ES2819309B2 - Compuestos agonistas nicotínicos y antioxidantes para el tratamiento de enfermedades neurodegenerativas - Google Patents
Compuestos agonistas nicotínicos y antioxidantes para el tratamiento de enfermedades neurodegenerativas Download PDFInfo
- Publication number
- ES2819309B2 ES2819309B2 ES201930908A ES201930908A ES2819309B2 ES 2819309 B2 ES2819309 B2 ES 2819309B2 ES 201930908 A ES201930908 A ES 201930908A ES 201930908 A ES201930908 A ES 201930908A ES 2819309 B2 ES2819309 B2 ES 2819309B2
- Authority
- ES
- Spain
- Prior art keywords
- compound
- ethyl
- formula
- quinuclidin
- indol
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Expired - Fee Related
Links
- 150000001875 compounds Chemical class 0.000 title claims description 131
- 208000015122 neurodegenerative disease Diseases 0.000 title claims description 15
- 230000004770 neurodegeneration Effects 0.000 title claims description 14
- 239000000181 nicotinic agonist Substances 0.000 title claims description 3
- 239000003963 antioxidant agent Substances 0.000 title description 6
- 230000003078 antioxidant effect Effects 0.000 title description 6
- ZMXDDKWLCZADIW-UHFFFAOYSA-N N,N-Dimethylformamide Chemical compound CN(C)C=O ZMXDDKWLCZADIW-UHFFFAOYSA-N 0.000 claims description 35
- 150000003839 salts Chemical class 0.000 claims description 28
- 238000000034 method Methods 0.000 claims description 23
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims description 21
- 239000012453 solvate Substances 0.000 claims description 18
- -1 2-isothiocyanatoethyl Chemical group 0.000 claims description 16
- 239000000460 chlorine Substances 0.000 claims description 16
- WKBOTKDWSSQWDR-UHFFFAOYSA-N Bromine atom Chemical compound [Br] WKBOTKDWSSQWDR-UHFFFAOYSA-N 0.000 claims description 14
- ZAMOUSCENKQFHK-UHFFFAOYSA-N Chlorine atom Chemical compound [Cl] ZAMOUSCENKQFHK-UHFFFAOYSA-N 0.000 claims description 14
- PXGOKWXKJXAPGV-UHFFFAOYSA-N Fluorine Chemical compound FF PXGOKWXKJXAPGV-UHFFFAOYSA-N 0.000 claims description 14
- GDTBXPJZTBHREO-UHFFFAOYSA-N bromine Substances BrBr GDTBXPJZTBHREO-UHFFFAOYSA-N 0.000 claims description 14
- 229910052794 bromium Inorganic materials 0.000 claims description 14
- 229910052801 chlorine Inorganic materials 0.000 claims description 14
- 239000011737 fluorine Substances 0.000 claims description 14
- 229910052731 fluorine Inorganic materials 0.000 claims description 14
- 125000000217 alkyl group Chemical group 0.000 claims description 13
- 125000000000 cycloalkoxy group Chemical group 0.000 claims description 13
- 239000000651 prodrug Substances 0.000 claims description 11
- 229940002612 prodrug Drugs 0.000 claims description 11
- IAZDPXIOMUYVGZ-UHFFFAOYSA-N Dimethylsulphoxide Chemical compound CS(C)=O IAZDPXIOMUYVGZ-UHFFFAOYSA-N 0.000 claims description 10
- 125000000449 nitro group Chemical group [O-][N+](*)=O 0.000 claims description 10
- 208000024827 Alzheimer disease Diseases 0.000 claims description 9
- 125000003545 alkoxy group Chemical group 0.000 claims description 9
- 239000008194 pharmaceutical composition Substances 0.000 claims description 9
- 125000003118 aryl group Chemical group 0.000 claims description 8
- 125000004093 cyano group Chemical group *C#N 0.000 claims description 8
- 239000003814 drug Substances 0.000 claims description 8
- 125000004435 hydrogen atom Chemical class [H]* 0.000 claims description 8
- 239000000203 mixture Substances 0.000 claims description 8
- 229910052760 oxygen Inorganic materials 0.000 claims description 8
- 239000001301 oxygen Substances 0.000 claims description 8
- 230000002265 prevention Effects 0.000 claims description 8
- QVGXLLKOCUKJST-UHFFFAOYSA-N atomic oxygen Chemical group [O] QVGXLLKOCUKJST-UHFFFAOYSA-N 0.000 claims description 7
- 229910052799 carbon Inorganic materials 0.000 claims description 7
- 238000006243 chemical reaction Methods 0.000 claims description 7
- 239000001257 hydrogen Substances 0.000 claims description 7
- 229910052739 hydrogen Inorganic materials 0.000 claims description 7
- 125000005913 (C3-C6) cycloalkyl group Chemical group 0.000 claims description 6
- 150000001721 carbon Chemical group 0.000 claims description 6
- 125000000753 cycloalkyl group Chemical group 0.000 claims description 6
- 238000002360 preparation method Methods 0.000 claims description 6
- 125000001072 heteroaryl group Chemical group 0.000 claims description 5
- 229910052757 nitrogen Inorganic materials 0.000 claims description 5
- 239000002904 solvent Substances 0.000 claims description 5
- 229910052717 sulfur Inorganic materials 0.000 claims description 5
- REUAXQZIRFXQML-UHFFFAOYSA-N 1-azabicyclo[2.2.2]octan-3-amine Chemical compound C1CC2C(N)CN1CC2 REUAXQZIRFXQML-UHFFFAOYSA-N 0.000 claims description 4
- 208000023105 Huntington disease Diseases 0.000 claims description 4
- 208000018737 Parkinson disease Diseases 0.000 claims description 4
- 206010002026 amyotrophic lateral sclerosis Diseases 0.000 claims description 4
- 125000005843 halogen group Chemical group 0.000 claims description 4
- 201000006417 multiple sclerosis Diseases 0.000 claims description 4
- 125000004433 nitrogen atom Chemical group N* 0.000 claims description 4
- 239000000546 pharmaceutical excipient Substances 0.000 claims description 4
- 230000008569 process Effects 0.000 claims description 4
- 125000004191 (C1-C6) alkoxy group Chemical group 0.000 claims description 3
- FWEZFGIJBIGAKG-QGZVFWFLSA-N O=C(NCCC(C1=C2)=CNC1=CC=C2Cl)N[C@H]1C(CC2)CCN2C1 Chemical compound O=C(NCCC(C1=C2)=CNC1=CC=C2Cl)N[C@H]1C(CC2)CCN2C1 FWEZFGIJBIGAKG-QGZVFWFLSA-N 0.000 claims description 3
- HQPYEOUYACONEJ-QGZVFWFLSA-N S=C(NCCC(C1=C2)=CNC1=CC=C2Cl)N[C@H]1C(CC2)CCN2C1 Chemical compound S=C(NCCC(C1=C2)=CNC1=CC=C2Cl)N[C@H]1C(CC2)CCN2C1 HQPYEOUYACONEJ-QGZVFWFLSA-N 0.000 claims description 3
- NINIDFKCEFEMDL-UHFFFAOYSA-N Sulfur Chemical group [S] NINIDFKCEFEMDL-UHFFFAOYSA-N 0.000 claims description 3
- 239000003795 chemical substances by application Substances 0.000 claims description 3
- 125000000956 methoxy group Chemical group [H]C([H])([H])O* 0.000 claims description 3
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 claims description 3
- 239000011593 sulfur Chemical group 0.000 claims description 3
- QKZZLSMOLMJMTM-GOSISDBHSA-N COC1=CC=C2NC=C(CCNC(N[C@H]3C(CC4)CCN4C3)=S)C2=C1 Chemical compound COC1=CC=C2NC=C(CCNC(N[C@H]3C(CC4)CCN4C3)=S)C2=C1 QKZZLSMOLMJMTM-GOSISDBHSA-N 0.000 claims description 2
- 229940123457 Free radical scavenger Drugs 0.000 claims description 2
- HQPYEOUYACONEJ-KRWDZBQOSA-N S=C(NCCC(C1=C2)=CNC1=CC=C2Cl)N[C@@H]1C(CC2)CCN2C1 Chemical compound S=C(NCCC(C1=C2)=CNC1=CC=C2Cl)N[C@@H]1C(CC2)CCN2C1 HQPYEOUYACONEJ-KRWDZBQOSA-N 0.000 claims description 2
- ZTIMMJAIGQJQEO-KRWDZBQOSA-N S=C(NCCC1=CNC2=CC=CC=C12)N[C@@H]1C(CC2)CCN2C1 Chemical compound S=C(NCCC1=CNC2=CC=CC=C12)N[C@@H]1C(CC2)CCN2C1 ZTIMMJAIGQJQEO-KRWDZBQOSA-N 0.000 claims description 2
- ZTIMMJAIGQJQEO-QGZVFWFLSA-N S=C(NCCC1=CNC2=CC=CC=C12)N[C@H]1C(CC2)CCN2C1 Chemical compound S=C(NCCC1=CNC2=CC=CC=C12)N[C@H]1C(CC2)CCN2C1 ZTIMMJAIGQJQEO-QGZVFWFLSA-N 0.000 claims description 2
- 150000007942 carboxylates Chemical class 0.000 claims description 2
- 125000004430 oxygen atom Chemical group O* 0.000 claims description 2
- 230000002093 peripheral effect Effects 0.000 claims description 2
- 239000002516 radical scavenger Substances 0.000 claims description 2
- 125000004434 sulfur atom Chemical group 0.000 claims description 2
- AAMZZFPOTFLQLQ-SFHVURJKSA-N COC1=CC=C2NC=C(CCNC(N[C@@H]3C(CC4)CCN4C3)=O)C2=C1 Chemical compound COC1=CC=C2NC=C(CCNC(N[C@@H]3C(CC4)CCN4C3)=O)C2=C1 AAMZZFPOTFLQLQ-SFHVURJKSA-N 0.000 claims 1
- AAMZZFPOTFLQLQ-GOSISDBHSA-N COC1=CC=C2NC=C(CCNC(N[C@H]3C(CC4)CCN4C3)=O)C2=C1 Chemical compound COC1=CC=C2NC=C(CCNC(N[C@H]3C(CC4)CCN4C3)=O)C2=C1 AAMZZFPOTFLQLQ-GOSISDBHSA-N 0.000 claims 1
- 229940123925 Nicotinic receptor agonist Drugs 0.000 claims 1
- FWEZFGIJBIGAKG-KRWDZBQOSA-N O=C(NCCC(C1=C2)=CNC1=CC=C2Cl)N[C@@H]1C(CC2)CCN2C1 Chemical compound O=C(NCCC(C1=C2)=CNC1=CC=C2Cl)N[C@@H]1C(CC2)CCN2C1 FWEZFGIJBIGAKG-KRWDZBQOSA-N 0.000 claims 1
- PVRPHYRCCGOKNT-KRWDZBQOSA-N O=C(NCCC1=CNC2=CC=CC=C12)N[C@@H]1C(CC2)CCN2C1 Chemical compound O=C(NCCC1=CNC2=CC=CC=C12)N[C@@H]1C(CC2)CCN2C1 PVRPHYRCCGOKNT-KRWDZBQOSA-N 0.000 claims 1
- PVRPHYRCCGOKNT-QGZVFWFLSA-N O=C(NCCC1=CNC2=CC=CC=C12)N[C@H]1C(CC2)CCN2C1 Chemical compound O=C(NCCC1=CNC2=CC=CC=C12)N[C@H]1C(CC2)CCN2C1 PVRPHYRCCGOKNT-QGZVFWFLSA-N 0.000 claims 1
- 150000001412 amines Chemical class 0.000 claims 1
- 230000006806 disease prevention Effects 0.000 claims 1
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 93
- JGFZNNIVVJXRND-UHFFFAOYSA-N N,N-Diisopropylethylamine (DIPEA) Chemical compound CCN(C(C)C)C(C)C JGFZNNIVVJXRND-UHFFFAOYSA-N 0.000 description 30
- 210000004027 cell Anatomy 0.000 description 25
- 102000019315 Nicotinic acetylcholine receptors Human genes 0.000 description 24
- 108050006807 Nicotinic acetylcholine receptors Proteins 0.000 description 24
- 150000003254 radicals Chemical class 0.000 description 16
- 230000000324 neuroprotective effect Effects 0.000 description 14
- 238000003818 flash chromatography Methods 0.000 description 12
- OKKJLVBELUTLKV-VMNATFBRSA-N methanol-d1 Chemical compound [2H]OC OKKJLVBELUTLKV-VMNATFBRSA-N 0.000 description 12
- 238000004007 reversed phase HPLC Methods 0.000 description 12
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 9
- 239000000556 agonist Substances 0.000 description 9
- 230000004044 response Effects 0.000 description 9
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 8
- 238000002474 experimental method Methods 0.000 description 8
- YJPIGAIKUZMOQA-UHFFFAOYSA-N Melatonin Natural products COC1=CC=C2N(C(C)=O)C=C(CCN)C2=C1 YJPIGAIKUZMOQA-UHFFFAOYSA-N 0.000 description 7
- 201000010099 disease Diseases 0.000 description 7
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 description 7
- DRLFMBDRBRZALE-UHFFFAOYSA-N melatonin Chemical compound COC1=CC=C2NC=C(CCNC(C)=O)C2=C1 DRLFMBDRBRZALE-UHFFFAOYSA-N 0.000 description 7
- 229960003987 melatonin Drugs 0.000 description 7
- 230000036542 oxidative stress Effects 0.000 description 7
- 230000000638 stimulation Effects 0.000 description 7
- 102000013498 tau Proteins Human genes 0.000 description 7
- 108010026424 tau Proteins Proteins 0.000 description 7
- REUAXQZIRFXQML-SSDOTTSWSA-N (3s)-1-azabicyclo[2.2.2]octan-3-amine Chemical compound C1CC2[C@H](N)CN1CC2 REUAXQZIRFXQML-SSDOTTSWSA-N 0.000 description 6
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 6
- GLEVLJDDWXEYCO-UHFFFAOYSA-N Trolox Chemical compound O1C(C)(C(O)=O)CCC2=C1C(C)=C(C)C(O)=C2C GLEVLJDDWXEYCO-UHFFFAOYSA-N 0.000 description 6
- OIPILFWXSMYKGL-UHFFFAOYSA-N acetylcholine Chemical compound CC(=O)OCC[N+](C)(C)C OIPILFWXSMYKGL-UHFFFAOYSA-N 0.000 description 6
- 229960004373 acetylcholine Drugs 0.000 description 6
- 230000000694 effects Effects 0.000 description 6
- 238000005259 measurement Methods 0.000 description 6
- 230000004112 neuroprotection Effects 0.000 description 6
- VEFJHAYOIAAXEU-UHFFFAOYSA-N okadaic acid Natural products CC(CC(O)C1OC2CCC3(CCC(O3)C=CC(C)C4CC(=CC5(OC(CC(C)(O)C(=O)O)CCC5O)O4)C)OC2C(O)C1C)C6OC7(CCCCO7)CCC6C VEFJHAYOIAAXEU-UHFFFAOYSA-N 0.000 description 6
- QNDVLZJODHBUFM-WFXQOWMNSA-N okadaic acid Chemical compound C([C@H](O1)[C@H](C)/C=C/[C@H]2CC[C@@]3(CC[C@H]4O[C@@H](C([C@@H](O)[C@@H]4O3)=C)[C@@H](O)C[C@H](C)[C@@H]3[C@@H](CC[C@@]4(OCCCC4)O3)C)O2)C(C)=C[C@]21O[C@H](C[C@@](C)(O)C(O)=O)CC[C@H]2O QNDVLZJODHBUFM-WFXQOWMNSA-N 0.000 description 6
- 230000004083 survival effect Effects 0.000 description 6
- CEIIEALEIHQDBX-UHFFFAOYSA-N 1-(5-chloro-2,4-dimethoxyphenyl)-3-(5-methyl-3-isoxazolyl)urea Chemical compound C1=C(Cl)C(OC)=CC(OC)=C1NC(=O)NC1=NOC(C)=C1 CEIIEALEIHQDBX-UHFFFAOYSA-N 0.000 description 5
- 238000001644 13C nuclear magnetic resonance spectroscopy Methods 0.000 description 5
- 238000005160 1H NMR spectroscopy Methods 0.000 description 5
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical group [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 5
- 206010029260 Neuroblastoma Diseases 0.000 description 5
- 238000002835 absorbance Methods 0.000 description 5
- 230000004913 activation Effects 0.000 description 5
- 230000032683 aging Effects 0.000 description 5
- 229910052791 calcium Inorganic materials 0.000 description 5
- 239000011575 calcium Substances 0.000 description 5
- 125000004432 carbon atom Chemical group C* 0.000 description 5
- 230000003833 cell viability Effects 0.000 description 5
- 238000007796 conventional method Methods 0.000 description 5
- 229940079593 drug Drugs 0.000 description 5
- 230000007760 free radical scavenging Effects 0.000 description 5
- 229910052736 halogen Inorganic materials 0.000 description 5
- 150000002367 halogens Chemical group 0.000 description 5
- 125000000623 heterocyclic group Chemical group 0.000 description 5
- 230000006951 hyperphosphorylation Effects 0.000 description 5
- 239000003642 reactive oxygen metabolite Substances 0.000 description 5
- 102000005962 receptors Human genes 0.000 description 5
- 108020003175 receptors Proteins 0.000 description 5
- MNULEGDCPYONBU-UIXCWHRQSA-N (1R,4E,5'S,6S,6'S,7R,8S,10R,11R,12S,14R,15S,16R,18Z,20Z,22R,25S,27R,28S,29R)-22-ethyl-7,11,14,15-tetrahydroxy-6'-[(2R)-2-hydroxypropyl]-5',6,8,10,12,14,16,28,29-nonamethylspiro[2,26-dioxabicyclo[23.3.1]nonacosa-4,18,20-triene-27,2'-oxane]-3,9,13-trione Polymers CC[C@@H]1CC[C@@H]2O[C@]3(CC[C@H](C)[C@H](C[C@@H](C)O)O3)[C@@H](C)[C@H](OC(=O)\C=C\[C@H](C)[C@@H](O)[C@H](C)C(=O)[C@H](C)[C@@H](O)[C@H](C)C(=O)[C@](C)(O)[C@@H](O)[C@H](C)C\C=C/C=C\1)[C@@H]2C MNULEGDCPYONBU-UIXCWHRQSA-N 0.000 description 4
- MNULEGDCPYONBU-CBLVMMTCSA-N (1R,4Z,5'S,6S,6'S,7R,8S,10R,11R,12S,14R,15S,16R,18Z,20Z,22R,25S,27R,28S,29R)-22-ethyl-7,11,14,15-tetrahydroxy-6'-[(2R)-2-hydroxypropyl]-5',6,8,10,12,14,16,28,29-nonamethylspiro[2,26-dioxabicyclo[23.3.1]nonacosa-4,18,20-triene-27,2'-oxane]-3,9,13-trione Polymers CC[C@@H]1CC[C@@H]2O[C@]3(CC[C@H](C)[C@H](C[C@@H](C)O)O3)[C@@H](C)[C@H](OC(=O)\C=C/[C@H](C)[C@@H](O)[C@H](C)C(=O)[C@H](C)[C@@H](O)[C@H](C)C(=O)[C@](C)(O)[C@@H](O)[C@H](C)C\C=C/C=C\1)[C@@H]2C MNULEGDCPYONBU-CBLVMMTCSA-N 0.000 description 4
- MNULEGDCPYONBU-WABYXMGOSA-N (1S,4E,5'R,6R,6'R,7S,8R,10S,11S,12R,14S,15R,16S,18E,22S,25R,27S,28R,29S)-22-ethyl-7,11,14,15-tetrahydroxy-6'-(2-hydroxypropyl)-5',6,8,10,12,14,16,28,29-nonamethylspiro[2,26-dioxabicyclo[23.3.1]nonacosa-4,18,20-triene-27,2'-oxane]-3,9,13-trione Polymers CC[C@H]1CC[C@H]2O[C@@]3(CC[C@@H](C)[C@@H](CC(C)O)O3)[C@H](C)[C@@H](OC(=O)\C=C\[C@@H](C)[C@H](O)[C@@H](C)C(=O)[C@@H](C)[C@H](O)[C@@H](C)C(=O)[C@@](C)(O)[C@H](O)[C@@H](C)C\C=C\C=C1)[C@H]2C MNULEGDCPYONBU-WABYXMGOSA-N 0.000 description 4
- MNULEGDCPYONBU-QECWTJOCSA-N (1r,4s,5e,5'r,6'r,7e,10s,11r,12s,14r,15s,16s,18r,19s,20r,21e,25s,26r,27s,29s)-4-ethyl-11,12,15,19-tetrahydroxy-6'-(2-hydroxypropyl)-5',10,12,14,16,18,20,26,29-nonamethylspiro[24,28-dioxabicyclo[23.3.1]nonacosa-5,7,21-triene-27,2'-oxane]-13,17,23-trione Polymers O([C@@H]1CC[C@@H](/C=C/C=C/C[C@H](C)[C@@H](O)[C@](C)(O)C(=O)[C@H](C)[C@@H](O)[C@H](C)C(=O)[C@H](C)[C@@H](O)[C@H](C)/C=C/C(=O)O[C@H]([C@H]2C)[C@H]1C)CC)[C@]12CC[C@@H](C)[C@@H](CC(C)O)O1 MNULEGDCPYONBU-QECWTJOCSA-N 0.000 description 4
- MNULEGDCPYONBU-WMBHJXFZSA-N (1r,4s,5e,5'r,6'r,7e,10s,11r,12s,14r,15s,16s,18r,19s,20r,21e,25s,26r,27s,29s)-4-ethyl-11,12,15,19-tetrahydroxy-6'-[(2s)-2-hydroxypropyl]-5',10,12,14,16,18,20,26,29-nonamethylspiro[24,28-dioxabicyclo[23.3.1]nonacosa-5,7,21-triene-27,2'-oxane]-13,17,23-trio Polymers O([C@@H]1CC[C@@H](/C=C/C=C/C[C@H](C)[C@@H](O)[C@](C)(O)C(=O)[C@H](C)[C@@H](O)[C@H](C)C(=O)[C@H](C)[C@@H](O)[C@H](C)/C=C/C(=O)O[C@H]([C@H]2C)[C@H]1C)CC)[C@]12CC[C@@H](C)[C@@H](C[C@H](C)O)O1 MNULEGDCPYONBU-WMBHJXFZSA-N 0.000 description 4
- MNULEGDCPYONBU-BOXGPLBDSA-N (1r,4s,5e,5'r,6'r,7e,10s,11s,12s,14r,15s,16s,18r,19s,20r,21e,25s,26r,27s,29s)-4-ethyl-11,12,15,19-tetrahydroxy-6'-[(2s)-2-hydroxypropyl]-5',10,12,14,16,18,20,26,29-nonamethylspiro[24,28-dioxabicyclo[23.3.1]nonacosa-5,7,21-triene-27,2'-oxane]-13,17,23-trio Polymers O([C@@H]1CC[C@@H](/C=C/C=C/C[C@H](C)[C@H](O)[C@](C)(O)C(=O)[C@H](C)[C@@H](O)[C@H](C)C(=O)[C@H](C)[C@@H](O)[C@H](C)/C=C/C(=O)O[C@H]([C@H]2C)[C@H]1C)CC)[C@]12CC[C@@H](C)[C@@H](C[C@H](C)O)O1 MNULEGDCPYONBU-BOXGPLBDSA-N 0.000 description 4
- REUAXQZIRFXQML-ZETCQYMHSA-N (3r)-1-azabicyclo[2.2.2]octan-3-amine Chemical compound C1CC2[C@@H](N)CN1CC2 REUAXQZIRFXQML-ZETCQYMHSA-N 0.000 description 4
- MNULEGDCPYONBU-YOKYSHDFSA-N (5'R,10S,11R,12S,14S,15R,16R,18R,19S,20R,26R,29S)-4-ethyl-11,12,15,19-tetrahydroxy-6'-[(2S)-2-hydroxypropyl]-5',10,12,14,16,18,20,26,29-nonamethylspiro[24,28-dioxabicyclo[23.3.1]nonacosa-5,7,21-triene-27,2'-oxane]-13,17,23-trione Polymers C([C@H](C)[C@@H](O)[C@](C)(O)C(=O)[C@@H](C)[C@H](O)[C@@H](C)C(=O)[C@H](C)[C@@H](O)[C@H](C)C=CC(=O)OC([C@H]1C)[C@H]2C)C=CC=CC(CC)CCC2OC21CC[C@@H](C)C(C[C@H](C)O)O2 MNULEGDCPYONBU-YOKYSHDFSA-N 0.000 description 4
- MNULEGDCPYONBU-VVXVDZGXSA-N (5e,5'r,7e,10s,11r,12s,14s,15r,16r,18r,19s,20r,21e,26r,29s)-4-ethyl-11,12,15,19-tetrahydroxy-6'-[(2s)-2-hydroxypropyl]-5',10,12,14,16,18,20,26,29-nonamethylspiro[24,28-dioxabicyclo[23.3.1]nonacosa-5,7,21-triene-27,2'-oxane]-13,17,23-trione Polymers C([C@H](C)[C@@H](O)[C@](C)(O)C(=O)[C@@H](C)[C@H](O)[C@@H](C)C(=O)[C@H](C)[C@@H](O)[C@H](C)/C=C/C(=O)OC([C@H]1C)[C@H]2C)\C=C\C=C\C(CC)CCC2OC21CC[C@@H](C)C(C[C@H](C)O)O2 MNULEGDCPYONBU-VVXVDZGXSA-N 0.000 description 4
- MNULEGDCPYONBU-MQLHLVDXSA-N CC[C@@H]1CC[C@@H]2O[C@]3(CC[C@H](C)[C@H](C[C@@H](C)O)O3)[C@@H](C)[C@H](OC(=O)\C=C\[C@H](C)[C@@H](O)[C@H](C)C(=O)[C@H](C)[C@@H](O)[C@H](C)C(=O)[C@](C)(O)[C@@H](O)[C@H](C)C\C=C\C=C\1)C2C Polymers CC[C@@H]1CC[C@@H]2O[C@]3(CC[C@H](C)[C@H](C[C@@H](C)O)O3)[C@@H](C)[C@H](OC(=O)\C=C\[C@H](C)[C@@H](O)[C@H](C)C(=O)[C@H](C)[C@@H](O)[C@H](C)C(=O)[C@](C)(O)[C@@H](O)[C@H](C)C\C=C\C=C\1)C2C MNULEGDCPYONBU-MQLHLVDXSA-N 0.000 description 4
- 210000004556 brain Anatomy 0.000 description 4
- 239000000872 buffer Substances 0.000 description 4
- 230000034994 death Effects 0.000 description 4
- 231100000517 death Toxicity 0.000 description 4
- MNULEGDCPYONBU-UHFFFAOYSA-N oligomycin A Natural products CC1C(C2C)OC(=O)C=CC(C)C(O)C(C)C(=O)C(C)C(O)C(C)C(=O)C(C)(O)C(O)C(C)CC=CC=CC(CC)CCC2OC21CCC(C)C(CC(C)O)O2 MNULEGDCPYONBU-UHFFFAOYSA-N 0.000 description 4
- MNULEGDCPYONBU-AWJDAWNUSA-N oligomycin A Polymers O([C@H]1CC[C@H](/C=C/C=C/C[C@@H](C)[C@H](O)[C@@](C)(O)C(=O)[C@@H](C)[C@H](O)[C@@H](C)C(=O)[C@@H](C)[C@H](O)[C@@H](C)/C=C/C(=O)O[C@@H]([C@@H]2C)[C@@H]1C)CC)[C@@]12CC[C@H](C)[C@H](C[C@@H](C)O)O1 MNULEGDCPYONBU-AWJDAWNUSA-N 0.000 description 4
- 239000013641 positive control Substances 0.000 description 4
- 230000009467 reduction Effects 0.000 description 4
- 229940080817 rotenone Drugs 0.000 description 4
- JUVIOZPCNVVQFO-UHFFFAOYSA-N rotenone Natural products O1C2=C3CC(C(C)=C)OC3=CC=C2C(=O)C2C1COC1=C2C=C(OC)C(OC)=C1 JUVIOZPCNVVQFO-UHFFFAOYSA-N 0.000 description 4
- 125000001424 substituent group Chemical group 0.000 description 4
- 231100000419 toxicity Toxicity 0.000 description 4
- 230000001988 toxicity Effects 0.000 description 4
- 231100000765 toxin Toxicity 0.000 description 4
- JILSJIQENRELKK-UHFFFAOYSA-N 3-(2-isothiocyanatoethyl)-1h-indole Chemical compound C1=CC=C2C(CCN=C=S)=CNC2=C1 JILSJIQENRELKK-UHFFFAOYSA-N 0.000 description 3
- VHUUQVKOLVNVRT-UHFFFAOYSA-N Ammonium hydroxide Chemical compound [NH4+].[OH-] VHUUQVKOLVNVRT-UHFFFAOYSA-N 0.000 description 3
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 3
- 241001465754 Metazoa Species 0.000 description 3
- NNHVUVXPQSROFS-UHFFFAOYSA-N N-[2-(1H-indol-3-yl)ethyl]imidazole-1-carboxamide Chemical class C=1NC2=CC=CC=C2C=1CCNC(=O)N1C=CN=C1 NNHVUVXPQSROFS-UHFFFAOYSA-N 0.000 description 3
- RWRDLPDLKQPQOW-UHFFFAOYSA-N Pyrrolidine Chemical compound C1CCNC1 RWRDLPDLKQPQOW-UHFFFAOYSA-N 0.000 description 3
- 238000000692 Student's t-test Methods 0.000 description 3
- 235000011114 ammonium hydroxide Nutrition 0.000 description 3
- 230000000903 blocking effect Effects 0.000 description 3
- 230000006378 damage Effects 0.000 description 3
- 238000004128 high performance liquid chromatography Methods 0.000 description 3
- RAXXELZNTBOGNW-UHFFFAOYSA-N imidazole Natural products C1=CNC=N1 RAXXELZNTBOGNW-UHFFFAOYSA-N 0.000 description 3
- 238000011534 incubation Methods 0.000 description 3
- 150000002500 ions Chemical class 0.000 description 3
- 230000014759 maintenance of location Effects 0.000 description 3
- 239000002609 medium Substances 0.000 description 3
- 210000002569 neuron Anatomy 0.000 description 3
- 230000037361 pathway Effects 0.000 description 3
- 238000011533 pre-incubation Methods 0.000 description 3
- 239000000243 solution Substances 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- YLQBMQCUIZJEEH-UHFFFAOYSA-N tetrahydrofuran Natural products C=1C=COC=1 YLQBMQCUIZJEEH-UHFFFAOYSA-N 0.000 description 3
- 239000003053 toxin Substances 0.000 description 3
- 125000002023 trifluoromethyl group Chemical group FC(F)(F)* 0.000 description 3
- SNICXCGAKADSCV-JTQLQIEISA-N (-)-Nicotine Chemical compound CN1CCC[C@H]1C1=CC=CN=C1 SNICXCGAKADSCV-JTQLQIEISA-N 0.000 description 2
- GHYOCDFICYLMRF-UTIIJYGPSA-N (2S,3R)-N-[(2S)-3-(cyclopenten-1-yl)-1-[(2R)-2-methyloxiran-2-yl]-1-oxopropan-2-yl]-3-hydroxy-3-(4-methoxyphenyl)-2-[[(2S)-2-[(2-morpholin-4-ylacetyl)amino]propanoyl]amino]propanamide Chemical compound C1(=CCCC1)C[C@@H](C(=O)[C@@]1(OC1)C)NC([C@H]([C@@H](C1=CC=C(C=C1)OC)O)NC([C@H](C)NC(CN1CCOCC1)=O)=O)=O GHYOCDFICYLMRF-UTIIJYGPSA-N 0.000 description 2
- QFLWZFQWSBQYPS-AWRAUJHKSA-N (3S)-3-[[(2S)-2-[[(2S)-2-[5-[(3aS,6aR)-2-oxo-1,3,3a,4,6,6a-hexahydrothieno[3,4-d]imidazol-4-yl]pentanoylamino]-3-methylbutanoyl]amino]-3-(4-hydroxyphenyl)propanoyl]amino]-4-[1-bis(4-chlorophenoxy)phosphorylbutylamino]-4-oxobutanoic acid Chemical compound CCCC(NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](Cc1ccc(O)cc1)NC(=O)[C@@H](NC(=O)CCCCC1SC[C@@H]2NC(=O)N[C@H]12)C(C)C)P(=O)(Oc1ccc(Cl)cc1)Oc1ccc(Cl)cc1 QFLWZFQWSBQYPS-AWRAUJHKSA-N 0.000 description 2
- ASJSXUWOFZATJM-UHFFFAOYSA-N 2-(3,5-diphenyl-1h-tetrazol-2-yl)-4,5-dimethyl-1,3-thiazole Chemical compound S1C(C)=C(C)N=C1N1N(C=2C=CC=CC=2)NC(C=2C=CC=CC=2)=N1 ASJSXUWOFZATJM-UHFFFAOYSA-N 0.000 description 2
- NXNJINJDHJRFFK-UHFFFAOYSA-N 3-(2-isothiocyanatoethyl)-5-methoxy-1h-indole Chemical compound COC1=CC=C2NC=C(CCN=C=S)C2=C1 NXNJINJDHJRFFK-UHFFFAOYSA-N 0.000 description 2
- MYTGFBZJLDLWQG-UHFFFAOYSA-N 5-chloro-1h-indole Chemical compound ClC1=CC=C2NC=CC2=C1 MYTGFBZJLDLWQG-UHFFFAOYSA-N 0.000 description 2
- DWAQDRSOVMLGRQ-UHFFFAOYSA-N 5-methoxyindole Chemical compound COC1=CC=C2NC=CC2=C1 DWAQDRSOVMLGRQ-UHFFFAOYSA-N 0.000 description 2
- KDCGOANMDULRCW-UHFFFAOYSA-N 7H-purine Chemical compound N1=CNC2=NC=NC2=C1 KDCGOANMDULRCW-UHFFFAOYSA-N 0.000 description 2
- 206010012289 Dementia Diseases 0.000 description 2
- 241000282412 Homo Species 0.000 description 2
- SIKJAQJRHWYJAI-UHFFFAOYSA-N Indole Chemical compound C1=CC=C2NC=CC2=C1 SIKJAQJRHWYJAI-UHFFFAOYSA-N 0.000 description 2
- YNAVUWVOSKDBBP-UHFFFAOYSA-N Morpholine Chemical compound C1COCCN1 YNAVUWVOSKDBBP-UHFFFAOYSA-N 0.000 description 2
- 238000005481 NMR spectroscopy Methods 0.000 description 2
- GLUUGHFHXGJENI-UHFFFAOYSA-N Piperazine Chemical compound C1CNCCN1 GLUUGHFHXGJENI-UHFFFAOYSA-N 0.000 description 2
- NQRYJNQNLNOLGT-UHFFFAOYSA-N Piperidine Chemical compound C1CCNCC1 NQRYJNQNLNOLGT-UHFFFAOYSA-N 0.000 description 2
- JUJWROOIHBZHMG-UHFFFAOYSA-N Pyridine Chemical compound C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 2
- KAESVJOAVNADME-UHFFFAOYSA-N Pyrrole Chemical compound C=1C=CNC=1 KAESVJOAVNADME-UHFFFAOYSA-N 0.000 description 2
- SMWDFEZZVXVKRB-UHFFFAOYSA-N Quinoline Chemical compound N1=CC=CC2=CC=CC=C21 SMWDFEZZVXVKRB-UHFFFAOYSA-N 0.000 description 2
- FFBCGXUTMXQAPN-UHFFFAOYSA-N S=C=NCCC(C1=C2)=CNC1=CC=C2Cl Chemical compound S=C=NCCC(C1=C2)=CNC1=CC=C2Cl FFBCGXUTMXQAPN-UHFFFAOYSA-N 0.000 description 2
- 238000003639 Student–Newman–Keuls (SNK) method Methods 0.000 description 2
- WYURNTSHIVDZCO-UHFFFAOYSA-N Tetrahydrofuran Chemical compound C1CCOC1 WYURNTSHIVDZCO-UHFFFAOYSA-N 0.000 description 2
- 238000009825 accumulation Methods 0.000 description 2
- 125000001931 aliphatic group Chemical group 0.000 description 2
- 125000004453 alkoxycarbonyl group Chemical group 0.000 description 2
- IOJUPLGTWVMSFF-UHFFFAOYSA-N benzothiazole Chemical compound C1=CC=C2SC=NC2=C1 IOJUPLGTWVMSFF-UHFFFAOYSA-N 0.000 description 2
- 230000004071 biological effect Effects 0.000 description 2
- 230000015572 biosynthetic process Effects 0.000 description 2
- 230000037396 body weight Effects 0.000 description 2
- 230000036755 cellular response Effects 0.000 description 2
- 230000001713 cholinergic effect Effects 0.000 description 2
- 230000003920 cognitive function Effects 0.000 description 2
- 238000002648 combination therapy Methods 0.000 description 2
- 229940125904 compound 1 Drugs 0.000 description 2
- 229940125773 compound 10 Drugs 0.000 description 2
- 229940125797 compound 12 Drugs 0.000 description 2
- 229940125782 compound 2 Drugs 0.000 description 2
- 229940126214 compound 3 Drugs 0.000 description 2
- 229940125898 compound 5 Drugs 0.000 description 2
- 239000003937 drug carrier Substances 0.000 description 2
- 239000012442 inert solvent Substances 0.000 description 2
- 238000007918 intramuscular administration Methods 0.000 description 2
- ZLVXBBHTMQJRSX-VMGNSXQWSA-N jdtic Chemical compound C1([C@]2(C)CCN(C[C@@H]2C)C[C@H](C(C)C)NC(=O)[C@@H]2NCC3=CC(O)=CC=C3C2)=CC=CC(O)=C1 ZLVXBBHTMQJRSX-VMGNSXQWSA-N 0.000 description 2
- 239000007788 liquid Substances 0.000 description 2
- 210000003470 mitochondria Anatomy 0.000 description 2
- 230000004065 mitochondrial dysfunction Effects 0.000 description 2
- 125000002950 monocyclic group Chemical group 0.000 description 2
- 230000035772 mutation Effects 0.000 description 2
- VMGAPWLDMVPYIA-HIDZBRGKSA-N n'-amino-n-iminomethanimidamide Chemical compound N\N=C\N=N VMGAPWLDMVPYIA-HIDZBRGKSA-N 0.000 description 2
- 210000002682 neurofibrillary tangle Anatomy 0.000 description 2
- 229960002715 nicotine Drugs 0.000 description 2
- SNICXCGAKADSCV-UHFFFAOYSA-N nicotine Natural products CN1CCCC1C1=CC=CN=C1 SNICXCGAKADSCV-UHFFFAOYSA-N 0.000 description 2
- 238000001543 one-way ANOVA Methods 0.000 description 2
- 230000008520 organization Effects 0.000 description 2
- 230000004792 oxidative damage Effects 0.000 description 2
- 230000001590 oxidative effect Effects 0.000 description 2
- 230000000144 pharmacologic effect Effects 0.000 description 2
- 230000001105 regulatory effect Effects 0.000 description 2
- BOLDJAUMGUJJKM-LSDHHAIUSA-N renifolin D Natural products CC(=C)[C@@H]1Cc2c(O)c(O)ccc2[C@H]1CC(=O)c3ccc(O)cc3O BOLDJAUMGUJJKM-LSDHHAIUSA-N 0.000 description 2
- 229920006395 saturated elastomer Polymers 0.000 description 2
- 230000019491 signal transduction Effects 0.000 description 2
- 239000007787 solid Substances 0.000 description 2
- 241000894007 species Species 0.000 description 2
- 238000003756 stirring Methods 0.000 description 2
- UCSJYZPVAKXKNQ-HZYVHMACSA-N streptomycin Chemical compound CN[C@H]1[C@H](O)[C@@H](O)[C@H](CO)O[C@H]1O[C@@H]1[C@](C=O)(O)[C@H](C)O[C@H]1O[C@@H]1[C@@H](NC(N)=N)[C@H](O)[C@@H](NC(N)=N)[C@H](O)[C@H]1O UCSJYZPVAKXKNQ-HZYVHMACSA-N 0.000 description 2
- 238000007920 subcutaneous administration Methods 0.000 description 2
- 239000000725 suspension Substances 0.000 description 2
- 230000005062 synaptic transmission Effects 0.000 description 2
- 238000003786 synthesis reaction Methods 0.000 description 2
- 238000012360 testing method Methods 0.000 description 2
- 230000001225 therapeutic effect Effects 0.000 description 2
- 231100000331 toxic Toxicity 0.000 description 2
- 230000002588 toxic effect Effects 0.000 description 2
- 102000038650 voltage-gated calcium channel activity Human genes 0.000 description 2
- 108091023044 voltage-gated calcium channel activity Proteins 0.000 description 2
- 125000006273 (C1-C3) alkyl group Chemical group 0.000 description 1
- 125000004169 (C1-C6) alkyl group Chemical group 0.000 description 1
- 102000010400 1-phosphatidylinositol-3-kinase activity proteins Human genes 0.000 description 1
- HYZJCKYKOHLVJF-UHFFFAOYSA-N 1H-benzimidazole Chemical compound C1=CC=C2NC=NC2=C1 HYZJCKYKOHLVJF-UHFFFAOYSA-N 0.000 description 1
- YBYIRNPNPLQARY-UHFFFAOYSA-N 1H-indene Natural products C1=CC=C2CC=CC2=C1 YBYIRNPNPLQARY-UHFFFAOYSA-N 0.000 description 1
- LXEKPEMOWBOYRF-QDBORUFSSA-N AAPH Chemical compound Cl.Cl.NC(=N)C(C)(C)\N=N\C(C)(C)C(N)=N LXEKPEMOWBOYRF-QDBORUFSSA-N 0.000 description 1
- 208000000044 Amnesia Diseases 0.000 description 1
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- 125000000882 C2-C6 alkenyl group Chemical group 0.000 description 1
- 125000003601 C2-C6 alkynyl group Chemical group 0.000 description 1
- QKZZLSMOLMJMTM-SFHVURJKSA-N COC1=CC=C2NC=C(CCNC(N[C@@H]3C(CC4)CCN4C3)=S)C2=C1 Chemical compound COC1=CC=C2NC=C(CCNC(N[C@@H]3C(CC4)CCN4C3)=S)C2=C1 QKZZLSMOLMJMTM-SFHVURJKSA-N 0.000 description 1
- 101000741929 Caenorhabditis elegans Serine/threonine-protein phosphatase 2A catalytic subunit Proteins 0.000 description 1
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 1
- 108091005462 Cation channels Proteins 0.000 description 1
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 1
- OUVXYXNWSVIOSJ-UHFFFAOYSA-N Fluo-4 Chemical compound CC1=CC=C(N(CC(O)=O)CC(O)=O)C(OCCOC=2C(=CC=C(C=2)C2=C3C=C(F)C(=O)C=C3OC3=CC(O)=C(F)C=C32)N(CC(O)=O)CC(O)=O)=C1 OUVXYXNWSVIOSJ-UHFFFAOYSA-N 0.000 description 1
- 239000007995 HEPES buffer Substances 0.000 description 1
- 206010020751 Hypersensitivity Diseases 0.000 description 1
- 206010061218 Inflammation Diseases 0.000 description 1
- 241000124008 Mammalia Species 0.000 description 1
- 108020005196 Mitochondrial DNA Proteins 0.000 description 1
- 229940123859 Nicotinic receptor antagonist Drugs 0.000 description 1
- 108091007960 PI3Ks Proteins 0.000 description 1
- 229930182555 Penicillin Natural products 0.000 description 1
- JGSARLDLIJGVTE-MBNYWOFBSA-N Penicillin G Chemical compound N([C@H]1[C@H]2SC([C@@H](N2C1=O)C(O)=O)(C)C)C(=O)CC1=CC=CC=C1 JGSARLDLIJGVTE-MBNYWOFBSA-N 0.000 description 1
- 108700019535 Phosphoprotein Phosphatases Proteins 0.000 description 1
- 102000045595 Phosphoprotein Phosphatases Human genes 0.000 description 1
- CZPWVGJYEJSRLH-UHFFFAOYSA-N Pyrimidine Chemical compound C1=CN=CN=C1 CZPWVGJYEJSRLH-UHFFFAOYSA-N 0.000 description 1
- 230000002292 Radical scavenging effect Effects 0.000 description 1
- ZTIMMJAIGQJQEO-UHFFFAOYSA-N S=C(NCCC1=CNC2=CC=CC=C12)NC1C(CC2)CCN2C1 Chemical compound S=C(NCCC1=CNC2=CC=CC=C12)NC1C(CC2)CCN2C1 ZTIMMJAIGQJQEO-UHFFFAOYSA-N 0.000 description 1
- MTCFGRXMJLQNBG-UHFFFAOYSA-N Serine Natural products OCC(N)C(O)=O MTCFGRXMJLQNBG-UHFFFAOYSA-N 0.000 description 1
- 230000002159 abnormal effect Effects 0.000 description 1
- 102000015296 acetylcholine-gated cation-selective channel activity proteins Human genes 0.000 description 1
- 108040006409 acetylcholine-gated cation-selective channel activity proteins Proteins 0.000 description 1
