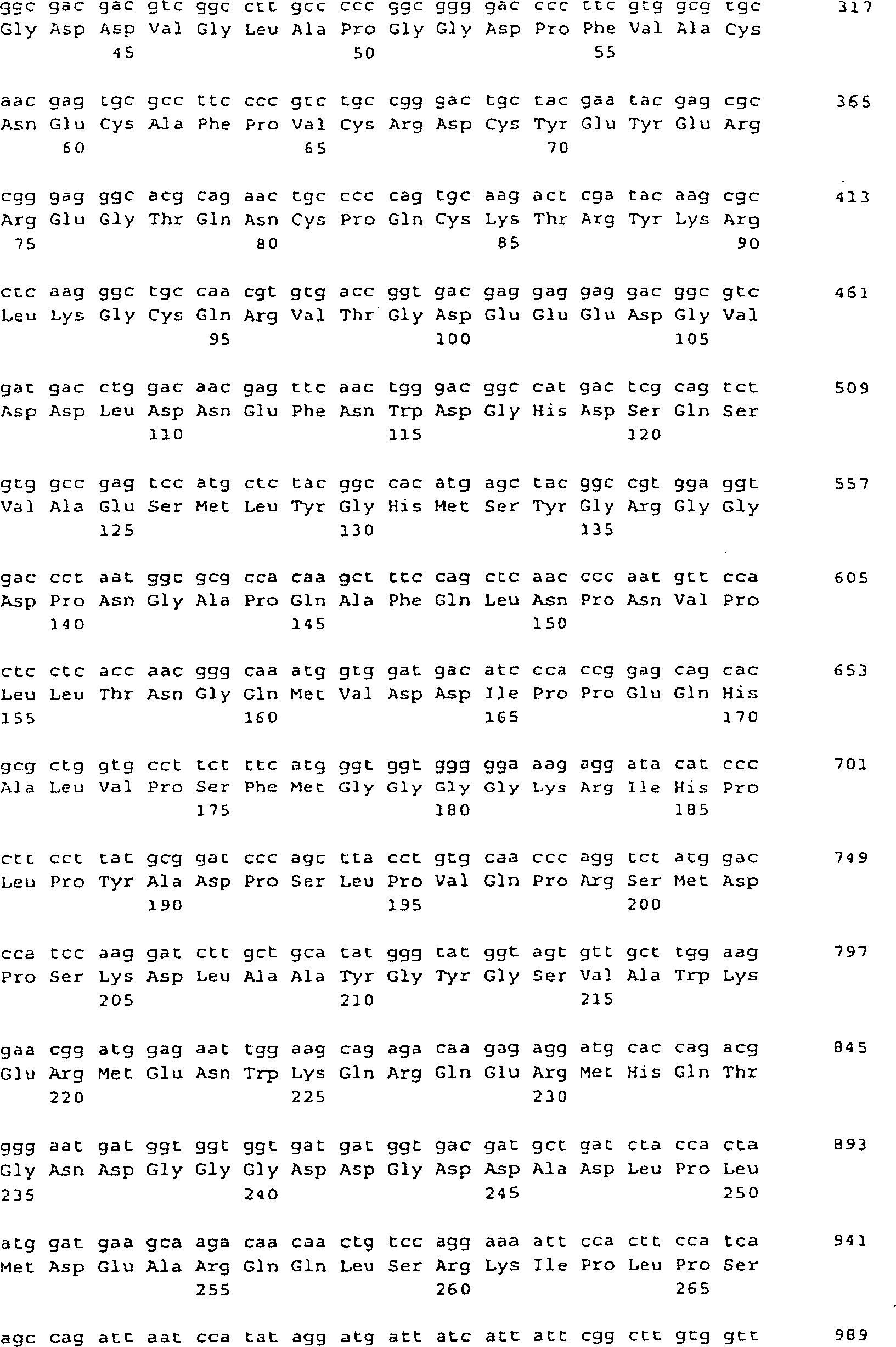
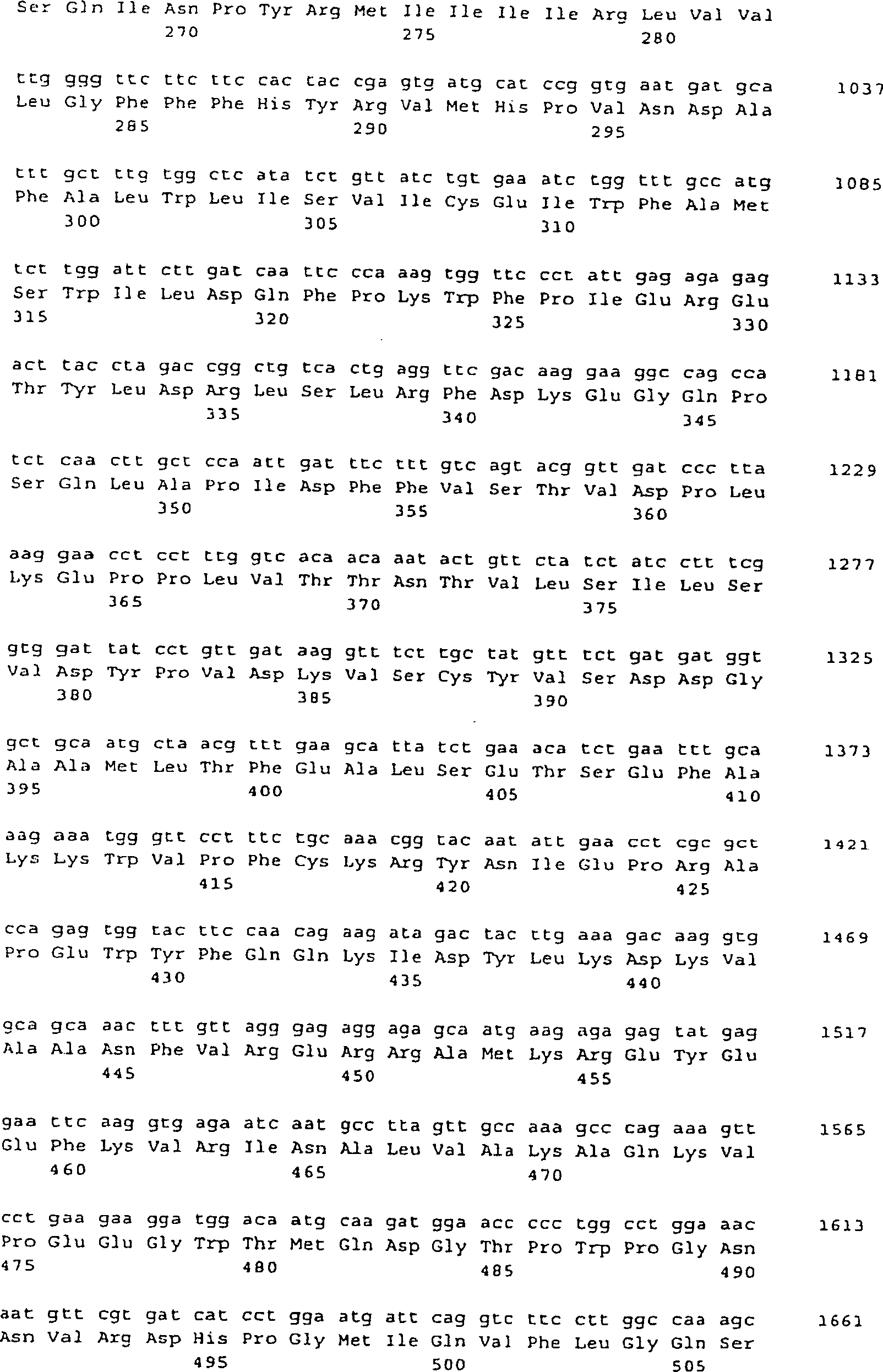
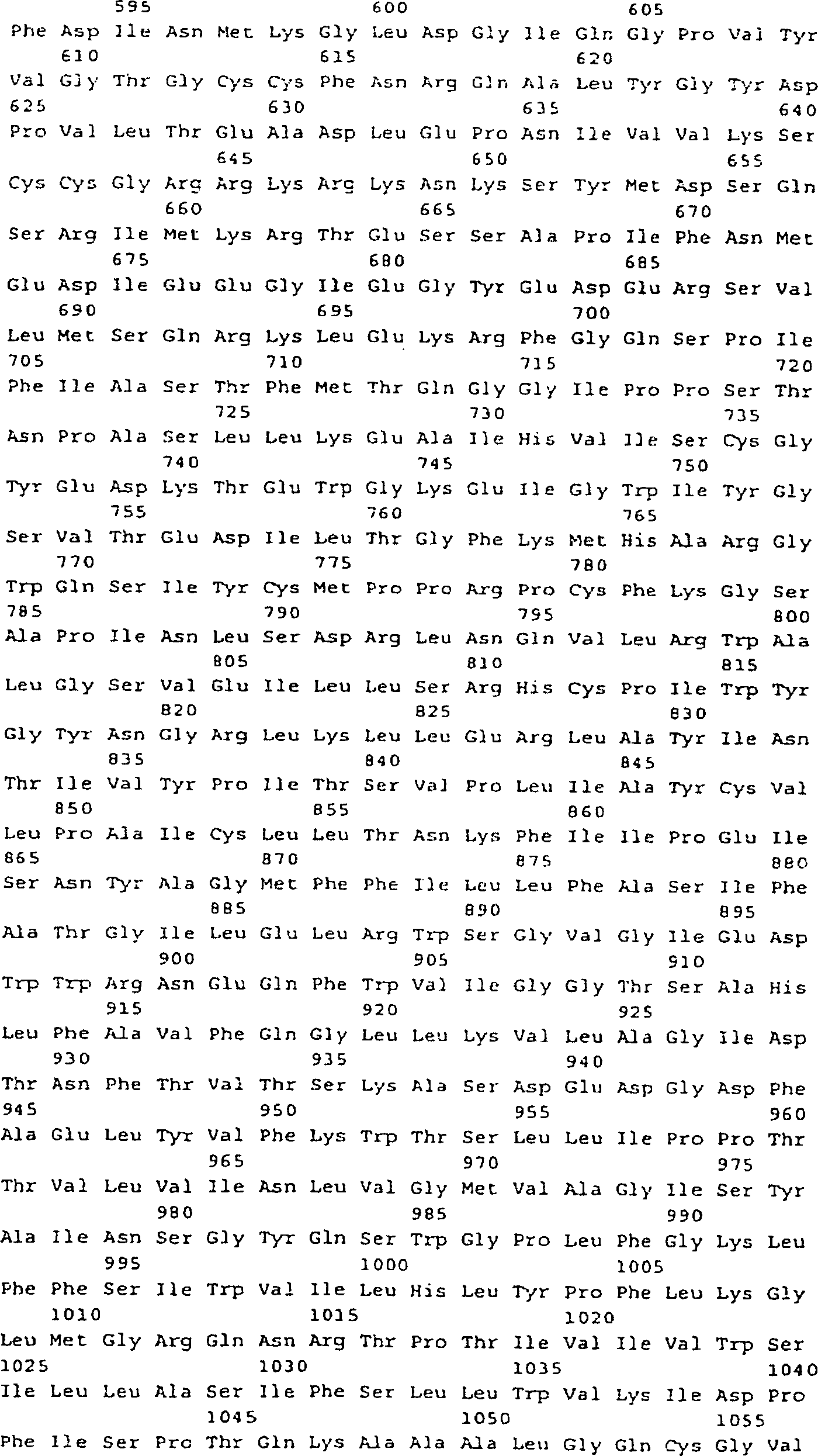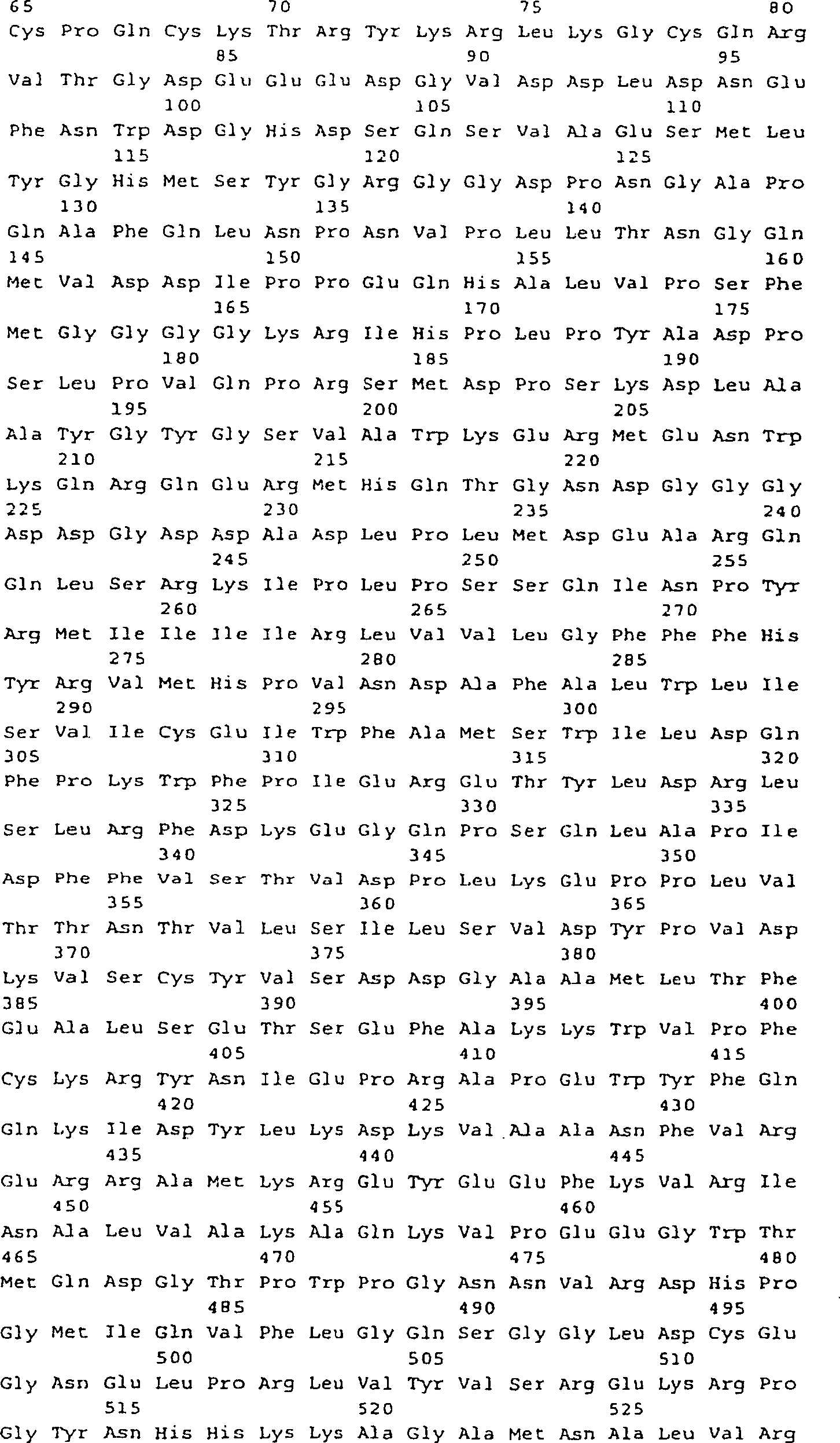-
TECHNISCHES GEBIET
-
Die
vorliegende Erfindung bezieht sich allgemein auf Pflanzenmolekularbiologie.
Sie bezieht sich spezifischer auf Nucleinsäuren und Verfahren zur Modulierung
ihrer Expression in Pflanzen.
-
HINTERGRUND DER ERFINDUNG
-
Polysaccaride
bilden die Masse der Pflanzenzellwände und werden traditionell
in drei Kategorien klassifiziert: Cellulose, Hemicellulose und Pectin.
Fry, S. C. (1988) The growing plant cell wall: Chemical and metabolic
analysis. New York: Longman Scientific & Technical. Während Cellulose an der Plasmamembran
hergestellt wird und direkt in die Zellwand abgelegt wird, werden
Hemicellulose- und Pectinpolymere zuerst im Golgi-Apparat hergestellt
und dann durch Exocytose zu der Zellwand exportiert. Ray, P. M.,
et al., (1976), Ber. Deutsch. Bot. Ges. Bd. 89, 121–146. Die
Vielzahl chemischer Verknüpfungen
in den Pectin- und Hemicellulosepolysacchariden zeigt, dass es im
Golgi-Apparat mehrere zehn Polysaccaridsynthasen geben muss. Darvill et
al., (1980). The primary cell walls of flowering plants. In The
Plant Cell (N. E. Tolbert, Herausg.), Bd. 1 in der Reihe: The biochemistry
of plants: A comprehensive treatise, Herausg., P. K. Stumpf und
E. E. Conn (New York: Academic Press), S. 91–162.
-
Cellulose
hat aufgrund ihrer Fähigkeit,
semikristalline Mikrofibrillen zu bilden, eine sehr hohe Zugfestigkeit,
die an die einiger Metalle heranreicht. Niklas, K. J. (1992). Plant
Biomechanis: An engineering approach to plant form and function,
The University of Chicago Press, S. 607. Es wurde festgestellt,
dass die Biegefestigkeit des Stengels von normaler Gerste und die
von Mutanten mit brüchigem
Stengel in direkter Korrelation mit der Cellulosekonzentration in
der Zellwand steht. Kokubo, et al., (1989), Plant Physiology 91,
876–882;
Kokubo, et al., (1991) Plant Physiology 97, 509–514.
-
Obwohl
Zucker- und Polysaccaridzusammensetzungen der Pflanzenzellwände gut
charakterisiert wurden, wurde ein sehr limitierter Fortschritt in
Richtung der Identifizierung der Enzyme, die bei der Bildung der
Polysaccaride involviert sind, gemacht, wobei der Grund ihre labile
Natur und ihre Widerspenstigkeit gegenüber einer Solubilisierung durch
verfügbare
Detergenzien ist. Gelegentliche Ansprüche auf die Identifizierung
von Cellulosesynthase aus Pflanzenquellen wurden über die
Jahre gestellt. Callaghan, T. und Benziman, M. (1984), Nature 311,
165–167;
Okuda, et al., (1993), Plant Physiol. 101, 1131–1142. Allerdings wurde diesen Ansprüchen mit
Skepsis begegnet. Callaghan, T. und Benziman, M. (1985), Nature
314. 383–384;
Delmer, et al., (1993), Plant Physiol. 103, 307–308. Erst kürzlich wurde
ein vermeintliches Gen für
Pflanzencellulosesynthasen (CelA) aus sich entwickelnden Baumwollfasern,
basierend auf Homolgie zu dem bakteriellen Gen, kloniert. Pear,
et al., Proc. Natl. Acad. Sci. (USA) 93, 12637–12642; Saxena, et al., (1990),
Plant Molecular Biology 15, 673–684;
siehe auch
WO 9818949 .
WO 9800549 stellt auch Gene
bereit, die für
Polypeptide codieren, welche bei der Cellulosebiosynthese in Pflanzen
involviert sind.
-
Ein
brüchiger
Halmknoten ist ein Hauptproblem bei der Getreidezüchtung bzw.
Maiszüchtung,
wobei auf dem Fachgebiet Zusammensetzungen und Verfahren zum Manipulieren
der Cellulosekonzentration in der Zellwand und dadurch Verleihen
von Pflanzenstengelqualität
für verbessertes
Standvermögen
oder verbesserte Silage benötigt
werden. Die vorliegende Erfindung stellt diese und andere Vorteile
bereit.
-
ZUSAMMENFASSUNG DER ERFINDUNG
-
Ganz
allgemein besteht die Aufgabe der vorliegenden Erfindung in der
Bereitstellung von Nucleinsäuren
und Proteinen, die sich auf Cellulosesynthasen beziehen. Eine Aufgabe
der vorliegenden Erfindung ist die Bereitstellung von: 1) Nucleinsäuren und
Proteinen, die sich auf Maiscellulosesynthasen beziehen; 2) transgenen
Pflanzen, die die Nucleinsäuren
der vorliegenden Erfindung umfassen; 3) Verfahren zum Modulieren
einer transgenen Pflanze, der Expression der Nucleinsäuren der
vorliegenden Erfindung.
-
Daher
stellt die vorliegende Erfindung in einem Aspekt eine isolierte
Cellulosesynthase-Nucleinsäure bereit,
die ein Polypeptid mit Cellulosesynthaseaktivität codiert, und umfasst:
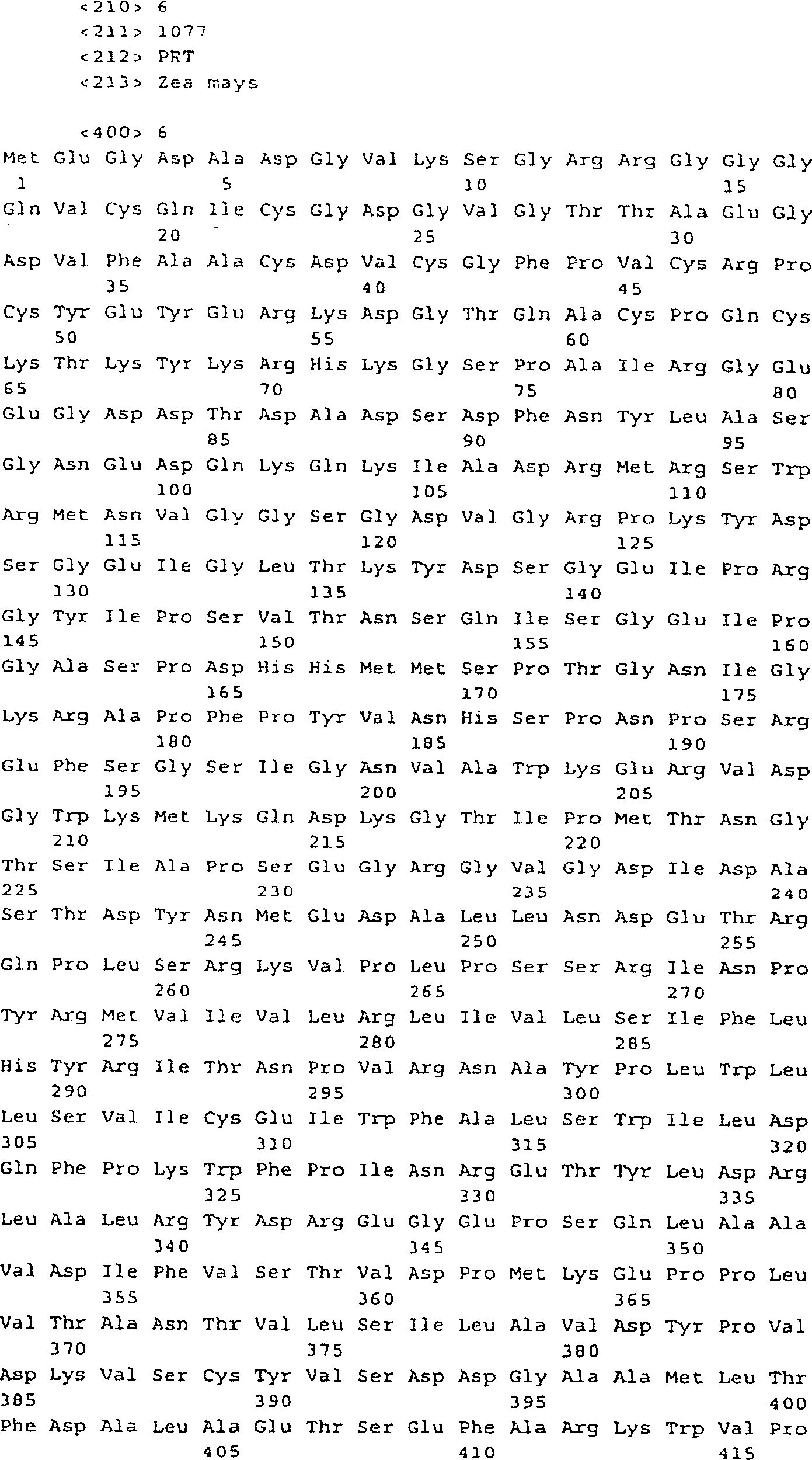
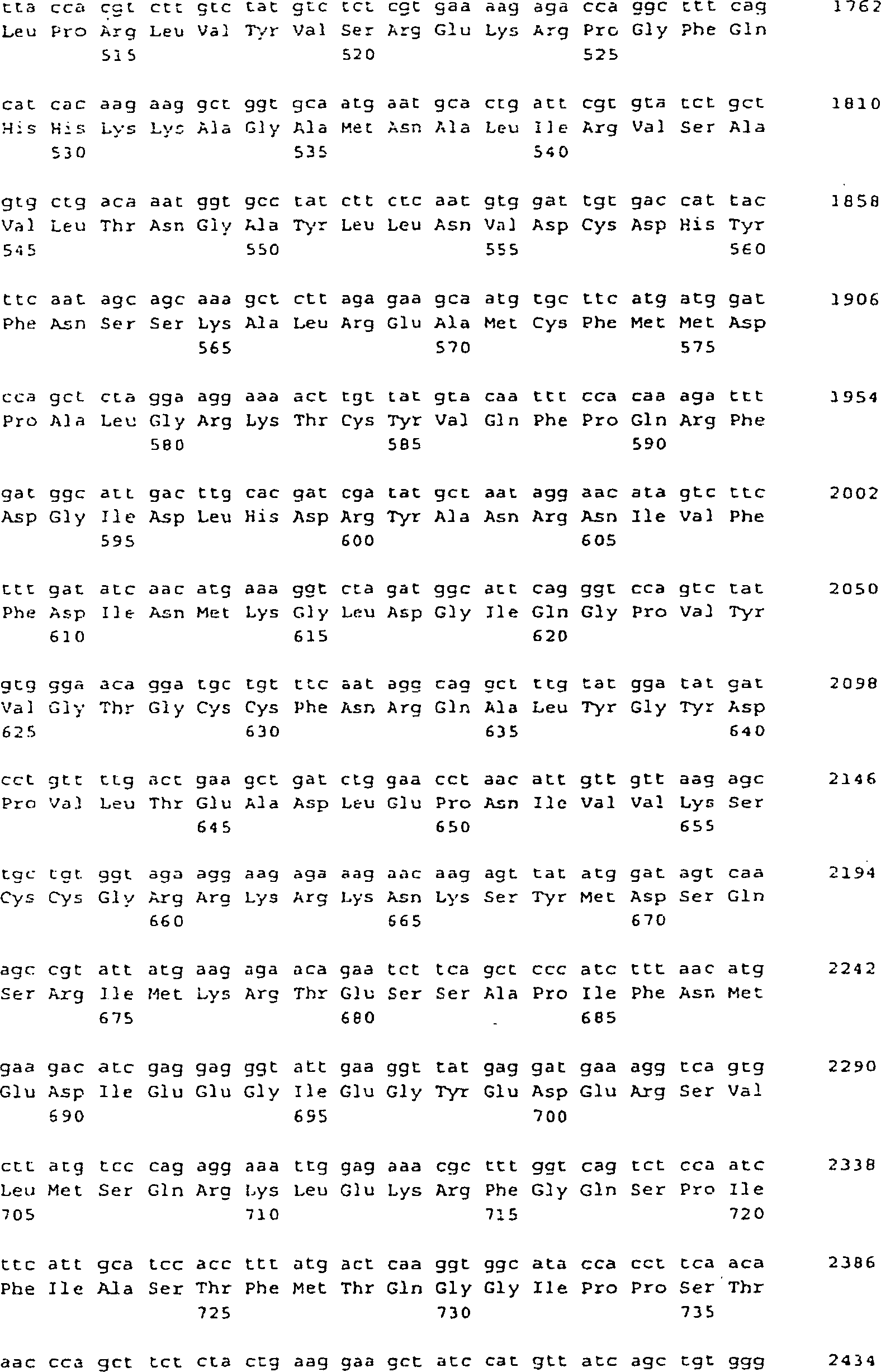
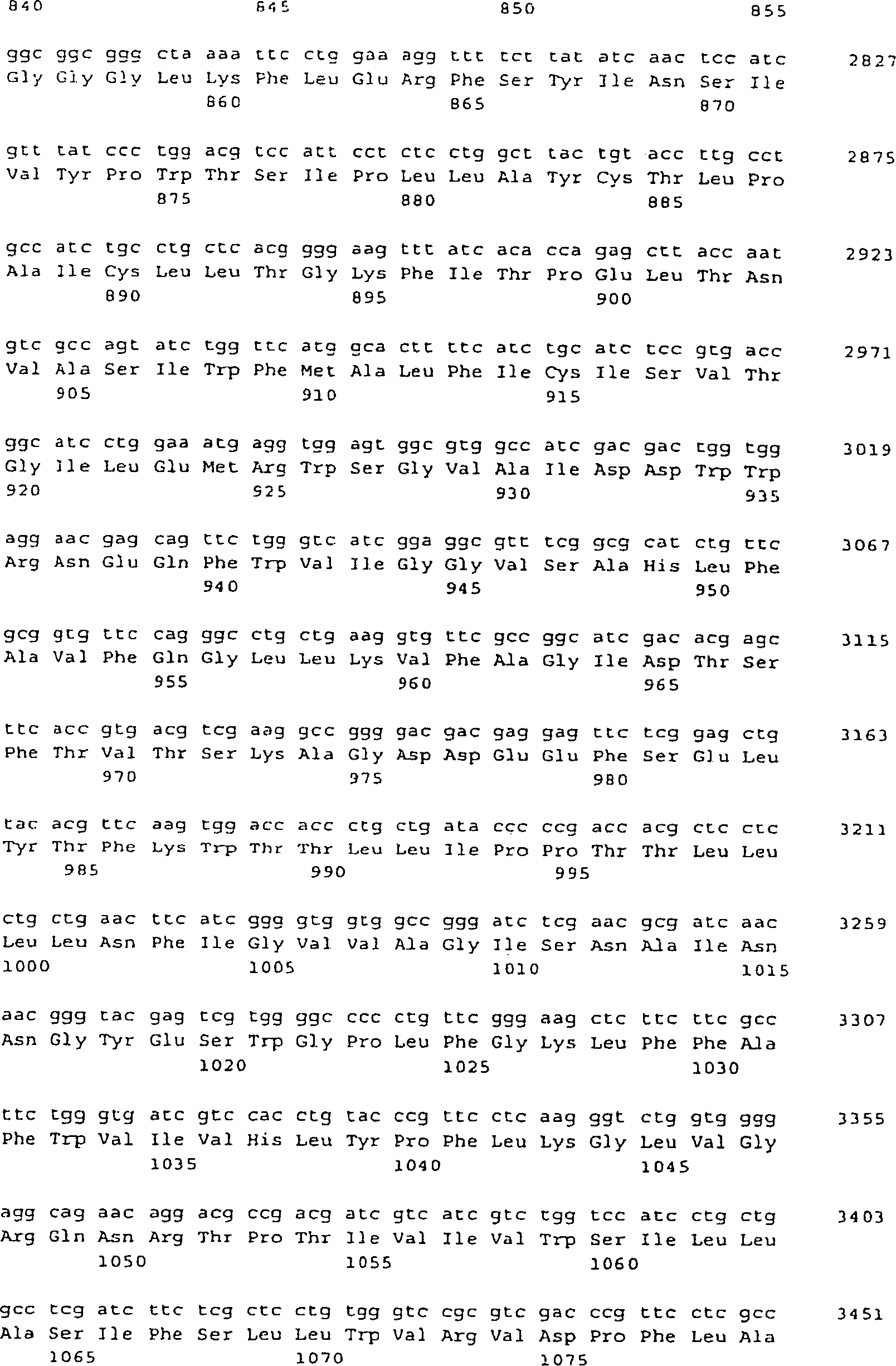
- (a) ein Polynucleotid von SEQ ID NO: 5, 9 oder
25;
- (b) ein Polynucleotid, das ein Polypeptid von SEQ ID NO: 6,
10 oder 26 codiert.
-
Die
isolierte Nucleinsäure
kann DNA oder RNA sein.
-
In
einem anderen Aspekt bezieht sich die vorliegende Erfindung auf
rekombinante Expressionskassetten, die eine Nucleinsäure der
vorliegenden Erfindung funktionell verknüpft mit einem Promotor umfassen.
In einigen Ausführungsformen
ist die Nucleinsäure
funktionell in Antisense-Orientierung mit dem Promotor verknüpft.
-
In
einem anderen Aspekt ist die vorliegende Erfindung auf eine Wirtszelle
gerichtet, die mit der rekombinanten Expressionskassette transfiziert
ist. Vorzugsweise ist die Zelle eine Pflanzenzelle.
-
In
einem weiteren Aspekt umfasst die vorliegende Erfindung ein isoliertes
Protein mit Cellulosesynthaseaktivität und ausgewählt aus:
- (a) einem Polypeptid von SEQ ID NO: 6, 10 oder
26; und
- (b) einem Polypeptid, das durch eine Nucleinsäure der
Erfindung codiert wird.
-
In
noch einem anderen Aspekt bezieht sich die vorliegende Erfindung
auf eine transgene Pflanze, die eine rekombinante Expressionskassette
umfasst, die einen Pflanzenpromotor funktionell verknüpft mit
einer der isolierten Nucleinsäuren
der vorliegenden Erfindung umfasst. In einigen Ausführungsformen
ist die transgene Pflanze Zea mays. Die vorliegende Erfindung stellt
auch Samen aus der transgenen Pflanze bereit.
-
In
einem weiteren Aspekt bezieht sich die vorliegende Erfindung auf
ein Verfahren zum Modulieren der Expression der Gene, die Proteine
der vorliegenden Erfindung codieren, in einer Pflanzenzelle, die
zur Pflanzenregeneration fähig
ist, umfassend die Schritte: (a) Transformieren einer Pflanzenzelle
mit einer rekombinanten Expressionskassette, umfassend ein Polynucleotid
der vorliegenden Erfindung, funktionell verknüpft mit einem Promotor; (b)
Wachsenlassen der Pflanzenzelle unter Pflanzenwachstumsbedingungen;
und (c) Induzieren der Expression des Polynucleotids für eine Zeit,
die ausreicht, um die Expression der Gene in der Pflanze zu modulieren.
In einigen Ausführungsformen
ist die Pflanze Mais. Eine Expression der Gene, die die Proteine
der vorliegenden Erfindung codieren, kann im Vergleich zu einer
nicht-transformierten Kontrollpflanze erhöht oder verringert sein.
