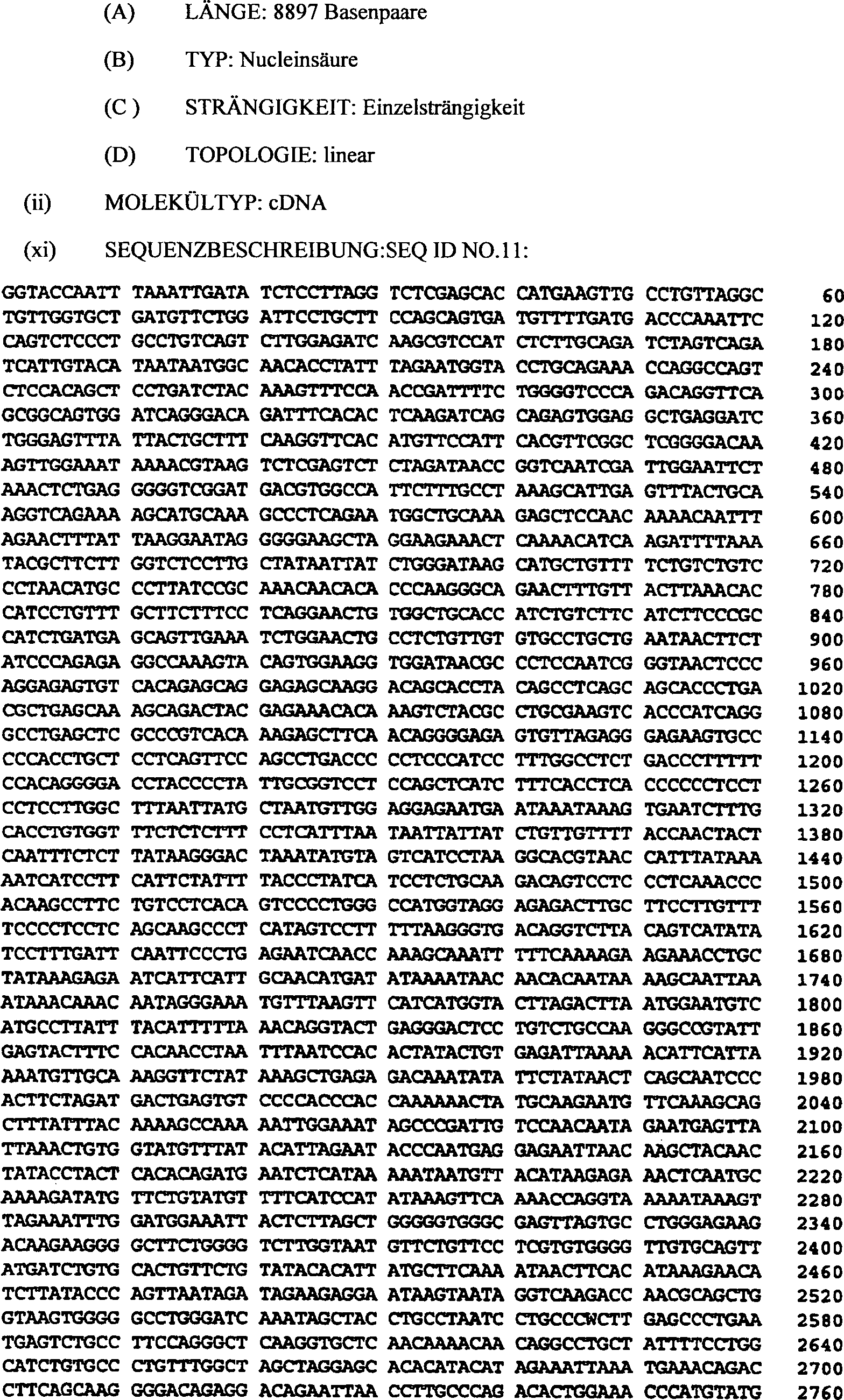-
TECHNISCHES GEBIET DER ERFINDUNG
-
Die
vorliegende Erfindung betrifft Methoden zum Hemmen oder Reduzieren
der durch Immunglobulin induzierten Toxizität, die aus der Therapie oder
der in vivo-Diagnose herrührt.
Spezifisch bietet die Erfindung statt der Verwendung unmodifizierter
Antikörper
oder rekombinanter Bindungsproteine zur Anwendung in vivo die Verwendung
modifizierter Antikörper
oder rekombinanter Bindungsproteine, die in der konstanten Domäne strukturell
so geändert
worden sind, dass die durch Immunglobulin induzierte Toxizität auf die
Verabreichung hin reduziert oder gehemmt wird.
-
HINTERGRUND DER ERFINDUNG
-
Im
Laufe der Jahre haben Forscher versucht, das Immunsystem für die therapeutische
Anwendung nutzbar zu machen. Immunglobulin-(Ig-)Moleküle, die
einen wichtigen Teil des Immunsystems darstellen, sind deshalb von
großem
Interesse, weil sie (1) mit einer vielfältigen Familie von Liganden
reagieren, (2) verschiedene Effektorfunktionen besitzen und (3)
von großer
biologischer Wichtigkeit sind. Trotz ihres Potentials hat ein hartnäckiges Problem
bei der Immuntherapie mit Immunglobulin neben anderen Problemen
aus der toxischen Auswirkung der Verwendung von Antikörpern, die
sowohl normale als auch kranke Zellen erkennen, auf normale Zellen
bestanden. Dieses Problem ist deshalb weitreichend, weil die meisten
Antikörper,
die zur Zeit zur Verfügung
stehen, ein Target erkennen, das sowohl an normalen als auch kranken
Zellen vorkommt (Slavin-Chiorini et al., Int. J. Cancer 53: 97–103 (1993)).
-
Die
Konstantregion kann den Zelltod durch antikörperabhängige zellvermittelte Zytotoxizität (ADCC) oder
durch komplementabhängige
Zytotoxizität
(CDC) fördern.
Trotz der Deletion von Anteilen der Konstantregion, insbesondere
der CH2-Domäne, kann die Antigenbindungsfunktion
beibehalten werden (D. Yelton, M. Scharf, Mutant monoclonal antibody
with alterations in biological functions – Mutanter monoklonaler Antikörper mit Änderungen
der biologischen Funktionen –,
J. Exp. Methods 156: 1131–1148
(1982)).
-
Andere
haben einen CH2-deletierten Antikörper erzeugt
(Mueller et al., Proc. Natl. Acad. Sci. USA 87: 5702–5705 (1990)).
Ihre Ergebnisse zeigen, dass der CH2-deletierte
Antikörper
aus dem Blut tumortragender Mäuse
viel schneller eliminiert wurde als der entsprechende intakte Antikörper. Andere
in vivo-Befunde haben ebenfalls bestätigt, dass ein CH2-deletierter
Antikörper,
ch14.18DCH2 genannt, ein potentiell nützliches Reagenz für die Radioimmundetektion
menschlicher Tumore aufgrund seiner reduzierten Immungenizität, erhöhten Targetspezifizität und schnellen
Entfernung aus dem Blutkreislauf ist (Mueller et al., Proc. Natl.
Acad. Sci. USA 87: 5702–5705
(1990)).
- • G.
J. Schreiber et al, Cancer Research, Band 52, 1992, S. 3262–3266, offenbaren
das antikarzinogene murine BR96-Antikörper F(ab')2 IgG3-Polypeptid
und eine klassengewechselte IgG1-Variante
des ursprünglichen
IgG3 BR96-Antikörpers,
der keine antikörperabhängige zelluläre Zytotoxizitäts-(ADCC)
und keine komplementabhängige
Zytotoxizitäts-(CDC)Effektorfunktionen
besitzt. S. D. Gillies et al., Human Antibodies and Hybridomas (Menschliche
Antikörper
und Hybridome), Band 1, 1990, S. 47–54, offenbaren Antigenbindungs-
und biologische Aktivitäten
gentechnisch veränderter
mutanter chimärer
Antikörper
mit menschlichen Tumorspezifizitäten.
Die Konstantregion der IgG-Kette wurde entweder durch Deletion der CH2-Domäne oder
durch Punktmutation von zwei Cysteinresten in der Scharnierregion
des IgG-Moleküls strukturell
geändert.
Die CH2-Deletion führte
zu einer drastisch reduzierten ADCC- und CDC-Aktivität. Die Cysteinmutation
führte
zu einer stark reduzierten ADCC-Aktivität und einer
reduzierten, jedoch immer noch signifikanten, Fähigkeit, CDC zu verursachen.
G. J. Weiner et al., Journal of Immunology, Band 152, 1994, S. 2385–2392, offenbaren
einen hybriden Anti-CD3 x Antitumor F(ab')2-Antikörper, der
für die
Immuntherapie verwendet wird. Der Fc-Teil
des Antikörpers
ist entfernt worden, um die toxischen Nebenwirkungen, denen man
bei einleitenden Studien an Subjekten begegnet ist, wie beispielsweise
die ADCC und unspezifische T-Zellaktivierung, zu reduzieren. A.
R. Duncan et al, Nature, Band 332, 1988, S. 738–740 offenbaren, dass gewisse
Punktmutationen an den Resten 318 (Glu), 320 (Lys) und 322 (Lys)
in der CH2-Domäne
verschiedener IgG-Isotypen die CDC reduzieren und dass es IgG-Isotypen,
wie beispielsweise IgG4, gibt, die nicht lytisch sind und daher
keine CDC-Reaktion
induzieren. Y. Xu et al., The Journal of Biological Chemistry, Band
269, 1994, S. 3469–3474,
offenbaren, dass die C-terminale Region der CH2-Domäne (Reste 292–340) für die isotypspezifischen
Unterschiede in der Komplementbindung und bei der Induktion der CDC-Reaktion
verantwortlich ist. Außerdem
wurde der Rest an Position 331 analysiert und es wurde gefunden,
dass er für
die Clq-Bindung und Komplementaktivierung im menschlichen IgG1 wichtig
ist. Eine Punktmutation von Pro331 zu Ser311 eliminiert diese Aktivität, während eine
Pro331-Punktmutation im IgG4-Isotyp, der
gewöhnlich
inaktiv ist, die lytische Aktivität des Isotyps wiederherstellte.
J. Lund et al, The Journal of Immunology, Band 147, 1991, S. 2657–2662 offenbaren,
dass die FcγRI-
und FcγRII-Bindungsstellen
in der CH2-Domäne
des menschlichen IgG3-Isotyp für
die toxische ADCC-Reaktion bei Subjekten, die mit Antikörpern behandelt
werden, verantwortlich ist. Die Bindungsstellen überlappen sich teilweise in der
Region der Reste 234 bis 239 an menschlichen IgG1- und IgG3-Isotypen.
Eine Bindung an menschliches IgG2 kann nicht festgestellt werden.
Des Weiteren führte
eine Punktmutation an Position 235 zur stärksten Reduktion der FcγRI-Bindung, und Punktmutationen
an den Positionen 234 und 237 führten
zur stärksten
Reduktion der FcγRII-Bindung. A. Morgan
et al., Immunology, 1995, Band 86, S. 319–324 offenbaren, dass der Austausch
des Leucins 235 in der CH2-Region von IgG1 die Lyse durch menschliches Komplement
eliminierte und das Ersetzen von Glycin bei 237 durch Alanin von
IgG1 die Lyse durch menschliches Komplement reduzierte. Das Ersetzen
der gesamten Region 233–236
durch die Sequenz, die im menschlichen IgG2 vorgefunden wird, eliminierte
die Lyse durch menschliches Komplement. Im Gegensatz dazu hatte
das Ersetzen von Lysin 320 im vorher beschriebenen Clq-Bindungsmotiv
durch Alanin keine Auswirkung auf die IgG1-vermittelte komplementabhängige Lyse.
S. M. Canfield et al., J. Exp. Med. Band 173, Juni 1991, S. 1483–1491 offenbaren
einen IgG-Antikörper
mit zwei Mutationen an den Positionen 234 und 331, der eine reduzierte
Fcγ-Rezeptorbindungsaktivität und daher
eine reduzierte ADCC-Reaktion aufweist.
-
Im
Allgemeinen bestehen ganze Antikörpermoleküle aus zwei
schweren (S) und zwei leichten (L) Ketten, die durch kovalente Bindungen
(Disulfid) und nichtkovalente Wechselwirkungen zusammengehalten
werden. Jede Kette enthält
eine variable Region (V) und eine Konstantregion (K). Die variablen
Regionen an den Aminotermini der beiden Ketten bilden die Antigenbindungsregion.
Die Konstantregion der S-Kette weist drei Komponenten oder Domänen auf.
Ab und zu tritt die erste Domäne
der Konstantregion (CH1) mit der K-Region der
L-Kette durch hydrophobe Wechselwirkungen und normalerweise eine
Disulfidbindung, je nach der Isotype, in Wechselwirkung. Der nächste Abschnitt
der K-Region ist
die als Scharnier wirkende Disulfidbindung, die auf stabile Weise
zwischen die S-Ketten eingeführt
ist. Die zweite Domäne
der Konstantregion (CH2) liegt neben der
Scharnierregion. CH2 enthält Sequenzen,
die für
Effektorfunktionen des Antikörpers
wichtig sind, wie beispielsweise die Sequenzen, die für die Komplementbindung
und die Fc-Rezeptorbindung verantwortlich sind. Die dritte Domäne der Konstantregion
(CH3) ist am Carboxylterminus der S-Kette
lokalisiert und wird als beim S-Kettenzusammenbau
sowie bei einigen Funktionen der C-Region eine wichtige Rolle spielend
betrachtet.
-
Heutzutage
werden viele Antikörper
in klinischen Versuchen gegen tumorassoziierte Antigene gerichtet.
Die meisten tumorassoziierten Antigene sind nicht tumorspezifisch,
sondern sind auch im Allgemeinen an der Zelloberfläche einiger
normaler, nicht tumorerzeugender Zellen anzutreffen. Die klinische
Verwendung einiger Antikörper,
die gegen tumorassoziierte Antigene gerichtet sind, ist wegen der
Toxizität,
die mit ihrer Verwendung verbunden ist, beschränkt. Aus diesem Grund besteht
ein Bedarf für
Methoden zum Hemmen der Toxizität,
die mit der Anwendung von Immunglobulin auf dem Gebiet der Krankheitstherapie
(z. B. Therapie gegen Tumore, Nierenerkrankung und dergleichen)
und in vivo-Diagnose
verbunden ist.
-
Wir
haben diesem Bedarf durch Entdecken von Methoden zum Hemmen oder
Reduzieren von Toxizität
für normale
Zellen, die allgemein mit der Immunglobulinimmuntherapie oder in
vivo-Diagnose verbunden ist, wobei das Immunglobulin sowohl kranke
als auch normale Zellen erkennt, entsprochen. Unsere Entdeckung
involviert die Erzeugung von Immunglobulinmolekülen oder Ig-Fusionsproteinen,
die strukturell geänderte
Konstantregionen aufweisen, die die durch Immunglobulin induzierte
Toxizität
hemmen oder reduzieren.
-
ZUSAMMENFASSUNG DER ERFINDUNG
-
Es
werden hier Methoden zum Hemmen der durch Immunglobulin induzierten
Toxizität
durch Verwendung bekannter Immunglobulin- oder Ig-Fusionsproteinmoleküle offenbart,
die in ihren Konstantregionen so strukturell geändert sind, dass die so gebildeten
strukturell geänderten
Immunglobulin- oder
Ig-Fusionsproteinmoleküle
eine reduzierte oder gehemmte Toxizität in vivo im Vergleich mit
ihren ursprünglichen
unmodifizierten Gegenstücken
aufweisen.
-
Die
vorliegende Erfindung bietet als Erstes einen BR96-Antikörper, der
eine menschliche IgG1-Konstantregion
aufweist, die in der CH2-Domäne
strukturell geändert
worden ist, wobei Leucin an der Aminosäureposition 235 zu Alanin mutiert
wird; Glycin an der Aminosäureposition
237 zu Alanin mutiert wird; Glutaminsäure an der Aminosäureposition
318 zu Serin mutiert wird; Lysin an der Aminosäureposition 320 zu Serin mutiert wird;
und Lysin an der Aminosäureposition
322 zu Serin mutiert wird.
-
Die
vorliegende Erfindung bietet auch einen BR96-Antikörper, der
eine menschliche IgG1-Konstantregion
aufweist, die in der CH2-Domäne
strukturell geändert
worden ist, wobei Leucin an der Aminosäureposition 235 zu Alanin mutiert
wird; Glycin an der Aminosäureposition
237 zu Alanin mutiert wird; und Prolin an der Aminosäureposition
331 zu Alanin mutiert wird.
-
Außerdem bietet
die vorliegende Erfindung einen BR96-Antikörper, der eine menschliche
IgG1-Konstantregion
aufweist, die in der CH2-Domäne
strukturell geändert
worden ist, wobei Leucin an der Aminosäureposition 235 zu Alanin mutiert
wird; Glycin an der Aminosäureposition
237 zu Alanin mutiert wird; Glutaminsäure an der Aminosäureposition
318 zu Serin mutiert wird; Lysin an der Aminosäureposition 320 zu Serin mutiert
wird; Lysin an der Aminosäureposition
322 zu Serin mutiert wird; und Prolin an der Aminosäureposition 331
zu Alanin mutiert wird.
