EP1805181B1 - Purinderivate zur verwendung als agonisten des adenosin-a-2a-rezeptors - Google Patents
Purinderivate zur verwendung als agonisten des adenosin-a-2a-rezeptors Download PDFInfo
- Publication number
- EP1805181B1 EP1805181B1 EP05797944A EP05797944A EP1805181B1 EP 1805181 B1 EP1805181 B1 EP 1805181B1 EP 05797944 A EP05797944 A EP 05797944A EP 05797944 A EP05797944 A EP 05797944A EP 1805181 B1 EP1805181 B1 EP 1805181B1
- Authority
- EP
- European Patent Office
- Prior art keywords
- formula
- compound
- optionally substituted
- amino
- purin
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Lifetime
Links
- 0 *c1nc(*)nc2c1nc[n]2[C@](C1)C=C[C@@]1N(*)C(OI)=O Chemical compound *c1nc(*)nc2c1nc[n]2[C@](C1)C=C[C@@]1N(*)C(OI)=O 0.000 description 29
- FSAWPQLVKUPBDB-HWKANZROSA-N C/C=N/C[NH+](C)[O-] Chemical compound C/C=N/C[NH+](C)[O-] FSAWPQLVKUPBDB-HWKANZROSA-N 0.000 description 1
- FSHYXLQHVHTYAY-HJWRWDBZSA-N CCC/C(/N)=C/N(CC)C=C Chemical compound CCC/C(/N)=C/N(CC)C=C FSHYXLQHVHTYAY-HJWRWDBZSA-N 0.000 description 1
- MXJYVLYENVWKQX-UHFFFAOYSA-N CCNCCNC Chemical compound CCNCCNC MXJYVLYENVWKQX-UHFFFAOYSA-N 0.000 description 1
- GDGPEKPCPUJDFU-YHMJZVADSA-N CNC(C[C@@H]1C=CC=CC1)C=[NH+][O-] Chemical compound CNC(C[C@@H]1C=CC=CC1)C=[NH+][O-] GDGPEKPCPUJDFU-YHMJZVADSA-N 0.000 description 1
- YAGGKOCHRIZNOA-UHFFFAOYSA-N CNCC1N(CC=C)CCC1 Chemical compound CNCC1N(CC=C)CCC1 YAGGKOCHRIZNOA-UHFFFAOYSA-N 0.000 description 1
- KCAUHAHOMIRXAN-UHFFFAOYSA-N CNCCN1CCCCC1 Chemical compound CNCCN1CCCCC1 KCAUHAHOMIRXAN-UHFFFAOYSA-N 0.000 description 1
- PHSPJQZRQAJPPF-UHFFFAOYSA-N CNCCc1c[nH]cn1 Chemical compound CNCCc1c[nH]cn1 PHSPJQZRQAJPPF-UHFFFAOYSA-N 0.000 description 1
- SGFJPSYHRUIWML-UHFFFAOYSA-N C[NH+](Cc1ccccc1)[O-] Chemical compound C[NH+](Cc1ccccc1)[O-] SGFJPSYHRUIWML-UHFFFAOYSA-N 0.000 description 1
- WGDHXHKTNGOFPW-QMMMGPOBSA-N N[C@@H](CC1)CN1c1ccccn1 Chemical compound N[C@@H](CC1)CN1c1ccccn1 WGDHXHKTNGOFPW-QMMMGPOBSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D473/00—Heterocyclic compounds containing purine ring systems
- C07D473/26—Heterocyclic compounds containing purine ring systems with an oxygen, sulphur, or nitrogen atom directly attached in position 2 or 6, but not in both
- C07D473/32—Nitrogen atom
- C07D473/34—Nitrogen atom attached in position 6, e.g. adenine
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D473/00—Heterocyclic compounds containing purine ring systems
- C07D473/26—Heterocyclic compounds containing purine ring systems with an oxygen, sulphur, or nitrogen atom directly attached in position 2 or 6, but not in both
- C07D473/32—Nitrogen atom
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/519—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim ortho- or peri-condensed with heterocyclic rings
- A61K31/52—Purines, e.g. adenine
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/06—Antiasthmatics
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P11/00—Drugs for disorders of the respiratory system
- A61P11/08—Bronchodilators
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P29/00—Non-central analgesic, antipyretic or antiinflammatory agents, e.g. antirheumatic agents; Non-steroidal antiinflammatory drugs [NSAID]
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P43/00—Drugs for specific purposes, not provided for in groups A61P1/00-A61P41/00
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D473/00—Heterocyclic compounds containing purine ring systems
- C07D473/02—Heterocyclic compounds containing purine ring systems with oxygen, sulphur, or nitrogen atoms directly attached in positions 2 and 6
- C07D473/16—Heterocyclic compounds containing purine ring systems with oxygen, sulphur, or nitrogen atoms directly attached in positions 2 and 6 two nitrogen atoms
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D473/00—Heterocyclic compounds containing purine ring systems
- C07D473/40—Heterocyclic compounds containing purine ring systems with halogen atoms or perhalogeno-alkyl radicals directly attached in position 2 or 6
Definitions
- This invention relates to organic compounds, their preparation and use as pharmaceuticals.
- the present invention provides compounds of formula I in free or salt form, wherein
- 5- or 6-membered heterocyclic ring containing at least one ring heteroatom selected from the group consisting of nitrogen, oxygen and sulphur may be, for example, furan, pyrrole, pyrrolidine, pyrazole, imidazole, triazole, isotriazole, tetrazole, thiadiazole, isothiazole, oxadiazole, pyridine, piperidine, pyrazine, oxazole, isoxazole, pyrazine, pyridazine, pyrimidine, piperazine, pyrrolidine, morpholino, triazine, oxazine or thiazole.
- Preferred heterocyclic rings include piperazine, pyrrolidine, morpholino, imidazole, isotriazole, pyrazole, tetrazole, thiazole, thiadiazole, pyridine, piperidine, pyrazine, furan, oxazole, isoxazole, oxadiazole and azetidine.
- the 5- or 6-membered heterocyclic ring can be unsubstituted or it can be substituted at one or more positions, preferably one or two positions, by halo, cyano, oxo, hydroxy, carboxy, amino, nitro, C 1 -C 8 -alkyl, C 1 -C 8 -alkylsulfonyl, aminocarbonyl, C 1 -C 8 -alkylcarbonyl or C 1 -C 8 -alkoxy optionally substituted at one or more positions, preferably one or two positions, by aminocarbonyl.
- Especially preferred substituents include methyl, ethyl, d propyl) and amino.
- R 5 is preferably unsubstituted imidazolyl, unsubstituted piperidinyl, or imidazolyl substituted at one position by C 1 -C 3 -alkyl.
- R 6 is preferably pyrrolidinyl, piperidinyl or piperazinyl and, where relevant, R 7 is preferably unsubstituted thiophenyl, unsubstituted pyridinyl, unsubstituted pyrrolidinyl, pyridinyl disubstituted by chloro, piperazinyl substituted Throughout this specification and in the claims that follow, unless the context requires otherwise, the word "comprise”, or variations such as “comprises” or “comprising”, will be understood to imply the inclusion of a stated integer or step or group of integers or steps but not the exclusion of any other integer or step or group of integers or steps.
- R 2 is hydrogen, unsubstituted C 1 -C 6 -alkyl or C 1 -C 5 -alkyl substituted at one position by C 6 -C 10 -aryl;
- R 3 is halo or C 2 -C 6 -alkynyl, or R 3 is amino optionally substituted at one position by C 3 -C 6 -cycloalkyl optionally substituted at one position by amino, or R 3 is C 1 -C 4 -alkylamino substituted at one or two positions by
- R 2 is hydrogen or C 1 -C 6 -alkyl optionally substituted by C 6 -C 10 -aryl;
- R 3 is halo or C 2 -C 5 -alkynyl, or R 3 is amino optionally substituted by C 3 -C 8 -cycloalkyl optionally substituted by amino, or R 3 is C 1 -C 4 -alkylamino optionally substituted by hydroxy, C 6 -C 8 -aryl or by R 5 , or R 3 is R 6 optionally
- the compounds represented by formula I are capable of forming acid addition salts, particularly pharmaceutically acceptable acid addition salts.
- R 2 is hydrogen or C 1 -C 8 -alkyl optionally substituted by C 6 -C 10 -aryl;
- R 3 is halo or C 2 -C 8 -alkynyl, or R 3 is amino optionally substituted by C 3 -C 8 -cycloalkyl optionally substituted by amino, or R 3 is C 1 -C 8
- R 2 is hydrogen or C 1 -C 6 -alkyl optionally substituted by C 6 -C 10 -aryl;
- R 3 is halo or C 2 -C 5 -alkynyl, or R 3 is amino optionally substituted by C 3 -C 8 -cycloalkyl optionally substituted by amino, or R 3 is C 1 -C 4 -alkylamino optionally substituted by hydroxy, C 6 -C 8 -aryl or by R 5 , or R 3 is R 6 optionally
- R 2 and R 3 are as hereinbefore defined, with a compound of formula III R 1 -X a III or a formula IIIa wherein R 1 is C 1 -C 8 -alkylcarbonyl, C 3 -C 8 -cycloalkylcarbonyl or C 7 -C 14 -aralkylcarbonyl, X a is a leaving group and K is hydrogen, C 1 -C 8 -alkyl or C 1 -C 8 -alkoxy, in the presence of a base;
- Process variant (A) may be carried out using known procedures for reacting amines with acid halides, acid anhydrides or mixed anhydrides e.g. carboxylic and carbonic anhydrides (or amide-forming derivatives thereof such as carboxylic acids) or sulfonyl halides e.g. mesyl halides, or analogously as hereinafter described in the Examples.
- the leaving group may be any suitable leaving group, for example halo, -SO 2 -C 1 -C 8 -alkyl or -SO 2 -C 6 -C 10 -aryl.
- reaction is conveniently carried out using an organic solvent, for example tetrahydrofuran (THF), in the presence of a base, for example diisopropylethylamine (DIPEA).
- a base for example diisopropylethylamine (DIPEA).
- Suitable reaction temperatures are from 10° C to 40° C, preferably room temperature.
- Process variant (B) may be carried out using known procedures for reacting halides, especially aromatic halides, with amines, or analogously as hereinafter described in the Examples.
- the reaction is conveniently carried out using an organic solvent, for example dichlorobenzene, dimethylsulfoxide, acetonitrile or N-methyl-pyrrolidone (NMP) or mixtures thereof op tionally in the presence of a catalyst, such as sodium iodide, and a base, such as triethylamine.
- Suitable reaction temperatures are from 100° C to 250° C, preferably between 120° C to 220° C, especially about 170° C, for example by heating with microwave radiation.
- Process variant (C) may be carried out using known procedures for reacting halides with amines, or analogously as hereinafter described in the Examples.
- the reaction is conveniently carried out using an organic solvent, for example tetrahydrofuran, preferably in an inert atmosphere, for example argon, optionally in the presence of a base, for example diisopropylethylamine.
- Suitable reaction temperatures from 0° C to 70° C, preferably between 40° C to 60° C, especially about 50° C.
- Process variant (D) may be carried out using known procedures for cleaving ester bonds, for example using a strong organic acid, such as trifluoroacetic acid.
- the reaction is conveniently carried out using an organic solvent, for example dichloromethane (DCM).
- Suitable reaction temperatures are from 0° C to 40° C, preferably room temperature.
- Process variant (E) may be carried out using known procedures for reacting amines with acyl-imidazoles or isocyanates, or analogously as hereinafter described in the Examples.
- T in formula X is preferably imidazolyl.
- the reaction is conveniently carried out using an organic solvent, for example toluene and/or isopropyl alcohol. Suitable reaction temperatures are from 0° C to 40° C, preferably room temperature.
- Process variant (F) may be carried out using known procedures for reacting halides with alkynes, or analogously as hereinafter described in the Examples.
- the catalyst is preferably a palladium catalyst (together with a CuI salt) and the base is preferably butylamine.
- the reaction is conveniently carried out using an organic solvent, such as dimethylformamide (DMF). Suitable reaction temperatures are from 40° C to 200° C, preferably 80° C to 160° C, especially about 120° C.
- Process variant (G) may be carried out using known procedures for reacting carboxylic acid alkyl esters with amines, or analogously as hereinafter described in the Examples.
- the base is preferably is preferably imidazole.
- the reaction is conveniently carried out using an organic solvent, such 1,2-dichloroethane, iso-propanol or a mixture thereof. Suitable reaction temperatures are from room temperature to 250° C, preferably 50° C to 100° C.
- Process variant (H) may be carried out using known procedures for sulfonylating heterocycles, or analogously as hereinafter described in the Examples.
- the sulphonylating agent is preferably an alkylsulfonylhalide, for example mesylchloride.
- the base is preferably triethylamine.
- the reaction is conveniently carried out using an organic solvent, such as dimethylformamide (DMF), preferably in an inert atmosphere. Suitable reaction temperatures are from 0° C to 40° C, preferably room temperature.
- DMF dimethylformamide
- Process variant (I) may be carried out using known procedures for reacting amines with acyl-imidazoles, isocyanates or arylcarbamates, or analogously as hereinafter described in the Examples.
- T in formula X is preferably imidazolyl.
- the reaction is conveniently carried out using an organic solvent, for example tetrahydrofuran or N-methyl-pyrrolidone (NMP), preferably in the presence of a base, for example triethylamine.
- NMP N-methyl-pyrrolidone
- suitable reaction temperatures are from 0° C to 40° C, preferably room temperature.
- suitable reaction temperatures are from room temperature to 120° C, preferably 80° C to 110° C, especially about 110° C.
- Process variant (J) may be carried out using known procedures for reacting N-heterocycles with acyl-imidazoles, isocyanates or arylcarbamates, or analogously as hereinafter described in the Examples.
- T in formula XIIe is preferably imidazolyl.
- the reaction is conveniently carried out using an organic solvent, for example tetrahydrofuran or N-methyl-pyrrolidone (NMP).
- NMP N-methyl-pyrrolidone
- suitable reaction temperatures are from 0° C to 40° C, preferably room temperature.
- suitable reaction temperatures are from room temperature to 120° C, preferably 80° C to 110° C, especially about 110° C.
- Process variant (K) may be carried out using known procedures for reacting amines with acyl-imidazoles, isocyanates or arylcarbamates, or analogously as hereinafter described in the Examples.
- the reaction is conveniently carried out using an organic solvent, for example tetrahydrofuran.
- suitable reaction temperatures are from 0° C to 40° C, preferably room temperature.
- suitable reaction temperatures are from room temperature to 120° C, preferably 80° C to 110° C, especially about 110° C.
- the protecting groups may be chosen in accordance with the nature of the functional group, for example as described in Protective Groups in Organic Synthesis, T.W. Greene and P.G.M. Wuts, John Wiley & Sons Inc, Third Edition, 1999 , which reference also describes procedures suitable for replacement of the protecting groups by hydrogen.
- Compounds of formula II may be prepared by deprotecting a compound of formula XIII where R 2 and R 3 are as hereinbefore defined, and each L is C 1 -C 8 -alkyl, using known procedures for cleaving ester bonds, or analogously as herein described in the Examples.
- the reaction is carried out using a strong organic acid, such as trifluoroacetic acid.
- Each L is preferably t-butyl.
- the reaction is conveniently carried out using an organic solvent, for example dichloromethane. Suitable reaction temperatures from 0° C to 40° C, preferably room temperature.
- Compounds of formula IV may be prepared by reacting a compound of formula II where R 3 is halo, with a compound of formula III or IIIa wherein R 1 is as hereinbefore defined, X is a leaving group, preferably halo, and K is hydrogen or C 1 -C 8 -alkyl, in the presence of a base, or analogously as herein described in the Examples.
- the reaction is conveniently carried out using an organic solvent, for example tetrahydrofuran.
- the base is preferably diisopropylethylamine. Suitable reaction temperatures from 0° C to 40° C, preferably room temperature.
- Compounds of formula VI may be prepared by reacting a compound of formula KIV where R 3 is as hereinbefore defined and X is halo, with a compound of formula III or IIIa, wherein R 1 is as hereinbefore defined, X is a leaving group, preferably halo, and K is hydrogen or C 1 -C 8 -alkyl, in the presence of a base, wherein R 1 is as hereinbefore defined and X is halo, or analogously as herein described in the Examples.
- the reaction is conveniently carried out using an organic solvent, for example tetrahydrofuran, preferably in the presence of a base, for example diisopropylethylamine. Suitable reaction temperatures from 0° C to 40° C, preferably room temperature.
- Compounds of formula VIII may be prepared by reacting a compound of formula XV where R 1 , R 2 and R 3 are as hereinbefore defined and L is C 1 -C 8 -alkyl, with a dihydroxylating agent, such as osmium tetroxide (OsO 4 ), either in a stoichiometrical amount or a catalytic amount, preferably together with a re-oxidant, such as N-methylmorpholine N-oxide (NMO), or alternatively using AD-mix- ⁇ or AD-mix- ⁇ , or analogously as herein described in the Examples.
- L is preferably t-butyl.
- the reaction is conveniently carried out using an organic solvent, for example THF. Suitable reaction temperatures from 0° C to 40° C, preferably room temperature.
- Compounds of formula IX may be prepared by reacting a compound of formula XVI where R 1 and R 2 are as hereinbefore defined and L is C 1 -C 8 -alkyl, is reacted with a compound of formula XVII wherein Y is C 1 -C 8 -alkyl or C 3 -C 8 -cycloalkyl, or analogously as herein described in the Examples.
- Suitable reaction temperatures from 80° C to 130° C, preferably 90° C to 120° C room temperature, especially about 105° C.
- Compounds of formula XIII may be prepared by reacting a compound of formula XVIII where R 2 and R 3 are as hereinbefore defined, and each L is C 1 -C 8 -alkyl or benzyl, with a hydroxylating agent, such as osmium tetroxide (OsO 4 ), either in a stoichiometrical amount or a catalytic amount, preferably together with a re-oxidant, such as N-methylmorpholine N-oxide (NMO), or alternatively using AD-mix- ⁇ or AD-mix- ⁇ , or analogously as herein described in the Examples.
- L 1 and L 2 are preferably t-butyl.
- the reaction is conveniently carried out using an organic solvent, for example tetrahydrofuran. Suitable reaction temperatures from 0° C to 40° C, preferably room temperature.
- Compounds of formula XIV may be prepared by reacting a compound of formula XIX where R 3 and X are as hereinbefore defined, and each L is C 1 -C 8 -alkyl or benzyl, with a strong organic acid, such as trifluoroacetic acid, or analogously as herein described in the Examples. Each L is preferably t-butyl.
- the reaction is conveniently carried out using an organic solvent, for example dichloromethane. Suitable reaction temperatures from 0° C to 40° C, preferably room temperature.
- Compounds of formula XV may be prepared by reacting a compound of formula XX where R 3 is as hereinbefore defined, X is halo and L is C 1 -C 8 -alkyl or benzyl, with a compound of formula VII, wherein R 2 is as hereinbefore defined, or analogously as herein described in the Examples.
- the reaction is conveniently carried out using an organic solvent, for example tetrahydrofuran, preferably in an inert atmosphere, for example in argon.
- Suitable reaction temperatures from 30° C to 70° C, preferably from 40° C to 60° C, especially about 50° C.
- Compounds of XVI may be prepared by reacting a compound of formula XXI where R 2 is as hereinbefore defined and L' is C 1 -C 8 -alkyl or benzyl but preferably methyl, with a compound of formula III or IIIa, wherein R 1 is as hereinbefore defined, X is a leaving group, preferably halo, and K is hydrogen or C 1 -C 8 -alkyl, or analogously as herein described in the Examples.
- the reaction is conveniently carried out using an organic solvent, for example tetrahydrofuran, preferably in the presence of a base, for example diisopropylethylamine. Suitable reaction temperatures from 0° C to 40° C, preferably room temperature.
- Compounds of formula XVIII may be prepared by reacting a compound of formula XXII where R 2 and R 3 are as hereinbefore defined, and L" is C 1 -C 8 -alkyl preferably methyl or ethyl, with a compound of formula XXIII where each L is C 1 -C 8 -alkyl or benzyl, preferably benzyl, and preferably in the presence of a catalyst, such as that generated from tetrakis(triphenylphosphine)palladium and triphenylphosphine, or analogously as herein described in the Examples.
- a catalyst such as that generated from tetrakis(triphenylphosphine)palladium and triphenylphosphine, or analogously as herein described in the Examples.
- each L is t-butyl or benzyl.
- the reaction is conveniently carried out in an inert environment, for example in argon, using an organic solvent, for example
- Compounds of formula XIX may be prepared by reacting a compound of formula XXIV where R 3 and X are as hereinbefore defined, and each L is C 1 -C 8 -alkyl or benzyl, with a hydroxylating agent, such as osmium tetroxide (OsO 4 ), either in a stoichiometrical amount or a catalytic amount, preferably together with a re-oxidant, such as N-methylmorpholine N-oxide (NMO), or alternatively using AD-mix- ⁇ or AD-mix- ⁇ , or analogously as herein described in the Examples.
- Each L is preferably t-butyl.
- the reaction is conveniently carried out using an organic solvent, for example tetrahydrofuran. Suitable reaction temperatures from 0° C to 40° C, preferably room temperature.
- Compounds of formula XX may be prepared by reacting a compound of formula XXV where R 3 is as hereinbefore defined, and L" is C 1 -C 8 -alkyl, with a compound of formula XXVa where R 1 is as hereinbefore defined, and L is C 1 -C 8 -alkyl or benzyl, preferably in the presence of a catalyst, such as that generated from tetrakis(triphenyl-phosphine)palladium and triphenylphosphine, or analogously as herein described in the Examples.
