.-% Lösung), Bicyclodiquat-Et4 2I– (N,N,N',N'-Tetraethyl-exo,exo-bicyclo[2.2.2]oct-7-en-2,3:5,6-dipyrrolidiniumdiiodid, fest) und destilliertes Wasser wurden in den folgenden Molverhältnissen kombiniert:
Si/Al2 30 H2O/Si 30 OH/Si 0,375 K+/Si 0,375 Bicyclodiquat-Et42I/Si 0,10 - Die kombinierte Mischung wurde in einen Autoklauen gegeben und 150 Stunden lang auf 160°C erhitzt. Das Produkt wurde dann filtriert und mit Wasser gewaschen und über Nacht unter einer IR-Lampe getrocknet. Der Feststoff wurde anschließend 8 Stunden lang in Luft bei einer Temperatur von 540°C calciniert, um das MCM-68 zu ergeben. Die Pulverröngtenbeugung des Endprodukts zeigte die Anwesenheit von Spurenmengen an Zeolith ZSM-12.
- Beispiel 7
- Synthese von Aluminiumsilikat MCM-68
- 7 g kolloidales Siliciumdioxid (30 Gew.-%), Al(OH)3 (Aluminiumhydroxid, fest), KOH (Kaliumhydroxid, 20 Gew.-% Lösung), Bicyclodiquat-Et42I (N,N,N',N'-Tetraethyl-exo,exo-bicyclo[2.2.2]oct-7-en-2,3:5,6-dipyrrolidiniumdiiodid, fest) und destilliertes Wasser wurden in den folgenden Molverhältnissen kombiniert:
Si/Al2 15 H2O/Si 30 OH/Si 0,375 K+/Si 0,375 Bicyclodiquat-Et42I/Si 0,10 - Die kombinierte Mischung wurde in einen Autoklauen gegeben und 240 Stunden lang auf 160°C erhitzt. Das Produkt wurde dann filtriert und mit Wasser gewaschen und über Nacht unter einer IR-Lampe getrocknet. Der Feststoff wurde anschließend 8 Stunden lang in Luft bei einer Temperatur von 540°C calciniert, um MCM-68 zu ergeben. Die Pulverröngtenbeugung des Endprodukts zeigte die Anwesenheit von Spurenmengen an Zeolith β an.
- Beispiel 8
- Synthese von Aluminiumsilikat MCM-68
- 14 g kolloidales Siliciumdioxid (30 Gew.-%), Al(OH)3 (Aluminiumhydroxid, fest), KOH (Kaliumhydroxid, 20 Gew.-% Lösung), Bicyclodiquat-Et42I (N,N,N',N'-Tetraethyl-exo,exo-bicyclo[2.2.2]oct-7-en-2,3:5,6-dipyrrolidiniumdiiodid, fest) und destilliertes Wasser wurden in den folgenden Molverhältnissen kombiniert:
Si/Al2 18 H2O/Si 30 OH/Si 0,375 K+/Si 0,375 Bicyclodiquat-Et42I/Si 0,10 - Die kombinierte Mischung wurde in einen Autoklauen gegeben und bei 200 UpM 200 Stunden lang auf 170°C erhitzt. Das Produkt wurde dann filtriert und mit Wasser gewaschen und über Nacht unter einer IR-Lampe getrocknet. Der Feststoff wurde anschließend 8 Stunden lang in Luft bei einer Temperatur von 540°C calciniert, um MCM-68 zu ergeben.
- Beispiel 9
- Synthese von Aluminiumsilikat MCM-68 mit 2 Gew.-% Keimen an MCM-68 wie synthetisiert
- 7 g kolloidales Siliciumdioxid (30 Gew.-%), Al(OH)3 (Aluminiumhydroxid, fest), KOH (Kaliumhydroxid, 20 Gew.-% Lösung), Bicyclodiquat-Et4 2I (N,N,N',N'-Tetraethyl-exo,exo-bicyclo[2.2.2]oct-7-en-2,3:5,6-dipyrrolidiniumdiiodid, fest) und destilliertes Wasser wurden in den folgenden Molverhältnissen kombiniert:
Si/Al2 18 H2O/Si 30 OH/Si 0,375 K+/Si 0,375 Bicyclodiquat-Et42I/Si 0,10 - Zu dieser Mischung wurden 2 Gew.-% Kristallkeime von MCM-68 wie synthetisiert aus Beispiel 5 gegeben. Die kombinierte Mischung wurde in einen Autoklaven gegeben und für 200 Stunden auf 160°C erhitzt. Das Produkt wurde dann filtriert und mit Wasser gewaschen und über Nacht unter einer IR-Lampe getrocknet. Der Feststoff wurde anschließend 8 Stunden lang in Luft bei einer Temperatur von 540°C erhitzt, um das als MCM-68 bezeichnete Material zu ergeben.
- Beispiel 10
- Hydrothermale Stabilität von MCM-68
- Die hydrothermale Stabilität des calcinierten H-MCM-68 aus Beispiel 5 wurde bestimmt, indem die Kristalle in einem Röhrenbedampfer bei 1500°F (815°C) 4 Stunden lang bedampft wurden und dann der Erhalt der Oberfläche gemessen wurde. Wie in Tabelle 4 gezeigt, weist MCM-68 exzellente hydrothermale Stabilität auf, da 77% der ursprünglichen Oberfläche nach der starken Bedampfung erhalten blieben. Tabelle 4
MCM-68 Nur calciniert Oberfläche in m2/g 547 Alpha 730 Bedampfung bei 1500°C für 4 Stunden Oberfläche in m2/g 421 % Oberflächenerhalt 77% - Beispiel 11
- Herstellung eines MCM-68/Siliciumdioxid-Ton-Katalysators
- Eine Mischung, die 40 Gew.-% H-Form MCM-68-Kristalle aus Beispiel 5, 30 Gew.-% wässriges kolloidales Siliciumdioxid und 30 Gew.-% Kaolin-Ton enthielt (auf 100% Aschebasis), wurde hergestellt. Während die Mischung leicht vermahlen wurde, wurde eine kleine Menge an Wasser zugegeben, um eine homogene Paste zu bilden. Die Paste wurde luftgetrocknet, um einen harten Kuchen zu bilden. Dann wurde der trockene Kuchen mit einem Mörser und Pistill klassiert und feine Teilchen, die mit Filtern zwischen 40 und 105 um abgetrennt wurden, wurden gesammelt. Dann wurde der 40 bis 105 um große fluidisierbare MCM-68-Katalysator in Luft bei 540°C (1000°F) 3 Stunden lang calciniert.
- Beispiel 12
- Herstellung eines Beta/Siliciumdioxid-Ton-Katalysators
- Ein Zeolith β-Katalysator wurde hergestellt, indem ein Beispiel 11 gemäßes Verfahren mit der Abweichung verfolgt wurde, dass H-Form-Zeolith β mit einem 35/1 SiO2/Al2O3 anstelle von MCM-68 verwendet wurde.
- Beispiel 13
- Herstellung eines USY/Siliciumdioxid-Ton-Katalysators
- Ein USY-Katalysator wurde hergestellt, indem das Verfahren von Beispiel 11 mit der Abweichung verwendet wurde, dass H-Form-USY mit einem 5,4 SiO2/Al2O3 und einer 24,54 × 10–10 m (Å) Einheitszellgröße anstelle von MCM-68 verwendet wurde.