-
Andere
Ausgestaltungen der Erfindung sind in den angehängten Ansprüchen 4–18 beansprucht. Weitere Ausführungsformen,
die durch die beanspruchte Erfindung nicht umfasst werden, sind
ebenfalls in der vorliegenden Beschreibung offenbart.
-
Eine
strukturelle Änderung
der Konstantregion kann auf eine Reihe verschiedener Arten und Weisen durchgeführt werden,
solange sie zum Reduzieren oder Hemmen durch Immunglobulin induzierter
Toxizität fürt.
-
Die
strukturelle Änderung
der Konstantregion wird durch Deletion der gesamten Konstantregion
bewirkt. In einer anderen Ausführungsform
wird nur die CH2-Domäne deletiert. In einer anderen
Ausführungsform wird
nur der Teil der CH2-Domäne deletiert, der den Fc-Rezeptor
bindet. In noch einer anderen Ausführungsform wird nur der Teil
der CH2-Domäne deletiert, der die Komplementkomponente
Clq bindet. Alternativ werden in einer anderen Ausführungsform
multiple Deletionen in einzelnen Fc-Rezeptor- und Komplementkomponentenbindungsdomänen bewirkt.
-
Alternativ
wird die strukturelle Änderung
durch Einfachmutation oder multiple Mutationen in der CH2-Domäne,
wie beispielsweise Aminosäureinsertionen
und -substitutionen, durchgeführt.
Die Mutation oder Mutationen müssen
zum Hemmen der durch Immunglobulin induzierten Toxizität führen. Beispielsweise
können
die Aminosäuren
in multiplen, mit Toxizität
verbundenen Domänen
in der Konstantregion so geändert
werden, dass die Konstantregion unfähig ist, eine ADCC-Reaktion
zu vermitteln oder Komplement zu aktivieren, wodurch die durch Immunglobulin
induzierte Toxizität,
die aus der Immuntherapie herrührt,
gehemmt wird. Alternativ können
multiple Aminosäuren
in einer einzigen mit Toxizität
verbundenen Domäne
in der Konstantregion geändert
werden.
-
Des
Weiteren kann aternativ eine strukturelle Änderung durch Isotypwechsel
bewirkt werden, der zu einem geänderten
Immunglobulinmolekül
führt,
das entweder keine Toxizität
oder eine beschränkte
Toxizität induziert,
jedoch keine schädliche
Wirkung ausübt.
Beispielsweise kann ein Isotypwechsel in der Konstantregion dazu
führen,
dass sie nicht in der Lage ist, eine CDC- oder ADCC-Reaktion oder
irgendeine andere Aktivität,
die Toxizität
vermittelt, zu vermitteln.
-
KURZE BESCHREIBUNG DER FIGUREN
-
1 ist
eine Koordinatengrafik, die die Ausscheidung am dem Plasma bei Hunden
mit hoher LeY-Expression mit Hilfe von chimärem BR96
im Vergleich mit dem Konstantregionenmutanten cBR96-2 zeigt.
-
2 ist
ein schematisches Diagramm eines pTWD-cJVK.L1 genannten Plasmids,
das die chimäre (c)BR96-Leichtkette
(SEQ ID NO. 11) umfasst.
-
3 ist
ein schematisches Diagramm eines pD16hJ1.L1 genannten Plasmids,
das die menschliche (h)BR96-Leichtkette (SEQ ID NO. 13) umfasst.
-
4 ist
ein schematisches Diagramm eines pD17-hJm14-dCH2.H1 genannten Plasmids
von hBR96-2A (d. h. dem menschlichen mutanten BR96 mit den H1-,
H2- und H3-Mutationen und der CH2-Deletion (PCT-Anmeldung
Nr. 95/305444, am 6. März
1996 veröffentlicht)).
-
5 ist
ein schematisches Diagramm eines pD17-cJ-dCH2.H1 genannten Plasmids
von cBR96-A (SEQ
ID No. 10) (d. h. chimären
BR96 mit der CH2-Deletion (PCT-Anmeldung
Nr. 95/305444, am 6. März
1996 veröffentlicht)).
-
6 ist
ein schematisches Diagramm eines pD17-cJ.h1 genannten Plasmids von
cBR96.
-
7 ist
eine Koordinatengrafik, die die Ergebnisse eines ELISA-Assays von
(1) hBR96-2A-Dox
gegen Ley (ausgefüllte Raute), (2) hBR96-2A gegen
Ley (96:0006A2 R/A) (ausgefülltes Viereck),
(3) hBR96-2A gegen Ley (96:0006B R/A) (ausgefülltes Dreieck)
und BR96-Dox gegen Ley (X) zeigt.
-
8 ist
eine Koordinatengrafik, die die Ergebnisse eines ELISA-Assays von
(1) BR96-A-Dox zu Ley (ausgefüllte Raute),
(2) chiBR96 zu Ley (ausgefülltes Viereck),
(3) cBR96-A zu Ley (96:0003 R/A) (ausgefülltes Dreieck)
und cBR96-Dox zu Ley (X) zeigt.
-
Die 9a–c sind
schematische Diagramme, die die Schritte zum Deletieren einer CH2-Domäne zeigen.
-
10a–c
sind schematische Diagramme, die die Konstruktion von BR96 IgG1
CH2-Domänenpunktmutationen
zeigen.
-
11 ist
ein schematisches Diagramm, das die Konstruktion des pNg1/14-Vektors
zeigt.
-
12 ist
ein schematisches Diagramm, das die Konstruktion von pD17-hBR96-2
zeigt.
-
13 ist
ein schematisches Diagramm, das die Konstruktion von pD17-hJmI4-dCH2.H1
zeigt.
-
Die 14A–J
sind die Nucleinsäuresequenz
von pD17-cJ-dCH2.H1, dem Plasmid, das in 5 gezeigt
ist, des chimären
BR96, das die CH2-Deletion aufweist.
-
15 ist
eine Koordinatengrafik, die die Ergebnisse eines ELISA-Assays zeigt,
bei dem ganzer chiBR96 und chiBR96 mit deletiertem CH2 an
Ley verglichen werden.
-
16 ist
eine Beschreibung der sieben strukturellen Änderungen.
-
17 ist
ein schematisches Diagramm eines pD17-hG1b genannten Plasmids.
-
Die 18A–F
sind die Nucleinsäuresequenz
von pD17-hJm14.H1.
-
Die 19A–N
sind die Nucleinsäuresequenz
von pD17-hG1b.
-
20 ist
eine Koordinatengrafik, die die komplementabhängige Zytotoxizität zeigt.
In der Beschreibung ist das ausgefüllte Quadrat hBR96-1, die ausgefüllte Raute
ist hBR96-2B, der ausgefüllte
Kreis ist hBR96-2C, das ausgefüllte
Dreieck ist hBR96-2D; das offene Quadrat ist hBR96-2H; der offene
Kreis ist hBR96-2A und das offene Dreieck ist 2B8 mAb gegen Flagella
des Typs b von Anti-Pseudonomas aeruginosa bestehende negative Kontrolle.
-
21 ist
eine Koordinatengrafik, die die antikörperabhängige, zellvermittelte Zytotoxizität zeigt.
In der Beschreibung ist das ausgefüllte Quadrat hBR96-1; die ausgefüllte Raute
ist hBR96-2B; der ausgefüllte Kreis
ist hBR96-2C; das ausgefüllte
Dreieck ist hBR96-2D; das offene Quadrat ist hBR96-2H; der offene
Kreis ist hBR96-2A und das offene Dreieck ist 2B8, monoklonalem
Antikörper
(mAb) gegen Flagella des Typs b von Pseudonomas aeruginosa, negative
Kontrolle.
-
22 ist
eine Koordinatengrafik, die die Bindungsaktivität von hBR96-2-Konstantregionmutanten
an LeY-HASA zeigt. In der Beschreibung ist die ausgefüllte Raute
hBR96-1; das ausgefüllte
Quadrat ist hBR96-2A (CH2-Deletion); das ausgefüllte Dreieck ist hBR96-2B (235-,
237-Mutationen);
das offene Quadrat ist hBR96-2C (318-, 320-, 322-Mutationen); der
offene Kreis ist hBR96-2D
(331-Mutation); und das offene Dreieck ist hBR96-2H (235-, 237-,
318-, 320-, 322-, 331-Mutationen).
-
23 ist eine Koordinatengrafik, die die Bindungsaktivität von hBR96-2-Konstantregionmutanten
an LNFPIII-RSA zeigt. LNFPIII ist eine Lacto-N-fucopentasose, ein
Lewis X-Trisaccharid
mit einem zusätzlichen Lactosespacer
(V Labs, Covington, LA). In der Beschreibung ist die ausgefüllte Raute
hBR96-1; das ausgefüllte
Quadrat ist hBR96-2A (CH2-Deletion); das ausgefüllte Dreieck ist hBR96-2B (235,
237-Mutationen); das offene Quadrat ist hBR96-2C (318-, 320-, 322-Mutationen);
der offene Kreis ist hBR96-2D (331-Mutation); und das offene Dreieck
ist hBR96-2H (235-, 237-, 318-, 320-, 322-, 331-Mutationen).
-
Die 24A und 24B bieten
eine Strategie für
das Einführen
multipler Mutationen durch RPCR. (A) Diagramm der 1,4 kpb IgG Schwerkettenregion,
das die Scharnier-CH2- und CH3-Domänen als
umrandete Regionen zeigt. Die stellenspezifischen Mutationen, die
in die CH2-Positionen L1, L2 und L3 eingeführt werden sollen,
werden durch komplementäre
Sätze mutanter
PCR-Primer (A1 und A2; B1 und B2; und C und C2) kodiert. Die Sternchen
(*) zeigen die Anzahl von Aminosäureänderungen
an, die an jeder L-Position eingeführt worden sind. Die beiden
PCR-Primer, nämlich
Rs (Rekombinations-sense) und Ra (Rekombinations-antisense), flankieren
die Eco-47-III-Restriktionsstellen und vermitteln die homologe Rekombination
mit Vektorenden. Die 3'-Enden
der Oligonuldeotide sind durch Pfeilköpfe dargestellt. (B) Ein homologes
Dreiwegrekombinationsgeschehen zwischen den Fragmenten RsA2, A1Ra
und dem linearisierten Vektor bildet das L1-mutante IgG. Zwei distal
gelegene Sätze
von Mutationen (L1 und L2) werden gleichzeitig durch Erhöhen der
Anzahl von rekombinierenden PCR-Produkten, die, wie gezeigt, in
der Vierweg-Rekombination von RsA2, A1B2, B1Ra mit dem Vektor gebildet
werden, eingeführt.
-
25 ist ein Gel, das die Eco-47-III-Restriktionsendonukleaseanalyse
von DNA zeigt, die aus Kolonien hergestellt werden, die durch multiple
PCR-Fragment-RPCR erzeugt wurden. Bahn M: 1kb-Leiter-DNA-Marker (GIBCO-BRL Life Science
Technology). Bahnen 1–12:
Zwölf willkürlich ausgewählte Kolonien, die
aus vierfachen homologen Rekombinationsereignissen resultieren,
wurden zum Herstellen des Plasmids verwendet und mit Eco47-III verdaut.
Die Klone 1, 2, 6 und 9 enthalten die vollständig zusammengebaute 1,4 kpb-Insertion.
-
26 bietet die Aminosäuresequenz für die variable
Schwerkettenregion von hBR96-2 und die menschliche IgG1-Konstantregion.
-
27 bietet die Aminosäuresequenz für die variable
Schwerkettenregion von hBR96-2A und die menschliche IgG1-Konstantregion
ohne die CH2-Domäne.
-
28 bietet die Aminosäuresequenz für die variable
Schwerkettenregion von chi BR96 und die menschliche IgG1-Konstantregion
ohne die CH2-Domäne.
-
GENAUE BESCHREIBUNG DER ERFINDUNG
DEFINITIONEN
-
Wie
hier verwendet bedeutet der Begriff „Hemmen der durch Immunglobulin
induzierten Toxizität" das Reduzieren oder
Milder von Symptomen, die allgemein mit der Toxizität verbunden
sind, die durch Immunglobulin- oder Ig-Fusionsproteintherapie verursacht
wird, z. B. der Toxizität,
die durch Effektorfunktionen des Fc-Rezeptors vermittelt werden.
Beispielsweise erkennt und bindet der BR96-Antikörper das BR96-Antigen, das
in gewissen Mengen im Magendarmkanal und in erhöhten Mengen in Tumoren (im
Vergleich mit dem normalen Gewebe des Magendarmkanals) anzutreffen
ist. Das Binden von BR96-Antikörper
an BR96-Antigen in vivo verursacht Symptome, die mit Magendarm-Toxizität verbunden
sind. Diese Symptome umfassen das schnelle Einsetzen von Erbrechen,
oft mit Blut, und Übelkeit.
Bei Menschen ist das Bluten auf den Magenfundus beschränkt, wobei
es eine Erosion der Oberflächenschleimhaut
des Magens verursacht.
-
Die
Pathologie der Wunde ist beschränkt
und sie bildet sich zurück.
Jedoch definiert die extreme Natur der Übelkeit und des Erbrechens,
die durch Antiemetika nicht gemildert werden, sie als die dosisbeschränkende Toxizität. Bei anderen
Antigenen, die in stark erhöhter
Konzentration im Zentralnervensystem (ZNS), in der Leber und an
anderen Stellen vorkommen, ist die Toxizität durch andere als die oben
beschriebenen Symptome gekennzeichnet.
-
Wie
der Begriff hier verwendet wird, kann das „Immunglobulinmolekül" durch B-Zellen gebildet
oder durch rekombinante Genmodifikation oder chemische Synthesemöglichkeiten
erzeugt werden. Beispiele von Immunglobulinmolekülen umfassen (1) Antikörper, z.
B. polyklonale und monoklonale Antikörper, chimäre oder humanisierte, und (2)
rekombinante, Ig-enthaltende Bindungsproteine, z. B. Ig-Fusionsproteine.
Rekombinante Ig-enthaltende Bindungsproteine umfassen Zelloberflächenproteine,
z. B. CD-Antigene (in einer Ausführungsform
CTLA4), an die ein Ig-Schwanz angehängt ist.
-
Wie
hier verwendet, bedeuten die Begriffe „strukturell geändert" oder „strukturelle Änderung" das Manipulieren
der Konstantregion derart, dass das so gebildete Molekül oder Protein
eine reduzierte Fähigkeit
zum Induzieren von Toxizität
aufweist. Die strukturelle Änderung
kann durch chemische Modifikation, proteolytische Änderung
oder durch rekombinante genetische Mittel durchgeführt werden.
Rekombinante genetische Mittel können
die Deletion, Insertion und Substitution von Aminosäureanteilen
umfassen, ist jedoch nicht darauf beschränkt.
-
Wie
hier verwendet, bedeutet der Begriff „multiple mit Toxizität verbundene
Domänen" mehr als eine einzelne
mit Toxizität
verbundene Domäne.
Da im Immunglobulinmolekül
mindestens zwei mit Toxizität
verbundene Domänen
vorzuliegen scheinen, wobei eine ungefähr bei Aminosäuren 231–238 und
die andere ungefähr
bei Aminosäuren
310–331
lokalisiert ist, umfasst ein Beispiel für die strukturelle Änderung
von multiplen mit Toxizität
verbundenen Domänen
die Insertion, Substitution oder Deletion von Aminosäureresten
in diesen beiden Domänen.
Diese Definition schließt
strukturelle Änderungen
aus, die auf eine einzige mit Toxizität verbundene Domäne gerichtet
sind.
-
Ausschließlich beispielsweise
kann die Konstantregion des Immunglobulinmoleküls so strukturell geändert werden,
dass das Molekül
keine CDC- oder ADCC-Reaktion mehr vermittelt. Jedoch umfassen die
erfindungsgemäßen Methoden
die Verwendung strukturell geänderter
Immunglobulinmoleküle,
gleichgültig,
ob sie eine CDC- oder ADCC-Antwort vermitteln. Das grundlegende
Erfordernis besteht darin, dass das geänderte Molekül eine durch
Immunglobulin induzierte Toxizität
hemmen muss.
-
Die
vorliegende Erfindung stellt als Erstes einen BR96-Antikörper bereit,
der eine menschliche IgG1-Konstantregion aufweist, die in der CH2-Domäne strukturell
geändert
worden ist, wobei Leucin an der Aminosäureposition 235 zu Alanin mutiert
wird; Glycin an der Aminosäureposition
237 zu Alanin mutiert wird; Glutaminsäure an der Aminosäureposition
318 zu Serin mutiert wird; Lysin an der Aminosäureposition 320 zu Serin mutiert
wird; und Lysin an der Aminosäureposition
322 zu Serin mutiert wird.
-
Die
vorliegende Erfindung stellt auch einen BR96-Antikörper bereit,
der eine menschliche IgG1-Konstantregion
aufweist, die in der CH2-Domäne
strukturell geändert
worden ist, wobei Leucin an der Aminosäureposition 235 zu Alanin mutiert
wird; Glycin an der Aminosäureposition
237 zu Alanin mutiert wird; und Prolin an der Aminosäureposition
331 zu Alanin mutiert wird.