- a catalyst such as that generated from tetrakis(triphenyl-phosphine)palladium and triphenylphosphine, or analogously as herein described in the Examples.
- L is t-butyl or benzyl.
- the reaction is conveniently carried out in an inert environment, for example in argon, using an organic solvent, for example deoxygenated tetrahydrofuran
- each L is C 1 -C 8 -alkyl or benzyl and L' is C 1 -C 4 -alkyl, is reacted with a strong acid, for example hydrochloric acid using known procedures for cleaving esters bonds, or analogously as herein described in the Examples.
- a strong acid for example hydrochloric acid
- each L is t-butyl or benzyl and L a is methyl or ethyl.
- the reaction is conveniently carried out in an inert environment, for example in argon, using an organic solvent, for example dioxane. Suitable reaction temperatures from 0° C to 40° C, preferably room temperature.
- Compounds of formula XXII may be prepared by reacting a compound of formula XXVII where R 2 and R 3 are as hereinbefore defined, with an acylating agent such as a carboxylic acid C 1 -C 8 -alkyl ester, for example 3-oxy-benzotriazole-1-carboxlic acid ethyl ester, in the presence of a base, such as diisoproplylamine, and a catalyst, such as 4-dimethylaminopyridine (DMAP), or analogously as herein described in the Examples.
- the reaction is conveniently carried out in an inert environment, for example in argon, using an organic solvent, for example deoxygenated THF. Suitable reaction temperatures from 0° C to 40° C, preferably room temperature.
- Compounds of formula XXIV may be prepared by reacting a compound of formula XXVIII where R 3 and X are as hereinbefore defined, and L" is C 1 -C 8 -alkyl, with a compound of formula XXIII where each L is C 1 -C 8 -alkyl or benzyl, preferably in the presence of a catalyst, such as that generated from tetrakis(triphenylphosphine)palladium and triphenylphosphine, or analogously as herein described in the Examples.
- a catalyst such as that generated from tetrakis(triphenylphosphine)palladium and triphenylphosphine, or analogously as herein described in the Examples.
- each L is t-butyl or benzyl.
- the reaction is conveniently carried out in an inert environment, for example in argon, using an organic solvent, for example deoxygenated tetrahydrofuran. Suitable reaction temperatures from
- Compounds of formula XXV may be prepared by reacting a compound of formula XXIX where R 3 and X are as hereinbefore defined, with an acylating agent such as a carboxylic acid C 1 -C 8 -alkyl ester, for example 3-oxy-benzotriazole-1-carboxlic acid ethyl ester, in the presence of a base, such as diisoproplylamine, and a catalyst, such as 4-dimethylaminopyridine (DMAP), or analogously as herein described in the Examples.
- the reaction is conveniently carried out in an inert environment, for example in argon, using an organic solvent, for example deoxygenated tetrahydrofuran.
- Suitable reaction temperatures from 0° C to 40° C, preferably room temperature.
- Compounds of formula XXVa are commercially available or may be obtained by known procedures for preparing such compounds, for example as described by Ken-ichi Takana et al in Chem. Pharm. Bull. 1988, 36, 3125 , or analogously as herein described in the Examples.
- Compounds of XXVI may be prepared by reacting a compound of formula XXX where R 2 is as hereinbefore defined, each L is C 1 -C 8 -alkyl and L' is C 1 -C 4 -alkyl or benzyl, preferably benzyl, is reacted with a hydroxylating agent, such as osmium tetroxide (OsO 4 ), either in a stoichiometrical amount or a catalytic amount, preferably together with a re-oxidant, such as N-methylmorpholine N-oxide (NMO), or alternatively using AD-mix- ⁇ or AD-mix- ⁇ , or analogously as herein described in the Examples.
- a hydroxylating agent such as osmium tetroxide (OsO 4 )
- OsO 4 osmium tetroxide
- NMO N-methylmorpholine N-oxide
- each L is t-butyl and L a is methyl or ethyl.
- the reaction is conveniently carried out using an organic solvent, for example tetrahydrofuran. Suitable reaction temperatures from 0° C to 40° C, preferably room temperature.
- Compounds of formula XXVII may be prepared by reacting a compound of formula XXXI where R 2 and R 3 are as hereinbefore defined, with (1S,4R)-cis 4-acetoxy-2-cyclopenten-1-ol in the presence of a base, such as sodium hydride, and a catalyst, such as that generated from tetrakis(triphenylphosphine)palladium and triphenylphosphine, or analogously as herein described in the Examples.
- the reaction is conveniently carried out in an inert environment, for example in argon, using an organic solvent, for example deoxygenated tetrahydrofuran or dimethylsulfoxide (DMSO). Suitable reaction temperatures from 40° C to 60° C, preferably about 50° C.
- Compounds of formula XXVIII may be prepared by reacting a compound of formula XXIX where R 3 and X are as hereinbefore defined, with an acylating agent such as a carboxylic acid C 1 -C 8 -alkyl ester, for example 3-oxy-benzotriazole-1-carboxlic acid ethyl ester, in the presence of a base, such as diisoproplylamine, and a catalyst, such as 4-dimethylaminopyridine (DMAP), or analogously as herein described in the Examples.
- the reaction is conveniently carried out in an inert environment, for example in argon, using an organic solvent, for example THF. Suitable reaction temperatures from 0° C to 40° C, preferably room temperature.
- Compounds of formula XXIX may be prepared by reacting a compound of formula XXXII where R 3 and X are as hereinbefore defined, with (1S,4R)-cis 4-Acetoxy-2-cyclopenten-1-ol in the presence of a base, such sodium hydride, and a catalyst, such as that generated from tetrakis(triphenylphosphine)palladium and triphenylphosphine, or analogously as herein described in the Examples.
- the reaction is conveniently carried out in an inert environment, for example in argon, using an organic solvent, for example deoxygenated tetrahydrofuran or dimethylsulfoxide (DMSO). Suitable reaction temperatures from 40° C to 60° C, preferably about 50° C.
- Compounds of formula XXX may be prepared by reacting a compound of formula XXXIII where R 2 is as hereinbefore defined, L" is C 1 -C 8 -alkyl or benzyl, and L' is C 1 -C 4 -alkyl, with a compound of formula XXIII where each L is C 1 -C 8 -alkyl, preferably in the presence of a catalyst, such as that generated from tetrakis(triphenylphosphine)palladium and triphenylphosphine, or analogously as herein described in the Examples.
- a catalyst such as that generated from tetrakis(triphenylphosphine)palladium and triphenylphosphine, or analogously as herein described in the Examples.
- each L" is t-butyl or benzyl and L' is methyl or ethyl.
- the reaction is conveniently carried out in an inert environment, for example in argon,
- Compounds of formula XXXI may be prepared by reacting a compound of formula XXXII where R 3 is as hereinbefore defined and X is halo, with a compound of formula VII where R 2 is as hereinbefore defined, or analogously as herein described in the Examples.
- the reaction is conveniently carried out in an inert environment, for example in argon, using an organic solvent, for example tetrahydrofuran. Suitable reaction temperatures from 40° C to 60° C, preferably about 50° C.
- Compounds of formula XXXIII may be prepared by reacting a compound of formula XXXIV where R 2 and L' are as hereinbefore defined, with a compound of formula XXXV where L" is C 1 -C 8 -alkyl, preferably methyl or ethyl, and X is halo, preferably chloro, or analogously as herein described in the Examples.
- the reaction is conveniently carried out in an inert environment, for example in argon, using an organic solvent, for example deoxygenated tetrahydrofuran, preferably in the presence of a base, for example pyridine. Suitable reaction temperatures from 0° C to 40° C, preferably room temperature.
- Compounds of formula XXXIV may be prepared by reacting a compound of formula XXXVI where R 2 is as hereinbefore defined and L' is C 1 -C 4 -alkyl, preferably methyl or ethyl, with (1S,4R)-cis 4-acetoxy-2-cyclopenten-1-ol in the presence of a base, such sodium hydride, and a catalyst, such as that generated from tetrakis(triphenylphosphine)palladium and triphenylphosphine, or analogously as herein described in the Examples.
- a base such sodium hydride
- a catalyst such as that generated from tetrakis(triphenylphosphine)palladium and triphenylphosphine, or analogously as herein described in the Examples.
- reaction is conveniently carried out in an inert environment, for example in argon, using an organic solvent, for example deoxygenated tetrahydrofuran or dimethyl sulfoxide.
- organic solvent for example deoxygenated tetrahydrofuran or dimethyl sulfoxide.
- Compounds of formula XXXVI may be prepared by reacting a salt compound of formula XXXVI where R 3 is as hereinbefore defined and L is C 1 -C 8 -alkyl, with a silating agent, for example (N,O-bis(trimethylsilyl)acetamide), or analogously as herein described in the Examples.
- a silating agent for example (N,O-bis(trimethylsilyl)acetamide), or analogously as herein described in the Examples.
- the reaction is conveniently carried out in an inert environment, for example in argon, using an organic solvent, for example dry chloroform. Suitable reaction temperatures from 60° C to 100° C, preferably about 80° C.
- the silylated intermediate thus formed is treated with methanol to give the free base.
- Compounds of formula I in free form may be converted into salt form, and vice versa, in a conventional manner.
- the compounds in free or salt form can be obtained in the form of hydrates or solvates containing a solvent used for crystallisation.
- Compounds of formula I can be recovered from reaction mixtures and purified in a conventional manner.
- Isomers, such as stereoisomers may be obtained in a conventional manner, e.g. by fractional crystallisation or asymmetric synthesis from correspondingly asymmetrically substituted, e.g. optically active, starting materials.
- Compounds of formula I and their pharmaceutically acceptable salts are useful as pharmaceuticals.
- they activate the adenosine A 2A receptor, i.e. they act as A 2A receptor agonists.
- Their properties as A 2A agonists may be demonstrated using the method described by L. J. Murphree et al in Molecular Pharmacology 61, 455-462 (2002 ).
- K i values below 1.0 ⁇ M in the above assay For example, the compounds of Examples 1, 2, 4, 6, 12, 14, 20, 33, 38, 39, 42, 47, 55 and 61 have K i values of 0.582, 0.018, 0.057, 0.008, 0.003, 0.690, 0.008, 0.052, 0.002, 0.003, 0.002, 0.002, 0.004 and 0.009 ⁇ M respectively.
- agents of the invention are useful in the treatment of conditions which respond to the activation of the adenosine A 2A receptor, particularly inflammatory or allergic conditions. Treatment in accordance with the invention may be symptomatic or prophylactic.
- agents of the invention are useful in the treatment of inflammatory or obstructive airways diseases, resulting, for example, in reduction of tissue damage, airways inflammation, bronchial hyperreactivity, remodelling or disease progression.
- Inflammatory or obstructive airways diseases and conditions to which the present invention is applicable include acute lung injury (ALI), adult/acute respiratory distress syndrome (ARDS), chronic obstructive pulmonary, airways or lung disease (COPD, COAD or COLD), including chronic bronchitis or dyspnea associated therewith, emphysema, as well as exacerbation of airways hyperreactivity consequent to other drug therapy, in particular other inhaled drug therapy.
- the invention is also applicable to the treatment of bronchitis of whatever type or genesis including, e.g., acute, arachidic, catarrhal, croupus, chronic or phthinoid bronchitis.
- inflammatory or obstructive airways diseases to which the present invention is applicable include pneumoconiosis (an inflammatory, commonly occupational, disease of the lungs, frequently accompanied by airways obstruction, whether chronic or acute, and occasioned by repeated inhalation of dusts) of whatever type or genesis, including, for example, aluminosis, anthracosis, asbestosis, chalicosis, ptilosis, siderosis, silicosis, tabacosis and byssinosis.
- asthma inflammatory or obstructive airways diseases to which the present inventi on is applicable
- asthma of whatever type or genesis including both intrinsic (non-allergic) asthma and extrinsic (allergic) asthma, mild asthma, moderate asthma, severe asthma, bronchitic asthma, exercise-induced asthma, occupational asthma and asthma induced following bacterial infection.
- Treatment of asthma is also to be understood as embracing treatment of subjects, e.g. of less than 4 or 5 years of age, exhibiting wheezing symptoms and diagnosed or diagnosable as "whezing symptoms and diagnosed or diagnosable as "whez infants", an established patient category of major medical concern and now often identified as incipient or early-phase asthmatics. (For convenience this particular asthmatic condition is referred to as "whez-infant syndrome”.)
- Prophylactic efficacy in the treatment of asthma will be evidenced by reduced frequency or severity of symptomatic attack, e.g. of acute asthmatic or bronchoconstrictor attack, improvement in lung function or improved airways hyperreactivity. It may further be evidenced by reduced requirement for other, symptomatic therapy, i.e. therapy for or intended to restrict or abort symptomatic attack when it occurs, for example anti-inflammatory (e.g. cortico-steroid) or bronchodilatory.
- Prophylactic benefit in asthma may in particular be apparent in subjects prone to "morning dipping". "Morning dipping" is a recognised asthmatic syndrome, common to a substantial percentage of asthmatics and cha-racterised by asthma attack, e.g. between the hours of about 4 to 6 am, i.e. at a time normally substantially distant from any previously administered symptomatic asthma therapy.
- agents of the invention are also useful in the treatment of eosinophil related disorders, e.g. eosinophilia, in particular eosinophil related disorders of the airways (e.g.
- eosinophilic infiltration of pulmonary tissues including hyper-eosinophilia as it effects the airways and/or lungs as well as, for example, eosinophil-related disorders of the airways consequential or concomitant to Löffler's syndrome, eosinophilic pneumonia, parasitic (in particular metazoan) infestation (including tropical eosinophilia), bronchopulmonary aspergillosis, polyarteritis nodosa (including Churg-Strauss syndrome), eosinophilic granuloma and eosinophil-related disorders affecting the airways occasioned by drug-reaction.
- Agents of the invention are also useful in the treatment of inflammatory or allergic conditions of the skin, for example psoriasis, contact dermatitis, atopic dermatitis, alopecia areata, erythema multiforma, dermatitis herpetiformis, scleroderma, vitili go, hypersensitivity angiitis, urticaria, bullous pemphigoid, lupus erythematosus, pemphisus, epidermolysis bullosa acquisita, and other inflammatory or allergic conditions of the skin.
- Agents of the invention may also be used for the treatment of other diseases or conditions, in particular diseases or conditions having an inflammatory component, for example, treatment of diseases and conditions of the eye such as conjunctivitis, keratoconjunctivitis sicca, and vernal conjunctivitis, diseases affecting the nose including allergic rhinitis, and inflammatory disease in which autoimmune reactions are implicated or having an autoimmune component or aetiology, including autoimmune haematological disorders (e.g.
- haemolytic anaemia haemolytic anaemia, aplastic anaemia, pure red cell anaemia and idiopathic thrombocytopenia
- systemic lupus erythematosus polychondritis, sclerodoma, Wegener granulamatosis, dermatomyositis, chronic active hepatitis, myasthenia gravis, Steven-Johnson syndrome, idiopathic sprue, autoimmune inflammatory bowel disease (e.g.
- ulcerative colitis and Crohn's disease endocrine opthalmopathy
- Grave's disease sarcoidosis, alveolitis, chronic hypersensitivity pneumonitis, multiple sclerosis, primary billiary cirrhosis, uveitis (anterior and posterior), keratoconjunct-ivitis sicca and vernal keratoconjunctivitis, interstitial lung fibrosis, psoriatic arthritis and glomerulonephritis (with and without nephrotic syndrome, e.g. including idiopathic nephrotic syndrome or minal change nephropathy).
- Other diseases or conditions which may be treated with agents of the invention include diabetes, e.g.
- diabetes mellitus type I (juvenile diabetes) and diabetes mellitus type II, diarrheal diseases, ischemia/reperfusion injuries, retinopathy, such as diabetic retinopathy or hyperbaric oxygen-induced retinopathy, conditions characterised by elevated intraocular pressure or secretion of ocular aqueous humor, such as glaucoma, ischemic tissue/organ damage from reperfusion and bedsores.
- retinopathy such as diabetic retinopathy or hyperbaric oxygen-induced retinopathy
- an agent of the invention in inhibiting inflammatory conditions, for example in inflammatory airways diseases, may be demonstrated in an animal model, e.g. a mouse or rat model, of airways inflammation or other inflammatory conditions, for example as described by Szarka et al, J. Immunol. Methods (1997) 202:49-57 ; Renzi et al, Am. Rev. Respir. Dis. (1993) 148:932-939 ; Tsuyuki et al., J. Clin. Invest. (1995) 96:2924-2931 ; Cernadas et al (1999) Am. J. Respir. Cell Mol. Biol. 20:1-8 ; and Fozard et al (2002) European Journal of Pharmacological 438, 183-188 .
- the agents of the invention are also useful as co-therapeutic agents for use in combination with other drug substances such as anti-inflammatory, bronchodilatory, antihistamine or antitussive drug substances, particularly in the treatment of obstructive or inflammatory airways diseases such as those mentioned hereinbefore, for example as potentiators of therapeutic activity of such drugs or as a means of reducing required dosaging or potential side effects of such drugs.
- An agent of the invention may be mixed with the other drug substance in a fixed pharmaceutical composition or it may be administered separately, before, simultaneously with or after the other drug substance.
- the invention includes a combination of an agent of the invention as hereinbefore described with an anti-inflammatory, bronchodilatory, antihistamine or anti-tussive drug substance, said agent of the invention and said drug substance being in the same or different pharmaceutical composition.
- Suitable anti-inflammatory drugs include steroids, in particular glucocorticosteroids such as budesonide, beclamethasone dipropionate, fluticasone propionate, ciclesonide or mometasone furoate, or steroids described in WO 02/88167 , WO 02/12266 , WO 02/100879 , WO 02/00679 (especially those of Examples 3, 11, 14, 17, 19, 26, 34, 37, 39, 51, 60, 67, 72, 73, 90, 99 and 101), WO 03/35668 , WO 03/48181 , WO 03/62259 , WO 03/64445 , WO 03/72592 , WO 04/39827 and WO 04/66920 ; non-steroidal glucocorticoid receptor agonists, such as those described in DE 10261874 , WO 00/00531 , WO 02/10143 , WO 03/82280 , WO 03/82787 , WO 03/862
- Suitable bronchodilatory drugs include anticholinergic or antimuscarinic agents, in particular ipratropium bromide, oxitropium bromide, tiotropium salts and CHF 4226 (Chiesi), and glycopyrrolate, but also those described in EP 424021 , US 3714357 , US 5171744 , US 2005/171147 , US 2005/182091 , WO 01/04118 , WO 02/00652 , WO 02/51841 , WO 02/53564 , WO 03/00840 , WO 03/33495 , WO 03/53966 , WO 03/87094 , WO 04/018422 , WO 04/05285 and WO 05/077361 .
- anticholinergic or antimuscarinic agents in particular ipratropium bromide, oxitropium bromide, tiotropium salts and CHF 4226 (Chiesi), and glycopyrrolate, but also those described
- Suitable dual anti-inflammatory and bronchodilatory drugs include dual beta-2 adrenoceptor agonist / muscarinic antagonists such as those disclosed in US 2004/0167167 , US 2004/0242622 , US 2005/182092 , WO 04/74246 and WO 04/74812 .
- Suitable antihistamine drug substances include cetirizine hydrochloride, acetaminophen, clemastine fumarate, promethazine, ioratidine, desloratidine, diphenhydramine and fexofenadine hydrochloride, activastine, astemizole, azelastine, ebastine, epinastine, mizolastine and tefenadine as well as those disclosed in JP 2004107299 , WO 03/099807 and WO 04/026841 .
