-
Querverweis
auf verwandte Anmeldungen
-
Diese
Anmeldung beansprucht die Priorität der vorläufigen US-Anmeldung Nr. 60/334
293, eingereicht am 30. November 2001.
-
Hintergrund
-
Diese
Erfindung betrifft substituierte 1,2,4-Triazolo[1,5-c]pyrimidin-Adenosin-A2a-Rezeptorantagonisten, die Verwendung der
Verbindungen zur Behandlung von Erkrankungen des zentralen Nervensystems,
insbesondere Morbus Parkinson, und pharmazeutische Zusammensetzungen,
die die Verbindungen enthalten.
-
Adenosin
ist als endogener Modulator einer Anzahl physiologischer Funktionen
bekannt. Auf der Ebene des cardiovaskulären Systems ist Adenosin ein
starker Vasodilator und ein Herzdepressor. Im zentralen Nervensystem
induziert Adenosin sedierende, anxiolytische und antiepileptische
Wirkungen. Im respiratorischen System induziert Adenosin Bronchokonstriktion.
Auf der Ebene der Nieren übt
es eine zweiphasige Wirkung aus, wobei es in niedrigen Konzentrationen
zu Vasokonstriktion führt
und in hohen Dosen zu Vasodilation führt. Adenosin wirkt als Lipolyseinhibitor
auf Fettzellen und als Antiaggregationsmittel für Thrombozyten.
-
Die
Wirkung von Adenosin wird durch die Wechselwirkung mit unterschiedlichen
membranspezifischen Rezeptoren vermittelt, die zu der mit G-Proteinen
gekoppelten Familie von Rezeptoren gehören. Biochemische und pharmakologische
Studien haben zusammen mit Fortschritten in der Molekularbiologie
die Identifizierung von mindestens vier Subtypen von Adenosinrezeptoren
ermöglicht:
A1, A2a, A2b und A3. A1 und A3 haben hohe
Affinität, sie
inhibieren die Aktivität
des Enzyms Adenylatcyclase, und A2a und
A2b haben niedrige Affinität, sie stimulieren
die Aktivität
desselben Enzyms. Es sind auch Analoga von Adenosin identifiziert worden,
die als Antagonisten mit den A1-, A2a-, A2b- und A3-Rezeptoren wirken können.
-
Selektive
Antagonisten für
den A2a-Rezeptor sind wegen ihrer geringeren
Nebenwirkungen von pharmakologischem Interesse. Im zentralen Nervensystem
können
A2a-Antagonisten antidepressive Eigenschaften haben
und kognitive Funktionen stimulieren.
-
Außerdem haben
die Daten gezeigt, dass A2a-Rezeptoren in
hoher Dichte in den Basalganglien vorhanden sind, die bekanntermaßen zur
Steuerung der Bewegung wichtig sind. A2a-Antagonisten
können
somit motorische Beeinträchtigung
infolge von neurodegenerativen Erkrankungen, wie Morbus Parkinson,
senile Demenz, wie bei Morbus Alzheimer, und Psychosen organischer
Herkunft verbessern.
-
Es
ist gefunden worden, dass einige mit Xanthin verwandte Verbindungen
selektive A1-Rezeptorantagonisten sind,
und es ist gefunden worden, dass Xanthin- und Nicht-Xanthinverbindungen
hohe A2a-Affinität mit unterschiedlichen Selektivitätsgraden
zu A2a gegenüber A1 haben.
Triazolopyrimidin-Adenosin-A2a-Rezeptorantagonisten
mit unterschiedlicher Substitution an der 7-Position sind bereits offenbart worden,
beispielsweise in WO-A-95/01356
US-A-5 565 460; WO-A-97/05138 und WO-A-98/52568. Pyrazolosubstituierte
Triazolopyrimidin-Adenosin-A2a-Rezeptorantagonisten
sind in US-A-09/207 143, eingereicht am 24. Mai 2001, EP-A-1 116
722 und EP-A-0 976 753 offenbart.
-
Zusammenfassung
der Erfindung
-
Die
Erfindung betrifft Verbindungen mit der Strukturformel I:
oder ein pharmazeutisch annehmbares
Salz oder Solvat davon, worin
R ausgewählt ist aus der Gruppe bestehend
aus R
4-Heteroaryl, R
5-Phenyl,
(C
4-C
6)-Cycloalkenyl, -C(=CH
2)CH
3, -C≡C-CH
3,
-CH=C(CH
3)
2,
und -CH=CH-CH
3;
R
2 -W-X ist;
R
3 ausgewählt ist
aus der Gruppe bestehend aus H, Halogen, Alkyl, Trifluormethyl,
Alkoxy, Alkoxyalkyl, Hydroxyalkyl, Alkylamino, Alkylaminoalkyl,
Dialkylamino, Dialkylaminoalkyl, Aminoalkyl, Aryl, Heteroaryl und
CN;
R
4 1 bis 3 Substituenten ist, die
gleich oder verschieden sein können
und unabhängig
ausgewählt
sind aus der Gruppe bestehend aus Wasserstoff, (C
1-C
6)-Alkyl, -CF
3, Halogen,
-NO
2, -NR
15R
16, (C
1-C
6)-Alkoxy, (C
1-C
6)-Alkylthio, (C
1-C
6)-Alkylsulfinyl, (C
1-C
6)-Alkylsulfonyl, -COOR
17 und
-C(O)NR
6R
7;
R
5 1 bis 5 Substituenten ist, die gleich oder
verschieden sein können
und unabhängig
ausgewählt
sind aus der Gruppe bestehend aus Wasserstoff, Halogen, (C
1-C
6)-Alkyl, Hydroxy,
(C
1-C
6)-Alkoxy, -CN, -NH
2,
(C
1-C
6)-Alkylamino,
Di-((C
1-C
6)alkyl)amino,
-CF
3, -OCF
3, -S(O)
0-2(C
1-C
6)-Alkyl
und -CH
2-SO
2-Phenyl;
R
6 und R
7, die gleich
oder verschieden sein können,
jeweils unabhängig
ausgewählt
sind aus der Gruppe bestehend aus Wasserstoff und (C
1-C
6)-Alkyl;
R
8 1
bis 5 Substituenten ist, die gleich oder verschieden sein können und
unabhängig
ausgewählt
sind aus der Gruppe bestehend aus Wasserstoff, Halogen, (C
1-C
6)-Alkyl, Hydroxy,
C
1-C
6-Alkoxy, -CN, Amino,
Di-((C
1-C
6)alkyl)amino,
-CF
3, -OCF
3, Acetyl,
-NO
2, Hydroxy(C
1-C
6)alkoxy, (C
1-C
6)-Alkoxy, (C
1-C
6)alkoxy, Di-((C
1-C
6)-alkoxy)(C
1-C
6)alkoxy, (C
1-C
6)-Alkoxy, (C
1-C
6)alkoxy-(C
1-C
6)alkoxy, Carboxy
(C
1-C
6)-alkoxy,
(C
1-C
6)-Alkoxycarbonyl(C
1-C
6)alkoxy, (C
3-C
6)Cycloalkyl(C
1-C
6)alkoxy, Di-((C
1-C
6)alkyl)amino(C
1-C
6)alkoxy, Morpholinyl, (C
1-C
6)-Alkyl-SO
2-, (C
1-C
6)-Alkyl-SO
2(C
1-C
6)alkoxy,
Tetrahydropyranyloxy, (C
1-C
6)-Alkylcarbonyl(C
1-C
6)alkoxy, (C
1-C
6)-Alkoxycarbonyl,
(C
1-C
6)-Alkylcarbonyloxy(C
1-C
6)alkoxy, -SO
2NH
2, Phenoxy,
-O-CH
2-P(O)(OR
6)
2- und -P(O)(OR
6)
2; oder
benachbarte
R
8-Substituenten zusammen -OCH
2-O-,
-O-CH
2CH
2-O-, -O-CF
2-O- oder -O-CF
2CF
2-O- sind und mit den Kohlenstoffatomen,
an die sie gebunden sind, einen Ring bilden;
R
9 ausgewählt ist
aus der Gruppe bestehend aus (C
1-C
6)-Alkyl, R
8-Aryl-,
R
8-Aryl(C
1-C
6)alkyl, Thienyl, Pyridyl, (C
3-C
6)Cycloalkyl, (C
1-C
6)-Alkyl-OC(O)-NH-(C
1-C
6)alkyl-, Di-((C
1-C
6)alkyl)aminomethyl, Cycloheteroalkyl(C
1-C
6)alkyl, Aryloxy(C
1-C
6)alkyl, Alkoxy(C
1-C
6)alkyl und
R
10 1
bis 2 Substituenten ist, die gleich oder verschieden sein können und
unabhängig
ausgewählt
sind aus der Gruppe bestehend aus Wasserstoff, (C
1-C
6)-Alkyl, R
5-Aryl
und R
4-Heteroaryl, oder zwei R
10-Substituenten an
dem gleichen Kohlenstoffatom =O bilden können;
R
11 Wasserstoff
oder (C
1-C
6)-Alkyl;
-C(O)-Alkyl ist; oder R
17 und R
11 zusammen
-(CH
2)
p-A-(CH
2)
q sind, worin p und
q jeweils unabhängig
2 oder 3 sind und A ausgewählt
ist aus der Gruppe bestehend aus einer Bindung, -CH
2-,
-S- oder -O- und mit dem Stickstoff, an den sie gebunden sind, einen
Ring bilden;
R
12 1 bis 2 Substituenten
ist, die gleich oder verschieden sind und unabhängig ausgewählt sind aus der Gruppe bestehend
aus Wasserstoff, (C
1-C
6)-Alkyl,
Hydroxy, (C
1-C
6)-Alkoxy,
Halogen und -CF
3;
R
13 ausgewählt ist
aus der Gruppe bestehend aus H, (C
1-C
6)-Alkyl,
Phenyl, Benzyl, (C
2-C
6)-Alkenyl, (C
1-C
6)-Alkoxy(C
1-C
6)alkyl, Di-((C
1-C
6)alkyl)amino(C
1-C
6)alkyl, Pyrrolidinyl
(C
1-C
6)alkyl und
Piperidino(C
1-C
6)alkyl;
R
14 ausgewählt
ist aus der Gruppe bestehend aus H, Halogen, (C
1-C
6)-Alkyl oder (C
1-C
6)-Alkoxy;
R
15 ausgewählt ist
aus der Gruppe bestehend aus H und (C
1-C
6)-Alkyl;
R
16 ausgewählt
ist aus der Gruppe bestehend aus H, (C
1-C
6)-Alkyl-C(O)-
und (C
1-C
6)-Alkyl-SO
2-;
R
17 ausgewählt ist
aus der Gruppe bestehend aus (C
1-C
6)-Alkyl, (C
1-C
6)-Hydroxyalkyl, (C
3-C
6)-Cycloalkyl, (C
1-C
6)-Alkoxy(C
1-C
6)alkoxy,
(C
1-C
6)-Alkoxy,
(C
1-C
6)-Alkoxy(C
1-C
6)alkyl, Allyl,
Propargyl, R
8-Heteroaryl-, R
8-Aryl-
und R
8-Aryl(C
1-C
6)alkyl-;
R
18 ausgewählt ist
aus der Gruppe bestehend aus einer Bindung, -CH
2,
-CH(OH)-, -CH(CH
3)-, -C(CH
3)
n-, -(CH
2)
n- und -O(CH
2)
n-;
Q und Q
1 gleich
oder verschieden sein können
und jeweils unabhängig
ausgewählt
sind aus der Gruppe bestehend aus
m und
n jeweils unabhängig
1 bis 3 sind;
p und q jeweils unabhängig 0 bis 2 sind;
s 0
bis 4 ist;
W Aryl oder Heteroaryl mit 1 bis 3 Heteroatomen
ist, die gleich oder verschieden sein können und unabhängig ausgewählt sind
aus der Gruppe bestehend aus N, O und S, und wobei das Aryl oder
Heteroaryl gegebenenfalls mit 1 bis 3 Substituenten substituiert
ist, die gleich oder verschieden sein können und unabhängig ausgewählt sind
aus der Gruppe bestehend aus Alkyl, Aryl, Alkylcycloalkyl, Halogen,
Hydroxy, Hydroxyalkyl, Alkoxy, Alkylalkoxy, Alkoxyalkoxy, -NR
6R
7, (C
2-C
6)-Alken und -CN;
X ausgewählt ist
aus der Gruppe bestehend aus H, NH
2, -N(R
6)(CH
2)
s-Aryl,
-N(R
6)(CH
2)
s-Heteroaryl, -N(R
6)(CH
2)
m+1-OH und -N(CH
3)
2 oder
X -R
18-Y-Z ist;
Y ausgewählt ist aus der Gruppe bestehend
aus -N(R
6)CH
2CH
2N(R
7)-, -N(R
6)(CH
2)
n-Aryl,
-OCH
2CH
2N(R
6), -O-, -S-, -CH
2S-,
-(CH
2)
2-3-N(R
6)-, R
8-zweiwertigem
Heteroaryl,
und
Z
ausgewählt
ist aus der Gruppe bestehend aus H, Alkyl, Alkoxyalkyl, R
8-Aryl-, R
8-Aryl
(C
1-C
6)alkyl-, R
8-Heteroaryl-, R
8-bicyclischem Alkyl-,
Aminoalkyl, Alkylamino, NH
2, -N-(R
6)(CH
2)
s-Aryl, -N(R
6)(CH
2)
s-Heteroaryl, -N(R
6)C(O)OR
17, Alkylcyclohe teroalkyl,
Cycloheteroalkyl, Cycloheteroalkylalkyl, Alkoxycycloheteroalkyl,
Heteroaryl; R
8-benzokondensiertem Heteroaryl-,
Diphenylmethyl und R
9-C(O)-; oder
wenn
ist,
Z auch -OH, R
9-SO
2-, R
17-N(R
11)(CH
2)
S-C(O)-, R
17-OC(O)-, R
17-O(CH
2)
nC(O)-; benzokondensiertes Heteroaryl(CH
2)
nC(O)-, benzokondensiertes
Heteroaryl (CH
2)
n-
oder R
17-N(R
11)-C(S)-
sein kann; oder
wenn
ist,
Z auch R
17R
11N-, Phenylamino
oder Pyridylamino sein kann; oder
Z und Y zusammengenommen
ausgewählt
sind aus der Gruppe bestehend aus:
worin,
wenn nicht anders definiert ist,
"Alkyl" (einschließlich der Alkylanteile von
Alkoxy, Alkylamino und Dialkylamino usw.) eine aliphatische Kohlenwasserstoffgruppe
bedeutet, die geradkettig oder verzweigt sein kann und 1 bis 20
Kohlenstoffatome enthält;
"Halo" Fluor-, Chlor-,
Brom- oder Iodgruppen bedeutet;
"Halogen" Fluor, Chlor, Brom oder Iod bedeutet;
"Alkoxy" eine Alkyl-O-Gruppe
bedeutet, in der die Alkylgruppe wie zuvor beschrieben ist;
"Alkenyl" eine aliphatische
Kohlenwasserstoffgruppe bedeutet, die mindestens eine Kohlenstoff-Kohlenstoff-Doppelbindung
enthält
und geradkettig oder verzweigt sein kann und 2 bis 15 Kohlenstoffatome
enthält;
"Alkanoyl" ein an ein Carbonyl
gebundenes Alkyl ist, wobei Alkyl die gleiche Bedeutung wie oben
definiert hat;
"Cycloalkyl" ein nicht-aromatisches,
mono- oder multicyclisches, kondensiertes Ringsystem bedeutet, das
3 bis 10 Ringkohlenstoffatome enthält;
"Cycloheteroalkyl" ein nicht-aromatisches, mono- oder
multicyclisches, kondensiertes Ringsystem bedeutet, das 3 bis 10
Ringkohlenstoffatome enthält,
wobei das Cycloheteroalkyl 1 oder 2 Heteroatome unabhängig ausgewählt aus
O, S oder N aufweist, wobei das Heteroatom/die Heteroatome eine
carbocyclische Ringstruktur unterbricht bzw. unterbrechen, mit der
Maßgabe,
dass die Ringe keine benachbarten Sauerstoff- und/oder Schwefelatome enthalten;
"Aryl" ein aromatisches
monocyclisches oder multicyclisches Ringsystem bedeutet, das 6 bis
14 Ringkohlenstoffatome enthält;
"Heteroaryl" cyclische aromatische
Gruppen mit 5 oder 6 Ringatomen oder bicyclische Gruppen mit 11
bis 12 Ringatomen mit einem oder zwei Heteroatomen bedeutet, die
unabhängig
ausgewählt
sind aus O, S oder N, wobei das Heteroatom/die Heteroatome eine
carbocyclische Ringstruktur unterbricht/unterbrechen und eine ausreichende
Anzahl delokalisierter n-Elektronen hat/haben, um aromatischen Charakter
zu liefern, mit der Maßgabe,
dass die Ringe keine benachbarten Sauerstoff- und/oder Schwefelatome enthalten.
-
Ein
weiterer Aspekt der Erfindung betrifft eine pharmazeutische Zusammensetzung,
die eine oder mehrere Verbindungen der Formel I und einen oder mehrere
pharmazeutisch annehmbare Träger
enthält.
-
Ein
weiterer Aspekt der Erfindung betrifft eine pharmazeutische Zusammensetzung,
die eine oder mehrere Verbindungen der Formel I und ein oder mehrere
Mittel in einem oder mehreren pharmazeutisch annehmbaren Träger(n) enthält, die
bekanntermaßen
zur Behandlung von Morbus Parkinson brauchbar sind.
-
Ein
weiterer Aspekt der Erfindung betrifft die Verwendung von einer
oder mehreren Verbindungen der Formel I zur Herstellung einer pharmazeutischen
Zusammensetzung zur Behandlung des Schlaganfalls oder von Erkrankungen
des zentralen Nervensystems. In diesem Aspekt der Erfindung schließen die
Erkrankungen des zentralen Nervensystems kognitive Erkrankungen
oder neurodegenerative Erkrankungen ein, wie Morbus Parkinson, senile
Demenz oder Psychosen organischer Herkunft. Die Menge der Verbindung,
die dem Patienten verabreicht wird, ist vorzugsweise eine therapeutisch
wirksame Menge.
-
Ein
weiterer Aspekt der Erfindung betrifft die Verwendung einer Verbindung
der Formel I zur Herstellung einer pharmazeutischen Zusammensetzung
zur Behandlung von Morbus Parkinson mit einer Kombination von einer
oder mehreren Verbindungen der Formel I und einem oder mehreren
Mitteln, die bekanntermaßen zur
Behandlung von Morbus Parkinson brauchbar sind, beispielsweise Dopamin,
einem dopaminergen Agonisten, einem Monoaminoxidase-Hemmer Typ B
(MAO-B), einem DOPA-Decarboxylaseinhibitor
(DCI) oder einem Catechol-O-methyltransferase-(COMT)-Hemmer.
Gemäß diesem
Aspekt der Erfindung können
eine oder mehrere Verbindungen der Formel I und ein oder mehrere
andere Anti-Parkinson-Mittel gleichzeitig oder sequentiell in getrennten
Dosierformen verabreicht werden.
-
Ein
weiterer Aspekt der Erfindung betrifft einen Kit, der in separaten
Behältern
in einer einzigen Verpackung pharmazeutische Zusammensetzungen zur
Verwendung in Kombination zur Behandlung von Morbus Parkinson enthält, wobei
ein Behälter
eine pharmazeutische Zusammensetzung enthält, die eine oder mehrere Verbindungen
der Formel B in einem oder mehreren pharmazeutisch annehmbaren Trägern enthält, und
wobei in separaten Behältern
eine oder mehrere pharmazeutische Zusammensetzungen enthalten sind,
die jeweils ein oder mehrere Mittel, die zur Behandlung von Morbus
Parkinson brauchbar sind, in einem oder mehreren pharmazeutisch
annehmbaren Trägern
enthalten.
-
Detaillierte
Beschreibung der Erfindung
-
Die
Erfindung betrifft Verbindungen mit der Strukturformel I:
oder ein pharmazeutisch annehmbares
Salz oder Solvat der Verbindung, worin R, R
2 und
R
3 wie oben definiert sind.
-
Wenn
nicht anders gesagt, gelten die folgenden Definitionen in der vorliegenden
Beschreibung und den Ansprüchen.
Diese Definitionen gelten unabhängig
davon, ob ein Begriff als solcher oder in Kombination mit anderen
Begriffen verwendet wird. Die Definition von "Alkyl" gilt somit für "Alkyl" sowie die "Alkyl"-Anteile von "Alkoxy", "Halogenalkyl" usw.
-
Wenn
irgendeine Variable (z. B. R2) mehr als
ein Mal in irgendeinem Bestandteil erscheint, ist seine Definition
bei jedem Vorkommen unabhängig
von seiner Definition in jedem anderen Vorkommen. Kombinationen
von Substituenten und/oder Variablen sind auch nur dann zulässig, wenn
solche Kombinationen zu stabilen Verbindungen führen.
-
"Alkyl" (einschließlich der
Alkylanteile von Alkoxy, Alkylamino und Dialkylamino) bedeutet eine
aliphatische Kohlenwasserstoffgruppe, die geradkettig oder verzweigt
sein kann und 1 bis etwa 20 Kohlenstoffatome in der Kette enthält. Bevorzugte
Alkylgruppen enthalten 1 bis etwa 12 Kohlenstoffatome in der Kette.
Besonders bevorzugte Alkylgruppen enthalten 1 bis etwa 6 Kohlenstoffatome
in der Kette. Verzweigtes Alkyl bedeutet, dass eine oder mehrere
niedere Alkylgruppen, wie Methyl, Ethyl oder Propyl, an eine lineare
Alkylkette gebunden sind. "Niederes
Alkyl" bedeutet
eine Gruppe mit etwa 1 bis etwa 6 Kohlenstoffatomen in der Kette,
die geradkettig oder verzweigt sein kann. Bevorzugte Alkylgruppen
in der vorliegenden Erfindung sind niedere Alkylgruppen. Nicht-einschränkende Bei spiele
für geeignete
Alkylgruppen schließen
Methyl, Ethyl, n-Propyl,
Isopropyl, n-Butyl, t-Butyl, n-Pentyl, Heptyl, Nonyl, Decyl, Trifluormethyl
und Cyclopropylmethyl ein. Alkylgruppen können mit einem oder mehreren
Substituenten substituiert sein, die gleich oder verschieden sind,
und sind ausgewählt
aus der Gruppe bestehend aus Alkyl, Aryl, Heteroaryl, Hydroxy, Alkoxy,
Halogen, Nitro, Cyano und Cycloalkyl.
-
"Halo" steht für Fluor-,
Chlor-, Brom- oder Iodgruppen. Bevorzugt sind Fluor, Chlor oder
Brom, und besonders bevorzugt sind Fluor und Chlor.
-
"Halogen" bedeutet Fluor,
Chlor, Brom oder Iod. Bevorzugt sind Fluor, Chlor oder Brom, und
besonders bevorzugt sind Fluor und Chlor.
-
"Alkoxy" bedeutet eine Alkyl-O-Gruppe,
in der die Alkylgruppe wie zuvor beschrieben ist. Nicht-einschränkende Beispiele
für geeignete
Alkoxygruppen sind Methoxy, Ethoxy, n-Propoxy und Isopropoxy. Die Alkylgruppe
ist über
den Ethersauerstoff an eine benachbarte Einheit gebunden.
-
Alkoxyalkyl
ist eine Einheit, die ein über
ein Alkyl an die Hauptgruppe gebundenes Alkoxy enthält.
-
"Alkoxycarbonyl" bedeutet eine Alkyl-O-C(O)-Gruppe.
Nicht-einschränkende
Beispiele für
geeignete Alkoxycarbonylgruppen schließen Methoxycarbonyl und Ethoxycarbonyl
ein. Das Alkoxy ist über
das Carbonyl an eine benachbarte Einheit gebunden.
-
"Alkylsulfonyl" bedeutet eine Alkyl-S(O)2-Gruppe. Bevorzugte Gruppen sind jene, in
denen die Alkylgruppe niederes Alkyl ist. Das Alkyl ist über das
Sulfonyl an eine benachbarte Einheit gebunden.
-
"Alkylsulfinyl" bedeutet eine Alkyl-S(O)-Gruppe.
Bevorzugte Gruppen sind jene, in denen die Alkylgruppe niederes
Al kyl ist. Das Alkyl ist über
das Sulfinyl an eine benachbarte Einheit gebunden.
-
"Alkenyl" bedeutet eine aliphatische
Kohlenwasserstoffgruppe, die mindestens eine Kohlenstoff-Kohlenstoff-Doppelbindung
enthält
und geradkettig oder verzweigt sein kann und 2 bis 15 Kohlenstoffatome
in der Kette enthält.
Bevorzugte Alkenylgruppen haben etwa 2 bis etwa 12 Kohlenstoffatome
in der Kette und insbesondere 2 bis 6 Kohlenstoffatome in der Kette.
Verzweigt bedeutet, dass eine oder mehrere niedere Alkylgruppen,
wie Methyl, Ethyl oder Propyl, an eine lineare Alkenylkette gebunden
sind. "Niederes
Alkenyl" bedeutet
2 bis 6 Kohlenstoffatome in der Kette, die geradkettig oder verzweigt
sein kann. Nicht-einschränkende
Beispiele für
geeignete Alkenylgruppen schließen
Ethenyl, Propenyl, n-Butenyl, 3-Methyl-but-2-enyl und n-Pentenyl ein.
-
Alkanoyl
ist ein an ein Carbonyl gebundenes Alkyl, wobei Alkyl die gleiche
Bedeutung wie oben definiert hat.
-
Alkylen
bezieht sich auf eine zweiwertige Alkylgruppe, wobei es sich ebenfalls
auf gerade oder verzweigte Ketten bezieht.
