ES2836733T3 - Antagonistas de integrina - Google Patents
Antagonistas de integrina Download PDFInfo
- Publication number
- ES2836733T3 ES2836733T3 ES19192402T ES19192402T ES2836733T3 ES 2836733 T3 ES2836733 T3 ES 2836733T3 ES 19192402 T ES19192402 T ES 19192402T ES 19192402 T ES19192402 T ES 19192402T ES 2836733 T3 ES2836733 T3 ES 2836733T3
- Authority
- ES
- Spain
- Prior art keywords
- compound
- compounds
- integrin
- pharmaceutically acceptable
- present
- Prior art date
- Legal status (The legal status is an assumption and is not a legal conclusion. Google has not performed a legal analysis and makes no representation as to the accuracy of the status listed.)
- Active
Links
- 102000006495 integrins Human genes 0.000 title description 53
- 108010044426 integrins Proteins 0.000 title description 53
- 239000005557 antagonist Substances 0.000 title description 12
- 150000001875 compounds Chemical class 0.000 claims abstract description 149
- 239000000203 mixture Substances 0.000 claims abstract description 51
- 150000003839 salts Chemical class 0.000 claims abstract description 25
- 125000002947 alkylene group Chemical group 0.000 claims abstract description 14
- 239000012453 solvate Substances 0.000 claims abstract description 11
- 125000002496 methyl group Chemical group [H]C([H])([H])* 0.000 claims abstract description 9
- 125000001495 ethyl group Chemical group [H]C([H])([H])C([H])([H])* 0.000 claims abstract description 7
- 229910052739 hydrogen Inorganic materials 0.000 claims abstract description 6
- 239000001257 hydrogen Substances 0.000 claims abstract description 6
- 125000004435 hydrogen atom Chemical class [H]* 0.000 claims abstract description 5
- 238000011282 treatment Methods 0.000 claims description 29
- 102000005789 Vascular Endothelial Growth Factors Human genes 0.000 claims description 17
- 108010019530 Vascular Endothelial Growth Factors Proteins 0.000 claims description 17
- 208000037265 diseases, disorders, signs and symptoms Diseases 0.000 claims description 17
- 238000002347 injection Methods 0.000 claims description 16
- 239000007924 injection Substances 0.000 claims description 16
- 239000008194 pharmaceutical composition Substances 0.000 claims description 15
- 230000002265 prevention Effects 0.000 claims description 14
- 208000035475 disorder Diseases 0.000 claims description 12
- 206010012689 Diabetic retinopathy Diseases 0.000 claims description 11
- 206010029113 Neovascularisation Diseases 0.000 claims description 11
- 230000000694 effects Effects 0.000 claims description 11
- 239000003937 drug carrier Substances 0.000 claims description 10
- 239000003814 drug Substances 0.000 claims description 8
- 230000002401 inhibitory effect Effects 0.000 claims description 8
- 108091008605 VEGF receptors Proteins 0.000 claims description 6
- 102000009484 Vascular Endothelial Growth Factor Receptors Human genes 0.000 claims description 6
- 206010012601 diabetes mellitus Diseases 0.000 claims description 6
- 230000005764 inhibitory process Effects 0.000 claims description 5
- 208000001344 Macular Edema Diseases 0.000 claims description 4
- 206010025415 Macular oedema Diseases 0.000 claims description 4
- 206010028980 Neoplasm Diseases 0.000 claims description 4
- 201000011510 cancer Diseases 0.000 claims description 4
- 201000010230 macular retinal edema Diseases 0.000 claims description 4
- 208000002691 Choroiditis Diseases 0.000 claims description 3
- 206010055665 Corneal neovascularisation Diseases 0.000 claims description 3
- 206010065630 Iris neovascularisation Diseases 0.000 claims description 3
- 208000003971 Posterior uveitis Diseases 0.000 claims description 3
- 208000000208 Wet Macular Degeneration Diseases 0.000 claims description 3
- 201000000159 corneal neovascularization Diseases 0.000 claims description 3
- 206010038848 Retinal detachment Diseases 0.000 claims description 2
- 230000004264 retinal detachment Effects 0.000 claims description 2
- 108010073929 Vascular Endothelial Growth Factor A Proteins 0.000 claims 4
- -1 blood pressure Substances 0.000 description 32
- 238000000034 method Methods 0.000 description 20
- YMWUJEATGCHHMB-UHFFFAOYSA-N Dichloromethane Chemical compound ClCCl YMWUJEATGCHHMB-UHFFFAOYSA-N 0.000 description 18
- ZMANZCXQSJIPKH-UHFFFAOYSA-N Triethylamine Chemical compound CCN(CC)CC ZMANZCXQSJIPKH-UHFFFAOYSA-N 0.000 description 18
- VEXZGXHMUGYJMC-UHFFFAOYSA-N Hydrochloric acid Chemical compound Cl VEXZGXHMUGYJMC-UHFFFAOYSA-N 0.000 description 15
- 206010064930 age-related macular degeneration Diseases 0.000 description 15
- 229940123038 Integrin antagonist Drugs 0.000 description 13
- 210000001508 eye Anatomy 0.000 description 13
- IMNFDUFMRHMDMM-UHFFFAOYSA-N N-Heptane Chemical compound CCCCCCC IMNFDUFMRHMDMM-UHFFFAOYSA-N 0.000 description 12
- 230000015572 biosynthetic process Effects 0.000 description 12
- 239000000243 solution Substances 0.000 description 12
- 238000003786 synthesis reaction Methods 0.000 description 12
- 239000004480 active ingredient Substances 0.000 description 11
- VYPSYNLAJGMNEJ-UHFFFAOYSA-N Silicium dioxide Chemical compound O=[Si]=O VYPSYNLAJGMNEJ-UHFFFAOYSA-N 0.000 description 10
- 239000002253 acid Substances 0.000 description 10
- 125000000999 tert-butyl group Chemical group [H]C([H])([H])C(*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 10
- 208000000318 vitreous detachment Diseases 0.000 description 10
- XEKOWRVHYACXOJ-UHFFFAOYSA-N Ethyl acetate Chemical compound CCOC(C)=O XEKOWRVHYACXOJ-UHFFFAOYSA-N 0.000 description 9
- 206010012688 Diabetic retinal oedema Diseases 0.000 description 8
- 108010049003 Fibrinogen Proteins 0.000 description 8
- 102000008946 Fibrinogen Human genes 0.000 description 8
- 108010067306 Fibronectins Proteins 0.000 description 8
- 102000016359 Fibronectins Human genes 0.000 description 8
- 230000033115 angiogenesis Effects 0.000 description 8
- 239000012062 aqueous buffer Substances 0.000 description 8
- 201000011190 diabetic macular edema Diseases 0.000 description 8
- 229940012952 fibrinogen Drugs 0.000 description 8
- 210000001525 retina Anatomy 0.000 description 8
- 230000008728 vascular permeability Effects 0.000 description 8
- 229940126062 Compound A Drugs 0.000 description 7
- NLDMNSXOCDLTTB-UHFFFAOYSA-N Heterophylliin A Natural products O1C2COC(=O)C3=CC(O)=C(O)C(O)=C3C3=C(O)C(O)=C(O)C=C3C(=O)OC2C(OC(=O)C=2C=C(O)C(O)=C(O)C=2)C(O)C1OC(=O)C1=CC(O)=C(O)C(O)=C1 NLDMNSXOCDLTTB-UHFFFAOYSA-N 0.000 description 7
- XBDQKXXYIPTUBI-UHFFFAOYSA-N Propionic acid Chemical compound CCC(O)=O XBDQKXXYIPTUBI-UHFFFAOYSA-N 0.000 description 7
- 210000002889 endothelial cell Anatomy 0.000 description 7
- 239000011541 reaction mixture Substances 0.000 description 7
- LFQSCWFLJHTTHZ-UHFFFAOYSA-N Ethanol Chemical compound CCO LFQSCWFLJHTTHZ-UHFFFAOYSA-N 0.000 description 6
- UIIMBOGNXHQVGW-UHFFFAOYSA-M Sodium bicarbonate Chemical compound [Na+].OC([O-])=O UIIMBOGNXHQVGW-UHFFFAOYSA-M 0.000 description 6
- 230000008485 antagonism Effects 0.000 description 6
- 238000003556 assay Methods 0.000 description 6
- 239000002585 base Substances 0.000 description 6
- 229910002091 carbon monoxide Inorganic materials 0.000 description 6
- 125000002915 carbonyl group Chemical group [*:2]C([*:1])=O 0.000 description 6
- 230000012292 cell migration Effects 0.000 description 6
- 239000003246 corticosteroid Substances 0.000 description 6
- 239000012043 crude product Substances 0.000 description 6
- 102000005962 receptors Human genes 0.000 description 6
- 108020003175 receptors Proteins 0.000 description 6
- 239000007864 aqueous solution Substances 0.000 description 5
- 210000004027 cell Anatomy 0.000 description 5
- 101150007330 cpdB gene Proteins 0.000 description 5
- 201000010099 disease Diseases 0.000 description 5
- 238000002474 experimental method Methods 0.000 description 5
- 230000003993 interaction Effects 0.000 description 5
- 239000003446 ligand Substances 0.000 description 5
- 230000001575 pathological effect Effects 0.000 description 5
- 239000000047 product Substances 0.000 description 5
- 125000004527 pyrimidin-4-yl group Chemical group N1=CN=C(C=C1)* 0.000 description 5
- 239000003826 tablet Substances 0.000 description 5
- QTBSBXVTEAMEQO-UHFFFAOYSA-M Acetate Chemical compound CC([O-])=O QTBSBXVTEAMEQO-UHFFFAOYSA-M 0.000 description 4
- 239000006144 Dulbecco’s modified Eagle's medium Substances 0.000 description 4
- 102000010834 Extracellular Matrix Proteins Human genes 0.000 description 4
- 108010037362 Extracellular Matrix Proteins Proteins 0.000 description 4
- PMZURENOXWZQFD-UHFFFAOYSA-L Sodium Sulfate Chemical compound [Na+].[Na+].[O-]S([O-])(=O)=O PMZURENOXWZQFD-UHFFFAOYSA-L 0.000 description 4
- 239000000427 antigen Substances 0.000 description 4
- 108091007433 antigens Proteins 0.000 description 4
- 102000036639 antigens Human genes 0.000 description 4
- 238000006243 chemical reaction Methods 0.000 description 4
- 239000003795 chemical substances by application Substances 0.000 description 4
- AMLYAMJWYAIXIA-VWNVYAMZSA-N cilengitide Chemical compound N1C(=O)[C@H](CC(O)=O)NC(=O)CNC(=O)[C@H](CCCN=C(N)N)NC(=O)[C@H](C(C)C)N(C)C(=O)[C@H]1CC1=CC=CC=C1 AMLYAMJWYAIXIA-VWNVYAMZSA-N 0.000 description 4
- 229950009003 cilengitide Drugs 0.000 description 4
- 238000004440 column chromatography Methods 0.000 description 4
- 229960001334 corticosteroids Drugs 0.000 description 4
- 239000003480 eluent Substances 0.000 description 4
- 238000001704 evaporation Methods 0.000 description 4
- 230000008020 evaporation Effects 0.000 description 4
- RWTNPBWLLIMQHL-UHFFFAOYSA-N fexofenadine Chemical compound C1=CC(C(C)(C(O)=O)C)=CC=C1C(O)CCCN1CCC(C(O)(C=2C=CC=CC=2)C=2C=CC=CC=2)CC1 RWTNPBWLLIMQHL-UHFFFAOYSA-N 0.000 description 4
- 239000012530 fluid Substances 0.000 description 4
- 239000012634 fragment Substances 0.000 description 4
- 230000012010 growth Effects 0.000 description 4
- 238000011534 incubation Methods 0.000 description 4
- 238000010232 migration assay Methods 0.000 description 4
- 239000000843 powder Substances 0.000 description 4
- 238000002360 preparation method Methods 0.000 description 4
- 125000002924 primary amino group Chemical group [H]N([H])* 0.000 description 4
- 201000007914 proliferative diabetic retinopathy Diseases 0.000 description 4
- 230000002207 retinal effect Effects 0.000 description 4
- 239000000377 silicon dioxide Substances 0.000 description 4
- 229910052938 sodium sulfate Inorganic materials 0.000 description 4
- 235000011152 sodium sulphate Nutrition 0.000 description 4
- 210000001519 tissue Anatomy 0.000 description 4
- XLYOFNOQVPJJNP-UHFFFAOYSA-N water Substances O XLYOFNOQVPJJNP-UHFFFAOYSA-N 0.000 description 4
- OZFAFGSSMRRTDW-UHFFFAOYSA-N (2,4-dichlorophenyl) benzenesulfonate Chemical compound ClC1=CC(Cl)=CC=C1OS(=O)(=O)C1=CC=CC=C1 OZFAFGSSMRRTDW-UHFFFAOYSA-N 0.000 description 3
- 239000012591 Dulbecco’s Phosphate Buffered Saline Substances 0.000 description 3
- 108010073385 Fibrin Proteins 0.000 description 3
- 102000009123 Fibrin Human genes 0.000 description 3
- BWGVNKXGVNDBDI-UHFFFAOYSA-N Fibrin monomer Chemical compound CNC(=O)CNC(=O)CN BWGVNKXGVNDBDI-UHFFFAOYSA-N 0.000 description 3
- 102100022337 Integrin alpha-V Human genes 0.000 description 3
- 241001465754 Metazoa Species 0.000 description 3
- OKKJLVBELUTLKV-UHFFFAOYSA-N Methanol Chemical compound OC OKKJLVBELUTLKV-UHFFFAOYSA-N 0.000 description 3
- 241001529936 Murinae Species 0.000 description 3
- 206010030113 Oedema Diseases 0.000 description 3
- YXFVVABEGXRONW-UHFFFAOYSA-N Toluene Chemical compound CC1=CC=CC=C1 YXFVVABEGXRONW-UHFFFAOYSA-N 0.000 description 3
- DTQVDTLACAAQTR-UHFFFAOYSA-M Trifluoroacetate Chemical compound [O-]C(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-M 0.000 description 3
- DTQVDTLACAAQTR-UHFFFAOYSA-N Trifluoroacetic acid Chemical compound OC(=O)C(F)(F)F DTQVDTLACAAQTR-UHFFFAOYSA-N 0.000 description 3
- 108010048673 Vitronectin Receptors Proteins 0.000 description 3
- 238000009825 accumulation Methods 0.000 description 3
- 230000008901 benefit Effects 0.000 description 3
- 210000004369 blood Anatomy 0.000 description 3
- 239000008280 blood Substances 0.000 description 3
- 125000004432 carbon atom Chemical group C* 0.000 description 3
- 230000000052 comparative effect Effects 0.000 description 3
- 238000011161 development Methods 0.000 description 3
- 230000018109 developmental process Effects 0.000 description 3
- 239000002552 dosage form Substances 0.000 description 3
- 229950003499 fibrin Drugs 0.000 description 3
- 239000001963 growth medium Substances 0.000 description 3
- 238000000338 in vitro Methods 0.000 description 3
- 238000011835 investigation Methods 0.000 description 3
- 239000011159 matrix material Substances 0.000 description 3
- 239000002609 medium Substances 0.000 description 3
- 235000017557 sodium bicarbonate Nutrition 0.000 description 3
- 229910000030 sodium bicarbonate Inorganic materials 0.000 description 3
- 239000002904 solvent Substances 0.000 description 3
- 208000010110 spontaneous platelet aggregation Diseases 0.000 description 3
- 239000000126 substance Substances 0.000 description 3
- 125000004213 tert-butoxy group Chemical group [H]C([H])([H])C(O*)(C([H])([H])[H])C([H])([H])[H] 0.000 description 3
- 238000012360 testing method Methods 0.000 description 3
- 230000001225 therapeutic effect Effects 0.000 description 3
- 230000008719 thickening Effects 0.000 description 3
- 229960004072 thrombin Drugs 0.000 description 3
- 208000029257 vision disease Diseases 0.000 description 3
- 230000004393 visual impairment Effects 0.000 description 3
- 238000005160 1H NMR spectroscopy Methods 0.000 description 2
- JWUJQDFVADABEY-UHFFFAOYSA-N 2-methyltetrahydrofuran Chemical compound CC1CCCO1 JWUJQDFVADABEY-UHFFFAOYSA-N 0.000 description 2
- 201000004569 Blindness Diseases 0.000 description 2
- 241000283690 Bos taurus Species 0.000 description 2
- KXDHJXZQYSOELW-UHFFFAOYSA-M Carbamate Chemical compound NC([O-])=O KXDHJXZQYSOELW-UHFFFAOYSA-M 0.000 description 2
- OKTJSMMVPCPJKN-UHFFFAOYSA-N Carbon Chemical compound [C] OKTJSMMVPCPJKN-UHFFFAOYSA-N 0.000 description 2
- BVKZGUZCCUSVTD-UHFFFAOYSA-L Carbonate Chemical compound [O-]C([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-L 0.000 description 2
- 102000019034 Chemokines Human genes 0.000 description 2
- 108010012236 Chemokines Proteins 0.000 description 2
- 108090000695 Cytokines Proteins 0.000 description 2
- 102000004127 Cytokines Human genes 0.000 description 2
- 238000012286 ELISA Assay Methods 0.000 description 2
- 206010061218 Inflammation Diseases 0.000 description 2
- 108700036276 KH902 fusion Proteins 0.000 description 2
- 206010025421 Macule Diseases 0.000 description 2
- 101100221809 Neurospora crassa (strain ATCC 24698 / 74-OR23-1A / CBS 708.71 / DSM 1257 / FGSC 987) cpd-7 gene Proteins 0.000 description 2
- KDLHZDBZIXYQEI-UHFFFAOYSA-N Palladium Chemical compound [Pd] KDLHZDBZIXYQEI-UHFFFAOYSA-N 0.000 description 2
- 208000034038 Pathologic Neovascularization Diseases 0.000 description 2
- QQONPFPTGQHPMA-UHFFFAOYSA-N Propene Chemical group CC=C QQONPFPTGQHPMA-UHFFFAOYSA-N 0.000 description 2
- UIIMBOGNXHQVGW-DEQYMQKBSA-M Sodium bicarbonate-14C Chemical compound [Na+].O[14C]([O-])=O UIIMBOGNXHQVGW-DEQYMQKBSA-M 0.000 description 2
- 108090000190 Thrombin Proteins 0.000 description 2
- 206010047571 Visual impairment Diseases 0.000 description 2
- 229960002833 aflibercept Drugs 0.000 description 2
- 108010081667 aflibercept Proteins 0.000 description 2
- 238000010171 animal model Methods 0.000 description 2
- 150000001450 anions Chemical group 0.000 description 2
- 229960000397 bevacizumab Drugs 0.000 description 2
- 230000031018 biological processes and functions Effects 0.000 description 2
- PFYXSUNOLOJMDX-UHFFFAOYSA-N bis(2,5-dioxopyrrolidin-1-yl) carbonate Chemical compound O=C1CCC(=O)N1OC(=O)ON1C(=O)CCC1=O PFYXSUNOLOJMDX-UHFFFAOYSA-N 0.000 description 2
- 229950000025 brolucizumab Drugs 0.000 description 2
- GZUXJHMPEANEGY-UHFFFAOYSA-N bromomethane Chemical compound BrC GZUXJHMPEANEGY-UHFFFAOYSA-N 0.000 description 2
- 239000002775 capsule Substances 0.000 description 2
- 239000000969 carrier Substances 0.000 description 2
- 150000001768 cations Chemical group 0.000 description 2
- 230000004663 cell proliferation Effects 0.000 description 2
- 239000003153 chemical reaction reagent Substances 0.000 description 2
- 210000003161 choroid Anatomy 0.000 description 2
- 229950005748 conbercept Drugs 0.000 description 2
- 101150025236 dmaW gene Proteins 0.000 description 2
- 230000003511 endothelial effect Effects 0.000 description 2
- 210000002744 extracellular matrix Anatomy 0.000 description 2
- 239000007943 implant Substances 0.000 description 2
- 230000004377 improving vision Effects 0.000 description 2
- 238000001727 in vivo Methods 0.000 description 2
- 230000002757 inflammatory effect Effects 0.000 description 2
- 230000004054 inflammatory process Effects 0.000 description 2
- 230000002452 interceptive effect Effects 0.000 description 2
- 230000004410 intraocular pressure Effects 0.000 description 2
- 150000002500 ions Chemical class 0.000 description 2
- 210000000265 leukocyte Anatomy 0.000 description 2
- 150000002632 lipids Chemical class 0.000 description 2
- 230000007246 mechanism Effects 0.000 description 2
- 229910052751 metal Inorganic materials 0.000 description 2
- 239000002184 metal Substances 0.000 description 2
- 230000005012 migration Effects 0.000 description 2
- 238000013508 migration Methods 0.000 description 2
- 231100000252 nontoxic Toxicity 0.000 description 2
- 230000003000 nontoxic effect Effects 0.000 description 2
- 230000037361 pathway Effects 0.000 description 2
- WEXRUCMBJFQVBZ-UHFFFAOYSA-N pentobarbital Chemical compound CCCC(C)C1(CC)C(=O)NC(=O)NC1=O WEXRUCMBJFQVBZ-UHFFFAOYSA-N 0.000 description 2
- HXITXNWTGFUOAU-UHFFFAOYSA-N phenylboronic acid Chemical compound OB(O)C1=CC=CC=C1 HXITXNWTGFUOAU-UHFFFAOYSA-N 0.000 description 2
- 239000006187 pill Substances 0.000 description 2
- 239000013641 positive control Substances 0.000 description 2
- 238000004445 quantitative analysis Methods 0.000 description 2
- 229960003876 ranibizumab Drugs 0.000 description 2
- 230000002829 reductive effect Effects 0.000 description 2
- 210000003786 sclera Anatomy 0.000 description 2
- 238000007789 sealing Methods 0.000 description 2
- 239000000725 suspension Substances 0.000 description 2
- 230000002194 synthesizing effect Effects 0.000 description 2
- 238000002560 therapeutic procedure Methods 0.000 description 2
- VZCYOOQTPOCHFL-UHFFFAOYSA-N trans-butenedioic acid Natural products OC(=O)C=CC(O)=O VZCYOOQTPOCHFL-UHFFFAOYSA-N 0.000 description 2
- 239000002525 vasculotropin inhibitor Substances 0.000 description 2
- MYZAXBZLEILEBR-RVFOSREFSA-N (2S)-1-[(2S,3R)-2-[[(2R)-2-[[2-[[(2S)-2-[(2-aminoacetyl)amino]-5-(diaminomethylideneamino)pentanoyl]amino]acetyl]amino]-3-sulfopropanoyl]amino]-3-hydroxybutanoyl]pyrrolidine-2-carboxylic acid Chemical compound C[C@@H](O)[C@H](NC(=O)[C@H](CS(O)(=O)=O)NC(=O)CNC(=O)[C@H](CCCN=C(N)N)NC(=O)CN)C(=O)N1CCC[C@H]1C(O)=O MYZAXBZLEILEBR-RVFOSREFSA-N 0.000 description 1
- YONLFQNRGZXBBF-ZIAGYGMSSA-N (2r,3r)-2,3-dibenzoyloxybutanedioic acid Chemical class O([C@@H](C(=O)O)[C@@H](OC(=O)C=1C=CC=CC=1)C(O)=O)C(=O)C1=CC=CC=C1 YONLFQNRGZXBBF-ZIAGYGMSSA-N 0.000 description 1
- 0 *c1c(N(CC2)CCC2c2ccc(CCCN3)c3n2)nc(*)nc1NCC(C(O)=O)NC(O*=CCC(O)=O)=O Chemical compound *c1c(N(CC2)CCC2c2ccc(CCCN3)c3n2)nc(*)nc1NCC(C(O)=O)NC(O*=CCC(O)=O)=O 0.000 description 1
- ZCZVGQCBSJLDDS-UHFFFAOYSA-N 1,2,3,4-tetrahydro-1,8-naphthyridine Chemical class C1=CC=C2CCCNC2=N1 ZCZVGQCBSJLDDS-UHFFFAOYSA-N 0.000 description 1
- RVGLEPQPVDUSOJ-UHFFFAOYSA-N 2-Methyl-3-hydroxypropanoate Chemical compound COC(=O)CCO RVGLEPQPVDUSOJ-UHFFFAOYSA-N 0.000 description 1
- DEOATLIOSSKIJL-UHFFFAOYSA-N 2-[1-(6-chloro-2,5-dimethylpyrimidin-4-yl)piperidin-4-yl]-1,8-naphthyridine Chemical compound ClC1=C(C(=NC(=N1)C)N1CCC(CC1)C1=NC2=NC=CC=C2C=C1)C DEOATLIOSSKIJL-UHFFFAOYSA-N 0.000 description 1
- KHMXVEUBFTUDOT-WSTBAARBSA-N 2-aminoacetic acid;(2s)-2-aminobutanedioic acid;(2s)-2-amino-5-(diaminomethylideneamino)pentanoic acid Chemical compound NCC(O)=O.OC(=O)[C@@H](N)CC(O)=O.OC(=O)[C@@H](N)CCCNC(N)=N KHMXVEUBFTUDOT-WSTBAARBSA-N 0.000 description 1
- FEWJPZIEWOKRBE-UHFFFAOYSA-M 3-carboxy-2,3-dihydroxypropanoate Chemical compound OC(=O)C(O)C(O)C([O-])=O FEWJPZIEWOKRBE-UHFFFAOYSA-M 0.000 description 1
