DE69827706T2 - 5-(Heteroaryl)alkyl)-3-oxo-pyrido(1,2-a)benzimidazol-4-carboxamid (PBI) Derivate zur Behandlung von Störungen des zentralen Nervensystems - Google Patents
5-(Heteroaryl)alkyl)-3-oxo-pyrido(1,2-a)benzimidazol-4-carboxamid (PBI) Derivate zur Behandlung von Störungen des zentralen Nervensystems Download PDFInfo
- Publication number
- DE69827706T2 DE69827706T2 DE69827706T DE69827706T DE69827706T2 DE 69827706 T2 DE69827706 T2 DE 69827706T2 DE 69827706 T DE69827706 T DE 69827706T DE 69827706 T DE69827706 T DE 69827706T DE 69827706 T2 DE69827706 T2 DE 69827706T2
- Authority
- DE
- Germany
- Prior art keywords
- alkyl
- methyl
- thienyl
- benzimidazole
- carboxamide
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- 125000000217 alkyl group Chemical group 0.000 title claims description 26
- 208000015114 central nervous system disease Diseases 0.000 title description 7
- JJDMKDXGNVJWCD-UHFFFAOYSA-N 1h-benzimidazole-4-carboxamide Chemical compound NC(=O)C1=CC=CC2=C1N=CN2 JJDMKDXGNVJWCD-UHFFFAOYSA-N 0.000 title description 3
- 125000001072 heteroaryl group Chemical group 0.000 title description 3
- 150000001875 compounds Chemical class 0.000 claims abstract description 65
- 210000003169 central nervous system Anatomy 0.000 claims abstract description 6
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims abstract description 6
- 239000008194 pharmaceutical composition Substances 0.000 claims abstract description 5
- -1 C 1 -C 8 -alkyl Chemical class 0.000 claims description 45
- 229940049706 benzodiazepine Drugs 0.000 claims description 17
- SIKJAQJRHWYJAI-UHFFFAOYSA-N Indole Chemical compound C1=CC=C2NC=CC2=C1 SIKJAQJRHWYJAI-UHFFFAOYSA-N 0.000 claims description 16
- 125000001424 substituent group Chemical group 0.000 claims description 16
- 125000000623 heterocyclic group Chemical group 0.000 claims description 14
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims description 13
- SVUOLADPCWQTTE-UHFFFAOYSA-N 1h-1,2-benzodiazepine Chemical compound N1N=CC=CC2=CC=CC=C12 SVUOLADPCWQTTE-UHFFFAOYSA-N 0.000 claims description 12
- 239000001257 hydrogen Substances 0.000 claims description 12
- 229910052739 hydrogen Inorganic materials 0.000 claims description 12
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 claims description 12
- FCEHBMOGCRZNNI-UHFFFAOYSA-N 1-benzothiophene Chemical compound C1=CC=C2SC=CC2=C1 FCEHBMOGCRZNNI-UHFFFAOYSA-N 0.000 claims description 8
- YLQBMQCUIZJEEH-UHFFFAOYSA-N Furan Chemical compound C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 claims description 8
- KYQCOXFCLRTKLS-UHFFFAOYSA-N Pyrazine Chemical compound C1=CN=CC=N1 KYQCOXFCLRTKLS-UHFFFAOYSA-N 0.000 claims description 8
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 claims description 8
- SMWDFEZZVXVKRB-UHFFFAOYSA-N Quinoline Chemical compound N1=CC=CC2=CC=CC=C21 SMWDFEZZVXVKRB-UHFFFAOYSA-N 0.000 claims description 8
- YTPLMLYBLZKORZ-UHFFFAOYSA-N Thiophene Chemical compound C=1C=CSC=1 YTPLMLYBLZKORZ-UHFFFAOYSA-N 0.000 claims description 8
- IOJUPLGTWVMSFF-UHFFFAOYSA-N benzothiazole Chemical compound C1=CC=C2SC=NC2=C1 IOJUPLGTWVMSFF-UHFFFAOYSA-N 0.000 claims description 8
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 8
- AWJUIBRHMBBTKR-UHFFFAOYSA-N isoquinoline Chemical compound C1=NC=CC2=CC=CC=C21 AWJUIBRHMBBTKR-UHFFFAOYSA-N 0.000 claims description 8
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 claims description 8
- 125000003545 alkoxy group Chemical group 0.000 claims description 7
- 239000000203 mixture Substances 0.000 claims description 7
- 150000003839 salts Chemical class 0.000 claims description 7
- 125000001541 3-thienyl group Chemical group S1C([H])=C([*])C([H])=C1[H] 0.000 claims description 6
- 125000004453 alkoxycarbonyl group Chemical group 0.000 claims description 6
- GLUUGHFHXGJENI-UHFFFAOYSA-N diethylenediamine Natural products C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 claims description 6
- PZOUSPYUWWUPPK-UHFFFAOYSA-N indole Natural products CC1=CC=CC2=C1C=CN2 PZOUSPYUWWUPPK-UHFFFAOYSA-N 0.000 claims description 6
- RKJUIXBNRJVNHR-UHFFFAOYSA-N indolenine Natural products C1=CC=C2CC=NC2=C1 RKJUIXBNRJVNHR-UHFFFAOYSA-N 0.000 claims description 6
- 150000004885 piperazines Chemical class 0.000 claims description 6
- 125000004198 2-fluorophenyl group Chemical group [H]C1=C([H])C(F)=C(*)C([H])=C1[H] 0.000 claims description 5
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 claims description 5
- 201000010099 disease Diseases 0.000 claims description 5
- 229910052731 fluorine Inorganic materials 0.000 claims description 5
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims description 5
- JYEUMXHLPRZUAT-UHFFFAOYSA-N 1,2,3-triazine Chemical compound C1=CN=NN=C1 JYEUMXHLPRZUAT-UHFFFAOYSA-N 0.000 claims description 4
- BAXOFTOLAUCFNW-UHFFFAOYSA-N 1H-indazole Chemical compound C1=CC=C2C=NNC2=C1 BAXOFTOLAUCFNW-UHFFFAOYSA-N 0.000 claims description 4
- 125000000175 2-thienyl group Chemical group S1C([*])=C([H])C([H])=C1[H] 0.000 claims description 4
- IJGRMHOSHXDMSA-UHFFFAOYSA-N Atomic nitrogen Chemical compound N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 claims description 4
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical group C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 claims description 4
- PCNDJXKNXGMECE-UHFFFAOYSA-N Phenazine Natural products C1=CC=CC2=NC3=CC=CC=C3N=C21 PCNDJXKNXGMECE-UHFFFAOYSA-N 0.000 claims description 4
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 claims description 4
- FZWLAAWBMGSTSO-UHFFFAOYSA-N Thiazole Chemical compound C1=CSC=N1 FZWLAAWBMGSTSO-UHFFFAOYSA-N 0.000 claims description 4
- 125000004414 alkyl thio group Chemical group 0.000 claims description 4
- 125000003710 aryl alkyl group Chemical group 0.000 claims description 4
- RFRXIWQYSOIBDI-UHFFFAOYSA-N benzarone Chemical compound CCC=1OC2=CC=CC=C2C=1C(=O)C1=CC=C(O)C=C1 RFRXIWQYSOIBDI-UHFFFAOYSA-N 0.000 claims description 4
- QRUDEWIWKLJBPS-UHFFFAOYSA-N benzotriazole Chemical compound C1=CC=C2N[N][N]C2=C1 QRUDEWIWKLJBPS-UHFFFAOYSA-N 0.000 claims description 4
- 239000012964 benzotriazole Substances 0.000 claims description 4
- 150000002431 hydrogen Chemical class 0.000 claims description 4
- 125000004435 hydrogen atom Chemical group [H]* 0.000 claims description 4
- 125000002887 hydroxy group Chemical group [H]O* 0.000 claims description 4
- CTAPFRYPJLPFDF-UHFFFAOYSA-N isoxazole Chemical compound C=1C=NOC=1 CTAPFRYPJLPFDF-UHFFFAOYSA-N 0.000 claims description 4
- PBMFSQRYOILNGV-UHFFFAOYSA-N pyridazine Chemical compound C1=CC=NN=C1 PBMFSQRYOILNGV-UHFFFAOYSA-N 0.000 claims description 4
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 claims description 4
- ILVXOBCQQYKLDS-UHFFFAOYSA-N pyridine N-oxide Chemical compound [O-][N+]1=CC=CC=C1 ILVXOBCQQYKLDS-UHFFFAOYSA-N 0.000 claims description 4
- VLLMWSRANPNYQX-UHFFFAOYSA-N thiadiazole Chemical compound C1=CSN=N1.C1=CSN=N1 VLLMWSRANPNYQX-UHFFFAOYSA-N 0.000 claims description 4
- 229930192474 thiophene Natural products 0.000 claims description 4
- 208000003870 Drug Overdose Diseases 0.000 claims description 3
- 206010033296 Overdoses Diseases 0.000 claims description 3
- 208000005392 Spasm Diseases 0.000 claims description 3
- 239000003937 drug carrier Substances 0.000 claims description 3
- 231100000725 drug overdose Toxicity 0.000 claims description 3
- 229910052736 halogen Inorganic materials 0.000 claims description 3
- 150000002367 halogens Chemical class 0.000 claims description 3
- 208000007101 Muscle Cramp Diseases 0.000 claims description 2
- 239000003085 diluting agent Substances 0.000 claims description 2
- 239000011737 fluorine Substances 0.000 claims description 2
- 229910052757 nitrogen Inorganic materials 0.000 claims description 2
- 125000001475 halogen functional group Chemical group 0.000 claims 3
- 239000012453 solvate Substances 0.000 claims 2
- 150000003852 triazoles Chemical class 0.000 claims 2
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 claims 1
- 206010040007 Sense of oppression Diseases 0.000 claims 1
- 208000013738 Sleep Initiation and Maintenance disease Diseases 0.000 claims 1
- 206010022437 insomnia Diseases 0.000 claims 1
- 238000000034 method Methods 0.000 abstract description 8
- 230000000694 effects Effects 0.000 description 16
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 15
- 239000000243 solution Substances 0.000 description 15
- 239000002904 solvent Substances 0.000 description 14
- 239000007787 solid Substances 0.000 description 13
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 12
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 description 11
- 102000004300 GABA-A Receptors Human genes 0.000 description 9
- 108090000839 GABA-A Receptors Proteins 0.000 description 9
- 229940079593 drug Drugs 0.000 description 9
- 239000003814 drug Substances 0.000 description 9
- 102000005962 receptors Human genes 0.000 description 9
- 108020003175 receptors Proteins 0.000 description 9
- 241001465754 Metazoa Species 0.000 description 8
- 239000001961 anticonvulsive agent Substances 0.000 description 8
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 8
- 239000003446 ligand Substances 0.000 description 7
- XKRFYHLGVUSROY-UHFFFAOYSA-N Argon Chemical compound [Ar] XKRFYHLGVUSROY-UHFFFAOYSA-N 0.000 description 6
- UHOVQNZJYSORNB-UHFFFAOYSA-N Benzene Chemical compound C1=CC=CC=C1 UHOVQNZJYSORNB-UHFFFAOYSA-N 0.000 description 6
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 6
- 238000005481 NMR spectroscopy Methods 0.000 description 6
- HEMHJVSKTPXQMS-UHFFFAOYSA-M Sodium hydroxide Chemical compound [OH-].[Na+] HEMHJVSKTPXQMS-UHFFFAOYSA-M 0.000 description 6
- 239000000556 agonist Substances 0.000 description 6
- 230000000949 anxiolytic effect Effects 0.000 description 6
- 229910052786 argon Inorganic materials 0.000 description 6
- 238000012360 testing method Methods 0.000 description 6
- 206010010904 Convulsion Diseases 0.000 description 5
- KFZMGEQAYNKOFK-UHFFFAOYSA-N Isopropanol Chemical compound CC(C)O KFZMGEQAYNKOFK-UHFFFAOYSA-N 0.000 description 5
- CWRVKFFCRWGWCS-UHFFFAOYSA-N Pentrazole Chemical compound C1CCCCC2=NN=NN21 CWRVKFFCRWGWCS-UHFFFAOYSA-N 0.000 description 5
- 229960003965 antiepileptics Drugs 0.000 description 5
- 239000002249 anxiolytic agent Substances 0.000 description 5
- 238000002474 experimental method Methods 0.000 description 5
- 230000035939 shock Effects 0.000 description 5
- 230000001225 therapeutic effect Effects 0.000 description 5
- 208000019901 Anxiety disease Diseases 0.000 description 4
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 4
- GELXFVQAWNTGPQ-UHFFFAOYSA-N [N].C1=CNC=N1 Chemical group [N].C1=CNC=N1 GELXFVQAWNTGPQ-UHFFFAOYSA-N 0.000 description 4
- 230000001773 anti-convulsant effect Effects 0.000 description 4
- 239000002585 base Substances 0.000 description 4
- 239000000969 carrier Substances 0.000 description 4
- 238000000262 chemical ionisation mass spectrometry Methods 0.000 description 4
- 229960003692 gamma aminobutyric acid Drugs 0.000 description 4
- BTCSSZJGUNDROE-UHFFFAOYSA-N gamma-aminobutyric acid Chemical compound NCCCC(O)=O BTCSSZJGUNDROE-UHFFFAOYSA-N 0.000 description 4
- 238000002360 preparation method Methods 0.000 description 4
- FODVQKYUAIWTKY-UHFFFAOYSA-N pyrido[1,2-a]benzimidazole Chemical class C1=CC=CN2C3=CC=CC=C3N=C21 FODVQKYUAIWTKY-UHFFFAOYSA-N 0.000 description 4
- RIOQSEWOXXDEQQ-UHFFFAOYSA-N triphenylphosphine Chemical compound C1=CC=CC=C1P(C=1C=CC=CC=1)C1=CC=CC=C1 RIOQSEWOXXDEQQ-UHFFFAOYSA-N 0.000 description 4
- 125000006479 2-pyridyl methyl group Chemical group [H]C1=C([H])C([H])=C([H])C(=N1)C([H])([H])* 0.000 description 3
- YYPNNBPPDFTQFX-UHFFFAOYSA-N 2-thiophen-3-ylethanol Chemical compound OCCC=1C=CSC=1 YYPNNBPPDFTQFX-UHFFFAOYSA-N 0.000 description 3
- QTBSBXVTEAMEQO-UHFFFAOYSA-N Acetic acid Chemical compound CC(O)=O QTBSBXVTEAMEQO-UHFFFAOYSA-N 0.000 description 3
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 3
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 description 3
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 3
- OFOBLEOULBTSOW-UHFFFAOYSA-N Malonic acid Chemical compound OC(=O)CC(O)=O OFOBLEOULBTSOW-UHFFFAOYSA-N 0.000 description 3
- 238000000585 Mann–Whitney U test Methods 0.000 description 3
- 241000699670 Mus sp. Species 0.000 description 3
- MUBZPKHOEPUJKR-UHFFFAOYSA-N Oxalic acid Chemical compound OC(=O)C(O)=O MUBZPKHOEPUJKR-UHFFFAOYSA-N 0.000 description 3
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 3
- 239000002253 acid Substances 0.000 description 3
- 150000001412 amines Chemical class 0.000 description 3