-
"Ringsystemsubstituent" bedeutet einen an
ein aromatisches oder nicht-aromatisches Ringsystem gebundenen Substituenten,
der beispielsweise einen verfügbaren
Wasserstoff an dem Ringsystem ersetzt. Ringsystemsubstituenten können gleich
oder verschieden sein, wobei jeder unabhängig ausgewählt ist aus der Gruppe bestehend
aus Alkyl, Aryl, Heteroaryl, Aralkyl, Alkylamino, Arylamino, Alkylaryl,
Aralkenyl, Heteroaralkyl, Alkylheteroaryl, Heteroaralkenyl, Hydroxy,
Hydroxyalkyl, Alkoxy, Aryloxy, Aralkoxy, Aralkyloxy, Acyl, Aroyl, Halo,
Nitro, Cyano, Carboxy, Alkoxycarbonyl, Aryloxycarbonyl, Aralkoxycarbonyl,
Alkylsulfonyl, Arylsulfonyl, Heteroarylsulfonyl, Alkylsulfinyl,
Arylsulfinyl, Heteroarylsulfinyl, Alkylthio, Aryl thio, Heteroarylthio,
Aralkylthio, Heteroaralkylthio, Cycloalkyl, Cycloalkenyl, Y1Y2N-, Y1Y2N-Alkyl-, Y1Y2NC(O)- und Y1Y2NSO2-, wobei Y1 und Y2 gleich oder
verschieden sein können
und unabhängig
ausgewählt
sind aus der Gruppe bestehend aus Wasserstoff, Alkyl, Aryl und Aralkyl.
-
Der
Begriff "gegebenenfalls
substituiert" bedeutet
optionale Substitution mit den angegebenen Gruppen, Resten oder
Einheiten.
-
"Cycloalkyl" bedeutet ein nicht-aromatisches
mono- oder multicyclisches kondensiertes Ringsystem, das 3 bis 10
Ringkohlenstoffatome, vorzugsweise 3 bis 7 Ringkohlenstoffatome,
insbesondere 3 bis 6 Ringkohlenstoffatome enthält. Das Cycloalkyl kann gegebenenfalls
mit einer oder mehreren "Ringsystemsubstituenten" substituiert sein,
die gleich oder verschieden sein können, und sie sind wie oben
definiert. Nichteinschränkende
Beispiele für
geeignete monocyclische Cycloalkyle schließen Cyclopropyl, Cyclobutyl,
Cyclopentyl, Cyclohexyl und dergleichen ein. Zu nicht-einschränkenden
Beispielen für
geeignete multicyclische Cycloalkyle gehören 1-Decalinyl, Norbornenyl,
Adamantyl und dergleichen. Das Cycloalkyl kann gegebenenfalls mit einer
oder mehreren "Ringsystemsubstituenten" substituiert sein,
die gleich oder verschieden sein können, und sie sind wie oben
definiert.
-
"Cycloheteroalkyl" bedeutet ein nicht-aromatisches,
mono- oder multicyclisches,
kondensiertes Ringsystem, das 3 bis 10 Ringkohlenstoffatome, vorzugsweise
3 bis 7 Ringkohlenstoffatome, insbesondere 3 bis 6 Ringkohlenstoffatome
enthält,
wobei das Cycloheteroalkyl 1 oder 2 Heteroatome unabhängig ausgewählt aus O,
S oder N aufweist, wobei das Heteroatom/die Heteroatome eine carbocyclische
Ringstruktur unterbricht bzw. unterbrechen, mit der Maßgabe, dass
die Ringe keine benachbarten Sauerstoff- und/oder Schwefelatome enthalten.
Das Cyclohe teroalkyl kann gegebenenfalls mit einer oder mehreren "Ringsystemsubstituenten" substituiert sein,
die gleich oder verschieden sein können, und sie sind wie oben
definiert.
-
"Aryl" bedeutet ein aromatisches
monocyclisches oder multicyclisches Ringsystem, das 6 bis 14 Kohlenstoffatome,
vorzugsweise 6 bis 10 Kohlenstoffatome enthält. Die Arylgruppe kann gegebenenfalls
mit einem oder mehreren "Ringsystemsubstituenten" substituiert sein,
die gleich oder verschieden sein können und wie oben definiert
sind. Nicht-einschränkende
Beispiele für
geeignete Arylgruppen schließen
Phenyl und Naphthyl ein.
-
"Heteroaryl" bedeutet cyclische
aromatische Gruppen mit 5 oder 6 Ringatomen oder bicyclische Gruppen
mit 11 bis 12 Ringatomen mit einem oder zwei Heteroatomen, die unabhängig ausgewählt sind
aus O, S oder N, wobei das Heteroatom/die Heteroatome eine carbocyclische
Ringstruktur unterbricht/unterbrechen und eine ausreichende Anzahl
delokalisierter n-Elektronen
hat/haben, um aromatischen Charakter zu liefern, mit der Maßgabe, dass
die Ringe keine benachbarten Sauerstoff- und/oder Schwefelatome enthalten, Bevorzugte
Heteroaryle enthalten 5 bis 6 Ringatome. Das "Heteroaryl" kann gegebenenfalls mit einem oder
mehreren "Ringsystemsubstituenten" substituiert sein,
die gleich oder verschieden sein können und wie oben definiert
sind. Der Präfix
Aza, Oxa oder Thia vor der Heteroarylstammbezeichnung bedeutet,
dass mindestens ein Stickstoff-, Sauerstoff- beziehungsweise Schwefelatom
als Ringatom vorhanden ist. Stickstoffatome können ein N-Oxid bilden. Es
kommen alle Regioisomere in Frage, z. B. 2-Pyridyl, 3-Pyridyl und
4-Pyridyl. Typische 6-gliedrige
Heteroarylgruppen sind Pyridyl, Pyrimidinyl, Pyrazinyl, Pyridazinyl
und dergleichen sowie deren N-Oxide. Brauchbare fünfgliedrige
Heteroarylringe sind Furyl, Thienyl, Pyrrolyl, Thiazolyl, Isothiazolyl,
Imidazolyl, Pyrazolyl, Isoxazolyl und dergleichen. Brauchbare bicyclische
Gruppen schließen
benzokondensierte Ringsysteme ein, die von den oben genannten Heteroarylgruppen
abgeleitet sind, z. B. Chinolyl, Phthalazinyl, Chinazolinyl, Benzofuranyl,
Benzothienyl, Indolyl und dergleichen.
-
Zweiwertiges
Heteroaryl bedeutet einen Heteroarylring, der an zwei verschiedene
Gruppen gebunden ist. Wenn Y zweiwertiges R8-Heteroaryl
ist, ist im Kontext dieser Erfindung ein Ringelement an die Variable
X gebunden und ein anderes Ringelement ist an die Variable Z gebunden,
wobei die R8-Substituenten an die restlichen
Ringelemente gebunden sind. Zweiwertige Heteroarylgruppen werden
benannt, indem "diyl" an den Namen des
Rings angehängt
wird, als Beispiel ist ein Pyridindiylring gezeigt:
-
-
Arylcarbonyl
ist ein über
ein Carbonyl an die Hauptgruppe gebundenes Aryl, wobei Aryl der
gleichen Definition wie oben beschrieben entspricht.
-
Alkylaryl
ist eine Einheit, die ein über
eine Arylgruppe an die Hauptgruppe oder den Ring gebundenes Alkyl
enthält.
-
Cycloalkylen
bezieht sich auf eine zweiwertige Cycloalkylgruppe.
-
Der
Begriff "Solvat" bedeutet hier ein
Aggregat, das aus einem gelösten
Ion oder Molekül
mit einem oder mehreren Lösungsmittelmolekülen besteht,
beispielsweise ein Hydrat, das derartige Ionen enthält.
-
Der
Begriff "Prodrug" bedeutet eine Verbindung,
die ein Arzneimittelvorläufer
ist, der nach der Verabreichung an einen Patienten in vivo über irgendeinen
chemischen oder physiologi schen Prozess den Wirkstoff freisetzt
(z. B. wird ein Prodrug dadurch, dass es in den physiologischen
pH-Wert gebracht wird, oder durch enzymatische Wirkung in die gewünschte Wirkstoffform
umgewandelt).
-
Der
Begriff "therapeutisch
wirksame Menge" bedeutet
hier eine ausreichende Menge, um Erkrankungen des zentralen Nervensystems
zu behandeln, wie Depression, kognitive Erkrankungen und neurodegenerative
Erkrankungen, wie Morbus Parkinson, senile Demenz und Psychosen
organischer Herkunft. Die therapeutisch wirksame Menge einer aktiven
Verbindung in einer Einheitsdosis der Zubereitung kann vorzugsweise im
Bereich von etwa 0,1 mg bis etwa 1000 mg, insbesondere etwa 1 mg
bis etwa 300 mg liegen.
-
Bestimmte
erfindungsgemäße Verbindungen
können
in unterschiedlichen stereoisomeren Formen vorliegen (z. B. Enantiomere,
Diastereoisomere und Atropisomere). Die Erfindung beinhaltet alle
derartigen Stereoisomere sowohl in reiner Form als auch gemischt
einschließlich
racemischer Mischungen.
-
Die
Verbindungen der Formel I können
Salze bilden, die auch innerhalb des Schutzumfangs dieser Erfindung
liegen. Die Bezugnahme auf eine Verbindung der Formel I soll hier
die Bezugnahme auf deren Salze einschließen, wenn nicht anders angegeben.
Der Begriff "Salz(e)" bezeichnet hier
saure Salze, die mit anorganischen und/oder organischen Säuren gebildet
sind, sowie basische Salze, die mit anorganischen und/oder organischen
Basen gebildet sind. Wenn eine Verbindung der Formel I zudem sowohl
eine basische Einheit wie, jedoch nicht begrenzt auf ein Pyridin
oder Imidazol, und eine saure Einheit enthält, wie eine Carbonsäure, jedoch
nicht darauf begrenzt, können
Zwitterionen ("innere
Salze") gebildet
werden und sie sind hier in den Begriff "Salz(e)" eingeschlossen. Pharmazeutisch annehmbare
(d. h. nicht-toxische, physiologisch annehmbare) Salze sind bevorzugt,
obwohl auch andere Salze brauchbar sind. Salze der Verbindungen
der Formel I können beispielsweise
gebildet werden, indem eine Verbindung der Formel I mit einer Menge
an Säure
oder Base, wie einer äquivalenten
Menge, in einem Medium umgesetzt werden, wie einem, in dem das Salz
ausfällt,
oder in einem wässrigen
Medium, gefolgt von Lyophilisierung.
-
Zu
beispielhaften Säureadditionssalzen
gehören
Acetate, Adipate, Alginate, Ascorbate, Aspartate, Benzoate, Benzolsulfonate,
Bisulfate, Borate, Butyrate, Citrate, Camphorate, Camphersulfonate,
Cyclopentanpropionate, Digluconate, Dodecylsulfate, Ethansulfonate,
Fumarate, Glucoheptanoate, Glycerophosphate, Hemisulfate, Heptanoate,
Hexanoate, Hydrochloride, wie die hier offenbarte Verbindung 174,
Hydrobromide, Hydroiodide, 2-Hydroxyethansulfonate, Lactate, Maleate,
Methansulfonate, 2-Naphthalinsulfonate,
Nicotinate, Nitrate, Oxalate, Pectinate, Persulfate, 3-Phenylpropionate,
Phosphate, Pikrate, Pivalate, Propionate, Salicylate, Succinate,
Sulfate, Sulfonate (wie die hier genannten), Tartrate, Thiocyanate,
Toluolsulfonate (auch als Tosylate bekannt), Undecanoate und dergleichen.
Säuren,
die allgemein für
die Bildung pharmazeutisch brauchbarer Salze aus basischen pharmazeutischen
Verbindungen als geeignet angesehen werden, sind zudem beispielsweise
in S. Berge et al, Journal of Pharmaceutical Sciences (1977) 66
(1) 1–19;
P. Gould, International J. of Pharmaceutics (1986) 33 201–217, und
Anderson et al, The Practice of Medicinal Chemistry (1996), Academic
Press, New York, erörtert.
-
Beispielhafte
basische Salze schließen
Ammoniumsalze, Alkalimetallsalze wie Natrium-, Lithium- und Kaliumsalze,
Erdalkalimetallsalze wie Calcium- und Magnesiumsalze, Salze mit
organischen Basen (beispielsweise organischen Aminen) ein, wie Benzathinen,
Dicyclohexylaminen, Hydrabaminen (mit N,N-Bis(de hydroabietyl)ethylendiamin
gebildet), N-Methyl-D-glucaminen, N-Methyl-D-glucamiden, t-Butylaminen
und Salzen mit Aminosäuren
wie Arginin, Lysin und dergleichen. Basische stickstoffhaltige Gruppen
können
mit Mitteln wie niederen Alkylhalogeniden (z. B. Methyl-, Ethyl-,
Propyl- und Butylchloriden, -bromiden und -iodiden), Dialkylsulfaten
(z. B. Dimethyl-, Diethyl-, Dibutyl- und Diamylsulfaten), langkettigen
Halogeniden (z. B. Decyl-, Lauryl-, Myristyl- und Stearylchloriden,
-bromiden und -iodiden), Aralkylhalogeniden (z. B. Benzyl- und Phenethylbromiden)
und anderen quaternisiert werden.
-
Alle
derartigen Säuresalze
und Basesalze sollen pharmazeutisch annehmbare Salze innerhalb des Schutzumfangs
der Erfindung sein, und alle Säure-
oder Basensalze werden für
erfindungsgemäße Zwecke als
zu den freien Formen der entsprechenden Verbindungen äquivalent
angesehen.
-
Diese
Verbindungen besitzen antagonistische Aktivität an A2a-Rezeptoren
und sind zur Behandlung von Morbus Parkinson und Depression brauchbar.
Sie können
allein oder in Kombination mit dopaminergen Mitteln verwendet werden,
wie L-DOPA oder Ropinrol. Sie können
auch zusammen mit bekannten antidepressiven therapeutischen Mitteln
verwendet werden. Die Verbindungen der Formel I werden nach den
in Reaktionsschemata 1 und 2 gezeigten Verfahren hergestellt. Die
durch Reaktionsschema 3 hergestellten Verbindungen fallen nicht
in den Bereich der Erfindung, und es ist zu erkennen, dass die Teile
des Schemas, die Verbindungen außerhalb des Bereichs der Erfindung
betreffen, bereitgestellt werden, um analoge Verfahren zu jenen zu
zeigen, nach denen erfindungsgemäße Verbindungen
hergestellt werden können.
-
-
In
Schema 1 ergeben palladiumkatalysierte Kupplungsreaktionen zwischen
2-Amino-4,6-dichlorpyrimidin (wobei X = Cl) II und einer Arylboronsäure in einer
Toluol, Ethanol, Na2CO3 (aq)-Lösung bei erhöhter Temperatur
Verbindungen mit der Formel III. Die Behandlung von III mit dem
entsprechenden Hydrazid in Butanol bei erhöhter Temperatur liefert ein
Hydrazid IV. Die Behandlung von Verbindungen der Formel IV mit N,O-Bis(trimethylsilyl)acetamid
bei erhöhter
Temperatur liefert Verbindungen der Formel I.
-
Wenn
Ausgangsmaterial II, 2-Amino-4,6-dichlorpyrimidin (wobei X = Cl)
mit einem geeigneten Hydrazid in Butanol bei erhöhter Temperatur behandelt wird,
wird alternativ das entsprechende Hydrazid V hergestellt. Die Behandlung
von Verbindungen mit der Struktur V in N,O-Bis(trimethylsilyl)acetamid
liefert Verbindungen mit der Formel VI. Die palladiumkatalysierten
Kupplungsreaktionen zwischen Verbindungen VI (wobei X = Cl) und
einer Arylboronsäure
in einer Toluol, Ethanol, Na2CO3 (aq)-Lösung bei
erhöhter
Temperatur ergibt Verbindungen der Formel I.
-
-
Verbindungen
der Formel I können
auch wie oben in Schema 2 gezeigt hergestellt werden. Der gewünschte Vorläufer IX
kann entweder durch Behandlung des geeigneten Ketons VII mit Alkylchlorformiat
in Gegenwart von Base oder durch Behandlung eines geeigneten β-Ketoesters
VIII mit R3X unter basischen Bedingungen
hergestellt werden. Der β-Ketoester
IX kann mit Guanidincarbonat bei erhöhter Temperatur in einem inerten
Lösungsmittel,
wie DMF, eine Kondensationsreaktion eingehen, um das Aminopyrimidin
X zu produzieren. Die Behandlung von X mit POCl3 bei
erhöhter
Temperatur ergibt das Chloranalogon XI. Die Behandlung von XI mit
dem entsprechenden Hydrazid in Butanol bei erhöhter Temperatur liefert ein
Hydrazid XII. Die Behand lung von Verbindungen der Formel XII mit
N,O-Bis(trimethylsilyl)acetamid bei erhöhter Temperatur liefert Verbindungen
der Formel I.
-
Alternativ
ergibt Behandlung von XI mit dem Boc-geschützten Hydrazin in einem inerten
Lösungsmittel,
wie DMF, bei erhöhter
Temperatur Verbindungen mit der Formel XIII, die wiederum mit einer
Säure,
wie TFA, bei Raumtemperatur entschützt werden können, um
das freie Hydrazin XIV zu ergeben. Die Behandlung von XIV mit der
passenden Carbonsäure
in Gegenwart eines Kupplungsmittels, wie EDCI, in einem inerten
Lösungsmittel,
wie DMF, bei Raumtemperatur ergibt Hydrazid XII. Schema
3
und R
19 wie oben definiert
ist.
-
Alternativ
können
analoge Verbindungen zu jenen der Formel I wie in Schema 3 gezeigt
hergestellt werden. Verbindung VI kann durch Behandlung mit K2CO3 in n-BuOH bei
erhöhten
Temperaturen nucleophile Substitutionsreaktionen mit Aminen mit
der Formel XV eingehen, um Verbindungen mit der Formel I zu ergeben.
-
Ein
weiterer Aspekt der Erfindung betrifft eine pharmazeutische Zusammensetzung,
die eine oder mehrere Verbindungen der Formel I und einen oder mehrere
pharmazeutisch annehmbare Träger
enthält.
-
Ein
weiterer Aspekt der Erfindung betrifft eine pharmazeutische Zusammensetzung,
die eine oder mehrere Verbindungen der Formel I und ein oder mehrere
Mittel in einem oder mehreren pharmazeutisch annehmbaren Träger(n) enthält, die
bekanntermaßen
zur Behandlung von Morbus Parkinson brauchbar sind.
-
Ein
weiterer Aspekt der Erfindung betrifft die Verwendung von einer
oder mehreren Verbindungen der Formel I zur Herstellung einer pharmazeutischen
Zusammensetzung zur Behandlung des Schlaganfalls oder von Erkrankungen
des zentralen Nervensystems. In diesem Aspekt der Erfindung schließen die
Erkrankungen des zentralen Nervensystems kognitive Erkrankungen
oder neurodegenerative Erkrankungen ein, wie Morbus Parkinson, senile
Demenz oder Psychosen organischer Herkunft. Die Erfindung betrifft
insbesondere die Verwendung von einer oder mehreren Verbindungen
mit der Formel I zur Herstellung einer pharmazeutischen Zusammensetzung
zur Behandlung von Morbus Parkinson. Die verabreichte Menge der
Verbindung ist vorzugsweise eine therapeutisch wirksame Menge.
-
Ein
weiterer Aspekt der Erfindung betrifft die Verwendung einer Verbindung
der Formel I zur Herstellung einer pharmazeutischen Zusammensetzung
zur Behandlung von Morbus Parkinson mit einer Kombination von einer
oder mehreren Verbindungen der Formel I und einem oder mehreren
Mitteln, die bekanntermaßen zur
Behandlung von Morbus Parkinson brauchbar sind, beispielsweise Dopamin,
einem dopaminergen Agonisten, einem Monoaminoxidase-Hemmer Typ B
(MAO-B), einem DOPA-Decarboxylaseinhibitor (DCI) oder einem Catechol-O-methyltransferase-(COMT)-Hemmer.
Gemäß diesem
Aspekt der Erfindung können
eine oder mehrere Verbindungen der Formel I und ein oder mehrere
andere Antiparkinsonmittel gleichzeitig oder sequentiell in getrennten
Dosierformen verabreicht werden.
-
Ein
weiterer Aspekt der Erfindung betrifft einen Kit, der in separaten
Behältern
in einer einzigen Verpackung pharmazeutische Zusammensetzungen zur
Verwendung in Kombination zur Behandlung von Morbus Parkinson enthält, wobei
ein Behälter
eine pharmazeutische Zusammensetzung enthält, die eine oder mehrere Verbindungen
der Formel I in einem oder mehreren pharmazeutisch annehmbaren Trägern enthält, und
wobei in separaten Behältern
eine oder mehrere pharmazeutische Zusammensetzungen enthalten sind,
die jeweils ein oder mehrere Mittel, die zur Behandlung von Morbus
Parkinson brauchbar sind, in einem oder mehreren pharmazeutisch
annehmbaren Trägern
enthalten.
-
Zur
Herstellung pharmazeutischer Zusammensetzungen aus den in dieser
Erfindung beschriebenen Verbindungen können inerte, pharmazeutisch
annehmbare Träger
fest oder flüssig
sein. Zubereitungen in fester Form schließen Pulver, Tabletten, dispergierbare
Körner,
Kapseln, Medizinalkapseln und Zäpfchen
ein. Die Pulver und Tabletten können
aus etwa 5 bis etwa 70 Gew.-% aktivem Bestandteil zusammengesetzt
sein, wozu Verbindungen der Formel I und gegebenenfalls andere Verbindungen
gehören,
die zur Behandlung von Morbus Parkinson brauchbar sind. Geeignete
feste Träger
sind in der Technik bekannt, z. B. Magnesiumcarbonat, Magnesiumstearat,
Talkum, Zucker, Lactose. Tabletten, Pulver, Kapseln und Medizinalkapseln
können
als feste Dosierformen verwendet werden, die für die orale Verabreichung geeignet
sind.
-
Zur
Herstellung von Zäpfchen
wird ein niedrig schmelzendes Wachs wie eine Mischung aus Fettsäureglyceriden
oder Kakaobutter zuerst geschmolzen und der aktive Bestandteil darin
homogen dispergiert, wie durch Rühren.
Die geschmolzene homogene Mischung wird dann in zweckmäßig bemessene
Formen gegossen, abkühlen
gelassen und verfestigt.
-
Zubereitungen
in flüssiger
Form schließen
Lösungen,
Suspensionen und Emulsionen ein. Als Beispiel hierfür können Wasser
oder Wasser/Propylenglykol-Lösungen
für die
parenterale Injektion genannt werden.
-
Zubereitungen
in flüssiger
Form können
auch Lösungen
für intranasale
Verabreichung einschließen.
-
Aerosolzubereitungen,
die zur Inhalation geeignet sind, können Lösungen und Feststoffe in Pulverform
einschließen,
die in Kombination mit einem pharmazeutisch annehmbaren Träger wie
inertem komprimiertem Gas vorliegen können.
-
Ebenfalls
eingeschlossen sind Zubereitungen in fester Form, die kurz vor Gebrauch
in Zubereitungen in flüssiger
Form für
orale oder parenterale Verabreichung überführt werden. Solche flüssigen Formen
schließen
Lösungen,
Suspensionen und Emulsionen ein.
-
Die
erfindungsgemäßen Verbindungen
können
auch transdermal verabreichbar sein. Die transdermalen Zusammensetzungen
können
die Form von Cremes, Lotionen, Aerosolen und/oder Emulsionen annehmen,
und können
in ein Transdermalpflaster vom Matrix- oder Reservoirtyp eingeschlossen werden,
wie in der Technik zu diesem Zweck konventionell ist.
-
Die
Verbindung wird vorzugsweise oral verabreicht.
-
Die
pharmazeutische Zubereitung liegt vorzugsweise in Einheitsdosisform
vor. In einer solchen Form wird die Zubereitung in Einheitsdosen
unterteilt, die geeignete Mengen der aktiven Komponente enthalten,
z. B. eine wirksame Menge, um den gewünschten Zweck zu erreichen.
-
Die
Menge an aktiver Verbindung in einer Einheitsdosis der Zubereitung
kann gemäß der speziellen Anwendung
auf etwa 0,1 mg bis etwa 1000 mg, vorzugsweise etwa 1 mg bis etwa
300 mg, variiert oder eingestellt werden.
-
Die
tatsächlich
verwendete Dosis kann gemäß den Erfordernissen
des Patienten und dem Schweregrad des behandelten Zustands variiert
werden. Die Bestimmung der richtigen Dosierung für eine spezielle Situation
liegt innerhalb des Wissens des Fachmanns. Die Behandlung wird allgemein
mit geringeren Dosierungen begonnen, die unter der optimalen Dosis
der Verbindung liegen. Nachfolgend wird die Dosierung in kleinen Schritten
erhöht,
bis die optimale Wirkung unter den Bedingungen erreicht wird. Der
Bequemlichkeit halber kann die gesamte Tagesdosis unterteilt und
auf Wunsch portionsweise über
den Tag verabreicht werden.
-
Die
Menge und Frequenz der Verabreichung der erfindungsgemäßen Verbindungen
und der pharmazeutisch annehmbaren Salze derselben werden gemäß der Beurteilung
des behandelnden Arztes unter Berücksichtigung von Faktoren wie
Alter, Zustand und Größe des Patienten
sowie des Schweregrads der zu behandelnden Symptome festgelegt.
Ein typisches empfohlenes Dosierschema für Verbindungen der Formel I ist
orale Verabreichung von etwa 10 mg bis etwa 2000 mg/Tag, vorzugsweise
etwa 10 bis etwa 1000 mg/Tag in zwei bis vier unterteilten Dosen,
um Erleichterung bei Erkrankungen des zentralen Nervensystems, wie
Morbus Parkinson, zu bewirken. Die Verbindungen sind bei Verabreichung
innerhalb dieses Dosierungsbereichs nicht giftig.
-
Beispiele
-
In
den folgenden Beispielen liegen mit einem Stern (*) markierte Verbindungen
nicht innerhalb des Bereichs der Erfindung und sie sind angegeben,
um zu den erfindungsgemäßen Verbindungen
analoge Verbindungen und analoge Verfahren, nach denen erfindungsgemäße Verbindungen
gebildet werden können,
zu illustrieren.
-
-
Stufe
1: Eine Mischung von 2-Amino-4-chlor-6-methylpyrimidin (1,44 g,
10,00 mmol) und 2-Furancarbonsäurehydrazid
(1,89 g, 15,0 mmol) in Butanol (50 ml) wurde 16 Stunden auf 90°C erwärmt. Nachdem
die Reaktionsmischung auf Raumtemperatur abgekühlt worden war, wurde der Rückstand
mit Methanol gewaschen und der resultierende Niederschlag abfiltriert,
um einen Feststoff zu ergeben.