- ALKYHXVLJMQRLQ-UHFFFAOYSA-M 3-carboxynaphthalen-2-olate Chemical compound C1=CC=C2C=C(C([O-])=O)C(O)=CC2=C1 ALKYHXVLJMQRLQ-UHFFFAOYSA-M 0.000 description 1
- QCQCHGYLTSGIGX-GHXANHINSA-N 4-[[(3ar,5ar,5br,7ar,9s,11ar,11br,13as)-5a,5b,8,8,11a-pentamethyl-3a-[(5-methylpyridine-3-carbonyl)amino]-2-oxo-1-propan-2-yl-4,5,6,7,7a,9,10,11,11b,12,13,13a-dodecahydro-3h-cyclopenta[a]chrysen-9-yl]oxy]-2,2-dimethyl-4-oxobutanoic acid Chemical compound N([C@@]12CC[C@@]3(C)[C@]4(C)CC[C@H]5C(C)(C)[C@@H](OC(=O)CC(C)(C)C(O)=O)CC[C@]5(C)[C@H]4CC[C@@H]3C1=C(C(C2)=O)C(C)C)C(=O)C1=CN=CC(C)=C1 QCQCHGYLTSGIGX-GHXANHINSA-N 0.000 description 1
- HBAQYPYDRFILMT-UHFFFAOYSA-N 8-[3-(1-cyclopropylpyrazol-4-yl)-1H-pyrazolo[4,3-d]pyrimidin-5-yl]-3-methyl-3,8-diazabicyclo[3.2.1]octan-2-one Chemical class C1(CC1)N1N=CC(=C1)C1=NNC2=C1N=C(N=C2)N1C2C(N(CC1CC2)C)=O HBAQYPYDRFILMT-UHFFFAOYSA-N 0.000 description 1
- 108010005094 Advanced Glycation End Products Proteins 0.000 description 1
- 239000005995 Aluminium silicate Substances 0.000 description 1
- 206010002329 Aneurysm Diseases 0.000 description 1
- 108010039627 Aprotinin Proteins 0.000 description 1
- IYMAXBFPHPZYIK-BQBZGAKWSA-N Arg-Gly-Asp Chemical class NC(N)=NCCC[C@H](N)C(=O)NCC(=O)N[C@@H](CC(O)=O)C(O)=O IYMAXBFPHPZYIK-BQBZGAKWSA-N 0.000 description 1
- BVKZGUZCCUSVTD-UHFFFAOYSA-M Bicarbonate Chemical compound OC([O-])=O BVKZGUZCCUSVTD-UHFFFAOYSA-M 0.000 description 1
- 102000004506 Blood Proteins Human genes 0.000 description 1
- 108010017384 Blood Proteins Proteins 0.000 description 1
- 108091003079 Bovine Serum Albumin Proteins 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-M Bromide Chemical compound [Br-] CPELXLSAUQHCOX-UHFFFAOYSA-M 0.000 description 1
- 238000006443 Buchwald-Hartwig cross coupling reaction Methods 0.000 description 1
- OYPRJOBELJOOCE-UHFFFAOYSA-N Calcium Chemical compound [Ca] OYPRJOBELJOOCE-UHFFFAOYSA-N 0.000 description 1
- 208000002177 Cataract Diseases 0.000 description 1
- 102000000844 Cell Surface Receptors Human genes 0.000 description 1
- 108010001857 Cell Surface Receptors Proteins 0.000 description 1
- VEXZGXHMUGYJMC-UHFFFAOYSA-M Chloride anion Chemical compound [Cl-] VEXZGXHMUGYJMC-UHFFFAOYSA-M 0.000 description 1
- KRKNYBCHXYNGOX-UHFFFAOYSA-K Citrate Chemical compound [O-]C(=O)CC(O)(CC([O-])=O)C([O-])=O KRKNYBCHXYNGOX-UHFFFAOYSA-K 0.000 description 1
- 206010053567 Coagulopathies Diseases 0.000 description 1
- 108010035532 Collagen Proteins 0.000 description 1
- 102000008186 Collagen Human genes 0.000 description 1
- RGHNJXZEOKUKBD-SQOUGZDYSA-M D-gluconate Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)C([O-])=O RGHNJXZEOKUKBD-SQOUGZDYSA-M 0.000 description 1
- FEWJPZIEWOKRBE-JCYAYHJZSA-N Dextrotartaric acid Chemical compound OC(=O)[C@H](O)[C@@H](O)C(O)=O FEWJPZIEWOKRBE-JCYAYHJZSA-N 0.000 description 1
- KCXVZYZYPLLWCC-UHFFFAOYSA-N EDTA Chemical compound OC(=O)CN(CC(O)=O)CCN(CC(O)=O)CC(O)=O KCXVZYZYPLLWCC-UHFFFAOYSA-N 0.000 description 1
- 108010041308 Endothelial Growth Factors Proteins 0.000 description 1
- 208000001351 Epiretinal Membrane Diseases 0.000 description 1
- PIICEJLVQHRZGT-UHFFFAOYSA-N Ethylenediamine Chemical compound NCCN PIICEJLVQHRZGT-UHFFFAOYSA-N 0.000 description 1
- KRHYYFGTRYWZRS-UHFFFAOYSA-M Fluoride anion Chemical compound [F-] KRHYYFGTRYWZRS-UHFFFAOYSA-M 0.000 description 1
- VZCYOOQTPOCHFL-OWOJBTEDSA-N Fumaric acid Chemical compound OC(=O)\C=C\C(O)=O VZCYOOQTPOCHFL-OWOJBTEDSA-N 0.000 description 1
- 208000010412 Glaucoma Diseases 0.000 description 1
- WQZGKKKJIJFFOK-GASJEMHNSA-N Glucose Natural products OC[C@H]1OC(O)[C@H](O)[C@@H](O)[C@@H]1O WQZGKKKJIJFFOK-GASJEMHNSA-N 0.000 description 1
- 239000012981 Hank's balanced salt solution Substances 0.000 description 1
- 241000282412 Homo Species 0.000 description 1
- 101001027128 Homo sapiens Fibronectin Proteins 0.000 description 1
- UFHFLCQGNIYNRP-UHFFFAOYSA-N Hydrogen Chemical compound [H][H] UFHFLCQGNIYNRP-UHFFFAOYSA-N 0.000 description 1
- CPELXLSAUQHCOX-UHFFFAOYSA-N Hydrogen bromide Chemical compound Br CPELXLSAUQHCOX-UHFFFAOYSA-N 0.000 description 1
- DGAQECJNVWCQMB-PUAWFVPOSA-M Ilexoside XXIX Chemical compound C[C@@H]1CC[C@@]2(CC[C@@]3(C(=CC[C@H]4[C@]3(CC[C@@H]5[C@@]4(CC[C@@H](C5(C)C)OS(=O)(=O)[O-])C)C)[C@@H]2[C@]1(C)O)C)C(=O)O[C@H]6[C@@H]([C@H]([C@@H]([C@H](O6)CO)O)O)O.[Na+] DGAQECJNVWCQMB-PUAWFVPOSA-M 0.000 description 1
- 238000012404 In vitro experiment Methods 0.000 description 1
- WHUUTDBJXJRKMK-VKHMYHEASA-N L-glutamic acid Chemical compound OC(=O)[C@@H](N)CCC(O)=O WHUUTDBJXJRKMK-VKHMYHEASA-N 0.000 description 1
- JVTAAEKCZFNVCJ-UHFFFAOYSA-M Lactate Chemical compound CC(O)C([O-])=O JVTAAEKCZFNVCJ-UHFFFAOYSA-M 0.000 description 1
- 108010085895 Laminin Proteins 0.000 description 1
- 102000007547 Laminin Human genes 0.000 description 1
- 206010024404 Leukostasis Diseases 0.000 description 1
- WHXSMMKQMYFTQS-UHFFFAOYSA-N Lithium Chemical compound [Li] WHXSMMKQMYFTQS-UHFFFAOYSA-N 0.000 description 1
- FYYHWMGAXLPEAU-UHFFFAOYSA-N Magnesium Chemical compound [Mg] FYYHWMGAXLPEAU-UHFFFAOYSA-N 0.000 description 1
- 206010063341 Metamorphopsia Diseases 0.000 description 1
- 206010027476 Metastases Diseases 0.000 description 1
- AFVFQIVMOAPDHO-UHFFFAOYSA-N Methanesulfonic acid Chemical compound CS(O)(=O)=O AFVFQIVMOAPDHO-UHFFFAOYSA-N 0.000 description 1
- 208000009857 Microaneurysm Diseases 0.000 description 1
- 208000005647 Mumps Diseases 0.000 description 1
- 241000699670 Mus sp. Species 0.000 description 1
- MBBZMMPHUWSWHV-BDVNFPICSA-N N-methylglucamine Chemical compound CNC[C@H](O)[C@@H](O)[C@H](O)[C@H](O)CO MBBZMMPHUWSWHV-BDVNFPICSA-N 0.000 description 1
- 241000283973 Oryctolagus cuniculus Species 0.000 description 1
- ZLMJMSJWJFRBEC-UHFFFAOYSA-N Potassium Chemical compound [K] ZLMJMSJWJFRBEC-UHFFFAOYSA-N 0.000 description 1
- 208000007135 Retinal Neovascularization Diseases 0.000 description 1
- 208000017442 Retinal disease Diseases 0.000 description 1
- 206010038923 Retinopathy Diseases 0.000 description 1
- 206010039729 Scotoma Diseases 0.000 description 1
- 229940122055 Serine protease inhibitor Drugs 0.000 description 1
- 101710102218 Serine protease inhibitor Proteins 0.000 description 1
- VMHLLURERBWHNL-UHFFFAOYSA-M Sodium acetate Chemical compound [Na+].CC([O-])=O VMHLLURERBWHNL-UHFFFAOYSA-M 0.000 description 1
- 229920002472 Starch Polymers 0.000 description 1
- 101100069223 Streptomyces avermitilis (strain ATCC 31267 / DSM 46492 / JCM 5070 / NBRC 14893 / NCIMB 12804 / NRRL 8165 / MA-4680) gpdQ gene Proteins 0.000 description 1
- QAOWNCQODCNURD-UHFFFAOYSA-L Sulfate Chemical compound [O-]S([O-])(=O)=O QAOWNCQODCNURD-UHFFFAOYSA-L 0.000 description 1
- 229920002253 Tannate Polymers 0.000 description 1
- 241001255741 Vanna Species 0.000 description 1
- 108010031318 Vitronectin Proteins 0.000 description 1
- 102100035140 Vitronectin Human genes 0.000 description 1
- HCHKCACWOHOZIP-UHFFFAOYSA-N Zinc Chemical compound [Zn] HCHKCACWOHOZIP-UHFFFAOYSA-N 0.000 description 1
- 230000002378 acidificating effect Effects 0.000 description 1
- 150000007513 acids Chemical class 0.000 description 1
- 239000013543 active substance Substances 0.000 description 1
- 230000002411 adverse Effects 0.000 description 1
- 208000038018 age-related macular disease Diseases 0.000 description 1
- 230000002776 aggregation Effects 0.000 description 1
- 238000004220 aggregation Methods 0.000 description 1
- 230000032683 aging Effects 0.000 description 1
- 150000001298 alcohols Chemical class 0.000 description 1
- 150000001335 aliphatic alkanes Chemical class 0.000 description 1
- 125000000217 alkyl group Chemical group 0.000 description 1
- IYABWNGZIDDRAK-UHFFFAOYSA-N allene Chemical group C=C=C IYABWNGZIDDRAK-UHFFFAOYSA-N 0.000 description 1
- 229910052782 aluminium Inorganic materials 0.000 description 1
- XAGFODPZIPBFFR-UHFFFAOYSA-N aluminium Chemical compound [Al] XAGFODPZIPBFFR-UHFFFAOYSA-N 0.000 description 1
- 235000012211 aluminium silicate Nutrition 0.000 description 1
- 150000001412 amines Chemical class 0.000 description 1
- 238000004458 analytical method Methods 0.000 description 1
- 230000002491 angiogenic effect Effects 0.000 description 1
- 230000003042 antagnostic effect Effects 0.000 description 1
- 238000011122 anti-angiogenic therapy Methods 0.000 description 1
- 230000003110 anti-inflammatory effect Effects 0.000 description 1
- 230000002137 anti-vascular effect Effects 0.000 description 1
- 229960004405 aprotinin Drugs 0.000 description 1
- 239000006286 aqueous extract Substances 0.000 description 1
- 210000001742 aqueous humor Anatomy 0.000 description 1
- 108010072041 arginyl-glycyl-aspartic acid Proteins 0.000 description 1
- 125000003118 aryl group Chemical group 0.000 description 1
- 125000004429 atom Chemical group 0.000 description 1
- 230000009286 beneficial effect Effects 0.000 description 1
- JUHORIMYRDESRB-UHFFFAOYSA-N benzathine Chemical compound C=1C=CC=CC=1CNCCNCC1=CC=CC=C1 JUHORIMYRDESRB-UHFFFAOYSA-N 0.000 description 1
- 229940077388 benzenesulfonate Drugs 0.000 description 1
- SRSXLGNVWSONIS-UHFFFAOYSA-M benzenesulfonate Chemical compound [O-]S(=O)(=O)C1=CC=CC=C1 SRSXLGNVWSONIS-UHFFFAOYSA-M 0.000 description 1
- WPYMKLBDIGXBTP-UHFFFAOYSA-N benzoic acid Chemical compound OC(=O)C1=CC=CC=C1 WPYMKLBDIGXBTP-UHFFFAOYSA-N 0.000 description 1
- 125000001797 benzyl group Chemical group [H]C1=C([H])C([H])=C(C([H])=C1[H])C([H])([H])* 0.000 description 1
- 239000011230 binding agent Substances 0.000 description 1
- 230000036772 blood pressure Effects 0.000 description 1
- 210000004204 blood vessel Anatomy 0.000 description 1
- 210000004155 blood-retinal barrier Anatomy 0.000 description 1
- 230000004378 blood-retinal barrier Effects 0.000 description 1
- 229940098773 bovine serum albumin Drugs 0.000 description 1
- 239000012267 brine Substances 0.000 description 1
- 210000005252 bulbus oculi Anatomy 0.000 description 1
- IAQRGUVFOMOMEM-UHFFFAOYSA-N butene Natural products CC=CC IAQRGUVFOMOMEM-UHFFFAOYSA-N 0.000 description 1
- DEGAKNSWVGKMLS-UHFFFAOYSA-N calcein Chemical compound O1C(=O)C2=CC=CC=C2C21C1=CC(CN(CC(O)=O)CC(O)=O)=C(O)C=C1OC1=C2C=C(CN(CC(O)=O)CC(=O)O)C(O)=C1 DEGAKNSWVGKMLS-UHFFFAOYSA-N 0.000 description 1
- BQRGNLJZBFXNCZ-UHFFFAOYSA-N calcein am Chemical compound O1C(=O)C2=CC=CC=C2C21C1=CC(CN(CC(=O)OCOC(C)=O)CC(=O)OCOC(C)=O)=C(OC(C)=O)C=C1OC1=C2C=C(CN(CC(=O)OCOC(C)=O)CC(=O)OCOC(=O)C)C(OC(C)=O)=C1 BQRGNLJZBFXNCZ-UHFFFAOYSA-N 0.000 description 1
- 239000011575 calcium Substances 0.000 description 1
- 229910052791 calcium Inorganic materials 0.000 description 1
- 230000005907 cancer growth Effects 0.000 description 1
- 230000015556 catabolic process Effects 0.000 description 1
- 239000003054 catalyst Substances 0.000 description 1
- 230000024245 cell differentiation Effects 0.000 description 1
- 239000006285 cell suspension Substances 0.000 description 1
- VDANGULDQQJODZ-UHFFFAOYSA-N chloroprocaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1Cl VDANGULDQQJODZ-UHFFFAOYSA-N 0.000 description 1
- 229960002023 chloroprocaine Drugs 0.000 description 1
- OEYIOHPDSNJKLS-UHFFFAOYSA-N choline Chemical compound C[N+](C)(C)CCO OEYIOHPDSNJKLS-UHFFFAOYSA-N 0.000 description 1
- 229960001231 choline Drugs 0.000 description 1
- 230000035602 clotting Effects 0.000 description 1
- 229920001436 collagen Polymers 0.000 description 1
- 230000004456 color vision Effects 0.000 description 1
- 230000001447 compensatory effect Effects 0.000 description 1
- 229940125904 compound 1 Drugs 0.000 description 1
- 229940125782 compound 2 Drugs 0.000 description 1
- 229940126214 compound 3 Drugs 0.000 description 1
- 210000000795 conjunctiva Anatomy 0.000 description 1
- 150000004696 coordination complex Chemical group 0.000 description 1
- 210000004087 cornea Anatomy 0.000 description 1
- 101150064194 cpdA gene Proteins 0.000 description 1
- 238000002425 crystallisation Methods 0.000 description 1
- 230000008025 crystallization Effects 0.000 description 1
- 125000000753 cycloalkyl group Chemical group 0.000 description 1
- USVZFSNDGFNNJT-UHFFFAOYSA-N cyclopenta-1,4-dien-1-yl(diphenyl)phosphane (2,3-dichlorocyclopenta-1,4-dien-1-yl)-diphenylphosphane iron(2+) Chemical compound [Fe++].c1cc[c-](c1)P(c1ccccc1)c1ccccc1.Clc1c(cc[c-]1Cl)P(c1ccccc1)c1ccccc1 USVZFSNDGFNNJT-UHFFFAOYSA-N 0.000 description 1
- 230000006378 damage Effects 0.000 description 1
- 230000003247 decreasing effect Effects 0.000 description 1
- 230000007850 degeneration Effects 0.000 description 1
- 230000001419 dependent effect Effects 0.000 description 1
- 238000001514 detection method Methods 0.000 description 1
- ACYGYJFTZSAZKR-UHFFFAOYSA-J dicalcium;2-[2-[bis(carboxylatomethyl)amino]ethyl-(carboxylatomethyl)amino]acetate Chemical compound [Ca+2].[Ca+2].[O-]C(=O)CN(CC([O-])=O)CCN(CC([O-])=O)CC([O-])=O ACYGYJFTZSAZKR-UHFFFAOYSA-J 0.000 description 1
- BSHICDXRSZQYBP-UHFFFAOYSA-N dichloromethane;palladium(2+) Chemical compound [Pd+2].ClCCl BSHICDXRSZQYBP-UHFFFAOYSA-N 0.000 description 1
- ZBCBWPMODOFKDW-UHFFFAOYSA-N diethanolamine Chemical compound OCCNCCO ZBCBWPMODOFKDW-UHFFFAOYSA-N 0.000 description 1
- 229940043237 diethanolamine Drugs 0.000 description 1
- 238000009792 diffusion process Methods 0.000 description 1
- GPAYUJZHTULNBE-UHFFFAOYSA-N diphenylphosphine Chemical compound C=1C=CC=CC=1PC1=CC=CC=C1 GPAYUJZHTULNBE-UHFFFAOYSA-N 0.000 description 1
- 239000001177 diphosphate Substances 0.000 description 1
- XPPKVPWEQAFLFU-UHFFFAOYSA-J diphosphate(4-) Chemical compound [O-]P([O-])(=O)OP([O-])([O-])=O XPPKVPWEQAFLFU-UHFFFAOYSA-J 0.000 description 1
- 235000011180 diphosphates Nutrition 0.000 description 1
- 238000002224 dissection Methods 0.000 description 1
- 238000001647 drug administration Methods 0.000 description 1
- 229940009662 edetate Drugs 0.000 description 1
- 229950008913 edisilate Drugs 0.000 description 1
- 239000000839 emulsion Substances 0.000 description 1
- 230000010595 endothelial cell migration Effects 0.000 description 1
- 230000008753 endothelial function Effects 0.000 description 1
- 229950007655 esilate Drugs 0.000 description 1
- 229950000206 estolate Drugs 0.000 description 1
- LZCLXQDLBQLTDK-UHFFFAOYSA-N ethyl 2-hydroxypropanoate Chemical compound CCOC(=O)C(C)O LZCLXQDLBQLTDK-UHFFFAOYSA-N 0.000 description 1
- 229940012017 ethylenediamine Drugs 0.000 description 1
- 238000011156 evaluation Methods 0.000 description 1
- 230000005284 excitation Effects 0.000 description 1
- 230000004438 eyesight Effects 0.000 description 1
- 230000003480 fibrinolytic effect Effects 0.000 description 1
- 230000003176 fibrotic effect Effects 0.000 description 1
- 230000001497 fibrovascular Effects 0.000 description 1
- 238000001914 filtration Methods 0.000 description 1
- 235000013305 food Nutrition 0.000 description 1
- 238000009472 formulation Methods 0.000 description 1
- 239000012458 free base Substances 0.000 description 1
- 238000004108 freeze drying Methods 0.000 description 1
- 229940050411 fumarate Drugs 0.000 description 1
- 229960001731 gluceptate Drugs 0.000 description 1
- KWMLJOLKUYYJFJ-VFUOTHLCSA-N glucoheptonic acid Chemical compound OC[C@@H](O)[C@@H](O)[C@H](O)[C@@H](O)[C@@H](O)C(O)=O KWMLJOLKUYYJFJ-VFUOTHLCSA-N 0.000 description 1
- 229940050410 gluconate Drugs 0.000 description 1
- 239000008103 glucose Substances 0.000 description 1
- 229930195712 glutamate Natural products 0.000 description 1
- 150000002334 glycols Chemical class 0.000 description 1
- 239000003102 growth factor Substances 0.000 description 1
- 210000003128 head Anatomy 0.000 description 1
- 125000005842 heteroatom Chemical group 0.000 description 1
- XGIHQYAWBCFNPY-AZOCGYLKSA-N hydrabamine Chemical compound C([C@@H]12)CC3=CC(C(C)C)=CC=C3[C@@]2(C)CCC[C@@]1(C)CNCCNC[C@@]1(C)[C@@H]2CCC3=CC(C(C)C)=CC=C3[C@@]2(C)CCC1 XGIHQYAWBCFNPY-AZOCGYLKSA-N 0.000 description 1
- 150000004677 hydrates Chemical class 0.000 description 1
- XMBWDFGMSWQBCA-UHFFFAOYSA-N hydrogen iodide Chemical compound I XMBWDFGMSWQBCA-UHFFFAOYSA-N 0.000 description 1
- 230000002209 hydrophobic effect Effects 0.000 description 1
- 238000011065 in-situ storage Methods 0.000 description 1
- 230000006698 induction Effects 0.000 description 1
- 239000003112 inhibitor Substances 0.000 description 1
- ZPNFWUPYTFPOJU-LPYSRVMUSA-N iniprol Chemical compound C([C@H]1C(=O)NCC(=O)NCC(=O)N[C@H]2CSSC[C@H]3C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@H](C(N[C@H](C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H](CC=4C=CC(O)=CC=4)C(=O)N[C@@H](CC=4C=CC=CC=4)C(=O)N[C@@H](CC=4C=CC(O)=CC=4)C(=O)N[C@@H](CC(N)=O)C(=O)N[C@@H](C)C(=O)N[C@@H](CCCCN)C(=O)N[C@@H](C)C(=O)NCC(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CSSC[C@H](NC(=O)[C@H](CC(O)=O)NC(=O)[C@H](CCC(O)=O)NC(=O)[C@H](C)NC(=O)[C@H](CO)NC(=O)[C@H](CCCCN)NC(=O)[C@H](CC=4C=CC=CC=4)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CC(N)=O)NC(=O)[C@H](CCCNC(N)=N)NC(=O)[C@H](CCCCN)NC(=O)[C@H](C)NC(=O)[C@H](CCCNC(N)=N)NC2=O)C(=O)N[C@@H](CCSC)C(=O)N[C@@H](CCCNC(N)=N)C(=O)N[C@@H]([C@@H](C)O)C(=O)N[C@@H](CSSC[C@H](NC(=O)[C@H](CC=2C=CC=CC=2)NC(=O)[C@H](CC(O)=O)NC(=O)[C@H]2N(CCC2)C(=O)[C@@H](N)CCCNC(N)=N)C(=O)N[C@@H](CC(C)C)C(=O)N[C@@H](CCC(O)=O)C(=O)N2[C@@H](CCC2)C(=O)N2[C@@H](CCC2)C(=O)N[C@@H](CC=2C=CC(O)=CC=2)C(=O)N[C@@H]([C@@H](C)O)C(=O)NCC(=O)N2[C@@H](CCC2)C(=O)N3)C(=O)NCC(=O)NCC(=O)N[C@@H](C)C(O)=O)C(=O)N[C@@H](CCC(N)=O)C(=O)N[C@H](C(=O)N[C@@H](CC=2C=CC=CC=2)C(=O)N[C@H](C(=O)N1)C(C)C)[C@@H](C)O)[C@@H](C)CC)=O)[C@@H](C)CC)C1=CC=C(O)C=C1 ZPNFWUPYTFPOJU-LPYSRVMUSA-N 0.000 description 1
- 150000007529 inorganic bases Chemical class 0.000 description 1
- 230000008611 intercellular interaction Effects 0.000 description 1
- 230000009545 invasion Effects 0.000 description 1
- 239000003456 ion exchange resin Substances 0.000 description 1
- 229920003303 ion-exchange polymer Polymers 0.000 description 1
- SUMDYPCJJOFFON-UHFFFAOYSA-N isethionic acid Chemical compound OCCS(O)(=O)=O SUMDYPCJJOFFON-UHFFFAOYSA-N 0.000 description 1
- VQTUBCCKSQIDNK-UHFFFAOYSA-N iso-butene Natural products CC(C)=C VQTUBCCKSQIDNK-UHFFFAOYSA-N 0.000 description 1
- NLYAJNPCOHFWQQ-UHFFFAOYSA-N kaolin Chemical compound O.O.O=[Al]O[Si](=O)O[Si](=O)O[Al]=O NLYAJNPCOHFWQQ-UHFFFAOYSA-N 0.000 description 1
- 229940001447 lactate Drugs 0.000 description 1
- 229940099584 lactobionate Drugs 0.000 description 1
- JYTUSYBCFIZPBE-AMTLMPIISA-N lactobionic acid Chemical compound OC(=O)[C@H](O)[C@@H](O)[C@@H]([C@H](O)CO)O[C@@H]1O[C@H](CO)[C@H](O)[C@H](O)[C@H]1O JYTUSYBCFIZPBE-AMTLMPIISA-N 0.000 description 1
- 238000013532 laser treatment Methods 0.000 description 1
- 239000007788 liquid Substances 0.000 description 1
- 229910052744 lithium Inorganic materials 0.000 description 1
- 208000018769 loss of vision Diseases 0.000 description 1
- 231100000864 loss of vision Toxicity 0.000 description 1
- 239000000314 lubricant Substances 0.000 description 1
- 239000011777 magnesium Substances 0.000 description 1
- 229910052749 magnesium Inorganic materials 0.000 description 1
- 229940049920 malate Drugs 0.000 description 1
- 238000013227 male C57BL/6J mice Methods 0.000 description 1
- VZCYOOQTPOCHFL-UPHRSURJSA-N maleic acid Chemical compound OC(=O)\C=C/C(O)=O VZCYOOQTPOCHFL-UPHRSURJSA-N 0.000 description 1
- BJEPYKJPYRNKOW-UHFFFAOYSA-N malic acid Chemical compound OC(=O)C(O)CC(O)=O BJEPYKJPYRNKOW-UHFFFAOYSA-N 0.000 description 1
- IWYDHOAUDWTVEP-UHFFFAOYSA-M mandelate Chemical compound [O-]C(=O)C(O)C1=CC=CC=C1 IWYDHOAUDWTVEP-UHFFFAOYSA-M 0.000 description 1
- 238000004519 manufacturing process Methods 0.000 description 1
- 239000000463 material Substances 0.000 description 1
- 230000001404 mediated effect Effects 0.000 description 1
- 229960003194 meglumine Drugs 0.000 description 1
- 239000012528 membrane Substances 0.000 description 1
- 230000009401 metastasis Effects 0.000 description 1
- 229940102396 methyl bromide Drugs 0.000 description 1
- LRMHVVPPGGOAJQ-UHFFFAOYSA-N methyl nitrate Chemical compound CO[N+]([O-])=O LRMHVVPPGGOAJQ-UHFFFAOYSA-N 0.000 description 1
- JZMJDSHXVKJFKW-UHFFFAOYSA-M methyl sulfate(1-) Chemical compound COS([O-])(=O)=O JZMJDSHXVKJFKW-UHFFFAOYSA-M 0.000 description 1
- 230000004899 motility Effects 0.000 description 1
- 238000010172 mouse model Methods 0.000 description 1
- 208000010805 mumps infectious disease Diseases 0.000 description 1
- 210000003205 muscle Anatomy 0.000 description 1