- 239000002775 capsule Substances 0.000 description 3
- 239000003795 chemical substances by application Substances 0.000 description 3
- KRKNYBCHXYNGOX-UHFFFAOYSA-N citric acid Chemical compound OC(=O)CC(O)(C(O)=O)CC(O)=O KRKNYBCHXYNGOX-UHFFFAOYSA-N 0.000 description 3
- 229940126214 compound 3 Drugs 0.000 description 3
- 230000036461 convulsion Effects 0.000 description 3
- FAMRKDQNMBBFBR-BQYQJAHWSA-N diethyl azodicarboxylate Substances CCOC(=O)\N=N\C(=O)OCC FAMRKDQNMBBFBR-BQYQJAHWSA-N 0.000 description 3
- 150000002148 esters Chemical class 0.000 description 3
- FAMRKDQNMBBFBR-UHFFFAOYSA-N ethyl n-ethoxycarbonyliminocarbamate Chemical compound CCOC(=O)N=NC(=O)OCC FAMRKDQNMBBFBR-UHFFFAOYSA-N 0.000 description 3
- 125000005843 halogen group Chemical group 0.000 description 3
- 230000000147 hypnotic effect Effects 0.000 description 3
- 239000005457 ice water Substances 0.000 description 3
- 238000011835 investigation Methods 0.000 description 3
- 239000007788 liquid Substances 0.000 description 3
- 238000001819 mass spectrum Methods 0.000 description 3
- 239000000463 material Substances 0.000 description 3
- 239000003158 myorelaxant agent Substances 0.000 description 3
- OQJBFFCUFALWQL-UHFFFAOYSA-N n-(piperidine-1-carbonylimino)piperidine-1-carboxamide Chemical compound C1CCCCN1C(=O)N=NC(=O)N1CCCCC1 OQJBFFCUFALWQL-UHFFFAOYSA-N 0.000 description 3
- 239000003921 oil Substances 0.000 description 3
- 239000000843 powder Substances 0.000 description 3
- 239000000741 silica gel Substances 0.000 description 3
- 229910002027 silica gel Inorganic materials 0.000 description 3
- 238000010561 standard procedure Methods 0.000 description 3
- 238000003756 stirring Methods 0.000 description 3
- 239000000725 suspension Substances 0.000 description 3
- TUQOTMZNTHZOKS-UHFFFAOYSA-N tributylphosphine Chemical compound CCCCP(CCCC)CCCC TUQOTMZNTHZOKS-UHFFFAOYSA-N 0.000 description 3
- VLSDXINSOMDCBK-UHFFFAOYSA-N 1,1'-azobis(N,N-dimethylformamide) Chemical compound CN(C)C(=O)N=NC(=O)N(C)C VLSDXINSOMDCBK-UHFFFAOYSA-N 0.000 description 2
- UYVFEZBIQHCLBF-UHFFFAOYSA-N 3h-imidazo[4,5-f]quinoline Chemical compound C1=CC=C2C(N=CN3)=C3C=CC2=N1 UYVFEZBIQHCLBF-UHFFFAOYSA-N 0.000 description 2
- 102000011045 Chloride Channels Human genes 0.000 description 2
- 108010062745 Chloride Channels Proteins 0.000 description 2
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 2
- 206010053398 Clonic convulsion Diseases 0.000 description 2
- 206010012373 Depressed level of consciousness Diseases 0.000 description 2
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 2
- AEMRFAOFKBGASW-UHFFFAOYSA-N Glycolic acid Chemical compound OCC(O)=O AEMRFAOFKBGASW-UHFFFAOYSA-N 0.000 description 2
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 2
- 241000124008 Mammalia Species 0.000 description 2
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 2
- OFBQJSOFQDEBGM-UHFFFAOYSA-N Pentane Chemical compound CCCCC OFBQJSOFQDEBGM-UHFFFAOYSA-N 0.000 description 2
- NBIIXXVUZAFLBC-UHFFFAOYSA-N Phosphoric acid Chemical compound OP(O)(O)=O NBIIXXVUZAFLBC-UHFFFAOYSA-N 0.000 description 2
- LCTONWCANYUPML-UHFFFAOYSA-N Pyruvic acid Chemical compound CC(=O)C(O)=O LCTONWCANYUPML-UHFFFAOYSA-N 0.000 description 2
- 241000700159 Rattus Species 0.000 description 2
- FAPWRFPIFSIZLT-UHFFFAOYSA-M Sodium chloride Chemical compound [Na+].[Cl-] FAPWRFPIFSIZLT-UHFFFAOYSA-M 0.000 description 2
- QAOWNCQODCNURD-UHFFFAOYSA-N Sulfuric acid Chemical compound OS(O)(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-N 0.000 description 2
- 239000004480 active ingredient Substances 0.000 description 2
- 239000000654 additive Substances 0.000 description 2
- 238000004458 analytical method Methods 0.000 description 2
- 239000005557 antagonist Substances 0.000 description 2
- 230000000573 anti-seizure effect Effects 0.000 description 2
- 230000036506 anxiety Effects 0.000 description 2
- 238000003556 assay Methods 0.000 description 2
- 150000001556 benzimidazoles Chemical class 0.000 description 2
- 150000001557 benzodiazepines Chemical class 0.000 description 2
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 2
- NDKBVBUGCNGSJJ-UHFFFAOYSA-M benzyltrimethylammonium hydroxide Chemical compound [OH-].C[N+](C)(C)CC1=CC=CC=C1 NDKBVBUGCNGSJJ-UHFFFAOYSA-M 0.000 description 2
- 238000004166 bioassay Methods 0.000 description 2
- 125000004432 carbon atom Chemical group C* 0.000 description 2
- 239000012043 crude product Substances 0.000 description 2
- 150000005690 diesters Chemical class 0.000 description 2
- XBDQKXXYIPTUBI-UHFFFAOYSA-N dimethylselenoniopropionate Natural products CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 2
- 229960002200 flunitrazepam Drugs 0.000 description 2
- 239000012458 free base Substances 0.000 description 2
- 238000010438 heat treatment Methods 0.000 description 2
- JVTAAEKCZFNVCJ-UHFFFAOYSA-N lactic acid Chemical compound CC(O)C(O)=O JVTAAEKCZFNVCJ-UHFFFAOYSA-N 0.000 description 2
- 238000002844 melting Methods 0.000 description 2
- 230000008018 melting Effects 0.000 description 2
- 239000012528 membrane Substances 0.000 description 2
- VNWKTOKETHGBQD-UHFFFAOYSA-N methane Chemical compound C VNWKTOKETHGBQD-UHFFFAOYSA-N 0.000 description 2
- 210000002569 neuron Anatomy 0.000 description 2
- 150000002825 nitriles Chemical class 0.000 description 2
- 239000006186 oral dosage form Substances 0.000 description 2
- VLTRZXGMWDSKGL-UHFFFAOYSA-N perchloric acid Chemical compound OCl(=O)(=O)=O VLTRZXGMWDSKGL-UHFFFAOYSA-N 0.000 description 2
- 239000013641 positive control Substances 0.000 description 2
- FGKGVVJVRCFGNJ-UHFFFAOYSA-N pyrido[1,2-a]benzimidazole-4-carboxamide Chemical compound C1=CC=C2N=C3C(C(=O)N)=CC=CN3C2=C1 FGKGVVJVRCFGNJ-UHFFFAOYSA-N 0.000 description 2
- 239000011541 reaction mixture Substances 0.000 description 2
- 238000010992 reflux Methods 0.000 description 2
- 230000001105 regulatory effect Effects 0.000 description 2
- YGSDEFSMJLZEOE-UHFFFAOYSA-N salicylic acid Chemical compound OC(=O)C1=CC=CC=C1O YGSDEFSMJLZEOE-UHFFFAOYSA-N 0.000 description 2
- 239000000932 sedative agent Substances 0.000 description 2
- 239000011734 sodium Substances 0.000 description 2
- 229910000104 sodium hydride Inorganic materials 0.000 description 2
- 235000000346 sugar Nutrition 0.000 description 2
- 230000001629 suppression Effects 0.000 description 2
- 239000003826 tablet Substances 0.000 description 2
- 239000008399 tap water Substances 0.000 description 2
- 235000020679 tap water Nutrition 0.000 description 2
- CZDYPVPMEAXLPK-UHFFFAOYSA-N tetramethylsilane Chemical compound C[Si](C)(C)C CZDYPVPMEAXLPK-UHFFFAOYSA-N 0.000 description 2
- JOXIMZWYDAKGHI-UHFFFAOYSA-N toluene-4-sulfonic acid Chemical compound CC1=CC=C(S(O)(=O)=O)C=C1 JOXIMZWYDAKGHI-UHFFFAOYSA-N 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- QBYIENPQHBMVBV-HFEGYEGKSA-N (2R)-2-hydroxy-2-phenylacetic acid Chemical compound O[C@@H](C(O)=O)c1ccccc1.O[C@@H](C(O)=O)c1ccccc1 QBYIENPQHBMVBV-HFEGYEGKSA-N 0.000 description 1
- VLSDXINSOMDCBK-BQYQJAHWSA-N (E)-1,1'-azobis(N,N-dimethylformamide) Chemical compound CN(C)C(=O)\N=N\C(=O)N(C)C VLSDXINSOMDCBK-BQYQJAHWSA-N 0.000 description 1
- BJEPYKJPYRNKOW-REOHCLBHSA-N (S)-malic acid Chemical compound OC(=O)[C@@H](O)CC(O)=O BJEPYKJPYRNKOW-REOHCLBHSA-N 0.000 description 1
- WBYWAXJHAXSJNI-VOTSOKGWSA-M .beta-Phenylacrylic acid Natural products [O-]C(=O)\C=C\C1=CC=CC=C1 WBYWAXJHAXSJNI-VOTSOKGWSA-M 0.000 description 1
- SCYULBFZEHDVBN-UHFFFAOYSA-N 1,1-Dichloroethane Chemical compound CC(Cl)Cl SCYULBFZEHDVBN-UHFFFAOYSA-N 0.000 description 1
- WSLDOOZREJYCGB-UHFFFAOYSA-N 1,2-Dichloroethane Chemical compound ClCCCl WSLDOOZREJYCGB-UHFFFAOYSA-N 0.000 description 1
- RYHBNJHYFVUHQT-UHFFFAOYSA-N 1,4-Dioxane Chemical compound C1COCCO1 RYHBNJHYFVUHQT-UHFFFAOYSA-N 0.000 description 1
- VZNCSZQPNIEEMN-UHFFFAOYSA-N 1-fluoro-2-isocyanatobenzene Chemical compound FC1=CC=CC=C1N=C=O VZNCSZQPNIEEMN-UHFFFAOYSA-N 0.000 description 1
- JPMRGPPMXHGKRO-UHFFFAOYSA-N 2-(chloromethyl)pyridine hydrochloride Chemical compound Cl.ClCC1=CC=CC=N1 JPMRGPPMXHGKRO-UHFFFAOYSA-N 0.000 description 1
- JKMHFZQWWAIEOD-UHFFFAOYSA-N 2-[4-(2-hydroxyethyl)piperazin-1-yl]ethanesulfonic acid Chemical compound OCC[NH+]1CCN(CCS([O-])(=O)=O)CC1 JKMHFZQWWAIEOD-UHFFFAOYSA-N 0.000 description 1
- 125000004493 2-methylbut-1-yl group Chemical group CC(C*)CC 0.000 description 1
- PKRSYEPBQPFNRB-UHFFFAOYSA-N 2-phenoxybenzoic acid Chemical compound OC(=O)C1=CC=CC=C1OC1=CC=CC=C1 PKRSYEPBQPFNRB-UHFFFAOYSA-N 0.000 description 1
- QBWKPGNFQQJGFY-QLFBSQMISA-N 3-[(1r)-1-[(2r,6s)-2,6-dimethylmorpholin-4-yl]ethyl]-n-[6-methyl-3-(1h-pyrazol-4-yl)imidazo[1,2-a]pyrazin-8-yl]-1,2-thiazol-5-amine Chemical compound N1([C@H](C)C2=NSC(NC=3C4=NC=C(N4C=C(C)N=3)C3=CNN=C3)=C2)C[C@H](C)O[C@H](C)C1 QBWKPGNFQQJGFY-QLFBSQMISA-N 0.000 description 1
- 125000005917 3-methylpentyl group Chemical group 0.000 description 1
- MVQVNTPHUGQQHK-UHFFFAOYSA-N 3-pyridinemethanol Chemical compound OCC1=CC=CN=C1 MVQVNTPHUGQQHK-UHFFFAOYSA-N 0.000 description 1
- WRFYIYOXJWKONR-UHFFFAOYSA-N 4-bromo-2-methoxyaniline Chemical compound COC1=CC(Br)=CC=C1N WRFYIYOXJWKONR-UHFFFAOYSA-N 0.000 description 1
- 125000004172 4-methoxyphenyl group Chemical group [H]C1=C([H])C(OC([H])([H])[H])=C([H])C([H])=C1* 0.000 description 1
- 125000000339 4-pyridyl group Chemical group N1=C([H])C([H])=C([*])C([H])=C1[H] 0.000 description 1
- NLHHRLWOUZZQLW-UHFFFAOYSA-N Acrylonitrile Chemical compound C=CC#N NLHHRLWOUZZQLW-UHFFFAOYSA-N 0.000 description 1
- 208000007848 Alcoholism Diseases 0.000 description 1
- BSYNRYMUTXBXSQ-UHFFFAOYSA-N Aspirin Chemical compound CC(=O)OC1=CC=CC=C1C(O)=O BSYNRYMUTXBXSQ-UHFFFAOYSA-N 0.000 description 1
- 239000005711 Benzoic acid Substances 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical group [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- 108091006146 Channels Proteins 0.000 description 1
- WBYWAXJHAXSJNI-SREVYHEPSA-N Cinnamic acid Chemical compound OC(=O)\C=C/C1=CC=CC=C1 WBYWAXJHAXSJNI-SREVYHEPSA-N 0.000 description 1
- 206010009346 Clonus Diseases 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- 102000005915 GABA Receptors Human genes 0.000 description 1
- 108010005551 GABA Receptors Proteins 0.000 description 1
- 208000034308 Grand mal convulsion Diseases 0.000 description 1
- 239000007995 HEPES buffer Substances 0.000 description 1
- 102000004310 Ion Channels Human genes 0.000 description 1
- 108090000862 Ion Channels Proteins 0.000 description 1
- 206010022998 Irritability Diseases 0.000 description 1
- 238000003820 Medium-pressure liquid chromatography Methods 0.000 description 1
- GRYLNZFGIOXLOG-UHFFFAOYSA-N Nitric acid Chemical compound O[N+]([O-])=O GRYLNZFGIOXLOG-UHFFFAOYSA-N 0.000 description 1
- CTQNGGLPUBDAKN-UHFFFAOYSA-N O-Xylene Chemical compound CC1=CC=CC=C1C CTQNGGLPUBDAKN-UHFFFAOYSA-N 0.000 description 1
- XYFCBTPGUUZFHI-UHFFFAOYSA-N Phosphine Natural products P XYFCBTPGUUZFHI-UHFFFAOYSA-N 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-N R-2-phenyl-2-hydroxyacetic acid Natural products OC(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-N 0.000 description 1
- PPTYJKAXVCCBDU-UHFFFAOYSA-N Rohypnol Chemical compound N=1CC(=O)N(C)C2=CC=C([N+]([O-])=O)C=C2C=1C1=CC=CC=C1F PPTYJKAXVCCBDU-UHFFFAOYSA-N 0.000 description 1
- KEAYESYHFKHZAL-UHFFFAOYSA-N Sodium Chemical compound [Na] KEAYESYHFKHZAL-UHFFFAOYSA-N 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-N Succinic acid Natural products OC(=O)CCC(O)=O KDYFGRWQOYBRFD-UHFFFAOYSA-N 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-N Tartaric acid Natural products [H+].[H+].[O-]C(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-N 0.000 description 1
- 241000906446 Theraps Species 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 201000007930 alcohol dependence Diseases 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 150000001350 alkyl halides Chemical class 0.000 description 1
- 230000003281 allosteric effect Effects 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N alpha-hydroxysuccinic acid Natural products OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- 229910000147 aluminium phosphate Inorganic materials 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 125000004103 aminoalkyl group Chemical group 0.000 description 1
- 150000001450 anions Chemical class 0.000 description 1
- 230000003556 anti-epileptic effect Effects 0.000 description 1
- 230000002243 anti-metrazol Effects 0.000 description 1
- 229940125681 anticonvulsant agent Drugs 0.000 description 1
- 239000000729 antidote Substances 0.000 description 1
- 229940075522 antidotes Drugs 0.000 description 1