-
Stufe
2: Der in Stufe 1 produzierte Feststoff (0,77 g, 3,30 mmol) in N,O-Bis(trimethylsilyl)acetamid
(5 ml) wurde über
Nacht auf 120°C
erwärmt.
Die Reaktionsmischung wurde abgekühlt und auf Eiswasser gegossen
und 4 Stunden gerührt.
Die Mischung wurde mit Ethylacetat extrahiert, über Natriumsulfat getrocknet,
filtriert und im Vakuum konzentriert. Der Rückstand wurde durch Silikagelchromatographie
gereinigt, um einen Feststoff zu produzieren. 1H-NMR
(DMSO-d6) δ 7,83 (br M, 3H), 7,10 (dd,
1H), 6,75 (s, 1H), 6,64 (dd, 1H), 2,30 (s, 3H). Massenspektrum (ESI):
216,0.
-
-
Stufe
1: Eine Mischung aus 2-Amino-4,6-dichlorpyrimidin (10,00 g, 60,98
mmol) und 2-Furancarbonsäurehydrazid
(7,68 g, 60,98 mmol) wurde in Butanol (200 ml) 20 Stunden auf 90°C erwärmt. Die
Reaktionsmischung wurde auf Raumtemperatur abgekühlt, die Mischung mit Ethylacetat
und Wasser extrahiert, die Ethylacetatschicht aufgefangen und über Natriumsulfat
getrocknet, filtriert und im Vakuum konzentriert. Der Rückstand
wurde durch Silikagelchromatographie gereinigt, um einen Feststoff
B zu produzieren. Massenspektrum (ESI): 254,0. 1H-NMR
(DMSO-d6) δ 5,76 (br s, 1H), 6,61 (br s,
2H), 6,64 (m, 1H), 7,20 (d, 1H), 7,88 (s, 1H), 9,01 (br s, 1H),
10,32 (br s, 1H).
-
Stufe
2: Das Produkt aus Stufe 1 B (9,20 g, 36,27 mmol) wurde in N,O-Bis(trimethylsilyl)acetamid
(49,4 g, 242,14 mmol) über
Nacht auf 120°C
erhitzt. Die Reaktionsmischung wurde abgekühlt und auf Eiswasser gegossen
und 4 Stunden gerührt.
Die Mischung wurde mit Ethylacetat extrahiert, über Natriumsulfat getrocknet,
filtriert und im Vakuum konzentriert. Der Rück stand wurde durch Silikagelchromatographie
gereinigt, um einen Feststoff C zu produzieren. Massenspektrum (ESI):
236,0. 1H-NMR (CDCl3) δ 6,18 (br
s, 2H), 6,61 (m, 1H), 7,04 (s, 1H), 7,24 (d, 1H), 7,64 (s, 1H).
-
Stufe
3: In einem geschlossenen Röhrchen
wurde das Produkt aus Stufe 2 C (50 mg, 0,21 mmol) mit 3,5-Dimethylbenzolboronsäure (63
mg, 0,42 mmol), Pd(PPh3)4 (24
mg, 0,02 mmol) und Natriumcarbonat (74 mg, 2,10 mmol) in einem Lösungsmittelsystem
von 3/1/1 Toluol/Ethanol/Wasser über
einen Zeitraum von 4 Stunden auf 103°C erwärmt. Nachdem die Reaktionsmischung
auf Raumtemperatur abgekühlt
worden war, wurde die Mischung mit EtOAc und Wasser extrahiert.
Der organische Anteil wurde aufgefangen, über Natriumsulfat getrocknet,
filtriert und im Vakuum konzentriert. Der Rückstand wurde durch Silikagelchromatographie
gereinigt, um einen Feststoff D zu produzieren. Massenspektrum (ESI):
306,1. 1H-NMR (CDCl3) δ 2,41 (s, 6H),
5,95 (br s, 2H), 7,10 (s, 1H), 7,24 (s, 1H), 7,39 (s, 1H), 7,60
(s, 2H), 7,64 (s, 1H).
-
Die
folgenden Verbindungen wurden in ähnlicher Weise hergestellt:
-
-
-
-
-
-
Die
ortho-(N-Morpholinomethyl)benzolboronsäure wurde nach einem bekannten
Literaturverfahren hergestellt (J. Am. Chem. Soc., Seite 3863, 1960)
und nachfolgend verwendet, um die Zielverbindung wie in Beispiel
2 beschrieben zu produzieren. Massenspektrum (ESI): 377,1. 1H-NMR (CDCl3) δ 2,36 (t,
4H), 3,56 (t, 4H), 6,04 (br s, 2H), 6,61 (m, 1H), 7,30 (s, 1H),
7,39 (m, 2H), 7,51 (m, 2H), 7,65 (d, 1H).
-
Die
folgenden Verbindungen wurden in ähnlicher Weise hergestellt:
-
-
-
-
Stufe
1: Eine Mischung von 2-Amino-4,6-dichlorpyrimidin (0,50 g, 3,048
mmol), 3-Isopropylbenzolboronsäure
(0,30 g, 1,572 mmol), Pd (PPh3)4 (0,09
g, 0,076 mmol) und 4 bis 10 Äquivalenten
Natriumcarbonat wurde in einem Lösungsmittelsystem
aus 1/1 Acetonitril/Wasser (15 ml) für einen Zeitraum von 4 Stunden
auf 90°C
erwärmt.
Die Reaktionsmischung wurde auf Raumtemperatur abgekühlt. Die
Mischung wurde mit EtOAc und Wasser extrahiert. Der organische Anteil
wurde aufgefangen, über
Natriumsulfat getrocknet, filtriert und im Vakuum konzentriert.
Der Rückstand
wurde durch Silikagelchromatographie gereinigt, um einen Feststoff zu
produzieren.
Massenspektrum (ESI): 248,0. 1H-NMR
(CDCl3) δ 1,30
(d, 6H), 2,98 (m, 1H), 5,93 (br s, 2H), 7,03 (s, 1H), 7,39 (m, 2H),
7,73 (d, 1H), 7,82 (s, 1H).
-
Stufe
2: Das Produkt aus Stufe 1 (0,39 g, 1,57 mmol) und 2-Furancarbonsäurehydrazid
(0,30 g, 2,36 mmol) in Butanol (10 ml) wurde 5 Stunden auf 120°C erwärmt. Die
Reaktionsmischung wurde auf Raumtemperatur abgekühlt und danach mit Ethylacetat
und Wasser extrahiert. Die Ethylacetatphase wurde aufgefangen, über Natriumsulfat
getrocknet, filtriert und im Vakuum konzentriert. Der Rückstand
wurde durch Silikagelchromatographie gereinigt, um einen Feststoff
zu produzieren. Massenspektrum (ESI): 338,1. 1H-NMR
(CDCl3) δ 1,22 (d, 6H), 2,89 (m, 1H), 5,42 (br s, 1H), 6,34
(s, 1H), 6,45 (br s, 2H), 7,18 (d, 1H), 7,23 (m, 2H), 7,44 (s, 1H),
7,53 (d, 1H), 7,67 (s, 1H).
-
Stufe
3: Das Produkt aus Stufe 2 (0,27 g, 0,80 mmol) wurde in N,O-Bis(trimethylsilyl)acetamid
(10 ml, 40,4 mmol) über
Nacht auf 120°C
erhitzt. Die Reaktionsmischung wurde abgekühlt und danach auf Eiswasser gegossen
und 4 Stunden gerührt.
Die Mischung wurde mit Ethylacetat extrahiert, über Natriumsulfat getrocknet,
filtriert und im Vakuum konzentriert. Der Rückstand wurde durch Silikagelchromatographie
gereinigt, um einen Feststoff zu produzieren. Massenspektrum (ESI):
320,0. 1H-NMR (CDCl3) δ 1,21 (d,
6H), 2,89 (m, 1H), 5,42 (br s, 2H), 6,60 (m, 1H), 7,33 (d, 1H),
7,41 (d, 1H), 7,43 (s, 1H), 7,64 (s, 1H).
-
Die
folgenden Verbindungen wurden in ähnlicher Weise hergestellt:
-
-
-
-
Beispiel
5
Verbindung
39*
-
Verbindung
37 aus Beispiel 4 (15 mg, 0,0165 mmol) wurde mit 10% Palladium auf
Kohle (5 mg) in 10 ml Lösungsmittelmischung
(9:1 EtOAc/EtOH) unter einer Wasserstoffatmosphäre für einen Zeitraum von 3 Stunden
bei Raumtemperatur gerührt.
Die Mischung wurde durch ein Kieselerdekissen geleitet, und der
organische Anteil wurde im Vakuum konzentriert. Der Rückstand
wurde durch Silikagelchromatographie gereinigt, um einen Feststoff
zu ergeben. Massenspektrum (ESI): 306,0. 1H-NMR
(CDCl3) δ 2,97
(t, 2H), 3,06 (t 2H), 5,97 (br s, 2H), 6,58 (m, 1H), 6,79 (s, 1H),
7,20 (m, 2H), 7,28 (m, 4H), 7,68 (m, 1H).
-
-
Stufe
1: Verbindung 38 aus Beispiel 4 (0,60 g, 1,95 mmol) wurde mit Triethylamin
(1,63 ml, 11,63 mmol) und Thionylchlorid (0,71 g, 9,76 mmol) 3 Stunden
bei 0°C
unter einer Stickstoffatmosphäre
kombiniert. Die Reaktionsmischung wurde im Vakuum konzentriert und
der Rückstand
B dann durch Silikagelchromatographie gereinigt. Massenspektrum
(ESI): 326,1.
-
Stufe
2: Das Produkt aus Stufe 1 (0,22 g, 0,66 mmol) wurde mit 1-(4-Methoxyethoxyphenyl)piperazin (0,31
g, 1,32 mmol) in Dimethylformamid (2,0 ml) in einem verschlossenen
Röhrchen
kombiniert und über Nacht
auf 110°C
erwärmt.
Die Reaktionsmischung wurde abgekühlt und mit Ethylacetat und
Salzlösung
extrahiert. Die Ethylacetatphase wurde aufgefangen, über Natriumsulfat
getrocknet, filtriert und im Vakuum konzentriert. Der Rückstand
C wurde durch Silikagelchromatographie gereinigt. Massenspektrum
(ESI): 526,1. 1H-NMR (CDCl3) δ 8,0 (S,
1H), 7,86 (m, 1H), 7,62 (dd, 1H), 7,41–7,45 (m, 3H), 7,24 (d, 1H),
6,82–6,85
(m, 4H), 6,58 (dd, 1H), 6,28 (br s, 2H), 4,06 (t, 2H), 3,72 (t,
2H), 3,66 (s, 2H), 3,43 (s, 3H), 3,00–3,12 (m, 4H), 2,65–2,67 (m,
4H).
-
Die
folgenden Verbindungen wurden in ähnlicher Weise hergestellt:
-
-
-
-
-
-
-
Beispiel
7
Verbindung
77*
-
Stufe
1: Zu 4-(4-Methoxyphenyl)buttersäure
(2,00 g, 10,3 mmol) in CH2Cl2 (10
ml) wurde SOCl2 (3,56 g, 30 mmol) gegeben.
Es wurde 3 Stunden gerührt
und konzentriert. 2,2-Dimethyl-1,3-dioxan-4,6-dion (1,78 g, 12,4 mmol),
Pyridin (2,37 g, 31 mmol) und CH2Cl2 (10 ml) wurden zugegeben. Es wurde 18 Stunden
gerührt, EtOH
(10 ml) zugegeben und 5 Stunden unter Rückfluss gerührt. Es wurde Wasser zugegeben,
mit EtOAc extrahiert und chromatographiert, um das Keton als Öl zu erhalten.
-
Stufe
2: Das Produkt aus Stufe 1 (0,366 g, 1,39 mmol) und Guanidincarbonat
(0,382 g, 2,12 mmol) wurden in EtOH (3 ml) kombiniert. Es wurde
18 Stunden auf Rückfluss
erwärmt.
Es wurde Wasser (20 ml) zugefügt,
in Eis gekühlt
und filtriert. Es wurde getrocknet, mit Hexan gewaschen und filtriert,
um das Pyrimidin als Feststoff zu erhalten.
-
Stufe
3: Das Produkt aus Stufe 2 (0,15 g, 0,58 mmol) wurde zu POCl3 (1,22 ml) gegeben. Es wurde eine Stunde
auf Rückfluss
erwärmt,
konzentriert, mit Eis behandelt, mit NH3 neutralisiert
und mit EtOAc extrahiert. Es wurde mittels PLC gereinigt, um das
Chlorpyrimidin als Feststoff zu erhalten.
-
Stufe
4: Das Produkt aus Stufe 3, 2-Furancarbonsäurehydrazid und 1,0 N HCl wurden
in EtOH kombiniert. Es wurde in einem verschlossenen Röhrchen 16
Stunden auf 90°C
erwärmt.
Es wurde mit NH3 basisch gemacht, mit EtOAc
extrahiert und mittels PLC gereinigt, um das Hydrazid als Feststoff
zu erhalten.
-
Stufe
5: Das Produkt aus Stufe 4 wurde zu BSA gegeben. Es wurde 18 Stunden
auf 120°C
erwärmt. Es
wurde in CH3OH gegossen, konzentriert und
mittels PLC gereinigt, um die Titelverbindung als Feststoff zu erhalten.
Massenspektrum (ESI): 350.
-
-
Stufe
1: Das Produkt B wurde in ähnlicher
Weise wie in Beispiel 4, Stufe 1, beschrieben synthetisiert. Massenspektrum
(ESI): 236,1.
-
Stufe
2: Zu einer Lösung
des Produkts aus Stufe 1 B (2,25 g, 9,53 mmol) in Dichlormethan
(100 ml), die unter einer inerten Atmosphäre bei 0°C gerührt wurde, wurde Triethylamin
(8,00 ml, 57,18 mmol) gegeben, gefolgt von Thionylchlorid (3,50
ml, 47,65 mmol), und die Mischung wurde eine weitere Stunde gerührt. Die Mischung
wurde auf Raumtemperatur erwärmt
und dann mit Dichlormethan und Salzlösung extrahiert. Der organische
Anteil wurde aufgefangen, über
Natriumsulfat getrocknet, filtriert und im Vakuum konzentriert.
Der Rückstand
wurde durch Silikagelchromatographie gereinigt, um das Produkt C
zu ergeben. Massenspektrum (ESI): 255,1.
-
Stufe
3: Eine Lösung
des Produkts aus Stufe 2 C (0,50 g, 1,97 mmol, 1-(2,4-Difluorphenyl)piperazin (0,39
g, 1,97 mmol), Kaliumiodid (0,33 g, 1,97 mmol) und Kaliumcarbonat
(0,82 g, 5,90 mmol) in Acetonitril (10 ml) wurde über Nacht
unter einer inerten Atmosphäre
bei 60°C
gerührt.
Die Mischung wurde auf Raumtemperatur abgekühlt und danach mit Ethylacetat,
und Salzlösung
extrahiert. Der organische Anteil wurde aufgefangen, über Natriumsulfat
getrocknet, filtriert und im Vakuum konzentriert. Der Rückstand
wurde durch Silikagelchromatographie gereinigt, um das Produkt D
zu ergeben. Massenspektrum (ESI): 416,1.
-
Stufe
4: Eine Lösung
des Produkts aus Stufe 3 D (0,84 g, 1,97 mmol) und Boc-geschütztem Hydrazin (0,31
g, 2,37 mmol) in DMF wurde über
Nacht unter einer inerten Atmosphäre bei 80°C gerührt. Die Mischung wurde auf
Raumtemperatur abgekühlt
und danach mit Ethylacetat und Salzlösung extrahiert. Der organische Anteil
wurde aufgefangen, über
Natriumsulfat getrocknet, filtriert und im Vakuum konzentriert.
Der Rückstand wurde durch
Silikagelchromatographie gereinigt, um das Produkt E zu ergeben.
Massenspektrum (ESI): 512,1.
-
Stufe
5: Zu einer Lösung
des Produkts aus Stufe 4 E (0,15 g, 0,29 mmol) in Dichlormethan
(5 ml), die bei Raumtemperatur gerührt wurde, wurde Trifluoressigsäure (5 ml)
gegeben und die Reaktionsmischung eine weitere Stunde gerührt. Die
Reaktionsmischung wurde im Vakuum konzentriert und in DMF (2 ml)
aufgenommen. Zu dieser Lösung
wurden Butincarbonsäure
(30 mg, 0,35 mmol), EDCI (68 mg, 0,35 mmol), HOBT (48 mg, 0,35 mmol),
NMM (41 μl,
0,35 mmol) gegeben und über
Nacht unter einer inerten Atmosphäre bei Raumtemperatur gerührt. Die
Mischung wurde mit Ethylacetat und Salzlösung extrahiert. Der organische
Anteil wurde aufgefangen, über
Natriumsulfat getrocknet, filtriert und im Vakuum konzentriert.
Der Rückstand
wurde durch Silikagelchromatographie gereinigt, um das Produkt F
zu ergeben. Massenspektrum (ESI): 478,1.
-
Stufe
6: Das Produkt aus Stufe 5 F (45 mg, 0,09 mmol) wurde in N,O-Bis(trimethylsilyl)acetamid
(2 ml) über
Nacht auf 120°C
erhitzt. Die Reaktionsmischung wurde abgekühlt, auf Eiswasser gegossen
und 4 Stunden gerührt.
Die Mischung wurde mit Ethylacetat extrahiert, über Natriumsulfat getrocknet,
filtriert und im Vakuum konzentriert. Der Rückstand wurde durch Silikagelchromatographie
gereinigt, um einen Feststoff G zu produzieren. Massenspektrum (ESI):
460,1, 1H-NMR (CDCl3) δ 7,98 (s,
1H), 7,88 (m, 1H), 7,45 (m, 2H), 7,39 (s, 1H), 6,90 (m, 1H), 6,79
(m, 2H), 5,97 (br s, 2H), 3,66 (s, 2H), 3,06 (t, 4H), 2,67 (t, 4H),
2,15 (s, 3H).
-
-
Stufe
1: Eine Lösung
von 5-Brom-3-(methanol)pyridin (6,69 g, 35,58 mmol), t-Butyldimethylsilylchlorid (4,71
g, 46,26 mmol) und Imidazol (7,25 g, 106,74 mmol) in Dichlormethan
(250 ml) wurde 3 Stunden bei Raumtemperatur gerührt. Die Mischung wurde mit
Dichlormethan und Salzlösung
extrahiert. Der organische Anteil wurde aufgefangen, über Natriumsulfat
getrocknet, filtriert und im Vakuum konzentriert. Der Rückstand wurde
durch Silikagelchromatographie gereinigt, um das Produkt B zu ergeben.
Massenspektrum (ESI): 304,1, 302,1.
-
Stufe
2: Zu einer gerührten
Lösung
des Produkts aus Stufe 1 B (7,35 g, 24,32 mmol) in Diethylether (125
ml) unter einer inerten Atmosphäre
bei –78°C wurde tropfenweise
eine 2,5 N Lösung
von n-Butyllithium in Hexanen (14,51 ml) gegeben. Nachdem 10 Minuten
gerührt
worden war, wurde Triisopropylborat (11,02 ml, 47,75 mmol) zugegeben
und die Lösung
auf Raumtemperatur erwärmt
und eine weitere Stunde gerührt.
Die Reaktion wurde mit Wasser gequencht. Die Reaktionsmischung wurde
im Vakuum konzentriert, und die resultierende feste Zwischenstufe
wurde in der nächsten
Stufe ohne weitere Reinigung verwendet.
-
Die
feste Zwischenstufe (6,50 g, 27,99 mmol) wurde in Dimethoxyethylen
(100 ml) aufgenommen, und 2-Amino-4,6-dichlorpyrimidin (9,18 g,
55,98 mmol), Natriumcarbonat (10,31 g, 97,26 mmol) und Tetrakis(triphenylphosphin)palladium
(1,40 g, 1,21 mmol) wurden zugegeben. Die Mischung wurde 4 Stunden
auf 90°C erwärmt. Die
Reaktionsmischung wurde abgekühlt
und mit Ethylacetat und Salzlösung
extrahiert. Die Ethylacetatphase wurde aufgefangen, über Natriumsulfat
getrocknet, filtriert und im Vakuum konzentriert. Der Rückstand
C wurde durch Silikagelchromatographie gereinigt. Massenspektrum
(ESI): 351,1.
-
Stufe
3: Eine Lösung
des Produkts aus Stufe 2 C (1,64 g, 4,67 mmol) und 2-Furancarbonsäurehydrazid
(0,92 g, 7,01 mmol) in 10 ml n-Butanol wurde über Nacht auf 90°C erwärmt. Die
Mischung wurde auf Raumtemperatur abgekühlt, mit Ethylacetat extrahiert
und mit Salzlösung
gewaschen. Der organische Anteil wurde aufgefangen, über Natriumsulfat
getrocknet, filtriert und im Vakuum konzentriert. Der Rückstand
D wurde in der nächsten
Stufe ohne weitere Reinigung verwendet. Massenspektrum (ESI): 441,1.
-
Stufe
4: Das Produkt aus Stufe 3 D (2,04 g, 4,63 mmol) wurde in N,O-Bis(trimethylsilyl)acetamid
(15 ml) über
Nacht auf 120°C
erhitzt. Die Reaktionsmischung wurde abgekühlt, auf Eiswasser gegossen
und 4 Stunden gerührt.
Die Mischung wurde mit Ethylacetat extrahiert, über Natriumsulfat getrocknet,
filtriert und im Vakuum konzentriert. Der Rückstand wurde durch Silikagelchromatographie
gereinigt, um einen Feststoff E zu produzieren. Massenspektrum (ESI):
309,1. 1H-NMR (CDCl3) δ 9,19 (s,
1H), 8,59 (s, 2H), 8,53 (s, 1H), 7,76 (s, 1H), 7,52 (s, 1H), 6,66
(m, 1H), 4,76 (s, 2H).
-
Die
folgenden Verbindungen wurden in ähnlicher Weise hergestellt:
-
-
Beispiel
10
Verbindung
82
-
Stufe
1: Eine Mischung von Bromid (7,0 g, 24,37 mmol), N-Boc-Piperazin (5,45
g, 29,24 mmol), Palladiumacetat (0,22 g, 0,97 mmol), Tris(tert.-butyl)phosphin
(0,79 g, 3,9 mmol) und Natrium-tert.-butoxid (3,28 g, 34,12 mmol)
in Toluol (50 ml) wurde 2 Stunden unter einer Stickstoffatmosphäre auf Rückfluss
erwärmt,
auf Raumtemperatur abgekühlt
und danach mit Wasser verdünnt.
Die resultierende Mischung wurde mit Ethylacetat ex trahiert, mit
Natriumsulfat getrocknet und filtriert. Diese wurde unter vermindertem
Druck eingedampft, wodurch ein sauberes Produkt B zurückblieb,
das für
seine Verwendung in Stufe 2 nicht gereinigt wurde. Massenspektrum
(ESI), M+1: 393,1, 337,1.
-
Stufe
2: Die Verbindung aus Stufe 1 B wurde mit Tetrabutylammoniumfluorid
(48,74 g, 48,74 mmol) 1,0 M Lösung
in THF) in THF (100 ml) eine Stunde bei Raumtemperatur behandelt,
mit Wasser verdünnt,
danach mit Ethylacetat extrahiert. Der resultierende Ethylacetatextrakt
wurde mit Natriumsulfat getrocknet und eingedampft, um ein Phenolderivat
C zu ergeben. Massenspektrum (ESI), M+1:
279,0, 242,0.
-
Stufe
3: Trifluormethansulfonsäureanhydrid
wurde unter N2 tropfenweise zu einer Mischung
des Phenols aus Stufe 2 C und Triethylamin (3,74 ml, 26,81 mmol).
in Dichlormethan (100 ml) gegeben, bei dieser Temperatur eine Stunde
gerührt
und danach auf Raumtemperatur erwärmt. Es wurde gesättigte Natriumbicarbonatlösung zugegeben
und mit Dichlormethan extrahiert. Diese wurde mit Natriumsulfat
getrocknet und auf einer kleinen Menge Silikagel adsorbiert, auf
eine Säule überführt und
mit Hexan/Ethylacetat (4:1) eluiert, um das Triflat D zu liefern.
Massenspektrum (ESI), M+1: 411,1, 355,1.
-
Stufe
4: Die Verbindung aus Stufe 3 D wurde über Nacht unter N2 bei
80°C mit
Bis(pinacolato)dibor in Gegenwart von PdCl2(dppf),
dppf und Kaliumacetat in 1,4-Dioxan (90 ml) behandelt, auf Raumtemperatur
abgekühlt,
mit Salzlösung
gewaschen, mit Natriumsulfat getrocknet und eingedampft. Der Rückstand
wurde durch Säulenchromatographie
an Silikagel gereinigt, um das Produkt E zu ergeben. Massenspektrum
(ESI), M+1: 389,1.
-
Stufe
5: Die Verbindung aus Stufe 4 E wurde mit 2-Amino-4,6-dichlorpyrimidin
wie in Stufe 1, Beispiel 1, behandelt, um Verbindung F zu bilden.
Massenspektrum (ESI), M+1: 390,1.
-
Stufe
6: Es wurde das gleiche Verfahren wie in Stufe 2 von Beispiel 4
durchgeführt,
um Verbindung G zu bilden. Massenspektrum (ESI), M+1:
480,1.
-
Stufe
7: Es wurde das gleiche Verfahren wie in Stufe 3 von Beispiel 4
durchgeführt,
um Verbindung H zu bilden. 1H-NMR (CDCl3) δ 7,61
(m, 2H), 7,43 (d, 1H), 7,38 (m, 2H), 7,12 (m, 1H), 7,02 (dd, 1H),
6,60 (dd, 1H), 6,02 (br, s, 2H), 3,62 (m, 4H), 3,21 (m, 4H), 1,50
(s, 9H), Massenspektrum (ESI), M+1: 462,1.
-
Die
folgenden Verbindungen wurden in ähnlicher Weise hergestellt:
-
-
-
Beispiel
11
Verbindung
92
-
Stufe
1: Das geschützte
Enoltriflat B wurde nach einem angepassten Literaturverfahren hergestellt (Synthesis,
Seite 993, 1991). 1H-NMR (CDCl3) δ 5,66 (t,
1H), 3,99 (s, 4H), 2,54 (m, 2H), 2,41 (m, 2H), 1,90 (t, 2H).
