ES2735310T3 - Un polímero basado en etileno - Google Patents
Un polímero basado en etileno Download PDFInfo
- Publication number
- ES2735310T3 ES2735310T3 ES13812379T ES13812379T ES2735310T3 ES 2735310 T3 ES2735310 T3 ES 2735310T3 ES 13812379 T ES13812379 T ES 13812379T ES 13812379 T ES13812379 T ES 13812379T ES 2735310 T3 ES2735310 T3 ES 2735310T3
- Authority
- ES
- Spain
- Prior art keywords
- hydrocarbyl
- atoms
- independently
- rcs
- formula
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 0 Cc1c(*)c(*2CC2)c(*I)c(*)c1NC Chemical compound Cc1c(*)c(*2CC2)c(*I)c(*)c1NC 0.000 description 6
- KJIBNMFWZIUIOK-UHFFFAOYSA-N CC(C)(C)c(cc1)ccc1-c1ccc(C(C)(C)C)cc1[N+]([O-])=O Chemical compound CC(C)(C)c(cc1)ccc1-c1ccc(C(C)(C)C)cc1[N+]([O-])=O KJIBNMFWZIUIOK-UHFFFAOYSA-N 0.000 description 1
- YAWXNJICXSLGGQ-UHFFFAOYSA-N CC(C)(C)c1ccc(c2ccc(C(C)(C)C)cc2[nH]2)c2c1 Chemical compound CC(C)(C)c1ccc(c2ccc(C(C)(C)C)cc2[nH]2)c2c1 YAWXNJICXSLGGQ-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F210/00—Copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond
- C08F210/16—Copolymers of ethene with alpha-alkenes, e.g. EP rubbers
-
- C—CHEMISTRY; METALLURGY
- C08—ORGANIC MACROMOLECULAR COMPOUNDS; THEIR PREPARATION OR CHEMICAL WORKING-UP; COMPOSITIONS BASED THEREON
- C08F—MACROMOLECULAR COMPOUNDS OBTAINED BY REACTIONS ONLY INVOLVING CARBON-TO-CARBON UNSATURATED BONDS
- C08F210/00—Copolymers of unsaturated aliphatic hydrocarbons having only one carbon-to-carbon double bond
- C08F210/14—Monomers containing five or more carbon atoms
-
- Y—GENERAL TAGGING OF NEW TECHNOLOGICAL DEVELOPMENTS; GENERAL TAGGING OF CROSS-SECTIONAL TECHNOLOGIES SPANNING OVER SEVERAL SECTIONS OF THE IPC; TECHNICAL SUBJECTS COVERED BY FORMER USPC CROSS-REFERENCE ART COLLECTIONS [XRACs] AND DIGESTS
- Y02—TECHNOLOGIES OR APPLICATIONS FOR MITIGATION OR ADAPTATION AGAINST CLIMATE CHANGE
- Y02P—CLIMATE CHANGE MITIGATION TECHNOLOGIES IN THE PRODUCTION OR PROCESSING OF GOODS
- Y02P20/00—Technologies relating to chemical industry
- Y02P20/50—Improvements relating to the production of bulk chemicals
- Y02P20/582—Recycling of unreacted starting or intermediate materials
Landscapes
- Chemical & Material Sciences (AREA)
- Health & Medical Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Medicinal Chemistry (AREA)
- Polymers & Plastics (AREA)
- Organic Chemistry (AREA)
- Transition And Organic Metals Composition Catalysts For Addition Polymerization (AREA)
- Engineering & Computer Science (AREA)
- Materials Engineering (AREA)
Abstract
Un proceso de polimerización de etileno que comprende: polimerizar etileno y 1-octeno en presencia de uno o más primeros sistemas de catalizador y, opcionalmente, uno o más segundos sistemas de catalizador en un solo reactor, en donde el primer sistema de catalizador comprende; (a) uno o más procatalizadores que comprenden un complejo de metal-ligando de Fórmula (I):**Fórmula** en donde: M es titanio, circonio o hafnio, estando cada uno, de manera independiente, en un estado de oxidación formal de +2, +3 o +4; y n es un número entero de 0 a 3 y en donde cuando n es 0, X está ausente; y cada X, de manera independiente, es un ligando monodentado que es neutro, monoaniónico o dianiónico; o dos X se toman en conjunto para formar un ligando bidentado que es neutro, monoaniónico o dianiónico; y X y n se eligen de tal manera que el complejo de metal-ligando de Fórmula (I) sea, en general, neutro; y cada Z, de manera independiente, es O, S, Nhidrocarbilo (C1-C40) o P-hidrocarbilo (C1-C40); y L es hidrocarbileno (C3-C40) o heterohidrocarbileno (C3-C40), en donde el hidrocarbileno (C3-C40) tiene una parte que comprende una cadena principal de enlazador de 3 átomos de carbono a 10 átomos de carbono que enlaza los átomos de Z en la Fórmula (I) (al que está enlazado L) y el heterohidrocarbileno (C3-C40) tiene una parte que comprende una cadena principal de enlazador de 3 átomos a 10 átomos que enlaza los átomos de Z en la Fórmula (I), en donde cada uno de los 3 a 10 átomos de la cadena principal de enlazador de 3 átomos a 10 átomos del heterohidrocarbileno (C3-C40), de manera independiente, es un átomo de carbono O, S, S(O), S(O)2, Si(RC)2, Ge(RC)2, P(RP), o N(RN), en donde, de manera independiente, cada RC es hidrocarbilo (C1-C30), cada RP es hidrocarbilo (C1-C30); y cada RN es hidrocarbilo (C1-C30) o está ausente; y R1-24 se seleccionan del grupo que consiste en un hidrocarbilo (C1-C40), heterohidrocarbilo (C1-C40), Si(RC)3, Ge(RC)3, P(RP)2, N(RN)2, ORC, SRC, NO2, CN, CF3, RCS(O)-, RCS(O)2-, (RC)2C=N-, RCC(O)O-, RCOC(O)-, RCC(O)N(R)-, (RC)2NC(O)-, un átomo de halógeno, un átomo de hidrógeno y una combinación de los mismos; y en donde al menos R1, R16 o ambos comprenden la Fórmula (II) y preferiblemente R1 y R16 son iguales;
Description
DESCRIPCIÓN
Un polímero basado en etileno
Campo de la invención
La presente invención se refiere a un proceso de polimerización de etileno.
Antecedentes de la invención
Los polímeros basados en olefinas, tales como polietileno y/o polipropileno, se producen a través de diversos sistemas de catalizador. La selección de tal sistema de catalizador usado en el proceso de polimerización de los polímeros basados en olefinas es un factor importante que contribuye a las características y propiedades de tales polímeros basados en olefinas.
El polietileno es conocido por su uso en la fabricación de una amplia diversidad de artículos. El proceso de polimerización de polietileno se puede variar en varios aspectos para producir una amplia diversidad de resinas de polietileno resultantes que tienen diferentes propiedades físicas que hacen que las diversas resinas sean adecuadas para su uso en diferentes aplicaciones. En general, se sabe que el polietileno se puede producir en reactores de bucle en fase de solución en los que el monómero de etileno y, opcionalmente, uno o más comonómeros de alfa olefina, que tienen, típicamente, de 3 a 10 átomos de carbono, se hacen circular en presencia de uno o más sistemas de catalizador a presión alrededor de un reactor de bucle mediante una bomba de circulación. Los monómeros de etileno y uno o más comonómeros opcionales están presentes en un diluyente líquido, tal como un alcano o isoalcano, por ejemplo, isobutano. También se puede añadir hidrógeno al reactor. Los sistemas de catalizador para la producción de polietileno pueden comprender, típicamente, un sistema de catalizador basado en cromo, un sistema de catalizador Ziegler Natta y/o un sistema de catalizador molecular (ya sea de metaloceno o no metaloceno). Los reactivos en el diluyente y el sistema de catalizador se hacen circular a una temperatura de polimerización elevada alrededor del reactor de bucle, produciendo de este modo un homopolímero y/o copolímero de polietileno, dependiendo de si están presentes o no uno o más comonómeros. Ya sea de manera periódica o continua, parte de la mezcla de reacción, incluyendo el producto de polietileno disuelto en el diluyente, junto con el etileno sin reaccionar y uno o más comonómeros opcionales, se retira del reactor de bucle. La mezcla de reacción, cuando se retira del reactor de bucle, se puede procesar para retirar el producto de polietileno del diluyente y los reactivos sin reaccionar, reciclándose el diluyente y los reactivos sin reaccionar típicamente de nuevo en el reactor de bucle. Como alternativa, la mezcla de reacción se puede enviar a un segundo reactor, por ejemplo, un reactor de bucle, conectado en serie al primer reactor de bucle donde se puede producir una segunda fracción de polietileno. El documento WO-A-2011/146044 describe un proceso que polimeriza, de manera selectiva, etileno en presencia de una alfa-olefina y un complejo de metal-ligando (precatalizador) y un catalizador útil en tales procesos; y composiciones relacionadas.
A pesar de los esfuerzos de investigación en el desarrollo de sistemas de catalizador adecuados para la polimerización de poliolefinas, tales como polietileno y/o polipropileno, sigue existiendo la necesidad de un procatalizador y un sistema de catalizador que presente una alta selectividad hacia el etileno a temperaturas de reacción más altas; lo que facilita, por tanto, la producción de polímeros de mayor peso molecular a temperaturas de reacción relativamente más altas. De manera adicional, a pesar de los esfuerzos de investigación en el desarrollo de poliolefinas, tales como polietileno y/o polipropileno, con propiedades mejoradas, sigue existiendo la necesidad de un polietileno que tenga propiedades mejoradas.
Compendio de la invención
La presente invención proporciona un proceso para la polimerización de etileno y 1-octeno.
En una realización, la presente invención proporciona un proceso de polimerización que comprende la polimerización de etileno con 1-octeno en presencia de uno o más primeros sistemas de catalizador y, opcionalmente, uno o más segundos sistemas de catalizador en un reactor individual, en donde el primer sistema de catalizador comprende;
(a) uno o más procatalizadores que comprenden un complejo de metal-ligando de Fórmula (I):

en donde:
M es titanio, circonio o hafnio, estando cada uno, de manera independiente, en un estado de oxidación formal de 2, 3 o 4; y n es un número entero de 0 a 3 y en donde cuando n es 0, X está ausente; y cada X, de manera independiente, es un ligando monodentado que es neutro, monoaniónico o dianiónico; o dos X se toman en conjunto para formar un ligando bidentado que es neutro, monoaniónico o dianiónico; y X y n se eligen de tal manera que el complejo de metal-ligando de Fórmula (I) sea, en general, neutro; y
cada Z, de manera independiente, es O, S, N-hidrocarbilo (Ci-C40) o P-hidrocarbilo (Ci-C40); y L es hidrocarbileno (C3-C40) o heterohidrocarbileno (C3-C40), en donde el hidrocarbileno (C3-C40) tiene una parte que comprende una cadena principal de enlazador de 3 átomos de carbono a 10 átomos de carbono que enlaza los átomos de Z en la Fórmula (I) (al que está enlazado L) y el heterohidrocarbileno (C3-C40) tiene una parte que comprende una cadena principal de enlazador de 3 átomos a 10 átomos que enlaza los átomos de Z en la Fórmula (I), en donde cada uno de los 3 a 10 átomos de la cadena principal de enlazador de 3 átomos a 10 átomos del heterohidrocarbileno (C3-C40), de manera independiente, es un átomo o heteroátomo de carbono, en donde cada heteroátomo, de manera independiente, es O, S, S(O), S(O)2, Si(RC)2, Ge(RC)2, P(RP), o N(RN), en donde, de manera independiente, cada RC es hidrocarbilo (C1-C30), cada RP es hidrocarbilo (C1-C30); y cada RN es hidrocarbilo (C1-C30) o está ausente; y
R1-24 se seleccionan del grupo que consiste en un hidrocarbilo (C1-C40), heterohidrocarbilo (C1-C40), Si(RC)3, Ge(RC)3, P(Rp)2, N(Rn)2, ORc, SRc, NO2, CN, CF3, RcS(O)-, RcS(O)2-, (Rc)2C=N-, RcC(O)O-, RcOC(O)-, RcC(O)N(R)-, (Rc)2NC(O)-, un átomo de halógeno, un átomo de hidrógeno y una combinación de los mismos; y en donde al menos R1, R16 o ambos comprenden la Fórmula (II) y preferiblemente R1 y R16 son iguales;

Cuando R22 es H, entonces R19 es un hidrocarbilo (C1-C40); heterohidrocarbilo (C1-C40); S¡(R°)3 , Ge(RC)3, P(RP)2, N(Rn)2, ORc, SRc, NO2 , CN, CF3, RCS(O)-, RCS(O)2-, (Rc)2C=N-, RCC(O)O-, RCOC(O)-, RCC(O)N(R)-, (RC)2 NC(O)- o un átomo de halógeno; y
cuando R19 es H, entonces R22 es un hidrocarbilo (C1-C40); heterohidrocarbilo (C1-C40); Si(RC)3 , Ge(RC)3, P(Rp)2 , N(Rn)2, ORc, SRc, NO2 , CN, CF3, RCS(O)-, RCS(O)2-, (Rc)2C=N-, RCC(O)O-, RCOC(O)-, RCC(O)N(R)-, (RC)2 NC(O)- o un átomo de halógeno; y, preferiblemente, R22 y R19 son, ambos, un hidrocarbilo (C1-C40); heterohidrocarbilo (C1-C40); Si(RC)3, Ge(RC)3 , P(Rp)2, N(Rn)2, ORc, SRc, NO2, CN, CF3, RCS(O)-, RCS(O)2-, (RC)2C=N-, RCC(O)O-, RCOC(O)-, RCC(O)N(R)-, (RC)2NC(O)- o un átomo de halógeno; y
cuando R8 es H, entonces R9 es un hidrocarbilo (C1-C40); heterohidrocarbilo (C1-C40); Si(RC)3 , Ge(RC)3, P(Rp)2, N(R)2, ORC, SRC, NO2 , CN, CF3, RCS(O)-, RCS(O)2-, (Rc)2C=N-, RCC(O)O-, RCOC(O)-, RCC(O)N(R)-, (RC)2NC(O)- o un átomo de halógeno; y
cuando R9 es H, entonces R8 es un hidrocarbilo (C1-C40); heterohidrocarbilo (C1-C40); Si(RC)3, Ge(RC)3, P(RP)2, N(Rn)2, ORc, SRc, NO2 , CN, CF3, RCS(O)-, RCS(O)2-, (Rc)2C=N-, RCC(O)O-, RCOC(O)-, RCC(O)N(R)-, (RC)2 NC(O)- o un átomo de halógeno; y
opcionalmente, dos o más grupos R (por ejemplo, de R9-15, R9-13, R9-12, R2-8, R4-8, R5-8) se pueden combinar en conjunto en estructuras de anillo, teniendo tales estructuras de anillo de 3 a 50 átomos en el anillo, excluyendo cualquier átomo de hidrógeno.
Cada uno de los grupos arilo, heteroarilo, hidrocarbilo, heterohidrocarbilo, Si(RC)3, Ge(RC)3, P(RP)2, N(RN)2, ORC,
SRC, RCS(O)-, RCS(O)2-, (Rc)2C=N-, RCC(O)O-, RCOC(O)-, RCC(O)N(R)-, (RC)2NC(O)-, hidrocarbileno y heterohidrocarbileno, de manera independiente, está no sustituido o sustituido con uno o más sustituyentes de RS. Cada RS, de manera independiente, es un átomo de halógeno, una sustitución de polifluoro, una sustitución de perfluoro, alquilo (Ci-Ci8) no sustituido, F3C-, FCH2O-, F2HCO-, F3CO-, R3Si-, R3Ge-, RO-, rS-, RS(O)-, RS(O)2-, R2P-, R2N-, R2C=N-, NC-, RC(O)O-, ROC(O)-, RC(O)N(R)- o R2NC(O)-, o dos del RS se toman en conjunto para formar un alquileno (C1-C18) no sustituido, en donde cada R, de manera independiente, es un alquilo (C1-C18) no sustituido; y (b) uno o más cocatalizadores; en donde la relación del número total de moles del uno o más complejos de metal-ligando de Fórmula (I) respecto al número total de moles del uno o más cocatalizadores es de 1:10.000 a 100:1.
Breve descripción de los dibujos
Con el fin de ilustrar la invención, se muestra, en los dibujos, una forma que es ejemplar; entendiéndose, sin embargo, que la presente invención no está limitada a las disposiciones e instrumentos precisos mostrados.
Las Figuras 1-20 ilustran las Fórmulas 1-20, respectivamente.
Descripción detallada de la invención
La presente invención proporciona un proceso para la polimerización de etileno y 1-octeno.
El sistema de catalizador comprende un componente de procatalizador y un componente de cocatalizador.
Componente de procatalizador
El componente de procatalizador comprende un complejo de metal-ligando de Fórmula (I):

en donde:
M es titanio, circonio o hafnio, estando cada uno, de manera independiente, en un estado de oxidación formal de 2, 3 o 4; y
n es un número entero de 0 a 3 y en donde cuando n es 0, X está ausente; y
cada X, de manera independiente, es un ligando monodentado que es neutro, monoaniónico o dianiónico; o dos X se toman en conjunto para formar un ligando bidentado que es neutro, monoaniónico o dianiónico; y
X y n se eligen de tal manera que el complejo de metal-ligando de Fórmula (I) sea, en general, neutro; y cada Z, de manera independiente, es O, S, N-hidrocarbilo (C1-C40) o P-hidrocarbilo (C1-C40); y
L es hidrocarbileno (C3-C40) o heterohidrocarbileno (C3-C40), en donde el hidrocarbileno (C3-C40) tiene una parte que comprende una cadena principal de enlazador de 3 átomos de carbono a 10 átomos de carbono que enlaza los átomos de Z en la Fórmula (I) (al que está enlazado L) y el heterohidrocarbileno (C3-C40) tiene una parte que comprende una cadena principal de enlazador de 3 átomos a 10 átomos que enlaza los átomos de Z en la Fórmula (I), en donde cada uno de los 3 a 10 átomos de la cadena principal de enlazador de 3 átomos a 10 átomos del heterohidrocarbileno (C3-C40), de manera independiente, es un átomo o heteroátomo de carbono, en donde cada heteroátomo, de manera independiente, es O, S, S(O), S(O)2, Si(RC)2, Ge(RC)2, P(RP), o N(RN), en donde, de manera independiente, cada RC es hidrocarbilo (C1-G30), cada RP es hidrocarbilo (C1-C30); y cada RN es hidrocarbilo (C1-C30) o está ausente; y
R1-24 se seleccionan del grupo que consiste en un hidrocarbilo (C1-C40), heterohidrocarbilo (C1-C40), Si(RC)3, Ge(RC)3, P(Rp)2, N(Rn)2, ORc, SRc, NO2, CN, CF3, RCS(O)-, RCS(O)2-, (Rc)2C=N-, RCC(O)O-, RCOC(O)-, RCC(O)N(R)-, (RC)2NC(O)-, un átomo de halógeno, un átomo de hidrógeno y una combinación de los mismos.
R1, R16 o ambos comprenden la Fórmula (II) y preferiblemente R1 y R16 son iguales; y

