ES2757591T3 - Estabilización de partículas virales - Google Patents
Estabilización de partículas virales Download PDFInfo
- Publication number
- ES2757591T3 ES2757591T3 ES15158279T ES15158279T ES2757591T3 ES 2757591 T3 ES2757591 T3 ES 2757591T3 ES 15158279 T ES15158279 T ES 15158279T ES 15158279 T ES15158279 T ES 15158279T ES 2757591 T3 ES2757591 T3 ES 2757591T3
- Authority
- ES
- Spain
- Prior art keywords
- virus
- viral
- sucrose
- lyophilization
- adenovirus
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
Classifications
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K35/00—Medicinal preparations containing materials or reaction products thereof with undetermined constitution
- A61K35/66—Microorganisms or materials therefrom
- A61K35/76—Viruses; Subviral particles; Bacteriophages
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
- A61K39/12—Viral antigens
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/16—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing nitrogen, e.g. nitro-, nitroso-, azo-compounds, nitriles, cyanates
- A61K47/18—Amines; Amides; Ureas; Quaternary ammonium compounds; Amino acids; Oligopeptides having up to five amino acids
- A61K47/183—Amino acids, e.g. glycine, EDTA or aspartame
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/20—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite containing sulfur, e.g. dimethyl sulfoxide [DMSO], docusate, sodium lauryl sulfate or aminosulfonic acids
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K47/00—Medicinal preparations characterised by the non-active ingredients used, e.g. carriers or inert additives; Targeting or modifying agents chemically bound to the active ingredient
- A61K47/06—Organic compounds, e.g. natural or synthetic hydrocarbons, polyolefins, mineral oil, petrolatum or ozokerite
- A61K47/26—Carbohydrates, e.g. sugar alcohols, amino sugars, nucleic acids, mono-, di- or oligo-saccharides; Derivatives thereof, e.g. polysorbates, sorbitan fatty acid esters or glycyrrhizin
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P31/00—Antiinfectives, i.e. antibiotics, antiseptics, chemotherapeutics
- A61P31/12—Antivirals
- A61P31/14—Antivirals for RNA viruses
- A61P31/16—Antivirals for RNA viruses for influenza or rhinoviruses
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P37/00—Drugs for immunological or allergic disorders
- A61P37/08—Antiallergic agents
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N7/00—Viruses; Bacteriophages; Compositions thereof; Preparation or purification thereof
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N7/00—Viruses; Bacteriophages; Compositions thereof; Preparation or purification thereof
- C12N7/02—Recovery or purification
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K39/00—Medicinal preparations containing antigens or antibodies
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/14—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles
- A61K9/19—Particulate form, e.g. powders, Processes for size reducing of pure drugs or the resulting products, Pure drug nanoparticles lyophilised, i.e. freeze-dried, solutions or dispersions
-
- C—CHEMISTRY; METALLURGY
- C12—BIOCHEMISTRY; BEER; SPIRITS; WINE; VINEGAR; MICROBIOLOGY; ENZYMOLOGY; MUTATION OR GENETIC ENGINEERING
- C12N—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA
- C12N2710/00—MICROORGANISMS OR ENZYMES; COMPOSITIONS THEREOF; PROPAGATING, PRESERVING, OR MAINTAINING MICROORGANISMS; MUTATION OR GENETIC ENGINEERING; CULTURE MEDIA dsDNA viruses
- C12N2710/00011—Details
- C12N2710/10011—Adenoviridae
- C12N2710/10311—Mastadenovirus, e.g. human or simian adenoviruses
- C12N2710/10351—Methods of production or purification of viral material
Landscapes
- Health & Medical Sciences (AREA)
- Life Sciences & Earth Sciences (AREA)
- Chemical & Material Sciences (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Veterinary Medicine (AREA)
- Public Health (AREA)
- Pharmacology & Pharmacy (AREA)
- Engineering & Computer Science (AREA)
- Chemical Kinetics & Catalysis (AREA)
- General Chemical & Material Sciences (AREA)
- Virology (AREA)
- Organic Chemistry (AREA)
- Epidemiology (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Oil, Petroleum & Natural Gas (AREA)
- Microbiology (AREA)
- Genetics & Genomics (AREA)
- Wood Science & Technology (AREA)
- Zoology (AREA)
- Biochemistry (AREA)
- Immunology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- Molecular Biology (AREA)
- Mycology (AREA)
- General Engineering & Computer Science (AREA)
- Biomedical Technology (AREA)
- Biotechnology (AREA)
- Oncology (AREA)
- Communicable Diseases (AREA)
- Pulmonology (AREA)
- Proteomics, Peptides & Aminoacids (AREA)
- Medicinal Preparation (AREA)
- Medicines Containing Antibodies Or Antigens For Use As Internal Diagnostic Agents (AREA)
- Micro-Organisms Or Cultivation Processes Thereof (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Medicines Containing Material From Animals Or Micro-Organisms (AREA)
- Food Preservation Except Freezing, Refrigeration, And Drying (AREA)
Abstract
Un método para conservar partículas virales durante liofilización que comprende: (a) proporcionar una solución acuosa de: (i) partículas virales de Adenoviridae, Orthomyxoviridae, Paramyxoviridae, Parvoviridae, Picornaviridae, Poxviridae, Herpesviridae, Togaviridae, Flaviviridae, Retroviridae, Filoviridae, Papillomaviridae, Caliciviridae, Coronaviridae, Reoviridae o Hepadnaviridae, (ii) sacarosa, o sacarosa y rafinosa, o manitol, a una concentración total de azúcar de 0,1 M a 3 M, (iii) de 0,1 a 1,5 M de N,N-dimetilglicina o N,N,N-trimetilglicina, o una sal o éster fisiológicamente aceptable de la misma, y (iv) de 0,1 a 1,5 M de metilsulfonilmetano; y (b) liofilizar la solución para formar una composición que incorpora dichas partículas virales.
Description
DESCRIPCIÓN
Estabilización de partículas virales
Campo de la invención
La presente invención hace referencia a la estabilización de partículas virales.
Antecedentes de la invención
Algunas moléculas biológicas son suficientemente estables, de tal manera que pueden ser aisladas, purificadas y a continuación almacenadas en solución a temperatura ambiente. Sin embargo, esto no es posible para muchos materiales, y por esta razón se han intentado técnicas que implican almacenaje a baja temperatura, adición de estabilizadores o crioprotectores, liofilización, desecación al vacío y secado al aire para asegurar la conservación durante el almacenamiento.
A pesar de la disponibilidad de estas técnicas, algunos materiales biológicos aún muestran niveles insatisfactorios de estabilidad durante el almacenamiento, y algunas técnicas acarrean inconvenientes y costes añadidos. Por ejemplo, el transporte y el almacenamiento refrigerado resulta costoso, y cualquier ruptura en el control de la temperatura puede dar como resultado una reducción de la eficacia de la molécula biológica. Además, el transporte refrigerado a menudo no está disponible durante el transporte de medicinas en países en vías de desarrollo.
Además, las tensiones de la criodesecación o liofilización pueden ser muy perjudiciales para algunos materiales biológicos. La liofilización de medicamentos biológicos implica la congelación de soluciones o suspensiones de biomateriales termosensibles, seguido de desecación primaria y secundaria. La técnica está basada en la sublimación del agua a temperaturas bajo cero en vacío sin fusión de la solución. La liofilización representa una etapa clave para la fabricación de productos farmacéuticos de proteínas sólidas y vacunas. La tasa de difusión de vapor de agua del biomaterial congelado es muy baja y por lo tanto, el proceso consume mucho tiempo. Adicionalmente, tanto la etapa de congelación como la de desecación introducen tensiones que son capaces de desdoblar o desnaturalizar las proteínas.
La patente WO 90/05182 describe un método para proteger las proteínas contra la desnaturalización durante la desecación. El método comprende las etapas de mezclar una solución acuosa de la proteína con un polielectrolito catiónico y poliol cíclico y eliminar el agua de la solución. Dietilaminoetil-Dextrano (DEAE-dextrano) y chitosán son los polielectrolitos catiónicos preferidos, aunque la polietilenimina se menciona también como adecuada.
La patente WO-A-2006/0850082 describe un producto desecado o conservado que comprende un azúcar, un material cargado tal como una proteína de histona y un componente biológico termosensible o de desecación. El azúcar forma una matriz de sólido amorfo. Sin embargo, la histona puede tener consecuencias inmunológicas si el componente biológico conservado se administra a humanos o animales.
La patente WO 2008/114021 describe un método para conservar partículas virales. El método comprende desecar una solución acuosa de uno o más azúcares, una polietilenimina y las partículas virales para formar una matriz de sólido amorfo que comprende las partículas virales. La solución acuosa contiene polietilenimina a una concentración de 15mM o menos en base a la masa molecular media numérica (Mn) de la polietilenimina y la concentración de azúcar o, si está presente más de un azúcar, la concentración de azúcar total es mayor de 0,1M.
El documento WO2008058035 enseña la liofilización de partículas VEE en presencia de un sacárido como sacarosa y de un plastificante como DMSO.
Resumen de la invención
Los presentes inventores han observado que las preparaciones virales se conserva de manera estable mediante (a) N,N-dimetilglicina o N,N,N-trimetilglicina, o sales o ésteres fisiológicamente aceptables de la misma, (b) metilsulfonilmetano y (c) sacarosa, o sacarosa y rafinosa, o manitol, durante la liofilización. La actividad vírica se conservó después de una prueba de exposición térmica posterior. La actividad vírica se conservó también durante pruebas de estabilidad a largo plazo. La actividad vírica puede también ser conservada en solución acuosa previamente a la desecación. Los virus fueron protegidos contra daños causados por liofilización.
Por consiguiente, la presente invención proporciona un método para conservar partículas virales durante la liofilización que comprende:
(a) proporcionar una solución acuosa de:
(i) partículas virales de Adenoviridae, Orthomyxoviridae, Paramyxoviridae, Parvoviridae, Picornaviridae, Poxviridae, Herpesviridae, Togaviridae, Flaviviridae, Retroviridae, Filoviridae, Papillomaviridae, Caliciviridae,
Coronaviridae, Reoviridae o Hepadnaviridae,
(ii) sacarosa, o sacarosa y rafinosa, o manitol, a una concentración de azúcar total de 0,1 M a 3 M,
(iii) de 0,1 a 1,5 M de N,N-dimetilglicina o N,N,N-trimetilglicina, o una sal o éster fisiológicamente aceptable de la misma, y
(iv) de 0,1 a 1,5 M de metilsulfonilmetano; y
(b) liofilizar la solución para formar una composición que incorpora dichas partículas virales.
La invención además comprende una composición que se encuentra sólida y liofilizada, que comprende partículas virales de Herpesviridae, Togaviridae, Flaviviridae, Retroviridae, Filoviridae, Papillomaviridae, Caliciviridae, Coronaviridae, Reoviridae, Hepadnaviridae, Adenoviridae, Orthomyxoviridae, Paramyxoviridae, Parvoviridae, Picornaviridae o Poxviridae y que se puede obtener mediante un método de la invención.
Breve descripción de las Figuras
La figura 1 muestra los resultados obtenidos en el Ejemplo de referencia 1. Se evaluó la capacidad de un excipiente para ayudar al adenovirus a resistir una variación cíclica entre 37°C y -20°C. Se utilizó dimetilsulfona (también denominada metilsulfonilmetano, MSM) como excipiente. Resumen del valor de p: **=p<0,01, *=p<0,05. Las barras de error muestran el error estándar de la media (n = 4).
La figura 2A muestra la temperatura establecida para la temperatura de almacenamiento del liofilizador VirTis Advantage utilizado en varios de los Ejemplos.
La figura 2B muestra la temperatura del condensador del liofilizador VirTis Advantage utilizado en varios de los Ejemplos.
La figura 3 muestra los resultados del experimento del Ejemplo de referencia 2 que investigó el efecto de los azúcares y el MSM sobre la conservación de adenovirus durante la liofilización. Las barras de error mostradas son el error estándar de la media (n = 3).
La figura 4 muestra los resultados obtenidos en el Ejemplo de referencia 3 de la infectividad del adenovirus analizada inmediatamente después de la descongelación, además de la de aquellas muestras liofilizadas después de la formulación con TMG (trimetilglicina) con o sin azúcares. La actividad adenoviral se expresó como pfu/ml, tal como se evaluó mediante el conteo de células positivas para la expresión de GFP. Las barras de error mostradas son el error estándar de la media (n = 3).
La figura 5 demuestra las condiciones de liofilización utilizadas en el Ejemplo de referencia 4.
La figura 6 muestra los gráficos de barra que demuestran los títulos de virus de muestras reconstituidas utilizadas en el Ejemplo de referencia 4 a continuación de la prueba de exposición térmica a 4°C o 37°C durante 7 días. También se muestra el título de partida del virus de entrada. Las barras de error representan el error estándar de la media (n=3).
La figura 7 muestra los resultados obtenidos en el Ejemplo de referencia 5 para muestras de adenovirus que se sometieron a prueba inmediatamente después de la descongelación ("Pre-Liofilización"), además de los de las muestras que fueron formuladas en PBS (solución tampón de fosfatos) a concentraciones de DMG de 0,00M, 0,07M, 0,23M y 0,70M, con o sin azúcares y que son posteriormente liofilizadas. La actividad del adenovirus expresada como pfu/ml fue evaluada mediante conteo de células positivas para la expresión de la GFP (Proteína verde fluorescente). Las barras de error mostradas son el error estándar de la media (n = 3).
La figura 8 muestra las condiciones de liofilización utilizadas en los Ejemplos de referencia 5 y 6.
La figura 9 muestra los resultados obtenidos en el Ejemplo de referencia 6 para muestras de adenovirus sometidas a prueba inmediatamente después de la descongelación, además de aquellas muestras liofilizadas después de la formulación con DMG con o sin azúcares y posteriormente sometidas a prueba de exposición térmica. (A) Actividad del adenovirus después de la liofilización y el almacenamiento a 4°C durante 7 días. (B) Actividad del adenovirus después de la liofilización y la prueba de exposición térmica a 37°C durante 7 días. Las barras de error indican el estándar de la media; n = 3 a menos que se exprese de otro modo.
La figura 10 muestra las temperaturas de almacenamiento, temperaturas del condensador y condiciones de vacío durante la liofilización en el liofilizador VirTis Advantage en el Ejemplo 7.
La figura 11 muestra los resultados obtenidos en el Ejemplo de referencia 7. La actividad del adenovirus expresada como pfu/ml se evaluó mediante conteo de las células positivas para GFP. Las barras de error indican el estándar de la media (n = 2). Se analizó la significancia utilizando un ANNOVA de una vía seguido de un post test de Bonferroni. Los resúmenes del valor de p son *=p<0,05 y **=p<0,01.
La figura 12 muestra la apariencia de las tortas liofilizadas obtenidas en el Ejemplo de referencia 8.
La figura 13 describe los resultados obtenidos en el Ejemplo de referencia 9. FD indica liofilización. Los viales que contienen adenovirus y PBS solo mostraron una pérdida mucho mayor de título de virus en comparación con los viales que contienen adenovirus, manitol y DMG. Las barras de error mostradas son el error estándar de la media (n = 2). Se analizó la significancia utilizando un ANOVA de una vía seguido de un post-test de Bonferroni. Todos los valores se compararon con el título de la solución madre o stock. Los resúmenes del valor de p son **=p<0,01 y ***=p<0,001.
La figura 14 muestra los resultados obtenidos en el Ejemplo 10 en el que se evaluó la capacidad de once formulaciones para estabilizar adenovirus a través de liofilización y prueba de exposición térmica.
La figura 15 muestra los resultados obtenidos en el Ejemplo 11 en el que se evaluó la capacidad de once formulaciones para estabilizar MVA a través de liofilización y prueba de exposición térmica.
La figura 16 muestra una representación en 3D del espacio de diseño en el Ejemplo de referencia 12. Las esferas representan las formulaciones dentro del espacio de diseño en el que fueron sometidas a prueba. Este diseño es un diseño de cribado factorial completo de tres factores.
Las figuras 17 y 18 muestran el programa de liofilización utilizado en el Ejemplo de referencia 12 y las lecturas de temperatura de los sensores durante ese programa.
La figura 19 muestra un gráfico de probabilidad normal residual para los datos de las formulaciones que contienen DMG en el Ejemplo de referencia 12.
La figura 20 muestra los coeficientes (efectos) de retención de los datos modelados de las formulaciones que contienen DMG en el Ejemplo de referencia 12. Las barras de error indican significancia si no cruzan el origen. La figura 21 muestra los coeficientes (efectos) de retención de los datos modelados de las formulaciones que contienen SMM en el Ejemplo de referencia 12. Las barras de error indican significancia si no cruzan el origen. La figura 22 muestra un gráfico de probabilidad normal residual para los datos de las formulaciones que contienen SMM en el Ejemplo de referencia 12.
La figura 23 muestra los coeficientes (efectos) de retención de los datos modelados de las formulaciones que contienen SMM en el Ejemplo de referencia 12 después de la inclusión de un término de 2° orden no específico. Las barras de error indican significancia si no cruzan el origen.
La figura 24 muestra un gráfico de probabilidad normal residual para los datos de las formulaciones que contienen SMM en el Ejemplo de referencia 12.
La figura 25 muestra los coeficientes (efectos) de retención de los datos modelados de las formulaciones que contienen TMG en el Ejemplo de referencia 12. Las barras de error indican significancia si no cruzan el origen. La figura 26 muestra un gráfico de probabilidad normal residual para los datos de las formulaciones que contienen TMG en el Ejemplo de referencia 12.
La figura 27 muestra una representación en 3D del espacio de diseño en el Ejemplo de referencia 13. Las esferas representan las formulaciones dentro del espacio de diseño en el que se sometieron a prueba. Este diseño es un diseño de superficie de respuesta o RSM de Doehlert.
La figura 28 muestra el programa de liofilización utilizado en el Ejemplo de referencia 13.
La figura 29 resume diversas estadísticas para el modelo obtenidas a partir de los datos en el Ejemplo de referencia 13.
La figura 30 muestra términos retenidos en el modelo en el Ejemplo de referencia 13 después del ajuste fino. Las barras de error que no cruzan el origen indican un factor significativo a un nivel de fiabilidad del 95%.
La figura 31 muestra un gráfico de superficie de respuesta del título viral previsto en las formulaciones de DMG y sacarosa, utilizando el modelo del Ejemplo de referencia 13 a tres niveles diferentes de rafinosa.- “Bajo” = rafinosa a 0mM, "Medio" = rafinosa a 150mM, "Alto" = rafinosa a 300mM.
La figura 32 muestra los ajustes y resultados de una predicción óptima basada en el modelo de los datos del Ejemplo de referencia 13 generados utilizando simulaciones de Monte-Carlo. Las óptimas previstas destacadas en este modelo son las concentraciones de sacarosa = 0,5M, DMG= 1M, rafinosa. =150mM.
Las figuras 33A y 33B muestran un gráfico de la región óptima de los datos del Ejemplo de referencia 13. Los gráficos se encuentran en los niveles estáticos de rafinosa = 0, 150, 272, 300mM. La variable representada es el título recuperado (pfu/ml). La figura 33A es un gráfico de contorno en el que una cruz marca el óptimo previsto. La figura 33B es una región del gráfico idéntica que destaca la región del modelo en la que la actividad recuperada prevista es mayor que o igual a la actividad inicial.
La figura 34 muestra el programa de liofilización utilizado en el Ejemplo de referencia 14.
La figura 35 muestra la actividad vírica recuperada en el Ejemplo de referencia 14 como un porcentaje del título de partida en la semana 15 posterior a la liofilización. Las barras de error son el error estándar de la media (n=2). La figura 36 muestra la actividad vírica recuperada en el tiempo a la temperatura de estabilidad acelerada (+25°C) en el Ejemplo de referencia 14.
La figura 37 muestra la actividad vírica recuperada en el tiempo en la temperatura de prueba de la tensión (+37°C) en el Ejemplo de referencia 14.
La figura 38 muestra una representación en 3D del espacio de diseño en el Ejemplo de referencia 15. Las esferas representan formulaciones dentro del espacio de diseño en el que se someten a prueba. Este es un diseño RSM de Doehlert.
La figura 39 muestra las condiciones de liofilización utilizadas en el Ejemplo de referencia 15.
La figura 40 resume la estadística del modelo en el Ejemplo de referencia 15 utilizada para representar los datos. La figura 41 muestra términos retenidos en el modelo del Ejemplo de referencia 15 después del ajuste fino. Las barras de error que no cruzan el origen indican un factor significativo en un nivel de fiabilidad del 95%.
La figura 42 muestra gráficos de contorno del título viral recuperado (TCID50/ml) con distintas formulaciones ene l Ejemplo de referencia 15.
La figura 43 muestra una representación del espacio de diseño en el Ejemplo de referencia 16. Los círculos numerados representan las formulaciones dentro del espacio de diseño en el que se someten a prueba.
La figura 44 muestra las condiciones de liofilización utilizadas en el Ejemplo de referencia 16.
La figura 45 resume la estadística del modelo en el Ejemplo de referencia 16 utilizada para representar los datos. La figura 46 muestra los términos retenidos en el modelo en el Ejemplo de referencia 16 después del ajuste fino. Las barras de error que no cruzan el origen indican un factor significativo en un nivel de fiabilidad del 95%.
La figura 47 muestra un gráfico de respuesta de superficie del título viral recuperado previsto en formulaciones de DMG y manitol utilizando el modelo del Ejemplo de referencia 16.
La figura 48 muestra una captura de pantalla de los ajustes y resultados de las predicciones óptimas basadas en
el modelo de los datos en el Ejemplo de referencia 16, generado utilizando simulaciones Monte-Carlo. La iteración 48 resaltada en gris es la formulación óptima (1,0107M DMG).
Descripción detallada de la invención
Resumen
La presente invención hace referencia a la conservación de partículas virales mediante (a) N,N-dimetilglicina o N,N,N-trimetilglicina, o sales o ésteres fisiológicamente aceptables de la misma, (b) metilsulfonilmetano y (c) sacarosa, o sacarosa y rafinosa, o manitol. Las partículas virales se ponen en contacto con (a) N,N-dimetilglicina o N,N,N-trimetilglicina, o sales o ésteres fisiológicamente aceptables de la misma, (b) metilsulfonilmetano y (c) sacarosa, o sacarosa y rafinosa, o manitol en una solución acuosa, y la solución resultante en la que las partículas virales están presentes se liofiliza a continuación para formar una composición que incorpora las partículas virales.
Las partículas virales pueden, por lo tanto, mezclarse con una solución acuosa (“mezcla de conservación”) de (a) N,N-dimetilglicina o N,N,N-trimetilglicina, o sales o ésteres fisiológicamente aceptables de la misma, (b) metilsulfonilmetano y (c) sacarosa, o sacarosa y rafinosa, o manitol. La solución resultante se liofiliza a continuación para formar una composición que incorpora las partículas virales. La composición liofilizada puede tomar la forma de una torta o polvo. La torta puede triturarse hasta formar un polvo si se requiere.
La invención permite que la estructura y función del virus sea conservada durante la etapa de liofilización. La actividad del virus a continuación de la desecación puede ser, por tanto, mantenida. La presencia de (a) N,N-dimetilglicina o N,N,N-trimetilglicina, o sales o ésteres fisiológicamente aceptables de la misma, (b) metilsulfonilmetano y (c) sacarosa, o sacarosa y rafinosa, o manitol, permite la conservación de la actividad viral.
Las partículas virales conservadas demuestran resistencia térmica mejorada, lo que permite una extensión de la vida en almacenamiento, facilidad de almacenaje y transporte y obviar la necesidad de una cadena de frío para su distribución. La invención puede, por tanto, proporcionar protección como un crioprotector (protección contra los daños por liofilización), lioprotector (protector durante la liofilización) y/o termoprotector (protección contra temperaturas superiores o inferiores a 4°C).
Además, las partículas virales se conservan en la solución acuosa previamente a la etapa de liofilización. Esto permite que la solución acuosa sea almacenada después de su preparación, hasta el mismo momento en que la etapa de desecación puede ser realizada, sin una pérdida indebida de la actividad viral.
Partículas virales
Las partículas virales utilizadas en la presente invención pueden ser virus completos tales como virus vivos, virus muertos, virus vivos atenuados, virus inactivos tales como virus químicamente inactivados o virus virulentos o no virulentos. Un virus vivo es capaz de infectar y replicarse dentro de la célula hospedadora. Un virus muerto está inactivado y no es capaz de replicarse dentro de la célula hospedadora. Las partículas pueden ser partículas similares a virus (VLP, por sus siglas en inglés) o nucleocápsides. El virus puede ser infeccioso para las células procariotas o eucariotas. El virus puede ser un virus humano o animal.
La partícula viral es, o se obtiene de, un virus de las siguientes familias:
- Adenoviridae tal como un adenovirus humano o adenovirus no humano, por ejemplo adenovirus humano A, B, C, D, E o F incluyendo los serotipos humanos Ad5, Ad2, Ad4, Ad6, Ad7, Ad11, Ad14, Ad24, Ad26, Ad35 y Ad36; - Caliciviridae tal como el virus Norwalk;
- Coronaviridae tal como coronavirus humano 299E u OC43 y SARS-coronavirus;
- Filoviridae tal como virus del ébola;
- Flaviviridae tal como virus de la fiebre amarilla, virus del Nilo occidental, virus dengue, virus de la hepatitis C; - Hepadnaviridae tal como virus de la hepatitis B;
- Herpesviridae tal como virus del herpes simple, por ejemplo, VHS1 o VHS2, herpesvirus humano 1, 3, 4, 5 o 6; - Orthomyxoviridae tal como el influenzavirus A, B, C incluyendo, pero sin limitarse a, los serotipos H1N1, H2N2, H3N2, H5N1, H7N7, H1N2, H9H2, H7N2, H7N3 y N10N7 del virus de la gripe A;
- Papillomaviridae tal como virus del papiloma humano;
- Paramyxoviridae tal como el virus parainfluenza humano 1, virus del sarampión y virus de las paperas;
- Parvoviridae tal como los virus adeno-asociados;
- Picornaviridae tal como poliovirus humano, virus de la fiebre aftosa (incluyendo los serotipos O, A, C, SAT-1, SAT-2, SAT-3 y Asia-1);
- Poxviridae tal como el virus vaccinia, virus de la viruela y virus de la viruela aviar (diftero-viruela aviar);
- Reoviridae tal como grupo del virus de la lengua azul;
- Retroviridae tal como lentivirus incluyendo virus de la inmunodeficiencia humana 1 y 2; y
- Togaviridae tal como virus de la rubéola.
En una realización preferida, la partícula viral puede ser o proceder de un virus Adenoviridae, Orthomyxoviridae, Paramyxoviridae, Parvoviridae, Picornaviridae o Poxviridae. En un modo de realización particularmente preferido, la partícula viral puede ser o puede obtenerse de un adenovirus, virus vaccinia, virus de la gripe, o virus del sarampión. El virus puede ser el virus Vaccinia modificado de Ankara (MVA) o una partícula viral obtenida a partir del MVA. Las partículas similares a virus (las VLP) incluyen proteínas virales obtenidas a partir de las proteínas estructurales de un virus, pero carecen del ácido nucleico viral. Cuando son sobre-expresadas, estas proteínas estructurales virales se auto-agrupan de forma espontánea formando partículas. Las VLP son incompetentes para la replicación. En algunos modos de realización, las VLP son proteínas virales incluidas en el interior de una bicapa lipídica. Ejemplos de VLP incluyen VLP obtenidas de fagos, VLP de proteína de la cápside mayor L1 del virus del papiloma humano (HPV), VLP de proteína de la cápside del virus Norwalk y VLP agrupados a partir de las proteínas estructurales del virus de la gripe tales como la proteína M1, proteína hemaglutinina HA y proteína neuraminidasa N1.
Las partículas virales pueden prepararse utilizando técnicas estándar bien conocidas para los expertos en el arte. Por ejemplo, un virus puede prepararse infectando células hospedadoras cultivadas con la cepa del virus que se va a utilizar, permitiendo que la infección progrese de tal forma que el virus se replique en las células cultivadas y que pueda ser liberado mediante métodos estándar conocidos en el arte para cosechar y purificar virus.
N,N-dimetilglicina o N,N,N-trimetilglicina o sales o ésteres fisiológicamente aceptable de la misma
La N,N-dimetilglicina o N,N,N-trimetilglicina o compuesto de la fórmula (II) puede estar presente como una sal o éster fisiológicamente aceptable de la misma.
La sal es habitualmente una sal con un ácido fisiológicamente aceptable y por tanto incluye aquellas formadas con un ácido inorgánico, tal como el ácido clorhídrico o sulfúrico, o un ácido orgánico tal como ácido cítrico, tartárico, málico, maleico, mandélico, fumárico o metanosulfónico. Se prefiere la sal clorhidrato.
El éster es habitualmente un éster de alquilo C1-C6, preferiblemente éster de alquilo C1-C4. El éster puede ser, por lo tanto, el éster de metilo, etilo, propilo, isopropilo, butilo, isobutilo o terc-butilo. Se prefiere el éster de etilo.
Tal como se utiliza en la presente patente, un grupo alquilo C1-C6 es preferiblemente un grupo alquilo C1-C4. Grupos alquilo preferidos se seleccionan de metilo, etilo, propilo, isopropilo, butilo, isobutilo y terc-butilo. Son particularmente preferidos metilo y etilo.
Para evitar cualquier duda, las definiciones incluyen compuestos en los que el anión carboxilato está protonado para proporcionar -COOH y el catión amonio está asociado con un anión farmacéuticamente aceptable. Además, para evita dudas, los compuestos definidos anteriormente pueden ser utilizados en cualquier forma tautomérica o enantiomérica.
Azúcares
Los azúcares usados en la presente invención son sacarosa, o sacarosa y rafinosa, o manitol. Cuando el manitol se utiliza, pueden obtenerse tortas de apariencia mejorada en la liofilización.
La presencia de azúcar puede actuar para mejorar la estabilidad. La adición de azúcar puede también proporcionar otros beneficios tales como una torta de liofilización modificada y una solubilidad mejorada para una reconstitución más rápida. Cuando se utiliza un azúcar, el azúcar es preferiblemente manitol.
La conservación de la actividad viral es particularmente efectiva cuando dos o más azúcares que son sacarosa y rafinosa se utilizan en la mezcla de conservación. La sacarosa es un disacárido de la glucosa y la fructosa. La rafinosa es un trisacárido compuesto de galactosa, fructosa y glucosa.
Procedimiento de conservación
En la presente invención, se deseca una solución acuosa que comprende las partículas virales, con (a) N,N-dimetilglicina o N,N,N-trimetilglicina, o sales o ésteres fisiológicamente aceptables de la misma, (b) metilsulfonilmetano y (c) sacarosa, o sacarosa y rafinosa, o manitol se liofiliza. Puede utilizarse cualquier solución acuosa adecuada. La solución puede estar tamponada. La solución puede ser una solución HEPES, solución tampón de fosfato, tampón Tris o de agua pura.
La solución puede tener un pH de 2 hasta aproximadamente 12 y puede ser una solución tampón. La solución puede ser tamponada con un tampón HEPES, tampón de fosfato, tampón Tris, tampón de citrato de sodio, tampón de bicina (es decir, tampón de N,N-bis(2-hidroxietil) glicina) o tampón MOPS (es decir, tampón de ácido 3-(N-morfolino) propanosulfónico). La solución puede o no contener NaCl. La solución puede por tanto ser una solución tampón salina de citrato de sodio (SSC).
Generalmente, una preparación de las partículas virales se mezcla con la mezcla de conservación, es decir, con una solución acuosa de un compuesto (a) N,N-dimetilglicina o N,N,N-trimetilglicina, o sales o ésteres fisiológicamente aceptables de la misma, (b) metilsulfonilmetano y (c) sacarosa, o sacarosa y rafinosa, o manitol. La mezcla de conservación puede en sí misma estar tamponada. Puede ser una solución HEPES, solución tampón de fosfato, tampón Tris o de agua pura.
De forma alternativa, la solución acuosa puede consistir habitualmente, o consistir esencialmente, en partículas virales, (a) N,N-dimetilglicina o N,N,N-trimetilglicina, o sales o ésteres fisiológicamente aceptables de la misma, (b) metilsulfonilmetano y (c) sacarosa, o sacarosa y rafinosa, o manitol.
Las concentraciones de (a) N,N-dimetilglicina o N,N,N-trimetilglicina, o sales o ésteres fisiológicamente aceptables de la misma, (b) metilsulfonilmetano y (c) sacarosa, o sacarosa y rafinosa, o manitol, pueden ser determinadas mediante experimentación rutinaria. También pueden seleccionarse de este modo las concentraciones optimizadas que dan como resultado la mejor estabilidad. La N,N-dimetilglicina o N,N,N-trimetilglicina, o sales o ésteres fisiológicamente aceptables de la misma, y (b) metilsulfonilmetano puede actuar de manera sinérgica para mejorar la estabilidad.
La concentración de azúcar (es decir, sacarosa, o sacarosa y rafinosa, o manitol) es de al menos 0,1M, al menos 0,2M o al menos 0,5M hasta 3M, 2,5M o 2M. La concentración de azúcar es preferentemente de 0,2M a 2M. De forma alternativa, la concentración de azúcar o la concentración de azúcar total si está presente más de un azúcar, puede encontrarse en un rango de 0,15M a 2M o de 0,2M a 1M.
La concentración de la N,N-dimetilglicina o N,N,N-trimetilglicina, o sales o ésteres fisiológicamente aceptables de la misma, en la solución acuosa para desecar, se encuentra en el rango de 1M a 1,5M, preferentemente de 5M a 1,25M.