-
Stufe
2: Das Produkt aus Stufe 1 B (5,70 g, 19,79 mol), 3-Hydroxyphenylboronsäure (6,10
g, 27,71 mmol), Lithiumchlorid (2,50 g, 58,98 mmol), eine wässrige 2
N Natriumcarbonatlösung
(27,70 ml) und Tetrakis(triphenylphosphin)palladium (1,14 g, 0,98
mmol) wurden in 100 ml Dimethoxyethan 2 Stunden auf Rückflusstemperatur
erwärmt.
Die Mischung wurde auf Raumtemperatur abgekühlt und im Vakuum konzentriert. Der
Rückstand
wurde mit Dichlormethan verdünnt
und mit 100 ml einer Mischung aus 6 Ammoniumhydroxid in 2 N Natriumcarbonatlösung gewaschen.
Der wässrige
Anteil wurde mit weiteren 100 ml Dichlormethan extrahiert. Die kombinierten
organischen Anteile wurde über
Natri umsulfat getrocknet, filtriert und im Vakuum konzentriert.
Der Rückstand
wurde durch Silikagelchromatographie gereinigt, um das Produkt C
zu ergeben. 1H-NMR (CDCl3) δ 7,16 (t,
1H), 6,97 (d, 1H), 6,85 (t, 1H), 6,69 (dd, 1H), 5,98 (m, 1H), 4,77
(s, 1H), 4,02 (s, 4H), 2,63 (m, 2H), 2,46 (m, 2H), 1,92 (t, 2H).
-
Stufe
3: Zu einer Lösung
des Produkts aus Stufe 2 C (2,20 g, 9,48 mmol) in Dichlormethan
(60 ml) wurde bei 0°C
unter einer inerten Atmosphäre
Triethylamin (1,45 ml, 10,43 mmol), danach Trifluormethansulfonsäureanhydrid
(1,75 ml, 10,43 mmol) gegeben. Die Mischung wurde unter Rühren auf
Raumtemperatur erwärmt
und weitere 2 Stunden gerührt.
Die Mischung wurde mit Wasser extrahiert. Der organische Anteil
wurde aufgefangen, über
Natriumsulfat getrocknet, filtriert und im Vakuum konzentriert.
Der Rückstand
(1,36 g, 3,74 mmol) wurde ohne weitere Reinigung verwendet und in
Dioxan (80 ml) aufgenommen. Zu dieser Lösung wurden Bis(pinacolato)dibor
(1,14 g, 4,49 mmol), PdCl2(dppf) (0,16 g,
0,22 mmol), dppf (0,12 g, 0,22 mmol) und Kaliumacetat (1,10 g, 11,22
mmol) gegeben, und die Mischung wurde unter einer inerten Atmosphäre über Nacht
auf 80°C
erwärmt.
Die Mischung wurde auf Raumtemperatur abgekühlt und danach mit Ethylacetat
und Salzlösung
extrahiert. Der organische Anteil wurde aufgefangen, über Natriumsulfat
getrocknet, filtriert und im Vakuum konzentriert. Der Rückstand
wurde durch Silikagelchromatographie gereinigt, um das Produkt D
zu ergeben. Massenspektrum (ESI): 343,1.
-
Stufe
4: Eine Lösung
des Produkts von Stufe 3 D (0,64 g, 1,87 mmol), 2-Amino-4,6-dichlorpyrimidin (0,31
g, 1,87 mmol), Natriumcarbonat (0,79 g, 7,48 mmol) und Tetrakis(triphenylphosphin)palladium
(0,11 g, 0,09 mmol) in 60 ml 1/1 Acetonitril/Wasser wurde 3 Stunden
auf 90°C
erwärmt.
Die Reaktionsmischung wurde abgekühlt und mit Ethylacetat und
Salzlösung
extrahiert. Die Ethylacetatphase wurde aufgefangen, über Natri umsulfat
getrocknet, filtriert und im Vakuum konzentriert. Der Rückstand
wurde durch Silikagelchromatographie gereinigt, was zu Verbindung
E führte.
Massenspektrum (ESI): 344,1.
-
Stufe
5: Eine Lösung
des Produkts aus Stufe 4 E (0,60 g, 1,75 mmol) und 2-Furancarbonsäurehydrazid
(0,33 g, 2,62 mmol) in 15 ml n-Butanol wurde 2 Stunden auf 90°C erwärmt. Die
Mischung wurde auf Raumtemperatur abgekühlt und im Vakuum konzentriert.
Der Rückstand
wurde in 5 ml N,O-Bis(trimethylsilyl)acetamid aufgenommen und unter
einer Stickstoffatmosphäre
3 Stunden auf 120°C
erwärmt.
Die Mischung wurde abgekühlt,
auf Eiswasser gegossen und danach 4 Stunden gerührt. Die Mischung wurde mit
Ethylacetat und Salzlösung
extrahiert, über
Natriumsulfat getrocknet, filtriert und im Vakuum konzentriert.
Der Rückstand
wurde durch Silikagelchromatographie gereinigt, um Verbindung F
zu ergeben. Massenspektrum (ESI): 416,1. 1H-NMR
(DMSO-d6) δ 8,10 (s, 1H), 7,92 (m, 2H),
7,87 (s, 1H), 7,51 (s, 1H), 7,38 (m, 1H), 7,15 (t, 1H), 6,03 (s, 1H),
3,86 (s, 1H), 4, 02 (s, 4H), 255 (m, 2H), 2,33 (m, 2H), 1,77 (t,
2H).
-
Beispiel
12
Verbindung
93
-
Stufe
1: Eine Lösung
von 3-Acetylbenzolboronsäure
(2,00 g, 12,20 mmol), 2-Amino-4,6-dichlorpyrimidin (4,00 g, 24,40
mmol), Natriumcarbonat (6,47 g, 61,00 mmol) und Tetrakis(tri phenylphosphin)palladium (0,70
g, 0,61 mmol) in 100 ml 1/1 Acetonitril/Wasser wurde 3 Stunden auf
90°C erwärmt. Die
Reaktionsmischung wurde abgekühlt
und mit Ethylacetat und Salzlösung
extrahiert. Die Ethylacetatphase wurde aufgefangen, über Natriumsulfat
getrocknet, filtriert und im Vakuum konzentriert. Der Rückstand
wurde durch Silikagelchromatographie gereinigt, um das Produkt B
zu ergeben. Massenspektrum (ESI): 248,0.
-
Stufe
2: Zu einer Lösung
des Ketonprodukts von Stufe 1 B (3,00 g, 12,11 mmol) in 75 ml Ethylalkohol wurde
bei 0°C
Natriumborhydrid (0,92 g, 24,22 mmol) gegeben, danach auf Raumtemperatur
erwärmt
und eine Stunde gerührt.
Die Mischung wurde mit Ethylacetat extrahiert und mit Salzlösung gewaschen.
Der organische Anteil wurde aufgefangen, über Natriumsulfat getrocknet,
filtriert und im. Vakuum konzentriert. Der Rückstand wurde durch Silikagelchromatographie
gereinigt, um das Produkt C zu ergeben. Massenspektrum (ESI): 250,0.
-
Stufe
3: Eine Lösung
des Produkts C aus Stufe 2 (0,27 g, 1,08 mmol) und 2-Furancarbonsäurehydrazid
(0,33 g, 2,62 mmol) in 10 ml n-Butanol wurde über Nacht auf 90°C erwärmt. Die
Mischung wurde auf Raumtemperatur abgekühlt, mit Ethylacetat extrahiert
und mit Salzlösung
gewaschen. Der organische Anteil wurde aufgefangen, über Natriumsulfat
getrocknet, filtriert und im Vakuum konzentriert. Der Rückstand
D wurde in der nächsten
Stufe ohne weitere Reinigung verwendet. Massenspektrum (ESI): 340,1.
-
Stufe
4: Das Produkt aus Stufe 3 D (0,37 mg, 1,08 mmol) wurde in 5 ml
N,O-Bis(trimethylsilyl)acetamid aufgenommen und unter einer Stickstoffatmosphäre 3 Stunden
auf 120°C
erwärmt.
Die Mischung wurde abgekühlt,
auf Eiswasser gegossen und danach 4 Stunden gerührt. Die Mischung wurde mit
Ethylacetat und Salzlösung
extrahiert, über
Natriumsulfat getrocknet, fil triert und im Vakuum konzentriert.
Der Rückstand
wurde durch Silikagelchromatographie gereinigt, um die Verbindung
E zu ergeben. Massenspektrum (ESI): 322,1. 1H-NMR
(CDCl3) δ 8,05
(s, 1H), 7,86 (m, 1H), 7,64 (d, 1H), 7,47 (m, 2H), 7,41 (s, 1H),
7,25 (s, 1H), 6,60 (m, 1H), 6,05 (br s, 2H), 5,01 (m, 1H), 3,49
(d, 3H).
-
Die
folgende Verbindung wurde in ähnlicher
Weise hergestellt:
-
-
Beispiel
13
Verbindung
95
-
Eine
Mischung des in ähnlicher
Weise wie in Beispiel 10 hergestellten Ketalprodukts (113 mg, 0,27 mmol),
20 ml wässriger
HCl-Lösung
und 20 ml Aceton wurde über
Nacht auf 100°C
erwärmt.
Die Mischung wurde auf Raumtemperatur abgekühlt, mit Ethylacetat extrahiert
und mit Wasser gewaschen. Der organische Anteil wurde aufgefangen, über Natriumsulfat
getrocknet, filtriert und im Vakuum konzentriert. Der Rückstand wurde
durch Silikagelchromatographie gereinigt, um das Produkt zu ergeben.
Massenspektrum (ESI): 375,1. 1H-NMR (CDCl3) δ 7,57
(d, 1H), 7,55 (t, 1H), 7,31 (m, 3H), 7,20 (d, 1H), 6,95 (dd, 1H),
6,65 (br s, 2H), 6, 53 (m, 1H), 3,60 (t, 4H), 2,51 (t, 4H).
-
Beispiel
14
Verbindung
96
-
Zu
einer Lösung
des Ketonprodukts aus Beispiel 13 (55 mg, 0,15 mmol), einer 70%
Lösung
von Ethylamin in Wasser (0,01 ml, 0,16 mmol) in 5 ml Tetrahydrofuran
wurde Natriumtriacetoxyborhydrid (46 mg, 0,22 mmol) gegeben und
2 Stunden bei Raumtemperatur gerührt.
Die Mischung wurde mir wässriger
3 N Natriumhydroxidlösung
gequencht, mit Ethylacetat extrahiert und mit Salzlösung gewaschen.
Der organische Anteil wurde aufgefangen, über Natriumsulfat getrocknet,
filtriert und im Vakuum konzentriert. Der Rückstand wurde durch Silikagelchromatographie
gereinigt und chromatographiert, um das Produkt zu ergeben. Massenspektrum
(ESI): 404,1. 1H-NMR (CDCl3) δ 7,63 (d,
1H), 7,60 (t, 1H), 7,40 (d, 2H), 7,34 (t, 1H), 7,03 (dd, 1H), 6,60 (m,
1H), 6,00 (br s, 2H), 3,77 (d von t, 2H), 2,85 (t von d, 2H), 2,74
(m, 2H), 2,03 (m, 2H), 1,55 (q von t, 3H), 1,15 (m, 4H).
-
Die
folgenden Verbindungen wurden in ähnlicher Weise hergestellt:
-
-
-
Beispiel
15
Verbindung
105
-
Zu
einer Lösung
des Ketonprodukts von Beispiel 13 (60 mg, 0,16 mmol) in 5 ml Ethylalkohol
wurde Natriumborhydrid (12 mg, 0,32 mmol) gegeben. Die Mischung
wurde bei Raumtemperatur 1 Stunde gerührt. Die Mischung wurde mit
Ethylacetat extrahiert und mit Salzlösung gewaschen. Der organische
Anteil wurde aufgefangen, über
Natriumsulfat getrocknet, filtriert und im Vakuum konzentriert.
Der Rückstand
wurde durch Silikagelchromatographie gereinigt, um das Produkt zu
ergeben. Massenspektrum (ESI): 377,1. 1H-NMR (CDCl3) δ 7,63
(s, 1H), 7,60 (t, 1H), 7,40 (m, 2H), 7,34 (t, 1H), 7,25 (m, 1H),
7,03 (d, 1H), 6,60 (m, 1H), 6,11 (br s, 2H), 3,88 (m, 2H), 3,65
(m, 2H), 3,01 (m, 2H), 2,04 (m, 2H), 1,17 (br s, 3H).
-
Beispiel
16
Verbindung
106
-
Zu
einer Lösung
des Produkts von Beispiel 14 (35 mg, 0,0868 mmol), Diisopropylethylamin
(0,02 ml, 0,0955 mmol) in 3 ml DMF wurde Ethylchlorformiat (0,01
ml, 0,0955 mmol) gegeben. Die Mischung wurde bei Raumtemperatur
3 Stunde gerührt.
Die Mischung wurde im Vakuum konzentriert und der Rückstand
durch Silikagelchromatographie gereinigt, um das Produkt zu ergeben.
Massenspektrum (ESI): 476,1. 1H-NMR (CDCl3) δ 7,61
(d, 1H), 7,58 (t, 1H), 7,39 (m, 2H), 7,33 (t, 1H), 7,22 (d, 1H),
7,00 (dd, 1H), 6,58 (m, 1H), 6,22 (br s, 2H), 4,15 (q, 3H), 3,82
(d, 2H), 3,46 (m, 4H), 3,01 (m, 2H), 2,05 (br s, 1H), 1,59 (br s,
1H), 1,26 (t, 3H), 1,12 (t, 3H).
-
Die
folgende Verbindung wurde in ähnlicher
Weise mit dem Acylchlorid hergestellt:
-
-
Beispiel
17
Verbindung
108
-
Zu
einer Lösung
des Produkts von Beispiel 11, Stufe 5 (80 mg, 0,15 mmol) in 5 ml
einer Lösung
von 9/1 Ethanol/Ethylacetat wurde 10% Palladium auf Kohle (160 mg)
gegeben. Die Mischung wurde in einer Hydrierapparatur unter 40 psi
eine Stunde bei Raumtemperatur geschüttelt. Die Mischung wurde durch
ein Celitekissen filtriert und im Vakuum konzentriert. Der Rückstand
wurde durch Silikagelchromatographie gereinigt, um das Produkt zu
ergeben. Massenspektrum (ESI): 418,1. 1H-NMR
(CDCl3) δ 7,90
(s, 1H), 7,77 (d, 1H), 7,63 (d, 1H), 7,48 (m, 2H), 7,40 (m, 1H),
6,59 (dd, 1H), 6,12 (br s, 2H), 4,00 (s, 4H), 1,67–1,96 (m,
9H).
-
Beispiel
18
Verbindung
109
-
Die
Verbindung aus Beispiel 10, Stufe 7, wurde durch Behandlung mit
einer 4,0 M Lösung
von HCl in Dioxan bei Raumtemperatur über Nacht oder mit einer 50%
TFA-Lösung
in Dichlormethan in 30 Minuten unter N2 entschützt, unter
vermindertem Druck eingedampft und ohne weitere Reinigung verwendet. 1H-NMR (DMSO-d6) δ 7,97 (br
s, 2H), 7,94 (s 1H), 7,64 (s, 1H), 7,50 (m, 2H), 7,30 (t, 1H), 7,08
(dd, 1H), 7,00 (dd, 1H), 6,70 (dd, 1H), 3,70 (br s, 1H), 3,08 (m,
4H), 2,90 (m, 4H). Massenspektrum (ESI), M+1:
362,1.
-
Beispiel
19
Verbindung
110
-
Zu
einer Lösung
des Produkts aus Beispiel 18 (0,10 g, 0,25 mmol) und Diisopropylethylamin
(0,097 g, 0,75 mmol) in DMF (5 ml) wurde tropfenweise bei Raumtemperatur
unter N2 Propionylchlorid (0,025 g, 0,28 mmol)
gegeben. Nach 2 Stunden wurde Wasser zugegeben und das Produkt mit
Ethylacetat extrahiert, mit Natriumsulfat getrocknet und eingedampft.
Reinigung mit präparativer
DC an Silikagel führte
zu dem Produkt. 1H-NMR (CDCl3) δ 7,63 (dd,
1H), 7,60 (m, 1H), 7,46 (d, 1H), 7,40 (s, 1H), 7,37 (d, 1H), 7,24
(m, 1H), 7,01 (dd, 1H), 6,60 (dd, 1H), 6,06 (br s, 2H), 3,82 (t,
2H), 3,65 (t, 2H), 3,25 (m, 4H), 2,40 (q, 2H), 1,19 (t, 3H). Massenspektrum
(ESI): 418,1.
-
Die
folgenden Verbindungen wurden in ähnlicher Weise hergestellt:
-
-
Beispiel
20
Verbindung
116
-
Zu
einer Lösung
des Produkts aus Beispiel 18 (0,125 g, 0,35 mmol) und Diisopropylethylamin
(0,134 g, 1,04 mmol) in DMF (5 ml) wurde tropfenweise bei Raumtemperatur
unter N2 Isopropylchlorformiat (0,7 ml, 0,7
mmol) gegeben. Nach 2 Stunden wurde Wasser zugegeben und das Produkt
mit Ethylacetat extrahiert, mit Natriumsulfat getrocknet und eingedampft.
Reinigung mit präparativer
DC an Silikagel führte
zu dem Produkt. 1H-NMR (CDCl3) δ 7,63 (m,
1H), 7,59 (m, 1H), 7,44 (d, 1H), 7,38 (s, 1H), 7,35 (d, 1H), 7,24
(m, 1H), 7,02 (dd, 1H), 6,58 (dd, 1H), 6,22 (br s, 1H), 4,95 (m,
1H), 3,65 (m, 4H), 3,22 (m, 4H), 1,26 (d, 6H); Massenspektrum (ESI),
M+1: 448,1.
-
Die
folgenden Verbindungen wurden in ähnlicher Weise hergestellt:
-
-
Beispiel
21
Verbindung
119
-
Zu
einer Lösung
des Produkts aus Beispiel 18 (0,11 g, 0,30 mmol) und Diisopropylethylamin
(0,043 g, 0,058 mmol) in DMF (5 ml) wurde tropfenweise bei Raumtemperatur
unter N2 Methylsulfonylchlorid (0,038 ml, 0,026
mmol) gegeben. Nach 3 Stunden wurde Wasser zugegeben und das Produkt
mit Ethylacetat extrahiert, mit Natriumsulfat getrocknet und eingedampft.
Reinigung mit präparativer
DC an Silikagel führte
zu dem Produkt. 1H-NMR (CDCl3) δ 7,62 (m,
2H), 7,50 (d, 1H), 7,40 (m, 2H), 7,23 (m 1H), 7,04 (dd, 1H), 6,62
(dd, 1H), 6,00 (br s, 2H), 3,37–3,42
(m, 8H), 2,82 (s, 3H). Massenspektrum (ESI): 440,1.
-
Die
folgende Verbindung wurde in ähnlicher
Weise hergestellt:
-
-
Beispiel
22
Verbindung
121
-
Stufe
1: Zu einer Lösung
des Produkts aus Beispiel 18 und Diisopropylethylamin in DMF (5
ml) wurde tropfenweise bei 0°C
unter N2 Chloracetylchlorid gegeben. Die
Mischung wurde auf Raumtemperatur erwärmt und über Nacht gerührt. Dann
wurde Wasser zugegeben und das Produkt mit Ethylacetat extrahiert,
mit Natriumsulfat getrocknet und eingedampft. Die Reinigung erfolgte
mit Säulenchromatographie
an Silikagel unter Verwendung von Ethylacetat, um die Zwischenstufe
B zu liefern. Massenspektrum (ESI), M+1:
438,1.
-
Stufe
2: Verbindung B aus Stufe 1 (0,11 g, 0,25 mmol) wurde mit Piperidin
im Überschuss
(10 Äquivalente)
in DMF (5 ml) über
Nacht bei Raumtemperatur unter N2 behandelt.
Die Mischung wurde unter vermindertem Druck eingedampft, und das
Produkt C wurde durch präparative
DC unter Verwendung von Ethylacetat/Methanol (9:1) gereinigt. 1H-NMR (CDCl3) δ 7,59 (m,
1H), 7,54 (m, 1H), 7,40 (d, 1H), 7,33 (m, 2H), 7,20 (m, 1H), 6,97
(dd, 1H), 6,55 (dd, 1H), 6,17 (br s, 2H), 3,80 (m, 2H), 3,74 (m,
2H), 3,20 (m, 4H), 3,13 (s, 2H), 2,38 (m, 4H), 1,49 (m, 4H), 1,38
(m, 2H). Massenspektrum (ESI), M+1: 487,1.
-
Die
folgenden Verbindungen wurden in ähnlicher Weise hergestellt:
-
-
-
-
Beispiel
23
Verbindung
130
-
Stufe
1: Zu einer Lösung
des Produkts aus Beispiel 18 (0,145 g, 0,4 mmol) und Benzaldehyd
(0,047 g, 0,44 mmol) in Dichlormethan (10 ml) wurde bei Raumtemperatur
unter N2 Natriumtriacetoxyborhydrid (0,127 g,
0,6 mmol) gegeben. Nach 5 Stunden wurde eine 2,0 M Lösung von
Natriumhydroxid zugegeben und das Produkt mit Dichlormethan extrahiert,
mit Natriumsulfat getrocknet und eingedampft. Reinigung mit präparativer
DC führte
zu dem Produkt. 1H-NMR (CDCl3) δ 7,63 (dd,
1H), 7,58 (m, 1H), 7,20–7,41
(m, 9H), 7,01 (dd, 1H), 6,59 (dd, 1H), 6,04 (br s, 2H), 3,59 (s,
2H), 3,29 (t, 4H), 2,64 (t, 4H). Massenspektrum (ESI), M+1: 452,1.
-
Die
folgenden Verbindungen wurden in ähnlicher Weise hergestellt:
-
-
Beispiel
24
Verbindung
135
-
Stufe
1: Zu einer Lösung
des Produkts aus Beispiel 18 (1,22 g, 3,38 mmol) und Chloracetaldehyd
(0,64 g, 4,06 mmol 50% Lösung
in Wasser) in Dichlormethan (60 ml) wurde bei Raumtemperatur unter
N2 Natriumtriacetoxyborhydrid (1,08 g, 5,07
mmol) gegeben. Nach 5 Stunden wurde eine 2,0 M Lösung von Natriumhydroxid zugegeben
und die resultierende Verbindung B mit Dichlormethan extrahiert,
mit Natriumsulfat getrocknet und eingedampft. Die Reinigung erfolgte
durch Säulenchromatographie.
Massenspektrum (ESI), M+1: 424,1.
-
Stufe
2: Die Verbindung aus Stufe 1 B (0,09, 0,21 mmol) wurde mit Morpholin
im Überschuss
(10 Äquivalente)
in DMF (5 ml) über
Nacht bei Raumtemperatur unter N2 behandelt.
Die Mischung wurde unter vermindertem Druck eingedampft und durch
präparative
DC unter Verwendung von Ethylacetat/Methanol (9:1) gereinigt, was
zu Verbindung C führte. 1H-NMR (CDCl3) δ 7,64 (dd,
1H), 7,58 (m, 1H), 7,30–7,44
(m, 3H), 7,25 (m, 2), 7,01 (dd, 1H), 6,60 (dd, 1H), 6,02 (br s,
2H), 3,75 (m, 4H), 3,33 (m, 4H), 2,75 (m, 2H), 2,65 (m, 2H), 2,57 (m,
4H), 2,11 (m, 4H). Massenspektrum (ESI), M+1:
475,1.
-
Die
folgenden Verbindungen wurden in ähnlicher Weise hergestellt:
-
-
Beispiel
25
Verbindung
139
-
Das
Produkt aus Beispiel 18 (0,10 g, 0,28 mmol) wurde für einen
Zeitraum von 12 Stunden unter Stickstoff mit 1-Fluor-2-nitrobenzol (0,079
g, 0,56 mmol) und Triethylamin (0,085 g, 0,84 mmol) in DMF (5 ml)
auf 100°C
erwärmt.
Die Mischung wurde unter vermindertem Druck eingedampft und durch
präparative
DC unter Verwendung von Ethylacetat/Hexan (7:3) gereinigt. 1H-NMR (CDCl3) δ 7,89 (dd,
1H), 7,65 (m, 2H), 7,36–7,63 (m,
4H), 7,19– 7,26
(m, 2H), 7,00–7,11
(m, 2H), 6,59 (dd, 1H), 6,11 (br s, 2H), 3,43 (m, 4H), 3,26 (m,
4H). Massenspektrum (ESI), M+1: 483,1.
-
Beispiel
26
Verbindung
140
-
Eine
Mischung des wie in Beispiel 4 hergestellten Phenolderivats (0,054
g, 0,18 mmol), Chlorethylmorpholinhydrochlorid (0,041 g, 0,22 mmol),
Kaliumcarbonat (0,076 g, 0,55 mmol) und Kaliumiodid (0,031 g, 0,18 mmol)
wurde unter N2 für einen Zeitraum von 19 Stunden
in Acetonitril (10 ml) auf 50°C
erwärmt.
Die Mischung wurde mit Ethylacetat verdünnt und filtriert, unter Vakuum
konzentriert und durch Chromatographie an Silikagel gereinigt. 1H-NMR (CD3OD) δ 7,76 (m,
1H), 7,71 (m, 1H), 7,66 (m, 1H), 7,38 (m, 2H), 7,24 (dd, 1H), 7,03
(dd, 1H), 6,66 (dd, 1H), 4,23 (t, 2H), 3,73 (t, 4H), 2,85 (t, 2H),
2,63 (t, 4H). Massenspektrum (ESI), Massenspektrum (ESI) M+1: 407,1.
-
Die
folgenden Verbindungen wurden in ähnlicher Weise hergestellt:
-
-
Beispiel
27
Verbindung
147
-
Stufe
1: Eine Mischung aus 2-Amino-6-chlor-4-pyrimidinolmonohydrat (2,0
g, 13,74 mmol) und 2-Furancarbonsäurehydrazid (1,91 g, 15,12
mmol) wurde in n-Butanol (50 ml) 20 Stunden auf 100°C erwärmt. Die Mischung
wurde im Vakuum konzentriert, um einen festen Rückstand B zu liefern, der ohne
Reinigung verwendet wurde. Massenspektrum (ESI), M+1:
236,1.