cuando R22 es H, entonces R19 es un hidrocarbilo (C1-C40); heterohidrocarbilo (C1-C40); Si(RC)3 , Ge(RC)3, P(RP)2 , N(Rn)2, ORc, SRc, NO2 , CN, CF3, RCS(O)-, RcS(O)2-, (Rc)2C=N-, RcC(O)O-, RcOC(O)-, RcC(O)N(R)-, (Rc)2 NC(O)- o un átomo de halógeno; y
cuando R19 es H, entonces R22 es un hidrocarbilo (C1-C40); heterohidrocarbilo (C1-C40); Si(RC)3, Ge(RC)3, P(RP)2, N(RN)2, ORc, SRc, NO2 , CN, CF3, RCS(O)-, RcS(O)2-, (RC)2C=N-, RcC(O)O-, RcOC(O)-, RCC(O)N(R)-, (Rc)2 NC(O)- o un átomo de halógeno; y
preferiblemente, R22 y R19 son, ambos, un hidrocarbilo (C1-C40); heterohidrocarbilo (C1-C40); Si(RC)3, Ge(RC)3, P(RP)2, N(RN)2, ORc, SRc, NO2 , CN, CF3, RCS(O)-, RcS(O)2-, (RC)2C=N-, RcC(O)O-, RcOC(O)-, RCC(O)N(R)-, (Rc)2 NC(O)- o un átomo de halógeno; y
cuando R8 es H, entonces R9 es un hidrocarbilo (C1-C40); heterohidrocarbilo (C1-C40); Si(RC)3, Ge(RC)3, P(RP)2, N(RN)2, ORc, SRc, NO2 , CN, CF3, RCS(O)-, RcS(O)2-, (RC)2C=N-, RcC(O)O-, RcOC(O)-, RCC(O)N(R)-, (Rc)2 NC(O)- o un átomo de halógeno; y cuando R9 es H, entonces R8 es un hidrocarbilo (C1-C40); heterohidrocarbilo (C1-C40); Si(RC)3 , Ge(RC)3 , P(Rp)2 , N(RN)2 , ORc, SRc, NO2 , CN, CF3, RCS(O)-, RCS(O)2-, (RC)2C=N-, RCC(O)O-, RCOC(O)-, RCC(O)N(R)-, (RC)2NC(O)- o un átomo de halógeno; y
preferiblemente, R8 y R9 son, ambos, un hidrocarbilo (C1-C40); heterohidrocarbilo (C1-C40); Si(RC)3, Ge(RC)3, P(RP)2, N(RN)2, ORc, SRc, NO2 , CN, CF3, RCS(O)-, RCS(O)2-, (RC)2C=N-, RCC(O)O-, RCOC(O)-, RCC(O)N(R)-, (RC)2 NC(O)- o un átomo de halógeno; y
opcionalmente, dos o más grupos R (de R9-13 o R4-8) se pueden combinar en conjunto en estructuras de anillo, teniendo tales estructuras de anillo de 3 a 50 átomos en el anillo, excluyendo cualquier átomo de hidrógeno.
Cada uno de los grupos arilo, heteroarilo, hidrocarbilo, heterohidrocarbilo, Si(RC)3 , Ge(RC)3, P(RP)2 , N(RN)2, ORC, SRC, RCS(O)-, RCS(O)2-, (RC)2C=N-, RCC(O)O-, RCOC(O)-, RCC(O)N(R)-, (RC)2NC(O)-, hidrocarbileno y heterohidrocarbileno, de manera independiente, está no sustituido o sustituido con uno o más sustituyentes de RS; y
cada RS, de manera independiente, es un átomo de halógeno, una sustitución de polifluoro, una sustitución de perfluoro, alquilo (C1-C18) no sustituido, F3C-, FCH2O-, F2HCO-, F3CO-, R3Si-, R3Ge-, RO-, RS-, RS(O)-, RS(O)2-, R2P-, R2N-, R2C=N-, NC-, RC(O)O-, ROC(O)-, RC(O)N(R)- o R2NC(O)-, o dos del RS se toman en conjunto para formar un alquileno (C1-C18) no sustituido, en donde cada R, de manera independiente, es un alquilo (C1-C18) no sustituido.
Opcionalmente, dos o más grupos R (de R20-24) se pueden combinar en conjunto en estructuras de anillo, teniendo tales estructuras de anillo de 3 a 50 átomos en el anillo, excluyendo cualquier átomo de hidrógeno.
Como se ha mencionado anteriormente, la presente invención emplea uno o más complejos de metal-ligando de Fórmula (I), que se describen en la presente memoria usando la terminología de grupos químicos convencional. Cuando se usa para describir determinados grupos químicos que contienen átomos de carbono (por ejemplo, (alquilo (C1-C40)), la expresión entre paréntesis (C1-C40) se puede representar mediante la forma "(Cx-Cy)", lo que significa que la versión no sustituida del grupo químico comprende desde un número x de átomos de carbono a un número y de átomos de carbono, en donde cada x e y, de manera independiente, es un número entero, como se describe para el grupo químico. La versión sustituida de RS del grupo químico puede contener más de y átomos de carbono, dependiendo de la naturaleza de RS. Por tanto, por ejemplo, un alquilo (C1-C40) no sustituido contiene de 1 a 40 átomos de carbono (x = 1 e y = 40). Cuando el grupo químico está sustituido con uno o más sustituyentes de RS que contienen átomos de carbono, el grupo químico (Cx-Cy) sustituido puede comprender más de y átomos de carbono totales; es decir, el número total de átomos de carbono del/de los sustituyente/s que contiene/n átomos de carbono del grupo químico (Cx-Cy) sustituido es igual a y más de la suma del número de átomos de carbono de cada uno de los sustituyentes que contienen átomos de carbono. Cualquier átomo de un grupo químico que no se especifique en la presente memoria se entiende como un átomo de hidrógeno.
En algunas realizaciones, cada uno de los grupos químicos (por ejemplo, X, L, R1-24, etc.) del complejo de metalligando de Fórmula (I) puede estar no sustituido, es decir, se puede definir sin el uso de un sustituyente de RS, siempre que se cumplan las condiciones mencionadas anteriormente. En otras realizaciones, al menos uno de los grupos químicos del complejo de metal-ligando de Fórmula (I) contiene, de manera independiente, uno o más de los sustituyentes de RS. Preferiblemente, teniendo en cuenta todos los grupos químicos, no hay más de un total de 20
RS, más preferiblemente no más de un total de 10 RS y todavía más preferiblemente no más de un total de 5 RS en el complejo de metal-ligando de Fórmula (I). En los casos en los que el compuesto de la invención contiene dos o más sustituyentes de RS, cada RS, de manera independiente, está enlazado a un grupo químico sustituido igual o diferente. Cuando dos o más RS están enlazados a un mismo grupo químico, estos, de manera independiente, están enlazados a un átomo o heteroátomo de carbono igual o diferente, según sea el caso, en el mismo grupo químico hasta e incluyendo la persustitución del grupo químico.
El término "persustitución" significa que cada átomo de hidrógeno (H) enlazado a un átomo o heteroátomo de carbono de un compuesto o grupo funcional no sustituido correspondiente, según sea el caso, se reemplaza con un sustituyente (por ejemplo, RS). El término "polisustitución" significa que cada uno de al menos dos, pero no todos, átomos de hidrógeno (H) enlazados a átomos o heteroátomos de carbono de un compuesto o grupo funcional no sustituido correspondiente, según sea el caso, se reemplaza con un sustituyente (por ejemplo, RS). Los sustituyentes de alquileno (C1-C18) y alquileno (C1-C8) son especialmente útiles para la formación de grupos químicos sustituidos, que son análogos bicíclicos o tricíclicos, según sea el caso, de los correspondientes grupos químicos monocíclicos o bicíclicos no sustituidos.
Como se usa en la presente memoria, la expresión "hidrocarbilo (C1-C40)" significa un radical hidrocarburo de 1 a 40 átomos de carbono y la expresión "hidrocarbileno (C1-C40)" significa un dirradical hidrocarburo de 1 a 40 átomos de carbono, en donde cada radical y dirradical hidrocarburo, de manera independiente, es aromático (6 átomos de carbono o más) o no aromático, saturado o insaturado, de cadena lineal o ramificada, cíclico (incluyendo mono- y poli-cíclico, poli-cíclico condensado y no condensado, incluyendo bicíclico; 3 átomos de carbono o más) o acíclico o una combinación de dos o más de los mismos; y cada radical y dirradical hidrocarburo, de manera independiente, es igual o diferente de otro radical y dirradical hidrocarburo, respectivamente, y, de manera independiente, está no sustituido o sustituido con uno o más de RS
Preferiblemente, un hidrocarbilo (C1-C40), de manera independiente, es un alquilo (C1-C40), cicloalquilo (C3-C40), cicloalquil (C3-C20)-alquileno (C1-C20), arilo (C6-C40) o aril (C6-C20)-alquileno (C1-C20) no sustituido o sustituido. Más preferiblemente, cada uno de los grupos hidrocarbilo (C1-C40) mencionados anteriormente, de manera independiente, tiene un máximo de 20 átomos de carbono (es decir, hidrocarbilo (C1-C20)) y todavía más preferiblemente un máximo de 12 átomos de carbono.
Las expresiones "alquilo (C1-C40)" y "alquilo (C1-C18)" significan un radical hidrocarburo lineal o ramificado saturado de 1 a 40 átomos de carbono o de 1 a 18 átomos de carbono, respectivamente, que está no sustituido o sustituido con uno o más de RS. Los ejemplos de alquilo (C1-C40) no sustituido son alquilo (C1-C20) no sustituido; alquilo (C1-C10) no sustituido; alquilo (C1-C5) no sustituido; metilo; etilo; 1 -propilo; 2-propilo; 1 -butilo; 2-butilo; 2-metilpropilo; 1,1-dimetiletilo; 1 -pentilo; 1-hexilo; 1 -heptilo; 1-nonilo; y 1-decilo. Los ejemplos de alquilo (C1-C40) sustituido son alquilo (C1-C20) sustituido, alquilo (C1-C10) sustituido, trifluorometilo y alquilo (C45). El alquilo (C45) es, por ejemplo, un alquilo (C27-C40) sustituido con un RS, que es un alquilo (C18-C5), respectivamente. Preferiblemente, cada alquilo (C1-C5), de manera independiente, es metilo, trifluorometilo, etilo, 1 -propilo, 1 -metiletilo o 1,1 -dimetiletilo.
La expresión "arilo (C6-C40)" significa un radical hidrocarburo aromático, mono-, bi- o tri-cíclico, no sustituido o sustituido (con uno o más de RS) de 6 a 40 átomos de carbono, de los que al menos de 6 a 14 de los átomos de carbono son átomos de carbono de anillos aromáticos y el radical mono-, bi- o tri-cíclico comprende 1, 2 o 3 anillos, respectivamente; en donde el 1 anillo es aromático y los 2 o 3 anillos, de manera independiente, están condensados o no condensados y al menos uno de los 2 o 3 anillos es aromático. Los ejemplos de arilo (C6-C40) no sustituido son arilo (C6-C20) no sustituido; arilo (C6-C18) no sustituido; 2-alquil (C1-C5)-fenilo; 2,4-bis-alquil (C1-C5)-fenilo; fenilo; fluorenilo; tetrahidrofluorenilo; indacenilo; hexahidroindacenilo; indenilo; dihidroindenilo; naftilo; tetrahidronaftilo; y fenantreno. Los ejemplos de arilo (C6-C40) sustituido son arilo (C6-C20) sustituido; arilo (C6-C18) sustituido; 2,4-bis[alquil (C20)]-fenilo; polifluorofenilo; pentafluorofenilo; y fluoren-9-ona-1-ilo.
La expresión "cicloalquilo (C3-C40)" significa un radical hidrocarburo cíclico saturado de 3 a 40 átomos de carbono que está no sustituido o sustituido con uno o más de RS Otros grupos cicloalquilo (por ejemplo, alquilo (C3-C12)) se definen de manera análoga. Los ejemplos de cicloalquilo (C3-C40) no sustituido son cicloalquilo (C3-C20) no sustituido, cicloalquilo (C3-C10) no sustituido, ciclopropilo, ciclobutilo, ciclopentilo, ciclohexilo, cicloheptilo, ciclooctilo, ciclononilo y ciclodecilo. Los ejemplos de cicloalquilo (C3-C40) sustituido son cicloalquilo (C3-C20) sustituido, cicloalquilo (C3-C10) sustituido, ciclopentanon-2-ilo y 1-fluorociclohexilo.
Los ejemplos de hidrocarbileno (C1-C40) son arileno (C6-C40), cicloalquileno (C3-C40) y alquileno (C1-C40) no sustituido o sustituido (por ejemplo, alquileno (C1-C20)). En algunas realizaciones, los dirradicales están sobre un mismo átomo de carbono (por ejemplo, -CH2-) o sobre átomos de carbono adyacentes (es decir, 1,2-dirradicales) o están separados por uno, dos o más átomos de carbono intermedios (por ejemplo, los respectivos 1,3-dirradicales, 1,4-dirradicales, etc.). Se prefiere un 1,2-, 1,3-, 1,4- o un alfa, omega-dirradical y más preferiblemente un 1,2-dirradical. El alfa, omega-dirradical es un dirradical que tiene una separación máxima de la cadena principal de carbono entre los carbonos radicales. Se prefiere más una versión de 1,2-dirradical, 1,3-dirradical o 1,4-dirradical de arileno (C6-C18), cicloalquileno (C3-C20) o alquileno (C2-C20).
La expresión "alquileno (C1-C40)" significa un dirradical de cadena lineal o cadena ramificada saturado (es decir, los
radicales no están sobre los átomos de anillo) de 1 a 40 átomos de carbono que está no sustituido o sustituido con uno o más de Rs. Los ejemplos de alquileno (C1-C40) no sustituido son alquileno (C1-C20) no sustituido, incluyendo l 1,2-alquileno (C2-C10) no sustituido; 1,3-alquileno (C3-C10); 1,4-alquileno (C4-C10); -CH2-, -CH2CH2-, -CH2CHCH3, -(CH2)4-, -(CH2)5-, -(CH2)6-, -(CH2)7-, -(CH2)8- y -(CH2)4C(H)(CH3)-. Los ejemplos de alquileno (C1-C40) sustituido son alquileno (C1-C20) sustituido, -CF2-, -C(O)- y -(CH2)14C(CH3)2(CH2)5-(es decir, un 1,20-eicosileno normal sustituido con 6,6-dimetilo). Como se ha mencionado anteriormente, dos RS se pueden tomar en conjunto para formar un alquileno (C1-C18), los ejemplos de alquileno (C1-C40) sustituido también incluyen 1,2-bis(metilen)ciclopentano, 1,2-bis(metilen)ciclohexano, 2,3-bis(metilen)-7,7-dimetil-biciclo[2.2.1]heptano y 2,3-bis (metilen)biciclo[2.2.2]octano.
La expresión "cicloalquileno (C3-C40)" significa un dirradical cíclico (es decir, los radicales están sobre los átomos de anillo) de 3 a 40 átomos de carbono que está no sustituido o sustituido con uno o más de RS Los ejemplos de cicloalquileno (C3-C40) no sustituido son 1,3-ciclopropileno, 1,1-ciclopropileno y 1,2-ciclohexileno. Los ejemplos de cicloalquileno (C3-C40) sustituido son 2-oxo-1,3-ciclopropileno y 1,2-dimetil-1,2-ciclohexileno.
La expresión "heterohidrocarbilo (C1-C40)" significa un radical heterohidrocarburo de 1 a 40 átomos de carbono y la expresión "heterohidrocarbileno (C1-C40)" significa un dirradical heterohidrocarburo de 1 a 40 átomos de carbono y cada heterohidrocarburo tiene, de manera independiente, uno o más heteroátomos O; S; S(O); S(O)2 ; Si(RC)2; Ge(RC)2; P(RP); y N(RN), en donde, de manera independiente, cada RC es hidrocarbilo (C1-C18) no sustituido, cada RP es hidrocarbilo (C1-C18) no sustituido; y cada RN es hidrocarbilo (C1-C18) no sustituido o está ausente (por ejemplo, ausente cuando N comprende -N= o N sustituido con tri-carbono). El radical heterohidrocarburo y cada uno de los dirradicales heterohidrocarburo está, de manera independiente, sobre un átomo de carbono o heteroátomo del mismo, aunque, preferiblemente, está sobre un átomo de carbono cuando está enlazado a un heteroátomo en la Fórmula (I) o a un heteroátomo de otro heterohidrocarbilo o heterohidrocarbileno. Cada heterohidrocarbilo (C1-C40) y heterohidrocarbileno (C1-C40), de manera independiente, está no sustituido o sustituido (con uno o más de RS), es aromático o no aromático, saturado o insaturado, de cadena lineal o de cadena ramificada, cíclico (incluyendo monoy poli-cíclico, poli-cíclico condensado y no condensado) o acrílico o una combinación de dos o más de los mismos; y cada uno es, respectivamente, igual o diferente de otro.
Preferiblemente, el heterohidrocarbilo (C1-C40), de manera independiente, es heteroalquilo (C1-C40), hidrocarbil (C1-C40)-O-, hidrocarbil (C1-C40)-S-, hidrocarbil (C1-C40)-S(O)-, hidrocarbil (C1-C40)-S(O)2-, hidrocarbil (C1-C40)-Si(RC)2-, hidrocarbil (C1-C40)-Ge(RC)2-, hidrocarbil (CrC 40)-N(RN)-, hidrocarbil (C1-C40)-P(Rp)-, heterocicloalquilo (C2-C40), heterocicloalquil (C2-C1g)-alquileno (C1-C20), cicloalquil (C3-C20)-heteroalquileno (C1-C19), heterocicloalquil (C2-C19)-heteroalquileno (C1-C20), heteroarilo (C1-C40), heteroaril (C1-C1g)-alquileno (C1-C20), aril (C6-C20)-heteroalquileno (C1-C19) o heteroaril (C1-C1g)-heteroalquileno (C1-C20) no sustituido o sustituido. La expresión "heteroarilo (C4-C40)" significa un radical hidrocarburo heteroaromático mono-, bi- o tri-cíclico no sustituido o sustituido (con uno o más de RS) de 1 a 40 átomos de carbono en total y de 1 a 4 heteroátomos y el radical mono-, bi- o tri-cíclico comprende 1, 2 o 3 anillos, respectivamente, en donde los 2 o 3 anillos, de manera independiente, están condensados o no condensados y al menos uno de los 2 o 3 anillos es heteroaromático. Otros grupos heteroarilo (por ejemplo, heteroarilo (C4-C12)) se definen de manera análoga. El radical hidrocarburo heteroaromático monocíclico es un anillo de 5 elementos o de 6 elementos. El anillo de 5 elementos tiene de 1 a 4 átomos de carbono y de 4 a 1 heteroátomos, respectivamente, siendo cada heteroátomo O, S, N o P y preferiblemente O, S o N. Los ejemplos de radical hidrocarburo heteroaromático de anillo de 5 elementos son pirrol-1-ilo; pirrol-2-ilo; furan-3-ilo; tiofen-2-ilo; pirazol-1-ilo; isoxazol-2-ilo; isotiazol-5-ilo; imidazol-2-ilo; oxazol-4-ilo; tiazol-2-ilo; 1,2,4-triazol-1-ilo; 1,3,4-oxadiazol-2-ilo; 1,3,4-tiadiazol-2-ilo; tetrazol-1-ilo; tetrazol-2-ilo; y tetrazol-5-ilo. El anillo de 6 elementos tiene 4 o 5 átomos de carbono y 2 o 1 heteroátomos, siendo los heteroátomos N o P y preferiblemente N. Los ejemplos de radical hidrocarburo heteroaromático de anillo de 6 elementos son piridin-2-ilo; pirimidin-2-ilo; y pirazin-2-ilo. El radical hidrocarburo heteroaromático bicíclico es preferiblemente un sistema de anillo-5,6 o -6,6 condensado. Los ejemplos de radical hidrocarburo heteroaromático bicíclico de sistema de anillo-5,6 condensado son indol-1-ilo; y bencimidazol-1-ilo. Los ejemplos de radical hidrocarburo heteroaromático bicíclico de sistema de anillo-6,6 condensado son quinolin-2-ilo; e isoquinolin-1-ilo. El radical hidrocarburo heteroaromático tricíclico es preferiblemente un sistema de anillo-5,6,5; -5,6,6; -6,5,6; o -6,6,6 condensado. Un ejemplo del sistema de anillo-5,6,5 condensado es 1,7-dihidropirrolo[3,2-/]indol-1-ilo. Un ejemplo del sistema de anillo-5,6,6 condensado es 1H-benzo[t]indol-1-ilo. Un ejemplo del sistema de anillo-6,5,6 condensado es 9H-carbazol-9-ilo. Un ejemplo del sistema de anillo-6,5,6 condensado es 9H-carbazol-9-ilo. Un ejemplo del sistema de anillo-6,6,6 condensado es acridin-9-ilo.
En algunas realizaciones, el heteroarilo (C4-C40) es carbazolilo 2,7-disustituido o carbazolilo 3,6-disustituido, más preferiblemente, en donde cada RS, de manera independiente, es fenilo, metilo, etilo, isopropilo o terc-butilo, todavía más preferiblemente 2,7-di(terc-butil)-carbazolilo, 3,6-di(terc-butil)-carbazolilo, 2,7-di(terc-octil)-carbazolilo, 3,6-di(terc-octil)-carbazolilo, 2,7-difenilcarbazolilo, 3,6-difenilcarbazolilo, 2,7-bis(2,4,6-trimetilfenil)-carbazolilo o 3,6-bis(2,4,6-trimetilfenil)-carbazolilo.
Los grupos heteroalquilo y heteroalquileno mencionados anteriormente son radicales o dirradicales de cadena lineal o ramificada saturados, respectivamente, que contienen átomos de carbono (C1-C40) o menos átomos de carbono, según sea el caso, y uno o más de los heteroátomos Si(RC)2, Ge(RC)2, P(RP), N(RN), N, O, S, S(O) y S(O)2, como se ha definido anteriormente, en donde cada uno de los grupos heteroalquilo y heteroalquileno, de manera independiente, están no sustituidos o sustituidos con uno o más de RS.
Los ejemplos de heterocicloalquilo (C2-C40) no sustituido son heterocicloalquilo (C2-C20) no sustituido, heterocicloalquilo (C2-C10) no sustituido, aziridin-1-ilo, oxetan-2-ilo, tetrahidrofuran-3-ilo, pirrolidin-1-ilo, tetrahidrotiofen-S,S-dióxido-2-ilo, morfolin-4-ilo, 1,4-dioxan-2-ilo, hexahidroazepin-4-ilo, 3-oxa-ciclooctilo, 5-tiociclononilo y 2-aza-ciclodecilo.
La expresión "átomo de halógeno" significa radical de átomo de flúor (F), átomo de cloro (Cl), átomo de bromo (Br) o átomo de yodo (I). Preferiblemente, cada átomo de halógeno es, de manera independiente, el radical Br, F o Cl y más preferiblemente el radical F o Cl. El término "haluro" significa anión de fluoruro (F-), cloruro (Cl-), bromuro (Br-) o yoduro (I-).
A menos que se indique de otro modo en la presente memoria, el término "heteroátomo" significa O, S, S(O), S(O)2, Si(RC)2 , Ge(RC)2, P(RP) o N(RN), en donde, de manera independiente, cada RC es hidrocarbilo (C1-C18) no sustituido, cada RP es hidrocarbilo (C1-C18) no sustituido; y cada RN es hidrocarbilo (C1-C18) no sustituido o está ausente (ausente cuando N comprende -N=). Preferiblemente, no existen átomos de germanio (Ge) en el compuesto o complejo de la invención.
Preferiblemente, no existen enlaces de O-O, S-S u O-S, diferentes de los enlaces de O-S en un grupo funcional dirradical S(O) o S(O)2, en el complejo de metal-ligando de Fórmula (I). Más preferiblemente, no existen enlaces de O-O, N-N, P-P, N-P, S-S u O-S, diferentes de los enlaces de O-S en un grupo funcional dirradical S(O) o S(O)2 , en el complejo de metal-ligando de Fórmula (I).
El término "saturado" significa que carece de enlaces dobles de carbono-carbono, enlaces triples de carbonocarbono y enlaces dobles de carbono-nitrógeno, carbono-fósforo y carbono-silicio (en grupos que contienen heteroátomos). En los casos en los que un grupo químico saturado está sustituido con uno o más sustituyentes de RS, uno o más enlaces dobles y/o triples, opcionalmente, pueden estar presentes o no en los sustituyentes de RS. El término "insaturado" significa que contiene uno o más enlaces dobles de carbono-carbono, enlaces triples de carbono-carbono y enlaces dobles de carbono-nitrógeno, carbono-fósforo y carbono-silicio (en grupos que contienen heteroátomos), sin incluir cualquiera de tales enlaces dobles que puedan estar presentes en los sustituyentes de RS, si los hubiera, o en anillos (hetero)aromáticos, si los hubiera.
M es titanio, circonio o hafnio. En una realización, M es circonio o hafnio y en otra realización M es hafnio. En algunas realizaciones, M está en un estado de oxidación formal de 2, 3 o 4. En algunas realizaciones, n es 0, 1, 2 o 3. Cada X, de manera independiente, es un ligando monodentado que es neutro, monoaniónico o dianiónico; o dos X se toman en conjunto para formar un ligando bidentado que es neutro, monoaniónico o dianiónico. X y n se eligen de tal manera que el complejo de metal-ligando de Fórmula (I) sea, en general, neutro. En algunas realizaciones, cada X, de manera independiente, es el ligando monodentado. En una realización, cuando existen dos o más ligandos monodentados X, cada X es igual. En algunas realizaciones, el ligando monodentado es el ligando monoaniónico. El ligando monoaniónico tiene un estado de oxidación formal neto de -1. Cada ligando monoaniónico puede ser, de manera independiente, hidruro, carbanión de hidrocarbilo (C1-C40), carbanión de heterohidrocarbilo (C1-C40), haluro, nitrato, carbonato, fosfato, sulfato, HC(O)O', hidrocarbil (CrC 40)-C(O)O‘, HC(O)N(H)- hidrocarbil (C1-C40)-C(O)N(H)-, hidrocarbil (C1-C40)-C(O)N(hidrocarbilo (C1-C20))-, RKRLN‘, RKO- RKS‘, RKRLP‘ o RMRKRLS¡-, en donde cada RK, RL y RM, de manera independiente, es hidrógeno, hidrocarbilo (C1-C40) o heterohidrocarbilo (C1-C40) o RK y RL se toman en conjunto para formar un hidrocarbileno (C2-C40) o heterohidrocarbileno (C1-C40) y RM es como se ha definido anteriormente.
En algunas realizaciones, al menos un ligando monodentado de X, de manera independiente, es el ligando neutro. En una realización, el ligando neutro es un grupo de base de Lewis neutro que es RXNRKRL, RKORL, RKSRL o RXPRKRL, en donde cada RX, de manera independiente, es hidrógeno, hidrocarbilo (C1-C40), [hidrocarbil (C1-C10)]3Si, [hidrocarbil (C1-C10)]3Si-hidrocarbilo (C1-C10) o heterohidrocarbilo (C1-C40) y cada RK y RL, de manera independiente, es como se ha definido anteriormente.
En algunas realizaciones, cada X es un ligando monodentado que, de manera independiente, es un átomo de halógeno, hidrocarbilo (C1-C20) no sustituido, hidrocarbil (C1-C20)-C(O)O- no sustituido o RKRLN- en donde cada uno de RK y RL, de manera independiente, es un hidrocarbilo (C1-C20) no sustituido. En algunas realizaciones, cada ligando monodentado X es un átomo de cloro, hidrocarbilo (C1-C10) (por ejemplo, alquilo (C1-C6) o bencilo), hidrocarbilo (C1-C10)-C(O)O- no sustituido o RKRLN- en donde cada uno de RK y RL, de manera independiente, es un hidrocarbilo (C1-C10) no sustituido.
En algunas realizaciones, existen al menos dos X y los dos X se toman en conjunto para formar el ligando bidentado. En algunas realizaciones, el ligando bidentado es un ligando bidentado neutro. En una realización, el ligando bidentado neutro es un dieno de Fórmula (RD)2C=C(RD)-C(RD)=C(RD)2, en donde cada RD, de manera independiente, es H, alquilo (C1-C6) no sustituido, fenilo o naftilo. En algunas realizaciones, el ligando bidentado es un monoligando (base de Lewis) monoaniónico. El monoligando (base de Lewis) monoaniónico puede ser un 1,3-dionato de Fórmula (D): RE-C(O')=CH-C(=O)-RE (D), en donde cada RD, de manera independiente, es H, alquilo (C1-C6) no sustituido, fenilo o naftilo. En algunas realizaciones, el ligando bidentado es un ligando dianiónico. El ligando dianiónico tiene un estado de oxidación formal neto de -2. En una realización, cada ligando dianiónico, de manera independiente, es carbonato, oxalato (es decir, -O2CC(O)O'), dicarbanión de hidrocarbileno (C2-C40), dicarbanión de
heterohidrocarbileno (C1-C40) fosfato o sulfato.
Como se ha mencionado anteriormente, el número y la carga (neutra, monoaniónica, dianiónica) de X se seleccionan dependiendo del estado de oxidación formal de M, de tal manera que el complejo de metal-ligando de Fórmula (I) sea, en general, neutro.
En algunas realizaciones, cada X es igual, en donde cada X es metilo; etilo; 1 -propilo; 2-propilo; 1 -butilo; 2,2-dimetilpropilo; trimetilsililmetilo; fenilo; bencilo; o cloro. En algunas realizaciones, n es 2 y cada X es igual.
En algunas realizaciones, al menos dos X son diferentes. En algunas realizaciones, n es 2 y cada X es uno diferente de metilo; etilo; 1 -propilo; 2-propilo; 1 -butilo; 2,2-dimetilpropilo; trimetilsililmetilo; fenilo; bencilo; y cloro.
El número entero n indica el número de X. En una realización, n es 2 o 3 y al menos dos X, de manera independiente, son ligandos monodentados monoaniónicos y un tercer X, si está presente, es un ligando monodentado neutro. En algunas realizaciones, n es 2 y dos X se toman en conjunto para formar un ligando bidentado. En algunas realizaciones, el ligando bidentado es 2,2-dimetil-2-silapropano-1,3-diílo o 1,3-butadieno.
Cada Z, de manera independiente, es O, S, N-hidrocarbilo (C1-C40) o P-hidrocarbilo (C1-C40). En algunas realizaciones, cada Z es diferente. En algunas realizaciones, un Z es O y un Z es NCH3. En algunas realizaciones, un Z es O y un Z es S. En algunas realizaciones, un Z es S y un Z es N-hidrocarbilo (C1-C40) (por ejemplo, NCH3). En algunas realizaciones, cada Z es igual. En algunas realizaciones, cada Z es O. En algunas realizaciones, cada Z es S. En algunas realizaciones, cada Z es N-hidrocarbilo (C1-C40) (por ejemplo, NCH3). En algunas realizaciones, al menos uno y, en algunas realizaciones, cada Z es P-hidrocarbilo (C1-C40) (por ejemplo, PCH3).
L es hidrocarbileno o heterohidrocarbileno (C3-C40) (de 3 a 40 átomos, en donde tal átomo no es H), en donde el hidrocarbileno tiene una parte que comprende una cadena principal de enlazador de 3 átomos de carbono a 10 átomos de carbono que enlaza los átomos de Z en la Fórmula (I) (al que está enlazado L) y el heterohidrocarbileno (C3-C40) (de 3 a 40 átomos, en donde tal átomo no es H) tiene una parte que comprende una cadena principal de enlazador de 3 átomos a 10 átomos que enlaza los átomos de Z en la Fórmula (I), en donde cada uno de los 3 a 10 átomos de la cadena principal de enlazador de 3 átomos a 10 átomos del heterohidrocarbileno (de 3 a 40 átomos, en donde tal átomo no es H), de manera independiente, es un átomo o heteroátomo de carbono, en donde cada heteroátomo, de manera independiente, e C(RC)2, O, S, S(O), S(O)2 , Si(RC)2 , Ge(RC)2, P(RP) o N(RN), en donde, de manera independiente, cada RC es hidrocarbilo (C1-C30), cada RP es hidrocarbilo (C1-C30); y cada RN es hidrocarbilo (C1-C30) o está ausente. En algunas realizaciones, L es el hidrocarbileno (C3-C40). Preferiblemente, la parte mencionada anteriormente que comprende una cadena principal de enlazador de 3 átomos de carbono a 10 átomos de carbono del hidrocarbileno (C3-C40) de L comprende una cadena principal de enlazador de 3 átomos de carbono a 10 átomos de carbono y, más preferiblemente, de 3 átomos de carbono o 4 átomos de carbono que enlaza los átomos de Z en la Fórmula (I) al que se enlaza L. En algunas realizaciones, L comprende la cadena principal de enlazador de 3 átomos de carbono (por ejemplo, L es -CH2CH2CH2-; -CH(CH3)CH2CH(CH3)-; -CH(CH3)CH(CH3)CH(CH3)-; -CH2C(CH3)2CH2-); 1,3-ciclopentano-diílo; o 1,3-ciclohexano-diílo. En algunas realizaciones, L comprende la cadena principal de enlazador de 4 átomos de carbono (por ejemplo, L es -CH2CH2CH2CH2-; -CH2C(CH3)2C(CH3)2CH2-; 1,2-bis(metileno)ciclohexano; o 2,3-bis(metileno)-biciclo[2.2.2]octano). En algunas realizaciones, L comprende la cadena principal de enlazador de 5 átomos de carbono (por ejemplo, L es -CH2CH2CH2CH2CH2- o 1,3-bis(metileno)ciclohexano). En algunas realizaciones, L comprende la cadena principal de enlazador de 6 átomos de carbono (por ejemplo, L es -CH2CH2CH2CH2CH2CH2- o 1,2-bis(etileno)ciclohexano).
En algunas realizaciones, L es el hidrocarbileno (C3-C40) y el hidrocarbileno (C3-C40) de L es un hidrocarbileno (C3-C12) y más preferiblemente hidrocarbileno (C3-C8). En algunas realizaciones, el hidrocarbileno (C3-C40) es un alquileno (C3-C40) no sustituido. En algunas realizaciones, el hidrocarbileno (C3-C40) es un alquileno (C3-C40) sustituido. En algunas realizaciones, el hidrocarbileno (C3-C40) es un cicloalquileno (C3-C40) no sustituido o cicloalquileno (C3-C40) sustituido, en donde cada sustituyente, de manera independiente, es RS, en donde preferiblemente el RS, de manera independiente, es alquilo (C1-C4).
En algunas realizaciones, L es el alquileno (C3-C40) no sustituido y, en algunas otras realizaciones, L es un alquileno (C3-C40) no sustituido acíclico y, todavía más preferiblemente, el alquileno (C2-C40) no sustituido acíclico es -CH2CH2CH2-, cis-CH(CH3)CH2CH(CH3)-, trans-CH(CH3)CH2CH(CH3)-, -CH(CH3)CH2CH(CH3)2-, -CH(CH3)CH(CH3)CH(CH3)-, -CH2C(CH3)2CH2-, -CH2CH2CH2CH2- o -CH2C(CH3)2C(CH3)2CH2-. En algunas realizaciones, L es trans-1,2-bis(metileno)ciclopentano, cis-1,2-bis(metileno)ciclopentano, trans-1,2-bis(metileno)ciclohexano o cis-1,2-bis(metileno)ciclohexano. En algunas realizaciones, el alquilen (C1-C40)-alquileno (C1-C40) sustituido es exo-2,3-bis(metileno)biciclo[2.2.2]octano o exo-2,3-bis(metileno)-7,7-dimetilbiciclo[2.2.1]heptano. En algunas realizaciones, L es el cicloalquileno (C3-C40) no sustituido y, en algunas otras realizaciones, L es cis-1,3-ciclopentano-diílo o cis-1,3-ciclohexano-diílo. En algunas realizaciones, L es el cicloalquileno (C3-C40) sustituido y, más preferiblemente, L es un alquilen (C1-C40)-cicloalquileno (C3-C40) sustituido y, en algunas otras realizaciones, L es el alquilen (C1-C40)-cicloalquileno (C3-C40) sustituido que es exobiciclo[2.2.2]octan-2,3-diílo.
En algunas realizaciones, L es el heterohidrocarbileno (de 3 a 40 átomos). En algunas realizaciones, la parte
mencionada anteriormente que comprende una cadena principal de enlazador de 3 átomos a 6 átomos del heterohidrocarbileno (de 3 a 40 átomos) de L comprende una cadena principal de enlazador de 3 átomos a 5 átomos y, en algunas otras realizaciones, de 3 átomos o 4 átomos que enlaza los átomos de Z en la Fórmula (I) al que se enlaza L. En algunas realizaciones, L comprende la cadena principal de enlazador de 3 átomos (por ejemplo, L es -CH2CH2CH(OCHs)-, -CH2Si(CHs)2CH2- o -CH2Ge(CH3)2CH2-). El "-CH2Si(CH3)2CH2-" puede hacer referencia en la presente memoria a un 1,3-dirradical de 2,2-dimetil-2-silapropano. En algunas realizaciones, L comprende la cadena principal de enlazador de 4 átomos (por ejemplo, L es -CH2CH2OCH2- o -CH2P(CH3)CH2c H2-). En algunas realizaciones, L comprende la cadena principal de enlazador de 5 átomos (por ejemplo, L es -CH2c H2OCH2CH2- o -CH2CH2N(CH3)CH2Ch2-). En algunas realizaciones, L comprende la cadena principal de enlazador de 6 átomos (por ejemplo, L es -CH2CH2C(OCH3)2CH2CH2CH2-, -CH2CH2CH2S(O)2CH2CH2- o -CH2CH2S(O)CH2CH2CH2-). En algunas realizaciones, cada uno de los 3 a 6 átomos de la cadena principal de enlazador de 3 átomos a 6 átomos es un átomo de carbono. En algunas realizaciones, al menos un heteroátomo es el C(RC)2. En algunas realizaciones, al menos un heteroátomo es el Si(RC)2. En algunas realizaciones, al menos un heteroátomo es el O. En algunas realizaciones, al menos un heteroátomo es el N(RN). En algunas realizaciones, no existen enlaces de O-O, S-S u O S, diferentes de los enlaces de O-S en el grupo difuncional dirradical S(O) o S(O)2, en -Z-L-Z-. En algunas realizaciones, no existen enlaces de O-O, N-N, P-P, N-P, S-S u O-S, diferentes de los enlaces de O-S en un grupo funcional dirradical S(O) o S(O)2, en -Z-L-Z-. En algunas realizaciones, el heterohidrocarbileno (de 3 a 40 átomos) es heterohidrocarbileno (de 3 a 11 átomos, excluyendo H) y, en algunas otras realizaciones, heterohidrocarbileno (de 3 a 7 átomos). En algunas realizaciones, el heterohidrocarbileno (de 3 a 7 átomos) de L es -CH2Sí(CH3)2Ch2-; -CH2CH2Sí(Ch3)2CH2-; o CH2Si(CH3)2CH2CH2-. En algunas realizaciones, el heterohidrocarbileno (Ci-C7) de L es -CH2Si(CH3)2CH2-, -CH2Si(CH2CHs)2CH2-, -CH2Si(isopropil)2CH2-, -CH2Si(tetrametileno)CH2- o -CH2Si(pentametileno)CH2-. El -CH2Si(tetrametileno)CH2- se denomina 1-silaciclopentan-1,1-dimetileno. El -CH2Si(pentametileno)CH2- se denomina 1-silaciclohexan-1,1-dimetileno.
En algunas realizaciones, el complejo de metal-ligando de Fórmula (I) es un complejo de metal-ligando de una cualquiera de las siguientes fórmulas:






En una realización, el complejo de metal-ligando de Fórmula (I) es un complejo de metal-ligando de uno cualquiera de los complejos de metal-ligando descritos anteriormente con la condición de que tal complejo de metal-ligando de Fórmula (I) excluya uno o más complejos de metal-ligando que contengan una cualquiera de las siguientes estructuras de ligando:

Componente de cocatalizador
El procatalizador que comprende el complejo de metal-ligando de Fórmula (I) se vuelve catalíticamente activo poniéndolo en contacto o combinándolo con el cocatalizador de activación o usando una técnica de activación, tal como aquellas que son conocidas en la técnica para su uso con reacciones de polimerización de olefinas basadas en metales. Los cocatalizadores de activación adecuados para su uso en la presente memoria incluyen alquil aluminios; alumoxanos poliméricos u oligoméricos (también conocidos como aluminoxanos); ácidos de Lewis neutros; y compuestos no poliméricos, no coordinantes, formadores de iones (incluyendo el uso de tales compuestos en condiciones de oxidación). Una técnica de activación adecuada es la culombimetría potenciostática. También se contemplan combinaciones de uno o más de los cocatalizadores y técnicas de activación anteriores. El término "alquil aluminio" significa un dihidruro de monoalquil aluminio o dihaluro de monoalquilaluminio, un hidruro de dialquil aluminio o haluro de dialquil aluminio o un trialquilaluminio. Los aluminoxanos y sus preparaciones se conocen, por ejemplo, en la patente estadounidense número (USPN) US 6.103.657. Los ejemplos de alumoxanos poliméricos u oligoméricos preferidos son metilalumoxano, metilalumoxano modificado con triisobutilaluminio e isobutilalumoxano. Los cocatalizadores de activación de ácido de Lewis ejemplares son compuestos de metales del Grupo 13 que contienen de 1 a 3 sustituyentes de hidrocarbilo, como se describe en la presente memoria. En algunas realizaciones, los compuestos de metales del Grupo 13 ejemplares son compuestos de aluminio sustituido con tri(hidrocarbilo) o tri(hidrocarbil)-boro. En algunas realizaciones, los compuestos de metales del Grupo 13 ejemplares son compuestos de aluminio sustituido con tri(hidrocarbilo) o tri(hidrocarbil)-boro, son compuestos de tri(alquil (C1-C10))aluminio o tri(aril (C6-C18))boro y derivados halogenados (incluyendo perhalogenados) de los mismos. En algunas otras realizaciones, los compuestos de metales del Grupo 13 ejemplares son tris(fenilo sustituido con fluoro)boranos, en otras realizaciones, tris(pentafluorofenil)borano. En algunas realizaciones, el cocatalizador de activación es un tris(hidrocarbil (Ci-C20))borato (por ejemplo, tetrafluoroborato de tritilo) o un tetra(hidrocarbil (C1-C20))borano de tri(hidrocarbil (C1-C20))amonio (por ejemplo, tetraquis(pentafluorofenil)borano de bis(octadecil)metilamonio). Como se usa en la presente memoria, el término "amonio" significa un catión de nitrógeno que es un (hidrocarbil (C1-C20))4N+, un (hidrocarbil(C1-C20))3N(H)+, un (hidrocarbil(C1-C20))2N(H)2+, hidrocarbil (C1-C20)N(H)3+ o N(H)4+, en donde cada hidrocarbilo (C1-C20) puede ser igual o diferente.
Las combinaciones ejemplares de cocatalizadores de activación de ácido de Lewis neutros incluyen mezclas que comprenden una combinación de un tri(alquil (C1-C4))aluminio y un compuesto de tri(aril (C6-C18))boro halogenado, especialmente un tris(pentafluorofenil)borano. Otras realizaciones ejemplares son combinaciones de tales mezclas
de ácido de Lewis neutras con un alumoxano polimérico u oligomérico y combinaciones de un ácido de Lewis neutro individual, especialmente tris(pentafluorofenil)borano con un alumoxano polimérico u oligomérico. Las relaciones de realizaciones ejemplares de números de moles de (complejo de metal-ligando):(tris(pentafluorofenilborano):(alumoxano) [por ejemplo, (complejo de metal-ligando del Grupo 4):(tris(pentafluorofenilborano):(alumoxano)] son de 1:1:1 a 1:10:30, otras realizaciones ejemplares son de 1:1:1,5 a 1:5:10.
Muchos cocatalizadores de activación y técnicas de activación se han enseñado previamente con respecto a diferentes complejos de metal-ligando en las siguientes USPN: US 5.064.802; US 5.153.157; US 5.296.433; US 5.321.106; US 5.350.723; US 5.425.872; US 5.625.087; US 5.721.185; US 5.783.512; US 5.883.204; US 5.919.983; US 6.696.379; y US 7.163.907. Los ejemplos de hidrocarbilóxidos se describen en el documento US 5.296.433. Los ejemplos de sales de ácido de Bronsted adecuadas para catalizadores de polimerización por adición se describen en los documentos US 5.064.802; US 5.919.983; US 5.783.512. Los ejemplos de sales adecuadas de un agente oxidante catiónico y un anión compatible no coordinante como cocatalizadores de activación para catalizadores de polimerización por adición se describen en el documento US 5.321.106. Los ejemplos de sales de carbenio adecuadas como cocatalizadores de activación para catalizadores de polimerización por adición se describen en el documento US 5.350.723. Los ejemplos de sales de sililio adecuadas como cocatalizadores de activación para catalizadores de polimerización por adición se describen en el documento US 5.625.087. Los ejemplos de complejos adecuados de alcoholes, mercaptanos, silanoles y oximas con tris(pentafluorofenil)borano se describen en el documento US 5.296.433. Algunos de estos catalizadores también se describen en una parte del documento US 6.515.155 B1, comenzando en la columna 50, en la línea 39, y pasando por la columna 56, en la línea 55.
En algunas realizaciones, el procatalizador que comprende el complejo de metal-ligando de Fórmula (I) se puede activar para formar una composición catalítica activa mediante la combinación con uno o más cocatalizadores, tales como un cocatalizador formador de cationes, un ácido de Lewis fuerte o una combinación de los mismos. Los cocatalizadores adecuados para su uso incluyen aluminoxanos poliméricos u oligoméricos, especialmente aluminoxano de metilo, así como compuestos formadores de iones inertes, compatibles, no coordinantes. Los cocatalizadores adecuados ejemplares incluyen, pero no se limitan a, aluminoxano de metilo modificado (MMAO), bis(alquil sebo hidrogenado)metilo, tetraquis(pentafluorofenil)borato(1-) amina (RIBS-2), trietilaluminio (TEA) y cualquier combinación de los mismos.
En algunas realizaciones, uno o más de los cocatalizadores de activación anteriores se usan en combinación entre sí. Una combinación especialmente preferida es una mezcla de un tri(hidrocarbil (C1-C4))aluminio, tri(hidrocarbil (C1-C4))borano o un borato de amonio con un compuesto de alumoxano oligomérico o polimérico.
La relación del número total de moles de uno o más complejos de metal-ligando de Fórmula (I) y el número total de moles de uno o más de los cocatalizadores de activación es de 1:10.000 a 100:1. En algunas realizaciones, la relación es de al menos 1:5.000, en algunas otras realizaciones, de al menos 1:1.000; y de 10:1 o menos y, en algunas otras realizaciones, de 1:1 o menos. Cuando se usa un alumoxano solo como cocatalizador de activación, preferiblemente el número de moles del alumoxano que se emplean es de al menos 100 veces el número de moles del complejo de metal-ligando de Fórmula (I). Cuando se usa tris(pentafluorofenil)borano solo como cocatalizador de activación, en algunas otras realizaciones, el número de moles del tris(pentafluorofenil)borano que se emplean respecto al número total de moles de uno o más complejos de metal-ligando de Fórmula (I) es de 0,5:1 a 10:1, en algunas otras realizaciones, de 1:1 a 6:1, en algunas otras realizaciones, de 1:1 a 5:1. Los cocatalizadores de activación restantes se emplean, en general, en cantidades aproximadamente en moles iguales a las cantidades en moles totales de uno o más complejos de metal-ligando de Fórmula (I).
Propiedades del sistema de catalizador
La composición de catalizador que comprende el procatalizador que comprende el complejo de metal-ligando de Fórmula (I) y uno o más cocatalizadores, como se describe en la presente memoria, tiene una relación de reactividad r-i, como se define adicionalmente en lo sucesivo en la presente memoria, en el intervalo de mayor de 100; por ejemplo, mayor de 150 o mayor de 200.
Se cree que las interacciones estéricas para los catalizadores dan como resultado la polimerización del etileno de manera más selectiva que la alfa-olefina estéricamente más grande (u otro comonómero de olefina más grande) durante el proceso de la invención (es decir, el catalizador polimeriza preferiblemente el etileno en presencia de la alfa-olefina). De nuevo, sin quedar limitados a ninguna teoría, se cree que tales interacciones estéricas hacen que el catalizador preparado con o a partir del complejo de metal-ligando de Fórmula (I) adopte una conformación que permita que el etileno acceda al M sustancialmente más fácilmente o adopte una conformación reactiva más fácilmente o ambas que el catalizador permita que la alfa-olefina lo haga. La diferencia resultante en las velocidades de polimerización (es decir, la selectividad) entre el etileno y la alfa-olefina con el catalizador en el proceso de la invención se puede caracterizar por la relación de reactividad n.
En los copolímeros aleatorios en los que la identidad del último monómero insertado dicta la velocidad a la que se insertan los monómeros posteriores, se emplea el modelo de copolimerización terminal. En este modelo las reacciones de inserción del tipo
■■■M 1.C* M J .— kjL^ ---M. 'M J £* (A)
donde C* representa el catalizador, Mi representa monómero/ y kij es la constante de velocidad que tiene la ecuación de velocidad

La fracción molar del comonómero (i=2) en los medios de reacción se define mediante la ecuación:
[m 2
Í2
[mJ+[
C)
m 2 (
Una ecuación simplificada para la composición de comonómero se puede derivar, como se describe en George Odian, Principles of Polymerization, Segunda Edición, John Wiley and Sons, 1970, de la siguiente manera:
F _ r1( l - / 2)2 ( l - / 2) /2
(D)
2 rx (l - f 2 )2 2(l - / 2) )/2 r j 2
A partir de esta ecuación, la fracción molar del comonómero en el polímero depende únicamente de la fracción molar del comonómero en los medios de reacción y dos relaciones de reactividad dependientes de la temperatura definidas en términos de las constantes de velocidad de inserción como:

Como alternativa, en el penúltimo modelo de copolimerización, las identidades de los dos últimos monómeros insertados en la cadena de polímero creciente dictan la velocidad de inserción de monómero posterior. Las reacciones de polimerización son de la forma

y las ecuaciones de velocidad individuales son:

El contenido de comonómero se puede calcular (de nuevo, como se describe en George Odian, mencionado anteriormente) como:

donde X se define como:
X = - L t i (J)
f i
y las relaciones de reactividad se definen como:

En este modelo, así como la composición de polímero, es una función únicamente de las relaciones de reactividad dependientes de la temperatura y la fracción molar del comonómero en el reactor. También se cumple lo mismo cuando se puede producir la inserción de comonómero o monómero inversa o en el caso de la interpolimerización de más de dos monómeros.
Las relaciones de reactividad para su uso en los modelos anteriores se pueden predecir usando técnicas teóricas
bien conocidas o derivadas empíricamente de datos de polimerización reales. Las técnicas teóricas adecuadas se describen, por ejemplo, en B. G. Kyle, Chemical and Process Thermodynamics, Tercera Edición, Prentice-Hall, 1999 y en Redlich-Kwong-Soave (RKS) Equation of State, Chemical Engineering Science, 1972, pág. 1197-1203. Los programas de soporte lógico disponibles en el mercado se pueden usar para ayudar a derivar relaciones de reactividad a partir de datos derivados experimentalmente. Un ejemplo de tal soporte lógico es Aspen Plus de Aspen Technology, Inc., Ten Canal Park, Cambridge, MA 02141-2201 Ee .Uu .
Por consiguiente, el proceso para la producción de polímeros basados en etileno según la presente invención proporciona, de manera selectiva, el segmento de polietileno rico (por ejemplo, un polietileno de alta densidad) o de polietileno rico del copolímero de poli(1-octeno de etileno) en presencia de 1-octeno, que está sustancialmente sin polimerizar de ese modo. El proceso para la producción de polímeros basados en etileno emplea condiciones de polimerización de olefinas. En algunas realizaciones, las condiciones de polimerización de olefinas producen, de manera independiente, un catalizador in situ que se forma mediante la reacción del procatalizador que comprende el complejo de metal-ligando de Fórmula (I) y uno o más cocatalizadores en presencia de uno o más ingredientes diferentes. Tales ingredientes diferentes incluyen, pero no se limitan a, (i) monómeros de olefina; (ii) otro complejo de metal-ligando de Fórmula (I); (iii) uno o más sistemas de catalizador; (iv) uno o más agentes transportadores de cadena; (v) uno o más estabilizadores de catalizador; (vi) uno o más disolventes; y (vii) una mezcla de dos o más cualesquiera de los mismos.
Un catalizador particularmente preferido es uno que puede lograr una alta selectividad para la polimerización del etileno en presencia del 1-octeno en el proceso para la producción de un polímero basado en etileno, en donde la alta selectividad se caracteriza por la relación de reactividad n descrita anteriormente. Preferiblemente, en el proceso de la invención, la relación de reactividad n es mayor de 50, más preferiblemente mayor de 100, todavía más preferiblemente mayor de150, todavía más preferiblemente mayor de 200. Cuando la relación de reactividad r-i, en el proceso de la invención, se aproxima al infinito, la incorporación del 1-octeno en (o sobre) el polietileno rico producido de este modo se aproxima al 0 por ciento en moles (% en moles).
La composición de catalizador que comprende el procatalizador y uno o más cocatalizadores, como se describe en la presente memoria, tiene una eficacia catalítica en el intervalo de mayor de 1.000.000 g de polímero por gramo de centro de metal activo; por ejemplo, de mayor de 2.000.000 g de polímero por gramo de centro de metal activo. La eficacia catalítica se mide en términos de cantidad de polímero producida con respecto a la cantidad de catalizador usada en el proceso de polimerización en solución, en donde la temperatura de polimerización es de al menos 130 °C, por ejemplo, en el intervalo de 170 a 195 °C, y la concentración de etileno es mayor de 5 g/l, por ejemplo, mayor de 6 g/l, y en donde la conversión de etileno es mayor del 70 por ciento, por ejemplo, mayor del 80 por ciento, o, como alternativa, mayor del 90 por ciento.
Proceso para la producción del procatalizador
En algunas realizaciones, los ligandos de la invención se pueden preparar usando procedimientos conocidos. De manera específica, los ligandos de la invención se pueden preparar usando una diversidad de rutas sintéticas, dependiendo de la variación deseada en el ligando. En general, se preparan bloques de construcción que, después, se unen entre sí con un grupo puente. Las variaciones en los sustituyentes del grupo R se pueden introducir en la síntesis de los bloques de construcción.
Se pueden introducir variaciones en el puente con la síntesis del grupo puente. Los ligandos específicos se pueden preparar según los esquemas generales mostrados a continuación, donde, en primer lugar, se preparan los bloques de construcción y, después, se acoplan entre sí. Hay varias formas diferentes de usar estos bloques de construcción. En una realización, en general, cada uno de los anillos de fenilo opcionalmente sustituidos se prepara como un bloque de construcción separado. Los fenilos opcionalmente sustituidos deseados se combinan después en bloques de construcción de bi-fenilo, que después forman puentes entre sí. En otra realización, los bloques de construcción de fenilo opcionalmente sustituidos forman puentes entre sí y, después, se añaden bloques de construcción de fenilo opcionalmente sustituidos adicionales para formar las estructuras de bi-arilo puenteadas. Los materiales de partida o reactivos usados, en general, están disponibles en el mercado o se preparan a través de medios sintéticos de rutina.
En los esquemas siguientes, el término ligando se refiere al precursor orgánico al procatalizador. El procatalizador se deriva de una reacción del ligando con un precursor metálico adecuado (titanio, circonio o hafnio).
Los sustituyentes orgánicos comunes se han abreviado como en el siguiente sistema clave:
Me = metilo
Et = etilo
Ph = fenilo
t-Bu = butilo terciario
i-Pr = isopropilo
n-Bu = butilo
Me2Si = dimetilsililo
MeaSi = trimetilsililo
Me2PhSi = dimetilfenilsililo
DME = dimetoxietano
THF = tetrahidrofurano
1. Preparación de nitro-1,1'-bifenilo sustituido

Al 1,1 '-bifenilo sustituido deseado, (aproximadamente 56 mmol) se añade anhídrido acético (aproximadamente 300 ml) en un matraz que se sumerge en un baño de agua a temperatura ambiente. A la suspensión se añade lentamente gota a gota una mezcla de ácido acético (aproximadamente 15 ml, 262 mmol) y ácido nítrico fumante (aproximadamente 9,0 ml, 191 mmol) durante el período de aproximadamente 10 minutos a través de un embudo de adición igualador de la presión. A continuación, la mezcla se deja agitar hasta que se complete la reacción, como se indica mediante el control por cromatografía de gases/espectroscopía de masas (CG/EM). A continuación, se añade la mezcla a aproximadamente 2,5 l de hielo-agua y se agita durante aproximadamente 1-2 horas. El precipitado se recoge mediante filtración al vacío y se lava con dos partes de aproximadamente 100 ml de hielo-agua. Este material bruto se disuelve en aproximadamente 250 ml de cloruro de metileno y se lava con agua (aproximadamente 250 ml) y, después, con NaOH acuoso 1 M (aproximadamente 250 ml). La fase orgánica se seca sobre sulfato de magnesio anhidro, se filtra y se concentra a alto vacío. El material bruto se purifica después mediante cromatografía ultrarrápida.
2. Preparación de 9H-carbazol sustituido

Al 2-nitro-1,1 '-bifenilo sustituido deseado (aproximadamente 25 mmol) en una caja de guantes se añade trietilfosfito (aproximadamente 31,0 ml, 180 mmol). La mezcla se retira de la caja de guantes y se lleva a la campana y se coloca en una atmósfera de nitrógeno y se calienta a reflujo suave (temperatura del manto de aproximadamente 175 °C), al tiempo que se controla el avance de la reacción mediante CG/Em . Una vez que se determina que se ha completado la reacción, se enfría y el condensador se retira de la reacción y el trietilfosfito se retira por destilación al vacío con una columna de trayectoria corta a aproximadamente 75 °C (temperatura del manto) hasta que quedan algunos ml de líquido. A continuación, el matraz se calienta adicionalmente hasta aproximadamente 125 °C hasta que no se produce ninguna destilación adicional. A continuación, el residuo se deja enfriar hasta temperatura ambiente, después se diluye y se lava con aproximadamente 100 ml de 1:1 de metanol:hielo-agua y se filtra. El precipitado se aísla mediante filtración al vacío y el residuo que queda en el matraz de reacción se disuelve en aproximadamente 300 ml de cloruro de metileno, se seca con sulfato de magnesio anhidro, se filtra y se concentra para dar el material bruto. Este material bruto se purifica, después, mediante cromatografía ultrarrápida.
3. Preparación de 9-(2-((tetrah¡dro-2H-p¡ran-2-N)ox¡)-5-(2,4,4-tr¡metMpentan-2-¡l)feml)-9H-carbazol sustituido:

A un matraz de fondo redondo de tres bocas de 250 ml en una caja de guantes se añade el 2-(2-yodofenoxi)tetrahidro-2H-pirano sustituido deseado (aproximadamente 52 mmol), el carbazol sustituido deseado (aproximadamente 29 mmol), K3PO4 (aproximadamente 23,40 g, 110,24 mmol), CuI anhidro (aproximadamente 0,22 g, 1,16 mmol), tolueno seco (aproximadamente 85 ml) y A/,W-dimetiletilendiamina (aproximadamente 0,45 ml, 4,18 mmol). El matraz se lleva de la caja de guantes a la campana y se calienta a reflujo. El avance de la reacción se controla mediante análisis de CG/EM y, en algunos casos, se añade CuI anhidro adicional (aproximadamente 0,2 g, 1,05 mmol) suspendido en tolueno seco (aproximadamente 0,9 ml) y A/,W-dimetiletilendiamina (aproximadamente 0,45 ml, 4,18 mmol) a la mezcla y se continúa calentando a reflujo hasta el momento en que se observa que la conversión se ha completado. A continuación, la reacción se deja enfriar hasta temperatura ambiente y se filtra a través de un pequeño tapón de sílice, se lava con tetrahidrofurano y se concentra para dar el producto bruto. Este material bruto se puede purificar mediante recristalización o cromatografía ultrarrápida.
4. Preparación de 2-((tetrah¡dro-2H-p¡ran-2-N)ox¡)-3-(4,4,5,5-tetramet¡l-1,3,2-d¡oxaborolan-2-No sustituido:

A un matraz de fondo redondo de tres bocas secado al horno a aproximadamente 0-10 °C en atmósfera de N2 se añade el 2-((tetrahidro-2H-piran-2-il)oxi)-5-(2,4,4-trimetilpentan-2-il)fenilo) deseado (aproximadamente 14 mmol) y tetrahidrofurano seco (aproximadamente 90 ml). Esta solución se enfría hasta aproximadamente 0-10 °C (baño de hielo-agua) durante aproximadamente 15 minutos y se añade lentamente n-butillitio 2,5 M en hexanos (aproximadamente 14 ml, 35,00 mmol). Después de agitar durante aproximadamente 4 horas, se añade lentamente 2-iso-propoxi-4,4,5,5-tetrametil-1,3,2-dioxaborolano (aproximadamente 7,0 ml, 34 mmol). La mezcla se agita durante una hora a aproximadamente 0-10 °C antes de dejar que la reacción se caliente hasta temperatura ambiente y, después, se agita durante aproximadamente 18 horas adicionales. A la mezcla de reacción se añade bicarbonato de sodio acuoso saturado frío (aproximadamente 75 ml). La mezcla se extrae con aproximadamente cuatro partes de 50 ml de cloruro de metileno. Las fases orgánicas se combinan y se lavan con bicarbonato de sodio acuoso saturado frío (aproximadamente 200 ml), salmuera (aproximadamente 200 ml), a continuación, se secan sobre sulfato de magnesio anhidro, se filtran y se concentran para dar el producto bruto, que se suspende en acetonitrilo (aproximadamente 75 ml) y se deja reposar durante una hora a temperatura ambiente antes de aislar el sólido mediante filtración al vacío. Los sólidos se lavan con una pequeña parte de acetonitrilo frío y se secan a alto vacío para proporcionar el producto.
5a. Preparación de ligando protegido (método 1, doble reacción de Suzuki simultánea).

A un matraz de fondo redondo en atmosfera de N2 se anade el 2-((tetrahidro-2H-piran-2-il)oxi)-3-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-ilo sustituido deseado (aproximadamente 9,9 mmol), dimetoxietano (aproximadamente 120 ml), una solución de NaOH (aproximadamente 1,30 g, 32,5 mmol) en agua (aproximadamente 35 ml), tetrahidrofurano (aproximadamente 60 ml) y las especies de bis-2-yodoarilo enlazadas deseadas (aproximadamente 4,7 mmol). El sistema se purga después con N2 durante aproximadamente 15 minutos y se añade Pd(PPh3)4 (aproximadamente 303 mg, 0,26 mmol). La mezcla se calienta a reflujo a aproximadamente 85 °C durante aproximadamente 48 horas y, después, se deja enfriar hasta temperatura ambiente. Una vez enfriado, se formó un precipitado en el matraz de reacción que se aísla mediante filtración al vacío y se seca a alto vacío durante una hora para proporcionar el ligando protegido bruto. Este ligando protegido se puede usar como tal en la siguiente etapa.
5b. Preparación de ligando protegido (método 2, reacciones de Suzuki secuenciales).

A un matraz de fondo redondo en atmósfera de N2 se añade el 2-((tetrahidro-2H-piran-2-il)oxi)-3-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-ilo sustituido deseado (4,7 mmol), dimetoxietano (aproximadamente 120 ml), una solución de NaOH (aproximadamente 1,30 g, 32,5 mmol) en agua (aproximadamente 35 ml), tetrahidrofurano (aproximadamente 60 ml) y las especies de bis-2-yodoarilo enlazadas deseadas (aproximadamente 4,7 mmol). El sistema se purga con N2 durante aproximadamente 15 minutos y se añade Pd(PPh3)4 (aproximadamente 303 mg, 0,26 mmol). La mezcla se calienta a reflujo a aproximadamente 85 °C durante aproximadamente 48 horas, momento en el que se añade el segundo 2-((tetrahidro-2H-piran-2-il)oxi)-3-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-ilo sustituido (aproximadamente 4,7 mmol), junto con Pd(PPh3)4 adicional (aproximadamente 303 mg, 0,26 mmol). La mezcla resultante se calienta de nuevo a reflujo a aproximadamente 85 °C durante aproximadamente 48 horas y, después, se deja enfriar hasta temperatura ambiente. Una vez enfriado, se forma un precipitado en el matraz de reacción que se aísla mediante filtración al vacío y se seca a alto vacío durante una hora para proporcionar el ligando protegido bruto. Este ligando protegido se puede usar como tal en la siguiente etapa.
6. Preparación de ligando

Al ligando protegido bruto se añade una mezcla de 1:1 de metanol/tetrahidrofurano (aproximadamente 200 ml) y aproximadamente 100 mg de monohidrato de ácido p-toluenosulfónico. La solución se calienta a aproximadamente
60 °C durante aproximadamente 8 horas, después se deja enfriar y se concentra. El residuo se disuelve en cloruro de metileno (aproximadamente 250 ml), se lava con salmuera (aproximadamente 250 ml), se seca sobre sulfato de magnesio anhidro, se filtra a través de una almohadilla de gel de sílice y, después, se concentra. Este material bruto se purifica mediante cromatografía ultrarrápida.
7. Ejemplo de preparación de procatalizador.