- QIQXTHQIDYTFRH-UHFFFAOYSA-N octadecanoic acid Chemical compound CCCCCCCCCCCCCCCCCC(O)=O QIQXTHQIDYTFRH-UHFFFAOYSA-N 0.000 description 1
- 229960002378 oftasceine Drugs 0.000 description 1
- 239000003921 oil Substances 0.000 description 1
- 210000001328 optic nerve Anatomy 0.000 description 1
- 238000005457 optimization Methods 0.000 description 1
- 239000006186 oral dosage form Substances 0.000 description 1
- 150000007530 organic bases Chemical class 0.000 description 1
- 150000002892 organic cations Chemical class 0.000 description 1
- 229940037201 oris Drugs 0.000 description 1
- 238000006213 oxygenation reaction Methods 0.000 description 1
- 238000004806 packaging method and process Methods 0.000 description 1
- 229910052763 palladium Inorganic materials 0.000 description 1
- 230000036961 partial effect Effects 0.000 description 1
- 230000008506 pathogenesis Effects 0.000 description 1
- 235000019371 penicillin G benzathine Nutrition 0.000 description 1
- 229960001412 pentobarbital Drugs 0.000 description 1
- 230000002085 persistent effect Effects 0.000 description 1
- 239000002831 pharmacologic agent Substances 0.000 description 1
- 230000000649 photocoagulation Effects 0.000 description 1
- 125000000587 piperidin-1-yl group Chemical group [H]C1([H])N(*)C([H])([H])C([H])([H])C([H])([H])C1([H])[H] 0.000 description 1
- 230000010118 platelet activation Effects 0.000 description 1
- 239000011148 porous material Substances 0.000 description 1
- 229910052700 potassium Inorganic materials 0.000 description 1
- 239000011591 potassium Substances 0.000 description 1
- 230000003389 potentiating effect Effects 0.000 description 1
- 229940071643 prefilled syringe Drugs 0.000 description 1
- MFDFERRIHVXMIY-UHFFFAOYSA-N procaine Chemical compound CCN(CC)CCOC(=O)C1=CC=C(N)C=C1 MFDFERRIHVXMIY-UHFFFAOYSA-N 0.000 description 1
- 229960004919 procaine Drugs 0.000 description 1
- 230000008569 process Effects 0.000 description 1
- 230000002062 proliferating effect Effects 0.000 description 1
- 230000035755 proliferation Effects 0.000 description 1
- 230000002035 prolonged effect Effects 0.000 description 1
- 230000001681 protective effect Effects 0.000 description 1
- 238000000746 purification Methods 0.000 description 1
- 239000012264 purified product Substances 0.000 description 1
- JUJWROOIHBZHMG-UHFFFAOYSA-N pyridine Substances C1=CC=NC=C1 JUJWROOIHBZHMG-UHFFFAOYSA-N 0.000 description 1
- UMJSCPRVCHMLSP-UHFFFAOYSA-N pyridine Natural products COC1=CC=CN=C1 UMJSCPRVCHMLSP-UHFFFAOYSA-N 0.000 description 1
- 150000003222 pyridines Chemical class 0.000 description 1
- 238000011002 quantification Methods 0.000 description 1
- 239000003642 reactive oxygen metabolite Substances 0.000 description 1
- 239000002464 receptor antagonist Substances 0.000 description 1
- 229940044551 receptor antagonist Drugs 0.000 description 1
- 230000009467 reduction Effects 0.000 description 1
- 230000033764 rhythmic process Effects 0.000 description 1
- 108700002400 risuteganib Proteins 0.000 description 1
- YGSDEFSMJLZEOE-UHFFFAOYSA-M salicylate Chemical compound OC1=CC=CC=C1C([O-])=O YGSDEFSMJLZEOE-UHFFFAOYSA-M 0.000 description 1
- 229960001860 salicylate Drugs 0.000 description 1
- 238000009094 second-line therapy Methods 0.000 description 1
- 238000000926 separation method Methods 0.000 description 1
- 239000003001 serine protease inhibitor Substances 0.000 description 1
- 210000002966 serum Anatomy 0.000 description 1
- 150000003384 small molecules Chemical class 0.000 description 1
- 229910052708 sodium Inorganic materials 0.000 description 1
- 239000011734 sodium Substances 0.000 description 1
- 239000001632 sodium acetate Substances 0.000 description 1
- 235000017281 sodium acetate Nutrition 0.000 description 1
- 239000007787 solid Substances 0.000 description 1
- 238000010186 staining Methods 0.000 description 1
- 235000019698 starch Nutrition 0.000 description 1
- KDYFGRWQOYBRFD-UHFFFAOYSA-L succinate(2-) Chemical compound [O-]C(=O)CCC([O-])=O KDYFGRWQOYBRFD-UHFFFAOYSA-L 0.000 description 1
- 235000000346 sugar Nutrition 0.000 description 1
- 150000008163 sugars Chemical class 0.000 description 1
- 239000000829 suppository Substances 0.000 description 1
- 230000004083 survival effect Effects 0.000 description 1
- 208000032598 susceptibility microvascular complications of diabetes Diseases 0.000 description 1
- 239000006188 syrup Substances 0.000 description 1
- 235000020357 syrup Nutrition 0.000 description 1
- 229940095064 tartrate Drugs 0.000 description 1
- 150000003892 tartrate salts Chemical class 0.000 description 1
- 210000001760 tenon capsule Anatomy 0.000 description 1
- WINGEFIITRDOLJ-UHFFFAOYSA-N tert-butyl 2-hydroxyacetate Chemical compound CC(C)(C)OC(=O)CO WINGEFIITRDOLJ-UHFFFAOYSA-N 0.000 description 1
- JAELLLITIZHOGQ-UHFFFAOYSA-N tert-butyl propanoate Chemical compound CCC(=O)OC(C)(C)C JAELLLITIZHOGQ-UHFFFAOYSA-N 0.000 description 1
- 238000010257 thawing Methods 0.000 description 1
- 229940124597 therapeutic agent Drugs 0.000 description 1
- 238000011200 topical administration Methods 0.000 description 1
- 230000000699 topical effect Effects 0.000 description 1
- 238000012546 transfer Methods 0.000 description 1
- 210000003606 umbilical vein Anatomy 0.000 description 1
- 210000003556 vascular endothelial cell Anatomy 0.000 description 1
- 230000000304 vasodilatating effect Effects 0.000 description 1
- 239000003981 vehicle Substances 0.000 description 1
- 230000004304 visual acuity Effects 0.000 description 1
- 210000004127 vitreous body Anatomy 0.000 description 1
- 235000012431 wafers Nutrition 0.000 description 1
- 230000029663 wound healing Effects 0.000 description 1
- 229910052725 zinc Inorganic materials 0.000 description 1
- 239000011701 zinc Substances 0.000 description 1
Classifications
-
- C—CHEMISTRY; METALLURGY
- C07—ORGANIC CHEMISTRY
- C07D—HETEROCYCLIC COMPOUNDS
- C07D471/00—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00
- C07D471/02—Heterocyclic compounds containing nitrogen atoms as the only ring hetero atoms in the condensed system, at least one ring being a six-membered ring with one nitrogen atom, not provided for by groups C07D451/00 - C07D463/00 in which the condensed system contains two hetero rings
- C07D471/04—Ortho-condensed systems
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K31/00—Medicinal preparations containing organic active ingredients
- A61K31/33—Heterocyclic compounds
- A61K31/395—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins
- A61K31/495—Heterocyclic compounds having nitrogen as a ring hetero atom, e.g. guanethidine or rifamycins having six-membered rings with two or more nitrogen atoms as the only ring heteroatoms, e.g. piperazine or tetrazines
- A61K31/505—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim
- A61K31/506—Pyrimidines; Hydrogenated pyrimidines, e.g. trimethoprim not condensed and containing further heterocyclic rings
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K45/00—Medicinal preparations containing active ingredients not provided for in groups A61K31/00 - A61K41/00
- A61K45/06—Mixtures of active ingredients without chemical characterisation, e.g. antiphlogistics and cardiaca
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0019—Injectable compositions; Intramuscular, intravenous, arterial, subcutaneous administration; Compositions to be administered through the skin in an invasive manner
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
- A61K9/0012—Galenical forms characterised by the site of application
- A61K9/0048—Eye, e.g. artificial tears
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P27/00—Drugs for disorders of the senses
- A61P27/02—Ophthalmic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61P—SPECIFIC THERAPEUTIC ACTIVITY OF CHEMICAL COMPOUNDS OR MEDICINAL PREPARATIONS
- A61P35/00—Antineoplastic agents
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K2300/00—Mixtures or combinations of active ingredients, wherein at least one active ingredient is fully defined in groups A61K31/00 - A61K41/00
-
- A—HUMAN NECESSITIES
- A61—MEDICAL OR VETERINARY SCIENCE; HYGIENE
- A61K—PREPARATIONS FOR MEDICAL, DENTAL OR TOILETRY PURPOSES
- A61K9/00—Medicinal preparations characterised by special physical form
Landscapes
- Health & Medical Sciences (AREA)
- Chemical & Material Sciences (AREA)
- Organic Chemistry (AREA)
- Veterinary Medicine (AREA)
- Pharmacology & Pharmacy (AREA)
- Public Health (AREA)
- General Health & Medical Sciences (AREA)
- Medicinal Chemistry (AREA)
- Animal Behavior & Ethology (AREA)
- Life Sciences & Earth Sciences (AREA)
- Epidemiology (AREA)
- Nuclear Medicine, Radiotherapy & Molecular Imaging (AREA)
- General Chemical & Material Sciences (AREA)
- Chemical Kinetics & Catalysis (AREA)
- Ophthalmology & Optometry (AREA)
- Engineering & Computer Science (AREA)
- Bioinformatics & Cheminformatics (AREA)
- Dermatology (AREA)
- Pharmaceuticals Containing Other Organic And Inorganic Compounds (AREA)
- Medicines That Contain Protein Lipid Enzymes And Other Medicines (AREA)
- Nitrogen Condensed Heterocyclic Rings (AREA)
Abstract
Un compuesto según la fórmula (I), o una sal farmacéuticamente aceptable, solvato farmacéuticamente aceptable, isómero o una mezcla de los mismos, **(Ver fórmula)** en donde R1 y R2 se seleccionan independientemente del grupo que consiste en hidrógeno, metilo y etilo; y Alk es un alquileno C1-4.
Description
DESCRIPCIÓN
Antagonistas de integrina
Campo de la invención
Esta presente invención se refiere a nuevos antagonistas de integrina y el uso de estos compuestos como medicamento, en particular para inhibir la neovascularización.
Antecedentes de la invención
El edema macular diabético (DME) es una acumulación de líquido en la mácula de pacientes con retinopatía diabética (DR), que puede ocurrir en cualquier etapa de la enfermedad. La DR es una de las complicaciones microvasculares más comunes de la diabetes. La DR que amenaza la visión (es decir, DME y la retinopatía diabética proliferativa (PDR) son la principal causa de la discapacidad visual y ceguera entre los adultos en edad laboral en todo el mundo. A nivel mundial, la prevalencia general de la DR se estima en el 34,6% de las personas con diabetes, mientras que la prevalencia general del DME se estima en aproximadamente el 20% de las personas con DR (Yau et al., 2012). Se espera que la prevalencia del DME aumente aún más debido al aumento de la prevalencia de la diabetes, el envejecimiento de la población y el aumento de la esperanza de vida de las personas con diabetes: el número de adultos con diabetes en todo el mundo se estimó en 425 millones en 2017 y se espera que aumente a 629 millones en 2045.
Aunque los mecanismos exactos por los cuales la diabetes causa retinopatía aún no están claros, varios estudios han demostrado la elevación de especies reactivas de oxígeno, productos finales de glicación avanzada y citoquinas y quimioquinas circulantes y vítreas en relación con la enfermedad. La inflamación en la retina es una importante característica patológica temprana de la DR. Los factores inflamatorios y vasodilatadores pueden modificar la función endotelial, lo que lleva a la ruptura de la barrera hemato-retiniana, lo que da como resultado la acumulación de proteínas y lípidos plasmáticos en la mácula. Cuando el engrosamiento afecta a la fóvea o amenaza con afectar a la fóvea, el paciente se vuelve sintomático con metamorfopsia y pérdida de la visión.
Se ha descubierto que la interacción entre el vítreo y la retina está implicada en el desarrollo del edema macular. En particular, cuando el vítreo y el área macular de la retina están estrechamente conjugados, el edema macular se puede promover en gran medida. Entre los pacientes con DR, el DME estuvo presente solo en el 20% de los pacientes con desprendimiento del vítreo posterior (PVD) en comparación con el 55% de los pacientes sin PVD, lo que sugiere un fuerte efecto protector del PVD. El mecanismo propuesto para el posible beneficio del PVD es el alivio de la tracción vitreomacular, sin embargo, también se ha sugerido que tanto la oxigenación transvítreal como la difusión mejorada del factor de crecimiento (lejos de la hialoide premacular) tiene efectos potencialmente beneficiosos.
Un buen control de la glucosa en sangre, la presión arterial y los lípidos en sangre es esencial y puede retrasar la aparición de la DR y ralentizar su progresión. En algunos casos, el tratamiento se realiza mediante fotocoagulación con láser focal/en rejilla utilizando pequeñas quemaduras de láser de intensidad de luz (50-100 pm) en microaneurismas o áreas difusas de engrosamiento. Sin embargo, este tratamiento podría dar como resultado complicaciones tales como la pérdida de visión central, escotomas centrales y disminución de la visión del color. Más recientemente, el láser de micropulso subumbral se ha desarrollado como un tratamiento que teóricamente evita dañar la retina neurosensorial interna, reduciendo así las posibles complicaciones.
Varios agentes farmacológicos se encuentran actualmente disponibles para el tratamiento del DME, incluyendo los agentes anti-factor de crecimiento del endotelio vascular (VEGF) y los corticosteroides. Los VEGF son un potente factor de vasopermeabilidad que contribuyen al engrosamiento macular y al deterioro visual asociado con el DME. Los compuestos anti-VEGF disminuyen la angiogénesis y la permeabilidad vascular, provocando la regresión de la neovascularización y la reducción del edema. Varios estudios clínicos han demostrado que el tratamiento con anti-VEGF es más eficaz que el tratamiento con láser focal/en rejilla para disminuir el grosor del subcampo central (CST) y mejorar la visión en pacientes con DME. Los sucesos adversos (AEs) relacionados con el tratamiento anti-VEGF son raros y se relacionan principalmente con la necesidad de inyecciones intravítreas repetidas (IVT) durante un periodo de tiempo prolongado.
La inflamación juega un importante papel en la patogénesis del DME. Las citoquinas y quimioquinas liberadas por los leucocitos en la sangre aumentan significativamente la permeabilidad vascular, lo que conduce a una mayor acumulación de líquido debajo de la retina. Las terapias con corticosteroides pueden inhibir los mediadores inflamatorios. Varios estudios clínicos han demostrado que los corticosteroides son efectivos para disminuir la CST y mejorar la visión en el DME. Si bien la carga de tratamiento de los implantes de corticosteroides es mucho menor que la de los agentes anti-VEGF, los corticosteroides intraoculares se asocian con mayores riesgos de desarrollo de cataratas y aumento de la presión intraocular. En general, el uso de corticosteroides IVT en pacientes con DME se reserva, por lo tanto, como terapia de segunda línea en aquellos que responden mal a la terapia con anti-VEGF IVT y está contraindicado en pacientes con glaucoma subyacente.
De los posibles tratamientos del DME mencionados anteriormente, los agentes anti-VEGF son actualmente el tratamiento estándar de primera línea para el DME. Sin embargo, la mayoría de los estudios hasta la fecha informan que una proporción sustancial de pacientes con DME, hasta un 40%, tienen edema persistente y pérdida de agudeza visual a pesar del tratamiento anti-VEGF. Estos hallazgos sugieren que otras vías, independientes del VEGF, contribuyen al desarrollo del DME y, por lo tanto, existe una necesidad clínica importante de tratamientos adicionales eficaces para el DME.
Las integrinas constituyen una familia de receptores transmembrana de la superficie celular que pueden mediar en las interacciones célula-célula y célula-matriz extracelular. Las integrinas están implicadas en varios procesos biológicos incluyendo la diferenciación celular, adhesión, forma, migración, motilidad, invasión, proliferación y supervivencia. Debido a su papel en estos procesos biológicos, las integrinas también se han asociado con diversas afecciones patológicas, tales como el cáncer y los trastornos oftálmicos. En el ojo, se ha demostrado que las integrinas juegan un papel importante en la neovascularización, la permeabilidad vascular y la adhesión vitreorretiniana.
Las integrinas son receptores heterodiméricos obligados que consisten en una subunidad a y p unidas de forma no covalente. Diferentes combinaciones de las subunidades 18 a y las 8 p conocidas constituyen la familia de 24 miembros heterodiméricos de integrinas reconocidos hasta ahora. La familia de receptores de la integrina se puede clasificar, en general, en 4 categorías diferentes dependiendo de su patrón de reconocimiento de ligando: 1) unión del tripéptido L-arginina-glicina-ácido aspártico (RGD), 2) unión de colágeno, 3) unión de laminina y 4) tipos de integrinas que se unen a leucocitos.
La interacción de integrinas con la matriz extracelular puede conducir a la neovascularización de la superficie retiniana, que eventualmente puede extenderse hacia la región vítrea. La tinción inmunohistológica en tejidos retinianos humanos derivados de pacientes con PDR ha demostrado que las células endoteliales vasculares que proliferan de manera activa expresan las integrinas avp3 y avp5, que no se expresan en gran medida en las células endoteliales inactivas (Friedlander et al., 1996; Ning et al., 2008). Además, se ha demostrado que avp3 y anbp3 se expresan en las membranas epirretinianas fibrovasculares de pacientes con PDR activa en la etapa fibrótica (Ning et al., 2008; Abu El-Asrar, Missotten y Geboes, 2010), mientras que a5p1 se ha demostrado que se sobreexpresa en un modelo de ratón de CNV inducido por láser (Umeda et al., 2006). En línea con esto, varios estudios no clínicos han demostrado que la inhibición de las integrinas atenúa la leucostasis y la permeabilidad vascular retiniana (Santulli et al., 2008; Iliaki et al., 2009; Rao et al., 2010). El antagonismo de avp3 y avp5 previno la neovascularización retiniana pero no dañó los vasos sanguíneos preexistentes (Friedlander et al., 1996; Hammes et al., 1996; Lahdenranta et al., 2007; Santulli et al., 2008), mientras que la inhibición de a5p1 inhibió la proliferación de células endoteliales y produjo la regresión de las membranas neovasculares coroideas en diferentes modelos animales (Ramakrishnan et al., 2006; Umeda et al., 2006).
El motivo RGD se encuentra muy comúnmente en muchos componentes de la matriz extracelular, incluyendo vitronectina, fibronectina y fibrinógeno. Por lo tanto, las integrinas están fuertemente ligadas a las proteínas de la matriz extracelular, por lo que median la adhesión célula-matriz extracelular, por ejemplo, en la interfaz vitreorretiniana. Se sabe que los análogos del motivo RGD compiten por el motivo RGD de las proteínas de la matriz extracelular para interrumpir las interacciones integrina-matiz extracelular y, por lo tanto, aflojar las uniones en experimentos in vitro (Gehlsen et al., 1988; Pierschbacher y Ruoslahti, 1987; Zhou, Zhang y Yue, 1996). En línea con esto, se ha demostrado que la inyección IVT de péptidos RGD solubles induce PVD en ojos de conejo (Oliveira et al., 2002). Además, en humanos, se ha demostrado que 3 inyecciones IVT del antagonista de la integrina ALG 1001 (Allegro Ophthalmics, LLC) inducen PVD total en 6 de 11 pacientes con DME con PVD parcial o nula al inicio del estudio en un estudio inicial de prueba de concepto (Kuppermann, 2013; Boyer et al., 2014).
Estas observaciones sustentan la preferencia para utilizar un antagonista de pan-integrina que se dirija a los diferentes tipos de integrinas que subyacen a diferentes aspectos de la enfermedad a tratar, más notablemente avp3, avp5 y a5p1. La complejidad es que dichos antagonistas de pan-integrina a menudo también antagonizarán otras integrinas que se unen a RGD, que pueden causar efectos secundarios. La más notable es la integrina plaquetaria, anbp3, cuyo antagonismo puede interferir con la activación y la agregación plaquetaria.
Las publicaciones de solicitud de patente WO2011/119282 A1, WO2011/094285 A1, US2006/0052398 A1 y US2008/058348 A1 describen compuestos como posibles antagonistas de integrina o como posibles antagonistas del receptor de vitronectina. Los compuestos descritos en estas solicitudes de Patente se pueden resumir como correspondientes generalmente a la Fórmula A

