ES2703541T3 - Método de purificación de factor VII transgénico - Google Patents
Método de purificación de factor VII transgénico Download PDFInfo
- Publication number
- ES2703541T3 ES2703541T3 ES13739975T ES13739975T ES2703541T3 ES 2703541 T3 ES2703541 T3 ES 2703541T3 ES 13739975 T ES13739975 T ES 13739975T ES 13739975 T ES13739975 T ES 13739975T ES 2703541 T3 ES2703541 T3 ES 2703541T3
- Authority
- ES
- Spain
- Prior art keywords
- human
- tgfvii
- tgfviia
- ligand
- affinity
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K1/00—General methods for the preparation of peptides, i.e. processes for the organic chemical preparation of peptides or proteins of any length
- C07K1/14—Extraction; Separation; Purification
- C07K1/16—Extraction; Separation; Purification by chromatography
- C07K1/22—Affinity chromatography or related techniques based upon selective absorption processes
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/14—Hydrolases (3)
- C12N9/48—Hydrolases (3) acting on peptide bonds (3.4)
- C12N9/50—Proteinases, e.g. Endopeptidases (3.4.21-3.4.25)
- C12N9/64—Proteinases, e.g. Endopeptidases (3.4.21-3.4.25) derived from animal tissue
- C12N9/6421—Proteinases, e.g. Endopeptidases (3.4.21-3.4.25) derived from animal tissue from mammals
- C12N9/6424—Serine endopeptidases (3.4.21)
- C12N9/6437—Coagulation factor VIIa (3.4.21.21)
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/14—Hydrolases (3)
- C12N9/24—Hydrolases (3) acting on glycosyl compounds (3.2)
-
- B—PERFORMING OPERATIONS; TRANSPORTING
- B01—PHYSICAL OR CHEMICAL PROCESSES OR APPARATUS IN GENERAL
- B01D—SEPARATION
- B01D15/00—Separating processes involving the treatment of liquids with solid sorbents; Apparatus therefor
- B01D15/08—Selective adsorption, e.g. chromatography
- B01D15/26—Selective adsorption, e.g. chromatography characterised by the separation mechanism
- B01D15/38—Selective adsorption, e.g. chromatography characterised by the separation mechanism involving specific interaction not covered by one or more of groups B01D15/265 and B01D15/30 - B01D15/36, e.g. affinity, ligand exchange or chiral chromatography
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K14/00—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof
- C07K14/435—Peptides having more than 20 amino acids; Gastrins; Somatostatins; Melanotropins; Derivatives thereof from animals; from humans
- C07K14/745—Blood coagulation or fibrinolysis factors
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K16/00—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies
- C07K16/18—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans
- C07K16/36—Immunoglobulins [IG], e.g. monoclonal or polyclonal antibodies against material from animals or humans against blood coagulation factors
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N9/00—Enzymes; Proenzymes; Compositions thereof; Processes for preparing, activating, inhibiting, separating or purifying enzymes
- C12N9/14—Hydrolases (3)
- C12N9/48—Hydrolases (3) acting on peptide bonds (3.4)
- C12N9/50—Proteinases, e.g. Endopeptidases (3.4.21-3.4.25)
- C12N9/64—Proteinases, e.g. Endopeptidases (3.4.21-3.4.25) derived from animal tissue
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12Y—ENZYMES
- C12Y304/00—Hydrolases acting on peptide bonds, i.e. peptidases (3.4)
- C12Y304/21—Serine endopeptidases (3.4.21)
- C12Y304/21021—Coagulation factor VIIa (3.4.21.21)
-
- G—PHYSICS
- G01—MEASURING; TESTING
- G01N—INVESTIGATING OR ANALYSING MATERIALS BY DETERMINING THEIR CHEMICAL OR PHYSICAL PROPERTIES
- G01N33/00—Investigating or analysing materials by specific methods not covered by groups G01N1/00 - G01N31/00
- G01N33/48—Biological material, e.g. blood, urine; Haemocytometers
- G01N33/50—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing
- G01N33/68—Chemical analysis of biological material, e.g. blood, urine; Testing involving biospecific ligand binding methods; Immunological testing involving proteins, peptides or amino acids
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/20—Immunoglobulins specific features characterized by taxonomic origin
- C07K2317/22—Immunoglobulins specific features characterized by taxonomic origin from camelids, e.g. camel, llama or dromedary
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07K—PEPTIDES
- C07K2317/00—Immunoglobulins specific features
- C07K2317/50—Immunoglobulins specific features characterized by immunoglobulin fragments
- C07K2317/56—Immunoglobulins specific features characterized by immunoglobulin fragments variable (Fv) region, i.e. VH and/or VL
- C07K2317/569—Single domain, e.g. dAb, sdAb, VHH, VNAR or nanobody®
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Organic Chemistry (AREA)
- Engineering & Computer Science (AREA)
- Genetics & Genomics (AREA)
- Zoology (AREA)
- Wood Science & Technology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Molecular Biology (AREA)
- Biochemistry (AREA)
- General Health & Medical Sciences (AREA)
- Biomedical Technology (AREA)
- Medicinal Chemistry (AREA)
- Microbiology (AREA)
- Biotechnology (AREA)
- General Engineering & Computer Science (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Hematology (AREA)
- Immunology (AREA)
- Biophysics (AREA)
- Analytical Chemistry (AREA)
- Urology & Nephrology (AREA)
- Cell Biology (AREA)
- Food Science & Technology (AREA)
- Physics & Mathematics (AREA)
- Pathology (AREA)
- General Physics & Mathematics (AREA)
- Toxicology (AREA)
- Gastroenterology & Hepatology (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Peptides Or Proteins (AREA)
- Preparation Of Compounds By Using Micro-Organisms (AREA)
Abstract
Un método de purificación de factor VII humano transgénico (TgFVII) y/o factor VII humano transgénico activado (TgFVIIa) producido en la leche de animal no humano transgénico, que comprende; (a) una etapa de cromatografía de afinidad que comprende las etapas de: i. poner en contacto la leche que contiene TgFVII humano y/o TgFVIIa humano con un ligando que es específico para TgFVII humano y/o TgFVIIa humano, en condiciones que permiten que TgFVII humano y/o TgFVIIa humano se una al ligando, y ii. recuperar TgFVII humano y/o TgFVIIa humano alterando de una forma no destructiva la interacción con dicho ligando, en el que dicho ligando es una proteína de unión al antígeno dirigida a al menos un TgFVIIa humano o epítope de TgFVII humano, o un fragmento funcional o su derivado, y posee una afinidad superior a 1 μM hacia TgFVII humano o TgFVIIa humano, (b) purificación cromatográfica de TgFVII humano y/o TgFVIIa humano eluido en un intercambiador iónico, en condiciones en las que TgFVII humano y/o TgFVIIa humano son retenidos sobre el intercambiador iónico, (c) separación cromatográfica de TgFVII humano y/o TgFVIIa humano eluido en la resina de pseudo-afinidad, en condiciones en las que TgFVII humano y/o TgFVIIa humano son retenidos sobre la resina de pseudoafinidad, (d) separación cromatográfica de TgFVII humano y/o TgFVIIa humano eluido en un soporte de exclusión por tamaño.
Description
DESCRIPCIÓN
Método de purificación de factor VII transgénico
CAMPO DE LA INVENCIÓN
La presente invención se refiere a un ligando de afinidad anti-factor VII que es particularmente útil para purificar factor VII activado humano recombinante de fuentes transgénicas. El ligando de afinidad combinado con otras etapas cromatográficas ortogonales permite la preparación de una disolución de FVII altamente purificada completamente activada libre de agregados con un bajo porcentaje de formas de FVII degradadas u oxidadas. ANTECEDENTES DE LA INVENCIÓN
El factor VII (FVII) es una glucoproteína dependiente de la vitamina K que en su forma activada (FVIIa) participa en el proceso de coagulación que activa el factor X y el factor IX en presencia de calcio y de factor tisular. FVII se secreta en forma de una única cadena peptídica de 406 restos, con un peso molecular de aproximadamente 50 kDa. El FVII contiene cuatro dominios estructurales distintivos: el dominio gamma-carboxílico del extremo N (Gla), dos dominios de tipo "factor de crecimiento epidérmico (EGF)" y un dominio de serina proteasa. La activación del FVII en FVIIa se caracteriza por la escisión del enlace del dominio de Arg152-Ile153 (Arginina 152-Isoleucina 153). FVIIa es, por tanto, un compuesto con una cadena ligera de 152 aminoácidos con un peso molecular de aproximadamente 20 kDa y con una cadena pesada de 254 aminoácidos con un peso molecular de aproximadamente 30 kDa unidas entre sí con un único puente disulfuro (Cys135-Cys262).
El FVIIa plasmático contiene varias modificaciones postraduccionales: los diez primeros ácidos glutámicos están [gamma]-carboxilados, Asp63 está parcialmente hidroxilada, Ser52 (Serina 52) y Ser60 (Serina 60) están O-glucosiladas y llevan los patrones de glucosa (Xilosa)0-2 y fucosa, respectivamente, Asn145 (Asparagina 145) y Asn322 (Asparagina 322) se N-glucosilan principalmente por estructuras complejas bisialiladas biantenarias.
FVII se usa para el tratamiento de pacientes que padecen hemofilia, que muestran una deficiencia de factor VIII (hemofilia de tipo A) o de factor IX (hemofilia de tipo B), como de pacientes que muestran otras deficiencias de factores de coagulación, por ejemplo, una deficiencia congénita de FVII. Es, por tanto, necesario que estén disponibles concentrados de FVIIa inyectable.
El método más antiguo para obtener concentrados de FVIIa consistió en la purificación de FVIIa de proteínas del plasma resultantes del fraccionamiento del plasma. Para ese fin, el documento EP 0346241 describe la preparación de una fracción enriquecida en FVlla, obtenida después de la adsorción y posterior elución de un producto secundario del fraccionamiento de proteínas plasmáticas que contienen FVII y FVIIa y otras proteínas tales como los factores IX (FIX), X (FX) y II (FII), particularmente el pre-eluato de PPSB (P=protrombina o FII, P=proconvertina o FVII, S=factor de Stuart o FX y B=factor B antihemófilo o FIX). El inconveniente de este proceso es que el FVII obtenido todavía contiene algunas trazas de los otros factores de coagulación.
Asimismo, el documento EP 0 547 932 describe un proceso de fabricación de un concentrado de FVIIa de alta pureza sustancialmente libre de factores dependientes de la vitamina K y de FVIII. El FVII obtenido por este proceso, a pesar de su pureza, muestra una actividad trombogénica residual.
En términos generales, uno de los principales inconvenientes de estos procesos es que dan solo pequeñas cantidades de productos. Además, todavía es difícil obtener un producto completamente libre de otras proteínas presentes en el plasma. Finalmente, aunque se están implementando varias precauciones en cada etapa de preparación de los factores de coagulación plasmáticos para garantizar su seguridad viral y bacteriana (seguimiento de donantes de sangre, pruebas para detectar contaminantes virales y bacterianos conocidos, purificación rigurosa y tratamientos inactivantes de virus para reducir en la medida de lo posible el riesgo de transmisión de los agentes patógenos transmitidos por la sangre), sin embargo, no se excluyen todos los riesgos de contaminación con agentes patógenos. Además, la aparición de una nueva variante de la enfermedad de Creutzfeldt-Jakob dio lugar a temores de transmisión de agentes patógenos no convencionales por hemoderivados. Además, el volumen de plasma recogido de los donantes sigue estando limitado.
Por tanto, desde los años 80 se aisló el ADN que codificaba el factor VII humano (Hagen et al. Proc. Natl. Acad. Sci., 1986, 83(8):2412-6) y se expresó en diversos sistemas de expresión.
Se han descrito adicionalmente diversos procesos para la purificación del factor VII recombinante (véase, por ejemplo, el documento WO2009/141418). Actualmente, los polipéptidos del factor VII recombinante normalmente se purifican por uno o dos procesos diferentes de múltiples etapas, ya sea empleando exclusivamente cromatografía en resinas convencionales, o incluyendo cromatografía de afinidad empleando una resina de afinidad con ligandos de proteína.
El factor VII recombinante se puede producir a partir de lotes de fermentación, o de mamíferos transgénicos. En particular, las composiciones de FVII producidas en las glándulas mamarias de conejo hembra se describen, por ejemplo, en la solicitud de patente WO2007/138199. Entonces, el FVII producido en la leche por mamíferos
transgénicos se puede purificar de la leche, por ejemplo, por filtración tangencial y cromatografía, o por precipitación con calcio, seguido por varias etapas de cromatografía.
Sin embargo, los métodos de purificación disponibles no carecen de inconvenientes. Todavía podrían estar presentes especialmente proteínas nativas del mamífero transgénico, y en particular factor VII nativo del mamífero transgénico, que provocarían inmunogenicidad tras la administración a un paciente humano.
SUMARIO DE LA INVENCIÓN
Es un objeto de la invención un método de purificación de factor VII transgénico (TgFVII) y/o factor VII transgénico activado (TgFVIIa) producido en la leche de animal no humano transgénico, que comprende:
(a) una etapa de cromatografía de afinidad que comprende las etapas de:
i. poner en contacto la leche que contiene TgFVII humano y/o TgFVIIa humano con un ligando que es específico para TgFVII humano y/o TgFVIIa humano, en condiciones que permiten que TgFVII humano y/o TgFVIIa humano se una al ligando, y
ii. recuperar TgFVII humano y/o TgFVIIa humano alterando de una forma no destructiva la interacción con dicho ligando, en la que dicho ligando es una proteína de unión al antígeno dirigida a al menos un TgFVII humano o epítope de TgFVIIa humano, o un fragmento funcional o su derivado y posee una afinidad superior a 1 pM hacia TgFVII humano o TgFVIIa humano, (b) purificación cromatográfica de TgFVII humano y/o TgFVIIa humano eluido en un intercambiador iónico, en condiciones en las que TgFVII humano y/o TgFVIIa humano son retenidos sobre el intercambiador iónico,
(c) separación cromatográfica de TgFVII humano y/o TgFVIIa humano eluido en resina de pseudo-afinidad, en condiciones en las que TgFVII humano y/o TgFVIIa humano son retenidos sobre la resina de pseudoafinidad,
(d) separación cromatográfica de TgFVII humano y/o TgFVIIa humano eluido en un soporte de exclusión de tamaño.
En una realización de la invención, el ligando es una proteína de unión al antígeno dirigida a al menos un epítope de TgFVII y/o TgFVIIa, o un fragmento funcional o su derivado. Preferentemente, el ligando comprende dos cadenas de polipéptidos pesadas que forman dos sitios de unión al antígeno completos y carecen de cadenas de polipéptidos ligeras, y deriva de cadenas de inmunoglobulina pesadas de Camelidae.
En una realización, el método de la invención es tal que la etapa (ii) comprende al menos una etapa de lavado para retirar el material no unido antes de eluir TgFVII o TgFVIIa del ligando, y además comprende opcionalmente una segunda etapa de lavado para retirar contaminantes unidos sobre la resina y que tienen una afinidad más débil que TgFVII o TgFVIIa
En una realización de la invención, el material biológico fuente usado para purificar TgFVII o TgFVIIa se obtiene de un animal transgénico que produce TgFVII y/o TgFVIIa. Preferentemente, el animal es un mamífero hembra, y preferentemente un conejo hembra, y el material biológico fuente es leche.
En una realización, el método de la invención comprende además las etapas preliminares, antes de la etapa (a), de recoger y clarificar la leche. Preferentemente, la etapa de clarificación de la leche se realiza por adición de sal de citrato, seguido por filtración.
En una realización, el método de la invención comprende una etapa b) de purificación cromatográfica de TgFVII y/o TgFVIIa eluidos sobre un intercambiador aniónico, en condiciones en las que TgFVII y/o TgFVIIa son retenidos sobre el intercambiador aniónico. Esta etapa se dedica preferentemente a la activación de TgFVII y al pulido de TgFVII y/o TgFVIIa.
En una realización, el método de la invención comprende una etapa c) de separación cromatográfica de TgFVII y/o TgFVIIa eluidos sobre resina de hidroxiapatita, en condiciones en las que TgFVII y/o TgFVIIa son retenidos sobre la resina de hidroxiapatita. Esta etapa se dedica preferentemente al pulido de TgFVII y/o TgFVIIa y al control de la degradación de TgFVII y/o TgFVIIa.
En una realización, el método de la invención comprende una etapa d) de separación cromatográfica de TgFVII y/o TgFVIIa eluidos sobre un soporte de exclusión por tamaño. Esta etapa se dedica preferentemente al pulido y formulación de TgFVII y/o TgFVIIa.
En una realización, el método de la invención comprende además al menos una etapa de eliminar y/o inactivar virus, preferentemente por nanofiltración y/o por tratamiento con disolvente/detergente.
En una realización, el método de la invención comprende además las etapas finales de formular, esterilizar y liofilizar TgFVII y/o TgFVIIa purificados.
En una realización, durante la etapa de cromatografía de afinidad, el método de la invención comprende además las etapas de:
(i) cargar una resina cromatográfica sobre la que se inmoviliza un ligando como se ha definido anteriormente, con leche de un conejo hembra transgénico que contiene TgFVII y/o TgFVIIa recombinantemente producidos, en condiciones que permiten que el ligando se una a TgFVII y/o TgFVIIa;
(111) lavar el material no unido;
(112) lavar los contaminantes que tienen una afinidad con el ligando más débil que TgFVII y/o TgFVIIa; y (113) eluir TgFVII y/o FVIIa de la resina.
En una realización, durante la etapa de cromatografía de afinidad del método de la invención, dicha segunda etapa de lavado para eliminar material unido débil se realiza con un tampón que comprende desde 10 % hasta 50 % de un agente hidrófobo, y cuya fuerza iónica varía desde 200 hasta 600 mM.
En una realización, durante la etapa de cromatografía de afinidad del método de la invención, dicha etapa de elución para eluir TgFVII y/o TgFVIIa de la resina se realiza con un tampón que comprende desde 20 % hasta 70 % de un agente hidrófobo, y cuya fuerza iónica varía desde 500 hasta 2500 mM.
Las características y ventajas adicionales de la invención surgirán de la siguiente descripción de realizaciones de la invención, dadas como ejemplos no limitantes, con referencia a los dibujos adjuntos enumerados a continuación. BREVE DESCRIPCIÓN DE LOS DIBUJOS
La Figura 1 muestra una representación esquemática de las formas de IgG1, IgG2 e IgG3 de inmunoglobulinas de Camelidae que tienen cadenas de polipéptidos pesadas y que carecen de cadenas ligeras.
La Figura 2 muestra la representación esquemática de la eliminación de RMP durante el proceso de la invención. El nivel de RMP se indica en ppm en función de la etapa del proceso.
La Figura 3 muestra la representación esquemática de la eliminación de ADN durante el proceso de la invención. El nivel de ADN se indica en ppb en función de la etapa del proceso.
La Figura 4 muestra la activación de TgFVII mediante el proceso de purificación. Los carriles 1-9 se corresponden respectivamente a: carriles 1 y 9: Peso molecular, carril 2: leche clarificada, carril 3: eluato seleccionado de FVII (después de la cromatografía de afinidad sobre el ligando de la invención), carril 4: producto intermedio 1 (después de ultrafiltración y diafiltración), carril 5: nanofiltrado, carril 6: eluato de QSXL, carril 7: eluato de CHT-I, carril 8: conjunto de SEC.
La Figura 5 muestra un histograma de la carga viral en muestras después de la etapa de prefiltración y de tratamiento con disolvente/detergente (muestra denominada Carga inicial) y después de la elución en cromatografía en Affinity FVII Select (muestra denominada Eluato A).
DESCRIPCIÓN DETALLADA DE LA INVENCIÓN
Factor VII o factor VlIa transgénico
El método de purificación de la invención se basa en particular en el uso de un ligando que muestra una afinidad específica por el factor VII transgénico (TgFVII) y/o el factor VII transgénico activado (TgFVIIa).
Se conocen las secuencias de aminoácidos del factor VII y factor VIIa transgénicos para su uso en la presente invención. La secuencia de aminoácidos del factor VII transgénico es en particular idéntica a la secuencia de aminoácidos del factor VII humano, tal como se desvela en la patente de EE.UU. N° 4.784.950.
Material biológico fuente
El método de la invención es especialmente útil para purificar FVII y/o FVIIa transgénicos obtenidos de diversas fuentes biológicas, que se denominan en el presente documento "material biológico fuente".
En una realización de la invención, el material biológico fuente contiene FVII y/o FVIIa transgénicos y se obtiene a partir del fraccionamiento de un líquido corporal de un animal transgénico que ha sido modificado (o es la progenie de un animal que ha sido modificado) para producir FVII y/o FVIIa transgénicos. El animal transgénico es preferentemente un mamífero, por ejemplo una vaca, conejo, cabra, oveja, etc. Preferentemente, el material biológico fuente que contiene TgFVII y/o TgFVIIa es una muestra de leche de mamífero, es decir, una muestra de leche de mamífero transgénico que produce FVII y/o FVIIa transgénicos en su leche.