- 229940005530 anxiolytics Drugs 0.000 description 1
- 125000002029 aromatic hydrocarbon group Chemical group 0.000 description 1
- 125000003118 aryl group Chemical group 0.000 description 1
- 150000005840 aryl radicals Chemical class 0.000 description 1
- 239000012298 atmosphere Substances 0.000 description 1
- 235000010233 benzoic acid Nutrition 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 230000004071 biological effect Effects 0.000 description 1
- 210000004556 brain Anatomy 0.000 description 1
- 239000012267 brine Substances 0.000 description 1
- 239000008366 buffered solution Substances 0.000 description 1
- 244000309464 bull Species 0.000 description 1
- KDYFGRWQOYBRFD-NUQCWPJISA-N butanedioic acid Chemical compound O[14C](=O)CC[14C](O)=O KDYFGRWQOYBRFD-NUQCWPJISA-N 0.000 description 1
- 125000000484 butyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 229910052799 carbon Inorganic materials 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 210000004027 cell Anatomy 0.000 description 1
- 210000000170 cell membrane Anatomy 0.000 description 1
- 238000000451 chemical ionisation Methods 0.000 description 1
- 238000006243 chemical reaction Methods 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 235000013985 cinnamic acid Nutrition 0.000 description 1
- 229930016911 cinnamic acid Natural products 0.000 description 1
- 235000015165 citric acid Nutrition 0.000 description 1
- 208000028502 clonic seizure Diseases 0.000 description 1
- 239000012612 commercial material Substances 0.000 description 1
- 230000006957 competitive inhibition Effects 0.000 description 1
- 229940125846 compound 25 Drugs 0.000 description 1
- 125000000753 cycloalkyl group Chemical group 0.000 description 1
- 238000006114 decarboxylation reaction Methods 0.000 description 1
- 230000018044 dehydration Effects 0.000 description 1
- 238000006297 dehydration reaction Methods 0.000 description 1
- 238000003795 desorption Methods 0.000 description 1
- 239000007884 disintegrant Substances 0.000 description 1
- 231100000673 dose–response relationship Toxicity 0.000 description 1
- 230000035622 drinking Effects 0.000 description 1
- 239000000975 dye Substances 0.000 description 1
- 238000000921 elemental analysis Methods 0.000 description 1
- ZAKAONRTRWRIJT-UHFFFAOYSA-N ethyl 3-ethoxy-3-iminopropanoate Chemical compound CCOC(=N)CC(=O)OCC ZAKAONRTRWRIJT-UHFFFAOYSA-N 0.000 description 1
- 230000002964 excitative effect Effects 0.000 description 1
- 239000000796 flavoring agent Substances 0.000 description 1
- 125000001153 fluoro group Chemical group F* 0.000 description 1
- 235000013305 food Nutrition 0.000 description 1
- 235000013355 food flavoring agent Nutrition 0.000 description 1
- 239000001530 fumaric acid Substances 0.000 description 1
- 235000011087 fumaric acid Nutrition 0.000 description 1
- XPFVYQJUAUNWIW-UHFFFAOYSA-N furfuryl alcohol Chemical compound OCC1=CC=CO1 XPFVYQJUAUNWIW-UHFFFAOYSA-N 0.000 description 1
- 239000003365 glass fiber Substances 0.000 description 1
- 150000002334 glycols Chemical class 0.000 description 1
- 239000003979 granulating agent Substances 0.000 description 1
- 125000004051 hexyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 238000004128 high performance liquid chromatography Methods 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- 150000003840 hydrochlorides Chemical class 0.000 description 1
- 229910000042 hydrogen bromide Inorganic materials 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- 229910000043 hydrogen iodide Inorganic materials 0.000 description 1
- 239000003326 hypnotic agent Substances 0.000 description 1
- 238000011534 incubation Methods 0.000 description 1
- 239000004615 ingredient Substances 0.000 description 1
- 108091008042 inhibitory receptors Proteins 0.000 description 1
- 238000002347 injection Methods 0.000 description 1
- 239000007924 injection Substances 0.000 description 1
- 125000000959 isobutyl group Chemical group [H]C([H])([H])C([H])(C([H])([H])[H])C([H])([H])* 0.000 description 1
- 239000012948 isocyanate Substances 0.000 description 1
- 150000002513 isocyanates Chemical class 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 239000004310 lactic acid Substances 0.000 description 1
- 235000014655 lactic acid Nutrition 0.000 description 1
- 238000012417 linear regression Methods 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- 239000011976 maleic acid Substances 0.000 description 1
- 239000001630 malic acid Substances 0.000 description 1
- 235000011090 malic acid Nutrition 0.000 description 1
- 229960002510 mandelic acid Drugs 0.000 description 1
- 229940098779 methanesulfonic acid Drugs 0.000 description 1
- WBYWAXJHAXSJNI-UHFFFAOYSA-N methyl p-hydroxycinnamate Natural products OC(=O)C=CC1=CC=CC=C1 WBYWAXJHAXSJNI-UHFFFAOYSA-N 0.000 description 1
- 239000002480 mineral oil Substances 0.000 description 1
- 235000010446 mineral oil Nutrition 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 229940035363 muscle relaxants Drugs 0.000 description 1
- VBEGHXKAFSLLGE-UHFFFAOYSA-N n-phenylnitramide Chemical class [O-][N+](=O)NC1=CC=CC=C1 VBEGHXKAFSLLGE-UHFFFAOYSA-N 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 125000001971 neopentyl group Chemical group [H]C([*])([H])C(C([H])([H])[H])(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 229910017604 nitric acid Inorganic materials 0.000 description 1
- 238000006053 organic reaction Methods 0.000 description 1
- 235000006408 oxalic acid Nutrition 0.000 description 1
- 239000001301 oxygen Substances 0.000 description 1
- 229910052760 oxygen Inorganic materials 0.000 description 1
- FJKROLUGYXJWQN-UHFFFAOYSA-N papa-hydroxy-benzoic acid Natural products OC(=O)C1=CC=C(O)C=C1 FJKROLUGYXJWQN-UHFFFAOYSA-N 0.000 description 1
- 239000004031 partial agonist Substances 0.000 description 1
- 125000001147 pentyl group Chemical group C(CCCC)* 0.000 description 1
- 230000000144 pharmacologic effect Effects 0.000 description 1
- 150000003003 phosphines Chemical class 0.000 description 1
- 229910000073 phosphorus hydride Inorganic materials 0.000 description 1
- 230000000704 physical effect Effects 0.000 description 1
- 238000000039 preparative column chromatography Methods 0.000 description 1
- 238000004321 preservation Methods 0.000 description 1
- 239000003755 preservative agent Substances 0.000 description 1
- 239000000047 product Substances 0.000 description 1
- 235000019260 propionic acid Nutrition 0.000 description 1
- 125000001436 propyl group Chemical group [H]C([*])([H])C([H])([H])C([H])([H])[H] 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- ANMYJKRGVYXJQX-UHFFFAOYSA-N pyrido[1,2-a]benzimidazole-1-carboxamide Chemical class NC(=O)c1cccc2nc3ccccc3n12 ANMYJKRGVYXJQX-UHFFFAOYSA-N 0.000 description 1
- 229940107700 pyruvic acid Drugs 0.000 description 1
- IUVKMZGDUIUOCP-BTNSXGMBSA-N quinbolone Chemical compound O([C@H]1CC[C@H]2[C@H]3[C@@H]([C@]4(C=CC(=O)C=C4CC3)C)CC[C@@]21C)C1=CCCC1 IUVKMZGDUIUOCP-BTNSXGMBSA-N 0.000 description 1
- 150000003254 radicals Chemical class 0.000 description 1
- 238000001953 recrystallisation Methods 0.000 description 1
- 230000002829 reductive effect Effects 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 230000004044 response Effects 0.000 description 1
- 238000002390 rotary evaporation Methods 0.000 description 1
- CVHZOJJKTDOEJC-UHFFFAOYSA-N saccharin Chemical compound C1=CC=C2C(=O)NS(=O)(=O)C2=C1 CVHZOJJKTDOEJC-UHFFFAOYSA-N 0.000 description 1
- 229940081974 saccharin Drugs 0.000 description 1
- 235000019204 saccharin Nutrition 0.000 description 1
- 239000000901 saccharin and its Na,K and Ca salt Substances 0.000 description 1
- 229960004889 salicylic acid Drugs 0.000 description 1
- 125000002914 sec-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 125000003548 sec-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- 229940125723 sedative agent Drugs 0.000 description 1
- 230000001624 sedative effect Effects 0.000 description 1
- 208000019116 sleep disease Diseases 0.000 description 1
- 239000011780 sodium chloride Substances 0.000 description 1
- QDRKDTQENPPHOJ-UHFFFAOYSA-N sodium ethoxide Chemical compound [Na+].CC[O-] QDRKDTQENPPHOJ-UHFFFAOYSA-N 0.000 description 1
- 239000012312 sodium hydride Substances 0.000 description 1
- 159000000000 sodium salts Chemical class 0.000 description 1
- HPALAKNZSZLMCH-UHFFFAOYSA-M sodium;chloride;hydrate Chemical compound O.[Na+].[Cl-] HPALAKNZSZLMCH-UHFFFAOYSA-M 0.000 description 1
- 238000004611 spectroscopical analysis Methods 0.000 description 1
- 238000001228 spectrum Methods 0.000 description 1
- 229910001220 stainless steel Inorganic materials 0.000 description 1
- 239000010935 stainless steel Substances 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- 238000007619 statistical method Methods 0.000 description 1
- 239000008223 sterile water Substances 0.000 description 1
- 238000007920 subcutaneous administration Methods 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- 150000003467 sulfuric acid derivatives Chemical class 0.000 description 1
- 239000000375 suspending agent Substances 0.000 description 1
- 238000003786 synthesis reaction Methods 0.000 description 1
- 239000011975 tartaric acid Substances 0.000 description 1
- 235000002906 tartaric acid Nutrition 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 210000001519 tissue Anatomy 0.000 description 1
- 238000012549 training Methods 0.000 description 1
- 230000001131 transforming effect Effects 0.000 description 1
- 238000003828 vacuum filtration Methods 0.000 description 1
- 239000003981 vehicle Substances 0.000 description 1
- 239000003643 water by type Substances 0.000 description 1
- 239000008096 xylene Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Biomedical Technology (AREA)
- Neurology (AREA)
- Neurosurgery (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Engineering & Computer Science (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Plural Heterocyclic Compounds (AREA)
Description
- HINTERGRUND DER ERFINDUNG
- Der Gamma-Aminobuttersäure-A-Rezeptor (GABA-A-Rezeptor) ist der am häufigsten vorkommende inhibitorische Rezeptor im Gehirn von Säugetieren. Er umfaßt eine heteropolymere Struktur, die einen Chloridionenkanal bildet und trägt vielfache Erkennungsstellen für die Bindung von modulatorischen Molekülen. Die Bindung von GABA an ihre spezifischen Erkennungstellen auf dem GABA-A-Rezeptor öffnet den Ionenkanal und läßt Chloridionen indie Nervenzellen fließen. Diese Aktion hyperpolarisiert die Zellmembran dieses Neurons und macht dadurch die Zelle weniger reaktiv gegenüber excitatorischen Reizen. Der Chloridionenstrom kann ebenso durch verschiedene Arzneien reguliert werden, die als positive oder negative Modulatoren des GABA-A-Rezeptors dienen (Smith and Olsen, Trends Pharm. Sci. 1995, 16, 162; Stephenson, Biochem. J., 1995, 310, l). Der sogenannte Benzodiazepin (BZD)-Rezeptor ist eine Stelle für solche allosterischen Modulatoren auf dem GABA-A-Rezeptor. Diese Stelle vermittelt zwei gegenteilige Effekte, einen, der die Wirkung von GABA amplifiziert („positive" Wirksamkeit) und der andere, der die Wirkung von GABA verringert („negative" Wirksamkeit). Mittel, die die GABA-Rezeptor/Chloridionenkanal-Funktionen über die BZD-Stelle erleichtern, werden als Agonisten bezeichnet, während Mittel, die eine solche Funktion verringern, als inverse Agonisten bezeichnet werden. Antagonisten an dieser Stelle blockieren die Effekte von Agonisten oder inversen Agonisten durch kompetitive Inhibition ihrer Bindung. Es ist daher möglich, eine Reihe von Verbindungen zu haben, von denen einige in gleicher Weise an die BZD-Stelle binden, aber gleiche und gegenteilige regulatorische Effekte auf den GABA-A-Rezeptor/Chloridinonenkanal haben. Ebenso ist innerhalb der Reihe ein Kontinuum an Aktivität möglich (Takada, S. et al. J. Med. Chem. 1988, 31, 1738). Daher können BZD-Rezeptor-Liganden ein breites Spektrum an pharmakologischen Effekten induzieren, die von muskelrelaxierenden, hypnotischen, beruhigenden, angstlösenden und anti-Krampf-Aktivitäten, erzeugt durch volle oder teilweise Agonisten („positiv"), bis zu krampffördernden, anti-Rausch-bewirkenden und Angst erzeugenden Aktivitäten, erzeugt durch inverse Agonisten („negativ"), reichen. (Ein besseres Verständnis von diesem Gebiet kann man erhalten aus: Mohler, H. Arzneim.-Forsch./Drug Res. 1992, 42 (2a), 211; Haefely, W. et al., Advances in Drug Research, Academic Press, Band 14, 1985, S. 165- 322; Skolnick, P. et al., GABA and Benzodiazepine Receptors, Squires, R., Ed., 1987, S. 99-102 und darin angegebene Literaturangaben.).
