-
Hintergrund
der Erfindung
-
Diese
Erfindung betrifft die Verwendung eines Phospholambaninhibitors
bei der Herstellung eines Medikaments zur Erhöhung des koronaren Flusses,
insbesondere zum Erreichen einer direkten Dilatation der Koronararterien.
-
Eine
Kontraktion der Muskelzelle wird gesteuert durch die Menge an freiem
cytosolischem Calcium, das mit Calmodulin in glatten Muskelzellen
oder mit Troponin C in Herzmuskelzellen und Skelettmuskelzellen wechselwirkt.
Diese calciumaktivierten Proteine lösen eine Kaskade von Ereignissen
aus, die zu einer Zellverkürzung
und Muskelkontraktion führen.
-
Eines
der bedeutendsten Enzyme, das dazu beiträgt, die Muskelkontraktion zu
beenden oder zu verhindern, ist die Ca2+-ATPase,
die im Innern der Zelle in dem sarkoplasmatischen Retikulum (SR)
lokalisiert ist. Dieses Enzym überträgt cytosolisches
Calcium gegen den Konzentrationsgradienten in die intrazellulären Calciumspeicher.
Die Funktion der SR Ca2+-ATPase (SERCA)
wird gesteuert durch ein kleines Protein, das Phospholamban genannt
wird. Wenn Phospholamban dephosphoryliert wird, inhibiert es die
SERCA. Im Gegensatz dazu inhibiert phospholyliertes Phospholamban
diese Calciumpumpe nicht. Die Entfernung des Inhibitoreffekts von
Phospholamban wird als eine Stimulation der Calciumaufnahme in das
SR angesehen, da einige der SERCA-Moleküle zu jeder Zeit unter der
Inhibitorsteuerung von Phospholamban stehen.
-
Es
zeigte sich, daß Phospholamban
eine bedeutende Rolle im Herzmuskel (Lindemann, J. P. et al., "Beta adrenergic stimulation
of phospholamban phosphorylation and Ca2+-ATPase
activity in guinea pig ventricles", J. Biol. Chem. 258: 464–471, 1983)
und in den langsamen Skelettmuskeln spielt, wobei der schnelle Skelettmuskel
keinerlei Phospholamban exprimiert (Hoh, J. F. Y., "Muscle fiber types
and function", Current Opinion
in Rheumatology, 4: 801–808,
1992). Darüber
hinaus wird Phospholamban in Mausaorta exprimiert (Lalli, J. et
al., "Targeted ablation
of the phospholamban gene is associated with a marked decrease in
sensitivity in aortic smooth muscle", Circ. Res. 80(4): 506–513, 1997),
und es wird daher angenommen, daß Phospholamban SERCA in peripherem
Gefäßgewebe
steuert.
-
Aufgrund
seiner Inhibitorwirkungen auf die SERCA, die im Herzgewebe vorhanden
ist, unterdrückt Phospholamban
sowohl die Relaxationsrate als auch die Kontraktionsrate im Herz
von Säugetieren.
Daher würde
eine Verbindung, die in der Lage ist, die Inhibitorwirkungen von
Phospholamban auf Herz-SERCA zu verringern, z. B. durch Unterbrechung
der Phospholamban-SERCA-Wechselwirkung, brauchbar sein bei der Behandlung
einer Herzinsuffizienz.
-
Erst
kürzlich
wurde ein Beweis erbracht, daß PLB
in aortalen Endothelialzellen vorhanden ist (Sutliff, R. L. et al., "Functional and biochemical
evidence for modulation of endothelial cell function by phospholamban", FASEB Journal 12(5):
A957, 1998), wo es die Aktivität
der Isoform der SERCA, die auf dem endoplasmatischen Retikulum vorhanden
ist, moduliert. Es wurde auch gezeigt, daß eine verringerte Aktivität des Calciumpumpens
des endoplasmatischen Retikulums in aortalen Endothelialzellen zu
einer mangelhaften Endothelium-abhängigen Relaxation von glatten
aortalen Gefäßzellen
führt (Liu,
L. H. et al., "Defective
endothelium-dependent relaxation of vascular smooth muscle and endothelial
cell Ca2+ signalling in mice lacking sarco(endo)plasmic
reticulum Ca2++-ATPase isoform 3", J. Biol. Chem.
272(48): 30538–30545,
1997).
-
Es
gibt keinen veröffentlichten
Beleg für
das Vorkommen von Phospholamban in Endothelialzellen von Koronararterien.
-
Zusammenfassung der Erfindung
-
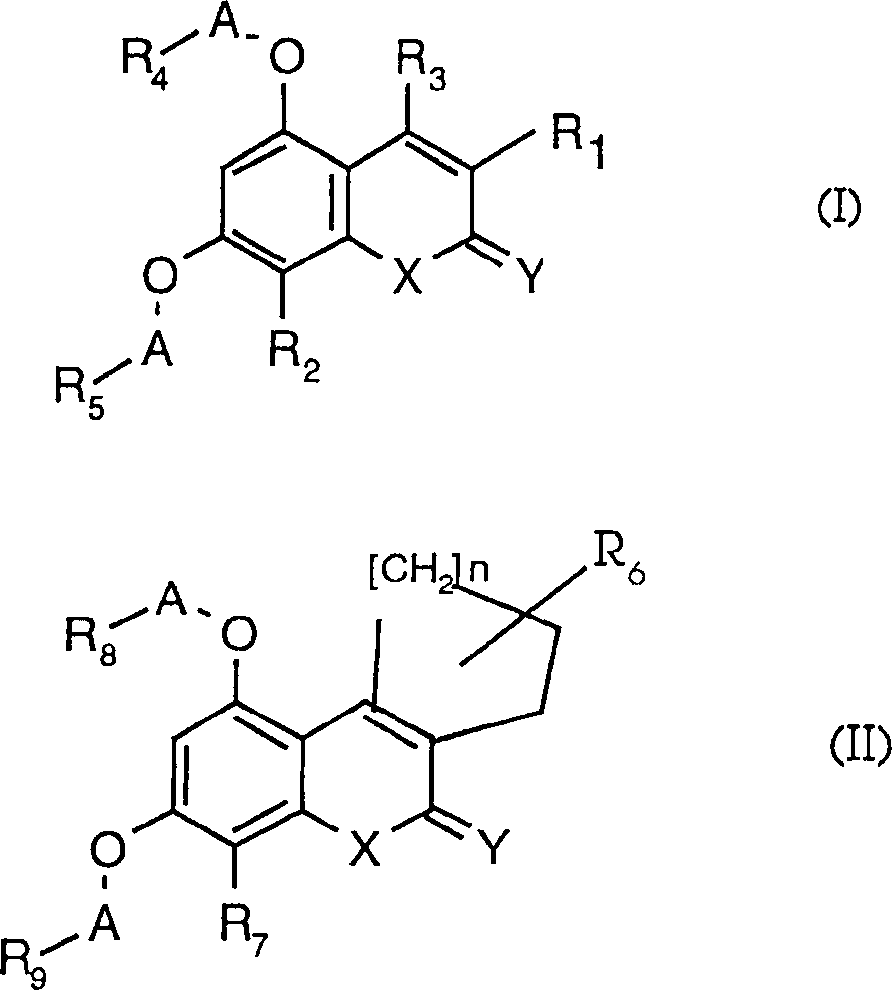
Es
wurde nun herausgefunden, daß Verbindungen
der Formeln (I) oder (II) wirksam sind bei der Abschwächung der
Inhibitoreffekte von Phospholamban auf Herz-SR-Ca
2+-ATPase (SERCA). Die
Verbindungen der Formeln (I) oder (II) wirken als Phospholambaninhibitoren
durch direktes Binden an das Phospholambanprotein. Dadurch eliminieren
die Verbindungen der Formeln (I) oder (II) die Inhibitorwirkung
von Phospholamban auf die SERCA, wie die Proteinkinasen, wenn sie
Phospholamban phosphorylieren.
worin
R
1 gleich
Wasserstoff, Alkyl, Alkenyl, Aryl, Arylalkyl, Hydroxyalkyl, Halogenalkyl,
Alkoxy, COR
10, CONR
10R
11, OR
10, S(O)
mR
10, NR
10COR
11 oder NR
10R
11 ist, wobei R
10 gleich
Wasserstoff, Alkyl, Alkenyl, Aryl, Arylalkyl, Hydroxyalkyl, Halogenalkyl,
Alkoxy oder Hydroxy ist und R
11 gleich Wasserstoff,
Alkyl, Aryl, Arylalkyl, Alkoxy, Aryloxy, Hydroxy oder Acyl ist oder
wobei im Fall, daß X
NR
11 ist, R
1 auch
Carboxylalkyl sein kann,
R
6 gleich
Wasserstoff, Alkyl, Alkenyl, Aryl, Arylalkyl ist,
R
2 und R
7 gleich Wasserstoff,
Alkyl, Aryl, Arylalkyl, Alkenyl, COR
10,
CONR
10R
11, Halogen,
Trifluormethyl, Nitro oder Cyano bedeuten, wobei R
10 und
R
11 wie oben definiert sind,
R
3 gleich Wasserstoff, Alkyl, Aryl oder Arylalkyl
ist,
A gleich Alkyl oder substituiertes Alkyl bedeutet,
m
gleich 0–2
ist und n gleich 1–3
ist,
Y gleich O, NR
11 oder S bedeutet,
wobei R
11 genauso wie oben ist,
X gleich
O, NR
11 oder S bedeutet, wobei R
11 genauso wie oben ist,
R
4,
R
5, R
8 und R
9 unabhängig
eine der folgenden Gruppen bedeuten:
oder in
dem Fall, wobei X gleich NR
11 ist, R
4, R
5, R
8 und
R
9 auch unabhängig HOOC-, R
12OOC-,
H
2NCO- oder HOHNCO- sein können, wobei
R
12 Alkyl, Arylalkyl oder Aryl bedeutet,
und
wobei jeder oben definierte Arylrest als solcher oder als Teil einer
anderen Gruppe substituiert sein kann,
und pharmazeutisch verträgliche Salze
davon.
-
Darüber hinaus
wurde überraschenderweise
herausgefunden, daß die
Phospholambaninhibitoren der Formeln (I) oder (II) den koronaren
Fluß in
isolierten schlagenden Herzen von Meerschweinchen, die mit konstantem
Druck durchströmt
wurden, erhöhten.
Die Größenordnung
des Effekts auf den koronaren Fluß überstieg deutlich die erhöhenden Effekte
der Verbindungen auf die Relaxations- und Kontraktionsgeschwindigkeiten
der linken Herzkammer, was anzeigt, daß die Verbindungen der Formeln
(I) oder (II) direkt die Koronararterien erweitern. Der Effekt der
Verbindungen (I) oder (II) auf den koronaren Fluß geht den anderen Effekten voraus,
was bestätigt,
daß die
Koronardilatation eine Folge war der direkten Wirkung der Verbindungen
auf die Koronarterien. Darüber
hinaus wurde herausgefunden, daß der
vasodilatorische Effekt der Phospholambaninhibitoren von (I) oder
(II) von einem Endothelial-mediierten Mechanismus herrührt. Wir
denken daher, daß Phospholambaninhibitoren
eine Vasodilatation induzie ren und den koronaren Fluß verstärken durch
Blockieren des Inhibitoreffekts von PLB auf SERCA auf dem endoplasmatischen
Retikulum der Endothelialzellen der Koronararterien.
-
Mit
der unerwarteten Fähigkeit
zur direkten Dilatation der Koronararterien sind Phospholambaninhibitoren,
wie die Verbindungen (I) oder (II), brauchbar bei der Behandlung
von Zuständen,
bei denen eine Erhöhung
des koronaren Flusses gewünscht
ist, z. B. bei koronaren Herzerkrankungen und bei einer hämodynamischen
Krise, bei der der niedrige aortale Blutdruck den koronaren Perfusionsdruck
senkt.
-
Kurze Beschreibung
der Zeichnungen
-
1 zeigt
CD-Spektren von 50 μM
an PLB[1–36
a. a.] (plb), PLB[1–36
a. a.] (Ser16PO3-, Thr17PO3-) (plbPP),
Verbindung des Beispiels 1c, und der Mischungen PLB[1–36 a. a.]
+ Verbindung des Beispiels 1c und PLB[1–36 a. a.] (Ser16PO3-, Thr17PO3-) +
Verbindung des Beispiels 1c in Wasser bei Raumtemperatur.
-
2A zeigt
die Wirkung der Verbindung des Beispiels 1c (50 und 100 μM) auf die
Ca2+-Aufnahmerate in die SR-Vesikel des
Herzmuskels.
-
2B zeigt
die Wirkung der Verbindung des Beispiels 1c (50 und 100 μM) auf die
Ca2+-Aufnahmerate in die SR-Vesikel des
schnellen Skelettmuskels.
-
3A zeigt
die Erhöhung
des koronaren Flusses, die mediiert wird durch den direkten Dilatationseffekt
der Verbindung des Beispiels 1c auf die Koronararterien, wel che
glatte Gefäßmuskelzellen
und Endothelialzellen enthalten.
-
3B zeigt
die Wirkung der Verbindung des Beispiels 1c auf die positive Ableitung
(positives dP/dt max) des linken Kammerdrucks.
-
3C zeigt
die Wirkung der Verbindung des Beispiels 1c auf die negative Ableitung
(negatives dP/dt max) des linken Kammerdrucks.
-
4A zeigt
die Wirkung der Verbindung des Beispiels 1c auf den koronaren Fluß (ml/min)
ohne Zugabe von Triton × 100.
-
4B zeigt
die Wirkung der Verbindung des Beispiels 1c auf den koronaren Fluß (ml/min)
mit Triton × 100,
das verwendet wird zur Zerstörung
der Endothelialzellen der Koronargefäße.
-
Ausführliche
Beschreibung der Erfindung
-
Die
vorliegende Erfindung stellt eine Verwendung eines Phospholambaninhibitors
zur Herstellung eines Medikaments zum Erreichen einer direkten Dilatation
der Koronararterien zur Verfügung.
Darüber
hinaus stellt die Erfindung eine Verwendung eines Phospholambaninhibitors
zur Herstellung eines Medikaments zur Behandlung von Koronarherzerkrankungen
zur Verfügung.
Die Erfindung stellt auch eine Verwendung eines Phospholambaninhibitors
zur Herstellung eines Medikaments zur Behandlung einer hämodynamischen
Krise zur Verfügung,
bei der der niedrige aortale Blutdruck den koronaren Perfusionsdruck
senkt.
-
Der
Begriff "Phospholambaninhibitor" bedeutet hier eine
Verbindung, welche den Inhibitoreffekt von Phospholamban auf SR
Ca2+-ATPase durch direktes Binden an das
Phospholambanprotein abschwächt.
-
Der
Inhibitoreffekt einer gegebenen Verbindung auf Phospholamban kann
dargestellt werden durch Messen des Effekts der Verbindung auf die
Calciumaufnahme in die SR-Vesikel,
die präpariert
wurden aus Herzgewebe, und in die SR-Vesikel, die präpariert
wurden aus schnellem Skelettmuskel (Psoas m.). Beide Arten an SR-Vesikeln
enthalten Ca2+-ATPase, jedoch enthalten
die Vesikel des schnellen Skelettmuskels kein Phospholamban (Hoh
JFY, "Muscle fiber
types and function",
Current Opinion in Rheumatology, 4: 801–808, 1992). Eine Erhöhung der
Calciumaufnahme in die SR-Vesikel, die präpariert wurden aus Herzgewebe,
jedoch nicht in die SR-Vesikel, die präpariert wurden aus schnellem
Skelettmuskel, zeigt an, daß die
Verbindung die Inhibitorwirkung von Phospholamban auf SR Ca2+-ATPase durch direktes Binden an das Phospholambanprotein
abschwächt
und daß die
Verbindung anwendbar ist als ein Phospholambaninhibitor in dem Verfahren
der Erfindung. Das direkte Binden einer Verbindung an das Phospholambanprotein
kann festgestellt werden durch die Circulardichroismus(CD)-Spektroskopie.
Die Verfahren zur Bestimmung, ob eine Verbindung die Inhibitorwirkung
von Phospholamban auf SR Ca2+-ATPase durch
direktes Binden an das Phospholambanprotein abschwächt, d.
h. ein Phospholambaninhibitor ist, werden im experimentellen Abschnitt
ausführlich
dargestellt.