- agents of the invention with anti-inflammatory drugs are those with antagonists of chemokine receptors, e.g. CCR-1, CCR-2, CCR-3, CCR-4, CCR-5, CCR-6, CCR-7, CCR-8, CCR-9 and CCR10, CXCR1, CXCR2, CXCR3, CXCR4, CXCR5, particularly CCR-5 antagonists such as Schering-Plough antagonists SC-351125, SCH-55700 and SCH-D, Takeda antagonists such as N-[[4-[[[[6,7-dihydro-2-(4-methylphenyl)-5H-benzocyclohepten-8-yl]carbonyl]amino]phenyl]-methyl]tetrahydro-N,N-dimethyl-2H-pyran-4-aminium chloride (TAK-770), and CCR-5 antagonists described in US 6166037 (particularly claims 18 and 19), WO 00/66558 (particularly claim 8), WO 00/665
- the invention also provides a method for the treatment of a condition responsive to activation of the adenosine A 2A receptor, for example an inflammatory or allergic condition, particularly an inflammatory or obstructive airways disease, which comprises administering to a subject, particularly a human subject, in need thereof a compound of formula I in free form or in the form of a pharmaceutically acceptable salt.
- a compound of formula I in free form or in the form of a pharmaceutically acceptable salt, for use in the manufacture of a medicament for the treatment of a condition responsive to activation of the adenosine A 2A receptor, particularly an inflammatory or obstructive airways disease.
- the agents of the invention may be administered by any appropriate route, e.g. orally, for example in the form of a tablet or capsule; parenterally, for example intravenously; by inhalation, for example in the treatment of inflammatory or obstructive airways disease; intranasally, for example in the treatment of allergic rhinitis; topically to the skin, for example in the treatment of atopic dermatitis; or rectally, for example in the treatment of inflammatory bowel disease.
- any appropriate route e.g. orally, for example in the form of a tablet or capsule; parenterally, for example intravenously; by inhalation, for example in the treatment of inflammatory or obstructive airways disease; intranasally, for example in the treatment of allergic rhinitis; topically to the skin, for example in the treatment of atopic dermatitis; or rectally, for example in the treatment of inflammatory bowel disease.
- the invention also provides a pharmaceutical composition
- a pharmaceutical composition comprising a compound of formula I in free form or in the form of a pharmaceutically acceptable salt, optionally together with a pharmaceutically acceptable diluent or carrier therefor.
- the composition may contain a co-therapeutic agent such as an anti-inflammatory, bronchodilatory, antihistamine or anti-tussive drug as hereinbefore described.
- Such compositions may be prepared using conventional diluents or excipients and techniques known in the galenic art.
- oral dosage forms may include tablets and capsules.
- Formulations for topical administration may take the form of creams, ointments, gels or transdermal delivery systems, e.g. patches.
- Compositions for inhalation may comprise aerosol or other atomizable formulations or dry powder formulations.
- the composition comprises an aerosol formulation
- it preferably contains, for example, a hydro-fluoro-alkane (HFA) propellant such as HFA134a or HFA227 or a mixture of these, and may contain one or more co-solvents known in the art such as ethanol (up to 20% by weight), and/or one or more surfactants such as oleic acid or sorbitan trioleate, and/or one or more bulking agents such as lactose.
- HFA hydro-fluoro-alkane
- the composition comprises a dry powder formulation, it preferably contains, for example, the compound of formula I having a particle diameter up to 10 microns, optionally together with a diluent or carrier, such as lactose, of the desired particle size distribution and a compound that helps to protect against product performance deterioration due to moisture e.g. magnesium stearate.
- a diluent or carrier such as lactose
- the composition comprises a nebulised formulation, it preferably contains, for example, the compound of formula I either dissolved, or suspended, in a vehicle containing water, a co-solvent such as ethanol or propylene glycol and a stabiliser, which may be a surfactant.
- the invention includes (A) a compound of formula I in inhalable form, e.g. in an aerosol or other atomisable composition or in inhalable particulate, e.g- micronised, form, (B) an inhalable medicament comprising a compound of formula I in inhalable form; (C) a pharmaceutical product comprising a compound of formula I in inhalable form in association with an inhalation device; and (D) an inhalation device containing a compound of formula I in inhalable form.
- A a compound of formula I in inhalable form, e.g. in an aerosol or other atomisable composition or in inhalable particulate, e.g- micronised, form
- B an inhalable medicament comprising a compound of formula I in inhalable form
- C a pharmaceutical product comprising a compound of formula I in inhalable form in association with an inhalation device
- an inhalation device containing a compound of formula I in inhalable form.
- Dosages of compounds of formula I employed in practising the present invention will of course vary depending, for example, on the particular condition to be treated, the effect desired and the mode of administration.
- suitable daily dosages for administration by inhalation are of the order of 0.005 to 10 mg
- suitable daily doses are of the order of 0.05 to 100 mg.
- Preferred compounds of formula I include those shown in Table 1 below. Methods for preparing such compounds are described hereinafter. The table also shows mass spectrometry, MH + (ESMS), data. The Examples are in free form, except for Examples 1-3, 7, 9-11 and 17-37, which are trifluoroacetate salts. TABLE 1 Ex.
- CDI 1,1'-carbonyldiimidazole
- DCM dichloromethane
- DIPEA diisopropylethylamine
- DMAP 4-dimethylaminopyridine
- DMF dimethylformamide
- DMSO dimethylsulfoxide
- LCMS liquid chromatographic mass spectroscopy
- TEA triethylamine
- TFA trifluoroacetic acid
- THF tetrahydrofuran
- TLC thinlayer chromatography.
- This compound is prepared from 2-isopropyl-5-oxo-5,6,7,8-tetrahydro-imidazo[1,5-c]pyrimidin-2-ium iodide by the procedure of Rahul Jain and Louis A. Cohen Tetrahedron 1996, 52, 5363 . 1 H nmr (MeOD, 400 MHz); 7.60(s, 1H), 6.95(s, 1H), 4.40(m, 1H), 2.90(t, 2H), 2.70(t, 2H), 1.45(d, 6H).
- the title compound is prepared from propyl-carbamic acid tert-butyl ester using the procedure described by Ken-ichi Takana et al in Chem. Pharm. Bull. 1988, 36, 3125 . 1 H nmr (CDCl 3 , 400 MHz); 7.25(br s, 1H), 2.75(q, 2H), 1.50(s, 9H), 1.15(t, 3H).
- 4,4'-Dimethoxybenzophenone (25 g, 103 mmol) is suspended in ethanol (150 ml) and pyridine (30 ml). Hydroxylamine hydrochloride (21.50 g, 310 mmol) is added and the reaction mixture is refluxed. The reaction is shown to be complete by TLC after 3 hours. The reaction mixture is allowed to cool and the solvent is removed in vacuo. The residue is partitioned between ethyl acetate (500 ml) and water (500 ml). The organic layer dried is over MgSO 4 , filtered and the solvent removed in vacuo. The title compound is obtained following crystallisation from ethylacetate/ cyclohexane.
- Bis-(4-methoxy-phenyl)-methanone oxime (20 g, 77.82 mmol) is suspended in ammonia .880 (450 ml) and ethanol (90 ml). Ammonium acetate (3.00 g, 38.91 mmol) is added followed by the portionwise addition of zinc dust (25.29 g, 389.10 mmol). Once the addition is complete the reaction mixture is slowly heated to 50°C. When the effervescence has ceased the reaction mixture is refluxed. The reaction is shown to be complete by TLC after 4 hours. The reaction mixture is allowed to cool and ethyl acetate is added (250 ml). The reaction mixture is filtered through CeliteTM and the phases are separated.
- the compound is used in solution in subsequent reactions.
- This solution consists of the imidazole-urea intermediate (C) together with variable amounts of the corresponding isocyanate and imidazole which result from reversible thermal elimination of imidazole under the reaction conditions.
- This solution is used in the subsequent steps since the imidazole-urea intermediate and isocyanate intermediate are equally suitable as precursors to ureas.
- the titled compound is prepared analogously to Intermediate D by replacing (S)-pyrrolidin-3-yl-carbamic acid tert-butyl ester with (R)-pyrrolidin-3-yl-carbamic acid tert-butyl ester and replacing 2-bromopyridine with 2-chloropyridine.
- the titled compound is prepared analogously to 9-[(1R,2S,3R,4S)-4-(tert-butoxycarbonylpropionyl-amino)-2,3-dihydroxy-cyclopentyl]-6-(2,2-diphenyl-ethylamino)-9H-purine-2-carboxylic acid methyl ester (Example 38) by replacing 9-[(1R,4S)-4-(tert-butoxycarbonylpropionyl-amino)-cyclopent-2-enyl]-6-(2,2-diphenyl-ethylamino)-9H-purine-2-carboxylic acid methyl ester with [(1S,4R)-4-(2,6-dichloro-purin-9-yl)-cyclopent-2-enyl]-propionyl-carbamic acid tert-butyl ester.
- 2,6-Dichloropurine (9.50 g, 50.29 mmol) is dissolved in THF (200 ml) under an atmosphere of argon.
- Diisopropylamine (7.14 g, 55.32 mmol) is added followed by C,C-bis-(4-methoxyphenyl)-methylamine (see preparation of intermediates) (12.22 g, 50.29 mmol) and the reaction mixture is stirred at 50°C.
- the reaction is shown to be complete by LCMS after 5 days.
- the solvent is removed in vacuo and replaced with MeOH (250 mL).
- the resulting precipitate is filtered off and dried to give the title compound.
- N-[(1S,2R,3S,4R)-4-(6-Amino-2-chloro-purin-9-yl)-2,3-dihydroxy-cyclopentyl]-propionamide (10.6mg, 31 ⁇ mol), 1-hexyne (25.4mg, 310 ⁇ mol), copper (I) iodide (1.5mg, 7.75 ⁇ mol), dichlorobis(triphenylphosphine)palladium(II) (5.5mg, 7.75 ⁇ mol), triphenylphosphine (4.0mg, 15.5 ⁇ mol), diethylamine (0.4mL) and DMF (0.2mL) are placed in a 0.5-2.5mL microwave vial.
- the reaction mixture is microwaved in a Personal Chemistry EmrysTM Optimizer microwave reactor at 120°C.
- the reaction is shown to be complete by LCMS after 1 hour.
- the solvent is removed in vacuo and the title compound is obtained after purification by reverse phase column chromatography (IsoluteTM C18, 0-100% acetonitrile in water - 0.1% TFA.).
- MS (ES+) m / e 387 (MH+).
- the title compound is prepared from di-Boc-[(1S,4R)-4-(2,6-dichloro-purin-9-yl)-cyclopent-2-enyl]-amine using a procedure analogous to that use to prepare (1R,2S,3R,SS)-3-(6- ⁇ [bis-(4-methoxy-phenyl)-methyl]-amino ⁇ -2-chloro-purin-9-yl)-5-(di-Boc-amino)-cyclopentane-1,2-diol.
- the title compound is prepared from (1S,2R,3S,5R)-3-(di-Boc-amino)-5-(2,6-dichloro-purin-9-yl)-cyclopentane-1,2-diol using a procedure analogous to that used to prepare (1S,2R,3S,5R)-3-amino-5-(6-amino-2-chloro-purin-9-yl)-cyclopentane-1,2-diol trifluoroacetate in Example 1.
- the title compound is prepared from (1S,2R,3S,SR)-3-amino-5-(2,6-dichloro-purin-9-yl)-cyclopentane-1,2-diol trifluoroacetate and propionyl chloride using a procedure analogous to that used to prepare N-[(1S,2R,3S,4R)-4-(6-amino-2-chloro-purin-9-yl)-2,3-dihydroxy-cyclopentyl]-propionamide trifluoroacetate in Example 2.
- MS (ES+) m / e 360 (MH+).
- N-[(1S,2R,3S,4R)-4-(2,6-Dichloro-purin-9-yl)-2,3-dihydroxy-cyclopentyl]-propionamide 160 mg, 0.44 mmol
- THF 5 ml
- Diisopropylamine 69 mg, 0.53 mmol
- 2,2-diphenylethylamine 96 mg, 0.49 mmol
- the reaction is shown to be complete by LCMS after 2 hours.
- the final compound of Example 4 may also be prepared using the following process:
- the title compound is prepared from ⁇ 2-chloro-9-[(1R,4S)-4-(di-Boc-amino)-cyclopent-2-enyl]-9H-purin-6-yl ⁇ -(2,2-diphenyl-ethyl)-amine using a procedure analogous to that of Prep. 11.
- 1 H nmr (MeOD, 400 MHz); 8.05(s, 1H), 7.35-7.15(m, 10H), 4.70-4.55(m, 4H), 4.50(m, 1H), 4.35(m, 1H), 4.20(m, 2H), 2.55(m, 1H), 2.45(m, 1H), 1.60(s, 18H).
- the free-base is formed as follows: N- ⁇ (1S,2R,3S,4R)-4-[2-(4-Amino-cyclohexylamino)-6-(2,2-diphenyl-ethylamino)-purin-9-yl]-2,3-dihydroxy-cyclopentyl ⁇ -propionamide trifluoroacetate (300 mg, 0.50 mmol) is loaded onto DOWEX® 50WX2-200 ion exchange resin (pre-washed with water). The resin is eluted with water until neutral pH and then with methanol: ammonia .880 (1: 1) to elute the free base.).
- the title compound is prepared from N-[(1S,2R,3S,4R)-4-(2,6-Dichloro-purin-9-yl)-2,3-dihydroxy-cyclopentyl]-propionamide using a procedure analogous to that used to prepare the compound of Example 3.
- This compound is prepared from N-[(1S,2R,3S,4R)-4-(2,6-Dichloro-purin-9-yl)-2,3-dihydroxy-cyclopentyl]-propionamide using histamine in a procedure analogous to that used to prepare the compound of Example 5.
- the title compound is prepared using N -(aminoethyl)piperidine in a procedure analogous to that used to prepare the compound of Example 5.
- This compound is prepared from N- ⁇ (1S,2R,35,4R)-4-[2-chloro-6-(2,2-diphenyl-ethylamino)-purin-9-yl]-2,3-dihydroxy-cyclopentyl ⁇ -propionamide (compound of Example 4) and 2-(1-ethyl-1H-imidazol-4-yl)-ethylarnine using a procedure analogous to that of Example 21.
- This compound is prepared from N- ⁇ (1S,2R,3S,4R)-4-[2-chloro-6-(2,2-diphenyl-ethylamino)-purin-9-yl]-2,3-dihydroxy-cyclopentyl ⁇ -propionamide (compound of Example 4) and 2-(1-isopropyl-1H-imidazol-4-yl)-ethylamine using a procedure analogous to that of Example 9 for the desired salt.
- Cyclopropanecarboxylic acid ⁇ (1S,2R,3S,4R)-4-[2-chloro-6-(2,2-diphenyl-ethylamino)-purin-9-yl]-2,3-dihydroxy-cyclopentyl ⁇ -amide and N- ⁇ (1S,2R,3S,4R)-4-[2-Chloro-6-(2,2-diphenylethylamino)-purin-9-yl]-2,3-dihydroxy-cyclopentyl ⁇ -butyramide are prepared using a procedure analogous to that of Example 4 in which propionyl chloride is replaces with the appropriate acylating agent.
- the title compound is prepared from carbonic acid (1S,4R)-4-(2,6-dichloro-purin-9-yl)-cyclopent-2-enyl ester ethyl ester (an intermediate for preparing the compound of Example 4) and propionyl-carbamic acid tert-butyl ester (see preparation of intermediates) using a procedure analogous to that of di-Boc-[(1S,4R)-4-(2,6-dichloro-purin-9-yl)-cyclopent-2-enyl]-amine (another intermediate for preparing the compound of Example 4).
- the title compound is prepared from ⁇ (1S,4R)-4-[2-chloro-6-(1-ethyl-propylamino)-purin-9-yl]-cyclopent-2-enyl ⁇ -propionyl-carbamic acid tert-butyl ester using a procedure analogous to that of (1R,2S,3R,5S)-3-(6- ⁇ [bis-(4-methoxy-phenyl)-methyl]-amino ⁇ -2-chloro-purin-9-yl)-5-(di-Boc-amino)-cyclopentane-1,2-diol (see Example 1).
- This compound is prepared from ⁇ (1S,2R,3S,4R)-4-[2-chloro-6-(1-ethyl-propylamino)-purin-9-yl]-2,3-dihydroxy-cyclopentyl ⁇ -propionyl-carbamic acid tert-butyl ester using a procedure analogous to that of Example 3.
- Cyclopropanecarboxylic acid ((1S,2R,3S,4R)-4- ⁇ 6-(2,2-diphenyl-ethylamino)-2-[2-(1-isopropyl-1H-imidazol-4-yl)-ethylamino]-purin-9-yl ⁇ -2,3-dihydroxy-cyclopentyl)-amide trifluoroacetate
- 2,6-Dichloropurine (20.00 g, 106 mmol) is dissolved in THF (250 ml) under an atmosphere of argon.
- Diisopropylamine (16.38 g, 127 mmol) is added followed by 2,2-diphenylethylamine (25.00 g, 127 mmol) and the reaction mixture is stirred at 50°C.
- the reaction is shown to be complete by LCMS after 6 hours. 50% of the solvent is removed in vacuo and replaced with MeOH. The resulting precipitate is filtered off and dried to give the title compound.
- the solvent is removed under reduced pressure and the residue is purified by reverse-phase chromatography eluting with a gradient system of acetonitrile (0.1 % TFA) : water (0.1% TFA) (0:100 by volume) gradually changing to acetonitrile (0.1 % TFA) : water (0.1% TFA) (100:0 by volume) to afford the title compound.
- Cyclopropanecarboxylic acid ((1S,2R,3S,4R)-4- ⁇ 6-(2,2-diphenyl-ethylamino)-2-[2-(1-isopropyl-1H-imidazol-4-yl)-ethylamino]-purin-9-yl ⁇ -2,3-dihydroxy-cyclopentyl)-amide trifluoroacetate
- reaction mixture is purified by reverse-phase chromatography eluting with a gradient system of acetonitrile (0.1% TFA) : water (0.1% TFA) (0:100 by volume) gradually changing to acetonitrile (0.1% TFA) : water (0.1% TFA) (100:0 by volume) to afford the title compound.
- LCMS electrorospray
- Cyclobutanecarboxylic acid ((1S,2R,3S,4R)-4- ⁇ 6-(2,2-diphenyl-ethylamino)-2-[2-(1-isopropyl-1H-imidazol-4-yl)-ethylamino]-purin-9-yl ⁇ -2,3-dihydroxy-cyclopentyl)-amide trifluoroacetate
- Cyclobutanecarboxylic acid ((1S,2R,3S,4R)-4- ⁇ 6-(2,2-diphenyl-ethylamino)-2-[2-(1-isopropyl-1H-imidazol-4-yl)-ethylamino]-purin-9-yl ⁇ -2,3-dihydroxy-cyclopentyl)-amide trifluoroacetate
- the title compound is prepared using a method that is analogous to that used to prepare the compound of Example 22 using cyclobutanecarboxylic acid ⁇ (1S,2R,3S,4R)-4-[2-chloro-6-(2,2-diphenyl-ethylamino)-purin-9-yl]-2,3-dihydroxycyclopentyl ⁇ -amide, 2-(1-isopropyl-1H-imidazol-4-yl)-ethylamine (see preparation of intermediates) (30 mg, 0.2 mmol) and sodium iodide (6 mg, 0.04 mmol).
- LCMS electrospray
- the title compound is prepared by the same method as cyclobutanecarboxylic acid ⁇ (1S,2R,3S,4R)-4-[2-chloro-6-(2,2-diphenyl-ethylamino)-purin-9-yl]-2,3-dihydroxycyclopentyl ⁇ -amide from (1S,2R,3S,5R)-3-amino-5-[2-chloro-6-(2,2-diphenyl-ethylamino)-purin-9-yl]-cyclopentane-1,2-diol hydrochloride (an intermediate for preparing the compound of Example 22) and butyryl chloride to afford the title compound (48 mg).
- the title compound is prepared using a method that is analogous to that used to prepare the compound of Example 22 using N- ⁇ (1S,2R,3S,4R)-4-[2-chloro-6-(2,2-diphenyl-ethylamino)-purin-9-yl]-2,3-dihydroxy-cyclopentyl ⁇ -butyramide, 2-(1-isopropyl-1H-imidazol-4-yl)-ethylamine (see preparation of intermediates) (30 mg, 0.2 mmol) and sodium iodide (6 mg, 0.04 mmol).
- LCMS electrospray
- the title compound is prepared by the same method as cyclobutanecarboxylic acid ⁇ (1S,2R,3S,4R) -4-[2-chloro-6-(2,2-diphenyl-ethylamino)-purin-9-yl]-2,3-dihydroxy-cyclopentyl ⁇ -amide from (1S,2R,3S,5R)-3-amino-5-[2-chloro-6-(2,2-diphenyl-ethylamino)-purin-9-yl]-cyclopentane-1,2-diol hydrochloride (an intermediate for preparing the compound of Example 22) and isobutyryl chloride to afford the title compound.
- the title compound is prepared using a method that is analogous to that used to prepare the compound of Example 22 using N- ⁇ (1S,2R,3S,4R)-4-[2-chloro-6-(2,2-diphenyl-ethylamino)-purin-9-yl]-2,3-dihydroxy-cyclopentyl ⁇ -isobutyramide, 2-(1-isopropyl-1H-imidazol-4-yl)-ethylamine (see preparation of intermediates) (30 mg, 0.2 mmol) and sodium iodide (6 mg, 0.04 mmol).
- LCMS electrospray
- the title compound is prepared using a method that is analogous to that used to prepare the compound of Example 22 using N- ⁇ (1S,2R,3S,4R)-4-[2-chloro-6-(2,2-diphenyl-ethylamino)-purin-9-yl]-2,3-dihydroxy-cyclopentyl ⁇ -2-phenyl-acetamide, 2-(1-isopropyl-1H-imidazol-4-yl)-ethylamine (see preparation of intermediates) (30 mg, 0.2 mmol) and sodium iodide (6 mg, 0.04 mmol).
- LCMS electrospray
- the title compound is prepared using a method that is analogous to that used to prepare the compound of Example 22 using cyclobutanecarboxylic acid ⁇ (1S,2R,3S,4R)-4-[2-chloro-6-(2,2-diphenyl-ethylamino)-purin-9-yl]-2,3-dihydroxycyclopentyl ⁇ -amide (an intermediate for preparing Example 23), 1-(2-aminoethyl)piperidine (0.057 ml, 0.4 mmol) and sodium iodide (6 mg, 0.04 mmol).
- LCMS electrospray
- the title compound is prepared using a method that is analogous to that used to prepare the compound of Example 22 using N- ⁇ (1S,2R,3S,4R)-4-[2-chloro-6-(2,2-diphenyl-ethylamino)-purin-9-yl]-2,3-dihydroxy-cyclopentyl ⁇ -butyramide (an intermediate for preparing Example 24), 1-(2-aminoethyl)-piperidine (0.057 ml, 0.4 mmol) and sodium iodide mg, 0.04 mmol).
- LCMS electrospray
- the title compound is prepared using a method that is analogous to that used to prepare the compound of Example 22 using N- ⁇ (1S,2R,3S,4R)-4-[2-chloro-6-(2,2-diphenyl-ethylamino)-purin-9-yl]-2,3-dihydroxy-cyclopentyl ⁇ -isobutyramide (an intermediate for preparing Example 25), 1-(2-aminoethyl)-piperidine (0.057 ml, 0.4 mmol) and sodium iodide (6 mg, 0.04 mmol).
- LCMS electrospray
- the title compound is prepared using a method that is analogous to that used to prepare the compound of Example 22 using N- ⁇ (1S,2R,3S,4R)-4-[2-chloro-6-(2,2-diphenyl-ethylamino)-purin-9-yl]-2,3-dihydroxy-cyclopentyl ⁇ -2-phenyl-acetamide (an intermediate of Example 26), 1-(2-aminoethyl)-piperidine (0.057 ml, 0.4 mmol) and sodium iodide (6 mg, 0.04 mmol).
- LCMS electrorospray
- the title compound is prepared by the same method as cyclobutanecarboxylic acid ⁇ (1S,2R,3S,4R)-4-[2-chloro-6-(2,2-diphenyl-ethylamino)-purin-9-yl]-2,3-dihydroxycyclopentyl ⁇ -amide from (1S,2R,3S,5R)-3-amino-5-[2-chloro-6-(2,2-diphenyl-ethylamino)-purin-9-yl]-cyclopentane-1,2-diol hydrochloride (an intermediate for preparing the compound of Example 22) and isoxazole-5-carbonyl chloride to afford the title compound.
- LCMS electrospray
- the title compound is prepared using a method that is analogous to that used to prepare the compound of Example 22 using isoxazole-5-carboxylic acid ⁇ (1S,2R,3S,4R)-4-[2-chloro-6-(2,2-diphenyl-ethylamino)-purin-9-yl]-2,3-dihydroxy-cyclopentyl ⁇ -amide, 1-(2-aminoethyl)-piperidine (51 mg, 0.4 mmol) and sodium iodide (6 mg, 0.04 mmol).