La concentración del metilsulfonilmetano en la solución acuosa para desecar se encuentra en el rango de 0,1M a 1,5M, preferentemente de 0,5M a 1,25M.
Se usa liofilización en la presente invención. Reduciendo el agua en el material y sellando el material en un vial, el material puede ser fácilmente almacenado, transportado y reconstituido posteriormente en su forma original. Las condiciones de desecación pueden ser optimizadas de forma adecuada mediante experimentación rutinaria.
En la desecación, se forma una composición que incorpora las partículas virales. Se produce una matriz que incorpora las partículas virales. La composición es habitualmente un sólido amorfo. Una matriz sólida, generalmente una matriz de un sólido amorfo, se forma generalmente de este modo. Por “amorfo” se entiende sin estructura y sin una organización de moléculas regular o repetida observable (es decir, no cristalina).
El azúcar o azúcares proporcionan la matriz amorfa en la composición desecada. La (a) N,N-dimetilglicina o N,N,N-trimetilglicina o sales o ésteres fisiológicamente aceptables de la misma, y (b) metilsulfonilmetano se dispersan en la matriz de azúcar. La (a) N,N-dimetilglicina o N,N,N-trimetilglicina o sales o ésteres fisiológicamente aceptables de la misma, y (b) metilsulfonilmetano se incorporan de este modo dentro de la matriz de azúcar. Las partículas virales se incorporan dentro de la matriz del azúcar también. El procedimiento de desecación puede, de este modo, ser efectuado mediante liofilización para formar una torta amorfa dentro de la cual las partículas virales se incorporan. La etapa de desecación se realiza en general tan pronto como la solución acuosa ha sido preparada o poco después de ello. De manera alternativa, la solución acuosa se almacena habitualmente previamente a la etapa de desecación. La partícula viral en la solución acuosa se conserva mediante la (a) N,N-dimetilglicina o N,N,N-trimetilglicina, o sales o ésteres fisiológicamente aceptables de la misma, (b) metilsulfonilmetano y (c) sacarosa, o sacarosa y rafinosa, o manitol, durante el almacenamiento.
La solución acuosa, o solución a granel intermedia, se almacena generalmente hasta 5 años, por ejemplo hasta 4 años, 3 años, 2 años o 1 año. Preferiblemente, la solución se almacena hasta 6 meses, más preferiblemente hasta 3 meses o hasta 2 meses, por ejemplo de 1 día a 1 mes o de 1 día a 1 semana. Previamente a la desecación, la solución se almacena habitualmente en un refrigerador o en un congelador. La temperatura de un refrigerador es habitualmente de 2 a 8 °C, preferiblemente de 4 a 6°C, o por ejemplo aproximadamente 4°C. La temperatura de un congelador es habitualmente de -10 a -80°C, preferiblemente -10 a - 30°C, por ejemplo aproximadamente -20°C. La solución se almacena habitualmente en un envase sellado, preferiblemente un envase de plástico inerte sellado, tal como una bolsa o una botella. El envase es habitualmente estéril. El volumen de la solución a granel intermedia es habitualmente de 0,1 a 100 litros, preferiblemente 0,5 a 100 litros, por ejemplo 0,5 a 50 litros, 1 a 20 litros o 5 a 10 litros. El envase tiene habitualmente un volumen de 0,1 a 100 litros, preferiblemente 0,5 a 100 litros, por ejemplo 0,5 a 50 litros, 1 a 20 litros o 5 a 10 litros.
Si la solución a granel intermedia almacenada se vierte habitualmente en una bandeja de liofilización previamente a
la etapa de liofilización.
El almacenamiento estable de la solución aumenta la flexibilidad del proceso de fabricación. De este modo, la solución puede ser fácilmente almacenada, transportada y posteriormente desecada.
Liofilización
La liofilización es un proceso de deshidratación utilizado habitualmente para conservar material perecedero o para hacer que el material sea más conveniente para el transporte. La liofilización representa una etapa clave para la fabricación de productos farmacéuticos de proteínas sólidas y vacunas. Sin embargo, los materiales biológicos están sujetos a tensiones tanto de liofilización como de desecación durante el proceso, que son capaces de desdoblar o desnaturalizar las proteínas. Además, la tasa de difusión de vapor de agua del material biológico congelado es muy baja y por lo tanto el proceso consume mucho tiempo. La técnica de conservación de la presente invención permite que los materiales biológicos sean protegidos contra las tensiones mecánicas por desecación y/o térmicas del procedimiento de liofilización.
Existen tres etapas principales para esta técnica, a saber, congelación, desecación primaria y desecación secundaria. La congelación se realiza habitualmente utilizando una máquina de liofilización. En esta etapa, es importante enfriar el material biológico por debajo de su punto eutéctico, (Teu) en el caso de productos cristalinos simples o temperatura de transición vítrea (Tg') en el caso de productos amorfos, es decir por debajo de la temperatura más baja a la que las fases sólida y líquida del material pueden coexistir. Esto asegura que tenga lugar una sublimación en lugar de una fusión en la siguiente etapa de desecación primaria.
Durante la desecación primaria, la presión es controlada mediante la aplicación de niveles apropiados de vacío mientras que se suministra suficiente calor para permitir que el agua se sublime. Al menos un 50%, habitualmente un 60 a un 70%, del agua en el material se sublima en esta etapa. La desecación primaria puede ser lenta ya que demasiado calor podría degradar o alterar la estructura del material biológico. Una cámara condensadora de frío y/o placas condensadoras proporciona superficies sobre las que el vapor de agua es atrapado por resolidificación. En el proceso de desecación secundaria, el agua de hidratación se elimina mediante la aplicación adicional de calor. Habitualmente, la presión también es reducida para fomentar un secado adicional. Después de completar el proceso de liofilización, el vacío puede romperse con un gas inerte tal como nitrógeno previamente al sellado o bien el material puede ser sellado bajo vacío.
Composición desecada
Puede obtenerse una composición con un contenido de humedad residual bajo. Se logra un nivel de contenido de humedad residual que ofrece una conservación a largo plazo a temperaturas mayores que las de refrigeración, por ejemplo dentro del rango de 4°C a 56°C o más, o más bajas que las temperaturas de refrigeración, por ejemplo dentro del rango de 0 a -70°C o por debajo. La composición desecada puede por tanto tener un contenido de humedad residual del 10% o menos, 5% o menos, 2% o menos o 1% o menos en peso. Preferiblemente, el contenido de humedad residual es del 0,5% o más, 1% o más. Habitualmente, una composición desecada tiene un contenido de humedad residual del 0,5 a 10% en peso y preferiblemente del 1 al 5% en peso.
La composición puede obtenerse en forma de polvos secos. Una torta resultante de la liofilización puede triturarse hasta tomar la forma de polvos. Una composición sólida de acuerdo a la invención puede, de este modo, tomar la forma de partículas de flujo libre. La composición sólida se proporciona habitualmente en forma de polvos en un vial, ampolla o jeringuilla sellados. Si es para su inhalación, los polvos pueden proporcionarse en un inhalador de polvos secos. La matriz sólida puede ser proporcionada de forma alternativa como un parche. Los polvos pueden comprimirse en forma de comprimidos.
La composición puede consistir habitualmente, o consistir esencialmente, en partículas virales, (a) N,N-dimetilglicina o N,N,N-trimetilglicina, o sales o ésteres fisiológicamente aceptables de la misma, (b) metilsulfonilmetano y (c) sacarosa, o sacarosa y rafinosa, o manitol.
Medición de la conservación de la partícula viral
La conservación en relación a las partículas virales hace referencia a la resistencia de la partícula viral a la degradación física o química y/o a la pérdida de actividad biológica, tal como la degradación del ácido nucleico o proteínas, pérdida de eficacia de transfección, pérdida de la capacidad para estimular una respuesta inmune celular o humoral, pérdida de infectividad viral, pérdida de inmunogenicidad, pérdida de título vírico, pérdida de la respuesta de la célula hospedadora o pérdida de la potencia de la vacuna, bajo la exposición a condiciones de desecación, congelación, temperaturas por debajo de 0°C o por debajo de -25°C, liofilización, temperatura ambiente, temperaturas por encima de 0°C, por encima de 25°C o por encima de 30°C. Preferiblemente, la conservación de acuerdo a la invención comprende crioprotección (protección contra el daño por congelación), lioprotección (protección durante la liofilización) y/o termoprotección (protección contra temperaturas superiores o inferiores a
4°C).
Los métodos de evaluación de la actividad viral tal como la infectividad y/o inmunogenicidad, son bien conocidos par los expertos en el arte e incluyen, pero no se limitan a, el crecimiento de un virus en un cultivo celular, la detección de anticuerpos específicos al virus en sangre, capacidad para provocar respuestas de linfocitos T y/o B, detección de antígenos virales, detección de ARN o ADN codificado por el virus, u observación de partículas del virus utilizando un microscopio.
Más aún, la presencia de un virus da lugar a cambios morfológicos en la célula hospedadora que pueden medirse para proporcionar una indicación de la actividad viral. Los cambios detectables como estos en la célula hospedadora debido a una infección viral se conocen como efecto citopatogénico. Los efectos citopatogénicos pueden consistir en redondeo celular, desorientación, inflamación o contracción, muerte y separación de la superficie. Muchos virus inducen la apoptosis (muerte celular programada) en las células infectadas, medible mediante técnicas tales como el ensayo TUNEL (del inglés “Terminal uridine deoxynucleotidyl transferase dUTP nick end labelling”) y otras técnicas bien conocidas para los expertos en el arte.
Los virus pueden también afectar a la regulación de la expresión de los genes de la célula hospedadora y puede ser analizada para obtener una indicación en relación a si la actividad viral está o no presente. Tales técnicas pueden implicar la adición de reactivos al cultivo celular para completar una reacción enzimática o química con un producto de expresión viral. Además, el genoma viral puede ser modificado para aumentar la detección de la infectividad viral. Por ejemplo, el genoma viral puede ser modificado genéticamente para expresar un marcador que pueda ser fácilmente detectado mediante microscopía de contraste de fases, microscopía de fluorescencia o mediante imagen radiológica. El marcador puede ser una proteína fluorescente expresada tal como la GFP (Proteína verde fluorescente) o una enzima expresada que puede estar implicada en una reacción de radiomarcaje o colorimétrica. El marcador puede además ser un producto génico que interrumpa o inhiba una función particular de las células que están siendo sometidas a ensayo.
Puede utilizarse un ensayo de unidades formadoras de placas para medir la infectividad viral y para indicar el título viral. En este ensayo, las células hospedadoras adecuadas se cultivan en una superficie plana hasta que forman una monocapa de células que cubren una botella o plato de plástico. La selección de una célula hospedadora en particular dependerá del tipo de virus. Ejemplos de células hospedadoras adecuadas incluyen, pero no se limitan a, células CHO, BHK, MDCK, 10T1/2, WEHI, células COS, BSC 1, BSC 40, BMT 10, VERO, WI38, MRC5, A549, HT1080,293, B-50, 3T3, NIH3T3, HepG2, Saos-2, Huh7, HEK293 y HeLa. La monocapa de células hospedadoras se infecta a continuación con las partículas virales. El medio líquido es reemplazado con uno semi-sólido, de manera que cualquier partícula vírica producida como resultado de una infección no pueda desplazarse lejos del sitio de su producción. Se produce una placa cuando una partícula vírica infecta una célula, se replica, y a continuación destruye esa célula. Una placa hace referencia a un área de células en la monocapa que muestra un efecto citopatológico, por ejemplo apareciendo redondeada y más oscuras que otras células vistas al microscopio, o como puntos blancos cuando se visualiza con el ojo; el centro de la placa puede carecer de células debido a la lisis inducida por el virus. El recién replicado virus infecta las células circundantes y éstas también son destruidas. Este proceso puede repetirse varias veces. Las células entonces se tiñen con un colorante tal como el azul de metileno, que colorea únicamente células vivas. No se produce tinción en las células muertas en la placa y aparecen como áreas no coloreadas sobre un fondo coloreado.
Cada placa es el resultado de la infección de una célula por parte de un virus, seguido de la replicación y propagación de ese virus. Sin embargo, los virus que no destruyen las células no pueden producir placas. Una placa hace referencia a un área de células en una monocapa que muestran un efecto citopatológico, por ejemplo apareciendo redondeadas o más oscuras que otras células vistas al microscopio, o como puntos blancos cuando son visualizadas ocularmente; el centro de la placa puede carecer de células debido a la lisis inducida por el virus. Se proporciona una indicación del título viral midiendo las “unidades formadoras de placas” (UFP). Los niveles de infectividad viral pueden medirse en una muestra de material biológico conservada de acuerdo a la presente invención, y compararse con las muestras de control, tal como muestras o virus recientemente cosechados sujetos a desecación y/o variación térmica sin adición de la mezcla de conservación como se usa en la presente invención. Algunos tipos de partículas virales para su uso en la invención, tales como proteínas virales, las VLP, o algunos virus inactivados no tienen la capacidad de formar placas en el ensayo de placas. En este caso, la conservación puede medirse mediante otros métodos, tales como métodos para determinar la inmunogenicidad, que son bien conocidos para los expertos en el arte. Por ejemplo, ensayos in vivo e in vitro para medir las respuestas inmunes del huésped mediadas por la célula o el anticuerpo son conocidos en el arte y adecuados para su uso en la presente invención. Por ejemplo, una respuesta inmune basada en un anticuerpo puede medirse comparando la cantidad, avidez y distribución isotópica de anticuerpos en suero en un modelo animal, antes y después de la inmunización utilizando la partícula viral conservada de la invención.
Uso de partículas virales conservadas de la invención
Vacunas
Las partículas virales conservadas de la presente invención pueden encontrar uso como vacuna. Por ejemplo, las partículas virales conservadas tales como los virus completamente muertos, virus vivos atenuados, virus químicamente inactivados, las VLP o los vectores virales vivos son adecuados para su uso como vacuna. Como vacuna, las partículas virales conservadas de la invención pueden ser utilizadas como antígenos o para codificar antígenos tales como proteínas virales para el tratamiento o prevención de una cantidad de condiciones que incluyen, pero no se limitan a, infección viral, secuelas de una infección viral incluyendo, pero sin limitarse a, toxicidad inducida por virus, cáncer y alergias. Tales antígenos contienen uno o más epítopos que estimularán el sistema inmune de un huésped para generar una respuesta específica al antígeno celular y/o humoral.
Las partículas virales conservadas puede ser utilizada para prevenir o tratar una infección por virus, tales como virus del papiloma humano (VPH), VIH, VHS2/VHS1, virus de la gripe (tipos A, B y C), virus para-influenza, poliovirus, virus RSV, rinovirus, rotavirus, virus de la hepatitis A, virus Norwalk, enterovirus, astrovirus, virus del sarampión, virus de las paperas, virus de varicela zóster, citomegalovirus, virus de Epstein-Barr, adenovirus, virus de la rubéola, virus del linfoma de linfocitos T humano de tipo I (HTLV-I), virus de la hepatitis B (VHB), virus de la hepatitis C (VHC), virus de la hepatitis D, virus de la viruela y virus vaccinia. La vacuna puede además ser utilizada para proporcionar una respuesta inmune adecuada contras numerosas enfermedades veterinarias tales como la fiebre aftosa (incluyendo los serotipos O, A, C, SAT-1, SAT-2, SAT-3 y Asia-1), coronavirus, lengua azul, virus de leucemia felina, la gripe aviar, el virus Hendra y Nipah, pestivirus, parvovirus canino y virus de la diarrea viral bovina. En un modo de realización, la vacuna es una subunidad, conjugado o vacuna multivalente. Por ejemplo, la vacuna conservada de la invención puede ser utilizada para tratar una infección por dos o más tipos de virus diferentes tales como sarampión, paperas y rubéola (por ejemplo vacuna MMR o triple vírica).
La partícula viral conservada puede estar presente en una composición que puede además comprender tampones adecuados y aditivos tales como antibióticos, adyuvantes u otras moléculas que aumentan la presentación de antígenos de la vacuna a células específicas del sistema inmune.
Una variedad de adyuvantes son bien conocidos en el arte y pueden ser utilizados para aumentar la potencia de la vacuna y/o para modular las respuestas inmunes celulares y humorales. Adyuvantes adecuados incluyen, sin limitarse a, sales minerales (por ejemplo, hidróxido de aluminio (“alum”), fosfato de aluminio, fosfato cálcico), adyuvantes particulados (por ejemplo, virosomas, ISCOMS (complejo estructurado de saponinas y lípidos)), derivados microbianos (por ejemplo, MPL (monofosforil lípido A), motivos CpG, toxinas modificadas que incluyen adyuvantes TLR tales como flagelina), derivados de plantas (por ejemplo, saponinas (QS-21)) y adyuvantes inmunoestimulantes endógenos (por ejemplo, citocinas y cualquier otra sustancia que actúe como agente inmunoestimulante para aumentar la efectividad de la vacuna).
La composición de la vacuna está en forma criodesecada (liofilizada) para proporcionar un almacenaje apropiado y maximizar la vida útil en almacenamiento de la preparación. Esto permitirá el almacenamiento de la vacuna durante periodos de tiempo prolongados y ayudará a mantener su inmunogenicidad, potencia y eficacia. La mezcla de conservación o uso en la presente invención está particularmente adaptada para conservar sustancias virales contra las tensiones por desecación y térmicas con las que se enfrenta durante los protocolos de criodesecación/liofilización. Por lo tanto, la mezcla de conservación es adecuada para añadirla al virus o partícula viral poco después de la cosecha y antes de someter la muestra al procedimiento de liofilización.
Para medir la conservación de una vacuna, la potencia de la vacuna puede medirse utilizando técnicas bien conocidas para los expertos en el arte. Por ejemplo, la generación de una respuesta inmune humoral o celular puede ser sometida a prueba en un modelo animal apropiado monitorizando la generación de anticuerpos o respuestas celulares inmunes ante la vacuna. La capacidad de las muestra de vacuna preparadas de acuerdo con el método de la presente invención de desencadenar una respuesta inmune puede compararse con vacunas no sujetas a la misma técnica de conservación.
Vectores virales
Un virus o vector viral conservado según el método de la presente invención, puede ser utilizado para transferir un gen heterólogo y otra secuencia de ácido nucleico a las células diana. De forma adecuada, la secuencia heteróloga (es decir, transgénica) codifica una proteína o producto génico que es capaz de ser expresado en la célula diana. Transgenes adecuados incluyen genes indicadores deseables, genes terapéuticos y genes que codifican polipéptidos inmunogénicos (para su uso en vacunas). La terapia génica, una aproximación para el tratamiento o la prevención de enfermedades asociadas con la expresión defectuosa de genes, implica la introducción de un gen terapéutico en las células, seguido de la expresión y producción de las proteínas requeridas. Esta aproximación permite el reemplazo de genes dañados o la inhibición de la expresión de genes no deseados. En particular, el vector vírico o viral conservado puede ser utilizado en la terapia génica para transferir a un paciente un transgén o gen terapéutico que codifica polipéptidos inmunogénicos.
En un modo de realización preferido, la partícula viral conservada es un vector viral vivo. Por “vector viral vivo” se entiende un vector viral vico que es no patogénico o de patogenicidad baja para las especies diana, y en los que ha
sido insertado uno o más genes que codifican antígenos que estimulan una respuesta inmune protectora contra otros virus o microorganismos, un gen indicador o una proteína terapéutica. En particular, se introduce ácido nucleico en el vector viral, de tal manera que aún sea capaz de replicarse, expresando por tanto un polipéptido codificado por la secuencia del ácido nucleico introducida y en el caso de una vacuna, provocando una respuesta inmune en el animal huésped infectado. En un modo de realización, el vector viral es un vector viral vivo atenuado, es decir está modificado para ser menos virulento (generador de enfermedad) que el virus de tipo silvestre.
La base de utilizar virus recombinantes como vacunas potenciales implica la incorporación de genes específicos de un organismo patogénico en el genoma de un virus no patogénico o atenuado. El virus recombinante puede entonces infectar células eucariotas específicas ya sea in vivo o in vitro, y hacer que expresen la proteína recombinante.
Las vacunas de vectores virales vivos obtenidas mediante la introducción de genes que codifican secuencias de organismos que originan enfermedad pueden ser preferidas sobre las aproximaciones con vacunas vivas atenuadas, de subunidades o ADN. Una de las más importantes características de seguridad de vectores virales vivos es que los receptores pueden quedar inmunizados contra antígenos específicos de organismos patógenos sin una exposición al propio factor de enfermedad. La seguridad se regula además mediante selección de un vector viral que está o bien atenuado para el huésped o que es incapaz de replicarse en el huésped, aunque todavía es capaz de expresar el antígeno heterólogo de interés. Una cepa de la vacuna que tiene un historial de seguridad en la especie diana ofrece una característica de seguridad adicional. Se han desarrollado diversos sistemas en los que al vector se le han eliminado genes esenciales, y la preparación de la vacuna se realiza en sistemas celulares que proporcionan la función que falta.
Una variedad de vectores tales como vectores retrovirales, lentivirales, del virus del herpes, del virus de la viruela, adenovirales y virales adenoasociados pueden ser utilizados para la administración de genes heterólogos a las células diana. El gen heterólogo de interés puede ser introducido en el vector viral. Los vectores virales conservados de la invención pueden comprender por ejemplo un vector vírico provisto en origen con capacidad de replicación, opcionalmente un promotor para la expresión del gen heterólogo y opcionalmente un regulador del promotor. Por ejemplo, los adenovirus útiles en la práctica de la presente invención pueden tener deleciones en la región E1 y/o E3 y/o E4, o pueden ser maximizados de otro modo para recibir ADN heterólogo.
El vector viral puede comprender un promotor constitutivo tal como un promotor de citomegalovirus (CMV), promotor del antígeno T grande SV40, promotor del virus LTR (repetición terminal larga) de tumor de mama de ratón, promotor de adenovirus principal tardío (MLP), promotor del virus LTR de tumor de mama de ratón, el promotor del virus LTR de tumor de mama de ratón, el promotor temprano de SV40, promotores de adenovirus tales como el promotor de adenovirus principal tardío (Ad MLP), promotores de HSV (tales como los promotores HSV IE), promotores de HPV tales como la región reguladora aguas arriba (upstream) de HPV (URR) o el promotor del virus del sarcoma de Rous, junto con otras secuencias de ácidos nucleicos virales ligadas de forma operativa al gen heterólogo de interés. Promotores inducibles o específicos al tejido pueden también ser utilizados para controlar la expresión del gen heterólogo de interés. Los promotores pueden también seleccionarse para ser compatibles con la célula hospedadora para la cual está diseñada la expresión.
El vector viral puede además comprender otros elementos moduladores de transcripción tales como potenciadores. Los potenciadores están ampliamente definidos como agentes que actúan en cis, los cuales cuando se encuentren ligados de forma operativa a una secuencia promotora/génica, aumentarán la transcripción de esa secuencia de genes. Los potenciadores pueden funcionar desde posiciones que se encuentran mucho más alejadas de una secuencia de interés que otros elementos de control de la expresión (por ejemplo los promotores), y pueden operar cuando están situados en cualquier orientación en relación a la secuencia de interés. Los potenciadores han sido identificados a partir de una serie de fuentes virales, incluyendo poliomavirus, virus BK, citomegalovirus (CMV), adenovirus, virus simio 40 (SV40), virus del sarcoma de Moloney, virus del papiloma bovino y virus del sarcoma de Rous. Ejemplos de potenciadores adecuados incluyen el potenciador génico temprano de SV40, el potenciador/ promotor obtenido a partir de repetición terminal larga del virus del sarcoma de Rous, y elementos obtenidos a partir de CMV humano o murino, por ejemplo, elementos incluidos en la secuencia A del intrón de CMV.
El vector viral que contiene un gen heterólogo de interés puede entonces ser conservado según el método de la invención antes de su almacenamiento, sometiéndolo a técnicas adicionales de conservación tales como liofilización, o su administración a un paciente o célula hospedadora.
Pueden incluirse en el vector viral conservado de la invención ácidos nucleicos que codifican polipéptidos conocidos por presentar actividad antiviral, moléculas inmunomoduladoras tales como citocinas (por ejemplo, TNF-alfa, interleucinas tales como la IL-6, y la IL-2, interferones, factores estimulantes de colonia tales como GMCSF), adyuvantes y moléculas coestimuladoras y accesorias. De forma alternativa, tales polipéptidos pueden proporcionarse por separado, por ejemplo en la mezcla de conservación adecuada para su uso en la invención, o pueden ser administrados simultáneamente, secuencialmente o por separado con vectores virales conservados de la invención.
Preferiblemente, el vector viral conservado de la invención puede ser introducido en células hospedadoras adecuadas utilizando una variedad de técnicas virales que son conocidas en el arte, tales como por ejemplo infección con vectores virales recombinantes tales como retrovirus, virus del herpes simple y adenovirus. Preferiblemente, la administración del vector viral conservado de la invención que contiene un gen de interés está mediada por la infección viral de una célula diana.
Una cantidad de sistemas basados en virus han sido desarrollados para transfectar células de mamíferos.
Por ejemplo, una molécula de ácido nucleico recombinante seleccionada puede ser introducida en un vector y empaquetada como partículas retrovirales utilizando técnica conocidas en el arte. El virus recombinante puede ser aislado entonces y administrado a las células del sujeto ya sea in vivo o ex vivo. Los vectores retrovirales pueden estar basados en el virus de la leucemia murina de Moloney (Mo-MLV). En un vector retroviral, uno o más de los genes virales (gag, pol & env) se reemplazan generalmente con el gen de interés.
Se conocen una cantidad de vectores de adenovirus. Los serotipos 2 y 5 del subgrupo C de adenovirus son utilizados comúnmente como vectores. El genoma de adenovirus de tipo silvestre es de aproximadamente 35kb de las que hasta 30kb pueden ser reemplazadas por ADN foráneo.
Existen cuatro unidades de transcripción temprana (E1, E2, E3 & E4), que tienen funciones reguladoras, y un transcrito tardío que codifica proteínas estructurales. Los vectores de adenovirus pueden tener el gen E1 y/o E3 inactivado. Los genes o gen que faltan pueden proporcionarse entonces en trans ya sea mediante un virus auxiliar, un plásmido o integrado en el genoma de una célula auxiliar. Los vectores de adenovirus pueden utilizar un mutante E2a sensible a la temperatura o una deleción de E4. Los vectores de adenovirus mínimos pueden contener únicamente las repeticiones terminales invertidas (ITR) y una secuencia de empaquetamiento alrededor del transgén, donde todos los genes virales necesarios se proporcionan en trans mediante un virus auxiliar. Entre los vectores adenovirales adecuados se incluyen, por tanto, vectores Ad5 y vectores de adenovirus de simio.
Los vectores virales pueden además obtenerse a partir de virus de la familia de la viruela, incluyendo virus vaccinia y virus de la viruela aviar, tal como las vacunas de la diftero-viruela aviar. Por ejemplo, el virus vaccinia modificado de Ankara (MVA) es una cepa del virus vaccinia que no se replica en la mayoría de los tipos de célula, incluyendo tejidos humanos normales. Un vector de MVA recombinante puede, por lo tanto, ser utilizado para administrar un polipéptido. Tipos de virus adicionales tales como los virus adenoasociados (AAV) y el virus del herpes simple (HSV) pueden también ser utilizados para desarrollar sistemas de vectores adecuados.
Administración
Las vacunas conservadas o partículas virales pueden ser administradas, en algunos casos, después de la reconstitución de un producto liofilizado, a un sujeto in vivo utilizando una variedad de vías y técnicas. Por ejemplo, las vacunas conservadas pueden proporcionarse en forma de una solución, suspensión o emulsión inyectable, y ser administradas mediante una inyección parenteral, subcutánea, oral, epidérmica, intradérmica, intramuscular, interarterial, intraperitoneal e intravenosa utilizando una aguja y jeringuilla convencionales, o utilizando un sistema de inyección líquida a presión. Las vacunas conservadas pueden ser administradas por vía tópica en la piel o tejido de la mucosa, tal como por vía nasal, intratraqueal, intestinal, sublingual, rectal o vaginal, o puede proporcionarse a modo de pulverización dividida finamente adecuada para su administración respiratoria o pulmonar.
Los métodos descritos en el presente documento pueden comprender adicionalmente la etapa de procesamiento de la mezcla en forma de una formulación adecuada para su administración como inyección líquida. Preferiblemente, el método además comprende la etapa de procesamiento de la mezcla en forma de una formulación adecuada para su administración mediante ingestión o por vía pulmonar.
El producto conservado se administra a un sujeto en una cantidad que es compatible con la formulación de dosificación y que será profiláctica y/o terapéuticamente efectiva. La administración del producto conservado o vacuna de la invención puede ser bien para un propósito “profiláctico” o bien “terapéutico”. Tal como se utiliza en la presente patente, el término “terapéutico” o “tratamiento” incluye cualquiera de lo siguiente: prevención de la infección o reinfección; reducción o eliminación de los síntomas; y reconducción o completa eliminación de un patógeno. El tratamiento puede ser efectuado de forma profiláctica (previamente a la infección) o terapéutica (a continuación de la infección).
Los siguientes ejemplos ilustran la invención. Se emplearon los siguientes materiales, equipo y técnicas a menos que se indique de otro modo en los Ejemplos:
Materiales
Células HEK-293 (ECACC 85120602)
Dimetilglicina DMG (Sigma D1156, Lote 077K1856)
Dimetilsulfona (MSM) (Sigma M81705, Lote 0001452516)
Sacarosa (Sigma 16104, Lote 70040)
Rafinosa (Sigma R0250, Lote 039K0016)
PBS (Sigma D8662, Lote 118K2339)
Agua (Sigma W3500, Lotes 8M0411 y RNBB1139)
Hydranal Metanol (Fluka 37817, Lote 8331D)
Hydranal Composite (Fluka 34805, Lote 8287a )
Viales de cristal de 5ml (Adelphi Tubes VCD005)
Tapones de liofilización de 14mm (Adelphi Tubes FDIA14WG/B)
Tapas de 14mm (Adelphi Tubes CWPP14)
Adenovirus GFP (Vector Biolabs cat. 1060)
Cepas 3A y 1A del virus del sarampión (obsequio proporcionado por P. Christian en NIBSC)
Medio de Eagle modificado por Dulbecco (DMEM) (Sigma D5796, Lote RNBB1139)
Suero fetal bovino (FBS) (Sigma F7524, Lote 109K3395)
Penicilina Estreptomicina (PS) (Sigma P4458, Lote 0409M00393)
Citrato de sodio salino (SSC) (Sigma S6639, Lote 020M8404)
Línea células BHK-21 (ECCAC CB2857)
HEK 293 (ECACC 85120602)
MVA (ATCC-VR-1508)
Viales de cristal de 2ml (Adelphi Tubes VCDIN2R)
Tapones de liofilización de 13mm (Adelphi Tubes FDW13)
Cierres de cápsula plegada (Adelphi Tubes COTW13)
Equipo
Liofilizador Advantage (VirTis)
Armario de seguridad HERAsafe™ clase II (Thermo Fisher)
Liofilizador VirTis Advantage (Biopharma Process Systems)
Incubadora de CO2 Binder (de Binder)
Cámara de ensayo de termociclado Binder APT line TM MK (de Binder)
Incubadora Thermo Scientific MaxQ 4450 (de Thermofiser)
Pesa KERN EW220-3NM (VWR)
Congelador Elcold -45°C (VWR)
Congelador Forma 900 series -80°C (Thermofisher)
Valorador volumétrico de Karl Fisher (Mettler Toldeo)
Microscopio invertido DMIL LED (Leica, EQP#062)
Pesa ATL-84-1 Atlion (Acculab, EQP#088)
Incubadora IP25037°C (LTE, EQP#016)
Protocolo de liofilización
Las muestras fueron liofilizadas con un liofilizador VirTis Advantage, utilizando el protocolo pre-programado que tiene una duración de 3 días aproximadamente. Las muestras se congelaron a - 40°C durante 1 hora antes de aplicar un vacío, inicialmente a 200 milliTorr con una bomba VLP Thermo Savant (Thermofisher, UK). La temperatura de almacenamiento y de vacío se ajustó a lo largo del proceso y el condensador se mantuvo a -80°C. La etapa 8 se alargó hasta que las muestras se taparon antes de liberar el vacío. El ciclo de desecación se muestra a continuación:

En la fase de desecación primaria la temperatura de almacenamiento se eleva hasta -32°C de - 45°C. La fase de desecación secundaria incluyó una rampa de valor de temperatura hasta 20°C hasta que la desecación se completa. La temperatura del condensador se ajustó para permanecer a una constante de -80°C. Las sondas registraron las temperaturas de almacenamiento y las temperaturas del condensador (ver las Figuras 2A y 2B).
Análisis estadístico
Se realizó un ANOVA de una vía, seguido de una comparación por pares de Tukey para analizar la significancia
entre diferentes excipientes, utilizando el software PRISM Graphpad, versión 4.00. Los resúmenes del valor de p son *=p< 0,10; **=p<0,05; ***=p < 0,005.
En algunos Ejemplos se calcularon los siguientes valores:
• R2= coeficiente de determinación. Una medida de bondad del ajuste. R2<0,5=modelo de significancia baja.
• Q2 = cálculo de la precisión de la predicción. Una medida de bondad de la predicción. Q2 debería ser >0,1 para un modelo significativo. Q2 debería ser >0,5 para un buen modelo. R2-Q2 < 0,2 a 0,3
• Validez del modelo (VM)= "una prueba de diversos problemas del modelo". Validez del modelo < 0,25 = indicador de problemas del modelo estadísticamente significativos, por ejemplo valores extremos, modelo incorrecto / transformación.
• Reproducibilidad (Rep) = medida de la variación entre las réplicas en comparación con la variabilidad total.
Reproducibilidad > 0,5 implica significancia.
Ejemplo de referencia 1
Liofilización
Cada tipo de excipiente (ver Tabla 1 más adelante) se preparó como una solución madre y se añadió 250pl a unos viales de cristal de 5ml debidamente etiquetados. A continuación, se añadió 50pl de adenovirus a cada vial. Después de vortizar, los viales se cargaron en el liofilizador VirTis Advantage y se liofilizaron de acuerdo al protocolo que se proporciona en la sección de técnicas experimentales generales anterior.
Tabla 1 - Concentraciones finales de mezclas de excipientes_____
PBS Sac (1M) Raf (100mM) | Sac (1M) Raf (100mM), Dimetil sulfona (1M) Prueba de exposición térmica
A continuación de la liofilización, las muestras fueron incubadas en una cámara de ensayo de temperatura APT line TM MK de Binder. Las muestras se sometieron a ciclos con una temperatura de 37°C durante 12 horas, una rampa de una hora hasta -20°C, 10 horas a -20°C seguido de una rampa de una hora hasta 37°C. Cada ciclo ascendió a 24 horas y se repitió durante un periodo de 2 semanas antes de realizar un ensayo con adenovirus según se ha descrito anteriormente.
Ensayo con adenovirus (GFP)
96 placas de cultivo celular de fondo plano (Jencons, UK) se sembraron con células HEK 293 (ECACC 85120602) a razón de 105 células por ml (100pl por pocillo) y se mantuvieron a 37°C con CO2 al 5%. Después de lograr una confluencia del 90%, los viales que contienen el adenovirus más el excipiente fueron reconstituidos en 300pl de PBS. Se realizó una etapa de dilución de 1 en 10 tomando 20 pl del vial reconstituido y añadiéndolo a 180 pl de medio de Eagle modificado por Dulbecco (DMEM). Una dilución de 1 en 100 adicional (de la muestra original) se realizó tomando 20pl de la dilución de 1 en 10 y añadiéndolo a 180pl de DMEM. 100pl de cada una de las diluciones resultantes (1 en 10 y 1 en 100) se añadió entonces a los pocillos de la placa que contiene células HEK 293. Adicionalmente, una muestra adicional de adenovirus, de la misma fuente y con la misma titulación (en almacenamiento a -80°C) utilizada en los tratamientos del excipiente, fue descongelada y utilizada para producir una dilución en serie de 1 en 10 (en DMEM). Las diluciones que están en un rango de 1 en 10 en 106 también se añadieron a pocillos individuales que contienen HEK 293s. A las 48 horas después de la inoculación, el número de células de GFP (Proteína verde fluorescente) por pocillo se contaron utilizando microscopio fluorescente, y este número se convirtió posteriormente a pfu/ml de las muestras tratadas, teniendo en cuenta el volumen aplicado y la dilución del inóculo.
Resultados y discusión
Este experimento fue diseñado para evaluar el efecto de enfriamiento y calentamiento sobre la recuperación viral en presencia de excipientes durante el almacenaje. Los resultados demuestran una recuperación deficiente en los excipientes que contienen solamente azúcares o PABS (Figura 1). En excipientes que contienen azúcares más dimetilsulfona, la recuperación fue significativamente mayor.
De manera adicional, los excipientes que contienen dimetilsulfona mostraron poco deterioro a continuación de la prueba de calor y congelación en comparación con las muestras de control. Los resultados indican que la termoprotección durante el proceso de FD (liofilización) es esencial, ya que los excipientes no adecuados tales como los azúcares solos o PBS, no producen ningún título vírico significativo después de la FD. Sin embargo, cuand los excipientes que contienen dimetil sulfona se utilizan en conjunción con azúcares, el título vírico permanece cerca del título original, incluso durante los ciclos de congelación descongelación.
Ejemplo de referencia 2
Cada tipo de excipiente más el virus (ver la Tabla 2), se preparó como una solución madre en PBS y se añadió 250|jl a unos viales de cristal de 5ml debidamente etiquetados. Todos los viales se prepararon por triplicado. Se añadió 50jl de adenovirus a cada vial. Después de vortizar, se introdujeron parcialmente tapones de goma y los viales se cargaron en el aparato VirTis advantage y fueron liofilizados (FD) de acuerdo al protocolo de liofilización proporcionado en la sección de técnicas experimentales generales anterior. Después de la liofilización, las muestras se analizaron para determinar el título vírico utilizando el ensayo con adenovirus descrito en el Ejemplo 1.