-
Stufe
2: Das Produkt aus Stufe 1 B (3,24 g, 13,74 mmol) wurde in N,O-Bis(trimethylsilyl)acetamid
(20,5 ml, 82,44 mmol) über
Nacht auf 120°C
erhitzt. Die Mischung wurde abgekühlt, und danach wurden langsam Methanol
und Wasser zugegeben und 4 Stunden auf Rückfluss erwärmt. Die resultierende Mischung
wurde auf Raumtemperatur abgekühlt
und der Niederschlag C durch Filtration aufgefangen. Massenspektrum
(ESI), M+1: 218,0.
-
Stufe
3: Trifluormethansulfonsäureanhydrid
(1,52 g, 5,37 mmol) wurde tropfenweise bei 0°C unter N2 zu
einer Lösung
des Produkts aus Stufe 2 C (1,06 g, 4,88 mmol) und Triethylamin
(0,54 g, 5,37 mmol) in Dichlormethan gegeben. Nach einer Stunde
wurde die Mischung auf Raumtemperatur erwärmt und eine gesättigte Lösung von
Natriumbicarbonat zugefügt.
Die resultierende Mischung wurde mit Dichlormethan extrahiert, mit
Natriumsulfat getrocknet und nach Filtration eingedampft. Säulen chromatographie
an Silikagel führte
dann zu dem Produkt D. Massenspektrum (ESI), M+1:
350,1.
-
Stufe
4: Eine Mischung des Produkts aus Stufe 3 D (0,25 g, 0,72 mmol),
2-Pyridyltributylzinn (0,32 g, 0,86 mmol) und Pd(dppf)Cl2 (0,029 g, 0,036 mmol) wurde in DMF (5 mL)
64 Stunden unter N2 auf 80°C erwärmt. Es
wurde Wasser zugegeben, anschließend mit Ethylacetat extrahiert,
mit Natriumsulfat getrocknet und danach filtriert. Der Rückstand
E wurde nach dem Eindampfen durch Chromatographie an Silikagel gereinigt. 1H-NMR (DMSO-d6) δ 8,70 (dd,
1H), 8,30 (d, 1H), 8,01 (br, S, 2H), 7,95 (m, 2H), 7,82 (s, 1H),
7,47 (m, 1H), 7,22 (dd, 1H), 6,72 (dd, 1H). Massenspektrum (ESI),
M+1: 279,0.
-
Beispiel
28
Verbindung
148
-
Stufe
1: Eine Mischung aus 2-Hydroxymethyl-5-brompyridin (2,17 g, 11,54
mmol), Bis(pinacolato)dibor (2,93 g, 11,54 mmol), PdCl2(dppf)
(0,57 g, 0,69 mmol) und Kaliumacetat (3,40 g, 34,62 mmol) wurde
unter N2 über Nacht in 1,4-Dioxan (65
ml) auf 80°C
erwärmt.
Die Mischung wurde auf Raumtemperatur abgekühlt, danach wurden 2-Amino-4,6-dichlorpyrimidin
(3,79 g, 23,08 mmol) und eine 2,0 M Lösung von Natriumbicarbonat (6,12 g
in 20 ml Wasser) zugegeben. Die resultierende Mischung wurde 20
Stunden auf 80°C
erwärmt,
abgekühlt
und danach mit Ethylacetat und Wasser verdünnt. Der organische Extrakt
wurde mit Salzlösung
gewaschen, mit Natriumsulfat getrocknet und eingedampft. Der Rückstand
wurde durch Säulenchromatographie
an Silikagel gereinigt, um das Produkt B zu ergeben. 1H-NMR
(CD3OD) δ 9,21
(d, 1H), 8,55 (dd, 1H), 7,76 (d, 1H), 7,75 (d, 1H), 7,49 (s, 1H),
7,25 (dd, 1H), 6,66 (dd, 1H), 4,78 (2H). Massenspektrum (ESI), M+1: 309,1.
-
Stufen
2 und 3: Wie die Stufen 2 und 3 von Beispiel 4. 1H-NMR (CD3OD) δ 9,21 (d,
1H), 8,55 (dd, 1H), 7,76 (d, 1H), 7,75 (d, 1H), 7,49 (s, 1H), 7,25
(dd, 1H), 6,66 (dd, 1H), 4,78 (2H). Massenspektrum (ESI), M+1: 309,1.
-
Beispiel
29
Verbindung
149
-
Stufe
1: Zu einer Lösung
von 3-Chlorphenol (0,062 g, 0,48 mmol) und Natriumhydrid (0,058
g, 1,44 mmol 60% NaH in Mineralöl)
in DMF (5 ml) wurde bei Raumtemperatur unter N2 das
Produkt aus Stufe 1 von Beispiel 22 (0,1 g, 0,24 mmol) gegeben,
und die Mischung wurde über
Nacht gerührt.
Dann wurde Wasser zugegeben und die Mischung mit Ethylacetat extrahiert,
mit Natriumsulfat getrocknet, filtriert und unter Vakuum konzentriert.
Reinigung durch präparative
DC an Silikagel unter Verwendung von Ethylacetat ergab das Produkt. 1H-NMR (CDCl3) δ 7,61 (s,
1H), 7,60 (m, 1H), 7,47 (m, 1H), 7,22 (m, 3H), 7,00 (m, 2H), 6,88
(dd, 1H), 6,60 (dd, 1H), 5,93 (br, s, 2H), 4,75 (s, 2H), 3,80 (m,
4H), 3,17 (m, 4H). Massenspektrum (ESI), M+1:
530,1.
-
Beispiel
30
Verbindung
150
-
Stufe
1: t-Butyllithium in Pentan (11,56 ml, 19,7 mmol 1,7 N in Pentan)
wurde tropfenweise zu einer Lösung
von 3-Bromphenol
(1,0 g, 5,8 mmol) in THF (86 ml) gegeben und unter N2 auf –78°C abgekühlt. Die Mischung
wurde 10 Minuten gerührt.
Bei –78°C wurde Benzyl-4-oxo-1-piperidincarboxylat
(1,35 g, 5,8 mmol) in THF (14 ml) zugegeben. Die Mischung wurde
auf Raumtemperatur erwärmt,
2 Stunden gerührt
und zwischen gesättigtem
NaHCO3 und EtOAc partitioniert. Die organischen
Materialien wurden mit H2O, Salzlösung gewaschen, über MgSO4 getrocknet, filtriert, im Vakuum konzentriert
und chromatographiert, um B zu ergeben. Massenspektrum (ESI): 328,1,
310,0.
-
Stufe
2: Triethylsilan (1,45 g, 12,5 mmol) und Trifluoressigsäure (1,42
g, 12,5 mmol) wurde zu einer Lösung
des Produkts aus Stufe 1 B (0,87 g, 2,66 mmol) in CH2Cl2 (23 ml) gegeben und unter N2 auf –78°C abgekühlt. Es
wurde weitere 2 Stun den bei –78°C und 20
Stunden bei Raumtemperatur gerührt.
Die Mischung wurde zwischen gesättigtem
NaHCO3 und CH2Cl2 partitioniert. Die organischen Materialien
wurden mit H2O, Salzlösung gewaschen, über MgSO4 getrocknet, filtriert, im Vakuum konzentriert
und chromatographiert, um C zu ergeben. Massenspektrum (ESI): 312,0.
-
Stufe
3: Das Produkt aus Stufe 2 C (458 mg, 1,47 mmol) wurde wie in Beispiel
11, Stufe 3 behandelt, um D zu ergeben. Massenspektrum (ESI): 444,1.
-
Stufe
4: Das Produkt aus Stufe 3 D (412 mg, 0,93 mmol) wurde wie in Beispiel
11, Stufe 4 behandelt, um E zu ergeben. Massenspektrum (ESI): 422,1.
-
Stufe
5: Das Produkt aus Stufe 4 E (250 mg, 0,59 mmol) wurde mit 2-Amino-4,6-dichlorpyrimidin
(195 mg, 1,18 mmol) wie in Beispiel 4, Stufe 1 behandelt (außer dass
4 Äquivalente
Na2CO3 verwendet
wurden), um F zu ergeben. Massenspektrum (ESI): 423,1.
-
Stufe
6: Das Produkt aus Stufe 5 F (155 mg, 0,37 mmol) wurde mit 2-Furancarbonsäurehydrazid
(60 mg, 0,48 mmol) in n-BuOH
(3 ml) kombiniert. Die Mischung wurde 20 Stunden gerührt und
auf 110°C
erwärmt. Die
Temperatur wurde auf Raumtemperatur sinken gelassen und im Vakuum
konzentriert, um einen Feststoff G zu ergeben, der ohne weitere
Reinigung weiterverarbeitet wurde. Massenspektrum (ESI): 513,1.
-
Stufe
7: Das Produkt aus Stufe 6 G (188 mg, 0,37 mmol) wurde mit N,O-Bis(trimethylsilyl)acetamid (1,65
g, 8,5 mmol) kombiniert. Die Mischung wurde 4 Stunden unter N2 gerührt
und auf 110°C
erwärmt.
Die Mischung wurde auf Raumtemperatur abgekühlt und im Vakuum konzentriert.
Der Rückstand
wurde in 2:1 H2O/MeOH aufgenommen, 2 Stunden
auf 100°C
erwärmt,
im Vakuum konzentriert und zwischen H2O
und EtOAc partitioniert. Die or ganischen Materialien wurden mit
H2O, Salzlösung gewaschen, über MgSO4 getrocknet, filtriert, im Vakuum konzentriert
und chromatographiert, um einen Feststoff H zu ergeben. Massenspektrum
(ESI): 495,1. 1H-NMR (CDCl3) δ 7,93 (s,
1H), 7,82 (d, 1H), 7,64 (m, 2H), 7,28–7,52 (m, 7H), 6,78 (bs, 1H), 6,63
(m, 1H); 5,19 (s, 2H), 4,38 (bs, 2H), 2,92 (bs, 2H), 2,80 (m, 1H),
1,91 (d, 2H), 1,70 (m, 2H).
-
Beispiel
31
Verbindung
151
-
Das
Produkt aus Beispiel 30, Stufe 7 (54 mg, 0,11 mmol) wurde mit Ammoniumacetat
(7 mg, 0,091 mmol) und 10% Pd/C (8 mg) in MeOH (3 ml) kombiniert.
Die Mischung wurde unter H2 bei atmosphärischem Druck
und Raumtemperatur 3 Stunden gerührt.
Die Mischung wurde durch Celite filtriert, und das Filtrat wurde im
Vakuum konzentriert. Das Filtrat wurde zwischen gesättigtem
NaHCO3 und CH2Cl2 partitioniert. Die organischen Materialien
wurden mit Salzlösung
gewaschen, über
MgSO4 getrocknet, filtriert und im Vakuum
konzentriert, um einen Feststoff zu ergeben. Massenspektrum. (ESI):
361,1. 1H-NMR (CD3OD) δ 8,00 (s,
1H), 7,94 (d, 1H), 7,76 (s, 1H), 7,34–7,44 (m, 3H), 7,25 (m, 1H),
6,67 (m, 1H), 3,23 (d, 2H), 2,82 (m, 3H), 1,93 (d, 2H), 1,80 (m,
2H).
-
Beispiel
32
Verbindung
152*
-
Eine
Mischung aus 2-(Aminomethyl)pyridin (92 mg, 0,85 mmol), dem Produkt
aus Beispiel 2, Stufe 2 (100 mg, 0,43 mmol) und K2CO3 (177 mg, 1,28 mmol) in nBuOH (2 ml) wurde
48 Stunden in einem verschlossenen Röhrchen auf 120°C erwärmt. Die
Mischung wurde auf Raumtemperatur abgekühlt, filtriert und im Vakuum
konzentriert. Der Rückstand
wurde an Silikagel chromatographiert, um einen Feststoff zu ergeben.
Massenspektrum (ESI): 308,1, 1H-NMR (CDCl3) δ 8,57
(m, 1H), 7,68 (m, 1H), 7,57 (m, 1H), 7,34 (d, 1H), 7,21 (m, 1H),
7,14 (dd, 1H), 6,55 (m, 1H), 6,39 (bs, 1H), 6,05 (s, 2H), 5,87 (s,
1H), 4,54 (d, 2H).
-
Die
folgenden Verbindungen wurden in ähnlicher Weise hergestellt:
-
-
Beispiel
33
Verbindung
156*
-
Stufe
1: 3-(Hydroxymethyl)benzonitril (2,0 g, 15 mmol) wurde mit Triethylamin
(9,11 g, 90 mmol) in CH2Cl2 (200
ml) kombiniert. Die Mischung wurde unter N2 auf
0°C abgekühlt und
SOCl2 (8,94 g, 75 mmol) wurde tropfenweise
zugegeben. Die Mischung wurde eine Stunde bei 0°C gerührt, mit Eis behandelt, mit
gesättigtem
NaHCO3 neutralisiert und mit CH2Cl2 extrahiert. Die organischen Materialien
wurden mit H2O und Salzlösung gewaschen, über MgSO4 getrocknet, filtriert und im Vakuum konzentriert,
um einen Feststoff B zu ergeben, mit dem ohne weitere Reinigung
weitergearbeitet wurde. 1H-NMR (CDCl3) δ 7,70
(s, 1H), 7,63 (m, 2H), 7,49 (t, 1H), 4,59 (s, 2H).
-
Stufe
2: Das Produkt von Stufe 1 B (1,04 g, 6,86 mmol), 2,4-Difluorphenylpiperazin
(1,24 g, 6,24 mmol), K2CO3 (2,59
g, 19 mmol) und KI (1,04 g, 6,24 mmol) wurde in CH3CN
(75 ml) kombiniert und 20 Stunden unter N2 unter
Rückfluss
gehalten. Die Mischung wurde filtriert, im Vakuum konzentriert und
an Silikagel chromatographiert, um C zu ergeben. Massenspektrum
(ESI): 314,1.
-
Stufe
3: Eine Lösung
des Produkts von Stufe 2 C (0,96 g, 3,06 mmol). in THF (5 ml) wurde
tropfenweise zu einer Suspension von LAH (0,128 g, 3,37 mmol) in
THF gegeben und unter N2 auf 0°C gekühlt. Die
Mischung wurde eine Stunde bei Raumtemperatur gerührt, auf
0°C abgekühlt, mit
Eis behandelt, mit 1 N NaOH gequencht und wieder auf Raumtemperatur
erwärmt.
Der resultierende Feststoff wurde filtriert und mit THF gewaschen.
Das Filtrat wurde im Vakuum konzentriert, um D zu ergeben, das ohne
weitere Reinigung weiterverarbeitet wurde. Massenspektrum (ESI):
318,1.
-
Stufe
4: Das Produkt aus Stufe 3 D (270 mg, 0,85 mmol) wurde mit dem Produkt
aus Beispiel 2, Stufe 2 (100 mg, 0,43 mmol) wie in Beispiel 32 kombiniert,
um einen Feststoff E zu ergeben. Massenspektrum (ESI): 517,1. 1H-NMR (CD3OD) δ 7,68 (s,
1H), 7,39 (s, 1H), 7,27 (m, 3H), 7,21 (m, 1H), 7,07 (d, 1H), 6,92
(m, 3H), 6,60 (dd, 1H), 5,69 (s, 1H), 4,54 (s, 2H), 3,61 (s, 2H),
2,92 (m, 4H), 2,60 (m, 4H).
-
Die
folgenden Verbindungen wurden in ähnlicher Weise hergestellt:
-
-
Beispiel
34
Verbindung
160*
-
Stufe
1: 3-(Chlormethyl)-5-cyanopyridiniumhydrochlorid [hergestellt wie
in Chem. Pharm. Bull. 38, 1990, 2446–58; Chem. Eur. J. 3, 1997,
410–16
beschrieben] (260 mg, 1,38 mmol), 2,4-Difluorphenylpiperazin (228
mg, 1,15 mmol) und Triethylamin (326 mg, 3,22 mmol) wurden in DMF
(7 mL) kombiniert. Die Mischung wurde bei Raumtemperatur 48 Stunde
gerührt.
Die Mischung wurde im Vakuum konzentriert und zwischen H2O und CH2Cl2 partitioniert. Die organischen Materialien
wurden mit Salzlösung
gewaschen, über
MgSO4 getrocknet, filtriert, im Vakuum konzentriert
und chromatographiert, um einen Feststoff B zu ergeben. Massenspektrum
(ESI): 315,1.
-
Stufe
2: Das Produkt aus Stufe 1 B (223 mg, 0,71 mmol), MeOH (3 ml), THF
(3 ml), 25% NH4OH (aq) und Raney-Nickel
wurden in einer Parr-Flasche kombiniert, mit EtOH (0,050 g) nass
gewaschen und 24 Stunden unter 50 psi hydriert. Die Mischung wurde über Celite
filtriert, und das Filtrat wurde und im Vakuum konzentriert, um
einen Feststoff C zu ergeben, der ohne weitere Reinigung weiterverarbeitet
wurde. Massenspektrum (ESI): 319,1.
-
Stufe
3: Das Produkt aus Stufe 2 C (230 mg, 0,72 mmol) wurde mit dem Produkt
aus Beispiel 2, Stufe 2 (85 mg, 0,36 mmol) wie in Beispiel 32 kombiniert,
um einen Feststoff D zu ergeben. Massenspektrum (ESI): 518,1, 1H-NMR (CDCl3) δ 8,47 (s,
1H), 8,37 (s, 1H), 7,88 (s, 1H), 7,69 (s, 1H), 7,08 (d, 1H), 6,86
(m, 4H), 6,60 (dd, 1H), 5,77 (s, 1H), 4,62 (s, 2H), 3,62 (s, 2H),
3,04 (m, 2H), 2,89 (m, 2H), 2,65 (m, 2H), 2,56 (m, 2H).
-
Beispiel
35
Verbindung
161*
-
Stufe
1: 4-(2-Chlorethyl)morpholinhydrochlorid (1,88 g, 10 mmol) und 2-Cyanophenol
(1,0 g, 8,4 mmol) wurden wie in Beispiel 33, Stufe 2 kombiniert,
um ein Öl
B zu ergeben. Massenspektrum (ESI): 233,0.
-
Stufe
2: Das Produkt aus Stufe 1 B (502 mg, 2,16 mmol) wurde wie in Beispiel
34, Stufe 2 hydriert, um ein Öl
C zu ergeben. Massenspektrum (ESI): 237,0.
-
Stufe
3: Das Produkt aus Stufe 2 C (202 mg, 0,85 mmol) wurde mit dem Produkt
aus Beispiel 2, Stufe 2 (100 mg, 0,43 mmol) wie in Beispiel 32 kombiniert,
um einen Feststoff D zu ergeben. Massenspektrum (ESI): 436,1. 1H-NMR (CD3OD) δ 7,69 (s,
1H), 7,28 (m, 2H), 7,10 (dd, 15 1H), 6,93 (m, 2H), 6,60 (dd, 1H),
5,75 (s, 1H), 4,48 (s, 2H), 4,21 (t, 2H), 3,70 (m, 4H), 2,86 (t,
2H), 2,61 (m, 4H).
-
Die
folgende Verbindung wurde in ähnlicher
Weise hergestellt:
-
-
Beispiel
36
Verbindung
163*
-
Stufe
1: Eine Lösung
von 2-Cyanophenol (1,0 g, 8,4 mmol) in DMF (30 ml) wurde tropfenweise
unter N2 zu einer Suspension von NaH (60%
in Öl,
502 mg, 12,6 mmol) in DMF (12 ml) gegeben, die auf 0°C gekühlt war.
Nachdem die Zugabe abgeschlossen war, wurde die Mischung 20 Minuten
bei Raumtemperatur gerührt. 2-Bromethylmethylether
(1,4 g, 10 mmol) wurde zugegeben und die Mischung 70 Stunden gerührt. Die
Mischung wurde im Vakuum konzentriert. Der resultierende Feststoff
wurde in Hexan suspendiert, welches abdekantiert wurde. Der nicht
gelöste
Feststoff wurde zwischen H2O und EtOAc partitioniert.
Die organischen Materialien wurden mit H2O,
Salzlösung
gewaschen, über
MgSO4 getrocknet, filtriert, im Vakuum konzentriert,
mit der obigen Hexanwäsche
kombiniert und chromatographiert, um ein Öl B zu ergeben. Massenspektrum
(ESI): 178,1.
-
Stufe
2: Das Produkt aus Stufe 1 (623 mg, 3,52 mmol) wurde wie in Beispiel
34, Stufe 2 hydriert, um ein Öl
C zu ergeben. Massenspektrum (ESI): 182,0.
-
Stufe
3: Das Produkt aus Stufe 2 (152 mg, 0,85 mmol) wurde mit dem Produkt
aus Beispiel 2, Stufe 2 (100 mg, 0,43 mmol) wie in Beispiel 32 kombiniert,
um ein Öl
D zu ergeben. Massenspektrum (ESI): 381,1. 1H-NMR
(CD3OD) δ 7,68
(s, 1H), 7,24 (m, 2H), 7,09 (dd, 1H), 6,93 (m, 2H), 6,60 (m, 1H),
5,75 (s, 1H), 4,50 (s, 2H), 4,19 (m, 2H), 3,80 (m, 2H), 3,45 (s,
3H).
-
Beispiel
37
Verbindung
164*
-
Stufe
1: 2-(2-Methoxyethoxy)benzaldehyd [hergestellt wie in Chem. Pharm.
Bull. 35, 1987, 1953–68 beschrieben]
(400 mg, 2,22 mmol) und 2-Methoxyethylamin (228 mg, 1,15 mmol) wurden
in MeOH (10 mL) kombiniert, und die Mischung wurde 20 Stunden bei
Raumtemperatur unter N2 gerührt. Die
Mischung wurde auf 0°C
abgekühlt,
und NaBH4 (134 mg, 3,55 mmol) wurde zugefügt. Die
Mischung wurde 20 Stunden bei Raumtemperatur unter N2 gerührt. Die
Mischung wurde zwischen gesättigtem
NaHCO3 und Et2O
partitioniert. Die organischen Materialien wurden mit H2O
und Salzlösung
gewaschen und über
MgSO4 getrocknet. Die Mischung wurde filtriert
und im Vakuum konzentriert, um ein Öl B zu ergeben, das ohne weitere
Reinigung weiterverarbeitet wurde. Massenspektrum (ESI): 240,1.
-
Stufe
2: Das Produkt aus Stufe 1 B (183 mg, 0,77 mmol) wurde mit dem Produkt
aus Beispiel 2, Stufe 2 (90 mg, 0,38 mmol) wie in Beispiel 32 kombiniert,
um einen Feststoff C zu ergeben. Massenspektrum (ESI): 439,1, 1H-NMR (CDCl3) δ 7,68 (s,
1H), 7,21 (t, 1H), 7,09 (m, 2H), 6,98 (d, 2H), 6,88 (t, 1H), 6,60
(m, 1H), 5,80 (s, 1H), 4,80 (s, 2H), 4,18 (m, 2H), 3,77 (m, 4H),
3,62 (t, 2H), 3,42 (s, 3H), 3,34 (s, 3H).
-
Die
folgenden Verbindungen wurden in ähnlicher Weise hergestellt:
-
-
Beispiel
38
Verbindung
171*
-
Stufe
1: 2-Cyanobenzaldehyd (500 mg, 3,8 mmol), Morpholin (365 mg, 4,2
mmol) und NaBH(OAc)3 (1,21 g, 5,72 mmol)
wurden in THF (20 ml) kombiniert. Die Mischung wurde 20 Stunden
bei Raumtemperatur unter N2 gerührt. Die
Mischung wurde mit 1 N NaOH gequencht und zwischen H2O
und EtOAc partitioniert. Die organischen Materialien wurden mit
H2O und Salzlösung gewaschen, danach über MgSO4 getrocknet, filtriert, im Vakuum konzentriert
und chromatographiert, um ein Öl
B zu ergeben. Massenspektrum (ESI): 203,0.
-
Stufe
2: Das Produkt aus Stufe 1 (172 mg, 0,85 mmol) wurde wie in Beispiel
34, Stufe 2 hydriert, um ein Öl
C zu ergeben. Massenspektrum (ESI): 207,0.
-
Stufe
3: Das Produkt aus Stufe 2 C (160 mg, 0,77 mmol) wurde mit dem Produkt
aus Beispiel 2, Stufe 2 (90 mg, 0,38 mmol) wie in Beispiel 32 kombiniert,
um einen Feststoff D zu ergeben. Massenspektrum (ESI): 406,1. 1H-NMR (CD3OD) δ 7,69 (s,
1H), 7,41 (d, 1H), 7,24 (m, 3H), 7,11 (s, 1H), 6,61 (m, 1H), 5,77
(s, 1H), 4,60 (s, 2H), 3,74 (t, 4H), 3,59 (s, 2H), 2,48 (s, 4H).
-
Beispiel
39
Verbindung
172*
-
Stufe
1: 2-Brombenzonitril (3 g, 16,5 mmol) und t-Butyl-1-piperazincarboxylat
(3,68 g, 19,8 mmol) wurden wie in Beispiel 10, Stufe 1 kombiniert
(außer
dass die Rückflusszeit
20 Stunden betrug und das Rohprodukt chromatographiert wurde), um
ein Öl
B zu ergeben. 1H-NMR (CD3OD) δ 7,60 (m,
2H), 7,14 (m, 2H), 3,62 (s, 4H), 3,14 (t, 4H), 1,49 (s, 9H).
-
Stufe
2: Das Produkt aus Stufe 1 (440 mg, 1,53 mmol) wurde wie in Beispiel
34, Stufe 2 hydriert, um ein Öl
C zu ergeben. Massenspektrum (ESI): 292,0.
-
Stufe
3: Das Produkt aus Stufe 2 C (477 mg, 1,64 mmol) und das Produkt
aus Beispiel 2, Stufe 2 (193 mg, 0,82 mmol) wurden wie in Beispiel
32 kombiniert, um einen Feststoff D zu ergeben.
Massenspektrum
(ESI): 491,1. 1H-NMR (CD3OD) δ 7,68 (s,
1H), 7,39 (d, 1H), 7,24–7,06
(m, 4H), 6,60 (m, 1H), 5,69 (5, 1H), 4,57 (s, 2H), 3,62 (bs, 4H),
2,91 (bs, 4H), 1,48 (s, 9H).
