-
1. GEBIET DER ERFINDUNG
-
Die
Erfindung betrifft kristalline Polymorphe eines polycyclischen Xanthinphosphodiesterase
("PDE") V-Inhibitors.
-
2. HINTERGRUND
-
WO 02/24698 lehrt eine
Klasse von Xanthin PDE V-Inhibitorverbindungen, die zur Behandlung
von Impotenz brauchbar sind. Es folgt ein allgemeines Verfahren,
das dort (Seite 75, Zeile 6, bis Seite 80, Zeile 2) zur Herstellung
von Xanthin PDE V-Inhibitorverbindungen mit der folgenden Formel
(I) offenbart ist:
- (i) Umsetzen einer Verbindung mit der Formel
(III) mit einem Alkylhalogenid in Gegenwart einer Base (Einführung von
R2 oder einer geschützten Form von R2);
- (ii) (a) Debenzylieren und anschließendes (b) Alkylieren der aus
Stufe (i) resultierenden Verbindung mit einem Alkylhalogenid, XCH2R3;
- (iii) (a) Deprotonieren und anschließendes (b) Halogenieren der
aus Stufe (ii) resultierenden Verbindung;
- (iv) Umsetzen der aus Stufe (iii) resultierenden Verbindung
mit einem Amin mit der Formel R4NH2, und
- (v) Entfernen eines schützenden
Anteils von R2, falls vorhanden, an der
aus Stufe (iv) resultierenden Verbindung, um die Verbindung mit
der Formel (I) zu bilden.
-
R
1, R
2, R
3 und
R
4 sind jeweils in
WO 02/24698 definiert.
-
WO-A-02/24698 (Seiten
44 und 68 bis 73) lehrt ferner eine Synthese für eine spezielle Xanthin PDE V-Inhibitorverbindung
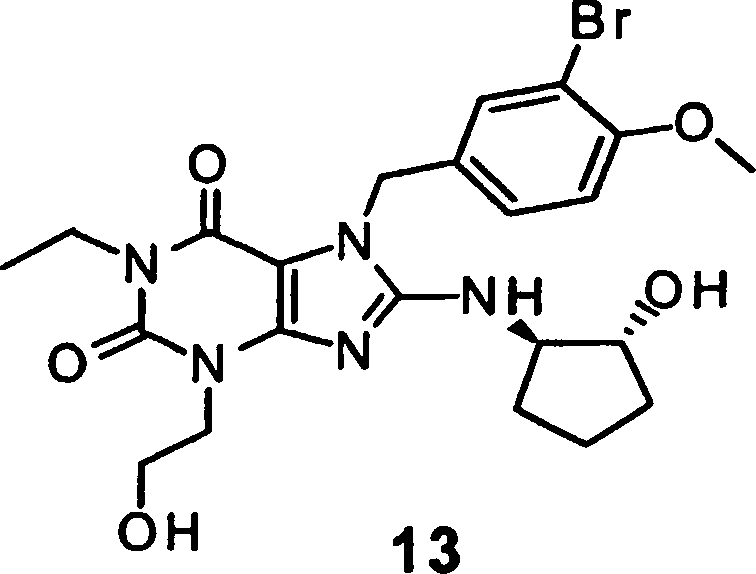
(die dort als Verbindung 13 oder Verbindung 114 in Tabelle II identifiziert
wird): Verbindung 13 kann als 1-Ethyl-3,7-dihydro-8-[(1R,2R)-(hydroxycyclopentyl)amino]-3-(2-hydroxyethyl)-7-[(3-brom-4-methoxyphenyl)methyl]-1H-purin-2,6-dion
bezeichnet werden:
Verbindung
13
-
Verbindung
13 zeigt gute PDE V-Inhibitoraktivität (Potenz) und Selektivität und ist
zur Behandlung der erektilen Dysfunktion brauchbar. Verbindung 13
kann jedoch, wenn sie nach dem in
WO
02/24698 beschriebenen Verfahren hergestellt ist, in Bezug
auf die thermodynamische Stabilität einige unerwünschte Eigenschaften
aufweisen.
-
Polymorphismus
kann als die Fähigkeit
einer Verbindung charakterisiert werden, in unterschiedlichen Kristallformen
zu kristallisieren, während
dieselbe chemische Formel erhalten bleibt. Polymorphe einer gegebenen
Wirkstoffsubstanz sind chemisch identisch, da sie die gleichen Atome
in der gleichen Weise aneinander gebunden enthalten, unterscheiden
sich jedoch in ihren Kristallformen, die eine oder mehrere physikalische Eigenschaften
beeinflussen können,
wie Löslichkeit,
Schmelzpunkt, Schüttdichte,
Fließeigenschaften,
usw.
-
Es
wäre vorteilhaft,
die thermodynamischen Eigenschaften von Verbindung 13 zu verbessern.
Es wäre ferner
vorteilhaft, Verbindung 13 in einer stabilen kristallinen Form herzustellen,
die konsistente physikalische Eigenschaften hat. Die Erfindung strebt
die Bereitstellung dieser und anderer Vorteile an, die sich im Verlauf der
Beschreibung deutlicher zeigen werden.
-
ZUSAMMENFASSUNG DER ERFINDUNG
-
Die
Erfindung liefert zwei kristalline Polymorphe von Verbindung 13.
Ein kristalliner Polymorph kann durch sein Pulverröntgenbeugungsspektrum
identifiziert werden, das in Form von "2θ-Winkeln
(°)" angegeben wird.
-
Ein
Aspekt der Erfindung liefert ein kristallines Polymorph Form 2 von
Verbindung 13:
das ein Röntgenpulverbeugungsmuster mit
charakteristischen Peak-Positionen von 8,1, 11,3, 17,2 und 22,2 Grad
2θ +/– 0,5 Grad
2θ zeigt.
-
Ein
weiterer Aspekt der Erfindung liefert ein kristallines Polymorph
Form 2 von Verbindung 13, das ein Röntgenpulverbeugungsmuster mit
charakteristischen Peak-Positionen von 8,1, 11,3, 13,1, 15,3, 16,1,
17,2, 17,6, 18,9, 20,9, 21,8, 22,2, 23,4, 24,1, 25,8 und 30,6 Grad
2θ +/– 0,5 Grad
2θ zeigt.
-
Ein
weiterer Aspekt der Erfindung liefert ein kristallines Polymorph
Form 2 von Verbindung 13, das ein Röntgenpulverbeugungsmuster zeigt,
welches im Wesentlichen dasselbe wie das in 5 gezeigte
Röntgenpulverbeugungsmuster
ist.
-
Ein
weiterer Aspekt der Erfindung liefert ein kristallines Polymorph
Form 2 von Verbindung 13, das ein Differentialthermoanalyse-Muster
zeigt, welches im Wesentlichen dasselbe wie das in 2 gezeigte
Differentialthermoanalyse-Muster ist.
-
Ein
weiterer Aspekt der Erfindung liefert ein kristallines Polymorph
Form 1 von Verbindung 13, das ein Röntgenpulverbeugungsmuster mit
charakteristischen Peak-Positionen von 7,3, 9,2 und 20,2° 2θ +/– 0,5° 2θ aufweist.
-
Ein
weiterer Aspekt der Erfindung liefert ein kristallines Polymorph
Form 1 von Verbindung 13, das ein Röntgenpulverbeugungsmuster mit
charakteristischen Peak-Positionen von 7,3, 8,4, 9,2, 12,7, 14,3,
15,0, 15,4, 16,5, 18,8, 20,2, 20,9, 24, 0, 25, 8, 26, 4, 27, 2,
27, 6, 29, 3, 31,9 und 34,6 Grad 2θ +/– 0,5 Grad 2θ zeigt.
-
Ein
weiterer Aspekt der Erfindung liefert ein kristallines Polymorph
Form 1 von Verbindung 13, das ein Röntgenpulverbeugungsmuster zeigt,
welches im Wesentlichen dasselbe wie das in 6 gezeigte
Röntgenpulverbeugungsmuster
ist.
-
Ein
weiterer Aspekt der Erfindung liefert ein kristallines Polymorph
Form 1 von Verbindung 13, das ein Differentialthermoanalyse-Muster
zeigt, welches im Wesentlichen dasselbe wie das in 4 gezeigte
Differentialthermoanalyse-Muster ist.
-
Andere
Aspekte der Erfindung beinhalten pharmazeutisch annehmbare Zusammensetzungen,
die aus den erfindungsgemäßen Polymorphen
hergestellt sind. Die erfindungsgemäßen Verbindungen können zur
Behandlung einer Vielfalt von Erkrankungen, Symptomen und physiologischen
Störungen
brauchbar sein, wie sexueller Dysfunktion (z. B. Impotenz).
-
Ein
weiteres Verständnis
der Erfindung ergibt sich aus der folgenden Zeichnungen, der Beschreibung sowie
den Ansprüchen.
-
KURZE BESCHREIBUNG DER ZEICHNUNGEN
-
1 ist
eine graphische Darstellung eines Röntgenpulverbeugungsspektrums
von kristallinem Polymorph Form 2 von Verbindung 13, die aus Acetonitril
kristallisiert ist. Die graphische Darstellung trägt die Intensität der Peaks,
definiert durch Zählwerte
pro Sekunde, gegen den Beugungswinkel 2θ in Grad auf. Die Probe war
nicht mikronisiert und nicht in den Probenhalter gepackt. Die Daten
wurden auf einem Rigaku MiniFlex Diffraktometer erfasst.
-
2 ist
eine graphische Darstellung eines Differentialthermoanalyse-Musters
von kristallinem Polymorph Form 2 von Verbindung 13, kristallisiert
aus Acetonitril. Die graphische Darstellung trägt den normalisierten Wärmefluss
in der Einheit Watt/Gramm ("W/g") gegen die gemessene
Probentemperatur in °C
auf.
-
3 ist
eine graphische Darstellung eines Röntgenpulverbeugungsspektrums
von kristallinem Polymorph Form 1 von Verbindung 13, kristallisiert
aus Methanol/Wasser. Die graphische Darstellung trägt die Intensität der Peaks,
definiert durch Zählwerte
pro Sekunde, gegen den Beugungswinkel 2θ in Grad auf. Die Probe war
nicht mikronisiert und nicht in den Probenhalter gepackt. Die Daten
wurden auf einem Rigaku MiniFlex Diffraktometer erfasst.
-
4 ist
eine graphische Darstellung eines Differentialthermoanalyse-Musters
von kristallinem Polymorph Form 1 von Verbindung 13, kristallisiert
aus Methanol/Wasser. Die graphische Darstellung trägt den normalisierten
Wärmefluss
in der Einheit Watt/Gramm ("W/g") gegen die gemessene
Probentemperatur in °C
auf.
-
5 ist
eine graphische Darstellung eines Röntgenpulverbeugungsspektrums
von kristallinem Polymorph Form 2 von Verbindung 13, kristallisiert
aus Acetonitril. Die graphische Darstellung trägt die Intensität der Peaks,
definiert durch Zählwerte
pro Sekunde, gegen den Beugungswinkel 2θ in Grad auf. Die Daten wurden
auf einem Bruker D8 Diffraktometer erfasst.
-
6 ist
eine graphische Darstellung eines Röntgenpulverbeugungsspektrums
von kristallinem Polymorph Form 1 von Verbindung 13, kristallisiert
aus Isopropanol/Wasser. Die graphische Darstellung trägt die Intensität der Peaks,
definiert durch Zählwerte
pro Sekunde, gegen den Beugungswinkel 2θ in Grad auf. Die Daten wurden
auf einem Bruker D8 Diffraktometer erfasst.
-
DETAILLIERTE BESCHREIBUNG
-
Die
folgenden Begriffe haben, wenn nicht anders angegeben, oben und
in der Beschreibung die folgenden Bedeutungen:
-
"Patient" schließt sowohl
Menschen als auch andere Tiere ein.
-
"Säuger" bedeutet Menschen und andere Säugetiere.
-
"Alkyl" bedeutet eine aliphatische
Kohlenwasserstoffgruppe, die geradkettig oder verzweigt sein kann und
1 bis etwa 20 Kohlenstoffatome in der Kette enthält. Bevorzugte Alkylgruppen
enthalten 1 bis etwa 12 Kohlenstoffatome in der Kette. Beson ders
bevorzugte Alkylgruppen enthalten 1 bis etwa 6 Kohlenstoffatome
in der Kette. Verzweigt bedeutet, dass eine oder mehrere niedere
Alkylgruppen, wie Methyl, Ethyl oder Propyl, an eine lineare Alkylkette
gebunden sind. Die Alkylgruppe kann mit einem oder mehreren Substituenten
substituiert sein, die gleich oder verschieden sein können. Nicht-einschränkende Beispiele
für geeignete
Alkylgruppen schließen
Methyl, Ethyl, n-Propyl,
Isopropyl, n-Butyl, t-Butyl, n-Pentyl, Heptyl, Nonyl, Decyl, Fluormethyl,
Trifluormethyl und Cyclopropylmethyl ein.