- Die Benzodiazepine sind eine Klasse von Verbindungen, die an den BZD-Rezeptor mit hoher Affinität binden. Die meisten der verwendeten Arzneien sind Liganden vom Agonisten-Typ für den Rezeptor. Solche Verbindungen sind im allgemeinen aufgrund ihrer krampflösenden, angstlösenden, beruhigenden und muskelentspannenden Effekte nützlich. Antagonisten der BZD-Bindungsstelle sind für die Behandlung einer Benzodiazepinarzneiüberdosis nützlich, und inverse Agonisten sind beim Behandeln von Alkoholismus nützlich.
- Die vorliegende Erfindung betrifft neue Stoffzusammensetzungen und ihre Verwendung. Verbindungen mit einer gewissen strukturellen Ähnlichkeit mit denjenigen der vorliegenden Erfindung werden in Rida, S. M. et al. J. Het. Chem. 1988, 25, 1087; Soliman, F.S.G. et al. Arch. Pharm. 1984, 317; Volovenko, Y. M. et al. U.S.S.R. Patent SU 1027166 (Chem Abs. 99(25) 212524t); Ohta, S. et al. Heterocycles 1991, 32, 1923; Ohta, S. et al. Chem. Pharm. Bull. 1991, 39, 2787 beschrieben.
- WO94/04532 beschreibt eine Klasse von Pyrido[1,2-a]benzimidazol-Verbindungen, die beim Behandeln von ZNS-Krankheiten nützlich sind. Die offenbarten Verbindungen tragen eine Wasserstoff-, Alkyl-, Cykloalkyl- oder Aralkyl-Gruppe an einem der Imidazol-Stickstoffatome.
-
US 5,521,200 offenbart dieselben Verbindungen wie WO94/04532. - J. Med. Chem, 38, 16 (1996) beschreibt eine Klasse von Pyrido[1,2-a]benzimidazol-Verbindungen, die beim Behandeln von ZNS-Krankheiten nützlich sind. Die offenbarten Verbindungen tragen eine Wasserstoff-, Methyl-, Ethyl- oder Benzylgruppe an einem der Imidazol-Stickstoffatome.
- Biorg. Med. Chem. Lett., 6, 333, (1996) beschreibt eine Klasse von Pyrido[1,2-a]benzimidazol-Verbindungen, die beim Behandeln von ZNS-Krankheiten nützlich sind. Die offenbarten Verbindungen enthalten ein nicht-substituiertes Imidazol-Stickstoffatom.
- WO98/15553 beschreibt eine Klasse von Pyrido[1,2-a]benzimidazol-Verbindungen, die beim Behandeln von ZNS-Krankheiten nützlich sind. Die offenbarten Verbindungen tragen eine Aminoalkylgruppe an einem der Imidazol-Stickstoffatome.
- ZUSAMMENFASSUNG DER ERFINDUNG
- Die vorliegende Erfindung ist auf Verbindungen der folgenden Formel 1 gerichtet:wobei R, Ar, und X definiert sind, wie hiernach angegeben. Die Verbindungen der Formel 1 sind bei der Behandlungen von Zentralnervensystemkrankheiten nützlich. Die Verbindungen sind Liganden für die BZD-Bindungsstelle an GABA-A-Rezeptoren und sind damit als Muskelrelaxantien, Hypnotika/Sedativa, einschließlich schlaffördernden Mitteln, angstlösenden Mitteln, krampflösenden Mitteln/Antiepileptika, Rausch-hemmenden Mitteln und Antidoten für Arznei-Überdosierungen (insbesondere einer Benzodiazepin-Überdosierung) nützlich.

- Die vorliegende Erfindung umfaßt ebenfalls pharmazeutische Zusammensetzungen, enthaltend ein oder mehrere Verbindungen der Formel 1 und Verfahren zur Behandlung von Krankheiten am zentralen Nervensystem, einschließlich Krämpfen, wie etwa epileptischen Anfällen, Angst, Muskelkrämpfen, Schlafstörungen und Benzodiazepin-Überdosierungen, wobei eine Verbindung der Formel 1 verwendet wird.
- DETAILIERTE BESCHREIBUNG DER ERFINDUNG
- Genauer gesagt, betrifft die Erfindung Verbindungen der folgenden Formel 1:wobei X unabhängig ausgewählt ist aus Wasserstoff, C1-C8-Alkyl, Halogen, Perfluor(C1-C8-Alkyl), Hydroxy, C1-C8-Alkoxy, Di(C1-C8-alkyl)-amino, C1-C8-Alkoxycarbonyl oder C1-C8-Alkylthio. Es kann bis zu vier unabhängige X-Substituenten an dem Phenyl geben. Bevorzugter ist X ausgewählt aus einem von C1-C8-Alkoxy, Wasserstoff, Halogen oder C1-C8-Alkyl. Bevorzugt gibt es nur einen X-Substituenten, der nicht Wasserstoff ist. Am bevorzugtesten ist ein solcher anderer X-Substituent Fluor.
- R ist ausgewählt aus allen (CH2)n-Heterozyklen, wobei n = 1–4 ist, und wobei der Heterozyklus ausgewählt ist aus Morpholin, Pyridin, Pyridin-N-oxid, Thiazol, Thiophen, Furan, Indol, Benzothiophen, Pyridazin, Pyrimidin, Indolin, Chinolin, Indazol, Imidazol, Benzofuran, Triazin, Pyrazin, Isochinolin, Isoxazol, Thiadiazol, Benzothiazol, Trazol, Benzotriazol, oder substituiertes Piperazin, wobei der Substituent Wasserstoff oder C1-C8-Alkyl ist; einem substituierten Heterozyklus, wobei es eine oder mehrere Substituenten gibt, die unabhängig ausgewählt sind aus einem von Halogen, Perfluor(C1-8)-alkyl, Nitro, C1-8-Alkylthio, C1-8-Alkoxy, C1-8-Alkyl, Di(C1-8-alkyl)-amino, Carboxy, oder C1-8Alkoxycarbonyl; vorausgesetzt, daß R nicht (CH2)nNR2R3 ist, wobei R2 und R3 mit dem Stickstoff zusammengenommen sind, um substituiertes Piperazin zu bilden, wobei das substituierte Piperazin mit C1-4-Alkyl oder Aralkyl-(C1-C4) substituiert ist. Bevorzugter ist R ausgewählt aus einem von (2-Furfuryl)-methyl, (4-Imidazo)-methyl; (3-Pyridyl-N-oxid)-methyl, (4-Pyridyl-N-oxid)-methyl, 2-(2-Pyridyl)-ethyl, (4-Pyridyl)-methyl, (3-Pyridyl)-methyl, 2-(2-Thienyl)-ethyl, (3-Thienyl)-methyl, 2-(3-Thienyl)-ethyl, (2-Thienyl)-methyl, (2-Pyridyl)-methyl, 2-(3-Thienyl)-ethyl, und (3-Furfuryl)-methyl. Am bevorzugtesten ist R ausgewählt aus einem von 2-(3-Thienyl)-ethyl, 3-(2-Thienyl)-ethyl, 2-and 3-(Thienyl)-methyl and 4-(Imidazo)-methyl.
- Ar ist ausgewählt aus einem aus Phenyl und substituiertem Phenyl, wobei die Phenylsubstituenten C1-8-Alkyl, Halogen, Perfluor(C1-8-Alkyl), Hydroxy, C1-8-Alkoxy, Di(C1-8-alkyl)-amino, C1-8-Alkoxycarbonyl oder C1-8-Alkylthio ist; einem Heterozyklus, wobei der Heterozyklus ausgewählt aus einem aus Pyridin, Pyridin-N-oxid, Thiazol, Thiophen, Furan, Indol, Benzothiophen, Pyridazin, Pyrimidin, Indol, Indolin, Chinolin, Indazol, Imidazol, Benzofuran, Triazin, Pyrazin, Isochinolin, Isoxazol, Thiadiazol, Benzothiazol, Trazol oder Benzotriazol; einem substituierten Heterozyklus, wobei es ein oder mehrere Substituenten gibt, die unabhängig ausgewählt sind aus einem aus Halogen, Perfluor(C1-8)-alkyl, Nitro, C1-8-Alkylthio, C1-8-Alkoxy, C1-8-Alkyl, Di(C1-8-alkyl)-amino, Carboxy, C1-8-Alkoxycarbonyl. Bevorzugter ist Ar ausgewählt aus einem aus 2-Fluorphenyl, 2,6-Difluorphenyl, 4-Methoxyphenyl, und 2-Fluor-4-Methoxyphenyl.