- QOMNQGZXFYNBNG-UHFFFAOYSA-N acetyloxymethyl 2-[2-[2-[5-[3-(acetyloxymethoxy)-2,7-difluoro-6-oxoxanthen-9-yl]-2-[bis[2-(acetyloxymethoxy)-2-oxoethyl]amino]phenoxy]ethoxy]-n-[2-(acetyloxymethoxy)-2-oxoethyl]-4-methylanilino]acetate Chemical compound CC(=O)OCOC(=O)CN(CC(=O)OCOC(C)=O)C1=CC=C(C)C=C1OCCOC1=CC(C2=C3C=C(F)C(=O)C=C3OC3=CC(OCOC(C)=O)=C(F)C=C32)=CC=C1N(CC(=O)OCOC(C)=O)CC(=O)OCOC(C)=O QOMNQGZXFYNBNG-UHFFFAOYSA-N 0.000 description 1
- 239000002253 acid Substances 0.000 description 1
- 230000009471 action Effects 0.000 description 1
- 230000003213 activating effect Effects 0.000 description 1
- 239000004480 active ingredient Substances 0.000 description 1
- 125000002252 acyl group Chemical group 0.000 description 1
- 150000004703 alkoxides Chemical group 0.000 description 1
- 125000004414 alkyl thio group Chemical group 0.000 description 1
- 208000030961 allergic reaction Diseases 0.000 description 1
- VREFGVBLTWBCJP-UHFFFAOYSA-N alprazolam Chemical compound C12=CC(Cl)=CC=C2N2C(C)=NN=C2CN=C1C1=CC=CC=C1 VREFGVBLTWBCJP-UHFFFAOYSA-N 0.000 description 1
- 150000001408 amides Chemical class 0.000 description 1
- 229940121363 anti-inflammatory agent Drugs 0.000 description 1
- 239000002260 anti-inflammatory agent Substances 0.000 description 1
- 230000003110 anti-inflammatory effect Effects 0.000 description 1
- 239000000935 antidepressant agent Substances 0.000 description 1
- 229940005513 antidepressants Drugs 0.000 description 1
- 230000006907 apoptotic process Effects 0.000 description 1
- 239000003125 aqueous solvent Substances 0.000 description 1
- 210000001130 astrocyte Anatomy 0.000 description 1
- 235000010290 biphenyl Nutrition 0.000 description 1
- 239000004305 biphenyl Substances 0.000 description 1
- 125000006267 biphenyl group Chemical group 0.000 description 1
- 239000008280 blood Substances 0.000 description 1
- 210000004369 blood Anatomy 0.000 description 1
- 210000000133 brain stem Anatomy 0.000 description 1
- 230000004094 calcium homeostasis Effects 0.000 description 1
- 150000004657 carbamic acid derivatives Chemical class 0.000 description 1
- 239000011203 carbon fibre reinforced carbon Substances 0.000 description 1
- 125000003178 carboxy group Chemical group [H]OC(*)=O 0.000 description 1
- 238000004113 cell culture Methods 0.000 description 1
- 239000003153 chemical reaction reagent Substances 0.000 description 1
- 239000000064 cholinergic agonist Substances 0.000 description 1
- 210000002932 cholinergic neuron Anatomy 0.000 description 1
- 238000004587 chromatography analysis Methods 0.000 description 1
- 208000037976 chronic inflammation Diseases 0.000 description 1
- 230000006020 chronic inflammation Effects 0.000 description 1
- 238000004440 column chromatography Methods 0.000 description 1
- 238000004891 communication Methods 0.000 description 1
- 230000000052 comparative effect Effects 0.000 description 1
- 239000013078 crystal Substances 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 125000004122 cyclic group Chemical group 0.000 description 1
- 125000001995 cyclobutyl group Chemical group [H]C1([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000000113 cyclohexyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C([H])([H])C1([H])[H] 0.000 description 1
- 125000001511 cyclopentyl group Chemical group [H]C1([H])C([H])([H])C([H])([H])C([H])(*)C1([H])[H] 0.000 description 1
- 125000001559 cyclopropyl group Chemical group [H]C1([H])C([H])([H])C1([H])* 0.000 description 1
- 210000000172 cytosol Anatomy 0.000 description 1
- 230000001086 cytosolic effect Effects 0.000 description 1
- 230000030609 dephosphorylation Effects 0.000 description 1
- 238000006209 dephosphorylation reaction Methods 0.000 description 1
- 230000008021 deposition Effects 0.000 description 1
- 230000002074 deregulated effect Effects 0.000 description 1
- 230000003831 deregulation Effects 0.000 description 1
- 239000003085 diluting agent Substances 0.000 description 1
- 208000002173 dizziness Diseases 0.000 description 1
- 239000002552 dosage form Substances 0.000 description 1
- 230000009977 dual effect Effects 0.000 description 1
- 230000008482 dysregulation Effects 0.000 description 1
- 210000002472 endoplasmic reticulum Anatomy 0.000 description 1
- 230000008995 epigenetic change Effects 0.000 description 1
- 230000001073 episodic memory Effects 0.000 description 1
- 239000003797 essential amino acid Substances 0.000 description 1
- 235000020776 essential amino acid Nutrition 0.000 description 1
- 150000002148 esters Chemical class 0.000 description 1
- 230000005284 excitation Effects 0.000 description 1
- 239000012091 fetal bovine serum Substances 0.000 description 1
- 239000007850 fluorescent dye Substances 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- 230000006870 function Effects 0.000 description 1
- 125000000524 functional group Chemical group 0.000 description 1
- 230000002496 gastric effect Effects 0.000 description 1
- ZDXPYRJPNDTMRX-UHFFFAOYSA-N glutamine Natural products OC(=O)C(N)CCC(N)=O ZDXPYRJPNDTMRX-UHFFFAOYSA-N 0.000 description 1
- 239000001963 growth medium Substances 0.000 description 1
- 125000001188 haloalkyl group Chemical group 0.000 description 1
- 125000005842 heteroatom Chemical group 0.000 description 1
- 210000001320 hippocampus Anatomy 0.000 description 1
- 230000013632 homeostatic process Effects 0.000 description 1
- 125000002887 hydroxy group Chemical group [H]O* 0.000 description 1
- 230000001976 improved effect Effects 0.000 description 1
- 238000010874 in vitro model Methods 0.000 description 1
- 125000003454 indenyl group Chemical group C1(C=CC2=CC=CC=C12)* 0.000 description 1
- PZOUSPYUWWUPPK-UHFFFAOYSA-N indole Natural products CC1=CC=CC2=C1C=CN2 PZOUSPYUWWUPPK-UHFFFAOYSA-N 0.000 description 1
- RKJUIXBNRJVNHR-UHFFFAOYSA-N indolenine Natural products C1=CC=C2CC=NC2=C1 RKJUIXBNRJVNHR-UHFFFAOYSA-N 0.000 description 1
- 230000001939 inductive effect Effects 0.000 description 1
- 230000004054 inflammatory process Effects 0.000 description 1
- 230000005764 inhibitory process Effects 0.000 description 1
- 230000003834 intracellular effect Effects 0.000 description 1
- 238000001990 intravenous administration Methods 0.000 description 1
- PNDPGZBMCMUPRI-UHFFFAOYSA-N iodine Chemical compound II PNDPGZBMCMUPRI-UHFFFAOYSA-N 0.000 description 1
- 230000001057 ionotropic effect Effects 0.000 description 1
- 125000001449 isopropyl group Chemical group [H]C([H])([H])C([H])(*)C([H])([H])[H] 0.000 description 1
- ZLTPDFXIESTBQG-UHFFFAOYSA-N isothiazole Chemical compound C=1C=NSC=1 ZLTPDFXIESTBQG-UHFFFAOYSA-N 0.000 description 1
- 231100001231 less toxic Toxicity 0.000 description 1
- 239000003446 ligand Substances 0.000 description 1
- 238000012417 linear regression Methods 0.000 description 1
- 230000003859 lipid peroxidation Effects 0.000 description 1
- 150000002632 lipids Chemical class 0.000 description 1
- 231100000863 loss of memory Toxicity 0.000 description 1
- 210000004324 lymphatic system Anatomy 0.000 description 1
- 239000003578 marine toxin Substances 0.000 description 1
- IMYZQPCYWPFTAG-IQJOONFLSA-N mecamylamine Chemical compound C1C[C@@H]2C(C)(C)[C@@](NC)(C)[C@H]1C2 IMYZQPCYWPFTAG-IQJOONFLSA-N 0.000 description 1
- 229960002525 mecamylamine Drugs 0.000 description 1
- 230000007246 mechanism Effects 0.000 description 1
- BUGYDGFZZOZRHP-UHFFFAOYSA-N memantine Chemical compound C1C(C2)CC3(C)CC1(C)CC2(N)C3 BUGYDGFZZOZRHP-UHFFFAOYSA-N 0.000 description 1
- 229960004640 memantine Drugs 0.000 description 1
- 230000006386 memory function Effects 0.000 description 1
- 230000004066 metabolic change Effects 0.000 description 1
- 230000002503 metabolic effect Effects 0.000 description 1
- 230000004060 metabolic process Effects 0.000 description 1
- 229910052751 metal Inorganic materials 0.000 description 1
- 239000002184 metal Substances 0.000 description 1
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 description 1
- 210000000274 microglia Anatomy 0.000 description 1
- 230000002438 mitochondrial effect Effects 0.000 description 1
- 238000002156 mixing Methods 0.000 description 1
- 125000004108 n-butyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000000740 n-pentyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000004123 n-propyl group Chemical group [H]C([H])([H])C([H])([H])C([H])([H])* 0.000 description 1
- 125000001624 naphthyl group Chemical group 0.000 description 1
- 229930014626 natural product Natural products 0.000 description 1
- 210000000478 neocortex Anatomy 0.000 description 1
- 230000008587 neuronal excitability Effects 0.000 description 1
- 230000007171 neuropathology Effects 0.000 description 1
- 239000004090 neuroprotective agent Substances 0.000 description 1
- 230000003957 neurotransmitter release Effects 0.000 description 1
- 239000003367 nicotinic antagonist Substances 0.000 description 1
- IJGRMHOSHXDMSA-UHFFFAOYSA-N nitrogen Substances N#N IJGRMHOSHXDMSA-UHFFFAOYSA-N 0.000 description 1
- QJGQUHMNIGDVPM-UHFFFAOYSA-N nitrogen group Chemical group [N] QJGQUHMNIGDVPM-UHFFFAOYSA-N 0.000 description 1
- 239000002777 nucleoside Substances 0.000 description 1
- 125000003835 nucleoside group Chemical group 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 239000003960 organic solvent Substances 0.000 description 1
- 238000012261 overproduction Methods 0.000 description 1
- 238000007911 parenteral administration Methods 0.000 description 1
- 230000007170 pathology Effects 0.000 description 1
- 230000001991 pathophysiological effect Effects 0.000 description 1
- 229940049954 penicillin Drugs 0.000 description 1
- 230000035699 permeability Effects 0.000 description 1
- 125000005561 phenanthryl group Chemical group 0.000 description 1
- 125000001997 phenyl group Chemical group [H]C1=C([H])C([H])=C(*)C([H])=C1[H] 0.000 description 1
- ZUOUZKKEUPVFJK-UHFFFAOYSA-N phenylbenzene Natural products C1=CC=CC=C1C1=CC=CC=C1 ZUOUZKKEUPVFJK-UHFFFAOYSA-N 0.000 description 1
- 150000003014 phosphoric acid esters Chemical class 0.000 description 1
- 229920001983 poloxamer Polymers 0.000 description 1
- 125000003367 polycyclic group Chemical group 0.000 description 1
- 229940126027 positive allosteric modulator Drugs 0.000 description 1
- 230000001242 postsynaptic effect Effects 0.000 description 1
- 230000003518 presynaptic effect Effects 0.000 description 1
- 230000003449 preventive effect Effects 0.000 description 1
- 230000003244 pro-oxidative effect Effects 0.000 description 1
- 108090000765 processed proteins & peptides Proteins 0.000 description 1
- 230000000750 progressive effect Effects 0.000 description 1
- 230000001737 promoting effect Effects 0.000 description 1
- 230000004224 protection Effects 0.000 description 1
- 108090000623 proteins and genes Proteins 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- SBYHFKPVCBCYGV-UHFFFAOYSA-N quinuclidine Chemical compound C1CC2CCN1CC2 SBYHFKPVCBCYGV-UHFFFAOYSA-N 0.000 description 1
- 239000007845 reactive nitrogen species Substances 0.000 description 1
- 229940075993 receptor modulator Drugs 0.000 description 1
- 238000011160 research Methods 0.000 description 1
- 230000035806 respiratory chain Effects 0.000 description 1
- 230000002441 reversible effect Effects 0.000 description 1
- 229910052702 rhenium Inorganic materials 0.000 description 1
- 229910052703 rhodium Inorganic materials 0.000 description 1
- 239000000523 sample Substances 0.000 description 1
- 239000003352 sequestering agent Substances 0.000 description 1
- 230000011664 signaling Effects 0.000 description 1
- 230000006886 spatial memory Effects 0.000 description 1
- 229960005322 streptomycin Drugs 0.000 description 1
- 238000006467 substitution reaction Methods 0.000 description 1
- 150000003871 sulfonates Chemical class 0.000 description 1
- 208000024891 symptom Diseases 0.000 description 1
- 230000007470 synaptic degeneration Effects 0.000 description 1
- 230000000946 synaptic effect Effects 0.000 description 1
- 230000016978 synaptic transmission, cholinergic Effects 0.000 description 1
- 210000003478 temporal lobe Anatomy 0.000 description 1
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 1
- 238000010257 thawing Methods 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- 125000003866 trichloromethyl group Chemical group ClC(Cl)(Cl)* 0.000 description 1
- 150000003672 ureas Chemical class 0.000 description 1
- 239000011782 vitamin Substances 0.000 description 1
- 229930003231 vitamin Natural products 0.000 description 1
- 229940088594 vitamin Drugs 0.000 description 1
- 235000013343 vitamin Nutrition 0.000 description 1
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D403/00—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00
- C07D403/02—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings
- C07D403/12—Heterocyclic compounds containing two or more hetero rings, having nitrogen atoms as the only ring hetero atoms, not provided for by group C07D401/00 containing two hetero rings linked by a chain containing hetero atoms as chain links
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/40—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil
- A61K31/403—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having five-membered rings with one nitrogen as the only ring hetero atom, e.g. sulpiride, succinimide, tolmetin, buflomedil condensed with carbocyclic rings, e.g. carbazole
- A61K31/404—Indoles, e.g. pindolol
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D209/00—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom
- C07D209/02—Heterocyclic compounds containing five-membered rings, condensed with other rings, with one nitrogen atom as the only ring hetero atom condensed with one carbocyclic ring
- C07D209/04—Indoles; Hydrogenated indoles
- C07D209/10—Indoles; Hydrogenated indoles with substituted hydrocarbon radicals attached to carbon atoms of the hetero ring
- C07D209/14—Radicals substituted by nitrogen atoms, not forming part of a nitro radical
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P25/00—Drugs for disorders of the nervous system
- A61P25/28—Drugs for disorders of the nervous system for treating neurodegenerative disorders of the central nervous system, e.g. nootropic agents, cognition enhancers, drugs for treating Alzheimer's disease or other forms of dementia
Landscapes
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Pharmacology & Pharmacy (AREA)
- Epidemiology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Animal Behavior & Ethology (AREA)
- General Health & Medical Sciences (AREA)
- Public Health (AREA)
- Veterinary Medicine (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
Description
DESCRIPCIÓN
COMPUESTOS AGONISTAS NICOTÍNICOS Y ANTIOXIDANTES PARA EL
TRATAMIENTO DE ENFERMEDADES NEURODEGENERATIVAS
CAMPO DE LA INVENCIÓN
La presente invención se encuadra principalmente en el sector farmacéutico con aplicaciones dirigidas a la prevención y/o tratamiento de enfermedades y cualquier tipo de afección o daño que implique una actividad anormal de receptores nicotínicos o que curse con altos niveles de estrés oxidativo y, en concreto, en la identificación de compuestos químicos útiles en el tratamiento preventivo y/o terapéutico de enfermedades neurodegenerativas tales como la enfermedad de Alzheimer (EA), enfermedad de Parkinson (EP), esclerosis lateral amiotrófica (ELA) y enfermedad de Huntington (EH), esclerosis múltiple (EM) o el ictus.
ANTECEDENTES DE LA INVENCIÓN
El envejecimiento se relaciona con la acumulación progresiva de cambios fisiológicos por mutaciones genéticas, cambios epigenéticos y metabólicos, que conducen a una mayor susceptibilidad a sufrir enfermedades relacionadas con la edad.
La población de 65 años o más, continúa aumentando con el consiguiente incremento del número de pacientes de Alzheimer y otras demencias. En 2016, estas enfermedades fueron la quinta causa principal de muerte mundial en 2016, representando 2.1 millones de muertes (3.7 %) (Organization, 2018a). En 2018, el número de personas que padecen EA y otras demencias se estimó en 50 millones, y se estima una prevalencia de 75 millones para 2030 y 152 millones para 2050 (Organization, 2018b).
La EA es una enfermedad neurodegenerativa relacionada con el envejecimiento. Se caracteriza fundamentalmente por pérdida de memoria y funciones cognitivas, debido, principalmente, a la muerte de neuronas colinérgicas en regiones del cerebro asociadas a memoria atencional, espacial y episódica, como son el neocórtex, los lóbulos temporales y el hipocampo (Mesulam, 2013, J Comp Neurol, 521: 4124-44). Además de esta pérdida sináptica, otros rasgos fisiopatológicos son la aparición de ovillos neurofibrilares intracelulares, que contienen la proteína tau hiperfosforilada; las placas seniles extracelulares, compuestas principalmente por agregados del péptido p-amiloide; la disfunción mitocondrial; la desregulación de la homeostasis del Ca2+; el estrés oxidativo (Sanabria-Castro y col., 2017,
Annals of Neurosciences, 24: 46-54) y la inflamación crónica (Glass y col., 2010, Cell, 140: 918-34).
Desde un punto de vista químico, los radicales libres son especies que contienen uno o más electrones no apareados altamente reactivos, capaces de oxidar a las moléculas presentes en el entorno (Halliwell, 2015, eLS). Desde un punto de vista biológico, los radicales libres de O2, conocidos como especies reactivas de oxígeno (ERO) o N2, especies reactivas de nitrógeno (ERN) ), son productos del metabolismo fisiológico (Di Carlo y col., 2012, Free Radical Research, 46: 1327-38). La principal fuente de ERO es la cadena de transporte electrónico para la generación de ATP en las mitocondrias. Por lo tanto, es necesario un control estricto de la homeostasis de los radicales libres. Sin embargo, durante el envejecimiento, el equilibrio mitocondrial de ERO se desregula induciendo un daño oxidativo exacerbado. El proceso de envejecimiento se asocia a la acumulación de radicales libres, generando un desequilibrio entre las especies pro-oxidantes y antioxidantes. Los radicales libres generados, dañan el ADN mitocondrial causando mutaciones acumuladas en los genes de la cadena de transporte electrónico, lo que resulta en una sobreproducción de ERO.
La neurotransmisión requiere grandes cantidades de energía, por lo tanto, el cerebro consume una gran cantidad de oxígeno. Este hecho, junto con la baja efectividad del sistema antioxidante y la alta concentración de lípidos en las neuronas, hace que el cerebro sea aún más susceptible al daño oxidativo (Salim, 2017, J Pharmacol Exp Ther, 360: 201-05). Considerando el envejecimiento el principal factor de riesgo de EA, el estrés oxidativo y la disfunción mitocondrial se consideran contribuyentes tempranos de la enfermedad. De hecho, hay evidencia de marcadores oxidativos como los nucleósidos oxidados derivados del ARN o la peroxidación lipídica, que aparecen antes de la formación de ovillos neurofibrilares o el depósito de p-amiloide (Ap) (Nunomura y col., 2001, Journal of Neuropathology & Experimental Neurology, 60: 759-67).
Como ya se ha mencionado, una de las principales características de la EA y la responsable de sus síntomas más notables, es la disminución de la actividad colinérgica. El receptor nicotínico de acetilcolina a7 (nAChR-a7) participa en la neurotransmisión colinérgica, siendo mediadores de la señalización colinérgica involucrada en la modulación de la memoria y las funciones cognitivas. Sin embargo, en la EA su expresión y función se altera, lo que lleva a la activación de vías pro y anti-supervivencia dependiendo de la etapa de la enfermedad, el tipo de célula implicada o el ligando utilizado.
El nAChR-a7 uno de los nAChR más predominantes expresados en el cerebro, junto con a4p2. Los nAChR-a7se encuentran en las neuronas, fuera de la hendidura sináptica en las ubicaciones pre y post-sinápticas donde promueven la excitabilidad neuronal y la liberación de neurotransmisores, así como de forma extra-sináptica, participando en la comunicación no excitable (Dineley y col., 2015, Trends Pharmacoí Sci, 36: 96-108). También se expresan en células no neuronales, incluidos los astrocitos y microglía, donde están implicadas en la inflamación y la neuroprotección (Shytle y col., 2004, Journal of Neurochemistry, 89: 337-43)(Egea y col., 2015, Biochem Pharmacoí, 97: 463-72).
El nAChR-a7 es único dentro de la familia nAChR porque tiene una naturaleza dual ionotrópica y metabotrópica. Como canal catiónico transmembrana inicia tres tipos de señales de calcio. En primer lugar, el influjo directo debido a su alta permeabilidad al Ca2+; en segundo lugar, indirectamente a través de la activación de canales de calcio dependientes de voltaje (VDCC) y, finalmente, la liberación de calcio inducida por calcio (CICR) desde el retículo endoplásmico (Shen y col., 2009, Acta Pharmacoí Sin, 30: 673-80). Todos ellos están involucrados en la neurotransmisión, no obstante, la señal de Ca2+ inducida puede conllevar la activación de mecanismos neuroprotectores (Parada y col., 2010, Free Radic Bioí Med, 49: 1815-21). Por otra parte, la vía metabotrópica puede desencadenar cascadas de señalización neuroprotectoras, como por ejemplo, la estimulación de nAChR-a7 con nicotina, que induce la activación de la ruta PI3K/Akt (Shaw y col., 2002, J Bioí Chem, 277: 44920-4).
En los documentos de patente WO2008/058096, WO2014/160783, WO2010/126605, WO2010/046710, WO2012/149478, WO2013/067036, WO2013/132124, WO2018/102885 se proponen distintas opciones de tratamiento de enfermedades neurodegenerativas basadas en uno o varios compuestos químicos y/o productos naturales cuya diana terapéutica es la actividad moduladora de receptores nicotínicos en el mismo sentido.
DESCRIPCIÓN DETALLADA DE LA INVENCIÓN
La invención se refiere al uso de compuestos con estructura 1-(2-(1H-indol-3-il)etil)-3-(quinuclidin-3-il)tiourea y 1-(2-(1 H-indol-3-il)etil)-3-(quinuclidin-3-il)urea con capacidad moduladora de receptores nicotínicos y capacidad secuestradora de radicales libres que ejercen, como consecuencia de ambas propiedades, efectos neuroprotectores.
En la presente invención se describe, por primera vez, la inclusión en una única molécula de la capacidad moduladora de receptores nicotínicos además de incluir la secuestradora de
radicales libres debido a la combinación de subestructuras a partir de las cuales se ha obtenido la estructura química planteada. Además, los compuestos objeto de la presente invención poseen capacidad neuroprotectora, por lo que pueden ser potencialmente útiles en la prevención y/o el tratamiento de enfermedades neurodegenerativas. Más específicamente, el objeto de la presente invención consiste en proporcionar nuevos compuestos útiles como ingredientes activos de un medicamento, que permitan la prevención y/o tratamiento de enfermedades neurodegenerativas.
Por lo tanto, en un aspecto, la invención se refiere a un compuesto de fórmula (I), sus sales, profármacos o solvatos. Dicho compuesto de fórmula (I) puede ser utilizado en el tratamiento de enfermedades neurodegenerativas y/o enfermedades que estén relacionadas con una desregulación de la actividad de receptores nicotínicos en relación a la neuroprotección asociada a la modulación de estos en distintas enfermedades neurodegenerativas.
En otro aspecto, la invención se refiere a una composición farmacéutica que comprende un compuesto de fórmula (I), o una sal, un profármaco o un solvato del mismo, y un vehículo farmacéuticamente aceptable.
En otro aspecto, la invención protege el uso de dicho compuesto de fórmula (I), o sus sales, profármacos o solvatos, farmacéuticamente aceptables, en la elaboración de una composición farmacéutica para la prevención o el tratamiento de enfermedades neurodegenerativas.
En el contexto de la presente invención, los siguientes términos tienen el significado detallado a continuación:
Cuando se usa el término “seleccionados independientemente”, los sustituyentes a los que se refiere (e.j. grupos R, como los grupos R1, R2, R3, R4, R5, R6 o X o Y o Z o variables como “n”) los grupos pueden ser idénticos o diferentes, o en su caso cuando sea especificado.
El término “alquilo” se refiere a un radical de cadena hidrocarbonada lineal o ramificado consistente solamente en átomos de carbono e hidrógeno que no contienen insaturaciones, teniendo de uno a ocho átomos de carbono y que está unido al resto de la molécula por un enlace sencillo. Preferiblemente, se refiere a un radical de cadena alifática lineal o ramificada que tiene entre 1 y 6, preferiblemente entre 1 y 3 (“alquiloC1-3”) átomos de carbono, y que está unido al resto de la molécula mediante un enlace sencillo. Este término incluye, por ejemplo y en un sentido no limitativo, metilo, etilo, n-propilo, i-propilo, n-butilo, t-butilo, n-pentilo, etc. Los
radicales alquilo pueden estar opcionalmente sustituidos por uno o más sustituyentes independientemente seleccionados del grupo consistente en halógenos, hidroxilo, alcóxidos, carboxi, ciano, carbonil, acil, alcoxicarbonil, amino, nitro, mercapto y alquiltio.
El término “alcoxilo” se refiere a un grupo -O-alquilo, donde alquilo es como se ha definido previamente. Preferiblemente alcoxilo es metoxilo.
El término “halógeno” se refiere a bromo, cloro, yodo o flúor. Preferiblemente, halógeno es flúor o cloro o bromo.
El término “haloalquil” se refiere a un radical alquilo, como ha sido definido previamente, que está sustituido por uno o más halógenos, como también han sido definidos previamente, incluyendo, por ejemplo, y en un sentido no limitativo, trifluorometil, triclorometil, 2,2,2,-trifluoroetil, 1 -fluorometil-2-fluoroetil, etc.
El término “alcoxicarbonil” se refiere a un radical de formula -C(O)OR donde R es un radical alquilo como se ha descrito previamente. Los radicales alcoxicarbonil pueden incluir, por ejemplo, y en un sentido no limitativo, metoxi, etoxi, propoxi, etc.
El término “cicloalquilo” se refiere a un grupo alifático mono o policíclico saturado o parcialmente saturado, que tiene entre 3 y 10, preferiblemente entre 3 y 6 átomos de carbono que está unido al resto de la molécula por medio de un enlace sencillo, incluyendo, por ejemplo, y en un sentido no limitativo, ciclopropilo, ciclobutilo, ciclohexilo, ciclopentilo, etc.
El término “amino” se refiere a un radical de fórmula -NH2.
El término “arilo” se refiere a un grupo aromático que tiene entre 6 y 18, preferiblemente entre 6 y 10 átomos de carbono, que comprende 1,2 ó 3 núcleos aromáticos, unidos por medio de un enlace carbono-carbono o condensados, incluyendo por ejemplo y en un sentido no limitativo fenilo, naftilo, difenilo, indenilo, fenantrilo, etc.
El término “heterociclo” se refiere a un radical de anillo de 3 a 10 miembros estable, preferiblemente un anillo de 5 ó 6 miembros, que consiste en átomos de carbono y desde uno hasta cinco heteroátomos seleccionados del grupo formado por nitrógeno, oxígeno y azufre, y que puede estar parcial o totalmente saturado, o puede ser aromático (“heteroarilo”). Para los fines de esta invención, el heterociclo puede ser un sistema de anillo monocíclico, bicíclico
o tricíclico, que puede incluir sistemas de anillos condensados. Los ejemplos de tales heterociclos incluyen, pero no se limitan a, pirrolidina, piperidina, piperazina, morfolina, tetrahidrofurano, bencimidazol, benzotiazol, furano, pirrol, piridina, pirimidina, isotiazol, imidazol, indol, purina, quinolina, tiadizol.
Tal como se entiende en esta área técnica, puede haber un cierto grado de sustitución en los radicales definidos anteriormente. Las referencias del presente documento con respecto a los grupos sustituidos indican que el radical especificado puede estar sustituido en una o más posiciones disponibles con uno o más sustituyentes. Dichos sustituyentes incluyen, por ejemplo, y en un sentido no limitativo, alquilo C1-6, alquenilo C2-6, alquinilo C2-6, cicloalquilo, arilo, heterociclo, halógeno, CN, NO2, CF3, -N(Ra)(Rb), -ORc, -SRd, -C(O)Re, -C(O)ORf, -C(O)N(Rg)(Rh), -OC(O)Ri; en los que Ra, Rb, Rc, Rd, Re, Rf, Rg, Rh y Ri se seleccionan independientemente de hidrógeno, alquilo, C1-C6, arilo, heterociclo y trifluorometilo.
El término “farmacéuticamente aceptable” se refiere, preferiblemente, a composiciones y entidades moleculares que son fisiológicamente tolerables y no producen, normalmente, una reacción alérgica o una reacción no favorable similar, tal como trastornos gástricos, mareo y similares, cuando se administra a un ser humano o animal. La expresión “farmacéuticamente aceptable” significa que está aprobado por una agencia reguladora como la agencia europea del medicamento o la agencia reguladora de EEUU, o que está incluido en la Farmacopea Estadounidense u otra farmacopea reconocida de modo general para su uso en animales y, de manera más particular, en seres humanos.
El término “sales” tal como aquí se utiliza se refiere a cualquier sal del compuesto de fórmula (I) que, cuando se administra a un sujeto, es capaz de proporcionar (directa o indirectamente) dicho compuesto de fórmula (I). El término “sujeto” incluye a cualquier animal, por ejemplo, un mamífero, incluyendo a los seres humanos. La preparación de dichas sales puede llevarse a cabo mediante métodos convencionales conocidos por los técnicos en la materia.
El término “profármaco” se emplea, en esta descripción, en el sentido más amplio, e incluye a cualquier compuesto derivado de un compuesto de fórmula (I) que, cuando se administra a un sujeto, es capaz de proporcionar, directa o indirectamente, un compuesto de fórmula (I), o una de sus sales farmacéuticamente aceptables, en dicho sujeto. Ventajosamente, dicho derivado es un compuesto que aumenta la biodisponibilidad del compuesto de fórmula (I) cuando se administra a un sujeto (por ejemplo, haciendo que un compuesto de fórmula (I) administrado por vía oral se absorba más fácilmente por la sangre), o que potencia la
liberación de un compuesto de fórmula (I) en un compartimento biológico (por ejemplo, el cerebro o el sistema linfático) con relación al compuesto original (sin derivatizar). La naturaleza de dicho derivado no es crítica, siempre y cuando pueda ser administrado a un sujeto y proporcione un compuesto de fórmula (I) en un compartimento biológico de dicho sujeto. Tales derivados serán evidentes para los técnicos en la materia, e incluyen, dependiendo de los grupos funcionales presentes en la molécula y sin limitación, los siguientes derivados de los compuestos presentes: ésteres, ésteres de aminoácido, ésteres de fosfato, ésteres de sulfonato de sales metálicas, carbamatos y amidas.
El término “solvato” tal como aquí se utiliza incluye cualquier compuesto formado por combinación de moléculas de un disolvente con moléculas o iones de un compuesto de fórmula (I) o de una sal del mismo; dicho disolvente puede ser un disolvente orgánico, por ejemplo, un alcohol, o un disolvente acuoso, por ejemplo, agua, en cuyo caso el solvato se denomina “hidrato”.
El término “excipiente farmacéuticamente aceptable” significa uno o más sólidos, o líquidos compatibles, diluyentes o sustancias de encapsulación que sean susceptibles de ser administradas a un sujeto.
El primer aspecto de la presente invención se refiere a un compuesto de fórmula (I):