- Beispiel 14
- Verhalten als FCC Additivkatalysatoren nach starkem Bedampfen
- Vor der Beurteilung in einer Pilotanlage wurden die Katalysatoren (Beispiele 11 bis 13) 4 Stunden lang bei 815°C (1500°F) bei 100% Dampf deaktiviert, um die Deaktivierung in einem FCC-Regenerator zu simulieren. Der USY/Siliciumdioxid-Ton-Katalysator (Beispiel 13) wurde nach der Dampfdeaktivierung als Basisfall allein untersucht. Die MCM-68 und β-Katalysatoren (Beispiele 11 und 12) wurden jeweils als ein Additivkatalysator in Verbindung mit dem USY/Siliciumdioxid-Ton-Katalysator untersucht. Fünfundzwanzig (25) Gew.-% eines Additivkatalysators wurden nach der Dampfdeaktivierung mit 75 Gew.-% dampfdeaktiviertem USY-Katalysator gemischt.
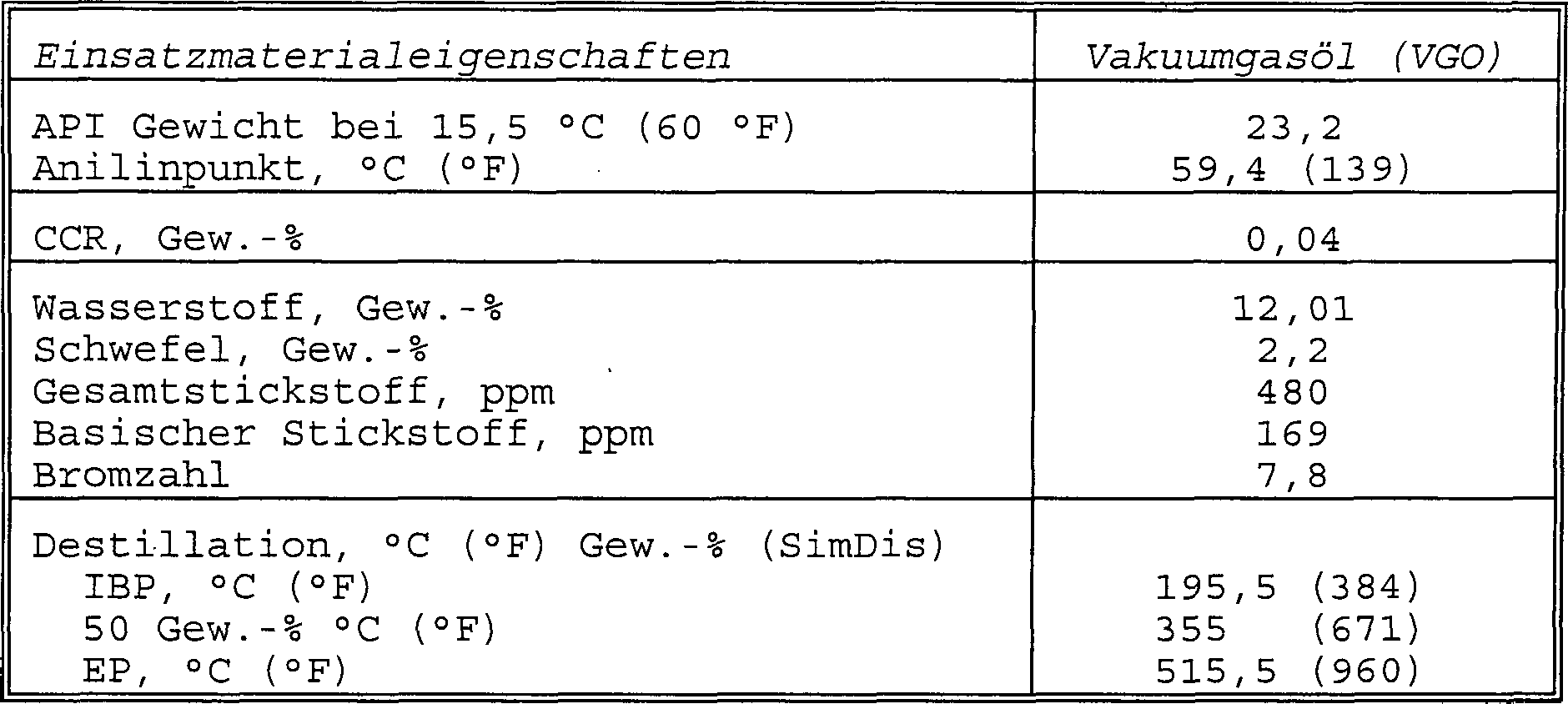
- Ein leichtes Vakuumgasöleinsatzmaterial mit den in Tabelle 5 gezeigten Eigenschaften wurde verwendet, um die Katalysatoren zu beurteilen.
- Tabelle 5
- Eine Mikrogrammmaßstabs-Crackeinheit bestehend aus einem Festbettkatalysatorreaktor in einem Pyrolyseerhitzer (Pyrojector II von SGE) wurde für die Beurteilung verwendet. Die Pyrolyse-Crackeinheit wurde an einen GC präparativen Maßstabs angeschlossen und das gecrackte Produkt wurde direkt zum GC geschickt. Die Einheit konnte die Koks und H2-Ausbeuten nicht messen, und daher wurde das Produkt exklusive H2 und Koks für Ausbeute und Umsatzungsberechnung auf 100% normalisiert. Dieses Verfahren der Datenanalyse war geeignet, um die Mehrertragsausbeuten, die mit einem zusätzlichen Katalysator verbunden sind, vergleichen zu können, da die Koks- und H2-Ausbeutedifferenzen unter den Katalysatormischungen gering sein würden. Die GC-Peaks für Propylen (C3") und n-Propan (n-C3) überlappten und konnten nicht getrennt werden, daher wurde nur die kombinierte C3-Ausbeute angegeben.