Los resultados se muestran en la Figura 3. Excipientes de PBS, solo azúcares y solo MSM proporcionaron una recuperación deficiente. La recuperación de virus aumentó significativamente cuando el excipiente incluyó MSM además de azúcares. Los resultados mostraron un efecto sinérgico entre el MSM y los azúcares, mientras que el MSM utilizado en solitario resultó ser un excipiente de estabilización deficiente.
Protocolo de humedad residual
Se tomaron algunos viales para la medición de humedad residual (ver Tabla 3 a continuación). La evaluación de la humedad residual fue realizada utilizando un valorador volumétrico Karl Fisher. El valorador (Mettler Toledo) trabaja en base al principio de que se consume un mol de I2 para cada mol de H2O. El valorador fue validado utilizando un estándar de 10mg/ml de agua (Sigma, UK).
La titulación fue realizada pesando los viales que contenían la mezcla del excipiente desecado utilizando una pesa (Kern, Germany). Se transfiere 1ml de líquido (hydranal metanol rapid e hydranal metanol composite, Fluka) desde la cámara de titulación al vial de cristal utilizando una jeringuilla de 5ml y una aguja. Una vez que el excipiente se ha disuelto en el líquido, se vuelve a recoger en la jeringuilla y el líquido se inyecta en la cámara de titulación. El vial se volvió a pesar y la diferencia de peso (el peso del excipiente) se introdujo en el valorador. El valorador calculó entonces la humedad residual.
Las mediciones indican que la presencia de MSM puede ayudar en la desecación de la torta durante la desecación secundaria.
T l h m r i l m r n l m z l l x i i n li filiz