donde
R se selecciona del grupo consistente en:
- hidrógeno
- Alquilo opcionalmente sustituido por uno, dos, o tres átomos de halógeno seleccionado entre flúor, cloro y bromo; cicloalquilo(C3-C6); alcoxilo(CrC6); cicloalcoxilo(C3-C6); ciano y nitro; y/o
- fenilo opcionalmente sustituido por uno, dos o tres grupos seleccionados independientemente entre flúor; cloro; bromo; alquilo opcionalmente sustituido por
uno, dos, o tres átomos de halógeno seleccionado entre flúor, cloro y bromo; cicloalquilo(C3-C6); alcoxilo(C1-Ce); cicloalcoxilo(C3-C6); ciano y nitro; o bien dos grupos pueden formar conjuntamente un grupo -O(CH2)oO-, -(CH2)p-, o -CH=CH-CH=CH-; o
- un grupo heteroarilo opcionalmente sustituido por uno, dos o tres grupos seleccionados independientemente entre flúor, cloro, bromo, alquilo, cicloalquilo(C3-C6), alcoxilo(Ci-C6), cicloalcoxilo(C3-C6), ciano y nitro;
Ri y R2 se seleccionan del grupo consistente en hidrógeno, alquilo, cicloalquilo, arilo o heteroarilo, opcionalmente sustituido por uno, dos o tres grupos seleccionados independientemente entre flúor, cloro, bromo, alquilo, alcoxilo, cicloalquilo(C3-C6), cicloalcoxilo(C3-C6), ciano, nitro y carboxilato o ambos grupos pueden formar conjuntamente un grupo -O(CH2)qO-, -(CH2V, o -CH=CH-CH=CH-;
X se selecciona entre un átomo de oxigeno o un átomo de azufre;
Y se selecciona entre un átomo de carbono o un átomo de nitrógeno;
Z se selecciona entre un átomo de carbono o un átomo de nitrógeno;
n es un número entero seleccionado entre 0, 1,2, 3, 4, 5 ó 6;
*3’’ indica la presencia de un centro quiral cuya configuración puede ser R o S; o sus estereoisomeros, sales, preferiblemente sales farmacéuticamente aceptables, profármacos o solvatos.
En una realización particular, dichas sales del compuesto de fórmula (I) son sales farmacéuticamente aceptables, es decir, sales que pueden ser administradas a un sujeto y proporcionan un compuesto de fórmula (I) en un compartimento biológico de dicho individuo.
En otra realización particular, dichas sales del compuesto de fórmula (I) son sales farmacéuticamente no aceptables, las cuales pueden ser útiles en la preparación de sales farmacéuticamente aceptables del compuesto de fórmula (I), o de sus profármacos o solvatos.
Los compuestos de fórmula (I), o sus sales, pueden estar en forma cristalina bien como compuestos libres o bien como solvatos, estando ambas formas incluidas dentro del ámbito de la presente invención.
En una realización particular, dicho solvato es un solvato farmacéuticamente aceptable, es decir, que puede ser administrado a un sujeto y proporcionar (directa o indirectamente) un compuesto de fórmula (I) o una sal del mismo.
En otra realización particular, dicho solvato no es farmacéuticamente aceptable, pero puede utilizarse en la preparación de solvatos farmacéuticamente aceptables del compuesto de fórmula (I) o de sus sales.
La preparación de dichos solvatos puede llevarse a cabo mediante métodos convencionales conocidos por los técnicos en la materia, poniendo en contacto el compuesto de fórmula (I) o una sal del mismo con el disolvente apropiado.
Los compuestos de la presente invención representados por la fórmula (I) descrita anteriormente pueden incluir enantiómeros por la presencia de centros quirales o isómeros geométricos, dependiendo de la presencia de enlaces múltiples (por ejemplo R, S). Los isómeros geométricos y enantiómeros de los compuestos de fórmula (I) y mezclas de los mismos se encuentran dentro del alcance de la presente invención.
En otra realización particular, los compuestos sujetos a esta invención dan composiciones farmacéuticamente aceptables que comprenden los compuestos de fórmula (I) con un portador farmacéuticamente aceptable, por ejemplo, composición farmacéutica que incluye uno o más compuestos de fórmula (I), solos o en combinación con uno o más agentes terapéuticos adicionales como una mezcla con excipientes farmacéuticamente aceptables.
En una realización particular preferida, la invención se refiere a compuestos de fórmula (I) en los que:
- R es un átomo de hidrógeno
- R1 se selecciona del grupo que comprende un alquilo opcionalmente sustituido por uno o dos grupos seleccionados independientemente entre flúor, cloro, y bromo; alcoxilo(C1-C6), nitro y amino; preferentemente R1 es -OCH3;
- R2 es hidrógeno;
- n es un entero seleccionado entre 0, 1 y 2, preferentemente n = 1 ;
- X es oxigeno o azufre;
*3’’ es un átomo de carbono con configuración R o S en cualquiera de sus posibles combinaciones
o sus estereoisomeros, sales, preferiblemente sales farmacéuticamente aceptables, profármacos o solvatos.
Compuestos de fórmula (I) particularmente preferidos de la presente invención son los siguientes:
• (R)-1 -(2-(5-metoxi-1 H-indol-3-il)etil)-3-(quinuclidin-3-il)tiourea
• (S)-1 -(2-(5-metoxi-1 H-indol-3-il)etil)-3-(quinuclidin-3-yl)tiourea
• (R)-1-(2-(5-cloro-1 H-indol-3-il)etil)-3-(quinuclidin-3-il)tiourea
• (S)-1 -(2-(5-cloro-1 H-indol-3-il)etil)-3-(quinuclidin-3-il)tiourea
• (R)- 1-(2-(1 H-indol-3-il)etil)-3-(quinuclidin-3-yl)tiourea
• (S)- 1-(2-(1H-indol-3-il)etil)-3-(quinuclidin-3-yl)tiourea
• (R)-1-(2-(5-metoxi-1 R-indol-3-il)etil)-3-(quinuclidin-3-il)urea
• (S)-1 -(2-(5-metoxi-1 R-indol-3-il)etil)-3-(quinuclidin-3-il)urea
• (R)-1-(2-(5-chloro-1 R-indol-3-yl)ethyl)-3-(quinuclidin-3-yl)urea
• (S)-1-(2-(5-cloro-1 H-indol-3-yl)etil)-3-(quinuclidin-3-il)urea
• (R)-1-(2-(1 R-indol-3-il)etil)-3-(quinuclidin-3-il)urea
• (S)-1-(2-(1 R-indol-3-il)etil)-3-(quinuclidin-3-il)urea
Los compuestos de fórmula (I) de la presente invención se pueden obtener mediante un procedimiento que implica hacer reaccionar 3-aminoquinuclidina (II) (Kume y col., Neurosci Lett, 443: 199-203) con configuración R o S.