- Die verwendeten Reaktionsbedingungen waren 524°C (975°F) Temperatur und ungefähr 1 bis 2 Sekunden an Dampfkontaktzeit. Ein Bereich an Umwandlungen wurde durch Variation der Katalysator : Öl-Verhältnisse untersucht. Die Ergebnisse der Katalysatoren sind in Tabelle 6 zusammengefasst, in der die Produktdaten auf eine konstante Umwandlung, 70 Gew.-% Umwandlung des Einsatzmaterials in 218°C (425°F)- oder weniger 218°C (425°F)-Material, interpoliert wurden. Als Benzinausbeute wurde das 52 bis 218°C (125 bis 425°F)-Bereich-Kohlenwasserstoffprodukt angesehen und als Leichttreibstofföl (LFO) das von 218 bis 349°C (425 bis 660°F). Für Mischungskatalysatoren sind die Ausbeuteverschiebungen gegenüber dem USY-Basisfall angegeben. Tabelle 6
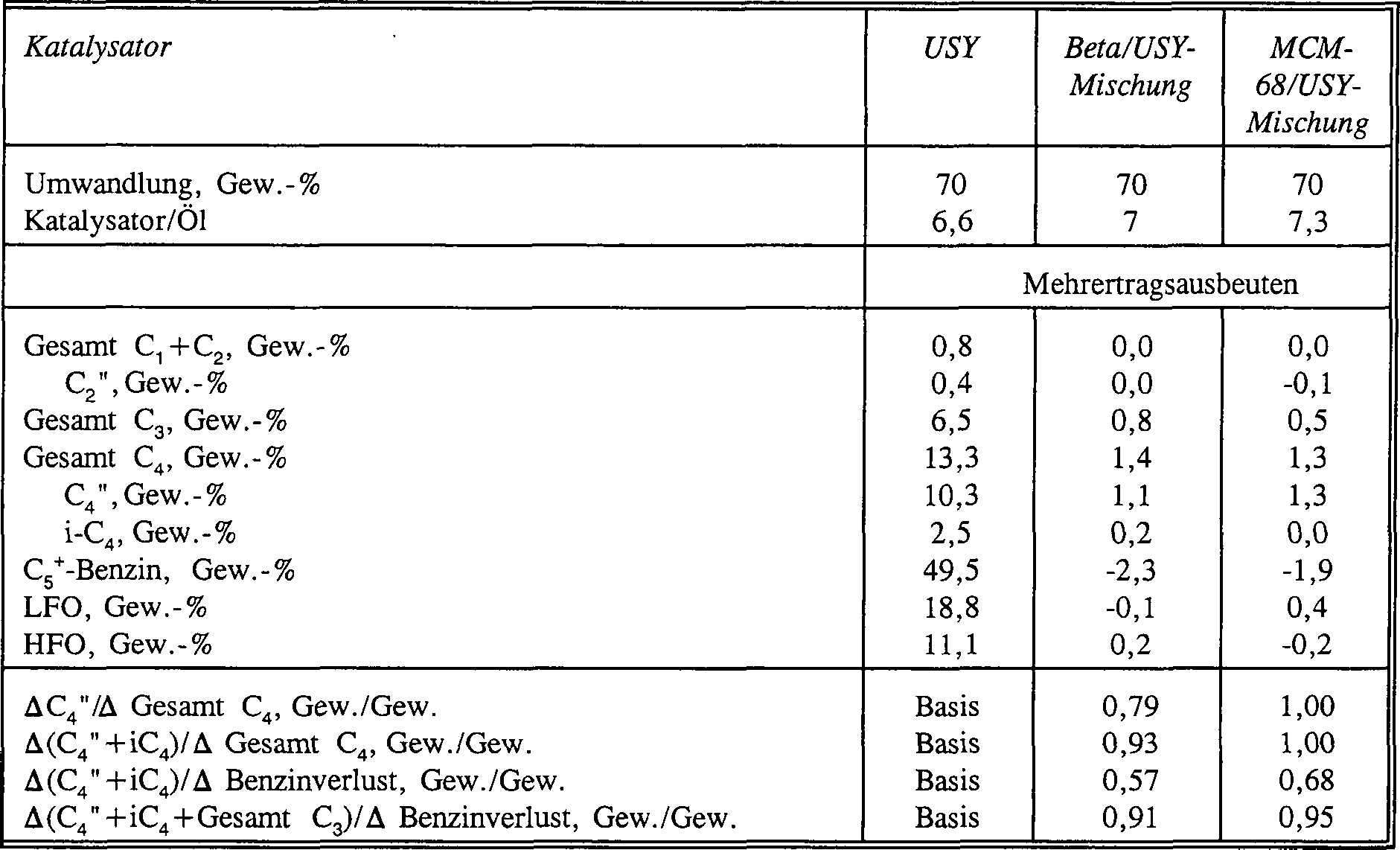
- Die Ergebnisse in Tabelle 6 zeigen an, dass MCM-68-Additivkatalysator zusätzliche C3- und C4-Olefine produziert. Benzinverlust ist ein direktes Anzeichen von Katalysatoraktivität, da der Additivkatalysator hauptsächlich die Schwerbenzinbereichskohlenwasserstoffmoleküle in Leichtbenzin und C3- bis C4-Paraffine und Olefine umwandelt. Der Verlust an Benzinausbeute wird durch die mit den C3- und C4-Olefinen hergestellten Alkylate mehr als ausgeglichen. Im Vergleich mit dem Zeolith β-Additivkatalysator erzeugt MCM-68 Katalysator weniger C3 und mehr C4", was zur Annahme führt, dass MCM-68 sogar noch C4-olefinselektiver ist als Beta. MCM-68 zeigte leicht niedrigeren Benzinvolumenverlust als Beta, aber produzierte mehr C4-Olefine. MCM-68 zeigte niedrigere H-Transferaktivität und der Mehrertrag an C4 verblieb als C4-Olefine.
- Beispiel 15
- Verhalten als FCC-Additivkatalysatoren nach zwischenzeitlichem Bedampfen
- MCM-68 wurde mit Zeolith β auch nach milderen Dampfdeaktivierungsbedingungen verglichen, z.B. bei 700°C (1300°F) Bedampfung, um zwischenzeitliche Katalysatordeaktivierung in einem FCC-Regenerator zu simulieren. Fünfundzwanzig (25) Gew.-% eines Additiv-Katalysators (Beispiele 11 und 12) wurden nach Dampfdeaktivierung bei 704°C (1300°F) für 4 Stunden mit 75 Gew.-% eines dampfdeaktivierten USY-Katalysators vermischt. Ein USY/Siliciumdioxid-Ton-Katalysator (Beispiel 13) wurde nach 815°C (1500°F) Dampfdeaktivierung allein als Basisfall untersucht. Das Verhalten der Katalysatoren ist in Tabelle 7 zusammengefasst, wo die Produktdaten auf eine konstante Umwandlung interpoliert sind, 74 Gew.-% Umwandlung an Einsatzmaterial zu 218°C (425°F) oder weniger als 218°C (425°F)-Material. Tabelle 7

- Die Ergebnisse, die in Tabelle 7 zusammengefasst sind, zeigen an, dass der MCM-68-Additivkatalysator Benzin und LCO crackt und sie hauptsächlich in C3-, C4-Olefine und Isobutan umwandelt. Der Zeolith β-Additivkatalysator und der MCM-68-Additivkatalysator zeigen vergleichbare C3-, C4" und iC4-Ausbeuten.
- Beispiel 16
- Verhalten von MCM-68-Katalysator als FCC-Basis-Crackkatalysator
- MCM-68-Katalysator (Beispiel 11) selbst wurde nach Dampfdeaktivierung bei 815°C (1500°F) für 4 Stunden bei 100% Dampf mit dem in Tabelle 4 angegebenen Vakuumgasöleinsatzmaterial untersucht. Das Verhalten des MCM-68 für das VGO-Cracken wurde mit dem des USY/Siliciumdioxid-Ton-Katalysators (Beispiel 13) nach Dampfdeaktivierung bei 815°C (1500°F) für 4 Stunden bei 100% Dampf verglichen. Die Ergebnisse des Verhaltens sind in Tabelle 8 zusammengefasst. Tabelle 8

- Die in Tabelle 8 zusammengefassten Ergebnisse zeigen, dass der MCM-68-Katalysator VGO effizient crackt. Verglichen mit USY hat MCM-68 eine niedrigere Crackaktivität, aber MCM-68 ist weitaus selektiver in der Herstellung von C3- , C4-Olefinen und Isobutan. USY ist ein benzinselektiver Katalysator, bei dem nur 19% des umgewandelten Materials C3 und C4 wurden. MCM-68 ist extrem olefinselektiv, da 38% des umgewandelten Materials C3 und C4 wurden. Außerdem zeigt MCM-68-Katalysator exzellente Selektivität hinsichtlich C4-Olefinen.