-
"Aryl" bedeutet ein aromatisches
monocyclisches oder multicyclisches Ringsystem, das etwa 6 bis etwa
14 Kohlenstoffatome, vorzugsweise etwa 6 bis etwa 10 Kohlenstoffatome
enthält.
Die Arylgruppe kann gegebenenfalls mit einem oder mehreren Ringsystemsubstituenten
substituiert sein, die gleich oder verschieden sein können. Nicht-einschränkende Beispiele
für geeignete
Arylgruppen schließen
Phenyl und Naphthyl ein.
-
"Polymorph" bedeutet eine kristalline
Form einer Substanz, die sich von einer anderen kristallinen Form
unterscheidet, mit dieser jedoch die gleiche chemische Formel gemeinsam
hat.
-
"Relative Intensität" bedeutet die Intensität eines
Peaks relativ zu der Intensität
des größten Peaks, gemessen
in einer Röntgenpulverbeugungsanalyse.
Die relative Intensität
kann entweder als Verhältnis
der Höhen
der Peaks (gemessen in Zählwerten
pro Sekunde) oder als Verhältnis
der Flächen
der Peaks berechnet werden. Die hier gezeigten relativen Intensitätsdaten
sind als Verhältnisse
der Höhen
der Peaks berechnet worden.
-
"Antilösungsmittel" bedeutet eine Substanz,
welche die Löslichkeit
eines gelösten
Stoffs in einem Lösungsmittel
reduziert.
-
"c-GMP" bedeutet cyclisches
Guanosinmonophosphat.
-
"Alkohol" bedeutet eine organische
Verbindung, die eine Hydroxylgruppe (-OH) enthält.
-
"Nitril" bedeutet eine organische
Verbindung, die eine -C≡N
Gruppe enthält.
-
"Ester" bedeutet eine organische
Verbindung, die eine RC(O)OR Gruppe enthält, wobei die R's unabhängig Alkyl
oder Aryl sind und die Klammern bedeuten, dass das eingeschlossene
O über
eine Doppelbindung an das C gebunden ist.
-
"Keton" bedeutet eine organische
Verbindung, die eine Carbonylgruppe (C=O) enthält, die an zwei Alkylgruppen
gebunden ist.
-
"Hilfsstoff" bedeutet eine im
Wesentlichen inerte Substanz, die als Verdünnungsmittel verwendet wird, oder
um einer Formulierung Form oder Konsistenz zu verleihen.
-
"Kohlenwasserstoff" bedeutet eine organische
Verbindung, die aus Kohlenstoff und Wasserstoff besteht.
-
Polymorphe von Verbindung 13
-
Verbindung
13 kann in mindestens zwei getrennten kristallinen polymorphen Formen
vorliegen, die jeweils eigene physikalische Eigenschaften haben.
Diese beiden unterschiedlichen kristallinen Polymorphe von Verbindung
13 sind als Form 1 und Form 2 identifiziert worden. Form 1 und 2
von Verbindung 13 können
durch Röntgenpulverbeugung
(1, 3, 5 und 6)
und/oder Differentialthermoanalyse (2 und 4)
charakterisiert werden.
-
Analytische Methodik zur chemischen Identifizierung
von Polymorphen
-
Proben
der beiden Polymorphe – Formen
1 und 2 von Verbindung 13 – wurden
als trockene Pulver mittels Röntgenpulverbeugung
("XRPD") und Differentialthermo-("DSC")-Analyse analy siert.
Die Proben wurden mit minimaler Vorbereitung analysiert, um jeglichen
Formveränderungen
vorzubeugen. Die Proben wurden leicht gerieben, um zu gewährleisten,
dass die Teilchen nicht miteinander verklumpt waren. Für diese
Analysen wurden keine Lösungsmittel,
Trocknungs- oder anderen Vorbereitungsstufen verwendet. Die XRPD-
und DSC-Daten können
Formen 1 und 2 von Verbindung 13 jeweils in einzigartiger Weise
identifizieren.
-
Es
wurden zahlreiche XRPD-Analysen mit einer Vielfalt von Analysegeräten durchgeführt. Einige
der Proben wurden mikronisiert, andere nicht. Ein Satz von Messungen
wurde mit einem Rigaku MiniFlex® Diffraktometer
(hergestellt 1999) durchgeführt,
das die Probe mit 54 Umdrehungen pro Minute ("UpM")
drehte, um bevorzugte Orientierungen der Kristalle zu reduzieren.
Die Polymorphproben wurden in Pulverform zugeführt und unter Verwendung eines
handgeführten
Dübels
mit minimalem Kraftaufwand auf eine Fläche einer Si-beschichteten
Aluminiumplatte mit wenig Hintergrundstreuung angeordnet. Zur Überprüfung der
Genauigkeit der Peak-Position wurde ein kristalliner Siliciumstandard
verwendet. Die Proben wurden Umgebungsbedingungen ausgesetzt. Die
in 1 und 3 gezeigten Röntgenspektren
sind mit einem Neun-Punkt-Savitzky-Golay-Parabolfilter gefiltert,
ansonsten jedoch im Wesentlichen rohe Spektrum ohne Hintergrundkorrektur
oder K-α2
Peak-Entfernung. Die auf den y-Achsen der 1 und 3 gezeigte
Zählwerte
sind in den Einheiten Zählwerte
pro Sekunde. Das Instrument verwendet einen variablen Divergenzschlitz
mit einer θ/2θ-Abtast-Achsenkonfiguration.
Die Intensität
der Peaks (y-Achse ist in Zählwerten
pro Sekunde) ist gegen den 2θ-Winkel
aufgetragen (die x-Achse ist in Grad 2θ). Die Daten in 1 und 3 wurden
mit Detektorzählwerten,
die auf die Erfassungszeit pro Schritt normalisiert waren, gegen
den 2θ-Winkel
aufgetragen.
-
Die
Daten wurden unter Verwendung von JADE® Musterverarbeitungssoftware
Version 5.0 von Materials Data Inc. ("MDI")
ausgewertet. Die Software führt
automatisch eine Endfilterung durch, passt einen Hintergrund an
und misst die Fläche
und Höhe
jedes Peaks. Die relativen Peak-Intensitäten werden unter Verwendung
eines Verhältnisses
der Höhe
jedes angegebenen Peaks zu der Höhe
des größten gemessenen Peaks
berechnet. Die verwendeten relativen Peak-Intensitäten entsprachen
direkt den gefilterten Zählwerten pro
Sekunde der Rohdaten. Form 2 von Verbindung 13 (1)
und Form 1 von Verbindung 13 (3) zeigten jeweils
eigene XRPD-Spektren. Die Röntgenpulverbeugung
ist in der Encyclopedia of Analytic Science, Herausgeber Alan Townshend,
Band 9, Seiten 5585–5593,
Academic Press, London (1995), erörtert, auf die hier Bezug genommen
wird.
-
Es
wurde unter Verwendung des Rigaku MiniFlex
® Diffraktometers
und der oben beschriebenen Verfahren gefunden, dass das kristalline
Polymorph Form 2 von Verbindung 13 ein Pulverröntgenbeugungsspektrum wie in
1 gezeigt
aufwies. Die relativen Intensitäten
und die 2θ-Winkelpositionen
der charakteristischen Peaks von
1 sind in
Tabelle 1 gezeigt: Tabelle 1: Form 2 von Verbindung 13
| 2θ Winkel
(°) | relative
Intensität
(% Höhe) | relative
Intensität
(Peakstärke) |
| 8,44 | 31,1 | S |
| 11,54 | 3,6 | VW |
| 13,36 | 13,9 | M |
| 15,56 | 5,2 | W |
| 16,42 | 100,0 | S |
| 17,44 | 28,3 | S |
| 17,92 | 20,3 | S |
| 19,18 | 15,2 | M |
| 21,20 | 12,8 | M |
| 22,12 | 10,1 | M |
| 22,50 | 13,9 | M |
| 23,06 | 2,8 | VWD |
| 23,70 | 15,3 | M |
| 24,46 | 50,1 | S |
| 25,70 | 16,5 | M |
| 26,04 | 18,4 | M |
| 26,40 | 12,3 | M |
| 27,34 | 5,1 | W |
| 27,86 | 3,0 | VW |
| 28,58 | 2,2 | VW |
| 29,08 | 6,4 | W |
| 29,74 | 11,2 | M |
| 30,48 | 5,5 | W |
| 30,88 | 43,2 | S |
| 31,62 | 2,2 | VW |
| 32,14 | 3,1 | W |
| 32,68 | 7,6 | W |
| 33,02 | 8,7 | W |
| 33,82 | 5,2 | WD |
| 34,68 | 4,3 | W |
| 35,78 | 4,2 | W |
| 36,30 | 3,9 | VW |
| 37,78 | 4,6 | W |
| 38,44 | 7,0 | WD |
| 38,86 | 3,4 | VW |
| 39,28 | 2,1 | VW |
| 40,04 | 1,1 | VWD |
| 40,48 | 1,9 | VW |
| 41,08 | 8,5 | W |
| 41,72 | 3,7 | W |
| 42,88 | 2,0 | WD |
| 43,76 | 6,2 | W |
| 44,76 | 4,1 | W |
| 45,40 | 2,3 | VWD |
| 45,82 | 3,2 | VWD |
| 46,72 | 3,0 | VWD |
| 47,44 | 3,5 | VWD |
| 48,68 | 1,0 | VWD |
| 49,60 | 8,9 | W |
wobei die Peak-Stärke die relativen Intensitäten gemäß dem folgenden
Schema kategorisiert: S ist stark (20,0–100,0%); M ist Mittel (9,0–19,9%);
W ist schwach (4,0–8,9%);
VW ist sehr schwach (0,1–3,9%)
und VWD ist sehr schwach und diffus (breit).
-
Es
wurde unter Verwendung des Rigaku MiniFlex
® Diffraktometers
und der oben beschriebenen Verfahren gefunden, dass das kristalline
Polymorph Form 1 von Verbindung 13 ein Pulverröntgenbeugungsspektrum wie in
3 gezeigt
aufwies. Die relativen Intensitäten
und die 2θ-Winkelpositionen
der charakteristischen Peaks von
3 sind in
Tabelle 2 gezeigt: Tabelle 2: Form 1 von Verbindung 13
| 2θ Winkel
(°) | relative
Intensität
(% Höhe) | relative
Intensität
(Peakstärke) |
| 7,48 | 100,0 | S |
| 8,52 | 0,9 | VW |
| 9,36 | 11,7 | M |
| 12,84 | 64,8 | S |
| 14,44 | 4,8 | WD |
| 15,10 | 2,7 | VWD |
| 15,52 | 2,2 | VWD |
| 16,58 | 13,2 | M |
| 19,02 | 35,8 | S |
| 20,34 | 14,4 | M |
| 21,00 | 4,7 | W |
| 21,94 | 4,1 | W |
| 22,70 | 3,1 | VWD |
| 22,98 | 4,5 | WD |
| 24,14 | 7,8 | W |
| 25,04 | 3,1 | VWD |
| 25,84 | 21,8 | S |
| 26,40 | 4,5 | W |
| 27,32 | 5,8 | W |
| 27,74 | 8,4 | W |
| 28,78 | 4,5 | WD |
| 29,20 | 9,9 | M |
| 30,40 | 1,2 | VWD |
| 32,08 | 3,4 | W |
| 33,02 | 4,3 | W |
| 33,66 | 5,1 | W |
| 34,63 | 5,0 | WD |
| 37,24 | 3,3 | VWD |
| 38,12 | 1,7 | VWD |
| 40,46 | 4,8 | W |
| 41,94 | 5,1 | W |
| 45,44 | 2,3 | WD |
| 47,52 | 2,3 | WD |
-
wobei
die Peakstärken
gemäß dem oben
beschriebenen Schema kategorisiert sind.
-
Die
XRPD-Analysen wurden unter Verwendung von unterschiedlichen Analysegeräten wiederholt.