-
Definitionen
-
Einheiten,
Präfixe
und Symbole können
in ihrer akzeptierten SI-Form angegeben werden. Wenn nichts anderes
angegeben ist, werden Nucleinsäuren
von links nach rechts in 5'-
zu 3'-Orientierung geschrieben; Aminosäuresequenzen
werden von links nach rechts in Amino-zu-Carboxy-Orientierung geschrieben. Numerische
Bereiche sind für
die Zahlen, die den Bereich definieren, einschließlich und
umfassen jede ganze Zahl innerhalb des definierten Bereichs. Aminosäuren können entweder
durch ihre allgemein bekannten Drei-Buchstaben-Symbole oder durch
die Ein-Buchstaben-Symbole, die durch die IUPAC-IUB-Biochemical
Nomenclature Commission empfohlen sind, benannt werden. Nucleotide
können
entsprechend mit ihren allgemein akzeptierten Ein-Buchstaben-Codes
bezeichnet werden. Wenn nichts anderes angegeben ist, sind Software-, elektrische
und elektronische Begriffe, wie sie hierin verwendet werden, wie
in The New IEEE Standard Dictionary of Electrical and electronics
Terms (5. Ausgabe, 1993) definiert. Die nachfolgend definierten
Begriffe werden unter Bezugnahme auf die Beschreibung als ganzes
vollständiger
definiert.
-
Mit „amplifiziert" ist die Konstruktion
von mehreren Kopien einer Nucleinsäuresequenz oder mehreren Kopien,
die komplementär
zu der Nucleinsäuresequenz
sind, gemeint, wobei wenigstens eine der Nucleinsäuresequenzen
als Matrize verwendet wird. Amplifikationssysteme umfassen das Polymerase-Ketten-Reaktions (PCR)-System,
das Ligase-Kettenreaktions (LCR)-System,
eine Amplifikation auf Nucleinsäurebasis
(NASBA, Cangene, Mississauga, Ontario), Q-Beta-Replikase-Systeme, ein Amplifikationssystem
auf Transkriptionsbasis (TAS) und eine Strand-Displacement-Amplification
(SDA). Siehe z. B., Diagnostic Molecular Microbiology: Principles
and Applications, D. H. Persing et al., Herausg., American Society
for Microbiology, Washington, D. C. (1993). Das Amplifikationsprodukt
wird als ein Amplicon bezeichnet.
-
Der
Ausdruck "Antikörper" umfasst eine Bezugnahme
auf Antigen bindende Formen von Antikörpern (z. B. Fab, F(ab)2). Der Ausdruck „Antikörper" bezieht sich häufig auf ein Polypeptid, das
im Wesentlichen durch ein Immunglobulingen oder durch Immunglobulingene
oder Fragmente davon codiert wird, das spezifisch an einen Analyten
(Antigen) bindet und diesen erkennt. Obgleich verschiedene Antikörperfragmente
bezüglich
des Verdaus eines intakten Antikörpers
definiert werden können,
wird ein Fachmann erkennen, dass solche Fragmente de novo entweder
chemisch oder unter Nutzung der DNA-Rekombinations-Methodologie synthetisiert
werden können.
So umfasst der Ausdruck Antikörper,
wie er hierin verwendet wird, auch Anti körperfragmente, z. B. Einzelketten-Fv,
chimäre
Antikörper
(d. h. die konstante und variable Regionen aus verschiedenen Spezies
umfassen), humanisierte Antikörper
(d. h. die eine Komplementarität
bestimmende Region (CDR) aus einer nicht-humanen Quelle umfassen)
und Heterokonjugat-Antikörper
(z. B. bispezifische Antikörper).
-
Der
Ausdruck „Antigen" umfasst die Bezeichnung
einer Substanz, gegen die ein Antikörper erzeugt werden kann und/oder
gegen die der Antikörper
spezifisch immunreaktiv ist. Die spezifischen immunreaktiven Stellen
im Antigen sind als Epitope oder antigene Determinanten bekannt.
Diese Epitope können
eine lineare Anordnung von Monomeren in einer polymeren Zusammensetzung
sein – z.
B. Aminosäuren
in einem Protein – oder
aus einer komplexeren Sekundär-
oder Tertiärstruktur
bestehen oder diese umfassen. Der Fachmann wird erkennen, dass alle
Immunogene (d. h. Substanzen, die fähig sind, eine Immunantwort
auszulösen)
Antigene sind; allerdings sind einige Antigene, z. B. Haptene, nicht
immunogen, können
aber durch Kupplung an ein Trägermolekül immunogen
gemacht werden. Ein Antikörper,
der immunologisch reaktiv mit einem bestimmten Antigen ist, kann
in vivo erzeugt werden oder durch rekombinante Verfahren, z. B.
Selektion von Bibliotheken rekombinanter Antikörper in Phagen oder ähnlichen
Vektoren, erzeugt werden. Siehe z. B. Huse et al., Science 246:
1275–1281
(1989); und Ward, et al., Nature 341: 544–546 (1989); und Vaughan et
al., Nature Biotech. 14: 309–314
(1996).
-
„Antisense-Orientierung" bzw. „Antisinn-Orientierung", wie der Begriff
hierin verwendet wird, umfasst eine Bezugnahme auf eine Duplex-Polynucleotidsequenz,
die funktionell mit einem Promotor in einer Orientierung verknüpft ist,
in der der Antisense-Strang transkribiert wird. Der Antisense-Strang
ist ausreichend komplementär
zu einem endogenen Transkriptionsprodukt, so dass eine Translation
des endogenen Transkriptionsproduktes oft inhibiert wird.
-
Der
Ausdruck „chromosomale
Region", wie er
hierin verwendet wird, umfasst eine Bezugnahme auf eine Länge eines
Chromosoms, die durch Bezugnahme auf das lineare DNA-Segment, das es umfasst,
gemessen werden kann. Die chromosomale Region kann durch Bezugnahme
auf zwei einzigartige DNA-Sequenzen, d. h. Marker, definiert werden.
-
Der
Ausdruck „konservativ
modifizierte Varianten" findet
sowohl auf Aminosäure-
als auch auf Nucleinsäuresequenzen
Anwendung. Für
bestimmte Nucleinsäuresequenzen
bezeichnen konservativ modifizierte Varianten solche Nucleinsäuren, die
für identische
oder konservativ modifizierte Varianten der Aminosäuresequenzen
codieren. Wegen der Degeneriertheit des genetischen Codes codiert
eine große
Zahl von funktionell identischen Nucleinsäuren für ein gegebenes Protein. Zum
Beispiel codieren die Codons GCA, GCC, GCG und GCU alle für die Aminosäure Alanin.
Demnach kann in jeder Position, in der ein Alanin durch ein Codon spezifiziert
wird, das Codon in ein beliebiges der entsprechenden Codons verändert werden,
ohne dass das codierte Polypeptid verändert wird. Solche Nucleinsäurevariationen
sind „stumme
Variationen" und
stellen eine Spezies einer konservativ modifizierten Variation dar.
Jede Nucleinsäuresequenz,
die für
ein Polypeptid codiert, beschreibt hierin auch jegliche mögliche stumme
Variation der Nucleinsäure.
Ein Fachmann wird erkennen, dass jedes Codon in einer Nucleinsäure (außer AUG,
das normalerweise das einzige Codon für Methionin ist, und UGG, das
normalerweise das einzige Codon für Tryptophan ist) modifiziert
werden kann, um ein funktionell identisches Molekül zu ergeben.
Dementsprechend ist jede stumme Variation einer Nucleinsäure, die für ein Polypeptid
der vorliegenden Erfindung codiert, in jeder beschriebenen Polypeptidsequenz
implizit und hierin durch Bezugnahme aufgenommen.
-
Was
Aminosäuresequenzen
angeht, wird ein Fachmann erkennen, dass einzelne Substitutionen,
Deletionen oder Additionen an einer Nucleinsäure-, Peptid-, Polypeptid-
oder Proteinsequenz, die eine einzelne Aminosäure oder einen geringen prozentualen
Anteil an Aminosäuren
in der codierten Sequenz verändert,
addiert oder deletiert, eine „konservativ
modifizierte Variante",
in der die Veränderung
in der Substitution einer Aminosäure
durch eine chemisch ähnlich
Aminosäure
resultiert. Demnach kann eine beliebige Anzahl von Aminosäureresten,
ausgewählt
aus der Gruppe von ganzen Zahlen, bestehend von 1 bis 15, verändert werden.
So können
z. B. 1, 2, 3, 4, 5, 7 oder 10 Veränderungen durchgeführt werden.
Konservativ modifizierte Varianten stellen typischerweise eine ähnliche
biologische Aktivität
wie die unmodifizierte Polypeptidsequenz, von der sie abgeleitet
sind, bereit. Zum Beispiel ist die Substratspezifität, Enzymaktivität oder Ligand/Rezeptor-Bindung
im Allgemeinen wenigstens 30%, 40%, 50%, 60%, 70%, 80% oder 90%
des nativen Proteins für sein
natives Substrat. Tabellen über
konservative Substitutionen, die funktionell ähnliche Aminosäuren bereitstellen,
sind auf dem Fachgebiet gut bekannt.
-
Die
folgenden sechs Gruppen enthalten jeweils Aminosäuren, die konservative Substitutionen
füreinander
sind:
- 1) Alanin (A), Serin (S), Threonin (T);
- 2) Asparaginsäure
(D), Glutaminsäure
(E);
- 3) Asparagin (N), Glutamin (Q);
- 4) Arginin (R), Lysin (K):
- 5) Isoleucin (I), Leucin (L), Methionin (M), Valin (V); und
- 6) Phenylalanin (F), Tyrosin (Y), Tryptophan (W).
-
Siehe
auch Creighton (1984) Proteins W. H. Freeman and Company.
-
Mit "codierend" oder "codiert" ist bei einer spezifizierten
Nucleinsäure
gemeint, umfassend die Information zur Translation in das spezifizierte
Protein. Eine Nucleinsäure,
die ein Protein codiert, kann nicht-translatierte Sequenzen (z.
B. Introns) innerhalb translatierter Regionen der Nucleinsäure umfassen
oder ein Fehlen solcher intervenierender nicht-translatierter Sequenzen
aufweisen (z. B. in cDNA). Die Information, durch welche ein Protein
codiert wird, wird durch die Verwendung von Codons spezifiziert.
Typischerweise ist die Aminosäuresequenz
durch die Nucleinsäure
unter Verwendung des „universellen" genetischen Codes
codiert. Allerdings können
Varianten des universellen Codes, wie sie in einigen Pflanzen-,
Tier- und Pilzmitochondrien, dem Bacterium Mycoplasma capricolum
(Proc. Natl. Acad. Sci. (USA), 82: 2306–2309 (1985)) oder der Ziliate Macronucleus
vorliegen, verwendet werden, wenn die Nucleinsäure unter Verwendung dieser
Organismen exprimiert wird.
-
Wenn
die Nucleinsäure
synthetisch hergestellt oder verändert
wird, können
Vorteile aus bekannten Codonpräferenzen
des vorgesehenen Wirts gezogen werden, in dem die Nucleinsäure exprimiert
werden soll. Obgleich Nucleinsäuresequenzen
der vorliegenden Erfindung sowohl in Monocotylen- als auch Dicotylen-Pflanzenspezies
exprimiert werden können,
können
Sequenzen so modifiziert werden, dass sie die spezifischen Codonpräferenzen
und GC-Gehalt-Präferenzen
von Monocotylen oder Dicotylen berücksichtigen, da sich diese
Präferenzen
als verschieden erwiesen haben (Murray et al. Nucl. Acids Res. 17:
477–498
(1989)). So kann das von Mais bevorzugte Codon für eine bestimmte Aminosäure aus
bekannten Gensequenzen aus Mais abgeleitet werden. Die Mais-Codonverwendung
für 28
Gene aus Maispflanzen ist in Tabelle 4 von Murray et al., oben,
aufgelistet.
-
Der
Ausdruck „Volllängen-Sequenz", wie er hierin verwendet
wird, bedeutet unter Bezugnahme auf ein spezifiziertes Polynucleotid
oder sein codiertes Protein die gesamte Aminosäuresequenz einer nativen (nicht-synthetischen),
endogenen, katalytisch aktiven Form des spezifizierten Proteins
aufweisend. Verfahren, um zu bestimmen, ob eine Sequenz eine Volllängen-Sequenz ist, sind
auf dem Fachgebiet gut bekannt; sie umfassen z. B. Techniken, wie
Northern- oder Western-Blots,
Primer-Verlängerung,
S1-Schutz und Ribonukleaseschutz. Siehe z. B. Plant Molecular Biology:
A Laboratory Manual, Clark, Herausg., Springer-Verlag, Berlin (1997).
Ein Vergleich mit bekannten homologen (orthologen und/oder paralogen)
Volllängen-Sequenzen kann
ebenfalls verwendet werden, um Volllängen-Sequenzen der vorliegenden
Erfindung zu identifizieren. Zusätzlich
unterstützen
Konsensus-Sequenzen, die typischerweise an den 5'- und 3'-untranslatierten
Regionen von mRNA vorliegen, die Identifizierung eines Polynucleotids
als Volllängen-Polynucleotid.
Zum Beispiel unterstützt
die Konsensus-Sequenz ANNNNAUGG,
in der das unterstrichene Codon das N-terminale Methionin darstellt,
die Bestimmung, ob das Polynucleotid ein vollständiges 5'-Ende hat. Konsensus-Sequenzen am 3'-Ende, z. B. Polyadenylierungssequenzen,
unterstützen
die Bestimmung, ob das Polynucleotid ein vollständiges 3'-Ende hat. Der Ausdruck „Genaktivität" bezieht sich auf
einen Schritt oder mehrere Schritte, der/die bei der Genexpresssion
involviert ist/sind, einschließlich
Transkription, Translation, und auch das Funktionieren des Proteins,
das durch das Gen codiert wird.
-
„Heterolog", wie der Ausdruck
für eine
Nucleinsäure
verwendet wird, ist eine Nucleinsäure, die von einer fremden
Spezies stammt oder, wenn sie von derselben Spezies stammt, im Vergleich
zu ihrer nativen Form in Zusammensetzung und/oder genomischem Ort
durch einen willkürlichen
menschlichen Eingriff wesentlich modifiziert ist. Zum Beispiel stammt
ein Promotor, der funktionell mit einem heterologen Strukturgen
verknüpft ist,
aus einer anderen Spezies als die, aus der das Strukturgen stammt,
oder, wenn sie aus der gleichen Spezies stammen, ist einer oder
beide im Vergleich zu ihrer ursprünglichen Form wesentlich modifiziert.
Ein heterologes Protein kann aus einer fremden Spezies stammen oder,
wenn es aus derselben Spezies stammt, ist es durch einen willkürlichen
menschlichen Eingriff im Vergleich zu seiner ursprünglichen
Form wesentlich modifiziert.
-
Mit „Wirtszelle" ist eine Zelle gemeint,
die einen Vektor enthält
und die Replikation und/oder Expression des Expressionsvektors stützt. Wirtszellen
können
prokaryotische Zellen, z. B. E. coli, oder eukaryotische Zellen,
z. B. Hefe-, Insekten-, Amphibien- oder Säugerzellen, sein. Wirtszellen
sind vorzugsweise Zellen einer monocotylen oder dicotylen Pflanze.
Eine besonders bevorzugte monocotyle Wirtszelle ist eine Mais-Wirtszelle.
-
Der
Ausdruck „Hybridisierungskomplex" umfasst die Bezugnahme
auf eine Duplex-Nucleinsäurestruktur,
die durch zwei einzelsträngige
Nucleinsäuresequenzen,
die selektiv miteinander hybridisiert sind, gebildet wird.
-
Mit „immunologisch
reaktive Bedingungen" oder „immunreaktive
Bedingungen" sind
Bedingungen gemeint, die es einem Antikörper, der gegen ein bestimmtes
Epitop erzeugt wurde, erlauben an das Epitop zu binden, und zwar
zu einem detektierbar höheren
Grad (z. B. wenigstens 2-fach stärker
als der Hintergrund) als der Antikörper an im Wesentlichen alle
anderen Epitope in einem Reaktiongemisch, das das bestimmte Epitop umfasst,
bindet. Immunologisch reaktive Bedingungen sind von dem Format der
Antikörperbindungsreaktion abhängig und
typischerweise sind sie solche, wie sie in Immunoassayprotokollen
verwendet werden. Siehe Harlow und Lane, Antibodies, A Laboratory
Manual, Cold Spring Harbor Publications, New York (1988), bezüglich einer
Beschreibung von Immunoassay-Formaten und -Bedingungen.
-
Der
Ausdruck „eingeführt" bedeutet im Kontext
mit Insertieren einer Nucleinsäure
in eine Zelle „Transfektion" oder „Transformation" oder „Transduktion" und umfasst eine
Bezugnahme auf einen Einbau einer Nucleinsäure in eine eukaryotische oder
prokaryotische Zelle, wo die Nucleinsäure in das Genom der Zelle
(z. B. Chromosom, Plasmid, Plastid oder Mitochondrien-DNA) eingebaut
werden kann, in ein autonomes Replikon umgewandelt werden kann oder
transient exprimiert werden kann (z. B. transfizierte mRNA).
-
Der
Ausdruck „isoliert" bezieht sich auf
Material, z. B. eine Nucleinsäure
oder ein Protein, das ist: (1) substantiell oder im Wesentlichen
frei von Komponenten, die normalerweise mit ihm assoziiert sind
oder mit ihm wechselwirken, wenn es in seiner natürlich vorkommenden
Umgebung gefunden wird. Das isolierte Material umfasst gegebenenfalls
Material, das mit dem Material in seiner natürlichen Umgebung nicht gefunden wird;
oder (2) wenn das Material in seiner natürlichen Umgebung ist, wurde
das Material synthetisch (nicht-natürlich) durch einen willkürlichen
menschlichen Eingriff bezüglich
der Zusammensetzung verändert
und/oder an eine Stelle in der Zelle (z. B. Genom oder subcelluläre Organelle)
platziert, die für
das in jener Umgebung gefundene Material nicht nativ ist. Die Veränderung
unter Erhalt des synthetischen Materials kann an dem Material in
seinem natürlichen
Zustand oder entfernt aus seinem natürlichen Zustand durchgeführt werden.
Zum Beispiel wird eine natürlich
vorkommende Nucleinsäure
eine isolierte Nucleinsäure,
wenn sie verändert
wird oder wenn sie aus DNA, die durch nicht-natürliche, synthetische (d. h. „von Menschen
gemachte") Verfahren, die
innerhalb der Zelle, aus der sie stammt, durchgeführt wurden,
verändert
wurde, transkribiert wird. Siehe z. B. Compounds and Methods for
Site Directed Mutagenesis in Eukaryotic Cells, Kmiec,
U.S. Patent Nr. 5,565,350 ; In Vivo Homologous
Sequence Targeting in Eukaryotic Cells; Zarling et al.,
PCT/US93/03868 . Entsprechend
wird eine natürlich
vorkommende Nucleinsäure
(z. B. ein Promotor) isoliert, wenn er durch nicht natürlich vorkommende
Mittel an eine Stelle des Genoms eingeführt wird, die für jene Nucleinsäure nicht
nativ ist. Nucleinsäuren,
die „isoliert" sind, wie der Begriff
hierin definiert ist, werden auch als „heterologe" Nucleinsäuren bezeichnet.
-
Wenn
nichts anderes angegeben ist, bezeichnet der Ausdruck „Cellulosesynthase-Nucleinsäure" eine Nucleinsäure der
vorliegenden Erfindung und meint eine Nucleinsäure, die ein Polynucleotid
der vorliegenden Erfindung (ein „Cellulosesynthase-Polynucleotid") umfasst, das für ein Cellulosesynthase-Polypeptid
codiert. Ein „Cellulosesynthase-Gen" ist ein Gen der
vorliegenden Erfindung und bezieht sich auf eine nicht-heterologe genomische
Form eines Volllängen-Cellulosesynthase-Polynucleotids.
-
So
wie der Ausdruck „lokalisiert
innerhalb der chromosomalen Region, definiert durch und umfassend" für bestimmte
Marker verwendet wird, bezieht er sich auf eine fortlaufende Länge eines
Chromosoms, die durch die angegebenen Marker begrenzt wird und diese
beinhaltet.
-
„Marker", wie der Ausdruck
hierin verwendet wird, bezieht sich auf einen Locus auf einem Chromosom,
der dazu dient, eine einzigartige Position auf dem Chromosom zu
identifizieren. Ein „polymorpher
Marker" umfasst
eine Bezugnahme auf einen Marker, der in mehreren Formen (Allelen)
auftritt, so dass unterschiedliche Formen des Markers, wenn sie
in einem homologen Paar vorliegen, die Übertragung von jedem der Chromosome
in das Paar erlauben, das verfolgt werden soll. Ein Genotyp kann
durch die Verwendung eines Markers oder einer Vielzahl von Markern
definiert werden.
-
Wie
der Ausdruck „Nucleinsäure" hierin verwendet
wird, bezieht er sich auf ein Desoxyribonucleotid- oder Ribonucleotidpolymer,
entweder in einzelsträngiger
oder doppelsträngiger
Form und, wenn nichts anderes angegeben ist, umfasst er Analoga,
die die essentielle Natur von natürlichen Nucleotiden dahingehend
haben, dass sie in ähnlicher
Weise wie natürlich
vorkommende Nucleotide an einzelsträngige Nucleinsäuren hybridisieren
(z. B. Peptid-Nucleinsäuren).
-
Mit „Nucleinsäurebibliothek" ist eine Sammlung
von isolierten DNA- oder RNA-Molekülen gemeint,
die die gesamte transkribierte Fraktion eines Genoms eines spezifizierten
Organismus umfasst und im Wesentlichen darstellt. Die Konstruktion
von beispielhaften Nucleinsäurebibliotheken,
z. B. genomischen und cDNA-Bibliotheken, wird in Standardwerken
der Molekularbiologie gelehrt, z. B. in Berger und Kimmel, Guide
to Molecular Cloning Techniques, Methods in Enzymology, Bd. 152,
Academic Press, Inc., San Diego, CA (Berger); Sambrook et al., Molecular
Cloning – A
Laboratory Manual, 2. Ausgabe, Band 1–3 (1989); und Current Protocols
in Molecular Biology, F. M. Ausubel et al., Herausg., Current Protocols,
ein Gemeinschaftsunternehmen zwischen Greene Publishing Associates,
Inc. und John Wiley & Sons,
Inc. (1994 Ergänzung).
-
Der
Ausdruck „funktionell
verknüpft", wie er hierin verwendet
wird, bezieht sich auf eine funktionelle Verknüpfung zwischen einem Promotor
und einer zweiten Sequenz, wobei die Pro motorsequenz eine Transkription
der DNA-Sequenz, die der zweiten Sequenz entspricht, initiiert und
vermittelt. Im Allgemeinen bedeutet funktionell verknüpft, dass
die Nucleinsäuresequenzen
fortlaufend verknüpft
sind und, wenn notwendig, um zwei Protein-codierende Regionen zu
verbinden, fortlaufend und in demselben Leseraster sind.
-
Der
Ausdruck „Pflanze", wie er hierin verwendet
wird, bezieht sich auf ganze Pflanzen, Pflanzenteile oder Organe
(z. B. Blätter,
Stengel, Wurzeln usw.), Pflanzenzellen, Samen und die Nachkommenschaft
derselben. Eine Pflanzenzelle, wie der Ausdruck hierin verwendet
wird, umfasst ohne Beschränkung
Zellen, die erhalten werden aus oder gefunden werden in: Samen,
Suspensionskulturen, Embryos, Merystemregionen, Callusgewebe, Blättern, Wurzeln,
Schösslingen,
Gametophyten, Sporophyten, Pollen und Mikrosporen. Pflanzenzellen
können
auch so verstanden werden, dass sie modifizierte Zellen, z. B. Protoplasten,
erhalten aus den vorher genannten Geweben, umfassen. Die Klasse
von Pflanzen, die in den Verfahren der Erfindung eingesetzt werden
kann, ist im Allgemeinen so breit wie die Klasse höherer Pflanzen,
die Transformationstechniken zugänglich
ist, einschließlich
monocotyler und dicotyler Pflanzen. Besonders bevorzugte Pflanzen
umfassen Mais, Sojabohnen, Sonnenblumen, Sorghum, Canola, Weizen,
Alfalfa, Baumwolle, Reis, Gerste und Hirse.
-
„Polynucleotid", wie der Ausdruck
hierin verwendet wird, bezieht sich auf ein Desoxyribopolynucleotid, Ribopolynucleotid
oder Analoga davon, die die essentielle Natur eines natürlichen
Ribonucleotids dahingehend haben, dass sie unter stringenten Hybridisierungsbedingungen
an im Wesentlichen dieselbe Nucleotidsequenz wie natürlich vorkommende
Nucleotide hybridisieren und/oder eine Translation in dieselbe(n)
Aminosäure(n)
wie das natürlich
vorkommende Nucleotid (die natürlich
vorkommenden Nucleotide) erlauben. Ein Polynucleotid kann ein Volllängen-Polynucleotid
oder eine Untersequenz eines nativen oder heterologen strukturellen
oder regulatorischen Gens sein. Wenn nichts anderes angegeben ist,
bezieht sich der Ausdruck auf die spezifizierte Sequenz sowie die
komplementäre
Sequenz derselben. Somit sind DNAs oder RNAs mit Grundgerüsten, die
aus Stabilitätsgründen oder
anderen Gründen
modifiziert sind, „Polynucleotide", wie der Ausdruck
hierin bestimmt ist. Darüber
hinaus sind DNAs oder RNAs, die ungewöhnliche Basen, z. B. Inosin,
oder modifizierte Basen, z. B. tritylierte Basen, um gerade zwei
Beispiele zu nennen, Polynucleotide, wie der Ausdruck hierin verwendet
wird. Es wird einzusehen sein, dass eine große Vielzahl von Modifikationen
an DNA und RNA durchgeführt
wurde, damit diese Ziele nützliche
Zwecke erfüllen,
wie es dem Fachmann bekannt ist. Der Ausdruck Polynucleotid, wie
er hierin verwendet wird, umfasst solche chemisch, enzymatisch oder
metabolisch modifizierten Formen von Polynucleotiden sowie die chemischen
Formen von DNA und RNA, die für Viren
und Zellen, einschließlich
unter anderem einfache und komplexe Zellen, charakteristisch sind.
-
Die
Ausdrücke „Polypeptid", „Peptid" und „Protein" werden hierin austauschbar
verwendet und beziehen sich auf ein Polymer aus Aminosäureresten.
Die Ausdrücke
finden Anwendung auf Aminosäurepolymere, in
denen ein Aminosäurerest
oder mehrere Aminosäurereste
ein künst liches
chemisches Analogon einer entsprechenden natürlich vorkommenden Aminosäure ist/sind,
sowie auf natürlich
vorkommende Aminosäurepolymere.
Die essentielle Natur solcher Analoga von natürlich vorkommenden Aminosäuren ist,
dass, wenn sie in ein Protein eingebaut sind, dieses Protein spezifisch
reaktiv gegenüber
Antikörpern
ist, die durch dasselbe Protein, das aber ganz aus natürlich vorkommenden
Aminosäuren
besteht, erzeugt werden. Die Ausdrücke „Polypeptid", „Peptid" und „Protein" schließen auch
Modifikationen, einschließlich,
aber nicht beschränkt
auf, Glycosylierung, Lipidanheftung, Sulfatierung, gamma-Carboxylierung
von Glutaminsäureresten,
Hydroxylierung und ADP-Ribosylierung, ein. Beispiele für Modifikationen
sind in den meisten Basiswerken, z. B. Proteins – Structure and Molecular Properties,
2. Ausg., T. E. Creighton, W. H. Freeman and Company, New York (1993) beschrieben. Über dieses
Thema gibt es viele detaillierte Übersichtsartikel, z. B. Wold,
F., Post-Translational Protein Modifications: Perspectives and Prospects,
S. 1–12
in Posttranslational Covalent Modification of Proteins, B. C. Johnson,
Herausg., Academic Press, New York (1983); Seifter et al., Meth.
Enzymol. 182: 626–646 (1990)
und Rattan et al., Protein Synthesis: Posttranslational Modifications
and Aging, Ann. N. Y. Acad. Sci. 663: 48–62 (1992). Es wird einzusehen
sein, wie es auch bekannt und oben angegeben wurde, dass Polypeptide
nicht immer ganz linear sind. Zum Beispiel können Polypeptide als Resultat
einer Ubiquitinierung verzweigt sein und können mit oder ohne Verzweigung
ringförmig
sein, im Allgemeinen als Resultat von Posttranslationsereignissen,
einschließlich
eines natürlichen
Prozessierungsereignisses und Ereignissen, die durch Manipulation
durch den Menschen entstanden sind, die nicht natürlich vorkommen.
Ringförmige,
verzweigte und verzweigte ringförmige
Polypeptide können
durch einen natürlichen
Nicht-Translations-Prozess und auch durch vollständig synthetische Verfahren
synthetisiert werden. Modifikationen können irgendwo in einem Polypeptid,
einschließlich
der Peptidhauptkette, der Aminosäureseitenketten
und der Amino- oder Carboxyl-Termini, auftreten. In der Tat ist
eine Blockierung der Amino- oder Carboxylgruppe in einem Polypeptid
oder beider durch eine kovalente Modifikation bei natürlich vorkommenden
und synthetischen Polypeptiden üblich
und solche Modifikationen können
ebenfalls in Polypeptiden der vorliegenden Erfindung vorliegen.
Zum Beispiel wird der aminoterminale Rest von Polypeptiden, die
in E. coli oder anderen Zellen hergestellt werden, vor einer proteolytischen
Prozessierung fast gleich bleibend N-Formylmethionin sein. Während einer
posttranslationalen Modifikation des Peptids kann ein Methioninrest
an dem NH2-Terminus deletiert werden. Dementsprechend zieht
diese Erfindung die Verwendung sowohl der Varianten des Proteins
der Erfindung mit Methionin am Aminoterminus als auch ohne Methionin
am Aminoterminus in Betracht. Im Allgemeinen umfasst der Ausdruck
Polypeptid, wie er hierin verwendet wird, alle derartigen Modifikationen,
insbesondere solche, die in Polypeptiden vorliegen, die durch Exprimieren
eines Polynucleotids in einer Wirtszelle synthetisiert werden.
-
Der
Ausdruck „Promotor", wie er hierin verwendet
wird, bezieht sich auf eine DNA-Region
stromaufwärts
vom Transkriptionsstart, die bei der Erkennung und Bindung von RNA-Polymerase und anderen
Proteinen unter Initiierung der Transkription involviert ist. Ein „Pflan zenpromotor" ist ein Promotor,
der fähig
ist, eine Transkription in Pflanzenzellen zu initiieren. Beispiele
für Pflanzenpromotoren
umfassen, sind aber nicht beschränkt
auf, solche, die aus Pflanzen, Pflanzenviren und Bakterien erhalten
werden, die Gene umfassen, die in Pflanzenzellen exprimiert werden,
z. B. Agrobacterium oder Rhizobium. Beispiele für Promotoren unter Entwicklungskontrolle
umfassen Promotoren, die bevorzugt eine Transkription in bestimmten
Geweben, z. B. Blättern,
Wurzeln oder Samen, initiieren. Solche Promotoren werden als „Gewebe-bevorzugt" bezeichnet. Promotoren,
welche eine Transkription nur in bestimmtem Gewebe initiieren, werden
als „Gewebe-spezifisch" bezeichnet. Ein „Zelltyp"-spezifischer Promotor
steuert in erster Linie eine Expression in bestimmten Zelltypen
in einem Organ oder mehreren Organen, z. B. Gefäßzellen in Wurzeln oder Blättern. Ein „induzierbarer" Promotor ist ein
Promotor, der unter Umweltkontrolle steht. Beispiele für Umweltbedingungen,
die eine Transkription durch induzierbare Promotoren bewirken, umfassen
anaerobe Bedingungen oder das Vorliegen von Licht. Gewebe-spezifische,
Gewebe-bevorzugte, Zelltyp-spezifische und induzierbare Promotoren
bilden die Klasse von „nicht-konstitutiven" Promotoren. Ein „konstitutiver" Promotor ist ein
Promotor, der unter den meisten Umweltbedingungen aktiv ist.
-
Der
Ausdruck „Cellulosesynthase-Polypeptid" ist ein Polypeptid
der vorliegenden Erfindung und bezieht sich auf eine Aminosäuresequenz
oder mehrere Aminosäuresequenzen
in glycosylierter oder nicht-glycosilierter Form. Der Ausdruck umfasst
auch Fragmente, Varianten, Homologe, Allele oder Vorläufer (z.
B. Preproproteine oder Proproteine) davon. Ein „Cellulosesynthase-Protein" ist ein Protein
der vorliegenden Erfindung und umfasst ein Cellulosesynthase-Polypeptid.
-
Der
Ausdruck „rekombinant", wie er hierin verwendet
wird, bezieht sich auf eine Zelle oder einen Vektor, die/der durch
die Einführung
einer heterologen Nucleinsäure
modifiziert wurde oder auf die Zelle, die aus einer so modifizierten
Zelle abgeleitet ist. So exprimieren z. B. rekombinante Zellen Gene,
die in identischer Form in der nativen (nicht-rekombinanten) Form
der Zelle nicht gefunden werden, oder exprimieren native Gene, die
ansonsten abnormal exprimiert, unter-exprimiert oder überhaupt
nicht exprimiert werden, als Resultat eines beabsichtigten menschlichen
Eingriffs. Der Ausdruck „rekombinant", wie er hierin verwendet
wird, umfasst die Veränderung
der Zelle oder des Vektors durch natürlich vorkommende Ereignisse
(z. B. spontane Mutation, natürliche
Transformation/Transduktion/Transposition), z. B. solche, die ohne
bewussten menschlichen Eingriff auftreten, nicht.
-
Der
Ausdruck „eine
rekombinante Expressionskassette",
wie er hierin verwendet wird, ist ein Nucleinsäurekonstrukt, das rekombinant
oder synthetisch erzeugt wurde, mit einer Reihe von spezifizierten
Nucleinsäureelementen,
welche die Transkription einer bestimmten Nucleinsäure in einer
Wirtszelle erlauben. Die rekombinante Expressionskassette kann in
ein Plasmid, Chromosom, in Mitochondrien-DNA, Plastid-DNA, Virus oder
ein Nucleinsäurefragment
eingebaut werden. Typischerweise umfasst der Teil der rekombinanten
Expressionskassette eines Expressions vektors unter anderen Sequenzen
eine Nucleinsäure,
die zu transkribieren ist, und einen Promotor.
-
Die
Ausdrücke „Rest" oder „Aminosäurerest" oder „Aminosäure" werden hierin austauschbar
verwendet, um eine Aminosäure
zu bezeichnen, die in ein Protein, Polypeptid oder Peptid (kollektiv „Protein") eingebaut ist.
