CN1596258A - 腺A2α受体拮抗剂 - Google Patents
腺A2α受体拮抗剂 Download PDFInfo
- Publication number
- CN1596258A CN1596258A CNA028239229A CN02823922A CN1596258A CN 1596258 A CN1596258 A CN 1596258A CN A028239229 A CNA028239229 A CN A028239229A CN 02823922 A CN02823922 A CN 02823922A CN 1596258 A CN1596258 A CN 1596258A
- Authority
- CN
- China
- Prior art keywords
- alkyl
- group
- mmol
- alkoxyl group
- compound
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Pending
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D487/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00
- C07D487/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, not provided for by groups C07D451/00 - C07D477/00 in which the condensed system contains two hetero rings
- C07D487/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/08—Antiepileptics; Anticonvulsants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/14—Drugs for disorders of the nervous system for treating abnormal movements, e.g. chorea, dyskinesia
- A61P25/16—Anti-Parkinson drugs
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/18—Antipsychotics, i.e. neuroleptics; Drugs for mania or schizophrenia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/24—Antidepressants
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P9/00—Drugs for disorders of the cardiovascular system
- A61P9/10—Drugs for disorders of the cardiovascular system for treating ischaemic or atherosclerotic diseases, e.g. antianginal drugs, coronary vasodilators, drugs for myocardial infarction, retinopathy, cerebrovascula insufficiency, renal arteriosclerosis
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Life Sciences & Earth Sciences (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Medicinal Chemistry (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Biomedical Technology (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Psychiatry (AREA)
- Pain & Pain Management (AREA)
- Urology & Nephrology (AREA)
- Vascular Medicine (AREA)
- Cardiology (AREA)
- Hospice & Palliative Care (AREA)
- Heart & Thoracic Surgery (AREA)
- Psychology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Nitrogen And Oxygen Or Sulfur-Condensed Heterocyclic Ring Systems (AREA)
Abstract
本发明涉及具有结构式I之化合物或其医药上可接受盐类或溶剂合物,其中R、R2及R3定义如说明书,其医药组合物,以及经由对需要治疗之病人投予本发明化合物来治疗中风或中枢神经系统疾病之方法。
Description
相关申请案的交叉参考
本案请求美国临时申请案60/334,293,申请日2001年11月30日的优先权。
技术领域
本发明涉及有取代1,2,4-三唑并[1,5-c]嘧啶腺苷A2a受体拮抗剂,该化合物用于治疗中枢神经系统疾病,特别帕金森病的用途,以及涉及含该化合物的医药组合物。
背景技术
已知腺苷为多种生理功能的内生性调理剂。在心血管系统层面上,腺苷为强力血管舒张剂及心脏抑制剂。在中枢神经系统,腺苷诱发镇定、抗焦虑及抗癫痫效果。在呼吸系统,腺苷诱发支气管收缩。在肾脏层面上,腺苷有双相作用,在低浓度时诱发血管收缩,而在高剂量时诱发血管舒张。腺苷对于脂肪细胞充当脂解抑制剂,以及对于血小板充当抗凝血剂。
腺苷作用系经由与属于与G蛋白偶合的受体家族的不同膜特异性受体的相互作用传递。生化及药理研究连同分子生物学之进展,使得能识别至少四亚型腺苷受体:A1、A2a、A2b及A3。 A1及A3有高亲和力,能抑制腺苷酸环化酶这种酶的活性,A2a及A2b有低亲和力,能刺激同种酶的活性。也已经识别能作为拮抗剂与A1、A2a、A2b及A3受体相互作用的腺苷类似物。
A2a受体的选择性拮抗剂由于副作用程度降低而具有药理学利益。A2a拮抗剂在中枢神经系统中具有抗抑郁性质且可刺激认知功能。此外,资料显示A2a受体以高密度存在于基底神经节中,已知基底神经节对运动的控制是重要的。如此,A2a拮抗剂可改善因神经退化疾病例如帕金森病、阿尔茨海默病等老年痴呆、以及器质性精神疾病造成的运动能力受损。
已发现若干与黄嘌呤有关的化合物是A1受体选择性拮抗剂,并发现黄嘌呤及非黄嘌呤化合物有高A2a亲和力,而且有不同程度的A2a相对于A1选择性。7位上有不同取代的三唑并嘧啶腺苷A2a受体拮抗剂以前已经公开于例如WO 95/01356;US 5,565,460;WO 97/05138;及WO98/52568。吡唑并有取代三唑并嘧啶腺苷A2a受体拮抗剂公开于2001年5月24日提交的US 09/207,143中。
发明内容
本发明涉及一种具有结构式I的化合物

或其医药上可接受盐或溶剂合物;其中:
R选自由R4-杂芳基、R5-苯基、(C4-C6)环烯基、-C(=CH2)CH3、-C≡C-CH3、 -CH=C(CH3)2、及-CH=CH-CH3组成的一组;
-CH=C(CH3)2、及-CH=CH-CH3组成的一组;
R2选自由-W-X、-NR19(CH2)m-W-X、及-NR19CH(CH3)-W-X组成的一组,或
R2选自由烷基、链烯基及-NR18R19组成的一组,其中该烷基、链烯基或-NR18R19是任选地有-W-X取代的;
R3选自由H、卤原子、烷基、三氟甲基、烷氧基、烷氧烷基、羟烷基、烷基氨基、烷基氨基烷基、二烷基氨基、二烷基氨基烷基、氨基烷基、芳基、杂芳基、及CN组成的一组;
R4为1至3个相同或相异的取代基且独立地选自由氢、(C1-C6)-烷基、-CF3、卤原子、-NO2、-NR15R16、(C1-C6)烷氧基、(C1-C6)烷硫基、(C1-C6)烷基亚磺酰基、(C1-C6)烷磺酰基、-COOR17及-C(O)NR6R7组成的一组;
R5为1至5个相同或相异的取代基且独立地选自由氢、卤原子、(C1-C6)烷基、羟基、(C1-C6)烷氧基、-CN、-NH2、(C1-C6)烷基氨基、二-((C1-C6)烷基)氨基、-CF3、-OCF3、-S(O)0-2(C1-C6)烷基及-CH2-SO2-苯基组成的一组;
R6及R7为相同或相异,且各自独立地选自由氢及(C1-C6)烷基组成的一组;
R8为1至5个相同或相异的取代基且独立地选自由氢、卤原子、(C1-C6)烷基、羟基、(C1-C6)烷氧基、-CN、氨基、二-((C1-C6)烷基)氨基、-CF3、-OCF3、乙酰基、-NO2、羟基(C1-C6)烷氧基、(C1-C6)-烷氧基(C1-C6)烷氧基、二-((C1-C6)-烷氧基)(C1-C6)烷氧基、(C1-C6)-烷氧基(C1-C6)烷氧基-(C1-C6)-烷氧基、羧基(C1-C6)-烷氧基、(C1-C6)-烷氧羰基(C1-C6)烷氧基、(C3-C6)环烷基(C1-C6)烷氧基、二-((C1-C6)烷基)氨基(C1-C6)烷氧基、吗啉基、(C1-C6)烷基-SO2-、(C1-C6)烷基-SO2-(C1-C6)烷氧基、四氢吡喃氧基、(C1-C6)烷基羰基(C1-C6)-烷氧基、(C1-C6)-烷氧羰基、(C1-C6)烷基羰氧基(C1-C6)-烷氧基、-SO2NH2、苯氧基、

-O-CH2-P(O)(OR6)2-、以及-P(O)(OR6)2组成的一组;或相邻的R8取代基共同形成为-O-CH2-O-、-O-CH2CH2-O-、-O-CF2-O-或-O-CF2CF2-O-,且与其所连接的碳原子一起形成一个环;
R9选自由(C1-C6)烷基、R8-芳基-、R8-芳基(C1-C6)烷基-、噻吩基、吡啶基、(C3-C6)-环烷基、(C1-C6)烷基-OC(O)-NH-(C1-C6)烷基-、二-((C1-C6)烷基)氨基甲基、环杂烷基(C1-C6)烷基、芳氧基(C1-C6)烷基、烷氧基(C1-C6)烷基以及

组成的一组;
R10为1-2个相同或相异的取代基且独立地选自由氢、(C1-C6)烷基、R5-芳基及R4-杂芳基组成的一组,或同一个碳上的两个R10取代基可形成=O;
R11为氢或(C1-C6)烷基;-C(O)烷基或R17与R11合在一起形成-(CH2)p-A-(CH2)q,其中p及q各自独立地为2或3,以及A选自由一个键、-CH2-、-S-及-O-组成的一组,以及与它们所连接的氮一起形成一个环;
R12为1-2个相同或相异的取代基且独立地选自由氢、(C1-C6)烷基、羟基、(C1-C6)烷氧基、卤原子及-CF3组成的一组;
R13选自由H、(C1-C6)烷基、苯基、苄基、(C2-C6)链烯基、(C1-C6)烷氧基(C1-C6)烷基、二-((C1-C6)烷基)氨基(C1-C6)烷基、吡咯烷基(C1-C6)烷基及哌啶子基(C1-C6)烷基组成的一组;
R14选自由H、卤原子、(C1-C6)烷基或(C1-C6)烷氧基组成的一组;
R15选自由H及(C1-C6)烷基组成的一组;
R16选自由H、(C1-C6)烷基-C(O)-及(C1-C6)烷基-SO2-组成的一组;
R17选自由(C1-C6)烷基、(C1-C6)羟烷基、(C3-C6)环烷基、(C1-C6)烷氧基(C1-C6)烷氧基、(C1-C6)烷氧基、(C1-C6)烷氧基(C1-C6)烷基、烯丙基、炔丙基、R8-杂芳基-、R8-芳基-及R8-芳基(C1-C6)烷基-组成的一组;
R18选自由一个键、-CH2-、-CH(OH)-、-CH(CH3)-、-C(CH3)n-、-(CH2)n-及-O(CH2)n-组成的一组;
R19选自由H、(C1-C6)烷基、(C1-C6)烷基(C1-C6)环烷基、(C1-C6)环烷基(C1-C6)烷基及(C1-C6)烷氧基(C1-C6)烷基组成的一组;
Q及Q1为相同或相异且各自独立地选自由

组成的一组;
m及n各自独立地为1-3;
p及q各自独立地为0-2;
s为0-4;
W为芳基或有1-3个可相同或相异且独立地选自由N、O和S组成的一组的杂原子的杂芳基,其中该芳基或杂芳基任选地有1-3个取代基取代,该取代基可相同或相异且独立地选自由烷基、芳基、烷基环烷基、卤原子、羟基、羟烷基、烷氧基、烷基烷氧基、烷氧基烷氧基、-NR6R7、(C2-C6)链烯基及-CN组成的一组,或
X选自由H、NH2、-N(R6)(CH2)s-芳基、-N(R6)(CH2)s-杂芳基、-N(R6)(CH2)m+1-OH、及-N(CH3)2,或
X为-R18-Y-Z,
Y选自由-N(R6)CH2CH2N(R7)-、-N(R6)(CH2)n芳基、-OCH2CH2N(R6)-、-O-、-S-、-CH2S-、-(CH2)2-3-N(R6)-、R8-二价杂芳基, 和
和

组成的一组;以及
Z选自由H、烷基、烷氧基烷基、R8-芳基-、R8-芳基(C1-C6)烷基-、R8-杂芳基-、R8-双环烷基-、氨基烷基、烷基氨基、NH2、-N-(R6)(CH2)s-芳基、-N(R6)(CH2)s-杂芳基、-N(R6)C(O)OR17、烷基环杂烷基、环杂烷基、环杂烷基烷基、烷氧基环杂烷基、杂芳基;R8-苯并稠合杂芳基-、二苯甲基及R9-C(O)-组成的一组;或
当Y为
时,Z也可为-OH、R9-SO2-、R17-N(R11)(CH2)s-C(O)-、R17-OC(O)-、R17-O(CH2)nC(O)-、苯并稠合杂芳基(CH2)nC(O)-、苯并稠合杂芳基(CH2)n-或R17-N(R11)-C(S)-;或
当Q为