Los métodos de producción de la proteína FVII transgénica en la leche de un animal transgénico pueden incluir las siguientes etapas: se transfiere a un embrión de un mamífero no humano una molécula de ADN sintética que contiene un gen que codifica el FVII transgénico (cuya secuencia de aminoácidos corresponde a la de FVII humano), gen que está controlado por un promotor de una proteína que es naturalmente secretada en la leche. El embrión se introduce entonces en un mamífero hembra de la misma especie, que entonces pare un animal transgénico. Una vez se ha desarrollado suficientemente el sujeto, se induce la lactancia del mamífero y se recoge la leche. La leche contiene entonces la proteína recombinante deseada. Un ejemplo de un proceso para la preparación de FVII transgénico (TgFVII) en la leche de un mamífero hembra distinto de un ser humano se proporciona en el documento EP 0 527 063. En dicha realización específica, se construye un plásmido que contiene un promotor de WAP mediante la introducción de una secuencia que contiene el promotor para el gen WAP, y este plásmido se crea de tal forma que pueda recibir un gen extraño que se vuelve dependiente del promotor WAP. Se incorpora el gen que codifica el FVII transgénico y se convierte en dependiente del promotor WAP. Se usan el plásmido que contiene el promotor y el gen que codifica la proteína FVII transgénica para obtener animales transgénicos, tales como conejos, mediante microinyección en el pronúcleo masculino de embriones de conejo. Los embriones se transfieren entonces al oviducto de hembras hormonalmente preparadas.
Métodos de purificación
La invención proporciona métodos de purificación de FVII y/o FVIIa transgénicos. Tales métodos implican en particular poner en contacto un material biológico fuente que contiene TgFVII y/o TgFVIIa con un ligando que es altamente específico para TgFVII y TgFVIIa.
La producción de FVII o FVIIa transgénicos (TgFVIIa) en la leche de conejos transgénicos resulta en la presencia de numerosas impurezas que se tienen que eliminar. El proceso de la presente invención se diseña especialmente para purificar FVII o FVIIa transgénico (TgFVIIa) de la leche de conejos transgénicos, mientras se eliminan las moléculas contaminantes acompañantes, las denominadas impurezas relacionadas con el hospedador (HRI), que incluyen proteínas de leche de conejo (RMP), pero también ADN de conejo y otras moléculas que podrían contaminar la leche de conejo como las proteínas del suero de conejo. Las proteínas residuales del suero (por ejemplo, albúmina, FVII de conejo, inmunoglobulinas y transferrina) se pueden introducir en el material de fuente de leche debido a la fuga pasiva de la corriente sanguínea en la leche.
La leche, de hecho, corresponde a una suspensión coloidal no estéril de proteínas solubles del suero de la leche, micelas insolubles de caseína y glóbulos de grasa de la leche. TgFVII, que se sintetiza y secreta por las células epiteliales mamarias, se encuentra en la fracción soluble del suero de la leche. Las posibles RMP contaminantes incluyen, pero no se limitan a: caseínas; lactoalbúmina, lactoglobulina y lactoferrina. Se mostró que cinco familias de proteínas: transferrina, caseínas, albúmina, proteína ácidas del suero de la leche e inmunoglobulina (Ig) representaban aproximadamente 90 % de las proteínas totales de la leche de conejo.
Además, en una realización de la presente invención, el proceso de purificación de la invención también se diseña para activar el zimógeno TgFVII expresado en la leche, en TgFVIIa. Desafortunadamente, una vez activado, TgFVIIa se vuelve sensible a la escisión suplementaria. La escisión de TgFVII y/o TgFVIIa conducirá a TgFVII con regiones ricas en Gla parcial o totalmente ausentes o conducirá a la escisión de la cadena ligera (LC) o pesada (HC) que podría limitar la potencia de la molécula. Por tanto, son posibles la degradación de TgFVIIa durante el proceso en productos escindidos no activos o la oxidación de TgFVII durante el proceso y se tienen que eliminar para garantizar la idoneidad terapéutica de FVII.
Clarificación
Se podría realizar opcionalmente la clarificación de la leche como tratamiento temprano de la leche mediante procedimientos como se describen en las solicitudes de patente WO2007/138198 o WO2008/099077, pero no existe limitación al tratamiento de la leche antes de aplicar la presente invención. Opcionalmente, la clarificación también se podría evitar, por ejemplo, si la primera etapa del proceso se realiza por una captura por afinidad en lecho fluidizado. Opcionalmente, también se podría realizar una decantación de grasa o separación de nata por cualquier tecnología disponible en la industria de la leche antes de la purificación. En una realización de la invención, preferentemente, la leche se mezcla con un tampón citrato para obtener rápidamente una fase de leche de tipo de plasma estabilizado, luego se obtiene la separación de la grasa por separación en fase líquida tal como centrifugación o decantación, seguido por filtración profunda. En una realización de la invención, preferentemente se realiza filtración profunda, ventajosamente para preparar moléculas activas biológicas de TgFVII y/o TgFVIIa por combinación de estrés de bajo cizallamiento, tecnología no oxidativa y la rápida disminución de la carga biológica y retención de tamaño de impurezas relacionadas que proceden de la recogida de la leche. En dicha realización, el tamaño de poro relativo del filtro profundo oscila desde 30 hasta 0,1 pm y la filtración profunda se implementa preferentemente por una filtración secuencial de 25 a 0,5 pm, seguida por filtración de 0,5 pm a 0,1 pm. Los filtros profundos basados en celulosa se adaptan bien a esta etapa con o sin adyuvante granular u otros compuestos de adyuvante de filtro. En una realización de la invención, la concentración de sales de citrato y la temperatura del proceso oscilan desde 0,15 M hasta sal saturada de citrato y desde 15 °C hasta 30 °C, y preferentemente desde 0,2 M hasta sal citrato 0,5 M y desde 22 °C hasta 27 °C. Estas combinaciones de sales de citrato y temperatura son necesarias para mantener las poblaciones de tamaño de las proteínas de la leche por debajo del orden de micrómetros y para obtener una clara
disolución de fase de tipo plasma. Por "clara" se indica que la leche clarificada en el proceso de la invención muestra una turbidez controlada inferior a 500 NTU (unidad de turbidez normalizada) o una lectura de densidad óptica en el espectrofotómetro a A400 nanómetros inferior a 1000 UA (unidad relativa de absorbancia).
En estas condiciones, se puede realizar ventajosamente la filtración en membrana con 0,2 pm de tamaño de poro para reducir la carga biológica en la fuente biológica de proteínas.
La disolución de carga biológica reducida y estabilizada resultante se podría almacenar opcionalmente a temperatura baja inferior a -20 °C durante varios meses.
Cromatografía de afinidad sobre un ligando específico
El ligando
El método de la invención para purificar TgFVII/TgFVIIa de un material biológico fuente comprende al menos una etapa de separación por cromatografía de afinidad en un ligando que es específico para FVII/FVIIa transgénico. En una realización de la invención, dicho ligando de afinidad es una proteína de unión al antígeno aislada que comprende dos cadenas de polipéptidos pesadas que forman un sitio de unión al antígeno completo o varios sitios de unión al antígeno, que carecen además de cadenas de polipéptidos ligeras. La proteína de unión al antígeno usada en el método de la invención está así normalmente compuesta de un dímero de cadenas pesadas y carece de cadenas ligeras. Las dos cadenas de polipéptidos pesadas son suficientes para formar uno o más sitios de unión al antígeno completos. Por "un sitio de unión al antígeno completo" se indica según la invención un sitio que solo permitirá el reconocimiento y la unión completa de un antígeno, que se puede verificar por cualquier método conocido con respecto a la prueba de la afinidad de unión.
Las cadenas pesadas de la proteína de unión al antígeno usadas en el método de la invención no tienen características especiales para interaccionar con las cadenas ligeras correspondientes (que están ausentes), y así son muy diferentes de las cadenas pesadas comunes de las inmunoglobulinas. A pesar de su estructura particular, dichas proteínas de unión al antígeno son, sin embargo, capaces de mostrar propiedades funcionales que son al menos equivalentes y preferentemente mejoradas cuando se compararon con inmunoglobulinas modelo estándar de cuatro cadenas. Las cadenas pesadas que forman dicha proteína de unión al antígeno parecen así ser intrínsecamente más adecuadas para la secreción por células procariotas y eucariotas inferiores.
En una realización particular, las proteínas de unión al antígeno usadas como ligandos de afinidad en el método de la invención se caracterizan porque sus cadenas de polipéptidos pesadas contienen una región variable (VH) y una región constante (CH), pero carecen del primer dominio de su región constante, denominado CH1. Estas proteínas de unión al antígeno que no tienen dominio CH1 son así tal que la región variable de sus cadenas se une directamente a la región bisagra en la parte del extremo C de la región variable. El documento WO 94/25591 desvela métodos para la preparación de dichos anticuerpos de cadenas pesadas, o sus fragmentos, a una gran escala que comprende transformar un moho o levadura con una secuencia de ADN expresable que codifica dicho anticuerpo o fragmento. En una realización de la invención, dicha proteína de unión al antígeno también se puede obtener como se describe en el documento WO 94/04678.
El dominio variable de una inmunoglobulina de la cadena pesada, como se usa en el método de la invención, no tiene sitios de interacción normales con el dominio VL o CH1, que no existen en las inmunoglobulinas de la cadena pesada. Dichas inmunoglobulinas también se designan comúnmente "VHH" en la bibliografía.
En una realización, las proteínas de unión al antígeno usadas como ligandos en el método de la invención se caracterizan porque sus regiones variables contienen en la posición 45 un aminoácido que es diferente de los restos de leucina, prolina o glutamina.
En una realización particular, las proteínas de unión al antígeno usadas como ligandos de afinidad en el método de la invención se caracterizan porque su región variable comprende regiones estructurales (FW) y regiones determinantes de la complementariedad (CDR), especialmente 4 regiones estructurales y 3 regiones de complementariedad. Así se distinguen de las inmunoglobulinas estándar de cuatro cadenas, especialmente por el hecho de que esta región variable puede ella misma contener un sitio de unión al antígeno o varios, y no requiere la contribución de la región variable de una cadena ligera.
Las secuencias de aminoácidos de las regiones estructurales 1 y 4 comprenden, entre otros, respectivamente, secuencias de aminoácidos que se pueden seleccionar de las siguientes:
Para el dominio de la región estructural 1:
GGSVQTGGSLRLSCEISGLTFD (SEQ ID NO: 1)
GGSVQTGGSLRLSCAVSGFSFS (SEQ ID NO: 2)
GGSEQGGGSLRLSCAISGYTYG (SEQ ID NO: 3)
GGSVQPGGSLTLSCTVSGATYS (SEQ ID NO: 4)
GGSVQAGGSLRLSCTGSGFPYS (SEQ ID NO: 5)
GGSVQAGGSLRLSCVAGFGTS (SEQ ID NO: 6)
GGSVQAGGSLRLSCVSFSPSS (SEQ ID NO: 7);
para el dominio de la región estructural 4:
WGQGTQVTVSS (SEQ ID NO: 8)
WGQGTLVTVSS (SEQ ID NO: 9)
WGQGAQVTVSS (SEQ ID NO: 10)
WGQGTQVTASS (SEQ ID NO: 11)
RGQGTQVTVSL (SEQ ID NO: 12) y/o,
para el dominio CDR3
ALQPGGYCGYGX........... CL (SEQ ID NO: 13)
VSLMDRISQH..............GC (SEQ ID NO: 14)
VPAHLGPGAILDLKKY...... KY (SEQ ID NO: 15)
FCYSTAGDGGSGE..........MY (SEQ ID NO: 16)
ELSGGSCELPLLF..........DY (SEQ ID NO: 17)
DWKYWTCGAQTGGYF........GQ (SEQ ID NO: 18)
RLTEMGACDARWATLATRTFAYNY (SEQ ID NO: 19)
QKKDRTRWAEPREW.........NN (SEQ ID NO: 20)
GSRFSSPVGSTSRLES-SDY-NY (SEQ ID NO: 21)
ADPSIYYSILXIEY.........KY (SEQ ID NO: 22)
DSPCYMPTMPAPPIRDSFGW-DD (SEQ ID NO: 23)
TSSFYWY CTTAPY.........NV (SEQ ID NO: 24)
TEIEWYGCNLRTTF TR (SEQ ID NO: 25)
NQLAGGWYLDPNYWLSVGAY-AI (SEQ ID NO: 26)
RLTEMGACDARWATLATRTFAYNY (SEQ ID NO: 27)
DGWTRKEGGIGLPWSVQCEDGYNY (SEQ ID NO: 28)
DSYPCHLL................DV (SEQ ID NO: 29)
VEYPIADMCS..............RY (SEQ ID NO: 30)
En una realización, la región constante de la proteína de unión al antígeno comprende los dominios CH2 y CH3 que comprenden una secuencia de aminoácidos seleccionada del siguiente grupo de secuencias:
para el dominio CH2:
APELLGGPTVFIFPPKPKDVLSITLTP (SEQ ID NO: 31)
APELPGGPSVFVFPTKPKDVLSISGRP (SEQ ID NO: 32)
APELPGGPSVFVFPPKPKDVLSISGRP (SEQ ID NO: 33)
APELLGGPSVFIFPPKPKDVLSISGRP (SEQ ID NO: 34)
para el dominio CH3:
GQTREPQVYTLA (SEQ ID NO: 35)
GQTREPQVYTLAPXRLEL (SEQ ID NO: 36)
GQPREPQVYTLPPSRDEL (SEQ ID NO: 37)
GQPREPQVYTLPPSREEM (SEQ ID NO: 38)
GQPREPQVYTLPPSQEEM (SEQ ID NO: 39)
Preferentemente, las proteínas de unión al antígeno se caracterizan por que su región bisagra comprende desde 0 hasta 50 aminoácidos. Las regiones particulares de las regiones bisagra de estas proteínas son las siguientes secuencias:
GTNEVCKCPKCP (SEQ ID NO: 40) o,
EPKIPQPQPKPQPQPQPQPKPQPKPEPECTCPKCP (SEQ ID NO: 41)
La región bisagra corta corresponde a una molécula de IgG3 y la secuencia de la bisagra larga corresponde a una molécula de IgG2.
En una realización particular, las proteínas de unión al antígeno usadas como ligando en el método de la invención se pueden aislar de animales, más específicamente de Camelidae, y preferentemente de la sangre de Camelidae. En una realización adicional, las proteínas de unión al antígeno derivan de inmunoglobulinas de Camelidae, y más específicamente de inmunoglobulinas de camélidos del viejo mundo (Camelus bactrianus y Camelus dromedarius) o camélidos del nuevo mundo (Lama pacos, Lama glama y Lama vicugna). Dependiendo del animal del que se originan, el peso molecular de las proteínas de unión al antígeno puede ser desde aproximadamente 43 kDa hasta aproximadamente 47 kDa, en particular 45 kDa.
En una realización, estas proteínas de unión al antígeno se pueden modificar y especialmente humanizar, mediante la sustitución de toda o parte de su región constante por toda o parte de una región constante de un anticuerpo humano. Por ejemplo, los dominios CH2 y/o CH3 de dichas proteínas de unión al antígeno se podrían sustituir por los dominios CH2 y/o CH3 de la inmunoglobulina humana IgG y3. En dichas proteínas de unión al antígeno humanizadas, una parte de las secuencias variables, y en particular uno o más de los restos de la región estructural que no intervienen en el sitio de unión, también se han sustituido por restos de anticuerpo humano de la región estructural. Dichas proteínas de unión al antígeno pueden comprender inmunoglobulinas de tipo G y especialmente inmunoglobulinas que se definen como inmunoglobulinas de clase 2 (IgG2) o inmunoglobulinas de clase 3 (IgG3). Las ventajas de la proteína de unión al antígeno anteriormente descrita se basan en el hecho de la ausencia de un dominio variable de la cadena ligera significa que no se presenta la dimensión adicional de variabilidad en el repertorio de anticuerpos resultante de la posibilidad de emparejar diferentes combinaciones de dominios variables, de manera que es posible preparar proteínas de unión al antígeno dirigidas a un antígeno específico (aquí FVII/FVIIa transgénicos) en evitar la generación de grandes números de proteínas de unión al antígeno irrelevantes. Una segunda ventaja es que se ha mostrado que los dominios VHH son significativamente más estables que los anticuerpos tradicionales, de manera que se espera que las separaciones cromatográficas basadas en dichas proteínas de unión al antígeno sean significativamente más robustas. Además, la ausencia de dependencia de la interacción con un dominio variable de la cadena ligera para mantener la integridad estructural y funcional da a estos dominios VHH una ventaja sustancial con respecto a otros fragmentos de anticuerpos pequeños, en términos de facilidad de producción y comportamiento en disolución. En particular, los fragmentos VHH son moléculas especialmente aptas para purificación por inmunoafinidad, debido a su estabilidad poco usual y su capacidad para replegarse eficientemente después de la desnaturalización completa, que frecuentemente ocurre durante la elución del antígeno.
Cuando el ligando de afinidad usado en el método de la invención es una proteína de unión al antígeno como se ha definido anteriormente, se puede dirigir contra cualquier epítope de FVII/FVIIa transgénico, a condición de que dicho epítope sea específico para FVII/FVIIa transgénico. Dichos epítopes comprenden cualquier estructura compuesta de aminoácidos, polipéptidos, hidratos de carbono, o su mezcla.
Por la expresión "ligando que es específico para FVII/FVIIa transgénico" se indica que el ligando usado para la etapa de cromatografía de afinidad de la invención no se une sustancialmente a, o restringe sustancialmente la unión de cualquier otra proteína, que incluye FVII o FVIIa de otras especies, tales como FVII o FVIIa de conejo o cabra. El ligando usado en la invención posee preferentemente una afinidad superior a 1 |jM hacia el factor VII o factor VIIa transgénico, y puede ventajosamente tener una afinidad de al menos 100 nM y más preferentemente de desde 1 hasta 10 nM para FVII o FVIIa transgénico. En una realización particular, el ligando usado en la invención se une a FVII o FVIIa transgénico con más de un logaritmo de diferencia cuando se compara con FVII o FVIIa de conejo. Por la expresión "restringe sustancialmente la unión de proteínas" se indica que el ligando usado en la invención permite al menos 4 logaritmos o 99,99 % de retirada de impurezas relacionadas con el hospedador (HRI) contenidas en la
disolución inicial cargada sobre el soporte cromatográfico, y al menos 3 logaritmos o 99,9 % de retirada de proteínas FVII o FVIIa endógenamente producidas por el mamífero transgénico usado para expresar FVII transgénico.
En una realización, la masa de factor VII o factor VIIa endógeno en la fracción eluida del soporte de afinidad se reduce 100 veces, preferentemente 1000 veces, o lo más preferentemente 10000 veces o más con respecto a la masa de estas proteínas dentro de la disolución inicial cargada sobre la columna. En comparación, la masa relativa del factor VII o factor VIIa transgénico cargado sobre el ligando específico de FVII y recuperado en la fracción de elución es al menos 30 %, preferentemente al menos 50 % o más de la masa de estas proteínas dentro de la disolución inicialmente cargada.
En una realización particular, la proteína de unión al antígeno de la invención (ligando) tiene las siguientes constantes cinéticas: Ka= 2,58x108 M y Kd= 9,88x10-9 M.
También se puede usar cualquier fragmento funcional o derivado funcional de las proteínas de unión al antígeno anteriormente descritas como ligando para la unión, específicamente factor VII transgénico y/o factor VII transgénico activado (FVIIa) en el método de la invención.
Un "fragmento funcional" de las proteínas de unión al antígeno anteriormente descritas es un fragmento que muestra una fuerte afinidad por el factor VII o factor VIIa humano, y preferentemente sustancialmente la misma afinidad con respecto a FVII o FVIIa humano que la proteína de unión al antígeno de la que se origina, o al menos aproximadamente 50 %, todavía preferentemente al menos aproximadamente 60, 70, 80 o 90 % de la afinidad mostrada por la proteína de unión al antígeno de la que se origina.
Los fragmentos englobados para uso como ligando en el método de la invención comprenden una cadena de polipéptidos pesada de una proteína de unión al antígeno que carece de cadenas ligeras, fragmentos obtenidos por digestión enzimática de las proteínas de unión al antígeno anteriormente descritas, especialmente las obtenidas por digestión parcial con papaína que conduce al fragmento Fc (fragmento constante) y que conduce al fragmento FVHh (que contiene los sitios de unión al antígeno de las cadenas pesadas) o su dímero F(VHh)2, o un fragmento obtenido por digestión adicional con papaína del fragmento Fc, que conduce al pFc correspondiente a la parte del extremo C del fragmento Fc, fragmentos homólogos obtenidos con otras enzimas proteolíticas, fragmentos de al menos 10, preferentemente 20 aminoácidos de la región variable de la proteína de unión al antígeno, o la región variable completa, especialmente un fragmento correspondiente a los dominios aislados VH o a los dímeros de VH unidos a la bisagra, y los fragmentos correspondientes a al menos 10, preferentemente 20 aminoácidos de la región constante o a la región constante completa de la proteína de unión al antígeno.