Die Aminosäure
kann eine natürlich
vorkommende Aminosäure
sein und kann, es sei denn, es gibt eine andere Beschränkung, bekannte
Analoga von natürlichen
Aminosäuren
umfassen, die in ähnlicher
Weise wie natürlich
vorkommende Aminosäuren
funktionieren.
-
Der
Ausdruck „hybridisiert
selektiv" bezieht
sich auf eine Hybridisierung einer Nucleinsäuresequenz unter stringenten
Hybridisierungsbedingungen an eine spezifizierte Target-Nucleinsäuresequenz
zu einem detektierbar höheren
Ausmaß (z.
B. wenigstens 2-fach gegenüber
dem Hintergrund) als ihre Hybridisierung an Nicht-Target-Nucleinsäuresequenzen
und unter substantiellem Ausschluss von Nicht-Target-Nucleinsäuren. Selektiv
hybridisierende Sequenzen haben typischerweise etwa wenigstens 80%
Sequenzidentität,
vorzugsweise 90% Sequenzidentität
und am bevorzugtesten 100% Sequenzidentität (d. h. komplementär) miteinander.
-
Der
Ausdruck „spezifisch
reaktiv" bezieht
sich auf eine Bindungsreaktion zwischen einem Antikörper und
einem Protein, das ein Epitop hat, das durch die Antigenbindungsstelle
des Antikörpers
erkannt wird. Diese Bindungsreaktion ist für das Vorliegen eines Proteins
mit dem erkannten Epitop bei Vorliegen einer heterogenen Population
von Proteinen und anderen biologischen Materialien determinativ.
Unter bezeichneten Immunoassaybedingungen binden demnach die spezifizierten
Antikörper
zu einem wesentlich höheren
Grad (wenigstens 2-fach gegenüber
dem Hintergrund) an einen Analyten als im Wesentlichen an alle anderen
in der Probe vorliegenden Analyten, denen das Epitop fehlt.
-
Die
Ausdrücke „stringente
Bedingungen" oder „stringente
Hybridisierungsbedingungen" beziehen
sich auf Bedingungen, unter denen eine Sonde zu einem detektierbar
größeren Grad
als andere Sequenzen (z. B. wenigstens 2-fach gegenüber dem
Hintergrund) an ihre Targetsequenz hybridisieren wird. Stringente
Bedingungen sind Sequenz-abhängig
und werden in verschiedenen Fällen
unterschiedlich sein. Indem die Stringenz der Hybridisierungs- und/oder
Waschbedingungen kontrolliert wird, können Targetsequenzen identifiziert
werden, die 100% komplementär
zu der Sonde sind (homologes Sondieren). Alternativ können Stringenzbedingungen
so eingestellt werden, dass sie eine gewisse Fehlpaarung in Sequenzen
zulassen, so dass niedrigere Similaritätsgrade detektiert werden (heterologes
Sondieren). Im Allgemeinen hat eine Sonde eine Länge von weniger als etwa 1000
Nucleotiden, vorzugsweise eine Länge
von weniger als 500 Nucleotiden.
-
Typischerweise
werden stringente Bedingungen solche sein, bei denen die Salzkonzentration
geringer als etwa 1,5 M Na-Ionen, typischerweise etwa 0,01 bis 1,0
M Na-Ionen-Konzentration
(oder an anderen Salzen), bei pH 7,0 bis 8,3 ist und die Temperatur
wenigstens etwa 30°C
für kurze
Sonden (z. B. 10 bis 50 Nucleotide) und wenigstens etwa 60°C für lange Sonden
(z. B. größer als
50 Nucleotide) ist. Stringente Bedingungen können auch durch die Zugabe
von destabilisierenden Mitteln, z. B. Formamid, erreicht werden.
Beispielhafte Bedingungen niederer Stringenz umfassen eine Hybridisierung
mit einer Pufferlösung
von 30 bis 35% Formamid, 1 M NaCl, 1% SDS (Natriumdodecylsulfat)
bei 37°C
und ein Waschen in 1 × bis
2 × SSC
(20 × SSC =
3,0 M NaC1/0,3 M Trinatriumcitrat) bei 50 bis 55°C. Beispielhafte Bedingungen
moderater Stringenz umfassen eine Hybridisierung in 40 bis 45% Formamid,
1 M NaCl, 1% SDS bei 37°C
und ein Waschen in 0,5 × bis
1 × SSC
bei 55 bis 60°C.
Beispielhafte Bedingungen hoher Stringenz umfassen Hybridisierung
in 50% Formamid, 1 M NaCl, 1% SDS bei 37°C und ein Waschen in 0,1 × SSC bei
60 bis 65°C.
-
Spezifität ist typischerweise
die Funktion von Nach-Hybridisierungs-Waschschritten, wobei die
kritischen Faktoren die Ionenstärke
und die Temperatur der Endwaschlösung
sind. Für
DNA-DNA-Hybride kann die Tm mit der Gleichung
von Meinkoth und Wahl, Anal. Biochem., 138: 267–284 (1984): Tm =
81,5°C +
16,6 (log M) + 0,41 (%GC) – 0,61
(% Form) – 500/L
näherungsweise
bestimmt werden; worin M die Molarität von einwertigen Kationen
ist, %GC der Prozentgehalt an Guanosin- und Cytosinnucleotiden in
der DNA ist, % Form der Prozentgehalt von Formamid in der Hybridisierungslösung ist
und L die Länge
des Hybrids in Basenpaaren ist. Die Tm ist
die Temperatur (unter definierter Ionenstärke und definiertem pH), bei
der 50% einer komplementären
Targetsequenz an eine perfekt passende Sonde hybridisieren. Tm wird um etwa 1°C für je 1% Fehlpaarung verringert;
demnach können
Tm, Hybridisierungs- und/oder Waschbedingungen
so eingestellt werden, dass eine Hybridisierung an Sequenzen der
gewünschten
Identität
erfolgt. Wenn z. B. Sequenzen mit ≥ 90% Identität gesucht
werden, kann die Tm 10°C gesenkt werden. Im Allgemeinen
werden stringente Bedingungen so gewählt, dass sie etwa 5°C unter dem
thermischen Schmelzpunkt (Tm) für die spezifische
Sequenz und ihr Komplement bei einer definierten Ionenstärke und
definiertem pH liegen. Allerdings können streng stringente Bedingungen
eine Hybridisierung und/oder ein Waschen bei 1, 2, 3, oder 4°C unter dem
thermischen Schmelzpunkt (Tm) verwenden;
moderat stringente Bedingungen können
eine Hybridisierung und/oder ein Waschen bei 6, 7, 8, 9 oder 10°C niedriger
als der thermische Schmelzpunkt (Tm) verwenden.
Bedingungen niedriger Stringenz können eine Hybridisierung und/oder
ein Waschen bei 11, 12, 13, 14, 15 oder 20°C unter dem thermischen Schmelzpunkt
(Tm) verwenden. Unter Verwendung der Gleichung,
der Hybridisierung und der Waschflüssigkeitenzusammensetzungen
und der gewünschten
Tm wird der Fachmann erkennen, dass Variationen
in der Stringenz der Hybridisierung und/oder der Waschlösungen inhärent beschrieben
sind. Wenn der gewünschte
Grad der Fehlpaarung in einer Tm von weniger
als 45°C
(wässrige
Lösung)
oder 32°C
(Formamidlösung)
resultiert, ist es bevorzugt, die SSC-Konzentration zu erhöhen, so
dass eine höhere
Temperatur verwendet werden kann. Eine ausgiebige Anleitung für die Hybridisierung
von Nucleinsäuren
wird in Tijssen, Laboratory Techniques in Biochemistry and Molecular
Biology-Hybridization
with Nucleic Acid Probes, Teil I, Kapitel 2 „Overview of principles of
hybridization and the strategy of nucleid acid probe assays", Elsevier, New York (1993);
und Current Pro tocols in Molecular Biology, Kapitel 2, Ausubel,
et al., Herausg., Greene Publishing and Wiley-Interscience, New York (1995) gefunden.
-
Der
Ausdruck "transgene
Pflanze", wie er
hierin verwendet wird, umfasst Bezugnahme auf eine Pflanze, die
in ihrem Genom ein heterologes Polynucleotid umfasst. Im Allgemeinen
ist das heterologe Polynucleotid stabil im Genom integriert, so
dass das Polynucleotid auf nachfolgende Generationen übergeht.
Das heterologe Polynucleotid kann allein oder als Teil einer rekombinanten
Expressionskassette in das Genom integriert werden. „Transgen" wird hierin verwendet,
um eine beliebige Zelle, Zelllinie, einen beliebigen Callus, ein beliebiges
Gewebe, einen beliebigen Pflanzenteil oder eine beliebige Pflanze,
deren/dessen Genotyp durch das Vorliegen einer heterologen Nucleinsäure verändert wurde,
wobei solche Transgenen eingeschlossen sind, die ursprünglich so
verändert
wurden, wie auch jene, die durch sexuelle Kreuzungen oder asexuelle
Vermehrung aus dem ursprünglichen
Transgen erzeugt wurden, zu beschreiben. Der Ausdruck „Transgen", wie er hierin verwendet
wird, umfasst nicht die Veränderung
des Genoms (chromosomales oder extra-chromosomales) durch herkömmliche
Pflanzenzüchtungsverfahren
oder durch natürlich
auftretende Ereignisse, z. B. statistische Fremdbefruchtung, nicht-rekombinante Virusinfektion,
nicht-rekombinante bakterielle Transformation, nicht-rekombinante
Transposition oder spontane Mutation.
-
Der
Ausdruck „Vektor", wie er hierin verwendet
wird, bezieht sich auf eine Nucleinsäure, die bei der Transfektion
einer Wirtszelle verwendet wird, und in welcher ein Polynucleotid
insertiert werden kann. Vektoren sind oft Replicons. Expressionsvektoren
erlauben eine Transkription einer darin insertierten Nucleinsäure.
-
Die
folgenden Ausdrücke
werden verwendet, um die Sequenzbeziehungen zwischen zwei oder mehr Nucleinsäuren oder
Polypeptiden zu beschreiben: (a) „Referenzsequenz", (b) „Vergleichsfenster", (c) „Sequenzidentität", (d) „Prozentwert
der Sequenzidentität" und (e) „substantielle
Identität".
- (a)
Der Ausdruck „Referenzsequenz", wie er hierin verwendet
wird, ist eine definierte Sequenz, die als Basis für einen
Sequenzvergleich verwendet wird. Eine Referenzsequenz kann ein Untersatz
oder die Gesamtheit einer spezifizierten Sequenz, zum Beispiel ein
Segment einer Volllängen-cDNA-
oder Gensequenz oder die vollständige
cDNA oder Gensequenz sein.
- (b) Der Ausdruck „Vergleichsfenster", wie er hierin verwendet
wird, bezieht sich auf ein fortlaufendes und spezifiziertes Segment
einer Polynucleotidsequenz, wobei die Polynucleotidsequenz mit einer
Referenzsequenz verglichen werden kann und wobei der Teil der Polynucleotidsequenz
im Vergleichsfenster Additionen oder Deletionen (d. h. Gaps) im
Vergleich zur Referenzsequenz (die keine Additionen oder Delektionen umfasst)
zur optimalen Anordnung der zwei Sequenzen umfassen kann. Im Allgemeinen
ist das Vergleichsfenster wenigstens 20 fortlaufende Nucleotide
lang und kann gegebenenfalls 30, 40, 50, 100 Nucleotide lang oder
länger
sein. Der Fachmann wird verstehen, dass zur Vermeidung einer hohen
Similarität
mit einer Referenzse quenz infolge des Einschlusses von Gaps in die
Polynucleotidsequenz typischerweise eine „gap penalty" eingeführt wird
und von der Zahl der Übereinstimmungen
subtrahiert wird.
-
Verfahren
zur Anordnung (alignment) von Sequenzen zum Vergleich sind auf dem
Fachgebiet gut bekannt. Eine optimale Anordnung von Sequenzen für einen
Vergleich kann durchgeführt
werden: durch den lokalen Homologiealgorithmus von Smith und Waterman,
Adv. Appl. Math. 2: 482 (1981); durch den Homologieanordnungsalgorithmus
von Needleman und Wunsch, J. Mol. Biol. 48: 443 (1970); durch die
Suche nach einem Similaritätsverfahren
von Pearson und Lipman, Proc. Natl. Acad. Sci. 85: 2444 (1988);
durch Durchführung
dieser Algorithmen mittels Computer, einschließlich, aber nicht beschränkt auf:
CLUSTAL im PC/Gene-Programm von Intelligenetics, Mountain View,
Kalifornien, GAP, BESTFIT, BLAST, FASTA und TFASTA im Wisconsin
Genetics Software Package, Genetics Computer Group (GCG), 575 Science
Dr., Madison, Wisconsin, USA; das CLUSTAL-Programm ist von Higgins
und Sharp, Gene 73: 237–244
(1988); Higgins und Sharp, CABIOS 5: 151–153 (1989); Corpet, et al.,
Nucleic Acids Research 16: 10881–90 (1988); Huang et al., Computer
Applications in the Biosciences 8: 155–65 (1992) und Pearson, et
al., Methods in Molecular Biology 24: 307–331 (1994) gut beschrieben.
Die BLAST-Familie von Programmen, die für Datenbank-Similaritätssuchen eingesetzt
werden kann, umfasst: BLASTN für
Nucleotid-Query-Sequenzen gegen Nucleotid-Datenbank-Sequenzen; BLASTX
für Nucleotid-Query-Sequenzen
gegen Protein-Datenbank-Sequenzen; BLASTP für Protein-Query-Sequenzen gegen
Protein-Datenbank-Sequenzen; TBLASTN für Protein-Query-Sequenzen gegen Nucleotid-Datenbank-Sequenzen
und TBLASTX für
Nucleotid-Query-Sequenzen
gegen Nucleotid-Datenbank-Sequenzen. Siehe Current Protocols in
Molecular Biology, Kapitel 19, Ausubel et al., Herausg., Greene Publishing
and Wiley-Interscience, New York (1995).
-
Wenn
nichts anderes angegeben ist, beziehen sich Sequenzidentitäts/Similaritäts-Werte,
die hierin bereitgestellt werden, auf den Wert, der unter Verwendung
des BLAST 2.0-Satzes von Programmen unter Verwendung von Default-Parametern
erhalten wurde. Altschul et al., Nucleic Acids Res. 25: 3389–3402 (1997) Software
zur Durchführung
von BLAST-Analysen ist allgemein verfügbar, z. B. vom National Center
for Biotechnology Information (http://www.ncbi.nlm.nih.gov/). Dieser
Algorithmus involviert zunächst
Identifizieren von High Scoring Sequence Pairs (HSPs) durch Identifizieren
kurzer Wörter
mit der Länge
W in der Query-Sequenz, die einem gewissen positiven Schwellenscore
T entsprechen oder genügen,
wenn sie mit einem Wort derselben Länge in der Datenbanksequenz
vergleichend angeordnet werden. T wird als die Nachbarschaftswort-Scoreschwelle
bezeichnet (Altschul et al, supra). Diese Anfangsnachbarschaftsworttreffer
wirken als Keime zur Initiierung von Suchen, um längere HSPs,
die sie enthalten, zu finden. Die Worttreffer werden dann in beiden
Richtungen entlang jeder Sequenz ausgedehnt, soweit der kumulative
Anordnungsscore erhöht
werden kann. Kumulative Scores werden errechnet, indem für Nucleotidsequenzen
die Parameter M (Belohnungsscore für ein Paar übereinstimmender Reste; immer > 0) und N (Penalty-Score
für Fehlpaarungsreste; im mer < 0) verwendet werden.
Für Aminosäuresequenzen
wird eine Scoring-Matrix verwendet, um den kumulativen Score zu
berechnen. Eine Extension der Worttreffer in jede Richtung wird
angehalten, wenn: der kumulative Anordnungsscore um die Menge X
von seinem maximal erreichten Wert abfällt; der kumulative Score gegen
null oder darunter geht, und zwar durch Akkumulation von einer oder
mehreren negativen Scoringrestanordnungen, oder das Ende jeder Sequenz
erreicht ist. Die BLAST-Algorithmus-Parameter W, T und X bestimmen
die Sensitivität
und die Geschwindigkeit der Anordnung. Das BLASTN-Programm (für Nucleotidsequenzen)
verwendet als Default eine Wortlänge
(W) von 11, eine Erwartung (E) von 10, einen Cutoff-Wert von 100, M
= 5, N = –4
und einen Vergleich beider Stränge.
Für Aminosäuresequenzen
verwendet das BLASTP-Programm als Default eine Wortlänge (W)
von 3, eine Erwartung (E) von 10 und die BLOSUM62-Scoring-Matrix (siehe
Henikoff & Henikoff
(1989) Proc. Natl. Acad. Sci. USA 89: 10915).
-
Der
BLAST-Algorithmus führt
zusätzlich
zur Berechnung des Prozentwerts der Sequenzidentität auch eine
statistische Analyse der Similarität zwischen zwei Sequenzen durch
(siehe z. B. Karlin & Altschul,
Proc. Natl. Acad. Sci. USA 90: 5873–5787 (1993)). Ein Maß für die Similarität, das durch
den BLAST-Algorithmus bereitgestellt wird, ist die kleinste Summenwahrscheinlichkeit
(P(N)), die einen Hinweis für
die Wahrscheinlichkeit liefert, mit der eine Übereinstimmung zwischen zwei
Nucleotid- oder Aminosäuresequenzen
zufällig
auftreten würde.
-
BLAST-Suchen
nehmen an, dass Proteine als statistische Sequenzen modelliert werden
können.
Allerdings umfassen viele reelle Proteine Regionen von nicht-statistischen
Sequenzen, die polymere Züge,
Wiederholungen kurzer Periode oder Regionen, die an einer Aminosäure oder
mehreren Aminosäuren
angereichert sind, sein können.
Solche Regionen mit niedriger Komplexität können zwischen nicht-verwandten
Proteinen angeordnet sein, selbst wenn andere Regionen des Proteins
vollständig
verschieden sind. Eine Reihe von Niedrigkomplexitäts-Filterprogrammen
können
verwendet werden, um solche Anordnungen mit niedriger Komplexität zu reduzieren.
Zum Beispiel können
die SEG (Wooten und Federhen, Comput. Chem., 17: 149–163 (1993))
und XNU (Claverie und States, Comput. Chem., 17: 191–201 (1993))-Niedrigkomplexitäts-Filter
allein oder in Kombination verwendet werden.
- (c) „Sequenzidentität" oder „Identität" im Kontext von zwei
Nucleinsäure-
oder Polypeptidsequenzen beziehen sich, wie die Ausdrücke hierin
verwendet werden, auf Reste in den zwei Sequenzen, die bei Anordnung auf
maximales Übereinstimmen über ein
spezifiziertes Vergleichsfenster gleich sind. Wenn der Prozentwert der
Sequenzidentität
bei Proteinen verwendet wird, wird anerkannt, dass Restpositionen,
die nicht identisch sind, sich oft durch konservative Aminosäuresubstitutionen
unterscheiden, in denen Aminosäurereste
für andere
Aminosäurereste
mit ähnlichen
chemischen Eigenschaften (z. B. Ladung oder Hydrophobie) eingesetzt
sind und daher die funktionellen Eigenschaften des Moleküls nicht
verändern.
Wenn Sequenzen sich in konservativen Substitutionen unterscheiden,
kann der Prozentwert der Sequenzidentität nach oben eingestellt werden,
um eine Korrektur bezüglich
der konservativen Natur der Substitution durchzufüh ren. Sequenzen,
die sich durch solche konservativen Substitutionen unterscheiden,
werden als mit „Sequenzsimilarität" oder „Similarität" bezeichnet. Mittel
zur Durchführung
dieser Einstellung sind dem Fachmann gut bekannt. Typischerweise
umfasst dies ein Scoring einer konservativen Substitution als eine
partielle anstelle einer vollständigen
Fehlpaarung, wodurch der Prozentwert der Sequenzidentität erhöht wird.
Wenn z. B. einer identischen Aminosäure ein Score von 1 gegeben
wird und einer nicht-konservativen Substitution ein Score von null
gegeben wird, wird einer konservativen Substitution ein Score zwischen
null und 1 gegeben. Das Scoring bzw. die Bewertung von konservativen
Substitutionen wird z. B. nach dem Algorithmus von Meyers und Miller,
Computer Applic. Biol. Sci., 4: 11–17 (1988) errechnet, z. B.
wie es in dem Programm PC/GENE (Intelligenetics, Mountain View,
Kalifornien, USA) durchgeführt
wird.
- (d) Wie der Ausdruck „Prozentwert
der Sequenzidentität" hierin verwendet
wird, bedeutet er den Wert, der durch Vergleich von zwei optimal
angeordneten Sequenzen über
ein Vergleichsfenster bestimmt wird, wobei der Teil der Polynucleotidsequenz
in dem Vergleichsfenster Additionen oder Deletionen (d. h. Gaps)
verglichen mit der Referenzsequenz (die keine Additionen oder Deletionen
umfasst) zur optimalen Anordnung der zwei Sequenzen umfassen kann.
Der Prozentwert wird errechnet, indem die Anzahl von Positionen
bestimmt wird, an denen die identische Nucleinsäurebase oder der Aminosäurerest
in beiden Sequenzen auftritt, um so die Zahl der übereinstimmenden
Positionen zu bestimmen, die Anzahl der übereinstimmenden Positionen
durch die Gesamtzahl der Positionen im Vergleichsfenster dividiert
wird und das Resultat mit 100 multipliziert wird, wodurch der Prozentwert
der Sequenzidentität
erhalten wird.
- (e) (i) Der Ausdruck „substantielle
Identität" von Polynucleotidsequenzen
bedeutet, dass ein Polynucleotid eine Sequenz umfasst, die wenigstens
70% Sequenzidentität,
vorzugsweise wenigstens 80%, bevorzugter wenigstens 90% und am bevorzugtesten
wenigstens 95% Sequenzidentität
zu einer Referenzsequenz hat, wobei eines der beschriebenen Anordnungsprogramme
unter Verwendung von Standardparametern verwendet wird. Ein Fachmann
wird erkennen, dass diese Werte geeigneterweise eingestellt werden
können, um
eine entsprechende Identität
von Proteinen zu bestimmen, die durch zwei Nucleotidsequenzen codiert werden,
indem die Codondegeneriertheit, die Aminosäuresimilarität, die Leserasterpositionierung
und dergleichen berücksichtigt
werden. Substantielle Identität
von Aminosäuresequenzen
bedeutet zu diesem Zweck normalerweise eine Sequenzidentität von wenigstens
60%, bevorzugter wenigstens 70%, 80%, 90% und am bevorzugtesten
wenigstens 95%.
-
Ein
anderer Hinweis, dass Nucleotidsequenzen im Wesentlichen identisch
sind, ist, wenn zwei Moleküle
unter stringenten Bedingungen aneinander hybridisieren. Allerdings
sind Nucleinsäuren,
die unter stringenten Bedingungen nicht aneinander hybridisieren,
noch im Wesentlichen identisch, wenn die Polypeptide, die sie codieren,
substantiell identisch sind. Dies kann auftreten, z. B. wenn eine
Kopie einer Nucleinsäure
unter Verwendung der maximalen Codondegeneriertheit, die durch den
genetischen Code zugelassen wird, erzeugt wird. Ein Hinweis, dass
zwei Nucleinsäuresequenzen
substantiell identisch sind, ist, dass das Polypeptid, das durch
die erste Nucleinsäure
codiert wird, immunologisch kreuzreaktiv mit dem Polypeptid ist,
das durch die zweite Nucleinsäure
codiert wird.
- (e) (ii) Der Ausdruck „substantielle
Identität" im Kontext eines
Peptids gibt an, dass ein Peptid eine Sequenz mit wenigstens 70%
Sequenzidentität
mit einer Referenzsequenz, vorzugsweise 80%, bevorzugter 85%, am
bevorzugtesten wenigstens 90% oder 95% Sequenzidentität mit einer
Referenzsequenz über
ein spezifiziertes Vergleichsfenster hat. Vorzugsweise wird eine
optimale Anordnung unter Verwendung des Homologie-Anordnungsalgorithmus
von Needleman und Wunsch, J. Mol. Biol. 48: 443 (1970) durchgeführt. Ein
Hinweis, dass zwei Peptidsequenzen substantiell identisch sind,
ist, dass ein Peptid immunologisch reaktiv mit Antikörpern ist,
die gegen das zweite Peptid entwickelt wurden. Somit ist ein Peptid
substantiell identisch mit einem zweiten Peptid, z. B. wenn die
zwei Peptide sich nur durch eine konservative Substitution unterscheiden.
Peptide, die „substantiell ähnlich" sind, teilen Sequenzen,
wie sie oben angegeben sind, außer
dass Restpositionen, die nicht identisch sind, sich durch konservative
Aminosäureänderungen
unterscheiden können.
-
Übersicht
-
Die
vorliegende Erfindung stellt unter anderem Zusammensetzungen und
Verfahren zum Modulieren (d. h. Erhöhen oder Senken) des Levels
von Polypeptiden der vorliegenden Erfindung in Pflanzen bereit.
Die Polypeptide der vorliegenden Erfindung können insbesondere in Entwicklungsstadien
in Geweben und/oder in Mengen, die für die nicht-rekombinant gentechnisch
manipulierten Pflanzen nicht charakteristisch sind, exprimiert werden.
Somit stellt die vorliegende Erfindung Verwendbarkeit in solchen
beispielhaften Anwendungen, wie die Verbesserung der Stengelqualität für einen
verbesserten Stand oder eine verbesserte Silage bereit. Außerdem sorgt
die vorliegende Erfindung für
eine erhöhte
Cellulosekonzentration im Pericarp für eine Härtung des Kerns bzw. des Korns
und somit für
eine Verbesserung seiner Handhabbarkeit.
-
Isolierte
Nucleinsäuren,
die Polynucleotide ausreichender Länge und Komplementarität zu einem
Gen der vorliegenden Erfindung umfassen, können als Sonden oder Amplifikationsprimer
bei der Detektion, quantitativen Bestimmung oder Isolierung von
Gentranskripten verwendet werden. Beispielsweise können isolierte Nucleinsäuren der
vorliegenden Erfindung als Sonden beim Detektieren von Defizienzien
auf dem Level von mRNA beim Screening auf gewünschte transgene Pflanzen,
zum Detektieren von Mutationen im Gen (z. B. Substitutionen, Deletionen
oder Additionen), zur Überwachung
der Hochregulierung der Expression oder von Änderungen bei der Enzymaktivität in Screening-Assays
von Verbindungen, zur Detektion einer Reihe von Allelvarianten (Polymorphismen)
des Gens oder zur Verwendung als molekulare Marker in Pflanzenzüchtungsprogrammen
verwendet werden. Die isolierten Nucleinsäuren der vorliegenden Erfindung
können
auch für
eine rekombinante Expression ihrer codierten Polypeptide oder zur
Verwendung als Immunogene bei der Herstellung und/oder dem Screening
von Antikörpern
eingesetzt werden. Die isolierten Nucleinsäuren der vorliegenden Erfindung
können
auch zur Verwendung bei der Sense- oder Antisense-Suppression eines
oder mehrerer Gen(e) der vorliegen den Erfindung in einer Wirtszelle,
in einem Gewebe oder einer Pflanze eingesetzt werden. Eine Bindung
chemischer Agenzien, welche an die isolierten Nucleinsäuren der
vorliegenden Erfindung binden, mit diesen interkalieren, sie spalten
und/oder vernetzen, können
ebenfalls eingesetzt werden, um eine Transkription oder Translation
zu modulieren.
-
Die
vorliegende Erfindung stellt auch isolierte Proteine bereit, die
ein Polypeptid der vorliegenden Erfindung umfassen (z. B. Preproenzym,
Proenzym oder Enzyme). Die vorliegende Erfindung stellt auch Proteine
bereit, die wenigstens ein Epitop aus einem Polypeptid der vorliegenden
Erfindung umfassen. Die Proteine der vorliegenden Erfindung können in
Assays auf Enzymagonisten oder Antagonisten der Enzymfunktion oder zur
Verwendung als Immunogene oder Antigene unter Erhalt von Antikörpern, die
spezifisch immunreaktiv mit einem Protein der vorliegenden Erfindung
sind, verwendet werden. Solche Antikörper können in Assays auf Expressionslevel,
zur Identifizierung und/oder Isolierung von Nucleinsäuren der
vorliegenden Erfindung aus Expressionsbibliotheken oder zur Reinigung
von Polypeptiden der vorliegenden Erfindung verwendet werden.
-
Die
isolierten Nucleinsäuren
und Proteine der vorliegenden Erfindung können bei einem breiten Bereich
von Pflanzentypen, insbesondere bei Monocotylen, wie den Spezies
der Graminiae-Familie, einschließlich Sorghum bicolor und Zea
mays, verwendet werden. Die isolierten Nucleinsäuren und Proteine der vorliegenden
Erfindung können
auch in Spezies aus den Gattungen: Cucurbita, Rosa, Vitis, Juglans,
Fragaria, Lotus, Medicago, Onobrychis, Trifolium, Trigonella, Vigna,
Citrus, Linum, Geranium, Manihot, Daucus, Arbidopsis, Brassica,
Raphanus, Sinapis, Atropa, Capsicum, Datura, Hyoscyamus, Lycopersicon,
Nicotiana, Solanum, Petunia, Digitalis, Majorana, Ciahorium, Helianthus,
Lactuca, Bromus, Asparagus, Antirrhinum, Heterocallis, Nemesis,
Pelargonium, Panieum, Pennisetum, Ranunculus, Senecio, Salpiglossis,
Cucumis, Browaalia, Glycine, Pisum, Phaseolus, Lolium, Oryza, Avena,
Hordeum, Secale, Triticum, Bambusa, Dendrocalamus und Melocanna
eingesetzt werden.
-
Nucleinsäuren
-
Die
vorliegende Erfindung stellt unter anderem isolierte Nucleinsäuren von
RNA, DNA und Analoga und/oder Chimären davon, die ein Polynucleotid
der vorliegenden Erfindung umfassen, bereit.
-
Ein
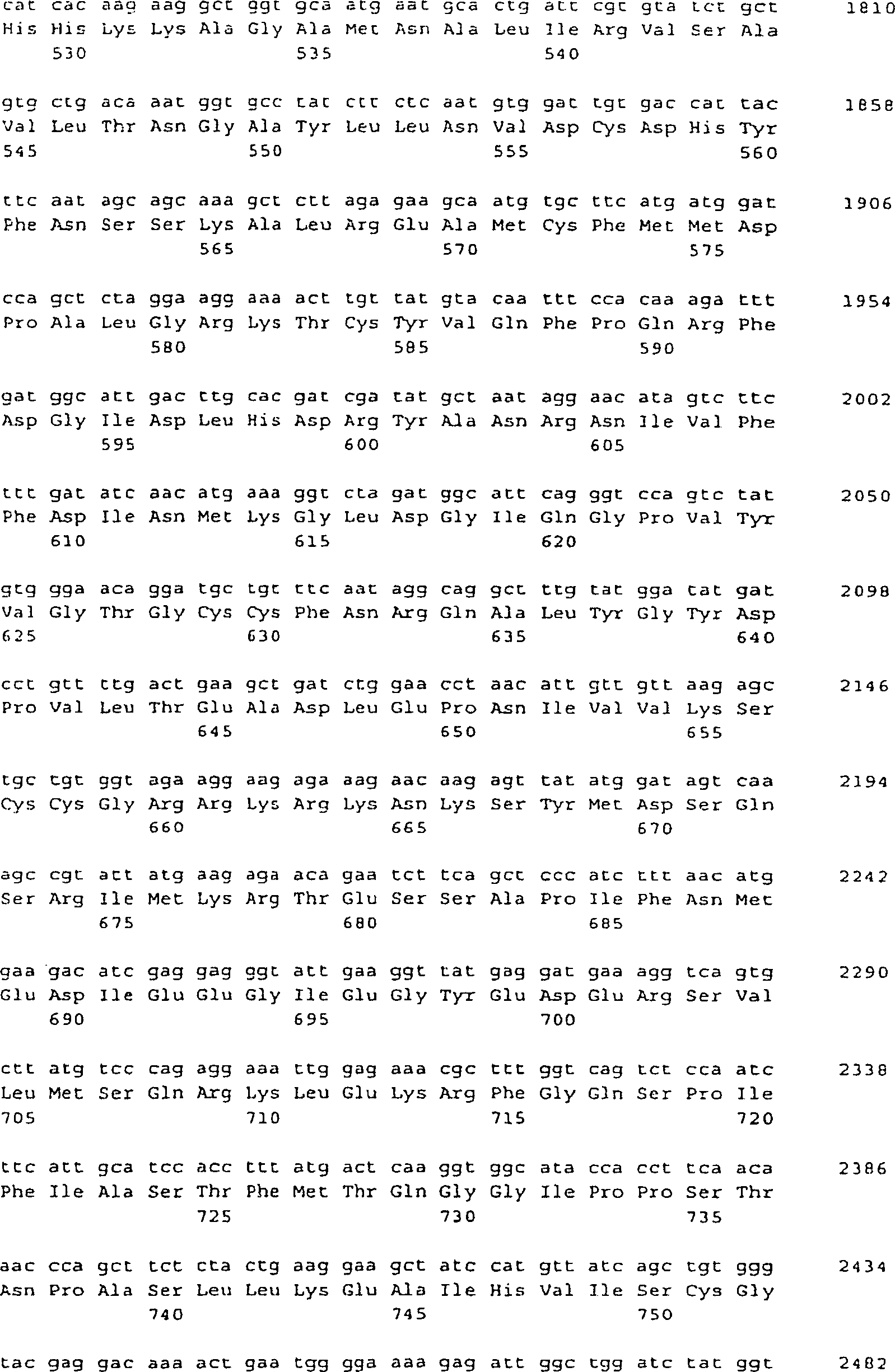
Polynucleotid der vorliegenden Erfindung umfasst:
- (a)
ein Polynucleotid, das ein Polypeptid von SEQ ID NO: 6, 10 oder
26 und konservativ modifizierte und polymorphe Varianten davon codiert,
einschließlich
z. B. der Polynucleotide von SEQ ID NO: 5, 9 oder 25.
-
A. Polynucleotide, die für ein Polypeptid
der vorliegenden Erfindung oder konservativ modifizierte oder polymorphe
Varianten davon codieren.
-
Wie
in (a) oben angegeben wurde, stellt die vorliegende Erfindung isolierte
Nucleinsäuren
bereit, die ein Polynucleotid der vorliegenden Erfindung umfassen,
wobei das Polynucleotid für
ein Polypeptid der vorliegenden Erfindung oder konservativ modifizierte
oder polymorphe Vari anten davon codiert. Der Fachmann wird erkennen,
dass die Degeneriertheit des genetischen Codes einer Vielzahl von
Polynucleotiden ermöglicht,
für die
identische Aminosäuresequenz
zu codieren. Solche „stummen
Variationen" können z.