en donde, generalmente Gi representa una piridina o tetrahidro naftiridina sustituida, Ri y R2 son hidrógeno, metilo o etilo, y R3 es un grupo terminal hidrófobo. En particular, en los compuestos ejemplificados con actividad confirmada, R3 comprende un alquilo, cicloalquilo o grupo terminal (hetero)aromático.
Existe la necesidad de nuevos antagonistas de integrinas, en particular antagonistas de integrinas que actúen simultáneamente sobre los diferentes tipos de integrinas que están implicadas en las vías de la enfermedad, tal como avP3, avP5 y a501, pero que tienen un riesgo reducido de interferir con la agregación plaquetaria a través de su efecto sobre anbP3.
Compendio de la invención
La presente invención proporciona compuestos según la fórmula (I) o la sal farmacéuticamente aceptable, el solvato farmacéuticamente aceptable, isómero o mezcla de los mismos.

en donde
R1 y R2 se seleccionan independientemente del grupo que consiste en hidrógeno, metilo o etilo; y Alk es un alquileno C1-4.
La presente invención se refiere además a una composición farmacéutica que comprende el compuesto de la presente invención, y uno o más vehículos farmacéuticamente aceptables.
La presente invención se refiere además a compuestos para su uso como un medicamento. Un objetivo adicional de la presente invención es proporcionar compuestos para inhibir la neovascularización patológica.
La presente invención se refiere además a compuestos para su uso en el tratamiento y/o prevención de trastornos oftálmicos, tales como el edema macular diabético y la retinopatía diabética.
Breve descripción de los dibujos
La Figura 1 muestra las proporciones de EC50 para cpd1, cpd2, cpd3 y los compuestos comparativos cpdA y cpdB para la integrina avP3 sobre anbP3 (los valores más bajos indican una mayor especificidad para avP3).
La Figura 2 muestra la inhibición de los vasos que aparecen por cpd3 en un ensayo de explante coroideo murino. Descripción detallada de la invención
Se entiende además que todas las definiciones y preferencias descritas para los compuestos de la invención anterior se aplican igualmente a esta realización y todas las realizaciones adicionales, como se describe a continuación.
Como se utiliza en lo anterior y en lo sucesivo, las siguientes definiciones se aplican a menos que se indique lo contrario.
El término “alquileno”, solo o en combinación significa un diradical derivado de alcano, que puede ser un alquileno de cadena lineal o un alquileno ramificado, que contiene de 1 a 4 átomos de carbono. El grupo alquileno de cadena lineal o ramificada se une en cualquier punto disponible para producir un compuesto estable. Según ciertas realizaciones, el alquileno Ca-b define un diradical de alquileno lineal o ramificado que tiene de A a B átomos de carbono, por ejemplo, un alquileno C1-4 define un dirradical de alquileno lineal o ramificado que tiene de 1 a 4 átomos de carbono, tal como, por ejemplo, metileno, etileno, 1-propileno, 2-propileno, I-butileno, 2-butileno, 2-metil-1-propileno.
Cuando se hace referencia a porcentajes, esto se refiere a porcentajes de peso a peso, a menos que el contexto indique claramente lo contrario.
Como se describió anteriormente en la presente memoria, la presente invención proporciona compuestos según la fórmula (I) o una sal farmacéuticamente aceptable, un solvato farmacéuticamente aceptable, un isómero o una mezcla de los mismos,