- LCMS electrospray
- the title compound is prepared using a method that is analogous to that used to prepare the compound of Example 22 using cyclopropanecarboxylic acid ⁇ (1S,2R,3S,4R)-4-[2-chloro-6-(2,2-diphenyl-ethylamino)-purin-9-yl]-2,3-dihydroxy-cyclopentyl ⁇ -amide (an intermediate for preparing Example 22), (R)-pyrrolidin-3-ylamine (34 mg, 0.4 mmol) and sodium iodide (6 mg, 0.04 mmol) to give a mixture of two regioisomers which are purified by reverse phase column chromatography (IsoluteTM C18, 0-100% acetonitrile in water - 0.1% TFA) to give a product which is predominantly cyclopropanecarboxylic acid ⁇ (1S,2R,3S,4R)-4-[2-((R)-3-aminopyrrolidin-1-yl)-6
- the title compound is prepared using a method that is analogous to that used to prepare the compound of Example 22 using cyclobutanecarboxylic acid ⁇ (1S,2R,3S,4R)-4-[2-chloro-6-(2,2-diphenyl-ethylamino)-purin-9-yl]-2,3-dihydroxy-cyclopentyl ⁇ -amide (an intermediate for preparing Example 23), (R)-pyrrolidin-3-ylamine (34 mg, 0.4 mmol) and sodium iodide (6 mg, 0.04 mmol) to give a mixture of two regioisomers which are purified by reverse phase column chromatography (IsoluteTM C18, 0-100% acetonitrile in water - 0.1 % TFA) to give a product which is predominantly cyclobutanecarboxylic acid ⁇ (1S,2R,3S,4R)-4-[2-((R)-3-aminopyrrolidin-1-yl
- the title compound is prepared using a method that is analogous to that used to prepare the compound of Example 22 using N- ⁇ (1S,2R,3S,4R)-4-[2-chloro-6-(2,2-diphenyl-ethylamino)-purin-9-yl]-2,3-dihydroxy-cyclopentyl ⁇ -2-phenyl-acetamide (an intermediate for preparing Example 26), (R)-pyrrolidin-3-ylamine (34 mg, 0.4 mmol) and sodium iodide (6 mg, 0.04 mmol) to give a mixture of two regioisomers which are purified by reverse phase column chromatography (IsoluteTM C18, 0-100% acetonitrile in water- 0.1% TFA) to give a product which is predominantly N- ⁇ (1S,2R,3S,4R)-4-[2-((R)-3-Amino-pyrrolidin-1-yl)-6-(2,2-diphenylethylamino
- the title compound is prepared using a method that is analogous to that used to prepare the compound of Example 22 using N- ⁇ (1S,2R,3S,4R)-4-[2-chloro-6-(2,2-diphenyl-ethylamino)-purin-9-yl] -2,3-dihydroxy-cyclopentyl ⁇ -3-phenyl-propionamide, (R)-pyrrolidin-3-ylamine (34 mg, 0.4 mmol) and sodium iodide (6 mg, 0.04 mmol) to give a mixture of two regioisomers which are purified by reverse phase column chromatography (IsoluteTM C18, 0-100% acetonitrile in water - 0.1 % TFA) to give a product which is predominantly N- ⁇ (1S,2R,3S,4R)-4-[2-((R)-3-Amino-pyrrolidin-1-yl)-6-(2,2-diphenyl-ethylamino)-
- N- ⁇ (1S,2R,3S,4R)-4-[2-((R)-3-Amino-pyrrolidin-1-yl)-6-(2,2-diphenyl-ethylamino)-purin-9-yl]-2,3-dihydroxy-cyclopentyl ⁇ -propionamide (30 mg, 0.04 mmol) is dissolved in toluene (2 ml) and i PrOH(1 ml).
- N -[1-(2-Pyridinyl)-4-piperidinyl]-1H-imidazole-1-carboxamide (prepared using the procedure described in international patent application WO 01/94368 ) (12 mg, 0.044 mmol) is added as a solution in dichloromethane. The reaction mixture is stirred at room temperature. The reaction is shown to be complete by LCMS after 24 hours. The solvent is removed in vacuo.
- Example 37a and 37b are assigned using secondary isotope effects in NMR Spectroscopy. Isotope effects are well established in NMR spectroscopy ( B. A. Bernheim and H. Batiz-Hernandez, Prog. Nucl. Magn. Reson. Spectrosc. 3, 63-85 [1967 ]). Primary isotope effects have been widely studied ( L.J. Altman et al. J. Am. Chem. Soc. 100, 8264-8266 [1978 ]), but it is the secondary isotope shift that has provided important structural information.
- the magnitude of the two- and three-bond effects vary with the configuration of the carbons, and also the substitution and hydrogen bonding of these exchangeable groups. It is these signal multiplet formations and magnitude of isotope effects are used to unambiguously assign and confirm the structures of Example 37a and Example 37b.
- Example 37a is bonded to two NH groups
- Example 37b is bonded to one NH group and to the fully substituted nitrogen of the proline ring.
- 6-(2,2-Diphenyl-ethylamino)-9-((1R,4S)-4-hydroxy-cyclopent-2-enyl)-9H-purine-2-carboxylic acid methyl ester (2.80 g, 6.14 mmol) is placed in an oven-dried flask under an atmosphere of argon. Dry tetrahydrofuran (30 ml) is added followed by dry pyridine (0.97 g, 12.3 mmol). Ethyl chloroformate (2.66 g, 24.6 mmol) is added slowly and the reaction mixture is stirred at room temperature. The reaction is shown to be complete by LCMS after 3 hours.
- 6-(2,2-Diphenyl-ethylamino)-9-((1R,4S)-4-ethoxycarbonyloxy-cyclopent-2-enyl)-9H-purine-2-carboxylic acid methyl ester (2.2 g, 4.2 mmol) is dissolved in deoxygenated tetrahydrofuran. The resultant solution is stirred under an atmosphere of argon at room temperature.
- the title compound is prepared from 9-((1R,4S)-4-di-tert-butoxycarbonylamino-cyclopent-2-enyl)-6-(2,2-diphenyl-ethylamino)-9H-purine-2-carboxylic acid methyl ester using a procedure analogous to that of (1R,2S,3R,5S)-3-[2-chloro-6-(2,2-diphenyl-ethylamino)-purin-9-yl]-5-(di-Boc-amino)-cyclopentane-1,2-diol.
- This compound which is the trifluoroacetate salt of the final compound of Example 37, is prepared using a method that is analogous to that used to prepare 9-((1R,4S)-4-Di-tert-butoxycarbonylamino-cyclopent-2-enyl)-6-(2,2-diphenyl-ethylamino)-9H-purine-2-carboxylic acid methyl ester by replacing di-t-butyl iminodicarboxylate with propionyl-carbamic acid tert-butyl ester.
- reaction mixture is partitioned between ethyl acetate and water and the organic portion is dried (MgSO 4 ) and concentrated in vacuo.
- the titled product is precipitated from methanol. Further product is derived from the mother liquor by chromatography on silica eluting with DCM : methanol (25:1).
- This compound is prepared analogously to 9-((1R,2S,3R,4S)-2,3-dihydroxy-4-propionylamino-cyclopentyl)-6-(2,2-diphenyl-ethylamino)-9H-purine-2-carboxylic acid (2-amino-ethyl)-amide by replacing 9-((1R,2S,3R,4S)-2,3-dihydroxy-4-propionylaminocyclopentyl)-6-(2,2-diphenyl-ethylamino)-9H-purine-2-carboxylic acid methyl ester with 9-[(1R,2S,3R,4S)-4-(tert-butoxy-carbonyl-propionyl-amino)-2,3-dihydroxy-cyclopentyl]-6-(2,2-diphenyl-ethylamino)-9H-purine-2-carboxylic acid methyl ester.
- This compound is prepared analogously to 9-((1R,2S,3R,4S)-2,3-dihydroxy-4-propionylamino-cyclopentyl)-6-(2,2-diphenyl-ethylamino)-9H-purine-2-carboxylic acid ⁇ 2-[3-(3,4,5,6-tetra-hydro-2H-[1,2]bipyridinyl-4-yl)ureido]-ethyl ⁇ -amide by replacing 9-((1R,2S,3R,4S)-2,3-dihydroxy-4-propionylamino-cyclopentyl)-6-(2,2-diphenyl-ethylamino)-9H-purine-2-carboxylic acid (2-amino-ethyl)-amide with ⁇ (1S,2R,3S,4R)-4-[2-(2-Aminoethylcarbamoyl)-6-(2,2-diphenyl-eth
- This compound is prepared analogously to 9-((1R,2S,3R,4S)-4-Amino-2,3-dihydroxycyclopentyl)-6-(2,2-diphenyl-ethylamino)-9H-purine-2-carboxylic acid methyl ester by replacing 9-((1R,-2S,3R,4S)-4-Di-tert-butoxycarbonylamino-2,3-dihydroxy-cyclopentyl)-6-(2,2-diphenyl-ethylamino)-9H-purine-2-carboxylic acid methyl ester with (1S,2R,3S,4R)-4-(6-(2,2-Diphenyl-ethylamino)-2- ⁇ 2-[3-(3,4,5,6-tetrahydro-2H-[1,2']bipyridinyl-4-yl)-ureido]-ethylcarbamoyl ⁇ -purin-9-yl)-2,
- This compound is prepared analogously to 9-((1R,2S,3R,4S)-2,3-dihydroxy-4-propionylamino-cyclopentyl)-6-(2,2-diphenyl-ethylamino)-9H-purine-2-carboxylic acid methyl ester by replacing 9-((1R,2S,3R,4S)-,4-amino-2,3-dihydroxy-cyclopentyl)-6-(2,2-diphenylethylami no)-9H-purine-2-carboxylic acid methyl ester hydrochloride with 9-((1R,2S,3R,4S)-4-Amino-2,3-dihydroxy-cyclopentyl)-6-(2,2-diphenyl-ethylamino)-9H-purine-2-carboxylic acid ⁇ 2-[3-(3,4,5,6-tetrahydro-2H-[1,2']bipyridinyl-4-yl)
- This compound is prepared analogously to Example 22 by replacing cyclopropane carboxylic acid ⁇ (1S,2R,3S,4R)-4-[2-chloro-6-(2,2-diphenyl-ethylamino)-purin-9-yl]-2,3-dihydroxycyclopentyl ⁇ -amide with N- ⁇ (1S,2R,3S,4R)-4-[2-chloro-6-(2,2-diphenyl-ethylamino)-purin-9-yl]-2,3-dihydroxy-cyclopentyl ⁇ -propionamide and by replacing 2-(1-isopropyl-1H-imidazol-4-yl)-ethylamine with 1,3-di(R)-pyrrolidin-3-yl-urea.
- This compound is prepared analogously to 9-((1R,2S,3R,4S)-2,3-Dihydroxy-4-propionylamino-cyclopentyl)-6-(2,2-diphenyl-ethylamino)-9H-purine-2-carboxylic acid ⁇ 2-[3-(3,4,5,6-tetrahydro-2H-[1,2]bipyridinyl-4-yl)ureido]-ethyl ⁇ -amide by replacing (1S,2R,3S,5R)-3-amino-5-[2-chloro-6-(2,2-diphenyl-ethylamino)-purin-9-yl]-cyclopentane-1,2-diol hydrochloride with 9-((1R,2S,3R,4S)-4-amino-2,3-dihydroxy-cyclopentyl)-6-(2,2-diphenyl-ethylamino)-9H-purine-2-carboxy
- a mixture comprising 9-((1R,2S,3R,4S)-4-amino-2,3-dihydroxy-cyclopentyl)-6-(2,2-diphenylethylamino)-9H-purine-2-carboxylic acid ⁇ 2-[3-(3,4,5,6-tetrahydro-2H-[1,2']bipyridinyl-4-yl)-ureido]-ethyl)-amide dihydrochloride (0.02 g, 25 ⁇ mol), TEA (0.013 g, 125 ⁇ mol) in THF (2 ml) is treated with acetyl chloride (0.003 g, 40 ⁇ mol).
- This compound is prepared analogously to cyclopropanecarboxylic acid ((1S,2R,3S,4R)-4- ⁇ 6-(2,2-diphenyl-ethylamino)-2-[2-(1-isopropyl-1H-imidazol-4-yl)-ethylamino]-purin-9-yl ⁇ -2,3-dihydroxy-cyclopentyl)-amide trifluoroacetate by replacing cyclopropanes-carboxylic acid ⁇ (1S,2R,3S,4R)-4-[2-chloro-6-(2,2-diphenyl-ethylamino)-purin-9-yl]-2,3-dihydroxycyclopentyl ⁇ -amide with N- ⁇ (1S,2R,3S,4R)-4-[2-chloro-6-(2,2-diphenyl-ethylamino)-purin-9-yl]-2,3-dihydroxy-cyclopentyl ⁇ -
- This compound is prepared analogously to 9-((1R,2S,3R,4S)-2,3-dihydroxy-4-propionylamino-cyclopentyl)-6-(2,2-diphenyl-ethylamino)-9H-purine-2-carboxylic acid ⁇ 2-[3-((R)-1-pyridin-2-yl-pyrrolidin-3-yl)-ureido]-ethyl ⁇ -amide trifluoroacetate by replacing 1-(2-amino-ethyl)-3-((R)-1-pyridin-2-yl-pyrrolidin-3-yl)-urea with 1-(2-amino-ethyl)-3-((S)-1-pyridin-2-yl-pyrrolidin-3-yl)-urea.
- This compound is prepared analogously to N-((1S,2R,3S,4R)-4- ⁇ 6-(1-Ethyl-propylamino)-2-[(R)-3-((R)-3-pyrrolidin-3-ylureido)-pyrrolidin-1-yl]-purin-9-yl ⁇ -2,3-dihydroxy-cyclopentyl)-propionamide trifluoroacetate by replacing N- ⁇ (1S,2R,3S,4R)-4-[2-chloro-6-(1-ethylpropylamino)-purin-9-yl]-2,3-dihydroxy-cyclopentyl ⁇ -propionamide with N-((1S,2R,3S,4R)-4- ⁇ 2-chloro-6-[(naphthalen-1-ylmethyl)-amino]-purin-9-yl ⁇ -2,3-dihydroxy-cyclopentyl)-propionamide trifluoroacetate.
- N- ⁇ (1S,2R,3S,4R)-4-[2-chloro-6-(1-ethyl-propylamino)-purin-9-yl]-2,3-dihydroxycyclopentyl ⁇ -propionamide (0.02 g, 0.03 mmol)
- (3R)-3-(BOC-amino)pyrrolidine 0.028 g, 0.15 mmol
- sodium iodide 0.004 g, 0.03 mmol
- Acetonitrile (0.25 ml) and NMP (0.25 ml) are added and the reaction mixture is heated using microwave radiation in a Personal Chemistry EmrysTM Optimizer microwave reactor at 160°C for 30 minutes.
- DCM (3 ml) and water (3 ml) are added to the reaction mixture and following agitation, the organic portion is separated and treated with TFA (0.5 ml). After standing at room temperature overnight purification is carried out using mass directed preparative LC-MS eluting with acetonitrile: water: trifluoroacetic acid to afford the titled compound.
- This compound is prepared analogously to N- ⁇ (1S,2R,3S,4R)-4-[2-((R)-3-Amino-pyrrolidin-1-yl)-6-(1-ethyl-propylamino)-purin-9-yl]-2,3-dihydroxy-cyclopentyl ⁇ -propionamide trifluoroacetate by replacing N- ⁇ (1S,2R,3S,4R)-4-[2-chloro-6-(1-ethyl-propylamino)-purin-9-yl]-2,3-dihydroxy-cyclopentyl ⁇ -propionamide with N-((1S,2R,3S,4R)-4- ⁇ 2-chloro-6-[(naphthalen-1-ylmethyl)-amino]-purin-9-yl ⁇ -2,3-dihydroxy-cyclopentyl)-propionamide trifluoroacetate.
- This compound is prepared analogously to N-((1S,2R,3S,4R)-4- ⁇ 6-(1-ethyl-propylamino)-2- ⁇ (R)-3-((R)-3-pyrrolidin-3-ylureido)-pyrrolidin-1-yl]-purin-9-yl ⁇ -2,3-dihydroxy-cyclopentyl)-propionamide trifluoroacetate by replacing N- ⁇ (1S,2R,3S,4R)-4-[2-chloro-6-(1-ethylpropylamino)-purin-9-yl]-2,3-dihydroxy-cyclopentyl ⁇ -propionamide with N- ⁇ (1S,2R,3S,4R)-4-[2-chloro-6-(3,3-dimethyl-butylamino)-purin-9-yl]-2,3-dihydroxy-cyclopentyl ⁇ -propionamide trifluoroacetate.
- This compound is prepared analogously to N-((1S,2R,3S,4R)-4- ⁇ 2-chloro-6-[(naphthalen-1-ylmethyl)-amino]-purin-9-yl ⁇ -2,3-dihydroxy-cyclopentyl)-propionamide trifluoroacetate (Example 50) by replacing 1-napthalenemethylamine with C-(9H-fluoren-9-yl)-methylamine.
- This compound is prepared analogously to N- ⁇ (1S,2R,3S,4R)-4-[6-(2,2-diphenyl-ethylamino)-2-((S)-1-hydroxymethyl-2-phenyl-ethylamino)-purin-9-yl]-2,3-dihydroxy-cyclopentyl ⁇ -propionamide (Example 16) by replacing N- ⁇ (1S,2R,3S,4R)-4-[2-chloro-6-(2,2-diphenylethylamino)-purin-9-yl]-2,3-dihydroxy-cyclopentyl ⁇ -propionamide (Example 4) with N-((1S,2R,3S,4R)-4- ⁇ 2-chloro-6-[(9H-fluoren-9-ylmethyl)-amino]-purin-9-yl ⁇ -2,3-dihydroxycyclopentyl)-propionamide.
- This compound is prepared analogously to N- ⁇ (1S,2R,3S,4R)-4-[2- ⁇ (R)-3-[3-(2,6-dichloropyridin-4-yl)-ureido]-pyrrolidin-1-yl ⁇ -6-(2,2-diphenyl-ethylamino)-purin-9-yl]-2,3-dihydroxycyclopentyl ⁇ -propionamide trifluoroacetate (Example 73) by replacing 2,6-dichloro-4-isocyanato-pyridine with 2-thienyl isocyanate.
- This compound is prepared analogously to 9-((1R,2S,3R,4S)-4-acetylamino-2,3-dihydroxycyclopentyl)-6-(2,2-diphenyl-ethylamino)-9H-purine-2-carboxylic acid ⁇ 2-[3-(3,4,5,6-tetrahydro-2H-[1,2']bipyridinyl-4-yl)-ureido]-ethyl ⁇ -amide trifluoroacetate (Ex.
- the compound exists as a mixture of the imidazole-urea intermediate together with variable amounts of the corresponding isocyanate and imidazole which are equally suitable as precursors to ureas.
- This compound is prepared analogously to 4-methyl-piperazine-1-carboxylic acid ⁇ (R)-1-[9-((1R,2S,3R,4S)-2,3-dihydroxy-4-propionylamino-cyclopentyl)-6-(2,2-diphenyl-ethylamino)-9H-purin-2-yl]-pyrrolidin-3-yl ⁇ -amide trifluoroacetate (Example 77) by replacing 1-methyl piperazine with 2-amino pyridine.
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Pharmacology & Pharmacy (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- Medicinal Chemistry (AREA)
- Life Sciences & Earth Sciences (AREA)
- General Chemical & Material Sciences (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Engineering & Computer Science (AREA)
- Pulmonology (AREA)
- Rheumatology (AREA)
- Pain & Pain Management (AREA)
- Immunology (AREA)
- Epidemiology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Saccharide Compounds (AREA)
Claims (12)
- Verbindung der Formel I
 R1 für C1-C8-Alkylcarbonyl, C3-C8-Cycloalkylcarbonyl, -SO2-C1-C8-Alkyl, C7-C14-Aralkylcarbonyl oder -C(=O)-C(=O)-NH-C1-C8-Alkyl, das gegebenenfalls durch R4 substituiert ist, steht;
R1 für C1-C8-Alkylcarbonyl, C3-C8-Cycloalkylcarbonyl, -SO2-C1-C8-Alkyl, C7-C14-Aralkylcarbonyl oder -C(=O)-C(=O)-NH-C1-C8-Alkyl, das gegebenenfalls durch R4 substituiert ist, steht;
R2 für Wasserstoff oder gegebenenfalls durch C6-C10-Aryl substituiertes C1-C8-Alkyl steht;
R3 für Halogen oder C2-C8-Alkinyl steht, oder R3 für gegebenenfalls durch C3-C8-Cycloalkyl, das gegebenenfalls durch Amino substituiert ist, substituiertes Amino steht,
oder R3 für gegebenenfalls durch Hydroxy, C6-C10-Aryl oder R5 substituiertes C1-C8-Alkylamino steht, oder R3 für R6, das gegebenenfalls durch Amino oder -NH-C(=O)-NH-R7 substituiert ist, steht,
oder R3 für -NH-R6, das gegebenenfalls durch -NH-C(=O)-NH-R7 substituiert ist, steht,
oder R3 für C1-C8-Alkylaminocarbonyl, das gegebenenfalls durch -NH-C(=O)-NH-R8 substituiert ist, steht;
R4, R5 und R6 jeweils unabhängig voneinander für einen 5- oder 6-gliedrigen heterocyclischen Ring, der mindestens ein Ringheteroatom aus der Gruppe bestehend aus Stickstoff, Sauerstoff und Schwefel enthält, stehen, wobei der 5- oder 6-gliedrige heterocyclische Ring gegebenenfalls durch C1-C8-Alkyl substituiert ist; und
R7 und R8 unabhängig voneinander für einen 5- oder 6-gliedrigen heterocyclischen Ring, der mindestens ein Ringheteroatom aus der Gruppe bestehend aus Stickstoff, Sauerstoff und Schwefel enthält, stehen, wobei der 5- oder 6-gliedrige heterocyclische Ring gegebenenfalls durch Halogen, C1-C8-Alkyl, C1-C8-Alkylsulfonyl oder einen 5- oder 6-gliedrigen heterocyclischen Ring, der mindestens ein Ringheteroatom aus der Gruppe bestehend aus Stickstoff, Sauerstoff und Schwefel enthält, substituiert ist. - Verbindung nach Anspruch 1, worin
R1 für C1-C4-Alkylcarbonyl, C3-C5-Cycloalkylcarbonyl, -SO2-C1-C4-Alkyl, C7-C10-Aralkylcarbonyl oder -C (=O) -C (=O) -NH-C1-C4-Alkyl, das gegebenenfalls in einer Position durch R4 substituiert ist, steht;
R2 für Wasserstoff, unsubstituiertes C1-C6-Alkyl oder gegebenenfalls in einer Position durch C6-C10-Aryl substituiertwa C1-C5-Alkyl steht;
R3 für Halogen oder C2-C6-Alkinyl steht,
oder R3 für gegebenenfalls in einer Position durch C3-C8-Cycloalkyl, das gegebenenfalls in einer Position durch Amino substituiert ist, substituiertes Amino steht,
oder R3 für gegebenenfalls in einer oder zwei Positionen durch Hydroxy, Phenyl oder R5 substituiertes C1-C4-Alkylamino steht,
oder R3 für R6, das gegebenenfalls in einer Position durch Amino oder -NH-C(=O)-NH-R7 substituiert ist, steht,
oder R3 für -NH-R6, das gegebenenfalls in einer Position durch -NH-C(=O)-NH-R7 substituiert ist, steht,
oder R3 für C1-C4-Alkylaminocarbonyl, das gegebenenfalls in einer Position durch -NH-C(=0)-NH-R8 substituiert ist, steht;
R4, R5 und R6 jeweils unabhängig voneinander für einen 5- oder 6-gliedrigen heterocyclischen Ring, der mindestens ein Ringheteroatom aus der Gruppe bestehend aus Stickstoff, Sauerstoff und Schwefel enthält, stehen, wobei der 5- oder 6-gliedrige heterocyclische Ring gegebenenfalls in einer Position durch C1-C4-Alkyl substituiert ist; und R7 und R8 unabhängig voneinander für einen 5- oder 6-gliedrigen heterocyclischen Ring, der mindestens ein Ringheteroatom aus der Gruppe bestehend aus Stickstoff, Sauerstoff und Schwefel enthält, stehen, wobei der 5- oder 6-gliedrige heterocyclische Ring gegebenenfalls in einer oder zwei Positionen durch Halogen, C1-C4-Alkyl, C1-C4-Alkylsulfonyl oder einen 5- oder 6-gliedrigen N-heterocyclischen Ring substituiert ist. - Verbindung nach Anspruch 1, worin
R1 für C1-C8-Alkylcarbonyl, C3-C8-Cycloalkylcarbonyl, -SO2-C1-C8-Alkyl, C7-C14-Aralkylcarbonyl oder -C(=O)-C(=O)-NH-C1-C8-Alkyl, das gegebenenfalls durch R4 substituiert ist, steht;
R2 für Wasserstoff oder gegebenenfalls durch C6-C10-Aryl substituiertes C1-C8-Alkyl steht;
R3 für Halogen oder C2-C8-Alkinyl steht,
oder R3 für gegebenenfalls durch C3-C8-Cycloalkyl, das gegebenenfalls durch Amino substituiert ist, substituiertes Amino steht,
oder R3 für gegebenenfalls durch Hydroxy, C6-C10-Aryl oder R5 substituiertes C1-C8-Alkylamino steht, oder R3 für R6, das gegebenenfalls durch Amino oder -NH-C(=O)-NH-R7 substituiert ist, steht,
oder R3 für C1-C8-Alkylaminocarbonyl, das gegebenenfalls durch -NH-C(=O)-NH-R8 substituiert ist, steht;
R4, R5 und R6 jeweils unabhängig voneinander für einen 5- oder 6-gliedrigen heterocyclischen Ring, der mindestens ein Ringheteroatom aus der Gruppe bestehend aus Stickstoff, Sauerstoff und Schwefel enthält, stehen; und
R7 und R8 unabhängig voneinander für einen 5- oder 6-gliedrigen heterocyclischen Ring, der mindestens ein Ringheteroatom aus der Gruppe bestehend aus Stickstoff, Sauerstoff und Schwefel enthält, stehen, wobei der 5- oder 6-gliedrige heterocyclische Ring gegebenenfalls durch einen 5- oder 6-gliedrigen heterocyclischen Ring, der mindestens ein Ringheteroatom aus der Gruppe bestehend aus Stickstoff, Sauerstoff und Schwefel enthält, substituiert ist. - Verbindung nach Anspruch 3, worin
R1 für C1-C4-Alkylcarbonyl, C3-C6-Cycloalkylcarbonyl, -SO2-C1-C4-Alkyl, C7-C10-Aralkylcarbonyl oder -C (=O) -C (=O) -NH-C1-C8-Alkyl, das gegebenenfalls durch R4 substituiert ist, steht;
R2 für Wasserstoff oder gegebenenfalls durch C6-C10-Aryl substituiertes C1-C6-Alkyl steht;
R3 für Halogen oder C2-C5-Alkinyl steht,
oder R3 für gegebenenfalls durch C3-C8-Cycloalkyl, das gegebenenfalls durch Amino substituiert ist, substituiertes Amino steht,
oder R3 für gegebenenfalls durch Hydroxy, C6-C10-Aryl oder R5 substituiertes C1-C4-Alkylamino steht, oder R3 für R6, das gegebenenfalls durch Amino oder -NH-C(=O)-NH-R7 substituiert ist, steht,
oder R3 für C1-C4-Alkylaminocarbonyl, das gegebenenfalls durch -NH-C(=O)-NH-R8 substituiert ist, steht;
R4, R5 und R6 jeweils unabhängig voneinander für einen 5- oder 6-gliedrigen heterocyclischen Ring, der mindestens ein Ringheteroatom aus der Gruppe bestehend aus Stickstoff, Sauerstoff und Schwefel enthält, stehen; und
R7 und R8 unabhängig voneinander für einen 5- oder 6-gliedrigen heterocyclischen Ring, der mindestens ein Ringheteroatom aus der Gruppe bestehend aus Stickstoff, Sauerstoff und Schwefel enthält, stehen, wobei der 5- oder 6-gliedrige heterocyclische Ring gegebenenfalls durch einen 5- oder 6-gliedrigen heterocyclischen Ring, der mindestens ein Ringheteroatom aus der Gruppe bestehend aus Stickstoff, Sauerstoff und Schwefel enthält, substituiert ist. - Verbindung der Formel I nach Anspruch 1, worin R1, R2 und R3 die in den folgenden Tabellen angegebenen Bedeutungen besitzen:
R1 R2 R3 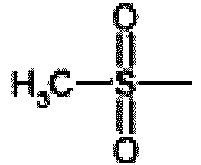
-H -Cl 
-H 