B. verwendet werden, um Allelvarianten von Polynucleotiden der vorliegenden
Erfindung selektiv zu hybridisieren und zu detektieren. Folglich
umfasst die vorliegende Erfindung Polynucleotide von SEQ ID NO:
5, 9 und 25 und stumme Variationen von Polynucleotiden, die ein
Polypeptid von SEQ ID NO: 6, 10 und 26 codieren. Die vorliegende Erfindung
stellt ferner isolierte Nucleinsäuren
bereit, die Polynucleotide umfassen, die für konservativ modifizierte
Varianten eines Polypeptids von SEQ ID NO: 6, 10 und 26 codieren.
Zusätzlich
stellt die vorliegende Erfindung isolierte Nucleinsäuren bereit,
die Polynucleotide umfassen, die für eine oder mehrere polymorphe
(allelische) Varianten von Polypeptiden/Polynucleotiden codieren.
Polymorphe Varianten werden häufig
verwendet, um die Segregation von chromosomalen Regionen in z. B.
Marker-unterstützen
Selektionsverfahren zur Ernteverbesserung zu verfolgen.
-
B. Polynucleotide, amplifiziert aus einer
Zea mays-Nucleinsäurebibliothek
-
Polynucleotide
der vorliegenden Erfindung können
aus einer Zea mays-Nucleinsäurebibliothek
als zusätzliche
Sequenzen amplifiziert werden. Die Zea mays-Linien B73, PHRE1, A632,
BMS-P2#10, W23 und Mo17 sind bekannt und allgemein verfügbar. Andere
allgemein bekannte und verfügbare
Maislinien können von
der Maize Genetics Cooperation (Urbana, IL) erhalten werden. Die
Nucleinsäurebibliothek
kann eine cDNA-Bibliothek, eine genomische Bibliothek oder eine
Bibliothek, die allgemein aus nuklearen Transkripten in einer beliebigen
Stufe der Intronprozessierung konstruiert wurde, sein. cDNA-Bibliotheken
können
normalisiert werden, um die Repräsentation
von relativ seltenen cDNAs zu erhöhen. In optionalen Ausführungsformen
wird die cDNA-Bibliothek unter Verwendung eines Volllängen-cDNA-Syntheseverfahrens
konstruiert. Beispiele für solche
Verfahren umfassen Oligo-Capping (Maruyama, K. und Sugano, S. Gene
138: 171–174,
1994), Biotinylierten CAP-Trapper (Carninci, P., Kvan, Ca. et al.
Genomics 37: 372–336,
1996) und CAP-Retentionsverfahren (Edery, E., Chu, L. L. et al.
Molecular and Cellular Biology 15: 3363–3371, 1995). Eine cDNA-Synthese wird
oft bei 50–55°C katalysiert,
um die Bildung von RNA-Sekundärdstruktur
zu verhindern. Beispiele für
reverse Transkriptasen, die bei diesen Temperaturen relativ stabil
sind, sind Superscript II-Reverse Transkriptase (Life Technologies,
Inc.), AMV-Reverse Transkriptase (Boehringer Mannheim) und Retro-Amp-Reverse-Transkriptase
(Epicenter). Als mRNA-Quellen werden vorzugsweise schnell wachsende
Gewebe oder sich schnell teilende Zellen verwendet, z. B. aus dem
sich streckenden Internodium von Getreidepflanzen bzw. Maispflanzen.
-
Polynucleotide
können
unter Verwendung der folgenden Primerpaare amplifiziert werden:
SEQ
ID NO: 3 und 4, welche ein Amplicon liefern, das eine Sequenz mit
substantieller Identität
zu SEQ ID NO: 1 umfasst;
SEQ ID NO: 7 und 8, die ein Amplicon
liefern, das eine Sequenz mit substantieller Identität zu SEQ
ID NO: 5 der Erfindung umfasst; und
SEQ ID NO: 11 und 12, die
ein Amplicon liefern, das eine Sequenz mit substantieller Identität zu SEQ
ID NO: 9 der Erfindung umfasst.
SEQ ID NO: 15 und 16, die ein
Amplicon liefern, das eine Sequenz mit substantieller Identität zu SEQ
ID NO: 13 umfasst.
SEQ ID NO: 19 und 20, die ein Amplicon liefern,
das eine Sequenz mit substantieller Identität zu SEQ ID NO: 17 umfasst;
SEQ
ID NO: 23 und 24, die ein Amplicon liefern, das eine Sequenz mit
substantieller Identität
zu SEQ ID NO: 21 umfasst;
SEQ ID NO: 27 und 28, die ein Amplicon
liefern, das eine Sequenz mit substantieller Identität zu SEQ
ID NO: 25 der Erfindung umfasst;
SEQ ID NO: 31 und 32, die
ein Amplicon liefern, das eine Sequenz mit substantieller Identität zu SEQ
ID NO: 29 umfasst.
SEQ ID NO: 35 und 36, die ein Amplicon liefern,
das eine Sequenz mit substantieller Identität zu SEQ ID NO: 33 umfasst;
SEQ
ID NO: 39 und 40, die ein Amplicon liefern, das eine Sequenz mit
substantieller Identität
zu SEQ ID NO: 37 umfasst; und
SEQ ID NO: 43 und 44, die ein
Amplicon liefern, das eine Sequenz mit substantieller Identität zu SEQ
ID NO: 41 umfasst.
SEQ ID NO: 47 und 48, die ein Amplicon liefern,
das eine Sequenz mit substantieller Identität zu SEQ ID NO: 45 umfasst.
SEQ
ID NO: 51 und 52, die ein Amplicon liefern, das eine Sequenz mit
substantieller Identität
zu SEQ ID NO: 49 umfasst;
SEQ ID NO: 55 und 56, die ein Amplicon
liefern, das eine Sequenz mit substantieller Identität zu SEQ
ID NO: 53 umfasst; und
SEQ ID NO: 59 und 60, die ein Amplicon
liefern, das eine Sequenz mit substantieller Identität zu SEQ
ID NO: 57 hat.
-
Die
vorliegende Erfindung bezieht sich auch auf Untersequenzen der Polynucleotide
der vorliegenden Erfindung. Eine Vielzahl von Untersequenzen kann
unter Verwendung von Primern erhalten werden, welche unter stringenten
Bedingungen selektiv an wenigstens zwei Stellen in einem Polynucleotid
der vorliegenden Erfindung oder an zwei Stellen in der Nucleinsäure, welche
ein Polynucleotid der Erfindung flankiert und umfasst, oder an eine
Stelle in einem Polynucleotid der Erfindung und eine Stelle in der
Nucleinsäure,
die sie umfasst, hybridisieren. Primer werden so ausgewählt, dass
sie unter stringenten Hybridisierungsbedingungen selektiv an ein
Polynucleotid der vorliegenden Erfindung hybridisieren. Im Allgemeinen
sind die Primer komplementär
zu einer Untersequenz der Zielnucleinsäure, welche sie amplifizieren.
Wie der Fachmann erkennen wird, werden die Stellen, an welche das
Primerpaar selektiv hybridisieren wird, so gewählt, dass unter den gewünschten
Amplifikationsbedingungen eine einzelne fortlaufende Nucleinsäure gebildet
werden kann.
-
Die
Primer können
so konstruiert sein, dass sie unter stringenten Bedingungen selektiv
an eine Sequenz (oder ihr Komplement) in der Zielnucleinsäure hybridisieren,
welche das Codon, das für
den Carboxy- oder Amino-terminalen Aminosäurerest codiert (d. h. die
3'-terminale codierende
Region bzw. die 5'-terminale codierende
Region) der Polynucleotide der vorliegenden Erfindung umfasst. Gegebenenfalls
werden die Primer so konstruiert sein, dass sie selektiv vollständig in
der codierenden Region des Targetpolynucleotids bzw. Zielpolynucleotids
der vorliegenden Erfindung hybridisieren, so dass das Amplifikationsprodukt
eines cDNA-Targets aus der codierenden Region jener cDNA bestehen
wird. Die Primerlänge
in Nucleotiden wird aus der Gruppe von ganzen Zahlen, bestehend
aus wenigstens 15 bis 50, ausgewählt.
So können
die Primer eine Länge
von wenigstens 15, 18, 20, 25, 30, 40 oder 50 Nucleotiden haben.
Der Fachmann wird erkennen, dass eine verlängerte Primersequenz verwendet
werden kann, um die Bindungsspezifität (d. h. ein Annealing) an eine
Targetsequenz zu erhöhen.
Eine Nicht-Annealing-Sequenz
am 5'-Ende eines
Primers (ein „Schwanz") kann addiert werden,
zum Beispiel um eine Klonierungsstelle an den terminalen Enden des
Amplicons einzuführen.
-
Die
Amplifikationsprodukte können
unter Verwendung von Expressionssystemen, die dem Fachmann auf dem
Fachgebiet gut bekannt sind und die unten diskutiert werden, translatiert
werden. Die resultierenden Translationsprodukte können als
Polypeptide der vorliegenden Erfindung bestätigt werden, indem zum Beispiel
auf die geeignete katalytische Aktivität (z. B. spezifische Aktivität und/oder
Substratspezifität)
analysiert wird oder das Vorliegen eines linearen Epitops oder mehrerer
linearer Epitope verifiziert wird, das/die für ein Polypeptid der vorliegenden
Erfindung spezifisch ist/sind. Verfahren zur Proteinsynthese aus
durch PCR abgeleitete Matrizen sind auf dem Fachgebiet bekannt und
im Handel erhältlich.
Siehe z. B. Amersham Life Sciences, Inc, Katalog '97, S. 354.
-
Verfahren
zum Erhalt von 5'-
und/oder 3'-Enden
eines Vektorinserts sind auf dem Fachgebiet gut bekannt. Siehe z.
B. RACE (Rapid Amplification of Complementary Ends), beschrieben
in Frohman, M. A., in PCR Protocols: A Guide to Methods and Applications,
M. A. Innis, D. H. Gelfand, J. J. Sninsky, T. J. White, Herausg.
(Academic Press, Inc., San Diego, (1990), S. 28–38.); siehe auch
U.S. Patent Nr. 5,470,722 und Current
Protocols in Molecular Biology, Unit 15.6, Ausubel et al., Herausg.,
Greene Publishing and Wiley-Interscience, New York (1995); Frohman
und Martin, Techniques 1: 165 (1989).
-
C. Polynucleotide, die selektiv an ein
Polynucleotid von (A) oder (B) hybridisieren
-
Die
vorliegende Erfindung bezieht sich auch auf isolierte Nucleinsäuren, die
Polynucleotide der vorliegenden Erfindung umfassen, wobei die Polynucleotide
unter selektiven Hybridisierungsbedingungen selektiv an ein Polynucleotid
der Abschnitte (A) oder (B), wie sie oben diskutiert wurden, hybridisieren.
Solche Polynucleotide können
zum Isolieren, Detektieren und/oder Quantifizieren von Nucleinsäuren, die
die Polynucleotide der Erfindung von (A) oder (B) umfassen, verwendet
werden. Solche Polynucleotide können
zum Beispiel verwendet werden, um Partial- oder Volllängenklone
in einer hinterlegten Bibliothek zu identifizieren, zu isolieren oder
zu amplifizieren. Solche Polynucleotide können genomische oder cDNA-Sequenzen
sein, die isoliert wurden oder ansonsten komplementär zu einer
cDNA aus einer Dicotylen- oder Monocotylen-Nucleinsäurebibliothek sind. Beispiele
für Spezies
von Monocotylen und Dicotylen umfassen, sind aber nicht beschränkt auf: Mais,
Canola, Sojabohne, Baumwolle, Weizen, Sorghum, Sonnenblume, Hafer,
Zuckerrohr, Hirse, Gerste und Reis. Gegebenenfalls umfasst die cDNA-Bibliothek wenigstens
80% Volllängensequenzen,
vorzugsweise wenigstens 85% oder 90% Volllängensequenzen, bevorzugter
wenigstens 95% Volllängensequenzen.
Die cDNA-Bibliotheken können
zur Erhöhung
der Repräsentation
von seltenen Sequenzen normalisiert werden. Hybridisierungsbedingungen
niedriger Stringenz werden typischerweise, aber nicht ausschließlich, mit
Sequenzen verwendet, die eine verringerte Sequenzidentität bezüglich komplementärer Sequenzen
haben. Bedingungen moderater und hoher Stringenz können gegebenenfalls
für Sequenzen
mit größerer Identität verwendet werden.
Bedingungen niedriger Stringenz erlauben eine selektive Hybridisierung
von Sequenzen, die etwa 70% Sequenzidentität haben, und können verwendet
werden, um orthologe oder paraloge Sequenzen zu identifizieren.
-
E. Polynucleotide, die für ein Protein
codieren, welches eine Untersequenz aus einem Prototyp-Polypeptid
hat und mit dem Prototyp-Polypeptid kreuzreaktiv ist
-
Die
vorliegende Erfindung bezieht sich auch auf isolierte Nucleinsäuren, die
Polynucleotide der vorliegenden Erfindung umfassen, wobei die Polynucleotide
ein Protein codieren, das eine Untersequenz von fortlaufenden Aminosäuren aus
einem Prototyp-Polypeptid der vorliegenden Erfindung, wie sie z.
B. in (a) oben bereitgestellt werden, hat. Die Länge der fortlaufenden Aminosäuren aus
dem Prototyp-Polypeptid wird aus der Gruppe von ganzen Zahlen ausgewählt, bestehend
aus wenigstens 10 bis zu der Zahl von Aminosäuren in der Prototyp-Sequenz.
So kann zum Beispiel ein derartiges Polynucleotid für ein Polypeptid
codieren, das eine Untersequenz mit wenigstens 10, 15, 20, 25, 30,
35, 40, 45 oder 50 fortlaufenden Aminosäuren aus dem Prototyp-Polypeptid hat. Außerdem kann
die Zahl solcher Untersequenzen, die durch ein Polynucleotid der vorliegenden
Ausführungsform
codiert werden, eine ganze Zahl, ausgewählt aus der Gruppe, bestehend
aus 1 bis 20, z. B. 2, 3, 4 oder 5, sein. Die Untersequenzen können durch
eine beliebige ganze Zahl von Nucleotiden von 1 bis zu der Anzahl
der Nucleotide in der Sequenz, z. B. wenigstens 5, 10, 15, 25, 50,
100 oder 200 Nucleotide, getrennt sein.
-
Wenn
solche Proteine als Immunogen präsentiert
werden, lösen
sie die Produktion von polyklonalen Antikörpern aus, die spezifisch an
ein Prototyp-Polypeptid binden, z. B., aber nicht beschränkt auf,
ein Polypeptid, das durch das Polynucleotid von (a) oder (b) oben
codiert wird. Im Allgemeinen bindet allerdings ein Protein, das
durch ein solches Polypeptid codiert wird, nicht an Antiseren, die
gegen das Prototyp-Polypeptid erzeugt wurden, wenn die Antiseren
vollständig
mit dem Prototyp-Polypeptid immunsorbiert wurden. Verfahren zur
Herstellung und zur Untersuchung auf Antikörperbindungsspezifität/affinität sind auf
dem Fachgebiet gut bekannt. Beispiele für Immunoassayformate umfassen
ELISA, kompetitive Immunoassays, Radioimmunoassays, Western-Blots,
indirekte Immunofluoreszenzassays und dergleichen.
-
In
einem bevorzugten Analysenverfahren bzw. Assayverfahren können vollständig immunsorbierte und
gepoolte Antiseren, die gegen das Prototyp-Polypeptid hervorgebracht
wurden, in einem kompetitiven Bindungsassay verwendet werden, um
das Protein zu untersuchen. Die erforderliche Konzentration des
Prototyp-Polypeptids zur Inhibierung von 50% der Bindung der Antiseren
an das Prototyp-Polypeptid wird bestimmt. Wenn die Menge des Proteins,
die erforderlich ist, um Bindung zu inhibieren, weniger als das
2-Fache der Menge des Prototyp-Proteins
ist, dann wird gesagt, dass es spezifisch an die auf das Immunogen
erzeugten Antiseren bindet. Dementsprechend umfassen die Proteine
der vorliegenden Erfindung allelische Varianten, konservativ modifizierte
Varianten und geringere rekombinante Modifikationen für ein Prototyp-Polypeptid.
-
Ein
solches Polynucleotid codiert gegebenenfalls ein Protein, das ein
Molekulargewicht als das nicht-glycosylierte Protein innerhalb von
20% des Molekulargewichts der nicht-glycosylierten Volllängen-Polypeptide der vorliegenden
Erfindung hat. Das Molekulargewicht kann in einfacher Weise durch
SDS-PAGE unter reduzierenden Bedingungen bestimmt werden. Vorzugsweise
liegt das Molekulargewicht innerhalb 15% eines Volllängen-Polypeptids
der vorliegenden Erfindung, bevorzugter innerhalb von 10 oder 5%
und am bevorzugtesten innerhalb von 3%, 2% oder 1% eines Volllängen-Polypeptids
der vorliegenden Erfindung. Eine Molekulargewichtsbestimmung eines
Proteins kann zweckmäßigerweise
durch SDS-PAGE unter denaturierenden Bedingungen durchgeführt werden.
-
Gegebenenfalls
werden solche Polynucleotide dieser Ausführungsform ein Protein codieren,
das eine spezifische Aktivität
von wenigstens 50%, 60%, 80% oder 90% des nativen, endogenen (d.
h. nicht-isolierten) Volllängen-Polypeptids
der vorliegenden Erfindung hat. Außerdem werden die Proteine,
die durch solche Polynucleotide codiert werden, gegebenenfalls eine
im Wesentlichen ähnliche
Affinitätskonstante
(Km) und/oder katalytische Aktivität (d. h.
die mikroskopische Ratenkonstante, kcat)
wie natives endogenes Volllängenprotein haben.
Der Fachmann wird erkennen, dass der kcat/Km-Wert die Spezifität für kompetitierende Substrate
bestimmt, und er wird oft als die Spezifitätskonstante bezeichnet. Solche
Proteine können
einen kcat/Km-Wert
von wenigstens 10% desjenigen eines nicht-isolierten Volllängen-Polypeptids
der vorliegenden Erfindung, wie er unter Verwendung des endogenen
Substrats jenes Polypeptids gemessen wird, haben. Gegebenenfalls
wird der kcat/Km-Wert
wenigstens 20%, 30%, 40%, 50% und am bevorzugtesten wenigstens 60%,
70%, 80%, 90% oder 95% des kcat/Km-Werts des nicht-isolierten Volllängen-Polypeptids
der vorliegenden Erfindung sein. Eine Bestimmung von kcat,
Km und kcat/Km kann durch eine Reihe von Mitteln, die
dem Fachmann gut bekannt sind, erfolgen. Beispielsweise können die
Anfangsraten (d. h. die ersten 5% oder weniger der Reaktion) unter
Verwendung von schnellen Misch- und Probenahmetechniken (z. B. Techniken
des kontinuierlichen Flusses, des gestoppten Flusses oder des raschen
Abschreckens), unter Verwendung von Flashphotolyse oder Relaxationsverfahren
(z. B. Temperatursprünge)
in Verbindung mit solchen beispielhaften Messverfahren, wie Spektralphotometrie,
Spektralfluorometrie, kernmagnetische Resonanz oder ra dioaktive
Verfahren, bestimmt werden. Kinetische Werte werden zweckdienlicherweise
unter Verwendung eines Lineweaver-Burk- oder Eadie-Hofstee-Plots
erhalten.
-
F. Polynucleotide, die zu den Polynucleotiden
von (A)–(E)
komplementär
sind
-
Die
vorliegende Erfindung bezieht sich auf isolierte Nucleinsäuren, die
Polynucleotide umfassen, welche zu den Polynucleotiden der Abschnitte
A–E oben
komplementär
sind. Wie der Fachmann erkennen wird, paaren sich Basen komplementärer Sequenzen über die
Gesamtheit ihrer Länge
mit den Polynucleotiden A–E (d.
h. sie haben über
ihre gesamte Länge
100% Sequenzidentität).
Komplementäre
Basen assoziieren durch Wasserstoffbindung in doppelsträngige Nucleinsäuren. Beispielsweise
sind die folgenden Basenpaare komplementär: Guanin und Cytosin; Adenin
und Thymin; und Adenin und Uracil.
-
G. Polynucleotide, die Untersequenzen
der Polynucleotide von (A)–(F)
sind
-
Die
vorliegende Erfindung bezieht sich auch auf isolierte Nucleinsäuren, welche
Polynucleotide umfassen, die wenigstens 15 fortlaufende Basen von
den Polynucleotiden von (A) bis (F), wie sie oben diskutiert wurden,
umfassen. Die Länge
des Polynucleotids wird als ganze Zahl angegeben, die aus der Gruppe,
bestehend aus wenigstens 15 bis zu der Länge der Nucleinsäuresequenz,
aus welcher das Polynucleotid eine Untersequenz ist, ausgewählt ist.
So können
z. B. solche Polynucleotide wenigstens 15, 20, 25, 30, 40, 50, 60, 75
oder 100 fortlaufende Nucleotide in der Länge aus den Polynucleotiden
von (A)–(F)
umfassen. Gegebenenfalls kann die Anzahl solcher Untersequenzen
eine beliebige ganze Zahl sein, die aus der Gruppe, bestehend aus
1 bis 20, z. B. 2, 3, 4 oder 5, ausgewählt ist. Die Untersequenzen
können
durch eine beliebige ganze Zahl von Nucleotiden von 1 bis zu der
Anzahl der Nucleotide in der Sequenz, z. B. wenigstens 5, 10, 15,
25, 50, 100 oder 200 Nucleotide, getrennt sein. Solche Untersequenzen
können
strukturelle Charakteristika der Sequenz, aus der sie abgeleitet
sind, umfassen. Alternativ können
den Untersequenzen bestimmte strukturelle Charakteristika der größeren Sequenz,
aus der sie abgeleitet sind, fehlen. Zum Beispiel kann eine Untersequenz
aus einem Polynucleotid, die für
ein Polypeptid codiert, das wenigstens ein lineares Epitop mit einer
Prototyp-Polypeptidsequenz, wie sie in (a) oben bereitgestellt wird,
gemeinsam hat, für
ein mit der Prototyp-Sequenz gemeinsames Epitop codieren. Alternativ
kann die Untersequenz nicht für
ein mit der Prototyp-Sequenz gemeinsames Epitop codieren, sondern
kann verwendet werden, um die größere Sequenz,
z. B. durch Nucleinsäurehybridisierung
mit der Sequenz, aus der es stammt, zu isolieren. Untersequenzen
können
verwendet werden, um eine Genexpression zu modulieren oder zu detektieren,
indem in die Untersequenzen Verbindungen eingeführt werden, die an die Nucleinsäuren binden,
mit diesen interkalieren, diese spalten und/oder vernetzen. Beispiele
für solche
Verbindungen umfassen Acridin-, Psoralen-, Phenanthrolin-, Naphthochinon-,
Daunomycin- oder Chlorethylaminoaryl-Konjugate.
-
Konstruktion von Nucleinsäuren
-
Die
isolierten Nucleinsäuren
der vorliegenden Erfindung können
unter Verwendung von (a) Standard-Rekombinationsverfahren, (b) Synthesetechniken
oder Kombinationen davon herge stellt werden. In einigen Ausführungsformen
werden die Polynucleotide der vorliegenden Erfindung kloniert, amplifiziert
oder in anderer Weise aus einer Monocotyle konstruiert werden. In
bevorzugten Ausführungsformen
ist die Monocotyle Zea mays.
-
Die
Nucleinsäuren
können
zweckdienlicherweise Sequenzen zusätzlich zu einem Polynucleotid
der vorliegenden Erfindung umfassen. Beispielsweise kann eine Multiklonierungsstelle,
die eine oder mehrere Endonuklease-Restriktionsstelle(n) umfasst,
in die Nucleinsäure
insertiert werden, um eine Isolierung des Polynucleotids zu unterstützen. Es
können
auch translatierbare Sequenzen insertiert werden, um die Isolierung
des translatierten Polynucleotids der vorliegenden Erfindung zu
unterstützen.
Eine Hexahistidinmarkersequenz stellt z. B. zweckdienliche Mittel
bereit, um die Proteine der vorliegenden Erfindung zu reinigen.
Ein Polynucleotid der vorliegenden Erfindung kann an einen Vektor,
Adapter oder Linker zur Klonierung und/oder Expression eines Polynucleotids
der vorliegenden Erfindung gebunden werden. Weitere Sequenzen können an
solche Klonierungs- und/oder Expressionssequenzen addiert werden,
um ihre Funktion bei einer Klonierung und/oder Expression zu optimieren,
eine Isolierung des Polynucleotids zu unterstützen oder die Einführung des
Polynucleotids in eine Zelle zu verbessern. Typischerweise ist die
Länge einer
Nucleinsäure
der vorliegenden Erfindung kleiner als die Länge ihres Polynucleotids der
vorliegenden Erfindung, ist weniger als 20 Kilobasenpaare, oft weniger
als 15 kb und häufig
weniger als 10 kb. Eine Verwendung von Klonierungsvektoren, Expressionsvektoren,
Adaptern und Linkern ist gut bekannt und auf dem Fachgebiet umfangreich
beschrieben. Für
eine Beschreibung von verschiedenen Nucleinsäuren siehe z. B. Stratagene
Cloning Systems, Kataloge 1995, 1996, 1997 (La Jolla, CA); und Amersham
Life Sciences, Inc., Katalog '97
(Arlington Heights, IL).
-
A. Rekombinante Verfahren zum Konstruieren
von Nucleinsäuren
-
Die
isolierten Nucleinsäurezusammensetzungen
dieser Erfindung, z. B. RNA, cDNA, genomische DNA oder ein Hybrid
davon, können
aus biologischen Pflanzenquellen unter Verwendung einer Reihe von
Klonierungsmethodologien, die dem Fachmann bekannt sind, erhalten
werden. In einigen Ausführungsformen werden
Oligonucleotidsonden, die unter stringenten Bedingungen selektiv
an die Polynucleotide der vorliegenden Erfindung hybridisieren,
verwendet, um die gewünschte
Sequenz in einer cDNA- oder genomischen DNA-Bibliothek zu identifizieren.
Obgleich eine Isolierung von RNA und die Konstruktion von cDNA und
genomischen Bibliotheken dem Fachmann gut bekannt ist, beleuchtet
das Folgende einige der verwendeten Verfahren.
-
A1. mRNA-Isolierung und -Reinigung
-
Die
Gesamt-RNA aus Pflanzenzellen umfasst solche Nucleinsäuren, wie
mitochondriale RNA, Chlorplasten-RNA, rRNA, tRNA, hnRNA und mRNA.
Eine Gesamt-RNA-Präparation
involviert typischerweise die Lyse von Zellen und die Entfernung
von Proteinen, gefolgt von einer Präzipitation von Nucleinsäuren. Eine
Extraktion von Gesamt-RNA aus Pflanzenzellen kann durch eine Reihe
von Mitteln erreicht werden. Häufig
umfassen Extraktionspuffer ein starkes Detergens, z. B. SDS, und
ein organisches Denaturierungsmittel, z. B. Guanidiniumisothiocyanat, Guanidinhydrochlorid
oder Phenol. Nach Gesamt-RNA-Isolierung wird Poly(A)
+-mRNA
typischerweise aus der verbleibenden RNA unter Verwendung von Oligo(dT)Cellulose
gereinigt. Beispielhafte Gesamt-RNA- und mRNA-Isolierungsprotokolle
sind in Plant Molecular Biology: A Laboratory Manual, Clark, Herausg.,
Springer-Verlag, Berlin (1997); und Current Protocols in Molecular
Biology, Ausubel, et al., Herausg., Greene Publishing and Wiley-Interscience,
New York (1995) beschrieben. Gesamt-RNA- und mRNA-Isolierungskits
sind im Handel erhältlich
von Lieferanten, z. B. Stratagene (La Jolla, CA), Clonetech (Palo
Alto, CA), Pharmacia (Piscataway, NJ) und 5'-3' (Paoli,
PA). Siehe auch die
U.S. Patente
Nr. 5,614,391 und
5,459,253 .
Die mRNA kann zu Populationen mit Größenbereichen von etwa 0,5,
1,0, 1,5, 2,0, 2,5 oder 3,0 kb fraktioniert werden. Die cDNA, die
für jede
dieser Fraktionen synthetisiert wurde, kann nach Größe so ausgewählt werden,
dass sie dieselbe Größe wie ihre
mRNA vor Vektorinsertion hat. Dieses Verfahren hilft, trunkierte
cDNA zu eliminieren, die durch unvollständig reverse transkribierte
mRNA gebildet wurde.
-
A2. Konstruktion einer cDNA-Bibliothek
-
Die
Konstruktion einer cDNA-Bibliothek zieht im Allgemeinen fünf Schritte
mit sich. Erstens, eine Erststrang-cDNA-Synthese wird aus einer
Poly(A)+-mRNA-Matrize unter Verwendung eines
Poly(dT)Primers oder statistischer Hexanucleotide initiiert. Zweitens,
das resultierende RNA-DNA-Hybrid wird in doppelsträngige cDNA
umgewandelt, typischerweise durch eine Kombination von RNAse H und
DNA-Polymerase I (oder Klenow-Fragment). Drittens, die Termini der
doppelsträngigen
cDNA werden an Adaptoren ligiert. Eine Ligation der Adaptoren wird
kohäsive
Enden zur Klonierung produzieren. Viertens, eine Größenselektion
der doppelsträngigen
cDNA eliminiert überschüssige Adaptoren
und Primerfragmente und eliminiert partielle cDNA-Moleküle durch
Abbau von mRNAs oder Versagen von reverser Transkriptase beim Synthetisieren
vollständiger erster
Stränge.
Fünftens,
die cDNAs werden in Klonierungsvektoren ligiert und gepackt. cDNA-Synthese-Protokolle
sind dem Fachmann gut bekannt und werden in Standardwerken beschrieben,
z. B. in: Plant Molecular Biology: A Laboratory Manual, Clark, Herausg.,
Springer-Verlag, Berlin (1997); und Current Protocols in Molecular
Biology, Ausubel et al., Herausg., Greene Publishing and Wiley-Interscience,
New York (1995). cDNA-Synthesekits
sind von einer Vielzahl kommerzieller Lieferanten, z. B. Stratagene
oder Pharmacia, erhältlich.
-
Es
wurde eine Reihe von cDNA-Syntheseprotokollen beschrieben, welche
im Wesentlichen reine Volllängen-cDNA-Bibliotheken
bereitstellen. Im Wesentlichen reine Volllängen-cDNA-Bibliotheken werden konstruiert,
so dass sie wenigstens 90% und bevorzugter wenigstens 93% oder 95%
Volllängeninserts
unter Klonen, die Inserts enthalten, umfassen. Die Insertlänge in solchen
Bibliotheken kann von 0 bis 8, 9, 10, 11, 12, 13 oder mehr Kilobasenpaare
sein. Vektoren zum Akkomodieren von Inserts dieser Größen sind
auf dem Fachgebiet bekannt und im Handel erhältlich. Siehe z. B. Stratagene's lambda ZAP Express
(cDNA-Klonierungsvektor mit 0 bis 12 kb Klonierungskapazität).
-
Ein
beispielhaftes Verfahren zum Konstruieren einer Bibliothek mit mehr
als 95% reiner Volllängen-cDNA
wird von Carninci et al., Genomics, 37: 327–336 (1996) beschrieben. In
diesem Protokoll wird die Cap-Struktur von eukaryotischer mRNA mit
Biotin chemisch markiert. Durch Verwendung von Streptavidin-beschichteten
magnetischen Perlen werden nur die Volllängen-Erststrang-cDNA/mRNA-Hybride selektiv
nach RNase I-Behandlung wiedergewonnen. Das Verfahren stellt eine
Bibliothek hoher Ausbeute mit einer zweifelsfreien Repräsentation
der Ausgangs-mRNA-Population bereit. Andere Verfahren für die Produktion
von Volllängenbibliotheken
sind auf dem Fachgebiet bekannt. Siehe z. B. Edery et al., Mol.
Cell Biol., 15(6): 3363–3371 (1995);
und die PCT-Anmeldung
WO 96/34981 .
-
A3. Normalisierte oder subtrahierte cDNA
Bibliotheken
-
Eine
nicht-normalisierte cDNA-Bibliothek stellt die mRNA-Population des
Gewebes dar, aus dem sie hergestellt ist. Da einzelne Klone durch
Klone, die von hoch exprimierten Genen stammen, ausgemustert werden,
kann ihre Isolierung mühsam
sein. Eine Normalisierung einer cDNA-Bibliothek ist das Verfahren
der Schaffung einer Bibliothek, in der jeder Klon gleichermaßen repräsentiert
ist.
-
Es
ist eine Reihe von Ansätzen
bekannt, um cDNA-Bibliotheken zu normalisieren. Ein Ansatz basiert auf
einer Hybridisierung an genomische DNA. Die Häufigkeit jeder hybridisierten
cDNA in der resultierenden normalisierten Bibliothek wäre proportional
zu der jedes korrespondierenden Gens in der genomischen DNA. Ein
anderer Ansatz basiert auf Kinetik. Wenn ein cDNA-Re-Annealing einer
Kinetik zweiter Ordnung folgt, lagern sich seltenere Spezies weniger
schnell an und die verbleibende einzelsträngige Fraktion von cDNA wird im
Verlauf der Hybridisierung fortschreitend stärker normalisiert. Ein spezifischer
Verlust einer cDNA-Spezies tritt ungeachtet ihrer Abundanz bei keinem
Cot-Wert auf. Eine Konstruktion von normalisierten Bibliotheken wird
beschrieben in: Ko, Nucl. Acids. Res., 18(19): 5705–5711 (1990);
Patanjali et al., Proc. Natl. Acad. U.S.A., 88: 1943–1947 (1991);
U.S. Patenten 5,482,685 und
5,637,685 . In einem beispielhaften
Verfahren, das von Soares et al. beschrieben wurde, resultierte
eine Normalisierung in einer Verringerung der Abundanz von Klonen
von einem Bereich einer vierstelligen Größenordnung zu einem engen Bereich
nur einer Größenordnung. Proc.
Natl. Acad. Sci. USA, 91: 9228–9232
(1994).
-
Subtrahierte
cDNA-Bibliotheken sind ein anderes Mittel, um den Verhältnisanteil
von weniger abundanten cDNA-Spezies zu erhöhen. Bei dieser Vorgehensweise
wird cDNA, die aus einem mRNA-Pool hergestellt wurde, durch Hybridisierung
an Sequenzen verarmt, die in einem zweiten mRNA-Pool vorliegen.