Ejemplo de referencia 3
Una mezcla de excipiente más virus se preparó y se procesó según se describe en el Ejemplo 2. El excipiente contenía TMG y opcionalmente azúcares. La concentración final de cada componente en el excipiente antes de la desecación se muestra en la Tabla 4 a continuación. Todos los viales se prepararon por triplicado.
T l 4

Los resultados que utilizan TMG (Trimetilglicina) en el excipiente se muestran en la Figura 4. La TMG parece aumentar la recuperación de la infectividad adenoviral de muestras liofilizadas. Sin embargo, la concentración más baja (0,07M) ofrece la mayor protección, y aumentar la concentración de TMG por encima de esta concentración reduce la protección ofrecida. El tratamiento con 0,07M de TMG ofreció una mayor protección que los azúcares por sí solos.
Ejemplo de referencia 4
El ejemplo 4 describe la experimentación para aclarar la interacción entre la S-metil-L-metionina (SMM), sacarosa y rafinosa como excipientes en una formulación de adenovirus liofilizada.
Preparación y liofilización del virus
El adenovirus recombinante (Vector Biolabs) que expresa la GFP aumentada bajo un promotor de CMV, y con un título (previo a la congelación) de 2x106 pfu/ml, se retiró del almacenamiento a -80°C y se dejó descongelar. Se diluyeron 50|jl de alícuotas del virus a 300|jl en PBS que contenía una concentración variable de cada uno de los excipientes. En la Tabla 5 puede verse una lista completa de las formulaciones de excipientes sometidas a ensayo.
Tabla 5 - Resumen de tratamientos de excipientes, cada tratamiento se realizó por trip licado Sacarosa (M) Rafinosa (mM) SMM (M) Prueba de exposición térmica


Cada tratamiento se preparó en 6 réplicas en viales. Estas muestras se prepararon en viales de cristal de 5ml, se introdujeron parcialmente tapones de goma, y después de vortizarlas se cargaron en el aparato VirTis advantage y se liofilizaron bajo las condiciones que se muestran en la Figura 5.
Prueba de exposición térmica con adenovirus liofilizado
Después de la liofilización, las muestras fueron retiradas inmediatamente y 3 réplicas de cada tratamiento se situaron a 37°C para la prueba de exposición térmica, mientras que las otras 3 se almacenaron a 4°C como controles post-liofilización. La prueba de exposición térmica se realizó durante 7 días, después de los cuales todos los viales se devolvieron a los viales de control y todos se mantuvieron a 4°C hasta que fue factible someterlos a ensayo.
Ensayo con virus infeccioso recuperado de las tortas rehidratadas
96 placas de cultivo celular de fondo plano (VWR, UK) se sembraron con células HEK 293 (ECACC 85120602) a razón de 105 células por ml (100|jl por pocillo) y se mantuvieron a 37°C con CO2 al 5%. Después de lograr una confluencia del 90%, los viales que contienen el adenovirus más el excipiente fueron reconstituidos en 300|jl de PBS. Las muestras reconstituidas fueron diluidas en serie 1:10 y 1:100 en DMEM más FBS al 5%. 100|jl de cada una de las muestras de virus diluidas resultantes se añadieron a continuación a pocillos individuales de la placa. Después de unas 48 horas adicionales, se contó el número de células de GFP por pocillo utilizando microscopio fluorescente.
Protección de infectividad adenoviral durante la liofilización (Figura 6)
Las muestras se sometieron a ensayo 33 días después de la liofilización, y en el caso de las muestras sometidas a la prueba de exposición al calor, 7 días a 37°C seguido de 26 días adicionales almacenados a 4°C. Los resultados se muestran en la Figura 6A.
La S-metil-L-metionina por sí sola muestra una protección dependiente de la concentración de adenovirus durante la liofilización. La S-metil-L-metionina aumentada en la formulación proporcionó un aumento en la infectividad viral recuperada de las muestras reconstituidas en este rango de concentración. Una co-formulación de S-metil-L-metionina con el tratamiento de baja concentración de azúcares (0,1M de sacarosa, 10mM Rafinosa) no alteró de manera significativa esta relación. Sin embargo, la co-formulación de S-metil-L-metionina con el tratamiento elevado en azúcar (1,0M sacarosa, 100mM Rafinosa) sí aumentó de forma significativa la recuperación de infectividad viral a bajas concentraciones de S-metil-L-metionina.
Ante esta evidencia, la formulación óptima para la protección de la infectividad viral durante la liofilización parecería ser bien una alta concentración de S-metil-L-metionina (>0,07M) sin azúcares, o bien una concentración de menos de 0,23M de S-metil-L-metionina en co-formulaciones con concentraciones altas de azúcares (1,0M de sacarosa, 100mM de Rafinosa).
Protección de infectividad adenoviral durante la liofilización y prueba de exposición térmica a 37°C (Figura 6) En ausencia de cualquier azúcar en la formulación, la S-metil-L-metionina ofrece solo una muy limitada retención de la infectividad viral durante la liofilización y posterior prueba de exposición térmica (Figure 6B). Incluso esta protección limitada solamente se observa a concentraciones de 0,07M y superiores. La co-formulación con una baja concentración de azúcares (0,1M sacarosa, 10mM Rafinosa) ofrece igualmente poca protección, aunque la eficacia puede ser aumentada a concentraciones bajas de S-metil-L-metionina.
La co-formulación de S-metil-L-metionina con una alta concentración de azúcares (1M de sacarosa, 100mM Rafinosa) demuestra un claro aumento de la protección entre 0,00M y 0,23M de S-metil-L-metionina, y este aumento se encuentra bastante por encima de un efecto aditivo, y por lo tanto podría ser considerado, posiblemente, como una verdadera sinergia tanto a 0,07M como a 0,23M. Las concentraciones óptimas parecen ser con S-metil-L-metionina entre 0,05 y 0,1M formulada con concentraciones altas de azúcares (1M sacarosa, 100mM Rafinosa). Sin embargo, en este rango la recuperación está alrededor de 2-3x105 pfu/ml, lo que representa casi una reducción logarítmica sobre el título sometido a ensayo del virus introducido.
Ejemplo de referencia 5
El adenovirus recombinante (Vector Biolabs) que expresa la GFP (proteína verde fluorescente) aumentada bajo un promotor de CMV, fue formulada con mezclas de excipientes de manera que, después de la liofilización, los niveles de adenovirus infeccioso recuperado pudieran analizarse fácilmente. Cada tipo de excipiente más virus (ver Tabla 6) se preparó como una solución madre en PBS y se añadieron 300pl a unos viales de cristal de 5ml debidamente etiquetados. Después de vortizarlos, se insertaron parcialmente tapones de goma y los viales se cargaron en el liofilizador VirTis Advantage y se liofilizaron (FD) según el protocolo de liofilización proporcionado anteriormente. Después de la liofilización, el título vírico de las muestras se sometió a ensayo en un ensayo con adenovirus según
se describe más adelante.
Tabla 6 - concentraciones finales de las mezclas de excipientes en el Ejemplo 5
DMG (M) Sacarosa (M) Rafinosa (mM) Virus

Las muestras se liofilizaron en el liofilizador VirTis Advantage de acuerdo al protocolo proporcionado en la sección de técnicas experimentales generales anterior. Después de la liofilización, las muestras se sometieron a ensayo en un ensayo con adenovirus tal como se describe en el Ejemplo 1.
Los resultados se muestran en la Figura 7. La recuperación del adenovirus liofilizado en PBS es habitualmente baja y este hecho se reprodujo en este experimento.
La DMG por sí sola proporcionó protección de la infectividad adenoviral durante la liofilización, y esto se compara favorablemente en relación al uso de los azúcares por sí solos. La formulación de azúcares y DMG demostró una protección dependiente de la dosis. La concentración más alta de DMG se muestra comparable con el adenovirus previamente a la liofilización.
Ejemplo de referencia 6
El experimento en este Ejemplo se desarrolla en base a la capacidad de la DMG de proteger el adenovirus durante la liofilización, en conjunto con la rafinosa y la sacarosa, explorando la capacidad de la DMG de proteger el adenovirus durante la prueba de exposición térmica. Se sometieron a prueba dos concentraciones de cada uno de los azúcares en una relación estática entre sí (Alta concentración de azúcares = 1M de sacarosa con 100mM de Rafinosa, baja concentración de azúcares = 0,1M de sacarosa con 10mM de Rafinosa), mientras que se estudiaron cinco concentraciones de DMG (0,007M, 0,023M, 0,070M, 0,230M, 0,700M).
Una cepa de adenovirus que expresa la GFP fue formulada con las mezclas del excipiente de manera que, después de la liofilización y del tratamiento térmico, los niveles de adenovirus infeccioso recuperado pudieran ser analizados fácilmente. El adenovirus fue formulado con los excipientes y liofilizados antes de su almacenamiento a 4°C y 37°C durante una semana. Las muestras se inocularon posteriormente contando el número de células que expresan la GFP- a las 48 horas posteriores a la inoculación.
Materiales y métodos
Preparación y liofilización del virus formulado
El adenovirus recombinante (Vector Biolabs) que expresa la GFP aumentada bajo un promotor de CMV, y con un título (previamente a la congelación) de 2x106 pfu/ml, se retiró del almacenamiento a -80°C y se dejó descongelar.
50|jl de alícuotas del virus se diluyeron a 300jl en PBS que contiene una concentración variable de cada uno de los excipientes. Una lista completa de las formulaciones de excipientes sometidas a prueba puede verse en la Tabla 7 a continuación. Cada tratamiento se preparó en 6 réplicas de viales de 5ml. Se introdujeron parcialmente tapones de goma, y después de vortizar los viales se cargaron en el liofilizador VirTis Advantage y se liofilizaron en el programa 10 (ver Figura 8).
Tabla 7 de tratamientos de excipjentes. cada tratamiento se realizó por trip licado
Sacarosa (M) Rafinosa (mM) DMG (M) Prueba de exposición térmica (°C)
00 0 0000 37