-H 


-Cl 




















-Cl 

-Cl 

-Cl 




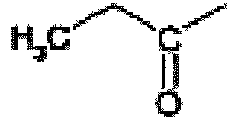



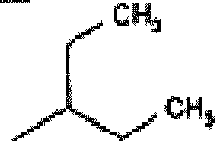








-H 








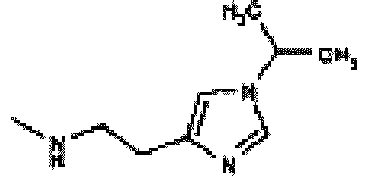


















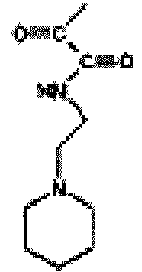


























R1 R2 R3 







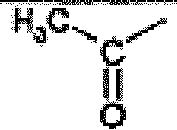







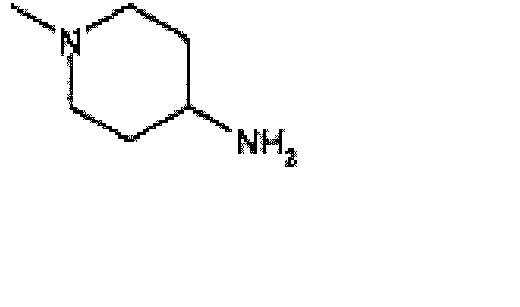




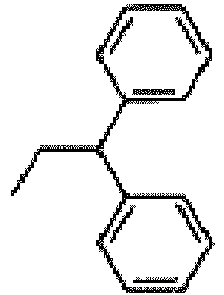



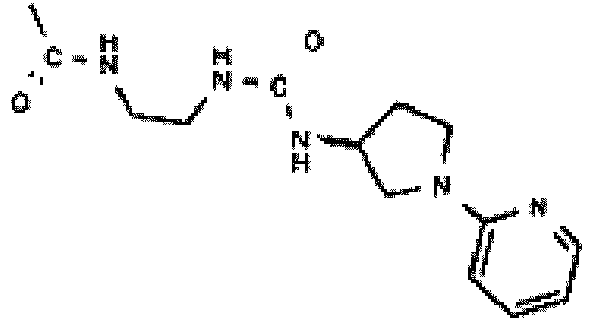






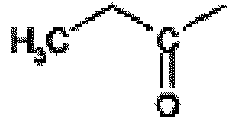

-Cl 

-Cl 

-Cl 

-Cl 







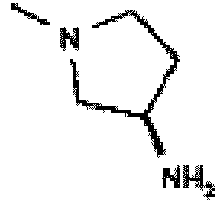








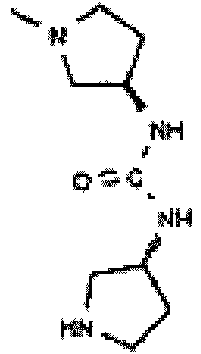
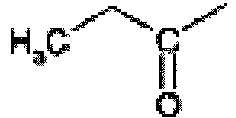







R1 R2 R3 























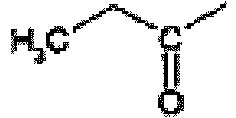






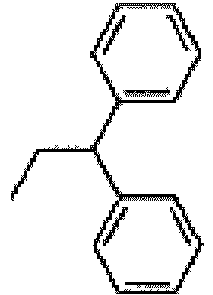

















-H 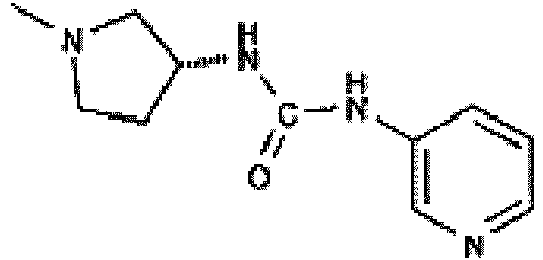
- Verbindung nach einem der vorhergehenden Ansprüche zur Verwendung als Pharmazeutikum.
- Verbindung nach einem der Ansprüche 1 bis 5 in Kombination mit einem entzündungshemmenden oder bronchodilatorischen Arzneistoff oder einem Arzneistoff mit Antihistaminwirkung oder antitussiver Wirkung, wobei die Verbindung und der Arzneistoff in der gleichen pharmazeutischen Zusammensetzung oder in verschiedenen pharmazeutischen Zusammensetzungen vorliegen.
- Pharmazeutische Zusammensetzung, umfassend als Wirkstoff eine Verbindung nach einem der Ansprüche 1 bis 5, gegebenenfalls zusammen mit einem pharmazeutisch unbedenklichen Verdünnungsmittel oder Träger.
- Pharmazeutische Zusammensetzung nach Anspruch 8, die ferner einen entzündungshemmenden oder bronchodilatorischen Arzneistoff oder einen Arzneistoff mit Antihistaminwirkung oder antitussiver Wirkung umfasst.
- Verwendung einer Verbindung nach einem der Ansprüche 1 bis 5 zur Herstellung eines Arzneimittels zur Behandlung eines Leidens, das durch Aktivierung des Adenosin-A2A-Rezeptors vermittelt wird.
- Verwendung einer Verbindung nach einem der Ansprüche 1 bis 5 zur Herstellung eines Arzneimittels zur Behandlung einer entzündlichen oder obstruktiven Atemwegserkrankung.
- Verfahren zur Herstellung einer Verbindung der Formel I gemäß Anspruch 1 in freier Form oder Salzform, bei dem man(i)(A) zur Herstellung von Verbindungen der Formel I eine Verbindung der Formel II