Die cDNA:mRNA-Hybride werden entfernt und der verbleibende unhybridisierte
cDNA-Pool wird bezüglich
Sequenzen, die in jenem Pool einmal vorhanden sind, angereichert.
Siehe Foote et al., in Plant Molecular Biology: A Laboratory Manual,
Clark, Herausg., Springer-Verlag, Berlin (1997); Kho und Zarbl,
Technique, 3(2): 58–63 (1991);
Sive und St. John, Nucl. Acid. Res., 16(22): 10937 (1988); Current
Protocols in Molecular Biology, Ausubel et al., Herausg., Greene
Publishing and Wiley-Interscience, New York (1995); und Swaroop
et al., Nucl. Acids Res., 19(8): 1954 (1991). cDNA-Subtraktions-Kits
sind im Handel erhältlich.
Siehe z. B. PCR-Select (Clontech).
-
A4. Konstruktion einer genomischen Bibliothek
-
Um
genomische Bibliotheken zu konstruieren, werden große Segmente
genomischer DNA durch Randomfragmentierung, z. B. unter Verwendung
von Restriktionsendonukleasen, erzeugt und mit Vektor-DNA unter
Bildung von Concatemeren, welche in den geeigneten Vektor gepackt
werden können,
ligiert. Methodologien zur Erreichung dieser Ziele und Sequenzierungsverfahren
zur Bestätigung
der Sequenz von Nucleinsäuren
sind auf dem Fachgebiet bekannt. Beispiele für geeignete molekularbiologische
Techniken und Instruktionen, die ausreichend sind, um Fachleute
durch viele Konstruktions-, Klonierungs- und Screening-Methodologien zu
leiten, werden bei Sambrook et al., Molecular Cloning: A Laboratory
Manual, 2. Ausgabe, Cold Spring Harbor Laboratory, Bd. 1–3 (1989),
Methods in Enzymology, Bd. 152: Guide to Molecular Cloning Techniques, Berger
und Kimmel, Herausg., San Diego: Academic Press, Inc. (1987), Current
Protocols in Molecular Biology, Ausubel, et al., Herausg., Greene
Publishing and Wiley-Interscience, New York (1995); Plant Molecular
Biology: A Laboratory Manual, Clark, Herausg., Springer-Verlag,
Berlin (1997) gefunden. Kits zur Konstruktion genomischer Bibliotheken
sind ebenfalls im Handel erhältlich.
-
A5. Nucleinsäure-Screening- und -Isolierungs-Verfahren
-
Die
cDNA- oder die genomische Bibliothek kann unter Verwendung einer
Sonde durchgemustert werden, welche auf der Sequenz eines Polynucleotids
der vorliegenden Erfindung, z. B. solchen, die hierin offenbart
sind, basiert. Sonden können
verwendet werden, um mit genomischen DNA- oder cDNA-Sequenzen zu hybridisiersen,
um homologene Gene in derselben oder einer unterschiedlichen Pflanzenspezies
zu isolieren. Der Fachmann wird erkennen, dass verschiedene Grade
der Hybridisierungsstringenz in dem Assay angewendet werden können und
ob die Hybridisierung oder das Waschmedium stringent sein können. Wenn
die Hybridisierungsbedingungen stringenter werden, muss es einen
größeren Komplementaritätsgrad zwischen
der Sonde und dem Target geben, damit eine Duplexbildung auftritt.
Der Stringenzgrad kann durch Temperatur, Ionenstärke, pH und das Vorliegen eines
partiell denaturierenden Lösungsmittels,
z. B. Formamid, kontrolliert werden. Beispielsweise wird die Hybridisierungsstringenz
zweckmäßigerweise
verändert,
indem die Polarität der
Reaktantenlösung
durch Manipulation der Formamidkonzentration innerhalb des Bereichs
von 0% bis 50% verändert
wird. Der Komplementaritätsgrad
(Sequenzidentität),
der für
eine detektierbare Bindung erforderlich ist, wird entsprechend der
Stringenz des Hybridisierungsmediums und/oder Waschmediums variieren.
Der Komplementaritätsgrad
wird optimal 100% sein; allerdings sollte einzusehen sein, dass
geringere Sequenzvariationen in den Sonden und Primern zur Reduzierung
der Stringenz des Hybridisierungs- und/oder Waschmediums kompensiert werden
können.
-
Die
Nucleinsäuren
von Interesse können
auch unter Anwendung von Amplifikationstechniken aus Nucleinsäureproben
amplifiziert werden. Beispielsweise kann die Polymerase-Ketten- Reaktions(PCR)-Technik eingesetzt
werden, um die Sequenzen von Polynucleotiden der vorliegenden Erfindung
und verwandten Genen direkt aus genomischen DNA- oder cDNA-Bibliotheken zu amplifizieren.
Eine PCR- und andere in vitro-Amplifikationsverfahren können auch
einsetzbar sein, um z. B. Nucleinsäuresequenzen, die für zu exprimierende
Proteine codieren, zu klonieren, um Nucleinsäuren zur Verwendung als Sonden
zum Detektieren des Vorliegens der gewünschten mRNA in Proben, zur
Nucleinsäuresequenzierung
oder für
andere Zwecke herzustellen. Beispiele für Techniken, die ausreichend
sind, um einen Fachmann durch in vitro-Amplifikationsverfahren zu leiten, werden
in Berger, Sambrook und Ausubel, sowie Mullis et al.,
U.S. Patent Nr. 4,683,202 (1987);
und PCR Protocols A Guide to Methods and Applications, Innis et
al., Herausg., Academic Press Inc., San Diego, CA (1990) gefunden.
Im Handel erhältliche
Kits zur genomischen PCR-Amplifikation sind auf dem Fachgebiet bekannt.
Siehe z. B. Advantage-GC Genomic PCR Kit (Clontech). Das T4-Gen-32-Protein
(Böhringer
Mannheim) kann eingesetzt werden, um die Ausbeute von langen PCR-Produkten
zu verbessern.
-
PCR-basierte
Screening-Verfahren wurden ebenfalls beschrieben. Wilfinger et al.
beschreiben ein PCR-basiertes Verfahren, bei dem die längste cDNA
im ersten Schritt identifiziert wird, so dass unvollständig Klone
aus einer Studie eliminiert werden können. BioTechniques, 22(3):
481–486
(1997). In diesem Verfahren wird ein Primerpaar mit einem Primer,
der an dem 5'-Ende
des Sense-Strangs der gewünschten
cDNA anlagert und der andere Primer an dem Vektor anlagert, synthetisiert.
Klone werden gepoolt, um ein Screening in großem Maßstab zu ermöglichen.
Durch dieses Verfahren wird der längstmögliche Klon unter Kandidatenklonen identifiziert.
Außerdem
wird das PCR-Produkt allein als ein Diagnostikum für das Vorliegen
der gewünschten cDNA
verwendet und es wird nicht das PCR-Produkt selbst verwendet. Solche
Verfahren sind insbesondere in Kombination mit einer Volllängen-cDNA-Konstruktionsmethodologie
wirksam.
-
B. Syntheseverfahren zum Konstruieren
von Nucleinsäuren
-
Die
isolierten Nucleinsäuren
der vorliegenden Erfindung können
auch durch direkte chemische Synthese nach z. B. den folgenden Verfahren
hergestellt werden: das Phosphotriesterverfahren von Narang et al., Meth.
Enzymol. 68: 90–99
(1979); das Phosphodiesterverfahren von Brown et al., Meth. Enzymol.
68: 109–151 (1979);
das Diethylphosphoramiditverfahren von Beaucage et al., Tetra. Lett.
22: 1859–1862
(1981); das Festphasen-Phosphoramidittriester-Verfahren, das von Beaucage und Caruthers,
Tetra. Letts. 22(20): 1859–1862 (1981)
beschrieben wurde, z. B. unter Verwendung eines automatischen Synthesizers,
z. B. wie er bei Needham-VanDevanter
et al., Nucleic Acids Res., 12: 6159–6168 (1984) beschrieben wurde,
und das Festphasenverfahren gemäß
U.S. Patent Nr. 4,458,066 .
Eine chemische Synthese produziert im Allgemeinen ein einzelsträngiges Oligonucleotid.
Dieses kann durch Hybridisierung mit einer komplementären Sequenz
oder durch Polymerisation mit einer DNA-Polymerase unter Verwendung
des Einzelstrangs als Matrize in doppelsträngige DNA umgewandelt werden.
Ein Fachmann wird erkennen, dass, da eine chemische Synthese von
DNA auf Sequenzen von etwa 100 Basen be schränkt ist, längere Sequenzen durch die Ligation
von kürzeren
Sequenzen erhalten werden können.
-
Rekombinante Expressionskassetten
-
Die
vorliegende Erfindung stellt ferner rekombinante Expressionskassetten
bereit, die eine Nucleinsäure
der vorliegenden Erfindung umfassen. Eine Nucleinsäuresequenz,
die für
das gewünschte
Polynucleotid der vorliegenden Erfindung codiert, z. B. eine cDNA
oder eine genomische Sequenz, die für ein Volllängenpolypeptid der vorliegenden
Erfindung codiert, kann verwendet werden, um eine rekombinante Expressionskassette
zu konstruieren, die in die gewünschte
Wirtszelle eingeführt
werden kann. Eine rekombinante Expressionskassette wird typischerweise
ein Polynucleotid der vorliegenden Erfindung funktionell verknüpft mit
Transkriptionsinitiationsregulatorsequenzen umfassen, welche die
Transkription des Polynucleotids in der vorgesehenen Wirtszelle,
z. B. Geweben einer transformierten Pflanze, steuern werden.
-
Pflanzenexpressionsvektoren
können
z. B. umfassen: (1) ein kloniertes Pflanzengen unter der Transkriptionskontrolle
von 5'- und 3'-regulatorischen
Sequenzen bzw. Regulatorsequenzen und (2) einen dominanten selektierbaren
Marker. Solche Pflanzenexpressionsvektoren können, wenn dies gewünscht wird,
auch eine Promotor-regulatorische Region (z. B. eine, die induzierbare
oder konstitutive, Umwelt- oder Entwicklungs-regulierte oder Zell-
oder Gewebe-spezifische/selektive
Expression verleiht), eine Transkriptionsinitiationsstartstelle,
eine Ribosomenbindungsstelle, ein RNA-Prozessierungssignal, eine
Transkriptionsterminationsstelle und/oder ein Polyadenylierungssignal
enthalten.
-
Es
kann ein Pflanzenpromotorfragment verwendet werden, welches die
Expression eines Polynucleotids der vorliegenden Erfindung in allen
Geweben einer regenerierten Pflanze steuern wird. Solche Promotoren
werden hierin als „konstitutive" Promotoren bezeichnet
und sind unter den meisten Umweltbedingungen und Zuständen der
Entwicklung oder Zelldifferenzierung aktiv. Beispiele für konstitutive
Promotoren umfassen die Blumenkohlmosaikvirus(CaMV)-35S-Transkriptionsinitiationsregion,
den 1'- oder 2'-Promotor, der aus T-DNA
von Agrobacterium tumefaciens abgeleitet ist, den Ubiquitin-1-Promotor,
den Smas-Promotor, den Cinnamylalkoholdehydrogenasepromotor (
U.S. Patent Nr. 5,683,439 ),
den Nos-Promotor, den pEMu-Promotor, den
Rubisco-Promotor, den GRP1-8-Promotor, den Actinpromotor, den F3.7-Promotor und andere
Transkriptionsinitiationsregionen aus verschiedenen Pflanzengenen,
die einem Fachmann bekannt sind.
-
Alternativ
kann der Pflanzenpromotor eine Expression eines Polynucleotids der
vorliegenden Erfindung in einem spezifischen Gewebe steuern oder
kann in anderer Weise unter genauerer Umwelt- oder Entwicklungskontrolle
sein. Solche Promotoren werden hier als „induzierbare" Promotoren bezeichnet.
Umweltbedingungen, die eine Transkription durch induzierbare Promotoren
bewirken können,
umfassen Angriff durch Pathogene, anaerobe Bedingungen oder das
Vorliegen von Licht. Beispiele für
induzierbare Promotoren sind der Adh1-Promotor, der durch Hypoxie
oder Kältestress
induzierbar ist, den Hsp70-Promotor, der durch Hitzestress induzierbar
ist, und der PPDK-Promotor, der durch Licht induzierbar ist.
-
Beispiele
für Promotoren
unter Entwicklungskontrolle umfassen Promotoren, die eine Transkription nur
oder vorzugsweise in bestimmten Geweben, z. B. Blättern, Wurzeln,
Früchten,
Samen oder Blüten,
initiieren. Das Wirken eines Promotors kann auch in Abhängigkeit
von seiner Lage im Genom variieren. So kann ein induzierbarer Promotor
in bestimmten Orten vollständig
oder partiell konstitutiv werden.
-
Sowohl
heterologe als auch nicht-heterologe (d. h. endogene) Promotoren
können
verwendet werden, um eine Expression der Nucleinsäuren der
vorliegenden Erfindung zu steuern. Diese Promotoren können zum Beispiel
auch in rekombinanten Expressionskassetten verwendet werden, um
eine Expression von Antisense-Nucleinsäuren zu steuern, um die Konzentration
und/oder die Zusammensetzung der Proteine der vorliegenden Erfindung
in einem gewünschten
Gewebe zu reduzieren, zu erhöhen
oder zu verändern.
So wird das Nucleinsäurekonstrukt
in einigen Ausführungsformen
einen Promotor, der in einer Pflanzenzelle, z. B. in Zea mays, funktionell
ist, funktionell mit einem Polynucleotid der vorliegenden Erfindung
verknüpft,
umfassen. Promotoren, die in diesen Ausführungsformen einsetzbar sind,
umfassen die endogenen Promotoren, die eine Expression eines Polypeptids
der vorliegenden Erfindung steuern.
-
In
einigen Ausführungsformen
können
isolierte Nucleinsäuren,
die als Promotor- oder Enhancer-Elemente dienen, in die geeignete
Position (im Allgemeinen stromaufwärts) einer nicht-heterologen Form
eines Polynucleotids der vorliegenden Erfindung eingeführt werden,
um so die Expression eines Polynucleotids der vorliegenden Erfindung
nach oben oder nach unten zu regulieren. Zum Beispiel können endogene
Promotoren in vivo durch Mutation, Deletion und/oder Substitution
verändert
werden (siehe Kmiec,
U.S. Patent
5,565,350 ; Zarling et al.,
PCT/US93/03868 )
oder isolierte Promotoren können
in der geeigneten Orientierung und im geeigneten Abstand von einem
Gen der vorliegenden Erfindung in eine Pflanzezelle eingeführt werden,
um so die Expression des Gens zu kontrollieren. Eine Genexpression
kann unter Bedingungen, die für
ein Pflanzenwachstum geeignet sind, moduliert werden, um so die
Gesamtkonzentration und/oder die Zusammensetzung der Polypeptide
der vorliegenden Erfindung in einer Pflanzenzelle zu verändern. Demnach
stellt die vorliegende Erfindung Zusammensetzungen und Verfahren
zur Herstellung von heterologen Promotoren und/oder Enhancern bereit,
die funktionell an eine native, endogene (d. h. nicht-heterologe)
Form eines Polynucleotids der vorliegenden Erfindung gebunden sind.
-
Methoden
zur Identifizierung von Promotoren mit einem besonderen Expressionsmuster
bezüglich
z. B. Gewebetyp, Zelltyp, Entwicklungsstufe und/oder Umweltbedingungen
sind auf dem Fachgebiet gut bekannt. Siehe z. B. The Maize Handbook,
Kapitel 114–115,
Freeling und Walbot, Herausg., Springer, New York (1994); Corn and
Corn Improvement, 3. Ausgabe, Kapitel 6, Sprague and Dudley, Herausg.,
American Society of Agronomy, Madison, Wisconsin (1988). Ein typischer
Schritt bei den Promotorisolierungsverfahren ist die Identifizierung
von Genprodukten, die mit einem gewissen Spezifitätsgrad im
Targetgewebe exprimiert werden. In dem Bereich von Methodologien
sind: differentielle Hybridisierung an cDNA-Bibliotheken; subtraktive
Hybridisierung; differentieller Display; differentielle 2-D-Proteingelelektrophorese;
DNA-Sondenarrays; und Isolierung von Proteinen, von denen bekannt
ist, dass sie mit gewisser Spezifität im Targetgewebe exprimiert
werden. Solche Verfahren sind dem Fachmann gut bekannt. Im Handel
verfügbare
Produkte zu Identifizierung von Promotoren sind auf dem Fachgebiet
bekannt, z. B. Clontech's
(Palo Alto, CA) Universal Genome Walker Kit.
-
Für die Protein-basierten
Verfahren ist es hilfreich, die Aminosäuresequenz für wenigstens
einen Teil des identifizierten Proteins zu erhalten und dann die
Proteinsequenz als die Basis zur Herstellung einer Nucleinsäure zu verwenden,
die als Sonde verwendet werden kann, um entweder genomische DNA
direkt oder vorzugsweise einen cDNA-Klon aus einer Bibliothek, die
aus dem Targetgewebe hergestellt wurde, zu identifizieren. Sobald
ein solcher cDNA-Klon identifiziert wurde, kann die Sequenz verwendet
werden, um die Sequenz am 5'-Ende
des Transkripts des angegebenen Gens zu identifizieren. Für eine differentielle
Hybridisierung, eine subtraktive Hybridisierung und einen differentiellen
Display wird die identifizierte Nucleinsäure, wie sie im Targetgewebe
angereichert ist, verwendet, um die Sequenz am 5'-Ende des Transkripts des angegebenen
Gens zu identifizieren. Sobald solche Sequenzen ausgehend entweder
von Proteinsequenzen oder Nucleinsäuresequenzen identifiziert
sind, kann eine beliebige dieser identifizierten Sequenzen, die
aus dem Gentranskript ist, verwendet werden, um eine genomische
Bibliothek, die aus dem Targetorganismus hergestellt wurde, durchzumustern.
Verfahren zur Identifizierung und Bestätigung der Transkriptionsstartstelle
sind auf dem Fachgebiet gut bekannt.
-
In
dem Verfahren zur Isolierung von Promotoren, die unter bestimmten
Umweltbedingungen oder bestimmtem Umweltstress oder in spezifischen
Geweben oder in bestimmten Entwicklungsstufen exprimiert werden,
wird eine Reihe von Genen identifiziert, die unter den gewünschten
Umständen,
in dem gewünschten
Gewebe oder in der gewünschten
Stufe exprimiert werden. Eine weitere Analyse wird eine Expression
jedes bestimmten Gens in einem anderen Gewebe oder in mehreren anderen
Geweben der Pflanze zeigen. Man kann einen Promotor mit Aktivität in dem
gewünschten
Gewebe oder bei dem gewünschten
Zustand identifizieren, jedoch hat dieser keine Aktivität in irgendeinem
anderen allgemeinen Gewebe.
-
Um
die Promotorsequenz zu identifizieren, werden die 5'-Teile der hier beschriebenen
Klone auf Sequenzen analysiert, die für Promotorsequenzen charakteristisch
sind. Beispielsweise umfassen Promotorsequenzelemente, die TATA-Box-Konsensus-Sequenz
(TATAAT), die üblicherweise
ein AT-reicher Abschnitt aus 5–10
bp ist, der etwa 20 bis 40 Basenpaare stromaufwärts der Transkriptionsstartstelle
lokalisiert ist. Die Identifizierung der TATA-Box ist auf dem Fachgebiet
gut bekannt. Ein Weg, um den Ort dieses Elements vorauszusagen,
besteht z. B. in der Identifizierung der Transkriptionsstartstelle
unter Verwendung von Standard-RNA-Kartierungstechniken, z. B. Primerextension,
S1-Analyse und/oder RNase-Schutz. Um das Vorliegen der AT-reichen
Sequenz zu bestätigen,
kann eine Struktur-Funktions-Analyse durchge führt werden, welche eine Mutagenese
der vermeintlichen Region und eine quantitative Bestimmung der Mutationswirkung
auf die Expression eines verknüpften
stromabwärts
gelegenen Reportergens involviert. Siehe z. B. The Maize Handbook,
Kapitel 114, Freeling und Walbot, Herausg., Springer, New York,
(1994).
-
In
Pflanzen gibt es außerdem
weiter stromaufwärts
von der TATA-Box in den Positionen –80 bis –100 typischerweise ein Promotorelement
(d. h. die CAAT-Box) mit einer Reihe von Adeninen, die das Trinucleotid G
(oder T) N G umgeben. J. Messing et al., in Genetic Engineering
in Plants, Kosage, Meredith und Hollaender, Herausg., S. 221–227 (1983).
In Mais gibt es keine gut konservierte CAAT-Box, aber es gibt einige
kurze, konservierte Protein-Bindungsmotive stromaufwärts der
TATA-Box. Diese umfassen Motive für die trans-wirkenden Transkriptionsfaktoren,
die bei der Lichtregulation, bei der anaeroben Induktion, der hormonalen
Regulierung oder der Anthocyanin-Biosynthese, wie es für jedes
Gen passend ist, involviert sind.
-
Sobald
Promotor- und/oder Gensequenzen bekannt sind, wird eine Region geeigneter
Größe aus der genomischen
DNA ausgewählt,
welche 5' zum Transkriptionsstart
oder der Translationsstartstelle ist, und solche Sequenzen werden
dann mit einer codierenden Sequenz verknüpft. Wenn die Transkriptionsstartstelle
als der Fusionspunkt verwendet wird, kann eine beliebige einer Reihe
von möglichen
5'-untranslatierten
Regionen zwischen der Transkriptionsstartstelle und der partiellen
codierenden Sequenz verwendet werden. Wenn die Translationsstartstelle
am 3'-Ende des spezifischen
Promotors verwendet wird, dann wird sie direkt mit dem Methionin-Startcodon
einer codierenden Sequenz verknüpft.
-
Wenn
eine Polypeptidexpression gewünscht
wird, ist sie im Allgemeinen wünschenswert,
um eine Polyadenylierungsregion am 3'-Ende einer codierenden Polynucleotidregion
einzuschließen.
Die Polyadenylierungsregion kann aus dem natürlichen Gen, aus einer Vielzahl
anderer Pflanzengene oder aus T-DNA stammen. Die 3'-Endsequenz, die
zu addieren ist, kann zum Beispiel aus den Nopalinsynthase- oder
Ocotpinsynthase-Genen stammen oder kann alternativ aus einem anderen
Pflanzengen oder weniger bevorzugt aus einem anderen eukaryotischen
Gen stammen.
-
Eine
Intronsequenz kann an die 5'-untranslatierte
Region oder die codierende Sequenz der partiellen codierenden Sequenz
addiert werden, um die Menge der reifen Message zu erhöhen, die
im Cytosol akkumuliert. Der Einschluss eines spleißbaren Introns
in die Transkriptionseinheit sowohl in Pflanzen- als auch Tierexpressionskonstrukten
führt erwiesenermaßen zur
Erhöhung
der Genexpression sowohl auf dem mRNA- als auch dem Protein-Level
bis zum 1000-fachen.
Buchman und Berg, Mol. Cell Biol. 8: 4395–4405 (1988); Callis et al.,
Genes Dev. 1: 1183–1200
(1987). Eine solche Intronverstärkung
der Genexpression ist typischerweise am größten, wenn die Platzierung
nahe dem 5'-Ende
der Transkriptionseinheit ist. Eine Verwendung von Maisintrons,
des Adh1-S-Introns 1, 2 und 6, des Bronze-1-Introns, ist auf dem
Fachgebiet bekannt. Siehe allgemein The Maize Handbook, Kapitel
116, Freeling und Walbot, Herausg. Springer, New York (1994).
-
Der
Vektor, der die Sequenzen aus einem Polynucleotid der vorliegenden
Erfindung umfasst, wird typischerweise ein Markergen umfassen, welches
einen selektierbaren Phänotyp
an Pflanzenzellen verleiht. Üblicherweise
wird das selektierbare Markergen für Antibiotikaresistenz codieren,
wobei geeignete Gene Gene umfassen, die für Resistenz gegen das Antibiotikum
Spectinomycin codieren (z. B. das aada-Gen), das Streptomycinphosphotransferase(SPT)-Gen,
das für
Streptomycinresistenz codiert, das Neomycinphosphotransferase(NPTII)-Gen,
das für
Kanamycin- oder Geniticinresistenz codiert, das Hygromycinphosphotransferase(HPT)-Gen,
das für
Hygromycinresistenz codiert, Gene, die für Resistenz gegenüber Herbiziden,
welche unter Inhibierung der Wirkung von Acetolactatsynthase (ALS)
wirken, insbesondere die Herbizide vom Sulfonylharnstofftyp (z.
B. das Acetolactatsynthase(ALS)-Gen, das Mutationen enthält, die
zu einer solchen Resistenz führen,
insbesondere die S4- und/oder Hra-Mutationen), codieren, Gene, die
für Resistenz
gegen Herbizide codieren, welche unter Inhibierung der Wirkung von
Glutaminsynthase wirken, z. B. Phosphinotricin oder Basta (z. B.
das bar-Gen), oder andere derartige Gene, die auf dem Fachgebiet
bekannt sind. Das bar-Gen codiert für Resistenz gegen das Herbizid
Basta, das nptII-Gen codiert für
Resistenz gegen die Antibiotika Kanamycin und Geniticin und das
ALS-Gen codiert für
Resistenz gegen das Herbizid Chlorsulforon.
-
Typische
Vektoren, die für
die Expression von Genen in höheren
Pflanzen verwendbar sind, sind auf dem Fachgebiet gut bekannt und
umfassen Vektoren, die von dem Tumor-induzierenden (Ti)-Plasmid von Agrobacterium
tumefaciens abgeleitet sind, die von Rogers et al., Meth. In Enzymol.,
153: 253–277
(1987) beschrieben wurden. Diese Vektoren sind in Pflanzen-integrierende
Vektoren derart, dass die Vektoren bei Transformation einen Teil
der Vektor-DNA in
das Genom der Wirtspflanze integrieren. Beispiele für A. tumefaciens-Vektoren,
die hier verwendbar sind, sind die Plasmide pKYLX6 und pKYLX7 von
Schardl et al., Gene, 61: 1–11
(1987) und Berger et al., Proc. Natl. Acad. Sci. USA, 86: 8402–8406 (1989).
Ein anderer hier verwendbarer Vektor ist das Plasmid pBI101.2, der
von Clontech Laboratories, Inc. (Palo Alto, CA) verfügbar ist.
-
Ein
Polynucleotid der vorliegenden Erfindung kann entweder in Sense-
oder Antisense-Orientierung, wie
es gewünscht
ist, exprimiert werden. Es wird einzusehen sein, dass die Kontrolle
der Genexpression entweder in Sense- oder Antisense-Orientierung
einen direkten Einfluss auf die beobachtbaren Pflanzencharakteristika
haben kann. Antisense-Technologie kann zweckmäßigerweise zur Genexpression
in Pflanzen verwendet werden. Um dies zu erreichen, wird ein Nucleinsäuresegment
aus dem gewünschten
Gen kloniert und funktionell mit einem Promotor verknüpft, so
dass der Antisense-Strang von RNA transkribiert werden wird. Das
Konstrukt wird dann in Pflanzen transformiert und der Antisense-Strang
von RNA wird produziert. Es wurde gezeigt, dass in Pflanzenzellen
Antisense-RNA eine Genexpression inhibiert, indem sie die Akkumulation von
mRNA verhindert, welche für
das Enzym von Interesse codiert, siehe z. B. Sheehy et al., Proc.
Nat'l. Acad. Sci.
(USA) 85: 8805–8809
(1988); und Hiatt et al.,
U.S
Patent Nr. 4,801,340 .
-
Ein
anderes Suppressionsverfahren ist Sense-Suppression. Die Einführung von
Nucleinsäure,
die in der Sense-Orientierung konfiguriert ist, erwies sich als
wirksames Mittel, durch welches die Transkription von Targetgenen
blockiert wird. Für
ein Beispiel der Verwendung dieses Verfahrens, um eine Expression
von endogenen Genen zu exprimieren, siehe Napoli et al., The Plant
Cell 2: 279–289
(1990) und
U.S Patent Nr. 5,034,323 .
-
Es
können
auch katalytische RNA-Moleküle
oder Ribozyme verwendet werden, um eine Expression von Pflanzengenen
zu inhibieren. Es ist möglich,
Ribozyme zu entwickeln, die sich spezifisch mit tatsächlich einer
beliebigen Target-RNA paaren und die Phosphodiesterhauptkette an
einer spezifischen Stelle spalten, wodurch die Target-RNA funktionell
inaktiviert wird. Bei der Durchführung
dieser Spaltung wird das Ribozym nicht selbst verändert und
ist somit zum Recycling und Spalten anderer Moleküle fähig, was
es zu einem echten Enzym macht. Der Einschluss von Ribozymsequenzen
in Antisense-RNAs verleiht diesen RNA-Spaltungsaktivität, wodurch
die Aktivität
der Konstrukte erhöht
wird. Die Entwicklung und die Verwendung von Target-RNA-spezifischen Ribozymen
wird bei Haseloff et al., Nature 334: 585–591 (1988) beschrieben.
-
Es
kann eine Vielzahl von Vernetzungsmitteln, Alkylierungsmitteln und
Radikale-erzeugenden
Spezies als vorstehende Gruppen an Polynucleotiden der vorliegenden
Erfindung verwendet werden, um Nucleinsäuren zu binden, zu markieren,
zu detektieren und/oder zu spalten. Beispielsweise beschreiben Vlassov,
V. V., et al., Nucleic Acids Res (1986) 14: 4065–4076 die kovalente Bindung
eines einzelsträngigen
DNA-Fragments mit alkylierenden Derivaten von Nucleotiden, die zu
Targetsequenzen komplementär
sind. Ein Bericht über eine ähnliche
Arbeit derselben Gruppe ist der von Knorre, D. G., et al., Biochimie
(1985) 67: 785–789.
Iverson und Dervan zeigten auch eine Sequenz-spezifische Spaltung
von einzelsträngiger
DNA, vermittelt durch Einbau eines modifizierten Nucleotids, welches
fähig war,
eine Spaltung zu aktivieren (J Am Chem Soc (1987) 109: 1241–1243).
Meyer, R. B., et al., J Am Chem Soc (1989) 111: 8517–8519 bewirkten
eine kovalente Vernetzung an ein Targetnucleotid unter Verwendung
eines Alkylierungsmittels, das komplementär zu der einzelsträngigen Targetnucleotidsequenz
war. Eine photoaktivierte Vernetzung an einzelsträngige Oligonucleotide, vermittelt
durch Psoralen, wurde von Lee, B. L., et al., Biochemistry (1988)
27: 3197–3203
offenbart. Die Verwendung einer Vernetzung in Tripelhelix-bildenden
Sonden wurde auch von Home, et al., J Am Chem Soc (1990) 112: 2435–2437 offenbart.
Eine Verwendung von N4, N4-Ethanocytosin als Alkylierungsmittel
zur Vernetzung an einzelsträngige
Oligonucleotide wurde auch von Webb und Matteucci, J Am Chem Soc
(1986) 108: 2764–2765;
Nucleic Acids Res (1986) 14: 7661–7674; Feteritz et al., J.
Am. Chem. Soc. 113: 4000 (1991) beschrieben. Auf dem Fachgebiet
sind verschiedene Verbindungen bekannt, um Nucleinsäuren zu
binden, zu detektieren, zu markieren und/oder zu spalten. Siehe
z. B. die
U.S.-Patente Nrn. 5,543,507 ;
5,672,593 ;
5,484,908 ;
5,256,648 ; und
5,681,941 .
-
Die
Erfindung stellt ein isoliertes Protein bereit, das Cellulosesynthaseaktivität hat und
ausgewählt
ist aus:
- (a) einem Polypeptid von SEQ ID NO:
6, 10 oder 26; und
- (b) einem Polypeptid, das durch eine Nucleinsäure der
Erfindung codiert wird.
-
Wie
der Fachmann erkennen wird, umfasst die vorliegende Erfindung katalytisch
aktive Polypeptide der vorliegenden Erfindung (d. h. Enzyme). Katalytisch
aktive Polypeptide haben eine spezifische Aktivität von wenigstens
20%, 30% oder 40% und vorzugsweise wenigstens 50%, 60% oder 70%
und am bevorzugtesten wenigstens 80%, 90% oder 95% der des nativen
(nicht-synthetischen)
endogenen Polypeptids. Darüber
hinaus ist die Substratspezifität
(kcat/Km) gegebenenfalls
im Wesentlichen ähnlich
der des nativen (nicht-synthetischen), endogenen Polypeptids. Typischerweise
wird die Km wenigstens 30%, 40% oder 50%
derjenigen des nativen (nicht-synthetischen),
endogenen Polypeptids sein und bevorzugter wenigstens 60%, 70%,
80% oder 90% sein. Verfahren zur Analyse und quantitativen Messung
der enzymatischen Aktivität
und der Substratspezifität
(kcat/Km) sind dem
Fachmann gut bekannt.
-
Im
Allgemeinen werden die Proteine der vorliegenden Erfindung, wenn
sie als ein Immunogen präsentiert
werden, die Produktion eines Antikörpers auslösen, der spezifisch reaktiv
auf ein Polypeptid der vorliegenden Erfindung ist. Ferner werden
die Proteine der vorliegenden Erfindung nicht an Antiserum binden,
das gegen ein Polypeptid der vorliegenden Erfindung erzeugt wurde,
welches mit demselben Polypeptid vollständig immunsorbiert wurde. Immunoassays
zur Bestimmung der Bindung sind dem Fachmann gut bekannt. Ein bevorzugter
Immunoassay ist ein kompetitiver Immunoassay, wie er unten diskutiert
wird. Somit können
die Proteine der vorliegenden Erfindung als Immunogene zur Konstruktion
von Antikörpern
verwendet werden, welche gegen ein Protein der vorliegenden Erfindung
immunreaktiv sind, und zwar für
solche beispielhaften Verwendbarkeiten, wie Immunoassays oder Proteinreinigungstechniken.
-
Expression von Proteinen in
Wirtszellen
-
Unter
Verwendung der Nucleinsäuren
der vorliegenden Erfindung kann man ein Protein der vorliegenden
Erfindung in einer rekombinant gentechnisch manipulierten Zelle,
z. B. Bakterien-, Hefe-, Insekten-, Säuger- oder vorzugsweise Pflanzenzellen,
exprimieren. Die Zellen produzieren das Protein in einem nicht-natürlichen
Zustand (z. B. bezüglich
Menge, Zusammensetzung, Ort und/oder Zeit), da sie durch menschlichen
Eingriff, um dies zu erreichen, gentechnisch verändert wurden.