El ligando (aproximadamente 0,38 mmol) y MCl4 (aproximadamente 0,38 mmol) se suspenden en aproximadamente 35 ml de tolueno frío (aproximadamente -30 °C). A esta mezcla se añaden aproximadamente 0,56 ml de solución de éter de dietilo 3 M de XMgBr. Después de aproximadamente 1-24 h de agitación, dependiendo del ligando particular, el disolvente se retira a presión reducida. Al residuo se añaden aproximadamente 20 ml de tolueno, seguido de aproximadamente 25 ml de hexano. A continuación, la suspensión se filtra y el disolvente se retira a presión reducida dando el procatalizador deseado.
Polímeros basados en olefinas
Las composiciones de catalizador que comprenden uno o más procatalizadores que comprenden el complejo de metal-ligando de Fórmula (I) y uno o más cocatalizadores se emplean para preparar interpolímeros (incluyendo copolímeros) de etileno y 1-octeno.
Polímeros basados en etileno
Los interpolímeros (incluyendo copolímeros) de etileno y 1-octeno tienen una densidad en el intervalo de 0,860 a 0,973 g/cm3.
Los interpolímeros (incluyendo copolímeros) de etileno y 1-octeno tienen una frecuencia de ramificación de cadena larga en el intervalo de 0,0 a 3 ramificaciones de cadena larga (LCB) por 1.000 C.
Los interpolímeros (incluyendo copolímeros) de etileno y 1-octeno tienen un peso molecular (Mw) en el intervalo de igual a o mayor de 20.000 g/mol, por ejemplo, en el intervalo de 20.000 a 350.000 g/mol.
Los interpolímeros (incluyendo copolímeros) de etileno y 1-octeno tienen un índice de fundición (I2) en el intervalo de 0,1 a 200 g/10 minutos.
Los interpolímeros (incluyendo copolímeros) de etileno y 1-octeno tienen una relación de flujo de fundición (Iio/I2) en el intervalo de 5 a 30.
Los interpolímeros (incluyendo copolímeros) de etileno y 1-octeno tienen una relación de viscosidad de cizallamiento cero (ZSVR) en el intervalo de igual a o mayor de 1,0; por ejemplo, de 1,0 a 10,0; o, como alternativa, de 1,0 a 8,0; o, como alternativa, de 1,0 a 7,0; o, como alternativa, de 1,0 a 5,0; o, como alternativa, de 1,0 a 4,0; o, como alternativa, de 1,0 a 3,0; o, como alternativa, de 1,0 a 2,0.
Los interpolímeros (incluyendo copolímeros) de etileno y 1-octeno pueden comprender, además, al menos 0,01 partes en peso de residuos de metales y/o residuos de óxidos de metales que quedan del sistema de catalizador de la invención por un millón de partes de los polímeros basados en etileno de la invención. Los residuos de metales y/o los residuos de óxidos de metales que quedan del sistema de catalizador en los polímeros basados en etileno se pueden medir mediante fluorescencia de rayos X (XRF), que se calibra con patrones de referencia.
Los interpolímeros (incluyendo copolímeros) de etileno y 1-octeno comprenden del 0,5 al 3 por ciento en moles de unidades derivadas de 1-octeno.
Los interpolímeros (incluyendo copolímeros) de etileno y 1-octeno comprenden del 97 al 99,5 por ciento en moles de unidades derivadas de etileno.
Se puede emplear cualquier proceso de polimerización convencional para producir los polímeros basados en etileno. Tales procesos de polimerización convencionales incluyen, pero no se limitan a, un proceso de polimerización en solución, un proceso de polimerización en fase de gas, un proceso de polimerización en fase de suspensión y combinaciones de los mismos, usando un solo reactor, por ejemplo, reactor isotérmico, reactor de bucle, reactor en fase de gas de lecho fluidizado, reactor de tanque agitado o reactor por lotes.
Los polímeros basados en etileno se pueden producir a través de un proceso de polimerización en fase de solución
usando un reactor de bucle o un reactor isotérmico.
En general, el proceso de polimerización en fase de solución se produce en un solo reactor bien agitado, tal como un reactor de bucle o un reactor isotérmico esférico, a una temperatura en el intervalo de 120 a 300 °C; por ejemplo, de 160 a 190 °C, y a presiones en el intervalo de 2,1 MPa a 10,5 MPa (de 300 a 1.500 psi); por ejemplo, de 2,8 a 5,25 MPa (de 400 a 750 psi). El tiempo de residencia en el proceso de polimerización en fase de solución se encuentra típicamente en el intervalo de 2 a 30 minutos; por ejemplo, de 10 a 20 minutos. Se añaden el etileno, uno o más disolventes, uno o más sistemas de catalizador, por ejemplo, un sistema de catalizador, opcionalmente, uno o más cocatalizadores, y 1-octeno.
Los disolventes ejemplares incluyen, pero no se limitan a, isoparafinas. Por ejemplo, tales disolventes están disponibles en el mercado con el nombre ISOPAR E a través de ExxonMobil Chemical Co., Houston, Texas. La mezcla resultante del polímero basado en etileno y el disolvente se retira después del reactor y el polímero basado en etileno se aísla. El disolvente se recupera típicamente a través de una unidad de recuperación de disolvente, es decir, intercambiadores de calor y un tambor de separador de líquido y vapor, y, después, se recicla de nuevo en el sistema de polimerización.
El procatalizador que comprende el complejo de metal-ligando de Fórmula (I) se puede activar para formar una composición catalítica activa mediante la combinación con uno o más cocatalizadores, como se ha descrito anteriormente, por ejemplo, un cocatalizador formador de cationes, un ácido de Lewis fuerte o una combinación de los mismos. Los cocatalizadores adecuados para su uso incluyen aluminoxanos poliméricos u oligoméricos, especialmente aluminoxano de metilo, así como compuestos formadores de iones inertes, compatibles, no coordinantes. Los cocatalizadores adecuados ejemplares incluyen, pero no se limitan a, aluminoxano de metilo modificado (MMAO), bis(alquil sebo hidrogenado)metilo, tetraquis(pentafluorofenil)borato(1-) amina (RIBS-2), trietilaluminio (TEA) y combinaciones de los mismos.
Los polímeros basados en etileno pueden comprender, además, uno o más aditivos. Tales aditivos incluyen, pero no se limitan a, agentes antiestáticos, potenciadores del color, colorantes, lubricantes, pigmentos, antioxidantes primarios, antioxidantes secundarios, adyuvantes de procesamiento, estabilizadores UV y combinaciones de los mismos. Los polímeros basados en etileno pueden contener cualquier cantidad de aditivos. Los polímeros basados en etileno pueden comprender de aproximadamente el 0 a aproximadamente el 10 por ciento por el peso combinado de tales aditivos, basado en el peso de los polímeros basados en etileno y el uno o más aditivos. Los polímeros basados en etileno pueden comprender, además, cargas, que pueden incluir, pero no se limitan a, cargas orgánicas o inorgánicas. Tales cargas, por ejemplo, carbonato de calcio, talco, Mg(OH)2, pueden estar presentes en niveles de aproximadamente 0 a aproximadamente 20, basados en el peso de los polímeros basados en etileno y el uno o más aditivos y/o cargas. Los polímeros basados en etileno se pueden mezclar, además, con uno o más polímeros para formar una mezcla.
Ejemplos
Los siguientes ejemplos ilustran la presente invención, pero no pretenden limitar el alcance de la invención. La preparación de los procatalizadores 2 y 3 comparativos se describe en los documentos WO 2007136496 y US 2011/0282018, respectivamente, en la medida en que se enseñan los procatalizadores 2 y 3 comparativos.
Realización específica para la síntesis real de catalizador
Preparación de 4,4'-di-ferc-butN-2-mtro-1,1'-bifemlo.

A 4,4'-di-terc-butilbifenilo (15,00 g, 56,30 mmol) se añadió anhídrido acético (300 ml) en un matraz que se sumergió en un baño de agua a temperatura ambiente. A la suspensión se añadió lentamente gota a gota una mezcla de ácido acético (15 ml, 261,81 mmol) y ácido nítrico fumante (9,0 ml, 191,43 mmol) durante el período de 10 minutos a través de un embudo de adición igualador de la presión. Se puso el sólido en solución y se volvió amarillo. La mezcla se dejó agitar durante 30 minutos y se comprobó mediante CG/EM, que mostró una reacción completa. La mezcla se añadió a 2,5 l de hielo-agua y se agitó durante 1 hora y 15 minutos. El precipitado de color amarillo se recogió mediante filtración al vacío y se lavó con dos partes de 100 ml de hielo-agua. Este sólido bruto se disolvió en 250 ml de cloruro de metileno. La solución se lavó con agua (250 ml) y NaOH acuoso 1 M (250 ml). La fase orgánica se secó sobre sulfato de magnesio anhidro, se filtró y se concentró a alto vacío para dar el producto bruto como un sólido de color amarillo. El sólido bruto se disolvió en una cantidad mínima de cloroformo para la carga en el cartucho de la columna. El producto en bruto se purificó mediante cromatografía ultrarrápida usando una columna P/N 5146135 Grace Reveleris de 330 g en un instrumento ISCO y eluyendo con un gradiente de cloroformo al 10 20 % en hexanos para proporcionar 11,04 g (63,0 %) del producto como un sólido de color amarillo claro.
RMN 1H (500 MHz, CDCls TMS) 57,80 (d, J = 2,0 Hz, 1H), 7,60 (dd, J = 8,1,2,0 Hz, 1H), 7,42 (d, J = 8,4 Hz, 2H),
7,36 (d, J = 8,1 Hz, 1H), 7,24 (d, J = 8,3 Hz, 2H), 1,38 (s, 9H), 1,35 (s, 9H). RMN 13C{1H} (126 MHz, CDCla TMS) 5 151,72, 150,93, 149,22, 134,24, 133,20, 131,55, 129,26, 127,55, 125,58, 120,85, 34,86, 34,59, 31,29, 31,05.
Preparación de 2,7-di-terc-butil-9H-carbazol.

A 4,4'-di-íerc-butil-2-nitro-1,1 '-bifenilo (8,00 g, 25,69 mmol) en una caja de guantes se añadió trietilfosfito (31,0 ml, 179,82 mmol). La mezcla se retiró de la caja de guantes y se llevó a la campana. La mezcla se colocó en una atmósfera de nitrógeno y se calentó a reflujo suave (temperatura del manto de 175 °C), al tiempo que se controlaba el avance de la reacción mediante CG/e M. Una vez que se determinó que se había completado la reacción (4 horas), se enfrió y el condensador se retiró de la reacción y el trietilfosfito se retiró por destilación al vacío con una columna de trayectoria corta a 75 °C (temperatura del manto) hasta que quedaron algunos ml de líquido. El matraz se calentó adicionalmente hasta 125 °C y no se produjo ninguna destilación adicional (el líquido restante puede ser trietilfosfato que hierve muy alto, subproducto esperado). El residuo se dejó enfriar hasta temperatura ambiente, después, se diluyó y se lavó con aproximadamente 100 ml de 1:1 de metanol:hielo-agua y se filtró. El precipitado aislado mediante filtración al vacío y el residuo adherente que quedaba en el matraz de reacción se disolvieron en aproximadamente 300 ml de cloruro de metileno, se secaron con sulfato de magnesio anhidro, se filtraron y se concentraron para dar 9,41 g de producto bruto como un aceite de color amarillo (aproximadamente el 80 % de producto de carbazol). Este producto bruto se absorbió en cloruro de metileno al 25 % en hexanos y se purificó mediante cromatografía ultrarrápida usando la misma concentración de eluyente y una columna Grace Reveleris de 330 g para proporcionar 4,70 g (66 %) de compuesto puro como polvo de color blanco.
RMN 1H (400 MHz, CDCla) 57,92 (d, J = 8,2 Hz, 2H), 7,76 (s, 1H), 7,37 (d, J = 1,3 Hz, 2H), 7,26 (dd, J = 8,3, 1,6 Hz, 2H), 1,40 (s, 18H). RMN 13C{1H} (126 MHz, C6D6) 5148,93, 140,04, 120,97, 119,48, 117,29, 107,01, 77,25, 77,00, 76,75, 35,05, 31,79.
Preparación de 2,7-di-terc-butil-9-(2-((tetrahidro-2H-piran-2-il)oxi)-5-(2,4,4-trimetilpentan-2-il)fenil)-9H-carbazol.

A un matraz de fondo redondo de tres bocas de 250 ml en una caja de guantes se añadió 2-(2-yodo-4-(2,4,4-trimetilpentan-2-il)fenoxi)tetrahidro-2H-pirano (21,74 g, 52,22 mmol), 2,7-di-f-butilcarbazol (8,03 g, 28,73 mmol), K3PO4 (23,40 g, 110,24 mmol), Cul anhidro (0,22 g, 1,16 mmol), tolueno seco (85 ml) y N,N'-dimetiletilendiamina (0,45 ml, 4,18 mmol). El matraz se llevó de la caja de guantes a la campana y se calentó en N2 a 125 °C (temperatura del manto de calentamiento). Después de 24 horas, el análisis de CG mostró una conversión de aproximadamente el 76 %, por lo tanto, se añadió Cul anhidro adicional (0,2 g, 1,05 mmol) suspendido en tolueno seco (0,9 ml) y N,N'-dimetiletilendiamina (0,45 ml, 4,18 mmol) y se continuó la agitación a 125 °C durante 72 horas adicionales. El análisis de CG después de un total de 96 horas mostró cantidades de traza de carbazol restante. La reacción se dejó enfriar hasta temperatura ambiente y se filtró a través de un pequeño tapón de sílice, se lavó con tetrahidrofurano y se concentró para dar 24,47 g de producto bruto como un aceite de color marrón oscuro. Este producto bruto se recristalizó a partir de hexanos calientes (50 ml) para proporcionar 13,48 g (90,9 %) del producto como un polvo blanquecino 98,12 % puro mediante CG.
RMN 1H (500 MHz, CDCh) 58,00 (dd, J = 8,2, 0,5 Hz, 2H), 7,44-7,49 (m, 2H), 7,45 (d, J = 2,5 Hz, 1H), 7,38 (d, J = 8.6 Hz, 1H), 7,30 (dt, J =8,2, 1,7 Hz, 2H), 7,19 (dd, J = 1,7, 0,5 Hz, 1H), 7,10 (dd, J = 1,7, 0,5 Hz, 1H), 5,25 (t, J = 2.7 Hz, 1H), 3,71 (td, J = 10,9, 2,9 Hz, 1H), 3,47 (dt, J = 11,2, 4,0 Hz, 1H), 1,76 (ABq, J = 14,6 Hz, 2H), 1,42 (s, 6H), 1,36 (s, 9H), 1,35 (s, 9H), 1,12-1,32 (m, 6H), 0,83 (s, 9H). RMN 13C{1H} (101 MHz, CDCh) 5151,18, 148,58, 148,51, 144,34, 142,00, 141,98, 127,78, 126,72, 126,44, 120,82, 120,73, 119,12, 119,08, 117,16, 117,10, 116,60, 106,88, 106,55, 97,19, 61,64, 57,13, 38,27, 35,10, 35,08, 32,48, 31,86, 31,81,31,74, 31,43, 30,10, 25,01, 17,86.
Preparación de 2,7-di-ferc-butil-9-(2-((tetrahidro-2W-piran-2-il)oxi)-3-(4,4,5,5-tetrametiM,3,2-dioxaborolan-2-il)-5-(2,4,4-trimetilpentan-2-il)feml)-9H-carbazol.

A un matraz de fondo redondo de tres bocas secado al horno a 0-10 °C en atmósfera de N2 se añadió 2,7-di-fercbutil-9-(2-((tetrahidro-2H-piran-2-il)oxi)-5-(2,4,4-trimetilpentan-2-il)fenil)-9H-carbazol (7,70 g, 13,56 mmol) y tetrahidrofurano seco (90 ml). Esta solución se enfrió hasta 0-10 °C (baño de hielo-agua) durante aproximadamente 15 minutos y se añadió lentamente n-butillitio 2,5 M en hexanos (14 ml, 35,00 mmol). Después de agitar durante 4 horas, se añadió lentamente 2-iso-propoxi-4,4,5,5-tetrametil-1,3,2-dioxaborolano (7,0 ml, 34,31 mmol). La mezcla se agitó durante una hora a 0-10 °C antes de dejar que la reacción se calentara hasta temperatura ambiente y se agitó durante 18 horas adicionales. A la mezcla de reacción se añadió bicarbonato de sodio acuoso saturado frío (75 ml). La mezcla se extrajo con cuatro partes de 50 ml de cloruro de metileno. Las fases orgánicas se combinaron y se lavaron con bicarbonato de sodio acuoso saturado frío (200 ml), salmuera (200 ml), después se secaron sobre sulfato de magnesio anhidro, se filtraron y se concentraron para dar 9,43 g de producto bruto como una espuma dorada. Este producto bruto se suspendió en acetonitrilo (75 ml) y se dejó reposar durante una hora a temperatura ambiente antes de aislar el sólido mediante filtración al vacío. Los sólidos se lavaron con una pequeña parte de acetonitrilo frío y se secaron a alto vacío para proporcionar 8,12 g (86,3 %) del producto como un polvo de color blanco.
RMN 1H (400 MHz, CDCls) 57,97 (dd, J = 8,2, 1,2 Hz, 2H), 7,81 (d, J = 2,6 Hz, 1H), 7,50 (d, J = 2,6 Hz, 1H), 7,29 (ddd, J = 8,2, 4,5, 1,7 Hz, 2H), 7,20 (dd, J = 12,9, 1,2 Hz, 2H), 5,02 (t, J = 2,8 Hz, 1H), 2,81 (td, J = 10,8, 2,8 Hz, 1H), 2,69 (dt, J = 10,2, 2,9 Hz, 1H), 1,75 (ABq, J = 14,6 Hz, 2H), 1,41 (s, 6H), 1,40 (s, 12H), 1,36 (s, 9H), 1,35 (s, 9H), 1,31 - 0,94 (m, 6H), 0,82 (s, 9H). RMN 13C (101 MHz, cdcls) 5 156,00, 148,68, 148,53, 145,66, 141,80, 141,74, 133,45, 130,47, 129,15, 120,86, 120,61, 118,93, 118,88, 117,04, 107,51, 107,14, 100,80, 83,59, 61,08, 57,08, 38,40, 35,09, 32,49, 31,93, 31,80, 31,53, 31,16, 29,95, 25,06, 25,03, 24,89, 17,99.
Preparación de 4-fluoro-2-yodo-6-metilfenol.

A un matraz de fondo redondo equipado con un embudo de adición en atmósfera de N2 a 0-10 °C se añadió metanol (150 ml), 4-fluoro-2-metilfenol (10,00 g, 79,28 mmol), Nal (14,29 g, 95,34 mmol) y NaOH (3,92 g, 98,00 mmol). Esta solución se dejó agitar durante ~ 15 minutos a 0-10 °C antes de añadir gota a gota NaOCl (155 ml del 5 % en v/v en blanqueador comercial, 104,11 mmol) durante el período de 2 horas. Una vez completada la adición de blanqueador, la reacción se dejó agitar durante una hora adicional a 0-10 °C. El análisis de CG mostró una conversión del ~50 %, por lo tanto, se añadió Nal adicional (7,16 g, 47,77 mmol) y blanqueador (75 ml, 50,38 mmol) (todos de una vez) y se agitó durante otra hora a 0-10 °C. Esta vez, el análisis de CG mostró una conversión completa, por lo tanto, se añadieron 50 ml de tiosulfato de sodio acuoso al 10 % en peso a la mezcla de reacción. La mezcla de reacción se acidificó después con HCl al 5 %, se extrajo en cloruro de metileno (500 ml), se lavó con 500 ml, cada uno, de tiosulfato de sodio acuoso al 10 % en peso, agua, después, salmuera, se secó sobre sulfato de magnesio anhidro, se filtró a través de un almohadilla de gel de sílice y después se concentró para dar un aceite de color rojo oscuro. Este producto bruto se purificó mediante cromatografía ultrarrápida usando una columna P/N 5146135 Grace Reveleris de 330 g en un instrumento Grace eluyendo con acetato de etilo al 2 % en hexanos para proporcionar 13,69 g (68,5 %) del producto bruto como un sólido blanquecino.
RMN 1H (400 MHz, CDCls) 57,19 (ddd, J = 7,5, 3,0, 0,6 Hz, 1H), 6,88 - 6,82 (m, 1H), 5,09 (d, J = 0,5 Hz, 1H), 2,28 (s, 4H). RMN 13C{1H} (101 MHz, CDCls) 5 156,12 (d, J = 242,5 Hz), 149,49 (d, J = 2,7 Hz), 125,59 (d, J = 7,8 Hz), 121,50 (d, J = 25,2 Hz), 118,08 (d, J = 22,4 Hz), 84,09 (d, J = 9,6 Hz), 17,38 (d, J = 1,2 Hz). RMN 19F (CDCls) 5 -123,15 (t, J = 8,2 Hz).
Preparación de propano-1,3-diil bis (4-metilbencenosulfonato).

A un matraz de fondo redondo en atmósfera de N2 se añadió gota a gota una solución de 1,3-propanodiol (19,25 g, 252,96 mmol) en piridina anhidra (50 ml) durante el período de 2 horas a una solución de cloruro de 4-metilbenceno-1 -sulfonilo (115,74 g, 607,10 mmol) en piridina anhidra (200 ml), que se enfrió a 0-10 °C. La mezcla de reacción se dejó agitar durante 4 horas adicionales a 0-10 °C y, después, se vertió en hielo-agua (500 ml), momento en el que había precipitado un sólido blanquecino. Este precipitado se recogió mediante filtración al vacío, se lavó con agua fría (200 ml), ácido sulfúrico diluido (10 % en peso, 200 ml), carbonato de sodio acuoso 1 M (200 ml) y nuevamente con agua (200 ml). Este producto húmedo se recristalizó en acetona para proporcionar 82,35 g (84,7 %) de un producto como cristales de color blanco.
RMN 1H (400 MHz, CDCls) 57,72 (d, J = 8,3 Hz, 4H), 7,33 (d, J = 8,5 Hz, 4H), 4,05 (t, J = 6,0 Hz, 4H), 2,43 (s, 6 H), 1,98 (p, J = 6,0 Hz, 2H). RMN 13C{1H} (101 MHz, CDCls) 5144,99, 132,59, 129,90, 127,79, 65,82, 28,62, 21,57.
Preparación de 1,3-bis (4-fluoro-2-yodo-6-metilfenoxi)propano.

A N,N-dimetilformamida (250 ml) se añadió 2-yodo-4-fluoro-6-metilfenol (13,09 g, 51,94 mmol), propano-1,3-diil bis(4-metilbencenosulfonato) (9,99 g, 25,98 mmol) y K2CO3 (15,08 g, 109,11 mmol). Esta mezcla se calentó a 100 °C durante 30 minutos y, después, se concentró hasta sequedad. El residuo se absorbió en una mezcla de 50/50 de cloruro de metileno/agua (200 ml) y se extrajo con cloruro de metileno (3 veces 100 ml). La fase orgánica se lavó con 500 ml, cada uno, de NaOH acuoso 2 N, agua, después, salmuera, se secó sobre sulfato de magnesio anhidro, se filtró a través de una almohadilla de gel de sílice y se concentró para dar 9,80 g (69,4 %) de un producto como un polvo de color blanco.
RMN 1H (400 MHz, CDCh) 57,31 (m, 2H), 6 , 8 8 (m, 2H), 4,08 (t, J = 6,5 Hz, 4H), 2,44 (p, J = 6,5 Hz, 2H), 2,34 (s, 6 H). RMN 13C (101 MHz, cdch) 5158,44 (d, J = 247,1 Hz), 153,56 (d, J = 3,0 Hz), 133,09 (d, J = 8,3 Hz), 123,39 (d, J = 24,8 Hz), 117,92 (d, J = 22,3 Hz), 91,35 (d, J = 9,5 Hz), 70,13 (d, J = 1,0 Hz), 31,04, 17,43 (d, J = 1,2 Hz). RMN 19F (376 MHz, CDCh) 5-118,17 (t, J = 8,1 Hz).
Preparación de 1,3-bis((3'-(2,7-di-ferc-butil-9W-carbazol-9-il)-5-fluoro-3-metil-2'-((tetrahidro-2W-piran-2-il)oxi)-5'-(2,4,4-trimetilpentan-2-il)-[1,1'-bifenil]-2-il)oxi)propano.

A un matraz de fondo redondo en atmósfera de N2 se añadió 2,7-di-fe/'c-butil-9-(2-((tetrahidro-2H-piran-2-il)oxi)-3-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-5-(2,4,4-trimetilpentan-2-il)fenil)-9H-carbazol (7,52 g, 9,89 mmol) (micromoles ajustados basándose en una pureza del 91,2 % mediante HPLC), dimetoxietano (120 ml), una solución de NaOH (1,30 g, 32,50 mmol) en agua (35 ml), tetrahidrofurano (60 ml) y 1,3-bis(4-fluoro-2-yodo-6-metilfenoxi)propano (2,56 g, 4,70 mmol). El sistema se purgó con N2 durante aproximadamente 15 minutos y se añadió Pd(PPh3)4 (303 mg, 0,26 mmol). La mezcla se calentó hasta reflujo a 85 °C durante 48 horas y, después, se dejó enfriar hasta temperatura ambiente. Una vez enfriado, se formó un precipitado en el matraz de reacción que se aisló mediante filtración al vacío y se secó a alto vacío durante una hora para proporcionar 6 , 1 0 g de ligando protegido bruto. Este ligando protegido se usó como tal en la siguiente etapa.
Preparación de 2',2"'-(propano-1,3-diMbis(oxi))bis(3-(2,7-di-ferc-butN-9H-carbazol-9-N)-5'-fluoro-3'-metM-5-(2,4,4-trimetilpentan-2-il)-[1,1'-bifenil]-2-ol) (ligando DOC-6156).

Al ligando protegido bruto se añadió una mezcla de 1:1 de metanol/tetrahidrofurano (200 ml) y aproximadamente 100 mg de monohidrato de ácido p-toluenosulfónico. La solución se calentó a 60°C durante 8 horas, después se dejó enfriar y se concentró. El residuo se disolvió en cloruro de metileno (250 ml), se lavó con salmuera (250 ml), se secó sobre sulfato de magnesio anhidro, se filtró a través de una almohadilla de gel de sílice y, después, se concentró para proporcionar 4,92 g de ligando bruto. Este producto bruto se purificó mediante cromatografía ultrarrápida usando una columna P/N 5146135 Grace Reveleris de 330 g en un instrumento ISCO eluyendo con acetato de etilo al 2 % en hexanos para proporcionar 4,23 g (71,7 %) del producto bruto como un sólido de color blanco.
RMN 1H (400 MHz, CDCls) 88,03 (dd, J= 8,2, 0,5 Hz, 4H), 7,44 (dd, J = 5,1, 2,4 Hz, 4H), 7,33 (dd, J = 8,3, 1,7 Hz, 4H), 7,00 (dd, J= 8,8, 3,0 Hz, 1H), 6,84 (ddd, J = 8,7, 3,1, 0,6 Hz, 1H), 6,18 (s, 2H), 3,66 (t, J = 6,4 Hz, 4H), 1,97 (s, 6H), 1,76 (s, 3H), 1,74 (pent, J = 6,4 Hz, 2H), 1,40 (s, 12H), 1,30 (s, 36H), 0,83 (s, 18H). RMN 13C (101 MHz, CDCla) 8 158,82 (d, J = 243,2 Hz), 150,16 (d, J = 2,5 Hz), 149,09, 147,76, 142,87, 141,68, 133,48 (d, J = 8,6 Hz), 132,89 (d, J = 8,7 Hz), 129,12, 127,50, 126,28 (d, J= 1,5 Hz), 124,99, 121,07, 119,51, 117,74, 117,18 (d, J = 22,5 Hz), 116,07 (d, J = 23,1 Hz), 106,20, 70,87, 57,17, 38,25, 35,06, 32,51, 31,91, 31,75, 31,66, 30,73, 16,44, 16,43. RMN 19F (376 MHz, CDCh) 8 -118,80 (t, J = 8,5 Hz). HRMS (ESI, M NH4+): (m/z) calculado para C85H108F2NaO4 1.272,830, hallado 1.272,830.
Preparación de procatalizador 1.