"Derivado funcional" significa un polipéptido que se diferencia de la proteína de unión al antígeno usada como ligando en el método de la invención por una o varias sustituciones, deleciones o adiciones de aminoácidos, hasta el punto de que el derivado muestre afinidad de unión y especificidad con respecto a FVII o FVIIa transgénico. El "derivado funcional" muestra sustancialmente preferentemente la misma afinidad que las proteínas de unión al antígeno anteriormente descritas o al menos aproximadamente 90 %, todavía preferentemente al menos aproximadamente 50, 60, 70 u 80 % de la afinidad mostrada por dicha proteína de unión al antígeno con respecto a FVIIa transgénico. Dicho derivado funcional incluye homólogos que muestran un alto porcentaje de identidad con las proteínas de unión al antígeno anteriormente descritas, con las que son al menos 80 %, 85 % 90 %, 95 % o 99 % idénticas, y preferentemente se diferencian de las proteínas de unión al antígeno anteriormente descritas por una o varias sustituciones conservativas.
En una realización de la invención, la proteína de unión al antígeno se inmoviliza sobre una fase sólida. La inmovilización se puede lograr por adsorción o por reticulación química.
Comúnmente, la unión de proteínas a superficies sólidas, tales como medios de cromatografía, se ha provocado exponiendo la superficie de una fase sólida a una disolución de la proteína de forma que la proteína se adsorba sobre la superficie sólida mediante mecanismos de unión no específica. Los procedimientos de inmovilización de proteínas sobre medios de cromatografía están bien establecidos en la bibliografía, véase, por ejemplo, en Boschetti E., Egly J.M., Monsigny M., 1983, Practical Guide for use in Affinity chromatography and related techniques, 2a edición, IBF Villeneuve-la-garenne, Francia, páginas 1-157. Si la superficie sólida se proporciona por un material hidrófobo tal como, por ejemplo, poliestireno, entonces la unión se provoca generalmente por la adsorción de regiones hidrófobas de la proteína sobre la superficie hidrófoba.
Están disponibles alternativas o mejoras al método de adsorción de proteínas en la preparación de superficies de proteínas inmovilizadas.
Un enfoque alternativo es usar reticulación química de restos en la proteína para la unión covalente a una superficie sólida activada usando químicas de acoplamiento convencionales, por ejemplo, como se describe en Boschetti E., Egly J.M., Monsigny M., 1983, Practical Guide for use in Affinity chromatography and related techniques, 2a edición, IBF Villeneuve-la-garenne, Francia, páginas 1-157. Los restos de aminoácidos que incorporan grupos sulfhidrilo, tales como cisteína, se pueden unir covalentemente usando un reactivo biespecífico tal como, por ejemplo, succinimidil-maleimidofenilbutirato (SMPB).
Alternativamente, se pueden acoplar grupos lisina localizados en la superficie del ligando a grupos carboxilo activados en la superficie sólida por acoplamiento de carbodiimida convencional usando 1-etil-3-[3-dimetilaminopropil]carbodiimida (EDC) y N-hidroxisuccinimida (NHS). También se puede unir el ligando usando una extensión en forma de una cola de péptido, inmovilizada a la fase sólida por adsorción no covalente o usando agentes de reticulación química convencional.
Según una realización de la invención, la proteína de unión al antígeno de la invención se une a una fase sólida por reticulación covalente usando químicas de acoplamiento convencionales. La fase sólida puede comprender naturalmente restos reticulables adecuados para la unión covalente o se puede recubrir o derivatizar para introducir grupos reticulables adecuados según métodos muy conocidos en la técnica.
La fase sólida sobre la que tiene lugar la inmovilización se puede proporcionar mediante una variedad de materiales y puede ser adecuadamente cualquier material de soporte de fase sólida convencionalmente usado en inmovilizar proteínas. El método de la invención es aplicable con cualquier material en fase sólida que sea susceptible a la inmovilización de proteínas o fragmentos de proteínas, tanto directamente como después del pretratamiento. Los materiales de soporte pueden estar en partículas (por ejemplo, perlas o gránulos, generalmente usados en columnas de extracción) o en forma de hoja (por ejemplo, membranas o filtros, portaobjetos de vidrio o plástico, placas de ensayo de microtitulación, tira reactiva, dispositivos de llenado capilar u otros) que pueden ser fibras o tubos planos, plegados o huecos. Las siguientes matrices se facilitan como ejemplos y no son exhaustivas, dichos ejemplos podrían incluir sílice (sílice amorfa porosa), por ejemplo, la serie FLASH de cartuchos que contienen sílice irregular de 60A (32-63 gm o 35-70 gm) suministrada por Biotage (una división de Dyax Corp.), soportes de agarosa o poliacrilamida, por ejemplo, la gama de Sepharose de productos suministrados por Amersham Pharmacia Biotech, o los soportes Affi-Gel suministrados por Bio-Rad. Además, hay polímeros macroporosos, tales como los soportes Affi-Prep estables a la presión como se suministran por Bio-Rad. Otros soportes que se podrían utilizar incluyen dextrano, colágeno, poliestireno, metacrilato, alginato de calcio, vidrio de poro controlado, aluminio, titanio y cerámica porosa. En una realización preferida se usan resinas como fase sólida, que incluyen resinas comerciales tales como Sephadex®, Sepharose®, Fractogel®, CIMGEL® Toyopearl®, HEMA®, Capto MP®, además de agarosa reticulada y poliestireno o poliacrilato macroporoso. La fase sólida también puede ser de naturaleza principalmente inorgánica, tal como vidrio macroporoso o minerales de arcilla, o combinaciones de resinas y extractos inorgánicos, tales como Ceramic HyperD® o gel de sílice.
Etapa de carga
El material biológico fuente que contiene FVII/FVIIa transgénico se pretrata opcionalmente (por ejemplo, por etapas de clarificación o de filtración) antes de la carga sobre el soporte de afinidad que comprende el ligando anteriormente descrito.
El material biológico fuente se ajusta preferentemente antes de la carga sobre el soporte de afinidad que comprende el ligando. En una realización de la invención, el pH del tampón de carga se ajusta desde aproximadamente 6,0 hasta 8,0. Para el fin de esta etapa cromatográfica de la invención, un tampón de carga adecuado normalmente corresponde a una disolución acuosa que comprende un agente de tamponamiento, normalmente fosfato, citrato o T ris. Sin embargo, también se podrían usar todos los tampones carboxílicos o de ión bipolar con un pK próximo a la neutralidad. Se podrían añadir iones divalentes, tales como calcio, magnesio, cinc, etc., al tampón de carga en el intervalo de desde 0 hasta 50 mM (concentración final).
La etapa de carga se realiza normalmente a una velocidad de flujo de 10 a 72 volúmenes de columna (VC) por hora, y preferentemente a 18 a 54 VC/hora. En algunas realizaciones, la velocidad de flujo de carga oscila desde 30 hasta 48 VC/hora, y preferentemente desde 36 VC/hora. Estos intervalos específicos permiten un tiempo de contacto suficiente de FVII/FVIIa transgénico con el ligando sin excesivo tiempo para formar un complejo no disociable.
En una realización de la invención, el material cargado se aclara después de la carga en la columna hasta que se hayan eluido los aglutinantes y contaminantes no específicos.
Primera etapa de lavado - Eliminación de material sin unir
En una realización de la invención, se realiza una primera etapa de lavado tal como eluir el material sin unir contenido en la disolución cargada. Dicho primer lavado se realiza preferentemente con un tampón de lavado que tiene un pH en el intervalo de desde 2 hasta 9. En algunas realizaciones, el pH del primer tampón de lavado oscila desde 3 hasta 10, desde 3 hasta 7, desde 5 hasta 9, desde 6,5 hasta 8,5 o desde 6,5 hasta 10,0, tras la aplicación a la resina de afinidad. En algunas realizaciones interesantes, el pH del primer tampón de lavado oscila desde 4,0 hasta 7,0, desde 7,0 hasta 9,0, o desde 4,5 hasta 8,5.
Durante la primera etapa de lavado, las débiles interacciones entre las proteínas contaminantes presentes en el material fuente y los ligandos inmovilizados se pueden reducir por dicho tampón en estos intervalos específicos de pH. Las interacciones por repulsión electrostática se usan de hecho ventajosamente para eluir las proteínas débilmente unidas.
La primera etapa del lavado normalmente se realiza a un caudal de 10 a 72 volúmenes de columna (VC) por hora, y preferentemente a 18 a 54 VC/hora. En algunas realizaciones, el caudal de lavado oscila desde 30 hasta 48 VC/hora, y es preferentemente de 36 VC/hora. En una realización de la invención, el primer tampón de lavado es una disolución acuosa que comprende un agente de tamponamiento, normalmente fosfato, o Tris. Sin embargo, también se podrían usar todos los tampones carboxílicos o de ión bipolar con un pK próximo a la neutralidad.
En una realización, el primer tampón de lavado comprende tampón Tris 0 a 100 mM, preferentemente 50 mM, a un pH entre 6,5 y 8,5, y lo más preferentemente a pH 7,5.
Se debe entender que la etapa de lavado se puede extender usando uno, dos o varios tampones de lavado diferentes, o por la aplicación de un tampón de lavado en gradiente.
Segunda etapa de lavado - Retirada de material unido débil
En una realización de la invención, se aplica ventajosamente un segundo tampón de lavado antes de la elución de TgFVM/TgFVNa, para eliminar contaminantes unidos sobre la resina pero que tienen una afinidad más débil que TgFVII o TgFVIIa. En una realización, el segundo tampón de lavado contiene un compuesto de éter diol y al menos una sal soluble.
En una realización de la invención, la concentración de iones del segundo tampón de lavado se aumenta con cloruro sódico o sales caotrópicas, como magnesio. En una realización, la fuerza iónica se aumenta con componentes iónicos tales como sal aniónica y/o catiónica monovalente o multivalente. En una realización, la fuerza iónica o la concentración iónica oscila 200 a 600 mM, o más preferentemente desde 300 hasta 500 o 350 hasta 450 mM. En el mismo tiempo, la hidrofobia del tampón se aumenta con agentes hidrófobos tales como etilenglicol o propilenglicol. En una realización, el porcentaje de agente hidrófobo en el segundo tampón de lavado oscila desde 10 % hasta 50 %, desde 20 % hasta 40 %, y preferentemente desde 25 % hasta 35 %.
Por "material unido débil" se indica que la segunda etapa de lavado desencadena la retirada de material que tiene una afinidad (Kd) de al menos 100 pM, preferentemente de al menos 10 pM, por el ligando de afinidad usado en el método de la invención.
En una realización, las concentraciones iónicas e hidrófobas del segundo tampón de lavado se aumentan juntas para eliminar tantos contaminantes unidos sobre la resina de afinidad como sea posible, y preferentemente todos. El segundo tampón de lavado comprende preferentemente 0 a 40 % de glicol éter y desde 0 hasta 0,5 M de una sal de sodio, en tampón Tris a un pH desde 6,5 hasta 8,5. En una realización preferida, el segundo tampón de lavado comprende 30 % de propilenglicol y NaCl 0,4 M, en tampón Tris a pH 7,5.
También se debe observar que la segunda etapa de lavado y la etapa de elución no necesitan ser etapas discretas, pero se pueden combinar, en particular si se usa un tampón de elución en gradiente en la etapa de elución.
Etapa de elución
Después de la(s) etapa(s) de lavado, TgFVM/TgFVNa unido sobre el ligando de la columna de afinidad se eluye con un tampón de elución, y se recoge una disolución purificada de TgFVII y/o TgFVIIa como un eluato. La desorción de FVII o FVIIa humano se logra preferentemente cambiando las condiciones fisicoquímicas de forma que el ligando de unión al antígeno ya no se una un FVII o FVIIa humano. La elución se puede lograr cambiando las condiciones con respecto al pH, sal, temperatura, o cualquier otra medida adecuada.
Por la expresión "alterar de una forma no destructiva la interacción con dicho ligando" se indica que la integridad y funcionalidad de TgFVII y/o TgFVIIa se preservan en el momento de eluir dichas formas transgénicas de FVII.
En una realización, la elución se realiza con un tampón que tiene un pH inferior a 3,0, pero es necesaria una rápida neutralización a pH superior a 6,0 para mantener la actividad biológica de la molécula eluida.
El tampón de elución normalmente es una disolución acuosa que comprende un agente de tamponamiento, normalmente fosfato, o Tris. Sin embargo, también se podrían usar todos los tampones carboxílicos o de ión bipolar con un pK próximo a la neutralidad. La etapa de elución se realiza preferentemente con un tampón de elución que tiene un pH desde 2 hasta 10. En algunas realizaciones, los intervalos de pH desde 3 hasta 9, desde 5 hasta 8 y lo más preferentemente desde 7 hasta 8. Las interacciones de repulsión electrostática no son suficientes para alterar las interacciones de afinidad específicas entre TgFVII o TgFVIIa y ligando. El tampón de elución puede comprender además ventajosamente cualquier componente adicional que permita el mantenimiento de TgFVII/TgFVIIa biológicamente activo sin pérdida de transformaciones postraduccionales como glucosilación, por ejemplo, o evitando su agregación u oxidación.
Preferentemente, la fuerza iónica del tampón de elución se aumenta con sales de cloruro sódico o caotrópicas como magnesio. En una realización, la fuerza iónica del tampón de elución oscila desde 500 hasta 2500 mM, desde 1000 hasta 1800 mM, o preferentemente desde 1200 hasta 1600 mM.
Preferentemente, la hidrofobia del tampón de elución aumenta con agentes hidrófobos como etilenglicol o propilenglicol. En una realización, el porcentaje del agente hidrófobo en el tampón de elución oscila desde 20 % hasta 70 %, desde 30 % hasta 50 %, o preferentemente desde 40 % hasta 50 %.
Lo más preferentemente, la fuerza iónica e hidrofobia del tampón de elución aumentan juntas en un tampón con un pH desde 7 hasta 8. En una realización, el porcentaje del agente hidrófobo en el tampón de elución oscila desde 20 % hasta 60 % y su fuerza iónica oscila desde 0,5 hasta 2,0 M. Preferentemente, el porcentaje del agente hidrófobo oscila desde 30 % hasta 50 % y la fuerza iónica oscila desde 1,0 hasta 1,8 M, o el porcentaje del agente hidrófobo oscila desde 40 % hasta 50 % y la fuerza iónica oscila desde 1,2 hasta 1,6 M.
En una realización preferida, la desorción de moléculas activas y estables de TgFVII y/o TgFVIIa se realiza con un tampón que comprende 45 % de propilenglicol y cloruro sódico 1,5 M en tampón T ris pH 7,5.
La etapa de elución normalmente se realiza a un caudal desde 10 hasta 72 volúmenes de columna (VC) por hora, y preferentemente a 18 a 54 VC/hora. En algunas realizaciones, el caudal de elución oscila desde 30 hasta 48 VC/hora, y es preferentemente de 36 VC/hora. Cuando aumenta el caudal, se reduce la posible reasociación para liberar el ligando inmovilizado y mejora la recuperación de TgFVII en la fracción de eluato. Este efecto está relacionado principalmente con la fuerte afinidad del ligando por TgFVII o TgFVIIa.
Después de la purificación por afinidad como se propone en la invención, se prepara más de 90 % de TgFVII/TgFVMa puro, que representa una reducción de proteínas de la leche de conejo (RMP) de más de 4 logaritmos decimales (reducción de 4 log10 o 99,99 %). Sin embargo, las RMP normalmente están por debajo de 500.000 ppm, preferentemente por debajo de 100.000 ppm, y lo más preferentemente por debajo de 50.000 ppm. Se ha mostrado que la transferrina de conejo es una de las RMP contaminantes concomitantes más importantes.
Para reducir RMP hasta un nivel muy bajo, normalmente inferior a 5 ppm, el esquema de purificación tiene que integrar diversos métodos ortogonales. "Métodos ortogonales" significa procedimientos de purificación multietapa que emplean mecanismos de separación que son distintos entre sí. En la presente invención, la purificación se basa en métodos cromatográficos debido a su selectividad, eficiencia y simplicidad de aumentar de escala. La combinación de los métodos se diseñó específicamente para garantizar una alta calidad final de TgFVII y/o TgFVIIa, libre de agregados y con niveles muy bajos de formas degradadas y oxidadas.
Además, el solicitante ha descubierto propiedades inesperadas y sorprendentes de la cromatografía de afinidad, específicamente su capacidad para mejorar la reducción del título viral para virus envuelto tal como el virus de la leucemia murina (MLV) y para virus no envuelto tal como el parvovirus porcino (PPV).
En un modo preferido particular, la etapa de cromatografía de afinidad se realiza para obtener un factor de reducción viral (RF) superior a 3 log10, preferentemente superior a 4 log10, más preferentemente superior a 5 log10.
"Factor de reducción viral" significa la resultante de la relación entre el valor de la cantidad viral (título x volumen) medida antes de la implementación de la etapa cromatográfica (es decir, el valor de título viral de la carga inicial) y el valor de cantidad viral (título x volumen) medido después de la implementación de la etapa cromatográfica (es decir, el valor del título medido en el eluato). El título viral se calcula usando la ecuación de Spearman Karber como se describe en Federal Gazette N° 84, 4 de mayo de 1994, y en Schmidt, N.J. & Emmons, R.W. (1989) en Diagnostic Procedures for Viral, Rickettsial and Chlamydial Infection, 6a Edición.
Ultrafiltración/diafiltración
Según la presente invención, el material purificado por afinidad se podría diafiltrar o diluir para reducir la fuerza iónica antes de purificarse adicionalmente. En una realización de la invención, se usa membrana de ultrafiltración con un intervalo de corte desde 5 hasta 50 kDa, preferentemente 10 hasta 30 kDa. Por intercambio de tampón, la fuerza iónica disminuye hasta un intervalo desde 1 hasta 50 mM y preferentemente 5 hasta 15 mM de tampón que contiene una sal de sodio y, en una realización de la invención una sal de citrato de trisodio.
En una realización de la invención, podría ser ventajoso realizar almacenamiento a largo plazo en esta etapa y preferentemente a baja temperatura, inferior a -20 °C.
En una realización de la invención, se podría purificar adicionalmente el material purificado por afinidad estabilizado por reducción de sal, por almacenamiento a largo plazo y por retirada de microorganismos.
Cromatografía de intercambio iónico
Según la invención, el proceso de la invención comprende al menos una etapa de cromatografía de intercambio iónico para purificar adicionalmente TgFVII y TgFVIIa.
Preferentemente, se realiza al menos una etapa de cromatografía en una resina de intercambio aniónico. Dicha cromatografía de intercambio aniónico se podrían implementar con un soporte cromatográfico de tipo base fuerte o débil. En alguna realización, se puede utilizar grupo funcionalizado con amonio cuaternario de diferentes resinas comerciales como Q-Sepharose de GE Healthcare, y lo más preferentemente como el tipo Q Sepharose XL. La lista
no es limitante y se podrían usar intercambiadores aniónicos fuertes de Bio-Rad, Pall Biosepra, Merck, Millipore u otros proveedores.
Ventajosamente, la activación de TgFVII en TgFVIIa aumenta durante la etapa de separación realizada en el intercambiador aniónico.
Etapa de carga
El material previamente purificado se adsorbe al soporte aniónico en primer lugar equilibrado en el mismo intervalo de conductividad y la misma sal como un uso.
En una realización de la invención, se introduce un cambio de sal justo después de la carga para reforzar la interacción de RMP con la carga aniónica. La sal de sodio, y más particularmente la sal de citrato de trisodio, se cambia a un tampón de imidazol con una conductividad más baja. En este tampón específico, el imidazol oscila desde 1 hasta 50 mM y preferentemente desde 5 hasta 30 mM y más preferentemente 20 mM. El pH del tampón también se mantiene en el intervalo desde 6,5 hasta 8,5 y preferentemente 7 hasta 8. En dichas condiciones, la transferrina y TgFVII o TgFVII no activo complejado con otras proteínas muestran una interacción reforzada con la resina de intercambio aniónico.
Primer lavado
En una realización de la invención, se aplica un primer lavado aumentando la concentración de sales y especialmente la concentración de sal de cloruro sódico en un intervalo entre 50 y 175 mM y más preferentemente entre 130 y 160 mM. Este lavado se podría aplicar por gradiente o por etapa. En una realización de la invención, este lavado muestra una elución específica de transferrina endógena de leche, mientras que, al mismo tiempo, en presencia de tampón imidazol, TgFVM/TgFVIIa permanece sobre el gel iónico y también se mantienen adsorbidas trazas de FVII transgénico complejado con otras proteínas.
Elución de TgFVII/TgFVIIa