-
Außerdem stellt
die vorliegende Erfindung einen BR96-Antikörper bereit, der eine menschliche IgG1-Konstantregion
aufweist, die in der CH2-Domäne
strukturell geändert
worden ist, wobei Leucin an der Aminosäureposition 235 zu Alanin mutiert
wird; Glycin an der Aminosäureposition
237 zu Alanin mutiert wird; Glutaminsäure an der Aminosäureposition
318 zu Serin mutiert wird; Lysin an der Aminosäureposition 320 zu Serin mutiert
wird; Lysin an der Aminosäureposition
322 zu Serin mutiert wird; und Prolin an der Aminosäureposition
331 zu Alanin mutiert wird.
-
Andere
Ausgestaltungen der Erfindung sind in den anhängenden Ansprüchen 4–18 beansprucht.
Weitere Ausführungsformen,
die die beanspruchte Erfindung nicht umfasst, sind ebenfalls in
der vorliegenden Beschreibung offenbart.
-
Eine
strukturelle Änderung
kann auf eine Reihe verschiedener Arten und Weisen bewirkt werden.
Beispielsweise kann die strukturelle Änderung durch Deletion der
gesamten Konstantregion bewirkt werden.
-
Alternativ
kann die strukturelle Änderung
durch Deletion der gesamten CH2-Domäne der Konstantregion
bewirkt werden. In diesem Fall kann die Deletion der gesamten CH2-Domäne
das Molekül
unfähig
machen, (1) einen Fc-Rezeptor zu binden, wodurch die Fähigkeit
des Moleküls,
die antikörperabhängige zelluläre Zytotoxizität (ADCC)
zu vermitteln, eliminiert wird, (2) Clq zu binden oder (3) das Komplement
zu aktivieren.
-
Alternativ
kann die strukturelle Änderung
durch Deletion nur desjenigen Teils der CH2-Domäne bewirkt werden,
der den Fc-Rezeptor oder das Komplement bindet.
-
Des
Weiteren kann alternativ eine einzige Mutation oder können multiple
Mutationen, wie Substitutionen und Insertionen, in der CH2-Domäne
durchgeführt
werden. Das grundlegende Erfordernis für irgendeine Mutation besteht
darin, dass sie die durch Immunglobulin induzierte Toxizität hemmen,
reduzieren oder blockieren muss. Beispielsweise lasst sich dies
durch Mutation der Konstantregion derart, dass das geänderte Molekül unfähig gemacht
wird, eine CDC-Reaktion oder eine ADCC-Reaktion zu vermitteln oder
Komplement zu aktivieren, erreichen.
-
Alternativ
kann eine strukturelle Änderung
durch Isotypwechsel (auch als Klassenwechsel bekannt) bewirkt werden,
derart, dass das geänderte
Molekül
im Subjekt keine Toxizität
induziert. In einer Ausführungsform
wird die Konstantregion des Immunglobulins strukturell so geändert, dass
es den Fc-Rezeptor
oder eine Komplementkomponente nicht mehr bindet, z. B. durch Wechseln
des ursprünglichen
IgG-Isotyps eines Moleküls
von IgG1 zu IgG4. Der Isotypwechsel kann durchgeführt werden,
gleichgültig
um welche Spezies es sich handelt, d. h. ein Isotyp aus einer nichtmenschlichen
Spezies kann durch einen Isotyp von einem Menschen ausgewechselt
werden (E. D. Finkelman et al. (1990), Annu. Rev. Immunol. 8: 303–333; T.
Honjo et al (1979) Cell 18: 559–568;
T. Honjo et al. in „Immunoglobulin
Genes (Immunoglobingene)" S.
124–149,
Academic Press, London).
-
Wie
hier verwendet, bedeutet der Begriff „Ig-Fusionsprotein" irgendein rekombinant
erzeugtes Antigen oder eine rekombinant erzeugte Ligandenbindungsdomäne, das/die
eine Konstantregion besitzt, die strukturell geändert werden kann.
-
Wie
hier verwendet, umfasst „zytotoxisches
Mittel" Antimetabolite,
Alkylierungsmittel, Anthracycline, Antibiotika, Antimitotika und
chemotherapeutische Mittel. Spezifische Beispiele innerhalb dieser
Gruppen umfassen, sind jedoch nicht darauf beschränkt, Ricin,
Doxorubicin, Daunorubicin, Taxol, Ethidiumbromid, Mitomycin, Etoposid,
Tenoposid, Vincristin, Vinblastin, Colchicin, Supporin, Gelonin,
PE40, Bryodin, Dihydroxyanthracindion, Actinomycin D und 1-Dehydrotestosteron.
-
Wie
hier verwendet, bezieht sich der Begriff „BR96" auf (1) den ganzen monoklonalen BR96-Antikörper, der
in PCT Nr. 95/305444, am 6. März
1996 veröffentlicht,
offenbart ist, (2) den monoklonalen chimären BR96-Antikörper, der
in PCT Nr. 95/305444, am 6. März
1996 veröffentlicht,
offenbart ist oder (3) mutante BR96-Moleküle, die in PCT Nr. 95/305444,
am 6. März
1996 veröffentlicht,
offenbart sind.
-
Wie
hier verwendet bedeutet „Behandeln" (1) das Herbeiführen einer
Tumorregression derart, dass der Tumor für eine Zeitspanne nicht wahrnehmbar
ist (Standardverfahren zur Messung von Tumoren können angewendet werden (A.
B. Miller et al. „Reporting
results of Cancer Treatment (Berichten der Ergebnisse der Krebsbehandlung)", Cancer 47: 207–214 (1981));
(2) die Stabilisation der Krankheit; oder (3) Herbeiführen irgendwelcher
klinisch vorteilhaften Wirkungen.
-
Wie
hier verwendet, ist eine „wirksame
Menge" eine Menge
des Antikörpers,
Immunkonjugats oder rekombinanten Moleküls, die Zellen abtötet oder
die Proliferation derselben hemmt.
-
Wie
hier verwendet, bedeutet „Verabreichen" die orale Verabreichung,
Verabreichung als Zäpfchen, durch
topischen Kontakt, die intravenöse,
intraperitoneale, intramuskuläre
oder subkutane Verabreichung oder die Implantation einer langsam
freisetzenden Vorrichtung, wie beispielsweise einer miniosmotischen
Pumpe, dem bzw. in das Subjekt.
-
Wie
hier verwendet umfasst „pharmazeutisch
akzeptabler Träger" irgendein Material,
das, wird es mit dem Antikörper
kombiniert, die Spezifizität
oder Wirksamkeit des Antikörpers
beibehält
und mit dem Immunsystem des Subjekts nicht reaktiv ist. Beispiele
umfassen, sind jedoch nicht darauf beschränkt, irgendwelche pharmazeutischen
Standardträger
wie beispielsweise eine phosphatgepufferte physiologische Kochsalzlösung, Wasser,
Emulsionen wie beispielsweise Öl-/Wasseremulsion
und verschiedene Typen von Benetzungsmitteln. Andere Träger können auch
sterile Lösungen,
Tabletten, einschließlich
dragierter Tabletten und Kapseln, umfassen.
-
Typischerweise
enthalten derartige Träger
Vehikel wie Starke, Milch, Zucker, gewisse Arten von Ton, Gelatine,
Stearinsäure
oder Salze derselben, Magnesium- oder Calciumstearat, Talkum, Pflanzenfette
oder -öle,
Gummiarten, Glykole oder andere bekannte Vehikel. Derartige Träger können auch
Geschmacksmittel und Färbemittel
oder andere Bestandteile umfassen. Zusammensetzungen, die derartige
Träger
umfassen, werden durch allgemein bekannte herkömmliche Verfahren formuliert.
-
Wie
hier verwendet bedeutet „Mutation" eine oder mehrere
Aminosäure-
oder Nucleinsäuremutation(en),
eingeführt
auf irgendeine Art und Weise, z. B. homologe Rekombination, fehleranfällige PCR
oder ortsgerichtete Mutagenese.
-
Zum
besseren Verständnis
der hier beschriebenen Erfindung wird die folgende Beschreibung
vorgelegt.
-
METHODEN DER VORLIEGENDEN
ERFINDUNG
-
Offenbart
ist hier eine Methode für
das Hemmen der durch Immunglobulin induzierten Toxizität, die aus der
Verwendung von Immunglobulin während
einer Therapie oder in vivo-Diagnose herrührt. Beispielsweise wären die
erfindungsgemäßen Methoden
zum Minimieren der Toxizität
nützlich,
die mit langanhaltender klinischer Exposition Immunglobulin gegenüber bei
seiner Anwendung während
oder nach Visualisierung des Tumors mit radioaktiv markierten Antikörpern verbunden
ist.
-
Der
Praxis dieser Erfindung gemäß schließt das Subjekt,
ist jedoch darauf beschränkt,
menschliche, Pferde-, Schweine-, Rinder-, Maus-, Kaninchen-, Katzen-
und Vogelsubjekte ein. Andere warmblütige Tiere sind ebenfalls in
dieser Erfindung eingeschlossen.
-
Die
Methode umfasst das Verabreichen eines Immunglobulimnoleküls dem Subjekt.
Das Immunglobulin kann IgG, IgM oder IgA sein. IgG ist bevorzugt.
-
In
einer Ausführungsform
der Erfindung erkennt und bindet das Immunglobulinmolekül Ley. In einer anderen Ausführungsform erkennt und bindet
das Immunglobulin Lex. In einer weiteren
Ausführungsform
ist das Immunglobulin ein monoklonaler BR96-Antikörper, der
von dem Hybridom produziert wird, das am 22. Februar 1989 bei der
American Type Culture Collection (ATCC), 12301 Parklaven Drive,
Rockville, MD 20852 hinterlegt und dem die ATCC-Zulassungsnummer
HB 10036 gewährt
worden ist. in noch einer anderen Ausführungsform ist das Immunglobulin
ein chimärer
Antikörper
ChiBR96, der von dem Hybridom produziert wird, das am 23. Mai 1990
bei ATCC, 12301 Parklaven Drive, Rockville, MD 20852 hinterlegt
worden ist und dem die ATCC-Zulassungsnummer HB 10460 gewahrt worden
ist.
-
Der
Praxis der Erfindung gemäß kann das
Immunglobulin ein hybrider Antikörper
mit Bindungsspezifizität
für zwei
verschiedene Antigene sein, wobei eines der Antigene dasjenige ist,
an das sich der monoklonale Antikörper BR96 sich bindet, der
von dem Hybridom produziert wird, das die identifizierenden Charakteristiken
von HB 10036 aufweist, wie es bei der ATCC hinterlegt worden ist.
Ebenfalls der Praxis der Erfindung gemäß kann das Immunglobulin ein
antiidiotypischer Antikörper
sein.
-
Wie
die Erfindung erfordert, wird mindestens ein Teil der Konstantregion
des Immunglobulinmoleküls strukturell
geändert.
Die strukturelle Änderung
kann durch verschiedene Möglichkeiten
bewirkt werden. In einer Ausführungsform
kann die gesamte Konstantregion, d. h. die CH1-,
CH2- und CH3-Domänen, deletiert
werden.
-
In
einer anderen Ausführungsform
wird nur die CH2-Domäne vom Immunglobulinmolekül (z. B. cBR96-A
(5), hBR96-2A (4)) deletiert.
In dieser Ausführungsform
kann die CH2-Deletion zu einem Molekül führen, das
nicht in der Lage ist, den Fc-Rezeptor oder eine Komplementkomponente
zu binden.
-
In
einer anderen Ausführungsform
wird nur der Teil der CH2-Domäne, die
die Komplementkomponente C1q bindet, deletiert. In noch einer anderen
Ausführungsform
werden Mutationen in spezifischen Teilen der CH2-Domäne durchgeführt. Beispielsweise
kann das Immunglobulinmolekül
durch strukturelles Ändern
multipler, mit Toxizität
verbundener Domänen
in der Konstantregion so modifiziert werden, dass die durch Immunglobulin
induzierte Toxizität
gehemmt wird. Eine Diskussion derartiger Mutationen ist des Weiteren
noch im Folgenden zu finden.
-
Gleichgültig, welche
Möglichkeit
benutzt wird, besteht das grundlegende Erfordernis für irgendeine strukturelle Änderung
der Konstantregion darin, dass die durch Immunglobulin induzierte
Toxizität
dadurch wesentlich reduziert oder gehemmt wird. In einer Ausführungsform
wird die durch Immunglobulin induzierte Toxizität durch strukturelles Ändern der
Konstantregion derart geändert,
dass die Fähigkeit
des Moleküls,
eine CDC-Reaktion oder ADCC-Reaktion zu vermitteln und/oder die
Komplementkaskade zu aktivieren, eliminiert oder gehemmt wird. Methoden
zum Bestimmen, ob das Molekül
in der Lage ist, eine CDC-Reaktion zu hemmen, sind allgemein bekannt,
z. B. involviert eine Methode einen 51Cr-Freisetztest
(H. Garrigues et al. Int. J. Cancer 29: 511 (1982); I. Hellström et al.
PNAS 82: 1499 (1985)). Methoden zum Bestimmen, ob das Molekül in der
Lage ist, eine ADCC-Reaktion zu hemmen, sind allgemein bekannt (I.
Hellström
et al. PNAS 82: 1499 (1985)). Methoden zum Bestimmen, ob das Molekül in der
Lage ist, eine Komplementkaskade zu aktivieren, sind allgemein bekannt.
-
Offenbart
ist hier eine Methode, die das Verabreichen einem Subjekt eines
Ig-Fusionsproteins umfasst, das eine strukturell geänderte Konstantregion
aufweist. Die strukturelle Änderung
der Konstantregion kann die Deletion der gesamten C-Region oder
von Teilen derselben, z. B. die Änderung
der CH2-Domäne derart, dass das geänderte Molekül den Fc-Rezeptor
oder eine Komplementkomponente nicht mehr bindet, umfassen.
-
Offenbart
ist hier eine Methode für
das Hemmen der durch Immunglobulin induzierten Toxizität, die aus einer
Immuntherapie bei einem Subjekt herrührt. Die Methode umfasst das
Verabreichen dem Subjekt eines Antikörpers, der so modifiziert worden
ist, dass mindestens ein Teil der Konstantregion, wie oben besprochen, strukturell
geändert
worden ist. In einer Ausführungsform
erkennt und bindet der Antikörper
Ley. In einer anderen Ausführungsform
erkennt und bindet der Antikörper
an Lex.
-
Der
Antikörper
kann der monoklonale Antikörper
BR96 sein, der von dem Hybridom erzeugt wird, das die identifizierenden
Charakteristiken von HB 10036 besitzt, wie es bei der ATCC hinterlegt
worden ist. Alternativ kann der Antikörper der chimäre Antikörper ChiBR96
sein, der von dem Hybridom erzeugt wird, das die identifizierenden
Charakteristiken von HB 10460 aufweist, wie es bei der ATCC hinterlegt
worden ist. Des Weiteren kann der Antikörper ein hybrider Antikörper mit
einer Bindungsspezifizität
für zwei
verschiedene Antigene sein, wobei eines der Antigene dasjenige ist,
mit dem sich der monoklonale Antikörper BR96 bindet, der von dem
Hybridom erzeugt wird, das die identifizierenden Charakteristiken
von HB 10036 aufweist, wie es bei der ATCC hinterlegt worden ist.
-
Außerdem wird
hier eine Methode für
das Hemmen der durch Immunglobulin induzierten Toxizität, die aus
einer Immuntherapie bei einer Krankheit in einem Subjekt herrührt, offenbart.
Die Krankheit wird je nach dem Antigen, das gebunden werden soll,
verschieden sein. Beispiele von Krankheiten umfassen, sind jedoch nicht
darauf beschränkt,
immunologische Krankheiten, Krebs, Herz-Kreislauf-Erkrankungen, neurologische Erkrankungen,
dermatologische Erkrankungen oder Nierenleiden.
-
Diese
Methode umfasst die folgenden Schritte. Schritt eins bietet das
Auswählen
eines Antikörpers
für ein
Target. Im Allgemeinen ist das Target mit der Krankheit verbunden
und der Antikörper,
der auf das Target gerichtet wird, ist bekannt. Beispielsweise kann
das Target das zu BR96-Antigen sein und der gewählte Antikörper ist BR96.
-
Schritt
zwei dieser Methode bietet das strukturelle Ändern der Konstantregion des
so gewählten
Antikörpers
derart, dass die durch Immunglobulin induzierte Toxizität gehemmt
wird. Die Inaktivierung kann irgendeine der oben besprochenen Möglichkeiten
umfassen. Beispielsweise kann die Inaktivierung durch strukturelles Ändern der
mit multipler Toxizität
verbundenen Domänen
in der CH2-Domäne der Konstantregion des so
gewählten
Ig-Proteins bewirkt werden.
-
Schritt
drei dieser Methode bietet das Verabreichen des strukturell geänderten
Antikörpers
aus Schritt zwei dem Subjekt unter Bedingungen, derart, dass der
strukturell geänderte
Antikörper
das Target erkennt und bindet und dass ein derartiges Binden Symptome,
die mit der Krankheit verbunden sind, direkt oder indirekt mildert.