continuación

Prueba de exposición térmica con el adenovirus liofilizado
Después de la liofilización, las muestras fueron inmediatamente retiradas y se colocaron 3 réplicas de cada tratamiento a 37°C para la prueba de exposición térmica mientras que las otras 3 fueron almacenadas a 4°C como controles post-liofilización. La prueba de exposición térmica tuvo una duración de 7 días, después los cuales todos los viales se devolvieron a los viales de control y se mantuvieron a 4°C hasta que fue factible someterlos a ensayo.
Ensayo con el virus infeccioso recuperado a partir de tortas rehidratadas
96 placas de cultivo celular de fondo plano (VWR, UK) se sembraron con células HEK 293 (ECACC 85120602) a razón de 105 células por ml (100|jl por pocillo) y se mantuvieron a 37°C con CO2 al 5%. Después de lograr una confluencia del 90%, los viales que contenían el adenovirus más excipiente fueron reconstituidos en 300ml de PBS. Las muestras reconstituidas fueron diluidas en serie de 1:10 y 1:100 en DMEM más FBS al 5%. 100ml de cada una de las muestras de virus diluidas resultantes se añadieron a continuación a pocillos individuales de la placa. Después de unas 48 horas adicionales, se contó el número de células de GFP por pocillo utilizando microscopio fluorescente. Los resultados se muestran en las Figuras 9A y 9B.
Resultados y discusión
Protección de la infectividad adenoviral durante la liofilización (ver Figura 9A)
Las muestras almacenadas a 4°C durante la duración del periodo de pruebas después de la liofilización, se sometieron a ensayo como una representación para los controles de liofilización y también como controles negativos para la prueba de exposición térmica. En ausencia de excipientes la liofilización de adenovirus durante este experimento redujo la infectividad de la muestra de 1,5x106pfu/ml hasta menos de 1,0x104pfu/ml.
El uso de la DMG como excipiente con adenovirus aumentó la recuperación de la infectividad viral después de la reconstitución de tortas liofilizadas. La concentración óptima de la DMG para la protección de adenovirus durante la liofilización en este experimento parece ser 0,07M o mayor. Utilizando excipientes, pueden lograrse fácilmente títulos recuperados de entre 7,5-8,5x105pfu/ml (en comparación con la introducción de un título de 1,5x106pfu/ml).
Protección de la infectividad adenoviral durante la liofilización y a continuación de la prueba de exposición térmica a 37°C (ver la Figura 9B)
No se observó una recuperación de la infectividad viral detectable a partir de los viales que no contenían excipientes
(adenovirus en PBS) después de la prueba de exposición térmica a 37°C. Esto representa una pérdida muy significativa de la infectividad viral sobre las muestras con una formulación equivalente mantenidas a 4°C.
Es posible recuperar la infectividad viral después de la prueba de exposición térmica con las muestras formuladas con azúcares únicamente (por ejemplo, 1,0M de sacarosa, 100mM de Rafinosa). Desafortunadamente, el título viral recuperado es tan solo 4,3x104pfu/ml, en comparación con 1,9x105pfu/ml de las muestras mantenidas a 4°C durante la duración del ensayo.
Cuando la DMG se utilizó como el único excipiente, la concentración óptima de DMG parecía ser 0,07M o mayor con una recuperación de alrededor de 7,5x105pfu/ml. A concentraciones de hasta 0,07M (0,007-0,07M), existe una correlación positiva entre la concentración de la DMG y el virus recuperado. Por encima de 0,07M de DMG su efecto parece saturarse.
La coformulación de adenovirus con la misma concentración de azúcares inferior y la DMG a 0,07M o superior, fue al menos tan buena como las concentraciones de DMG equivalentes en ausencia de cualquier azúcar, y posiblemente proporcionó un ligero aumento del efecto protector.
La coformulación de DMG a 0,023M o menos, con una mayor concentración de azúcar (1,0M de sacarosa, 100mM de Rafinosa), aumentó la recuperación a niveles comparables a los tratamientos en los que el efecto de la DMG se creía saturado. Sin embargo, a concentraciones de DMG de 0,07M o por encima, la adición de una elevada concentración de azúcar no tuvo un beneficio obvio. Estas observaciones sugieren que la adición de azúcares a las formulaciones de DMG en una concentración más elevada (1,0M de sacarosa, 100mM de Rafinosa) reduce la cantidad de DMG requerida para saturar su efecto.
Ejemplo de referencia 7
El adenovirus recombinante (Vector Biolabs) que expresa la GFP aumentada bajo un promotor de CMV, fue formulada con mezclas de excipientes de manera que, después de la liofilización, los niveles de adenovirus infeccioso recuperado pudieran analizarse fácilmente. Cada tipo de excipiente (ver Tabla 8 a continuación) se preparó como una solución madre y se añadieron 250|jl a unos viales de cristal de 2ml debidamente etiquetados. Se añadieron 50|jl de adenovirus a cada vial de las soluciones madre. Después de vortizarlos, los viales se cargaron en el liofilizador VirTis Advantage y se liofilizaron.
Tabla 8 - concentraciones finales de las mezclas de excipiente para el Ejemplo 7
0,7M de DMG | 0,07M de Manitol | 0,7M de DMG, 0,07M de Manitol |
Protocolo de liofilización
Las muestras fueron liofilizadas con un liofilizador VirTis Advantage, utilizando el protocolo pre-programado que tiene una duración de 3 días aproximadamente. Las muestras se congelaron a - 40°C durante 1 hora antes de que se aplicara vacío, inicialmente a 300 milliTorr con una bomba VLP Thermo Savant (Thermofisher, UK). La temperatura de almacenamiento y de vacío se ajustó a lo largo del proceso y el condensador se mantuvo a -80°C. La etapa 9 se alargó hasta que las muestras se taparon antes de liberar el vacío. El ciclo de desecación se muestra en la Tabla 9 a continuación:
T l - n i i n l li filiz r

En la fase de desecación primaria, la temperatura de almacenamiento se mantiene a -34°C. La fase de desecación secundaria incluyó una rampa hasta 20°C hasta que la desecación se completó. La temperatura del condensador se ajustó para permanecer a una constante de -80°C. Las sondas registraron las temperaturas de almacenamiento y las temperaturas del condensador (ver Figura 10).
Ensayo con adenovirus
96 placas de cultivo celular de fondo plano (Jencons, UK) se sembraron con células HEK 293 (ECACC 85120602) a razón de 105 células por ml (100|jl por pocillo) y se mantuvieron a 37°C con CO2 al 5%. Después de lograr una confluencia del 90%, los viales que contenían el adenovirus más el excipiente fueron reconstituidos en 1ml medio Esencial Mínimo de Dulbecco (DMEM) más suero fetal bovino al 5% (FBS). Se realizó una etapa de dilución de 1:10 tomando 100 j l del vial reconstituido y añadiéndolo a 900 j l de DMEM. 100jl del virus diluido resultante se añadió entonces a la primera fila en la placa y una dilución de 1:2 se extendió por la placa. El proceso se repitió con el siguiente excipiente. Después de 48 horas, el número de células de GFP por pocillo se contaron utilizando microscopio fluorescente.
Análisis estadístico
Se realizó un ANOVA de una vía, seguido por un post-test de Bonferroni para analizar la significancia entre diferentes excipientes utilizando el software PRISM Graphpad versión 4.00. Los resúmenes del valor de p son *=p< 0,05; **=p<0,01; ***=p < 0,001.
Resultados y discusión
La Figura 11 muestra el beneficio de una combinación de manitol y DMG en la conservación del título de adenovirus después de la liofilización. Después de la liofilización hubo una caída de aproximadamente medio logaritmo en el título vírico cuando se utilizó DMG como excipiente por sí mismo. Cuando el manitol fue el único excipiente la pérdida en el título fue más significativa que con la DMG, siendo el título vírico reducido en 2 logaritmos. Cuando, sin embargo, tanto el manitol como la DMG se utilizaron, no hubo una pérdida significativa en el título y la apariencia de la torta liofilizada mejoró.
Ejemplo de referencia 8
El objetivo del experimento en este Ejemplo era evaluar la formación de la torta. El ejemplo 8 fue realizado de la misma manera que el Ejemplo 7, excepto que se empleó un panel más amplio de mezclas de excipientes. Cada tipo de excipiente (ver Tabla 10 a continuación) se preparó como una solución madre y se añadieron 300jl a unos viales de k2 ml debidamente etiquetados. Después de vortizarlos, los viales se cargaron en el liofilizador VirTis Advantage que se hizo funcionar según el protocolo de liofilización proporcionado en la Tabla 9. Después de la liofilización, las muestras fueron fotografiadas y evaluadas en cuanto a la formación de tortas.
-

Se examinó la apariencia de las tortas liofilizadas después de la liofilización. Los resultados se muestran en la Figura 12. Diversas concentraciones de manitol se utilizaron en presencia de 0,7M de DMG. La concentración de manitol más elevada fue de 0,58M y la más baja fue de 0,018M. A la concentración más elevada de manitol (0,58M), se formó una torta opaca blanca. A medida que la concentración disminuyó, se formó una espuma clara transparente menos deseable.
Ejemplo de referencia 9
El Ejemplo 9 fue realizado de la misma manera que el Ejemplo 7, excepto en que se prepararon únicamente dos tipos de excipientes. El primer excipiente fue el adenovirus en PBS que se preparó hasta un volumen final de 300ul. El segundo excipiente fue manitol (0,58M) y DMG (0,7M) con el adenovirus en viales de cristal de 2ml. Después de vortizarlos, los viales se cargaron en el liofilizador VirTis Advantage y se liofilizaron de acuerdo con el protocolo proporcionado en la Tabla 9. Después de la liofilización, las muestras o bien fueron sometidas a ensayo para determinar el título vírico, o bien tratadas con calor durante una semana a 37°C y a continuación sometidas a ensayo.
Los resultados se muestran en la Figura 13. Después de la liofilización, hubo una caída en el título vírico de más de medio logaritmo en los controles con PBS. No se observó una pérdida significativa en el título vírico en las muestras que contenían DMG y manitol, en comparación con la solución madre del virus original. Después del tratamiento con calor a 37°C, hubo de nuevo una caída en el título vírico mayor de medio logaritmo en los controles con PBS. El título vírico en las muestras que contenían DMG y manitol disminuyó en aproximadamente un logaritmo de 0,3, en comparación con el título de la solución madre original.
Ejemplo10: Estabilización de adenovirus
Preparación y liofilización de virus
El adenovirus recombinante humano Ad5 (Vector Biolabs) que expresa la GFP aumentada (proteína verde
fluorescente) bajo un promotor de CMV, y con un título (previamente a la congelación) de 6,7x105 pfu/ml en SSC, se retiró del almacenamiento a -80°C y se dejó descongelar. Se añadieron 50jl de alícuotas del virus a viales de liofilización de 2 ml. A estas muestras de virus de 50jl se añadió 250jl de una mezcla de formulación compuesta de DMG, MSM y opcionalmente sacarosa. Cada mezcla de formulación se preparó en SSC. La concentración de DMG, MSM y sacarosa en cada formulación después de la adición a la muestra del virus se muestra en la Tabla 11:
T l 11 - F rm l i n m i r

Se introdujeron parcialmente tapones de goma. Después de vortizarlos, los viales se cargaron en un liofilizador Virtis Advantage Plus EL85 y se fueron liofilizados en el programa 4. Por tanto, las muestras fueron liofilizadas utilizando los ciclos de desecación mostrados en la Tabla 12 a continuación. Las muestras se congelaron a -45°C durante 1 hora antes de que se aplicara un vacío, inicialmente a 300 milli Torr con una bomba VLP Thermo Savant (Thermofisher, UK). Se ajustó la temperatura de almacenamiento y la de vacío a lo largo del proceso y el condensador se mantuvo a -42°C. La etapa 11 se alargó hasta que las muestras se taparon antes de liberar el vacío.
T l 12 - i l i n

En el tratamiento térmico, la temperatura de almacenamiento cayó a -40°C.
Prueba de exposición térmica con virus liofilizado
Después de la liofilización, los viales se taparon inmediatamente, se retiraron, se plegaron sobre el recipiente y a continuación se colocaron a 37°C para la prueba de exposición térmica. La prueba de exposición térmica tuvo una duración de 7 días, después de los cuales todos los viales se volvieron a poner a 4°C hasta que fue factible someterlos a ensayo.
Ensayo de adenovirus infeccioso recuperado a partir de tortas rehidratadas
96 placas de cultivo celular de fondo plano (VWR, UK) se sembraron con células HEK 293 (ECACC 85120602) a razón de 105 células por ml (100jl por pocillo) y se mantuvieron a 37°C con CO2 al 5%. Después de lograr una confluencia del 90%, las células fueron inoculadas.
Los viales que contenían adenovirus más excipiente fueron reconstituidos en 300jl de SSC. Se realizó entonces una etapa de dilución de 1 en 10 tomando 20jl del vial reconstituido y añadiendo a 180jl de Medio de Eagle modificado por Dulbecco (DMEM). Se realizó una dilución adicional de 1 en 100 (de la muestra original) tomando 20jl de la dilución de 1 en 10 y añadiéndola a 180jl de DMEM. Se añadieron entonces 100jl de cada dilución resultante (1 en 10 y 1 en 100), a los pocillos de la placa que contenía células HEK 293.
Además, una muestra adicional de adenovirus, de la misma fuente y con el mismo título (en almacenamiento a -80°C) utilizada en los tratamientos de los excipientes, fue descongelada y utilizada para producir una dilución en serie de 1 en 10 (en DMEM 10% FBS). Las diluciones en un rango de 1 en 10 a 1 en 106 también fueron añadidas
a pocilios individuales que contenían células HEK 293. A las 48 horas después de la inoculación, el número de células de GFP (Proteína verde fluorescente) por pocillo se contó utilizando microscopio fluorescente, y este número fue posteriormente convertido a pfu/ml de las muestras tratadas teniendo en cuenta el volumen aplicado y la dilución del inóculo.
Resultados
Los resultados se muestra en la Figura 14. Cuando los datos se analizaron mediante análisis de regresión lineal múltiple (MLR, por sus siglas en inglés), utilizando el programa MODDE 9.0 (Umetrics, Suecia), se observó un efecto sinérgico cuando se utilizaron MSM y DMG en combinación y cuando se utilizaron DMG y sacarosa en combinación. Ejemplo 11: Estabilización de MVA
Preparación y liofilización del virus
Se recuperó MVA del almacenamiento a -80°C y se descongeló. 50|jl de alícuotas se añadieron a viales de liofilización de 2 ml. A estas muestras de virus se añadieron 250|jl de una mezcla de formulación detallada en la Tabla 11 anterior. Se introdujeron parcialmente tapones de goma. Después de vortizarlos, los viales se cargaron en un liofilizador Virtis Advantage Plus EL85 y se liofilizaron en el programa 4, tal como se describe en el Ejemplo 10. Prueba de exposición térmica del virus liofilizado
Después de la liofilización, los viales se taparon inmediatamente, se retiraron, se plegaron sobre el recipiente y después se colocaron a 37°C para la prueba de exposición térmica. La prueba de exposición térmica tuvo una duración de 7 días, después de los cuales todos los viales se volvieron a poner a 4°C hasta que fue factible someterlos a ensayo.
Ensayo de MVA infeccioso a partir de tortas rehidratadas
El MVA más excipiente fueron reconstituidos en 300ml de SSC. Las muestras reconstituidas fueron diluidas y sometidas a ensayo.
Las placas de ensayo (96 pocillos) se sembraron con células BHK-21 (100|jl por pocillo, 105 células/ml). Las células fueron diluidas en DMEM complementado con FBS al 10%, y PS al 1%. Las placas se colocaron a 37°C, 5% CO2 durante 1 a 2 horas.
Mientras tanto, se preparó una dilución en serie de las muestras de MVA formuladas (en los mismos medios de cultivo) en un rango de 10-1 a 10-4. Cada dilución en serie se preparó 4 veces. Se aplicó 35|jl de cada dilución a pocillos individuales que contenían células BHK-21 y los pocillos se llenaron con 65|jl adicionales de los medios. El día 6 después de la inoculación, los pocillos se evaluaron en cuanto a la presencia o ausencia de efecto citopatológico (CPE) y se calculó el TCID50. Estos se utilizaron a continuación para calcular la concentración de MVA infeccioso por ml en los viales sometidos a prueba de exposición térmica.
Resultados
Los resultados se muestran en la Figura 15. El rango de respuestas en este estudio de cribado fue del 0,6-60,5% del título de partida (ver la Figura 15). Esto se evaluó en relación a un segundo alícuota del virus mantenido a -80°C hasta el ensayo. La figura 15 muestra la respuesta a cada tratamiento de la formulación como un porcentaje de un control positivo. La formulación con el mejor rendimiento comprendía 0,15M de sacarosa, 1M de d Mg , 1M de MSM. En general, los resultados sugieren fuertemente que esta combinación de excipientes tiene un potencial significativo para la estabilización de virus en un entorno liofilizado.
Ejemplo de referencia 12
Materiales
Químicos
Proveedor Código de producto N° de lote
20x SSC Sigma S6639 020M8404 Betaína Sigma B2629 069K1514 Dimetil glicina Sigma D1156 077K1856 Dimetil sulfona Sigma M81705 0001452516 Medio de Eagle modificado por Dulbecco Sigma D5796 RNBB1139 Suero Fetal bovino Sigma F7524 109K3395
(continuación)
Proveedor Código de producto N° de lote Penicilina estreptomicina Sigma P4458 0409M0093 Rafinosa Sigma R0250 050M0053
S-metil metionina Sigma 64382
Sacarosa Sigma 16104 SZB90120 Agua Sigma W3500 8M0411
X-Gen 500 P-Lin Fermentas R0521 00064973 Biológicos
Proveedor Código de producto
Adenovirus Vector Biolabs Ad-CMV-GFP
Línea celular BHK-21 ECACC CB2857
HEK 293 ECACC 85120602
Otros
Fabricante Código de producto
Viales de cristal de 5ml Adelphi Tubes VCD005
Tapones de liofilización de 14mm Adelphi Tubes FDIA14WG/B
Tapas de 14 mm Adelphi Tubes CWPP14
Viales de cristal de 2 ml Adelphi Tubes VCDIN2R
Tapones de liofilización de 13 mm Adelphi Tubes FDW13
Cierres de cápsula plegada Adelphi Tubes COTW13
Equipo
Fabricante Código de producto Liofilizador Virtis Advantage Plus EL85 Virtis EQP#084
Armario de seguridad HERAsafe™ clase II Thermo Fisher EQP#011&012
Microscopio invertido DMIL LED Leica EQP#062
Incubadora de CO2 Binder Binder EQP#014
Congelador Forma 900 series -80°C Thermofisher EQP#015
Pesa ATL-84-1 Atlion Acculab EQP#088
Incubadora IP25037°C LTE EQP#016
Métodos
Diseño del Experimento
Se utilizó MODDE 9.0 para generar un diseño de cribado factorial completo de tres factores a dos niveles (ver Figura 16 que muestra valores codificados, y la Tabla 13 que muestra las concentraciones reales aplicadas). Este diseño implica probar las combinaciones de los excipientes a niveles altos y bajos del rango sometido a prueba, además de puntos centrales replicados. Los puntos centrales replicados ofrecen una indicación de error en el experimento. El diseño puede modelar efectos de 1er orden de cada factor sometido a prueba (excipiente) y las interacciones entre ellos, es decir, determinar el impacto de la presencia de los excipientes para la formulación. No puede modelar efectos mayores de 2° orden, pero puede dar una indicación de si están presentes (curvatura en los datos). Los efectos de segundo orden son el resultado de la covarianza dentro de los datos, es decir, dos o más variables son dependientes entre sí. Aunque los efectos de 2° orden son de esperar, la intención es utilizar este simple estudio de cribado, con tratamientos mínimos, para detectar cualquier efecto del excipiente y a continuación continuar con cualquier excipiente que tenga un efecto en un estudio más sofisticado que pueda modelar los efectos de forma más precisa.
Estabilidad de adenovirus en un entorno liofilizado
Prueba de exposición térmica y preparación de adenovirus formulado en un entorno liofilizado
El adenovirus recombinante que expresa la GFP aumentada bajo un promotor de CMV, con un título (previamente a la congelación) de 6,7x105 pfu/ml en citrato de sodio salino (SSC), se retiró del almacenamiento a -80°C y se dejó descongelar a temperatura ambiente. Posteriormente, se añadieron 50|jl de alícuotas del virus a 15 viales de liofilización individuales de 2 ml. A cada vial se añadió 250|jl de una mezcla de excipiente. Las formulaciones de la mezcla de excipiente una vez mezcladas con el virus se describen en la Tabla 13 y se prepararon en SSC.
Tabla 13