Zur Erfassung der XRPD-Daten wurden Rigaku DMAX 2200 und Bruker
D8 Diffraktometer verwen det. Bei diesen Analysen wurden die Proben
in einer solchen Weise in die Probenhalter gepackt, dass Messfehler
minimiert wurden, die aus unebenen Probenoberflächen oder inkonsistenten Probedicken
resultieren könnten.
-
Das
Rigaku DMAX-2200 Diffraktometer (hergestellt 1998) wurde mit einem
Startwinkel von 6 Grad und automatischen variablen Divergenzschlitzen
betrieben. Die Strahlbreite betrug 20 mm. Die Vorrichtung verwendete
einen Graphit-Monochromator und einen Szintillationsdetektor. Die
Schrittgröße betrug
während
des Abtastens 0,02 Grad über
eine Schrittdauer von 0,3 Sekunden. Die Abtastgeschwindigkeit betrug
4 Grad pro Minute. Die Probendrehgeschwindigkeit betrug 40 UpM.
-
Das
Bruker D8 Diffraktometer (hergestellt 2002) hatte eine Paralleloptikkonfiguration
mit einem GOBEL-Strahlfokussierspiegel, und es wurde ein PSD-Detektor,
der mit einem festen radialen Soller-Schlitz ausgestattet war, mit
einer Anton Paar TTK450 Temperaturstufe verwendet. Die Divergenzschlitze
wurden auf 0,6 mm fixiert. Der Probenhalter war ein von oben beladbarer
Messingblock. Die Proben wurden mit einem Mikroskop-Objektträger aus
Glas nivelliert. Die Probenkammer wurde nicht gespült, wurde
nicht über
30°C erwärmt und
befand sich nicht unter Vakuum. Die Instrumentenkalibrierung wurde
mittels Glimmer-Standards verifiziert. Die Schrittgröße betrug
während
des Abtastens 0,013 Grad über
Schrittdauern von 0,1 und 0,5 Sekunden. Die Datenglättung erfolgte
unter Verwendung von EVA-Analysesoftware, Version 7.0, erhalten
von Bruker®,
geschrieben von SOCABIN®. Die Daten wurden mit
einem Fast Fourier-Glättungsprogramm
(20,000 × 1)
gefiltert. Die Strahlungsquellen waren für alle drei Diffraktometer
Kupfer (Kα).
-
Beispiele
für XRPD-Daten,
die mit dem Bruker D8 erfasst wurden, sind in den
5 und
6 gezeigt,
die XRPD-Spektren für
Form 2 beziehungsweise 1 sind. Die Peak-Positionen aus Spektren,
die mit den drei oben beschriebenen Instrumenten erzeugt wurden,
sind in den Tabellen 3 und 4 gezeigt. Tabelle 3 liefert Peakpositionsdaten
aus fünf
Beispielen von XRPD-Spektren,
die mit Proben von Form 1 erzeugt wurden. Für jedes Beispiel sind die Positionen
von neunzehn charakteristischen Peaks gezeigt. Der Durchschnittswert
und die Standardabweichungen der Peakpositionsdaten jedes charakteristischen
Peaks wurden außerdem
analysiert. Tabelle 4 liefert Peakpositionsdaten aus sechs Beispielen
von XRPD-Spektren, die mit Proben von Form 2 erzeugt wurden. Die
Variation von Probe zu Probe beträgt allgemein etwa +/– 0,5° 2θ, vorzugsweise
etwa +/– 0,3° 2θ. TABELLE 3. FORM 1 – PULVER RÖNTGENBEUGUNGSDATEN (PEAK-POSITIONEN ° 2θ)
| Beispiel Nr. | 1 | 2 | 3 | 4 | 5 | Mittelwert | gerundeter Mittelwert | σ | 2 σ | gerundet,
2 σ | Maximalwert
2 σ |
| Instrument | Rigaku
MiniFlex | Bruker D8 | Rigaku
MiniFlex | Rigaku
MiniFlex | Rigaku DMA X 2200 | | | | | | |
| Peak Nr. ↓ | | | | | | | | | | | |
| 1. | 7,341 | 7,224 | 7,481 | 7,401 | 7,26 | 7,341 | 7,3 | 0,10 | 0,21 | 0,2 | 0,3 |
| 2. | 8,419 | 8,293 | 8,523 | 8,518 | 8,34 | 8,419 | 8,4 | 0,10 | 0,21 | 0,2 | |
| 3. | 9,220 | 9,106 | 9,361 | 9,299 | 9,14 | 9,225 | 9,2 | 0,11 | 0,21 | 0,2 | |
| 4. | 12,719 | 12,633 | 12,841 | 12,820 | 12,64 | 12,731 | 12,7 | 0,10 | 0,20 | 0,2 | |
| 5. | 14,299 | 14,205 | 14,440 | 14,380 | 14,26 | 14,317 | 14,3 | 0,09 | 0,19 | 0,2 | |
| 6. | 14,960 | 14,872 | 15,100 | 15,020 | 14,90 | 14,970 | 15,0 | 0,09 | 0,18 | 0,2 | |
| 7. | 15,360 | 15,272 | 15,519 | 15,419 | 15,30 | 15,374 | 15,4 | 0,10 | 0,20 | 0,2 | |
| 8. | 16,439 | 16,362 | 16,580 | 16,520 | 16,38 | 16,456 | 16,5 | 0,09 | 0,19 | 0,2 | |
| 9. | 18,880 | 18,700 | 19,019 | 18,860 | 18,72 | 18,836 | 18,8 | 0,13 | 0,26 | 0,3 | |
| 10. | 20,200 | 20,150 | 20,340 | 20,340 | 20,14 | 20,234 | 20,2 | 0,10 | 0,20 | 0,2 | |
| 11. | 20,861 | 20,803 | 21,001 | 20,960 | 20,82 | 20,889 | 20,9 | 0,09 | 0,17 | 0,2 | |
| 12. | 23,980 | 23,900 | 24,140 | 23,980 | 23,92 | 23,984 | 24,0 | 0,09 | 0,19 | 0,2 | |
| 13. | 25,720 | 25,749 | 25,840 | 25,880 | 25,76 | 25,790 | 25,8 | 0,07 | 0,13 | 0,1 | |
| 14. | 26,261 | 26,443 | 26,400 | 26,520 | 26,44 | 26,413 | 26,4 | 0,10 | 0,19 | 0,2 | |
| 15. | 27,200 | 27,184 | 27,320 | 27,341 | 27,18 | 27,245 | 27,2 | 0,08 | 0,16 | 0,2 | |
| 16. | 27,620 | 27,545 | 27,740 | 27,700 | 27,56 | 27,633 | 27,6 | 0,09 | 0,17 | 0,2 | |
| 17. | 29,060 | 29,320 | 29,200 | 29,419 | 29,28 | 29,256 | 29,3 | 0,13 | 0,27 | 0,3 | |
| 18. | 31,920 | 31,862 | 32,079 | 31,960 | 31,86 | 31,936 | 31,9 | 0,09 | 0,18 | 0,2 | |
| 19. | 34,579 | 34,508 | 34,640 | 34,640 | 34,54 | 34,581 | 34,6 | 0,06 | 0,12 | 0,1 | |
TABELLE 4. FORM 2 – RÖNTGENBEUGUNGSDATEN (PEAK-POSITIONEN
2θ)
| Beispiel Nr. | 6 | 7 | 8 | 9 | 10 | 11 | Mittelwert | gerundeter Mittelwert | σ | 2 σ | gerundet,
2 σ | Maximal wert 2 σ |
| Instru ment | Rigaku MiniFlex | Bruker D8 | Rigaku MiniFlex | Rigaku MiniFlex | Rigaku DMA X 2200 | Bruker D8 | | | | | | |
| Peak Nr. ↓ | | | | | | | | | | | | |
| 1. | 8,179 | 7,977 | 8,139 | 8,439 | 8,200 | 7,954 | 8,148 | 8,1 | 0,176 | 0,352 | 0,4 | 0,4 |
| 2. | 11,261 | 11,175 | 11,240 | 11,540 | 11,320 | 11,068 | 11,267 | 11,3 | 0,159 | 0,318 | 0,3 | |
| 3. | 13,081 | 12,851 | 13,059 | 13,359 | 13,120 | 12,886 | 13,059 | 13,1 | 0,183 | 0,366 | 0,4 | |
| 4. | 15,279 | 15,164 | 15,260 | 15,560 | 15,340 | 15,107 | 15,285 | 15,3 | 0,159 | 0,317 | 0,3 | |
| 5. | 16,141 | 15,993 | 16,100 | 16,420 | 16,180 | 15,942 | 16,129 | 16,1 | 0,168 | 0,337 | 0,3 | |
| 6. | 17,179 | 17,028 | 17,140 | 17,440 | 17,220 | 16,973 | 17,163 | 17,2 | 0,164 | 0,329 | 0,3 | |
| 7. | 17,659 | 17,430 | 17,600 | 17,919 | 17,680 | 17,442 | 17,622 | 17,6 | 0,180 | 0,361 | 0,4 | |
| 8. | 18,920 | 18,726 | 18,899 | 19,180 | 18,980 | 18,723 | 18,905 | 18,9 | 0,171 | 0,343 | 0,3 | |
| 9. | 20,900 | 20,948 | 20,880 | 21,200 | 20,940 | 20,735 | 20,934 | 20,9 | 0,151 | 0,303 | 0,3 | |
| 10. | 21,840 | 21,732 | 21,820 | 22,120 | 21,900 | 21,669 | 21,847 | 21,8 | 0,157 | 0,314 | 0,3 | |
| 11. | 22,221 | 22,039 | 22,219 | 22,500 | 22,280 | 22,050 | 22,218 | 22,2 | 0,169 | 0,339 | 0,3 | |
| 12. | 23,439 | 23,353 | 23,420 | 23,699 | 23,480 | 23,250 | 23,440 | 23,4 | 0,150 | 0,300 | 0,3 | |
| 13. | 24,200 | 24,095 | 24,000 | 24,461 | 24,100 | 23,854 | 24,118 | 24,1 | 0,204 | 0,409 | 0,4 | |
| 14. | 25,780 | 25,655 | 25,720 | 26,039 | 25,800 | 25,562 | 25,759 | 25,8 | 0,162 | 0,325 | 0,3 | |
| 15. | 30,640 | 30,547 | 30,600 | 30,880 | 30,680 | 30,450 | 30,633 | 30,6 | 0,145 | 0,290 | 0,3 | |
-
Wie
aus Tabelle 3 hervorgeht, sind Peak-Nummern 1, 3 und 10 mit durchschnittlichen
Peak-Positionen bei 7,3, 9,2 beziehungsweise 20,2 repräsentativ
für Form
1. Wie aus Tabelle 4 hervorgeht, sind Peak-Nummern 1, 2, 6 und 11
mit durchschnittlichen Peak-Positionen bei 8,1, 11,3, 17,2 beziehungsweise
22,2 repräsentativ für Form 2.
Die Peak-Nummern 7, 9 und 12 von Form 1 haben durchschnittliche
Peak-Positionen von 15,4, 18,8 und 24,0. Diese scheinen ungefähr mit den
Peak-Nummern 4, 8 und 13 von Form 2 zusammenzufallen.
-
Das
zum Testen der Polymorph-Proben verwendete DSC-Instrument war ein Perkin-Elmer® Modell Pryis
1 (hergestellt 1999), das mit einem gekühlten Kühlsystem ausgestattet war.
Die DSC-Zelle/Probenkammer wurde mit 40 ml ultrahochreinem Stickstoffgas
pro Minute gespült.
Das Instrument wurde mit hochreinem Indium kalibriert. Die Genauigkeit
der gemessenen Probentemperatur lag bei diesem Verfahren innerhalb
von etwa +/– 1°C, und die
Schmelzwärme
konnte mit einem relativen Fehler von etwa +/– 5% gemessen werden. Die Proben
wurden in einer Standard-Aluminium-DSC-Pfanne ohne Deckel von Perkin-Elmer
angeordnet. Zwischen etwa 3 mg und etwa 6 mg des Polymorph-Probenpulvers wurde
in den Boden der Pfanne gegeben und sanft heruntergedrückt, um
den Kontakt mit der Pfanne herzustellen. Das Gewicht jeder Probe
wurde genau gemessen und bis auf etwa ein Hundertstel Milligramm
aufgezeichnet. Das Instrument verwendete eine leere Referenzpfanne.