时,Z也可为R17R11N-、苯基氨基或吡啶基氨基;或
Z与Y合在一起选自由下列基团组成的一组



和

本发明的另一方面涉及一种医药组合物,包含一种或多种式I化合物以及一种或多种医药上可接受载体。
本发明的另一方面涉及一种医药组合物,包含一种或多种式I化合物以及一种或多种已知可用于治疗帕金森病的药剂于一种或多种医药上可接受载体中。
本发明的另一方面涉及一种治疗中风或中枢神经系统疾病的方法,包含对需要此种治疗的患者给药一种或多种式I化合物。在本发明的这个方面,中枢神经系统疾病包括认知疾病或神经退化疾病例如帕金森病、老年痴呆或器质性来源的精神病。较佳对患者给药的化合物量是治疗有效量。
本发明的另一方面涉及一种治疗帕金森病的方法,该方法系使用一种或多种式I化合物与一种或多种可用于帕金森病治疗药剂的组合,该可用于帕金森病治疗的药剂包含例如多巴胺;多巴胺能兴奋剂;B型单胺氧化酶(MAO-B)抑制剂;DOPA脱羧酶抑制剂(DCI);或儿茶酚-O-甲基转移酶(COMT)抑制剂。在本发明的这个方面,一种或多种式I化合物与一种或多种其它抗帕金森病药剂可以以分开剂型同时或循序给药。
本发明的又另一方面涉及一种药剂盒,包含在单一包装内的分开容器中组合用于治疗帕金森病的医药组合物,其中一个容器含有一种包含一种或多种式I化合物于一种或多种医药上可接受载体中的医药组合物;且其中在各分开容器中,一种或多种医药组合物各自包含一种或多种可用于帕金森病治疗的药剂于一种或多种医药上可接受载体中。
发明的详细说明
本发明涉及具有结构式I的化合物
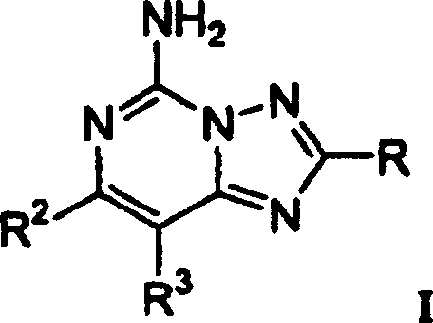
或该化合物的医药上可接受盐或溶剂合物,其中R、R2及R3定义如前。
除非另行陈述,否则下列定义适用于本说明书和权利要求书全文。这些定义的适用与某一术语本身单独使用还是与其它术语组合使用无关。因此,「烷基」的定义适用于「烷基」以及「烷氧基」、「卤烷基」等的「烷基」部分。
当任何变量(例如R2)在任何成分中出现不止一次时,其在每次出现中的定义独立于其在每个另一次出现中的定义。此外,只有当取代基和/或变量的组合导致稳定化合物时,这样的组合才是允许的。
烷基(包括烷氧基、烷基氨基及二烷基氨基的烷基部分)表示可以是链中含有1个至约20个碳原子的直链或分支链的脂肪族烃基。较佳烷基在链中含有1个至12个碳原子。更佳烷基在链中含有1个至约6个碳原子。分支链烷基表示一个或多个低级烷基如甲基、乙基或丙基连接到直链烷基链上。「低级烷基」表示一种在链中含有约1个至约6个碳原子且可为直链或分支链的基团。本发明的较佳烷基为低级烷基。适用烷基之非限制性实例包括甲基、乙基、正丙基、异丙基、正丁基、叔丁基、正戊基、庚基、壬基、癸基、三氟甲基及环丙基甲基。烷基可以有一个或多个取代基取代,取代基可相同或相异且选自烷基、芳基、杂芳基、羟基、烷氧基、卤原子、硝基、氰基及环烷基组成的一组。
「卤原子」表示氟、氯、溴或碘基。较佳为氟、氯或溴,及更佳为氟及氯。
「卤素」包含氟、氯、溴或碘。较佳为氟、氯或溴,及更佳为氟及氯。
「烷氧基」表示烷基-O-基,其中烷基定义如前。适当烷氧基之非限制性实例包括甲氧基、乙氧基、正丙氧基及异丙氧基。烷基是经由醚氧连接至毗邻片段。
烷氧烷基为一种含有一个经由烷基连接至主基的烷氧基的片段。
「烷氧羰基」表示烷基-O-C(O)-基。适当烷氧羰基的非限制性实例包括甲氧羰基及乙氧羰基。烷氧基经由羰基连接至相邻的片段。
「烷基磺酰基」表示烷基-S(O)2-基。较佳基团是其中烷基为低级烷基的烷基磺酰基。烷基经由磺酰基连接至相邻的片段。
「烷基亚磺酰基」表示烷基-S(O)-基。较佳基团是其中烷基为低级烷基的烷基亚磺酰基。烷基经由亚磺酰基连接至相邻的片段。
「链烯基」表示含有至少一个碳-碳双键之脂肪族烃基,该脂肪族烃基可为直链或分支链,且链中包含2至15个碳原子。较佳链烯基在链中含有2至12个碳原子;更佳在链中含有2至6个碳原子。分支链表示一个或多个低级烷基如甲基、乙基或丙基连接至直链烯基链上。「低级链烯基」表示链中含有2至6个碳原子且该链可为直链或分支链。适当链烯基的非限制性实例包括乙烯基、丙烯基、正丁烯基、3-甲基丁-2-烯基及正戊烯基。
链烷酰基为一个附着于羰基之烷基,其中烷基定义同前。
亚烷基表示二价烷基,同样指直链或分支链。
「环系取代基」表示连接于芳香族或非芳香族环系的取代基,该取代基例如置换环系上一个可供利用的氢。环系取代基可相同或相异,各自独立地选自烷基、芳基、杂芳基、芳烷基、烷基氨基、芳氨基、烷芳基、芳烯基、杂芳烷基、烷杂芳基、杂芳烯基、羟基、羟烷基、烷氧基、芳氧基、芳烷氧基、芳烷基氧基、酰基、芳酰基、卤原子、硝基、氰基、羧基、烷氧羰基、芳氧羰基、芳烷氧羰基、烷基磺酰基、芳基磺酰基、杂芳基磺酰基、烷基亚磺酰基、芳基亚磺酰基、杂芳基亚磺酰基、烷硫基、芳硫基、杂芳硫基、芳烷硫基、杂芳烷硫基、环烷基、环烯基、Y1Y2N-、Y1Y2N-烷基-、Y1Y2NC(O)-及Y1Y2NSO2-(其中Y1及Y2可以相同或相异且独立地选自氢、烷基、芳基及芳烷基组成的一组)组成的一组。
「任选地有取代」这一术语表示以特定基团、基或片段的任选取代。
「环烷基」表示含3至10个环碳原子,较佳3至7个环碳原子,更佳3至6个环碳原子之非芳香族单环或多环稠合环系。环烷基可以任选地有一个或多个「环系取代基」取代,该环系取代基可相同或相异且定义同上。适当单环环烷基之非限制性实例包括环丙基、环丁基、环戊基、环己基等。适当多环系环烷基之非限制性实例包括1-十氢萘基、降冰片基、金刚烷基等。环烷基可以任选地有一个或多个「环系取代基」取代,该环系取代基可相同或相异且定义同上。
「环杂烷基」表示包含3至10个环碳原子,较佳3至7个环碳原子,及更佳3至6个环碳原子之非芳香族单环或多环稠合环系,其中该环杂烷基含1或2个独立地选自O、S或N的杂原子,该杂原子插入碳环结构中,但环不含毗邻氧原子和/或硫原子。环杂烷基可任选地有一个或多个「环系取代基」取代,该环系取代基可相同或相异且定义同上。
「芳基」表示包含6至14个环碳原子,且较佳6至10个环碳原子的芳香族单环或多环环系。芳基可任选地有一个或多个「环系取代基」取代,该环系取代基可相同或相异且定义如本文。适当芳基的非限制性实例包括苯基及萘基。
「杂芳基」表示5或6个环原子的环状芳香族基、或11至12个环原子的双环基,其1个或2个杂原子独立地选自O、S或N,该杂原子插入碳环系环结构中,且有足够数目的非定域π电子来提供芳香族特性,但该环不含毗邻氧和/或硫原子。较佳杂芳基含有5至6个环原子。「杂芳基」可任选地有一个或多个「环系取代基」取代,该环系取代基可相同或相异,且定义如本文。杂芳基词根名称前的前缀氮杂、氧杂或硫杂分别表示至少存在一个氮、氧或硫原子作为环原子。氮原子可形成N-氧化物。意图涵盖全部区域异构体例如2-吡啶基、3-吡啶基及4-吡啶基。有用的6-员杂芳基包括吡啶基、嘧啶基、吡嗪基、哒嗪基等及其N-氧化物。有用的5-员杂芳基包括呋喃基、噻吩基、吡咯基。噻唑基、异噻唑基、咪唑基、吡唑基、异噁唑基等。有用的双环基包括衍生自上述杂芳基之苯并稠合环系,例如喹啉基、2,3-二氮杂萘基、喹唑啉基、苯并呋喃基、苯并噻吩基、吲哚基等。
二价杂芳基表示键合至两个不同基团的杂芳基。在本发明范围内,当Y为二价R8-杂芳基时,一个环员连接至变量X,另一个环员连接至变量Z;R8取代基连接至其余环员上。二价杂芳基是对环名加上「二基」来命名,例如吡啶二基环显示为:
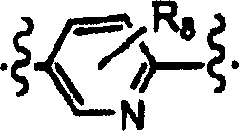
芳羰基为芳基经由羰基连接至主基,其中芳基定义如前。
烷芳基为一种含有烷基经由芳基连接至主基或环的片段。
亚环烷基表示二价环烷基。
「溶剂合物」一词在本文中用来表示由一个溶质离子或分子与一个或多个溶剂分子组成的聚集体,例如含有此种离子的水合物。
「药物前体」一词表示一种属于药物先质的化合物,该化合物在对病人给药后经由一些化学或生理过程而于活体内释放出药物(例如一种药物前体在达到生理pH或经由酶作用时被转化成所希望的药物形式)。
「治疗有效量」一词在本文中用来表示足够治疗中枢神经系统疾病之用量,该等疾病例如抑郁症、认知疾病以及神经退化疾病如帕金森病、老年痴呆及器质性来源之精神病。较好一单位制剂剂量中活性化合物的治疗有效量的范围可为约0.1毫克至约1000毫克,更佳约1毫克至约300毫克。
本发明的某些化合物可以不同立体异构形式(例如对映异构体、非对映异构体及阻转异构体)存在。本发明意图涵盖全部此等立体异构体的纯粹形式和混合物,包括外消旋混合物。
式I化合物可形成也属于本发明之范围的盐。本文中对式I化合物的提法,除非另有指示,否则要理解成包括提到其盐。「盐」一词在本文中用来表示与无机酸和/或有机酸生成之酸性盐,以及与无机碱和/或有机碱生成之碱性盐。此外,当式I化合物含有碱性片段例如但不限于吡啶或咪唑,及酸性片段例如但不限于羧酸时可形成两性离子(「内盐」),也含括在此处「盐」一词的范围内。以医药上可接受(即无毒、生理上可接受)盐为佳,尽管其它盐也有用。式I化合物之盐例如可经由式I化合物与定量例如当量酸或碱于一种介质例如盐会沉淀于其中的介质、或于水性介质中反应、随后冻干而生成。
酸加成盐实例包括乙酸盐、己二酸盐、藻酸盐、抗坏血酸盐、天冬氨酸盐、苯甲酸盐、苯磺酸盐、硫酸氢盐、硼酸盐、丁酸盐、柠檬酸盐、樟脑酸盐、樟脑磺酸盐、环戊烷丙酸盐、二聚葡糖酸盐、硫酸十二烷酯盐、乙烷磺酸盐、富马酸盐、葡庚酸盐、甘油磷酸盐、半硫酸盐、庚酸盐、己酸盐、盐酸盐例如本文中公开的化合物174、氢溴酸盐、氢碘酸盐、2-羟基乙烷磺酸盐、乳酸盐、马来酸盐、甲烷磺酸盐、2-萘磺酸盐、烟碱酸盐、硝酸盐、草酸盐、果胶酯酸盐、过硫酸盐、3-苯基丙酸盐、磷酸盐、苦味酸盐、新戊酸盐、丙酸盐、水杨酸盐、琥珀酸盐、硫酸盐、磺酸盐(例如前述)、酒石酸盐、硫氰酸盐、甲苯磺酸盐(也称为tosylates)、十一烷酸盐等。此外,一般视为适合用于从碱性医药化合物生成医药上可接受盐的酸论述于例如S.Berge等人,医药科学杂志(1977)
66(1)1-19;P.Gould,国际药学杂志(1986)33 201-217;以及Anderson等人,医药化学实践(1996年,学术出版社,纽约)。这些公开内容均列为本文参考文献。
碱性盐实例包括铵盐;碱金属盐如钠、锂及钾盐;碱土金属盐如钙及镁盐;与有机碱(例如有机胺)例如为苄星、二环己基胺类、海巴明(与N,N-二(脱氢枞酸基)乙二胺生成)、N-甲基-D-葡糖胺类、N-甲基-D-葡糖酰胺类、叔丁胺类生成的盐;以及与氨基酸如精氨酸、赖氨酸等生成的盐。碱性含氮基可使用下列试剂季铵化:例如,低级烷基卤(例如甲基、乙基、丙基、及丁基氯、溴及碘)、硫酸二烷酯(例如硫酸二甲酯、二乙酯、二丁酯及二戊酯)、长链卤(例如癸基、月桂基、肉豆蔻基及硬脂基氯、溴及碘)、芳烷基卤(例如苄基及苯乙基溴)等。
所有这样的酸盐及碱盐皆意在成为本发明范围内的医药上可接受盐,全部酸盐及碱盐皆视为等效于本发明目的之对应化合物的游离形式。
这些化合物在A2a受体上有拮抗活性,可用于治疗帕金森病及抑郁症。它们可单独或与多巴胺能剂如L-DOPA或罗匹尼罗组合使用。它们也可与已知抗抑郁治疗剂配合使用。
式I化合物是用下列三种反应方案中所示方法制备的:
方案1:

式中X=Cl,Br,I
在方案1中,2-氨基-4,6-二氯-嘧啶(式中X=Cl)II与芳基硼酸之间在甲苯、乙醇、碳酸钠(水)溶液中在高温下的钯催化偶合反应产生式III化合物。III使用适当酰肼在丁醇中在升高温下处理,获得酰肼IV。式IV化合物用N,O-二(三甲基甲硅烷基)乙酰胺在高温下处理,获得式I化合物。
另外,当起始物料II 2-氨基-4,6-二氯-嘧啶(式中X=Cl)用适当酰肼在丁醇中在高温下处理时,产生了对应酰肼V。结构式V化合物在N,O-二(三甲基甲硅烷基)乙酰胺处理获得式VI化合物。化合物VI(式中X=Cl)与芳基硼酸之间在甲苯、乙醇、碳酸钠(水)溶液中在高温下的钯催化偶合反应,获得式I化合物。
方案2
式I化合物也可如以上方案2所示那样制备。所希望的先质IX即可使用氯甲酸烷酯于碱存在下处理适当酮VII制备;也可在碱性条件下使用R3X处理适当β-酮酯VIII制备。β-酮酯IX可与碳酸胍盐在高温下于惰性溶剂(例如DMF)中进行缩合反应制造氨基嘧啶X。X在高温下用POCl3处理获得氯类似物XI。XI使用适当酰肼在丁醇中在高温下处理,获得酰肼XII。式XII化合物使用N,O-二(三甲基甲硅烷基)乙酰胺处理,获得式I化合物。
另外,XI使用有Boc-保护之肼于惰性溶剂(如DMF)中在高温下处理获得式XIII化合物,后者又可使用酸如TFA于室温脱去保护获得游离肼XIV。XIV使用适当羧酸在偶合剂如EDCl存在下在惰性溶剂如DMF中在室温处理,制造酰肼XII。
方案3
式中

且R10如以上定义
另外,式I化合物可如方案3所示那样制备。化合物VI可通过使用碳酸钾在正丁醇中在高温下处理而与式XV胺进行亲核置换反应来制造式I化合物。
本发明的另一方面涉及一种医药组合物,包含一种或多种式I化合物以及一种或多种医药上可接受载体。
本发明的另一方面涉及一种医药组合物,包含一种或多种式I化合物和一种或多种已知可用于治疗帕金森病的药剂以及一种或多种医药上可接受载体。
本发明的另一方面涉及一种治疗中风或中枢神经系统疾病之方法,包含对需要此种治疗的患者给药一种或多种式I化合物。在本发明的这一方面,中枢神经系统疾病包括认知疾病或神经退化疾病例如帕金森病、老年痴呆或器质性来源的精神病。具体地说,本发明是针对治疗帕金森病的方法,包含对需要此种治疗的患者给药一种或多种式I化合物。较佳所给药的化合物量是治疗有效量。
本发明的另一方面涉及一种治疗帕金森病的方法,该方法并用一种或多种式I化合物与一种或多种可用于帕金森病治疗的药剂,例如多巴胺;多巴胺能兴奋剂;B型单胺氧化酶抑制剂(MAO-B);DOPA脱羧酶抑制剂(DCI);或儿茶酚-O-甲基转移酶(COMT)抑制剂。在本发明的这一方面,一种或多种式I化合物与一种或多种其它抗帕金森剂可以以分开剂型同时或顺序给药。
本发明的又另一方面涉及一种药剂盒,包含在单一包装内若干分开容器中可组合用于治疗帕金森病的医药组合物,其中一个容器包含一种医药组合物,该组合物含有一种或多种式I化合物与一种或多种医药上可接受载体;以及其中在若干分开容器中,一种或多种医药组合物各自包含一种或多种帕金森病治疗用药剂和一种或多种医药上可接受载体。
为了从本发明所述化合物制备医药组合物,惰性医药上可接受载体可为固体或液体。固体剂型制剂包括散剂、片剂、可分散颗粒剂、胶囊剂、扁胶囊剂及栓剂。散剂及片剂可包含约5至约70重量%有效成分,包括式I化合物以及任选地其它可用于治疗帕金森病的化合物。适当固体载体是业内已知的,例如碳酸镁、硬脂酸镁、滑石、糖、乳糖。片剂、散剂、扁胶囊剂及胶囊剂可用作为适合经口给药的固体剂型。
为制备栓剂,首先熔化低熔点蜡,如脂肪酸甘油酯类或可可脂的混合物,将有效成分搅拌均匀分散于其中。然后将熔融均匀混合物倾入方便尺寸的模具内,让其冷却及固化。
液体剂型制剂也可以包括溶液剂、悬浮液剂及乳液剂。例如非经肠注射用水溶液剂或水-丙二醇溶液剂。
液体剂型制剂也包括经鼻内给药用溶液剂。
适合于吸入用气雾剂制剂可以包括溶液及粉末形式的固体剂型,这可以与医药上可接受载体例如惰性压缩气体组合。
也包括意图在临使用前转化成经口或非经肠给药用液体剂型制剂的固体剂型制剂,此种液体剂型包括溶液剂、悬浮液剂及乳液剂。
本发明的化合物也可经皮输送。经皮组合物可呈霜剂、洗剂、气雾剂和/或乳液剂剂型,而且可包括在惯常用于此项目的基体型或贮器型经皮贴剂。
较好将化合物经口给药。
较好该医药制剂呈单元剂型。以这样的剂型,将制剂细分成含适量有效成分的单元剂量,例如可达到所希望目的之有效量。
活性化合物在制剂单元剂量中的数量可根据特定用途改变或调整为约0.1毫克至约1000毫克,更佳约1毫克至约300毫克。
所采用的实际剂量可依据患者需要以及所治疗病情的严重程度改变。特定情况的恰当剂量的确定属于业内已知范围。通常治疗始于较小剂量,即低于该化合物的最佳剂量。随后,该剂量以小增量递增,直至达到该种情况下的最佳效果为止。为方便起见,若希望可将总日剂量细分并于该日内分批给药。
本发明化合物及其医药上可接受盐的给药量及给药频率将由主治临床医师考虑到例如患者年龄、情况及身材以及所治疗症状严重程度等因素的判断来调整。式I化合物的典型推荐剂量体制为每日口服约10毫克至约2000毫克,且较佳约10毫克至约1000毫克,分成二剂至四剂,以提供中枢神经系统疾病如帕金森病的缓解。当在这样的剂量范围内给药时,该化合物是无毒的。
实施例
下列实施例用来提供对本发明的进一步了解,但无论如何不意味着限制本发明的有效范围。
实施例1
步骤1:2-氨基-4-氯-6-甲基嘧啶(1.44克,10.00毫摩尔)及2-呋喃甲酰肼(1.89克,15.0毫摩尔)在丁醇(50毫升)中的混合物在90℃加热16小时。反应混合物冷却至室温后,残留物以甲醇洗涤,所得沉淀过滤,获得固体。
步骤2:步骤1制造的固体(0.77克,3.30毫摩尔)在N,O-二(三甲基甲硅烷基)乙酰胺(5毫升)中于120℃加热过夜。反应混合物冷却及倾入冰水中,搅拌4小时。使用乙酸乙酯萃取该混合物,以硫酸钠干燥,过滤及真空浓缩。残留物用硅胶色谱法精制获得固体。
1H NMR(DMSO-d6)δ7.83(br.M,3H),7.10(dd,1H),6.75(s,1H),6.64(dd,1H),2.30(s,3H).质谱(ESI):216.0.
实施例2

步骤1:2-氨基-4,6-二氯-嘧啶(10.00克,60.98毫摩尔)及2-呋喃甲酰肼(7.68克,60.98摩尔)的混合物于丁醇(200毫升)中在90℃加热20小时。反应混合物冷却至室温,混合物以乙酸乙酯及水萃取,将乙酸乙酯层收集、用硫酸钠干燥、过滤及真空浓缩。残留物用硅胶色谱法精制获得固体B。
质谱(ESI):254.0,1H NMR(DMSO-d6)δ5.76(br s,1H),6.61(br s,2H),6.64(m,1H),7.20(d,1H),7.88(s,1H),9.01(br s,1H),10.32(br s,1H).
步骤2:步骤1产物B(9.20克,36.27毫摩尔)在N,O-二(三甲基甲硅烷基)乙酰胺(49.4克,242.14毫摩尔)中于120℃加热过夜。反应混合物冷却并倾入冰水中,搅拌4小时。混合物使用乙酸乙酯萃取,以硫酸钠干燥、过滤及真空浓缩。残留物用硅胶色谱法精制,获得固体C。
质谱(ESI):236.0,1H NMR(CDCl3)δ6.18(br s,2H),6.61(m,1H),7.04(s,1H),7.24(d,1H),7.64(s,1H).
步骤3:在封管内,步骤2产物C(50毫克,0.21毫摩尔)与3,5-二甲基苯硼酸(63毫克,0.42毫摩尔),Pd(PPh3)4(24毫克,0.02毫摩尔)及碳酸钠(74毫克,2.10毫摩尔)于3/1/1的甲苯/乙醇/水的溶剂系统中在103℃加热4小时时间。反应混合物冷却至室温后,混合物以乙酸乙酯及水萃取。将有机部分收集、以硫酸钠干燥、过滤及真空浓缩。残留物用硅胶色谱法精制,产生固体D。
质谱(ESI):306.1,1H NMR(CDCl3)δ2.41(s,6H),5.95(br s,2H),7.10(s,1H),7.24(s,1H),7.39(s,1H),7.60(s,2H),7.64(s,1H).
下列化合物以类似方式制备:


实施例3
化合物19
邻-(N-吗啉代甲基)-苯硼酸是用一种已知文献程序(美国化学会志,3863页,1960年)制备的,随后用来如实施例2所述那样制造目标化合物。质谱
(ESI):377.1,1H NMR(CDCl3)δ2.36(t,4H),3.56(t,4H),6.04(br s,2H),6.61(m,1H),7.30(s,1H),7.39(m,2H),7.51(m,2H),7.65(d,1H).
下列化合物以类似方式制备:

实施例4

化合物26
步骤1:2-氨基-4,6-二氯-嘧啶(0.50克,3.048毫摩尔),3-异丙基苯硼酸(0.30克,1.572毫摩尔),Pd(PPh3)4(0.09克,0.076毫摩尔)及4-10当量碳酸钠的混合物于1/1的乙腈/水的溶剂系统(15毫升)中于90℃加热4小时。反应混合物冷却至室温。混合物以乙酸乙酯及水萃取。将有机部分收集、用硫酸钠干燥、过滤及真空浓缩。残留物用硅胶色谱法精制,产生一种固体。质谱
(ESI)248.0,1H NMR(CDCl3)δ1.30(d,6H),2.98(m,1H),5.93(br s,2H),7.03(s,1H),7.39(m,2H),7.73(d,1H),7.82(s,1H).
步骤2:步骤1产物(0.39克,1.57毫摩尔)及2-呋喃甲酰肼(0.30克,2.36毫摩尔)在丁醇(10毫升)中于120℃加热5小时。反应混合物冷却至室温,然后用乙酸乙酯及水萃取。将乙酸乙酯层收集、用硫酸钠干燥、过滤及真空浓缩。残留物用硅胶色谱法精制,产生一种固体。质谱
(ESI):338.1,1H NMR(CDCl3)δ1.22(d,6H),2.89(m,1H),5.42(br s,1H),6.34(s,1H),6.45(br s,2H),7.18(d,1H),7.23(m,2H),7.44(s,1H),7.53(d,1H),7.67(s,1H).
步骤3:步骤2产物(0.27克,0.80毫摩尔)在N,O-二(三甲基甲硅烷基)乙酰胺(10毫升,40.4毫摩尔)中于120℃加热过夜。反应混合物冷却,然后倾至冰水中搅拌4小时。混合物用乙酸乙酯萃取、用硫酸钠干燥、过滤及真空浓缩。残留物用硅胶色谱法精制,产生一种固体。质谱
(ESI):320.0,1H NMR(CDCl3)δ1.21(d,6H),2.89(m,1H),5.42(br s,2H),6.60(m,1H),7.33(d,1H),7.41(d,1H),7.43(s,1H),7.64(s,1H).
下列化合物以类似方式制备:
实施例5
化合物39
实施例4化合物37(15毫克,0.0165毫摩尔)与10%钯/碳(5毫克)在10毫升溶剂混合物(9∶1乙酸乙酯/乙醇)中在氢气气氛下于室温搅拌3小时。让混合物通过硅藻土层,有机部分真空浓缩。残留物用硅胶色谱法精制获得一种固体。质谱
(ESI):306.0,1H NMR(CDCl3)δ2.97(t,2H),3.06(t,2H),5.97(br s,2H),6.58(m,1H),6.79(s,1H),7.20(m,2H),7.28(m,4H),7.68(m,1H).
实施例6

化合物40
步骤1:实施例4化合物38(0.60克,1.95毫摩尔)与三乙胺(1.63毫升,11.63毫摩尔)及亚磺酰氯(0.71克,9.76毫摩尔)于0℃在氮气气氛下化合3小时。反应混合物真空浓缩,然后残留物B用硅胶色谱法精制。质谱(ESI):326.1。
步骤2:步骤1产物(0.22克,0.66毫摩尔)在封管内与1-(4-甲氧乙氧基苯基)哌嗪(0.31克,1.32毫摩尔)在二甲基甲酰胺(2.0毫升)中化合,于110℃加热过夜。反应混合物冷却,用乙酸乙酯及食盐水萃取。将乙酸乙酯层收集、用硫酸钠干燥、过滤及真空浓缩。残留物C用硅胶色谱法精制。质谱
(ESI):526.1,1H NMR(CDCl3)δ8.0(S,1H),7.86(m,1H),7.62(dd,1H),7.41-7.45(m,3H),7.24(d,1H),6.82-6.85(m,4H),6.58(dd,1H),6.28(br.s,2H),4.06(t,2H),3.72(t,2H),3.66(s,2H),3.43(S,3H),3.00-3.12(m,4H),2.65-2.67(m,4H).
下列化合物以类似方式制备:



实施例7
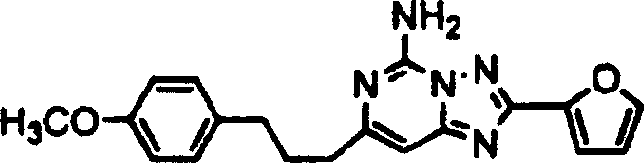
化合物77
步骤1:向4-(4-甲氧基苯基)丁酸(2.00克,10.3毫摩尔)的二氯甲烷(10毫升)溶液中加入亚磺酰氯(3.56克,30毫摩尔)。搅拌3小时及浓缩。加入2,2-二甲基-1,3-二噁烷-4,6-二酮(1.78克,12.4毫摩尔),吡啶(2.37克,31毫摩尔)及二氯甲烷(10毫升)。搅拌18小时,加入乙醇(10毫升),及回流加热5小时。加水后,用乙酸乙酯萃取及色谱法精制获得酮,呈油状。
步骤2:将步骤1产物(0.366克,1.39毫摩尔)及碳酸胍盐(0.382克,2.12毫摩尔)合并于乙醇(3毫升)中。回流加热18小时。加水(20毫升),以冰冷却及过滤。干燥,用己烷洗涤及过滤,获得嘧啶,呈固体状。
步骤3:将步骤2产物(0.15克,0.58毫摩尔)添加至磷酰氯(1.22毫升)中。回流加热1小时,浓缩,以冰处理,以氨中和,及以乙酸乙酯萃取。用PLC纯化,获得氯嘧啶,呈固体状。
步骤4:合并步骤3产物、2-糠酰肼及1.0N HCl乙醇溶液。在封管中于90℃加热16小时。用氨碱化,用乙酸乙酯萃取,用PLC纯化,获得酰肼,呈固体状。
步骤5:添加步骤4产物至BSA中。于120℃加热18小时。倾入甲醇中,浓缩,用PLC纯化,获得标题化合物,呈固体状,质谱(ESI):350。
实施例8
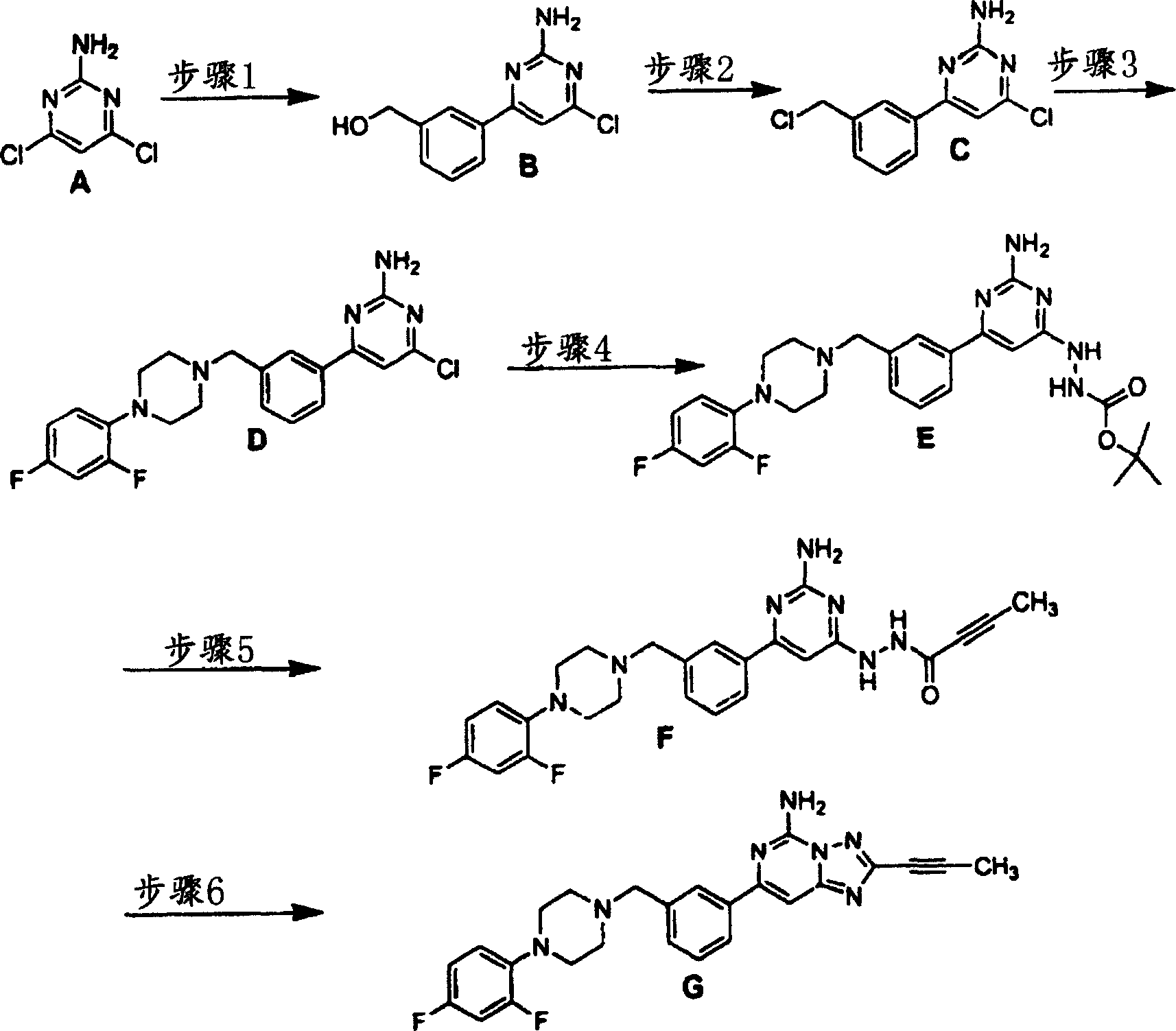
化合物78
步骤1:产物B系以如实施例4步骤1所述的类似方式合成。质谱(ESI):236.1。
步骤2:向步骤1产物B于(2.25克,9.53毫摩尔)的二氯甲烷(100毫升)溶液中,边在惰性气氛下于0℃搅拌,边加入三乙胺(8.00毫升,57.18毫摩尔),接着加入亚磺酰氯(3.50毫升,47.65毫摩尔),混合物再搅拌1小时。将混合物温热至室温,然后用二氯甲烷及食盐水萃取。将有机部分收集、用硫酸钠干燥、过滤及真空浓缩。残留物用硅胶色谱法纯化,获得产物C。质谱(ESI):255.1。
步骤3:步骤2产物C(0.50克,1.97毫摩尔)、1-(2,4-二氟苯基)-哌嗪(0.39克,1.97毫摩尔)、碘化钾(0.33克,1.97毫摩尔)、及碳酸钾(0.82克,5.90毫摩尔)的乙腈(10毫升)溶液在惰性气氛下于60℃搅拌过夜。混合物冷却至室温,然后以乙酸乙酯及食盐水萃取。将有机部分收集、用硫酸钠干燥、过滤及真空浓缩。残留物用硅胶色谱法纯化,获得产物D。质谱(ESI):416.1。
步骤4:步骤3产物D(0.84克,1.97毫摩尔)及Boc保护之肼(0.31克,2.37毫摩尔)的DMF溶液在惰性气氛于80℃搅拌过夜。混合物冷却至室温,然后用乙酸乙酯及食盐水萃取。将有机部分收集、用硫酸钠干燥、过滤及真空浓缩。残留物用硅胶色谱法纯化,获得产物E。质谱(ESI):512.1。
步骤5:向步骤4产物E(0.15克,0.29毫摩尔)的二氯甲烷(5毫升)溶液中,在室温搅拌下加入三氟乙酸(5毫升),反应混合物又搅拌1小时。将反应混合物真空浓缩并收集于DMF(2毫升)中。向此溶液中加入丁酸(30毫克,0.35毫摩尔)、EDCl(68毫克,0.35毫摩尔)、HOBT(48毫克,0.35毫摩尔)、NMM(41微升,0.35毫摩尔),在惰性气氛下于室温搅拌过夜。混合物用乙酸乙酯及食盐水萃取。将有机部分收集、用硫酸钠干燥、过滤及真空浓缩。残留物用硅胶色谱法纯化,获得产物F。质谱(ESI):478.1。
步骤6:步骤5产物F(45毫克,0.09毫摩尔)在N,O-二(三甲基甲硅烷基)乙酰胺(2毫升)中于120℃加热过夜。反应混合物冷却,倾入冰水中及搅拌4小时。混合物用乙酸乙酯萃取、用硫酸钠干燥、过滤及真空浓缩。残留物用硅胶色谱法纯化,制造一种固体G。质谱(ESI):
460.1,1H NMR(CDCl3)δ7.98(s,1H),7.88(m,1H),7.45(m,2H),7.39(s,1H),6.90(m,1H),6.79(m,2H),5.97(br s,2H),3.66(s,2H),3.06(t,4H),2.67(t,4H),2.15(s,3H).
实施例9
化合物79
步骤1:5-溴-3-(甲醇)-吡啶(6.69克,35.58毫摩尔)、叔丁基二甲基甲硅烷基氯(4.71克,46.26毫摩尔)、及咪唑(7.25克,106.74毫摩尔)的二氯甲烷(250毫升)溶液在室温搅拌3小时。混合物用二氯甲烷及食盐水萃取。将有机部分收集、用硫酸钠干燥、过滤及真空浓缩。残留物用硅胶色谱法纯化,获得产物B。质谱(ESI):304.1,302.1。
步骤2:向步骤1产物B(7.35克,24.32毫摩尔)的乙醚(125毫升)搅拌溶液中,在惰性气氛下于-78℃滴加2.5N正丁基锂的己烷(14.51毫升)溶液。搅拌10分钟后,加入硼酸三异丙酯(11.02毫升,47.75毫摩尔),溶液温热至室温、又搅拌1小时。反应以水猝灭。反应混合物真空浓缩,所得固体中间体未进一步纯化就用于下一步骤。
固体中间物(6.50克,27.99毫摩尔)收集于二甲氧乙烯(100毫升)中,加入2-氨基-4,6-二氯嘧啶(9.18克,55.98毫摩尔)、碳酸钠(10.31克,97.26毫摩尔)及四(三苯膦)钯(1.40克,1.21毫摩尔)。混合物于90℃加热4小时。反应混合物冷却、用乙酸乙酯和食盐水萃取。将乙酸乙酯层收集、用硫酸钠干燥、过滤及真空浓缩。残留物C用硅胶色谱法纯化。质谱(ESI):351.1。
步骤3:步骤2产物C(1.64克,4.67毫摩尔)及2-糠酰肼(0.92克,7.01毫摩尔)的10毫升正丁醇溶液于90℃加热过夜。混合物冷却至室温、用乙酸乙酯萃取、以食盐水洗涤。将有机部分收集、用硫酸钠干燥、过滤及真空浓缩。残留物D未进一步纯化就用于下一步骤。质谱(ESI):441.1。
步骤4:步骤3产物D(2.04克,4.63毫摩尔)在N,O-二(三甲基甲硅烷基)乙酰胺(15毫升)中于120℃加热过夜。反应混合物冷却,倾入冰水中、搅拌4小时。混合物用乙酸乙酯萃取、用硫酸钠干燥、过滤及真空浓缩。残留物用硅胶色谱法纯化,制造一种固体E。质谱(ESI):309.1,1H NMR(CDCl3)δ9.19(s,1H),8.59(s,2H),8.53(s,1H),7.76(s,1H),7.52(s,1H),6.66(m,1H),4.76(s,2H)。
下列化合物以类似方式制备。
实施例10
化合物82
步骤1:溴化物(7.0克,24.37毫摩尔)、N-Boc哌嗪(5.45克,29.24毫摩尔)、乙酸钯(0.22克,0.97毫摩尔)、三(叔丁基)膦(0.79克,3.9毫摩尔)及叔丁醇钠(3.28克,34.12毫摩尔)在甲苯(50毫升)中的混合物于氮气气氛下回流加热2小时,冷却至室温,然后以水稀释。所得混合物用乙酸乙酯萃取,用硫酸钠干燥及过滤。滤液于减压下蒸发,留下洁净产物B,后者未纯化就用于步骤2。质谱(ESI),M+1:393.1,337.1。
步骤2:步骤1产物B用氟化四丁铵(48.74克,48.74毫摩尔,1.0MTHF溶液)在THF(100毫升)中于室温处理1小时,以水稀释,然后用乙酸乙酯萃取。所得乙酸乙酯萃取物以硫酸钠干燥及蒸发,获得苯酚衍生物C。质谱(ESI),M+1:279.0,242.0。
步骤3:在0℃于氮下将三氟甲烷磺酐滴加至步骤2所得苯酚C及三乙胺(3.74毫升,26.81毫摩尔)在二氯甲烷(100毫升)中的混合物中,于此温度搅拌1小时,然后温热至室温。加入饱和碳酸氢钠溶液及以二氯甲烷萃取。萃取物使用硫酸钠干燥,及吸附于少量硅胶上,转移至一柱中且使用己烷/乙酸乙酯(4∶1)洗脱,获得三氟甲烷磺酸盐D。质谱(ESI),M+1:411.1,355.1。
步骤4:步骤3化合物D用二(邻二叔醇)二硼在PdCl2(dppf)、dppf及乙酸钾存在下在1,4-二噁烷(90毫升)中于80℃氮气下处理过夜,冷却至室温,以食盐水洗涤,以硫酸钠干燥及蒸发。残留物用硅胶柱色谱法纯化,获得产物E。质谱(ESI),M+1:389.1。
步骤5:步骤4化合物E同实施例4步骤1一样用2-氨基-4,6-二氯嘧啶处理,生成化合物F。质谱(ESI),M+1:390.1。
步骤6:执行与实施例4步骤2相同的程序,生成化合物G。质谱(ESI),M+1:480.1。
步骤7:执行与实施例4步骤3相同的程序,生成化合物H。
1H NMR(CDCl3)δ7.61(m,2H),7.43(d,1H),7.38(m,2H),7.12(m,1H),7.02(dd,1H),6.60(dd,1H),6.02(br.s,2H),3.62(m,4H),3.21(m,4H),1.50(s,9H).质谱(ESI),M+1:462.1
下列化合物以类似方式制备:
实施例11
化合物92
步骤1:受保护三氟甲烷磺酸烯醇酯B系采用参考文献程序(Synthesis,993页,1991年)制备。1H NMR(CDCl3)δ5.66(t,1H),3.99(s,4H),2.54(m,2H),2.41(m,2H),1.90(t,2H)。
步骤2:步骤1产物B(5.70克,19.79毫摩尔),3-羟基苯基硼酸(6.10克,27.71毫摩尔),氯化锂(2.50克,58.98毫摩尔),2N碳酸钠水溶液(27.70毫升),及四(三苯膦)钯(1.14克,0.98毫摩尔)于100毫升二甲氧乙烷中在回流温度加热2小时。混合物冷却至室温及真空浓缩。残留物以二氯甲烷稀释,以100毫升6%氢氧化铵与2N碳酸钠水溶液的混合物洗涤。含水部分使用另外100毫升二氯甲烷萃取。合并有机部分用硫酸钠干燥、过滤及真空浓缩。残留物用硅胶色谱法纯化,获得产物C。1H NMR(CDCl3)δ7.16(t,1H),6.97(d,1H),6.85(t,1H),6.69(dd,1H),5.98(m,1H),4.77(s,1H),4.02(s,4H),2.63(m,2H),2.46(m,2H),1.92(t,2H)。
步骤3:向步骤2产物C(2.20克,9.48毫摩尔)的二氯甲烷(60毫升)溶液中,于0℃于惰性气氛下,加入三乙胺(1.45毫升,10.43毫摩尔),然后加入三氟甲烷磺酐(1.75毫升,10.43毫摩尔)。混合物经搅拌温热至室温,又搅拌2小时。混合物以水萃取。将有机部分收集、用硫酸钠干燥、过滤及真空浓缩。残留物(1.36克,3.74毫摩尔)未进一步纯化即供使用,并收集于二噁烷(80毫升)中。向此溶液中加入二(二甲基吡啶酸基)二硼(1.14克,4.49毫摩尔)、PdCl2(dppf)(0.16克,0.22毫摩尔)、dppf(0.12克,0.22毫摩尔)及乙酸钾(1.10克,11.22毫摩尔),混合物于惰性气氛下加热至80℃过夜。混合物冷却至室温,然后用乙酸乙酯和食盐水萃取。将有机部分收集,用硫酸钠干燥、过滤及真空浓缩。残留物用硅胶色谱法纯化,获得产物D。质谱(ESI):343.1。
步骤4:步骤3产物D(0.64克,1.87毫摩尔),2-氨基-4,6-二氯嘧啶(0.31克,1.87毫摩尔),碳酸钠(0.79克,7.48毫摩尔)及四(三苯膦)钯(0.11克,0.09毫摩尔)在60毫升1/1的乙腈/水中的溶液于90℃加热3小时。反应混合物冷却,用乙酸乙酯及食盐水萃取。将乙酸乙酯层收集,用硫酸钠干燥、过滤及真空浓缩。残留物以硅胶色谱法纯化,获得化合物E。质谱(ESI):344.1。
步骤5:步骤4产物E(0.60克,1.75毫摩尔)及2-糠酰肼(0.33克,2.62毫摩尔)在15毫升正丁醇中的溶液于90℃加热2小时。混合物冷却至室温及真空浓缩。残留物收集于5毫升N,O-二(三甲基甲硅烷基)乙酰胺中,于氮气气氛下加热至120℃和3小时。将混合物冷却,倾入冰水中然后搅拌4小时。混合物用乙酸乙酯及食盐水萃取,用硫酸钠干燥、过滤及真空浓缩。残留物用硅胶色谱法纯化,获得化合物F。质谱(ESI):416.1。1H NMR(DMSO-d6)δ8.10(s,1H),7.92(m,2H),7.87(s,1H),7.51(s,1H),7.38(m,1H),7.15(t,1H),6.03(s,1H),3.86(s,1H),4.02(s,4H),255(m,2H),2.33(m,2H),1.77(t,2H)
实施例12