Claims (10)
- Verfahren zum katalytischen Cracken eines Kohlenwasserstoffeinsatzmaterials, bei dem das Einsatzmaterial mit einer Katalysatorzusammensetzung kontaktiert wird, die ein poröses kristallines Material umfasst, das mindestens ein Kanalsystem, in dem jeder Kanal durch einen 12-gliedrigen Ring von vierflächig koordinierten Atomen definiert ist, und mindestens zwei weitere, unabhängige Kanalsysteme enthält, in denen jeweils jeder Kanal durch einen 10-gliedrigen Ring von vierflächig koordinierten Atomen definiert ist, wobei die Anzahl an einzelnen Kanälen mit 10-gliedrigen Ringen doppelt so groß wie die Zahl der Kanäle mit 12-gliedrigen Ringen ist.
- Verfahren nach Anspruch 1, bei dem das poröse kristalline Material ein Kanalsystem mit 12-gliedrigen Ringen und zwei Kanalsysteme mit 10-gliedrigen Ringen enthält.
- Verfahren nach Anspruch 2, bei dem die Kanäle in jedem Kanalsystem mit 10-gliedrigen Ringen des kristallinen Materials sich in einer Richtung ausdehnen, die im Wesentlichen senkrecht zu den Kanälen in dem anderen Kanalsystem mit 10-gliedrigen Ringen und dem Kanalsystem mit 12-gliedrigen Ringen ist.
- Verfahren nach Anspruch 1, bei dem das poröse kristalline Material ein Gerüst von vierflächigen Atomen umfasst, die durch Sauerstoffatome verbrückt sind, wobei das vierflächige Gerüst der Atome definiert ist durch eine Einheitszelle mit folgenden Atomkoordinaten in Nanometern und einem Variationsbereich von ±0,05 Nanometer



- Verfahren nach Anspruch 1, bei dem das poröse kristalline Material eine Zusammensetzung aufweist, die das molare Verhältnis
d (10–10m) (d (Å)) Relative Intensität [100 × I/I(o)] 13,60+/–0,39 S 13,00+/–0,37 VS 10,92+/–0,31 M 10,10+/–0,29 M 9,18+/–0,26 VS 8,21+/–0,23 W 4,58+/–0,13 W 4,54+/–0,13 W 4,45+/–0,13 VW-W 4,32+/–0,12 VW 4,22+/–0,12 VW 4,10+/–0,12 VS 4,05+/–0,11 M 3,94+/–0,11 M 3,85+/–0,11 M 3,80+/–0,11 VW 3,40+/–0,10 W 3,24+/–0,09 W 2,90+/–0,08 VW - Verfahren nach Anspruch 5, bei dem X ein dreiwertiges Element ausgewählt aus der Gruppe bestehend aus Bor, Eisen, Indium, Gallium, Aluminium und einer Kombination davon ist und Y ein vierwertiges Element ausgewählt aus der Gruppe bestehend aus Silicium, Zinn, Titan, Germanium und einer Kombination davon ist.
- Verfahren nach Anspruch 5, bei dem X Aluminium umfasst und Y Silicium umfasst.
- Verfahren nach Anspruch 5, bei dem die Katalysatorzusammensetzung außerdem ein großporiges Molekularsieb mit einer Porengröße von mehr als 7 × 10–10 m (Angstrom) umfasst.
- Verfahren nach Anspruch 8, bei dem das großporige Molekularsieb Zeolith Y ist.
- Verfahren nach Anspruch 8, bei dem das Gewichtsverhältnis des synthetischen porösen kristallinen Materials zu dem großporigen Molekularsieb 0,005 bis 0,50 ist.
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US09/234,544 US6049018A (en) | 1999-01-21 | 1999-01-21 | Synthetic porous crystalline MCM-68, its synthesis and use |
| US234544 | 1999-01-21 | ||
| PCT/US2000/001448 WO2000043466A1 (en) | 1999-01-21 | 2000-01-21 | Catalytic cracking process using an mcm-68 catalyst |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| DE60009030D1 DE60009030D1 (de) | 2004-04-22 |
| DE60009030T2 true DE60009030T2 (de) | 2004-09-02 |
Family
ID=22881810
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| DE60009030T Expired - Lifetime DE60009030T2 (de) | 1999-01-21 | 2000-01-21 | Katalytisches crack-verfahren mit mcm-68-katalysator |
| DE60006964T Expired - Lifetime DE60006964T2 (de) | 1999-01-21 | 2000-01-21 | Synthetisches poröses kristallines mcm-68, synthese und verwendung desselben |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| DE60006964T Expired - Lifetime DE60006964T2 (de) | 1999-01-21 | 2000-01-21 | Synthetisches poröses kristallines mcm-68, synthese und verwendung desselben |
Country Status (16)
| Country | Link |
|---|---|
| US (1) | US6049018A (de) |
| EP (2) | EP1137739B1 (de) |
| JP (1) | JP4689046B2 (de) |
| KR (1) | KR100815658B1 (de) |
| CN (2) | CN100473609C (de) |
| AR (1) | AR022332A1 (de) |
| AT (1) | ATE255536T1 (de) |
| AU (3) | AU768270B2 (de) |
| CA (2) | CA2360030C (de) |
| DE (2) | DE60009030T2 (de) |
| ES (1) | ES2211504T3 (de) |
| NO (1) | NO20013602L (de) |
| RU (1) | RU2223912C2 (de) |
| TW (1) | TW527414B (de) |
| WO (3) | WO2000043335A1 (de) |
| ZA (1) | ZA200105984B (de) |
Families Citing this family (126)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US6419819B1 (en) * | 1998-12-30 | 2002-07-16 | Exxonmobil Oil Corporation | Synthetic porous crystalline MCM-67, its synthesis and use |
| US6310265B1 (en) * | 1999-11-01 | 2001-10-30 | Exxonmobil Chemical Patents Inc. | Isomerization of paraffins |
| US7198711B1 (en) * | 2000-01-21 | 2007-04-03 | Exxonmobil Research And Engineering Company | Catalytic cracking processing using an MCM-68 catalyst |
| US6506953B1 (en) * | 2000-01-24 | 2003-01-14 | Exxonmobil Oil Corporation | Hydroalkylation of aromatic hydrocarbons |
| ES2325981T3 (es) | 2000-09-25 | 2009-09-28 | Exxonmobil Chemical Patents Inc. | Hidrogenacion de los efluentes de escision en la produccion de fenol. |
| US6645462B1 (en) * | 2000-11-03 | 2003-11-11 | Exxon Mobil Oil Corporation | Synthetic porous crystalline MCM-71, its synthesis and use |
| US6656268B2 (en) | 2001-01-26 | 2003-12-02 | Exxonmobil Oil Corporation | Synthetic porous crystalline MCM-70, its synthesis and use |