Das Instrument war so programmiert, dass es die Probe etwa 1 Minute
auf etwa 30°C
hielt, bevor mit dem dynamischen Aufheizen mit 10°C/Minute
auf etwa 300°C
begonnen wurde. Die Daten wurden in der Einheit "Watt/Gramm" angegeben, wodurch der auf ein Probengewicht
normalisierte Wärmefluss
wiedergegeben wird. Der normalisierte Wärmefluss wurde gegen die gemessene
Probentemperatur aufgetragen. Die Auftragungen erfolgten mit aufwärts weisenden
endothermen Peaks. Die endothermen Schmelzpeaks wurden in diesen
Analysen hinsichtlich extrapolierten Temperaturen für Beginn- und Endtemperaturen,
Spitzentemperatur (Peak-Temperatur) und Schmelzwärme bewertet. Die Schmelztemperatur
und die zum Schmelzen einer Probe erforderliche Wärme waren
für Form
2 von Verbindung 13 (2) und Form 1 von Verbindung 13
(4) besonders. Differentialthermoanalyse ist in
der Encyclopedia of Analytic Science, Herausgeber Alan Townshend,
Band 9, Seiten 5155–5160,
Academic Press, London (1995), erörtert.
-
2 zeigt
eine graphische Darstellung eines DSC-Musters für Form 2 von Verbindung 13.
Diese graphische Darstellung zeigt eine Endotherme, die bei 165,300°C beginnt
und bei 171,729°C
endet, was dem Schmelzpunkt des Polymorphs entspricht.
-
4 zeigt
eine graphische Darstellung eines DSC-Musters für Form 1 von Verbindung 13.
Diese graphische Darstellung zeigt eine Endotherme, die bei 178,092°C beginnt
und bei 181,022°C
endet, was dem Schmelzpunkt des Polymorphs entspricht.
-
Die
Herstellung von Verbindung 13 ist in
WO
02/24698 gelehrt. Ein alternatives Verfahren zur Herstellung
von Verbindung 13 wird in einer gleichzeitig anhängigen US-Patentanmeldung mit dem Titel Process
for Preparing Xanthine Phosphodiesterase V Inhibitors and Precursors
Thereof (Verbindung 13 wird in der gleichzeitig anhängigen Anmeldung
als Verbindung 13A bezeichnet) gelehrt, die am gleichen Tag wie
die vorliegende Anmeldung eingereicht wurde. Dieses Verfahren ist
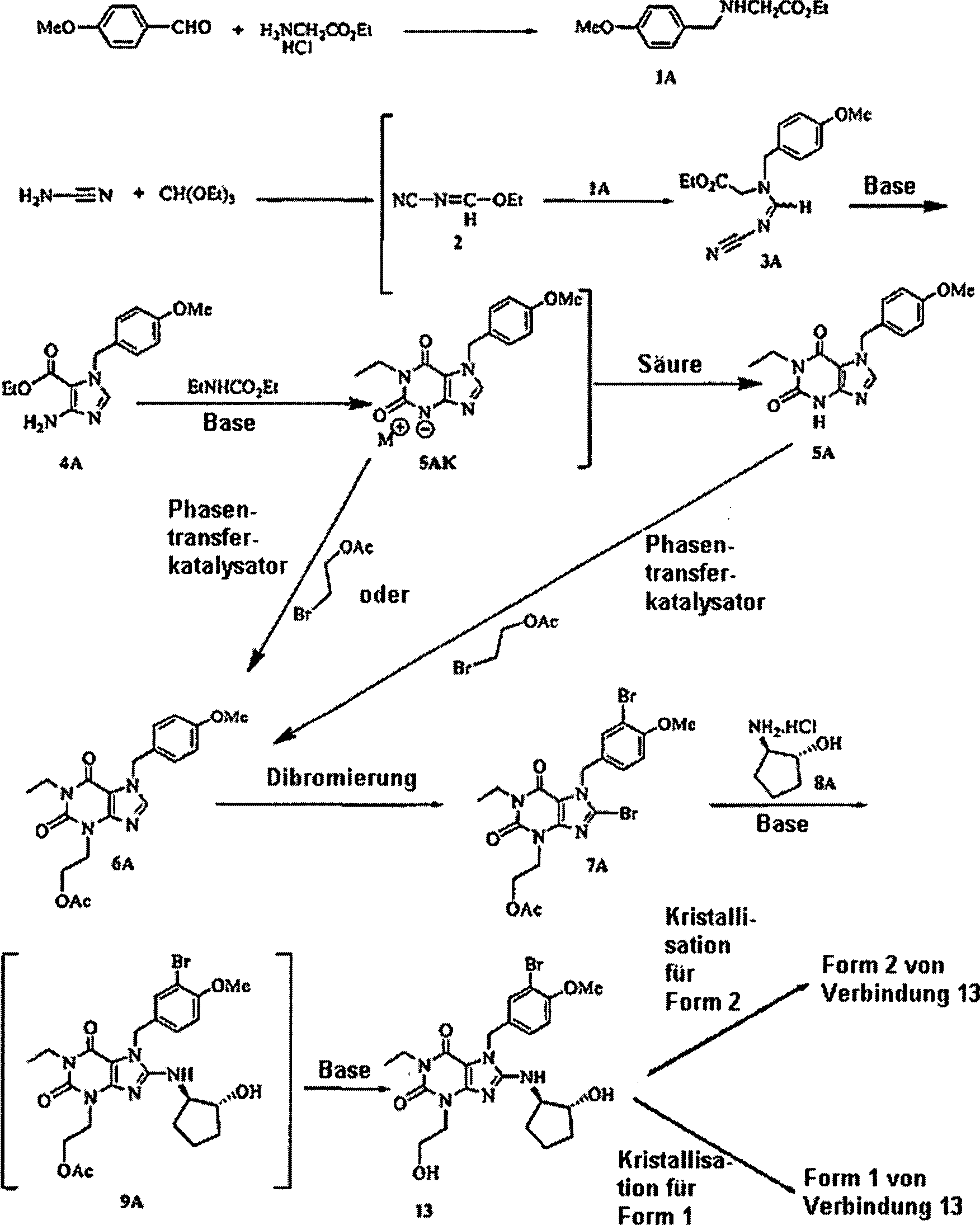
in Schema I gezeigt, das die folgenden Abkürzungen verwendet: Me ist Methyl;
Et ist Ethyl, OMe ist Methoxy, M
+ ist ein
Metallion und OAc ist Acetat: Schema
I: Allgemeine Synthese für
die Formen 1 und 2 von Verbindung 13
-
Die
Verwendung des in Schema I abgebildeten Verfahrens erzeugt eine
rohe Form 1 von Verbindung 13 vor der letzten Kristallisationsstufe.
Man kann in Abhängigkeit
von dem Kristal lisationslösungsmittel,
in dem die letzte Stufe durchgeführt
wird, reine Formen 1 oder 2 von Verbindung 13 herstellen.
-
Die
Kristallisation von irgendeiner Form von Verbindung 13 zu Form 2
von Verbindung 13 wird vorzugsweise in einem organischen Lösungsmittel
ausgewählt
aus der Gruppe bestehend aus Alkoholen (z. B. Methanol, Ethanol,
n-Propylalkohol, Isopropylalkohol, usw.), Nitrilen (z. B. Acetonitril,
Propionitril, Butyronitril, Valeronitril, Benzonitril, p-Tolunitril,
usw.), Estern (z. B. Methylacetat, Ethylacetat, n-Propylacetat,
Isopropylacetat, usw.), Ketonen (z. B. Methylisobutylketon, Aceton,
usw.) und Mischungen davon bewirkt. Höhere Homologe der beispielhaften
Alkohole, Nitrile, Ester und Ketone wandeln Verbindung 13 auch in
Form 2 von Verbindung 13 um. Zu bevorzugteren Lösungsmitteln gehören Isopropylalkohol,
Acetonitril und Mischungen davon. Die Kristallisationsstufe von
Form 2 wird in einer im Wesentlichen nicht-wässrigen Lösungsmittelmischung durchgeführt, die
für diese
Stufe eine Kristallisationslösungsmittelmischung
bedeutet, die weniger als oder gleich etwa 5 Gew.-%, vorzugsweise
weniger als oder gleich etwa 2 Gew.-% Wassergehalt bedeutet, bezogen auf
das Gewicht der Kristallisationslösungsmittelmischung.
-
Die
Kristallisation kann ohne Wärmezufuhr
erfolgen, es ist jedoch bevorzugt, dass sie durch Kühlen einer
erwärmten
gesättigten
Lösung
von Verbindung 13 initiiert wird, die in einem Kristallisationslösungsmittel gelöst ist.
Verbindung 13 wird im Allgemeinen in ein Kristallisationslösungsmittel
gegeben, und es wird Wärme zugeführt, bis
Verbindung 13 sich in der Lösung
löst. Die
zugeführte
Wärme kann
in Abhängigkeit
von den Verfahrensbedingungen und der Konzentration von Verbindung
13 in dem Kristallisationslösungsmittel
variieren (z. B. ausreichende Wärme,
um die Lösungsmitteltemperatur
auf etwa 30 bis 100°C
zu erhöhen).
Nachdem sich die Lösung
gebildet hat, wird die Wärmezufuhr
fortgesetzt, um die Lösung
zu konzentrieren (z. B. bis zu ihrem Übersättigungspunkt). Die konzentrierte
Lösung
wird dann abgekühlt,
um die gewünschten
Kristalle zu liefern.
-
Es
ist auch bevorzugt, beim Kühlen
einer übersättigten
Lösung
von Verbindung 13 in dem Kristallisationslösungsmittel für Form 2
mit Impfen zu arbeiten, um die Krustenbildung von Produkt an den
Reaktorwänden
(das Kleben von kristallisierten Teilchen an Reaktorwänden) zu
minimieren und/oder zu verhindern, welche schwierig zu entfernen
sein können.
Es ist bevorzugt, dass die Kristallisationslösung von Form 2 mit einer geringen
Menge (z. B. etwa 0,2% Gew./Gew. bis etwa 1% Gew./Gew.) von Form
2 von Verbindung 13 geimpft wird, um dazu beizutragen, die Umwandlung
in Form 2 zu erleichtern, die Ausbeute der Charge zu erhöhen und
die mögliche
Krustenbildung von Produkt an den Reaktorwänden zu vermeiden. Die Krustenbildung
von Produkt an Reaktorwänden
führt zu
Ausbeuteverlust und Einschluss von Lösungsmittel in die isolierte
kristallisierte Produktsubstanz. Das eingeschlossene Lösungsmittel
kann oft nicht auf ein bevorzugtes Level von etwa 0,1% Gew./Gew.
bis etwa 0,2% Gew./Gew. verringert werden, nicht einmal durch längeres Trocknen.
Das Impfen der Charge zu einem geeigneten Zeitpunkt während der
Kristallisation minimiert und/oder umgeht dieses Problem. Die Charge
wird vorzugsweise am oder um den Übersättigungspunkt herum geimpft;
bei Acetonitril als Kristallisationslösungsmittel liegt der Übersättigungspunkt
um eine Konzentration von etwa 7 Volumina bis etwa 8 Volumina Lösungsmittel
(1 g Feststoff auf etwa 1 ml bis etwa 8 ml Lösungsmittel).
-
Die
Kristallisation von Verbindung 13 zu Form 1 der Verbindung 13 wird
vorzugsweise erreicht, indem Verbindung 13 in einem organischen
Lösungsmittel
gelöst
wird und dann Wasser zugegeben wird. Bevorzugte organische Lösungsmittel
beinhalten beliebige der oben beschriebenen Kristallisationslösungsmittel
für Form 2
(d. h. Alkohole, Nitrile, Ester und Ketone). Besonders bevorzugte
organische Lösungsmittel
beinhalten Methanol und Isopropanol. Es ist wie bei den oben beschriebenen
Kristallisationen für
Form 2 bevorzugt, Verbindung 13 in einem organischen Kristallisationslösungsmittel
für Form
1 zu lösen,
indem die Mischung erwärmt wird,
bis sich Verbindung 13 in Lösung
löst, und
das Erwärmen
fortzusetzen, bis etwa der Übersättigungspunkt erreicht
ist. Dann wird Wasser zugefügt,
um die Kristalle der Form 1 von Verbindung 13 auszufällen.
-
Alternativ
kann Form 1 von Verbindung 13 erhalten werden, indem ein Antilösungsmittel
(statt Wasser) zu einer Lösung
von Verbindung 13 in einem Kristallisationslösungsmittel gegeben wird. Bevorzugte
Antilösungsmittel
sind Kohlenwasserstoffe, wie Hexan, Heptan, Toluol, Xylol und dergleichen.