En la realización de la invención, se eluye TgFVN/TgFVNa del intercambiador aniónico disminuyendo la concentración de sales y especialmente la concentración de sal de cloruro sódico en un intervalo entre 0 y 75 mM y más preferentemente entre 40 y 60 mM y al mismo tiempo, introduciendo una sal de calcio a concentración muy baja, en el intervalo desde 2 hasta 9 mM y más preferentemente entre 5 y 8 mM. Se ha estudiado durante un largo tiempo la interacción específica de calcio con las proteínas que contienen el dominio gla, tales como la molécula de FVII. El cambio conformacional en la molécula de FVII conduce a un cambio de carga global que permite la elución a baja conductividad. En una realización de la invención, se mantienen adsorbidas en presencia de imidazol trazas de TgFVII complejadas con otras proteínas. Otras sales como tris-hidroximetilaminometano o citrato de trisodio muestran una co-elución de TgFVII libre y/o con TgFVII complejado con otras proteínas
TgFVII eluido del intercambiador aniónico comprende desde 50 % hasta 100 % de formas activadas de TgFVIIa y más preferentemente desde 80 hasta 100 %.
Además, el solicitante ha descubierto propiedades inesperadas y sorprendentes de la cromatografía de intercambio iónico, específicamente su capacidad para mejorar la reducción de título viral para virus envuelto tal como el virus de la leucemia murina (MLV) y para virus no envuelto tal como el parvovirus porcino (PPV).
En un modo preferido particular, la etapa de cromatografía de intercambio iónico se realiza para obtener un factor de reducción viral (RF) superior a 3 log10, preferentemente superior a 4 log10, más preferentemente superior a 5 log 10. Cromatografía de pseudo-afinidad, preferentemente sobre hidroxiapatita
Según la invención, el eluato recogido de la cromatografía de intercambio aniónico y que contiene TgFVII y/o TgFVIIa se purifica por cromatografía de pseudo-afinidad ventajosamente sobre un gel de hidroxiapatita (Ca10(PO4)6(OH)2).
Esta cromatografía de pseudo-afinidad adicional se realiza preferentemente bajo las siguientes condiciones: la columna se equilibra con un tampón A acuoso que comprende fosfato de potasio o sodio desde 10 hasta 30 mM o una mezcla de los mismos, a un pH entre 7,5 y 8,5.
Las moléculas de TgFVII/TgFVIIa formuladas con calcio son retenidas sobre el soporte, mientras que Des-gla-FVII y otras escisiones se retiran en el flujo a través ("Des-Gla-FVII" significa un FVII cuyo dominio g La se ha retirado, posiblemente por proteólisis).
La percolación del tampón A, justo después de la etapa de carga, eluye una parte de TgFVII y/o TgFVIIa parcialmente escindido durante el proceso y esto permite una buena eliminación de formas de FVII no deseables, tales como TgFVIIa de baja actividad o inactivo.
La elución de TgFVIIa se realiza con un tampón que comprende una sal de fosfato, tal como fosfato de sodio o potasio, o una mezcla de las mismas, a una concentración predeterminada, que preferentemente representa un tampón B que comprende fosfato de sodio desde 0,1 M hasta 0,5 M y preferentemente desde 0,1 M hasta 0,2 M con un pH entre 7,5 y 8,5. El eluato resultante contiene TgFVIIa completamente activo, con un bajo porcentaje de formas escindidas.
Cromatografía de exclusión por tamaño
Las trazas finales de RMP solo se podrían identificar por espectrometría de masas o por ELISA específica. Antes de la etapa de pulido final, RMP varían entre 50 y 500 ppm y lo más preferentemente inferior o igual a 300 ppm.
Según la invención, la etapa d) se realiza por cromatografía de exclusión por tamaño. La altura de la columna de exclusión por tamaño, porosidad de la resina y tampón de cromatografía se tienen que seleccionar para alcanzar la mejor selectividad. Se requieren resinas de alto rendimiento equivalentes a Superdex S75 prep grad o más preferentemente Superdex S200 prep grad para la etapa de pulido. La altura del lecho de gel superior a 60 cm e inferior a 120 cm y más preferentemente superior a 80 cm e inferior a 110 cm permite la resolución suficiente para retirar 2 logaritmos adicionales de trazas de RMP.
En una realización, el intercambio de tampón aplicado durante la SEC contiene un detergente no iónico a una concentración entre 20 y 500 ppm, o preferentemente desde 50 hasta 200 ppm, en combinación con aminoácidos como arginina o glicina o azúcar sin limitación. La presencia del detergente iónico en el tampón ayuda ventajosamente a mantener el estado monomérico de TgFVII/TgFVIIa durante SEC, que conduce a una separación óptima entre FVII y moléculas contaminantes. En una realización de la invención, el tampón de formulación contiene mezclas de diferentes aminoácidos con monolaurato de sorbitano o/y monooleato como se describe en la patente de EE.UU. 2012/0087908. Se aplica un caudal lineal entre 10 y 40 cm/h, más preferentemente entre 20 y 25 cm/h, combinado con un volumen de carga que varía desde 1 % hasta 10 % del volumen de columna de SEC, más preferentemente desde 2 hasta 5 % del volumen de columna de SEC para obtener un pico monomérico de TgFVII y/o TgFVIIa inferior a 5 ppm de RMP o una pureza superior a 99,9995 %.
Además, el solicitante ha descubierto propiedades inesperadas y sorprendentes de la cromatografía de exclusión por tamaño, específicamente su capacidad para mejorar la reducción del título viral para virus envuelto tal como el virus de la leucemia murina (MLV) y para el virus no envuelto tal como el parvovirus porcino (PPV).
En un modo preferido particular, la etapa de cromatografía de exclusión por tamaño se realiza para obtener un factor de reducción (RF) viral superior a 3 log10, preferentemente superior a 4 log10, más preferentemente superior a 5 log10.
Así, la etapa de cromatografía de afinidad y/o la etapa de cromatografía de intercambio iónico y/o la etapa de cromatografía de exclusión por tamaño permiten obtener un factor de reducción (RF) viral superior a 3 log10, preferentemente superior a 4 log10, más preferentemente superior a 5 log10.
Eliminación/Inactivación de virus
El método de la invención también puede comprender al menos una etapa dedicada a la inactivación/retirada viral, pero lo más preferentemente se tienen que incluir al menos dos etapas dedicadas a la inactivación/retirada viral ortogonal. La leche clarificada podría ser viralmente inactivada por tratamiento químico con disolvente y/o tratamiento con detergente u otros tipos de tratamiento eficientes. En una realización de la invención, se usa disolvente/detergente, en particular en presencia de una mezcla de Tween® 80 (1 % a 1,5 % en peso/volumen) y de TnBP (fosfato de tri-n-butilo 0,3 % a 0,5 % v/v), para inactivar virus envueltos. Otros detergentes no iónicos como Octoxinol100 o Triton X114 o colato de sodio o Tween® 20 sin limitación también podrían sustituir o complementar Tween® 80.
Además, el eluato resultante de cualquiera de las etapas cromatográficas descritas se puede someter a una etapa de nanofiltración para eliminar eficientemente los virus, en particular los virus no envueltos, tales como el parvovirus B19. Es en particular posible usar los filtros ASAHI PLANOVA™15 que retienen los virus con un tamaño superior a 15 nm.
En una realización de la invención, se realiza nanofiltración, preferentemente después de la primera etapa cromatográfica, donde la pureza de TgFVII es superior a 90 % y la parte principal de altos tamaños moleculares de proteína como micelas de caseínas se eliminan suficientemente para permitir la filtración en dichos nanofiltros de tamaño de poro. La concentración de FVII de partida es preferentemente entre 1,0 y 2,5 g/l y más preferentemente entre 1,3 y 2,2 g/l y un caudal típico es entre 5 y 15 LMH, preferentemente entre 8 y 12 LMH ("LMH" significa litros por metro cuadrado por hora, o L/m2/h).
Además de la retirada viral, la nanofiltración muestra también la ventajosa reducción del ADN endógeno por un mecanismo de tamizado a través de la anchura de los nanofiltros.
En una realización de la invención, se podrían usar filtros de nanofiltración equivalentes al tamaño de poro medio en nanómetros de Planova15. El filtro de tamaño de poro de 20 nanómetros podría sustituir el filtro de 15 nanómetros y el filtro de 15 nanómetros se podría acoplar al filtro con porosidad más alta. En una realización de la invención, un nanofiltro secuencial 0,2 pm/0,1 pm seguido por un Planova 20 seguido por un nanofiltro Planova 15 podría reducir ventajosamente cualquier riesgo al mismo tiempo (biocarga, micoplasmas, virus, agentes adventicios, TSE) sin pérdida de la actividad biológica de TgFVII/TgFVIIa.
Formulación, esterilización, liofilización
Una vez se recupera el último eluato que contiene TgFVII y TgFVIIa purificados, dicho eluato se podría enviar a una etapa de filtración sobre filtros de 0,22 pm, cargar en recipientes y luego congelar/secar.
Preferentemente, la membrana de PVDF modificada se usa ventajosamente en la invención debido a la baja unión de proteínas y la baja adsorción de detergente no iónico descrito por los proveedores. Los filtros Durapore® Membrane de Millipore se adaptan bien a esta etapa sin limitación de otros filtros comerciales.
Los ejemplos a continuación ilustran la invención sin limitar su alcance.
EJEMPLOS
Ejemplo 1: Preparación de ligandos anti-FVII/FVIIa específicos
Se inmunizó una llama con antígeno FVIIa humano altamente puro. Tras el segundo refuerzo, se observó un título incrementado más de 100 veces, que indica un enriquecimiento específico contra los antígenos FVIIa/FVII humanos. Se recogió ARN de la llama y se generaron bibliotecas de expresión de VHH. Se probaron los clones en ELISA para la expresión de VHH de llama con la capacidad para unirse a FVIIa humano. Se seleccionaron 23 clones VHH y se probaron adicionalmente para su capacidad para unirse a FVII y FVIIa humano derivado de plasma y FVII de conejo recombinante. Se encontró que todos los clones eran capaces de unirse a FVII y FVIIa humano, y no se observó unión cruzada hacia FVII de conejo recombinante. Los clones se probaron adicionalmente para sus capacidades de unión y elución con respecto a FVII/FVIIa, así como para la estabilidad de sus propiedades de unión a pH ácido. Se reclonó un candidato seleccionado en el sistema de expresión de levadura de BAC y se expresaron ligandos correspondientes, entonces se acoplaron sobre NHS-Sepharose a una densidad de ligando de 2,5 mg por ml de matriz.
Ejemplo 2: Purificación de TgFVIIa de leche
El presente ejemplo describe la preparación de una disolución de TgFVII activada altamente purificada a partir de 150 litros de leche de conejo.
2.1 Recogida de leche
El conjunto compuesto inicial de leche consistió en un ciclo completo de lactancia de mini-conjuntos de leche recogida 6 veces al día, envasada junta en una botella de 1 l, se muestreó para los controles de seguridad biológica (biocarga, endotoxinas y virus endógeno), luego se guardó y se congeló a temperatura muy baja (<-60 °C) durante un almacenamiento a largo plazo (varios meses a <-20 °C). Se recogió leche desde Día 4 hasta Día 24 correspondiente a un ciclo de ordeño natural.
2.2 Clarificación
Botellas congeladas que contenían leche se dispusieron en un gran tanque de descongelación mantenido a 32 37 °C. El tiempo de descongelación fue alrededor de 1 hora para obtener el último trozo de hielo en cada botella. Se transfirieron 147,4 kg de material fuente (SM) descongelado y se reunieron en una bolsa de sistema de mezcla de 500 ml. Se añadió tampón citrato de trisodio 0,315 M para obtener una concentración de proteína en el intervalo 20 a 40 g/l. La temperatura de la mezcla se ajustó a 24-26 °C. Se detuvo el sistema de mezclado para permitir la decantación de grasa en la capa superior del SM diluido y citrado.
Se equilibraron preliminarmente en tampón filtros profundos con intervalo de tamaño de poro entre 15 y 0,5 pm (PDH4) y entre 0,3 y 0,1 pm (PEKS) manipulados con un dispositivo STAX (Pall Corporation). Se filtró la leche tratada con tampón citrato y se cargaron 444 Kg de filtrado en una bolsa de un solo uso antes de realizar una filtración de 0,2 pm con filtro Sartobran® de 0,45/0,2 pm (Sartorius Corporation). Se adaptó la capacidad de filtración de los diferentes filtros para filtrar sin obstrucción desde aproximadamente 2 hasta 15 kg de proteínas por metro cuadrado dependiendo del tamaño de poro medio progresivo.
2.3 Tratamiento con S/D
Se hizo la adición de disoluciones de polisorbato 80 y fosfato de tri-n-butilo a la fuente clarificada de material (CSM) para obtener 509,8 kg de disolución que contenía 0,8-1,5 % en peso/volumen de detergente y 0,2-0,5 % v/v de disolvente. La temperatura se ajustó a 25 ± 2 °C y se mantuvo al menos 2 horas de tiempo de contacto en dichas condiciones para inactivar cualquier posible virus no envuelto en la fuente biológica. Las trazas de lípidos residuales
o triglicéridos procedentes de la leche también se disolvieron químicamente mediante la adición de detergente y disolvente, haciendo una disolución más adecuada para las etapas cromatográficas adicionales.
2.4 Cromatografía de afinidad en ligando específico de FVII/FVIIa transgénico
Se equilibró la columna con tampón Tris 50 mM a pH 7,5, luego se cargó la disolución que contenía las proteínas a 220 l/h. 6 litros de gel de afinidad fueron lo suficientemente grandes como para adsorber todo el TgFVII del conjunto de leche tratado. Después de la carga, el primer lavado se hizo con tampón Tris. Fueron necesarios 90 litros para garantizar un retorno inicial estable. Durante este gran lavado se retiraron proteínas unidas por interacciones no específicas.
Se realizó una etapa de lavado optimizada para maximizar la elución de RMP y minimizar la desorción de FVII aumentando la fuerza iónica hasta 500 mM de cloruro sódico y la hidrofobia relativa con 30 % v/v de propilenglicol hasta el tampón Tris inicial.
Se realizó una etapa de elución optimizada para minimizar la desorción de RMP y maximizar la elución de FVII aumentando la fuerza iónica hasta 1500 mM de cloruro sódico y la hidrofobia relativa con 45 % v/v de propilenglicol. Se recogieron 50,6 litros en la fracción de eluato de afinidad. La relación FVII amidolítico/FVII antigénico del eluato de afinidad fue próxima a 1 correspondiente a 1 unidad de factor de coagulación para 1 unidad de molécula de antígeno. La etapa de afinidad permitió la retirada de la parte principal de RMP con una eliminación de 4 log.
De esta forma, se inmunopurificó satisfactoriamente TgFVM/TgFVNa coagulable.
2.5 Ultrafiltración
Como alternativa, se fabricó un producto intermedio que se podría almacenar usando la tecnología de ultrafiltración. Se concentró y estabilizó el eluato tratado con S/D por afinidad sobre membrana de 30 kDa de poliétersulfona, TangenX Sius-LS® de un solo uso de Novasep. La presión transmembranaria se mantuvo a 0,14 MPa (20 psig) durante la fase de concentración por ajuste de la velocidad de la bomba de recirculación. Cuando se obtuvo una concentración de proteína de 2 g/l, se realizó diafiltración en tampón citrato para retirar compuesto hidrófilo, sales y disolventes químicos.
2.6 Filtración y congelación estériles - Producto intermedio 1
Se filtraron asépticamente 10,4 l del producto intermedio concentrado con una actividad específica de 2638 unidades por mg con cápsulas Sartobran® de 0,45/0,2 gm bajo una campana de flujo laminar en recipientes de 1 l.
Las botellas se almacenaron <-60 °C para constituir el producto intermedio para el proceso aguas abajo.
2.7 Filtración 0,2/0,1 gm y nanofiltración sobre filtros Planova de 20N y 15 N
Fueron necesarias más purificaciones para un uso humano clínico y farmacéutico del producto TgFVN/TgFVNa. En primer lugar, para reducir la posible carga viral, el producto intermedio se filtró a través del tren de filtración:
• Cápsula desechable Sartopore 2® de 0,2/0,1 gm de Sartorius
• Planova® 201 m2 seguido por Planova® 15 de 1 m2 de ASAHI KASEI
Se bombearon directamente 9,45 l de producto intermedio a través del tren de filtración a 12 LMH de caudal inicial y se monitorizó la presión transmembranaria durante toda la etapa para mantener un máximo de 0,08 MPa (12 psig) en cada nanofiltro. En esta etapa del proceso, la pureza se calculó alrededor de 95 % y se identificó la transferrina endógena como uno de los principales contaminantes.
En esta etapa del proceso, se evalúa la activación de TgFVII entre 20 y 50 % por cuantificación de análisis de SDS-PAGE en condiciones reducidas. La activación parcial se atribuye al calcio de la leche liberado durante la fase de clarificación del proceso de purificación.
2.8 Cromatografía de intercambio iónico sobre Q Sepharose XL
Se potenció la pureza y activación de TgFVII por cromatografía de intercambio aniónico y elución dependiente de calcio. Se cargó la disolución nanofiltrada en un intercambiador aniónico Q-Sepharose XL (GE Healthcare) previamente equilibrado con citrato de sodio 10 mM a pH 7,0.
Se realizó una primera etapa de lavado con imidazol 20 mM a pH 7,5 y conductividad bajo 5 mS/cm a 25 °C. Se realizó una segunda etapa de lavado introduciendo 150 mM de cloruro sódico, además de imidazol 20 mM, para alcanzar una conductividad de 18 mS/cm a 25 °C. Se identificó que el pico de lavado estaba principalmente compuesto de transferrina libre. Entonces se eluyó TgFVII/TgFVIIa disminuyendo la sal de cloruro sódico hasta 50 mM y en el mismo tiempo añadiendo cloruro de calcio hasta 7 mM, mientras se mantenía el imidazol a 20 mM. La
fracción de elución contuvo más de 90 % de forma activada de TgFVIIa evaluado por SDS-PAGE en condición reducida. Se midieron los niveles de RMP por ELISA y se compararon con TgFVII, se calculó RMP residual inferior a 500 ppm (véase la Tabla 3).
La cromatografía QSXL contribuyó a la reducción de los niveles de HRI, con una puntuación media de 2 log10, de la que 1,6 log10 fueron transferrina.
Durante esta etapa, la activación en FVIIa se potenció por la presencia de calcio ionizado en el tampón de elución. Entonces se almacenó el eluato de Q-Sepharose XL a 2/+8 °C durante la noche.
2.9 Cromatografía de pseudo-afinidad sobre hidroxiapatita
Durante el almacenamiento, podrían ocurrir algunas escisiones de FVII, especialmente la escisión en la cadena pesada, siendo la cadena ligera protegida por iones de calcio. Los adsorbentes de hidroxiapatita y fosfato de calcio son bien conocidos por adsorber factores de coagulación tales como FVII/FVIIa y más ampliamente proteínas que contienen el dominio Gla. Se usó esta pseudo-afinidad para la etapa de pulido del proceso.
El eluato de Q Sepharose XL que contiene FVII/FVIIa transgénico se concentró en una columna de tipo I de hidroxiapatita CHT Ceramic (BioRad), previamente equilibrada con fosfato de potasio 25 mM a pH 8,0.
Se realizó una etapa de lavado isocrático con fosfato de potasio 25 mM a pH 8,0. Esta etapa de lavado eluyó específicamente formas proteolizadas, especialmente TgFVII/TgFVIIa que contienen una o más escisiones sobre su cadena pesada, y se monitorizó para limitar la mínima pérdida de formas activas completas en un modo isocrático. Entonces se eluyó TgFVII/TgFVIIa como un concentrado de TgFVIIa con fosfato de potasio 150 mM a pH 8,0.
2.10 Cromatografía de exclusión por tamaño (SEC) en Superdex 200
Se hizo la estabilización de TgFVII/TgFVIIa monomérico por la última etapa de pulido del proceso. La SEC permitió la retirada de fosfato de potasio y la formulación en al menos polisorbato 80 y arginina.
Se cargó la fracción eluida de la columna cromatográfica de hidroxiapatita y que contenía TgFVII/TgFVIIa sobre una columna de Superdex 200 (GE Healthcare), previamente equilibrada con citrato de sodio 5 mM, HCl de arginina 114 mM, isoleucina 46 mM, glicina16 mM, lisina 6,5 mM, 0,07 % (v/v) de Tween 80, pH 6,0.
Las proteínas con mayor radio dinámico o mayor tamaño molecular como la transferrina (70 kDa) o FVII complejado con otras proteínas se eluyeron justo antes de TgFVIIa monomérico (45 kDa) por exclusión por tamaño.
Las proteínas con menor radio dinámico o menor tamaño molecular como las caseínas o péptidos se eluyeron justo después de TgFVIIa monomérico (45 kDa).
La fracción que contenía TgFVII/TgFVIIa se eluyó con el mismo tampón que se usa para equilibrar la columna. 2.11 Estabilización y formulación
Se formuló la disolución final de TgFVII/TgFVIIa a una concentración de 1 g de TgFVIIa/l de tampón de formulación que comprende citrato de sodio 5 mM, HCI de arginina 114 mM, isoleucina 46 mM, glicina 16 mM, lisina 6,5 mM, 0,07 % (v/v) de Tween 80, pH 6,0.
Se evaluó por SDS-PAGE en condición reducida 93,5 % de forma activada de TgFVIIa.
2.12 Esterilización
Entonces se filtraron asépticamente sobre una membrana Millipak 100 de 0,2 gm 5243 ml de disolución de TgFVII/TgFVIIa estabilizada formulada.
Ejemplo 3: Resultados analíticos durante el proceso
Se analizaron las muestras generadas durante el proceso de purificación como se describe en el Ejemplo 2.
3.1 Reproducibilidad del proceso de purificación.
Los rendimientos de FVII de cada etapa, seguido con el ensayo amidolítico de FVII o absorbancia en 8 lotes, se resumen en la Tabla 1 y 2.
Tabla 1: Rendimientos amidolíticos de FVII hasta la ultrafiltración