-
In
einer Ausführungsform
bietet der Schritt eins das Auswählen
eines Ig-Fusionsproteins für
ein Target. Des Weiteren bietet die Methode das Mutieren des so
gewählten
Ig-Fusionsproteins durch strukturelles Ändern der CH2-Domäne der Konstantregion
des Ig-Proteins durch die gleiche Möglichkeit wie oben besprochen.
-
Offenbart
sind hier Methoden zum Behandeln menschlicher Karzinome. Beispielsweise
können
das Immunglobulin, der Antikörper
oder das Ig-Fusionsprotein, die oben besprochen worden sind, in
Kombination mit Standard- oder herkömmlichen Behandlungsmethoden,
wie beispielsweise Chemotherapie, Strahlungstherapie, verwendet
werden, oder sie können
mit einem therapeutischen Arzneimittel oder Toxin sowie einem Lymphokin
oder einem tumorhemmenden Wachstumsfaktor zur Abgabe des therapeutischen
Mittels an die Stelle des Karzinoms konjugiert oder daran geknüpft werden.
-
Techniken
für das
Konjugieren therapeutischer Mittel an Immunglobuline sind allgemein
bekannt (vergleiche z. B. Arnon et al., „Monoklonal Antibodies for
Immunotargeting of Drugs In Cancer Therapy", in Monoklonal Antibodies and Cancer
Therapy (Monoklonale Antikörper
für die
gezielte immunologische Abgabe von Arzneimitteln bei der Krebstherapie,
in Monoklonalen Antikörpern
und Krebstherapie), Reisfeld et al. (Verfasser), S. 243–56 (Alan
R. Liss, Inc., 1985); Hellström
et al., „Antibodies
for Drug Delivery",
in Controlled Drug Delivery (Antikörper zur Arzneimittelabgabe,
in gezielte Arzneimittelabgabe) (zweite Ausgabe), Robinson et al. (Verfasser),
S. 623–53
(Marcel Dekker, Inc., 1987); Thorpe, „Antibody Carriers of Zytotoxic
Agents in Cancer Therapy: A Review", in Monoklonal Antibodies 1984: Biological
And Clinical Applications, (Antikörper als Träger von zytotoxischen Mitteln
bei der Krebstherapie: eine Übersicht,
in Monoklonalen Antikörpern
1984: Biologische und klinische Anwendungen) Pinchera et al. (Verfasser),
S. 475–506
(1985); und Thorpe et al., „The Preparation
and Zytotoxic Properties Of Antibody-Toxin Conjugates (Die Herstellung
und zytotoxischen Eigenschaften von Antikörpertoxinkonjugaten)", Immunol. Rev.,
62: 119–58
(1982)).
-
Alternativ
kann der strukturell geänderte
Antikörper
oder das strukturell geänderte
Ig-Fusionsprotein an
einer Hochenergie-Strahlungsquelle, z. B. einen Radioisotop wie
beispielsweise 131I gekoppelt werden, der, ist
er an der Tumorstelle lokalisiert, zum Abtöten von Zellen in einer Entfernung
von mehreren Zelldurchmessern führt
(vergleiche z. B. Order, „Analysis,
Results, And Future Prospective Of The Therapeutic Use Of Radiolabeled
Antibody in Cancer Therapy",
in Monoklonal Antibodies For Cancer Detection And Therapy (Analyse,
Ergebnisse und zukünftige
Aussichten der therapeutischen Verwendung von radioaktiv markiertem
Antikörper
bei der Krebstherapie, in monoklonale Antikörper für die Krebserfassung und -therapie),
Baldwin et al. (Verfasser), S. 303–16 (Academic Press 1985)).
Noch einer anderen Ausführungsform
gemäß kann der
strukturell geänderte
BR96-Antikörper
an einen zweiten Antikörper
zur Bildung eines Antikörperheterokonjugats
für die
Behandlung von Tumorzellen, wie von Segal im Patent der Vereinigten
Staaten 4676980 beschrieben, konjugiert werden.
-
Noch
andere therapeutische Anwendungen für den erfindungsgemäßen strukturell
geänderten
Antikörper
oder das erfindungsgemäße strukturell
geänderte
Ig-Fusionsprotein umfassen das Konjugieren oder Verknüpfen, z.
B. durch rekombinante DNA-Techniken oder proteinchemische Techniken,
an ein Enzym, das dazu fähig
ist, ein Prodrug zu einem zytotoxischen Arzneimittel umzuwandeln
und die Anwendung dieses Antikörperenzymkonjugats
in Kombination mit dem Prodrug zum Umwandeln des Prodrugs zu einem
zytotoxischen Mittel an der Tumorstelle (vergleiche z. B. Senter
et al., „Anti-Tumor
Effects Of The Antibody-alkaline Phosphatase (Antitumorwirkungen
von antikörperalkalischer
Phosphatase)", Proc.
Natl. Acad. Scie. USA, 85: 4842–46
(1988); „Enhancement
of the in vitro and in vivo Antitumor Activities of Phosphorylated
Mitomycin C and Etoposide Derivatives by Monoklonal Antibody-Alkaline Phosphatase
Conjugates (Verbesserung der Antitumoraktivitäten in vitro und in vivo von
phosphoryliertem Mitomycin C und Etoposidderivaten durch monoklonale
antikörperalkalische
Phosphatasekonjugate)",
Cancer Research 49: 5789–5792
(1989); and Senter, „Activation
of Prodrugs by Antibody-Enzyme Conjugates: A New Approach to Cancer
Therapy (Aktivierung von Prodrugs durch Antikörperenzymkonjugate: ein neuer
Ansatz bei der Krebstherapie)",
FASEB J. 4: 188–193 (1990)).
-
Es
ist daher offensichtlich, dass die vorliegende Erfindung pharmazeutische
Zusammensetzungen, die Immunglobulinmolekale, Antikörper und
Ig-Fusionsproteine enthalten, die alle strukturell geänderte CH2-Domänen
aufweisen, und ihre Verwendung bei Methoden zum Behandeln menschlicher
Karzinome einschließt. Beispielsweise
umfasst die Erfindung pharmazeutische Zusammensetzungen zur Verwendung
bei der Behandlung von menschlichen Karzinomen, die eine pharmazeutisch
wirksame Menge eines strukturell geänderten BR96 und eines pharmazeutisch
akzeptablen Trägers
umfassen.
-
Die
Zusammensetzungen können
den strukturell geänderten
Antikörper
oder das strukturell geänderte Ig-Fusionsprotein
oder strukturell geänderte
Antikörperfragmente
entweder unmodifiziert, an ein therapeutisches Mittel (z. B. ein
Arzneimittel, Toxin, Enzym oder einen zweiten Antikörper) konjugiert
enthalten. Die Zusammensetzungen können außerdem andere Antikörper oder
Konjugate zum Behandeln von Karzinomen (z. B. einen Antikörpercocktail)
umfassen.
-
Die
erfindungsgemäßen Zusammensetzungen
können
mit Hilfe herkömmlicher
Verabreichungsmodi, einschließlich,
jedoch nicht darauf beschränkt,
intrathekaler, intravenöser,
intraperitonealer, oraler, intralymphatischer, oder durch direkte
Verabreichung in den Tumor verabreicht werden. Die intravenöse Verabreichung ist
bevorzugt.
-
Die
erfindungsgemäße Zusammensetzung
kann in einer Reihe verschiedener Dosierformen vorliegen, die flüssige Lösungen oder
Suspensionen, Tabletten, Pillen, Pulver, Zäpfchen, polymere Mikrokapseln oder
Mikrovesikeln, Liposome und injizierbare oder durch Infusion einführbare Lösungen umfassen,
jedoch nicht darauf beschränkt
sind. Die bevorzugte Form hängt
vom Verabreichungsmodus und der therapeutischen Anwendung ab.
-
Die
erfindungsgemäßen Zusammensetzungen
umfassen auch bevorzugt herkömmliche
pharmazeutisch akzeptable Träger
und Hilfsmittel, die im Stand der Technik bekannt sind, wie beispielsweise
menschliches Serumalbumin, Ionenaustauscher, Aluminiumoxid, Lecithin,
Puffersubstanzen wie Phosphate, Glycin, Sorbinsäure, Kaliumsorbat und Salze
oder Elektrolyte wie beispielsweise Protaminsulfat.
-
Der
Praxis der Erfindung gemäß kann der
pharmazeutische Träger
ein Lipidträger
sein. Der Lipidträger kann
ein Phospholipid sein. Des Weiteren kann der Lipidträger eine
Fettsäure
sein. Auch kann der Lipidträger ein
Detergenz sein. Wie hier verwendet, ist ein Detergenz irgendeine
Substanz, die die Oberflächenspannung einer
Flüssigkeit ändert, indem
es sie üblicherweise
reduziert.
-
In
einem Beispiel der Erfindung kann das Detergenz ein nichtionisches
Detergenz sein. Beispiele nichtionischer Detergenzien umfassen,
sind jedoch nicht darauf beschränkt,
Polysorbat 80 (auch als Tween® 80 oder (Polyoxyethylensorbitanmonoleat)
bekannt, Brij® und
Triton® (beispielsweise
Triton® WR-1339
und Triton® A-20).
-
Alternativ
kann das Detergenz ein ionisches Detergenz sein. Ein Beispiel eines
ionischen Detergenz umfasst, ist jedoch nicht darauf beschränkt, Alkyltrimethylammoniumbromid.
-
Außerdem kann
erfindungsgemäß der Lipidträger ein
Liposom sein. Wie bei dieser Anwendung verwendet, ist ein „Liposom" irgendein membrangebundenes
Vesikel, das irgendwelche erfindungsgemäße Moleküle oder Kombinationen derselben
enthält.
-
Der
wirksamste Verabreichungsmodus und die wirksamsten Dosiermöglichkeiten
für die
erfindungsgemäßen Zusammensetzungen
hängen
von der Schwere und dem Verlauf der Krankheit, der Gesundheit des Subjekts
und der Reaktion auf die Behandlung und der Beurteilung durch den
behandelnden Arzt ab.
-
Der
Zusammenhang zwischen den Dosierungen für Tiere verschiedener Größen und
Spezies und Menschen, auf mg/m2 Oberflächenbereich
bezogen, wird von Freireich, E. J., et al., Cancer Chemother. (Krebschemotherapie),
Rep. 50 (4): 219–244
(1966) beschrieben. Änderungen
des Dosierplans können
zum Optimieren des Hemmens des Wachstums der Tumorzellen und der
Abtötungsreaktion
gemacht werden, z. B. können
Dosen geteilt und auf täglicher
Basis verabreicht werden oder die Dosis kann je nach der Situation
im Verhältnis
reduziert werden (z. B. mehrere geteilte Dosen können täglich verabreicht oder je nach
der spezifischen therapeutischen Situation im Verhältnis reduziert
werden).
-
DIE MOLEKÜLE DER ERFINDUNG
-
Die
vorliegende Erfindung stellt als Erstes einen BR96-Antikörper bereit,
der eine menschliche IgG1-Konstantregion aufweist, die in der CH2-Domäne strukturell
geändert
worden ist, wobei Leucin an der Aminosäureposition 235 zu Alanin mutiert
wird; Glycin an der Ammnosäureposition
237 zu Alanin mutiert wird; Glutaminsäure an der Aminosäureposition
318 zu Serin mutiert wird; Lysin an der Aminosäureposition 320 zu Serin mutiert
wird; und Lysin an der Aminosäureposition
322 zu Serin mutiert wird.
-
Die
vorliegende Erfindung stellt auch einen BR96-Antikörper bereit,
der eine menschliche IgG1-Konstantregion
aufweist, die in der CH2-Domäne
strukturell geändert
worden ist, wobei Leucin an der Aminosäureposition 235 zu Alanin mutiert
wird; Glycin an der Aminosäureposition
237 zu Alanin mutiert wird; und Prolin an der Aminosäureposition
331 zu Alanin mutiert wird.
-
Außerdem stellt
die vorliegende Erfindung einen BR96-Antikörper bereit, der eine menschliche IgG1-Konstantregion
aufweist, die in der CH2-Domäne
strukturell geändert
worden ist, wobei Leucin an der Aminosäureposition 235 zu Alanin mutiert
wird; Glycin an der Aminosäureposition
237 zu Alanin mutiert wird; Glutaminsäure an der Aminosäureposition
318 zu Serin mutiert wird; Lysin an der Aminosäureposition 320 zu Serin mutiert
wird; Lysin an der Aminosäureposition
322 zu Serin mutiert wird; und Prolin an der Aminosäureposition
331 zu Alanin mutiert wird.
-
Andere
Ausgestaltungen der Erfindung sind in den anhängenden Ansprüchen 4–18 beansprucht.
Weitere Ausführungsformen,
die durch die beanspruchte Erfindung nicht umfasst werden, sind
ebenfalls in der vorliegenden Beschreibung offenbart.
-
Offenbart
sind hier strukturell geänderte
BR96 oder BR96-Ig-Fusionsproteine. Strukturell geänderte BR96-Antikörper oder
-Ig-Fusionsproteine weisen die variable Region von BR96 und eine
modifizierte Konstantregion auf. Diese Modifikation stellt strukturell
geänderte
BR96-Antikörper
oder Ig-Fusionsproteine
mit der Fähigkeit
bereit, die durch Immunglobulin induzierte Toxizität zu hemmen.
-
Verschiedene
Ausführungsformen
strukturell geänderter
BR96 oder BR96-Ig-Fusionsproteine sind bereits hergestellt worden.
-
In
einer Ausführungsform,
cBR96-A genannt, wurde die gesamte CH2-Domäne von cBR96
deletiert. cBR96-A wird von dem Plasmid exprimiert, das die in SEQ.
ID. NO. 10 gezeigte Sequenz aufweist. cBR96 wird von einem Plasmid
exprimiert, das die Sequenz in SEQ ID NO 9 aufweist.
-
In
einer anderen Ausführungsform,
hBR96-2A genannt, wurde die gesamte CH2-Domäne von hBR96 deletiert.
hBR96-A wird von dem Plasmid exprimiert, das die in SEQ. ID. NO.
12 gezeigte Sequenz aufweist. hBR96 ist ein mutanter BR96, der die
H1-, H2- und H3-Mutationen, die in der PCT-Anmeldung Nr. 95/305444, am
6. März
1996 veröffentlicht,
beschrieben sind, aufweist.
-
In
noch einer anderen Ausführungsform,
hBR96-2B genannt, ist der Leucinrest, der sich an der Aminosäureposition
235 befindet, zu Alanin mutiert. Außerdem ist der Glycinrest,
der sich an der Aminosäureposition
237 befindet, zu Alanin mutiert. Die verwendete Nummerierung der
Aminosäurepositionen
ist bei Kabat et al. Sequences of Proteins of Immunological Interest
(Sequenzen von Proteinen, die von immunologischem Interesse sind),
5. Ausgabe (1991), United States Department of Health and Human
Services, beschrieben.
-
In
einer weiteren Ausführungsform,
hBR96-2C genannt, ist der Glutaminsäurerest an Position 318 zu Serin
mutiert; der Lyuinrest, der sich an Position 320 befindet, ist zu
Serin mutiert; und der Lysinrest, der sich an Position 322 befindet,
ist zu Serin unter Anwendung von Standardprotokollen (Alexander
R. Duncan and Greg Winter „The
binding site of Clq an IgG" (Die
Bindungsstelle für
Clq an IgG), Nature 332: 738 (1988)) mutiert.
-
In
einer anderen Ausführungsform,
hBR96-2D genannt, ist der Prolinrest an Position 331 zu Alanin mutiert
(m-H. Tao et al., „Structural
features of human Immunglobulin G that determine isotypespecific
differences in complement activation (Strukturelle Merkmale von
menschlichem Immunglobulin G, die die isotypspezifischen Unterschiede
in der Komplementaktivierung bestimmen)", J. Exp. Med 178: 661–667 (1993);
Y. Xu et al., „Residue
at position 331 in the IgG1 and IgG4 domains contributes to their
differential ability to bind and activate complement (Der Rest an
Position 331 in den IgG1- und IgG4-Domänen
ist mitbestimmend für ihre
unterschiedliche Fähigkeit
bei, Komplement zu binden und aktivieren)" J. Biol. Chem. 269: 3469–3474 (1994)).
-
In
einer zusätzlichen
Ausführungsform,
hBR96-2E genannt, ist der Leucinrest an Position 235 zu Alanin mutiert;
der Glycinrest, der sich an Position 237 befindet, ist zu Alanin
mutiert; der Glutaminsäurerest,
der sich an Position 318 befindet, ist zu Serin mutiert; der Lysinrest,
der sich an Position 320 befindet, ist zu Serin mutiert; und der
Lysinrest, der sich an Position 322 befindet, ist zu Seren mutiert
(A. Morgan et al., „The
N-terminal end of the CH2 domain of chimeric
human IgG1 anti-HLA-DR is necessary for C1q, Fc(gamma)RI and Fc(gamma)RIII
binding (Das N-terminale Ende der CH2-Domäne von chimärem menschlichem
IgG1-anti-HLA-DR ist für
die Bindung von Clq, Fc(gamma)RI- und Fc(gamma)RIII notwendig)" Immunol. 86: 319–324 (1995)).