-
Es
wird davon ausgegangen, dass dem Fachmann zahlreiche Expressionssysteme
geläufig
sind, die zur Expression einer Nucleinsäure, die für ein Protein der vorliegenden
Erfindung codiert, verfügbar
sind. Es wird kein Versuch unternommen, die verschiedenen Verfahren,
die für
die Expression von Proteinen in Prokaryoten oder Eukaryoten bekannt
sind, zu beschreiben.
-
Kurz
zusammengefasst, die Expression von isolierten Nucleinsäuren, die
für ein
Protein der vorliegenden Erfindung codieren, wird typischerweise
erreicht, indem z. B. die DNA oder cDNA an einen Promotor (der entweder
konstitutiv oder induzierbar ist) funktionell verknüpft wird,
gefolgt vom Einbau in einen Expressionsvektor. Die Vektoren können zur
Replikation und Integration entweder in Prokaryoten oder Eukaryoten
geeignet sein. Typische Expressionsvekto ren enthalten Transkriptions-
und Translationsterminatoren, Initiationssequenzen und Promotoren,
die für
eine Regulation der Expression der DNA, die für ein Protein der vorliegenden Erfindung
codiert, verwendbar sind. Um hohe Expressionslevel eines klonierten
Gens zu erhalten, ist es wünschenswert,
Expressionsvektoren zu konstruieren, die mindestens einen starken
Promotor zur Steuerung der Transkription, eine Ribosomenbindungsstelle
zur Translationsinitiation und einen Transkriptions/Translationsterminator
enthalten. Ein Fachmann wird erkennen, dass Modifikationen an einem
Protein der vorliegenden Erfindung durchgeführt werden können, ohne
seine biologische Aktivität
zu vermindern. Einige Modifikationen können durchgeführt werden,
um die Klonierung, Expression oder den Einbau des Targetingmoleküls in ein Fusionsprotein
zu erleichtern. Solche Modifikationen sind dem Fachmann gut bekannt
und umfassen z. B. ein Methionin, das an den Aminoterminus addiert
wird, um eine Initiationsstelle bereitzustellen, oder zusätzliche Aminosäuren (z.
B. Poly-His), die an einem Terminus platziert sind, um zweckdienlich
lokalisierte Reinigungssequenzen zu schaffen. Es können auch
Restriktionsstellen oder Terminationscodons eingeführt werden.
-
A. Expression in Prokaryoten
-
Prokaryotische
Zellen können
als Wirte zur Expression verwendet werden. Prokaryoten werden am häufigsten
durch verschiedene Stämme
von E. coli repräsentiert;
allerdings können
auch andere mikrobielle Stämme
verwendet werden. Üblicherweise
verwendete prokaryotische Kontrollsequenzen, welche hierin so definiert
sind, dass sie Promotoren zur Transkriptionsinitiation, gegebenenfalls
mit einem Operator, zusammen mit Ribosomenbindungsstellensequenzen
umfassen, umfassen solche üblicherweise
verwendeten Promotoren, wie die beta-Lactamase (Penicillinase)-
und Lactose (lac)-Promotorsysteme (Chang et al., Nature 198: 1056
(1977)), das Trypotphan (trp)-Promotorsystem (Goeddel et al., Nucleic
Acids Res. 8: 4057 (1980)) und den von lambda stammenden P-L-Promotor
und die N-Gen-Ribosomenbindungsstelle (Schimatake et al., Nature
292: 128 (1981)). Der Einschluss von Selektionsmarkern in DNA-Vektoren,
die in E. coli transfiziert sind, ist ebenfalls verwendbar. Beispiele
für solche
Marker umfassen Gene, die Resistenz gegen Ampicillin, Tetracyclin
oder Chloramphenicol spezifizieren.
-
Der
Vektor wird so gewählt,
dass er eine Einführung
in die geeignete Wirtszelle erlaubt. Bakterielle Vektoren stammen
typischerweise aus Plasmid oder Phage. Geeignete Bakterienzellen
werden mit Phage-Vektor-Partikel infiziert oder mit nackter Phagen-Vektor-DNA
transfiziert. Wenn ein Plasmivektor verwendet wird, werden die Bakterienzellen
mit der Plasmidvektor-DNA transfiziert. Expressionssysteme zum Exprimieren
eines Proteins der vorliegenden Erfindung sind unter Verwendung
von Bacillus sp. und Salmonella (Palva, et al., Gene 22: 229–235 (1983);
Mosbach, et al., Nature 302: 543–545 (1983)), verfügbar.
-
B. Expression in Eukaryoten
-
Eine
Vielfalt von eukaryotischen Expressionssystemen, z. B. Hefe, Insektenzelllinien,
Pflanzen- und Säugerzellen,
ist dem Fachmann bekannt. Wie kurz unten erläutert wurde, kann eine Sequenz
der vorliegenden Erfindung in diesen eukaryotischen Systemen exprimiert
werden. In einigen Ausführungsformen
werden transformierte/transfizierte Pflanzenzellen, wie sie unten
diskutiert wurden, als Expressionssysteme zur Produktion der Proteine
der vorliegenden Erfindung verwendet.
-
Eine
Synthese von heterologen Proteinen in Hefe ist gut bekannt. Sherman,
F., et al., Methods in Yeast Genetics, Cold Spring Harbor Laboratory
(1982) ist eine gut bekannte Arbeit, die die verschiedenen Verfahren beschreibt,
die zur Herstellung des Proteins in Hefe verfügbar sind. Zwei umfangreich
verwendete Hefen zur Produktion von eukaryotischen Proteinen sind
Saccharomyces cervisisiae und Pichia pastoris. Vektoren, Stämme und
Protokolle zur Expression in Saccharomyces und Pichia sind auf dem
Fachgebiet bekannt und von kommerziellen Lieferanen (z. B. Invitrogen)
verfügbar.
Geeignete Vektoren haben üblicherweise
Expressionskontrollsequenzen, z. B. Promotoren, einschließlich 3-Phosphoglyceratkinase
oder Alkoholoxidase, und einen Replikationsursprung, Terminierungssequenzen
und dergleichen, wenn es gewünscht
ist.
-
Ein
Protein der vorliegenden Erfindung kann, sobald es exprimiert ist,
aus Hefe isoliert werden, indem die Zellen lysiert werden und Standardprotein-Isolierungstechniken
auf die Lysate angewendet werden. Die Überwachung des Reinigungsprozesses
kann unter Verwendung von Western-Blot-Techniken oder Radioimmunoassay
oder anderen Standard-Immunoassaytechniken erreicht werden.
-
Die
Sequenzen, die Proteine der vorliegenden Erfindung codieren, können zur
Verwendung beim Transfizieren von Zellkulturen von zum Beispiel
Säuger-,
Insekten- oder Pflanzenherkunft an verschiedene Expressionsvektoren
ligiert werden. Beispiele für
Zellkulturen, die für
die Produktion der Peptide einsetzbar sind, sind Säugerzellen.
Säugerzellsysteme
werden oft in der Form von Zellmonolayern sein, obgleich auch Säugerzellsuspensionen
verwendet werden können.
Eine Reihe geeigneter Wirtszelllinien, die fähig sind, intakte Proteine
zu exprimieren, wurden auf dem Fachgebiet entwickelt und umfassen
die HEK293-, BHK21- und CHO-Zelllinien. Expressionsvektoren für diese
Zellen können
Expressionskontrollsequenzen umfassen, z. B. einen Replikationsursprung,
einen Promotor (z. B. den CMV-Promotor, einen HSV-tk-Promotor, oder
pgk (Phosphoglyceratkinase)-Promotor, einen Enhancer (Queen et al.,
Immunol. Rev. 89: 49 (1986)) und notwendige Prozessierungsinformationsstellen,
z. B. Ribosomenbindungsstellen, RNA-Spleißstellen, Polyadenylierungsstellen
(z. B. eine SV40-large T Ag-Poly-A-Additionsstelle) und Transkriptionsterminatorsequenzen.
Andere Tierzellen, die zur Produktion von Proteinen der vorliegenden
Erfindung nützlich
sind, sind zum Beispiel von der American Type Culture Collection
Catalogue of Cell Lines and Hybridomas (7. Ausgabe, 1992) erhältlich.
-
Geeignete
Vektoren zum Exprimieren von Proteinen der vorliegenden Erfindung
in Insektenzellen stammen üblicherweise
aus dem SF9-Baculovirus. Geeignete Insektenzelllinien umfassen Mosquitolarven-, Seidenwurm-,
Heerwurm-, Motten- und Drosophila-Zelllinien, zum Beispiel eine
Schneider-Zelllinie (Siehe Schneider, J. Embryol. Exp. Morphol.
27: 353–365
(1987)).
-
Wenn
höhere
Tier- oder Pflanzen-Wirtszellen verwendet werden, werden typischerweise
wie bei Hefe Polyadenylierungs- oder Transkriptionsterminatorsequenzen
in den Vektor einge baut. Ein Beispiel für eine Terminatorsequenz ist
die Polyadenylierungssequenz aus dem Rinderwachstumshormongen. Sequenzen
für ein akurates
Spleißen
des Transkripts können
ebenfalls enthalten sein. Ein Beispiel für eine Spleißsequenz
ist das VP1-Intron aus SV40 (Sprague, et al., J. Virol. 45: 773–781 (1983)).
Zusätzlich
können
Gensequenzen zur Kontrolle einer Replikation in der Wirtszelle in
den Vektor eingearbeitet werden, z. B. die, die in Vektoren des Rinderpapillomavirus-Typs
gefunden werden. Saveria-Campo, M., Bovine Papilloma Virus DNA a
Eukaryotic Cloning Vektor in DNA Cloning Bd. II a Practical Approach,
D. M Glover, Herausg.; IRL Press, Arlington, Virginia S. 213–238 (1985).
-
Transfektion/Transformation von Zellen
-
Das
Verfahren der Transformation/Transfektion ist für die vorliegende Erfindung
nicht kritisch. Derzeit sind verschiedene Verfahren zur Transformation
oder Transfektion verfügbar.
Wenn neuere Verfahren zum Transformieren von Nutzpflanzen oder anderen
Wirtszellen verfügbar
sind, können
sie direkt angewendet werden. Dementsprechend wurde eine weite Vielzahl
von Verfahren entwickelt, um eine DNA-Sequenz in das Genom einer
Wirtszelle zu insertieren, um die Transkription und/oder Translation
der Sequenz zu erzielen, um phänotypische
Veränderungen
in dem Organismus durchzuführen.
Auf diese Weise kann ein beliebiges Verfahren, das für eine effiziente
Transformation/Transfektion sorgt, eingesetzt werden.
-
A. Pflanzentransformation
-
Eine
DNA-Sequenz, die das gewünschte
Polynucleotid der vorliegenden Erfindung codiert, zum Beispiel eine
cDNA oder eine genomische Sequenz, die für ein Volllängenprotein codiert, wird verwendet
werden, um eine rekombinante Expressionskassette zu konstruieren,
welche in die gewünschte
Pflanze eingeführt
werden kann.
-
Isolierte
Nucleinsäuren
der vorliegenden Erfindung können
nach Techniken, die auf dem Fachgebiet bekannt sind, in Pflanzen
eingeführt
werden. Im Allgemeinen werden rekombinante Expressionskassetten,
wie sie oben beschrieben wurden und die zur Transformation von Pflanzenzellen
geeignet sind, hergestellt. Techniken zum Transformieren einer weiten
Vielzahl von höheren
Pflanzenspezies sind gut bekannt und in der technischen, wissenschaftlichen
und Patentliteratur beschrieben. Siehe zum Beispiel Weising et al.,
Ann. Rev. Genet. 22: 421–477
(1988). Zum Beispiel kann das DNA-Konstrukt direkt in die genomische
DNA der Pflanzenzelle eingeführt
werden, indem Techniken wie z. B. Elektroporation, PEG-Poration,
Partikelbeschuss, Siliciumfaserabgabe oder Mikroinjektion von Pflanzenzellenprotoplasten
oder embryogenem Callus verwendet werden. Siehe z. B. Tomes et al.,
Direct DNA Transfer into Intact Plant Cells via Microprojectile
Bombardment, S. 197–213
in Plant Cell, Tissue and Organ Culture, Fundamental Methods, (Herausg.
O. L. Gamborg und G. C. Phillips, Springer-Verlag Berlin Heidelberg
New York, 1995). Alternativ können
die DNA-Konstrukte mit geeigneten T-DNA-flankierenden Regionen kombiniert
werden und in einen herkömmlichen
Agrobacterium tumefaciens-Wirtsvektor eingeführt werden. Die Virulenzfunktionen
des Agrobacterium tumefaciens-Wirts werden die Insertion des Konstrukts
und angrenzender Marker in die Pflanzenzell-DNA steuern, wenn die
Zelle durch die Bakterien infiziert wird. Siehe
U.S. Patent Nr. 5,591,616 .
-
Die
Einführung
von DNA-Konstrukten unter Verwendung einer Polyethylenglykolpräzipitation
ist in Paszkowski et al., Embo J. 3: 2717–2722 (1984) beschrieben. Elektroporationstechniken
werden in Fromm et al., Proc. Natl. Acad. Sci. 82: 5824 (1985) beschrieben.
Ballistische Transformationstechniken sind in Klein et al., Nature
327: 70–73
(1987) beschrieben.
-
Agrobacterium
tumefaciens-vermittelte Transformationstechniken sind in der wissenschaftlichen
Literatur gut beschrieben. Siehe zum Beispiel Horsch et al., Science
233: 496–498
(1984), und Fraley et al., Proc. Natl. Acad. Sci. 80: 4803 (1983).
Obgleich Agrobacterium in erster Linie in Dicotylen einsetzbar ist,
können bestimmte
Monocotylen durch Agrobacterium transformiert werden. Zum Beispiel
wird eine Agrobacterium-Transformation von Mais im
U.S. Patent Nr. 5,550,318 beschrieben.
-
Andere
Verfahren zur Transfektion oder Transformation umfassen (1) Agrobacterium
rhizogenes-vermittelte Transformation (siehe z. B. Lichtenstein
und Fuller in: Genetic Engineering, Bd. 6, PWJ Rigby, Herausg.,
London, Academic Press, 1987; und Lichtenstein, C. P., und Draper,
J. in: DNA Cloning, Bd. II, D. M. Glover, Herausg., Oxford, IRI
Press, 1985), PCT-Anmeldung/US87/02512
(
WO 88/02405 , veröffentlicht
am 7. 4. 1988) beschreiben die Verwendung von A. rhizogenes-Stamm
A4 und seines Ri-Plasmids zusammen mit A. tumefaciens-Vektoren pARC8 oder
pARC16); (2) Liposomen-vermittelte DNA-Aufnahme (siehe z. B. Freeman
et al., Plant Cell Physiol, 25: 1353, 1984), (3) das Vortex-Verfahren
(siehe z. B. Kindle, Proc. Natl. Acad. Sci. USA 87: 1228, (1990)).
-
DNA
kann auch durch direkten DNA-Transfer in Pollen eingeführt werden,
wie es bei Zhou et al., Methods in Enzymology, 101: 433 (1983);
D. Hess, Intern Rev. Cytol., 107: 367 (1987); Luo et al., Plane
Mol. Biol. Reporter, 6: 165 (1988) beschrieben ist. Eine Expression
von für
Polypeptid codierenden Genen kann durch Injektion der DNA in Fortpflanzungsorgane
einer Pflanze erhalten werden, wie es von Pena et al., Nature, 325: 274
(1987) beschrieben wurde. DNA kann auch direkt in die Zellen von
unreifen Embryos und die Rehydratation von getrockneten Embryos
injiziert werden, wie es von Neuhaus et al., Theor. Appl. Genet.,
75: 30 (1987); und Benbrook et al., in Proceedings Bio Expo 1986,
Butterworth, Stoneham, Mass., S. 27–54 (1986) beschrieben wurde.
Auf dem Fachgebiet ist eine Vielzahl von Pflanzenviren bekannt,
die als Vektoren verwendet werden können, und diese umfassen Blumenkohlmosaikvirus
(CaMV), Geminivirus, Trespenmosaikvirus und Tabakmosaikvirus.
-
B. Transfektion von Prokaryoten, niederen
Eukaryoten und Tierzellen
-
Tier-
und niedere eukaryotische (z. B. Hefe-) Wirtszellen sind für eine Transfektion
durch verschiedene Mittel kompetent oder kompetent gemacht. Es gibt
mehrere gut bekannte Verfahren zur Einführung von DNA in Tierzellen.
Diese umfassen Calciumphosphatpräzipitation,
Fusion der Rezipientenzellen mit Bakterienprotoplasten, die die
DNA enthalten, Behandlung der Rezipientenzellen mit Liposomen, die
die DNA enthalten, DEAE-Dextran, Elektroporation, Biolistika und
Mikroinjektion der DNA direkt in die Zellen. Die transfizierten Zellen
werden durch Mittel, die auf dem Fachgebiet gut bekannt sind, kultiviert.
Kuchler, R. J., Biochemical Methods in Cell Culture and Virology,
Dowden, Hutchinson und Ross, Inc. (1977).
-
Synthese von Proteinen
-
Die
Proteine der vorliegenden Erfindung können unter Verwendung von nicht-cellulären Syntheseverfahren
konstruiert werden. Eine Festphasensynthese von Proteinen mit weniger
als etwa 50 Aminosäuren
Länge kann
durchgeführt
werden, indem die C-terminale Aminosäure der Sequenz an einen unlöslichen
Träger gebunden
wird, gefolgt von einer sequentiellen Addition der restlichen Aminosäuren in
der Sequenz. Techniken zur Festphasensynthese sind in Barany und
Merrifield, Solid-Phase Peptide Synthesis, S. 3–284, in The Peptides: Analysis,
Synthesis, Biologe. Bd. 2: Special Methods in Peptide Synthesis,
Teil A.; Merrifield, et al., J. Am. Chem. Soc. 85: 2149–2156 (1963)
und Stewart et al., Solid Phase Peptide Synthesis, 2. Ausgabe, Pierce Chem.
Co., Rockford, Ill. (1984) beschrieben. Proteine größerer Länge können durch
Kondensation der Amino- und Carboxytermini kürzerer Fragmente synthetisiert
werden. Verfahren zur Bildung von Peptidbindungen durch Aktivierung
eines carboxyterminalen Endes (z. B. durch die Verwendung des Kopplungsreagenses N,N'-Dicyclohexylcarbodiimid)
sind dem Fachmann bekannt.
-
Reinigung von Proteinen
-
Die
Proteine der vorliegenden Erfindung können durch Standardtechniken,
die dem Fachmann gut bekannt sind, gereinigt werden. Rekombinant
produzierte Proteine der vorliegenden Erfindung können direkt
exprimiert werden oder als Fusionsprotein exprimiert werden. Das
rekombinante Protein wird durch eine Kombination von Zelllyse (z.
B. Beschallung, French-Press) und Affinitätschromatographie gereinigt.
Für Fusionsprodukte
setzt ein anschließender
Verdau des Fusionsproteins mit einem geeigneten proteolytischen
Enzym das gewünschte
rekombinante Protein frei.
-
Die
Proteine dieser Erfindung, rekombinant oder synthetisch, können durch
Standardtechniken, die auf dem Fachgebiet gut bekannt sind, zu substantieller
Reinheit gereinigt werden; diese Techniken umfassen Detergent-Solubilisierung,
selektive Präzipitation
mit solchen Substanzen wie Ammoniumsulfat, Säulenchromatographie, Immunreinigungsverfahren
und andere. Siehe z. B. R. Scopes, Protein Purification: Principles and
Practice, Springer-Verlag: New York (1982); Deutscher, Guide to
Protein Purification, Academic Press (1990). Beispielsweise können Antikörper gegen
die Proteine erzeugt werden, wie es hierin beschrieben wird. Eine
Reinigung aus E. coli kann nach den Verfahren, die im
U.S. Patent Nr. 4,511,503 beschrieben
sind, erreicht werden. Das Protein kann dann aus Zellen, die das
Protein exprimieren, isoliert werden und außerdem durch hierin beschriebene
Standardproteinchemietechniken gereinigt werden. Eine Detektion
des exprimierten Proteins wird durch Verfahren erreicht, die auf
dem Fachgebiet bekannt sind und z. B. Radioimmunoassays, Western-Blotting-Techniken
oder Immunopräzipitation
umfassen.
-
Transgene Pflanzen-Regeneration
-
Transformierte
Pflanzenzellen, die aus einer beliebigen der obigen Transformationstechniken
stammen, können
kultiviert werden, um eine ganze Pflanze zu regenerieren, welche
den transformierten Genotyp besitzt. Solche Regenerationstechniken
basieren oft auf einer Manipulation von bestimmten Phytohormonen in
einem Gewebekulturwachstumsmedium, die typischerweise auf einem
Biozid- und/oder Herbizidmarker basiert, der zusammen mit einem
Polynucleotid der vorliegenden Erfindung eingeführt wurde. Zur Transformation und
Regeneration von Mais siehe Gordon-Kamm et al., The Plant Cell,
2: 603–618
(1990).
-
Pflanzenzellen,
die mit einem Pflanzenexpressionsvektor transformiert wurden, können z.
B. aus Einzelzellen, Callusgewebe oder Blattscheiben nach Standardpflanzengewebekulturtechniken
regeneriert werden. Auf dem Fachgebiet ist gut bekannt, dass verschiedene
Zellen, Gewebe und Organe aus fast jeder beliebigen Pflanze erfolgreich
unter Regeneration einer ganzen Pflanze kultiviert werden können. Eine
Pflanzenregeneration aus kultivierten Protoplasten wird in Evans
et al., Protoplasts Isolation and Culture, Handbook of Plant Cell
Culture, Macmillilan Publishing Company, New York, S. 124–176 (1983);
und Binding, Regeneration of Plants, Plant Protoplasts, CRC Press,
Boca Raton, S. 21–73
(1985) beschrieben.
-
Die
Regeneration von Pflanzen, die das Fremdgen durch Agrobacterium
eingeführt
enthalten, aus Blattexplantaten kann erreicht werden, wie es von
Horsch et al., Science, 227: 1229–1231 (1985) beschrieben wurde.
In diesem Verfahren werden Transformanten in Gegenwart eines Selektionsmittels
und in einem Medium, das die Regeneration von Schösslingen
in der Pflanzespezies induziert, die transformiert wurde, wachsen gelassen,
wie es bei Fraley et al., Proc. Natl. Acad. Sci. USA, 80: 4803 (1983)
beschrieben wurde. Dieses Verfahren produziert typischerweise Schösslinge
in zwei bis vier Wochen und diese Transformantenschösslinge werden
dann in ein geeignetes Wurzel-induzierendes Medium, das das Selektionsmittel
und ein Antibiotikum zur Verhinderung von bakteriellem Wachstum
enthält,
transferiert. Transgene Pflanzen der Erfindung können fertil oder steril sein.
-
Eine
Regeneration kann auch aus Pflanzencallus, Explantaten, Organen
oder Teilen davon erreicht werden. Solche Regenerationstechniken
sind allgemein in Klee et al., Ann. Rev. of Plant Phys. 38. 467–486 (1987)
beschrieben. Die Regeneration von Pflanzen entweder aus Einzelpflanzenprotoplasten
oder verschiedenen Explantaten ist auf dem Fachgebiet gut bekannt.
Siehe zum Beispiel Methods for Plant Molecular Biology, A. Weissbach
und H. Weissbach, Herausg., Academic Press, Inc., San Diego, Kalif.
(1988). Dieses Regenerations- und Wachstumsverfahren beinhaltet
die Schritte einer Selektion von Transformantenzellen und Schösslingen,
Bewurzeln der Transformantenschösslinge
und Wachstum der Pflänzchen
in Erde. Für
eine Maiszellkultur und -regeneration siehe allgemein The Maize
Handbook, Freeling und Walbot, Herausg., Springer, New York (1994);
Corn and Corn Improvement, 3. Ausgabe, Sprague und Dudley Herausg.,
American Society of Agronomy, Madison, Wisconsin (1988).
-
Ein
Fachmann wird erkennen, dass, nachdem die rekombinante Expressionskassette
stabil in transgene Pflanzen eingebaut wurde und als funktionell
bestätigt
wurde, sie durch sexuelle Kreuzung in andere Pflanzen eingeführt werden
kann. In Abhängigkeit
von der zu kreuzenden Spezies kann eine beliebige einer Reihe von
Standardzüchtungstechniken
verwendet werden.
-
Bei
vegetativ vermehrten Nutzpflanzen können reife transgene Pflanzen
durch die Entnahme von Schnitten oder durch Gewebekulturtechniken
vermehrt werden, um mehrere identische Pflanzen zu erzeugen. Es
wird eine Selektion von wünschenswerten
Transgenen durchgeführt
und neue Varietäten
werden erhalten und zur kommerziellen Verwendung vegetativ vermehrt.
Bei durch Säen
vermehrten Nutzpflanzen können
reife transgene Pflanzen geselbstet werden, um eine homozygote Inzuchtpflanze
zu erzeugen. Die Inzuchtpflanze produziert Samen, die die neu eingeführte heterologe
Nucleinsäure
enthalten. Diese Samen können
wachsen gelassen werden, um Pflanzen zu produzieren, die den ausgewählten Phänotyp produzieren.
-
Teile,
die von der regenerierten Pflanze erhalten wurden, z. B. Blüten, Samen,
Blätter,
Zweige, Früchte und
dergleichen, werden von der Erfindung mit umfasst, vorausgesetzt,
dass diese Teile Zellen umfassen, die die isolierte Nucleinsäure der
vorliegenden Erfindung umfassen. Nachkommenschaft und Varianten
und Mutanten der regenerierten Pflanzen sind im Rahmen der Erfindung
ebenfalls eingeschlossen, vorausgesetzt, dass diese Teile die eingeführten Nucleinsäuresequenzen
umfassen.
-
Transgene
Pflanzen, die den selektierbaren Marker exprimieren, können bezüglich einer
Transmission der Nucleinsäure
der vorliegenden Erfindung beispielsweise durch Standard-Immunoblot- und -DNA-Detektions-Techniken
durchgemustert werden. Transgene Linien werden typischerweise auch
bezüglich
der Expressionslevel der heterologen Nucleinsäure evaluiert. Eine Expression
auf dem RNA-Level kann anfangs bestimmt werden, um Expressions-positive
Pflanzen zu identifizieren und zu quantifizieren. Standardtechniken für die RNA-Analyse
können
verwendet werden und umfassen PCR-Amplifikationsassays, die Oligonucleotidprimer
verwenden, die so konzipiert sind, dass sie nur die heterologen
RNA-Matrizen amplifizieren, und Lösungshybridisierungsassays,
die heterologe Nucleinsäure-spezifische
Sonden verwenden. Die RNA-positiven Pflanzen können dann durch Western-Immunoblot-Analyse
auf Proteinexpression analysiert werden, wobei die spezifisch reaktiven
Antikörper
der vorliegenden Erfindung verwendet werden. Außerdem kann eine in situ-Hybridisierung
und Immuncytochemie nach Standardprotokollen durchgeführt werden,
wobei Nucleinsäure-spezifische
Polynucleotidsonden bzw. Antikörper
verwendet werden, um Expressionsstellen innerhalb eines transgenen
Gewebes zu lokalisieren. Im Allgemeinen wird üblicherweise eine Reihe von
transgenen Linien bezüglich
der eingebauten Nucleinsäure
durchgemustert, um Pflanzen mit den geeignetesten Expressionsprofilen
zu identifizieren und auszuwählen.
-
Eine
bevorzugte Ausführungsform
ist eine transgene Pflanze, die für die addierte heterologe Nucleinsäure homozygot
ist, d. h. eine transgene Pflanze, die zwei addierte Nucleinsäuresequenzen
enthält,
ein Gen an demselben Locus auf jedem Chromosom eines Chromosomenpaars. Eine
homozygote transgene Pflanze kann durch sexuelle Kreuzung (Selbstung)
einer heterozygoten transgenen Pflanze, die eine einzelne addierte heterologe
Nucleinsäure
enthält,
Keimen einiger der Samen, die produziert wurden, und Analysieren
der resultierenden produzierten Pflanzen auf veränderte Expression eines Polynucleotids
der vorliegenden Erfindung im Vergleich zu einer Kontrollpflanze
(d. h. eine native, nicht-transgene Pflanze) erhalten werden. Eine Rückkreuzung
zu einer Elternpflanze und eine Auskreuzung mit einer nicht-transgenen
Pflanze werden ebenfalls in Betracht gezogen.
-
Modulierung der Polypeptidlevel und/oder
-zusammensetzung
-
Die
vorliegende Erfindung stellt ferner ein Verfahren zur Modulierung
(d. h. Erhöhung
oder Verminderung) der Konzentration oder Zusammensetzung der Polypeptide
der vorliegenden Erfindung in einer Pflanze oder einem Teil davon
bereit. Eine Modulierung kann durch Erhöhen oder Vermindern der Konzentration und/oder
der Zusammensetzung (d. h. das Verhältnis der Polypeptide der vorliegenden
Erfindung) in einer Pflanze durchgeführt werden. Das Verfahren umfasst
Transformieren einer Pflanzenzelle mit einer rekombinanten Expressionskassette,
umfassend ein Polynucleotid der vorliegenden Erfindung, wie es oben
beschrieben wurde, unter Erhalt einer transformierten Pflanzenzelle,
Kultivieren der transformierten Pflanzenzelle unter Pflanzenbildungsbedingungen
und Induzieren einer Expression eines Polynucleotids der vorliegenden
Erfindung in der Pflanze für
eine Zeit, die ausreichend ist, um die Konzentration und/oder die
Zusammensetzung in der Pflanze oder einem Pflanzenteil zu modulieren.
-
In
einigen Ausführungsformen
kann der Gehalt und/oder die Zusammensetzung von Polypeptiden der vorliegenden
Erfindung in einer Pflanze moduliert werden, indem in vivo oder
in vitro der Promotor eines nicht-isolierten Gens der vorliegenden
Erfindung verändert
wird, um die Genexpression hoch oder runter zu regulieren. In einigen
Ausführungsformen
können
die codierenden Regionen von nativen Genen der vorliegenden Erfindung
durch Substitution, Addition, Insertion oder Deletion verändert werden,
um die Aktivität
des codierten Enzyms zu senken. Siehe z. B. Kmiec,
U.S. Patent 5,565,350 ; Zarling et
al.,
PCT/US93/03868 .
Und in einigen Ausführungsformen
wird eine isolierte Nucleinsäure
(z. B. ein Vektor), die eine Promotorsequenz umfasst, in eine Pflanzenzelle
transfiziert. Anschließend
wird eine Pflanzenzelle, die den Promotor funktionell mit einem
Polynucleotid der vorliegenden Erfindung verknüpft umfasst, ausgewählt und
zwar durch Mittel, die dem Fachmann bekannt sind, z. B., aber nicht
beschränkt
auf, Southern-Blot-DNA-Sequenzierung oder PCR-Analyse, wobei Primer
verwendet werden, die für
den Promotor und das Gen spezifisch sind, und daraus produzierte
Amplicons detektiert werden. Eine Pflanze oder ein Pflanzenteil,
die/der durch die vorstehende Ausführungsform verändert oder
modifiziert wurde, wird unter Pflanzenbildungsbedingungen für eine Zeit
wachsen gelassen, die ausreichend ist, um die Konzentration und/oder
Zusammensetzung von Polypeptiden der vorliegenden Erfindung in der
Pflanze zu modulieren. Pflanzenbildungsbedingungen sind auf dem
Fachgebiet gut bekannt und oben kurz diskutiert.
-
Im
Allgemeinen wird die Konzentration oder Zusammensetzung im Vergleich
zu einer nativen Kontrollpflanze, einem nativen Pflanzenteil oder
einer Zelle, denen die vorstehend genannte rekombinante Expressionskassette
fehlt, um wenigstens 5%, 10%, 20%, 30%, 40%, 50%, 60%, 70%, 80%
oder 90% erhöht
oder verringert. Eine Modulation bzw. Modulierung in der vorliegenden
Erfindung kann während
des Wachstums und/oder anschließend
an das Wachstum der Pflanze zu der gewünschten Entwicklungsstufe erfolgen.
Eine Modulierung der Nucleinsäureexpression
temporär
und/oder in bestimmten Geweben kann durch Verwendung des geeigneten
Promotors, der funktionell mit dem Polynucleotid der vorliegenden
Erfindung in z. B. Sense- oder Antisense-Orientierung verknüpft ist,
wie es detaillierter oben diskutiert ist, kontrolliert werden. Die
Induktion der Expression eines Polynucleotids der vorliegenden Erfindung
kann auch durch exogene Verabreichung einer wirksamen Menge an induzierender
Verbindung kontrolliert werden. Induzierbare Promotoren und induzierende
Verbindungen, welche die Expression aus diesen Promotoren aktivieren,
sind auf dem Fachgebiet gut bekannt. In bevorzugten Ausführungsformen
werden die Polypeptide der vorliegenden Erfindung in Monocotylen,
insbesondere Mais, moduliert.
-
Molekulare Marker
-
Die
vorliegende Erfindung stellt ein Verfahren zur Genotypisierung einer
Pflanze, die ein Polynucleotid der vorliegenden Erfindung umfasst,
bereit. Die Pflanze ist vorzugsweise eine Monocotyle, z. B. Mais
oder Sorghum. Eine Genotypisierung stellt ein Mittel zur Unterscheidung
von Homologen eines Chromosomenpaars bereit und kann eingesetzt
werden, um Segreganten in einer Pflanzenpopulation zu differenzieren.
Molekulare Marker-Methoden können
für phylogenetische
Studien, eine Charakterisierung einer genetischen Beziehung unter
Nutzpflanzenvarietäten,
zur Identifizierung von Kreuzungen oder somatischen Hybriden, zur
Lokalisierung von chromosomalen Segmenten, die monogene Merkmale
beeinträchtigen,
zur kartenbasierten Klonierung und zur Studie einer quantitativen
Vererbung verwendet werden. Siehe z. B. Plant Molecular Biology:
A Laboratory Manual, Kapitel 7, Clar, Herausg., Springer-Verlag,
Berlin (1997). Für
molekulare Marker-Methoden siehe allgemein The DNA Revolution von
Andrew H. Paterson 1996 (Kapitel 2) in: Genome Mapping in Plants (Herausg.
Andrew H. Paterson) von Academic Press/R. G. Landis Company, Austin,
Texas, S. 7–21.
-
Das
besondere Verfahren der Genotypisierung in der vorliegenden Erfindung
kann eine beliebige Anzahl von analytischen Techniken mit molekularem
Marker verwenden, z. B., aber nicht beschränkt auf, Restriktionsfragmentlängenpolymorphismen
(RFLPs). RFLPs sind das Produkt von allelischen Unterschieden zwischen
DNA-Restriktionsfragmenten, verursacht durch Nucleotidsequenzvariabilität. Wie es
dem Fachmann bekannt ist, werden RFLPs typischerweise durch Extraktion
von genomischer DNA und Verdau mit einem Restriktionsenzym detektiert.