con el derivado correspondiente de 3-(2-isotiocianatoetil)-5-R-1 H-indol (III) o N-(2-(5-R-1 H-indol-3-il)etil)-1 H-imidazol-1 -carboxamida (IV):

(III) (IV)
donde R, R1, R2, X, Y, Z y n tienen el significado ya indicado.
Por tanto, en otro aspecto, la presente invención describe un procedimiento para la obtención de un compuesto de fórmula (I) que comprende hacer reaccionar dicho compuesto de fórmula (II) con un compuesto de fórmula (III) o fórmula (IV).
Dicha reacción se lleva a cabo en un disolvente inerte apropiado, a la temperatura adecuada. En una realización particular, la reacción se lleva a cabo inicialmente mezclando la quinuclidina correspondiente y el derivado del compuesto (III) o (IV) a 0 °C, manteniéndolos con agitación en un en presencia de una base orgánica tal como diisopropil etil amina (DIPEA) y en disolvente inerte, tal como el constituido por un polar no prótico como por ejemplo dimetilformamida, dimetilsulfoxido, etc., o sus mezclas, resultando especialmente util la dimetilformamida.
Los compuestos de fórmula (II), (III) y (IV) son compuestos conocidos y pueden obtenerse comercialmente o bien se pueden preparar por métodos convencionales.
Los compuestos de fórmula (I) obtenidos por el procedimiento anterior, si se desea, pueden ser purificados por métodos convencionales, tales como cristalización o cromatografía.
Los compuestos de fórmula (I) de la presente invención presentan actividad moduladora de receptores nicotínicos y actividad secuestradora de radicales libres.
En una realización particular, la invención comprende un compuesto de fórmula (I) para su uso en el tratamiento de enfermedades que mejoran con la administración de un agente secuestrador de radicales libres y/o un agente neuroprotector y/o un modulador de receptores nicotínicos.
Por tanto, en otros aspectos, la invención incluye un compuesto de fórmula (I) para su uso en la prevención o el tratamiento de una enfermedad neurodegenerativa central y/o periférica.
Por tanto, la invención comprende un compuesto de fórmula (I) para su uso en el tratamiento de enfermedades neurodegenerativas, ej., la EA, la EP, ELA, EH, EM.
Para su administración a un sujeto en necesidad de tratamiento, los compuestos de fórmula (I) de la presente invención se administran convenientemente formulados con los excipientes adecuados para su administración por cualquier vía apropiada, por ejemplo, por vía oral,
parenteral, subcutánea, intramuscular, intravascular o rectal, siendo preferiblemente por vía oral.
En una realización particular, la composición farmacéutica de la invención se presenta en una forma farmacéutica de administración por vía oral, bien en forma sólida o líquida. La composición farmacéutica de la invención también puede ser adaptada para su administración parenteral (ej., vía intramuscular, intravenosa, etc.), La composición farmacéutica de la invención también puede ser adaptada para su administración subcutánea en forma de, por ejemplo, soluciones o suspensiones estériles, en la forma de dosificación apropiada; Las formulaciones se pueden preparar según métodos convencionales tales como los que se describen en las farmacopeas Española, Europea o de Estados Unidos de América.
El compuesto de fórmula (I) de la invención se administrará en una cantidad terapéuticamente efectiva que generalmente dependerá de la eficacia del compuesto de fórmula (I) elegido, de la gravedad de la patología a tratar, etc. No obstante, típicamente se administrará a dosis diarias comprendidas entre 0,1 y 500 mg de compuesto de fórmula (I) por kg de peso corporal, más preferentemente las dosis diarias estarán comprendidas entre 25 y 250 mg/kg peso corporal.
La administración de los compuestos de fórmula (I) de la invención, sus sales, profármacos o solvatos, farmacéuticamente aceptables, se puede llevar a cabo en solitario o en combinación con fármacos adicionales, tales como fármacos útiles para el tratamiento de una enfermedad neurodegenerativa, para proporcionar una terapia combinada; dichos fármacos adicionales pueden formar parte de la misma composición farmacéutica de la invención que comprende el compuesto de fórmula (I) y/o sus sales, profármacos o solvatos farmacéuticamente aceptables, o no, en cuyo caso, se administrarán de forma simultánea o secuencial a la administración de la composición farmacéutica de la invención. Ejemplos ilustrativos, no limitativos, de dichos fármacos adicionales que pueden emplearse para proporcionar una terapia de combinación incluyen agentes tales como la memantina, vitaminas, antiinflamatorios o antidepresivos.
MODO DE REALIZACIÓN DE LA INVENCIÓN
La presente invención se ilustra adicionalmente mediante los siguientes ejemplos, los cuales no pretenden ser limitativos de su alcance.
1. OBTENCIÓN DE LOS COMPUESTOS DE LA INVENCIÓN
Los compuestos cuya actividad biológica es objeto de la presente invención se sintetizaron siguiendo los siguientes procedimientos en síntesis orgánica.
Procedimiento general para la síntesis de los derivados 1-(2-(1fl-indol-3-il)etil)-3-(quinuclidin-3-il)tiourea/urea
A una suspensión del derivado 3-(2-isotiocianatoetil)-1H-indol o N-(2-(1 H-indol-3-il)etil)-1 H-imidazol-1-carboxamida (1 eq) correspondiente y DIPEA (3.5 eq) en DMF (10 mL/mmol) a 0 °C, se añadió (R)- or (S)-3-aminoquinuclidina (1.5 eq). La reacción se mantuvo con agitación incrementando su temperatura a temperatura ambiente hasta que la reacción se completó. A continuación, se evaporó el disolvente y se purificó el crudo de reacción mediante cromatografía en columna para obtener el derivado correspondiente.
Ejemplo 1: (fí)-1-(2-(5-metoxi-1H-indol-3-il)etil)-3-(quinuclidin-3-yl)tiourea (Compuesto 1 ).