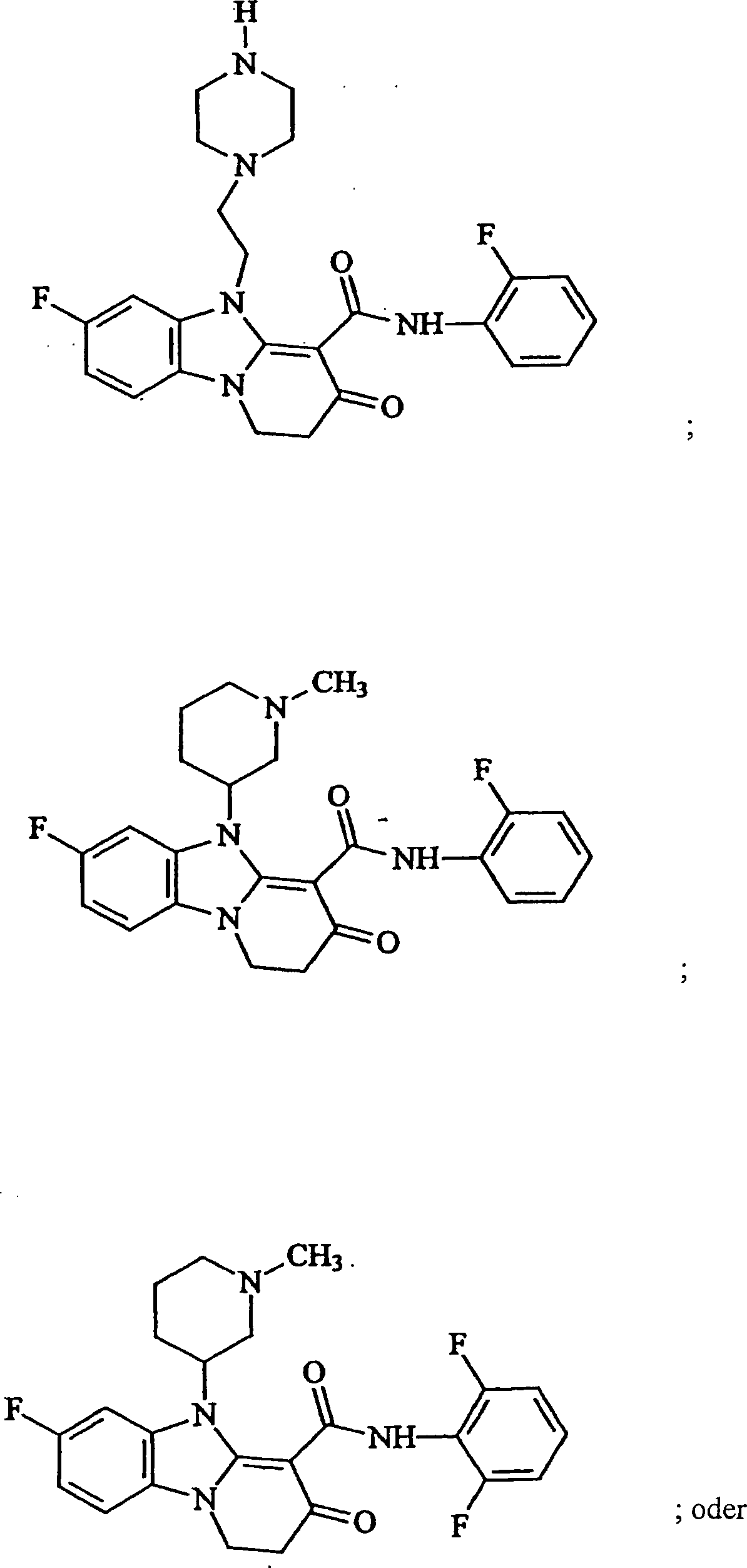
- Jedoch ist die Verbindung der Formel (I) nicht:

- Wie hierin verwendet, sofern nicht anderweitig festgestellt, schließen Alkyl und Alkoxy, sei es in alleiniger Verwendung oder als Teil einer Substituentengruppe, gerade und verzweigte Ketten ein. Zum Beispiel schließen Alkylradikale Methyl, Ethyl, Propyl, Isopropyl, Butyl, Isobutyl, sec-Butyl, t-Butyl, Pentyl, 2-Methyl-3-butyl, 1-Methylbutyl, 2-Methylbutyl, Neopentyl, Hexyl, 1-Methylpentyl, 3-methylpentyl ein. Alkoxy-Radikale sind Sauerstoffether, gebildet aus den zuvor beschriebenen geradkettigen oder verzweigtkettigen Alkylgruppen. Sofern nicht anderweitig festgestellt, bedeutet „kurzkettig" ("lower") bei Verwendung mit Alkyl und Alkoxy eine Kohlenstoffkettenzusammensetzung aus 1–8 Kohlenstoffatomen. Natürlich müssen, wenn der Alkyl oder Alkoxy-Substituent verzweigt ist, wenigstens 3 Kohlenstoffatome vorhanden sein.
- Der Begriff „Aryl", wie hierin alleine oder in Kombination mit anderen Begriffen verwendet, bezeichnet aromatische Kohlenwasserstoffgruppen, wie etwa Phenyl oder Naphthyl. Der Begriff „Aralkyl" bedeutet ein Radikal, enthaltend eine kurzkettige Alkylgruppe, die mit einem Arylradikal substituiert ist. Unter Bezug auf Substituenten bedeutet der Begriff unabhängig, daß, wenn mehr als einer eines solchen Substituenten möglich ist, solche Substituenten dieselben oder unterschiedlich voneinander sein können.
- Beispiele für besonders bevorzugte Verbindungen der Formel 1 schließen ein:
7-Fluor-1,2-dihydro-5-[2-(3-thienyl)ethyl]-3-oxo-N-(2-fluorphenyl)pyrido-[1,2-a]benzimidazol-4-carboxamid,
7-Fluor-1,2-dihydro-5-[2-(2-thienyl)ethyl]-3-oxo-N-(2-fluorphenyl)pyrido-[1,2-a]benzimidazol-4-carboxamid,
7-Fluor-1,2-dihydro-5-[(2-thienyl)methyl]-3-oxo-N-(2-fluorphenyl)pyrido-1,2-a]benzimidazol-4-carboxamid,
7-Fluor-1,2-dihydro-5-[(3-thienyl)methyl]-3-oxo-N-(2-fluorphenyl)pyrido-[1,2-a]benzimidazol-4-carboxamid, und
7-Fluor-1,2-dihydro-5-[(4-imidazo)-methyl]-3-oxo-N-(2-fluorphenyl)pyrido-[1,2-a]-benzimidazol-4-carboxamid. - Wenn Verbindungen eine basische Gruppe enthalten, können Säureadditionssalze hergestellt werden und können ausgewählt sein aus dem Salz der Salzsäure, Bromwasserstoff, Jodwasserstoff, Perchlorsäure, Schwefelsäure, Salpetersäure, Phosphorsäure, Essigsäure, Propionsäure, Glykolsäure, Milchsäure, Brenztraubensäure, Oxasäure, Malonsäure, Bernsteinsäure, Maleinsäure, Fumarsäure, Äpfelsäure, Weinsäure, Zitronensäure, Benzoesäure, Zimtsäure, Mandelsäure, Methansulfonsäure, p-Toluolsulfonsäure, Cyclohexansulfamidsäure, Salicylsäure, 2-Phenoxybenzoesäure, 2-Acetoxybenzoesäure oder Saccharin und ähnliches. Solche Salze können hergestellt werden durch Umsetzen der freien Base der Verbindungen der Formel 1 mit der Säure und durch Isolieren des Salzes.
- Hydrate und andere Sulfate der Verbindung der Formel 1 sind ebenso vom Umfang dieser Erfindung umfaßt und innerhalb der Definition der Formel 1 eingeschlossen.
- Die Verbindungen der Formel 1 werden hergestellt, wie in dem folgenden Schema 1 dargestellt.
- Schema 1

- Genauer gesagt, wird das Pyridobenzimidazolesterderivat 2 gemäß dem folgenden Schema 2 hergestellt.
- Schema 2
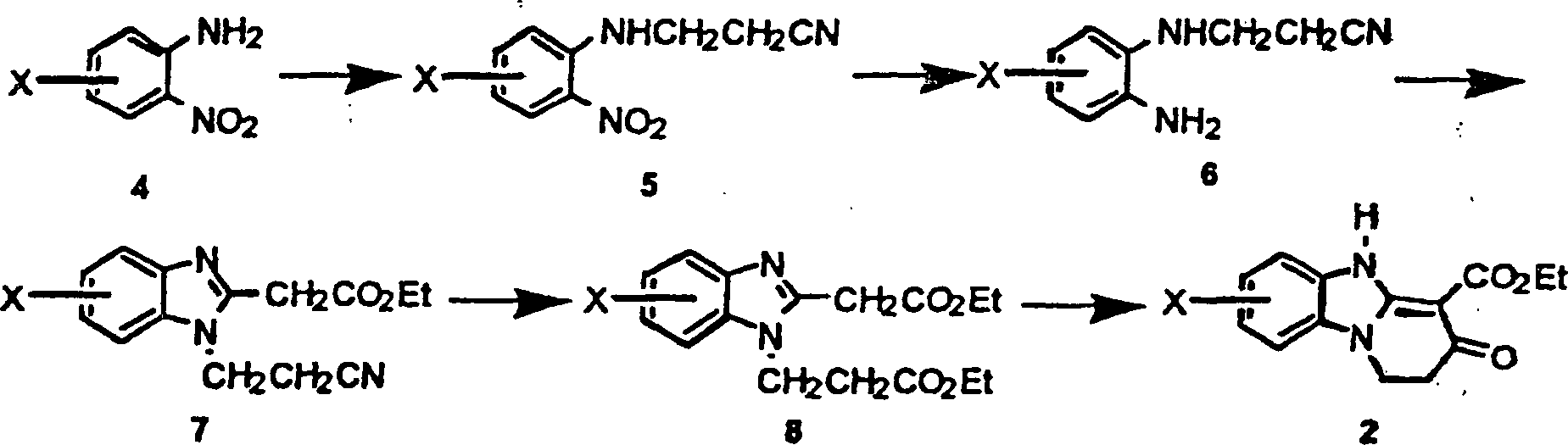
- Genauer gesagt, wird das substituierte Nitroanilinderivat 4, kommerziell erhältlich (z. B.: Aldrich Chemical Co.) oder mittels auf dem Gebiet bekannter Standardverfahren hergestellt, mit einer Mischung aus Acrylonitril und einer geeigneten Base, wie etwa Triton B (N-Benzyltrimethylammoniumhydroxid), in einem geeigneten Lösungsmittel, wie etwa Dioxan, bei Zimmertemperatur für 1–4 Tage behandelt, um das erwünschte Nitrilderivat 5 zu ergeben. Die Nitrogruppe des Nitrilderivats 5 wird reduziert, um das Aminoderivat 6 durch Behandlung des Derivats mit einem geeigneten Reduktionskatalysator, wie etwa Pd/C, in einem geeigneten Lösungsmittel, wie etwa Ethylacetat, unter einer Wasserstoffatmosphäre von ungefähr 50–60 psig für 3–12 h zu ergeben. Das Benzimidazolderivat 7 wird durch Erhitzen des Aminoderivats 6 mit Ethyl-ethoxycarbonylacetimidat·HCl in einem geeigneten Lösungsmittel, wie etwa EtOH, für ungefähr 4–24h hergestellt. Die Behandlung des Benzimidazolderivats 7 mit einer wasserfreien Säure, wie etwa HCl(g) in einem geeigneten Lösungsmittel, wie etwa EtOH, unter Rückfluß für ungefähr 4–24h, ergibt das Diesterderivat 8. Der Diester wird mit einer geeigneten Base, wie etwa Natriumethoxid, in einem geeigneten Lösungsmittel, wie etwa EtOH, für ungefähr 12–24h bei Zimmertemperatur behandelt, gefolgt von einer Behandlung mit ethanolischer HCl, um Pyridobenzimidazol 2 zu ergeben.
- Das Pyridobenzimidazolesterderivat 2 wird dann unter Rückfluß mit einem geeigneten substituierten Aminderivat in einem geeigneten Lösungsmittel, wie etwa Xylol oder Dimethylformamid, für ungefähr 1–24 h erhitzt, um das Pyrido[1,2-a]benzimidazolamidderivat 3 zu ergeben.
- Das Pyridobenzimidazolderivat 3 wird selektiv an der N5-Position unter Verwendung des Verfahrens von Mitsunobu alkyliert (siehe Hughes, D. Organic Reactions, 42, 355–656) oder den vor kurzem berichteten modifizierten Prozeduren (siehe Tsunoda Tetrahedron Letters 1993, 34 1639–1642 und Tsunoda Chemistry Letters 1994, 539–542). Eine Behandlung des Pyridobenzimidazolderivats 3 mit einem geeigneten heterozyklischen Alkanol, wie etwa 2-(3-Thienyl)-ethanol und 2-Furanylmethanol und 1–5 Äquivalenten eines geeigneten aktivierenden Mittels, wie etwa Diethylazodicarboxylat (DEAD), Azodicarbonyldipiperidin (ADDP), oder 1,1-Azobis(N,N-dimethylfomamid) (TMAD) und einem geeigneten trisubstituierten Phosphin, wie etwa Triphenylphosphin oder Tributylphosphin, in einem geeigneten Lösungsmittel, wie etwa Benzol, THF, oder DMF bei ungefähr 0 °C auf Zimmertemperatur für ungefähr 1–24 h ergab die erwünschten N5-(Heteroaryl)-alkyl-pyridobenzimidazol-derivative 1.
- Alternativ könnten die Verbindungen der Erfindung gemäß Schema 3 hergestellt werden, indem Ester 2 genommen wird und N5 mit einer Base deprotoniert wird, wie etwa NaH oder K2CO3, und indem das darauffolgende Anion mit einem Alkylhalogenid, wie etwa 2-Picolylhalogenid in einem geeigneten Lösungsmittel, wie etwa DMF oder DMSO, umgesetzt wird, um Ester vom Typ 9 zu ergeben. Diese werden dann mit einer Base, wie etwa Na-OH (3N in Ethanol) unter Erhitzen behandelt, um Enaminone vom Typ 10 zu ergeben. Eine Reaktion der Verbindungen vom Typ 10 mit Isocyanaten typischerweise bei Zimmertemperatur in einem Lösungsmittel, wie etwa 1,2-Dichlorethan, ergibt die Zielverbindungen vom Typ 1.