-
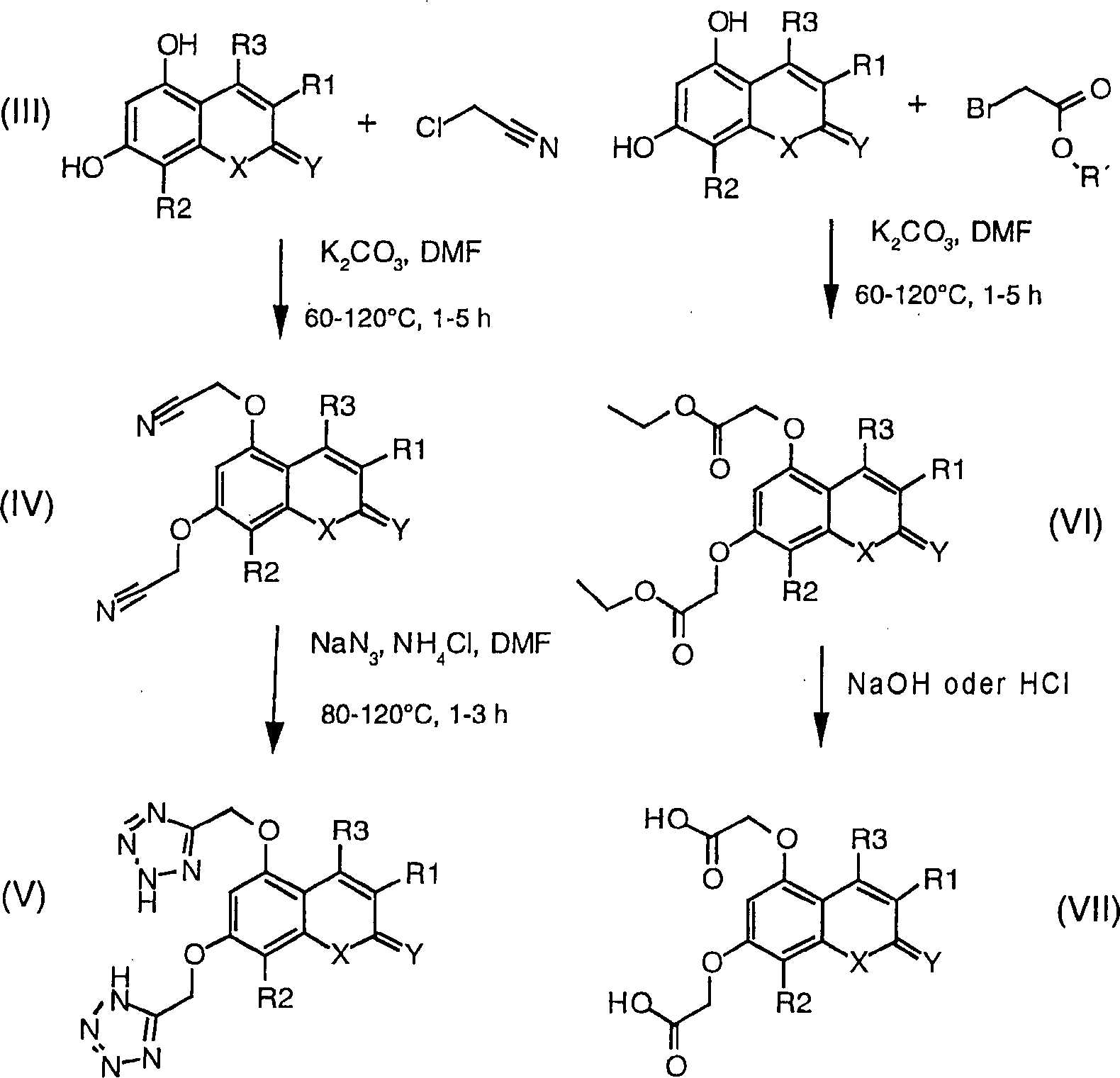
Die
Verbindungen der Formeln (I) oder (II) können hergestellt werden aus
den 1,3-Dihydroxy-substituierten heteroaromatischen Verbindungen
durch Alkylieren der Dihydroxyverbindungen mittels geeigneter Alkylierungsmittel,
zum Beispiels mittels Chloracetonitril oder Bromes sigsäureester
gemäß dem folgenden
Schema 1, wobei R1, R2,
R3, X und Y genauso wie oben definiert sind,
R' eine Schutzgruppe
für das
Hydroxyl, z. B. Methyl, Benzyl oder Tetrahydropyranyl, ist.
-
-
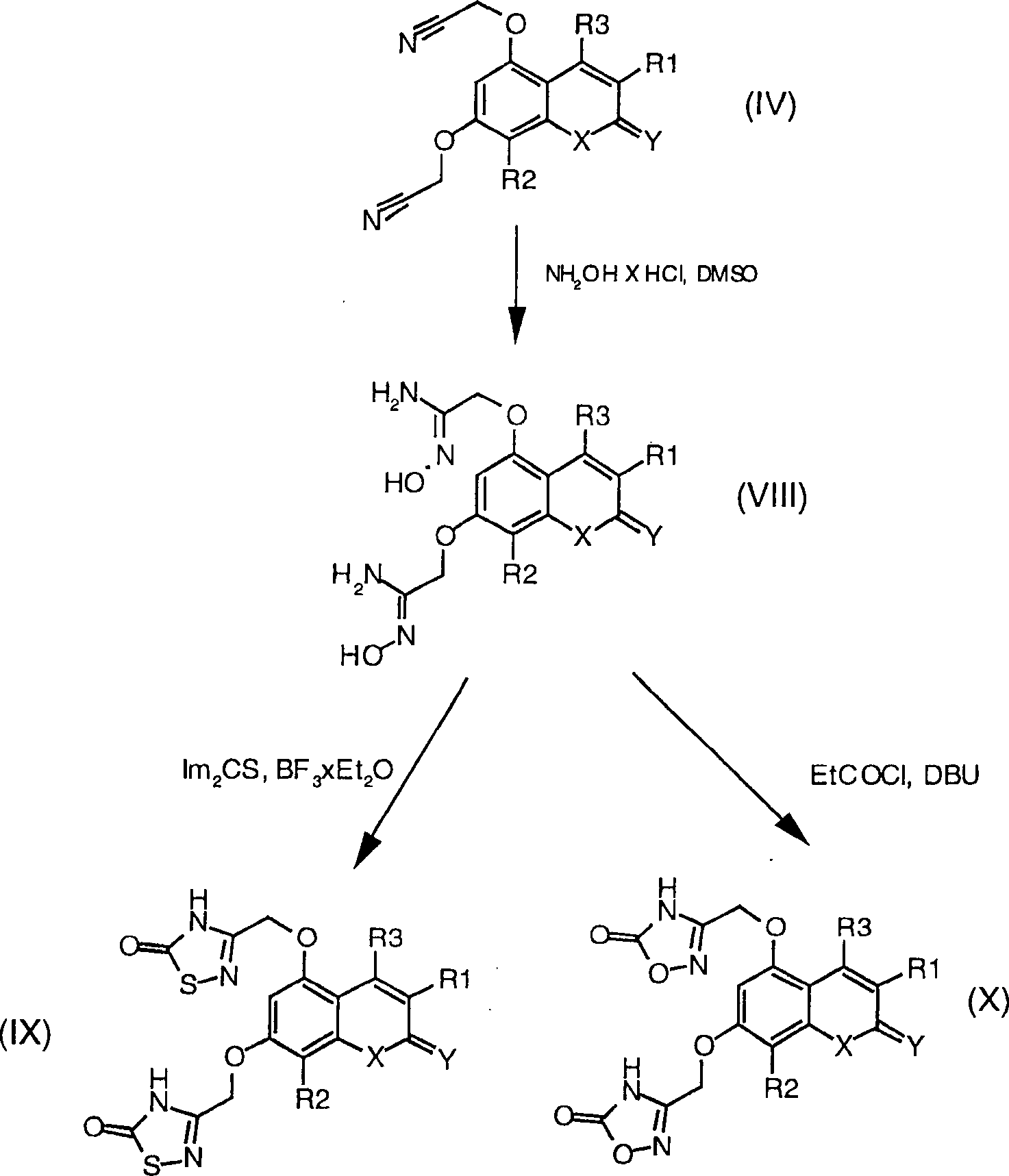
Die
oben beschriebene Cyanoverbindung (IV) wird verwendet zur Herstellung
der 1,2,4-Oxadiazol- und 1,2,4-Thiadiazol-Derivaten,
wobei die Verfahren verwendet werden, die beschrieben sind in J.
Med. Chem. 1996, 39, 5228–5235.
-
Die
Synthesen werden in Schema 2 aufgezeigt, wobei R1,
R2, R3, X und Y
genauso wie oben definiert sind.
-
-
Die
anderen Heterocyclen wie Gruppen R4, R5, R8 und R9 werden hergestellt wie in Bioorg. Med. Chem.
Lett., 1994, 4, 45–50,
beschrieben
-
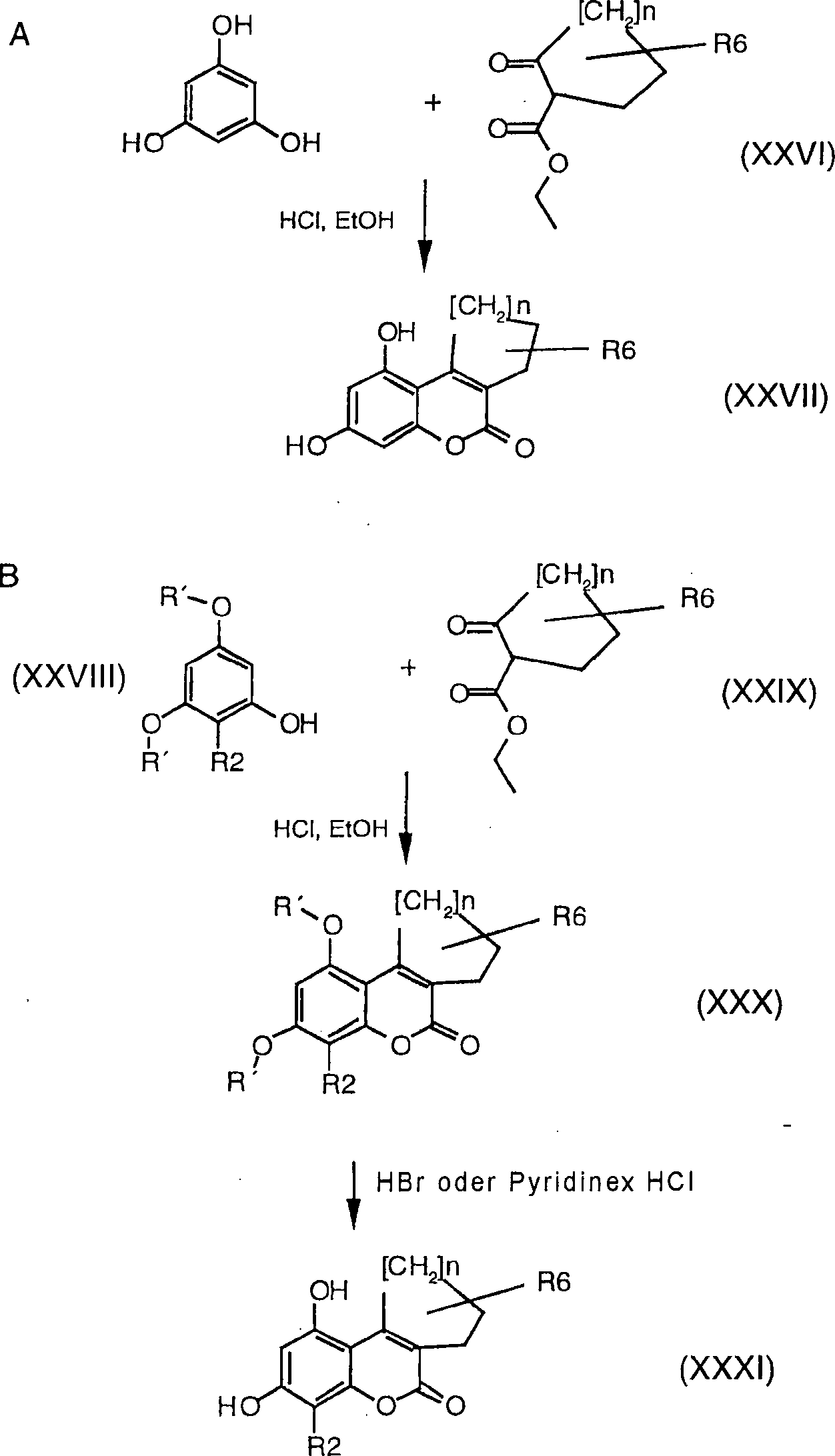
Die
Dihydroxyaromaten (III) werden hergestellt unter Verwendung der
Literaturverfahren. Die Cumarine (XIV), (XVI) und (XX) werden hergestellt
durch die Verwendung der Knoevenagel-Kondensation oder der von-Pechmann-Reaktion,
wie in Schema 3 und 4 dargestellt, wobei R1,
R2 und R3 genauso
wie oben definiert sind, Z gleich Alkyl, Aryl, Arylalkyl oder Alkenyl
ist und R' eine
Schutzgruppe für
die Hydroxyle, z. B. Methyl, Benzyl oder Tetrahydropyranyl, ist.
-
-
-
Die
Chinolinone werden hergestellt durch die Knorr-Reaktion, wie in Schema 5 beschrieben,
wobei R1, R11 und
R3 genauso wie oben definiert sind, X ein
Halogen ist.
-
-
Die
cyclischen Verbindungen (II) können
entsprechend hergestellt werden aus Verbindung (XXXI), die gemäß Sche ma
6 hergestellt werden kann, wobei R2 und
R6 genauso wie oben definiert sind, R' ein Schutzgruppe
für die
Hydroxyle, z. B. Methyl, Benzyl oder Tetrahydropyranyl, ist.
-
-
Cyclische
Chinolinverbindungen (II) können
unter Verwendung von Schema 5 entsprechend hergestellt werden aus
(XXVI).
-
Salze
und Ester der Verbindungen können,
wenn anwendbar, hergestellt werden durch bekannte Verfahren. Physiologisch
verträgliche
Salze sind brauchbar als aktive Medikamente, jedoch sind Salze mit
Alkali- oder Erdalkalimetallen bevorzugt. Physiologisch verträgliche Ester
sind ebenfalls als aktive Medikamente brauchbar. Beispiele sind
die Ester mit aliphatischen oder aromatischen Alkoholen.
-
Der
Begriff "Alkyl", wie er hierin als
solches oder als Teil einer anderen Gruppe verwendet wird, umfaßt sowohl
geradkettige, verzweigtkettige und cyclische Reste von bis zu 18
Kohlenstoffatomen, vorzugsweise 1 bis 8 Kohlenstoffatomen, am meisten
bevorzugt 1 bis 4 Kohlenstoffatomen. Der Begriff "niederes Alkyl", wie er hierin als
solches oder als Teil einer anderen Gruppe verwendet wird, umfaßt geradkettige,
verzweigtkettige und cyclische Reste von 1 bis 7, vorzugsweise 1
bis 4, am meisten bevorzugt 1 oder 2 Kohlenstoffatomen. Spezifische
Beispiele für
die Alkyl- und niederen Alkylreste sind jeweils Methyl, Ethyl, Propyl,
Isopropyl, Butyl, tert-Butyl, Pentyl, Cyclopentyl, Hexyl, Cyclohexyl,
Octyl, Decyl und Dodecyl, einschließlich der zahlreichen verzweigtkettigen
Isomere davon.
-
Der
Begriff "Acyl", wie er hierin als
solches oder als Teil einer anderen Gruppe verwendet wird, bezeichnet
eine Alkylcarbonyl- oder Alkenylcarbonylgruppe, wobei die Alkyl-
und Alkenylgruppen wie oben definiert sind.
-
Der
Begriff "Aryl", wie er hierin als
solches oder als Teil einer anderen Gruppe verwendet wird, bezeichnet eine
monocyclische oder bicyclische Gruppe, die von 6 bis 10 Kohlenstoffatome
im Ringabschnitt enthält. Spezifische
Beispiele für
Arylgruppen sind Phenyl, Naphthyl und dergleichen. "Aroyl" bedeutet auf eine
entsprechende Weise eine Arylcarbonylgruppe.
-
Der
Begriff "Alkoxy", wie er hierin als
solches oder als Teil einer anderen Gruppe verwendet wird, umfaßt eine
Alkylgruppe, wie sie oben definiert ist, verknüpft mit einem Sauerstoffatom. "Aryloxy" bedeutet auf entsprechende
Weise eine mit einem Sauerstoffatom verbundene Arylgruppe.
-
Der
Begriff "substituiert", wie er hierin in
Zusammenhang mit verschiedenen Resten verwendet wird, bezeichnet
Halogensubstituenten wie Fluor, Chlor, Brom, Iod oder Trifluormethylgruppe,
Amino-, Alkyl-, Alkoxy-, Aryl-, Alkylaryl-, Halogenaryl-, Cycloalkyl-,
Alkylcycloalkyl-, Hydroxy-, Alkylamino-, Alkanoylamino-, Arylcarbonylamino-,
Nitro-, Cyano-, Thiol- oder Alkylthio-Substituenten.
-
Die "substituierten" Gruppen können 1 bis
3, vorzugsweise 1 oder 2, am meisten bevorzugt 1 der oben erwähnten Substituenten
enthalten.
-
Phospholambaninhibitoren
wie die Verbindungen der Formeln (I) oder (II) können einem Patienten in therapeutisch
wirksamen Mengen verabreicht werden, die im allgemeinen von ungefähr 0,1 bis
500 mg, üblicherweise
von ungefähr
0,5 bis 50 mg, pro Tag reichen in Abhängigkeit von dem Alter, dem
Gewicht, dem Zustand des Patienten, der Verabreichungsroute und
dem verwendeten Phospholambaninhibitor. Der Begriff "therapeutisch wirksame
Menge" bedeutet
hierbei eine Menge, die eine direkte Dilatation der Koronararterien eines
Patienten erzeugt. Die in der Erfindung verwendete aktive Verbindung,
welche eine Ver bindung der Formel (I) oder (II) oder irgendeine
Verbindung ist, die eine wie oben definierte Phospholamban-inhibierende
Aktivität
besitzt, kann unter den im Stand der Technik bekannten Prinzipien
zu Dosierungsformen formuliert werden. Sie kann einem Patienten
als solches oder in Kombination mit geeigneten pharmazeutischen
Hilfsstoffen in der Form von Tabletten, Dragees, Kapseln, Suppositorien,
Emulsionen, Suspensionen oder Lösungen
gegeben werden. Die Wahl geeigneter Bestandteile für die Zusammensetzung
ist Routine für
einen Durchschnittsfachmann auf dem Gebiet. Es ist offensichtlich,
daß geeignete
Träger,
Lösungsmittel,
gelbildende Bestandteile, Dispersionen-bildende Bestandteile, Antioxidationsmittel,
Farbstoffe, Süßstoffe,
Benetzungsmittel und andere Bestandteile, die normalerweise auf
diesem Gebiet der Technologie verwendet werden, ebenfalls eingesetzt
werden können.