Se suspendieron el ligando (0,4778 g, 0,38 mmol) y HfCU (0,122 g, 0,38 mmol) en 35 ml de tolueno frío (-30 °C). A esta mezcla se añadieron 0,56 ml de solución de éter de dietilo 3 M de MeMgBr. La mezcla de reacción se mantuvo de color amarillo pálido durante aproximadamente 20 min y, después, comenzó a oscurecerse. Después de 1,5 h de agitación, el disolvente se retiró a presión reducida. Al residuo se añadieron 20 ml de tolueno, seguido de 25 ml de hexano. La suspensión se filtró dando una solución incolora. El disolvente se retiró a presión reducida dando 0,367 g de un sólido de color blanco. Rendimiento del 66,0 %. Los cristales para el análisis de rayos X se dejaron crecer a partir de C6re6 en el tubo de RMN.
RMN 1H (400 MHz, tolueno) 88,14 (d, J = 8,2 Hz, 2H), 7,98 (d, J = 8,2 Hz, 2H), 7,85 (d, J = 1,6 Hz, 2H), 7,79 (d, J = 2.5 Hz, 2H), 7,61 (d, J = 1,6 Hz, 2H), 7,46 (dd, J = 8,2, 1,6 Hz, 2H), 7,32 (d, J = 2,5 Hz, 2H), 7,30 (dd, J = 8,2, 1,6 Hz, 2H), 6,86 (dd, J = 8,9, 3,2 Hz, 2H), 6,12 (d, J = 5,1 Hz, 2H), 3,49 (dt, J = 9,9, 4,9 Hz, 2H), 3,27 (dt, J = 10,5, 5,5 Hz, 2H), 1,72 (d, J = 14,4 Hz, 1H), 1,59 (d, J = 14,4 Hz, 11H), 1,57 (s, 18H), 1,36 - 1,31 (m, 2H), 1,27 (s, 6H), 1,26 (s, 6H), 1,25 (s, 18H), 1,12 (s, 6H), 0,87 (s, 18H), -0,93 (s, 6H). RMN 13C{1H} (101 MHz, tolueno) 8 160,47 (d, J = 246,3 Hz), 153,83, 149,41 (d, J = 2,7 Hz), 149,38, 147,86, 142,19, 141,51, 140,54, 135,89 (d, J = 8,6 Hz), 135,11 (d, J = 8,9 Hz), 130,45 (d, J = 1,4 Hz), 128,34, 127,81, 126,82, 123,46, 120,93, 120,27, 118,93, 117,48, 117,34 (d, J = 23.5 Hz), 117,21 (d, J = 22,5 Hz), 109,65, 107,68, 76,14, 57,86, 50,94, 38,28, 35,48, 35,24, 33,08, 32,76, 32,40, 32,02, 31,68, 30,32, 29,96, 16,45. RMN 19F (376 MHz, benceno-d6) 8-115,22 (t, J = 8,6 Hz).
Preparación de 4-fluoro-2-yodofenol.

Al metanol (200 ml) a 0-10 °C se añadió 4-fluorofenol (8,00 g, 71,37 mmol), Nal (12,84 g, 85,64 mmol) y NaOH (3,43 g, 85,64 mmol). Esta solución se dejó agitar durante ~ 15 minutos a 0-10 °C antes de añadir gota a gota NaOCl (133 ml de solución al 5% en peso de blanqueador comercial, 92,77 mmol) durante el período de 1 hora, después se dejó agitar durante una hora adicional a 0-10 °C. La reacción se inactivó con tiosulfato de sodio acuoso al 10 % en peso (50 ml), después, la mezcla de reacción se acidificó después con HCl al 10 %. La solución orgánica se extrajo en cloruro de metileno (300 ml), se lavó con 500 ml, cada uno, de tiosulfato de sodio al 10 % en peso, agua, después, salmuera, se secó sobre sulfato de magnesio anhidro, se filtró a través de un almohadilla de gel de sílice y, después, se concentró para dar un compuesto bruto. Este producto bruto se purificó mediante recristalización en hexanos para proporcionar 11,52 g (67,8 %) de compuesto como cristales de color blanco.
RMN 1H (500 MHz, CDCls) 57,36 (dd, J = 7,6, 2,9 Hz, 1H), 6,97 (ddd, J = 8,9, 7,7, 2,9 Hz, 2H), 6,92 (dd, J = 9,0, 4,9 Hz, 1H), 5,10 (s, 1H). RMN 13C (101 MHz, cloroformo-d) 5 156,42 (d, J = 243,0 Hz), 151,45 (d, J = 2,6 Hz), 124,34 (d, J = 25,3 Hz), 116,83 (d, J = 23,1 Hz), 115,08 (d, J = 7,8 Hz), 84,23 (d, J = 9,0 Hz). RMN 19F (376 MHz, CDCls) 5- 122,52 (td, J = 7,6, 4,9 Hz).
Preparación de 1-(3-bromopropoxi)-4-fluoro-2-yodobenceno).

Un matraz de fondo redondo de tres bocas se equipó con una barra de agitación magnética, septos, un condensador y una entrada de gas nitrógeno. El matraz se cargó con 4-fluoro-2-yodofenol (7,0020 g, 29,420 mmol), carbonato de potasio (8,2954 g, 60,020 mmol), 1,3-dibromopropano (59,00 ml, 581,262 mmol) y acetona (200 ml). La mezcla se agitó hasta una disolución completa y se calentó a reflujo durante una noche. Se muestreó la solución para el análisis de CG/EM (0,1 ml de muestra diluida en acetona y se filtró) para determinar la terminación de la reacción. Después de 16,5 horas, la reacción se dejó enfriar hasta temperatura ambiente y se filtró mediante filtración al vacío. El matraz de fondo redondo se lavó con acetona (2 veces 20 ml) y también se filtró. El filtrado se concentró mediante evaporación rotatoria para retirar la acetona. La solución de color amarillo que quedó se destiló al vacío (temperatura del manto de calentamiento de 80 - 100 °C) para retirar el 1,3-dibromopropano restante. Se dejó un aceite de color marrón bruto que se analizó mediante RMN 1H. El aceite de color marrón se disolvió en una pequeña cantidad de hexanos y se purificó mediante cromatografía de columna en el sistema Isco CombiFlash usando una columna Grace de 330 g y un gradiente de acetato de etilo al 0-5 % en hexanos para 2 volúmenes de columna, aumentando después a acetato de etilo al 5 % en hexanos hasta que eluyó el producto. Las fracciones se analizaron mediante TLC y CG/EM. Las fracciones puras se combinaron y se concentraron mediante evaporación rotatoria para proporcionar el producto como un aceite de color amarillo. El aceite de color amarillo se secó a alto vacío para proporcionar 8,99 g (85,1 %).
RMN 1H (500 MHz, CDCh) 57,47 (dd, J = 7,6, 3,0 Hz, 1H), 6,99 (ddd, J = 9,0, 7,8, 3,0 Hz, 1H), 6,73 (dd, J = 9,0, 4,6 Hz, 1H), 4,07 (t, J = 5,7 Hz, 2H), 3,68 (t, J = 6,4 Hz, 2H), 2,32 (p, J = 6,2 Hz, 2H). RMN 13C (126 MHz, CDCh) 5 156,64 (d, J = 243,6 Hz), 153,60 (d, J = 2,6 Hz),
125,81 (d, J = 24,9 Hz), 115,49 (d, J = 22,5 Hz), 112,22 (d, J = 8,2 Hz), 67,02, 32,08, 30,15. RMN 19F (376 MHz, CDCh) 5-121,86 --121,97 (m).
Preparación de 5-fluoro-2-(3-(4-fluoro-2-yodofenoxi)propoxi)-1-yodo-3-metilbenceno.

Un matraz de fondo redondo de tres bocas se equipó con una barra de agitación magnética, septos, un condensador y una entrada de gas nitrógeno. El matraz se cargó con 1-(3-bromopropoxi)-4-fluoro-2-yodobenceno (8,9856 g, 25,032 mmol), 4-fluoro-2-yodo-6-metilfenol (6,3096 g, 25,036 mmol), carbonato de potasio (7,400 g, 53,542 mmol) y
acetona (165 ml). La mezcla se agitó hasta una disolución completa y se calentó a reflujo durante una noche. Se muestreó la solución para el análisis de CG/EM (0,1 ml de muestra diluida en acetona y se filtró) para determinar la terminación. Después de 16 horas, la reacción se dejó enfriar hasta temperatura ambiente y se filtró mediante filtración al vacío. El matraz de fondo redondo se lavó con acetona (2 veces 20 ml) y también se filtró. El filtrado se concentró mediante evaporación rotatoria para proporcionar el producto bruto como un aceite de color marrón oscuro. El producto bruto se analizó mediante RMN 1H. El aceite de color marrón oscuro se disolvió en una pequeña cantidad de hexanos y se purificó mediante cromatografía de columna en el sistema Isco CombiFlash usando una columna Grace de 330 g y un gradiente de acetato de etilo al 0-5 % en hexanos para 2 volúmenes de columna, aumentando después a acetato de etilo al 5 % en hexanos hasta que eluyó el producto. Las fracciones se analizaron mediante TLC y Cg /EM. Las fracciones puras se combinaron y se concentraron mediante evaporación rotatoria para proporcionar un producto bruto como un sólido de color amarillo. El sólido de color amarillo se secó a alto vacío para proporcionar 11,55 g (87,1 %).
RMN 1H (400 MHz, CDCls) 57,49 (dd, J = 7,6, 3,0 Hz, 1H), 7,29 (ddd, J = 7.5, 3,0, 0,7 Hz, 1H), 7,01 (ddd, J = 9,0, 7,8, 3,0 Hz, 1H), 6,85 (ddd, J = 8,6, 3,0, 0,8 Hz, 1H), 6,76 (dd, J = 9,0, 4,6 Hz, 1H), 4,25 (t, J = 5,9 Hz, 2H), 4,07 (t, J = 6,0 Hz, 2H), 2,34 (p, J = 5,9 Hz, 2H), 2,27 (d, J = 0,7 Hz, 3H). RMN 13C (101 MHz, CDCls) 5 158,73 (d, J = 181,2 Hz), 156,28 (d, J = 178,1 Hz), 153,85 (d, J = 2,1 Hz), 153,05 (d, J = 3,1 Hz), 133,14 (d, J = 8,2 Hz), 125,99 (d, J = 25,1 Hz), 123,26 (d, J = 24,8 Hz), 117,89 (d, J = 22,2 Hz), 115,55 (d, J = 22,4 Hz), 111,75 (d, J = 8,1 Hz), 91,33 (d, J = 9,3 Hz), 85,81 (d, J = 8,2 Hz), 68,89 (d, J = 1,3 Hz), 65,82, 29,86, 17,22 (d, J = 1,3 Hz). RMN 19F (376 MHz, CDCh) 5 -117,93 - - 118,11 (m), -122,39 - -122,55 (m).
Preparación de 3-(2,7-di-ferc-butil-9W-carbazol-9-N)-2'-(3-((3'-(2,7-di-ferc-butil-9W-carbazol-9-N)-5-fluoro-2'-hidroxi-5'-(2,4,4-trimetilpentan-2-il)-[1,1'-bifenil]-2-il)oxi)propoxi)-5'-fluoro-3'-metil-5-(2,4,4-trimetilpentan-2-il)-[1,1 '-bifenil]-2-ol.

Un matraz de fondo redondo de tres bocas se equipó con una barra de agitación magnética, septos, un condensador y una entrada de gas nitrógeno. El matraz se cargó con 2,7-di-fe/'c-butil-9-(2-((tetrahidro-2H-piran-2-il)oxi)-3-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-5-(2,4,4-trimetilpentan-2-il)fenil)-9H-carbazol (9,4182 g, 13,575 mmol), 1,2-DME (170 ml), una solución de NaoH (1,8145 g, 45,438 mmol) en agua (49 ml), THF (57 ml) y 5-fluoro-2-(3-(4-fluoro-2-yodofenoxi)propoxi)-1-yodo-3-metilbenceno (3,4233 g, 6,458 mmol). La solución se agitó y se purgó con nitrógeno durante aproximadamente 15 minutos, después se añadió Pd(PPh3)4 (0,5432 g, 0,470 mmol). La mezcla se calentó hasta reflujo a 85 °C durante 19 horas y se comprobó la terminación mediante TLC (acetato de etilo al 5% en hexanos). Después de 19 horas, la reacción se dejó enfriar hasta temperatura ambiente. La mezcla se transfirió a un embudo de separación para una separación de fases. La fase orgánica se secó sobre sulfato de magnesio, se filtró mediante filtración al vacío y se concentró mediante evaporación rotatoria para proporcionar un sólido de color naranja dorado espumoso (22,73 g) como un ligando protegido bruto. El ligando bruto se analizó mediante RMN 1H. El ligando protegido bruto se disolvió en una mezcla de tetrahidrofurano (250 ml) y metanol (250 ml) y, después, se calentó hasta 60 °C. A la solución se añadió monohidrato de ácido p-toluenosulfónico (3,0380 g, 15,971 mmol) hasta que la solución se volvió ácida. La reacción se agitó a 60 °C durante una noche y se comprobó la terminación mediante TLC (acetato de etilo al 5 % en hexanos). La mezcla de reacción se dejó enfriar hasta temperatura ambiente y, después, se concentró mediante evaporación rotatoria para proporcionar un sólido adherente de color marrón (15,13 g). El sólido se analizó mediante r Mn 1H. El producto bruto se disolvió en cloroformo y se añadió gel de sílice. La suspensión se concentró mediante evaporación rotatoria para proporcionar una mezcla en polvo seca. La mezcla en polvo se cargó en el sistema Isco CombiFlash y se circuló usando una columna Grace de 330 g y un gradiente de acetato de etilo al 2-5 % en hexanos hasta que eluyó el producto. Las fracciones se analizaron mediante TLC. Las fracciones puras se combinaron y se concentraron mediante evaporación rotatoria para proporcionar el producto como un sólido cristalino de color amarillo claro. A fin de retirar las trazas de acetato de etilo, el sólido se disolvió en diclorometano y se concentró mediante evaporación rotatoria para proporcionar un sólido cristalino de color amarillo claro (se repitió dos veces). El sólido se secó a alto vacío para proporcionar 6,17 g
(77,0 %).
RMN 1H (400 MHz, CDCI3) 88,12 (d, J = 8,3 Hz, 2H), 8,08 (d, J = 8,2 Hz, 2H), 7,55 (d, J = 2,4 Hz, 1H), 7,51 (d, J = 2,2 Hz, 1H), 7,43 (q, J = 2,4 Hz, 2H), 7,40 (t, J = 1,9 Hz, 2H), 7,38 (t, J = 1,9 Hz, 2H), 7,19 (dd, J = 8,9, 3,2 Hz, 1H), 7,17 (dd, J = 1,6, 0,7 Hz, 2H), 7,15 (d, J = 1,0 Hz, 2H), 7,09 (dd, J = 8,8, 3,4 Hz, 1H), 6,88 (ddd, J = 8,6, 3,1, 0,9 Hz, 1H), 6,79 (ddd, J = 8,9, 7,8, 3,1 Hz, 1H), 6,63 (s, 1H), 6,48 (dd, J = 9,1, 4,5 Hz, 1H), 5,71 (s,1H), 3,96 (t, J = 6,7 Hz, 2H), 3,69 (t, J = 5,5 Hz, 2H), 2,01 (s, 3H), 1,88 (p, J = 6,0 Hz, 2H), 1,83 (s, 2H), 1,79 (s, 2H), 1,49 (s, 6H), 1,44 (s, 6H), 1,37 (s, 18H), 1,36 (s, 18H), 0,89 (s, 9H), 0,87 (s, 9H). RMN19F (376 MHz, CDCI3) 8 -118,16 (t, J = 8,7 Hz), -122,85 - - 122,93 (m).
Preparación de procatalizador 4

La reacción se ajustó en una caja de guantes en atmósfera de nitrógeno. Un frasco se cargó con HfCU (0,1033 g, 0,3225 mmol) y tolueno (20 ml). La suspensión se enfrió hasta -25 °C en el refrigerador de la caja de guantes durante 30 minutos. A la suspensión fría en agitación se añadió bromuro de metilmagnesio 3,0 M en éter de dietilo (0,45 ml, 1,35 mmol). La mezcla se agitó enérgicamente durante 2 minutos. El sólido se puso en solución, pero la solución de reacción estaba turbia y amarillenta. A la mezcla se añadió el ligando (0,4000 g, 0,3221 mmol) como un sólido. El vial que contenía el sólido se enjuagó con tolueno (2,0 ml). El disolvente de enjuague se añadió a la mezcla de reacción. La reacción fue seguida por RMN. Después de agitar durante 1,5 horas, la mezcla de reacción se filtró (embudo de medio fritado). La torta se lavó con dos partes de 10 ml de tolueno. A la solución de filtrado incolora se añadieron hexanos (5 ml) y se concentró al vacío para proporcionar un sólido de color blanco. Al sólido se añadió tolueno (30 ml) y se agitó hasta que casi todo el sólido estaba en solución. Después se añadieron hexanos (25 ml). La solución turbia de color amarillento se filtró (filtro de jeringa) y se concentró a alto vacío para proporcionar 0,4317 g (92,5 %) del complejo de Hf como un sólido de color bronce.
La muestra analítica para rayos X se obtuvo mediante recristalización en benceno-cfe.
RMN 1H (400 MHz, C6D6) 88,20 (dd, J = 8,2, 0,5 Hz, 1H), 8,15 (dt, J = 8,3, 0,6 Hz, 2H), 8,04 (dd, J = 8,3, 0,6 Hz, 1H), 7,92 (d, J = 1,3 Hz, 1H), 7,81 (d, J = 2,5 Hz, 1H), 7,73 (ddd, J = 13,7, 1,7, 0,6 Hz, 2H), 7,68 (d, J = 2,3 Hz, 2H), 7,46 (dd, J = 8,2, 1,7 Hz, 1H), 7,41 (dd, J = 3,2, 1,6 Hz, 1H), 7,39 (dd, J = 3,2, 1,9 Hz, 2H), 7,35 (dd, J = 8,3, 1,7 Hz, 1H), 7,24 (d, J = 2,5 Hz, 1H), 6,94 (dt, J = 9,1, 3,2 Hz, 2H), 6,26 (ddd, J = 8,9, 7,4, 3,2 Hz, 1H), 6,13 (dd, J = 8,7, 3,1 Hz, 1H), 5,69 (dd, J = 8,9, 5,0 Hz, 1H), 3,79 (dt, J = 10,0, 5,2 Hz, 1H), 3,66 (dt, J = 10,2, 4,9 Hz, 1H), 3,52 (dt, J = 9,7, 5,6 Hz, 1H), 3,16 (dt, J = 10,5, 5,2 Hz, 1H), 1,64 - 1,56 (m, 2H), 1,49 (s, 9H), 1,44 (s, 9H), 1,37 - 1,29 (m, 2H), 1,26 (s, 10H), 1,25 (s, 6H), 1,20 - 1,17 (m, 6H), 0,89 (s, 9H), 0,80 (s, 9H), -0,69 (s, 2H), -1,10 (s, 2H). RMN 19F (376 MHz, C6D6) 8-113,82 (ddd, J = 9,0, 7,3, 5,0 Hz), -115,71 (t, J = 8,4 Hz).
Preparación de bis(clorometil)dietilsilano:
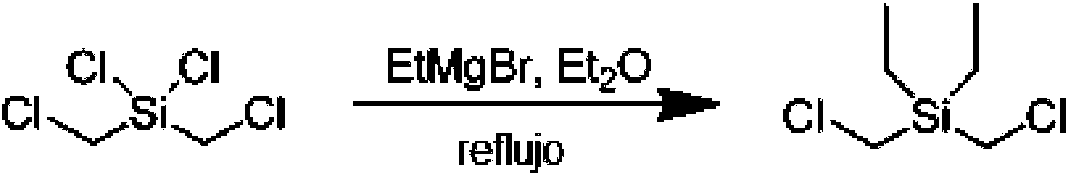
Un matraz de fondo redondo de tres bocas se equipó con una barra de agitación magnética, dos septos, un condensador y una entrada de gas nitrógeno. El matraz se colocó en atmósfera de nitrógeno y se cargó con bromuro de etilmagnesio (40 ml, 120 mmol) y éter de dietilo (60 ml). A la solución se añadió bis(clorometildiclorosilano) (9,5002 g, 47,993 mmol) a través de una jeringa. La mezcla se calentó hasta reflujo. Después de unos pocos minutos, la mezcla de color blanco turbia se volvió transparente y se observó un precipitado de color blanco. La reacción se calentó a reflujo durante 5 horas y, después, se dejó reposar durante una noche a temperatura ambiente. La mezcla se filtró y la torta se lavó con dos partes de 30 ml de éter de dietilo. El filtrado se agitó lentamente, se enfrió hasta 0 °C (baño de hielo-agua) y se añadió lentamente HCl acuoso 0,1 M (29 ml) a través de un embudo de adición. Mientras se añadía el HCl 0,1 M, comenzaron a formarse sólidos. La mezcla se transfirió a un embudo de separación y los sólidos de color blanco se dejaron atrás. Las fases se separaron y la fase acuosa brumosa de color blanco se extrajo con dos partes de 15 ml de éter de dietilo. Las fases orgánicas se combinaron, se secaron sobre sulfato de magnesio, se filtraron mediante filtración al vacío y se concentraron mediante
evaporación rotatoria para proporcionar un aceite de color amarillo claro como un producto bruto. El aceite bruto se secó a alto vacío durante 1 hora para proporcionar 7,6709 g (86,3 %) del producto.
RMN 1H (500 MHz, CDCls) 52,94 (d, J = 0,4 Hz, 4H), 1,03 (t, J = 7,8 Hz, 4H), 0,80 (q, J = 7,8 Hz, 3H). Referencia:
Anderson, W. K.; Kasliwal, R.; Houston, D. M.; Wamg, Y.; Narayanan, V. L.; Haugwitz, R. D.; Plowman, J. J. Med. Chem. 1995, 38, 3789-3797.
Preparación de dietilbis((4-fluoro-2-yodo-6-metilfenoxi)metil)silano:

Un matraz de fondo redondo de tres bocas se equipó con un condensador, dos septos, una barra de agitación magnética y una entrada de gas nitrógeno. El matraz se colocó en atmósfera de nitrógeno y se cargó con hidruro de sodio (95 %, 0,4137 g, 16,376 mmol) y N,N-dimetilformamida anhidra (8,5 ml). La suspensión se enfrió hasta 0 °C y una solución de 4-fluoro-2-yodo-6-metilfenol (4,2466 g, 16,850 mmol) en N,Ñ-dimetilformamida anhidra (8,5 ml) se añadió lentamente a través de una jeringa a una velocidad para mantener el control de la reacción (desprendimiento de hidrógeno). El baño de hielo se retiró y la mezcla rojiza resultante se agitó durante 30 minutos. Después, se añadió una solución de bis(clorometil)dietilsilano (1,3002 g, 7,022 mmol) en N,N-dimetilformamida anhidra (4,5 ml) a través de una jeringa. La mezcla de reacción se calentó hasta 60 °C durante 17 horas. La reacción se dejó enfriar hasta temperatura ambiente y, después, se enfrió hasta 0 °C (baño de hielo-agua). A la solución enfriada se añadió lentamente agua (21,5 ml). Se dejó un lodo espeso en el fondo del matraz cuando se transfirió la mezcla a un embudo de separación. El matraz se lavó con algo de acetato de etilo para disolver el lodo y la solución se colocó en el embudo de separación. Las fases se separaron y la fase acuosa se extrajo con tres partes de 25 ml de acetato de etilo. Las fases orgánicas se combinaron y se lavaron con hidróxido de sodio 1 M (35 ml), después, con salmuera (21,5 ml). La fase orgánica se secó sobre sulfato de magnesio anhidro, se filtró mediante filtración al vacío y se concentró mediante evaporación rotatoria para proporcionar el producto bruto como un aceite de color marrón rojizo. El aceite se disolvió en una pequeña cantidad de hexanos y se purificó mediante cromatografía de columna usando una columna Grace de 330 g y un gradiente de diclorometano al 0-5 % en hexanos sobre 2 volúmenes de columna, el resto en diclorometano al 5 % en hexanos hasta que eluyó el producto. Las fracciones puras se combinaron y se concentraron mediante evaporación rotatoria para proporcionar el producto como un aceite incoloro. A fin de retirar las trazas de hexanos, el aceite se disolvió en diclorometano y se concentró mediante evaporación rotatoria para proporcionar un aceite incoloro (se repitió dos veces). El aceite se secó a alto vacío para proporcionar 3,1112 g (71,9 %) del producto como un sólido de color blanco opacificado.
RMN 1H (400 MHz, CDCls) 57,29 (ddd, J = 7,7, 3,1, 0,8 Hz, 2H), 6,85 (ddd, J = 8,8, 3,1, 0,9 Hz, 2H), 3,86 (s, 4H), 2,32 (s, 6H), 1,21 (t, J = 7,9 Hz, 6H), 1,10 -0,99 (m, 4H). RMN 19F (376 MHz, CDCh) 5-118,50 (t, J = 8,1 Hz).
Preparación de 2',2"'-(((dietilsilanodiil)bis(metileno))bis(oxi))bis(3-(2,7-di-ferc-butil-9tf-carbazol-9-il)-5'-fluoro-3'-metil-5-(2,4,4-trimetilpentan-2-il)-[1,1 '-bifenil]-2-ol):

Un matraz de fondo redondo de tres bocas se equipó con una barra de agitación magnética, dos septos, un condensador y una entrada de gas nitrógeno. El matraz se cargó con 2,7-di-ferc-butil-9-(2-((tetrahidro-2H-piran-2-il)oxi)-3-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-5-(2,4,4-trimetilpentan-2-il)fenil)-9H-carbazol (7,4057 g, 10,674 mmol), una solución de hidróxido de sodio (1,3599 g, 33,998 mmol) en agua (33 ml), tetrahidrofurano (165 ml) y dietilbis((4-fluoro-2-yodo-6-metilfenoxi)metil)silano (2,9865 g, 4,846 mmol). La solución se agitó y se purgó con nitrógeno durante aproximadamente 45 minutos, después se añadió tetraquis(trifenilfosfino)paladio(0) (0,4079 g, 0,3529 mmol). La mezcla se calentó hasta reflujo a 60 °C durante 23 horas y se analizó mediante HPLC en cuanto a la terminación. Después de 24 horas, la reacción se dejó enfriar hasta temperatura ambiente. La fase orgánica se
separó, se secó sobre sulfato de magnesio y se filtró mediante filtración al vacío. Los sólidos se enjuagaron con diclorometano. El filtrado se concentró mediante evaporación rotatoria para proporcionar un sólido de color naranja dorado adherente como un ligando protegido bruto (10,9973 g). El ligando se disolvió en cloroformo y se añadió gel de sílice. La suspensión se concentró mediante evaporación rotatoria para proporcionar una mezcla seca en polvo de gel de sílice y ligando. La mezcla en polvo se cargó en el sistema Isco CombiFlash y se circuló usando una columna Grace de 330 g y un gradiente de cloroformo al 30 % en hexanos para 6 volúmenes de columna (VC), aumentando a cloroformo al 50 % en hexanos sobre 3 VC, después el resto en cloroformo al 50 % en hexanos hasta que eluyó el producto. Las fracciones puras se combinaron y se concentraron mediante evaporación rotatoria para proporcionar un sólido cristalino de color blanco (6,4445 g). El sólido se disolvió en una mezcla de tetrahidrofurano (33 ml) y metanol (33 ml) y, después, se calentó hasta 60 °C. A la solución se añadió monohidrato de ácido para-toluenosulfónico (0,1858 g, 0,9767 mmol). La reacción se agitó a 60 °C durante una noche y se dejó enfriar hasta temperatura ambiente. La mezcla se concentró mediante evaporación rotatoria para proporcionar un sólido cristalino de color amarillo claro bruto (5,9845 g). El sólido se disolvió en cloroformo y se añadió gel de sílice. La suspensión se concentró mediante evaporación rotatoria para proporcionar una mezcla seca en polvo de gel de sílice y ligando.
La mezcla en polvo se cargó en el sistema Isco CombiFlash y se circuló usando una columna Grace de 330 g y un gradiente de acetato de etilo al 2 % en heptano hasta que eluyó el producto. Las fracciones puras se combinaron y se concentraron mediante evaporación rotatoria para proporcionar un sólido cristalino de color amarillo. A fin de retirar las trazas de heptano, el sólido se disolvió en diclorometano y se concentró mediante evaporación rotatoria para proporcionar un sólido cristalino de color amarillo (se repitió dos veces). El sólido se secó a alto vacío para proporcionar 3,9614 g (61,6 %) de un sólido cristalino de color amarillo.
RMN 1H (500 MHz, CDCls) 8 7,99 (d, J = 8,2 Hz, 4H), 7,47 (d, J = 2,4 Hz, 2H), 7,39 (d, J = 2,4 Hz, 2H), 7,30 (dd, J = 8,2, 1,6 Hz, 4H), 7,15 (amplio s, 4H), 6,94 (dd, J = 8,9, 3,1 Hz, 2H), 6,87 (dd, J = 8 ,6 , 3,2 Hz, 2H), 6,48 (amplio s, 2H), 3,45 (s, 4H), 2,08 (s, 6 H), 1,73 (s, 4H), 1,39 (s, 12H), 1,29 (s, 36H), 0,79 (s, 18H), 0,35 (amplio s, 10H).
RMN 19F (376 MHz, CDCla) 8-118,32 --119,14 (amplio s).
Preparación de procatalizador 5:

La reacción se ajustó en una caja de guantes en atmósfera de nitrógeno. Un frasco se cargó con HfCU (0,1258 g, 0,3928 mmol) y tolueno (24 ml). La suspensión se enfrió hasta -25 °C en el refrigerador de la caja de guantes durante 30 minutos. A la suspensión fría en agitación se añadió bromuro de metilmagnesio 3,0 M en éter de dietilo (0,55 ml, 1,65 mmol). La mezcla se agitó enérgicamente durante 2 minutos. El sólido se puso en solución, pero la solución de reacción estaba turbia y amarillenta. A la mezcla se añadió el ligando (0,5023 g, 0,3783 mmol) como un sólido. El matraz que contenía el sólido se enjuagó con tolueno (3,0 ml). El disolvente de enjuague se añadió a la mezcla de reacción. La mezcla de reacción se agitó a temperatura ambiente durante 5,5 horas. A la mezcla de color amarillo se añadieron hexanos (12 ml) y la suspensión se filtró. La solución de color amarillo transparente se concentró al vacío durante una noche para proporcionar 0,412 g (71,0%) del producto como un sólido de color amarillo.
RMN 1H (400 MHz, C6D6) 8 8,19 (d, J = 8,2 Hz, 2H), 8,10 (d, J = 8,3 Hz, 2H), 7,83 (d, J = 1,6 Hz, 2H), 7,76 (d, J = 2,5 Hz, 2H), 7,74 (d, J = 1,6 Hz, 2H), 7,49 (dd, J = 8,2, 1,6 Hz, 2H), 7,41 - 7,33 (m, 4H), 6,93 (dd, J = 8,9, 3,2 Hz, 2H), 6,14 (dd, J = 8,2, 3,3 Hz, 2H), 3,91 (d, J = 14,1 Hz, 2H), 3,47 (d, J = 14,1 Hz, 2H), 1,62 (d, J = 14,6 Hz, 2H), 1,57 (d, J = 14,4 Hz, 2H), 1,53 (s, 18H), 1,26 (d, J = 2,5 Hz, 30H), 1,13 (s, 6 H), 0,82 (s, 18H), 0,56 (t, J = 8,0 Hz, 6 H), 0,26 - 0,06 (m, 4H), -0,72 (s, 6 H).
RMN 19F (376 MHz, C6D6) 8 -116,35 (t, J = 8,3 Hz).
Preparación de (dorometil)dietil((4-fluoro-2-yodo-6-metilfenoxi)metil)silano:

Un matraz de fondo redondo de tres bocas se equipó con dos septos y una entrada de gas nitrógeno. El matraz se colocó en atmósfera de nitrógeno y se cargó con hidruro de sodio (95 %, 0,2496 g, 10,400 mmol) y se añadió N,N-dimetilformamida anhidra (10,0 ml) a través de una jeringa. La suspensión se enfrió hasta 0 °C a través de un baño de hielo-agua. A la suspensión se añadió una solución de 4-fluoro-2-yodo-6-metilfenol (2,4753 g, 9,822 mmol) en N,N-dimetilformamida anhidra (10,0 ml) a través de una jeringa para mantener el control de la reacción (desprendimiento de hidrógeno). Se retiró el baño de hielo-agua y la solución de color marrón resultante se agitó durante 30 minutos a temperatura ambiente. Otro matraz de fondo redondo de tres bocas se equipó con una barra de agitación magnética, dos septos, un embudo de adición y una entrada de gas nitrógeno. El matraz se colocó en atmósfera de nitrógeno y se cargó con una solución de bis(clorometil)dietilsilano (5,4571 g, 29,471 mmol) en N,N-dimetilformamida anhidra (12,5 ml) que se añadió a través de una jeringa. La solución de fenóxido anterior de la reacción de 4-fluoro-2-yodo-6-metilfenol e hidruro de sodio en N,N-dimetilformamida anhidra se añadió al embudo de adición a través de una jeringa. La solución se añadió gota a gota a la solución de bis(clorometil)dietilsilano en N,N-dimetilformamida anhidra a temperatura ambiente. Después de 1 hora, se determinó que la reacción se había completado. Los sólidos en el fondo del matraz se filtraron mediante filtración al vacío y se lavaron con dos partes de 5 ml de acetato de etilo. El filtrado se transfirió a un matraz de fondo redondo y se enfrió hasta 0 °C (baño de hieloagua). A la solución enfriada se añadió lentamente HCl acuoso 1 M (16,5 ml) a través de un embudo de adición (a una velocidad para mantener el control de la reacción). La reacción se concentró mediante evaporación rotatoria (temperatura de baño = 60-75 °C) para retirar tanta N,N-dimetilformamida como fuera posible. La solución restante se absorbió en agua (33 ml), se transfirió a un embudo de separación y, después, se añadió acetato de etilo (33 ml). Las fases se separaron. La fase acuosa se extrajo con acetato de etilo (4 veces 33 ml). Las fases orgánicas combinadas se lavaron con agua (33 ml). Se formó una pequeña emulsión entre las dos fases. Se añadió un chorro de agua y el embudo se agitó con movimientos circulares (se repitió hasta que desapareció la emulsión). Las fases se separaron y la fase orgánica se secó sobre sulfato de magnesio anhidro, se filtró mediante filtración al vacío y se concentró mediante evaporación rotatoria para proporcionar un aceite bruto de color marrón rojizo. El aceite se purificó mediante cromatografía de columna usando una columna Grace de 330 g y un gradiente de hexanos al 100 % hasta que eluyó el producto. Las fracciones puras se combinaron y se concentraron mediante evaporación rotatoria para proporcionar el producto como un aceite de color amarillo pálido. A fin de retirar las trazas de acetato de etilo y hexanos, el aceite se disolvió en diclorometano y se concentró mediante evaporación rotatoria para proporcionar un aceite de color amarillo pálido (se repitió dos veces). El aceite se secó a alto vacío para proporcionar 2,0211 g (51,4 %) del producto como un aceite de color amarillo pálido.
RMN 1H (400 MHz, CDCls) 57,29 (ddq, J = 7,5, 3,1, 0,6 Hz, 1H), 6,85 (ddq, J = 8,7, 3,1, 0,7 Hz, 1H), 3,74 (s, 2H), 3,09 (s, 2H), 2,31 (t, J = 0,6 Hz, 3H), 1,14 - 1,08 (m, 6 H), 0,94 - 0,86 (m, 4H).
RMN 19F (376 MHz, CDCls) 5 -118,34 (t, J = 8,0 Hz).
Preparación de dietil((4-fluoro-2-yodo-6-metilfenoxi)metil)((4-fluoro-2-yodofenoxi)metil)silano:

Un matraz de fondo redondo de tres bocas se equipó con una barra de agitación magnética, dos septos, un condensador y una entrada de gas nitrógeno. El matraz se colocó en atmósfera de nitrógeno y se cargó con hidruro de sodio (0,2750 g, 11,458 mmol) y N,N-dimetilformamida anhidra (10 ml). La solución se enfrió hasta 0 °C (baño de hielo-agua). Una solución de 4-fluoro-2-yodofenol (2,4893 g, 10,459 mmol) en N,N-dimetilformamida anhidra (10 ml) se añadió lentamente a través de una jeringa para mantener el control de la reacción (desprendimiento de hidrógeno). La mezcla resultante se agitó durante 30 minutos a temperatura ambiente. Una solución de (clorometil)dietil((4-fluoro-2-yodo-6-metilfenoxi)metil)silano (3,4893 g, 8,707 mmol) en N,N-dimetilformamida anhidra (4,5 ml) se añadió lentamente a través de una jeringa a temperatura ambiente. La solución de color marrón resultante se agitó a 60 °C. Después de 18,5 horas, la reacción se dejó enfriar hasta temperatura ambiente. La reacción se enfrió adicionalmente hasta 0 °C (baño de hielo-agua) y se añadió lentamente agua (25 ml) (a una velocidad para mantener el control de la reacción). Los sólidos se formaron durante la adición y permanecieron después de la adición. La mezcla se transfirió a un matraz de fondo redondo de 1 boca. Los sólidos se disolvieron en diclorometano y se transfirieron al matraz. La mezcla se concentró mediante evaporación rotatoria (temperatura de baño = 60-75 °C) para retirar tanta N,N-dimetilformamida como fuera posible. La solución restante se absorbió en agua (30 ml) y se transfirió a un embudo de separación y, después, se añadió acetato de etilo (30 ml). Las fases se
separaron. La fase acuosa se extrajo con cuatro partes de 30 ml de acetato de etilo. Las fases orgánicas combinadas se lavaron con dos partes de 21 ml de NaOH acuoso 1 M. La fase orgánica se lavó con salmuera (25 ml), se secó sobre sulfato de magnesio anhidro, se filtró mediante filtración al vacío y se concentró mediante evaporación rotatoria para proporcionar un aceite de color naranja bruto (4,7914 g). El aceite bruto se purificó mediante cromatografía de columna usando una columna Grace de 330 g y un gradiente de hexanos al 100 % para 1 volumen de columna (VC), aumentando a acetato de etilo al 5 % en hexanos sobre 1 VC, el resto en acetato de etilo al 5 % en hexanos hasta que eluyó el producto. Las fracciones puras se combinaron y se concentraron mediante evaporación rotatoria para proporcionar el producto como un aceite de color amarillo. A fin de retirar las trazas de acetato de etilo y hexanos, el aceite se disolvió en diclorometano y se concentró mediante evaporación rotatoria para proporcionar un aceite de color amarillo (se repitió dos veces). El aceite se secó a alto vacío para proporcionar 3,7015 g (70,6 %) del producto como un aceite de color amarillo. RMN 1H (400 MHz, CDCh) 57,48 (dd, J = 7,6, 3,0 Hz, 1H), 7,29 (ddd, J = 7,6, 3,0, 0,7 Hz, 1H), 7,03 (ddd, J = 9,1, 7,8, 3,0 Hz, 1H), 6 , 8 8 (dd, J = 9,1, 4,6 Hz, 1H), 6,83 (ddd, J = 8,7, 3,1, 0,8 Hz, 1H) 3,91 (s, 2H), 3,88 (s, 2H), 2,27 (t, J = 0,7 Hz, 3H), 1,21 - 1,14 (m, 6 H), 1,03 -0,95 (m, 4H).
RMN 19F (376 MHz, CDCls) 5 -118,35 (dd, J = 8,4, 7,7 Hz), -123,07 (td, J = 7,7, 4,6 Hz).
Preparación de 3-(2,7-di-terc-butil-9H-carbazol-9-il)-2'-(((((3'-(2,7-di-terc-butil-9H-carbazol-9-il)-5-fluoro-2'-hidroxi-5'-(2,4,4-trimetilpentan-2-il)-[1,1'-bifenil]-2-il)oxi)metil)dietilsilil)metoxi)-5'-fluoro-3'-metil-5-(2,4,4-trimetilpentan-2-il)-[1,1'-bifenil]-2-ol:
Un matraz de fondo redondo de tres bocas se equipó con una barra de agitación magnética, dos septos, un condensador y una entrada de gas nitrógeno. El matraz se colocó en atmósfera de nitrógeno y se cargó con 2,7-diferc-butil-9-(2-((tetrahidro-2H-piran-2-il)oxi)-3-(4,4,5,5-tetrametil-1,3,2-dioxaborolan-2-il)-5-(2,4,4-trimetilpentan-2-il)fenil)-9H-carbazol (5,2419 g, 7,555 mmol), 1,2-dimetoxietano (85 ml), una solución de hidróxido de sodio (0,9106 g, 22,765 mmol) en agua (25 ml), tetrahidrofurano (30 ml) y dietil((4-fluoro-2-yodo-6-metilfenoxi)metil)((4-fluoro-2-yodofenoxi)metil)silano (1,9770 g, 3,283 mmol). La solución se agitó y se purgó con nitrógeno durante aproximadamente 15 minutos, después se añadió tetraquis(trifenilfosfino)paladio(0) (0,2755 g, 0,2384 mmol). La mezcla se calentó hasta reflujo a 85 °C. Después de 20 horas, la reacción se dejó enfriar hasta temperatura ambiente. La mezcla se transfirió a un embudo de separación para una separación de fases. La fase orgánica se secó sobre sulfato de magnesio y se filtró mediante filtración al vacío. Los sólidos se enjuagaron con diclorometano (2 veces 20 ml). El filtrado se concentró mediante evaporación rotatoria para proporcionar un sólido de color naranja dorado adherente como un ligando protegido bruto (15,6875 g). El ligando protegido bruto se disolvió en una mezcla de tetrahidrofurano (65 ml) y metanol (65 ml) y, después, se calentó hasta 60 °C. Se añadió monohidrato de ácido para-toluenosulfónico (0,2492 g, 1,310 mmol) a la solución. La reacción se agitó a 60 °C durante una noche y se comprobó mediante TLC en cuanto a la terminación. El ligando se concentró hasta obtener un sólido de color naranja dorado adherente (15,3856 g). El ligando se disolvió en cloroformo y se añadió gel de sílice. La suspensión se concentró mediante evaporación rotatoria para proporcionar una mezcla seca en polvo de gel de sílice y ligando. La mezcla en polvo se dividió en dos columnas separadas. Ambas columnas se cargaron en el sistema Isco CombiFlash y se circularon usando una columna Grace de 330 g. La primera columna se circuló usando un gradiente de diclorometano al 30 % en hexanos hasta que eluyó el producto. Las fracciones se analizaron mediante TLC y todas las fracciones que contenían solo el producto se concentraron mediante evaporación rotatoria para proporcionar un sólido cristalino blanquecino. El sólido se secó a alto vacío para proporcionar 1,4 g. La segunda columna se circuló usando un gradiente de diclorometano al 30 % en hexanos para 2 volúmenes de columna, después aumentando a diclorometano al 35 % en hexanos hasta que eluyó el producto. Las fracciones se analizaron mediante TLC, que mostró una combinación del ligando con otras impurezas. Todas las fracciones que contenían la mayor parte del producto se concentraron mediante evaporación rotatoria para proporcionar 2,1863 g de un sólido cristalino blanquecino. El sólido se disolvió en cloroformo y se añadió gel de sílice. La suspensión se concentró mediante evaporación rotatoria para proporcionar una mezcla seca en polvo de gel de sílice y ligando. La mezcla en polvo se cargó en el sistema Isco CombiFlash y se circuló usando una columna Grace de 330 g y un gradiente de diclorometano al 30 % en hexanos y, después, aumentando a diclorometano al 35 % en hexanos hasta que eluyó el
producto. Las fracciones que contenían el ligando se concentraron mediante evaporación rotatoria para proporcionar un sólido cristalino blanquecino. El sólido se secó a alto vacío para proporcionar 0,4672 g de un sólido cristalino blanquecino. El rendimiento global fue de 1,8672 g (23,1 %) del producto como un sólido cristalino blanquecino. RMN 1H (400 MHz, CDCls) 8 8,05 (dd, J = 8,2, 0,6 Hz, 2H), 7,99 (dd, J = 8,3, 0,7 Hz, 2H), 7,48 (d, J = 2,4 Hz, 1H), 7,40 (d, J = 2,4 Hz, 1H), 7,35 (d, J = 2,4 Hz, 1H), 7,32 -7,28 (m, 4H), 7,24 (d, J = 2,4 Hz, 1H), 7,09 (d, J = 1,6 Hz, 2H), 7,00 (dd, J = 8,9, 3,2 Hz, 1H), 6,95 (dd, J = 9,0, 3,2 Hz, 1H), 6,82 (dd, J = 8,7, 3,0 Hz, 1H), 6,75 (s, 1H), 6,65 (ddd, J = 8,9, 7,8, 3,1 Hz, 1H), 6,18 (dd, J = 9,1, 4,5 Hz, 1H), 5,64 (s, 1H), 3,60 - 3,47 (amplio m, 2H), 3,38 (s, 2H), 1,90 (s, 3H), 1,74 (amplio s, 2H), 1,69 (s, 2H), 1,40 (s, 6 H), 1,35 - 1,33 (m, 6 H), 1,30 (s, 18H), 1,28 (s, 18H), 0,79 (s, 9H), 0,77 (s, 9H), 0,43 (t, J = 7,7 Hz, 6 H), 0,36 - 0,31 (amplio m, 4H). RMN 19F (376 MHz, CDCla) 8 -118,46 (t, J = 8,9 Hz), -123,65 (m).
Preparación de procatalizador 6:

La reacción se ajustó en una caja de guantes en atmósfera de nitrógeno. Un frasco se cargó con HfCU (0,0489 g, 0,1522 mmol) y tolueno (12 ml). La suspensión se enfrió hasta -25 °C en el refrigerador de la caja de guantes durante 30 minutos a -25 °C. A la suspensión fría en agitación se añadió bromuro de metilmagnesio 3,0 M frío en éter de dietilo (0,20 ml, 0,60 mmol). La mezcla se agitó enérgicamente durante 2 minutos. El sólido se puso en solución. La solución de reacción estaba turbia y era de color amarillo pálido. A la mezcla se añadió el ligando (0,2000 g, 0,1522 mmol) como un sólido a una velocidad para mantener el control de la reacción. El matraz que contenía el sólido se enjuagó con tolueno (aproximadamente 2 ml). El disolvente de enjuague se añadió a la mezcla de reacción. La mezcla de reacción se agitó a temperatura ambiente durante 3 horas. A la mezcla de color marrón se añadieron hexanos (12 ml). La mezcla se filtró. El filtrado se concentró a alto vacío para proporcionar 0,2341 g (101,1 %) del producto deseado. El exceso de masa se atribuyó a la trampa de tolueno residual en el sólido.
RMN 1H (400 MHz, C6D6) 8 8,19 (dd, J = 8,2, 0,5 fHz, 1H), 8,18 - 8,15 (m, 2H), 8,04 (dd, J = 8,3, 0,6 Hz, 1H), 7,82 (ddd, J = 2,4, 1,7, 0,6 Hz, 2H), 7,76 (d, J = 2,5 Hz, 1H), 7,72 (dd, J = 1,7, 0,6 Hz, 1H), 7,66 (dd, J = 1,8, 0,6 Hz, 1H), 7,63 (d, J = 2,6 Hz, 1H), 7,47 - 7,44 (dos m, 2H), 7,41 (ddd, J = 8,3, 6,7, 1,7 Hz, 2H), 7,35 (dd, J = 8,3, 1,7 Hz, 1H), 7,21 (d, J = 2,6 Hz, 1H), 6,99 - 6,95 (dos m, 2H), 6,55 (ddd, J = 9,1, 7,3, 3,2 Hz, 1H), 6,11 (ddd, J = 8,4, 3,2, 0,7 Hz, 1H), 5,44 (dd, J = 9,1, 4,8 Hz, 1H), 4,51 (d, J = 13,7 Hz, 1H), 4,37 (d, J = 14,5 Hz, 1H), 3,41 (d, J = 13,7 Hz, 1H), 3,28 (d, J = 14,5 Hz, 1H), 1,60 (s, 2H), 1,54 (s, 2H), 1,45 (s, 8 H), 1,41 (s, 8 H), 1,33 (d, J =1,1 Hz, 4H), 1,28 (d, J = 0,4 Hz, 17H), 1,23 (s, 3H), 1,19 (s, 3H), 0,92 (s, 0H), 0,83 (s, 9H), 0,82 (s, 8 H), 0,54 (td, J = 7,9, 2,7 Hz, 6 H), 0,25 -0,09 (m, 3H), 0,10 - -0,06 (m, 1H), -0,58 (d, J= 0,5 Hz, 3H), -1,07 (d, J = 0,5 Hz, 3H). RMN 19F (376 MHz, C6D6) 8 -116,21(m), -116,30 (t, J = 8 , 8 Hz).
Preparación de polímeros basados en etileno en un solo reactor
Todas las materias primas (etileno, 1-octeno) y el disolvente del proceso (un disolvente isoparafínico de alta pureza de intervalo de ebullición estrecho Isopar E disponible en el mercado a través de ExxonMobil Corporation) se purifican con tamices moleculares antes de la introducción en el entorno de reacción. El hidrógeno se suministra en cilindros presurizados como una calidad de alta pureza y no se purifica adicionalmente. La corriente de alimentación de monómero del reactor (etileno) se presuriza a través de un compresor mecánico hasta una presión por encima de la presión de reacción a 3,68 MPa (525 psig). La alimentación de disolvente y comonómero (1-octeno) se presuriza a través de una bomba mecánica de desplazamiento positivo hasta una presión por encima de la presión de reacción a 3,68 MPa (525 psig). Los componentes individuales de catalizador se diluyen manualmente por lotes hasta las concentraciones de componentes especificadas con disolvente purificado (Isopar E) y se presurizan hasta una presión por encima de la presión de reacción a 3,68 MPa (525 psig). Todos los flujos de alimentación de la reacción se miden con medidores de flujo de masa y se controlan, de manera independiente, con sistemas de control de válvulas automatizados por ordenador.
El reactor de polimerización en solución continua consiste en un bucle lleno de líquido, no adiabático, isotérmico, en circulación y controlado de manera independiente. El reactor tiene un control independiente de todas las alimentaciones de disolvente, monómero, comonómero, hidrógeno y componente de catalizador nuevas. La alimentación de disolvente, monómero, comonómero e hidrógeno combinada al reactor se realiza con control de temperatura a cualquiera entre 5 °C y 50 °C y, de manera típica, 25 °C, mediante el paso de la corriente de
alimentación a través de un intercambiador de calor. La alimentación de comonómero nueva al reactor de polimerización se alimenta con la alimentación de disolvente. La alimentación nueva total a cada reactor de polimerización se inyecta en el reactor en dos localizaciones con volúmenes de reactor bastantes iguales entre cada localización de inyección. La alimentación nueva se controla, de manera típica, con cada inyector que recibe la mitad del flujo de masa de alimentación nueva total. Los componentes de catalizador se inyectan en el reactor de polimerización a través de guías de inyección especialmente diseñadas y cada uno se inyecta por separado en la misma localización relativa en el reactor sin tiempo de contacto antes con el reactor. La alimentación de componente de catalizador principal se controla por ordenador para mantener la concentración de monómero del reactor en una diana especificada. Los componentes de cocatalizador se alimentan basándose en las relaciones molares especificadas calculadas para el componente de catalizador principal. Inmediatamente después de cada localización de inyección nueva (alimentación o catalizador), las corrientes de alimentación se mezclan con el contenido del reactor de polimerización en circulación con elementos de mezclado estático de Kenics. El contenido de cada reactor se hace circular continuamente a través de intercambiadores de calor responsables de retirar gran parte del calor de reacción y con la temperatura del lado del refrigerante responsable de mantener el entorno de reacción isotérmico a la temperatura especificada. La circulación alrededor de cada bucle del reactor se proporciona mediante una bomba de tornillo.
El efluente del primer reactor de polimerización (que contiene disolvente, monómero, comonómero, hidrógeno, componentes de catalizador y polímero fundido) sale del bucle del primer reactor y pasa a través de una válvula de control (responsable de mantener la presión del primer reactor en una diana especificada). A medida que sale la corriente del reactor, esta se pone en contacto con agua para detener la reacción. Además, en este punto se pueden añadir diversos aditivos, tales como antioxidantes. A continuación, la corriente pasa a través de otro conjunto de elementos de mezclado estático de Kenics para dispersar uniformemente el eliminador de catalizador y los aditivos. Después de la adición de los aditivos, el efluente (que contiene disolvente, monómero, comonómero, hidrógeno, componentes de catalizador y polímero fundido) pasa a través de un intercambiador de calor para elevar la temperatura de la corriente en la preparación para la separación del polímero de los otros componentes de reacción de ebullición más baja. Después, la corriente entra en un sistema de separación y desvolatización de dos etapas donde el polímero se retira del disolvente, el hidrógeno y el monómero y comonómero sin reaccionar. La corriente reciclada se purifica antes de entrar de nuevo en el reactor. El polímero fundido separado y desvolatizado se bombea a través de un troquel especialmente diseñado para la peletización bajo el agua, se corta en pellas sólidas uniformes, se seca y se transfiere a una tolva. Después de la validación de las propiedades iniciales del polímero, las pellas de polímero sólidas se vierten manualmente en una caja para su almacenamiento. Cada caja contiene, de manera típica, ~544 kg (1.200 libras) de pellas de polímero.
Las partes no poliméricas retiradas en la etapa de desvolatilización pasan a través de diversas etapas del proceso que separan la mayor parte del etileno que se retira del sistema a una unidad de destrucción de ventilación (se recicla en unidades de fabricación). La mayor parte del disolvente se recicla de nuevo de vuelta al reactor después del paso a través de los lechos de purificación. Este disolvente todavía puede tener un comonómero sin reaccionar que se enriquezca con comonómero nuevo antes de volver a entrar en el reactor. Este enriquecimiento del comonómero es una parte esencial del método de control de la densidad del producto. Este disolvente de reciclaje aún puede tener algo de hidrógeno que se enriquezca después con hidrógeno nuevo para alcanzar la diana de peso molecular del polímero. Una cantidad muy pequeña de disolvente sale del sistema como coproducto debido al vehículo de disolvente en las corrientes de catalizador y una pequeña cantidad de disolvente que es parte de los comonómeros de calidad comercial.
Las condiciones de polimerización en un solo reactor, como se ha descrito anteriormente, para PE-A comparativo, PE-B comparativo, PE-C comparativo, PE-D comparativo, PE-1 de la invención, PE-2 de la invención y PE-3 de la invención se indican en la Tabla 1. Las propiedades para PE-A comparativo, PE-B comparativo, PE-C comparativo, PE-D comparativo, PE-1 de la invención, PE-2 de la invención y PE-3 de la invención se sometieron a ensayo y se indican en la Tabla 1.
Tabla 1



Preparación de polímeros basados en etileno en un reactor por lotes de 2 l
Las reacciones de polimerización se llevaron a cabo a 140 °C y 190 °C. Se usó un reactor Parr de 2 litros en las polimerizaciones. Todas las alimentaciones se hicieron pasar a través de columnas de alúmina y catalizador Q-5™ (disponible a través de Engelhard Chemicals Inc.) antes de la introducción en el reactor. Las soluciones de procatalizador y activador se manipularon en la caja de guantes. A 140 °C, se cargó un reactor de 2 litros agitado con aproximadamente 605 g de alcanos mezclados y 300 g de comonómero de 1-octeno. Mientras el reactor alcanzaba la temperatura de polimerización, se añadieron 10 pmol de MMAO al reactor como eliminador de trazas de O2 y agua. Una vez a la temperatura, el reactor se saturó con etileno a 2 MPa (288 psig). A 190 °C, se cargó un reactor de 2 litros con aproximadamente 520 g de alcanos mezclados y 300 g de comonómero de 1-octeno. Mientras el reactor alcanzaba la temperatura de polimerización, se añadieron 10 pmol de MMAO al reactor como eliminador de trazas de O2 y agua. Una vez a la temperatura, el reactor se saturó con etileno a 2 , 8 MPa (400 psig). Los procatalizadores y el activador, como soluciones diluidas en tolueno, se mezclaron y transfirieron a un tanque de adición de catalizador y se inyectaron en el reactor. Las condiciones de polimerización se mantuvieron durante 10 minutos con etileno añadido a voluntad. El calor se retiró continuamente del recipiente de reacción a través de un serpentín de enfriamiento interno. La solución resultante se retiró del reactor y se estabilizó mediante la adición de 10 ml de una solución de tolueno que contenía aproximadamente 67 mg de un antioxidante de fenol impedido (Irganox™ 1010 de Ciba Geigy Corporation) y 133 mg de un estabilizador de fósforo (Irgafos™ 168 de Ciba Geigy
Corporation). Entre los ciclos de polimerización, se realizó un ciclo de lavado en el que se añadieron al reactor 850 g de alcanos mezclados y el reactor se calentó hasta 160 °C. Después, se vació el reactor del disolvente calentado inmediatamente antes de comenzar un nuevo ciclo de polimerización. Los polímeros se recuperaron mediante el secado durante aproximadamente 12 h en un horno de vacío con incremento de temperatura con un punto de ajuste final de 140 °C.
Los ejemplos de reactores por lotes 1-12 (BRE 1-12) se prepararon según el proceso anterior según las condiciones indicadas en las Tablas 4 y 5 y los BRE 1-12 se sometieron a ensayo para determinar sus propiedades y aquellas propiedades se enumeran en las Tablas 4 y 5.