| EP1894909B1 (de) | 2001-02-07 | 2017-04-05 | Badger Licensing LLC | Herstellung von alkylaromatischen Verbindungen |
| ES2628745T3 (es) | 2001-02-07 | 2017-08-03 | Badger Licensing Llc | Producción de compuestos alquilaromáticos |
| US6933419B2 (en) * | 2001-04-27 | 2005-08-23 | Exxonmobil Oil Corporation | Production of diisopropylbenzene |
| US6518471B1 (en) * | 2001-06-25 | 2003-02-11 | Exxonmobil Chemical Patents Inc. | Selective production of meta-diisopropylbenzene |
| US6555089B1 (en) * | 2001-07-13 | 2003-04-29 | Chevron U.S.A. Inc. | Zeolite SSZ-58 composition of matter and synthesis thereof |
| ES2296989T3 (es) * | 2001-07-13 | 2008-05-01 | Chevron U.S.A. Inc. | Zeolita ssz-58. |
| WO2003074452A1 (en) | 2002-02-28 | 2003-09-12 | Stone & Webster, Inc. | Production of alkyl aromatic compounds |
| WO2003097531A1 (en) * | 2002-05-01 | 2003-11-27 | Exxonmobil Chemical Patents Inc. | Synthetic porous crystalline mcm-71, its synthesis and use |
| US7279018B2 (en) | 2002-09-06 | 2007-10-09 | Fortum Oyj | Fuel composition for a diesel engine |
| US6753453B2 (en) | 2002-11-19 | 2004-06-22 | Exxonmobil Chemical Patents Inc. | Production of meta-diisopropylbenzene |
| FR2850099B1 (fr) * | 2003-01-16 | 2005-03-04 | Inst Francais Du Petrole | Solide cristallise im-9 et son procede de preparation |
| TWI335239B (en) * | 2003-03-21 | 2011-01-01 | Stone & Webster Inc | Production of alkyl aromatic compounds with catalyst reactivation |
| US7361798B2 (en) * | 2003-10-03 | 2008-04-22 | Exxonmobil Chemical Patents Inc. | Production of dialkylbenzenes |
| WO2007001934A2 (en) * | 2005-06-23 | 2007-01-04 | Chevron U.S.A. Inc. | Molecular sieve ssz-56 composition of matter and synthesis thereof |
| US7226569B2 (en) * | 2005-06-23 | 2007-06-05 | Chevron U.S.A. Inc. | Reduction of oxides of nitrogen in a gas stream using molecular sieve SSZ-56 |
| US7226576B2 (en) * | 2005-06-23 | 2007-06-05 | Chevron U.S.A. Inc. | Molecular sieve SSZ-56 composition of matter and synthesis thereof |
| US8562941B2 (en) * | 2005-12-20 | 2013-10-22 | Exxonmobil Research And Engineering Company | Perturbed synthesis of materials |
| ES2284379B1 (es) * | 2006-02-28 | 2008-11-01 | Universidad Politecnica De Valencia | Un material cristalino microporoso, zeolita itq-37, procedimiento de preparacion y uso. |
| JP4923248B2 (ja) * | 2006-08-23 | 2012-04-25 | 国立大学法人横浜国立大学 | チタノシリケート及びその製法 |
| JP5017642B2 (ja) * | 2006-09-08 | 2012-09-05 | 国立大学法人横浜国立大学 | Mcm−68のトポロジーを持つ結晶性多孔質シリケート及びその製法 |
| CA2584876A1 (en) * | 2007-02-02 | 2008-08-02 | Albemarle Netherlands Bv | A crystalline microporous material of zeolitic nature |
| EP2110368A1 (de) | 2008-04-18 | 2009-10-21 | Total Petrochemicals France | Alkylierung aromatischer Substrate und Transalkylierungsverfahren |
| US8025863B2 (en) * | 2008-06-20 | 2011-09-27 | Exxonmobil Research And Engineering Company | Synthesis and use of MSE-framework type molecular sieves |
| US7922997B2 (en) * | 2008-09-30 | 2011-04-12 | Uop Llc | UZM-35 aluminosilicate zeolite, method of preparation and processes using UZM-35 |
| US8048403B2 (en) * | 2008-12-16 | 2011-11-01 | Uop Llc | UZM-26 family of crystalline aluminosilicate compositions and method of preparing the compositions |
| US7575737B1 (en) * | 2008-12-18 | 2009-08-18 | Uop Llc | UZM-27 family of crystalline aluminosilicate compositions and a method of preparing the compositions |
| JP5131624B2 (ja) * | 2009-03-05 | 2013-01-30 | 国立大学法人横浜国立大学 | パラフィンの接触分解法 |
| US8157985B2 (en) * | 2009-06-22 | 2012-04-17 | Uop Llc | Process for catalytic cracking of hydrocarbons using UZM-35HS |
| US7981273B2 (en) * | 2009-06-22 | 2011-07-19 | Uop Llc | Process for catalytic cracking of hydrocarbons using UZM-35 |
| US7982081B2 (en) * | 2009-06-29 | 2011-07-19 | Uop Llc | Process for alkylation of aromatic hydrocarbons using UZM-35 |
| US8071830B1 (en) | 2009-06-29 | 2011-12-06 | Uop Llc | Process for alkylation of aromatic hydrocarbons using UZM-35 |
| WO2011017183A2 (en) * | 2009-08-04 | 2011-02-10 | Uop Llc | Uzm-29 family of crystalline zeolitic compositions and a method of preparing the compositions |
| US9321710B2 (en) | 2010-01-25 | 2016-04-26 | Exxonmobil Chemical Patents Inc. | Process for producing phenol |
| WO2012145029A2 (en) | 2011-04-19 | 2012-10-26 | Exxonmobil Chemical Patents Inc. | Process for producing phenol |
| US8138385B2 (en) | 2010-03-31 | 2012-03-20 | Uop Llc | Process for xylene and ethylbenzene isomerization using UZM-35HS |
| US8058496B2 (en) * | 2010-03-31 | 2011-11-15 | Uop Llc | Process for xylene and ethylbenzene isomerization using UZM-35 |
| US7982082B1 (en) | 2010-06-21 | 2011-07-19 | Uop Llc | Process for alkylation of aromatic hydrocarbons using UZM-35 |
| US8071831B1 (en) | 2010-06-21 | 2011-12-06 | Uop Llc | Process for xylene and ethylbenzene isomerization using UZM-35 |
| US8247631B2 (en) | 2010-06-21 | 2012-08-21 | Uop Llc | Process for catalytic cracking of hydrocarbons using UZM-35 |
| US8053618B1 (en) | 2010-06-21 | 2011-11-08 | Uop Llc | UZM-35 zeolitic composition, method of preparation and processes |
| US8022262B1 (en) | 2010-06-21 | 2011-09-20 | Uop Llc | UZM-35 zeolitic composition method of preparation and processes |
| RU2525417C2 (ru) * | 2010-06-21 | 2014-08-10 | Юоп Ллк | Цеолитная композиция uzm-35, способ получения и способы применения |