Siguiendo el procedimiento general descrito, (R)-3-aminoquinuclidina (90 mg, 0.44 mmol), DIPEA (0.15 mL, 0.88 mmol) y 3-(2-isotiocianatoetil)-5-metoxi-1 H-indol (0.100 mg, 0.44 mmol) en DMF (4.4 mL), durante 24 h, produjo tras cromatografía flash (CH2Cl2/MeOH/NH4OH, 96:3:1-90:9:1), el compuesto 1 (98 mg, 62 %). Rf 0.06 (CH2Ch/MeOH 90:10). p.f. 125 - 127 °C. 1H NMR (300 MHz, MeOD) 57,22 (dd, J = 9,0, 5,6 Hz, 1H, H7’), 7,18 (d, J = 3,2 Hz, 1H, H4’), 7,06 (d, J = 5,1 Hz, 1H, H2’), 6,75 (dd, J = 9,0, 3,2 Hz, 1H, H6’), 4,37 - 4.15 (m, 1H, H3’’), 3,84 (s, 3H, OCHs), 3,83 - 3,79 (m, 2H, 2xH1), 2,99 (t, J = 6,6 Hz, 2H, 2xH2), 2,85 (q, J = 10,9, 8,7 Hz, 4H, 2xH6’’, H7’’, H8’’), 2,54 (d, J = 10,7 Hz, 1H, H2’’a), 1,96 (s, 1H, H4’’), 1,80 - 1,63 (m, 4H, H2’’b, H5’’a, H7’’, H8’’), 1,52 (dd, J = 15,5, 8,6 Hz, 1H, H5’’b) ppm. 13C NMR (76 MHz, MeOD) 5183,6, 155,0, 133,4, 129,3, 124,4, 113,0, 112,9, 112,6, 101,7, 56,4, 55,8, 51,3, 47,7, 47,2, 46,1,26,5, 26,2, 25,6, 20,3 ppm. HRMS (ES+): Masa teórica calculada para C19H26N4OS, 358,1827; encontrada [(M)+], 358,1823. IR (film) u/cm-13237, 3053, 2926, 2869, 2321, 1993, 1656, 1616, 1539, 1482, 1452, 1438, 1379, 1341, 1309, 1213, 1167, 1025, 920, 792. [a]D4=
13,60 ° (c. 1,25 mg/mL, MeOH) ee 97 %. Los enantiómeros se separaron mediante HPLC OVM analítica quiral en fase inversa, tampón 20 mM KH2PO4 pH 5,9/ AcCN 95:5, tiempo de retención del enantiómero (R): 18,497 (menor), 22,357 (mayor). La pureza fue determinada del 100 % mediante HPLC en fase inversa C18 acoplado a espectrómetro de masas midiendo la corriente iónica total (TIC).
Ejemplo 2: (S)-1-(2-(5-metoxi-1rtLindol-3-il)etil)-3-(quinuclidin-3-il)tiourea (Compuesto 2).