Hexan kann beispielsweise zu einer Lösung von Verbindung 13 in einem
Esterlösungsmittel
gegeben werden (z. B. Ethylacetat, Isopropylacetat und dergleichen),
und Form 1 der Verbindung 13 fällt
aus. Die Antilösungsmitteltechnik
ist allgemein zum Isolieren der kinetischen Form 1 von Verbindung
13 bevorzugt. Es ist in Bezug auf die Technik von organischem Lösungsmittel,
gefolgt von Wasser, allgemein bevorzugt, die Kristallisationsbedingungen
zu kontrollieren, um die kinetische Form 1 von Verbindung 13 zu
isolieren. Dies kann bewirkt werden, indem das Produkt so rasch
wie möglich
(vorzugsweise sofort) filtriert wird, nachdem die Kristallisation
erfolgt ist.
-
Formen
1 und 2 von Verbindung 13 können
aus einer amorphen Form von Verbindung 13 oder aus einer anderen
Form von Verbindung 13 erhalten werden, indem das geeignete Kristallisationsverfahren
gewählt
wird. Form 2 von Verbindung 13 kann beispielsweise zu Form 1 von
Verbindung 13 kristallisiert werden, werden, indem die erstere Substanz
in einem organischen Lösungsmittel
gelöst
wird und der Lösung
Wasser zugefügt
wird, bis Form 1 von Verbindung 13 ausfällt. In ähnlicher Weise kann Form 2
von Verbindung 13 aus Form 1 von Verbindung 13 durch Kristallisation
aus Kristallisationslösungsmittel
für Form
2 von Verbindung 13 erhalten werden.
-
Wie
sich aus einem Vergleich der 1 und 2 mit 3 beziehungsweise
4 ergibt, zeigen Formen 1 und 2 andere DSC- und XRPD-Graphen. Die
beiden Polymorphe unterscheiden sich ferner auch in ihren Wasserlöslichkeiten
(Form 1: etwa 50 μg/ml
gegenüber
Form 2: etwa 30 μg/ml).
Form 2 von Verbindung 13 ist bei den Verarbeitungstemperaturen thermodynamisch
stabiler als Form 1 von Verbindung 13. Es kann sich aus Form 1 ein
Gleichgewicht zugunsten von Form 2 einstellen, wenn sie in einem
der Kristallisationslösungsmittel
für Form
2 suspendiert wird (z. B. Alkohol, Nitril, Ester, usw.). Wenn beispielsweise
eine Mischung aus Form 1 von Verbindung 13 und Form 2 von Verbindung
13 in einem organischen Kristallisationslösungsmittel (z. B. Ethylacetat,
Isopropanol, Acetonitril und dergleichen) suspendiert wird und einen
längeren
Zeitraum (z. B. mehr als oder gleich etwa 10 Stunden) gelagert wird,
wandelt sich die Form 1-Komponente der Mischung in Form 2 von Verbindung
13 um.
-
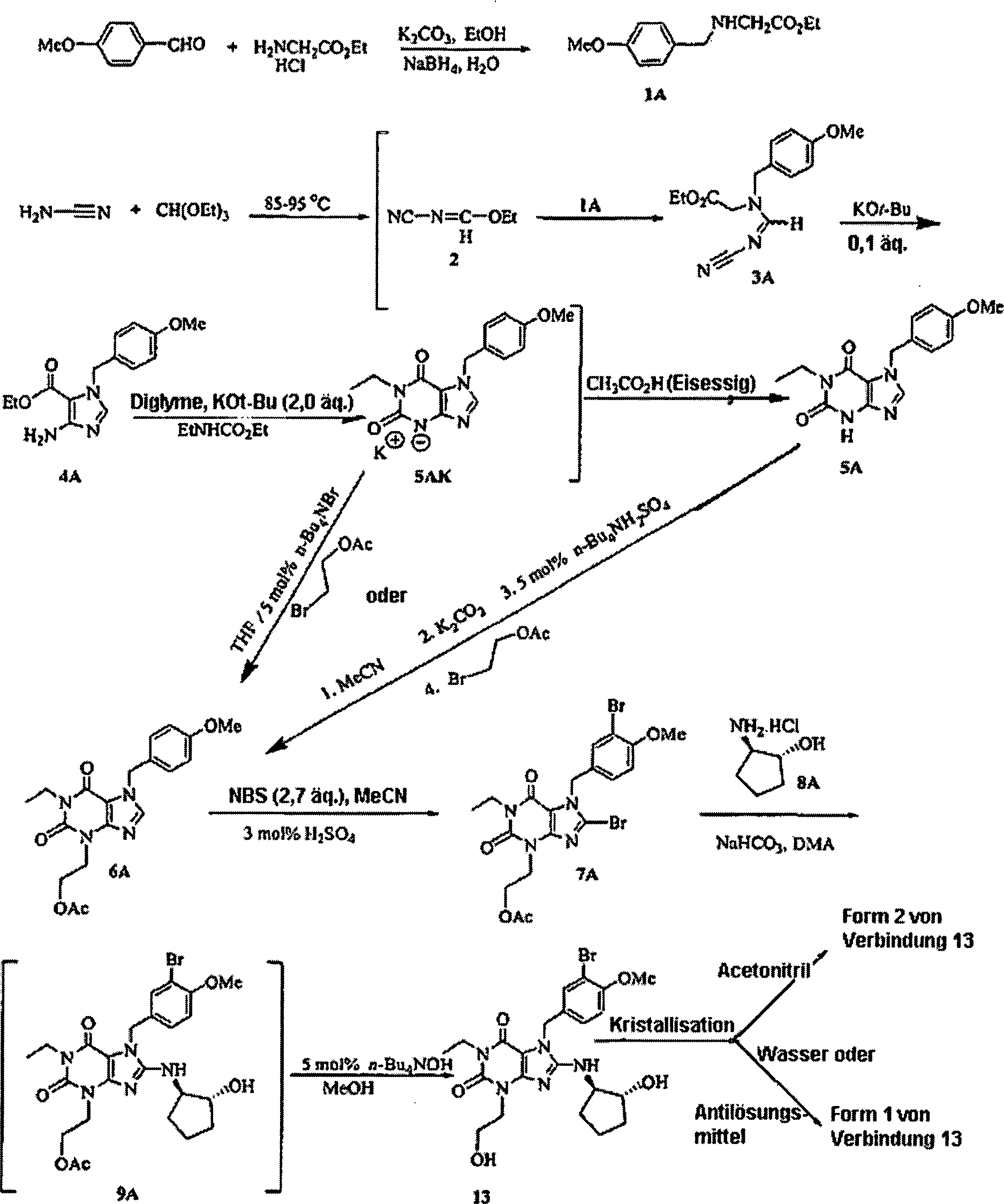
Schema
II zeigt bevorzugte Reaktionsbedingungen für die Stufen aus Schema I,
die zur Herstellung der Formen 1 und 2 von Verbindung 13 verwendet
werden. Schema II wird auch in einer gleichzeitig anhängigen US-Patentanmeldung
mit dem Titel Process for Preparing Xanthine Phosphodiesterase V
Inhibitors and Precursors Thereof gelehrt (Verbindung 13 wird in
der gleichzeitig anhängigen
Anmeldung als Verbindung 13A bezeichnet). Schema II ermöglicht eine
effiziente Herstellung der Formen 1 und 2 von Verbindung 13 im kommerziellen
Maßstab,
ohne dass die Zwischenprodukte chromatographisch gereinigt wer den
müssen.
Die hier offenbarten experimentellen Bedingungen sind bevorzugte
Bedingungen, und ein Durchschnittsfachmann kann sie nach Bedarf
modifizieren, um die gleichen Produkte zu erhalten. Die folgenden
Abkürzungen
werden in Schema II verwendet: EtOH ist Ethanol; Me ist Methyl;
Et ist Ethyl; Bu ist Butyl; n-Bu ist normal-Butyl, t-Bu ist tert.-Butyl,
OAc ist Acetat; KOt-Bu ist Kalium-tert.-butoxid; NBS ist N-Bromsuccinimid; NMP
ist 1-Methyl-2-pyrrolidinon; DMA ist N,N-Dimethylacetamid; n-Bu
4NBr
ist Tetrabutylammoniumbromid; n-Bu
4NOH ist Tetrabutylammoniumhydroxid und äq. ist Äquivalente. Schema
11: Spezifische Synthesen der Formen 1 und 2 von Verbindung 13
-
Aktivität der Verbindung, Pharmazeutische
Zusammensetzungen und Anwendungsmethoden
-
Die
Formen 1 und 2 von Verbindung 13 sind jeweils zum Inhibieren von
PDE V Isoenzymen brauchbar. Ihre Isoenzymaktivitäten (Potenzen) und Isoenzymselektivitäten können durch
den PDE V IC50-Wert gemessen werden, der
die Konzentration der Verbindung (in nM) ist, die erforderlich ist,
um 50% Inhibierung des PDE V-Isoenzyms zu liefern. Je niedriger
der Wert von PDE V IC50 ist, um so aktiver
ist die Verbindung zur Inhibierung des PDE V Isoenzyms. Ein IC50-Wert kann in ähnlicher Weise für andere
PDE-Isoenzyme erhalten werden, wie das PDE VI-Isoenzym. Die Isoenzymselektivität kann in
dieser Hinsicht als die Aktivität
einer PDE-Inhibitorverbindung für
ein spezielles PDE-Isoenzym
im Unterschied zu einem anderen PDE-Isoenzym definiert werden, beispielsweise
die Aktivität
einer Verbindung zur Inhibierung der PDE V Isoenzym, verglichen
mit der Aktivität
derselben Verbindung zur Inhibierung eines PDE VI-Isoenzyms. Nachdem
die PDE V-IC50- und PDE VI-IC50-Werte
gemessen wurden, kann man ein Selektionsverhältnis von PDE VI IC50/PDE V IC50 berechnen, das
ein Indikator für
die Isoenzymselektivität
ist – je
größer das
Selektionsverhältnis
ist, um so selektiver ist die Verbindung bei der Inhibierung des
PDE V-Isoenzyms relativ zu PDE VI-Isoenzym.
-
Formen
1 und 2 von Verbindung 13 haben jeweils eine PDE V IC50 zwischen
etwa 2 nM und etwa 3 nM. Diese Verbindungen sind relativ hochpotente
Inhibitoren des PDE V-Isoenzyms. Im Unterschied dazu haben Formen
1 und 2 von Verbindung 13 jeweils eine PDE VI IC50 von
mehr als etwa 350 nM, was bedeutet, dass sie eine relativ niedrige
Potenz zur Inhibierung des PDE VI Isoenzyms aufweisen. Die IC50-Daten für PDE V und VI ermöglichen
das Berechnen eines Indikators für
die Isoenzymselektivität – das Verhältnis von
PDE VI IC50/PDE V IC50 (hier
als "PDE VI/PDE
V" bezeichnet).
Je höher
das Verhältnis
von PDE VI/PDE V ist, um so selektiver ist die Verbindung zur Inhibierung
des PDE V-Isoenzyms
relativ zu PDE VI-Isoenzym. Die Formen 1 und 2 von Verbindung 13
haben jeweils ein PDE VI/PDE V-Verhältnis von mehr als etwa 140,
was bedeutet, dass sie jeweils eine relativ hohe Selektivität zur Inhibierung
des PDE V-Isoenzyms (relativ zu dem PDE VI-Isoenzym) aufweisen.
-
Wie
aus diesen Daten hervorgeht, sind die Formen 1 und 2 von Verbindung
13 potente (gemessen durch PDE V IC50) und
selektive (gemessen durch PDE VI IC50/PDE
V IC50) PDE V-Isoenzym-Inhibitoren. Ein Fachmann würde die
biologischen Daten als signifikant ansehen und zusammen mit den
pharmazeutischen Eigenschaften der Zusammensetzungen, welche die
erfindungsgemäßen Verbindungen
enthalten, therapeutische Anwendungen für die erfindungsgemäßen Verbindungen
in zahlreichen Anwendungen finden, von denen hier einige spezifiziert
werden.
-
Die
Formen 1 und 2 von Verbindung 13 haben jeweils mindestens ein asymmetrisches
Kohlenstoffatom. Alle Isomere einschließlich Stereoisomeren, Enantiomeren,
Tautomeren und Rotationsisomeren werden hier als Teil der Erfindung
angesehen. Die Erfindung beinhaltet d- und 1-Isomere sowohl in reiner
Form als auch gemischt einschließlich racemischer Mischungen.