-
In
noch einer weiteren Ausführungsform,
hBR96-2F genannt, ist der Leucinrest, der sich an Position 235 befindet,
zu Alanin mutiert; der Glycinrest, der sich an Position 237 befindet,
ist zu Alanin mutiert; und der Prolinrest, der sich an Position
331 befindet, ist zu Alanin mutiert.
-
In
noch einer anderen Ausführungsform,
hBR96-2G genannt, ist der Glutaminsäurerest, der sich an Position
318 befindet, zu Seren mutiert; der Lysinrest, der sich an Position
320 befindet, ist zu Serin mutiert; der Lysinrest, der sich an Position
322 befindet, ist zu Seren mutiert; und der Prolinrest, der sich
an Position 331 befindet, ist zu Alanin mutiert.
-
In
einer anderen Ausführugsform,
hBR96-2H genannt, ist der Leucinrest, der sich an Position 235 befindet,
zu Alanin mutiert; der Glycinrest, der sich an Position 237 befindet,
ist zu Alanin mutiert; der Glutaminsäurerest, der sich an Position
318 befindet, ist zu Serin mutiert; der Lysinrest, der sich an Position
320 befindet, ist zu Seren mutiert; der Lysinrest, der sich an Position
322 befindet, ist zu Serin mutiert; und der Prolinrest, der sich
an Position 331 befindet, ist zu Alanin mutiert.
-
In
Abhängigkeit
von seiner Form kann ein strukturell geänderter BR96-Antikörper oder
ein strukturell geändertes
Fusionsprotein ein monofunktioneller Antikörper, wie beispielsweise ein
monoklonaler Antikörper oder
ein bifunktioneller Antikörper,
wie beispielsweise ein hybrider Antikörper oder ein Heteroantikörper sein. Die
Verwendung von strukturell geändertem
BR96, d. h. als therapeutisches oder diagnostisches Mittel, bestimmt,
welche der verschiedenen Formen von strukturell geändertem
BR96 gebildet wird.
-
Es
bestehen mehrere Möglichkeiten
für die
Antikörperexpression.
Immunexpressionsbibliotheken können
mit Transfektomtechnologie kombiniert werden, d. h. die Gene für die Fab-Moleküle, die
aus der Immunglobulin-Genexpressionsbibliothek stammen, können mit
den erwünschten
Konstantdomänenexonen fusioniert
werden. Diese rekombinanten Gene können dann transfiziert und
in einem Transfektom exprimiert werden, das ein Antikörpermolekül sekretieren
würde.
-
Sind
sie einmal hergestellt, so können
die erfindungsgemäßen Polypeptide
modifiziert, d. h. durch Aminosäuremodifikationen
innerhalb des Moleküls
zur Bildung von Derivatmolekülen
modifiziert werden. Derartige Derivatmoleküle würden die funktionelle Eigenschaft
des Polypeptids beibehalten, nämlich
würde das Molekül, das derartige
Substitutionen aufweist, die Bindung des Polypeptids an das BR96-Antigen oder Teile davon
immer noch gestatten.
-
Es
ist ein allgemein akzeptiertes Prinzip der Proteinchemie, dass gewisse
Aminosäuresubstitutionen, „konservative
Aminosäuresubstitutionen" genannt, häufig in
einem Protein ohne Ändern
entweder der Konformation oder der Funktion des Proteins durchgeführt werden
können.
-
Aminosäuresubstitutionen
umfassen, sind jedoch nicht notwendigerweise darauf beschränkt, Aminosäuresubstitutionen,
die im Stand der Technik als „konservativ" bekannt sind.
-
Derartige Änderungen
umfassen das Substituieren von irgendeinem unter Isoleucin (I),
Valin (V) und Leucin (L) für
irgendeine andere dieser hydrophoben Aminosäuren; Asparaginsäure (D)
für Glutaminsäure (E) und
umgekehrt; Glutamin (Q) für
Asparagin (N) und umgekehrt; und Serin (S) für Threonin (T) und umgekehrt.
-
Andere
Substitutionen können
ebenfalls, je nach der Umgebung der spezifischen Aminosäure und
ihrer Rolle in der dreidimensionalen Struktur des Proteins als konservativ
betrachtet werden. Beispielsweise können Glycin (G) und Alanin
(A) häufig
untereinander austauschbar sein, wie auch Alanin und Valin (V).
-
Methionin
(M), das relativ hydrophob ist, kann häufig mit Leucin und Isoleucin
und manchmal mit Valin getauscht werden. Lysin (K) und Arginin (R)
werden häufig
an Stellen ausgetauscht, an denen das signifikante charakteristische
Merkmal des Aminosäurerests
dessen Ladung ist und die unterschiedlichen pK-Werte dieser beiden
Aminosäurereste
nicht signifikant sind. Noch andere Änderungen können als „konservativ" in spezifischen
Umgebungen betrachtet werden.
-
In
einer erfindungsgemäßen Ausführungsform
ist das Polypeptid im Wesentlichen rein, d. h. von anderen Aminosäureresten
frei, die die Bindung des Polypeptids an sein Target hemmen oder
reduzieren und die Magendarm-Toxizität, die normalerweise während oder
nach der Antikörpertherapie
auftritt, hemmen oder reduzieren würden.
-
NUCLEINSÄUREMOLEKÜLE, DIE DIE VORLIEGENDE ERFINDUNG
KODIEREN
-
Die
Nukleotidsequenzen und Aminosäuresequenzen
der variablen und Konstantregionen von BR96 sind bekannt. Die Sequenz
für die
Immunglobulinkonstantregion ist bekannt und in 18 dargestellt.
Es wurden spezifische Mutationen in der Konstantregion des BR96-Antikörpers durchgeführt. Nucleinsäuremoleküle, die
die sieben oben beschriebenen Mutanten (hBR96-2B bis hBR96-2H) kodieren,
sind wie folgt:
- In hBR96-2B ist Alarm in an den Aminosäurepositionen
235 und 237 durch die Kodone GCU, GCC, GCA oder GCT kodiert.
- In hBR96-2C ist Serin an den Positionen 318, 320 und 322 durch
UCU, UCC, UCA oder UGG kodiert.
- In hBR96-2D ist Alanin in Position 331 durch die Kodone GCU,
GCC, GCA oder GCG kodiert.
- In hBR96-2E ist Alanin an den Positionen 235 und 237 durch die
Kodone GCU, GCC, GCA oder GCG kodiert. Serin an den Positionen 318,
320 und 322 ist durch UCU, UCC, UCA oder UGG kodiert.
- In hBR96-2F ist Alanin an den Positionen 235, 237 und 331 durch
die Kodone GCU, GCC, GCA oder GCG kodiert.
- In hBR96-2G ist Serin an den Positionen 318, 320, 322 durch
UCU, UCC, UCA oder UGG kodiert. Des Weiteren ist Alanin an der Position
331 durch die Kodone GCU, GCC, GCA oder GCG kodiert.
- In hBR96-2H ist Alanin an den Positionen 235, 237 und 331 durch
die Kodone GCU, GCC, GCA oder GCG kodiert. Außerdem ist Serin an den Positionen
318, 320, 322 durch UCU, UCC, UCA oder UGG kodiert.
- Irgendwelche der obigen können
Desoxyribonucleinsäure
(DNA), z. B. eine komplementäre
DNA (cDNA) oder eine Ribonucleinsäure (RNA) sein.
-
IMMUNKONJUGATE
-
Immunkonjugate
(die ganze Antikörper
oder Ig-Fusionsproteine aufweisen) können unter Anwendung einer
umfangreichen Reihe verschiedener chemotherapeutischer Mittel wie
Folsäure
und Anthracycline (Peterson et al., „Transport And Storage of
Anthracyclines In Experimental Systems and Human Leukemia", in Anthracycline
Antibiotics in Cancer Therapy (Transport und Lagerung von Anthracyclinen
in Versuchssystemen und bei der menschlichen Leukämie, in
Anthracyclinantibiotika bei der Krebstherapie), Muggia et al. (Verfasser),
S. 132 (Martinus Nijhoff Publishers (1982); Smyth et al., „Specific
Targeting of Chlorambucil to Tumors With the Use of Monoklonal Antibodies
(Spezifisches Targeting von Chlorambucil auf Tumore unter Verwendung
monoklonaler Antikörper)", J. Natl. Cancer
Inst., 76: 503–510
(1986)) einschließlich
Doxorubicin (DOX) (Yang und Reisfeld „Doxorubicin Conjugated with
a Monoklonal Antibody Directed to a Human Melanoms-Associated Proteoglycan
Suppresses Growth of Established Tumor xenografts in Nude Mice PNAS
(USA) (Doxorubicin, mit einem monoklonalen Antikörper konjugiert, der gegen
ein mit menschlichem Melanom assoziiertes Proteoglycan gerichtet
ist, unterdrückt
das Wachstum von vorhandenen Tumorxenotransplantaten in Nacktmäuse-PNAS
(USA)" 85: 1189–1193 (1988)),
Daunomycin (Arnon und Sela „In
Vitro and in vivo Efficacy of Conjugates of Daunomycin With Anti-Tumor
Antibodies (In vitro- und in vivo-Wirksamkeit von Konjugaten von
Daunomycin mit Antitumor-Antikörpern)" Immunol. Rev., 65:
5–27 (1982))
und Morpholinodoxorubicin (Mueller et al., „Antibody Conjugates With
Morpholinodoxorubicin and Acid-Cleavable Linken (Antikörperkonjugate mit
Morpholinodoxorubicin und säurespaltbaren
Linkem)", Bioconjugate
Chem., 1: 325–330
(1990)) konstruiert werden.
-
BR96
ist bereits an Doxorubicin konjugiert worden und hat sich als bei
der Therapie gewisser Krebsarten oder Karzinome wirksam erwiesen
(Trail, P. A., Willner, D., Lasch, S. J., Henderson, A. J., Casazza,
A. M., Firestone, R. A., Hellström,
I., und Hellström,
K. E. Cure of xenografted human karzinomas by BR96-doxorubicin immunoconjugates
(Heilung von menschlichen Xenotransplantatkarzinomen durch BR96-Doxorubicinimmunkonjugate),
Science, 261: 212–215,
1993).
-
Der
Praxis der Erfindung gemäß können strukturell
geänderte
BR96 in Form von, einschließlich,
unreduziertem IgG, reduziertem strukturell geändertem IgG und Fusionsproteinen
(PCT-Anmeldung Nr. 95/305444, am 6. März 1996 veröffentlicht) verwendet worden.
-
Geeignete
therapeutische Mittel zur Verwendung beim Herstellen des Immunkonjugats
umfassen Pseunomonas exotoxin A (PE) in entweder der nativen PE-
oder der LysPE40-Form. LysPE40 ist eine gestutzte Form, die einen
genetisch modifizierten Aminoterminus aufweist, der für Konjugationszwecke
einen Lysinrest umfasst. Doxorubicin ist ebenfalls ein geeignetes
therapeutisches Mittel.
-
Zusätzliche
Beispiele therapeutischer Mittel umfassen, sind jedoch nicht darauf
beschränkt,
Antimetabolite, Alkylierungsmittel, Anthracycline, Antibiotika und
Antimitotika.
-
Antimetabolite
umfassen Methotrexat, 6-Mercaptopurin, 6-Thioguanin, Cytabarin,
5-Fluorouracildecarbazin.
-
Alkylierungsmittel
umfassen Mechlorethamin, Thiotepachlorambucil, Melphalan, Carmustin
(BSNU) und Lomustin (CCNU), Cyclothosphamid, Busulfan, Dibrommannit,
Streptozotocin, Mitomycin C und cis-Dichlordiaminplatin(II)(DDP-)Cisplatin.
-
Anthracycline
umfassen Daunorubicin (früher
Daunomycin) und Doxorubicin (hier auch als Adriamycin bezeichnet).
Zusätzliche
Beispiele umfassen Mitozantron und Bisantren.
-
Antibiotika
umfassen Dactinomycin (früher
Actinomycin), Bleomycin, Mithramycin und Anthramycin (AMC).
-
Antimitotika
umfassen Vincristin und Vinblastin (die allgemein als Vincaalkaloide
bezeichnet werden).
-
Andere
zytotoxische Mittel umfassen Procarbazin, Hydroxyharnstoff, Asparaginase,
Corticosteroide, Mytotan (O,P'-(DDD)),
Interferone.
-
Weitere
Beispiele zytotoxischer Mittel umfassen, sind jedoch nicht darauf
beschränkt,
Ricin, Bryodin, Gelonin, Supporin, Doxorubicin, Taxol, Zytochalasin
B, Gramicidin D, Ethidiumbromid, Etoposid, Tenoposid, Colchicin,
Dihydroxyanthracindion, 1-Dehydrotestosteron und Glukocorticoid.
-
Offensichtlich
sind Analoge und Homologe derartiger therapeutischer und zytotoxischer
Mittel in der vorliegenden Erfindung eingeschlossen. Beispielsweise
gibt es zum chemotherapeutischen Mittel Aminopterin einen korrelativ
verbesserten Analog, nämlich
Methotrexat.
-
Des
Weiteren ist der verbesserte Analog von Doxorubicin ein Fe-Chelat.
Außerdem
ist der verbesserte Analog von 1-Methylnitrosoharnstoff Lomustin.
Des Weiteren ist der verbesserte Analog von Vinblastin Vincristin.
Auch ist der verbesserte Analog von Mechlorethamin Cyclophosphamid.
-
METHODEN ZUM HERSTELLEN ERFINDUNGSGEMÄSSER MOLEKÜLE
-
Es
gibt mehrere Ansätze
zum Herstellen ortsspezifischer Mutation in der CH2-Domäne eines
Immunglobulinmoleküls.
Ein Ansatz umfasst die PCR-Amplifikation der CH2-Domäne mit den
Mutationen, mit anschließender
homologer Rekombination der mutierten CH2 in
den Vektor, der das erwünschte
Immunglobulin, z. B. hBR96-2, enthält. Beispielsweise sind hBR96-2B
und hBR96-2D durch diese Methode hergestellt worden.
-
Ein
anderer Ansatz wäre
das Einführen
von Mutationen durch ortsgerichtete Mutagenese von einzelsträngiger DNA.
Beispielsweise weist der Vektor pD17-hG1b, der nur die Konstantregion
von IgG1 und nicht die V-Domäne
von hBR96 enthält,
den f1-Ursprung der Replikation auf. Dies verleiht dem Vektor die
Eigenschaften eines Phagemids und es können ortsgerichtete Mutageneseversuche
der Methode von Kunkel, et al. entsprechend durchgeführt werden
(Kunkel, T. A., J. D. Roberts und R. A. Zakour, 1987 Methods Enzymol. 154:
367–383)
wie in dem Muta-Gene®-Kit zur in-vitro-Mutagenese
von Phagemiden, Version 2, von Bio-Rad bereitgestellt. Beispielsweise
wurden hBR96-2B, -C, -D, -E, -F, -G und -H mit dieser Methode hergestellt.
-
Zum
vollständigeren
Verständnis
der hier beschriebene Erfindung werden folgende Beispiele aufgeführt. Man
sollte sich im Klaren darüber
sein, dass diese Beispiele nur veranschaulichenden Zwecken dienen und
nicht als den Umfang dieser Erfindung in irgendeiner Weise einschränkend ausgelegt
werden sollen.
-
BEISPIEL 1
-
Es
wurde das folgende Standard-ELISA-Protokoll benutzt.
-
Materialien:
Imqmulon2 96-Mikrotiterplatten und Probeverdünnungskonzentrat von Genetic
Systems (10x); das Antikörperkonjugat
war Ziege-anti-Mensch-Kappa-HRP, an Maus adsorbiert, Southern Biotech,
in einer Verdünnung
von 1:10.000 mit Konjugatverdünnungsmittel
von Genetic Systems (1x); EIA Chromogen-Reagenzmittel (TMB) von
Genetic Systems (1:100); EIA-gepuffertes Substrat von Genetic Systems
(1x); der primäre
Antikörper
oder das Antigen waren Affinipure F(ab')2 Fragment-Ziege-anti-Mensch-IgG, Fc-fragmentspezifisch
(Jackson Immuno Research), Ziege-anti-Mensch-Kappa-UNLB (Southern
Biotechnology Associates), Ley-HSA (Alberts
Research Council).
-
Methoden:
Der primäre
Antikörper
oder das Antigen in 0,05 M Carbonat/Bicarbonatpuffer auf 1,0 μg/ml verdünnen. 100 μl der verdünnten Lösung pro
Vertiefung in Immulon 2-Platten eingeben. Die Platten dicht verschließen und
bei 4°C über Nacht
inkubieren.
-
Lösungen entfernen
durch Invertieren der Platten und blotten auf Papiertüchern. Zum
Blockieren 200 μl/Vertiefung
Probeverdünnugsmittelkonzentrat
von Genetic Systems (1x) hinzugeben. Mindestens 1 Stunde bei Raumtemperatur
inkubieren und die Inhalte der Platten dann wegschütten. Die
Platten 3x in physiologischer Kochsalzlösung/Tween waschen. Platten
trockenblotten. Die Platten bei Raumtemperatur trocknen lassen (45
min bis 1 Stunde). Die Platten dicht verschließen und bei 4°C lagern.