-
Die
folgende Verbindung wurde in ähnlicher
Weise hergestellt:
-
-
Beispiel
40
Verbindung
174*
-
Das
Produkt aus Beispiel 39, Stufe 3 (150 mg, 0,31 mmol), 4 M HCl/Dioxan
(1 ml) und Dioxan wurden kombiniert (2 ml). Die Mischung wurde 20
Stunden bei Raumtemperatur unter N2 gerührt und
danach im Vakuum konzentriert. Der Rückstand wurde in Et2O suspendiert, erneut im Vakuum konzentriert,
und dies mehrfach wiederholt. Der resultierende Feststoff wurde
in Et2O aufgenommen, filtriert und getrocknet
(Vakuumofen, 50°C),
um einen Feststoff zu ergeben. Massenspektrum (ESI): 391,1. 1H-NMR (CD3OD) δ 7,91 (s,
1H), 7,43–7,17
(m, 6H), 6,78 (m, 1H), 3,66 (s, 2H), 3,43 (bs, 4H), 3,20 (m, 4H).
-
Beispiel
41
Verbindung
175*
-
Stufe
1: Das Produkt aus Beispiel 39, Stufe 1 (1,11 g, 4,1 mmol) wurde
wie in Beispiel 40 entschützt, um
einen Feststoff B zu ergeben. Massenspektrum (ESI): 188,0.
-
Stufe
2: Die freie Base des Produkts aus Stufe 1 B (200 mg, 1,1 mmol),
Triethylamin (130 mg, 1,3 mmol) und Essigsäureanhydrid (4 ml) wurden kombiniert.
Die Mischung wurde 20 Stunden bei Raumtemperatur unter N2 gerührt.
Die Mischung wurde im Vakuum konzentriert und zwischen gesättigtem
NaHCO3 und CH2Cl2 partitioniert. Die organischen Materialien
wurden mit H2O, Salzlösung gewaschen und über MgSO4 getrocknet, filtriert und im Vakuum konzentriert,
um ein Öl
C zu ergeben, mit dem ohne weitere Reinigung weitergearbeitet wurde.
Massenspektrum (ESI): 230,0.
-
Stufe
3: Das Produkt aus Stufe 2 C (230 mg, 1,0 mmol) wurde wie in Beispiel
34, Stufe 2 hydriert, um ein Öl
D zu ergeben. Massenspektrum (ESI): 234,0.
-
Stufe
4: Das Produkt aus Stufe 3 D (235 mg, 1,0 mmol) wurde mit dem Produkt
aus Beispiel 2, Stufe 2 (119 mg, 0,50 mmol) wie in Beispiel 32 kombiniert,
um einen Feststoff E zu ergeben. Massenspektrum (ESI): 433,1. 1H-NMR (CD3OD) δ 7,68 (s,
1H), 7,40 (d, 1H), 7,24–7,10
(m, 4H), 6,60 (m, 1H), 5,70 (s, 1H), 4,60 (s, 2H), 3,78 (bs, 2H),
3,73 (t, 2H), 2,96 (dt, 4H), 2,15 (S, 3H).
-
Die
folgenden Verbindungen wurden in ähnlicher Weise hergestellt:
-
-
Zu
bevorzugten erfindungsgemäßen Verbindungen
gehören
die folgenden Verbindungen, jedoch nicht auf diese begrenzt, die
ausgewählt
sind aus der Gruppe bestehend aus:
-
-
-
-
Beispiel 42
-
Die
pharmakologische Aktivität
der erfindungsgemäßen Verbindungen
wurde durch die folgenden in vitro- und in vivo-Assays ermittelt, um die A2a-Rezeptoraktivität zu messen.
-
Protokoll des kompetitiven
Bindungsassay zwischen humanem Adenosin A2a und
A1-Rezeptor
-
Membranquellen:
-
A2a: Humane A2a Adenosinrezeptormembranen,
Katalog Nr. RB-HA2a,
Receptor Biology, Inc., Beltsville, MD, USA. Es wurde in Membranverdünnungspuffer
(siehe unten) auf 17 μg/100 μl verdünnt.
-
Assaypuffer:
-
Membranverdünnungspuffer:
Dulbecco's phosphatgepufferte
Salzlösung
(Gibco/BRL) + 10 mM MgCl2.
-
Verbindungsverdünnungspuffer:
-
Dulbecco's phosphatgepufferte
Salzlösung
(Gibco/BRL) + 10 mM MgCl2 ergänzt mit
1,6 mg/ml Methylcellulose und 16 DMSO. Wurde täglich frisch hergestellt.
-
Liganden:
-
A2a: [3H]-SCH 58261, kundenspezifische Synthese,
Amersham Pharmacia Biotech, Piscataway, NJ, USA. Der Vorrat wurde
in 1 nM in Membranverdünnungspuffer
hergestellt. Endassaykonzentration war 0,5 nM.
-
A1: [3H]-DPCPX, Amersham Pharmacia Biotech,
Piscataway, NJ, USA. Der Vorrat wurde in 2 nM in Membranverdünnungspuffer
hergestellt. Endassaykonzentration war 1 nM.
-
Unspezifische Bindung:
-
A2a: Zur Bestimmung der unspezifischen Bindung
wurden 100 μM
CGS 15923 (RBI, Natick, MA, USA) zugegeben. Der Arbeitsvorrat wurde
mit 400 nM in Verbindungsverdünnungspuffer
hergestellt.
-
A1: Zur Bestimmung der unspezifischen Bindung
wurden 100 μM
NECA (RBI, Natick, MA, USA) zugegeben. Der Arbeitsvorrat wurde mit
400 μM in
Verbindungsverdünnungspuffer
hergestellt. Verbindungsverdünnung:
1
mM Vorratslösungen
der Verbindungen wurden in 100% DMSO hergestellt. Es wurde in Verbindungsverdünnungspuffer
verdünnt.
Es wurde in 10 Konzentrationen im Bereich von 3 μM bis 30 pM getestet. Es wurden Arbeitslösungen mit
der 4-fachen Endkonzentration in Verbindungsverdünnungspuffer hergestellt.
-
Assayverfahren:
-
Assays
wurden in Tiefmulden-96-Muldenplatten durchgeführt. Das Gesamtassayvolumen
war 200 μl. 50 μl Verbindungsverdün nungspuffer
(Gesamtligandenbindung) oder 50 μl
CGS 15923 Arbeitslösung
(A2a unspezifische Bindung) oder 50 μl NECA Arbeitslösung (A1 unspezifische Bindung) oder 50 μl Arzneimittelarbeitslösung wurden
zugefügt.
50 μl Ligandenvorratslösung ([3H]-SCH
58261 für
A2a, [3H]-DPCPX für A1)
wurden zugefügt.
100 μl verdünnte Membranen
wurden zugefügt,
die den entsprechenden Rezeptor enthielten. Es wurde gemischt und
bei Raumtemperatur 90 Minuten inkubiert. Es wurde mit einem Brandel-Zellernter auf Packard
GF/B Filterplatten geerntet. Es wurden 45 μl Microscint 20 (Packard) zugefügt und mit
dem Packard Top-Count
Microszintillationszähler
gezählt.
Die IC50-Werte wurden bestimmt, indem die
Verdrängungskurven mit
einem iterativen Kurvenanpassungsprogramm (Excel) angepasst wurden.
Die Ki-Werte wurden mit der Cheng-Prusoff-Gleichung bestimmt.
-
Haloperidol-induzierte
Katalepsie bei der Ratte
-
Es
wurden männliche
Sprague-Dawley Ratten (Charles River, Calco, Italien) verwendet,
die 175 bis 200 g wogen. Der kataleptische Zustand wurde durch subkutane
Verabreichung des Dopaminrezeptorantagonisten Haloperidol (1 mg/kg,
sc) 90 Minuten vor dem Testen der Tiere mit dem vertikalen Gittertest
induziert. Bei diesem Test wurden die Ratten auf der Drahtmaschenabdeckung
eines 25 × 43
Plexiglaskäfigs
angeordnet, der in einem Winkel von etwa 70 Grad zu dem Versuchstisch
angeordnet war. Die Ratte wurde so auf dem Gitter angeordnet, dass
alle vier Beine abgespreizt und gestreckt waren ("Froschstellung"). Die Verwendung einer
derartigen unnatürlichen
Körperhaltung
ist für
die Spezifität
dieses Tests auf Katalepsie wesentlich. Die Zeitspanne von der Platzierung
der Pfoten bis zur ersten vollständigen
Entfernung einer Pfote (dezente Latenz) wurde maximal 120 Sekunden
gemessen.
-
Die
untersuchten selektiven A2a Adenosinantagonisten
wurden oral in Dosen im Bereich zwischen 0,03 und 3 mg/kg 1 und
4 Stunden vor der Bewertung der Tiere verabreicht.
-
In
separaten Experimenten wurden die antikataleptischen Wirkungen der
Referenzverbindung, L-DOPA (25, 50 und 100 mg/kg, ip) untersucht.
-
6-OHDA Läsion des
mittleren Vorderhirnbündels
bei Ratten
-
In
allen Experimenten wurden erwachsene männliche Sprague-Dowley Ratten verwendet
(Charles River, Calco, Como, Italien), die 275–300 g wogen. Die Ratten wurden
in Gruppen von 4 pro Käfig
mit freiem Zugang zu Nahrung und Wasser unter kontrollierter Temperatur
und 12 Stunden Hell/Dunkel-Zyklus untergebracht. Am Tag vor dem
chirurgischen Eingriff wurden die Ratten über Nacht mit freiem Zugang
zu Wasser fasten gelassen.
-
Die
unilaterale 6-Hydroxydopamin-(6-OHDA)-Läsion des mittleren Vorderhirnbündels wurde
mit geringen Veränderungen
nach dem Verfahren durchgeführt,
das von Ungerstedt et al. (Brain Research. 1971, 6-OHDA and Cathecolamine
Neurons, North Holland, Amsterdam, 101–127) beschrieben wurde. Kurz
gesagt wurden die Tiere mit Chloralhydrat anästhesiert (400 mg/kg, ip) und
30 Minuten vor der 6-OHDA-Injektion mit Desipramin (10 mpk, ip)
behandelt, um die Aufnahme des Toxins an den noradrenergischen Enden
zu blockieren. Danach wurden die Tiere in einem Stereotaxierahmen
angeordnet. Die Haut über
der Hirnschale wurde umgeschlagen, und die Stereotaxiekoordinaten
(–2,2
posterior vom Bregma (AP), +1,5 lateral vom Bregma (ML), 7,8 ventral
von der Dura (DV)) wurden gemäß dem Atlas
von Pellegrino et al. genommen (L. J. Pellegrino, A. S. Pellegrino
und A. J. Cushman, A Stereotaxic Atlas of the Rat Brain, 1979, New
York, Plenum Press). Ein Trepanationsloch wurde in dem Schädel über der
Läsionsstelle
positioniert und eine an einer Hamilton-Spritze befestigte Nadel
wurde in das linke MFB abgesenkt. Danach wurden 8 μg 6-OHDA-HCl
in 4 μl
Salzlösung
mit 0,05% Ascorbinsäure
als Antioxidans gelöst
und mit einer Infusionspumpe mit einer konstanten Durchflussrate
von 1 μl/1
Min infundiert. Die Nadel wurde nach weiteren 5 Minuten gezogen,
die chirurgische Wunde verschlossen und die Tiere sich 2 Wochen
erholen gelassen.
-
Zwei
Wochen nach der Läsion
wurden den Ratten L-DOPA (50 mg/kg, ip) plus Benserazid (25 mg/kg, ip)
verabreicht und auf Basis der Anzahl vollständiger kontralateraler Drehungen
selektiert, die in dem zweistündigen
Testzeitraum durch automatische Rotameter quantifiziert wurden (Vorbereitungstest).
Jede Ratte, die nicht mindestens 200 vollständige Umdrehungen/2 Stunden
zeigte, wurde nicht in die Studie eingeschlossen.
-
Ausgewählte Ratten
erhielten das Testarzneimittel 3 Tage nach dem Vorbereitungstest
(maximale Dopaminrezeptorüberempfindlichkeit).
Die neuen A2a-Rezeptorantagonisten wurden