Im Allgemeinen werden die resultierenden Fragmente nach Größe getrennt
und mit einer Sonde hybridisiert; Einzelkopiesonden sind bevorzugt.
Restriktionsfragmente aus homologen Chromosomen werden gezeigt.
Differenzen in der Fragmentgröße unter
Allelen stellen einen RFLP dar. Somit stellt die vorliegende Erfindung
außerdem
ein Mittel zur Verfolgung der Segregation eines Gens oder einer
Nucleinsäure
der vorliegenden Erfindung sowie chromosomale Sequenzen, die genetisch
mit diesen Genen oder Nucleinsäuren
verknüpft
sind, bereit, wobei solche Techniken wie RFLP-Analyse verwendet
werden. Verknüpfte
chromosomale Sequenzen sind innerhalb von 50 centiMorgans (cM),
oft innerhalb von 40 oder 30 cM, vorzugsweise innerhalb von 20 oder
10 cM, bevorzugter innerhalb von 5, 3, 2 oder 1 cM eines Gens der
vorliegenden Erfindung.
-
In
der vorliegenden Erfindung hybridisieren die Nucleinsäuresonden,
die zum molekularen Marker-Kartieren von Pflanzenkerngenomen verwendet
werden, unter selektiven Hybridisierungsbedingungen selektiv an
ein Gen, das ein Polynucleotid der vorliegenden Erfindung codiert.
In bevorzugten Ausführungsformen
werden die Sonden aus Polynucleotiden der vorliegenden Erfindung
ausgewählt.
Typischerweise sind diese Sonden cDNA-Sonden oder genomische PstI-Klone. Die Länge der
Sonden ist oben detaillierter diskutiert, allerdings haben sie typischerweise
eine Länge
von wenigstens 15 Basen, bevorzugter wenigstens 20, 25, 30, 35,
40 oder 50 Basen. Im Allgemeinen sind die Sonden allerdings kleiner
als etwa 1 Kilobase in der Länge.
Vorzugsweise sind die Sonden Einzelkopiesonden, die an einen einmal
vorkommenden Locus in einem haploiden Chromosomenkomplement hybridisieren.
Einige Beispiele für
Restriktionsenzyme, die bei der RFLP-Kartierung verwendet werden,
sind EcoRI, EcoRv und SstI. Der Ausdruck „Restriktionsenzym", wie er hierin verwendet
wird, bezieht sich auf eine Zusammensetzung, die eine spezifische
Nucleotidsequenz erkennt und allein oder in Verbindung mit einer
anderen Zusammensetzung an einer spezifischen Nucleotidsequenz spaltet.
-
Das
Verfahren zum Detektieren eines RFLP umfasst die Schritte: (a) Verdauen
genomischer DNA einer Pflanze mit einem Restriktionsenzym; (b) Hybridisieren
einer Nucleinsäuresonde
unter selektiven Hybridisierungsbedingungen an eine Sequenz eines
Polynucleotids der vorliegenden Erfindung der genomischen DNA; (c)
Detektieren eines RFLP daraus. Andere Verfahren zur Differenzierug
von polymorphen (allelischen) Varianten von Polynucleotiden der
vorliegenden Erfindung können
unter Verwendung von Techniken mit molekularem Marker, die dem Fachmann
gut bekannt sind, erhalten werden; diese umfassen Techniken wie:
1) Einzelstrangkonformationsanalyse (SSCP); 2) denaturierende Gradienten-Gelelektrophorese
(DGGE); 3) RNase-Schutzassays;
4) Allel-spezifische Oligonucleotide (ASOs); 5) die Verwendung von
Proteinen, die Nucleotidfehlpaarungen erkennen, z. B. das E. coli-mutS-Protein;
und 6) Allel-spezifische PCR. Andere Ansätze, die auf der Detektion
von Fehlpaarungen zwischen den zwei komplementären DNA-Strängen basieren, umfassen „clamped" denaturierende Gelelektrophorese
(CDGE); Heteroduplexanalyse (HA); und chemische Fehlpaarungsspaltung
(CMC). Beispiele für
polymorphe Varianten werden in Tabelle I oben bereitgestellt. Demnach
stellt die vorliegende Erfindung außerdem ein Verfahren zur Genotypisierung
bereit, umfassend die Schritte In-Kontakt-Bringen einer Probe, von
der angenommen wird, dass sie ein Polynucleotid der vorliegenden
Erfindung umfasst, unter stringenten Bedingungen mit einer Nucleinsäuresonde.
Im Allgemeinen ist die Probe eine Pflanzenprobe, vorzugsweise eine
Probe, von der angenommen wird, dass sie ein Maispolynucleotid der
vorliegenden Erfindung umfasst (z. B. Gen, mRNA). Die Nucleinsäuresonde
hybridisiert unter stringenten Bedingungen selektiv an eine Untersequenz
eines Polynucleotids der vorliegenden Erfindung, das einen polymorphen
Marker umfasst. Selektive Hybridisierung der Nucleinsäuresonde
an die polymorphe Marker-Nucleinsäuresequenz führt zu einem
Hybridisierungskomplex. Eine Detektion des Hybridisierungskomplexes zeigt
das Vorliegen jenes polymorphen Markers in der Probe an. In bevorzugten
Ausführungsformen
umfasst die Nucleinsäuresonde
ein Polynucleotid der vorliegenden Erfindung.
-
UTRs und Codonpräferenz
-
Es
wurde gefunden, dass eine translationale Effizienz im Allgemeinen
durch spezifische Sequenzelemente in der 5'-nicht-codierenden oder untranslatierten
Region (5'-UTR)
der RNA reguliert wird. Positive Sequenzmotive umfassen translationale
Initiationskonsensussequenzen (Kozak, Nucleic Acids Res. 15: 8125 (1987))
und die 7-Methylguanosin-Cap-Struktur (Drummond et al., Nucleic
Acids Res. 13: 7375 (1985)). Negative Elemente umfassen stabile
intramolekulare 5'-UTR-Stem-Loop-Strukturen
(Muesing et al., Cell 48: 691 (1987)) und AUG-Sequenzen oder kurze offene Leseraster,
denen ein geeignetes AUG in der 5'-UTR vorausgeht (Kozak, oben, Rao et
al., Mol. and Cell. Biol. 8: 284 (1988)). Dementsprechend stellt
die vorliegende Erfindung 5'-
und/oder 3'-UTR-Regionen
zur Modulation der Translation von heterologen codierenden Sequenzen
bereit.
-
Außerdem können die
Polypeptid-codierenden Segmente der Polynucleotide der vorliegenden
Erfindung unter Änderung
der Codonverwendung modifiziert werden. Eine veränderte Codonverwendung kann
verwendet werden, um eine translationale Effizienz zu verändern und/oder
die codierende Sequenz zur Expression in einem gewünschten
Wirt zu optimieren oder die Codonverwendung in einer heterologen
Sequenz zur Expression in Mais zu optimieren. Eine Codonverwendung
in den codierenden Regionen der Polynucleotide der vorliegenden
Erfindung kann statistisch analysiert werden, wobei im Handel verfügbare Software-Packages,
z. B. „Codon
Preference", erhältlich von
der University of Wisconsin Genetics Computer Group (siehe Devereux
et al., Nucleic Acids Res. 12: 387–395 (1984)) oder Mac-Vektor
4.1 (Eastman Kodak Co., New Haven, Conn.), verwendet werden. Somit
stellt die vorliegende Erfindung ein Häufigkeitscharakteristikum der
Codonverwendung für
die codierende Region wenigstens eines der Polynucleotide der vorliegenden
Erfindung bereit. Die Anzahl der Polynucleotide, die verwendet werden
können,
um die Häufigkeit
der Codonverwendung zu bestimmen, kann eine beliebige ganze Zahl
von 1 bis zu der Zahl der Polynucleotide der vorliegenden Erfindung,
wie sie hierin bereitgestellt werden, sein. Gegebenenfalls werden
die Polynucleotide Volllängensequenzen
sein. Eine beispielhafte Zahl der Sequenzen zur statistischen Analyse
kann wenigstens 1, 5, 10, 20, 50 oder 100 sein.
-
Sequenz-Shuffling
-
Die
vorliegende Erfindung bezieht sich auf Verfahren zum Sequenz-Shuffling
unter Verwendung von Polynucleotiden der vorliegenden Erfindung
und auf Zusammensetzungen, die dar aus resultieren. Sequenz-Shuffling
wird in der PCT-Veröffentlichung
Nr. 96/19256 beschrieben. Siehe auch Zhang, J.-H, et al., Proc.
Natl. Acad. Sci. USA 94: 4504–4509
(1997). Im Allgemeinen stellt Sequenz-Shuffling ein Mittel zur Erzeugung
von Bibliotheken von Polynucleotiden mit einem gewünschten
Charakteristikum, welches selektiert werden kann oder auf welches
durchgemustert werden kann, bereit. Bibliotheken rekombinanter Polynucleotide
werden aus einer Gruppe von Polynucleotiden mit verwandter Sequenz
erzeugt, welche Sequenzregionen umfassen, die substantielle Sequenzidentität haben
und in vitro oder in vivo homolog rekombiniert werden können. Die
Population von Sequenz-rekombinierten Polynucleotiden umfasst eine
Subpopulation von Polynucleotiden, die gewünschte oder vorteilhafte Charakteristika
besitzen und die durch ein geeignetes Selektions- oder Screeningverfahren
selektiert werden können.
Die Charakteristika können
eine beliebige Eigenschaft oder ein beliebiges Attribut sein, die/das
in einem Screeningsystem selektiert oder detektiert werden kann;
diese können
folgende Eigenschaften umfassen: ein codiertes Protein, ein Transkriptionselement,
eine Sequenz, die die Transkription kontrolliert, RNA-Prozessierung,
RNA-Stabilität,
Chromatinkonformation, Translation oder andere Eigenschaften eines
Gens oder Transgens, ein replikatives Element, ein Protein-bindendes
Element oder dergleichen, z. B. ein beliebiges Merkmal, welches
eine selektierbare oder detektierbare Eigenschaft verleiht. Das
ausgewählte
Charakteristikum kann ein verringertes Km und/oder erhöhtes Kcat gegenüber dem
Wildtyp-Protein sein, wie es hierin bereitgestellt wird. In anderen
Ausführungsformen
wird ein Protein oder ein Polynucleotid, das mit Sequenz-Shuffling
erzeugt wird, eine Ligandenbindungsaffinität haben, die größer ist
als die des Wildtyp-Polynucleotids
ohne Shuffling. Die Verstärkung
solcher Eigenschaften kann wenigstens 110%, 120%, 130%, 140% oder
wenigstens 150% des Wildtyp-Werts sein.
-
Generische und Konsensus-Sequenzen
-
Die
vorliegende Erfindung bezieht sich auch auf Polynucleotide und Polypeptide
mit: (a) einer generischen Sequenz von wenigstens zwei homologen
Polynucleotiden bzw. Polypeptiden der vorliegenden Erfindung und
(b) einer Konsensussequenz aus wenigstens drei homologen Polynucleotiden
bzw. Polypeptiden der vorliegenden Erfindung. Ein Polynucleotid
mit einer Aminosäure-
oder Nucleinsäure-Konsensussequenz
kann verwendet werden, um Antikörper
zu erzeugen oder Nucleinsäuresonden
oder Primer zu erzeugen, um auf Homologe in anderen Arten, Gattungen,
Familien, Ordnungen, Klassen, Phyla oder Reichen durchzumustern. Zum
Beispiel kann ein Polynucleotid mit Konsensussequenzen aus einer
Genfamilie von Zea mays verwendet werden, um Antikörper oder
Nucleinsäuresonden
oder Primer für
andere Gramineae-Spezies, z. B. Weizen, Reis oder Sorghum, zu erzeugen.
Alternativ kann ein Polynucleotid mit einer Konsensussequenz, die
aus orthologen Genen erzeugt wurde, verwendet werden, um Orthologe
anderer Taxa zu identifizieren oder zu isolieren. Typischerweise
wird ein Polynucleotid mit einer Konsensussequenz eine Länge von
wenigstens 9, 10, 15, 20, 25, 30 oder 40 Aminosäuren oder eine Länge von
20, 30, 40, 50, 100 oder 150 Nucleotiden haben. Wie der Fachmann
weiß,
kann eine konservative Aminosäuresubstitution
für Aminosäuren verwendet
werden, die sich in der ange ordneten Sequenz unterscheiden, aber
aus derselben konservativen Substitutionsgruppe sind, wie sie oben
diskutiert wurde. Gegebenenfalls sind für jede 10 Aminosäuren-Länge einer
Konsensussequenz nicht mehr als ein oder zwei konservative Aminosäuren substituiert.
-
Ähnliche
Sequenzen, die für
die Erzeugung einer Konsensus- oder generischen Sequenz verwendet werden,
umfassen eine beliebige Anzahl an Kombination von allelischen Varianten
desselben Gens, orthologe oder paraloge Sequenzen, wie sie hierin
bereitgestellt werden. Gegebenenfalls werden ähnliche Sequenzen, die bei
der Erzeugung einer Konsensus- oder generischen Sequenz verwendet
werden, unter Verwendung der kleinsten Summenwahrscheinlichkeit
des BLAST-Algorithmus (P(N)) identifiziert. In Kapitel 7 von Current
Protocols in Molecular Biology, F. M. Ausubel et al., Herausg.,
Current Protocols, ein Gemeinschaftsunternehmen von Greene Publishing
Associates, Inc., und John Wiley & Sons,
Inc. (Ergänzung
30) werden verschiedene Lieferanten für Sequenz-Analyse-Software
aufgelistet. Eine Polynucleotidsequenz wird als ähnlich zu einer Referenzsequenz
angesehen, wenn die kleinste Summenwahrscheinlichkeit in einem Vergleich
der Testnucleinsäure
zu der Referenznucleinsäure
kleiner als etwa 0,1, bevorzugter kleiner als etwa 0,01 oder 0,001
und am wahrscheinlichsten kleiner als etwa 0,0001 oder 0,00001 ist. Ähnliche
Polynucleotide können
angeordnet werden und eine Konsensus- oder generische Sequenz erzeugt
werden, wobei Mehrsequenzanordnungs-Software verwendet wird, die
von einer Reihe kommerzieller Lieferanten verfügbar ist, z. B. Genetics Computer
Group's (Madison,
WI) PILEUP-Software, Vector NTIs (North Bethesda, MD) ALIGNX oder
Genecode's (Ann
Arbor, MI) SEQUENCHER. Zweckmäßigerweise
können
Default-Parameter solcher Software eingesetzt werden, um Konsensus-
oder generische Sequenzen zu erzeugen.
-
Detektion von Nucleinsäuren
-
Die
vorliegende Erfindung bezieht sich außerdem auf Verfahren zum Detektieren
eines Polynucleotids der vorliegenden Erfindung in einer Nucleinsäureprobe,
von der angenommen wird, dass sie ein Polynucleotid der vorliegenden
Erfindung umfasst, z. B. ein Pflanzenzelllysat, insbesondere ein
Lysat von Mais bzw. Getreide. In einigen Ausführungsformen kann ein Gen der
vorliegenden Erfindung oder ein Teil davon vor dem Schritt des In-Kontakt-Bringens
der Nucleinsäureprobe
mit einem Polynucleotid der vorliegenden Erfindung amplifiziert
werden. Die Nucleinsäureprobe
wird mit dem Polynucleotid unter Bildung eines Hybridisierungskomplexes
in Kontakt gebracht. Das Polynucleotid hybridisiert unter stringenten
Bedingungen an ein Gen, das ein Polypeptid der vorliegenden Erfindung
codiert. Die Bildung des Hybridisierungskomplexes wird verwendet,
um ein Gen, das für
ein Polypeptid der vorliegenden Erfindung codiert, in der Nucleinsäureprobe
zu detektieren. Der Fachmann wird erkennen, dass einer isolierten
Nucleinsäure,
die ein Polynucleotid der vorliegenden Erfindung umfasst, Kreuzhybridisierungssequenzen
in üblichen
Nicht-Target-Genen, die ein falsch positives Resultat liefern würden, fehlen
sollten.
-
Eine
Detektion des Hybridisierungskomplexes kann unter Verwendung einer
Reihe von gut bekannten Verfahren erreicht werden. Beispielsweise
kann die Nucleinsäureprobe
oder ein Teil davon durch Hybridisierungsformate, einschließlich, aber
nicht beschränkt
auf, Lösungspha sen-,
Festphasen-, Mischphasen- oder In situ-Hybridisierungsassays, analysiert
werden. Kurz ausgedrückt,
in Lösungs
(oder Flüssigkeits)-Phasenhybridisierungen
sind sowohl die Targetnucleinsäure
als auch die Sonde oder der Primer frei, um im Reaktionsgemisch
zu wechselwirken. In Festphasenhybridisierungsassays sind Sonden
oder Primer typischerweise an einen festen Träger gebunden, wo sie für eine Hybridisierung
mit Targetnucleinsäure
in Lösung
verfügbar
sind. In einer gemischten Phase hybridisieren Nucleinsäure-Intermediate
in Lösung
an Targetnucleinsäuren
in Lösung
wie auch an eine Nucleinsäure,
die an einen festen Träger
gebunden ist. Bei einer In situ-Hybridisierung wird die Targetnucleinsäure aus
ihrer cellulären
Umgebung freigesetzt, so dass sie zur Hybridisierung in der Zelle
verfügbar
ist, während
die celluläre
Morphologie für
eine anschließende
Interpretation und Analyse konserviert wird. Die folgenden Artikel
liefern eine Übersicht über die
verschiedenen Hybridisierungsassayformate: Singer et al., Biotechniques
4(3): 230–250
(1986); Haase et al., Methods in Virology, Bd. VII, S. 189–226 (1984);
Wilkinson, The theory and practice of in situ hybridization in:
In situ Hybridization, D. G. Wilkinson, Herausg., IRL Press, Oxford
University Press, Oxford: und Nucleic Acid Hybridization: A Practical
Approach, Hames; B. D. und Higgins, S. J., Herausg., ILR Press (1987).
-
Nucleinsäure-Markierungen und Detektionsverfahren
-
Die
Mittel, durch welche Nucleinsäuren
der vorliegenden Erfindung markiert werden, sind kein kritischer
Aspekt der vorliegenden Erfindung und eine Markierung kann durch
eine Vielzahl von Verfahren, die derzeit bekannt sind oder später entwickelt
werden, erfolgen. Detektierbare Markierungen, die zur Verwendung
in der vorliegenden Erfindung geeignet sind, umfassen eine beliebige
Zusammensetzung, die durch spektroskopische Radioisotopen-, photochemische,
biochemische, immunchemische, elektrische, optische oder chemische
Mittel detektierbar sind. Nützliche
Markierungen in der vorliegenden Erfindung umfassen Biotin zur Färbung mit
markiertem Streptavidinkonjugat, magnetische Perlen, Fluoreszenzfarbstoffe
(z. B. Fluorescein, Texasrot, Rhodamin, grün fluoreszierendes Protein
und dergleichen), radioaktive Markierungen (z. B. 3H, 125I, 35S, 14C oder 32P), Enzyme
(z. B. Meerrettichperoxidase, alkalische Phosphatase und andere,
die üblicherweise
in einem ELISA verwendet werden) und kolorimetrische Markierungen,
z. B. kolloidales Gold oder gefärbte
Glas- oder Kunststoff (z. B. Polystyrol, Polypropylen, Latex usw.)
-Perlen.
-
Nucleinsäuren der
vorliegenden Erfindung können
durch ein beliebiges von mehreren Verfahren, die typischerweise
verwendet werden, um das Vorliegen von hybridisierten Nucleinsäuren zu
detektieren, markiert werden. Ein übliches Detektionsverfahren
ist die Verwendung von Autoradiographie unter Verwendung von Sonden,
die mit 3H, 125I, 35S, 14C oder 32P oder dergleichen markiert sind. Die Wahl
eines radioaktiven Isotops hängt
von Forschungspräferenzen,
bedingt durch die Einfachheit der Synthese, die Stabilität und die
Halbwertszeit der ausgewählten
Isotope ab. Andere Markierungen umfassen Liganden, die an Antikörper binden, die
mit Fluorphoren, chemilumineszierenden Mitteln und Enzymen markiert
sind. Alternativ können
Sonden direkt mit Markierungen, z. B. Fluorophoren, chemilumineszierenden
Mitteln oder Enzy men, konjugiert werden. Die Wahl der Markierung
hängt von
der verlangten Empfindlichkeit, der Einfachheit der Konjugation
mit der Sonde, Stabilitätsanforderungen
und verfügbaren
Instrumenten ab. Eine Markierung der Nucleinsäuren der vorliegenden Erfindung
wird in einfacher Weise durch Verwendung von markierten PCR-Primern
erreicht.
-
In
einigen Ausführungsformen
wird die Markierung gleichzeitig während des Amplifikationsschrittes bei
der Herstellung der Nucleinsäuren
eingebaut. So wird zum Beispiel eine Polymerasekettenreaktion (PCR) mit
markierten Primern oder markierten Nucleotiden ein markiertes Amplifikationsprodukt
bereitstellen. In einer anderen Ausführungsform baut eine Transkriptionsamplifikation
unter Verwendung eines markierten Nucleotids (z. B. Fluorescein-markiertes
UTP und/oder CTP) eine Markierung in die transkribierten Nucleinsäuren ein.
-
Nicht-radioaktive
Sonden werden oft durch indirekte Mittel markiert. Zum Beispiel
wird ein Ligandenmolekül
kovalent an die Sonde gebunden. Der Ligand bindet dann an ein Antiligandenmolekül, welches
inhärent
detektierbar ist oder kovalent an ein detektierbares Signalsystem,
zum Beispiel ein Enzym, ein Fluorophor oder eine chemilumineszierende
Verbindung, gebunden ist. Enzyme von Interessse als Markierungen
werden in erster Linie Hydrolasen, z. B. Phosphatasen, Esterasen
und Glycosidasen, oder Oxidoreduktasen, insbesondere Peroxidasen,
sein. Fluoreszierende Verbindungen umfassen Fluorescein und seine
Derivate, Rhodamin und seine Derivate, Dansyl, Umbelliferon usw.
Chemilumineszierende Verbindungen umfassen Luciferin und 2,3-Dihydrophthalazindione,
z. B. Luminol. Liganden und Antiliganden können in großem Umfang variiert werden.
Wenn ein Ligand einen natürlichen
Antiliganden hat, nämlich
Liganden wie Biotin, Tyroxin und Cortisol, kann er in Verbindung
mit seinen markierten, natürlich
auftretenden Antiliganden verwendet werden. Alternativ kann eine
beliebige Hapten- oder Antigen-Verbindung in Kombination mit einem
Antikörper
verwendet werden.
-
Sonden
können
auch durch direkte Konjugation mit einer Markierung markiert werden.
Beispielsweise wurden klonierte DNA-Sonden direkt an Meerrettichperoxidase
oder alkalische Phosphatase gekoppelt (Renz, M. und Kurz, K., A
Colorimetric Method for DNA Hybridization, Nucl. Acids Res. 12:
3435–3444
(1984)) und synthetische Oligonucleotide wurden direkt mit alkalischer
Phosphatase gekoppelt (Jablonski, E., et al., Preparation of Oligodeoxynucleotide-Alkaline Phosphatase
Conjugates and Their Use as Hybridization Probes, Nuc. Acids. Res.
14: 6115–6128
(1986); und Li P., et al., Enzyme-linked Synthetic Oligonucleotide
Probes: Non-Radioactive
Detection of Enterotoxigenic Escherichia Coli in Faeca Specimens,
Nucl. Acids Res. 15: 5275–5287
(1987)).
-
Mittel
zur Detektion solcher Markierungen sind dem Fachmann gut bekannt.
So können
z. B. radioaktive Markierungen unter Verwendung eines photographischen
Films oder eines Szintillationszählers
detektiert werden, fluoreszierende Marker können unter Verwendung eines
Photodetektors zum Detektieren von emittiertem Licht detektiert
werden. Enzymatische Markierungen werden typischerweise detektiert,
indem das Enzym mit einem Substrat versehen wird und das Reaktionsprodukt,
das durch die Wirkung des Enzyms auf das Substrat produziert wird,
de tektiert wird, und kolorimetrische Markierungen werden durch einfaches
Sichtbarwerden der gefärbten
Markierung detektiert.
-
Antikörper
gegen Proteine
-
Antikörper können gegen
ein Protein der vorliegenden Erfindung, einschließlich individueller,
allelischer, Stamm- oder Spezies-Varianten und Fragmente davon,
sowohl in ihren natürlich
auftretenden (Volllängen-)Formen
und in rekombinanten Formen, entwickelt werden. Außerdem werden
Antikörper
gegen diese Proteine in entweder ihren nativen Konfigurationen oder
in nicht-nativen
Konfigurationen entwickelt. Es können auch
anti-idiotypische Antikörper
erzeugt werden. Den Fachleuten sind viele Verfahren zur Herstellung
von Antikörpern
bekannt. Die folgende Diskussion wird als allgemeine Übersicht
der verfügbaren
Techniken präsentiert;
allerdings wird ein Fachmann erkennen, dass viele Variationen bei
den folgenden Verfahren bekannt sind.
-
Es
wird eine Reihe von Immunogenen verwendet, um Antikörper zu
produzieren, die mit einem Protein der vorliegenden Erfindung spezifisch
reaktiv sind. Ein isoliertes rekombinantes, synthetisches oder natives Polynucleotid
der vorliegenden Erfindung ist das bevorzugte Immunogen (Antigen)
für die
Produktion von monoklonalen oder polyklonalen Antikörpern. Der
Fachmann wird leicht verstehen, dass die Proteine der vorliegenden
Erfindung vor einer Bildung von Antikörpern zum Screening von Expressions-Bibliotheken
oder für
andere Assays, in denen ein vermeintliches Protein der vorliegenden
Erfindung exprimiert oder in einer nicht-nativen Sekundär, Tertiär- oder
Quarternärstruktur
denaturiert wird, typischerweise denaturiert und gegebenenfalls
reduziert werden. Nicht-isolierte Polypeptide der vorliegenden Erfindung
können
entweder in reiner Form oder in unreiner Form eingesetzt werden.
-
Das
Protein der vorliegenden Erfindung wird dann in ein Tier injiziert,
welches fähig
ist, Antikörper
zu produzieren. Es können
entweder monoklonale oder polyklonale Antikörper für eine anschließende Verwendung
in Immunoassays zur Messung des Vorliegens und der Menge des Proteins
der vorliegenden Erfindung erzeugt werden. Verfahren zum Produzieren
von polyklonalen Antikörpern
sind dem Fachmann bekannt. Kurz ausgedrückt, ein Immunogen (Antigen),
vorzugsweise ein gereinigtes Protein, ein Protein, das an einen
geeigneten Träger
gekoppelt ist (z. B. GST, Schlüssellochnapfschneckenhämatocyanin
usw.) oder ein Protein, das in einen Immunisierungsvektor, z. B.
ein rekombinantes Vacciniavirus eingebaut ist (siehe
U.S. Patent Nr. 4,722,848 ) wird mit
einem Adjuvans vermischt und Tiere werden mit dem Gemisch immunisiert.
Die Immunantwort des Tieres auf die Immunogenpräparation wird überwacht,
indem Testblutproben genommen werden und der Reaktivitätstiter
auf das Protein von Interesse bestimmt wird. Wenn geeignet hohe
Antikörpertiter
auf das Immunogen erhalten werden, wird Blut aus dem Tier gesammelt
und es werden Antiseren hergestellt. Eine weitere Fraktionierung
der Antiseren, um auf Antikörper,
die zu dem Protein reaktiv sind, anzureichern, wird auf Wunsch durchgeführt (siehe
z. B. Coligan, Current Protocols in Immunology, Wiley/Greene, NY
(1991); und Harlow und Lane, Antibodies: A Laboratory Manual, Cold
Spring Harbor Press, NY (1989)).
-
Antikörper, einschließlich Bindungsfragmente
und rekombinante Einzelkettenversionen davon, gegen vorbestimmte
Fragmente eines Proteins der vorliegenden Erfindung werden durch
Immunisieren von Tieren, z. B. mit Konjugaten der Fragmente mit
Trägerproteinen,
wie oben beschrieben, erzeugt. Typischerweise ist das Immunogen
von Interesse ein Protein mit wenigstens etwa 5 Aminosäuren, typischerweise
ist das Protein 10 Aminosäuren
lang, vorzugsweise 15 Aminosäuren
lang und bevorzugter ist das Protein 20 Aminosäuren lang oder größer. Die
Peptide werden typischerweise an ein Trägerprotein (z. B. als Fusionsprotein)
gekoppelt oder werden rekombinant in einem Immunisierungsvektor
exprimiert. Antigene Determinanten an Peptiden, an welche Antikörper binden,
haben typischerweise eine Länge
von 3 bis 10 Aminosäuren.
-
Monoklonale
Antikörper
werden aus Zellen hergestellt, die den gewünschten Antikörper sezernieren. Monoklonale
Antikörper
werden auf Bindung an ein Protein, von dem das Immunogen abgeleitet
ist, durchgemustert. Spezifische monoklonale und polyklonale Antikörper werden üblicherweise
eine Antikörperbindungsstelle
mit einer Affinitätskonstante
für ihr
verwandtes monovalentes Antigen von wenigstens zwischen 106–107, üblicherweise
wenigstens 108, vorzugsweise wenigstens
109, bevorzugter wenigstens 1010 und
am bevorzugtesten 1011 Liter/mol, haben.
-
In
einigen Fällen
ist es wünschenswert,
monoklonale Antikörper
aus verschiedenen Säugerwirten,
z. B. Mäuse,
Nager, Primaten, Menschen usw., herzustellen. Eine Beschreibung
von Techniken zur Herstellung solcher monoklonaler Antikörper wird
in Basic and Clinical Immunology, 4. Ausgabe, Sites et al., Herausg., Lange
Medical Publications, Los Altos, CA und darin zitierten Literaturstellen;
Harlow und Lane, Supra; Goding, Monoclonal Antibodies: Principles
and Practice, 2. Ausgabe, Academic Press, New York, NY (1986); und Kohler
und Milstein, Nature 256: 495–497
(1975) gefunden. Kurz zusammengefasst, dieses Verfahren läuft ab, indem
einem Tier ein Immunogen, das ein Protein der vorliegenden Erfindung
umfasst, injiziert wird. Das Tier wird dann getötet und Zellen werden aus seiner
Milz entnommen, welche mit Myelomzellen fusioniert werden. Das Resultat
ist eine Hybridzelle oder ein „Hybridom", das zur Reproduktion
in vitro fähig
ist. Die Population von Hybridomen wird dann durchgemustert, um
einzelne Klone zu isolieren, von denen jeder eine einzelne Antikörperspezies
gegen das Immunogen sezerniert. Auf diese Weise sind die individuellen
Antikörperspezies, die
erhalten werden, die Produkte von immortalisierten und klonierten
einzelnen B-Zellen aus dem immunen Tier, erzeugt als Reaktion auf
eine spezifische Stelle, die an der immunogenen Substanz erkannt
wird.
-
Andere
geeignete Techniken involvieren eine Selektion von Bibliotheken
von rekombinanten Antikörpern
in einem Phagen oder ähnlichen
Vektoren (siehe z. B. Huse et al., Science 246: 1275–1281 (1989);
und Ward et al., Nature 341: 544–546 (1989); und Vaughan et
al., Nature Biotechnology, 14: 309–314 (1996)). Alternativ können humane
monoklonale Antikörper
hoher Affinität
aus transgenen Mäusen
erhalten werden, die Fragmente der nicht-umgelagerten humanen schwere-
und leichte-Kette-Ig-Loci umfassen (d. h. transgene Minilocus-Mäuse). Fishwild
et al., Nature Biotech., 14: 845–851 (1996). Es können auch
rekombinante Immunglobuline produziert werden. Siehe Cabilly,
U.S. Patent Nr. 4,816,567 ;
und Queen et al., Proc. Natl. Acad. Sci. 86: 10029–10033 (1989).
-
Die
Antikörper
dieser Erfindung werden auch für
eine Affinitätschromatographie
bei der Isolierung von Proteinen der vorliegenden Erfindung verwendet.
Säulen
werden hergestellt, z. B. mit den Antikörpern, die an einen festen
Träger,
z. B. Partikel, wie Agarose, Sephadex, oder dergleichen gebunden
sind, wobei ein Zelllysat durch die Säule geführt wird, gewaschen wird und
mit steigenden Konzentrationen eines milden Denaturierungsmittels
behandelt wird, wodurch gereinigtes Protein freigesetzt wird.
-
Die
Antikörper
können
verwendet werden, um Expressions-Bibliotheken nach bestimmten Expressionsprodukten,
z. B. normales oder abnormales Protein, durchzumustern. Üblicherweise
werden die Antikörper in
einem solchen Verfahren mit einer Gruppierung markiert, die eine
einfache Detektion des Vorliegens von Antigen durch Antikörperbindung
erlaubt.
-
Antikörper, die
gegen ein Protein der vorliegenden Erfindung entwickelt wurden,
können
auch verwendet werden, um anti-idiotypische Antikörper zu
erzeugen. Sie sind zum Detektieren oder Diagnostizieren von verschiedenen
pathologischen Zuständen,
die mit dem Vorliegen der entsprechenden Antigene verbunden sind,
einsetzbar.
-
Häufig werden
die Proteine und Antikörper
der vorliegenden Erfindung markiert, indem sie entweder kovalent
oder nicht-kovalent mit einer Substanz verbunden werden, die für ein detektierbares
Signal sucht. Es ist ein weiter Bereich von Markierungen und Konjugationstechniken
bekannt und ausgiebig in der wissenschaftlichen und Patentliteratur
beschrieben. Geeignete Markierungen umfassen Radionucleotide, Enzyme, Substrate,
Cofaktoren, Inhibitoren, fluoreszierende Gruppierungen, chemilumineszierende
Gruppierungen, magnetische Partikel und dergleichen.
-
Proteinimmunoassays
-
Mittel
zum Detektieren der Proteine der vorliegenden Erfindung sind keine
kritischen Aspekte der vorliegenden Erfindung. In einer bevorzugten
Ausführungsform
werden die Proteine detektiert und/oder quantitativ bestimmt, indem
ein beliebiger einer Reihe von gut anerkannten immunologischen Bindungsassays
verwendet wird (siehe z. B.
U.S.