Siguiendo el procedimiento general descrito, (S)-3-aminoquinuclidina (86 mg, 0,42 mmol), DIPEA (0,25 mL, 1,5 mmol) y 3-(2-isotiocianatoetil)-5-metoxi-1 H-indol (0,90 mg, 0,42 mmol) en DMF (4,2 mL), durante 24 h, produjo tras cromatografía flash (CH2Ch/MeOH/NH4OH, 96:3:1-90:9:1), el compuesto 2 (127 mg, 85 %). [a]B4= - 12,12 o (c. 1,32 mg/mL, MeOH) ee 94 %. Los enantiómeros fueron separados mediante HPLC OVM analítica quiral en fase reversa, tampón 20 mM KH2PO4 pH 5,9/ AcCN 95:5, tiempo de retención del enantiómero (S): 18,593 (mayor), 24,977 (menor). La pureza fue determinada del 99 % mediante HPLC en fase inversa C18 acoplado a espectrómetro de masas midiendo la corriente iónica total (TIC).
Ejemplo 3: (fí)-1-(2-(5-cloro-1H-indol-3-il)etil)-3-(quinuclidin-3-il)tiourea (Compuesto 3).

Siguiendo el procedimiento general descrito, (R)-3-aminoquinuclidina (57 mg, 0,45 mmol), DIPEA (0,18 mL, 1,03 mmol) y 3-(2-isotiocianatoetil)-5-cloro-1 H-indol (70 mg, 0,30 mmol) en DMF (3 mL), durante 41 h, produjo tras cromatografía flash (CH2Ch/MeOH/NH4OH, 99:0:1 -90:9:1), el compuesto 3 (84 mg, 78 %). Rf 0,03 (CH2Ch/MeOH 90:5). p.f. 122 - 124 °C. 1H
NMR (250 MHz, MeOD) 57,57 (dd, J = 2,1, 0,6 Hz, 1H, H4’), 7,27 (dd, J = 8,6, 0,4 Hz, 1H, H7’), 7,13 (s, 1 H, H2’), 7,02 (dd, J = 8,6, 2,0 Hz, 1H, H6’), 4,48 - 4,29 (m, 1H, H3’’), 3,75 (t, J = 6,8 Hz, 2H, 2xH1), 3,50 (dd, J = 13,5, 9,6 Hz, 1H, H2’’a), 2,98 (t, J = 6,8 Hz, 6H, 2xH2, 2xH6’’, H7’’, H8’’), 2,73 (d, J = 13,9 Hz, 1H, H2’’b), 2,05 (d, J = 3,9 Hz, 1H, H4’’), 1,81 (dd, J = 8,5, 5,5 Hz, 3H, H5’’a, H7’’, H8’’), 1,61 (dd, J = 12,0, 6,8 Hz, 1H, H5’’b) ppm. 13C NMR (63 MHz, MeOD) 5183,9, 160,0, 136,5, 130,2, 125,4, 122,4, 119,1, 113,5, 113,4, 55,7, 50,6, 47,6, 47,1,46,3, 26,3, 25,7, 24,7, 19,7 ppm. Masa teórica calculada para C18H23ClN4S, 362,1332; encontrada [(M+H)+], 363,1407. IR (film) u/cm-13246, 2922, 2867, 2110, 1736, 1652, 1557, 1544, 1479, 1454, 1031, 862, 792. [a]D 4 = 4,59 o (c.1,09 mg/mL, MeOH). La pureza fue determinada del 97 % mediante HPLC en fase inversa C18 acoplado a espectrómetro de masas midiendo la corriente iónica total (TIC).
Ejemplo 4: (S)-1-(2-(5-chloro-1 H-indol-3-yl)ethyl)-3-(quinuclidin-3-yl)thiourea (Compuesto 4).

Siguiendo el procedimiento general descrito, (H)-3-aminoquinuclidina (57 mg, 0,45 mmol), DIPEA (0,18 mL, 1,03 mmol) y 3-(2-isotiocianatoetil)-5-cloro-1 H-indol (70 mg, 0,30 mmol) en DMF (3 mL), durante 64 h, produjo tras cromatografía flash (CH2Ch/MeOH/NH4OH, 99:0:1 -90:9:1), el compuesto 4 (68 mg, 63 %). [a]D4= - 4,67 o (c.1,07 mg/mL, MeOH). La pureza fue determinada del 100 % mediante HPLC en fase inversa C18 acoplado a espectrómetro de masas midiendo la corriente iónica total (TIC).
Ejemplo 5: (fí)-1-(2-(1H-indol-3-il)etil)-3-(quinuclidin-3-yl)tiourea (Compuesto 5).

Siguiendo el procedimiento general descrito, (H)-3-aminoquinuclidina (30 mg, 0,24 mmol), DIPEA (0,15 mL, 0,84 mmol) y 3-(2-isotiocianatoetil)-1 H-indol (49 mg, 0.24 mmol) en DMF (1
mL), durante 62 h, produjo tras cromatografía flash (CH2Cl2/MeOH/Et3N, 98:1:1-90:9:1), el compuesto 5 (25 mg, 32 %). Rf: 0,06 (CH2Ch/MeOH 90:10). P.f. 131-133 °C. 1H NMR (250 MHz, MeOD) 57,58 (dd, J = 7,6, 1,3 Hz, 1H, H4’), 7,30 (dt, J = 8,0, 1,0 Hz, 1H, H7’), 7,06 (dd, J = 8,6, 6,4 Hz, 2H, H2’, H6’), 6,97 (ddd, J = 8,0, 7,0, 1,2 Hz, 1H, H5’), 4,30 (dd, J = 8,0, 4,8 Hz, 1H, H3’’), 3,76 (t, J = 7,8, 4,9 Hz, 2H, 2xH1), 3,47 - 3,35 (m, 1H, H2’’b), 3,00 (t, J = 6,9 Hz, 2H, 2xH2), 2,87 (t, J = 7,9, 7,5 Hz, 4H, 2xH6’’, H7’’, H8’’), 2,59 (dd, J = 13,8, 7,5 Hz, 1H, H2’’a), 1,98 (s, 1 H, H4’’), 1.75 (q, J = 10,2, 7,0, 5,6 Hz, 3H, H5’’a, H7’’, H8’’), 1,56 (t, J = 15,3, 8,2 Hz, 1H, H5’’b) ppm 13C NMR (63 MHz, MeOD) 5183,4, 138,2, 128,9, 123,7, 122,4, 119,7, 119,5, 113,2, 112,3, 55,6, 51,0, 47,7, 47,1,46,2, 26,4, 26,1,25,2, 20,0 ppm. Masa teórica calculada para C18H24N4S, 328,1722; encontrada [(M+H)+], 329,1804. IR (film) u/cm-13232, 3053, 2923, 2866, 2320, 1538, 1454, 1227, 1093, 1050, 985, 741. [a]D4= 6,67 ° (c. 1,20 mg/ml, MeOH) ee 99 %. Los enantiómeros fueron separados mediante HPLC OVM analítica quiral en fase reversa, tampón 5 mM KH2PO4 pH 5,1/ MeOH 90:10, tiempo de retención del enantiómero (R): 7,583 min. La pureza fue determinada del 100 % mediante HPLC en fase inversa C18 acoplado a espectrómetro de masas midiendo la corriente iónica total (TIC).
Ejemplo 6: (S)-1-(2-(1rtLindol-3-il)etil)-3-(quinuclidin-3-il)tiourea (Compuesto 6).

Siguiendo el procedimiento general descrito, (S)-3-aminoquinuclidina (62 mg, 0.49 mmol), DIPEA (0.31 mL, 1.71 mmol) y 3-(2-isotiocianatoetil)-1 H-indol (100 mg, 0.49 mmol) en DMF (2 mL), durante 64 h, produjo tras cromatografía flash (CH2Ch/MeOH/Et3N, 98:1:1 -90:9:1), el compuesto 6 (75 mg, 47 %). [a]D4= - 6.67° (c. 1.20 mg/ml, MeOH). La pureza fue determinada del 98 % mediante HPLC en fase inversa C18 acoplado a espectrómetro de masas midiendo la corriente iónica total (TIC).
Ej emplo 7: (fí)-1-(2-(5-metoxi-1rtLindol-3-il)etil)-3-(quinuclidin-3-il)urea (Compuesto 7).

Siguiendo el procedimiento general descrito, (fi)-3-aminoquinuclidina (30 mg, 0,24 mmol), DIPEA (0,15 mL, 0,84 mmol) y N-(2-(5-metoxi-1 H-indol-3-il)etil)-1 H-imidazol-1-carboxamida (65 mg, 0,24 mmol) en DMF (1 mL), durante 16 h, produjo tras cromatografía flash (CH2Cl2/MeOH/Et3N, 95:5:1-80:19:1), el compuesto 7 (25 mg, 30 %). Rf 0,03 (CH2Ch/MeOH 90:10). P.f. 117 - 119 °C. 1H NMR (250 MHz, MeOD) 57,19 (d, J = 8,7 Hz, 1H, H7’), 7,02 (d, J = 2,6 Hz, 2H, H2’, H4’), 6,73 (dd, J = 8,8, 2,5 Hz, 1H, H6’), 3,90 - 3,81 (m, 1H, H3’’), 3,80 (s, 3H, OCHs), 3,41 (t, J = 6,6 Hz, 3H, H2’’b, 2xH1), 2,87 (q, J = 7,0 Hz, 2H, H7’’, H8’’), 3,05 - 2,92 (m, 2H, 2xH6’’), 2,88 (q, J = 7,0 Hz, 2H, 2xH2), 2,53 (dd, J = 13,8, 5,0 Hz, 1H, H2’’a), 1,89 (q, J = 3,0 Hz, 2H, H4’’, H5’’a), 1,82 (dd, J = 11,6, 3,8 Hz, 2H, H7’’, H8’’), 1,61 (m, 1H, H5’’b) ppm.
13C NMR (63 MHz, MeOD) 5 160,8, 155,0, 133,5, 129,2, 124,4, 113,2, 112,9, 112,5, 101,5, 56,3, 56,0, 47,7, 47,3, 47,1, 41,8, 27,1, 26,9, 25,1, 19,7 ppm. Masa teórica calculada para C19H26N4OS, 342,2056; encontrada [(M+H)+], 343,2147. IR (film) u/cm-13247, 2929, 2875, 1648, 1556, 1483, 1440, 1213, 1172, 1027, 920, 795. [a ]2D5= + 4,90 o (c. 1,02 mg/mL, MeOH). La pureza fue determinada del 97 % mediante HPLC en fase inversa C18 acoplado a espectrómetro de masas midiendo la corriente iónica total (TIC).
Ejemplo 8: (S)-1-(2-(5-methoxy-1rt-indol-3-yl)ethyl)-3-(quinuclidin-3-yl)urea (Compuesto 8).