| JP5351216B2 (ja) | 2010-07-01 | 2013-11-27 | 日本化学工業株式会社 | ゼオライトの製造方法 |
| US8981170B2 (en) | 2010-08-20 | 2015-03-17 | Exxonmobil Chemical Patents Inc. | Process for producing cycloalkylaromatic compounds |
| US9169175B2 (en) | 2010-09-14 | 2015-10-27 | Exxonmobil Chemical Patents Inc. | Cyclohexylbenzene compositions |
| WO2012036821A1 (en) | 2010-09-14 | 2012-03-22 | Exxonmobil Chemical Patents Inc. | Phenol compositions |
| SG189126A1 (en) * | 2010-10-15 | 2013-05-31 | Exxonmobil Chem Patents Inc | Selecting an improved catalyst composition and hydrocarbon conversion process using same |
| JP6014943B2 (ja) | 2010-12-17 | 2016-10-26 | エクソンモービル ケミカル パテンツ インコーポレイテッド | 脱水素触媒及び方法 |
| US9233887B2 (en) | 2010-12-21 | 2016-01-12 | Exxonmobil Chemical Patents Inc. | Process for producing a monocycloalkyl-substituted aromatic compound |
| JP5711393B2 (ja) | 2011-02-18 | 2015-04-30 | エクソンモービル ケミカル パテンツ インコーポレイテッド | シクロヘキシルベンゼンを生成するための方法 |
| US9017641B2 (en) | 2011-02-21 | 2015-04-28 | Exxonmobil Chemical Patents Inc. | Hydrogen purification process |
| WO2012118542A1 (en) | 2011-02-28 | 2012-09-07 | Exxonmobil Chemical Patents Inc. | Process for producing phenol |
| CN103443060B (zh) | 2011-03-28 | 2016-01-20 | 埃克森美孚化学专利公司 | 脱氢方法 |
| US8763364B2 (en) | 2011-04-18 | 2014-07-01 | Chevron U.S.A. Inc. | Treatment of cold start engine exhaust |
| JP5793792B2 (ja) | 2011-04-19 | 2015-10-14 | エクソンモービル ケミカル パテンツ インコーポレイテッド | フェノールの製造方法 |
| US9388102B2 (en) | 2011-04-19 | 2016-07-12 | Exxonmobil Chemical Patents Inc. | Process for producing phenol |
| KR101579359B1 (ko) | 2011-04-19 | 2015-12-21 | 엑손모빌 케미칼 패턴츠 인코포레이티드 | 페놀 및/또는 시클로헥사논의 제조 방법 |
| JP5766067B2 (ja) * | 2011-08-29 | 2015-08-19 | 日揮触媒化成株式会社 | アルミナ含有メソポーラス多孔体の合成方法およびアルミナ含有メソポーラス多孔体 |
| US9067870B2 (en) | 2011-09-23 | 2015-06-30 | Exxonmobil Chemical Patents Inc. | Process for producing phenol |
| CA2851798C (en) | 2011-10-12 | 2018-01-02 | Exxonmobil Research And Engineering Company | Synthesis of mse-framework type molecular sieves |
| US8916130B2 (en) * | 2011-10-12 | 2014-12-23 | Exxonmobil Research And Engineering Company | Synthesis of MSE-framework type molecular sieves |
| CN104203824B (zh) * | 2011-11-25 | 2017-10-17 | UniZeo株式会社 | 沸石及所述沸石的制造方法以及石蜡的接触分解催化剂 |
| US9090528B2 (en) | 2012-02-27 | 2015-07-28 | Exxonmobil Chemical Patents Inc. | Hydroalkylation process |
| WO2013130151A1 (en) | 2012-02-29 | 2013-09-06 | Exxonmobil Chemical Patents Inc. | Process for producing phenol comprising a step of hydroalkylation of benzene to cyclohexylbenzene |
| WO2013130153A1 (en) | 2012-03-01 | 2013-09-06 | Exxonmobil Chemical Patents Inc. | Process of producing cyclohexylbenzene |
| US8545801B1 (en) | 2012-04-12 | 2013-10-01 | Chevron U.S.A. Inc. | Method for preparing molecular sieve SSZ-87 |
| WO2013154671A1 (en) * | 2012-04-12 | 2013-10-17 | Chevron U.S.A. Inc. | Processes using molecular sieve ssz-87 |
| US8545800B1 (en) * | 2012-04-12 | 2013-10-01 | Chevron U.S.A. Inc. | Molecular sieve SSZ-87 |
| WO2013165656A1 (en) | 2012-05-02 | 2013-11-07 | Exxonmobil Chemical Patents Inc. | Process for separating a mixture comprising cyclohexanone and phenol |
| WO2013165659A1 (en) | 2012-05-02 | 2013-11-07 | Exxonmobil Chemical Patents Inc. | Process for producing phenol and cyclohexanone |
| CN104271539B (zh) | 2012-05-03 | 2016-08-24 | 埃克森美孚化学专利公司 | 氢化方法 |
| SG11201502384WA (en) | 2012-11-16 | 2015-04-29 | Exxonmobil Res & Eng Co | Synthesis of mse-framework type molecular sieves |
| US8940941B2 (en) | 2012-12-06 | 2015-01-27 | Exxonmobil Chemical Patents Inc. | Process for producing phenol and method for regenerating catalyst deactivated in the process |
| US10035762B2 (en) | 2012-12-21 | 2018-07-31 | Exxonmobil Chemical Patents Inc. | Synthesis of succinimides and quaternary ammonium ions for use in making molecular sieves |
| KR101755964B1 (ko) | 2013-03-04 | 2017-07-07 | 엑손모빌 케미칼 패턴츠 인코포레이티드 | 페놀 및/또는 사이클로헥사논의 제조 시스템 및 방법 |
| WO2014137628A1 (en) | 2013-03-04 | 2014-09-12 | Exxonmobil Chemical Patents Inc. | System and process for making cyclohexylbenzene |
| EP2978526A1 (de) | 2013-03-25 | 2016-02-03 | ExxonMobil Chemical Patents Inc. | Verfahren zur herstellung einer alkylierten aromatischen verbindung |
| WO2014158978A1 (en) | 2013-03-25 | 2014-10-02 | Exxonmobil Chemical Patents Inc. | Process for making alkylated aromatic compound |
| CN105164093A (zh) | 2013-04-02 | 2015-12-16 | 埃克森美孚化学专利公司 | 制造苯酚和/或环己酮的方法与装置 |
| CN104109075A (zh) * | 2013-04-16 | 2014-10-22 | 中国石油化工股份有限公司 | 具有二甲苯高选择性的甲苯歧化方法 |
| CN104107717B (zh) * | 2013-04-16 | 2016-12-28 | 中国石油化工股份有限公司 | 提高二甲苯选择性的甲苯歧化催化剂及其用途 |
| US20160368844A1 (en) | 2013-07-24 | 2016-12-22 | Exxonmobil Chemical Patents Inc. | Process for Producing Mixtures of Cyclohexanone and Cyclohexanol |
| US9335285B2 (en) | 2013-10-18 | 2016-05-10 | Exxonmobil Chemical Patents Inc. | Method for measuring acid strength in reaction medium using trimethylphosphine oxide and 31P NMR |
| US9144792B2 (en) | 2013-10-23 | 2015-09-29 | Exxonmobil Chemical Patents Inc. | Hydroalkylating process |
| EP3083530A1 (de) | 2013-12-20 | 2016-10-26 | ExxonMobil Chemical Patents Inc. | Cyclohexylbenzolzusammensetzung |
| WO2015094530A2 (en) | 2013-12-20 | 2015-06-25 | Exxonmobil Chemical Patents Inc. | Process for making phenol and/or cyclohexanone |