-
Die
Proben wie folgt prüfen.
Die Proben und Standards in Probeverdümmungsmittel im Verhältnis von 1:10
verdünnen.
Serienverdünnungen
in einzelnen Rundbodenplatten durchführen. Von den endgültigen Verdünnungen
100 μl/Vertiefung
in mit Antigen beschichtete Assayplatten übertragen; dann bei 4°C über Nacht inkubieren.
Die Platten 3x mit physiologischer Kochsalzlösung/Tween waschen.
-
Zur
Konjugation, 100 μl/Vertiefung
Antikörper-HRP-Konjugat
in Konjugatverdünnungsmittel
von Genetic Systems (1x) hinzugeben. Die Platten 60 min lang bei
Raumtemperatur inkubieren. Die Platten 3x in physiologischer Kochsalzlösung/Tween
waschen.
-
100 μl/Vertiefung
EIA-Chromogenreagenz von Genetic Systems (TMB), 1:100, in EIA-gepuffertem Substrat
(1x) hinzugeben. 15 min lang bei Raumtemperatur inkubieren und die
Reaktion mit 100 μl/Vertiefung 1
N H2SO4 stoppen.
Die Platte bei 450/630 nm im EIA-Plattenableser auswerten.
-
BEISPIEL 2
-
KONSTRUKTION VON CH2-DELETIERTEN
BR96-MOLEKÜLEN
-
Strategie
für das
Deletieren von CH2-Domänen: Um CH2-deletierte
BR96-Moleküle
zu konstruieren, wurden die Scharnier-, CH2-
und CH3-Domänen von chimärem BR96
und humanisiertem BR9696-2-IgG1-Molekülen entfernt durch Restriktionsverdau
mit Eco47-III in nichtkodierenden Regionen humanisiert. Die Scharnier-
und CH3-Domänen wurden durch Polymerasekettenreaktion
(PCR) aus einem menschlichen IgG1-(pNγ1.14-)Molekül amplifiziert, dem die CH2-Domäne
fehlt. Zwei Oligonukleotide (sense 49mer, antisense 50mer), die
zu den Sequenzen zu beiden Seiten der IgG1-Konstantregion homolog
waren, wodurch Eco47-III-Stellen konserviert wurden, wurden synthetisiert.
Die amplifizierten Scharnier- und
CH3-Domänen-PCR-Fragmente
wurden in Eco47-III-Stellen in BR96-IgG1-Molekülen durch homologe in vivo-Rekombination
eingesetzt (P. Subeck et al, Nucleic Acid Research (1993) 21: 3601–3602).
Die neuen BR96-IgG1-Moleküle
wurden durch Restriktionskartierung und Sequenzierung verifiziert.
-
Es
wurde eine Sewing-PCR-Strategie für die Konstruktion des CH2-deletierten menschlichen IgG1 (pNγ1.14) benutzt
(Robert M. Horton, et al. (1990) Biotech 8 (5) S. 528).
-
Die
CH1-Domäne
wurde als 580 bp-Fragment mit einem sense-Oligonuldeotid (5' TGG CAC CGA AAG CTT
TCT GGG GCA GGC CAG GCC TGA 3')
(Primer A) und einem antisense-Oligonukleotid
(5' TCC GAG CAT
GTT GGT ACC CAC GTG GTG GTC GAC GCT GAG CCT GGC TTC GAG CAG ACA
3') (Primer B) aus einem
linearisierten menschlichen IgG1-Konstantregionsvektor
(pNγ1.7)
amplifiziert. Das PCR-Fragment erstreckt sich vom 5'-Ende der Hind-III-Stelle (fett gedruckt) über den
Cel-II-, Sal-I-, Dra-III-, Kpn-I-, 6 bp-Nukleotidspacer und Mro-I-Stellen
(fettgedruckt) am 3'-Ende
der CH1-Domäne.
-
Die
CH3-Domäne
wurde dann teilweise (zur Xba-I-Stelle) mit einem sense-Primer (5' GTC GAC CAC CAC
GTG GGT ACC AAC ATG TCC GGA GCC ACA TGG ACA GAG GCC GGC T 3') (Primer C) und
einem antisense-Primer (5' CTG
GTT CTT GTT CAT CTC CTC TCT AGA TGG 3') (Primer D) aus einem lirearisierten menschlichen
IgG1-Konstantregionsvektor (pNγ1.7)
teilweise amplifiziert. Ein PCR-Fragment (etwa 150 bp) mit Sal-I-,
Dra-III-, Kpn-I-, 6 Nukleotidspacer und Mro-1-Stellen (fettgedruckt) an seinem 5'-Ende erstreckt sich nur
bis einschließlich
zur Xba-1-Stelle (fettgedruckt) innerhalb der CH3-Domäne.
-
Die
CH1- und CH3-Partial-PRC-Fragmente
wurden in einer PCR ohne irgendwelchen Primer kombiniert. Die Reaktion
wurde für
zwei volle Zyklen von Denaturierung und Reassoziierung durchgeführt, um
es den Fragmenten zu erlauben, sich an der homologen Region an den
3'-Enden zu kombinieren.
Die (oben beschriebenen) Primer A und D wurden der Reaktion hinzugegeben
und der PCR-Zyklus
wurde abgeschlossen. Die Polymerase verlängert die DNA mit dem Primer
A und Primer D, wobei sich ein PCR-Fragment voller Länge (660
bp) ergibt. Das neu verlängerte
PCR-Fragment ist in folgender Reihenfolge vom 5'-Ende bis zum 3'-Ende angeordnet: Hind-III-CH1-Cel-II-Sal-I-Dra-III-Kpn-I-6bp-Spacer-Mro-1-CH3-partial-Xba-1.
-
Das
kombinierte PCR-Fragment, mit den CH1- und
CH3-Partialdomänen, wurde dann durch eine blunt-end-Ligation
in die Sma-I-Stelle von einem pEMBL18-Vektor kloniert und die Sequenz
wurde durch Didesoxysequenzierung bestätigt (Sanger et al. (1977)
PNAS (USA) 74: 5463–5466).
-
Um
das CH1 und Partial-CH3 in
einen Säugerexpressionsvektor
zu überführen, wurden
sowohl der pEMBL18- als auch der pNγ1.7-Vektor mit Hind-III und
Xba-I verdaut. Die Hind-III- und Xba-I-Fragmente wurden an den entsprechenden
Stellen von dem einem linearisierten pNγ1.7-Vektor ligiert. Das neue
Konstrukt mit CH1 und einer vollen CH3-Domäne
wurde pNγ1.10-Vektor
genannt.
-
Das
Scharnierfragment wurde aus einem mit Hind-III verdauten pNγ1.7-Vektor
amplifiziert, wobei die Primer so gestaltet waren, dass sie das
Scharnierexon mit einer Sal-I- und einer Dra-III-Klonierstelle an
jedem Ende flankierten. Solche Stellen befinden sich auch zwischen
den CH1- und CH3-Domänen des
pNγ1.10-Konstrukts.
Das sense-Oligonukleotid (5' ACC
ATG GTC GAC CTC AGA CCT GCC AAG AGC CAT ATC 3') mit einem 6 bp-Spacer und einer Sal-I-Klonierstelle
(fettgedruckt) und das antisense-Oligonukleotid
(5' CAT GGT CAC
GTG GTG TGT CCC TGG ATG CAG GCT ACT CTA G 3') mit einem 6 bp-Spacer und einer Dra-III-Klonierstelle
(fettgedruckt) wurden zur Amplifikation des Scharnierfragments (250
bp) eingesetzt.
-
Das
Scharnierregions-PCR-Fragment wurde in eine SMA-I-Stelle am pEMBL18
durch blunt-end-Ligation
kloniert. Sowohl der pEMBL18 mit der Scharnierdomäne als auch
der pNγ1.10
mit den CH2- und CH3-Domänen wurden
mit Sal-I und Dra-III verdaut. Das so verdaute Scharnierfragment
wurde in die entsprechenden Stellen des mit Sal-I und Dra-III lirearisierten
pNγ1.10-Vektors
kloniert. Das neue Konstrukt, das nun die CH1-, Scharnier-
und CH3-Domänen trägt, wurde pNγ1.11 genannt.
-
Um
das endgültige
CH2-deletierte menschliche IgG1-Konstrukt
herzustellen, wurden sowohl das pNγ1.11-Konstrukt als auch der
pNγ1.11-Vektor
mit BamH1 und HindIII verdaut. Ein Fragment, das die CH1-, Scharnier-
und CH3-Domänen enthielt, wurde in den
linearisierten pNγ1.1
1-Vektor kloniert. In dem neuen Konstantregion-IgG1-Konstrukt fehlt
die CH2-Domäne und es wird pNγ1.14 genannt
(11).
-
Zum
Verdauen von BR96 IgG1 mit Eco47-III wurde ein Restriktionsfragment
mit Scharnier-, CH2- und CH3-Domänen an der
Konstantregionssequenz des BR96 IgG1-Vektors sowohl in chimären als
auch humanisierten Molekülen
identifiziert. Das 5'-Ende
dieses Fragments liegt innerhalb des Introns zwischen CH1 und dem Scharnier und das 3'-Ende befindet sich
innerhalb des CH3-Introns des BR96 IgG1-Moleküls. Die
Scharnier-, CH2- und CH3-Domänen (1,368
kb-Fragment) wurden aus den BR96 IgG1-Molekülen durch Restriktionsverdau
mit Eco47-III- an entfernt. Eco47-III ist ein blunt-end cutter.
Die mit diesem Enzym verdaute BR96 IgG1-DNA erfordert keine Vorbehandlung
vor dem Klonieren. 12 zeigt eine schematische Darstellung
des pD17-hBR96-2-Vektors, die die Eco47-III-Stellen, die beim Klonieren
verwendet wurden, zeigt.
-
Das
CH2-deletierte BR96 IgG1 wurde dann wie folgt konstruiert. Die Scharnier-
und CH3-Domänen wurden
aus einem CH2-deletierten L6 IgG1-(pNγ1.14-)Konstrukt
mit einem sense-Oligonukleotid
(5' CAGGGAGGGAGGGTGTCTGCTGGAAGCCAGGCTCAGCGCTGACCTCAG
A 3') homolog zur
Konstantregionsequenz von IgG1 am 5'-Ende der Eco47-III-Stelle (fettgedruckt)
und einem antisense-Oligonukleotid (5' GGAAAGAACCATCACAGTCTCGCAGGGGCCCAGGGCAGCGC
TGGGTGCTT 3') homolog
zur Konstantregion von IgG1 am 3'-Ende
der Eco47-III-Stelle (fettgedruckt) amplifiziert. Die Eco47-III-Stelle
am 3'-Ende des pNγ1.14-Konstrukts
wird durch den Klonierungsvorgang modifiziert. Die Eco47-III-Stelle
wird so in einen Antisenseprimer eingeführt und bei der Amplifikation
der Scharnier- und CH3-Domänen benutzt.
-
Der
pD17-BR96 IgG1-Vektor wurde mit Eco47-III verdaut und die Scharnier-,
CH2 und CH3-Domänen wurden
entfernt. Der lirearisierte pD17-BR96 IgG1-Vektor wurde mit äquimolaren
Mengen von Scharnier- und CH3-PCR-Fragmenten
gemischt. Die Kotransformation des PCR-Fragments mit linearisierter
DNA in kompetente E. coli DH5a Zellen führte zu einem rekombinanten
Molekül,
durch homologe Rekombination in Bakterien vermittelt. In diesem
Konstrukt fehlt die CH2-Domäne der BR96
IgG1-Moleküle
und wird pD17-BR96-dCH2 genannt (13).
-
1,9
Gramm CH2-deletierter chimärer BR96
wurde als Rohmaterial aus 89 1 überstehender
Kulturlösung
erhalten.
-
BEISPIEL 3
-
Die
Toxizität,
Lokalisierung und Ausscheidung von CH2-deletiertem
chimärem
BR96 wurde in vivo wie folgt getestet.
-
Drei
Hunde erhielten 400 mg/m2 cBR96-A, dem CH2-Deletionsmutanten von chimärem BR96
und zwei erhielten chimären
BR96. Beide Moleküle
waren unter milden Bedingungen reduziert und alkyliert worden. Dies
ist notwendig, um die Dimerisierung der Deletionsmutante zu einer
vierwertige Form zu verhindern. Beide Kontrollhunde erfuhren die
typische GI-Toxizität
und keine der drei, die die Mutante erhielten, wiesen irgendeine
Toxizität
auf. Die Kontrollhunde und zwei der Testhunde wurden nach 1 h getötet, um
Gewebe des Zwölffingerdarms
zum Messen der Antikörperposition
zu erhalten. Beide Kontrollhunde wiesen eine auffallend sichtbare
Magendarmpathologie auf und die Testhunde hatten ein normal aussehendes
Magendarmgewebe. Der dritte Hund zeigt weiterhin keine Anzeichen
von Toxizität.
-
Ergebnisse:
Lokalisation des CH2-deletierten cBR96 (cBR96-A) zum Magendarmkanal
erfolgte in signifikantem Maße
bei Hunden, die mit 400 mg/m
2 behandelt
worden waren, obwohl das intakte chiBR96 etwas besser lokalisierte.
Die Lokalisationsdaten zeigen, dass ungefähr äquivalente Mengen von intaktem
und CH
2-deletiertem cBR96 bei diesen Hunden
dem Magendarmkanal zugeführt
wurden. TABELLE 5. LOKALISATION VON CBR96 IM MAGENDARMGEWEBE
| Gruppe | Tier | Spezifische
Lokalisierung | Durchschnittswert |
| cBR96 | Nr.
271
Nr. 272 | 155
114 | 135 |
| cBR96-A | Nr.
273
Nr. 274 | 126
52 | 89 |
-
Unter
Berücksichtigung
des durchschnittlichen Grades der spezifischen Lokalisation wurde
eine Menge von cBR96-A, die zumindest 66% der Menge an cBR96 entsprach,
dem Zielorgan der Toxizität,
nämlich dem
Zwölffingerdarm,
zugeliefert. Auf der Basis der Ermittlung der Dosisbereiche, die
mit cBR96 bei Hunden erfolgte (einige klinische Anzeichen von Toxizität wurden
bei Dosen von 10 mg/m2 beobachtet), könnte dieser Unterschied,
selbst wenn es sich um einen realen Unterschied handeln sollte,
den Unterschied zwischen signifikanter Toxizität und keiner Toxizität nicht
erklären;
vorbehaltlich einer mikroskopischen histopathologischen Untersuchung
zeigte die bisherige Beurteilung, dass Hunde, die mit cBR96-A behandelt
worden sind, keine Toxizität
aufweisen. Diese Beurteilung beruhte auf der Analyse von 2 gefrorenen
Blöcken
pro Hund und 2 Schnitten pro Block. Die Replikate waren ziemlich
gut. Wir untersuchten auch aufbewahrte gefrorene Gewebe aus Hunden,
die mit nativem cBR96 oder F(ab)2BR96 behandelt worden waren, und
die Lokalisationsniveaus bei diesen Geweben betrugen 110 bzw. 0,
im Einklang mit unseren früheren
Daten.
-
Angenommen,
dass bei nur leicht erhöhten
(2X) Dosen von cBR96-A keine Toxizität besteht, so zeigen diese
Daten an, dass die CH2-Domäne mit der
Induktion akuter Gastroenteropathie verbunden ist und dass die Entfernung
dieser Domäne
die Induktion von durch BR96 vermittelter Gastroenteropathie verhindert.
-
Diese
Studie bestätigt
die Ergebnisse, die zeigen, dass F(ab')2 im Hundemodell nicht toxisch ist
und dass die Toxizität
durch die Konstantregion vermittelt wird. Die CH2-Deletionsmutante
ist ein Kandidat für
das klinische Targeting von Wirkstoffen. Aufgrund der sehr langen
Halbwertzeit von chimärem
BR96 sollte eine gewisse Abnahme der Halbwertzeit bei der Mutante
hinnehmbar sein.
-
1 zeigt
die Messwerte für
die Ausscheidung von cBR96-A in Hunden mit hoher Ley Expression. Bei
der Studie wurde ein chimärer
cBR96-2 zum Vergleich mit der Konstantregionsmutante von verwendet.
-
CBR96-2
wurde schneller ausgeschieden als der chimäre BR96. Die Lokalisation von
cBR96-A am Magendarmepithel wird nicht signifikant durch diese schnellere
Ausscheidung beeinflusst. Mehr als genug cBR96 wurde lokalisiert,
um eine etwaige Toxizität
hervorrufen zu können.
-
Diskussion:
Die Konstantregion von chimärem
igG ist für
die Magendarm-Toxizität,
die bei klinischen Versuchen zu sehen war, z. B. bei chiBR96-dox,
verantwortlich. Die Magendarm-Toxizität, die im Hundemodell zu sehen
war, ist der klinischen Toxizität
sehr ähnlich.