Patente 4,366,241 ;
4,376,100 ;
4,517,288 ; und
4,837,168 ). Für eine Übersicht der allgemeinen Immunoassays
siehe auch Methods in Cell Biology, Bd. 37: Antibodies in Cell Biology,
Asai, Herausg., Academic Press, Inc. New York (1993); Basic and
Clinical Immunology, 7. Ausgabe, Stites & Terr, Herausg. (1991). Darüber hinaus
können
die Immunoassays der vorliegenden Erfindung in einer beliebigen
von mehreren Konfigurationen durchgeführt werden, z. B. solche, für die eine Übersicht
in Enzyme Immunoassay, Maggio, Herausg., CRC Press, Boca Raton,
Florida (1980); Tijan, Practice and Theory of Enzyme Immunoassays,
Laboratory Techniques in Biochemistry and Molecular Biology, Elsevier
Science Publishers B. V., Amsterdam (1985); Harlow und Lane, oben;
Immunoassays: A Practical Guide, Chan, Herausg, Academic Press, Orlando,
FL (1987); Principles and Practice of Immunoassaysm, Price und Newman,
Herausg., Stockton Press, NY (1991); und Non-isotopic Immunoassays,
Ngo, Herausg., Plenum Press, NY (1988) gegeben wird. Immuno logische
Bindungsassays (oder Immunoassays) verwenden typischerweise ein „Fangmittel", um spezifisch an
den Analyten (in diesem Fall ein Protein der vorliegenden Erfindung)
spezifisch zu binden und oft zu immobilisieren. Das Fangmittel ist
eine Gruppierung, die spezifisch an den Analyten bindet. In einer
bevorzugten Ausführungsform
ist das Fangmittel ein Antikörper,
der spezifisch ein Protein (Proteine) der vorliegenden Erfindung
bindet. Der Antikörper
kann durch ein beliebiges einer Reihe von Mitteln, die dem Fachmann
auf dem Gebiet, wie es hierin beschrieben wird, bekannt sind, produziert
werden.
-
Immunoassays
verwenden oft auch ein Markierungsmittel, um spezifisch an den Bindungskomplex, der
durch das Fangmittel und den Analyten gebildet wird, zu binden und
diesen zu markieren. Das Markierungsmittel kann selbst eine der
Gruppierungen sein, die den Antikörper-Analytenkomplex umfassen.
Demnach kann das Markierungsmittel ein markiertes Protein der vorliegenden
Erfindung oder ein markierter Antikörper, der spezifisch reaktiv
zu einem Protein der vorliegenden Erfindung ist, sein. Alternativ
kann das Markierungsmittel eine dritte Gruppierung, z. B. ein anderer
Antikörper,
der spezifisch an den Antikörper/Protein-Komplex
bindet, sein.
-
In
einer bevorzugten Ausführungsform
ist das Markierungsmittel ein zweiter Antikörper, der eine Markierung trägt. Alternativ
kann dem zweiten Antikörper
eine Markierung fehlen, aber er kann wiederum an einen markierten
dritten Antikörper
gebunden sein, der für
Antikörper
der Spezies spezifisch ist, aus dem der zweite Antikörper abgeleitet
ist. Der zweite kann mit einer detektierbaren Gruppierung, z. B.
Biotin, modifiziert sein, an welches ein drittes markiertes Molekül spezifisch
binden kann, z. B. Enzym-markiertes Streptavidin.
-
Andere
Proteine, die fähig
sind, spezifisch konstante Immunglobulinregionen zu binden, z. B.
Protein A oder Protein G, können
ebenfalls als das Markierungsmittel verwendet werden. Diese Proteine
sind normale Bestandteile der Zellwände von Streptococcenbakterien.
Sie weisen eine starke nicht-immunogene Reaktivität mit konstanten
Immunglobulinregionen aus einer Vielzahl von Spezies auf (Siehe
allgemein Kronval, et al., J. Immunol. 111: 1401–1406 (1973) und Akerstrom
et al., J. Immunol. 135: 2589–2542
(1985)).
-
Durch
die Assays können
Inkubations- und/oder Waschschritte nach jeder Kombination von Reagenzien
erforderlich sein. Inkubationsschritte können von etwa 5 Sekunden bis
mehrere Stunden, vorzugsweise von etwa 5 Minuten bis etwa 24 Stunden,
variieren. Allerdings wird die Inkubationszeit vom Assayformat,
Analyten, Lösungsvolumen,
von Konzentrationen und dergleichen abhängen. Üblicherweise werden die Assays bei
Umgebungstemperatur durchgeführt
werden, obgleich sie über
einen Temperaturbereich, z. B. 10°C
bis 40°C,
durchgeführt
werden können.
-
Obgleich
die Details der Immunoassays der vorliegenden Erfindung mit dem
bestimmten verwendeten Format variieren können, umfasst das Verfahren
zum Detektieren eines Proteins der vorliegenden Erfindung in einer
biologischen Probe im Allgemeinen die Schritte des In-Kontakt-Bringens der biologischen
Probe mit einem Antikörper,
der unter immunologisch reaktiven Be dingungen spezifisch mit einem
Protein der vorliegenden Erfindung reagiert. Der Antikörper wird
unter immunologisch reaktiven Bedingungen an das Protein binden gelassen
und das Vorliegen des gebundenen Antikörpers wird direkt oder indirekt
detektiert.
-
A. Nicht-kompetitive Assay-Formate
-
Immunoassays
zum Detektieren von Proteinen der vorliegenden Erfindung umfassen
kompetitive und nicht-kompetitive Formate. Nicht-kompetitive Immunoassays
sind Assays, in denen die Menge an gefangenem Analyten (d. h. ein
Protein der vorliegenden Erfindung) direkt gemessen wird. In einem
bevorzugten „Sandwich"-Assay kann das Fangmittel
(z. B. ein Antikörper,
der unter immunreaktiven Bedingungen mit einem Protein der vorliegenden
Erfindung spezifisch reaktiv ist) direkt an ein festes Substrat
gebunden werden, wo sie immobilisiert werden. Diese immobilisierten
Antikörper
fangen dann das Protein, das in der Testprobe vorliegt. Das so immobilisierte
Protein wird dann durch ein Markierungsmittel, z. B. einen zweiten
Antikörper,
der eine Markierung trägt,
gebunden. Alternativ kann dem zweiten Antikörper eine Markierung fehlen,
er kann aber wiederum an einen markierten dritten Antikörper gebunden
werden, der für
die Antikörper
der Spezies, aus dem der zweite Antikörper stammt, spezifisch ist.
Der zweite kann mit einer detektierbaren Gruppierung, z. B. Biotin, modifiziert
sein, an welche ein drittes markiertes Molekül spezifisch binden kann, z.
B. Enzym-markiertes Streptavidin.
-
B. Kompetitive Assay-Formate
-
In
kompetitiven Assays wird die Menge an Analyt, der in der Probe vorliegt,
indirekt gemessen, indem die Menge eines zugesetzten (exogenen)
Analyten (z. B. ein Protein der vorliegenden Erfindung) von einem Fangmittel
(z. B. ein Antikörper,
der unter immunreaktiven Bedingungen mit dem Protein spezifisch
reaktiv ist) durch den in der Probe vorliegenden Analyten verdrängt wird
(durch Wettbewerb entfernt wird). In einem kompetitiven Assay wird
eine bekannte Menge an Analyt zu der Probe gegeben und die Probe
wird dann mit einem Fangmittel, das spezifisch ein Protein der vorliegenden
Erfindung bindet, in Kontakt gebracht. Die Menge an Protein, die
an das Fangmittel gebunden ist, ist umgekehrt proportional zu der
Konzentration an Analyt, die in der Probe vorliegt.
-
In
einer besonders bevorzugten Ausführungsform
wird der Antikörper
an einem festen Substrat immobilisiert. Die Menge an Protein, die
an dem Antikörper
gebunden ist, kann entweder durch Messen der Proteinmenge, die in
einem Protein/Antikörper-Komplex
vorliegt oder alternativ durch Messen der Menge an restlichem unkomplexiertem
Protein bestimmt werden. Die Proteinmenge kann detektiert werden,
indem ein markiertes Protein bereitgestellt wird.
-
Ein
Hapteninhibierungsassay ist ein anderer bevorzugter kompetitiver
Assay. In diesem Assay wird ein bekannter Analyt (z. B. ein Protein
der vorliegenden Erfindung) an einem festen Substrat immobilisiert.
Eine bekannte Menge an Antikörper,
der unter immunreaktiven Bedingungen mit dem Protein spezifisch
reaktiv ist, wird der Probe zugesetzt und die Probe wird dann mit
dem immobilisierten Protein in Kontakt gebracht. In diesem Fall
ist die Menge an Antikörper,
die an das immobilisierte Protein gebunden ist, umgekehrt proportional zu
der Menge an Protein, die in der Probe vorliegt. Wiederum kann die
Menge an immobilisiertem Antikörper detektiert
werden, indem entweder die immobilisierte Antikörperfraktion oder die Fraktion
des Antikörpers,
die in Lösung
bleibt, detektiert wird. Eine Detektion kann direkt sein, wenn der
Antikörper
markiert ist, oder durch die anschließende Zugabe einer markierten
Gruppierung, die spezifisch an den Antikörper bindet, wie es oben beschrieben
wurde, indirekt sein.
-
C. Erzeugung von gepoolten Antiseren zur
Verwendung in Immunoassays
-
Ein
Protein, das spezifisch an einen Antikörper bindet oder das spezifisch
immunreaktiv mit einem Antikörper
ist, der gegen ein definiertes Immunogen erzeugt wurde, wird in
einem Immunoassay bestimmt. Der Immunoassay verwendet ein polyklonales
Antiserum, welches gegen ein Polypeptid der vorliegenden Erfindung
(d. h. das immunogene Polypeptid) entwickelt wurde. Dieses Antiserum
wird so gewählt,
dass es eine niedrige Kreuzreaktivität gegen andere Proteine hat
und eine beliebige solche Kreuzreaktivität durch Immunoabsorption vor
Verwendung im Immunoassay (z. B. durch Immunsorbtion der Antiseren
mit einem Protein mit unterschiedlicher Substratspezifität (z. B.
ein anderes Enzym) und/oder einem Protein mit derselben Substratspezifität, aber
in anderer Form) entfernt wird.
-
Um
Antisera zur Verwendung in einem Immunoassay zu produzieren, wird
ein Polypeptid der vorliegenden Erfindung isoliert, wie es hierin
beschrieben ist. Beispielsweise kann ein rekombinantes Protein in
einer Säuger-
oder einer anderen eukaryotischen Zelllinie produziert werden. Ein
Inzuchtstamm von Mäusen
wird unter Verwendung eines Standardadjuvans, z. B. Freund's-Adjuvans, und unter Verwendung eines
Standard-Mausimmunisierungsprotokolls (siehe Harlow und Lane, oben)
mit dem Protein immunisiert. Alternativ wird ein synthetisches Polypeptid,
das von den hierin offenbarten Sequenzen abgeleitet ist und an ein
Trägerprotein
konjugiert ist, als Immunogen verwendet. Polyklonale Seren werden
gesammelt und gegen das immunogene Polypeptid in einem Immunoassay,
z. B. in einem Festphasenimmunoassay, mit dem an einem festen Träger immobilisierten
Immunogen getitert. Polyklonale Antisera mit einem Titer von 104 oder größer werden gesammelt
und auf ihre Kreuzreaktivität
gegen Polypeptide unterschiedlicher Formen oder Substratspezifität getestet,
wobei ein kompetitiver Bindungsimmunoassay verwendet wird, z. B.
der, der bei Harlow und Lane, oben, auf den Seiten 570–573 beschrieben
ist. Vorzugsweise werden zwei oder mehr unterschiedliche Formen von
Polypeptiden bei dieser Bestimmung verwendet. Diese unterschiedlichen
Typen von Polypeptiden werden als Kompetitoren eingesetzt, um Antikörper zu
identifizieren, die durch das Polypeptid, auf das analysiert wird, spezifisch
gebunden werden. Die kompetitiven Polypeptide können als rekombinante Proteine
produziert werden und unter Verwendung von Standard-Molekularbiologie-
und Standard-Proteinchemie-Techniken,
wie sie hierin beschrieben werden, isoliert werden.
-
Immunoassays
im kompetitiven Bindungsformat werden für Kreuzreaktivitätsbestimmungen
verwendet. Beispielsweise wird das immunogene Polypeptid an einem
festen Träger
immobilisiert. Proteine, die dem Assay zugesetzt werden, kompetitieren
mit der Bindung der Antiseren gegen das immobilisierte Antigen.
Die Fähigkeit
der obigen Proteine, um die Bindung der Antise ren gegen das immobilisierte
Protein zu kompetitieren, wird mit dem immunogenen Polypeptid verglichen.
Die prozentuale Kreuzreaktivität
für die
obigen Proteine wird errechnet, wobei Standardberechnungen verwendet
werden. Solche Antiseren mit weniger als 10% Kreuzreaktivität mit einer
unterschiedlichen Form eines Polypeptids werden ausgewählt und
gepoolt. Die kreuzreagierenden Antikörper werden dann durch Immunabsorption
mit einer unterschiedlichen Form eines Polypeptids aus den gepoolten
Antiseren entfernt.
-
Die
immunoabsorbierten und gepoolten Antiseren werden dann in einem
kompetitiven Bindungsimmunoassay, wie er hierin beschrieben wird,
eingesetzt, um ein zweites „Target"-Polypeptid mit dem immunogenen Polypeptid
zu vergleichen. Um diesen Vergleich durchzuführen, werden die zwei Polypeptide
jeweils in einem weiten Konzentrationsbereich analysiert und die
Menge jedes Polypeptids, die erforderlich ist, um 50% der Bindung
der Antiseren an das immobilisierte Protein zu inhibieren, wird
unter Verwendung von Standardtechniken bestimmt. Wenn die Menge
des Target-Polypeptids, die erforderlich ist, weniger als die zweifache
Menge des immunogenen Polypeptids, die erforderlich ist, ist, dann
wird gesagt, dass das Target-Polypeptid
spezifisch an einen Antikörper,
der gegen das immunogene Protein erzeugt wurde, bindet. Als abschließende Spezifitätsbestimmung
wird das gepoolte Antiserum vollständig mit dem immunogenen Polypeptid
immunsorbiert, bis keine Bindung an das verwendete Polypeptid bei
der Immunsorbtion detektierbar ist. Das vollständig immunsorbierte Antiserum
wird dann auf Reaktivität
mit dem Test-Polypeptid untersucht. Wenn keine Reaktivität beobachtet wird,
dann wird das Test-Polypeptid durch das Antiserum, das durch das
immunogene Protein hervorgerufen wurde, spezifisch gebunden.
-
D. Andere Assay-Formate
-
In
einer besonders bevorzugten Ausführungsform
wird eine Western-Blot (Immunoblot)-Analyse verwendet, um das Vorliegen
von Protein der vorliegenden Erfindung in der Probe zu detektieren
und zu quantifizieren. Die Technik umfasst im Allgemeinen ein Trennen
von Probenproteinen durch Gelelektrophorese auf der Basis des Molekulargewichts,
Transferieren der getrennten Proteine auf einen geeigneten festen
Träger (z.
B. ein Nitrocellulosefilter, ein Nylonfilter oder ein derivatisiertes
Nylonfilter) und Inkubieren der Probe mit den Antikörpern, die
ein Protein der vorliegenden Erfindung spezifisch binden. Die Antikörper binden
spezifisch an das Protein auf dem festen Träger. Diese Antikörper können direkt
markiert sein oder können
alternativ anschließend
unter Verwendung von markierten Antikörpern (z. B. markierte Schaf-Anti-Maus-Antikörper), die spezifisch
an die Antikörper
binden, detektiert werden.
-
E. Quantifizierung von Proteinen
-
Die
Proteine der vorliegenden Erfindung können durch ein beliebiges einer
Reihe von Mitteln, die dem Fachmann gut bekannt sind, detektiert
und quantifiziert werden. Diese umfassen analytische biochemische Verfahren,
wie z. B. Elektrophorese, Kapillarelektrophorese, Hochleistungsflüssigkeitschromatographie (HPLC),
Dünnschichtchromatographie
(TLC), Hyperdiffusionschromatographie und dergleichen, und verschiedene
immunologische Verfahren, z. B. Flüs sigkeits- oder Gelprecipitinreaktionen,
Immunodiffusion (einzelne oder doppelte), Immunoelektrophorese,
Radioimmunoassays (RIAs), enzymgekoppelte Immunadsorptionsassays
(ELISA), Immunfluoreszenzassays und dergleichen.
-
F. Reduktion einer nicht-spezifischen
Bindung
-
Ein
Fachmann wird erkennen, dass es oft wünschenswert ist, eine nicht-spezifische
Bindung in Immunoassays und während
einer Analytenreinigung zu reduzieren. Wenn der Assay ein Antigen,
einen Antikörper oder
ein anderes Fangmittel, immobilisiert auf einem festen Träger involviert,
ist es wünschenswert,
die Menge an nicht-spezifischer Bindung an dem Substrat zu minimieren.
Mittel zur Reduzierung einer solchen nicht-spezifischen Bindung
sind dem Fachmann gut bekannt. Typischerweise involviert dies ein
Beschichten des Substrats mit einer proteinartigen Zusammensetzung.
In großem
Umfang werden insbesondere Proteinzusammensetzungen, z. B. Rinderserumalbumin
(BSA), Nicht-Fett-Milchpulver und Gelatine verwendet.
-
G. Immunoassay Markierungen
-
Das
Markierungsmittel kann z. B. ein monoklonaler Antikörper, ein
polyklonaler Antikörper,
ein Bindungsprotein oder ein Komplex oder ein Polymer, z. B. eine
Affinitätsmatrix,
Kohlenhydrat oder Lipid sein. Detektierbare Markierungen, die zur
Verwendung in der vorliegenden Erfindung geeignet sind, umfassen
eine beliebige Zusammensetzung, die durch spektroskopische, Radioisotopen-,
photochemische, biochemische, immunochemische, elektrische, optische
oder chemische Mittel detektierbar ist. Eine Detektion kann durch
ein beliebiges bekanntes Verfahren, z. B. Immunoblotting, Western-Analyse,
Gelmobilitätsverschiebungsassays, Fluoreszenz-in
situ-Hybridisierungsanalyse
(FISH), Verfolgung von radioaktiven oder biolumineszierenden Markern,
kernmagnetische Resonanz, elektronenparamagnetische Resonanz, Spektroskopie
mit gestopptem Fluss, Säulenchromatographie,
Kapillarelektrophorese oder durch andere Verfahren, die ein Molekül auf der Basis
einer Änderung
in Größe und/oder
Ladung verfolgen, erfolgen. Die bestimmte Markierung oder die bestimmte
detektierbare Gruppe, die in dem Assay verwendet wird, ist kein
kritischer Aspekt der Erfindung. Die detektierbare Gruppe kann ein
beliebiges Material mit einer detektierbaren physikalischen oder
chemischen Eigenschaft sein. Solche detektierbaren Markierungen
sind auf dem Gebiet der Immunoassays gut entwickelt und im Allgemeinen
kann eine beliebige Markierung, die in solchen Verfahren verwendbar
ist, auf die vorliegende Erfindung angewendet werden. Somit ist
eine Markierung eine beliebige Zusammensetzung, die durch spektroskopische,
photochemische, biochemische, immunochemische, elektrische, optische
oder chemische Mittel detektierbar ist. Verwendbare Markierungen
in der vorliegenden Erfindung umfassen magnetische Perlen, Fluoreszenzfarbstoffe,
radioaktive Markierungen, Enzyme und kolorimetrische Markierungen
oder gefärbte
Glas- oder Kunststoffperlen, wie sie oben für Nucleinsäuremarkierungen diskutiert
wurden.
-
Die
Markierung kann nach Verfahren, die auf dem Fachgebiet gut bekannt
sind, direkt oder indirekt an die gewünschte Komponente des Assays
gekoppelt werden. Wie oben angegeben wurde, kann eine weite Vielzahl
von Markierungen verwendet werden, wobei die Wahl der Markierung
von der erforderlichen Sensitivität, der Einfachheit der Konjugation
der Verbindung, Stabilitätsanforderungen,
verfügbaren
Instrumenten und Entsorgungsvorschriften abhängt.
-
Nicht-radioaktive
Markierungen werden oft durch indirekte Mittel gebunden. Im Allgemeinen
wird ein Ligandenmolekül
(z. B. Biotin) kovalent an das Molekül gebunden. Der Ligand bindet
dann an ein Anti-Liganden (z. B. Streptavidin)-Molekül, welches
entweder inhärent
detektierbar ist oder kovalent an ein Signalsystem, z. B. ein detektierbares
Enzym, eine fluoreszierende Verbindung oder eine chemilumineszierende
Verbindung, gebunden ist. Es kann eine Reihe von Liganden und Anti-Liganden
verwendet werden. Wenn ein Ligand einen natürlichen Anti-Liganden hat, z.
B. Biotin, Thyroxin und Cortisol, kann er in Verbindung mit dem
markierten, natürlich
vorkommenden Anti-Liganden verwendet werden. Alternativ kann eine
Hapten- oder Antigenverbindung in Kombination mit einem Antikörper verwendet
werden.
-
Die
Moleküle
können
auch direkt mit Signal erzeugenden Verbindungen konjugiert werden,
z. B. durch Konjugation mit einem Enzym oder Fluorophor. Enzyme
von Interesse als Markierungen werden in erster Linie Hydrolasen,
insbesondere Phosphatasen, Esterasen und Glycosidasen oder Oxidoreduktasen,
insbesondere Peroxidasen, sein. Fluoreszierende Verbindungen umfassen
Fluorescein und seine Derivate, Rhodamin und seine Derivate, Dansyl,
Umbelliferon usw. Chemilumineszierende Verbindungen umfassen Luceriferin
und 2,3-Dihydrophathalazindione, z. B. Luminol. Für eine Übersicht über verschiedene
Markierungs- oder Signal-produzierende
Systeme, die verwendet werden können,
siehe
U.S. Patent Nr. 4,391,904 ,
welches hier durch Bezugnahme aufgenommen wird.
-
Mittel
zum Detektieren von Markierungen sind dem Fachmann gut bekannt.
Wenn die Markierung z. B. eine radioaktive Markierung ist, so umfassen
Mittel zur Detektion einen Szintillationszähler oder einen photographischen
Film, wie bei Autoradiographie. Wenn die Markierung eine fluoreszierende
Markierung ist, kann sie durch Anregen des Fluorochroms mit dem
Licht geeigneter Wellenlänge
und Detektieren der resultierenden Fluoreszenz, z. B. durch Mikroskopie,
Untersuchung mit den Augen, über
photographischen Film, durch die Verwendung von elektronischen Detektoren,
z. B. mit Ladung gekoppelten Vorrichtungen (CCDs) oder Photomultiplier
und dergleichen detektiert werden. In ähnlicher Weise können enzymatische
Markierungen durch Bereitstellung geeigneter Substrate für das Enzym
und Detektieren des resultierenden Reaktionsprodukts detektiert
werden. Schließlich
können
einfache kolorimetrische Markierungen in einfacher Weise durch Betrachten
der Farbe, die mit der Markierung assoziiert ist, detektiert werden.
So erscheint in verschiedenen Dipstick-Assays gold oft pinkfarben,
während
verschiedene konjugierte Perlen die Farbe der Perlen zeigen.
-
Einige
Assay-Formate erfordern keine Verwendung von markierten Komponenten.
Beispielsweise können
Agglutinationsassays verwendet werden, um das Vorliegen der Target-Antikörper zu
detektieren. In diesem Fall werden mit Antigen überzogene Partikel durch Proben,
die die Target-Antikörper
umfassen, agglutiniert. In diesem Format muss keine der Komponenten markiert
werden und das Vorliegen des Target-Antikörpers wird durch einfache visuelle
Inspektion detektiert.
-
Assays auf Verbindungen, die die enzymatische
Aktivität
oder die Expression modulieren
-
Die
vorliegende Erfindung bezieht sich auch auf Mittel zur Identifizierung
von Verbindungen, die an (z. B. Substrate) katalytisch aktive Polypeptide
der vorliegenden Erfindung binden und/oder die enzymatische Aktivität von katalytisch
aktiven Polypeptiden der vorliegenden Erfindung erhöhen oder
verringern (d. h. modulieren). Das Verfahren umfasst In-Kontakt-Bringen
eines Polypeptids der vorliegenden Erfindung mit einer Verbindung,
deren Fähigkeit,
Enzymaktivität
zu binden oder zu modulieren, bestimmt werden soll. Das verwendete
Polypeptid wird wenigstens 20%, vorzugsweise wenigstens 30% oder
40%, bevorzugter wenigstens 50% oder 60% und am bevorzugtesten wenigstens
70% oder 80% der spezifischen Aktivität des nativen Volllängen-Polypeptids
der vorliegenden Erfindung (z. B. Enzym) haben. Im Allgemeinen wird
das Polypeptid in einem Bereich vorliegen, der ausreichend ist,
um die Wirkung der Verbindung zu bestimmen, typischerweise etwa
1 nM bis 10 μM.
Entsprechend wird die Verbindung in einer Konzentration von etwa
1 nM bis 10 μM
vorliegen. Der Fachmann wird einsehen, dass solche Faktoren wie
Enzymkonzentration, Ligandenkonzentrationen (d. h. Substrate, Produkte,
Inhibitoren, Aktivatoren), pH, Ionenstärke und Temperatur so kontrolliert
werden, dass verwendbare Kinetikdaten erhalten werden und das Vorliegen
oder Fehlen einer Verbindung, die Polypeptidaktivität bindet
oder moduliert, bestimmen. Verfahren zur Messung der Enzymkinetik
sind auf dem Fachgebiet gut bekannt. Siehe z. B. Segel, Biochemial
Calculations, 2. Ausgabe, John Wiley and Sons, New York (1976).
-
Obgleich
die vorliegende Erfindung detailliert durch Erläuterungen und Beispiele zu
Zwecken des Verständnisses
beschrieben wurde, wird es klar sein, dass bestimmte Änderungen
und Modifikationen im Rahmen der beigefügten Ansprüche durchgeführt werden
können.
-
Beispiel 1
-
Dieses
Beispiel beschreibt die Konstruktion von cDNA-Bibliotheken
-
Gesamt-RNA-Isolierung
-
Gesamt-RNA
wurde aus Maisgeweben mit TRIzol-Reagens (Life Technology Inc. Gaithersburg,
MD) unter Verwendung einer Modifikation des Guanidinisocyanat/Säure-Phenol-Verfahrens, beschrieben
von Chomczynski und Sacchi (Chomczynski, P. und Sacchi, N. Anal.
Biochem. 162, 156 (1987)), isoliert. Kurz ausgedrückt, Pflanzengewebeproben
wurden in flüssigem
Stickstoff vor Zugabe des TRIzol-Reagenses pulverisiert und dann
mit einem Mörser
und Pistill weiter homogenisiert. Der Zusatz von Chloroform, gefolgt
von einer Zentrifugation, wurde zur Trennung einer wässrigen
Phase und einer organischen Phase durchgeführt. Die Gesamt-RNA wurde durch Präzipitation
mit Isopropylalkohol aus der wässrigen
Phase gewonnen.
-
Poly(A)+ RNA-Isolierung
-
Die
Auswahl von Poly(A)+RNA aus Gesamt-RNA wurde unter Verwendung eines
PolyATact-Sytems (Promega Corporation, Madison, WI) durchgeführt. Kurz
ausgedrückt,
biotinylierte Oligo(dT)-Primer wurden verwendet, um an die 3'-Poly(A)-Tails an
mRNA zu hybridisieren. Die Hybride wurden unter Verwendung von Streptavidin,
gekoppelt an paramagnetische Partikel, und eines magnetischen Trennungsstands
gefangen. Die mRNA wurde unter Bedingungen hoher Stringenz gewaschen
und mit RNase-freiem entionisiertem Wasser eluiert.
-
Konstruktion einer cDNA-Bibliothek
-
Eine
cDNA-Synthese wurde durchgeführt
und unidirektionale cDNA-Bibliotheken wurden unter Verwendung des
SuperScript-Plasmidsystems (Life Technology Inc. Gaithersburg, MD)
konstruiert. Der erste cDNA-Strang wurde synthetisiert, indem ein
Oligo(dT)-Primer, der eine NotI-Stelle enthielt, geprimt wurde.
Die Reaktion wurde durch SuperScript-reverse Transkriptase II bei
45°C katalysiert.
Der zweite cDNA-Strang wurde mit alpha-32P-dCTP
markiert und ein Teil der Reaktion wurde durch Agarosegelelektrophorese
analysiert, um die cDNA-Größen zu bestimmen.
cDNA-Moleküle,
die kleiner als 500 Basenpaare waren und nicht-ligierte Adapter
wurden durch Sephacryl-S400-Chromatographie entfernt. Die ausgewählten cDNA-Moleküle wurden zwischen
NotI- und SalI-Stellen in den pSPORT1-Vektor ligiert.
-
Beispiel 2
-
Dieses
Beispiel beschreibt eine cDNA-Sequenzierung und eine Bibliothekssubtraktion
-
Sequenzierung einer Matrizenpräparation
-
Einzelne
Kolonien wurden herausgenommen bzw. angeimpft und DNA wurde entweder
durch PCR mit M13-Vorwärtsprimer
und M13-Rückwärtsprimern
oder durch Plasmidisolierung hergestellt. Alle cDNA-Klone wurden
unter Verwendung von M13-Rückwärtsprimern
sequenziert.
-
Q-bot-Subtraktionsverfahren
-
cDNA-Bibliotheken,
die einem Subtraktionsverfahren unterzogen worden waren, wurden
auf einer 22 × 22
cm2-Agarplatte in einer Dichte von etwa
3000 Kolonien pro Platte ausplattiert. Die Platten wurden für 12–24 Stunden
in einem 37°C-Inkubator
inkubiert. Kolonien wurden in 384-Well-Platten mit einem Kolonien-Picker-Roboter,
Q-bot (GENETIX Limited), aufgenommen. Diese Platten wurden über Nacht
bei 37°C
inkubiert.
-
Sobald
ausreichend Kolonien abgeimpft waren, wurden sie auf 22 × 22 cm2-Nylonmembranen unter Verwendung des Q-bot
gebracht. Jede Membran enthielt 9216 Kolonien oder 36864 Kolonien.
Diese Membranen wurden auf eine Agarplatte mit geeignetem Antibiotikum
gelegt. Die Platten wurden über
Nacht bei 37°C inkubiert.
-
Nachdem
die Kolonien am zweiten Tag isoliert worden waren, wurden diese
Filter auf mit Denaturierungslösung
vorbefeuchtetes Filterpapier für
vier Minuten gelegt, dann wurden sie auf einem siedenden Wasserbad
für weitere
vier Minuten inkubiert. Die Filter wurden dann für vier Minuten auf mit Neutralisierungslösung vorbefeuchtetes
Filterpapier gelegt. Die überschüssige Lösung wurde
entfernt, indem die Filter für
eine Minute auf trockene Filterpapiere gelegt wurden, die Kolonienseite
der Filter wurde in Proteinase K-Lösung gelegt, wurde für 40–50 Minuten
bei 37°C
inkubiert. Die Filter wurden auf trockene Filterpapiere gelegt,
um über Nacht
zu trocknen. DNA wurde durch UV-Licht-Behandlung an eine Nylonmembran
vernetzt.
-
Eine
Koloniehybridisierung wurde durchgeführt, wie sie von Sambrook,
J., Fritsch, E. F. und Maniatis, T., (in Molecular Cloning: A laboratory
Manual, 2. Auflage), beschrieben ist. Die folgenden Sonden wurden
bei der Kolonienhybridisierung verwendet:
- 1.
cDNA des ersten Strangs aus demselben Gewebe wie die Bibliothek
wurde hergestellt, um die meisten redundanten Klone zu entfernen.
- 2. 48–192
am stärksten
redundante cDNA-Klone aus derselben Bibliothek, basierend auf früheren Sequenzierungsdaten.
- 3. 192 stark redundante cDNA-Klone in der gesamten Mais-Partialsequenz-Datenbank.
- 4. Ein Sal-A20-Oligonucleotid: TCG ACC CAC GCG TCC GAA AAA AAA
AAA AAA AAA AAA entfernt Klone, die einen Poly A-Tail, aber keine
cDNA enthalten.
- 5. cDNA-Klone, die von rRNA abgeleitet sind.
-
Das
Bild der Autoradiographie wurde in einen Computer gescannt und die
Signalintensität
und die Bezeichnungen der kalten Kolonie wurden für jede Kolonie
analysiert. Ein Rearraying von kalten Kolonien aus 384 Well-Platten
zu 96 Well-Platten wurde unter Verwendung des Q-bot durchgeführt.
-
Beispiel 3
-
Dieses
Beispiel beschreibt die Identifizierung des Gens aus einer Computer-Homologiesuche.
Genidentitäten
wurden durch Durchführung
von BLAST (Basic Local Alignment Search Tool; Altschul, S. F., et
al., (1993) J. Mol. Biol. 215: 403–410; Siehe auch www.ncbi.nlm.nih.gov/BLAST-Suchen
unter Default-Parametern für
Similarität
für Sequenzen,
die in der BLAST-„nr"-Datenbank (umfasst
alle nicht-redundanten GenBank-CDS-Translationen, Sequenzen, die
aus der dreidimensionalen Struktur-Brookhaven Protein-Datenbank,
der letzten Hauptausgabe der SWISS-PROT-Proteinsequenz-Datenbank,
EMBL und DDBJ-Datenbanken stammen) enthalten sind, durchgeführt. Die
cDNA-Sequenzen wurden auf Ähnlichkeit
mit allen öffentlich zugänglichen
DNA-Sequenzen, die in der „nr"-Datenbank enthalten
sind, analysiert, wobei der BLASTN-Algorithmus verwendet wurde.
Die DNA-Sequenzen wurden in alle Leseraster translatiert und auf
Similarität
mit allen öffentlich
zugänglichen
Proteinsequenzen, die in der „nr"-Datenbank enthalten
sind, unter Verwendung des BLASTX-Algorithmus (Gish, W. und States,
D. J. (1993) Nature Genetics 3: 266–272), bereitgestellt von NCBI, verglichen.
In einigen Fällen
wurden die Sequenzierungsdaten aus zwei oder mehr Klonen, die überlappende DNA-Segmente enthielten,
verwendet, um fortlaufende DNA-Sequenzen zu konstruieren.
-
Die
obigen Beispiele werden bereitgestellt, um die Erfindung zu erläutern, aber
nicht um ihren Umfang zu beschränken.
Einem Fachmann auf dem Fachgebiet werden andere Varianten der Erfindung
leicht klar werden und werden durch die beigefügten Ansprüche umfasst.
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-
-