Die Zusammensetzungen, welche die aktive Verbindung enthalten, können enteral oder
parenteral gegeben werden, wobei die orale Route der bevorzugte
Weg ist. Der Gehalt der aktiven Verbindung in der Zusammensetzung
reicht von ungefähr
0,5 bis 100%, vorzugsweise von ungefähr 0,5 bis ungefähr 20%,
bezogen auf das Gewicht der Gesamtzusammensetzung.
-
Experimente
-
Der
Effekt der in der Erfindung verwendeten Verbindungen auf Phospholamban
wurde verifiziert durch Messen ihrer Effekte auf die Calciumaufnahme
in die SR-Vesikel, die präpariert
wurden aus Herzgewebe, und in die Vesikel, die präpariert
wurden aus schnellem Skelettmuskel (Psoas m.). Beide Arten an SR-Vesikel
enthalten Ca2+-ATPase, jedoch enthalten
die Vesikel aus dem schnellen Skelettmuskel kein Phospholamban (Hoh JFY, "Muscle fiber types
and function", Current
Opinion in Rheumatology, 4: 801– 808,
1992). Diese Experimente der Calciumaufnahme zeigten, daß die Verbindung
des Beispiels 1c aktiv war in lediglich den Vesikeln, die aus Herzgewebe
präpariert
wurden, was vermuten ließ,
daß der
Effekt sich auf die Abschwächung
einer Phospholambaninhibition begründete. Darüber hinaus wurde das direkte
Binden der Verbindung des Beispiels 1c an Phospholamban demonstriert
durch Anwendung der Circulardichroismus-Spektroskopie. Ferner wurde
die Exprimierung von Phospholamban in Schweinekoronararterien belegt
mittels mRNA-Messungen und unter Verwendung spezifischer Antikörper gegen
Phospholamban. Ferner wurden die Effekte der Verbindung des Beispiels
1c auf den koronaren Fluß und
auf die Ableitungen des linken Kammerdrucks in isolierten schlagenden
Herzen von Meerschweinchen, die mit konstantem Druck durchspült wurden,
demonstriert. Schließlich wurde
der Endothelial-mediierte Mechanismus des vasodilatorischen Effekts
der Verbindungen der Erfindung demonstriert. Die Verfahren werden
nachfolgend ausführlich
beschrieben.
-
Experiment 1. Effekte
auf die Calciumaufnahme in die SR-Vesikel, die aus Herz- und schnellem
Skelettmuskel präpariert
wurden
-
Herstellung
der SR-Vesikel
-
Meerschweinchen
(10–12)
wurden enthauptet. Ihre Herzen oder die Psoas-Muskel wurden entnommen,
in eiskaltem 0,9%igem NaCl gewaschen und in einem Puffer, der 20
mM Tris-Maleat, 0,3 M Sucrose, pH 7,0, enthielt, in Stücke geschnitten.
Danach wurden Gewebestücke
mit Polytron und ferner mit Potter (10 Hübe) homogenisiert. Die Homogenisate
wurden während
15 min bei 4°C
bei 1000 g zentrifugiert. Der Überstand wurde
gesammelt, und das Pellet wurde in 5 ml an Puffer (20 mM Tris-Maleat,
0,3 M Sucro se, pH 7,0) resuspendiert und erneut während 10
min bei 4°C
bei 1000 g zentrifugiert. Der erhaltene Überstand wurde mit dem früher gesammelten Überstand
vereinigt und nochmals während
20 min bei 4°C
bei 10.000 g zentrifugiert. Der abschließende Überstand wurde in eine Flasche
filtriert, die mit einem Magnetrührer
ausgestattet war. Es wurde KCl zu dem filtrierten Überstand
gegeben, um die Endkonzentration von 0,6 M (bei 4°C) zu ergeben.
Die erhaltene Lösung
wurde während
60 min bei 4°C
bei 100.000 g zentrifugiert. Das Pellet wurde in 5 ml des Puffers,
der 20 mM Tris-Maleat, 0,3 M Sucrose, pH 7,0, enthielt, suspendiert
und während
60 min bei 4°C
bei 100.000 g zentrifugiert. Das erhaltene Pellet wurde in 5 ml
an Puffer, der 20 mM Tris-Maleat, 0,3 M Sucrose, 0,1 M KCl, pH 7,0,
enthielt, suspendiert und bis zur Verwendung bei –80°C gelagert.
Die Proteinkonzentration wurde ebenfalls gemessen, um die getrennt
hergestellten Vesikelzubereitungen zu standardisieren.
-
Calciumaufnahmeassay
-
In
dem Calciumaufnahmeassay wurde der Fluoreszenzindikator Fluo-3 verwendet,
um die Abnahme der extravesikulären
Ca2+-Konzentration zu erfassen, wenn die
SR-Ca2+-ATPase
Ca2+ von dem extravesikulären Raum
in die SR-Vesikel überträgt.
-
Die
oben erhaltenen SR-Vesikel (50 μg
Protein/ml) wurden mit oder ohne der Testverbindung während 5
min bei 37°C
in dem Assaypuffer, der 40 mM Imidazol, 95 mM KCl, 5 mM NaN3, 5 mM MgCl2, 0,5
mM EGTA, 5 mM Kaliumoxalat, 2 μM
Ruthenium-Rot, 5 μM
Fluo-3, pH 7,0, enthielt, vorinkubiert. Das freie Calcium wurde mittels
CaCl2 auf 0,1 μM oder auf 0,04 μM eingestellt.
Die Reaktion wurde initiiert durch Zugabe von ATP (5 mM). Das endgültige Reaktionsvolumen
betrug 1,5 ml. Die Fluoreszenz der Reak tionsmischung wurde während 3
min unter Verwendung der Anregungs- und Emissionswellenlängen von
510 nm bzw. 530 nm gemessen.
-
Ergebnisse
-
Die 2A und 2B zeigen
die Wirkung der Verbindung des Beispiels 1c (50 und 100 μM) auf die Ca2+-Aufnahmerate in die Herz-SR-Vesikel (A)
und die SR-Vesikel des schnellen Skelettmuskels (B). Angegeben werden
Mittelwerte ± SEM
der drei Experimente pro Gruppe. Es ist ersichtlich, daß die Verbindung
des Beispiels 1c die Calciumaufnahme in die Herz-SR-Vesikel beschleunigt,
aber die Calciumaufnahme in die SR-Vesikel, die aus dem schnellen
Skelettmuskel präpariert
wurden, nicht verändert.
-
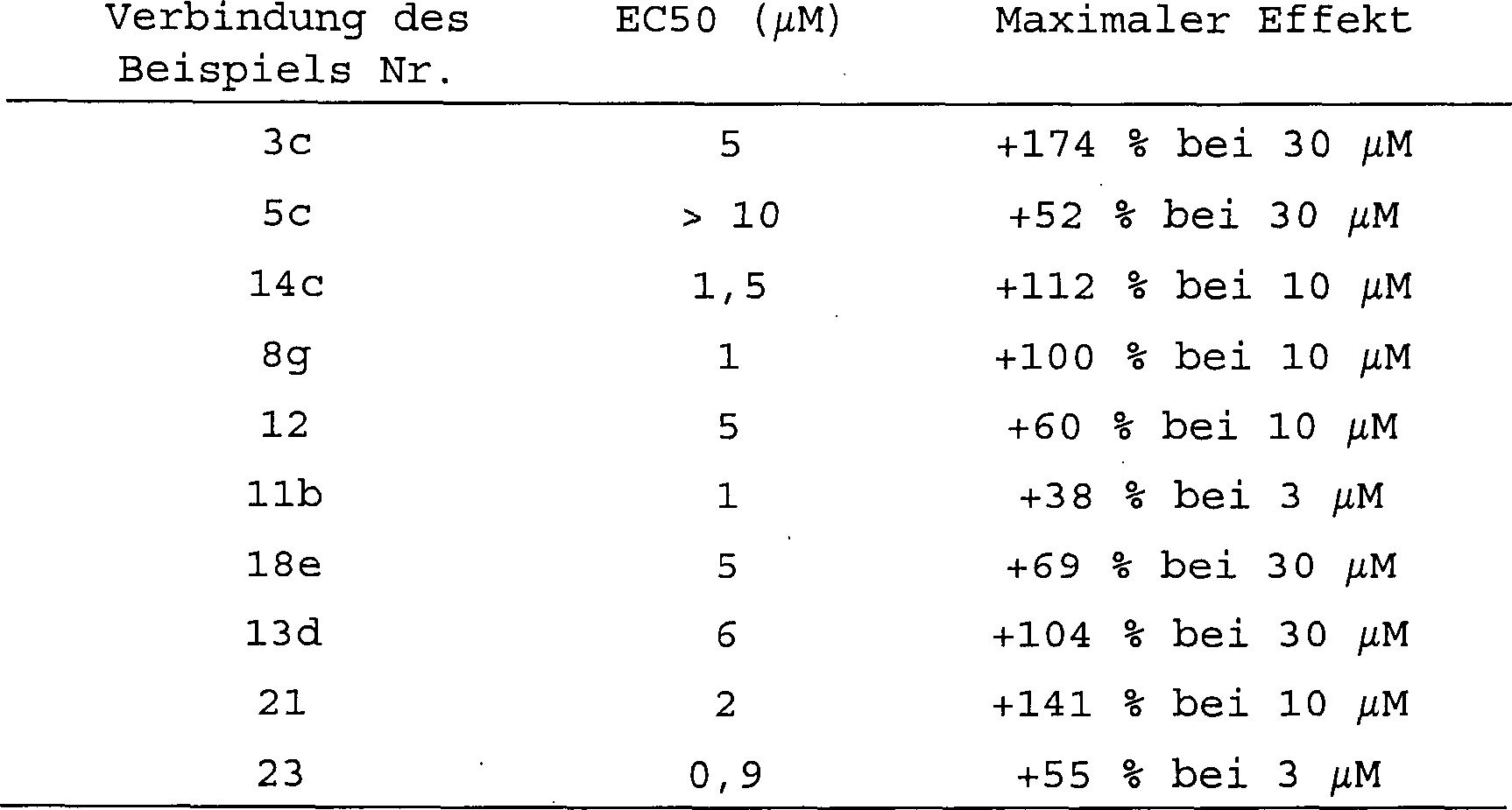
Tabelle
1 zeigt die Wirkungen verschiedener anderer Phospholambaninhibitoren
der Formeln (I) oder (II) auf die Ca
2+-Aufnahmerate
in die Herz-SR-Vesikel (A) und die SR-Vesikel des schnellen Skelettmuskels (B).
Die Experimente wurden bei 0,1 μM
bzw. bei 0,04 μM
an freien Calciumkonzentrationen durchgeführt. TABELLE
1. Stimulation (%) der Ca
2+-Aufnahme in
die Vesikelpräparationen,
die erhalten wurden aus dem ventrikulären Myocardium (A) und dem
schnellen Skelettmuskel (B) des Herzens von Meerschweinchen
- nb
- nicht bestimmt
-
Experiment 2. Bindung
der Verbindung des Beispiels 1c an den cytosolischen Teil von Phospholamban,
demonstriert mittels Circulardichroismus(CD)-Spektroskopie
-
Durch
Peptidsystesen wurden sowohl das N-terminale Fragment mit 36 Aminosäuren des
Human-Phospholambans (PLB[1–36
a. a.]) als auch das N-terminale Fragment mit 36 Aminosäuren des
doppelt phosphorylierten Human-Phospholambans (PLB[1–36 a. a.]
(Ser16PΟ3-, Thr17PΟ3-)) erhalten. Die Peptide wurden mittels
Umkehrphasen-HPLC gereinigt, mittels Massenspektroskopie hinsichtlich
der Homogenität analysiert,
und es wurde eine Reinheit von 97% gefunden. Die Peptide wurden
lyophilisiert und dann für
eine CD-Analyse
in Wasser auf eine Endkonzentration von 0,1 mM resuspendiert. Der
pH-Wert beider Lösungen war
zwischen 3 und 4 und wurde nicht weiter eingestellt. Die Verbindung
des Beispiels 1c wurde mit einer Endkonzentration von 0,1 mM in
Wasser gelöst.
Der pH-Wert wurde durch Zugabe von 1 N NaOH auf 7,2 eingestellt.
-
Es
wurden Circulardichroismusspektren von Proben mit 100 μl bei 24°C erhalten.
Die Spektren wurden aufgezeichnet auf einem Jasco J-720 Spektropolarimeter
unter Verwendung einer Quarzküvette
mit einer Pfadlänge
von 1 mm. Die Bandbreite betrug 1 nm, die Empfindlichkeit 20 mdeg,
die Schrittauflösung
0,5 nm, die Antwortzeit 0,5 s und die Scangeschwindigkeit 20 nm/min
(von 250 bis 190 nm). Die Spektren wurden dargestellt in [θ) × 10–3 × Grad × cm2 × dmol–1.
-
Die
CD-Spektren von PLB[1–36
a. a.] und der Mischungen PLB[1–36
a. a.] + Verbindung des Beispiels 1c zeigen, daß eine dramatische Änderung
in der durchschnittlichen Struktur des Peptids stattgefunden hat nach
Zugabe der Verbindung des Beispiels 1c. Es ist eine deutliche Zunahme
des α-helikalen
Beitrags zu sehen (1). Ein solches Verhalten wurde
für viele
Calmodulin-bindende Peptide gezeigt, welche Helices in dem gebundenen
Zustand bilden. Die CD-Studien zeigten, daß, wenn solche Peptide Calmodulin
binden, eine Erhöhung
der Helizität
des Komplexes über
die Summe der beiden einzelnen nicht wechselwirkenden Komponenten
auftritt (zur Übersicht
siehe: O'Neil, K.
T. und DeGrado, W. F. "How
calmodulin binds its targets: sequence independent recognition of
amphiphilic α-helices", TIBS 15: 59–64, 1990).
Darüber
hinaus wurde früher
mittels NMR demonstriert, daß das
N-terminale Fragment von PLB[aa.1–25] direkt mit Calmodulin
wechselwirkt (Gao, Y. et al. "Interaction
of calmodulin with phospholamban and caldesmon: comparative studies
by 1H-NMR
spectroscopy", Biochim.
Biophys. Acta 1160: 22–34,
1992). Das vorliegende Experiment verifiziert somit, daß die Verbindung
des Beispiels 1c einen Komplex mit PLB an seiner N-terminalen Domäne bildet.
-
Die
Verbindung des Beispiels 1c, zugegeben zu PLB[1–36 a. a.] (Ser16PO3-, Thr17PO3-), besitzt
keinen Einfluß auf
die Struktur des phosphorylierten Peptids in dem Maße wie für das nichtphosphorylierte.
Die CD-Messungen zeigen, daß die
Verbindungen des Beispiels 1c mit dem cytosolischen Teil des Phospholambans
PLB[1–36
a. a.] wechselwirken und nicht wechselwirken oder schwach wechselwirken
mit dem phosphorylierten Phospholamban (PLB[1–36 a. a.] (Ser16PO3-, Thr17PO3-)).
Somit ist die Wechselwirkung spezifisch für das nichtphosphorylierte
Phospholamban.
-
Experiment 3. Erfassung
von Phospholamban und Ca2+-ATPase-Polypeptiden
durch Immunoblotten aus Schweineherzgewebe
-
Verfahren
-
Gewebeproben
des Myocardium der linken Herzkammer, der Aorta, der proximalen
Koronararterie (die ersten 2 cm der Koronararterie ausgehend von
der Aorta) ebenso wie der distaleren Koronararterie (die nächsten 2
cm), die von drei Schweinen erhalten und in einen Pool gegeben wurden,
wurden unter flüssigem Stickstoff
pulverisiert und die Pulver in Homogenisationspuffer (50 mM K+-Phosphatpuffer pH 7,0, 10 mM NaF, 1 mM
EDTA, 0,3 mM PMSF, 0,5 mM DTT, 0,3 M Sucrose) suspendiert. Jede
Gewebeprobe wurde mit einem Potter-Elvehjelm-Homogenisator 20 mal
bei +4°C
homogenisiert (1100 Upm). Die Proteinkonzentration wurde gemessen
gemäß Bradford,
M., (1976), Anal. Biochem., 72, 248–254. Zur Bestimmung der Ca2+-ATPase und der Phospholamban(PLB)-Proteine
wurden 30 μg
an Gesamtprotein einer jeden Gewebeprobe mittels SDS-PAGE (13%)
analysiert gemäß Laemmli,
U., (1970), Nature, 227, 680–685.
Die Proteine wurden mittels Western-Blotting (Towbin, H. et al.,
(1979), Proc. Natl. Acad. Sci., USA, 76, 4350–4354) auf einer 0,45 μm-Nitrocellulosemembran
(Bio-Rad Laboratories) erfaßt.
Die Membranen zur Erfassung von Ca2+-ATPase-Protein wurden
inkubiert mit anti-SR Ca2+-ATPase-polyklonalem
Antikörper
(1 : 500), gefolgt von einem sekundären anti-Kaninchen-IgG-POD-Antikörper. Zur
Erfassung der Ca2+-ATPase wurde alternativ
ein monoklonaler anti-SR-Ca2+-ATPase-Antikörper (1
: 1000; Affinity Bioreagents), gefolgt von einem anti-Maus-IgG-POD,
verwendet.