-
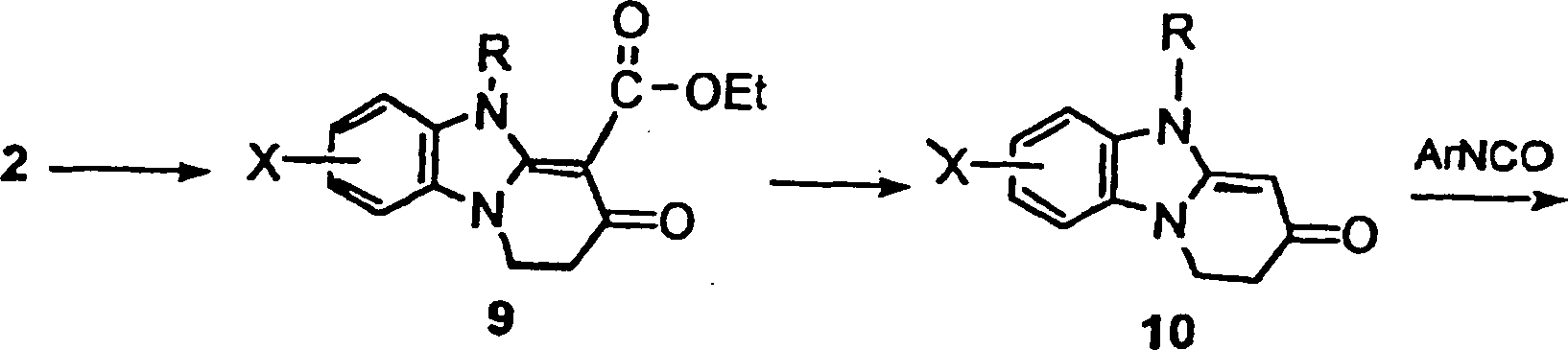
- Die Verbindungen dieser Erfindung wurden auf ihre Affinität für die Benzodiazepinstellen des GABA-A-Rezeptors getestet. Da die Verbindungen, die an diesen Rezeptor binden, bei der Behandlung von Krankheiten des Zentralen Nervensystems nützlich sind, wurden die Verbindungen ebenso in geeigneten Untersuchungen („screens") getestet, um spezifische Aktivitäten zu bewerten. Die Ergebnisse der verschiedenen Untersuchungen werden in Tabelle 1 gezeigt.
- Benzodiazepin-Rezeptor-Bindungsagssay
- Ausgewählte Verbindungen, die gemäß den experimentellen Details hergestellt wurden, die in den folgenden Beispielen angegeben sind, wurden auf ihre Bindung an die Benzodiazepinstelle des GABA-A-Rezeptors getestet (Williams, M. et al., J. Pharm. Exper. Therap. 1988, 248, 89). Die Fähigkeit der Verbindungen der Erfindung, die Bindung von Flunitrazepam an hergestellten Rezeptoren zu inhibieren, wurde beurteilt. Für jede Probe wurden Membranen aus ca. 10 mg Gewebe in einem K2HPO4-gepufferten Inkubationsmedium inkubiert (Endkonzentration = 2.0 mL). Die Konzentration des Liganden (3H-flunitrazepam) war ca. 3nM. Die Proben wurden für 10–20 min bei 25 °C inkubiert, wonach das Membranmaterial und gebundener Ligan auf Glasfaserfilterblättern unter Vakuumfiltration gesammelt wurde. Das gesammelte Material wurde mit 10 mM HEPES-gepufferter Lösung gewaschen, und die mit jeder Probe assoziierte Radioaktivität wurde mittels Flüssigszintillationsspektrometrie gemessen. Die Bindung der Testarznei an den Rezeptor wurde bestimmt, indem die Menge an radioaktiv markiertem Liganden, der in Kontrollproben gebunden war, mit der Menge an Liganden verglichen wurde, die in der Anwesenheit der Arznei gebunden war. Die Konzentrations-Antwort-Daten wurden auf eine Vielzahl von Weisen analysiert. Der IC50 wurde gewöhnlich durch Transformieren der Daten in ein log-logit-Format, dann durch Durchführung einer linearen Regressionsanalyse berechnet. Diese Prozedur liefert einen Hill-Koeffizienten ebenso wie den IC50-Wert. Der IC50-Wert ist für alle getesteten Verbindungen in Tabelle 1 aufgelistet. Ein IC50-Wert von über 10 000 für eine jeweilige Verbindung würde anzeigen, daß die Verbindung in dieser Untersuchung nicht aktiv war. Dies ist eine allgemeine Untersuchung, und Verbindungen, die hier aktiv sind, haben eine potentielle Nützlichkeit für die Behandlung von einer oder mehreren Krankheiten des Zentralen Nervensystems.
- Assay zum Bestimmen der Unterdrückung von Metrazol-induzierten Krämpfen in erwachsenen männlichen Mäusen
- Verbindungen der Erfindung wurden auf ihre Fähigkeit getestet, mit Metrazol-induzierte Krämpfe in Mäusen zu reduzieren (Swinyard, E. A. J. Am. Phar. Assoc. 1949, 38, 201). Männliche CD1-Mäuse mußten für wenigstens 16 Stunden fasten, wurden in gleiche Gruppen aufgeteilt, und Testverbindungen oder ein Träger wurden parenteral verabreicht. Wasser wurde nicht verwehrt, außer während der Beobachtungsperioden. Zum Zeitpunkt einer vermuteten Spitzenaktivität wurde die Anti-Pentylentetrazol (anti-Metrazol)-Aktivität durch die subkutane Verabreichung der CD90-Dosis von Metrazol bewertet (die Dosis von Metrazol wurde anhand der Dosis-Antwortkurve bestimmt, die klonische Krämpfe in 90% der Tiere verursachte, die den entsprechenden Träger für dieses Experiment erhielten). Metrazol wurde in 0,9% Natriumchloridlösung aufgelöst, und sein Dosisvolumen betrug 10 mL/kg. Tiere wurden zur Beobachtung der klonischen Krämpfe, tonischen Krämpfe und des Todes für eine Periode von 30 min. individuell untergebracht. Testverbindungen, die den klonischen Anfallanteil des Krampfs in wenigstens 50% der Tiere blockierten, wurde als aktiv angesehen. Der biologische Assay wurde als gültig angesehen, wenn die Effekte eines bekannten Antikrampfmittels (positive Kontrolle) innerhalb desselben Experiments nachgewiesen wurden. Die Aktivität wurde als eine prozentuale Verringerung von klonischen Krämpfen aus der Trägergruppe berichtet. Die ED50-Werte der aktiven Verbindungen wurden mittels dem Verfahren der probits berechnet (Finney, D. J. 1971. Probit Analysis. London: Cambridge Univeristy Press) und werden in Tabelle 1 aufgelistet. Ein ED50-Wert größer als 30 zeigt an, daß eine aktive Dosis für die getestete Verbindung nicht bestimmt worden war. Verbindungen, die in dieser Untersuchung aktiv sind, werden als aktive anti-Krampf/antiepileptische Mittel angesehen.
- Assay zum Messen der Unterdrückung von Angst in der erwachsenen männlichen Ratte
- Die angstlösende Aktivität von ausgewählten Verbindungen der Erfindung wurde durch Bestimmen ihrer Fähigkeit beurteilt, ein Verhalten auszulösen (zu enthemmen), das durch Bestrafung unterdrückt worden war (Vogel, J. R. et al. Psychopharmacology 1971, 21,1). Männliche Ratten erhielten kein Wasser für 48 h und erhielten keine Nahrung für 24 h vor dem Testen. Nach den ersten 24 Stunden des Wasserentzugs wurden sie in die Konfliktkammer für eine Trainingsperiode eingebracht, wo sie 200 mal unbestraft an einer Flasche, enthaltend Leitungswasser, lecken durften. Das Experiment wurde am nächsten Tag durchgeführt. Zum erwarteten Zeitpunkt der Spitzenaktivität wurden die Tiere in die Kammer gebracht, und es wurde ihnen Zugang zum Leitungswaser erlaubt. Wenn sie nicht tranken, wurde das Ex periment nach 5 min. abgebrochen, und die Tiere wurden auf Zeichen einer ZNS-Unterdrückung („CNS depression") bewertet. Ihr erstes Lecken initiiert einen 3-min-Testabschnitt. Darauffolgend wurde jedes zwanzigste Lecken durch einen 0,2s-Schock bestraft, der über das Trinkrohr aus rostfreiem Stahl abgegeben wurde. Kontrolltiere, die mit Träger behandelt worden waren, waren im allgemeinen willig, eine mittlere Anzahl von 3–8 Schocks pro Testabschnitt zu akzeptieren. Tiere, die mit einer aktiven angstlösenden Arznei behandelt wurden, ertrugen signifikant mehr Schocks als die Kontrolltiere. Der Wilcoxonrank-Summentest (Mann-Whitney-U-test), wurde verwendet, um auf ein Anwachsen (p < 0,05, einseitig) hinsichtlich der mittleren Anzahl („median number") von Schocks in den mit Arznei behandelten Gruppen zu testen, im Vergleich mit einer zeitgleich laufenden Trägerbehandelten Gruppe. Der biologische Assay wird als gültig angesehen, wenn die Effekte eines bekannten angstlösenden Mittels (positive Kontrolle) innerhalb des selben Experiments nachgewiesen werden. Eine Verbindung wurde als aktiv angesehen, wenn es einen signifikanten Unterschied hinsichtlich der mittleren Anzahl an tolerierten Schocks zwischen der Arzneibehandelten Gruppe und der Kontrollgruppe gibt. Die minimalen wirksamen Dosen (MED) für die aktiven Verbindungen der Erfindung werden in Tabellen 1 bis 5 aufgelistet. Die MED wurde als die minimale Dosis der Arzneibehandlung definiert, analysiert unter Verwendung des Wilcoxon-Rank-Summen-Test (SAS; Statistical Analysis System, Version 5.16). Wenn der MED-Wert größer als 10 ist, war eine aktive Dosis der getesteten Verbindung nicht bestimmt worden.
- Tabelle 1 Biologische Aktivität der Verbindungen der Formel I:

- Um die pharmazeutischen Zusammensetzungen dieser Erfindung herzustellen wird eine oder mehrere Verbindungen oder Salze davon als der aktive Inhaltsstoff intensiv mit einem pharmazeutischen Träger gemäß herkömmlichen pharmazeutischen Mischungstechniken gemischt, wobei der Träger eine große Vielzahl von Formen annehmen kann, abhängig von der zur Verabreichung gewünschten Form der Herstellung, z.B. oral oder parenteral. Bei der Herstellung der Zusammensetzungen in oraler Dosierungsform kann jedes der gewöhnlichen pharmazeutischen Medien verwendet werden. Für flüssige orale Zubereitungen, wie z.B. Suspensionen, Elixiere und Lösungen, schließen daher geeignete Träger und Zusatzstoffe Wasser, Glykole, Öle, Alkohole, Aromastoffe, Konservierungsmittel, Farbstoffe und ähnliches ein; für feste orale Zubereitungen, wie etwa z.B. Pulver, Kapseln und Tabletten, schließen geeignete Träger und Zusatzstoffe Stärken, Zucker, Verdünnungsmittel, Granuliermittel, Schmiermittel, Bindemittel, Zersetzungsmittel und ähnliches ein. Wegen der Leichtigkeit ihrer Verabreichung stellen Tabletten und Kapseln die vorteilhafteste orale Dosierungsform dar, bei der offensichtlich feste pharmazeutische Träger verwendet werden. Sofern erwünscht, können Tabletten mittels Standardtechniken zuckerbeschichtet oder enterisch beschichtet sein. Für Parenteralia wird der Träger gewöhnlich steriles Wasser umfassen, obwohl andere Inhaltsstoffe, z.B. um die Löslichkeit zu erhöhen oder aus Konservierungszwecken, eingeschlossen sein können. Injizierbare Suspensionen können ebenso hergestellt werden, wobei geeignete flüssige Träger, Suspendiermittel und ähnliches verwendet werden kann. Die pharmazeutischen Zusammensetzungen hierin werden bevorzugt pro Dosierungseinheit, z.B. Tablette, Kapsel, Pulver, Injektion, Teelöffel und ähnliches, ungefähr 5 bis ungefähr 500 mg des aktiven Inhaltsstoffes enthalten, obwohl andere Einheitsdosierungen verwendet werden können.
- Bei der therapeutischen Verwendung bei der Behandlung von Krankheiten des zentralen Nervensystems in Säugetieren können die Verbindungen dieser Erfindung in einer Menge von ungefähr 0,2 bis 25 mg/kg pro Tag verabreicht werden. Bei einer therapeutischen Verwendung als ein angstlösendes Mittel können die Verbindungen der Erfindung in einer Menge von ungefähr 0,2 bis 25 mg/kg pro Tag verabreicht werden. Bei der therapeutischen Verwendung als ein Antikrampfmittel/antiepileptisches Mittel können die Verbindungen der Erfindung in einer Menge von ungefähr 0,2 bis 25 mg/kg pro Tag verabreicht werden. Bei der therapeutischen Verwendung als ein Mittel zum Behandeln von Benzodiazepin-Überdosierungen können die Verbindungen der Erfindung in einer Menge von 0,2 bis 25 mg/kg pro Tag verabreicht werden. Bei der therapeutischen Verwendung als ein Beruhigungsmittel/Hypnotikum beträgt eine therapeutisch wirksame Menge ungefähr 0,2 bis 25 mg/kg pro Tag. Als ein Muskelrelaxans können ungefähr 0,2 bis 25 mg/kg pro Tag der Verbindungen dieser Erfindung verwendet werden. Die Bestimmung der optimalen Dosierung für eine individuelle Situation liegt innerhalb des Könnens eines Fachmanns.
- BEISPIELE
- Die folgenden Beispiele beschreiben die Erfindung in größerem Detail und sollen die Erfindung veranschaulichen.
- Schmelzpunktbestimmungen wurden auf einem Thomas Hoover-oder Mel-Temp-Schmelzpunktapparat durchgeführt und sind nicht korrigiert. Jede Verbindung hat wenigstens zwei analytische Resultate (Elementaranalyse, Schmp.) hier angegeben. Kernspinresonanz (NMR)-Spektren für Wasserstoffatome wurden in dem angezeigten Lösungsmittel mit Tetramethylsilan (TMS) als internem Standard auf einem Brüker-AM-360 (360 MHz) oder einem AT-300 (300 MHz)-Spektrometer gemessen. Die Werte werden in parts per million feldabwärts von TMS ausgedrückt. Elementaranalysen wurden mit Robertson Microlit (Madison, NJ) gemessen und werden in Gewichtsprozent jeden Elements pro Gesamtmolekulargewicht ausgedrückt. Die Massenspektren (MS) wurden auf einem Finnigan 3300-Spektrometer (Methan) unter Verwendung von Desorption-chemischer Ionisierung-Techniken bestimmt. Die gesamte preparative Säulenchromatographie wurde unter Verwendung einer Waters Prep 500A HPLC (Kieselgel) durchgeführt, wobei das geeignete kommerziell erhältliche Lösungsmittel verwendet wurde. Sofern nicht anderweitig festgestellt, wurden die in den Beispielen verwendeten Materialien von leicht verfügbaren kommerziellen Lieferanten erhalten oder mit Standardverfahren synthetisiert, die jedem Fachmann auf dem Gebiet der chemischen Synthese bekannt sind. Die Substituentengruppen, die zwischen den Beispielen variieren, sind Wasserstoff, sofern nicht anderweitig festgestellt.