en donde
R1 y R2 se seleccionan independientemente del grupo que consiste en hidrógeno, metilo o etilo; y Alk es alquileno C1-4.
Según una cierta realización de la presente invención, tanto R1 como R2 son metilo, o R1 es hidrógeno y R2 es etilo. Según una realización preferida, la presente invención proporciona aquellos compuestos en los que R1 y R2 son ambos metilos.
Según una determinada realización de la presente invención, Alk es alquileno C1-2.
Según una realización preferida de la presente invención, el compuesto se selecciona del grupo que consiste en

o una sal farmacéuticamente aceptable, un solvato farmacéuticamente aceptable, un isómero o una mezcla de los mismos.
El compuesto de la presente invención, como se detalló anteriormente, puede tener un centro de quiralidad y existir como formas estereoquímicamente isoméricas. El término “formas estereoquímicamente isoméricas” como se utiliza en la presente memoria define todos los posibles compuestos formados por los mismos átomos unidos mediante la misma secuencia de enlaces pero que tienen diferentes estructuras tridimensionales que no son intercambiables, que el compuesto antiinflamatorio como se especifica en la presente memoria, puede poseer.
A menos que se mencione o se indique lo contrario, la designación química de los compuestos, como se detalló anteriormente, abarca la mezcla de todas las formas estereoquímicamente isoméricas posibles, que dichos compuestos pueden poseer. Dicha mezcla puede contener todos los diastereómeros y/o enantiómeros de la estructura molecular básica de dichos compuestos para su uso. Se pretende que todas las formas estereoquímicamente isoméricas de los compuestos de la presente invención, tanto en forma pura como mezcladas entre sí, se incluyan dentro del alcance de la presente invención.
En una realización preferida, la presente invención proporciona las formas enantioméricas (S) de los compuestos según la Fórmula I. Por lo tanto, en una realización particular, la presente invención proporciona un compuesto de acuerdo con la Fórmula II,

en donde R1, R2 y Alk son como se definen en la solicitud.
En una realización adicional, la presente invención proporciona un compuesto seleccionado del grupo que consiste en