-
Die
Membranen zur Erfassung des PLB-Proteins wurden mit monoklonalem
anti-PLB-Antikörper
(Upstate Biotechnology, Inc.) bei einer Verdünnung von 1 : 1000 inkubiert,
die Membranen wurden ferner mit anti-Maus-IgG-POD gewaschen und
inkubiert. Die Membranen wurden entwickelt mittels ECL-Immunodetektion (Amersham
Life Science), gefolgt von einem TMB-stabilisierten Substrat für HPR (Promega
Corp.).
-
Ergebnisse
-
Gemäß den Western-Blots
wurde die Ca2+-ATPase in allen Gewebeproben
detektiert. Die Western-Blots ergaben auch, daß PLB in dem linken ventrikulären Myocardium
und in der distalen Koronararterie gefunden wurde. Basierend auf
unseren Western-Blots konnte kein PLB aus Gewebeproben detektiert
werden, die von der Aorta und dem proximalen Teil der Koronararterie
stammen.
-
Experiment 4. Analyse
der Anwesenheit von Phospholamban-mRNA im Schweineherzgewebe
-
Verfahren
-
Zur
Beantwortung der Frage, ob der glatte Muskel oder die Endothelialzellen
einer Schweinekoronararterie Phospholamban(PLB)-mRNA enthielten,
wurden Northern-Analysen mit den Proben dieser drei gepoolten Herzen
durchgeführt
im Hinblick auf die Anwesenheit von PLB-mRNA, wobei auf dem Gebiet
gut bekannte Verfahren verwendet wurden. Es wurde eine RNA-Reinigung
in kleinem Maßstab
mit Gewebe des linken ventrikulären
Myocardiums, der Aorta und der Koronararterie (proximaler und distalerer
Bereich) des Schweins durchgeführt
durch Pulverisieren von 200–800
mg des Gewebes unter flüssigem
Stickstoff in einem Mörser.
Das Pulver wurde in einer Denaturierungslösung (4 M Guanidiniumthiocyanat,
25 mM Natriumcitrat, pH 7, 0,5% N-Lauroylsarcosin, 0,1 M 2-Mercaptoethanol)
suspendiert. Nach einer sauren Phenol/Chloroform-Extraktion wurde
die gesamte RNA zweimal mit Isopropanol ausgefällt und anschließend in
Wasser gelöst.
Die Konzentration der RNAs in den resultierenden Zubereitungen wurde
spektrofotometrisch bestimmt, und deren Integrität wurde in nicht denaturierendem
Agarosegel, das mit Ethidiumbromid angefärbt war, analysiert.
-
Die
Northern-Analyse von PLB-mRNA in diesen Zubereitungen wurden gemäß herkömmlichen
Verfahren durchgeführt
(Sambrook, J., Fritsch, E. F., Maniatis, T. (1989), Molecular Cloning:
A laboratoy Manual, 2. Ausg., Cold Spring Harbor Laboratory Press,
Cold Spring Harbor, NY). Kurz zusammengefaßt wurden 10 μg und 20 μg an Gesamt-RNA
von jedem Gewebe in 1% Formaldehyd/Agarose-Gel geladen. Als eine
Kontrolle wurden 10 μg
an RNA-Zubereitung, die von einem menschlichen Herzmuskelgewebe
gewonnen wurde, verwendet, und es wurden im Handel erhältliche
RNA-Ladder (BRL, Gaithersburg, MD) verwendet, um die Molekulargewichte
der RNAs zu bestimmen. Nach dem Transfer der separierten RNAs auf
einen Nylonfilter (Hybond-N, Amersham, UK) wurde der Filter mit
dem 32P-markierten Codierbereich des menschlichen
PLB-Gens hybridisiert. Nach der Hybridisierung wurde der Filter
in stringenten Bedingungen, die dem Durchschnittsfachmann auf dem
Gebiet gut bekannt sind, gewaschen und auf Kodak X-Omat-Film belichtet.
-
Ergebnisse
-
Die
Ergebnisse zeigen, daß das
linke ventrikuläre
Gewebe des Schweineherzens RNA enthält, welche mit der PLB-Hybridisierungssonde
reagiert. In den Proben von Geweben der Aorta und der Koronararterie
des Schweins wurde, wenn auch mit deutlich geringerer Intensität, ein Signal
an derselben Position detektiert. Diese Ergebnisse unterstützen somit
die Ansicht, daß das
PLB-Gen in dem Koronararteriengewebe des Schweins aktiv ist.
-
Experiment 5. Die Wirkungen
auf die Ableitungen des linken ventrikulären Drucks und des koronaren
Flusses
-
Verfahren
-
In
dieser Studie wurden Meerschweinchen beiderlei Geschlechts mit einem
Gewicht von 300–400
g verwendet. Nachdem die Meerschweinchen durch einen Schlag auf
den Kopf getötet
und enthauptet wurden, wurde das Herz schnell entnommen. Das Herz
wurde dann in kaltem oxygenierten Perfusionspuffer gespült. In die
Aorta wurde eine Kanüle
eingesetzt und mit einer Binde gesichert. Eine Gegenperfusion begann,
sobald das Herz in eine thermostati sch gesteuerte Feuchtigkeitskammer
der Langendorff-Apparatur gegeben wurde. Als ein Perfusionspuffer
wurde eine modifizierte Tyrode-Lösung
(37°C) verwendet,
die äquilibriert
wurde in einem thermostatisch gesteuerten Blasenoxygenator mit Carbogen
(95% O2 und 5% CO2).
Die Zusammensetzung der Tyrode-Lösung
betrug (in mM): NaCl 135; MgCl2 × 6H2O 1; KCl 5; CaCl2 × 2H2O 2; NaHCO3 15; Na2HPO4 × 2H2O 1; Glucose 10; pH 7,3–7,4. Die Experimente wurden
unter Bedingungen eines konstanten Drucks (50 mmHg) durchgeführt. Nach
einer kurzen Vorstabilisierung (10 min) wurde durch die linke Pulmonalvene
und den linken Vorhof ein Latexballon (Größe 4) sorgfältig in der linken Kammer plaziert.
Der Latexballon wurde an einer Edelstahlkanüle befestigt, die mit einem
Druckwandler gekoppelt war. Der Latexballon, die Kanüle und die
Kammer des Druckwandlers waren mit einer Mischung aus Ethylenglycol/Wasser
(1 : 1) gefüllt,
wobei jegliche Luftblasen vermieden wurden. Durch den Druckwandler
wurde der isovolumetrische linke Ventrikulardruck aufgezeichnet.
Die maximalen positiven und negativen Ableitungen des linken Ventrikulardrucks
wurden unter Verwendung der digitalisierten Drucksignale berechnet.
Am Anfang des Experiments wurde das Volumen des Ballons eingestellt,
um einen diastolischen Druck von ungefähr 5 mmHg zu erhalten. Die koronare
Fluß (ml/min)
wurde kontinuierlich mittels eines elektromagnetischen Durchflußmessers
mit einer oberhalb der Aortakanüle
eingesetzten Durchflußsonde
aufgezeichnet. Man ließ das
Herz mit einer koaxialen Elektrode, die auf der Oberfläche des
rechten Vorhofs angeordnet war, mit 250 Schlägen/min schlagen. Vor dem Starten
des Experiments ließ man
das Herz weitere 30–50
min mit einem Vehikel (0,1% DMSO) sich stabilisieren, um einen stabilen
koronaren Fluß zu
erreichen.
-
Nach
einem Aufzeichnen der Basislinie während 15 min wurden in Intervallen
von 15 min verschiedene Konzentra tionen der Testverbindung des Beispiels
1c zu dem Perfusionspuffer gegeben. Es wurde der Konzentrationsbereich
von 0,3–30 μM getestet.
Die Vehikelkonzentration (0,1% DMSO) wurde während des Experiments konstant
gehalten.
-
Ergebnisse
-
3A zeigt
die Zunahme des koronaren Flusses, der durch den direkten Dilatationseffekt
der Verbindung des Beispiels 1c auf die Koronararterien, welche
vaskuläre
glatte Muskelzellen und Endothelialzellen enthielten, mediiert wurde.
Die 3B und 3C zeigen
die Wirkung der Verbindung des Beispiels 1c auf die positiven und
negativen Ableitungen (positives und negatives dP/dt max) des linken
Ventrikulardrucks. Gegeben sind Mittelwerte ± SEM aus sechs Herzen, die
mit 250 Schlägen/min
schlugen.
-
Aus
den obigen Experimenten folgt, daß die Zunahme des koronaren
Flusses mediiert wird durch den direkten Dilatationseffekt auf die
Koronararterien und daß der
Dilatationseffekt von der Inhibierung des in den Koronararterien
vorhandenen Phospholambans herrührt.
Die Ergebnisse lassen daher eine potentielle Verwendung von Phospholambaninhibitoren
bei der Behandlung von Patienten als möglich erscheinen, welche unter
einer Abnahme des koronaren Flusses leiden.
-
Tabelle
2 zeigt die EC50-Werte und maximalen Effekte verschiedener anderer
Phospholambaninhibitoren der Formel (I) oder (II) auf den koronaren
Fluß.
-
TABELLE
2. EC50-Werte und maximale Effekte (%-Änderung von der Basislinie)
auf den koronaren Fluß
-
Experiment 6
-
Dieses
Experiment zeigt auf, daß der
Vasodilatoreffekt des Phospholambaninhibitors des Beispiels 1c von
einem Endothelial-mediierten Mechanismus herrührt. Die Substanz P, ein Endothelial-abhängiger Vasodilator,
wurde als eine Referenzverbindung verwendet. Triton × 100 wurde
verwendet zur Zerstörung
der Endothelialzellen der Koronargefäße.
-
Das
Experiment wurde durchgeführt
unter einem Zustand des konstanten Drucks (50 mmHg) an einem freischlagenden
Herzen. Der koronare Fluß (ml/min)
wurde kontinuierlich mittels eines elektromagnetischen Durchflußmessers
mit einer oberhalb der Aortakanüle
eingesetzten Durchflußsonde
aufgezeichnet.
-
Nach
15 min der Aufzeichnung der Grundlinie wurde ein Bolusdosis der
Substanz P (0,1 μM/0,3
ml) über
einen Sei tenzweig der Aortakanüle
(4B) dem Perfusionspuffer zugegeben. Die Zugabe
der Substanz P wurde zweimal wiederholt, bevor die Injektion von
Triton × 100
(0,05%/0,2 ml) über
eine Nadel, die unmittelbar oberhalb der Aorta in die Kanüle eingesetzt
wurde, gegeben wurde. Nach 85 min der Triton × 100-Injektion wurde eine
Kontrolldosis der Substanz P zweimal zu dem Perfusionspuffer gegeben,
um zu überprüfen, ob
die Endothelialzellen zerstört
worden sind. Danach wurde der Konzentrationsbereich von 0,3–30 μM der Verbindung
des Beispiels 1c getestet. Die Vehikelkonzentration (0,1% DMSO)
wurde im Verlauf des Experiments konstant gehalten. In 4A ist
ein Kontrollexperiment gezeigt, wobei der Effekt des Beispiels 1c
bei demselben Konzentrationsbereich zu sehen ist, wenn das Endothelin
nicht durch Triton × 100
zerstört
wird.
-
Die
folgenden nichteinschränkenden
Beispiels veranschaulichen die Herstellung von Phospholambaninhibitoren.
-
BEISPIELE
Beispiel
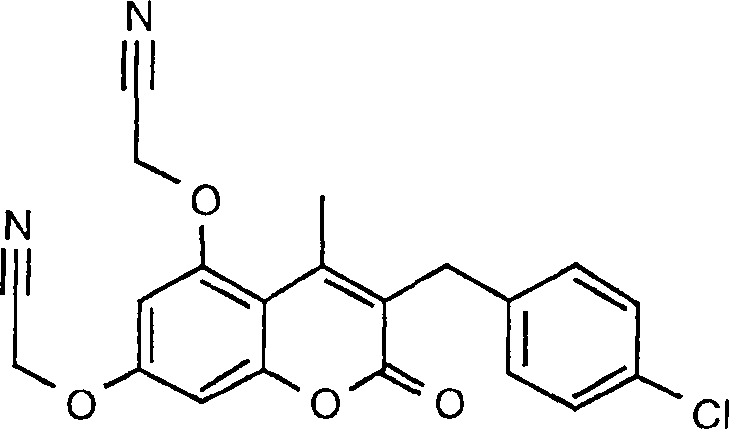
1. Herstellung von 3-Benzyl-5,7-bis[(1H-tetrazol-5-yl)methoxy]-4-methyl-2H-1-benzopyran-2-on
a)
3-Benzyl-5,7-dihydroxy-4-methyl-2H-1-benzopyran-2-on
-
Eine
Lösung
von Phloroglucinoldihydrat (20 g) und Ethyl-2-benzylacetoacetat
(27,5 ml) in Ethanol (320 ml) wurde mit trockenem HCl bei 0°C während fünf Stunden
behandelt, und die Lösung
wurde über
Nacht auf der Tempera tur gehalten. Die gelbe Lösung wurde konzentriert und
mit Wasser verrieben, die Feststoffe filtriert, mit Wasser gewaschen
und getrocknet. Das resultierende Hydrat wurde dreimal aus Toluol
bis zur Trockne eingedampft, mit Petrolether (Siedepunkt 40–60°C) verrieben
und filtriert. Ausbeute: 33,4 g (96%). Schmelzpunkt: 258–260°C.
1H-NMR (DMSO-d6,
400 MHz): 2,525 (s, 3H, CH3), 3,887 (s,
2H, CH2Ph), 6,171 (d, 1H, J = 2,4 Hz), 6,274
(d, 1H, J = 2,4 Hz), 7,167–7,279
(m, 5H, Ph), 10,2 (s, 1H, OH), 10,47 (s, 1H, OH).
-
b)
3-Benzyl-5,7-bis(cyanomethoxy)-4-methyl-2H-1-benzopyran-2-on
-
Chloracetonitril
(6,86 g), Kaliumcarbonat (23,9 g) und 12,2 g des Produkts aus Beispiel
1a wurden während
zwei Stunden bei 100°C
unter Stickstoff in 120 ml an DMF gerührt. Die Reaktionsmischung
wurde gekühlt
und in Eiswasser gegossen. Die Feststoffe wurden filtriert und mit
Wasser gewaschen. Ausbeute: 13,8 g (88%). Schmelzpunkt: 147–154°C.
1H-NMR (DMSO-d6,
400 MHz): 2,525 (s, 3H, CH3), 3,969 (s,
2H, CH2Ph), 5,307 (s, 2H, OCH2CN),
5,314 (s, 2H, OCH2CN), 6,814 (d, 1H, J =
2,5 Hz), 6,940 (d, 1H, J = 2,5 Hz), 7,18–7,292 (m, 5H, Ph).
-
c)
3-Benzyl-5,7-bis[(1H-tetrazol-5-yl)methoxy]-4-methyl-2H-1-benzopyran-2-on
-
Das
Produkt aus Beispiel 1b (1 g), Natriumazid (0,42 g) und Ammoniumchlorid
(0,34 g) wurden während
fünf Stunden
bei 100°C
unter Stickstoff in DMF (5 ml) gerührt. Man ließ die Reaktionsmischung
abkühlen und
sie wurde dann in Eiswasser gegossen. Der pH-Wert der Lösung wurde
auf 10–11
eingestellt und die Lösung
entweder einmal mit Ethylacetat extrahiert oder durch CELITE filtriert.
Die Lösung
wurde mit Chlorwasserstoffsäure
auf pH 2 angesäuert,
auf 5°C
gehalten und filtriert. Ausbeute: 0,96 g (81%). Schmelzpunkt: 229–233°C.
1H-NMR (DMSO-d6,
400 MHz): 2,468 (s, 3H, CH3), 3,937 (s,
2H, CH2Ph), 5,596 (s, 2H, OCH2Tet),
5,602 (s, 2H, OCH2Tet), 6,832 (d, 1H, J
= 2,4 Hz), 6,851 (d, 1H, J = 2,4 Hz), 7,171–7,283 (m, 5H, Ph).
-
Beispiel
2. Herstellung von 7,8,9,10-Tetrahydro-1,3-bis[(1H-tetrazol-5-yl)methoxy]-7-phenyl-6H-dibenzo[b,d]pyran-6-on
a)
7,8,9,10-Tetrahydro-1,3-dihydroxy-7-phenyl-6H-dibenzo[b,d]pyran-6-on
-
Eine
Lösung
von Phloroglucinol (0,7 g) und 2-Ethoxycarbonyl-3-phenylcyclohexanon
(1,5 g) in Ethanol wurde wie in Beispiel 1a beschrieben mit trockenem
HCl behandelt. Das Produkt wurde zuerst aus Ethanol-Wasser (1 :
1) rekristallisiert und dann mit Ether verrieben. Ausbeute: 0,61
g.
1H-NMR (DMSO-d6,
400 MHz): 1,38–1,52
(m, 1H), 1,57–1,66
(m, 1H), 1,69–1,78
(m, 1H), 1,86–1,96
(m, 1H), 2,9–3,02
(m, 1H), 3,3–3,4
(m, 1H), 4,050 (b, 1H), 6,157 (d, 1H, J = 2,4 Hz), 6,297 (d, 1H,
J = 2,4 Hz), 7,076–7,265
(m, 5H), 10,153 (s, 1H), 10,456 (s, 1H).
-
b)
7,8,9,10-Tetrahydro-1,3-bis(cyanomethoxy)-7-phenyl-6H-dibenzo[b,d]pyran-6-on
-
Das
Produkt aus Beispiel 2a (0,5 g) wurde mit Chloracetonitril (0,25
g) und Kaliumcarbonat (1,12 g) wie in Beispiel 1b beschrieben in
DMF (5 ml) behandelt. Ausbeute: 0,6 g.