Se introdujeron parcialmente tapones de goma, y después de vortizar los viales se cargaron en un liofilizador y se liofilizaron en el programa 4 (ver la Figura 17). Después de la liofilización las muestras fueron inmediatamente tapadas bajo vacío, retiradas, cerradas con cierres plegados sobre el recipiente y colocadas a 37°C para la prueba de exposición térmica. La prueba de exposición térmica tuvo una duración de 7 días, después de los cuales todos los viales se mantuvieron a 4°C hasta que fue factible someterlos a ensayo. Las muestras liofilizadas fueron reconstituidas en 300|jl de SSC inmediatamente antes del ensayo.
Ensayo con adenovirus
Se prepararon células HEK 293 en placas de cultivo celular de 96 pocillos de fondo plano para la inoculación mediante sembrado a razón de 105 células por ml (100|jl por pocillo) y se mantuvieron a 37°C con CO2 al 5%. Después de 2 horas las células se inocularon tal como sigue a continuación.
Las muestras de virus sometidas a la prueba de exposición térmica fueron diluidas a una relación de 1 en 10, y 1 en 100 en DMEM 10% FBS. Se añadieron a continuación 100|jl de cada muestra del virus diluido resultante a pocillos individuales de la placa de ensayo. De forma adicional, se descongeló un segundo alícuota del adenovirus original en SSC a partir de -80°C, y también se preparó una dilución en serie de 10 veces (de 1 en 10 a 1 en 100.000) en DMEM 10% FBS. Se inocularon dos repeticiones de esta dilución en serie de control positivo a cada placa de 96 pocillos utilizada. Después de 48 horas adicionales, se contó el número de células de GFP por pocillo se contó utilizando microscopio fluorescente.
Resultados
General
Se observó un buen rango de respuestas en cada experimento. La mayoría produjo un rango de actividad viral recuperada de entre tan solo un pequeño porcentaje y un 32-46% (ver Tabla 14).
T l 14 - r m r l i n l m l r n r r x i i n m i r ^

Esta dispersión de respuestas fue suficiente para permitir que se aplicara un modelo adecuado. También es indicativo de un efecto protector de las formulaciones.
En diversos modelos la respuesta más baja estaba por debajo del umbral de detección del ensayo. Para facilitar la transformación de los grupos de datos (transformaciones logarítmicas) se asignó a la respuesta, en estos casos, el nivel del umbral de detección, que en este caso (teniendo en cuenta el nivel contable y a continuación tomando en consideración los factores de dilución, etc.), es de 6x102 pfu/ml.
Se exponen análisis de modelización adicionales en la Tabla 15 a la 17.
Tabla 15 - parámetros de evaluación del modelo a partir de modelos en los que se permite una interacción R2  | Rep.
| Rep.
Sin curvatura
0,95 | 0,66 | 0,89 | 0,88


Tabla 16 - coeficientes de retención en el modelo después del ajuste fino
Factores / Interacciones
NE Sac Raf NExS NExR SxR Curvatura

Las siguientes abreviaturas/columnas están presentes en la Tabla 16
NE = nuevo excipiente, Sac = sacarosa, Raf= rafinosa (todos efectos de 1er orden).
NExS = interacción entre el NE y sacarosa.
NExR = interacción entre el NE y rafinosa.
SxR = interacción entre sacarosa y rafinosa.
Curvatura = indicación de un efecto de 2° orden.
* = término no significativo retenido en el modelo para la estabilidad del modelo.
Tabla 17 - coeficientes de retención en el modelo después del ajuste fino y la inclusión de un efecto hi i 2° r n

Las siguientes abreviaturas/columnas están presentes en la Tabla 17
NE = nuevo excipiente, Sac = sacarosa, Raf = rafinosa (todos efectos de 1 er orden).
NExS = interacción entre el NE y sacarosa.
NExR = interacción entre el NE y rafinosa.
SxR = interacción entre sacarosa y rafinosa.
Curvatura = indicación de efecto de 2° orden.
Término de 2° orden = un término de 2° orden previsto por curvatura en los datos, que refuerza los modelos. El diseño experimental es incapaz de identificar efectos de 2° orden específicos.
DMG
Se observó una buena dispersión de respuestas en este grupo de datos (recuperación de 0,36-32,84%) y de forma notable la respuesta más baja se encuentra por encima del límite de detección (ver Tabla 14). Un punto de datos, muestra ID 11 (ver Tabla 13) fue eliminado del análisis después de ser identificado como un resultado de valor atípico obtenido durante el ajuste fino del modelo. La razón de este resultado extremo no se conoce pero se cree que se debe a un error de un operador. Todos los cuatro indicadores de la fuerza del modelo son elevados (ver Tabla 14), y se observó ninguna curvatura en los datos (ver la Figura 19). Solamente dos factores críticos fueron identificados por el modelo, se observó que la DMG y la sacarosa son efectos de 1er orden positivos significativos (ver Tabla 16 y la Figura 20). No se observó ningún otro efecto o interacción. La rafinosa no mostró indicación de tener un efecto en el modelo, y por tanto recuperación viral, en el rango sometido a prueba.
SMM
Se observó un buen rango de respuestas en este grupo de datos (recuperación 0,09-40, 30%) (ver Tabla 14), aunque el rango más bajo estuvo por debajo del umbral de detección. Durante el ajuste fino del modelo un punto de datos (muestra ID 7) se eliminó del análisis como un valor atípico obvio (ver Tabla 13). El primer modelo generado identificó únicamente la sacarosa como un factor crítico (ver la Figura 21), sin embargo, fue necesario retener los factores no significativos (SMM, rafinosa, y SMM*sacarosa) dentro del modelo para alcanzar cualquier tipo de significancia relevante.
Incluso así, el modelo tiene un valor relativamente pobre en R2 y Q2 (0,5 y 0,4 respectivamente). La figura 22 muestra evidencia de curvatura en el modelo. Después de esta observación, se desarrolló un nuevo modelo con la inclusión de un efecto de 2° orden. Como en ejemplos previos el efecto de 2° orden específico no puede ser identificado con este diseño experimental. El nuevo modelo mostró un valor más elevado en todos los cuatro parámetros de evaluación del modelo. Este modelo identificó la sacarosa, y la rafinosa como efectos de 1er orden además de una interacción entre SMM y sacarosa, y el efecto putativo de 2° orden de un excipiente. (ver la Figura 23). Este nuevo modelo no mostró evidencia alguna de curvatura dentro del modelo (ver la Figura 24).
TMG
Se observó una buena difusión de respuestas en este grupo de datos (0,72-46,27%) y todos los puntos de datos estuvieron por encima del umbral detectable (ver Tabla 13). Se generaron valores aceptables para todos los cuatro parámetros de evaluación del modelo (ver Tabla 14). El modelo identifica un efecto de 1er orden de TMG y una interacción entre TMG y la sacarosa (ver Tabla 16). La rafinosa y la sacarosa se identifican como factores no significativos, pero la sacarosa se retiene en el modelo para conservar el modelo jerárquico (ver la Figura 25). La Figura 26 sugiere curvatura en el modelo; sin embargo, el modelo no resultó mejorado por la inclusión de una interacción de 2° orden, lo que sugiere alguna otra causa para la curvatura.
Ejemplo de referencia 13
Materiales
Químicos
Proveedor Código de producto N° de lote
20x SSC Sigma S6639 020M8404 Dimetil glicina Sigma D1156 077K1856
Medio de Eagle modificado por Dulbecco Sigma D5796 RNBB1139 Suero Fetal bovino Sigma F7524 109K3395 Penicilina estreptomicina Sigma P4458 0409M0093 Rafinosa Sigma R0250 050M0053 Sacarosa Sigma 16104 SZB90120
Agua Sigma W3500 8M0411 Biológicos
Proveedor Código de producto
Adenovirus Vector Biolabs Ad-CMV-GFP
HEK 293 ECACC 85120602
Otros
Fabricante Código de producto
Viales de cristal de 2 ml Adelphi Tubes VCDIN2R
Tapones de liofilización de 13 mm Adelphi Tubes FDW13
Cierres de cápsula plegada Adelphi Tubes COTW13
Equipo
Fabricante Código de producto Liofilizador Virtis Advantage Plus EL85 Virtis EQP#084
Armario de seguridad HERAsafe™ clase II Thermo Fisher EQP#011&012
Incubadora de CO2 Binder Binder EQP#014
Congelador Forma 900 series -80°C Thermofisher EQP#015
Pesa ATL-84-1 Atlion Acculab EQP#088
Incubadora IP25037°C LTE EQP#016
Métodos
Diseño del Experimento
Se utilizó MODDE 9.0 (Umetrics) para generar un diseño experimental de Doehlert (ver la Figura 27). Los diseños de Doehlert son diseños de modelización de superficie de respuesta construidos a partir de simplex regular. Estos son fácilmente extensibles en diferentes direcciones, y pueden añadirse nuevos factores a un diseño existente. A diferencia de diseños de formulaciones regulares, los factores no significativos pueden ser eliminados del análisis y por tanto no se convierten en un factor de confusión. Además, se someten a prueba diferentes factores dentro del diseño en una cantidad de niveles diferentes, por lo que es posible asignar más niveles de prueba para factores que se sospechan son de mayor importancia. Por tanto, los excipientes se sometieron a prueba en 7 niveles, mientras que la sacarosa se sometió a prueba en 5 niveles y la rafinosa solo en 3 niveles. Este modelo mantiene la capacidad de modelar efectos e interacciones de segundo orden. El diseño incluyó 3 factores y 3 réplicas de puntos centrales, lo que dio como resultado 15 muestras de ensayo.
Se probó la sacarosa entre 0 y 1M. Tel nivel superior de sacarosa se estableció en 1M porque ha demostrado estar cerca del límite para una liofilización aceptable. También ha demostrado ser un nivel altamente exitoso en estudios previos, y en general las concentraciones más altas de sacarosa no son deseables en preparaciones parenterales. El nivel más bajo de sacarosa se estableció en 0 M. La rafinosa se probó en un rango de 0 a 300nM, aunque la naturaleza del diseño Doehlert significaba que los niveles no incluían 0mM, en su lugar se probaron las siguientes concentraciones; 27,5, 150,0, y 272,5mM.
La DMG se sometió a prueba en un rango lineal de 0 a 2M. Fue posible limitar este rango en base a experimentos previos en los que la concentración óptima estaba frecuentemente entre 0,5 y 1,5M en un entorno liofilizado.
Estabilidad de adenovirus en un entorno liofilizado
Prueba de exposición térmica y preparación del adenovirus formulado en un entorno liofilizado
El adenovirus recombinante que expresa la GFP aumentada bajo un promotor de CMV, con un título (previamente a la congelación) de 6,7x105 pfu/ml en SSC, se retiró del almacenamiento a -80°C y se dejó descongelar a temperatura ambiente. Posteriormente, se añadieron 50|jl de alícuotas del virus a 15 viales de cristal de 2 ml. A cada vial se añadió 250|jl de una mezcla de excipiente. Las formulaciones de la mezcla de excipiente una vez mezcladas con el virus, se describen en la Tabla 18 y se prepararon en SSC.

Se introdujeron parcialmente tapones de goma, y después de vortizar los viales se cargaron en un liofilizador Virtis advantage y se liofilizaron en el programa 4 (ver la Figura 28). Después de la liofilización las muestras fueron inmediatamente tapadas bajo vacío, retiradas, cerradas con cierres plegados sobre el recipiente y colocadas a 37°C para la prueba de exposición térmica. La prueba de exposición térmica tuvo una duración de 7 días, después de los cuales todos los viales se mantuvieron a 4°C hasta que fue factible someterlos a ensayo. Las muestras liofilizadas fueron reconstituidas en 300|jl de SSC inmediatamente antes del ensayo.
Ensayo de adenovirus
Se prepararon células HEK 293 en placas de cultivo celular de 96 pocillos de fondo plano para su inoculación por sembrado a razón de 105 células por ml (100|jl por pocillo) y se mantuvieron a 37°C con CO2 al 5%.Después de 2 horas las células fueron inoculadas tal como sigue a continuación.
Las muestras de virus sometidas a prueba de exposición térmica fueron diluidas en una relación de 1 en 10, y 1 en 100 en DMEM 10% FBS. 100|jl de cada una de las muestras de virus diluidas resultantes se añadieron entonces a pocilios individuales de la placa de ensayo. Adicionalmente, un segundo alícuota del adenovirus original en SSC, fue descongelado a partir de -80°C, y también se preparó una dilución en serie de 10 veces (de 1 en 10 a 1 en 100.000) en DMEM 10% FBS. Se inocularon dos repeticiones de esta dilución en serie de control positivo a cada una de las placas de 96 pocillos utilizadas. Después de 48 horas adicionales, se contó el número de células de GFP por pocillo utilizando microscopio fluorescente.
Resultados
Se produjo un modelo fuerte en el que todos los cuatro indicadores sugerían una buena significancia (R2=0,93, Q2=0,79, Validez del Modelo=0,98, Reproducibilidad=0,68) (ver la Figura 29). De éstas, la cifra para la reproducibilidad es la única que es ligeramente baja, aunque está bastante por encima de 0,5. La razón para que este valor esté ligeramente más bajo de lo que ha sido habitual podría ser la variación ligeramente más alta entre los puntos centrales replicados, o más bien que el nivel de variación entre estos es proporcionalmente más grande en comparación con la variación total en el ensayo.
El modelo identificó (ver la Figura 30) efectos de 1er orden tanto de la sacarosa como de la DMG, además de un efecto de 2° orden de la DMG. No se observaron efectos de 1er o 2° orden de la rafinosa. Sin embargo, la rafinosa no tiene una interacción con la DMG y por tanto el coeficiente de rafinosa de 1er orden debe ser retenido en el modelo para conservar la estructura jerárquica de los modelos. Además, el efecto de la rafinosa de 2° orden fue retenido, ya que dio como resultado un modelo más fuerte (según se evalúa mediante los indicadores mostrados en la Figura 29 y tratados anteriormente). En cualquier el efecto de la rafinosa de 2° orden estuvo cerca de la significancia en un nivel de fiabilidad del 90% y puede ser un efecto genuino que simplemente no puede ser detectado de forma concluyente en el rango probado.
La Figura 31 muestra una serie de gráficos en 3D de la actividad vírica recuperada (Eje Y) contra las concentraciones de sacarosa variable (eje X) y la DMG (eje Z). “Bajo” indica una concentración de rafinosa de 0mM, "Med" indica una concentración de rafinosa de 150mM y "Alto" indica una concentración de rafinosa de 300mM. Cada gráfico muestra el modelo a una concentración estática y variable de rafinosa. La conservación mejorada de adenovirus se logra aumentando la concentración de sacarosa. Si esta tendencia continúa, más allá del rango probado el experimento no es capaz de identificar un verdadero valor óptimo de sacarosa. En contraste, la concentración óptima de DMG se encuentra claramente dentro del rango probado. Aumentar la concentración de rafinosa parece disminuir la concentración óptima de DMG.
Se utilizaron simulaciones Monte-Carlo para predecir una formulación óptima (ver la Figura 32). Se predijo que un valor óptimo de 0,5M de sacarosa, 1M de DMG, 150mM de Rafinosa produce un título vírico de 4,2x105 pfu/ml o un 98% de ese valor de entrada antes de la prueba de exposición térmica (en base a un control positivo que tenía un título de 4,3x105 pfu/ml).
El valor óptimo previsto se muestra en un gráfico de contorno (Figura 33a) que pone el valor óptimo en contexto. El modelo predice las regiones completas del espacio de diseño en el que las formulaciones producirían un 100% o más de actividad vírica recuperada. Esta región necesita ser vista como una meseta en los datos dentro de la cual sería de esperar una pérdida de la actividad vírica cerca de cero. La Figura 33b destaca esta región. La figura muestra que a medida que la concentración de rafinosa es aumentada, la región baja el eje Y (concentración de DMG) y sube el eje X (concentración de sacarosa).
Conclusiones
Una formulación de DMG, sacarosa y rafinosa ha sido identificada con un potencial significativo para la conservación del adenovirus a través de la liofilización y la prueba de exposición al calor. Los modelos basados en los datos predicen que una recuperación del 100% de la actividad viral es posible. En este modelo, se identificó una concentración óptima de DMG de entre 0,5 y 1,5M. La concentración óptima de sacarosa está más allá del rango probado y también más allá de otras restricciones de la concentración de sacarosa. La rafinosa no parece ser un factor crítico en este modelo.
Ejemplo de referencia 14
Materiales
Químicos
Proveedor Código de producto N° de lote
20x SSC Sigma S6639 020M8404
(continuación)
Proveedor Código de producto N° de lote
Betaína Sigma B2629 069K1514
Dimetil glicina Sigma D1156 077K1856
Medio de Eagle modificado por Dulbecco Sigma D5796 RNBB1139
Suero Fetal bovino Sigma F7524 109K3395
Penicilina estreptomicina Sigma P4458 0409M0093
Rafinosa Sigma R0250 050M0053
Sacarosa Sigma 16104 SZB90120
Agua Sigma W3500 8M0411
Biológicos
Proveedor Código de producto
Adenovirus Vector Biolabs Ad-CMV-GFP
HEK 293 ECACC 85120602
Otros
Fabricante Código de producto
Viales de cristal de 2 ml Adelphi Tubes VCDIN2R
Tapones de liofilización de 13 mm Adelphi Tubes FDW13
Cierres de cápsula plegada Adelphi Tubes COTW13
Equipo
Fabricante Código de producto
Liofilizador Virtis Advantage Plus EL85 Virtis EQP#084
Armario de seguridad HERAsafe™ clase II Thermo Fisher EQP#011&012
Microscopio invertido DMIL LED Leica EQP#062
Incubadora de CO2 Binder Binder EQP#014
Congelador Forma 900 series -80°C Thermofisher EQP#015
Pesa ATL-84-1 Atlion Acculab EQP#088
Incubadora IP25037°C LTE EQP#016
4°C congelador de muestras de largo plazo LEC EQP#090
Incubadora KB115 25°C Binder EQP#008
Diseño del experimento
Se planeó una estabilidad a largo plazo para someter a prueba la supuesta formulación óptima de DMG, sacarosa y rafinosa para adenovirus en un entorno liofilizado. Se sometieron a prueba tres formulaciones:
B Adenovirus en tampón de SSC solo;
B Adenovirus en 0,5M de sacarosa y 150mM de rafinosa en SSC; y
B Adenovirus en 0,5M de sacarosa, 150mM rafinosa y 1M de DMG también en SSC.
Se seleccionó una temperatura de ensayo de estabilidad a largo plazo de 4°C±3. Esto es ampliamente consistente con las directrices estándar de la industria para los ensayos a largo plazo de productos pensados para su almacenamiento refrigerado (+5°C±3). Se adoptó una temperatura de estabilidad acelerada de 25°C y una prueba de exposición térmica de 37°C para representar una temperatura de la prueba de tensiones, o una temperatura adicional de estabilidad térmica acelerada elevada.
Las muestras se probaron a 25°C y a 37°C, 1, 2, 5 y 15 semanas después de la liofilización. Las muestras a 4°C se sometieron a prueba a las 15 semanas después de la liofilización.
Preparación de una prueba de exposición térmica del adenovirus formulado en un entorno liofilizado
El adenovirus recombinante que expresa la GFP aumentada bajo un promotor de CMV, con un título (previamente a la congelación) de 6,7x105 pfu/ml en SSC, se retiró del almacenamiento a -80°C y se dejó descongelar. Posteriormente, se añadieron 50pl de alícuotas del virus a viales de liofilización de 2 ml. A cada vial se añadió 250pl de una mezcla de excipiente. Las formulaciones utilizadas de la mezcla de excipientes fueron tal como se ha descrito anteriormente, concretamente (i) un tampón solo (SSC), (ii) azúcares (0,5M de sacarosa, 150mM de Rafmosa en SSC), y (iii) una formulación óptima putativa (0,5M de sacarosa, 150mM de Rafinosa, 1M de DMG, también en SSC).
Se introdujeron tapones de goma, y después de vortizar se cargaron en un liofilizador VirTis Advantage y liofilizados
en el programa 1 (ver la Figura 34). Después de la liofilización las muestras fueron inmediatamente tapadas bajo vacío, retiradas, cerradas con cierres plegados sobre el recipiente y divididas entre tres tratamientos térmicos. Posteriormente, en cada punto de tiempo se recuperaron 2 viales de cada formulación de acuerdo a la programación anterior y se reconstituyeron en 300jl de SSC inmediatamente antes del ensayo.
Ensayo de adenovirus
Se prepararon células HEK 293 en placas de cultivo celular de 96 pocillos de fondo plano para su inoculación mediante sembrado a razón de 105 células por ml (100jl por pocillo) y se mantuvieron a 37°C con CO2 al 5%. Después de 2 horas fueron inoculadas como sigue a continuación.
Las muestras de virus sometidas a prueba de exposición térmica fueron recuperadas de la prueba de exposición térmica tal como se ha descrito anteriormente diluidas en una relación de 1 en 10, y 1 en 100 en DMEM 10% FBS. A continuación 100jl de cada muestra de virus diluida resultante se añadió a pocillos individuales de la placa de ensayo.
Adicionalmente, un segundo alícuota del adenovirus original en SSC, se descongeló a partir de -80°C y también se preparó una dilución en serie de 10 veces (de 1 en 10 a 1 en 100.000) en DMEM 10% FBS. Dos repeticiones de esta dilución en serie de control positivo fueron inoculadas a cada placa de 96 pocillos utilizada. Después de 48 horas adicionales, se contó el número de células de GFP por pocillo utilizando un microscopio fluorescente.
Resultados
A las 15 semanas, (ver la Figura 35) no se recuperó ninguna actividad vírica delas muestras formuladas en el tampón solo (SSC). La formulación con azúcares evita parte de esta pérdida. Sin embargo, únicamente en las muestras almacenadas a 4°C las pérdidas son menores de una caída de un logaritmo completo, es decir, actividades recuperadas sobre un 10% del título de partida. En este tratamiento en este punto de tiempo las pérdidas son progresivamente peores con la temperatura en aumento. Ya que la temperatura elevada es un modo estándar de simular estudios de estabilidad térmica de mayor duración (estabilidad acelerada), se sugiere que las pérdidas de azúcares no han alcanzado su punto final a 4°C, y que pueden esperarse pérdidas adicionales en el tiempo.
Utilizando el valor óptimo putativo de la formulación, las pérdidas se reducen adicionalmente. De hecho, en todas las tres temperaturas de almacenamiento las pérdidas son aproximadamente una pérdida de medio logaritmo (33% de actividad recuperada). Las respuestas en todas las tres temperaturas están entre una recuperación de un 27,84 y un 30,00%, lo que representa una pérdida de 0,52-0,54 logaritmos. No parece haber diferencias significativas entre las tres temperaturas (+4°C, 25°C y 37°C) con esta formulación. Esto implica que o bien (a) la degradación ha alcanzado un punto final y no puede esperarse más degradación en el tiempo, o (b) que la tasa de disminución ha sido tan lenta que la diferencia en los estudios de temperatura acelerada no pueden ser detectadas.
Las Figuras 36 y 37 respaldan más aún estas observaciones. A ambas temperaturas, 25°C y 37°C, no se recupera actividad vírica alguna de las muestras almacenadas solo en tampón en cualquier punto de tiempo. Las formuladas solo en azúcares retienen algo de la actividad de principio a fin. Su actividad disminuye en un grado ligeramente mayor y ligeramente más rápido a mayor temperatura (+37°C). En la formulación putativa óptima hay un descenso pronunciado en la actividad viral a 37°C, pero ambas temperaturas descienden a niveles similares en el tiempo.
Tanto a 25°C como a 37°C, con todas las formulaciones probadas tiene lugar la mayor parte del descenso en la actividad vírica entre t=0 y t=5 semanas. De hecho, en el caso de formulaciones de un tampón solo y de azúcares, una gran parte de la degradación tiene lugar entre t=0 y t=1 semana. El valor de la respuesta utilizada como el t=0 fue el título del virus previamente a la liofilización y a la prueba de exposición térmica, mientras que los puntos de tiempo están representados como semanas después de la prueba de exposición térmica. Consecuentemente, las diferencias observadas entre el t=0 y t=1 semana son la suma de la degradación durante la liofilización y durante la primera semana de la prueba de exposición térmica.
Ejemplo de referencia 15
Materiales
Químicos
Proveedor Codigo de producto N° de lote
20x SSC Sigma S6639 020M8404 Betaína Sigma B2629 069K1514 Medio de Eagle modificado por Dulbecco Sigma D5796 RNBB1139 Suero Fetal bovino Sigma F7524 109K3395
(continuación)
Proveedor Código de producto N° de lote
Penicilina estreptomicina Sigma P4458 0409M0093
Trimetil glicina Sigma
Agua Sigma W3500 8M0411
Biológicos
Proveedor Código de producto
Línea celular BHK-21 ECACC CB2857
MVA ATCC VR-1508
Otros
Fabricante Código de producto
Viales de cristal de 2 ml Adelphi Tubes VCDIN2R
Tapones de liofilización de 13 mm Adelphi Tubes FDW13
Cierres de cápsula plegada Adelphi Tubes COTW13
Equipo
Fabricante Código de producto
Liofilizador Virtis Advantage Plus EL85 Virtis EQP#084
Armario de seguridad HERAsafe™ clase II Thermo Fisher EQP#011&012
Microscopio invertido DMIL LED Leica EQP#062
Incubadora de CO2 Binder Binder EQP#014
Congelador Forma 900 series -80°C Thermofisher EQP#015
Pesa ATL-84-1 Atlion Acculab EQP#088
Incubadora IP25037°C LTE EQP#016
Métodos
Diseño del Experimento
Se utilizó MODDE 9.0 (Umetrics) para generar un diseño experimental de Doehlert (ver la Figura 38), según se describe en el Ejemplo 13. Por tanto, la TMG se sometió a prueba en siete niveles, mientras que la sacarosa se sometió a prueba en cinco niveles y la rafinosa en tres niveles. Este modelo mantiene la capacidad de modelar efectos e interacciones de segundo orden. El diseño incluyó tres factores y tres réplicas de puntos centrales, lo que dio como resultado quince muestras de ensayo.
Se sometió a prueba la sacarosa entre 0 y 1M. La rafinosa se probó en un rango de 0 a 300mM, aunque la naturaleza del diseño de Doehlert significó que los niveles probados no incluían 0mM. En su lugar se sometieron a prueba los siguientes rangos: 27,5, 150,0, y 272,5mM. La TMG se probó en un rango lineal de 0 a 2M.
Prueba de exposición térmica y preparación de MVA formulado en un entorno liofilizado
Se recuperó MVA del almacenamiento a -80°C y se descongeló. Se añadieron 50|jl de alícuotas del MVA a viales de cristal de liofilización de 2 ml, posteriormente se añadió a cada vial 250|jl de una mezcla de excipiente. Las formulaciones de la mezcla de excipiente una vez mezcladas con el virus, se describen en la Tabla 19 y se prepararon en SSC.
Tabla 19