化合物93
步骤1:3-乙酰基苯硼酸(2.00克,12.20毫摩尔),2-氨基-4,6-二氯嘧啶(4.00克,24.40毫摩尔),碳酸钠(6.47克,61.00毫摩尔)及四(三苯膦)钯(0.70克,0.61毫摩尔)在100毫升1/1的乙腈/水中的溶液于90℃加热3小时。反应混合物冷却,用乙酸乙酯及食盐水萃取。将乙酸乙酯层收集、用硫酸钠干燥、过滤及真空浓缩。残留物以硅胶色谱法纯化,获得产物B。质谱(ESI):248.0。
步骤2:向步骤1酮产物B(3.00克,12.11毫摩尔)的75毫升乙醇溶液中,于0℃加入硼氢化钠(0.92克,24.22毫摩尔),然后温热至室温及搅拌1小时。混合物用乙酸乙酯萃取,以食盐水洗涤。将有机部分收集、以硫酸钠干燥、过滤及真空浓缩。残留物用硅胶色谱法纯化,获得产物C。质谱(ESI):250.0。
步骤3:步骤2产物C(0.27克,1.08毫摩尔)及2-糠酰肼(0.33克,2.62毫摩尔)在10毫升正丁醇中的溶液于90℃加热过夜。混合物冷却至室温,用乙酸乙酯萃取,及以食盐水洗涤。将有机部分收集,用硫酸钠干燥、过滤及真空浓缩。残留物D未进一步纯化就用于下一步骤。质谱(ESI):340.1。
步骤4:步骤3产物D(0.37毫克,1.08毫摩尔)收集于5毫升N,O-二(三甲基甲硅烷基)乙酰胺中,于氮气气氛下加热至120℃历3小时。将混合物冷却,倾入冰水中,然后搅拌4小时。混合物用乙酸乙酯及食盐水萃取,用硫酸钠干燥、过滤及真空浓缩。残留物用硅胶色谱法纯化,获得化合物E。质谱
(ESI):322.1.1H NMR(CDCl3)δ8.05(s,1H),7.86(m,1H),7.64(d,1H),7.47(m,2H),7.41(s,1H),7.25(s,1H),6.60(m,1H),6.05(br s,2H),5.01(m,1H),3.49(d,3H).
下示化合物以类似方式制备:
化合物94
实施例13
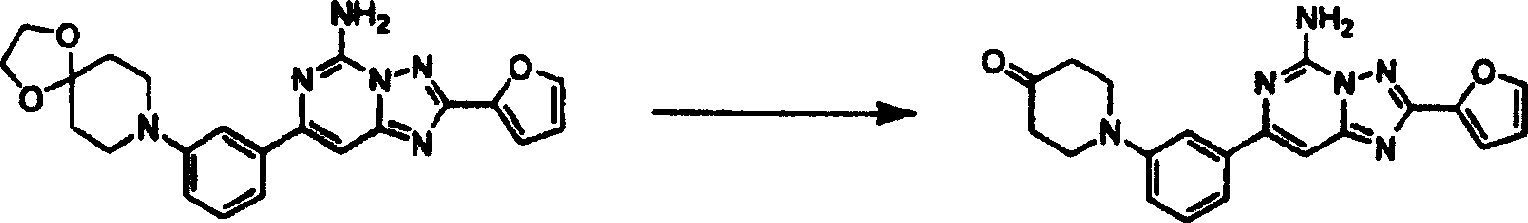
化合物95
以类似于实施例10的方式制备的缩酮产物(113毫克,0.27毫摩尔),20毫升5%盐酸水溶液及20毫升丙酮的混合物于100℃加热过夜。混合物冷却至室温,用乙酸乙酯萃取及以水洗涤。将有机部分收集,用硫酸钠干燥、过滤及真空浓缩。残留物用硅胶色谱法纯化,获得产物。质谱(ESI):375.1。1H NMR(CDCl3)δ7.57(d,1H),7.55(t,1H),7.31(m,3H),7.20(d,1H),6.95(dd,1H),6.65(brs,2H),6.53(m,1H),3.60(t,4H),2.51(t,4H).
实施例14
化合物96
向实施例13酮产物(55毫克,0.15毫摩尔),70%乙胺水溶液(0.01毫升,0.16毫摩尔)在5毫升四氢呋喃中的溶液中,加入三乙酰氧基硼氢化钠(46毫克,0.22毫摩尔)及于室温搅拌2小时。混合物以3N氢氧化钠水溶液猝灭,用乙酸乙酯萃取,及以食盐水洗涤。将有机部分收集、用硫酸钠干燥、过滤及真空浓缩。残留物用硅胶色谱法纯化,获得产物。质谱
(ESI):404.1.1H NMR(CDCl3)δ7.63(d,1H),7.60(t,1H),7.40(d,2H),7.34(t,1H),7.03(dd,1H),6.60(m,1H),6.00(br s,2H),3.77(d of t,2H),2.85(t of d,2H),2.74(m,2H),2.03(m,2H),1.55(q of t,3H),1.15(m,4H).
下列化合物以类似方式制备。
| 化合物 | 结构 | M+1(ES1) |

实施例15

化合物105
向实施例13酮产物(60毫克,0.16毫摩尔)的5毫升乙醇溶液中,加入硼氢化钠(12毫克,0.32毫摩尔)。混合物于室温搅拌1小时。混合物用乙酸乙酯萃取,及以食盐水洗涤。将有机部分收集、用硫酸钠干燥、过滤及真空浓缩。残留物用硅胶色谱法纯化,获得产物。质谱
(ESI):377.1.1H NMR(CDCl3)δ7.63(s,1H),7.60(t,1H),7.40(m,2H),7.34(t,1H),7.25(m,1H),7.03(d,1H),6.60(m,1H),6.11(br s,2H),3.88(m,2H),3.65(m,2H),3.01(m,2H),2.04(m,2H),1.17(br s,3H).
实施例16

化合物106
向实施例14产物(35毫克,0.0868毫摩尔)、二异丙基乙胺(0.02毫升,0.0955毫摩尔)的3毫升DMF溶液中加入氯甲酸乙酯(0.01毫升,0.0955毫摩尔)。混合物于室温搅拌3小时。混合物真空浓缩,残留物用硅胶色谱法纯化,获得产物。质谱
(ESI):476.1.1H NMR(CDCl3)δ7.61(d,1H),7.58(t,1H),7.39(m,2H),7.33(t,1H),7.22(d,1H),7.00(dd,1H),6.58(m,1H),6.22(br s,2H),4.15(q,3H),3.82(d,2H),3.46(m,4H),3.01(m,2H),2.05(br s,1H),1.59(br s,1H),1.26(t,3H),1.12(t,3H).
下列化合物使用酰氯以类似方式制备。

实施例17
化合物108
向实施例11步骤5产物(80毫克,0.15毫摩尔)在5毫升9/1的乙醇/乙酸乙酯溶液中的溶液中加入10%钯/碳(160毫克)。混合物在一台氢化装置中在室温下于40psi振摇1小时。混合物经硅藻土(celite)层过滤及真空浓缩。残留物用硅胶色谱法纯化,获得产物。质谱
(ESI):418.1.1H NMR(CDCl3)δ7.90(s,1H),7.77(d,1H),7.63(d,1H),7.48(m,2H),7.40(m,1H),6.59(dd,1H),6.12(br s,2H),4.00(s,4H),1.67-1.96(m,9H).
实施例18

化合物109
实施例10步骤7化合物通过在室温下用4.0M HCl二噁烷溶液处理过夜、或在氮气下使用50%TFA二氯甲烷溶液处理30分钟脱保护,减压下蒸发,及未经进一步纯化即供使用。
1H NMR(DMSO-d6)δ7.97(br.s,2H),7.94(s,1H),7.64(s,1H),7.50(m,2H),7.30(t,1H),7.08(dd,1H),7.00(dd,1H),6.70(dd,1H),3.70(br.s,1H),3.08(m,4H),2.90(m,4H).质谱(ESI),M+1:362.1
实施例19
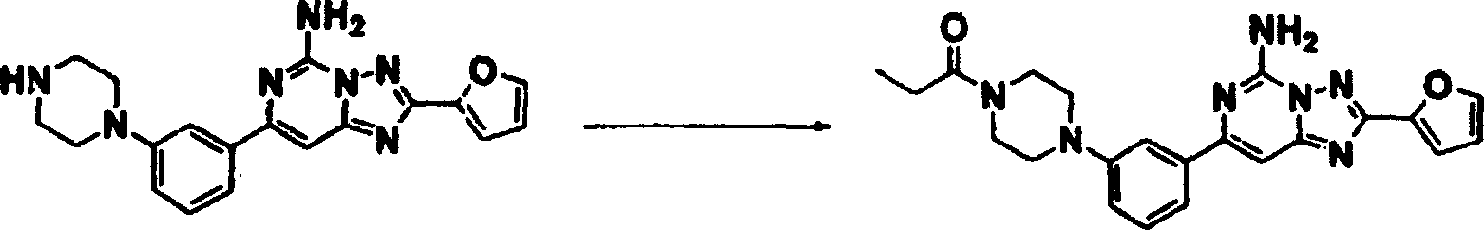
化合物110
向实施例18产物(0.10克,0.25毫摩尔)及二异丙基乙基胺(0.097克,0.75毫摩尔)的DMF(5毫升)溶液中于室温在氮气下滴加丙酰氯(0.025克,0.28毫摩尔)。2小时后加水,所得产物用乙酸乙酯萃取,用硫酸钠干燥及蒸发。使用硅胶制备型TLC纯化,获得产物。
1H NMR(CDCl3)δ7.63(dd,1H),7.60(m,1H),7.46(d,1H),7.40(s,1H),7.37(d,1H),7.24(m,1H),7.01(dd,1H),6.60(dd,1H),6.06(br.S,2H),3.82(t,2H),3.65(t,2H),3.25(m,4H),2.40(q,2H),1.19(t,3H).质谱(ESI)418.1.
下列化合物以类似方式制备。
实施例20
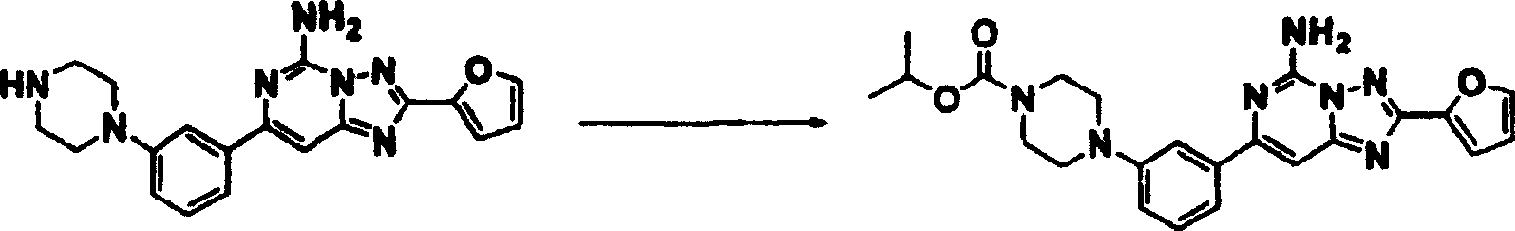
化合物116
向实施例18产物(0.125克,0.35毫摩尔)及二异丙基乙基胺(0.134克,1.04毫摩尔)的DMF(5毫升)溶液中于室温在氮气下滴加氯甲酸异丙酯(0.7毫升,0.7毫摩尔)。2小时后加水,所得产物用乙酸乙酯萃取,用硫酸钠干燥及蒸发。使用硅胶制备型TLC进行纯化,获得产物。
1H NMR(CDCl3)δ7.63(m,1H),7.59(m,1H),7.44(d,1H),7.38(s,1H),7.35(d,1H),7.24(m,1H),7.02(dd,1H),6.58(dd,1H),6.22(br.S,1H),4.95(m,1H),3.65(m,4H),3.22(m,4H),1.26(d,6H);质谱(ESI),M+1:448.1
下列化合物以类似方式制备:
实施例21
化合物119
向实施例18产物(0.11克,0.30毫摩尔)及二异丙基乙基胺(0.043克,0.058毫摩尔)的DMF(5毫升)溶液中于室温在氮气下滴加甲基磺酰氯(0.038毫升,0.026毫摩尔)。3小时后加水,所得产物用乙酸乙酯萃取,用硫酸钠干燥及蒸发。使用硅胶制备型TLC进行纯化,获得产物。
1H NMR(CDCl3)δ7.62(m,2H),7.50(d,1H),7.40(m,2H),7.23(m,1H),7.04(dd,1H),6.62(dd,1H),6.00(br.S,2H),3.37-3.42(m,8H),2.82(s,3H).质谱(ESI)440.1.
下列化合物以类似方式制备:
实施例22
化合物121
步骤1:向实施例18产物及二异丙基乙基胺的DMF(5毫升)溶液中于0℃在氮气下滴加氯乙酰氯。混合物温热至室温及搅拌过夜。然后加水,所得产物用乙酸乙酯萃取,用硫酸钠干燥及蒸发。以使用乙酸乙酯的硅胶柱色谱法进行纯化,获得中间物B。质谱(ESI),M+1:438.1。
步骤2:步骤1化合物B(0.11克,0.25毫摩尔)于室温在氮气下用过量哌啶(10当量)的DMF(5毫升)溶液处理过夜。混合物在减压下蒸发,产物C用制备型TLC以乙酸乙酯/甲醇(9∶1)纯化。
1H NMR(CDCl3)δ7.59(m,1H),7.54(m,1H),7.40(d,1H),7.33(m,2H),7.20(m,1H),6.97(dd,1H),6.55(dd,1H),6.17(br.S,2H),3.80(m,2H),3.74(m,2H),3.20(m,4H),3.13(s,2H),2.38(m,4H),1.49(m,4H),1.38(m,2H).质谱(ESI),M+1:487.1.
下列化合物以类似方式制备:
| 化合物 | 结构 | M+1(ES1) |