1H-NMR
(DMSO-d6, 400 MHz): 1,38–1,58 (m, 1H), 1,6–1,7 (m,
1H), 1,7–1,76
(m, 1H), 1,89–1,99
(m, 1H), 2,9–3,03
(m, 1H), 3,2–3,28
(m 1H), 4,111 (b, 1H), 5,314 (s, 2H), 5,349 (s, 2H), 6,840 (d, 1H,
J = 2,5 Hz), 6,925 (d, 1H, J = 2,5 Hz), 7,108–7,274 (m, 5H).
-
c)
7,8,9,10-Tetrahydro-1,3-bis[(1H-tetrazol-5-yl)methoxy]-7-phenyl-6H-dibenzo[b,d]pyran-6-on
-
Das
Produkt aus Beispiel 2b (0,6 g) wurde wie in Beispiel 1c mit Natriumazid
(0,2 g) und Ammoniumchlorid (0,17 g) in DMF (5 ml) behandelt. Das
Produkt wurde aus einer Mischung aus DMF, Ethanol und Wasser (ungefähr 1 : 2
: 3) rekristallisiert. Ausbeute: 0,41 g. Schmelzpunkt: 153–154°C.
1H-NMR (DMSO-d6,
400 MHz): 1,38–1,5
(m, 1H), 1,5–1,6
(m, 1H), 1,69–1,76
(m, 1H) 1,87–1,96
(m, 1H), 2,9–3,05
(m, 1H), 3,2–3,3
(m, 1H), 4,094 (b, 1H), 5,602 (s, 2H), 5,643 (s, 2H), 6,832 (d,
1H, J = 2,3 Hz), 6,851 (d, 1H, J = 2,3 Hz), 7,089–7,212 (m,
5H).
-
Beispiel
3. Herstellung von 3-Benzyl-5,7-bis[(2,5-dihydro-5-oxo-4H-1,2,4-oxadiazol-3-yl)methoxy]-4-methyl-2H-1-benzopyran-2-on
a)
3-Benzyl-5,7-bis[(hydroxyamidino)methoxy]-4-methyl-2H-1-benzopyran-2-on
-
Zu
einer Suspension aus Hydroxylaminhydrochlorid (0,97 g) in DMSO (2
ml) wurde Triethylamin (1,94 ml) ge geben und die resultierende Mischung
während
30 min bei Raumtemperatur gerührt.
Die Kristalle wurden filtriert und mit THF gewaschen. Das Filtrat
wurde konzentriert und das Produkt aus Beispiel 1b (0,5 g) zugegeben.
Diese Lösung
wurde über
Nacht bei 75°C
gehalten. Die Reaktionsmischung wurde mit Eiswasser behandelt, der
pH-Wert auf 11 eingestellt und die Feststoffe filtriert, mit Wasser
gewaschen und getrocknet. Ausbeute: 0,5 g. Schmelzpunkt: 155–160°C.
1H-NMR (DMSO-d6,
400 MHz): 2,56 (s, 3H, CH3), 3,938 (s, 2H),
4,466 (s, 2H), 4,486 (s, 2H), 5,565 (s, H, NH2), 5,709
(s, 2H, NH2), 6,658 (d, 1H, J = 2,4 Hz),
6,692 (d, 1H, J = 2,4 Hz), 7,168–7,284 (m, 5H, Ph), 9,346 (s,
1H, OH), 9,362 (s, 1H, OH).
-
b)
3-Benzyl-5,7-bis[(ethoxycarbonyloxyamidino)methoxy]-4-methyl-2H-1-benzopyran-2-on
-
Ethylchlorformiat
(0,45 ml) wurde zu einer Lösung
des Produkts aus Beispiel 3a (1 g) und Pyridin (0,38 ml) in DMF
(5 ml) bei 0°C
gegeben. Die Reaktionsmischung wurde während zusätzlicher 30 Minuten bei dieser Temperatur
gehalten, und dann wurde Eiswasser zugegeben. Die Feststoffe wurden
filtriert und mit Wasser gewaschen. Ausbeute: 1,63 g. Schmelzpunkt:
87–92°C.
1H-NMR (DMSO-d6,
400 MHz): 1,215–1,256
(m, 6H), 2,553 (s, 3H), 3,947 (s, 2H), 4,140–4,198 (m, 4H), 4,566 (s, 2H),
4,599 (s, 2H), 6,688 (d, 1H, J = 2,4 Hz), 6,718 (d, 1H, J = 2,4
Hz), 6,792 (b, 2H, NH2), 6,818 (b, 2H, NH2), 7,171–7,285 (m, 5H).
-
c)
3-Benzyl-5,7-bis[(2,5-dihydro-5-oxo-4H-1,2,4-oxadiazol-3-yl)methoxy]-4-methyl-2H-1-benzopyran-2-on
-
Das
Produkt des vorherigen Beispiels (1,5 g) und DBU (0,8 ml) in DMF
(5 ml) wurde bei Raumtemperatur über
Nacht gerührt.
Die Reaktionsmischung wurde mit Eiswasser behandelt und angesäuert. Die
Feststoffe wurden filtriert und mit Wasser gewaschen. Die resultierende
feste Masse wurde in 0,1 N Natriumhydroxidlösung aufgenommen, mit Aktivkohle
behandelt und schließlich
angesäuert.
Ausbeute: 0,64 g. Schmelzpunkt: 130–136°C.
1H-NMR
(DMSO-d6, 400 MHz): 2,524 (s, 3H), 3,954
(s, 2H), 5,187 (s, 2H), 5,215 (s, 2H), 6,748 (d, 1H, J = 2,4 Hz),
6,834 (d, 1H, J = 2,4 Hz), 7,158–7,289 (m, 5H), 12,8 (b, 2H).
-
Beispiel
4. Herstellung von 7,8,9,10-Tetrahydro-bis[(1H-tetrazol-5-yl)methoxy]-1,3-dihydroxy-6H-dibenzo[b,d]pyran-6-on
a)
7,8,9,10-Tetrahydro-1,3-dihydroxy-6H-dibenzo[b,d]pyran-6-on
-
Phloroglucinol
(1 g) und Ethyl-2-oxocyclohexancarboxylat (1,32 g) wurden in 75%iger
Schwefelsäure (10
ml) über
Nacht gerührt,
die Mischung in Eiswasser gegossen und filtriert. Ausbeute: 1,55
g.
1H-NMR (DMSO-d6,
400 MHz): 1,65 (b, 4H), 2,345 (b, 2H), 3,037 (b, 2H), 6,138 (d,
1H, J = 2,4 Hz), 6,245 (d, 1H, J = 2,4 Hz), 10,069 (b, 1H, OH),
10,322 (s, 1H, OH).
-
b)
7,8,9,10-Tetrahydro-bis(cyanomethoxy)-1,3-dihydroxy-6H-dibenzo[b,d]pyran-6-on
-
Das
Produkt des vorherigen Beispiels (0,5 g), Chloracetonitril (0,34
g) und Kaliumcarbonat (1,5 g) in DMF (5 ml) wurde wie in Beispiel
1b umgesetzt. Ausbeute: 0,44 g.
1H-NMR
(DMSO-d6, 400 MHz): 1,68 (b, 4H), 2,41 (b,
2H), 3,00 (b, 2H), 5,297 (s, 2H), 5,309 (s, 2H), 6,797 (d, 1H, J
= 2,4 Hz), 6,899 (d, 1H, J = 2,4 Hz).
-
c)
7,8,9,10-Tetrahydro-bis[(1H-tetrazol-5-yl)methoxy]-1,3-dihydroxy-6H-dibenzo[b,d]pyran-6-on
-
Das
Produkt des vorherigen Beispiels (0,4 g) wurde wie in Beispiel 1c
mit Natriumazid (0,18 g) und Ammoniumchlorid (0,14 g) in DMF (2,5
ml) behandelt. Das Produkt wurde aus Ethanol-DMF (1 : 1) rekristallisiert.
Ausbeute: 0,17 g. Schmelzpunkt: 283–286°C.
1H-NMR
(DMSO-d6, 400 MHz): 1,626 (b, 4H), 2,393
(b, 2H), 2,971 (b, 2H), 5,583 (s, 2H), 5,599 (s, 2H), 6,811 (s,
2H).
-
Beispiel
5. Herstellung von 5,7-Bis[(1H-tetrazol-5-yl)methoxy]-4-phenyl-2H-1-benzopyran-2-on
a)
5,7-Dihydroxy-4-phenyl-2H-1-benzopyran-2-on
-
Eine
Lösung
von Phloroglucinol (2,00 g) und Ethylbenzoylacetat (3,05 g) in Ethanol
(30 ml) wurde wie in Beispiel 1a beschrieben mit trockenem HCl behandelt.
Das Produkt wurde aus Ethanol-Wasser (1 : 1) rekristallisiert. Ausbeute:
3,0 g (75%).
1H-NMR (DMSO-d6, 300 MHz): 5,739 (s, 1H, CH=C), 6,155 (d,
1H, J = 2,3 Hz), 6,263 (d, 1H, J = 2,3 Hz), 7,305–7,381 (m,
5H, Ph), 10,084 (s, 1H, OH), 10,368 (s, 1H, OH).
-
b)
5,7-Bis(cyanomethoxy)-4-phenyl-2H-1-benzopyran-2-on
-
Das
Produkt des vorherigen Beispiels (1,00 g) wurde wie in Beispiel
1b beschrieben mit Chloracetonitril (0,62 g) und Kaliumcarbonat
(2,72 g) in DMF (5 ml) behandelt. Die Reaktionsmischung wurde in
Eiswasser gegossen und die Mischung mit Ethylacetat extrahiert.
Das Ethylacetat wurde mit 1 M NaOH gewaschen, mit Natriumsulfat
getrocknet und verdampft. Das Produkt wurde aus Isopropanol rekristallisiert.
Ausbeute: 0,41 g (31%).
1H-NMR (DMSO-d6, 300 MHz): 4,845 (s, 2H, OCH2CN),
5,344 (s, 2H, OCH2CN), 6,086 (s, 1H, CH=C),
6,770 (d, 1H, J = 2,4 Hz), 7,040 (d, 1H, J = 2,4 Hz), 7,320–7,443 (m,
5H, Ph).
-
c)
5,7-Bis[(1H-tetrazol-5-yl)methoxy]-4-phenyl-2H-1-benzopyran-2-on
-
Das
Produkt des vorherigen Beispiels (0,40 g) wurde während 2
Stunden bei 100°C
mit Natriumazid (0,16 g) und Ammoniumchlorid (0,14 g) in DMF (2
ml) behandelt. Das Produkt wurde wie in Beispiel 1c beschrieben
isoliert. Ausbeute: 0,40 g (79%). Schmelzpunkt: 222–224°C.
1H-NMR (DMSO-d6,
400 MHz): 5,148 (s, 2H, OCH2Tet), 5,649
(s, 2H, OCH2Tet), 5,968 (s, 1H, CH=C), 6,811 (d, 1H,
J = 2,3 Hz), 6,962 (d, 1H, J = 2,3 Hz), 6,994–7,185 (m, 5H, Ph).
-
Beispiel
6. Herstellung von 7,8,9,10-Tetrahydro-1,3-bis[(1H-tetrazol-5-yl)methoxy]-8-phenyl-6H-dibenzo[b,d]pyran-6-on
a)
7,8,9,10-Tetrahydro-1,3-dihydroxy-8-phenyl-6H-dibenzo[b,d]pyran-6-on
-
Eine
Lösung
von Phloroglucinol (1,56 g) und Ethyl-2-oxo-5-phenylcyclohexancarboxylat (2,52
g) in Ethanol (25 ml) wurde wie in Beispiel 1a beschrieben mit trockenem
HCl behandelt. Der Niederschlag wurde filtriert und mit Wasser und
EtOH gewaschen. Ausbeute: 1,0 g (32%).
1H-NMR
(DMSO-d6, 400 MHz): 1,72–1,82 (m, 1H), 2,01 (b, 1H),
2,317–2,387
(m, 1H), 2,707–2,763
(m, 1H), 2,830 (b, 1H), 3,041 (b, 1H), 3,35 und 3,40 (b, 1H), 6,174
(d, 1H, J = 2,3 Hz), 6,277 (d, 1H, J = 2,3 Hz), 7,200–7,350 (m,
5H, Ph), 10,131 (s, 1H, OH), 10,401 (s, 1H, OH).
-
b)
7,8,9,10-Tetrahydro-1,3-bis(cyanomethoxy)-8-phenyl-6H-dibenzo[b,d]pyran-6-on
-
Das
Produkt des vorherigen Beispiels (1,0 g) wurde wie in Beispiel 1b
beschrieben mit Chloracetonitril (0,57 g) und Kaliumcarbonat (1,0
g) in DMF (5 ml) behandelt. Das DMF wurde verdampft und der Rückstand in
EtOAc gelöst.
Das Ethylacetat wurde mit 1 M NaOH gewaschen, mit Natriumsulfat
getrocknet und verdampft. Das Produkt wurde aus Aceton-Isopropanol
(1 : 3) rekristallisiert. Ausbeute: 0,50 g (40%).
1H-NMR
(DMSO-d6, 300 MHz): 1,75–1,88 (m, 1H), 2,05 (b, 1H),
2,38–2,48
(m, 1H), 2,77–2,85
(m, 1H), 2,90 (b, 1H), 3,07 (b, 1H), 3,22 und 3,28 (b, 1H), 5,316
(s, 2H, OCH2CN), 5,331 (s, 2H, OCH2CN), 6,829 (d, 1H, J = 2,5 Hz), 6,939 (d,
1H, J = 2,5 Hz), 7,210–7,380
(m, 5H, Ph).
-
c)
7,8,9,10-Tetrahydro-1,3-bis[(1H-tetrazol-5-yl)methoxy]-8-phenyl-6H-dibenzo[b,d]pyran-6-on
-
Das
Produkt des vorherigen Beispiels (0,30 g) wurde mit Natriumazid
(0,10 g) und Ammoniumchlorid (0,09 g) in DMF (2 ml) während 3,5
Stunden bei 100°C
behandelt. Das Produkt wurde auf dieselbe Weise wie in Beispiel
1c isoliert. Ausbeute: 0,30 g (82%). Schmelzpunkt: 235–245°C.
1H-NMR (DMSO-d6,
400 MHz): 1,70–1,80
(m, 1H), 1,96 (b, 1H), 2,38–2,446
(m, 1H), 2,836 (m, 2H), 3,052 (b, 1H), 3,252 und 3,301 (b, 1H),
5,604 (s, 2H, OCH2CN), 5,632 (s, 2H, OCH2CN), 6,827 (d, 1H, J = 2,5 Hz), 6,858 (d,
1H, J = 2,5 Hz), 7,209–7,351
(m, 5H, Ph).
-
Beispiel
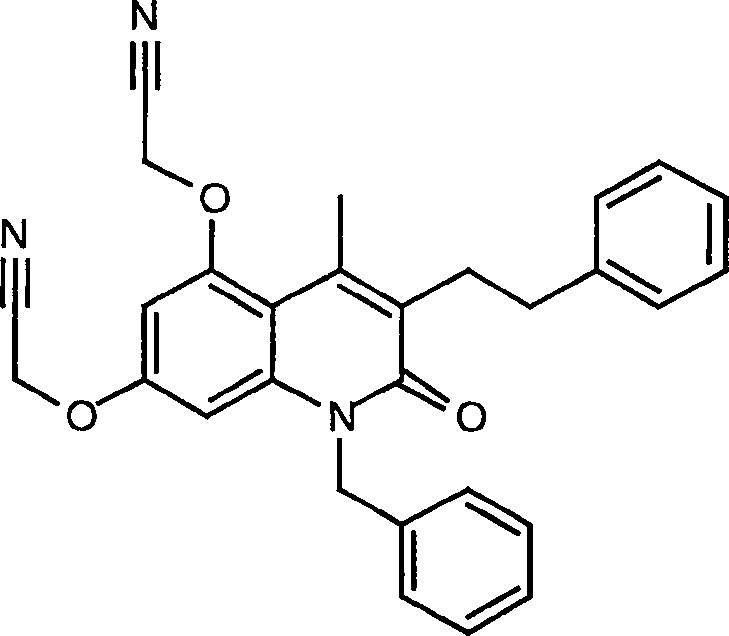
7. Herstellung von 5,7-Bis[(1H-tetrazol-5-yl)methoxy]-4-methyl-3-(2-phenylethyl)-2H-1-benzopyran-2-on
a) 5,7-Dihydroxy-4-methyl-3-(2-phenylethyl)-2H-1-benzopyran-2-on
-
Eine
Lösung
von Phloroglucinol (0,87 g) und Ethyl-2-(2-phenylethyl)acetoacetat (1,62 g)
in Ethanol (30 ml) wurde wie in Beispiel 1a beschrieben mit trockenem
HCl behandelt. Ausbeute: 1,77 g (87%). Schmelzpunkt: 248–252°C.
1H-NMR (DMSO-d6,
300 MHz): 2,413 (s, 3H, CH3), 2,652–2,782 (m,
4H, CH2CH2), 6,151
(d, 1H, J = 2,4 Hz), 6,256 (d, 1H, J = 2,4 Hz), 7,183–7,304 (m,
5H, Ph), 10,137 (s, 1H, OH), 10,369 (s, 1H, OH).