o una sal farmacéuticamente aceptable, un solvato farmacéuticamente aceptable, un isómero o una mezcla de los mismos.
Para uso terapéutico, las sales de los compuestos para uso de la presente invención, como se detalla anteriormente, son aquellas en las que el contraión es farmacéuticamente aceptable, cuyas sales pueden denominarse sales de adición de ácido y base farmacéuticamente aceptables. Sin embargo, las sales de ácidos y bases que no son farmacéuticamente aceptables también pueden encontrar uso, por ejemplo, en la preparación o purificación de un compuesto farmacéuticamente aceptable. Todas las sales, farmacéuticamente aceptables o no, se incluyen dentro ámbito de la presente invención.
Las sales de adición de ácido y base farmacéuticamente aceptables como se mencionan anteriormente en la presente memoria pretenden comprender las formas de sal de adición de ácido y base no tóxicas terapéuticamente activas que los compuestos para uso de la presente invención, como se detalla anteriormente, pueden formar. Las sales de adición de ácido farmacéuticamente aceptables se pueden obtener convenientemente tratando la forma básica con dicho ácido apropiado en una forma de anión. Los aniones apropiados comprenden, por ejemplo, trifluoroacetato, acetato, bencenosulfonato, benzoato, bicarbonato, bitartrato, bromuro, edetato de calcio, camsiato, carbonato, cloruro, citrato, diclorhidrato, edetato, edisilato, estolato, esilato, fumarato, gluceptato, gluconato, glutamato, glicolilarsanilato, hexilresorcinato, hidrabamina, bromhidrato, clorhidrato, hidroxinaftoato, ioduro, isetionato, lactato, lactobionato, malato, maleato, mandelato, mesilato, metilbromuro, metilnitrato, metilsulfato, mucato, napsilato, nitrato, pamoato (embonato), pantotenato, fosfato/difosfato, poligalacturonato, salicilato, estearato, subacetato, succinato, sulfato, tanato, tartrato, teoclato, trietioduro, y similares. El contraión de elección se puede introducir utilizando resinas de intercambio iónico. Por el contrario, dichas formas de sal se pueden convertir
mediante tratamiento con una base apropiada en la forma de base libre. Una sal preferida es la sal de clorhidrato de los compuestos descritos en la presente memoria.
Los compuestos para uso como se especifica en la presente memoria, que contienen un protón ácido también pueden convertirse en sus formas de sal de adición de amina o metal no tóxicas mediante el tratamiento con bases orgánicas e inorgánicas apropiadas en una forma de catión. Las sales básicas apropiadas comprenden aquellas formadas con cationes orgánicos tales como benzatina, cloroprocaina, colina, dietanolamina, etilendiamina, meglumina, procaina, y similares; y aquellas formadas con cationes metálicos tales como aluminio, calcio, litio, magnesio, potasio, sodio, zinc, y similares. Por el contrario, dichas formas de sal pueden convertirse por tratamiento con un ácido apropiado en la forma libre.
El término sal de adición, como se utiliza anteriormente en la presente memoria, también comprende los solvatos que pueden formar los compuestos para su uso, como se especifica en la presente memoria, así como las sales de los mismos. Dichos solvatos son, por ejemplo, hidratos, alcoholatos y similares.
Como se entenderá a partir de las descripciones de la presente memoria, la presente invención proporciona particularmente compuestos aislados y composiciones aisladas. Los compuestos se obtienen en particular mediante síntesis in vitro, tal como mediante síntesis química. En una realización adicional, los compuestos de la invención tienen una pureza de al menos el 85%, en particular al menos el 90%, más en particular al menos el 95%. En una realización particular, la presente invención proporciona un método para proporcionar un compuesto de la invención, comprendiendo el método sintetizar químicamente el compuesto de la invención y envasar el compuesto sintetizado en un recipiente estéril. En una realización adicional, la presente invención proporciona un método para proporcionar un compuesto de la invención, comprendiendo el método sintetizar químicamente el compuesto con una pureza de al menos el 85%, en particular al menos el 90%, más en particular al menos el 95%. Preferiblemente, el compuesto sintetizado se envasa posteriormente en un envase estéril.
Composiciones
La presente invención se refiere además a una composición farmacéutica, comprendiendo la composición un compuesto como se define anteriormente y como se define en una cualquiera de las realizaciones presentadas en la presente memoria.
En el resto del texto, se entiende la expresión “compuesto” o “compuesto según la invención”, para los fines de la presente invención, tanto en plural como en singular, es decir que la composición de la invención puede comprender uno o más de un “compuesto según la invención”.
En una realización particular, la presente invención proporciona una composición farmacéutica que comprende el compuesto según una cualquiera de las reivindicaciones, y uno o más vehículos farmacéuticamente aceptables. Ejemplos de formulaciones farmacéuticamente aceptables, así como los métodos para prepararlas se pueden encontrar en, por ejemplo, Remington’s Pharmaceutical Sciences (por ejemplo, 20th Edition; Lippincott, Williams & Wilkins, 2000) o en cualquier manual de Farmacopea (por ejemplo, US-, European- o International Pharmacopeia). En otra realización, la presente invención proporciona una composición que comprende un tampón acuoso en el que se ha disuelto un compuesto de la invención. En una realización adicional, la presente invención proporciona una composición farmacéutica que comprende un tampón acuoso en el que se ha disuelto un compuesto de la invención y uno o más vehículos farmacéuticamente aceptables. En otra realización adicional, la presente invención proporciona una composición farmacéutica que consiste en un tampón acuoso, un compuesto de la invención y uno o más vehículos farmacéuticamente aceptables.
En una realización particular, la presente invención proporciona una composición que comprende un antagonista de la integrina, en donde al menos el 90% de los compuestos antagonistas de la integrina de la composición es un compuesto según la invención. Preferiblemente al menos el 95%, especialmente al menos el 99% de los compuestos antagonistas de la integrina de la invención es un compuesto según la invención. En otra realización particular, las composiciones de la invención están sustancialmente libres de otro antagonista de la integrina, tal como un compuesto A. En una realización adicional, la proporción del compuesto de la invención con respecto a otros compuestos antagonistas de integrina, tal como el compuesto A, es más del 98:2, particularmente más del 99:1, más particularmente más del 99,9:0,1. En otra realización más, la presente invención proporciona una composición como se describe en la presente memoria, en la que la composición está sustancialmente libre de compuesto A, en particular en la que la composición comprende menos del 3%, especialmente menos del 2%, preferiblemente menos del 1% del compuesto A, más en particular con la condición de que la composición no comprenda el compuesto A.
En otra realización, la presente invención proporciona una composición que comprende un primer principio activo y un segundo principio activo, en donde el primer principio activo es un antagonista de la integrina según la invención, y en donde la composición está sustancialmente libre de un antagonista de la integrina distinto del primer principio activo. En una realización adicional, el segundo principio activo es un compuesto que se une a e inhibe la actividad de VEGF o un receptor de VEGF.
Antagonista de la integrina o compuesto antagonista de la integrina en la presente solicitud se refiere preferiblemente a un compuesto que tiene una concentración efectiva media máxima (EC50) frente a receptores de la integrina de menos de 1 pM, en particular una EC50 frente al receptor de la integrina avp3. Los expertos en la técnica conocen métodos adecuados para determinar los valores de EC50. Un método particular para determinar el valor de un compuesto en particular es un ensayo ELISA de competición en donde el receptor de la integrina bajo investigación (en particular el receptor de la integrina avp3 humano) se recubre en una placa de múltiples pocillos utilizando una disolución de 4 pg/mL y los pocillos se bloquean con albúmina de suero bovino al 5%. La concentración de fibronectina que se va a utilizar en el ensayo se determina experimentalmente en un experimento separado ensayando la unión de varias concentraciones de fibronectina a la integrina recubierta bajo investigación, siendo la concentración de fibronectina para el ensayo la que da el 80% de unión máxima. La concentración determinada de fibronectina humana se añade después a los pocillos recubiertos y bloqueados en presencia de concentraciones crecientes del compuesto. Después de 2 h de incubación a 37°C, se lavan los pocillos y se detecta la fibronectina unida con la ayuda de un reactivo específico, tal como un anticuerpo anti-fibronectina detectable. A continuación, se realiza un análisis cuantitativo de los datos para determinar los valores de EC50, es decir, la concentración del compuesto que reduce la unión de fibronectina al receptor recubierto en un 50%.
En otra realización, la presente invención proporciona una composición que comprende un tampón acuoso y un compuesto de la invención. En una realización adicional, la presente invención proporciona una composición farmacéutica que comprende un tampón acuoso, un compuesto de la invención y uno o más vehículos farmacéuticamente aceptables. En otra realización adicional, la presente invención proporciona una composición farmacéutica que consiste en un tampón acuoso, un compuesto de la invención y uno o más vehículos farmacéuticamente aceptables.
En una realización particular, la presente invención proporciona una composición que comprende entre 0,1 y 1000 mg de un compuesto de la invención por ml de la composición, en particular entre 1 y 500 mg/ml, más en particular entre 1 y 100 mg/ml. En una realización adicional, la composición comprende entre 10 y 100 mg/ml, tal como entre 20 y 75 mg/ml.
Compuestos y/o composiciones para su uso como medicamento
La presente invención se refiere además a un compuesto para su uso como medicamento.
En una realización particular, la presente invención se refiere a un compuesto para su uso en la inhibición de la neovascularización.
En una realización adicional, la presente invención se refiere a un compuesto para su uso en el tratamiento y/o prevención del cáncer. En particular, para inhibir el crecimiento y/o metástasis del cáncer.
En otra realización, la presente invención se refiere a un compuesto para su uso en el tratamiento y/o prevención de un trastorno oftálmico. En particular para tratar y/o prevenir la neovascularización en el ojo. Más en general, la presente invención proporciona compuestos y composiciones farmacéuticas para tratar, reducir, mejorar, o inhibir la progresión de la neovascularización ocular (patológica). En otro aspecto, la presente invención proporciona compuestos y composiciones farmacéuticas para tratar, reducir, mejorar, o inhibir la progresión de un trastorno oftálmico resultante de, o que tiene una etiología en, la neovascularización ocular.
En una realización adicional, la presente invención se refiere a un compuesto para su uso en el tratamiento y/o prevención de trastornos oftálmicos, en donde dicho trastorno oftálmico se selecciona del grupo que consiste en degeneración macular húmeda relacionada con la edad, desprendimiento de retina, uveítis posterior, neovascularización de la córnea, neovascularización del iris, edema macular diabético y retinopatía diabética.
En otra realización más, los compuestos de la invención se proporcionan para su uso como antagonista de la integrina, en particular, un antagonista del receptor de vitronectina, in vitro o in vivo. En una realización particular, la integrina es una integrina de unión a RGD. En una realización adicional, la integrina es una integrina que comprende una subunidad av, a5 o a8. En una realización más particular, la integrina es un receptor avp3, avp5 o asp1.
En otra realización, los compuestos de la invención se utilizan para reducir la permeabilidad vascular, preferiblemente para reducir la permeabilidad vascular retiniana. En una realización adicional, los compuestos de la invención se utilizan en el tratamiento y/o prevención de un edema, en particular de un edema macular, más en particular de un edema macular diabético.
En otra realización, los compuestos de la invención se proporcionan para su uso en la inducción del desprendimiento de vítreo posterior (PVD). En otra realización, los compuestos de la invención se proporcionan para su uso en el tratamiento de la tracción vitreomacular.
Como se mencionó anteriormente, la presente invención se refiere a compuestos nuevos, sus sales farmacéuticamente aceptables, solvatos farmacéuticamente aceptables, isómeros o mezclas de los mismos. El compuesto de la presente invención puede formularse también en diversas formas farmacéuticas para fines de administración. Como composiciones apropiadas, se pueden citar todas las composiciones empleadas
habitualmente para la administración sistemática de fármacos. Para preparar las composiciones farmacéuticas de esta invención, una cantidad eficaz del compuesto particular, opcionalmente en forma de sal de adición o complejo metálico, como el principio activo se combina en una mezcla estrecha con un vehículo farmacéuticamente aceptable, cuyo vehículo puede tomar una amplia variedad de formas dependiendo de la forma de preparación deseada para la administración. Estas composiciones farmacéuticas se desean en forma de dosificación unitaria adecuada, particularmente, para la administración por vía oral, rectal, percutánea, por inyección parenteral o intravítrea. Por ejemplo, al preparar las composiciones en forma de dosificación oral, se puede emplear cualquiera de los medios farmacéuticos habituales tal como, por ejemplo, agua, glicoles, aceites, alcoholes y similares en el caso de preparaciones líquidas orales tal como suspensiones, jarabes, elixires, emulsiones y soluciones; o vehículos sólidos tal como almidones, azúcares, caolín, lubricantes, aglutinantes, agentes disgregantes y similares en el caso de polvos, píldoras, cápsulas y comprimidos.
Es especialmente ventajoso formular las composiciones mencionadas anteriormente en forma de dosificación unitaria para facilitar la administración y uniformidad de la dosificación. La forma de dosificación unitaria como se utiliza en la presente memoria se refiere a unidades físicamente discretas adecuadas como dosificaciones unitarias, conteniendo cada unidad una cantidad predeterminada de principio activo calculada para producir el efecto terapéutico deseado en relación con el vehículo farmacéutico requerido. Ejemplos de dichas formas de dosificación unitaria son comprimidos (incluyendo comprimidos ranurados o recubiertos), cápsulas, píldoras, supositorios, sobres de polvo, obleas, soluciones o suspensiones inyectables y similares, y múltiplos segregados de los mismos.
En una realización particular, la presente invención proporciona un envase que comprende un compuesto de la invención y un prospecto con instrucciones para administrar el compuesto a un paciente que tiene un trastorno oftálmico como se enumera en la presente memoria, en particular un paciente que tiene degeneración macular húmeda relacionada con la edad, retinopatía diabética o edema macular diabético, más en particular retinopatía diabética o edema macular diabético.
Para llevar a cabo el tratamiento y/o la prevención, los compuestos o las composiciones como se describen en la presente memoria se pueden administrar a un paciente mediante cualquier método que conduzca a la administración del agente terapéutico al sitio de la afección oftálmica, tal como la administración en el ojo. En otra realización, el uso, tratamiento y/o prevención comprende poner en contacto el humor vítreo y/o acuoso con una cantidad eficaz de una composición que comprende un compuesto de la invención. La administración puede ser por vía ocular, tal como tópica, subconjuntival, sub-Tenon, intraocular, implantes oculares, etcétera. La administración tópica puede comprender la administración de una o unas pocas gotas de una composición que comprende un compuesto de la invención al ojo. La administración a áreas dentro del ojo, in situ, se puede lograr mediante inyección, cánula u otro dispositivo invasivo diseñado para introducir cantidades medidas con precisión de una composición oftálmica deseada en un compartimento o tejido particular dentro del ojo (por ejemplo, cámara posterior o retina). Una inyección intraocular puede ser en el vítreo (intravítreo), o debajo de la conjuntiva (subconjuntival), o detrás del ojo (retrobulbar), en la esclerótica o debajo de la Cápsula de Tenon (sub-Tenon). También se contemplan otras vías de administración intraocular y sitios y formas de inyección y están dentro del alcance de la invención. En una realización preferida, el tratamiento y/o prevención comprende la administración del compuesto mediante inyección intravítrea. Preferiblemente, esto se realiza a través de agujas de calibre autosellantes u otro dispositivo de suministro adecuadamente calibrado. La inyección en el ojo puede realizarse a través de la pars plana mediante la aguja autosellante.
Cuando se administra la composición mediante inyección intravítrea, los agentes activos deben concentrarse para minimizar el volumen de inyección. Preferiblemente, el volumen para la inyección es menor de aproximadamente 5 mL. Volúmenes como este pueden requerir un drenaje compensatorio del líquido vítreo para evitar aumentos en la presión intraocular y fugas del líquido inyectado a través de la abertura formada por la aguja de suministro. Más preferiblemente, el volumen inyectado está entre aproximadamente 10 y 200 pL. Lo más preferiblemente, el volumen para la inyección está entre 30 y 100 pL, en particular aproximadamente 50 pL.
Con respecto a las vías de administración preferidas, la sal farmacéuticamente aceptable, el solvato farmacéuticamente aceptable, el isómero o la mezcla de los mismos es también una sal ópticamente aceptable, un solvato ópticamente aceptable, un isómero o una mezcla de los mismos. En una realización preferida, la composición farmacéutica comprende el compuesto y uno o más vehículos ópticamente aceptables. Curiosamente, se encontró que los compuestos de la invención tienen una alta solubilidad en agua de más de 100 mg/ml, mientras que los compuestos de la técnica anterior tienen solubilidades en agua de un solo dígito o más bajas en mg/ml. En una realización preferida, una composición de la invención como se describe en la presente memoria es una disolución acuosa que comprende un compuesto de la invención. En otra realización, la presente invención proporciona un kit de piezas que comprende un envase que comprende un compuesto de la invención y otro envase que comprende un tampón acuoso para disolver el compuesto de la invención.
Como entenderá el experto en la materia, una composición como se describe en la presente memoria es preferiblemente una composición estéril. En una realización particular, la presente invención proporciona un envase estéril que comprende un compuesto o composición de la invención. En una realización adicional, el envase estéril es un vial que comprende un compuesto o composición de la invención. El vial puede comprender el compuesto como un polvo o como una solución, preferiblemente una solución acuosa. En una realización más particular, la
presente invención proporciona un vial estéril que comprende una solución acuosa del compuesto de la invención. En una realización aún más particular, un vial como se describe en la presente memoria comprende una parte que está diseñada para ser perforada por una aguja de jeringa. En una realización alternativa, la presente invención proporciona un kit que comprende un vial con el compuesto de la invención en forma de polvo y un envase que comprende una solución acuosa, particularmente una solución tampón acuosa. En otra realización, el envase estéril es una jeringa precargada con una composición farmacéutica de la invención, particularmente una solución acuosa que comprende el compuesto de la invención. En otra realización más, el envase estéril es un envase que comprende al menos una tableta que comprende una composición farmacéutica de la invención. En particular, un blíster o envase que comprende múltiples comprimidos que comprenden un compuesto de la invención y uno o más vehículos farmacéuticamente aceptables. En una realización particular, un vial como se describe comprende menos de 5 ml, en particular menos de 4 ml, más en particular menos de 3 ml de una solución que comprende el compuesto de la invención.
En una realización preferida, la presente invención se refiere además al compuesto para su uso, en el que el tratamiento, inhibición y/o prevención de los trastornos mencionados anteriormente comprende además administrar un compuesto que se une a e inhibe la actividad de VEGF o un receptor de VEGF. Este compuesto puede ser un inhibidor de VEGF, un anticuerpo del receptor de VEGF o un fragmento de unión a antígeno del mismo, o un anticuerpo de VEGF o un fragmento de unión a antígeno del mismo. Ejemplos adecuados de dichos compuestos incluyen bevacizumab, ranibizumab, brolucizumab o conbercept. Otro posible compuesto adicional es una trampa de VEGF, que comprende un dominio de unión a VEGF, tal como aflibercept.
La presente invención se refiere además a una combinación que comprende el compuesto de la presente invención y un compuesto adicional que se une a e inhibe la actividad de VEGF o un receptor de VEGF. Como se especificó anteriormente, este compuesto adicional puede ser un inhibidor de VEGF, un anticuerpo del receptor de VEGF o un fragmento de unión a antígeno del mismo, o un anticuerpo de VEGF o un fragmento de unión a antígeno del mismo, tal como bevacizumab, ranibizumab, brolucizumab o conbercept. Otro posible compuesto adicional es una trampa de VEGF, que comprende un dominio de unión de VEGF, como aflibercept.
Siempre que se haga referencia a una combinación de principios activos, los principios activos se pueden proporcionar en una composición única o en una composición separada. Además, una combinación de principios activos también abarca principios activos que están enlazados de forma no covalente o covalente.
La presente invención se refiere además a una combinación como se especificó anteriormente para su uso como un medicamento o para inhibir, tratar y/o prevenir la neovascularización patológica, el cáncer o un trastorno oftálmico tal como degeneración macular húmeda relacionada con la edad, desprendimiento de retina, uveítis posterior, neovascularización de la córnea, neovascularización del iris, edema macular diabético y retinopatía diabética.
La presente invención también proporciona un método para antagonizar una integrina, en particular un receptor de vitronectina, en un sujeto que lo necesite, comprendiendo el método administrar el compuesto de la invención. Más en particular, la invención proporciona métodos para inhibir la angiogénesis en un sujeto. La presente invención se refiere además a un método para inhibir la neovascularización (patológica) en un sujeto que lo necesite, comprendiendo el método administrar el compuesto de la presente invención. En una realización adicional, la presente invención proporciona un método para inhibir la neovascularización ocular en un sujeto, comprendiendo el método administrar un compuesto de la invención al sujeto. En otra realización más particular, el método comprende la administración del compuesto o composición de la invención al ojo de un sujeto, tal como mediante inyección intravítrea. En particular, el método comprende la inyección intravítrea de una composición como se describe en la presente memoria.
La presente invención proporciona además los compuestos y composiciones descritos en la presente memoria para su uso en la fabricación de un medicamento para la prevención y/o el tratamiento de un trastorno como se describe en la presente memoria.
Ejemplos
Ejemplo 1 - Síntesis de los compuestos de la invención
Síntesis del N-[(2S)-1 -(terc-butoxi)-3-({2,5-dimetil-6-[4-(1,8-naftiridin-2-il)piperidin-1 -il]pirimidin-4-il}amino)-1 -hidroxipropan-2-il]carbamato de bencilo

Se prepararon 2-[1 -(6-cloro-2,5-dimetilpirimidin-4-il)piperidin-4-il]-1,8-naftiridina y (2S)-3-amino-2-{[(benziloxi)carbonil] amino}propanoato de terc-butilo según el método descrito en el documento US2008/0058348 A1, que se incorpora en la presente memoria como referencia. Ambas moléculas se acoplaron en una reacción de aminación de Buchwald-Hartwig catalizada por el complejo dicloro[1,1’-bis(difenilfosfino) ferroceno] paladio (II) diclorometano (1:1), 1, 1 ’-ferrocenodiil-bis difenilfosfina, ácido fenilborónico, trietilamina y fluoruro de serio en 2-metiltetrahidrofurano. La mezcla de reacción se filtró a través de Celita y se extrajo con ácido clorhídrico acuoso. El extracto acuoso se neutralizó con acetato de sodio acuoso y se extrajo con diclorometano, el cual se lavó con bicarbonato de sodio acuoso y se secó sobre sulfato de sodio para dar un producto crudo que se purificó mediante cromatografía en columna de sílice utilizando tolueno/acetato de etilo como eluyente. El material cromatografiado se disolvió en acetato de etilo y se purificó adicionalmente mediante cristalización como la sal del ácido (-)-dibenzoil-L-tartárico. Se eliminó la sal del ácido tartárico utilizando bicarbonato de sodio acuoso para dar el producto purificado.
Síntesis del (2S)-2-amino-3-({2,5-dimetil-6-[4-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)piperidin-1-il]pirimidin-4-il}amino) propanoato de terc-butilo

Se hidrogenó N-[(2S)-1-(terc-butoxi)-3-({2,5-dimetil-6-[4-(1,8-naftiridin-2-il)piperidin-1-il]pirimidin-4-il}amino)-1 -hidroxipropan-2-il]carbamato de bencilo sobre paladio en carbón activado en 2-metiltetrahidrofurano. El catalizador se retiró mediante filtración a través de Celita. El disolvente se eliminó por evaporación para dar el producto deseado.
Síntesis de (2S)-3-({2,5-dimetil-6-[4-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)piperidin-1-il]pirimidin-4-il}amino)-2-{[(3-metoxi-3-oxopropoxi)carbonil] amino}n-propanoato de terc-butilo
Se hizo reaccionar carbonato de N,N-disuccinimilo, en presencia de trietilamina, con 3-hidroxipropanoato de metilo en diclorometano para dar 2-({[(2,5-dioxopirrolidin-1-il)oxi]carbonil} oxi)acetato de metilo. Se añadió (2S)-2-amino-3-({2,5-dimetil-6-[4-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)piperidin-1 -il]pirimidin-4-il}amino)propanoato de terc-butilo para dar el producto deseado. La mezcla de reacción se lavó con ácido clorhídrico acuoso y bicarbonato de sodio acuoso y después se secó sobre sulfato de sodio para dar el producto crudo que se purificó mediante cromatografía en columna en sílice utilizando acetato de etilo/n-heptano/trietilamina como eluyente.
Síntesis de trifluoroacetato del ácido (2S)-3-({2,5-dimetil-6-[4-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)piperidin-1-il]pirimidin-4-il}amino)-2-{[(3-metoxi-3-oxopropoxi)carbonil]amino]propanoico

(2S)-3-({2,5-dimetil-6-[4-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)piperidin-1-il]pirimidin-4-il}amino)-2-{[(2-metoxi-2-oxoetoxi)carbonil]amino}propanoato de terc-butilo se disolvió en diclorometano y se trató con ácido trifluoroacético. La mezcla de reacción se lavó con brine (disolución saturada de cloruro de sodio) y el disolvente se eliminó por evaporación para dar el producto deseado como una sal del ácido trifluoroacético. El compuesto resultante se denomina además como compuesto comparativo A (cpd A).
MS de baja resolución mediante electrospray (ES) m/z: 556 [ (M+1), 100%]
Síntesis del ácido (2S)-2-{[(2-carboxietoxi)carbonil]amino}-3-({2,5-dimetil-6-[4-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)piperidin-1 -il]pirimidin-4-il}amino)propanoico