continuación

Se introdujeron parcialmente tapones de goma, y después de vortizar los viales se cargaron en un liofilizador Virtis advantage y se liofilizaron según se describe en la Figura 39. Después de la liofilización las muestras fueron inmediatamente tapadas bajo vacío, retiradas, cerradas con cierres plegados sobre el recipiente y colocadas a 37°C para la prueba de exposición térmica. La prueba de exposición térmica tuvo una duración de 7 días, después de los cuales todos los viales se devolvieron a los viales de control y se mantuvieron a 4°C hasta que fue factible someterlos a ensayo. Las muestras liofilizadas fueron reconstituidas en 300|jl de SSC inmediatamente antes del ensayo.
Ensayo de MVA
Las placas de ensayo (96 pocillos) se sembraron con células BHK-21 (100|jl por pocillo, 105 células/ml). Las células fueron diluidas en DMEM complementado con FBS al 10%, y PS al 1%. Las placas se colocaron a 37°C, 5% CO2 durante 1- 2 horas.
Mientras tanto, se preparó una dilución en serie de 10 veces de las muestras de MVA (en los mismos medios de cultivo) en un rango de 1 en 10 a 1 en 10.000. Cada dilución en serie se preparó 5 veces. Se aplicó 100|jl de cada dilución a pocillos individuales que contenían células BHK-21 (según se ha descrito anteriormente).
El día 6 p.i. (después de la inoculación), los pocillos se evaluaron en cuanto a la presencia o ausencia de CPE y se calculó el TCID50. Estos se utilizaron a continuación para calcular la concentración de MVA infeccioso por ml en los viales sometidos a prueba de exposición térmica.
Resultados
Los datos de este estudio se muestran en la tabla 19. Las respuestas variaron de un 6 a un 92% del título de partida. Durante el análisis, la formulación número 14 fue identificada como un claro valor atípico y se excluyó de este análisis. Esto aumentó los parámetros de evaluación del modelo.
El modelo descrito aquí (ver las Figuras 40 y 41) demuestra un efecto de 1er orden para la sacarosa. Se observó que la rafinosa no tenía ningún efecto de 1er orden pero sí demostró una interacción de 2° orden. No se observó que la TMG tuviera un efecto de 1er orden es este estudio, pero se identificó un efecto de 2° orden. Finalmente, se identificó una interacción entre la sacarosa y la TMG.
La Figura 42 muestra un gráfico de contorno del modelo. El valor óptimo de concentración de la TMG se encuentra cerca del centro del modelo (alrededor de 1M), aunque éste varía ligeramente a medida que varían los otros excipientes. La Rafinosa también muestra un valor óptimo. Finalmente, como regla general, a mayor concentración de sacarosa mejor conservación del MVA.
Simulaciones Monte-Carlo identificaron un valor óptimo de 1M de sacarosa, 1,14M de TMG y 141,76mM de rafinosa y proporcionaron una recuperación prevista de 1,14x106 pfu/ml o 87,7% del título de partida.
Ejemplo de referencia 16
Materiales
Químicos
Proveedor Codigo de producto N° de lote
20x SSC Sigma S6639 020M8404
Dimetil glicina Sigma D1156 077K1856
Medio de Eagle modificado por Dulbecco Sigma D5796 RNBB1139
Suero Fetal bovino Sigma F7524 109K3395
Penicilina estreptomicina Sigma P4458 0409M0093
Agua Sigma W3500 8M0411
Biológicos
Proveedor Código de producto
Línea celular BHK-21 ECACC CB2857
MVA ATCC VR-1508
Otros
Fabricante Código de producto
Viales de cristal de 2 ml Adelphi Tubes VCDIN2R
Tapones de liofilización de 13 mm Adelphi Tubes FDW13
Cierres de cápsula plegada Adelphi Tubes COTW13
Equipo
Fabricante Código de producto
Liofilizador Virtis Advantage Plus EL85 Virtis EQP#084
Armario de seguridad HERAsafe™ clase II Thermo Fisher EQP#011&012
Microscopio invertido DMIL LED Leica EQP#062
Incubadora de CO2 Binder Binder EQP#014
Congelador Forma 900 series -80°C Thermofisher EQP#015
Pesa ATL-84-1 Atlion Acculab EQP#088
Incubadora IP25037°C LTE EQP#016
Métodos
Se utilizó MODDE 9.0 para generar un diseño central compuesto de cara centrada (CCF, por sus siglas en inglés) (ver la Figura 43). Los diseños CCF son una forma de modelización de superficie de respuesta (RSM) que somete prueba únicamente tres niveles de cada factor, pero sigue soportando un modelo cuadrático. A diferencia de diseños de formulaciones regulares, los factores no significativos pueden ser eliminados del análisis y por tanto no se convierten en un factor de confusión.
Prueba de exposición térmica y preparación del MVA formulada en un entorno liofilizado
Se recuperó MVA del almacenamiento a -80°C y se descongeló. Se añadieron 50|jl de alícuotas del MVA a viales de cristal de liofilización de 2 ml. Posteriormente se añadió a cada vial 250|jl de una mezcla de excipiente. Las formulaciones de la mezcla de excipiente una vez mezcladas con el virus, se describen en la Tabla 20 y se prepararon en SSC.
Tabla 20
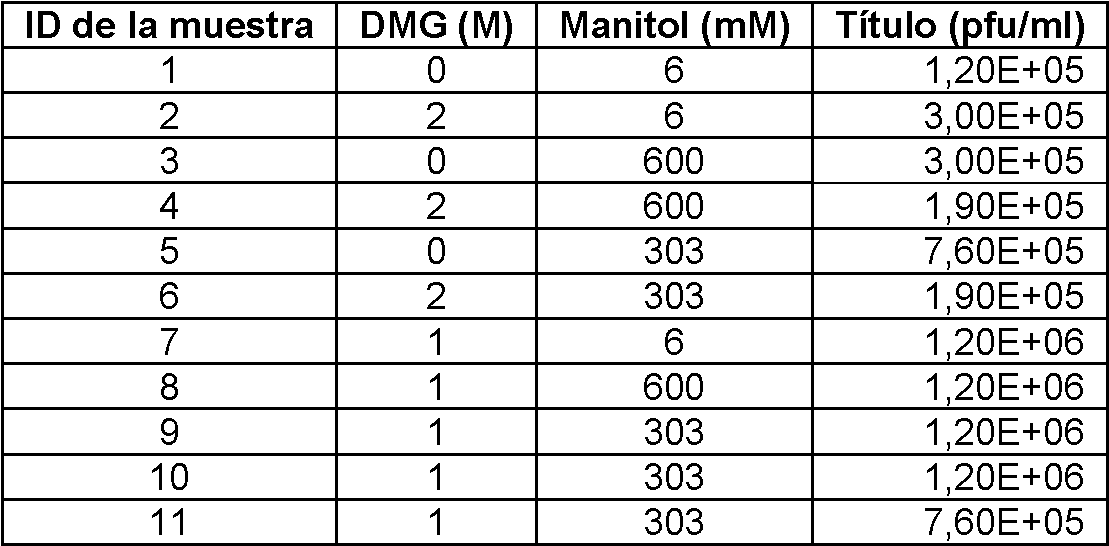
Se introdujeron parcialmente tapones de goma, y después de vortizar los viales se cargaron en un liofilizador Virtis advantage y se liofilizaron según se describe en la Figura 44. Después de la liofilización las muestras fueron inmediatamente tapadas bajo vacío, retiradas, cerradas con cierres plegados sobre el recipiente y colocadas a 37°C para la prueba de exposición térmica. La prueba de exposición térmica tuvo una duración de 7 días, después de los cuales todos los viales se devolvieron a los viales de control y se mantuvieron a 4°C hasta que fue factible someterlos a ensayo. Las muestras liofilizadas fueron reconstituidas en 300|jl de SSC inmediatamente antes del ensayo.
Ensayo de MVA
Las placas de ensayo (96 pocillos) se sembraron con células BHK-21 (100|jl por pocillo, 105 células/ml). Las células fueron diluidas en DMEM complementado con FBS al 10%, y PS al 1%. Las placas se colocaron a 37°C, 5% CO2 durante 1- 2 horas.
Mientras tanto, se preparó una dilución en serie de 10 veces de las muestras de MVA en los mismos medios de cultivo, en un rango de 1 en 10 a 1 en 10.000. Cada dilución en serie se preparó 5 veces. Se aplicó 100|jl de cada dilución a pocillos individuales que contenían células BHK-21 (según se ha descrito anteriormente).
El día 6 p.i. (después de la inoculación), los pocillos se evaluaron en cuanto a la presencia o ausencia de CPE y se calculó el TCID50. Estos se utilizaron a continuación para calcular la concentración de MVA infeccioso por ml en los viales sometidos a prueba de exposición térmica.
Posteriormente, se preparó una dilución en serie de dos veces del MVA formulado en un rango de 1 en 2.000 a 1 en 32.000. Estas diluciones se sometieron a ensayo por separado pero de la misma forma que antes.
Resultados
El ensayo del primer pase de estas muestras (intervalo logarítmico = 1) produjo únicamente cinco niveles de respuesta y uno de estos estaba por debajo del umbral de detección. Más importante, seis de los once tratamientos se encontraban por encima del umbral de detección máximo. Estas muestras se volvieron a someter a ensayo (intervalo logarítmico = 0,3). Las muestras se mantuvieron en forma líquida a 4°C entre los dos ensayos. Algunas muestras ofrecieron un valor significativo (entre los umbrales máximo y mínimo) en ambos ensayos. Esto permitió la determinación de la pérdida entre los dos ensayos.
Después del ensayo de segundo pase ningún tratamiento produjo un título por debajo del umbral de detección. Para facilidad de transformación, se asignó a este tratamiento una respuesta igual al umbral de detección mínimo.
El modelo generado a partir de estos datos es relativamente fuerte. Tres de cuatro parámetros tiene un valor de validez del modelo de 0,9 (R2=0,82, Q2=0,70, Validez del Modelo =0,91, Reproducibilidad =0,70) (ver Figura 45). El modelo identificó únicamente un factor significativo. Se observó que la DMG tenía un efecto de segundo orden (no-lineal / cuadrático) (ver la Figura 46).
La Figura 47 muestra el modelo de RSM generado. Es efectivamente una curva de respuesta de la dosis de DMG simple que no se ve modificada por el manitol dentro del rango de concentración probado. La curva de respuesta de la dosis identifica un claro valor óptimo de concentración de DMG, como ocurre con las simulaciones monte-carlo (ver la Figura 48). El valor óptimo de concentración de DMG previsto es de 1,00M y la recuperación prevista de actividad viral es de un 117% del título de partida.
Claims (6)
1. Un método para conservar partículas virales durante liofilización que comprende:
(a) proporcionar una solución acuosa de:
(i) partículas virales de Adenoviridae, Orthomyxoviridae, Paramyxoviridae, Parvoviridae, Picornaviridae, Poxviridae, Herpesviridae, Togaviridae, Flaviviridae, Retroviridae, Filoviridae, Papillomaviridae, Caliciviridae, Coronaviridae, Reoviridae o Hepadnaviridae,
(ii) sacarosa, o sacarosa y rafinosa, o manitol, a una concentración total de azúcar de 0,1 M a 3 M,
(iii) de 0,1 a 1,5 M de N,N-dimetilglicina o N,N,N-trimetilglicina, o una sal o éster fisiológicamente aceptable de la misma, y
(iv) de 0,1 a 1,5 M de metilsulfonilmetano; y
(b) liofilizar la solución para formar una composición que incorpora dichas partículas virales.
2. El método según la reivindicación 1, en el que la concentración total de azúcar es de 0,2 M a 2 M.
3. El método según la reivindicación 1 o 2, en el que la solución acuosa es liofilizada en viales o ampollas que se sellan a continuación.
4. El método según la reivindicación 1 o 2, en el que las partículas virales están compuestas de un virus vivo o un virus muerto.
5. El método según la reivindicación 4, en el que el virus vivo es virus completo o virus vivo atenuado.
6. Una composición que es sólida y liofilizada, que comprende partículas virales procedentes de Herpesviridae, Togaviridae, Flaviviridae, Retroviridae, Filoviridae, Papillomaviridae, Caliciviridae, Coronaviridae, Reoviridae, Hepadnaviridae, Adenoviridae, Orthomyxoviridae, Paramyxoviridae, Parvoviridae, Picornaviridae o Poxviridae, y que se puede obtener mediante un método según se define en una cualquiera de las reivindicaciones 1 a 5.
Applications Claiming Priority (7)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| GBGB1005497.1A GB201005497D0 (en) | 2010-03-31 | 2010-03-31 | Method for stabilising viruses |
| GBGB1005521.8A GB201005521D0 (en) | 2010-03-31 | 2010-03-31 | Stabilised liquid formulations of virus particles |
| GBGB1005520.0A GB201005520D0 (en) | 2010-03-31 | 2010-03-31 | Stabilisation of viral particles |
| EP10250706 | 2010-03-31 | ||
| GBGB1014962.3A GB201014962D0 (en) | 2010-09-08 | 2010-09-08 | Mehtod for stabilising viruses |
| GBGB1017648.5A GB201017648D0 (en) | 2010-10-19 | 2010-10-19 | Liquid preparation containing stabilised viruses |
| GBGB1017647.7A GB201017647D0 (en) | 2010-10-19 | 2010-10-19 | Method for stabilising viruses |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2757591T3 true ES2757591T3 (es) | 2020-04-29 |
Family
ID=43903999
Family Applications (2)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES11714373.5T Active ES2543795T3 (es) | 2010-03-31 | 2011-03-31 | Estabilización de partículas virales |
| ES15158279T Active ES2757591T3 (es) | 2010-03-31 | 2011-03-31 | Estabilización de partículas virales |
Family Applications Before (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES11714373.5T Active ES2543795T3 (es) | 2010-03-31 | 2011-03-31 | Estabilización de partículas virales |
Country Status (11)
| Country | Link |
|---|---|
| US (2) | US10206960B2 (es) |
| EP (2) | EP2552465B1 (es) |
| JP (1) | JP5960120B2 (es) |
| CN (1) | CN102892424B (es) |
| CA (1) | CA2795013C (es) |
| DK (2) | DK2552465T3 (es) |
| ES (2) | ES2543795T3 (es) |
| GB (1) | GB2499479A (es) |
| HU (2) | HUE026885T2 (es) |
| IL (1) | IL221858A (es) |
| WO (1) | WO2011121306A1 (es) |
Families Citing this family (18)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| EP2552410B1 (en) | 2010-03-31 | 2018-10-24 | Stabilitech Biopharma Ltd | Method for preserving alum adjuvants and alum-adjuvanted vaccines |
| BR112012025047A2 (pt) * | 2010-03-31 | 2016-06-21 | Stabilitech Ltd | formulações líquidas estabilizadas |
| HUE026885T2 (en) | 2010-03-31 | 2016-08-29 | Stabilitech Ltd | Stabilize virus fragments |
| GB201117233D0 (en) | 2011-10-05 | 2011-11-16 | Stabilitech Ltd | Stabilisation of polypeptides |
| TWI690322B (zh) | 2012-10-02 | 2020-04-11 | 法商傳斯堅公司 | 含病毒的調配物及其使用 |
| RU2720808C2 (ru) * | 2013-09-03 | 2020-05-13 | Джорджия Тек Рисёч Корпорейшн | Термостабильные препараты вакцин и микроиглы |
| GB201406569D0 (en) * | 2014-04-11 | 2014-05-28 | Stabilitech Ltd | Vaccine compositions |
| BR112019014004A2 (pt) * | 2017-01-06 | 2020-02-11 | Stabilitech Biopharma Ltd | Método de fornecimento de transgene, comprimido ou cápsula, método de preparação de partículas virais e produto |
| GB2562241B (en) | 2017-05-08 | 2022-04-06 | Stabilitech Biopharma Ltd | Vaccine compositions |
| AR113996A1 (es) * | 2017-12-22 | 2020-07-08 | Intervet Int Bv | Vacunas líquidas de virus envueltos vivos |
| EP3847246A1 (en) | 2018-09-06 | 2021-07-14 | Bavarian Nordic A/S | Storage improved poxvirus compositions |
| MX2021014912A (es) * | 2019-06-07 | 2022-01-18 | Intervet Int Bv | Celula mutante mdbk con inactivacion de irf3/irf7 y su uso para producir vacunas. |
| US20210284716A1 (en) | 2020-03-11 | 2021-09-16 | Immunitybio, Inc. | ACE2-Fc Trap |
| US11857620B2 (en) | 2020-03-11 | 2024-01-02 | Immunitybio, Inc. | Method of inducing immunity against SARS-CoV-2 using spike (s) and nucleocapsid (N)-ETSD immunogens delivered by a replication-defective adenovirus |
| US12285480B2 (en) | 2020-03-11 | 2025-04-29 | Nantcell, Inc. | Method of inducing immunity against SARS-CoV-2 using spike and nucleocapsid-ETSD immunogens delivered by RNA and replication-defective adenoviruses |
| US20230114464A1 (en) | 2020-03-12 | 2023-04-13 | Bavarian Nordic A/S | Conditions Improving Poxvirus Stability |
| WO2021215952A1 (ru) | 2020-04-24 | 2021-10-28 | Общество С Ограниченной Ответственностью "Индженик" | Способ получения частиц бактериофагов семейства levivirus |
| AR127944A1 (es) | 2021-12-23 | 2024-03-13 | Intervet Int Bv | Vacunas líquidas estabilizadas de virus vivos |
Family Cites Families (89)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| US3927208A (en) | 1973-05-14 | 1975-12-16 | Rit Rech Ind Therapeut | Live bovine adenovirus vaccines, preparation thereof and method of vaccination using them |
| US4631189A (en) | 1980-03-10 | 1986-12-23 | Da Vinci Laboratories, a division of FoodScience Corporation | N,N-dimethylglycine and use in immune response |
| JPS6013718A (ja) | 1983-07-05 | 1985-01-24 | Chemo Sero Therapeut Res Inst | B型肝炎ワクチン |
| JPS60193925A (ja) | 1984-03-13 | 1985-10-02 | Chemo Sero Therapeut Res Inst | 凍結乾燥製剤化ワクチン |
| US4808700A (en) | 1984-07-09 | 1989-02-28 | Praxis Biologics, Inc. | Immunogenic conjugates of non-toxic E. coli LT-B enterotoxin subunit and capsular polymers |
| JPS61189228A (ja) | 1985-02-19 | 1986-08-22 | Nippon Sekijiyuujishiya | 血液凝固第8因子製剤の製法 |
| US4950596A (en) | 1985-03-04 | 1990-08-21 | The Dow Chemical Company | Stabilization of intracellular enzymes |
| JPS6238173A (ja) | 1985-08-13 | 1987-02-19 | 三菱レイヨン株式会社 | 抗血液凝固剤 |
| US5169758A (en) | 1986-05-02 | 1992-12-08 | Boehringer Mannheim Gmbh | Stabilized, NAD(P)H-dependent, soluble nitrate reductase, a process for the preparation thereof and a reagent containing it |
| US4816568A (en) | 1986-05-16 | 1989-03-28 | International Minerals & Chemical Corp. | Stabilization of growth hormones |
| NZ221306A (en) | 1986-08-15 | 1990-02-26 | Commw Scient Ind Res Org | 2-component immunoadjuvant comprising a mycobacterial free immunoadjuvant oil and a polycationic polyelectrolyte immunoadjuvant and vaccines thereof |
| US4980277A (en) | 1987-10-16 | 1990-12-25 | Cultor Ltd. | Cryoprotectant solution and method |
| WO1989011297A1 (en) | 1988-05-27 | 1989-11-30 | Centocor, Inc. | Freeze-dried formulation for antibody products |
| GB8826429D0 (en) | 1988-11-11 | 1988-12-14 | Univ Leeds Ind Service Ltd | Enzyme stabilisation systems |
| CA2065023A1 (en) | 1989-08-15 | 1991-02-16 | Brent Dorval | Stabilized vaccine compositions |
| CA2051092C (en) | 1990-09-12 | 2002-07-23 | Stephen A. Livesey | Method and apparatus for cryopreparation, dry stabilization and rehydration of biological suspensions |
| AU2309692A (en) * | 1991-07-03 | 1993-02-11 | Cryolife, Inc. | Method for stabilization of biomaterials |
| RU2099418C1 (ru) | 1991-11-21 | 1997-12-20 | Лонца Аг | Фрагмент днк, обеспечивающий использование бетаина, рекомбинантная плазмидная днк с фрагментом днк, обеспечивающим использование бетаина, штамм бактерий rhizobium/agrobacterium, предназначенный для стабильного хранения плазмиды с фрагментом днк, способ получения штамма бактерий |
| ZA936095B (en) | 1992-08-21 | 1994-03-14 | Univ Melbourne | Cytokine applications. |
| FR2698879B1 (fr) | 1992-12-09 | 1995-01-13 | Rhone Poulenc Biochimie | gellules modifiées au niveau du catabolisme de la bétaïne, préparation et utilisations, notamment pour la production de métabolites ou d'enzymes. |
| GB9320782D0 (en) | 1993-10-08 | 1993-12-01 | Univ Leeds Innovations Ltd | Stabilising of proteins on solution |
| WO1995011700A1 (en) | 1993-10-29 | 1995-05-04 | Pharmos Corp. | Submicron emulsions as vaccine adjuvants |
| US5580856A (en) | 1994-07-15 | 1996-12-03 | Prestrelski; Steven J. | Formulation of a reconstituted protein, and method and kit for the production thereof |
| US5965438A (en) | 1995-06-07 | 1999-10-12 | Phyton, Inc. | Cryopreservation of plant cells |
| CA2226575C (en) | 1995-07-27 | 2011-10-18 | Genentech, Inc. | Stabile isotonic lyophilized protein formulation |
| GB9521806D0 (en) | 1995-10-25 | 1996-01-03 | Cortecs Ltd | Preservation methods |
| FR2746109B1 (fr) | 1996-03-12 | 1998-04-17 | Rhone Poulenc Rorer Sa | Milieu pour la conservation de materiel biologique |
| US20070179096A1 (en) | 1996-04-24 | 2007-08-02 | Novo Nordisk A/S | Pharmaceutical Formulation |
| US6037116A (en) * | 1996-06-14 | 2000-03-14 | Biostore New Zealand, Ltd. | Compositions comprising betaine, sodium citrate and sodium chloride and methods for the preservation of biological materials |
| CA2242035C (en) | 1997-07-01 | 2004-11-23 | Transgene S.A. | Compositions useful for transferring therapeutically active substances into a target cell, and their use in gene therapy |
| AU5459698A (en) | 1997-11-26 | 1999-06-15 | Universal Preservation Technologies, Inc. | Preservation of sensitive biological samples by vitrification |
| DE69940354D1 (de) | 1998-11-16 | 2009-03-12 | Introgen Therapeutics Inc | Adenovirus-formulierungen zur gentherapie |
| US6689600B1 (en) | 1998-11-16 | 2004-02-10 | Introgen Therapeutics, Inc. | Formulation of adenovirus for gene therapy |
| AUPQ347199A0 (en) | 1999-10-15 | 1999-11-11 | Csl Limited | Novel polypeptide fragments |
| EP1289494A2 (en) | 2000-06-08 | 2003-03-12 | Powderject Vaccines, Inc. | Powder compositions |
| DE10041515A1 (de) | 2000-08-24 | 2002-03-14 | Gerold Schuler | Verfahren zur Herstellung gebrauchsfertiger, Antigen-beladener oder -unbeladener, kryokonservierter reifer dendritischer Zellen |
| US20040110267A1 (en) | 2000-12-15 | 2004-06-10 | Stratagene | Room temperature stable competent cells |
| CA2449593A1 (en) | 2001-06-08 | 2002-12-19 | Powderject Vaccines, Inc. | Spray freeze-dried compositions |
| US7101693B2 (en) | 2001-09-07 | 2006-09-05 | Brigham Young University | Plasticized hydrophilic glasses for improved stabilization of biological agents |
| JP4936618B2 (ja) | 2001-09-21 | 2012-05-23 | 株式会社ノエビア | 微細エマルション組成物 |
| US20040161776A1 (en) | 2001-10-23 | 2004-08-19 | Maddon Paul J. | PSMA formulations and uses thereof |
| DE60329070D1 (de) | 2001-12-28 | 2009-10-15 | Kyowa Hakko Kirin Co Ltd | Antikörper gegen den fibroblastenwachstumsfaktor 23 |
| EP3210624A1 (en) | 2002-02-27 | 2017-08-30 | Immunex Corporation | Stabilized tnfr-fc composition comprising arginine |
| JP2003261591A (ja) | 2002-03-08 | 2003-09-19 | Koji Hayade | 凍結乾燥安定剤 |
| EP1539244A1 (en) | 2002-06-27 | 2005-06-15 | Novo Nordisk A/S | Use of dimethyl sulfone as isotonicity agent |
| US20060228334A1 (en) * | 2002-07-10 | 2006-10-12 | Manuel Rosa-Calatrava | Modified adenoviral fiber with ablated to cellular receptors |
| WO2004035818A1 (en) | 2002-10-21 | 2004-04-29 | Biotage Ab | Use of osmolytes to heat stabilise luciferase and/or apyrase |
| US7462700B2 (en) | 2002-12-10 | 2008-12-09 | Schering-Plough Animal Health Corporation | Canine RANKL and methods for preparing and using the same |
| EP1633391B1 (en) | 2003-06-03 | 2011-10-19 | Novo Nordisk A/S | Stabilized pharmaceutical peptide compositions |
| EP1638993A4 (en) | 2003-06-10 | 2007-05-09 | Univ Melbourne | IMMUNOMODULATING COMPOSITIONS, APPLICATIONS THEREOF AND METHOD FOR THE PRODUCTION THEREOF |
| US20060247167A1 (en) | 2003-09-01 | 2006-11-02 | Novo Nordisk A/S | Stable formulations of peptides |
| US7642077B2 (en) | 2003-12-08 | 2010-01-05 | Genencor International, Inc. | Biocomposite comprising co-precipitate of enzyme, silicate and polyamine |
| EP1711619A4 (en) | 2003-12-30 | 2007-04-11 | Virumar Pituach Tarkivin Ltd | INTEGRATED VIRUS COMPLEXES, METHOD FOR THE MANUFACTURE THEREOF, VACCINES CONTAINING THEM AND METHOD FOR THE USE THEREOF |
| CA2558676C (en) | 2004-03-03 | 2019-04-16 | Essentia Biosystems, Inc. | Compositions and methods for topical diagnostic and therapeutic transport |
| US20060058238A1 (en) | 2004-09-15 | 2006-03-16 | Lee Laurent-Applegate | Fetal skin cell protein compositions for the treatment of skin conditions, disorders or diseases and methods of making and using the same |
| EP2452670A1 (en) | 2004-10-01 | 2012-05-16 | Ramscor, Inc. | Conveniently implantable sustained release drug compositions |
| CA2586846C (en) * | 2004-11-10 | 2013-12-31 | Qlt Usa Inc. | A stabilized polymeric delivery system |
| BRPI0606867A2 (pt) | 2005-01-28 | 2009-07-21 | Wyeth Corp | formulação lìquida; forma de dosagem unitária farmacêutica; kit; recipiente; método para aumentar a estabilidade de um polipeptìdeo de ligação a antìgeno numa formulação; método para preparar uma forma de dosagem unitária farmacêutica; formulação estável; formulação; e produto terapêutico |
| GB0502661D0 (en) | 2005-02-09 | 2005-03-16 | Stabilitech Ltd | A desiccated product |
| TWI398272B (zh) * | 2005-03-08 | 2013-06-11 | Intervet Int Bv | 化學定義的安定劑 |
| CN101155914B (zh) | 2005-04-08 | 2013-03-06 | 阿戈斯治疗公司 | 树状细胞组合物和方法 |
| PT1954308E (pt) | 2005-09-16 | 2011-11-03 | Merial Ltd | Estabilizadores para vacinas liofilizadas |
| EP1933867A1 (en) | 2005-10-04 | 2008-06-25 | Alk-Abello A/S | Solid vaccine formulation |
| CN101360821A (zh) | 2005-11-21 | 2009-02-04 | 圣诺菲·帕斯图尔有限公司 | 重组病毒的稳定制剂 |
| ES2352855T3 (es) | 2005-11-30 | 2011-02-23 | International Institute Of Cancer Immunology, Inc. | Nuevos compuestos peptídicos del tumor de wilms. |
| WO2008051245A2 (en) | 2005-12-02 | 2008-05-02 | Novartis Ag | Nanoparticles for use in immunogenic compositions |
| AU2007215012A1 (en) | 2006-02-15 | 2007-08-23 | Imclone Llc | Antibody formulation |
| US8021659B2 (en) | 2006-04-28 | 2011-09-20 | Naidu Lp | Coenzyme Q10, lactoferrin and angiogenin compositions and uses thereof |
| ES2286951B1 (es) | 2006-05-26 | 2008-10-01 | Instituto Cientifico Y Tecnologico De Navarra, S.A | Nanoparticulas bioadhesivas para la administracion de moleculas biologicamente activas. |
| US20090053197A1 (en) | 2006-06-14 | 2009-02-26 | Ramaekers Joseph C | Transfer Factor Compositions and Methods |
| NZ576563A (en) * | 2006-11-03 | 2012-09-28 | Alphavax Inc | Alphavirus and alphavirus replicon particle formulations and methods |
| GB0705245D0 (en) * | 2007-03-19 | 2007-04-25 | Stabilitech Ltd | Stable biological products |
| CA2681567C (en) | 2007-03-22 | 2016-07-19 | The Regents Of The University Of Colorado, A Body Corporate | Method of preparing an immunologically-active adjuvant-bound dried vaccine composition |
| JP2010530846A (ja) | 2007-06-01 | 2010-09-16 | アコロジックス インコーポレイティッド | 高温で安定なペプチド製剤 |
| WO2009006097A1 (en) | 2007-06-29 | 2009-01-08 | Johnson & Johnson Regenerative Therapeutics, Llc | Liquid protein formulations comprising gdf-5 for use at elevated temperatures |
| EP2192908A4 (en) | 2007-07-25 | 2010-09-01 | Bioe Inc | DIFFERENTIATION OF PRECURSOR CELLS OF MULTIPLE ABSTRACTS AGAINST CHONDROCYTES |
| US20090123436A1 (en) | 2007-11-08 | 2009-05-14 | Surmodics, Inc. | Cryopreservative compositions and methods |
| BRPI0911048A2 (pt) | 2008-04-14 | 2015-12-29 | Atrm Llc | formulações líquidas tamponadas de gdf-5 |
| US20110212127A1 (en) | 2008-09-24 | 2011-09-01 | Stabilitech Ltd. | Method for Preserving Polypeptides Using a Sugar and Polyethyleneimine |
| BRPI0918988A2 (pt) | 2008-09-24 | 2015-12-01 | Stabilitech Ltd | métodos para preservar um polipeptídeo e um imunógeno de vacina, e para preparar uma vacina, pó seco, produto preservado, frasco selado, ampola ou seringa, uso de um excipiente, e, vacina. |
| US8795686B2 (en) | 2008-11-07 | 2014-08-05 | Serum Institute Of India | Stable, dried rotavirus vaccine, compositions and process for preparation thereof |
| EP2430195B1 (en) * | 2009-05-11 | 2019-01-23 | Biomatrica, INC. | Compositions and methods for biological sample storage |
| CN101670104A (zh) | 2009-09-24 | 2010-03-17 | 浙江大学 | 一种含人参皂甙的油佐剂及其制备方法 |
| WO2011109415A2 (en) | 2010-03-02 | 2011-09-09 | Amgen Inc. | Reducing viscosity of pharmaceutical formulations |
| BR112012025047A2 (pt) | 2010-03-31 | 2016-06-21 | Stabilitech Ltd | formulações líquidas estabilizadas |
| HUE026885T2 (en) | 2010-03-31 | 2016-08-29 | Stabilitech Ltd | Stabilize virus fragments |
| EP2552410B1 (en) * | 2010-03-31 | 2018-10-24 | Stabilitech Biopharma Ltd | Method for preserving alum adjuvants and alum-adjuvanted vaccines |
| GB201117233D0 (en) | 2011-10-05 | 2011-11-16 | Stabilitech Ltd | Stabilisation of polypeptides |
| GB201406569D0 (en) | 2014-04-11 | 2014-05-28 | Stabilitech Ltd | Vaccine compositions |
-
2011
- 2011-03-31 HU HUE11714373A patent/HUE026885T2/en unknown
- 2011-03-31 DK DK11714373.5T patent/DK2552465T3/en active
- 2011-03-31 JP JP2013501933A patent/JP5960120B2/ja active Active
- 2011-03-31 GB GB1216469.5A patent/GB2499479A/en not_active Withdrawn
- 2011-03-31 HU HUE15158279A patent/HUE047521T2/hu unknown
- 2011-03-31 WO PCT/GB2011/000498 patent/WO2011121306A1/en not_active Ceased
- 2011-03-31 US US13/637,817 patent/US10206960B2/en active Active
- 2011-03-31 EP EP11714373.5A patent/EP2552465B1/en active Active
- 2011-03-31 ES ES11714373.5T patent/ES2543795T3/es active Active
- 2011-03-31 DK DK15158279T patent/DK2898890T3/da active
- 2011-03-31 CN CN201180018127.0A patent/CN102892424B/zh not_active Expired - Fee Related
- 2011-03-31 CA CA2795013A patent/CA2795013C/en active Active
- 2011-03-31 ES ES15158279T patent/ES2757591T3/es active Active
- 2011-03-31 EP EP15158279.8A patent/EP2898890B1/en active Active
-
2012
- 2012-09-09 IL IL221858A patent/IL221858A/en active IP Right Grant
-
2019
- 2019-01-02 US US16/237,889 patent/US20190307819A1/en not_active Abandoned
Also Published As
| Publication number | Publication date |
|---|---|
| HUE047521T2 (hu) | 2020-04-28 |
| HUE026885T2 (en) | 2016-08-29 |
| CA2795013C (en) | 2018-10-16 |
| EP2898890A1 (en) | 2015-07-29 |
| JP5960120B2 (ja) | 2016-08-02 |
| CN102892424B (zh) | 2015-04-01 |
| IL221858A (en) | 2017-04-30 |
| WO2011121306A1 (en) | 2011-10-06 |
| ES2543795T3 (es) | 2015-08-21 |
| EP2898890B1 (en) | 2019-08-21 |
| US20130129685A1 (en) | 2013-05-23 |
| GB2499479A (en) | 2013-08-21 |
| US20190307819A1 (en) | 2019-10-10 |
| DK2552465T3 (en) | 2015-07-27 |
| AU2011234269A1 (en) | 2012-10-04 |
| EP2552465B1 (en) | 2015-04-22 |
| CA2795013A1 (en) | 2011-10-06 |
| DK2898890T3 (da) | 2019-11-25 |
| GB201216469D0 (en) | 2012-10-31 |
| JP2013524782A (ja) | 2013-06-20 |
| US10206960B2 (en) | 2019-02-19 |
| CN102892424A (zh) | 2013-01-23 |
| EP2552465A1 (en) | 2013-02-06 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2757591T3 (es) | Estabilización de partículas virales | |
| DK2552478T3 (en) | EXCIPIENTS FOR STABILIZING VIRUS PARTICLES | |
| US8313897B2 (en) | Method for preserving viral particles | |
| Dumpa et al. | Stability of vaccines | |
| ES2897659T3 (es) | Formulaciones de vacunas térmicamente estables, procesos y microagujas que incluyen las formulaciones de vacunas | |
| ES2673823T3 (es) | Formulaciones de alfavirus y de partículas de replicones de alfavirus y métodos relacionados | |
| KR20080070866A (ko) | 재조합 바이러스용 안정화 제형 | |
| CN110430867B (zh) | 从水性混合物冷冻干燥的组合物及其制备方法 | |
| US20230277654A1 (en) | Stable formulations of cytomegalovirus | |
| AU2011234269B2 (en) | Stabilisation of viral particles | |
| EP3953457A1 (en) | Broad and long-lasting influenza vaccine | |
| CN108619521A (zh) | 一种动物非灭活疫苗冷冻干燥保护剂及其使用方法 |