Isomere können
unter Verwendung konventioneller Techniken hergestellt werden, entweder
indem optisch reine oder optisch angereicherte Ausgangsmaterialien umgesetzt
werden oder indem Isomere der erfindungsgemäßen Verbindungen getrennt werden.
-
Die
Formen 1 und 2 von Verbindung 13 können in unsolvatisierten sowie
solvatisierten Formen einschließlich
hydratisierten Formen vorliegen. Im Allgemeinen sind die solvatisierten
Formen mit pharmazeutisch annehmbaren Lösungsmitteln, wie Wasser, Ethanol
und dergleichen, für
erfindungsgemäße Zwecke
den unsolvatisierten Formen äquivalent.
-
Die
Erfindung beinhaltet Formen 1 und/oder 2 von Verbindung 13, ein
Verfahren zur Herstellung jeder erfindungsgemäßen Verbindung und Verfahren
zur Herstellung und Verwendung einer pharmazeutischen Zusammensetzung,
die mindestens eine erfindungsgemäße Verbindung und mindestens
einen pharmazeutisch annehmbaren Hilfsstoff oder Träger enthält, um eine
Vielfalt von Störungen,
Symptomen und Erkrankungen zu behandeln. Die erfindungsgemäßen Verbindungen
zeigen unerwartet vorteilhafte Eigenschaften in Bezug auf PDE V-Isoenzymaktivität und -selektivität, wodurch
sie zur Behandlung von Urogenitalerkrankungen besonders nützlich sind,
wie der männlichen
und weiblichen sexuellen Dysfunktion, z. B. der erektilen Dysfunktion.
-
Formen
1 und 2 von Verbindung 13 können
zusammen mit einem pharmazeutisch annehmbaren Hilfsstoff oder Träger formuliert
werden. Die resultierenden Zusammensetzungen können in vivo an Säuger (z.
B. Männer
oder Frauen) und Nicht-Säuger
verabreicht werden, um eine Vielfalt von Erkrankungszuständen (Störungen,
Symptome und Erkrankungen) zu behandeln. Die erfindungsgemäßen Verbindungen
und Zusammensetzungen können
beispielsweise zur Behandlung von Erkrankungen des Urogenitalsystems
verwendet werden, insbesondere der erektilen Dysfunktion des Mannes
(z. B. Impotenz) und der weiblichen sexuellen Dysfunktion. Die männliche
erektile Dysfunktion kann definiert werden als Unfähigkeit
des Mannes, eine Peniserektion zu erhalten, zu erreichen und/oder
aufrechtzuerhalten, die ausreicht, um den Verkehr mit seinem Partner/seiner
Partnerin durchzuführen.
Es wird angenommen, dass die erfindungsgemäßen PDE V-Inhibitoren bei der
Behandlung der erektilen Dysfunktion vorteilhafte therapeutische
Mittel sind, da sie die cGMP-Level im menschlichen Körper erhöhen. Eine
derartige Wirkung kann die Relaxation des glatten Muskels des Corpus cavernosum
erleichtern, was zu einem erhöhten
Blutfluss darin führen
würde,
der zu einer Erek tion führt.
Hierdurch werden die erfindungsgemäßen Verbindungen zur Behandlung
von Impotenz und anderen Erkrankungstypen besonders brauchbar, die
durch cGMP-Level beeinflusst werden.
-
Ein
weiterer Aspekt der Erfindung ist daher ein Verfahren zur Behandlung
der erektilen Dysfunktion bei einem Säuger, der dieser Behandlung
bedarf, wobei dem dem Säuger
mindestens eine Form 1 von Verbindung 13 und/oder mindestens eine
Form 2 von Verbindung 13 oder eine pharmazeutische Zusammensetzung
davon in einer wirksamen Menge verabreicht wird, um ein oder mehrere
der mit der erektilen Dysfunktion verbundenen Symptome in ausreichendem
Maße zu
lindern und/oder zu reduzieren, so dass der Patient den Geschlechtsverkehr
durchführen
und vollziehen kann.
-
Viagra®,
das 1998 zur Behandlung der Impotenz eingeführt wurde, ist momentan das
am häufigsten zur
Behandlung der physiologisch verursachten (männlichen) erektilen Dysfunktion
("MED" oder "ED") verschriebene Medikament.
Bestimmte Patienten können
jedoch bei der Einnahme von Viagra® unerwünschte Nebenwirkungen
erfahren. Es ist beispielsweise berichtet worden, dass Viagra® eine
Nebenwirkungen auf das Sehvermögen
haben kann, indem die Farbunterscheidung (blau/grün) des Patienten
beeinträchtigt
wird, was zu einer Visusänderung
des "blauen Halos" des Lichts führt. Diese
Nebenwirkung ist vermutlich auf die Inhibierung des PDE VI-Isoenzyms
(das sich in der Retina befindet) zurückzuführen. Siehe Physicians' Desk Reference®,
55. Auflage, Seiten 2534–37
(2001).
-
Ein
Vorteil der Formen 1 und 2 von Verbindung 13 liegt darin, dass sie
verglichen mit anderen Typen von PDE-Isoenzymen, wie dem PDE VI-Isoenzym,
besonders selektiv für
das PDE V-Isoenzym sein können. Es
wird angenommen, dass diese erhöhte
Selektivität
die mit der Verwendung von Viagra® zusammenhängenden
Nebenwirkungen lindern wird. Die hohe Selektivi tät der erfindungsgemäßen Verbindungen
sollte insbesondere das Auftreten der Visusänderung des "blauen Halos" des Lichts minimieren
und möglicherweise
sogar verhindern. Es wird angenommen, dass die erhöhte Isoenzymselektivität bei der
Inhibierung des PDE V-Isoenzyms (das sich in einem Penis befindet)
gegenüber
dem PDE VI-Isoenzym (das sich in einer Retina befindet) dazu führt, dass
die Visusnebenwirkung des "blauen
Halos" vermieden
wird.
-
Die
Formen 1 und 2 von Verbindung 13 können allein oder in Kombination
mit anderen Wirkstoffen verwendet werden, insbesondere anderen Typen
von PDE-Inhibitoren (insbesondere cGMP PDE V-Inhibitoren), Prostanoiden, α-adrenergen
Rezeptoragonisten, Dopaminrezeptoragonisten, Melanocortinrezeptoragonisten,
Endothelinrezeptorantagonisten, Endothelin-Konversionsenzym-Inhibitoren, Angiotensin
II-Rezeptorantagonisten, Angiotensin-Konversionsenzym-Inhibitoren, Neutralmetalloendopeptidase-Inhibitoren, Renininhibitoren,
Serotonin 5-HT2c-Rezeptoragonisten, Nociceptinrezeptoragonisten,
Rho-Kinaseinhibitoren, Kaliumkanalmodulatoren und Mehrfachwirkstoffresistenz-Protein
5-Inhibitoren. Beispiele für
therapeutische Mittel, die in Kombination mit den Formen 1 und 2
von Verbindung 13 verwendet werden, sind die Folgenden: andere Typen
von PDE V-Inhibitoren,
wie Sildenafilcitrat (Viagra®, Pfizer, Connecticut,
USA), VardenafilTM (Bayer, Deutschland)
und IC-351 (CialisTM, Lilly-ICOS, Washington
und Indiana, USA); Prostanoide wie Prostaglandin E1; α-adrenerge
Agonisten, wie Phentolaminmesylat; Dopaminrezeptoragonisten, wie
Apomorphin; Angiotensin II-Antagonisten, wie Losartan, Irbesartan,
Valsartan und Candesartan, und ETA-Antagonisten,
wie Bosentan und ABT-627.
-
Es
sei darauf hingewiesen, dass andere Kombinationen als die oben beschriebenen
durch einen Fachmann mit Routineexperimenten zusammengestellt werden
können,
um Erkrankungszustände bei
Säugern
zu behandeln, während
man in dem Umfang der Erfindung bleibt. Obwohl die Formen 1 und
2 von Verbindung 13 jeweils zur Anwendung einer Monotherapie eines
Patienten verwendet werden können,
können
sie auch in einer Kombinationstherapie verwendet werden, bei der
eine oder beide hiervon in Kombination mit einer oder mehreren anderen
pharmazeutischen Verbindungen (entweder getrennt oder physikalisch
zu einer Einzelform kombiniert) verabreicht wird. Die Kombinationstherapie
ist zur Behandlung einer Vielfalt von Störungen, Symptomen und Erkrankungen
geeignet, wie einem oder mehreren der oben beschriebenen Erkrankungszuständen bei
Säugern.
-
Die
Formen 1 und 2 von Verbindung 13 sind wegen ihrer cGMP-PDE V-inhibierenden
Wirkungen (wie bereits erörtert)
zur Behandlung von urologischen Störungen brauchbar, insbesondere
sexuellen Dysfunktionen von Mann und Frau. Andere physiologische
Störungen,
Symptome und Erkrankungen können
auch von der cGMP-PDE V-Inhibierung profitieren. Die erfindungsgemäßen Verbindungen
und pharmazeutischen Zusammensetzungen daraus können spezieller zur Behandlung
von kardiovaskulären
und zerebrovaskulären
Erkrankungen, Angina pectoris, Hypertonie, Restenose nach Angioplastie,
Endarteriektomie, Stenteinsetzung, peripher vaskulären Erkrankungen,
Hirnschlag, die Atemwege betreffenden Störungen, wie reversibler Obstruktion
der Luftwege, chronischem Asthma und Bronchitis, allergischen Störungen,
die mit Atopie assoziiert sind, wie Urtikaria, Ekzem und Rhinitis,
pulmonaler Hypertonie, ischämischer
Herzkrankheit, gestörter
Glucosetoleranz, Diabetes und ihre einhergehenden Komplikationen,
Insulinresistenzsyndrom, Hyperglykämie, polyzystischem Ovarialsyndrom,
glomerulären
Erkrankungen, Niereninsuffizienz, Nephritis, tubulär interstitieller Erkrankung,
autoimmunologischen Erkrankungen, Glaukom, Darmmotilitätsstörungen,
Kachexie und Krebs verwendet werden.
-
Ein
weiterer Aspekt der Erfindung ist die Bereitstellung eines Kits,
der separate Behälter
in einer einzigen Packung aufweist, wobei die erfindungsgemäßen pharmazeutischen
Verbindungen und/oder Zusammensetzungen in Kombination mit pharmazeutisch
annehmbaren Hilfsstoffen oder Trägern
zur Behandlung einer Vielfalt von physiologischen Störungen und
Erkrankungen verwendet werden, bei denen die cGMP-PDE V-Inhibierung
eine Rolle spielt.
-
Pharmazeutisch annehmbare
Dosierformen
-
Formen
1 und 2 von Verbindung 13 können
Menschen oder anderen Säugern
auf vielen Wegen verabreicht werden, einschließlich oraler Dosierformen und
Injektionen (intravenös,
intramuskulär,
intraperitoneal, subkutan und dergleichen). Zahlreiche andere Dosierformen,
die die erfindungsgemäßen Verbindungen
enthalten, können
durch den Fachmann unter Verwendung der geeigneten pharmazeutischen
Hilfsstoffe und Träger,
wie nachfolgend definiert, leicht formuliert werden. Unter Berücksichtigung
der Compliance des Patienten sind allgemein die oralen Dosierformen
am meisten bevorzugt.
-
Die
Rate der systemischen Abgabe kann durch den Fachmann in befriedigender
Weise kontrolliert werden, indem irgendeine oder mehrere der Folgenden
manipuliert werden:
- (a) der spezielle Wirkstoff/die
speziellen Wirkstoffe;
- (b) der pharmazeutisch annehmbare Hilfsstoff oder Träger bzw.
die pharmazeutisch annehmbaren Hilfsstoffe oder Träger, solange
die Varianten die Aktivität
des speziellen gewählten
Wirkstoffs/der speziellen gewählten
Wirkstoffe nicht stören;
- (c) der Typ des Hilfsstoffs/der Hilfsstoffe oder des Trägers/der
Träger
und die damit verbundene erwünschte Dicke
und Permeabilität
(Quelleigenschaften) des Hilfsstoffs/der Hilfsstoffe oder des Trägers/der
Träger;
- (d) die zeitabhängigen
Bedingungen des Hilfsstoffs/der Hilfsstoffe oder des Trägers/der
Träger;
- (e) die Teilchengröße des Wirkstoffs
und
- (f) die pH-abhängigen
Bedingungen des Hilfsstoffs/der Hilfsstoffe oder des Trägers/der
Träger.