-
b)
5,7-Bis(cyanomethoxy)-4-methyl-3-(2-phenylethyl)-2H-1-benzopyran-2-on
-
Das
Produkt des vorherigen Beispiels (0,90 g) wurde mit Chloracetonitril
(0,48 g) und Kaliumcarbonat (2,1 g) während 0,5 Stunden bei 100°C in DMF
(5 ml) behandelt. Das Produkt wurde wie in Beispiel 1b beschrieben
isoliert. Ausbeute: 1,00 g (88%). Schmelzpunkt: 179–183°C.
1H-NMR (DMSO-d6,
300 MHz): 2,384 (s, 3H, CH3), 2,699–2,754 (m,
2H, CH2CH2), 2,805–2,841 (m,
2H, CH2CH2), 5,302
(s, 4H, OCH2CN), 6,790 (d, 1H, J = 2,5 Hz),
6,909 (d, 1H, J = 2,5 Hz), 7,190–7,307 (m, 5H, Ph).
-
c)
5,7-Bis[(1H-tetrazol-5-yl)methoxy]-4-methyl-3-(2-phenylethyl)-2H-1-benzopyran-2-on
-
Das
Produkt des vorherigen Beispiels (0,40 g) wurde mit Natriumazid
(0,15 g) und Ammoniumchlorid (0,12 g) während 2,5 Stunden bei 100°C in DMF
(2 ml) behandelt. Das Produkt wurde wie in Beispiel 1c isoliert. Ausbeute:
0,385 g (78%). Schmelzpunkt: 248–250°C.
1H-NMR
(DMSO-d6, 400 MHz): 2,368 (s, 3H, CH3), 2,668–2,707 (m, 2H, CH2CH2), 2,783–2,822 (m, 2H, CH2CH2), 5,593 (s, 2H, OCH2Tet),
5,604 (s, 2H, OCH2Tet), 6,819 (d, 1H, J
= 2,3 Hz), 6,834 (d, 1H, J = 2,3 Hz), 7,161–7,291 (m, 5H, Ph).
-
Beispiel
8. Herstellung von 5,7-Bis[(1H-tetrazol-5-yl)methoxy]-1,3-dibenzyl-4-methyl-2(1H)-chinolinon
a)
2-Benzyl-3-oxobutansäure-3,5-dimethoxyanilid
-
3,5-Dimethoxyanilin
(5 g) wurde portionsweise zu vorerwärmtem (160°C) Ethyl-2-benzylacetoacetat (15
ml) unter Stickstoff gegeben und während 60 Minuten bei der Temperatur
gehalten. Die gekühlte
Lösung wurde
mit Hepta nethylether verdünnt
und filtriert. Ausbeute: 5,2 g (49%).
1H-NMR
(DMSO-d6, 300 MHz): 2,183 (s, 3H), 3,069
(d, 2H, J = 7,2 Hz), 3,923 (t, 1H, J = 7,2 Hz), 6,616 (dd. 1H, J
= 2,3 Hz), 6,765 (d, 2H, J = 2,3 Hz), 7,13–7,3 (m, 5H), 10,123 (s, 1H).
-
b)
3-Benzyl-5,7-dimethoxy-4-methyl-2(1H)-chinolinon
-
Das
Produkt des vorherigen Beispiels (1,2 g) wurde zu vorerwärmter (85°C) Methansulfonsäure (3,5 ml)
gegeben und während
15 Minuten bei der Temperatur gehalten. Man ließ die Lösung abkühlen und behandelte sie dann
mit Eiswasser. Das Produkt wurde filtriert, mit Natriumbicarbonat
und Wasser gewaschen. Ausbeute: 1,08 g (95%).
1H-NMR
(300 MHz): 2,486 (s, 3H), 3,785 (s, 3H), 3,808 (s, 3H), 3,985 (s,
2H), 6,315 (d, 1H, J = 2,4 Hz), 6,472 (d, 1H, J = 2,4 Hz), 7,1–7,3 (m,
5H), 11,52 (s, H).
-
c)
3-Benzyl-5,7-dihydroxy-4-methyl-2(1H)-chinolinon
-
Das
Produkt des vorherigen Beispiels (1 g) wurde während 20 Minuten unter Stickstoff
in Pyridinhydrochlorid (5 g) unter Rückfluß gehalten. Die Reaktionsmischung
wurde mit Wasser behandelt und das Produkt filtriert. Ausbeute:
0,9 g (100%). Schmelzpunkt: 307–312°C.
1H-NMR (300 MHz): 2,503 (s, 3H), 3,942 (s,
2H), 6,102 (d, 1H, J = 2,3 Hz), 6,187 (d, 1H, J = 2,3 Hz), 7,1– 7,25 (m,
5H), 9,725 (s, 1H), 9,984 (s, 1H), 11,285 (s, 1H).
-
d)
1,3-Dibenzyl-5,7-dimethoxy-4-methyl-2(1H)-chinolinon
-
Das
Produkt des Beispiels 8b (1 g), Kalium-t-butoxid (0,62 g) und Benzylbromid
(0,68 ml) wurden während
4 Stunden bei 60°C
in DMSO (10 ml) gerührt.
Die Reaktionsmischung wurde mit Wasser behandelt, mit Toluol extrahiert
und verdampft. Das Produkt wurde mit Ethylether verrieben und filtriert.
Ausbeute: 0,5 g (39%).
1H-NMR (400
MHz): 2,537 (s, 3H), 3,708 (s, 3H), 3,826 (s, 3H), 4,124 (s, 2H),
5,56 (b, 2H), 6,413–6,434
(m, 2H), 7,154–7,332
(m, 10H).
-
e)
1,3-Dibenzyl-5,7-dihydroxy-4-methyl-2(1H)-chinolinon
-
Das
Produkt des vorherigen Beispiels (2 g) wurde wie in Beispiel 8c
beschrieben mit Pyridinhydrochlorid (10 g) behandelt. Das Produkt
wurde mit Ethylacetat extrahiert und eingedampft. Ausbeute: 1,4
g (75%).
1H-NMR (400 MHz): 2,570 (s,
3H), 4,076 (s, 2H), 5,450 (b, 2H), 6,135 (d, 1H, J = 2,2 Hz), 6,199
(d, 1H, J = 2,2 Hz), 7,128–7,333
(m, 10H), 9,83 (b, 1H), 10,166 (s, 1H).
-
f)
5,7-Bis(cyanomethoxy)-1,3-dibenzyl-4-methyl-2(1H)-chinolinon
-
Das
Produkt des vorherigen Beispiels (1,4 g) wurde wie in Beispiel 1b
beschrieben mit Chloracetonitril (0,76 g) und K2CO3 (2,5 g) in DMF (20 ml) behandelt. Ausbeute:
1,5 g (89%).
1H-NMR (400 MHz): 2,555
(s, 3H), 4,146 (s, 2H), 5,214 (s, 2H), 5,275 (s, 2H), 5,578 (s,
2H), 6,735 (s, 2H), 7,13–7,33
(m, 10H).
-
g)
5,7-Bis[(1H-tetrazol-5-yl)methoxy]-1,3-dibenzyl-4-methyl-2(1H)-chinolinon
-
Das
Produkt des vorherigen Beispiels (1,3 g) wurde wie in Beispiel 1c
beschrieben mit Natriumazid (0,41 g) und Ammoniumchlorid (0,34 g)
behandelt. Ausbeute: 0,69 g (45%).
1H-NMR
(400 MHz): 2,471 (s, 3H), 4,113 (s, 2H), 5,477 (s, 2H), 5,55 (b,
2H), 5,574 (s, 2H), 6,670 (d, 1H, J = 2,1 Hz), 6,775 (d, 1H, J =
2,1 Hz), 7,13–7,32
(m, 10H).
-
Beispiel
9. Herstellung von 5,7-Bis[(1H-tetrazol-5-yl)methoxy]-3-benzyl-1,4-dimethyl-2(1H)-chinolinon
a)
3-Benzyl-5,7-dimethoxy-1,4-dimethyl-2(1H)-chinolinon
-
Das
Produkt des Beispiels 8b (0,5 g), t-BuOK (0,2 g) und Methyliodid
(0,4 ml) wurden während
zwei Tagen bei 35°C
in DMSO (5 ml) gerührt.
Die Reaktionsmischung wurde mit Wasser behandelt und mit Toluol extrahiert.
Das Produkt wurde durch Säulenchromatographie
unter Verwendung von Toluol-Ethylacetat-Essigsäure 8 : 2 : 1 als dem Eluenten
gereinigt. Ausbeute: 0,24 g (46%).
1H-NMR
(300 MHz): 2,51 (s, 3H), 3,632 (s, 2H), 3,846 (s, 3H), 3,896 (s,
3H), 4,047 (s, 2H), 6,468 (d, 1H, J = 2,3 Hz), 6,558 (d, 1H, J =
2,3 Hz), 7,1–7,26
(m, 5H).
-
b)
3-Benzyl-5,7-dihydroxy-1,4-dimethyl-2(1H)-chinolinon
-
Das
Produkt des vorherigen Beispiels (0,2 g) wurde wie in Beispiel 8c
beschrieben mit Pyridinhydrochlorid (2 g) behandelt und das Produkt
mit Ethylacetat extrahiert. Ausbeute: 0,16 g (89%).
1H-NMR (400 MHz): 2,567 (s, 3H), 3,515 (s,
3H), 4,005 (5, 2H), 6,244 (d, 1H, J = 2,3 Hz), 6,268 (d, 1H, J =
2,3 Hz), 7,08–7,25
(m. 5H), 9,879 (s, 1H), 10,113 (s, 1H).
-
c)
5,7-Bis(cyanomethoxy)-3-benzyl-1,4-dimethyl-2(1H)-chinolinon
-
Das
Produkt des vorherigen Beispiels (0,15 g), Chloracetonitril (0,08
g) und K2CO3 (0,28
g) wurden wie in Beispiel 1b beschrieben in DMF (2 ml) umgesetzt.
Ausbeute: 0,16 g (84%).
1H-NMR (400
MHz): 2,524 (s, 3H), 3,658 (s, 3H), 4,079 (s, 2H), 5,292 (s, 2H),
5,379 (s, 2H), 6,766 (d, 1H, J = 2,3 Hz), 6,855 (d, 1H, J = 2,3
Hz), 7,13–7,24
(m 5H).
-
d)
5,7-Bis[(1H-tetrazol-5-yl)methoxy]-3-benzyl-1,4-dimethyl-2(1H)-chinolinon
-
Das
Produkt des vorherigen Beispiels (0,15 g) wurde wie in Beispiel
1c beschrieben mit NaN3 (57 mg) und NH4Cl (47 mg) in DMF (2 ml) behandelt. Ausbeute:
0,115 g. Schmelzpunkt: 250–253°C.
1H-NMR (400 MHz): 2,451 (s, 3H), 3,649 (s,
3H), 4,042 (s, 2H), 6,792 (d, 1H, J = 2,2 Hz), 6,833 (d, 1H, J =
Hz), 7,1–7,25
(m, 5H).
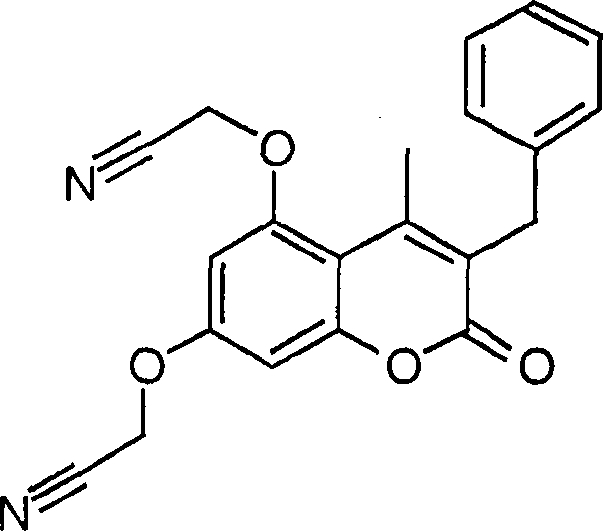
-
Beispiel
10. Herstellung von 3-Benzyl-5,7-bis[(2-methyl-1H-tetrazol-5yl)methoxy]-4-methyl-2H-1-benzopyran-2-on und der drei Isomeren
-
0,07
ml an Methyliodid wurden zu einer Lösung von 0,2 g des Produkts
aus Beispiel 1c und 0,31 g an K2CO3 in 2 ml an DMF gegeben, und die Mischung
wurde während
4 Stunden bei Raumtemperatur gerührt. Die
Reaktionsmischung wurde in Eiswasser gegossen und filtriert. Ausbeute:
0,2 g als eine Mischung von vier Regioisomeren. Schmelzpunkt: 71–76°C.
1H-NMR (DMSO-d6,
400 MHz): 2,47 (s, CH3), 2,48 (s, CH3), 3,93 (s, CH2Ph),
4,11 (s, NCH3), 4,12 (s, NCH3), 4,15
(s, NCH3), 4,38 (s, NCH3),
4,40 (s, NCH3), 5,51 (s, OCH2),
5,52 (s, OCH2), 5,62 (s, OCH2),
5,67 (s, OCH2), 6,84–6,91 (m, 2H), 7,16–7,28 (m,
5H, Ph).
-
Beispiel
11. Herstellung von 3-Benzyl-5,7-bis[1-(1H-tetrazol-5-yl)ethoxy]-4-methyl-2H-1-benzopyran-2-on,
Mischung aus Stereoisomeren
a) 3-Benzyl-5,7-bis-[(1-cyano)ethoxy)-4-methyl-2H-1-benzopyran-2-on
-
Das
Produkt des Beispiels 1a (1 g), 2-Chlorpropionitril (0,7 g) und
Kaliumcarbonat (2 g) wurden während
60 Minuten unter Stickstoff bei 110°C in DMF (15 ml) erwärmt. Die
Mischung wurde mit Wasser behandelt, filtriert und mit 1 N NaOH
und Wasser gewaschen. Ausbeute: 1,2 g.
1H-NMR
(DMSO-d6, 300 MHz): 1,74–1,78 (t + t, 6H, CH-CH3), 2,53 (s, 3H), 3,97 (s, 2H), 5,58–5,66 (m,
2H, CH-CH3), 6,87 (m, 1H), 6,99 (d, 1H), 7,18–7,31 (m,
5H).
-
b)
3-Benzyl-5,7-bis[1-(1H-tetrazol-5-yl)ethoxy]-4-methyl-2H-1-benzopyran-2-on, Mischung
aus Stereoisomeren
-
Das
Produkt des vorherigen Beispiels (0,5 g), Natriumazid (0,18 g) und
Ammoniumchlorid (0,15 g) wurden während 90 Minuten bei 100°C in DMF
(7 ml) erwärmt.
Das Produkt wurde mit Wasser behandelt, mit Ethylacetat extrahiert
und eingedampft. Ausbeute: 0,57 g. Schmelzpunkt: 91–104°C.
1H-NMR (DMSO-d6,
300 MHz): 1,69–1,77
(m, 6H, CH-CH3), 2,54 (s, 3H), 3,94 (s,
2H), 6,10–6,17
((m, 2H, CH-CH3), 6,65 (dd, 1H), 6,74 (dd, 1H), 7,13–7,30 (m,
5H).
-
Beispiel
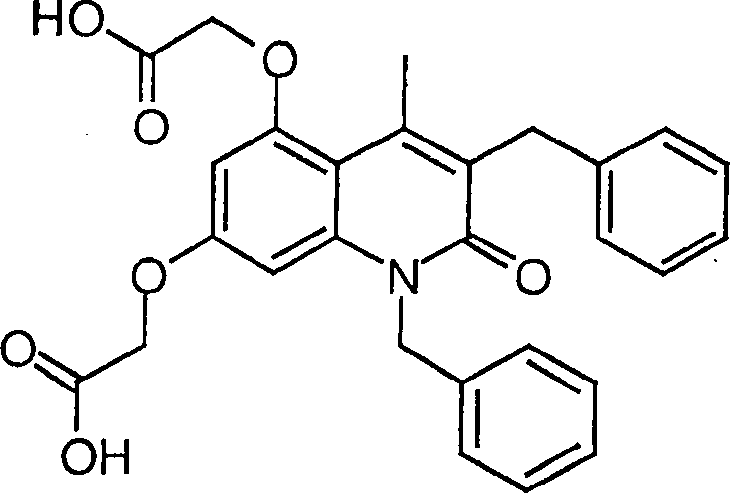
12. Herstellung von 5,7-Bis(carboxymethoxy)-1,3-dibenzyl-4-methyl-2(1H)-chinolinon
-
Das
Produkt des Beispiels 8f (0,2 g) wurde während einer Stunde in einer
Lösung
von konzentrierter Chlorwasserstoffsäure (3 ml) und Essigsäure (2 ml)
unter Rückfluß gehalten.
Das Produkt wurde bei 25°C
filtriert. Ausbeute: 0,14 g.
1H-NMR
(300 Mhz, DMSO-d6): 2,63 (s, CH3),
4,14 (s, 2H, CH2Ph), 4,66 (s, 2H, OCH2COOH), 4,79 (s, 2H, OCH2COOH),
5,53 (s, 2H, NCH2Ph), 6,41 (d, 1H, J = 2,2
Hz), 6,45 (d, 1H, J = 2,2 Hz), 7,13–7,34 (m, 10H, Ph).
-
Beispiel
13. Herstellung von 3-Benzyl-5,7-bis[(1H-tetrazol-5-yl)methoxy]-1-(4-fluorbenzyl)-4-methyl-2(1H)-chinolinon
a)
1-Benzyl-5,7-dimethoxy-3-(4-fluorbenzyl)-4-methyl-2(1H)-chinolinon
-
Das
Produkt des Beispiels 8b (2 g), Kalium-tert-butoxid (0,87 g) und
4-Fluorbenzylchlorid (1,12 g) wurden wie in Beispiel 8d während drei
Stunden bei 60°C
in DMSO (20 ml) erwärmt.