Métodos de ensayo
Los métodos de ensayo incluyen lo siguiente:
Densidad
Las muestras que se miden en cuanto a la densidad se preparan según la norma ASTM D-1928. Las mediciones se realizan en el período de una hora de prensado de la muestra usando el Método B de la norma ASTM D-792.
Índice de fundición
El Índice de fundición (I2) se mide según la ASTM-D 1238, en condiciones a 190 °C/2,16 kg, y se indica en gramos eluidos por 10 minutos. El caudal de fundición (I10) se mide según la norma ASTM-D 1238, en condiciones a 190 °C/10 kg, y se indica en gramos eluidos por 10 minutos.
Cristalinidad de DSC
La calorimetría diferencial de barrido (DSC) se puede usar para medir el comportamiento en la fundición y cristalización de un polímero en un amplio intervalo de temperatura. Por ejemplo, el Q1000 DSC de TA Instruments, equipado con un RCS (sistema de enfriamiento refrigerado) y un automuestreador se usa para realizar este análisis. Durante el ensayo, se usa un flujo de gas de purga de nitrógeno de 50 ml/min. Cada muestra se presiona en estado fundido hasta dar una película delgada a aproximadamente 175 °C; después, la muestra en estado fundido se enfría al aire hasta temperatura ambiente (~25 °C). Una muestra para ensayo de 3-10 mg, de 6 mm de diámetro, se extrae del polímero enfriado, se pesa, se coloca en un recipiente de aluminio ligero (aprox. 50 mg) y se aplasta. Después, se realiza un análisis para determinar sus propiedades térmicas.
El comportamiento térmico de la muestra se determina incrementando la temperatura de la muestra hacia arriba y hacia abajo para crear un flujo de calor frente al perfil de temperatura. En primer lugar, la muestra se calienta rápidamente hasta 180 °C y se mantiene isotérmica durante 3 minutos con el fin de retirar su historial térmico. A continuación, la muestra se enfría hasta -40 °C a una velocidad de enfriamiento de 10°C/minuto y se mantiene isotérmica a -40 °C durante 3 minutos. Después, la muestra se calienta hasta 150 °C (este es el incremento del "segundo calentamiento") a una velocidad de calentamiento de 10 °C/minuto. Se registran las curvas de enfriamiento y segundo calentamiento. La curva de enfriamiento se analiza mediante el ajuste de los criterios de valoración de referencia desde el inicio de la cristalización hasta -20 °C. La curva de calentamiento se analiza mediante el ajuste de los criterios de valoración de referencia desde -20 °C hasta el final de la fundición. Los valores determinados son temperatura de pico de fundición (Tm), temperatura de pico de cristalización (Tc), calor de fusión (Hf) (en Julios por gramo) y el % de cristalinidad calculado para las muestras usando la ecuación adecuada, por ejemplo, para el interpolímero de etileno/alfa-olefina usando la Ecuación 1, como se muestra en la Figura 1.
El calor de fusión (Hf) y la temperatura de pico de fundición se indican a partir de la segunda curva de calentamiento. La temperatura de pico de cristalización se determina a partir de la curva de enfriamiento.
Barrido de frecuencia por espectroscopía dinámica mecánica (DMS)
Los barridos de frecuencia a temperatura constante, reología de fundición, se realizaron usando un reómetro de Sistema de Expansión Reométrica Avanzado (ARES) de TA Instruments equipado con placas paralelas de 25 mm con una purga de nitrógeno. Los barridos de frecuencia se realizaron a 190 °C para todas las muestras con una abertura de 2,0 mm y una tensión constante del 10 %. El intervalo de frecuencia fue de 0,1 a 100 radianes/segundo. La respuesta a la tensión se analizó en términos de amplitud y fase, a partir de lo que se calcularon el módulo de almacenamiento (G'), el módulo de pérdida (G") y la viscosidad de fundición dinámica (n*).
Cromatografía de permeación en gel (GPC)
Los interpolímeros de etileno/alfa-olefina se sometieron a ensayo para determinar sus propiedades a través de GPC, según el siguiente procedimiento. El sistema de GPC consiste en un cromatógrafo de alta temperatura Waters (Milford, MA) a 150 °C (otros instrumentos de GPC de alta temperatura adecuados incluyen Polymer Laboratories (Shropshire, Reino Unido) Modelo 210 y Modelo 220) equipado con un refractómetro diferencial integrado (RI). Los detectores adicionales pueden incluir un detector de infrarrojos IR4 de Polymer ChAR (Valencia, España), un detector de dispersión de luz láser de 2 ángulos de Precision Detectors (Amherst, MA) Modelo 2040 y un viscosímetro de solución capilar 150R 4 de Viscotek (Houston, TX). Un GPC con los dos últimos detectores independientes y al menos uno de los primeros detectores se conoce a veces como "GPC en 3D", mientras que el término "GPC" solo se refiere, en general, al GPC convencional. Dependiendo de la muestra, se usa el ángulo de 15 grados o el ángulo de 90 grados del detector de dispersión de luz para fines de cálculo. La recogida de datos se realiza usando el soporte lógico Viscotek TriSEC, Versión 3, y un gestor de datos Viscotek de 4 canales DM400. El sistema también está equipado con un dispositivo de desgasificación de disolvente en línea de Polymer Laboratories (Shropshire, Reino Unido). Se pueden usar columnas de GPC de alta temperatura adecuadas, tales como cuatro columnas de 30 cm de largo Shodex HT803 de 13 pm (micrómetros) o cuatro columnas de 30 cm de Polymer Labs de empaquetamiento de tamaño de poro mixto de 20 pm (micrómetros) (MixA LS, Polymer Labs). El compartimento
de alimentador rotatorio de muestra se opera a 140 °C y el compartimento de columna se opera a 150 °C. Las muestras se preparan a una concentración de 0,1 gramos de polímero en 50 mililitros de disolvente. El disolvente cromatográfico y el disolvente de preparación de muestra contienen 200 ppm de hidroxitolueno butilado (BHT). Ambos disolventes se pulverizan con nitrógeno. Las muestras de polietileno se agitan suavemente a 160 °C durante cuatro horas. El volumen de inyección es de 200 microlitros. El caudal a través del GPC se ajusta en 1 ml/minuto.
El conjunto de columna de GPC se calibra antes de realizar los Ejemplos mediante la circulación de veintiún patrones de poliestireno de estrecha distribución de peso molecular. El peso molecular (MW) de los patrones varía de 580 a 8.400.000 gramos por mol y los patrones están contenidos en 6 mezclas de "cóctel". Cada mezcla de patrón tiene al menos una decena de separación entre pesos moleculares individuales. Las mezclas de patrón se adquieren a través de Polymer Laboratories (Shropshire, Reino Unido). Los patrones de poliestireno se preparan a 0,025 g en 50 ml de disolvente para pesos moleculares iguales a o mayores de 1.000.000 gramos por mol y 0,05 g en 50 ml de disolvente para pesos moleculares menores de 1.000.000 gramos por mol. Los patrones de poliestireno se disolvieron a 80 °C con agitación suave durante 30 minutos. En primer lugar, las mezclas de patrones estrechos se circulan y con el fin de disminuir el componente de peso molecular más alto para minimizar la degradación. Los pesos moleculares de pico de patrones de poliestireno se convierten en Mw de polietileno usando los valores de K y a (a veces denominados como a) de Mark-Houwink mencionados posteriormente para el poliestireno y polietileno. Véase la sección de Ejemplos para una demostración de este procedimiento.
Con GPC en 3D, también se obtienen, de manera independiente, el peso molecular promedio en peso absoluto ("Mw, Abs") y la viscosidad intrínseca de patrones de polietileno estrechos adecuados usando las mismas condiciones mencionadas anteriormente. Estos patrones de polietileno lineal estrechos se pueden obtener a través de Polymer Laboratories (Shropshire, Reino Unido; Parte n.° PL2650-0101 y PL2650-0102).
Este enfoque sistemático para la determinación de compensaciones de multidetectores se realiza de manera consistente con lo publicado por Balke, Mourey, y col. (Mourey and Balke, Chromatography Polym., Chapter 12, (1992)) (Balke, Thitiratsakul, Lew, Cheung, Mourey, Chromatography Polym., Chapter 13, (1992)), optimizando los resultados de log de triple detector (MW y viscosidad intrínseca) de poliestireno amplio de Dow 1683 (American Polymer Standards Corp.; Mentor, OH) o su equivalente para los resultados de calibración de columna de patrón estrecho de la curva de calibración de patrones de poliestireno estrechos. Los datos de peso molecular, considerando la determinación de compensación de volumen de detector, se obtienen de manera consistente con lo publicado por Zimm (Zimm, B.H., J. Chem. Phys., 16, 1099 (1948)) y Kratochvil (Kratochvil, P., Classical Light Scattering from Polymer Solutions, Elsevier, Oxford, NY (1987)). La concentración inyectada global usada en la determinación del peso molecular se obtiene a partir del área de detector de masa y la constante de detector de masa derivadas de un homopolímero de polietileno lineal adecuado o uno de los patrones de polietileno. Los pesos moleculares calculados se obtienen usando una constante de dispersión de luz derivada de uno o más de los patrones de polietileno mencionados y un coeficiente de concentración de índice de refracción, dn/dc, de 0,104. En general, la respuesta del detector de masa y la constante de dispersión de luz se deben determinar a partir de un patrón lineal con un peso molecular superior a aproximadamente 50.000 Daltons. La calibración del viscosímetro se puede realizar usando los métodos descritos por el fabricante o, como alternativa, usando los valores publicados de patrones lineales adecuados, tales como los materiales de referencia certificados (SRM) 1475a, 1482a, 1483 o 1484a. Se supone que las concentraciones cromatográficas son suficientemente bajas para eliminar el abordaje de los efectos del 2 ° coeficiente viral (efectos de la concentración sobre el peso molecular).
g' mediante GPC en 3D
El índice (g') para el polímero de muestra se determina mediante la calibración, en primer lugar, de los detectores de dispersión de luz, viscosidad y concentración descritos en el método de cromatografía de permeación en gel supra con polietileno de homopolímero SRM 1475a (o una referencia equivalente). Las compensaciones del detector de dispersión de luz y viscosímetro se determinan con respecto al detector de concentración, como se describe en la calibración. Las referencias se sustraen de los cromatogramas de dispersión de luz, viscosímetro y concentración, a continuación, se ajustan las ventanas de integración asegurando que se integra todo el intervalo de volumen de retención de bajo peso molecular en los cromatogramas de dispersión de luz y viscosímetro que indican la presencia de polímero detectable a partir del cromatograma de índice de refracción. Se usa un polietileno de homopolímero lineal para establecer una línea de referencia lineal de Mark-Houwink (MH) mediante la inyección de una referencia de polietileno de amplio peso molecular, tal como patrón de SRM1475a, el cálculo del archivo de datos y el registro de la viscosidad intrínseca (IV) y el peso molecular (Mw), cada uno derivado de los detectores de dispersión de luz y de viscosidad, respectivamente, y la concentración, como se determina a partir de la constante de masa del detector de RI para cada porción cromatográfica. Para el análisis de las muestras, se repite el procedimiento para cada porción cromatográfica para obtener una línea de Mark-Houwink de muestra. Cabe destacar que, en algunas muestras, los pesos moleculares inferiores, la viscosidad intrínseca y los datos de peso molecular pueden requerir que se extrapolen de tal manera que el peso molecular y la viscosidad intrínseca medidos se aproximen de manera asintótica a una curva de calibración de GPC de homopolímero lineal. Para este fin, muchas muestras de polímero basado en etileno altamente ramificado requieren que la línea de referencia lineal se desplace ligeramente para considerar la contribución de ramificaciones de cadena corta antes de proceder al cálculo del índice de ramificación de cadena larga (g').
Se calcula una g prima (gf) para cada porción (i) cromatografía de muestra ramificada y se mide el peso molecular (Mi) según la Ecuación 2, como se muestra en la Figura 2, donde el cálculo utiliza la IVreferencia Nnealj a peso molecular equivalente, Mj, en la muestra de referencia lineal. En otras palabras, la porción (i) IV de muestra y la porción (j) IV de referencia tienen el mismo peso molecular (Mi = Mj). Para simplificar, se calculan las porciones IVreferencia lineal,j a partir de un ajuste polinómico de quinto orden de la representación gráfica de Mark-Houwink de referencia. La relación de IV o gi' se obtiene únicamente a pesos moleculares mayores de 3.500 debido a las limitaciones de señal a ruido en los datos de dispersión de luz. El número de ramificaciones a lo largo del polímero de muestra (Bn) en cada porción de datos (i) se puede determinar mediante el uso de la Ecuación 3, como se muestra en la Figura 3, lo que supone un factor épsilon de apantallamiento de viscosidad de 0,75.
Finalmente, la cantidad de LCBf promedio por 1.000 carbonos en el polímero por todas las porciones (i) se puede determinar usando la Ecuación 4, como se muestra en la Figura 4.
Índice de ramificación gpcBR mediante GPC en 3D
En la configuración de GPC en 3D se pueden usar los patrones de polietileno y poliestireno para medir las constantes de Mark-Houwink, K y a, de manera independiente para cada uno de los dos tipos de polímeros, poliestireno y polietileno. Estos se pueden usar para refinar los pesos moleculares equivalentes de polietileno de Williams and Ward en la aplicación de los siguientes métodos.
El índice de ramificación gpcBR se determina mediante la calibración, en primer lugar, de los detectores de dispersión de luz, viscosidad y concentración, como se ha descrito anteriormente. Las referencias se sustraen después de los cromatogramas de dispersión de luz, viscosímetro y concentración. Después, se ajustan las ventanas de integración para garantizar la integración de todo el intervalo de volumen de retención de bajo peso molecular en los cromatogramas de dispersión de luz y viscosímetro que indican la presencia de polímero detectable a partir del cromatograma de índice de refracción. Después, se usan patrones de polietileno lineal para establecer las constantes de Mark-Houwink de polietileno y poliestireno, como se ha descrito anteriormente. En la obtención de las constantes, se usan los dos valores para construir dos calibraciones convencionales de referencia lineales ("cc") para el peso molecular del polietileno y la viscosidad intrínseca del polietileno en función del volumen de elución, como se muestra en las Ecuaciones 5 y 6 , Figuras 5 y 6 , respectivamente.
El índice de ramificación gpcBR es un método robusto para la caracterización de ramificación de cadena larga. Véase Yau, Wallace W., "Examples of Using 3D-GPC - TREF for Polyolefin Characterization", Macromol. Symp., 2007, 257, 29-45. El índice evita los cálculos de GPC en 3D porción a porción usados tradicionalmente en la determinación de valores de g' y los cálculos de frecuencia de ramificación a favor de áreas completas de detector de polímeros y productos de puntos del área. A partir de los datos de GPC en 3D, se puede obtener el Mw volumétrico de la muestra mediante el detector de dispersión de luz (LS) usando el método del área de pico. El método evita la relación porción a porción de la señal del detector de dispersión de luz sobre la señal del detector de concentración, como se requiere en la determinación de g'.
El cálculo del área en la Ecuación 7, mostrado en la Figura 7, ofrece más precisión debido a que, como área de muestra global, esta es mucho menos sensible a la variación producida por el ruido del detector y los ajustes de GPC sobre la referencia y los límites de integración. Mayormente, el cálculo del área de pico no se ve afectado por las compensaciones de volumen del detector. De manera similar, la viscosidad intrínseca (IV) de la muestra de alta precisión se obtiene mediante el método del área mostrado en la Ecuación 8 , como se muestra en la Figura 8 , donde DPi representa la señal de presión diferencial controlada directamente del viscosímetro en línea.
A fin de determinar el índice de ramificación gpcBR, se usa el área de elución de dispersión de luz para el polímero de muestra para determinar el peso molecular de la muestra. El área de elución del detector de viscosidad para el polímero de muestra se usa para determinar la viscosidad intrínseca (IV o [r|]) de la muestra.
Inicialmente, el peso molecular y la viscosidad intrínseca para una muestra de patrón de polietileno lineal, tal como SRM1475a o un equivalente, se determinan usando las calibraciones convencionales tanto para el peso molecular como la viscosidad intrínseca en función del volumen de elución, según las Ecuaciones 9 y 10, como se muestra en las Figuras 9 y 10, respectivamente.
La Ecuación 11, como se muestra en la Figura 11, se usa para determinar el índice de ramificación gpcBR, donde [r|] es la viscosidad intrínseca medida, [r|]cc es la viscosidad intrínseca de la calibración convencional, Mw es el peso molecular promedio en peso medido y Mw,cc es el peso molecular promedio en peso de la calibración convencional. El Mw mediante dispersión de luz (LS) usando la Ecuación 7, como se muestra en la Figura 7, se denomina comúnmente Mw absoluto; mientras que el Mw,cc de la Ecuación 9, como se muestra en la Figura 9, usando la curva de calibración de peso molecular de GPC convencional, a menudo se denomina Mw de cadena de polímero. Todos los valores estadísticos con el subíndice "cc" se determinan usando sus respectivos volúmenes de elución, la correspondiente calibración convencional, como se ha descrito anteriormente, y la concentración (Ci) derivada de la respuesta del detector de masa. Los valores sin subíndices son valores medidos basándose en las áreas del detector de masa, LALLS, y del viscosímetro. El valor de Kpe se ajusta de manera iterativa hasta que la muestra de referencia lineal tenga un valor medido de gpcBR de cero. Por ejemplo, los valores finales para a y Log K para la
determinación de gpcBR, en este caso particular, son 0,725 y -3,355, respectivamente, para el polietileno, y 0,722 y 3,993 para el poliestireno, respectivamente.
Una vez que se han determinado los valores de K y a, se repite el procedimiento usando las muestras ramificadas. Se analizan las muestras ramificadas usando las constantes de Mark-Houwink finales como los mejores valores de calibración "cc" y aplicando las Ecuaciones 7-11, como se muestra en las Figuras 7-11, respectivamente.
La interpretación de gpcBR es directa. En los polímeros lineales, el gpcBR calculado de la Ecuación 11, como se muestra en la Figura 11, será próximo a cero, puesto que los valores medidos mediante LS y viscosimetría serán próximos al patrón de calibración convencional. En los polímeros ramificados, el gpcBR será mayor de cero, especialmente con altos niveles de LCB, debido a que el Mw del polímero medido será mayor que el Mw,cc calculado y la IVcc calculada será mayor que la viscosidad intrínseca (IV) del polímero medido. De hecho, el valor de gpcBR representa el cambio de IV fraccionada debido al efecto de contracción del tamaño molecular como resultado de la ramificación del polímero. Un valor de gpcBR de 0,5 o 2,0 significaría un efecto de contracción del tamaño molecular de la IV al nivel del 50 % y 200 %, respectivamente, frente a una molécula de polímero lineal de peso equivalente. En estos ejemplos particulares, la ventaja de usar el gpcBR en comparación con los cálculos del índice g' y la frecuencia de ramificación se debe a la mayor precisión de gpcBR. Todos los parámetros usados en la determinación del índice de gpcBR se obtienen con buena precisión y no se ven afectados adversamente por la baja respuesta del detector de GPC en 3D a alto peso molecular del detector de concentración. Los errores en la alineación del volumen del detector tampoco afectan a la precisión de la determinación del índice de gpcBR. En otros casos particulares, pueden resultar preferibles otros métodos para la determinación de momentos de Mw a la técnica mencionada anteriormente.
Método de CEF
El análisis de distribución de comonómeros se realiza con el fraccionamiento por elución y cristalización (CEF) (PolymerChar en España) (B Monrabal y col., Macromol. Symp. 257, 71-79 (2007)). Se usa, como disolvente, ortodiclorobenceno (ODCB) con 600 ppm de antioxidante de hidroxitolueno butilado (BHT). La preparación de la muestra se realiza con automuestreador a 160 °C durante 2 horas con agitación a 4 mg/ml (a menos que se especifique de otro modo). El volumen de inyección es de 300 pl. El perfil de temperatura de CEF es: la cristalización a 3 °C/min de 110 °C a 30 °C, el equilibrio térmico a 30 °C durante 5 minutos, la elución a 3 °C/min de 30 °C a 140 °C. El caudal durante la cristalización es de 0,052 ml/min. El caudal durante la elución es de 0,50 ml/min. Los datos se recogen en un punto de datos/segundo.
Se empaqueta la columna de CEF mediante la Dow Chemical Company con perlas de vidrio a 125 pm ± 6 % (MO-SCI Specialty Products) con tubo inoxidable de 0,32 cm (1/8 pulg ). Las perlas de vidrio se lavan con ácido mediante MO-SCI Specialty con el requerimiento de la Dow Chemical Company. El volumen de columna es de 2,06 ml. Se realiza la calibración de la temperatura de columna mediante el uso de una mezcla de polietileno lineal de material de referencia de patrón de1475a de NIST (1,0 mg/ml) e Eicosano (2 mg/ml) en ODCB. La temperatura se calibra mediante el ajuste de la velocidad de calentamiento de elución de tal manera que el polietileno lineal 1475a de NIST tenga una temperatura de pico a 101,0 °C y el Eicosano tenga una temperatura de pico de 30,0 °C. Se calcula la resolución de la columna de CEF con una mezcla de polietileno lineal 1475a de NISt (1,0 mg/ml) y hexacontano (Fluka, de pureza, >97,0 %, 1 mg/ml). Se consigue una separación de referencia de hexacontano y polietileno 1475a de NIST. El área de hexacontano (de 35,0 °C a 67,0 °C) al área de 1475a de NIST de 67,0 °C a 110,0 °C es de 50 a 50, la cantidad de fracción soluble por debajo de 35,0 °C es del <1,8 % en peso. La resolución de la columna de CEF se define en la Ecuación 12, como se muestra en la Figura 12, donde la resolución de la columna es de 6,0. Método de CDC
Se calcula la constante de distribución de comonómeros (CDC) a partir del perfil de distribución de comonómeros mediante CEF. Se define la CDC como el índice de distribución de comonómeros dividido por el factor de la forma de distribución de comonómeros multiplicando por 100, como se muestra en la Ecuación 13, Figura 13.
El índice de distribución de comonómeros representa la fracción de peso total de cadenas de polímero con el contenido de comonómero variando entre 0,5 de contenido de comonómero de la mediana (Cmediana) y 1,5 de Cmediana de 35,0 °C a 119,0 °C. El factor de la forma de distribución de comonómeros se define como la relación de la mitad del ancho del perfil de distribución de comonómeros dividida por la desviación típica del perfil de distribución de comonómeros de la temperatura de pico (Tp).
Se calcula la CDC del perfil de distribución de comonómeros mediante CEF y la CDC se define como el índice de distribución de comonómeros dividido por el factor de la forma de distribución de comonómeros multiplicando por 100, como se muestra en la Ecuación 13, Figura 13, y en donde el índice de distribución de comonómeros representa la fracción de peso total de cadenas de polímero con el contenido de comonómero variando entre 0,5 de contenido de comonómero de la mediana (Cmediana) y 1,5 de Cmediana de 35,0 °C a 119,0 °C y en donde el factor de la forma de distribución de comonómeros se define como una relación de la mitad del ancho del perfil de distribución de comonómeros dividida por la desviación típica del perfil de distribución de comonómeros de la temperatura de pico (Tp).
La CDC se calcula según las siguientes etapas:
(A) obtener una fracción de peso a cada temperatura (T) (wt (T)) de 35,0 °C a 119,0 °C con un aumento de etapa de temperatura de 0,200 °C de CEF según la Ecuación 14, como se muestra en la Figura 14;
(B) calcular la temperatura de la mediana (Tmediana) en una fracción de peso acumulativa de 0,500, según la Ecuación 15, como se muestra en la Figura 15;
(C) calcular el correspondiente contenido de comonómero de la mediana en % en moles (Cmediana) a la temperatura de la mediana (Tmediana) mediante el uso de la curva de calibración del contenido de comonómero según la Ecuación 16, como se muestra en la Figura 16;
(D) construir una curva de calibración del contenido de comonómero mediante el uso de una serie de materiales de referencia con una cantidad conocida de contenido de comonómero, es decir, once materiales de referencia con una distribución de comonómero estrecha (distribución de comonómero monomodal en CEF de 35,0 °C a 119,0 °C) con un Mw promedio en peso de 35.000 a 115.000 (medido a través de GPC convencional) a un contenido de comonómero que varía del 0,0% en moles al 7,0% en moles se analizas con CEF en las mismas condiciones experimentales especificadas en las secciones experimentales de CEF;
(E) calcular la calibración de contenido de comonómero mediante el uso de la temperatura de pico (Tp) de cada material de referencia y su contenido de comonómero; la calibración se calcula a partir de cada material de referencia, como se muestra en la Fórmula 16, Figura 16, en donde: R2 es la constante de correlación;
(F) calcular el índice de distribución de comonómeros a partir de la fracción de peso total con el contenido de comonómero variando de 0,5* Cmediana a 1,5* Cmediana y, si la Tmediana es mayor de 98,0 °C, el índice de distribución de comonómeros se define como 0,95;
(G) obtener la altura máxima de pico del perfil de distribución de comonómeros de CEF mediante la búsqueda de cada punto de datos para el pico más alto de 35,0 °C a 119,0 °C (si los dos picos son idénticos, entonces se selecciona el pico de temperatura menor); la mitad del ancho se define como la diferencia de temperatura entre la temperatura delantera y la temperatura trasera a la mitad de la altura máxima de pico, la temperatura delantera a la mitad del pico máximo se busca hacia adelante de 35,0 °C, mientras que la temperatura trasera a la mitad del pico máximo se busca hacia atrás de 119,0 °C, en el caso de una distribución bimodal definida donde la diferencia en las temperaturas de pico es igual a o mayor de 1 ,1 veces la suma de la mitad del ancho de cada pico, la mitad del ancho de la composición de polímero basado en etileno de la invención se calcula como el promedio aritmético de la mitad del ancho de cada pico; y
(H) calcular la desviación típica de la temperatura (Desv.típ.) según la Ecuación 17, como se muestra en la Figura 17.
Método de viscosidad de cizaMamiento cero y deformación
Las viscosidades de cizallamiento cero se obtienen a través de ensayos de deformación que se realizan en un reómetro con control de tensión AR-G2 (TA Instruments; New Castle, Del) usando placas paralelas de 25 mm de diámetro a 190 °C. El horno del reómetro se ajusta a la temperatura de ensayo durante al menos 30 minutos antes de la puesta a cero de los accesorios. A la temperatura de prueba, se inserta un disco de muestra moldeado por compresión entre las placas y se deja que se equilibre durante 5 minutos. Después, se desciende la placa superior hasta 50 pm por encima de la abertura de ensayo deseada (1,5 mm). Se retira por recorte cualquier material superfluo se recorta y la placa superior se desciende hasta la abertura deseada. Las mediciones se realizan con purga de nitrógeno a un caudal de 5 l/min. El tiempo de deformación predeterminado se ajusta en 2 horas.
Se aplica una tensión de cizallamiento bajo constante de 20 Pa en todas las muestras para garantizar que la velocidad de cizallamiento de estado estacionario sea suficientemente baja para que esté en la región Newtoniana. Las velocidades de cizallamiento de estado estacionario resultantes son del orden de 10-3 s-1 en las muestras de este estudio. El estado estacionario se determina tomando una regresión lineal para todos los datos en la última ventana de tiempo del 10 % de la representación gráfica de log(J(t)) frente a log(t), donde J(t) es la deformación a tensión constante y t es el tiempo de deformación. Si la pendiente de la regresión lineal es mayor de 0,97, se considera que se ha alcanzado el estado estacionario, entonces se detiene el ensayo de deformación. En todos los casos, en este estudio, la pendiente cumple con el criterio en 30 minutos. Se determina la velocidad de cizallamiento de estado estacionario de la pendiente de la regresión lineal de todos los puntos de datos en la última ventana de tiempo del 10% de la representación gráfica frente a t, donde £ es tensión. Se determina la viscosidad a cizallamiento cero a partir de la relación de la tensión aplicada con respecto a la velocidad de cizallamiento de estado estacionario.
Con el fin de determinar si se degrada la muestra durante el ensayo de deformación, se realiza un ensayo de cizallamiento oscilatorio de pequeña amplitud antes y después del ensayo de deformación sobre la misma muestra para ensayo de 0,1 rad/s a 100 rad/s. Se comparan los valores de viscosidad compleja de los dos ensayos. Si la diferencia de los valores de viscosidad a 0,1 rad/s es mayor del 5 %, se considera que la muestra se ha degradado durante el ensayo de deformación y se descarta el resultado.
Relación de viscosidad de cizallamiento cero
Se define la relación de viscosidad a cizallamiento cero (ZSVR) como la relación de la viscosidad de cizallamiento cero (ZSV) del polímero de la invención con respecto a la ZSV de un material de polietileno lineal al peso molecular promedio en peso equivalente (Mw-gpc), como se muestra en la Ecuación 18, como se muestra en la Figura 18.
El valor r|0 (en Pa.s) se obtiene a partir del ensayo de deformación a 190 °C a través del método descrito anteriormente. Se sabe que la ZSV del polietileno lineal de r|0L tiene una dependencia de la ley de potencia de su Mw cuando el Mw está por encima del peso molecular crítico Mc. Un ejemplo de tal relación se describe en Karjala y col. (Annual Technical Conference - Society of Plastics Engineers (2008), 6 6 a, 887-891), como se muestra en la Ecuación 19, como se muestra en la Figura 19, para calcular los valores de ZSVR. Con referencia a la Ecuación 19, como se muestra en la Figura 19, el valor de Mw-gpc (g/mol) se determina mediante el uso del método de GPC, como se define inmediatamente a continuación en la presente memoria.
Determinación de Mw-gpc
A fin de obtener valores de Mw-gpc, el sistema cromatográfico consiste en un Modelo PL-210 de Polymer Laboratories o un Modelo PL-220 de Polymer Laboratories. Los compartimentos de columna y alimentador rotatorio se operan a 140 °C. Se usan tres columnas Mixed B de 10 ym de Polymer Laboratories con un disolvente de 1,2,4-triclorobenceno. Las muestras se preparan a una concentración de 0,1 g de polímero en 50 ml de disolvente. El disolvente usado para preparar las muestras contenía 200 ppm del antioxidante de hidroxitolueno butilado (BHT). Las muestras se prepararon mediante la agitación ligera durante 4 horas a 160 °C. El volumen de inyección usado es de 100 microlitros y el caudal es de 1,0 ml/min. Se lleva a cabo la calibración del conjunto de columnas de GPC con veintiún patrones de poliestireno de distribución estrecha de peso molecular adquiridos a través de Polymer Laboratories. Los pesos moleculares de pico de patrón de poliestireno se convierten en pesos moleculares de polietileno usando la Ecuación 20, como se muestra en la Figura 20.
Con referencia a la Ecuación 20, como se muestra en la Figura 20, M es el peso molecular, A tiene el valor de 0,4316 y B es igual a 1,0. Se determina un polinomio de tercer orden para construir la calibración logarítmica de peso molecular en función del volumen de elución. Los cálculos de pesos moleculares equivalentes de polietileno se realizan usando el soporte lógico Viscotek TriSEC Versión 3.0. La precisión del peso molecular promedio en peso Mw es excelente al < 2 , 6 %.
Caracterización del polímero. Se midieron las temperaturas de fundición (Tm) y de transición vítrea (Tg) de polímeros mediante calorimetría diferencial de barrido (Q2000 DSC, TA Instruments, Inc.). En primer lugar, se calentaron las muestras de temperatura ambiente a 200 °C usando la característica "cambiar a". Después de mantenerse a esta temperatura durante 4 min, se enfriaron las muestras hasta -90 °C a 10 °C/min, se mantuvieron durante 4 min y se calentaron después de nuevo hasta 200 °C. Se determinó la información de la distribución de peso molecular (Mw, Mn) mediante análisis en un cromatógrafo de permeación en gel de alta temperatura de dilución asistida por robótica, construido por Dow, tradicional (RAD-GPC). Se disolvieron las muestras de polímero durante 90 minutos a 160 °C a una concentración de 5-7 mg/ml en 1,2,4-triclorobenceno (TCB) estabilizado con 300 ppm de BHT en viales tapados mientras se agitaba. Después, estas se diluyeron a 1 mg/ml inmediatamente antes de que se inyectara una alícuota de 400 pl de la muestra. La GPC utilizó dos (2) columnas MIXED-B de 10 pm de gel de Polymer Labs PL (300 mm x 10 mm) a un caudal de 2,0 ml/minuto a 150 °C. La detección de muestras se realizó usando un detector PolyChar IR4 en modo de concentración. Se utilizó una calibración convencional de patrones de poliestireno (PS) estrecho, con unidades aparentes ajustadas a homopolietileno (PE) usando coeficientes de Mark-Houwink conocidos para PS y PE en TCB a esta temperatura. A fin de determinar la incorporación de 1-octeno, se disolvieron muestras de polímeros a una concentración de 30 mg/ml en 1,2,4-triclorobenceno a 160 °C durante 1 h mientras se agitaba. Se depositó una alícuota de 100 pl de cada solución de polímero/TCB en celdas individuales en una oblea de silicio tradicional a 160 °C en medio inerte de nitrógeno. Se mantuvo la oblea a 160 °C durante 45 minutos y, después, se retiró del calor y se dejó enfriar hasta temperatura ambiente. Después, se analizó la oblea usando un espectrómetro infrarrojo Nicolet Nexus 670 FT-IR ESP. Se determinó el % en moles de 1-octeno en cada muestra tomando una relación del área de CH3 (números de onda de 1.382,7-1.373,5) con respecto al área de CH2 (números de onda de 1.525-1.400) y normalizando a una curva de patrón generada a través de análisis de RMN de patrones de polímeros de etileno-co-1 -octeno.
Claims (2)
1. Un proceso de polimerización de etileno que comprende:
polimerizar etileno y 1 -octeno en presencia de uno o más primeros sistemas de catalizador y, opcionalmente, uno o más segundos sistemas de catalizador en un solo reactor, en donde el primer sistema de catalizador comprende;
(a) uno o más procatalizadores que comprenden un complejo de metal-ligando de Fórmula (I):