| CN104944442B (zh) * | 2014-03-26 | 2017-10-03 | 中国石油化工股份有限公司 | 一种合成b‑mcm‑68杂原子分子筛的方法 |
| CN106459779A (zh) | 2014-05-30 | 2017-02-22 | 埃克森美孚化学专利公司 | 用于从有机液体中除去水和/或氧气的方法和装置 |
| EP3180303B1 (de) | 2014-08-15 | 2018-07-18 | ExxonMobil Chemical Patents Inc. | Verfahren und system zur herstellung von cyclohexanon |
| SG11201700415UA (en) | 2014-08-15 | 2017-02-27 | Exxonmobil Chemical Patents Inc | Process and system for making cyclohexanone |
| EP3180305A1 (de) | 2014-08-15 | 2017-06-21 | ExxonMobil Chemical Patents Inc. | Verfahren und system zur herstellung von cyclohexanon |
| US9868687B2 (en) | 2014-09-30 | 2018-01-16 | Exxonmobil Chemical Patents Inc. | Process for making cyclohexanone |
| WO2016060820A1 (en) | 2014-10-14 | 2016-04-21 | Exxonmobil Research And Engineering Company | Removal of occluded alkali metal cations from mse-framework type molecular sieves |
| US10183285B2 (en) * | 2015-01-30 | 2019-01-22 | Exxonmobil Research And Engineering Company | Process for preparing a molecular sieve |
| JP6430303B2 (ja) * | 2015-03-16 | 2018-11-28 | 国立大学法人横浜国立大学 | Afx型ゼオライトの製法 |
| US10214486B2 (en) | 2015-06-30 | 2019-02-26 | Exxonmobil Chemical Patents Inc. | Process and reactor system for oxidizing cycloalkylbenzene |
| WO2017003644A1 (en) | 2015-06-30 | 2017-01-05 | Exxonmobil Chemical Patents Inc. | Gas distribution in oxidation reactions |
| JP6621281B2 (ja) * | 2015-09-18 | 2019-12-18 | 国立大学法人横浜国立大学 | チタノシリケートの製造方法 |
| WO2017052856A1 (en) | 2015-09-25 | 2017-03-30 | Exxonmobil Chemical Patents Inc. | Catalyst and its use in dehydrocyclization processes |
| US10202318B2 (en) | 2015-09-25 | 2019-02-12 | Exxonmobil Chemical Patents Inc. | Catalyst and its use in hydrocarbon conversion process |
| CN109310999A (zh) | 2016-04-25 | 2019-02-05 | 埃克森美孚化学专利公司 | 催化芳构化 |
| JP6734692B2 (ja) | 2016-04-26 | 2020-08-05 | 三井金属鉱業株式会社 | Mse型ゼオライトの製造方法 |
| KR20190009806A (ko) | 2016-05-24 | 2019-01-29 | 엑손모빌 케미칼 패턴츠 인코포레이티드 | 촉매 금속을 포함하는 합성 제올라이트 |
| WO2018071184A1 (en) | 2016-10-10 | 2018-04-19 | Exxonmobil Chemical Patents Inc. | Heavy aromatics to btx conversion process and catalyst compositions used |
| WO2018071185A1 (en) | 2016-10-10 | 2018-04-19 | Exxonmobil Chemical Patents Inc. | Heavy aromatics to btx conversion process and dual bed catalyst systems used |
| KR102524765B1 (ko) | 2017-02-16 | 2023-04-25 | 엑손모빌 테크놀로지 앤드 엔지니어링 컴퍼니 | 경질 파라핀 전환을 위한 고정층 방사류 반응기 |
| WO2018183009A1 (en) | 2017-03-29 | 2018-10-04 | Exxonmobil Chemical Patents Inc. | Catalyst compositions and their use in aromatic alkylation processes |
| WO2018183012A1 (en) | 2017-03-29 | 2018-10-04 | Exxonmobil Chemical Patents Inc. | Methods for removing impurities from a hydrocarbon stream and their use in aromatic alkylation processes |
| WO2019036119A1 (en) | 2017-08-18 | 2019-02-21 | Exxonmobil Chemical Patents Inc. | METHOD FOR CYCLOHEXYLBENZENE HYDROPEROXIDATION AND METHOD FOR OPERATING OXIDATION REACTOR |
| US10427993B2 (en) | 2017-08-31 | 2019-10-01 | Uop Llc | Process for recovering benzene and fuel gas in an aromatics complex |
| US10793493B2 (en) | 2017-08-31 | 2020-10-06 | Uop Llc | Process for recovering benzene and fuel gas in an aromatics complex |
| CN112888658B (zh) | 2018-08-24 | 2024-04-16 | 罗地亚经营管理公司 | 微孔铝钛硅酸盐结晶沸石,其制备方法和应用 |
| WO2020080165A1 (ja) | 2018-10-17 | 2020-04-23 | 国立大学法人大阪大学 | 化合物、及びその製造方法 |
| JP7449948B2 (ja) | 2019-01-16 | 2024-03-14 | エクソンモービル テクノロジー アンド エンジニアリング カンパニー | ゼオライトの中の焼結抵抗性金属種 |
| US10703986B1 (en) | 2019-01-30 | 2020-07-07 | Exxonmobil Research And Engineering Company | Selective oxidation using encapsulated catalytic metal |
| EP3941621A1 (de) | 2019-03-18 | 2022-01-26 | ExxonMobil Research and Engineering Company | Mesoporöse katalysatorverbindungen und ihre verwendungen |
| CN116323872A (zh) * | 2020-11-04 | 2023-06-23 | 巴斯夫公司 | 包括磷稳定的mse框架型沸石的催化剂、其制备和其在流化催化应用中的用途 |
| US20240190881A1 (en) * | 2021-03-25 | 2024-06-13 | Mitsui Mining & Smelting Co., Ltd. | Pyrrolidine derivative, production method therefor, mse-type zeolite, and production method therefor |
| WO2025230715A1 (en) | 2024-05-03 | 2025-11-06 | ExxonMobil Technology and Engineering Company | Catalyst for alkane dehydrogenation |
Family Cites Families (15)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US2882243A (en) * | 1953-12-24 | 1959-04-14 | Union Carbide Corp | Molecular sieve adsorbents |
| JPS6035284B2 (ja) * | 1981-01-27 | 1985-08-14 | 東レ株式会社 | ペンタシル型ゼオライトの製造法 |
| US4419220A (en) * | 1982-05-18 | 1983-12-06 | Mobil Oil Corporation | Catalytic dewaxing process |
| US4828679A (en) * | 1984-03-12 | 1989-05-09 | Mobil Oil Corporation | Octane improvement with large size ZSM-5 catalytic cracking |
| US4919788A (en) * | 1984-12-21 | 1990-04-24 | Mobil Oil Corporation | Lubricant production process |
| US4740292A (en) * | 1985-09-12 | 1988-04-26 | Mobil Oil Corporation | Catalytic cracking with a mixture of faujasite-type zeolite and zeolite beta |
| US4983276A (en) * | 1988-10-06 | 1991-01-08 | Mobil Oil Corp. | Octane improvement in catalytic cracking and cracking catalyst composition therefor |
| WO1990004567A1 (en) * | 1988-10-20 | 1990-05-03 | Chevron Research Company | New zeolite ssz-31 |
| US5236575A (en) * | 1991-06-19 | 1993-08-17 | Mobil Oil Corp. | Synthetic porous crystalline mcm-49, its synthesis and use |