实施例23

化合物130
步骤1:向实施例18产物(0.145克,0.4毫摩尔)及苯甲醛(0.047克,0.44毫摩尔)的二氯甲烷(10毫升)溶液中于室温在氮气下加入三乙酰氧基硼氢化钠(0.127克,0.6毫摩尔)。5小时后,加入2.0M氢氧化钠溶液,所得产物用二氯甲烷萃取,用硫酸钠干燥及蒸发。用制备型TLC纯化,获得产物。
1H NMR(CDCl3)δ7.63(dd,1H),7.58(m,1H),7.20-7.41(m,9H),7.01(dd,1H),6.59(dd,1H),6.04(br.S,2H),3.59(s,2H),3.29(t,4H),2.64(t,4H).质谱(ESI),M+1:452.1.
下列化合物以类似方式制备。
实施例24
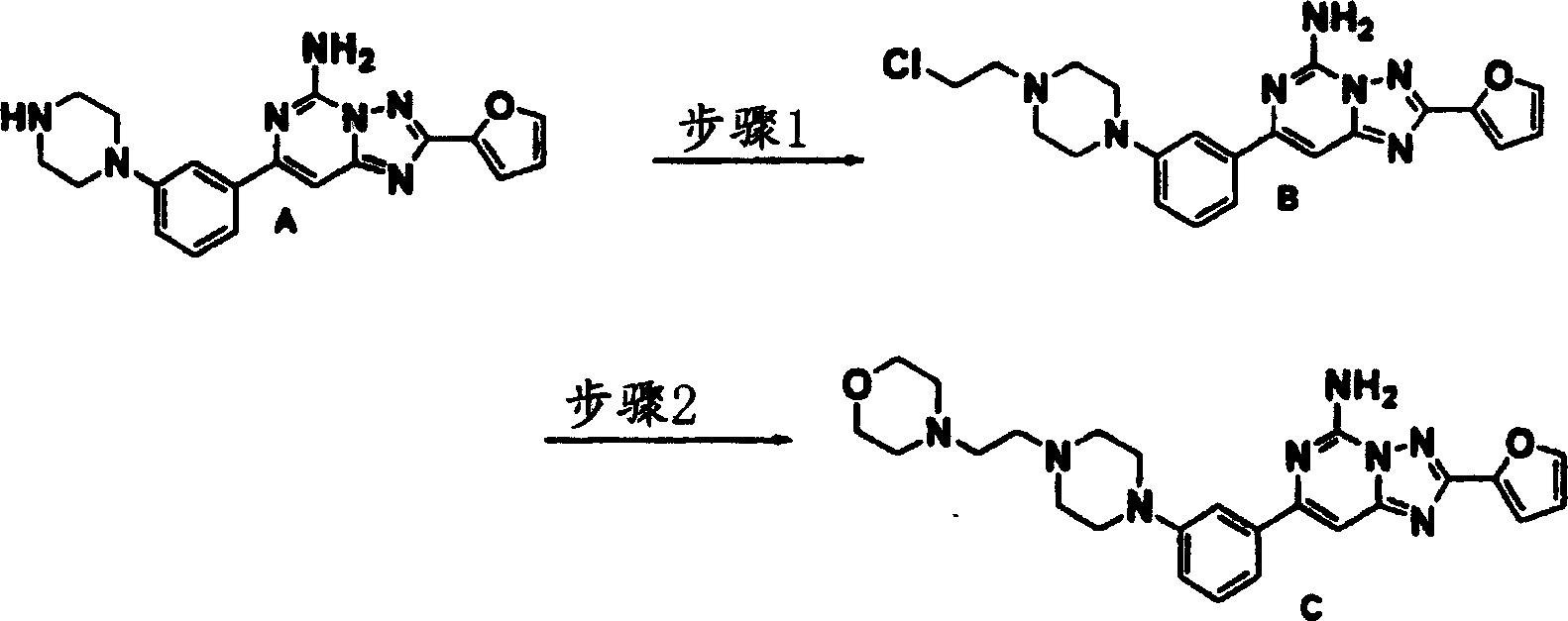
化合物135
步骤1:向实施例18产物(1.22克,3.38毫摩尔)及氯乙醛(0.64克,4.06毫摩尔50%水溶液)的二氯甲烷(60毫升)溶液中于室温在氮气下加入三乙酰氧基硼氢化钠(1.08克,5.07毫摩尔)。5小时后,加入2.0M氢氧化钠溶液,所得化合物B用二氯甲烷萃取,用硫酸钠干燥及蒸发。用柱色谱法进行纯化。质谱(ESI),M+1:424.1。
步骤2:步骤1化合物B(0.09克,0.21毫摩尔)使用过量吗啉(10当量)的DMF(5毫升)溶液于室温在氮气下处理过夜。混合物减压蒸发并使用制备型TLC以乙酸乙酯/甲醇(9∶1)纯化,获得化合物C。
1H NMR(CDCl3)δ7.64(dd,1H),7.58(m,1H),7.30-7.44(m,3H),7.25(m,2),7.01(dd,1H),6.60(dd,1H),6.02(br.S,2H),3.75(m,4H),3.33(m,4H),2.75(m,2H),2.65(m,2H),2.57(m,4H),2.11(m,4H).质谱(ESI),M+1:475.1
下列化合物以类似方式制备。
实施例25

化合物139
实施例18产物(0.10克,0.28毫摩尔)与1-氟-2-硝基苯(0.079克,0.56毫摩尔)及三乙胺(0.085克,0.84毫摩尔)的DMF(5毫升)溶液于100℃在氮气下加热12小时时间。混合物于减压下蒸发及用制备型TLC以乙酸乙酯/己烷(7∶3)纯化。
1H NMR(CDCl3)δ7.89(dd,1H),7.65(m,2H),7.36-7.63(m,4H),7.19-7.26(m,2H),7.00-7.11(m,2H),6.59(dd,1H),6.11(br,S,2H),3.43(m,4H),3.26(m,4H).
质谱(ESI),M+1:483.1
实施例26

化合物140
如实施例4那样制备的苯酚衍生物(0.054克,0.18毫摩尔),氯乙基吗啉盐酸盐(0.041克,0.22毫摩尔),碳酸钾(0.076克,0.55毫摩尔)及碘化钾(0.031克,0.18毫摩尔)的混合物于50℃于乙腈(10毫升)中在氮气下加热19小时时间。混合物用乙酸乙酯稀释及过滤,于减压下浓缩及用硅胶色谱法纯化。
1H NMR(CD3OD)δ7.76(m,1H),7.71(m,1H),7.66(m,1H),7.38(m,2H),7.24(dd,1H),7.03(dd,1H),6.66(dd,1H),4.23(t,2H),3.73(t,4H),2.85(t,2H),2.63(t,4H).质谱(ESI),M+1:407.1.
下列化合物以类似方式制备:
实施例27
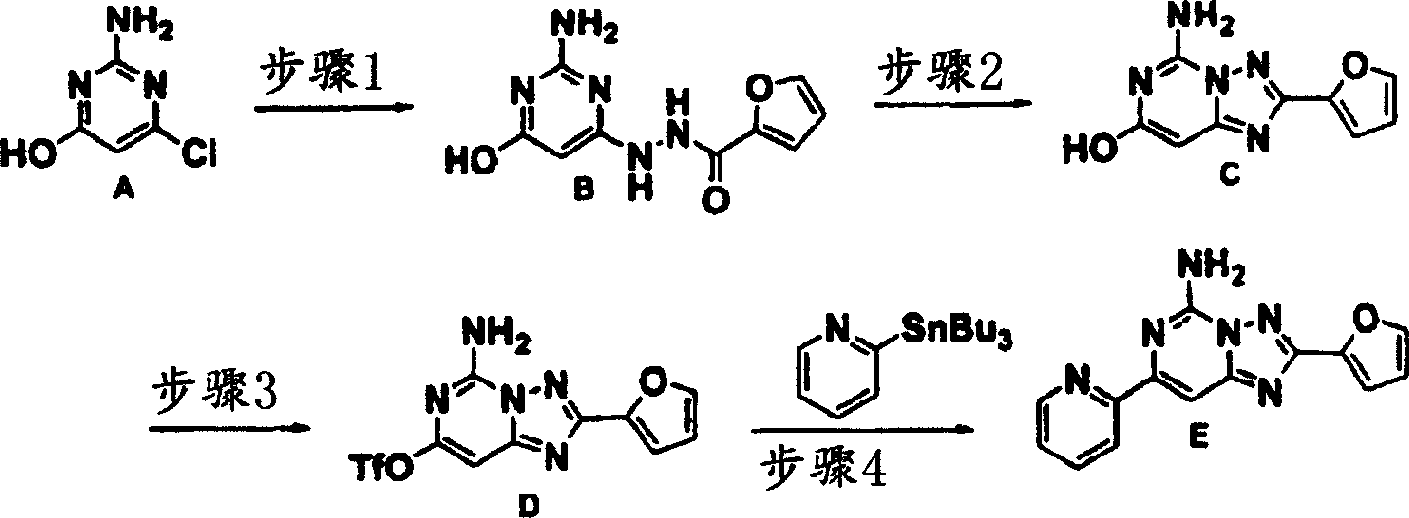
化合物147
步骤1:2-氨基-6-氯-4-嘧啶醇一水合物(2.0克,13.74毫摩尔)及2-糠酰肼(1.91克,15.12毫摩尔)的混合物在正丁醇(50毫升)中于100℃加热20小时。混合物真空浓缩,获得固体残留物B,后者未经纯化即供使用。
质谱(ESI),M+1:236.1。
步骤2:步骤1产物B(3.24克,13.74毫摩尔)在N,O-二(三甲基甲硅烷基)乙酰胺(20.5毫升,82.44毫摩尔)中于120℃加热过夜。混合物冷却,然后缓慢加入甲醇及水,回流加热4小时。所得混合物冷却至室温,过滤收集沉淀C。质谱(ESI),M+1:218.0。
步骤3:三氟甲烷磺酐(1.52克,5.37毫摩尔)于0℃在氮气下滴加至步骤2产物C(1.06克,4.88毫摩尔)及三乙胺(0.54克,5.37毫摩尔)的二氯甲烷(20毫升)溶液中。1小时后,混合物温热至室温及加入饱和碳酸氢钠溶液。所得混合物用二氯甲烷萃取,用硫酸钠干燥及过滤后蒸发。硅胶柱色谱法获得产物D。
质谱(ESI),M+1:350.1。
步骤4:步骤3产物D(0.25克,0.72毫摩尔),2-吡啶基三丁基锡(0.32克,0.86毫摩尔)及Pd(dppf)Cl2(0.029克,0.036毫摩尔)的混合物在DMF(5毫升)中于80℃在氮气下加热64小时。加水后用乙酸乙酯萃取,用硫酸钠干燥然后过滤。蒸发后残留物E用硅胶色谱法纯化。
1H NMR(DMSO-d6)δ8.70(dd,1H),8.30(d,1H),8.01(br.S,2H),7.95(m,2H),7.82(s,1H),7.47(m,1H),7.22(dd,1H),6.72(dd,1H)。质谱(ESI),M+1:279.0。
实施例28

化合物148
步骤1:2-羟甲基-5-溴吡啶(2.17克,11.54毫摩尔),二(邻二叔醇基)二硼(2.93克,11.54毫摩尔),PdCl2(dppf)(0.57克,0.69毫摩尔)及乙酸钾(3.40克,34.62毫摩尔)的混合物在1,4-二噁烷(65毫升)中于80℃在氮气下加热过夜。混合物冷却至室温,然后加入2-氨基-4,6-二氯-嘧啶(3.79克,23.08毫摩尔)及2.0M碳酸氢钠溶液(6.12克于20毫升水中)。所得混合物于80℃加热20小时,冷却,然后用乙酸乙酯及水稀释。有机萃取物用食盐水洗涤,用硫酸钠干燥及蒸发。残留物用硅胶柱色谱法纯化,获得产物B。
1H NMR(CD3OD)δ9.21(d,1H),8.55(dd,1H),7.76(d,1H),7.75(d,1H),7.49(s,1H),7.25(dd,1H),6.66(dd,1H),4.78(2H)。质谱(ESI),M+1:309.1。
步骤2及3:同实施例4步骤2及3。1H NMR(CD3OD)δ9.21(d,1H),8.55(dd,1H),7.76(d,1H),7.75(d,1H),7.49(s,1H),7.25(dd,1H),6.66(dd,1H),4.78(2H)。质谱(ESI),M+1:309.1。
实施例29
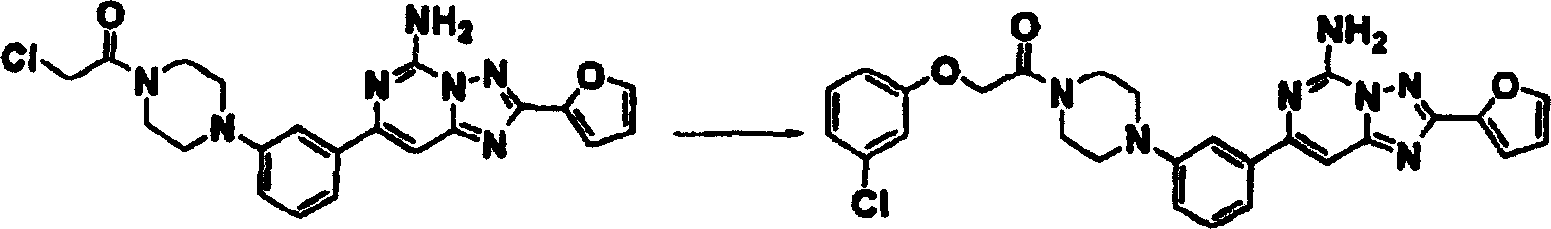
化合物149
步骤1:向3-氯苯酚(0.062克,0.48毫摩尔)及氢化钠(0.058克,1.44毫摩尔60%NaH于矿物油中)的DMF(5毫升)溶液中于室温在氮气下加入实施例22步骤1产物(0.11克,0.24毫摩尔),混合物搅拌过夜。然后加水,混合物用乙酸乙酯萃取,用硫酸钠干燥、过滤及真空浓缩。以使用乙酸乙酯的制备型硅胶TLC纯化,获得产物。1HNMR(CDCl3)δ7.61(s,1H),7.60(m,1H),7.47(m,1H),7.22(m,3H),7.00(m,2H),6.88(dd,1H),6.60(dd,1H),5.93(br.s,2H),4.75(s,2H),3.80(m,4H),3.17(m,4H).质谱(ESI),M+1:530.1
实施例30
化合物150
步骤1:叔丁基锂戊烷溶液(11.56毫升,19.7毫摩尔,1.7N于戊烷中)滴加至3-溴苯酚(1.0克,5.8毫摩尔)的THF(86毫升)溶液中,在氮气下冷却至-78℃。混合物搅拌10分钟。于-78℃加入4-氧代-1-哌啶羧酸苄基酯(1.35克,5.8毫摩尔)的THF(14毫升)溶液。混合物温热至室温,搅拌2小时,分配于饱和碳酸氢钠及乙酸乙酯之间。有机相用水、食盐水洗涤,用硫酸镁干燥、过滤、真空浓缩及色谱分离,获得B。质谱(ESI):328.1,310.0。
步骤2:将三乙基甲硅烷(1.45克,12.5毫摩尔)及三氟乙酸(1.42克,12.5毫摩尔)添加至步骤1产物B(0.87克,2.66毫摩尔)的二氯甲烷(23毫升)溶液中,在氮气下冷却至-78℃。于-78℃继续搅拌2小时,然后在室温继续搅拌20小时。混合物分配于饱和碳酸氢钠及二氯甲烷之间。有机相用水、食盐水洗涤,用硫酸镁干燥、过滤、真空浓缩及色谱分离,获得C。质谱(ESI):312.0。
步骤3:步骤2产物C(458毫克,1.47毫摩尔)如实施例11步骤3那样处理,获得D。质谱(ESI):444.1。
步骤4:步骤3产物D(412毫克,0.93毫摩尔)如实施例11步骤4那样处理,获得E。质谱(ESI):422.1。
步骤5:步骤4产物E(250毫克,0.59毫摩尔)如实施例4步骤1那样(但使用4当量碳酸钠)使用2-氨基-4,6-二氯嘧啶(195毫克,1.18毫摩尔)处理,获得F。质谱(ESI):423.1。
步骤6:步骤5产物F(155毫克,0.37毫摩尔)与2-糠酰肼(60毫克,0.48毫摩尔)合并于正丁醇(3毫升)中。混合物搅拌并于110℃加热20小时。温度冷却至室温,真空浓缩获得固体G,固体G未进一步纯化即继续进行反应。质谱(ESI):513.1。
步骤7:步骤6产物G(188毫克,0.37毫摩尔)与N,O-二(三甲基甲硅烷基)乙酰胺(1.65克,8.5毫摩尔)合并。混合物在氮气下于110℃搅拌及加热4小时。混合物冷却至室温及真空浓缩。残留物收集于2∶1水/甲醇中,于100℃加热2小时,真空浓缩及分配于水及乙酸乙酯之间。有机相用食盐水洗涤,用硫酸钠干燥、过滤、真空浓缩及色谱分离,获得固体H。质谱(ESI):495.1,1H NMR(CDCl3)δ7.93(s,1H),7.82(d,1H),7.64(m,2H),7.28-7.52(m,7H),6.78(bs,1H),6.63(m,1H),5.19(s,2H),4.38(bs,2H),2.92(bs,2H),2.80(m,1H),1.91(d,2H),1.70(m,2H)。
实施例31

化合物151
实施例30步骤7产物(54毫克,0.11毫摩尔)与乙酸铵(7毫克,0.091毫摩尔)及10%Pd/C(8毫克)合并于甲醇(3毫升)中。混合物在氢气下于大气压及室温搅拌3小时。混合物以硅藻土过滤,滤液真空浓缩。滤液分配于饱和碳酸氢钠及二氯甲烷之间。有机相用食盐水洗涤,用硫酸镁干燥、过滤、及真空浓缩,获得固体。质谱
(ESI):361.1,1H NMR(CD3OD)δ8.00(s,1H),7.94(d,1H),7.76(s,1H),7.34-7.44(m,3H),7.25(m,1H),6.67(m,1H),3.23(d,2H),2.82(m,3H),1.93(d,2H),1.80(m,2H)
实施例32