en donde:
M es titanio, circonio o hafnio, estando cada uno, de manera independiente, en un estado de oxidación formal de 2, 3 o 4; y n es un número entero de 0 a 3 y en donde cuando n es 0, X está ausente; y cada X, de manera independiente, es un ligando monodentado que es neutro, monoaniónico o dianiónico; o dos X se toman en conjunto para formar un ligando bidentado que es neutro, monoaniónico o dianiónico; y X y n se eligen de tal manera que el complejo de metal-ligando de Fórmula (I) sea, en general, neutro; y cada Z, de manera independiente, es O, S, N-hidrocarbilo (Ci-C40) o P-hidrocarbilo (Ci-C40); y L es hidrocarbileno (C3-C40) o heterohidrocarbileno (C3-C40), en donde el hidrocarbileno (C3-C40) tiene una parte que comprende una cadena principal de enlazador de 3 átomos de carbono a 10 átomos de carbono que enlaza los átomos de Z en la Fórmula (I) (al que está enlazado L) y el heterohidrocarbileno (C3-C40) tiene una parte que comprende una cadena principal de enlazador de 3 átomos a 10 átomos que enlaza los átomos de Z en la Fórmula (I), en donde cada uno de los 3 a 10 átomos de la cadena principal de enlazador de 3 átomos a 10 átomos del heterohidrocarbileno (C3-C40), de manera independiente, es un átomo de carbono O, S, S(O), S(O)2, Si(RC)2 , Ge(RC)2, P(RP), o N(RN), en donde, de manera independiente, cada RC es hidrocarbilo (C1-C30), cada RP es hidrocarbilo (C1-C30); y cada RN es hidrocarbilo (C1-C30) o está ausente; y
R1-24 se seleccionan del grupo que consiste en un hidrocarbilo (Ci-C40), heterohidrocarbilo (Ci-C40), Si(RC)3, Ge(RC)3, P(Rp)2, N(Rn)2, ORc, SRc, NO2, CN, CF3, RcS(O)-, RcS(O)2-, (Rc)2C=N-, RcC(O)O-, RcOC(O)-, RcC(O)N(R)-, (Rc)2NC(O)-, un átomo de halógeno, un átomo de hidrógeno y una combinación de los mismos; y en donde al menos R1, R16 o ambos comprenden la Fórmula (II) y preferiblemente R1 y R16 son iguales;

cuando R22 es H, entonces R19 es un hidrocarbilo (C1-C40); heterohidrocarbilo (C1-C40); Si(RC)3, Ge(RC)3, P(RP)2, N(Rn)2, ORc, SRc, NO2 , CN, CF3, RCS(O)-, RCS(O)2-, (RC)2C=N-, RCC(O)O-, RCOC(O)-, RCC(O)N(R)-, (R^NC(O)- o un átomo de halógeno;
cuando R19 es H, entonces R22 es un hidrocarbilo (C1-C40); heterohidrocarbilo (C1-C40); Si(RC)3 , Ge(RC)3, P(RP)2 , N(Rn)2, ORc, SRc, NO2 , CN, CF3, RCS(O)-, RcS(O)2-, (Rc)2C=N-, RcC(O)O-, RcOC(O)-, RcC(O)N(R)-, (Rc)2 NC(O)- o un átomo de halógeno;
preferiblemente, R22 y R19 son, ambos, un hidrocarbilo (C1-C40);
heterohidrocarbilo (C1-C40); Si(RC)3 , Ge(RC)3, P(RP)2, N(RN)2 , ORC, SRC, NO2 , CN, CF3 , RCS(O)-, RCS(O)2-, (Rc)2C=N-, RcC(O)O-, RcOC(O)-, RcC(O)N(R)-, (Rc)2NC(O)- o un átomo de halógeno; y
cuando R8 es H, entonces R9 es un hidrocarbilo (C1-C40); heterohidrocarbilo (C1-C40); Si(RC)3, Ge(RC)3, P(RP)2, N(Rn)2, ORc, SRc, NO2 , CN, CF3, RCS(O)-, RcS(O)2-, (Rc)2C=N-, RcC(O)O-, RcOC(O)-, RcC(O)N(R)-, (Rc)2 NC(O)- o
un átomo de halógeno;
cuando R9 es H, entonces R8 es un hidrocarbilo (C1-C40); heterohidrocarbilo (C1-C40); Si(RC)3 , Ge(RC)3 , P(Rp)2, N(Rn)2, ORc, SRc, NO2, CN, CF3, RCS(O)-, RcS(O)2-, (Rc)2C=N-, RcC(O)O-, RcOC(O)-, RcC(O)N(R)-, (Rc)2NC(O)- o un átomo de halógeno;
opcionalmente, dos o más grupos R (por ejemplo, de R9-15, R9'13, R9'12, R2'8, R4-8, R5-8) se pueden combinar en conjunto en estructuras de anillo, teniendo tales estructuras de anillo de 3 a 50 átomos en el anillo, excluyendo cualquier átomo de hidrógeno;
cada uno de los grupos arilo, heteroarilo, hidrocarbilo, heterohidrocarbilo, Si(RC)3, Ge(RC)3 , P(RP)2, N(RN)2, ORC, SRC, RCS(O)-, RcS(O)2-, (RC)2C=N-, RcC(O)O-, RcOC(O)-, RCC(O)N(R)-, (Rc)2NC(O)-, hidrocarbileno y heterohidrocarbileno, de manera independiente, está no sustituido o sustituido con uno o más sustituyentes de RS; cada RS, de manera independiente, es un átomo de halógeno, una sustitución de polifluoro, una sustitución de perfluoro, alquilo (C1-C18) no sustituido, F3C-, FCH2O-, F2HCO-, F3CO-, R3Si-, R3Ge-, RO-, RS-, RS(O)-, RS(O)2-, R2P-, R2N-, R2C=N-, NC-, RC(O)O-, ROC(O)-, RC(O)N(R)- o R2NC(O)-, o dos del RS se toman en conjunto para formar un alquileno (C1-C18) no sustituido, en donde cada R, de manera independiente, es un alquilo (C1-C18) no sustituido; y
(b) uno o más cocatalizadores; en donde la relación del número total de moles de uno o más complejos de metalligando de Fórmula (I) respecto al número total de moles de uno o más cocatalizadores es de 1:10.000 a 100:1, produciendo de este modo un copolímero de etileno/octeno que comprende del 0,5 al 3 por ciento en moles de unidades derivadas de octeno y del 97 al 99,5 por ciento en moles de unidades derivadas de etileno.
2. El proceso de polimerización de etileno según la reivindicación 1, en donde Z es O.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| US201261746173P | 2012-12-27 | 2012-12-27 | |
| PCT/US2013/073977 WO2014105412A1 (en) | 2012-12-27 | 2013-12-10 | An ethylene based polymer |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2735310T3 true ES2735310T3 (es) | 2019-12-17 |
Family
ID=49881079
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES13812379T Active ES2735310T3 (es) | 2012-12-27 | 2013-12-10 | Un polímero basado en etileno |
Country Status (10)
| Country | Link |
|---|---|
| US (1) | US20150337062A1 (es) |
| EP (1) | EP2938645B1 (es) |
| JP (1) | JP6653173B2 (es) |
| KR (1) | KR20150100844A (es) |
| CN (1) | CN104854144B (es) |
| AR (1) | AR094323A1 (es) |
| BR (1) | BR112015015544A2 (es) |
| ES (1) | ES2735310T3 (es) |
| SG (2) | SG11201505060QA (es) |
| WO (1) | WO2014105412A1 (es) |
Families Citing this family (33)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| SG11201505069VA (en) | 2012-12-27 | 2015-07-30 | Dow Global Technologies Llc | A polymerization process for producing ethylene based polymers |
| BR112015015548B1 (pt) | 2012-12-27 | 2021-05-18 | Dow Global Technologies Llc | processo de polimerização para a produção de polímeros a base de etileno |
| EP2938643B1 (en) * | 2012-12-27 | 2018-01-31 | Dow Global Technologies LLC | Catalyst systems for olefin polymerization |
| CN113201084A (zh) | 2013-06-28 | 2021-08-03 | 陶氏环球技术有限责任公司 | 使用卤化双苯基苯氧基催化剂控制聚烯烃的分子量 |
| WO2017004456A1 (en) * | 2015-06-30 | 2017-01-05 | Dow Global Technologies Llc | A polymerization process for producing ethylene based polymers |
| CN107787336B (zh) * | 2015-06-30 | 2021-05-28 | 陶氏环球技术有限责任公司 | 用于制备基于乙烯的聚合物的聚合方法 |
| BR112018006183B1 (pt) | 2015-09-30 | 2022-06-14 | Dow Global Technologies Llc | Pró-catalisador e processo de polimerização |
| BR112018005909B1 (pt) | 2015-09-30 | 2022-05-03 | Dow Global Technologies Llc | Pró-catalisador para a polimerização de etileno e processo de polimerização para a produção de polímeros à base de etileno |
| US10766975B2 (en) * | 2016-07-28 | 2020-09-08 | Exxonmobil Chemical Patents Inc. | Catalyst compositions and use thereof |
| SG11201900653RA (en) | 2016-07-29 | 2019-02-27 | Dow Global Technologies Llc | Silyl-bridged bis-biphenyl-phenoxy catalysts for olefin polymerization |
| BR112019001249B1 (pt) | 2016-07-29 | 2022-10-18 | Dow Global Technologies Llc | Elastômero multimodal, olefina termoplástica e método para produzir um elastômero multimodal |
| US10221260B2 (en) | 2016-07-29 | 2019-03-05 | Exxonmobil Chemical Patents Inc. | Phenolate transition metal complexes, production and use thereof |
| WO2018022279A1 (en) | 2016-07-29 | 2018-02-01 | Exxonmobil Chemical Patents Inc. | Phenolate transition metal complexes, production and use thereof |
| SG11201908414YA (en) * | 2017-03-15 | 2019-10-30 | Dow Global Technologies Llc | Catalyst system for multi-block copolymer formation |
| WO2018183056A1 (en) * | 2017-03-31 | 2018-10-04 | Dow Global Technologies Llc | Germanium-bridged bis-biphenyl-phenoxy catalysts for olefin polymerization |
| JP7173983B2 (ja) | 2017-03-31 | 2022-11-17 | ダウ グローバル テクノロジーズ エルエルシー | オレフィン重合のためのビス-ビフェニル-フェノキシ触媒 |
| CN110678488B (zh) * | 2017-06-20 | 2023-06-27 | 陶氏环球技术有限责任公司 | 用于烯烃聚合的联芳基苯氧基第iv族过渡金属催化剂 |
| WO2018236863A1 (en) * | 2017-06-20 | 2018-12-27 | Dow Global Technologies Llc | Biaryl phenoxy group iv transition metal catalysts for olefin polymerization |
| ES2911503T3 (es) | 2017-09-29 | 2022-05-19 | Dow Global Technologies Llc | Catalizadores de bis-fenil-fenoxi-poliolefina que tienen un ligando de metilentrialquilsilicio en el metal para mejorar la solubilidad |
| US11066489B2 (en) | 2017-09-29 | 2021-07-20 | Dow Global Technologies Llc | Bis-phenyl-phenoxy polyolefin catalysts having two methylenetrialkylsilicon ligands on the metal for improved solubility |
| US11242415B2 (en) | 2017-09-29 | 2022-02-08 | Dow Global Technologies Llc | Bis-phenyl-phenoxy polyolefin catalysts having an alkoxy- or amido-ligand on the metal for improved solubility |
| KR102100142B1 (ko) | 2017-12-21 | 2020-04-14 | 사빅 에스케이 넥슬렌 컴퍼니 피티이 엘티디 | 금속-리간드 착체, 이를 포함하는 에틸렌계 중합용 촉매 조성물 및 이를 이용한 에틸렌계 중합체의 제조방법 |
| WO2019123028A1 (ko) * | 2017-12-21 | 2019-06-27 | 사빅 에스케이 넥슬렌 컴퍼니 피티이 엘티디 | 금속-리간드 착체, 이를 포함하는 에틸렌계 중합용 촉매 조성물 및 이를 이용한 에틸렌계 중합체의 제조방법 |
| SG11202101332PA (en) | 2018-08-30 | 2021-04-29 | Dow Global Technologies Llc | Bis-phenyl-phenoxy polyolefin procatalysts having two anthracenyl ligands |
| US11066494B2 (en) | 2018-09-20 | 2021-07-20 | Exxonmobil Chemical Patents Inc. | Amine bridged anilide phenolate catalyst compounds |
| ES2967889T3 (es) * | 2019-10-01 | 2024-05-06 | Nova Chem Int Sa | Polimerización de alta productividad con catalizador de ligando de ariloxi éter |
| CA3150990A1 (en) * | 2019-10-01 | 2021-04-08 | Nova Chemicals Corporation | Control of unsaturation in polymers produced in solution process |
| KR20220097924A (ko) * | 2019-11-04 | 2022-07-08 | 다우 글로벌 테크놀로지스 엘엘씨 | 바이모달 촉매 시스템 |
| EP4097152B1 (en) | 2020-01-31 | 2025-09-03 | Dow Global Technologies LLC | Polymerization processes that include group iii and lanthanide bis-phenyl-phenoxy metal-ligand complexes and chain transfer agents |
| JP7627274B2 (ja) | 2020-01-31 | 2025-02-05 | ダウ グローバル テクノロジーズ エルエルシー | グループiii及びランタニドビス-フェニル-フェノキシ金属-配位子錯体 |
| CN115698104B (zh) | 2020-05-29 | 2025-06-20 | 陶氏环球技术有限责任公司 | 用于制备乙烯基共聚物的含有锗桥连双-联苯-苯氧基有机金属化合物的喷雾干燥的催化剂 |
| US20230303597A1 (en) * | 2020-12-28 | 2023-09-28 | Sabic Sk Nexlene Company Pte. Ltd. | Metal-ligand complex, catalyst composition for preparing ethylene-based polymer containing the same, and preparation method of ethylene -based polymer using the same |
| CA3207940A1 (en) * | 2021-02-15 | 2022-08-18 | Joseph F. DEWILDE | Biphenylphenol polymerization catalysts having improved kinetic induction times |
Family Cites Families (23)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US7163907B1 (en) | 1987-01-30 | 2007-01-16 | Exxonmobil Chemical Patents Inc. | Aluminum-free monocyclopentadienyl metallocene catalysts for olefin polymerization |
| US5153157A (en) | 1987-01-30 | 1992-10-06 | Exxon Chemical Patents Inc. | Catalyst system of enhanced productivity |
| US5064802A (en) | 1989-09-14 | 1991-11-12 | The Dow Chemical Company | Metal complex compounds |
| JP2545006B2 (ja) | 1990-07-03 | 1996-10-16 | ザ ダウ ケミカル カンパニー | 付加重合触媒 |
| US5721185A (en) | 1991-06-24 | 1998-02-24 | The Dow Chemical Company | Homogeneous olefin polymerization catalyst by abstraction with lewis acids |
| US5296433A (en) | 1992-04-14 | 1994-03-22 | Minnesota Mining And Manufacturing Company | Tris(pentafluorophenyl)borane complexes and catalysts derived therefrom |
| US5350723A (en) | 1992-05-15 | 1994-09-27 | The Dow Chemical Company | Process for preparation of monocyclopentadienyl metal complex compounds and method of use |
| US5372682A (en) | 1993-06-24 | 1994-12-13 | The Dow Chemical Company | Electrochemical preparation of addition polymerization catalysts |
| US5625087A (en) | 1994-09-12 | 1997-04-29 | The Dow Chemical Company | Silylium cationic polymerization activators for metallocene complexes |
| JP2000517349A (ja) | 1996-03-27 | 2000-12-26 | ザ ダウ ケミカル カンパニー | 分散した触媒活性剤による溶液重合法 |
| RU2178422C2 (ru) | 1996-03-27 | 2002-01-20 | Дзе Дау Кемикал Компани | Активатор катализаторов полимеризации олефинов, каталитическая система и способ полимеризации |
| US5783512A (en) | 1996-12-18 | 1998-07-21 | The Dow Chemical Company | Catalyst component dispersion comprising an ionic compound and solid addition polymerization catalysts containing the same |
| US6103657A (en) | 1997-07-02 | 2000-08-15 | Union Carbide Chemicals & Plastics Technology Corporation | Catalyst for the production of olefin polymers |
| US6696379B1 (en) | 1997-09-19 | 2004-02-24 | The Dow Chemical Company | Supported modified alumoxane catalyst activator |
| ES2379243T3 (es) | 1999-12-10 | 2012-04-24 | Dow Global Technologies Llc | Complejos de metal del grupo 4 sustituidos, catalizadores y procedimiento de polimerización de olefinas |
| US6897276B2 (en) * | 2002-04-24 | 2005-05-24 | Symyx Technologies, Inc. | Bridged bi-aromatic ligands, catalysts, processes for polymerizing and polymers therefrom |
| ES2534469T3 (es) | 2006-05-17 | 2015-04-23 | Dow Global Technologies Llc | Procedimiento de polimerización de polietileno en disolución a alta temperatura |
| WO2011109563A2 (en) * | 2010-03-02 | 2011-09-09 | Dow Global Technologies Llc | Ethylene-based polymer compositions |
| WO2011041696A1 (en) * | 2009-10-02 | 2011-04-07 | Dow Global Technologies Inc. | Block composites and impact modified compositions |
| US8686087B2 (en) * | 2009-10-02 | 2014-04-01 | Dow Global Technologies Llc | Block composites in soft compounds |
| CN102906129B (zh) * | 2010-05-17 | 2015-02-25 | 陶氏环球技术有限责任公司 | 选择性聚合乙烯的方法及其催化剂 |
| WO2011146044A1 (en) * | 2010-05-17 | 2011-11-24 | Dow Global Technologies Llc | Process for selectively polymerizing ethylene and catalyst therefor |
| US20120116034A1 (en) * | 2010-11-08 | 2012-05-10 | Dow Global Technologies, Inc. | Solution polymerization process and procatalyst carrier systems useful therein |
-
2013
- 2013-12-10 CN CN201380065848.6A patent/CN104854144B/zh active Active
- 2013-12-10 JP JP2015550431A patent/JP6653173B2/ja active Active
- 2013-12-10 ES ES13812379T patent/ES2735310T3/es active Active
- 2013-12-10 EP EP13812379.9A patent/EP2938645B1/en active Active
- 2013-12-10 SG SG11201505060QA patent/SG11201505060QA/en unknown
- 2013-12-10 BR BR112015015544A patent/BR112015015544A2/pt not_active Application Discontinuation
- 2013-12-10 WO PCT/US2013/073977 patent/WO2014105412A1/en not_active Ceased
- 2013-12-10 KR KR1020157019930A patent/KR20150100844A/ko not_active Ceased
- 2013-12-10 SG SG10201705303SA patent/SG10201705303SA/en unknown
- 2013-12-10 US US14/654,858 patent/US20150337062A1/en not_active Abandoned
- 2013-12-27 AR ARP130105080A patent/AR094323A1/es active IP Right Grant
Also Published As
| Publication number | Publication date |
|---|---|
| JP6653173B2 (ja) | 2020-02-26 |
| CN104854144A (zh) | 2015-08-19 |
| JP2016506443A (ja) | 2016-03-03 |
| CN104854144B (zh) | 2018-08-07 |
| EP2938645A1 (en) | 2015-11-04 |
| US20150337062A1 (en) | 2015-11-26 |
| KR20150100844A (ko) | 2015-09-02 |
| AR094323A1 (es) | 2015-07-29 |
| SG10201705303SA (en) | 2017-07-28 |
| WO2014105412A1 (en) | 2014-07-03 |
| BR112015015544A2 (pt) | 2017-07-11 |
| EP2938645B1 (en) | 2019-05-15 |
| SG11201505060QA (en) | 2015-07-30 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2735310T3 (es) | Un polímero basado en etileno | |
| ES2720207T3 (es) | Un proceso de polimerización para producir polímeros a base de etileno | |
| ES2665586T3 (es) | Sistemas catalizadores para polimerización de olefinas | |
| ES2731574T3 (es) | Un proceso de polimerización para producir polímeros a base de etileno | |
| ES2750524T3 (es) | Procedimiento para la polimerización de etileno y octeno | |
| US10239974B2 (en) | Ethylene-based polymers | |
| ES2811135T3 (es) | Un procedimiento de polimerización para producir polímeros a base de etileno |