| US5266541A (en) * | 1991-12-20 | 1993-11-30 | Mobil Oil Corp. | Crystalline oxide material |
| US5225179A (en) * | 1992-08-27 | 1993-07-06 | Chevron Research And Technology Company | Method of making molecular sieves |
| US5362697A (en) * | 1993-04-26 | 1994-11-08 | Mobil Oil Corp. | Synthetic layered MCM-56, its synthesis and use |
| US5501848A (en) * | 1994-02-08 | 1996-03-26 | Chevron U.S.A. Inc. | Method for preparing crystalline aluminophosphate materials using azapolycyclic templating agents |
| US5591421A (en) * | 1994-07-11 | 1997-01-07 | Chevron U.S.A. Inc. | Zeolite SSZ-41 |
| US5472594A (en) * | 1994-07-18 | 1995-12-05 | Texaco Inc. | FCC process for producing enhanced yields of C4 /C5 olefins |
-
1999
- 1999-01-21 US US09/234,544 patent/US6049018A/en not_active Expired - Lifetime
-
2000
- 2000-01-21 AU AU26223/00A patent/AU768270B2/en not_active Expired
- 2000-01-21 EP EP00904469A patent/EP1137739B1/de not_active Expired - Lifetime
- 2000-01-21 WO PCT/US2000/001458 patent/WO2000043335A1/en not_active Ceased
- 2000-01-21 RU RU2001122712/15A patent/RU2223912C2/ru active
- 2000-01-21 CA CA002360030A patent/CA2360030C/en not_active Expired - Lifetime
- 2000-01-21 ES ES00904471T patent/ES2211504T3/es not_active Expired - Lifetime
- 2000-01-21 AR ARP000100282A patent/AR022332A1/es active IP Right Grant
- 2000-01-21 AU AU25132/00A patent/AU2513200A/en not_active Abandoned
- 2000-01-21 JP JP2000594736A patent/JP4689046B2/ja not_active Expired - Lifetime
- 2000-01-21 CA CA2333986A patent/CA2333986C/en not_active Expired - Fee Related
- 2000-01-21 AU AU26221/00A patent/AU764939B2/en not_active Ceased
- 2000-01-21 CN CNB008039577A patent/CN100473609C/zh not_active Expired - Lifetime
- 2000-01-21 DE DE60009030T patent/DE60009030T2/de not_active Expired - Lifetime
- 2000-01-21 AT AT00904471T patent/ATE255536T1/de not_active IP Right Cessation
- 2000-01-21 CN CN2009100066281A patent/CN101544649B/zh not_active Expired - Lifetime
- 2000-01-21 WO PCT/US2000/001459 patent/WO2000043316A1/en not_active Ceased
- 2000-01-21 WO PCT/US2000/001448 patent/WO2000043466A1/en not_active Ceased
- 2000-01-21 KR KR1020017009204A patent/KR100815658B1/ko not_active Expired - Lifetime
- 2000-01-21 DE DE60006964T patent/DE60006964T2/de not_active Expired - Lifetime
- 2000-01-21 EP EP00904471A patent/EP1194375B1/de not_active Expired - Lifetime
- 2000-02-01 TW TW089100957A patent/TW527414B/zh not_active IP Right Cessation
-
2001
- 2001-07-20 ZA ZA200105984A patent/ZA200105984B/en unknown
- 2001-07-20 NO NO20013602A patent/NO20013602L/no not_active Application Discontinuation
Also Published As
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| DE60009030T2 (de) | Katalytisches crack-verfahren mit mcm-68-katalysator | |
| DE69819989T2 (de) | Zusammensetzung, die Molekularsiebe vom Pentasil-Typ enthält, sowie ihre Herstellung und Verwendung | |
| EP0034727B1 (de) | Kristalline isotaktische Zeolithe, Verfahren zur Herstellung derselben sowie deren Verwendung als Katalysatoren | |
| DE4310792C2 (de) | Zeolithisches Material, dessen Verwendung sowie Verfahren zum Herstellen eines solchen zeolithhaltigen Materials | |
| DE69904027T2 (de) | Erhöhung der wirkung eines zeolitischen katalysators mit aluminiumphosphat und phosphor | |
| DE60130758T2 (de) | Zeolith ssz-53 | |
| EP0007081B1 (de) | Verfahren zur Herstellung von stickstoffhaltigen kristallinen Metallsilikaten mit Zeolithstruktur, nach dem Verfahren hergestellte Metallsilikate sowie deren Verwendung als Katalysatoren | |
| DE2560441C2 (de) | Verfahren zur Umwandlung von Paraffinen zu einem Olefine enthaltenden Produkt | |
| DE60127852T2 (de) | Kristalline zeolithische aluminosilikatzusammentsetzung: uzm-4 und diese verwendende verfahren | |
| DE69006266T2 (de) | Krackkatalysator und Verfahren. | |
| DE69432667T9 (de) | Modifizierte mikrokugelformige fcc katalysatoren | |
| DD298379A5 (de) | Verfahren zur herstellung eines ethers | |
| DE1567714B2 (de) | Kristalliner Aluminosilicatzeolith | |
| DE3119160A1 (de) | Caesiumhaltiger zeolith und verfahren zu dessen herstellung | |
| DE69021780T2 (de) | Verfahren zur Herstellung eines Mischvorrats für Benzin mit hoher Oktanzahl. | |
| DE2748278A1 (de) | Zeolithartiges material, verfahren zur herstellung desselben und dessen verwendung | |
| DE69312162T2 (de) | Verfahren zur Herstellung von Zeolithen mit vermindertem Oberflächensäuregehalt | |
| DE69903562T2 (de) | Delaminierter mikroporöser feststoff | |
| DE69801932T2 (de) | CON-typ phosphorhaltiger Zeolith und dessen Anwendung im katalytischen Crackverfahren | |
| DE2749024C2 (de) | Kristalline Aluminosilicat-Zeolithe, Verfahren zu ihrer Herstellung und ihre Verwendung | |
| DE69805016T2 (de) | Katalytisches Krackverfahren von Kohlenwasserstoffeinsätzen mit einem Katalysator, der einen IM-5 Zeolit enthäle, der eventuel entaluminiert kann sein | |
| DE4114532B4 (de) | Einen β-Zeolith, einen Y-Zeolith und eine Matrix umfassender Crackkatalysator für Kohlenwasserstoffchargen | |
| DE2321399A1 (de) | Verfahren zur dampfphasenalkylierung in gegenwart eines kristallinen aluminosilikatkatalysators | |
| DE69323325T2 (de) | Krackverfahren und dazu hergestellter zsm-5-katalysator | |
| DE68928078T2 (de) | Neues zeolith ssz-31 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 8364 | No opposition during term of opposition |