化合物152
2-(氨基甲基)吡啶(92毫克,0.85毫摩尔),实施例2步骤2产物(100毫克,0.43毫摩尔)及碳酸钾(177毫克,1.28毫摩尔)于正丁醇(2毫升)中的混合物于120℃在封管中加热48小时。混合物冷却至室温,过滤及真空浓缩。残留物用硅胶色谱法获得固体。质谱
(ESI):308.1,1H NMR(CDCl3)δ8.57(m,1H),7.68(m,1H),7.57(s,1H),7.34(d,1H),7.21(m,1H),7.14(dd,1H),6.55(m,1H),6.39(bs,1H),6.05(s,2H),5.87(s,1H),4.54(d,2H)
下列化合物以类似方式制备:
实施例33
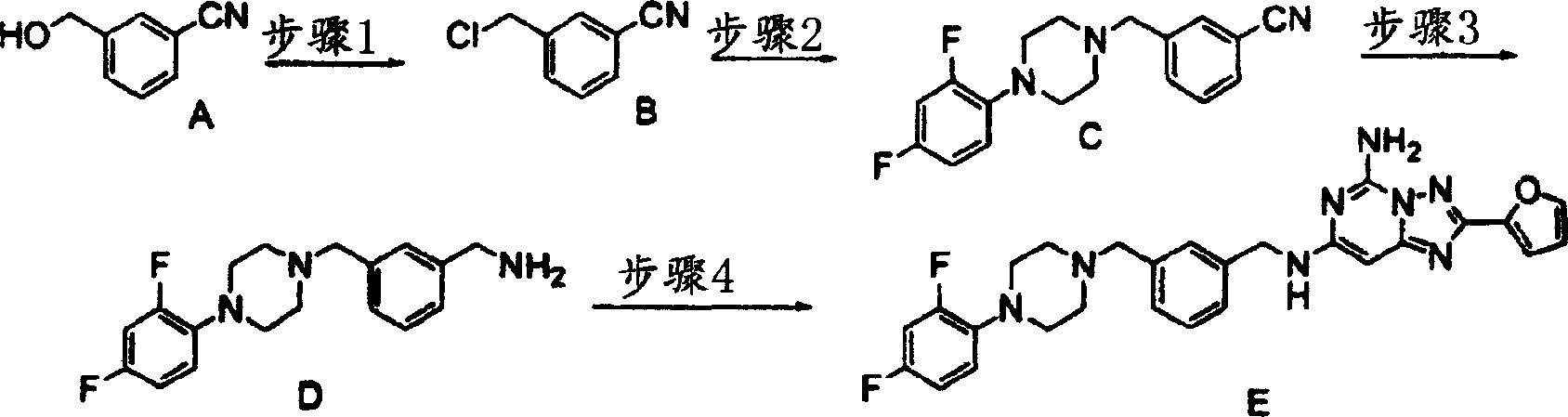
化合物156
步骤1:3-(羟甲基)苄腈(2.0克,15毫摩尔)与三乙胺(9.11克,90毫摩尔)合并于二氯甲烷(200毫升)中。混合物在氮气下冷却至0℃,滴加亚磺酰氯(8.94克,75毫摩尔)。混合物于0℃搅拌1小时,以冰处理,以饱和碳酸氢钠中和及以乙酸乙酯萃取。有机相用水及食盐水洗涤,用硫酸镁干燥,过滤及真空浓缩,获得固体B,未进一步纯化即继续进行反应。1H NMR(CDCl3)δ7.70(s,1H),7.63(m,2H),7.49(t,1H),4.59(s,2H)。
步骤2:步骤1产物B(1.04克,6.86毫摩尔),2,4-二氟苯基哌嗪(1.24克,6.24毫摩尔),碳酸钾(2.59克,19毫摩尔)及碘化钾(1.04克,6.24毫摩尔)合并于乙腈(75毫升)中,在氮气下回流20小时。混合物过滤,真空浓缩及硅胶色谱法纯化,获得C。质谱(ESI):314.1。
步骤3:步骤2产物C(0.96克,3.06毫摩尔)的THF(5毫升)溶液滴加至LAH(0.128克,3.37毫摩尔)的THF(7毫升)悬浮液中,在氮气下冷却至0℃。混合物于室温搅拌1小时,冷却至0℃,以冰处理,以1N氢氧化钠猝灭,温热回升至室温。所得固体经过滤及以THF洗涤。滤液真空浓缩,获得D,未进一步纯化即继续反应。质谱(ESI):318.1。
步骤4:步骤3产物D(270毫克,0.85毫摩尔)如同实施例32一样与实施例2步骤2(100毫克,0.43毫摩尔)化合,获得固体E。质谱
(ESI):517.1,1H NMR(CD3OD)δ7.68(s,1H),7.39(s,1H),7.27(m,3H),7.21(m,1H),7.07(d,1H),6.92(m,3H),6.60(dd,1H),5.69(s,1H),4.54(s,2H),3.61(s,2H),2.92(m,4H),2.60(m,4H)
下列化合物以类似方式制备:
实施例34
化合物160
步骤1:3-(氯甲基)-5-氰基吡啶鎓盐酸盐[如Chem,Pharm.Bull.38,1990,2446-58;Chem.Eur.J.3,1997,410-16中所述那样制备](260毫克,1.38毫摩尔),2,4-二氟苯基哌嗪(228毫克,1.15毫摩尔)及三乙胺(326毫克,3.22毫摩尔)合并于DMF(7毫升)中。混合物于室温搅拌48小时。混合物真空浓缩及分配于水及二氯甲烷之间。有机相用食盐水洗涤,用硫酸镁干燥、过滤、真空浓缩及色谱法纯化,获得固体B。质谱(ESI):315.1。
步骤2:步骤1产物B(223毫克,0.71毫摩尔),甲醇(3毫升),THF(3毫升),25%氢氧化铵(水溶液)(3毫升)及阮内镍合并,在帕尔瓶内用乙醇(0.050克)洗湿且于50psi加氢24小时。混合物经硅藻土过滤,滤液真空浓缩,获得固体C,未经进一步纯化即继续进行反应。质谱(ESI):319.1。
步骤3:步骤2产物C(230毫克,0.72毫摩尔)及实施例2步骤2产物(85毫克,0.36毫摩尔)如实施例32那样化合,获得固体D。质谱
(ESI):518.1,1H NMR(CD3OD)δ8.47(s,1H),8.37(s,1H),7.88(s,1H),7.69(s,1H),7.08(d,1H),6.86(m,4H),6.60(dd,1H),5.77(s,1H),4.62(s,2H),3.62(s,2H),3.04(m,2H),2.89(m,2H),2.65(m,2H),2.56(m,2H)
实施例35

化合物161
步骤1:4-(2-氯乙基)吗啉盐酸盐(1.88克,10毫摩尔)及2-氰基苯酚(1.0克,8.4毫摩尔)如实施例33步骤2那样化合,获得油状物B。质谱(ESI):233.0。
步骤2:步骤1产物B(502毫克,2.16毫摩尔)如实施例34步骤2那样加氢,获得油状物C。质谱(ESI):237.0。
步骤3:步骤2产物C(202毫克,0.85毫摩尔)和实施例2步骤2产物(100毫克,0.43毫摩尔)如实施例32那样化合,获得固体D。质谱
(ESI):436.1,1H NMR(CD3OD)δ7.69(s,1H),7.28(m,2H),7.10(dd,1H),6.93(m,2H),6.60(dd,1H),5.75(s,1H),4.48(s,2H),4.21(t,2H),3.70(m,4H),2.86(t,2H),2.61(m,4H)
下列化合物以类似方式制备:

实施例36

化合物163
步骤1:2-氰基苯酚(1.0克,8.4毫摩尔)的DMF(30毫升)溶液滴加至在氮气下冷却至0℃的氢化钠(60%于油中,502毫克,12.6毫摩尔)的DMF(12毫升)悬浮液中。添加完成后,混合物于室温搅拌20分钟。加入2-溴乙基甲基醚(1.4克,10毫摩尔),混合物搅拌70小时。混合物真空浓缩。所得固体悬浮于己烷中,并倾析出。未溶解的固体分配于水及乙酸乙酯之间。有机相用水、食盐水洗涤,用硫酸镁干燥、过滤、真空浓缩,与上述己烷洗液合并及色谱法纯化,获得油状物B。质谱(ESI):178.1。
步骤2:步骤1产物(623毫克,3.52毫摩尔)如实施例34步骤2那样加氢,获得油状物C。质谱(ESI):182.0。
步骤3:步骤2产物(152毫克,0.85毫摩尔)及实施例2步骤2产物(100毫克,0.43毫摩尔)如实施例32那样化合,获得油状物D。质谱
(ESI):381.1,1H NMR(CD3OD)δ7.68(s,1H),7.24(m,2H),7.09(dd,1H),6.93(m,2H),6.60(m,1H),5.75(s,1H),4.50(s,2H),4.19(m,2H),3.80(m,2H),3.45(s,3H)
实施例37
化合物164
步骤1:2-(2-甲氧基乙氧基)苯甲醛[如Chem.Pharm.Bull.35,1987,1953-68所述那样制备](400毫克,2.22毫摩尔)和2-甲氧乙基胺(228毫克,1.15毫摩尔)合并于甲醇(10毫升)中,混合物于室温在氮气下搅拌20小时。混合物冷却至0℃,加入硼氢化钠(134毫克,3.55毫摩尔)。混合物于室温在氮气下搅拌20小时。混合物分配于饱和碳酸氢钠及乙醚之间。有机相用水及食盐水洗涤,用硫酸镁干燥。混合物过滤及真空浓缩,获得油状物B,未经进一步纯化即继续反应。质谱(ESI):240.1。
步骤2:步骤1产物B(183毫克,0.77毫摩尔)及实施例2步骤2产物(90毫克,0.38毫摩尔)如实施例32那样化合,获得固体C。质谱
(ESI):439.1,1H NMR(CD3OD)δ7.68(s,1H),7.21(t,1H),7.09(m,2H),6.98(d,2H),6.88(t,1H),6.60(m,1H),5.80(s,1H),4.80(s,2H),4.18(m,2H),3.77(m,4H),3.62(t,2H),3.42(s,3H),3.34(s,3H)
下列化合物以类似方式制备:

实施例38
化合物171
步骤1:2-氰基苯甲醛(500毫克,3.8毫摩尔),吗啉(365毫克,4.2毫摩尔)及NaBH(OAc)3(1.21克,5.72毫摩尔)合并于THF(20毫升)中。混合物于室温在氮气下搅拌20小时。混合物以1N氢氧化钠猝灭及分配于水及乙酸乙酯之间。有机相用水及食盐水洗涤,然后用硫酸镁干燥、过滤、真空浓缩及色谱分离,获得油状物B。质谱(ESI):203.0。
步骤2:步骤1产物(172毫克,0.85毫摩尔)如实施例34步骤2那样加氢,获得油状物C。质谱(ESI):207.0。
步骤3:步骤2产物C(160毫克,0.77毫摩尔)与实施例2步骤2产物(90毫克,0.38毫摩尔)如同实施例32那样化合,获得固体D。质谱
(ESI):406.1 1H NMR(CD3OD)δ7.69(s,1H),7.41(d,1H),7.24(m,3H),7.11(s,1H),6.61(m,1H),5.77(s,1H),4.60(s,2H),3.74(t,4H),3.59(s,2H),2.48(s,4H)
实施例39
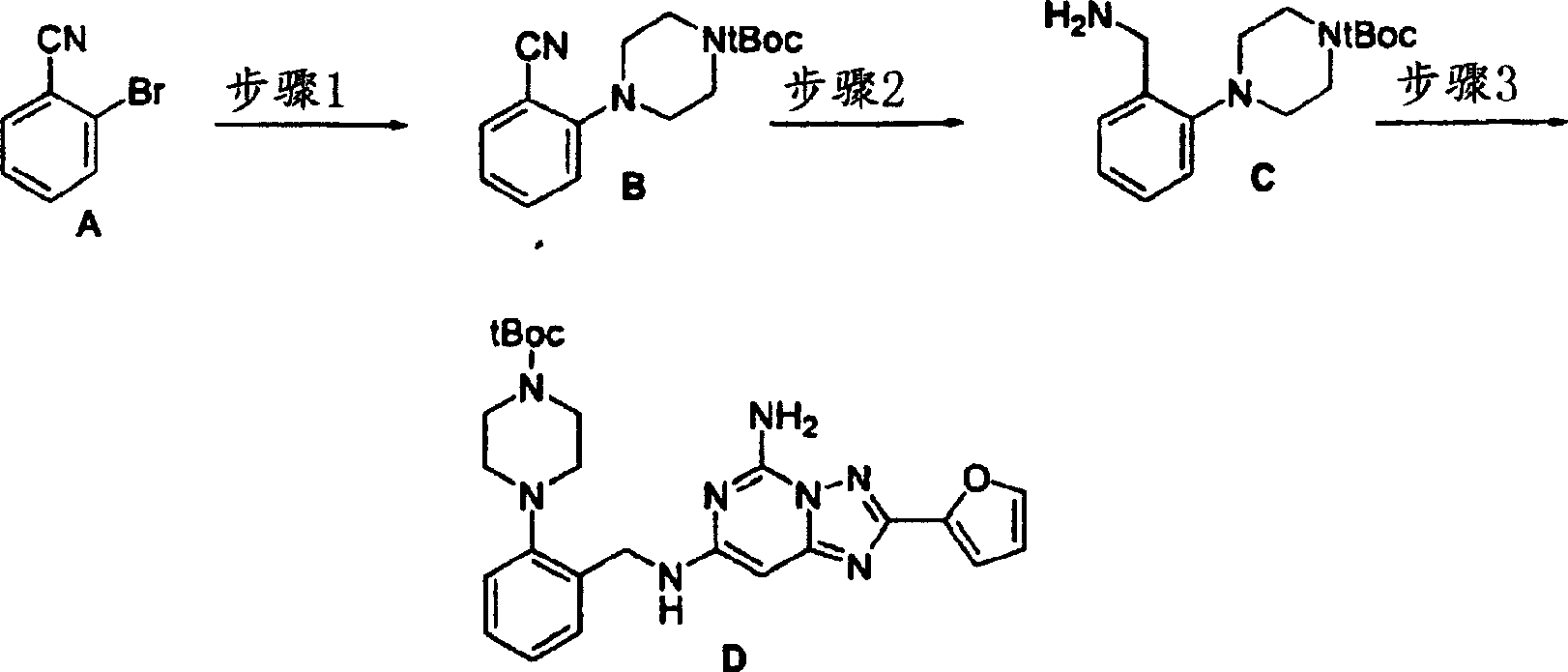
化合物172
步骤1:2-溴苄腈(3克,16.5毫摩尔)及1-哌嗪羧酸叔丁酯(3.68克,19.8毫摩尔)如实施例10步骤1那样化合(但回流时间为20小时,粗产物经色谱法纯化)获得油状物B。1H NMR(CD3OD)δ7.60(m,2H),7.14(m,2H),3.62(s,4H),3.14(t,4H),1.49(s,9H)。
步骤2:步骤1产物(440毫克,1.53毫摩尔)如实施例34步骤2那样加氢,获得油状物C。质谱(ESI):292.0。
步骤3:步骤2产物C(477毫克,1.64毫摩尔)及实施例2步骤2产物(193毫克,0.82毫摩尔)如实施例32那样化合,获得固体D。质谱
(ESI):491.1,1H NMR(CD3OD)δ7.68(s,1H),7.39(d,1H),7.24-7.06(m,4H),6.60(m,1H),5.69(s,1H),4.57(s,2H),3.62(bs,4H),2.91(bs,4H),1.48(s,9H)
下列化合物以类似方式制备:
实施例40
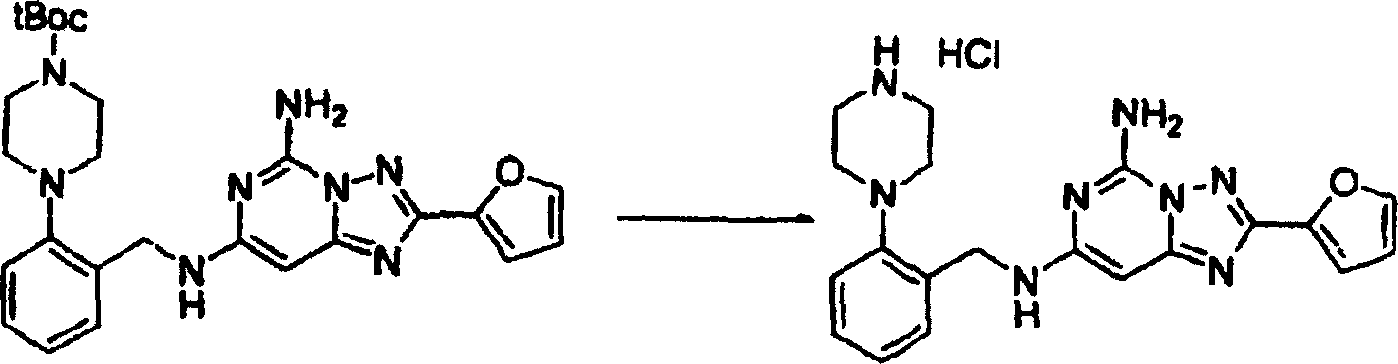
化合物174
实施例39步骤3产物(150毫克,0.31毫摩尔),4M盐酸/二噁烷(1毫升)与二噁烷(2毫升)化合。混合物于室温在氮气下搅拌20小时,然后真空浓缩。残留物悬浮于乙醚中,再度真空浓缩,重复数次。所得固体收集于乙醚中,过滤及干燥(真空烘箱,50℃),获得固体。质谱(ESI):391.1,1H NMR(CD3OD)δ7.91(s,1H),7.43-7.17(m,6H),6.78(m,1H),3.66(s,2H),3.43(bs,4H),3.20(m,4H)。
实施例41
化合物175
步骤1:实施例39步骤1产物(1.11克,4.1毫摩尔)如实施例40那样脱保护,获得固体B。质谱(ESI):188.0。
步骤2:步骤1产物B的游离碱(200毫克,1.1毫摩尔),三乙胺(130毫克,1.3毫摩尔)及乙酐(4毫升)合并。混合物于室温在氮气下搅拌20小时。混合物经真空浓缩及分配于饱和碳酸氢钠及二氯甲烷之间。有机相用水、食盐水洗涤及用硫酸镁干燥,过滤及真空浓缩,获得油状物C,未经进一步纯化即继续反应。质谱(ESI):230.0。
步骤3:步骤2产物C(230毫克,1.0毫摩尔)如实施例34步骤2那样加氢,获得油状物D。质谱(ESI):234.0。
步骤4:步骤3产物D(235毫克,1.0毫摩尔)及实施例2步骤2产物(119毫克,0.50毫摩尔)如实施例32那样化合。获得固体E。质谱
(ESI):433.1,1H NMR(CD3OD)δ7.68(s,1H),7.40(d,1H),7.24-7.10(m,4H),6.60(m,1H),5.70(s,1H),4.60(s,2H),3.78(bs,2H),3.73(t,2H),2.96(dt,4H),2.15(s,3H)
下列化合物以类似方式制备:
本发明的较好化合物包括但不限于选自下列组成的一组的化合物:
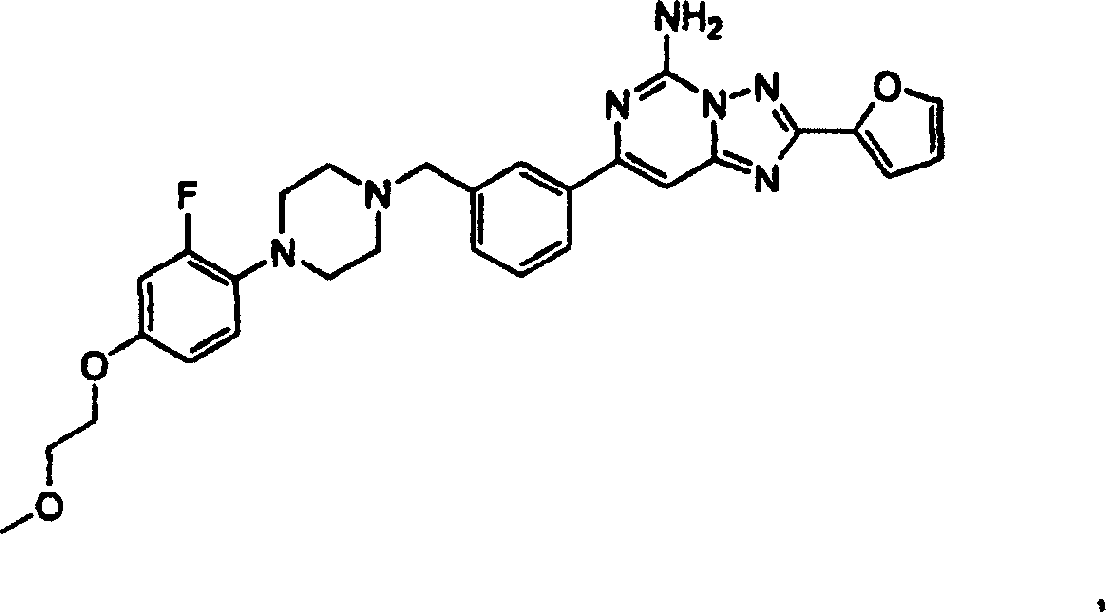
和