Tabla 2: Rendimientos de la absorbancia de FVII hasta la filtración por esterilización

Los rendimientos de FVII mostraron resultados coherentes con menos de 10 % de variación con respecto al valor medio para cada etapa.
3.2 Eliminación de RMP
Se determinó la eliminación media de RMP para los lotes probados y se muestra en la Figura 2 (eliminación de RMP).
La eliminación de RMP durante el proceso de purificación fue perfectamente reproducible. Las etapas que contribuyeron más a la eliminación de RMP fueron:
- la cromatografía de afinidad en un ligando que es específico para FVII/FVIIa
- la cromatografía en Q Sepharose XL, y
- la cromatografía en Superdex200.
Tabla 3: Eliminación de RMP durante el proceso de purificación.

La eliminación global de RMP resultante del proceso fue entre 8 y 9 log10.
3.3 Eliminación de ADN de conejo
También se evaluó la eliminación de ADN de conejo ensayando su contenido en los diferentes productos intermedios del proceso por una técnica de Q-PCR. Los resultados correspondientes se presentan en la Figura 3 (eliminación de ADN). Los resultados se expresan en ppb con respecto a la concentración de TgFVII/TgFVIIa ensayada por ELISA en la leche y por DO280 para los otros productos intermedios.
Tabla 4: Eliminación de ADN de conejo durante el proceso de purificación.
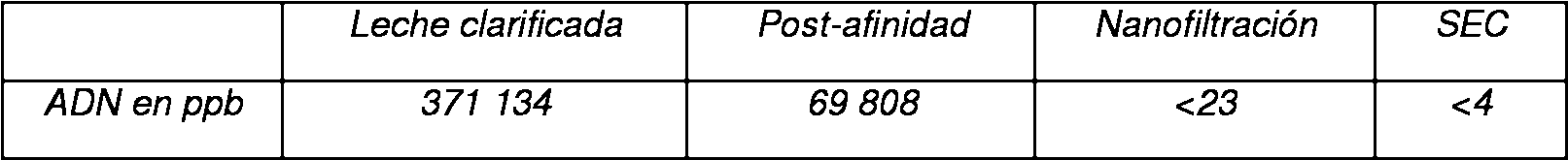
La etapa que contribuyó más fue la nanofiltración. Después de la cromatografía en Q Sepharose XL, todos los lotes estuvieron por debajo del nivel de cuantificación (LOQ).
La eliminación total de ADN resultante del proceso estuvo entre 5 y 6 log10.
3.4 Cinética de la activación de FVII
Como se desveló anteriormente, TgFVII se activa en TgFVIIa durante el proceso, y la activación se potencia principalmente con cromatografía en Q Sepharose XL. Como se detalla en la Tabla 5 a continuación, al final del proceso, la relación de activación definida por la relación de la unidad de coagulación por unidad de antígeno estuvo entre 17 y 25.
Tabla 5: Cuantificación de TgFVIIa mediante el proceso de purificación