oral in Dosierniveaus im Bereich zwischen 0,1 und 3 mg/kg zu unterschiedlichen
Zeiten (d. h. 1, 6, 12 h) vor der Injektion einer unter dem Schwellenwert
liegenden Dosis von L-DOPA (4 mpk, ip) plus Benserazid (4 mpk, ip)
und der Bewertung des Drehverhaltens verabreicht.
-
Beispiel 43
-
Es
folgen Beispiele für
pharmazeutische Dosierformen, die eine erfindungsgemäße Verbindung
enthalten.
-
Beispiele
für pharmazeutische
Dosierungsformen Tabletten
-
Herstellungsverfahren
-
Positionen
Nr. 1 und 2 wurden in einem geeigneten Mischer 10 bis 15 Minuten
gemischt. Die Mischung wurde mit Position Nr. 3 granuliert. Die
feuchten Körner
wurden nach Bedarf durch ein grobes Sieb (z. B. 1/4'', 0,63 cm) gemahlen. Die feuchten Körner wurden
getrocknet. Die getrockneten Körner
wurden nach Bedarf gesiebt und mit Position Nr. 4 gemischt und 10
bis 15 Minuten gemischt. Position Nr. 5 wurde zugegeben und 1 bis
3 Minuten gemischt. Die Mischung wurde mit einer geeigneten Tablettiermaschine
auf geeignete Größe und geeignetes
Gewicht gepresst.
-
-
Herstellungsverfahren
-
Positionen
Nr. 1, 2 und 3 wurden in einem geeigneten Mischer 10 bis 15 Minuten
gemischt. Position Nr. 4 wurde zugegeben und 1 bis 3 Minuten gemischt.
Die Mischung wurden auf einer geeigneten Verkapselungsmaschine in
geeignete zweiteilige Hartgelatinekapseln gefüllt.
-
Obwohl
die vorliegende Erfindung im Zusammenhang mit den hier beschriebenen
spezifischen Ausführungsformen
beschrieben worden ist, ergeben sich Durchschnittsfachleuten viele
Alternativen, Modifikationen und Varianten davon von selbst.
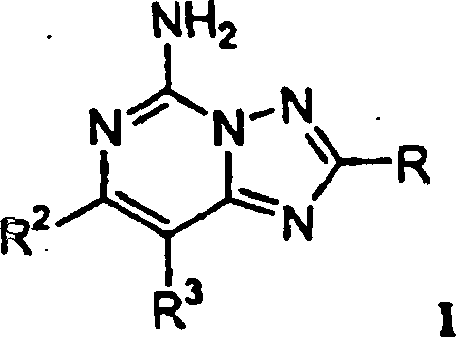
 und -CH=CH-CH3;
und -CH=CH-CH3;




 worin, wenn nicht anders definiert ist,
worin, wenn nicht anders definiert ist,
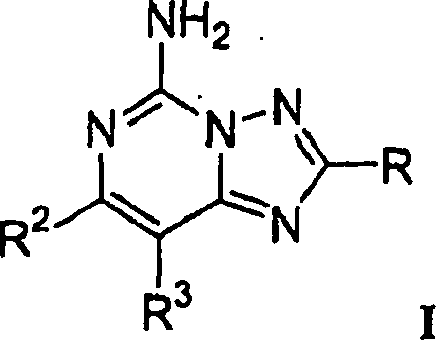
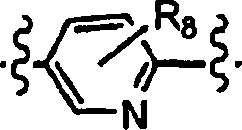
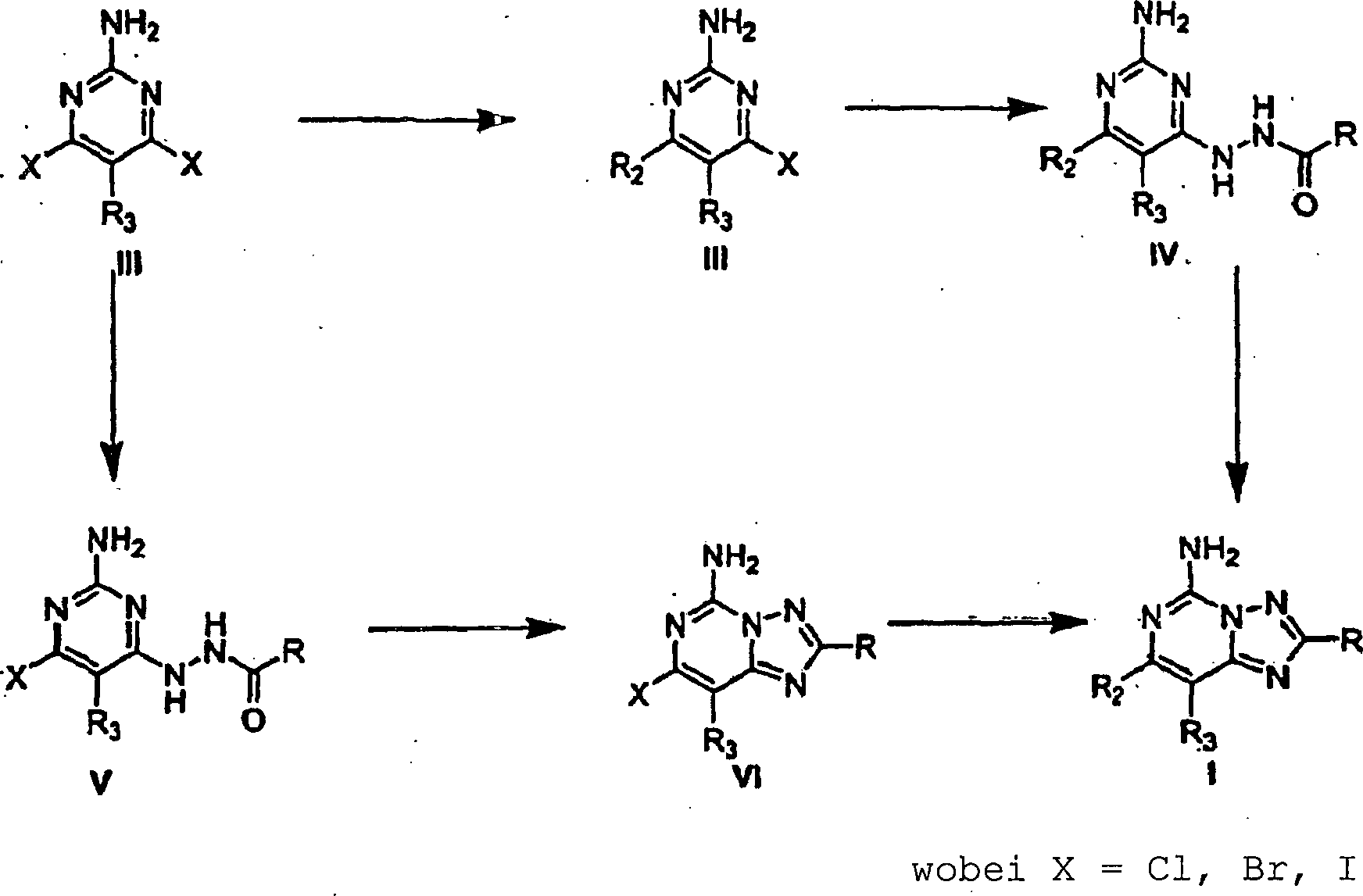

























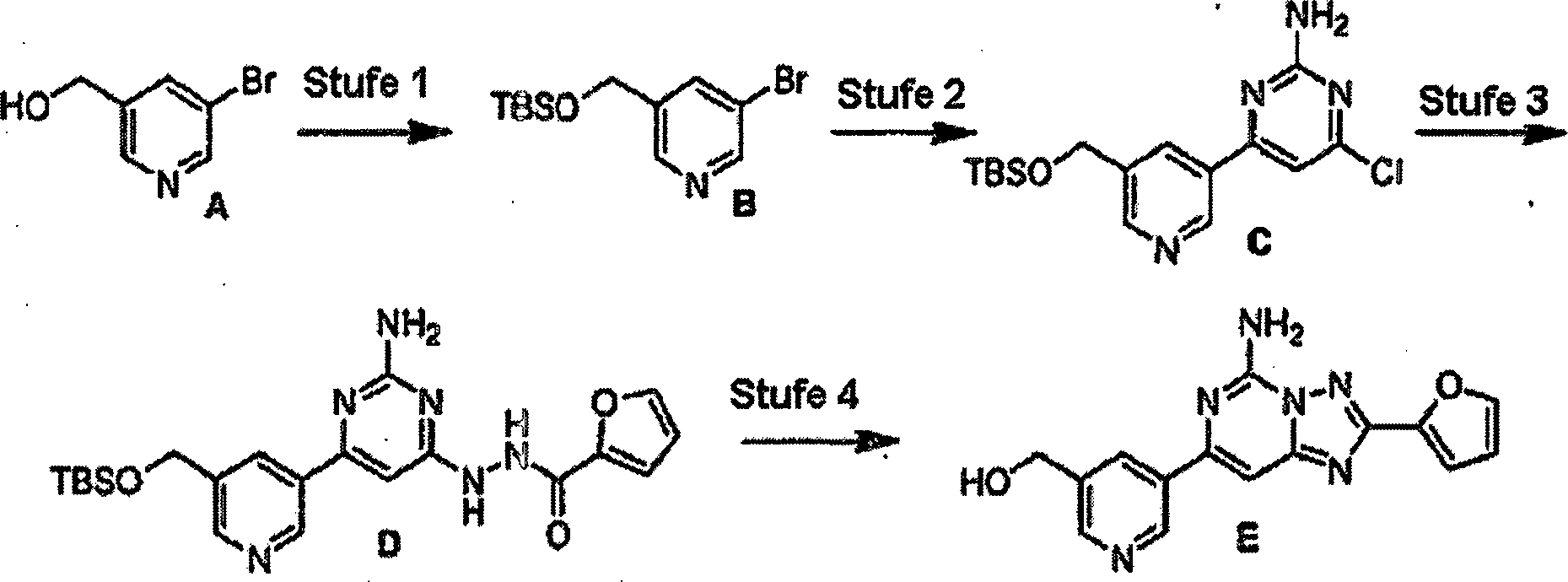









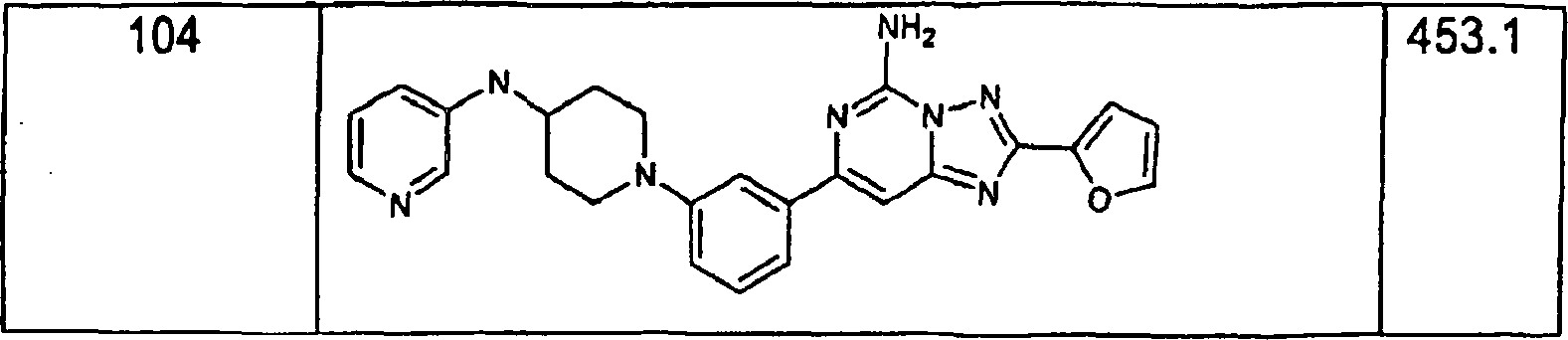







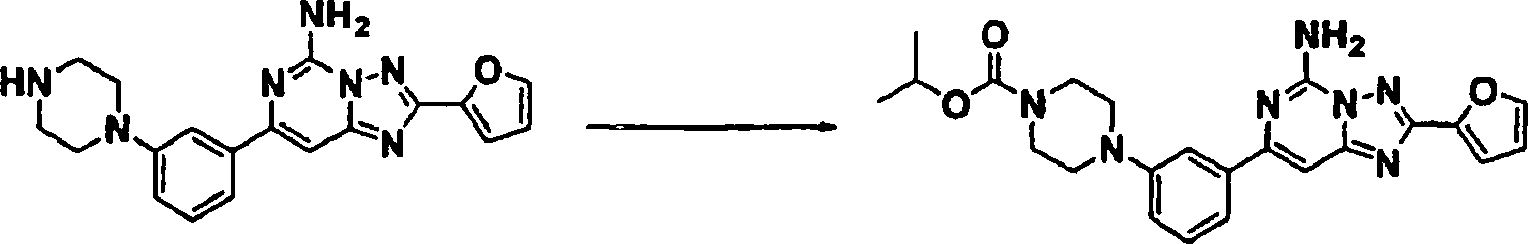



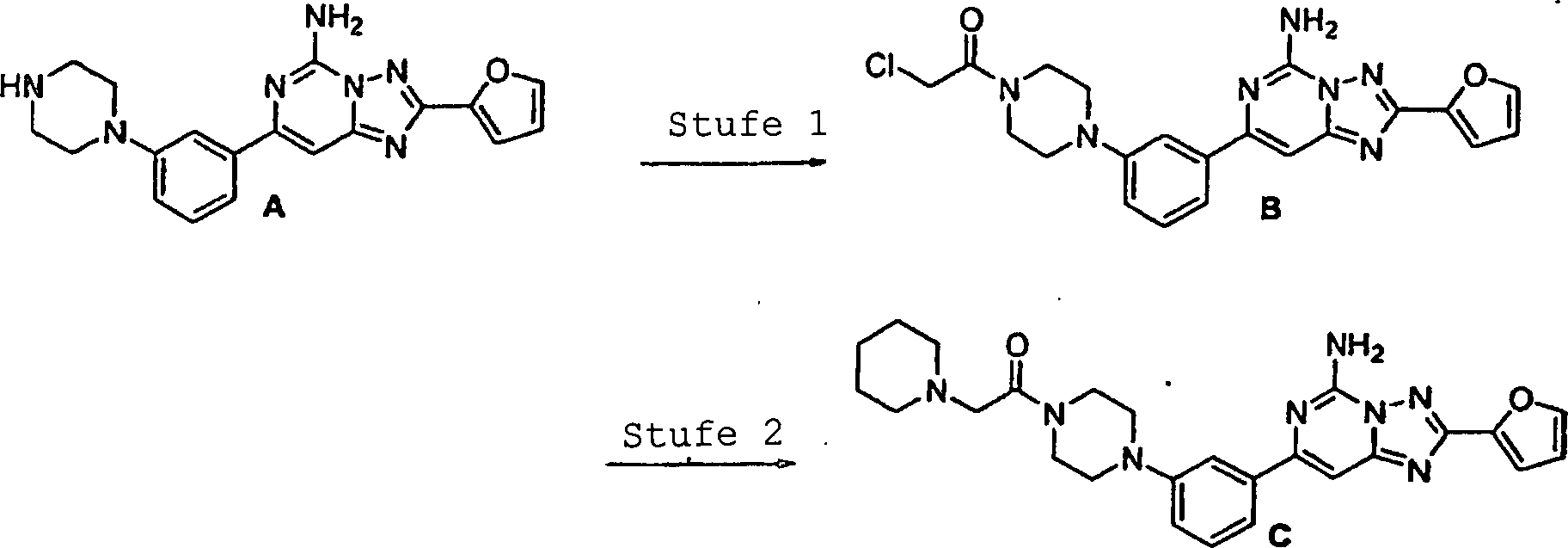


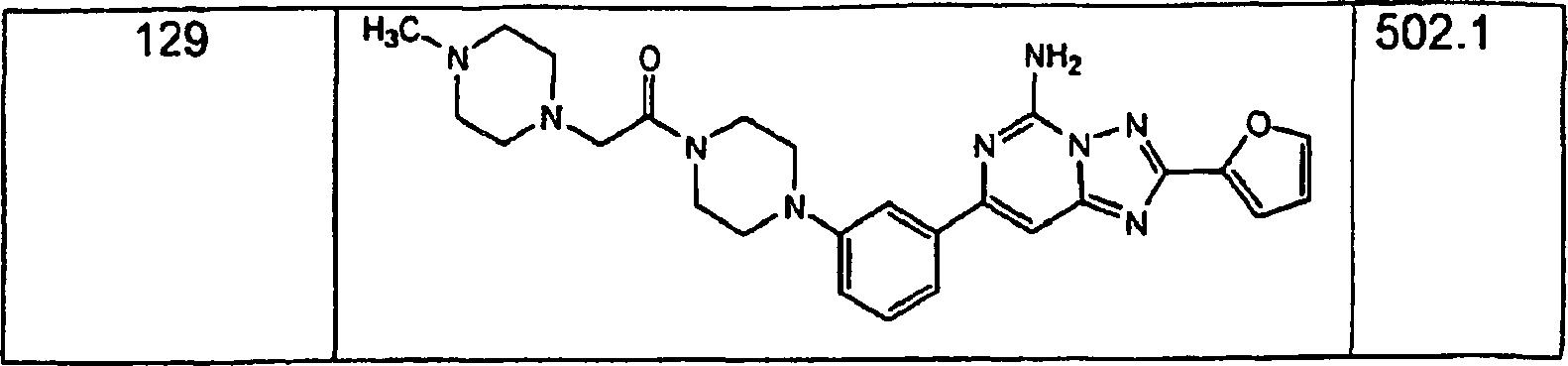
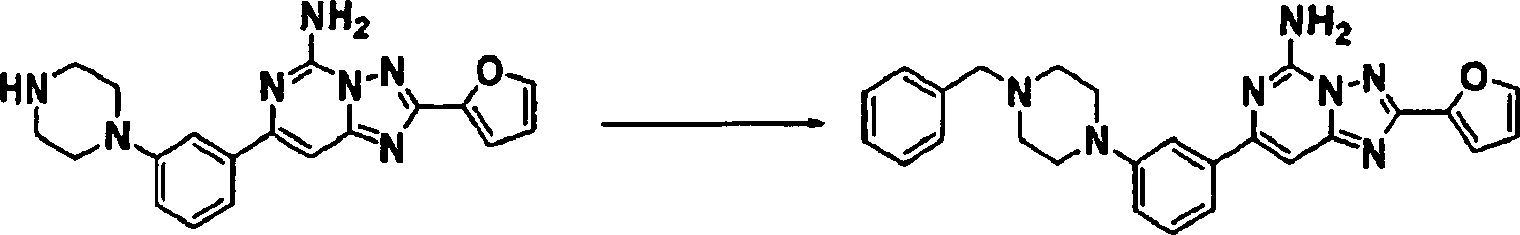

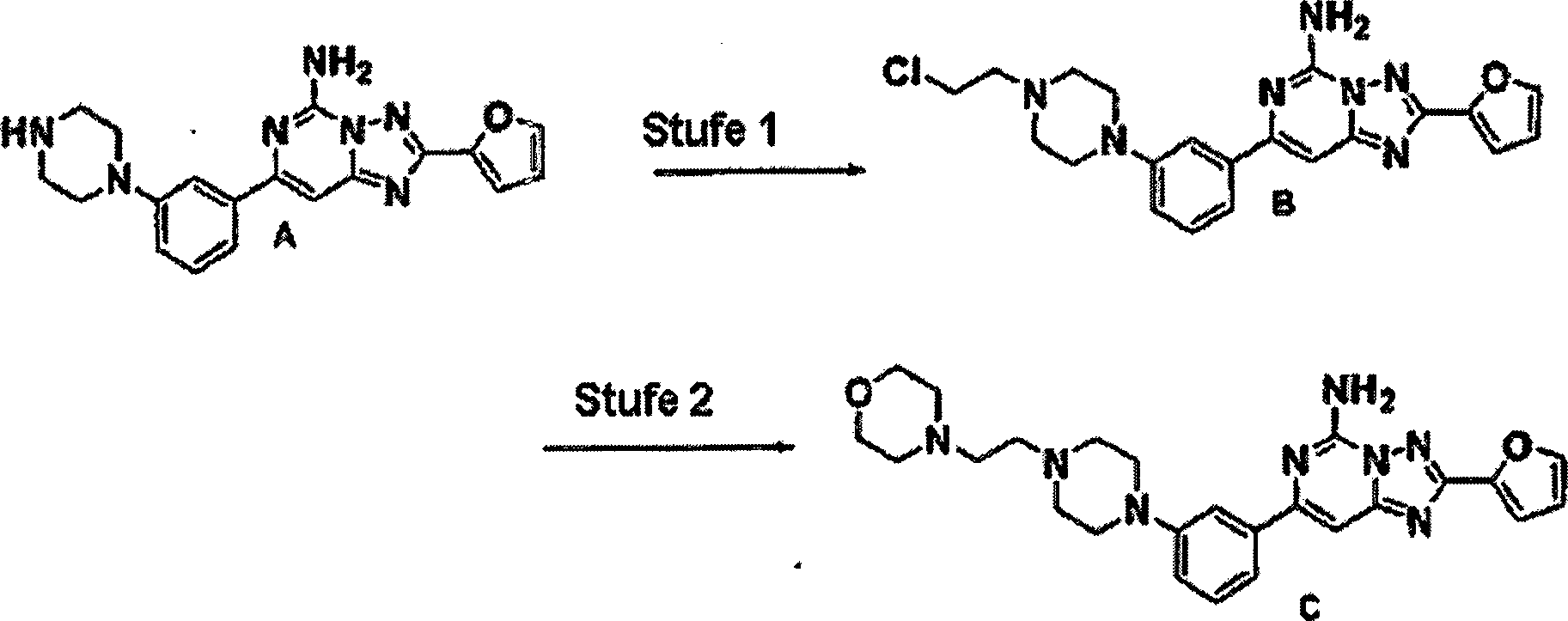









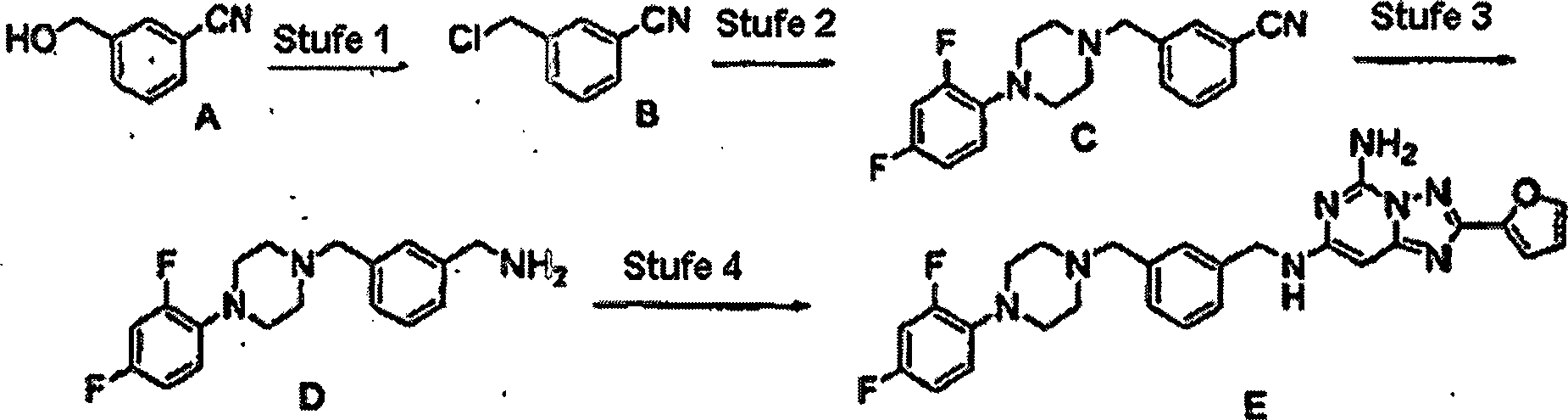

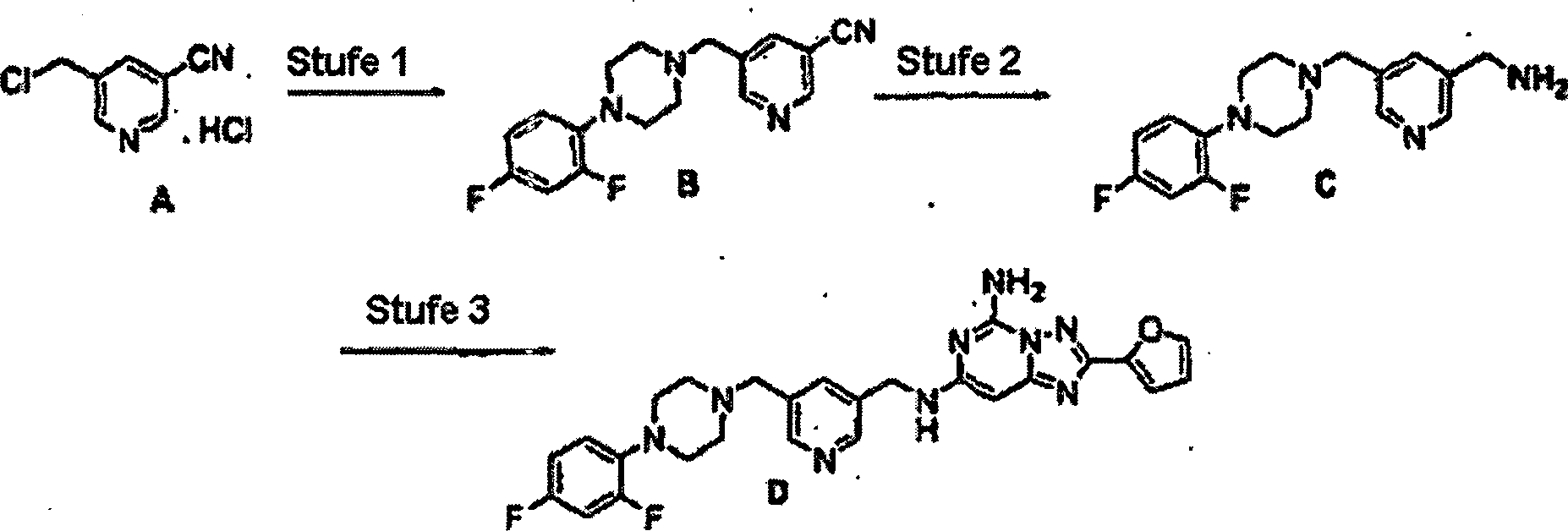
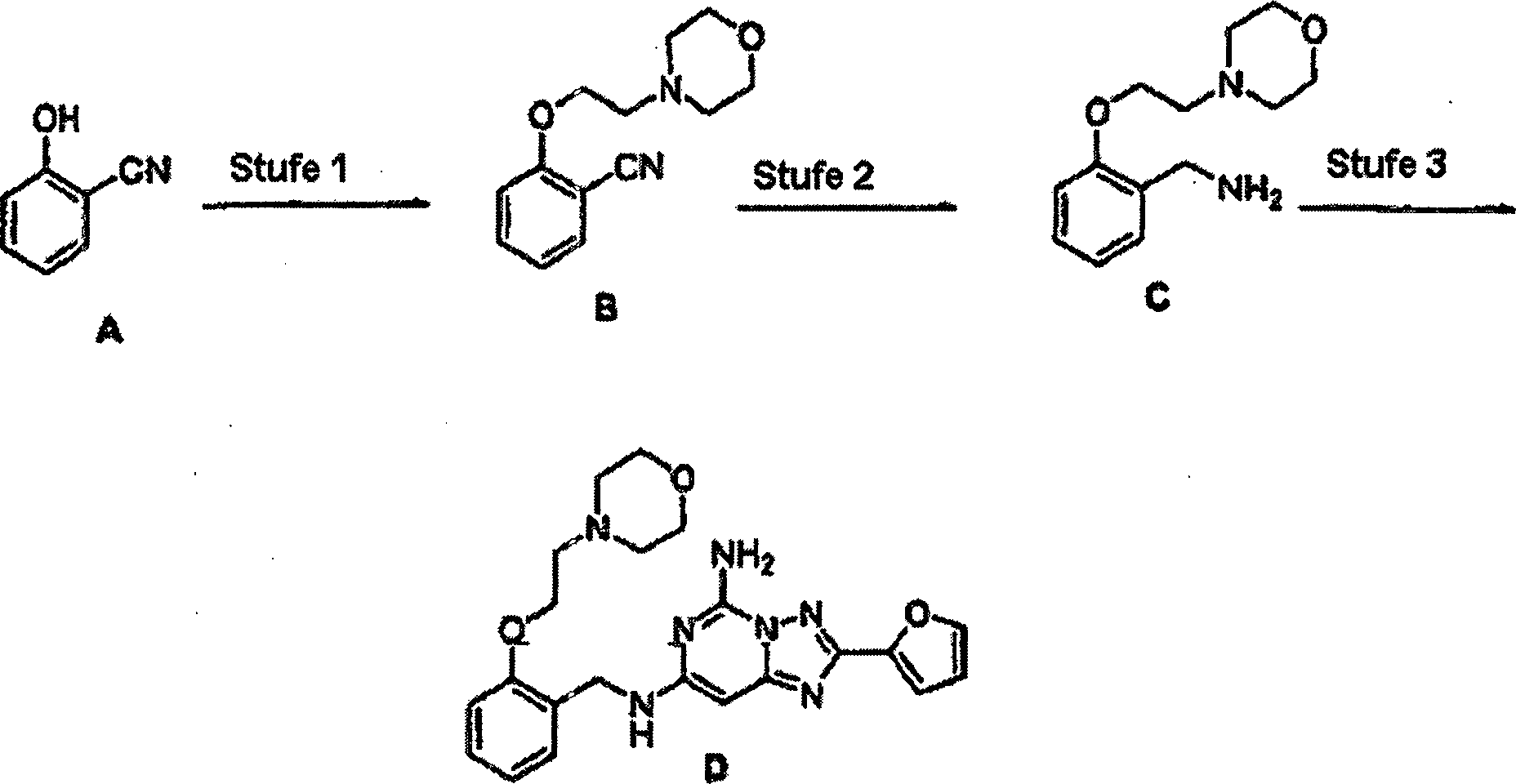



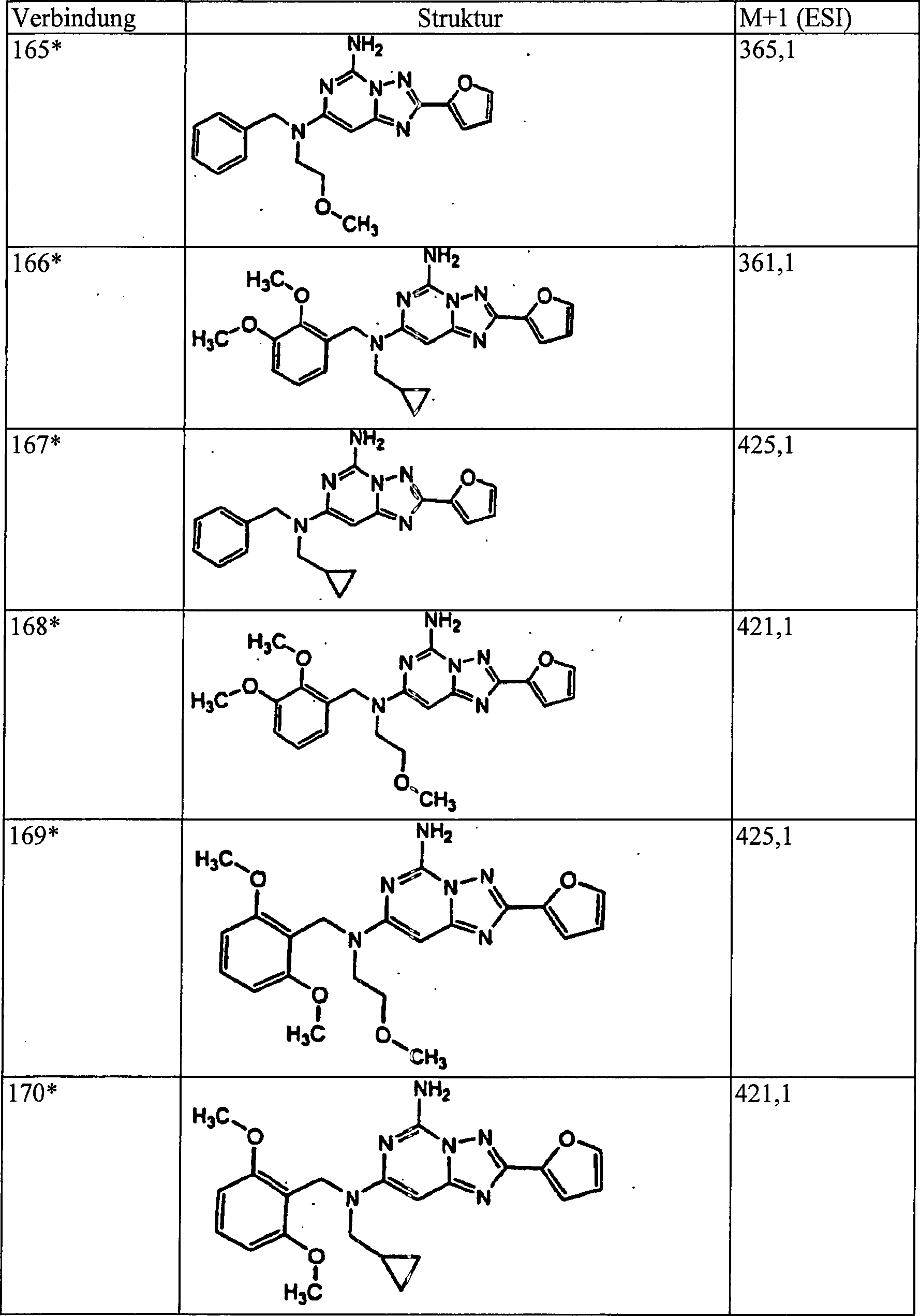


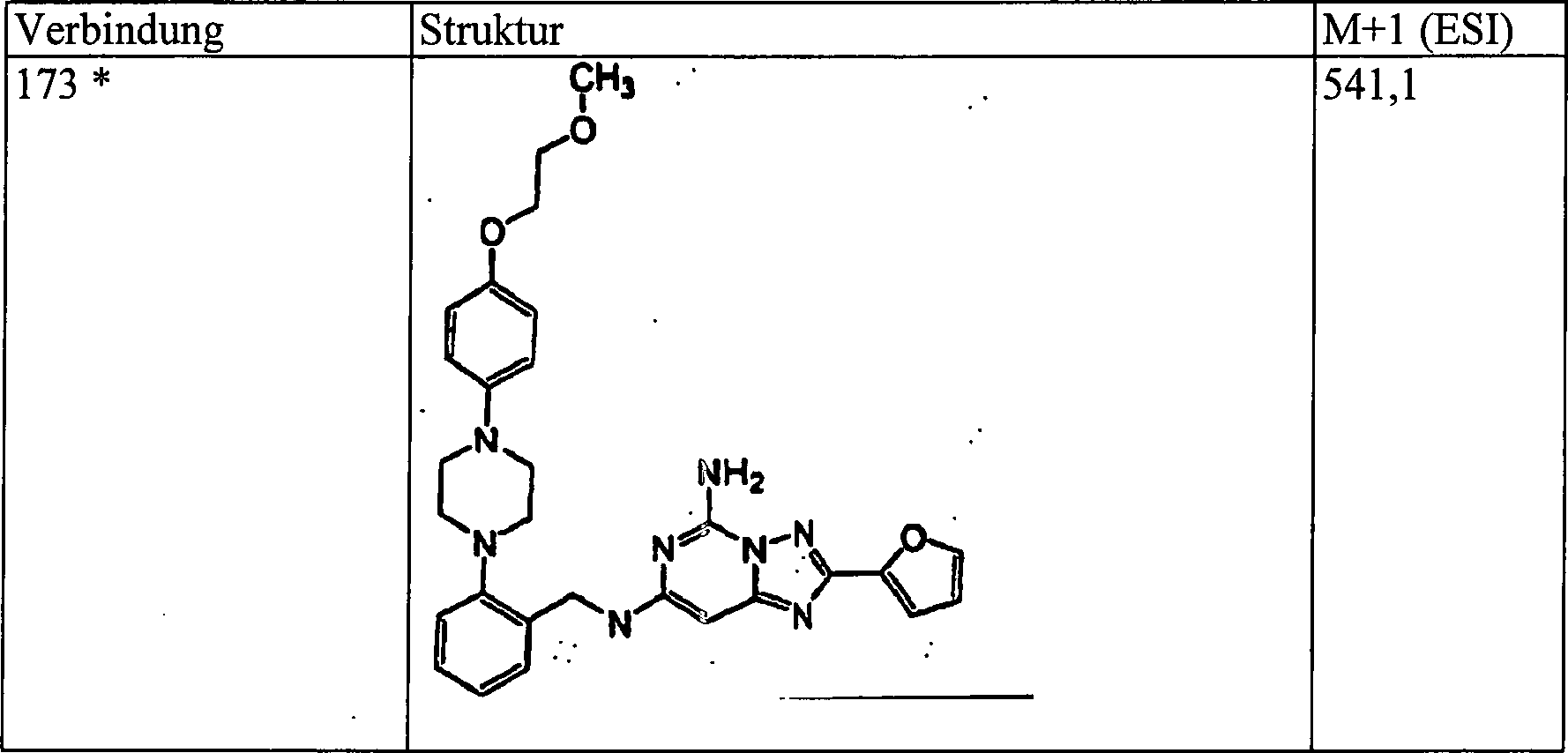
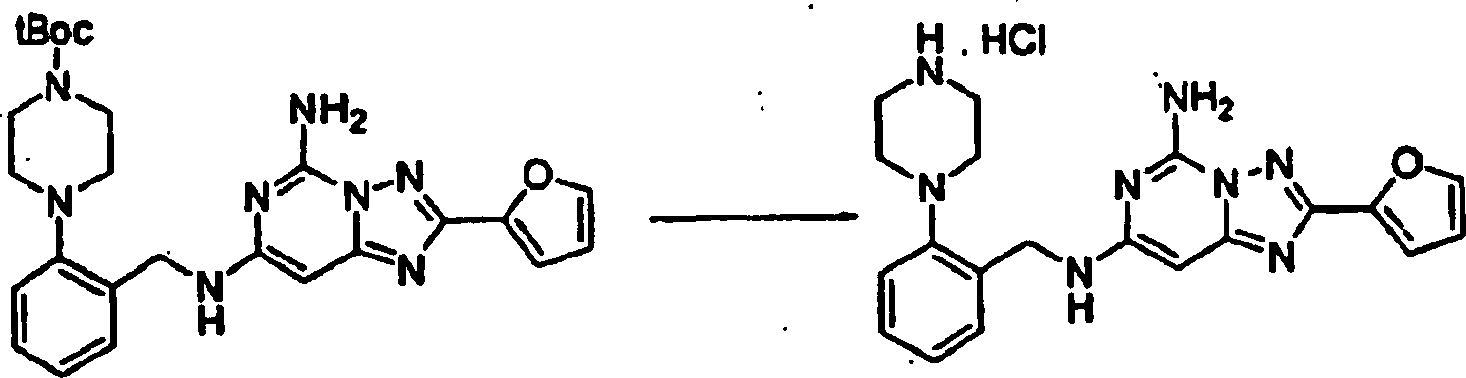







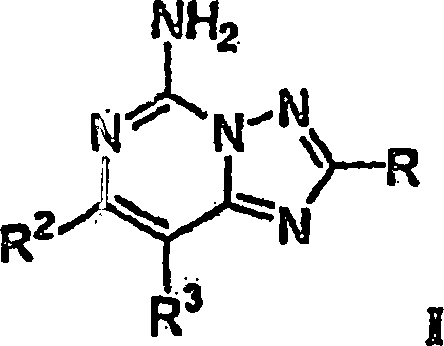 -CH=C(CH3)2,
-CH=C(CH3)2, und -CH=CH-CH3; R2 -W-X ist; R3 ausgewählt ist aus der Gruppe bestehend aus H, Halo, Alkyl, Trifluormethyl, Alkoxy, Alkoxyalkyl, Hydroxyalkyl, Alkylamino, Alkylaminoalkyl, Dialkylamino, Dialkylaminoalkyl, Aminoalkyl, Aryl, Heteroaryl und CN; R4 1 bis 3 Substituenten ist, die gleich oder verschieden sein können und unabhängig ausgewählt sind aus der Gruppe bestehend aus Wasserstoff, C1-C6-Alkyl, -CF3, Halogen, -NO2, -NR15R16, (C1-C6)-Alkoxy, (C1-C6)-Alkylthio, (C1-C6)-Alkylsulfinyl, (C1-C6)-Alkylsulfonyl, -COOR17 und -C(O)NR6R7; R5 1 bis 5 Substituenten ist, die gleich oder verschieden sein können und unabhängig ausgewählt sind aus der Gruppe bestehend aus Wasserstoff, Halogen, (C1-C6)-Alkyl, Hydroxy, (C1-C6)-Alkoxy, -CN, -NH2, (C1-C6)-Alkylamino, Di-((C1-C6)alkyl)amino, -CF3, -OCF3, -S(O)0-2(C1-C6)-Alkyl und -CH2-SO2-Phenyl; R6 und R7, die gleich oder verschieden sein können, jeweils unabhängig ausgewählt sind aus der Gruppe bestehend aus Wasserstoff und (C1-C6)-Alkyl; R8 1 bis 5 Substituenten ist, die gleich oder verschieden sein können und unabhängig ausgewählt sind aus der Gruppe bestehend aus Wasserstoff, Halogen, (C1-C6)-Alkyl, Hydroxy, C1-C6-Alkoxy, -CN, Amino, Di-((C1-C6)alkyl)amino, -CF3, -OCF3, Acetyl, -NO2, Hydroxy(C1-C6)alkoxy, (C1-C6)-Alkoxy(C1-C6)alkoxy, Di-((C1-C6)-alkoxy)(C1-C6)alkoxy, (C1-C6)-Alkoxy(C1-C6)alkoxy-(C1-C6)-alkoxy, Carboxy(C1-C6)-alkoxy, (C1-C6)-Alkoxycarbonyl(C1-C6)alkoxy, (C3-C6)Cycloalkyl(C1-C6)alkoxy, Di-((C1-C6)alkyl)amino(C1-C6)alkoxy, Morpholinyl, (C1-C6)-Alkyl-SO2-, (C1-C6)-Alkyl-SO2(C1-C6)alkoxy, Tetrahydropyranyloxy, (C1-C6)-Alkylcarbonyl(C1-C6)alkoxy, (C1-C6-Alkoxycarbonyl, (C1-C6)-Alkylcarbonyloxy(C1-C6)alkoxy, -SO2NH2, Phenoxy,
und -CH=CH-CH3; R2 -W-X ist; R3 ausgewählt ist aus der Gruppe bestehend aus H, Halo, Alkyl, Trifluormethyl, Alkoxy, Alkoxyalkyl, Hydroxyalkyl, Alkylamino, Alkylaminoalkyl, Dialkylamino, Dialkylaminoalkyl, Aminoalkyl, Aryl, Heteroaryl und CN; R4 1 bis 3 Substituenten ist, die gleich oder verschieden sein können und unabhängig ausgewählt sind aus der Gruppe bestehend aus Wasserstoff, C1-C6-Alkyl, -CF3, Halogen, -NO2, -NR15R16, (C1-C6)-Alkoxy, (C1-C6)-Alkylthio, (C1-C6)-Alkylsulfinyl, (C1-C6)-Alkylsulfonyl, -COOR17 und -C(O)NR6R7; R5 1 bis 5 Substituenten ist, die gleich oder verschieden sein können und unabhängig ausgewählt sind aus der Gruppe bestehend aus Wasserstoff, Halogen, (C1-C6)-Alkyl, Hydroxy, (C1-C6)-Alkoxy, -CN, -NH2, (C1-C6)-Alkylamino, Di-((C1-C6)alkyl)amino, -CF3, -OCF3, -S(O)0-2(C1-C6)-Alkyl und -CH2-SO2-Phenyl; R6 und R7, die gleich oder verschieden sein können, jeweils unabhängig ausgewählt sind aus der Gruppe bestehend aus Wasserstoff und (C1-C6)-Alkyl; R8 1 bis 5 Substituenten ist, die gleich oder verschieden sein können und unabhängig ausgewählt sind aus der Gruppe bestehend aus Wasserstoff, Halogen, (C1-C6)-Alkyl, Hydroxy, C1-C6-Alkoxy, -CN, Amino, Di-((C1-C6)alkyl)amino, -CF3, -OCF3, Acetyl, -NO2, Hydroxy(C1-C6)alkoxy, (C1-C6)-Alkoxy(C1-C6)alkoxy, Di-((C1-C6)-alkoxy)(C1-C6)alkoxy, (C1-C6)-Alkoxy(C1-C6)alkoxy-(C1-C6)-alkoxy, Carboxy(C1-C6)-alkoxy, (C1-C6)-Alkoxycarbonyl(C1-C6)alkoxy, (C3-C6)Cycloalkyl(C1-C6)alkoxy, Di-((C1-C6)alkyl)amino(C1-C6)alkoxy, Morpholinyl, (C1-C6)-Alkyl-SO2-, (C1-C6)-Alkyl-SO2(C1-C6)alkoxy, Tetrahydropyranyloxy, (C1-C6)-Alkylcarbonyl(C1-C6)alkoxy, (C1-C6-Alkoxycarbonyl, (C1-C6)-Alkylcarbonyloxy(C1-C6)alkoxy, -SO2NH2, Phenoxy, O-CH2-P(O)(OR6)2-, und -P(O)(OR6)2; oder benachbarte R8-Substituenten zusammen -O-CH2-O-, -O-CH2CH2-O-, -O-CF2-O- oder -O-CF2CF2-O- sind und mit den Kohlenstoffatomen, an die sie gebunden sind, einen Ring bilden; R9 ausgewählt ist aus der Gruppe bestehend aus (C1-C6)-Alkyl, R8-Aryl-, R8-Aryl (C1-C6)alkyl, Thienyl, Pyridyl, (C3-C6)-Cycloalkyl, (C1-C6)-Alkyl-OC(O)-NH-(C1-C6)alkyl-, Di-((C1-C6)alkyl)aminomethyl, Cycloheteroalkyl(C1-C6)alkyl, Aryloxy(C1-C6)alkyl, Alkoxy(C1-C6)alkyl und
O-CH2-P(O)(OR6)2-, und -P(O)(OR6)2; oder benachbarte R8-Substituenten zusammen -O-CH2-O-, -O-CH2CH2-O-, -O-CF2-O- oder -O-CF2CF2-O- sind und mit den Kohlenstoffatomen, an die sie gebunden sind, einen Ring bilden; R9 ausgewählt ist aus der Gruppe bestehend aus (C1-C6)-Alkyl, R8-Aryl-, R8-Aryl (C1-C6)alkyl, Thienyl, Pyridyl, (C3-C6)-Cycloalkyl, (C1-C6)-Alkyl-OC(O)-NH-(C1-C6)alkyl-, Di-((C1-C6)alkyl)aminomethyl, Cycloheteroalkyl(C1-C6)alkyl, Aryloxy(C1-C6)alkyl, Alkoxy(C1-C6)alkyl und R10 1 bis 2 Substituenten ist, die gleich oder verschieden sein können und unabhängig ausgewählt sind aus der Gruppe bestehend aus Wasserstoff, (C1-C6)-Alkyl, R5-Aryl und R4-Heteroaryl, oder zwei R10-Substituenten an dem gleichen Kohlenstoffatom =O bilden können; R11 Wasserstoff oder (C1-C6) -Alkyl; -C(O)-Alkyl ist; oder R17 und R11 zusammen -(CH2)p-A-(CH2)q sind, worin p und q jeweils unabhängig 2 oder 3 sind und A ausgewählt ist aus der Gruppe bestehend aus einer Bindung, -CH2-, -S- oder -O- und mit dem Stickstoff, an den sie gebunden sind, einen Ring bilden; R12 1 bis 2 Substituenten ist, die gleich oder verschieden sind und unabhängig ausgewählt sind aus der Gruppe bestehend aus Wasserstoff, (C1-C6)-Alkyl, Hydroxy, (C1-C6)-Alkoxy, Halogen und -CF3; R13 ausgewählt ist aus der Gruppe bestehend aus H, (C1-C6)-Alkyl, Phenyl, Benzyl, (C2-C6)-Alkenyl, (C1-C6)-Alkoxy(C1-C6)alkyl, Di-((C1-C6)alkyl)amino(C1-C6)alkyl, Pyrrolidinyl(C1-C6)alkyl und Piperidino(C1-C6)alkyl; R14 ausgewählt ist aus der Gruppe bestehend aus H, Halogen, (C1-C6)-Alkyl oder (C1-C6)-Alkoxy; R15 ausgewählt ist aus der Gruppe bestehend aus H und (C1-C6)-Alkyl; R16 ausgewählt ist aus der Gruppe bestehend aus H, (C1-C6)-Alkyl-C(O)- und (C1-C6)-Alkyl-SO2-; R17 ausgewählt ist aus der Gruppe bestehend aus (C1-C6)-Alkyl, (C1-C6)-Hydroxyalkyl, (C3-C6)-Cycloalkyl, (C1-C6)-Alkoxy(C1-C6)alkoxy, (C1-C6)-Alkoxy, (C1-C6)-Alkoxy(C1-C6)alkyl, Allyl, Propargyl, R8-Heteroaryl-, R8-Aryl- und R8-Aryl(C1-C6)alkyl-; R18 ausgewählt ist aus der Gruppe bestehend aus einer Bindung, -CH2, -CH(OH)-, -CH(CH3)-, -C(CH3)n-, -(CH2)n- und -O(CH2)n-: Q und Q1 gleich oder verschieden sein können und jeweils unabhängig ausgewählt sind aus der Gruppe bestehend aus