实施例42
本发明化合物的药理活性是用下列离体试验及活体试验测量A2a受体活性而确定的。
人类腺苷A2a及A1受体竞争结合试验方案
膜来源:
A2a:人类A2a腺苷受体膜,类别#RB-HA2a,受体生物公司,美国马里兰州贝茨维尔。用膜稀释缓冲液稀释成17微克/100微升(参见下文)。
试验缓冲液:
膜稀释缓冲液:杜别克磷酸盐缓冲食盐水(Gibco/BRL)+10mM氯化镁。
化合物稀释缓冲液:杜别克磷酸盐缓冲食盐水(Cibco/BRL)+补充1.6毫克/毫升甲基纤维素及16%DMSO的10mM氯化镁。
每日新鲜制备。
配体:
A2a:[3H]-SCH 58261,照客户需求合成,艾莫森法马西亚生物技术公司,美国纽约州匹斯卡塔威。储备液是以膜稀释缓冲液制备成1nM。最终试验浓度为0.5nM。
A1:[3H]-DPCPX,艾莫森法马西亚生物技术公司,美国纽约州匹斯卡塔威。储备液以膜稀释缓冲液制备成2nM。最终试验浓度为1nM。
非特异性结合:
A2a:为确定非特异性结合,添加100nM CGS 15923(RBI公司,麻省拿提克)。工作储备液系以化合物稀释缓冲液制备成400nM。A1:为确定非特异性结合,添加100μM NECA(RBI公司,麻省拿提克)。工作储备液是以化合物稀释缓冲液制备成400μM。
化合物稀释:
制备1mM化合物的100%DMSO储备溶液。以化合物稀释缓冲液稀释。以3μM至30pM范围内的10种浓度试验。以化合物稀释缓冲液制备4X最终浓度工作溶液。
试验程序:
以深孔之96孔孔板进行试验。总试验容积为200微升。添加50微升化合物稀释缓冲液(总配体结合)或50微升CGS 15923工作溶液(A2a非特异性结合)或50微升NECA工作溶液(A1非特异性结合)或50微升药物工作溶液。添加50微升配体储备液([3H]-SCH 58261用于A2a,[3H]-DPCPX用于A1)。添加100微升含适当受体的稀释膜。混合。于室温培育90分钟。使用Brandel细胞收集器用Packard GF/B过滤板收集。加入45微升Microscint 20(帕克公司),使用Packard TopCount微闪烁计数器计数。使用迭代曲线拟合程序(Excel)拟合置换曲线确定IC50值。使用Cheng-Prusoff方程确定Ki值。
氟哌啶醇诱发大鼠强直性昏厥
使用体重175-200克的雄性Sprague-Dawley大鼠(意大利凯尔可查尔斯河)。动物进行垂直格栅试验前90分钟,经皮下给药多巴胺受体拮抗剂氟哌啶醇(1毫克/千克,皮下注射)诱发强直性昏厥状态。为了本试验,大鼠置于25×43有机玻璃笼的金属丝网盖上,笼子相对于工作台以70度角放置。大鼠以四肢全部外展且伸开(「青蛙姿」)置于格栅上。使用此种不自然的姿势为本强直性昏厥试验之特异性所必需。测量放置足掌直至初次完全移开一足当经历的时间(体面潜伏期)最长经历120秒。
所评估之选择性A2a腺苷拮抗剂是以对动物评分前1及4小时以0.03及3毫克/千克范围内的剂量口服给药的。
在单独实验中,确定了参考化合物L-DOPA(25、50及100毫克/千克,经腹膜内给药)的抗强直性昏厥效果。
大鼠中前脑束之6-OHDA损伤
全部实验使用体重275-300克之成年雄性Sprague-Dowley大鼠(意大利可莫凯尔可查尔斯河)。大鼠以每笼4头圈养,自由取食及饮水,处于受控制温度及12小时明/暗周期。手术前当天,大鼠空腹过夜,任意给水。
根据Ungerstedt等人所述方法(
Brain Research,1971,6-OHDA及儿茶酚胺神经元,阿姆斯特丹北荷兰,101-127)但有微小改变,进行中前脑束单侧6-羟基多巴胺(6-OHDA)损伤。简言之,动物使用氯醛水合物(400毫克/千克,经腹膜内给药)麻醉,于6-OHDA注射前30分钟使用地昔帕明(10毫克/千克,经腹膜内给药)处理,以期阻断毒素由去甲肾上腺素能终端吸收。然后,动物置于立体定位架上。反映头颅上的皮肤,根据Pellegrino等人的图集(Pellegrino L.J.,PellegrinoA.S.及Cushman A.J.,
大鼠脑立体定位图集,1979年,约纽:普雷南(Plenum)出版社)取立体定位坐标[-2.2前囟门后(AP),+1.5前囟门侧边(ML),7.8硬脑膜前侧(DV)]。然后于损伤部位上方颅骨设一钻孔,附着于汉尔顿注射器的针头下降至左MFB。然后将8微克6-OHDA-HCl溶解于4微升食盐水,含0.05%抗坏血酸作为抗氧化剂,使用输注泵以1微升/1分钟恒定流速输注。又经5分钟后抽出针头,关闭手术伤口,让动物复原2周。
损伤后2周,对大鼠给药L-DOPA(50毫克/千克,经腹膜内给药)加苄丝肼(25毫克/千克,经腹膜内给药),根据用自动旋转仪在2小时试验时间内计算的完全对侧旋转次数选择大鼠(初步试验)。任何未显示2小时至少旋转200圈的大鼠均不包括在本实验中。
选用的大鼠于初步试验后3日接受试验药物(最大多巴胺受体超敏感度)。在注射亚阈限剂量的L-DOPA(4毫克/千克,经腹膜内给药)加苄丝肼(4毫克/千克,经腹膜内给药)前不同时点(即1、6、12小时),以0.1至3毫克/千克范围内的剂量水平经口给药新颖A2a受体拮抗剂,评比大鼠转圈表现。
实施例43
以下为含有本发明化合物之医药剂型实施例。
医药剂型实施例
片剂
| 编号 | 组分 | 毫克/片 | 毫克/片 |
| 1. | 活性化合物 | 100 | 500 |
| 2. | 乳糖USP | 122 | 113 |
| 3. | 玉米淀粉,食品级,呈10%纯水糊剂 | 30 | 40 |
| 4. | 玉米淀粉,食品级 | 45 | 40 |
| 5. | 硬脂酸镁 | 3 | 7 |
| 总量 | 300 | 700 | |
制备方法
在适当混合器内混合第1及2项10-15分钟。混合物使用第3项造粒。必要时通过粗筛(例如1/4英寸,0.63厘米)研磨湿粒。使湿粒干燥。必要时将干燥颗粒过筛,混合第4项且混合10-15分钟。加入第5项且混合1-3分钟。在适当压片机上将混合物压缩成适当尺寸和重量。
胶囊剂
| 编号 | 组分 | 毫克/胶囊 | 毫克/胶囊 |
| 1. | 活性化合物 | 100 | 500 |
| 2. | 乳糖USP | 106 | 123 |
| 3. | 玉米淀粉,食品级 | 40 | 70 |
| 4. | 硬脂酸镁NF | 7 | 7 |
| 总量 | 253 | 700 | |
制造方法
在适当掺混机中将第1、2及3项混合10-15分钟。加入第4项且混合1-3分钟。在适当胶囊机上将混合物灌装于适当两件式硬明胶胶囊中。
虽然前文已经结合特定实施方案说明了本发明,但其许多替代、修改及变化对业内一般技能人士是显而易见的。全部此类替代、修改及变化意图皆落入本发明的精神和范围内。
Claims (15)
1.一种具有结构式I的化合物

或其医药上可接受盐或溶剂合物;其中:
R选自由R4-杂芳基、R5-苯基、(C4-C6)环烯基、-C(=CH2)CH3、-C≡C-CH3、
-CH=C(CH3)2、

R2选自由-W-X、-NR19(CH2)m-W-X、及-NR19CH(CH3)-W-X组成的一组,或
R2选自由烷基、链烯基及-NR18R19组成的一组,其中该烷基、链烯基或-NR18R19是任选地有-W-X取代的;
R3选自由H、卤原子、烷基、三氟甲基、烷氧基、烷氧烷基、羟烷基、烷基氨基、烷基氨基烷基、二烷基氨基、二烷基氨基烷基、氨基烷基、芳基、杂芳基、及CN组成的一组;
R4为1至3个相同或相异的取代基且独立地选自由氢、(C1-C6)-烷基、-CF3、卤原子、-NO2、-NR15R16、(C1-C6)烷氧基、(C1-C6)烷硫基、(C1-C6)烷基亚磺酰基、(C1-C6)烷磺酰基、-COOR17及-C(O)NR6R7组成的一组;
R5为1至5个相同或相异的取代基且独立地选自由氢、卤原子、(C1-C6)烷基、羟基、(C1-C6)烷氧基、-CN、-NH2、(C1-C6)烷基氨基、二-((C1-C6)烷基)氨基、-CF3、-OCF3、-S(O)0-2(C1-C6)烷基及-CH2-SO2-苯基组成的一组;
R6及R7为相同或相异,且各自独立地选自由氢及(C1-C6)烷基组成的一组;
R8为1至5个相同或相异的取代基且独立地选自由氢、卤原子、(C1-C6)烷基、羟基、(C1-C6)烷氧基、-CN、氨基、二-((C1-C6)烷基)氨基、-CF3、-OCF3、乙酰基、-NO2、羟基(C1-C6)烷氧基、(C1-C6)-烷氧基(C1-C6)烷氧基、二-((C1-C6)-烷氧基)(C1-C6)烷氧基、(C1-C6)-烷氧基(C1-C6)烷氧基-(C1-C6)-烷氧基、羧基(C1-C6)-烷氧基、(C1-C6)-烷氧羰基(C1-C6)烷氧基、(C3-C6)环烷基(C1-C6)烷氧基、二-((C1-C6)烷基)氨基(C1-C6)烷氧基、吗啉基、(C1-C6)烷基-SO2-、(C1-C6)烷基-SO2-(C1-C6)烷氧基、四氢吡喃氧基、(C1-C6)烷基羰基(C1-C6)-烷氧基、(C1-C6)-烷氧羰基、(C1-C6)烷基羰氧基(C1-C6)-烷氧基、-SO2NH2、苯氧基、

-O-CH2-P(O)(OR6)2-、以及-P(O)(OR6)2组成的一组;或相邻的R8取代基共同形成为-O-CH2-O-、-O-CH2CH2-O-、-O-CF2-O-或-O-CF2CF2-O-,且与其所连接的碳原子一起形成一个环;
R9选自由(C1-C6)烷基、R8-芳基-、R8-芳基(C1-C6)烷基-、噻吩基、吡啶基、(C3-C6)-环烷基、(C1-C6)烷基-OC(O)-NH-(C1-C6)烷基-、二-((C1-C6)烷基)氨基甲基、环杂烷基(C1-C6)烷基、芳氧基(C1-C6)烷基、烷氧基(C1-C6)烷基以及
组成的一组;
R10为1-2个相同或相异的取代基且独立地选自由氢、(C1-C6)烷基、R5-芳基及R4-杂芳基组成的一组,或同一个碳上的两个R10取代基可形成=O;
R11为氢或(C1-C6)烷基;-C(O)烷基或R17与R11合在一起形成-(CH2)p-A-(CH2)q,其中p及q各自独立地为2或3,以及A选自由一个键、-CH2-、-S-及-O-组成的一组,以及与它们所连接的氮一起形成一个环;
R12为1-2个相同或相异的取代基且独立地选自由氢、(C1-C6)烷基、羟基、(C1-C6)烷氧基、卤原子及-CF3组成的一组;
R13选自由H、(C1-C6)烷基、苯基、苄基、(C2-C6)链烯基、(C1-C6)烷氧基(C1-C6)烷基、二-((C1-C6)烷基)氨基(C1-C6)烷基、吡咯烷基(C1-C6)烷基及哌啶子基(C1-C6)烷基组成的一组;
R14选自由H、卤原子、(C1-C6)烷基或(C1-C6)烷氧基组成的一组;
R15选自由H及(C1-C6)烷基组成的一组;
R16选自由H、(C1-C6)烷基-C(O)-及(C1-C6)烷基-SO2-组成的一组;
R17选自由(C1-C6)烷基、(C1-C6)羟烷基、(C3-C6)环烷基、(C1-C6)烷氧基(C1-C6)烷氧基、(C1-C6)烷氧基、(C1-C6)烷氧基(C1-C6)烷基、烯丙基、炔丙基、R8-杂芳基-、R8-芳基-及R8-芳基(C1-C6)烷基-组成的一组;
R18选自由一个键、-CH2-、-CH(OH)-、-CH(CH3)-、-C(CH3)n-、-(CH2)n-及-O(CH2)n-组成的一组;
R19选自由H、(C1-C6)烷基、(C1-C6)烷基(C1-C6)环烷基、(C1-C6)环烷基(C1-C6)烷基及(C1-C6)烷氧基(C1-C6)烷基组成的一组;
Q及Q1为相同或相异且各自独立地选自由


组成的一组;
m及n各自独立地为1-3;
p及q各自独立地为0-2;
s为0-4;
W为芳基或有1-3个可相同或相异且独立地选自由N、O和S组成的一组的杂原子的杂芳基,其中该芳基或杂芳基任选地有1-3个取代基取代,该取代基可相同或相异且独立地选自由烷基、芳基、烷基环烷基、卤原子、羟基、羟烷基、烷氧基、烷基烷氧基、烷氧基烷氧基、-NR6R7、(C2-C6)链烯基及-CN组成的一组,或
X选自由H、NH2、-N(R6)(CH2)s-芳基、-N(R6)(CH2)s-杂芳基、-N(R6)(CH2)m+1-OH、及-N(CH3)2,或
X为-R18-Y-Z,
Y选自由-N(R6)CH2CH2N(R7)-、-N(R6)(CH2)n芳基、-OCH2CH2N(R6)-、-O-、-S-、-CH2S-、-(CH2)2-3-N(R6)-、R8-二价杂芳基,
和
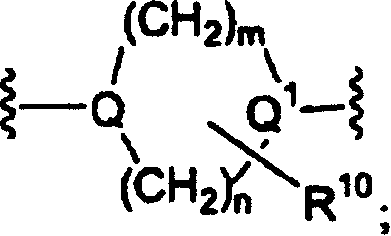
组成的一组;以及
Z选自由H、烷基、烷氧基烷基、R8-芳基-、R8-芳基(C1-C6)烷基-、R8-杂芳基-、R8-双环烷基-、氨基烷基、烷基氨基、NH2、-N-(R6)(CH2)s-芳基、-N(R6)(CH2)s-杂芳基、-N(R6)C(O)OR17、烷基环杂烷基、环杂烷基、环杂烷基烷基、烷氧基环杂烷基、杂芳基;R8-苯并稠合杂芳基-、二苯甲基及R9-C(O)-组成的一组;或
当Y为

时,Z也可为-OH、R9-SO2-、R17-N(R11)(CH2)s-C(O)-、R17-OC(O)-、R17-O(CH2)nC(O)-、苯并稠合杂芳基(CH2)nC(O)-、苯并稠合杂芳基(CH2)n-或R17-N(R11)-C(S)-;或
当Q为
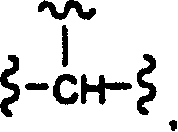
时,Z也可为R17R11N-、苯基氨基或吡啶基氨基;或
Z与Y合在一起选自由下列基团组成的一组
或其N-氧化物,
和
2.按照权利要求1的化合物,其中R为
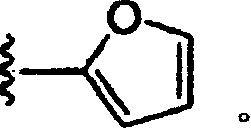
3.按照权利要求1的化合物,其中R3为H。
4.按照权利要求1的化合物,其中R为
以及R3为H。
5.按照权利要求1的化合物,选自由得自实施例1-41的化合物1-66、77-86及88-177中任何一种组成的一组。
6.按照权利要求5的化合物,选自由下列组成的一组:

和
7.一种医药组合物,包含一种或多种权利要求1的化合物以及一种或多种可接受载体。
8.按照权利要求7的医药组合物,进一步包含一种或多种其它可用于治疗帕金森病的药剂。
9.按照权利要求8的医药组合物,其中所述一种或多种其它可用于治疗帕金森病的药剂选自由L-DOPA、多巴胺能激动剂、MAO-B抑制剂、DOPA脱羧酶抑制剂以及COMT抑制剂组成的一组。
10.一种治疗中枢神经系统疾病或中风的方法,包含对需要此种治疗的患者给药一种或多种权利要求1的化合物。
11.按照权利要求10的方法,其中所述中枢神经系统疾病为认知疾病或神经退化疾病。
12.按照权利要求10的方法,其中所述中枢神经系统疾病为帕金森病、老年痴呆或器质性来源的精神病。
13.按照权利要求12的方法,其中所述中枢神经系统疾病为帕金森病。
14.按照权利要求13的方法,进一步包含给药一种或多种其它可用于治疗帕金森病的药剂,该药剂可相同或相异,且独立地选自L-DOPA、多巴胺能激动剂、MAO-B抑制剂、DOPA脱羧酶抑制剂以及COMT抑制剂组成的一组。
15.一种药剂盒,包含在单一包装内的分开容器中组合用于治疗帕金森病的医药组合物,其中一个容器含有一种包含一种或多种式I化合物于一种或多种医药上可接受载体中的医药组合物;且其中在各分开容器中,一种或多种医药组合物各自包含一种或多种可用于帕金森病治疗的药剂于一种或多种医药上可接受载体中。
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US33429301P | 2001-11-30 | 2001-11-30 | |
| US60/334,293 | 2001-11-30 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| CN1596258A true CN1596258A (zh) | 2005-03-16 |
Family
ID=23306533
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| CNA028239229A Pending CN1596258A (zh) | 2001-11-30 | 2002-11-26 | 腺A2α受体拮抗剂 |
Country Status (18)
| Country | Link |
|---|---|
| US (1) | US7041666B2 (zh) |
| EP (1) | EP1453835B1 (zh) |
| JP (2) | JP4284181B2 (zh) |
| KR (1) | KR20050044607A (zh) |
| CN (1) | CN1596258A (zh) |
| AR (1) | AR038366A1 (zh) |
| AT (1) | ATE317844T1 (zh) |
| AU (1) | AU2002346572A1 (zh) |
| CA (1) | CA2468681C (zh) |
| DE (1) | DE60209251T2 (zh) |
| ES (1) | ES2258164T3 (zh) |
| HU (1) | HUP0402270A3 (zh) |
| IL (1) | IL161572A0 (zh) |
| MX (1) | MXPA04005156A (zh) |
| PE (1) | PE20030739A1 (zh) |
| TW (1) | TW200300686A (zh) |
| WO (1) | WO2003048164A2 (zh) |
| ZA (1) | ZA200404160B (zh) |
Cited By (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| WO2019206336A1 (zh) * | 2018-04-28 | 2019-10-31 | 南京明德新药研发有限公司 | 一种三唑并嘧啶类化合物的晶型、盐型及其制备方法 |
| WO2020052631A1 (en) * | 2018-09-12 | 2020-03-19 | Dizal (Jiangsu) Pharmaceutical Co., Ltd. | Triazolo-pyrimidine compounds and uses thereof |
| CN112384515A (zh) * | 2018-02-27 | 2021-02-19 | 因赛特公司 | 作为a2a/a2b抑制剂的咪唑并嘧啶和三唑并嘧啶 |
| CN113906022A (zh) * | 2019-01-29 | 2022-01-07 | 因赛特公司 | 作为a2a/a2b抑制剂的吡唑并吡啶和三唑并吡啶 |
Families Citing this family (26)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP1544200A1 (en) * | 2002-09-24 | 2005-06-22 | Kyowa Hakko Kogyo Co., Ltd. | 1,2,4 -TRIAZOLO 1,5-c PYRIMIDINE DERIVATIVE |
| JP2006514697A (ja) | 2002-12-19 | 2006-05-11 | シェーリング コーポレイション | アデノシンA2aレセプターアンタゴニストの使用 |
| US20060106040A1 (en) | 2002-12-19 | 2006-05-18 | Michael Grzelak | Adenosine A2a receptor antagonists for the treatment of extra-pyramidal syndrome and other movement disorders |
| US20070010522A1 (en) * | 2003-04-09 | 2007-01-11 | Chi Vu | Triazolo[1,5-c]pyrimidines and pyrazolo[1,5-c]pyrimidines useful as a2a adenosine receptor antagonists |
| JP4800216B2 (ja) * | 2003-10-24 | 2011-10-26 | エグゼリクシス, インコーポレイテッド | p70S6キナーゼモジュレーターおよび使用方法 |
| AR050926A1 (es) * | 2004-09-03 | 2006-12-06 | Astrazeneca Ab | Derivados de benzamida como inhibidores de la histonadesacetilasa(hdac) |
| WO2006129626A1 (ja) * | 2005-05-30 | 2006-12-07 | Kyowa Hakko Kogyo Co., Ltd. | [1,2,4]トリアゾロ[1,5-c]ピリミジン誘導体の製造法 |
| ES2273599B1 (es) | 2005-10-14 | 2008-06-01 | Universidad De Barcelona | Compuestos para el tratamiento de la fibrilacion auricular. |
| US8835631B2 (en) | 2007-05-24 | 2014-09-16 | Mitsubishi Tanabe Pharma Corporation | Therapeutic agent for cerebral infarction |
| JP2010531364A (ja) * | 2007-06-25 | 2010-09-24 | ニューロジェン・コーポレーション | ピペラジニルオキソアルキルテトラヒドロ−β−カルボリンおよび関連類似体 |
| US20100093756A1 (en) * | 2008-10-13 | 2010-04-15 | Berbay J Kent | HETEROARYL SUBSTITUTED THIENO[2,3-d]PYRIMIDINE AND THEIR USE AS ADENOSINE A2a RECEPTOR ANTAGONISTS |
| US20100093764A1 (en) * | 2008-10-13 | 2010-04-15 | Devraj Chakravarty | AMINES AND SULFOXIDES OF THIENO[2,3-d]PYRIMIDINE AND THEIR USE AS ADENOSINE A2a RECEPTOR ANTAGONISTS |
| KR101211338B1 (ko) | 2008-11-25 | 2012-12-11 | 닛산 지도우샤 가부시키가이샤 | 도전 부재 및 이것을 사용한 고체 고분자형 연료 전지 |
| EP2424840B1 (en) * | 2009-04-27 | 2014-08-06 | Boehringer Ingelheim International GmbH | Cxcr3 receptor antagonists |
| US8952004B2 (en) | 2010-01-07 | 2015-02-10 | Boehringer Ingelheim International Gmbh | CXCR3 receptor antagonists |
| CN103261202B (zh) * | 2010-09-24 | 2016-01-20 | 阿迪维纳斯疗法有限公司 | 作为腺苷受体拮抗剂的稠合三环化合物 |
| WO2012127472A1 (en) * | 2011-03-22 | 2012-09-27 | Mapi Pharma Ltd. | Process and intermediates for the preparation of preladenant and related compounds |
| PL3611174T3 (pl) * | 2017-04-07 | 2022-08-08 | Medshine Discovery Inc. | Pochodne [1,2,4]triazolo[1,5-c]pirymidyny jako inhibitor receptora a2a |
| WO2019222677A1 (en) * | 2018-05-18 | 2019-11-21 | Incyte Corporation | Fused pyrimidine derivatives as a2a / a2b inhibitors |
| GEP20237548B (en) | 2018-07-05 | 2023-10-10 | Incyte Corp | Fused pyrazine derivatives as a2a /a2b inhibitors |
| WO2020106560A1 (en) | 2018-11-20 | 2020-05-28 | Merck Sharp & Dohme Corp. | Substituted amino triazolopyrimidine and amino triazolopyrazine adenosine receptor antagonists, pharmaceutical compositions and their use |
| KR20210093964A (ko) * | 2018-11-20 | 2021-07-28 | 머크 샤프 앤드 돔 코포레이션 | 치환된 아미노 트리아졸로피리미딘 및 아미노 트리아졸로피라진 아데노신 수용체 길항제, 제약 조성물 및 그의 용도 |
| JP2022511778A (ja) | 2018-11-30 | 2022-02-01 | メルク・シャープ・アンド・ドーム・コーポレーション | アデノシン受容体拮抗薬としての7-、8-及び10-置換されたアミノトリアゾロキナゾリン誘導体、医薬組成物及びそれらの使用 |
| TW202039496A (zh) | 2018-11-30 | 2020-11-01 | 美商默沙東藥廠 | 做為腺苷受體拮抗劑之9-經取代胺基三唑喹唑啉衍生物、醫藥組合物及其用途 |
| CA3124088A1 (en) | 2018-12-20 | 2020-06-25 | Incyte Corporation | Imidazopyridazine and imidazopyridine compounds as inhibitors of activin receptor-like kinase-2 |
| WO2020128036A1 (en) * | 2018-12-21 | 2020-06-25 | Ryvu Therapeutics S.A. | Modulators of the adenosine a2a receptor |
Family Cites Families (10)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU622330B2 (en) | 1989-06-23 | 1992-04-02 | Takeda Chemical Industries Ltd. | Condensed heterocyclic compounds having a nitrogen atom in the bridgehead for use as fungicides |
| IT1264901B1 (it) | 1993-06-29 | 1996-10-17 | Schering Plough S P A | Analoghi eterociclici di 1,2,4-triazolo(15-c)pirimidine ad attivita' antagonista per il recettore a2 dell'adenosina |
| EP0666079B1 (en) | 1993-07-27 | 2001-11-07 | Kyowa Hakko Kogyo Co., Ltd. | Remedy for parkinson's disease |
| IT1277392B1 (it) | 1995-07-28 | 1997-11-10 | Schering Plough S P A | Analoghi eterociclici di 1,2,4-triazolo(1,5-c]pirimidine ad attivita' antagonista per il recettore a2a dell'adenosina |
| JP4195729B2 (ja) * | 1997-03-24 | 2008-12-10 | 協和醗酵工業株式会社 | [1,2,4]トリアゾロ[1,5−c]ピリミジン誘導体 |
| IT1291372B1 (it) | 1997-05-21 | 1999-01-07 | Schering Plough S P A | Uso di analoghi eterociclici di 1,2,4-triazolo (1,5-c) pirimidine per la preparazione di medicamenti utili per il trattamento delle malattie |
| IL142128A0 (en) * | 1998-09-22 | 2002-03-10 | Kyowa Hakko Kogyo Kk | [1,2,4] triazolo [1,5-c] pyrimidine derivatives |
| SI1283839T1 (zh) | 2000-05-26 | 2005-08-31 | Schering Corp | |
| JPWO2002079204A1 (ja) * | 2001-03-28 | 2004-07-22 | 協和醗酵工業株式会社 | 8−チアゾリル[1,2,4]トリアゾロ[1,5−c]ピリミジン誘導体 |
| ES2283625T3 (es) | 2001-11-30 | 2007-11-01 | Schering Corporation | Antagonistas del receptopr a2a de adenosina de (1,2,4)-triazol biciclicos. |
-
2002
- 2002-11-25 PE PE2002001128A patent/PE20030739A1/es not_active Application Discontinuation
- 2002-11-25 TW TW091134178A patent/TW200300686A/zh unknown
- 2002-11-25 AR ARP020104528A patent/AR038366A1/es unknown
- 2002-11-26 AU AU2002346572A patent/AU2002346572A1/en not_active Abandoned
- 2002-11-26 IL IL16157202A patent/IL161572A0/xx unknown
- 2002-11-26 HU HU0402270A patent/HUP0402270A3/hu unknown
- 2002-11-26 DE DE60209251T patent/DE60209251T2/de not_active Expired - Lifetime
- 2002-11-26 CN CNA028239229A patent/CN1596258A/zh active Pending
- 2002-11-26 US US10/304,504 patent/US7041666B2/en not_active Expired - Lifetime
- 2002-11-26 KR KR1020047008031A patent/KR20050044607A/ko not_active Withdrawn
- 2002-11-26 CA CA2468681A patent/CA2468681C/en not_active Expired - Fee Related
- 2002-11-26 JP JP2003549354A patent/JP4284181B2/ja not_active Expired - Fee Related
- 2002-11-26 AT AT02784641T patent/ATE317844T1/de not_active IP Right Cessation
- 2002-11-26 MX MXPA04005156A patent/MXPA04005156A/es active IP Right Grant
- 2002-11-26 ES ES02784641T patent/ES2258164T3/es not_active Expired - Lifetime
- 2002-11-26 WO PCT/US2002/038134 patent/WO2003048164A2/en not_active Ceased
- 2002-11-26 EP EP02784641A patent/EP1453835B1/en not_active Expired - Lifetime
-
2004
- 2004-05-27 ZA ZA200404160A patent/ZA200404160B/en unknown
-
2008
- 2008-06-04 JP JP2008147394A patent/JP2008303217A/ja active Pending
Cited By (13)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| CN112384515A (zh) * | 2018-02-27 | 2021-02-19 | 因赛特公司 | 作为a2a/a2b抑制剂的咪唑并嘧啶和三唑并嘧啶 |
| CN112384515B (zh) * | 2018-02-27 | 2024-12-10 | 因赛特公司 | 作为a2a/a2b抑制剂的咪唑并嘧啶和三唑并嘧啶 |
| CN112105617B (zh) * | 2018-04-28 | 2022-04-05 | 南京明德新药研发有限公司 | 一种三唑并嘧啶类化合物的晶型、盐型及其制备方法 |
| WO2019206336A1 (zh) * | 2018-04-28 | 2019-10-31 | 南京明德新药研发有限公司 | 一种三唑并嘧啶类化合物的晶型、盐型及其制备方法 |
| CN112105617A (zh) * | 2018-04-28 | 2020-12-18 | 南京明德新药研发有限公司 | 一种三唑并嘧啶类化合物的晶型、盐型及其制备方法 |
| US10858365B2 (en) | 2018-09-12 | 2020-12-08 | Dizal (Jiangsu) Pharmaceutical Co., Ltd. | Triazolo-pyrimidine compounds and uses thereof |
| CN112279852A (zh) * | 2018-09-12 | 2021-01-29 | 迪哲(江苏)医药股份有限公司 | 三唑并-嘧啶化合物和其用途 |
| CN111635408A (zh) * | 2018-09-12 | 2020-09-08 | 迪哲(江苏)医药有限公司 | 三唑并-嘧啶化合物和其用途 |
| CN111635408B (zh) * | 2018-09-12 | 2022-07-22 | 迪哲(江苏)医药股份有限公司 | 三唑并-嘧啶化合物和其用途 |
| US11629147B2 (en) | 2018-09-12 | 2023-04-18 | Dizal (Jiangsu) Pharmaceutical Co., Ltd. | Triazolo-pyrimidine compounds and uses thereof |
| TWI820209B (zh) * | 2018-09-12 | 2023-11-01 | 大陸商迪哲(江蘇)醫藥股份有限公司 | 三唑并-嘧啶化合物及其用途 |
| WO2020052631A1 (en) * | 2018-09-12 | 2020-03-19 | Dizal (Jiangsu) Pharmaceutical Co., Ltd. | Triazolo-pyrimidine compounds and uses thereof |
| CN113906022A (zh) * | 2019-01-29 | 2022-01-07 | 因赛特公司 | 作为a2a/a2b抑制剂的吡唑并吡啶和三唑并吡啶 |
Also Published As
| Publication number | Publication date |
|---|---|
| CA2468681C (en) | 2011-01-25 |
| AR038366A1 (es) | 2005-01-12 |
| TW200300686A (en) | 2003-06-16 |
| IL161572A0 (en) | 2004-09-27 |
| US20030212080A1 (en) | 2003-11-13 |
| ES2258164T3 (es) | 2006-08-16 |
| EP1453835B1 (en) | 2006-02-15 |
| JP4284181B2 (ja) | 2009-06-24 |
| US7041666B2 (en) | 2006-05-09 |
| KR20050044607A (ko) | 2005-05-12 |
| EP1453835A2 (en) | 2004-09-08 |
| HUP0402270A3 (en) | 2008-09-29 |
| ATE317844T1 (de) | 2006-03-15 |
| DE60209251D1 (de) | 2006-04-20 |
| JP2005511698A (ja) | 2005-04-28 |
| CA2468681A1 (en) | 2003-06-12 |
| ZA200404160B (en) | 2005-04-08 |
| WO2003048164A3 (en) | 2003-10-16 |
| JP2008303217A (ja) | 2008-12-18 |
| WO2003048164A2 (en) | 2003-06-12 |
| AU2002346572A1 (en) | 2003-06-17 |
| DE60209251T2 (de) | 2006-11-09 |
| HUP0402270A2 (hu) | 2005-02-28 |
| PE20030739A1 (es) | 2003-08-28 |
| MXPA04005156A (es) | 2004-08-11 |
| HK1064100A1 (zh) | 2005-01-21 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| CN1596258A (zh) | 腺A2α受体拮抗剂 | |
| CN1247588C (zh) | 腺苷A2a受体拮抗剂 | |
| CN1261433C (zh) | 用作神经激肽受体拮抗剂的1,3,8-三氮杂-螺[4.5]癸-4-酮衍生物 | |
| CN1064362C (zh) | 嘧啶并[5,4-d]嘧啶,含这种化合物的药物,其用途和其制备方法 | |
| CN1026588C (zh) | 新的1-烷基取代苯并咪唑衍生物的制备方法 | |
| CN1274676C (zh) | 喹啉和喹唑啉衍生物 | |
| CN1198825C (zh) | 8-苯基-6,9-二氢-[1,2,4]三唑并[3,4-i]嘌呤-5-酮衍生物 | |
| CN1020906C (zh) | 2-(杂环烷基)咪唑并-吡啶-嘧啶化合物的制备方法 | |
| CN1823068A (zh) | 作为糖原合酶激酶3抑制剂的三唑并嘧啶衍生物 | |
| CN1582285A (zh) | 用作糖原合酶激酶3β抑制剂(GSK3抑制剂)的杂芳胺化合物 | |
| CN1950371A (zh) | 用作组胺h3受体配体的四氢萘啶衍生物 | |
| CN1150165C (zh) | 用于治疗中枢神经系统紊乱的1-芳基磺酰基-2-芳基-吡咯烷衍生物 | |
| CN1630657A (zh) | 脱氮嘌呤及其用途 | |
| CN1688581A (zh) | [1,2,4]-三唑二环腺苷A2a受体拮抗剂 | |
| CN1400973A (zh) | 新的联芳基甲酰胺 | |
| CN1212695A (zh) | 4-氨基-嘧啶衍生物,含该化合物的药物制剂,其用途及其制造方法 | |
| CN1741999A (zh) | 用作GSK-3β抑制剂的哒嗪酮衍生物 | |
| CN1288464A (zh) | 作为orl-1受体激动剂的4-(2-酮-1-苯并咪唑啉基)哌啶化合物 | |
| CN1860118A (zh) | 作为蛋白激酶抑制剂的化合物和组合物 | |
| CN1503793A (zh) | N-取代的非芳基-杂环nmda/nr2b拮抗剂 | |
| CN1278819A (zh) | 作为促肾上腺皮质素释放激素拮抗剂、可用于治疗cns障碍和紧张相关性疾病的杂环基取代的环稠合吡啶和嘧啶 | |
| CN1503673A (zh) | 消炎药 | |
| CN1845915A (zh) | 作为亲代谢谷氨酸受体-5的调节剂的联吡啶胺和醚类化合物 | |
| CN1777609A (zh) | 2-炔基-及2-链烯基-吡唑并-[4,3,-e]-1,2,4-三唑并[1,5-c]-嘧啶腺苷A2a受体拮抗剂 | |
| CN1816551A (zh) | 吡咯并嘧啶A2b选择性拮抗剂化合物,它们的合成及用途 |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| C06 | Publication | ||
| PB01 | Publication | ||
| C10 | Entry into substantive examination | ||
| SE01 | Entry into force of request for substantive examination | ||
| AD01 | Patent right deemed abandoned | ||
| C20 | Patent right or utility model deemed to be abandoned or is abandoned |