R1-xa III
oder der Formel IIIa (B) zur Herstellung von Verbindungen der Formel I, wobei R3 für durch C3-C8-Cycloalkyl, das gegebenenfalls durch Amino substituiert ist, substituiertes Amino steht oder R3 für gegebenenfalls durch Hydroxy, C6-C10-Aryl oder R5 substituiertes C1-C8-Alkylamino steht oder R3 für R6, das gegebenenfalls durch Amino oder -NH-C(=O)-NH-R7 substituiert ist, steht, eine Verbindung der Formel IV
(B) zur Herstellung von Verbindungen der Formel I, wobei R3 für durch C3-C8-Cycloalkyl, das gegebenenfalls durch Amino substituiert ist, substituiertes Amino steht oder R3 für gegebenenfalls durch Hydroxy, C6-C10-Aryl oder R5 substituiertes C1-C8-Alkylamino steht oder R3 für R6, das gegebenenfalls durch Amino oder -NH-C(=O)-NH-R7 substituiert ist, steht, eine Verbindung der Formel IV
 (C) zur Herstellung von Verbindungen der Formel I eine Verbindung der Formel VI
(C) zur Herstellung von Verbindungen der Formel I eine Verbindung der Formel VI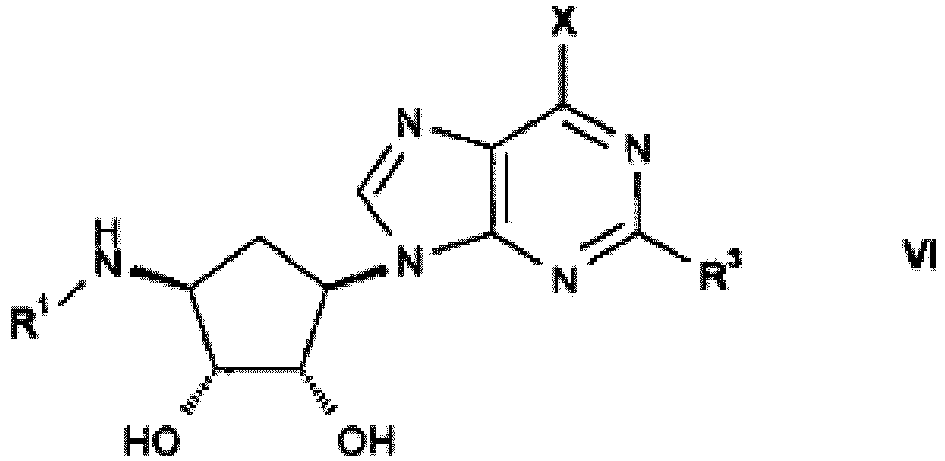
H2N-R2 VII
worin R2 die in Anspruch 1 angegebene Bedeutung besitzt, umsetzt;(D) zur Herstellung von Verbindungen der Formel I eine Verbindung der Formel VIII (E) zur Herstellung von Verbindungen der Formel I, worin R3 für C1-C9-Alkylamino-carbonyl oder C3-C8-Cycloalkylaminocarbonyl, das durch -NH-C(=O)-NH-R8 substituiert ist, steht, wobei R8 die in Anspruch 1 angegebene Bedeutung besitzt, eine Verbindung der Formel IX
(E) zur Herstellung von Verbindungen der Formel I, worin R3 für C1-C9-Alkylamino-carbonyl oder C3-C8-Cycloalkylaminocarbonyl, das durch -NH-C(=O)-NH-R8 substituiert ist, steht, wobei R8 die in Anspruch 1 angegebene Bedeutung besitzt, eine Verbindung der Formel IX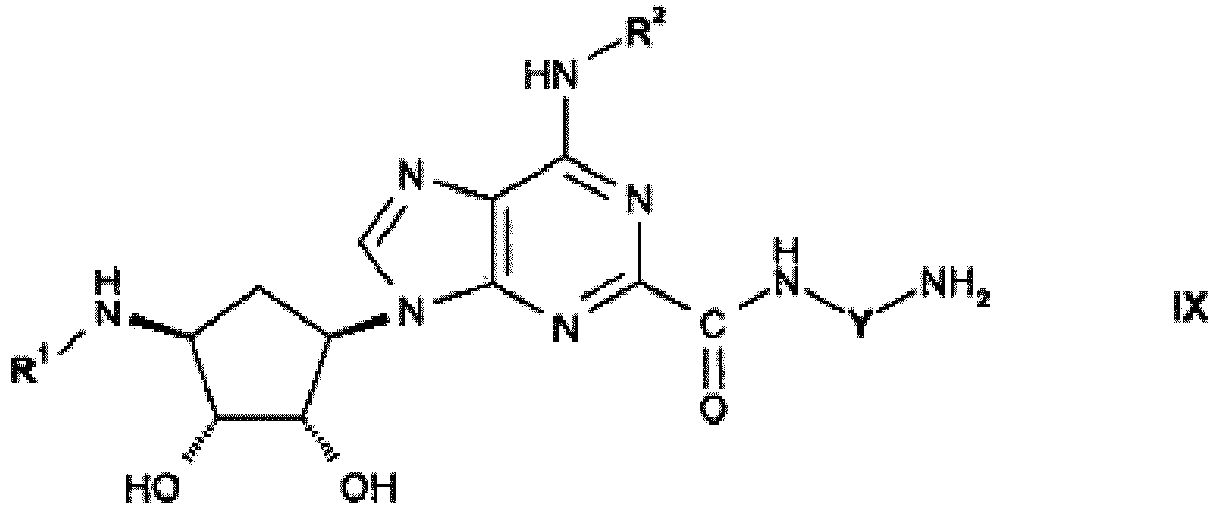

O=C=N—R8 XI
worin T für C6-C10-Aryloxy oder einen 5- oder 6-gliedrigen heterocyclischen Ring, der mindestens ein Ringheteroatom aus der Gruppe bestehend aus Stickstoff, Sauerstoff und Schwefel enthält, steht und R8 die in Anspruch 1 angegebene Bedeutung besitzt, umsetzt;(F) zur Herstellung von Verbindungen der Formel I, worin R3 für C2-C8-Alkinyl steht, eine Verbindung der Formel IV, wobei R1 und R2 die in Anspruch 1 angegebene Bedeutung besitzen, in Gegenwart einer Base und eines Katalysators mit einer Verbindung der Formel XII
Rx-C≡C-H XII
worin Rx für C1-C8-Alkyl steht, umsetzt;(G) zur Herstellung von Verbindungen der Formel I, worin R3 für gegebenenfalls durch -NH-C(=O)-NH-R8 substituiertes C1-C8-Alkylaminocarbonyl steht, eine Verbindung der Formel XIIa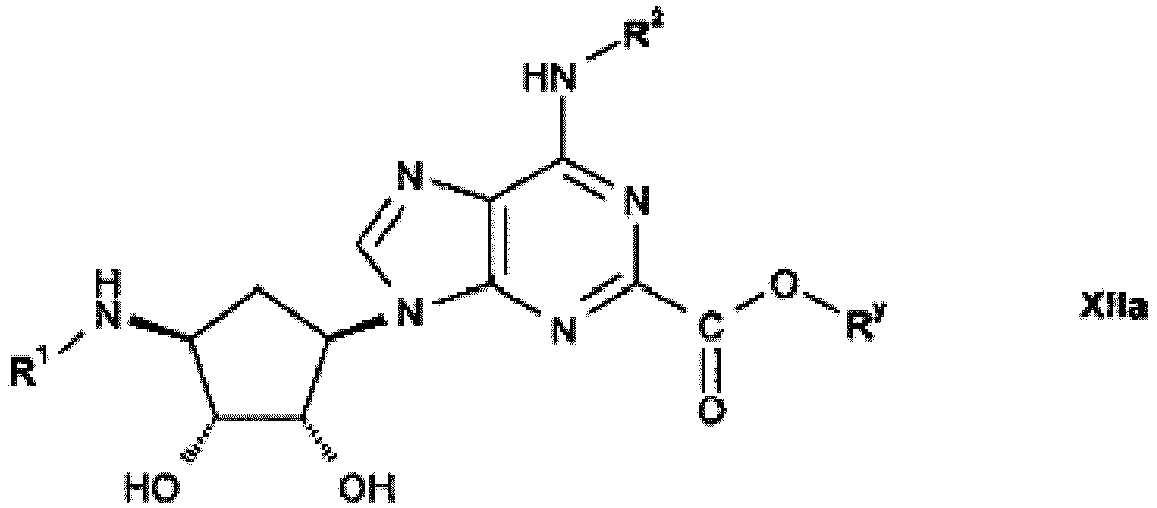
 (H) zur Herstellung von Verbindungen der Formel I, worin R3 für gegebenenfalls durch -NH-C(=O)-NH-R8 substituiertes C1-C8-Alkylaminocarbonyl steht, wobei R8 für einen 5-oder 6-gliedrigen heterocyclischen Ring, der mindestens ein Ringheteroatom aus der Gruppe bestehend aus Stickstoff, Sauerstoff und Schwefel enthält, steht, wobei der Ring durch C1-C8-Alkylsulfonyl substituiert ist, eine Verbindung der Formel I, worin R3 für gegebenenfalls durch -NH-C(=O)-NH-R8 substituiertes C1-C8-Alkylaminocarbonyl steht, wobei R8 für einen 5- oder 6-gliedrigen heterocyclischen Ring, der mindestens ein Ringheteroatom aus der Gruppe bestehend aus Stickstoff, Sauerstoff und Schwefel enthält, steht, in Gegenwart einer Base mit einem Sulfonylierungsmittel umsetzt;(I) zur Herstellung von Verbindungen der Formel I, worin R3 für durch -NH-C (=O) -NH-R7 substituiertes R6 steht, wobei R7 die in Anspruch 1 angegebene Bedeutung besitzt, eine Verbindung der Formel XIIc
(H) zur Herstellung von Verbindungen der Formel I, worin R3 für gegebenenfalls durch -NH-C(=O)-NH-R8 substituiertes C1-C8-Alkylaminocarbonyl steht, wobei R8 für einen 5-oder 6-gliedrigen heterocyclischen Ring, der mindestens ein Ringheteroatom aus der Gruppe bestehend aus Stickstoff, Sauerstoff und Schwefel enthält, steht, wobei der Ring durch C1-C8-Alkylsulfonyl substituiert ist, eine Verbindung der Formel I, worin R3 für gegebenenfalls durch -NH-C(=O)-NH-R8 substituiertes C1-C8-Alkylaminocarbonyl steht, wobei R8 für einen 5- oder 6-gliedrigen heterocyclischen Ring, der mindestens ein Ringheteroatom aus der Gruppe bestehend aus Stickstoff, Sauerstoff und Schwefel enthält, steht, in Gegenwart einer Base mit einem Sulfonylierungsmittel umsetzt;(I) zur Herstellung von Verbindungen der Formel I, worin R3 für durch -NH-C (=O) -NH-R7 substituiertes R6 steht, wobei R7 die in Anspruch 1 angegebene Bedeutung besitzt, eine Verbindung der Formel XIIc

O=C=N-R7 XIa
worin T für C6-C10-Aryloxy oder einen 5- oder 6-gliedrigen heterocyclischen Ring, der mindestens ein Ringheteroatom aus der Gruppe bestehend aus Stickstoff, Sauerstoff und Schwefel enthält, steht und R8 die in Anspruch 1 angegebene Bedeutung besitzt, umsetzt;(J) zur Herstellung von Verbindungen der Formel I, worin R3 für durch -NH-C (=O) -NH-R7 substituiertes R6 steht, wobei R7 die in Anspruch 1 angegebene Bedeutung besitzt, eine Verbindung der Formel XIId oder XIIe oder eine geschützte Form davon