TgFVII podría ser aproximadamente y fácilmente seguido por SDS-PAGE (coloración con azul de Coomasie, Figura 4)
También se podrían clasificar las isoformas de FVII con una mayor exactitud por análisis de mapeo de proteínas (RP(C4)-HPLC-EM). Se determinaron las formas escindidas del sitio de activación (cadena pesada y cadena ligera) para generar TgFVIIa biológicamente activo. La cadena sin escindir correspondió a TgFVII no activado con actividad biológica muy baja y las isoformas escindidas de las cadenas ligeras y pesadas correspondieron a la degradación molecular e inactivación de la actividad biológica (% de LC: Escisión de cadena ligera o % de Des-Gla, % de HC: Escisión de cadena pesada).
El método analítico no fue capaz de cuantificar específicamente las formas oxidadas. Las formas se incluyeron en la cuantificación de la forma no activada.
El TgFVII no activado no degradado disminuyó progresivamente durante el proceso, un signo de la activación de TgFVII en TgFVIIa. Para los 3 lotes probados, la activación fue progresiva y, al final del proceso, permaneció un promedio de 3,6 % de la forma no activada. Esta forma fue no degradada y retuvo su capacidad para ser activada
después en el proceso (operación de formulación) y durante la administración intravenosa. Retuvo sus posibilidades de activación en plasma humano.
3.5 Degradación de TgFVIIa en el proceso
Para este ejemplo, se concentró la fracción de eluato de hidroxiapatita (CHT) hasta 3,9 g/l de TgFVIIa y los resultados de RP-HPLC se resumieron en la Tabla 6 en comparación con los resultados de SEC mostrados en la Tabla 7.
Tabla 6: Reparto de la forma de FVII en el eluato de CHT (RP-HPLC):