Sowohl beim Menschen als auch beim Hund vermittelt die Verabreichung
des unkonjugierten Antikörpers
eine akute Magendarm-Toxizität,
die durch das schnelle Einsetzen von Erbrechen, oft mit Blut, gekennzeichnet
ist.
-
Beim
Menschen ist die Blutung auf den Magenfundus begrenzt, wobei sie
die Erosion der Oberflächenschleimhaut
des Magens verursacht. Obwohl die Pathologie der Wunde beschränkt ist
und sie sich wieder zurückbildet,
definiert die extreme Natur der Übelkeit
und des Erbrechens, die durch Antiemetika nicht gemildert wird,
sie als die dosisbegrenzende Toxizität.
-
Diese
Toxizität
wird bei Mensch und Hund durch das Antikörpermolekül als solches vermittelt. Bei
höheren
Dosen des Antikörper-dox-Konjugats
ist im Hundemodell wahrscheinlich aufgrund von Doxorubicin noch eine
zusätzliche
Toxizität
zu erkennen. Obwohl das intakte IgG von BR96 beim Hund und beim
Menschen Toxizität
verursacht, ist das F(ab')2-Molekül (zweiwertig,
und nur ohne Konstantregion) bei Hunden nicht toxisch. Dieser Befund
hat unsere Versuche mit hohen Dosen motiviert und diese verbessern
die Affinität
und Spezifizität
von BR96 zum Tumorartigen.
-
Bekanntlich
vermittelt die CH2-Domäne die Komplement- und die
FcR-Bindung. Es war bisher nicht bekannt, dass die strukturelle Änderung
der CH2-Domäne zu einer Hemmung der durch
Immunglobulin induzierten Toxizität führen würde.
-
TOXIKOLOGIESTUDIE VON hBR96-2B
-
Die
Toxizitätsstudie
an hBR96-2B in Hunden mit starker Expression von Lewis Y (n = 2)
zeigte, dass eine Dosis von 400 mg/m2 weder
Bluterbrechen noch blutigen Stuhl verursachte, im Gegensatz zu BR96,
das stets ein oder beide Indikationen verursacht. Ein nach 24 h
getöteter
Hund wies ein im Ganzen normales makroskopisches Aussehen des Magendarmkanals
auf, wiederum im deutlichen Gegensatz zu chimärem BR96, der hämorrhagische
Läsionen
und Schleimhauterosionen hervorruft.
-
BEISPIEL 4
-
Die
Polymerasekettenreaktion (PCR) ist eine vielseitige Technik, die
weitverbreitete Anwendung findet, für die Amplifizierung und darauffolgende
Modifizierung von Immunglobulingenen. Die Schnelligkeit und Genauigkeit,
mit der Antikörpergene
in vitro modifiziert werden können,
hat zur Bildung einer Reihe verschiedener neuartiger Antikörpergene
geführt.
Beispielsweise sind PCR-Methoden zum gentechnischen Herstellen von
Antikörpern
mit einer erhöhten
Affinität
zum Antigen, zum „Humanisieren" von Antikörpern und
zum Modulieren der Effektorfunktion verwendet worden (Marks, J.
D., A. D. Griffiths, M. Malmqvist, T. Clackson, J. M. Bye und G.
Winter. 1992. Bypassing immunization: high affinity human antibodies
by chain shuffling (Umgehen des Immunisierens: menschliche Hochaffinitätsantikörper durch
Kettenmischen). Bio/Technology 10: 779–783; Rosok, M. J., D. E. Yelton,
L. J. Harris, J. Bajorath, K. -E. Hellström, I. Hellström, G. A.
Cruz, K. Kristensson, H. Lin, W. D. Huse und S. M. Glaser. 1996.
A combinatorial library strategy for the rapid humanization of antikarzinoma
BR96 (Kombinatorische Bibliotheksstrategie für die schnelle Humanisierung
von Antikarzinom-BR96) Fab. J. Biol. Chem. 271: 22611–22618;
Morgan, A. N., D. Jones, A. M. Nesbitt, L. Chaplin, M. W. Bodmer
und S. Emtage. 1995. The N-terminal end of the CH2 domain of chimeric
human IgG1 anti-HLA-DR is necessary for Clq, FcγRI and FcγRIII binding (Das N-terminale
Ende der CH2-Domäne
von chimärem menschlichen
IgG-Anti-HLA-DR
ist für
die Clq-, FcγRI-
und FcγRIII-Bindung
erforderlich). Immunology. 86: 319–324) Als Teil einer umfangreicheren
Studie wollten wir verschiedene ortsspezifische Mutationen in die CH2-Konstantdomäne von menschlichem IgG1 einführen. Es
wurden sechs spezifische Aminosäurereste,
die über
die ganze CH2-Domäne verteilt sind, denen vorher
Rollen bei der Immuneffektorfunktion zugeschrieben worden waren,
als Targets für
die Mutagenese ausgewählt
(Morgan, A. N., D. Jones, A. M. Nesbitt, L. Chaplin, M. W. Bodmer
und S. Emtage. 1995. The N-terminal end of the CH2 domain of chimeric
human IgG1 anti-HLA-DR is necessary for Clq, FcγRI and FcγRIII binding (Das N-terminale
Ende der CH2-Domäne
von chimärem
menschlichen IgG-Anti-HLA-DR ist für die Clq-, FcγRI- und FcγRIII-Bindung
erforderlich). Immunology. 86: 319–324; Duncan, A. R. und G.
Winter. 1988. The binding site for Clq an IgG (Die Bindungsstelle
für C1q an
IgG). Nature 332: 738–740;
Tao, M. -H., R. I. F. Smith und S. L. Morrison. 1993. Structural
features of human Immunglobulin G that determine isotype-specific
differences in complement activation (Strukturelle Merkmale von
menschlichem Immunglobulin G, die isotypspezifische Unterschiede
der Komplementaktivierung bestimmen) J. Exp. Med. 178: 661–667). Fünf der sechs
Reste wurden in zwei Clustern gruppiert – wobei ein Cluster aus zwei
Resten besteht, die zwei Aminosäuren
von einander entfernt liegen (Position 1 oder L1); und ein zweiter
Cluster aus drei Resten besteht, der sich über eine Sequenz von fünf Aminosäuren (L2)
erstreckt. Die überbleibende
Aminosäureposition
(L3) ergab die Gesamtzahl von sechs Resten. Wir waren daran interessiert,
einen Satz mutanter CH2-Domänen-IgG
zu konstruieren, bestehend aus jeder L-Mutation für sich,
aber auch in Kombination mit anderen L-Mutanten (z. B. L1; L1; und
L2; L1, L2 und L3; usw.) bestehen.
-
Es
sind verschiedene in vitro-Methoden beschrieben worden, wobei die
PCR dazu verwendet wird, distal positionierte ortspezifische Mutationen
innerhalb einer Gensequenz gleichzeitig einzuführen (Ho, S. N., H. D. Hunt,
R. M. Horton, J. K. Pullen und L. R. Pease. 1989. Site-directed
mutagenesis by overlap extension (Ortsgerichtete Mutagenese durch Überlappungsverlängerung).
Gene 77: 51–59;
Ge, L. und P. Rudolpf. 1996. Simultaneous introduction of multiple
mutations using overlap extention PCR (Simultane Einführung multipler Mutationen
mit Hilfe von Überlappungsverlängerungs-PCR).
BioTechniques 22: 28–30).
Alternativ dazu ist ein Rekombinations-PCR (RPCR) genanntes in vivo-Verfahren
ebenfalls erfolgreich zum schnellen und effizienten Bilden von distal
positionierten ortsspezifischen Mutationen verwendet worden (Jones,
D. H. und S. C. Winistorfer. 1993. Use of polymerase chain reaction
for making recombinant constructs (Anwendung der Polymerasekettenreaktion
zum Bilden rekombinanter Konstrukte). S. 241–250. In B. A. White (Verfasser),
Methods in Molecular Biology (Methoden der Molekularbiologie), Band
15, Humana Press Inc., Totowa, NJ, Jones, D. H. und B. H. Howard,
1991. A rapid method for recombination and site-specific mutagenesis
by placing homologous ends an DNA using polymerase chain reaction
(Eine Schnellmethode für
die Rekombination und ortsspezifische Mutagenese durch Positionieren
von homologen Enden an DNA unter Anwendung der Polymerasekettenreaktion).
BioTechniques 10: 62–66.)
Bei der RPCR wird die Rekombinationsmaschinerie von E. coli zum
Bilden intakter kreisförmiger
rekombinanter Plasmide aus einer transfizierten Mischung von linearem durch
PCR gebildetem Produkt und linearisiertem Vektor verwendet. Die
in vivo-Rekombination wird durch das Verbinden von Nukleotidsequenzen
vermittelt, die in die 5'-Enden
beider PCR-Primer eingeführt
werden, und die zu DNA-Sequenzen, die durch den Vektor kodiert werden,
homolog sind. In diesem Bericht beschreiben wir eine Erweiterung
des RPCR-Verfahrens für
das gleichzeitige Einführen
komplexer Kombinationen von Mutationen in eine Antikörper-CH2-Domäne.
-
Humanisierte
BR96-Variabelregion-Schwer- und Leichtkettengene, die vorher als
zusammengebautes aktives Fab-Fragment in einen M13-Phagenexpressionsvektor
kloniert und coexprimiert worden waren, stellten das Ausgangsmaterial
dar (Rosok, M. J., D. E. Yelton, L. J. Harris, J. Bajorath, K. -E.
Hellström,
I. Hellström, G.
A. Cruz, K. Kristensson, H. Lin, W. D. Huse und S. M. Glaser. 1996.
A combinatorial library strategy for the rapid humanization of antikarzinoma
BR96 (Kombinatorielle Bibliotheksstrategie für die schnelle Humanisierung
eines Antikarzinom-BR96) Fab. J. Biol. Chem. 271: 22611–22618).
Die Schwer- und Leichtketten-V-Gene wurden durch PCR aus einer einzelsträngigen M13-DNA-Template
amplifiziert und durch in vivo-Rekombination subldoniert (Jones,
D. H. und B. H. Howard, 1991. A rapid method for recombination and
site-specific mutagenesis by placing homologous ends an DNA using
polymerase chain reaction (Eine Schnellmethode für die Rekombination und ortsspezifische
Mutagenese durch Positionieren von homologen Enden an DNA unter
Anwendung der Polymerasekettenreaktion). BioTechniques 10: 62–66) in
die Vektoren pD17-hG1a und pD16-hCκ, um pBR96-hG1a bzw. pBR96-hCκ zu bilden.
pD17-hG1a und pD16-hCκ sind
eukaryotische Immunglobulinexpressionsvektoren, die von pcDNA3 deriviert
sind (Invitrogen, San Diego, CA). Das Plasmid pD17-hG1a wurde noch
weiter durch ortsspezifische Mutagenese weitermodifiziert, um mit
Hilfe von Standardverfahren zwei Eco47-III-Restriktionsstellen,
die die Immunglobulinscharnier-CH2-CH3- Domänen flankieren, einzuführen. Der
Empfangsvektor wurde dann durch Verdauen von pBR96-hG1a mit Eco47-III,
Isolieren des Rückgrats
des Vektors durch Agarosegelelektrophorese, gefolgt vom Extrahieren
der Vektor-DNA aus der ausgeschnittenen Gelscheibe mit Hilfe des
Qiagen-Gelextraktionskits (Qiagen, Chatsworth, CA) hergestellt.
-
Die
Strategie für
das Einführen
multipler Mutationen innerhalb des Immunglobulin-CH2-Gens,
in 24 gezeigt, gründet sich auf die homologe
in vivo-Rekombination mehrerer unabhängig amplifizierter PCR-Produkte
miteinander sowie mit der pBR96-hG1a-Vektor-DNA. Für das Einführen von
Mutationen an zwei distalen Stellen werden zwei PCR-Produkte synthetisiert
(24B). Das eine Ende jedes PCR-Produkts dient dem Rekombinieren mit
einem homologen Ende des linearen Vektors und das andere Ende, das
die Mutation bzw. Mutationen, die von Interesse ist bzw. sind, kodiert,
dem Rekombinieren mit dem benachbarten PCR-Produkt. Wie in 24B gezeigt, können
zusätzliche
distal positionierte Mutationen in eine Targetsequenz durch anteilmäßiges Erhöhen der
Anzahl der beteiligten PCR-Produkte eingeführt werden. Die Rekombination
benachbarter PCR-Produkte erfolgt immer über die Regionen hinweg, die
die erwünschten
Mutationen enthalten, weshalb die Oligonukleotidprimer, die diese
Enden (z. B. A1, A2) kodieren, komplementäre Mutantenreste enthalten.
Diese mutagenen PCR-Primer enthalten an jeder Seite der mutanten
Reste mindestens 15 flankierte Nukleotide der Wildtyp-Sequenz entweder
zum Primen der Polymerisationsreaktion oder zum Vermitteln der Rekombination
flankieren. Zwei 49 Nukleotide lange PCR-Sense- und Antisenseprimer
(Rs und Ra) für
PCR enthalten Sequenzen für
die Rekombination mit den Endregionen des mit Eco47-III verdauten pBR96-hG1a-Vektors.
-
Jede
L-Mutation wurde in einer getrennten PCR-Reaktion amplifiziert.
Die Reaktionsbedingungen waren 250 ng intakte pBR96-hG1a-DNA-Template,
10 μl 1X
Pfu-Puffer (Stratagene, Inc., San Diego, CA), 10 nmol dNTPs, jeweils
200 ng der geeigneten PCR-Primer, 10% Dimethylsulfoxid (ATCC, Rockville,
MD) und 2,5 Einheiten klonierte Pfu-DNA-Polymerase in einem Reaktionsvolumen
von 100 μl.
Proben wurden zuerst 5 min lang bei 95°C denaturiert, 5 min lang auf
45°C abgekühlt und
1 min lang bei 72°C
elongiert, darauf folgten von 25 Zyklen von Denaturierung bei 94°C für 45 sec,
Annealing bei 45°C
für 45
sec, und Elongieren für
1 min/kb bei 72°C,
gefolgt von einem letzten Elongieren für 7 min bei 72°C in einem
DNA-Thermocycler von Perkin-Elmer (Norwalk, CT). Die amplifizierten
Produkte wurden aus 1% Agarosegel gereinigt, mit dem Qiagen-Gelextraktionskit
extrahiert und die gewonnene DNA wurde quantifiziert. 50 ng von
jedem PCR-Produkt wurden mit 25 ng von dem mit Eco47-III verdauten
pBR96-hG1a-Vektors
gemischt, der Verfahrensweise des Herstellers (GIBCO BRL/Life Technologies,
Gaithersburg, MD) gemäß in kompetene
Max E. Coli DH5α transfiziert
und die gesamte Transfektionsreaktionsmischung wurde auf selektive
LB-Agarplatten, die 100 μg/ml
Ampicillin enthielten, ausplattiert.
-
Die
Ergebnisse von mehreren Klonierversuchen sind in der Tabelle, die
folgt, zusammengefasst. Typischerweise führten die Transformationen
zur Bildung von 80 bis 200 Bakterienkolonien. Einzelne Kolonien wurden
ausgewählt
und über
Nacht in 2 ml Flüssigkulturen
angezüchtet
zum Isolieren von Miniprep-Plasmid-DNA (Qiagen) und Analysieren
durch Kartierung mit Restriktionsendonuklease Eco47-III. Unter 24 unabhängigen Transformanten,
die aus dreifachen homologen Rekombinationsereignissen (zwei PCR-Produkte plus
Vektor), die analysiert wurden, enthielten 11 Klone die vorausgesagte
1,4 kpb-DNA-Insertion.
-
25 zeigt eine repräsentative diagnostische Restriktionsanalyse
von DNA aus Klonen, die aus vierfachen homologen Rekombinationsereignissen
(drei PCR-Produkten plus Vektor) hervorgegangen sind. Zusätzliche
Probenahme aus Klonen, die von vierfacher Rekombination herrühren, ergaben
eine Klonierungseffizienz von 29% (7 von 24 untersuchten Klonen
enthielten Insertionen). Aufgrund der geringen Anzahl von untersuchten
Proben wissen wir allerdings nicht, ob die Unterschiede in der Klonierungseffizienz,
die zwischen den dreifachen und vierfachen Rekombinationsgeschehnissen
beobachten worden sind, von Bedeutung sind.
-
Um
die Expression der Leγ-Bindungsaktivität der CH2-mutanten IgG zu beurteilen, wurden Miniprep-DNA
aus 6 Klonen, die aus der dreifachen Rekombinationsreaktion stammten
und 6 Klonen, die aus der vierfachen Rekombinationsreaktion stammten,
die alle die vorausgesagten diagnostischen Eco47-III-Restriktionsmuster aufwiesen, isoliert,
mit pBR96-hCκ-DNA
gemischt und zum Kotransfizieren von COS7-Zellen verwendet. 48 Stunden
alte gebrauchte Überstände aus
3 ml-Kulturen wurden auf die Produktion von IgG insgesamt und Leγ-Bindungsaktivität hin mittels
Enzym-Immunosorbent-Assay (EIA), wie beschrieben (Yelton, D. E., M.