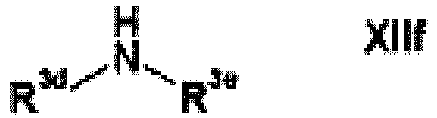 (K) zur Herstellung von Verbindungen der Formel I, wobei R3 für durch -NH-C(=O)-NH-R7 substituiertes R6 steht, wobei R7 die oben angegebene Bedeutung besitzt, eine Verbindung der Formel XIId oder XIIe, wobei R1, R2 und R6 die in Anspruch 1 angegebene Bedeutung besitzen und T für C6-C10-Aryloxy oder einen 5- oder 6-gliedrigen heterocyclischen Ring, der mindestens ein Ringheteroatom aus der Gruppe bestehend aus Stickstoff, Sauerstoff und Schwefel enthält, steht, mit einer Verbindung der Formel XIIg
(K) zur Herstellung von Verbindungen der Formel I, wobei R3 für durch -NH-C(=O)-NH-R7 substituiertes R6 steht, wobei R7 die oben angegebene Bedeutung besitzt, eine Verbindung der Formel XIId oder XIIe, wobei R1, R2 und R6 die in Anspruch 1 angegebene Bedeutung besitzen und T für C6-C10-Aryloxy oder einen 5- oder 6-gliedrigen heterocyclischen Ring, der mindestens ein Ringheteroatom aus der Gruppe bestehend aus Stickstoff, Sauerstoff und Schwefel enthält, steht, mit einer Verbindung der Formel XIIg
H2N-R7 XIIg
wobei R7 die in Anspruch 1 angegebene Bedeutung besitzt, umsetzt; und(ii) jegliche Schutzgruppen abspaltet und die erhaltene Verbindung der Formel Ia in freier Form oder Salzform gewinnt.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP10178514A EP2292619A1 (de) | 2004-10-22 | 2005-10-21 | Purinderivate und ihre Verwendung als Adenosin A-2A Rezeptor Agonisten |
Applications Claiming Priority (3)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GB0423551A GB0423551D0 (en) | 2004-10-22 | 2004-10-22 | Organic compounds |
| GB0514619A GB0514619D0 (en) | 2005-07-15 | 2005-07-15 | Organic compounds |
| PCT/EP2005/011344 WO2006045552A1 (en) | 2004-10-22 | 2005-10-21 | Purine derivatives for use as adenosin a-2a receptor agonists |
Related Child Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP10178514A Division-Into EP2292619A1 (de) | 2004-10-22 | 2005-10-21 | Purinderivate und ihre Verwendung als Adenosin A-2A Rezeptor Agonisten |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| EP1805181A1 EP1805181A1 (de) | 2007-07-11 |
| EP1805181B1 true EP1805181B1 (de) | 2012-08-29 |
Family
ID=35589070
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP05797944A Expired - Lifetime EP1805181B1 (de) | 2004-10-22 | 2005-10-21 | Purinderivate zur verwendung als agonisten des adenosin-a-2a-rezeptors |
| EP10178514A Withdrawn EP2292619A1 (de) | 2004-10-22 | 2005-10-21 | Purinderivate und ihre Verwendung als Adenosin A-2A Rezeptor Agonisten |
Family Applications After (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| EP10178514A Withdrawn EP2292619A1 (de) | 2004-10-22 | 2005-10-21 | Purinderivate und ihre Verwendung als Adenosin A-2A Rezeptor Agonisten |
Country Status (21)
| Country | Link |
|---|---|
| US (1) | US8163754B2 (de) |
| EP (2) | EP1805181B1 (de) |
| JP (1) | JP4904279B2 (de) |
| KR (1) | KR20070073798A (de) |
| AR (1) | AR051642A1 (de) |
| AU (2) | AU2005298878B2 (de) |
| BR (1) | BRPI0517455A (de) |
| CA (1) | CA2582434A1 (de) |
| EC (1) | ECSP077404A (de) |
| ES (1) | ES2394453T3 (de) |
| GT (1) | GT200500281A (de) |
| IL (1) | IL182528A0 (de) |
| MA (1) | MA28931B1 (de) |
| MX (1) | MX2007004735A (de) |
| NO (1) | NO20072122L (de) |
| NZ (1) | NZ554546A (de) |
| PE (2) | PE20061129A1 (de) |
| RU (1) | RU2403253C2 (de) |
| TN (1) | TNSN07154A1 (de) |
| TW (1) | TW200630366A (de) |
| WO (1) | WO2006045552A1 (de) |
Families Citing this family (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GT200500281A (es) | 2004-10-22 | 2006-04-24 | Novartis Ag | Compuestos organicos. |
| GB0500785D0 (en) * | 2005-01-14 | 2005-02-23 | Novartis Ag | Organic compounds |
| GB0607954D0 (en) * | 2006-04-21 | 2006-05-31 | Novartis Ag | Organic compounds |
| GB0607950D0 (en) * | 2006-04-21 | 2006-05-31 | Novartis Ag | Organic compounds |
| GB0607945D0 (en) * | 2006-04-21 | 2006-05-31 | Novartis Ag | Organic compounds |
| EP2322525B1 (de) | 2006-04-21 | 2013-09-18 | Novartis AG | Purinderivate als Adenosin A2A Rezeptoragonisten |
| GB0607953D0 (en) * | 2006-04-21 | 2006-05-31 | Novartis Ag | Organic compounds |
| GB0607944D0 (en) * | 2006-04-21 | 2006-05-31 | Novartis Ag | Organic compounds |
| GB0607948D0 (en) * | 2006-04-21 | 2006-05-31 | Novartis Ag | Organic compounds |
| EP1889846A1 (de) * | 2006-07-13 | 2008-02-20 | Novartis AG | Purinderivate als A2a Agonisten |
| EP1903044A1 (de) | 2006-09-14 | 2008-03-26 | Novartis AG | Adenosinderivate als Agonisten des A2A-Rezeptors |
| CA2669108A1 (en) * | 2006-11-10 | 2008-05-15 | Novartis Ag | Cyclopentene diol monoacetate derivatives |
| TW200837055A (en) * | 2007-03-13 | 2008-09-16 | Arete Therapeutics Inc | Soluble epoxide hydrolase inhibitors |
| WO2009050199A1 (en) * | 2007-10-17 | 2009-04-23 | Novartis Ag | Purine derivatives as adenosine al receptor ligands |
| US20090181934A1 (en) * | 2007-10-17 | 2009-07-16 | Novartis Ag | Organic Compounds |
| EA201270102A1 (ru) | 2009-06-30 | 2012-08-30 | Форест Лэборетериз Холдингз Лимитед | Соединения алкокси-карбонил-амино-алкинил-аденозина и их производные в качестве агонистов ar |
| EP3039126B1 (de) | 2013-08-26 | 2019-10-09 | The J. David Gladstone Institutes, A Testamentary Trust Established under The Will of J. David Gladstone | Kleinmolekülige zelluläre reprogrammierung zur erzeugung neuronaler zellen |
Family Cites Families (194)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GB1219606A (en) | 1968-07-15 | 1971-01-20 | Rech S Et D Applic Scient Soge | Quinuclidinol derivatives and preparation thereof |
| GB1528382A (en) | 1974-12-26 | 1978-10-11 | Teijin Ltd | Cyclopentene diols and acyl esters thereof and processes for their preparation |
| JPS5481295A (en) * | 1977-12-13 | 1979-06-28 | Suami T | Adenosin analogue compounds |
| FR2459240A1 (fr) | 1979-06-21 | 1981-01-09 | Cm Ind | Aminopiperidines anorexigenes, procede pour leur preparation, intermediaires dans ledit procede et medicaments qui les contiennent |
| EP0147716A3 (de) | 1983-12-24 | 1987-10-28 | ANT Nachrichtentechnik GmbH | Verfahren und Anordnung zur verschlüsselbaren Übertragung einer Nachrichten-Binärzeichenfolge mit Authentizitätsprüfung |
| JPS6235216A (ja) | 1985-08-09 | 1987-02-16 | Noritoshi Nakabachi | 不均質物質層の層厚非破壊測定方法および装置 |
| US4738954A (en) | 1985-11-06 | 1988-04-19 | Warner-Lambert Company | Novel N6 -substituted-5'-oxidized adenosine analogs |
| US4873360A (en) | 1986-07-10 | 1989-10-10 | Board Of Governors Of Wayne State University | Process for the preparation of cyclopentanoids and novel intermediates produced thereby |
| IL84414A0 (en) | 1986-11-14 | 1988-04-29 | Ciba Geigy Ag | N9-cyclopentyl-substituted adenine derivatives |
| US4954504A (en) | 1986-11-14 | 1990-09-04 | Ciba-Geigy Corporation | N9 -cyclopentyl-substituted adenine derivatives having adenosine-2 receptor stimulating activity |
| JP2586897B2 (ja) | 1987-03-09 | 1997-03-05 | 富士薬品工業株式会社 | 光学活性なシス−シクロペンテン−3,5−ジオ−ルモノエステルの製造法 |
| GB8923590D0 (en) | 1989-10-19 | 1989-12-06 | Pfizer Ltd | Antimuscarinic bronchodilators |
| US5561134A (en) * | 1990-09-25 | 1996-10-01 | Rhone-Poulenc Rorer Pharmaceuticals Inc. | Compounds having antihypertensive, cardioprotective, anti-ischemic and antilipolytic properties |
| SG80526A1 (en) | 1990-09-25 | 2001-05-22 | Rhone Poulenc Rorer Int | Compounds having antihypertensive and anti- ischemic properies |
| PT100441A (pt) | 1991-05-02 | 1993-09-30 | Smithkline Beecham Corp | Pirrolidinonas, seu processo de preparacao, composicoes farmaceuticas que as contem e uso |
| US5451700A (en) | 1991-06-11 | 1995-09-19 | Ciba-Geigy Corporation | Amidino compounds, their manufacture and methods of treatment |
| WO1993018007A1 (fr) | 1992-03-13 | 1993-09-16 | Tokyo Tanabe Company Limited | Nouveau derive de carbostyrile |
| JP3192424B2 (ja) | 1992-04-02 | 2001-07-30 | スミスクライン・ビーチャム・コーポレイション | アレルギーまたは炎症疾患の治療用化合物 |
| EP0636026B1 (de) | 1992-04-02 | 2001-12-05 | Smithkline Beecham Corporation | Verbindungen zur behandlung von entzündlichen erkrankungen und zur hemmung der produktion von tumornekrosefaktor |
| HU225869B1 (en) | 1992-04-02 | 2007-11-28 | Smithkline Beecham Corp | Compounds with antiallergic and antiinflammatory activity and pharmaceutical compns. contg. them |
| IT1254915B (it) | 1992-04-24 | 1995-10-11 | Gloria Cristalli | Derivati di adenosina ad attivita' a2 agonista |
| US5688774A (en) | 1993-07-13 | 1997-11-18 | The United States Of America As Represented By The Department Of Health And Human Services | A3 adenosine receptor agonists |
| US5691188A (en) | 1994-02-14 | 1997-11-25 | American Cyanamid Company | Transformed yeast cells expressing heterologous G-protein coupled receptor |
| US6143749A (en) * | 1995-06-07 | 2000-11-07 | Abbott Laboratories | Heterocyclic substituted cyclopentane compounds |
| EA000884B1 (ru) | 1996-01-02 | 2000-06-26 | Рон-Пуленк Рорер Фармасьютикалз Инк. | Способ получения 2,4-дигидроксипиридина и 2,4-дигидрокси-3-нитропиридина |
| US6376472B1 (en) | 1996-07-08 | 2002-04-23 | Aventis Pharmaceuticals, Inc. | Compounds having antihypertensive, cardioprotective, anti-ischemic and antilipolytic properties |
| GB9622386D0 (en) | 1996-10-28 | 1997-01-08 | Sandoz Ltd | Organic compounds |
| AU750322B2 (en) | 1997-05-09 | 2002-07-18 | Trustees Of The University Of Pennsylvania, The | Methods and compositions for reducing ischemic injury of the heart by administering adenosine receptor agonists and antagonists |
| US6166037A (en) | 1997-08-28 | 2000-12-26 | Merck & Co., Inc. | Pyrrolidine and piperidine modulators of chemokine receptor activity |
| AU9281298A (en) | 1997-10-01 | 1999-04-23 | Kyowa Hakko Kogyo Co. Ltd. | Benzodioxole derivatives |
| US6541669B1 (en) | 1998-06-08 | 2003-04-01 | Theravance, Inc. | β2-adrenergic receptor agonists |
| FR2780057B1 (fr) | 1998-06-18 | 2002-09-13 | Sanofi Sa | Phenoxypropanolamines, procede pour leur preparation et compositions pharmaceutiques les contenant |
| GB9813535D0 (en) * | 1998-06-23 | 1998-08-19 | Glaxo Group Ltd | Chemical compounds |
| GB9813554D0 (en) | 1998-06-23 | 1998-08-19 | Glaxo Group Ltd | Chemical compounds |
| AU750462B2 (en) | 1998-06-23 | 2002-07-18 | Glaxo Group Limited | 2-(purin-9-yl)-tetrahydrofuran-3,4-diol derivatives |
| GB9813540D0 (en) | 1998-06-23 | 1998-08-19 | Glaxo Group Ltd | Chemical compounds |
| KR100563395B1 (ko) | 1998-06-30 | 2006-03-23 | 다우 글로벌 테크놀로지스 인크. | 중합체 폴리올 및 이의 제조방법 |
| AU5879299A (en) | 1998-10-16 | 2000-05-08 | Monaghan, Sandra Marina | Adenine derivatives |
| CN1383427A (zh) | 1998-12-31 | 2002-12-04 | 阿温蒂斯药物公司 | 制备n6-取代的脱氮杂一腺苷衍生物的方法 |
| US7427606B2 (en) | 1999-02-01 | 2008-09-23 | University Of Virginia Patent Foundation | Method to reduce inflammatory response in transplanted tissue |
| GB9913083D0 (en) | 1999-06-04 | 1999-08-04 | Novartis Ag | Organic compounds |
| CA2371583C (en) | 1999-05-04 | 2005-09-13 | Schering Corporation | Piperazine derivatives useful as ccr5 antagonists |
| HK1039330B (en) | 1999-05-04 | 2005-12-09 | Schering Corporation | Piperidine derivatives useful as ccr5 antagonists |
| US6683115B2 (en) | 1999-06-02 | 2004-01-27 | Theravance, Inc. | β2-adrenergic receptor agonists |
| US6322771B1 (en) | 1999-06-18 | 2001-11-27 | University Of Virginia Patent Foundation | Induction of pharmacological stress with adenosine receptor agonists |
| US6403567B1 (en) | 1999-06-22 | 2002-06-11 | Cv Therapeutics, Inc. | N-pyrazole A2A adenosine receptor agonists |
| US6214807B1 (en) | 1999-06-22 | 2001-04-10 | Cv Therapeutics, Inc. | C-pyrazole 2A A receptor agonists |
| US6841549B1 (en) * | 1999-07-02 | 2005-01-11 | Eisai Co., Ltd. | Condensed imidazole compounds and a therapeutic agent for diabetes mellitus |
| ES2165768B1 (es) | 1999-07-14 | 2003-04-01 | Almirall Prodesfarma Sa | Nuevos derivados de quinuclidina y composiciones farmaceuticas que los contienen. |
| EP1212089B1 (de) | 1999-08-21 | 2006-03-22 | ALTANA Pharma AG | Synergistische kombination von roflumilast und salmeterol |
| US6586413B2 (en) | 1999-11-05 | 2003-07-01 | The United States Of America As Represented By The Department Of Health And Human Services | Methods and compositions for reducing ischemic injury of the heart by administering adenosine receptor agonists and antagonists |
| OA11558A (en) | 1999-12-08 | 2004-06-03 | Advanced Medicine Inc | Beta 2-adrenergic receptor agonists. |
| GB0003960D0 (en) | 2000-02-18 | 2000-04-12 | Pfizer Ltd | Purine derivatives |
| BR0110331A (pt) | 2000-04-27 | 2003-01-07 | Boehringer Ingelheim Pharma | Substâncias que mimetizam beta que têm uma atividade de longa duração, processos para prepará-las e o uso das mesmas como medicamentos |
| TWI227240B (en) * | 2000-06-06 | 2005-02-01 | Pfizer | 2-aminocarbonyl-9H-purine derivatives |
| CZ294251B6 (cs) | 2000-06-27 | 2004-11-10 | Laboratorios S. A. L. V. A. T., S. A. | Karbamáty a jejich použití pro výrobu farmaceutického prostředku |
| GB0015876D0 (en) | 2000-06-28 | 2000-08-23 | Novartis Ag | Organic compounds |
| DE10038639A1 (de) | 2000-07-28 | 2002-02-21 | Schering Ag | Nichtsteroidale Entzündungshemmer |
| WO2002012266A1 (en) | 2000-08-05 | 2002-02-14 | Glaxo Group Limited | 17.beta.-carbothioate 17.alpha.-arylcarbonyloxyloxy androstane derivative as anti-inflammatory agents |
| GB0022695D0 (en) | 2000-09-15 | 2000-11-01 | Pfizer Ltd | Purine Derivatives |
| GB0028383D0 (en) | 2000-11-21 | 2001-01-03 | Novartis Ag | Organic compounds |
| JP4445704B2 (ja) | 2000-12-22 | 2010-04-07 | アルミラル・ソシエダッド・アノニマ | キヌクリジンカルバメート誘導体およびm3アンダゴニストとしてのそれらの使用 |
| IL156558A0 (en) | 2000-12-28 | 2004-01-04 | Almirall Prodesfarma Ag | Novel quinuclidine derivatives and medicinal compositions containing the same |
| JP4012070B2 (ja) | 2001-01-16 | 2007-11-21 | カン−フィテ・バイオファーマ・リミテッド | ウイルスの複製を阻害するためのアデノシンa3受容体アゴニストの使用 |
| GB0103630D0 (en) | 2001-02-14 | 2001-03-28 | Glaxo Group Ltd | Chemical compounds |
| GB2372741A (en) | 2001-03-03 | 2002-09-04 | Univ Leiden | C2,8-Disubstituted adenosine derivatives and their different uses |
| EP1366025B1 (de) | 2001-03-08 | 2007-06-27 | Glaxo Group Limited | Agonisten von beta-adrenorezeptoren |
| US20040162422A1 (en) * | 2001-03-20 | 2004-08-19 | Adrian Hall | Chemical compounds |
| JP4143413B2 (ja) | 2001-03-22 | 2008-09-03 | グラクソ グループ リミテッド | β2−アドレナリン受容体アゴニストとしてのホルムアニリド誘導体 |
| PT1383786E (pt) | 2001-04-30 | 2008-12-30 | Glaxo Group Ltd | Derivados anti-inflamatórios de éster 17.beta-carbotioato de androstano com um grupo éster ciclíco na posição 17.alfa |
| EP1258247A1 (de) | 2001-05-14 | 2002-11-20 | Aventis Pharma Deutschland GmbH | Adenosin-Analoga für die Behandlung Von Insulinresistenz und Diabetes |
| US20040248867A1 (en) | 2001-06-12 | 2004-12-09 | Keith Biggadike | Novel anti-inflammatory 17.alpha.-heterocyclic-esters of 17.beta.carbothioate androstane derivatives |
| EP2319919B1 (de) | 2001-06-21 | 2015-08-12 | BASF Enzymes LLC | Nitralasen |
| ES2438985T3 (es) | 2001-09-14 | 2014-01-21 | Glaxo Group Limited | Formulación de inhalación que comprende derivados de fenetanolamina para el tratamiento de enfermedades respiratorias |
| EP1434782A2 (de) | 2001-10-01 | 2004-07-07 | University of Virginia Patent Foundation | 2-propynyladenosine-analoge mit a2a-agonistischer aktivität and diese enthaltende gemische |
| JP2005512974A (ja) | 2001-10-17 | 2005-05-12 | ユ セ ベ ソシエテ アノニム | キヌクリジン誘導体、その調製方法、及びm2及び/又はm3ムスカリン受容体阻害剤としてのその使用 |
| GB0125259D0 (en) | 2001-10-20 | 2001-12-12 | Glaxo Group Ltd | Novel compounds |
| AR037517A1 (es) | 2001-11-05 | 2004-11-17 | Novartis Ag | Derivados de naftiridinas, un proceso para su preparacion, composicion farmaceutica y el uso de los mismos para la preparacion de un medicamento para el tratamiento de una enfermedad inflamatoria |
| US6653323B2 (en) | 2001-11-13 | 2003-11-25 | Theravance, Inc. | Aryl aniline β2 adrenergic receptor agonists |
| TWI249515B (en) | 2001-11-13 | 2006-02-21 | Theravance Inc | Aryl aniline beta2 adrenergic receptor agonists |
| WO2003048181A1 (en) | 2001-12-01 | 2003-06-12 | Glaxo Group Limited | 17.alpha. -cyclic esters of 16-methylpregnan-3,20-dione as anti-inflammatory agents |
| EP1461336B1 (de) | 2001-12-20 | 2013-05-22 | CHIESI FARMACEUTICI S.p.A. | 1-alkyl-1-azoniabicyclo (2.2.2) octan carbamatderivate und deren verwendung als muscarinische rezeptor antagonisten |
| WO2003072592A1 (en) | 2002-01-15 | 2003-09-04 | Glaxo Group Limited | 17.alpha-cycloalkyl/cycloylkenyl esters of alkyl-or haloalkyl-androst-4-en-3-on-11.beta.,17.alpha.-diol 17.beta.-carboxylates as anti-inflammatory agents |
| WO2003062259A2 (en) | 2002-01-21 | 2003-07-31 | Glaxo Group Limited | Non-aromatic 17.alpha.-esters of androstane-17.beta.-carboxylate esters as anti-inflammatory agents |
| US7414036B2 (en) | 2002-01-25 | 2008-08-19 | Muscagen Limited | Compounds useful as A3 adenosine receptor agonists |
| GB0202216D0 (en) | 2002-01-31 | 2002-03-20 | Glaxo Group Ltd | Novel compounds |
| GB0204719D0 (en) | 2002-02-28 | 2002-04-17 | Glaxo Group Ltd | Medicinal compounds |
| CA2477764A1 (en) | 2002-03-26 | 2003-10-09 | Boehringer Ingelheim Pharmaceuticals, Inc. | Glucocorticoid mimetics, methods of making them, pharmaceutical compositions, and uses thereof |
| UA80120C2 (en) | 2002-03-26 | 2007-08-27 | Boehringer Ingelheim Pharma | Glucocorticoid mimetics, pharmaceutical composition based thereon |
| AU2003234716A1 (en) | 2002-04-10 | 2003-10-27 | Joel M. Linden | Use of a2a adenosine receptor agonists for the treatment of inflammatory diseases |
| DE60335869D1 (de) | 2002-04-11 | 2011-03-10 | Merck Sharp & Dohme | 1h-benzo(f)indazol-5-yl-derivate als selektive glucocorticoid-rezeptor-modulatoren |
| ES2206021B1 (es) | 2002-04-16 | 2005-08-01 | Almirall Prodesfarma, S.A. | Nuevos derivados de pirrolidinio. |
| AU2003222841A1 (en) | 2002-04-25 | 2003-11-10 | Glaxo Group Limited | Phenethanolamine derivatives |
| EP1507754A1 (de) | 2002-05-28 | 2005-02-23 | Theravance, Inc. | Alkoxy aryl beta-2 adrenergen rezeptoragonisten |
| US7186864B2 (en) | 2002-05-29 | 2007-03-06 | Boehringer Ingelheim Pharmaceuticals, Inc. | Glucocorticoid mimetics, methods of making them, pharmaceutical compositions, and uses thereof |
| ES2201907B1 (es) | 2002-05-29 | 2005-06-01 | Almirall Prodesfarma, S.A. | Nuevos derivados de indolilpiperidina como potentes agentes antihistaminicos y antialergicos. |
| DE10224888A1 (de) | 2002-06-05 | 2003-12-24 | Merck Patent Gmbh | Pyridazinderivate |
| US7074806B2 (en) | 2002-06-06 | 2006-07-11 | Boehringer Ingelheim Pharmaceuticals, Inc. | Glucocorticoid mimetics, methods of making them, pharmaceutical compositions, and uses thereof |
| DE10225574A1 (de) | 2002-06-10 | 2003-12-18 | Merck Patent Gmbh | Aryloxime |
| DE10227269A1 (de) | 2002-06-19 | 2004-01-08 | Merck Patent Gmbh | Thiazolderivate |
| EP1517895B1 (de) | 2002-06-25 | 2007-03-14 | Merck Frosst Canada Ltd. | 8-(biaryl)chinolin-pde4-inhibitoren |
| ES2204295B1 (es) | 2002-07-02 | 2005-08-01 | Almirall Prodesfarma, S.A. | Nuevos derivados de quinuclidina-amida. |
| EP1519922A1 (de) | 2002-07-02 | 2005-04-06 | Merck Frosst Canada & Co. | Diaryl-substituierte ethanpyridone als pde4 inhibitoren |
| SI1521733T1 (sl) | 2002-07-08 | 2014-10-30 | Pfizer Products Inc. | Modulatorji glukokortikoidnega receptorja |
| GB0217225D0 (en) | 2002-07-25 | 2002-09-04 | Glaxo Group Ltd | Medicinal compounds |
| PE20050130A1 (es) | 2002-08-09 | 2005-03-29 | Novartis Ag | Compuestos organicos |
| WO2004018451A1 (en) | 2002-08-10 | 2004-03-04 | Altana Pharma Ag | Pyridazinone-derivatives as pde4 inhibitors |
| AU2003255376A1 (en) | 2002-08-10 | 2004-03-11 | Altana Pharma Ag | Piperidine-derivatives as pde4 inhibitors |
| CA2494613C (en) | 2002-08-10 | 2011-06-28 | Altana Pharma Ag | Pyrrolidinedione substituted piperidine-phthalazones as pde4 inhibitors |
| WO2004018450A1 (en) | 2002-08-10 | 2004-03-04 | Altana Pharma Ag | Piperidine-n-oxide-derivatives |
| AU2003253408A1 (en) | 2002-08-17 | 2004-03-11 | Nycomed Gmbh | Novel phenanthridines |
| EP1581533A2 (de) | 2002-08-17 | 2005-10-05 | ALTANA Pharma AG | Benzonaphthyridine mit pde 3/4 hemmender wirkung |
| SE0202483D0 (sv) | 2002-08-21 | 2002-08-21 | Astrazeneca Ab | Chemical compounds |
| CA2496175A1 (en) | 2002-08-21 | 2004-03-04 | Boehringer Ingelheim Pharmaceuticals, Inc. | Substituted hihydroquinolines as glucocorticoid mimetics, methods of making them, pharmaceutical compositions, and uses thereof |
| JP2006501236A (ja) | 2002-08-23 | 2006-01-12 | ランバクシー ラボラトリーズ リミテッド | ムスカリン性レセプター拮抗薬としてのフルオロおよびスルホニルアミノ含有3,6−二置換アザビシクロ(3.1.0)ヘキサン誘導体 |
| CA2495827C (en) | 2002-08-29 | 2012-05-08 | Altana Pharma Ag | 2-hydroxy-6-phenylphenanthridines as pde-4 inhibitors |
| EA011095B1 (ru) | 2002-08-29 | 2008-12-30 | Бёрингер Ингельхайм Фармасьютиклз, Инк. | Производные 3-(сульфонамидоэтил)индола, предназначенные для использования в качестве миметиков глюкокортикоидов при лечении воспалительных, аллергических и пролиферативных заболеваний |
| ES2281658T3 (es) | 2002-08-29 | 2007-10-01 | Nycomed Gmbh | 3-hidroxi-6-fenilfenantridinas como inhibidores de pde-4. |