Ausbeute: 1,28 g.
1H-NMR (400 Mhz,
DMSO-d6): 2,53 (s, 3H), 3,73 (s, 3H), 3,83
(s, 3H), 5,55 (s, 2H), 6,43 (s, 2H), 7,12–7,2 (m, 5H), 7,26–7,28 (m,
4H).
-
b)
3-Benzyl-5,7-dihydroxy-1-(4-fluorbenzyl)-4-methyl-2(1H)-chinolinon
-
Das
Produkt des vorherigen Beispiels (1,25 g) wurde während 9
Minuten bei ungefähr
225°C in
Pyridinhydrochlorid (12,5 g) erwärmt.
Ausbeute: 1 g.
1H-NMR (300 Mhz, DMSO-d6): 2,56 (s, 3H), 4,07 (s, 2H), 5,4 (b, 2H),
6,13 (d, 1H, J = 2,1 Hz), 6,20 (d, 1H, J = 2,1 Hz), 7,12–7,28 (m,
9H), 9,88 (s, 1H), 10,22 (s, 1H).
-
c)
3-Benzyl-5,7-Bis(cyanomethoxy)-1-(4-fluorbenzyl)-4-methyl-2(1H)-chinolinon
-
Das
Produkt des vorherigen Beispiels (1 g), ClCH2CN
(0,43 g) und K2CO3 (1,42
g) wurden während einer
Stunde bei 120°C
in DMF (8 ml) erwärmt.
Ausbeute: 0,94 g.
1H-NMR (300 Mhz,
DMSO-d6): 2,55 (s, 3H), 4,14 (s, 2H), 5,25
(s, 2H), 5,28 (s, 2H), 5,57 (s, 2H), 6,74 (s, 2H, ArH), 7,1–7,3 (m,
9H).
-
d)
3-Benzyl-5,7-bis[(1H-tetrazol-5-yl)methoxy]-1-(4-fluorbenzyl)-4-methyl-2(1H)-chinolinon
-
Das
Produkt des vorherigen Beispiels (0,5 g), Natriumazid (0,14 g) und
Ammoniumchlorid (0,12 g) wurden während 90 Minuten bei 120°C in DMF
(5 ml) erwärmt.
Das Produkt wurde mit Acetonitril verrieben. Ausbeute: 0,28 g. Schmelzpunkt:
126–132°C.
1H-NMR (300 Mhz, DMSO-d6):
2,48 (s, 3H), 4,11 (s, 2H), 5,51 (s, 2H), 5,55 (s, 2H), 5,58 (s,
2H), 6,67 (d, 1H, J = 2,1 Hz), 6,78 (d, 1H, J = 2,1 Hz).
-
Beispiel
14. Herstellung von 5,7-Bis[(1H-tetrazol-5-yl)methoxy]-3-(4-chlorbenzyl)-4-methyl-2H-1-benzopyran-2-on
a) 3-(4-Chlorbenzyl)-5,7-dihydroxy-4-methyl-2H-1-benzopyran-2-on
-
Eine
Lösung
von Phloroglucinol (1,57 g) und Ethyl-2-(4-chlorbenzyl)acetoacetat (3,18 g)
in Ethanol (25 ml) wurde während
1,5 Stunden bei 0°C
mit trockenem HCl behandelt und die Lösung über Nacht bei der Temperatur
gehalten. Das Lösungsmittel
wurde verdampft und der Rückstand
mit Wasser verrieben. Ausbeute: 3,87 g (98%). Schmelzpunkt: 270–278°C.
1H-NMR (DMSO-d6,
300 MHz): 2,52 (s, 3H, CH3), 3,87 (s, 2H,
CH2), 6,17 (d, 1H, J = 2,4 Hz), 6,28 (d,
1H, J = 2,4 Hz), 7,18–7,34
(m, 4H, Ph), 10,21 (s, 1H, OH), 10,48 (s, 1H, OH).
-
b)
5,7-Bis(cyanomethoxy)-3-(4-chlorbenzyl)-4-methyl-2H-1-benzopyran-2-on
-
Das
Produkt des vorherigen Beispiels (1,00 g), Chloracetonitril (0,50
g) und Kaliumcarbonat (2,18 g) wurden während 30 Minuten bei 100°C in DMF
(5 ml) erwärmt.
Das Produkt wurde wie in Beispiel 1b beschrieben isoliert. Ausbeute:
0,90 g (72%).
1H-NMR (DMSO-d6, 300 MHz): 2,52 (s, 3H, CH3),
3,95 (s, 2H, CH2), 5,308 (s, 2H, OCH2CN), 5,312 (s, 2H, OCH2CN),
6,81 (d, 1H, J = 2,5 Hz), 6,94 (d, 1H, J = 2,5 Hz), 7,22–7,33 (m,
4H, Ph).
-
c)
5,7-Bis[(1H-tetrazol-5-yl)methoxy]-3-(4-chlorbenzyl)-4-methyl-2H-1-benzopyran-2-on
-
Das
Produkt des vorherigen Beispiels (0,40 g), Natriumazid (0,14 g)
und Ammoniumchlorid (0,11 g) wurden während 2 Stunden bei 100°C in DMF
(2 ml) erwärmt.
Das Produkt wurde wie in Beispiel 1c isoliert. Ausbeute: 0,40 g
(82%).
1H-NMR (DMSO-d6,
300 MHz): 2,46 (s, 3H, CH3), 3,92 (s, 2H,
CH2), 5,602 (s, 2H, OCH2Tet),
5,609 (s, 2H, OCH2Tet), 6,83 (d, 1H, J =
2,5 Hz), 6,85 (d, 1H, J = 2,5 Hz), 7,20–7,33 (m, 4H, Ph).
-
Beispiel
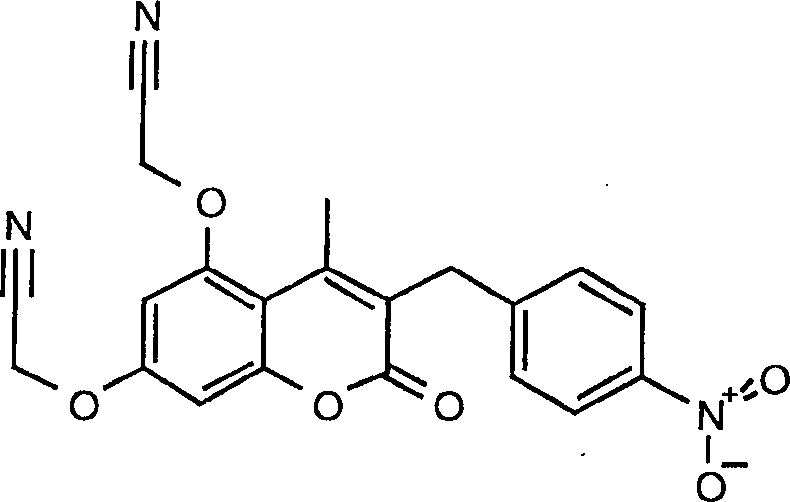
15. Herstellung von 5,7-Bis[(1H-tetrazol-5-yl)methoxy]-3-(4-nitrobenzyl)-4-methyl-2H-1-benzopyran-2-on
a) 5,7-Dihydroxy-4-methyl-3-(4-nitrobenzyl)-1-benzopyran-2-on
-
Eine
Lösung
von Phloroglucinol (0,48 g) und Ethyl-2-(4-nitrobenzyl)acetoacetat (1,00 g)
in Ethanol (150 ml) wurde während
7,5 Stunden bei 0°C
mit trockener HCl behandelt und die Lösung über Nacht bei der Temperatur
gehalten. Das Lösungsmittel
wurde verdampft und der Rückstand
mit Wasser verrieben. Ausbeute: 0,63 g (51%). Schmelzpunkt: 280–285°C.
1H-NMR (DMSO-d6,
300 MHz): 2,53 (s, 3H, CH3), 4,03 (s, 2H,
CH2), 6,19 (d, 1H, J = 2,4 Hz), 6,29 (d,
1H, J = 2,4 Hz), 7,40–7,51
und 8,11–8,17
(m, 4H, Ph), 10,25 (s, 1H, OH), 10,52 (s, 1H, OH).
-
b)
5,7-Bis(cyanomethoxy)-3-(4-nitrobenzyl)-4-methyl-2H-1-benzopyran-2-on
-
Das
Produkt des vorherigen Beispiels (0,57 g), Chloracetonitril (0,27
g) und Kaliumcarbonat (1,20 g) wurden während 50 Minuten bei 100°C in DMF
(2 ml) erwärmt.
Das Produkt wurde wie in Beispiel 1b beschrieben isoliert. Ausbeute:
0,47 g (67%). Schmelzpunkt: 178–185°C.
1H-NMR (DMSO-d6,
400 MHz): 2,53 (s, 3H, CH3), 4,11 (s, 2H,
CH2), 5,319 (s, 2H, OCH2CN),
5,323 (s, 2H, OCH2CN), 6,83 (d, 1H, J =
2,4 Hz), 6,96 (d, 1H, J = 2,4 Hz), 7,48–7,53 und 8,12–8,16 (m,
4H, Ph).
-
c)
5,7-Bis[(1H-tetrazol-5-yl)methoxy]-3-(4-nitrobenzyl)-4-methyl-2H-1-benzopyran-2-on
-
Das
Produkt des vorherigen Beispiels (0,38 g), Natriumazid (0,12 g)
und Ammoniumchlorid (0,11 g) wurden während 2 Stunden bei 100°C in DMF
(3 ml) erwärmt.
Das Produkt wurde wie in Beispiel 1c beschrieben isoliert. Ausbeute:
0,25 g (54%). Schmelzpunkt: 240–244°C.
1H-NMR (DMSO-d6,
400 MHz): 2,47 (s, 3H, CH3), 4,08 (s, 2H,
CH2), 5,611 (s, 2H, OCH2Tet),
5,623 (s, 2H, OCH2Tet), 6,85 (d, 1H, J =
2,4 Hz), 6,87 (d, 1H, J = 2,4 Hz), 7,46–7,50 und 8,12–8,16 (m,
4H, Ph).
-
Beispiel
16. Herstellung von 5,7-Bis[(1H-tetrazol-5-yl)methoxy]-3-cyclopentyl-4-methyl-2H-1-benzopyran-2-on
a) 3-Cyclopentyl-5,7-dihydroxy-4-methyl-2H-1-benzopyran-2-on
-
Eine
Lösung
von Phloroglucinol (2,00 g) und Ethyl-2-cyclopentylacetoacetat (3,14 g) in Ethanol
(40 ml) wurde während
2,5 Stunden bei 0°C
mit trockenem HCl behandelt und die Lösung über Nacht auf der Temperatur
gehalten. Das Lösungsmittel
wurde verdampft und der Rückstand
mittels Flashchromatographie gereinigt, wobei mit Toluol-EtOAc-AcOH (8 : 1
: 1) eluiert wurde. Ausbeute: 1,22 g (29%).
1H-NMR
(DMSO-d6, 300 MHz): 1,50–1,88 (m, 8H, -(CH2)4-), 2,57 (s, 3H, CH3),
3,25 (m, 1H, CH), 6,11 (d, 1H, J = 2,4 Hz), 6,25 (d, 1H, J = 2,4
Hz), 10,25 (b, 2H, OH).
-
b)
5,7-Bis(cyanomethoxy)-3-cyclopentyl-4-methyl-2H-1-benzopyran-2-on
-
Das
Produkt des vorherigen Beispiels (0,50 g), Chloracetonitril (0,31
g) und Kaliumcarbonat (0,61 g) wurden während 40 Minuten bei 80°C in DMF
(2 ml) erwärmt.
Das Produkt wurde wie in Beispiel 1b beschrieben isoliert. Ausbeute:
0,56 g (86%).
1H-NMR (DMSO-d6, 300 MHz): 1,55–1,90 (m, 8H, -(CH2)4-), 2,56 (s, 3H, CH3),
3,37 (m, 1H, CH), 5,29 (s, 2H, OCH2CN),
5,31 (s, 2H, OCH2CN), 6,75 (d, 1H, J = 2,5
Hz), 6,88 (d, 1H, J = 2,5 Hz).
-
c)
5,7-Bis[(1H-tetrazol-5-yl)methoxy]-3-cyclopentyl-4-methyl-2H-1benzopyran-2-on
-
Das
Produkt des vorherigen Beispiels (0,30 g), Natriumazid (0,13 g)
und Ammoniumchlorid (0,11 g) wurden während 1,5 Stunden bei 100°C in DMF
(1 ml) erwärmt.
Das Produkt wurde wie in Beispiel 1c beschrieben isoliert. Ausbeute:
0,30 g (80%). Schmelzpunkt: 248–252°C.
1H-NMR (DMSO-d6,
400 MHz): 1,53–1,89
(m, 8H, -(CH2)4-),
2,51 (s, 3H, CH3), 3,34 (m, 1H, CH), 5,59
(s, 2H, OCH2Tet), 5,61 (s, 2H, OCH2Tet), 6,80 (s, 2H).
-
Beispiel
17. Herstellung von 5,7-Bis[(1H-tetrazol-5-yl)methoxy]-4-methyl-3-(1-naphtylmethyl)-2H-1-benzopyran-2-on
a)
5,7-dihydroxy-4-methyl-3-(1-naphtylmethyl)-2H-1-benzopyran-2-on
-
Eine
Lösung
von Phloroglucinol (0,47 g) und Ethyl-2-(1-naphthylmethyl)acetoacetat (1,00
g) in Ethanol (20 ml) wurde während
3 Stunden bei 0°C
mit trockenem HCl behandelt und die Lösung über Nacht auf der Temperatur
gehalten. Das Lösungsmittel
wurde verdampft und der Rückstand
mit Wasser verrieben und aus Isopropanol-Wasser (1 : 1) rekristallisiert.
Ausbeute: 0,96 g (78%). Schmelzpunkt: 275–280°C.
1H-NMR
(DMSO-d6, 400 MHz): 2,45 (s, 3H, CH3), 4,32 (s, 2H, CH2),
6,23 (d, 1H, J = 2,5 Hz), 6,32 (d, 1H, J = 2,5 Hz), 6,97–8,25 (m,
7H, Naph), 10,26 (s, 1H, OH), 10,53 (s, 1H, OH).
-
b)
5,7-Bis(cyanomethoxy)-4-methyl-3-(1-naphtylmethyl)-2H-1-benzopyran-2-on
-
Das
Produkt des vorherigen Beispiels (0,80 g), Chloracetonitril (0,36
g) und Kaliumcarbonat (0,66 g) wurden während 1 Stunde bei 100°C in DMF
(4 ml) erwärmt.
Das Produkt wurde wie in Beispiel 1b beschrieben isoliert. Ausbeute:
0,30 g (30%).
1H-NMR (DMSO-d6, 300 MHz): 2,45 (s, 3H, CH3),
4,40 (s, 2H, CH2), 5,34 (s, 2H, OCH2CN), 5,36 (s, 2H, OCH2CN),
6,86 (d, 1H, J = 2,5 Hz), 7,010 (d, 1H, J = 2,5 Hz), 7,016–8,27 (m,
7H, Naph).
-
c)
5,7-Bis[(1H-tetrazol-5-yl)methoxy]-4-methyl-3-(1-naphtylmethyl)-2H-1-benzopyran-2-on
-
Das
Produkt des vorherigen Beispiels (0,25 g), Natriumazid (0,080 g)
und Ammoniumchlorid (0,072 g) wurden während 2,5 Stunden bei 100°C in DMF
(2 ml) erwärmt.
Das Produkt wurde wie in Beispiel 1c beschrieben isoliert. Ausbeute:
0,11 g (36%). Schmelzpunkt: 164–174°C.
1H-NMR (DMSO-d6,
300 MHz): 2,40 (s, 3H, CH3), 4,37 (s, 2H,
CH2), 5,63 (s, 2H, OCH2Tet),
5,65 (s, 2H, OCH2Tet), 6,87 (d, 1H, J =
2,5 Hz), 6,92 (d, 1H, J = 2,5 Hz), 6,98–8,26 (m, 7H, Naph).
-
Beispiel
18. Herstellung von 1-Benzyl-5,7-bis-[(1H-tetrazol-5-yl)-methoxy]-4-methyl-2(1H)-chinolinon
a)
5,7-Dimethoxy-4-methyl-2(1H)-chinolinon
-
tert-Butyl-acetoacetat
(1,58 g) wurde auf 120°C
erwärmt,
und es wurde in Xylol (4 ml) gelöstes
Dimethoxyanilin (1,53 g) zugegeben. Die Mischung wurde während 20
Minuten bei 120–130°C erwärmt und
dann auf Raumtemperatur gekühlt.
Es wurde Methansulfonsäure
(2 ml) zugegeben und die Mischung während 10 Minuten bei Umgebungstemperatur
gerührt.
Es wurde Wasser (40 ml) zugegeben und der Niederschlag filtriert und
getrocknet. Ausbeute: 1,31 g (60%).
1H-NMR
(DMSO-d6, 300 MHz): 2,50 (s, 3H, CH3), 3,79 (s, 3H, OCH3),
3,83 (s, 3H, OCH3), 6,03 (s, 1H, CH=C), 6,31
(d, 1H, J = 2,3 Hz), 6,45 (d, 1H, J = 2,3 Hz), 11,4 (b, 1H, NH).