Siguiendo el procedimiento general descrito, (S)-3-aminoquinuclidina (28 mg, 0,22 mmol), DIPEA (0,14 mL, 0,80 mmol) y N-(2-(5-metoxi-1 H-indol-3-il)etil)-1 H-imidazol-1-carboxamida (60 mg, 0,20 mmol) en DMF (0,8 mL), durante 16 h, produjo tras cromatografía flash (C^Ch/MeOH/EtsN, 95:5:1-80:19:1), el compuesto 8 (45 mg, 66 %). [a]D4= - 19,0o (c. 1,00 mg/mL, MeOH). La pureza fue determinada del 100 % mediante HPLC en fase inversa Cía acoplado a espectrómetro de masas midiendo la corriente iónica total (TIC).
Ejemplo 9: (fí)-1-(2-(5-cloro-1rtLindol-3-il)etil)-3-(quinuclidin-3-il)urea (Compuesto 9).

Siguiendo el procedimiento general descrito, (R)-3-aminoquinuclidina (40 mg, 0,31 mmol), DIPEA (0,2 mL, 0,84 mmol) y N-(2-(5-cloro-1 H-indol-3-il)etil)-1 H-imidazol-1-carboxamida (80 mg, 0,28 mmol) en DMF (1,1 mL), durante 5 días, produjo tras cromatografía flash (CH2Cl2/MeOH/Et3N, 95:4:1-80:19:1), el compuesto 9 (30 mg, 31 %). Rf 0,03 (CH2Cl2/MeOH/Et3N, 95:5:1-80:19:1). P.f. 139 - 141 °C. 1H NMR (250 MHz, MeOD) 57,50 (dd, J = 2,1,0,6 Hz, 1H, H4’), 7,26 (dd, J = 8,6, 0,6 Hz, 1H, H7’), 7,09 (s, 1H, H2’), 7,00 (dd, J = 8,6, 2,1 Hz, 1H, H6’), 3,79 (dd, J = 8,3, 4,4 Hz, 1H, H3’’), 3,37 (t, J = 6,8 Hz, 2H, 2xH1), 3,34 (dt, J = 3,3, 1,6 Hz, 2H, H2’’b, H7’’), 3,01 - 2,79 (m, 6H, 2xH2, H4’’, H6’’a, H7’’, H8’’), 2,60 -2,45 (dd, J = 14,0, 1,8, 1H, H2’’a), 1,85 (d, J = 3,1 Hz, 1H, H6’’b), 1,75 (dd, J = 13,2, 6,9 Hz, 2H, H5’’a, H8’’), 1,62 - 1,44 (m, 1H, H5’’b) ppm. 13C NMR (63 MHz, MeOD) 5 160,7, 136,5, 130,2, 125,3, 122,4, 118,9, 113,5, 113,4, 56,1, 47,8, 47,4, 47,1,42,0, 27,0, 26,9, 25,3 ppm. Masa teórica calculada para C18H23ClN4O, 346,1560; encontrada [(M+H)+], 347,1645. IR (film) u/cm-13250, 2930, 2871, 1638, 1556, 1454, 1314, 1251, 1227, 1098, 1052, 891, 793. [a]2Ds= + 13,86 o (c. 1,01 mg/mL, MeOH). La pureza fue determinada del 100 % mediante HPLC en fase inversa C18 acoplado a espectrómetro de masas midiendo la corriente iónica total (TIC).
Ejemplo 10: (S)-1-(2-(5-cloro-1H-indol-3-il)etil)-3-(quinuclidin-3-il)urea (Compuesto 10).

Siguiendo el procedimiento general descrito, (S)-3-aminoquinuclidina (32 mg, 0,25 mmol), DIPEA (0,12 mL, 0,68 mmol) y N-(2-(5-cloro-1 H-indol-3-il)etil)-1 H-imidazol-1-carboxamida (50 mg, 0,17 mmol) en DMF (0,7 mL), durante 14 h, produjo tras cromatografía flash
(ChhCh/MeOH/EtsN, 95:5:1-80:19:1), el compuesto 10 (40 mg, 68 %). [a]2D5= - 12,750 (c. 1,02 mg/mL, MeOH). La pureza fue determinada del 96 % mediante HPLC en fase inversa C18 acoplado a espectrómetro de masas midiendo la corriente iónica total (TIC).
Ejemplo 11: (fi)-1-(2-(1rtLindol-3-yl)etil)-3-(quinuclidin-3-il)urea (Compuesto 11).

Siguiendo el procedimiento general descrito, (fi)-3-aminoquinuclidina (55 mg, 0,26 mmol), DIPEA (0,13 mL, 0,84 mmol) y N-(2-(1 H-indol-3-il)etil)-1 H-imidazol-1-carboxamida (34 mg, 0,18 mmol) en DMF (1,1 mL), durante 24 h, produjo tras cromatografía flash (CH2Cl2/MeOH/Et3N, 95:5:1-90:9:1), el compuesto 11 (50 mg, 62 %). Rf 0,03 (CH2Ch/MeOH 90:10). P.f. 161 - 163 °C. 1H NMR (250 MHz, MeOD) 57,55 (dd, J = 8,2, 1,3 Hz, 1H, H4’), 7,30 (dt, J = 8,2, 2,8 Hz, 1H, H7’), 7,04 (m, 2H, H2’, H6’), 6,97 (tdd, J = 7,4, 2,3, 1,0 Hz, 1H, H5’), 4,06 - 3,91 (m, 1H, H3’’), 3,79 - 3,63 (m, 1H, H2’’b), 3,49 - 3,28 (m, 4H, 2xH1, H6’’b, H7’’), 3,01 (d, J = 9,3 Hz, 1H, H4’’), 2,95 - 2,82 (m, 4H, 2xH2, H6’’a, H8’’), 2,75 (t, J = 7,7 Hz, 1H, H5’’a), 2,37 (dd, J = 13,5, 5,4 Hz, 1H, H2’’a), 2,14 - 1,96 (m, 2H, H7’’, H8’’), 1,67 (m, 1H, H5’’b) ppm.13C NMR (63 MHz, MeOD) 5 159,5, 137,2, 127,9, 122,6, 121,3, 118,6, 118,4, 112,3, 111,3, 55,4, 46,9, 46,2, 45,9, 40,9, 26,3, 26,1, 23,0, 19,5 ppm. Masa teórica calculada para C18H24N4O, 312.1950; encontrada [(M+H)+], 313.2017; encontrada [(M+H+CHs)+], 327.2171. IR (film) u/cm-13235, 2941, 2311, 1738, 1624, 1544, 1484, 1452, 1439, 1378, 1351, 1213, 1172, 1064, 1022, 923, 797. [a]D5 = 7,84 o (c. 1,02 mg/mL, MeOH). La pureza fue determinada del 99 % mediante HPLC en fase inversa C18 acoplado a espectrómetro de masas midiendo la corriente iónica total (TIC).
Ejemplo 12: (S)-1-(2-(1rt-indol-3-il)etil)-3-(quinuclidin-3-il)urea (Compuesto 12).

Siguiendo el procedimiento general descrito, (S)-3-aminoquinuclidina (55 mg, 0,26 mmol), DIPEA (0,18 mL, 1,04 mmol) y N-(2-(1H-indol-3-il)etil)-1H-imidazol-1-carboxamida (44 mg,
0,26 mmol) en DMF (1,1 mL), durante 24 h, produjo tras cromatografía flash (ChhCh/MeOH/EtsN, 95:5:1-90:9:1), el compuesto 12 (20 mg, 25 %). [a]2Ds= - 12,75o (c. 1,02 mg/mL, MeOH). La pureza fue determinada del 96 % mediante HPLC en fase inversa C18 acoplado a espectrómetro de masas midiendo la corriente iónica total (TIC).
2. ACTIVIDADES BIOLÓGICAS ESTUDIADAS EN LOS COMPUESTOS DE LA INVENCIÓN.
Ejemplo 13
Estudio de la capacidad moduladora de receptores nicotínicos por los compuestos objeto de la invención medida como la variación de la concentración intracitosólica de calcio ([Ca2+1c inducida por la estimulación de acetilcolina.
Cultivo de células de neuroblastoma SH-SY5Y
Las células SH-SY5Y [ECACC 94030304], procedentes de pases entre el 5 y el 16 tras su descongelación, se mantuvieron en un medio Eagle modificado por Dulbecco (DMEM) conteniendo 15 aminoácidos no esenciales y suplementado con suero bovino fetal al 10 %, glutamina 1 mM, 50 unidades/mL de penicilina y 50 pg/mL de estreptomicina (reactivos de GIBCO, Madrid, España). Las células se sembraron en recipientes que contenían medio suplementado y se mantuvieron en un incubador a 37 °C en atmósfera húmeda con un 5 % de CO2 haciendo pases 1:4 una vez por semana. Para los experimentos, las células se cultivaron en placas de 48 pocillos a una densidad de 2x105 células /pocillo, o en placas de 96 pocillos a una densidad de 8x104 células /pocillo.
Medida de la respuesta celular a la estimulación de receptores nicotínicos con acetilcolina.
Se cultivaron las células de neuroblastoma SH-SY5Y a confluencia en placas negras de 96 pocillos. Se cargaron las células con la sonda fluorescente fluo-4/AM a la concentración de 5 pM durante 1 h a 37 °C en DMEM, añadiendo la mitad de volumen de ácido plurónico del 20 % en DMSO. Seguidamente se lavaron dos veces con una solución Krebs-HEPES y se mantuvieron a temperatura ambiente durante 30 min antes de comenzar el experimento.
La fluorescencia se midió en un lector de fluorescencia en placas (FLUOstar Optima, BMG, Germany), siendo las longitudes de onda de excitación y emisión 485 y 520 nm, respectivamente. La Tabla 1 recoge el efecto de los diferentes compuestos de la invención a
la concentración 10 pM sobre el incremento de la concentración citosólica de calcio ([Ca2+]c) inducida por la estimulación de receptores nicotínicos con acetilcolina a la concentración de 100 pM en células SH-SY5Y en forma de porcentaje de inhibición respecto a un control sin tratamiento. Una reducción de la respuesta relativa indica la acción de los compuestos objeto de la patente sobre los receptores estimulados por acetilcolina, es decir, todos los subtipos de receptores nicotínicos presentes en estas células.
Con objeto de conocer su selectividad sobre el receptor nicotínico del subtipo a7, se utilizó el compuesto PNU282989 (agonista selectivo del receptor a7) en combinación con el compuesto PNU120596 (modulador alostérico positivo selectivo del receptor a7) ambos a la concentración de 10 pM. La estimulación con esta combinación de compuestos produce una activación selectiva del receptor nicotínico a7, por tanto, un bloqueo de la señal de los compuestos objeto de la presente invención indicaría actividad sobre este subtipo especifico de receptor. Los resultados se muestran en la Tabla 1.
Tabla 1
Porcentajes de respuesta relativa al agonista acetilcolina o la combinación de PNU282989 / PNU120596.

Los datos se muestran como media ± SEM de duplicados en cuatro experimentos distintos. Test t de Student respecto a la respuesta del agonista *p < 0.05; **p < 0.01 ;***p < 0.001.
Los resultados indican que los compuestos objeto de la invención son capaces de bloquear la respuesta de los receptores nicotínicos inducida por su agonista natural, acetilcolina, entre un 20 y un 50 %. Además, los compuestos objeto de la invención inhibieron la estimulación selectiva de receptores nicotínicos del subtipo a7. Este resultado demuestra que los compuestos objeto de la invención actúan de forma selectiva sobre este subtipo de receptor. La modulación de la señal nicotínica se relaciona con efectos neuroprotectores y comparados con el compuesto control, mecamilamina, todos ellos mostraron mejor capacidad de bloqueo. Además, este bloqueo fue similar al del antagonista selectivo de receptores nicotínicos a7, metilcaconitina (MLA).
Medida de la respuesta celular a la estimulación de receptores nicotínicos con los compuestos objeto de la invención.
El bloqueo de la respuesta de agonistas nicotínicos (ACh y la combinación de PNU282989 y PNU120596) indica que los compuestos objeto de la presente invención modulan selectivamente el receptor nicotínico a7. Con objeto de demostrar su capacidad agonista se estimularon células SH-SY5Y, previamente cargadas con la sonda Fluo-4AM en las mismas condiciones anteriores, con cada uno de los compuestos a concentraciones crecientes en presencia del modulador selectivo de receptores nicotínicos a7, PNU120596 (10 pM). Las respuestas obtenidas se normalizaron respecto a la respuesta de la combinación de agonista y modulador a7 (PNU282989, 10 pM y PNU120596, 10 pM) se representaron frente a la concentración de cada uno de los compuestos, y las curvas resultantes se ajustaron mediante ajuste no lineal. Posteriormente se calculó la concentración a la cual se obtenía el 50 % de la respuesta máxima normalizada. Los datos obtenidos se representan en la Tabla 2 como valores de concentración efectiva 50 % (CE50).
Tabla 2
Capacidad agonista selectiva de receptores nicotínicos del subtipo a7 expresados como valores de CE
50
normalizando la respuesta al 100 % respecto a PNU282989
.


Los datos se expresan como valor de la media ± SEM de tres experimentos en duplicado en células SH-SY5Y.
Todos los compuestos objeto de la invención mostraron capacidad agonista de receptores nicotínicos a7 con valores de CE50 en el rango micromolar con valores entre 1,1 y 11,1 pM. Este resultado demuestra que todos los compuestos objeto de la invención son agonistas selectivos del receptor nicotínico a7, promoviendo la entrada del ion Ca2+ al citosol.
Ejemplo 14
Medida de la capacidad de captación de radicales libres de oxígeno
Para estudiar la capacidad de captación de radicales libres de oxígeno de los compuestos objeto de la invención se empleó el test ORAC-FL (capacidad de absorbancia de los radicales de oxígeno) desarrollado por Ou y col. (Ou y col., 2001, J Agrie Food Chem, 49: 4619-26). Los compuestos fueron estudiados a seis concentraciones (0,03, 0,1, 0,3, 1, 3 y 5 pM). Las diferentes disoluciones de Trolox (1, 2, 4, 6, 8 pM) y los compuestos, así como melatonina como control positivo a las mismas concentraciones que los compuestos, se realizaron usando como disolvente el tampón PBS (10 mM, pH 7,4) a 37 °C.
Las medidas se llevaron a cabo a 37°C. En primer lugar, se realizó una medida de fluorescencia mediante el lector multipocillo FluoStar Optima (BMG Labtech) (Ex.485 nm, Em.
520 nm) para determinar la señal basal. A continuación, se añadieron 25 pl de AAPH (12 mM) con pipeta multicanal. La fluorescencia fue medida durante 90 min a 37 °C. A partir del área bajo la curva y las concentraciones de los compuestos, se obtuvieron las pendientes de las regresiones lineales que se dividen por la pendiente de la recta del Trolox, obteniendo así los resultados de cada compuesto como equivalentes de Trolox (E.T.). Los datos se muestran en la Tabla 2 como la media ± SEM de al menos tres experimentos por duplicado a seis concentraciones distintas.
La capacidad captadora de radicales libres de los compuestos objeto de la invención ha mejorado con respecto al compuesto de referencia Trolox, un conocido antioxidante, en la misma magnitud que el control positivo melatonina. En este estudio, destacan los compuestos 9, 10 y 11.
Tabla 3
Capacidad de captación de radicales de oxígeno en equivalentes de Trolox por los compuestos de la invención, así como melatonina como control positivo

Los datos se muestran como media ± SEM de cuatro experimentos en duplicado a 5 concentraciones diferentes. Test t Student para datos apareados *p < 0.05, *p < 0.01, ***p < 0.001 con respecto a trolox. Test t Student para datos desapareados #p < 0.05, ##p < 0.01, comparado con melatonina; $p < 0.05, $$p < 0.01 entre enantiómeros.
Ejemplo 15
Estudio de la capacidad neuroprotectora de los compuestos objeto de la invención frente a hiperfosforilación de tau y estrés oxidativo
Medida de la viabilidad celular: 3-(4,5-dimetiltiazol-2-ilo)-2,5-difeniltetrazol, MTT
El parámetro que se usó para medir la viabilidad celular fue la reducción metabólica del bromuro de 3-(4,5-dimetiltiazol-2-il)-2,5-difeniltetrazol (MTT). La cantidad de células vivas es proporcional a la cantidad de formazán formada (Mosmann, 1983, J Immunol Methods, 65: 55-63). Para determinar la viabilidad celular en células de neuroblastoma humano, SH-SY5Y, se añaden 10 pl/pocillo de MTT (5 mg/mL) y, tras 2 h, el medio es retirado sin perder los
cristales de formazán, que se disuelven en 100 ^L de DMSO. Posteriormente, se mide la absorbancia de las muestras a 570 nm con el lector FluoStar Optima (BMG Labtech). Los valores de absorbancia obtenidos con el tóxico solo y con cada compuesto en presencia del tóxico se restaron del valor de absorbancia obtenido en condiciones basales, sin tratamiento. El valor obtenido de la resta de los valores de absorbancia basal menos tóxico sólo, se consideró el 100 % de muerte y los valores obtenidos con los compuestos en presencia de tóxico, se normalizaron como porcentajes de dicho valor. Para calcular el porcentaje de supervivencia, se restaron estos valores a 100.
Neuroprotección frente a hiperfosforilación de tau inducida por ácido okadaico (20 nM), protocolo de pre-incubación y co-incubación:
Estudiamos la capacidad neuroprotectora de los compuestos objeto de esta patente en un modelo in vitro de hiperfosforilación de tau. Se evaluó el efecto neuroprotector de los compuestos en células de neuroblastoma humano, frente a la hiperfosforilación de tau producida por el ácido okadaico (AO), una toxina marina inhibidora de las serin/treonin fosfatasas PP1 y PP2A, lo que impide la desfosforilación de tau (Kamat y col., 2014, Mol Neurobiol, 50: 852-65), realizando el siguiente protocolo:
En este protocolo, las células fueron pre-incubadas con cada uno de los derivados estudiados a la concentración 1 |iM durante 24 h. Tras el periodo de pre-incubación, el medio se retiró y fue sustituido por medio de cultivo con los compuestos y ácido okadaico, a la concentración de 20 nM. Transcurridas 18 h, la viabilidad celular fue evaluada por el método de la reducción de MTT. En todos los ensayos farmacológicos se utilizó un control positivo con fines comparativos y para evaluar la bondad del método empleado. Para ello se utilizó melatonina (1 ^M) que ha demostrado capacidad neuroprotectora en diversos modelos.
Los resultados obtenidos para los compuestos objeto de la presente invención se muestran en la Tabla 4, y vienen expresados en porcentaje de supervivencia celular y en porcentaje de la actividad neuroprotectora.
En este protocolo, todos los compuestos objeto de la invención son capaces de mostrar efecto neuroprotector, favoreciendo así la supervivencia celular, y por tanto destacando 4, 9 y 2. Por tanto, la combinación de actividades farmacológicas de los compuestos objeto de la invención se traduce en un efecto neuroprotector frente a la toxicidad inducida por la hiperfosforilación de la proteína Tau.
Tabla 4
Porcentaje de neuroprotección producido por los compuestos de la invención y melatonina, a la concentración de 1 pM, frente a la toxicidad inducida por ácido okadaico.