Se disuelve trifluoroacetato del ácido (2S)-3-({2,5-dimetil-6-[4-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)piperidin-1-il]pirimidin-4-il}amino)-2-{[(3-metoxi-3-oxopropoxi)carbonil]amino}propanoico en ácido clorhídrico acuoso 2M. Una vez completada la reacción el disolvente se elimina mediante liofilización para dar el producto deseado. El ácido (2S)-2-{[(2-carboxietoxi)carbonil]amino}-3-({2,5-dimetil-6-[4-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)piperidin-1 -il]pirimidin-4-il}amino)propanoico, se denomina además como compuesto 1 (cpd1).
MS de baja resolución mediante electrospray (ES) m/z: 542 [ (M+1), 100%]
Síntesis de (2S)-2-({[2-(terc-butoxi)-2-oxoetoxi] carbonil}amino)-3-({2,5-dimetil-6-[4-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)piperidin-1 -il]pirimidin-4-il}amino)propanoato de terc-butilo

Se hizo reaccionar carbonato de N,N-disuccinimidilo, en presencia de trietilamina, con 2-hidroxiacetato de terc-butilo en diclorometano para dar 2-({[(2,5-d¡oxop¡rrol¡d¡n-1-¡l) oxi] carbonil} oxi) acetato de terc-butilo. Se añadió (2S)-2-am¡no-3-({2,5-d¡met¡l-6-[4-(5,6,7,8-tetrah¡dro-1,8-naftiridin-2-il)piperidin-1 -il]pirimidin-4-il}amino) propanoato de tercbutilo. Una vez completada la reacción la mezcla de reacción se lavó con ácido clorhídrico acuoso y bicarbonato de sodio acuoso y se secó después sobre sulfato de sodio para dar el producto crudo. El producto crudo se purificó mediante cromatografía en columna en sílice utilizando acetato de etilo/n-heptano/trietilamina como eluyente.
Síntesis de diclorhidrato del ácido (2S)-2-([(carboximetoxi)carbonil]amino)-3-({2,5-dimetil-6-[4-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)piperidin-1-il]pirimidin-4-il}amino)propanoico (THR-687)

(2S)-2-({[2-(terc-butoxi)-2-oxoetoxi] carbon¡l}am¡no)-3-({2,5-d¡met¡l-6-[4-(5,6,7,8-tetrah¡dro-1,8-naftiridin-2-il)piperidin-1-il]pirimidin-4-il}amino)propanoato de terc-butilo se hidrolizó con ácido clorhídrico acuoso. La mezcla de reacción se lavó con n-heptano para dar el compuesto 2 (cpd2) después de la evaporación.
1H RMN (400 MHz, DMSO-ds) 6: 7,64 (d 1H), 6,71 (d 1H), 4,63 (d 1H), 4,46 (d 1H), 4,57 (dd 1H), 4,06 (dd 1H), 3,85 (dd 1H), 3,77 (m 2H), 3,53 (dd 2H), 3,27 (m, 2H), 3,01 (tt 1H), 2,84 (dd 2H), 2,58, (s 3H), 2,10 (dd, 2H), 2,07, (s 3H), 1,99 (t, 2H), 1,95 (td 2H)
Síntesis del (2S)-3-({2,5-dimetil-6-[4-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)piperidin-1 -il]pirimidin-4-il}amino)-2-({[(1 -etoxi-1 -oxopropan-2-il)oxi] carbonil}amino)propanoato de terc-butilo
Se hizo reaccionar carbonato de N,N-disuccinimidilo, en presencia de trietilamina, con 2-hidroxipropanoato de etilo en diclorometano para dar 2-({[(2,5-dioxopirrolidin-1-il) oxi] carbonil} oxi) propanoato de etilo. Se añadió (2S)-2-amino-3-({2,5-dimetil-6-[4-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)piperidin-1 -il]pirimidin-4-il}amino)propanoato de tercbutilo. Una vez completada la reacción la mezcla de reacción se lavó con ácido clorhídrico acuoso y bicarbonato de sodio acuoso y se secó después sobre sulfato de sodio para dar el producto crudo. El producto crudo se purificó mediante cromatografía en columna en sílice utilizando diclorometano/metanol como eluyente.
Síntesis del diclorhidrato del ácido (2S)-2-{[(1-carboxietoxi)carbonil] amino}-3-({2,5-dimetil-6-[4-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)piperidin-1 -il]pirimidin-4-il}amino)propanoico

(2S)-3-({2,5-dimetil-6-[4-(5,6,7,8-tetrahidro-1,8-naftiridin-2-il)piperidin-1 -il]pirimidin-4-il}amino)-2-({[(1 -etoxi-1 -oxopropan-2-il)oxi]carbonil}amino)propanoato de terc-butilo se hidrolizó con ácido clorhídrico acuoso. La mezcla de reacción se lavó con n-heptano para dar el producto después de la evaporación, denominado además como compuesto 3 (cpd3).
1H RMN (400 MHz, D2O) 5: 7,5 (d 1H), 6,5 (d 1 H), 4,5 (m 1H), 4,2 (m 1 H), 3,9 (dd 1H), 3,5-3,7 (m 3H), 3,4 (t 2H), 3,1 (t 2H), 2,8 (m 1 H), 2,7 (t 2H), 2,4 (s 3H), 1,9 (s 3H), 1,9-2,0 (m 3H), 1,7-1,9 (m 4H), 1,3 (d 3H).
Por consiguiente, se han preparado los siguientes compuestos de la invención:


Otros ejemplos de compuestos según la invención son:

El Compuesto A (cpd A) se sintetizó como se describió anteriormente:

El Compuesto B (CpdB) se sintetizó según los procedimientos del documento US2008058348 A1:
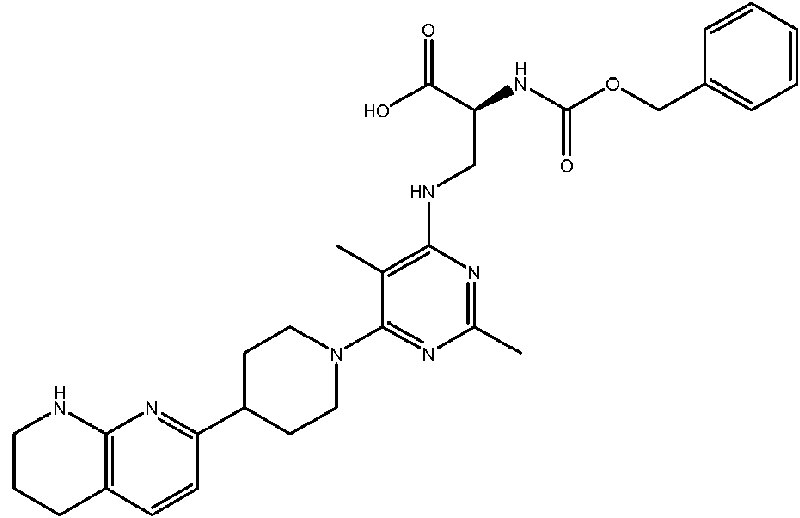
Ejemplo 2: antagonismo de integrina
El principio de los ensayos ELISA de competición utilizados en la presente memoria fue el siguiente: (1) el receptor de integrina bajo investigación se recubrió en placas de 96 pocillos y los pocillos se bloquearon, (2) después se añadió un ligando de integrina (fibronectina) a los pocillos relevantes en presencia de concentraciones crecientes de un antagonista de integrina dado, (3) después de un período de tiempo definido, se lavaron los pocillos y se detectó el ligando unido con la ayuda de un reactivo específico. Después se realizó un análisis cuantitativo de los datos para determinar los valores de EC50, es decir, la concentración del antagonista de la integrina que reduce la unión del ligando al receptor recubierto en un 50%.
Tabla 1: EC50 para los diferentes antagonistas de integrinas e integrina humana

++: entre 1 y 10 nM; +: entre 10 y 25 nM; : >25 nM
El compuesto según la fórmula (I) inhibe las interacciones entre las integrinas y sus ligandos. El compuesto según la fórmula (I) se ha demostrado que antagoniza varios receptores de integrina, incluyendo av03, av05 y a501, con una afinidad nanomolar de un solo dígito. Estos receptores de integrina se ha demostrado que juegan un papel en la angiogénesis (av03, av05 y a501), la permeabilidad vascular (av03 y av05) y la adhesión vitreorretiniana (a501).
En particular, los compuestos de la invención muestran un antagonismo significativamente reducido hacia la integrina plaquetaria anbP3, que está implicada en la agregación plaquetaria. La Figura 1 establece las proporciones determinadas de valores de EC50 para av03 con respecto anbP3. Como se deriva de estos resultados, los compuestos de la presente invención muestran una actividad mucho menor hacia la integrina plaquetaria en comparación con la integrina av03. Por el contrario, los compuestos como se describen en la técnica anterior muestran una actividad significativamente mayor hacia la integrina plaquetaria en comparación con la integrina av03.
Ejemplo 3 - ensayo de migración celular
La cicatrización de heridas de células endoteliales está mediada por la migración y proliferación de células endoteliales, ambos componentes críticos del proceso de angiogénesis, y se utiliza de forma rutinaria como modelo sustituto del potencial angiogénico in vitro. El ensayo de migración celular ORISTM se utilizó con células endoteliales de la vena umbilical humana (HUVECs).
Se cultivaron células HUVEC (SCCE-001, N° Cat 1000519, Merck Chemicals) en matraces T175 (N° Cat 353112, falcon) en un medio de crecimiento EndoGro (SCME-003, Merck Chemicals) y se dividieron cuando alcanzaron una confluencia del 90-100%.
Para el ensayo, las células se utilizaron en los pasos 2 a 4 después de la descongelación y al 80-90% de confluencia. Al comienzo del día, las placas de ensayo de migración celular OrisTM recubiertas con fibronectina (N° Cat #CMAFN.l01, AMS Biotechnology) se retiraron de la refrigeración y se colocaron a temperatura ambiente durante 1 hora. Las Oris Detection Masks se lavaron con etanol y se aplicaron al fondo de las placas de 96 pocillos, alineando la esquina de Al de las carátulas con la esquina de Al de las placas. Las células se recogieron mediante tripsinización (N° Cat 25300, Gibco), se centrifugaron a 250 g durante 5 minutos y se suspendieron a 0,4.106 por mL en medio de crecimiento EndoGro completo. 100 pL de la suspensión celular se añadieron a los pocillos y las placas se incubaron durante 6 horas a 37°C y 5% de CO2 para permitir que las células se adhieran.
Al final de la incubación, la herramienta de tapón se lavó con etanol y se utilizó para quitar los tapones de todos los pocillos excepto los de referencia. Se añadieron a los pocillos 100 pL de medio de cultivo que contenía diferentes concentraciones de los compuestos (cpd2, cpd3, cpdB, cilengitide) y las placas se transfirieron durante 24 horas a la incubadora a 37°C y 5% de CO2 para permitir la migración.
Justo antes del final de la incubación, se preparó una disolución de 4 pg/mL de calceína AM (N° Cat 354217, Beckton Dickinson) en HBSS precalentado (N° Cat 14025, Gibco). Se retiraron los tapones de los pocillos de referencia y el medio se retiró cuidadosamente de todos los pocillos con un multicanal (sin aspiración al vacío). Los pocillos se lavaron con 100 pL de DPBS. Después, se añadieron 100 pL de la disolución de calceína AM y las placas se incubaron durante 1 hora a 37°C y 5% de CO2. Después de la incubación, se leyó la fluorescencia a 494 nm de excitación y longitudes de onda de emisión de 517 nm utilizando Flexstation y el software SofiMax Pro 5.4.5 (Molecular Devices). La fluorescencia se leyó desde el fondo de las placas con la tapa puesta para mantenerlas estériles. Después se retiraron las carátulas y se tomaron fotografías con el microscopio de fluorescencia Axiovert (Zeiss) utilizando luz reflejada, GFP y un objetivo de 2,5x.
Los valores de IC50 para cpdB, cpd2 y cpd3 estuvieron todos por debajo de 1 pM. La IC50 para el control positivo Cilengitide fue de 100 pM. Se puede concluir que los compuestos de fórmula (I) retienen una fuerte actividad e inhiben la migración celular con valores de IC50 en el rango nanomolar.
Ejemplo 4: Ensayo de explante coroideo murino
El ensayo se basa en un modelo de explante de angiogénesis microvascular murina, que se asemeja mucho a la angiogénesis coroidea in vivo. La principal ventaja de este modelo de cultivo organotípico es que las células se mantienen dentro de su entorno normal, preservando así las interacciones célula-célula, mientras se mantiene un mayor nivel de control experimental que en los modelos animales. El modelo de explante coroideo o brevemente el modelo EMCA permite una evaluación rápida de nuevas terapias anti-angiogénicas, con el beneficio adicional de excluir la posible falta de exposición que se encuentra a menudo al probar moléculas pequeñas en ojos de animales. Los explantes de RPE-esclerótica coroidea (en lo sucesivo denominados explantes coroideos) se incrustan en un gel de fibrinógeno/fibrina tridimensional. La adición de un medio que contiene suero induce un extenso crecimiento de células endoteliales en la matriz después de 3 a 4 días en cultivo.
Animales
Todos los experimentos se realizaron utilizando ratones macho C57BL/6J de 4 a 5 semanas de edad (Charles River Laboratories France), alojados en condiciones estándar de laboratorio según un ritmo normal de día/noche con acceso ad libitum a comida y agua. Todos los experimentos con animales fueron aprobados por el Institutional Ethical Committee de KU Leuven.
Disección del tejido coroideo y preparación del cultivo de explante
Después de la optimización inicial, se estableció un protocolo general. Brevemente, después de la eutanasia de los ratones utilizando pentobarbital (200 mg/Kg, IP, Dolethal, Vetoquinol) seguido de dislocación cervical, los ojos se enuclearon inmediatamente con unas pinzas curvas y se mantuvieron en DPBS helado (N° Cat 14190-136, Gihco). Bajo un microscopio estereoscópico, el ojo se colocó en una gota de DPBS helado y se mantuvo en su lugar con unas pinzas en una placa de Petri helada. La córnea y el cristalino se extrajeron mediante una incisión circunferencial en el limbo (límite córnea-esclerótica) utilizando tijeras de resorte Vannas (Fine Science Tools). A continuación, se extrajeron los músculos redundantes del globo ocular restante y se hizo un corte desde el borde de la cámara posterior hacia la cabeza del nervio óptico. Después de una cuidadosa separación del complejo RPEcoroides-esclerótica de la retina, se hicieron de 6 a 8 explantes en la periferia de este complejo utilizando una aguja perforada (750 gm de diámetro, Mediquip Surgical). Los explantes se transfirieron a una placa de 48 pocillos (BD Biosciences), precargada con 350 gL de DMEM sin suero (N° Cat 11995-065, Gibco) y 1% (v/v) de Pen/Strep (N° Cat 15140-122, Gibco), y se incubaron durante la noche a 37°C en 5% de CO2.
Explante de coroides incrustado en gel de fibrinógeno/fibrina y condiciones de cultivo
Se disolvió fibrinógeno bovino (3,1 mg/ml, N° Cat F863, Sigma-Aldrich) en DMEM (N° Cat 11995-065, Gibco) que contenía 1 % (v/v) de Pen/Strep (N° Cat 15140-122, Gibco), y se filtró posteriormente a través de un filtro de poros de 0,22 gm (Corning). A continuación, se añadieron 10 gg/mL de aprotina inhibidor de la serina proteasa (N° Cat A6279, Sigma-Aldrich) a la disolución de fibrinógeno para bloquear la actividad fibrinolítica. Después de la adición de 400 mU/mL de trombina bovina (SRP6555, Sigma-Aldrich), esta disolución de fibrinógeno-aprotinina-trombina se dispensó rápidamente en placas de 48 pocillos (100 gL por pocillo) y se dejó solidificar a 37°C durante al menos 5 minutos. En la parte superior de los pocillos recubiertos de fibrina, se añadieron 175 gL de disolución de fibrinógeno más aprotinina, seguido de la transferencia de explanes de RPE-coroides individuales en 175 gL de medio DMEM utilizando una punta cortada. Finalmente, se añadieron 1,4 gL de trombina (400 mU/nit) a cada pocillo y esta mezcla se dejó gelificar a 37°C y 5% de CO2 durante 1 hora. Después de la coagulación, se añadieron 350 gL de DMEM que contenía FBS al 10% (SH30071.03, inactivado por calor. HyClone) y la(s) molécula(s) bajo prueba (cpd3, cpdB y cilengitide) en la parte superior del gel y las placas se incubaron a 37°C en 5% de CO2.
Cuantificación de la aparición de vasos
Después de 3 a 4 días, se tomaron imágenes de campo brillante a 2,5x aumentos con el microscopio invertido Axio Vert. A1 (Zeiss) utilizando el software de microscopio ZEN lite 2012 (Zeiss). El análisis del área de crecimiento del vaso se realizó manualmente en imágenes ZVI, más específicamente delineando el cuerpo del explante, así como el área de crecimiento total del vaso (incluyendo el cuerpo del explante) utilizando el software Axiovision Rel 4.8 (“Measure>Outline”, Zeiss). Se realizaron análisis adicionales en Microsoft Excel, donde se cuantificó el área de aparición del cultivo organotípico endotelial restando el área del explante de tejido del área total ocupada por los brotes. Como las áreas absolutas de brotación entre experimentos independientes son ligeramente diferentes, todos los datos dentro de cada experimento se normalizaron a la condición tratada con vehículo y se calcularon los valores de IC50 (GraphPad Prism 5).
Resultados
Todos los antagonistas de la integrina bloquearon la formación de brotes endoteliales de una manera dependiente de la concentración. Esto está en línea con los estudios que muestran que las células endoteliales pueden asociarse con el fibrinógeno a través de la especificidad de reconocimiento de ROD. De hecho, el fibrinógeno contiene múltiples sitios de unión para las integrinas avp3 y a5p1, que se sabe que son expresadas por células endoteliales y reguladores importantes de la angiogénesis (patológica). Cpd3 y cpdB inhibieron el crecimiento de vasos con valores de IC50 alrededor de 1 gM, mientras que para el control positivo Cilengitide, la IC50 medida fue de alrededor de 300 gM. Imágenes de campo brillantes representativas para cpd3 se muestran en la Figura 2. Estos hallazgos están en línea con los resultados del ensayo de migración celular, discutido en el ejemplo 3.
En conclusión, los compuestos de la presente invención son inhibidores de pan-integrina que muestran un fuerte antagonismo frente a múltiples receptores de integrina que se ha demostrado que juegan un papel en la angiogénesis (avp3, avp5 y a5p1), permeabilidad vascular (avp3 y avp5) y adhesión vitreorretiniana (a5p1). Por otro lado, relacionado con los compuestos comparativos, los compuestos de la invención muestran un antagonismo mucho más bajo de la integrina plaquetaria (anbp3), reduciendo por lo tanto el riesgo de interferir con la agregación plaquetaria y aumentar la ventana terapéutica. Además, se ha demostrado que los compuestos de la invención inhiben la migración celular y reducen la aparición de vasos. La invención proporciona nuevos antagonistas de integrina que son útiles para, entre otros, el tratamiento de las enfermedades neovasculares.
Claims (14)
1. Un compuesto según la fórmula (I), o una sal farmacéuticamente aceptable, solvato farmacéuticamente aceptable, isómero o una mezcla de los mismos,