Tabla 7: Reparto de la forma de FVII en el producto purificado (RP-HPLC):

El total de formas de TgFVII ni escindidas ni oxidadas fue superior a 90 % de TgFVII total para tanto el eluato de CHT como el producto purificado final (eluato de SEC). Esto demuestra claramente la capacidad de la etapa de CHT para controlar la degradación de Tg FVII a pesar de la activación de Tg FVII.
Ejemplo 4: Calidad de la preparación de TgFVIIa altamente purificado
4.1 Cuantificación de RMP residual en la disolución purificada.
El uso de un ligando altamente específico de FVII derivado de un anticuerpo de llama en combinación con técnicas cromatográficas más clásicas (intercambio iónico, pseudo-afinidad en hidroxiapatita y exclusión por tamaño) permitió así optimizar la purificación de TgFVII/TgFVIIa de leche, eliminando más particularmente RMP en la medida de lo posible.
Los niveles de RMP fueron de aproximadamente 10 ppm para lotes piloto (15 l de leche) como se representa en la Tabla 8 y por debajo del nivel inferior de cuantificación (LLOQ) o inferior a 6 ng/ml para los lotes producidos a gran escala (150 l de leche).
La transferrina se ensayó por ELISA en el producto purificado y su contenido se encontró próximo a 3 ppm para los lotes piloto.
Tabla 8: Contaminación por RMP y transferrina en el producto purificado


4.2 Cuantificación de ADN de conejo residual en la disolución purificada.
El proceso como se describe alcanza así un ADN residual diana inferior a 4 ppb.
4.3 Factor de coagulación VII de conejo
Puesto que los conejos tienen un factor VII endógeno en su circulación y existe cierta fuga pasiva de proteínas séricas de la corriente sanguínea en la leche, se evaluó la capacidad del proceso de purificación para separar humano de FVII de conejo. Se desarrolló internamente un ELISA específico para la investigación de la presencia de dicha posible proteína concomitante en el producto purificado. Los resultados fueron inferiores a LLOQ (<45 pg/ml para todas las preparaciones analizadas).
Ejemplo 5: Estudios de eliminación viral para virus envuelto (MLV, virus de la leucemia murina) después de implementar una etapa de cromatografía de afinidad en ligando específico de Tg FVIIa
Las características del virus de la leucemia murina (MLV) se describen en la tabla a continuación:

5.1 Ensayo de valoración de la infectividad
5.1.1 Principio de la valoración de la infectividad
El método de valoración es un ensayo cuantitativo en el que la medición del título de virus se basa en la detección de la producción de virus en las células infectadas, por observación de un efecto citopático específico.
El ensayo de valoración se hará en dos placas de 96 pocillos diferentes como se describe a continuación:
• Se diluye la muestra de prueba con medio de cultivo por diluciones triples en serie (se realizan ocho duplicados para cada dilución) a través de la placa de 96 pocillos ("placa de dilución de muestras").
• Cada pocillo de la "placa de dilución de muestras" se inocula entonces en el pocillo correspondiente de una nueva placa de 96 pocillos ("placa de valoración de muestras").
Se añade suspensión de células a cada pocillo de la "placa de valoración de muestras" y las placas se incuban entonces a temperatura apropiada con o sin atmósfera de CO2 (dependiendo de los virus).
Después de un periodo de incubación que permite la replicación e infección viral de células adyacentes, dependiendo de los virus,
• se cuentan los pocillos con focos después de la infección por observación en microscopio óptico invertido. • o se añade una sobrecapa de tinción (cristal violeta 1p-galactosidasa) y los pocillos se examinan para el efecto citopático. Los pocillos infectados aparecen como áreas claras, mientras que los pocillos no infectados se tiñen (o al revés, dependiendo de los virus).
Se calcula el título infeccioso expresado como 50 % de dosis infecciosa de cultivo de tejido por mililitro (TCID50/ml) usando la fórmula apropiada.
5.1.2 Controles de valoración
* Control negativo
Durante cada ensayo de valoración, las células se preparan como control de referencia de células. Estas células se preparan en las mismas condiciones que las usadas para la valoración de las muestras generadas durante los ensayos preliminares, excepto que se inoculan con medio de cultivo no añadido (4 - 8 pocillos en cada placa de 96 pocillos).
* Control de valoración positivo (control de referencia del virus)
Durante cada ensayo de valoración, se valora un control de referencia del virus de aproximadamente 103 - 105 TCID50/ml en las mismas condiciones que las usadas para la valoración de las muestras generadas durante los ensayos preliminares.
5.1.3 Criterios de aceptación del ensayo de valoración
Se conserva el ensayo de valoración y se valida cuando:
• El control de referencia de células para cada placa de valoración es conforme al resultado esperado,
• El título infeccioso del control de referencia del virus obtenido está en el intervalo esperado.
5.1.4. Determinación del título viral
El título viral se determina según Schwartz D., 1993 (Schwartz D., "Methodes statistiques a I’usage des medecins et des biologistes", Flammarion Medecine-Sciences, cuarta edición, 1993); Kaplan M. y Koprowski H., "Laboratory techniques in rabies, tercera edición, editado por la Organización Mundial de la Salud, Ginebra, 1973.
Se calcula la TCID50 (50 % de dosis infecciosa de cultivo de tejido) usando la fórmula de Spearman Karber. La TCID50 se evalúa por ensayo cuántico y se define como la dosis de virus capaz de infectar 50 % de los cultivos inoculados.
5.2 Protocolo experimental
Las etapas implementadas en el proceso son las siguientes:
- Descongelación del material fuente clarificado;
- Infección del material fuente clarificado descongelado con 1 % del virus envuelto MLV;
- Prefiltración con filtros 0,2 |um;
- Carga sobre la columna FVII Select;
- Lavado de la columna;
- Elución.
La Tabla 9 proporciona las condiciones de proceso implementadas durante la etapa de cromatografía en Affinity FVII Select.
Tabla 9:


5.3 Resultados
Se ha medido la eliminación viral en una muestra de material fuente descongelado y clarificado después de la etapa de prefiltración (muestra llamada Carga 2) y después de la elución en cromatografía en Affinity FVII Select (muestra llamada Eluato). Los resultados se muestran en la Tabla 10.
Tabla 10: Medición de la eliminación viral antes (Carga 2) y después de la cromatografía en Affinity FVII Select
(Eluato)

Los resultados de la Tabla 10 muestran que la etapa de cromatografía en Affinity FVII Select reduce el título viral de un factor de reducción (RF) superior a 4,37 log10.
Ejemplo 6: Estudios de eliminación viral para el virus no envuelto (PPV, parvovirus porcino) después de implementar una etapa de cromatografía de afinidad en ligando específico de Tg FVIIa.
Las características del parvovirus porcino (PPV) se describen en la tabla a continuación:

6.1 Protocolo experimental
Las etapas implementadas en el proceso son las siguientes:
- Descongelación del material fuente clarificado;
- Infección del material fuente clarificado descongelado con 1 % del virus no envuelto MLV;
- Prefiltración con filtros 0,65/0,45 gm y 0,45/0,2 gm;
- Tratamiento con disolvente/detergente (1 hora);
- Carga sobre la columna FVII Select;
- Lavado;
- Elución.
La Tabla 11 proporciona las condiciones de proceso implementadas durante la etapa de cromatografía en Affinity FVII Select.

6.2 Resultados
Se ha medido la eliminación viral en una muestra de material fuente descongelado y clarificado después de la etapa de prefiltración y de tratamiento con disolvente/detergente (muestra llamada Carga inicial) y después de la elución en cromatografía en Affinity FVII Select (muestra llamada Eluato A). Los resultados se muestran en forma de histograma en la Figura 5.
El histograma de la Figura 5 muestra que la etapa de cromatografía en Affinity FVII Select reduce el título viral de un factor de reducción (RF) superior a 3,8 log10.
Ejemplo 7: Estudios de eliminación viral para el virus envuelto (MLV) y virus no envuelto (PPV) después de implementar una etapa de cromatografía de exclusión por tamaño Superdex S200 prep grad
7.1 Protocolo experimental
Las etapas implementadas en el proceso son las siguientes:
- Descongelación del material de partida muestreado de los lotes clínicos (eluato de hidroxiapatita-1 cerámica (CHT-1));
- Infección del material de partida con 1 % de adición (1 % de MLV o 1 % de PPV);
- Prefiltración con filtros 0,2 gm (MLV) o filtros 0,1 gm (PPV);
- Carga de 2,3 % de volumen de columna (VC) en la columna S200;
- Recogida del eluato S200 (fracción correspondiente a FVII).
La Tabla 12 proporciona condiciones de proceso implementadas durante la etapa de cromatografía de exclusión por tamaño Superdex S200 prep grad.


7.2 Resultados
Se ha medido la eliminación viral en una muestra de eluato de CHT-1 después de la etapa de prefiltración en filtros 0,2 pm (para virus MLV) o en filtros 0,1 pm (para virus PPV) (muestra llamada Carga 2), y después de elución en cromatografía de exclusión por tamaño Superdex S200 (muestra llamada Eluato). Los resultados se muestran en la Tabla 13.
Tabla 13: Medición de la eliminación viral antes (Carga 2) y después de la cromatografía de exclusión por tamaño Superdex S200 (Eluato)

Los resultados de la Tabla 10 muestran que la cromatografía de exclusión por tamaño la cromatografía Superdex S200 reduce el título viral de un factor de reducción igual a 3,01 log10 para virus envuelto (MLV) y un factor de reducción igual a 3,00 log10 para virus no envuelto (PPV).
En resumen:
- Se obtienen factores de reducción altos de la eliminación viral con la cromatografía en Affinity FVII Select, que son superiores a 4,37 log10 para virus envuelto (MLV) e iguales a 3,8 log10 para virus no envuelto (PPV);
- Se obtienen factores de reducción medios de la eliminación viral con la cromatografía de exclusión por tamaño Superdex S200, que son iguales a 3,0 log10 para virus envuelto (MLV) y virus no envuelto (PPV); la cromatografía en Affinity FVII Select es más eficiente que la cromatografía de exclusión por tamaño en Superdex S200 para reducir el título viral de virus envueltos y no envueltos.
Ejemplo 8: Estudios de eliminación viral para virus envuelto y virus no envuelto después de implementar varias técnicas
8.1 Protocolo experimental
Se realizaron por separado los estudios de validación viral de cromatografía de afinidad FVII Select, nanofiltración y Q Sepharose XL con 1 % de adición de diversos virus en el material de partida para cada operación de la unidad de purificación y la valoración del virus con adición en la fracción de interés (uno era FVII recuperado). Se añadieron el factor de reducción log10 obtenido para cada operación de la unidad de purificación para cada virus y se estimó virus por virus un factor de reducción global.
8.2 Resultados
Los resultados se dan en la Tabla 14.
Tabla 14:

Claims (1)
- REIVINDICACIONES1. Un método de purificación de factor VII humano transgénico (TgFVII) y/o factor VII humano transgénico activado (TgFVIIa) producido en la leche de animal no humano transgénico, que comprende;(a) una etapa de cromatografía de afinidad que comprende las etapas de:i. poner en contacto la leche que contiene TgFVII humano y/o TgFVIIa humano con un ligando que es específico para TgFVII humano y/o TgFVIIa humano, en condiciones que permiten que TgFVII humano y/o TgFVIIa humano se una al ligando, yii. recuperar TgFVII humano y/o TgFVIIa humano alterando de una forma no destructiva la interacción con dicho ligando, en el que dicho ligando es una proteína de unión al antígeno dirigida a al menos un TgFVIIa humano o epítope de TgFVII humano, o un fragmento funcional o su derivado, y posee una afinidad superior a 1 pM hacia TgFVII humano o TgFVIIa humano,(b) purificación cromatográfica de TgFVII humano y/o TgFVIIa humano eluido en un intercambiador iónico, en condiciones en las que TgFVII humano y/o TgFVIIa humano son retenidos sobre el intercambiador iónico, (c) separación cromatográfica de TgFVII humano y/o TgFVIIa humano eluido en la resina de pseudo-afinidad, en condiciones en las que TgFVII humano y/o TgFVIIa humano son retenidos sobre la resina de pseudoafinidad,(d) separación cromatográfica de TgFVII humano y/o TgFVIIa humano eluido en un soporte de exclusión por tamaño.2. El método según la reivindicación 1, en el que dicho ligando comprende dos cadenas pesadas de polipéptidos que forman dos sitios de unión al antígeno completos y carece de cadenas ligeras de polipéptidos.3. El método según las reivindicaciones 1 a 2, en el que dicho ligando deriva de cadenas de inmunoglobulina pesadas de Camelidae y posee una afinidad de al menos 100 nM hacia TgFVII humano o TgFVIIa humano.4. El método según las reivindicaciones 1 a 3, en el que la etapa (ii) comprende al menos una etapa de lavado para eliminar el material no unido antes de eluir TgFVII humano o TgFVIIa humano del ligando, y además opcionalmente comprende una segunda etapa de lavado para eliminar el material unido débil.5. El método según la reivindicación 1, en el que el animal es un mamífero hembra, y preferentemente un conejo hembra.6. El método según la reivindicación 1, que comprende además las etapas preliminares, antes de la etapa (a), de recoger y clarificar leche.7. El método según la reivindicación 6, en el que la etapa de clarificación de la leche se realiza por adición de sal de citrato, seguido por filtración.8. El método según la reivindicación 1, en el que el intercambiador iónico de la etapa b) es un intercambiador aniónico.9. El método según la reivindicación 1, en el que la resina de pseudo-afinidad de la etapa c) es hidroxiapatita.10. El método según las reivindicaciones 1 a 9, que comprende además al menos una etapa de eliminar y/o inactivar virus, preferentemente por nanofiltración y/o por tratamiento con disolvente/detergente.11. El método según las reivindicaciones 1 a 10, que comprende además las etapas finales de formular, esterilizar y liofilizar TgFVII humano y/o TgFVIIa humano liofilizados.12. El método según las reivindicaciones 1 a 11, en el que la etapa de cromatografía de afinidad comprende las etapas de:(i) cargar una resina cromatográfica sobre la que se inmoviliza un ligando como se define en cualquiera de las reivindicaciones 1 a 3, con leche de un conejo hembra no transgénico que contiene TgFVII humano y/o TgFVIIa humano recombinantemente producidos, en condiciones que permiten que el ligando se una a TgFVII humano y/o TgFVIIa humano;(111) lavar el material no unido;(112) lavar el material unido débil; y(113) eluir TgFVII humano y/o TgFVIIa humano de la resina.13. El método según las reivindicaciones 4 a 12, en el que, durante dicha etapa de cromatografía de afinidad, dicha segunda etapa de lavado para eliminar material unido débil se realiza con un tampón que comprende desde 10 % hasta 50 % de un agente hidrófobo, y cuya fuerza iónica varía desde 200 hasta 600 mM.14. El método según las reivindicaciones 4 a 13, en el que, durante dicha etapa de cromatografía de afinidad, dicha etapa de elución para eluir TgFVII humano y/o TgFVIIa humano de la resina se realiza con un tampón que comprende desde 20 % hasta 70 % de un agente hidrófobo, y cuya fuerza iónica varía desde 500 hasta 2500 mM.15. El método según las reivindicaciones 1 a 14, en el que el FVII transgénico humano purificado resultante es un FVII de alta pureza casi completamente activado que comprende un bajo porcentaje de formas degradadas, proteolizadas u oxidadas.16. El método según las reivindicaciones 1 a 15, caracterizado por que la etapa de cromatografía de afinidad y/o la etapa de cromatografía de intercambio iónico y/o la etapa de cromatografía de exclusión por tamaño permiten obtener un factor de reducción viral superior a 3 log10, preferentemente superior a 4 log10, más preferentemente superior a 5 log10.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP12305882.8A EP2687595B1 (en) | 2012-07-19 | 2012-07-19 | Method for purifying transgenic factor VII |
| PCT/EP2013/065205 WO2014013024A1 (en) | 2012-07-19 | 2013-07-18 | Method for purifying transgenic factor vii |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2703541T3 true ES2703541T3 (es) | 2019-03-11 |
Family
ID=48856605
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES13739975T Active ES2703541T3 (es) | 2012-07-19 | 2013-07-18 | Método de purificación de factor VII transgénico |
Country Status (19)
| Country | Link |
|---|---|
| US (1) | US9982247B2 (es) |
| EP (2) | EP2687595B1 (es) |
| JP (1) | JP6219947B2 (es) |
| KR (2) | KR20140129015A (es) |
| CN (1) | CN104640980B (es) |
| AR (1) | AR091836A1 (es) |
| AU (1) | AU2013291969B2 (es) |
| CA (1) | CA2879109C (es) |
| DK (1) | DK2875129T3 (es) |
| ES (1) | ES2703541T3 (es) |
| IL (1) | IL236755B (es) |
| MX (1) | MX358139B (es) |
| MY (1) | MY172995A (es) |
| PH (1) | PH12015500083A1 (es) |
| PL (1) | PL2875129T3 (es) |
| SG (1) | SG11201500254PA (es) |
| TR (1) | TR201818506T4 (es) |
| TW (1) | TWI531576B (es) |
| WO (1) | WO2014013024A1 (es) |
Families Citing this family (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| JP4874806B2 (ja) | 2003-12-01 | 2012-02-15 | ノボ ノルディスク ヘルス ケア アクチェンゲゼルシャフト | 液体因子vii組成物のウイルス濾過 |
| ES2811526T3 (es) | 2010-12-30 | 2021-03-12 | Lab Francais Du Fractionnement | Glicoles como agentes de inactivación de patógenos |
| FR3006591B1 (fr) | 2013-06-11 | 2016-05-06 | Lab Francais Du Fractionnement | Composition de facteur vii presentant un point isoelectrique substantiellement homogene |
| FR3038517B1 (fr) | 2015-07-06 | 2020-02-28 | Laboratoire Francais Du Fractionnement Et Des Biotechnologies | Utilisation de fragments fc modifies en immunotherapie |
| ES2991988T3 (es) | 2017-10-30 | 2024-12-05 | Takeda Pharmaceuticals Co | Detergentes compatibles con el medio ambiente para la inactivación de virus con envoltura lipídica |
| FR3082427B1 (fr) * | 2018-06-14 | 2020-09-25 | Lab Francais Du Fractionnement | Combinaison de facteur vii et d'un anticorps bispecifique anti-facteurs ix et x |
| CN114438061B (zh) * | 2020-10-30 | 2023-10-20 | 北京双鹭立生医药科技有限公司 | 用于纯化凝血因子vii的方法 |
Family Cites Families (17)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| GR860984B (en) | 1985-04-17 | 1986-08-18 | Zymogenetics Inc | Expression of factor vii and ix activities in mammalian cells |
| FR2632524B1 (fr) | 1988-06-09 | 1992-03-13 | Fondation Nale Transfusion San | Procede de preparation d'une fraction concentree en facteur viia et son application a titre de medicament |
| FR2677652B1 (fr) | 1991-06-12 | 2005-05-27 | Agronomique Inst Nat Rech | Procede de preparation d'une proteine d'interet dans le lait d'un animal transgenique, produit obtenu et cellule eucaryote utilisee. |
| FR2684999A1 (fr) | 1991-12-16 | 1993-06-18 | Aquitaine Dev Transf Sanguine | Procede de fabrication d'un concentre de facteur vii active de haute purete essentiellement depourvu des facteurs vitamine k dependants et des facteurs viiic et viiicag. |
| DK1621554T4 (da) | 1992-08-21 | 2012-12-17 | Univ Bruxelles | Immunoglobuliner blottet for lette kæder |
| DK38293D0 (da) | 1993-03-31 | 1993-03-31 | Novo Nordisk As | Fremstilling af proteiner |
| ES2162863T3 (es) | 1993-04-29 | 2002-01-16 | Unilever Nv | Produccion de anticuerpos o fragmentos (funcionalizados) de los mismos derivados de inmunoglobulinas de cadena pesada de camelidae. |
| US6268487B1 (en) | 1996-05-13 | 2001-07-31 | Genzyme Transgenics Corporation | Purification of biologically active peptides from milk |
| TWI268933B (en) | 2003-06-10 | 2006-12-21 | Animal Technology Inst Taiwan | Method for separating protein from animal milk |
| FR2901707B1 (fr) | 2006-05-31 | 2017-09-29 | Lab Francais Du Fractionnement | Composition de facteur vii recombinant ou transgenique, chaque molecule de facteur vii possedant deux sites de n-glycosylation a motifs glycanniques definis |
| FR2901796A1 (fr) | 2006-05-31 | 2007-12-07 | Lab Francais Du Fractionnement | Procede d'extraction d'une ou de plusieurs proteines presentes dans du lait |
| FR2910786B1 (fr) * | 2006-12-29 | 2017-08-11 | Laboratoire Francais Du Fractionnement Et Des Biotechnologies (Lfb) | "procede d'extraction d'une proteine presente dans du lait" |
| US20110059510A1 (en) | 2008-03-25 | 2011-03-10 | Pierre-Jean Ripoll | Transgenic rabbits producing human factor vii |
| WO2009141418A1 (en) | 2008-05-23 | 2009-11-26 | Novo Nordisk A/S | Formulations of peg-functionalised serine proteases with high concentrations of an aromatic preservative |
| FR2942233B1 (fr) | 2009-02-19 | 2015-03-13 | Lfb Biotechnologies | Moyens pour la purification d'une proteine du plasma sanguin, et procedes pour sa mise en oeuvre |
| FR2947181B1 (fr) * | 2009-06-26 | 2012-05-04 | Lfb Biotechnologies | Composition de facteur vii |
| CN102161701A (zh) * | 2011-01-28 | 2011-08-24 | 上海兴迪金生物技术有限公司 | 从细胞培养液或血浆组分中分离纯化高纯度活化凝血七因子的方法 |
-
2012
- 2012-07-19 EP EP12305882.8A patent/EP2687595B1/en not_active Not-in-force
-
2013
- 2013-07-18 CA CA2879109A patent/CA2879109C/en active Active
- 2013-07-18 SG SG11201500254PA patent/SG11201500254PA/en unknown
- 2013-07-18 KR KR1020147022570A patent/KR20140129015A/ko not_active Ceased
- 2013-07-18 US US14/415,254 patent/US9982247B2/en active Active
- 2013-07-18 DK DK13739975.4T patent/DK2875129T3/en active
- 2013-07-18 EP EP13739975.4A patent/EP2875129B1/en active Active
- 2013-07-18 JP JP2015522099A patent/JP6219947B2/ja not_active Expired - Fee Related
- 2013-07-18 KR KR1020177018813A patent/KR101842412B1/ko not_active Expired - Fee Related
- 2013-07-18 TR TR2018/18506T patent/TR201818506T4/tr unknown
- 2013-07-18 MY MYPI2015000126A patent/MY172995A/en unknown
- 2013-07-18 ES ES13739975T patent/ES2703541T3/es active Active
- 2013-07-18 CN CN201380048473.2A patent/CN104640980B/zh active Active
- 2013-07-18 TW TW102125684A patent/TWI531576B/zh not_active IP Right Cessation
- 2013-07-18 AR ARP130102563A patent/AR091836A1/es unknown
- 2013-07-18 MX MX2015000778A patent/MX358139B/es active IP Right Grant
- 2013-07-18 WO PCT/EP2013/065205 patent/WO2014013024A1/en not_active Ceased
- 2013-07-18 PL PL13739975T patent/PL2875129T3/pl unknown
- 2013-07-18 AU AU2013291969A patent/AU2013291969B2/en not_active Ceased
-
2015
- 2015-01-13 PH PH12015500083A patent/PH12015500083A1/en unknown
- 2015-01-15 IL IL236755A patent/IL236755B/en active IP Right Grant
Also Published As
| Publication number | Publication date |
|---|---|
| JP6219947B2 (ja) | 2017-10-25 |
| DK2875129T3 (en) | 2019-01-07 |
| EP2687595B1 (en) | 2018-05-30 |
| EP2875129B1 (en) | 2018-09-05 |
| JP2015522600A (ja) | 2015-08-06 |
| SG11201500254PA (en) | 2015-02-27 |
| MX2015000778A (es) | 2015-10-14 |
| WO2014013024A1 (en) | 2014-01-23 |
| KR20170083166A (ko) | 2017-07-17 |
| PL2875129T4 (pl) | 2019-03-29 |
| CN104640980A (zh) | 2015-05-20 |
| PL2875129T3 (pl) | 2019-03-29 |
| EP2875129A1 (en) | 2015-05-27 |
| CA2879109C (en) | 2019-03-05 |
| KR101842412B1 (ko) | 2018-03-26 |
| TW201416373A (zh) | 2014-05-01 |
| PH12015500083A1 (en) | 2015-03-02 |
| CA2879109A1 (en) | 2014-01-23 |
| CN104640980B (zh) | 2019-01-08 |
| TR201818506T4 (tr) | 2019-01-21 |
| AR091836A1 (es) | 2015-03-04 |
| MX358139B (es) | 2018-08-06 |
| MY172995A (en) | 2019-12-18 |
| US9982247B2 (en) | 2018-05-29 |
| US20150175983A1 (en) | 2015-06-25 |
| KR20140129015A (ko) | 2014-11-06 |
| AU2013291969A1 (en) | 2015-02-05 |
| EP2687595A1 (en) | 2014-01-22 |
| AU2013291969B2 (en) | 2018-07-12 |
| TWI531576B (zh) | 2016-05-01 |
| IL236755B (en) | 2018-03-29 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2703541T3 (es) | Método de purificación de factor VII transgénico | |
| JP7774384B2 (ja) | 治療タンパク質の精製方法 | |
| ES2229784T3 (es) | Proceso de produccion de inmunoglobulinas para administracion intravenosa y otros productos de inmunoglobulina. | |
| JP5730211B2 (ja) | ウイルスのアルギニン不活化 | |
| CN102939107B (zh) | 抗体制剂 | |
| ES2295308T3 (es) | Procedimiento para la produccion de gammaglobulina g humana inactivada de virus. | |
| ES2704487T3 (es) | Método de purificación de un anticuerpo | |
| US20150210737A1 (en) | Integrated process for the production of therapeutics (human albumin, intravenous immunoglobulins, clotting factor viii and clotting factor ix) from human plasma | |
| ES2833100T3 (es) | Procedimiento | |
| ES2625030T3 (es) | Procedimiento de preparación de un concentrado de inmunoglobulinas polivalentes | |
| US20180305401A1 (en) | Processes for purifying proteins from plasma | |
| JP2024527980A (ja) | 免疫グロブリンgを精製する方法およびその使用 | |
| Bertolini | The purification of plasma proteins for therapeutic use | |
| Brinkman et al. | The purification of plasma proteins for therapeutic use | |
| KR20150084836A (ko) | 벤조나아제 부재 하의 시나지스 분리 방법 | |
| US20210024616A1 (en) | Method for Purifying Antibodies from Raw Milk | |
| CA2833631C (en) | Novel process for the preparation of a virus-inactivated fv concentrate starting from human plasma, scalable to industrial level | |
| AU2024351533A1 (en) | IMMUNOGLOBULIN FORMULATIONS DEPLETED IN IgA | |
| CN117769570A (zh) | 纯化免疫球蛋白g的方法及其用途 | |
| RU2559576C1 (ru) | Способ получения вирусбезопасного полного протромбинового комплекса |