- Beispiel 1 (20)
- 7-Fluor-1,2-dihydro-5-[2-(3-thienyl)ethyl]-oxo-N-(2-fluorphenyl)pyrido[1,2-a]benzimidazol-4-carboxamid
- Die Verbindung 3 (X=7-F, Ar=2-FPh; 1,0 g, 2,92 mmol) wurde in 75 mL trockenem THF unter Rühren unter Argon aufgelöst. Zu dieser Lösung wurde 2-(3-Thienyl)ethanol (1,12 g, 8,76 mmol) und Triphenylphosphin (2,29 g, 8,76 mmol) zugegeben. Die Lösung wurde für 10 min gerührt und dann auf 0 °C mit einem Eiswasserbad abgekühlt. Diethylazodicarboxylat (1,52 g, 8,76 mmol) wurde tropfenweise zugegeben. Das Eisbad wurde entfernt, die Lösung wurde bei Zimmertemperatur über Nacht gerührt. Die Reaktionsmischung wurde in vacuo aufkonzentriert, um ein dunkles gelbes Öl zu ergeben. Das Öl wurde mittels MPLC-Chromatographie aufgereinigt, (Kieselgel; 1,5:98,5 Methanol/Methylenchlorid). Der resultierende Feststoff wurde umkristallisiert aus Isopropanol, um 20 (0,69 g, 52,4%) als einen weißen kristallinen Feststoff zu ergeben; Schmp. 222,8–224,4 °C. H-1 NMR (Me2SO-d6) δ 12,38–12,20 (s, 1H), 8,54–8,45 (t, 1H), 7,90–7,75 (m, 1H), 7,73–7,61 (m, 1H), 7,4–7,32 (m, 1H), 7,3–6.92 (m, 5-H), 6,9–6,81 (d, 1H), 4,75–4,61 (m, 2H), 4,38–4,30 (m, 2H), 3,05–2,92 (m, 2H), 2,71–2,59 (m, 2H). CI-MS m/e 452 (MH+).
- Auf ähnliche Weise wurden die folgenden Verbindungen der vorliegenden Erfindung hergestellt:

- Beispiel 2 (#17)
- 7-Fluor-1,2-dihydro-5-(3-pyridylmethyl)-3-oxo-N-(2-fluorphenyl)pyrido[1,2-a]benzimidazol-4-carboxamid
- Verbindung 3 (X=7-F, Ar=2-FPh; 1,0 g, 2,92 mmol) wurde in 75 ml trockenem THF unter Rühren unter Argon aufgelöst. Zur Lösung wurden 3-Pyridylcarbinol (2, 0,647g, 5,84 mmol) und Tri-n-butylphosphin, (1,18 g, 5,84 mmol) zugegeben. Die Lösung wurde für 10 Minuten gerührt, dann auf 0 °C mit einem Eisbad abgekühlt. 1,1-Azo-bis(N,N-dimethylformamid) (1,00 g, 5,84 mmol) wurde in einem Teil zugegeben. Das Eisbad wurde entfernt, und die Lösung wurde über Nacht bei Zimmertemperatur gerührt. Das Lösungsmittel wurde mittels Ro weißen kristallinen Feststoff zu ergeben, Schmp. 158,6–159,6 °C. H-1 NMR (Me2SO-d6) δ 12,21–12,15 (s, 1H, CONH), 8,5–8,4 (m, 3H), 7,80-7,71 (m, 1H), 7,69–7,62 (m, 1H, 4-Pyridyl), 7,51–7,47 (m, 1H), 7,3–6.92 (m, 5-H), 5,88–5,71 (s, 2H), 5,45–5,41 (s, 2H), 4,3–4,30 (m, 2H), 3,42–3,39 (s, 1H), 2,78–2,69 (m, 2H). CI-MS: m/e 424 (MH+).
- Auf die gleiche Weise wurden die folgenden Verbindungen der vorliegenden Erfindung hergestellt:
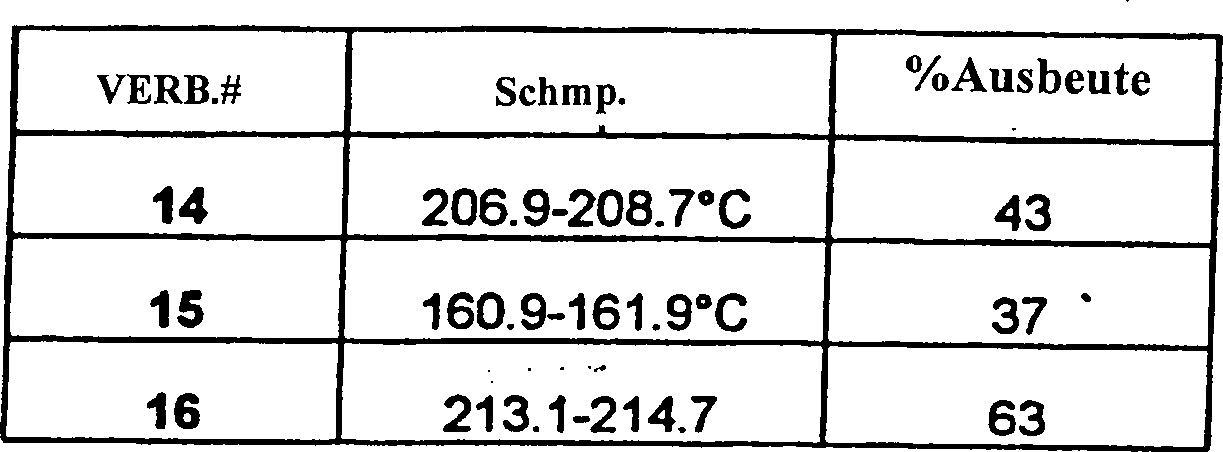
- Beispiel 3 (#25)
- 7-Fluor-1,2-dihydro-5-[2-(3-thienyl)ethyl]-3-oxo-N-(2-fluorphenyl)pyrido[1,2-a]benzimidazol-4-carboxamid
- Verbindung 3 (X=F, Ar=2,6-F2Ph; 1,0 g, 2,78 mmol) wurden in 70 ml trockenes Benzol unter Rühren unter Argon aufgelöst. Zur Lösung wurde 2-(3-Thienyl)ethanol (0,5343 g, 4,17 mmol) und Tri-n-butylphosphin (0,843 g, 4,168 mmol) zugegeben. Die Lösung wurde für 10 min gerührt, darin auf 0 °C mit einem Eiswasserbad abgekühlt. Azodicarbonyldipiperidin (1,051 g, 4,168 mmol) wurde in einer Portion zugegeben. Das Eisbad wurde entfernt, und die Lösung bei Zimmertemperatur über Nacht gerührt. Das Lösungsmittel wurde mittels Rotationsverdampfung in vacuo entfernt, um einen dunkelgelben Feststoff zu ergeben. Der Feststoff wurde aus Isopropanol umkristallisiert, um Verbindung 25 (0,4157 g, 21%) als einen weißen kristallinen Feststoff zu ergeben, Schmp. 166,6–167,2 °C. H-1 NMR (Me2SO-d6) δ 11,52–11,42 (s, 1H, CONH), 8,54–8,45 (t, 1H), 7,90–7,75 (m, 1H), 7,79–7,61 (m, 1H), 7,4–7,32 (m, 1H), 7,3–6.92 (m, 4-H), 6,9–6,85 (d, 1H), 4,75–4,61 (m, 2H), 4,38–4,30 (m, 2H), 3,05–2,92 (m, 2H), 2,71–2,59 (m, 2H). CI-MS: m/e 470 (MH+).
- Auf die gleiche Weise wurden die folgenden Verbindungen der vorliegenden Erfindung hergestellt:

- Beispiel 4 (28)
- 1,2-Dihydro-5-(2-pyridylmethyl)-3-oxo-N-(2-fluorphenyl)pyrido[1,2-a]-benzimidazol-4-carboxamid
- Natriumhydrid (60% in Mineralöl; 9,80 g, 245 mmol) wurde mit Pentan gespült (2 × 100mL) und mit wasserfreiem DMF (175 mL) abgedeckt. Diese Suspension wurde in einem Eiswasserbad gekühlt (10 °C). Dann wurde der Pyridobenzimidazolester (2, X=H, 10,32 g, 40,0 mmol) in einer Portion zugegeben. Das resultierende unlösliche Natriumsalz des PBI-Ester wurde mit zusätzlichem DMF (40 ml) abgedeckt und darauffolgend mit 2-Picolylchloridhydrochlorid (32,81 g, 200 mmol) behandelt. Die Reaktionsmischung wurde mit H2O (200 ml) verdünnt und mit CHCl3 (3 × 200 ml) extrahiert. Die CHCl3-Lösung wurde mit wäßrigem 1N NaOH (3 × 150 ml), H2O (4 × 250 ml) gewaschen, getrocknet (Na2SO4) und aufkonzentriert, um 20,87 g eines rohen, dunklen, rötlichen Feststoffs zu ergeben (9, X=H, R=2-Pyridylmethyl). Dieses rohe Produkt wurde weiter bis zur Decarbethoxylierung ohne weitere Aufreinigung geführt. MS(CI-CH4) m/e 350 (MH+). Eine Lösung der Probe von 9, die oben hergestellt wurde (20,77 g), und wäßrige 3N NaOH (200 ml) in Ethanol (300 ml) wurde unter Rückfluß für 7 h erhitzt und dann bei Zimmertemperatur für 3 d gehalten. Der Ethanol wurde in vacuo entfernt, was einen braunen Rückstand lieferte, der mit H2O (100 ml) verdünnt und mit CHCl3 (3 × 150 ml) extrahiert wurde. Die kombinierte CHCl3-Lösung wurde mit H2O (150 ml), Salzlake (2 × 150 ml) gewaschen, getrocknet (Na2SO4) und aufkonzentriert, um einen braunen feuchten Feststoff (12,7g) zu ergeben. Dieser Feststoff wurde mit Et2O abgedeckt und gefiltert, um 5,67 g des Enaminonprodukts 10 (X=H, R=2-Pyridylmethyl) als ein braunes Pulver zu ergeben. CI-MS (CH4) m/e 278 (MH+). Eine Lösung von dem oben hergestellten Enaminon 10 (1,39 g 5,00 mmol) und 2-(Fluor)phenylisocyanat (0,70 ml, 6,23 mmol) in Dichlorethan (50 ml) wurde bei Zimmertemperatur für 40 h gerührt. Das Lösungsmittel wurde in vacuo entfernt, und das Rohprodukt wurde auf Kieselgel (2:98 MeOH/CHCl3) chromatographiert, um 1,84 g eines hellbraunen Feststoffs zu ergeben. Diese freie Base wurde in 75 mL 2:1 CHCl3/MeOH-Mischung aufgelöst, gefiltert und mit konzentrierter HCl in iPrOH angesäuert, bis der pH 3 betrug. Eine Umkristallisierung des HCl-Salzes von 28 aus McOH/Et2O lieferte 1,15 g (51%) eines weißen amorphen Feststoffs, Schmp. 213–216 °C (dec.). H-1 NMR (300 MHz, DMSO-d6) δ 12,2 (s, 1H, Amid NH), 8,6 (d, 1H), 8,4 (t, 1H), 7,9 (t, 1H), 7,75 (d, 1H), 7,5 (m, 4H), 7,35 (m, 1H), 7,25 (m, 1H), 7,10 (m, 1H), 5,9 (s, 2H), 4,4 (t, 2H). 2,75 (s, 2H).
- Auf gleiche Weise wurden die folgenden Verbindungen der vorliegenden Erfindung hergestellt:

- Tabelle 2: Physikalische Eigenschaften von N-5 (Heteroaryl)alkyl-PBI-Derivaten.