R10 1 bis 2 Substituenten ist, die gleich oder verschieden sein können und unabhängig ausgewählt sind aus der Gruppe bestehend aus Wasserstoff, (C1-C6)-Alkyl, R5-Aryl und R4-Heteroaryl, oder zwei R10-Substituenten an dem gleichen Kohlenstoffatom =O bilden können; R11 Wasserstoff oder (C1-C6) -Alkyl; -C(O)-Alkyl ist; oder R17 und R11 zusammen -(CH2)p-A-(CH2)q sind, worin p und q jeweils unabhängig 2 oder 3 sind und A ausgewählt ist aus der Gruppe bestehend aus einer Bindung, -CH2-, -S- oder -O- und mit dem Stickstoff, an den sie gebunden sind, einen Ring bilden; R12 1 bis 2 Substituenten ist, die gleich oder verschieden sind und unabhängig ausgewählt sind aus der Gruppe bestehend aus Wasserstoff, (C1-C6)-Alkyl, Hydroxy, (C1-C6)-Alkoxy, Halogen und -CF3; R13 ausgewählt ist aus der Gruppe bestehend aus H, (C1-C6)-Alkyl, Phenyl, Benzyl, (C2-C6)-Alkenyl, (C1-C6)-Alkoxy(C1-C6)alkyl, Di-((C1-C6)alkyl)amino(C1-C6)alkyl, Pyrrolidinyl(C1-C6)alkyl und Piperidino(C1-C6)alkyl; R14 ausgewählt ist aus der Gruppe bestehend aus H, Halogen, (C1-C6)-Alkyl oder (C1-C6)-Alkoxy; R15 ausgewählt ist aus der Gruppe bestehend aus H und (C1-C6)-Alkyl; R16 ausgewählt ist aus der Gruppe bestehend aus H, (C1-C6)-Alkyl-C(O)- und (C1-C6)-Alkyl-SO2-; R17 ausgewählt ist aus der Gruppe bestehend aus (C1-C6)-Alkyl, (C1-C6)-Hydroxyalkyl, (C3-C6)-Cycloalkyl, (C1-C6)-Alkoxy(C1-C6)alkoxy, (C1-C6)-Alkoxy, (C1-C6)-Alkoxy(C1-C6)alkyl, Allyl, Propargyl, R8-Heteroaryl-, R8-Aryl- und R8-Aryl(C1-C6)alkyl-; R18 ausgewählt ist aus der Gruppe bestehend aus einer Bindung, -CH2, -CH(OH)-, -CH(CH3)-, -C(CH3)n-, -(CH2)n- und -O(CH2)n-: Q und Q1 gleich oder verschieden sein können und jeweils unabhängig ausgewählt sind aus der Gruppe bestehend aus m und n jeweils unabhängig 1 bis 3 sind; p und q jeweils unabhängig 0 bis 2 sind; s 0 bis 4 ist: W Aryl oder Heteroaryl mit 1 bis 3 Heteroatomen ist, die gleich oder verschieden sein können und unabhängig ausgewählt sind aus der Gruppe bestehend aus N, O und S, und wobei das Aryl oder Heteroaryl gegebenenfalls mit 1 bis 3 Substituenten substituiert ist, die gleich oder verschieden sein können und unabhängig ausgewählt sind aus der Gruppe bestehend aus Alkyl, Aryl, Alkylcycloalkyl, Halogen, Hydroxy, Hydroxyalkyl, Alkoxy, Alkylalkoxy, Alkoxyalkoxy, -NR6R7, (C2-C6)-Alken und -CN; X ausgewählt ist aus der Gruppe bestehend aus H, NH2, -N(R6)(CH2)s-Aryl, -N(R6)(CH2)s-Heteroaryl, -N(R6)(CH2)m+1-OH und -N(CH3)2 oder X -R18-Y-Z ist; Y ausgewählt ist aus der Gruppe bestehend aus -N(R6)CH2CH2N(R7)-, -N(R6)(CH2)n-Aryl, -OCH2CH2N(R6), -O-, -S-, -CH2S-, -(CH2)2-3-N(R6)-, R8-zweiwertigem Heteroaryl,
m und n jeweils unabhängig 1 bis 3 sind; p und q jeweils unabhängig 0 bis 2 sind; s 0 bis 4 ist: W Aryl oder Heteroaryl mit 1 bis 3 Heteroatomen ist, die gleich oder verschieden sein können und unabhängig ausgewählt sind aus der Gruppe bestehend aus N, O und S, und wobei das Aryl oder Heteroaryl gegebenenfalls mit 1 bis 3 Substituenten substituiert ist, die gleich oder verschieden sein können und unabhängig ausgewählt sind aus der Gruppe bestehend aus Alkyl, Aryl, Alkylcycloalkyl, Halogen, Hydroxy, Hydroxyalkyl, Alkoxy, Alkylalkoxy, Alkoxyalkoxy, -NR6R7, (C2-C6)-Alken und -CN; X ausgewählt ist aus der Gruppe bestehend aus H, NH2, -N(R6)(CH2)s-Aryl, -N(R6)(CH2)s-Heteroaryl, -N(R6)(CH2)m+1-OH und -N(CH3)2 oder X -R18-Y-Z ist; Y ausgewählt ist aus der Gruppe bestehend aus -N(R6)CH2CH2N(R7)-, -N(R6)(CH2)n-Aryl, -OCH2CH2N(R6), -O-, -S-, -CH2S-, -(CH2)2-3-N(R6)-, R8-zweiwertigem Heteroaryl, Z ausgewählt ist aus der Gruppe bestehend aus H, Alkyl, Alkoxyalkyl, R8-Aryl-, R8-Aryl(C1-C6)alkyl-, R8-Heteroaryl-, R8-bicyclischem Alkyl-, Aminoalkyl, Alkylamino, NH2, -N-(R6)(CH2)s-Aryl, -N(R6)(CH2)s-Heteroaryl, -N(R6)C(O)OR17, Alkylcycloheteroalkyl, Cycloheteroalkyl, Cycloheteroalkylalkyl, Alkoxycycloheteroalkyl, Heteroaryl; R8-benzokondensiertem Heteroaryl-, Diphenylmethyl und R9-C(O)-; oder wenn Y
Z ausgewählt ist aus der Gruppe bestehend aus H, Alkyl, Alkoxyalkyl, R8-Aryl-, R8-Aryl(C1-C6)alkyl-, R8-Heteroaryl-, R8-bicyclischem Alkyl-, Aminoalkyl, Alkylamino, NH2, -N-(R6)(CH2)s-Aryl, -N(R6)(CH2)s-Heteroaryl, -N(R6)C(O)OR17, Alkylcycloheteroalkyl, Cycloheteroalkyl, Cycloheteroalkylalkyl, Alkoxycycloheteroalkyl, Heteroaryl; R8-benzokondensiertem Heteroaryl-, Diphenylmethyl und R9-C(O)-; oder wenn Y ist, Z auch -OH, R9-SO2-, R17-N(R11)(CH2)S-C(O)-, R17-OC(O)-, R17-O(CH2)nC(O)-, benzokondensiertes Heteroaryl(CH2)nC(O)-, benzokondensiertes Heteroaryl (CH2)n- oder R17-N(R11)-C(S)- sein kann; oder wenn Q
ist, Z auch -OH, R9-SO2-, R17-N(R11)(CH2)S-C(O)-, R17-OC(O)-, R17-O(CH2)nC(O)-, benzokondensiertes Heteroaryl(CH2)nC(O)-, benzokondensiertes Heteroaryl (CH2)n- oder R17-N(R11)-C(S)- sein kann; oder wenn Q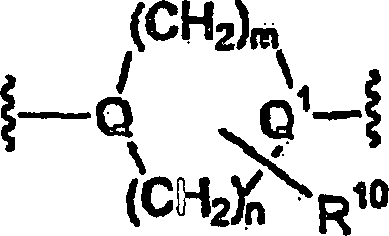 ist, Z auch R17R11N-, Phenylamino oder Pyridylamino sein kann; oder Z und Y zusammengenommen ausgewählt sind aus der Gruppe bestehend aus:
ist, Z auch R17R11N-, Phenylamino oder Pyridylamino sein kann; oder Z und Y zusammengenommen ausgewählt sind aus der Gruppe bestehend aus:
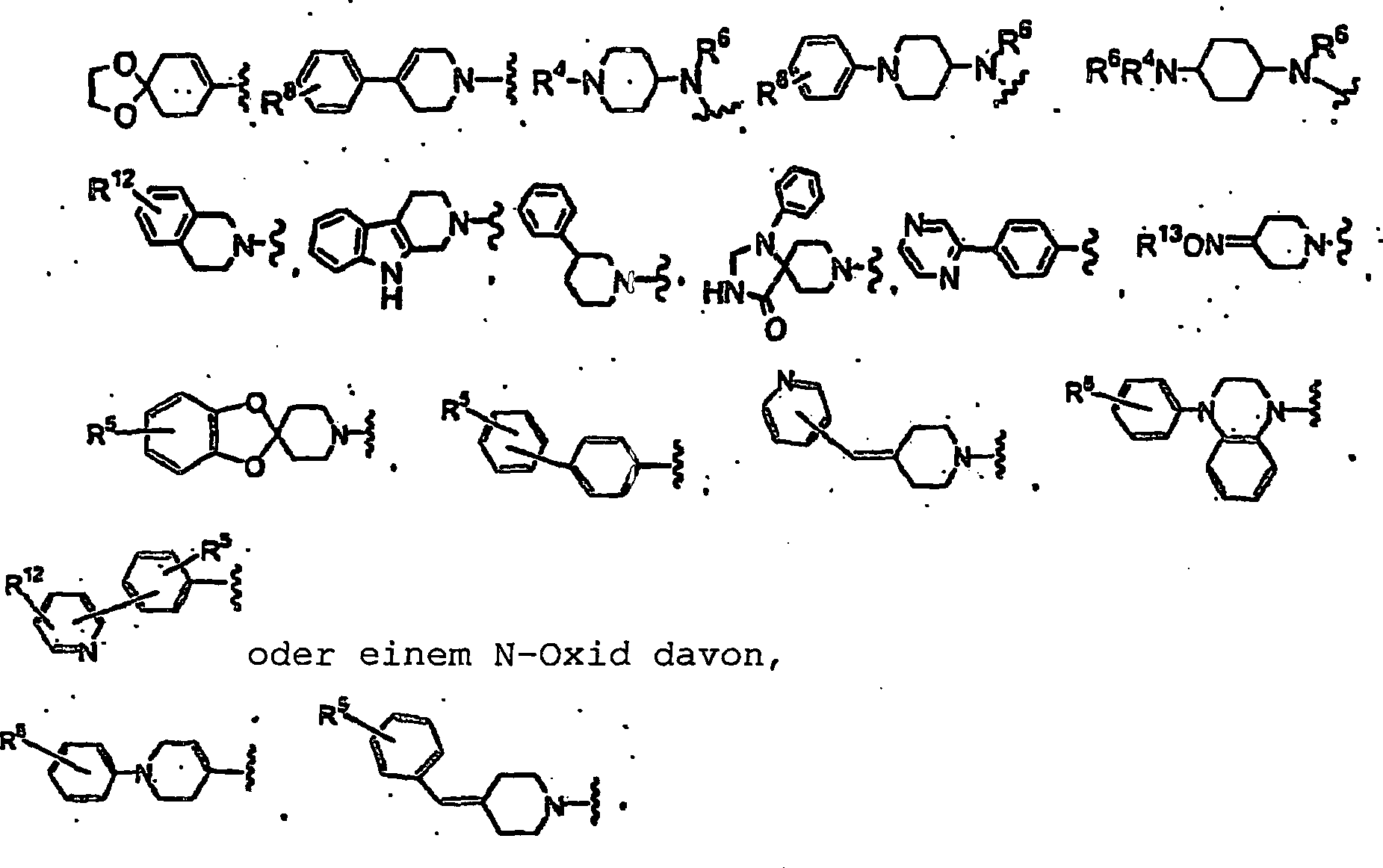 worin, wenn nicht anders definiert ist, "Alkyl" (einschließlich der Alkylanteile von Alkoxy, Alkylamino und Dialkylamino usw.) eine aliphatische Kohlenwasserstoffgruppe bedeutet, die geradkettig oder verzweigt sein kann und 1 bis 20 Kohlenstoffatome enthält; "Halo" Fluor-, Chlor-, Brom- oder Iodgruppen bedeutet; "Halogen" Fluor, Chlor, Brom oder Iod bedeutet; "Alkoxy" eine Alkyl-O-Gruppe bedeutet, in der die Alkylgruppe wie zuvor beschrieben ist; "Alkenyl" eine aliphatische Kohlenwasserstoffgruppe bedeutet, die mindestens eine Kohlenstoff-Kohlenstoff-Doppelbindung enthält und geradkettig oder verzweigt sein kann und 2 bis 15 Kohlenstoffatome enthält; "Alkanoyl" ein an ein Carbonyl gebundenes Alkyl ist, wobei Alkyl die gleiche Bedeutung wie oben definiert hat; "Cycloalkyl" ein nicht-aromatisches mono- oder multicyclisches kondensiertes Ringsystem bedeutet, das 3 bis 10 Ringkohlenstoffatome enthält; "Cycloheteroalkyl" ein nicht-aromatisches mono- oder multicyclisches kondensiertes Ringsystem bedeutet, das 3 bis 10 Ringkohlenstoffatome enthält, wobei das Cycloheteroalkyl 1 oder 2 Heteroatome unabhängig ausgewählt aus O, S oder N aufweist, wobei das Heteroatom/die Heteroatome eine carbocyclische Ringstruktur unterbricht bzw. unterbrechen, mit der Maßgabe, dass die Ringe keine benachbarten Sauerstoff- und/oder Schwefelatome enthalten; "Aryl" ein aromatisches monocyclisches oder multicyclisches Ringsystem bedeutet, das 6 bis 14 Ringkohlenstoffatome enthält; "Heteroaryl" cyclische aromatische Gruppen mit 5 oder 6 Ringatomen oder bicyclische Gruppen mit 11 bis 12 Ringatomen mit einem oder zwei Heteroatomen bedeutet, die unabhängig ausgewählt sind aus O, S oder N, wobei das Heteroatom/die Heteroatome eine carbocyclische Ringstruktur unterbricht/unterbrechen und eine ausreichende Anzahl delokalisierter n-Elektronen hat/haben, um aromatischen Charakter zu liefern, mit der Maßgabe, dass die Ringe keine benachbarten Sauerstoff- und/oder Schwefelatome enthalten.
worin, wenn nicht anders definiert ist, "Alkyl" (einschließlich der Alkylanteile von Alkoxy, Alkylamino und Dialkylamino usw.) eine aliphatische Kohlenwasserstoffgruppe bedeutet, die geradkettig oder verzweigt sein kann und 1 bis 20 Kohlenstoffatome enthält; "Halo" Fluor-, Chlor-, Brom- oder Iodgruppen bedeutet; "Halogen" Fluor, Chlor, Brom oder Iod bedeutet; "Alkoxy" eine Alkyl-O-Gruppe bedeutet, in der die Alkylgruppe wie zuvor beschrieben ist; "Alkenyl" eine aliphatische Kohlenwasserstoffgruppe bedeutet, die mindestens eine Kohlenstoff-Kohlenstoff-Doppelbindung enthält und geradkettig oder verzweigt sein kann und 2 bis 15 Kohlenstoffatome enthält; "Alkanoyl" ein an ein Carbonyl gebundenes Alkyl ist, wobei Alkyl die gleiche Bedeutung wie oben definiert hat; "Cycloalkyl" ein nicht-aromatisches mono- oder multicyclisches kondensiertes Ringsystem bedeutet, das 3 bis 10 Ringkohlenstoffatome enthält; "Cycloheteroalkyl" ein nicht-aromatisches mono- oder multicyclisches kondensiertes Ringsystem bedeutet, das 3 bis 10 Ringkohlenstoffatome enthält, wobei das Cycloheteroalkyl 1 oder 2 Heteroatome unabhängig ausgewählt aus O, S oder N aufweist, wobei das Heteroatom/die Heteroatome eine carbocyclische Ringstruktur unterbricht bzw. unterbrechen, mit der Maßgabe, dass die Ringe keine benachbarten Sauerstoff- und/oder Schwefelatome enthalten; "Aryl" ein aromatisches monocyclisches oder multicyclisches Ringsystem bedeutet, das 6 bis 14 Ringkohlenstoffatome enthält; "Heteroaryl" cyclische aromatische Gruppen mit 5 oder 6 Ringatomen oder bicyclische Gruppen mit 11 bis 12 Ringatomen mit einem oder zwei Heteroatomen bedeutet, die unabhängig ausgewählt sind aus O, S oder N, wobei das Heteroatom/die Heteroatome eine carbocyclische Ringstruktur unterbricht/unterbrechen und eine ausreichende Anzahl delokalisierter n-Elektronen hat/haben, um aromatischen Charakter zu liefern, mit der Maßgabe, dass die Ringe keine benachbarten Sauerstoff- und/oder Schwefelatome enthalten.