-
Pharmazeutisch
annehmbare Hilfsstoffe oder Träger
beinhalten Aromatisierungsmittel, Farbstoffe oder Pigmente von pharmazeutischer
Qualität,
Lösungsmittel,
Colösungsmittel,
Puffersysteme, Tenside, Konservierungsmittel, Süßungsmittel, Viskositätsmittel,
Füllstoffe,
Schmiermittel, Gleitmittel, Sprengmittel, Bindemittel und Harze.
-
Es
können
konventionelle Aromatisierungsmittel verwendet werden, wie jene,
die in Remington's Pharmaceutical
Sciences, 18. Auflage, Mack Publishing Co., 1288–1300 (1990), beschrieben sind,
worauf hier Bezug genommen wird. Die erfindungsgemäßen pharmazeutischen
Zusammensetzungen enthalten allgemein 0% bis etwa 2% Aromatisierungsmittel.
-
Es
können
auch konventionelle Farbstoffe und/oder Pigmente verwendet werden,
wie jene, die im Handbook of Pharmaceutical Excipients, von der
American Pharmaceutical Association & the Pharmaceutical Society of Great
Britain, 81–90
(1986), beschrieben sind, worauf hier Bezug genommen wird. Die erfindungsgemäßen pharmazeutischen
Zusammensetzungen enthalten allgemein 0% bis etwa 2% Farbstoff(e)
und/oder Pigment(e).
-
Die
erfindungsgemäßen pharmazeutischen
Zusammensetzungen enthalten allgemein etwa 0,1% bis etwa 99,9% Lösungsmittel.
Ein bevorzugtes Lösungsmittel
ist Wasser. Zu bevorzugten Colösungsmitteln
gehören
Ethanol, Glycerin, Propylenglykol, Polyethylenglykol und dergleichen.
Die erfindungsgemäßen pharmazeutischen
Zusammensetzungen können
0% bis etwa 50% Colösungsmittel
enthalten.
-
Zu
bevorzugten Puffersystemen gehören
Essig-, Bor-, Kohlen-, Phosphor-, Bernstein-, Äpfel-, Wein-, Zitronen-, Essig-,
Benzoe-, Milch-, Glycerin-, Glucon-, Glutar- und Glutaminsäuren und
ihre Natrium-, Kalium- und Ammoniumsalze. Besonders bevorzugte Puffer
sind Phosphor-, Wein-, Zitronen- und Essigsäuren und ihre Salze. Die erfindungsgemäßen pharmazeutischen
Zusammensetzungen enthalten allgemein 0 bis etwa 5% Puffer.
-
Bevorzugte
Tenside beinhalten Polyoxyethylensorbitan-Fettsäureester, Polyoxyethylenmonoalkylether,
Sucrosemonoester und Lanolinester und -ether, Alkylsulfatsalze und
Natrium-, Kalium- und Ammoniumsalze von Fettsäuren. Die erfindungsgemäßen pharmazeutischen
Zusammensetzungen enthalten allgemein 0 bis etwa 2% Tensid(e).
-
Zu
bevorzugten Konservierungsmitteln gehören Phenol, Alkylester von
para-Hydroxybenzoesäure, o-Phenylphenolbenzoesäure und
deren Salze, Borsäure
und deren Salze, Sorbinsäure
und deren Salze, Chlorbutanol, Benzylalkohol, Thimerosal, Phenylquecksilber(II)acetat
und -nitrat, Nitromersol, Benzalkoniumchlorid, Cetylpyridiniumchlorid,
Methylparaben und Propylparaben. Besonders bevorzugte Konservierungsmittel
sind die Salze von Benzoesäure,
Cetylpyridiniumchlorid, Methylparaben und Propylparaben. Die erfindungsgemäßen pharmazeutischen
Zusammensetzungen enthalten allgemein 0 bis etwa 2% Konservierungsmittel.
-
Bevorzugte
Süßungsmittel
sind Sucrose, Glucose, Saccharin, Sorbit, Mannit und Aspartame.
Besonders bevorzugte Süßungsmittel
sind Sucrose und Saccharin. Die erfindungsgemäßen pharmazeutischen Zusammensetzungen
enthalten allgemein 0 bis etwa 5% Süßungsmittel.
-
Bevorzugte
Viskositätsmittel
sind Methylcellulose, Natrium-Carboxymethylcellulose, Hydroxypropylmethylcellulose,
Hydroxypropylcellulose, Natriumalginat, Carbomer, Povidon, Aka ziengummi,
Guar-Gummi, Xanthan-Gummi und Tragakanth. Besonders bevorzugte Viskositätsmittel
sind Methylcellulose, Carbomer, Xanthan-Gummi, Guar-Gummi, Povidon,
Natrium-Carboxymethylcellulose und Magnesiumaluminiumsilikat. Die
erfindungsgemäßen pharmazeutischen
Zusammensetzungen enthalten allgemein 0% bis etwa 5% Viskositätsmittel.
-
Zu
bevorzugten Füllstoffen
gehören
Lactose, Mannit, Sorbit, Tricalciumphosphat, Dicalciumphosphat, komprimierbarer
Zucker, Stärke,
Calciumsulfat, Dextro- und mikrokristalline Cellulose. Die erfindungsgemäßen pharmazeutischen
Zusammensetzungen enthalten allgemein 0% bis etwa 75% Füllstoff(e).
-
Bevorzugte
Schmiermittel/Gleitmittel sind Magnesiumstearat, Stearinsäure und
Talkum. Die bevorzugten erfindungsgemäßen pharmazeutischen Zusammensetzungen
enthalten 0% bis 7%, vorzugsweise etwa 1% bis etwa 5% Schmiermittel/Gleitmittel.
-
Bevorzugte
Sprengmittel sind Stärke,
Natrium-Stärkeglykolat,
Crospovidon und Croscarmelose-Natrium und mikrokristalline Cellulose.
Die bevorzugten erfindungsgemäßen pharmazeutischen
Zusammensetzungen enthalten 0% bis etwa 20%, vorzugsweise etwa 4%
bis etwa 15% Sprengmittel.
-
Zu
bevorzugten Bindemitteln gehören
Akaziengummi, Tragakanth, Hydroxyropylcellulose, vorgelatinisierte
Stärke,
Gelatine, Povidon, Hydroxypropylcellulose, Hydroxypropylmethylcellulose,
Methylcellulose, Zuckerlösungen,
wie Sucrose und Sorbitol, und Ethylcellulose. Die bevorzugten erfindungsgemäßen pharmazeutischen
Zusammensetzungen enthalten allgemein 0% bis etwa 12%, vorzugsweise
etwa 1% bis etwa 10% Bindemittel.
-
Zusätzliche
Mittel, die dem versierten Formulierungshersteller bekannt sind,
können
mit den erfindungsgemäßen Verbindungen
kombiniert werden, um eine Einzeldosierform zu erzeu gen. Alternativ
können zusätzliche
Mittel einem Säuger
separat als Teil einer Mehrfachdosierform verabreicht werden.
-
Eine
pharmazeutische Zusammensetzung enthält in der Regel etwa 0,1% bis
etwa 99,9% (bezogen auf das Gewicht oder Volumen, vorzugsweise Gew./Gew.)
des Wirkstoffs (Formen 1 und/oder 2 von Verbindung 13), vorzugsweise
etwa 5% bis etwa 95%, insbesondere etwa 20% bis etwa 80%. Inerte,
pharmazeutisch annehmbare Hilfsstoffe oder Träger zur Herstellung pharmazeutischer
Zusammensetzungen, die die erfindungsgemäßen Verbindungen enthalten,
können
fest oder flüssig
sein. Zubereitungen in fester Form schließen Pulver, Tabletten, dispergierbare
Körner,
Kapseln, Oblatenkapseln und Zäpfchen
ein. Geeignete feste Hilfsstoffe oder Träger sind in der Technik bekannt,
z. B. Magnesiumcarbonat, Magnesiumstearat, Talkum, Zucker und Lactose.
Tabletten, Pulver, Kapseln und Oblatenkapseln können als feste Dosierungsformen
verwendet werden, die für
die orale Verabreichung geeignet sind. Beispiele für pharmazeutisch
annehmbare Hilfsstoffe oder Träger
und Fertigungsverfahren für
verschiedene Zusammensetzungen finden sich in Remington's Pharmaceutical
Sciences, 18. Auflage, Mack Publishing Co. (1990), worauf hier in
vollem Umfang Bezug genommen wird.
-
Zubereitungen
in flüssiger
Form schließen
Lösungen,
Suspensionen und Emulsionen ein. Übliche Zubereitungen in flüssiger Form
beinhalten Wasser oder Wasser-Propylenglykol-Lösungen für die parenterale Injektion
oder unter Zugabe von Süßungsmitteln
und Opazifizierungsmitteln für
orale Lösungen,
Suspensionen und Emulsionen. Zubereitungen in flüssiger Form können auch
Lösungen
für intranasale
Verabreichung einschließen.
-
Aerosolzubereitungen,
die zur Inhalation geeignet sind, können Lösungen und Feststoffe in Pulverform
einschließen,
die in Kombination mit einem pharmazeutisch annehmbaren Hilfsstoff oder
Träger
wie inertem komprimiertem Gas, z. B. Stickstoff, vorliegen können.
-
Ebenfalls
eingeschlossen sind Zubereitungen in fester Form, die kurz vor Gebrauch
in Zubereitungen in flüssiger
Form für
orale oder parenterale Verabreichungen überführt werden sollen. Solche flüssigen Formen schließen Lösungen,
Suspensionen und Emulsionen ein.
-
Die
erfindungsgemäßen Verbindungen
können
auch transdermal verabreicht werden. Die transdermalen Zusammensetzungen
können
die Form von Cremes, Lotionen, Aerosolen und/oder Emulsionen annehmen,
und können
in ein Transdermalpflaster vom Matrix- oder Reservoirtyp eingeschlossen werden,
wie in der Technik zu diesem Zweck konventionell ist.
-
Der
bevorzugte Verabreichungsmodus der erfindungsgemäßen Verbindungen ist oral.
Die pharmazeutische Zubereitung liegt vorzugsweise in Einheitsdosisform
vor. In einer solchen Form wird die Zubereitung in geeignet bemessene
Einheitsdosen unterteilt, die geeignete Mengen der aktiven Komponente
enthalten, z. B. eine wirksame Menge, um den gewünschten Zweck zu erreichen.
-
Die
Menge an Wirkstoff (Formen 1 und/oder 2 von Verbindung 13) kann
in einer Einzeldosis der Zubereitung gemäß einer speziellen Anwendung
von etwa 0,01 mg bis etwa 4000 mg, vorzugsweise etwa 0,02 mg bis
etwa 2000 mg, insbesondere etwa 0,03 mg bis etwa 1000 mg, bevorzugter
etwa 0,04 mg bis etwa 500 mg und am meisten bevorzugt etwa 0,05
mg bis etwa 250 mg variiert oder eingestellt werden. Ein typisches empfohlenes
Tagesdosierschema für
die orale Verabreichung kann im Bereich von etwa 0,02 mg/kg/Tag
bis etwa 2000 mg/kg/Tag in zwei bis vier unterteilten Dosen liegen.
Der Bequemlichkeit halber kann die gesamte Tagesdosis unterteilt
und nach Bedarf portionsweise über
den Tag verabreicht werden. Die erfindungsgemäßen pharma zeutischen Zusammensetzungen
können
typischerweise etwa 1 bis etwa 5 Mal pro Tag oder alternativ als
kontinuierliche Infusion verabreicht werden Diese Verabreichung
kann als chronische oder akute Therapie verwendet werden. Die Menge
an Wirkstoff, die mit den Hilfsstoff- oder Trägermaterialien zur Herstellung einer
Einzeldosierform kombiniert werden kann, variiert in Abhängigkeit
von dem behandelten Wirt und dem speziellen Verabreichungsmodus.
Eine typische Zubereitung enthält,
wie bereits offenbart, etwa 0,1% bis etwa 99,9% aktive Verbindung,
vorzugsweise etwa 5% bis etwa 95%, insbesondere etwa 20% bis etwa
80%. Die tatsächlich
verwendete Dosis kann gemäß den Erfordernissen
des Patienten und dem Schweregrad des behandelten Zustands variiert
werden. Die Bestimmung des richtigen Dosierschemas für eine spezielle
Situation liegt innerhalb des Wissens des Fachmanns. Der Bequemlichkeit
halber kann die gesamte Tagesdosis unterteilt und nach Bedarf portionsweise über den
Tag verabreicht werden.