| GB0220730D0 (en) | 2002-09-06 | 2002-10-16 | Glaxo Group Ltd | Medicinal compounds |
| JP2006096662A (ja) | 2002-09-18 | 2006-04-13 | Sumitomo Pharmaceut Co Ltd | 新規6−置換ウラシル誘導体及びアレルギー性疾患の治療剤 |
| EP1541574A4 (de) | 2002-09-18 | 2007-06-20 | Ono Pharmaceutical Co | Triazaspiro 5.5 undecanderivate und arzneimittel, die diese als wirkstoff enthalten |
| JP2004107299A (ja) | 2002-09-20 | 2004-04-08 | Japan Energy Corp | 新規1−置換ウラシル誘導体及びアレルギー性疾患の治療剤 |
| EP1542996A4 (de) | 2002-09-20 | 2009-11-18 | Merck & Co Inc | OCTAHYDRO-2H-NAPHTOç1,2-F INDOL-4-CARBONSûUREAMIDDERIVATE ALS SELEKTIVE MODULATOREN DES GLUCOCORTICOIDREZEPTORS |
| DE10246374A1 (de) | 2002-10-04 | 2004-04-15 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | Neue Betamimetika mit verlängerter Wirkungsdauer, Verfahren zu deren Herstellung und deren Verwendung als Arzneimittel |
| EP1440966A1 (de) | 2003-01-10 | 2004-07-28 | Pfizer Limited | Zur Behandlung von Krankheiten geeignete Indolderivate |
| EP1556034B1 (de) | 2002-10-11 | 2008-04-16 | Pfizer Limited | Indolderivate als beta-2 agonisten |
| WO2004037807A2 (en) | 2002-10-22 | 2004-05-06 | Glaxo Group Limited | Medicinal arylethanolamine compounds |
| EA010408B1 (ru) | 2002-10-23 | 2008-08-29 | Гленмарк Фармасьютикалс Лтд. | Трициклические соединения для лечения воспалительных и аллергических нарушений, способы их приготовления и содержащие их фармацевтические составы |
| ATE390407T1 (de) | 2002-10-28 | 2008-04-15 | Glaxo Group Ltd | Phenethanolamin-derivate zur behandlung von atemwegserkrankungen |
| GB0225030D0 (en) | 2002-10-28 | 2002-12-04 | Glaxo Group Ltd | Medicinal compounds |
| GB0225287D0 (en) | 2002-10-30 | 2002-12-11 | Glaxo Group Ltd | Novel compounds |
| GB0225535D0 (en) | 2002-11-01 | 2002-12-11 | Glaxo Group Ltd | Medicinal compounds |
| GB0225540D0 (en) | 2002-11-01 | 2002-12-11 | Glaxo Group Ltd | Medicinal compounds |
| DE10253220A1 (de) | 2002-11-15 | 2004-05-27 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | Neue Dihydroxy-Methyl-Phenyl-Derivate, Verfahren zu deren Herstellung und deren Verwendung als Arzneimittel |
| DE10253282A1 (de) | 2002-11-15 | 2004-05-27 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | Neue Arzneimittel zur Behandlung von chronisch obstruktiver Lungenerkrankung |
| DE10253426B4 (de) | 2002-11-15 | 2005-09-22 | Elbion Ag | Neue Hydroxyindole, deren Verwendung als Inhibitoren der Phosphodiesterase 4 und Verfahren zu deren Herstellung |
| DE10261874A1 (de) | 2002-12-20 | 2004-07-08 | Schering Ag | Nichtsteroidale Entzündungshemmer |
| WO2004066920A2 (en) | 2003-01-21 | 2004-08-12 | Merck & Co. Inc. | 17-carbamoyloxy cortisol derivatives as selective glucocorticoid receptor modulators |
| PE20040950A1 (es) | 2003-02-14 | 2005-01-01 | Theravance Inc | DERIVADOS DE BIFENILO COMO AGONISTAS DE LOS RECEPTORES ADRENERGICOS ß2 Y COMO ANTAGONISTAS DE LOS RECEPTORES MUSCARINICOS |
| EP1460064A1 (de) | 2003-03-14 | 2004-09-22 | Pfizer Limited | Indole-2-carboxamide als beta-2 Agonisten |
| JP4767842B2 (ja) | 2003-04-01 | 2011-09-07 | セラヴァンス, インコーポレーテッド | β2アドレナリン作用性レセプターアゴニスト活性およびムスカリン性レセプターアンタゴニスト活性を有するジアリールメチル化合物および関連化合物 |
| WO2004087142A1 (en) | 2003-04-04 | 2004-10-14 | Novartis Ag | Quinoline-2-one-derivatives for the treatment of airways diseases |
| US7268147B2 (en) | 2003-05-15 | 2007-09-11 | Pfizer Inc | Compounds useful for the treatment of diseases |
| DE602004021921D1 (de) | 2003-05-28 | 2009-08-20 | Theravance Inc | Azabicycloalkanverbindungen als muscarinrezeptor antagonisten |
| GB0312832D0 (en) | 2003-06-04 | 2003-07-09 | Pfizer Ltd | 2-amino-pyridine derivatives useful for the treatment of diseases |
| DE602004011513T2 (de) | 2003-06-04 | 2009-01-29 | Pfizer Inc. | 2-amino-pyridin-derivate als beta-2 adrenoreceptor agonisten |
| DE10332239B3 (de) | 2003-07-16 | 2005-03-03 | Framatome Anp Gmbh | Zirkoniumlegierung und Bauteile für den Kern von leichtwassergekühlten Kernreaktoren |
| WO2005033121A2 (en) | 2003-10-03 | 2005-04-14 | King Pharmaceuticals Research & Development, Inc. | Synthesis of 2-aralkyloxyadenosines, 2-alkoxyadenosines, and their analogs |
| GB0323701D0 (en) | 2003-10-09 | 2003-11-12 | Glaxo Group Ltd | Formulations |
| GB0324654D0 (en) | 2003-10-22 | 2003-11-26 | Glaxo Group Ltd | Medicinal compounds |
| GB0324886D0 (en) | 2003-10-24 | 2003-11-26 | Glaxo Group Ltd | Medicinal compounds |
| GB0329182D0 (en) | 2003-12-17 | 2004-01-21 | Glaxo Group Ltd | Chemical compounds |
| US8506794B2 (en) | 2003-12-19 | 2013-08-13 | Shell Oil Company | Systems, methods, and catalysts for producing a crude product |
| DE602004006895T2 (de) | 2003-12-29 | 2008-01-31 | Can-Fite Biopharma Ltd. | Verfahren zur behandlung von multipler sklerose |
| DE102004001413A1 (de) | 2004-01-09 | 2005-08-18 | Boehringer Ingelheim Pharma Gmbh & Co. Kg | 3-Hydroxymethyl-4-Hydroxy-Phenyl-Derivate zur Behandlung von chronisch obstruktiver Lungenerkrankung |
| PT1708992E (pt) | 2004-01-22 | 2007-11-16 | Pfizer | Derivados de sulfonamida para o tratamento de doenças |
| EP1708991B1 (de) | 2004-01-22 | 2007-10-17 | Pfizer Limited | Sulfonamidderivate zur behandlung von krankheiten |
| US7320990B2 (en) | 2004-02-13 | 2008-01-22 | Theravance, Inc. | Crystalline form of a biphenyl compound |
| ATE479433T1 (de) | 2004-02-14 | 2010-09-15 | Boehringer Ingelheim Int | Neue langwirksame beta-2-agonisten, und deren verwendung als arzneimittel |
| CA2557285A1 (en) | 2004-03-05 | 2005-09-15 | Cambridge Biotechnology Limited | Adenosine receptor agonists |
| WO2005107463A1 (en) | 2004-05-03 | 2005-11-17 | University Of Virginia Patent Foundation | Agonists of a2a adenosine receptors for treatment of diabetic nephropathy |
| AR049384A1 (es) | 2004-05-24 | 2006-07-26 | Glaxo Group Ltd | Derivados de purina |
| EP1778239B1 (de) | 2004-07-28 | 2013-08-21 | Can-Fite Biopharma Ltd. | Adenosin-a3-rezeptoragonisten zur behandlung von trockenem auge, einschliesslich sjögren-syndrom |
| US7825102B2 (en) | 2004-07-28 | 2010-11-02 | Can-Fite Biopharma Ltd. | Treatment of dry eye conditions |
| JP2008512457A (ja) | 2004-09-09 | 2008-04-24 | アメリカ合衆国 | A3及びa1アデノシン受容体作用薬としてのプリン誘導体 |
| GT200500281A (es) | 2004-10-22 | 2006-04-24 | Novartis Ag | Compuestos organicos. |
| US20080051364A1 (en) | 2004-11-08 | 2008-02-28 | Pninna Fishman | Therapeutic Treatment of Accelerated Bone Resorption |
| GB0500785D0 (en) | 2005-01-14 | 2005-02-23 | Novartis Ag | Organic compounds |
| RS52458B (sr) * | 2005-02-04 | 2013-02-28 | Millennium Pharmaceuticals Inc. | Inhibitori e1 aktivirajućih enzima |
| GB0505219D0 (en) | 2005-03-14 | 2005-04-20 | Novartis Ag | Organic compounds |
| GB0514809D0 (en) | 2005-07-19 | 2005-08-24 | Glaxo Group Ltd | Compounds |
| JP5339916B2 (ja) | 2005-11-30 | 2013-11-13 | キャン−ファイト・バイオファーマ・リミテッド | 骨関節炎の治療におけるa3アデノシン受容体アゴニストの使用 |
| CN101410114B (zh) | 2006-01-26 | 2012-07-04 | 美国政府卫生与公共服务部 | A3腺苷受体别构调节剂 |
| EA024006B1 (ru) * | 2006-02-02 | 2016-08-31 | Миллениум Фармасьютикалз, Инк. | Ингибиторы е1 активирующих ферментов |
| WO2007092936A2 (en) | 2006-02-08 | 2007-08-16 | University Of Virginia Patent Foundation | Method to treat gastric lesions |
| GB0607944D0 (en) | 2006-04-21 | 2006-05-31 | Novartis Ag | Organic compounds |
| GB0607948D0 (en) | 2006-04-21 | 2006-05-31 | Novartis Ag | Organic compounds |
| GB0607954D0 (en) | 2006-04-21 | 2006-05-31 | Novartis Ag | Organic compounds |
| GB0607951D0 (en) | 2006-04-21 | 2006-05-31 | Novartis Ag | Organic compounds |
| GB0607953D0 (en) * | 2006-04-21 | 2006-05-31 | Novartis Ag | Organic compounds |
| GB0607950D0 (en) | 2006-04-21 | 2006-05-31 | Novartis Ag | Organic compounds |
| EP2322525B1 (de) | 2006-04-21 | 2013-09-18 | Novartis AG | Purinderivate als Adenosin A2A Rezeptoragonisten |
| GB0607945D0 (en) | 2006-04-21 | 2006-05-31 | Novartis Ag | Organic compounds |
| EP1889846A1 (de) | 2006-07-13 | 2008-02-20 | Novartis AG | Purinderivate als A2a Agonisten |
| US8008307B2 (en) * | 2006-08-08 | 2011-08-30 | Millennium Pharmaceuticals, Inc. | Heteroaryl compounds useful as inhibitors of E1 activating enzymes |
| EP1903044A1 (de) | 2006-09-14 | 2008-03-26 | Novartis AG | Adenosinderivate als Agonisten des A2A-Rezeptors |
| CA2669108A1 (en) | 2006-11-10 | 2008-05-15 | Novartis Ag | Cyclopentene diol monoacetate derivatives |
| WO2008124150A1 (en) | 2007-04-09 | 2008-10-16 | University Of Virginia Patent Foundation | Method of treating enteritis, intestinal damage, and diarrhea from c. difficile with an a2a adenosine receptor agonist |
| US20080262001A1 (en) | 2007-04-23 | 2008-10-23 | Adenosine Therapeutics, Llc | Agonists of a2a adenosine receptors for treating recurrent tumor growth in the liver following resection |
| EP2170401A1 (de) | 2007-06-29 | 2010-04-07 | Government of the United States of America, Represented by the Secretary, Department of Health and Human Services | Dendrimer-konjugate von agonisten und antagonisten der gpcr-superfamilie |
| US20090181934A1 (en) * | 2007-10-17 | 2009-07-16 | Novartis Ag | Organic Compounds |
| WO2009050199A1 (en) | 2007-10-17 | 2009-04-23 | Novartis Ag | Purine derivatives as adenosine al receptor ligands |
| WO2009061516A1 (en) | 2007-11-08 | 2009-05-14 | New York University School Of Medicine | Medical implants containing adenosine receptor agonists and methods for inhibiting medical implant loosening |
| JP2011509305A (ja) | 2008-01-09 | 2011-03-24 | ピージーエックスヘルス、リミテッド、ライアビリティー、カンパニー | A2arアゴニストによる神経障害性疼痛の髄腔内治療 |
-
2005
- 2005-10-07 GT GT200500281A patent/GT200500281A/es unknown
- 2005-10-20 PE PE2005001232A patent/PE20061129A1/es not_active Application Discontinuation
- 2005-10-20 AR ARP050104383A patent/AR051642A1/es not_active Application Discontinuation
- 2005-10-20 PE PE2010000097A patent/PE20100266A1/es not_active Application Discontinuation
- 2005-10-21 MX MX2007004735A patent/MX2007004735A/es active IP Right Grant
- 2005-10-21 WO PCT/EP2005/011344 patent/WO2006045552A1/en not_active Ceased
- 2005-10-21 RU RU2007118729/04A patent/RU2403253C2/ru not_active IP Right Cessation
- 2005-10-21 CA CA002582434A patent/CA2582434A1/en not_active Abandoned
- 2005-10-21 EP EP05797944A patent/EP1805181B1/de not_active Expired - Lifetime
- 2005-10-21 AU AU2005298878A patent/AU2005298878B2/en not_active Ceased
- 2005-10-21 EP EP10178514A patent/EP2292619A1/de not_active Withdrawn
- 2005-10-21 KR KR1020077008995A patent/KR20070073798A/ko not_active Ceased
- 2005-10-21 BR BRPI0517455-4A patent/BRPI0517455A/pt not_active IP Right Cessation
- 2005-10-21 NZ NZ554546A patent/NZ554546A/en not_active IP Right Cessation
- 2005-10-21 TW TW094136805A patent/TW200630366A/zh unknown
- 2005-10-21 US US11/576,607 patent/US8163754B2/en not_active Expired - Fee Related
- 2005-10-21 JP JP2007537224A patent/JP4904279B2/ja not_active Expired - Fee Related
- 2005-10-21 ES ES05797944T patent/ES2394453T3/es not_active Expired - Lifetime
-
2007
- 2007-04-12 MA MA29825A patent/MA28931B1/fr unknown
- 2007-04-12 IL IL182528A patent/IL182528A0/en unknown
- 2007-04-19 EC EC2007007404A patent/ECSP077404A/es unknown
- 2007-04-20 TN TNP2007000154A patent/TNSN07154A1/fr unknown
- 2007-04-24 NO NO20072122A patent/NO20072122L/no not_active Application Discontinuation
-
2009
- 2009-10-30 AU AU2009233613A patent/AU2009233613A1/en not_active Abandoned
Also Published As
| Publication number | Publication date |
|---|---|
| BRPI0517455A (pt) | 2008-10-07 |
| AU2005298878B2 (en) | 2009-07-30 |
| NO20072122L (no) | 2007-07-20 |
| JP4904279B2 (ja) | 2012-03-28 |
| AU2005298878A1 (en) | 2006-05-04 |
| WO2006045552A1 (en) | 2006-05-04 |
| RU2007118729A (ru) | 2008-11-27 |
| TW200630366A (en) | 2006-09-01 |
| MA28931B1 (fr) | 2007-10-01 |
| TNSN07154A1 (en) | 2008-11-21 |
| AU2009233613A1 (en) | 2009-11-26 |
| NZ554546A (en) | 2010-10-29 |
| PE20100266A1 (es) | 2010-04-21 |
| JP2008517027A (ja) | 2008-05-22 |
| AR051642A1 (es) | 2007-01-31 |
| ECSP077404A (es) | 2007-05-30 |
| RU2403253C2 (ru) | 2010-11-10 |
| CA2582434A1 (en) | 2006-05-04 |
| KR20070073798A (ko) | 2007-07-10 |
| MX2007004735A (es) | 2007-06-18 |
| EP2292619A1 (de) | 2011-03-09 |
| ES2394453T3 (es) | 2013-01-31 |
| US20080200483A1 (en) | 2008-08-21 |
| EP1805181A1 (de) | 2007-07-11 |
| GT200500281A (es) | 2006-04-24 |
| US8163754B2 (en) | 2012-04-24 |
| PE20061129A1 (es) | 2006-12-13 |
| IL182528A0 (en) | 2007-09-20 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| US20090105476A1 (en) | Organic Compounds | |
| EP2322525B1 (de) | Purinderivate als Adenosin A2A Rezeptoragonisten | |
| EP2066669B1 (de) | Adenosinderivate als agonisten am a2a-rezeptor | |
| EP2044070B1 (de) | Purinderivate als a2a-agonisten | |
| US20090099214A1 (en) | Organic Compounds | |
| AU2009233613A1 (en) | Purine derivatives for use as adenosin A-2A receptor agonists | |
| US20090281127A1 (en) | Organic Compounds | |
| CN101044141B (zh) | 用作腺苷a-2a受体激动剂的嘌呤衍生物 | |
| HK1131392B (en) | Adenosine derivatives as a2a receptor agonists | |
| HK1125642B (en) | Purine derivatives for use as adenosin a2a receptor agonists |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| PUAI | Public reference made under article 153(3) epc to a published international application that has entered the european phase |
Free format text: ORIGINAL CODE: 0009012 |
|
| 17P | Request for examination filed |
Effective date: 20070522 |
|
| AK | Designated contracting states |
Kind code of ref document: A1 Designated state(s): AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IS IT LI LT LU LV MC NL PL PT RO SE SI SK TR |
|
| AX | Request for extension of the european patent |
Extension state: HR |
|
| RAX | Requested extension states of the european patent have changed |
Extension state: HR Payment date: 20070522 |
|
| REG | Reference to a national code |
Ref country code: HK Ref legal event code: DE Ref document number: 1106521 Country of ref document: HK |
|
| 17Q | First examination report despatched |
Effective date: 20090618 |
|
| RAP1 | Party data changed (applicant data changed or rights of an application transferred) |
Owner name: NOVARTIS AG |
|
| GRAP | Despatch of communication of intention to grant a patent |
Free format text: ORIGINAL CODE: EPIDOSNIGR1 |
|
| GRAS | Grant fee paid |
Free format text: ORIGINAL CODE: EPIDOSNIGR3 |
|
| GRAA | (expected) grant |
Free format text: ORIGINAL CODE: 0009210 |
|
| AK | Designated contracting states |
Kind code of ref document: B1 Designated state(s): AT BE BG CH CY CZ DE DK EE ES FI FR GB GR HU IE IS IT LI LT LU LV MC NL PL PT RO SE SI SK TR |
|
| AX | Request for extension of the european patent |
Extension state: HR |
|
| REG | Reference to a national code |
Ref country code: GB Ref legal event code: FG4D |
|
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: EP |
|
| REG | Reference to a national code |
Ref country code: AT Ref legal event code: REF Ref document number: 573030 Country of ref document: AT Kind code of ref document: T Effective date: 20120915 |
|
| REG | Reference to a national code |
Ref country code: IE Ref legal event code: FG4D |
|
| REG | Reference to a national code |
Ref country code: DE Ref legal event code: R096 Ref document number: 602005035912 Country of ref document: DE Effective date: 20121025 |
|
| REG | Reference to a national code |
Ref country code: AT Ref legal event code: MK05 Ref document number: 573030 Country of ref document: AT Kind code of ref document: T Effective date: 20120829 |
|
| REG | Reference to a national code |
Ref country code: NL Ref legal event code: VDEP Effective date: 20120829 |
|
| REG | Reference to a national code |
Ref country code: LT Ref legal event code: MG4D Effective date: 20120829 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: LT Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20120829 Ref country code: IS Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20121229 Ref country code: CY Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20120829 Ref country code: FI Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20120829 Ref country code: AT Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20120829 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: FR Payment date: 20121018 Year of fee payment: 8 Ref country code: DE Payment date: 20121017 Year of fee payment: 8 |
|
| REG | Reference to a national code |
Ref country code: ES Ref legal event code: FG2A Ref document number: 2394453 Country of ref document: ES Kind code of ref document: T3 Effective date: 20130131 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: GR Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20121130 Ref country code: SE Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20120829 Ref country code: LV Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20120829 Ref country code: SI Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20120829 Ref country code: PT Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20121231 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: ES Payment date: 20121031 Year of fee payment: 8 Ref country code: IT Payment date: 20121013 Year of fee payment: 8 Ref country code: GB Payment date: 20121017 Year of fee payment: 8 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: EE Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20120829 Ref country code: CZ Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20120829 Ref country code: DK Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20120829 Ref country code: RO Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20120829 Ref country code: NL Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20120829 |
|
| PGFP | Annual fee paid to national office [announced via postgrant information from national office to epo] |
Ref country code: BE Payment date: 20121203 Year of fee payment: 8 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: PL Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20120829 Ref country code: SK Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20120829 Ref country code: MC Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20121031 |
|
| REG | Reference to a national code |
Ref country code: CH Ref legal event code: PL |
|
| PLBE | No opposition filed within time limit |
Free format text: ORIGINAL CODE: 0009261 |
|
| STAA | Information on the status of an ep patent application or granted ep patent |
Free format text: STATUS: NO OPPOSITION FILED WITHIN TIME LIMIT |
|
| REG | Reference to a national code |
Ref country code: IE Ref legal event code: MM4A |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: LI Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20121031 Ref country code: CH Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20121031 Ref country code: BG Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20121129 Ref country code: IE Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20121021 |
|
| 26N | No opposition filed |
Effective date: 20130530 |
|
| REG | Reference to a national code |
Ref country code: DE Ref legal event code: R097 Ref document number: 602005035912 Country of ref document: DE Effective date: 20130530 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: TR Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20120829 |
|
| REG | Reference to a national code |
Ref country code: HK Ref legal event code: WD Ref document number: 1106521 Country of ref document: HK |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: LU Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20121021 |
|
| GBPC | Gb: european patent ceased through non-payment of renewal fee |
Effective date: 20131021 |
|
| REG | Reference to a national code |
Ref country code: DE Ref legal event code: R119 Ref document number: 602005035912 Country of ref document: DE Effective date: 20140501 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: GB Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20131021 Ref country code: HU Free format text: LAPSE BECAUSE OF FAILURE TO SUBMIT A TRANSLATION OF THE DESCRIPTION OR TO PAY THE FEE WITHIN THE PRESCRIBED TIME-LIMIT Effective date: 20051021 |
|
| REG | Reference to a national code |
Ref country code: FR Ref legal event code: ST Effective date: 20140630 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: IT Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20131021 Ref country code: DE Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20140501 Ref country code: FR Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20131031 |
|
| REG | Reference to a national code |
Ref country code: ES Ref legal event code: FD2A Effective date: 20150709 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: ES Free format text: LAPSE BECAUSE OF NON-PAYMENT OF DUE FEES Effective date: 20131022 |
|
| PG25 | Lapsed in a contracting state [announced via postgrant information from national office to epo] |
Ref country code: BE Free format text: THE PATENT HAS BEEN ANNULLED BY A DECISION OF A NATIONAL AUTHORITY Effective date: 20120829 |