Los datos se muestran como media ± SEM de cuatro experimentos en triplicado. ANOVA de una vía, posthoc test de Newman-Keuls. ###p < 0,001 con respecto a condiciones basales; *p < 0,05; **p < 0,01, ***p < 0,001 comparado con AO.
Neuroprotección frente a estrés oxidativo inducido por la combinación de rotenona (30 uM) y oligomicina A (10 uM):
Se evaluó el efecto neuroprotector de los compuestos en células de neuroblastoma humano, frente a estrés oxidativo producido por rotenona y oligomicina A, bloqueantes de la cadena respiratoria de la mitocondria.
Tabla 5
Porcentaje de neuroprotección producido por los compuestos de la invención y melatonina, a la concentración de 1 pM, frente a la toxicidad inducida por ácido okadaico.


Los datos se muestran como media ± SEM de cuatro experimentos en triplicado. ANOVA de una vía, posthoc test de Newman-Keuls. ###p < 0,001 con respecto a condiciones basales; *p < 0,05; **p < 0,01, *** p < 0,001 comparado con R/O.
En este estudio, se realizó una pre-incubación de 24 h con los compuestos sintetizados a una concentración de 1 pM y una co-incubación de 24 h de éstos en presencia rotenona/oligomicina A (30 pM/10 pM, respectivamente). Transcurridas 24h, la viabilidad celular fue evaluada por el método de la reducción de MTT.
Con este protocolo se obtiene información sobre todas las potenciales actividades biológicas presentes en la estructura objeto de estudio.
En este modelo, al estar presente durante las 24 horas previas a la incubación del tóxico, el compuesto es capaz de mostrar su capacidad activadora de receptores nicotínicos a7, los cuales desencadenan una cascada de señalización favoreciendo así la supervivencia celular. Además, también está presente a la vez que se exponen las células al estímulo tóxico, la mezcla de rotenona/oligomicina A que provoca la aparición de una gran cantidad de radicales libres en el interior celular. Estos radicales dañan la célula e inducen la apoptosis celular. Los porcentajes de supervivencia y protección se resumen en la Tabla 5.
Los compuestos objeto de la presente invención mostraron carácter neuroprotector frente a la toxicidad inducida por estrés oxidativo. El carácter antioxidante demostrado por los compuestos se traduce en un aumento de la supervivencia celular tras un insulto que provoca la aparición de radicales libres, destacan los compuestos 7, 8 y 11.
Claims (13)
1. Un compuesto de formula (I):

donde
R se selecciona del grupo consistente en:
- hidrógeno
- Alquilo opcionalmente sustituido por uno, dos, o tres átomos de halógeno seleccionado entre flúor, cloro y bromo; cicloalquilo(C3-C6); alcoxilo(Ci-C6); cicloalcoxilo(C3-C6); ciano y nitro; y/o
- fenilo opcionalmente sustituido por uno, dos o tres grupos seleccionados independientemente entre flúor; cloro; bromo; alquilo opcionalmente sustituido por uno, dos, o tres átomos de halógeno seleccionado entre flúor, cloro y bromo; cicloalquilo(C3-C6); alcoxilo(C1-C6); cicloalcoxilo(C3-C6); ciano y nitro; o bien dos grupos pueden formar conjuntamente un grupo -O(CH2)oO-, -(CH2V , o -CH=CH-CH=CH-; o
- un grupo heteroarilo opcionalmente sustituido por uno, dos o tres grupos seleccionados independientemente entre flúor, cloro, bromo, alquilo, cicloalquilo(C3-C6), alcoxilo(Ci-C6), cicloalcoxilo(C3-C6), ciano y nitro;
R1 y R2 se seleccionan del grupo consistente en hidrógeno, alquilo, cicloalquilo, arilo o heteroarilo, opcionalmente sustituido por uno, dos o tres grupos seleccionados independientemente entre flúor, cloro, bromo, alquilo, alcoxilo, cicloalquilo(C3-C6), cicloalcoxilo(C3-C6), ciano, nitro y carboxilato o ambos grupos pueden formar conjuntamente un grupo -O(CH2)qO-, -(CH2V, o -CH=CH-CH=CH-;
X se selecciona entre un átomo de oxígeno o un átomo de azufre;
Y se selecciona entre un átomo de carbono o un átomo de nitrógeno;
Z se selecciona entre un átomo de carbono o un átomo de nitrógeno;
n es un número entero seleccionado entre 0, 1,2, 3, 4, 5 ó 6;
*3’’ indica la presencia de un centro quiral cuya configuración puede ser R o S;
o sus estereoisomeros, sales, preferiblemente sales farmacéuticamente aceptables, profármacos o solvatos.
2. Un compuesto según la reivindicación 1 donde
R es un átomo de hidrógeno
R1 se selecciona del grupo que comprende un alquilo opcionalmente sustituido por uno o dos grupos seleccionados independientemente entre flúor, cloro, y bromo; alcoxilo(C1-C6), nitro y amino; preferentemente R1 es -OCH3;
R2 es hidrógeno;
n es un entero seleccionado entre 0, 1 y 2, preferentemente n = 1;
X es oxígeno o azufre;
*3’’ es un átomo de carbono con configuración R o S en cualquiera de sus posibles combinaciones
o sus estereoisómeros, sales, preferiblemente sales farmacéuticamente aceptables, profármacos o solvatos.
3. Un compuesto según las reivindicaciones 1 o 2 caracterizado por que consiste en:
• R)-1 -(2-(5-metoxi-1 H-indol-3-il)etil)-3-(quinuclidin-3-il)tiourea
• (S)-1-(2-(5-metoxi-1 H-indol-3-il)etil)-3-(quinuclidin-3-yl)tiourea
• (R)-1-(2-(5-cloro-1 H-indol-3-il)etil)-3-(quinuclidin-3-il)tiourea
• (S)-1 -(2-(5-cloro-1 H-indol-3-il)etil)-3-(quinuclidin-3-il)tiourea
• (R)- 1-(2-(1 H-indol-3-il)etil)-3-(quinuclidin-3-yl)tiourea
• (S)- 1-(2-(1H-indol-3-il)etil)-3-(quinuclidin-3-yl)tiourea
• (R)-1-(2-(5-metoxi-1 H-indol-3-il)etil)-3-(quinuclidin-3-il)urea
• (S)-1 -(2-(5-metoxi-1 H-indol-3-il)etil)-3-(quinuclidin-3-il)urea
• (R)-1-(2-(5-cloro-1 H-indol-3-yl)ethyl)-3-(quinuclidin-3-yl)urea
• (S)-1 -(2-(5-cloro-1 H-indol-3-yl)etil)-3-(quinuclidin-3-il)urea
• (R)-1-(2-(1 H-indol-3-il)etil)-3-(quinuclidin-3-il)urea
• (S)-1-(2-(1 H-indol-3-il)etil)-3-(quinuclidin-3-il)urea
4. Un procedimiento para la preparación de un compuesto de fórmula (I) según reivindicación 1 caracterizado por hacer reaccionar 3-aminoquinuclidina (II) con configuración R o S.
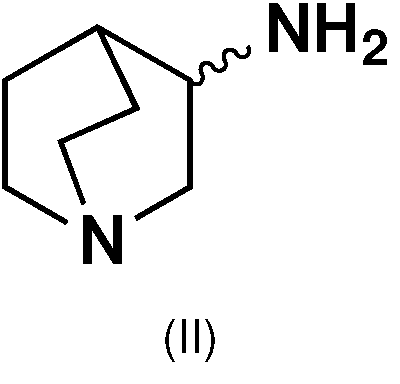
con el derivado correspondiente de 3-(2-isotiocianatoetil)-5-R-1H-indol (III) o N-(2-(5-R-1 H-indol-3-il)etil)-1 H-imidazol-1 -carboxamida (IV):

(III) (IV)
donde R, R1, R2, X, Y, Z y n tienen el significado ya indicado.
5. Procedimiento según la reivindicación 4, en el que la reacción entre el compuesto de fórmula (II) y el compuesto determinado de fórmula (III) en presencia de una amina orgánica, en un disolvente seleccionado entre dimetilformamida, dimetilsulfoxido y sus mezclas.
6. Composición farmacéutica que comprende al menos un compuesto de fórmula (I) según cualquiera de las reivindicaciones 1 a 3 y un excipiente adecuado.
7. Compuesto de fórmula (I) según cualquiera de las reivindicaciones 1 a 3 para su uso en medicina.
8. Compuesto de fórmula (I) según reivindicación 8 para su uso en el tratamiento o prevención de enfermedades que mejoran con la administración de un agonista de receptores nicotínicos y/o un agente secuestrador de radicales libres.
9. Compuesto de fórmula (I) según reivindicación 9 para su uso en el tratamiento o prevención de enfermedades neurodegenerativas.
10. Compuesto de fórmula (I) según reivindicación 10 para su uso en el tratamiento o prevención de una enfermedad neurodegenerativa central y/o periférica.
11. Compuesto de fórmula (I) según la reivindicación 10 para su uso en el tratamiento o prevención de la enfermedad de Alzheimer, la enfermedad de Parkinson, la enfermedad de Huntington, la esclerosis lateral amiotrófica y la esclerosis múltiple.
12. Compuesto de fórmula (I) según cualquiera de las reivindicaciones 7-10 caracterizado por que se administra en dosis diarias comprendidas entre 0,1 y 500 mg.
13. Compuesto de fórmula (I) según reivindicación 12 caracterizado por que se administra en dosis diarias comprendidas entre 25 y 250 mg.
Priority Applications (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| ES201930908A ES2819309B2 (es) | 2019-10-14 | 2019-10-14 | Compuestos agonistas nicotínicos y antioxidantes para el tratamiento de enfermedades neurodegenerativas |
Applications Claiming Priority (1)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| ES201930908A ES2819309B2 (es) | 2019-10-14 | 2019-10-14 | Compuestos agonistas nicotínicos y antioxidantes para el tratamiento de enfermedades neurodegenerativas |
Publications (2)
| Publication Number | Publication Date |
|---|---|
| ES2819309A1 ES2819309A1 (es) | 2021-04-15 |
| ES2819309B2 true ES2819309B2 (es) | 2021-11-17 |
Family
ID=75420282
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES201930908A Expired - Fee Related ES2819309B2 (es) | 2019-10-14 | 2019-10-14 | Compuestos agonistas nicotínicos y antioxidantes para el tratamiento de enfermedades neurodegenerativas |
Country Status (1)
| Country | Link |
|---|---|
| ES (1) | ES2819309B2 (es) |
Family Cites Families (2)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| SE0401971D0 (sv) * | 2004-08-02 | 2004-08-02 | Astrazeneca Ab | Piperidne derivatives |
| DE102006033109A1 (de) * | 2006-07-18 | 2008-01-31 | Grünenthal GmbH | Substituierte Heteroaryl-Derivate |
-
2019
- 2019-10-14 ES ES201930908A patent/ES2819309B2/es not_active Expired - Fee Related
Also Published As
| Publication number | Publication date |
|---|---|
| ES2819309A1 (es) | 2021-04-15 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| JP7838004B2 (ja) | Polybromo-1(pbrm1)の小分子分解剤 | |
| CN109071528B (zh) | 用于诊断和治疗的双环化合物 | |
| ES2820827T3 (es) | Formas sólidas de un compuesto modulador de quinasas | |
| US10358443B2 (en) | Heteroaryl amides as inhibitors of protein aggregation | |
| CA3173262A1 (en) | Tropomyosin receptor kinase (trk) degradation compounds and methods of use | |
| ES2360333A1 (es) | Derivados de bis (aralquil) amino y sistemas (hetero) aromaticos de seis miembros y su uso en el tratamiento de patologias neurodegenerativas, incluida la enfermedad de alzheimer. | |
| ES2696708T3 (es) | Uso de derivados de bencimidazol-prolina | |
| ES2882285T3 (es) | Derivados de bis-heteroarilo como moduladores de la agregación de proteínas | |
| ES2926918T3 (es) | Tratamiento de síntomas cognitivos y del estado de ánimo en trastornos neurodegenerativos y neuropsiquiátricos con agonistas del receptor de GABAA que contienen alfa5 | |
| BR112012033075B1 (pt) | Composto piperidina substituído antagonista de receptor c5a, composição farmacêutica, e, uso do composto | |
| KR20200096570A (ko) | 알츠하이머병과 같은 타우 응집체와 연관된 장애의 치료, 완화 또는 예방을 위한 1,3,4,5-테트라히드로-2h-피리도[4,3-b]인돌 유도체 | |
| BR112019011924A2 (pt) | derivados de bis-heteroarila bicíclicos como moduladores de agregação de proteína | |
| WO2021238007A1 (zh) | 可诱导prc2蛋白复合物核心亚基降解的双功能化合物和药物组合物及应用 | |
| US11091474B2 (en) | Alkoxy bis-heteroaryl derivatives as modulators of protein aggregation | |
| ES2924950T3 (es) | Novedosos compuestos para el tratamiento, alivio o prevención de trastornos asociados con los agregados Tau | |
| ES2819309B2 (es) | Compuestos agonistas nicotínicos y antioxidantes para el tratamiento de enfermedades neurodegenerativas | |
| ES2976567T3 (es) | Formas cristalinas de (1-pirimidin-2-il-ciclopropil)-amida del ácido (3S,4S)-1-ciclopropilmetil-4-{[5-(2,4-difluoro-fenil)-isoxazol-3-carbonil]-amino}-piperidin-3-carboxílico | |
| ES2998532T3 (en) | Quinone reductase 2 inhibitor compounds and uses thereof | |
| ES2781377B2 (es) | Compuestos derivados de 2-iodo-7-(((2-(5-metoxi-1h-indol-3-il)etil)amino)metil)-1-alquil/aril-6,7,7a,8-tetrahidro-3h-pirrolo[2,1-j]quinolina-3,9(5h)-diona como agentes antioxidantes, inductores de nrf2 y moduladores nicotinicos para el tratamiento de enfermedades neurodegenerativas | |
| ES2749512T3 (es) | Derivados de tetrahidroquinolina y su uso como inhibidores de Epac | |
| ES2360435B1 (es) | Derivados de biss (aralquil) amino y sistemas (6+5)-heteroaromaticos y su uso en el tratamiento de patologias neurodegenerativas, incluida la enfermedad de alzheimer | |
| Teichmann | Photoswitchable Kinase Inhibitors to Reversibly Control Kinase Activity | |
| TW202535872A (zh) | 四氫異喹啉異雙功能bcl-xl降解劑 | |
| WO2025101575A1 (en) | Tetrahydroisoquinoline heterobifunctional bcl-x l degraders | |
| WO2025101588A1 (en) | Tetrahydroisoquinoline heterobifunctional bcl-xl degraders |
Legal Events
| Date | Code | Title | Description |
|---|---|---|---|
| BA2A | Patent application published |
Ref document number: 2819309 Country of ref document: ES Kind code of ref document: A1 Effective date: 20210415 |
|
| FG2A | Definitive protection |
Ref document number: 2819309 Country of ref document: ES Kind code of ref document: B2 Effective date: 20211117 |
|
| FD2A | Announcement of lapse in spain |
Effective date: 20240404 |