-
Die
pharmazeutisch annehmbaren Hilfsstoffe oder Träger, die zusammen mit den erfindungsgemäßen Verbindungen
verwendet werden, werden in einer ausreichenden Konzentration verwendet,
um eine praktische Größe-zu-Dosis-Beziehung
zu liefern. Die pharmazeutisch annehmbaren Hilfsstoffe oder Träger können insgesamt
etwa 0,1% bis etwa 99,9% (bezogen auf das Gewicht oder Volumen,
vorzugsweise Gew./Gew.) der erfindungsgemäßen pharmazeutischen Zusammensetzungen
ausmachen, vorzugsweise etwa 5 Gew.-% bis etwa 95 Gew.-%, insbesondere
etwa 20 Gew.-% bis etwa 80 Gew.-%.
-
Es
kann bei Besserung des Zustands des Patienten, wenn gewünscht oder
erforderlich, eine Erhaltdosis einer Verbindung, Zusammensetzung
oder Kombination der Erfindung verabreicht werden. Die Dosis oder
Frequenz der Verabreichung oder beide können danach in Abhängigkeit
von den Symptomen auf ein Niveau reduziert werden, bei der der gebesserte
Zustand erhalten bleibt. Wenn die Symptome auf das gewünschte Niveau
gelindert worden sind, sollte die Behandlung enden. Die Patienten
können
bei jedem Wiederauftreten der Krankheitssymptome jedoch auf Langzeitbasis
intermittierende Behandlung benötigen.
-
Die
spezifischen Dosier- und Behandlungsschemata für jeden speziellen Patienten
können
variiert werden und hängen
von vielen Faktoren ab, zu denen die Aktivität der speziellen verwendeten
Verbindung, das Alter, Körpergewicht,
der allgemeine Gesundheitsstatus, das Geschlecht und die Ernährung des
Patienten, die Verabreichungszeit, die Ausscheidungsgeschwindigkeit,
die spezifische Wirkstoffkombination, die Schwere und der Verlauf
der behandelten Symptome, die Disposition des Patienten für den zu
behandelnden Zustand und die Beurteilung durch den behandelnden
Arzt gehören.
Die Bestimmung des richtigen Dosierschemas für eine spezielle Situation
liegt innerhalb des Wissens des Fachmanns. Die Menge und Frequenz der
Verabreichung der Formen 1 und/oder 2 von Verbindung 13 oder ihrer
pharmazeutischen Zusammensetzungen können gemäß dem Urteil des behandelnden
Arztes bezogen auf die oben genannten Faktoren reguliert werden.
Wie ein Fachmann erkennen wird, können niedrigere oder höhere Dosen
als die gerade genannten erforderlich sein.
-
Es
ist beispielsweise oft der Fall, dass sich ein richtiges Dosierniveau
auf das Gewicht des Patienten bezieht. Beispielsweise sind bei der
erfindungsgemäßen Verbindung
bzw. den erfindungsgemäßen Verbindungen
und den hier beschriebenen Zusammensetzungen Dosierniveaus zwischen
etwa 0,01 mg/kg und etwa 100 mg/kg Körpergewicht pro Tag, vorzugsweise
zwischen etwa 0,5 mg/kg und etwa 75 mg/kg Körpergewicht pro Tag und insbesondere
zwischen etwa 1 mg/kg und etwa 50 mg/kg Körpergewicht pro Tag zur Behandlung einer
Vielfalt von biologischen Störun gen,
insbesondere der sexuelle Dysfunktion von Mann und Frau, therapeutisch
nützlich.
Bei zwei unterschiedlich schweren Patienten wird für den schwereren
Patienten eine höhere Dosierung
verwendet, wobei alles andere gleich ist.
-
Formen
1 und/oder 2 von Verbindung 13 sollen eine wirksame Behandlung der
(männlichen)
erektilen Dysfunktion liefern, einschließlich einer vernünftigen
Zeit bis zum Eintreten der Wirkung nach der Verabreichung und einer
vernünftigen
Dauer nach der Verabreichung. Bei der Behandlung der erektilen Dysfunktion kann
beispielsweise etwa eine Stunde vor dem vorgesehenen Geschlechtsverkehr
eine Dosis der erfindungsgemäßen Verbindung(en)
genommen werden. Spezielle Dosen wirken innerhalb von etwa dreißig Minuten nach
ihrer Verabreichung. Ideale Dosen beeinflussen einen Patienten innerhalb
von etwa fünfzehn
Minuten nach ihrer Verabreichung. Während Nahrungsmittel, Diät, vorbestehende
Zustände,
Alkohol und andere systemische Zustände die Zeitverzögerung in
die Länge
ziehen können,
die das erfindungsgemäße Arzneimittel nach
seiner Verabreichung zum Wirken braucht, sei darauf hingewiesen,
dass optimale Dosen in Kombination mit sexueller Stimulation innerhalb
einer vernünftigen
Zeitspanne und für
eine vernünftige
Zeit zu einer wirksamen Arzneimittelbehandlung führen werden.
-
Reinheit des Polymorphen
-
Die
erfindungsgemäßen Polymorphe
von Verbindung 13, Formen 1 und 2, sind jeweils im Wesentlichen
frei von chemischen Verunreinigungen (z. B. Nebenprodukten, die
während
der Herstellung der Formen 1 oder 2 von Verbindung 13 erzeugt wurden). "Im wesentlichen frei" von chemischen Verunreinigungen
bedeutet für
erfindungsgemäße Zwecke
weniger als oder gleich etwa 5% Gew./Gew. chemische Verunreinigungen, vorzugsweise
weniger als oder gleich etwa 3% Gew./Gew. chemische Verunreinigungen,
insbesondere weniger als oder gleich etwa 2% Gew./Gew. chemi sche
Verunreinigungen und bevorzugter weniger als oder gleich etwa 1
Gew./Gew. chemische Verunreinigungen.
-
Die
erfindungsgemäßen Polymorphe
von Verbindung 13 sind vorzugsweise im Wesentlichen frei von anderen
Formen von Verbindung 13. "Im
Wesentlichen frei" von
anderen Formen von Verbindung 13 bedeutet für erfindungsgemäße Zwecke
weniger als oder gleich etwa 15 (Gew./Gew.) der anderen Formen von
Verbindung 13, vorzugsweise weniger als oder gleich etwa 10% Gew./Gew.
von anderen Formen von Verbindung 13, insbesondere weniger als oder
gleich etwa 5 Gew./Gew. von anderen Formen von Verbindung 13, und
besonders bevorzugt weniger als oder gleich etwa 2 Gew./Gew. von
anderen Formen von Verbindung 13.
-
Herstellung von Verbindung 13 in Form
1 und Form 2
-
Präparation
1: Form 1 von Verbindung 13
-
Etwa
1 g von Verbindung 13 (in beliebiger Form, sowohl kristallin als
auch nicht-kristallin) wurden in Lösung gelöst, indem es in etwa 5 bis
10 Volumina eines Alkohols (z. B. Methanol oder Isopropanol) auf
etwa den Siedepunkt der Lösung
erwärmt
wurde, und die Lösung
wurde dann filtriert, um jegliches Teilchenmaterial zu entfernen.
Gewünschtenfalls
kann in der Auflösungsstufe
Darco zugefügt
werden, um jegliche Farbverunreinigungen aus der Charge zu entfernen.
Die Lösung
wurde auf etwa die Hälfte
des ursprünglichen
Volumens konzentriert, auf etwa Raumtemperatur abgekühlt und
mit etwa dem gleichen Volumen an Wasser verdünnt. Der ausgefällte Feststoff
wurde gekühlt,
filtriert, mit etwa einer 25 wässrigen
Alkohollösung
gewaschen und bei etwa 70 bis 80°C
unter Vakuum getrocknet, um Form 1 von Verbindung 13 zu liefern.
- Ausbeute: etwa 90–95%
- Morphologie: Nadeln.
- Schmelzpunkt: etwa 175–182°C
- Durchschnittliche DSC-Schmelzwärme: etwa 70 J/g. Siehe 4,
die 71,112 J/g zeigt.
- Röntgenpulverbeugungswinkel
[in Grad]: Siehe Tabelle 2 und 3.
-
Präparation
2A: Form 2 von Verbindung 13 ohne Impfen
-
Etwa
1 g von Verbindung 13 (in jeglicher Form, sowohl kristallin als
auch nicht kristallin) wurde gelöst, indem
es in etwa 10–20
Volumina eines Kristallisationslösungsmittels
für Form
2 (z. B. Alkohol, Nitril, Ester oder Keton) erwärmt wurde. Dann wurde die Lösung filtriert,
um jegliches Teilchenmaterial zu entfernen. Gewünschtenfalls kann in der Auflösungsstufe
Darco zugefügt
werden, um jegliche Farbverunreinigungen aus der Charge zu entfernen.
Die Lösung
wurde auf etwa die Hälfte
des ursprünglichen
Volumens konzentriert und auf etwa Raumtemperatur abgekühlt. Dann
wurde die Charge etwa 18 Stunden bei Raumtemperatur gerührt, um
die im Gleichgewicht befindliche reine Form 2 der Verbindung 13
zu erhalten.
- Ausbeute: etwa 75–85% Morphologie: Plättchen.
- Schmelzpunkt: etwa 164–172°C
- Durchschnittliche DSC-Schmelzwärme etwa 100 J/g. Siehe 2,
die 98,521 J/g zeigt.
- Röntgenpulverbeugungswinkel
[in Grad]: Siehe Tabelle 1 und 1.
-
Präparation
2B: Form 2 von Verbindung 13 mit Impfen
-
Die
Charge wurde in der gleichen Weise wie oben für die Herstellung von 2A beschrieben
bis zum Kühlen
der Lösung
auf etwa Raumtemperatur behandelt. An diesem Punkt wurde die Lösung mit
einer kleinen Menge der festen Form 2 von Verbindung 13 (z. B. etwa
0,2% Gew./Gew. bis etwa 1% Gew./Gew., bezogen auf das Gewicht des
Ausgangsmaterials) geimpft. Der kristallisierte Feststoff wurde
dann gekühlt,
filtriert, mit Kristallisationslösungsmittel
gewaschen und bei etwa 70 bis 80°C
unter Vakuum getrocknet, um Form 2 von Verbindung 13 zu liefern.
Die erhaltene Ausbeute (etwa 90–95%)
ist etwas mehr, als in der obigen Präparation 2A erhalten wurde
(weil die Krustenbildung des Produkts vermieden werden konnte, die
während
der Präparation
2A auftrat).
-
Die
Morphologie, der Schmelzpunkt, die DSC-Schmelzwärme und die Röntgenpulverbeugungsdaten sind
die gleichen, wie sie nachfolgend für die durch Präparation
2A hergestellte Form 2 gezeigt sind.
-
Beispiel
-
Etwa
10 g von Form 1 von Verbindung 13 wurden zu etwa 17 Volumina Acetonitril
gegeben und gelöst, indem
die Charge auf etwa 80–85°C erwärmt wurde.
Die Charge wurde mit Darco behandelt, um jegliche Farbverunreinigungen
zu entfernen. Die heiße
Lösung
wurde filtriert, um jegliches Teilchenmaterial zu entfernen, und
die Charge wurde atmosphärisch
auf ein Endvolumen von etwa 6–7
Volumina konzentriert. Etwa 0,05 g Impfmaterial von Form 2 von Verbindung
13 (was etwa 0,5 Gew.-% der Anfangscharge von Form 1 von Verbindung
13 ist) wurden als Aufschlämmung
in Acetonitril zugegeben. Die Charge wurde allmählich auf Raumtemperatur abgekühlt, dort
etwa 3 Stunden gehalten und danach auf etwa 0–5°C abgekühlt. Die resultierende Suspension
wurde filtriert, mit Acetonitril gewaschen und bei etwa 70–80°C im Vakuum
getrocknet, um in etwa 90–95%
Ausbeute Form 2 von Verbindung 13 zu liefern.
-
Die
Morphologie, der Schmelzpunkt, die DSC-Schmelzwärme und die Röntgenpulverbeugungsdaten sind
die gleichen, wie sie zuvor für
die durch Präparation
2B hergestellte Form 2 gezeigt wurden.