Claims (10)
- Verbindung der folgenden Formel I:wobei X unabhängig ausgewählt ist aus Wasserstoff, C1-C8-Alkyl, Halogen, Perfluor(C1-C8-Alkyl), Hydroxy, C1-C8-Alkoxy, di(C1-C8-Alkyl)-amino, C1-C8-Alkoxycarbonyl oder C1-C8-Alkylthio; wobei R ausgewählt ist aus jedem (CH2)n-Heterozyklus, wo n = 1–4 und wobei der Heterozyklus ausgewählt ist aus Morpholin, Pyridin, Pyridin-N-oxid, Thiazol, Thiophen, Furan, Indol, Benzothiophen, Pyridazin, Pyrimidin, Indolin, Chinolin, Indazol, Imidazol, Benzofuran, Triazin, Pyrazin, Isochinolin, Isoxazol, Thiadiazol, Benzothiazol, Triazol, Benzotriazol oder substituiertes Piperazin, wo der Substituent Wasserstoff oder C1-8-Alkyl ist; ein substituierter Heterozyklus, wo ein oder mehrere Substituenten vorhanden sind, welche unabhängig ausgewählt sind aus Halogen, Perfluor(C1-8)-Alkyl, Nitro, C1-8-Alkythio, C1-8-Alkoxy, C1-8-Alkyl, di(C1-8-Alkyl)-amino, Carboxy oder C1-8-Alkoxycarbonyl; vorausgesetzt, daß R nicht (CH2)nNR2R3 ist, wo R2 und R3 mit dem Stickstoff zusammengenommen sind, um substituiertes Piperazin zu bilden, wobei das substituierte Piperazin mit C1-4-Alkyl oder Aralkyl(C1-C4) substituiert ist; wobei Ar ausgewählt ist aus Phenyl und substituiertem Phenyl, wobei die Phenylsubstituenten C1-C8-Alkyl, Halogen, Perfluor(C1-8-Alkyl), Hydroxy, C1-8-Alkoxy, di(C1-8-Alkyl)-amino, C1-8-Alkoxycarbonyl oder C1-8-Alkylthio sind; ein Heterozyklus, wobei der Heterozyklus ausgewählt ist aus Pyridin, Pyridin-N-oxid, Thiazol, Thiophen, Furan, Indol, Benzothiophen, Pyridazin, Pyrirmidin, Indol, Indolin, Chinolin, Indazol, Imidazol, Benzofuran, Triazin, Pyrazin, Isochinolin, Isoxazol, Thiadiazol, Benzothiazol, Triazol oder Benzotriazol; ein substituierter Heterozyklus, wo ein oder mehrere Substituenten vorhanden sind, die unabhängig ausgewählt sind aus Halogen, Perfluor(C1-8)-Alkyl, Nitro, C1-8-Alkylthio, C1-8-Alkoxy, C1-8-Alkyl, di(C1-8-Alkyl)-amino, Carboxy, C1-8-Alkoxycarbonyl; oder ein pharmazeutisch akzeptables Salz, Solvat, Hydrat, Tautomer oder Rotomer derselben ist; vorausgesetzt, daß die Verbindung der Formel (I) nicht ist:



- Verbindung nach Anspruch 1, wobei nur ein X-Substituent außer Wasserstoff vorhanden ist.
- Verbindung nach Anspruch 2, wobei der X-Substituent außer Wasserstoff Fluor ist.
- Verbindung nach einem der Ansprüche 1 bis 3, wobei R ausgewählt ist aus (2-Furfuryl)methyl; (4-Imidazo)methyl; (3-Pyridyl-N-oxid)methyl; (4-Pyridyl-N-oxid)methyl; 2-(2-Pyridyl)ethyl; (4-Pyridyl)methyl; (3-Pyridyl)methyl; 2-(2-Thienyl)ethyl; (3-Thienyl)methyl; 2-(3-Thienyl)ethyl; (2-Thienyl)methyl; (2-Pyridyl)methyl; 2-(3-Thienyl)ethyl; und (3-Furfuryl)methyl.
- Verbindung nach Anspruch 4, wobei R 4-(Imidazo)methyl ist.
- Verbindung nach einem der Ansprüche 1 bis 5, wobei Ar 2-Fluorphenyl oder 2,6-Difluorphenyl ist.
- Verbindung nach Anspruch 1, ausgewählt aus 7-Fluor-1,2-dihydro-5-[2-(3-thienyl)ethyl]-3-oxo-N-(2-fluorphenyl)pyrido-[1,2-a]benzimidazol-4-carboxamid, 7-Fluor-1,2-dihydro-5-[2-(2-thienyl)ethyl]-3-oxo-N-(2-fluorphenyl)pyrido-[1,2-a]benzimidazol-4-carboxamid, 7-Fluor-1,2-dihydro-5-[(2-thienyl)methyl]-3-oxo-N-(2-fluorphenyl)pyrido-[1,2-a]benzimidazol-4-carboxamid, 7-Fluor-1,2-dihydro-5-[(3-thienyl)methyl]-3-oxo-N-(2-fluorphenyl)pyrido-[1,2-a]benzimidazol-4-carboxamid und 7-Fluor-1,2-dihydro-5-[(4-imidazo)methyl]-3-oxo-N-(2-fluorphenyl)pyrido-[1,2-a]benzimidazol-4-carboxamid.
- Pharmazeutische Zusammensetzung, die eine Verbindung der Formel I umfaßt, wie in einem der Ansprüche 1 bis 7 definiert ist oder ein pharmazeutisch akzeptables Salz, Solvat, Hydrat, Tautomer oder Rotomer desselben in einer wirksamen Menge zur Behandlung von Krankheiten des zentralen Nervensystems und ein pharmazeutisch akzeptabler Träger oder Verdünnungsmittel.
- Verbindung nach einem der Ansprüche 1 bis 7 oder die Zusammensetzung nach Anspruch 8 zur Behandlung von Krankheiten des zentralen Nervensystems wie beispielsweise Beklemmung, Krampf, Schlaflosigkeit, Muskelspasmus oder eine Arzneiüberdosis an Benzodiazepin.
- Verbindung oder Zusammensetzung nach Anspruch 9 zur Verwendung einer wirksamen Menge von 0,2 bis 25 mg/kg pro Tag.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US08/884,804 US5922731A (en) | 1997-06-30 | 1997-06-30 | 5- (heteroaryl)alkyl!-3-oxo-pyrido 1,2-a!benzimidazole-4-carboxamide derivatives useful in treating central nervous system disorders |
| US884804 | 1997-06-30 |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| DE69827706D1 DE69827706D1 (de) | 2004-12-30 |
| DE69827706T2 true DE69827706T2 (de) | 2005-10-06 |
Family
ID=25385433
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| DE69827706T Expired - Fee Related DE69827706T2 (de) | 1997-06-30 | 1998-06-29 | 5-(Heteroaryl)alkyl)-3-oxo-pyrido(1,2-a)benzimidazol-4-carboxamid (PBI) Derivate zur Behandlung von Störungen des zentralen Nervensystems |
Country Status (12)
| Country | Link |
|---|---|
| US (1) | US5922731A (de) |
| EP (1) | EP0889044B1 (de) |
| JP (1) | JPH11100380A (de) |
| AT (1) | ATE283269T1 (de) |
| AU (2) | AU7589298A (de) |
| CA (1) | CA2242261A1 (de) |
| DE (1) | DE69827706T2 (de) |
| ES (1) | ES2234076T3 (de) |
| MX (1) | MXPA98005355A (de) |
| NZ (1) | NZ330832A (de) |
| PT (1) | PT889044E (de) |
| WO (1) | WO1999000389A1 (de) |
Families Citing this family (4)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| AU1224499A (en) * | 1997-12-09 | 1999-06-28 | Neurosearch A/S | Chemical compounds for use as anxiolytic agents and a method for the identification of anxiolytic compounds |
| US6762294B2 (en) | 1999-02-19 | 2004-07-13 | The United States Of America As Represented By The Department Of Health And Human Services | Polynucleotides encoding polymorphic human GABAA receptor α-6 subunit |
| ES2254157T3 (es) * | 1999-02-19 | 2006-06-16 | The Government Of The Usa, As Represented By The Secretary, Department Of Health And Human Services | Subunidad alfa-6 del receptor poliformico humano gabaa. |
| JP2003506373A (ja) * | 1999-08-02 | 2003-02-18 | オーソ−マクニール・フアーマシユーチカル・インコーポレーテツド | 中枢神経系疾患の治療で有用なC−6環置換ピリド[1,2−a]ベンズイミダゾール誘導体 |
Family Cites Families (3)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP0656002B1 (de) * | 1992-08-19 | 2000-11-08 | Mcneilab, Inc. | 3-oxo-pyrido [1,2-a] benzimidazol-4-carboxyl und 4-oxo-azepino [1,2-a] benzimidazol-5-carboxyl derivate geeignet für die behandlung von zentralnervensystemerkrankungen |
| US5521200A (en) * | 1993-12-30 | 1996-05-28 | Ho; Winston | 2-oxo-pyrrolo[1,2-A]benzimidazole-3-carboxyl derivatives useful in treating central nervous system disorders |
| PT935598E (pt) * | 1996-10-07 | 2004-03-31 | Ortho Mcneil Pharm Inc | Derivados de 3-oxo-pirido (1,2-a)benzimidazole-4-carboxamida alquilo substituidos contendo 5 heteroatomos uteis no tratamento de disturbios do sistema nervoso central |
-
1997
- 1997-06-30 US US08/884,804 patent/US5922731A/en not_active Expired - Lifetime
-
1998
- 1998-05-19 AU AU75892/98A patent/AU7589298A/en not_active Abandoned
- 1998-05-19 WO PCT/US1998/010456 patent/WO1999000389A1/en not_active Ceased
- 1998-06-29 ES ES98305119T patent/ES2234076T3/es not_active Expired - Lifetime
- 1998-06-29 JP JP10196485A patent/JPH11100380A/ja active Pending
- 1998-06-29 EP EP98305119A patent/EP0889044B1/de not_active Expired - Lifetime
- 1998-06-29 PT PT98305119T patent/PT889044E/pt unknown
- 1998-06-29 DE DE69827706T patent/DE69827706T2/de not_active Expired - Fee Related
- 1998-06-29 AT AT98305119T patent/ATE283269T1/de not_active IP Right Cessation
- 1998-06-29 NZ NZ330832A patent/NZ330832A/xx unknown
- 1998-06-30 MX MXPA98005355A patent/MXPA98005355A/es not_active IP Right Cessation
- 1998-06-30 AU AU73950/98A patent/AU757320B2/en not_active Ceased
- 1998-06-30 CA CA002242261A patent/CA2242261A1/en not_active Abandoned
Also Published As
| Publication number | Publication date |
|---|---|
| WO1999000389A1 (en) | 1999-01-07 |
| MXPA98005355A (es) | 2004-10-28 |
| AU757320B2 (en) | 2003-02-13 |
| DE69827706D1 (de) | 2004-12-30 |
| AU7395098A (en) | 1999-01-07 |
| JPH11100380A (ja) | 1999-04-13 |
| ES2234076T3 (es) | 2005-06-16 |
| US5922731A (en) | 1999-07-13 |
| NZ330832A (en) | 2000-01-28 |
| EP0889044A1 (de) | 1999-01-07 |
| ATE283269T1 (de) | 2004-12-15 |
| CA2242261A1 (en) | 1998-12-30 |
| EP0889044B1 (de) | 2004-11-24 |
| PT889044E (pt) | 2005-01-31 |
| AU7589298A (en) | 1999-01-19 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| DE69209679T2 (de) | Aryl(oder Heteroaryl)-piperazinyl-alkyl-azol-derivate, ihre Herstellung und ihre Anwendung als Medikament | |
| DE60216115T2 (de) | BENZIMIDAZO 4,5-föISOCHINOLINON-DERIVATE | |
| DE60204718T2 (de) | 3-substituierte oxindol beta 3 agonisten | |
| EP1177178B1 (de) | Heterozyklisch substituierte benzimidazole, deren herstellung und anwendung | |
| EP0944631B1 (de) | Neue substituierte pyrazolderivate zur behandlung von herzkreislauferkrankungen | |
| DE69911448T2 (de) | Pyrazolopyrimidinone, cgmp pde5 inhibitoren, zur behandlung von sexuellen funktionsstörungen | |
| DE60315615T2 (de) | Tricyclische verbindungen basierend auf thiophen und arzneimittel, die diese umfassen | |
| DE60316013T2 (de) | Heteroaryl-pyrimidinderivate als jak-inhibitoren | |
| DE19834044A1 (de) | Neue substituierte Pyrazolderivate | |
| EP1222191A2 (de) | Benzodiazepin-derivate, deren herstellung und anwendung | |
| DE10219294A1 (de) | Substituierte N-(1,4,5,6-Tetrahydro-cyclopentapyrazol-3-yl)-Derivate, deren Herstellung und Verwendung als Arzneimittel | |
| DE69824195T2 (de) | Dipyridoimidazolderivate zur behandlung von störungen des zentralen nervensystems | |
| DE69329654T2 (de) | 3-oxo-pyrido [1,2-a] benzimidazol-4-carboxyl und 4-oxo-azepino [1,2-a] benzimidazol-5-carboxyl derivate geeignet für die behandlung von zentralnervensystemerkrankungen | |
| DE69726749T2 (de) | 5-heteroatom-alkyl substituierte 3-oxo-pyrido(1,2-a)benzimidazol-4-carboxamid derivate zur behandlung zentral nervöser system störungen | |
| AU690906B2 (en) | Novel polycyclic systems, and derivatives thereof, as neurotransmitter release enhancers useful in the treatment of cognitive disoders | |
| DE69827706T2 (de) | 5-(Heteroaryl)alkyl)-3-oxo-pyrido(1,2-a)benzimidazol-4-carboxamid (PBI) Derivate zur Behandlung von Störungen des zentralen Nervensystems | |
| DE3887417T2 (de) | Thienocinnolinverbindungen und deren verwendung als heilmittel. | |
| DE60003550T2 (de) | Substituierte 3-pyridyl-4-arylpyrrole und deren einsatz in verfahren zur therapeutischen und prophylaktischen behandlung | |
| DE69810340T2 (de) | Naphtho-imidazo(1,2-a)pyridin derivate, deren herstellung und deren verwendung in der behandlung von störungen des zentralnervensystems | |
| US5521200A (en) | 2-oxo-pyrrolo[1,2-A]benzimidazole-3-carboxyl derivatives useful in treating central nervous system disorders | |
| DE69413112T2 (de) | 5-HT3 Pyrrolopyrazinderivate | |
| DE60107852T2 (de) | Thienopyrrolidinone | |
| DE60002395T2 (de) | Metalloproteaseinhibitoren, Verfahren zu ihrer Herstellung und diese enthaltende pharmazeutische Zubereitungen | |
| DE68913167T2 (de) | Indol-Derivate, Verfahren zu ihrer Herstellung und sie enthaltende pharmazeutische Präparate. | |
| DE60026848T2 (de) | C-6 ringsubstituierte pyridoä1,2-aübenzimidazolderivate zur behandlung von störungen des zentralen nervensystems |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| 8364 | No opposition during term of opposition | ||
| 8328 | Change in the person/name/address of the agent |
Representative=s name: BOEHMERT & BOEHMERT, 28209 BREMEN |
|
| 8339 | Ceased/non-payment of the annual fee |