en donde
R1 y R2 se seleccionan independientemente del grupo que consiste en hidrógeno, metilo y etilo; y Alk es un alquileno C1-4.
2. El compuesto según la reivindicación 1, en donde tanto R1 como R2 son metilos.
3. El compuesto según una cualquiera de las reivindicaciones 1 o 2, en donde Alk es un alquileno C1 o C2.
4. El compuesto según una cualquiera de las reivindicaciones anteriores, en donde el compuesto se selecciona del grupo que consiste en:
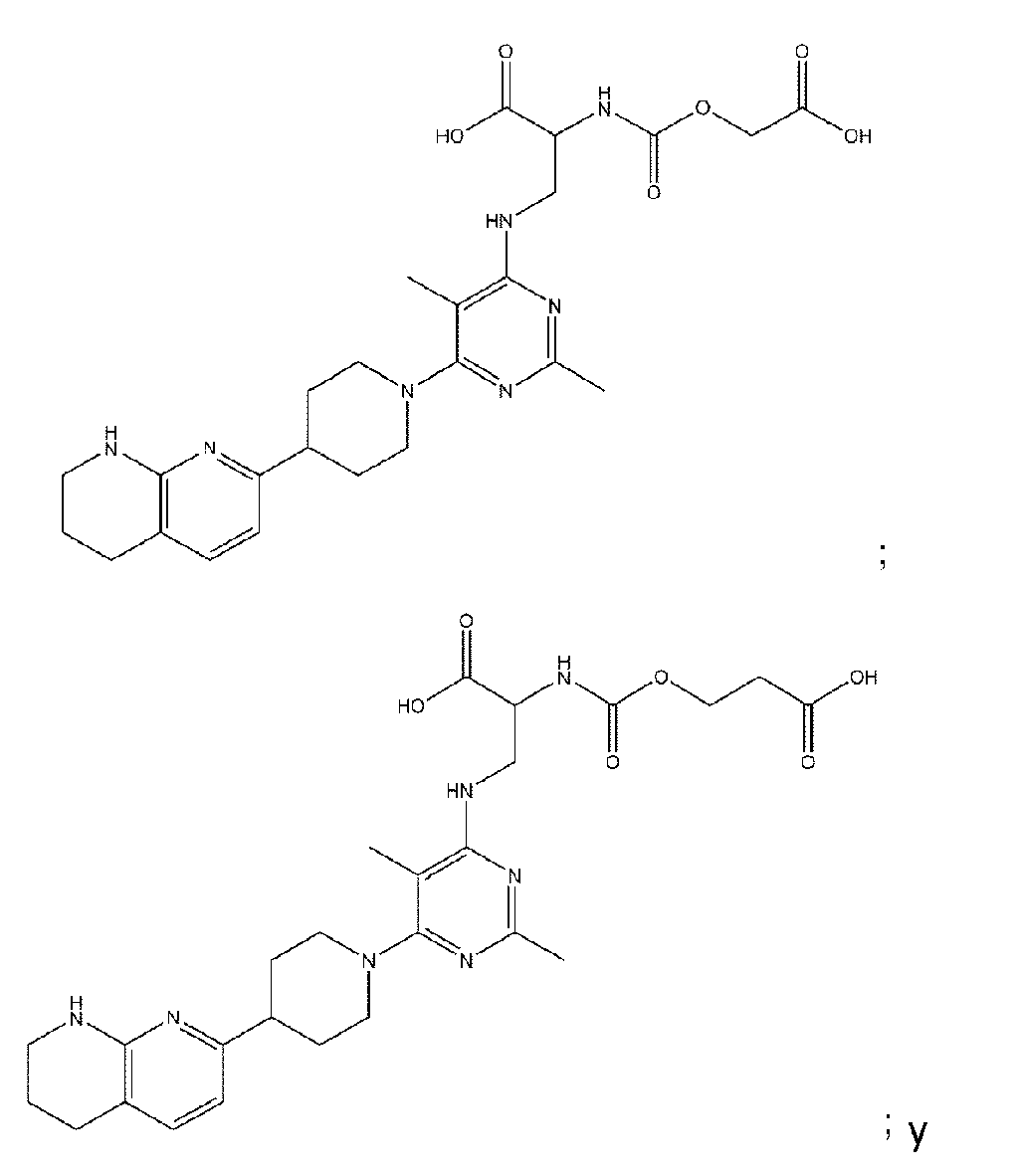

5. Una composición farmacéutica que comprende el compuesto según una cualquiera de las reivindicaciones anteriores, y uno o más de vehículos farmacéuticamente aceptables.
6. El compuesto según una cualquiera de las reivindicaciones 1 a 4, para su uso como medicamento.
7. El compuesto para su uso según la reivindicación 6, para su uso en la inhibición de la neovascularización.
8. El compuesto para su uso según la reivindicación 6, para su uso en el tratamiento y/o prevención del cáncer.
9. El compuesto para su uso según la reivindicación 6, para su uso en el tratamiento y/o prevención de un trastorno oftálmico.
10. El compuesto para su uso según la reivindicación 9, en donde dicho trastorno oftálmico se selecciona del grupo que consiste en degeneración macular húmeda relacionada con la edad, desprendimiento de retina, uveítis posterior, neovascularización de la córnea, neovascularización del iris, edema macular diabético y retinopatía diabética.
11. El compuesto para su uso según la reivindicación 9 o 10, en donde el tratamiento y/o prevención comprende la administración del compuesto mediante una inyección intravítrea.
12. El compuesto para su uso según una cualquiera de las reivindicaciones 7 a 10, en donde la inhibición, tratamiento o prevención comprende además la administración de un compuesto que se une a e inhibe la actividad del factor de crecimiento endotelial vascular (VEGF) o un receptor de VEGF.
13. Una combinación que comprende el compuesto según una cualquiera de las reivindicaciones 1 a 4; y un compuesto que se une a e inhibe la actividad del factor de crecimiento endotelial vascular (VEGF) o un receptor de VEGF.
14. La combinación de la reivindicación 13, para su uso como se define en una cualquiera de las reivindicaciones 6 a 11.
Applications Claiming Priority (2)
| Application Number | Priority Date | Filing Date | Title |
|---|---|---|---|
| EP18189615 | 2018-08-17 | ||
| EP18194258 | 2018-09-13 |
Publications (1)
| Publication Number | Publication Date |
|---|---|
| ES2836733T3 true ES2836733T3 (es) | 2021-06-28 |
Family
ID=67659335
Family Applications (1)
| Application Number | Title | Priority Date | Filing Date |
|---|---|---|---|
| ES19192402T Active ES2836733T3 (es) | 2018-08-17 | 2019-08-19 | Antagonistas de integrina |
Country Status (24)
| Country | Link |
|---|---|
| US (2) | US10703752B2 (es) |
| EP (2) | EP3837257A1 (es) |
| JP (1) | JP7083073B2 (es) |
| KR (1) | KR20210057735A (es) |
| CN (1) | CN112805284A (es) |
| AU (1) | AU2019326932A1 (es) |
| BR (1) | BR112021002839A2 (es) |
| CA (1) | CA3109326A1 (es) |
| CO (1) | CO2021002395A2 (es) |
| CY (1) | CY1123871T1 (es) |
| DK (1) | DK3613739T3 (es) |
| ES (1) | ES2836733T3 (es) |
| HU (1) | HUE051802T2 (es) |
| IL (1) | IL280857A (es) |
| LT (1) | LT3613739T (es) |
| MA (1) | MA53218A (es) |
| MX (1) | MX2021001786A (es) |
| PL (1) | PL3613739T3 (es) |
| RS (1) | RS61182B1 (es) |
| SG (1) | SG11202101424XA (es) |
| SI (1) | SI3613739T1 (es) |
| SM (1) | SMT202000623T1 (es) |
| WO (1) | WO2020043533A1 (es) |
| ZA (1) | ZA202101406B (es) |
Families Citing this family (7)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FI3873884T3 (fi) | 2018-10-30 | 2025-02-24 | Gilead Sciences Inc | 3-(kinolin-8-yyli)-1,4-dihydropyrido[3,4-d]pyrimidiini-2,4-dionijohdannaisia alfa-4-beta-7-integriinin estäjinä tulehdussairauksien hoidossa |
| JP7189368B2 (ja) | 2018-10-30 | 2022-12-13 | ギリアード サイエンシーズ, インコーポレイテッド | アルファ4ベータ7インテグリンの阻害のための化合物 |
| ES3013256T3 (en) | 2018-10-30 | 2025-04-11 | Gilead Sciences Inc | Imidazo[1,2-a]pyridine derivatives as alpha4beta7 integrin inhibitors for the treatment of inflammatory diseases |
| EP3873605B1 (en) | 2018-10-30 | 2024-10-23 | Gilead Sciences, Inc. | Compounds for inhibition of alpha4beta7 integrin |
| WO2021030438A1 (en) | 2019-08-14 | 2021-02-18 | Gilead Sciences, Inc. | Compounds for inhibition of alpha 4 beta 7 integrin |
| WO2023001288A1 (zh) * | 2021-07-23 | 2023-01-26 | 百奥泰生物制药股份有限公司 | 整合素GPIIb/IIIa拮抗剂及其和抗VEGF抗体联合用药的应用 |
| CN118356501A (zh) * | 2023-01-19 | 2024-07-19 | 百奥泰生物制药股份有限公司 | 一种复方制剂及其应用 |
Family Cites Families (5)
| Publication number | Priority date | Publication date | Assignee | Title |
|---|---|---|---|---|
| FR2847254B1 (fr) | 2002-11-19 | 2005-01-28 | Aventis Pharma Sa | Nouveaux derives antagonistes du recepteur de la vitronectine, leur procede de preparation, leur application comme medicaments et les compositions pharmaceutiques les refermant |
| FR2870541B1 (fr) * | 2004-05-18 | 2006-07-14 | Proskelia Sas | Derives de pyrimidines antigonistes du recepteur de la vitronectine |
| US20110189174A1 (en) | 2010-02-01 | 2011-08-04 | Afshin Shafiee | Compositions and methods for treating, reducing, ameliorating, alleviating, or inhibiting progression of, pathogenic ocular neovascularization |
| US20110237605A1 (en) | 2010-03-26 | 2011-09-29 | Eric Phillips | Molecular Crystal of (4-(1,8-Naphthyridin-2-YL)Piperidin-1-YL)Pyrimidine Derivative |
| WO2015175954A1 (en) * | 2014-05-16 | 2015-11-19 | Scifluor Life Sciences, Inc. | Alpha v integrin antagonist compositions |
-
2019
- 2019-08-19 ES ES19192402T patent/ES2836733T3/es active Active
- 2019-08-19 EP EP19755380.3A patent/EP3837257A1/en not_active Withdrawn
- 2019-08-19 BR BR112021002839-0A patent/BR112021002839A2/pt not_active IP Right Cessation
- 2019-08-19 SM SM20200623T patent/SMT202000623T1/it unknown
- 2019-08-19 AU AU2019326932A patent/AU2019326932A1/en not_active Abandoned
- 2019-08-19 PL PL19192402.6T patent/PL3613739T3/pl unknown
- 2019-08-19 US US16/544,259 patent/US10703752B2/en not_active Expired - Fee Related
- 2019-08-19 DK DK19192402.6T patent/DK3613739T3/da active
- 2019-08-19 LT LTEP19192402.6T patent/LT3613739T/lt unknown
- 2019-08-19 MX MX2021001786A patent/MX2021001786A/es unknown
- 2019-08-19 RS RS20201460A patent/RS61182B1/sr unknown
- 2019-08-19 KR KR1020217007569A patent/KR20210057735A/ko not_active Ceased
- 2019-08-19 WO PCT/EP2019/072185 patent/WO2020043533A1/en not_active Ceased
- 2019-08-19 SG SG11202101424XA patent/SG11202101424XA/en unknown
- 2019-08-19 CN CN201980054419.6A patent/CN112805284A/zh active Pending
- 2019-08-19 JP JP2021532523A patent/JP7083073B2/ja not_active Expired - Fee Related
- 2019-08-19 US US17/268,406 patent/US20210198258A1/en not_active Abandoned
- 2019-08-19 SI SI201930011T patent/SI3613739T1/sl unknown
- 2019-08-19 CA CA3109326A patent/CA3109326A1/en active Pending
- 2019-08-19 HU HUE19192402A patent/HUE051802T2/hu unknown
- 2019-08-19 EP EP19192402.6A patent/EP3613739B1/en active Active
- 2019-08-19 MA MA053218A patent/MA53218A/fr unknown
-
2020
- 2020-11-25 CY CY20201101113T patent/CY1123871T1/el unknown
-
2021
- 2021-02-14 IL IL280857A patent/IL280857A/en unknown
- 2021-02-24 CO CONC2021/0002395A patent/CO2021002395A2/es unknown
- 2021-03-01 ZA ZA2021/01406A patent/ZA202101406B/en unknown
Also Published As
| Publication number | Publication date |
|---|---|
| PL3613739T3 (pl) | 2021-03-22 |
| SG11202101424XA (en) | 2021-03-30 |
| JP7083073B2 (ja) | 2022-06-09 |
| CN112805284A (zh) | 2021-05-14 |
| JP2021534252A (ja) | 2021-12-09 |
| CO2021002395A2 (es) | 2021-07-30 |
| SI3613739T1 (sl) | 2021-02-26 |
| BR112021002839A2 (pt) | 2021-05-11 |
| EP3613739A1 (en) | 2020-02-26 |
| HUE051802T2 (hu) | 2021-03-29 |
| MA53218A (fr) | 2022-04-20 |
| MX2021001786A (es) | 2021-07-02 |
| EP3837257A1 (en) | 2021-06-23 |
| CA3109326A1 (en) | 2020-03-05 |
| WO2020043533A1 (en) | 2020-03-05 |
| RS61182B1 (sr) | 2021-01-29 |
| EP3613739B1 (en) | 2020-11-04 |
| US20200055849A1 (en) | 2020-02-20 |
| ZA202101406B (en) | 2022-07-27 |
| US20210198258A1 (en) | 2021-07-01 |
| CY1123871T1 (el) | 2022-03-24 |
| SMT202000623T1 (it) | 2021-01-05 |
| DK3613739T3 (en) | 2020-12-07 |
| LT3613739T (lt) | 2021-02-25 |
| AU2019326932A1 (en) | 2021-03-18 |
| US10703752B2 (en) | 2020-07-07 |
| IL280857A (en) | 2021-04-29 |
| KR20210057735A (ko) | 2021-05-21 |
Similar Documents
| Publication | Publication Date | Title |
|---|---|---|
| ES2836733T3 (es) | Antagonistas de integrina | |
| KR20130093093A (ko) | 이관능성 rho 키나아제 저해제 화합물,조성물 및 용도 | |
| JP6868014B2 (ja) | 翼状片を治療するための組成物及び方法 | |
| ES2763556T3 (es) | Antagonistas fluorados de integrina | |
| US10485786B2 (en) | Pharmaceutical composition for preventing or treating macular degeneration | |
| JP2007527417A (ja) | 白内障、黄斑変性および他の眼疾患の改善 | |
| WO2023069255A1 (en) | Methods and compositions comprising peptidomimitics for treating, preventing, inhibiting, ameliorating or delaying the onset of ophthalmic conditions | |
| JP2021100984A (ja) | 濾過胞を維持するための組成物 | |
| JP2019534269A (ja) | Csf−1rの阻害剤を使用して眼疾患を治療するための方法 | |
| HK40051528A (en) | Integrin antagonists | |
| WO2021165206A1 (en) | Treatment of dry amd with integrin antagonists | |
| EP4687896A1 (en) | Lats kinase inhibitor to treat retinal degeneration | |
| CN118434434A (zh) | 用于治疗、预防、抑制、改善眼科病状或延缓其发作的方法和包括肽模拟物的组合物 | |
| EP4508043A2 (en) | Methods of treating ocular fibrotic pathologies | |
| KR20200074179A (ko) | Ret9 및 vegfr2 억제제 | |
| JP2014533732A (ja) | 網膜神経保護のための7−(1h−イミダゾール−4−イルメチル)−5,6,7,8−テトラヒドロ−キノリンを含む医薬組成物 |