-
b)
1-Benzyl-5,7-dimethoxy-4-methyl-2(1H)-chinolinon
-
Das
Produkt des vorherigen Beispiels (1,20 g) wurde in DMSO (15 ml)
suspendiert, und es wurden t-BuOK (0,68 g) und Benzylbromid (1,03
g) zugegeben. Die Reaktionsmischung wurde über Nacht bei Umgebungstemperatur
gerührt.
Es wurde Wasser zugegeben und das Produkt mit EtOAc extrahiert.
EtOAc wurde getrocknet und zur Trockne eingedampft. Der Rückstand
wurde aus Toluol rekristallisiert. Ausbeute: 0,80 g (47%).
1H-NMR (DMSO-d6,
300 MHz): 2,55 (d, 3H, J = 1,1 Hz, CH3),
3,71 (s, 3H, OCH3), 3,84 (s, 3H, OCH3), 5,48 (b, 2H, NCH2),
6,29 (d, 1H, J = 1,1 Hz, CH=C), 6,4 (s, 2H), 7,18–7,33 (m,
5H, Ph).
-
c)
1-Benzyl-5,7-dihydroxy-4-methyl-2(1H)-chinolinon
-
Das
Produkt des vorherigen Beispiels (0,69 g) wurde in CH2Cl2 (14 ml) gelöst und die Reaktionsmischung
auf –20°C gekühlt. Es
wurde BBr3 (2,4 g) in CH2Cl2 (1 M Lösung)
zugegeben und man ließ die
Mischung sich über
Nacht auf Umgebungstemperatur erwärmen. Der Niederschlag wurde
filtriert, mit CH2Cl2 gewaschen
und in EtOAc gelöst.
EtOAc wurde mit verdünnter
HCl gewaschen, getrocknet und zur Trockne eingedampft. Ausbeute:
0,34 g (54%).
1H-NMR (DMSO-d6, 300 MHz): 2,56 (d, 3H, J = Hz, CH3), 5,33 (b, 2H, NCH2),
6,11 (d, 1H, J = 2,1 Hz), 6,13 (d, 1H, J = 1,0 Hz, CH=C), 6,17 (d,
1H, J = 2,1 Hz), 7,12–7,34
(m, 5H, Ph), 9,90 (b, 1H, OH), 10,22 (s, 1H, OH).
-
d)
1-Benzyl-5,7-bis(cyanomethoxy)-4-methyl-2(1H)-chinolinon
-
Das
Produkt des vorherigen Beispiels (0,34 g), Chloracetonitril (0,13
g) und Kaliumcarbonat (0,34 g) wurden während 1,5 Stunden bei 100°C in DMF
(2 ml) erwärmt.
Es wurde Wasser zugegeben und der Niederschlag filtriert und getrocknet.
Das Produkt wurde aus Isopropanol rekristallisiert. Ausbeute: 0,20
g (46%).
1H-NMR (DMSO-d6,
400 MHz): 2,57 (s, 3H, CH3), 5,22 (s, 2H,
OCH2CN), 5,30 (s, 2H, OCH2CN),
5,50 (b, 2H, NCH2), 6,42 (s, 1H, CH=C),
6,70 (d, 1H, J = 2,1 Hz), 6,73 (d, 1H, J = 2,1 Hz), 7,21–7,32 (m,
5H, Ph).
-
e)
1-Benzyl-5,7-bis-[(1H-tetrazol-5-yl)methoxy]-4-methyl-2(1H)-chinolinon
-
Das
Produkt des vorherigen Beispiels (0,20 g), Natriumazid (0,072 g)
und Ammoniumchlorid (0,060 g) wurden während 3 Stunden bei 100°C in DMF
(2 ml) erwärmt.
Das Produkt wurde wie in Beispiel 1c beschrieben isoliert. Ausbeute:
0,21 g (85%). Schmelzpunkt: 246–249°C.
1H-NMR (DMSO-d6,
400 MHz): 2,50 (s, 3H, CH3), 5,48 (b, 4H,
OCH2Tet, NCH2),
5,60 (s, 2H, OCH2Tet), 6,34 (s, 1H, CH=C),
6,64 (d, 1H, J = 1,9 Hz), 6,77 (d, 1H, J = 1,9 Hz), 7,18–7,32 (m,
5H, Ph).
-
Beispiel
19. Herstellung von 1-Benzyl-5,7-bis[1H-tetrazol-5-yl)methoxy]-3-(2-fluorbenzyl)-4-methyl-2(1H)-chinolinon
a)
5,7-Dimethoxy-3-(2-fluorbenzyl)-4-methyl-2(1H)-chinolinon
-
Ethyl-2-(2-fluorbenzyl)acetoacetat
(2,5 g) in Xylol (1 ml) wurde auf 150°C erwärmt, und es wurde 3,5-Dimethoxyanilin
(1,46 g) in Xylol (4 ml) in kleinen Portionen während 30 Minuten zugegeben.
Die Reaktionsmischung wurde während
3 Stunden bei 160°C
erwärmt
und dann auf Raumtemperatur gekühlt.
Es wurde Methansulfonsäure
(1,7 ml) zugegeben und die Mischung während 30 Minuten bei Umgebungstemperatur
gerührt.
Es wurde Wasser zugegeben und der Niederschlag filtriert und getrocknet.
Das Produkt wurde mit warmem Ethanol verrieben. Ausbeute: 0,64 g
(21%).
1H-NMR (DMSO-d6,
300 MHz): 2,45 (s, 3H), 3,79 (s, 3H), 3,82 (s, 3H), 3,97 (s, 2H),
6,33 (d, 1H, J = 2,4 Hz), 6,48 (d, 1H, J = 2,4 Hz), 6,90–7,25 (m,
4H), 11,61 (s, 1H).
-
b)
1-Benzyl-5,7-dimethoxy-3-(2-fluorbenzyl)-4-methyl-2(1H)-chinolinon
-
Das
Produkt des vorherigen Beispiels (0,62 g) wurde mit t-BuOK (0,23
g) und Benzylbromid (0,36 g) während
2,6 Stunden bei 60°C
in DMSO (12 ml) behandelt. Das Produkt wurde wie in Beispiel 18b
beschrieben isoliert. Ausbeute: 0,39 g (49%).
1H-NMR
(DMSO-d6, 400 MHz): 2,51 (s, 3H), 3,72 (s,
3H), 3,84 (s, 3H), 4,11 (s, 2H), 5,55 (b, 2H), 6,433 (d, 1H, J =
2,1 Hz), 6,443 (d, 1H, J = 2,1 Hz), 6,97–7,33 (m, 9H).
-
c)
1-Benzyl-5,7-dihydroxy-3-(2-fluorbenzyl)-4-methyl-2(1H)-chinolinon
-
Das
Produkt des vorherigen Beispiels (0,34 g) wurde wie in Beispiel
18c mit BBr3 (8,48 g) in CH2Cl2 (7 ml) behandelt. Ausbeute: 0,30 g (82%).
1H-NMR (DMSO-d6,
400 MHz): 2,55 (s, 3H), 4,06 (s, 2H), 5,40 (b, 2H), 6,13 (d, 1H,
J = 2,1 Hz), 6,22 (d, 1H, J = 2,1 Hz), 6,97–7,33 (m, 9H), 10,3 (b, 2H).
-
d)
1-Benzyl-5,7-bis(cyanomethoxy)-3-(2-fluorbenzyl)-4-methyl-2(1H)-chinolinon
-
Das
Produkt des vorherigen Beispiels (0,21 g), Chloracetonitril (0,086
g) und Kaliumcarbonat (0,37 g) wurden während 2 Stunden bei 100°C in DMF
(2 ml) er wärmt.
Das Produkt wurde wie in Beispiel 1b beschrieben isoliert. Ausbeute:
0,18 g (71%).
1H-NMR (DMSO-d6, 400 MHz): 2,53 (s, 3H), 4,13 (s, 2H),
5,23 (s, 2H), 5,29 (s, 2H), 5,57 (b, 2H), 6,746 (d, 1H, J = 2,3
Hz), 6,756 (d, 1H, J = 2,3 Hz), 7,00–7,32 (m, 9H).
-
e)
1-Benzyl-5,7-bis[1H-tetrazol-5-yl)methoxy]-3-(2-fluorbenzyl)-4-methyl-2(1H)-chinolinon
-
Das
Produkt des vorherigen Beispiels (0,17 g), Natriumazid (0,051 g)
und Ammoniumchlorid (0,042 g) wurden während 3 Stunden bei 100°C in DMF
erwärmt.
Das Produkt wurde wie in Beispiel 1c beschrieben isoliert. Ausbeute:
0,17 g (85%). Schmelzpunkt: 135–140°C.
1H-NMR (DMSO-d6,
400 MHz): 2,46 (s, 3H), 4,10 (s, 2H), 5,48 (s, 2H), 5,51 (b, 2H),
5,59 (s, 2H), 6,68 (d, 1H, J = 2,2 Hz), 6,79 (d, 1H, J = 2,2 Hz),
6,99–7,32
(m, 9H).
-
Beispiel
20. Herstellung von 1-Benzyl-5,7-bis[1H-tetrazol-5-yl)methoxy]-4-methyl-3-(2-phenylethyl)-2(1H)-chinolinon
a)
5,7-Dimethoxy-4-methyl-3-(2-phenylethyl)-2(1H)-chinolinon
-
Ethyl-2-(2-phenylethyl)acetoacetat
(2,70 g) in Xylol (5 ml) wurde wie in Beispiel 19a beschrieben mit 3,5-Dimethoxyanilin
(1,60 g) bei 150°C
behandelt. Es wurde bei Raumtemperatur Methansulfonsäure (4,0
ml) zugegeben und die Mischung während
1 Stunde bei 80°C
erwärmt.
Das Produkt wurde wie in Beispiel 19a beschrieben isoliert. Ausbeute:
1,38 g (41%).
1H-NMR (DMSO-d6, 400 MHz): 2,45 (s, 3H), 2,64–2,68 (m,
2H), 2,82–2,86
(m, 2H), 3,78 (s, 3H), 3,81 (s, 3H), 6,30 (d, 1H, J = 2,3 Hz), 6,45
(d, 1H, J = 2,3 Hz), 7,18–7,30
(m, 5H), 11,45 (s, H).
-
b)
1-Benzyl-5,7-dimethoxy-4-methyl-3-(2-phenylethyl)-2(1H)-chinolinon
-
Das
Produkt des vorherigen Beispiels (0,61 g), t-BuOK (0,24 g) und Benzylbromid
(0,36 g) wurden während
2 Stunden bei 60°C
in DMSO (12 ml) erwärmt.
Das Produkt wurde wie in Beispiel 18b beschrieben isoliert. Ausbeute:
0,31 g (40%).
1H-NMR (DMSO-d6, 400 MHz): 2,51 (s, 3H), 2,73–2,77 (m,
2H), 2,96–3,00
(m, 2H), 3,70 (s, 3H), 3,83 (s, 3H), 5,55 (b, 2H), 6,40 (s, 2H),
7,17–7,33
(m, 10H).
-
c)
1-Benzyl-5,7-dihydroxy-4-methyl-3-(2-phenylethyl)-2(1H)-chinolinon
-
Das
Produkt des vorherigen Beispiels (0,31 g) wurde mit BBr3 (0,75
g) in CH2Cl2 (5
ml) wie in Beispiel 18c beschrieben behandelt. Ausbeute: 0,26 g
(89%).
1H-NMR (DMSO-d6,
300 MHz): 2,56 (s, 3H), 2,69–2,75
(m, 2H), 2,90–2,95
(m, 2H), 5,39 (b, 2H), 6,08 (d, 1H, J = 2,0 Hz), 6,19 (d, 1H, J
= 2,0 Hz), 7,11–7,33
(m, 10H), 10,2 (b, 2H).
-
d)
1-Benzyl-5,7-bis(cyanomethoxy)-4-methyl-3-(2-phenylethyl)-2(1H)-chinolinon
-
Das
Produkt des vorherigen Beispiels (0,22 g), Chloracetonitril (0,091
g) und Kaliumcarbonat (0,39 g) wurden während 2 Stunden bei 100°C erwärmt. Das
Produkt wurde wie in Beispiel 1b beschrieben isoliert. Ausbeute:
0,20 g (76%).
1H-NMR (DMSO-d6, 400 MHz): 2,50 (s, 3H), 2,73–2,77 (m,
2H), 2,98–3,02
(m, 2H), 5,21 (s, 2H), 5,29 (s, 2H), 5,56 (b, 2H), 6,70 (d, 1H,
J = 2,1 Hz), 6,72 (d, 1H, J = 2,1 Hz), 7,18–7,33 (m, 10H).
-
e)
1-Benzyl-5,7-bis[1H-tetrazol-5-yl)methoxy]-4-methyl-3-(2-phenylethyl)-2(1H)-chinolinon
-
Das
Produkt des vorherigen Beispiels (0,19 g), Nariumazid (0,057 g)
und Ammoniumchlorid (0,047 g) wurden während 3 Stunden bei 100°C in DMF
erwärmt.
Das Produkt wurde wie in Beispiel 1c beschrieben isoliert. Ausbeute:
0,18 g (78%). Schmelzpunkt: 215–218°C.
1H-NMR (DMSO-d6,
400 MHz): 2,46 (s, 3H), 2,70–2,74
(m, 2H), 2,95–2,99
(m, 2H), 5,47 (s, 2H), 5,54 (b, 2H), 5,57 (s, 2H), 6,64 (d, 1H,
J = 2,0 Hz), 6,77 (d, 1H, J = 2,0 Hz), 7,16–7,33 (m, 10H).
-
Beispiel
21. Herstellung von 5,7-Bis(aminocarbonylmethoxy)-1 3-dibenzyl-4-methyl-2(1H)-chinolinon
-
Die
Mischung aus 5,7-Dihydroxy-1,3-dibenzyl-4-methyl-2(1H)-chinolinon (0,5 g), Kaliumcarbonat
(0,9 g) und 2-Chloracetamid
(0,25 g) wurde während
2 Stunden bei 100°C
in DMF (6,5 ml) umgesetzt. Die Reaktionsmischung wurde mit Eiswasser
behandelt und filtriert. Das Produkt wurde mit heißem Ethanol
verrieben. Ausbeute: 0,32 g. Schmelzpunkt: 252–253°C.
1H-NMR
(400 MHz, DMSO-d6): 2,63 (s, 3H, CH3), 4,13 (s, 2H, PhCH2),
4,37 (s, 2H, OCH2), 4,55 (s, 2H, OCH2), 5,54 (s, 2H, NCH2Ph),
6,40 (d, 1H, J = 2 Hz, ArH), 6,53 (d, 1H, J = 2 Hz, ArH), 7,13–7,33 (m,
10H, Ph), 7,44 (d, 2H, J = 65 Hz, CONH2),
7,47 (d, 2H, J = 68 Hz, CONH2)
-
Beispiel
22. Herstellung von 5,7-Bis(ethoxycarbonylmethoxy)-1,3-dibenzyl-4-methyl-2(1H)-chinolinon
-
Die
Mischung aus 5,7-Dihydroxy-1,3-dibenzyl-4-methyl-2(1H)-chinolinon (1 g), Ethyl-2-bromacetat (0,63
ml) und Kaliumcarbonat (1,49 g) wurde während 3 Stunden unter Stickstoff
bei 110°C
in DMF (5 ml) erwärmt,
in Eiswasser gegossen und filtriert. Das resultierende Feststoffmaterial
wurde mit Ether verrieben und erneut filtriert. Ausbeute: 1,03 g.
Schmelzpunkt: 113–116°C.
1H-NMR (400 MHz, DMSO-d6):
1,15 (t, 3H, CH3CH2,
J = 7,1 Hz), 1,20 (t, 3H, CH3CH2,
J = 7,1 Hz), 2,63 (s, 3H, CH3), 4,03 (q,
2H, CH2CH3, J =
7,1 Hz), 4,13 (s, 2H, CH2Ph), 4,17 (q, 2H,
CH2CH3, J = 7,1
Hz), 4,78 (s, 2H, OCH2), 4,90 (s, 2H, OCH2), 6,41 (d, 1H, J = 2,2 Hz), 6,44 (d, 1H,
J = 2,2 Hz), 7,13–7,33
(m, 10H, Ph).
-
Beispiel
23. Herstellung von 5,7-Bis(hydroxyaminocarbonylmethoxy)-1,3-dibenzyl-4-methyl-2(1H)-chinolinon
-
Das
Produkt des vorherigen Beispiels (0,3 g), Hydroxylaminhydrochlorid
(0,32 g) und 5 N NaOH (1,05 ml) wurden während 6 Stunden bei 50°C in Ethanol
(8 ml) umgesetzt. Die Reaktionsmischung wurde mit Wasser behandelt
und basisch gemacht (pH 10) und filtriert. Das Filtrat wurde auf
pH 2 angesäuert
und filtriert. Ausbeute: 0,2 g. Schmelzpunkt: 121–127°C.
1H-NMR (400 MHz, DMSO-d6):
die tautomeren Formen der Hydroxamsäure sind in den OCH2-Signalen zu sehen: 2,63 (s, 3H, CH3), 4,13 (S, 2H, CH2Ph),
4,41 (s, 2H, OCH2), 4,54 (s, 2H, OCH2), 4,64 (s, 2H, HON=C(OH)CH2O),
4,65 (s, 2H, HON=C(OH)CH2O), 4,77 (s, 2H,
HON=C(OH)CH2O), 4,78 ((s, 2H, HON=C(OH)CH2O), 5,54 (s, 2H, NCH2Ph),
6,38–6,54
(m, 2H, ArH), 7,14–7,34
(m, 10H, Ph), 9,05 (b, 2H, NOH), 10,84 (s, 1H, HONHCO), 10,88 (s,
1H, HONHCO).