J. Rosok, G. A. Cruz, W. L. Cosand, J. Bajorath, I. Hellstom, K.
-E. Hellstorm, W. D. Huse und S. M. Glaser. 1995. Affinity maturation
of the BR96 anti-karzinoma antibody by codon-based mutagenesis (Affinitätsreifung des
BR96-Antikarzinomantikörpers
durch Mutagenese auf Kodonbasis). J. Immunol. 155: 1994–2004) untersucht.
Es zeigte sich, dass alle zwölf
Kulturen ungefähr
2–3 μg/ml Leγ-reaktives
IgG sezernierten. Die Spektren der Leγ-Bindungsaktivitäten waren
alle denjenigen von nativem humanisiertem BR96-IgG ähnlich,
was anzeigt, dass die homolog rekombinierten Antikörper keine
ausgedehnten Mutationen erwarben, die die Antigenbindung beeinflussen
könnten.
Um zu bestätigen,
dass die erwünschten
CH2-Mutationen eingebaut worden sind, und
um die rekombinierten Gene auf falsch eingebaute Nukleotide hin
zu beurteilen, wurden vier der Klone, die funktionellen Antikörper bilden,
mit Hilfe des Sequenase-DNA-Sequenzierkits, Version 2 (United States
Biochemical) sequenziert. Es zeigte sich, dass ein Klon eine einzige
Nukleotidänderung
innerhalb des Vorwärts-PCR-Primers,
der zum Vermitteln der Rekombination mit Vektor-DNA verwendet wird,
enthielt. Wir sind unsicher, ob dieser Fehler während der chemischen Synthese
des Oligonukleotidprimers oder aufgrund eines falschen Einbaus während der
PCR-Reaktion trotz der Tatsache erfolgte, dass wir eine wärmebeständige Polymerase
mit Proofreadingaktivität
verwendet haben.
-
Ein
RPCR-Verfahren für
das homologe Rekombinieren von bis zu drei einzelnen, durch PCR
gebildeten mutierten Antikörpersequenzprodukten
in einen eukaryotischen Expressionsvektor für die schnelle Konstruktion
von gentechnisch veränderten
IgG-Molekülen
wird hier beschrieben. Der Vorteil dieses Ansatzes besteht in der
Möglichkeit,
gleichzeitig multiple distal positionierte Mutationen mit PCR-Produkten einzuführen, die
in einem einzigen PCR-Ansatz synthetisiert worden sind. Rekombinante
DNA werden mit einer angemessen hohen Klonierungseffizienz und Treue
korrekter Nukleotidsequenzen erzeugt. Die Fähigkeit, mehrere einzelne PCR-Produkte
effizient zu verbinden, sollte Kombinationsstrategien für das Konstruieren
komplex mutierter Proteindomänen
sowie für
das Erweitern der Anzahl und Position erwünschter Mutationen erlauben. Analyse von Transformanten, die durch
Mehrfachfragment-RPCR gebildet worden sind
| Konstruiertes
mutantes IgG | PCR-Fragmente
in Reaktion | HRa-Ereignisse | Analysierte
Kolonien | Klonierungseffizienzb |
| 2 | 2 | Dreifach | 24 | 45% |
| 2 | 3 | Vierfach | 24 | 33% |
| aHR-homologe Rekombination |
| bKlonierungseffizienz (Anzahl von Klonen,die
1,4 kbp Insertionen/Gesamtanzahl von Kolonien enthalten. |
-
BEISPIEL 5
-
Dieses
Beispiel bietet zwei Methoden zum Einführen von ortsspezifischen Mutationen
in die CH2-Domäne von menschliche
IgG1-Konstantregionen enthaltenden Vektoren.
-
Eine
Methode involviert die PCR-Amplifikation eines Segments oder von
Segmenten der Konstantregion, wobei Mutationen mit Hilfe von in
geeigneter Weise konstruierten Oleonukleotiden eingeführt werden. Der
das bzw. die Fragment(e) aufnehmende Vektor wird mit einem Restriktionsenzym
zum Linearisieren des Vektors verdaut. PCR-Amplifikationsprimer
sind so gestaltet, dass sie die 5'-Enden der PCR-Fragmente an die DNA-Sequenz
der Vektoren hybridisieren können.
Sollte mehr als ein PCR-Fragment
amplifiziert werden, so werden den beiden Fragmenten gemeinsame
Sequenzen durch Oligonukleotide eingeführt. Es werden Bakterien mit
den PCR-Fragmenten und mit dem verdauten Vektor transfiziert. Die
Fragmente und der Vektor können
durch homologe Rekombination mit Hilfe der Rekombinationsmaschinerie
der Bakterien rekombiniert werden. Es werden Bakterienkolonien ausgewählt und
die DNA wird bezüglich
ihrer Größe und ihrer
Restriktionskarte als vorläufige
Bestimmung analysiert, dass der Vektor und das bzw. die Fragmente
richtig rekombiniert sind, analysiert. Die korrekte Insertion von
Fragmenten mit den Mutationen wird durch Didesoxynukleotidsequenzanalyse
bestätigt.
Die DNA wird dann in Säugerzellen,
wie für
den CH2-deletierten Antikörper
beschrieben, eingeführt
und der exprimierte Antikörper
hinsichtlich der Bindungs- und funktionellen Aktivität analysiert.
-
Als
Beispiel wurden die Mutationen Leu zu Ala am Rest 235 in CH2 und
Gly zu Ala am Rest 237 durch das in Beispiel 4 offenbarte Verfahren
eingeführt.
Der Schwerkettenvektor, der bei diesem Verfahren verwendet wurde,
war pD17-hG1a, der dem hier beschriebenen pD17-BR96-Vektor ähnlich,
mit der Ausnahme, dass humanisierte V-Regionen (Rosok, M. J., D.
E. Yelton, L. J. Harris, J. Bajorath, K. E. Hellström, I. Hellstrom,
G. A. Cruz, K. Kristensson, H. Lin, W. D. Huse und S. M. Glaser,
1996, J. Biol. Chem 271 37: 22611–22618) durch drei Affinitätsmutationen
(H1-, H2- und H3-Mutationen) substituiert wurden.
-
pBR96-hG1a
enthält
zwei Eco47-III-Restriktionsstellen, die die Ig-Scharnier-CH2-CH3-Domänen flankieren.
Der Aufnahmevektor wurde durch (1) den Verdau von pBR96-hG1a mit
Eco47-III, (2) Isolieren des Vektors durch Agarosegelelektrophorese
und (3) Extrahieren der Vektor-DNA aus der ausgeschnittenen Gelscheibe
mit Hilfe des Qiagen-Gelextraktionskits (Qiagen, Chatsworth, CA)
hergestellt. Um Mutationen an einem einzigen Ort, wie beispielsweise
den Positionen 235 und 237, einzuführen, wurden zwei PCR-Produkte synthetisiert.
-
Um
zwei distal positionierte Mutationen, wie beispielsweise bei der
Mutante F (hier auch als hBR96-2F bezeichnet) mit Mutationen bei
235, 237, 331, sind 3 PCR-Produkte erforderlich. Die Rekombination
benachbarter PCR-Produkte erfolgt über die Regionen hinweg, die
die erwünschten Mutationen
enthalten; daher enthalten die Oligonukleotidprimer, die diese Enden
kodieren, komplementäre
mutante Reste. Die mutagenen PCR-Primer enthalten mindestens 15
flankierende Nukleotide der Wildtyp-Sequenz an jeder Seite der mutanten
Reste entweder zum Starten der Polymerisationsreaktion oder zum
Vermitteln der Rekombination. Zwei 49-Nukleotide lange PCR-sense-
und antisense-Primer enthalten Sequenzen für das Rekombinieren mit den Endregionen
des mit Eco47-III verdauten pBR96-hG1a-Vektors.
-
Bei
der PCR-Amplifikation wurden 250 ng intakte pBR96-hG1a-DNA-Template,
10 μl 10X
Pfu-Puffer (Stratagene,
Inc., San Diego, CA), 10 nmol dNTP, jeweils 200 ng der entsprechenden
PCR-Primer, 10% Dimethylsulfoxid (ATCC, Rockville, MD) und 2,5 Einheiten
klonierte Pfu-DNA-Polymerase (Stratagene, Inc., San Diego, CA) in
100 μl Reaktionsmischung
verwendet. Es wurden Proben 5 min lang bei 95°C denaturiert, 5 min lang bei
45°C einem
Annealing und 1 min lang bei 72°C
einer Elongation unterzogen, gefolgt von 25 Denaturierungszyklen
für 45
sec bei 94°C,
Annealing für
45 sec bei 45°C,
Elongation für
1 min/kb bei 72°C
und einer letzten Elongation für
7 min bei 72°C.
Die amplifizierten Produkte wurden aus einem 1%-igen Agarosegel
gereinigt, mit Hilfe des Qiagen-Gelextraktionskits extrahiert und
quantifiziert. Jeweils 50 mg PCR-Produkt wurden mit 25 ng des mit
Eco47-III verdauten pBR96-hG1a-Vektors gemischt und in MAX Efficiency
E. Coli DH5αWZ den Anweisungen des Herstellers (GIBCO
BRL/Life Technologies, Gaithersburg, MD) entsprechend transfiziert.
Die gesamte Transfektionsreaktionsmischung wurde auf LB-Agarplatten,
die 100 μg/ml
Ampicillin enthielten, ausplattiert.
-
Es
wurden Bakterienkolonien ausgewählt
und über
Nacht in 2 ml Flüssigkulturen
bei 37°C
gezüchtet. DNA
wurde isoliert und durch Kartierung mit Eco47-III-Restriktionsendonuklease
analysiert. Klone mit einer Insertion von der richtigen Größe wurden
sequenziert (Sequenase Version 2, U. S. Biochemical Corp., Cleveland,
OH).
-
Die
zweite Methode für
das Einführen
ortsspezifischer Mutationen in die CH2-Domäne von menschlichem
IgG1 involvierte die Methode von Kunkel (1987 Methods Enzymology,
supra). Für
diesen Vorgang wurde pD17-hG1b-DNA mit dem F1-Replikationsursprung
in elektrokompetentes E. coli CJ236 dut-ung-(Bio-Rad Laboratories,
Hercules, CA) mittels Elektroporation den Anleitungen des Herstellers
gemäß eingeführt. PD17-hG1b
ist ein Vektor, der eine Konstantregion, jedoch keine variable Region
aufweist. Der F1-Replikationsursprung gestattet die Behandlung dieses
Vektors wie ein Phagemid.
-
Bakterien,
die das Plasmid enthalten, wurden durch Ampicillinresistenz ausgewählt. Uridinylierte
Einzelstrang-DNA wurde mit Hilfe des Protokolls der Muta-Gene Phagemid
in vitro Mutagenese, Version 2 (Bio-Rad) hergestellt. Es wurden
Mutationen durch ortsgerichtete Mutagenese mit einem geeigneten
antisense-Oligonukleotid eingeführt.
Bei Molekülen
mit Mutationen an mehr als einer Stelle wurden Mutationen durch eine
der beiden oben besprochenen Methoden eingeführt. Eine Methode wäre, (1)
eine Mutante, beispielsweise die Mutante 2C (hier auch als BR96-2C
bezeichnet) mit Mutationen an den Resten 318, 320, 322 herzustellen,
(2) ssDNA davon zu isolieren und (3) einen zweiten Satz Mutationen
mit entsprechendem antisense-Oligonukleotid einzuführen. Die
zweite Methode wäre,
ein Annealing von zwei antisense-Oligonukleotiden mit derselben
uridinylierten ssDNA durchzuführen
und mit beiden Änderungssätzen nach
Mutante zu screenen. Die Mutante 2H (hBR96-2H) wurde auch durch
eine Kombination dieser Methoden hergestellt.
-
Die
V-Region der humanisierten BR96-2-Schwerkette wurde durch die oben
beschriebene homologe Rekombinationsmethode in pD17-hJm14.H1 eingeführt. Das
pD17-hJm14.H1-Plasmid enthält
die humanisierte variable Region des BR96 mit H1/H2/H3-Mutationen
und dieses Plasmid wurde zum Transfizieren mutanter Sequenzen in
Säugerzellen
verwendet. Der pD17G1b-Vektor, der die Fc-Mutation(en) enthält, wurde mit NheI 3 h lang
bei 37°C
verdaut und die DNA mit den oben beschriebenen Methoden isoliert.
Die Insertion der V-Region in den Vektor wurde aus der Größe und durch
Restriktionsenzymkartierung bestimmt und durch Sequenzanalyse bestätigt.
-
Die
transiente Expression ganzer Antikörper wurde mittels Transfektion
von COS-Zellen durchgeführt. Zur
Bildung von Antikörpern
wurden stabile Transfektionen von CHO-Zellen durchgeführt (vergleiche
die Beschreibung der deletierten CH2-Mutante). Alle Mutanten wurden
aus CHO-Kulturüberständen durch
Protein-A-Chromatografie gereinigt.
-
Die
dem Vektor homologen Oligonukleotidprimer, die zum Einführen der
Konstantregionmutationen verwendet wurden, waren wie folgt:
-
Vektorsequenzen homologe Oligonukleotide:
-
- Sens(sense)CH2 E47-3-5: CAG GGA GGG AGG GTG TCT GCT GGA
AGC CAG GCT CAG CGC TGA CCT CAGA
- D CH2 E47-3 A (Antisense): GGA AAG AAC CAT CAC AGT CTC GCA GGG
GCC CAG GGC AGC GCT GGG TGC TT
-
Oligonukleotide zum Mutieren von Leu235
zu Ala und Gly237 zu Ala (die unterstrichenen Sequenzen zeigen die
Mutationsorte an):
-
Antisense
CH2 L235-G237/aa: GAA GAG GAA GAC TGA CGG TGC CCC CGC GAG TTC AGG
TGC TGA GG
-
SensCH2
L235-G237/AA: CCT CAG CAC CTG AAC TCG CGG GGG CAC CGT CAG TCT TCC
TCT TC
-
Oligonukleotide zum Mutieren von Glu318,
Lys320, Lys322 zu Ser
-
- Antis(Antisense)CH2 EKK/SSS-2: CTG GGA GGG CTT TGT TGG AGA
CCG AGC ACG AGT ACG ACT TGC CAT TCA GCC
-
Oligonukleotide zum Mutieren von Pro331
zu Ala:
-
- Antis CH2 P331/A/3: GAT GGT TTT CTC GAT GGC GGC TGG GAG
GGC
- Sense CH2 P33/A: GCC CTC CCA GCC GCC ATC GAG AAA ACC ATC
-
Alternatives antisense-Oligo zum Einführen von
Ala an 331 durch ortsgerichtete Mutation:
-
- CH2P331A: GAT GGT TTT CTC GAT AGC GGC TGG GAG GGC TTT G
-
Oligonukleotide zum Mutieren von Glu318
zu Ser, Lys320 zu Ser, Lys322 zu Ser und Pro331 zu Ala:
-
- Antis CH2 EKK/SSA-6: GAT GGT TTT CTC GAT GGC GGC TGG GAG
GGC TTT GTT GGA GAC CGA GCA CGA GTA CGA CTT GCC ATT CAG CCA GTC
CTG GTG
- Sense CH2 EKKP/SSA-6: CAC CAG GAC TGG CTG AAT GGC AAG TCG TAC
TCG TGC TCG GTC TCC AAC AAA GCC CTC CCA GCC GCC ATC GAG AAA ACC
ATC
-
IN VITRO ASSAYS DER MUTANTEN
-
Die
Ergebnisse des CDC-Assays zeigen, dass mutanter hBR96-2B eine etwa
10 mal geringere Aktivität
aufweist als das Kontroll-hBR96-1 (zwei Affinitätsmutationen, eine in H2 und
eine in H3, vergleiche das oben erwähnte Patent (20)).
Die Mutanten, die die geringste Fähigkeit aufweisen, Zellen in
Gegenwart des Komplements zu töten,
sind hBR96-2C mit den Dreifachmutationen an den Positionen 318,
320 und 322 und die hBR96-2H-Mutante (die am wenigsten zytotoxischen
Antikörper
in der Reihe), die alle sechs Mutationen an drei verschiedenen Orten
enthält.
Die ADCC-Aktivität
wurde durch das CH2-deletierte hBR96-2-Molekül am stärksten beeinflusst (21).
-
hBR96-2B
und -2H zeigten 100- bis 1000-fach weniger Abtötungsaktivität in Gegenwart
von Effektorzellen. Im ADCC-Assay zeigte das hBR96-2B-Molekül auch etwa
10-fach weniger Aktivität
(21).
-
Die 26–28 stellen
die Aminosäuresequenzen
bereit für
die variable Schwerkettenregion für sowohl chimären als
auch humanisierten BR96, der die H1-, H2- und H3-Mutationen aufweist.
Die Aminosäuresequenz
für die
variable Leichtkettenregion ist bekannt und Verfahren zum Erzeugen
derselben sind in der PCT-Anmeldung Nr. 95/305444 zu finden. Außerdem ist
die Aminosäuresequenz
für die
IgG1-Konstantregion bereitgestellt.
Die Mutationen in der Konstantregion sind hervorgehoben.
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-